
樹脂粒子及び樹脂分散体
【課題】 溶融特性と高温高湿度下での耐ブロッキング性に優れ、粒径が均一である樹脂粒子を提供する。
【解決手段】 樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られた、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
【解決手段】 樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られた、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は樹脂分散体および樹脂粒子に関する。さらに詳しくは、粉体塗料、電子写真トナー、静電記録トナー等の各種用途に有用な、樹脂粒子およびその水性分散体に関する。
【背景技術】
【0002】
粒径が均一で、かつ、電気的特性、熱的特性、化学的安定性等に優れた樹脂粒子として、ポリマー微粒子を分散安定剤として得られた樹脂粒子が知られている(特許文献1参照)。
【特許文献1】特開2002−284881号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
しかしながら、樹脂粒子の溶融特性を向上させる(より低温で溶融させる)ためには、分子量やガラス転移温度(以下Tgと略す)を下げる必要があるが、そうした場合、高温高湿度下での樹脂粒子の耐ブロッキング性が劣るという問題点を有していた。
本発明は従来技術における上記の事情に鑑みてなされたものである。すなわち、溶融特性と耐ブロッキング性に優れ、粒径が均一である樹脂粒子を提供することを目的とする。
【課題を解決するための手段】
【0004】
本発明者らは、上記の問題点を解決するべく鋭意検討した結果、本発明に到達した。
すなわち本発明は、下記〔I〕〜〔III〕である。
〔I〕 樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られる、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
Ti(−X)m(−OR)n (I)
[式中、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、他のカルボキシル基が同一分子内のOR基と分子内で重縮合し環構造を形成していてもよく、または、別の分子のOR基と分子間で重縮合し2〜5個のTi原子を含む構造を形成していてもよい。m=1〜3、n=1〜3であり、mとnの和は4である。]
〔II〕 上記水性樹脂分散体(X1)中において、樹脂粒子(B)に付着された樹脂粒子(A)が、有機溶剤に溶解される、および/または、溶融されることにより、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されて得られる樹脂粒子(C2)からなる水性樹脂分散体(X2)。
〔III〕 〔I〕または〔II〕の水性樹脂分散体から水性媒体が除去されてなる樹脂粒子。
【発明の効果】
【0005】
本発明の樹脂分散体およびそれから得られる樹脂粒子は以下の効果を有する。
1.溶融特性、耐ブロッキング性に優れ、粒径が均一である。
2.環境上問題あるスズ化合物を触媒として使用しなくとも、良好な樹脂性能を有する。
3.帯電特性、粉体流動性に優れる。
4.粒子表面の平滑性に優れる(特に皮膜化されたシェル層を有する場合)。
5.水中で分散により得ることが可能な樹脂粒子であるため、低コストで製造できる。
6.加熱溶融した塗膜の機械的物性も良好である。
【発明を実施するための最良の形態】
【0006】
以下、本発明について詳細に説明する。
本発明に用いるチタン含有触媒(e)は、前記式(I)で表される化合物であり、式(I)の構造のものを2種以上併用してもよい。
一般式(I)において、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。炭化水素基の炭素数は、好ましくは1〜6、さらに好ましくは1〜4である。
炭素数1〜24の炭化水素基の具体例としては、脂肪族炭化水素基並びにエーテル結合および/もしくは水酸基を含む脂肪族炭化水素基(メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、n−ヘキシル基、n−オクチル基、β−メトキシエチル基、β−エトキシエチル基、およびβ−ヒドロキシエチル基など)、芳香族炭化水素基並びにエーテル結合および/もしくは水酸基を含む芳香族炭化水素基[フェニル基;ヒドロキシフェニル基;ビスフェノールA、ビスフェノールFおよびビスフェノールADなどの炭素数2〜4のアルキレンオキシド(以下、特記しない限り、AOと略記する)〔エチレンオキシド(以下EOと略記する)、プロピレンオキシド(以下POと略記する)、およびブチレンオキシド(以下BOと略記する)など〕付加物(付加モル数1〜3)から1個のOHを除いた残基など]が挙げられる。
これらRのうち好ましくは、炭素数1〜6の炭化水素基であり、さらに好ましくは、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、およびn−ヘキシル基であり、とくに好ましくは、n−プロピル基、イソプロピル基、およびn−ブチル基である。
【0007】
Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、Ti原子に結合し残基を形成するのと別のカルボキシル基が、同一分子内のOR基{Ti原子に直接結合した水酸基(RがHの場合)、アルコキシ基(Rが炭化水素基の場合)、またはRが1〜2個の水酸基を含む炭化水素基の場合の該水酸基}と分子内で重縮合し環構造を形成していてもよく、チタン含有触媒(a)の別の分子のOR基(上記と同様)と分子間で重縮合し、複数のTi原子を含む繰り返し構造を形成していてもよい。
上記芳香族カルボン酸としては、炭素数7〜50のものが好ましく、安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸など)、ナフタレンモノカルボン酸などの芳香族モノカルボン酸;フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸など)、ナフタレンジカルボン酸、トリメリット酸、およびピロメリット酸などの2〜6価またはそれ以上の芳香族ポリカルボン酸;が挙げられる。
芳香族ポリカルボン酸の場合、前述のようにその複数のカルボキシル基により、複数のTi原子を含む繰り返し構造を形成していてもよいが、この場合の1分子内のTi原子数は2〜5である。1分子内のTi原子数が6以上の場合、触媒活性が低下し好ましくない。
Xとして好ましいものは、フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸など)の残基、および安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸など)の残基であり、特に好ましいものはテレフタル酸、イソフタル酸、およびオルトフタル酸の残基である。
【0008】
式(I)中、m=1〜3、n=1〜3であり、mとnの和、すなわちTi原子の結合価数は4である。好ましくは、m=1〜2、n=2〜3である。mが3を超えると触媒活性が低下し、nが3を超えると耐加水分解性が低下し、いずれもポリエステル製造上好ましくない。mが1または2の場合、触媒活性が特に高く好ましい。Ti原子の結合価数が4以外の場合は、式(I)と類似の構造でも触媒活性が劣るか副反応が起き好ましくない。
【0009】
一般式(I)で表される化合物の具体例としては、チタントリイソプロポキシベンゼンカルボキシレート、チタントリブトキシベンゼンカルボキシレート、チタントリイソプロポキシテレフタレート、チタントリブトキシテレフタレート、チタントリイソプロポキシイソフタレート、チタントリイソプロポキシフタレート、チタンジイソプロポキシジベンゼンカルボキシレート、チタンジブトキシジベンゼンカルボキシレート、チタンジイソプロポキシジテレフタレート、チタンジブトキシジテレフタレート、チタンジイソプロポキシジイソフタレート、チタンジイソプロポキシジフタレート、チタンジヒドロキシジベンゼンカルボキシレート、チタンジヒドロキシジテレフタレート、チタンジヒドロキシジイソフタレート、チタンジヒドロキシジフタレート、チタンイソプロポキシトリテレフタレート、およびこれらの分子内または分子間重縮合物などが挙げられる。
【0010】
本発明に用いるチタン含有触媒(e)は、ポリエステル重合時の触媒活性の観点から、30℃の水への溶解度が[5g/100ml]以下であることが好ましく、[2g/100ml]以下であることがさらに好ましく、[1g/100ml]以下であることがとくに好ましい。溶解度が[5g/100ml]以下であると、重合反応時に触媒が加水分解を受けにくく、触媒活性の持続性の観点から好ましい。
【0011】
これらのチタン含有触媒(e)は、例えば、市販されているチタンテトラアルコキシドと芳香族カルボン酸を、酢酸エチル中で70〜90℃にて反応させることで得ることができる。
【0012】
本発明に用いるポリエステル樹脂(b1)を構成する重縮合ポリエステル樹脂としては、ポリオールとポリカルボン酸との重縮合物であるポリエステル樹脂(bX)、(bX)にさらにポリエポキシド(f)などを反応させて得られる変性ポリエステル樹脂(bY)などが挙げられる。(bX)、(bY)などは単独で使用してもよいし、2種以上を組み合わせて混合物として使用してもよい。
ポリオールとしては、ジオール(g)および3価以上のポリオール(h)が、ポリカルボン酸としては、ジカルボン酸(i)および3価以上のポリカルボン酸(j)が挙げられ、それぞれ2種以上を併用してもよい。
ポリエステル樹脂(bX)および(bY)としては、以下のものなどが挙げられ、これらのものを併用することもできる。
(bX1):(g)および(i)を用いた線状のポリエステル樹脂
(bX2):(g)および(i)とともに(h)および/または(j)を用いた非線状のポリエステル樹脂
(bY1):(bX2)に(f)を反応させた変性ポリエステル樹脂
【0013】
ジオール(g)としては、水酸基価180〜1900(mgKOH/g、以下同様)のものが好ましい。具体的には、アルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、オクタンジオール、デカンジオール、ドデカンジオール、テトラデカンジオール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);アルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコールおよびポリブチレングリコールなど);脂環式ジオール(1,4−シクロヘキサンジメタノールおよび水素添加ビスフェノールAなど);上記脂環式ジオールの炭素数2〜4のアルキレンオキシド〔エチレンオキシド(以下EOと略記する)、プロピレンオキシド(以下POと略記する)およびブチレンオキシド(以下BOと略記する)など〕付加物(付加モル数1〜30);ビスフェノール類(ビスフェノールA、ビスフェノールFおよびビスフェノールSなど)の炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数2〜30)などが挙げられる。
これらのうち好ましいものは、炭素数2〜12のアルキレングリコール、ビスフェノール類のアルキレンオキシド付加物およびこれらの併用であり、とくに好ましいものはビスフェノール類のアルキレンオキシド付加物、炭素数2〜4のアルキレングリコールおよびこれらの2種以上の併用であり、もっとも好ましいものは、炭素数2〜4のアルキレングリコールである。
なお、上記および以下において水酸基価および酸価は、JIS K 0070に規定の方法で測定される。
【0014】
ジオール(g)としては、上記のヒドロキシル基以外の官能基を有しないジオール以外に、他の官能基を有するジオール(g1)を用いてもよい。(g1)としては、カルボキシル基を有するジオール、スルホン酸基もしくはスルファミン酸基を有するジオール、およびこれらの塩等が挙げられる。
カルボキシル基を有するジオールとしては、ジアルキロールアルカン酸[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸(DMPA)、2,2−ジメチロールブタン酸、2 ,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸など]が挙げられる。
スルホン酸基もしくはスルファミン酸基を有するジオールとしては、スルファミン酸ジオール[N,N−ビス(2−ヒドロキシアルキル)スルファミン酸(アルキル基のC1〜6)またはそのAO付加物(AOとしてはEOまたはPOなど、AOの付加モル数1〜6):例えばN,N−ビス(2−ヒドロキシエチル)スルファミン酸およびN,N−ビス(2−ヒドロキシエチル)スルファミン酸PO2モル付加物など];ビス(2−ヒドロキシエチル)ホスフェートなどが挙げられる。
これらの中和塩基を有するジオールの中和塩基としては、例えば前記炭素数3〜30の3級アミン(トリエチルアミンなど)および/またはアルカリ金属(ナトリウム塩など)が挙げられる。
これらのうち好ましいものは、炭素数2〜12のアルキレングリコール、カルボキシル基を有するジオール、ビスフェノール類のAO付加物、およびこれらの併用である。
【0015】
3価以上(3〜8価またはそれ以上)のポリオール(h)としては、水酸基価150〜1900のものが好ましい。具体的には、炭素数3〜36の3〜8価またはそれ以上の脂肪族多価アルコール(アルカンポリオールおよびその分子内もしくは分子間脱水物、例えば、グリセリン、トリエチロールエタン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール、ソルビタン、ポリグリセリン、およびジペンタエリスリトール;糖類およびその誘導体、例えば蔗糖およびメチルグルコシド;など);上記脂肪族多価アルコールの炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数1〜30);トリスフェノール類(トリスフェノールPAなど)の炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数2〜30);ノボラック樹脂(フェノールノボラックおよびクレゾールノボラックなど:平均重合度3〜60)の炭素数2〜4のアルキレンオキシド(EO、PO、BOなど)付加物(付加モル数2〜30)などが挙げられる。
これらのうち好ましいものは、3〜8価またはそれ以上の脂肪族多価アルコールおよびノボラック樹脂のアルキレンオキシド付加物(付加モル数2〜30)であり、とくに好ましいものはノボラック樹脂のアルキレンオキシド付加物である。
【0016】
ジカルボン酸(i)としては、酸価180〜1250(mgKOH/g、以下同様)のものが好ましい。具体的には、アルキレンジカルボン酸(コハク酸、アジピン酸、セバシン酸、ドデセニルコハク酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸など);アルケニレンジカルボン酸(マレイン酸、フマール酸など);炭素数8以上の分岐アルキレンジカルボン酸[ダイマー酸、アルケニルコハク酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸など)、アルキルコハク酸(デシルコハク酸、ドデシルコハク酸、オクタデシルコハク酸など);芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、ナフタレンジカルボン酸など)などが挙げられる。これらのうち好ましいものは、炭素数4〜20のアルケニレンジカルボン酸および炭素数8〜20の芳香族ジカルボン酸である。なお、(i)としては、上述のものの酸無水物または低級アルキル(炭素数1〜4)エステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0017】
3価以上(3〜6価またはそれ以上)のポリカルボン酸(j)としては、酸価150〜1250のものが好ましい。具体的には、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など);不飽和カルボン酸のビニル重合体[数平均分子量(以下Mnと記載、ゲルパーミエーションクロマトグラフィー(GPC)による):450〜10000](スチレン/マレイン酸共重合体、スチレン/アクリル酸共重合体、α−オレフィン/マレイン酸共重合体、スチレン/フマル酸共重合体など)などが挙げられる。これらのうち好ましいものは、炭素数9〜20の芳香族ポリカルボン酸であり、とくに好ましいものはトリメリット酸、およびピロメリット酸である。なお、3価以上のポリカルボン酸(j)としては、上述のものの酸無水物または低級アルキル(炭素数1〜4)エステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0018】
また、(g)、(h)、(i)および(j)とともに炭素数4〜20の脂肪族または芳香族ヒドロキシカルボン酸(k)、炭素数6〜12のラクトン(l)を共重合することもできる。
ヒドロキシカルボン酸(k)としては、ヒドロキシステアリン酸、硬化ヒマシ油脂肪酸などが挙げられる。ラクトン(l)としては、カプロラクトンなどが挙げられる。
【0019】
ポリエポキシド(f)としては、ポリグリシジルエーテル〔エチレングリコールジグリシジルエーテル、テトラメチレングリコールジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールFジグリシジルエーテル、グリセリントリグリシジルエーテル、ペンタエリスリトールテトラグリシジルエーテル、フェノールノボラック(平均重合度3〜60)グリシジルテーテル化物など〕;ジエンオキサイド(ペンタジエンジオキサイド、ヘキサジエンジオキサイドなど)などが挙げられる。これらの中で好ましくは、ポリグリシジルエーテルであり、さらに好ましくは、エチレングリコールジグリシジルエーテルおよびビスフェノールAジグリシジルエーテルである。
(f)の1分子当たりのエポキシ基数は、好ましくは2〜8、さらに好ましくは2〜6、とくに好ましくは2〜4である。
(f)のエポキシ当量は、好ましくは50〜500である。下限は、さらに好ましく70、とくに好ましくは80であり、上限は、さらに好ましく300、とくに好ましくは200である。エポキシ基数とエポキシ当量が上記範囲内であると、現像性と定着性が共に良好である。上述の1分子当たりのエポキシ基数およびエポキシ当量の範囲を同時に満たせばさらに好ましい。
【0020】
ポリオールとポリカルボン酸の反応比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/2、さらに好ましくは1.5/1〜1/1.3、とくに好ましくは1.3/1〜1/1.2である。また使用するポリオールとポリカルボン酸の種類は、最終的に調整される樹脂粒子のガラス転移点が45〜85℃となるよう分子量調整も考慮して選択される。
【0021】
本発明において、樹脂(b)中の分子量1500以下の成分の比率は、1.8%以下が好ましく、さらに好ましくは1.3%以下、とくに好ましくは1.1%以下である。分子量1500以下の成分の比率が1.8%以下になることで、保存安定性がより良好となる。上記および以下において、%は特に記載のない限り、重量%を意味する。
【0022】
上記および以下において、本発明における樹脂(ポリウレタン樹脂を除く)の、ピークトップ分子量(Mp)、Mn、重量平均分子量(Mw)、および分子量1500以下の成分の比率は、THF可溶分についてGPCを用いて以下の条件で測定される。
装置(一例) : 東ソー製 HLC−8120
カラム(一例): TSKgelGMHXL(2本)
TSKgelMultiporeHXL−M(1本)
試料溶液 : 0.25%のTHF溶液
溶液注入量 : 100μl
流量 : 1ml/分
測定温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
得られたクロマトグラム上最大のピーク高さを示す分子量をピークトップ分子量(Mp)と称する。さらに分子量1500で分割したときのピーク面積の比率で低分子量物の存在比を評価する。
なお、ポリウレタン樹脂のMp、Mn、Mw、および分子量1500以下の成分の比率は、GPCを用いて以下の条件で測定される。
装置(一例) : 東ソー製 HLC−8220GPC
カラム(一例): Guardcolumn α
TSKgel α−M
試料溶液 : 0.125%のジメチルホルムアミド溶液
溶液注入量 : 100μl
流量 : 1ml/分
温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
【0023】
ポリエステル樹脂(b1)の酸価は、好ましくは0.1〜60、さらに好ましくは0.2〜50、特に0.5〜40である。酸価が0.1〜60の範囲では、帯電性が良好である。
ポリエステル樹脂(b1)の水酸基価は、好ましくは1〜70、さらに好ましくは3〜60、特に5〜55である。酸価が1〜70の範囲では、環境安定性が良好である。
ポリエステル樹脂(b1)のTgは、好ましくは40〜90℃、さらに好ましくは50〜80℃、とくに55〜75℃である。Tgが40℃〜90℃の範囲では耐熱保存安定性と低温定着性が良好である。
なお、上記および以下においてポリエステル樹脂(b1)のTgは、セイコー電子工業(株)製DSC20,SSC/580を用いてASTM D3418−82に規定の方法(DSC法)で測定される。
【0024】
本発明においてポリエステル樹脂(b1)は、通常のポリエステルの製造法と同様にして製造することができる。例えば、不活性ガス(窒素ガス等)雰囲気中で、チタン含有触媒(e)の存在下、反応温度が好ましくは150〜280℃、さらに好ましくは160〜250℃、とくに好ましくは170〜240℃で反応させることにより行うことができる。また反応時間は、重縮合反応を確実に行う観点から、好ましくは30分以上、とくに2〜40時間である。反応末期の反応速度を向上させるために減圧する(例えば1〜50mmHg)ことも有効である。
(e)の添加量としては、重合活性などの観点から、得られる重合体の重量に対して、好ましくは0.0001〜0.8%、さらに好ましくは0.0002〜0.6%、とくに好ましくは0.0015〜0.55%である。
また、(e)の触媒効果を損なわない範囲で他のエステル化触媒を併用することもできる。他のエステル化触媒の例としては、スズ含有触媒(例えばジブチルスズオキシド)、三酸化アンチモン、(e)以外のチタン含有触媒〔例えばチタンアルコキシド、シュウ酸チタニルカリウム、テレフタル酸チタン、およびチタンジヒドロキシビス(トリエタノールアミネート)とその分子内重縮合物〕、ジルコニウム含有触媒(例えば酢酸ジルコニル)、ゲルマニウム含有触媒、アルカリ(土類)金属触媒(例えばアルカリ金属もしくはアルカリ土類金属のカルボン酸塩:酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、安息香酸ナトリウム、および安息香酸カリウムなど)、および酢酸亜鉛等が挙げられる。これらの他の触媒の添加量としては、得られる重合体に対して、0〜0.6%が好ましい。0.6%以内とすることで、ポリエステル樹脂の着色が少なくなり、カラー用のトナーに用いるのに好ましい。添加された全触媒中の(e)の含有率は、50〜100%が好ましい。
【0025】
線状のポリエステル樹脂(bX1)の製造方法としては、例えば、得られる重合体の重量に対して0.0001〜0.8%の触媒(e)と、必要により他の触媒の存在下、ジオール(g)、およびジカルボン酸(i)を、180℃〜260℃に加熱し、常圧および/または減圧条件で脱水縮合させて、(bX1)を得る方法が挙げられる。
【0026】
非線状のポリエステル樹脂(bX2)の製造方法としては、例えば、得られる重合体の重量に対して0.0001〜0.8%の触媒(e)と、必要により他の触媒の存在下、ジオール(g)、ジカルボン酸(i)、および3価以上のポリオール(h)を、180℃〜260℃に加熱し、常圧および/または減圧条件で脱水縮合させた後、さらに3価以上のポリカルボン酸(j)を反応させて、(bX2)を得る方法が挙げられる。(j)を、(g)、(i)および(h)と同時に反応させることもできる。
【0027】
変性ポリエステル樹脂(bY1)の製造方法としては、ポリエステル樹脂(bX2)にポリエポキシド(f)を加え、180℃〜260℃でポリエステルの分子伸長反応を行うことで、(bY1)を得る方法が挙げられる。
(f)と反応させる(bX2)の酸価は、好ましくは1〜60、さらに好ましくは5〜50である。酸価が1以上であると、(f)が未反応で残存して樹脂の性能に悪影響を及ぼす恐れがなく、60以下であると、樹脂の熱安定性が良好である。
また、(bY1)を得るのに用いる(f)の量は、低温定着性および耐ホットオフセット性の観点から、(bX2)に対して、好ましくは0.01〜10%、さらに好ましくは0.05〜5%である。
【0028】
ポリエステル樹脂を2種以上併用する場合、および少なくとも1種のポリエステル樹脂と他の樹脂を混合する場合、粉体混合または溶融混合してもよい。
溶融混合する場合の温度は、好ましくは80〜180℃、さらに好ましくは100〜170℃、とくに好ましくは120〜160℃である。
混合温度が低すぎると充分に混合できず、不均一となることがある。2種以上のポリエステル樹脂を混合する場合、混合温度が高すぎると、エステル交換反応による平均化などが起こるため、必要な樹脂物性が維持できなくなる場合がある。
【0029】
溶融混合する場合の混合時間は、好ましくは10秒〜30分、さらに好ましくは20秒〜10分、とくに好ましくは30秒〜5分である。2種以上のポリエステル樹脂を混合する場合、混合時間が長すぎると、エステル交換反応による平均化などが起こるため、必要な樹脂物性が維持できなくなる場合がある。
溶融混合する場合の混合装置としては、反応槽などのバッチ式混合装置、および連続式混合装置が挙げられる。適正な温度で短時間で均一に混合するためには、連続式混合装置が好ましい。連続式混合装置としては、エクストルーダー、コンティニアスニーダー、3本ロールなどが挙げられる。これらのうちエクストルーダーおよびコンティニアスニーダーが好ましい。
粉体混合する場合は、通常の混合条件および混合装置で混合することができる。
粉体混合する場合の混合条件としては、混合温度は、好ましくは0〜80℃、さらに好ましくは10〜60℃である。混合時間は、好ましくは3分以上、さらに好ましくは5〜60分である。混合装置としては、ヘンシェルミキサー、ナウターミキサー、およびバンバリーミキサー等が挙げられる。好ましくはヘンシェルミキサーである。
【0030】
また、ポリエステル樹脂(b1)を構成単位として含有する樹脂(b2)としては、(b1)を構成単位として含有するものであればとくに限定されないが、例えば、ポリウレタン樹脂の場合、後述する樹脂(a)の項で述べたポリウレタン樹脂において、活性水素基含有化合物(D)の少なくとも一部として、(b1)を用いることにより得られる。(D)中の(b1)の含有量は、好ましくは60%以上、さらに好ましくは80%以上、とくに好ましくは100%である。
【0031】
また、(b)中に、(b1)および(b2)以外に、必要により、他の樹脂を含有させることもできる。
他の樹脂としては、(e)以外の触媒の存在下に形成されたポリエステル樹脂、ビニル系樹脂、エポキシ樹脂、ポリウレタン樹脂などが挙げられる。その具体例については、後述する(a)と同様のものが使用できる。
他の樹脂のMwは、好ましくは1000〜200万である。
樹脂(b)における他の樹脂の含有量は、好ましくは0〜40%、さらに好ましくは0〜30%、とくに好ましくは0〜20%である。また、(b)中のポリエステル以外の樹脂の含有量は、樹脂物性の点から、好ましくは30%以下、さらに好ましくは20%以下、とくに好ましくは10%以下、最も好ましくは0%である。
【0032】
樹脂(b)のMn、Mw、融点、Tg、sp値は、用途によって好ましい範囲に適宜調整すればよい。
樹脂(b)のsp値は、樹脂(a)とのsp値差が上記の範囲内にあるのが好ましいが、好ましくは7〜18、さらに好ましくは8〜14、とくに好ましくは9〜14である。
例えば、樹脂粒子(C1)または(C2)〔以下(C1)または(C2)を(C)と記載することもある〕、樹脂粒子(B)をスラッシュ成形用樹脂、粉体塗料として用いる場合、(b)のMnは、好ましくは2,000〜50万、さらに好ましくは4,000〜20万である。
(b)の融点(DSCにて測定、以下融点はDSCでの測定値)、好ましくは0℃〜200℃、さらに好ましくは35℃〜150℃である。(b)のTgは、好ましくは−60℃〜100℃、さらに好ましくは−30℃〜60℃である。
液晶ディスプレイ等の電子部品製造用スペーサー、電子測定機の標準粒子として用いる場合、(b)のMnは、好ましくは2万〜1,000万、さらに好ましくは4万〜200万である。(b)の融点は、好ましくは40℃〜300℃、さらに好ましくは70℃〜250℃である。(b)のTgは好ましくは−0℃〜250℃、さらに好ましくは50℃〜200℃である。
電子写真、静電記録、静電印刷などに使用されるトナーとして用いる場合、(b)のMnは、好ましくは1,000〜500万、さらに好ましくは2,000〜50万である。(b)の融点は、好ましくは20℃〜300℃、さらに好ましくは80℃〜250℃である。(b)のTgは、好ましくは20℃〜200℃、さたに好ましくは40℃〜150℃である。(b)のsp値は、好ましくは8〜16、さらに好ましくは9〜14である。溶融性と画像定着性の両立という観点から、(b)の50〜90%(とくに60〜80%)が、テトラヒドロフラン可溶分のMwが2000〜12000である線状ポリエステルであることが好ましい。線状ポリエステルのMwは、さらに好ましくは4000〜11000、とくに好ましくは7000〜10000である。線状ポリエステルのMwがこの範囲内であると、溶融性と画像密着性のバランスが特に良好である。
【0033】
本発明においては、樹脂(a)としては水性分散体を形成しうる樹脂であればいかなる樹脂であっても使用でき、熱可塑性樹脂であっても熱硬化性樹脂であってもよい。(a)としては例えば、ビニル系樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂、ポリアミド樹脂、ポリイミド樹脂、ケイ素系樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、アニリン樹脂、アイオマー樹脂、ポリカーボネート樹脂等が挙げられる。(a)としては上記樹脂の2種以上を併用しても差し支えない。このうち好もしいのは、微細球状樹脂粒子の水性分散体が得られやすいという観点からポリウレタン樹脂、ビニル系樹脂、エポキシ樹脂、ポリエステル樹脂、およびそれらの併用である。
【0034】
ビニル系樹脂は、ビニル系モノマーを単独重合または共重合したポリマーである。ビニル系モノマーとしては、下記(1)〜(10)が挙げられる。
【0035】
(1)ビニル系炭化水素:
(1−1)脂肪族ビニル系炭化水素:アルケン類、例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン、前記以外のα−オレフィン等;アルカジエン類、例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン、1,7−オクタジエン。
(1−2)脂環式ビニル系炭化水素:モノ−もしくはジ−シクロアルケンおよびアルカジエン類、例えばシクロヘキセン、(ジ)シクロペンタジエン、ビニルシクロヘキセン、エチリデンビシクロヘプテン等;テルペン類、例えばピネン、リモネン、インデン等。
(1−3)芳香族ビニル系炭化水素:スチレンおよびそのハイドロカルビル(アルキル、シクロアルキル、アラルキルおよび/またはアルケニル)置換体、例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン、トリビニルベンゼン等;およびビニルナフタレン。
【0036】
(2)カルボキシル基含有ビニル系モノマーおよびその金属塩:
炭素数3〜30の不飽和モノカルボン酸、不飽和ジカルボン酸ならびにその無水物およびそのモノアルキル(炭素数1〜24)エステル、例えば(メタ)アクリル酸、(無水)マレイン酸、マレイン酸モノアルキルエステル、フマル酸、フマル酸モノアルキルエステル、クロトン酸、イタコン酸、イタコン酸モノアルキルエステル、イタコン酸グリコールモノエーテル、シトラコン酸、シトラコン酸モノアルキルエステル、桂皮酸等のカルボキシル基含有ビニル系モノマー。
【0037】
(3)スルホン基含有ビニル系モノマー、ビニル系硫酸モノエステル化物およびこれらの塩:炭素数2〜14のアルケンスルホン酸、例えばビニルスルホン酸、(メタ)アリルスルホン酸、メチルビニルスルホン酸、スチレンスルホン酸;およびその炭素数2〜24のアルキル誘導体、例えばα−メチルスチレンスルホン酸等;スルホ(ヒドロキシ)アルキル−(メタ)アクリレートもしくは(メタ)アクリルアミド、例えば、スルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸、3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸、アルキル(炭素数3〜18)アリルスルホコハク酸、ポリ(n=2〜30)オキシアルキレン(エチレン、プロピレン、ブチレン:単独、ランダム、ブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[ポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル等]、ポリオキシエチレン多環フェニルエーテル硫酸エステル、および下記一般式(1−1)〜(1−3)で示される硫酸エステルもしくはスルホン酸基含有モノマー;ならびそれらの塩等。
【0038】
O−(AO)nSO3H
|
CH2=CHCH2−OCH2CHCH2O−Ar−R (1−1)
CH=CH−CH3
|
R−Ar−O−(AO)nSO3H (1−2)
CH2COOR’
|
HO3SCHCOOCH2CH(OH)CH2OCH2CH=CH2 (1−3)
(式中、Rは炭素数1〜15のアルキル基、Aは炭素数2〜4のアルキレン基を示し、nが複数の場合同一でも異なっていてもよく、異なる場合はランダムでもブロックでもよい。Arはベンゼン環を示し、nは1〜50の整数を示し、R’はフッ素原子で置換されていてもよい炭素数1〜15のアルキル基を示す。)
【0039】
(4)燐酸基含有ビニル系モノマーおよびその塩:
(メタ)アクリロイルオキシアルキル(C1〜C24)燐酸モノエステル、例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート、フェニル−2−アクリロイロキシエチルホスフェート、(メタ)アクリロイルオキシアルキル(炭素数1〜24)ホスホン酸類、例えば2−アクリロイルオキシエチルホスホン酸。
【0040】
なお、上記(2)〜(4)の塩としては、金属塩、アンモニウム塩、およびアミン塩(4級アンモニウム塩を含む)が挙げられる。金属塩を形成する金属としては、Al、Ti、Cr、Mn、Fe、Zn、Ba、Zr、Ca、Mg、Na、およびK等が挙げられる。
好ましくはアルカリ金属塩、およびアミン塩であり、さらに好ましくは、ナトリウム塩および炭素数3〜20の3級モノアミンの塩である。
【0041】
(5)ヒドロキシル基含有ビニル系モノマー:
ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等
【0042】
(6)含窒素ビニル系モノマー:
(6−1)アミノ基含有ビニル系モノマー:アミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、t−ブチルアミノエチルメタクリレート、N−アミノエチル(メタ)アクリルアミド、(メタ)アリルアミン、モルホリノエチル(メタ)アクリレート、4ービニルピリジン、2ービニルピリジン、クロチルアミン、N,N−ジメチルアミノスチレン、メチルα−アセトアミノアクリレート、ビニルイミダゾール、N−ビニルピロール、N−ビニルチオピロリドン、N−アリールフェニレンジアミン、アミノカルバゾール、アミノチアゾール、アミノインドール、アミノピロール、アミノイミダゾール、アミノメルカプトチアゾール、これらの塩等
(6−2)アミド基含有ビニル系モノマー:(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド、N−ブチルアクリルアミド、ジアセトンアクリルアミド、N−メチロール(メタ)アクリルアミド、N,N’−メチレン−ビス(メタ)アクリルアミド、桂皮酸アミド、N,N−ジメチルアクリルアミド、N,N−ジベンジルアクリルアミド、メタクリルホルムアミド、N−メチルN−ビニルアセトアミド、N−ビニルピロリドン等
(6−3)ニトリル基含有ビニル系モノマー:(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等
(6−4)4級アンモニウムカチオン基含有ビニル系モノマー:ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリルアミド、ジエチルアミノエチル(メタ)アクリルアミド、ジアリルアミン等の3級アミン基含有ビニル系モノマーの4級化物(メチルクロライド、ジメチル硫酸、ベンジルクロライド、ジメチルカーボネート等の4級化剤を用いて4級化したもの)
(6−5)ニトロ基含有ビニル系モノマー:ニトロスチレン等
【0043】
(7)エポキシ基含有ビニル系モノマー:
グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等
【0044】
(8)ハロゲン元素含有ビニル系モノマー:
塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等
【0045】
(9)ビニルエステル、ビニル(チオ)エーテル、ビニルケトン、ビニルスルホン類:
(9−1)ビニルエステル、例えば酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ポリ(メタ)アリロキシアルカン類[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]等、ポリアルキレングリコール鎖を有するビニル系モノマー[ポリエチレングリコール(分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(分子量500)モノアクリレート、メチルアルコールエチレンオキサイド(エチレンオキサイドを以下EOと略記する)10モル付加物(メタ)アクリレート、ラウリルアルコールEO30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート類[多価アルコール類のポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等
(9−2)ビニル(チオ)エーテル、例えばビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル、ビニルブチルエーテル、ビニル2−エチルヘキシルエーテル、ビニルフェニルエーテル、ビニル2−メトキシエチルエーテル、メトキシブタジエン、ビニル2−ブトキシエチルエーテル、3,4−ジヒドロ1,2−ピラン、2−ブトキシ−2’−ビニロキシジエチルエーテル、ビニル2−エチルメルカプトエチルエーテル、アセトキシスチレン、フェノキシスチレン等
(9−3)ビニルケトン、例えばビニルメチルケトン、ビニルエチルケトン、ビニルフェニルケトン;
ビニルスルホン、例えばジビニルサルファイド、p−ビニルジフェニルサルファイド、ビニルエチルサルファイド、ビニルエチルスルフォン、ジビニルスルフォン、ジビニルスルフォキサイド等
【0046】
(10)その他のビニル系モノマー:
イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等
【0047】
本発明において、(a)として有機酸金属塩(m)の構成単位を含有するビニル系樹脂を用いる場合、この樹脂は、例えば、モノマーの少なくとも一部として、上記モノマー(2)〜(4)のうち、Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる金属の塩を、1種以上用いることにより得られる。これらの有機酸金属塩モノマー(m)の、重合に用いる全モノマー中の使用量は、好ましくは5〜60%である。下限はさらに好ましくは10%であり、上限はさらに好ましくは50%である。
