
樹脂粒子およびその製造方法
【課題】 従来にない優れた耐熱保存性と溶融特性を両立できる樹脂粒子を得る製造方法を提供する。
【解決手段】 ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
【解決手段】 ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂粒子およびその製造方法に関する。
【背景技術】
【0002】
従来より結晶性の高い樹脂粒子の形成法として、有機溶媒中から結晶性の樹脂を析出させる方法(例えば、特許文献1参照)、相分離溶媒を使用する方法(例えば、特許文献2参照)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2005−15589号公報
【特許文献2】特開平8−176310号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、上記の方法で得られた樹脂粒子は、耐熱保存性と溶融特性の両立の点で不十分であった。
本発明の課題は、耐熱保存性と溶融特性を従来になく両立できる結晶性樹脂粒子を得る製造方法を提供することである。
【課題を解決するための手段】
【0005】
発明者らは鋭意検討した結果、液状または超臨界状態の二酸化炭素で処理することにより樹脂粒子の耐熱保存性と溶融特性を両立できることを見出し、本発明に至った。
すなわち、本発明は下記2発明である。
(I) ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
(II) 上記の製造方法により得られる樹脂粒子(X)。
【発明の効果】
【0006】
本発明の製造方法で得られる本発明の樹脂粒子(X)は、耐熱保存性及び溶融特性を両立できる。
【図面の簡単な説明】
【0007】
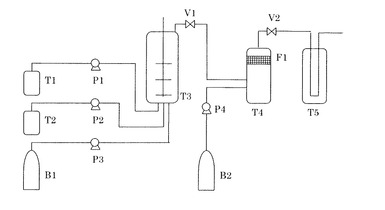
【図1】本発明における樹脂粒子の作成に用いる実験装置のフローチャートである。
【発明を実施するための形態】
【0008】
以下、本発明を詳細に説明する。
本第1発明は、樹脂粒子の製造方法に関する発明であって、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)〔以下、二酸化炭素(C)と記載する場合がある。〕で処理した後に、この二酸化炭素(C)を除去することにより樹脂粒子(X)を得る工程を含む製造方法である。
【0009】
本発明において、得られる樹脂粒子(X)の示差走査熱量(DSC)測定による融解熱が下記関係式を満足することが必要である。
0≦H2/H1≦0.9 (1)
H1はDSC測定による初回昇温時の融解熱(J/g)を表し、H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。
【0010】
ここで、融解熱の測定はJIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して測定される。
具体的には、試料(5mg)を採取してアルミパンに入れ、示差走査熱量測定装置(DSC)(例えば、「エスアイアイナノテクノロジー(株)製 RDC220」、「セイコー電子工業(株)製 DSC20」など)により、昇温速度毎分10℃で、溶融による吸熱ピークの温度〔融点(m)〕(℃)を求めることができる。また、吸熱ピークの面積より融解熱を求めることができる。なお、初回昇温後、2回目昇温前の冷却は、冷却速度90℃/分で、0℃まで冷却する。
【0011】
関係式(1)に示したように、H2/H1は0以上であり、好ましくは0.01以上、さらに好ましくは0.1以上である。
また、H2/H1は0.9以下であり、好ましくは0.85以下、さらに好ましくは0.81以下である。0.9を超えると耐熱保存性が悪化し、好ましくない。
【0012】
本発明の樹脂粒子(X)中に含有される樹脂(A)は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と非結晶性部(b)から構成される樹脂であり、具体的には結晶性部(a)を構成する樹脂と非結晶性部(b)を構成する樹脂を結合することにより得られる。
以下に、結晶性部(a)を構成する樹脂について詳細に説明する。
結晶性部(a)を構成する樹脂は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とし、結晶性を有していれば特に制限はない。耐熱保存性の観点から、融点(m)が30〜120℃の範囲であることが好ましく、さらに好ましくは40〜100℃である。
ここで、融点(m)は、前記の融解熱の測定法と同様に、JIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して、示差走査熱量測定装置(DSC)で測定した、初回昇温時の溶融による吸熱ピークの温度(℃)を意味する。
【0013】
ポリウレタンおよび/またはポリウレア(p)は、ポリウレタン、ポリウレア、ポリウレタン・ポリウレアのいずれかを意味し、ジオール(c)を含有するポリオール成分および/またはジアミン(d)を含有するポリアミン成分と、ジイソシアネート(e)を含有するポリイソシアネート成分から合成されて得られるポリウレタンおよび/またはポリウレアである。ただし、必要に応じて3官能以上のポリオール(f)や3官能以上のポリアミン(g)、3官能以上のポリイソシアネート(h)を併用してもよい。
【0014】
ポリウレタンおよび/またはポリウレア(p)を構成するポリオール成分としては、脂肪族ジオール(c1)が好ましく、炭素数が2〜36の範囲であることが好ましい。さらに好ましくは、直鎖型脂肪族ジオール(c11)である。また、直鎖型脂肪族ポリエステルジオール(c12)および直鎖型脂肪族ポリエーテルジオール(c13)も好ましい。
脂肪族ジオール、脂肪族ポリエステルジオール、脂肪族ポリエーテルジオールが分岐型では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、脂肪族ジオールは炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0015】
ジオール(c)および必要により3官能以上(3〜8価またはそれ以上)のポリオール(f)で構成されるポリオール成分としては、直鎖型脂肪族ジオール(c11)、直鎖型脂肪族ポリエステルジオール(c12)および直鎖型脂肪族ポリエーテルジオール(c13)の合計含有量がポリオール成分の70モル%以上であることが好ましく、より好ましくは80モル%以上である。必要に応じてその他のジオール(c2)が含まれても構わない。
直鎖型脂肪族ジオール、直鎖型脂肪族ポリエステルジオールおよび直鎖型脂肪族ポリエーテルジオールの合計含有量が70モル%未満では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0016】
直鎖型脂肪族ジオール(c11)としては、具体的には、例えば、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,7−ヘプタンジオール、1,8−オクタンジオール、1,9−ノナンジオール、1,10−デカンジオール、1,11−ウンデカンジオール、1,12−ドデカンジオール、1,13−トリデカンジオール、1,14−テトラデカンジオール、1,18−オクタデカンジオール、1,20−エイコサンジオールなどが挙げられるが、これらに限定されるものではない。
これらのうち、入手容易性を考慮するとエチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,9−ノナンジオール、1,10−デカンジオールが好ましい。
【0017】
直鎖型脂肪族ポリエステルジオール(c12)としては、上記直鎖型脂肪族ジオールと直鎖型脂肪族ジカルボン酸(コハク酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸、デシルコハク酸など)からなるポリエステルジオールなどが挙げられる。
【0018】
直鎖型脂肪族ポリエーテルジオール(c13)としては、上記直鎖型脂肪族ジオールのエチレンオキサイド(以下EOと略記する)、テトラヒドロフラン(以下THFと略記する)など〕付加物(付加モル数1〜300)などが挙げられる。
【0019】
その他、必要に応じて組み合わせて使用される(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)としては、炭素数2〜36の上記以外の脂肪族ジオール(1,2−プロピレングリコール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);(c13)以外の炭素数4〜36のアルキレンエーテルグリコール(ジプロピレングリコール、ポリプロピレングリコールなど);炭素数4〜36の脂環式ジオール(1,4−シクロヘキサンジメタノール、水素添加ビスフェノールAなど);上記脂環式ジオールのアルキレンオキサイド(以下AOと略記する)〔EO、プロピレンオキサイド(以下POと略記する)、ブチレンオキサイド(以下BOと略記する)など〕付加物(付加モル数1〜30);ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど)のAO〔EO、PO、BOなど〕付加物(付加モル数2〜30);分岐型脂肪族ポリエステルジオール;芳香族ポリエステルジオール;分岐型脂肪族ポリエーテルジオール;ポリラクトンジオール(ポリε−カプロラクトンジオールなど);およびポリブタジエンジオールなどが挙げられる。
【0020】
さらに、その他のジオール(c2)としては、上記のヒドロキシル基以外の末端官能基を有しないジオール以外に、他の官能基を有するジオールを用いてもよい。
このような官能基を有するジオールとしては、カルボキシル基を有するジオール(c21)、スルホン酸基もしくはスルファミン酸基を有するジオール(c22)、およびこれらの塩等が挙げられる。
カルボキシル基を有するジオール(c21)としては、ジアルキロールアルカン酸[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸(DMPA)、2,2−ジメチロールブタン酸、2 ,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸など]が挙げられる。
【0021】
スルホン酸基もしくはスルファミン酸基を有するジオール(c22)としては、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸、スルホイソフタル酸ジ(エチレングリコール)エステル、スルファミン酸ジオール[N,N−ビス(2−ヒドロキシアルキル)スルファミン酸(アルキル基のC1〜6)またはそのAO付加物(AOとしてはEOまたはPOなど、AOの付加モル数1〜6):例えばN,N−ビス(2−ヒドロキシエチル)スルファミン酸およびN,N−ビス(2−ヒドロキシエチル)スルファミン酸PO2モル付加物など];ビス(2−ヒドロキシエチル)ホスフェートなどが挙げられる。
これらのジオールの中和塩基としては、例えば炭素数3〜30の3級アミン(トリエチルアミンなど)および/またはアルカリ金属(ナトリウム塩など)が挙げられる。
これらの、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)として好ましいものは、ビスフェノール類のAO付加物、炭素数2〜12のアルキレングリコール、カルボキシル基を有するジオール、およびこれらの併用である。
【0022】
ポリウレタンおよび/またはポリウレア(p)を構成するポリアミン成分としては、脂肪族ジアミン(d1)が好ましく、炭素数が2〜36の範囲であることが好ましい。さらに好ましくは、直鎖型脂肪族ジアミン(d11)である。
脂肪族ジアミン(d1)が分岐型では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0023】
ジアミン(d)および必要により3官能以上(3〜8価またはそれ以上)のポリアミン(g)で構成されるポリアミン成分としては、直鎖型脂肪族ジアミン(d11)の含有量がポリアミン成分の70モル%以上であることが好ましく、より好ましくは80モル%以上である。必要に応じてその他のジアミン(d2)が含まれても構わない。
直鎖型脂肪族ジアミン(d11)の含有量が70モル%未満では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0024】
直鎖型脂肪族ジアミン(d11)としては、具体的には、例えば、エチレンジアミン、1,3−プロパンジアミン、1,4−ブタンジアミン、1,5−ペンタンジアミン、1,6−ヘキサンジアミン、1,7−ヘプタンジジアミン、1,8−オクタンジアミン、1,9−ノナンジアミン、1,10−デカンジアミン、1,11−ウンデカンジアミン、1,12−ドデカンジアミン、1,13−トリデカンジアミン、1,14−テトラデカンジアミン、1,18−オクタデカンジアミン、1,20−エイコサンジアミンなどが挙げられるが、これらに限定されるものではない。
これらのうち、入手容易性を考慮するとエチレンジアミン、1,3−プロパンジアミン、1,4−ブタンジアミン、1,5−ペンタンジアミン、1,6−ヘキサンジアミン、1,9−ノナンジアミン、1,10−デカンジアミンが好ましい。
【0025】
その他、直鎖型脂肪族ジアミン(d11)と、必要に応じて組み合わせて使用される(d11)以外の脂肪族ジアミン(d1)およびその他のジアミン(d2)としては、上記以外の脂肪族ポリアミン類(C2〜C18)および芳香族ポリアミン類(C6〜C20)が挙げられる。
上記以外の脂肪族ポリアミン類(C2〜C18)としては、〔1〕分岐脂肪族ジアミン{C2〜C6 アルキレンジアミン(1,2−プロピレンジアミンなど)、ポリアルキレン(C2〜C6)ジアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミンなど〕};〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミンなど〕;〔3〕脂環または複素環含有脂肪族ジアミン{脂環式ジアミン(C4〜C15)〔1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など〕、複素環式ジアミン(C4〜C15)〔ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジン、3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕;〔4〕芳香環含有脂肪族アミン類(C8〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど);等が挙げられる。
【0026】
芳香族ポリアミン類(C6〜C20)としては、〔1〕非置換芳香族ジアミン〔1,2−、1,3−および1,4−フェニレンジアミン、2,4´−および4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4”−トリアミン、ナフチレンジアミンなど;〔2〕核置換アルキル基〔メチル,エチル,n−およびi−プロピル、ブチルなどのC1〜C4アルキル基〕を有する芳香族ジアミン、たとえば2,4−および2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、3,3´,5,5´−テトラメチルベンジジン、3,3´,5,5´−テトラメチル−4,4´−ジアミノジフェニルメタン、3,5−ジエチル−3´−メチル−2´,4−ジアミノジフェニルメタン、3,3´−ジエチル−2,2´−ジアミノジフェニルメタン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、3,3´,5,5´−テトラエチル−4,4´−ジアミノベンゾフェノン、3,3´,5,5´−テトラエチル−4,4´−ジアミノジフェニルエーテル、3,3´,5,5´−テトライソプロピル−4,4´−ジアミノジフェニルスルホンなど〕、およびこれらの異性体の種々の割合の混合物;〔3〕核置換電子吸引基(Cl,Br,I,Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ジアミン〔メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン;4,4´−ジアミノ−3,3´−ジメチル−5,5´−ジブロモ−ジフェニルメタン、3,3´−ジクロロベンジジン、3,3´−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキシド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4´−メチレンビス(2−ヨードアニリン)、4,4´−メチレンビス(2−ブロモアニリン)、4,4´−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリンなど〕;〔4〕2級アミノ基を有する芳香族ジアミン〔上記〔1〕〜〔3〕の芳香族ジアミンの−NH2の一部または全部が−NH−R´(R´はアルキル基たとえばメチル,エチルなどの低級アルキル基)で置き換ったもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕が挙げられる。
【0027】
ポリアミン成分としては、これらの他、ポリアミドポリアミン〔ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど〕、ポリエーテルポリアミン〔ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物など〕等が挙げられる。
【0028】
ジイソシアネート(e)および必要に応じて併用される3官能以上のポリイソシアネート(h)としては、炭素数(NCO基中の炭素を除く。以下同様。)6〜20の芳香族ジイソシアネート、炭素数2〜18の脂肪族ジイソシアネート、炭素数4〜15の脂環式ジイソシアネート、炭素数8〜15の芳香脂肪族ジイソシアネート、これらのジイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。また、必要により3官能以上(3〜8価またはそれ以上)のポリイソシアネート(h)を併用してもよい。
【0029】
上記芳香族ジイソシアネートおよび3価以上の芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)またはその混合物との縮合生成物;ジアミノジフェニルメタンと少量(たとえば5〜20重量%)の3官能以上のポリアミンとの混合物〕のホスゲン化物:ポリアリルポリイソシアネート(PAPI)]、1,5−ナフチレンジイソシアネート、4,4’,4”−トリフェニルメタントリイソシアネート、m−およびp−イソシアナトフェニルスルホニルイソシアネートなどが挙げられる。
【0030】
上記脂肪族ジイソシアネートおよび3価以上の脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエートなどが挙げられる。
【0031】
上記脂環式ジイソシアネートおよび3価以上の脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−および/または2,6−ノルボルナンジイソシアネートなどが挙げられる。
【0032】
上記芳香脂肪族ジイソシアネートおよび3価以上の芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
【0033】
また、上記ジイソシアネートおよび3価以上のポリイソシアネートの変性物としては、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。
具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのジイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]などが挙げられる。
【0034】
