
樹脂改質用添加剤および樹脂組成物
【課題】発泡成形性を改善し、高い発泡倍率の成型体が得られる樹脂改質添加剤の提供。
【解決手段】無水マレイン化等による酸素含有基、エポキシ変性体のアミノ化等による窒素含有基、および不飽和ポリオレフィンとヒドロシランとの反応等によるケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。添加剤を含む樹脂組成物は伸長粘度に優れ、特に発泡用途に好適に用いることが出来る。
【解決手段】無水マレイン化等による酸素含有基、エポキシ変性体のアミノ化等による窒素含有基、および不飽和ポリオレフィンとヒドロシランとの反応等によるケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。添加剤を含む樹脂組成物は伸長粘度に優れ、特に発泡用途に好適に用いることが出来る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂改質用添加剤および添加剤を含む樹脂組成物に関する。本発明は特に、該樹脂改質用添加剤を含む発泡性樹脂組成物に関する。
【背景技術】
【0002】
微細な発泡セルを有するポリオレフィン発泡体として、さまざまなものが知られている。特に発泡ポリプロピレンは、耐熱性、柔軟性、耐衝撃性、軽量性などの特性に優れることから様々な用途に用いられている。しかしながら、ポリプロピレンは発泡成形性が悪く、特に発泡倍率の改善が求められている。
特許文献1〜3では、樹脂の発泡成形性の改善において、伸長粘度の値が発泡成形性の指標にできることが示されている。即ち、樹脂の伸長粘度の増大が発泡成形性を改善させる。しかしながら、ポリプロピレンの発泡成形には特別な組成のポリプロピレンの作製が必要であった(特許文献1、3)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開平11−060835
【特許文献2】特開2008−201866
【特許文献3】特開2009−275119
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、ポリオレフィン特にポリプロピレンの伸長時間および伸長粘度を向上させ、例えば発泡成形性を改善し、高い発泡倍率の成型体が得られる樹脂改質添加剤を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは鋭意検討を行い、末端に特定の極性基が結合したポリオレフィンが前記課題を解決できることを見出して本発明を完成した。
すなわち本発明の要旨は以下にある:
[1] 下記一般式(I)、(II)、(III)または(IV)で表される酸素含有基、窒素含有基およびケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。
【0006】
【化1】

【0007】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表し、Raは水素原子、ハロゲン原子、炭素数1〜10の炭化水素基、窒素含有基、酸素含有基またはケイ素含有基を表しそれぞれ同一でも異なっていてもよく、RbおよびRcはそれぞれ独立に、水素原子、ハロゲン原子、炭化水素基、−OR基、−SR基または−NR2基を表し(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)、環構造を形成してもよく、RdおよびReは一方が水酸基、ポリアルキレングリコール基またはアシルオキシ基を表し、他方は下記一般式(V)、一般式(VI) 、一般式(VII) のいずれかで示される基、シアノ基、カルボキシル基、エステル基またはアミド基を表す。nは平均官能基数を表し、0.1〜1.0である。)
【0008】
【化2】
【0009】
( 式中、Xは酸素原子または硫黄原子を表し、Rf は水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、下記一般式(VIII)
【0010】
【化3】
【0011】
( 式中、R1aは m+1 価の炭化水素基を表し、G は同一または相異なり、-OR1b 、-NR1 c R1d (R1b 、R1 c、R1dはポリアルキレングリコール基を表す) で表される基を表し、m は1〜 5 0 の整数を表す)で表される基を表す。)
【0012】
【化4】
【0013】
( 式中、Rg,Rh は同一または相異なり、水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、上記一般式(VII)で表される基を表す。)
【0014】
【化5】
【0015】
( 式中、Ri、Rj、Rkは同一または相異なり、水素原子、炭化水素基、アシル基、シアノ基、カルボキシル基、エステル基、アミド基を表す。)
【0016】
[2] 前記極性基含有ポリオレフィン重合体を含有する樹脂組成物の伸長粘度最大値が、180℃恒温下、毎秒伸長速度0.01の測定で100kPa・s以上を示すことを特徴とする[1]に記載の樹脂改質用添加剤。
[3] 前記極性基含有ポリオレフィン重合体中の重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることを特徴とする[1]〜2に記載の樹脂改質用添加剤。
[4] 前記樹脂改質用途が発泡性樹脂用であることを特徴とする[2]〜[3]に記載の樹脂改質用添加剤。
[5][4]に記載の発泡性樹脂用添加剤0.1〜15重量%と、熱可塑性樹脂70〜99.8重量%と、発泡剤0.1〜15重量%とを含む発泡性樹脂組成物。
[6] 前記熱可塑性樹脂が、ポリエチレン、ポリプロピレン、ポリスチレン、ポリウレタンおよびポリカーボネートからなる群から選ばれる1種以上の樹脂であることを特徴とする[5]に記載の発泡性樹脂組成物。
[7][4]〜[6]に記載の発泡性樹脂組成物から得られる発泡成形体。
【発明を実施するための形態】
【0017】
(樹脂改質用添加剤)
本発明の樹脂改質用添加剤は、前記した一般式(I)、(II)、(III)または(IV)で表される。
一般式(I)、(II)、(III)または(IV)で表される化合物は、まず末端不飽和ポリオレフィンを得た後に、末端不飽和基を以下に示す方法でそれぞれ官能基に変換することによって調製出来る。
【0018】
一般式(I)で表される化合物の中でも、Raとして酸素含有基が好ましく、さらにアルコキシ基が好ましく、特にエトキシ基・メトキシ基が好ましい。
一般式(III)で表される化合物の中でも、RbおよびRcとして酸素含有基が好ましく、さらに水酸基またはアルコキシ基が好ましく、特に水酸基が好ましい。
一般式(IV)で表される化合物の中でも、RdおよびReとして式(V)ないし(VI)で表される構造が好ましい。
式(V)のXとして酸素原子が好ましく、Rfとしては水素原子またはポリアルキレングリコールが好ましく、さらに水素原子が好ましい。式(VI)のRgおよびRhとしてポリアルキレングリコールもしくはアルカノール基が好ましく、特に−CH2CH2OHが好ましい。
【0019】
(末端不飽和ポリオレフィン(X))
末端不飽和ポリオレフィンの製造方法は、特開2003−73412に公開されている。
R1−CH=CH2 (X)
上記一般式(X)中、R1は、エチレン単独重合鎖、またはエチレン、プロピレン、ブテン、ビニルノルボルネン、ジエンを含む炭素数2〜20のオレフィンから選択される少なくとも2種以上の共重合鎖である。
【0020】
炭素数2〜20のオレフィンとしては、具体的には、エチレン、プロピレン、1−ブテン、1−ペンテン、3−メチル−1−ブテン、1−ヘキセン、4−メチル−1−ペンテン、3−メチル−1−ペンテン、3,4−ジメチル−1−ペンテン、4−メチル−1−ヘキセン、3−エチル−1−ペンテン、3−エチル−4−メチル−1−ペンテン、3,4−ジメチル−1−ヘキセン、4−メチル−1−ヘプテン、3,4−ジメチル−1−ヘプテン、1−オクテン、1−デセン、1−ドデセン、1−テトラデセン、1−ヘキサデセン、1−オクタデセン、1−エイコセン、ビニルシクロヘキサンなどのα−オレフィン; シス−2−ブテン、トランス−2−ブテン、などの内部二重結合を含むオレフィン; イソブテン、2−メチル−1−ペンテン、2,4−ジメチル−1−ペンテン、2,4−ジメチル−1−ヘキセン、2,4,4−トリメチル−1−ペンテン、2,4−ジメチル−1−ヘプテン、2−メチル−1−ブテン、2−メチル−1−ヘキセン、2−メチル−1−ヘプテン、2−メチル−1−オクテン、2,3−ジメチル−1−ブテン、2,3−ジメチル−1−ペンテン、2,3−ジメチル−1−ヘキセン、2,3−ジメチル−1−オクテン、2,3,3−トリメチル−1−ブテン、2,3,3−トリメチル−1−ペンテン、2,3,3−トリメチル−1−ヘキセン、2,3,3−トリメチル−1−オクテン、2,3,4−トリメチル−1−ペンテン、2,3,4−トリメチル−1−ヘキセン、2,3,4−トリメチル−1−オクテン、2,4,4−トリメチル−1−ヘキセン、2,4,4−トリメチル−1−オクテン、2−メチル−3−シクロヘキシル−1−プロピレン、ビニリデンシクロペンタン、ビニリデンシクロヘキサン、ビニリデンシクロオクタン、2−メチルビニリデンシクロペンタン、3−メチルビニリデンシクロペンタン、4−メチルビニリデンシクロペンタンなどのビニリデン化合物; スチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、2,4−ジメチルスチレン、o−エチルスチレン、m−エチルスチレン、p−エチルスチレンなどのアリールビニル化合物; α−メチルスチレン、α−エチルスチレン、2−メチル−3−フェニルプロピレンなどのアリールビニリデン化合物; メタクリル酸メチル、メタクリル酸エチル、メタクリル酸−n−プロピル、メタクリル酸イソプロピル、メタクリル酸−n−ブチル、メタクリル酸イソブチル、メタクリル酸−tert−ブチル、2−シアノプロピレン、2−アミノプロピレン、2−ヒドロキシメチルプロピレン、2−フルオロプロピレン、2−クロロプロピレンなどの官能基置換ビニリデン化合物; シクロブテン、シクロペンテン、1−メチル−1−シクロペンテン、3−メチル−1−シクロペンテン、2−メチル−1−シクロペンテン、シクロヘキセン、1−メチル−1−シクロヘキセン、3−メチル−1−シクロヘキセン、2−メチル−1−シクロヘキセン、シクロヘプテン、シクロオクテン、ノルボルネン、5−メチル−2−ノルボルネン、テトラシクロドデセン、5,6−ジヒドロジシクロペンタジエン、3a,4,5,6,7,7a−ヘキサヒドロ−1Hインデン、トリシクロ[6.2.1.02,7]ウンデカ−4−エン、シクロペンタジエン、ジシクロペンタジエンなどの内部二重結合を含む脂肪族環状オレフィン; シクロペンタ−2−エニルベンゼン、シクロペンタ−3−エニルベンゼン、シクロヘキサ−2−エニルベンゼン、シクロヘキサ−3−エニルベンゼン、インデン、1,2−ジヒドロナフタレン、1,4−ジヒドロナフタレン、1,4−メチノ1,4,4a,9aテトラヒドロフルオレンなどの芳香環を含有する環状オレフィン; ブタジエン、イソプレン、4−メチル−1,3−ペンタジエン、4−メチル−1,4−ペンタジエン、1,3−ペンタジエン、1,4−ペンタジエン、1,5−ヘキサジエン、1,4−ヘキサジエン、1,3−ヘキサジエン、1,3−オクタジエン、1,4−オクタジエン、1,5−オクタジエン、1,6−オクタジエン、1,7−オクタジエン、エチリデンノルボルネン、ビニルノルボルネン、ジシクロペンタジエン、7−メチル−1,6−オクタジエン、4−エチリデン−8−メチル−1,7−ノナジエン、5,9−ジメチル−1,4,8−デカトリエンなどの二個以上の二重結合を有する環状、又は鎖状のジエン、又はポリエンなどが挙げられる。
【0021】
また、オレフィンは、酸素、窒素、硫黄等の原子を含んだ官能基を有していてもよい。例えばアクリル酸、フマル酸、イタコン酸、ビシクロ[2.2.1]ヘプタ−5−エン−2,3−ジカルボン酸などの不飽和カルボン酸およびこれらのナトリウム塩、カリウム塩、リチウム塩、亜鉛塩、マグネシウム塩、カルシウム塩などの不飽和カルボン酸金属塩; 無水マレイン酸、無水イタコン酸、ビシクロ[2.2.1]ヘプタ−5−エン−2,3−ジカルボン酸無水物などの不飽和カルボン酸無水物; アクリル酸メチル、アクリル酸エチル、アクリル酸−n−プロピル、アクリル酸イソプロピル、アクリル酸−n−ブチル、アクリル酸イソブチル、アクリル酸−tert−ブチル、アクリル酸−2−エチルヘキシル、などの不飽和カルボン酸エステル; 酢酸ビニル、プロピオン酸ビニル、カプロン酸ビニル、カプリン酸ビニル、ラウリン酸ビニル、ステアリン酸ビニル、トリフルオロ酢酸ビニルなどのビニルエステル類; アクリル酸グリシジル、メタクリル酸グリシジル、イタコン酸モノグリシジルエステルなどの不飽和グリシジルエステル; 塩化ビニル、フッ化ビニル、フッ化アリルなどのハロゲン化オレフィン; アクリロニトリル、2−シアノ−ビシクロ[2.2.1]ヘプタ−5−エンなどの不飽和シアノ化合物; メチルビニルエーテル、エチルビニルエーテルなどの不飽和エーテル化合物; アクリルアミド、メタクリルアミド、N,N−ジメチルアクリルアミド等の不飽和アミド; メトキシスチレン、エトキシスチレン、ビニル安息香酸、ビニル安息香酸メチル、ビニルベンジルアセテート、ヒドロキシスチレン、o−クロロスチレン、p−クロロスチレン、ジビニルベンゼンなどの官能基含有スチレン誘導体; N−ビニルピロリドンなどが挙げられる。
【0022】
上記のうち、R1としては、エチレン単独重合鎖、プロピレン単独重合鎖またはエチレンとプロピレンとの共重合鎖が好ましい。
【0023】
上記一般式(X)中、R1の数平均分子量は、100〜50,000であることが好ましく、100〜10,000であることがより好ましい。これより基が短くなると、樹脂中やオイル中における分散性が悪くなるし、長くなると添加剤としての性能が出難くなるので好ましくない。
【0024】
(I)シリル化ポリオレフィンの製造方法
前記一般式(I)で表されるシリル化ポリオレフィンは、下記の[工程1]および[工程2]を順次実施することにより、製造できる。
【0025】
[工程1]白金触媒組成物(C)を得る工程
[工程1]では、下記一般式(IX)で表されるヒドロシランと二塩化白金とを混合攪拌し、得られた懸濁溶液を濾過して濾液として白金触媒組成物(C)を得る。
Ra3−SiH (IX)
【0026】
上記一般式(IX)中、Raは、水素原子、ハロゲン原子、炭化水素基、酸素含有基、またはケイ素含有基を表し、それぞれ同一であっても異なっていてもよい。
ハロゲン原子としては、フッ素、塩素、臭素、ヨウ素が挙げられる。
【0027】
炭化水素基としては、アルキル基、アルケニル基、アリール基が挙げられる。
アルキル基としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基、ヘキシル基、2−エチルヘキシル基、オクチル基、デシル基、オクタデシル基等の直鎖状または分岐状アルキル基; シクロペンチル基、シクロヘキシル基、ノルボルニル基等のシクロアルキル基; ベンジル基、フェニルエチル基、フェニルプロピル基等のアリールアルキル基が挙げられる。
アルケニル基としては、ビニル基、プロペニル基、シクロヘキセニル基が挙げられる。
アリール基としては、フェニル基、トリル基、ジメチルフェニル基、トリメチルフェニル基、エチルフェニル基、プロピルフェニル基、ビフェニル基、ナフチル基、メチルナフチル基、アントリル基、フェナントリル基等が挙げられる。
【0028】
酸素含有基としては、アルコキシ基、アリールオキシ基が挙げられる。
アルコキシ基としては、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、ヘキシルオキシ基、オクチルオキシ基、ベンジルオキシ基、2−フェニルエトキシ基等が挙げられる。
アリールオキシ基としては、フェノキシ基、トリルオキシ基、ビフェニルオキシ基、ナフチルオキシ基等が挙げられる。
【0029】
ケイ素含有基としては、アルキルシリル基、アルケニルシリル基、アリールシリル基、アルキルシロキシ基、アルケニルシロキシ基、アリールシロキシ基、アルコキシシリル基、アリールオキシシリル基、アルコキシシロキシ基、アリールオキシシロキシ基等が挙げられる。
【0030】
また上記の炭化水素基、酸素含有基、およびケイ素含有基は、1つ以上のヘテロ原子を含んでいてもよい。具体的には、これらの基の少なくとも一つの水素が、ハロゲン原子、酸素、窒素、ケイ素、リン、イオウを含む基で置換された基が含まれる。
【0031】
一般式(IX)のRaとして上記に挙げた原子または基のうち、水素原子、アルキル基、アルコキシ基、アリール基、およびケイ素含有基からなる群から選ばれる1種以上の原子または基であることが好ましく、なかでも、水素原子; メチル基、エチル基、プロピル基、ブチル基等の炭素原子数1〜4のアルキル基; メトキシ基、エトキシ基、プロポキシ基、ブトキシ基等の炭素原子数1〜4のアルコキシ基; フェニル基、ナフチル基等のアリール基; で表される基からなる群から選ばれる1種以上の原子または基であることがより好ましい。この中でも、メトキシ基およびエトキシ基が特に好ましい。
【0032】
[工程1]で使用する二塩化白金は、特に制限されないが粒径は1000μm以下が好ましく、更には500μm以下が好ましい。粒径が大きくなると調製時間が長くなる。
[工程1]におけるヒドロシランと二塩化白金の使用量は、ヒドロシラン量が二塩化白金に対し等量以上であれば特に制限されないが、好ましくは2倍当量以上である。ヒドロシランの量が少ないと、触媒調製上必要な攪拌が困難になる。
【0033】
[工程1]におけるヒドロシランと二塩化白金との混合攪拌は、これが可能であれば手段は問わないが、窒素気流下、攪拌機を備えた反応容器中に二塩化白金を適当量仕込み、予め設定したヒドロシランを添加して攪拌を行う。少量の場合はサンプル管にスターラーチップを入れ、同様に仕込んで攪拌しても良い。
【0034】
ヒドロシランと二塩化白金との混合攪拌時間は、通常60時間以上であり、70時間以上が好ましく、80時間以上が更に好ましい。反応時間が短いと、[工程2]で得られるシリル化ポリオレフィン(I)中の不純物である異性体のビニレン誘導体の生成割合が増大するため好ましくない。混合攪拌時間の上限は特に無いが、経済的な観点から概ね1ヶ月以内である。
【0035】
ヒドロシランと二塩化白金との混合攪拌の温度は、ヒドロシランの沸点以下であれば特に制限は無いが、通常0〜50℃の範囲、好ましくは10〜30℃の範囲である。また圧力は、通常は常圧で行うことができるが、必要に応じて加圧下または減圧下で行うこともできる。
【0036】
[工程1]においては反応で得られた懸濁溶液を濾過して固形分を除去し、濾液として白金触媒組成物(C)を得る。濾過で使用するフィルターは10μmより小さな目のフィルターを使用することが好ましく、1μm以下の目のフィルターを使用することが更に好ましい。これより大きな目のフィルターを使用すると、未反応の二塩化白金の固形分が触媒中に混入し、触媒が不均一化するため、合成目的物の不純物であるビニレン誘導体の生成量が増大する原因となる。また濾過の際、上記の溶媒を使用して固形分を洗浄することもできる。
濾過で除去される固形分、すなわち未反応の二塩化白金の量は、使用した二塩化白金の量に対して50%以下、好ましくは10%以下である。二塩化白金の反応率は、調製時間によって調節することができる。
【0037】
このようにして調製した白金触媒組成物(C)には、ナノコロイド状になった白金化合物、ヒドロシランが含まれる。この白金触媒組成物(C)は、そのままで次の[工程2]に用いることができるが、前記一般式(IX)のヒドロシランを添加して希釈し、触媒濃度を調整することもできる。
【0038】
[工程2]末端不飽和ポリオレフィンとヒドロシランとを反応させる工程
[工程2]では、前記[工程1]で得られた白金触媒組成物(C)の存在下、一般式(X)で表される末端不飽和ポリオレフィンと一般式(IX)で表されるヒドロシランとを反応させ、一般式(I)で表されるシリル化ポリオレフィンを得る。
【0039】
一般式(X)で表される末端不飽和ポリオレフィンと反応させるヒドロシランは、前記一般式(IX)で表される化合物である。
前記一般式(IX)中、Raは、[工程1]で示したものと同様の原子または基である。[工程2]で用いる一般式(IX)で表されるヒドロシランは、前記[工程1]で用いたヒドロシランと異なるものを用いることもできるが、好ましくは[工程1]で用いたものと同一のものを用いる。
【0040】
一般式(X)で表される末端不飽和ポリオレフィンと一般式(IX)で表されるヒドロシランとを反応させる際の量比は、目的によって異なるが、0.01〜10当量倍の範囲であり、好ましくは0.1〜2当量倍の範囲である。ここでヒドロシランの量とは、前記[工程1]で用い、白金触媒組成物(C)中に含まれる部分と、[工程2]で新たに追加する部分との合算量である。前記[工程1]において必要なヒドロシラン量の全量を用いた場合には、[工程2]ではヒドロシランを追加することなく実施することもできる。