【0048】
ビニル系モノマーの共重合体としては、上記(1)〜(10)の任意のモノマー同士を、2元またはそれ以上の個数で、好ましくは樹脂粒子(A)中のカルボキシル基の含量が0.1〜10%になるように、任意の割合で共重合したポリマーが挙げられるが、例えば、スチレン−(メタ)アクリル酸エステル−(メタ)アクリル酸共重合体、スチレン−ブタジエン−(メタ)アクリル酸共重合体、(メタ)アクリル酸−アクリル酸エステル共重合体、スチレン−アクリロニトリル−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸−ジビニルベンゼン共重合体、スチレン−スチレンスルホン酸−(メタ)アクリル酸エステル共重合体、およびこれらの共重合体の塩などが挙げられる。
【0049】
樹脂(a)は、水性分散体中で樹脂粒子(A)形成することが必要であることから、少なくとも水性樹脂分散体(X1)を形成する条件下で水に完全に溶解していないことが必要である。そのため、ビニル系樹脂が共重合体である場合には、ビニル系樹脂を構成する疎水性モノマーと親水性モノマーの比率は、選ばれるモノマーの種類によるが、一般に疎水性モノマーが10%以上であることが好ましく、30%以上であることがより好ましい。疎水性モノマーの比率が、10%以下になるとビニル系樹脂が水溶性になり、(C1)または(C2)の粒径均一性が損なわれる。ここで、親水性モノマーとは水に任意の割合で溶解するモノマーをいい、疎水性モノマーとは、それ以外のモノマー(基本的に水に混和しないモノマー)をいう。
【0050】
ポリエステル樹脂としては、ポリオールと、ポリカルボン酸またはその酸無水物またはその低級アルキルエステルとの重縮合物、およびこれらの重縮合物の金属塩などが挙げられる。ポリオールとしては前述のジオール(g)〔(g1)を含む〕および3価以上のポリオール(h)が、ポリカルボン酸またはその酸無水物またはその低級アルキルエステルとしては、ジカルボン酸(i)および3価以上のポリカルボン酸(j)およびこれらの酸無水物または低級アルキルエステルが挙げられる。
ポリオールとポリカルボン酸の比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/1.2、さらに好ましくは1.5/1〜1/1.1、とくに好ましくは1.3/1〜1/1.05である。
【0051】
本発明において、(a)として有機酸金属塩(m)の構成単位を含有するポリエステル樹脂を用いる場合、この樹脂は、例えば、COOHの残基を有するポリエステル(酸価が好ましくは1〜100、さらに好ましくは5〜50)を合成し、その少なくとも1部のCOOH基を、Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる少なくとも1種の金属の塩とすることにより得られる。
金属塩とする方法としては、例えば、COOH基を有するポリエステルと該当する金属の水酸化物とを反応することにより得られる。
【0052】
ポリウレタン樹脂としては、ポリイソシアネート(11)と活性水素含有化合物(D){水、ポリオール[ジオール(g)〔ヒドロキシル基以外の官能基を有するジオール(g1)を含む〕、および3〜8価またはそれ以上のポリオール(h)]、ポリカルボン酸[ジカルボン酸(i)、および3〜6価またはそれ以上のポリカルボン酸(j)]、ポリオールとポリカルボン酸の重縮合により得られるポリエステルポリオール、炭素数6〜12のラクトンの開環重合体、ポリアミン(12)、ポリチオール(13)、およびこれらの併用等}の重付加物、並びに(11)と活性水素含有化合物を反応させてなる末端イソシアネート基プレポリマーと、該プレポリマーのイソシアネート基に対して当量の1級および/または2級モノアミン(16)とを反応させて得られる、アミノ基含有ポリウレタン樹脂が挙げられる。
ポリウレタン樹脂中のカルボキシル基の含有量は、0.1〜10%が好ましい。
【0053】
ポリイソシアネート(11)としては、炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ポリイソシアネート、炭素数2〜18の脂肪族ポリイソシアネート、炭素数4〜15の脂環式ポリイソシアネート、炭素数8〜15の芳香脂肪族ポリイソシアネートおよびこれらのポリイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。
【0054】
上記芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)またはその混合物との縮合生成物;ジアミノジフェニルメタンと少量(たとえば5〜20%)の3官能以上のポリアミンとの混合物〕のホスゲン化物:ポリアリルポリイソシアネート(PAPI)]、1,5−ナフチレンジイソシアネート、4,4’,4”−トリフェニルメタントリイソシアネート、m−およびp−イソシアナトフェニルスルホニルイソシアネートなどが挙げられる。
上記脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエートなどの脂肪族ポリイソシアネートなどが挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−および/または2,6−ノルボルナンジイソシアネートなどが挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
また、上記ポリイソシアネートの変性物には、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。
具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのポリイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が含まれる。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、および炭素数4〜15の脂環式ポリイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0055】
ポリアミン(12)の例としては、脂肪族ポリアミン類(C2 〜C18):〔1〕脂肪族ポリアミン{C2〜C6 アルキレンジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン、ヘキサメチレンジアミンなど)、ポリアルキレン(C2〜C6)ポリアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミンなど〕};〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミンなど〕;〔3〕脂環または複素環含有脂肪族ポリアミン〔3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕;〔4〕芳香環含有脂肪族アミン類(C8 〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど)、脂環式ポリアミン(C4 〜C15):1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など、複素環式ポリアミン(C4 〜C15):ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジンなど、芳香族ポリアミン類(C6 〜C20):〔1〕非置換芳香族ポリアミン〔1,2−、1,3−および1,4−フェニレンジアミン、2,4´−および4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4”−トリアミン、ナフチレンジアミンなど;〔2〕核置換アルキル基〔メチル,エチル,n−およびi−プロピル、ブチルなどのC1〜C4アルキル基)を有する芳香族ポリアミン、たとえば2,4−および2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、3,3´,5,5´−テトラメチルベンジジン、3,3´,5,5´−テトラメチル−4,4´−ジアミノジフェニルメタン、3,5−ジエチル−3´−メチル−2´,4−ジアミノジフェニルメタン、3,3´−ジエチル−2,2´−ジアミノジフェニルメタン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、3,3´,5,5´−テトラエチル−4,4´−ジアミノベンゾフェノン、3,3´,5,5´−テトラエチル−4,4´−ジアミノジフェニルエーテル、3,3´,5,5´−テトライソプロピル−4,4´−ジアミノジフェニルスルホンなど〕、およびこれらの異性体の種々の割合の混合物;〔3〕核置換電子吸引基(Cl,Br,I,Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ポリアミン〔メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン;4,4´−ジアミノ−3,3´−ジメチル−5,5´−ジブロモ−ジフェニルメタン、3,3´−ジクロロベンジジン、3,3´−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキシド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4´−メチレンビス(2−ヨードアニリン)、4,4´−メチレンビス(2−ブロモアニリン)、4,4´−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリンなど〕;〔4〕2級アミノ基を有する芳香族ポリアミン〔上記〔1〕〜〔3〕の芳香族ポリアミンの−NH2の一部または全部が−NH−R´(R´はアルキル基たとえばメチル,エチルなどの低級アルキル基)で置き換ったもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕、ポリアミドポリアミン:ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど、ポリエーテルポリアミン:ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物などが挙げられる。
【0056】
ポリチオール(17)としては、炭素数2〜36のアルカンジチオール(エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなど)等が挙げられる。
【0057】
1級および/または2級モノアミン(18)としては、炭素数2〜24のアルキルアミン(エチルアミン、n−ブチルアミン、イソブチルアミンなど)等が挙げられる。
【0058】
本発明において、(a)として前記有機酸金属塩(m)の構成単位を含有するポリウレタン樹脂を用いる場合、この樹脂は、例えば、ジオール(g)、(g1)および/または3価以上のポリオール(h)の少なくとも一部として、カルボン酸基、スルホン酸基、リン酸基、およびこれらの酸の上記金属塩基から選ばれる1種以上の基を有するジオールおよび/または3価以上のポリオールを用いることにより得られる。カルボン酸基、スルホン酸基、またはリン酸基を有するジオールおよび/またはポリオールを用いる場合は、ポリウレタン樹脂形成後に、例えば、さらにこれらの酸基と該当する金属の水酸化物とを反応することにより、上記有機酸金属塩(m)の構成単位を含有するポリウレタン樹脂が得られる。
カルボン酸(塩)基を有するジオールまたはポリオールとしては、ジアルキロールアルカン酸(塩)[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸、2,2−ジメチロールブタン酸、2,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸、およびこれらの酸の前記金属塩など]等が挙げられる。
スルホン酸(塩)基を有するジオールまたはポリオールとしては、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸、スルホイソフタル酸ジ(エチレングリコール)エステル、およびこれらの酸の前記金属塩などが挙げられる。
リン酸(塩)基を有するジオールまたはポリオールとしては、ビス(2−ヒドロキシエチル)ホスフェートおよびその前記金属塩などが挙げられる。
これらの有機酸金属(塩)基を有するジオールまたは3価以上のポリオールの、(g)、(g1)および(h)の合計量に対する使用量は、好ましくは10〜90モル%である。下限は、さらに好ましくは20モル%であり、上限は、さらに好ましくは60モル%である。
【0059】
エポキシ樹脂としては、ポリエポキシド(14)の開環重合物、ポリエポキシド(14)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(g)、(g1)および3価以上のポリオール(h)]、ジカルボン酸(i)、3価以上のポリカルボン酸(j)、ポリアミン(12)、ポリチオール(13)等}との重付加物、またはポリエポキシド(14)とジカルボン酸(i)または3価以上のポリカルボン酸(j)の酸無水物との硬化物などが挙げられる。
【0060】
本発明に用いるポリエポキシド(14)は、分子中に2個以上のエポキシ基を有していれば、特に限定されない。ポリエポキシド(14)として好ましいものは、硬化物の機械的性質の観点から分子中にエポキシ基を2〜6個有するものである。ポリエポキシド(14)のエポキシ当量(エポキシ基1個当たりの分子量)は、好ましくは65〜1000であり、さらに好ましくは90〜500である。エポキシ当量が1000を超えると、架橋構造がルーズになり硬化物の耐水性、耐薬品性、機械的強度等の物性が悪くなり、一方、エポキシ当量が65未満のものを合成するのは困難である。
【0061】
ポリエポキシド(14)の例としては、芳香族系ポリエポキシ化合物、複素環系ポリエポキシ化合物、脂環族系ポリエポキシ化合物あるいは脂肪族系ポリエポキシ化合物が挙げられる。芳香族系ポリエポキシ化合物としては、多価フェノール類のグリシジルエーテル体およびグリシジルエステル体、グリシジル芳香族ポリアミン、並びに、アミノフェノールのグリシジル化物等が挙げられる。多価フェノールのグリシジルエーテル体としては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールBジグリシジルエーテル、ビスフェノールADジグリシジルエーテル、ビスフェノールSジグリシジルエーテル、ハロゲン化ビスフェノールAジグリシジル、テトラクロロビスフェノールAジグリシジルエーテル、カテキンジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ハイドロキノンジグリシジルエーテル、ピロガロールトリグリシジルエーテル、1,5−ジヒドロキシナフタリンジグリシジルエーテル、ジヒドロキシビフェニルジグリシジルエーテル、オクタクロロ−4,4’−ジヒドロキシビフェニルジグリシジルエーテル、テトラメチルビフェニルジグリシジルエーテル、ジヒドロキシナフチルクレゾールトリグリシジルエーテル、トリス(ヒドロキシフェニル)メタントリグリシジルエーテル、ジナフチルトリオールトリグリシジルエーテル、テトラキス(4−ヒドロキシフェニル)エタンテトラグリシジルエーテル、p−グリシジルフェニルジメチルトリールビスフェノールAグリシジルエーテル、トリスメチル−tret−ブチル−ブチルヒドロキシメタントリグリシジルエーテル、9,9’−ビス(4−ヒドキシフェニル)フロオレンジグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)テトラクレゾールグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)フェニルグリシジルエーテル、ビス(ジヒドロキシナフタレン)テトラグリシジルエーテル、フェノールまたはクレゾールノボラック樹脂のグリシジルエーテル体、リモネンフェノールノボラック樹脂のグリシジルエーテル体、ビスフェノールA2モルとエピクロロヒドリン3モルの反応から得られるジグリシジルエーテル体、フェノールとグリオキザール、グルタールアルデヒド、またはホルムアルデヒドの縮合反応によって得られるポリフェノールのポリグリシジルエーテル体、およびレゾルシンとアセトンの縮合反応によって得られるポリフェノールのポリグリシジルエーテル体等が挙げられる。多価フェノールのグリシジルエステル体としては、フタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、テレフタル酸ジグリシジルエステル等が挙げられる。グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。さらに、本発明において前記芳香族系として、P−アミノフェノールのトリグリシジルエーテル、トリレンジイソシアネートまたはジフェニルメタンジイソシアネートとグリシドールの付加反応によって得られるジグリシジルウレタン化合物、前記2反応物にポリオールも反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマーおよびビスフェノールAのアルキレンオキシド(エチレンオキシドまたはプロピレンオキシド)付加物のジグリシジルエーテル体も含む。複素環系ポリエポキシ化合物としては、トリスグリシジルメラミンが挙げられる;脂環族系ポリエポキシ化合物としては、ビニルシクロヘキセンジオキシド、リモネンジオキシド、ジシクロペンタジエンジオキシド、ビス(2,3−エポキシシクロペンチル)エーテル、エチレングリコールビスエポキシジシクロペンチルエール、3,4−エポキシ−6−メチルシクロヘキシルメチル−3’,4’−エポキシ−6’−メチルシクロヘキサンカルボキシレート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)アジペート、およびビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)ブチルアミン、ダイマー酸ジグリシジルエステル等が挙げられる。また、脂環族系としては、前記芳香族系ポリエポキシド化合物の核水添化物も含む;脂肪族系ポリエポキシ化合物としては、多価脂肪族アルコールのポリグリシジルエーテル体、多価脂肪酸のポリグリシジルエステル体、およびグリシジル脂肪族アミンが挙げられる。多価脂肪族アルコールのポリグリシジルエーテル体としては、エチレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、テトラメチレングリコールジグリシジルエーテル、1,6−ヘキサンジオールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、ポリテトラメチレングリコールジグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、グリセロールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ソルビトールポリグリシジルエーテルおよびポリグリセロールンポリグリシジルエーテル等が挙げられる。多価脂肪酸のポリグリシジルエステル体としては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート、ジグリシジルピメレート等が挙げられる。グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミンが挙げられる。また、本発明において脂肪族系としては、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含む。これらのうち、好ましいのは、脂肪族系ポリエポキシ化合物および芳香族系ポリエポキシ化合物である。本発明のポリエポキシドは、2種以上併用しても差し支えない。
【0062】
本発明においては、帯電特性の点から、樹脂(a)の構成単位として前記Al、Ti、Cr、Mn、Fe、Zn、Ba、Zrから選ばれる金属の有機酸金属塩(m)の単位を含有する、および/または、樹脂粒子(A)中に前記有機酸金属塩(m)を含有することが好ましい。有機酸金属塩(m)を構成する有機酸としてはカルボン酸が好ましい。金属の中では、ZnおよびFeが好ましく、さらに好ましくはZnである。
これらのうち、(A)中に有機酸金属塩(m)を含有することが、調整が容易な点から好ましい。
【0063】
有機酸金属塩(m)のうち、カルボン酸金属塩の具体例としては、(アルキル置換)安息香酸、(アルキル置換)サルチル酸、ナフテン酸等の炭素数7〜35の芳香族脂肪酸、およびステアリル酸等の炭素数12〜35の高級脂肪酸、並びに、前記の樹脂(a)を構成するカルボキシル基含有ビニルモノマーの金属塩として例示したものが好ましい。スルホン酸金属塩の具体例としては、炭素数7〜30のアルキルベンゼンスルホン酸、および(a)を構成するスルホン基含有ビニルモノマーの金属塩として例示したものが好ましい。リン酸金属塩としては(a)を構成するリン酸基含有ビニルモノマーの金属塩として例示したものが好ましい。これらの中でも、油溶性の金属塩が、樹脂粒子(C)の帯電性の効果を発揮する上で、さらに好ましい。組成の面からは、カルボン酸金属塩がさらに好ましく、カルボン酸のZn塩およびFe塩が特に好ましい。
(A)中に有機酸金属塩(m)を含有させる方法としては、樹脂(a)の前駆体と反応性がなければ(a)の作成前に投入してもよいが、(a)の作成後に(a)と混合する方が好ましい。
これらの有機酸金属塩(m)の構成単位、および有機酸金属塩(m)の使用割合に特に制限はないが、得られる樹脂粒子(C)に対するこれらの合計量が0.01〜10%であることが好ましい。下限は、さらに好ましくは0.05%であり、上限は、さらに好ましくは1%である。
【0064】
本発明において、微細な球状樹脂粒子(A)の水性分散液(W)を得るため、かつ耐熱保存安定性、帯電特性に優れ、粒径が均一な樹脂粒子(C1)の水性分散体(X1)を得るために、樹脂(a)は、スルホン酸アニオン基(−SO3-)を含有することが好ましい。スルホン酸アニオン基(−SO3-)の合計含有量は、(a)の重量に基づいて0.001〜10%が好ましい。下限は、さらに好ましくは0.002%であり、上限は、さらに好ましくは5%、とくに好ましくは2%、最も好ましくは1%である。また、樹脂を形成するスルホン酸アニオン基(−SO3-)を含有するモノマーの好ましい炭素数は3〜50、更に好ましくは3〜30、特に好ましくは4〜15である。
スルホン酸アニオン基(−SO3-)基含有量が上記範囲の下限以上や樹脂を形成するスルホン酸アニオン基(−SO3-)を含有するモノマーの炭素数が上記範囲の上限以下であると、樹脂(a)が水系媒体中に分散しやすく、微細な球状の樹脂粒子(A)の水性分散体(W)を容易に得ることができる。また、得られる樹脂粒子(C1)または(C2)の耐ブロッキング性、及び帯電特性が向上する。
【0065】
また、本発明において、微細な球状樹脂粒子(A)の水性分散体(W)を得るために、樹脂(a)は、カルボキシル基を含有することが好ましい。カルボキシル基はその少なくとも一部が塩基で中和されていてもよい。カルボキシル基の塩基中和率は、20〜100当量%が好ましく、40〜100当量%がさらに好ましい。
カルボキシル基の含有量〔塩基で中和されている場合は、カルボキシル基(−COOH基)に換算した含有量〕は、(a)の重量に基づいて0.1〜10%が好ましい。下限は、さらに好ましくは0.5%、とくに好ましくは1%、最も好ましくは3%であり、上限は、さらに好ましくは9.5%、とくに好ましくは9%、最も好ましくは8%である。
塩基中和率や、カルボキシル基含有量が上記範囲の下限以上であると、樹脂(a)が水系媒体中に分散しやすく、微細な球状の樹脂粒子(A)の水性分散体(W)を容易に得ることができる。また、得られる樹脂粒子(C)の帯電特性が向上する。
【0066】
上記の中和塩を形成する塩基としては、アンモニア、炭素数1〜30のモノアミン、後述のポリアミン(21)、4級アンモニウム、アルカリ金属(ナトリウム、カリウム等)、およびアルカリ土類金属(カルシウム塩、マグネシウム塩等)などが挙げられる。
上記炭素数1〜30のモノアミンとしては、炭素数1〜30の1級および/または2級アミン(エチルアミン、n−ブチルアミン、イソブチルアミン等)、炭素数3〜30の3級アミン(トリメチルアミン、トリエチルアミン、ラウリルジメチルアミン等)が挙げられる。4級アンモニウムとしては炭素数4〜30のトリアルキルアンモニウム(ラウリルトリメチルアンモニウム等)などが挙げられる。
これらの中で、好ましくは、アルカリ金属、4級アンモニウム、モノアミン、およびポリアミンであり、さらに好ましくは、ナトリウム、および炭素数1〜20のモノアミンであり、とくに好ましくは、炭素数3〜20の3級モノアミンである。
また、ビニル系樹脂、およびポリエステル樹脂を形成するカルボキシル基またはその塩を含有するモノマーの好ましい炭素数は3〜30であり、さらに好ましくは3〜15、とくに好ましくは3〜8である。
【0067】
本発明においては、樹脂(a)と樹脂(b)のsp値差(Δsp)をKとし、樹脂(a)の重量平均分子量(Mw)の自然対数値をln(Mw)を Hとした時、点(K、H)が、図1に示す下記4点A、B、C、Dからなる四角形ABCDの辺上を含む内部に含まれるように、(a)および(b)が選択されるのが好ましい。
A(0.3、ln3000)、B(1.5、ln1000)、
C(1.3、ln200000)、D(0.1、ln200000)
点(K、H)は、以下の4点A’、B’、C’、D’からなる四角形A’B’C’D’の辺上を含む内部に含まれることがさらに好ましく、以下の4点A”、B”、C”、D”からなる四角形A”B”C”D”の辺上を含む内部に含まれることがとくに好ましい。
A’(0.3、ln3200)、B’(1.45、ln1500)、
C’(1.3、ln100000)、D’(0.15、ln100000)
A”(0.3、ln3400)、B”(1.4、ln2000)、
C”(1.3、ln50000)、D”(0.2、ln50000)
【0068】
点(K,H)が直線ABより下になる場合、造粒時、樹脂粒子(A)が有機溶剤等に溶解しやすく、造粒が上手く出来ない場合がある。また点(K,H)が直線CDより上になる場合、樹脂粒子(A)が有機溶剤等に全く膨潤せず、また熱溶融もしにくくなることから樹脂粒子(C1)の平滑性が不十分となり粉体流動性の低下につながることがある。また、点(K,H)が直線ADより左になる場合、樹脂(a)と樹脂(b)のsp値差が小さいため、樹脂粒子(A)が樹脂(b)の溶液に溶解して造粒が上手く出来ない場合がある。また、点(K,H)が直線BCより右になる場合樹脂(a)と樹脂(b)のsp値差が大きく、樹脂粒子(A)が有機溶剤等に全く膨潤せず粒子の平滑性が不十分となり粉体流動性の低下につながり、さらに点(K,H)が右に位置する場合、つまり樹脂(a)と樹脂(b)のsp値差がより開く場合、樹脂(a)と樹脂(b)の吸着力が減少し、造粒困難となることがある。
【0069】
本発明においては、樹脂粒子(A)の樹脂粒子(B)に対するsp値(sp値の計算方法はPolymer Engineering and Science,Feburuary,1974,Vol.14,No.2 P.147〜154による)、また樹脂粒子(A)の分子量を制御することで樹脂粒子(C1)の粒子表面を平滑にすることができる。
【0070】
樹脂粒子(A)が水や分散時に用いる有機溶剤に対してに、溶解したり、膨潤したりするのを低減する観点から、樹脂(a)の分子量、sp値、結晶性、架橋点間分子量等を適宜調整するのが好ましい。
【0071】
樹脂(a)のMnは、好ましくは100〜500万、さらに好ましくは200〜500万、とくに好ましくは500〜500,000であり、sp値は、好ましくは7〜18、さらに好ましくは8〜14である。樹脂(a)の融点(DSCにて測定)は、好ましくは50℃以上、さらに好ましくは80〜200℃である。また、樹脂粒子(C)の、耐熱性、耐水性、耐薬品性、粒径の均一性等を向上させたい場合には、樹脂(a)に架橋構造を導入させてもよい。かかる架橋構造は、共有結合性、配位結合性、イオン結合性、水素結合性等、いずれの架橋形態であってもよい。樹脂(a)に架橋構造を導入する場合の架橋点間分子量は、好ましくは50以上、さらに好ましくは500以上、とくに好ましくは1000以上である。
【0072】
樹脂(a)のガラス転移温度(Tg)は、樹脂粒子(C)の粒径均一性、粉体流動性、保存時の耐熱性、耐ストレス性の観点から、好ましくは20℃〜250℃、さらに好ましくは30℃〜230℃、より好ましくは40℃〜200℃、とくに好ましくは50℃〜120℃ある。水性樹脂分散体(X1)を作成する温度よりTgが低いと、合一を防止したり、分裂を防止したりする効果が小さくなり、粒径の均一性を高める効果が小さくなる。
また、(a)と必要により(m)からなる樹脂粒子(A)のTgは、同様の理由で、好ましくは20〜200℃、さらに好ましくは30〜120℃、とくに好ましくは40〜80℃である。
なお、本発明におけるTgは、前記DSC測定またはフローテスター測定(DSCで測定できない場合)から求められる値である。
【0073】
フローテスター測定には、島津製作所製の高架式フローテスターCFT500型を用いる。フローテスター測定の条件は下記のとおりであり、以下測定は全てこの条件で行われる。
(フローテスター測定条件)
荷重:30kg/cm2、昇温速度:3.0℃/min、
ダイ口径:0.50mm、ダイ長さ:10.0mm
【0074】
また図2に示すフローチャートにあるA点(試料が圧縮荷重を受け変形し始める温度)をガラス転移温度(Tg)とし、B点(内部空隙が消失し不均一な応力の分布を持ったまま外観均一な1個の透明体あるいは相になる点)の温度を軟化開始温度(Ts)、C点(試料の熱膨張によるピストンのわずかな上昇が行われた後、再びピストンが明らかに下降し始める点)の温度を流出開始温度(Tfb)、そしてD点(図において流出終了点Smaxと最低値Sminの差の1/2(X)を求め、XとSminを加えた点)の温度を流出温度(T1/2)とする。
【0075】
樹脂(a)の軟化開始温度(Ts)は、保存時の耐熱性、耐ストレス性、紙面などへの定着特性の観点から、好ましくは40℃〜270℃、さらに好ましくは50℃〜250℃、とくに好ましくは60℃〜220℃、最も好ましくは70℃〜160℃あり、また流出温度(T1/2)は、好ましくは60℃〜300℃、さらに好ましくは65℃〜280℃、とくに好ましくは70℃〜250℃、最も好ましくは80℃〜185℃ある。トナーなどとして用いる場合、軟化開始温度(Ts)、流出温度(T1/2)が高温であると低温定着性や高光沢性などの悪化因子となることがある。なお、本発明における軟化開始温度、流出温度は、上記フローテスター測定から求められる値である。
【0076】
樹脂(a)のガラス転移温度(Tg)と流出温度(T1/2)との温度差は、好ましくは0℃〜130℃、さらに好ましくは0℃〜125℃、とくに好ましくは0℃〜120℃、最も好ましくは0℃〜105℃である。このガラス転移温度と軟化開始温度の温度差が上記範囲内であると、樹脂粒子をトナーとして用いる場合、樹脂粒子の耐熱保存と高光沢の両立が容易である。
【0077】
また、樹脂(a)のガラス転移温度(Tg)と軟化開始温度(Ts)との好ましい温度差は、0℃〜100℃、より好ましくは0℃〜70℃、さらに好ましくは0℃〜50℃、とくに好ましくは0℃〜35℃である。このガラス転移温度と軟化開始温度の温度差が上記範囲内であると、樹脂粒子をトナーとして用いる場合、樹脂粒子の耐熱保存と高光沢の両立が容易である。
【0078】
樹脂(a)の、ガラス転移温度(Tg)、軟化開始温度(Ts)および流出温度(T1/2)は、(a)の分子量および/または(a)を構成する単量体組成を変更することで、容易に調整できる。(a)の分子量(分子量が大きくなるほど、これらの温度は高くなる。)を調整する方法としては、公知の方法でよく、例えば、ポリウレタン樹脂やポリエステル樹脂のような逐次反応で重合する場合には、単量体の仕込み比の調整が挙げられ、ビニル樹脂のような連鎖反応で重合する場合には、重合開始剤量および連鎖移動剤量の調整、反応温度、反応濃度の調整が挙げられる。ガラス転移温度(Tg)と流出温度(T1/2)との温度差を好ましい範囲に調整するには、(a)の分子量と(a)を構成する単量体組成との組み合わせを適切に選択すればよい。
【0079】
樹脂粒子(A)の水性分散液(W)中に、水以外に後述の有機溶剤(u)のうち水と混和性の有機溶剤(アセトン、メチルエチルケトン等)が含有されていてもよい。この際、含有される有機溶剤は、樹脂粒子(A)の凝集を引き起こさないもの、樹脂粒子(A)を溶解しないもの、および樹脂粒子(C1)の造粒を妨げることがないものであればどの種であっても、またどの程度の含有量であってもかまわないが、水との合計量の40%以下用いて、乾燥後の樹脂粒子(C)中に残らないものが好ましい。
【0080】
樹脂(a)を樹脂粒子(A)の水性分散液にする方法は、とくに限定されないが、以下の〔1〕〜〔8〕が挙げられる。
〔1〕ポリエステル樹脂、ポリウレタン樹脂等の重付加あるいは縮合系樹脂の場合において、前駆体(モノマー、オリゴマー等)またはその有機溶剤溶液を必要であれば適当な分散剤存在下で水性媒体中に分散させ、その後に加熱したり、硬化剤を加えたりして前躯体を硬化させて樹脂粒子(A)の水性分散体を製造する方法。
〔2〕ポリエステル樹脂、ポリウレタン樹脂等の重付加あるいは縮合系樹脂の場合において、前駆体(モノマー、オリゴマー等)またはその有機溶剤溶液(液体であることが好ましい。加熱により液状化してもよい)中に適当な乳化剤を溶解させた後、水を加えて転相乳化し、硬化剤を加えたりして前躯体を硬化させて樹脂粒子(A)の水性分散体を製造する方法。
〔3〕ビニル系樹脂の場合において、モノマーを出発原料として、懸濁重合法、乳化重合法、シード重合法または分散重合法等の重合反応により、直接、樹脂粒子(A)の水性分散液を製造する方法。
〔4〕あらかじめ重合反応(付加重合、開環重合、重付加、付加縮合、縮合重合等いずれの重合反応様式であってもよい。以下同様。)により作成した樹脂を機械回転式またはジェット式等の微粉砕機を用いて粉砕し、次いで、分級するすることによって樹脂粒子を得た後、適当な分散剤存在下で水中に分散させる方法。
〔5〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液を霧状に噴霧することにより樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水中に分散させる方法。
〔6〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液に貧溶剤を添加するか、またはあらかじめ有機溶剤に加熱溶解した樹脂溶液を冷却することにより樹脂粒子を析出させ、次いで、有機溶剤を除去して樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水中に分散させる方法。
〔7〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液を、適当な分散剤存在下で水性媒体中に分散させ、これを加熱または減圧等によって有機溶剤を除去する方法。
〔8〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液中に適当な乳化剤を溶解させた後、水を加えて転相乳化する方法。
【0081】
上記〔1〕〜〔8〕の方法において、併用する乳化剤または分散剤としては、公知の界面活性剤(s)、水溶性ポリマー(t)等を用いることができる。また、乳化または分散の助剤として有機溶剤(u)、可塑剤(v)等を併用することができる。
【0082】
界面活性剤(s)としては、アニオン界面活性剤(s−1)、カチオン界面活性剤(s−2)、両性界面活性剤(s−3)、非イオン界面活性剤(s−4)などが挙げられる。界面活性剤(s)は2種以上の界面活性剤を併用したものであってもよい。(s)の具体例としては、以下に述べるものの他特開2002−284881号公報に記載のものが挙げられる。
【0083】
アニオン界面活性剤(s−1)としては、カルボン酸またはその塩、硫酸エステル塩、カルボキシメチル化物の塩、スルホン酸塩およびリン酸エステル塩等が用いられる。
【0084】
カルボン酸またはその塩としては、炭素数8〜22の飽和または不飽和脂肪酸またはその塩が使用でき、例えば、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、アラキジン酸、ベヘン酸、オレイン酸、リノール酸およびリシノール酸並びにヤシ油、パーム核油、米ぬか油および牛脂などをケン化して得られる高級脂肪酸の混合物等が挙げられる。
その塩としては、これらのナトリウム塩、カリウム塩、アミン塩、アンモニウム塩、4級アンモニウム塩およびアルカノールアミン塩(モノエタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩等)などの塩があげられる。
【0085】
硫酸エステル塩としては、高級アルコール硫酸エステル塩(炭素数8〜18の脂肪族アルコールの硫酸エステル塩)、高級アルキルエーテル硫酸エステル塩(炭素数8〜18の脂肪族アルコールのEOまたはPO1〜10モル付加物の硫酸エステル塩)、硫酸化油(炭素数12〜50の天然の不飽和油脂または不飽和のロウをそのまま硫酸化して中和したもの)、硫酸化脂肪酸エステル(不飽和脂肪酸(炭素数6〜40)の低級アルコール(炭素数1〜8)エステルを硫酸化して中和したもの)および硫酸化オレフィン(炭素数12〜18のオレフィンを硫酸化して中和したもの)等が使用できる。
塩としては、ナトリウム塩、カリウム塩、アミン塩、アンモニウム塩、4級アンモニウム塩およびアルカノールアミン塩(モノエタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩等)等が挙げられる。
【0086】
高級アルコール硫酸エステル塩としては、例えば、オクチルアルコール硫酸エステル塩、デシルアルコール硫酸エステル塩、ラウリルアルコール硫酸エステル塩、ステアリルアルコール硫酸エステル塩、チーグラー触媒を用いて合成されたアルコール(例えば、商品名:ALFOL 1214:CONDEA社製)の硫酸エステル塩およびオキソ法で合成されたアルコール(例えば、商品名:ドバノール23、25、45、ダイヤドール115−L、115H、135:三菱化学製:、商品名:トリデカノール:協和発酵製、商品名:オキソコール1213、1215、1415:日産化学製)の硫酸エステル塩等が挙げられる。
【0087】
高級アルキルエーテル硫酸エステル塩としては、例えば、ラウリルアルコールEO2モル付加物硫酸エステル塩およびオクチルアルコールEO3モル付加物硫酸エステル塩等が挙げられる。
硫酸化油としては、例えば、ヒマシ油、落花生油、オリーブ油、ナタネ油、牛脂および羊脂などの硫酸化物の塩等が挙げられる。
硫酸化脂肪酸エステルとしては、例えば、オレイン酸ブチルおよびリシノレイン酸ブチル等の硫酸化物の塩等が挙げられる。
硫酸化オレフィンとしては、例えば、商品名:ティーポール(シェル社製)等が挙げられる。
【0088】
カルボキシメチル化物の塩としては、炭素数8〜16の脂肪族アルコールのカルボキシメチル化物の塩および炭素数8〜16の脂肪族アルコールのEOまたはPO1〜10モル付加物のカルボキシメチル化物の塩等が使用できる。
【0089】
脂肪族アルコールのカルボキシメチル化物の塩としては、例えば、オクチルアルコールカルボキシメチル化ナトリウム塩、ラウリルアルコールカルボキシメチル化ナトリウム塩、ドバノール23のカルボキシメチル化ナトリウム塩、トリデカノールカルボキシメチル化ナトリウム塩等が挙げられる。
【0090】
脂肪族アルコールのEO1〜10モル付加物のカルボキシメチル化物の塩としては、例えば、オクチルアルコールEO3モル付加物カルボキシメチル化ナトリウム塩、ラウリルアルコールEO4モル付加物カルボキシメチル化ナトリウム塩、およびトリデカノールEO5モル付加物カルボキシメチル化ナトリウム塩などが挙げられる。
【0091】
スルホン酸塩としては、アルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、スルホコハク酸ジエステル塩、α−オレフィンスルホン酸塩、イゲポンT型およびその他芳香環含有化合物のスルホン酸塩等が使用できる。
アルキルベンゼンスルホン酸塩としては、例えば、ドデシルベンゼンスルホン酸ナトリウム塩等が挙げられる。
【0092】
