
樹脂組成物、及び電子装置の製造方法

【課題】電極端子の接続信頼性が高い樹脂組成物、及びこの樹脂組成物を用いて製造する電子装置の製造方法を提供する。
【解決手段】第一基板110の半田バンプ111と第二基板120の半田バンプ121とを半田接合する際に、第一基板110の半田バンプ111と第二基板120の半田バンプ121との間に導入され、半田接合した後に熱硬化する樹脂組成物130であって、前記樹脂組成物は、熱硬化性樹脂を含み、180℃、及び250℃におけるゲルタイムが100秒以上である。
【解決手段】第一基板110の半田バンプ111と第二基板120の半田バンプ121とを半田接合する際に、第一基板110の半田バンプ111と第二基板120の半田バンプ121との間に導入され、半田接合した後に熱硬化する樹脂組成物130であって、前記樹脂組成物は、熱硬化性樹脂を含み、180℃、及び250℃におけるゲルタイムが100秒以上である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂組成物、及び電子装置の製造方法に関する。
【背景技術】
【0002】
近年、半導体パッケージや電子部品が軽薄短小化されている。このような半導体パッケージ製品の中でも、従来のリードフレーム接合に代わり、直接垂直接続できるフリップチップ型実装方式が重要視されている。
【0003】
そこで、半導体パッケージにおける電子情報のやり取りを行う配線の微細化により、端子も微細化されている。一方で、環境問題に対応するため、端子接合等に用いられる半田等は無鉛化が進んでいる。そのため、接合用の接着材などの樹脂材料中にボイドが発生しやすくなり、いかにボイドを少なくするかという課題が生じている。このボイドを減らす方法として、樹脂組成物材料や実装方式の改善が行われている(例えば特許文献1,2)。
【0004】
また、樹脂材料中のボイドを減らす方法として、加圧流体中での硬化方法なども提案されている(例えば特許文献3,4)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2003−158154号公報
【特許文献2】特表2006−505674号公報
【特許文献3】特開2002−57175号公報
【特許文献4】特開2008−98608号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、上記特許文献記載の技術では、樹脂材料を硬化する際に発生するボイドを十分に抑制するものではなかった。そのため、より確実な端子接合を実現する点で依然として改善の余地があった。
【0007】
本発明は、電極端子の接続信頼性が高い樹脂組成物、及びこの樹脂組成物を用いた電子装置の製造方法を提供するものである。
【課題を解決するための手段】
【0008】
本発明者らは、特定のゲルタイムを有する樹脂組成物を用いることによって、上記課題を解決できることを見出した。
【0009】
すなわち、本発明によれば、基板の電極端子と半導体チップの電極端子とを半田接合する際に、前記基板の電極端子と前記半導体チップの電極端子との間に導入され、半田接合した後に熱硬化する樹脂組成物であって、前記樹脂組成物は、熱硬化性樹脂を含み180℃、及び250℃におけるゲルタイムが100秒以上であることを特徴とする樹脂組成物が提供される。
【0010】
また、本発明によれば、基板の電極端子と半導体チップの電極端子とを、半田接合する電子装置の製造方法であって、前記基板の前記電極端子と、前記半導体チップの前記電極端子との間に、樹脂組成物を導入して、前記基板の前記電極端子と前記半導体チップの前記電極端子とを溶融接合しつつ、前記樹脂組成物を前記基板と前記半導体チップとの間に充填する工程と、前記樹脂組成物を熱硬化する工程と、を含み、前記樹脂組成物は、上記に特定される樹脂組成物であることを特徴とする電子装置の製造方法が提供される。
【0011】
本発明において、樹脂組成物は180℃、及び250℃におけるゲルタイムが100秒以上である。すなわち、250℃におけるゲルタイムが100秒以上であることにより、半田の融点よりも高い温度領域において、樹脂組成物の硬化速度が遅くなるため、半田接合後も樹脂組成物の流動性が得られ、最終的に得られる電子装置でのボイド形成が抑制され、接続信頼性が向上できる。一方、180℃におけるゲルタイムが100秒以上であることにより、半田の融点よりも低い温度領域において、樹脂組成物の硬化速度が遅くなるため、半田接合前にゲル化を始まりにくくし、流動性が保持されることによって、ボイドの形成を抑制できる。
【発明の効果】
【0012】
本発明によれば、電極端子の接続信頼性が高い樹脂組成物、及びこの樹脂組成物を用いて製造する電子装置の製造方法が提供される。
【図面の簡単な説明】
【0013】
【図1】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【図2】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【図3】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【発明を実施するための形態】
【0014】
以下、本実施形態について説明する。
【0015】
[電子装置]
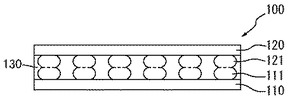
図3に示すように、電子装置100は、第一基板110の一面に形成されている半田バンプ111と第二基板120の一面に形成されている半田バンプ121とが個々に接合され、第一基板110と第二基板120との間に樹脂層130が充填されている。
【0016】
(樹脂組成物)
まず、樹脂層130に用いられる樹脂組成物について説明する。本発明における樹脂組成物は、第一基板110の一面に形成されている半田バンプ111と第二基板120の一面に形成されている半田バンプ121とを半田接合する際に、第一基板110の一面に形成されている半田バンプ111と、第二基板120の一面に形成されている半田バンプ121との間に導入され、半田接合した後に熱硬化する樹脂組成物である。すなわち、最終的に得られる電子装置100において、第一基板110と第二基板120との間のすき間を埋めるように樹脂層130が形成されればよい。なお、本実施形態において、第二基板120は半導体チップである。
【0017】
本発明における樹脂組成物は、180℃、及び250℃におけるゲルタイムが100秒以上である。すなわち、本発明の樹脂組成物のゲルタイムは、半田バンプ111,121の半田の融点より、低い領域及び高い領域の両領域において、100秒以上であることを意味する。
【0018】
ゲルタイムとは、樹脂組成物を所定の温度で加熱し始めてから、樹脂組成物が流動性を失い、粘性が急激に増加し、ゲル化するまでの時間である。またゲル化とは、樹脂組成物すべてが完全に流動性を失ってゲル化される場合に限られない。
【0019】
ここで、通常の半田接続までの工程を考えた場合、半田融点以上の温度でさらされた際、およそ10秒あれば接続には十分な時間であると考えられる。そこで、半田の融点より高い温度、例えば250℃でのゲルタイムが100秒以上であることにより、半田接合時に樹脂組成物の流動性が維持でき、樹脂組成物中にボイドが形成されにくくなる。また、例えば、半田の融点より10℃〜50℃高い温度におけるゲルタイムが100秒以上であってもよい。
【0020】
一方、例えば、リフローというコンベア式の表面実装法でも、半田を融点以上に加熱した220℃での時間を一般的には50秒から80秒としている。そこで、半田の融点より高い温度、例えば180℃でのゲルタイムが100秒以上であることにより、半田を融解させる前のゲル化を始まりにくくし、流動性が保持されるといえるため、ボイドの形成を抑制できる。また、例えば、半田の融点より10℃〜50℃低い温度におけるゲルタイムが100秒以上であってもよい。
【0021】
このようなゲルタイムは、樹脂組成物の反応性を制御することにより、適宜調整される。
【0022】
また、本発明における樹脂組成物の120℃での溶融粘度が、0.01Pa・s以上10,000Pa・s以下であることが好ましい。これにより、樹脂組成物の流動性が維持されるため、樹脂組成物中にボイドが形成されにくくなる。またさらに好ましくは、樹脂組成物の120℃での溶融粘度は、0.01以上5,000以下である。溶融粘度は、構成原料を低粘度化させたり、希釈剤などを使用したりすることにより適宜調整される。
【0023】
また、本発明における樹脂組成物の反応開始温度が100℃以上160℃以下であることが好ましい。反応開始温度とは、樹脂組成物の硬化が開始した温度であり、例えば熱分析装置(DSC)を用いて測定することができる。実装プロセス中におけるフロアライフを長くすることができる観点から、110℃以上がより好ましい。また、後硬化の際に低温処理ができる観点から、150℃以下がより好ましい。反応開始温度は、硬化促進剤の種類や量を調整したり、構成成分の反応性を制御したりすることにより適宜調整される。
【0024】
または樹脂組成物のピーク温度が130℃以上300℃以下であることが好ましい。実装プロセス中に反応が進行しないように抑制できる観点から、150℃以上がより好ましい。また、後硬化の際に効率的に反応終了させること、半導体装置の制限温度以下で効果を進行させることができる観点から、260℃以下がより好ましい。ピーク温度とは、反応が最も活発化する温度である。ピーク温度は、硬化促進剤の種類や量を調整したり、構成成分の反応性を制御したりすることにより調整することができる。
【0025】
本実施形態において、樹脂組成物の充填方法は、例えば、(i)本発明に係る樹脂組成物が常温で液状の場合には、そのまま第一基板110および半田バンプ111といった支持体又は被着体に塗布して樹脂層を形成する方法、又は本発明に係る樹脂組成物が常温で固体状の場合には、いったん溶剤に溶解又は分散させて樹脂ワニスにした後、この樹脂ワニスを支持体又は被着体に塗布して樹脂層を形成する方法、及び(ii)本発明に係る樹脂組成物をフィルム状に成形し、このフィルムを支持体又は被着体にラミネートする等の方法が挙げられる。
【0026】
なお、以下に説明する電子装置100の製造方法では、(ii)フィルム状に成型した樹脂層130を用いた場合について説明するが、上記(i)の方法を用いてもよい。
【0027】
(i)樹脂層を形成する場合
本発明に係る樹脂組成物に含有される熱硬化性樹脂としては、特に限定されず、例えば、エポキシ樹脂、オキセタン樹脂、フェノール樹脂、(メタ)アクリレート樹脂、不飽和ポリエステル樹脂、ジアリルフタレート樹脂、マレイミド樹脂等が挙げられ、これらの中でも、エポキシ樹脂が好ましい。エポキシ樹脂は、硬化性と保存性、硬化物の耐熱性、耐湿性、耐薬品性等に優れることから、好適に用いられる。
【0028】
本発明に係る樹脂組成物に含有されるエポキシ樹脂は、室温で固形のエポキシ樹脂と、室温で液状のエポキシ樹脂のうち、いずれでもよいし、これらの両方でもよい。本発明に係る樹脂組成物が、エポキシ樹脂を含有することにより、樹脂層の溶融挙動の設計の自由度をさらに高めることができる。
【0029】
本発明に係る樹脂組成物に含有されるエポキシ樹脂のうち、室温で固形のエポキシ樹脂としては、特に限定されないが、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、3官能エポキシ樹脂、4官能エポキシ樹脂等が挙げられる。さらに具体的には、固形3官能エポキシ樹脂とクレゾールノボラック型エポキシ樹脂との双方を含むものが挙げられ、これらは1種単独又は2種以上の組み合わせであってもよい。
【0030】
本発明に係る樹脂組成物に含有されるエポキシ樹脂のうち、室温で液状のエポキシ樹脂としては、特に限定されないが、ビスフェノールA型エポキシ樹脂およびビスフェノールF型エポキシ樹脂等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。室温で液状のエポキシ樹脂のエポキシ当量は、好ましくは150〜300であり、より好ましくは160〜250であり、更に好ましくは170〜220である。これにより、樹脂層の硬化物における収縮率が大きくなるのを防止して、電子装置に反りが生じるのを確実に防止することができるとともに、ポリイミド樹脂との反応性が低下するのが確実に防止される。
【0031】
本発明に係る樹脂組成物中、熱硬化性樹脂の配合量は、樹脂組成物の構成材料の25〜75重量%が好ましく、45〜70重量%が特に好ましい。樹脂組成物中の熱硬化性樹脂の配合量が上記範囲にあることにより、熱硬化性樹脂を硬化させる際に、良好な硬化性が得られると共に、樹脂層の良好な溶融挙動の設計が可能となる。
【0032】
本発明に係る樹脂組成物は、フラックス活性剤を含有してもよい。本実施形態において、フラックス活性剤とは、フラックス作用を有する化合物である。これにより、半田接合工程において、半田の表面を覆っている酸化被膜が除去されるので、良好な半田接合を行うことができる。フラックス作用を有する化合物としては、特に限定されないが、カルボキシル基又はフェノール性水酸基のいずれか、あるいは、カルボキシル基及びフェノール性水酸基の両方を備える化合物が好ましい。
【0033】
カルボキシル基を備えるフラックス作用を有する化合物とは、分子中にカルボキシル基が1つ以上存在するものをいい、液状であっても固体であってもよい。また、フェノール性水酸基を備えるフラックス作用を有する化合物とは、分子中にフェノール性水酸基が1つ以上存在するものをいい、液状であっても固体であってもよい。また、カルボキシル基及びフェノール性水酸基を備えるフラックス作用を有する化合物とは、分子中にカルボキシル基及びフェノール性水酸基がそれぞれ1つ以上存在するものをいい、液状であっても固体であってもよい。
【0034】
これらのうち、カルボキシル基を備えるフラックス作用を有する化合物としては、脂肪族酸無水物、脂環式酸無水物、芳香族酸無水物、脂肪族カルボン酸、芳香族カルボン酸等が挙げられる。
【0035】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂肪族酸無水物としては、無水コハク酸、ポリアジピン酸無水物、ポリアゼライン酸無水物、ポリセバシン酸無水物等が挙げられる。
【0036】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂環式酸無水物としては、メチルテトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、無水メチルハイミック酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、トリアルキルテトラヒドロ無水フタル酸、メチルシクロヘキセンジカルボン酸無水物等が挙げられる。
【0037】