これらのポリイソシアネート成分のうちで好ましいものは、炭素数6〜15の芳香族ジイソシアネート、炭素数4〜12の脂肪族ジイソシアネート、および炭素数4〜15の脂環式ジイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0035】
ポリウレタンおよび/またはポリウレア(p)の分子量は、ゲルパーミエーションクロマトグラフィー(GPC)によって求めた重量平均分子量(Mw)が2000〜80000のものが好ましい。ガラス転移温度(Tg)は、好ましくは−100〜40℃、さらに好ましくは−80〜0℃である。融点(m)は、好ましくは40〜100℃である。
【0036】
結晶性部(a)を構成する樹脂は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とし、結晶性を有していれば特に制限はなく、ポリウレタンおよび/またはポリウレア(p)の単独樹脂であっても、結晶性を有する他の樹脂との複合樹脂であってもよい。
複合樹脂に使用される結晶性を有する他の樹脂としては、ポリウレタンおよび/またはポリウレア(p)のブロックを導入しやすいことから、ポリエステル樹脂およびポリアミド樹脂から選ばれる1種以上の樹脂が好ましい。
複合樹脂は、例えば、結晶性ポリエステル樹脂、結晶性ポリアミド樹脂、およびこれらの混合物とポリウレタンおよび/またはポリウレア(p)とを結合することで得られる。
【0037】
結晶性ポリエステル樹脂は、ジオール(c)を含有するポリオール成分とジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリエステル樹脂が好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリカルボン酸(j)を併用してもよい。
結晶性ポリアミド樹脂は、ジアミン(d)を含有するポリアミンと、ジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリアミド樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリカルボン酸(j)を併用してもよい。
【0038】
結晶性ポリエステル樹脂、結晶性ポリアミド樹脂に用いられるポリオール成分、ポリカルボン酸成分、およびポリアミン成分(それぞれ3官能以上のものを含む)についてそれぞれ示す。
【0039】
ポリオール成分のうち、ジオール(c)としては、脂肪族ジオール(c1)が好ましく、炭素数が2〜36の範囲であることが好ましい。また直鎖型脂肪族ジオール(c11)がより好ましい。
脂肪族ジオール(c1)が分岐型では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0040】
ジオール(c)および必要により3官能以上(3〜8価またはそれ以上)のポリオール(f)で構成されるポリオール成分としては、直鎖型脂肪族ジオール(c11)の含有量がポリオール成分の80モル%以上であることが好ましく、より好ましくは90モル%以上である。必要に応じてその他のジオール(c2)が含まれても構わない。
直鎖型脂肪族ジオール(c11)の含有量が80モル%未満では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0041】
直鎖型脂肪族ジオール(c11)としては、具体的には、前記のものが挙げられ、好ましいものも同様である。
その他、直鎖型脂肪族ジオール(c11)と必要に応じて組み合わせて使用される、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)としては、前記の、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)、カルボキシル基を有するジオール(c21)、スルホン酸基もしくはスルファミン酸基を有するジオール(c22)、およびこれらの塩として例示したものが挙げられ、好ましいものも同様である。
必要により用いられる3官能以上(3〜8価またはそれ以上)のポリオール(f)としては、前記のものが挙げられ、好ましいものも同様である。
【0042】
ポリカルボン酸成分のうち、ジカルボン酸(i)としては、炭素数4〜36のアルカンジカルボン酸(コハク酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸、デシルコハク酸など)、炭素数6〜40の脂環式ジカルボン酸〔ダイマー酸(2量化リノール酸)など〕、炭素数4〜36のアルケンジカルボン酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸などのアルケニルコハク酸、マレイン酸、フマール酸、シトラコン酸など)、炭素数8〜36の芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、t−ブチルイソフタル酸、2,6−ナフタレンジカルボン酸、4,4’−ビフェニルジカルボン酸など)などが挙げられる。
また、必要により、3官能以上(3〜6価またはそれ以上)のポリカルボン酸(j)を併用してもよい。
3官能以上のポリカルボン酸(j)としては、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など)、および炭素数6〜36の脂肪族または脂環式ポリカルボン酸(ヘキサントリカルボン酸など)などが挙げられる。
なお、ジカルボン酸(i)、および3官能以上のポリカルボン酸(j)としては、上述のものの酸無水物または炭素数1〜4の低級アルキルエステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
結晶性や入手容易性を考慮すると、アジピン酸、セバシン酸、ドデカンジカルボン酸、テレフタル酸、およびイソフタル酸が特に好ましい。
【0043】
ポリアミン成分のうち、ジアミン(d)としては、脂肪族ジアミン(d1)が好ましく、炭素数が2〜36の範囲であることが好ましい。また直鎖型脂肪族ジアミン(d11)がより好ましい。
脂肪族ジアミン(d1)が分岐型では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0044】
ジアミン(d)および必要により3官能以上(3〜8価またはそれ以上)のポリアミン(g)で構成されるポリアミン成分としては、直鎖型脂肪族ジアミン(d11)の含有量がポリアミン成分の80モル%以上であることが好ましく、より好ましくは90モル%以上である。必要に応じてその他のジアミン(d2)が含まれても構わない。
直鎖型脂肪族ジアミン(d11)の含有量が80モル%未満では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0045】
ポリアミン成分の例としては、前記のものが挙げられる。
【0046】
結晶性部(a)を構成する樹脂中のポリウレタンおよび/またはポリウレア(p)の割合は、好ましくは20重量%以上、さらに好ましくは30重量%以上、とくに好ましくは40〜95重量%である。
【0047】
ポリウレタンおよび/またはポリウレア(p)と結晶性ポリエステル樹脂、および結晶性ポリアミド樹脂を結合する方法は、それぞれが含有する官能基の反応性を考慮して、結合剤(カップリング剤)を使用するかしないかを選択し、また使用する場合は、含有する官能基にあった結合剤を選択し、結合させ複合樹脂とすることができる。
【0048】
複合樹脂を作製する時に結合剤を使わない場合、必要により加熱減圧しつつ、ポリウレタンおよび/またはポリウレア(p)が含有する官能基とそれ以外の樹脂が含有する官能基との反応を進める方法が挙げられる。反応温度は180℃〜230℃で行うのが好ましい。
【0049】
結合剤を使う場合は、末端の官能基の種類に合わせて、種々の結合剤が使用できる。
多価カルボン酸、多価アルコール、多価イソシアネート、酸無水物、多官能エポキシ等を結合剤として用いて、脱水反応や、付加反応を行うことで、複合樹脂である結晶性部(a)が得られる。
多価カルボン酸および酸無水物としては、前記ポリカルボン酸成分と同様のものが挙げられる。多価アルコールとしては、前記ポリオール成分と同様のものが挙げられる。多価イソシアネートとしては、前記ポリイソシアネート成分と同様のものが挙げられる。
多官能エポキシとしては、ビスフェノールA型および−F型エポキシ化合物、フェノールノボラック型エポキシ化合物、クレゾールノボラック型エポキシ化合物、水添ビスフェノールA型エポキシ化合物、ビスフェノールAまたは−FのAO付加体のジグリシジルエーテル、水添ビスフェノールAのAO付加体のジグリシジルエーテル、ジオール(エチレングリコール、プロピレングリコール、ネオペンチルグリコール、ブタンジオール、ヘキサンジオール、シクロヘキサンジメタノール、ポリエチレングリコールおよびポリプロピレングリコール等)のジグリシジルエーテル、トリメチロールプロパンジおよび/またはトリグリシジルエーテル、ペンタエリスリトールトリおよび/またはテトラグリシジルエーテル、ソルビトールヘプタおよび/またはヘキサグリシジルエーテル、レゾルシンジグリシジルエーテル、ジシクロペンタジエン・フェノール付加型グリシジルエーテル、メチレンビス(2,7−ジヒドロキシナフタレン)テトラグリシジルエーテル、1,6−ジヒドロキシナフタレンジグリシジルエーテル、ポリブタジエンジグリシジルエーテルなどが挙げられる。
【0050】
複合樹脂を作製する方法のうち、脱水反応の例としては、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂が共に水酸基を有する場合には、これらの水酸基を結合剤(例えば多価カルボン酸)で結合する反応が挙げられる。この場合、例えば、無溶剤下、反応温度180℃〜230℃で反応し、複合樹脂である結晶性部(a)が得られる。
【0051】
付加反応の例としては、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂が共に末端に水酸基を有する場合であり、これらを結合剤(例えば多価イソシアネート)で結合する反応や、またポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂の片方が末端に水酸基を有する場合で、もう一方が末端にイソシアネート基を有する樹脂の場合、結合剤を用いずにこれらを結合する反応が挙げられる。この場合、例えば、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂ともに溶解可能な溶剤に溶解させ、これに必要であるなら結合剤を投入し、反応温度80℃〜150℃で反応し、複合樹脂である結晶性部(a)が得られる。
【0052】
非結晶性部(b)を構成する樹脂としては、ポリエステル樹脂(ラクトン開環重合物を含む)、ポリウレタン樹脂、ポリウレア樹脂、ポリアミド樹脂、ポリスチレン樹脂、スチレンアクリル系ポリマー等が挙げられる。
これらのうち、ポリエステル樹脂、ポリウレタン樹脂、ポリウレア樹脂、ポリアミド樹脂、およびそれらの複合樹脂が好ましく、ポリウレタン樹脂およびポリエステル樹脂がさらに好ましい。
【0053】
前記結晶性部(a)と同様に、ポリエステル樹脂は、ジオール(c)を含有するポリオール成分とジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリエステル樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリカルボン酸(j)を併用できる。
ポリウレタン樹脂は、ジオール(c)を含有するポリオール成分と、ジイソシアネート(e)を含有するポリイソシアネート成分とから合成されるポリウレタン樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリイソシアネート(h)を併用できる。
ポリウレア樹脂は、ジアミン(d)を含有するポリアミンと、ジイソシアネート(e)を含有するポリイソシアネート成分とから合成されるポリウレア樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリイソシアネート(h)を併用できる。
ポリアミド樹脂は、ジアミン(d)を含有するポリアミンと、ジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリアミド樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリカルボン酸(j)を併用できる。
非結晶性ポリエステル樹脂、非結晶性ポリウレタン樹脂、非結晶性ポリアミド樹脂、非結晶性ポリウレア樹脂を構成するモノマーとしては、前記ポリオール成分、前記ポリカルボン酸成分、前記ポリイソシアネート成分、および前記ポリアミン成分の具体例として示したものと同様のものが挙げられ、非結晶性樹脂となるものであればいかなる組合せでも構わない。
【0054】
非結晶性部(b)を構成する樹脂のガラス転移温度(Tg)は、耐熱保存性の観点から、好ましくは40〜250℃、さらに好ましくは50〜240℃、とくに好ましくは60〜230℃である。
ここで、ガラス転移温度は、JIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して、示差走査熱量測定装置(セイコー電子工業(株)製 DSC20、エスアイアイナノテクノロジー(株)製 RDC220等)を用いて、測定される。
【0055】
結晶性部(a)を構成する樹脂と非結晶性部(b)を構成する樹脂との結合は、(a)および(b)を構成する樹脂のそれぞれの末端官能基の反応性を考慮して、結合剤(カップリング剤)を使用するかしないかを選択し、また使用する場合は、末端官能基にあった結合剤を選択し、(a)と(b)を結合させ、ブロックポリマーである樹脂(A)とすることが出来る。
なお、上記方法で、樹脂(A)と未反応の(a)および/または(b)の混合物〔好ましくは(A)と(a)の混合物〕が得られる場合、混合物をそのまま本発明の樹脂粒子の製造方法に使用してもよい。
【0056】
結合剤を使わない場合、必要により加熱減圧しつつ、(a)を形成する樹脂の末端官能基と(b)を形成する樹脂の末端官能基の反応を進める。特に末端の官能基がカルボキシル基と水酸基との反応や、カルボキシル基とアミノ基との反応の場合、片方の樹脂の酸価が高く、もう一方の樹脂の水酸基価やアミン価が高い場合、反応がスムーズに進行する。反応温度は180℃〜230℃で行うのが好ましい。
【0057】
結合剤を使う場合は、末端の官能基の種類に合わせて、種々の結合剤が使用できる。
多価カルボン酸、多価アルコール、多価イソシアネート、酸無水物、多官能エポキシ等を結合剤として用いて、脱水反応や、付加反応を行うことで、結晶性部(a)と非結晶性部(b)を結合させて、樹脂(A)が得られる。
これらの結合剤の具体例としては、前記のものが挙げられる。
【0058】
(a)と(b)を結合させる方法のうち、脱水反応の例としては、結晶性部(a)、非結晶部(b)とも末端に水酸基を有する樹脂の場合には、これらの水酸基を結合剤(例えば多価カルボン酸)で結合する反応が挙げられる。この場合、例えば、無溶剤下、反応温度180℃〜230℃で反応し樹脂(A)が得られる。
付加反応の例としては、結晶性部(a)、非結晶性部(b)とも末端に水酸基を有する樹脂であり、これらを結合剤(例えば多価イソシアネート)で結合する反応や、また結晶性部(a)、非結晶性部(b)の片方が末端に水酸基を有する樹脂で、もう一方が末端にイソシアネート基を有する樹脂の場合、結合剤を用いずにこれらを結合する反応が挙げられる。この場合、例えば、結晶性部(a)、非結晶性部(b)ともに溶解可能な溶剤に溶解させ、これに必要であるなら結合剤を投入し、反応温度80℃〜150℃で反応し樹脂(A)が得られる。
【0059】
結晶性部(a)と非結晶性部(b)から構成される樹脂(A)は、結晶性樹脂であることが好ましく、耐熱保存性の観点から、融点(m)は、好ましくは40℃以上、さらに好ましくは50℃以上、特に好ましくは55℃以上である。また、溶融特性の観点から、110℃以下が好ましく、さらに好ましくは100℃以下、特に好ましくは90℃以下である。ここで、融点(m)は、前記の方法で測定される。
【0060】
樹脂(A)の軟化点(s)[℃]と融点(m)[℃]との比(s/m)は、好ましくは0.8〜1.55であり、より好ましくは0.8〜1.2、特に好ましくは0.85〜1.15である。この範囲内であると、劣化しにくい樹脂粒子となる。なお、軟化点(s)は、次のように測定される値である。
<軟化点>
降下式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、プランジャーの降下量の最大値の1/2に対応する温度をグラフから読み取り、この値(測定試料の半分が流出したときの温度)を軟化点とする。
【0061】
樹脂(A)の粘弾性特性において、下記〔条件1〕を満たすのが好ましく、下記〔条件1−2〕を満たすのがさらに好ましい。
〔条件1〕 G’(m+20)=50〜1×106[Pa]
〔条件1−2〕 G’(m+20)=100〜5×105[Pa]
[G’:貯蔵弾性率[Pa]]
(m+20)℃におけるG’が50Pa以上であると、樹脂粒子を静電塗装する場合、低温塗布時でも均一に塗布しやすく、塗布温度領域が広くなる。また、1×106[Pa]以下であると、低温での溶融性が向上する。
動的粘弾性測定値(貯蔵弾性率G’、損失弾性率G”)は、Rheometric Scientific社製 動的粘弾性測定装置 RDS−2を用い周波数1Hz条件下で測定される。測定温度範囲は30℃〜200℃で、この温度間の溶融粘弾性を測定することによって、温度−G’、温度−G”の曲線として得ることができる。
〔条件1〕を満たす樹脂(A)は、(A)を構成する組成中の結晶性成分の比率を調整することや樹脂分子量を調整すること等により得ることができる。例えば、結晶性部(a)の比率や結晶性成分の比率を増加させると、G’(m+20)の値は小さくなる。結晶性成分としては、直鎖構造を有するポリオール、ポリイソシアネート等が挙げられる。また樹脂分子量を低下させることでもG’(m+20)の値は小さくなる。
【0062】
樹脂(A)の溶融開始温度(x)は、好ましくは(m±20)℃(mは融点)の温度範囲内であり、さらに好ましくは(m±15)℃の温度範囲内、特に好ましくは(m±10)℃の温度範囲内である。(x)は、具体的には30〜100℃が好ましく、さらに好ましくは40〜80℃である。なお、溶融開始温度(x)は、次のように測定される値である。
<溶融開始温度>
降下式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、試料の熱膨張によるピストンのわずかな上昇が行われた後、再びピストンが明らかに下降し始める点の温度をグラフから読み取り、この値を溶融開始温度とする。
【0063】
また、樹脂(A)は、損失弾性率G”[Pa]と溶融開始温度(x)[℃]に関して、以下の〔条件2〕を満たすことが好ましく、〔条件2−2〕を満たすことがさらに好ましく、〔条件2−3〕を満たすことが特に好ましく、〔条件2−4〕を満たすことが最も好ましい。
〔条件2〕 |LogG”(x+20)−LogG”(x)|>2.0
〔条件2−2〕 |LogG”(x+20)−LogG”(x)|>2.5
〔条件2−3〕 |LogG”(x+15)−LogG”(x)|>2.5
〔条件2−4〕 |LogG”(x+10)−LogG”(x)|>2.5
樹脂(A)の溶融開始温度(x)が上記範囲内であり、かつ〔条件2〕を満たすと、樹脂の低粘性化速度が速く、樹脂粒子を静電塗装する場合、塗布温度領域の低温側、高温側で同等の表面平滑性を得ることができる。また、溶融開始から塗布可能粘性に至るまでが速く、優れた低温溶融性を得るのに有利である。〔条件2〕は、どれだけ早く、少ない熱で溶融できるかという、樹脂のシャープメルト性の指標であり、実験的に求めたものである。
溶融開始温度(x)の好ましい範囲、および〔条件2〕を満たす樹脂(A)は、(A)の構成成分中の結晶性成分の比率を調整すること等により得ることができる。例えば、結晶性成分の比率を大きくすると、(m)と(x)の温度差が小さくなる。
【0064】