【0041】
上記末端不飽和ポリオレフィンとヒドロシランとの反応は、前記[工程1]で調製した白金触媒組成物(C)の存在下で行う。白金触媒組成物(C)と末端不飽和ポリオレフィンとの量比は、白金換算で10-10〜10-1当量倍の範囲であり、好ましくは10-7〜10-3当量倍の範囲である。
【0042】
上記末端不飽和ポリオレフィンとヒドロシランとの反応における反応方法としては、最終的に反応すればよく、その方法は限定されるものではないが、例えば以下のように行う。反応容器中に上記末端不飽和ポリオレフィンを装入し、窒素雰囲気下、ヒドロシランと白金触媒組成物を装入する。予め内温を上記末端不飽和ポリオレフィンの融点以上に昇温しておいた油浴中に、上記反応器をセットし攪拌する。反応後油浴を除いて室温に冷却し、得られた反応混合物をメタノールまたはアセトンなどの貧溶媒中に取り出し2時間攪拌する。その後、得られた固体をろ取し上記貧溶媒で洗浄し、60℃、2hPa以下の減圧下で乾燥させ、目的物を得ることが出来る。
【0043】
[工程2]における末端不飽和ポリオレフィンとヒドロシランとの反応は、反応温度を100〜200℃の範囲とすることが好ましく、反応させる末端不飽和ポリオレフィンの融点より高い温度で行うことがより好ましい。反応温度が100℃より低いと、反応効率が低下することがあるので好ましくない。また圧力は、通常は常圧で行うことができるが、必要に応じて加圧下または減圧下で行うこともできる。
【0044】
[工程2]においては、必要に応じて溶媒を使用することもできる。使用する溶媒は、原料のヒドロシランおよび末端不飽和ポリオレフィンに対して不活性なものが使用できる。常圧下で反応させる場合、反応させる末端不飽和ポリオレフィンの融点以上の沸点を有するものを使用するのが好ましい。使用できる溶媒の具体例は、例えばn -ヘキサン等の脂肪族炭化水素類、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、酢酸エチル等のエステル類、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン、メチルプロピルケトン等のケトン類、テトラヒドロフラン、1,4−ジオキサン等のエーテル類、クロロホルム、ジクロルエタン、トリクロルエタン、パークロルエタン等のハロゲン化炭化水素などが挙げられる。これらのうち、特にトルエン、キシレン等の芳香族炭化水素が好ましい。
【0045】
溶媒を使用する場合は溶媒の使用量は原料の溶解性に作用するが、原料に対し100質量倍以下が好ましく、より好ましくは20質量倍以下である。本発明では、無溶媒で実施することが最も好ましい。
【0046】
以上のように、白金触媒組成物(C)の存在下、末端不飽和ポリオレフィンとヒドロシランとを反応させることにより、一般式(I)で表されるシリル化ポリオレフィンを含む反応混合物が得られる。
上記反応後のシリル化ポリオレフィン(I)を含む反応混合物には、シリル化ポリオレフィン(I)の他に、未反応の末端不飽和ポリオレフィン、副生物であるビニレン誘導体が含まれている。および微量の未反応シランが含まれている。
【0047】
本発明の方法においては、[工程1]で得られた非常に高活性で高選択性の白金触媒組成物(C)を用いるため、[工程2]の末端不飽和ポリオレフィンとヒドロシランとの反応が効率よく進行する。このため、末端不飽和ポリオレフィンの二重結合の反応率は、通常90%以上、好ましくは95%以上であり、副生物であるビニレン誘導体の生成量は、シリル化ポリオレフィン(I)に対して、通常10重量%以下、好ましくは5重量%以下である。
【0048】
シリル化ポリオレフィン(I)は、上記反応混合物から、貧溶媒への再沈、またはスラッジングにより取り出すことができる。貧溶媒はシリル化ポリオレフィン(I)の溶解度が小さいものであれば選択することができ、好ましくは上記不純物が除けるものが良い。
【0049】
(II)無水マレイン化ポリオレフィンの製造方法
本発明の一般式(II)で示される重合体は、一般式(X)で表される末端不飽和ポリオレフィン、無水マレイン酸と(b)重合禁止剤とを、溶媒系または無溶媒系で反応させて製造できる。
【0050】
(b)重合禁止剤とは、特に限定されるものではないが、例えば、フェノール系重合禁止剤、ニトロソアミン系重合禁止剤、フェノチアジン等が挙げられる。
【0051】
フェノール系重合禁止剤としては、例えば、2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン、4−メトキシフェノール、2,5−ジ−t−ブチルハイドロキノン、メチルハイドロキノン、p−ベンゾキノン、t−ブチル−p−ベンゾキノン、2,5−ジフェニル−p−ベンゾキノン等が挙げられる。前記ニトロソ系重合禁止剤としては、例えば、N−ニトロソフェニルヒドロキシルアミン、トリス(N−ニトロソ−N−フェニルヒドロキシルアミナト)アルミニウム等が挙げられる。
【0052】
これらの中で、好ましくはフェノール系重合禁止剤であり、より好ましくは2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン、4−メトキシフェノール、2,5−ジ−t−ブチルハイドロキノン、メチルハイドロキノン及びp−ベンゾキノンであり、さらに好ましくは、2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン及び4−メトキシフェノールである。
【0053】
また、一般式(II)で示される重合体の製造方法は、工業的にも使用可能であり、且つ、例えば一般式(X)で表される末端不飽和ポリオレフィンは、無水マレイン酸とほぼ定量的に反応する。本発明において、末端不飽和ポリオレフィンの変性率は80%以上である。また、該変性率は85%以上であることが好ましく、95%以上であることがより好適である。
【0054】
(III)マレイン化ポリオレフィンの製造方法
本発明の前記一般式(III)で示される重合体は、前記一般式(X)で示される末端不飽和ポリオレフィンと、重合禁止剤と、下記一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかとを、溶媒系または無溶媒系で反応させて製造できる。
【0055】
【化6】
【0056】
【化7】
【0057】
上記式中、Rb及びRcは、前記一般式(III)におけるものと同様である。
さらに、一般式(XI)及び一般式(XII)で示される化合物は、反応性の二重結合を有する化合物であり、且つ、互いにシス−トランスの関係にある幾何異性体である。一般式(XI)および一般式(XII)におけるRb及びRcが環構造を形成する時、下記一般式(XIII)で示される構造となる。
【0058】
【化8】
【0059】
上記式中、Rsは、酸素原子、−NR基を表す(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)。
【0060】
これら一般式(XI)及び一般式(XII)で示される化合物は、単独で用いても、あるいは2種類以上混合して用いてもよい。これら化合物の使用量は、特に制限されるものではないが、好ましくは用いる末端不飽和ポリオレフィンの質量の0.01倍乃至50倍の範囲、より好ましくは、0.1倍乃至20倍の範囲、さらに好ましくは、0.5倍乃至5倍の範囲である。
【0061】
一般式(III)で示される重合体を製造するに際し、反応は無溶媒でも、あるいは溶媒を用いてもどちらでもよい。用いる溶媒としては本発明を阻害しない限り特に限定されるものではないが、例えば、n−ヘキサン等の脂肪族炭化水素類;シクロヘキサン等の脂環式炭化水素類;トルエン、キシレン、メシチレン等の芳香族炭化水素類;酢酸エチル等のエステル類;アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン等のケトン類;テトラヒドロフラン、1,4−ジオキサン等のエーテル類;クロロホルム、ジクロルエタン、トリクロロエタン、パークロロエタン等のハロゲン化炭化水素類;クロロベンゼン、o−ジクロロベンゼン等のハロゲン化芳香族類;ニトロベンゼン等の置換芳香族類;などが挙げられる。
【0062】
これらの中で、好ましくは、脂肪族炭化水素類、脂環式炭化水素類、芳香族炭化水素類、ハロゲン化炭化水素類、ハロゲン化芳香族類であり、より好ましくは、芳香族炭化水素類、ハロゲン化芳香族類であり、さらに好ましくはメシチレン、o−ジクロロベンゼンが挙げられる。これら溶媒は、単独で用いても、二種類以上混合して用いてもよい。
【0063】
上記溶媒の使用量は、原料の溶解性によるが、好ましくは原料として用いる末端不飽和ポリオレフィンの質量の0.1倍乃至100倍の範囲、より好ましくは0.5倍乃至50倍の範囲、さらに好ましくは1倍乃至10倍の範囲である。
【0064】
反応は、例えば次のようにして行うことができる。反応器に前記一般式(X)で示される末端不飽和ポリオレフィン、重合禁止剤、一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかを仕込み昇温する。原料は一括で仕込んでも、または分割して適宜添加してもよい。この時、反応を促進させるために触媒等の添加物を使用してもよい。反応温度は、本発明を阻害しない限り特に制限されるものではないが、好ましくは室温乃至300℃の範囲、より好ましくは100℃乃至250℃の範囲、より好ましくは150℃乃至230℃の範囲で行われる。使用する化合物、溶媒によっては反応温度が沸点を超える場合があるためオートクレーブ等適切な反応装置を選択する。反応時間は、使用する末端不飽和ポリオレフィン、溶媒、重合禁止剤の量や反応性により変わるが、通常20時間乃至120時間である。
【0065】
反応の進行は、1H−NMRで確認できる。すなわち、測定サンプル管中で重合体を、ロック溶媒と溶媒を兼ねた重水素化−1,1,2,2−テトラクロロエタンに完全に溶解させた後、120℃において測定した。ケミカルシフトは、重水素化−1,1,2,2−テトラクロロエタンのピークを5.91ppmとして、他のピークのケミカルシフト値を決定した。
【0066】
例えば、エチレンのみからなる片末端二重結合含有重合体の場合、飽和末端におけるメチル基の3プロトン分のピークが0.80ppm乃至0.95ppm、末端二重結合の3プロトン分のピークが4.88ppm乃至5.05ppmに2プロトン分のピーク(x)、5.75ppm乃至5.90ppmに1プロトン分のピークとして観測される。反応が進行すると、4.88ppm乃至5.05ppmに観測される2プロトン分のピーク(x)と、5.75ppm乃至5.90ppmに観測される1プロトン分のピークとが消失し、反応進行に伴い二重結合のシフトが起こり、新たに5.20ppm乃至5.40ppmに1プロトン分のピーク(y)、5.50ppm乃至5.70ppmに1プロトン分のピークが観測され、二重結合が一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかで変性されたポリオレフィン重合体が生成する。
【0067】
この時、反応の転化率(U%)(本発明における「変性率」に相当)は、ピーク(x)およびピーク(y)のピーク積分値を各々SBおよびSDとすれば、下記式(α)にて算出される。
式(α):U(%)=SD×100/[(SB/2)+SD]
【0068】
前記製造方法は、工業的にも使用可能であり、且つ、原料の一般式(X)で表される末端不飽和ポリオレフィンは、一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかとほぼ定量的に反応する。本発明において、上記式(α)から求められる前記二重結合の極性基(一般式(XI)及び一般式(XII)で示される構造の基)による変性率は80%以上である。また、該変性率は85%以上であることが好ましく、95%以上であることがより好適である。
【0069】
反応後は、晶析操作、洗浄等の簡単な操作により、過剰の原料、溶媒、重合禁止剤等を除去して、目的とする本発明の第2の重合体(第1の重合体を得るための中間体化合物)を得ることができる。また、上記反応において、原料の末端不飽和ポリオレフィンの製造工程から単離せずに上記反応を実施することもできる。
【0070】
(IV)アミノ化ポリオレフィンの製造方法
本発明の一般式(IV)で示される重合体は、一般式(X)で表される末端不飽和ポリオレフィンをエポキシ化して、すなわち上記ポリオレフィンの末端の二重結合を酸化して、一般式(XIII)で示される末端にエポキシ基を含有する重合体を得る。
【0071】
【化9】
【0072】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表す)
かかるエポキシ化方法は特に限定されるものではないが、以下の方法を例示することができる。
【0073】
(1)過ギ酸、過酢酸、過安息香酸などの過酸による酸化
(2)チタノシリケートおよび過酸化水素による酸化
(3)メチルトリオキソレニウム等のレニウム酸化物触媒と過酸化水素による酸化
(4)マンガンポルフィリンまたは鉄ポルフィリン等のポルフィリン錯体触媒と過酸化水素または次亜塩素酸塩による酸化
(5)マンガンSalen等のSalen錯体と過酸化水素または次亜塩素酸塩による酸化
(6)マンガン−トリアザシクロノナン(TACN)錯体等のTACN錯体と過酸化水素による酸化
(7)タングステン化合物などのVI族遷移金属触媒と相間移動触媒存在下、過酸化水素による酸化
上記(1)〜(7)の方法の中でも、活性面で特に(1)および(7)の方法が好ましい。
【0074】
末端エポキシ基含有重合体の全片末端中のエポキシ含有率は1H-NMRによって決定される。例えば、エチレンのみからなる片末端二重結合含有重合体をエポキシ化して得られた末端エポキシ基含有重合体の場合、飽和末端におけるメチル基の3プロトン分のピーク(j)が0.65〜0.9ppm、エポキシ基付け根の3プロトン分のピーク(k)が1プロトンずつ2.3〜2.4ppm、2.6〜2.7ppm、2.8〜2.9ppmに観測される。エポキシ変性が十分でない場合は、末端二重結合の3プロトン分のピーク(l)が4.70〜5.0ppmに2プロトン、5.5〜5.8ppmに1プロトン観測される。各ピーク(j)、(k)および(l)のピーク面積を各々SC、SDおよびSEとすれば、エポキシ基含有率(Ep(%))は下記式にて算出される。
Ep(%)=SD×200/(SC+SD+SE)
【0075】
上記の方法で得られた末端エポシキシ基含有重合体を開環重合し一般式(IV)の構造単位を有する重合体を製造することができる。触媒、重合条件などについては、公知のアルキレンオキサイドの開環重合方法を利用することができ、例えば、大津隆行著,「改訂高分子合成の化学」,株式会社化学同人,1971年1月,p.172−180には、種々の単量体を重合してポリオールを得る例が開示されている。開環重合に用いられる触媒としては、上記文献に開示されたように、カチオン重合向けにAlCl3、SbCl5、BF3、FeCl3のようなルイス酸、アニオン重合向けにアルカリ金属の水酸化物またはアルコキシド、アミン類、フォスファゼン触媒、配位アニオン重合向けにアルカリ土類金属の酸化物、炭酸塩、アルコキシドあるいは、Al、Zn、Feなどのアルコキシドを用いることができる。ここで、フォスファゼン触媒としては、例えば、特開平10−77289号公報に開示された化合物、具体的には市販のテトラキス[トリス(ジメチルアミノ)フォスフォラニリデンアミノ]フォスフォニウムクロリドのアニオンをアルカリ金属のアルコキシドを用いてアルコキシアニオンとしたものなどが利用できる。
【0076】
上記触媒存在下、開始剤として水、アミン類、ジオール類、ポリオール類等の活性水素化合物を使用して、末端エポキシ基含有重合体のみを開環重合させることにより末端エポキシ基含有重合体のホモ重合体が得られ、末端エポキシ基含有重合体と他のアルキレンオキシドを開環重合させることにより共重合体を得ることができる。
【0077】
反応溶媒としては、末端エポキシ基含有重合体、アルキレンオキサイドに対して不活性なものが使用でき、n−ヘキサン、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、ジオキサン等のエーテル類、ジクロロベンゼン等のハロゲン化炭化水素などが挙げられる。
【0078】
触媒の使用量はホスファゼン触媒以外については原料の末端エポキシ基含有重合体eの1モルに対して、0.05〜5モルが好ましく、より好ましくは0.1〜3モルの範囲である。ホスファゼン触媒の使用量は、重合速度、経済性等の点から、末端エポキシ基含有重合体の1モルに対して1×10−4〜5×10−1モルが好ましく、より好ましくは5×10−4〜1×10−1モルである。
【0079】
反応温度は通常25〜150℃、好ましくは50〜110℃とし、反応時間は使用する触媒の量、反応温度、オレフィン類の反応性等の反応条件により変わるが、通常数分〜50時間である。
【0080】
開始剤として水またはジオール類を用いることにより、両末端に水酸基を有する重合体を得ることができる。また、予めアルキレンオキサイドを重合して得た特定の分子量のポリエーテルポリオールを開始剤として用いると所定の分子量の親水性単位を導入することが可能になり所望の物性を有する両末端に水酸基を有するブロック共重合体の製造が容易となる。
【0081】
ポリエーテルポリオールとしてはポリエチレングルコール、ポリプロピレングリコール、ポリテトラエチレングリコール等を挙げることができ中でもポリエチレングルコール、ポリプロピレングリコールが望ましい。
【0082】
本発明の一般式(IV)の官能基含有率は、全片末端の50%以上が好ましく、より好ましくは70%以上、更に好ましくは80%以上、更により好ましくは90%以上である。一般式(IV)の官能基含有率は1H-NMRによって決定される。例えば、エチレンのみからなる末端エポキシ基含有重合体を官能基化して得られた一般式(IV)化合物の場合、置換基付け根の3プロトン分のピーク(j)が1プロトンずつ3.2〜3.4ppm、3.4〜3.5ppm、3.6〜3.8ppmに観測される。官能基化が十分でない場合は、エポキシ基付け根の3プロトン分のプロトンピーク(k)が1プロトンずつ2.3〜2.4ppm、2.6〜2.7ppm、2.8〜2.9ppmに観測される。各ピーク(j)および(k)のピーク面積を各々SDおよびSEとし、原料の末端エポキシ基含有重合体のエポキシ基含有率をEp(%)とすれば、一般式(IV)の官能基含有率(Fc(%))は下記式にて算出される。
Fc(%)=SD/(SD+SE)×Ep(%)
【0083】
(1)一般式(IV)においてRe、Rdの一方が水酸基である重合体の製造方法を示す:
[(1a)Re、Rdの一方が水酸基で、他方が一般式(V)で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体に、酸または塩基触媒存在下、一般式(XIV)
【0084】
【化10】
【0085】
(式中、X、Rfは一般式(V)と同様の原子または基を表す)で示される化合物(以下、反応剤Aと表記する)を反応させることにより、Re、Rdの一方が水酸基で、他方が一般式(V)で示される基である重合体を得ることができる。
一般式(XIV)としては、例えば、水、メタノール、エタノール、プロパノール、オクタノール、アリルアルコール、シクロヘキサノール、プロペニルアルコール、ヘキセノール、ブロモデカノール、トリフルオロエタノール、ヘキサフルオロ-2-プロパノール、パーフルオロオクタノール、ベンジルアルコール、ジフルオロベンジルアルコール、ペンタフルオロフェニルメタノール、ビス(4-メトキシフェニル)メタノール、フェネチルアルコール、フェニルプロピルアルコール、フェノール、ジクロロフェノール、メトキシフェノール、メトキシカルボニルフェノール、ニトロフェノール、ヘキサフルオロフェノール、メチルフェノール、ジメチルフェノール、ナフチルアルコール等のアルコール類、グリセリン、ブタントリオール、ペンタエリスリトール等の多価アルコール類、チオメタノール、チオエタノール等のチオアルコール類、酢酸、プロピオン酸、マレイン酸、コハク酸、マロン酸、ブタン酸、ヘキサン酸、オクタン酸、アクリル酸、メタクリル酸、安息香酸、トリフルオロメチル安息香酸、ニトロ安息香酸、フタル酸、ナフチル酸、パーフルオロヘプタン酸、パーフルオロオクタン酸等のカルボン酸類、モノエチレングリコール、ジエチレングリコール、トリエチレングリコール、ポリエチレングリコール、フェニルエチレングリコール、モノプロピレングリコール、ジプロピレングリコール、トリプロピレングリコール、ポリプロピレングリコール、プロパンジオール、クロロプロパンジオール、ブロモプロパンジオール、メトキシプロパンジオール、アリルオキシプロパンジオール、ブタンジオール、ヘキサンジオール、シクロヘキサンメタンジオール等のポリアルキレングリコール類を挙げることができる。これらは単独で用いてもよく2種以上の混合物でもよい。ポリエチレングリコール、ポリプロピレングリコールについては、二官能性、三官能性、四官能性の化合物を全て含むものとする。