カルボキシル基を備えるフラックス作用を有する化合物に係る芳香族酸無水物としては、無水フタル酸、無水トリメリット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸無水物、エチレングリコールビストリメリテート、グリセロールトリストリメリテート等が挙げられる。
【0038】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂肪族カルボン酸としては、下記一般式(1)で示される化合物や、蟻酸、酢酸、プロピオン酸、酪酸、吉草酸、ピバル酸カプロン酸、カプリル酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、アクリル酸、メタクリル酸、クロトン酸、オレイン酸、フマル酸、マレイン酸、シュウ酸、マロン酸、琥珀酸等が挙げられる。
【0039】
HOOC−(CH2)n−COOH (1)
(式(1)中、nは、0以上20以下の整数を表す。)
【0040】
カルボキシル基を備えるフラックス作用を有する化合物に係る芳香族カルボン酸としては、安息香酸、フタル酸、イソフタル酸、テレフタル酸、ヘミメリット酸、トリメリット酸、トリメシン酸、メロファン酸、プレーニト酸、ピロメリット酸、メリット酸、トリイル酸、キシリル酸、ヘメリト酸、メシチレン酸、プレーニチル酸、トルイル酸、ケイ皮酸、サリチル酸、2,3−ジヒドロキシ安息香酸、2,4−ジヒドロキシ安息香酸、ゲンチジン酸(2,5−ジヒドロキシ安息香酸)、2,6−ジヒドロキシ安息香酸、3,5−ジヒドロキシ安息香酸、浸食子酸(3,4,5−トリヒドロキシ安息香酸)、1,4−ジヒドロキシ−2−ナフトエ酸、3,5−ジヒドロキシ−2−ナフトエ酸等のナフトエ酸誘導体、フェノールフタリン、ジフェノール酸等が挙げられる。
【0041】
これらのカルボキシル基を備えるフラックス作用を有する化合物のうち、フラックス作用を有する化合物が有する活性度、樹脂層の硬化時におけるアウトガスの発生量、及び硬化後の樹脂層の弾性率やガラス転移温度等のバランスが良い点で、前記一般式(1)で示される化合物が好ましい。そして、前記一般式(1)で示される化合物のうち、式(1)中のnが3〜10である化合物が、硬化後の樹脂層における弾性率が増加するのを抑制することができるとともに、支持体と被着体の接着性を向上させることができる点で、特に好ましい。
【0042】
前記一般式(1)で示される化合物のうち、式(1)中のnが3〜10である化合物としては、例えば、n=3のグルタル酸(HOOC−(CH2)3−COOH)、n=4のアジピン酸(HOOC−(CH2)4−COOH)、n=5のピメリン酸(HOOC−(CH2)5−COOH)、n=8のセバシン酸(HOOC−(CH2)8−COOH)及びn=10のHOOC−(CH2)10−COOH等が挙げられる。
【0043】
フェノール性水酸基を備えるフラックス作用を有する化合物としては、フェノール類が挙げられ、具体的には、例えば、フェノール、o−クレゾール、2,6−キシレノール、p−クレゾール、m−クレゾール、o−エチルフェノール、2,4−キシレノール、2,5キシレノール、m−エチルフェノール、2,3−キシレノール、メジトール、3,5−キシレノール、p−ターシャリブチルフェノール、カテコール、p−ターシャリアミルフェノール、レゾルシノール、p−オクチルフェノール、p−フェニルフェノール、ビスフェノールA、ビスフェノールF、ビスフェノールAF、ビフェノール、ジアリルビスフェノールF、ジアリルビスフェノールA、トリスフェノール、テトラキスフェノール等のフェノール性水酸基を含有するモノマー類、フェノールノボラック樹脂、o−クレゾールノボラック樹脂、ビスフェノールFノボラック樹脂、ビスフェノールAノボラック樹脂等が挙げられる。
【0044】
上述したようなカルボキシル基又はフェノール性水酸基のいずれか、あるいは、カルボキシル基及びフェノール性水酸基の両方を備える化合物は、エポキシ樹脂のような熱硬化性樹脂との反応で三次元的に取り込まれる。
【0045】
そのため、硬化後のエポキシ樹脂の三次元的なネットワークの形成を向上させるという観点からは、フラックス作用を有する化合物としては、フラックス作用を有し且つエポキシ樹脂の硬化剤として作用する化合物、すなわち、フラックス活性硬化剤が好ましい。フラックス活性硬化剤としては、例えば、1分子中に、エポキシ樹脂に付加することができる2つ以上のフェノール性水酸基と、フラックス作用(還元作用)を示す芳香族に直接結合した1つ以上のカルボキシル基とを備える化合物が挙げられる。このようなフラックス活性硬化剤としては、2,3−ジヒドロキシ安息香酸、2,4−ジヒドロキシ安息香酸、ゲンチジン酸(2,5−ジヒドロキシ安息香酸)、2,6−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、没食子酸(3,4,5−トリヒドロキシ安息香酸)等の安息香酸誘導体;1,4−ジヒドロキシ−2−ナフトエ酸、3,5−ジヒドロキシ−2−ナフトエ酸、3,7−ジヒドロキシ−2−ナフトエ酸等のナフトエ酸誘導体;フェノールフタリン;及びジフェノール酸等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。
【0046】
フラックス作用を有する化合物の含有量は、樹脂組成物に対して、1〜30重量%が好ましく、3〜20重量%が特に好ましい。樹脂層中のフラックス作用を有する化合物の配合量が、上記範囲であることにより、樹脂層のフラックス活性を向上させることができるとともに、樹脂層中に、熱硬化性樹脂と未反応のフラックス作用を有する化合物が残存するのが抑制できる。なお、未反応のフラックス作用を有する化合物が残存すると、マイグレーションが発生する場合がある。
【0047】
また、熱硬化性樹脂の硬化剤として作用する化合物の中には、フラックス作用も有する化合物がある。例えば、エポキシ樹脂の硬化剤として作用するフェノールノボラック樹脂、クレゾールノボラック樹脂、脂肪族ジカルボン酸、芳香族ジカルボン酸等は、フラックス作用も有している。
【0048】
また、本発明に係る樹脂組成物中には、フラックス作用を有する化合物以外の硬化剤が含まれているのが好ましい。これにより、熱硬化性樹脂の硬化性をより向上させることができる。
【0049】
本発明に係る樹脂組成物に含有される硬化剤としては、特に限定されず、例えば、フェノール類、アミン類、チオール類が挙げられる。本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合は、硬化剤は、好ましくはフェノール類である。本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合に、硬化剤がフェノール類であることにより、樹脂層において、エポキシ樹脂との良好な反応性を得ることができ、さらには、この樹脂層中に含まれるエポキシ樹脂の硬化時の低寸法変化および硬化後の適切な物性(例えば、耐熱性、耐湿性等)を得ることができる。
【0050】
本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合に、硬化剤として含有されるフェノール類としては、特に限定されないが、エポキシ樹脂と反応し得る官能基を2以上有するものが好ましい。これにより、樹脂層におけるエポキシ樹脂の硬化物の特性(例えば、耐熱性、耐湿性等)の向上を図ることができる。
【0051】
このようなエポキシ樹脂と反応し得る官能基を2以上有するフェノール類としては、具体的には、例えば、ビスフェノールA、テトラメチルビスフェノールA、ジアリルビスフェノールA、ビフェノール、ビスフェノールF、ジアリルビスフェノールF、トリスフェノール、テトラキスフェノール、フェノールノボラック類、クレゾールノボラック類等が挙げられる。中でも、フェノールノボラック類およびクレゾールノボラック類が好ましい。これにより、樹脂層の溶融粘度を好適なものとすることができ、エポキシ樹脂との反応性を向上させることができる。さらに、樹脂層におけるエポキシ樹脂の硬化物の特性(例えば、耐熱性、耐湿性等)をより優れたものとすることができる。
【0052】
本発明に係る樹脂組成物に含有される硬化剤として、フェノールノボラック類を用いる場合、本発明に係る樹脂組成物中、硬化剤の配合量は、樹脂組成物の構成材料の5〜30重量%が好ましく、10〜25重量%が特に好ましい。樹脂組成物中の硬化剤の配合量が上記範囲にあることにより、樹脂層において、熱硬化性樹脂を確実に硬化させることができると共に、樹脂層中において、熱硬化性樹脂と未反応の硬化剤が残存するのが防止され、この残存物が存在することによるマイグレーションの発生を好適に防止することができる。
【0053】
なお、本発明に係る樹脂組成物に含有される熱硬化性樹脂がエポキシ樹脂である場合、フェノールノボラック樹脂の配合量は、エポキシ樹脂に対する当量比で規定されてもよい。具体的には、エポキシ樹脂に対するフェノールノボラック類の当量比は、0.5〜1.2であるのが好ましく、0.6〜1.1であるのが特に好ましく、0.7〜0.98であるのが更に好ましい。エポキシ樹脂に対するフェノールノボラック樹脂の配合量が上記範囲にあることにより、樹脂層において、熱硬化性樹脂を確実に硬化させることができると共に、樹脂層中において、熱硬化性樹脂と未反応の硬化剤の残存が防止され、この残存物が存在することによるマイグレーションの発生を好適に防止することができる。
【0054】
本発明に係る樹脂組成物は、さらに、上述した硬化剤の他、硬化促進剤として、例えば、融点が150℃以上のイミダゾール化合物を含有することができる。これにより、樹脂層の硬化を確実に行うことができ、電子装置の信頼性を高めることができる。このような融点が150℃以上のイミダゾール化合物としては、2−フェニルヒドロキシイミダゾール、2−フェニル−4−メチルヒドロキシイミダゾール等が挙げられる。なお、イミダゾール化合物の融点の上限に特に制限はなく、例えば、樹脂層の硬化の際の加熱温度に応じて適宜設定される。
【0055】
本発明に係る樹脂組成物が、硬化促進剤として、このようなイミダゾール化合物を含有する場合、樹脂組成物中の硬化剤の配合量は、樹脂組成物の構成材料の0.005〜10重量%が好ましく、0.01〜5重量%が特に好ましい。樹脂組成物中の硬化剤の配合量が上記範囲にあることにより、熱硬化性樹脂の硬化促進剤としての機能をさらに効果的に発揮させて、樹脂層において、熱硬化性樹脂の硬化性を向上させることができると共に、樹脂層中において、半田が溶融する温度において樹脂層の溶融粘度が高くなり過ぎず、良好な半田接合体を得ることができる。
【0056】
なお、上述したような硬化促進剤は、1種単独又は2種類以上の組み合わせでもよい。
【0057】
また、本発明に係る樹脂組成物は、フラックス作用を有する化合物及び熱硬化性樹脂の他に、フィラー、カップリング剤や、フラックス作用を有する化合物の活性を高めるためのフラックス活性剤や、樹脂の相溶性、安定性、作業性等の各種特性を向上させるための各種添加剤を適宜含有してもよい。
【0058】
本発明に係る樹脂組成物が、カップリング剤を含有することにより、樹脂層の支持体および被着体への密着性をさらに高めることができる。
【0059】
カップリング剤としては、エポキシシランカップリング剤、芳香族含有アミノシランカップリング剤のようなシランカップリング剤等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。
【0060】
本発明に係る樹脂組成物中、シランカップリング剤の配合量は、樹脂組成物の構成材料の0.01〜5重量%が好ましい。
【0061】
本発明に係る樹脂組成物を樹脂ワニスにして用いる場合、溶剤としては、特に限定されないが、上述したような樹脂組成物の構成材料に対して、不活性なものが好ましく、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジイソブチルケトン(DIBK)、シクロヘキサノン、ジアセトンアルコール(DAA)等のケトン類;ベンゼン、キシレン、トルエン等の芳香族炭化水素類;メチルアルコール、エチルアルコール、イソプロピルアルコール、n−ブチルアルコール等のアルコール類;メチルセロソルブ、エチルセロソルブ、ブチルセロソルブ、メチルセロソルブアセテート、エチルセロソルブアセテート、ブチロセルソルブアセテート(BCSA)等のセロソルブ系;N−メチル−2−ピロリドン(NMP)、テトラヒドロフラン(THF)、ジメチルホルムアミド(DMF)、二塩基酸エステル(DBE)、3−エトキシプロピオン酸エチル(EEP)、ジメチルカーボネート(DMC)等が挙げられる。また、樹脂ワニス中、溶剤の含有量は、溶媒に混合した固形成分の含有量が10〜60重量%となることが好ましい。
【0062】
(ii)フィルムを形成する場合
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、上記フラックス作用を有する化合物及び熱硬化性樹脂の他に、更に、フィルム形成性樹脂を含有するのが好ましい。本発明に係る樹脂組成物が、フィルム形成性樹脂を含有することにより、確実にフィルムとすることができる。
【0063】
フィルム形成性樹脂としては、例えば、(メタ)アクリル系樹脂、フェノキシ樹脂、ポリエステル樹脂、ポリウレタン樹脂、ポリイミド樹脂、シロキサン変性ポリイミド樹脂、ポリブタジエン、ポリプロピレン、スチレン−ブタジエン−スチレン共重合体、スチレン−エチレン−ブチレン−スチレン共重合体、ポリアセタール樹脂、ポリビニルブチラール樹脂、ポリビニルアセタール樹脂、ブチルゴム、クロロプレンゴム、ポリアミド樹脂、アクリロニトリル−ブタジエン共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体、アクリロニトリル−ブタジエン−スチレン共重合体、ポリ酢酸ビニル、ナイロン等が挙げられ、その中でも、(メタ)アクリル系樹脂、フェノキシ樹脂が好ましい。(メタ)アクリル系樹脂またはフェノキシ樹脂を適用することにより、フィルム形成性と支持体および被着体に対する密着性を両立することができる。フィルム形成樹脂は、1種単独又は2種以上の組み合わせでもよい。
【0064】
なお、フィルム形成性樹脂において、(メタ)アクリル系樹脂とは、(メタ)アクリル酸及びその誘導体の重合体、あるいは(メタ)アクリル酸及びその誘導体と他の単量体との共重合体を意味する。ここで、(メタ)アクリル酸などと表記するときは、アクリル酸又はメタクリル酸を意味する。
【0065】