また、樹脂(A)の粘弾性特性において、(m+30)℃の損失弾性率G”と(m+70)℃の損失弾性率G”の比〔G”(m+30)/G”(m+70)〕が0.05〜50であることが好ましく、より好ましくは0.1〜10である〔m:(A)の融点〕。
損失弾性率の比が上記の範囲で維持されることによって、樹脂粒子を静電塗装する場合、塗布温度領域でより安定した表面平滑性を得ることができる。
上記のG”の比の条件を満たす樹脂(A)は、(A)を構成する組成中の結晶性成分の比率や結晶性部(a)の分子量を調整すること等により得ることができる。例えば、結晶性部(a)の比率や結晶性成分の比率を増加させると、〔G”(m+30)/G”(m+70)〕の値は小さくなる。また結晶性部(a)の分子量を増加させると〔G”(m+30)/G”(m+70)〕の値は小さくなる。結晶性成分としては、直鎖構造を有するポリオール、ポリイソシアネート等が挙げられる。
【0065】
樹脂(A)の重量平均分子量(Mw)は、溶融特性の観点から5000〜100000が好ましく、さらに好ましくは6000〜80000、特に好ましくは8000〜50000である。
(A)を構成する結晶性部(a)のMwは、2000〜80000が好ましく、さらに好ましくは3000〜60000、特に好ましくは4000〜30000である。
また、(A)を構成する非結晶性部(b)のMwは、500〜50000が好ましく、さらに好ましくは750〜20000であり、特に好ましくは1000〜10000である。
結晶性部(a)及び非結晶性部(b)のMwは、結合させる前にそれぞれ(a)を構成する樹脂及び(b)を構成する樹脂のMwを測定することで得られる。
なお、本発明において樹脂の重量平均分子量(Mw)は、ゲルパーミエーションクロマトグラフイー(GPC)を用いて以下の条件で測定される。
【0066】
装置(一例) :東ソー(株)製 HLC−8120
カラム(一例):TSK GEL GMH6 2本 〔東ソー(株)製〕
測定温度 :40℃
試料溶液 :0.25重量%のTHF溶液
溶液注入量 :100μL
検出装置 :屈折率検出器
基準物質 :東ソー製 標準ポリスチレン(TSKstandard POLY
STYRENE)12点(分子量 500 1050 2800
5970 9100 18100 37900 96400
190000 355000 1090000 2890000)
【0067】
結晶性部(a)が樹脂(A)中に占める割合は、30〜95重量%が好ましく、さらに好ましくは40〜90重量%であり、特に好ましくは50〜85重量%である。この範囲であると樹脂(A)の結晶性が損なわれず、耐熱保存性が良好である。
【0068】
樹脂(A)は、結晶性部(a)と非結晶性部(b)から構成されるブロック樹脂であるが、(a)と(b)とが、下記の形式で線状に結合された両末端が(a)の樹脂であり、{−(b)−(a)}の単位の繰り返し数の平均値nが0.5〜3.5であることが好ましく、さらに好ましくはn=0.7〜2.0、とくに好ましくはn=0.9〜1.5である。
(a){−(b)−(a)}n
上記式は、具体的には、結晶性部(a)と非結晶性部(b)とが、
(a)〔n=0〕、
(a)−(b)−(a)〔n=1〕、
(a)−(b)−(a)−(b)−(a)〔n=2〕、
(a)−(b)−(a)−(b)−(a)−(b)−(a)〔n=3〕
等の形式で線状に結合された樹脂、およびこれらの混合物〔n=0のみからなるものを除く〕を意味する。なお、nが0のものを含有するということは、樹脂(A)と共に結晶性部(a)を構成する樹脂を含有することを意味する。
nが3.5以下であると、樹脂(A)の結晶性が損なわれない。またnが0.5以上であると(A)の溶融後の弾性が良好であり、樹脂粒子を静電塗装に用いる場合、塗布可能温度領域がより広くなる。なお、nは原料の使用量〔(a)と(b)のモル比〕から求めた計算値である。また、樹脂(A)の結晶化度の観点から(A)の両末端は結晶性部(a)であることが好ましい。
なお、両末端が非結晶性部(b)である場合は、結晶化度が落ちるため、樹脂(A)に結晶性を持たせるために、(A)中の結晶性部(a)の比率を75重量%以上にするのが好ましい。
【0069】
本発明において、樹脂粒子(B)を液状又は超臨界状態の二酸化炭素(C)中で処理し、その後に、二酸化炭素(C)を除去することにより、目的の樹脂粒子(X)を得ることができる。ここで、処理とは樹脂粒子(B)を(C)中に分散し、一定時間(C)と接触させ、(B)を膨潤させることをいう。この処理を行うことにより、H2/H1の比が1または1以上である樹脂粒子(B)でも、0.9以下である目的の樹脂粒子(X)に変性することができる場合がある。
上記一定時間〔樹脂粒子(B)が形成されるに要する時間も含む。〕は、通常10秒〜180分、好ましくは30秒〜60分である。
膨潤は、一定時間、樹脂粒子(B)を二酸化炭素(C)と接触させることにより、二酸化炭素が樹脂粒子(B)中に浸透することで起こる。従って、膨潤の度合いは(C)との接触時間、(C)の圧力、温度によって調節できる。
【0070】
樹脂粒子(B)を(C)中に分散する方法としては、以下の(1)〜(4)が挙げられる。
(1)溶剤(S)に溶解させた樹脂(A)を(C)中に分散し、樹脂粒子(B)分散体を得る方法
(2)溶融させた樹脂(A)を(C)中に分散し、樹脂粒子(B)分散体を得る方法
(3)別法にて樹脂粒子(B)を作成し、(B)を攪拌や超音波照射等により直接(C)中に分散する方法
(4)別法にて作成された樹脂粒子(B)を溶剤(T)中に分散し、該分散液を(C)中に導入する方法
なお、(1)及び(2)の分散方法において、分散用微粒子(D)を使用することが好ましい。(D)を(C)中に分散させることで、(D)は(B)の表面に吸着し、(B)を(C)中に安定に分散させることができる。
これらの方法のうち、樹脂粒子の粒子径の調整が容易で、粒度分布(体積平均粒子径Dvと個数平均粒子径Dnの比Dv/Dn)を小さくすることができる点から(1)が好ましい。
【0071】
溶剤(S)としては、例えば、ケトン溶剤(アセトン、メチルエチルケトン等)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、環状エーテル等)、エステル溶剤(酢酸エステル、ピルビン酸エステル、2−ヒドロキシイソ酪酸エステル、乳酸エステル等)、アミド溶剤(ジメチルホルムアミド等)、アルコール類(メタノール、エタノール、フッ素含有アルコール等)、芳香族炭化水素溶剤(トルエン、キシレン等)、および脂肪族炭化水素溶剤(オクタン、デカン等)などが挙げられる。これらの溶剤の2種以上の混合溶剤、または、これらの有機溶剤と水との混合溶剤を用いることもできる。
好ましくは、溶剤除去の観点から、混合溶剤(特に、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、およびアセトンと水の混合溶剤)である。
【0072】
樹脂(A)の溶液(L)は、樹脂(A)を溶剤(S)に溶解させて製造する。
溶液(L)の40℃における粘度は、好ましくは10〜100万mPa・sであり、更に好ましくは、50〜50万mPa・sであり、特に好ましくは、100〜20万mPa・sである。この範囲であれば、樹脂粒子(B)の分散性が向上する。
また、溶液(L)中の樹脂(A)の重量比率は、好ましくは5〜95重量%であり、更に好ましくは、10〜90重量%であり、特に好ましくは、15〜85重量%である。この範囲であれば、効率よく樹脂粒子(B)を形成することができる。
【0073】
必要により用いる分散用微粒子(D)としては、樹脂粒子(B)を分散できれば、特に制限はなく、無機微粒子(シリカ、チタニア等)、有機微粒子(アクリル樹脂、ポリエステル樹脂など)などが挙げられる。
微粒子(D)の体積平均粒子径は、樹脂粒子(B)を分散できれば、特に制限はないが、好ましくは30〜1000nmであり、さらに好ましくは50〜500nmである。この範囲であれば、(C)中での樹脂粒子(B)の分散性が向上する。
微粒子(D)と樹脂粒子(B)の重量比率(重量%)は、特に制限はないが、樹脂粒子(B)に対して、0.1〜20%が好ましく、更に好ましくは、0.5〜15%である。この範囲であれば、樹脂粒子(B)の分散性が向上する。
微粒子(D)としては、(C)に溶解せず、(C)中に安定分散するものが好ましい。
【0074】
本発明において、微粒子(D)を二酸化炭素(C)中に分散する方法はいかなる方法でもよく、例えば、容器内に(D)及び(C)を仕込み、攪拌や超音波照射等により、(D)を直接(C)中に分散する方法や、微粒子(D)が溶剤中に分散された分散液を(C)中に導入する方法等が挙げられる。
【0075】
上記溶剤としては、溶剤(S)と同様のものが挙げられる。微粒子(D)の分散性から、好ましくは、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)、及びエステル溶剤(酢酸エチル、酢酸ブチルなど)である。
【0076】
微粒子(D)と溶剤の重量比率(重量%)は、特に制限はないが、溶剤に対して、微粒子(D)が50以下が好ましく、更に好ましくは30以下であり、特に好ましくは20以下である。この範囲であれば、効率よく微粒子(D)を(C)中に導入することができる。
微粒子(D)を溶剤中に分散する方法としては特に制限はないが、微粒子(D)を溶剤に仕込み、攪拌や超音波照射等により直接分散する方法や、微粒子を高温下で溶剤に溶解させて晶析する方法などが挙げられる。
【0077】
樹脂粒子(B)を二酸化炭素(C)中に分散する方法(1)において、樹脂(A)の溶液(L)は、(C)中に分散するため、(C)と混合する際の温度において適度な粘度であることが好ましく、粒度分布の観点から、好ましくは10万mPa・s以下、さらに好ましくは5万mPa・s以下である。樹脂(A)の(C)への溶解度は、好ましくは3%以下、さらに好ましくは1%以下である。
樹脂(A)のSP値は、好ましくは8〜16、さらに好ましくは9〜14である。SP値とは、下記に示した様に、凝集エネルギー密度と分子容の比の平方根で表されるものである。
SP=(△E/V)1/2
ここで△Eは凝集エネルギー密度を表す。Vは分子容を表し、その値は、ロバート エフ.フェドールス(Robert F.Fedors)らの計算によるもので、例えばポリマー エンジニアリング アンド サイエンス(Polymer engineering and science)第14巻、147〜154頁に記載されている。
【0078】
本発明において、樹脂粒子(B)および樹脂粒子(X)中に他の添加剤(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を含有しても差し支えない。樹脂粒子(B)および(X)中に他の添加剤を含有させる方法としては、あらかじめ樹脂(A)と添加剤を混合した後、(C)中にその混合物を加えて分散させるのが好ましい。
【0079】
方法(1)において、樹脂(A)の溶液(L)を、二酸化炭素(C)中に分散する方法はいかなる方法を用いてもよい。具体例としては、樹脂(A)の溶液(L)を攪拌機や分散機等で分散する方法、樹脂(A)の溶液(L)を二酸化炭素(C)中にスプレーノズルを介して噴霧して液滴を形成し、液滴中の樹脂を過飽和状態とし、樹脂粒子を析出させる方法(ASES:Aerosol Solvent Extraction Systemとして知られている)、同軸の多重管(2重管、3重管等)から溶液(L)を高圧ガス、エントレーナ等とともにそれぞれ別の管から同時に噴出させて、液滴に外部応力を加え分裂を促進させて、粒子を得る方法(SEDS:Solution Enhanced Dispersion by Supercritical Fluidsとして知られている)、超音波を照射する方法等が挙げられる。
【0080】
このようにして二酸化炭素(C)中に樹脂(A)の溶液(L)を分散し、必要により微粒子(D)を表面に吸着させることにより、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)が(C)中に分散した分散体が形成される。なお、微粒子(D)を用いた場合、樹脂粒子(B)の表面には、微粒子(D)が固着しているか、(D)が被膜化した被膜が形成される。
分散体は単一相であることが好ましい。すなわち、(B)が分散している二酸化炭素(C)を含む相の他に、溶剤(S)相が分離する状態は好ましくない。したがって、溶剤相が分離しないように、(C)に対する(A)の溶液(L)の量を設定することが好ましい。溶液(L)の量は、例えば(C)に対して90重量%以下が好ましく、さらに好ましくは5〜80重量%、特に好ましくは10〜70重量%である。
なお、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)中に含有する(S)の量は、好ましくは10〜90重量%、さらに好ましくは20〜70重量%である。
また、樹脂(A)と二酸化炭素(C)の重量比は、好ましくは(A):(C)が、1:(0.1〜100)、さらに好ましくは1:(0.5〜50)、特に好ましくは1:(1〜20)である。
【0081】
上記方法(2)は、樹脂(A)を溶剤(S)に溶解させた溶液(L)の代わりに、溶融させた樹脂(A)を用いる以外は、上記方法(1)と同様であり、樹脂(A)を含有する樹脂粒子(B)が二酸化炭素(C)中に分散した分散体が形成される。
【0082】
上記方法(3)及び(4)において、液状、塊状、粒状、ペレット状、または粗粉末状の樹脂(A)から樹脂粒子(B)を作成する方法として、以下の方法が挙げられる。
(5)液状にした樹脂(A)を水性媒体に分散し、取り出す方法。
(6)固状の樹脂(A)を破砕または粉砕する方法。
【0083】
方法(5)について説明する。
固状の樹脂であっても、樹脂(A)を融点以上に加熱することや、樹脂(A)を有機溶剤に溶解することにより液状にすることができる。
有機溶剤としては、前記の溶剤(S)等を用いることが出来る。
水性媒体としては、水を必須構成成分とする液体であれば制限なく使用でき、水、並びに界面活性剤を含む水溶液等を用いることができる。
界面活性剤としては、公知の界面活性剤(たとえば、特開2004−124059号公報に記載の界面活性剤)等を使用することができる。
分散装置としては、一般に乳化機、分散機として使用されているものであれば特に限定されず、公知の分散装置(たとえば、特開2002−284881号公報に記載の分散装置)等を使用することができる。
粒子化された樹脂粒子(B)を水性媒体から取り出す方法としては、固液分離法(遠心分離器、スパクラフィルター及び/又はフィルタープレス等)により樹脂粒子(B)を分離した後、水洗する方法等が適用できる。
得られる樹脂粒子(B)は、必要により乾燥する。乾燥としては、樹脂粒子の融点未満で行うことが好ましく、必要により減圧下で行う。乾燥機としては、公知の乾燥装置(例えば、流動層式乾燥機、減圧乾燥機及び循風乾燥機)が用いられる。
【0084】
次に上記の方法(6)について説明する。
破砕又は粉砕に用いることができる破砕機としては、公知の破砕機{例えば、乳化分散の理論と実際(特殊機化(株)製、1997年4月17日発行)の80〜86頁}等が使用できる。
【0085】
このようにして得られた樹脂粒子(B)は、必要に応じて、風力分級器又はふるい等を用いて分級し、体積平均粒子径、体積平均粒子径と個数平均粒子径の比を調整することができる。
【0086】
上記方法(4)において、溶剤(T)としては、樹脂粒子(B)を分散できれば、特に制限はないが、単一溶媒(例えば、アセトン、エタノール、ジメチルホルムアミド、イソプロパノールなど)、又は混合溶媒[例えば水、アルコール溶剤(メタノール、エタノール等)、アミド溶剤(ジメチルホルムアミド等)、ケトン溶剤(アセトン、メチルエチルケトンなど)、エステル溶剤(酢酸エチル、酢酸ブチルなど)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル等)、芳香族炭化水素溶剤(トルエン、キシレン等)、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)等の2種以上の混合溶剤]が挙げられる。
【0087】
樹脂粒子(B)と溶剤(T)の重量比率は、特に制限はないが、溶剤(T)に対して、樹脂粒子(B)が55重量%以下が好ましく、更に好ましくは50重量%以下であり、特に好ましくは20〜45重量%である。この範囲であれば、効率よく樹脂粒子(B)を(C)中に導入することができる。
ここで、二酸化炭素(C)と溶剤(T)の重量比率は、特に制限はないが、二酸化炭素(C)に対して、(T)が1〜50重量%が好ましく、更に好ましくは、5〜40重量%である。この範囲であれば、樹脂粒子(B)の分散性が向上する。
【0088】
方法(3)および(4)において、二酸化炭素(C)の重量に対する樹脂粒子(B)の重量比率としては、60重量%以下が好ましく、更に好ましくは55重量%以下であり、特に好ましくは20〜50重量%である。この範囲であれば、効率よく処理して樹脂粒子(X)を製造できる。
【0089】
本発明において、樹脂粒子(B)の処理に用いる二酸化炭素(C)としては、液状のものと超臨界状態のものが使用できるが、超臨界状態が好ましい。
ここで、液状の二酸化炭素とは、二酸化炭素の温度軸と圧力軸とで表す相図上において、二酸化炭素の三重点(温度=−57℃、圧力=0.5MPa)と二酸化炭素の臨界点(温度=31℃、圧力=7.4MPa)を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件である二酸化炭素を表す。一方、超臨界状態の二酸化炭素とは、臨界温度以上の温度・圧力条件である二酸化炭素を表す。なお、本発明における圧力とは、2成分以上の混合ガスの場合、全圧を示す。
【0090】
二酸化炭素(C)中には、分散媒としての物性値(粘度、拡散係数、誘電率、溶解度、界面張力等)を調整するために、他の物質を適宜含んでよく、例えば、窒素、ヘリウム、アルゴン、空気等の不活性気体等が挙げられる。
(C)と他の物質の合計中の(C)の重量分率は、好ましくは70%以上、さらに好ましくは80%以上、とくに好ましくは90%以上である。
【0091】
樹脂粒子(B)を(C)中で処理する際、下記の温度で行うことが好ましい。
減圧時に配管内で二酸化炭素が固体に相転移し、流路を閉塞させないようにするために、30℃以上が好ましく、また、樹脂粒子(B)の熱劣化を防止するために、200℃以下が好ましい。さらに30〜150℃が好ましく、より好ましくは34〜130℃、特に好ましくは35〜100℃、最も好ましくは40℃〜80℃である。
【0092】
また、該処理は下記の圧力で行うことが好ましい。
樹脂粒子(B)を(C)に良好に分散させるために好ましくは3MPa以上であり、設備コスト、運転コストの観点から、好ましくは40MPa以下である。さらに好ましくは3.5〜35MPa、より好ましくは4〜30MPa、特に好ましくは4.5〜25MPa、最も好ましくは5〜20MPaである。
【0093】
二酸化炭素(C)中で処理する際の温度及び圧力は、樹脂粒子(B)が(C)中に溶解せず、且つ二酸化炭素(C)が(B)に浸透可能な範囲内で設定することが好ましい。通常、低温・低圧ほど(B)が(C)中に溶解しない傾向となり、高温・高圧ほど二酸化炭素(C)が樹脂粒子(B)に浸透しやすい傾向となる。
【0094】
樹脂粒子(B)が分散した分散体から、通常、減圧により、二酸化炭素(C)を除去して、本発明の樹脂粒子(X)を得る。その際、独立に圧力制御された容器を多段に設けることにより段階的に減圧してもよく、また一気に常温常圧まで減圧してもよい。
樹脂粒子(X)の捕集方法は特に限定されず、フィルターでろ別する方法や、サイクロン等により遠心分離する方法が例として挙げられる。
樹脂粒子(X)は減圧後に捕集してもよく、また減圧前に一旦高圧中で捕集した後、減圧してもよい。高圧下で捕集した後に減圧する場合の、高圧下からの樹脂粒子(X)の取り出し方としては、バッチ操作で捕集容器を減圧してもよく、またロータリーバルブを使用して連続的取り出し操作を行ってもよい。
【0095】
前記の樹脂粒子(B)を二酸化炭素(C)中に分散する方法(1)を用いて、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)が(C)中に分散した分散体を得た場合、樹脂粒子(B)を形成させ処理した後、溶剤(S)を除去又は減少させる必要がある。
