
樹脂粒子及びその製造方法
【課題】 液状又は超臨界状態の流体を使用して得られる、粒度分布が十分狭い樹脂粒子、及び、液状又は超臨界状態の流体を使用して粒度分布が十分狭い樹脂粒子を得る製造方法を提供する。
【解決手段】 微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として特定組成の結晶性樹脂(a1)を用いる樹脂粒子(C) 。
【解決手段】 微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として特定組成の結晶性樹脂(a1)を用いる樹脂粒子(C) 。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、粒度分布が狭い樹脂粒子、及び、液状又は超臨界状態の流体を用いる該樹脂粒子の製造方法に関するものである。
【背景技術】
【0002】
従来より非水媒体中における粒子形成法として、超臨界流体中に樹脂溶液を噴霧する方法(例えば、特許文献1、2参照)、有機顔料、酸化ケイ素などの微粒子分散剤の存在下において超臨界流体中に加熱溶融させた樹脂を機械的に分散させて微粒子化した後、減圧して樹脂粒子を得る方法(例えば、特許文献3参照)、微粒子分散剤及び活性剤の存在下において超臨界流体中に樹脂溶液を機械的に分散させて、微粒子化した後、減圧して樹脂粒子を得る方法(例えば特許文献4,5)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】WO97/31691号パンフレット
【特許文献2】WO95/01221号パンフレット
【特許文献3】特開2005−107405号公報
【特許文献4】特開2006−321830号公報
【特許文献5】特開2007−277511号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
上記の超臨界流体中に樹脂溶液を噴霧する方法によれば、親水性成分を必要とせず、界面活性物質が樹脂粒子中または表面に残存しないため、粉体特性、電気特性等に優れた粒子を得ることができるが、シャープな粒度分布が得難い問題がある。上記の超臨界流体中に加熱溶融した樹脂を分散する方法によれば、粉体特性、電気特性等に優れ、粒度分布の狭い粒子を得ることができるとされているが、実用的観点からみて粒子の粒度分布が十分狭いとは、言い難い。また、上記文献の技術においては有機顔料、酸化ケイ素等により樹脂粒子表面が被覆されているため、電子写真トナー用樹脂粒子として使用した場合、低温定着性が劣る問題があった。上記の微粒子分散剤及び活性剤の存在下において超臨界流体中に樹脂溶液を分散させる方法によれば、樹脂粒子が凝集してしまうため、粒度分布の狭い粒子を得ることが困難であった。
【0005】
本発明の課題は、液状又は超臨界状態の流体を使用して得られる、粒度分布が十分狭い樹脂粒子、及び、液状又は超臨界状態の流体を使用して粒度分布が十分狭い樹脂粒子を得る製造方法を見出すことである。
【課題を解決するための手段】
【0006】
本発明は、従来技術における上記の事情に鑑みてなされたものである。すなわち、本発明は、下記6発明である。
(I) 微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として結晶性樹脂(a1)を用い、結晶性樹脂(a1)が、下記(a12)〜(a14)からなる群から選ばれる少なくとも1種である樹脂粒子。
(a12) 炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア。
(a13) アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂。
(a14) (メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂。
(II) (I)の樹脂粒子を含有する電子写真トナー用樹脂粒子。
(III) (II)の電子写真トナー用樹脂粒子を含有する電子写真トナー。
(IV) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
(V) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
(VI) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
【発明の効果】
【0007】
本発明の樹脂粒子、及び、本発明の製造方法により得られる樹脂粒子は、粒度分布が十分狭く、かつ樹脂粒子の耐湿耐熱保存性が良好である。
【図面の簡単な説明】
【0008】
【図1】樹脂粒子の作成に用いた実験装置である。
【発明を実施するための形態】
【0009】
以下に本発明を詳述する。微粒子(A)は、そのガラス転移温度(以下、Tgと記載する場合がある。)又は融点未満の温度において、液状又は超臨界状態の二酸化炭素(X)〔以下、二酸化炭素(X)と記載する場合がある。〕による膨潤度(以下、膨潤度と記載する。)が16%以下の微粒子であり、好ましくは10%以下、さらに好ましくは5%以下である。膨潤度が16%を超える微粒子を使用した場合は、樹脂粒子の凝集を抑制できなくなり、樹脂粒子の粒度分布が悪化する。
【0010】
膨潤度の測定方法は、磁気浮遊天秤を用いて測定することができる。なお、膨潤度の測定方法の詳細はJ.Supercritical Fluids.19、187−198(2001)に記載されている。
【0011】
微粒子(A)としては、結晶性樹脂(a1)、非結晶性樹脂(a2)、及び無機化合物(a3)からなる群から選ばれる少なくとも1種を用いる。非結晶性樹脂(a2)としては、架橋性の非結晶性樹脂がより好ましい。これらの中では、結晶性樹脂(a1)および非結晶性樹脂(a2)が好ましく、結晶性樹脂(a1)がさらに好ましい。
【0012】
結晶性樹脂(a1)の融点は、50〜110℃が好ましく、さらに好ましくは55〜100℃、とくに好ましくは60〜90℃である。結晶性樹脂(a1)の融点が50℃以上であれば本発明の樹脂粒子(C)が長期間の保管でもブロッキングしにくい。110℃以下であれば電子写真用トナーとして用いた場合には低温定着性が良好である。融点の測定は示差走査熱量測定(以下、DSCと記載する。)における吸熱ピークより求めることができる。
【0013】
結晶性樹脂(a1)の結晶化度は、二酸化炭素(X)による膨潤抑制、及び樹脂粒子(B)への吸着性の観点より、好ましくは20〜95%であり、より好ましくは30〜80%である。結晶化度は、DSCを用いて吸熱ピークの面積から融解熱量(ΔHm(J/g))を求め、測定されたΔHmに基づき以下の式により結晶化度(%)を算出する。
結晶化度=(ΔHm/a)×100
上式中、aは結晶化度が100%となるように外挿した場合の融解熱量である。
【0014】
結晶性樹脂(a1)の数平均分子量は、キャリア汚染性の観点より、好ましくは1000以上であり、更に好ましくは1500以上、特に好ましくは2000以上である。また、溶融粘度の観点より、好ましくは1000000以下であり、更に好ましくは500000以下、特に好ましくは300000以下である。
【0015】
結晶性樹脂(a1)の組成は特に限定されないが、好ましい具体例としては、例えば、脂肪族もしくは芳香族ポリエステル、脂肪族ポリウレタン及び/又はポリウレア、アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂、(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂(a14)、結晶性ポリオレフィン(a15)等が挙げられる。
【0016】
脂肪族もしくは芳香族ポリエステルとしては、後述のジオール(11)、ジカルボン酸(13)を使用することができ、特に炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールと炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジカルボン酸を必須構成単位とし、かつ、該ジオールのアルキレン鎖の炭素数と該ジカルボン酸のアルキレン鎖の炭素数の合計数が10〜52であり、必要により炭素数6〜30の芳香族ジカルボン酸を構成単位とする結晶性ポリエステル(a11)が好ましい。
保存安定性の観点から、上記ジオールのアルキレン鎖の炭素数と上記ジカルボン酸のアルキレン鎖の炭素数の合計数が、10以上が好ましく、更に好ましくは12以上であり、特に好ましくは14以上である。また、定着性の観点から、52以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。
【0017】
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジオールとして好ましいものは、1,4−ブタンジオール、1,6−ヘキサンジオール、及び1,10−デカンジオールである。
炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジカルボン酸のアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジカルボン酸として好ましいものは、アジピン酸、セバシン酸、ドデカンジカルボン酸、及びオクタデカンジカルボン酸である。
また、芳香族ポリエステルの保存安定性の観点から、芳香族ジカルボン酸の炭素数は6〜30が好ましく、更に好ましくは8〜24あり、特に好ましくは8〜20である。炭素数6〜30の芳香族ジカルボン酸として好ましいものは、フタル酸、イソフタル酸、テレフタル酸、及びナフタレンジカルボン酸である。
【0018】
また、芳香族ポリエステルの場合は、樹脂強度の観点から、ジカルボン酸は直鎖脂肪族ジカルボン酸と芳香族ジカルボン酸の併用が好ましく、直鎖脂肪族ジカルボン酸と芳香族ジカルボン酸の合計に対する芳香族ジカルボン酸の比率は、好ましくは90重量以下、更に好ましくは1〜85重量%、特に好ましくは3〜80重量%である。
【0019】
脂肪族ポリウレタン及び/又はポリウレアとしては、後述のジオール(11)、ジアミン〔後述のポリアミン(16)のうち2価のもの〕、及びジイソシアネート〔後述のポリイソシアネート(15)のうち2価のもの〕を使用することができ、特に炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア(a12)が好ましい。
なお、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールと後述のジカルボン酸(13)とを反応させて得られるポリエステルジオールと炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートから得られるポリウレタンも(a12)に含まれる。
脂肪族ポリウレタン及び/又はポリウレアは、保存安定性の観点から、ジオール及び/又はジアミンのアルキレン鎖の炭素数(ジオールとジアミンの混合物を使用する場合は、その重量比で平均されたアルキレン鎖の炭素数)とジイソシアネートのアルキレン鎖の炭素数の合計数が、10以上が好ましく、更に好ましくは12以上であり、特に好ましくは14以上である。また、定着性の観点から、52以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。
【0020】
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールの、アルキレン鎖の好ましい炭素数、及び好ましい具体例は、結晶性ポリエステル(a11)における場合と同様である。
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジアミンとして好ましいものは、テトラメチレンジアミン、及びヘキサメチレンジアミンである。
また、上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジイソシアネートとして好ましいものは、テトラメチレンジイソシアネート、及びヘキサメチレンジイソシアネートである。
【0021】
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂としては、アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂(a13)が好ましい。保存安定性の観点から、そのアルキル基の炭素数は、12以上であることが好ましく、更に好ましくは14以上であり、特に好ましくは18以上である。また、定着性の観点から、50以下が好ましく、更に好ましくは40以下であり、特に好ましくは30以下である。保存安定性の観点からアルキル基は直鎖が好ましい。アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートとして好ましいものは、オクタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート、及びベヘニルアクリレートである。
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂は、アルキル(メタ)アクリレートの単独重合体でも、他の単量体との共重合体でもよい。他の単量体としては、後述のビニルモノマーを適宜選択することができる。
結晶性ビニル樹脂(a13)中のアルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートの構成単位の含有量は、好ましくは40重量%以上、更に好ましくは45重量%以上、とくに好ましくは60重量%以上である。
【0022】
(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂(a14)としては、樹脂粒子への付着性の観点から、(メタ)アクリロニトリルの構成単位の含有量が0.01〜40重量%であることが好ましく、更に好ましくは0.05〜35重量%であり、特に好ましくは0.1〜30重量%である。
併用する結晶性ビニルモノマーとしては、結晶性のビニル樹脂が形成され得るものであれば特に限定されないが、上記のアルキル基の炭素数が12〜50であるアルキル(メタ)アクリレート、及びエチレン等が挙げられる。
【0023】
結晶性ポリオレフィン(a15)としては、ポリエチレン、ポリプロピレン等が挙げられる。
【0024】
脂肪族もしくは芳香族ポリエステルの製造方法としては、低分子ポリオールおよび/または数平均分子量1000以下のポリアルキレンエーテルジオールとポリカルボン酸とを反応させる方法、ラクトンの開環重合による方法、低分子ジオールと低級アルコール(メタノールなど)の炭酸ジエステルとを反応させる方法などの公知の製造方法が挙げられる。
【0025】
脂肪族ポリウレタン及び/又はポリウレアの製造方法としては、低分子ポリオール(上記の方法で得られるポリエステルポリオールを含む)及び/又は低分子量ジアミンとジイソシアネートを反応させる方法などの公知の製造方法が挙げられる。
【0026】
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂、および(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂の製造方法としては、溶液重合、塊状重合、懸濁重合などの公知のビニルモノマーの重合法が挙げられる。
【0027】
ポリオレフィンの製造方法としては、付加重合等の公知の重合法が挙げられる。
【0028】
結晶性樹脂(a1)の中で、特に好ましいものは、(a11)、(a12)、(a13)、および(a14)であり、最も好ましくは(a13)である。
【0029】
非結晶性樹脂(a2)としては、例えばビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂、ポリアミド、ポリイミド、シリコーン樹脂、フッ素樹脂、フェノール樹脂、メラミン樹脂、ポリカーボネート、セルロース及びこれらの混合物等が挙げられる。非結晶性樹脂(a2)としては、架橋性の非結晶性樹脂が好ましい。
(a2)の組成は膨潤度が前記の範囲となるものであれば得に限定されず、通常用いられている樹脂でよい。
例えば、架橋性ビニル樹脂としては、2個以上のビニル重合性官能基を有するビニルモノマー(ジビニルベンゼン等)を含むビニルモノマーの共重合体等が挙げられる。
架橋性ポリエステル樹脂としては、ポリオールとポリカルボン酸の重縮合物であって、ポリオール及び/又はポリカルボン酸の少なくとも一部として、後述の3価以上のポリオール(12)及び/又は3価以上のポリカルボン酸(14)を用いて得られるポリエステル樹脂等が挙げられる。
同様に、他の樹脂の場合も架橋性のモノマーを少なくとも一部用いて得られる樹脂がより好ましい。
【0030】
微粒子(A)として、結晶性樹脂(a1)と非結晶性樹脂(a2)を併用してもよい。(a1)と(a2)の混合物の融点は、50〜150℃であることが好ましい。(a2)の含有量は、(a1)と(a2)の合計重量に対して、0〜50重量%であることが好ましい。また非結晶性樹脂(a2)を結晶性樹脂(a1)で被覆した微粒子であってもよい。
【0031】
結晶性樹脂(a1)および/または非結晶性樹脂(a2)を含有する微粒子(A)の製法はいかなる製法であってもよいが、具体例としては、乾式で製造する方法〔微粒子(A)を構成する材料(a)をジェットミル等の公知の乾式粉砕機により乾式粉砕する方法〕、湿式で製造する方法〔(a)の粉末を有機溶剤中に分散し、ビーズミルやロールミル等の公知の湿式分散機により湿式粉砕する方法、(a)の溶剤溶液をスプレードライヤー等により噴霧乾燥する方法、(a)の溶剤溶液を貧溶媒添加や冷却によって過飽和させ析出させる方法、(a)の溶剤溶液を水あるいは有機溶剤中に分散する方法、(a)の前駆体を水中で乳化重合法、ソープフリー乳化重合法、シード重合法、懸濁重合法等により重合させる方法、(a)の前駆体を有機溶剤中で分散重合等により重合させる方法〕が挙げられる。また上記方法により非結晶性樹脂(a2)の微粒子(A’)を合成した後、公知のコーティング法、シード重合法、メカノケミカル法等により、結晶性樹脂(a1)を(A’)表面に形成してもよい。これらのうち、微粒子(A)の製造しやすさの観点から、湿式で製造する方法が好ましく、さらに好ましくは、析出させる方法、乳化重合法、分散重合である。
【0032】
微粒子(A)はそのまま用いてもよく、また樹脂粒子(B)への吸着性を持たせたり、本発明の樹脂粒子(C)の粉体特性や電気特性を改質するために、例えばシラン系、チタネート系、アルミネート系等のカップリング剤による表面処理、各種界面活性剤による表面処理、ポリマーによるコーティング処理等により表面改質されていてもよい。微粒子(A)及び樹脂粒子(B)のいずれか一方が、少なくともその表面に酸性官能基を有し、他の一方が少なくともその表面に塩基性官能基を有することが好ましい。
【0033】
微粒子(A)及び樹脂粒子(B)はその内部に酸性官能基又は塩基性官能基を有していてもよい。酸性官能基としてはカルボン酸基、スルホン酸基等が挙げられる。塩基性官能基としては第1級アミノ基、第2級アミノ基、第3級アミノ基等が挙げられる。
【0034】
微粒子(A)及び樹脂粒子(B)は少なくともその表面に酸性官能基又は塩基性官能基を付与するために、結晶性樹脂(a1)、樹脂(b)として酸性官能基又は塩基性官能基を有する樹脂を使用してもよいし、微粒子(A)及び樹脂粒子(B)にこれら官能基を付与するために表面処理してもよい。
【0035】
酸性官能基を有する結晶性樹脂(a1)としては、酸価を有する脂肪族ポリエステル、酸性官能基を有する単量体(例えば、後述のカルボキシル基含有ビニルモノマー、スルホン基含有ビニルモノマーなど)を共重合したビニル樹脂等が挙げられる。
【0036】
塩基性官能基を有する結晶性樹脂(a1)としては、塩基性官能基を有する単量体(例えば、後述のアミノ基含有ビニルモノマーなど)を共重合したビニル樹脂等が挙げられる。
【0037】
無機化合物(a3)としては、例えば、珪藻土、アルミナ、酸化亜鉛、チタニア、ジルコニア、酸化カルシウム、酸化マグネシウム、酸化鉄、酸化銅、酸化スズ、酸化クロム、酸化アンチモン、酸化イットリウム、酸化セリウム、酸化サマリウム、酸化ランタン、酸化タンタル、酸化テルビウム、酸化ユーロピウム、酸化ネオジウム、フェライト類等の金属酸化物、水酸化カルシウム、水酸化マグネシウム、水酸化アルミニウム、塩基性炭酸マグネシウム等の金属水酸化物、重質炭酸カルシウム、軽質炭酸カルシウム、炭酸亜鉛、炭酸バリウム、ドーソナイト、ハイドロタルイサイト等の金属炭酸塩、硫酸カルシウム、硫酸バリウム、石膏繊維等の金属硫酸塩、シリカ、珪酸カルシウム(ウォラストナイト、ゾノトライト)、カオリン、クレー、タルク、マイカ、モンモリロナイト、ベントナイト、活性白土、セピオライト、イモゴライト、セリサイト、ガラス繊維、ガラスビーズ、ガラスフレーク等の金属珪酸塩、窒化アルミニウム、窒化ホウ素、窒化珪素等の金属窒化物、チタン酸カリウム、チタン酸カルシウム、チタン酸マグネシウム、チタン酸バリウム、チタン酸ジルコン酸鉛アルミニウムボレート等の金属チタン酸塩、ホウ酸亜鉛、ホウ酸アルミニウム等の金属ホウ酸塩、リン酸三カルシウム等の金属燐酸塩、硫化モリブデン等の金属硫化物、炭化珪素等の金属炭化物、カーボンブラック、グラファイト、炭素繊維等の炭素類、金、銀その他の無機粒子が挙げられる。これらの中で好ましくは、シリカおよび金属炭酸塩である。
【0038】
樹脂粒子(B)は樹脂(b)より構成される。本発明において、樹脂(b)としては、熱可塑性樹脂(b1)、又は該熱可塑性樹脂を微架橋した樹脂(b2)、又は熱可塑性樹脂を海成分、硬化樹脂を島成分とするポリマーブレンド(b3)が挙げられ、2種以上を併用してもよい。熱可塑性樹脂(b1)としては、例えばビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂等が挙げられる。このうち好ましいのは、微細球状樹脂粒子の分散体が得られやすいという観点からビニル樹脂、ポリウレタン樹脂、ポリエステル樹脂およびそれらの併用である。
【0039】
ビニル樹脂は、ビニルモノマーを単独重合または共重合したポリマーである。ビニルモノマーとしては、下記(1)〜(10)が挙げられる。
(1)ビニル炭化水素:
(1−1)脂肪族ビニル炭化水素:アルケン類、例えばエチレン、プロピレン、前記以外のα−オレフィン等;アルカジエン類、例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン、1,7−オクタジエン。
(1−2)脂環式ビニル炭化水素:モノ−もしくはジ−シクロアルケンおよびアルカジエン類、例えば(ジ)シクロペンタジエン等;テルペン類、例えばピネン等。
(1−3)芳香族ビニル炭化水素:スチレンおよびそのハイドロカルビル(アルキル、シクロアルキル、アラルキルおよび/またはアルケニル)置換体、例えばα−メチルスチレン、2,4−ジメチルスチレン等;およびビニルナフタレン。
(2)カルボキシル基含有ビニルモノマー及びその塩:炭素数3〜30の不飽和モノカルボン酸、不飽和ジカルボン酸ならびにその無水物およびそのモノアルキル(炭素数1〜24)エステル、例えば(メタ)アクリル酸、(無水)マレイン酸、マレイン酸モノアルキルエステル、フマル酸、フマル酸モノアルキルエステル、クロトン酸、イタコン酸、イタコン酸モノアルキルエステル、イタコン酸グリコールモノエーテル、シトラコン酸、シトラコン酸モノアルキルエステル、桂皮酸等のカルボキシル基含有ビニルモノマー。
(3)スルホン基含有ビニルモノマー、ビニル硫酸モノエステル化物及びこれらの塩:炭素数2〜14のアルケンスルホン酸、例えばビニルスルホン酸;およびその炭素数2〜24のアルキル誘導体、例えばα−メチルスチレンスルホン酸等;スルホ(ヒドロキシ)アルキル−(メタ)アクリレートもしくは(メタ)アクリルアミド、例えば、スルホプロピル(メタ)アクリレート、および硫酸エステルもしくはスルホン酸基含有ビニルモノマー;ならびそれらの塩等。
(4)燐酸基含有ビニルモノマー及びその塩:(メタ)アクリロイルオキシアルキル(C1〜C24)燐酸モノエステル、例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート、フェニル−2−アクリロイロキシエチルホスフェート、(メタ)アクリロイルオキシアルキル(炭素数1〜24)ホスホン酸類、例えば2−アクリロイルオキシエチルホスホン酸。なお、上記(2)〜(4)の塩としては、例えばアルカリ金属塩(ナトリウム塩、カリウム塩等)、アルカリ土類金属塩(カルシウム塩、マグネシウム塩等)、アンモニウム塩、アミン塩もしくは4級アンモニウム塩が挙げられる。
(5)ヒドロキシル基含有ビニルモノマー:ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等。
(6)含窒素ビニルモノマー:
(6−1)アミノ基含有ビニルモノマー:アミノエチル(メタ)アクリレート等、
(6−2)アミド基含有ビニルモノマー:(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド等、
(6−3)ニトリル基含有ビニルモノマー:(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等、
(6−4)4級アンモニウムカチオン基含有ビニルモノマー:ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリルアミド、ジエチルアミノエチル(メタ)アクリルアミド、ジアリルアミン等の3級アミン基含有ビニルモノマーの4級化物(メチルクロライド、ジメチル硫酸、ベンジルクロライド、ジメチルカーボネート等の4級化剤を用いて4級化したもの)等、
(6−5)ニトロ基含有ビニルモノマー:ニトロスチレン等。
(7)エポキシ基含有ビニルモノマー:グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等。
(8)ハロゲン元素含有ビニルモノマー:塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等。
(9)ビニルエステル、ビニル(チオ)エーテル、ビニルケトン、ビニルスルホン類:
(9−1)ビニルエステル、例えば酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、オクタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート、ベヘニル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ポリ(メタ)アリロキシアルカン類[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]等、ポリアルキレングリコール鎖を有するビニルモノマー[ポリエチレングリコール(分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(分子量500)モノアクリレート、メチルアルコールエチレンオキサイド10モル付加物(メタ)アクリレート、ラウリルアルコールエチレンオキサイド30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート類[多価アルコール類のポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等、
(9−2)ビニル(チオ)エーテル、例えばビニルメチルエーテル等、
(9−3)ビニルケトン、例えばビニルメチルケトン等。
(10)その他のビニルモノマー:イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等。
【0040】
ビニルモノマーの共重合体としては、上記(1)〜(10)の任意のモノマー同士を任意の割合で共重合したポリマーが挙げられるが、例えばスチレン−(メタ)アクリル酸エステル共重合体、スチレン−ブタジエン共重合体、(メタ)アクリル酸−アクリル酸エステル共重合体、スチレン−アクリロニトリル共重合体、スチレン−無水マレイン酸共重合体、スチレン−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸、ジビニルベンゼン共重合体、スチレン−スチレンスルホン酸−(メタ)アクリル酸エステル共重合体等が挙げられる。
【0041】
ポリエステル樹脂としては、ポリオールと、ポリカルボン酸(その酸無水物、その低級アルキルエステルを含む)との重縮合物などが挙げられる。ポリオールとしてはジオール(11)および3価以上のポリオール(12)が挙げられ、ポリカルボン酸としては、ジカルボン酸(13)および3価以上のポリカルボン酸(14)が挙げられる。
ポリオールとポリカルボン酸の反応比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/1、さらに好ましくは1.5/1〜1/1、とくに好ましくは1.3/1〜1.02/1である。
【0042】
ジオール(11)としては、アルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、オクタンジオール、デカンジオール、ドデカンジオール、テトラデカンジオール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);アルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレンエーテルグリコールなど);脂環式ジオール(1,4−シクロヘキサンジメタノール、水素添加ビスフェノールAなど);ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど);上記脂環式ジオールのアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;上記ビスフェノール類のアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;その他、ポリラクトンジオール(ポリε−カプロラクトンジオールなど)、ポリブタジエンジオールなどが挙げられる。これらのうち好ましいものは、炭素数2〜12のアルキレングリコールおよびビスフェノール類のアルキレンオキサイド付加物であり、特に好ましいものはビスフェノール類のアルキレンオキサイド付加物、およびこれと炭素数2〜12のアルキレングリコールとの併用である。
【0043】
3価以上のポリオール(12)としては、3〜8価またはそれ以上の多価脂肪族アルコール(グリセリン、トリメチロールエタン、トリメチロールプロパン、ペンタエリスリトール、ソルビトールなど);トリスフェノール類(トリスフェノールPAなど);ノボラック樹脂(フェノールノボラック、クレゾールノボラックなど);上記トリスフェノール類のアルキレンオキサイド付加物;上記ノボラック樹脂のアルキレンオキサイド付加物、アクリルポリオール[ヒドロキシエチル(メタ)アクリレートと他のビニルモノマーの共重合物など]などが挙げられる。
【0044】
ジカルボン酸(13)としては、アルキレンジカルボン酸(コハク酸、アジピン酸、セバシン酸、ドデセニルコハク酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸など);アルケニレンジカルボン酸(マレイン酸、フマール酸など);炭素数8以上の分岐アルキレンジカルボン酸[ダイマー酸、アルケニルコハク酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸など)、アルキルコハク酸(デシルコハク酸、ドデシルコハク酸、オクタデシルコハク酸など);芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、ナフタレンジカルボン酸など)などが挙げられる。これらのうち好ましいものは、炭素数4〜20のアルケニレンジカルボン酸および炭素数8〜20の芳香族ジカルボン酸である。
【0045】
3価以上(3〜6価又はそれ以上)のポリカルボン酸(14)としては、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など)などが挙げられる。
【0046】
なお、ジカルボン酸(13)または3価以上のポリカルボン酸(14)としては、上述のものの酸無水物または低級アルキルエステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0047】
ポリウレタン樹脂としては、ポリイソシアネート(15)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(11)および3価以上のポリオール(12)]、ジカルボン酸(13)、3価以上のポリカルボン酸(14)、ポリアミン(16)、ポリチオール(17)等}との重付加物などが挙げられる。
【0048】
ポリイソシアネート(15)としては、炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ポリイソシアネート、炭素数2〜18の脂肪族ポリイソシアネート、炭素数4〜15の脂環式ポリイソシアネート、炭素数8〜15の芳香脂肪族ポリイソシアネートおよびこれらのポリイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。
上記芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)などが挙げられる。
上記脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)などが挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)などが挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