フィルム形成性樹脂として用いられるアクリル系樹脂としては、具体的には、ポリアクリル酸、ポリメタクリル酸、ポリアクリル酸メチル、ポリアクリル酸エチル、ポリアクリル酸ブチル、ポリアクリル酸−2−エチルヘキシル等のポリアクリル酸エステル;ポリメタクリル酸メチル、ポリメタクリル酸エチル、ポリメタクリル酸ブチル等のポリメタクリル酸エステル;ポリアクリロニトリル、ポリメタクリロニトリル、ポリアクリルアミド、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリロニトリル−ブタジエン共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体、アクリロニトリル−ブタジエン−スチレン共重合体、アクリロニトリル−スチレン共重合体、メタクリル酸メチル−スチレン共重合体、メタクリル酸メチル−アクリロニトリル共重合体、メタクリル酸メチル−α−メチルスチレン共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート−メタクリル酸共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート−アクリル酸共重合体、アクリル酸ブチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート共重合体、アクリル酸ブチル−アクリロニトリル−アクリル酸共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリル酸エチル−アクリロニトリル−N,Nジメチルアクリルアミド共重合体等が挙げられる。中でも、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリル酸エチル−アクリロニトリル−N,Nジメチルアクリルアミド共重合体が好ましい。
【0066】
なお、フィルム形成性樹脂として用いられるアクリル系樹脂として、ニトリル基、エポキシ基、水酸基、カルボキシル基等の官能基を有する単量体を共重合させてなる(メタ)アクリル系樹脂を用いることにより、フィルム状の樹脂層の支持体および被着体への密着性、および熱硬化性樹脂等との相溶性を向上させることができる。このような(メタ)アクリル系樹脂において、前記官能基を有する単量体の使用量は特に限定されないが、(メタ)アクリル系樹脂の全重量に対し、0.1〜50mol%程度であることが好ましく、0.5〜45mol%程度であるのがより好ましく、1〜40mol%程度であるのがさらに好ましい。かかる範囲内に設定することにより、支持体および被着体の密着性を優れたものとしつつ、フィルム状の樹脂層の粘着力が強くなりすぎるのを好適に防止して、作業性の向上を図ることができる。
【0067】
前記アクリル系樹脂の重量平均分子量は、例えば1000以上100万以下であり、3000以上90万以下が好ましい。前記アクリル系樹脂の重量平均分子量が上記範囲にあることにより、樹脂組成物の成膜性をさらに向上させることができるとともに硬化時の流動性を確保することが可能となる。
【0068】
また、フィルム形成性樹脂として、フェノキシ樹脂を用いる場合、その数平均分子量は5000〜15000のフェノキシ樹脂が好ましい。かかる数平均分子量のフェノキシ樹脂を用いることにより、フィルム状の樹脂層の流動性を抑制し、フィルム状の樹脂層の厚みを均一なものとすることができる。
【0069】
フェノキシ樹脂の骨格は、特に限定されるものではないが、例えば、ビスフェノールAタイプ、ビスフェノールFタイプ、ビフェニル骨格タイプ等が挙げられる。これらの中でも、飽和吸水率が1%以下であるフェノキシ樹脂であるのが好ましい。これにより、フィルム状の樹脂層に起因する発泡や剥離などの発生を抑制することができる。
【0070】
なお、飽和吸水率は、フェノキシ樹脂を25μm厚のフィルムに加工し、100℃雰囲気中で1時間乾燥(絶乾状態)し、さらに、そのフィルムを40℃90%RH雰囲気の恒温恒湿槽に放置し、重量変化を24時間おきに測定し、重量変化が飽和した時点の重量を用いて、下記式(2)により算出することができる。
【0071】
飽和吸水率(%)={(飽和した時点の重量−絶乾時点の重量)/絶乾時点の重量}×100 (2)
【0072】
また、フィルム形成性樹脂として、ポリイミド樹脂を用いる場合、ポリイミド樹脂としては、繰り返し単位中にイミド結合を持つものが挙げられる。このようなポリイミド樹脂としては、例えば、ジアミンと酸二無水物を反応させ、得られたポリアミド酸を加熱、脱水閉環することにより得られるものが挙げられる。ジアミンとしては、芳香族ジアミンである、3,3'−ジメチル−4,4'ジアミノジフェニル、4,6−ジメチル−m−フェニレンジアミン、2,5−ジメチル−p−フェニレンジアミン、シロキサンジアミンである、1,3−ビス(3−アミノプロピル)−1,1,3,3−テトラメチルジシロキサン等が挙げられる。これらは1種単独又は2種以上の組み合わせでもよい。また、酸二無水物としては、3,3,4,4'−ビフェニルテトラカルボン酸、ピロメリット酸二無水物、4,4'−オキシジフタル酸二無水物等が挙げられる。
【0073】
なお、このようなポリイミド樹脂は、後述する溶剤に可溶なものでも、不溶なものでもよいが、溶剤に可溶なものが好ましい。ポリイミド樹脂が溶剤に可溶であることにより、溶液材料に含まれる構成材料との相溶解性が向上することから、取り扱いに優れる。特に、シロキサン変性ポリイミド樹脂は、様々な溶媒に溶かすことができるため好適に用いられる。
【0074】
また、フィルム形成性樹脂は、市販品であってもよい。
【0075】
更に、本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、効果を損ねない範囲で、各種可塑剤、安定剤、帯電防止剤や顔料等の添加剤を含有することができる。
【0076】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物中、フィルム形成性樹脂の配合量は、樹脂組成物の構成材料の5〜45重量%が好ましい。本発明に係る樹脂組成物中のフィルム形成性樹脂の配合量が上記範囲にあることにより、フィルム状の樹脂層の成膜性低下を抑制しつつ、硬化後のフィルム状の樹脂層における弾性率の増加を抑制することができる。その結果、フィルム状の樹脂層と支持体および被着体の密着性をさらに向上させることができる。更に、フィルム状の樹脂層の溶融粘度の増加を抑制することができる。
【0077】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、熱硬化性樹脂を含有し、そのような熱硬化性樹脂としては、特に限定されるものではないが、エポキシ樹脂を含むことが好ましい。エポキシ樹脂は、エポキシ基を有するモノマー、オリゴマーおよびポリマーのいずれかをいう。エポキシ樹脂の具体例としては、例えば、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂等のノボラック型エポキシ樹脂;ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂等のビスフェノール型エポキシ樹脂;ハイドロキノン型エポキシ樹脂;ビフェニル型エポキシ樹脂;スチルベン型エポキシ樹脂;トリフェノールメタン型エポキシ樹脂;トリアジン核含有エポキシ樹脂;ジシクロペンタジエン変性フェノール型エポキシ樹脂;ナフトール型エポキシ樹脂、およびフェニレンおよび/またはビフェニレン骨格を有するフェノールアラルキル型エポキシ樹脂、フェニレンおよび/またはビフェニレン骨格を有するナフトールアラルキル型エポキシ樹脂等のアラルキル型エポキシ樹脂、その他3官能以上のエポキシ樹脂等が挙げられる。
【0078】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物に含有されるエポキシ樹脂の含有量は、特に限定されるものではないが、10〜90重量%が好ましく、20〜80重量%が特に好ましい。樹脂組成物に含有されるエポキシ樹脂の含有量が上記範囲にあることにより、フィルム状の樹脂層の硬化後の低い線膨張係数と靭性を両立することができる。
【0079】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物に含有されるエポキシ樹脂の軟化点は、フィルム形成性樹脂との相溶性を有するものであれば、特に限定されるものではないが、40〜100℃が好ましく、50〜90℃が特に好ましい。上記下限値以上とすることで、フィルム状の樹脂層のタック性を低減することができるため、フィルム状の樹脂層の作業性を向上することができる。また、上記上限値以下とすることで、フィルム状の樹脂層の溶融粘度の上昇を抑えることができる。
【0080】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物は、特に限定されるものではないが、硬化剤を含有することが好ましい。このような硬化剤は、エポキシ樹脂の硬化剤として作用するものであればよく、適宜選択されている。具体的には、ジエチレントリアミン、トリエチレンテトラミン、メタキシレリレンジアミン、などの脂肪族ポリアミン、ジアミノジフェニルメタン、m−フェニレンジアミン、ジアミノジフェニルスルフォン、などの芳香族ポリアミン、ジシアンジアミド、有機酸ジヒドラジドなどを含むポリアミン化合物等のアミン系硬化剤、ヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、などの脂肪族酸無水物、無水トリトメット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸、などの芳香族酸無水物等の酸無水物系硬化剤、フェノールノボラック樹脂、クレゾールノボラック樹脂、フェノールアラルキル(フェニレン、ビフェニレン骨格を含む)樹脂、ナフトールアラルキル(フェニレン、ビフェニレン骨格を含む)樹脂、トリフェノールメタン樹脂、ジシクロペンタジエン型フェノール樹脂、ビス(モノまたはジt−ブチルフェノール)プロパン、メチレンビス(2−プロペニル)フェノール、プロピレンビス(2−プロペニル)フェノール、ビス[(2−プロペニルオキシ)フェニル]メタン、ビス[(2−プロペニルオキシ)フェニル]プロパン、4,4'−(1−メチルエチリデン)ビス[2−(2−プロペニル)フェノール]、4,4'−(1−メチルエチリデン)ビス[2−(1−フェニルエチル)フェノール]、4,4'−(1−メチルエチリデン)ビス[2−メチル−6−ヒドロキシメチルフェノール]、4,4'−(1−メチルエチリデン)ビス[2−メチル−6−(2−プロペニル)フェノール]、4,4'−(1−メチルテトラデシリデン)ビスフェノールなどのフェノール系硬化剤等が挙げられる。
【0081】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物中、硬化剤の含有量は、エポキシ樹脂のエポキシ当量と硬化剤の当量比を計算して求められる。硬化剤がフェノール樹脂の場合、エポキシ樹脂のエポキシ当量と硬化剤の官能基の当量比は、0.5〜1.5が好ましく、0.7〜1.3が特に好ましい。上記範囲とすることで、フィルムの耐熱性と保存性を両立することができる。
【0082】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物は、硬化促進剤を含有してもよい。このような硬化促進剤は、エポキシ樹脂と硬化剤との硬化反応を促進させるものであればよく、適宜選択される。具体的には、イミダゾール類、1,8−ジアザビシクロ(5,4,0)ウンデセン等のアミン系触媒、トリフェニルホスフィンやテトラ置換ホスホニウムと多官能フェノール化合物との塩等のリン化合物が挙げられる。これらの中でも、フィルム状の樹脂層の速硬化性、保存性、半導体素子上のアルミパッド腐食性を両立するイミダゾール類、リン化合物が好ましい。
【0083】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明の樹脂組成物中、硬化促進剤の含有量は、0.001〜10重量%が好ましく、0.01〜5重量%が特に好ましい。上記範囲とすることで、フィルム状の樹脂層の速硬化性および保存性、硬化後の物性のバランスを保つことが可能となる。
【0084】
硬化促進剤としてのイミダゾール類としては、融点が150℃以上のイミダゾール化合物が好ましく、例えば2−フェニルヒドロキシイミダゾール、2−フェニル−4−メチルヒドロキシイミダゾール、2−フェニル−4−メチルイミダゾール等が挙げられる。
【0085】
硬化促進剤としてのリン化合物の中でも、フィルム状の樹脂層の速硬化性、半導体素子のアルミパッドへの腐食性、さらにはフィルム状の樹脂層の保存性により優れる、テトラ置換ホスホニウムと多官能フェノール化合物との塩が特に好ましい。
【0086】
テトラ置換ホスホニウムと多官能フェノール化合物との塩は、単なる混合物ではなく、塩構造、超分子構造等の構造を有する化合物である。テトラ置換ホスホニウムと多官能フェノール化合物との塩のテトラ置換ホスホニウムは、フィルムの硬化性と保存性のバランスから、アルキル基や芳香族化合物がリン原子に4つ配位している化合物が好ましい。
【0087】
テトラ置換ホスホニウムの置換基は、特に限定されるものではなく、互いに同一であっても異なっていてもよく、置換又は無置換のアリール基やアルキル基を置換基として有するテトラ置換ホスホニウムイオンが、熱や加水分解に対して安定であり好ましい。具体的なテトラ置換ホスホニウムとしては、テトラフェニルホスホニウム、テトラトリルホスホニウム、テトラエチルフェニルホスホニウム、テトラメトキシフェニルホスホニウム、テトラナフチルホスホニウム、テトラベンジルホスホニウム、エチルトリフェニルホスホニウム、n−ブチルトリフェニルホスホニウム、2−ヒドロキシエチルトリフェニルホスホニウム、トリメチルフェニルホスホニウム、メチルジエチルフェニルホスホニウム、メチルジアリルフェニルホスホニウム、テトラ−n−ブチルホスホニウム等が例示でき、これらの中でもテトラフェニルホスホニウムがフィルムの速硬化性と保存性のバランスから好ましい。
【0088】
テトラ置換ホスホニウムと多官能フェノール化合物との分子化合物の多官能フェノール化合物とは、フェノール性の水酸基を有するもので少なくともその1つの水酸基の水素が外れてフェノキシド型の化合物となっているものであり、具体的には、ヒドロキシベンゼン化合物、ビフェノール化合物、ビスフェノール化合物、ヒドロキシナフタレン化合物、フェノールノボラック樹脂、フェノールアラルキル樹脂等が挙げられる。
【0089】