【0086】
酸触媒としては例えば、塩酸、硫酸、リン酸等の鉱酸類、p-トルエンスルホン酸等のスルホン酸類、アンバーリスト-15(登録商標)等の固体酸類、三フッ化ホウ素エーテル錯体、三塩化ホウ素、三臭化ホウ素、三塩化アルミニウム、三臭化アルミニウム、四塩化スズ、二塩化亜鉛等のルイス酸を挙げることができる。
【0087】
塩基触媒としては例えば、リチウム、ナトリウム、カリウム、セシウム等のアルカリ金属の水酸化物、炭酸塩、炭酸水素塩、マグネシウム、カルシウム等のアルカリ土類金属の水酸化物、炭酸塩、炭酸水素塩、ピリジン、4-ジメチルアミノピリジン、トリエチルアミン等の有機アミン類、アンバーリスト-21(登録商標)、アンバーリスト-93(登録商標)等の弱塩基性イオン交換樹脂等が挙げられる。
酸または塩基触媒の使用量は、末端エポキシ基含有重合体に対して、0.01〜10質量倍が好ましく、より好ましくは0.1〜5質量倍、最も好ましくは0.5〜2質量倍である。これらの酸または塩基触媒は単独で用いてもよいし、2種以上を混合して用いても構わない。
【0088】
反応溶媒としては、原料の末端エポキシ基含有重合体に対して不活性なものが使用でき、例えばn-ヘキサン等の脂肪族炭化水素類、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、酢酸エチル等のエステル類、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン、メチルプロピルケトン等のケトン類、テトラヒドロフラン、1,4-ジオキサン等のエーテル類、クロロホルム、ジクロルエタン、トリクロルエタン、ジクロロベンゼン等のハロゲン化炭化水素などが挙げられる。原料の末端エポキシ基含有重合体がその溶媒に対して不溶でない限り、トルエン、キシレン等の芳香族炭化水素が好ましい。溶媒の使用量は原料の溶解性に作用するが、原料の末端エポキシ基含有重合体に対し0.8〜100質量倍が好ましく、より好ましくは1〜50質量倍、更に好ましくは2〜20質量倍である。
【0089】
反応は、例えば次のようにして行うことができる。反応器に、末端エポキシ基含有重合体、反応剤A、酸または塩基触媒を入れて混合し、均一に溶解するまで昇温する。ここで反応剤Aをあらかじめアルカリ金属またはアルカリ土類金属の塩として使用してもよい。反応温度は用いる末端エポキシ基含有重合体が溶解する温度が好ましい。反応温度は、25〜300℃が好ましく、より好ましくは50〜250℃、更に好ましくは80〜200℃である。使用する化合物、溶媒によっては反応温度が沸点を超える場合があるためオートクレーブ等適切な反応装置を選択する。反応時間は使用する触媒の量、反応温度、重合体類の反応性等の反応条件により変わるが、通常数分から50時間である。
【0090】
反応後は晶析操作、洗浄等の簡単な操作により、過剰の触媒、反応剤A、反応溶媒を除去して目的とする重合体を得ることができる。上記反応において、原料の末端エポキシ基含有重合体の製造工程から単離精製せずに上記反応を実施することもできる。
また、一般式(IV)においてRd、Reともに水酸基である重合体の製造方法としては、アルコール等の相溶化溶媒の共存下、末端エポキシ基含有重合体と水を反応させる方法が好ましい。
【0091】
[(1b)Rd、Reの一方が水酸基で、他方が一般式(VI)で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体に、一般式(XV)
【0092】
【化11】
【0093】
(式中、Rg,Rhは一般式(VI)と同様の原子または基を表す)で示される化合物を反応させることにより、Rg,Rhの一方が水酸基で、他方が一般式(VI)で示される基である重合体を得ることができる。反応は、酸または塩基触媒を共存させてもよい。
一般式(XV)としては、例えば、アンモニア、メチルアミン、エチルアミン、メチルプロピルアミン、エタノールアミン、ジエタノールアミン、エチルプロピルアミン、ブチルアミン、デシルアミン、オクタデシルアミン、ピロリジン、ピペリジン、ピペラジン、ヘキサメチレンイミン、エチレンジアミン、ジアミノプロパン、ジアミノブタン、ジエチレントリアミン、N-(アミノエチル)プロパンジアミン、イミノビスプロピルアミン、スペルミジン、スペルミン、トリエチレンテトラミン、シクロプロピルアミン、シクロブチルアミン、N-メチルシクロヘキシルアミン、ジアミノシクロヘキサン、ベンジルアミン、トリス(アミノプロピル)アミン、トリス(アミノエチル)アミン、アミノメチルヘプタンジアミン、アニリン、クロロアニリン、トルイジン、アミノフェノール、メチレンジアニリン、フェニレンジアミン、アミノナフタレン、ジェファーミン類(登録商標)等を挙げることができる。ジェファーミン(登録商標)としては末端にアミノ基を含有するポリアルキレングリコール類全てを含むものとする。ジエタノールアミンが中でも好ましい
酸、塩基触媒とその使用量、反応溶媒とその使用量については、(1a)と同様である。
反応方法は、(1a)の場合と同様に行うことができるが、酸、塩基触媒の非存在下でも反応は進行する。
【0094】
[(1c)Rd、Reの一方が水酸基で、他方がシアノ基、カルボキシル基、エステル基、アミド基で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体にシアノ化剤を反応させることにより、Rd、Reの一方が水酸基で、他方がシアノ基の重合体が得られる。
得られたシアノ基を含む重合体を加水分解によりカルボキシル基に誘導できる。更にこのカルボキシル基をエステル化することによりエステル基に誘導でき、アミド化することによりアミド基に誘導できる。これらの加水分解、エステル化、アミド化は、一般的な方法を用いる事ができる。
【0095】
シアノ化剤としてはシアン化ナトリウム、シアン化カリウム、トリメチルシリルシアニド、ジエチルアルミニウムシアニド、アセトンシアノヒドリン等を挙げることができる。シアノ化剤の使用量は原料の末端エポキシ基含有重合体の0.9〜20質量倍が好ましく、より好ましくは1〜10質量倍、更に好ましくは1.1〜10質量倍である。
反応方法は、(1a)の場合と同様に行うことができる。この場合、酸、塩基触媒を用いないで反応を行うことができる。
反応溶媒とその使用量については(1a)と同様である。
【0096】
[(2)一般式(IV)においてRd、Reの一方がポリエチレングリコール基である重合体の製造方法]
一般式(IV)においてRdまたはReの一方が水酸基の官能基含有重合体(以下、重合体Hと表記する)を原料とし、該水酸基にエポキシ化合物を反応させることにより、一般式(IV)においてRd、Reの一方がポリエチレングリコール基である重合体を得ることができる。
上記水酸基に付加重合するエポキシ化合物としては、プロピレンオキシド、エチレンオキシド、1,2-ブチレンオキシド、2,3-ブチレンオキシド、スチレンオキシド、シクロヘキセンオキシド、エピクロロヒドリン、エピブロモヒドリン、メチルグリシジルエーテル、アリルグリシジルエーテル等が挙げられる。これらは2種以上併用してもよい。これらの中で、好ましくは、プロピレンオキシド、エチレンオキシド、1,2-ブチレンオキシド、2,3-ブチレンオキシド、スチレンオキシドである。より好ましくはプロピレンオキシド、及びエチレンオキシドである。
【0097】
本反応に用いる触媒としては、例えばアルカリ金属水酸化物が挙げられる。また、ホスファゼニウム化合物、ホスフィンオキシド化合物、及びホスファゼン化合物(以下、P=N結合を有する化合物と表記する)を用いることもできる。
アルカリ金属水酸化物としては、例えば水酸化カルシウム、水酸化ナトリウム、水酸化リチウム、水酸化ルビシウム、水酸化セシウム等が挙げられる。
【0098】
ホスファゼニウム化合物としては、例えば、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムヒドロキシド、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムメトキシド、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムエトキシド、テトラキス[トリ(ピロリジン-1-イル)ホスフォラニリデンアミノ]ホスフォニウムtert-ブトキシド等が挙げられる。
ホスフィンオキシド化合物としては、例えば、トリス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフィンオキシド、又はトリス[トリス(ジエチルアミノ)ホスフォラニリデンアミノ]ホスフィンオキシド等が挙げられる。
ホスファゼン化合物としては、例えば、1-tert-ブチル-2,2,2-トリメチルホスファゼン、1-(1,1,3,3-テトラメチルブチル)-2,2,4,4,4-ペンタイソプロピル-2λ5,4λ5-カテナジ(ホスファゼン)、1-tert-ブチル-2,2,2-トリアリルホスファゼン、1-シクロヘキシル-2,2,4,4,4-ペンタアリル-2λ5,4λ5-カテナジ(ホスファゼン)、1-エチル-2,4,4,4-トリベンジル-2-トリベンジルホスフォラニリデンアミノ-2λ5,4λ5-カテナジ(ホスファゼン)、1-メチル-2,2,2-トリシクロペンチルホスファゼンまたは1-プロピル-2,2,4,4,4-シクロヘキシル-2λ5,4λ5-カテナジ(ホスファゼン)等が挙げられる。
【0099】
触媒であるアルカリ金属水酸化物の使用量は原料の重合体Aの1モルに対して、0.05〜0.5モルが好ましく、より好ましくは0.1〜0.3モルの範囲である。
触媒であるP=N結合を有する化合物の使用量は、重合速度、経済性等の点から、原料の重合体Hの1モルに対して1×10-4〜5×10-1モルが好ましい。より好ましくは5×10-4〜1×10-1モル、更に好ましくは1×10-3〜1×10-2モルである。
【0100】
原料である重合体Hにエポキシ化合物を付加重合する温度は、重合速度、副反応抑制の点から、15〜130℃が好ましい。より好ましくは40〜120℃、更に好ましくは50〜110℃の範囲である。エポキシ化合物の付加重合温度を上記範囲内の低い温度で行う場合は、原料の重合体Hに対するP=N結合を有する化合物の濃度を先に述べた範囲内で高めることが好ましい。
エポキシ化合物の付加重合反応の圧力は、副反応抑制の点から、882kPa以下が好ましい。通常、耐圧反応器内でエポキシ化合物の付加重合が行われる。エポキシ化合物の反応は減圧状態から開始しても、大気圧の状態から開始してもよい。大気圧の状態から開始する場合には、窒素、又は、ヘリウム等の不活性気体存在下で行うことが望ましい。反応圧力は、より好ましくは686kPa以下、更に好ましくは490kPa以下である。エポキシ化合物としてプロピレンオキシドを用いる場合には、反応圧力は490kPa以下が好ましい。
【0101】
反応におけるエポキシ化合物の供給方法は、必要量のエポキシ化合物の一部を一括して供給し、残部を連続的に供給する方法、又は、全てのエポキシ化合物を連続的に供給する方法等が用いられる。必要量のエポキシ化合物の一部を一括して供給する方法においては、エポキシ化合物の重合反応初期の反応温度は、上記温度範囲内でより低温側とし、エポキシ化合物の装入後に、次第に反応温度を上昇する方法が好ましい。
エポキシ化合物としてプロピレンオキシド及びエチレンオキシドを併用する場合の重合方法には、(a)プロピレンオキシドを重合した後、エチレンオキシドをブロックで共重合するエチレンオキシドキャップ反応、(b)プロピレンオキシドとエチレンオキシドをランダムに共重合するランダム反応、(c)プロピレンオキシドを重合した後、エチレンオキシドを重合し、次いで、プロピレンオキシドを重合するトリブロック共重合反応が挙げられる。これらの中で好ましい重合方法は、エチレンオキシドキャップ反応とトリブロック共重合反応である。
【0102】
付加重合器の最大圧力は、エポキシ化合物の装入速度、重合温度、触媒量等に影響される。エポキシ化合物の装入速度は、付加重合機の最大圧力が882kPaを超えないように制御することが好ましい。エポキシ化合物の装入が完了すると、付加重合器の内圧は徐々に低下する。内圧の変化が認められなくなるまで付加重合反応を継続することが好ましい。ポリアルキレングリコール基を含有する重合体の水酸基価(OHV)を基準とすると、OHVが2〜200mgKOH/gとなるまで付加重合を継続することが好ましい。
エポキシ化合物の付加重合反応に際して、溶媒を使用することもできる。溶媒としては、例えば、ペンタン、ヘキサン、ペプタン等の脂肪族炭化水素類、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン等のエーテル類、ジメチルスルホキシド、N,N-ジメチルホルムアミド等の非プロトン性極性溶媒等が挙げられる。
【0103】
次に、上記のようにして製造されたポリアルキレングリコール基を含有する重合体の精製方法について説明する。ポリアルキレングリコール基を含有する粗製重合体中に残存するアルカリ金属水酸化物またはP=N結合を有する化合物は、塩酸、リン酸等の鉱酸類、酢酸等の有機酸、炭酸ガス等による中和、吸着剤による吸着除去、水あるいは水/有機溶媒を用いた水洗、イオン交換樹脂によるイオン交換等の方法により除去することができる。
一般式(IV)においてRd、Reの一方がポリエチレングリコール基であり、他方が一般式(V)で表され、かつRfが一般式(VIII)で表される重合体の製造方法としては、まず、原料となる末端エポキシ基含有重合体と一般式(XVI)
【0104】
【化12】
【0105】
(式中、Eは酸素原子または硫黄原子を表し、R1aはm+1価の炭化水素基を表し、Tは同一または相異なり水酸基、アミノ基を表し、mは1〜50の整数を表す)
で示される反応剤A’とを(1a)に記載した方法により反応させ、一般式(IV)においてRd、Reの一方が水酸基、他方が下記一般式(XVII)で示される基で表される重合体を得る。
【0106】
【化13】
(式中、E、R1a、T、mは一般式(XVI)の定義と同様である)
【0107】
得られた重合体の水酸基およびTで表される水酸基またはアミノ基に、上述の方法によりエポキシ化合物を重合させることにより得ることができる。
一般式(IV)においてRd、Reの一方がポリエチレングリコール基であり、他方が一般式(VI)で表され、かつRg、Rhが一般式(VIII)で表される重合体の製造方法としては、まず、原料となる末端エポキシ基含有重合体と一般式(XVIII)
【0108】
【化14】
【0109】
(式中、R1aは同一または相異なりm+1価の炭化水素基を表し、Tは同一または相異なり水酸基、アミノ基を表し、mは1〜50の整数を表す)
で示される反応剤A’’とを(1a)に記載した方法により反応させ、一般式(IV)においてRd、Reの一方が水酸基、他方が下記一般式(XIX)で示される基で表される重合体を得る。
【0110】
【化15】
(式中、R1a、T、mは一般式(XVIII)の定義と同様である)
【0111】
得られた重合体の水酸基およびTで表される水酸基またはアミノ基に、上述の方法によりエポキシ化合物を重合させることにより得ることができる。
反応試剤A’としては、グリセリン、ペンタエリスリトール、ブタントリオール、ジペンタエリスリトール、ポリペンタエリスリトール、ジヒドロキシベンゼン、トリヒドロキシベンゼン等を挙げることができる。
反応試剤A’’としては、アミノフェノール、ヘキサメチレンイミン、エチレンジアミン、ジアミノプロパン、ジアミノブタン、ジエチレントリアミン、N-(アミノエチル)プロパンジアミン、イミノビスプロピルアミン、スペルミジン、スペルミン、トリエチレンテトラミン、ポリエチレンイミン等を挙げることができる。
【0112】
[(3)一般式(IV)においてRd、Reの一方がアシルオキシ基である重合体の製造方法]
前記重合体Hを原料とし、該水酸基等をアシル化することにより、一般式(IV)においてRd、Reの一方がアシルオキシ基である重合体を得ることができる。アシル化は、重合体Hと、対応する酸ハロゲン化物あるいは酸無水物を塩基触媒存在下反応させる一般的な方法で実施できる。
【0113】
酸ハロゲン化物としては、例えば塩化アセチル、臭化プロピオニル、塩化アクリロイル、塩化メタクリロイル、臭化ヘキサノイル、ヨウ化オクタノイル、塩化ベンゾイル、ヨウ化4-トリフルオロメチルベンゾイル、臭化3-ニトロベンゾイル、塩化ナフトイル、臭化パーフルオロヘプテノイル、ヨウ化パーフルオロオクテノイル等を挙げることができる。
酸無水物としては、無水酢酸、無水プロピオン酸、無水アクリル酸、無水メタクリル酸、無水フタル酸、無水マレイン酸、無水コハク酸等を挙げることができる。
塩基触媒としては(1a)で例示した触媒を挙げることができる。
前記式のPOで表される重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることが好ましい。
【0114】
(樹脂組成物)
本発明の樹脂改質用添加剤は、各種熱可塑性樹脂に添加して用いることができる。本発明の樹脂改質用添加剤は、熱可塑性樹脂の伸長粘度を改善することができる
ベースとなる熱可塑性樹脂としてはポリオレフィンが好ましく、ポリプロピレンもしくはポリエチレンが好ましく、とりわけポリプロピレンが好ましい。
【0115】
本発明の樹脂改質用添加剤は、各種用途に用いることができるが、発泡性樹脂の改質用添加剤として特に好適に用いられる。本発明の樹脂改質用添加剤を用いると、均一かつ発泡倍率の大きい発泡セルが得られる点で好ましい。
添加処方としては、熱可塑性樹脂99〜60重量%に対し、樹脂用添加剤1〜40重量%が好ましい。
【0116】
発泡性樹脂の改質用添加剤として用いるに際しては、さらに発泡剤を含むことが出来る。発泡剤としては、二酸化炭素、窒素等のガス、プロパン、ブタン、ペンタン、ヘキサンおよびそれらの各構造異性体等の脂肪族炭化水素、塩化メチル、塩化エチル、テトラフロロエタン、ジフロロエタンなどのハロゲン化炭化水素、メタノール、エタノール、プロパノールなどのアルコール、ジメチルエーテル、ジエチルエーテル、メチルエチルエーテル等のエーテル、アルゴン、水等が挙げられる。また、本発明に前述の発泡剤の他に、気泡調節剤または発泡核剤を用いても良い。気泡調節剤または発泡核剤としては、例えば炭酸アンモニウム、重曹、重炭酸アンモニウム、亜硝酸アンモニウム等の無機系分解性発泡剤、アゾビスイソブチロニトリル、ジアゾアミノベンゼン、アゾジカルボンアミド等のアゾ化合物、N,N′−ジメチル−N,N′−ジニトロソテレフタルアミド、N,N′−ジニトロソペンタンメチレンテトラミン等のニトロソ化合物、p−トルエンスルホニルヒドラジド、p−トルエンスルホニルセミカルバジド、バリウムアゾジカルボキシレート等の分解性発泡剤、タルク、シリカ等の無機粉末、多価カルボン酸等の酸性塩、多価カルボン酸と炭酸ナトリウム又は重曹との反応混合物等が挙げられ、これらは、単独でも組み合わすこともできる。この場合、発泡性樹脂用添加剤0.1〜35重量%と、熱可塑性樹脂50〜99.8重量%と、発泡剤0.1〜15重量%とを含む組成が好ましい。組成はさらに発泡性樹脂用添加剤9〜30重量%と、熱可塑性樹脂重量60〜90%と、発泡剤1〜10重量%が好ましい
【0117】
熱可塑性樹脂、樹脂用添加剤、更に任意で発泡剤を含む組成物は、各成分をミキサー等で混合して得ることが出来る。得られた組成物を、押し出し成型など公知の成型法により、シート・フィルム・発泡シートなどに成型すればよい。
また、各成分所定量をそれぞれ押出し器に直接投入し、押出し機内で溶融混錬しても良い。更に発泡剤を発泡時に導入する場合は、密閉した系に溶融混練した組成物を導入し、発泡剤を圧入して射出成型、または押し出し成型しても良い。
【実施例】
【0118】
以下、実施例等により本発明をさらに具体的に説明するが、本発明の範囲はこれらの実施例等に限定されるものではない。
〔測定および計算方法〕
分子量、融点(Tm)、粒子径、収率、転化率および異性化率、伸長粘度、CO2含浸率、発泡倍率、発泡平均セル径等は以下に記載の方法で測定・計算した。
【0119】
[m1]分子量の測定方法
重量平均分子量(Mw)及び分子量分布(Mw/Mn)は、ミリポア社製GPC−150を用い以下のようにして測定した。すなわち、分離カラムは、TSK GNH HTであり、カラムサイズは直径7.5mm、長さ300mmのものを使用した。カラム温度は140℃とし、移動相にはオルトジクロロベンゼン(和光純薬)及び酸化防止剤としてBHT(武田薬品)0.025質量%を用い、1.0ml/分で移動させた。試料濃度は0.1質量%とし、試料注入量は500マイクロリットルとした。検出器として示差屈折計を用いた。標準ポリスチレンは東ソー社製を用いた。
【0120】
数平均分子量Mn、および重量平均分子量Mwは、市販の単分散標準ポリエチレンを用いて検量線を作成し、下記の換算法に基づいて求めた。
分子量換算 : PE換算/汎用較正法