アルキルナフタレンスルホン酸塩としては、例えば、ドデシルナフタレンスルホン酸ナトリウム塩等が挙げられる。
スルホコハク酸ジエステル塩としては、例えば、スルホコハク酸ジ−2−エチルヘキシルエステルナトリウム塩などが挙げられる。
芳香環含有化合物のスルホン酸塩としては、アルキル化ジフェニルエーテルのモノまたはジスルホン酸塩およびスチレン化フェノールスルホン酸塩などが挙げられる。
【0093】
リン酸エステル塩としては、高級アルコールリン酸エステル塩および高級アルコールEO付加物リン酸エステル塩等が使用できる。
高級アルコールリン酸エステル塩としては、例えば、ラウリルアルコールリン酸モノエステルジナトリウム塩およびラウリルアルコールリン酸ジエステルナトリウム塩等が挙げられる。
高級アルコールEO付加物リン酸エステル塩としては、例えば、オレイルアルコールEO5モル付加物リン酸モノエステルジナトリウム塩等が挙げられる。
【0094】
カチオン界面活性剤(s−2)としては、第4級アンモニウム塩型界面活性剤およびアミン塩型界面活性剤等が使用できる。
第4級アンモニウム塩型界面活性剤としては、炭素数3〜40の3級アミンと4級化剤(例えば、メチルクロライド、メチルブロマイド、エチルクロライド、ベンジルクロライドおよびジメチル硫酸などのアルキル化剤並びにEOなど)との反応等で得られ、例えば、ラウリルトリメチルアンモニウムクロライド、ジデシルジメチルアンモニウムクロライド、ジオクチルジメチルアンモニウムブロマイド、ステアリルトリメチルアンモニウムブロマイド、ラウリルジメチルベンジルアンモニウムクロライド(塩化ベンザルコニウム)、セチルピリジニウムクロライド、ポリオキシエチレントリメチルアンモニウムクロライドおよびステアラミドエチルジエチルメチルアンモニウムメトサルフェートなどが挙げられる。
【0095】
アミン塩型界面活性剤としては、1〜3級アミンを無機酸(例えば、塩酸、硝酸、硫酸、ヨウ化水素酸、リン酸および過塩素酸など)または有機酸(酢酸、ギ酸、蓚酸、乳酸、グルコン酸、アジピン酸、炭素数2〜24のアルキルリン酸、リンゴ酸およびクエン酸など)で中和すること等により得られる。
第1級アミン塩型界面活性剤としては、例えば、炭素数8〜40の脂肪族高級アミン(例えば、ラウリルアミン、ステアリルアミン、セチルアミン、硬化牛脂アミンおよび、ロジンアミンなどの高級アミン)の無機酸塩または有機酸塩および低級アミン(炭素数2〜6)の高級脂肪酸(炭素数8〜40、ステアリン酸、オレイン酸など)塩などが挙げられる。
【0096】
第2級アミン塩型界面活性剤としては、例えば炭素数4〜40の脂肪族アミンのEO付加物などの無機酸塩または有機酸塩が挙げられる。
また、第3級アミン塩型界面活性剤としては、例えば、炭素数4〜40の脂肪族アミン(例えば、トリエチルアミン、エチルジメチルアミン、N,N,N’,N’−テトラメチルエチレンジアミンなど)、脂肪族アミン(炭素数2〜40)のEO(2モル以上)付加物、炭素数6〜40の脂環式アミン(例えば、N−メチルピロリジン、N−メチルピペリジン、N−メチルヘキサメチレンイミン、N−メチルモルホリンおよび1,8−ジアザビシクロ(5,4,0)−7−ウンデセンなど)、炭素数5〜30の含窒素ヘテロ環芳香族アミン(例えば、4−ジメチルアミノピリジン、N−メチルイミダゾールおよび4,4’−ジピリジルなど)の無機酸塩または有機酸塩およびトリエタノールアミンモノステアレート、ステアラミドエチルジエチルメチルエタノールアミンなどの3級アミンの無機酸塩または有機酸塩などが挙げられる。
【0097】
両性界面活性剤(s−3)としては、カルボン酸塩型両性界面活性剤、硫酸エステル塩型両性界面活性剤、スルホン酸塩型両性界面活性剤およびリン酸エステル塩型両性界面活性剤などが使用できる。
【0098】
カルボン酸塩型両性界面活性剤は、アミノ酸型両性界面活性剤、ベタイン型両性界面活性剤およびイミダゾリン型両性界面活性剤などが用いられる。アミノ酸型両性界面活性剤は、分子内にアミノ基とカルボキシル基を持っている両性界面活性剤であり、例えば、一般式(2)で示される化合物等が挙げられる。
【0099】
[R−NH−(CH2)n−COO]mM (2)
[式中、Rは1価の炭化水素基;nは1または2;mは1または2;Mは水素イオン、アルカリ金属イオン、アルカリ土類金属イオン、アンモニウムカチオン、アミンカチオン、アルカノールアミンカチオンなどである。]
【0100】
一般式(2)で表される両面活性剤としては、例えば、アルキル(炭素数6〜40)アミノプロピオン酸型両性界面活性剤(ステアリルアミノプロピオン酸ナトリウム、ラウリルアミノプロピオン酸ナトリウムなど);アルキル(炭素数4〜24)アミノ酢酸型両性界面活性剤(ラウリルアミノ酢酸ナトリウムなど)などが挙げられる。
【0101】
ベタイン型両性界面活性剤は、分子内に第4級アンモニウム塩型のカチオン部分とカルボン酸型のアニオン部分を持っている両性界面活性剤であり、例えば、アルキル(炭素数6〜40)ジメチルベタイン(ステアリルジメチルアミノ酢酸ベタイン、ラウリルジメチルアミノ酢酸ベタインなど)、炭素数6〜40のアミドベタイン(ヤシ油脂肪酸アミドプロピルベタインなど)、アルキル(炭素数6〜40)ジヒドロキシアルキル(炭素数6〜40)ベタイン(ラウリルジヒドロキシエチルベタインなど)などが挙げられる。
【0102】
イミダゾリン型両性界面活性剤としては、イミダゾリン環を有するカチオン部分とカルボン酸型のアニオン部分を持っている両性界面活性剤であり、例えば、2−ウンデシル−N−カルボキシメチル−N−ヒドロキシエチルイミダゾリニウムベタインなどが挙げられる。
【0103】
その他の両性界面活性剤として、例えば、ナトリウムラウロイルグリシン、ナトリウムラウリルジアミノエチルグリシン、ラウリルジアミノエチルグリシン塩酸塩、ジオクチルジアミノエチルグリシン塩酸塩などのグリシン型両性界面活性剤;ペンタデシルスルホタウリンなどのスルホベタイン型両性界面活性剤、スルホン酸塩型両性界面活性剤およびリン酸エステル塩型両性界面活性剤などが挙げられる。
【0104】
非イオン界面活性剤(s−4)としては、AO付加型非イオン界面活性剤および多価アルコ−ル型非イオン界面活性剤などが使用できる。
AO付加型非イオン界面活性剤は、炭素数8〜40の高級アルコ−ル、炭素数8〜40の高級脂肪酸または炭素数8〜40のアルキルアミン等に直接AO(炭素数2〜20)を付加させるか、グリコ−ルにAOを付加させて得られるポリアルキレングリコ−ルに高級脂肪酸などを反応させるか、あるいは多価アルコ−ルに高級脂肪酸を反応して得られたエステル化物にAOを付加させるか、高級脂肪酸アミドにAOを付加させることにより得られる。
【0105】
AOとしては、たとえばEO、POおよびBOが挙げられる。
これらのうち好ましいものは、EOおよびEOとPOのランダムまたはブロック付加物である。
AOの付加モル数としては10〜50モルが好ましく、該AOのうち50〜100%がEOであるものが好ましい。
【0106】
AO付加型非イオン界面活性剤としては、例えば、オキシアルキレンアルキルエ−テル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)(例えば、オクチルアルコールEO20モル付加物、ラウリルアルコールEO20モル付加物、ステアリルアルコールEO10モル付加物、オレイルアルコールEO5モル付加物、ラウリルアルコールEO10モルPO20モルブロック付加物など);ポリオキシアルキレン高級脂肪酸エステル(アルキレンの炭素数2〜24、高級脂肪酸の炭素数8〜40)(例えば、ステアリル酸EO10モル付加物、ラウリル酸EO10モル付加物など);ポリオキシアルキレン多価アルコ−ル高級脂肪酸エステル(アルキレンの炭素数2〜24、多価アルコールの炭素数3〜40、高級脂肪酸の炭素数8〜40)(例えば、ポリエチレングリコール(重合度20)のラウリン酸ジエステル、ポリエチレングリコール(重合度20)のオレイン酸ジエステルなど);ポリオキシアルキレンアルキルフェニルエーテル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)(例えば、ノニルフェノールEO4モル付加物、ノニルフェノールEO8モルPO20モルブロック付加物、オクチルフェノールEO10モル付加物、ビスフェノールA・EO10モル付加物、スチレン化フェノールEO20モル付加物など);ポリオキシアルキレンアルキルアミノエ−テル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)および(例えば、ラウリルアミンEO10モル付加物、ステアリルアミンEO10モル付加物など);ポリオキシアルキレンアルカノ−ルアミド(アルキレンの炭素数2〜24、アミド(アシル部分)の炭素数8〜24)(例えば、ヒドロキシエチルラウリン酸アミドのEO10モル付加物、ヒドロキシプロピルオレイン酸アミドのEO20モル付加物など)が挙げられる。
【0107】
多価アルコ−ル型非イオン界面活性剤としては、多価アルコール脂肪酸エステル、多価アルコール脂肪酸エステルAO付加物、多価アルコールアルキルエーテルおよび多価アルコールアルキルエーテルAO付加物等が使用できる。多価アルコールの炭素数としては3〜24、脂肪酸の炭素数としては8〜40、AOの炭素数としては2〜24である。
【0108】
多価アルコール脂肪酸エステルとしては、例えば、ペンタエリスリトールモノラウレート、ペンタエリスリトールモノオレート、ソルビタンモノラウレート、ソルビタンモノステアレート、ソルビタンモノラウレート、ソルビタンジラウレート、ソルビタンジオレートおよびショ糖モノステアレートなどが挙げられる。
【0109】
多価アルコール脂肪酸エステルAO付加物としては、例えば、エチレングリコールモノオレートEO10モル付加物、エチレングリコールモノステアレートEO20モル付加物、トリメチロールプロパンモノステアレートEO20モルPO10モルランダム付加物、ソルビタンモノラウレートEO10モル付加物、ソルビタンジステアレートEO20モル付加物およびソルビタンジラウレートEO12モルPO24モルランダム付加物などが挙げられる。
【0110】
多価アルコールアルキルエーテルとしては、例えば、ペンタエリスリトールモノブチルエーテル、ペンタエリスリトールモノラウリルエーテル、ソルビタンモノメチルエーテル、ソルビタンモノステアリルエーテル、メチルグリコシドおよびラウリルグリコシドなどが挙げられる。
【0111】
多価アルコールアルキルエーテルAO付加物としては、例えば、ソルビタンモノステアリルエーテルEO10モル付加物、メチルグリコシドEO20モルPO10モルランダム付加物、ラウリルグリコシドEO10モル付加物およびステアリルグリコシドEO20モルPO20モルランダム付加物などが挙げられる。
【0112】
水溶性ポリマー(t)としては、セルロース系化合物(例えば、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、エチルヒドロキシエチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルセルロースおよびそれらのケン化物など)、ゼラチン、デンプン、デキストリン、アラビアゴム、キチン、キトサン、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレングリコール、ポリエチレンイミン、ポリアクリルアミド、アクリル酸(塩)含有ポリマー(ポリアクリル酸ナトリウム、ポリアクリル酸カリウム、ポリアクリル酸アンモニウム、ポリアクリル酸の水酸化ナトリウム部分中和物、アクリル酸ナトリウム−アクリル酸エステル共重合体)、スチレン−無水マレイン酸共重合体の水酸化ナトリウム(部分)中和物、水溶性ポリウレタン(ポリエチレングリコール、ポリカプロラクトンジオール等とポリイソシアネートの反応生成物等)などが挙げられる。
【0113】
本発明に用いる有機溶剤(u)は、乳化分散の際に必要に応じて水性媒体中に加えても、被乳化分散体中[樹脂(b)を含む油相中]に加えてもよい。
有機溶剤(u)の具体例としては、トルエン、キシレン、エチルベンゼン、テトラリン等の芳香族炭化水素系溶剤;n−ヘキサン、n−ヘプタン、ミネラルスピリット、シクロヘキサン等の脂肪族または脂環式炭化水素系溶剤;塩化メチル、臭化メチル、ヨウ化メチル、メチレンジクロライド、四塩化炭素、トリクロロエチレン、パークロロエチレンなどのハロゲン系溶剤;酢酸エチル、酢酸ブチル、メトキシブチルアセテート、メチルセロソルブアセテート、エチルセロソルブアセテートなどのエステル系またはエステルエーテル系溶剤;ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチルセロソルブ、ブチルセロソルブ、プロピレングリコールモノメチルエーテルなどのエーテル系溶剤;アセトン、メチルエチルケトン、メチルイソブチルケトン、ジ−n−ブチルケトン、シクロヘキサノンなどのケトン系溶剤;メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、t−ブタノール、2−エチルヘキシルアルコール、ベンジルアルコールなどのアルコール系溶剤;ジメチルホルムアミド、ジメチルアセトアミドなどのアミド系溶剤;ジメチルスルホキシドなどのスルホキシド系溶剤、N−メチルピロリドンなどの複素環式化合物系溶剤、ならびにこれらの2種以上の混合溶剤が挙げられる。
【0114】
可塑剤(v)は、乳化分散の際に必要に応じて水性媒体中に加えても、被乳化分散体中[樹脂(b)を含む油相中]に加えてもよい。
可塑剤(v)としては、何ら限定されず、以下のものが例示される。
(v1)フタル酸エステル[フタル酸ジブチル、フタル酸ジオクチル、フタル酸ブチルベンジル、フタル酸ジイソデシル等];
(v2)脂肪族2塩基酸エステル[アジピン酸ジ−2−エチルヘキシル、セバシン酸−2−エチルヘキシル等];
(v3)トリメリット酸エステル[トリメリット酸トリ−2−エチルヘキシル、トリメリット酸トリオクチル等];
(v4)燐酸エステル[リン酸トリエチル、リン酸トリ−2−エチルヘキシル、リン酸トリクレジール等];
(v5)脂肪酸エステル[オレイン酸ブチル等];
(v6)およびこれらの2種以上の混合物が挙げられる。
【0115】
本発明において用いる樹脂粒子(A)の粒径は、通常、形成される樹脂粒子(B)の粒径よりも小さく、粒径均一性の観点から、粒径比[樹脂粒子(A)の体積平均粒径]/[樹脂粒子(B)の体積平均粒径]の値が0.001〜0.3の範囲であるのが好ましい。粒径比の下限は、さらに好ましくは0.003であり、上限は、さらに好ましくは0.25である。粒径比が、0.3より大きいと(A)が(B)の表面に効率よく吸着しないため、得られる(C)の粒度分布が広くなる傾向がある。
【0116】
樹脂粒子(A)の体積平均粒径は、所望の粒径の樹脂粒子(C)を得るのに適した粒径になるように、上記粒径比の範囲で適宜調整することができる。
(A)の体積平均粒径は、一般的には、0.0005〜30μmが好ましい。上限は、さらに好ましくは20μm、とくに好ましくは10μmであり、下限は、さらに好ましくは0.01μm、とくに好ましくは0.02μm、最も好ましくは0.04μmである。ただし、例えば、体積平均粒径1μmの樹脂粒子(C)を得たい場合には、好ましくは0.0005〜0.3μm、とくに好ましくは0.001〜0.2μmの範囲、10μmの樹脂粒子(C)を得た場合には、好ましくは0.005〜3μm、とくに好ましくは0.05〜2μm、100μmの粒子(C)を得たい場合には、好ましくは0.05〜30μm、とくに好ましくは0.1〜20μmである。
なお、体積平均粒径は、レーザー式粒度分布測定装置LA−920(堀場製作所製)やマルチサイザーIII(コールター社製)、光学系としてレーザードップラー法を用いるELS−800(大塚電子社製)などで測定できる。もし、各測定装置間で粒径の測定値に差を生じた場合は、ELS−800での測定値を採用する。
なお、上記粒径比が得やすいことから、後述する樹脂粒子(B)の体積平均粒径は、0.1〜300μmが好ましい。さらに好ましくは0.5〜250μm、特に好ましくは1〜200μmである。
【0117】
本発明の水性樹脂分散体および樹脂粒子(C)の製造方法においては、水性樹脂分散体(X1)は、樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)またはその有機溶剤溶液とを混合し、(W)中に(b)またはその有機溶剤溶液を分散させて、(A)の水性分散液中で、(b)からなる樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着されてなる構造の樹脂粒子(C1)の水性分散体として得ることができる。
【0118】
上記製造方法においては、水性分散液(W)中に、樹脂(b)もしくはその有機溶剤溶液を分散させて、(b)からなる樹脂粒子(B)が形成される際に、樹脂粒子(B)の表面に樹脂粒子(A)を吸着させることで樹脂粒子(C1)同士が合一するのを防ぎ、また、高剪断条件下で(C1)が分裂され難くする。これにより、(C1)の粒径を一定の値に収斂させ、粒径の均一性を高める効果を発揮する。そのため、樹脂粒子(A)は、分散する際の温度において、剪断により破壊されない程度の強度を有すること、水に溶解したり、膨潤したりしにくいこと、(b)もしくはその有機溶剤溶液に溶解しにくいことが好ましい特性としてあげられる
【0119】
樹脂(b)もしくはその有機溶剤溶液を分散させる場合には、分散装置を用いることができる。
本発明で使用する分散装置は、一般に乳化機、分散機として市販されているものであればとくに限定されず、例えば、ホモジナイザー(IKA社製)、ポリトロン(キネマティカ社製)、TKオートホモミキサー(特殊機化工業社製)等のバッチ式乳化機、エバラマイルダー(荏原製作所社製)、TKフィルミックス、TKパイプラインホモミキサー(特殊機化工業社製)、コロイドミル(神鋼パンテック社製)、スラッシャー、トリゴナル湿式微粉砕機(三井三池化工機社製)、キャピトロン(ユーロテック社製)、ファインフローミル(太平洋機工社製)等の連続式乳化機、マイクロフルイダイザー(みずほ工業社製)、ナノマイザー(ナノマイザー社製)、APVガウリン(ガウリン社製)等の高圧乳化機、膜乳化機(冷化工業社製)等の膜乳化機、バイブロミキサー(冷化工業社製)等の振動式乳化機、超音波ホモジナイザー(ブランソン社製)等の超音波乳化機等が挙げられる。このうち粒径の均一化の観点で好ましいものは、APVガウリン、ホモジナイザー、TKオートホモミキサー、エバラマイルダー、TKフィルミックス、TKパイプラインホモミキサーが挙げられる。
【0120】
樹脂(b)を樹脂粒子(A)の水性分散液(W)に分散させる際、樹脂(b)は液体であることが好ましい。樹脂(b)が常温で固体である場合には、融点以上の高温下で液体の状態で分散させたり、(b)の有機溶剤溶液を用いてもよい。
樹脂(b)もしくはその有機溶剤溶液の粘度は、は、粒径均一性の観点から、好ましくは10〜5万mPa・s(B型粘度計で測定)、さらに好ましくは100〜1万mPa・sである。
分散時の温度としては、好ましくは0〜150℃(加圧下)、さらに好ましくは5〜98℃である。分散体の粘度が高い場合は、高温にして粘度を上記好ましい範囲まで低下させて、乳化分散を行うのが好ましい。
樹脂(b)の有機溶剤溶液に用いる溶剤は、樹脂(b)を常温もしくは加熱下で溶解しうる溶剤であればとくに限定されず、具体的には、有機溶剤(u)と同様のものが例示される。好ましいものは樹脂(b)の種類によって異なるが、(b)とのsp値差が3以下であるのが好適である。また、樹脂粒子(C)の粒径均一性の観点からは、樹脂(b)を溶解させるが、樹脂(a)からなる樹脂粒子(A)を溶解・膨潤させにくい溶剤が好ましい。
【0121】
樹脂(b)100部に対する水性分散液(W)の使用量は、好ましくは50〜2,000重量部、さらに好ましくは100〜1,000重量部である。50重量部以上では(b)の分散状態が良好であり、2,000重量部以下であると経済的である。
【0122】
本発明においては、前記の点(K,H)が特定範囲内に含まれるような樹脂(a)と(b)、あるいは前記好ましい範囲の(Ts)、(Tg)等の物性を有する樹脂(a)を用いる場合、とくに(b)の有機溶剤溶液(とくに下記の好ましい溶剤)を用いる場合、有機溶剤を水性樹脂分散体(X1)中に好ましくは10〜50%(とくに20〜40%)用い、40℃以下で好ましくは1%以下(とくに0.5%以下)となるまで脱溶剤するのみで、樹脂粒子(A)が有機溶剤に溶解されて膜状化し、(B)の表面に(A)の被膜が形成されてなる樹脂粒子(C2)の水性樹脂分散体(X2)が得られる場合が多い。しかし、(A)の被膜が形成されていない場合、あるいは(A)の少なくとも一部からの被膜が形成されている場合でも、さらに樹脂粒子(C)表面の被膜の平滑性をより良好にするため、以下の操作を行うと、(B)で構成されるコア層(Q)の表面の少なくとも一部、好ましくは全面に(A)から形成された表面が平滑な被膜〔シェル層(P)〕を有する樹脂粒子(C2)の水性樹脂分散体(X2)が得られ、それから得られる(C2)の保存安定性が優れる点から好ましい。
上記の方法としては、(B)に付着された(A)を有機溶剤に溶解させる方法、および、水性樹脂分散体(X1)を加熱して(A)を溶融し被膜化させる方法が挙げられ、これらの方法を併用してもよい。
【0123】
樹脂粒子(A)を有機溶剤に溶解させて被膜化させる場合に用いる溶剤は、被膜化する際に(X1)中に添加してもよいが、(X1)を得る際の原料として、樹脂(b)の有機溶剤溶液を用い、その溶剤を、樹脂粒子(B)の形成後も直ちに除去せずにそれを用いる方が、(B)中に溶剤が含有されるため(A)の溶解が容易であり、樹脂の凝集が起こりにくく好ましい。
有機溶剤としては、(b)との親和性が高いものが好ましく、具体例としては、前記の有機溶剤(u)と同様のものが挙げられる。(u)の中で好ましいものは、被膜化の点から、テトラヒドロフラン、トルエン、アセトン、メチルエチルケトン、および酢酸エチルであり、さらに好ましくは酢酸エチルである。
(A)を有機溶剤に溶解させる際の、水性樹脂分散体中の溶剤濃度は、好ましくは3〜60%、さらに好ましくは10〜45%、とくに好ましくは15〜30%である。また、溶解は、水性樹脂分散体を、例えば1〜10時間攪拌することにより行い、溶解時の温度は、15〜45℃が好ましく、15〜30℃がさらに好ましい。
【0124】
(A)を溶融して(B)の表面に被膜化させる場合、水性樹脂分散体(X1)中の固形分含量〔水および有機溶剤以外の成分の含量〕を、好ましくは1〜50%、さらに好ましくは5〜30%に調製する。また、このときの溶剤含有量は、好ましくは2%以下、さらに好ましくは1%以下、とくに好ましくは0.5%以下である。(X1)中の固形分含量が多かったり、溶剤含有量が2%を越える場合、(X1)を60℃以上に昇温すると凝集物が発生することがある。溶融時の加熱の条件は、(A)が溶融される条件であればとくに限定されないが、例えば、撹拌下、好ましくは40〜100℃、さらに好ましくは60〜90℃、とくに好ましくは60〜80℃で、好ましくは1〜300分間加熱する方法が挙げられる。
なお、被膜化処理の方法とし、溶剤含有量が2%以下の樹脂粒子(C1)の水性分散体(X1)を加熱処理し、(A)をコア(Q)上で溶融させることでより表面が平滑な樹脂粒子(C2)を得る際の好ましい加熱処理温度は、(P)のTg以上であり、また80℃以下の温度範囲が好ましい。加熱処理温度が(P)のTg未満であると得られる樹脂粒子(C2)の表面平滑性はほとんど変化がない。また80℃を越える温度で加熱処理するとシェル(P)がコアから剥がれる場合がある。
これらの(A)の被膜化方法の中で、好ましい方法は、(A)を溶融させる方法、および(A)を溶解させる方法と(A)を溶融させる方法の併用である。
【0125】
樹脂粒子(C1)あるいは(C2)は、樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)または(b)の有機溶剤溶液とが混合され、(W)中に(b)または(b)の有機溶剤溶液が分散され、樹脂(b)からなる樹脂粒子(B)の表面に樹脂粒子(A)が付着してなる構造の樹脂粒子(C1)の水性分散体(X1)、および/または、(B)で構成されるコア層(Q)の表面に(A)から形成されたシェル層(P)を有する構造の樹脂粒子(C2)の水性分散体(X2)を形成させた後、水性樹脂分散体(X1)または(X2)〔以下、「(X1)または(X2)」のことを、(X)と記載することもある。〕から水性媒体を除去することにより得られる。
【0126】
水性媒体を除去する方法としては、
〔1〕水性樹脂分散体を減圧下または常圧下で乾燥する方法
〔2〕遠心分離器、スパクラフィルター、フィルタープレスなどにより固液分離し、得られた粉末を乾燥する方法
〔3〕水性樹脂分散体を凍結させて乾燥させる方法(いわゆる凍結乾燥)
等が例示される。
上記〔1〕、〔2〕において、得られた粉末を乾燥する際、流動層式乾燥機、減圧乾燥機、循風乾燥機など公知の設備を用いて行うことができる。
また、必要に応じ、風力分級器などを用いて分級し、所定の粒度分布とすることもできる。
【0127】
樹脂粒子(C1)は、実質的に、相対的に小さい樹脂粒子(A)と相対的に大きい樹脂粒子(B)から構成され、(A)が(B)の表面に付着された形で存在する。また、樹脂粒子(C2)は、(A)が(B)に付着後、溶解および/または溶融され、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されたものである。
両粒子の付着力をさらに強めたい場合には、水性媒体中に分散した際に、(A)と(B)が正負逆の電荷を持つようにしたり、(A)(B)が同一の電荷持つ場合には、界面活性剤(s)または水溶性ポリマー(t)のうち、(A)および(B)と逆電荷を持つものを使用したり、また樹脂(a)と樹脂(b)のsp値差を前記の範囲内でできるだけ小さく(例えば2以下)したりすることが有効である。
ある。
【0128】
また、本発明の水性樹脂分散体および樹脂粒子(C)を製造する際、樹脂粒子(A)の樹脂粒子(B)に対する吸着力は、以下のような方法で制御することができる。
〔1〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が正負逆の電荷を持つようにすると吸着力が発生し、この場合、樹脂粒子(A)、樹脂粒子(B)各々の電荷を大きくするほど、吸着力が強くなり樹脂粒子(A)の樹脂粒子(B)に対する被覆率が大きくなる。
〔2〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が同極性(どちらも正、またはどちらも負)の電荷を持つようにすると、被覆率は下がる傾向にある。この場合、一般に活性剤(s)および/または水溶性ポリマー(t)[とくに樹脂粒子(A)および樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。
〔3〕水性分散体(X1)を製造する際に、樹脂(a)がカルボキシル基、リン酸基、スルホン酸基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが低いほど被覆率が大きくなる。逆に、pHを高くするほど被覆率が小さくなる。
〔4〕水性分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム塩基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが高いほど被覆率が大きくなる。逆に、pHを低くするほど被覆率が小さくなる。
〔5〕樹脂粒子(A)と樹脂粒子(B)のΔsp値を小さくすると被覆率が大きくなる。
【0129】
樹脂粒子(C)の粒径均一性、保存安定性等の観点から、樹脂粒子(C)は、0.01〜60%の(A)と40〜99.99%の(B)からなるのが好ましく、さらに好ましくは0.1〜50%の(A)と50〜99.9%の(B)、とくに好ましくは1〜45%の(A)と55〜99%の(B)からなる。
【0130】
樹脂粒子(C1)の粒径均一性、粉体流動性、保存安定性等の観点からは、樹脂粒子(B)の表面の5%以上、好ましくは30%以上、さらに好ましくは50%以上、とくに好ましくは80%以上が樹脂粒子(A)で覆われているのがよい。(C1)の表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[樹脂粒子(A)に覆われている部分の面積/樹脂粒子(A)に覆われている部分の面積+樹脂粒子(B)が露出している部分の面積]×100
【0131】
樹脂粒子(C2)においての下式で求められる表面被覆率はいかなる率であってもかまわないが、粒径均一性、粉体流動性、保存安定性等の観点からは、コア層(Q)の表面の70%以上、好ましくは80%以上、さらに好ましくは90%以上、とくに好ましくは95%以上がシェル層(P)で覆われているのがよい。なお、表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[(P)に覆われている部分の面積/(P)に覆われている部分の面積+(Q)が露出している部分の面積]×100
【0132】
粒径均一性の観点から、樹脂粒子(C)の体積分布の変動係数は、30%以下であるのが好ましく、0.1〜15%であるのがさらに好ましい。
また、粒径均一性から、樹脂粒子(C)の[体積平均粒径/個数平均粒径](Dv/Dn)の値は、1.0〜1.4であるのが好ましく、1.0〜1.2であるのがさらに好ましい。
(C)の体積平均粒径は、用途により異なるが、一般的には0.1〜300μmが好ましい。上限は、さらに好ましくは250μm、特に好ましくは200μmであり、下限は、さらに好ましくは0.5μm、特に好ましくは1μmである。
なお、体積平均粒径および個数平均粒径は、マルチサイザーIII(コールター社製)で同時に測定することができる。
【0133】
本発明の樹脂粒子(C2)の形状の制御は、以下の方法で行うことが出来る。
樹脂粒子(A)と樹脂粒子(B)のsp値差、また樹脂粒子(A)の分子量を制御することで粒子形状や粒子表面性を制御することができる。sp値差が小さいといびつな形で表面平滑な粒子が得られやすく、また、sp値差が大きいと球形で表面はザラつきのある粒子が得られやすい。また、(A)の分子量が大きいと表面はザラつきのある粒子が得られやすく、分子量が小さいと表面平滑な粒子が得られやすい。ただし、(A)と(B)のsp値差は小さすぎても大きすぎても造粒困難になる。また樹脂粒子(A)の分子量も小さすぎると造粒困難になる。このことから、好ましい(A)と(B)のsp値差は0.01〜5.0でより好ましくは0.1〜3.0、さらに好ましくは0.2〜2.0である。また、好ましい樹脂粒子(A)のMwは100〜100万で、より好ましくは1000〜50万、さらに好ましくは2000〜20万、特に好ましくは3000〜10万である。
【0134】
本発明の樹脂粒子(C)は、樹脂粒子(A)と樹脂粒子(B)の粒径、および、樹脂粒子(A)による樹脂粒子(B)表面の被覆率あるいはシェル層(P)によるコア層(Q)表面の被覆率を変えることで粒子表面に所望の凹凸を付与することができる。粉体流動性を向上させたい場合には、(C)のBET値比表面積が0.5〜5.0m2/gであるのが好ましい。本発明のBET比表面積は、比表面積計例えばQUANTASORB(ユアサアイオニクス製)を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)したものである。
同様に粉体流動性の観点から、(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
【0135】
樹脂粒子(C)の形状は、粉体流動性、溶融レベリング性等の観点から球状であるのが好ましい。その場合、粒子(A)および粒子(B)も球状であるのが好ましい。(C)は平均円形度が0.95〜1.00であるのが好ましい。平均円形度は、さらに好ましくは0.96〜1.0、とくに好ましくは0.97〜1.0である。なお、平均円形度は、光学的に粒子を検知して、投影面積の等しい相当円の周囲長で除した値である。具体的には、フロー式粒子像分析装置(FPIA−2000;シスメックス社製)を用いて測定する。所定の容器に、予め不純固形物を除去した水100〜150mlを入れ、分散剤として界面活性剤(ドライウエル;富士写真フィルム社製)0.1〜0.5mlを加え、さらに測定資料0.1〜9.5g程度を加える。試料を分散した懸濁液を超音波分散器(ウルトラソニッククリーナ モデル VS−150;ウエルボクリア社製)で約1〜3分間分散処理を行ない、分散濃度を3,000〜10,000個/μLにして樹脂粒子の形状および分布を測定する。
【0136】
本発明の水性樹脂分散体および樹脂粒子(C)の製造方法において、樹脂粒子(C)は、樹脂粒子(A)の樹脂粒子(B)に対する粒径比、および、水性樹脂分散体(X1)中における樹脂粒子(A)による樹脂粒子(B)表面の被覆率、水性樹脂分散体(X1)中における樹脂粒子(B)/水性媒体界面上で樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さ、を変えることで粒子表面を平滑にしたり、粒子表面に所望の凹凸を付与したりすることができる。
樹脂粒子(A)による樹脂粒子(B)表面の被覆率や樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さは、以下のような方法で制御することができる。
〔1〕水性樹脂分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が正負逆の電荷を持つようにすると被覆率、深さが大きくなる。この場合、樹脂粒子(A)、樹脂粒子(B)各々の電荷を大きくするほど、被覆率、深さが大きくなる。
〔2〕水性樹脂分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が同極性(どちらも正、またはどちらも負)の電荷を持つようにすると、被覆率は下がり、深さが小さくなる傾向にある。この場合、一般に界面活性剤(s)および/または水溶性ポリマー(t)[とくに樹脂粒子(A)および樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。また、水溶性ポリマー(t)を使用する場合には、水溶性ポリマー(t)の分子量が大きいほど深さが小さくなる。
〔3〕水性樹脂分散体(X1)を製造する際に、樹脂(a)がカルボキシル基、リン酸基、スルホン酸基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが低いほど被覆率、深さが大きくなる。逆に、pHを高くするほど被覆率、深さが小さくなる。
〔4〕水性樹脂分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム塩基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが高いほど被覆率、深さが大きくなる。逆に、pHを低くするほど被覆率、深さが小さくなる。
〔5〕樹脂(a)と樹脂(b)のSP値差を小さくするほど被覆率、深さが大きくなる。
【0137】
樹脂粒子(C)を構成する樹脂粒子(A)および/または(B)中に、添加剤(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を混合しても差し支えない。(A)または(B)中に添加剤を添加する方法としては、水系媒体中で水性樹脂分散体(X1)を形成させる際に混合してもよいが、あらかじめ樹脂(a)または樹脂(b)と添加剤を混合した後、水系媒体中にその混合物を加えて分散させたほうがより好ましい。
また、本発明においては、添加剤は、必ずしも、水系媒体中で粒子を形成させる時に混合しておく必要はなく、粒子を形成せしめた後、添加してもよい。たとえば、着色剤を含まない粒子を形成させた後、公知の染着の方法で着色剤を添加したり、有機溶剤(u)および/または可塑剤(v)とともに上記添加剤を含浸させることもできる。
添加剤として、前記有機酸塩(m)からなる荷電制御剤を樹脂粒子(A)中に含有させると、帯電特性が向上し好ましい。
(m)としては前述のものが挙げられ、使用量も同様である。
【0138】
また、本発明において、添加剤として、樹脂粒子(B)中に、樹脂(b)と共に、ワックス(c)、およびビニル系ポリマー鎖がグラフトした変性ワックス(d)を含有すると、耐熱保存安定性がより向上し好ましい。
(B)中の(c)の含有量は、好ましくは20%以下、さらに好ましくは1〜15%である。(d)の含有量は、好ましくは10%以下、さらに好ましくは0.5〜8%である。(c)と(d)の合計含有量は、好ましくは25%以下、さらに好ましくは1〜20%である。
【0139】
ワックス(c)はあらかじめ変性ワックス(d)と有機溶剤不存在下の溶融混練処理および/または有機溶剤(u)存在下加熱溶解混合処理した後に樹脂(b)に分散される。
ワックス(c)としては、ポリオレフィンワックス、パラフィンワックス、カルボニル基含有ワックスおよびこれらの混合物等が挙げられるが、このうち、とくに好ましいのはパラフィンワックス(c1)である。(c1)としては、融点50〜90℃で炭素数20〜36の直鎖飽和炭化水素を主成分とする石油系ワックスが挙げられる。
また、離型性の観点から、(c)のMnは、好ましくは400〜5000、さらに好ましくは1000〜3000、とくに1500〜2000である。尚、上記および以下においてワックスのMnは、GPCを用いて測定される(溶媒:オルソジクロロベンゼン、基準物質:ポリスチレン)。
【0140】
ワックス(c)は、ビニル系ポリマー鎖がグラフトした変性ワックス(d)と無溶媒下溶融混練処理および/または前記の有機溶剤(u)存在下の加熱溶解混合処理した後に、樹脂(b)に分散されるのが好ましい。この方法により、ワックス分散処理時に変性ワックス(d)を共存させることにより、(d)のワックス基部分が効率よく(c)表面に吸着、あるいはワックスのマトリクス構造内に一部絡みあうことにより、ワックス(c)表面と樹脂(b)との親和性が良好になり、(c)をより均一に樹脂粒子(B)中に内包することができ、分散状態の制御が容易になる。
【0141】
変性ワックス(d)は、ワックスにビニル系ポリマー鎖がグラフトしたものである。(d)に用いられるワックスとしては上記ワックス(c)と同様のものが挙げられ、好ましいものも同様である。(d)のビニル系ポリマー鎖を構成するビニル系モノマーとしては、前記ビニル樹脂を構成するモノマー(1)〜(10)と同様のものが挙げられるが、この中でとくに好ましいのは(1)、(2)、および(6)である。ビニル系ポリマー鎖はビニル系モノマーの単独重合体でもよいし、共重合体でもよい。
【0142】
変性ワックス(d)におけるワックス成分の量(未反応ワックスを含む)は、0.5〜99.5%が好ましく、さらに好ましくは1〜80%、とくに好ましくは5〜50%、最も好ましくは10〜30%である。また(d)のTgは、樹脂粒子(C)の耐熱保存安定性の観点から、好ましくは40〜90℃、さらに好ましくは50〜80℃である。
(d)のMnは、好ましくは1500〜10000、とくに1800〜9000である。Mnが1500〜10000の範囲では、樹脂粒子(C)の機械強度が良好である。
【0143】
変性ワックス(d)は、例えばワックス(c)を有機溶剤(例えばトルエンまたはキシレン)に溶解または分散させ、100〜200℃に加熱した後、ビニル系モノマーをパーオキサイド系開始剤(ベンゾイルパーオキサイド、ジターシャリーブチルパーオキサイド、ターシャリブチルパーオキサイドベンゾエート等)とともに滴下して重合後、有機溶剤を留去することにより得られる。
変性ワックス(d)の合成におけるパーオキサイド系開始剤の量は、(d)の原料の合計重量に基づいて、好ましくは0.2〜10%、さらに好ましくは0.5〜5%である。
【0144】
パーオキサイド重合開始剤としては、油溶性パーオキサイド重合開始剤および水溶性パーオキサイド重合開始剤等が用いられる。
油溶性パーオキサイド重合開始剤としては、アセチルシクロヘキシルスルホニルパーオキサイド、イソブチリルパーオキサイド、ジイソプロピルパーオキシジカーボネート、ジ−2−エチルヘキシルパーオキシジカーボネート、2,4−ジクロロベンゾイルパーオキサイド、t−ブチルパーオキシビバレート、3,5,5−トリメチルヘキサノニルパーオキサイド、オクタノイルパーオキサイド、デカノイルパーオキサイド、ラウロイルパーオキサイド、ステアロイルパーオキサイド、プロピオニトリルパーオキサイド、サクシニックアシッドパーオキサイド、アセチルパーオキサイド、t−ブチルパーオキシ−2−エチルヘキサノエート、ベンゾイルパーオキサイド、パラクロロベンゾイルパーオキサイド、t−ブチルパーオキシイソブチレート、t−ブチルパーオキシマレイックアシッド、t−ブチルパーオキシラウレート、シクロヘキサノンパーオキサイド、t−ブチルパーオキシイソプロピルカーボネート、2,5−ジメチル−2,5−ジベンゾイルパーオキシヘキサン、t−ブチルパーオキシアセテート、t−ブチルパーオキシベンゾエート、ジイソブチルジパーオキシフタレート、メチルエチルケトンパーオキサイド、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジt−ブチルパーオキシヘキサン、t−ブチルクミルパーオキサイド、t−ブチルヒドロパーオキサイド、ジt−ブチルパーオキサイド、ジイソプロピルベンゼンヒドロパーオキサイド、パラメンタンヒドロパーオキサイド、ピナンヒドロパーオキサイド、2,5−ジメチルヘキサン−2,5−ジヒドロパーオキサイドおよびクメンパーオキサイド等が挙げられる。