多官能フェノール化合物としては、例えば、ビス(4−ヒドロキシ−3,5−ジメチルフェニル)メタン(通称テトラメチルビスフェノールF)、4,4'−スルホニルジフェノール及び、4,4'−イソプロピリデンジフェノール(通称ビスフェノールA)、ビス(4−ヒドロキシフェニル)メタン、ビス(2−ヒドロキシフェニル)メタン、(2−ヒドロキシフェニル)(4−ヒドロキシフェニル)メタン及びこれらのうちビス(4−ヒドロキシフェニル)メタン、ビス(2−ヒドロキシフェニル)メタン、(2−ヒドロキシフェニル)(4−ヒドロキシフェニル)メタンの3種の混合物(例えば、本州化学工業株式会社製、ビスフェノールF−D)等のビスフェノール類、1,2−ベンゼンジオール、1,3−ベンゼンジオール、1,4−ベンゼンジオール等のジヒドロキシベンゼン類、1,2,4−ベンゼントリオール等のトリヒドロキシベンゼン類、1,6−ジヒドロキシナフタレン等のジヒドロキシナフタレン類の各種異性体、2,2'−ビフェノール、4,4'−ビフェノール等のビフェノール類の各種異性体等の化合物が挙げられるが、速硬化性と保存性のバランスに優れる1,2−ジヒドロキシナフタレン、4,4'−スルホニルジフェノールが好ましい。
【0090】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、このようなフィルム状の樹脂層は、例えば、フラックス作用を有する化合物及び熱硬化性樹脂と、必要に応じて、フィルム形成性樹脂や、その他の成分とを溶剤中に溶解させてフィルム状の樹脂層形成用材料(液状材料)を調製し、その後、このフィルム状の樹脂層形成用材料を、ポリエステルシート等の剥離処理が施された基材上に塗布し、所定の温度で、溶剤を除去し、乾燥させることにより得られる。
【0091】
なお、フィルム状の樹脂層形成用材料の調製に用いられる溶剤としては、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、DIBK(ジイソブチルケトン)、シクロヘキサノン、DAA(ジアセトンアルコール)等のケトン類、ベンゼン、キシレン、トルエン等の芳香族炭化水素類、メチルアルコール、エチルアルコール、イソプロピルアルコール、n−ブチルアルコール等のアルコール類、メチルセロソルブ、エチルセロソルブ、ブチルセロソルブ、メチルセロソルブアセテート、エチルセロソルブアセテート、BCSA(ブチロセルソルブアセテート)等のセロソルブ系、NMP(N−メチル−2−ピロリドン)、THF(テトラヒドロフラン)、DMF(ジメチルホルムアミド)、DBE(二塩基酸エステル)、EEP(3−エトキシプロピオン酸エチル)、DMC(ジメチルカーボネート)等が挙げられる。
【0092】
また、フィルム状の樹脂層の厚さ(平均)は、特に限定されないが、3〜100μm程度であるのが好ましく、5〜50μm程度であるのがより好ましい。
【0093】
[電子装置の製造方法]
本実施の形態の電子装置100の製造方法を以下に順番に説明する。
【0094】
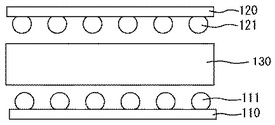
まず、図1に示すように、半田バンプ111を第一基板110の一面に形成し、半田バンプ121を第二基板120の一面に形成する。
【0095】
半田バンプ111,121との接合に用いられる半田としては、特に制限されず、錫、銀、鉛、亜鉛、ビスマス、インジウム及び銅からなる群から選択される少なくとも2種以上を含む合金等が挙げられる。半田バンプ111,121の融点は、100〜350℃である。
【0096】
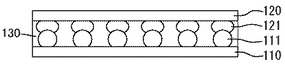
つぎに、第一基板110の半田バンプ111と第二基板120の半田バンプ121との間に樹脂層130を導入し、図2に示すようにして、第一基板110の半田バンプ111と、第二基板120の半田バンプ121とを溶融接合しつつ、樹脂層130を第一基板110と第二基板120との間に充填する。
【0097】
すなわち、本実施形態において、第一基板110の半田バンプ111と第二基板120の半田バンプ121とで樹脂層130を挟むようにして積層するとともに、半田バンプ121と半田バンプ111とを溶融接合することで、樹脂層130が溶融して広がり半田接合しつつ第一基板110と第二基板120との間を樹脂層130によって充填できる。
【0098】
半田接合は、半田バンプ111,121の融点以上の温度であればよく、例えば、150〜380℃である。
【0099】
つぎに、樹脂層130を硬化する。樹脂層130が熱硬化性樹脂を含むことにより、熱硬化させることができる(図3)。
【0100】
加熱温度は、樹脂層130の硬化温度以上の温度であればよく、適宜選択されるが、通常100〜250℃、好ましくは150〜200℃である。加熱時間は、樹脂層130の種類により、適宜選択されるが、通常、0.5〜3時間、好ましくは1〜2時間である。
【0101】
また、樹脂層130を硬化させるとき、加圧してもよい。加圧は、好ましくは0.1MPa以上10MPa以下が好ましく、0.5MPa以上5MPa以下がより好ましい。これにより、樹脂組成物の硬化物中に空隙(ボイド)が発生し難くなる。加圧は、流体を用いて行われることが好ましく、例えば、窒素ガス、アルゴンガス、空気等のガスが挙げられる。安価な点で、空気が特に好ましい。
【0102】
加圧流体により加圧しながら、樹脂層130を硬化させる方法としては、例えば、圧力容器内に、加熱する処理対象物を設置し、次いで、圧力容器内に、加圧流体を導入して加圧しつつ、処理対象物を加熱する方法、更に、具体的には、加圧オーブン中に、処理対象物を設置し、加圧オーブン内に加圧用のガスを導入しつつ、加圧オーブンで処理対象物を加熱する方法が挙げられる。
【0103】
ここで、硬化後の樹脂組成物の反応率(B)に対する、溶融接合直後の樹脂組成物の反応率(A)が、0.01≦A/B≦0.7 であることが好ましい。
【0104】
本実施形態において反応率とは、以下の手順により求められる。
(i)加熱処理を行っていない樹脂層(未硬化の樹脂層)について、示差走査熱量計(DSC)を用いて、測定温度範囲25〜300℃、昇温速度:10℃/分の測定条件で加熱し、その時の発熱量(「全て反応した時に得られる熱量」とする)を測定する。
(ii)溶融接合と同じ条件(加熱温度、加熱時間など)で加熱された樹脂層について、(i)と同様の条件で、その時の発熱量を測定し、「全て反応した時に得られる熱量」に対する比率を反応率(A)とする。
同様にして、硬化された樹脂層について、(i)と同様の条件で、発熱量を測定し、「全て反応した時に得られる熱量」に対する比率を反応率(B)とする。
【0105】
上記のようにして求められた反応率(B)に対する反応率(A)は、0.01以上0.7以下であることにより、溶融接続直後の樹脂組成物が固化前の状態であるため、再加熱及び加圧を行うことによってボイドを除去することができる。
【0106】
上述のような構成において、本実施の形態の電子装置100の製造方法によれば、半田の融点より10℃高い温度でのゲルタイムが100秒以上であるという特定の樹脂組成物を用いた樹脂層130により、樹脂組成物の硬化速度が低下し、ボイドが発生しにくくなる。また、半田接合後も樹脂組成物の流動性が得られる観点からも、最終的に得られる電子装置100でのボイド形成が抑制され、接続信頼性が向上できる。
【0107】
なお、本発明は本実施の形態に限定されるものではなく、その要旨を逸脱しない範囲で各種の変形を許容する。たとえば、上記形態では半田バンプ111,121の両方が球状に形成されていることを例示した。しかし、その一方ないし両方が多段状に形成されていてもよい(図示せず)。
【0108】
なお、上述した実施の形態および複数の変形例は、その内容が相反しない範囲で組み合わせることができる。また、上述した実施の形態および変形例では、各部の構造などを具体的に説明したが、その構造などは本願発明を満足する範囲で各種に変更することができる。
【実施例】
【0109】
次に、本発明の実施例について説明する。
得られた樹脂組成物またはその硬化物について以下のような評価を行った。評価結果をそれぞれ表に示した。
【0110】
[ゲルタイム(秒)]
180℃、及び250℃としたホットプレート上に樹脂組成物をおき、樹脂組成物がゲル化するまでの時間(秒)をそれぞれ測定した。なお、本実施例及び比較例で用いた半田バンプの融点は217℃であった。
【0111】
[120℃での溶融粘度(Pa・s)]
樹脂組成物を15℃/minの速度で昇温し、Haake製レオメーターを用いて、温度に対する粘度挙動を測定し、120℃での粘度値を読み取った。
条件:20mmΦ,6.3rad/s
【0112】
[半田接合工程直後の反応率と、硬化後の反応率の比]
樹脂組成物の未硬化物をDSC(10℃/min)で測定し、発熱量を得た。この発熱量を「全て反応した時に得られる熱量」とした。
半田接合工程における熱処理後の樹脂組成物と、硬化後の樹脂組成物それぞれについて、同様にDSCで測定して発熱量を得た。得られた発熱量からそれぞれについての反応率(%)を計算し、その比を求めた。
【0113】
[実装後のボイド性]
150℃で基板と接触させ、設定温度350℃まで一気に加熱して5秒間保持したサンプル樹脂組成物を超音波探傷(SAT:scanning acoustic tomograph)により観察し、ボイドの有無について以下のように判定した。
判定
○:ボイドなし
△:バンプにまたがるボイドが発生
×:ボイドが多数発生
【0114】
[加圧硬化後のボイド性]
150℃/2h、5atmで硬化した後の樹脂組成物をSATにより観察し、ボイドの有無について以下のように判定した。
判定
○:ボイドなし
△:バンプにまたがるボイドが発生
×:ボイドが多数発生
【0115】
(実施例1)
・樹脂組成物の作成
ビスフェノールF型エポキシ樹脂(DIC株式会社製、EXA−830LVP、エポキシ当量160)19.2重量%と、ビフェニル型エポキシ樹脂(日本化薬株式会社製、NC−3000、エポキシ当量272)57.6重量%と、ゲンチジン酸(東京化成工業株式会社製、2,5−ジヒドロキシ安息香酸、融点202℃)23.1重量%と、2−フェニル−4−メチルイミダゾール(四国化成工業株式会社製、2P4MZ)0.1重量%とを秤量し、3本ロールにて分散混練してから、真空下脱泡処理をしてペースト状の樹脂組成物を得た。
・半導体装置の作製
得られたペースト状の樹脂組成物を、回路パターンが形成された回路基板(コア材として、住友ベークライト株式会社製、ELC−4785GS)に、半田バンプを有する半導体素子(サイズ15×15×0.65mm)をフリップチップボンダーで280℃、10秒間加熱したものの、半導体チップと基板の間にできる空隙(0.05mm)の間に充填させた。その後に、150℃で120分間加熱して樹脂組成物を硬化させ、半導体装置を得た。
【0116】
(実施例2、3)
表1に示す組成の樹脂組成物を、実施例1と同様の方法で、それぞれ作成した。得られた樹脂組成物を用い、実施例1と同様の方法で、それぞれ半導体装置を得た。
【0117】
(実施例4)
・フィルム状樹脂層の作製
EPICLON 840−S(大日本インキ社株式会社製)、45重量%、YX6954(ジャパンエポキシレジン社製)24.9重量%、フェノールフタリン(東京化成工業株式会社製)15重量%、PR−55617(住友ベークライト株式会社製)15重量%、2P4MZ(四国化成工業株式会社製)0.1重量%をあらかじめ混合させ、基材上に塗布し、フィルム状樹脂層を得た。
・半導体装置の作製
得られたフィルム状樹脂層を、高さが60μm、ピッチが150μmの半田バンプを有する半導体部品(サイズ10×10×0.2mm)に、真空ラミネーター(名機株式会社製、MVLP)を用い、100℃、0.8MPa、30秒でラミネートし、樹脂層付きチップをフリップチップボンダー(Panasonic株式会社製、FCB3)を用いて回路パターンが形成された回路基板(コア材として、住友ベークライト株式会社製、ELC−4785GS)に、280℃で10秒間加熱して半田接続を行った。そして、180℃で60分間加熱して、フィルム状樹脂層を熱硬化し、半導体装置を得た。
【0118】
(実施例5〜7)および(比較例1〜5)
表1に示す組成の樹脂組成物を、実施例1と同様の方法で、それぞれ作成した。得られた樹脂組成物を用い、実施例1と同様の方法で、それぞれ半導体装置を得た。
【0119】
実施例および比較例で得られた樹脂組成物またはフィルム状樹脂層、および半導体装置についてそれぞれ評価を行った。その結果を表に示す。
【0120】
【表1】

【0121】
なお、表1中の記載は、それぞれ以下を示す。
・EXA−830LVP DIC株式会社製、エポキシ当量160
・NC−3000 日本化薬株式会社製、エポキシ当量272
・EPICLON 840S 大日本インキ株式会社製、エポキシ当量185
・RE−810NM 日本化薬株式会社製、エポキシ当量210
・PR55617 住友ベークライト株式会社製、水酸基当量104
・NH2200R 日立化成工業株式会社、分子量166
・2P4MZ 四国化成工業株式会社製、融点174〜184℃
・2MZ 四国化成工業株式会社製、融点140〜148℃
・C05−MB 住友ベークライト株式会社製、融点272℃
・ゲンチジン酸 東京化成工業株式会社製、2,5−ジヒドロキシ安息香酸、融点202℃
・フェノールフタリン 東京化成工業株式会社製、融点235℃
・セバシン酸 東京化成工業株式会社製、融点134℃
・SO25H アドマテックス株式会社製
・YX6954 ジャパンエポキシレジン株式会社製
【符号の説明】
【0122】
100 電子装置
110 第一基板
111 半田バンプ
120 第二基板
121 半田バンプ
130 樹脂層
【技術分野】
【0001】
本発明は、樹脂組成物、及び電子装置の製造方法に関する。
【背景技術】
【0002】
近年、半導体パッケージや電子部品が軽薄短小化されている。このような半導体パッケージ製品の中でも、従来のリードフレーム接合に代わり、直接垂直接続できるフリップチップ型実装方式が重要視されている。
【0003】
そこで、半導体パッケージにおける電子情報のやり取りを行う配線の微細化により、端子も微細化されている。一方で、環境問題に対応するため、端子接合等に用いられる半田等は無鉛化が進んでいる。そのため、接合用の接着材などの樹脂材料中にボイドが発生しやすくなり、いかにボイドを少なくするかという課題が生じている。このボイドを減らす方法として、樹脂組成物材料や実装方式の改善が行われている(例えば特許文献1,2)。
【0004】
また、樹脂材料中のボイドを減らす方法として、加圧流体中での硬化方法なども提案されている(例えば特許文献3,4)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2003−158154号公報
【特許文献2】特表2006−505674号公報
【特許文献3】特開2002−57175号公報
【特許文献4】特開2008−98608号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、上記特許文献記載の技術では、樹脂材料を硬化する際に発生するボイドを十分に抑制するものではなかった。そのため、より確実な端子接合を実現する点で依然として改善の余地があった。
【0007】