また、上記ポリイソシアネートの変性物には、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのポリイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が含まれる。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、および炭素数4〜15の脂環式ポリイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0049】
ポリアミン(16)の例としては、下記のものが挙げられる。
・脂肪族ポリアミン類(C2〜C18):
〔1〕脂肪族ポリアミン{C2〜C6アルキレンジアミン(エチレンジアミン、テトラメチレンジアミン、及びヘキサメチレンジアミンなど)、ポリアルキレン(C2〜C6)ポリアミン〔ジエチレントリアミンなど〕}
〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミンなど〕
〔3〕脂環または複素環含有脂肪族ポリアミン〔3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕
〔4〕芳香環含有脂肪族アミン類(C8〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど)、
・脂環式ポリアミン(C4〜C15):1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など、
・芳香族ポリアミン類(C6〜C20):
〔1〕非置換芳香族ポリアミン〔1,2−、1,3−および1,4−フェニレンジアミンなど;核置換アルキル基〔メチル、エチル、n−およびi−プロピル、ブチルなどのC1〜C4アルキル基)を有する芳香族ポリアミン、たとえば2,4−および2,6−トリレンジアミンなど〕、およびこれらの異性体の種々の割合の混合物
〔2〕核置換電子吸引基(Cl、Br、I、Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ポリアミン〔メチレンビス−o−クロロアニリンなど〕
〔3〕2級アミノ基を有する芳香族ポリアミン〔上記(4)〜(6)の芳香族ポリアミンの−NH2の一部または全部が−NH−R´(R´はメチル、エチルなどの低級アルキル基で置換したもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕、
・複素環式ポリアミン(C4〜C15):ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジンなど、
・ポリアミドポリアミン:ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど、
・ポリエーテルポリアミン:ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物など。
【0050】
ポリチオール(17)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなどが挙げられる。
【0051】
エポキシ樹脂としては、ポリエポキシド(18)の開環重合物、ポリエポキシド(18)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(11)および3価以上のポリオール(12)]、ジカルボン酸(13)、3価以上のポリカルボン酸(14)、ポリアミン(16)、ポリチオール(17)等}との重付加物、またはポリエポキシド(18)とジカルボン酸(13)または3価以上のポリカルボン酸(14)の酸無水物との硬化物などが挙げられる。
【0052】
ポリエポキシド(18)としては、分子中に2個以上のエポキシ基を有していれば、特に限定されない。ポリエポキシド(18)として好ましいものは、硬化物の機械的性質の観点から分子中にエポキシ基を2〜6個有するものである。ポリエポキシド(18)のエポキシ当量(エポキシ基1個当たりの分子量)は、好ましくは65〜1000であり、さらに好ましくは90〜500である。エポキシ当量が1000以下であると、架橋構造が密になり硬化物の耐水性、耐薬品性、機械的強度等の物性が向上し、一方、エポキシ当量が65以上のものは、合成するのが容易である。
【0053】
ポリエポキシド(18)の例としては、芳香族系ポリエポキシ化合物、複素環系ポリエポキシ化合物、脂環族系ポリエポキシ化合物あるいは脂肪族系ポリエポキシ化合物が挙げられる。芳香族系ポリエポキシ化合物としては、多価フェノール類のグリシジルエーテル体およびグリシジルエステル体、グリシジル芳香族ポリアミン、並びに、アミノフェノールのグリシジル化物等が挙げられる。多価フェノールのグリシジルエーテル体としては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル等が挙げられる。多価フェノールのグリシジルエステル体としては、フタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、テレフタル酸ジグリシジルエステル等が挙げられる。グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。さらに、本発明において前記芳香族系ポリエポキシ化合物として、P−アミノフェノールのトリグリシジルエーテル、トリレンジイソシアネートまたはジフェニルメタンジイソシアネートとグリシドールとの付加反応によって得られるジグリシジルウレタン化合物、前記2反応物にポリオールも反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマー、およびビスフェノールAのアルキレンオキシド(エチレンオキシドまたはプロピレンオキシド)付加物のジグリシジルエーテル体も含む。複素環系ポリエポキシ化合物としては、トリスグリシジルメラミンが挙げられる。脂環族系ポリエポキシ化合物としては、ビニルシクロヘキセンジオキシド等が挙げられる。また、脂環族系ポリエポキシ化合物としては、前記芳香族系ポリエポキシド化合物の核水添化物も含む。脂肪族系ポリエポキシ化合物としては、多価脂肪族アルコールのポリグリシジルエーテル体、多価脂肪酸のポリグリシジルエステル体、およびグリシジル脂肪族アミンが挙げられる。多価脂肪族アルコールのポリグリシジルエーテル体としては、エチレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル等が挙げられる。多価脂肪酸のポリグリシジルエステル体としては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート、ジグリシジルピメレート等が挙げられる。グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミンが挙げられる。また、本発明において脂肪族系ポリエポキシ化合物としては、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含む。これらのうち、好ましいのは、脂肪族系ポリエポキシ化合物および芳香族系ポリエポキシ化合物である。ポリエポキシドは、2種以上併用しても差し支えない。
【0054】
熱可塑性樹脂を微架橋した樹脂(b2)とは、架橋構造を導入させ樹脂(b)のTgが20〜200℃である樹脂を言うものとする。かかる架橋構造は、共有結合性、配位結合性、イオン結合性、水素結合性等、いずれの架橋形態であってもよい。具体例としては、例えば樹脂(b2)としてポリエステルを選択する場合、重合時にポリオールとポリカルボン酸のいずれか、あるいは両方に3官能以上の官能基数を有するものを使用することにより架橋構造を導入することができる。また樹脂(b2)としてビニル樹脂を選択する場合、重合時に二重結合を2つ以上有するモノマーを添加することにより、架橋構造を導入することができる。
【0055】
熱可塑性樹脂を海成分、硬化樹脂を島成分とするポリマーブレンド(b3)としては、Tgが20〜200℃、且つ軟化開始温度が40〜220℃であるもの、具体的にはビニル樹脂、ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂及びこれらの混合物が挙げられる。
【0056】
樹脂(b)の数平均分子量(GPCにて測定、以下Mnと略記する場合がある。)は、好ましくは1000〜500万、より好ましくは2,000〜500,000、溶解性パラメーター(SP値、詳細は後述する。)は、好ましくは7〜18、より好ましくは8〜14である。また、本発明の樹脂粒子(C)の熱特性を改質したい場合には、樹脂(b2)又は樹脂(b3)を使用するとよい。
【0057】
樹脂(b)のガラス転移温度(Tg)は好ましくは20℃〜200℃、より好ましくは40℃〜150℃である。20℃以上では粒子の保存安定性が良好である。なお、本発明におけるTgは、DSC測定から求められる値である。
【0058】
樹脂(b)の軟化開始温度は、好ましくは40℃〜220℃、より好ましくは50℃〜200℃である。40℃以上では長期の保存性が良好である。220℃以下では定着温度が上昇せず問題がない。なお、本発明における軟化開始温度は、フローテスター測定から求められる値である。
【0059】
樹脂粒子(B)の体積平均粒径は、好ましくは1〜10μmであり、より好ましくは2〜8μmである。
【0060】
本発明の樹脂粒子(C)は、微粒子(A)が樹脂粒子(B)の表面に固着されてなるか、又は樹脂粒子(B)の表面に、微粒子(A)が皮膜化された皮膜が形成されてなる粒子である。微粒子(A)が樹脂粒子(B)の表面に固着されてなるとは、(A)が単に(B)の表面に付着し容易に脱離するような場合は含まないものとする。
【0061】
微粒子(A)の粒径は、樹脂粒子(B)の粒径よりも小さい。粒径比[微粒子(A)の体積平均粒径/[本発明の樹脂粒子(C)の体積平均粒径]の値は、好ましくは0.001〜0.3、より好ましくは0.002〜0.2、さらに好ましくは0.003〜0.1、特に好ましくは0.01〜0.08である。上記範囲内であると(A)が(B)の表面に効率よく吸着するため、得られる本発明の樹脂粒子(C)の粒度分布が狭くなる。
【0062】
微粒子(A)の体積平均粒径は、好ましくは0.01〜0.5μm、特に好ましくは0.015〜0.4μmである。なお、体積平均粒径は、動的光散乱式粒度分布測定装置(例えば LB−550:堀場製作所製)、レーザー式粒度分布測定装置(例えば LA−920:堀場製作所製)、マルチサイザーIII(ベックマン・コールター社製)等で測定できる。
【0063】
本発明の樹脂粒子(C)の体積平均粒径は、好ましくは1〜10μmであり、より好ましくは2〜8μm、さらに好ましくは3〜6μmである。1μm以上であると、粉体としてのハンドリング性が向上する。10μm以下であると、電子写真用トナーとした時の画像の解像度が向上する。
【0064】
本発明の樹脂粒子(C)の体積平均粒径DVと、本発明の樹脂粒子(C)の個数平均粒径DNの比:DV/DNは、好ましくは1.0〜1.5、より好ましくは1.0〜1.4、特に好ましくは1.0〜1.3である。1.5以下であると粉体特性(流動性、帯電均一性等)、画像の解像度が著しく向上する。
【0065】
本発明の樹脂粒子(C)は、の粒径均一性、粉体流動性、保存安定性等の観点からは、樹脂粒子(B)の表面の5%以上が、微粒子(A)もしくは(A)由来の皮膜で覆われているのが好ましく、更に好ましくは30%以上である。なお、表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[(A)もしくは(A)由来の皮膜に覆われている部分の(B)の表面積/{(A)もしくは(A)由来の皮膜に覆われている部分の(B)の表面積+(B)の表面が露出している部分の面積}]×100
【0066】
本発明の樹脂粒子(C)は、微粒子(A)を構成する材料(a)と樹脂粒子(B)を構成する樹脂(b)の重量比率が、好ましくは(0.1:99.9)〜(30:70)であり、さらに好ましくは(0.2:99.8)〜(20:80)である。材料(a)と樹脂(b)の重量比率がこの範囲内であると、低温定着性と長期の保存安定性が両立し好ましい。
本発明の樹脂粒子(C)中の(a)が結晶性樹脂(a1)である場合、公知の方法、例えばDSCにより(a1)に固有な吸熱ピークの吸熱量から結晶性樹脂(a1)の重量比率を算出する方法により測定することがでる。
【0067】
本発明の樹脂粒子(C)は、液状又は超臨界状態の二酸化炭素(X)中で製造する以下の製造方法で得ることが好ましい。
製造方法(1)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで二酸化炭素(X)を除去することにより樹脂粒子を得る製造方法。
製造方法(2)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子を得る製造方法。
製造方法(3)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子を得る製造方法。
【0068】
製造方法(2)について詳細に説明する。
23℃、0.1MPaの標準状態における、溶剤(S)と樹脂(b)との等重量混合物における、溶剤(S)に対する樹脂(b)の不溶分は、樹脂(b)の重量に対して、好ましくは20重量%以下、さらに好ましくは15重量%以下である。不溶分重量(重量%)が20重量%以下であれば得られる樹脂粒子の粒度分布が狭くなる。
製造方法(3)において樹脂(b)の代わりに前躯体(b0)を用いる場合、および樹脂(b)と前駆体(b0)の混合物を用いる場合も同様である。
【0069】
また、溶剤(S)の溶解性パラメーター(SP値)は9〜16が好ましく、さらに好ましくは10〜15である。SP値とは、下記に示した様に、凝集エネルギー密度と分子容の比の平方根で表されるものである。
SP=(△E/V)1/2
ここで△Eは凝集エネルギー密度を表す。Vは分子容を表し、その値は、ロバート エフ.フェドールス(Robert F.Fedors)らの計算によるもので、例えばポリマー エンジニアリング アンド サイエンス(Polymer engineering and science)第14巻、147〜154頁に記載されている。
【0070】
溶剤(S)の具体例としては、例えば、ケトン溶剤(アセトン、メチルエチルケトン等)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、環状エーテル等)、エステル溶剤(酢酸エステル、ピルビン酸エステル、2−ヒドロキシイソ酪酸エステル、乳酸エステル等)、アミド溶剤(ジメチルホルムアミド等)、アルコール類(メタノール、エタノール、フッ素含有アルコール等)、芳香族炭化水素溶剤(トルエン、キシレン等)、および脂肪族炭化水素溶剤(オクタン、デカン等)などが挙げられる。これらの溶剤の2種以上の混合溶剤、または、これらの有機溶剤と水との混合溶剤を用いることもできる。
【0071】
粒子形成のし易さの観点から、単一溶剤としては、環状エーテル、ピルビン酸エステル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、2−ヒドロキシイソ酪酸エステル、乳酸エステル、フッ素含有アルコールが好ましい。
上記環状エーテルとしては、1,4−ジオキサン、1,3−ジオキソラン等が挙げられる。
ピルビン酸エステルとしては、ピルビン酸メチル、ピルビン酸エチル等が挙げられる。
エチレングリコールモノアルキルエーテルとしては、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル等が挙げられる。
プロピレングリコールモノアルキルエーテルとしては、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル等が挙げられる。
2−ヒドロキシイソ酪酸エステルとしては、2−ヒドロキシイソ酪酸メチル等が挙げられる。
乳酸エステルとしては、乳酸メチル、乳酸エチル等が挙げられる。
フッ素含有アルコールとしては、2,2,3,3−テトラフルオロプロパノール、トリフルオロエタノール等が挙げられる。
また、混合溶剤としては、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、アセトンと水の混合溶剤、メチルエチルケトンと水の混合溶剤が好ましい。
製造方法(3)における溶剤(S)も同様である。
【0072】
樹脂(b)の溶液(L)は、樹脂(b)を溶剤(S)に溶解させて製造する。溶液(L)の重量に対して樹脂(b)の濃度は好ましくは10〜90重量%、さらに好ましくは20〜80重量%である。
製造方法(3)における溶液(L0)中の前駆体(b0)の濃度も同様である。
【0073】
製造方法(2)の樹脂(b)の溶液(L)を二酸化炭素(X)中に分散させる分散工程では、下記の分散安定剤(E)を使用することが出来る。分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有する化合物である。さらには、二酸化炭素に親和性を有するジメチルシロキサン基、含フッ素基と共に、樹脂(b)に親和性を有する化学構造を有することが好ましい。
【0074】
例えば樹脂(b)がビニル樹脂である場合、分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有するモノマーを構成単位とするビニル樹脂であることが好ましい。
ジメチルシロキサン基を有するモノマー(あるいは反応性オリゴマー)(M1−1)としては、メタクリル変性シリコーンが好ましく、次式に示す構造を持つ。
(CH3)3SiO((CH3)2SiO)aSi(CH3)2R
但しaは、平均値で15〜45であり、Rはメタクリル基を含む有機変性基である。Rの例としては、−C3H6OCOC(CH3)=CH2が挙げられる。
【0075】
また、フッ素を含有するモノマー(M1−2)の具体例としては、テトラフルオロエチレン(TFE)、ヘキサフルオロプロピレン(HFP)、クロロトリフルオロエチレン(CTFE)等のパーフルオロオレフィン;パーフルオロ(アルキルビニルエーテル)(PFAVE)、パーフルオロ(1,3−ジオキソール)、パーフルオロ(2,2−ジメチル−1,3−ジオキソール)(PFDD)、パーフルオロ−(2−メチレン−4−メチル−1,3−ジオキソラン)(MMD)、パーフルオロブテニルビニルエーテル(PFBVE)等のパーフルオロビニルエーテル;ビニリデンフルオライド(VdF)、トリフルオロエチレン、1,2−ジフルオロエチレン、フッ化ビニル、トリフルオロプロピレン、3,3,3−トリフルオロ−2−トリフルオロメチルプロペン、3,3,3−トリフルオロプロペン、パーフルオロ(ブチル)エチレン(PFBE)等の水素原子含有フルオロオレフィン;1,1−ジヒドロパーフルオロオクチルアクリレート(DPFOA)、1,1−ジヒドロパーフルオロオクチルメタクリレート(DPFOMA)、2−(パーフルオロオクチル)エチルアクリレート(PFOEA)、2−(パーフルオロオクチル)エチルメタクリレート(PFOEMA)、2−(パーフルオロヘキシル)エチルメタクリレート(PFHEMA)、2−(パーフルオロブチル)エチルメタクリレート(PFBEMA)等のポリフルオロアルキル(メタ)アクリレート;α−フルオロスチレン、β−フルオロスチレン、α,β−ジフルオロスチレン、β,β−ジフルオロスチレン、α,β,β−トリフルオロスチレン、α−トリフルオロメチルスチレン、2,4,6−トリ(トリフルオロメチル)スチレン、2,3,4,5,6−ペンタフルオロスチレン、2,3,4,5,6−ペンタフルオロ−α−メチルスチレン、2,3,4,5,6−ペンタフルオロ−β−メチルスチレン等のフルオロスチレン等が挙げられる。
【0076】
また樹脂(b)がウレタン樹脂である場合、分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有するモノマーを構成単位とするウレタン樹脂であることが好ましい。
(M1−1)としてはアミノ変性シリコーン、カルボキシル変性シリコーン、カルビノール変性シリコーン、メルカプト変性シリコーン等の活性水素を含む官能基を有するポリシロキサンが好ましい。(M1−2)としては、2,2ビス(4−ヒドロキシフェニル)ヘキサフルオロプロパン、3,3,4,4−テトラフルオロ−1,6−ヘキサンジオール等の含フッ素基ポリオール、含フッ素基(ポリ)アミン、含フッ素基(ポリ)チオール等の活性水素を含む官能基を有するフッ素化合物、ビス(イソシアナトメチル)パーフルオロプロパン、ビス(イソシアナトメチル)パーフルオロブタン、ビス(イソシアナトメチル)パーフルオロペンタン及びビス(イソシアナトメチル)パーフルオロヘキサン等の含フッ素基(ポリ)イソシアネートが好ましい。
【0077】
また樹脂(b)が酸価を有する場合、分散性の観点より分散安定剤(E)はアミノ基を有することが好ましい。樹脂(b)の酸価は1〜50が好ましく、さらに好ましくは3〜40、最も好ましくは5〜30である。アミノ基は1級、2級、3級のいずれでもよく、また含フッ素基、ジメチルシロキサン基を含む化合物の側鎖、片末端、両末端、側鎖両末端いずれの位置に導入されたものを使用してもよい。
【0078】
分散安定剤(E)としては、例えばジメチルシロキサン基を有するモノマー(あるいは反応性オリゴマー)(M1−1)、及び/又はフッ素を含有するモノマー(M1−2)と、前述の樹脂(b)を構成するモノマーとの共重合体(例えば、メタクリル変性シリコーンとメタクリル酸メチルとの共重合体、メタクリル酸ヘプタフルオロブチルとメタクリル酸メチルとの共重合体等)が好ましい。共重合の形態はランダム、ブロック、グラフトのいずれでもよいが、ブロックあるいはグラフトが好ましい。
【0079】
また樹脂(b)が酸価を有する場合、分散安定性の観点より微粒子(A)は粒子表面にアミノ基を有することが好ましい。アミノ基は1級、2級、3級のいずれでもよく、またアミノ基を含有させる形態は特に限定されず、例えばアミノ基を有する化合物を微粒子(A)中に分散、含浸等の方法により含有させる方法、微粒子(A)を構成する成分にアミノ基を有する化合物を使用する方法、微粒子(A)表面にアミノ基含有カップリング剤等を反応させる方法、微粒子(A)表面にアミノ基含有化合物を吸着させる方法等が挙げられる。
【0080】
分散安定剤(E)の添加量は、分散安定性の観点から、樹脂(b)の重量に対し0.01〜50重量%が好ましく、さらに好ましくは0.02〜40重量%、特に好ましくは0.03〜30重量%である。分散安定剤(E)の好ましい重量平均分子量の範囲は100〜10万であり、さらに好ましくは200〜5万、特に好ましくは500〜3万である。この範囲内にすると、(E)の分散安定効果が向上する。
なお、製造方法(1)及び(3)においても、分散工程で、分散安定剤(E)を使用することができる。
【0081】
本発明において、微粒子(A)を二酸化炭素(X)中に分散する方法はいかなる方法でもよく、例えば、容器内に(A)及び(X)を仕込み、攪拌や超音波照射等により、(A)を直接(X)中に分散する方法や、微粒子(A)が溶剤(T)中に分散された分散液を(X)中に導入する方法等が挙げられる。
【0082】
二酸化炭素(X)の重量に対する微粒子(A)の重量比率(重量%)としては、50以下が好ましく、更に好ましくは30以下であり、特に好ましくは0.1〜20である。この範囲であれば、効率よく樹脂粒子(C1)を製造できる。
【0083】
溶剤(T)としては、溶剤(S)と同様のものが挙げられる。微粒子(A)の分散性から、好ましくは、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)、及びエステル溶剤(酢酸エチル、酢酸ブチルなど)である。
【0084】
微粒子(A)と溶剤(T)の重量比率(重量%)は、特に制限はないが、溶剤(T)に対して、微粒子(A)が50以下が好ましく、更に好ましくは30以下であり、特に好ましくは20以下である。この範囲であれば、効率よく微粒子(A)を(X)中に導入することができる。
【0085】
微粒子(A)を溶剤(T)中に分散する方法としては特に制限はないが、微粒子(A)を溶剤(T)に仕込み、攪拌や超音波照射等により直接分散する方法や微粒子を高温下で溶剤(T)に溶解させて晶析する方法などが挙げられる。
【0086】
このようにして二酸化炭素(X)中に(A)が分散している分散体(X0)が得られる。微粒子(A)としては、膨潤度が前記の範囲であって、(X)に溶解せず、(X)中に安定分散するものが好ましい。
【0087】
樹脂(b)の溶液(L)は、(X)中に分散するため、適度な粘度であることが好ましく、粒度分布の観点から、好ましくは100Pa・s以下、さらに好ましくは10Pa・s以下である。樹脂(b)の(X)への溶解度は、好ましくは3%以下、さらに好ましくは1%以下である。
【0088】
樹脂(b)のSP値は、好ましくは8〜16、さらに好ましくは9〜14である。
【0089】
本発明において樹脂(b)を含有する樹脂粒子(B)中に他の添加物(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を含有しても差し支えない。樹脂粒子(B)中に他の添加物を含有させる方法としては、あらかじめ樹脂(b)と添加物を混合した後、(X)中にその混合物を加えて分散させるのが好ましい。
【0090】
本発明において、樹脂(b)の溶液(L)を、(X)中に微粒子(A)が分散している分散体(X0)中に分散する方法はいかなる方法を用いてもよい。具体例としては、樹脂(b)の溶液(L)を攪拌機や分散機等で分散する方法、樹脂(b)の溶液(L)を二酸化炭素(X)中に(A)が分散している分散体(X0)中にスプレーノズルを介して噴霧して液滴を形成し、液滴中の樹脂を過飽和状態とし、樹脂粒子を析出させる方法(ASES:Aerosol Solvent Extraction Systemとして知られている)、同軸の多重管(2重管、3重管等)から溶液(L)、溶液(L0)、樹脂(b)の前駆体(b0)、分散体(X0)を高圧ガス、エントレーナ等とともにそれぞれ別の管から同時に噴出させて、液滴に外部応力を加え分裂を促進させて、粒子を得る方法(SEDS:Solution Enhanced Dispersion by Supercritical Fluidsとして知られている)、超音波を照射する方法等が挙げられる。製造方法(3)及び(1)における、樹脂(b)の前駆体(b0)の溶液(L0)及び樹脂(b)の前駆体(b0)の場合も同様である。
【0091】
このようにして二酸化炭素(X)中に(A)が分散している分散体(X0)中に樹脂(b)の溶液(L)を分散し、微粒子(A)を表面に吸着させながら、分散された樹脂(b)を粒子成長させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成する。(C1)が(X)中に分散したものを分散体(X1)とする。
分散体(X1)は単一相であることが好ましい。すなわち、樹脂(b)の溶液(L)を使用する場合、(C1)が分散している二酸化炭素(X)を含む相の他に、溶剤(S)相が分離する状態は好ましくない。したがって、溶剤相が分離しないように、分散体(X0)に対する(b)の溶液(L)の量を設定することが好ましい。例えば(X0)に対して90重量%以下が好ましく、さらに好ましくは5〜80重量%、特に好ましくは10〜70重量%である。
なお、樹脂(b)の溶液(L)、または製造方法(3)の前駆体(b0)の溶液(L0)を用いた場合に、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)中に含有する(S)の量は、好ましくは10〜90重量%、さらに好ましくは20〜70重量%である。
また、樹脂(b)と二酸化炭素(X)の重量比は、好ましくは(b):(X)が、1:(0.1〜100)、さらに好ましくは1:(0.5〜50)、特に好ましくは1:(1〜20)である。製造方法(1)及び(3)における前駆体体(b0)と二酸化炭素(X)の重量比も同様である。
【0092】
本発明において、液状の二酸化炭素とは、二酸化炭素の温度軸と圧力軸とで表す相図上において、二酸化炭素の三重点(温度=−57℃、圧力=0.5MPa)と二酸化炭素の臨界点(温度=31℃、圧力=7.4MPa)を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件である二酸化炭素を表し、超臨界状態の二酸化炭素とは、臨界温度以上の温度・圧力条件である二酸化炭素を表す(ただし、圧力は、2成分以上の混合ガスの場合、全圧を表す)。
【0093】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作は、以下に述べる温度で行うことが好ましい。すなわち、減圧時に配管内で二酸化炭素が固体に相転移し、流路を閉塞させないようにするために、30℃以上が好ましく、また、微粒子(A)、樹脂粒子(B1)、樹脂粒子(C1)の熱劣化を防止するために、200℃以下が好ましい。さらに30〜150℃が好ましく、より好ましくは34〜130℃、特に好ましくは35〜100℃、最も好ましくは40℃〜80℃である。分散体(X0)、分散体(X1)の温度も同様である。また、製造方法(1)、(3)の場合も同様である。本発明の製造方法(1)〜(3)において、二酸化炭素(X)中で行う操作は、微粒子(A)のTg又は融点以上の温度でも未満の温度でも行うことができるが、Tg又は融点未満の温度において行うことが好ましい。
【0094】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作は以下に述べる圧力で行うことが好ましい。すなわち、樹脂粒子(C1)を(X)中に良好に分散させるために、好ましくは7MPa以上であり、設備コスト、運転コストの観点から、好ましくは40MPa以下である。さらに好ましくは7.5〜35MPa、より好ましくは8〜30MPa、特に好ましくは8.5〜25MPa、最も好ましくは9〜20MPaである。分散体(X0)及び分散体(X1)を形成する容器内の圧力も同様である。製造方法(1)、(3)の場合も同様である。
【0095】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作の温度及び圧力は、樹脂(b)が(X)中に溶解せず、且つ(b)が凝集・合一可能な範囲内で設定することが好ましい。通常、低温・低圧ほど目的分散物が(X)中に溶解しない傾向となり、高温・高圧ほど(b)が凝集・合一し易い傾向となる。分散体(X0)、分散体(X1)についても同様である。製造方法(1)、(3)の場合も同様である。
【0096】
本発明における二酸化炭素(X)中には、分散媒としての物性値(粘度、拡散係数、誘電率、溶解度、界面張力等)を調整するために、他の物質(e)を適宜含んでよく、例えば、窒素、ヘリウム、アルゴン、空気等の不活性気体等が挙げられる。
【0097】
本発明における(X)と他の物質(e)の合計中の二酸化炭素(X)の重量分率は、好ましくは70%以上、さらに好ましくは80%以上、とくに好ましくは90%以上である。
【0098】
樹脂粒子(C1)の分散した分散体(X1)から、通常、減圧により二酸化炭素(X)を除去し、本発明の樹脂粒子(C)を得る。その際、独立に圧力制御された容器を多段に設けることにより段階的に減圧してもよく、また一気に常温常圧まで減圧してもよい。得られる樹脂粒子の捕集方法は特に限定されず、フィルターでろ別する方法や、サイクロン等により遠心分離する方法が例として挙げられる。樹脂粒子は減圧後に捕集してもよく、また減圧前に一旦高圧中で捕集した後、減圧してもよい。高圧下で捕集した後に減圧する場合の、高圧下からの樹脂粒子の取り出し方としては、バッチ操作で捕集容器を減圧してもよく、またロータリーバルブを使用して連続的取り出し操作を行ってもよい。
【0099】
微粒子(A)が結晶性樹脂(a1)を含有する場合、樹脂粒子(C1)を形成させた後、必要に応じて、さらなる工程として、結晶性樹脂(a1)の、好ましくは、融点マイナス50℃以上、より好ましくは融点マイナス10℃以上、さらに好ましくは融点以上、に加熱することにより、樹脂粒子(B)の表面に付着した微粒子(A)を溶融させて、微粒子(A)を樹脂粒子(B)の表面に固着、又は微粒子(A)由来の皮膜を形成して樹脂粒子(C2)を形成する工程を行うこともできる。(C2)の凝集を抑制するという観点から、加熱する時間は0.01〜1時間が好ましく、さらに好ましくは、0.05〜0.7である。
【0100】
本発明の製造方法(1)〜(3)により得られる本発明の樹脂粒子(C)は、樹脂粒子(B)又は(B1)の表面に一旦微粒子(A)が固着されるが、(a)として結晶性樹脂(a1)を用いた場合、(a)と樹脂(b)の組成、溶剤(S)又は(T)の種類によっては、製造工程中に、微粒子(A)が皮膜化されて、(B)の表面に(A)が皮膜化された皮膜が形成される場合がある。
本発明の樹脂粒子(C)は、樹脂粒子(B)の表面に、微粒子(A)が固着されたもの、(A)由来の皮膜が形成されたもの、(A)の一部が皮膜化されたもののいずれであってもよい。
なお、本発明の樹脂粒子(C)の表面状態及び形状は、例えば、走査電子顕微鏡(SEM)を用い、樹脂粒子の表面を1万倍または3万倍拡大した写真にて観察できる。
【0101】
樹脂粒子(C1)〔樹脂粒子(C2)の場合も含む〕を形成させた後、必要に応じて、さらなる工程として、溶剤(S)を除去又は減少させる工程を行うことが好ましい。すなわち、(C1)が(X)中に分散した分散体(X1)中に溶剤(S)を含む場合、そのまま容器を減圧にすると、(X1)中に溶解した溶剤が凝縮し、樹脂粒子(C1)を再溶解してしまったり、樹脂粒子(C1)を捕集する際に樹脂粒子(C1)同士が合一してしまう等の問題が生じる場合がある。溶剤を除去又は減少させる方法としては、例えば、樹脂(b)の溶剤(S)の溶液(L)を分散して得られた分散体(X1)に、さらに二酸化炭素〔好ましくは二酸化炭素(X)〕を混合して樹脂粒子(C1)から溶剤(S)を二酸化炭素の相に抽出し、つぎに、溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素〔好ましくは二酸化炭素(X)〕で置換し、その後に減圧することが好ましい。
【0102】