溶剤(S)を除去又は減少させる方法として、そのまま容器を減圧にする方法があるが、(B)中に溶解した溶剤が凝縮し、樹脂粒子(B)を再溶解してしまったり、樹脂粒子(X)を捕集する際に樹脂粒子(X)同士が合一してしまう等の問題が生じる場合がある。
好ましい方法としては、例えば、樹脂粒子(B)が(C)中に分散した分散体に、さらに二酸化炭素(C)を混合して樹脂粒子(B)から溶剤(S)を二酸化炭素の相に抽出し、つぎに、溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素(C)で置換し、その後に減圧する方法が挙げられる。
【0096】
二酸化炭素の混合方法は、分散体より高い圧力の二酸化炭素を加えてもよく、また該分散体を分散体より低い圧力の二酸化炭素中に加えてもよいが、連続操作の容易性の観点からより好ましくは後者である。該分散体と混合する二酸化炭素の量は、樹脂粒子(B)の合一防止の観点から、分散体の体積の1〜50倍が好ましく、さらに好ましくは1〜40倍、最も好ましくは1〜30倍である。上記のように樹脂粒子(B)中に含有される溶剤を除去ないし減少させ、その後、二酸化炭素を除去することにより、樹脂粒子(B)同士が合一することを防ぐことができる。
【0097】
溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素で置換する方法としては、樹脂粒子(B)を一旦フィルターやサイクロンで補足した後、圧力を保ちながら、溶剤(S)が完全に除去されるまで二酸化炭素を流通させる方法が挙げられる。流通させる二酸化炭素の量は、該分散体からの溶剤除去の観点から、分散体の体積に対して1〜100倍が好ましく、さらに好ましくは1〜70倍、最も好ましくは1〜50倍である。
【0098】
上記の製造方法で得られた本発明の樹脂粒子(X)は、液状又は超臨界状態の二酸化炭素(C)での処理により結晶化度が向上し、融点の融解熱が増加する。
【0099】
本発明の製造方法により得られた本発明の樹脂粒子(X)の体積平均粒子径は、好ましくは1〜12μmであり、より好ましくは2〜10μm、さらに好ましくは3〜8μmである。1μm以上であると粉体としてのハンドリング性が向上する。12μm以下であると溶融特性が向上する。
【0100】
樹脂粒子(X)の体積平均粒子径Dvと樹脂粒子(X)の個数平均粒子径Dnの比Dv/Dnは、好ましくは1.0〜1.5、より好ましくは1.0〜1.4、さらに好ましくは1.0〜1.3である。1.5以下であると粉体としてのハンドリング性、溶融特性が著しく向上する。
【0101】
樹脂粒子(X)の体積平均粒子径、および体積平均粒子径Dvと樹脂粒子(X)の個数平均粒子径Dnの比(Dv/Dn)は、前記の樹脂粒子(B)を(C)中に分散する方法(3)あるいは(4)を用いる場合は、樹脂粒子(B)の製造段階で調整する。方法(1)の場合、溶剤(S)に溶解させた樹脂(A)及び必要により微粒子(D)を(C)中に分散する際の、攪拌速度、および樹脂(A)に対する微粒子(D)の比率により調整することができる。攪拌速度を上げれば体積平均粒子径が小さくなり、また樹脂(A)に対する微粒子(D)の比率を多くすれば体積平均粒子径が小さくなる。
樹脂粒子(X)のDv/Dnについても同様であり、攪拌速度を上げればDv/Dnが小さくなり、また樹脂(A)に対する微粒子(D)の比率を多くすればDv/Dnが小さくなる。
このようにして得られた樹脂粒子(X)は、必要に応じて、風力分級器又はふるい等を用いて分級し、体積平均粒子径、体積平均粒子径と個数平均粒子径の比をさらに調整することができる。
【0102】
なお、体積平均粒子径及び個数平均粒子径は、レーザー式粒度分布測定装置LA−920(堀場製作所製)やマルチサイザーIII(コールター社製)、光学系としてレーザードップラー法を用いるELS−800(大塚電子社製)などで測定できる。
【実施例】
【0103】
以下、実施例及び比較例により本発明をさらに説明するが、本発明はこれらに限定されるものではない。以下、特に定めない限り、%は重量%、部は重量部を示す。
【0104】
製造例1
反応容器に、メチルエチルケトン1000部、1,6−ヘキサンジオール430部、ヘキサメチレンジイソシアネート570部を投入し、80℃で7時間反応を行い、80℃、20kPaで脱溶剤し、ポリウレタン樹脂[結晶性部(a−1)]を得た。[結晶性部(a−1)]はMw7500、融点(m)75℃、水酸基価34であった。
【0105】
製造例2
メチルエチルケトン1000部、1,6−ヘキサンジオール210部、水酸基価56の1,6−ヘキサンジオールとセバシン酸からなるポリエステルジオール(豊国製油(株)製、商品名「HS 2H−200S」)500部、ヘキサメチレンジイソシアネート290部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[結晶性部(a−2)]得た。[結晶性部(a−2)]はMw9000、融点(m)60℃、水酸基価26であった。
【0106】
製造例3
メチルエチルケトン1000部、1,6−ヘキサンジオール350部、1,6−ヘキサンジアミン90部、ヘキサメチレンジイソシアネート560部を使用する以外は、製造例1と同様に行い、ポリウレタンウレア樹脂[結晶性部(a−3)]を得た。[結晶性部(a−3)]はMw6000、融点(m)80℃、水酸基価44であった。
【0107】
製造例4
テトラヒドロフラン500部、1,2−プロピレングリコール360部、トルエンジイソシアネート640部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[非晶性部(b−1)]を得た。[非晶性部(b−1)]はMw3000であった。
【0108】
製造例5
メチルエチルケトン500部、1,4−シクロヘキサンジメタノール210部、トルエンジイソシアネート290部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[非晶性部(b−2)]を得た。[非晶性部(b−2)]はMw5000であった。
【0109】
製造例6
反応容器に、テレフタル酸270部、ビスフェノールAのPO2モル付加物730部、ジブチルチンオキサイド5部を投入し、常圧、230℃で5時間脱水反応を行った後、0.5kPaの減圧下で5時間脱水反応を行い、ポリエステル樹脂[非結晶性部(b−3)]を得た。[非結晶性部(b−3)]は、Mw5000、水酸基価55であった。
【0110】
製造例7
反応容器に、メチルエチルケトン1150部、前記の[非結晶性部(b−1)]200部、トルエンジイソシアネート50部を投入し、80℃で5時間反応させた後、前記の[結晶性部(a−1)]900部を投入し、80℃で5時間反応を行い、80℃、20kPaで脱溶剤し、樹脂(A−1)を得た。(A−1)は、Mw18000、軟化点(s)75℃、融点(m)72℃、軟化点(s)と融点(m)の比(s/m)1.04であった。
また、G’(m+20)は7×103Paであり、溶融開始温度(x)は70℃、|LogG”(x+20)−LogG”(x)|の値は3.4であった。
(m+30)℃における損失弾性率G”と(m+70)℃における損失弾性率G”の比〔G”(m+30)/G”(m+70)〕は2.7であった。
(b)と(a)の結合形式の式におけるn=1.08であった。上記物性値は表1に記載した。
反応容器に、この樹脂(A−1)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−1)得た。
【0111】
製造例8
メチルエチルケトン700部、前記の[非結晶性部(b−1)]100部、およびトルエンジイソシアネート25部を投入し反応させた後、前記の[結晶性部(a−2)]575部を投入する以外は、製造例7と同様に行い、樹脂(A−2)を得た。樹脂(A−2)の物性値は表1に記載した。
反応容器に、この樹脂(A−2)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−2)得た。
【0112】
製造例9
メチルエチルケトン1030部、前記の[非結晶性部(b−1)]200部、およびトルエンジイソシアネート50部を投入し反応させた後、前記の[結晶性部(a−3)]780部を投入する以外は、製造例7と同様に行い、樹脂(A−3)を得た。樹脂(A−3)の物性値は表1に記載した。
反応容器に、この樹脂(A−3)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−3)得た。
【0113】
製造例10
反応容器に、メチルエチルケトン780部、前記の[非結晶性部(b−2)]200部、前記の[結晶性部(a−1)]580部を投入し、80℃で5時間反応を行い、80℃、20kPaで脱溶剤し、樹脂(A−4)を得た。樹脂(A−4)の物性値は表1に記載した。
反応容器に、この樹脂(A−4)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−4)得た。
【0114】
製造例11
反応容器に、テトラヒドロフラン500部、前記の[非結晶性部(b−3)]200部、およびm−キシリレンジイソシアネート38部を投入し反応させた後、前記の[結晶性部(a−1)]600部を投入する以外は、製造例7と同様に行い、樹脂(A−5)を得た。樹脂(A−5)の物性値は表1に記載した。
反応容器に、この樹脂(A−5)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−5)得た。
【0115】
【表1】

【0116】
<分散用微粒子分散液の調製>
製造例12
滴下ロートを備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート(炭素数22個の直鎖アルキル基を有するアルコールのアクリレート:ブレンマーVA(日油製))150部、アゾビスイソブチロニトリル(AIBN)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、アクリル系結晶性樹脂を得た。この樹脂の融点は65℃、数平均分子量50000であった。
ノルマルヘキサン700部、上記のアクリル系結晶性樹脂300部を混合した後、ビーズミル(ダイノーミルマルチラボ:シンマルエンタープライゼス製)で粒径0.3mmのジルコニアビーズを用いて粉砕を行い、乳白色の分散用微粒子(D−1)が分散したヘキサン分散液を得た。この分散液の体積平均粒径は0.3μmであった。
【0117】
製造例13
製造例12のアクリル系結晶性樹脂の微粒子(D−1)分散体の調整において、ヘキサン700部をイオン交換水700部に変更した以外は同様にして、比較例用の分散用微粒子(D’−1)の水分散液を得た。この水分散液の体積平均粒径は0.3μmであった。
【0118】
実施例1
図1の実験装置において、まずバルブV1、V2を閉じ、ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素(純度99.99%)を導入し、14MPa、40℃に調整した。また樹脂溶液タンクT1に製造例7で得られた樹脂溶液(L−1)を、微粒子分散液タンクT2には製造例12で作成した分散用微粒子(D−1)のヘキサン分散液を仕込んだ。
次に、液状の二酸化炭素のボンベB1から、ポンプP3を用いて液状の二酸化炭素を分散槽T3に仕込み、超臨界状態(9MPa、40℃)に調整し、さらにタンクT2から、ポンプP2を用いて微粒子(D−1)ヘキサン分散液を導入した。
次に分散槽T3の内部を2000rpmで攪拌しながら、タンクT1から、ポンプP1を用いて樹脂溶液(L−1)を分散槽T3内に導入した。導入後T3の内部の圧力は14MPaとなった。
なお、分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
分散用微粒子(D−1)のヘキサン分散液 45部
二酸化炭素 550部
【0119】
なお、上記の導入した二酸化炭素の重量は、二酸化炭素の温度(40℃)、及び圧力(15MPa)から二酸化炭素の密度を下記文献に記載の状態式より算出し、これに分散槽T3の体積を乗じることにより算出した。
文献:Journal of Physical and Chemical Refarence data、vol.25、P.1509〜1596
【0120】
樹脂溶液(L−1)を導入後、1分間攪拌し、超臨界状態の二酸化炭素に樹脂粒子(B−1)が分散した分散体を得た。
次に、バルブV1を開き、B1からP3を用いてT3及びT4内に超臨界状態の二酸化炭素を導入することで、(B−1)の分散体をT3からT4内に移送した。(B−1)の分散体をT3からT4に移送する間、圧力が一定に保たれるように、V2の開度を調節した。この操作を30秒間行い、V1を閉めた。
次に、圧力調整バルブV2の開度を調整することで、圧力を14MPaに保持しながら、圧力ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素を導入した。
この操作により、溶剤を含む二酸化炭素を溶剤トラップ槽T5に排出すると共に、樹脂粒子(B−1)をフィルターF1に捕捉した。圧力ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素を導入する操作は、上記の分散槽T3に導入した二酸化炭素重量の5倍量を粒子回収槽T4に導入した時点で停止した。この停止の時点で、溶剤を含む二酸化炭素を、溶剤を含まない二酸化炭素で置換すると共に樹脂粒子(B−1)をフィルターF1に捕捉する操作は完了した。さらに、圧力調整バルブV2を少しずつ開き、粒子回収槽内を大気圧まで減圧することで、フィルターF1に補足されている本発明の樹脂粒子(X−1)を得た。
【0121】
実施例2〜5
樹脂溶液(L−1)を製造例8〜11で得られた樹脂溶液(L−2)〜(L−5)に変更する以外は実施例1と同様にし、本発明の樹脂粒子(X−2)〜(X−5)を得た。
【0122】
比較例1
ビーカー内にイオン交換水97部、製造例13で得られた分散用微粒子(D’−1)の水分散液15.4部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)10部を入れ均一に溶解した。ついで25℃で、TK式ホモミキサーを10,000rpmに撹拌しながら、製造例7で得られた樹脂溶液(L−1)75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまでアセトンを留去し、樹脂粒子の水性樹脂分散体を得た。
次いで濾別し40℃×18時間乾燥を行い、揮発分を0.5%以下として、比較のための樹脂粒子(R−1)を得た。この後、超臨界の二酸化炭素による処理は行わず、物性評価した。
【0123】
比較例2
ビーカー内にイオン交換水97部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)15部を入れ均一に溶解した。
ついで25℃で、TK式ホモミキサーを12,000rpmに撹拌しながら、製造例7で得られた樹脂溶液(L−1)75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまでアセトンを留去し、樹脂粒子の水性樹脂分散体を得た。
次いで濾別し40℃×18時間乾燥を行い、揮発分を0.5%以下として、比較のための樹脂粒子(R−2)を得た。この後、超臨界の二酸化炭素による処理は行わず、物性評価した。
【0124】
<融点(m)、融解熱の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃で、溶融による吸熱ピークの温度(℃)を求め、これを融点(m)とした。
また、吸熱ピークの面積より融解熱を求めた。求めた融解熱を用いて、下記計算式より、融解熱の比を求めた。
融解熱の比=H2/H1
【0125】
<体積平均粒径の評価>
樹脂粒子をドデシルベンゼンスルホン酸ナトリウム水溶液(濃度0.1%)に分散して体積平均粒径をコールターカウンター[マルチサイザーIII(ベックマン・コールター社製)]で測定した。
【0126】
<耐熱保存性の評価>
樹脂粒子の耐熱保存性を下記の方法で評価した。
直径が約3cmの30mlのガラス製スクリュー管に樹脂粒子を10g採取した。この樹脂粒子が入ったガラス製スクリュー管を50℃に温調された恒温器に15時間静置し、ブロッキングの程度により下記の基準で評価した。
○: ブロッキングが発生しない。
△: ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×: ブロッキングが発生し、簡単に指などで力を加えても分散しない。
【0127】
<低温溶融性の評価>
溶融温度は、以下の方法により評価した。
日本テストパネル社製リン酸亜鉛処理鋼板標準板に市販のコロナ帯電方式スプレーガンを用いて膜圧が20〜40μmになるように樹脂粒子を静電塗装し、焼き付け温度を変化させて評価を行ったとき、5分間焼き付けた後の目視確認による表面平滑性が良好となる最低温度を測定した。
【0128】
実施例1〜5で作成した本発明の樹脂粒子(X−1)〜(X−5)、および比較例1、2で作成した比較のための樹脂粒子(R−1)、(R−2)の評価結果を表2に示す。
【0129】
【表2】
【0130】
実施例1〜5の樹脂粒子は、耐熱保存性及び低温溶融性に優れていたのに対し、比較例1の樹脂粒子は、低温溶融性が著しく悪化し、耐熱保存性も悪化した。また、比較例2の樹脂粒子は、低温溶融性は良好であるが、耐熱保存性が著しく悪化した。
【産業上の利用可能性】
【0131】
本発明の製造方法により得られる樹脂粒子は、耐熱保存性と低温溶融性に優れているため、電子写真トナーの母体粒子、塗料用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成型用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、電気粘性用粒子、その他成型用樹脂粒子として有用である。
【符号の説明】
【0132】
T1:樹脂溶液タンク
T2:溶液タンク
T3:分散槽(最高使用圧力20MPa、最高使用温度200℃、攪拌機つき)
T4:粒子回収槽(最高使用圧力20MPa、最高使用温度100℃)
T5:溶剤トラップ
F1:セラミックフィルター(メッシュ:0.5μm)
B1、B2:二酸化炭素ボンベ
P1、P2:溶液ポンプ
P3、P4:二酸化炭素ポンプ
V1:バルブ
V2:圧力調整バルブ
【技術分野】
【0001】
本発明は、樹脂粒子およびその製造方法に関する。
【背景技術】
【0002】
従来より結晶性の高い樹脂粒子の形成法として、有機溶媒中から結晶性の樹脂を析出させる方法(例えば、特許文献1参照)、相分離溶媒を使用する方法(例えば、特許文献2参照)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2005−15589号公報
【特許文献2】特開平8−176310号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかし、上記の方法で得られた樹脂粒子は、耐熱保存性と溶融特性の両立の点で不十分であった。
本発明の課題は、耐熱保存性と溶融特性を従来になく両立できる結晶性樹脂粒子を得る製造方法を提供することである。
【課題を解決するための手段】
【0005】
発明者らは鋭意検討した結果、液状または超臨界状態の二酸化炭素で処理することにより樹脂粒子の耐熱保存性と溶融特性を両立できることを見出し、本発明に至った。
すなわち、本発明は下記2発明である。
(I) ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
(II) 上記の製造方法により得られる樹脂粒子(X)。
【発明の効果】
【0006】
本発明の製造方法で得られる本発明の樹脂粒子(X)は、耐熱保存性及び溶融特性を両立できる。
【図面の簡単な説明】
【0007】
【図1】本発明における樹脂粒子の作成に用いる実験装置のフローチャートである。
【発明を実施するための形態】
【0008】
以下、本発明を詳細に説明する。