【0145】
水溶性パーオキサイド重合開始剤としては、例えば、過酸化水素、過酢酸、過硫酸アンモニウム、過硫酸カリウムおよび過硫酸ナトリウム等が挙げられる。
【0146】
ワックス(c)と変性ワックス(d)を混合する方法としては、〔1〕それぞれの融点以上の温度で溶融混練する方法、〔2〕(c)と(d)を有機溶剤(u)中に溶解あるいは懸濁させた後、冷却晶析、溶剤晶析等により液中に析出、あるいはスプレードライ等により気体中に析出させる方法、〔3〕(c)と(d)を有機溶剤(u)中に溶解あるいは懸濁させた後、分散機により機械的に湿式粉砕させる方法、等が挙げられる。これらの中では、〔2〕の方法が好ましい。
ワックス(c)および変性ワックス(d)を(b)中に分散させる方法としては、(c)および(d)と、(b)とを、それぞれ有機溶剤溶液もしくは分散液とした後、それら同士を混合する方法等が挙げられる。
【実施例】
【0147】
以下実施例により本発明をさらに説明するが、本発明はこれに限定されるものではない。以下の記載において「部」は重量部を示す。
【0148】
製造例1(樹脂粒子(A)の水性分散液の製造)
攪拌棒および温度計をセットした反応容器に、アジピン酸と1,4−ブタンジオール(モル比1:1)から得られたポリエステル(数平均分子量1000)177部、1,2−プロピレングリコール(以下プロピレングリコールと記載)7部、ジメチロールプロピオン酸72部、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸4部、およびアセトン500部を仕込む。この溶液にイソホロンジイソシアネート(IPDI)246部を仕込み55℃で11時間反応し、[ウレタンプレポリマー1]を得た。このプレポリマーにジーtーブチルサリチル酸Zn14部を加え、さらにトリエチルアミンを加え、ジメチロールプロピオン酸由来のカルボン酸を100当量%アミン中和し、均一溶液にする。この溶液を攪拌下、水1500部に加え、乳化する。さらに水320部、エチレンジアミン9部、n−ブチルアミン6部を加え、50℃、4時間伸長反応を行いウレタン系樹脂の水性分散液[微粒子分散液W1]を得た。[微粒子分散液W1]をELS−800で測定した体積平均粒径は0.09μmであった。[微粒子分散液W1]の一部を乾燥して樹脂分を単離し、該樹脂分のフローテスター測定によるガラス転移温度は62℃、軟化開始温度は105℃であり、流出温度は180℃であった。
【0149】
製造例2(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、スチレン80部、メタクリル酸85部、アクリル酸ブチル120部、過酸化ベンゾイル(25%含水品)62部の混合溶液を、120分間かけて滴下した。この重合溶液50部をさらに撹拌下のイオン水60部に滴下して、水性分散液[微粒子分散液W2]を得た。[微粒子分散液W2]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.09μmであった。[微粒子分散液W2]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは72℃、軟化開始温度は98℃であり、流出温度は175℃であった。
【0150】
製造例3(樹脂粒子(A)の水性分散液の製造)
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール377部、テレフタル酸ジメチルエステル350部、アジピン酸40部、および重縮合触媒としてテトラブトキシチタネート0.3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、フローテスター測定にて1000Pa・sが85℃になったになった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し[ポリエステルb0]を得た。[ポリエステルb0]のMwは3200であった。次にこの[ポリエステルb0]30部に対しアセトン30部を加え溶解させ、さらにトリエチルアミン2.5部を加えた。よく攪拌されているこの溶液に、水90部を加えることでポリエステル系樹脂の水性分散液[微粒子分散液W3]を得た。[微粒子分散液W3]をELS−800で測定した体積平均粒径は0.05μmであった。[微粒子分散液W3]の一部を乾燥して樹脂分を単離し、該樹脂分のフローテスター測定によるガラス転移温度は60℃、軟化開始温度は90℃であり、流出温度は155℃であった。
【0151】
製造例4(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、メタクリル酸メチル71部、酢酸ビニル143部、アクリル酸73部、過酸化ベンゾイル(25%含水品)25部の混合溶液を、120分間かけて滴下した。この重合溶液50部をさらに撹拌下のイオン交換水60部に滴下して、水性分散液[微粒子分散液W4]を得た。[微粒子分散液W4]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.05μmであった。[微粒子分散液W4]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは50℃、軟化開始温度は60℃であり、流出温度は120℃であった。
【0152】
製造例5(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、アクリル酸ブチル30部、スチレン25部、メタクリル酸45部、アルキルアリルスルホコハク酸のナトリウム塩(エレミノールJS−2、三洋化成工業製)8部、過酸化ベンゾイル(25%含水品)25部の混合溶液を、120分間かけて滴下した。この重合溶液29部をさらに撹拌下のイオン交換水60部に滴下して、水性分散液[微粒子分散液W5]を得た。[微粒子分散液W5]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.05μmであった。[微粒子分散液W5]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは120℃、軟化開始温度は160℃であり、流出温度は250℃であった。
【0153】
製造例6[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル126部とテレフタル酸200部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド1617部を滴下しながら60℃で4時間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタントリイソプロポキシテレフタレートと未反応のテレフタル酸の混合物(e1)(チタントリイソプロポキシテレフタレートの濃度65%)を得た。(e1)の水への溶解度は[0.6g/100ml]、さらに精製して得たチタントリイソプロポキシテレフタレートの水への溶解度は[0.9g/100ml]であった。
【0154】
製造例7[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル520部とイソフタル酸340部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド284部を滴下しながら60℃で5時間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタンジイソプロポキシジイソフタレートと未反応のイソフタル酸の混合物(e2)(チタンジイソプロポキシジイソフタレートの濃度95%)を得た。
(e2)の水への溶解度は[0.3g/100ml]、さらに精製して得たチタンジイソプロポキシジイソフタレートの水への溶解度も[0.3g/100ml]であった。
【0155】
製造例8[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル520部、安息香酸244部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド284部を滴下しながら60℃で5時間反応させた後、イオン交換水を50部入れ、30分間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタンジヒドロキシジベンゼンカルボキシレート(e3)を得た。(e3)の水への溶解度は[0.1g/100ml]であった。
【0156】
製造例9[線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物430部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部、無水マレイン酸10部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が5になった時点で取り出し、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−1)を得た。
(bX1−1)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は11、Tgは61℃、Mnは7040、Mwは17100、Mpは18900であった。分子量1500以下の成分の比率は1.0%であった。
【0157】
製造例10[非線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのEO2モル付加物350部、ビスフェノールAのPO3モル付加物326部、テレフタル酸278部、無水フタル酸40部及び重縮合触媒として(e1)2部を入れ、230℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が2以下になった時点で180℃に冷却し、無水トリメリット酸62部を加え、常圧密閉下2時間反応後取り出し、室温まで冷却後、粉砕して非線状ポリエステル樹脂(bX2−1)を得た。
(bX2−1)はTHF不溶分を含有しておらず、その酸価は34、水酸基価は18、Tgは68℃、Mnは3820、Mpは11100であった。分子量1500以下の成分の比率は0.8%であった。
【0158】
製造例11[線状ポリエステル樹脂の合成]
重縮合触媒を前記(e2)に代える以外は製造例9の(bX1−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−2)を得た。
(bX1−2)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は10、Tgは60℃、Mnは6690、Mwは17250、Mpは20080であった。分子量1500以下の成分の比率は0.8%であった。
【0159】
製造例12[非線状ポリエステル樹脂の合成]
重縮合触媒を前記(e2)に代える以外は製造例10の(bX2−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX2−2)を得た。
(bX2−2)はTHF不溶分を含有しておらず、その酸価は32、水酸基価は16、Tgは69℃、Mnは4220、Mpは11780であった。分子量1500以下の成分の比率は0.7%であった。
【0160】
製造例13[線状ポリエステル樹脂の合成]
重縮合触媒を(e3)に代える以外は製造例9の(bX1−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−3)を得た。
(bX1−3)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は10、Tgは61℃、Mnは6810、Mwは17400、Mpは20170であった。分子量1500以下の成分の比率は1.1%であった。
【0161】
製造例14[非線状ポリエステル樹脂の合成]
重縮合触媒を(e3)に代える以外は製造例10の(bX2−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX2−3)を得た。
(bX2−3)はTHF不溶分を含有しておらず、その酸価は31、水酸基価は14、Tgは66℃、Mnは4250、Mpは11920であった。分子量1500以下の成分の比率は0.7%であった。
【0162】
製造例15[ウレタン変性ポリエステル樹脂(ポリウレタン樹脂)の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物430部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部、無水マレイン酸10部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら6時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が12になった時点で80℃まで冷却し、酢酸エチル1000部を入れ、1時間攪拌した。その後イソホロンジイソシアネート60部を入れ2時間反応を行った後、室温まで冷却後粉砕してウレタン変性ポリエステル樹脂(bZ−1)の溶剤溶液(樹脂溶液i)を得た。
(bZ−1)はTHF不溶分を含有しておらず、その酸価は5、水酸基価は9、Tgは61℃、Mnは8790、Mpは13200であった。分子量1500以下の成分の比率は0.9%であった。
【0163】
製造例16[線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール701部、テレフタル酸ジメチルエステル716部、アジピン酸180部、および重縮合触媒として(e1)3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が150℃になった時点で取り出した。回収されたプロピレングリコールは316部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し線状ポリエステル樹脂(bX1−4)を得た。(bX1−4)はTHF不溶分を含有しておらず、その酸価は15、水酸基価は26、Tgは61℃、Mnは8800、Mwは27600、Mpは32200であった。分子量1500以下の成分の比率は4.0%であった。
【0164】
製造例17[非線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール557部、テレフタル酸ジメチルエステル569部、アジピン酸184部、および重縮合触媒として(e1)2部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に1時間反応させた。回収されたプロピレングリコールは175部であった。次いで180℃まで冷却し、無水トリメリット酸121部を加え、常圧密閉下2時間反応後、220℃、常圧で反応させ、軟化点が180℃になった時点で取り出し、室温まで冷却後、粉砕し粒子化し非線状ポリエステル樹脂(bX2−4)を得た。(bX2−4)はTHF不溶分を含有しておらず、その酸価は5、水酸基価は24、Tgは66℃、Mnは7200、Mpは41900であった。分子量1500以下の成分の比率は2.3%であった。
【0165】
製造例18[線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物420部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が13になった時点で取り出し、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−5)を得た。
(bX1−5)はTHF不溶分を含有しておらず、その酸価は15、水酸基価は13、Tgは55℃、Mnは2830、Mwは7100、Mpは6810であった。分子量1500以下の成分の比率は1.0%であった。
【0166】
製造例19[線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール700部、テレフタル酸ジメチルエステル700部、アジピン酸90部、および重縮合触媒として(e1)3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が90℃になった時点で取り出した。回収されたプロピレングリコールは316部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し線状ポリエステル樹脂(bX1−6)を得た。(bX1−6)はTHF不溶分を含有しておらず、その酸価は17、水酸基価は23、Tgは54℃、Mnは3300、Mwは9100、Mpは9400であった。分子量1500以下の成分の比率は3.8%であった。
【0167】
製造例20[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例9の(bX1−1)と同様に反応させた。触媒失活のために反応が途中で停止してしまい、生成水が留出しなくなる問題が生じたため、反応途中でチタンテトライソプロポキシド2部を4回追加し、比較用線状ポリエステル樹脂(CbX1−1)を得た。
(CbX1−1)は、THF不溶分を含有しておらず、その酸価は8、水酸基価は12、Tgは59℃、Mnは6180、Mwは13700、Mpは18600であった。分子量1500以下の成分の比率は2.4%であった。
【0168】
製造例21[比較用非線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例10の(bX2−1)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX2−1)を得た。
(CbX2−1)は、THF不溶分を含有しておらず、その酸価は35、水酸基価は14、Tgは67℃、Mnは3520、Mpは11800であった。分子量1500以下の成分の比率は2.3%であった。
【0169】
製造例22[比較用ウレタン変性ポリエステル樹脂(ポリウレタン樹脂)の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例15の(bZ−1)と同様に反応させ比較用ウレタン変性ポリエステル樹脂(CbZ−1)の溶剤溶液(樹脂溶液ii)を得た。
(CbZ−1)は、THF不溶分を含有しておらず、その酸価は4、水酸基価は11、Tgは61℃、Mnは7800、Mpは13000であった。分子量1500以下の成分の比率は1.2%であった。
【0170】
製造例23[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例16の(bX1−4)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX1−2)を得た。
(CbX1−2)は、THF不溶分を含有しておらず、その酸価は13、水酸基価は28、Tgは61℃、Mnは7830、Mwは27400、Mpは33900であった。分子量1500以下の成分の比率は4.6%であった。
【0171】
製造例24[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例17の(bX2−4)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX2−2)を得た。
(CbX2−2)は、THF不溶分を含有しておらず、その酸価は13、水酸基価は28、Tgは61℃、Mnは7800、Mpは33900であった。分子量1500以下の成分の比率は4.6%であった。
【0172】
製造例25(着色剤分散液の製造)
ビーカー内に銅フタロシアニン20部と着色剤分散剤(ソルスパーズ28000;アビシア株式会社製)4部、および酢酸エチル76部を入れ、攪拌して均一分散させた後、ビーズミルによって銅フタロシアニンを微分散して、[着色剤分散液1]を得た。[着色剤分散液1]をLA−920で測定した体積平均粒径は0.3μmであった。
【0173】
製造例26(変性ワックスの製造)
温度計および撹拌機の付いたオートクレーブ反応槽中に、キシレン454部、低分子量ポリエチレン(三洋化成工業(株)製 サンワックス LEL−400:軟化点128℃)150部を投入し、窒素置換後170℃に昇温して十分溶解し、スチレン595部、メタクリル酸メチル255部、ジ−t−ブチルパーオキシヘキサヒドロテレフタレート34部およびキシレン119部の混合溶液を170℃で3時間で滴下して重合し、さらにこの温度で30分間保持した。次いで脱溶剤を行い、[変性ワックス 1]を得た。[変性ワックス 1]のグラフト鎖のsp値は 10.35(cal/cm3)1/2、Mnは1872、Mwは5194、Tgは56.9℃であった。
【0174】
製造例27(ワックス分散液の製造)
温度計および撹拌機の付いた反応容器中に、パラフィンワックス(融点73℃)10部、[変性ワックス1]1部、酢酸エチル33部を投入し、78℃に加熱して充分溶解し、1時間で30℃まで冷却を行いワックスを微粒子状に晶析させ、さらにウルトラビスコミル(アイメックス製)で湿式粉砕し、[ワックス分散液1]を得た。
【0175】
製造例28(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−1)40部とポリエステル樹脂(bX2−1)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液1]を得た
【0176】
製造例29(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−2)40部とポリエステル樹脂(bX2−2)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液2]を得た
【0177】
製造例30(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−3)40部とポリエステル樹脂(bX2−3)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液3]を得た
【0178】
製造例31(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−1)40部とウレタン変性ポリエステル樹脂(bZ−1)の溶剤溶液[樹脂溶液i]120部および酢酸エチル40部を入れ、攪拌して均一分散させ、[樹脂溶液4]を得た
【0179】
製造例32(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−4)60部とポリエステル樹脂(bX2−4)40部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液5]を得た
【0180】
製造例33(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−5)60部とポリエステル樹脂(bX2−1)40部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液6]を得た。
【0181】
製造例34(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−6)80部とポリエステル樹脂(bX2−1)20部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液7]を得た。
【0182】
製造例35(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−1)40部と(CbX2−1)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液8]を得た
【0183】
製造例36(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−1)40部とウレタン変性ポリエステル樹脂(CbZ−1)の溶剤溶液[樹脂溶液ii]120部および酢酸エチル40部を入れ、攪拌して均一分散させ、[樹脂溶液9]を得た
【0184】
製造例37(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−2)40部とポリエステル樹脂(CbX2−2)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液10]を得た
【0185】
実施例1
ビーカー内に[樹脂溶液1]60部、[ワックス分散液1]27部、および[着色剤分散液1]10部を入れ、25℃にてTK式ホモミキサーで8,000rpmで撹拌し、均一に溶解、分散させて[樹脂溶液1A]を得た。
ビーカー内にイオン交換水92部、[微粒子分散液W1]30.8部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)10部を入れ均一に溶解した。ついで25℃で、TK式ホモミキサーを10,000rpmに撹拌しながら、[樹脂溶液1A]75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまで酢酸エチルを留去し、(B)で構成されたコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成された樹脂粒子の水性樹脂分散体(XF1)を得た。次いで濾別し、40℃×18時間乾燥を行い、揮発分を0.5%以下として、樹脂粒子(F1)を得た。
【0186】
実施例2
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF2)および、樹脂粒子(F2)を得た。
【0187】
実施例3
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W3]27.2部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF3)および、樹脂粒子(F3)を得た。
【0188】
実施例4
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液2]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF4)および、樹脂粒子(F4)を得た。
【0189】
実施例5
実施例4の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、実施例4と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF5)および、樹脂粒子(F5)を得た。
【0190】
実施例6
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液3]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF6)および、樹脂粒子(F6)を得た。
【0191】
実施例7
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液4]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF7)および、樹脂粒子(F7)を得た。
【0192】
実施例8
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液5]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF8)および、樹脂粒子(F8)を得た。
【0193】
実施例9
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W4]15部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF9)および、樹脂粒子(F9)を得た。
【0194】
実施例10
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W5]70部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF10)および、樹脂粒子(F10)を得た。
【0195】
実施例11
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液6]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF11)および、樹脂粒子(F11)を得た。
【0196】
実施例12
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液7]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF12)および、樹脂粒子(F12)を得た。
【0197】
比較例1
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液8]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF13)および、樹脂粒子(F13)を得た。
【0198】
比較例2
比較例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、比較例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF14)および、樹脂粒子(F14)を得た。
【0199】
比較例3
比較例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W3]27.2部を用いる他は、比較例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF15)および、樹脂粒子(F15)を得た。
【0200】
比較例4
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液9]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF16)および、樹脂粒子(F16)を得た。
【0201】
比較例5
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液10]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF17)および、樹脂粒子(F17)を得た。
【0202】
物性測定例
実施例1〜12および比較例1〜5で得た樹脂粒子(F1)〜(F17)を水に分散して粒度分布をコールターカウンターで測定した。また、樹脂粒子の平均円形度、帯電特性、耐熱保存安定性、および低温定着性を測定した。その結果を表1に示す。なお、○は請求項4の四角形ABCDの内部、×は外部を意味する。
【0203】
【表1】

【0204】
帯電特性、耐熱保存安定性、溶融性、および画像密着性の測定方法は以下の通りである。なお、これら以外の表1記載の物性の測定方法は、いずれも前記方法による。
【0205】
〔帯電特性〕(帯電量)
50ccの共栓付ガラス瓶に、樹脂粒子0.5g、鉄粉(日本鉄粉株式会社製「F−150」)10gを精秤し、共栓をして23℃、50%RHの雰囲気下でターブラシェーカミキサー(ウイリー・ア・バショッフェン社製)にセットし、回転数90rpmで2分攪拌する。攪拌後の混合粉体0.2gを目開き20μmステンレス金網がセットされたブローオフ粉体帯電量測定装置(京セラケミカル株式会社製TB−203)に装填し、ブロー圧10KPa,吸引圧5KPaの条件で、残存鉄粉の帯電量を測定し、定法により樹脂粒子の帯電量を算出する。なお、トナー用としてはマイナス帯電量が高いほど帯電特性が優れている。
【0206】
〔耐熱保存安定性1〕
50℃に温調された乾燥機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○ : ブロッキングが発生しない。
△ : ブロッキングが発生するが、力を加えると容易に分散する。
× : ブロッキングが発生し、力を加えても分散しない。
〔耐熱保存安定性2〕
50℃に温調された乾燥機に樹脂粒子2gを入れた100ml容量のプラスチックカップを72時間静置し、ブロッキングの程度により下記の基準で評価した。
◎ : 試験後のプラスチックカップを45度に傾けると樹脂粒子が流動する。
○ : 試験後のプラスチックカップを45度に傾けても樹脂粒子は流動しないが、4
5度に傾けた状態でカップ底に軽く叩くなどして弱い刺激を与えると樹脂粒子
が流動する。
△ : 以上◎、○の条件を満たさないが、指で軽く摘むなどして力を加えると容易に
分散する。
× : ブロッキングが発生し、力を加えても試験前の樹脂粒子の状態に戻らない。
【0207】
〔溶融性〕
樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、よく混ぜて均一にした後、この粉体を紙面上に0.6mg/cm2となるよう均一に載せる(このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通した時のコールドオフセットの発生温度を測定した。
【0208】
〔画像密着性〕
樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、よく混ぜて均一にした後、この粉体を紙面上に0.6mg/cm2となるよう均一に載せる(このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通して作成した定着画像を、一定荷重(500g分銅)で折り曲げ、折り曲げ部での画像濃度を測定し(グレタグ・マクベス社製反射濃度計「RD−19」使用)、画像濃度(折り曲げ後)/画像濃度(折り曲げ前)が60%となる温度を画像密着温度とした。
【産業上の利用可能性】
【0209】
本発明の樹脂分散体および樹脂粒子は、粒径が均一で、帯電特性、耐熱保存安定性等に優れるため、スラッシュ成形用樹脂、粉体塗料、液晶等の電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真、静電記録、静電印刷などに用いられるトナー、各種ホットメルト接着剤、その他成形材料等に用いる樹脂粒子として極めて有用である。
【図面の簡単な説明】
【0210】
【図1】本発明における、(a)と(b)のsp値差(K)と、(a)の重量平均分子量(Mw)の自然対数値ln(Mw)(H)の関係を示すグラフである。
【図2】樹脂粒子のフローテスター測定におけるフローチャートを示す概念図である。
【技術分野】
【0001】
本発明は樹脂分散体および樹脂粒子に関する。さらに詳しくは、粉体塗料、電子写真トナー、静電記録トナー等の各種用途に有用な、樹脂粒子およびその水性分散体に関する。
【背景技術】
【0002】
粒径が均一で、かつ、電気的特性、熱的特性、化学的安定性等に優れた樹脂粒子として、ポリマー微粒子を分散安定剤として得られた樹脂粒子が知られている(特許文献1参照)。
【特許文献1】特開2002−284881号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
しかしながら、樹脂粒子の溶融特性を向上させる(より低温で溶融させる)ためには、分子量やガラス転移温度(以下Tgと略す)を下げる必要があるが、そうした場合、高温高湿度下での樹脂粒子の耐ブロッキング性が劣るという問題点を有していた。
本発明は従来技術における上記の事情に鑑みてなされたものである。すなわち、溶融特性と耐ブロッキング性に優れ、粒径が均一である樹脂粒子を提供することを目的とする。
【課題を解決するための手段】
【0004】
本発明者らは、上記の問題点を解決するべく鋭意検討した結果、本発明に到達した。
すなわち本発明は、下記〔I〕〜〔III〕である。
〔I〕 樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られる、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
Ti(−X)m(−OR)n (I)
[式中、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、他のカルボキシル基が同一分子内のOR基と分子内で重縮合し環構造を形成していてもよく、または、別の分子のOR基と分子間で重縮合し2〜5個のTi原子を含む構造を形成していてもよい。m=1〜3、n=1〜3であり、mとnの和は4である。]
〔II〕 上記水性樹脂分散体(X1)中において、樹脂粒子(B)に付着された樹脂粒子(A)が、有機溶剤に溶解される、および/または、溶融されることにより、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されて得られる樹脂粒子(C2)からなる水性樹脂分散体(X2)。
〔III〕 〔I〕または〔II〕の水性樹脂分散体から水性媒体が除去されてなる樹脂粒子。
【発明の効果】
【0005】
本発明の樹脂分散体およびそれから得られる樹脂粒子は以下の効果を有する。
1.溶融特性、耐ブロッキング性に優れ、粒径が均一である。
2.環境上問題あるスズ化合物を触媒として使用しなくとも、良好な樹脂性能を有する。
3.帯電特性、粉体流動性に優れる。
4.粒子表面の平滑性に優れる(特に皮膜化されたシェル層を有する場合)。
5.水中で分散により得ることが可能な樹脂粒子であるため、低コストで製造できる。
6.加熱溶融した塗膜の機械的物性も良好である。
【発明を実施するための最良の形態】
【0006】
以下、本発明について詳細に説明する。
本発明に用いるチタン含有触媒(e)は、前記式(I)で表される化合物であり、式(I)の構造のものを2種以上併用してもよい。
一般式(I)において、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。炭化水素基の炭素数は、好ましくは1〜6、さらに好ましくは1〜4である。
炭素数1〜24の炭化水素基の具体例としては、脂肪族炭化水素基並びにエーテル結合および/もしくは水酸基を含む脂肪族炭化水素基(メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、n−ヘキシル基、n−オクチル基、β−メトキシエチル基、β−エトキシエチル基、およびβ−ヒドロキシエチル基など)、芳香族炭化水素基並びにエーテル結合および/もしくは水酸基を含む芳香族炭化水素基[フェニル基;ヒドロキシフェニル基;ビスフェノールA、ビスフェノールFおよびビスフェノールADなどの炭素数2〜4のアルキレンオキシド(以下、特記しない限り、AOと略記する)〔エチレンオキシド(以下EOと略記する)、プロピレンオキシド(以下POと略記する)、およびブチレンオキシド(以下BOと略記する)など〕付加物(付加モル数1〜3)から1個のOHを除いた残基など]が挙げられる。
これらRのうち好ましくは、炭素数1〜6の炭化水素基であり、さらに好ましくは、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、およびn−ヘキシル基であり、とくに好ましくは、n−プロピル基、イソプロピル基、およびn−ブチル基である。
【0007】
Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、Ti原子に結合し残基を形成するのと別のカルボキシル基が、同一分子内のOR基{Ti原子に直接結合した水酸基(RがHの場合)、アルコキシ基(Rが炭化水素基の場合)、またはRが1〜2個の水酸基を含む炭化水素基の場合の該水酸基}と分子内で重縮合し環構造を形成していてもよく、チタン含有触媒(a)の別の分子のOR基(上記と同様)と分子間で重縮合し、複数のTi原子を含む繰り返し構造を形成していてもよい。