なお、汎用較正の計算には、以下に示すMark−Houwink粘度式の係数を用いた。
ポリエチレン(PE)の係数 : KPE=5.06×10-4, aPE=0.70
【0121】
[m2]融点の測定方法
融点(Tm)はDSCを用い測定して得られたピークトップ温度を採用した。装置は島津製作所製DSC−60Aを使用した。対照セルはアルミナを使用し、窒素流量は50ml/分の設定で行った。また10℃/分で(30)℃から300℃までの昇温条件で測定した。この昇温測定の前に、一旦、樹脂を200℃程度まで昇温し、5分間保持した後、20℃/分で常温(25℃)まで降温する操作を行い、樹脂の熱履歴を統一することが望ましい。
【0122】
[m3]NMR解析による収率、転化率、異性化率、末端不飽和率、炭素千個あたりの二重結合数の測定・計算方法
シリル化ポリオレフィン[A]の収率、転化率、異性化率、末端不飽和率、炭素千個あたりの二重結合数は1H−NMRによって決定される。収率は原料の末端不飽和重合体のモル数に対して得られたシリル化ポリオレフィン[A]のモル数の割合、転化率は原料の末端不飽和重合体のモル数に対する同消費モル数の割合、異性化率は原料の末端不飽和重合体のモル数に対して生成したビニレン体のモル数の割合、末端不飽和率は末端二重結合と末端メチルの合計に対する末端二重結合の割合、炭素千個あたりの二重結合数はプロトン数から導き出される炭素数に対する二重結合数の割合を炭素千個あたりの二重結合数に補正したものと定義される。
【0123】
例えば、エチレンのみからなる末端不飽和重合体をトリエトキシシランでヒドロシリル化して得られたシリル化ポリオレフィン[A]のエトキシ基メチレンの6プロトン分のピーク(C)が3.8ppm、異性化したビニレン基の2プロトン分のピーク(D)が5.4ppmに観測される。ヒドロシリル化が十分でない場合は、未反応ビニル基の2プロトン分のピーク(E)が4.8〜5.1ppmに、1プロトン分のピーク(F)が5.6〜5.8ppmに観測される。原料の二重結合含有重合体については、2プロトン分の主鎖メチレン(G)が1.0〜1.5ppmに観測され、末端に二重結合を持たないものは3プロトン分の末端メチル(H)が0.8ppmに観測される。さらに二重結合に隣接した炭素上の2プロトン分のピーク(I)が1.9ppmに観測される。
【0124】
各ピーク(C)、(D)、(E)、(F)、(G)、(H)および(I)のピーク面積を各々SC、SD、SE、SF、SG、SHおよびSIとすれば、収率(YLD(%))、転化率(CVS(%))、異性化率(ISO(%))、末端不飽和率(VE(%))、炭素千個あたりの二重結合数(VN(個/1000C))は下記式にて算出される。
YLD(%)=(SC/3)/(SC/3+SD+SE)×100
CVS(%)={1−SE/(SC/3+SD+SE)}×100
ISO(%)=SD/(SC/3+SD+SE)×100
VE(%)=(SE/2)/(SE/2+SH/3)×100
VN(個/1000C)=(SE+SF)/3×1000/{(SD+SE+SF+SG+SH+SI)/2}
【0125】
[m4]伸長粘度測定方法
伸長粘度測定は、Rheometric Scientific社製 RMEエロンゲーショーナルレオメーター(RME−SS−RC−SD−BH)を使用し、付属の解析装置にて行った。
評価サンプルは、対象樹脂を110×7×2mmの形状に熱プレスにより成型し用いた。
【0126】
[m5]CO2含浸率測定方法
熱可塑性樹脂シ−ト(5cm×5cm×2mm厚み)を内容積1Lの高圧容器に入れ、液化炭酸ガスを注入し、超臨界状態(40℃×20MPa)で3時間放置後、高圧容器から熱可塑性樹脂シ−トを取り出し、電子天秤で経時による重量変化を測定する。重量変化のグラフから、取り出し直後の炭酸ガス含浸の熱可塑性樹脂シ−ト重量を求め、次式により算出する。
(2)−(1)/(1)×100%
(1)=未発泡の熱可塑性樹脂シ−ト重量
(2)=炭酸ガス含浸の熱可塑性樹脂シ−ト重量
【0127】
[m6]発泡倍率測定方法
熱可塑性樹脂シ−トの比重を電子比重計(MD-200S:アルファ-ミラ-シ゛ュ製)を用いて測定し、発泡熱可塑性樹脂の比重を測定して、熱可塑性樹脂シ−トの比重に対する割合を算出し、小数点以下第2位を四捨五入した値を発泡倍率とした。
【0128】
[m7]発泡平均セル径測定方法
走査型電子顕微鏡により撮影した発泡熱可塑性樹脂の断面写真について、発泡セル径の画像処理により円相当径を算出し、その値を平均セル径とした。
【0129】
[m8]フィルムの機械物性(引張強度、引張伸度及び引張弾性率)測定方法
引張強度、引張伸度及び引張弾性率は引張試験機により評価した。引張試験機は、島津製作所社製;EZ−S 100N型を用いた。試験対象のフィルムを、全体長さ50mm、つかみ部分巾10mm、試験片部分巾5mm、試験片部分長さ30mmのダンベル型に打抜いた。この試験フィルムを引張試験のチャックにはさみ、30mm/分の速度で試験を行った。
【0130】
〔使用原料〕
実施例、比較例に使用した原料については、以下のものを使用した。なお、原料ポリマーのモル数はすべてMnに基づいた値で表した。
(1)塩化白金および塩化白金酸(試薬): アルドリッチ社製
(2)トリエトキシシラン(試薬): 関東化学社製
(3)無水マレイン酸(試薬): 和光純薬社製
(4)片末端ビニルを有するポリエチレン(P1): 特開2003−73412の実施例1記載の方法に準じて合成したもの(Mn=730、Mw/Mn=1.9、135℃デカリン中で測定した極限粘度[η]=0.08dl/g、融点(Tm)116℃、末端不飽和率96mol%(NMR基準))
【0131】
〔フィルム作製〕
実施例、比較例の引張試験に使用したフィルムについては、以下の通りに作製した。
マイクロコンパウンダー(DSM社製)を使用し、所定の混合比率のポリマーを、混練温度230℃で5分間混練、次いでTダイより射出し付帯のフィルム巻き取り機にて巻き取った。上記で得られたポリマー混合物からフィルムを作製した。この結果、厚さ約50μm、巾3cmのフィルムが得られた。
【0132】
[合成例1]
〔白金触媒組成物(C−1)の調製〕
マグネットスターラーチップを入れた50mlサンプル管中、塩化白金(II)0.50gをトリエトキシシラン(10ml)中に懸濁し、窒素気流下、室温で24時間攪拌した。所定時間攪拌した後、シリンジにて反応液を約0.4ml採取し、0.45μmPTFEフィルターを用いて濾過して10mlサンプル管中に濾液を採取した。得られた濾液をマイクロピペットにて10μl秤取って10mlサンプル管中に分取した後、トリエトキシシラン1.99mlを加えて200倍希釈し、白金触媒組成物(C−1)を得た。
【0133】
〔末端ビニルを有するポリエチレンへのアルコキシシラン導入〕
還流管の付いた1000mlの反応器に末端ビニルを有するポリエチレン(P1)200g(0.27mol)を装入し、窒素雰囲気下、トリエトキシシラン56ml(262mmol)と白金触媒組成物(C−1)3ml(Pt換算で2.8×10−3mmol(トリエトキシシラン14mmolを含む))を装入した。予め内温130℃に昇温しておいた油浴中に、上記反応器をセットし攪拌した。約20分後ポリマーは融解した。次いで1時間後に冷却し、アセトン約1500ml中に反応物を投入、2時間攪拌した。その後、固体をろ取しアセトンで洗浄し、60℃、2hPa以下の減圧下で乾燥させることにより、白色固体のポリマー(PE−TES)217gを得た。NMR解析の結果、得られたポリマーは収率88.2%、オレフィン転化率100%、異性化率11.8%だった。
【0134】
[合成例2]
〔末端ビニルを有するポリエチレンへのマレイン酸導入〕
還流管の付いた1000mlの反応器に、末端ビニルを有するポリエチレン(P1)200g(0.27mol)、無水マレイン酸134g(1.37mol)、及び塩化アルミニウム(III)0.5g(3.75mmol)を仕込み、195℃に昇温し、30時間加熱攪拌した。反応終了後、120℃に冷却した反応ポリマーにトルエン200gを加えて溶解させた後、アセトン(1000ml)に投入し晶析させた。その後、アセトン洗浄(1000ml×2回)し、減圧乾燥機で80℃、9時間乾燥させ、白色固体のポリマー(PE−MAH)203gを得た。NMR解析の結果、得られたポリマーはオレフィン転化率99.0%だった。
【0135】
[合成例3]
特開2006−131870号公報の合成例2に従って、Mw=2058、Mn=1118、Mw/Mn=1.84(GPC)の末端エポキシ基含有エチレン重合体(E1)(Mn 1118)を合成し、原料として用いた。
1000mLフラスコに、末端エポキシ基含有エチレン重合体(E1) 84 g(75mmol)、ジエタノールアミン39.4g(375mmol)、トルエン150g を仕込み、150℃にて4時間撹拌した。その後、冷却しながらアセトンを加え、反応生成物を析出させ、固体をろ取した。得られた固体をアセトン水溶液で1回、更にアセトンで3回撹拌洗浄した後、固体をろ取した。その後、室温にて減圧下乾燥させることにより、白色固体のポリマー(PE−DEA)87gを得た。
【0136】
[実施例1]
前記合成例1によって合成されたPE−TESとポリプロピレン(プライムポリマー社製;VP103W)について、PE−TESを全量の5重量%になるように混合し、マイクロコンパウンダー(DSM社製;DSM−Xplore)を使用し、温度230℃で5分混練、次いで900Nのトルク制御にて押し出してストランドを作製した後測定サンプルを成型した。次いで、[m4]伸長粘度測定法に従い伸長粘度を測定した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は489kPas(232s時)であった。また、別の測定サンプルを株式会社AKICO社製高圧容器(型式:特注型000819)中に静置し、CO2を20MPa、温度40℃に3時間保持(バッチ式発泡試験装置)を行った。次いで初期[m5]CO2含浸率を測定したところ、6.8%だった(表1)。
【0137】
[実施例2]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例1と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は7,470kPas(683s時)、初期CO2含浸率は4.8%だった(表1)。
【0138】
[比較例1]
ベースポリマーのポリプロピレン(プライムポリマー社製;VP103W)について、実施例1と同様にマイクロコンパウンダーを使用し、ストランドを作製した後測定サンプルを成型した。伸長粘度を測定した結果、180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は14kPas(79s時)、初期CO2含浸率は3.5%だった(表1)。
【0139】
[実施例3]
ベースポリマーのポリプロピレンをプライムポリマー社製;F102Wに換えた以外には、実施例1と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は876kPas(570s時)であった。また、別の測定サンプルを使用して、バッチ式発泡試験装置に従い、発泡倍率を測定したところ4.3倍だった(表1)。
【0140】
[実施例4]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は6,585kPas(683s時)、発泡倍率は3.8倍だった(表1)。
【0141】
[実施例5]
PE−TESを前記合成例3によって合成されたPE−DEAに換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は514kPas(476s時)、発泡倍率は3.9倍だった(表1)。
【0142】
[比較例2]
PE−TESを前記ポリプロピレン(プライムポリマー社製;VP103W)に換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は52kPas(46s時)、発泡倍率は3.2倍だった(表1)。
【0143】
【表1】
【0144】
[実施例6]
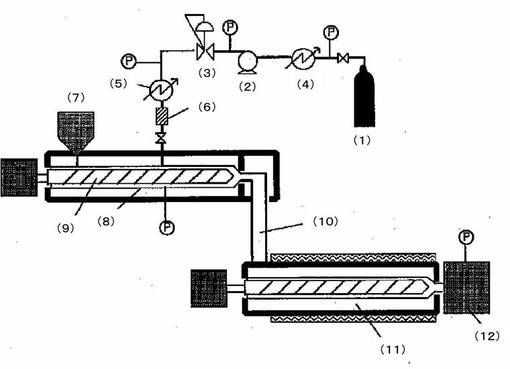
前記合成例1によって合成されたPE−TESとポリプロピレン(プライムポリマー社製;F102W)について、PE−TESを全量の5重量%になるように混合し、図1に示すタンデム型の押出機に投入した。装置としては図1に示したスクリュウ径30mmφの第1押出機(8)とスクリュウ径40mmφの第2押出機(11)を有する。二酸化炭素添加部は、第1押出機の中央付近に設けた。該材料をホッパー(7)より第1押出機(8)に投入し、220℃で加熱溶融させた。二酸化炭素は、サイホン式の液化二酸化炭素ボンベ(1)を使用し、液相部分から直接取り出せるようにした。液化二酸化炭素ボンベ(1)からプランジャーポンプ(2)までの流路を冷媒循環機(4)を用いて、−12℃に調節したエチレングリコール水溶液で冷却し、二酸化炭素を液体状態でプランジャーポンプ(2)まで注入できるようにした。この時二酸化炭素の温度は、−5℃であった。次に注入した液状二酸化炭素を200g〜500g/時間となるようプランジャーポンプ(2)を制御し、プランジャーポンプ(2)の吐出圧力を30MPaとなるよう保圧弁(3)にて調整した。次に保圧弁(3)から第1押出機(8)の二酸化炭素添加部までのラインを50℃となるようヒーターで加熱し、二酸化炭素を第1押出機(8)内の溶融した該材料に添加した。このときの添加部の溶融樹脂圧力は20MPaであった。つまり、該溶融材料に溶解する直前の二酸化炭素は、温度が50℃以上、圧力が20MPaである超臨界状態の二酸化炭素となっている。このようにして、該溶融材料100重量部に対して超臨界二酸化炭素を5重量部の割合で第1押出機(8)内に添加し、スクリュウで均一に溶解させた。次にこの溶融物を第2押出機(11)へ送り、樹脂温度を165℃に調整し、2.1kg/時間の押出量でダイス(12)より押し出した。このときのダイス(12)圧力は、4.8MPaであった。
【0145】
ダイス(12)としては、出口隙間が、直径2.0mmφのストランドダイス(12)を使用した。押し出された該材料は、ダイス(12)から出たと同時に発泡し、ストランドとして放出される。二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率は15.9倍だった。
【0146】
また、ストランドダイス(12)の径を1.0mmφに換えて発泡させた場合、二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率は29.4倍だった(表2)。発泡セルの観察結果は他の試料よりもサイズが小さく比較的均一サイズで等方的な円または多角形だった。
【0147】
[実施例7]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率はそれぞれの場合で、16.9倍、27.6倍だった(表2)。発泡セルの観察結果は大きなセルが点在し、等方的な円または多角形だった。
【0148】
[実施例8]
PE−TESを前記合成例3によって合成されたPE−DEAに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率はそれぞれの場合で、12.1倍、24.6倍だった(表2)。発泡セルの観察結果はセルサイズが大きく比較的均一サイズで等方的な円または多角形だった。
【0149】
[比較例3]
材料を前記ポリプロピレン(プライムポリマー社製;F102W)のみに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動は小さかったが、径が1mmφの場合出口で詰まり気味であり、安定したストランドを得るのが困難であった。得られたストランドの発泡倍率はそれぞれの場合で、9.4倍、21.0倍だった(表2)。発泡セルの観察結果は断面の内側と外側でセルサイズ、形状共に不均一でやや扁平気味だった。
【表2】
【0150】
[実施例9]
前記合成例1によって合成されたPE−TES(変性率85%)とポリプロピレン(プライムポリマー社製;J715M)について、PE−TESを全量の10重量%になるようにマイクロコンパウンダー(DSM社製;DSM−Xplore)を使用し、温度230℃で5分混練、次いで前記〔フィルム作製〕方法に従い、900Nのトルク制御にて押し出しフィルムを作製した。 さらに[m8]フィルムの機械物性測定方法に従い、引張弾性率、引張伸度、破断点応力を測定した。引張弾性率1.0GPa、引張伸度480%以上、破断点応力37MPa以上だった。(表3)
【0151】
[実施例10]
PE−TES(変性率78%)に、含量を30%に換えた以外は実施例9と同様に操作した。引張弾性率1.0GPa、引張伸度490%以上、破断点応力31MPa以上だった。(表3)
【0152】
[実施例11]
PE−TES(変性率87%)10.7部と、未変性の末端ビニルを有するポリエチレン(P1)89.3部を混合して変性率10%のPE−TESとし、含量を10%として実施例9と同様に操作した。引張弾性率1.0GPa、引張伸度480%以上、破断点応力25MPa以上だった。(表3)
【0153】
[比較例9]
ポリプロピレン(プライムポリマー社製;J715M)のみを実施例9と同様に操作した。引張弾性率0.5GPa、引張伸度460%、破断点応力18MPaだった。(表3)
【0154】
[比較例10〜11]
PE−TESを未変性PEに、含量をそれぞれ10%、30%に換えた以外は実施例9と同様に操作した。引張弾性率、引張伸度、破断点応力を表3に示す。
【0155】
【表3】
【図面の簡単な説明】
【0156】
【図1】本発明の樹脂用添加剤を含む組成物を用い、発泡成型体を得る方法を示す
【符号の説明】
【0157】
(1)液化二酸化炭素ボンベ
(2)定量ポンプ
(3)保圧弁
(4)冷媒循環器
(5)ヒーター
(6)流量計
(7)ホッパー
(8)第1押出機
(9)スクリュウ
(10)連結部
(11)第2押出機
(12)ダイス
【技術分野】
【0001】
本発明は、樹脂改質用添加剤および添加剤を含む樹脂組成物に関する。本発明は特に、該樹脂改質用添加剤を含む発泡性樹脂組成物に関する。
【背景技術】
【0002】
微細な発泡セルを有するポリオレフィン発泡体として、さまざまなものが知られている。特に発泡ポリプロピレンは、耐熱性、柔軟性、耐衝撃性、軽量性などの特性に優れることから様々な用途に用いられている。しかしながら、ポリプロピレンは発泡成形性が悪く、特に発泡倍率の改善が求められている。
特許文献1〜3では、樹脂の発泡成形性の改善において、伸長粘度の値が発泡成形性の指標にできることが示されている。即ち、樹脂の伸長粘度の増大が発泡成形性を改善させる。しかしながら、ポリプロピレンの発泡成形には特別な組成のポリプロピレンの作製が必要であった(特許文献1、3)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開平11−060835
【特許文献2】特開2008−201866
【特許文献3】特開2009−275119
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、ポリオレフィン特にポリプロピレンの伸長時間および伸長粘度を向上させ、例えば発泡成形性を改善し、高い発泡倍率の成型体が得られる樹脂改質添加剤を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは鋭意検討を行い、末端に特定の極性基が結合したポリオレフィンが前記課題を解決できることを見出して本発明を完成した。
すなわち本発明の要旨は以下にある:
[1] 下記一般式(I)、(II)、(III)または(IV)で表される酸素含有基、窒素含有基およびケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。
【0006】
【化1】
【0007】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表し、Raは水素原子、ハロゲン原子、炭素数1〜10の炭化水素基、窒素含有基、酸素含有基またはケイ素含有基を表しそれぞれ同一でも異なっていてもよく、RbおよびRcはそれぞれ独立に、水素原子、ハロゲン原子、炭化水素基、−OR基、−SR基または−NR2基を表し(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)、環構造を形成してもよく、RdおよびReは一方が水酸基、ポリアルキレングリコール基またはアシルオキシ基を表し、他方は下記一般式(V)、一般式(VI) 、一般式(VII) のいずれかで示される基、シアノ基、カルボキシル基、エステル基またはアミド基を表す。nは平均官能基数を表し、0.1〜1.0である。)
【0008】
【化2】
【0009】
( 式中、Xは酸素原子または硫黄原子を表し、Rf は水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、下記一般式(VIII)