樹脂粒子(C1)が二酸化炭素(X)中に分散した分散体(X1)と二酸化炭素の混合方法は、(X1)より高い圧力の二酸化炭素を加えてもよく、また(X1)を(X1)より低い圧力の二酸化炭素中に加えてもよいが、連続操作の容易性の観点からより好ましくは後者である。(X1)と混合する二酸化炭素の量は、樹脂粒子(C1)の合一防止の観点から、(X1)の体積の1〜50倍が好ましく、さらに好ましくは1〜40倍、最も好ましくは1〜30倍である。上記のように樹脂粒子(C1)中に含有される溶剤を除去ないし減少させ、その後、二酸化炭素を除去することにより、樹脂粒子(C1)同士が合一することを防ぐことができる。
【0103】
溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素で置換する方法としては、樹脂粒子(C1)を一旦フィルターやサイクロンで補足した後、圧力を保ちながら、溶剤(S)が完全に除去されるまで二酸化炭素を流通させる方法が挙げられる。流通させる二酸化炭素の量は、分散体(X1)からの溶剤除去の観点から、(X1)の体積に対して1〜100倍が好ましく、さらに好ましくは1〜50倍、最も好ましくは1〜30倍である。
【0104】
次に製造方法(1)について詳細に説明する。
本発明において、樹脂(b)の前駆体(b0)としては、化学反応により樹脂(b)になりうるものであれば特に限定されず、例えば、樹脂(b)がビニル樹脂である場合は、(b0)は、先述のビニルモノマー(単独で用いても、混合して用いてもよい)が挙げられ、樹脂(b)が縮合系樹脂(例えば、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂)である場合は、(b0)は、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせが例示される。
【0105】
ビニルモノマーを前駆体(b0)として用いた場合、(b0)は通常用いられる開始剤を含有してもよい。上記開始剤としては、パーオキサイド系重合開始剤(I)、アゾ系重合開始剤(II)等が挙げられる。また、パーオキサイド系重合開始剤(I)と還元剤とを併用してレドックス系重合開始剤(III)を形成してもよい。更には、(I)〜(III)のうちから2種以上を併用してもよい。
【0106】
上記開始剤を用いる場合、二酸化炭素(X)中に(b0)を分散する前に、予めモノマーと混合しておくことが好ましい。重合温度は好ましくは40〜100℃、さらに好ましくは60〜90℃である。
【0107】
前駆体(b0)としては、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせを用いることもできる。ここで「反応性基」とは硬化剤(β)と反応可能な基のことをいう。反応性基含有プレポリマー(α)が有する反応性基と、硬化剤(β)の組み合わせとしては、下記(1)、(2)などが挙げられる。
(1):反応性基含有プレポリマー(α)が有する反応性基が、活性水素化合物と反応可能な官能基(α1)であり、硬化剤(β)が活性水素基含有化合物(β1)であるという組み合わせ。
(2):反応性基含有プレポリマー(α)が有する反応性基が活性水素含有基(α2)であり、硬化剤(β)が活性水素含有基と反応可能な化合物(β2)であるという組み合わせ。
【0108】
上記組合せ(1)において、活性水素化合物と反応可能な官能基(α1)としては、イソシアネート基(α1a)、ブロック化イソシアネート基(α1b)、エポキシ基(α1c)、酸無水物基(α1d)および酸ハライド基(α1e)などが挙げられる。これらのうち好ましいものは、(α1a)、(α1b)および(α1c)であり、特に好ましいものは、(α1a)および(α1b)である。ブロック化イソシアネート基(α1b)は、ブロック化剤によりブロックされたイソシアネート基のことをいう。上記ブロック化剤としては、オキシム類[アセトオキシム、メチルイソブチルケトオキシム、ジエチルケトオキシム、シクロペンタノンオキシム、シクロヘキサノンオキシム、メチルエチルケトオキシム等];ラクタム類[γ−ブチロラクタム、ε−カプロラクタム、γ−バレロラクタム等];炭素数1〜20の脂肪族アルコール類[エタノール、メタノール、オクタノール等];フェノール類[フェノール、m−クレゾール、キシレノール、ノニルフェノール等];活性メチレン化合物[アセチルアセトン、マロン酸エチル、アセト酢酸エチル等];塩基性窒素含有化合物[N,N−ジエチルヒドロキシルアミン、2−ヒドロキシピリジン、ピリジンN−オキサイド、2−メルカプトピリジン等];およびこれらの2種以上の混合物が挙げられる。これらのうち好ましいのはオキシム類であり、特に好ましいものはメチルエチルケトオキシムである。
【0109】
反応性基含有プレポリマー(α)の骨格としては、ポリエーテル(αw)、ポリエステル(αx)、エポキシ樹脂(αy)およびポリウレタン(αz)などが挙げられる。これらのうち好ましいものは、(αx)、(αy)および(αz)であり、特に好ましいものは(αx)および(αz)である。ポリエーテル(αw)としては、ポリエチレンオキサイド、ポリプロピレンオキサイド、ポリブチレンオキサイド、ポリテトラメチレンオキサイドなどが挙げられる。ポリエステル(αx)としては、ジオール(11)とジカルボン酸(13)の重縮合物、ポリラクトン(ε−カプロラクトンの開環重合物)などが挙げらる。エポキシ樹脂(αy)としては、ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど)とエピクロルヒドリンとの付加縮合物などが挙げられる。ポリウレタン(αz)としては、ジオール(11)とポリイソシアネート(15)の重付加物、ポリエステル(αx)とポリイソシアネート(15)の重付加物などが挙げられる。
【0110】
ポリエステル(αx)、エポキシ樹脂(αy)、ポリウレタン(αz)などに反応性基を含有させる方法としては、(1):二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させる方法、(2):二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させ、さらに残存した該官能基と反応可能な官能基及び反応性基を含有する化合物を反応させる方法などが挙げられる。上記方法(1)では、水酸基含有ポリエステルプレポリマー、カルボキシル基含有ポリエステルプレポリマー、酸ハライド基含有ポリエステルプレポリマー、水酸基含有エポキシ樹脂プレポリマー、エポキシ基含有エポキシ樹脂プレポリマー、水酸基含有ポリウレタンプレポリマー、イソシアネート基含有ポリウレタンプレポリマーなどが得られる。構成成分の比率は、例えば、水酸基含有ポリエステルプレポリマーの場合、ポリオールとポリカルボン酸の比率が、水酸基[OH]とカルボキシル基[COOH]のモル比[OH]/[COOH]として、好ましくは2/1〜1/1、さらに好ましくは1.5/1〜1/1、とくに好ましくは1.3/1〜1.02/1である。他の骨格、他の末端基のプレポリマーの場合も、構成成分が変わるだけで比率は同様である。上記方法(2)では、上記方法(1)で得られたプレプリマーに、ポリイソシアネートを反応させることでイソシアネート基含有プレポリマーが得られ、ブロック化ポリイソシアネートを反応させることでブロック化イソシアネート基含有プレポリマーが得られ、ポリエポキサイドを反応させることでエポキシ基含有プレポリマーが得られ、ポリ酸無水物を反応させることで酸無水物基含有プレポリマーが得られる。官能基および反応性基を含有する化合物の使用量は、例えば、水酸基含有ポリエステルにポリイソシアネートを反応させてイソシアネート基含有ポリエステルプレポリマーを得る場合、ポリイソシアネートの比率が、イソシアネート基[NCO]と、水酸基含有ポリエステルの水酸基[OH]のモル比[NCO]/[OH]として、好ましくは5/1〜1/1、さらに好ましくは4/1〜1.2/1、とくに好ましくは2.5/1〜1.5/1である。他の骨格、他の末端基を有するプレポリマーの場合も、構成成分が変わるだけで比率は同様である。
【0111】
反応性基含有プレポリマー(α)中の1分子当たりに含有する反応性基は、通常1個以上、好ましくは、平均1.5〜3個、さらに好ましくは、平均1.8〜2.5個である。上記範囲にすることで、硬化剤(β)と反応させて得られる硬化物の分子量が高くなる。反応性基含有プレポリマー(α)の数平均分子量は、好ましくは500〜30,000、さらに好ましくは1,000〜20,000、とくに好ましくは2,000〜10,000である。反応性基含有プレポリマー(α)の重量平均分子量は、1,000〜50,000、好ましくは2,000〜40,000、さらに好ましくは4,000〜20,000である。反応性基含有プレポリマー(α)の粘度は、100℃において、好ましくは2,000ポイズ以下、さらに好ましくは1,000ポイズ以下である。2,000ポイズ以下にすることで、少量の溶剤で粒度分布のシャープな樹脂粒子(C)が得られる点で好ましい。
【0112】
活性水素基含有化合物(β1)としては、脱離可能な化合物でブロック化されていてもよいポリアミン(β1a)、ポリオール(β1b)、ポリメルカプタン(β1c)および水(β1d)などが挙げられる。これらのうち好ましいものは、(β1a)、(β1b)および(β1d)であり、さらに好ましいものは、(β1a)および(β1d)であり、特に好ましいものは、ブロック化されたポリアミン(β1a)および(β1d)である。(β1a)としては、ポリアミン(16)と同様のものが例示される。(β1a)として好ましいものは、4,4’−ジアミノジフェニルメタン、キシリレンジアミン、イソホロンジアミン、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、ヘキサメチレンジアミンおよびそれらの混合物である。
【0113】
(β1a)が脱離可能な化合物でブロック化されたポリアミンである場合の例としては、前記ポリアミン類と炭素数3〜8のケトン類(アセトン、メチルエチルケトン、メチルイソブチルケトンなど)から得られるケチミン化合物、炭素数2〜8のアルデヒド化合物(ホルムアルデヒド、アセトアルデヒド)から得られるアルジミン化合物、エナミン化合物、およびオキサゾリジン化合物などが挙げられる。
【0114】
ポリオール(β1b)としては、前記のジオール(11)およびポリオール(12)と同様のものが例示される。ジオール(11)単独、またはジオール(11)と少量のポリオール(12)の混合物が好ましい。
【0115】
ポリメルカプタン(β1c)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなどが挙げられる。
【0116】
必要により活性水素基含有化合物(β1)と共に反応停止剤(βs)を用いることができる。反応停止剤を(β1)と一定の比率で併用することにより、(b)を所定の分子量に調整することが可能である。反応停止剤(βs)としては、モノアミン(ジエチルアミン、ジブチルアミン、ブチルアミン、ラウリルアミン、モノエタノールアミン、ジエタノールアミンなど);モノアミンをブロックしたもの(ケチミン化合物など);モノオール(メタノール、エタノール、イソプロパノール、ブタノール、フェノール;モノメルカプタン(ブチルメルカプタン、ラウリルメルカプタンなど);モノイソシアネート(ラウリルイソシアネート、フェニルイソシアネートなど);モノエポキサイド(ブチルグリシジルエーテルなど)などが挙げられる。
【0117】
上記組合せ(2)における反応性基含有プレポリマー(α)が有する活性水素含有基(α2)としては、アミノ基(α2a)、水酸基(アルコール性水酸基およびフェノール性水酸基)(α2b)、メルカプト基(α2c)、カルボキシル基(α2d)およびそれらが脱離可能な化合物でブロック化された有機基(α2e)などが挙げられる。これらのうち好ましいものは、(α2a)、(α2b)およびアミノ基が脱離可能な化合物でブロック化された有機基(α2e)であり、特に好ましいものは、(α2b)である。アミノ基が脱離可能な化合物でブロック化された有機基としては、前記(β1a)の場合と同様のものが例示できる。
【0118】
活性水素含有基と反応可能な化合物(β2)としては、ポリイソシアネート(β2a)、ポリエポキシド(β2b)、ポリカルボン酸(β2c)、ポリ酸無水物(β2d)およびポリ酸ハライド(β2e)などが挙げられる。これらのうち好ましいものは、(β2a)および(β2b)であり、さらに好ましいものは、(β2a)である。
【0119】
ポリイソシアネート(β2a)としては、ポリイソシアネート(15)と同様のものが例示され、好ましいものも同様である。
【0120】
ポリエポキシド(β2b)としては、ポリエポキシド(18)と同様のものが例示され、好ましいものも同様である。
【0121】
ポリカルボン酸(β2c)としては、ジカルボン酸(β2c−1)および3価以上のポリカルボン酸(β2c−2)が挙げられ、(β2c−1)単独、および(β2c−1)と少量の(β2c−2)の混合物が好ましい。ジカルボン酸(β2c−1)としては、前記ジカルボン酸(13)と、ポリカルボン酸としては、前記ポリカルボン酸(14)と、それぞれ、同様のものが例示され、好ましいものも同様である。
【0122】
ポリカルボン酸無水物(β2d)としては、ピロメリット酸無水物などが挙げられる。ポリ酸ハライド類(β2e)としては、前記(β2c)の酸ハライド(酸クロライド、酸ブロマイド、酸アイオダイド)などが挙げられる。さらに、必要により(β2)と共に反応停止剤(βs)を用いることができる。
【0123】
硬化剤(β)の比率は、反応性基含有プレポリマー(α)中の反応性基の当量[α]と、硬化剤(β)中の活性水素含有基[β]の当量の比[α]/[β]として、好ましくは1/2〜2/1、さらに好ましくは1.5/1〜1/1.5、とくに好ましくは1.2/1〜1/1.2である。なお、硬化剤(β)が水(β1d)である場合は水は2価の活性水素化合物として取り扱う。
【0124】
前駆体(b0)として反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせを用いる場合、二酸化炭素(X)中に微粒子(A)が分散している分散体(X0)において(b0)を反応させる方法は特に限定されないが、(X0)中に(b0)を分散する直前に(α)と(β)を混合し、分散すると同時に反応させる方法が好ましい。反応時間は、プレポリマー(α)の有する反応性基の構造と硬化剤(β)の組み合わせによる反応性により選択されるが、好ましくは5分〜24時間である。反応は減圧前に(X0)中で完結させてもよく、また(X0)である程度反応させ、減圧し(C)を取り出した後、恒温槽などで熟成させ完結させてもよい。また、必要に応じて公知の触媒を使用することができる。具体的には、例えばイソシアネートと活性水素化合物の反応の場合には、ジブチルチンラウレート、ジオクチルチンラウレートなどが挙げられる。反応温度は好ましくは30〜100℃、さらに好ましくは40〜80℃である。
【0125】
反応性基含有プレポリマー(α)と硬化剤(β)からなる前駆体(b0)を反応させた樹脂(b)が樹脂粒子(B)および樹脂粒子(C)の構成成分となる。反応性基含有プレポリマー(α)と硬化剤(β)を反応させた樹脂(b)の重量平均分子量は、好ましくは3,000以上、さらに好ましくは3,000〜1000万、とくに好ましくは,5000〜100万である。
【0126】
また、反応性基含有プレポリマー(α)と硬化剤(β)との反応時に、反応性基含有プレポリマー(α)および硬化剤(β)と反応しないポリマー[いわゆるデッドポリマー]を系内に含有させることもできる。この場合(b)は、反応性基含有プレポリマー(α)と硬化剤(β)を反応させて得られた樹脂と、反応させていない樹脂の混合物となる。
製造方法(1)の場合、上述した事項及び、樹脂(b)を溶剤(S)に溶解させた溶液(L)の代わりに樹脂(b)の前駆体(b0)を用いて分散時に前駆体(b0)を反応させる以外は、製造方法(2)と同様である。
【0127】
製造方法(3)について詳細に説明する。
樹脂(b)の溶剤(S)の溶液(L)の代わりに、樹脂(b)の前駆体(b0)の溶剤(S)の溶液(L0)を用い、分散時に前駆体(b0)を反応させる以外は製造方法(2)と同様である。
【0128】
上記製造方法(1)〜(3)によれば、親水性基を有する界面活性物質を実質的に含有しない本発明の樹脂粒子(C)を製造することができる。ここで、親水性基を有する界面活性物質とは、アニオン界面活性剤(S−1)、カチオン界面活性剤(S−2)、両性界面活性剤(S−3)、非イオン界面活性剤(S−4)などが挙げられる。
【0129】
これら界面活性剤の具体例としては、国際公開WO03/106541号パンフレットに記載のものが挙げられる。通常、水溶剤中で親水性基を有する界面活性物質を用いて製造された樹脂粒子は、親水性基を有する界面活性物質を実質的に含有している。
【0130】
樹脂粒子が親水性基を有する界面活性物質を実質的に含有しないことを分析する方法としては、公知の表面濡れ性評価(色材協会誌、第73[3]号、2000年、P132〜138による。)が挙げられる。表面濡れ性の評価法は次の通りである。すなわち100mlビーカーに樹脂粒子0.1gを入れ、そこにイオン交換水を20ml添加し、マグネティックスターラーで攪拌し、液面に樹脂粒子を浮かべた後、アセトンを少しづつ滴下し、表面に浮かぶ樹脂粒子が無くなるアセトン重量(Wa)と水の重量(Ww)を有効数字3桁で求め、(1)式より、樹脂粒子表面の溶解度パラメータ(δm)を算出する。
δm=(9.75×Wa+23.43×Ww)/(Wa+Ww) (1)
【0131】
樹脂粒子表面の溶解度パラメータ(δm)が、9.8〜21、好ましくは9.8〜20であれば、樹脂粒子が親水性基を有する界面活性物質を実質的に含有しないものと判断される。δmが、9.8〜21であれば、樹脂粒子の耐湿保存性は良好であり、高湿下におけるトナーとしての電気特性、流動性、定着性が良好である。本測定方法では9.8未満は測定できない。
【0132】
樹脂粒子(C)は、微粒子(A)と樹脂粒子(B)の粒径、及び、微粒子(A)又は(A)由来の皮膜による樹脂粒子(B)表面の被覆率を変えることで粒子表面に所望の凹凸を付与することができる。さらに減圧時の温度・圧力をコントロールすることにより内部に気泡を有する多孔質体が得られ、比表面積を大きくすることができる。粉体流動性を向上させたい場合には、樹脂粒子のBET値比表面積が0.5〜5.0m2/gであるのが好ましい。BET比表面積は、比表面積計、例えば、QUANTASORB(ユアサアイオニクス製)を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)したものである。同様に粉体流動性の観点から、本発明の樹脂粒子(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
【0133】
本発明の製造方法で得られる樹脂粒子(C)は粒度分布がシャープであり、且つ通常、水溶性の界面活性物質やイオン性物質を含まないため、疎水性である。したがって本発明の樹脂粒子(C)は電子写真トナー用として有用である。またその他の用途として、塗料用添加剤、接着剤用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成形用樹脂、粉体塗料、電子部品製造用スペーサー、触媒用担体、静電記録トナー、静電印刷トナー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、クロマトグラフ充填剤、電気粘性流体用粒子等としても有用である。
【0134】
本発明の樹脂粒子(C)において、微粒子(A)が、融点が50〜110℃である、結晶性樹脂(a1)を含有する場合は、低温定着性に優れ、耐熱保存性にも優れるという効果を有することから、特に電子写真トナー用として有用である。
融点が50〜110℃である結晶性樹脂(a1)の中でも、(a11)、(a12)、(a13)、及び(a14)が特に好ましい。
【0135】
電子写真プロセスにおいて使用される電子写真トナーは、その現像工程において、例えば、静電荷像が形成されている感光体等の像担持体に一旦付着され、次に転写工程において、感光体から転写紙等の転写媒体に転写された後、定着工程において紙面に定着される。電子写真トナーとしては、通常、流動特性を付与するため、トナー用樹脂粒子と各種金属酸化物等の無機粉末等を混合して使用されており、この無機粉末等は外添剤と呼ばれている。本発明の電子写真トナーは、本発明の樹脂粒子に外添剤を添加したものが好ましい。
【0136】
外添剤としては、例えば、二酸化珪素(シリカ)、二酸化チタン(チタニア)、酸化アルミニウム、酸化亜鉛、酸化マグネシウム、酸化セリウム、酸化鉄、酸化銅、酸化錫等が知られている。特に、シリカや酸化チタン微粒子とジメチルジクロロシラン、ヘキサメチルジシラザン、シリコーンオイル等の有機珪素化合物とを反応させ、シリカ微粒子表面のシラノール基を有機基で置換し疎水化したシリカ微粒子が好ましく用いられる。
【0137】
外添剤の使用量(重量%)としては、特に制限はないが、定着性と流動性の両立の観点から、電子写真トナー用樹脂粒子の重量に対して0.01〜5が好ましく、さらに好ましくは、0.1〜4、特に好ましくは0.5〜3である。
【0138】
本発明の電子写真トナーは、本発明の電子写真トナー用樹脂粒子に外添剤を添加し、混合することにより製造することができる。
【実施例】
【0139】
以下実施例により本発明をさらに説明するが、本発明はこれに限定されるものではない。以下の記載において「部」は重量部、「%」は重量%を示す。
なお、実施例1、5、8、9、10、19、および20は参考例である。
【0140】
下記の膨潤度、結晶化度、数平均分子量、融点、ガラス転移温度、体積平均粒径は以下の方法で測定した。
<膨潤度の測定方法>
試料(5mg)を採取して磁気浮遊天秤(MSB−SCC・SCW 日本ベル社製)を用いて40℃、10MPaにおける超臨界状態の二酸化炭素が試料に浸透する重量を測定し、試料の重量で除することで、膨潤度(%)を求めた。
<結晶化度の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)を用いて室温から昇温速度20℃/minにて温度を変化させながら、吸熱ピークの面積より求めた融解熱量(ΔHm(J/g))を求めた。測定されたΔHmに基づき以下の式により結晶化度(%)を算出した。
結晶化度=(融解熱量/a)×100
上式中、aは以下のようにして測定する。
測定しようとする樹脂と同組成の標品となる樹脂の融解熱量をDSCで測定し、JISK0131(1996年)(X線回折分析通則 13結晶化度測定 (2)絶対法)に準じた測定方法で結晶化度を測定する。縦軸に融解熱量、横軸に結晶化度を座標にとり、標品のデータをプロットし、その点と原点の2点から直線を引き、結晶化度が100%となるように外挿した場合の融解熱量を求めた値がaである。
【0141】
<数平均分子量(Mn)の測定方法>
試料をそれぞれ濃度2.5g/Lでテトラヒドロフランに溶解させ、ポリスチレンを標準物質として、GPCにより測定した。
GPC機種:HLC−8120GPC、東ソー(株)製
カラム :TSKgel GMHXL)2本+TSKgel Multipore HXL−M(東ソー(株)製)
<融点の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃で、結晶溶融による吸熱ピークの温度(℃)を求めた。
<ガラス転移温度(Tg)の測定方法>
試料をそれぞれ5mg秤り取り、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃でガラス転移温度を測定した。
<体積平均粒径の測定方法>
試料5mgをイオン交換水10gに分散させた後、マルチサイザーIII(コールター社製)により測定した。
【0142】
製造例1<樹脂(b−1)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、1.2−プロピレングリコール(以下、プロピレングリコールと記載)831部、テレフタル酸703部、アジピン酸47部、および縮合触媒としてテトラブトキシチタネート0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が87℃になった時点で180℃まで冷却し、さらに無水トリメリット酸24部、テトラブトキシチタネート0.5部を投入し90分反応させた後、取り出した。回収されたプロピレングリコールは442部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、ポリエステル樹脂(b−1)を得た。この樹脂のMnは1900、Tgは45℃であった。
【0143】
製造例2<樹脂(b−2)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール729部、テレフタル酸683部、アジピン酸67部、無水トリメリット酸38部および縮合触媒としてテトラブトキシチタネート0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは172部であった。軟化点が160℃になった時点で取り出し、室温まで冷却後、粉砕し粒子化し、ポリエステル樹脂(b−2)を得た。この樹脂のMnは5700、Tgは63℃であった。
【0144】
製造例3<樹脂(b−3)の調製>
撹拌棒および温度計をセットしたオートクレーブに、キシレン24部を投入し、アクリル酸ブチル/メタクリル酸メチル/スチレン/アクリル酸2−エチルヘキシル(25重量%/33重量%/40重量%/2重量%)の混合モノマー2,000部と重合触媒1部を、170℃で3時間かけて滴下重合をおこなった。180℃まで昇温しながら常圧で脱揮し、180℃になったところで減圧に切り替え、2時間かけて減圧で脱揮をおこない、ビニル樹脂(b−3)を得た。この樹脂のMnは10,500、Tgは62℃であった。
【0145】
製造例4<樹脂溶液(L−1)の調製>
攪拌装置のついた容器に、アセトン490部、メタノール175部、イオン交換水35部からなる混合溶剤である溶剤(S−1)に、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)と樹脂(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−1)を得た。溶剤(S−1)は、標準状態の樹脂(b)と溶剤(S−1)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−1)のSP値は11.8であった。
【0146】
製造例5<樹脂溶液(L−2)の調製>
攪拌装置のついた容器に、アセトン450部、イオン交換水50部からなる混合溶剤である溶剤(S−2)に、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−2)を得た。溶剤(S−2)は、標準状態の樹脂(b)と溶剤(S−2)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−2)のSP値は11.3であった。
【0147】
製造例6<樹脂溶液(L−3)の調製>
攪拌装置のついた容器に、アセトン490部、メタノール210部からなる混合溶剤である溶剤(S−3)に、製造例3で得られた樹脂(b−3)280部、及びカーボンブラック15部を仕込み、樹脂(b−3)が完全に溶解するまで攪拌し、樹脂溶液(L−3)を得た。溶剤(S−3)は、標準状態の樹脂(b)と溶剤(S−3)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−3)のSP値は11.3であった。
【0148】
製造例7<樹脂溶液(L−4)の調製>
攪拌装置のついた容器に、アセトン560部、イオン交換水70部、デカン70部からなる混合溶剤である溶剤(S−4)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−4)を得た。溶剤(S−4)は、標準状態の樹脂(b)と溶剤(S−4)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−4)のSP値は10.3であった。
【0149】
製造例8<樹脂前駆体(b0−1)の調整>
オートクレーブに、製造例1で得られた樹脂(b−1)407部、イソホロンジイソシアネート(IPDI)54部、アセトン485部を投入し、密閉状態で100℃、5時間反応を行い、分子末端にイソシアネート基を有する樹脂前駆体(b0−1)を得た。樹脂前駆体(b0−1)のNCO含量は0.8%であった。
【0150】
製造例9<硬化剤(β)の調整>
撹拌機、脱溶剤装置、および温度計をセットした反応容器に、イソホロンジアミン50部とメチルエチルケトン300部を投入し、50℃で5時間反応を行った後、脱溶剤してケチミン化合物である硬化剤(β)(ウレタンプレポリマーの鎖伸長剤)を得た。硬化剤(β)の全アミン価は415であった。
【0151】
製造例10<樹脂前駆体溶液(L−5)の調整>
攪拌装置のついた容器に、ジメチルホルムアミドである溶剤(S−5)700部、製造例1で得られた樹脂(b−1)228部、製造例8で得られた樹脂前駆体(b0−1)57部、硬化剤(β)1.5部、及びカーボンブラック15部を仕込み、樹脂前駆体(b0−1)及び硬化剤(β)が完全に溶解するまで攪拌し、樹脂溶液(L−5)を得た。溶剤(S−5)は、標準状態の上記重量比の樹脂(b)および前駆体(b0)と溶剤(S−5)の等重量混合物における樹脂(b)および前駆体(b0)の重量に対する(b)および(b0)の不溶分重量は0.1重量%以下、溶剤(S−5)のSP値は12.0であった。
【0152】
製造例11<樹脂溶液(L−6)の調製>
攪拌装置のついた容器に、アセトン490部、イオン交換水210部からなる混合溶剤である溶剤(S−6)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−6)を得た。溶剤(S−6)は、標準状態の樹脂(b)と溶剤(S−6)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は15重量%、溶剤(S−6)のSP値は14.0であった。
【0153】
製造例12<樹脂溶液(L−7)の調製>
攪拌装置のついた容器に、メチルエチルケトン665部、イオン交換水35部からなる混合溶剤である溶剤(S−7)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−7)を得た。溶剤(S−7)は、標準状態の樹脂(b)と溶剤(S−7)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−7)のSP値は9.7であった。
【0154】
製造例13<樹脂溶液(L−8)の調製>
攪拌装置のついた容器に、1,3−ジオキソラン(S−8)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−8)を得た。溶剤(S−8)は、標準状態の樹脂(b)と溶剤(S−8)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.5重量%、溶剤(S−8)のSP値は9.4であった。
【0155】
製造例14<樹脂溶液(L−9)の調製>
攪拌装置のついた容器に、ピルビン酸メチル(S−9)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−9)を得た。溶剤(S−9)は、標準状態の樹脂(b)と溶剤(S−9)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は1重量%、溶剤(S−9)のSP値は10.6であった。
【0156】
製造例15<樹脂溶液(L−10)の調製>
攪拌装置のついた容器に、プロピレングリコールモノメチルエーテル(S−10)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−10)を得た。溶剤(S−10)は、標準状態の樹脂(b)と溶剤(S−10)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は3重量%、溶剤(S−10)のSP値は11.3であった。
【0157】
製造例16<樹脂溶液(L−11)の調製>
攪拌装置のついた容器に、2−ヒドロキシイソ酪酸メチル(S−11)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−11)を得た。溶剤(S−11)は、標準状態の樹脂(b)と溶剤(S−11)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は7重量%、溶剤(S−11)のSP値は11.8であった。
【0158】
製造例17<樹脂溶液(L−12)の調製>
攪拌装置のついた容器に、乳酸メチル(S−12)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−12)を得た。溶剤(S−12)は、標準状態の樹脂(b)と溶剤(S−12)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は10重量%、溶剤(S−12)のSP値は12.4であった。
【0159】
製造例18<樹脂溶液(L−13)の調製>
攪拌装置のついた容器に、トリフルオロエタノール(S−13)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−13)を得た。溶剤(S−13)は、標準状態の樹脂(b)と溶剤(S−13)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は18重量%、溶剤(S−13)のSP値は15.1であった。
【0160】
製造例19<結晶性樹脂(a1−1)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸230部、1,6−ヘキサンジオール195部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−1)を得た。この樹脂の結晶化度は60%、融点は60℃、Mnは8,000であった。
【0161】