上記芳香族カルボン酸としては、炭素数7〜50のものが好ましく、安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸など)、ナフタレンモノカルボン酸などの芳香族モノカルボン酸;フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸など)、ナフタレンジカルボン酸、トリメリット酸、およびピロメリット酸などの2〜6価またはそれ以上の芳香族ポリカルボン酸;が挙げられる。
芳香族ポリカルボン酸の場合、前述のようにその複数のカルボキシル基により、複数のTi原子を含む繰り返し構造を形成していてもよいが、この場合の1分子内のTi原子数は2〜5である。1分子内のTi原子数が6以上の場合、触媒活性が低下し好ましくない。
Xとして好ましいものは、フタル酸類(テレフタル酸、イソフタル酸、オルトフタル酸など)の残基、および安息香酸類(安息香酸、パラヒドロキシ安息香酸、パラメチル安息香酸など)の残基であり、特に好ましいものはテレフタル酸、イソフタル酸、およびオルトフタル酸の残基である。
【0008】
式(I)中、m=1〜3、n=1〜3であり、mとnの和、すなわちTi原子の結合価数は4である。好ましくは、m=1〜2、n=2〜3である。mが3を超えると触媒活性が低下し、nが3を超えると耐加水分解性が低下し、いずれもポリエステル製造上好ましくない。mが1または2の場合、触媒活性が特に高く好ましい。Ti原子の結合価数が4以外の場合は、式(I)と類似の構造でも触媒活性が劣るか副反応が起き好ましくない。
【0009】
一般式(I)で表される化合物の具体例としては、チタントリイソプロポキシベンゼンカルボキシレート、チタントリブトキシベンゼンカルボキシレート、チタントリイソプロポキシテレフタレート、チタントリブトキシテレフタレート、チタントリイソプロポキシイソフタレート、チタントリイソプロポキシフタレート、チタンジイソプロポキシジベンゼンカルボキシレート、チタンジブトキシジベンゼンカルボキシレート、チタンジイソプロポキシジテレフタレート、チタンジブトキシジテレフタレート、チタンジイソプロポキシジイソフタレート、チタンジイソプロポキシジフタレート、チタンジヒドロキシジベンゼンカルボキシレート、チタンジヒドロキシジテレフタレート、チタンジヒドロキシジイソフタレート、チタンジヒドロキシジフタレート、チタンイソプロポキシトリテレフタレート、およびこれらの分子内または分子間重縮合物などが挙げられる。
【0010】
本発明に用いるチタン含有触媒(e)は、ポリエステル重合時の触媒活性の観点から、30℃の水への溶解度が[5g/100ml]以下であることが好ましく、[2g/100ml]以下であることがさらに好ましく、[1g/100ml]以下であることがとくに好ましい。溶解度が[5g/100ml]以下であると、重合反応時に触媒が加水分解を受けにくく、触媒活性の持続性の観点から好ましい。
【0011】
これらのチタン含有触媒(e)は、例えば、市販されているチタンテトラアルコキシドと芳香族カルボン酸を、酢酸エチル中で70〜90℃にて反応させることで得ることができる。
【0012】
本発明に用いるポリエステル樹脂(b1)を構成する重縮合ポリエステル樹脂としては、ポリオールとポリカルボン酸との重縮合物であるポリエステル樹脂(bX)、(bX)にさらにポリエポキシド(f)などを反応させて得られる変性ポリエステル樹脂(bY)などが挙げられる。(bX)、(bY)などは単独で使用してもよいし、2種以上を組み合わせて混合物として使用してもよい。
ポリオールとしては、ジオール(g)および3価以上のポリオール(h)が、ポリカルボン酸としては、ジカルボン酸(i)および3価以上のポリカルボン酸(j)が挙げられ、それぞれ2種以上を併用してもよい。
ポリエステル樹脂(bX)および(bY)としては、以下のものなどが挙げられ、これらのものを併用することもできる。
(bX1):(g)および(i)を用いた線状のポリエステル樹脂
(bX2):(g)および(i)とともに(h)および/または(j)を用いた非線状のポリエステル樹脂
(bY1):(bX2)に(f)を反応させた変性ポリエステル樹脂
【0013】
ジオール(g)としては、水酸基価180〜1900(mgKOH/g、以下同様)のものが好ましい。具体的には、アルキレングリコール(エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール、オクタンジオール、デカンジオール、ドデカンジオール、テトラデカンジオール、ネオペンチルグリコール、2,2−ジエチル−1,3−プロパンジオールなど);アルキレンエーテルグリコール(ジエチレングリコール、トリエチレングリコール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコールおよびポリブチレングリコールなど);脂環式ジオール(1,4−シクロヘキサンジメタノールおよび水素添加ビスフェノールAなど);上記脂環式ジオールの炭素数2〜4のアルキレンオキシド〔エチレンオキシド(以下EOと略記する)、プロピレンオキシド(以下POと略記する)およびブチレンオキシド(以下BOと略記する)など〕付加物(付加モル数1〜30);ビスフェノール類(ビスフェノールA、ビスフェノールFおよびビスフェノールSなど)の炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数2〜30)などが挙げられる。
これらのうち好ましいものは、炭素数2〜12のアルキレングリコール、ビスフェノール類のアルキレンオキシド付加物およびこれらの併用であり、とくに好ましいものはビスフェノール類のアルキレンオキシド付加物、炭素数2〜4のアルキレングリコールおよびこれらの2種以上の併用であり、もっとも好ましいものは、炭素数2〜4のアルキレングリコールである。
なお、上記および以下において水酸基価および酸価は、JIS K 0070に規定の方法で測定される。
【0014】
ジオール(g)としては、上記のヒドロキシル基以外の官能基を有しないジオール以外に、他の官能基を有するジオール(g1)を用いてもよい。(g1)としては、カルボキシル基を有するジオール、スルホン酸基もしくはスルファミン酸基を有するジオール、およびこれらの塩等が挙げられる。
カルボキシル基を有するジオールとしては、ジアルキロールアルカン酸[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸(DMPA)、2,2−ジメチロールブタン酸、2 ,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸など]が挙げられる。
スルホン酸基もしくはスルファミン酸基を有するジオールとしては、スルファミン酸ジオール[N,N−ビス(2−ヒドロキシアルキル)スルファミン酸(アルキル基のC1〜6)またはそのAO付加物(AOとしてはEOまたはPOなど、AOの付加モル数1〜6):例えばN,N−ビス(2−ヒドロキシエチル)スルファミン酸およびN,N−ビス(2−ヒドロキシエチル)スルファミン酸PO2モル付加物など];ビス(2−ヒドロキシエチル)ホスフェートなどが挙げられる。
これらの中和塩基を有するジオールの中和塩基としては、例えば前記炭素数3〜30の3級アミン(トリエチルアミンなど)および/またはアルカリ金属(ナトリウム塩など)が挙げられる。
これらのうち好ましいものは、炭素数2〜12のアルキレングリコール、カルボキシル基を有するジオール、ビスフェノール類のAO付加物、およびこれらの併用である。
【0015】
3価以上(3〜8価またはそれ以上)のポリオール(h)としては、水酸基価150〜1900のものが好ましい。具体的には、炭素数3〜36の3〜8価またはそれ以上の脂肪族多価アルコール(アルカンポリオールおよびその分子内もしくは分子間脱水物、例えば、グリセリン、トリエチロールエタン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール、ソルビタン、ポリグリセリン、およびジペンタエリスリトール;糖類およびその誘導体、例えば蔗糖およびメチルグルコシド;など);上記脂肪族多価アルコールの炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数1〜30);トリスフェノール類(トリスフェノールPAなど)の炭素数2〜4のアルキレンオキシド(EO、POおよびBOなど)付加物(付加モル数2〜30);ノボラック樹脂(フェノールノボラックおよびクレゾールノボラックなど:平均重合度3〜60)の炭素数2〜4のアルキレンオキシド(EO、PO、BOなど)付加物(付加モル数2〜30)などが挙げられる。
これらのうち好ましいものは、3〜8価またはそれ以上の脂肪族多価アルコールおよびノボラック樹脂のアルキレンオキシド付加物(付加モル数2〜30)であり、とくに好ましいものはノボラック樹脂のアルキレンオキシド付加物である。
【0016】
ジカルボン酸(i)としては、酸価180〜1250(mgKOH/g、以下同様)のものが好ましい。具体的には、アルキレンジカルボン酸(コハク酸、アジピン酸、セバシン酸、ドデセニルコハク酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸、オクタデカンジカルボン酸など);アルケニレンジカルボン酸(マレイン酸、フマール酸など);炭素数8以上の分岐アルキレンジカルボン酸[ダイマー酸、アルケニルコハク酸(ドデセニルコハク酸、ペンタデセニルコハク酸、オクタデセニルコハク酸など)、アルキルコハク酸(デシルコハク酸、ドデシルコハク酸、オクタデシルコハク酸など);芳香族ジカルボン酸(フタル酸、イソフタル酸、テレフタル酸、ナフタレンジカルボン酸など)などが挙げられる。これらのうち好ましいものは、炭素数4〜20のアルケニレンジカルボン酸および炭素数8〜20の芳香族ジカルボン酸である。なお、(i)としては、上述のものの酸無水物または低級アルキル(炭素数1〜4)エステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0017】
3価以上(3〜6価またはそれ以上)のポリカルボン酸(j)としては、酸価150〜1250のものが好ましい。具体的には、炭素数9〜20の芳香族ポリカルボン酸(トリメリット酸、ピロメリット酸など);不飽和カルボン酸のビニル重合体[数平均分子量(以下Mnと記載、ゲルパーミエーションクロマトグラフィー(GPC)による):450〜10000](スチレン/マレイン酸共重合体、スチレン/アクリル酸共重合体、α−オレフィン/マレイン酸共重合体、スチレン/フマル酸共重合体など)などが挙げられる。これらのうち好ましいものは、炭素数9〜20の芳香族ポリカルボン酸であり、とくに好ましいものはトリメリット酸、およびピロメリット酸である。なお、3価以上のポリカルボン酸(j)としては、上述のものの酸無水物または低級アルキル(炭素数1〜4)エステル(メチルエステル、エチルエステル、イソプロピルエステルなど)を用いてもよい。
【0018】
また、(g)、(h)、(i)および(j)とともに炭素数4〜20の脂肪族または芳香族ヒドロキシカルボン酸(k)、炭素数6〜12のラクトン(l)を共重合することもできる。
ヒドロキシカルボン酸(k)としては、ヒドロキシステアリン酸、硬化ヒマシ油脂肪酸などが挙げられる。ラクトン(l)としては、カプロラクトンなどが挙げられる。
【0019】
ポリエポキシド(f)としては、ポリグリシジルエーテル〔エチレングリコールジグリシジルエーテル、テトラメチレングリコールジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールFジグリシジルエーテル、グリセリントリグリシジルエーテル、ペンタエリスリトールテトラグリシジルエーテル、フェノールノボラック(平均重合度3〜60)グリシジルテーテル化物など〕;ジエンオキサイド(ペンタジエンジオキサイド、ヘキサジエンジオキサイドなど)などが挙げられる。これらの中で好ましくは、ポリグリシジルエーテルであり、さらに好ましくは、エチレングリコールジグリシジルエーテルおよびビスフェノールAジグリシジルエーテルである。
(f)の1分子当たりのエポキシ基数は、好ましくは2〜8、さらに好ましくは2〜6、とくに好ましくは2〜4である。
(f)のエポキシ当量は、好ましくは50〜500である。下限は、さらに好ましく70、とくに好ましくは80であり、上限は、さらに好ましく300、とくに好ましくは200である。エポキシ基数とエポキシ当量が上記範囲内であると、現像性と定着性が共に良好である。上述の1分子当たりのエポキシ基数およびエポキシ当量の範囲を同時に満たせばさらに好ましい。
【0020】
ポリオールとポリカルボン酸の反応比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/2、さらに好ましくは1.5/1〜1/1.3、とくに好ましくは1.3/1〜1/1.2である。また使用するポリオールとポリカルボン酸の種類は、最終的に調整される樹脂粒子のガラス転移点が45〜85℃となるよう分子量調整も考慮して選択される。
【0021】
本発明において、樹脂(b)中の分子量1500以下の成分の比率は、1.8%以下が好ましく、さらに好ましくは1.3%以下、とくに好ましくは1.1%以下である。分子量1500以下の成分の比率が1.8%以下になることで、保存安定性がより良好となる。上記および以下において、%は特に記載のない限り、重量%を意味する。
【0022】
上記および以下において、本発明における樹脂(ポリウレタン樹脂を除く)の、ピークトップ分子量(Mp)、Mn、重量平均分子量(Mw)、および分子量1500以下の成分の比率は、THF可溶分についてGPCを用いて以下の条件で測定される。
装置(一例) : 東ソー製 HLC−8120
カラム(一例): TSKgelGMHXL(2本)
TSKgelMultiporeHXL−M(1本)
試料溶液 : 0.25%のTHF溶液
溶液注入量 : 100μl
流量 : 1ml/分
測定温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
得られたクロマトグラム上最大のピーク高さを示す分子量をピークトップ分子量(Mp)と称する。さらに分子量1500で分割したときのピーク面積の比率で低分子量物の存在比を評価する。
なお、ポリウレタン樹脂のMp、Mn、Mw、および分子量1500以下の成分の比率は、GPCを用いて以下の条件で測定される。
装置(一例) : 東ソー製 HLC−8220GPC
カラム(一例): Guardcolumn α
TSKgel α−M
試料溶液 : 0.125%のジメチルホルムアミド溶液
溶液注入量 : 100μl
流量 : 1ml/分
温度 : 40℃
検出装置 : 屈折率検出器
基準物質 : 東ソー製 標準ポリスチレン(TSKstandard POLYSTYRENE)12点(分子量 500 1050 2800 5970 9100 18100 37900 96400 190000 355000 1090000 2890000)
【0023】
ポリエステル樹脂(b1)の酸価は、好ましくは0.1〜60、さらに好ましくは0.2〜50、特に0.5〜40である。酸価が0.1〜60の範囲では、帯電性が良好である。
ポリエステル樹脂(b1)の水酸基価は、好ましくは1〜70、さらに好ましくは3〜60、特に5〜55である。酸価が1〜70の範囲では、環境安定性が良好である。
ポリエステル樹脂(b1)のTgは、好ましくは40〜90℃、さらに好ましくは50〜80℃、とくに55〜75℃である。Tgが40℃〜90℃の範囲では耐熱保存安定性と低温定着性が良好である。
なお、上記および以下においてポリエステル樹脂(b1)のTgは、セイコー電子工業(株)製DSC20,SSC/580を用いてASTM D3418−82に規定の方法(DSC法)で測定される。
【0024】
本発明においてポリエステル樹脂(b1)は、通常のポリエステルの製造法と同様にして製造することができる。例えば、不活性ガス(窒素ガス等)雰囲気中で、チタン含有触媒(e)の存在下、反応温度が好ましくは150〜280℃、さらに好ましくは160〜250℃、とくに好ましくは170〜240℃で反応させることにより行うことができる。また反応時間は、重縮合反応を確実に行う観点から、好ましくは30分以上、とくに2〜40時間である。反応末期の反応速度を向上させるために減圧する(例えば1〜50mmHg)ことも有効である。
(e)の添加量としては、重合活性などの観点から、得られる重合体の重量に対して、好ましくは0.0001〜0.8%、さらに好ましくは0.0002〜0.6%、とくに好ましくは0.0015〜0.55%である。
また、(e)の触媒効果を損なわない範囲で他のエステル化触媒を併用することもできる。他のエステル化触媒の例としては、スズ含有触媒(例えばジブチルスズオキシド)、三酸化アンチモン、(e)以外のチタン含有触媒〔例えばチタンアルコキシド、シュウ酸チタニルカリウム、テレフタル酸チタン、およびチタンジヒドロキシビス(トリエタノールアミネート)とその分子内重縮合物〕、ジルコニウム含有触媒(例えば酢酸ジルコニル)、ゲルマニウム含有触媒、アルカリ(土類)金属触媒(例えばアルカリ金属もしくはアルカリ土類金属のカルボン酸塩:酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸カルシウム、安息香酸ナトリウム、および安息香酸カリウムなど)、および酢酸亜鉛等が挙げられる。これらの他の触媒の添加量としては、得られる重合体に対して、0〜0.6%が好ましい。0.6%以内とすることで、ポリエステル樹脂の着色が少なくなり、カラー用のトナーに用いるのに好ましい。添加された全触媒中の(e)の含有率は、50〜100%が好ましい。
【0025】
線状のポリエステル樹脂(bX1)の製造方法としては、例えば、得られる重合体の重量に対して0.0001〜0.8%の触媒(e)と、必要により他の触媒の存在下、ジオール(g)、およびジカルボン酸(i)を、180℃〜260℃に加熱し、常圧および/または減圧条件で脱水縮合させて、(bX1)を得る方法が挙げられる。
【0026】
非線状のポリエステル樹脂(bX2)の製造方法としては、例えば、得られる重合体の重量に対して0.0001〜0.8%の触媒(e)と、必要により他の触媒の存在下、ジオール(g)、ジカルボン酸(i)、および3価以上のポリオール(h)を、180℃〜260℃に加熱し、常圧および/または減圧条件で脱水縮合させた後、さらに3価以上のポリカルボン酸(j)を反応させて、(bX2)を得る方法が挙げられる。(j)を、(g)、(i)および(h)と同時に反応させることもできる。
【0027】
変性ポリエステル樹脂(bY1)の製造方法としては、ポリエステル樹脂(bX2)にポリエポキシド(f)を加え、180℃〜260℃でポリエステルの分子伸長反応を行うことで、(bY1)を得る方法が挙げられる。
(f)と反応させる(bX2)の酸価は、好ましくは1〜60、さらに好ましくは5〜50である。酸価が1以上であると、(f)が未反応で残存して樹脂の性能に悪影響を及ぼす恐れがなく、60以下であると、樹脂の熱安定性が良好である。
また、(bY1)を得るのに用いる(f)の量は、低温定着性および耐ホットオフセット性の観点から、(bX2)に対して、好ましくは0.01〜10%、さらに好ましくは0.05〜5%である。
【0028】
ポリエステル樹脂を2種以上併用する場合、および少なくとも1種のポリエステル樹脂と他の樹脂を混合する場合、粉体混合または溶融混合してもよい。
溶融混合する場合の温度は、好ましくは80〜180℃、さらに好ましくは100〜170℃、とくに好ましくは120〜160℃である。
混合温度が低すぎると充分に混合できず、不均一となることがある。2種以上のポリエステル樹脂を混合する場合、混合温度が高すぎると、エステル交換反応による平均化などが起こるため、必要な樹脂物性が維持できなくなる場合がある。
【0029】
溶融混合する場合の混合時間は、好ましくは10秒〜30分、さらに好ましくは20秒〜10分、とくに好ましくは30秒〜5分である。2種以上のポリエステル樹脂を混合する場合、混合時間が長すぎると、エステル交換反応による平均化などが起こるため、必要な樹脂物性が維持できなくなる場合がある。
溶融混合する場合の混合装置としては、反応槽などのバッチ式混合装置、および連続式混合装置が挙げられる。適正な温度で短時間で均一に混合するためには、連続式混合装置が好ましい。連続式混合装置としては、エクストルーダー、コンティニアスニーダー、3本ロールなどが挙げられる。これらのうちエクストルーダーおよびコンティニアスニーダーが好ましい。
粉体混合する場合は、通常の混合条件および混合装置で混合することができる。
粉体混合する場合の混合条件としては、混合温度は、好ましくは0〜80℃、さらに好ましくは10〜60℃である。混合時間は、好ましくは3分以上、さらに好ましくは5〜60分である。混合装置としては、ヘンシェルミキサー、ナウターミキサー、およびバンバリーミキサー等が挙げられる。好ましくはヘンシェルミキサーである。
【0030】
また、ポリエステル樹脂(b1)を構成単位として含有する樹脂(b2)としては、(b1)を構成単位として含有するものであればとくに限定されないが、例えば、ポリウレタン樹脂の場合、後述する樹脂(a)の項で述べたポリウレタン樹脂において、活性水素基含有化合物(D)の少なくとも一部として、(b1)を用いることにより得られる。(D)中の(b1)の含有量は、好ましくは60%以上、さらに好ましくは80%以上、とくに好ましくは100%である。
【0031】
また、(b)中に、(b1)および(b2)以外に、必要により、他の樹脂を含有させることもできる。
他の樹脂としては、(e)以外の触媒の存在下に形成されたポリエステル樹脂、ビニル系樹脂、エポキシ樹脂、ポリウレタン樹脂などが挙げられる。その具体例については、後述する(a)と同様のものが使用できる。
他の樹脂のMwは、好ましくは1000〜200万である。
樹脂(b)における他の樹脂の含有量は、好ましくは0〜40%、さらに好ましくは0〜30%、とくに好ましくは0〜20%である。また、(b)中のポリエステル以外の樹脂の含有量は、樹脂物性の点から、好ましくは30%以下、さらに好ましくは20%以下、とくに好ましくは10%以下、最も好ましくは0%である。
【0032】
樹脂(b)のMn、Mw、融点、Tg、sp値は、用途によって好ましい範囲に適宜調整すればよい。
樹脂(b)のsp値は、樹脂(a)とのsp値差が上記の範囲内にあるのが好ましいが、好ましくは7〜18、さらに好ましくは8〜14、とくに好ましくは9〜14である。
例えば、樹脂粒子(C1)または(C2)〔以下(C1)または(C2)を(C)と記載することもある〕、樹脂粒子(B)をスラッシュ成形用樹脂、粉体塗料として用いる場合、(b)のMnは、好ましくは2,000〜50万、さらに好ましくは4,000〜20万である。
(b)の融点(DSCにて測定、以下融点はDSCでの測定値)、好ましくは0℃〜200℃、さらに好ましくは35℃〜150℃である。(b)のTgは、好ましくは−60℃〜100℃、さらに好ましくは−30℃〜60℃である。
液晶ディスプレイ等の電子部品製造用スペーサー、電子測定機の標準粒子として用いる場合、(b)のMnは、好ましくは2万〜1,000万、さらに好ましくは4万〜200万である。(b)の融点は、好ましくは40℃〜300℃、さらに好ましくは70℃〜250℃である。(b)のTgは好ましくは−0℃〜250℃、さらに好ましくは50℃〜200℃である。
電子写真、静電記録、静電印刷などに使用されるトナーとして用いる場合、(b)のMnは、好ましくは1,000〜500万、さらに好ましくは2,000〜50万である。(b)の融点は、好ましくは20℃〜300℃、さらに好ましくは80℃〜250℃である。(b)のTgは、好ましくは20℃〜200℃、さたに好ましくは40℃〜150℃である。(b)のsp値は、好ましくは8〜16、さらに好ましくは9〜14である。溶融性と画像定着性の両立という観点から、(b)の50〜90%(とくに60〜80%)が、テトラヒドロフラン可溶分のMwが2000〜12000である線状ポリエステルであることが好ましい。線状ポリエステルのMwは、さらに好ましくは4000〜11000、とくに好ましくは7000〜10000である。線状ポリエステルのMwがこの範囲内であると、溶融性と画像密着性のバランスが特に良好である。
【0033】
本発明においては、樹脂(a)としては水性分散体を形成しうる樹脂であればいかなる樹脂であっても使用でき、熱可塑性樹脂であっても熱硬化性樹脂であってもよい。(a)としては例えば、ビニル系樹脂、ポリウレタン樹脂、エポキシ樹脂、ポリエステル樹脂、ポリアミド樹脂、ポリイミド樹脂、ケイ素系樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、アニリン樹脂、アイオマー樹脂、ポリカーボネート樹脂等が挙げられる。(a)としては上記樹脂の2種以上を併用しても差し支えない。このうち好もしいのは、微細球状樹脂粒子の水性分散体が得られやすいという観点からポリウレタン樹脂、ビニル系樹脂、エポキシ樹脂、ポリエステル樹脂、およびそれらの併用である。
【0034】
ビニル系樹脂は、ビニル系モノマーを単独重合または共重合したポリマーである。ビニル系モノマーとしては、下記(1)〜(10)が挙げられる。
【0035】
(1)ビニル系炭化水素:
(1−1)脂肪族ビニル系炭化水素:アルケン類、例えばエチレン、プロピレン、ブテン、イソブチレン、ペンテン、ヘプテン、ジイソブチレン、オクテン、ドデセン、オクタデセン、前記以外のα−オレフィン等;アルカジエン類、例えばブタジエン、イソプレン、1,4−ペンタジエン、1,6−ヘキサジエン、1,7−オクタジエン。
(1−2)脂環式ビニル系炭化水素:モノ−もしくはジ−シクロアルケンおよびアルカジエン類、例えばシクロヘキセン、(ジ)シクロペンタジエン、ビニルシクロヘキセン、エチリデンビシクロヘプテン等;テルペン類、例えばピネン、リモネン、インデン等。
(1−3)芳香族ビニル系炭化水素:スチレンおよびそのハイドロカルビル(アルキル、シクロアルキル、アラルキルおよび/またはアルケニル)置換体、例えばα−メチルスチレン、ビニルトルエン、2,4−ジメチルスチレン、エチルスチレン、イソプロピルスチレン、ブチルスチレン、フェニルスチレン、シクロヘキシルスチレン、ベンジルスチレン、クロチルベンゼン、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン、トリビニルベンゼン等;およびビニルナフタレン。
【0036】
(2)カルボキシル基含有ビニル系モノマーおよびその金属塩:
炭素数3〜30の不飽和モノカルボン酸、不飽和ジカルボン酸ならびにその無水物およびそのモノアルキル(炭素数1〜24)エステル、例えば(メタ)アクリル酸、(無水)マレイン酸、マレイン酸モノアルキルエステル、フマル酸、フマル酸モノアルキルエステル、クロトン酸、イタコン酸、イタコン酸モノアルキルエステル、イタコン酸グリコールモノエーテル、シトラコン酸、シトラコン酸モノアルキルエステル、桂皮酸等のカルボキシル基含有ビニル系モノマー。
【0037】
(3)スルホン基含有ビニル系モノマー、ビニル系硫酸モノエステル化物およびこれらの塩:炭素数2〜14のアルケンスルホン酸、例えばビニルスルホン酸、(メタ)アリルスルホン酸、メチルビニルスルホン酸、スチレンスルホン酸;およびその炭素数2〜24のアルキル誘導体、例えばα−メチルスチレンスルホン酸等;スルホ(ヒドロキシ)アルキル−(メタ)アクリレートもしくは(メタ)アクリルアミド、例えば、スルホプロピル(メタ)アクリレート、2−ヒドロキシ−3−(メタ)アクリロキシプロピルスルホン酸、2−(メタ)アクリロイルアミノ−2,2−ジメチルエタンスルホン酸、2−(メタ)アクリロイルオキシエタンスルホン酸、3−(メタ)アクリロイルオキシ−2−ヒドロキシプロパンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、3−(メタ)アクリルアミド−2−ヒドロキシプロパンスルホン酸、アルキル(炭素数3〜18)アリルスルホコハク酸、ポリ(n=2〜30)オキシアルキレン(エチレン、プロピレン、ブチレン:単独、ランダム、ブロックでもよい)モノ(メタ)アクリレートの硫酸エステル[ポリ(n=5〜15)オキシプロピレンモノメタクリレート硫酸エステル等]、ポリオキシエチレン多環フェニルエーテル硫酸エステル、および下記一般式(1−1)〜(1−3)で示される硫酸エステルもしくはスルホン酸基含有モノマー;ならびそれらの塩等。
【0038】
O−(AO)nSO3H
|
CH2=CHCH2−OCH2CHCH2O−Ar−R (1−1)
CH=CH−CH3
|
R−Ar−O−(AO)nSO3H (1−2)
CH2COOR’
|
HO3SCHCOOCH2CH(OH)CH2OCH2CH=CH2 (1−3)
(式中、Rは炭素数1〜15のアルキル基、Aは炭素数2〜4のアルキレン基を示し、nが複数の場合同一でも異なっていてもよく、異なる場合はランダムでもブロックでもよい。Arはベンゼン環を示し、nは1〜50の整数を示し、R’はフッ素原子で置換されていてもよい炭素数1〜15のアルキル基を示す。)
【0039】
(4)燐酸基含有ビニル系モノマーおよびその塩:
(メタ)アクリロイルオキシアルキル(C1〜C24)燐酸モノエステル、例えば、2−ヒドロキシエチル(メタ)アクリロイルホスフェート、フェニル−2−アクリロイロキシエチルホスフェート、(メタ)アクリロイルオキシアルキル(炭素数1〜24)ホスホン酸類、例えば2−アクリロイルオキシエチルホスホン酸。
【0040】
なお、上記(2)〜(4)の塩としては、金属塩、アンモニウム塩、およびアミン塩(4級アンモニウム塩を含む)が挙げられる。金属塩を形成する金属としては、Al、Ti、Cr、Mn、Fe、Zn、Ba、Zr、Ca、Mg、Na、およびK等が挙げられる。
好ましくはアルカリ金属塩、およびアミン塩であり、さらに好ましくは、ナトリウム塩および炭素数3〜20の3級モノアミンの塩である。
【0041】
(5)ヒドロキシル基含有ビニル系モノマー:
ヒドロキシスチレン、N−メチロール(メタ)アクリルアミド、ヒドロキシエチル(メタ)アクリレート、ヒドロキシプロピル(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、(メタ)アリルアルコール、クロチルアルコール、イソクロチルアルコール、1−ブテン−3−オール、2−ブテン−1−オール、2−ブテン−1,4−ジオール、プロパルギルアルコール、2−ヒドロキシエチルプロペニルエーテル、庶糖アリルエーテル等
【0042】
(6)含窒素ビニル系モノマー:
(6−1)アミノ基含有ビニル系モノマー:アミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、t−ブチルアミノエチルメタクリレート、N−アミノエチル(メタ)アクリルアミド、(メタ)アリルアミン、モルホリノエチル(メタ)アクリレート、4ービニルピリジン、2ービニルピリジン、クロチルアミン、N,N−ジメチルアミノスチレン、メチルα−アセトアミノアクリレート、ビニルイミダゾール、N−ビニルピロール、N−ビニルチオピロリドン、N−アリールフェニレンジアミン、アミノカルバゾール、アミノチアゾール、アミノインドール、アミノピロール、アミノイミダゾール、アミノメルカプトチアゾール、これらの塩等
(6−2)アミド基含有ビニル系モノマー:(メタ)アクリルアミド、N−メチル(メタ)アクリルアミド、N−ブチルアクリルアミド、ジアセトンアクリルアミド、N−メチロール(メタ)アクリルアミド、N,N’−メチレン−ビス(メタ)アクリルアミド、桂皮酸アミド、N,N−ジメチルアクリルアミド、N,N−ジベンジルアクリルアミド、メタクリルホルムアミド、N−メチルN−ビニルアセトアミド、N−ビニルピロリドン等
(6−3)ニトリル基含有ビニル系モノマー:(メタ)アクリロニトリル、シアノスチレン、シアノアクリレート等
(6−4)4級アンモニウムカチオン基含有ビニル系モノマー:ジメチルアミノエチル(メタ)アクリレート、ジエチルアミノエチル(メタ)アクリレート、ジメチルアミノエチル(メタ)アクリルアミド、ジエチルアミノエチル(メタ)アクリルアミド、ジアリルアミン等の3級アミン基含有ビニル系モノマーの4級化物(メチルクロライド、ジメチル硫酸、ベンジルクロライド、ジメチルカーボネート等の4級化剤を用いて4級化したもの)
(6−5)ニトロ基含有ビニル系モノマー:ニトロスチレン等
【0043】
(7)エポキシ基含有ビニル系モノマー:
グルシジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート、p−ビニルフェニルフェニルオキサイド等
【0044】
(8)ハロゲン元素含有ビニル系モノマー:
塩化ビニル、臭化ビニル、塩化ビニリデン、アリルクロライド、クロルスチレン、ブロムスチレン、ジクロルスチレン、クロロメチルスチレン、テトラフルオロスチレン、クロロプレン等
【0045】
(9)ビニルエステル、ビニル(チオ)エーテル、ビニルケトン、ビニルスルホン類:
(9−1)ビニルエステル、例えば酢酸ビニル、ビニルブチレート、プロピオン酸ビニル、酪酸ビニル、ジアリルフタレート、ジアリルアジペート、イソプロペニルアセテート、ビニルメタクリレート、メチル4−ビニルベンゾエート、シクロヘキシルメタクリレート、ベンジルメタクリレート、フェニル(メタ)アクリレート、ビニルメトキシアセテート、ビニルベンゾエート、エチルα−エトキシアクリレート、炭素数1〜50のアルキル基を有するアルキル(メタ)アクリレート[メチル(メタ)アクリレート、エチル(メタ)アクリレート、プロピル(メタ)アクリレート、ブチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、ドデシル(メタ)アクリレート、ヘキサデシル(メタ)アクリレート、ヘプタデシル(メタ)アクリレート、エイコシル(メタ)アクリレート等]、ジアルキルフマレート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ジアルキルマレエート(2個のアルキル基は、炭素数2〜8の、直鎖、分枝鎖もしくは脂環式の基である)、ポリ(メタ)アリロキシアルカン類[ジアリロキシエタン、トリアリロキシエタン、テトラアリロキシエタン、テトラアリロキシプロパン、テトラアリロキシブタン、テトラメタアリロキシエタン等]等、ポリアルキレングリコール鎖を有するビニル系モノマー[ポリエチレングリコール(分子量300)モノ(メタ)アクリレート、ポリプロピレングリコール(分子量500)モノアクリレート、メチルアルコールエチレンオキサイド(エチレンオキサイドを以下EOと略記する)10モル付加物(メタ)アクリレート、ラウリルアルコールEO30モル付加物(メタ)アクリレート等]、ポリ(メタ)アクリレート類[多価アルコール類のポリ(メタ)アクリレート:エチレングリコールジ(メタ)アクリレート、プロピレングリコールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、ポリエチレングリコールジ(メタ)アクリレート等]等
(9−2)ビニル(チオ)エーテル、例えばビニルメチルエーテル、ビニルエチルエーテル、ビニルプロピルエーテル、ビニルブチルエーテル、ビニル2−エチルヘキシルエーテル、ビニルフェニルエーテル、ビニル2−メトキシエチルエーテル、メトキシブタジエン、ビニル2−ブトキシエチルエーテル、3,4−ジヒドロ1,2−ピラン、2−ブトキシ−2’−ビニロキシジエチルエーテル、ビニル2−エチルメルカプトエチルエーテル、アセトキシスチレン、フェノキシスチレン等
(9−3)ビニルケトン、例えばビニルメチルケトン、ビニルエチルケトン、ビニルフェニルケトン;
ビニルスルホン、例えばジビニルサルファイド、p−ビニルジフェニルサルファイド、ビニルエチルサルファイド、ビニルエチルスルフォン、ジビニルスルフォン、ジビニルスルフォキサイド等
【0046】
(10)その他のビニル系モノマー:
イソシアナトエチル(メタ)アクリレート、m−イソプロペニル−α,α−ジメチルベンジルイソシアネート等
【0047】
本発明において、(a)として有機酸金属塩(m)の構成単位を含有するビニル系樹脂を用いる場合、この樹脂は、例えば、モノマーの少なくとも一部として、上記モノマー(2)〜(4)のうち、Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる金属の塩を、1種以上用いることにより得られる。これらの有機酸金属塩モノマー(m)の、重合に用いる全モノマー中の使用量は、好ましくは5〜60%である。下限はさらに好ましくは10%であり、上限はさらに好ましくは50%である。
【0048】
ビニル系モノマーの共重合体としては、上記(1)〜(10)の任意のモノマー同士を、2元またはそれ以上の個数で、好ましくは樹脂粒子(A)中のカルボキシル基の含量が0.1〜10%になるように、任意の割合で共重合したポリマーが挙げられるが、例えば、スチレン−(メタ)アクリル酸エステル−(メタ)アクリル酸共重合体、スチレン−ブタジエン−(メタ)アクリル酸共重合体、(メタ)アクリル酸−アクリル酸エステル共重合体、スチレン−アクリロニトリル−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸共重合体、スチレン−(メタ)アクリル酸−ジビニルベンゼン共重合体、スチレン−スチレンスルホン酸−(メタ)アクリル酸エステル共重合体、およびこれらの共重合体の塩などが挙げられる。
【0049】
樹脂(a)は、水性分散体中で樹脂粒子(A)形成することが必要であることから、少なくとも水性樹脂分散体(X1)を形成する条件下で水に完全に溶解していないことが必要である。そのため、ビニル系樹脂が共重合体である場合には、ビニル系樹脂を構成する疎水性モノマーと親水性モノマーの比率は、選ばれるモノマーの種類によるが、一般に疎水性モノマーが10%以上であることが好ましく、30%以上であることがより好ましい。疎水性モノマーの比率が、10%以下になるとビニル系樹脂が水溶性になり、(C1)または(C2)の粒径均一性が損なわれる。ここで、親水性モノマーとは水に任意の割合で溶解するモノマーをいい、疎水性モノマーとは、それ以外のモノマー(基本的に水に混和しないモノマー)をいう。
【0050】
ポリエステル樹脂としては、ポリオールと、ポリカルボン酸またはその酸無水物またはその低級アルキルエステルとの重縮合物、およびこれらの重縮合物の金属塩などが挙げられる。ポリオールとしては前述のジオール(g)〔(g1)を含む〕および3価以上のポリオール(h)が、ポリカルボン酸またはその酸無水物またはその低級アルキルエステルとしては、ジカルボン酸(i)および3価以上のポリカルボン酸(j)およびこれらの酸無水物または低級アルキルエステルが挙げられる。
ポリオールとポリカルボン酸の比率は、水酸基[OH]とカルボキシル基[COOH]の当量比[OH]/[COOH]として、好ましくは2/1〜1/1.2、さらに好ましくは1.5/1〜1/1.1、とくに好ましくは1.3/1〜1/1.05である。
【0051】
本発明において、(a)として有機酸金属塩(m)の構成単位を含有するポリエステル樹脂を用いる場合、この樹脂は、例えば、COOHの残基を有するポリエステル(酸価が好ましくは1〜100、さらに好ましくは5〜50)を合成し、その少なくとも1部のCOOH基を、Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる少なくとも1種の金属の塩とすることにより得られる。
金属塩とする方法としては、例えば、COOH基を有するポリエステルと該当する金属の水酸化物とを反応することにより得られる。
【0052】
ポリウレタン樹脂としては、ポリイソシアネート(11)と活性水素含有化合物(D){水、ポリオール[ジオール(g)〔ヒドロキシル基以外の官能基を有するジオール(g1)を含む〕、および3〜8価またはそれ以上のポリオール(h)]、ポリカルボン酸[ジカルボン酸(i)、および3〜6価またはそれ以上のポリカルボン酸(j)]、ポリオールとポリカルボン酸の重縮合により得られるポリエステルポリオール、炭素数6〜12のラクトンの開環重合体、ポリアミン(12)、ポリチオール(13)、およびこれらの併用等}の重付加物、並びに(11)と活性水素含有化合物を反応させてなる末端イソシアネート基プレポリマーと、該プレポリマーのイソシアネート基に対して当量の1級および/または2級モノアミン(16)とを反応させて得られる、アミノ基含有ポリウレタン樹脂が挙げられる。
ポリウレタン樹脂中のカルボキシル基の含有量は、0.1〜10%が好ましい。
【0053】
ポリイソシアネート(11)としては、炭素数(NCO基中の炭素を除く、以下同様)6〜20の芳香族ポリイソシアネート、炭素数2〜18の脂肪族ポリイソシアネート、炭素数4〜15の脂環式ポリイソシアネート、炭素数8〜15の芳香脂肪族ポリイソシアネートおよびこれらのポリイソシアネートの変性物(ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物など)およびこれらの2種以上の混合物が挙げられる。