【0010】
【化3】
【0011】
( 式中、R1aは m+1 価の炭化水素基を表し、G は同一または相異なり、-OR1b 、-NR1 c R1d (R1b 、R1 c、R1dはポリアルキレングリコール基を表す) で表される基を表し、m は1〜 5 0 の整数を表す)で表される基を表す。)
【0012】
【化4】
【0013】
( 式中、Rg,Rh は同一または相異なり、水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、上記一般式(VII)で表される基を表す。)
【0014】
【化5】
【0015】
( 式中、Ri、Rj、Rkは同一または相異なり、水素原子、炭化水素基、アシル基、シアノ基、カルボキシル基、エステル基、アミド基を表す。)
【0016】
[2] 前記極性基含有ポリオレフィン重合体を含有する樹脂組成物の伸長粘度最大値が、180℃恒温下、毎秒伸長速度0.01の測定で100kPa・s以上を示すことを特徴とする[1]に記載の樹脂改質用添加剤。
[3] 前記極性基含有ポリオレフィン重合体中の重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることを特徴とする[1]〜2に記載の樹脂改質用添加剤。
[4] 前記樹脂改質用途が発泡性樹脂用であることを特徴とする[2]〜[3]に記載の樹脂改質用添加剤。
[5][4]に記載の発泡性樹脂用添加剤0.1〜15重量%と、熱可塑性樹脂70〜99.8重量%と、発泡剤0.1〜15重量%とを含む発泡性樹脂組成物。
[6] 前記熱可塑性樹脂が、ポリエチレン、ポリプロピレン、ポリスチレン、ポリウレタンおよびポリカーボネートからなる群から選ばれる1種以上の樹脂であることを特徴とする[5]に記載の発泡性樹脂組成物。
[7][4]〜[6]に記載の発泡性樹脂組成物から得られる発泡成形体。
【発明を実施するための形態】
【0017】
(樹脂改質用添加剤)
本発明の樹脂改質用添加剤は、前記した一般式(I)、(II)、(III)または(IV)で表される。
一般式(I)、(II)、(III)または(IV)で表される化合物は、まず末端不飽和ポリオレフィンを得た後に、末端不飽和基を以下に示す方法でそれぞれ官能基に変換することによって調製出来る。
【0018】
一般式(I)で表される化合物の中でも、Raとして酸素含有基が好ましく、さらにアルコキシ基が好ましく、特にエトキシ基・メトキシ基が好ましい。
一般式(III)で表される化合物の中でも、RbおよびRcとして酸素含有基が好ましく、さらに水酸基またはアルコキシ基が好ましく、特に水酸基が好ましい。
一般式(IV)で表される化合物の中でも、RdおよびReとして式(V)ないし(VI)で表される構造が好ましい。
式(V)のXとして酸素原子が好ましく、Rfとしては水素原子またはポリアルキレングリコールが好ましく、さらに水素原子が好ましい。式(VI)のRgおよびRhとしてポリアルキレングリコールもしくはアルカノール基が好ましく、特に−CH2CH2OHが好ましい。
【0019】
(末端不飽和ポリオレフィン(X))
末端不飽和ポリオレフィンの製造方法は、特開2003−73412に公開されている。
R1−CH=CH2 (X)
上記一般式(X)中、R1は、エチレン単独重合鎖、またはエチレン、プロピレン、ブテン、ビニルノルボルネン、ジエンを含む炭素数2〜20のオレフィンから選択される少なくとも2種以上の共重合鎖である。
【0020】
炭素数2〜20のオレフィンとしては、具体的には、エチレン、プロピレン、1−ブテン、1−ペンテン、3−メチル−1−ブテン、1−ヘキセン、4−メチル−1−ペンテン、3−メチル−1−ペンテン、3,4−ジメチル−1−ペンテン、4−メチル−1−ヘキセン、3−エチル−1−ペンテン、3−エチル−4−メチル−1−ペンテン、3,4−ジメチル−1−ヘキセン、4−メチル−1−ヘプテン、3,4−ジメチル−1−ヘプテン、1−オクテン、1−デセン、1−ドデセン、1−テトラデセン、1−ヘキサデセン、1−オクタデセン、1−エイコセン、ビニルシクロヘキサンなどのα−オレフィン; シス−2−ブテン、トランス−2−ブテン、などの内部二重結合を含むオレフィン; イソブテン、2−メチル−1−ペンテン、2,4−ジメチル−1−ペンテン、2,4−ジメチル−1−ヘキセン、2,4,4−トリメチル−1−ペンテン、2,4−ジメチル−1−ヘプテン、2−メチル−1−ブテン、2−メチル−1−ヘキセン、2−メチル−1−ヘプテン、2−メチル−1−オクテン、2,3−ジメチル−1−ブテン、2,3−ジメチル−1−ペンテン、2,3−ジメチル−1−ヘキセン、2,3−ジメチル−1−オクテン、2,3,3−トリメチル−1−ブテン、2,3,3−トリメチル−1−ペンテン、2,3,3−トリメチル−1−ヘキセン、2,3,3−トリメチル−1−オクテン、2,3,4−トリメチル−1−ペンテン、2,3,4−トリメチル−1−ヘキセン、2,3,4−トリメチル−1−オクテン、2,4,4−トリメチル−1−ヘキセン、2,4,4−トリメチル−1−オクテン、2−メチル−3−シクロヘキシル−1−プロピレン、ビニリデンシクロペンタン、ビニリデンシクロヘキサン、ビニリデンシクロオクタン、2−メチルビニリデンシクロペンタン、3−メチルビニリデンシクロペンタン、4−メチルビニリデンシクロペンタンなどのビニリデン化合物; スチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、2,4−ジメチルスチレン、o−エチルスチレン、m−エチルスチレン、p−エチルスチレンなどのアリールビニル化合物; α−メチルスチレン、α−エチルスチレン、2−メチル−3−フェニルプロピレンなどのアリールビニリデン化合物; メタクリル酸メチル、メタクリル酸エチル、メタクリル酸−n−プロピル、メタクリル酸イソプロピル、メタクリル酸−n−ブチル、メタクリル酸イソブチル、メタクリル酸−tert−ブチル、2−シアノプロピレン、2−アミノプロピレン、2−ヒドロキシメチルプロピレン、2−フルオロプロピレン、2−クロロプロピレンなどの官能基置換ビニリデン化合物; シクロブテン、シクロペンテン、1−メチル−1−シクロペンテン、3−メチル−1−シクロペンテン、2−メチル−1−シクロペンテン、シクロヘキセン、1−メチル−1−シクロヘキセン、3−メチル−1−シクロヘキセン、2−メチル−1−シクロヘキセン、シクロヘプテン、シクロオクテン、ノルボルネン、5−メチル−2−ノルボルネン、テトラシクロドデセン、5,6−ジヒドロジシクロペンタジエン、3a,4,5,6,7,7a−ヘキサヒドロ−1Hインデン、トリシクロ[6.2.1.02,7]ウンデカ−4−エン、シクロペンタジエン、ジシクロペンタジエンなどの内部二重結合を含む脂肪族環状オレフィン; シクロペンタ−2−エニルベンゼン、シクロペンタ−3−エニルベンゼン、シクロヘキサ−2−エニルベンゼン、シクロヘキサ−3−エニルベンゼン、インデン、1,2−ジヒドロナフタレン、1,4−ジヒドロナフタレン、1,4−メチノ1,4,4a,9aテトラヒドロフルオレンなどの芳香環を含有する環状オレフィン; ブタジエン、イソプレン、4−メチル−1,3−ペンタジエン、4−メチル−1,4−ペンタジエン、1,3−ペンタジエン、1,4−ペンタジエン、1,5−ヘキサジエン、1,4−ヘキサジエン、1,3−ヘキサジエン、1,3−オクタジエン、1,4−オクタジエン、1,5−オクタジエン、1,6−オクタジエン、1,7−オクタジエン、エチリデンノルボルネン、ビニルノルボルネン、ジシクロペンタジエン、7−メチル−1,6−オクタジエン、4−エチリデン−8−メチル−1,7−ノナジエン、5,9−ジメチル−1,4,8−デカトリエンなどの二個以上の二重結合を有する環状、又は鎖状のジエン、又はポリエンなどが挙げられる。
【0021】
また、オレフィンは、酸素、窒素、硫黄等の原子を含んだ官能基を有していてもよい。例えばアクリル酸、フマル酸、イタコン酸、ビシクロ[2.2.1]ヘプタ−5−エン−2,3−ジカルボン酸などの不飽和カルボン酸およびこれらのナトリウム塩、カリウム塩、リチウム塩、亜鉛塩、マグネシウム塩、カルシウム塩などの不飽和カルボン酸金属塩; 無水マレイン酸、無水イタコン酸、ビシクロ[2.2.1]ヘプタ−5−エン−2,3−ジカルボン酸無水物などの不飽和カルボン酸無水物; アクリル酸メチル、アクリル酸エチル、アクリル酸−n−プロピル、アクリル酸イソプロピル、アクリル酸−n−ブチル、アクリル酸イソブチル、アクリル酸−tert−ブチル、アクリル酸−2−エチルヘキシル、などの不飽和カルボン酸エステル; 酢酸ビニル、プロピオン酸ビニル、カプロン酸ビニル、カプリン酸ビニル、ラウリン酸ビニル、ステアリン酸ビニル、トリフルオロ酢酸ビニルなどのビニルエステル類; アクリル酸グリシジル、メタクリル酸グリシジル、イタコン酸モノグリシジルエステルなどの不飽和グリシジルエステル; 塩化ビニル、フッ化ビニル、フッ化アリルなどのハロゲン化オレフィン; アクリロニトリル、2−シアノ−ビシクロ[2.2.1]ヘプタ−5−エンなどの不飽和シアノ化合物; メチルビニルエーテル、エチルビニルエーテルなどの不飽和エーテル化合物; アクリルアミド、メタクリルアミド、N,N−ジメチルアクリルアミド等の不飽和アミド; メトキシスチレン、エトキシスチレン、ビニル安息香酸、ビニル安息香酸メチル、ビニルベンジルアセテート、ヒドロキシスチレン、o−クロロスチレン、p−クロロスチレン、ジビニルベンゼンなどの官能基含有スチレン誘導体; N−ビニルピロリドンなどが挙げられる。
【0022】
上記のうち、R1としては、エチレン単独重合鎖、プロピレン単独重合鎖またはエチレンとプロピレンとの共重合鎖が好ましい。
【0023】
上記一般式(X)中、R1の数平均分子量は、100〜50,000であることが好ましく、100〜10,000であることがより好ましい。これより基が短くなると、樹脂中やオイル中における分散性が悪くなるし、長くなると添加剤としての性能が出難くなるので好ましくない。
【0024】
(I)シリル化ポリオレフィンの製造方法
前記一般式(I)で表されるシリル化ポリオレフィンは、下記の[工程1]および[工程2]を順次実施することにより、製造できる。
【0025】
[工程1]白金触媒組成物(C)を得る工程
[工程1]では、下記一般式(IX)で表されるヒドロシランと二塩化白金とを混合攪拌し、得られた懸濁溶液を濾過して濾液として白金触媒組成物(C)を得る。
Ra3−SiH (IX)
【0026】
上記一般式(IX)中、Raは、水素原子、ハロゲン原子、炭化水素基、酸素含有基、またはケイ素含有基を表し、それぞれ同一であっても異なっていてもよい。
ハロゲン原子としては、フッ素、塩素、臭素、ヨウ素が挙げられる。
【0027】
炭化水素基としては、アルキル基、アルケニル基、アリール基が挙げられる。
アルキル基としては、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基、ヘキシル基、2−エチルヘキシル基、オクチル基、デシル基、オクタデシル基等の直鎖状または分岐状アルキル基; シクロペンチル基、シクロヘキシル基、ノルボルニル基等のシクロアルキル基; ベンジル基、フェニルエチル基、フェニルプロピル基等のアリールアルキル基が挙げられる。
アルケニル基としては、ビニル基、プロペニル基、シクロヘキセニル基が挙げられる。
アリール基としては、フェニル基、トリル基、ジメチルフェニル基、トリメチルフェニル基、エチルフェニル基、プロピルフェニル基、ビフェニル基、ナフチル基、メチルナフチル基、アントリル基、フェナントリル基等が挙げられる。
【0028】
酸素含有基としては、アルコキシ基、アリールオキシ基が挙げられる。
アルコキシ基としては、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、ヘキシルオキシ基、オクチルオキシ基、ベンジルオキシ基、2−フェニルエトキシ基等が挙げられる。
アリールオキシ基としては、フェノキシ基、トリルオキシ基、ビフェニルオキシ基、ナフチルオキシ基等が挙げられる。
【0029】
ケイ素含有基としては、アルキルシリル基、アルケニルシリル基、アリールシリル基、アルキルシロキシ基、アルケニルシロキシ基、アリールシロキシ基、アルコキシシリル基、アリールオキシシリル基、アルコキシシロキシ基、アリールオキシシロキシ基等が挙げられる。
【0030】
また上記の炭化水素基、酸素含有基、およびケイ素含有基は、1つ以上のヘテロ原子を含んでいてもよい。具体的には、これらの基の少なくとも一つの水素が、ハロゲン原子、酸素、窒素、ケイ素、リン、イオウを含む基で置換された基が含まれる。
【0031】
一般式(IX)のRaとして上記に挙げた原子または基のうち、水素原子、アルキル基、アルコキシ基、アリール基、およびケイ素含有基からなる群から選ばれる1種以上の原子または基であることが好ましく、なかでも、水素原子; メチル基、エチル基、プロピル基、ブチル基等の炭素原子数1〜4のアルキル基; メトキシ基、エトキシ基、プロポキシ基、ブトキシ基等の炭素原子数1〜4のアルコキシ基; フェニル基、ナフチル基等のアリール基; で表される基からなる群から選ばれる1種以上の原子または基であることがより好ましい。この中でも、メトキシ基およびエトキシ基が特に好ましい。
【0032】
[工程1]で使用する二塩化白金は、特に制限されないが粒径は1000μm以下が好ましく、更には500μm以下が好ましい。粒径が大きくなると調製時間が長くなる。
[工程1]におけるヒドロシランと二塩化白金の使用量は、ヒドロシラン量が二塩化白金に対し等量以上であれば特に制限されないが、好ましくは2倍当量以上である。ヒドロシランの量が少ないと、触媒調製上必要な攪拌が困難になる。
【0033】
[工程1]におけるヒドロシランと二塩化白金との混合攪拌は、これが可能であれば手段は問わないが、窒素気流下、攪拌機を備えた反応容器中に二塩化白金を適当量仕込み、予め設定したヒドロシランを添加して攪拌を行う。少量の場合はサンプル管にスターラーチップを入れ、同様に仕込んで攪拌しても良い。
【0034】
ヒドロシランと二塩化白金との混合攪拌時間は、通常60時間以上であり、70時間以上が好ましく、80時間以上が更に好ましい。反応時間が短いと、[工程2]で得られるシリル化ポリオレフィン(I)中の不純物である異性体のビニレン誘導体の生成割合が増大するため好ましくない。混合攪拌時間の上限は特に無いが、経済的な観点から概ね1ヶ月以内である。
【0035】
ヒドロシランと二塩化白金との混合攪拌の温度は、ヒドロシランの沸点以下であれば特に制限は無いが、通常0〜50℃の範囲、好ましくは10〜30℃の範囲である。また圧力は、通常は常圧で行うことができるが、必要に応じて加圧下または減圧下で行うこともできる。
【0036】
[工程1]においては反応で得られた懸濁溶液を濾過して固形分を除去し、濾液として白金触媒組成物(C)を得る。濾過で使用するフィルターは10μmより小さな目のフィルターを使用することが好ましく、1μm以下の目のフィルターを使用することが更に好ましい。これより大きな目のフィルターを使用すると、未反応の二塩化白金の固形分が触媒中に混入し、触媒が不均一化するため、合成目的物の不純物であるビニレン誘導体の生成量が増大する原因となる。また濾過の際、上記の溶媒を使用して固形分を洗浄することもできる。
濾過で除去される固形分、すなわち未反応の二塩化白金の量は、使用した二塩化白金の量に対して50%以下、好ましくは10%以下である。二塩化白金の反応率は、調製時間によって調節することができる。
【0037】
このようにして調製した白金触媒組成物(C)には、ナノコロイド状になった白金化合物、ヒドロシランが含まれる。この白金触媒組成物(C)は、そのままで次の[工程2]に用いることができるが、前記一般式(IX)のヒドロシランを添加して希釈し、触媒濃度を調整することもできる。
【0038】
[工程2]末端不飽和ポリオレフィンとヒドロシランとを反応させる工程
[工程2]では、前記[工程1]で得られた白金触媒組成物(C)の存在下、一般式(X)で表される末端不飽和ポリオレフィンと一般式(IX)で表されるヒドロシランとを反応させ、一般式(I)で表されるシリル化ポリオレフィンを得る。
【0039】
一般式(X)で表される末端不飽和ポリオレフィンと反応させるヒドロシランは、前記一般式(IX)で表される化合物である。
前記一般式(IX)中、Raは、[工程1]で示したものと同様の原子または基である。[工程2]で用いる一般式(IX)で表されるヒドロシランは、前記[工程1]で用いたヒドロシランと異なるものを用いることもできるが、好ましくは[工程1]で用いたものと同一のものを用いる。
【0040】
一般式(X)で表される末端不飽和ポリオレフィンと一般式(IX)で表されるヒドロシランとを反応させる際の量比は、目的によって異なるが、0.01〜10当量倍の範囲であり、好ましくは0.1〜2当量倍の範囲である。ここでヒドロシランの量とは、前記[工程1]で用い、白金触媒組成物(C)中に含まれる部分と、[工程2]で新たに追加する部分との合算量である。前記[工程1]において必要なヒドロシラン量の全量を用いた場合には、[工程2]ではヒドロシランを追加することなく実施することもできる。
【0041】
上記末端不飽和ポリオレフィンとヒドロシランとの反応は、前記[工程1]で調製した白金触媒組成物(C)の存在下で行う。白金触媒組成物(C)と末端不飽和ポリオレフィンとの量比は、白金換算で10-10〜10-1当量倍の範囲であり、好ましくは10-7〜10-3当量倍の範囲である。
【0042】
上記末端不飽和ポリオレフィンとヒドロシランとの反応における反応方法としては、最終的に反応すればよく、その方法は限定されるものではないが、例えば以下のように行う。反応容器中に上記末端不飽和ポリオレフィンを装入し、窒素雰囲気下、ヒドロシランと白金触媒組成物を装入する。予め内温を上記末端不飽和ポリオレフィンの融点以上に昇温しておいた油浴中に、上記反応器をセットし攪拌する。反応後油浴を除いて室温に冷却し、得られた反応混合物をメタノールまたはアセトンなどの貧溶媒中に取り出し2時間攪拌する。その後、得られた固体をろ取し上記貧溶媒で洗浄し、60℃、2hPa以下の減圧下で乾燥させ、目的物を得ることが出来る。
【0043】
[工程2]における末端不飽和ポリオレフィンとヒドロシランとの反応は、反応温度を100〜200℃の範囲とすることが好ましく、反応させる末端不飽和ポリオレフィンの融点より高い温度で行うことがより好ましい。反応温度が100℃より低いと、反応効率が低下することがあるので好ましくない。また圧力は、通常は常圧で行うことができるが、必要に応じて加圧下または減圧下で行うこともできる。
【0044】
[工程2]においては、必要に応じて溶媒を使用することもできる。使用する溶媒は、原料のヒドロシランおよび末端不飽和ポリオレフィンに対して不活性なものが使用できる。常圧下で反応させる場合、反応させる末端不飽和ポリオレフィンの融点以上の沸点を有するものを使用するのが好ましい。使用できる溶媒の具体例は、例えばn -ヘキサン等の脂肪族炭化水素類、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、酢酸エチル等のエステル類、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン、メチルプロピルケトン等のケトン類、テトラヒドロフラン、1,4−ジオキサン等のエーテル類、クロロホルム、ジクロルエタン、トリクロルエタン、パークロルエタン等のハロゲン化炭化水素などが挙げられる。これらのうち、特にトルエン、キシレン等の芳香族炭化水素が好ましい。
【0045】
溶媒を使用する場合は溶媒の使用量は原料の溶解性に作用するが、原料に対し100質量倍以下が好ましく、より好ましくは20質量倍以下である。本発明では、無溶媒で実施することが最も好ましい。
【0046】
以上のように、白金触媒組成物(C)の存在下、末端不飽和ポリオレフィンとヒドロシランとを反応させることにより、一般式(I)で表されるシリル化ポリオレフィンを含む反応混合物が得られる。
上記反応後のシリル化ポリオレフィン(I)を含む反応混合物には、シリル化ポリオレフィン(I)の他に、未反応の末端不飽和ポリオレフィン、副生物であるビニレン誘導体が含まれている。および微量の未反応シランが含まれている。
【0047】
本発明の方法においては、[工程1]で得られた非常に高活性で高選択性の白金触媒組成物(C)を用いるため、[工程2]の末端不飽和ポリオレフィンとヒドロシランとの反応が効率よく進行する。このため、末端不飽和ポリオレフィンの二重結合の反応率は、通常90%以上、好ましくは95%以上であり、副生物であるビニレン誘導体の生成量は、シリル化ポリオレフィン(I)に対して、通常10重量%以下、好ましくは5重量%以下である。
【0048】
シリル化ポリオレフィン(I)は、上記反応混合物から、貧溶媒への再沈、またはスラッジングにより取り出すことができる。貧溶媒はシリル化ポリオレフィン(I)の溶解度が小さいものであれば選択することができ、好ましくは上記不純物が除けるものが良い。