本発明は、電極端子の接続信頼性が高い樹脂組成物、及びこの樹脂組成物を用いた電子装置の製造方法を提供するものである。
【課題を解決するための手段】
【0008】
本発明者らは、特定のゲルタイムを有する樹脂組成物を用いることによって、上記課題を解決できることを見出した。
【0009】
すなわち、本発明によれば、基板の電極端子と半導体チップの電極端子とを半田接合する際に、前記基板の電極端子と前記半導体チップの電極端子との間に導入され、半田接合した後に熱硬化する樹脂組成物であって、前記樹脂組成物は、熱硬化性樹脂を含み180℃、及び250℃におけるゲルタイムが100秒以上であることを特徴とする樹脂組成物が提供される。
【0010】
また、本発明によれば、基板の電極端子と半導体チップの電極端子とを、半田接合する電子装置の製造方法であって、前記基板の前記電極端子と、前記半導体チップの前記電極端子との間に、樹脂組成物を導入して、前記基板の前記電極端子と前記半導体チップの前記電極端子とを溶融接合しつつ、前記樹脂組成物を前記基板と前記半導体チップとの間に充填する工程と、前記樹脂組成物を熱硬化する工程と、を含み、前記樹脂組成物は、上記に特定される樹脂組成物であることを特徴とする電子装置の製造方法が提供される。
【0011】
本発明において、樹脂組成物は180℃、及び250℃におけるゲルタイムが100秒以上である。すなわち、250℃におけるゲルタイムが100秒以上であることにより、半田の融点よりも高い温度領域において、樹脂組成物の硬化速度が遅くなるため、半田接合後も樹脂組成物の流動性が得られ、最終的に得られる電子装置でのボイド形成が抑制され、接続信頼性が向上できる。一方、180℃におけるゲルタイムが100秒以上であることにより、半田の融点よりも低い温度領域において、樹脂組成物の硬化速度が遅くなるため、半田接合前にゲル化を始まりにくくし、流動性が保持されることによって、ボイドの形成を抑制できる。
【発明の効果】
【0012】
本発明によれば、電極端子の接続信頼性が高い樹脂組成物、及びこの樹脂組成物を用いて製造する電子装置の製造方法が提供される。
【図面の簡単な説明】
【0013】
【図1】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【図2】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【図3】本発明の実施の形態の電子装置の製造方法の一工程を示す工程図である。
【発明を実施するための形態】
【0014】
以下、本実施形態について説明する。
【0015】
[電子装置]
図3に示すように、電子装置100は、第一基板110の一面に形成されている半田バンプ111と第二基板120の一面に形成されている半田バンプ121とが個々に接合され、第一基板110と第二基板120との間に樹脂層130が充填されている。
【0016】
(樹脂組成物)
まず、樹脂層130に用いられる樹脂組成物について説明する。本発明における樹脂組成物は、第一基板110の一面に形成されている半田バンプ111と第二基板120の一面に形成されている半田バンプ121とを半田接合する際に、第一基板110の一面に形成されている半田バンプ111と、第二基板120の一面に形成されている半田バンプ121との間に導入され、半田接合した後に熱硬化する樹脂組成物である。すなわち、最終的に得られる電子装置100において、第一基板110と第二基板120との間のすき間を埋めるように樹脂層130が形成されればよい。なお、本実施形態において、第二基板120は半導体チップである。
【0017】
本発明における樹脂組成物は、180℃、及び250℃におけるゲルタイムが100秒以上である。すなわち、本発明の樹脂組成物のゲルタイムは、半田バンプ111,121の半田の融点より、低い領域及び高い領域の両領域において、100秒以上であることを意味する。
【0018】
ゲルタイムとは、樹脂組成物を所定の温度で加熱し始めてから、樹脂組成物が流動性を失い、粘性が急激に増加し、ゲル化するまでの時間である。またゲル化とは、樹脂組成物すべてが完全に流動性を失ってゲル化される場合に限られない。
【0019】
ここで、通常の半田接続までの工程を考えた場合、半田融点以上の温度でさらされた際、およそ10秒あれば接続には十分な時間であると考えられる。そこで、半田の融点より高い温度、例えば250℃でのゲルタイムが100秒以上であることにより、半田接合時に樹脂組成物の流動性が維持でき、樹脂組成物中にボイドが形成されにくくなる。また、例えば、半田の融点より10℃〜50℃高い温度におけるゲルタイムが100秒以上であってもよい。
【0020】
一方、例えば、リフローというコンベア式の表面実装法でも、半田を融点以上に加熱した220℃での時間を一般的には50秒から80秒としている。そこで、半田の融点より高い温度、例えば180℃でのゲルタイムが100秒以上であることにより、半田を融解させる前のゲル化を始まりにくくし、流動性が保持されるといえるため、ボイドの形成を抑制できる。また、例えば、半田の融点より10℃〜50℃低い温度におけるゲルタイムが100秒以上であってもよい。
【0021】
このようなゲルタイムは、樹脂組成物の反応性を制御することにより、適宜調整される。
【0022】
また、本発明における樹脂組成物の120℃での溶融粘度が、0.01Pa・s以上10,000Pa・s以下であることが好ましい。これにより、樹脂組成物の流動性が維持されるため、樹脂組成物中にボイドが形成されにくくなる。またさらに好ましくは、樹脂組成物の120℃での溶融粘度は、0.01以上5,000以下である。溶融粘度は、構成原料を低粘度化させたり、希釈剤などを使用したりすることにより適宜調整される。
【0023】
また、本発明における樹脂組成物の反応開始温度が100℃以上160℃以下であることが好ましい。反応開始温度とは、樹脂組成物の硬化が開始した温度であり、例えば熱分析装置(DSC)を用いて測定することができる。実装プロセス中におけるフロアライフを長くすることができる観点から、110℃以上がより好ましい。また、後硬化の際に低温処理ができる観点から、150℃以下がより好ましい。反応開始温度は、硬化促進剤の種類や量を調整したり、構成成分の反応性を制御したりすることにより適宜調整される。
【0024】
または樹脂組成物のピーク温度が130℃以上300℃以下であることが好ましい。実装プロセス中に反応が進行しないように抑制できる観点から、150℃以上がより好ましい。また、後硬化の際に効率的に反応終了させること、半導体装置の制限温度以下で効果を進行させることができる観点から、260℃以下がより好ましい。ピーク温度とは、反応が最も活発化する温度である。ピーク温度は、硬化促進剤の種類や量を調整したり、構成成分の反応性を制御したりすることにより調整することができる。
【0025】
本実施形態において、樹脂組成物の充填方法は、例えば、(i)本発明に係る樹脂組成物が常温で液状の場合には、そのまま第一基板110および半田バンプ111といった支持体又は被着体に塗布して樹脂層を形成する方法、又は本発明に係る樹脂組成物が常温で固体状の場合には、いったん溶剤に溶解又は分散させて樹脂ワニスにした後、この樹脂ワニスを支持体又は被着体に塗布して樹脂層を形成する方法、及び(ii)本発明に係る樹脂組成物をフィルム状に成形し、このフィルムを支持体又は被着体にラミネートする等の方法が挙げられる。
【0026】
なお、以下に説明する電子装置100の製造方法では、(ii)フィルム状に成型した樹脂層130を用いた場合について説明するが、上記(i)の方法を用いてもよい。
【0027】
(i)樹脂層を形成する場合
本発明に係る樹脂組成物に含有される熱硬化性樹脂としては、特に限定されず、例えば、エポキシ樹脂、オキセタン樹脂、フェノール樹脂、(メタ)アクリレート樹脂、不飽和ポリエステル樹脂、ジアリルフタレート樹脂、マレイミド樹脂等が挙げられ、これらの中でも、エポキシ樹脂が好ましい。エポキシ樹脂は、硬化性と保存性、硬化物の耐熱性、耐湿性、耐薬品性等に優れることから、好適に用いられる。
【0028】
本発明に係る樹脂組成物に含有されるエポキシ樹脂は、室温で固形のエポキシ樹脂と、室温で液状のエポキシ樹脂のうち、いずれでもよいし、これらの両方でもよい。本発明に係る樹脂組成物が、エポキシ樹脂を含有することにより、樹脂層の溶融挙動の設計の自由度をさらに高めることができる。
【0029】
本発明に係る樹脂組成物に含有されるエポキシ樹脂のうち、室温で固形のエポキシ樹脂としては、特に限定されないが、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、3官能エポキシ樹脂、4官能エポキシ樹脂等が挙げられる。さらに具体的には、固形3官能エポキシ樹脂とクレゾールノボラック型エポキシ樹脂との双方を含むものが挙げられ、これらは1種単独又は2種以上の組み合わせであってもよい。
【0030】
本発明に係る樹脂組成物に含有されるエポキシ樹脂のうち、室温で液状のエポキシ樹脂としては、特に限定されないが、ビスフェノールA型エポキシ樹脂およびビスフェノールF型エポキシ樹脂等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。室温で液状のエポキシ樹脂のエポキシ当量は、好ましくは150〜300であり、より好ましくは160〜250であり、更に好ましくは170〜220である。これにより、樹脂層の硬化物における収縮率が大きくなるのを防止して、電子装置に反りが生じるのを確実に防止することができるとともに、ポリイミド樹脂との反応性が低下するのが確実に防止される。
【0031】
本発明に係る樹脂組成物中、熱硬化性樹脂の配合量は、樹脂組成物の構成材料の25〜75重量%が好ましく、45〜70重量%が特に好ましい。樹脂組成物中の熱硬化性樹脂の配合量が上記範囲にあることにより、熱硬化性樹脂を硬化させる際に、良好な硬化性が得られると共に、樹脂層の良好な溶融挙動の設計が可能となる。
【0032】
本発明に係る樹脂組成物は、フラックス活性剤を含有してもよい。本実施形態において、フラックス活性剤とは、フラックス作用を有する化合物である。これにより、半田接合工程において、半田の表面を覆っている酸化被膜が除去されるので、良好な半田接合を行うことができる。フラックス作用を有する化合物としては、特に限定されないが、カルボキシル基又はフェノール性水酸基のいずれか、あるいは、カルボキシル基及びフェノール性水酸基の両方を備える化合物が好ましい。
【0033】
カルボキシル基を備えるフラックス作用を有する化合物とは、分子中にカルボキシル基が1つ以上存在するものをいい、液状であっても固体であってもよい。また、フェノール性水酸基を備えるフラックス作用を有する化合物とは、分子中にフェノール性水酸基が1つ以上存在するものをいい、液状であっても固体であってもよい。また、カルボキシル基及びフェノール性水酸基を備えるフラックス作用を有する化合物とは、分子中にカルボキシル基及びフェノール性水酸基がそれぞれ1つ以上存在するものをいい、液状であっても固体であってもよい。
【0034】
これらのうち、カルボキシル基を備えるフラックス作用を有する化合物としては、脂肪族酸無水物、脂環式酸無水物、芳香族酸無水物、脂肪族カルボン酸、芳香族カルボン酸等が挙げられる。
【0035】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂肪族酸無水物としては、無水コハク酸、ポリアジピン酸無水物、ポリアゼライン酸無水物、ポリセバシン酸無水物等が挙げられる。
【0036】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂環式酸無水物としては、メチルテトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、無水メチルハイミック酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、トリアルキルテトラヒドロ無水フタル酸、メチルシクロヘキセンジカルボン酸無水物等が挙げられる。
【0037】
カルボキシル基を備えるフラックス作用を有する化合物に係る芳香族酸無水物としては、無水フタル酸、無水トリメリット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸無水物、エチレングリコールビストリメリテート、グリセロールトリストリメリテート等が挙げられる。
【0038】
カルボキシル基を備えるフラックス作用を有する化合物に係る脂肪族カルボン酸としては、下記一般式(1)で示される化合物や、蟻酸、酢酸、プロピオン酸、酪酸、吉草酸、ピバル酸カプロン酸、カプリル酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、アクリル酸、メタクリル酸、クロトン酸、オレイン酸、フマル酸、マレイン酸、シュウ酸、マロン酸、琥珀酸等が挙げられる。
【0039】
HOOC−(CH2)n−COOH (1)
(式(1)中、nは、0以上20以下の整数を表す。)
【0040】
カルボキシル基を備えるフラックス作用を有する化合物に係る芳香族カルボン酸としては、安息香酸、フタル酸、イソフタル酸、テレフタル酸、ヘミメリット酸、トリメリット酸、トリメシン酸、メロファン酸、プレーニト酸、ピロメリット酸、メリット酸、トリイル酸、キシリル酸、ヘメリト酸、メシチレン酸、プレーニチル酸、トルイル酸、ケイ皮酸、サリチル酸、2,3−ジヒドロキシ安息香酸、2,4−ジヒドロキシ安息香酸、ゲンチジン酸(2,5−ジヒドロキシ安息香酸)、2,6−ジヒドロキシ安息香酸、3,5−ジヒドロキシ安息香酸、浸食子酸(3,4,5−トリヒドロキシ安息香酸)、1,4−ジヒドロキシ−2−ナフトエ酸、3,5−ジヒドロキシ−2−ナフトエ酸等のナフトエ酸誘導体、フェノールフタリン、ジフェノール酸等が挙げられる。
【0041】
これらのカルボキシル基を備えるフラックス作用を有する化合物のうち、フラックス作用を有する化合物が有する活性度、樹脂層の硬化時におけるアウトガスの発生量、及び硬化後の樹脂層の弾性率やガラス転移温度等のバランスが良い点で、前記一般式(1)で示される化合物が好ましい。そして、前記一般式(1)で示される化合物のうち、式(1)中のnが3〜10である化合物が、硬化後の樹脂層における弾性率が増加するのを抑制することができるとともに、支持体と被着体の接着性を向上させることができる点で、特に好ましい。
【0042】
前記一般式(1)で示される化合物のうち、式(1)中のnが3〜10である化合物としては、例えば、n=3のグルタル酸(HOOC−(CH2)3−COOH)、n=4のアジピン酸(HOOC−(CH2)4−COOH)、n=5のピメリン酸(HOOC−(CH2)5−COOH)、n=8のセバシン酸(HOOC−(CH2)8−COOH)及びn=10のHOOC−(CH2)10−COOH等が挙げられる。
【0043】