【0054】
上記芳香族ポリイソシアネートの具体例としては、1,3−および/または1,4−フェニレンジイソシアネート、2,4−および/または2,6−トリレンジイソシアネート(TDI)、粗製TDI、2,4’−および/または4,4’−ジフェニルメタンジイソシアネート(MDI)、粗製MDI[粗製ジアミノフェニルメタン〔ホルムアルデヒドと芳香族アミン(アニリン)またはその混合物との縮合生成物;ジアミノジフェニルメタンと少量(たとえば5〜20%)の3官能以上のポリアミンとの混合物〕のホスゲン化物:ポリアリルポリイソシアネート(PAPI)]、1,5−ナフチレンジイソシアネート、4,4’,4”−トリフェニルメタントリイソシアネート、m−およびp−イソシアナトフェニルスルホニルイソシアネートなどが挙げられる。
上記脂肪族ポリイソシアネートの具体例としては、エチレンジイソシアネート、テトラメチレンジイソシアネート、ヘキサメチレンジイソシアネート(HDI)、ドデカメチレンジイソシアネート、1,6,11−ウンデカントリイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、リジンジイソシアネート、2,6−ジイソシアナトメチルカプロエート、ビス(2−イソシアナトエチル)フマレート、ビス(2−イソシアナトエチル)カーボネート、2−イソシアナトエチル−2,6−ジイソシアナトヘキサノエートなどの脂肪族ポリイソシアネートなどが挙げられる。
上記脂環式ポリイソシアネートの具体例としては、イソホロンジイソシアネート(IPDI)、ジシクロヘキシルメタン−4,4’−ジイソシアネート(水添MDI)、シクロヘキシレンジイソシアネート、メチルシクロヘキシレンジイソシアネート(水添TDI)、ビス(2−イソシアナトエチル)−4−シクロヘキセン−1,2−ジカルボキシレート、2,5−および/または2,6−ノルボルナンジイソシアネートなどが挙げられる。
上記芳香脂肪族ポリイソシアネートの具体例としては、m−および/またはp−キシリレンジイソシアネート(XDI)、α,α,α’,α’−テトラメチルキシリレンジイソシアネート(TMXDI)などが挙げられる。
また、上記ポリイソシアネートの変性物には、ウレタン基、カルボジイミド基、アロファネート基、ウレア基、ビューレット基、ウレトジオン基、ウレトイミン基、イソシアヌレート基、オキサゾリドン基含有変性物などが挙げられる。
具体的には、変性MDI(ウレタン変性MDI、カルボジイミド変性MDI、トリヒドロカルビルホスフェート変性MDIなど)、ウレタン変性TDIなどのポリイソシアネートの変性物およびこれらの2種以上の混合物[たとえば変性MDIとウレタン変性TDI(イソシアネート含有プレポリマー)との併用]が含まれる。
これらのうちで好ましいものは6〜15の芳香族ポリイソシアネート、炭素数4〜12の脂肪族ポリイソシアネート、および炭素数4〜15の脂環式ポリイソシアネートであり、とくに好ましいものはTDI、MDI、HDI、水添MDI、およびIPDIである。
【0055】
ポリアミン(12)の例としては、脂肪族ポリアミン類(C2 〜C18):〔1〕脂肪族ポリアミン{C2〜C6 アルキレンジアミン(エチレンジアミン、プロピレンジアミン、トリメチレンジアミン、テトラメチレンジアミン、ヘキサメチレンジアミンなど)、ポリアルキレン(C2〜C6)ポリアミン〔ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン,トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミンなど〕};〔2〕これらのアルキル(C1〜C4)またはヒドロキシアルキル(C2〜C4)置換体〔ジアルキル(C1〜C3)アミノプロピルアミン、トリメチルヘキサメチレンジアミン、アミノエチルエタノールアミン、2,5−ジメチル−2,5−ヘキサメチレンジアミン、メチルイミノビスプロピルアミンなど〕;〔3〕脂環または複素環含有脂肪族ポリアミン〔3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなど〕;〔4〕芳香環含有脂肪族アミン類(C8 〜C15)(キシリレンジアミン、テトラクロル−p−キシリレンジアミンなど)、脂環式ポリアミン(C4 〜C15):1,3−ジアミノシクロヘキサン、イソホロンジアミン、メンセンジアミン、4,4´−メチレンジシクロヘキサンジアミン(水添メチレンジアニリン)など、複素環式ポリアミン(C4 〜C15):ピペラジン、N−アミノエチルピペラジン、1,4−ジアミノエチルピペラジン、1,4ビス(2−アミノ−2−メチルプロピル)ピペラジンなど、芳香族ポリアミン類(C6 〜C20):〔1〕非置換芳香族ポリアミン〔1,2−、1,3−および1,4−フェニレンジアミン、2,4´−および4,4´−ジフェニルメタンジアミン、クルードジフェニルメタンジアミン(ポリフェニルポリメチレンポリアミン)、ジアミノジフェニルスルホン、ベンジジン、チオジアニリン、ビス(3,4−ジアミノフェニル)スルホン、2,6−ジアミノピリジン、m−アミノベンジルアミン、トリフェニルメタン−4,4´,4”−トリアミン、ナフチレンジアミンなど;〔2〕核置換アルキル基〔メチル,エチル,n−およびi−プロピル、ブチルなどのC1〜C4アルキル基)を有する芳香族ポリアミン、たとえば2,4−および2,6−トリレンジアミン、クルードトリレンジアミン、ジエチルトリレンジアミン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、4,4´−ビス(o−トルイジン)、ジアニシジン、ジアミノジトリルスルホン、1,3−ジメチル−2,4−ジアミノベンゼン、1,3−ジメチル−2,6−ジアミノベンゼン、1,4−ジイソプロピル−2,5−ジアミノベンゼン、2,4−ジアミノメシチレン、1−メチル−3,5−ジエチル−2,4−ジアミノベンゼン、2,3−ジメチル−1,4−ジアミノナフタレン、2,6−ジメチル−1,5−ジアミノナフタレン、3,3´,5,5´−テトラメチルベンジジン、3,3´,5,5´−テトラメチル−4,4´−ジアミノジフェニルメタン、3,5−ジエチル−3´−メチル−2´,4−ジアミノジフェニルメタン、3,3´−ジエチル−2,2´−ジアミノジフェニルメタン、4,4´−ジアミノ−3,3´−ジメチルジフェニルメタン、3,3´,5,5´−テトラエチル−4,4´−ジアミノベンゾフェノン、3,3´,5,5´−テトラエチル−4,4´−ジアミノジフェニルエーテル、3,3´,5,5´−テトライソプロピル−4,4´−ジアミノジフェニルスルホンなど〕、およびこれらの異性体の種々の割合の混合物;〔3〕核置換電子吸引基(Cl,Br,I,Fなどのハロゲン;メトキシ、エトキシなどのアルコキシ基;ニトロ基など)を有する芳香族ポリアミン〔メチレンビス−o−クロロアニリン、4−クロロ−o−フェニレンジアミン、2−クロル−1,4−フェニレンジアミン、3−アミノ−4−クロロアニリン、4−ブロモ−1,3−フェニレンジアミン、2,5−ジクロル−1,4−フェニレンジアミン、5−ニトロ−1,3−フェニレンジアミン、3−ジメトキシ−4−アミノアニリン;4,4´−ジアミノ−3,3´−ジメチル−5,5´−ジブロモ−ジフェニルメタン、3,3´−ジクロロベンジジン、3,3´−ジメトキシベンジジン、ビス(4−アミノ−3−クロロフェニル)オキシド、ビス(4−アミノ−2−クロロフェニル)プロパン、ビス(4−アミノ−2−クロロフェニル)スルホン、ビス(4−アミノ−3−メトキシフェニル)デカン、ビス(4−アミノフェニル)スルフイド、ビス(4−アミノフェニル)テルリド、ビス(4−アミノフェニル)セレニド、ビス(4−アミノ−3−メトキシフェニル)ジスルフイド、4,4´−メチレンビス(2−ヨードアニリン)、4,4´−メチレンビス(2−ブロモアニリン)、4,4´−メチレンビス(2−フルオロアニリン)、4−アミノフェニル−2−クロロアニリンなど〕;〔4〕2級アミノ基を有する芳香族ポリアミン〔上記〔1〕〜〔3〕の芳香族ポリアミンの−NH2の一部または全部が−NH−R´(R´はアルキル基たとえばメチル,エチルなどの低級アルキル基)で置き換ったもの〕〔4,4´−ジ(メチルアミノ)ジフェニルメタン、1−メチル−2−メチルアミノ−4−アミノベンゼンなど〕、ポリアミドポリアミン:ジカルボン酸(ダイマー酸など)と過剰の(酸1モル当り2モル以上の)ポリアミン類(上記アルキレンジアミン,ポリアルキレンポリアミンなど)との縮合により得られる低分子量ポリアミドポリアミンなど、ポリエーテルポリアミン:ポリエーテルポリオール(ポリアルキレングリコールなど)のシアノエチル化物の水素化物などが挙げられる。
【0056】
ポリチオール(17)としては、炭素数2〜36のアルカンジチオール(エチレンジチオール、1,4−ブタンジチオール、1,6−ヘキサンジチオールなど)等が挙げられる。
【0057】
1級および/または2級モノアミン(18)としては、炭素数2〜24のアルキルアミン(エチルアミン、n−ブチルアミン、イソブチルアミンなど)等が挙げられる。
【0058】
本発明において、(a)として前記有機酸金属塩(m)の構成単位を含有するポリウレタン樹脂を用いる場合、この樹脂は、例えば、ジオール(g)、(g1)および/または3価以上のポリオール(h)の少なくとも一部として、カルボン酸基、スルホン酸基、リン酸基、およびこれらの酸の上記金属塩基から選ばれる1種以上の基を有するジオールおよび/または3価以上のポリオールを用いることにより得られる。カルボン酸基、スルホン酸基、またはリン酸基を有するジオールおよび/またはポリオールを用いる場合は、ポリウレタン樹脂形成後に、例えば、さらにこれらの酸基と該当する金属の水酸化物とを反応することにより、上記有機酸金属塩(m)の構成単位を含有するポリウレタン樹脂が得られる。
カルボン酸(塩)基を有するジオールまたはポリオールとしては、ジアルキロールアルカン酸(塩)[C6〜24のもの、例えば2,2−ジメチロールプロピオン酸、2,2−ジメチロールブタン酸、2,2−ジメチロールヘプタン酸、2,2−ジメチロールオクタン酸、およびこれらの酸の前記金属塩など]等が挙げられる。
スルホン酸(塩)基を有するジオールまたはポリオールとしては、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸、スルホイソフタル酸ジ(エチレングリコール)エステル、およびこれらの酸の前記金属塩などが挙げられる。
リン酸(塩)基を有するジオールまたはポリオールとしては、ビス(2−ヒドロキシエチル)ホスフェートおよびその前記金属塩などが挙げられる。
これらの有機酸金属(塩)基を有するジオールまたは3価以上のポリオールの、(g)、(g1)および(h)の合計量に対する使用量は、好ましくは10〜90モル%である。下限は、さらに好ましくは20モル%であり、上限は、さらに好ましくは60モル%である。
【0059】
エポキシ樹脂としては、ポリエポキシド(14)の開環重合物、ポリエポキシド(14)と活性水素基含有化合物(D){水、ポリオール[前記ジオール(g)、(g1)および3価以上のポリオール(h)]、ジカルボン酸(i)、3価以上のポリカルボン酸(j)、ポリアミン(12)、ポリチオール(13)等}との重付加物、またはポリエポキシド(14)とジカルボン酸(i)または3価以上のポリカルボン酸(j)の酸無水物との硬化物などが挙げられる。
【0060】
本発明に用いるポリエポキシド(14)は、分子中に2個以上のエポキシ基を有していれば、特に限定されない。ポリエポキシド(14)として好ましいものは、硬化物の機械的性質の観点から分子中にエポキシ基を2〜6個有するものである。ポリエポキシド(14)のエポキシ当量(エポキシ基1個当たりの分子量)は、好ましくは65〜1000であり、さらに好ましくは90〜500である。エポキシ当量が1000を超えると、架橋構造がルーズになり硬化物の耐水性、耐薬品性、機械的強度等の物性が悪くなり、一方、エポキシ当量が65未満のものを合成するのは困難である。
【0061】
ポリエポキシド(14)の例としては、芳香族系ポリエポキシ化合物、複素環系ポリエポキシ化合物、脂環族系ポリエポキシ化合物あるいは脂肪族系ポリエポキシ化合物が挙げられる。芳香族系ポリエポキシ化合物としては、多価フェノール類のグリシジルエーテル体およびグリシジルエステル体、グリシジル芳香族ポリアミン、並びに、アミノフェノールのグリシジル化物等が挙げられる。多価フェノールのグリシジルエーテル体としては、ビスフェノールFジグリシジルエーテル、ビスフェノールAジグリシジルエーテル、ビスフェノールBジグリシジルエーテル、ビスフェノールADジグリシジルエーテル、ビスフェノールSジグリシジルエーテル、ハロゲン化ビスフェノールAジグリシジル、テトラクロロビスフェノールAジグリシジルエーテル、カテキンジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ハイドロキノンジグリシジルエーテル、ピロガロールトリグリシジルエーテル、1,5−ジヒドロキシナフタリンジグリシジルエーテル、ジヒドロキシビフェニルジグリシジルエーテル、オクタクロロ−4,4’−ジヒドロキシビフェニルジグリシジルエーテル、テトラメチルビフェニルジグリシジルエーテル、ジヒドロキシナフチルクレゾールトリグリシジルエーテル、トリス(ヒドロキシフェニル)メタントリグリシジルエーテル、ジナフチルトリオールトリグリシジルエーテル、テトラキス(4−ヒドロキシフェニル)エタンテトラグリシジルエーテル、p−グリシジルフェニルジメチルトリールビスフェノールAグリシジルエーテル、トリスメチル−tret−ブチル−ブチルヒドロキシメタントリグリシジルエーテル、9,9’−ビス(4−ヒドキシフェニル)フロオレンジグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)テトラクレゾールグリシジルエーテル、4,4’−オキシビス(1,4−フェニルエチル)フェニルグリシジルエーテル、ビス(ジヒドロキシナフタレン)テトラグリシジルエーテル、フェノールまたはクレゾールノボラック樹脂のグリシジルエーテル体、リモネンフェノールノボラック樹脂のグリシジルエーテル体、ビスフェノールA2モルとエピクロロヒドリン3モルの反応から得られるジグリシジルエーテル体、フェノールとグリオキザール、グルタールアルデヒド、またはホルムアルデヒドの縮合反応によって得られるポリフェノールのポリグリシジルエーテル体、およびレゾルシンとアセトンの縮合反応によって得られるポリフェノールのポリグリシジルエーテル体等が挙げられる。多価フェノールのグリシジルエステル体としては、フタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、テレフタル酸ジグリシジルエステル等が挙げられる。グリシジル芳香族ポリアミンとしては、N,N−ジグリシジルアニリン、N,N,N’,N’−テトラグリシジルキシリレンジアミン、N,N,N’,N’−テトラグリシジルジフェニルメタンジアミン等が挙げられる。さらに、本発明において前記芳香族系として、P−アミノフェノールのトリグリシジルエーテル、トリレンジイソシアネートまたはジフェニルメタンジイソシアネートとグリシドールの付加反応によって得られるジグリシジルウレタン化合物、前記2反応物にポリオールも反応させて得られるグリシジル基含有ポリウレタン(プレ)ポリマーおよびビスフェノールAのアルキレンオキシド(エチレンオキシドまたはプロピレンオキシド)付加物のジグリシジルエーテル体も含む。複素環系ポリエポキシ化合物としては、トリスグリシジルメラミンが挙げられる;脂環族系ポリエポキシ化合物としては、ビニルシクロヘキセンジオキシド、リモネンジオキシド、ジシクロペンタジエンジオキシド、ビス(2,3−エポキシシクロペンチル)エーテル、エチレングリコールビスエポキシジシクロペンチルエール、3,4−エポキシ−6−メチルシクロヘキシルメチル−3’,4’−エポキシ−6’−メチルシクロヘキサンカルボキシレート、ビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)アジペート、およびビス(3,4−エポキシ−6−メチルシクロヘキシルメチル)ブチルアミン、ダイマー酸ジグリシジルエステル等が挙げられる。また、脂環族系としては、前記芳香族系ポリエポキシド化合物の核水添化物も含む;脂肪族系ポリエポキシ化合物としては、多価脂肪族アルコールのポリグリシジルエーテル体、多価脂肪酸のポリグリシジルエステル体、およびグリシジル脂肪族アミンが挙げられる。多価脂肪族アルコールのポリグリシジルエーテル体としては、エチレングリコールジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、テトラメチレングリコールジグリシジルエーテル、1,6−ヘキサンジオールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、ポリテトラメチレングリコールジグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、グリセロールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ソルビトールポリグリシジルエーテルおよびポリグリセロールンポリグリシジルエーテル等が挙げられる。多価脂肪酸のポリグリシジルエステル体としては、ジグリシジルオキサレート、ジグリシジルマレート、ジグリシジルスクシネート、ジグリシジルグルタレート、ジグリシジルアジペート、ジグリシジルピメレート等が挙げられる。グリシジル脂肪族アミンとしては、N,N,N’,N’−テトラグリシジルヘキサメチレンジアミンが挙げられる。また、本発明において脂肪族系としては、ジグリシジルエーテル、グリシジル(メタ)アクリレートの(共)重合体も含む。これらのうち、好ましいのは、脂肪族系ポリエポキシ化合物および芳香族系ポリエポキシ化合物である。本発明のポリエポキシドは、2種以上併用しても差し支えない。
【0062】
本発明においては、帯電特性の点から、樹脂(a)の構成単位として前記Al、Ti、Cr、Mn、Fe、Zn、Ba、Zrから選ばれる金属の有機酸金属塩(m)の単位を含有する、および/または、樹脂粒子(A)中に前記有機酸金属塩(m)を含有することが好ましい。有機酸金属塩(m)を構成する有機酸としてはカルボン酸が好ましい。金属の中では、ZnおよびFeが好ましく、さらに好ましくはZnである。
これらのうち、(A)中に有機酸金属塩(m)を含有することが、調整が容易な点から好ましい。
【0063】
有機酸金属塩(m)のうち、カルボン酸金属塩の具体例としては、(アルキル置換)安息香酸、(アルキル置換)サルチル酸、ナフテン酸等の炭素数7〜35の芳香族脂肪酸、およびステアリル酸等の炭素数12〜35の高級脂肪酸、並びに、前記の樹脂(a)を構成するカルボキシル基含有ビニルモノマーの金属塩として例示したものが好ましい。スルホン酸金属塩の具体例としては、炭素数7〜30のアルキルベンゼンスルホン酸、および(a)を構成するスルホン基含有ビニルモノマーの金属塩として例示したものが好ましい。リン酸金属塩としては(a)を構成するリン酸基含有ビニルモノマーの金属塩として例示したものが好ましい。これらの中でも、油溶性の金属塩が、樹脂粒子(C)の帯電性の効果を発揮する上で、さらに好ましい。組成の面からは、カルボン酸金属塩がさらに好ましく、カルボン酸のZn塩およびFe塩が特に好ましい。
(A)中に有機酸金属塩(m)を含有させる方法としては、樹脂(a)の前駆体と反応性がなければ(a)の作成前に投入してもよいが、(a)の作成後に(a)と混合する方が好ましい。
これらの有機酸金属塩(m)の構成単位、および有機酸金属塩(m)の使用割合に特に制限はないが、得られる樹脂粒子(C)に対するこれらの合計量が0.01〜10%であることが好ましい。下限は、さらに好ましくは0.05%であり、上限は、さらに好ましくは1%である。
【0064】
本発明において、微細な球状樹脂粒子(A)の水性分散液(W)を得るため、かつ耐熱保存安定性、帯電特性に優れ、粒径が均一な樹脂粒子(C1)の水性分散体(X1)を得るために、樹脂(a)は、スルホン酸アニオン基(−SO3-)を含有することが好ましい。スルホン酸アニオン基(−SO3-)の合計含有量は、(a)の重量に基づいて0.001〜10%が好ましい。下限は、さらに好ましくは0.002%であり、上限は、さらに好ましくは5%、とくに好ましくは2%、最も好ましくは1%である。また、樹脂を形成するスルホン酸アニオン基(−SO3-)を含有するモノマーの好ましい炭素数は3〜50、更に好ましくは3〜30、特に好ましくは4〜15である。
スルホン酸アニオン基(−SO3-)基含有量が上記範囲の下限以上や樹脂を形成するスルホン酸アニオン基(−SO3-)を含有するモノマーの炭素数が上記範囲の上限以下であると、樹脂(a)が水系媒体中に分散しやすく、微細な球状の樹脂粒子(A)の水性分散体(W)を容易に得ることができる。また、得られる樹脂粒子(C1)または(C2)の耐ブロッキング性、及び帯電特性が向上する。
【0065】
また、本発明において、微細な球状樹脂粒子(A)の水性分散体(W)を得るために、樹脂(a)は、カルボキシル基を含有することが好ましい。カルボキシル基はその少なくとも一部が塩基で中和されていてもよい。カルボキシル基の塩基中和率は、20〜100当量%が好ましく、40〜100当量%がさらに好ましい。
カルボキシル基の含有量〔塩基で中和されている場合は、カルボキシル基(−COOH基)に換算した含有量〕は、(a)の重量に基づいて0.1〜10%が好ましい。下限は、さらに好ましくは0.5%、とくに好ましくは1%、最も好ましくは3%であり、上限は、さらに好ましくは9.5%、とくに好ましくは9%、最も好ましくは8%である。
塩基中和率や、カルボキシル基含有量が上記範囲の下限以上であると、樹脂(a)が水系媒体中に分散しやすく、微細な球状の樹脂粒子(A)の水性分散体(W)を容易に得ることができる。また、得られる樹脂粒子(C)の帯電特性が向上する。
【0066】
上記の中和塩を形成する塩基としては、アンモニア、炭素数1〜30のモノアミン、後述のポリアミン(21)、4級アンモニウム、アルカリ金属(ナトリウム、カリウム等)、およびアルカリ土類金属(カルシウム塩、マグネシウム塩等)などが挙げられる。
上記炭素数1〜30のモノアミンとしては、炭素数1〜30の1級および/または2級アミン(エチルアミン、n−ブチルアミン、イソブチルアミン等)、炭素数3〜30の3級アミン(トリメチルアミン、トリエチルアミン、ラウリルジメチルアミン等)が挙げられる。4級アンモニウムとしては炭素数4〜30のトリアルキルアンモニウム(ラウリルトリメチルアンモニウム等)などが挙げられる。
これらの中で、好ましくは、アルカリ金属、4級アンモニウム、モノアミン、およびポリアミンであり、さらに好ましくは、ナトリウム、および炭素数1〜20のモノアミンであり、とくに好ましくは、炭素数3〜20の3級モノアミンである。
また、ビニル系樹脂、およびポリエステル樹脂を形成するカルボキシル基またはその塩を含有するモノマーの好ましい炭素数は3〜30であり、さらに好ましくは3〜15、とくに好ましくは3〜8である。
【0067】
本発明においては、樹脂(a)と樹脂(b)のsp値差(Δsp)をKとし、樹脂(a)の重量平均分子量(Mw)の自然対数値をln(Mw)を Hとした時、点(K、H)が、図1に示す下記4点A、B、C、Dからなる四角形ABCDの辺上を含む内部に含まれるように、(a)および(b)が選択されるのが好ましい。
A(0.3、ln3000)、B(1.5、ln1000)、
C(1.3、ln200000)、D(0.1、ln200000)
点(K、H)は、以下の4点A’、B’、C’、D’からなる四角形A’B’C’D’の辺上を含む内部に含まれることがさらに好ましく、以下の4点A”、B”、C”、D”からなる四角形A”B”C”D”の辺上を含む内部に含まれることがとくに好ましい。
A’(0.3、ln3200)、B’(1.45、ln1500)、
C’(1.3、ln100000)、D’(0.15、ln100000)
A”(0.3、ln3400)、B”(1.4、ln2000)、
C”(1.3、ln50000)、D”(0.2、ln50000)
【0068】
点(K,H)が直線ABより下になる場合、造粒時、樹脂粒子(A)が有機溶剤等に溶解しやすく、造粒が上手く出来ない場合がある。また点(K,H)が直線CDより上になる場合、樹脂粒子(A)が有機溶剤等に全く膨潤せず、また熱溶融もしにくくなることから樹脂粒子(C1)の平滑性が不十分となり粉体流動性の低下につながることがある。また、点(K,H)が直線ADより左になる場合、樹脂(a)と樹脂(b)のsp値差が小さいため、樹脂粒子(A)が樹脂(b)の溶液に溶解して造粒が上手く出来ない場合がある。また、点(K,H)が直線BCより右になる場合樹脂(a)と樹脂(b)のsp値差が大きく、樹脂粒子(A)が有機溶剤等に全く膨潤せず粒子の平滑性が不十分となり粉体流動性の低下につながり、さらに点(K,H)が右に位置する場合、つまり樹脂(a)と樹脂(b)のsp値差がより開く場合、樹脂(a)と樹脂(b)の吸着力が減少し、造粒困難となることがある。
【0069】
本発明においては、樹脂粒子(A)の樹脂粒子(B)に対するsp値(sp値の計算方法はPolymer Engineering and Science,Feburuary,1974,Vol.14,No.2 P.147〜154による)、また樹脂粒子(A)の分子量を制御することで樹脂粒子(C1)の粒子表面を平滑にすることができる。
【0070】
樹脂粒子(A)が水や分散時に用いる有機溶剤に対してに、溶解したり、膨潤したりするのを低減する観点から、樹脂(a)の分子量、sp値、結晶性、架橋点間分子量等を適宜調整するのが好ましい。
【0071】
樹脂(a)のMnは、好ましくは100〜500万、さらに好ましくは200〜500万、とくに好ましくは500〜500,000であり、sp値は、好ましくは7〜18、さらに好ましくは8〜14である。樹脂(a)の融点(DSCにて測定)は、好ましくは50℃以上、さらに好ましくは80〜200℃である。また、樹脂粒子(C)の、耐熱性、耐水性、耐薬品性、粒径の均一性等を向上させたい場合には、樹脂(a)に架橋構造を導入させてもよい。かかる架橋構造は、共有結合性、配位結合性、イオン結合性、水素結合性等、いずれの架橋形態であってもよい。樹脂(a)に架橋構造を導入する場合の架橋点間分子量は、好ましくは50以上、さらに好ましくは500以上、とくに好ましくは1000以上である。
【0072】
樹脂(a)のガラス転移温度(Tg)は、樹脂粒子(C)の粒径均一性、粉体流動性、保存時の耐熱性、耐ストレス性の観点から、好ましくは20℃〜250℃、さらに好ましくは30℃〜230℃、より好ましくは40℃〜200℃、とくに好ましくは50℃〜120℃ある。水性樹脂分散体(X1)を作成する温度よりTgが低いと、合一を防止したり、分裂を防止したりする効果が小さくなり、粒径の均一性を高める効果が小さくなる。
また、(a)と必要により(m)からなる樹脂粒子(A)のTgは、同様の理由で、好ましくは20〜200℃、さらに好ましくは30〜120℃、とくに好ましくは40〜80℃である。
なお、本発明におけるTgは、前記DSC測定またはフローテスター測定(DSCで測定できない場合)から求められる値である。
【0073】
フローテスター測定には、島津製作所製の高架式フローテスターCFT500型を用いる。フローテスター測定の条件は下記のとおりであり、以下測定は全てこの条件で行われる。
(フローテスター測定条件)
荷重:30kg/cm2、昇温速度:3.0℃/min、
ダイ口径:0.50mm、ダイ長さ:10.0mm
【0074】
また図2に示すフローチャートにあるA点(試料が圧縮荷重を受け変形し始める温度)をガラス転移温度(Tg)とし、B点(内部空隙が消失し不均一な応力の分布を持ったまま外観均一な1個の透明体あるいは相になる点)の温度を軟化開始温度(Ts)、C点(試料の熱膨張によるピストンのわずかな上昇が行われた後、再びピストンが明らかに下降し始める点)の温度を流出開始温度(Tfb)、そしてD点(図において流出終了点Smaxと最低値Sminの差の1/2(X)を求め、XとSminを加えた点)の温度を流出温度(T1/2)とする。
【0075】
樹脂(a)の軟化開始温度(Ts)は、保存時の耐熱性、耐ストレス性、紙面などへの定着特性の観点から、好ましくは40℃〜270℃、さらに好ましくは50℃〜250℃、とくに好ましくは60℃〜220℃、最も好ましくは70℃〜160℃あり、また流出温度(T1/2)は、好ましくは60℃〜300℃、さらに好ましくは65℃〜280℃、とくに好ましくは70℃〜250℃、最も好ましくは80℃〜185℃ある。トナーなどとして用いる場合、軟化開始温度(Ts)、流出温度(T1/2)が高温であると低温定着性や高光沢性などの悪化因子となることがある。なお、本発明における軟化開始温度、流出温度は、上記フローテスター測定から求められる値である。
【0076】
樹脂(a)のガラス転移温度(Tg)と流出温度(T1/2)との温度差は、好ましくは0℃〜130℃、さらに好ましくは0℃〜125℃、とくに好ましくは0℃〜120℃、最も好ましくは0℃〜105℃である。このガラス転移温度と軟化開始温度の温度差が上記範囲内であると、樹脂粒子をトナーとして用いる場合、樹脂粒子の耐熱保存と高光沢の両立が容易である。
【0077】
また、樹脂(a)のガラス転移温度(Tg)と軟化開始温度(Ts)との好ましい温度差は、0℃〜100℃、より好ましくは0℃〜70℃、さらに好ましくは0℃〜50℃、とくに好ましくは0℃〜35℃である。このガラス転移温度と軟化開始温度の温度差が上記範囲内であると、樹脂粒子をトナーとして用いる場合、樹脂粒子の耐熱保存と高光沢の両立が容易である。
【0078】
樹脂(a)の、ガラス転移温度(Tg)、軟化開始温度(Ts)および流出温度(T1/2)は、(a)の分子量および/または(a)を構成する単量体組成を変更することで、容易に調整できる。(a)の分子量(分子量が大きくなるほど、これらの温度は高くなる。)を調整する方法としては、公知の方法でよく、例えば、ポリウレタン樹脂やポリエステル樹脂のような逐次反応で重合する場合には、単量体の仕込み比の調整が挙げられ、ビニル樹脂のような連鎖反応で重合する場合には、重合開始剤量および連鎖移動剤量の調整、反応温度、反応濃度の調整が挙げられる。ガラス転移温度(Tg)と流出温度(T1/2)との温度差を好ましい範囲に調整するには、(a)の分子量と(a)を構成する単量体組成との組み合わせを適切に選択すればよい。
【0079】
樹脂粒子(A)の水性分散液(W)中に、水以外に後述の有機溶剤(u)のうち水と混和性の有機溶剤(アセトン、メチルエチルケトン等)が含有されていてもよい。この際、含有される有機溶剤は、樹脂粒子(A)の凝集を引き起こさないもの、樹脂粒子(A)を溶解しないもの、および樹脂粒子(C1)の造粒を妨げることがないものであればどの種であっても、またどの程度の含有量であってもかまわないが、水との合計量の40%以下用いて、乾燥後の樹脂粒子(C)中に残らないものが好ましい。
【0080】
樹脂(a)を樹脂粒子(A)の水性分散液にする方法は、とくに限定されないが、以下の〔1〕〜〔8〕が挙げられる。
〔1〕ポリエステル樹脂、ポリウレタン樹脂等の重付加あるいは縮合系樹脂の場合において、前駆体(モノマー、オリゴマー等)またはその有機溶剤溶液を必要であれば適当な分散剤存在下で水性媒体中に分散させ、その後に加熱したり、硬化剤を加えたりして前躯体を硬化させて樹脂粒子(A)の水性分散体を製造する方法。
〔2〕ポリエステル樹脂、ポリウレタン樹脂等の重付加あるいは縮合系樹脂の場合において、前駆体(モノマー、オリゴマー等)またはその有機溶剤溶液(液体であることが好ましい。加熱により液状化してもよい)中に適当な乳化剤を溶解させた後、水を加えて転相乳化し、硬化剤を加えたりして前躯体を硬化させて樹脂粒子(A)の水性分散体を製造する方法。
〔3〕ビニル系樹脂の場合において、モノマーを出発原料として、懸濁重合法、乳化重合法、シード重合法または分散重合法等の重合反応により、直接、樹脂粒子(A)の水性分散液を製造する方法。
〔4〕あらかじめ重合反応(付加重合、開環重合、重付加、付加縮合、縮合重合等いずれの重合反応様式であってもよい。以下同様。)により作成した樹脂を機械回転式またはジェット式等の微粉砕機を用いて粉砕し、次いで、分級するすることによって樹脂粒子を得た後、適当な分散剤存在下で水中に分散させる方法。
〔5〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液を霧状に噴霧することにより樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水中に分散させる方法。
〔6〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液に貧溶剤を添加するか、またはあらかじめ有機溶剤に加熱溶解した樹脂溶液を冷却することにより樹脂粒子を析出させ、次いで、有機溶剤を除去して樹脂粒子を得た後、該樹脂粒子を適当な分散剤存在下で水中に分散させる方法。
〔7〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液を、適当な分散剤存在下で水性媒体中に分散させ、これを加熱または減圧等によって有機溶剤を除去する方法。
〔8〕あらかじめ重合反応により作成した樹脂を有機溶剤に溶解した樹脂溶液中に適当な乳化剤を溶解させた後、水を加えて転相乳化する方法。
【0081】
上記〔1〕〜〔8〕の方法において、併用する乳化剤または分散剤としては、公知の界面活性剤(s)、水溶性ポリマー(t)等を用いることができる。また、乳化または分散の助剤として有機溶剤(u)、可塑剤(v)等を併用することができる。
【0082】
界面活性剤(s)としては、アニオン界面活性剤(s−1)、カチオン界面活性剤(s−2)、両性界面活性剤(s−3)、非イオン界面活性剤(s−4)などが挙げられる。界面活性剤(s)は2種以上の界面活性剤を併用したものであってもよい。(s)の具体例としては、以下に述べるものの他特開2002−284881号公報に記載のものが挙げられる。
【0083】
アニオン界面活性剤(s−1)としては、カルボン酸またはその塩、硫酸エステル塩、カルボキシメチル化物の塩、スルホン酸塩およびリン酸エステル塩等が用いられる。
【0084】
カルボン酸またはその塩としては、炭素数8〜22の飽和または不飽和脂肪酸またはその塩が使用でき、例えば、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、アラキジン酸、ベヘン酸、オレイン酸、リノール酸およびリシノール酸並びにヤシ油、パーム核油、米ぬか油および牛脂などをケン化して得られる高級脂肪酸の混合物等が挙げられる。
その塩としては、これらのナトリウム塩、カリウム塩、アミン塩、アンモニウム塩、4級アンモニウム塩およびアルカノールアミン塩(モノエタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩等)などの塩があげられる。
【0085】
硫酸エステル塩としては、高級アルコール硫酸エステル塩(炭素数8〜18の脂肪族アルコールの硫酸エステル塩)、高級アルキルエーテル硫酸エステル塩(炭素数8〜18の脂肪族アルコールのEOまたはPO1〜10モル付加物の硫酸エステル塩)、硫酸化油(炭素数12〜50の天然の不飽和油脂または不飽和のロウをそのまま硫酸化して中和したもの)、硫酸化脂肪酸エステル(不飽和脂肪酸(炭素数6〜40)の低級アルコール(炭素数1〜8)エステルを硫酸化して中和したもの)および硫酸化オレフィン(炭素数12〜18のオレフィンを硫酸化して中和したもの)等が使用できる。
塩としては、ナトリウム塩、カリウム塩、アミン塩、アンモニウム塩、4級アンモニウム塩およびアルカノールアミン塩(モノエタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩等)等が挙げられる。
【0086】
高級アルコール硫酸エステル塩としては、例えば、オクチルアルコール硫酸エステル塩、デシルアルコール硫酸エステル塩、ラウリルアルコール硫酸エステル塩、ステアリルアルコール硫酸エステル塩、チーグラー触媒を用いて合成されたアルコール(例えば、商品名:ALFOL 1214:CONDEA社製)の硫酸エステル塩およびオキソ法で合成されたアルコール(例えば、商品名:ドバノール23、25、45、ダイヤドール115−L、115H、135:三菱化学製:、商品名:トリデカノール:協和発酵製、商品名:オキソコール1213、1215、1415:日産化学製)の硫酸エステル塩等が挙げられる。
【0087】
高級アルキルエーテル硫酸エステル塩としては、例えば、ラウリルアルコールEO2モル付加物硫酸エステル塩およびオクチルアルコールEO3モル付加物硫酸エステル塩等が挙げられる。
硫酸化油としては、例えば、ヒマシ油、落花生油、オリーブ油、ナタネ油、牛脂および羊脂などの硫酸化物の塩等が挙げられる。
硫酸化脂肪酸エステルとしては、例えば、オレイン酸ブチルおよびリシノレイン酸ブチル等の硫酸化物の塩等が挙げられる。
硫酸化オレフィンとしては、例えば、商品名:ティーポール(シェル社製)等が挙げられる。
【0088】
カルボキシメチル化物の塩としては、炭素数8〜16の脂肪族アルコールのカルボキシメチル化物の塩および炭素数8〜16の脂肪族アルコールのEOまたはPO1〜10モル付加物のカルボキシメチル化物の塩等が使用できる。
【0089】
脂肪族アルコールのカルボキシメチル化物の塩としては、例えば、オクチルアルコールカルボキシメチル化ナトリウム塩、ラウリルアルコールカルボキシメチル化ナトリウム塩、ドバノール23のカルボキシメチル化ナトリウム塩、トリデカノールカルボキシメチル化ナトリウム塩等が挙げられる。
【0090】
脂肪族アルコールのEO1〜10モル付加物のカルボキシメチル化物の塩としては、例えば、オクチルアルコールEO3モル付加物カルボキシメチル化ナトリウム塩、ラウリルアルコールEO4モル付加物カルボキシメチル化ナトリウム塩、およびトリデカノールEO5モル付加物カルボキシメチル化ナトリウム塩などが挙げられる。
【0091】
スルホン酸塩としては、アルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、スルホコハク酸ジエステル塩、α−オレフィンスルホン酸塩、イゲポンT型およびその他芳香環含有化合物のスルホン酸塩等が使用できる。
アルキルベンゼンスルホン酸塩としては、例えば、ドデシルベンゼンスルホン酸ナトリウム塩等が挙げられる。
【0092】
アルキルナフタレンスルホン酸塩としては、例えば、ドデシルナフタレンスルホン酸ナトリウム塩等が挙げられる。
スルホコハク酸ジエステル塩としては、例えば、スルホコハク酸ジ−2−エチルヘキシルエステルナトリウム塩などが挙げられる。
芳香環含有化合物のスルホン酸塩としては、アルキル化ジフェニルエーテルのモノまたはジスルホン酸塩およびスチレン化フェノールスルホン酸塩などが挙げられる。
【0093】
リン酸エステル塩としては、高級アルコールリン酸エステル塩および高級アルコールEO付加物リン酸エステル塩等が使用できる。
高級アルコールリン酸エステル塩としては、例えば、ラウリルアルコールリン酸モノエステルジナトリウム塩およびラウリルアルコールリン酸ジエステルナトリウム塩等が挙げられる。
高級アルコールEO付加物リン酸エステル塩としては、例えば、オレイルアルコールEO5モル付加物リン酸モノエステルジナトリウム塩等が挙げられる。
【0094】
カチオン界面活性剤(s−2)としては、第4級アンモニウム塩型界面活性剤およびアミン塩型界面活性剤等が使用できる。
第4級アンモニウム塩型界面活性剤としては、炭素数3〜40の3級アミンと4級化剤(例えば、メチルクロライド、メチルブロマイド、エチルクロライド、ベンジルクロライドおよびジメチル硫酸などのアルキル化剤並びにEOなど)との反応等で得られ、例えば、ラウリルトリメチルアンモニウムクロライド、ジデシルジメチルアンモニウムクロライド、ジオクチルジメチルアンモニウムブロマイド、ステアリルトリメチルアンモニウムブロマイド、ラウリルジメチルベンジルアンモニウムクロライド(塩化ベンザルコニウム)、セチルピリジニウムクロライド、ポリオキシエチレントリメチルアンモニウムクロライドおよびステアラミドエチルジエチルメチルアンモニウムメトサルフェートなどが挙げられる。
【0095】
アミン塩型界面活性剤としては、1〜3級アミンを無機酸(例えば、塩酸、硝酸、硫酸、ヨウ化水素酸、リン酸および過塩素酸など)または有機酸(酢酸、ギ酸、蓚酸、乳酸、グルコン酸、アジピン酸、炭素数2〜24のアルキルリン酸、リンゴ酸およびクエン酸など)で中和すること等により得られる。