【0049】
(II)無水マレイン化ポリオレフィンの製造方法
本発明の一般式(II)で示される重合体は、一般式(X)で表される末端不飽和ポリオレフィン、無水マレイン酸と(b)重合禁止剤とを、溶媒系または無溶媒系で反応させて製造できる。
【0050】
(b)重合禁止剤とは、特に限定されるものではないが、例えば、フェノール系重合禁止剤、ニトロソアミン系重合禁止剤、フェノチアジン等が挙げられる。
【0051】
フェノール系重合禁止剤としては、例えば、2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン、4−メトキシフェノール、2,5−ジ−t−ブチルハイドロキノン、メチルハイドロキノン、p−ベンゾキノン、t−ブチル−p−ベンゾキノン、2,5−ジフェニル−p−ベンゾキノン等が挙げられる。前記ニトロソ系重合禁止剤としては、例えば、N−ニトロソフェニルヒドロキシルアミン、トリス(N−ニトロソ−N−フェニルヒドロキシルアミナト)アルミニウム等が挙げられる。
【0052】
これらの中で、好ましくはフェノール系重合禁止剤であり、より好ましくは2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン、4−メトキシフェノール、2,5−ジ−t−ブチルハイドロキノン、メチルハイドロキノン及びp−ベンゾキノンであり、さらに好ましくは、2,6−ジ−t−ブチルフェノール、2,6−ジ−t−ブチル−4−メチルフェノール、ハイドロキノン及び4−メトキシフェノールである。
【0053】
また、一般式(II)で示される重合体の製造方法は、工業的にも使用可能であり、且つ、例えば一般式(X)で表される末端不飽和ポリオレフィンは、無水マレイン酸とほぼ定量的に反応する。本発明において、末端不飽和ポリオレフィンの変性率は80%以上である。また、該変性率は85%以上であることが好ましく、95%以上であることがより好適である。
【0054】
(III)マレイン化ポリオレフィンの製造方法
本発明の前記一般式(III)で示される重合体は、前記一般式(X)で示される末端不飽和ポリオレフィンと、重合禁止剤と、下記一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかとを、溶媒系または無溶媒系で反応させて製造できる。
【0055】
【化6】
【0056】
【化7】
【0057】
上記式中、Rb及びRcは、前記一般式(III)におけるものと同様である。
さらに、一般式(XI)及び一般式(XII)で示される化合物は、反応性の二重結合を有する化合物であり、且つ、互いにシス−トランスの関係にある幾何異性体である。一般式(XI)および一般式(XII)におけるRb及びRcが環構造を形成する時、下記一般式(XIII)で示される構造となる。
【0058】
【化8】
【0059】
上記式中、Rsは、酸素原子、−NR基を表す(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)。
【0060】
これら一般式(XI)及び一般式(XII)で示される化合物は、単独で用いても、あるいは2種類以上混合して用いてもよい。これら化合物の使用量は、特に制限されるものではないが、好ましくは用いる末端不飽和ポリオレフィンの質量の0.01倍乃至50倍の範囲、より好ましくは、0.1倍乃至20倍の範囲、さらに好ましくは、0.5倍乃至5倍の範囲である。
【0061】
一般式(III)で示される重合体を製造するに際し、反応は無溶媒でも、あるいは溶媒を用いてもどちらでもよい。用いる溶媒としては本発明を阻害しない限り特に限定されるものではないが、例えば、n−ヘキサン等の脂肪族炭化水素類;シクロヘキサン等の脂環式炭化水素類;トルエン、キシレン、メシチレン等の芳香族炭化水素類;酢酸エチル等のエステル類;アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン等のケトン類;テトラヒドロフラン、1,4−ジオキサン等のエーテル類;クロロホルム、ジクロルエタン、トリクロロエタン、パークロロエタン等のハロゲン化炭化水素類;クロロベンゼン、o−ジクロロベンゼン等のハロゲン化芳香族類;ニトロベンゼン等の置換芳香族類;などが挙げられる。
【0062】
これらの中で、好ましくは、脂肪族炭化水素類、脂環式炭化水素類、芳香族炭化水素類、ハロゲン化炭化水素類、ハロゲン化芳香族類であり、より好ましくは、芳香族炭化水素類、ハロゲン化芳香族類であり、さらに好ましくはメシチレン、o−ジクロロベンゼンが挙げられる。これら溶媒は、単独で用いても、二種類以上混合して用いてもよい。
【0063】
上記溶媒の使用量は、原料の溶解性によるが、好ましくは原料として用いる末端不飽和ポリオレフィンの質量の0.1倍乃至100倍の範囲、より好ましくは0.5倍乃至50倍の範囲、さらに好ましくは1倍乃至10倍の範囲である。
【0064】
反応は、例えば次のようにして行うことができる。反応器に前記一般式(X)で示される末端不飽和ポリオレフィン、重合禁止剤、一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかを仕込み昇温する。原料は一括で仕込んでも、または分割して適宜添加してもよい。この時、反応を促進させるために触媒等の添加物を使用してもよい。反応温度は、本発明を阻害しない限り特に制限されるものではないが、好ましくは室温乃至300℃の範囲、より好ましくは100℃乃至250℃の範囲、より好ましくは150℃乃至230℃の範囲で行われる。使用する化合物、溶媒によっては反応温度が沸点を超える場合があるためオートクレーブ等適切な反応装置を選択する。反応時間は、使用する末端不飽和ポリオレフィン、溶媒、重合禁止剤の量や反応性により変わるが、通常20時間乃至120時間である。
【0065】
反応の進行は、1H−NMRで確認できる。すなわち、測定サンプル管中で重合体を、ロック溶媒と溶媒を兼ねた重水素化−1,1,2,2−テトラクロロエタンに完全に溶解させた後、120℃において測定した。ケミカルシフトは、重水素化−1,1,2,2−テトラクロロエタンのピークを5.91ppmとして、他のピークのケミカルシフト値を決定した。
【0066】
例えば、エチレンのみからなる片末端二重結合含有重合体の場合、飽和末端におけるメチル基の3プロトン分のピークが0.80ppm乃至0.95ppm、末端二重結合の3プロトン分のピークが4.88ppm乃至5.05ppmに2プロトン分のピーク(x)、5.75ppm乃至5.90ppmに1プロトン分のピークとして観測される。反応が進行すると、4.88ppm乃至5.05ppmに観測される2プロトン分のピーク(x)と、5.75ppm乃至5.90ppmに観測される1プロトン分のピークとが消失し、反応進行に伴い二重結合のシフトが起こり、新たに5.20ppm乃至5.40ppmに1プロトン分のピーク(y)、5.50ppm乃至5.70ppmに1プロトン分のピークが観測され、二重結合が一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかで変性されたポリオレフィン重合体が生成する。
【0067】
この時、反応の転化率(U%)(本発明における「変性率」に相当)は、ピーク(x)およびピーク(y)のピーク積分値を各々SBおよびSDとすれば、下記式(α)にて算出される。
式(α):U(%)=SD×100/[(SB/2)+SD]
【0068】
前記製造方法は、工業的にも使用可能であり、且つ、原料の一般式(X)で表される末端不飽和ポリオレフィンは、一般式(XI)及び一般式(XII)で示される化合物の少なくともいずれかとほぼ定量的に反応する。本発明において、上記式(α)から求められる前記二重結合の極性基(一般式(XI)及び一般式(XII)で示される構造の基)による変性率は80%以上である。また、該変性率は85%以上であることが好ましく、95%以上であることがより好適である。
【0069】
反応後は、晶析操作、洗浄等の簡単な操作により、過剰の原料、溶媒、重合禁止剤等を除去して、目的とする本発明の第2の重合体(第1の重合体を得るための中間体化合物)を得ることができる。また、上記反応において、原料の末端不飽和ポリオレフィンの製造工程から単離せずに上記反応を実施することもできる。
【0070】
(IV)アミノ化ポリオレフィンの製造方法
本発明の一般式(IV)で示される重合体は、一般式(X)で表される末端不飽和ポリオレフィンをエポキシ化して、すなわち上記ポリオレフィンの末端の二重結合を酸化して、一般式(XIII)で示される末端にエポキシ基を含有する重合体を得る。
【0071】
【化9】
【0072】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表す)
かかるエポキシ化方法は特に限定されるものではないが、以下の方法を例示することができる。
【0073】
(1)過ギ酸、過酢酸、過安息香酸などの過酸による酸化
(2)チタノシリケートおよび過酸化水素による酸化
(3)メチルトリオキソレニウム等のレニウム酸化物触媒と過酸化水素による酸化
(4)マンガンポルフィリンまたは鉄ポルフィリン等のポルフィリン錯体触媒と過酸化水素または次亜塩素酸塩による酸化
(5)マンガンSalen等のSalen錯体と過酸化水素または次亜塩素酸塩による酸化
(6)マンガン−トリアザシクロノナン(TACN)錯体等のTACN錯体と過酸化水素による酸化
(7)タングステン化合物などのVI族遷移金属触媒と相間移動触媒存在下、過酸化水素による酸化
上記(1)〜(7)の方法の中でも、活性面で特に(1)および(7)の方法が好ましい。
【0074】
末端エポキシ基含有重合体の全片末端中のエポキシ含有率は1H-NMRによって決定される。例えば、エチレンのみからなる片末端二重結合含有重合体をエポキシ化して得られた末端エポキシ基含有重合体の場合、飽和末端におけるメチル基の3プロトン分のピーク(j)が0.65〜0.9ppm、エポキシ基付け根の3プロトン分のピーク(k)が1プロトンずつ2.3〜2.4ppm、2.6〜2.7ppm、2.8〜2.9ppmに観測される。エポキシ変性が十分でない場合は、末端二重結合の3プロトン分のピーク(l)が4.70〜5.0ppmに2プロトン、5.5〜5.8ppmに1プロトン観測される。各ピーク(j)、(k)および(l)のピーク面積を各々SC、SDおよびSEとすれば、エポキシ基含有率(Ep(%))は下記式にて算出される。
Ep(%)=SD×200/(SC+SD+SE)
【0075】
上記の方法で得られた末端エポシキシ基含有重合体を開環重合し一般式(IV)の構造単位を有する重合体を製造することができる。触媒、重合条件などについては、公知のアルキレンオキサイドの開環重合方法を利用することができ、例えば、大津隆行著,「改訂高分子合成の化学」,株式会社化学同人,1971年1月,p.172−180には、種々の単量体を重合してポリオールを得る例が開示されている。開環重合に用いられる触媒としては、上記文献に開示されたように、カチオン重合向けにAlCl3、SbCl5、BF3、FeCl3のようなルイス酸、アニオン重合向けにアルカリ金属の水酸化物またはアルコキシド、アミン類、フォスファゼン触媒、配位アニオン重合向けにアルカリ土類金属の酸化物、炭酸塩、アルコキシドあるいは、Al、Zn、Feなどのアルコキシドを用いることができる。ここで、フォスファゼン触媒としては、例えば、特開平10−77289号公報に開示された化合物、具体的には市販のテトラキス[トリス(ジメチルアミノ)フォスフォラニリデンアミノ]フォスフォニウムクロリドのアニオンをアルカリ金属のアルコキシドを用いてアルコキシアニオンとしたものなどが利用できる。
【0076】
上記触媒存在下、開始剤として水、アミン類、ジオール類、ポリオール類等の活性水素化合物を使用して、末端エポキシ基含有重合体のみを開環重合させることにより末端エポキシ基含有重合体のホモ重合体が得られ、末端エポキシ基含有重合体と他のアルキレンオキシドを開環重合させることにより共重合体を得ることができる。
【0077】
反応溶媒としては、末端エポキシ基含有重合体、アルキレンオキサイドに対して不活性なものが使用でき、n−ヘキサン、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、ジオキサン等のエーテル類、ジクロロベンゼン等のハロゲン化炭化水素などが挙げられる。
【0078】
触媒の使用量はホスファゼン触媒以外については原料の末端エポキシ基含有重合体eの1モルに対して、0.05〜5モルが好ましく、より好ましくは0.1〜3モルの範囲である。ホスファゼン触媒の使用量は、重合速度、経済性等の点から、末端エポキシ基含有重合体の1モルに対して1×10−4〜5×10−1モルが好ましく、より好ましくは5×10−4〜1×10−1モルである。
【0079】
反応温度は通常25〜150℃、好ましくは50〜110℃とし、反応時間は使用する触媒の量、反応温度、オレフィン類の反応性等の反応条件により変わるが、通常数分〜50時間である。
【0080】
開始剤として水またはジオール類を用いることにより、両末端に水酸基を有する重合体を得ることができる。また、予めアルキレンオキサイドを重合して得た特定の分子量のポリエーテルポリオールを開始剤として用いると所定の分子量の親水性単位を導入することが可能になり所望の物性を有する両末端に水酸基を有するブロック共重合体の製造が容易となる。
【0081】
ポリエーテルポリオールとしてはポリエチレングルコール、ポリプロピレングリコール、ポリテトラエチレングリコール等を挙げることができ中でもポリエチレングルコール、ポリプロピレングリコールが望ましい。
【0082】
本発明の一般式(IV)の官能基含有率は、全片末端の50%以上が好ましく、より好ましくは70%以上、更に好ましくは80%以上、更により好ましくは90%以上である。一般式(IV)の官能基含有率は1H-NMRによって決定される。例えば、エチレンのみからなる末端エポキシ基含有重合体を官能基化して得られた一般式(IV)化合物の場合、置換基付け根の3プロトン分のピーク(j)が1プロトンずつ3.2〜3.4ppm、3.4〜3.5ppm、3.6〜3.8ppmに観測される。官能基化が十分でない場合は、エポキシ基付け根の3プロトン分のプロトンピーク(k)が1プロトンずつ2.3〜2.4ppm、2.6〜2.7ppm、2.8〜2.9ppmに観測される。各ピーク(j)および(k)のピーク面積を各々SDおよびSEとし、原料の末端エポキシ基含有重合体のエポキシ基含有率をEp(%)とすれば、一般式(IV)の官能基含有率(Fc(%))は下記式にて算出される。
Fc(%)=SD/(SD+SE)×Ep(%)
【0083】
(1)一般式(IV)においてRe、Rdの一方が水酸基である重合体の製造方法を示す:
[(1a)Re、Rdの一方が水酸基で、他方が一般式(V)で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体に、酸または塩基触媒存在下、一般式(XIV)
【0084】
【化10】
【0085】
(式中、X、Rfは一般式(V)と同様の原子または基を表す)で示される化合物(以下、反応剤Aと表記する)を反応させることにより、Re、Rdの一方が水酸基で、他方が一般式(V)で示される基である重合体を得ることができる。
一般式(XIV)としては、例えば、水、メタノール、エタノール、プロパノール、オクタノール、アリルアルコール、シクロヘキサノール、プロペニルアルコール、ヘキセノール、ブロモデカノール、トリフルオロエタノール、ヘキサフルオロ-2-プロパノール、パーフルオロオクタノール、ベンジルアルコール、ジフルオロベンジルアルコール、ペンタフルオロフェニルメタノール、ビス(4-メトキシフェニル)メタノール、フェネチルアルコール、フェニルプロピルアルコール、フェノール、ジクロロフェノール、メトキシフェノール、メトキシカルボニルフェノール、ニトロフェノール、ヘキサフルオロフェノール、メチルフェノール、ジメチルフェノール、ナフチルアルコール等のアルコール類、グリセリン、ブタントリオール、ペンタエリスリトール等の多価アルコール類、チオメタノール、チオエタノール等のチオアルコール類、酢酸、プロピオン酸、マレイン酸、コハク酸、マロン酸、ブタン酸、ヘキサン酸、オクタン酸、アクリル酸、メタクリル酸、安息香酸、トリフルオロメチル安息香酸、ニトロ安息香酸、フタル酸、ナフチル酸、パーフルオロヘプタン酸、パーフルオロオクタン酸等のカルボン酸類、モノエチレングリコール、ジエチレングリコール、トリエチレングリコール、ポリエチレングリコール、フェニルエチレングリコール、モノプロピレングリコール、ジプロピレングリコール、トリプロピレングリコール、ポリプロピレングリコール、プロパンジオール、クロロプロパンジオール、ブロモプロパンジオール、メトキシプロパンジオール、アリルオキシプロパンジオール、ブタンジオール、ヘキサンジオール、シクロヘキサンメタンジオール等のポリアルキレングリコール類を挙げることができる。これらは単独で用いてもよく2種以上の混合物でもよい。ポリエチレングリコール、ポリプロピレングリコールについては、二官能性、三官能性、四官能性の化合物を全て含むものとする。
【0086】
酸触媒としては例えば、塩酸、硫酸、リン酸等の鉱酸類、p-トルエンスルホン酸等のスルホン酸類、アンバーリスト-15(登録商標)等の固体酸類、三フッ化ホウ素エーテル錯体、三塩化ホウ素、三臭化ホウ素、三塩化アルミニウム、三臭化アルミニウム、四塩化スズ、二塩化亜鉛等のルイス酸を挙げることができる。
【0087】
塩基触媒としては例えば、リチウム、ナトリウム、カリウム、セシウム等のアルカリ金属の水酸化物、炭酸塩、炭酸水素塩、マグネシウム、カルシウム等のアルカリ土類金属の水酸化物、炭酸塩、炭酸水素塩、ピリジン、4-ジメチルアミノピリジン、トリエチルアミン等の有機アミン類、アンバーリスト-21(登録商標)、アンバーリスト-93(登録商標)等の弱塩基性イオン交換樹脂等が挙げられる。
酸または塩基触媒の使用量は、末端エポキシ基含有重合体に対して、0.01〜10質量倍が好ましく、より好ましくは0.1〜5質量倍、最も好ましくは0.5〜2質量倍である。これらの酸または塩基触媒は単独で用いてもよいし、2種以上を混合して用いても構わない。
【0088】
反応溶媒としては、原料の末端エポキシ基含有重合体に対して不活性なものが使用でき、例えばn-ヘキサン等の脂肪族炭化水素類、シクロヘキサン等の脂環式炭化水素類、トルエン、キシレン等の芳香族炭化水素類、酢酸エチル等のエステル類、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジエチルケトン、メチルプロピルケトン等のケトン類、テトラヒドロフラン、1,4-ジオキサン等のエーテル類、クロロホルム、ジクロルエタン、トリクロルエタン、ジクロロベンゼン等のハロゲン化炭化水素などが挙げられる。原料の末端エポキシ基含有重合体がその溶媒に対して不溶でない限り、トルエン、キシレン等の芳香族炭化水素が好ましい。溶媒の使用量は原料の溶解性に作用するが、原料の末端エポキシ基含有重合体に対し0.8〜100質量倍が好ましく、より好ましくは1〜50質量倍、更に好ましくは2〜20質量倍である。
【0089】
反応は、例えば次のようにして行うことができる。反応器に、末端エポキシ基含有重合体、反応剤A、酸または塩基触媒を入れて混合し、均一に溶解するまで昇温する。ここで反応剤Aをあらかじめアルカリ金属またはアルカリ土類金属の塩として使用してもよい。反応温度は用いる末端エポキシ基含有重合体が溶解する温度が好ましい。反応温度は、25〜300℃が好ましく、より好ましくは50〜250℃、更に好ましくは80〜200℃である。使用する化合物、溶媒によっては反応温度が沸点を超える場合があるためオートクレーブ等適切な反応装置を選択する。反応時間は使用する触媒の量、反応温度、重合体類の反応性等の反応条件により変わるが、通常数分から50時間である。
【0090】
反応後は晶析操作、洗浄等の簡単な操作により、過剰の触媒、反応剤A、反応溶媒を除去して目的とする重合体を得ることができる。上記反応において、原料の末端エポキシ基含有重合体の製造工程から単離精製せずに上記反応を実施することもできる。
また、一般式(IV)においてRd、Reともに水酸基である重合体の製造方法としては、アルコール等の相溶化溶媒の共存下、末端エポキシ基含有重合体と水を反応させる方法が好ましい。
【0091】
[(1b)Rd、Reの一方が水酸基で、他方が一般式(VI)で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体に、一般式(XV)
【0092】
【化11】
【0093】
(式中、Rg,Rhは一般式(VI)と同様の原子または基を表す)で示される化合物を反応させることにより、Rg,Rhの一方が水酸基で、他方が一般式(VI)で示される基である重合体を得ることができる。反応は、酸または塩基触媒を共存させてもよい。