製造例20<結晶性樹脂(a1−2)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート(炭素数22個の直鎖アルキル基を有するアルコールのアクリレートプレンマーVA〔日油(株)製〕)150部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−2)を得た。この樹脂の結晶化度は42%、融点は65℃、Mnは50,000であった。
【0162】
製造例21<結晶性樹脂(a1−3)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート120部、2−デシルテトラデシルメタクリレート30部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−3)を得た。この樹脂の結晶化度は36%、融点は62℃、Mnは50,000であった。
【0163】
製造例22<結晶性樹脂(a1−4)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート150部、ブチルアクリレート50部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−4)を得た。この樹脂の結晶化度は20%、融点は50℃、Mnは40,000であった。
【0164】
製造例23<結晶性樹脂(a1−5)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸460部、1,6−ヘキサンジオール230部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−5)を得た。この樹脂の結晶化度は65%、融点は70℃、Mnは10,000であった。
【0165】
製造例24<結晶性樹脂(a1−6)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸460部、1,6−ヘキサンジオール230部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させた。得られた樹脂を50℃に冷却後、メチルエチルケトン300部に溶解させ、ヘキサメチレンジイソシアネート20部をいれ、3時間反応させ、20mmHgの減圧下で脱溶剤後、取り出した。取り出した樹脂を粉砕、粒子化し、結晶性ポリウレタン樹脂(a1−6)を得た。この樹脂の結晶化度は50%、融点は70℃、Mnは15,000であった。
【0166】
製造例25<結晶性樹脂(a1−7)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート135部、アクリロニトリル 15部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−7)を得た。この樹脂の結晶化度は41%、融点は62℃、Mnは50,000であった。
【0167】
製造例26<結晶性樹脂(a1−8)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、テレフタル酸90部、セバシン酸340部、1,6−ヘキサンジオール310部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−8)を得た。この樹脂の結晶化度は60%、融点は67℃、Mnは9,000であった。
【0168】
製造例27<結晶性樹脂(a1−9)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ステアリルアクリレート(炭素数18個の直鎖アルキル基を有するアルコールのアクリレートプレンマーSA〔日油(株)製〕)150部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−9)を得た。この樹脂の結晶化度は32%、融点は54℃、Mnは50,000であった。
【0169】
製造例28<微粒子(A−1)分散液〜微粒子(A−9)分散液の調製>
ノルマルヘキサン700部、結晶性樹脂(a1−1)〜(a1−9)の各々300部を混合した後、ビーズミル(ダイノーミルマルチラボ:シンマルエンタープライゼス製)で粒径0.3mmのジルコニアビーズを用いて粉砕を行い、乳白色の微粒子(A−1)分散液〜微粒子(A−9)分散液を得た。分散液の体積平均粒径は表1及び表2に記載のように0.2〜0.4μmであった。また、微粒子(A−1)〜(A−9)の膨潤度は、表1及び表2に記載のとおりであった。
【0170】
製造例29<微粒子(A−10)分散液の調製>
製造例28において、結晶性樹脂(a1−1)〜(a1−9)の各々の代わりに、ポリオレフィン樹脂(サンワックス161−P(三洋化成工業製)、結晶化度は75%、融点は107℃、Mnは5,000)(a1−10)を用いた以外は製造例28と同様にして、乳白色の微粒子(A−10)分散液を得た。この分散液の体積平均粒径は0.4μmであった。また、微粒子(A−10)の膨潤度は15%であった。
【0171】
製造例30<微粒子(A−11)の調製>
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン139部、ジビニルベンゼン20部、メタクリル酸138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成して非結晶性ビニル樹脂(a2−1)(スチレン−メタクリル酸−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩−ジビニルベンゼンの共重合体、架橋性)の微粒子(A−11)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、微粒子(A−11)を得た。非結晶性ビニル樹脂(a2−1)の結晶化度は0%、ガラス転移温度は69℃であった。また、微粒子(A−11)の膨潤度は1%であった。
【0172】
製造例31<微粒子(A−12)分散液の調製>
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン139部、メタクリル酸メチル138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成して非結晶性ビニル樹脂(a2−2)(スチレン−メタクリル酸メチル−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の微粒子(A−12)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、微粒子(A−12)を得た。非結晶性ビニル樹脂(a2−2)の結晶化度は0%、ガラス転移温度は56℃であった。また、微粒子(A−12)の膨潤度は10%であった。
【0173】
製造例32<分散安定剤(E)溶液の調製>
攪拌機を備えた反応容器内にTHF700部を仕込み、反応容器内の空気を窒素置換した後、加熱して還流温度とした。次に、メタクリル酸メチル150部、メタクリル変性シリコーン(官能基等量:12,000g/mol、Mn12,000、信越化学工業製:X22−2426、)150部、アゾビスイソブチロニトリル1.5部の混合物を反応基内に2時間で適下後、還流温度で6時間熟成し、分散安定剤(E−1)溶液を得た。(E−1)の重量平均分子量は20,000であった。
【0174】
実施例1
図1の実験装置において、まずバルブV1、V2を閉じ、ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素(純度99.99%)を導入し、14MPa、40℃に調整した。また樹脂溶液タンクT1に樹脂溶液(L−1)、微粒子分散液タンクT2に微粒子(A−1)分散液を仕込んだ。次にボンベB1、ポンプP3より二酸化炭素を分散槽T3に導入し、9MPa、40℃に調整し、さらにタンクT2、ポンプP2より微粒子(A−1)分散液を導入した。次に分散槽T3の内部を2000rpmで攪拌しながら、タンクT1、ポンプP1より樹脂溶液(L−1)を分散槽T3内に導入した。導入後T3の内部の圧力は14MPaとなった。
【0175】
なお分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−1)分散液 45部
二酸化炭素 550部
なお導入した二酸化炭素の重量は、二酸化炭素の温度(40℃)、及び圧力(15MPa)から二酸化炭素の密度を下記文献2に記載の状態式より算出し、これに分散槽T3の体積を乗じることにより算出した。
文献2:Journal of Physical and Chemical Refarence data、vol.25、P.1509〜1596
【0176】
樹脂溶液(L−1)を導入後、1分間攪拌し分散体(X1)を得た。バルブV1を開き、P3よりT4内に二酸化炭素を導入した後、分散体(X1)をT4内に導入し、この間圧力が一定に保たれるように、V2の開度を調節した。この操作を30秒間行い、V1を閉めた。この操作によりT4内に導入された樹脂溶液からの溶剤の抽出を行った。さらにT4を60℃に加熱し、15分間保持した。この操作により、微粒子(A−1)を樹脂溶液(L−1)から形成された樹脂粒子(B−1)の表面に固着させ、樹脂粒子(C−1)を生成した。次に圧力ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素を導入しつつ圧力調整バルブV2により圧力を14MPaに保持することにより、抽出された溶剤を含む二酸化炭素を溶剤トラップ槽T5に排出すると共に、樹脂粒子(C−1)をフィルターF1に捕捉した。圧力ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素を導入する操作は、上記の分散槽T3に導入した二酸化炭素重量の5倍量を粒子回収槽T4に導入した時点で二酸化炭素の導入を停止した。この停止の時点で、溶剤を含む二酸化炭素を、溶剤を含まない二酸化炭素で置換すると共に樹脂粒子(C−1)をフィルターF1に捕捉する操作は完了した。さらに、圧力調整バルブV2を少しずつ開き、粒子回収槽内を大気圧まで減圧し、フィルターF1に補足されている、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−1)を得た。
【0177】
実施例2
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−2)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−2)を得た。実施例2における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−2) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0178】
実施例3
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−3)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−3)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−3)を得た。実施例3における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−3) 270部
微粒子(A−3)分散液 45部
二酸化炭素 550部
【0179】
実施例4
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−4)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−4)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−4)を得た。実施例4における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−4) 270部
微粒子(A−4)分散液 45部
二酸化炭素 550部
【0180】
実施例5
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−5)分散液を使用したこと、疎水性分散安定剤(E−1)を14部追加したこと、樹脂溶液(L−1)の代わりに(L−5)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−5)を得た。実施例5における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−5) 270部
微粒子(A−5)分散液 45部
分散安定剤(E−1)溶液 14部
二酸化炭素 550部
【0181】
実施例6
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−6)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−6)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−6)を得た。実施例6における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−6) 270部
微粒子(A−6)分散液 45部
二酸化炭素 550部
【0182】
実施例7
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−7)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−7)を得た。実施例7における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−7)分散液 45部
二酸化炭素 550部
【0183】
実施例8
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−8)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−8)を得た。実施例8における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−8)分散液 45部
二酸化炭素 550部
【0184】
実施例9
実施例1において、微粒子(A−1)分散液の代わりに、膨潤度が0%である疎水性シリカ(RX−50、日本アエロジル製、体積平均粒径150nm)(a3−1)からなる微粒子(A−13)を用いたこと、樹脂溶液(L−1)の代わりに樹脂前駆体(b0−1)及びイオン交換水20部を使用したこと、以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)が固着された樹脂粒子(C−9)を得た。実施例9における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂前駆体(b0−1) 270部
イオン交換水(ケチミン伸長のための水) 20部
疎水性シリカ(A−9) 7部
二酸化炭素 550部
【0185】
実施例10
実施例1において、微粒子(A−1)分散液の代わりに、T3に予め微粒子(A−11)を仕込んだこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)が固着された樹脂粒子(C−10)を得た。実施例10における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−10) 13.5部
二酸化炭素 550部
【0186】
実施例11
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−7)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−11)を得た。実施例11における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−7) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0187】
実施例12
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−8)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−12)を得た。実施例12における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−8) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0188】
実施例13
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−9)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−13)を得た。実施例13における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−9) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0189】
実施例14
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−10)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−14)を得た。実施例14における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−10) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0190】
実施例15
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−11)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−15)を得た。実施例15における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−11) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0191】
実施例16
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−12)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−16)を得た。実施例16における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−12) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0192】
実施例17
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−13)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−17)を得た。実施例17における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−13) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0193】
実施例18
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−9)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−2)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−18)を得た。実施例18における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−2) 270部
微粒子(A−9)分散液 45部
二酸化炭素 550部
【0194】
実施例19
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−10)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−19)を得た。実施例19における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−10)分散液 45部
二酸化炭素 550部
【0195】
実施例20
ビーカー内に微粒子(A−12)の水性分散液(固形分濃度20重量%)11部、ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液80部、イオン交換水300部を混合攪拌し、樹脂溶液(L−1)150部を混合した後、TKホモミキサー(特殊機化製)を使用し、回転数12,000rpmで10分間混合した。混合後、撹拌棒および温度計をセットした反応容器に混合液を投入し、50℃で2時間で脱溶剤を行い、次いで濾別、乾燥を行い、樹脂粒子(C−20)を得た。実施例20におけるビーカーへの仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 150部
微粒子(A−12)の水性分散液 11部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液 80部
イオン交換水 300部
【0196】
比較例1
実施例1において微粒子(A−1)分散液を仕込まない以外は実施例1と同様にして、比較樹脂粒子(C−1’)を得た。比較例1における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
二酸化炭素 550部
【0197】
比較製造例1
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン100部、メタクリル酸メチル138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(a−1’)(スチレン−メタクリル酸メチル−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、比較樹脂微粒子(A−1’)を得た。(A−1’)の膨潤度は18%であった。
【0198】
比較例2
実施例1において微粒子(A−1)分散液の代わりに、T3に予め比較樹脂微粒子(A−1’)を仕込んだこと以外は実施例1と同様にして、比較樹脂粒子(C−2’)を得た。比較例2における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
比較樹脂微粒子(A−1’) 15部
二酸化炭素 550部
【0199】
比較製造例2
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン111部、メタクリル酸メチル128部、アクリル酸ブチル164部、2−エチルヘキシルメタクリレート58部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(a−2’)(スチレン−メタクリル酸メチル−アクリル酸ブチル−2−エチルヘキシルメタクリレート−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の比較樹脂粒子(A−2’)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。また、(A−2’)の膨潤度は21%であった。
【0200】
比較例3
ビーカー内に比較樹脂粒子(A−2’)の水性分散液11部、ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液80部、イオン交換水300部を混合攪拌し、樹脂溶液(L−1)150部を混合した後、TKホモミキサー(特殊機化製)を使用し、回転数12,000rpmで10分間混合した。混合後、撹拌棒および温度計をセットした反応容器に混合液を投入し、50℃で2時間で脱溶剤を行い、次いで濾別、乾燥を行い、樹脂粒子(C−3’)を得た。比較例3におけるビーカーへの仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 150部
比較樹脂粒子(A−2’)の水性分散液 11部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液 80部
イオン交換水 300部
【0201】
比較例4
実施例1において、樹脂溶液(L−1)の代わりに樹脂(b−1)228部及び(b−2)57部を使用し、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用した点及び、タンクT1、ポンプP1より樹脂(b−1)を分散槽T3内に導入する際に、100℃に昇温し、溶融させた点以外は同様にして、樹脂粒子(C−4’)を得た。比較例4における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂(b−1) 228部
樹脂(b−2) 57部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0202】
本発明の樹脂粒子及び比較の樹脂粒子の原料とその物性値を表1〜3に示した。
【0203】
評価結果
実施例1〜20、比較例1〜4で得られた樹脂粒子について、以下に記載した評価方法で表面濡れ性、粒度分布、耐熱保存性、耐湿耐熱保存性、低温定着温度を評価し、結果を表4〜6に記載した。
<表面濡れ性評価>
100mlビーカーに樹脂粒子0.1gを入れ、そこにイオン交換水20mlを添加し、マグネティックスターラーで攪拌し、液面に樹脂粒子を浮かべた後、アセトンを少しづつ滴下し、表面に浮かぶ樹脂粒子が無くなるアセトン重量(Wa)と水の重量(Ww)を有効数字3桁で求め、(1)式より、樹脂粒子表面の溶解度パラメータ(δm)を算出した。
δm=(9.75×Wa+23.43×Ww)/(Wa+Ww) (1)
<粒度分布の評価>
樹脂粒子をドデシルベンゼンスルホン酸ナトリウム水溶液(濃度0.1%)に分散して樹脂粒子(表中ではCと表記)の体積平均粒径/個数平均粒径をコールターカウンター[マルチサイザーIII(ベックマン・コールター社製)]で測定した。体積平均粒径/個数平均粒径が小さいほど、粒度分布がシャープであることを示す。
【0204】
<耐熱保存性の評価>
樹脂粒子の耐熱保存性を下記の方法で評価した。即ち、50℃に温調された乾燥機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○:ブロッキングが発生しない。
△:ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×:ブロッキングが発生し、簡単に指などで力を加えても分散しない。
<耐湿耐熱保存性の評価>
樹脂粒子の耐湿耐熱保存性を下記の方法で評価した。即ち、50℃、湿度80%に温調された恒温恒湿機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○:ブロッキングが発生しない。
△:ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×:ブロッキングが発生し、簡単に指などで力を加えても分散しない。
【0205】
本発明の樹脂粒子を用いて、以下の方法で本発明の電子写真用トナーを作成した。即ち、本発明の電子写真トナー用樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、ミキサーを用いてよく混ぜてアエロジルR972が樹脂粒子表面に均一に付着した電子写真用トナーを作成した。トナーの評価方法は以下のとおり。
【0206】
<低温定着温度の評価>
低温定着温度は、以下の方法により評価した。上記で得た電子写真用トナーを紙面上に0.6mg/cm2となるよう均一に載せる。このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通した時のコールドオフセットの発生温度を測定した。
【0207】
【表1】

【0208】
【表2】
【0209】
【表3】
【0210】
【表4】
【0211】
【表5】
【0212】
【表6】
【0213】
実施例1〜20で得られた樹脂粒子は体積平均粒径/個数平均粒径が小さく、粒度分布がシャープになったのに対し、比較例1、2、4で得られた樹脂粒子は粒子が凝集し、粒度分布が著しく悪化した。また、実施例1〜20で得られた樹脂粒子は耐湿耐熱保存性に優れるが、比較例3で得られた樹脂粒子は耐湿耐熱保存性に劣っていた。また、実施例1〜8、11〜19で得られた樹脂粒子は電子写真用トナーとして用いた場合には、低温定着性に優れる(低温定着温度が低い。)という効果も有していた。
【産業上の利用可能性】
【0214】
本発明の樹脂粒子は電子写真トナーとしてきわめて有用である。また、塗料用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成型用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、電気粘性用粒子、その他成型用樹脂粒子としても有用である。
【符号の説明】
【0215】
T1:樹脂溶液タンク
T2:微粒子分散液タンク
T3:分散槽(最高使用圧力20MPa、最高使用温度100℃、攪拌機つき)
T4:粒子回収槽(最高使用圧力20MPa、最高使用温度100℃)
F1:セラミックフィルター(メッシュ:0.5μm)
T5:溶剤トラップ
B1、B2:二酸化炭素ボンベ
P1、P2:溶液ポンプ
P3、P4:二酸化炭素ポンプ
V1:バルブ
V2:圧力調整バルブ
【技術分野】
【0001】
本発明は、粒度分布が狭い樹脂粒子、及び、液状又は超臨界状態の流体を用いる該樹脂粒子の製造方法に関するものである。
【背景技術】
【0002】
従来より非水媒体中における粒子形成法として、超臨界流体中に樹脂溶液を噴霧する方法(例えば、特許文献1、2参照)、有機顔料、酸化ケイ素などの微粒子分散剤の存在下において超臨界流体中に加熱溶融させた樹脂を機械的に分散させて微粒子化した後、減圧して樹脂粒子を得る方法(例えば、特許文献3参照)、微粒子分散剤及び活性剤の存在下において超臨界流体中に樹脂溶液を機械的に分散させて、微粒子化した後、減圧して樹脂粒子を得る方法(例えば特許文献4,5)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】WO97/31691号パンフレット
【特許文献2】WO95/01221号パンフレット
【特許文献3】特開2005−107405号公報
【特許文献4】特開2006−321830号公報
【特許文献5】特開2007−277511号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
上記の超臨界流体中に樹脂溶液を噴霧する方法によれば、親水性成分を必要とせず、界面活性物質が樹脂粒子中または表面に残存しないため、粉体特性、電気特性等に優れた粒子を得ることができるが、シャープな粒度分布が得難い問題がある。上記の超臨界流体中に加熱溶融した樹脂を分散する方法によれば、粉体特性、電気特性等に優れ、粒度分布の狭い粒子を得ることができるとされているが、実用的観点からみて粒子の粒度分布が十分狭いとは、言い難い。また、上記文献の技術においては有機顔料、酸化ケイ素等により樹脂粒子表面が被覆されているため、電子写真トナー用樹脂粒子として使用した場合、低温定着性が劣る問題があった。上記の微粒子分散剤及び活性剤の存在下において超臨界流体中に樹脂溶液を分散させる方法によれば、樹脂粒子が凝集してしまうため、粒度分布の狭い粒子を得ることが困難であった。
【0005】
本発明の課題は、液状又は超臨界状態の流体を使用して得られる、粒度分布が十分狭い樹脂粒子、及び、液状又は超臨界状態の流体を使用して粒度分布が十分狭い樹脂粒子を得る製造方法を見出すことである。
【課題を解決するための手段】
【0006】
本発明は、従来技術における上記の事情に鑑みてなされたものである。すなわち、本発明は、下記6発明である。
(I) 微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として結晶性樹脂(a1)を用い、結晶性樹脂(a1)が、下記(a12)〜(a14)からなる群から選ばれる少なくとも1種である樹脂粒子。
(a12) 炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア。
(a13) アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂。
(a14) (メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂。
(II) (I)の樹脂粒子を含有する電子写真トナー用樹脂粒子。
(III) (II)の電子写真トナー用樹脂粒子を含有する電子写真トナー。
(IV) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
(V) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
(VI) 微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む(I)の樹脂粒子の製造方法。
【発明の効果】
【0007】
本発明の樹脂粒子、及び、本発明の製造方法により得られる樹脂粒子は、粒度分布が十分狭く、かつ樹脂粒子の耐湿耐熱保存性が良好である。
【図面の簡単な説明】
【0008】
【図1】樹脂粒子の作成に用いた実験装置である。
【発明を実施するための形態】
【0009】