第1級アミン塩型界面活性剤としては、例えば、炭素数8〜40の脂肪族高級アミン(例えば、ラウリルアミン、ステアリルアミン、セチルアミン、硬化牛脂アミンおよび、ロジンアミンなどの高級アミン)の無機酸塩または有機酸塩および低級アミン(炭素数2〜6)の高級脂肪酸(炭素数8〜40、ステアリン酸、オレイン酸など)塩などが挙げられる。
【0096】
第2級アミン塩型界面活性剤としては、例えば炭素数4〜40の脂肪族アミンのEO付加物などの無機酸塩または有機酸塩が挙げられる。
また、第3級アミン塩型界面活性剤としては、例えば、炭素数4〜40の脂肪族アミン(例えば、トリエチルアミン、エチルジメチルアミン、N,N,N’,N’−テトラメチルエチレンジアミンなど)、脂肪族アミン(炭素数2〜40)のEO(2モル以上)付加物、炭素数6〜40の脂環式アミン(例えば、N−メチルピロリジン、N−メチルピペリジン、N−メチルヘキサメチレンイミン、N−メチルモルホリンおよび1,8−ジアザビシクロ(5,4,0)−7−ウンデセンなど)、炭素数5〜30の含窒素ヘテロ環芳香族アミン(例えば、4−ジメチルアミノピリジン、N−メチルイミダゾールおよび4,4’−ジピリジルなど)の無機酸塩または有機酸塩およびトリエタノールアミンモノステアレート、ステアラミドエチルジエチルメチルエタノールアミンなどの3級アミンの無機酸塩または有機酸塩などが挙げられる。
【0097】
両性界面活性剤(s−3)としては、カルボン酸塩型両性界面活性剤、硫酸エステル塩型両性界面活性剤、スルホン酸塩型両性界面活性剤およびリン酸エステル塩型両性界面活性剤などが使用できる。
【0098】
カルボン酸塩型両性界面活性剤は、アミノ酸型両性界面活性剤、ベタイン型両性界面活性剤およびイミダゾリン型両性界面活性剤などが用いられる。アミノ酸型両性界面活性剤は、分子内にアミノ基とカルボキシル基を持っている両性界面活性剤であり、例えば、一般式(2)で示される化合物等が挙げられる。
【0099】
[R−NH−(CH2)n−COO]mM (2)
[式中、Rは1価の炭化水素基;nは1または2;mは1または2;Mは水素イオン、アルカリ金属イオン、アルカリ土類金属イオン、アンモニウムカチオン、アミンカチオン、アルカノールアミンカチオンなどである。]
【0100】
一般式(2)で表される両面活性剤としては、例えば、アルキル(炭素数6〜40)アミノプロピオン酸型両性界面活性剤(ステアリルアミノプロピオン酸ナトリウム、ラウリルアミノプロピオン酸ナトリウムなど);アルキル(炭素数4〜24)アミノ酢酸型両性界面活性剤(ラウリルアミノ酢酸ナトリウムなど)などが挙げられる。
【0101】
ベタイン型両性界面活性剤は、分子内に第4級アンモニウム塩型のカチオン部分とカルボン酸型のアニオン部分を持っている両性界面活性剤であり、例えば、アルキル(炭素数6〜40)ジメチルベタイン(ステアリルジメチルアミノ酢酸ベタイン、ラウリルジメチルアミノ酢酸ベタインなど)、炭素数6〜40のアミドベタイン(ヤシ油脂肪酸アミドプロピルベタインなど)、アルキル(炭素数6〜40)ジヒドロキシアルキル(炭素数6〜40)ベタイン(ラウリルジヒドロキシエチルベタインなど)などが挙げられる。
【0102】
イミダゾリン型両性界面活性剤としては、イミダゾリン環を有するカチオン部分とカルボン酸型のアニオン部分を持っている両性界面活性剤であり、例えば、2−ウンデシル−N−カルボキシメチル−N−ヒドロキシエチルイミダゾリニウムベタインなどが挙げられる。
【0103】
その他の両性界面活性剤として、例えば、ナトリウムラウロイルグリシン、ナトリウムラウリルジアミノエチルグリシン、ラウリルジアミノエチルグリシン塩酸塩、ジオクチルジアミノエチルグリシン塩酸塩などのグリシン型両性界面活性剤;ペンタデシルスルホタウリンなどのスルホベタイン型両性界面活性剤、スルホン酸塩型両性界面活性剤およびリン酸エステル塩型両性界面活性剤などが挙げられる。
【0104】
非イオン界面活性剤(s−4)としては、AO付加型非イオン界面活性剤および多価アルコ−ル型非イオン界面活性剤などが使用できる。
AO付加型非イオン界面活性剤は、炭素数8〜40の高級アルコ−ル、炭素数8〜40の高級脂肪酸または炭素数8〜40のアルキルアミン等に直接AO(炭素数2〜20)を付加させるか、グリコ−ルにAOを付加させて得られるポリアルキレングリコ−ルに高級脂肪酸などを反応させるか、あるいは多価アルコ−ルに高級脂肪酸を反応して得られたエステル化物にAOを付加させるか、高級脂肪酸アミドにAOを付加させることにより得られる。
【0105】
AOとしては、たとえばEO、POおよびBOが挙げられる。
これらのうち好ましいものは、EOおよびEOとPOのランダムまたはブロック付加物である。
AOの付加モル数としては10〜50モルが好ましく、該AOのうち50〜100%がEOであるものが好ましい。
【0106】
AO付加型非イオン界面活性剤としては、例えば、オキシアルキレンアルキルエ−テル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)(例えば、オクチルアルコールEO20モル付加物、ラウリルアルコールEO20モル付加物、ステアリルアルコールEO10モル付加物、オレイルアルコールEO5モル付加物、ラウリルアルコールEO10モルPO20モルブロック付加物など);ポリオキシアルキレン高級脂肪酸エステル(アルキレンの炭素数2〜24、高級脂肪酸の炭素数8〜40)(例えば、ステアリル酸EO10モル付加物、ラウリル酸EO10モル付加物など);ポリオキシアルキレン多価アルコ−ル高級脂肪酸エステル(アルキレンの炭素数2〜24、多価アルコールの炭素数3〜40、高級脂肪酸の炭素数8〜40)(例えば、ポリエチレングリコール(重合度20)のラウリン酸ジエステル、ポリエチレングリコール(重合度20)のオレイン酸ジエステルなど);ポリオキシアルキレンアルキルフェニルエーテル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)(例えば、ノニルフェノールEO4モル付加物、ノニルフェノールEO8モルPO20モルブロック付加物、オクチルフェノールEO10モル付加物、ビスフェノールA・EO10モル付加物、スチレン化フェノールEO20モル付加物など);ポリオキシアルキレンアルキルアミノエ−テル(アルキレンの炭素数2〜24、アルキルの炭素数8〜40)および(例えば、ラウリルアミンEO10モル付加物、ステアリルアミンEO10モル付加物など);ポリオキシアルキレンアルカノ−ルアミド(アルキレンの炭素数2〜24、アミド(アシル部分)の炭素数8〜24)(例えば、ヒドロキシエチルラウリン酸アミドのEO10モル付加物、ヒドロキシプロピルオレイン酸アミドのEO20モル付加物など)が挙げられる。
【0107】
多価アルコ−ル型非イオン界面活性剤としては、多価アルコール脂肪酸エステル、多価アルコール脂肪酸エステルAO付加物、多価アルコールアルキルエーテルおよび多価アルコールアルキルエーテルAO付加物等が使用できる。多価アルコールの炭素数としては3〜24、脂肪酸の炭素数としては8〜40、AOの炭素数としては2〜24である。
【0108】
多価アルコール脂肪酸エステルとしては、例えば、ペンタエリスリトールモノラウレート、ペンタエリスリトールモノオレート、ソルビタンモノラウレート、ソルビタンモノステアレート、ソルビタンモノラウレート、ソルビタンジラウレート、ソルビタンジオレートおよびショ糖モノステアレートなどが挙げられる。
【0109】
多価アルコール脂肪酸エステルAO付加物としては、例えば、エチレングリコールモノオレートEO10モル付加物、エチレングリコールモノステアレートEO20モル付加物、トリメチロールプロパンモノステアレートEO20モルPO10モルランダム付加物、ソルビタンモノラウレートEO10モル付加物、ソルビタンジステアレートEO20モル付加物およびソルビタンジラウレートEO12モルPO24モルランダム付加物などが挙げられる。
【0110】
多価アルコールアルキルエーテルとしては、例えば、ペンタエリスリトールモノブチルエーテル、ペンタエリスリトールモノラウリルエーテル、ソルビタンモノメチルエーテル、ソルビタンモノステアリルエーテル、メチルグリコシドおよびラウリルグリコシドなどが挙げられる。
【0111】
多価アルコールアルキルエーテルAO付加物としては、例えば、ソルビタンモノステアリルエーテルEO10モル付加物、メチルグリコシドEO20モルPO10モルランダム付加物、ラウリルグリコシドEO10モル付加物およびステアリルグリコシドEO20モルPO20モルランダム付加物などが挙げられる。
【0112】
水溶性ポリマー(t)としては、セルロース系化合物(例えば、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、エチルヒドロキシエチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルセルロースおよびそれらのケン化物など)、ゼラチン、デンプン、デキストリン、アラビアゴム、キチン、キトサン、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレングリコール、ポリエチレンイミン、ポリアクリルアミド、アクリル酸(塩)含有ポリマー(ポリアクリル酸ナトリウム、ポリアクリル酸カリウム、ポリアクリル酸アンモニウム、ポリアクリル酸の水酸化ナトリウム部分中和物、アクリル酸ナトリウム−アクリル酸エステル共重合体)、スチレン−無水マレイン酸共重合体の水酸化ナトリウム(部分)中和物、水溶性ポリウレタン(ポリエチレングリコール、ポリカプロラクトンジオール等とポリイソシアネートの反応生成物等)などが挙げられる。
【0113】
本発明に用いる有機溶剤(u)は、乳化分散の際に必要に応じて水性媒体中に加えても、被乳化分散体中[樹脂(b)を含む油相中]に加えてもよい。
有機溶剤(u)の具体例としては、トルエン、キシレン、エチルベンゼン、テトラリン等の芳香族炭化水素系溶剤;n−ヘキサン、n−ヘプタン、ミネラルスピリット、シクロヘキサン等の脂肪族または脂環式炭化水素系溶剤;塩化メチル、臭化メチル、ヨウ化メチル、メチレンジクロライド、四塩化炭素、トリクロロエチレン、パークロロエチレンなどのハロゲン系溶剤;酢酸エチル、酢酸ブチル、メトキシブチルアセテート、メチルセロソルブアセテート、エチルセロソルブアセテートなどのエステル系またはエステルエーテル系溶剤;ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチルセロソルブ、ブチルセロソルブ、プロピレングリコールモノメチルエーテルなどのエーテル系溶剤;アセトン、メチルエチルケトン、メチルイソブチルケトン、ジ−n−ブチルケトン、シクロヘキサノンなどのケトン系溶剤;メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、t−ブタノール、2−エチルヘキシルアルコール、ベンジルアルコールなどのアルコール系溶剤;ジメチルホルムアミド、ジメチルアセトアミドなどのアミド系溶剤;ジメチルスルホキシドなどのスルホキシド系溶剤、N−メチルピロリドンなどの複素環式化合物系溶剤、ならびにこれらの2種以上の混合溶剤が挙げられる。
【0114】
可塑剤(v)は、乳化分散の際に必要に応じて水性媒体中に加えても、被乳化分散体中[樹脂(b)を含む油相中]に加えてもよい。
可塑剤(v)としては、何ら限定されず、以下のものが例示される。
(v1)フタル酸エステル[フタル酸ジブチル、フタル酸ジオクチル、フタル酸ブチルベンジル、フタル酸ジイソデシル等];
(v2)脂肪族2塩基酸エステル[アジピン酸ジ−2−エチルヘキシル、セバシン酸−2−エチルヘキシル等];
(v3)トリメリット酸エステル[トリメリット酸トリ−2−エチルヘキシル、トリメリット酸トリオクチル等];
(v4)燐酸エステル[リン酸トリエチル、リン酸トリ−2−エチルヘキシル、リン酸トリクレジール等];
(v5)脂肪酸エステル[オレイン酸ブチル等];
(v6)およびこれらの2種以上の混合物が挙げられる。
【0115】
本発明において用いる樹脂粒子(A)の粒径は、通常、形成される樹脂粒子(B)の粒径よりも小さく、粒径均一性の観点から、粒径比[樹脂粒子(A)の体積平均粒径]/[樹脂粒子(B)の体積平均粒径]の値が0.001〜0.3の範囲であるのが好ましい。粒径比の下限は、さらに好ましくは0.003であり、上限は、さらに好ましくは0.25である。粒径比が、0.3より大きいと(A)が(B)の表面に効率よく吸着しないため、得られる(C)の粒度分布が広くなる傾向がある。
【0116】
樹脂粒子(A)の体積平均粒径は、所望の粒径の樹脂粒子(C)を得るのに適した粒径になるように、上記粒径比の範囲で適宜調整することができる。
(A)の体積平均粒径は、一般的には、0.0005〜30μmが好ましい。上限は、さらに好ましくは20μm、とくに好ましくは10μmであり、下限は、さらに好ましくは0.01μm、とくに好ましくは0.02μm、最も好ましくは0.04μmである。ただし、例えば、体積平均粒径1μmの樹脂粒子(C)を得たい場合には、好ましくは0.0005〜0.3μm、とくに好ましくは0.001〜0.2μmの範囲、10μmの樹脂粒子(C)を得た場合には、好ましくは0.005〜3μm、とくに好ましくは0.05〜2μm、100μmの粒子(C)を得たい場合には、好ましくは0.05〜30μm、とくに好ましくは0.1〜20μmである。
なお、体積平均粒径は、レーザー式粒度分布測定装置LA−920(堀場製作所製)やマルチサイザーIII(コールター社製)、光学系としてレーザードップラー法を用いるELS−800(大塚電子社製)などで測定できる。もし、各測定装置間で粒径の測定値に差を生じた場合は、ELS−800での測定値を採用する。
なお、上記粒径比が得やすいことから、後述する樹脂粒子(B)の体積平均粒径は、0.1〜300μmが好ましい。さらに好ましくは0.5〜250μm、特に好ましくは1〜200μmである。
【0117】
本発明の水性樹脂分散体および樹脂粒子(C)の製造方法においては、水性樹脂分散体(X1)は、樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)またはその有機溶剤溶液とを混合し、(W)中に(b)またはその有機溶剤溶液を分散させて、(A)の水性分散液中で、(b)からなる樹脂粒子(B)を形成させることにより、(B)の表面に(A)が付着されてなる構造の樹脂粒子(C1)の水性分散体として得ることができる。
【0118】
上記製造方法においては、水性分散液(W)中に、樹脂(b)もしくはその有機溶剤溶液を分散させて、(b)からなる樹脂粒子(B)が形成される際に、樹脂粒子(B)の表面に樹脂粒子(A)を吸着させることで樹脂粒子(C1)同士が合一するのを防ぎ、また、高剪断条件下で(C1)が分裂され難くする。これにより、(C1)の粒径を一定の値に収斂させ、粒径の均一性を高める効果を発揮する。そのため、樹脂粒子(A)は、分散する際の温度において、剪断により破壊されない程度の強度を有すること、水に溶解したり、膨潤したりしにくいこと、(b)もしくはその有機溶剤溶液に溶解しにくいことが好ましい特性としてあげられる
【0119】
樹脂(b)もしくはその有機溶剤溶液を分散させる場合には、分散装置を用いることができる。
本発明で使用する分散装置は、一般に乳化機、分散機として市販されているものであればとくに限定されず、例えば、ホモジナイザー(IKA社製)、ポリトロン(キネマティカ社製)、TKオートホモミキサー(特殊機化工業社製)等のバッチ式乳化機、エバラマイルダー(荏原製作所社製)、TKフィルミックス、TKパイプラインホモミキサー(特殊機化工業社製)、コロイドミル(神鋼パンテック社製)、スラッシャー、トリゴナル湿式微粉砕機(三井三池化工機社製)、キャピトロン(ユーロテック社製)、ファインフローミル(太平洋機工社製)等の連続式乳化機、マイクロフルイダイザー(みずほ工業社製)、ナノマイザー(ナノマイザー社製)、APVガウリン(ガウリン社製)等の高圧乳化機、膜乳化機(冷化工業社製)等の膜乳化機、バイブロミキサー(冷化工業社製)等の振動式乳化機、超音波ホモジナイザー(ブランソン社製)等の超音波乳化機等が挙げられる。このうち粒径の均一化の観点で好ましいものは、APVガウリン、ホモジナイザー、TKオートホモミキサー、エバラマイルダー、TKフィルミックス、TKパイプラインホモミキサーが挙げられる。
【0120】
樹脂(b)を樹脂粒子(A)の水性分散液(W)に分散させる際、樹脂(b)は液体であることが好ましい。樹脂(b)が常温で固体である場合には、融点以上の高温下で液体の状態で分散させたり、(b)の有機溶剤溶液を用いてもよい。
樹脂(b)もしくはその有機溶剤溶液の粘度は、は、粒径均一性の観点から、好ましくは10〜5万mPa・s(B型粘度計で測定)、さらに好ましくは100〜1万mPa・sである。
分散時の温度としては、好ましくは0〜150℃(加圧下)、さらに好ましくは5〜98℃である。分散体の粘度が高い場合は、高温にして粘度を上記好ましい範囲まで低下させて、乳化分散を行うのが好ましい。
樹脂(b)の有機溶剤溶液に用いる溶剤は、樹脂(b)を常温もしくは加熱下で溶解しうる溶剤であればとくに限定されず、具体的には、有機溶剤(u)と同様のものが例示される。好ましいものは樹脂(b)の種類によって異なるが、(b)とのsp値差が3以下であるのが好適である。また、樹脂粒子(C)の粒径均一性の観点からは、樹脂(b)を溶解させるが、樹脂(a)からなる樹脂粒子(A)を溶解・膨潤させにくい溶剤が好ましい。
【0121】
樹脂(b)100部に対する水性分散液(W)の使用量は、好ましくは50〜2,000重量部、さらに好ましくは100〜1,000重量部である。50重量部以上では(b)の分散状態が良好であり、2,000重量部以下であると経済的である。
【0122】
本発明においては、前記の点(K,H)が特定範囲内に含まれるような樹脂(a)と(b)、あるいは前記好ましい範囲の(Ts)、(Tg)等の物性を有する樹脂(a)を用いる場合、とくに(b)の有機溶剤溶液(とくに下記の好ましい溶剤)を用いる場合、有機溶剤を水性樹脂分散体(X1)中に好ましくは10〜50%(とくに20〜40%)用い、40℃以下で好ましくは1%以下(とくに0.5%以下)となるまで脱溶剤するのみで、樹脂粒子(A)が有機溶剤に溶解されて膜状化し、(B)の表面に(A)の被膜が形成されてなる樹脂粒子(C2)の水性樹脂分散体(X2)が得られる場合が多い。しかし、(A)の被膜が形成されていない場合、あるいは(A)の少なくとも一部からの被膜が形成されている場合でも、さらに樹脂粒子(C)表面の被膜の平滑性をより良好にするため、以下の操作を行うと、(B)で構成されるコア層(Q)の表面の少なくとも一部、好ましくは全面に(A)から形成された表面が平滑な被膜〔シェル層(P)〕を有する樹脂粒子(C2)の水性樹脂分散体(X2)が得られ、それから得られる(C2)の保存安定性が優れる点から好ましい。
上記の方法としては、(B)に付着された(A)を有機溶剤に溶解させる方法、および、水性樹脂分散体(X1)を加熱して(A)を溶融し被膜化させる方法が挙げられ、これらの方法を併用してもよい。
【0123】
樹脂粒子(A)を有機溶剤に溶解させて被膜化させる場合に用いる溶剤は、被膜化する際に(X1)中に添加してもよいが、(X1)を得る際の原料として、樹脂(b)の有機溶剤溶液を用い、その溶剤を、樹脂粒子(B)の形成後も直ちに除去せずにそれを用いる方が、(B)中に溶剤が含有されるため(A)の溶解が容易であり、樹脂の凝集が起こりにくく好ましい。
有機溶剤としては、(b)との親和性が高いものが好ましく、具体例としては、前記の有機溶剤(u)と同様のものが挙げられる。(u)の中で好ましいものは、被膜化の点から、テトラヒドロフラン、トルエン、アセトン、メチルエチルケトン、および酢酸エチルであり、さらに好ましくは酢酸エチルである。
(A)を有機溶剤に溶解させる際の、水性樹脂分散体中の溶剤濃度は、好ましくは3〜60%、さらに好ましくは10〜45%、とくに好ましくは15〜30%である。また、溶解は、水性樹脂分散体を、例えば1〜10時間攪拌することにより行い、溶解時の温度は、15〜45℃が好ましく、15〜30℃がさらに好ましい。
【0124】
(A)を溶融して(B)の表面に被膜化させる場合、水性樹脂分散体(X1)中の固形分含量〔水および有機溶剤以外の成分の含量〕を、好ましくは1〜50%、さらに好ましくは5〜30%に調製する。また、このときの溶剤含有量は、好ましくは2%以下、さらに好ましくは1%以下、とくに好ましくは0.5%以下である。(X1)中の固形分含量が多かったり、溶剤含有量が2%を越える場合、(X1)を60℃以上に昇温すると凝集物が発生することがある。溶融時の加熱の条件は、(A)が溶融される条件であればとくに限定されないが、例えば、撹拌下、好ましくは40〜100℃、さらに好ましくは60〜90℃、とくに好ましくは60〜80℃で、好ましくは1〜300分間加熱する方法が挙げられる。
なお、被膜化処理の方法とし、溶剤含有量が2%以下の樹脂粒子(C1)の水性分散体(X1)を加熱処理し、(A)をコア(Q)上で溶融させることでより表面が平滑な樹脂粒子(C2)を得る際の好ましい加熱処理温度は、(P)のTg以上であり、また80℃以下の温度範囲が好ましい。加熱処理温度が(P)のTg未満であると得られる樹脂粒子(C2)の表面平滑性はほとんど変化がない。また80℃を越える温度で加熱処理するとシェル(P)がコアから剥がれる場合がある。
これらの(A)の被膜化方法の中で、好ましい方法は、(A)を溶融させる方法、および(A)を溶解させる方法と(A)を溶融させる方法の併用である。
【0125】
樹脂粒子(C1)あるいは(C2)は、樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)または(b)の有機溶剤溶液とが混合され、(W)中に(b)または(b)の有機溶剤溶液が分散され、樹脂(b)からなる樹脂粒子(B)の表面に樹脂粒子(A)が付着してなる構造の樹脂粒子(C1)の水性分散体(X1)、および/または、(B)で構成されるコア層(Q)の表面に(A)から形成されたシェル層(P)を有する構造の樹脂粒子(C2)の水性分散体(X2)を形成させた後、水性樹脂分散体(X1)または(X2)〔以下、「(X1)または(X2)」のことを、(X)と記載することもある。〕から水性媒体を除去することにより得られる。
【0126】
水性媒体を除去する方法としては、
〔1〕水性樹脂分散体を減圧下または常圧下で乾燥する方法
〔2〕遠心分離器、スパクラフィルター、フィルタープレスなどにより固液分離し、得られた粉末を乾燥する方法
〔3〕水性樹脂分散体を凍結させて乾燥させる方法(いわゆる凍結乾燥)
等が例示される。
上記〔1〕、〔2〕において、得られた粉末を乾燥する際、流動層式乾燥機、減圧乾燥機、循風乾燥機など公知の設備を用いて行うことができる。
また、必要に応じ、風力分級器などを用いて分級し、所定の粒度分布とすることもできる。
【0127】
樹脂粒子(C1)は、実質的に、相対的に小さい樹脂粒子(A)と相対的に大きい樹脂粒子(B)から構成され、(A)が(B)の表面に付着された形で存在する。また、樹脂粒子(C2)は、(A)が(B)に付着後、溶解および/または溶融され、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されたものである。
両粒子の付着力をさらに強めたい場合には、水性媒体中に分散した際に、(A)と(B)が正負逆の電荷を持つようにしたり、(A)(B)が同一の電荷持つ場合には、界面活性剤(s)または水溶性ポリマー(t)のうち、(A)および(B)と逆電荷を持つものを使用したり、また樹脂(a)と樹脂(b)のsp値差を前記の範囲内でできるだけ小さく(例えば2以下)したりすることが有効である。
ある。
【0128】
また、本発明の水性樹脂分散体および樹脂粒子(C)を製造する際、樹脂粒子(A)の樹脂粒子(B)に対する吸着力は、以下のような方法で制御することができる。
〔1〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が正負逆の電荷を持つようにすると吸着力が発生し、この場合、樹脂粒子(A)、樹脂粒子(B)各々の電荷を大きくするほど、吸着力が強くなり樹脂粒子(A)の樹脂粒子(B)に対する被覆率が大きくなる。
〔2〕水性分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が同極性(どちらも正、またはどちらも負)の電荷を持つようにすると、被覆率は下がる傾向にある。この場合、一般に活性剤(s)および/または水溶性ポリマー(t)[とくに樹脂粒子(A)および樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。
〔3〕水性分散体(X1)を製造する際に、樹脂(a)がカルボキシル基、リン酸基、スルホン酸基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが低いほど被覆率が大きくなる。逆に、pHを高くするほど被覆率が小さくなる。
〔4〕水性分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム塩基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが高いほど被覆率が大きくなる。逆に、pHを低くするほど被覆率が小さくなる。
〔5〕樹脂粒子(A)と樹脂粒子(B)のΔsp値を小さくすると被覆率が大きくなる。
【0129】
樹脂粒子(C)の粒径均一性、保存安定性等の観点から、樹脂粒子(C)は、0.01〜60%の(A)と40〜99.99%の(B)からなるのが好ましく、さらに好ましくは0.1〜50%の(A)と50〜99.9%の(B)、とくに好ましくは1〜45%の(A)と55〜99%の(B)からなる。
【0130】
樹脂粒子(C1)の粒径均一性、粉体流動性、保存安定性等の観点からは、樹脂粒子(B)の表面の5%以上、好ましくは30%以上、さらに好ましくは50%以上、とくに好ましくは80%以上が樹脂粒子(A)で覆われているのがよい。(C1)の表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[樹脂粒子(A)に覆われている部分の面積/樹脂粒子(A)に覆われている部分の面積+樹脂粒子(B)が露出している部分の面積]×100
【0131】
樹脂粒子(C2)においての下式で求められる表面被覆率はいかなる率であってもかまわないが、粒径均一性、粉体流動性、保存安定性等の観点からは、コア層(Q)の表面の70%以上、好ましくは80%以上、さらに好ましくは90%以上、とくに好ましくは95%以上がシェル層(P)で覆われているのがよい。なお、表面被覆率は、走査電子顕微鏡(SEM)で得られる像の画像解析から下式に基づいて求めることができる。
表面被覆率(%)=[(P)に覆われている部分の面積/(P)に覆われている部分の面積+(Q)が露出している部分の面積]×100
【0132】
粒径均一性の観点から、樹脂粒子(C)の体積分布の変動係数は、30%以下であるのが好ましく、0.1〜15%であるのがさらに好ましい。
また、粒径均一性から、樹脂粒子(C)の[体積平均粒径/個数平均粒径](Dv/Dn)の値は、1.0〜1.4であるのが好ましく、1.0〜1.2であるのがさらに好ましい。
(C)の体積平均粒径は、用途により異なるが、一般的には0.1〜300μmが好ましい。上限は、さらに好ましくは250μm、特に好ましくは200μmであり、下限は、さらに好ましくは0.5μm、特に好ましくは1μmである。
なお、体積平均粒径および個数平均粒径は、マルチサイザーIII(コールター社製)で同時に測定することができる。
【0133】
本発明の樹脂粒子(C2)の形状の制御は、以下の方法で行うことが出来る。
樹脂粒子(A)と樹脂粒子(B)のsp値差、また樹脂粒子(A)の分子量を制御することで粒子形状や粒子表面性を制御することができる。sp値差が小さいといびつな形で表面平滑な粒子が得られやすく、また、sp値差が大きいと球形で表面はザラつきのある粒子が得られやすい。また、(A)の分子量が大きいと表面はザラつきのある粒子が得られやすく、分子量が小さいと表面平滑な粒子が得られやすい。ただし、(A)と(B)のsp値差は小さすぎても大きすぎても造粒困難になる。また樹脂粒子(A)の分子量も小さすぎると造粒困難になる。このことから、好ましい(A)と(B)のsp値差は0.01〜5.0でより好ましくは0.1〜3.0、さらに好ましくは0.2〜2.0である。また、好ましい樹脂粒子(A)のMwは100〜100万で、より好ましくは1000〜50万、さらに好ましくは2000〜20万、特に好ましくは3000〜10万である。
【0134】
本発明の樹脂粒子(C)は、樹脂粒子(A)と樹脂粒子(B)の粒径、および、樹脂粒子(A)による樹脂粒子(B)表面の被覆率あるいはシェル層(P)によるコア層(Q)表面の被覆率を変えることで粒子表面に所望の凹凸を付与することができる。粉体流動性を向上させたい場合には、(C)のBET値比表面積が0.5〜5.0m2/gであるのが好ましい。本発明のBET比表面積は、比表面積計例えばQUANTASORB(ユアサアイオニクス製)を用いて測定(測定ガス:He/Kr=99.9/0.1vol%、検量ガス:窒素)したものである。
同様に粉体流動性の観点から、(C)の表面平均中心線粗さRaが0.01〜0.8μmであるのが好ましい。Raは、粗さ曲線とその中心線との偏差の絶対値を算術平均した値のことであり、例えば、走査型プローブ顕微鏡システム(東陽テクニカ製)で測定することができる。
【0135】
樹脂粒子(C)の形状は、粉体流動性、溶融レベリング性等の観点から球状であるのが好ましい。その場合、粒子(A)および粒子(B)も球状であるのが好ましい。(C)は平均円形度が0.95〜1.00であるのが好ましい。平均円形度は、さらに好ましくは0.96〜1.0、とくに好ましくは0.97〜1.0である。なお、平均円形度は、光学的に粒子を検知して、投影面積の等しい相当円の周囲長で除した値である。具体的には、フロー式粒子像分析装置(FPIA−2000;シスメックス社製)を用いて測定する。所定の容器に、予め不純固形物を除去した水100〜150mlを入れ、分散剤として界面活性剤(ドライウエル;富士写真フィルム社製)0.1〜0.5mlを加え、さらに測定資料0.1〜9.5g程度を加える。試料を分散した懸濁液を超音波分散器(ウルトラソニッククリーナ モデル VS−150;ウエルボクリア社製)で約1〜3分間分散処理を行ない、分散濃度を3,000〜10,000個/μLにして樹脂粒子の形状および分布を測定する。
【0136】
本発明の水性樹脂分散体および樹脂粒子(C)の製造方法において、樹脂粒子(C)は、樹脂粒子(A)の樹脂粒子(B)に対する粒径比、および、水性樹脂分散体(X1)中における樹脂粒子(A)による樹脂粒子(B)表面の被覆率、水性樹脂分散体(X1)中における樹脂粒子(B)/水性媒体界面上で樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さ、を変えることで粒子表面を平滑にしたり、粒子表面に所望の凹凸を付与したりすることができる。
樹脂粒子(A)による樹脂粒子(B)表面の被覆率や樹脂粒子(A)が樹脂粒子(B)側に埋め込まれている深さは、以下のような方法で制御することができる。
〔1〕水性樹脂分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が正負逆の電荷を持つようにすると被覆率、深さが大きくなる。この場合、樹脂粒子(A)、樹脂粒子(B)各々の電荷を大きくするほど、被覆率、深さが大きくなる。
〔2〕水性樹脂分散体(X1)を製造する際に、樹脂粒子(A)と樹脂粒子(B)が同極性(どちらも正、またはどちらも負)の電荷を持つようにすると、被覆率は下がり、深さが小さくなる傾向にある。この場合、一般に界面活性剤(s)および/または水溶性ポリマー(t)[とくに樹脂粒子(A)および樹脂粒子(B)と逆電荷を有するもの]を使用すると被覆率が上がる。また、水溶性ポリマー(t)を使用する場合には、水溶性ポリマー(t)の分子量が大きいほど深さが小さくなる。
〔3〕水性樹脂分散体(X1)を製造する際に、樹脂(a)がカルボキシル基、リン酸基、スルホン酸基等の酸性官能基を有する樹脂(一般に酸性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが低いほど被覆率、深さが大きくなる。逆に、pHを高くするほど被覆率、深さが小さくなる。
〔4〕水性樹脂分散体(X1)を製造する際に、樹脂(a)が1級アミノ基、2級アミノ基、3級アミノ基、4級アンモニウム塩基等の塩基性官能基を有する樹脂(一般に塩基性官能基1個当たりの分子量が1,000以下であるのが好ましい)である場合に、水性媒体のpHが高いほど被覆率、深さが大きくなる。逆に、pHを低くするほど被覆率、深さが小さくなる。
〔5〕樹脂(a)と樹脂(b)のSP値差を小さくするほど被覆率、深さが大きくなる。
【0137】
樹脂粒子(C)を構成する樹脂粒子(A)および/または(B)中に、添加剤(顔料、充填剤、帯電防止剤、着色剤、離型剤、荷電制御剤、紫外線吸収剤、酸化防止剤、ブロッキング防止剤、耐熱安定剤、難燃剤など)を混合しても差し支えない。(A)または(B)中に添加剤を添加する方法としては、水系媒体中で水性樹脂分散体(X1)を形成させる際に混合してもよいが、あらかじめ樹脂(a)または樹脂(b)と添加剤を混合した後、水系媒体中にその混合物を加えて分散させたほうがより好ましい。
また、本発明においては、添加剤は、必ずしも、水系媒体中で粒子を形成させる時に混合しておく必要はなく、粒子を形成せしめた後、添加してもよい。たとえば、着色剤を含まない粒子を形成させた後、公知の染着の方法で着色剤を添加したり、有機溶剤(u)および/または可塑剤(v)とともに上記添加剤を含浸させることもできる。
添加剤として、前記有機酸塩(m)からなる荷電制御剤を樹脂粒子(A)中に含有させると、帯電特性が向上し好ましい。
(m)としては前述のものが挙げられ、使用量も同様である。
【0138】
また、本発明において、添加剤として、樹脂粒子(B)中に、樹脂(b)と共に、ワックス(c)、およびビニル系ポリマー鎖がグラフトした変性ワックス(d)を含有すると、耐熱保存安定性がより向上し好ましい。
(B)中の(c)の含有量は、好ましくは20%以下、さらに好ましくは1〜15%である。(d)の含有量は、好ましくは10%以下、さらに好ましくは0.5〜8%である。(c)と(d)の合計含有量は、好ましくは25%以下、さらに好ましくは1〜20%である。
【0139】
ワックス(c)はあらかじめ変性ワックス(d)と有機溶剤不存在下の溶融混練処理および/または有機溶剤(u)存在下加熱溶解混合処理した後に樹脂(b)に分散される。
ワックス(c)としては、ポリオレフィンワックス、パラフィンワックス、カルボニル基含有ワックスおよびこれらの混合物等が挙げられるが、このうち、とくに好ましいのはパラフィンワックス(c1)である。(c1)としては、融点50〜90℃で炭素数20〜36の直鎖飽和炭化水素を主成分とする石油系ワックスが挙げられる。
また、離型性の観点から、(c)のMnは、好ましくは400〜5000、さらに好ましくは1000〜3000、とくに1500〜2000である。尚、上記および以下においてワックスのMnは、GPCを用いて測定される(溶媒:オルソジクロロベンゼン、基準物質:ポリスチレン)。
【0140】
ワックス(c)は、ビニル系ポリマー鎖がグラフトした変性ワックス(d)と無溶媒下溶融混練処理および/または前記の有機溶剤(u)存在下の加熱溶解混合処理した後に、樹脂(b)に分散されるのが好ましい。この方法により、ワックス分散処理時に変性ワックス(d)を共存させることにより、(d)のワックス基部分が効率よく(c)表面に吸着、あるいはワックスのマトリクス構造内に一部絡みあうことにより、ワックス(c)表面と樹脂(b)との親和性が良好になり、(c)をより均一に樹脂粒子(B)中に内包することができ、分散状態の制御が容易になる。
【0141】
変性ワックス(d)は、ワックスにビニル系ポリマー鎖がグラフトしたものである。(d)に用いられるワックスとしては上記ワックス(c)と同様のものが挙げられ、好ましいものも同様である。(d)のビニル系ポリマー鎖を構成するビニル系モノマーとしては、前記ビニル樹脂を構成するモノマー(1)〜(10)と同様のものが挙げられるが、この中でとくに好ましいのは(1)、(2)、および(6)である。ビニル系ポリマー鎖はビニル系モノマーの単独重合体でもよいし、共重合体でもよい。
【0142】
変性ワックス(d)におけるワックス成分の量(未反応ワックスを含む)は、0.5〜99.5%が好ましく、さらに好ましくは1〜80%、とくに好ましくは5〜50%、最も好ましくは10〜30%である。また(d)のTgは、樹脂粒子(C)の耐熱保存安定性の観点から、好ましくは40〜90℃、さらに好ましくは50〜80℃である。
(d)のMnは、好ましくは1500〜10000、とくに1800〜9000である。Mnが1500〜10000の範囲では、樹脂粒子(C)の機械強度が良好である。
【0143】
変性ワックス(d)は、例えばワックス(c)を有機溶剤(例えばトルエンまたはキシレン)に溶解または分散させ、100〜200℃に加熱した後、ビニル系モノマーをパーオキサイド系開始剤(ベンゾイルパーオキサイド、ジターシャリーブチルパーオキサイド、ターシャリブチルパーオキサイドベンゾエート等)とともに滴下して重合後、有機溶剤を留去することにより得られる。
変性ワックス(d)の合成におけるパーオキサイド系開始剤の量は、(d)の原料の合計重量に基づいて、好ましくは0.2〜10%、さらに好ましくは0.5〜5%である。
【0144】
パーオキサイド重合開始剤としては、油溶性パーオキサイド重合開始剤および水溶性パーオキサイド重合開始剤等が用いられる。
油溶性パーオキサイド重合開始剤としては、アセチルシクロヘキシルスルホニルパーオキサイド、イソブチリルパーオキサイド、ジイソプロピルパーオキシジカーボネート、ジ−2−エチルヘキシルパーオキシジカーボネート、2,4−ジクロロベンゾイルパーオキサイド、t−ブチルパーオキシビバレート、3,5,5−トリメチルヘキサノニルパーオキサイド、オクタノイルパーオキサイド、デカノイルパーオキサイド、ラウロイルパーオキサイド、ステアロイルパーオキサイド、プロピオニトリルパーオキサイド、サクシニックアシッドパーオキサイド、アセチルパーオキサイド、t−ブチルパーオキシ−2−エチルヘキサノエート、ベンゾイルパーオキサイド、パラクロロベンゾイルパーオキサイド、t−ブチルパーオキシイソブチレート、t−ブチルパーオキシマレイックアシッド、t−ブチルパーオキシラウレート、シクロヘキサノンパーオキサイド、t−ブチルパーオキシイソプロピルカーボネート、2,5−ジメチル−2,5−ジベンゾイルパーオキシヘキサン、t−ブチルパーオキシアセテート、t−ブチルパーオキシベンゾエート、ジイソブチルジパーオキシフタレート、メチルエチルケトンパーオキサイド、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジt−ブチルパーオキシヘキサン、t−ブチルクミルパーオキサイド、t−ブチルヒドロパーオキサイド、ジt−ブチルパーオキサイド、ジイソプロピルベンゼンヒドロパーオキサイド、パラメンタンヒドロパーオキサイド、ピナンヒドロパーオキサイド、2,5−ジメチルヘキサン−2,5−ジヒドロパーオキサイドおよびクメンパーオキサイド等が挙げられる。
【0145】
水溶性パーオキサイド重合開始剤としては、例えば、過酸化水素、過酢酸、過硫酸アンモニウム、過硫酸カリウムおよび過硫酸ナトリウム等が挙げられる。
【0146】
ワックス(c)と変性ワックス(d)を混合する方法としては、〔1〕それぞれの融点以上の温度で溶融混練する方法、〔2〕(c)と(d)を有機溶剤(u)中に溶解あるいは懸濁させた後、冷却晶析、溶剤晶析等により液中に析出、あるいはスプレードライ等により気体中に析出させる方法、〔3〕(c)と(d)を有機溶剤(u)中に溶解あるいは懸濁させた後、分散機により機械的に湿式粉砕させる方法、等が挙げられる。これらの中では、〔2〕の方法が好ましい。
ワックス(c)および変性ワックス(d)を(b)中に分散させる方法としては、(c)および(d)と、(b)とを、それぞれ有機溶剤溶液もしくは分散液とした後、それら同士を混合する方法等が挙げられる。
【実施例】
【0147】
以下実施例により本発明をさらに説明するが、本発明はこれに限定されるものではない。以下の記載において「部」は重量部を示す。
【0148】
製造例1(樹脂粒子(A)の水性分散液の製造)