一般式(XV)としては、例えば、アンモニア、メチルアミン、エチルアミン、メチルプロピルアミン、エタノールアミン、ジエタノールアミン、エチルプロピルアミン、ブチルアミン、デシルアミン、オクタデシルアミン、ピロリジン、ピペリジン、ピペラジン、ヘキサメチレンイミン、エチレンジアミン、ジアミノプロパン、ジアミノブタン、ジエチレントリアミン、N-(アミノエチル)プロパンジアミン、イミノビスプロピルアミン、スペルミジン、スペルミン、トリエチレンテトラミン、シクロプロピルアミン、シクロブチルアミン、N-メチルシクロヘキシルアミン、ジアミノシクロヘキサン、ベンジルアミン、トリス(アミノプロピル)アミン、トリス(アミノエチル)アミン、アミノメチルヘプタンジアミン、アニリン、クロロアニリン、トルイジン、アミノフェノール、メチレンジアニリン、フェニレンジアミン、アミノナフタレン、ジェファーミン類(登録商標)等を挙げることができる。ジェファーミン(登録商標)としては末端にアミノ基を含有するポリアルキレングリコール類全てを含むものとする。ジエタノールアミンが中でも好ましい
酸、塩基触媒とその使用量、反応溶媒とその使用量については、(1a)と同様である。
反応方法は、(1a)の場合と同様に行うことができるが、酸、塩基触媒の非存在下でも反応は進行する。
【0094】
[(1c)Rd、Reの一方が水酸基で、他方がシアノ基、カルボキシル基、エステル基、アミド基で示される基である重合体の製造方法]
原料となる末端エポキシ基含有重合体にシアノ化剤を反応させることにより、Rd、Reの一方が水酸基で、他方がシアノ基の重合体が得られる。
得られたシアノ基を含む重合体を加水分解によりカルボキシル基に誘導できる。更にこのカルボキシル基をエステル化することによりエステル基に誘導でき、アミド化することによりアミド基に誘導できる。これらの加水分解、エステル化、アミド化は、一般的な方法を用いる事ができる。
【0095】
シアノ化剤としてはシアン化ナトリウム、シアン化カリウム、トリメチルシリルシアニド、ジエチルアルミニウムシアニド、アセトンシアノヒドリン等を挙げることができる。シアノ化剤の使用量は原料の末端エポキシ基含有重合体の0.9〜20質量倍が好ましく、より好ましくは1〜10質量倍、更に好ましくは1.1〜10質量倍である。
反応方法は、(1a)の場合と同様に行うことができる。この場合、酸、塩基触媒を用いないで反応を行うことができる。
反応溶媒とその使用量については(1a)と同様である。
【0096】
[(2)一般式(IV)においてRd、Reの一方がポリエチレングリコール基である重合体の製造方法]
一般式(IV)においてRdまたはReの一方が水酸基の官能基含有重合体(以下、重合体Hと表記する)を原料とし、該水酸基にエポキシ化合物を反応させることにより、一般式(IV)においてRd、Reの一方がポリエチレングリコール基である重合体を得ることができる。
上記水酸基に付加重合するエポキシ化合物としては、プロピレンオキシド、エチレンオキシド、1,2-ブチレンオキシド、2,3-ブチレンオキシド、スチレンオキシド、シクロヘキセンオキシド、エピクロロヒドリン、エピブロモヒドリン、メチルグリシジルエーテル、アリルグリシジルエーテル等が挙げられる。これらは2種以上併用してもよい。これらの中で、好ましくは、プロピレンオキシド、エチレンオキシド、1,2-ブチレンオキシド、2,3-ブチレンオキシド、スチレンオキシドである。より好ましくはプロピレンオキシド、及びエチレンオキシドである。
【0097】
本反応に用いる触媒としては、例えばアルカリ金属水酸化物が挙げられる。また、ホスファゼニウム化合物、ホスフィンオキシド化合物、及びホスファゼン化合物(以下、P=N結合を有する化合物と表記する)を用いることもできる。
アルカリ金属水酸化物としては、例えば水酸化カルシウム、水酸化ナトリウム、水酸化リチウム、水酸化ルビシウム、水酸化セシウム等が挙げられる。
【0098】
ホスファゼニウム化合物としては、例えば、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムヒドロキシド、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムメトキシド、テトラキス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフォニウムエトキシド、テトラキス[トリ(ピロリジン-1-イル)ホスフォラニリデンアミノ]ホスフォニウムtert-ブトキシド等が挙げられる。
ホスフィンオキシド化合物としては、例えば、トリス[トリス(ジメチルアミノ)ホスフォラニリデンアミノ]ホスフィンオキシド、又はトリス[トリス(ジエチルアミノ)ホスフォラニリデンアミノ]ホスフィンオキシド等が挙げられる。
ホスファゼン化合物としては、例えば、1-tert-ブチル-2,2,2-トリメチルホスファゼン、1-(1,1,3,3-テトラメチルブチル)-2,2,4,4,4-ペンタイソプロピル-2λ5,4λ5-カテナジ(ホスファゼン)、1-tert-ブチル-2,2,2-トリアリルホスファゼン、1-シクロヘキシル-2,2,4,4,4-ペンタアリル-2λ5,4λ5-カテナジ(ホスファゼン)、1-エチル-2,4,4,4-トリベンジル-2-トリベンジルホスフォラニリデンアミノ-2λ5,4λ5-カテナジ(ホスファゼン)、1-メチル-2,2,2-トリシクロペンチルホスファゼンまたは1-プロピル-2,2,4,4,4-シクロヘキシル-2λ5,4λ5-カテナジ(ホスファゼン)等が挙げられる。
【0099】
触媒であるアルカリ金属水酸化物の使用量は原料の重合体Aの1モルに対して、0.05〜0.5モルが好ましく、より好ましくは0.1〜0.3モルの範囲である。
触媒であるP=N結合を有する化合物の使用量は、重合速度、経済性等の点から、原料の重合体Hの1モルに対して1×10-4〜5×10-1モルが好ましい。より好ましくは5×10-4〜1×10-1モル、更に好ましくは1×10-3〜1×10-2モルである。
【0100】
原料である重合体Hにエポキシ化合物を付加重合する温度は、重合速度、副反応抑制の点から、15〜130℃が好ましい。より好ましくは40〜120℃、更に好ましくは50〜110℃の範囲である。エポキシ化合物の付加重合温度を上記範囲内の低い温度で行う場合は、原料の重合体Hに対するP=N結合を有する化合物の濃度を先に述べた範囲内で高めることが好ましい。
エポキシ化合物の付加重合反応の圧力は、副反応抑制の点から、882kPa以下が好ましい。通常、耐圧反応器内でエポキシ化合物の付加重合が行われる。エポキシ化合物の反応は減圧状態から開始しても、大気圧の状態から開始してもよい。大気圧の状態から開始する場合には、窒素、又は、ヘリウム等の不活性気体存在下で行うことが望ましい。反応圧力は、より好ましくは686kPa以下、更に好ましくは490kPa以下である。エポキシ化合物としてプロピレンオキシドを用いる場合には、反応圧力は490kPa以下が好ましい。
【0101】
反応におけるエポキシ化合物の供給方法は、必要量のエポキシ化合物の一部を一括して供給し、残部を連続的に供給する方法、又は、全てのエポキシ化合物を連続的に供給する方法等が用いられる。必要量のエポキシ化合物の一部を一括して供給する方法においては、エポキシ化合物の重合反応初期の反応温度は、上記温度範囲内でより低温側とし、エポキシ化合物の装入後に、次第に反応温度を上昇する方法が好ましい。
エポキシ化合物としてプロピレンオキシド及びエチレンオキシドを併用する場合の重合方法には、(a)プロピレンオキシドを重合した後、エチレンオキシドをブロックで共重合するエチレンオキシドキャップ反応、(b)プロピレンオキシドとエチレンオキシドをランダムに共重合するランダム反応、(c)プロピレンオキシドを重合した後、エチレンオキシドを重合し、次いで、プロピレンオキシドを重合するトリブロック共重合反応が挙げられる。これらの中で好ましい重合方法は、エチレンオキシドキャップ反応とトリブロック共重合反応である。
【0102】
付加重合器の最大圧力は、エポキシ化合物の装入速度、重合温度、触媒量等に影響される。エポキシ化合物の装入速度は、付加重合機の最大圧力が882kPaを超えないように制御することが好ましい。エポキシ化合物の装入が完了すると、付加重合器の内圧は徐々に低下する。内圧の変化が認められなくなるまで付加重合反応を継続することが好ましい。ポリアルキレングリコール基を含有する重合体の水酸基価(OHV)を基準とすると、OHVが2〜200mgKOH/gとなるまで付加重合を継続することが好ましい。
エポキシ化合物の付加重合反応に際して、溶媒を使用することもできる。溶媒としては、例えば、ペンタン、ヘキサン、ペプタン等の脂肪族炭化水素類、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン等のエーテル類、ジメチルスルホキシド、N,N-ジメチルホルムアミド等の非プロトン性極性溶媒等が挙げられる。
【0103】
次に、上記のようにして製造されたポリアルキレングリコール基を含有する重合体の精製方法について説明する。ポリアルキレングリコール基を含有する粗製重合体中に残存するアルカリ金属水酸化物またはP=N結合を有する化合物は、塩酸、リン酸等の鉱酸類、酢酸等の有機酸、炭酸ガス等による中和、吸着剤による吸着除去、水あるいは水/有機溶媒を用いた水洗、イオン交換樹脂によるイオン交換等の方法により除去することができる。
一般式(IV)においてRd、Reの一方がポリエチレングリコール基であり、他方が一般式(V)で表され、かつRfが一般式(VIII)で表される重合体の製造方法としては、まず、原料となる末端エポキシ基含有重合体と一般式(XVI)
【0104】
【化12】
【0105】
(式中、Eは酸素原子または硫黄原子を表し、R1aはm+1価の炭化水素基を表し、Tは同一または相異なり水酸基、アミノ基を表し、mは1〜50の整数を表す)
で示される反応剤A’とを(1a)に記載した方法により反応させ、一般式(IV)においてRd、Reの一方が水酸基、他方が下記一般式(XVII)で示される基で表される重合体を得る。
【0106】
【化13】
(式中、E、R1a、T、mは一般式(XVI)の定義と同様である)
【0107】
得られた重合体の水酸基およびTで表される水酸基またはアミノ基に、上述の方法によりエポキシ化合物を重合させることにより得ることができる。
一般式(IV)においてRd、Reの一方がポリエチレングリコール基であり、他方が一般式(VI)で表され、かつRg、Rhが一般式(VIII)で表される重合体の製造方法としては、まず、原料となる末端エポキシ基含有重合体と一般式(XVIII)
【0108】
【化14】
【0109】
(式中、R1aは同一または相異なりm+1価の炭化水素基を表し、Tは同一または相異なり水酸基、アミノ基を表し、mは1〜50の整数を表す)
で示される反応剤A’’とを(1a)に記載した方法により反応させ、一般式(IV)においてRd、Reの一方が水酸基、他方が下記一般式(XIX)で示される基で表される重合体を得る。
【0110】
【化15】
(式中、R1a、T、mは一般式(XVIII)の定義と同様である)
【0111】
得られた重合体の水酸基およびTで表される水酸基またはアミノ基に、上述の方法によりエポキシ化合物を重合させることにより得ることができる。
反応試剤A’としては、グリセリン、ペンタエリスリトール、ブタントリオール、ジペンタエリスリトール、ポリペンタエリスリトール、ジヒドロキシベンゼン、トリヒドロキシベンゼン等を挙げることができる。
反応試剤A’’としては、アミノフェノール、ヘキサメチレンイミン、エチレンジアミン、ジアミノプロパン、ジアミノブタン、ジエチレントリアミン、N-(アミノエチル)プロパンジアミン、イミノビスプロピルアミン、スペルミジン、スペルミン、トリエチレンテトラミン、ポリエチレンイミン等を挙げることができる。
【0112】
[(3)一般式(IV)においてRd、Reの一方がアシルオキシ基である重合体の製造方法]
前記重合体Hを原料とし、該水酸基等をアシル化することにより、一般式(IV)においてRd、Reの一方がアシルオキシ基である重合体を得ることができる。アシル化は、重合体Hと、対応する酸ハロゲン化物あるいは酸無水物を塩基触媒存在下反応させる一般的な方法で実施できる。
【0113】
酸ハロゲン化物としては、例えば塩化アセチル、臭化プロピオニル、塩化アクリロイル、塩化メタクリロイル、臭化ヘキサノイル、ヨウ化オクタノイル、塩化ベンゾイル、ヨウ化4-トリフルオロメチルベンゾイル、臭化3-ニトロベンゾイル、塩化ナフトイル、臭化パーフルオロヘプテノイル、ヨウ化パーフルオロオクテノイル等を挙げることができる。
酸無水物としては、無水酢酸、無水プロピオン酸、無水アクリル酸、無水メタクリル酸、無水フタル酸、無水マレイン酸、無水コハク酸等を挙げることができる。
塩基触媒としては(1a)で例示した触媒を挙げることができる。
前記式のPOで表される重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることが好ましい。
【0114】
(樹脂組成物)
本発明の樹脂改質用添加剤は、各種熱可塑性樹脂に添加して用いることができる。本発明の樹脂改質用添加剤は、熱可塑性樹脂の伸長粘度を改善することができる
ベースとなる熱可塑性樹脂としてはポリオレフィンが好ましく、ポリプロピレンもしくはポリエチレンが好ましく、とりわけポリプロピレンが好ましい。
【0115】
本発明の樹脂改質用添加剤は、各種用途に用いることができるが、発泡性樹脂の改質用添加剤として特に好適に用いられる。本発明の樹脂改質用添加剤を用いると、均一かつ発泡倍率の大きい発泡セルが得られる点で好ましい。
添加処方としては、熱可塑性樹脂99〜60重量%に対し、樹脂用添加剤1〜40重量%が好ましい。
【0116】
発泡性樹脂の改質用添加剤として用いるに際しては、さらに発泡剤を含むことが出来る。発泡剤としては、二酸化炭素、窒素等のガス、プロパン、ブタン、ペンタン、ヘキサンおよびそれらの各構造異性体等の脂肪族炭化水素、塩化メチル、塩化エチル、テトラフロロエタン、ジフロロエタンなどのハロゲン化炭化水素、メタノール、エタノール、プロパノールなどのアルコール、ジメチルエーテル、ジエチルエーテル、メチルエチルエーテル等のエーテル、アルゴン、水等が挙げられる。また、本発明に前述の発泡剤の他に、気泡調節剤または発泡核剤を用いても良い。気泡調節剤または発泡核剤としては、例えば炭酸アンモニウム、重曹、重炭酸アンモニウム、亜硝酸アンモニウム等の無機系分解性発泡剤、アゾビスイソブチロニトリル、ジアゾアミノベンゼン、アゾジカルボンアミド等のアゾ化合物、N,N′−ジメチル−N,N′−ジニトロソテレフタルアミド、N,N′−ジニトロソペンタンメチレンテトラミン等のニトロソ化合物、p−トルエンスルホニルヒドラジド、p−トルエンスルホニルセミカルバジド、バリウムアゾジカルボキシレート等の分解性発泡剤、タルク、シリカ等の無機粉末、多価カルボン酸等の酸性塩、多価カルボン酸と炭酸ナトリウム又は重曹との反応混合物等が挙げられ、これらは、単独でも組み合わすこともできる。この場合、発泡性樹脂用添加剤0.1〜35重量%と、熱可塑性樹脂50〜99.8重量%と、発泡剤0.1〜15重量%とを含む組成が好ましい。組成はさらに発泡性樹脂用添加剤9〜30重量%と、熱可塑性樹脂重量60〜90%と、発泡剤1〜10重量%が好ましい
【0117】
熱可塑性樹脂、樹脂用添加剤、更に任意で発泡剤を含む組成物は、各成分をミキサー等で混合して得ることが出来る。得られた組成物を、押し出し成型など公知の成型法により、シート・フィルム・発泡シートなどに成型すればよい。
また、各成分所定量をそれぞれ押出し器に直接投入し、押出し機内で溶融混錬しても良い。更に発泡剤を発泡時に導入する場合は、密閉した系に溶融混練した組成物を導入し、発泡剤を圧入して射出成型、または押し出し成型しても良い。
【実施例】
【0118】
以下、実施例等により本発明をさらに具体的に説明するが、本発明の範囲はこれらの実施例等に限定されるものではない。
〔測定および計算方法〕
分子量、融点(Tm)、粒子径、収率、転化率および異性化率、伸長粘度、CO2含浸率、発泡倍率、発泡平均セル径等は以下に記載の方法で測定・計算した。
【0119】
[m1]分子量の測定方法
重量平均分子量(Mw)及び分子量分布(Mw/Mn)は、ミリポア社製GPC−150を用い以下のようにして測定した。すなわち、分離カラムは、TSK GNH HTであり、カラムサイズは直径7.5mm、長さ300mmのものを使用した。カラム温度は140℃とし、移動相にはオルトジクロロベンゼン(和光純薬)及び酸化防止剤としてBHT(武田薬品)0.025質量%を用い、1.0ml/分で移動させた。試料濃度は0.1質量%とし、試料注入量は500マイクロリットルとした。検出器として示差屈折計を用いた。標準ポリスチレンは東ソー社製を用いた。
【0120】
数平均分子量Mn、および重量平均分子量Mwは、市販の単分散標準ポリエチレンを用いて検量線を作成し、下記の換算法に基づいて求めた。
分子量換算 : PE換算/汎用較正法
なお、汎用較正の計算には、以下に示すMark−Houwink粘度式の係数を用いた。
ポリエチレン(PE)の係数 : KPE=5.06×10-4, aPE=0.70
【0121】
[m2]融点の測定方法
融点(Tm)はDSCを用い測定して得られたピークトップ温度を採用した。装置は島津製作所製DSC−60Aを使用した。対照セルはアルミナを使用し、窒素流量は50ml/分の設定で行った。また10℃/分で(30)℃から300℃までの昇温条件で測定した。この昇温測定の前に、一旦、樹脂を200℃程度まで昇温し、5分間保持した後、20℃/分で常温(25℃)まで降温する操作を行い、樹脂の熱履歴を統一することが望ましい。
【0122】
[m3]NMR解析による収率、転化率、異性化率、末端不飽和率、炭素千個あたりの二重結合数の測定・計算方法
シリル化ポリオレフィン[A]の収率、転化率、異性化率、末端不飽和率、炭素千個あたりの二重結合数は1H−NMRによって決定される。収率は原料の末端不飽和重合体のモル数に対して得られたシリル化ポリオレフィン[A]のモル数の割合、転化率は原料の末端不飽和重合体のモル数に対する同消費モル数の割合、異性化率は原料の末端不飽和重合体のモル数に対して生成したビニレン体のモル数の割合、末端不飽和率は末端二重結合と末端メチルの合計に対する末端二重結合の割合、炭素千個あたりの二重結合数はプロトン数から導き出される炭素数に対する二重結合数の割合を炭素千個あたりの二重結合数に補正したものと定義される。
【0123】
例えば、エチレンのみからなる末端不飽和重合体をトリエトキシシランでヒドロシリル化して得られたシリル化ポリオレフィン[A]のエトキシ基メチレンの6プロトン分のピーク(C)が3.8ppm、異性化したビニレン基の2プロトン分のピーク(D)が5.4ppmに観測される。ヒドロシリル化が十分でない場合は、未反応ビニル基の2プロトン分のピーク(E)が4.8〜5.1ppmに、1プロトン分のピーク(F)が5.6〜5.8ppmに観測される。原料の二重結合含有重合体については、2プロトン分の主鎖メチレン(G)が1.0〜1.5ppmに観測され、末端に二重結合を持たないものは3プロトン分の末端メチル(H)が0.8ppmに観測される。さらに二重結合に隣接した炭素上の2プロトン分のピーク(I)が1.9ppmに観測される。
【0124】
各ピーク(C)、(D)、(E)、(F)、(G)、(H)および(I)のピーク面積を各々SC、SD、SE、SF、SG、SHおよびSIとすれば、収率(YLD(%))、転化率(CVS(%))、異性化率(ISO(%))、末端不飽和率(VE(%))、炭素千個あたりの二重結合数(VN(個/1000C))は下記式にて算出される。
YLD(%)=(SC/3)/(SC/3+SD+SE)×100
CVS(%)={1−SE/(SC/3+SD+SE)}×100
ISO(%)=SD/(SC/3+SD+SE)×100
VE(%)=(SE/2)/(SE/2+SH/3)×100
VN(個/1000C)=(SE+SF)/3×1000/{(SD+SE+SF+SG+SH+SI)/2}
【0125】
[m4]伸長粘度測定方法
伸長粘度測定は、Rheometric Scientific社製 RMEエロンゲーショーナルレオメーター(RME−SS−RC−SD−BH)を使用し、付属の解析装置にて行った。
評価サンプルは、対象樹脂を110×7×2mmの形状に熱プレスにより成型し用いた。
【0126】
[m5]CO2含浸率測定方法
熱可塑性樹脂シ−ト(5cm×5cm×2mm厚み)を内容積1Lの高圧容器に入れ、液化炭酸ガスを注入し、超臨界状態(40℃×20MPa)で3時間放置後、高圧容器から熱可塑性樹脂シ−トを取り出し、電子天秤で経時による重量変化を測定する。重量変化のグラフから、取り出し直後の炭酸ガス含浸の熱可塑性樹脂シ−ト重量を求め、次式により算出する。
(2)−(1)/(1)×100%
(1)=未発泡の熱可塑性樹脂シ−ト重量
(2)=炭酸ガス含浸の熱可塑性樹脂シ−ト重量
【0127】
[m6]発泡倍率測定方法
熱可塑性樹脂シ−トの比重を電子比重計(MD-200S:アルファ-ミラ-シ゛ュ製)を用いて測定し、発泡熱可塑性樹脂の比重を測定して、熱可塑性樹脂シ−トの比重に対する割合を算出し、小数点以下第2位を四捨五入した値を発泡倍率とした。