以下に本発明を詳述する。微粒子(A)は、そのガラス転移温度(以下、Tgと記載する場合がある。)又は融点未満の温度において、液状又は超臨界状態の二酸化炭素(X)〔以下、二酸化炭素(X)と記載する場合がある。〕による膨潤度(以下、膨潤度と記載する。)が16%以下の微粒子であり、好ましくは10%以下、さらに好ましくは5%以下である。膨潤度が16%を超える微粒子を使用した場合は、樹脂粒子の凝集を抑制できなくなり、樹脂粒子の粒度分布が悪化する。
【0010】
膨潤度の測定方法は、磁気浮遊天秤を用いて測定することができる。なお、膨潤度の測定方法の詳細はJ.Supercritical Fluids.19、187−198(2001)に記載されている。
【0011】
微粒子(A)としては、結晶性樹脂(a1)、非結晶性樹脂(a2)、及び無機化合物(a3)からなる群から選ばれる少なくとも1種を用いる。非結晶性樹脂(a2)としては、架橋性の非結晶性樹脂がより好ましい。これらの中では、結晶性樹脂(a1)および非結晶性樹脂(a2)が好ましく、結晶性樹脂(a1)がさらに好ましい。
【0012】
結晶性樹脂(a1)の融点は、50〜110℃が好ましく、さらに好ましくは55〜100℃、とくに好ましくは60〜90℃である。結晶性樹脂(a1)の融点が50℃以上であれば本発明の樹脂粒子(C)が長期間の保管でもブロッキングしにくい。110℃以下であれば電子写真用トナーとして用いた場合には低温定着性が良好である。融点の測定は示差走査熱量測定(以下、DSCと記載する。)における吸熱ピークより求めることができる。
【0013】
結晶性樹脂(a1)の結晶化度は、二酸化炭素(X)による膨潤抑制、及び樹脂粒子(B)への吸着性の観点より、好ましくは20〜95%であり、より好ましくは30〜80%である。結晶化度は、DSCを用いて吸熱ピークの面積から融解熱量(ΔHm(J/g))を求め、測定されたΔHmに基づき以下の式により結晶化度(%)を算出する。
結晶化度=(ΔHm/a)×100
上式中、aは結晶化度が100%となるように外挿した場合の融解熱量である。
【0014】
結晶性樹脂(a1)の数平均分子量は、キャリア汚染性の観点より、好ましくは1000以上であり、更に好ましくは1500以上、特に好ましくは2000以上である。また、溶融粘度の観点より、好ましくは1000000以下であり、更に好ましくは500000以下、特に好ましくは300000以下である。
【0015】
結晶性樹脂(a1)の組成は特に限定されないが、好ましい具体例としては、例えば、脂肪族もしくは芳香族ポリエステル、脂肪族ポリウレタン及び/又はポリウレア、アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂、(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂(a14)、結晶性ポリオレフィン(a15)等が挙げられる。
【0016】
脂肪族もしくは芳香族ポリエステルとしては、後述のジオール(11)、ジカルボン酸(13)を使用することができ、特に炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールと炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジカルボン酸を必須構成単位とし、かつ、該ジオールのアルキレン鎖の炭素数と該ジカルボン酸のアルキレン鎖の炭素数の合計数が10〜52であり、必要により炭素数6〜30の芳香族ジカルボン酸を構成単位とする結晶性ポリエステル(a11)が好ましい。
保存安定性の観点から、上記ジオールのアルキレン鎖の炭素数と上記ジカルボン酸のアルキレン鎖の炭素数の合計数が、10以上が好ましく、更に好ましくは12以上であり、特に好ましくは14以上である。また、定着性の観点から、52以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。
【0017】
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジオールとして好ましいものは、1,4−ブタンジオール、1,6−ヘキサンジオール、及び1,10−デカンジオールである。
炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジカルボン酸のアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジカルボン酸として好ましいものは、アジピン酸、セバシン酸、ドデカンジカルボン酸、及びオクタデカンジカルボン酸である。
また、芳香族ポリエステルの保存安定性の観点から、芳香族ジカルボン酸の炭素数は6〜30が好ましく、更に好ましくは8〜24あり、特に好ましくは8〜20である。炭素数6〜30の芳香族ジカルボン酸として好ましいものは、フタル酸、イソフタル酸、テレフタル酸、及びナフタレンジカルボン酸である。
【0018】
また、芳香族ポリエステルの場合は、樹脂強度の観点から、ジカルボン酸は直鎖脂肪族ジカルボン酸と芳香族ジカルボン酸の併用が好ましく、直鎖脂肪族ジカルボン酸と芳香族ジカルボン酸の合計に対する芳香族ジカルボン酸の比率は、好ましくは90重量以下、更に好ましくは1〜85重量%、特に好ましくは3〜80重量%である。
【0019】
脂肪族ポリウレタン及び/又はポリウレアとしては、後述のジオール(11)、ジアミン〔後述のポリアミン(16)のうち2価のもの〕、及びジイソシアネート〔後述のポリイソシアネート(15)のうち2価のもの〕を使用することができ、特に炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア(a12)が好ましい。
なお、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールと後述のジカルボン酸(13)とを反応させて得られるポリエステルジオールと炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートから得られるポリウレタンも(a12)に含まれる。
脂肪族ポリウレタン及び/又はポリウレアは、保存安定性の観点から、ジオール及び/又はジアミンのアルキレン鎖の炭素数(ジオールとジアミンの混合物を使用する場合は、その重量比で平均されたアルキレン鎖の炭素数)とジイソシアネートのアルキレン鎖の炭素数の合計数が、10以上が好ましく、更に好ましくは12以上であり、特に好ましくは14以上である。また、定着性の観点から、52以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。
【0020】
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオールの、アルキレン鎖の好ましい炭素数、及び好ましい具体例は、結晶性ポリエステル(a11)における場合と同様である。
上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジアミンとして好ましいものは、テトラメチレンジアミン、及びヘキサメチレンジアミンである。
また、上記炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートのアルキレン鎖の炭素数は、結晶性の観点から、2以上が好ましく、更に好ましくは3以上であり、特に好ましくは4以上である。また、定着性の観点から50以下が好ましく、更に好ましくは45以下であり、特に好ましくは40以下、最も好ましくは30以下である。直鎖脂肪族ジイソシアネートとして好ましいものは、テトラメチレンジイソシアネート、及びヘキサメチレンジイソシアネートである。
【0021】
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂としては、アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂(a13)が好ましい。保存安定性の観点から、そのアルキル基の炭素数は、12以上であることが好ましく、更に好ましくは14以上であり、特に好ましくは18以上である。また、定着性の観点から、50以下が好ましく、更に好ましくは40以下であり、特に好ましくは30以下である。保存安定性の観点からアルキル基は直鎖が好ましい。アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートとして好ましいものは、オクタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート、及びベヘニルアクリレートである。
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂は、アルキル(メタ)アクリレートの単独重合体でも、他の単量体との共重合体でもよい。他の単量体としては、後述のビニルモノマーを適宜選択することができる。
結晶性ビニル樹脂(a13)中のアルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートの構成単位の含有量は、好ましくは40重量%以上、更に好ましくは45重量%以上、とくに好ましくは60重量%以上である。
【0022】
(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂(a14)としては、樹脂粒子への付着性の観点から、(メタ)アクリロニトリルの構成単位の含有量が0.01〜40重量%であることが好ましく、更に好ましくは0.05〜35重量%であり、特に好ましくは0.1〜30重量%である。
併用する結晶性ビニルモノマーとしては、結晶性のビニル樹脂が形成され得るものであれば特に限定されないが、上記のアルキル基の炭素数が12〜50であるアルキル(メタ)アクリレート、及びエチレン等が挙げられる。
【0023】
結晶性ポリオレフィン(a15)としては、ポリエチレン、ポリプロピレン等が挙げられる。
【0024】
脂肪族もしくは芳香族ポリエステルの製造方法としては、低分子ポリオールおよび/または数平均分子量1000以下のポリアルキレンエーテルジオールとポリカルボン酸とを反応させる方法、ラクトンの開環重合による方法、低分子ジオールと低級アルコール(メタノールなど)の炭酸ジエステルとを反応させる方法などの公知の製造方法が挙げられる。
【0025】
脂肪族ポリウレタン及び/又はポリウレアの製造方法としては、低分子ポリオール(上記の方法で得られるポリエステルポリオールを含む)及び/又は低分子量ジアミンとジイソシアネートを反応させる方法などの公知の製造方法が挙げられる。
【0026】
アルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂、および(メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂の製造方法としては、溶液重合、塊状重合、懸濁重合などの公知のビニルモノマーの重合法が挙げられる。
【0027】
ポリオレフィンの製造方法としては、付加重合等の公知の重合法が挙げられる。
【0028】
結晶性樹脂(a1)の中で、特に好ましいものは、(a11)、(a12)、(a13)、および(a14)であり、最も好ましくは(a13)である。
【0029】
非結晶性樹脂(a2)としては、例えばビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂、ポリアミド、ポリイミド、シリコーン樹脂、フッ素樹脂、フェノール樹脂、メラミン樹脂、ポリカーボネート、セルロース及びこれらの混合物等が挙げられる。非結晶性樹脂(a2)としては、架橋性の非結晶性樹脂が好ましい。
(a2)の組成は膨潤度が前記の範囲となるものであれば得に限定されず、通常用いられている樹脂でよい。
例えば、架橋性ビニル樹脂としては、2個以上のビニル重合性官能基を有するビニルモノマー(ジビニルベンゼン等)を含むビニルモノマーの共重合体等が挙げられる。
架橋性ポリエステル樹脂としては、ポリオールとポリカルボン酸の重縮合物であって、ポリオール及び/又はポリカルボン酸の少なくとも一部として、後述の3価以上のポリオール(12)及び/又は3価以上のポリカルボン酸(14)を用いて得られるポリエステル樹脂等が挙げられる。
同様に、他の樹脂の場合も架橋性のモノマーを少なくとも一部用いて得られる樹脂がより好ましい。
【0030】
微粒子(A)として、結晶性樹脂(a1)と非結晶性樹脂(a2)を併用してもよい。(a1)と(a2)の混合物の融点は、50〜150℃であることが好ましい。(a2)の含有量は、(a1)と(a2)の合計重量に対して、0〜50重量%であることが好ましい。また非結晶性樹脂(a2)を結晶性樹脂(a1)で被覆した微粒子であってもよい。
【0031】
結晶性樹脂(a1)および/または非結晶性樹脂(a2)を含有する微粒子(A)の製法はいかなる製法であってもよいが、具体例としては、乾式で製造する方法〔微粒子(A)を構成する材料(a)をジェットミル等の公知の乾式粉砕機により乾式粉砕する方法〕、湿式で製造する方法〔(a)の粉末を有機溶剤中に分散し、ビーズミルやロールミル等の公知の湿式分散機により湿式粉砕する方法、(a)の溶剤溶液をスプレードライヤー等により噴霧乾燥する方法、(a)の溶剤溶液を貧溶媒添加や冷却によって過飽和させ析出させる方法、(a)の溶剤溶液を水あるいは有機溶剤中に分散する方法、(a)の前駆体を水中で乳化重合法、ソープフリー乳化重合法、シード重合法、懸濁重合法等により重合させる方法、(a)の前駆体を有機溶剤中で分散重合等により重合させる方法〕が挙げられる。また上記方法により非結晶性樹脂(a2)の微粒子(A’)を合成した後、公知のコーティング法、シード重合法、メカノケミカル法等により、結晶性樹脂(a1)を(A’)表面に形成してもよい。これらのうち、微粒子(A)の製造しやすさの観点から、湿式で製造する方法が好ましく、さらに好ましくは、析出させる方法、乳化重合法、分散重合である。
【0032】
微粒子(A)はそのまま用いてもよく、また樹脂粒子(B)への吸着性を持たせたり、本発明の樹脂粒子(C)の粉体特性や電気特性を改質するために、例えばシラン系、チタネート系、アルミネート系等のカップリング剤による表面処理、各種界面活性剤による表面処理、ポリマーによるコーティング処理等により表面改質されていてもよい。微粒子(A)及び樹脂粒子(B)のいずれか一方が、少なくともその表面に酸性官能基を有し、他の一方が少なくともその表面に塩基性官能基を有することが好ましい。
【0033】
微粒子(A)及び樹脂粒子(B)はその内部に酸性官能基又は塩基性官能基を有していてもよい。酸性官能基としてはカルボン酸基、スルホン酸基等が挙げられる。塩基性官能基としては第1級アミノ基、第2級アミノ基、第3級アミノ基等が挙げられる。
【0034】
微粒子(A)及び樹脂粒子(B)は少なくともその表面に酸性官能基又は塩基性官能基を付与するために、結晶性樹脂(a1)、樹脂(b)として酸性官能基又は塩基性官能基を有する樹脂を使用してもよいし、微粒子(A)及び樹脂粒子(B)にこれら官能基を付与するために表面処理してもよい。
【0035】
酸性官能基を有する結晶性樹脂(a1)としては、酸価を有する脂肪族ポリエステル、酸性官能基を有する単量体(例えば、後述のカルボキシル基含有ビニルモノマー、スルホン基含有ビニルモノマーなど)を共重合したビニル樹脂等が挙げられる。
【0036】
塩基性官能基を有する結晶性樹脂(a1)としては、塩基性官能基を有する単量体(例えば、後述のアミノ基含有ビニルモノマーなど)を共重合したビニル樹脂等が挙げられる。
【0037】
無機化合物(a3)としては、例えば、珪藻土、アルミナ、酸化亜鉛、チタニア、ジルコニア、酸化カルシウム、酸化マグネシウム、酸化鉄、酸化銅、酸化スズ、酸化クロム、酸化アンチモン、酸化イットリウム、酸化セリウム、酸化サマリウム、酸化ランタン、酸化タンタル、酸化テルビウム、酸化ユーロピウム、酸化ネオジウム、フェライト類等の金属酸化物、水酸化カルシウム、水酸化マグネシウム、水酸化アルミニウム、塩基性炭酸マグネシウム等の金属水酸化物、重質炭酸カルシウム、軽質炭酸カルシウム、炭酸亜鉛、炭酸バリウム、ドーソナイト、ハイドロタルイサイト等の金属炭酸塩、硫酸カルシウム、硫酸バリウム、石膏繊維等の金属硫酸塩、シリカ、珪酸カルシウム(ウォラストナイト、ゾノトライト)、カオリン、クレー、タルク、マイカ、モンモリロナイト、ベントナイト、活性白土、セピオライト、イモゴライト、セリサイト、ガラス繊維、ガラスビーズ、ガラスフレーク等の金属珪酸塩、窒化アルミニウム、窒化ホウ素、窒化珪素等の金属窒化物、チタン酸カリウム、チタン酸カルシウム、チタン酸マグネシウム、チタン酸バリウム、チタン酸ジルコン酸鉛アルミニウムボレート等の金属チタン酸塩、ホウ酸亜鉛、ホウ酸アルミニウム等の金属ホウ酸塩、リン酸三カルシウム等の金属燐酸塩、硫化モリブデン等の金属硫化物、炭化珪素等の金属炭化物、カーボンブラック、グラファイト、炭素繊維等の炭素類、金、銀その他の無機粒子が挙げられる。これらの中で好ましくは、シリカおよび金属炭酸塩である。
【0038】
樹脂粒子(B)は樹脂(b)より構成される。本発明において、樹脂(b)としては、熱可塑性樹脂(b1)、又は該熱可塑性樹脂を微架橋した樹脂(b2)、又は熱可塑性樹脂を海成分、硬化樹脂を島成分とするポリマーブレンド(b3)が挙げられ、2種以上を併用してもよい。熱可塑性樹脂(b1)としては、例えばビニル樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂等が挙げられる。このうち好ましいのは、微細球状樹脂粒子の分散体が得られやすいという観点からビニル樹脂、ポリウレタン樹脂、ポリエステル樹脂およびそれらの併用である。
【0039】
ビニル樹脂は、ビニルモノマーを単独重合または共重合したポリマーである。ビニルモノマーとしては、下記(1)〜(10)が挙げられる。
(1)ビニル炭化水素:
(1−1)脂肪族ビニル炭化水素:アルケン類、例えばエチレン、プロピレン、前記以外のα−オレフィン等;アルカジエン類、例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン、1,7−オクタジエン。
(1−2)脂環式ビニル炭化水素:モノ−もしくはジ−シクロアルケンおよびアルカジエン類、例えば(ジ)シクロペンタジエン等;テルペン類、例えばピネン等。
(1−3)芳香族ビニル炭化水素:スチレンおよびそのハイドロカルビル(アルキル、シクロアルキル、アラルキルおよび/またはアルケニル)置換体、例えばα−メチルスチレン、2,4−ジメチルスチレン等;およびビニルナフタレン。
(2)カルボキシル基含有ビニルモノマー及びその塩:炭素数3〜30の不飽和モノカルボン酸、不飽和ジカルボン酸ならびにその無水物およびそのモノアルキル(炭素数1〜24)エステル、例えば(メタ)アクリル酸、(無水)マレイン酸、マレイン酸モノアルキルエステル、フマル酸、フマル酸モノアルキルエステル、クロトン酸、イタコン酸、イタコン酸モノアルキルエステル、イタコン酸グリコールモノエーテル、シトラコン酸、シトラコン酸モノアルキルエステル、桂皮酸等のカルボキシル基含有ビニルモノマー。
(3)スルホン基含有ビニルモノマー、ビニル硫酸モノエステル化物及びこれらの塩:炭素数2〜14のアルケンスルホン酸、例えばビニルスルホン酸;およびその炭素数2〜24のアルキル誘導体、例えばα−メチルスチレンスルホン酸等;スルホ(ヒドロキシ)アルキル−(メタ)アクリレートもしくは(メタ)アクリルアミド、例えば、スルホプロピル(メタ)アクリレート、および硫酸エステルもしくはスルホン酸基含有ビニルモノマー;ならびそれらの塩等。
(4)燐酸基含有ビニルモノマー及びその塩:(メタ)アクリロイルオキシアルキル(C1〜C24)燐酸モノエステル、例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート、フェニル−2−アクリロイロキシエチルホスフェート、(メタ)アクリロイルオキシアルキル(炭素数1〜24)ホスホン酸類、例えば2−アクリロイルオキシエチルホスホン酸。なお、上記(2)〜(4)の塩としては、例えばアルカリ金属塩(ナトリウム塩、カリウム塩等)、アルカリ土類金属塩(カルシウム塩、マグネシウム塩等)、アンモニウム塩、アミン塩もしくは4級アンモニウム塩が挙げられる。
(5)ヒドロキシル基含有ビニルモノマー:ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等。
(6)含窒素ビニルモノマー:
(6−1)アミノ基含有ビニルモノマー:アミノエチル(メタ)アクリレート等、
(6−2)アミド基含有ビニルモノマー:(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド等、
(6−3)ニトリル基含有ビニルモノマー:(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等、
(6−4)4級アンモニウムカチオン基含有ビニルモノマー:ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリルアミド、ジエチルアミノエチル(メタ)アクリルアミド、ジアリルアミン等の3級アミン基含有ビニルモノマーの4級化物(メチルクロライド、ジメチル硫酸、ベンジルクロライド、ジメチルカーボネート等の4級化剤を用いて4級化したもの)等、
(6−5)ニトロ基含有ビニルモノマー:ニトロスチレン等。
(7)エポキシ基含有ビニルモノマー:グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等。
(8)ハロゲン元素含有ビニルモノマー:塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等。
(9)ビニルエステル、ビニル(チオ)エーテル、ビニルケトン、ビニルスルホン類:
(9−1)ビニルエステル、例えば酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、オクタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート、ベヘニル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ポリ(メタ)アリロキシアルカン類[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]等、ポリアルキレングリコール鎖を有するビニルモノマー[ポリエチレングリコール(分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(分子量500)モノアクリレート、メチルアルコールエチレンオキサイド10モル付加物(メタ)アクリレート、ラウリルアルコールエチレンオキサイド30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート類[多価アルコール類のポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等、
(9−2)ビニル(チオ)エーテル、例えばビニルメチルエーテル等、
(9−3)ビニルケトン、例えばビニルメチルケトン等。
(10)その他のビニルモノマー:イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等。
【0040】
ビニルモノマーの共重合体としては、上記(1)〜(10)の任意のモノマー同士を任意の割合で共重合したポリマーが挙げられるが、例えばスチレン−(メタ)アクリル酸エステル共重合体、スチレン−ブタジエン共重合体、(メタ)アクリル酸−アクリル酸エステル共重合体、スチレン−アクリロニトリル共重合体、スチレン−無水マレイン酸共重合体、スチレン−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸、ジビニルベンゼン共重合体、スチレン−スチレンスルホン酸−(メタ)アクリル酸エステル共重合体等が挙げられる。
【0041】
ポリエステル樹脂としては、ポリオールと、ポリカルボン酸(その酸無水物、その低級アルキルエステルを含む)との重縮合物などが挙げられる。ポリオールとしてはジオール(11)および3価以上のポリオール(12)が挙げられ、ポリカルボン酸としては、ジカルボン酸(13)および3価以上のポリカルボン酸(14)が挙げられる。
ポリオールとポリカルボン酸の反応比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/1、さらに好ましくは1.5/1〜1/1、とくに好ましくは1.3/1〜1.02/1である。
【0042】
ジオール(11)としては、アルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、オクタンジオール、デカンジオール、ドデカンジオール、テトラデカンジオール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);アルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレンエーテルグリコールなど);脂環式ジオール(1,4−シクロヘキサンジメタノール、水素添加ビスフェノールAなど);ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど);上記脂環式ジオールのアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;上記ビスフェノール類のアルキレンオキサイド(エチレンオキサイド、プロピレンオキサイド、ブチレンオキサイドなど)付加物;その他、ポリラクトンジオール(ポリε−カプロラクトンジオールなど)、ポリブタジエンジオールなどが挙げられる。これらのうち好ましいものは、炭素数2〜12のアルキレングリコールおよびビスフェノール類のアルキレンオキサイド付加物であり、特に好ましいものはビスフェノール類のアルキレンオキサイド付加物、およびこれと炭素数2〜12のアルキレングリコールとの併用である。
【0043】
3価以上のポリオール(12)としては、3〜8価またはそれ以上の多価脂肪族アルコール(グリセリン、トリメチロールエタン、トリメチロールプロパン、ペンタエリスリトール、ソルビトールなど);トリスフェノール類(トリスフェノールPAなど);ノボラック樹脂(フェノールノボラック、クレゾールノボラックなど);上記トリスフェノール類のアルキレンオキサイド付加物;上記ノボラック樹脂のアルキレンオキサイド付加物、アクリルポリオール[ヒドロキシエチル(メタ)アクリレートと他のビニルモノマーの共重合物など]などが挙げられる。
【0044】
ジカルボン酸(13)としては、アルキレンジカルボン酸(コハク酸、アジピン酸、セバシン酸、ドデセニルコハク酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸など);アルケニレンジカルボン酸(マレイン酸、フマール酸など);炭素数8以上の分岐アルキレンジカルボン酸[ダイマー酸、アルケニルコハク酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸など)、アルキルコハク酸(デシルコハク酸、ドデシルコハク酸、オクタデシルコハク酸など);芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、ナフタレンジカルボン酸など)などが挙げられる。これらのうち好ましいものは、炭素数4〜20のアルケニレンジカルボン酸および炭素数8〜20の芳香族ジカルボン酸である。
【0045】
3価以上(3〜6価又はそれ以上)のポリカルボン酸(14)としては、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など)などが挙げられる。
【0046】
なお、ジカルボン酸(13)または3価以上のポリカルボン酸(14)としては、上述のものの酸無水物または低級アルキルエステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0047】
ポリウレタン樹脂としては、ポリイソシアネート(15)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(11)および3価以上のポリオール(12)]、ジカルボン酸(13)、3価以上のポリカルボン酸(14)、ポリアミン(16)、ポリチオール(17)等}との重付加物などが挙げられる。
【0048】
ポリイソシアネート(15)としては、炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ポリイソシアネート、炭素数2〜18の脂肪族ポリイソシアネート、炭素数4〜15の脂環式ポリイソシアネート、炭素数8〜15の芳香脂肪族ポリイソシアネートおよびこれらのポリイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。
上記芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)などが挙げられる。
上記脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)などが挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)などが挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
また、上記ポリイソシアネートの変性物には、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのポリイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が含まれる。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、および炭素数4〜15の脂環式ポリイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0049】
ポリアミン(16)の例としては、下記のものが挙げられる。
・脂肪族ポリアミン類(C2〜C18):
〔1〕脂肪族ポリアミン{C2〜C6アルキレンジアミン(エチレンジアミン、テトラメチレンジアミン、及びヘキサメチレンジアミンなど)、ポリアルキレン(C2〜C6)ポリアミン〔ジエチレントリアミンなど〕}
〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミンなど〕
〔3〕脂環または複素環含有脂肪族ポリアミン〔3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕
〔4〕芳香環含有脂肪族アミン類(C8〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど)、
・脂環式ポリアミン(C4〜C15):1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など、
・芳香族ポリアミン類(C6〜C20):
〔1〕非置換芳香族ポリアミン〔1,2−、1,3−および1,4−フェニレンジアミンなど;核置換アルキル基〔メチル、エチル、n−およびi−プロピル、ブチルなどのC1〜C4アルキル基)を有する芳香族ポリアミン、たとえば2,4−および2,6−トリレンジアミンなど〕、およびこれらの異性体の種々の割合の混合物
〔2〕核置換電子吸引基(Cl、Br、I、Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ポリアミン〔メチレンビス−o−クロロアニリンなど〕
〔3〕2級アミノ基を有する芳香族ポリアミン〔上記(4)〜(6)の芳香族ポリアミンの−NH2の一部または全部が−NH−R´(R´はメチル、エチルなどの低級アルキル基で置換したもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕、
・複素環式ポリアミン(C4〜C15):ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジンなど、
・ポリアミドポリアミン:ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど、
・ポリエーテルポリアミン:ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物など。
【0050】
ポリチオール(17)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなどが挙げられる。
【0051】
エポキシ樹脂としては、ポリエポキシド(18)の開環重合物、ポリエポキシド(18)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(11)および3価以上のポリオール(12)]、ジカルボン酸(13)、3価以上のポリカルボン酸(14)、ポリアミン(16)、ポリチオール(17)等}との重付加物、またはポリエポキシド(18)とジカルボン酸(13)または3価以上のポリカルボン酸(14)の酸無水物との硬化物などが挙げられる。
【0052】
ポリエポキシド(18)としては、分子中に2個以上のエポキシ基を有していれば、特に限定されない。ポリエポキシド(18)として好ましいものは、硬化物の機械的性質の観点から分子中にエポキシ基を2〜6個有するものである。ポリエポキシド(18)のエポキシ当量(エポキシ基1個当たりの分子量)は、好ましくは65〜1000であり、さらに好ましくは90〜500である。エポキシ当量が1000以下であると、架橋構造が密になり硬化物の耐水性、耐薬品性、機械的強度等の物性が向上し、一方、エポキシ当量が65以上のものは、合成するのが容易である。
【0053】