攪拌棒および温度計をセットした反応容器に、アジピン酸と1,4−ブタンジオール(モル比1:1)から得られたポリエステル(数平均分子量1000)177部、1,2−プロピレングリコール(以下プロピレングリコールと記載)7部、ジメチロールプロピオン酸72部、3−(2,3−ジヒドロキシプロポキシ)−1−プロパンスルホン酸4部、およびアセトン500部を仕込む。この溶液にイソホロンジイソシアネート(IPDI)246部を仕込み55℃で11時間反応し、[ウレタンプレポリマー1]を得た。このプレポリマーにジーtーブチルサリチル酸Zn14部を加え、さらにトリエチルアミンを加え、ジメチロールプロピオン酸由来のカルボン酸を100当量%アミン中和し、均一溶液にする。この溶液を攪拌下、水1500部に加え、乳化する。さらに水320部、エチレンジアミン9部、n−ブチルアミン6部を加え、50℃、4時間伸長反応を行いウレタン系樹脂の水性分散液[微粒子分散液W1]を得た。[微粒子分散液W1]をELS−800で測定した体積平均粒径は0.09μmであった。[微粒子分散液W1]の一部を乾燥して樹脂分を単離し、該樹脂分のフローテスター測定によるガラス転移温度は62℃、軟化開始温度は105℃であり、流出温度は180℃であった。
【0149】
製造例2(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、スチレン80部、メタクリル酸85部、アクリル酸ブチル120部、過酸化ベンゾイル(25%含水品)62部の混合溶液を、120分間かけて滴下した。この重合溶液50部をさらに撹拌下のイオン水60部に滴下して、水性分散液[微粒子分散液W2]を得た。[微粒子分散液W2]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.09μmであった。[微粒子分散液W2]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは72℃、軟化開始温度は98℃であり、流出温度は175℃であった。
【0150】
製造例3(樹脂粒子(A)の水性分散液の製造)
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール377部、テレフタル酸ジメチルエステル350部、アジピン酸40部、および重縮合触媒としてテトラブトキシチタネート0.3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、フローテスター測定にて1000Pa・sが85℃になったになった時点で取り出した。取り出した樹脂を室温まで冷却後、粉砕し粒子化し[ポリエステルb0]を得た。[ポリエステルb0]のMwは3200であった。次にこの[ポリエステルb0]30部に対しアセトン30部を加え溶解させ、さらにトリエチルアミン2.5部を加えた。よく攪拌されているこの溶液に、水90部を加えることでポリエステル系樹脂の水性分散液[微粒子分散液W3]を得た。[微粒子分散液W3]をELS−800で測定した体積平均粒径は0.05μmであった。[微粒子分散液W3]の一部を乾燥して樹脂分を単離し、該樹脂分のフローテスター測定によるガラス転移温度は60℃、軟化開始温度は90℃であり、流出温度は155℃であった。
【0151】
製造例4(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、メタクリル酸メチル71部、酢酸ビニル143部、アクリル酸73部、過酸化ベンゾイル(25%含水品)25部の混合溶液を、120分間かけて滴下した。この重合溶液50部をさらに撹拌下のイオン交換水60部に滴下して、水性分散液[微粒子分散液W4]を得た。[微粒子分散液W4]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.05μmであった。[微粒子分散液W4]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは50℃、軟化開始温度は60℃であり、流出温度は120℃であった。
【0152】
製造例5(樹脂粒子(A)の水性分散液の製造)
撹拌棒および温度計をセットした反応容器に、イソプロパノール130部を仕込み、攪拌下、アクリル酸ブチル30部、スチレン25部、メタクリル酸45部、アルキルアリルスルホコハク酸のナトリウム塩(エレミノールJS−2、三洋化成工業製)8部、過酸化ベンゾイル(25%含水品)25部の混合溶液を、120分間かけて滴下した。この重合溶液29部をさらに撹拌下のイオン交換水60部に滴下して、水性分散液[微粒子分散液W5]を得た。[微粒子分散液W5]をLA−920およびELS−800で測定した体積平均粒径は、いずれも0.05μmであった。[微粒子分散液W5]の一部を乾燥して樹脂分を単離した。該樹脂分のDSC測定によるTgは120℃、軟化開始温度は160℃であり、流出温度は250℃であった。
【0153】
製造例6[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル126部とテレフタル酸200部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド1617部を滴下しながら60℃で4時間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタントリイソプロポキシテレフタレートと未反応のテレフタル酸の混合物(e1)(チタントリイソプロポキシテレフタレートの濃度65%)を得た。(e1)の水への溶解度は[0.6g/100ml]、さらに精製して得たチタントリイソプロポキシテレフタレートの水への溶解度は[0.9g/100ml]であった。
【0154】
製造例7[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル520部とイソフタル酸340部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド284部を滴下しながら60℃で5時間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタンジイソプロポキシジイソフタレートと未反応のイソフタル酸の混合物(e2)(チタンジイソプロポキシジイソフタレートの濃度95%)を得た。
(e2)の水への溶解度は[0.3g/100ml]、さらに精製して得たチタンジイソプロポキシジイソフタレートの水への溶解度も[0.3g/100ml]であった。
【0155】
製造例8[チタン含有触媒(e)の合成]
冷却管、撹拌機及び液中バブリング可能な窒素導入管の付いた反応槽中に、酢酸エチル520部、安息香酸244部を入れ、窒素にて液中バブリング下、60℃まで徐々に昇温し、チタンテトライソプロポキシド284部を滴下しながら60℃で5時間反応させた後、イオン交換水を50部入れ、30分間反応させスラリー状物である反応混合物を得た。反応混合物をろ紙でろ別し40℃/20kPa・sで乾燥させることで、チタンジヒドロキシジベンゼンカルボキシレート(e3)を得た。(e3)の水への溶解度は[0.1g/100ml]であった。
【0156】
製造例9[線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物430部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部、無水マレイン酸10部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が5になった時点で取り出し、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−1)を得た。
(bX1−1)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は11、Tgは61℃、Mnは7040、Mwは17100、Mpは18900であった。分子量1500以下の成分の比率は1.0%であった。
【0157】
製造例10[非線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのEO2モル付加物350部、ビスフェノールAのPO3モル付加物326部、テレフタル酸278部、無水フタル酸40部及び重縮合触媒として(e1)2部を入れ、230℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が2以下になった時点で180℃に冷却し、無水トリメリット酸62部を加え、常圧密閉下2時間反応後取り出し、室温まで冷却後、粉砕して非線状ポリエステル樹脂(bX2−1)を得た。
(bX2−1)はTHF不溶分を含有しておらず、その酸価は34、水酸基価は18、Tgは68℃、Mnは3820、Mpは11100であった。分子量1500以下の成分の比率は0.8%であった。
【0158】
製造例11[線状ポリエステル樹脂の合成]
重縮合触媒を前記(e2)に代える以外は製造例9の(bX1−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−2)を得た。
(bX1−2)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は10、Tgは60℃、Mnは6690、Mwは17250、Mpは20080であった。分子量1500以下の成分の比率は0.8%であった。
【0159】
製造例12[非線状ポリエステル樹脂の合成]
重縮合触媒を前記(e2)に代える以外は製造例10の(bX2−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX2−2)を得た。
(bX2−2)はTHF不溶分を含有しておらず、その酸価は32、水酸基価は16、Tgは69℃、Mnは4220、Mpは11780であった。分子量1500以下の成分の比率は0.7%であった。
【0160】
製造例13[線状ポリエステル樹脂の合成]
重縮合触媒を(e3)に代える以外は製造例9の(bX1−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−3)を得た。
(bX1−3)はTHF不溶分を含有しておらず、その酸価は8、水酸基価は10、Tgは61℃、Mnは6810、Mwは17400、Mpは20170であった。分子量1500以下の成分の比率は1.1%であった。
【0161】
製造例14[非線状ポリエステル樹脂の合成]
重縮合触媒を(e3)に代える以外は製造例10の(bX2−1)と同様に反応させ、室温まで冷却後粉砕して線状ポリエステル樹脂(bX2−3)を得た。
(bX2−3)はTHF不溶分を含有しておらず、その酸価は31、水酸基価は14、Tgは66℃、Mnは4250、Mpは11920であった。分子量1500以下の成分の比率は0.7%であった。
【0162】
製造例15[ウレタン変性ポリエステル樹脂(ポリウレタン樹脂)の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物430部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部、無水マレイン酸10部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら6時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が12になった時点で80℃まで冷却し、酢酸エチル1000部を入れ、1時間攪拌した。その後イソホロンジイソシアネート60部を入れ2時間反応を行った後、室温まで冷却後粉砕してウレタン変性ポリエステル樹脂(bZ−1)の溶剤溶液(樹脂溶液i)を得た。
(bZ−1)はTHF不溶分を含有しておらず、その酸価は5、水酸基価は9、Tgは61℃、Mnは8790、Mpは13200であった。分子量1500以下の成分の比率は0.9%であった。
【0163】
製造例16[線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール701部、テレフタル酸ジメチルエステル716部、アジピン酸180部、および重縮合触媒として(e1)3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が150℃になった時点で取り出した。回収されたプロピレングリコールは316部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し線状ポリエステル樹脂(bX1−4)を得た。(bX1−4)はTHF不溶分を含有しておらず、その酸価は15、水酸基価は26、Tgは61℃、Mnは8800、Mwは27600、Mpは32200であった。分子量1500以下の成分の比率は4.0%であった。
【0164】
製造例17[非線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール557部、テレフタル酸ジメチルエステル569部、アジピン酸184部、および重縮合触媒として(e1)2部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に1時間反応させた。回収されたプロピレングリコールは175部であった。次いで180℃まで冷却し、無水トリメリット酸121部を加え、常圧密閉下2時間反応後、220℃、常圧で反応させ、軟化点が180℃になった時点で取り出し、室温まで冷却後、粉砕し粒子化し非線状ポリエステル樹脂(bX2−4)を得た。(bX2−4)はTHF不溶分を含有しておらず、その酸価は5、水酸基価は24、Tgは66℃、Mnは7200、Mpは41900であった。分子量1500以下の成分の比率は2.3%であった。
【0165】
製造例18[線状ポリエステル樹脂の合成]
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、ビスフェノールAのPO2モル付加物420部、ビスフェノールAのPO3モル付加物300部、テレフタル酸257部、イソフタル酸65部及び重縮合触媒として(e1)2部を入れ、220℃で窒素気流下に生成する水を留去しながら10時間反応させた。次いで5〜20mmHgの減圧下に反応させ、酸価が13になった時点で取り出し、室温まで冷却後粉砕して線状ポリエステル樹脂(bX1−5)を得た。
(bX1−5)はTHF不溶分を含有しておらず、その酸価は15、水酸基価は13、Tgは55℃、Mnは2830、Mwは7100、Mpは6810であった。分子量1500以下の成分の比率は1.0%であった。
【0166】
製造例19[線状ポリエステル樹脂の合成]
冷却管、撹拌機および窒素導入管の付いた反応槽中に、プロピレングリコール700部、テレフタル酸ジメチルエステル700部、アジピン酸90部、および重縮合触媒として(e1)3部を入れ、180℃で窒素気流下に、生成するメタノールを留去しながら8時間反応させた。次いで230℃まで徐々に昇温しながら、窒素気流下に、生成するプロピレングリコール、水を留去しながら4時間反応させ、さらに5〜20mmHgの減圧下に反応させ、軟化点が90℃になった時点で取り出した。回収されたプロピレングリコールは316部であった。取り出した樹脂を室温まで冷却後、粉砕し粒子化し線状ポリエステル樹脂(bX1−6)を得た。(bX1−6)はTHF不溶分を含有しておらず、その酸価は17、水酸基価は23、Tgは54℃、Mnは3300、Mwは9100、Mpは9400であった。分子量1500以下の成分の比率は3.8%であった。
【0167】
製造例20[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例9の(bX1−1)と同様に反応させた。触媒失活のために反応が途中で停止してしまい、生成水が留出しなくなる問題が生じたため、反応途中でチタンテトライソプロポキシド2部を4回追加し、比較用線状ポリエステル樹脂(CbX1−1)を得た。
(CbX1−1)は、THF不溶分を含有しておらず、その酸価は8、水酸基価は12、Tgは59℃、Mnは6180、Mwは13700、Mpは18600であった。分子量1500以下の成分の比率は2.4%であった。
【0168】
製造例21[比較用非線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例10の(bX2−1)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX2−1)を得た。
(CbX2−1)は、THF不溶分を含有しておらず、その酸価は35、水酸基価は14、Tgは67℃、Mnは3520、Mpは11800であった。分子量1500以下の成分の比率は2.3%であった。
【0169】
製造例22[比較用ウレタン変性ポリエステル樹脂(ポリウレタン樹脂)の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例15の(bZ−1)と同様に反応させ比較用ウレタン変性ポリエステル樹脂(CbZ−1)の溶剤溶液(樹脂溶液ii)を得た。
(CbZ−1)は、THF不溶分を含有しておらず、その酸価は4、水酸基価は11、Tgは61℃、Mnは7800、Mpは13000であった。分子量1500以下の成分の比率は1.2%であった。
【0170】
製造例23[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例16の(bX1−4)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX1−2)を得た。
(CbX1−2)は、THF不溶分を含有しておらず、その酸価は13、水酸基価は28、Tgは61℃、Mnは7830、Mwは27400、Mpは33900であった。分子量1500以下の成分の比率は4.6%であった。
【0171】
製造例24[比較用線状ポリエステル樹脂の合成]
重縮合触媒をチタンテトライソプロポキシドに代える以外は製造例17の(bX2−4)と同様に反応させた。常圧下で16時間、減圧下で8時間反応させた。反応速度が遅かったため、反応途中でチタンテトラプロポキシド2部を3回追加し、比較用非線状ポリエステル樹脂(CbX2−2)を得た。
(CbX2−2)は、THF不溶分を含有しておらず、その酸価は13、水酸基価は28、Tgは61℃、Mnは7800、Mpは33900であった。分子量1500以下の成分の比率は4.6%であった。
【0172】
製造例25(着色剤分散液の製造)
ビーカー内に銅フタロシアニン20部と着色剤分散剤(ソルスパーズ28000;アビシア株式会社製)4部、および酢酸エチル76部を入れ、攪拌して均一分散させた後、ビーズミルによって銅フタロシアニンを微分散して、[着色剤分散液1]を得た。[着色剤分散液1]をLA−920で測定した体積平均粒径は0.3μmであった。
【0173】
製造例26(変性ワックスの製造)
温度計および撹拌機の付いたオートクレーブ反応槽中に、キシレン454部、低分子量ポリエチレン(三洋化成工業(株)製 サンワックス LEL−400:軟化点128℃)150部を投入し、窒素置換後170℃に昇温して十分溶解し、スチレン595部、メタクリル酸メチル255部、ジ−t−ブチルパーオキシヘキサヒドロテレフタレート34部およびキシレン119部の混合溶液を170℃で3時間で滴下して重合し、さらにこの温度で30分間保持した。次いで脱溶剤を行い、[変性ワックス 1]を得た。[変性ワックス 1]のグラフト鎖のsp値は 10.35(cal/cm3)1/2、Mnは1872、Mwは5194、Tgは56.9℃であった。
【0174】
製造例27(ワックス分散液の製造)
温度計および撹拌機の付いた反応容器中に、パラフィンワックス(融点73℃)10部、[変性ワックス1]1部、酢酸エチル33部を投入し、78℃に加熱して充分溶解し、1時間で30℃まで冷却を行いワックスを微粒子状に晶析させ、さらにウルトラビスコミル(アイメックス製)で湿式粉砕し、[ワックス分散液1]を得た。
【0175】
製造例28(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−1)40部とポリエステル樹脂(bX2−1)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液1]を得た
【0176】
製造例29(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−2)40部とポリエステル樹脂(bX2−2)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液2]を得た
【0177】
製造例30(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−3)40部とポリエステル樹脂(bX2−3)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液3]を得た
【0178】
製造例31(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−1)40部とウレタン変性ポリエステル樹脂(bZ−1)の溶剤溶液[樹脂溶液i]120部および酢酸エチル40部を入れ、攪拌して均一分散させ、[樹脂溶液4]を得た
【0179】
製造例32(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−4)60部とポリエステル樹脂(bX2−4)40部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液5]を得た
【0180】
製造例33(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−5)60部とポリエステル樹脂(bX2−1)40部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液6]を得た。
【0181】
製造例34(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(bX1−6)80部とポリエステル樹脂(bX2−1)20部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液7]を得た。
【0182】
製造例35(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−1)40部と(CbX2−1)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液8]を得た
【0183】
製造例36(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−1)40部とウレタン変性ポリエステル樹脂(CbZ−1)の溶剤溶液[樹脂溶液ii]120部および酢酸エチル40部を入れ、攪拌して均一分散させ、[樹脂溶液9]を得た
【0184】
製造例37(樹脂溶液の製造)
温度計および撹拌機の付いた反応容器中に、ポリエステル樹脂(CbX1−2)40部とポリエステル樹脂(CbX2−2)60部および酢酸エチル100部を入れ、攪拌して均一分散させ、[樹脂溶液10]を得た
【0185】
実施例1
ビーカー内に[樹脂溶液1]60部、[ワックス分散液1]27部、および[着色剤分散液1]10部を入れ、25℃にてTK式ホモミキサーで8,000rpmで撹拌し、均一に溶解、分散させて[樹脂溶液1A]を得た。
ビーカー内にイオン交換水92部、[微粒子分散液W1]30.8部、カルボキシメチルセルロースナトリウム1部、およびドデシルジフェニルエーテルジスルホン酸ナトリウムの48.5%水溶液(三洋化成工業製、「エレミノールMON−7」)10部を入れ均一に溶解した。ついで25℃で、TK式ホモミキサーを10,000rpmに撹拌しながら、[樹脂溶液1A]75部を投入し2分間撹拌した。ついでこの混合液を撹拌棒および温度計付のコルベンに移し、昇温して35℃で濃度が0.5%以下となるまで酢酸エチルを留去し、(B)で構成されたコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成された樹脂粒子の水性樹脂分散体(XF1)を得た。次いで濾別し、40℃×18時間乾燥を行い、揮発分を0.5%以下として、樹脂粒子(F1)を得た。
【0186】
実施例2
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF2)および、樹脂粒子(F2)を得た。
【0187】
実施例3
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W3]27.2部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF3)および、樹脂粒子(F3)を得た。
【0188】
実施例4
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液2]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF4)および、樹脂粒子(F4)を得た。
【0189】
実施例5
実施例4の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、実施例4と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF5)および、樹脂粒子(F5)を得た。
【0190】
実施例6
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液3]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF6)および、樹脂粒子(F6)を得た。
【0191】
実施例7
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液4]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF7)および、樹脂粒子(F7)を得た。
【0192】
実施例8
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液5]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF8)および、樹脂粒子(F8)を得た。
【0193】
実施例9
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W4]15部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF9)および、樹脂粒子(F9)を得た。
【0194】
実施例10
実施例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W5]70部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF10)および、樹脂粒子(F10)を得た。
【0195】
実施例11
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液6]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF11)および、樹脂粒子(F11)を得た。
【0196】
実施例12
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液7]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF12)および、樹脂粒子(F12)を得た。
【0197】
比較例1
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液8]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF13)および、樹脂粒子(F13)を得た。
【0198】
比較例2
比較例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W2]20部を用いる他は、比較例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF14)および、樹脂粒子(F14)を得た。
【0199】
比較例3
比較例1の[微粒子分散液W1]30.8部の代りに、[微粒子分散液W3]27.2部を用いる他は、比較例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF15)および、樹脂粒子(F15)を得た。
【0200】
比較例4
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液9]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF16)および、樹脂粒子(F16)を得た。
【0201】
比較例5
実施例1の[樹脂溶液1]60部の代りに、[樹脂溶液10]60部を用いる他は、実施例1と同様にして、(Q)の表面に(P)が形成された樹脂粒子の水性樹脂分散体(XF17)および、樹脂粒子(F17)を得た。
【0202】
物性測定例
実施例1〜12および比較例1〜5で得た樹脂粒子(F1)〜(F17)を水に分散して粒度分布をコールターカウンターで測定した。また、樹脂粒子の平均円形度、帯電特性、耐熱保存安定性、および低温定着性を測定した。その結果を表1に示す。なお、○は請求項4の四角形ABCDの内部、×は外部を意味する。
【0203】
【表1】
【0204】
帯電特性、耐熱保存安定性、溶融性、および画像密着性の測定方法は以下の通りである。なお、これら以外の表1記載の物性の測定方法は、いずれも前記方法による。
【0205】
〔帯電特性〕(帯電量)
50ccの共栓付ガラス瓶に、樹脂粒子0.5g、鉄粉(日本鉄粉株式会社製「F−150」)10gを精秤し、共栓をして23℃、50%RHの雰囲気下でターブラシェーカミキサー(ウイリー・ア・バショッフェン社製)にセットし、回転数90rpmで2分攪拌する。攪拌後の混合粉体0.2gを目開き20μmステンレス金網がセットされたブローオフ粉体帯電量測定装置(京セラケミカル株式会社製TB−203)に装填し、ブロー圧10KPa,吸引圧5KPaの条件で、残存鉄粉の帯電量を測定し、定法により樹脂粒子の帯電量を算出する。なお、トナー用としてはマイナス帯電量が高いほど帯電特性が優れている。
【0206】
〔耐熱保存安定性1〕
50℃に温調された乾燥機に樹脂粒子を15時間静置し、ブロッキングの程度により下記の基準で評価した。
○ : ブロッキングが発生しない。
△ : ブロッキングが発生するが、力を加えると容易に分散する。
× : ブロッキングが発生し、力を加えても分散しない。
〔耐熱保存安定性2〕
50℃に温調された乾燥機に樹脂粒子2gを入れた100ml容量のプラスチックカップを72時間静置し、ブロッキングの程度により下記の基準で評価した。
◎ : 試験後のプラスチックカップを45度に傾けると樹脂粒子が流動する。
○ : 試験後のプラスチックカップを45度に傾けても樹脂粒子は流動しないが、4
5度に傾けた状態でカップ底に軽く叩くなどして弱い刺激を与えると樹脂粒子
が流動する。
△ : 以上◎、○の条件を満たさないが、指で軽く摘むなどして力を加えると容易に
分散する。
× : ブロッキングが発生し、力を加えても試験前の樹脂粒子の状態に戻らない。
【0207】
〔溶融性〕
樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、よく混ぜて均一にした後、この粉体を紙面上に0.6mg/cm2となるよう均一に載せる(このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通した時のコールドオフセットの発生温度を測定した。
【0208】
〔画像密着性〕
樹脂粒子にアエロジルR972(日本アエロジル社製)を1.0%添加し、よく混ぜて均一にした後、この粉体を紙面上に0.6mg/cm2となるよう均一に載せる(このとき粉体を紙面に載せる方法は、熱定着機を外したプリンターを用いる(上記の重量密度で粉体を均一に載せることができるのであれば他の方法を用いてもよい)。この紙を加圧ローラーに定着速度(加熱ローラ周速)213mm/sec、定着圧力(加圧ローラ圧)10kg/cm2の条件で通して作成した定着画像を、一定荷重(500g分銅)で折り曲げ、折り曲げ部での画像濃度を測定し(グレタグ・マクベス社製反射濃度計「RD−19」使用)、画像濃度(折り曲げ後)/画像濃度(折り曲げ前)が60%となる温度を画像密着温度とした。
【産業上の利用可能性】
【0209】
本発明の樹脂分散体および樹脂粒子は、粒径が均一で、帯電特性、耐熱保存安定性等に優れるため、スラッシュ成形用樹脂、粉体塗料、液晶等の電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真、静電記録、静電印刷などに用いられるトナー、各種ホットメルト接着剤、その他成形材料等に用いる樹脂粒子として極めて有用である。
【図面の簡単な説明】
【0210】
【図1】本発明における、(a)と(b)のsp値差(K)と、(a)の重量平均分子量(Mw)の自然対数値ln(Mw)(H)の関係を示すグラフである。
【図2】樹脂粒子のフローテスター測定におけるフローチャートを示す概念図である。
【特許請求の範囲】
【請求項1】
樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られる、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
Ti(−X)m(−OR)n (I)
[式中、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、他のカルボキシル基が同一分子内のOR基と分子内で重縮合し環構造を形成していてもよく、または、別の分子のOR基と分子間で重縮合し2〜5個のTi原子を含む構造を形成していてもよい。m=1〜3、n=1〜3であり、mとnの和は4である。]
【請求項2】
(a)が、40〜270℃の軟化開始温度、20〜250℃のガラス転移温度、60〜300℃の流出温度、および0〜130℃のガラス転移温度と流出温度の差を有する請求項1記載の水性樹脂分散体。
【請求項3】
(a)が、ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂およびビニル系樹脂から選ばれる少なくとも1つの樹脂である請求項1または2記載の水性樹脂分散体。
【請求項4】
(a)と(b)のsp値差(Δsp)をKとし、(a)の重量平均分子量の自然対数値〔ln(Mw)〕をHとした時、点(K、H)が以下の4点A、B、C、Dからなる四角形ABCDの辺上を含む内部に含まれる請求項1〜3いずれか記載の水性樹脂分散体。
A(0.3、ln3000)、B(1.5、ln1000)、
C(1.3、ln200000)、D(0.1、ln200000)
【請求項5】
(a)がスルホン酸アニオン基(−SO3-)を(a)の重量に基づいて0.001〜10重量%含有する請求項1〜4いずれか記載の水性樹脂分散体。
【請求項6】
(a)が下記有機酸金属塩(m)の構成単位を含有、および/または、(A)が下記有機酸金属塩(m)を含有する請求項1〜5いずれか記載の水性樹脂分散体。
有機酸金属塩(m):Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる金属のカルボン酸塩、スルホン酸塩、およびリン酸塩から選ばれる1種以上の塩。
【請求項7】
請求項1〜6いずれか記載の水性樹脂分散体(X1)中において、樹脂粒子(B)に付着された樹脂粒子(A)が、有機溶剤に溶解される、および/または、溶融されることにより、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されて得られる樹脂粒子(C2)からなる水性樹脂分散体(X2)。
【請求項8】
(b)の50〜90重量%が、テトラヒドロフラン可溶分の重量平均分子量が2000〜12000の線状ポリエステルである請求項1〜7いずれか記載の水性樹脂分散体。
【請求項9】
請求項1〜8いずれか記載の水性樹脂分散体から水性媒体が除去されてなる樹脂粒子。
【請求項10】
スラッシュ成形用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真トナー、静電記録トナー、静電印刷トナーまたはホットメルト接着剤用である請求項9記載の樹脂粒子。
【請求項1】
樹脂(a)からなる樹脂粒子(A)の水性分散液(W)と、樹脂(b)もしくはその有機溶剤溶液とが混合され、(W)中に(b)もしくはその有機溶剤溶液が分散され、(A)の水性分散液中で(b)からなる樹脂粒子(B)が形成されることにより得られる、(B)の表面に(A)が付着されてなる樹脂粒子(C1)の水性分散体(X1)であって、(b)が下記一般式(I)で表される少なくとも1種のチタン含有触媒(e)の存在下に形成されてなるポリエステル樹脂(b1)および/または(b1)を構成単位として含む樹脂(b2)からなることを特徴とする水性樹脂分散体。
Ti(−X)m(−OR)n (I)
[式中、RはH、または1〜3個のエーテル結合および/もしくは1〜2個の水酸基を含んでいてもよい炭素数1〜24の炭化水素基である。Xは芳香族モノもしくはポリカルボン酸から1個のカルボキシル基のHを除いた残基であり、ポリカルボン酸の場合、他のカルボキシル基が同一分子内のOR基と分子内で重縮合し環構造を形成していてもよく、または、別の分子のOR基と分子間で重縮合し2〜5個のTi原子を含む構造を形成していてもよい。m=1〜3、n=1〜3であり、mとnの和は4である。]
【請求項2】
(a)が、40〜270℃の軟化開始温度、20〜250℃のガラス転移温度、60〜300℃の流出温度、および0〜130℃のガラス転移温度と流出温度の差を有する請求項1記載の水性樹脂分散体。
【請求項3】
(a)が、ポリエステル樹脂、ポリウレタン樹脂、エポキシ樹脂およびビニル系樹脂から選ばれる少なくとも1つの樹脂である請求項1または2記載の水性樹脂分散体。
【請求項4】
(a)と(b)のsp値差(Δsp)をKとし、(a)の重量平均分子量の自然対数値〔ln(Mw)〕をHとした時、点(K、H)が以下の4点A、B、C、Dからなる四角形ABCDの辺上を含む内部に含まれる請求項1〜3いずれか記載の水性樹脂分散体。
A(0.3、ln3000)、B(1.5、ln1000)、
C(1.3、ln200000)、D(0.1、ln200000)
【請求項5】
(a)がスルホン酸アニオン基(−SO3-)を(a)の重量に基づいて0.001〜10重量%含有する請求項1〜4いずれか記載の水性樹脂分散体。
【請求項6】
(a)が下記有機酸金属塩(m)の構成単位を含有、および/または、(A)が下記有機酸金属塩(m)を含有する請求項1〜5いずれか記載の水性樹脂分散体。
有機酸金属塩(m):Al、Ti、Cr、Mn、Fe、Zn、Ba、およびZrから選ばれる金属のカルボン酸塩、スルホン酸塩、およびリン酸塩から選ばれる1種以上の塩。
【請求項7】
請求項1〜6いずれか記載の水性樹脂分散体(X1)中において、樹脂粒子(B)に付着された樹脂粒子(A)が、有機溶剤に溶解される、および/または、溶融されることにより、(B)で構成されるコア層(Q)の表面に(A)が被膜化されたシェル層(P)が形成されて得られる樹脂粒子(C2)からなる水性樹脂分散体(X2)。
【請求項8】
(b)の50〜90重量%が、テトラヒドロフラン可溶分の重量平均分子量が2000〜12000の線状ポリエステルである請求項1〜7いずれか記載の水性樹脂分散体。
【請求項9】
請求項1〜8いずれか記載の水性樹脂分散体から水性媒体が除去されてなる樹脂粒子。
【請求項10】
スラッシュ成形用樹脂、粉体塗料、電子部品製造用スペーサー、電子測定機器の標準粒子、電子写真トナー、静電記録トナー、静電印刷トナーまたはホットメルト接着剤用である請求項9記載の樹脂粒子。
【図1】

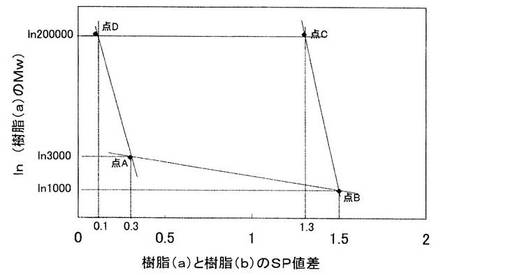
【図2】
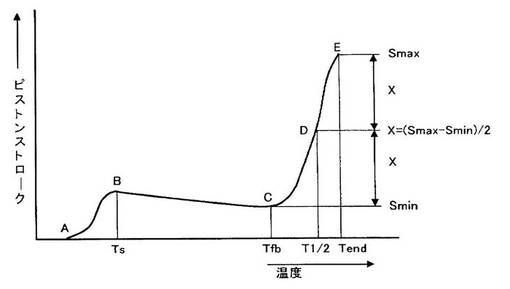
【図2】
【公開番号】特開2008−56915(P2008−56915A)
【公開日】平成20年3月13日(2008.3.13)
【国際特許分類】
【出願番号】特願2007−200131(P2007−200131)
【出願日】平成19年7月31日(2007.7.31)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
【公開日】平成20年3月13日(2008.3.13)
【国際特許分類】
【出願日】平成19年7月31日(2007.7.31)
【出願人】(000002288)三洋化成工業株式会社 (1,719)
【Fターム(参考)】
[ Back to top ]