フェノール性水酸基を備えるフラックス作用を有する化合物としては、フェノール類が挙げられ、具体的には、例えば、フェノール、o−クレゾール、2,6−キシレノール、p−クレゾール、m−クレゾール、o−エチルフェノール、2,4−キシレノール、2,5キシレノール、m−エチルフェノール、2,3−キシレノール、メジトール、3,5−キシレノール、p−ターシャリブチルフェノール、カテコール、p−ターシャリアミルフェノール、レゾルシノール、p−オクチルフェノール、p−フェニルフェノール、ビスフェノールA、ビスフェノールF、ビスフェノールAF、ビフェノール、ジアリルビスフェノールF、ジアリルビスフェノールA、トリスフェノール、テトラキスフェノール等のフェノール性水酸基を含有するモノマー類、フェノールノボラック樹脂、o−クレゾールノボラック樹脂、ビスフェノールFノボラック樹脂、ビスフェノールAノボラック樹脂等が挙げられる。
【0044】
上述したようなカルボキシル基又はフェノール性水酸基のいずれか、あるいは、カルボキシル基及びフェノール性水酸基の両方を備える化合物は、エポキシ樹脂のような熱硬化性樹脂との反応で三次元的に取り込まれる。
【0045】
そのため、硬化後のエポキシ樹脂の三次元的なネットワークの形成を向上させるという観点からは、フラックス作用を有する化合物としては、フラックス作用を有し且つエポキシ樹脂の硬化剤として作用する化合物、すなわち、フラックス活性硬化剤が好ましい。フラックス活性硬化剤としては、例えば、1分子中に、エポキシ樹脂に付加することができる2つ以上のフェノール性水酸基と、フラックス作用(還元作用)を示す芳香族に直接結合した1つ以上のカルボキシル基とを備える化合物が挙げられる。このようなフラックス活性硬化剤としては、2,3−ジヒドロキシ安息香酸、2,4−ジヒドロキシ安息香酸、ゲンチジン酸(2,5−ジヒドロキシ安息香酸)、2,6−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、没食子酸(3,4,5−トリヒドロキシ安息香酸)等の安息香酸誘導体;1,4−ジヒドロキシ−2−ナフトエ酸、3,5−ジヒドロキシ−2−ナフトエ酸、3,7−ジヒドロキシ−2−ナフトエ酸等のナフトエ酸誘導体;フェノールフタリン;及びジフェノール酸等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。
【0046】
フラックス作用を有する化合物の含有量は、樹脂組成物に対して、1〜30重量%が好ましく、3〜20重量%が特に好ましい。樹脂層中のフラックス作用を有する化合物の配合量が、上記範囲であることにより、樹脂層のフラックス活性を向上させることができるとともに、樹脂層中に、熱硬化性樹脂と未反応のフラックス作用を有する化合物が残存するのが抑制できる。なお、未反応のフラックス作用を有する化合物が残存すると、マイグレーションが発生する場合がある。
【0047】
また、熱硬化性樹脂の硬化剤として作用する化合物の中には、フラックス作用も有する化合物がある。例えば、エポキシ樹脂の硬化剤として作用するフェノールノボラック樹脂、クレゾールノボラック樹脂、脂肪族ジカルボン酸、芳香族ジカルボン酸等は、フラックス作用も有している。
【0048】
また、本発明に係る樹脂組成物中には、フラックス作用を有する化合物以外の硬化剤が含まれているのが好ましい。これにより、熱硬化性樹脂の硬化性をより向上させることができる。
【0049】
本発明に係る樹脂組成物に含有される硬化剤としては、特に限定されず、例えば、フェノール類、アミン類、チオール類が挙げられる。本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合は、硬化剤は、好ましくはフェノール類である。本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合に、硬化剤がフェノール類であることにより、樹脂層において、エポキシ樹脂との良好な反応性を得ることができ、さらには、この樹脂層中に含まれるエポキシ樹脂の硬化時の低寸法変化および硬化後の適切な物性(例えば、耐熱性、耐湿性等)を得ることができる。
【0050】
本発明に係る樹脂組成物が熱硬化性樹脂としてエポキシ樹脂を含有する場合に、硬化剤として含有されるフェノール類としては、特に限定されないが、エポキシ樹脂と反応し得る官能基を2以上有するものが好ましい。これにより、樹脂層におけるエポキシ樹脂の硬化物の特性(例えば、耐熱性、耐湿性等)の向上を図ることができる。
【0051】
このようなエポキシ樹脂と反応し得る官能基を2以上有するフェノール類としては、具体的には、例えば、ビスフェノールA、テトラメチルビスフェノールA、ジアリルビスフェノールA、ビフェノール、ビスフェノールF、ジアリルビスフェノールF、トリスフェノール、テトラキスフェノール、フェノールノボラック類、クレゾールノボラック類等が挙げられる。中でも、フェノールノボラック類およびクレゾールノボラック類が好ましい。これにより、樹脂層の溶融粘度を好適なものとすることができ、エポキシ樹脂との反応性を向上させることができる。さらに、樹脂層におけるエポキシ樹脂の硬化物の特性(例えば、耐熱性、耐湿性等)をより優れたものとすることができる。
【0052】
本発明に係る樹脂組成物に含有される硬化剤として、フェノールノボラック類を用いる場合、本発明に係る樹脂組成物中、硬化剤の配合量は、樹脂組成物の構成材料の5〜30重量%が好ましく、10〜25重量%が特に好ましい。樹脂組成物中の硬化剤の配合量が上記範囲にあることにより、樹脂層において、熱硬化性樹脂を確実に硬化させることができると共に、樹脂層中において、熱硬化性樹脂と未反応の硬化剤が残存するのが防止され、この残存物が存在することによるマイグレーションの発生を好適に防止することができる。
【0053】
なお、本発明に係る樹脂組成物に含有される熱硬化性樹脂がエポキシ樹脂である場合、フェノールノボラック樹脂の配合量は、エポキシ樹脂に対する当量比で規定されてもよい。具体的には、エポキシ樹脂に対するフェノールノボラック類の当量比は、0.5〜1.2であるのが好ましく、0.6〜1.1であるのが特に好ましく、0.7〜0.98であるのが更に好ましい。エポキシ樹脂に対するフェノールノボラック樹脂の配合量が上記範囲にあることにより、樹脂層において、熱硬化性樹脂を確実に硬化させることができると共に、樹脂層中において、熱硬化性樹脂と未反応の硬化剤の残存が防止され、この残存物が存在することによるマイグレーションの発生を好適に防止することができる。
【0054】
本発明に係る樹脂組成物は、さらに、上述した硬化剤の他、硬化促進剤として、例えば、融点が150℃以上のイミダゾール化合物を含有することができる。これにより、樹脂層の硬化を確実に行うことができ、電子装置の信頼性を高めることができる。このような融点が150℃以上のイミダゾール化合物としては、2−フェニルヒドロキシイミダゾール、2−フェニル−4−メチルヒドロキシイミダゾール等が挙げられる。なお、イミダゾール化合物の融点の上限に特に制限はなく、例えば、樹脂層の硬化の際の加熱温度に応じて適宜設定される。
【0055】
本発明に係る樹脂組成物が、硬化促進剤として、このようなイミダゾール化合物を含有する場合、樹脂組成物中の硬化剤の配合量は、樹脂組成物の構成材料の0.005〜10重量%が好ましく、0.01〜5重量%が特に好ましい。樹脂組成物中の硬化剤の配合量が上記範囲にあることにより、熱硬化性樹脂の硬化促進剤としての機能をさらに効果的に発揮させて、樹脂層において、熱硬化性樹脂の硬化性を向上させることができると共に、樹脂層中において、半田が溶融する温度において樹脂層の溶融粘度が高くなり過ぎず、良好な半田接合体を得ることができる。
【0056】
なお、上述したような硬化促進剤は、1種単独又は2種類以上の組み合わせでもよい。
【0057】
また、本発明に係る樹脂組成物は、フラックス作用を有する化合物及び熱硬化性樹脂の他に、フィラー、カップリング剤や、フラックス作用を有する化合物の活性を高めるためのフラックス活性剤や、樹脂の相溶性、安定性、作業性等の各種特性を向上させるための各種添加剤を適宜含有してもよい。
【0058】
本発明に係る樹脂組成物が、カップリング剤を含有することにより、樹脂層の支持体および被着体への密着性をさらに高めることができる。
【0059】
カップリング剤としては、エポキシシランカップリング剤、芳香族含有アミノシランカップリング剤のようなシランカップリング剤等が挙げられ、これらは1種単独又は2種以上の組み合わせでもよい。
【0060】
本発明に係る樹脂組成物中、シランカップリング剤の配合量は、樹脂組成物の構成材料の0.01〜5重量%が好ましい。
【0061】
本発明に係る樹脂組成物を樹脂ワニスにして用いる場合、溶剤としては、特に限定されないが、上述したような樹脂組成物の構成材料に対して、不活性なものが好ましく、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、ジイソブチルケトン(DIBK)、シクロヘキサノン、ジアセトンアルコール(DAA)等のケトン類;ベンゼン、キシレン、トルエン等の芳香族炭化水素類;メチルアルコール、エチルアルコール、イソプロピルアルコール、n−ブチルアルコール等のアルコール類;メチルセロソルブ、エチルセロソルブ、ブチルセロソルブ、メチルセロソルブアセテート、エチルセロソルブアセテート、ブチロセルソルブアセテート(BCSA)等のセロソルブ系;N−メチル−2−ピロリドン(NMP)、テトラヒドロフラン(THF)、ジメチルホルムアミド(DMF)、二塩基酸エステル(DBE)、3−エトキシプロピオン酸エチル(EEP)、ジメチルカーボネート(DMC)等が挙げられる。また、樹脂ワニス中、溶剤の含有量は、溶媒に混合した固形成分の含有量が10〜60重量%となることが好ましい。
【0062】
(ii)フィルムを形成する場合
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、上記フラックス作用を有する化合物及び熱硬化性樹脂の他に、更に、フィルム形成性樹脂を含有するのが好ましい。本発明に係る樹脂組成物が、フィルム形成性樹脂を含有することにより、確実にフィルムとすることができる。
【0063】
フィルム形成性樹脂としては、例えば、(メタ)アクリル系樹脂、フェノキシ樹脂、ポリエステル樹脂、ポリウレタン樹脂、ポリイミド樹脂、シロキサン変性ポリイミド樹脂、ポリブタジエン、ポリプロピレン、スチレン−ブタジエン−スチレン共重合体、スチレン−エチレン−ブチレン−スチレン共重合体、ポリアセタール樹脂、ポリビニルブチラール樹脂、ポリビニルアセタール樹脂、ブチルゴム、クロロプレンゴム、ポリアミド樹脂、アクリロニトリル−ブタジエン共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体、アクリロニトリル−ブタジエン−スチレン共重合体、ポリ酢酸ビニル、ナイロン等が挙げられ、その中でも、(メタ)アクリル系樹脂、フェノキシ樹脂が好ましい。(メタ)アクリル系樹脂またはフェノキシ樹脂を適用することにより、フィルム形成性と支持体および被着体に対する密着性を両立することができる。フィルム形成樹脂は、1種単独又は2種以上の組み合わせでもよい。
【0064】
なお、フィルム形成性樹脂において、(メタ)アクリル系樹脂とは、(メタ)アクリル酸及びその誘導体の重合体、あるいは(メタ)アクリル酸及びその誘導体と他の単量体との共重合体を意味する。ここで、(メタ)アクリル酸などと表記するときは、アクリル酸又はメタクリル酸を意味する。
【0065】
フィルム形成性樹脂として用いられるアクリル系樹脂としては、具体的には、ポリアクリル酸、ポリメタクリル酸、ポリアクリル酸メチル、ポリアクリル酸エチル、ポリアクリル酸ブチル、ポリアクリル酸−2−エチルヘキシル等のポリアクリル酸エステル;ポリメタクリル酸メチル、ポリメタクリル酸エチル、ポリメタクリル酸ブチル等のポリメタクリル酸エステル;ポリアクリロニトリル、ポリメタクリロニトリル、ポリアクリルアミド、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリロニトリル−ブタジエン共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体、アクリロニトリル−ブタジエン−スチレン共重合体、アクリロニトリル−スチレン共重合体、メタクリル酸メチル−スチレン共重合体、メタクリル酸メチル−アクリロニトリル共重合体、メタクリル酸メチル−α−メチルスチレン共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート−メタクリル酸共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート−アクリル酸共重合体、アクリル酸ブチル−アクリロニトリル−2−ヒドロキシエチルメタクリレート共重合体、アクリル酸ブチル−アクリロニトリル−アクリル酸共重合体、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリル酸エチル−アクリロニトリル−N,Nジメチルアクリルアミド共重合体等が挙げられる。中でも、アクリル酸ブチル−アクリル酸エチル−アクリロニトリル共重合体、アクリル酸エチル−アクリロニトリル−N,Nジメチルアクリルアミド共重合体が好ましい。
【0066】
なお、フィルム形成性樹脂として用いられるアクリル系樹脂として、ニトリル基、エポキシ基、水酸基、カルボキシル基等の官能基を有する単量体を共重合させてなる(メタ)アクリル系樹脂を用いることにより、フィルム状の樹脂層の支持体および被着体への密着性、および熱硬化性樹脂等との相溶性を向上させることができる。このような(メタ)アクリル系樹脂において、前記官能基を有する単量体の使用量は特に限定されないが、(メタ)アクリル系樹脂の全重量に対し、0.1〜50mol%程度であることが好ましく、0.5〜45mol%程度であるのがより好ましく、1〜40mol%程度であるのがさらに好ましい。かかる範囲内に設定することにより、支持体および被着体の密着性を優れたものとしつつ、フィルム状の樹脂層の粘着力が強くなりすぎるのを好適に防止して、作業性の向上を図ることができる。
【0067】
前記アクリル系樹脂の重量平均分子量は、例えば1000以上100万以下であり、3000以上90万以下が好ましい。前記アクリル系樹脂の重量平均分子量が上記範囲にあることにより、樹脂組成物の成膜性をさらに向上させることができるとともに硬化時の流動性を確保することが可能となる。
【0068】
また、フィルム形成性樹脂として、フェノキシ樹脂を用いる場合、その数平均分子量は5000〜15000のフェノキシ樹脂が好ましい。かかる数平均分子量のフェノキシ樹脂を用いることにより、フィルム状の樹脂層の流動性を抑制し、フィルム状の樹脂層の厚みを均一なものとすることができる。