本第1発明は、樹脂粒子の製造方法に関する発明であって、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)〔以下、二酸化炭素(C)と記載する場合がある。〕で処理した後に、この二酸化炭素(C)を除去することにより樹脂粒子(X)を得る工程を含む製造方法である。
【0009】
本発明において、得られる樹脂粒子(X)の示差走査熱量(DSC)測定による融解熱が下記関係式を満足することが必要である。
0≦H2/H1≦0.9 (1)
H1はDSC測定による初回昇温時の融解熱(J/g)を表し、H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。
【0010】
ここで、融解熱の測定はJIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して測定される。
具体的には、試料(5mg)を採取してアルミパンに入れ、示差走査熱量測定装置(DSC)(例えば、「エスアイアイナノテクノロジー(株)製 RDC220」、「セイコー電子工業(株)製 DSC20」など)により、昇温速度毎分10℃で、溶融による吸熱ピークの温度〔融点(m)〕(℃)を求めることができる。また、吸熱ピークの面積より融解熱を求めることができる。なお、初回昇温後、2回目昇温前の冷却は、冷却速度90℃/分で、0℃まで冷却する。
【0011】
関係式(1)に示したように、H2/H1は0以上であり、好ましくは0.01以上、さらに好ましくは0.1以上である。
また、H2/H1は0.9以下であり、好ましくは0.85以下、さらに好ましくは0.81以下である。0.9を超えると耐熱保存性が悪化し、好ましくない。
【0012】
本発明の樹脂粒子(X)中に含有される樹脂(A)は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と非結晶性部(b)から構成される樹脂であり、具体的には結晶性部(a)を構成する樹脂と非結晶性部(b)を構成する樹脂を結合することにより得られる。
以下に、結晶性部(a)を構成する樹脂について詳細に説明する。
結晶性部(a)を構成する樹脂は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とし、結晶性を有していれば特に制限はない。耐熱保存性の観点から、融点(m)が30〜120℃の範囲であることが好ましく、さらに好ましくは40〜100℃である。
ここで、融点(m)は、前記の融解熱の測定法と同様に、JIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して、示差走査熱量測定装置(DSC)で測定した、初回昇温時の溶融による吸熱ピークの温度(℃)を意味する。
【0013】
ポリウレタンおよび/またはポリウレア(p)は、ポリウレタン、ポリウレア、ポリウレタン・ポリウレアのいずれかを意味し、ジオール(c)を含有するポリオール成分および/またはジアミン(d)を含有するポリアミン成分と、ジイソシアネート(e)を含有するポリイソシアネート成分から合成されて得られるポリウレタンおよび/またはポリウレアである。ただし、必要に応じて3官能以上のポリオール(f)や3官能以上のポリアミン(g)、3官能以上のポリイソシアネート(h)を併用してもよい。
【0014】
ポリウレタンおよび/またはポリウレア(p)を構成するポリオール成分としては、脂肪族ジオール(c1)が好ましく、炭素数が2〜36の範囲であることが好ましい。さらに好ましくは、直鎖型脂肪族ジオール(c11)である。また、直鎖型脂肪族ポリエステルジオール(c12)および直鎖型脂肪族ポリエーテルジオール(c13)も好ましい。
脂肪族ジオール、脂肪族ポリエステルジオール、脂肪族ポリエーテルジオールが分岐型では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、脂肪族ジオールは炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0015】
ジオール(c)および必要により3官能以上(3〜8価またはそれ以上)のポリオール(f)で構成されるポリオール成分としては、直鎖型脂肪族ジオール(c11)、直鎖型脂肪族ポリエステルジオール(c12)および直鎖型脂肪族ポリエーテルジオール(c13)の合計含有量がポリオール成分の70モル%以上であることが好ましく、より好ましくは80モル%以上である。必要に応じてその他のジオール(c2)が含まれても構わない。
直鎖型脂肪族ジオール、直鎖型脂肪族ポリエステルジオールおよび直鎖型脂肪族ポリエーテルジオールの合計含有量が70モル%未満では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0016】
直鎖型脂肪族ジオール(c11)としては、具体的には、例えば、エチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,7−ヘプタンジオール、1,8−オクタンジオール、1,9−ノナンジオール、1,10−デカンジオール、1,11−ウンデカンジオール、1,12−ドデカンジオール、1,13−トリデカンジオール、1,14−テトラデカンジオール、1,18−オクタデカンジオール、1,20−エイコサンジオールなどが挙げられるが、これらに限定されるものではない。
これらのうち、入手容易性を考慮するとエチレングリコール、1,3−プロパンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,9−ノナンジオール、1,10−デカンジオールが好ましい。
【0017】
直鎖型脂肪族ポリエステルジオール(c12)としては、上記直鎖型脂肪族ジオールと直鎖型脂肪族ジカルボン酸(コハク酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸、デシルコハク酸など)からなるポリエステルジオールなどが挙げられる。
【0018】
直鎖型脂肪族ポリエーテルジオール(c13)としては、上記直鎖型脂肪族ジオールのエチレンオキサイド(以下EOと略記する)、テトラヒドロフラン(以下THFと略記する)など〕付加物(付加モル数1〜300)などが挙げられる。
【0019】
その他、必要に応じて組み合わせて使用される(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)としては、炭素数2〜36の上記以外の脂肪族ジオール(1,2−プロピレングリコール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);(c13)以外の炭素数4〜36のアルキレンエーテルグリコール(ジプロピレングリコール、ポリプロピレングリコールなど);炭素数4〜36の脂環式ジオール(1,4−シクロヘキサンジメタノール、水素添加ビスフェノールAなど);上記脂環式ジオールのアルキレンオキサイド(以下AOと略記する)〔EO、プロピレンオキサイド(以下POと略記する)、ブチレンオキサイド(以下BOと略記する)など〕付加物(付加モル数1〜30);ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど)のAO〔EO、PO、BOなど〕付加物(付加モル数2〜30);分岐型脂肪族ポリエステルジオール;芳香族ポリエステルジオール;分岐型脂肪族ポリエーテルジオール;ポリラクトンジオール(ポリε−カプロラクトンジオールなど);およびポリブタジエンジオールなどが挙げられる。
【0020】
さらに、その他のジオール(c2)としては、上記のヒドロキシル基以外の末端官能基を有しないジオール以外に、他の官能基を有するジオールを用いてもよい。
このような官能基を有するジオールとしては、カルボキシル基を有するジオール(c21)、スルホン酸基もしくはスルファミン酸基を有するジオール(c22)、およびこれらの塩等が挙げられる。
カルボキシル基を有するジオール(c21)としては、ジアルキロールアルカン酸[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸(DMPA)、2,2−ジメチロールブタン酸、2 ,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸など]が挙げられる。
【0021】
スルホン酸基もしくはスルファミン酸基を有するジオール(c22)としては、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸、スルホイソフタル酸ジ(エチレングリコール)エステル、スルファミン酸ジオール[N,N−ビス(2−ヒドロキシアルキル)スルファミン酸(アルキル基のC1〜6)またはそのAO付加物(AOとしてはEOまたはPOなど、AOの付加モル数1〜6):例えばN,N−ビス(2−ヒドロキシエチル)スルファミン酸およびN,N−ビス(2−ヒドロキシエチル)スルファミン酸PO2モル付加物など];ビス(2−ヒドロキシエチル)ホスフェートなどが挙げられる。
これらのジオールの中和塩基としては、例えば炭素数3〜30の3級アミン(トリエチルアミンなど)および/またはアルカリ金属(ナトリウム塩など)が挙げられる。
これらの、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)として好ましいものは、ビスフェノール類のAO付加物、炭素数2〜12のアルキレングリコール、カルボキシル基を有するジオール、およびこれらの併用である。
【0022】
ポリウレタンおよび/またはポリウレア(p)を構成するポリアミン成分としては、脂肪族ジアミン(d1)が好ましく、炭素数が2〜36の範囲であることが好ましい。さらに好ましくは、直鎖型脂肪族ジアミン(d11)である。
脂肪族ジアミン(d1)が分岐型では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0023】
ジアミン(d)および必要により3官能以上(3〜8価またはそれ以上)のポリアミン(g)で構成されるポリアミン成分としては、直鎖型脂肪族ジアミン(d11)の含有量がポリアミン成分の70モル%以上であることが好ましく、より好ましくは80モル%以上である。必要に応じてその他のジアミン(d2)が含まれても構わない。
直鎖型脂肪族ジアミン(d11)の含有量が70モル%未満では、ポリウレタンおよび/またはポリウレアの結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0024】
直鎖型脂肪族ジアミン(d11)としては、具体的には、例えば、エチレンジアミン、1,3−プロパンジアミン、1,4−ブタンジアミン、1,5−ペンタンジアミン、1,6−ヘキサンジアミン、1,7−ヘプタンジジアミン、1,8−オクタンジアミン、1,9−ノナンジアミン、1,10−デカンジアミン、1,11−ウンデカンジアミン、1,12−ドデカンジアミン、1,13−トリデカンジアミン、1,14−テトラデカンジアミン、1,18−オクタデカンジアミン、1,20−エイコサンジアミンなどが挙げられるが、これらに限定されるものではない。
これらのうち、入手容易性を考慮するとエチレンジアミン、1,3−プロパンジアミン、1,4−ブタンジアミン、1,5−ペンタンジアミン、1,6−ヘキサンジアミン、1,9−ノナンジアミン、1,10−デカンジアミンが好ましい。
【0025】
その他、直鎖型脂肪族ジアミン(d11)と、必要に応じて組み合わせて使用される(d11)以外の脂肪族ジアミン(d1)およびその他のジアミン(d2)としては、上記以外の脂肪族ポリアミン類(C2〜C18)および芳香族ポリアミン類(C6〜C20)が挙げられる。
上記以外の脂肪族ポリアミン類(C2〜C18)としては、〔1〕分岐脂肪族ジアミン{C2〜C6 アルキレンジアミン(1,2−プロピレンジアミンなど)、ポリアルキレン(C2〜C6)ジアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミンなど〕};〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミンなど〕;〔3〕脂環または複素環含有脂肪族ジアミン{脂環式ジアミン(C4〜C15)〔1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など〕、複素環式ジアミン(C4〜C15)〔ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジン、3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕;〔4〕芳香環含有脂肪族アミン類(C8〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど);等が挙げられる。
【0026】
芳香族ポリアミン類(C6〜C20)としては、〔1〕非置換芳香族ジアミン〔1,2−、1,3−および1,4−フェニレンジアミン、2,4´−および4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4”−トリアミン、ナフチレンジアミンなど;〔2〕核置換アルキル基〔メチル,エチル,n−およびi−プロピル、ブチルなどのC1〜C4アルキル基〕を有する芳香族ジアミン、たとえば2,4−および2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、3,3´,5,5´−テトラメチルベンジジン、3,3´,5,5´−テトラメチル−4,4´−ジアミノジフェニルメタン、3,5−ジエチル−3´−メチル−2´,4−ジアミノジフェニルメタン、3,3´−ジエチル−2,2´−ジアミノジフェニルメタン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、3,3´,5,5´−テトラエチル−4,4´−ジアミノベンゾフェノン、3,3´,5,5´−テトラエチル−4,4´−ジアミノジフェニルエーテル、3,3´,5,5´−テトライソプロピル−4,4´−ジアミノジフェニルスルホンなど〕、およびこれらの異性体の種々の割合の混合物;〔3〕核置換電子吸引基(Cl,Br,I,Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ジアミン〔メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン;4,4´−ジアミノ−3,3´−ジメチル−5,5´−ジブロモ−ジフェニルメタン、3,3´−ジクロロベンジジン、3,3´−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキシド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4´−メチレンビス(2−ヨードアニリン)、4,4´−メチレンビス(2−ブロモアニリン)、4,4´−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリンなど〕;〔4〕2級アミノ基を有する芳香族ジアミン〔上記〔1〕〜〔3〕の芳香族ジアミンの−NH2の一部または全部が−NH−R´(R´はアルキル基たとえばメチル,エチルなどの低級アルキル基)で置き換ったもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕が挙げられる。
【0027】
ポリアミン成分としては、これらの他、ポリアミドポリアミン〔ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど〕、ポリエーテルポリアミン〔ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物など〕等が挙げられる。
【0028】
ジイソシアネート(e)および必要に応じて併用される3官能以上のポリイソシアネート(h)としては、炭素数(NCO基中の炭素を除く。以下同様。)6〜20の芳香族ジイソシアネート、炭素数2〜18の脂肪族ジイソシアネート、炭素数4〜15の脂環式ジイソシアネート、炭素数8〜15の芳香脂肪族ジイソシアネート、これらのジイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。また、必要により3官能以上(3〜8価またはそれ以上)のポリイソシアネート(h)を併用してもよい。
【0029】
上記芳香族ジイソシアネートおよび3価以上の芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)またはその混合物との縮合生成物;ジアミノジフェニルメタンと少量(たとえば5〜20重量%)の3官能以上のポリアミンとの混合物〕のホスゲン化物:ポリアリルポリイソシアネート(PAPI)]、1,5−ナフチレンジイソシアネート、4,4’,4”−トリフェニルメタントリイソシアネート、m−およびp−イソシアナトフェニルスルホニルイソシアネートなどが挙げられる。
【0030】
上記脂肪族ジイソシアネートおよび3価以上の脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエートなどが挙げられる。
【0031】
上記脂環式ジイソシアネートおよび3価以上の脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−および/または2,6−ノルボルナンジイソシアネートなどが挙げられる。
【0032】
上記芳香脂肪族ジイソシアネートおよび3価以上の芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
【0033】
また、上記ジイソシアネートおよび3価以上のポリイソシアネートの変性物としては、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。
具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのジイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]などが挙げられる。
【0034】
これらのポリイソシアネート成分のうちで好ましいものは、炭素数6〜15の芳香族ジイソシアネート、炭素数4〜12の脂肪族ジイソシアネート、および炭素数4〜15の脂環式ジイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0035】
ポリウレタンおよび/またはポリウレア(p)の分子量は、ゲルパーミエーションクロマトグラフィー(GPC)によって求めた重量平均分子量(Mw)が2000〜80000のものが好ましい。ガラス転移温度(Tg)は、好ましくは−100〜40℃、さらに好ましくは−80〜0℃である。融点(m)は、好ましくは40〜100℃である。
【0036】
結晶性部(a)を構成する樹脂は、ポリウレタンおよび/またはポリウレア(p)を必須構成成分とし、結晶性を有していれば特に制限はなく、ポリウレタンおよび/またはポリウレア(p)の単独樹脂であっても、結晶性を有する他の樹脂との複合樹脂であってもよい。
複合樹脂に使用される結晶性を有する他の樹脂としては、ポリウレタンおよび/またはポリウレア(p)のブロックを導入しやすいことから、ポリエステル樹脂およびポリアミド樹脂から選ばれる1種以上の樹脂が好ましい。
複合樹脂は、例えば、結晶性ポリエステル樹脂、結晶性ポリアミド樹脂、およびこれらの混合物とポリウレタンおよび/またはポリウレア(p)とを結合することで得られる。
【0037】
結晶性ポリエステル樹脂は、ジオール(c)を含有するポリオール成分とジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリエステル樹脂が好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリカルボン酸(j)を併用してもよい。
結晶性ポリアミド樹脂は、ジアミン(d)を含有するポリアミンと、ジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリアミド樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリカルボン酸(j)を併用してもよい。
【0038】