ポリエポキシド(18)の例としては、芳香族系ポリエポキシ化合物、複素環系ポリエポキシ化合物、脂環族系ポリエポキシ化合物あるいは脂肪族系ポリエポキシ化合物が挙げられる。芳香族系ポリエポキシ化合物としては、多価フェノール類のグリシジルエーテル体およびグリシジルエステル体、グリシジル芳香族ポリアミン、並びに、アミノフェノールのグリシジル化物等が挙げられる。多価フェノールのグリシジルエーテル体としては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル等が挙げられる。多価フェノールのグリシジルエステル体としては、フタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、テレフタル酸ジグリシジルエステル等が挙げられる。グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。さらに、本発明において前記芳香族系ポリエポキシ化合物として、P−アミノフェノールのトリグリシジルエーテル、トリレンジイソシアネートまたはジフェニルメタンジイソシアネートとグリシドールとの付加反応によって得られるジグリシジルウレタン化合物、前記2反応物にポリオールも反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマー、およびビスフェノールAのアルキレンオキシド(エチレンオキシドまたはプロピレンオキシド)付加物のジグリシジルエーテル体も含む。複素環系ポリエポキシ化合物としては、トリスグリシジルメラミンが挙げられる。脂環族系ポリエポキシ化合物としては、ビニルシクロヘキセンジオキシド等が挙げられる。また、脂環族系ポリエポキシ化合物としては、前記芳香族系ポリエポキシド化合物の核水添化物も含む。脂肪族系ポリエポキシ化合物としては、多価脂肪族アルコールのポリグリシジルエーテル体、多価脂肪酸のポリグリシジルエステル体、およびグリシジル脂肪族アミンが挙げられる。多価脂肪族アルコールのポリグリシジルエーテル体としては、エチレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル等が挙げられる。多価脂肪酸のポリグリシジルエステル体としては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート、ジグリシジルピメレート等が挙げられる。グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミンが挙げられる。また、本発明において脂肪族系ポリエポキシ化合物としては、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含む。これらのうち、好ましいのは、脂肪族系ポリエポキシ化合物および芳香族系ポリエポキシ化合物である。ポリエポキシドは、2種以上併用しても差し支えない。
【0054】
熱可塑性樹脂を微架橋した樹脂(b2)とは、架橋構造を導入させ樹脂(b)のTgが20〜200℃である樹脂を言うものとする。かかる架橋構造は、共有結合性、配位結合性、イオン結合性、水素結合性等、いずれの架橋形態であってもよい。具体例としては、例えば樹脂(b2)としてポリエステルを選択する場合、重合時にポリオールとポリカルボン酸のいずれか、あるいは両方に3官能以上の官能基数を有するものを使用することにより架橋構造を導入することができる。また樹脂(b2)としてビニル樹脂を選択する場合、重合時に二重結合を2つ以上有するモノマーを添加することにより、架橋構造を導入することができる。
【0055】
熱可塑性樹脂を海成分、硬化樹脂を島成分とするポリマーブレンド(b3)としては、Tgが20〜200℃、且つ軟化開始温度が40〜220℃であるもの、具体的にはビニル樹脂、ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂及びこれらの混合物が挙げられる。
【0056】
樹脂(b)の数平均分子量(GPCにて測定、以下Mnと略記する場合がある。)は、好ましくは1000〜500万、より好ましくは2,000〜500,000、溶解性パラメーター(SP値、詳細は後述する。)は、好ましくは7〜18、より好ましくは8〜14である。また、本発明の樹脂粒子(C)の熱特性を改質したい場合には、樹脂(b2)又は樹脂(b3)を使用するとよい。
【0057】
樹脂(b)のガラス転移温度(Tg)は好ましくは20℃〜200℃、より好ましくは40℃〜150℃である。20℃以上では粒子の保存安定性が良好である。なお、本発明におけるTgは、DSC測定から求められる値である。
【0058】
樹脂(b)の軟化開始温度は、好ましくは40℃〜220℃、より好ましくは50℃〜200℃である。40℃以上では長期の保存性が良好である。220℃以下では定着温度が上昇せず問題がない。なお、本発明における軟化開始温度は、フローテスター測定から求められる値である。
【0059】
樹脂粒子(B)の体積平均粒径は、好ましくは1〜10μmであり、より好ましくは2〜8μmである。
【0060】
本発明の樹脂粒子(C)は、微粒子(A)が樹脂粒子(B)の表面に固着されてなるか、又は樹脂粒子(B)の表面に、微粒子(A)が皮膜化された皮膜が形成されてなる粒子である。微粒子(A)が樹脂粒子(B)の表面に固着されてなるとは、(A)が単に(B)の表面に付着し容易に脱離するような場合は含まないものとする。
【0061】
微粒子(A)の粒径は、樹脂粒子(B)の粒径よりも小さい。粒径比[微粒子(A)の体積平均粒径/[本発明の樹脂粒子(C)の体積平均粒径]の値は、好ましくは0.001〜0.3、より好ましくは0.002〜0.2、さらに好ましくは0.003〜0.1、特に好ましくは0.01〜0.08である。上記範囲内であると(A)が(B)の表面に効率よく吸着するため、得られる本発明の樹脂粒子(C)の粒度分布が狭くなる。
【0062】
微粒子(A)の体積平均粒径は、好ましくは0.01〜0.5μm、特に好ましくは0.015〜0.4μmである。なお、体積平均粒径は、動的光散乱式粒度分布測定装置(例えば LB−550:堀場製作所製)、レーザー式粒度分布測定装置(例えば LA−920:堀場製作所製)、マルチサイザーIII(ベックマン・コールター社製)等で測定できる。
【0063】
本発明の樹脂粒子(C)の体積平均粒径は、好ましくは1〜10μmであり、より好ましくは2〜8μm、さらに好ましくは3〜6μmである。1μm以上であると、粉体としてのハンドリング性が向上する。10μm以下であると、電子写真用トナーとした時の画像の解像度が向上する。
【0064】
本発明の樹脂粒子(C)の体積平均粒径DVと、本発明の樹脂粒子(C)の個数平均粒径DNの比:DV/DNは、好ましくは1.0〜1.5、より好ましくは1.0〜1.4、特に好ましくは1.0〜1.3である。1.5以下であると粉体特性(流動性、帯電均一性等)、画像の解像度が著しく向上する。
【0065】
本発明の樹脂粒子(C)は、の粒径均一性、粉体流動性、保存安定性等の観点からは、樹脂粒子(B)の表面の5%以上が、微粒子(A)もしくは(A)由来の皮膜で覆われているのが好ましく、更に好ましくは30%以上である。なお、表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[(A)もしくは(A)由来の皮膜に覆われている部分の(B)の表面積/{(A)もしくは(A)由来の皮膜に覆われている部分の(B)の表面積+(B)の表面が露出している部分の面積}]×100
【0066】
本発明の樹脂粒子(C)は、微粒子(A)を構成する材料(a)と樹脂粒子(B)を構成する樹脂(b)の重量比率が、好ましくは(0.1:99.9)〜(30:70)であり、さらに好ましくは(0.2:99.8)〜(20:80)である。材料(a)と樹脂(b)の重量比率がこの範囲内であると、低温定着性と長期の保存安定性が両立し好ましい。
本発明の樹脂粒子(C)中の(a)が結晶性樹脂(a1)である場合、公知の方法、例えばDSCにより(a1)に固有な吸熱ピークの吸熱量から結晶性樹脂(a1)の重量比率を算出する方法により測定することがでる。
【0067】
本発明の樹脂粒子(C)は、液状又は超臨界状態の二酸化炭素(X)中で製造する以下の製造方法で得ることが好ましい。
製造方法(1)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで二酸化炭素(X)を除去することにより樹脂粒子を得る製造方法。
製造方法(2)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子を得る製造方法。
製造方法(3)
微粒子(A)が分散された、二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子を得る製造方法。
【0068】
製造方法(2)について詳細に説明する。
23℃、0.1MPaの標準状態における、溶剤(S)と樹脂(b)との等重量混合物における、溶剤(S)に対する樹脂(b)の不溶分は、樹脂(b)の重量に対して、好ましくは20重量%以下、さらに好ましくは15重量%以下である。不溶分重量(重量%)が20重量%以下であれば得られる樹脂粒子の粒度分布が狭くなる。
製造方法(3)において樹脂(b)の代わりに前躯体(b0)を用いる場合、および樹脂(b)と前駆体(b0)の混合物を用いる場合も同様である。
【0069】
また、溶剤(S)の溶解性パラメーター(SP値)は9〜16が好ましく、さらに好ましくは10〜15である。SP値とは、下記に示した様に、凝集エネルギー密度と分子容の比の平方根で表されるものである。
SP=(△E/V)1/2
ここで△Eは凝集エネルギー密度を表す。Vは分子容を表し、その値は、ロバート エフ.フェドールス(Robert F.Fedors)らの計算によるもので、例えばポリマー エンジニアリング アンド サイエンス(Polymer engineering and science)第14巻、147〜154頁に記載されている。
【0070】
溶剤(S)の具体例としては、例えば、ケトン溶剤(アセトン、メチルエチルケトン等)、エーテル溶剤(テトラヒドロフラン、ジエチルエーテル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、環状エーテル等)、エステル溶剤(酢酸エステル、ピルビン酸エステル、2−ヒドロキシイソ酪酸エステル、乳酸エステル等)、アミド溶剤(ジメチルホルムアミド等)、アルコール類(メタノール、エタノール、フッ素含有アルコール等)、芳香族炭化水素溶剤(トルエン、キシレン等)、および脂肪族炭化水素溶剤(オクタン、デカン等)などが挙げられる。これらの溶剤の2種以上の混合溶剤、または、これらの有機溶剤と水との混合溶剤を用いることもできる。
【0071】
粒子形成のし易さの観点から、単一溶剤としては、環状エーテル、ピルビン酸エステル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、2−ヒドロキシイソ酪酸エステル、乳酸エステル、フッ素含有アルコールが好ましい。
上記環状エーテルとしては、1,4−ジオキサン、1,3−ジオキソラン等が挙げられる。
ピルビン酸エステルとしては、ピルビン酸メチル、ピルビン酸エチル等が挙げられる。
エチレングリコールモノアルキルエーテルとしては、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル等が挙げられる。
プロピレングリコールモノアルキルエーテルとしては、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル等が挙げられる。
2−ヒドロキシイソ酪酸エステルとしては、2−ヒドロキシイソ酪酸メチル等が挙げられる。
乳酸エステルとしては、乳酸メチル、乳酸エチル等が挙げられる。
フッ素含有アルコールとしては、2,2,3,3−テトラフルオロプロパノール、トリフルオロエタノール等が挙げられる。
また、混合溶剤としては、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、アセトンと水の混合溶剤、メチルエチルケトンと水の混合溶剤が好ましい。
製造方法(3)における溶剤(S)も同様である。
【0072】
樹脂(b)の溶液(L)は、樹脂(b)を溶剤(S)に溶解させて製造する。溶液(L)の重量に対して樹脂(b)の濃度は好ましくは10〜90重量%、さらに好ましくは20〜80重量%である。
製造方法(3)における溶液(L0)中の前駆体(b0)の濃度も同様である。
【0073】
製造方法(2)の樹脂(b)の溶液(L)を二酸化炭素(X)中に分散させる分散工程では、下記の分散安定剤(E)を使用することが出来る。分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有する化合物である。さらには、二酸化炭素に親和性を有するジメチルシロキサン基、含フッ素基と共に、樹脂(b)に親和性を有する化学構造を有することが好ましい。
【0074】
例えば樹脂(b)がビニル樹脂である場合、分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有するモノマーを構成単位とするビニル樹脂であることが好ましい。
ジメチルシロキサン基を有するモノマー(あるいは反応性オリゴマー)(M1−1)としては、メタクリル変性シリコーンが好ましく、次式に示す構造を持つ。
(CH3)3SiO((CH3)2SiO)aSi(CH3)2R
但しaは、平均値で15〜45であり、Rはメタクリル基を含む有機変性基である。Rの例としては、−C3H6OCOC(CH3)=CH2が挙げられる。
【0075】
また、フッ素を含有するモノマー(M1−2)の具体例としては、テトラフルオロエチレン(TFE)、ヘキサフルオロプロピレン(HFP)、クロロトリフルオロエチレン(CTFE)等のパーフルオロオレフィン;パーフルオロ(アルキルビニルエーテル)(PFAVE)、パーフルオロ(1,3−ジオキソール)、パーフルオロ(2,2−ジメチル−1,3−ジオキソール)(PFDD)、パーフルオロ−(2−メチレン−4−メチル−1,3−ジオキソラン)(MMD)、パーフルオロブテニルビニルエーテル(PFBVE)等のパーフルオロビニルエーテル;ビニリデンフルオライド(VdF)、トリフルオロエチレン、1,2−ジフルオロエチレン、フッ化ビニル、トリフルオロプロピレン、3,3,3−トリフルオロ−2−トリフルオロメチルプロペン、3,3,3−トリフルオロプロペン、パーフルオロ(ブチル)エチレン(PFBE)等の水素原子含有フルオロオレフィン;1,1−ジヒドロパーフルオロオクチルアクリレート(DPFOA)、1,1−ジヒドロパーフルオロオクチルメタクリレート(DPFOMA)、2−(パーフルオロオクチル)エチルアクリレート(PFOEA)、2−(パーフルオロオクチル)エチルメタクリレート(PFOEMA)、2−(パーフルオロヘキシル)エチルメタクリレート(PFHEMA)、2−(パーフルオロブチル)エチルメタクリレート(PFBEMA)等のポリフルオロアルキル(メタ)アクリレート;α−フルオロスチレン、β−フルオロスチレン、α,β−ジフルオロスチレン、β,β−ジフルオロスチレン、α,β,β−トリフルオロスチレン、α−トリフルオロメチルスチレン、2,4,6−トリ(トリフルオロメチル)スチレン、2,3,4,5,6−ペンタフルオロスチレン、2,3,4,5,6−ペンタフルオロ−α−メチルスチレン、2,3,4,5,6−ペンタフルオロ−β−メチルスチレン等のフルオロスチレン等が挙げられる。
【0076】
また樹脂(b)がウレタン樹脂である場合、分散安定剤(E)は、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有するモノマーを構成単位とするウレタン樹脂であることが好ましい。
(M1−1)としてはアミノ変性シリコーン、カルボキシル変性シリコーン、カルビノール変性シリコーン、メルカプト変性シリコーン等の活性水素を含む官能基を有するポリシロキサンが好ましい。(M1−2)としては、2,2ビス(4−ヒドロキシフェニル)ヘキサフルオロプロパン、3,3,4,4−テトラフルオロ−1,6−ヘキサンジオール等の含フッ素基ポリオール、含フッ素基(ポリ)アミン、含フッ素基(ポリ)チオール等の活性水素を含む官能基を有するフッ素化合物、ビス(イソシアナトメチル)パーフルオロプロパン、ビス(イソシアナトメチル)パーフルオロブタン、ビス(イソシアナトメチル)パーフルオロペンタン及びビス(イソシアナトメチル)パーフルオロヘキサン等の含フッ素基(ポリ)イソシアネートが好ましい。
【0077】
また樹脂(b)が酸価を有する場合、分散性の観点より分散安定剤(E)はアミノ基を有することが好ましい。樹脂(b)の酸価は1〜50が好ましく、さらに好ましくは3〜40、最も好ましくは5〜30である。アミノ基は1級、2級、3級のいずれでもよく、また含フッ素基、ジメチルシロキサン基を含む化合物の側鎖、片末端、両末端、側鎖両末端いずれの位置に導入されたものを使用してもよい。
【0078】
分散安定剤(E)としては、例えばジメチルシロキサン基を有するモノマー(あるいは反応性オリゴマー)(M1−1)、及び/又はフッ素を含有するモノマー(M1−2)と、前述の樹脂(b)を構成するモノマーとの共重合体(例えば、メタクリル変性シリコーンとメタクリル酸メチルとの共重合体、メタクリル酸ヘプタフルオロブチルとメタクリル酸メチルとの共重合体等)が好ましい。共重合の形態はランダム、ブロック、グラフトのいずれでもよいが、ブロックあるいはグラフトが好ましい。
【0079】
また樹脂(b)が酸価を有する場合、分散安定性の観点より微粒子(A)は粒子表面にアミノ基を有することが好ましい。アミノ基は1級、2級、3級のいずれでもよく、またアミノ基を含有させる形態は特に限定されず、例えばアミノ基を有する化合物を微粒子(A)中に分散、含浸等の方法により含有させる方法、微粒子(A)を構成する成分にアミノ基を有する化合物を使用する方法、微粒子(A)表面にアミノ基含有カップリング剤等を反応させる方法、微粒子(A)表面にアミノ基含有化合物を吸着させる方法等が挙げられる。
【0080】
分散安定剤(E)の添加量は、分散安定性の観点から、樹脂(b)の重量に対し0.01〜50重量%が好ましく、さらに好ましくは0.02〜40重量%、特に好ましくは0.03〜30重量%である。分散安定剤(E)の好ましい重量平均分子量の範囲は100〜10万であり、さらに好ましくは200〜5万、特に好ましくは500〜3万である。この範囲内にすると、(E)の分散安定効果が向上する。
なお、製造方法(1)及び(3)においても、分散工程で、分散安定剤(E)を使用することができる。
【0081】
本発明において、微粒子(A)を二酸化炭素(X)中に分散する方法はいかなる方法でもよく、例えば、容器内に(A)及び(X)を仕込み、攪拌や超音波照射等により、(A)を直接(X)中に分散する方法や、微粒子(A)が溶剤(T)中に分散された分散液を(X)中に導入する方法等が挙げられる。
【0082】
二酸化炭素(X)の重量に対する微粒子(A)の重量比率(重量%)としては、50以下が好ましく、更に好ましくは30以下であり、特に好ましくは0.1〜20である。この範囲であれば、効率よく樹脂粒子(C1)を製造できる。
【0083】
溶剤(T)としては、溶剤(S)と同様のものが挙げられる。微粒子(A)の分散性から、好ましくは、脂肪族炭化水素溶剤(デカン、ヘキサン、ヘプタンなど)、及びエステル溶剤(酢酸エチル、酢酸ブチルなど)である。
【0084】
微粒子(A)と溶剤(T)の重量比率(重量%)は、特に制限はないが、溶剤(T)に対して、微粒子(A)が50以下が好ましく、更に好ましくは30以下であり、特に好ましくは20以下である。この範囲であれば、効率よく微粒子(A)を(X)中に導入することができる。
【0085】
微粒子(A)を溶剤(T)中に分散する方法としては特に制限はないが、微粒子(A)を溶剤(T)に仕込み、攪拌や超音波照射等により直接分散する方法や微粒子を高温下で溶剤(T)に溶解させて晶析する方法などが挙げられる。
【0086】
このようにして二酸化炭素(X)中に(A)が分散している分散体(X0)が得られる。微粒子(A)としては、膨潤度が前記の範囲であって、(X)に溶解せず、(X)中に安定分散するものが好ましい。
【0087】
樹脂(b)の溶液(L)は、(X)中に分散するため、適度な粘度であることが好ましく、粒度分布の観点から、好ましくは100Pa・s以下、さらに好ましくは10Pa・s以下である。樹脂(b)の(X)への溶解度は、好ましくは3%以下、さらに好ましくは1%以下である。
【0088】
樹脂(b)のSP値は、好ましくは8〜16、さらに好ましくは9〜14である。
【0089】
本発明において樹脂(b)を含有する樹脂粒子(B)中に他の添加物(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を含有しても差し支えない。樹脂粒子(B)中に他の添加物を含有させる方法としては、あらかじめ樹脂(b)と添加物を混合した後、(X)中にその混合物を加えて分散させるのが好ましい。
【0090】
本発明において、樹脂(b)の溶液(L)を、(X)中に微粒子(A)が分散している分散体(X0)中に分散する方法はいかなる方法を用いてもよい。具体例としては、樹脂(b)の溶液(L)を攪拌機や分散機等で分散する方法、樹脂(b)の溶液(L)を二酸化炭素(X)中に(A)が分散している分散体(X0)中にスプレーノズルを介して噴霧して液滴を形成し、液滴中の樹脂を過飽和状態とし、樹脂粒子を析出させる方法(ASES:Aerosol Solvent Extraction Systemとして知られている)、同軸の多重管(2重管、3重管等)から溶液(L)、溶液(L0)、樹脂(b)の前駆体(b0)、分散体(X0)を高圧ガス、エントレーナ等とともにそれぞれ別の管から同時に噴出させて、液滴に外部応力を加え分裂を促進させて、粒子を得る方法(SEDS:Solution Enhanced Dispersion by Supercritical Fluidsとして知られている)、超音波を照射する方法等が挙げられる。製造方法(3)及び(1)における、樹脂(b)の前駆体(b0)の溶液(L0)及び樹脂(b)の前駆体(b0)の場合も同様である。
【0091】
このようにして二酸化炭素(X)中に(A)が分散している分散体(X0)中に樹脂(b)の溶液(L)を分散し、微粒子(A)を表面に吸着させながら、分散された樹脂(b)を粒子成長させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成する。(C1)が(X)中に分散したものを分散体(X1)とする。
分散体(X1)は単一相であることが好ましい。すなわち、樹脂(b)の溶液(L)を使用する場合、(C1)が分散している二酸化炭素(X)を含む相の他に、溶剤(S)相が分離する状態は好ましくない。したがって、溶剤相が分離しないように、分散体(X0)に対する(b)の溶液(L)の量を設定することが好ましい。例えば(X0)に対して90重量%以下が好ましく、さらに好ましくは5〜80重量%、特に好ましくは10〜70重量%である。
なお、樹脂(b)の溶液(L)、または製造方法(3)の前駆体(b0)の溶液(L0)を用いた場合に、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)中に含有する(S)の量は、好ましくは10〜90重量%、さらに好ましくは20〜70重量%である。
また、樹脂(b)と二酸化炭素(X)の重量比は、好ましくは(b):(X)が、1:(0.1〜100)、さらに好ましくは1:(0.5〜50)、特に好ましくは1:(1〜20)である。製造方法(1)及び(3)における前駆体体(b0)と二酸化炭素(X)の重量比も同様である。
【0092】
本発明において、液状の二酸化炭素とは、二酸化炭素の温度軸と圧力軸とで表す相図上において、二酸化炭素の三重点(温度=−57℃、圧力=0.5MPa)と二酸化炭素の臨界点(温度=31℃、圧力=7.4MPa)を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件である二酸化炭素を表し、超臨界状態の二酸化炭素とは、臨界温度以上の温度・圧力条件である二酸化炭素を表す(ただし、圧力は、2成分以上の混合ガスの場合、全圧を表す)。
【0093】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作は、以下に述べる温度で行うことが好ましい。すなわち、減圧時に配管内で二酸化炭素が固体に相転移し、流路を閉塞させないようにするために、30℃以上が好ましく、また、微粒子(A)、樹脂粒子(B1)、樹脂粒子(C1)の熱劣化を防止するために、200℃以下が好ましい。さらに30〜150℃が好ましく、より好ましくは34〜130℃、特に好ましくは35〜100℃、最も好ましくは40℃〜80℃である。分散体(X0)、分散体(X1)の温度も同様である。また、製造方法(1)、(3)の場合も同様である。本発明の製造方法(1)〜(3)において、二酸化炭素(X)中で行う操作は、微粒子(A)のTg又は融点以上の温度でも未満の温度でも行うことができるが、Tg又は融点未満の温度において行うことが好ましい。
【0094】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作は以下に述べる圧力で行うことが好ましい。すなわち、樹脂粒子(C1)を(X)中に良好に分散させるために、好ましくは7MPa以上であり、設備コスト、運転コストの観点から、好ましくは40MPa以下である。さらに好ましくは7.5〜35MPa、より好ましくは8〜30MPa、特に好ましくは8.5〜25MPa、最も好ましくは9〜20MPaである。分散体(X0)及び分散体(X1)を形成する容器内の圧力も同様である。製造方法(1)、(3)の場合も同様である。
【0095】
本発明の製造方法(2)において、二酸化炭素(X)中で行う操作の温度及び圧力は、樹脂(b)が(X)中に溶解せず、且つ(b)が凝集・合一可能な範囲内で設定することが好ましい。通常、低温・低圧ほど目的分散物が(X)中に溶解しない傾向となり、高温・高圧ほど(b)が凝集・合一し易い傾向となる。分散体(X0)、分散体(X1)についても同様である。製造方法(1)、(3)の場合も同様である。
【0096】
本発明における二酸化炭素(X)中には、分散媒としての物性値(粘度、拡散係数、誘電率、溶解度、界面張力等)を調整するために、他の物質(e)を適宜含んでよく、例えば、窒素、ヘリウム、アルゴン、空気等の不活性気体等が挙げられる。
【0097】
本発明における(X)と他の物質(e)の合計中の二酸化炭素(X)の重量分率は、好ましくは70%以上、さらに好ましくは80%以上、とくに好ましくは90%以上である。
【0098】
樹脂粒子(C1)の分散した分散体(X1)から、通常、減圧により二酸化炭素(X)を除去し、本発明の樹脂粒子(C)を得る。その際、独立に圧力制御された容器を多段に設けることにより段階的に減圧してもよく、また一気に常温常圧まで減圧してもよい。得られる樹脂粒子の捕集方法は特に限定されず、フィルターでろ別する方法や、サイクロン等により遠心分離する方法が例として挙げられる。樹脂粒子は減圧後に捕集してもよく、また減圧前に一旦高圧中で捕集した後、減圧してもよい。高圧下で捕集した後に減圧する場合の、高圧下からの樹脂粒子の取り出し方としては、バッチ操作で捕集容器を減圧してもよく、またロータリーバルブを使用して連続的取り出し操作を行ってもよい。
【0099】
微粒子(A)が結晶性樹脂(a1)を含有する場合、樹脂粒子(C1)を形成させた後、必要に応じて、さらなる工程として、結晶性樹脂(a1)の、好ましくは、融点マイナス50℃以上、より好ましくは融点マイナス10℃以上、さらに好ましくは融点以上、に加熱することにより、樹脂粒子(B)の表面に付着した微粒子(A)を溶融させて、微粒子(A)を樹脂粒子(B)の表面に固着、又は微粒子(A)由来の皮膜を形成して樹脂粒子(C2)を形成する工程を行うこともできる。(C2)の凝集を抑制するという観点から、加熱する時間は0.01〜1時間が好ましく、さらに好ましくは、0.05〜0.7である。
【0100】
本発明の製造方法(1)〜(3)により得られる本発明の樹脂粒子(C)は、樹脂粒子(B)又は(B1)の表面に一旦微粒子(A)が固着されるが、(a)として結晶性樹脂(a1)を用いた場合、(a)と樹脂(b)の組成、溶剤(S)又は(T)の種類によっては、製造工程中に、微粒子(A)が皮膜化されて、(B)の表面に(A)が皮膜化された皮膜が形成される場合がある。
本発明の樹脂粒子(C)は、樹脂粒子(B)の表面に、微粒子(A)が固着されたもの、(A)由来の皮膜が形成されたもの、(A)の一部が皮膜化されたもののいずれであってもよい。
なお、本発明の樹脂粒子(C)の表面状態及び形状は、例えば、走査電子顕微鏡(SEM)を用い、樹脂粒子の表面を1万倍または3万倍拡大した写真にて観察できる。
【0101】
樹脂粒子(C1)〔樹脂粒子(C2)の場合も含む〕を形成させた後、必要に応じて、さらなる工程として、溶剤(S)を除去又は減少させる工程を行うことが好ましい。すなわち、(C1)が(X)中に分散した分散体(X1)中に溶剤(S)を含む場合、そのまま容器を減圧にすると、(X1)中に溶解した溶剤が凝縮し、樹脂粒子(C1)を再溶解してしまったり、樹脂粒子(C1)を捕集する際に樹脂粒子(C1)同士が合一してしまう等の問題が生じる場合がある。溶剤を除去又は減少させる方法としては、例えば、樹脂(b)の溶剤(S)の溶液(L)を分散して得られた分散体(X1)に、さらに二酸化炭素〔好ましくは二酸化炭素(X)〕を混合して樹脂粒子(C1)から溶剤(S)を二酸化炭素の相に抽出し、つぎに、溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素〔好ましくは二酸化炭素(X)〕で置換し、その後に減圧することが好ましい。
【0102】
樹脂粒子(C1)が二酸化炭素(X)中に分散した分散体(X1)と二酸化炭素の混合方法は、(X1)より高い圧力の二酸化炭素を加えてもよく、また(X1)を(X1)より低い圧力の二酸化炭素中に加えてもよいが、連続操作の容易性の観点からより好ましくは後者である。(X1)と混合する二酸化炭素の量は、樹脂粒子(C1)の合一防止の観点から、(X1)の体積の1〜50倍が好ましく、さらに好ましくは1〜40倍、最も好ましくは1〜30倍である。上記のように樹脂粒子(C1)中に含有される溶剤を除去ないし減少させ、その後、二酸化炭素を除去することにより、樹脂粒子(C1)同士が合一することを防ぐことができる。
【0103】
溶剤(S)を含む二酸化炭素を溶剤(S)を含まない二酸化炭素で置換する方法としては、樹脂粒子(C1)を一旦フィルターやサイクロンで補足した後、圧力を保ちながら、溶剤(S)が完全に除去されるまで二酸化炭素を流通させる方法が挙げられる。流通させる二酸化炭素の量は、分散体(X1)からの溶剤除去の観点から、(X1)の体積に対して1〜100倍が好ましく、さらに好ましくは1〜50倍、最も好ましくは1〜30倍である。
【0104】
次に製造方法(1)について詳細に説明する。
本発明において、樹脂(b)の前駆体(b0)としては、化学反応により樹脂(b)になりうるものであれば特に限定されず、例えば、樹脂(b)がビニル樹脂である場合は、(b0)は、先述のビニルモノマー(単独で用いても、混合して用いてもよい)が挙げられ、樹脂(b)が縮合系樹脂(例えば、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂)である場合は、(b0)は、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせが例示される。
【0105】
ビニルモノマーを前駆体(b0)として用いた場合、(b0)は通常用いられる開始剤を含有してもよい。上記開始剤としては、パーオキサイド系重合開始剤(I)、アゾ系重合開始剤(II)等が挙げられる。また、パーオキサイド系重合開始剤(I)と還元剤とを併用してレドックス系重合開始剤(III)を形成してもよい。更には、(I)〜(III)のうちから2種以上を併用してもよい。
【0106】
上記開始剤を用いる場合、二酸化炭素(X)中に(b0)を分散する前に、予めモノマーと混合しておくことが好ましい。重合温度は好ましくは40〜100℃、さらに好ましくは60〜90℃である。
【0107】
前駆体(b0)としては、反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせを用いることもできる。ここで「反応性基」とは硬化剤(β)と反応可能な基のことをいう。反応性基含有プレポリマー(α)が有する反応性基と、硬化剤(β)の組み合わせとしては、下記(1)、(2)などが挙げられる。
(1):反応性基含有プレポリマー(α)が有する反応性基が、活性水素化合物と反応可能な官能基(α1)であり、硬化剤(β)が活性水素基含有化合物(β1)であるという組み合わせ。
(2):反応性基含有プレポリマー(α)が有する反応性基が活性水素含有基(α2)であり、硬化剤(β)が活性水素含有基と反応可能な化合物(β2)であるという組み合わせ。
【0108】
上記組合せ(1)において、活性水素化合物と反応可能な官能基(α1)としては、イソシアネート基(α1a)、ブロック化イソシアネート基(α1b)、エポキシ基(α1c)、酸無水物基(α1d)および酸ハライド基(α1e)などが挙げられる。これらのうち好ましいものは、(α1a)、(α1b)および(α1c)であり、特に好ましいものは、(α1a)および(α1b)である。ブロック化イソシアネート基(α1b)は、ブロック化剤によりブロックされたイソシアネート基のことをいう。上記ブロック化剤としては、オキシム類[アセトオキシム、メチルイソブチルケトオキシム、ジエチルケトオキシム、シクロペンタノンオキシム、シクロヘキサノンオキシム、メチルエチルケトオキシム等];ラクタム類[γ−ブチロラクタム、ε−カプロラクタム、γ−バレロラクタム等];炭素数1〜20の脂肪族アルコール類[エタノール、メタノール、オクタノール等];フェノール類[フェノール、m−クレゾール、キシレノール、ノニルフェノール等];活性メチレン化合物[アセチルアセトン、マロン酸エチル、アセト酢酸エチル等];塩基性窒素含有化合物[N,N−ジエチルヒドロキシルアミン、2−ヒドロキシピリジン、ピリジンN−オキサイド、2−メルカプトピリジン等];およびこれらの2種以上の混合物が挙げられる。これらのうち好ましいのはオキシム類であり、特に好ましいものはメチルエチルケトオキシムである。
【0109】