【0069】
フェノキシ樹脂の骨格は、特に限定されるものではないが、例えば、ビスフェノールAタイプ、ビスフェノールFタイプ、ビフェニル骨格タイプ等が挙げられる。これらの中でも、飽和吸水率が1%以下であるフェノキシ樹脂であるのが好ましい。これにより、フィルム状の樹脂層に起因する発泡や剥離などの発生を抑制することができる。
【0070】
なお、飽和吸水率は、フェノキシ樹脂を25μm厚のフィルムに加工し、100℃雰囲気中で1時間乾燥(絶乾状態)し、さらに、そのフィルムを40℃90%RH雰囲気の恒温恒湿槽に放置し、重量変化を24時間おきに測定し、重量変化が飽和した時点の重量を用いて、下記式(2)により算出することができる。
【0071】
飽和吸水率(%)={(飽和した時点の重量−絶乾時点の重量)/絶乾時点の重量}×100 (2)
【0072】
また、フィルム形成性樹脂として、ポリイミド樹脂を用いる場合、ポリイミド樹脂としては、繰り返し単位中にイミド結合を持つものが挙げられる。このようなポリイミド樹脂としては、例えば、ジアミンと酸二無水物を反応させ、得られたポリアミド酸を加熱、脱水閉環することにより得られるものが挙げられる。ジアミンとしては、芳香族ジアミンである、3,3'−ジメチル−4,4'ジアミノジフェニル、4,6−ジメチル−m−フェニレンジアミン、2,5−ジメチル−p−フェニレンジアミン、シロキサンジアミンである、1,3−ビス(3−アミノプロピル)−1,1,3,3−テトラメチルジシロキサン等が挙げられる。これらは1種単独又は2種以上の組み合わせでもよい。また、酸二無水物としては、3,3,4,4'−ビフェニルテトラカルボン酸、ピロメリット酸二無水物、4,4'−オキシジフタル酸二無水物等が挙げられる。
【0073】
なお、このようなポリイミド樹脂は、後述する溶剤に可溶なものでも、不溶なものでもよいが、溶剤に可溶なものが好ましい。ポリイミド樹脂が溶剤に可溶であることにより、溶液材料に含まれる構成材料との相溶解性が向上することから、取り扱いに優れる。特に、シロキサン変性ポリイミド樹脂は、様々な溶媒に溶かすことができるため好適に用いられる。
【0074】
また、フィルム形成性樹脂は、市販品であってもよい。
【0075】
更に、本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、効果を損ねない範囲で、各種可塑剤、安定剤、帯電防止剤や顔料等の添加剤を含有することができる。
【0076】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物中、フィルム形成性樹脂の配合量は、樹脂組成物の構成材料の5〜45重量%が好ましい。本発明に係る樹脂組成物中のフィルム形成性樹脂の配合量が上記範囲にあることにより、フィルム状の樹脂層の成膜性低下を抑制しつつ、硬化後のフィルム状の樹脂層における弾性率の増加を抑制することができる。その結果、フィルム状の樹脂層と支持体および被着体の密着性をさらに向上させることができる。更に、フィルム状の樹脂層の溶融粘度の増加を抑制することができる。
【0077】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、本発明に係る樹脂組成物は、熱硬化性樹脂を含有し、そのような熱硬化性樹脂としては、特に限定されるものではないが、エポキシ樹脂を含むことが好ましい。エポキシ樹脂は、エポキシ基を有するモノマー、オリゴマーおよびポリマーのいずれかをいう。エポキシ樹脂の具体例としては、例えば、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂等のノボラック型エポキシ樹脂;ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂等のビスフェノール型エポキシ樹脂;ハイドロキノン型エポキシ樹脂;ビフェニル型エポキシ樹脂;スチルベン型エポキシ樹脂;トリフェノールメタン型エポキシ樹脂;トリアジン核含有エポキシ樹脂;ジシクロペンタジエン変性フェノール型エポキシ樹脂;ナフトール型エポキシ樹脂、およびフェニレンおよび/またはビフェニレン骨格を有するフェノールアラルキル型エポキシ樹脂、フェニレンおよび/またはビフェニレン骨格を有するナフトールアラルキル型エポキシ樹脂等のアラルキル型エポキシ樹脂、その他3官能以上のエポキシ樹脂等が挙げられる。
【0078】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物に含有されるエポキシ樹脂の含有量は、特に限定されるものではないが、10〜90重量%が好ましく、20〜80重量%が特に好ましい。樹脂組成物に含有されるエポキシ樹脂の含有量が上記範囲にあることにより、フィルム状の樹脂層の硬化後の低い線膨張係数と靭性を両立することができる。
【0079】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物に含有されるエポキシ樹脂の軟化点は、フィルム形成性樹脂との相溶性を有するものであれば、特に限定されるものではないが、40〜100℃が好ましく、50〜90℃が特に好ましい。上記下限値以上とすることで、フィルム状の樹脂層のタック性を低減することができるため、フィルム状の樹脂層の作業性を向上することができる。また、上記上限値以下とすることで、フィルム状の樹脂層の溶融粘度の上昇を抑えることができる。
【0080】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物は、特に限定されるものではないが、硬化剤を含有することが好ましい。このような硬化剤は、エポキシ樹脂の硬化剤として作用するものであればよく、適宜選択されている。具体的には、ジエチレントリアミン、トリエチレンテトラミン、メタキシレリレンジアミン、などの脂肪族ポリアミン、ジアミノジフェニルメタン、m−フェニレンジアミン、ジアミノジフェニルスルフォン、などの芳香族ポリアミン、ジシアンジアミド、有機酸ジヒドラジドなどを含むポリアミン化合物等のアミン系硬化剤、ヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、などの脂肪族酸無水物、無水トリトメット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸、などの芳香族酸無水物等の酸無水物系硬化剤、フェノールノボラック樹脂、クレゾールノボラック樹脂、フェノールアラルキル(フェニレン、ビフェニレン骨格を含む)樹脂、ナフトールアラルキル(フェニレン、ビフェニレン骨格を含む)樹脂、トリフェノールメタン樹脂、ジシクロペンタジエン型フェノール樹脂、ビス(モノまたはジt−ブチルフェノール)プロパン、メチレンビス(2−プロペニル)フェノール、プロピレンビス(2−プロペニル)フェノール、ビス[(2−プロペニルオキシ)フェニル]メタン、ビス[(2−プロペニルオキシ)フェニル]プロパン、4,4'−(1−メチルエチリデン)ビス[2−(2−プロペニル)フェノール]、4,4'−(1−メチルエチリデン)ビス[2−(1−フェニルエチル)フェノール]、4,4'−(1−メチルエチリデン)ビス[2−メチル−6−ヒドロキシメチルフェノール]、4,4'−(1−メチルエチリデン)ビス[2−メチル−6−(2−プロペニル)フェノール]、4,4'−(1−メチルテトラデシリデン)ビスフェノールなどのフェノール系硬化剤等が挙げられる。
【0081】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物中、硬化剤の含有量は、エポキシ樹脂のエポキシ当量と硬化剤の当量比を計算して求められる。硬化剤がフェノール樹脂の場合、エポキシ樹脂のエポキシ当量と硬化剤の官能基の当量比は、0.5〜1.5が好ましく、0.7〜1.3が特に好ましい。上記範囲とすることで、フィルムの耐熱性と保存性を両立することができる。
【0082】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明に係る樹脂組成物は、硬化促進剤を含有してもよい。このような硬化促進剤は、エポキシ樹脂と硬化剤との硬化反応を促進させるものであればよく、適宜選択される。具体的には、イミダゾール類、1,8−ジアザビシクロ(5,4,0)ウンデセン等のアミン系触媒、トリフェニルホスフィンやテトラ置換ホスホニウムと多官能フェノール化合物との塩等のリン化合物が挙げられる。これらの中でも、フィルム状の樹脂層の速硬化性、保存性、半導体素子上のアルミパッド腐食性を両立するイミダゾール類、リン化合物が好ましい。
【0083】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合で、本発明に係る樹脂組成物がエポキシ樹脂を含有する場合、本発明の樹脂組成物中、硬化促進剤の含有量は、0.001〜10重量%が好ましく、0.01〜5重量%が特に好ましい。上記範囲とすることで、フィルム状の樹脂層の速硬化性および保存性、硬化後の物性のバランスを保つことが可能となる。
【0084】
硬化促進剤としてのイミダゾール類としては、融点が150℃以上のイミダゾール化合物が好ましく、例えば2−フェニルヒドロキシイミダゾール、2−フェニル−4−メチルヒドロキシイミダゾール、2−フェニル−4−メチルイミダゾール等が挙げられる。
【0085】
硬化促進剤としてのリン化合物の中でも、フィルム状の樹脂層の速硬化性、半導体素子のアルミパッドへの腐食性、さらにはフィルム状の樹脂層の保存性により優れる、テトラ置換ホスホニウムと多官能フェノール化合物との塩が特に好ましい。
【0086】
テトラ置換ホスホニウムと多官能フェノール化合物との塩は、単なる混合物ではなく、塩構造、超分子構造等の構造を有する化合物である。テトラ置換ホスホニウムと多官能フェノール化合物との塩のテトラ置換ホスホニウムは、フィルムの硬化性と保存性のバランスから、アルキル基や芳香族化合物がリン原子に4つ配位している化合物が好ましい。
【0087】
テトラ置換ホスホニウムの置換基は、特に限定されるものではなく、互いに同一であっても異なっていてもよく、置換又は無置換のアリール基やアルキル基を置換基として有するテトラ置換ホスホニウムイオンが、熱や加水分解に対して安定であり好ましい。具体的なテトラ置換ホスホニウムとしては、テトラフェニルホスホニウム、テトラトリルホスホニウム、テトラエチルフェニルホスホニウム、テトラメトキシフェニルホスホニウム、テトラナフチルホスホニウム、テトラベンジルホスホニウム、エチルトリフェニルホスホニウム、n−ブチルトリフェニルホスホニウム、2−ヒドロキシエチルトリフェニルホスホニウム、トリメチルフェニルホスホニウム、メチルジエチルフェニルホスホニウム、メチルジアリルフェニルホスホニウム、テトラ−n−ブチルホスホニウム等が例示でき、これらの中でもテトラフェニルホスホニウムがフィルムの速硬化性と保存性のバランスから好ましい。
【0088】
テトラ置換ホスホニウムと多官能フェノール化合物との分子化合物の多官能フェノール化合物とは、フェノール性の水酸基を有するもので少なくともその1つの水酸基の水素が外れてフェノキシド型の化合物となっているものであり、具体的には、ヒドロキシベンゼン化合物、ビフェノール化合物、ビスフェノール化合物、ヒドロキシナフタレン化合物、フェノールノボラック樹脂、フェノールアラルキル樹脂等が挙げられる。
【0089】
多官能フェノール化合物としては、例えば、ビス(4−ヒドロキシ−3,5−ジメチルフェニル)メタン(通称テトラメチルビスフェノールF)、4,4'−スルホニルジフェノール及び、4,4'−イソプロピリデンジフェノール(通称ビスフェノールA)、ビス(4−ヒドロキシフェニル)メタン、ビス(2−ヒドロキシフェニル)メタン、(2−ヒドロキシフェニル)(4−ヒドロキシフェニル)メタン及びこれらのうちビス(4−ヒドロキシフェニル)メタン、ビス(2−ヒドロキシフェニル)メタン、(2−ヒドロキシフェニル)(4−ヒドロキシフェニル)メタンの3種の混合物(例えば、本州化学工業株式会社製、ビスフェノールF−D)等のビスフェノール類、1,2−ベンゼンジオール、1,3−ベンゼンジオール、1,4−ベンゼンジオール等のジヒドロキシベンゼン類、1,2,4−ベンゼントリオール等のトリヒドロキシベンゼン類、1,6−ジヒドロキシナフタレン等のジヒドロキシナフタレン類の各種異性体、2,2'−ビフェノール、4,4'−ビフェノール等のビフェノール類の各種異性体等の化合物が挙げられるが、速硬化性と保存性のバランスに優れる1,2−ジヒドロキシナフタレン、4,4'−スルホニルジフェノールが好ましい。
【0090】
本発明に係る樹脂組成物をフィルム状に成形して用いる場合、このようなフィルム状の樹脂層は、例えば、フラックス作用を有する化合物及び熱硬化性樹脂と、必要に応じて、フィルム形成性樹脂や、その他の成分とを溶剤中に溶解させてフィルム状の樹脂層形成用材料(液状材料)を調製し、その後、このフィルム状の樹脂層形成用材料を、ポリエステルシート等の剥離処理が施された基材上に塗布し、所定の温度で、溶剤を除去し、乾燥させることにより得られる。
【0091】
なお、フィルム状の樹脂層形成用材料の調製に用いられる溶剤としては、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、DIBK(ジイソブチルケトン)、シクロヘキサノン、DAA(ジアセトンアルコール)等のケトン類、ベンゼン、キシレン、トルエン等の芳香族炭化水素類、メチルアルコール、エチルアルコール、イソプロピルアルコール、n−ブチルアルコール等のアルコール類、メチルセロソルブ、エチルセロソルブ、ブチルセロソルブ、メチルセロソルブアセテート、エチルセロソルブアセテート、BCSA(ブチロセルソルブアセテート)等のセロソルブ系、NMP(N−メチル−2−ピロリドン)、THF(テトラヒドロフラン)、DMF(ジメチルホルムアミド)、DBE(二塩基酸エステル)、EEP(3−エトキシプロピオン酸エチル)、DMC(ジメチルカーボネート)等が挙げられる。
【0092】
また、フィルム状の樹脂層の厚さ(平均)は、特に限定されないが、3〜100μm程度であるのが好ましく、5〜50μm程度であるのがより好ましい。
【0093】
[電子装置の製造方法]
本実施の形態の電子装置100の製造方法を以下に順番に説明する。
【0094】
まず、図1に示すように、半田バンプ111を第一基板110の一面に形成し、半田バンプ121を第二基板120の一面に形成する。
【0095】
半田バンプ111,121との接合に用いられる半田としては、特に制限されず、錫、銀、鉛、亜鉛、ビスマス、インジウム及び銅からなる群から選択される少なくとも2種以上を含む合金等が挙げられる。半田バンプ111,121の融点は、100〜350℃である。
【0096】