結晶性ポリエステル樹脂、結晶性ポリアミド樹脂に用いられるポリオール成分、ポリカルボン酸成分、およびポリアミン成分(それぞれ3官能以上のものを含む)についてそれぞれ示す。
【0039】
ポリオール成分のうち、ジオール(c)としては、脂肪族ジオール(c1)が好ましく、炭素数が2〜36の範囲であることが好ましい。また直鎖型脂肪族ジオール(c11)がより好ましい。
脂肪族ジオール(c1)が分岐型では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0040】
ジオール(c)および必要により3官能以上(3〜8価またはそれ以上)のポリオール(f)で構成されるポリオール成分としては、直鎖型脂肪族ジオール(c11)の含有量がポリオール成分の80モル%以上であることが好ましく、より好ましくは90モル%以上である。必要に応じてその他のジオール(c2)が含まれても構わない。
直鎖型脂肪族ジオール(c11)の含有量が80モル%未満では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0041】
直鎖型脂肪族ジオール(c11)としては、具体的には、前記のものが挙げられ、好ましいものも同様である。
その他、直鎖型脂肪族ジオール(c11)と必要に応じて組み合わせて使用される、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)としては、前記の、(c11)以外の脂肪族ジオール(c1)およびその他のジオール(c2)、カルボキシル基を有するジオール(c21)、スルホン酸基もしくはスルファミン酸基を有するジオール(c22)、およびこれらの塩として例示したものが挙げられ、好ましいものも同様である。
必要により用いられる3官能以上(3〜8価またはそれ以上)のポリオール(f)としては、前記のものが挙げられ、好ましいものも同様である。
【0042】
ポリカルボン酸成分のうち、ジカルボン酸(i)としては、炭素数4〜36のアルカンジカルボン酸(コハク酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸、デシルコハク酸など)、炭素数6〜40の脂環式ジカルボン酸〔ダイマー酸(2量化リノール酸)など〕、炭素数4〜36のアルケンジカルボン酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸などのアルケニルコハク酸、マレイン酸、フマール酸、シトラコン酸など)、炭素数8〜36の芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、t−ブチルイソフタル酸、2,6−ナフタレンジカルボン酸、4,4’−ビフェニルジカルボン酸など)などが挙げられる。
また、必要により、3官能以上(3〜6価またはそれ以上)のポリカルボン酸(j)を併用してもよい。
3官能以上のポリカルボン酸(j)としては、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など)、および炭素数6〜36の脂肪族または脂環式ポリカルボン酸(ヘキサントリカルボン酸など)などが挙げられる。
なお、ジカルボン酸(i)、および3官能以上のポリカルボン酸(j)としては、上述のものの酸無水物または炭素数1〜4の低級アルキルエステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
結晶性や入手容易性を考慮すると、アジピン酸、セバシン酸、ドデカンジカルボン酸、テレフタル酸、およびイソフタル酸が特に好ましい。
【0043】
ポリアミン成分のうち、ジアミン(d)としては、脂肪族ジアミン(d1)が好ましく、炭素数が2〜36の範囲であることが好ましい。また直鎖型脂肪族ジアミン(d11)がより好ましい。
脂肪族ジアミン(d1)が分岐型では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。また、炭素数が36を超えると、実用上の材料の入手が困難な場合がある。
【0044】
ジアミン(d)および必要により3官能以上(3〜8価またはそれ以上)のポリアミン(g)で構成されるポリアミン成分としては、直鎖型脂肪族ジアミン(d11)の含有量がポリアミン成分の80モル%以上であることが好ましく、より好ましくは90モル%以上である。必要に応じてその他のジアミン(d2)が含まれても構わない。
直鎖型脂肪族ジアミン(d11)の含有量が80モル%未満では、樹脂の結晶性が低下し、融点が降下するため、耐熱保存性が悪化してしまう場合がある。
【0045】
ポリアミン成分の例としては、前記のものが挙げられる。
【0046】
結晶性部(a)を構成する樹脂中のポリウレタンおよび/またはポリウレア(p)の割合は、好ましくは20重量%以上、さらに好ましくは30重量%以上、とくに好ましくは40〜95重量%である。
【0047】
ポリウレタンおよび/またはポリウレア(p)と結晶性ポリエステル樹脂、および結晶性ポリアミド樹脂を結合する方法は、それぞれが含有する官能基の反応性を考慮して、結合剤(カップリング剤)を使用するかしないかを選択し、また使用する場合は、含有する官能基にあった結合剤を選択し、結合させ複合樹脂とすることができる。
【0048】
複合樹脂を作製する時に結合剤を使わない場合、必要により加熱減圧しつつ、ポリウレタンおよび/またはポリウレア(p)が含有する官能基とそれ以外の樹脂が含有する官能基との反応を進める方法が挙げられる。反応温度は180℃〜230℃で行うのが好ましい。
【0049】
結合剤を使う場合は、末端の官能基の種類に合わせて、種々の結合剤が使用できる。
多価カルボン酸、多価アルコール、多価イソシアネート、酸無水物、多官能エポキシ等を結合剤として用いて、脱水反応や、付加反応を行うことで、複合樹脂である結晶性部(a)が得られる。
多価カルボン酸および酸無水物としては、前記ポリカルボン酸成分と同様のものが挙げられる。多価アルコールとしては、前記ポリオール成分と同様のものが挙げられる。多価イソシアネートとしては、前記ポリイソシアネート成分と同様のものが挙げられる。
多官能エポキシとしては、ビスフェノールA型および−F型エポキシ化合物、フェノールノボラック型エポキシ化合物、クレゾールノボラック型エポキシ化合物、水添ビスフェノールA型エポキシ化合物、ビスフェノールAまたは−FのAO付加体のジグリシジルエーテル、水添ビスフェノールAのAO付加体のジグリシジルエーテル、ジオール(エチレングリコール、プロピレングリコール、ネオペンチルグリコール、ブタンジオール、ヘキサンジオール、シクロヘキサンジメタノール、ポリエチレングリコールおよびポリプロピレングリコール等)のジグリシジルエーテル、トリメチロールプロパンジおよび/またはトリグリシジルエーテル、ペンタエリスリトールトリおよび/またはテトラグリシジルエーテル、ソルビトールヘプタおよび/またはヘキサグリシジルエーテル、レゾルシンジグリシジルエーテル、ジシクロペンタジエン・フェノール付加型グリシジルエーテル、メチレンビス(2,7−ジヒドロキシナフタレン)テトラグリシジルエーテル、1,6−ジヒドロキシナフタレンジグリシジルエーテル、ポリブタジエンジグリシジルエーテルなどが挙げられる。
【0050】
複合樹脂を作製する方法のうち、脱水反応の例としては、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂が共に水酸基を有する場合には、これらの水酸基を結合剤(例えば多価カルボン酸)で結合する反応が挙げられる。この場合、例えば、無溶剤下、反応温度180℃〜230℃で反応し、複合樹脂である結晶性部(a)が得られる。
【0051】
付加反応の例としては、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂が共に末端に水酸基を有する場合であり、これらを結合剤(例えば多価イソシアネート)で結合する反応や、またポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂の片方が末端に水酸基を有する場合で、もう一方が末端にイソシアネート基を有する樹脂の場合、結合剤を用いずにこれらを結合する反応が挙げられる。この場合、例えば、ポリウレタンおよび/またはポリウレア(p)、複合樹脂を形成するその他の樹脂ともに溶解可能な溶剤に溶解させ、これに必要であるなら結合剤を投入し、反応温度80℃〜150℃で反応し、複合樹脂である結晶性部(a)が得られる。
【0052】
非結晶性部(b)を構成する樹脂としては、ポリエステル樹脂(ラクトン開環重合物を含む)、ポリウレタン樹脂、ポリウレア樹脂、ポリアミド樹脂、ポリスチレン樹脂、スチレンアクリル系ポリマー等が挙げられる。
これらのうち、ポリエステル樹脂、ポリウレタン樹脂、ポリウレア樹脂、ポリアミド樹脂、およびそれらの複合樹脂が好ましく、ポリウレタン樹脂およびポリエステル樹脂がさらに好ましい。
【0053】
前記結晶性部(a)と同様に、ポリエステル樹脂は、ジオール(c)を含有するポリオール成分とジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリエステル樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリカルボン酸(j)を併用できる。
ポリウレタン樹脂は、ジオール(c)を含有するポリオール成分と、ジイソシアネート(e)を含有するポリイソシアネート成分とから合成されるポリウレタン樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリオール(f)や、3官能以上のポリイソシアネート(h)を併用できる。
ポリウレア樹脂は、ジアミン(d)を含有するポリアミンと、ジイソシアネート(e)を含有するポリイソシアネート成分とから合成されるポリウレア樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリイソシアネート(h)を併用できる。
ポリアミド樹脂は、ジアミン(d)を含有するポリアミンと、ジカルボン酸(i)を含有するポリカルボン酸成分とから合成されるポリアミド樹脂であることが好ましい。ただし、必要に応じて、3官能以上のポリアミン(g)や、3官能以上のポリカルボン酸(j)を併用できる。
非結晶性ポリエステル樹脂、非結晶性ポリウレタン樹脂、非結晶性ポリアミド樹脂、非結晶性ポリウレア樹脂を構成するモノマーとしては、前記ポリオール成分、前記ポリカルボン酸成分、前記ポリイソシアネート成分、および前記ポリアミン成分の具体例として示したものと同様のものが挙げられ、非結晶性樹脂となるものであればいかなる組合せでも構わない。
【0054】
非結晶性部(b)を構成する樹脂のガラス転移温度(Tg)は、耐熱保存性の観点から、好ましくは40〜250℃、さらに好ましくは50〜240℃、とくに好ましくは60〜230℃である。
ここで、ガラス転移温度は、JIS K7122(1987)「プラスチックの転移熱測定方法」に準拠して、示差走査熱量測定装置(セイコー電子工業(株)製 DSC20、エスアイアイナノテクノロジー(株)製 RDC220等)を用いて、測定される。
【0055】
結晶性部(a)を構成する樹脂と非結晶性部(b)を構成する樹脂との結合は、(a)および(b)を構成する樹脂のそれぞれの末端官能基の反応性を考慮して、結合剤(カップリング剤)を使用するかしないかを選択し、また使用する場合は、末端官能基にあった結合剤を選択し、(a)と(b)を結合させ、ブロックポリマーである樹脂(A)とすることが出来る。
なお、上記方法で、樹脂(A)と未反応の(a)および/または(b)の混合物〔好ましくは(A)と(a)の混合物〕が得られる場合、混合物をそのまま本発明の樹脂粒子の製造方法に使用してもよい。
【0056】
結合剤を使わない場合、必要により加熱減圧しつつ、(a)を形成する樹脂の末端官能基と(b)を形成する樹脂の末端官能基の反応を進める。特に末端の官能基がカルボキシル基と水酸基との反応や、カルボキシル基とアミノ基との反応の場合、片方の樹脂の酸価が高く、もう一方の樹脂の水酸基価やアミン価が高い場合、反応がスムーズに進行する。反応温度は180℃〜230℃で行うのが好ましい。
【0057】
結合剤を使う場合は、末端の官能基の種類に合わせて、種々の結合剤が使用できる。
多価カルボン酸、多価アルコール、多価イソシアネート、酸無水物、多官能エポキシ等を結合剤として用いて、脱水反応や、付加反応を行うことで、結晶性部(a)と非結晶性部(b)を結合させて、樹脂(A)が得られる。
これらの結合剤の具体例としては、前記のものが挙げられる。
【0058】
(a)と(b)を結合させる方法のうち、脱水反応の例としては、結晶性部(a)、非結晶部(b)とも末端に水酸基を有する樹脂の場合には、これらの水酸基を結合剤(例えば多価カルボン酸)で結合する反応が挙げられる。この場合、例えば、無溶剤下、反応温度180℃〜230℃で反応し樹脂(A)が得られる。
付加反応の例としては、結晶性部(a)、非結晶性部(b)とも末端に水酸基を有する樹脂であり、これらを結合剤(例えば多価イソシアネート)で結合する反応や、また結晶性部(a)、非結晶性部(b)の片方が末端に水酸基を有する樹脂で、もう一方が末端にイソシアネート基を有する樹脂の場合、結合剤を用いずにこれらを結合する反応が挙げられる。この場合、例えば、結晶性部(a)、非結晶性部(b)ともに溶解可能な溶剤に溶解させ、これに必要であるなら結合剤を投入し、反応温度80℃〜150℃で反応し樹脂(A)が得られる。
【0059】
結晶性部(a)と非結晶性部(b)から構成される樹脂(A)は、結晶性樹脂であることが好ましく、耐熱保存性の観点から、融点(m)は、好ましくは40℃以上、さらに好ましくは50℃以上、特に好ましくは55℃以上である。また、溶融特性の観点から、110℃以下が好ましく、さらに好ましくは100℃以下、特に好ましくは90℃以下である。ここで、融点(m)は、前記の方法で測定される。
【0060】
樹脂(A)の軟化点(s)[℃]と融点(m)[℃]との比(s/m)は、好ましくは0.8〜1.55であり、より好ましくは0.8〜1.2、特に好ましくは0.85〜1.15である。この範囲内であると、劣化しにくい樹脂粒子となる。なお、軟化点(s)は、次のように測定される値である。
<軟化点>
降下式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、プランジャーの降下量の最大値の1/2に対応する温度をグラフから読み取り、この値(測定試料の半分が流出したときの温度)を軟化点とする。
【0061】
樹脂(A)の粘弾性特性において、下記〔条件1〕を満たすのが好ましく、下記〔条件1−2〕を満たすのがさらに好ましい。
〔条件1〕 G’(m+20)=50〜1×106[Pa]
〔条件1−2〕 G’(m+20)=100〜5×105[Pa]
[G’:貯蔵弾性率[Pa]]
(m+20)℃におけるG’が50Pa以上であると、樹脂粒子を静電塗装する場合、低温塗布時でも均一に塗布しやすく、塗布温度領域が広くなる。また、1×106[Pa]以下であると、低温での溶融性が向上する。
動的粘弾性測定値(貯蔵弾性率G’、損失弾性率G”)は、Rheometric Scientific社製 動的粘弾性測定装置 RDS−2を用い周波数1Hz条件下で測定される。測定温度範囲は30℃〜200℃で、この温度間の溶融粘弾性を測定することによって、温度−G’、温度−G”の曲線として得ることができる。
〔条件1〕を満たす樹脂(A)は、(A)を構成する組成中の結晶性成分の比率を調整することや樹脂分子量を調整すること等により得ることができる。例えば、結晶性部(a)の比率や結晶性成分の比率を増加させると、G’(m+20)の値は小さくなる。結晶性成分としては、直鎖構造を有するポリオール、ポリイソシアネート等が挙げられる。また樹脂分子量を低下させることでもG’(m+20)の値は小さくなる。
【0062】
樹脂(A)の溶融開始温度(x)は、好ましくは(m±20)℃(mは融点)の温度範囲内であり、さらに好ましくは(m±15)℃の温度範囲内、特に好ましくは(m±10)℃の温度範囲内である。(x)は、具体的には30〜100℃が好ましく、さらに好ましくは40〜80℃である。なお、溶融開始温度(x)は、次のように測定される値である。
<溶融開始温度>
降下式フローテスター{たとえば、(株)島津製作所製、CFT−500D}を用いて、1gの測定試料を昇温速度6℃/分で加熱しながら、プランジャーにより1.96MPaの荷重を与え、直径1mm、長さ1mmのノズルから押し出して、「プランジャー降下量(流れ値)」と「温度」とのグラフを描き、試料の熱膨張によるピストンのわずかな上昇が行われた後、再びピストンが明らかに下降し始める点の温度をグラフから読み取り、この値を溶融開始温度とする。
【0063】
また、樹脂(A)は、損失弾性率G”[Pa]と溶融開始温度(x)[℃]に関して、以下の〔条件2〕を満たすことが好ましく、〔条件2−2〕を満たすことがさらに好ましく、〔条件2−3〕を満たすことが特に好ましく、〔条件2−4〕を満たすことが最も好ましい。
〔条件2〕 |LogG”(x+20)−LogG”(x)|>2.0
〔条件2−2〕 |LogG”(x+20)−LogG”(x)|>2.5
〔条件2−3〕 |LogG”(x+15)−LogG”(x)|>2.5
〔条件2−4〕 |LogG”(x+10)−LogG”(x)|>2.5
樹脂(A)の溶融開始温度(x)が上記範囲内であり、かつ〔条件2〕を満たすと、樹脂の低粘性化速度が速く、樹脂粒子を静電塗装する場合、塗布温度領域の低温側、高温側で同等の表面平滑性を得ることができる。また、溶融開始から塗布可能粘性に至るまでが速く、優れた低温溶融性を得るのに有利である。〔条件2〕は、どれだけ早く、少ない熱で溶融できるかという、樹脂のシャープメルト性の指標であり、実験的に求めたものである。
溶融開始温度(x)の好ましい範囲、および〔条件2〕を満たす樹脂(A)は、(A)の構成成分中の結晶性成分の比率を調整すること等により得ることができる。例えば、結晶性成分の比率を大きくすると、(m)と(x)の温度差が小さくなる。
【0064】
また、樹脂(A)の粘弾性特性において、(m+30)℃の損失弾性率G”と(m+70)℃の損失弾性率G”の比〔G”(m+30)/G”(m+70)〕が0.05〜50であることが好ましく、より好ましくは0.1〜10である〔m:(A)の融点〕。
損失弾性率の比が上記の範囲で維持されることによって、樹脂粒子を静電塗装する場合、塗布温度領域でより安定した表面平滑性を得ることができる。
上記のG”の比の条件を満たす樹脂(A)は、(A)を構成する組成中の結晶性成分の比率や結晶性部(a)の分子量を調整すること等により得ることができる。例えば、結晶性部(a)の比率や結晶性成分の比率を増加させると、〔G”(m+30)/G”(m+70)〕の値は小さくなる。また結晶性部(a)の分子量を増加させると〔G”(m+30)/G”(m+70)〕の値は小さくなる。結晶性成分としては、直鎖構造を有するポリオール、ポリイソシアネート等が挙げられる。
【0065】
樹脂(A)の重量平均分子量(Mw)は、溶融特性の観点から5000〜100000が好ましく、さらに好ましくは6000〜80000、特に好ましくは8000〜50000である。
(A)を構成する結晶性部(a)のMwは、2000〜80000が好ましく、さらに好ましくは3000〜60000、特に好ましくは4000〜30000である。
また、(A)を構成する非結晶性部(b)のMwは、500〜50000が好ましく、さらに好ましくは750〜20000であり、特に好ましくは1000〜10000である。
結晶性部(a)及び非結晶性部(b)のMwは、結合させる前にそれぞれ(a)を構成する樹脂及び(b)を構成する樹脂のMwを測定することで得られる。
なお、本発明において樹脂の重量平均分子量(Mw)は、ゲルパーミエーションクロマトグラフイー(GPC)を用いて以下の条件で測定される。
【0066】
装置(一例) :東ソー(株)製 HLC−8120
カラム(一例):TSK GEL GMH6 2本 〔東ソー(株)製〕
測定温度 :40℃
試料溶液 :0.25重量%のTHF溶液
溶液注入量 :100μL
検出装置 :屈折率検出器
基準物質 :東ソー製 標準ポリスチレン(TSKstandard POLY