反応性基含有プレポリマー(α)の骨格としては、ポリエーテル(αw)、ポリエステル(αx)、エポキシ樹脂(αy)およびポリウレタン(αz)などが挙げられる。これらのうち好ましいものは、(αx)、(αy)および(αz)であり、特に好ましいものは(αx)および(αz)である。ポリエーテル(αw)としては、ポリエチレンオキサイド、ポリプロピレンオキサイド、ポリブチレンオキサイド、ポリテトラメチレンオキサイドなどが挙げられる。ポリエステル(αx)としては、ジオール(11)とジカルボン酸(13)の重縮合物、ポリラクトン(ε−カプロラクトンの開環重合物)などが挙げらる。エポキシ樹脂(αy)としては、ビスフェノール類(ビスフェノールA、ビスフェノールF、ビスフェノールSなど)とエピクロルヒドリンとの付加縮合物などが挙げられる。ポリウレタン(αz)としては、ジオール(11)とポリイソシアネート(15)の重付加物、ポリエステル(αx)とポリイソシアネート(15)の重付加物などが挙げられる。
【0110】
ポリエステル(αx)、エポキシ樹脂(αy)、ポリウレタン(αz)などに反応性基を含有させる方法としては、(1):二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させる方法、(2):二以上の構成成分のうちの一つを過剰に用いることで構成成分の官能基を末端に残存させ、さらに残存した該官能基と反応可能な官能基及び反応性基を含有する化合物を反応させる方法などが挙げられる。上記方法(1)では、水酸基含有ポリエステルプレポリマー、カルボキシル基含有ポリエステルプレポリマー、酸ハライド基含有ポリエステルプレポリマー、水酸基含有エポキシ樹脂プレポリマー、エポキシ基含有エポキシ樹脂プレポリマー、水酸基含有ポリウレタンプレポリマー、イソシアネート基含有ポリウレタンプレポリマーなどが得られる。構成成分の比率は、例えば、水酸基含有ポリエステルプレポリマーの場合、ポリオールとポリカルボン酸の比率が、水酸基[OH]とカルボキシル基[COOH]のモル比[OH]/[COOH]として、好ましくは2/1〜1/1、さらに好ましくは1.5/1〜1/1、とくに好ましくは1.3/1〜1.02/1である。他の骨格、他の末端基のプレポリマーの場合も、構成成分が変わるだけで比率は同様である。上記方法(2)では、上記方法(1)で得られたプレプリマーに、ポリイソシアネートを反応させることでイソシアネート基含有プレポリマーが得られ、ブロック化ポリイソシアネートを反応させることでブロック化イソシアネート基含有プレポリマーが得られ、ポリエポキサイドを反応させることでエポキシ基含有プレポリマーが得られ、ポリ酸無水物を反応させることで酸無水物基含有プレポリマーが得られる。官能基および反応性基を含有する化合物の使用量は、例えば、水酸基含有ポリエステルにポリイソシアネートを反応させてイソシアネート基含有ポリエステルプレポリマーを得る場合、ポリイソシアネートの比率が、イソシアネート基[NCO]と、水酸基含有ポリエステルの水酸基[OH]のモル比[NCO]/[OH]として、好ましくは5/1〜1/1、さらに好ましくは4/1〜1.2/1、とくに好ましくは2.5/1〜1.5/1である。他の骨格、他の末端基を有するプレポリマーの場合も、構成成分が変わるだけで比率は同様である。
【0111】
反応性基含有プレポリマー(α)中の1分子当たりに含有する反応性基は、通常1個以上、好ましくは、平均1.5〜3個、さらに好ましくは、平均1.8〜2.5個である。上記範囲にすることで、硬化剤(β)と反応させて得られる硬化物の分子量が高くなる。反応性基含有プレポリマー(α)の数平均分子量は、好ましくは500〜30,000、さらに好ましくは1,000〜20,000、とくに好ましくは2,000〜10,000である。反応性基含有プレポリマー(α)の重量平均分子量は、1,000〜50,000、好ましくは2,000〜40,000、さらに好ましくは4,000〜20,000である。反応性基含有プレポリマー(α)の粘度は、100℃において、好ましくは2,000ポイズ以下、さらに好ましくは1,000ポイズ以下である。2,000ポイズ以下にすることで、少量の溶剤で粒度分布のシャープな樹脂粒子(C)が得られる点で好ましい。
【0112】
活性水素基含有化合物(β1)としては、脱離可能な化合物でブロック化されていてもよいポリアミン(β1a)、ポリオール(β1b)、ポリメルカプタン(β1c)および水(β1d)などが挙げられる。これらのうち好ましいものは、(β1a)、(β1b)および(β1d)であり、さらに好ましいものは、(β1a)および(β1d)であり、特に好ましいものは、ブロック化されたポリアミン(β1a)および(β1d)である。(β1a)としては、ポリアミン(16)と同様のものが例示される。(β1a)として好ましいものは、4,4’−ジアミノジフェニルメタン、キシリレンジアミン、イソホロンジアミン、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、ヘキサメチレンジアミンおよびそれらの混合物である。
【0113】
(β1a)が脱離可能な化合物でブロック化されたポリアミンである場合の例としては、前記ポリアミン類と炭素数3〜8のケトン類(アセトン、メチルエチルケトン、メチルイソブチルケトンなど)から得られるケチミン化合物、炭素数2〜8のアルデヒド化合物(ホルムアルデヒド、アセトアルデヒド)から得られるアルジミン化合物、エナミン化合物、およびオキサゾリジン化合物などが挙げられる。
【0114】
ポリオール(β1b)としては、前記のジオール(11)およびポリオール(12)と同様のものが例示される。ジオール(11)単独、またはジオール(11)と少量のポリオール(12)の混合物が好ましい。
【0115】
ポリメルカプタン(β1c)としては、エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなどが挙げられる。
【0116】
必要により活性水素基含有化合物(β1)と共に反応停止剤(βs)を用いることができる。反応停止剤を(β1)と一定の比率で併用することにより、(b)を所定の分子量に調整することが可能である。反応停止剤(βs)としては、モノアミン(ジエチルアミン、ジブチルアミン、ブチルアミン、ラウリルアミン、モノエタノールアミン、ジエタノールアミンなど);モノアミンをブロックしたもの(ケチミン化合物など);モノオール(メタノール、エタノール、イソプロパノール、ブタノール、フェノール;モノメルカプタン(ブチルメルカプタン、ラウリルメルカプタンなど);モノイソシアネート(ラウリルイソシアネート、フェニルイソシアネートなど);モノエポキサイド(ブチルグリシジルエーテルなど)などが挙げられる。
【0117】
上記組合せ(2)における反応性基含有プレポリマー(α)が有する活性水素含有基(α2)としては、アミノ基(α2a)、水酸基(アルコール性水酸基およびフェノール性水酸基)(α2b)、メルカプト基(α2c)、カルボキシル基(α2d)およびそれらが脱離可能な化合物でブロック化された有機基(α2e)などが挙げられる。これらのうち好ましいものは、(α2a)、(α2b)およびアミノ基が脱離可能な化合物でブロック化された有機基(α2e)であり、特に好ましいものは、(α2b)である。アミノ基が脱離可能な化合物でブロック化された有機基としては、前記(β1a)の場合と同様のものが例示できる。
【0118】
活性水素含有基と反応可能な化合物(β2)としては、ポリイソシアネート(β2a)、ポリエポキシド(β2b)、ポリカルボン酸(β2c)、ポリ酸無水物(β2d)およびポリ酸ハライド(β2e)などが挙げられる。これらのうち好ましいものは、(β2a)および(β2b)であり、さらに好ましいものは、(β2a)である。
【0119】
ポリイソシアネート(β2a)としては、ポリイソシアネート(15)と同様のものが例示され、好ましいものも同様である。
【0120】
ポリエポキシド(β2b)としては、ポリエポキシド(18)と同様のものが例示され、好ましいものも同様である。
【0121】
ポリカルボン酸(β2c)としては、ジカルボン酸(β2c−1)および3価以上のポリカルボン酸(β2c−2)が挙げられ、(β2c−1)単独、および(β2c−1)と少量の(β2c−2)の混合物が好ましい。ジカルボン酸(β2c−1)としては、前記ジカルボン酸(13)と、ポリカルボン酸としては、前記ポリカルボン酸(14)と、それぞれ、同様のものが例示され、好ましいものも同様である。
【0122】
ポリカルボン酸無水物(β2d)としては、ピロメリット酸無水物などが挙げられる。ポリ酸ハライド類(β2e)としては、前記(β2c)の酸ハライド(酸クロライド、酸ブロマイド、酸アイオダイド)などが挙げられる。さらに、必要により(β2)と共に反応停止剤(βs)を用いることができる。
【0123】
硬化剤(β)の比率は、反応性基含有プレポリマー(α)中の反応性基の当量[α]と、硬化剤(β)中の活性水素含有基[β]の当量の比[α]/[β]として、好ましくは1/2〜2/1、さらに好ましくは1.5/1〜1/1.5、とくに好ましくは1.2/1〜1/1.2である。なお、硬化剤(β)が水(β1d)である場合は水は2価の活性水素化合物として取り扱う。
【0124】
前駆体(b0)として反応性基を有するプレポリマー(α)と硬化剤(β)の組み合わせを用いる場合、二酸化炭素(X)中に微粒子(A)が分散している分散体(X0)において(b0)を反応させる方法は特に限定されないが、(X0)中に(b0)を分散する直前に(α)と(β)を混合し、分散すると同時に反応させる方法が好ましい。反応時間は、プレポリマー(α)の有する反応性基の構造と硬化剤(β)の組み合わせによる反応性により選択されるが、好ましくは5分〜24時間である。反応は減圧前に(X0)中で完結させてもよく、また(X0)である程度反応させ、減圧し(C)を取り出した後、恒温槽などで熟成させ完結させてもよい。また、必要に応じて公知の触媒を使用することができる。具体的には、例えばイソシアネートと活性水素化合物の反応の場合には、ジブチルチンラウレート、ジオクチルチンラウレートなどが挙げられる。反応温度は好ましくは30〜100℃、さらに好ましくは40〜80℃である。
【0125】
反応性基含有プレポリマー(α)と硬化剤(β)からなる前駆体(b0)を反応させた樹脂(b)が樹脂粒子(B)および樹脂粒子(C)の構成成分となる。反応性基含有プレポリマー(α)と硬化剤(β)を反応させた樹脂(b)の重量平均分子量は、好ましくは3,000以上、さらに好ましくは3,000〜1000万、とくに好ましくは,5000〜100万である。
【0126】
また、反応性基含有プレポリマー(α)と硬化剤(β)との反応時に、反応性基含有プレポリマー(α)および硬化剤(β)と反応しないポリマー[いわゆるデッドポリマー]を系内に含有させることもできる。この場合(b)は、反応性基含有プレポリマー(α)と硬化剤(β)を反応させて得られた樹脂と、反応させていない樹脂の混合物となる。
製造方法(1)の場合、上述した事項及び、樹脂(b)を溶剤(S)に溶解させた溶液(L)の代わりに樹脂(b)の前駆体(b0)を用いて分散時に前駆体(b0)を反応させる以外は、製造方法(2)と同様である。
【0127】
製造方法(3)について詳細に説明する。
樹脂(b)の溶剤(S)の溶液(L)の代わりに、樹脂(b)の前駆体(b0)の溶剤(S)の溶液(L0)を用い、分散時に前駆体(b0)を反応させる以外は製造方法(2)と同様である。
【0128】
上記製造方法(1)〜(3)によれば、親水性基を有する界面活性物質を実質的に含有しない本発明の樹脂粒子(C)を製造することができる。ここで、親水性基を有する界面活性物質とは、アニオン界面活性剤(S−1)、カチオン界面活性剤(S−2)、両性界面活性剤(S−3)、非イオン界面活性剤(S−4)などが挙げられる。
【0129】
これら界面活性剤の具体例としては、国際公開WO03/106541号パンフレットに記載のものが挙げられる。通常、水溶剤中で親水性基を有する界面活性物質を用いて製造された樹脂粒子は、親水性基を有する界面活性物質を実質的に含有している。
【0130】
樹脂粒子が親水性基を有する界面活性物質を実質的に含有しないことを分析する方法としては、公知の表面濡れ性評価(色材協会誌、第73[3]号、2000年、P132〜138による。)が挙げられる。表面濡れ性の評価法は次の通りである。すなわち100mlビーカーに樹脂粒子0.1gを入れ、そこにイオン交換水を20ml添加し、マグネティックスターラーで攪拌し、液面に樹脂粒子を浮かべた後、アセトンを少しづつ滴下し、表面に浮かぶ樹脂粒子が無くなるアセトン重量(Wa)と水の重量(Ww)を有効数字3桁で求め、(1)式より、樹脂粒子表面の溶解度パラメータ(δm)を算出する。
δm=(9.75×Wa+23.43×Ww)/(Wa+Ww) (1)
【0131】
樹脂粒子表面の溶解度パラメータ(δm)が、9.8〜21、好ましくは9.8〜20であれば、樹脂粒子が親水性基を有する界面活性物質を実質的に含有しないものと判断される。δmが、9.8〜21であれば、樹脂粒子の耐湿保存性は良好であり、高湿下におけるトナーとしての電気特性、流動性、定着性が良好である。本測定方法では9.8未満は測定できない。
【0132】
樹脂粒子(C)は、微粒子(A)と樹脂粒子(B)の粒径、及び、微粒子(A)又は(A)由来の皮膜による樹脂粒子(B)表面の被覆率を変えることで粒子表面に所望の凹凸を付与することができる。さらに減圧時の温度・圧力をコントロールすることにより内部に気泡を有する多孔質体が得られ、比表面積を大きくすることができる。粉体流動性を向上させたい場合には、樹脂粒子のBET値比表面積が0.5〜5.0m2/gであるのが好ましい。BET比表面積は、比表面積計、例えば、QUANTASORB(ユアサアイオニクス製)を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)したものである。同様に粉体流動性の観点から、本発明の樹脂粒子(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
【0133】
本発明の製造方法で得られる樹脂粒子(C)は粒度分布がシャープであり、且つ通常、水溶性の界面活性物質やイオン性物質を含まないため、疎水性である。したがって本発明の樹脂粒子(C)は電子写真トナー用として有用である。またその他の用途として、塗料用添加剤、接着剤用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成形用樹脂、粉体塗料、電子部品製造用スペーサー、触媒用担体、静電記録トナー、静電印刷トナー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、クロマトグラフ充填剤、電気粘性流体用粒子等としても有用である。
【0134】
本発明の樹脂粒子(C)において、微粒子(A)が、融点が50〜110℃である、結晶性樹脂(a1)を含有する場合は、低温定着性に優れ、耐熱保存性にも優れるという効果を有することから、特に電子写真トナー用として有用である。
融点が50〜110℃である結晶性樹脂(a1)の中でも、(a11)、(a12)、(a13)、及び(a14)が特に好ましい。
【0135】
電子写真プロセスにおいて使用される電子写真トナーは、その現像工程において、例えば、静電荷像が形成されている感光体等の像担持体に一旦付着され、次に転写工程において、感光体から転写紙等の転写媒体に転写された後、定着工程において紙面に定着される。電子写真トナーとしては、通常、流動特性を付与するため、トナー用樹脂粒子と各種金属酸化物等の無機粉末等を混合して使用されており、この無機粉末等は外添剤と呼ばれている。本発明の電子写真トナーは、本発明の樹脂粒子に外添剤を添加したものが好ましい。
【0136】
外添剤としては、例えば、二酸化珪素(シリカ)、二酸化チタン(チタニア)、酸化アルミニウム、酸化亜鉛、酸化マグネシウム、酸化セリウム、酸化鉄、酸化銅、酸化錫等が知られている。特に、シリカや酸化チタン微粒子とジメチルジクロロシラン、ヘキサメチルジシラザン、シリコーンオイル等の有機珪素化合物とを反応させ、シリカ微粒子表面のシラノール基を有機基で置換し疎水化したシリカ微粒子が好ましく用いられる。
【0137】
外添剤の使用量(重量%)としては、特に制限はないが、定着性と流動性の両立の観点から、電子写真トナー用樹脂粒子の重量に対して0.01〜5が好ましく、さらに好ましくは、0.1〜4、特に好ましくは0.5〜3である。
【0138】
本発明の電子写真トナーは、本発明の電子写真トナー用樹脂粒子に外添剤を添加し、混合することにより製造することができる。
【実施例】
【0139】
以下実施例により本発明をさらに説明するが、本発明はこれに限定されるものではない。以下の記載において「部」は重量部、「%」は重量%を示す。
なお、実施例1、5、8、9、10、19、および20は参考例である。
【0140】
下記の膨潤度、結晶化度、数平均分子量、融点、ガラス転移温度、体積平均粒径は以下の方法で測定した。
<膨潤度の測定方法>
試料(5mg)を採取して磁気浮遊天秤(MSB−SCC・SCW 日本ベル社製)を用いて40℃、10MPaにおける超臨界状態の二酸化炭素が試料に浸透する重量を測定し、試料の重量で除することで、膨潤度(%)を求めた。
<結晶化度の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)を用いて室温から昇温速度20℃/minにて温度を変化させながら、吸熱ピークの面積より求めた融解熱量(ΔHm(J/g))を求めた。測定されたΔHmに基づき以下の式により結晶化度(%)を算出した。
結晶化度=(融解熱量/a)×100
上式中、aは以下のようにして測定する。
測定しようとする樹脂と同組成の標品となる樹脂の融解熱量をDSCで測定し、JISK0131(1996年)(X線回折分析通則 13結晶化度測定 (2)絶対法)に準じた測定方法で結晶化度を測定する。縦軸に融解熱量、横軸に結晶化度を座標にとり、標品のデータをプロットし、その点と原点の2点から直線を引き、結晶化度が100%となるように外挿した場合の融解熱量を求めた値がaである。
【0141】
<数平均分子量(Mn)の測定方法>
試料をそれぞれ濃度2.5g/Lでテトラヒドロフランに溶解させ、ポリスチレンを標準物質として、GPCにより測定した。
GPC機種:HLC−8120GPC、東ソー(株)製
カラム :TSKgel GMHXL)2本+TSKgel Multipore HXL−M(東ソー(株)製)
<融点の測定方法>
試料(5mg)を採取してアルミパンに入れ、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃で、結晶溶融による吸熱ピークの温度(℃)を求めた。
<ガラス転移温度(Tg)の測定方法>
試料をそれぞれ5mg秤り取り、DSC(示差走査熱量測定)(測定装置:RDC220、エスアイアイナノテクノロジー(株)製)により、昇温速度毎分10℃でガラス転移温度を測定した。
<体積平均粒径の測定方法>
試料5mgをイオン交換水10gに分散させた後、マルチサイザーIII(コールター社製)により測定した。
【0142】
製造例1<樹脂(b−1)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、1.2−プロピレングリコール(以下、プロピレングリコールと記載)831部、テレフタル酸703部、アジピン酸47部、および縮合触媒としてテトラブトキシチタネート0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が87℃になった時点で180℃まで冷却し、さらに無水トリメリット酸24部、テトラブトキシチタネート0.5部を投入し90分反応させた後、取り出した。回収されたプロピレングリコールは442部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し、ポリエステル樹脂(b−1)を得た。この樹脂のMnは1900、Tgは45℃であった。
【0143】
製造例2<樹脂(b−2)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール729部、テレフタル酸683部、アジピン酸67部、無水トリメリット酸38部および縮合触媒としてテトラブトキシチタネート0.5部を入れ、180℃で窒素気流下に、生成する水を留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させた。回収されたプロピレングリコールは172部であった。軟化点が160℃になった時点で取り出し、室温まで冷却後、粉砕し粒子化し、ポリエステル樹脂(b−2)を得た。この樹脂のMnは5700、Tgは63℃であった。
【0144】
製造例3<樹脂(b−3)の調製>
撹拌棒および温度計をセットしたオートクレーブに、キシレン24部を投入し、アクリル酸ブチル/メタクリル酸メチル/スチレン/アクリル酸2−エチルヘキシル(25重量%/33重量%/40重量%/2重量%)の混合モノマー2,000部と重合触媒1部を、170℃で3時間かけて滴下重合をおこなった。180℃まで昇温しながら常圧で脱揮し、180℃になったところで減圧に切り替え、2時間かけて減圧で脱揮をおこない、ビニル樹脂(b−3)を得た。この樹脂のMnは10,500、Tgは62℃であった。
【0145】
製造例4<樹脂溶液(L−1)の調製>
攪拌装置のついた容器に、アセトン490部、メタノール175部、イオン交換水35部からなる混合溶剤である溶剤(S−1)に、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)と樹脂(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−1)を得た。溶剤(S−1)は、標準状態の樹脂(b)と溶剤(S−1)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−1)のSP値は11.8であった。
【0146】
製造例5<樹脂溶液(L−2)の調製>
攪拌装置のついた容器に、アセトン450部、イオン交換水50部からなる混合溶剤である溶剤(S−2)に、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−2)を得た。溶剤(S−2)は、標準状態の樹脂(b)と溶剤(S−2)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−2)のSP値は11.3であった。
【0147】
製造例6<樹脂溶液(L−3)の調製>
攪拌装置のついた容器に、アセトン490部、メタノール210部からなる混合溶剤である溶剤(S−3)に、製造例3で得られた樹脂(b−3)280部、及びカーボンブラック15部を仕込み、樹脂(b−3)が完全に溶解するまで攪拌し、樹脂溶液(L−3)を得た。溶剤(S−3)は、標準状態の樹脂(b)と溶剤(S−3)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−3)のSP値は11.3であった。
【0148】
製造例7<樹脂溶液(L−4)の調製>
攪拌装置のついた容器に、アセトン560部、イオン交換水70部、デカン70部からなる混合溶剤である溶剤(S−4)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−4)を得た。溶剤(S−4)は、標準状態の樹脂(b)と溶剤(S−4)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−4)のSP値は10.3であった。
【0149】
製造例8<樹脂前駆体(b0−1)の調整>
オートクレーブに、製造例1で得られた樹脂(b−1)407部、イソホロンジイソシアネート(IPDI)54部、アセトン485部を投入し、密閉状態で100℃、5時間反応を行い、分子末端にイソシアネート基を有する樹脂前駆体(b0−1)を得た。樹脂前駆体(b0−1)のNCO含量は0.8%であった。
【0150】
製造例9<硬化剤(β)の調整>
撹拌機、脱溶剤装置、および温度計をセットした反応容器に、イソホロンジアミン50部とメチルエチルケトン300部を投入し、50℃で5時間反応を行った後、脱溶剤してケチミン化合物である硬化剤(β)(ウレタンプレポリマーの鎖伸長剤)を得た。硬化剤(β)の全アミン価は415であった。
【0151】
製造例10<樹脂前駆体溶液(L−5)の調整>
攪拌装置のついた容器に、ジメチルホルムアミドである溶剤(S−5)700部、製造例1で得られた樹脂(b−1)228部、製造例8で得られた樹脂前駆体(b0−1)57部、硬化剤(β)1.5部、及びカーボンブラック15部を仕込み、樹脂前駆体(b0−1)及び硬化剤(β)が完全に溶解するまで攪拌し、樹脂溶液(L−5)を得た。溶剤(S−5)は、標準状態の上記重量比の樹脂(b)および前駆体(b0)と溶剤(S−5)の等重量混合物における樹脂(b)および前駆体(b0)の重量に対する(b)および(b0)の不溶分重量は0.1重量%以下、溶剤(S−5)のSP値は12.0であった。
【0152】
製造例11<樹脂溶液(L−6)の調製>
攪拌装置のついた容器に、アセトン490部、イオン交換水210部からなる混合溶剤である溶剤(S−6)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−6)を得た。溶剤(S−6)は、標準状態の樹脂(b)と溶剤(S−6)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は15重量%、溶剤(S−6)のSP値は14.0であった。
【0153】
製造例12<樹脂溶液(L−7)の調製>
攪拌装置のついた容器に、メチルエチルケトン665部、イオン交換水35部からなる混合溶剤である溶剤(S−7)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−7)を得た。溶剤(S−7)は、標準状態の樹脂(b)と溶剤(S−7)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.1重量%以下、溶剤(S−7)のSP値は9.7であった。
【0154】
製造例13<樹脂溶液(L−8)の調製>
攪拌装置のついた容器に、1,3−ジオキソラン(S−8)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−8)を得た。溶剤(S−8)は、標準状態の樹脂(b)と溶剤(S−8)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は0.5重量%、溶剤(S−8)のSP値は9.4であった。
【0155】
製造例14<樹脂溶液(L−9)の調製>
攪拌装置のついた容器に、ピルビン酸メチル(S−9)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−9)を得た。溶剤(S−9)は、標準状態の樹脂(b)と溶剤(S−9)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は1重量%、溶剤(S−9)のSP値は10.6であった。
【0156】
製造例15<樹脂溶液(L−10)の調製>
攪拌装置のついた容器に、プロピレングリコールモノメチルエーテル(S−10)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−10)を得た。溶剤(S−10)は、標準状態の樹脂(b)と溶剤(S−10)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は3重量%、溶剤(S−10)のSP値は11.3であった。
【0157】
製造例16<樹脂溶液(L−11)の調製>
攪拌装置のついた容器に、2−ヒドロキシイソ酪酸メチル(S−11)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−11)を得た。溶剤(S−11)は、標準状態の樹脂(b)と溶剤(S−11)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は7重量%、溶剤(S−11)のSP値は11.8であった。
【0158】
製造例17<樹脂溶液(L−12)の調製>
攪拌装置のついた容器に、乳酸メチル(S−12)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−12)を得た。溶剤(S−12)は、標準状態の樹脂(b)と溶剤(S−12)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は10重量%、溶剤(S−12)のSP値は12.4であった。
【0159】
製造例18<樹脂溶液(L−13)の調製>
攪拌装置のついた容器に、トリフルオロエタノール(S−13)700部、製造例1で得られた樹脂(b−1)228部、製造例2で得られた樹脂(b−2)57部、及びカーボンブラック15部を仕込み、樹脂(b−1)、(b−2)が完全に溶解するまで攪拌し、樹脂溶液(L−13)を得た。溶剤(S−13)は、標準状態の樹脂(b)と溶剤(S−13)の等重量混合物における樹脂(b)の重量に対する樹脂(b)の不溶分重量は18重量%、溶剤(S−13)のSP値は15.1であった。
【0160】
製造例19<結晶性樹脂(a1−1)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸230部、1,6−ヘキサンジオール195部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−1)を得た。この樹脂の結晶化度は60%、融点は60℃、Mnは8,000であった。
【0161】
製造例20<結晶性樹脂(a1−2)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート(炭素数22個の直鎖アルキル基を有するアルコールのアクリレートプレンマーVA〔日油(株)製〕)150部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−2)を得た。この樹脂の結晶化度は42%、融点は65℃、Mnは50,000であった。
【0162】
製造例21<結晶性樹脂(a1−3)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート120部、2−デシルテトラデシルメタクリレート30部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−3)を得た。この樹脂の結晶化度は36%、融点は62℃、Mnは50,000であった。
【0163】
製造例22<結晶性樹脂(a1−4)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート150部、ブチルアクリレート50部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−4)を得た。この樹脂の結晶化度は20%、融点は50℃、Mnは40,000であった。
【0164】
製造例23<結晶性樹脂(a1−5)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸460部、1,6−ヘキサンジオール230部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−5)を得た。この樹脂の結晶化度は65%、融点は70℃、Mnは10,000であった。
【0165】
製造例24<結晶性樹脂(a1−6)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、ドデカン2酸460部、1,6−ヘキサンジオール230部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させた。得られた樹脂を50℃に冷却後、メチルエチルケトン300部に溶解させ、ヘキサメチレンジイソシアネート20部をいれ、3時間反応させ、20mmHgの減圧下で脱溶剤後、取り出した。取り出した樹脂を粉砕、粒子化し、結晶性ポリウレタン樹脂(a1−6)を得た。この樹脂の結晶化度は50%、融点は70℃、Mnは15,000であった。
【0166】
製造例25<結晶性樹脂(a1−7)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ベヘニルアクリレート135部、アクリロニトリル 15部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−7)を得た。この樹脂の結晶化度は41%、融点は62℃、Mnは50,000であった。
【0167】
製造例26<結晶性樹脂(a1−8)の調製>
冷却管、撹拌機および窒素導入管の付いた反応槽中に、テレフタル酸90部、セバシン酸340部、1,6−ヘキサンジオール310部、縮合触媒としてテトラブトキシチタネート0.5部を入れ、230℃まで徐々に昇温しながら、窒素気流下に、生成する水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し結晶性ポリエステル樹脂(a1−8)を得た。この樹脂の結晶化度は60%、融点は67℃、Mnは9,000であった。
【0168】
製造例27<結晶性樹脂(a1−9)の調製>
撹拌装置、加熱冷却装置、温度計、滴下ロート、および窒素吹き込み管を備えた反応容器に、トルエン500部を仕込み、別のガラス製ビーカーに、トルエン350部、ステアリルアクリレート(炭素数18個の直鎖アルキル基を有するアルコールのアクリレートプレンマーSA〔日油(株)製〕)150部、AIBN(アゾビスイソブチロニトリル)7.5部を仕込み、20℃で撹拌、混合して単量体溶液を調製し、滴下ロートに仕込んだ。反応容器の気相部の窒素置換を行った後に密閉下80℃で2時間かけて単量体溶液を滴下し、滴下終了から2時間、85℃で熟成した後、トルエンを130℃で3時間減圧除去して、結晶性ビニル樹脂(a1−9)を得た。この樹脂の結晶化度は32%、融点は54℃、Mnは50,000であった。
【0169】
製造例28<微粒子(A−1)分散液〜微粒子(A−9)分散液の調製>