【0128】
[m7]発泡平均セル径測定方法
走査型電子顕微鏡により撮影した発泡熱可塑性樹脂の断面写真について、発泡セル径の画像処理により円相当径を算出し、その値を平均セル径とした。
【0129】
[m8]フィルムの機械物性(引張強度、引張伸度及び引張弾性率)測定方法
引張強度、引張伸度及び引張弾性率は引張試験機により評価した。引張試験機は、島津製作所社製;EZ−S 100N型を用いた。試験対象のフィルムを、全体長さ50mm、つかみ部分巾10mm、試験片部分巾5mm、試験片部分長さ30mmのダンベル型に打抜いた。この試験フィルムを引張試験のチャックにはさみ、30mm/分の速度で試験を行った。
【0130】
〔使用原料〕
実施例、比較例に使用した原料については、以下のものを使用した。なお、原料ポリマーのモル数はすべてMnに基づいた値で表した。
(1)塩化白金および塩化白金酸(試薬): アルドリッチ社製
(2)トリエトキシシラン(試薬): 関東化学社製
(3)無水マレイン酸(試薬): 和光純薬社製
(4)片末端ビニルを有するポリエチレン(P1): 特開2003−73412の実施例1記載の方法に準じて合成したもの(Mn=730、Mw/Mn=1.9、135℃デカリン中で測定した極限粘度[η]=0.08dl/g、融点(Tm)116℃、末端不飽和率96mol%(NMR基準))
【0131】
〔フィルム作製〕
実施例、比較例の引張試験に使用したフィルムについては、以下の通りに作製した。
マイクロコンパウンダー(DSM社製)を使用し、所定の混合比率のポリマーを、混練温度230℃で5分間混練、次いでTダイより射出し付帯のフィルム巻き取り機にて巻き取った。上記で得られたポリマー混合物からフィルムを作製した。この結果、厚さ約50μm、巾3cmのフィルムが得られた。
【0132】
[合成例1]
〔白金触媒組成物(C−1)の調製〕
マグネットスターラーチップを入れた50mlサンプル管中、塩化白金(II)0.50gをトリエトキシシラン(10ml)中に懸濁し、窒素気流下、室温で24時間攪拌した。所定時間攪拌した後、シリンジにて反応液を約0.4ml採取し、0.45μmPTFEフィルターを用いて濾過して10mlサンプル管中に濾液を採取した。得られた濾液をマイクロピペットにて10μl秤取って10mlサンプル管中に分取した後、トリエトキシシラン1.99mlを加えて200倍希釈し、白金触媒組成物(C−1)を得た。
【0133】
〔末端ビニルを有するポリエチレンへのアルコキシシラン導入〕
還流管の付いた1000mlの反応器に末端ビニルを有するポリエチレン(P1)200g(0.27mol)を装入し、窒素雰囲気下、トリエトキシシラン56ml(262mmol)と白金触媒組成物(C−1)3ml(Pt換算で2.8×10−3mmol(トリエトキシシラン14mmolを含む))を装入した。予め内温130℃に昇温しておいた油浴中に、上記反応器をセットし攪拌した。約20分後ポリマーは融解した。次いで1時間後に冷却し、アセトン約1500ml中に反応物を投入、2時間攪拌した。その後、固体をろ取しアセトンで洗浄し、60℃、2hPa以下の減圧下で乾燥させることにより、白色固体のポリマー(PE−TES)217gを得た。NMR解析の結果、得られたポリマーは収率88.2%、オレフィン転化率100%、異性化率11.8%だった。
【0134】
[合成例2]
〔末端ビニルを有するポリエチレンへのマレイン酸導入〕
還流管の付いた1000mlの反応器に、末端ビニルを有するポリエチレン(P1)200g(0.27mol)、無水マレイン酸134g(1.37mol)、及び塩化アルミニウム(III)0.5g(3.75mmol)を仕込み、195℃に昇温し、30時間加熱攪拌した。反応終了後、120℃に冷却した反応ポリマーにトルエン200gを加えて溶解させた後、アセトン(1000ml)に投入し晶析させた。その後、アセトン洗浄(1000ml×2回)し、減圧乾燥機で80℃、9時間乾燥させ、白色固体のポリマー(PE−MAH)203gを得た。NMR解析の結果、得られたポリマーはオレフィン転化率99.0%だった。
【0135】
[合成例3]
特開2006−131870号公報の合成例2に従って、Mw=2058、Mn=1118、Mw/Mn=1.84(GPC)の末端エポキシ基含有エチレン重合体(E1)(Mn 1118)を合成し、原料として用いた。
1000mLフラスコに、末端エポキシ基含有エチレン重合体(E1) 84 g(75mmol)、ジエタノールアミン39.4g(375mmol)、トルエン150g を仕込み、150℃にて4時間撹拌した。その後、冷却しながらアセトンを加え、反応生成物を析出させ、固体をろ取した。得られた固体をアセトン水溶液で1回、更にアセトンで3回撹拌洗浄した後、固体をろ取した。その後、室温にて減圧下乾燥させることにより、白色固体のポリマー(PE−DEA)87gを得た。
【0136】
[実施例1]
前記合成例1によって合成されたPE−TESとポリプロピレン(プライムポリマー社製;VP103W)について、PE−TESを全量の5重量%になるように混合し、マイクロコンパウンダー(DSM社製;DSM−Xplore)を使用し、温度230℃で5分混練、次いで900Nのトルク制御にて押し出してストランドを作製した後測定サンプルを成型した。次いで、[m4]伸長粘度測定法に従い伸長粘度を測定した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は489kPas(232s時)であった。また、別の測定サンプルを株式会社AKICO社製高圧容器(型式:特注型000819)中に静置し、CO2を20MPa、温度40℃に3時間保持(バッチ式発泡試験装置)を行った。次いで初期[m5]CO2含浸率を測定したところ、6.8%だった(表1)。
【0137】
[実施例2]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例1と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は7,470kPas(683s時)、初期CO2含浸率は4.8%だった(表1)。
【0138】
[比較例1]
ベースポリマーのポリプロピレン(プライムポリマー社製;VP103W)について、実施例1と同様にマイクロコンパウンダーを使用し、ストランドを作製した後測定サンプルを成型した。伸長粘度を測定した結果、180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は14kPas(79s時)、初期CO2含浸率は3.5%だった(表1)。
【0139】
[実施例3]
ベースポリマーのポリプロピレンをプライムポリマー社製;F102Wに換えた以外には、実施例1と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は876kPas(570s時)であった。また、別の測定サンプルを使用して、バッチ式発泡試験装置に従い、発泡倍率を測定したところ4.3倍だった(表1)。
【0140】
[実施例4]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は6,585kPas(683s時)、発泡倍率は3.8倍だった(表1)。
【0141】
[実施例5]
PE−TESを前記合成例3によって合成されたPE−DEAに換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は514kPas(476s時)、発泡倍率は3.9倍だった(表1)。
【0142】
[比較例2]
PE−TESを前記ポリプロピレン(プライムポリマー社製;VP103W)に換えた以外は実施例4と同様に操作した。180℃の恒温下、伸長速度0.01/s時の伸長粘度の最大値は52kPas(46s時)、発泡倍率は3.2倍だった(表1)。
【0143】
【表1】
【0144】
[実施例6]
前記合成例1によって合成されたPE−TESとポリプロピレン(プライムポリマー社製;F102W)について、PE−TESを全量の5重量%になるように混合し、図1に示すタンデム型の押出機に投入した。装置としては図1に示したスクリュウ径30mmφの第1押出機(8)とスクリュウ径40mmφの第2押出機(11)を有する。二酸化炭素添加部は、第1押出機の中央付近に設けた。該材料をホッパー(7)より第1押出機(8)に投入し、220℃で加熱溶融させた。二酸化炭素は、サイホン式の液化二酸化炭素ボンベ(1)を使用し、液相部分から直接取り出せるようにした。液化二酸化炭素ボンベ(1)からプランジャーポンプ(2)までの流路を冷媒循環機(4)を用いて、−12℃に調節したエチレングリコール水溶液で冷却し、二酸化炭素を液体状態でプランジャーポンプ(2)まで注入できるようにした。この時二酸化炭素の温度は、−5℃であった。次に注入した液状二酸化炭素を200g〜500g/時間となるようプランジャーポンプ(2)を制御し、プランジャーポンプ(2)の吐出圧力を30MPaとなるよう保圧弁(3)にて調整した。次に保圧弁(3)から第1押出機(8)の二酸化炭素添加部までのラインを50℃となるようヒーターで加熱し、二酸化炭素を第1押出機(8)内の溶融した該材料に添加した。このときの添加部の溶融樹脂圧力は20MPaであった。つまり、該溶融材料に溶解する直前の二酸化炭素は、温度が50℃以上、圧力が20MPaである超臨界状態の二酸化炭素となっている。このようにして、該溶融材料100重量部に対して超臨界二酸化炭素を5重量部の割合で第1押出機(8)内に添加し、スクリュウで均一に溶解させた。次にこの溶融物を第2押出機(11)へ送り、樹脂温度を165℃に調整し、2.1kg/時間の押出量でダイス(12)より押し出した。このときのダイス(12)圧力は、4.8MPaであった。
【0145】
ダイス(12)としては、出口隙間が、直径2.0mmφのストランドダイス(12)を使用した。押し出された該材料は、ダイス(12)から出たと同時に発泡し、ストランドとして放出される。二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率は15.9倍だった。
【0146】
また、ストランドダイス(12)の径を1.0mmφに換えて発泡させた場合、二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率は29.4倍だった(表2)。発泡セルの観察結果は他の試料よりもサイズが小さく比較的均一サイズで等方的な円または多角形だった。
【0147】
[実施例7]
PE−TESを前記合成例2によって合成されたPE−MAHに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率はそれぞれの場合で、16.9倍、27.6倍だった(表2)。発泡セルの観察結果は大きなセルが点在し、等方的な円または多角形だった。
【0148】
[実施例8]
PE−TESを前記合成例3によって合成されたPE−DEAに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動が小さく、二酸化炭素添加量、発泡倍率のいずれも変化なく、安定したストランドを得た。得られたストランドの発泡倍率はそれぞれの場合で、12.1倍、24.6倍だった(表2)。発泡セルの観察結果はセルサイズが大きく比較的均一サイズで等方的な円または多角形だった。
【0149】
[比較例3]
材料を前記ポリプロピレン(プライムポリマー社製;F102W)のみに換えた以外は実施例6と同様に操作した。ストランドダイス(12)の径を2.0mmφの場合と1.0mmφの場合、いずれも発泡時の二酸化炭素添加部の樹脂圧力の変動は小さかったが、径が1mmφの場合出口で詰まり気味であり、安定したストランドを得るのが困難であった。得られたストランドの発泡倍率はそれぞれの場合で、9.4倍、21.0倍だった(表2)。発泡セルの観察結果は断面の内側と外側でセルサイズ、形状共に不均一でやや扁平気味だった。
【表2】
【0150】
[実施例9]
前記合成例1によって合成されたPE−TES(変性率85%)とポリプロピレン(プライムポリマー社製;J715M)について、PE−TESを全量の10重量%になるようにマイクロコンパウンダー(DSM社製;DSM−Xplore)を使用し、温度230℃で5分混練、次いで前記〔フィルム作製〕方法に従い、900Nのトルク制御にて押し出しフィルムを作製した。 さらに[m8]フィルムの機械物性測定方法に従い、引張弾性率、引張伸度、破断点応力を測定した。引張弾性率1.0GPa、引張伸度480%以上、破断点応力37MPa以上だった。(表3)
【0151】
[実施例10]
PE−TES(変性率78%)に、含量を30%に換えた以外は実施例9と同様に操作した。引張弾性率1.0GPa、引張伸度490%以上、破断点応力31MPa以上だった。(表3)
【0152】
[実施例11]
PE−TES(変性率87%)10.7部と、未変性の末端ビニルを有するポリエチレン(P1)89.3部を混合して変性率10%のPE−TESとし、含量を10%として実施例9と同様に操作した。引張弾性率1.0GPa、引張伸度480%以上、破断点応力25MPa以上だった。(表3)
【0153】
[比較例9]
ポリプロピレン(プライムポリマー社製;J715M)のみを実施例9と同様に操作した。引張弾性率0.5GPa、引張伸度460%、破断点応力18MPaだった。(表3)
【0154】
[比較例10〜11]
PE−TESを未変性PEに、含量をそれぞれ10%、30%に換えた以外は実施例9と同様に操作した。引張弾性率、引張伸度、破断点応力を表3に示す。
【0155】
【表3】
【図面の簡単な説明】
【0156】
【図1】本発明の樹脂用添加剤を含む組成物を用い、発泡成型体を得る方法を示す
【符号の説明】
【0157】
(1)液化二酸化炭素ボンベ
(2)定量ポンプ
(3)保圧弁
(4)冷媒循環器
(5)ヒーター
(6)流量計
(7)ホッパー
(8)第1押出機
(9)スクリュウ
(10)連結部
(11)第2押出機
(12)ダイス
【特許請求の範囲】
【請求項1】
下記一般式(I)、(II)、(III)または(IV)で表される酸素含有基、窒素含有基およびケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。
【化1】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表し、Raは水素原子、ハロゲン原子、炭素数1〜10の炭化水素基、窒素含有基、酸素含有基またはケイ素含有基を表しそれぞれ同一でも異なっていてもよく、RbおよびRcはそれぞれ独立に、水素原子、ハロゲン原子、炭化水素基、−OR基、−SR基または−NR2基を表し(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)、環構造を形成してもよく、RdおよびReは一方が水酸基、ポリアルキレングリコール基またはアシルオキシ基を表し、他方は下記一般式(V)、一般式(VI) 、一般式(VII) のいずれかで示される基、シアノ基、カルボキシル基、エステル基またはアミド基を表す。nは平均官能基数を表し、0.1〜1.0である。)
【化2】
( 式中、Xは酸素原子または硫黄原子を表し、Rf は水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、下記一般式(VIII)
【化3】
( 式中、R1aは m+1 価の炭化水素基を表し、G は同一または相異なり、-OR1b 、-NR1 c R1d (R1b 、R1 c、R1dはポリアルキレングリコール基を表す) で表される基を表し、m は1〜 5 0 の整数を表す)で表される基を表す。)
【化4】
( 式中、Rg,Rh は同一または相異なり、水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、上記一般式(VIII)で表される基を表す。)
【化5】
( 式中、Ri、Rj、Rkは同一または相異なり、水素原子、炭化水素基、アシル基、シアノ基、カルボキシル基、エステル基、アミド基を表す。)
【請求項2】
前記極性基含有ポリオレフィン重合体を含有する樹脂組成物の伸長粘度最大値が、180℃恒温下、毎秒伸長速度0.01の測定で100kPa・s以上を示すことを特徴とする請求項1に記載の樹脂改質用添加剤。
【請求項3】
前記極性基含有ポリオレフィン重合体中の重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることを特徴とする請求項2に記載の樹脂改質用添加剤。
【請求項4】
前記樹脂改質用途が発泡性樹脂用であることを特徴とする請求項2ないし3に記載の樹脂改質用添加剤。
【請求項5】
請求項4に記載の発泡性樹脂用添加剤0.1〜15重量%と、熱可塑性樹脂70〜99.8重量%と、発泡剤0.1〜15重量%とを含む発泡性樹脂組成物。
【請求項6】
前記熱可塑性樹脂が、ポリエチレン、ポリプロピレン、ポリスチレン、ポリウレタンおよびポリカーボネートからなる群から選ばれる1種以上の樹脂であることを特徴とする請求項5に記載の発泡性樹脂組成物。
【請求項7】
請求項5ないし6に記載の発泡性樹脂組成物から得られる発泡成形体。
【請求項1】
下記一般式(I)、(II)、(III)または(IV)で表される酸素含有基、窒素含有基およびケイ素含有基からなる極性基が結合した構造を有することを特徴とする極性基含有ポリオレフィン重合体からなる樹脂改質用添加剤。
【化1】
(式中、POはエチレンおよび炭素数3〜20のオレフィンからなる群から選ばれる1種以上の単量体を重合して得られる重合鎖を表し、Raは水素原子、ハロゲン原子、炭素数1〜10の炭化水素基、窒素含有基、酸素含有基またはケイ素含有基を表しそれぞれ同一でも異なっていてもよく、RbおよびRcはそれぞれ独立に、水素原子、ハロゲン原子、炭化水素基、−OR基、−SR基または−NR2基を表し(Rは水素原子または1つ以上の置換基を有していても良い炭化水素基を表す)、環構造を形成してもよく、RdおよびReは一方が水酸基、ポリアルキレングリコール基またはアシルオキシ基を表し、他方は下記一般式(V)、一般式(VI) 、一般式(VII) のいずれかで示される基、シアノ基、カルボキシル基、エステル基またはアミド基を表す。nは平均官能基数を表し、0.1〜1.0である。)
【化2】
( 式中、Xは酸素原子または硫黄原子を表し、Rf は水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、下記一般式(VIII)
【化3】
( 式中、R1aは m+1 価の炭化水素基を表し、G は同一または相異なり、-OR1b 、-NR1 c R1d (R1b 、R1 c、R1dはポリアルキレングリコール基を表す) で表される基を表し、m は1〜 5 0 の整数を表す)で表される基を表す。)
【化4】
( 式中、Rg,Rh は同一または相異なり、水素原子、炭化水素基、アシル基、ポリアルキレングリコール基、上記一般式(VIII)で表される基を表す。)
【化5】
( 式中、Ri、Rj、Rkは同一または相異なり、水素原子、炭化水素基、アシル基、シアノ基、カルボキシル基、エステル基、アミド基を表す。)
【請求項2】
前記極性基含有ポリオレフィン重合体を含有する樹脂組成物の伸長粘度最大値が、180℃恒温下、毎秒伸長速度0.01の測定で100kPa・s以上を示すことを特徴とする請求項1に記載の樹脂改質用添加剤。
【請求項3】
前記極性基含有ポリオレフィン重合体中の重合鎖が、エチレンもしくはプロピレンの単独重合鎖、またはエチレンと炭素数3〜8のα−オレフィンとの共重合鎖であることを特徴とする請求項2に記載の樹脂改質用添加剤。
【請求項4】
前記樹脂改質用途が発泡性樹脂用であることを特徴とする請求項2ないし3に記載の樹脂改質用添加剤。
【請求項5】
請求項4に記載の発泡性樹脂用添加剤0.1〜15重量%と、熱可塑性樹脂70〜99.8重量%と、発泡剤0.1〜15重量%とを含む発泡性樹脂組成物。
【請求項6】
前記熱可塑性樹脂が、ポリエチレン、ポリプロピレン、ポリスチレン、ポリウレタンおよびポリカーボネートからなる群から選ばれる1種以上の樹脂であることを特徴とする請求項5に記載の発泡性樹脂組成物。
【請求項7】
請求項5ないし6に記載の発泡性樹脂組成物から得られる発泡成形体。
【図1】

【公開番号】特開2012−67211(P2012−67211A)
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願番号】特願2010−213700(P2010−213700)
【出願日】平成22年9月24日(2010.9.24)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願日】平成22年9月24日(2010.9.24)
【出願人】(000005887)三井化学株式会社 (2,318)
【Fターム(参考)】
[ Back to top ]