STYRENE)12点(分子量 500 1050 2800
5970 9100 18100 37900 96400
190000 355000 1090000 2890000)
【0067】
結晶性部(a)が樹脂(A)中に占める割合は、30〜95重量%が好ましく、さらに好ましくは40〜90重量%であり、特に好ましくは50〜85重量%である。この範囲であると樹脂(A)の結晶性が損なわれず、耐熱保存性が良好である。
【0068】
樹脂(A)は、結晶性部(a)と非結晶性部(b)から構成されるブロック樹脂であるが、(a)と(b)とが、下記の形式で線状に結合された両末端が(a)の樹脂であり、{−(b)−(a)}の単位の繰り返し数の平均値nが0.5〜3.5であることが好ましく、さらに好ましくはn=0.7〜2.0、とくに好ましくはn=0.9〜1.5である。
(a){−(b)−(a)}n
上記式は、具体的には、結晶性部(a)と非結晶性部(b)とが、
(a)〔n=0〕、
(a)−(b)−(a)〔n=1〕、
(a)−(b)−(a)−(b)−(a)〔n=2〕、
(a)−(b)−(a)−(b)−(a)−(b)−(a)〔n=3〕
等の形式で線状に結合された樹脂、およびこれらの混合物〔n=0のみからなるものを除く〕を意味する。なお、nが0のものを含有するということは、樹脂(A)と共に結晶性部(a)を構成する樹脂を含有することを意味する。
nが3.5以下であると、樹脂(A)の結晶性が損なわれない。またnが0.5以上であると(A)の溶融後の弾性が良好であり、樹脂粒子を静電塗装に用いる場合、塗布可能温度領域がより広くなる。なお、nは原料の使用量〔(a)と(b)のモル比〕から求めた計算値である。また、樹脂(A)の結晶化度の観点から(A)の両末端は結晶性部(a)であることが好ましい。
なお、両末端が非結晶性部(b)である場合は、結晶化度が落ちるため、樹脂(A)に結晶性を持たせるために、(A)中の結晶性部(a)の比率を75重量%以上にするのが好ましい。
【0069】
本発明において、樹脂粒子(B)を液状又は超臨界状態の二酸化炭素(C)中で処理し、その後に、二酸化炭素(C)を除去することにより、目的の樹脂粒子(X)を得ることができる。ここで、処理とは樹脂粒子(B)を(C)中に分散し、一定時間(C)と接触させ、(B)を膨潤させることをいう。この処理を行うことにより、H2/H1の比が1または1以上である樹脂粒子(B)でも、0.9以下である目的の樹脂粒子(X)に変性することができる場合がある。
上記一定時間〔樹脂粒子(B)が形成されるに要する時間も含む。〕は、通常10秒〜180分、好ましくは30秒〜60分である。
膨潤は、一定時間、樹脂粒子(B)を二酸化炭素(C)と接触させることにより、二酸化炭素が樹脂粒子(B)中に浸透することで起こる。従って、膨潤の度合いは(C)との接触時間、(C)の圧力、温度によって調節できる。
【0070】
樹脂粒子(B)を(C)中に分散する方法としては、以下の(1)〜(4)が挙げられる。
(1)溶剤(S)に溶解させた樹脂(A)を(C)中に分散し、樹脂粒子(B)分散体を得る方法
(2)溶融させた樹脂(A)を(C)中に分散し、樹脂粒子(B)分散体を得る方法
(3)別法にて樹脂粒子(B)を作成し、(B)を攪拌や超音波照射等により直接(C)中に分散する方法
(4)別法にて作成された樹脂粒子(B)を溶剤(T)中に分散し、該分散液を(C)中に導入する方法
なお、(1)及び(2)の分散方法において、分散用微粒子(D)を使用することが好ましい。(D)を(C)中に分散させることで、(D)は(B)の表面に吸着し、(B)を(C)中に安定に分散させることができる。
これらの方法のうち、樹脂粒子の粒子径の調整が容易で、粒度分布(体積平均粒子径Dvと個数平均粒子径Dnの比Dv/Dn)を小さくすることができる点から(1)が好ましい。
【0071】
溶剤(S)としては、例えば、ケトン溶剤(アセトン、メチルエチルケトン等)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、環状エーテル等)、エステル溶剤(酢酸エステル、ピルビン酸エステル、2−ヒドロキシイソ酪酸エステル、乳酸エステル等)、アミド溶剤(ジメチルホルムアミド等)、アルコール類(メタノール、エタノール、フッ素含有アルコール等)、芳香族炭化水素溶剤(トルエン、キシレン等)、および脂肪族炭化水素溶剤(オクタン、デカン等)などが挙げられる。これらの溶剤の2種以上の混合溶剤、または、これらの有機溶剤と水との混合溶剤を用いることもできる。
好ましくは、溶剤除去の観点から、混合溶剤(特に、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、およびアセトンと水の混合溶剤)である。
【0072】
樹脂(A)の溶液(L)は、樹脂(A)を溶剤(S)に溶解させて製造する。
溶液(L)の40℃における粘度は、好ましくは10〜100万mPa・sであり、更に好ましくは、50〜50万mPa・sであり、特に好ましくは、100〜20万mPa・sである。この範囲であれば、樹脂粒子(B)の分散性が向上する。
また、溶液(L)中の樹脂(A)の重量比率は、好ましくは5〜95重量%であり、更に好ましくは、10〜90重量%であり、特に好ましくは、15〜85重量%である。この範囲であれば、効率よく樹脂粒子(B)を形成することができる。
【0073】
必要により用いる分散用微粒子(D)としては、樹脂粒子(B)を分散できれば、特に制限はなく、無機微粒子(シリカ、チタニア等)、有機微粒子(アクリル樹脂、ポリエステル樹脂など)などが挙げられる。
微粒子(D)の体積平均粒子径は、樹脂粒子(B)を分散できれば、特に制限はないが、好ましくは30〜1000nmであり、さらに好ましくは50〜500nmである。この範囲であれば、(C)中での樹脂粒子(B)の分散性が向上する。
微粒子(D)と樹脂粒子(B)の重量比率(重量%)は、特に制限はないが、樹脂粒子(B)に対して、0.1〜20%が好ましく、更に好ましくは、0.5〜15%である。この範囲であれば、樹脂粒子(B)の分散性が向上する。
微粒子(D)としては、(C)に溶解せず、(C)中に安定分散するものが好ましい。
【0074】
本発明において、微粒子(D)を二酸化炭素(C)中に分散する方法はいかなる方法でもよく、例えば、容器内に(D)及び(C)を仕込み、攪拌や超音波照射等により、(D)を直接(C)中に分散する方法や、微粒子(D)が溶剤中に分散された分散液を(C)中に導入する方法等が挙げられる。
【0075】
上記溶剤としては、溶剤(S)と同様のものが挙げられる。微粒子(D)の分散性から、好ましくは、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)、及びエステル溶剤(酢酸エチル、酢酸ブチルなど)である。
【0076】
微粒子(D)と溶剤の重量比率(重量%)は、特に制限はないが、溶剤に対して、微粒子(D)が50以下が好ましく、更に好ましくは30以下であり、特に好ましくは20以下である。この範囲であれば、効率よく微粒子(D)を(C)中に導入することができる。
微粒子(D)を溶剤中に分散する方法としては特に制限はないが、微粒子(D)を溶剤に仕込み、攪拌や超音波照射等により直接分散する方法や、微粒子を高温下で溶剤に溶解させて晶析する方法などが挙げられる。
【0077】
樹脂粒子(B)を二酸化炭素(C)中に分散する方法(1)において、樹脂(A)の溶液(L)は、(C)中に分散するため、(C)と混合する際の温度において適度な粘度であることが好ましく、粒度分布の観点から、好ましくは10万mPa・s以下、さらに好ましくは5万mPa・s以下である。樹脂(A)の(C)への溶解度は、好ましくは3%以下、さらに好ましくは1%以下である。
樹脂(A)のSP値は、好ましくは8〜16、さらに好ましくは9〜14である。SP値とは、下記に示した様に、凝集エネルギー密度と分子容の比の平方根で表されるものである。
SP=(△E/V)1/2
ここで△Eは凝集エネルギー密度を表す。Vは分子容を表し、その値は、ロバート エフ.フェドールス(Robert F.Fedors)らの計算によるもので、例えばポリマー エンジニアリング アンド サイエンス(Polymer engineering and science)第14巻、147〜154頁に記載されている。
【0078】
本発明において、樹脂粒子(B)および樹脂粒子(X)中に他の添加剤(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を含有しても差し支えない。樹脂粒子(B)および(X)中に他の添加剤を含有させる方法としては、あらかじめ樹脂(A)と添加剤を混合した後、(C)中にその混合物を加えて分散させるのが好ましい。
【0079】
方法(1)において、樹脂(A)の溶液(L)を、二酸化炭素(C)中に分散する方法はいかなる方法を用いてもよい。具体例としては、樹脂(A)の溶液(L)を攪拌機や分散機等で分散する方法、樹脂(A)の溶液(L)を二酸化炭素(C)中にスプレーノズルを介して噴霧して液滴を形成し、液滴中の樹脂を過飽和状態とし、樹脂粒子を析出させる方法(ASES:Aerosol Solvent Extraction Systemとして知られている)、同軸の多重管(2重管、3重管等)から溶液(L)を高圧ガス、エントレーナ等とともにそれぞれ別の管から同時に噴出させて、液滴に外部応力を加え分裂を促進させて、粒子を得る方法(SEDS:Solution Enhanced Dispersion by Supercritical Fluidsとして知られている)、超音波を照射する方法等が挙げられる。
【0080】
このようにして二酸化炭素(C)中に樹脂(A)の溶液(L)を分散し、必要により微粒子(D)を表面に吸着させることにより、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)が(C)中に分散した分散体が形成される。なお、微粒子(D)を用いた場合、樹脂粒子(B)の表面には、微粒子(D)が固着しているか、(D)が被膜化した被膜が形成される。
分散体は単一相であることが好ましい。すなわち、(B)が分散している二酸化炭素(C)を含む相の他に、溶剤(S)相が分離する状態は好ましくない。したがって、溶剤相が分離しないように、(C)に対する(A)の溶液(L)の量を設定することが好ましい。溶液(L)の量は、例えば(C)に対して90重量%以下が好ましく、さらに好ましくは5〜80重量%、特に好ましくは10〜70重量%である。
なお、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)中に含有する(S)の量は、好ましくは10〜90重量%、さらに好ましくは20〜70重量%である。
また、樹脂(A)と二酸化炭素(C)の重量比は、好ましくは(A):(C)が、1:(0.1〜100)、さらに好ましくは1:(0.5〜50)、特に好ましくは1:(1〜20)である。
【0081】
上記方法(2)は、樹脂(A)を溶剤(S)に溶解させた溶液(L)の代わりに、溶融させた樹脂(A)を用いる以外は、上記方法(1)と同様であり、樹脂(A)を含有する樹脂粒子(B)が二酸化炭素(C)中に分散した分散体が形成される。
【0082】
上記方法(3)及び(4)において、液状、塊状、粒状、ペレット状、または粗粉末状の樹脂(A)から樹脂粒子(B)を作成する方法として、以下の方法が挙げられる。
(5)液状にした樹脂(A)を水性媒体に分散し、取り出す方法。
(6)固状の樹脂(A)を破砕または粉砕する方法。
【0083】
方法(5)について説明する。
固状の樹脂であっても、樹脂(A)を融点以上に加熱することや、樹脂(A)を有機溶剤に溶解することにより液状にすることができる。
有機溶剤としては、前記の溶剤(S)等を用いることが出来る。
水性媒体としては、水を必須構成成分とする液体であれば制限なく使用でき、水、並びに界面活性剤を含む水溶液等を用いることができる。
界面活性剤としては、公知の界面活性剤(たとえば、特開2004−124059号公報に記載の界面活性剤)等を使用することができる。
分散装置としては、一般に乳化機、分散機として使用されているものであれば特に限定されず、公知の分散装置(たとえば、特開2002−284881号公報に記載の分散装置)等を使用することができる。
粒子化された樹脂粒子(B)を水性媒体から取り出す方法としては、固液分離法(遠心分離器、スパクラフィルター及び/又はフィルタープレス等)により樹脂粒子(B)を分離した後、水洗する方法等が適用できる。
得られる樹脂粒子(B)は、必要により乾燥する。乾燥としては、樹脂粒子の融点未満で行うことが好ましく、必要により減圧下で行う。乾燥機としては、公知の乾燥装置(例えば、流動層式乾燥機、減圧乾燥機及び循風乾燥機)が用いられる。
【0084】
次に上記の方法(6)について説明する。
破砕又は粉砕に用いることができる破砕機としては、公知の破砕機{例えば、乳化分散の理論と実際(特殊機化(株)製、1997年4月17日発行)の80〜86頁}等が使用できる。
【0085】
このようにして得られた樹脂粒子(B)は、必要に応じて、風力分級器又はふるい等を用いて分級し、体積平均粒子径、体積平均粒子径と個数平均粒子径の比を調整することができる。
【0086】
上記方法(4)において、溶剤(T)としては、樹脂粒子(B)を分散できれば、特に制限はないが、単一溶媒(例えば、アセトン、エタノール、ジメチルホルムアミド、イソプロパノールなど)、又は混合溶媒[例えば水、アルコール溶剤(メタノール、エタノール等)、アミド溶剤(ジメチルホルムアミド等)、ケトン溶剤(アセトン、メチルエチルケトンなど)、エステル溶剤(酢酸エチル、酢酸ブチルなど)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル等)、芳香族炭化水素溶剤(トルエン、キシレン等)、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)等の2種以上の混合溶剤]が挙げられる。
【0087】
樹脂粒子(B)と溶剤(T)の重量比率は、特に制限はないが、溶剤(T)に対して、樹脂粒子(B)が55重量%以下が好ましく、更に好ましくは50重量%以下であり、特に好ましくは20〜45重量%である。この範囲であれば、効率よく樹脂粒子(B)を(C)中に導入することができる。
ここで、二酸化炭素(C)と溶剤(T)の重量比率は、特に制限はないが、二酸化炭素(C)に対して、(T)が1〜50重量%が好ましく、更に好ましくは、5〜40重量%である。この範囲であれば、樹脂粒子(B)の分散性が向上する。
【0088】
方法(3)および(4)において、二酸化炭素(C)の重量に対する樹脂粒子(B)の重量比率としては、60重量%以下が好ましく、更に好ましくは55重量%以下であり、特に好ましくは20〜50重量%である。この範囲であれば、効率よく処理して樹脂粒子(X)を製造できる。
【0089】
本発明において、樹脂粒子(B)の処理に用いる二酸化炭素(C)としては、液状のものと超臨界状態のものが使用できるが、超臨界状態が好ましい。
ここで、液状の二酸化炭素とは、二酸化炭素の温度軸と圧力軸とで表す相図上において、二酸化炭素の三重点(温度=−57℃、圧力=0.5MPa)と二酸化炭素の臨界点(温度=31℃、圧力=7.4MPa)を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件である二酸化炭素を表す。一方、超臨界状態の二酸化炭素とは、臨界温度以上の温度・圧力条件である二酸化炭素を表す。なお、本発明における圧力とは、2成分以上の混合ガスの場合、全圧を示す。
【0090】
二酸化炭素(C)中には、分散媒としての物性値(粘度、拡散係数、誘電率、溶解度、界面張力等)を調整するために、他の物質を適宜含んでよく、例えば、窒素、ヘリウム、アルゴン、空気等の不活性気体等が挙げられる。
(C)と他の物質の合計中の(C)の重量分率は、好ましくは70%以上、さらに好ましくは80%以上、とくに好ましくは90%以上である。
【0091】
樹脂粒子(B)を(C)中で処理する際、下記の温度で行うことが好ましい。
減圧時に配管内で二酸化炭素が固体に相転移し、流路を閉塞させないようにするために、30℃以上が好ましく、また、樹脂粒子(B)の熱劣化を防止するために、200℃以下が好ましい。さらに30〜150℃が好ましく、より好ましくは34〜130℃、特に好ましくは35〜100℃、最も好ましくは40℃〜80℃である。
【0092】
また、該処理は下記の圧力で行うことが好ましい。
樹脂粒子(B)を(C)に良好に分散させるために好ましくは3MPa以上であり、設備コスト、運転コストの観点から、好ましくは40MPa以下である。さらに好ましくは3.5〜35MPa、より好ましくは4〜30MPa、特に好ましくは4.5〜25MPa、最も好ましくは5〜20MPaである。
【0093】
二酸化炭素(C)中で処理する際の温度及び圧力は、樹脂粒子(B)が(C)中に溶解せず、且つ二酸化炭素(C)が(B)に浸透可能な範囲内で設定することが好ましい。通常、低温・低圧ほど(B)が(C)中に溶解しない傾向となり、高温・高圧ほど二酸化炭素(C)が樹脂粒子(B)に浸透しやすい傾向となる。
【0094】
樹脂粒子(B)が分散した分散体から、通常、減圧により、二酸化炭素(C)を除去して、本発明の樹脂粒子(X)を得る。その際、独立に圧力制御された容器を多段に設けることにより段階的に減圧してもよく、また一気に常温常圧まで減圧してもよい。
樹脂粒子(X)の捕集方法は特に限定されず、フィルターでろ別する方法や、サイクロン等により遠心分離する方法が例として挙げられる。
樹脂粒子(X)は減圧後に捕集してもよく、また減圧前に一旦高圧中で捕集した後、減圧してもよい。高圧下で捕集した後に減圧する場合の、高圧下からの樹脂粒子(X)の取り出し方としては、バッチ操作で捕集容器を減圧してもよく、またロータリーバルブを使用して連続的取り出し操作を行ってもよい。
【0095】
前記の樹脂粒子(B)を二酸化炭素(C)中に分散する方法(1)を用いて、樹脂(A)と溶剤(S)を含有する樹脂粒子(B)が(C)中に分散した分散体を得た場合、樹脂粒子(B)を形成させ処理した後、溶剤(S)を除去又は減少させる必要がある。
溶剤(S)を除去又は減少させる方法として、そのまま容器を減圧にする方法があるが、(B)中に溶解した溶剤が凝縮し、樹脂粒子(B)を再溶解してしまったり、樹脂粒子(X)を捕集する際に樹脂粒子(X)同士が合一してしまう等の問題が生じる場合がある。
好ましい方法としては、例えば、樹脂粒子(B)が(C)中に分散した分散体に、さらに二酸化炭素(C)を混合して樹脂粒子(B)から溶剤(S)を二酸化炭素の相に抽出し、つぎに、溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素(C)で置換し、その後に減圧する方法が挙げられる。
【0096】
二酸化炭素の混合方法は、分散体より高い圧力の二酸化炭素を加えてもよく、また該分散体を分散体より低い圧力の二酸化炭素中に加えてもよいが、連続操作の容易性の観点からより好ましくは後者である。該分散体と混合する二酸化炭素の量は、樹脂粒子(B)の合一防止の観点から、分散体の体積の1〜50倍が好ましく、さらに好ましくは1〜40倍、最も好ましくは1〜30倍である。上記のように樹脂粒子(B)中に含有される溶剤を除去ないし減少させ、その後、二酸化炭素を除去することにより、樹脂粒子(B)同士が合一することを防ぐことができる。
【0097】
溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素で置換する方法としては、樹脂粒子(B)を一旦フィルターやサイクロンで補足した後、圧力を保ちながら、溶剤(S)が完全に除去されるまで二酸化炭素を流通させる方法が挙げられる。流通させる二酸化炭素の量は、該分散体からの溶剤除去の観点から、分散体の体積に対して1〜100倍が好ましく、さらに好ましくは1〜70倍、最も好ましくは1〜50倍である。
【0098】
上記の製造方法で得られた本発明の樹脂粒子(X)は、液状又は超臨界状態の二酸化炭素(C)での処理により結晶化度が向上し、融点の融解熱が増加する。
【0099】
本発明の製造方法により得られた本発明の樹脂粒子(X)の体積平均粒子径は、好ましくは1〜12μmであり、より好ましくは2〜10μm、さらに好ましくは3〜8μmである。1μm以上であると粉体としてのハンドリング性が向上する。12μm以下であると溶融特性が向上する。