ノルマルヘキサン700部、結晶性樹脂(a1−1)〜(a1−9)の各々300部を混合した後、ビーズミル(ダイノーミルマルチラボ:シンマルエンタープライゼス製)で粒径0.3mmのジルコニアビーズを用いて粉砕を行い、乳白色の微粒子(A−1)分散液〜微粒子(A−9)分散液を得た。分散液の体積平均粒径は表1及び表2に記載のように0.2〜0.4μmであった。また、微粒子(A−1)〜(A−9)の膨潤度は、表1及び表2に記載のとおりであった。
【0170】
製造例29<微粒子(A−10)分散液の調製>
製造例28において、結晶性樹脂(a1−1)〜(a1−9)の各々の代わりに、ポリオレフィン樹脂(サンワックス161−P(三洋化成工業製)、結晶化度は75%、融点は107℃、Mnは5,000)(a1−10)を用いた以外は製造例28と同様にして、乳白色の微粒子(A−10)分散液を得た。この分散液の体積平均粒径は0.4μmであった。また、微粒子(A−10)の膨潤度は15%であった。
【0171】
製造例30<微粒子(A−11)の調製>
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン139部、ジビニルベンゼン20部、メタクリル酸138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成して非結晶性ビニル樹脂(a2−1)(スチレン−メタクリル酸−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩−ジビニルベンゼンの共重合体、架橋性)の微粒子(A−11)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、微粒子(A−11)を得た。非結晶性ビニル樹脂(a2−1)の結晶化度は0%、ガラス転移温度は69℃であった。また、微粒子(A−11)の膨潤度は1%であった。
【0172】
製造例31<微粒子(A−12)分散液の調製>
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン139部、メタクリル酸メチル138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成して非結晶性ビニル樹脂(a2−2)(スチレン−メタクリル酸メチル−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の微粒子(A−12)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、微粒子(A−12)を得た。非結晶性ビニル樹脂(a2−2)の結晶化度は0%、ガラス転移温度は56℃であった。また、微粒子(A−12)の膨潤度は10%であった。
【0173】
製造例32<分散安定剤(E)溶液の調製>
攪拌機を備えた反応容器内にTHF700部を仕込み、反応容器内の空気を窒素置換した後、加熱して還流温度とした。次に、メタクリル酸メチル150部、メタクリル変性シリコーン(官能基等量:12,000g/mol、Mn12,000、信越化学工業製:X22−2426、)150部、アゾビスイソブチロニトリル1.5部の混合物を反応基内に2時間で適下後、還流温度で6時間熟成し、分散安定剤(E−1)溶液を得た。(E−1)の重量平均分子量は20,000であった。
【0174】
実施例1
図1の実験装置において、まずバルブV1、V2を閉じ、ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素(純度99.99%)を導入し、14MPa、40℃に調整した。また樹脂溶液タンクT1に樹脂溶液(L−1)、微粒子分散液タンクT2に微粒子(A−1)分散液を仕込んだ。次にボンベB1、ポンプP3より二酸化炭素を分散槽T3に導入し、9MPa、40℃に調整し、さらにタンクT2、ポンプP2より微粒子(A−1)分散液を導入した。次に分散槽T3の内部を2000rpmで攪拌しながら、タンクT1、ポンプP1より樹脂溶液(L−1)を分散槽T3内に導入した。導入後T3の内部の圧力は14MPaとなった。
【0175】
なお分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−1)分散液 45部
二酸化炭素 550部
なお導入した二酸化炭素の重量は、二酸化炭素の温度(40℃)、及び圧力(15MPa)から二酸化炭素の密度を下記文献2に記載の状態式より算出し、これに分散槽T3の体積を乗じることにより算出した。
文献2:Journal of Physical and Chemical Refarence data、vol.25、P.1509〜1596
【0176】
樹脂溶液(L−1)を導入後、1分間攪拌し分散体(X1)を得た。バルブV1を開き、P3よりT4内に二酸化炭素を導入した後、分散体(X1)をT4内に導入し、この間圧力が一定に保たれるように、V2の開度を調節した。この操作を30秒間行い、V1を閉めた。この操作によりT4内に導入された樹脂溶液からの溶剤の抽出を行った。さらにT4を60℃に加熱し、15分間保持した。この操作により、微粒子(A−1)を樹脂溶液(L−1)から形成された樹脂粒子(B−1)の表面に固着させ、樹脂粒子(C−1)を生成した。次に圧力ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素を導入しつつ圧力調整バルブV2により圧力を14MPaに保持することにより、抽出された溶剤を含む二酸化炭素を溶剤トラップ槽T5に排出すると共に、樹脂粒子(C−1)をフィルターF1に捕捉した。圧力ボンベB2、ポンプP4より粒子回収槽T4に二酸化炭素を導入する操作は、上記の分散槽T3に導入した二酸化炭素重量の5倍量を粒子回収槽T4に導入した時点で二酸化炭素の導入を停止した。この停止の時点で、溶剤を含む二酸化炭素を、溶剤を含まない二酸化炭素で置換すると共に樹脂粒子(C−1)をフィルターF1に捕捉する操作は完了した。さらに、圧力調整バルブV2を少しずつ開き、粒子回収槽内を大気圧まで減圧し、フィルターF1に補足されている、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−1)を得た。
【0177】
実施例2
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−2)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−2)を得た。実施例2における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−2) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0178】
実施例3
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−3)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−3)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−3)を得た。実施例3における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−3) 270部
微粒子(A−3)分散液 45部
二酸化炭素 550部
【0179】
実施例4
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−4)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−4)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−4)を得た。実施例4における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−4) 270部
微粒子(A−4)分散液 45部
二酸化炭素 550部
【0180】
実施例5
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−5)分散液を使用したこと、疎水性分散安定剤(E−1)を14部追加したこと、樹脂溶液(L−1)の代わりに(L−5)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−5)を得た。実施例5における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−5) 270部
微粒子(A−5)分散液 45部
分散安定剤(E−1)溶液 14部
二酸化炭素 550部
【0181】
実施例6
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−6)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−6)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−6)を得た。実施例6における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−6) 270部
微粒子(A−6)分散液 45部
二酸化炭素 550部
【0182】
実施例7
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−7)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−7)を得た。実施例7における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−7)分散液 45部
二酸化炭素 550部
【0183】
実施例8
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−8)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−8)を得た。実施例8における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−8)分散液 45部
二酸化炭素 550部
【0184】
実施例9
実施例1において、微粒子(A−1)分散液の代わりに、膨潤度が0%である疎水性シリカ(RX−50、日本アエロジル製、体積平均粒径150nm)(a3−1)からなる微粒子(A−13)を用いたこと、樹脂溶液(L−1)の代わりに樹脂前駆体(b0−1)及びイオン交換水20部を使用したこと、以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)が固着された樹脂粒子(C−9)を得た。実施例9における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂前駆体(b0−1) 270部
イオン交換水(ケチミン伸長のための水) 20部
疎水性シリカ(A−9) 7部
二酸化炭素 550部
【0185】
実施例10
実施例1において、微粒子(A−1)分散液の代わりに、T3に予め微粒子(A−11)を仕込んだこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)が固着された樹脂粒子(C−10)を得た。実施例10における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−10) 13.5部
二酸化炭素 550部
【0186】
実施例11
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−7)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−11)を得た。実施例11における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−7) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0187】
実施例12
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−8)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−12)を得た。実施例12における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−8) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0188】
実施例13
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−9)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−13)を得た。実施例13における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−9) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0189】
実施例14
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−10)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−14)を得た。実施例14における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−10) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0190】
実施例15
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−11)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−15)を得た。実施例15における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−11) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0191】
実施例16
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−12)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−16)を得た。実施例16における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−12) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0192】
実施例17
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−13)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−17)を得た。実施例17における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−13) 270部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0193】
実施例18
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−9)分散液を使用し、樹脂溶液(L−1)の代わりに樹脂溶液(L−2)を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−18)を得た。実施例18における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−2) 270部
微粒子(A−9)分散液 45部
二酸化炭素 550部
【0194】
実施例19
実施例1において、微粒子(A−1)分散液の代わりに、微粒子(A−10)分散液を使用したこと以外は実施例1と同様にして、樹脂粒子(B)の表面に微粒子(A)由来の皮膜が形成された樹脂粒子(C−19)を得た。実施例19における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
微粒子(A−10)分散液 45部
二酸化炭素 550部
【0195】
実施例20
ビーカー内に微粒子(A−12)の水性分散液(固形分濃度20重量%)11部、ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液80部、イオン交換水300部を混合攪拌し、樹脂溶液(L−1)150部を混合した後、TKホモミキサー(特殊機化製)を使用し、回転数12,000rpmで10分間混合した。混合後、撹拌棒および温度計をセットした反応容器に混合液を投入し、50℃で2時間で脱溶剤を行い、次いで濾別、乾燥を行い、樹脂粒子(C−20)を得た。実施例20におけるビーカーへの仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 150部
微粒子(A−12)の水性分散液 11部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液 80部
イオン交換水 300部
【0196】
比較例1
実施例1において微粒子(A−1)分散液を仕込まない以外は実施例1と同様にして、比較樹脂粒子(C−1’)を得た。比較例1における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
二酸化炭素 550部
【0197】
比較製造例1
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン100部、メタクリル酸メチル138部、アクリル酸ブチル184部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(a−1’)(スチレン−メタクリル酸メチル−アクリル酸ブチル−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。さらに水性分散液を凍結乾燥し、比較樹脂微粒子(A−1’)を得た。(A−1’)の膨潤度は18%であった。
【0198】
比較例2
実施例1において微粒子(A−1)分散液の代わりに、T3に予め比較樹脂微粒子(A−1’)を仕込んだこと以外は実施例1と同様にして、比較樹脂粒子(C−2’)を得た。比較例2における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 270部
比較樹脂微粒子(A−1’) 15部
二酸化炭素 550部
【0199】
比較製造例2
撹拌棒および温度計をセットした反応容器に、水683部、メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製)11部、スチレン111部、メタクリル酸メチル128部、アクリル酸ブチル164部、2−エチルヘキシルメタクリレート58部、過硫酸アンモニウム1部を仕込み、400回転/分で15分間撹拌したところ、白色の乳濁液が得られた。加熱して、系内温度75℃まで昇温し5時間反応させた。更に、1%過硫酸アンモニウム水溶液30部加え、75℃で5時間熟成してビニル樹脂(a−2’)(スチレン−メタクリル酸メチル−アクリル酸ブチル−2−エチルヘキシルメタクリレート−メタクリル酸EO付加物硫酸エステルのナトリウム塩の共重合体)の比較樹脂粒子(A−2’)の水性分散液を得た。水性分散液をLA−920で測定した体積平均粒径は、0.15μmであった。また、(A−2’)の膨潤度は21%であった。
【0200】
比較例3
ビーカー内に比較樹脂粒子(A−2’)の水性分散液11部、ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液80部、イオン交換水300部を混合攪拌し、樹脂溶液(L−1)150部を混合した後、TKホモミキサー(特殊機化製)を使用し、回転数12,000rpmで10分間混合した。混合後、撹拌棒および温度計をセットした反応容器に混合液を投入し、50℃で2時間で脱溶剤を行い、次いで濾別、乾燥を行い、樹脂粒子(C−3’)を得た。比較例3におけるビーカーへの仕込み組成の重量比は次の通りである。
樹脂溶液(L−1) 150部
比較樹脂粒子(A−2’)の水性分散液 11部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液 80部
イオン交換水 300部
【0201】
比較例4
実施例1において、樹脂溶液(L−1)の代わりに樹脂(b−1)228部及び(b−2)57部を使用し、微粒子(A−1)分散液の代わりに、微粒子(A−2)分散液を使用した点及び、タンクT1、ポンプP1より樹脂(b−1)を分散槽T3内に導入する際に、100℃に昇温し、溶融させた点以外は同様にして、樹脂粒子(C−4’)を得た。比較例4における分散槽T3への仕込み組成の重量比は次の通りである。
樹脂(b−1) 228部
樹脂(b−2) 57部
微粒子(A−2)分散液 45部
二酸化炭素 550部
【0202】
本発明の樹脂粒子及び比較の樹脂粒子の原料とその物性値を表1〜3に示した。
【0203】
評価結果
実施例1〜20、比較例1〜4で得られた樹脂粒子について、以下に記載した評価方法で表面濡れ性、粒度分布、耐熱保存性、耐湿耐熱保存性、低温定着温度を評価し、結果を表4〜6に記載した。
<表面濡れ性評価>
100mlビーカーに樹脂粒子0.1gを入れ、そこにイオン交換水20mlを添加し、マグネティックスターラーで攪拌し、液面に樹脂粒子を浮かべた後、アセトンを少しづつ滴下し、表面に浮かぶ樹脂粒子が無くなるアセトン重量(Wa)と水の重量(Ww)を有効数字3桁で求め、(1)式より、樹脂粒子表面の溶解度パラメータ(δm)を算出した。
δm=(9.75×Wa+23.43×Ww)/(Wa+Ww) (1)
<粒度分布の評価>
樹脂粒子をドデシルベンゼンスルホン酸ナトリウム水溶液(濃度0.1%)に分散して樹脂粒子(表中ではCと表記)の体積平均粒径/個数平均粒径をコールターカウンター[マルチサイザーIII(ベックマン・コールター社製)]で測定した。体積平均粒径/個数平均粒径が小さいほど、粒度分布がシャープであることを示す。
【0204】
<耐熱保存性の評価>
樹脂粒子の耐熱保存性を下記の方法で評価した。即ち、50℃に温調された乾燥機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○:ブロッキングが発生しない。
△:ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×:ブロッキングが発生し、簡単に指などで力を加えても分散しない。
<耐湿耐熱保存性の評価>
樹脂粒子の耐湿耐熱保存性を下記の方法で評価した。即ち、50℃、湿度80%に温調された恒温恒湿機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○:ブロッキングが発生しない。
△:ブロッキングが発生するが、簡単に指などで力を加えると容易に分散する。
×:ブロッキングが発生し、簡単に指などで力を加えても分散しない。
【0205】
本発明の樹脂粒子を用いて、以下の方法で本発明の電子写真用トナーを作成した。即ち、本発明の電子写真トナー用樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、ミキサーを用いてよく混ぜてアエロジルR972が樹脂粒子表面に均一に付着した電子写真用トナーを作成した。トナーの評価方法は以下のとおり。
【0206】
<低温定着温度の評価>
低温定着温度は、以下の方法により評価した。上記で得た電子写真用トナーを紙面上に0.6mg/cm2となるよう均一に載せる。このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通した時のコールドオフセットの発生温度を測定した。
【0207】
【表1】
【0208】
【表2】
【0209】
【表3】
【0210】
【表4】
【0211】
【表5】
【0212】
【表6】
【0213】
実施例1〜20で得られた樹脂粒子は体積平均粒径/個数平均粒径が小さく、粒度分布がシャープになったのに対し、比較例1、2、4で得られた樹脂粒子は粒子が凝集し、粒度分布が著しく悪化した。また、実施例1〜20で得られた樹脂粒子は耐湿耐熱保存性に優れるが、比較例3で得られた樹脂粒子は耐湿耐熱保存性に劣っていた。また、実施例1〜8、11〜19で得られた樹脂粒子は電子写真用トナーとして用いた場合には、低温定着性に優れる(低温定着温度が低い。)という効果も有していた。
【産業上の利用可能性】
【0214】
本発明の樹脂粒子は電子写真トナーとしてきわめて有用である。また、塗料用添加剤、化粧品用添加剤、紙塗工用添加剤、スラッシュ成型用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子ペーパー用粒子、医療診断用担体、電気粘性用粒子、その他成型用樹脂粒子としても有用である。
【符号の説明】
【0215】
T1:樹脂溶液タンク
T2:微粒子分散液タンク
T3:分散槽(最高使用圧力20MPa、最高使用温度100℃、攪拌機つき)
T4:粒子回収槽(最高使用圧力20MPa、最高使用温度100℃)
F1:セラミックフィルター(メッシュ:0.5μm)
T5:溶剤トラップ
B1、B2:二酸化炭素ボンベ
P1、P2:溶液ポンプ
P3、P4:二酸化炭素ポンプ
V1:バルブ
V2:圧力調整バルブ
【特許請求の範囲】
【請求項1】
微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として結晶性樹脂(a1)を用い、結晶性樹脂(a1)が、下記(a12)〜(a14)からなる群から選ばれる少なくとも1種である樹脂粒子。
(a12) 炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア。
(a13) アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂。
(a14) (メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂。
【請求項2】
結晶性樹脂(a1)の融点が50〜110℃である請求項1に記載の樹脂粒子。
【請求項3】
親水性基を有する界面活性物質を実質的に含有しない請求項1または2に記載の樹脂粒子。
【請求項4】
請求項1〜3のいずれか1項に記載の樹脂粒子を含有する電子写真トナー用樹脂粒子。
【請求項5】
請求項4に記載の電子写真トナー用樹脂粒子を含有する電子写真トナー。
【請求項6】
該電子写真トナー用樹脂粒子にさらに外添剤が添加されてなる請求項5に記載の電子写真トナー。
【請求項7】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項8】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項9】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項10】
樹脂粒子(C1)を形成させた後、溶剤(S)を含有する液状又は超臨界状態の二酸化炭素(X)を、溶剤(S)を含有しない二酸化炭素で置換し、(C1)中に含有される溶剤(S)を除去又は減少させ、その後、減圧して二酸化炭素を除去する請求項8又は9記載の製造方法。
【請求項11】
23℃、0.1MPaの標準状態における、溶剤(S)と、樹脂(b)又は前駆体(b0)との等重量混合物における樹脂(b)又は前駆体(b0)の不溶分の重量が、樹脂(b)又は前駆体(b0)の重量に対して20重量%以下であり、溶剤(S)の溶解性パラメーターが9〜16である請求項8〜10のいずれか1項に記載の製造方法。
【請求項12】
溶剤(S)が、環状エーテル、ピルビン酸エステル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、2−ヒドロキシイソ酪酸エステル、乳酸エステル、フッ素含有アルコール、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、アセトンと水の混合溶剤、又はメチルエチルケトンと水の混合溶剤である請求項8〜11のいずれか1項に記載の製造方法。
【請求項13】
液状又は超臨界状態の二酸化炭素(X)中への分散工程において、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有する分散安定剤(E)を使用する請求項7〜12のいずれか1項に記載の製造方法。
【請求項1】
微粒子(A)が、樹脂(b)を含有する樹脂粒子(B)の表面に固着され又は皮膜化されてなる樹脂粒子(C)であり、微粒子(A)のガラス転移温度又は融点未満の温度における、液状又は超臨界状態の二酸化炭素(X)による微粒子(A)の膨潤度が16%以下であって、微粒子(A)として結晶性樹脂(a1)を用い、結晶性樹脂(a1)が、下記(a12)〜(a14)からなる群から選ばれる少なくとも1種である樹脂粒子。
(a12) 炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジオール及び/又は炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジアミンと、炭素数2〜50のアルキレン鎖を有する直鎖脂肪族ジイソシアネートを必須構成単位とし、かつ、該ジオール及び/又はジアミンのアルキレン鎖の平均炭素数と該ジイソシアネートのアルキレン鎖の炭素数の合計数が10〜52である結晶性ポリウレタン及び/又はポリウレア。
(a13) アルキル基の炭素数が12〜50であるアルキル(メタ)アクリレートを必須構成単位とする結晶性ビニル樹脂。
(a14) (メタ)アクリロニトリルと結晶性ビニルモノマーを必須構成単位とする結晶性ビニル樹脂。
【請求項2】
結晶性樹脂(a1)の融点が50〜110℃である請求項1に記載の樹脂粒子。
【請求項3】
親水性基を有する界面活性物質を実質的に含有しない請求項1または2に記載の樹脂粒子。
【請求項4】
請求項1〜3のいずれか1項に記載の樹脂粒子を含有する電子写真トナー用樹脂粒子。
【請求項5】
請求項4に記載の電子写真トナー用樹脂粒子を含有する電子写真トナー。
【請求項6】
該電子写真トナー用樹脂粒子にさらに外添剤が添加されてなる請求項5に記載の電子写真トナー。
【請求項7】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)を含有する樹脂粒子(B)の表面に微粒子(A)が固着した樹脂粒子(C)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項8】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)を溶剤(S)に溶解させた溶液(L)を分散させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項9】
微粒子(A)が分散された、液状又は超臨界状態の二酸化炭素(X)中に、樹脂(b)の前駆体(b0)を溶剤(S)に溶解させた溶液(L0)を分散させ、さらに前駆体(b0)を反応させることにより、樹脂(b)と溶剤(S)を含有する樹脂粒子(B1)の表面に微粒子(A)が固着した樹脂粒子(C1)を形成させ、次いで液状又は超臨界状態の二酸化炭素(X)と溶剤(S)を除去することにより樹脂粒子(C)を得る工程を含む請求項1〜3のいずれか1項に記載の樹脂粒子の製造方法。
【請求項10】
樹脂粒子(C1)を形成させた後、溶剤(S)を含有する液状又は超臨界状態の二酸化炭素(X)を、溶剤(S)を含有しない二酸化炭素で置換し、(C1)中に含有される溶剤(S)を除去又は減少させ、その後、減圧して二酸化炭素を除去する請求項8又は9記載の製造方法。
【請求項11】
23℃、0.1MPaの標準状態における、溶剤(S)と、樹脂(b)又は前駆体(b0)との等重量混合物における樹脂(b)又は前駆体(b0)の不溶分の重量が、樹脂(b)又は前駆体(b0)の重量に対して20重量%以下であり、溶剤(S)の溶解性パラメーターが9〜16である請求項8〜10のいずれか1項に記載の製造方法。
【請求項12】
溶剤(S)が、環状エーテル、ピルビン酸エステル、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、2−ヒドロキシイソ酪酸エステル、乳酸エステル、フッ素含有アルコール、アセトンとメタノールと水の混合溶剤、アセトンとメタノールの混合溶剤、アセトンとエタノールの混合溶剤、アセトンと水の混合溶剤、又はメチルエチルケトンと水の混合溶剤である請求項8〜11のいずれか1項に記載の製造方法。
【請求項13】
液状又は超臨界状態の二酸化炭素(X)中への分散工程において、ジメチルシロキサン基及びフッ素を含有する官能基の少なくとも一方の基を有する分散安定剤(E)を使用する請求項7〜12のいずれか1項に記載の製造方法。
【図1】

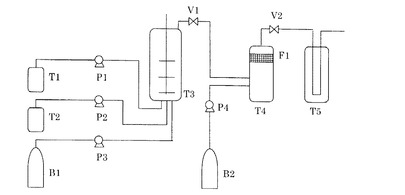
【公開番号】特開2013−49855(P2013−49855A)
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願番号】特願2012−217266(P2012−217266)
【出願日】平成24年9月28日(2012.9.28)
【分割の表示】特願2009−70240(P2009−70240)の分割
【原出願日】平成21年3月23日(2009.3.23)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願日】平成24年9月28日(2012.9.28)
【分割の表示】特願2009−70240(P2009−70240)の分割
【原出願日】平成21年3月23日(2009.3.23)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
[ Back to top ]