【0100】
樹脂粒子(X)の体積平均粒子径Dvと樹脂粒子(X)の個数平均粒子径Dnの比Dv/Dnは、好ましくは1.0〜1.5、より好ましくは1.0〜1.4、さらに好ましくは1.0〜1.3である。1.5以下であると粉体としてのハンドリング性、溶融特性が著しく向上する。
【0101】
樹脂粒子(X)の体積平均粒子径、および体積平均粒子径Dvと樹脂粒子(X)の個数平均粒子径Dnの比(Dv/Dn)は、前記の樹脂粒子(B)を(C)中に分散する方法(3)あるいは(4)を用いる場合は、樹脂粒子(B)の製造段階で調整する。方法(1)の場合、溶剤(S)に溶解させた樹脂(A)及び必要により微粒子(D)を(C)中に分散する際の、攪拌速度、および樹脂(A)に対する微粒子(D)の比率により調整することができる。攪拌速度を上げれば体積平均粒子径が小さくなり、また樹脂(A)に対する微粒子(D)の比率を多くすれば体積平均粒子径が小さくなる。
樹脂粒子(X)のDv/Dnについても同様であり、攪拌速度を上げればDv/Dnが小さくなり、また樹脂(A)に対する微粒子(D)の比率を多くすればDv/Dnが小さくなる。
このようにして得られた樹脂粒子(X)は、必要に応じて、風力分級器又はふるい等を用いて分級し、体積平均粒子径、体積平均粒子径と個数平均粒子径の比をさらに調整することができる。
【0102】
なお、体積平均粒子径及び個数平均粒子径は、レーザー式粒度分布測定装置LA−920(堀場製作所製)やマルチサイザーIII(コールター社製)、光学系としてレーザードップラー法を用いるELS−800(大塚電子社製)などで測定できる。
【実施例】
【0103】
以下、実施例及び比較例により本発明をさらに説明するが、本発明はこれらに限定されるものではない。以下、特に定めない限り、%は重量%、部は重量部を示す。
【0104】
製造例1
反応容器に、メチルエチルケトン1000部、1,6−ヘキサンジオール430部、ヘキサメチレンジイソシアネート570部を投入し、80℃で7時間反応を行い、80℃、20kPaで脱溶剤し、ポリウレタン樹脂[結晶性部(a−1)]を得た。[結晶性部(a−1)]はMw7500、融点(m)75℃、水酸基価34であった。
【0105】
製造例2
メチルエチルケトン1000部、1,6−ヘキサンジオール210部、水酸基価56の1,6−ヘキサンジオールとセバシン酸からなるポリエステルジオール(豊国製油(株)製、商品名「HS 2H−200S」)500部、ヘキサメチレンジイソシアネート290部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[結晶性部(a−2)]得た。[結晶性部(a−2)]はMw9000、融点(m)60℃、水酸基価26であった。
【0106】
製造例3
メチルエチルケトン1000部、1,6−ヘキサンジオール350部、1,6−ヘキサンジアミン90部、ヘキサメチレンジイソシアネート560部を使用する以外は、製造例1と同様に行い、ポリウレタンウレア樹脂[結晶性部(a−3)]を得た。[結晶性部(a−3)]はMw6000、融点(m)80℃、水酸基価44であった。
【0107】
製造例4
テトラヒドロフラン500部、1,2−プロピレングリコール360部、トルエンジイソシアネート640部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[非晶性部(b−1)]を得た。[非晶性部(b−1)]はMw3000であった。
【0108】
製造例5
メチルエチルケトン500部、1,4−シクロヘキサンジメタノール210部、トルエンジイソシアネート290部を使用する以外は、製造例1と同様に行い、ポリウレタン樹脂[非晶性部(b−2)]を得た。[非晶性部(b−2)]はMw5000であった。
【0109】
製造例6
反応容器に、テレフタル酸270部、ビスフェノールAのPO2モル付加物730部、ジブチルチンオキサイド5部を投入し、常圧、230℃で5時間脱水反応を行った後、0.5kPaの減圧下で5時間脱水反応を行い、ポリエステル樹脂[非結晶性部(b−3)]を得た。[非結晶性部(b−3)]は、Mw5000、水酸基価55であった。
【0110】
製造例7
反応容器に、メチルエチルケトン1150部、前記の[非結晶性部(b−1)]200部、トルエンジイソシアネート50部を投入し、80℃で5時間反応させた後、前記の[結晶性部(a−1)]900部を投入し、80℃で5時間反応を行い、80℃、20kPaで脱溶剤し、樹脂(A−1)を得た。(A−1)は、Mw18000、軟化点(s)75℃、融点(m)72℃、軟化点(s)と融点(m)の比(s/m)1.04であった。
また、G’(m+20)は7×103Paであり、溶融開始温度(x)は70℃、|LogG”(x+20)−LogG”(x)|の値は3.4であった。
(m+30)℃における損失弾性率G”と(m+70)℃における損失弾性率G”の比〔G”(m+30)/G”(m+70)〕は2.7であった。
(b)と(a)の結合形式の式におけるn=1.08であった。上記物性値は表1に記載した。
反応容器に、この樹脂(A−1)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−1)得た。
【0111】
製造例8
メチルエチルケトン700部、前記の[非結晶性部(b−1)]100部、およびトルエンジイソシアネート25部を投入し反応させた後、前記の[結晶性部(a−2)]575部を投入する以外は、製造例7と同様に行い、樹脂(A−2)を得た。樹脂(A−2)の物性値は表1に記載した。
反応容器に、この樹脂(A−2)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−2)得た。
【0112】
製造例9
メチルエチルケトン1030部、前記の[非結晶性部(b−1)]200部、およびトルエンジイソシアネート50部を投入し反応させた後、前記の[結晶性部(a−3)]780部を投入する以外は、製造例7と同様に行い、樹脂(A−3)を得た。樹脂(A−3)の物性値は表1に記載した。
反応容器に、この樹脂(A−3)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−3)得た。
【0113】
製造例10
反応容器に、メチルエチルケトン780部、前記の[非結晶性部(b−2)]200部、前記の[結晶性部(a−1)]580部を投入し、80℃で5時間反応を行い、80℃、20kPaで脱溶剤し、樹脂(A−4)を得た。樹脂(A−4)の物性値は表1に記載した。
反応容器に、この樹脂(A−4)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−4)得た。
【0114】
製造例11
反応容器に、テトラヒドロフラン500部、前記の[非結晶性部(b−3)]200部、およびm−キシリレンジイソシアネート38部を投入し反応させた後、前記の[結晶性部(a−1)]600部を投入する以外は、製造例7と同様に行い、樹脂(A−5)を得た。樹脂(A−5)の物性値は表1に記載した。
反応容器に、この樹脂(A−5)300部、アセトン630部、イオン交換水70部を加えて、溶解させ、樹脂溶液(L−5)得た。
【0115】
【表1】
【0116】
<分散用微粒子分散液の調製>
製造例12
滴下ロートを備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート(炭素数22個の直鎖アルキル基を有するアルコールのアクリレート:ブレンマーVA(日油製))150部、アゾビスイソブチロニトリル(AIBN)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、アクリル系結晶性樹脂を得た。この樹脂の融点は65℃、数平均分子量50000であった。
ノルマルヘキサン700部、上記のアクリル系結晶性樹脂300部を混合した後、ビーズミル(ダイノーミルマルチラボ:シンマルエンタープライゼス製)で粒径0.3mmのジルコニアビーズを用いて粉砕を行い、乳白色の分散用微粒子(D−1)が分散したヘキサン分散液を得た。この分散液の体積平均粒径は0.3μmであった。
【0117】
製造例13
製造例12のアクリル系結晶性樹脂の微粒子(D−1)分散体の調整において、ヘキサン700部をイオン交換水700部に変更した以外は同様にして、比較例用の分散用微粒子(D’−1)の水分散液を得た。この水分散液の体積平均粒径は0.3μmであった。
【0118】
実施例1
図1の実験装置において、まずバルブV1、V2を閉じ、ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素(純度99.99%)を導入し、14MPa、40℃に調整した。また樹脂溶液タンクT1に製造例7で得られた樹脂溶液(L−1)を、微粒子分散液タンクT2には製造例12で作成した分散用微粒子(D−1)のヘキサン分散液を仕込んだ。
次に、液状の二酸化炭素のボンベB1から、ポンプP3を用いて液状の二酸化炭素を分散槽T3に仕込み、超臨界状態(9MPa、40℃)に調整し、さらにタンクT2から、ポンプP2を用いて微粒子(D−1)ヘキサン分散液を導入した。
次に分散槽T3の内部を2000rpmで攪拌しながら、タンクT1から、ポンプP1を用いて樹脂溶液(L−1)を分散槽T3内に導入した。導入後T3の内部の圧力は14MPaとなった。
なお、分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
分散用微粒子(D−1)のヘキサン分散液 45部
二酸化炭素 550部
【0119】
なお、上記の導入した二酸化炭素の重量は、二酸化炭素の温度(40℃)、及び圧力(15MPa)から二酸化炭素の密度を下記文献に記載の状態式より算出し、これに分散槽T3の体積を乗じることにより算出した。
文献:Journal of Physical and Chemical Refarence data、vol.25、P.1509〜1596
【0120】
樹脂溶液(L−1)を導入後、1分間攪拌し、超臨界状態の二酸化炭素に樹脂粒子(B−1)が分散した分散体を得た。
次に、バルブV1を開き、B1からP3を用いてT3及びT4内に超臨界状態の二酸化炭素を導入することで、(B−1)の分散体をT3からT4内に移送した。(B−1)の分散体をT3からT4に移送する間、圧力が一定に保たれるように、V2の開度を調節した。この操作を30秒間行い、V1を閉めた。
次に、圧力調整バルブV2の開度を調整することで、圧力を14MPaに保持しながら、圧力ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素を導入した。
この操作により、溶剤を含む二酸化炭素を溶剤トラップ槽T5に排出すると共に、樹脂粒子(B−1)をフィルターF1に捕捉した。圧力ボンベB2から、ポンプP4を用いて粒子回収槽T4に二酸化炭素を導入する操作は、上記の分散槽T3に導入した二酸化炭素重量の5倍量を粒子回収槽T4に導入した時点で停止した。この停止の時点で、溶剤を含む二酸化炭素を、溶剤を含まない二酸化炭素で置換すると共に樹脂粒子(B−1)をフィルターF1に捕捉する操作は完了した。さらに、圧力調整バルブV2を少しずつ開き、粒子回収槽内を大気圧まで減圧することで、フィルターF1に補足されている本発明の樹脂粒子(X−1)を得た。
【0121】
実施例2〜5
樹脂溶液(L−1)を製造例8〜11で得られた樹脂溶液(L−2)〜(L−5)に変更する以外は実施例1と同様にし、本発明の樹脂粒子(X−2)〜(X−5)を得た。
【0122】
比較例1
ビーカー内にイオン交換水97部、製造例13で得られた分散用微粒子(D’−1)の水分散液15.4部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)10部を入れ均一に溶解した。ついで25℃で、TK式ホモミキサーを10,000rpmに撹拌しながら、製造例7で得られた樹脂溶液(L−1)75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまでアセトンを留去し、樹脂粒子の水性樹脂分散体を得た。
次いで濾別し40℃×18時間乾燥を行い、揮発分を0.5%以下として、比較のための樹脂粒子(R−1)を得た。この後、超臨界の二酸化炭素による処理は行わず、物性評価した。
【0123】
比較例2
ビーカー内にイオン交換水97部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)15部を入れ均一に溶解した。
ついで25℃で、TK式ホモミキサーを12,000rpmに撹拌しながら、製造例7で得られた樹脂溶液(L−1)75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまでアセトンを留去し、樹脂粒子の水性樹脂分散体を得た。
次いで濾別し40℃×18時間乾燥を行い、揮発分を0.5%以下として、比較のための樹脂粒子(R−2)を得た。この後、超臨界の二酸化炭素による処理は行わず、物性評価した。
【0124】
<融点(m)、融解熱の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃で、溶融による吸熱ピークの温度(℃)を求め、これを融点(m)とした。
また、吸熱ピークの面積より融解熱を求めた。求めた融解熱を用いて、下記計算式より、融解熱の比を求めた。
融解熱の比=H2/H1
【0125】
<体積平均粒径の評価>
樹脂粒子をドデシルベンゼンスルホン酸ナトリウム水溶液(濃度0.1%)に分散して体積平均粒径をコールターカウンター[マルチサイザーIII(ベックマン・コールター社製)]で測定した。
【0126】
<耐熱保存性の評価>
樹脂粒子の耐熱保存性を下記の方法で評価した。
直径が約3cmの30mlのガラス製スクリュー管に樹脂粒子を10g採取した。この樹脂粒子が入ったガラス製スクリュー管を50℃に温調された恒温器に15時間静置し、ブロッキングの程度により下記の基準で評価した。
○: ブロッキングが発生しない。
△: ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×: ブロッキングが発生し、簡単に指などで力を加えても分散しない。
【0127】
<低温溶融性の評価>
溶融温度は、以下の方法により評価した。
日本テストパネル社製リン酸亜鉛処理鋼板標準板に市販のコロナ帯電方式スプレーガンを用いて膜圧が20〜40μmになるように樹脂粒子を静電塗装し、焼き付け温度を変化させて評価を行ったとき、5分間焼き付けた後の目視確認による表面平滑性が良好となる最低温度を測定した。
【0128】
実施例1〜5で作成した本発明の樹脂粒子(X−1)〜(X−5)、および比較例1、2で作成した比較のための樹脂粒子(R−1)、(R−2)の評価結果を表2に示す。
【0129】
【表2】
【0130】
実施例1〜5の樹脂粒子は、耐熱保存性及び低温溶融性に優れていたのに対し、比較例1の樹脂粒子は、低温溶融性が著しく悪化し、耐熱保存性も悪化した。また、比較例2の樹脂粒子は、低温溶融性は良好であるが、耐熱保存性が著しく悪化した。
【産業上の利用可能性】
【0131】
本発明の製造方法により得られる樹脂粒子は、耐熱保存性と低温溶融性に優れているため、電子写真トナーの母体粒子、塗料用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成型用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、電気粘性用粒子、その他成型用樹脂粒子として有用である。
【符号の説明】
【0132】
T1:樹脂溶液タンク
T2:溶液タンク
T3:分散槽(最高使用圧力20MPa、最高使用温度200℃、攪拌機つき)
T4:粒子回収槽(最高使用圧力20MPa、最高使用温度100℃)
T5:溶剤トラップ
F1:セラミックフィルター(メッシュ:0.5μm)
B1、B2:二酸化炭素ボンベ
P1、P2:溶液ポンプ
P3、P4:二酸化炭素ポンプ
V1:バルブ
V2:圧力調整バルブ
【特許請求の範囲】
【請求項1】
ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
【請求項2】
樹脂粒子(B)が樹脂(A)及び溶剤(S)を含有し、(C)及び(S)を除去する工程を含む請求項1記載の製造方法。
【請求項3】
樹脂(A)が、40〜110℃の融点(m)を有し、0.8〜1.55の軟化点(s)[℃]と融点(m)[℃]の比(s/m)を有し、(m±20)℃の温度範囲内に溶融開始温度(x)を有し、かつ以下の条件を満たす樹脂である請求項1または2記載の製造方法。
〔条件1〕G’(m+20)=50〜1×106[Pa]
〔条件2〕|LogG”(x+20)−LogG”(x)|>2.0
[G’:貯蔵弾性率[Pa]、G”:損失弾性率[Pa]]
【請求項4】
樹脂(A)の、(m+30)℃における損失弾性率G”と、(m+70)℃における損失弾性率G”の比〔G”(m+30)/G”(m+70)〕[mは樹脂(A)の融点]が、0.05〜50である請求項1〜3のいずれか記載の製造方法。
【請求項5】
樹脂(A)が、結晶性部(a)と非結晶性部(b)とが下記の形式で線状に結合された樹脂であり、nが0.5〜3.5である請求項1〜4のいずれか記載の製造方法。
(a){−(b)−(a)}n
【請求項6】
結晶性部(a)の樹脂(A)中に占める重量比率が30〜95%である請求項1〜5のいずれか記載の製造方法。
【請求項7】
請求項1〜6のいずれか記載の製造方法により得られる樹脂粒子(X)。
【請求項1】
ポリウレタンおよび/またはポリウレア(p)を必須構成成分とする結晶性部(a)と、非結晶性部(b)から構成される樹脂(A)を含有する樹脂粒子(B)を液状または超臨界状態の二酸化炭素(C)で処理し、次いで(C)を除去する工程を含む樹脂粒子(X)の製造方法であって、得られる(X)の示差走査熱量(DSC)測定による融解熱が下記関係式(1)を満足する樹脂粒子(X)の製造方法。
0≦H2/H1≦0.9 (1)
[関係式(1)中、H1はDSC測定による初回昇温時の融解熱(J/g);H2はDSC測定による2回目昇温時の融解熱(J/g)の測定値を表す。]
【請求項2】
樹脂粒子(B)が樹脂(A)及び溶剤(S)を含有し、(C)及び(S)を除去する工程を含む請求項1記載の製造方法。
【請求項3】
樹脂(A)が、40〜110℃の融点(m)を有し、0.8〜1.55の軟化点(s)[℃]と融点(m)[℃]の比(s/m)を有し、(m±20)℃の温度範囲内に溶融開始温度(x)を有し、かつ以下の条件を満たす樹脂である請求項1または2記載の製造方法。
〔条件1〕G’(m+20)=50〜1×106[Pa]
〔条件2〕|LogG”(x+20)−LogG”(x)|>2.0
[G’:貯蔵弾性率[Pa]、G”:損失弾性率[Pa]]
【請求項4】
樹脂(A)の、(m+30)℃における損失弾性率G”と、(m+70)℃における損失弾性率G”の比〔G”(m+30)/G”(m+70)〕[mは樹脂(A)の融点]が、0.05〜50である請求項1〜3のいずれか記載の製造方法。
【請求項5】
樹脂(A)が、結晶性部(a)と非結晶性部(b)とが下記の形式で線状に結合された樹脂であり、nが0.5〜3.5である請求項1〜4のいずれか記載の製造方法。
(a){−(b)−(a)}n
【請求項6】
結晶性部(a)の樹脂(A)中に占める重量比率が30〜95%である請求項1〜5のいずれか記載の製造方法。
【請求項7】
請求項1〜6のいずれか記載の製造方法により得られる樹脂粒子(X)。
【図1】

【公開番号】特開2010−196038(P2010−196038A)
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願番号】特願2009−299081(P2009−299081)
【出願日】平成21年12月29日(2009.12.29)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願日】平成21年12月29日(2009.12.29)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
[ Back to top ]