つぎに、第一基板110の半田バンプ111と第二基板120の半田バンプ121との間に樹脂層130を導入し、図2に示すようにして、第一基板110の半田バンプ111と、第二基板120の半田バンプ121とを溶融接合しつつ、樹脂層130を第一基板110と第二基板120との間に充填する。
【0097】
すなわち、本実施形態において、第一基板110の半田バンプ111と第二基板120の半田バンプ121とで樹脂層130を挟むようにして積層するとともに、半田バンプ121と半田バンプ111とを溶融接合することで、樹脂層130が溶融して広がり半田接合しつつ第一基板110と第二基板120との間を樹脂層130によって充填できる。
【0098】
半田接合は、半田バンプ111,121の融点以上の温度であればよく、例えば、150〜380℃である。
【0099】
つぎに、樹脂層130を硬化する。樹脂層130が熱硬化性樹脂を含むことにより、熱硬化させることができる(図3)。
【0100】
加熱温度は、樹脂層130の硬化温度以上の温度であればよく、適宜選択されるが、通常100〜250℃、好ましくは150〜200℃である。加熱時間は、樹脂層130の種類により、適宜選択されるが、通常、0.5〜3時間、好ましくは1〜2時間である。
【0101】
また、樹脂層130を硬化させるとき、加圧してもよい。加圧は、好ましくは0.1MPa以上10MPa以下が好ましく、0.5MPa以上5MPa以下がより好ましい。これにより、樹脂組成物の硬化物中に空隙(ボイド)が発生し難くなる。加圧は、流体を用いて行われることが好ましく、例えば、窒素ガス、アルゴンガス、空気等のガスが挙げられる。安価な点で、空気が特に好ましい。
【0102】
加圧流体により加圧しながら、樹脂層130を硬化させる方法としては、例えば、圧力容器内に、加熱する処理対象物を設置し、次いで、圧力容器内に、加圧流体を導入して加圧しつつ、処理対象物を加熱する方法、更に、具体的には、加圧オーブン中に、処理対象物を設置し、加圧オーブン内に加圧用のガスを導入しつつ、加圧オーブンで処理対象物を加熱する方法が挙げられる。
【0103】
ここで、硬化後の樹脂組成物の反応率(B)に対する、溶融接合直後の樹脂組成物の反応率(A)が、0.01≦A/B≦0.7 であることが好ましい。
【0104】
本実施形態において反応率とは、以下の手順により求められる。
(i)加熱処理を行っていない樹脂層(未硬化の樹脂層)について、示差走査熱量計(DSC)を用いて、測定温度範囲25〜300℃、昇温速度:10℃/分の測定条件で加熱し、その時の発熱量(「全て反応した時に得られる熱量」とする)を測定する。
(ii)溶融接合と同じ条件(加熱温度、加熱時間など)で加熱された樹脂層について、(i)と同様の条件で、その時の発熱量を測定し、「全て反応した時に得られる熱量」に対する比率を反応率(A)とする。
同様にして、硬化された樹脂層について、(i)と同様の条件で、発熱量を測定し、「全て反応した時に得られる熱量」に対する比率を反応率(B)とする。
【0105】
上記のようにして求められた反応率(B)に対する反応率(A)は、0.01以上0.7以下であることにより、溶融接続直後の樹脂組成物が固化前の状態であるため、再加熱及び加圧を行うことによってボイドを除去することができる。
【0106】
上述のような構成において、本実施の形態の電子装置100の製造方法によれば、半田の融点より10℃高い温度でのゲルタイムが100秒以上であるという特定の樹脂組成物を用いた樹脂層130により、樹脂組成物の硬化速度が低下し、ボイドが発生しにくくなる。また、半田接合後も樹脂組成物の流動性が得られる観点からも、最終的に得られる電子装置100でのボイド形成が抑制され、接続信頼性が向上できる。
【0107】
なお、本発明は本実施の形態に限定されるものではなく、その要旨を逸脱しない範囲で各種の変形を許容する。たとえば、上記形態では半田バンプ111,121の両方が球状に形成されていることを例示した。しかし、その一方ないし両方が多段状に形成されていてもよい(図示せず)。
【0108】
なお、上述した実施の形態および複数の変形例は、その内容が相反しない範囲で組み合わせることができる。また、上述した実施の形態および変形例では、各部の構造などを具体的に説明したが、その構造などは本願発明を満足する範囲で各種に変更することができる。
【実施例】
【0109】
次に、本発明の実施例について説明する。
得られた樹脂組成物またはその硬化物について以下のような評価を行った。評価結果をそれぞれ表に示した。
【0110】
[ゲルタイム(秒)]
180℃、及び250℃としたホットプレート上に樹脂組成物をおき、樹脂組成物がゲル化するまでの時間(秒)をそれぞれ測定した。なお、本実施例及び比較例で用いた半田バンプの融点は217℃であった。
【0111】
[120℃での溶融粘度(Pa・s)]
樹脂組成物を15℃/minの速度で昇温し、Haake製レオメーターを用いて、温度に対する粘度挙動を測定し、120℃での粘度値を読み取った。
条件:20mmΦ,6.3rad/s
【0112】
[半田接合工程直後の反応率と、硬化後の反応率の比]
樹脂組成物の未硬化物をDSC(10℃/min)で測定し、発熱量を得た。この発熱量を「全て反応した時に得られる熱量」とした。
半田接合工程における熱処理後の樹脂組成物と、硬化後の樹脂組成物それぞれについて、同様にDSCで測定して発熱量を得た。得られた発熱量からそれぞれについての反応率(%)を計算し、その比を求めた。
【0113】
[実装後のボイド性]
150℃で基板と接触させ、設定温度350℃まで一気に加熱して5秒間保持したサンプル樹脂組成物を超音波探傷(SAT:scanning acoustic tomograph)により観察し、ボイドの有無について以下のように判定した。
判定
○:ボイドなし
△:バンプにまたがるボイドが発生
×:ボイドが多数発生
【0114】
[加圧硬化後のボイド性]
150℃/2h、5atmで硬化した後の樹脂組成物をSATにより観察し、ボイドの有無について以下のように判定した。
判定
○:ボイドなし
△:バンプにまたがるボイドが発生
×:ボイドが多数発生
【0115】
(実施例1)
・樹脂組成物の作成
ビスフェノールF型エポキシ樹脂(DIC株式会社製、EXA−830LVP、エポキシ当量160)19.2重量%と、ビフェニル型エポキシ樹脂(日本化薬株式会社製、NC−3000、エポキシ当量272)57.6重量%と、ゲンチジン酸(東京化成工業株式会社製、2,5−ジヒドロキシ安息香酸、融点202℃)23.1重量%と、2−フェニル−4−メチルイミダゾール(四国化成工業株式会社製、2P4MZ)0.1重量%とを秤量し、3本ロールにて分散混練してから、真空下脱泡処理をしてペースト状の樹脂組成物を得た。
・半導体装置の作製
得られたペースト状の樹脂組成物を、回路パターンが形成された回路基板(コア材として、住友ベークライト株式会社製、ELC−4785GS)に、半田バンプを有する半導体素子(サイズ15×15×0.65mm)をフリップチップボンダーで280℃、10秒間加熱したものの、半導体チップと基板の間にできる空隙(0.05mm)の間に充填させた。その後に、150℃で120分間加熱して樹脂組成物を硬化させ、半導体装置を得た。
【0116】
(実施例2、3)
表1に示す組成の樹脂組成物を、実施例1と同様の方法で、それぞれ作成した。得られた樹脂組成物を用い、実施例1と同様の方法で、それぞれ半導体装置を得た。
【0117】
(実施例4)
・フィルム状樹脂層の作製
EPICLON 840−S(大日本インキ社株式会社製)、45重量%、YX6954(ジャパンエポキシレジン社製)24.9重量%、フェノールフタリン(東京化成工業株式会社製)15重量%、PR−55617(住友ベークライト株式会社製)15重量%、2P4MZ(四国化成工業株式会社製)0.1重量%をあらかじめ混合させ、基材上に塗布し、フィルム状樹脂層を得た。
・半導体装置の作製
得られたフィルム状樹脂層を、高さが60μm、ピッチが150μmの半田バンプを有する半導体部品(サイズ10×10×0.2mm)に、真空ラミネーター(名機株式会社製、MVLP)を用い、100℃、0.8MPa、30秒でラミネートし、樹脂層付きチップをフリップチップボンダー(Panasonic株式会社製、FCB3)を用いて回路パターンが形成された回路基板(コア材として、住友ベークライト株式会社製、ELC−4785GS)に、280℃で10秒間加熱して半田接続を行った。そして、180℃で60分間加熱して、フィルム状樹脂層を熱硬化し、半導体装置を得た。
【0118】
(実施例5〜7)および(比較例1〜5)
表1に示す組成の樹脂組成物を、実施例1と同様の方法で、それぞれ作成した。得られた樹脂組成物を用い、実施例1と同様の方法で、それぞれ半導体装置を得た。
【0119】
実施例および比較例で得られた樹脂組成物またはフィルム状樹脂層、および半導体装置についてそれぞれ評価を行った。その結果を表に示す。
【0120】
【表1】
【0121】
なお、表1中の記載は、それぞれ以下を示す。
・EXA−830LVP DIC株式会社製、エポキシ当量160
・NC−3000 日本化薬株式会社製、エポキシ当量272
・EPICLON 840S 大日本インキ株式会社製、エポキシ当量185
・RE−810NM 日本化薬株式会社製、エポキシ当量210
・PR55617 住友ベークライト株式会社製、水酸基当量104
・NH2200R 日立化成工業株式会社、分子量166
・2P4MZ 四国化成工業株式会社製、融点174〜184℃
・2MZ 四国化成工業株式会社製、融点140〜148℃
・C05−MB 住友ベークライト株式会社製、融点272℃
・ゲンチジン酸 東京化成工業株式会社製、2,5−ジヒドロキシ安息香酸、融点202℃
・フェノールフタリン 東京化成工業株式会社製、融点235℃
・セバシン酸 東京化成工業株式会社製、融点134℃
・SO25H アドマテックス株式会社製
・YX6954 ジャパンエポキシレジン株式会社製
【符号の説明】
【0122】
100 電子装置
110 第一基板
111 半田バンプ
120 第二基板
121 半田バンプ
130 樹脂層
【特許請求の範囲】
【請求項1】
基板の電極端子と半導体チップの電極端子とを半田接合する際に、前記基板の電極端子と前記半導体チップの電極端子との間に導入され、半田接合した後に熱硬化する樹脂組成物であって、
前記樹脂組成物は、熱硬化性樹脂を含み、180℃、及び250℃におけるゲルタイムが100秒以上であることを特徴とする樹脂組成物。
【請求項2】
請求項1に記載の樹脂組成物において、
前記樹脂組成物の120℃での溶融粘度が、0.01Pa・s以上10,000Pa・s以下であることを特徴とする樹脂組成物。
【請求項3】
請求項1または2に記載の樹脂組成物において、
前記樹脂組成物の反応開始温度が100℃以上160℃以下であることを特徴とする樹脂組成物。
【請求項4】
請求項1乃至3いずれか一項に記載の樹脂組成物において、
前記熱硬化性樹脂は、エポキシ樹脂であることを特徴とする樹脂組成物。
【請求項5】
請求項1乃至3いずれか一項に記載の樹脂組成物において、
前記樹脂組成物は、フラックス活性剤を含むことを特徴とする樹脂組成物。
【請求項6】
請求項5に記載の樹脂組成物において、
前記フラックス活性剤は、カルボキシル基を有する化合物であることを特徴とする樹脂組成物。
【請求項7】
基板の電極端子と半導体チップの電極端子とを、半田接合する電子装置の製造方法であって、
前記基板の前記電極端子と、前記半導体チップの前記電極端子との間に、樹脂組成物を導入して、前記基板の前記電極端子と前記半導体チップの前記電極端子とを溶融接合しつつ、前記樹脂組成物を前記基板と前記半導体チップとの間に充填する工程と、
前記樹脂組成物を熱硬化する工程と、
を含み、
前記樹脂組成物は、請求項1乃至6いずれか一項に記載の樹脂組成物であることを特徴とする電子装置の製造方法。
【請求項8】
請求項7に記載の電子装置の製造方法において、
前記樹脂組成物を硬化する前記工程は、加圧流体を用いて前記樹脂組成物を加圧しながら硬化することを特徴とする電子装置の製造方法。
【請求項9】
請求項7または8に記載の電子装置の製造方法において、
前記加圧流体は、ガス及び/又は空気であることを特徴とする電子装置の製造方法。
【請求項10】
請求項7乃至9いずれか一項に記載の電子装置の製造方法において、
前記硬化後の樹脂組成物の反応率(B)に対する、前記溶融接合直後の樹脂組成物の反応率(A)が、0.01≦A/B≦0.7 であることを特徴とする電子装置の製造方法。
【請求項11】
請求項7乃至10いずれか一項に記載の電子装置の製造方法において、
前記加圧流体による加圧が、0.1MPa以上10MPa以下であることを特徴とする電子装置の製造方法。
【請求項12】
請求項7乃至11いずれか一項に記載の電子装置の製造方法において、
前記樹脂組成物を硬化する前記工程は、圧力容器内で行われ、前記圧力容器を前記加圧流体により加圧することを特徴とする電子装置の製造方法。
【請求項13】
請求項7乃至12いずれか一項に記載の電子装置の製造方法において、
前記樹脂組成物の硬化は、100℃以上250℃以下で行われることを特徴とする電子装置の製造方法。
【請求項1】
基板の電極端子と半導体チップの電極端子とを半田接合する際に、前記基板の電極端子と前記半導体チップの電極端子との間に導入され、半田接合した後に熱硬化する樹脂組成物であって、
前記樹脂組成物は、熱硬化性樹脂を含み、180℃、及び250℃におけるゲルタイムが100秒以上であることを特徴とする樹脂組成物。
【請求項2】
請求項1に記載の樹脂組成物において、
前記樹脂組成物の120℃での溶融粘度が、0.01Pa・s以上10,000Pa・s以下であることを特徴とする樹脂組成物。
【請求項3】
請求項1または2に記載の樹脂組成物において、
前記樹脂組成物の反応開始温度が100℃以上160℃以下であることを特徴とする樹脂組成物。
【請求項4】
請求項1乃至3いずれか一項に記載の樹脂組成物において、
前記熱硬化性樹脂は、エポキシ樹脂であることを特徴とする樹脂組成物。
【請求項5】
請求項1乃至3いずれか一項に記載の樹脂組成物において、
前記樹脂組成物は、フラックス活性剤を含むことを特徴とする樹脂組成物。
【請求項6】
請求項5に記載の樹脂組成物において、
前記フラックス活性剤は、カルボキシル基を有する化合物であることを特徴とする樹脂組成物。
【請求項7】
基板の電極端子と半導体チップの電極端子とを、半田接合する電子装置の製造方法であって、
前記基板の前記電極端子と、前記半導体チップの前記電極端子との間に、樹脂組成物を導入して、前記基板の前記電極端子と前記半導体チップの前記電極端子とを溶融接合しつつ、前記樹脂組成物を前記基板と前記半導体チップとの間に充填する工程と、
前記樹脂組成物を熱硬化する工程と、
を含み、
前記樹脂組成物は、請求項1乃至6いずれか一項に記載の樹脂組成物であることを特徴とする電子装置の製造方法。
【請求項8】
請求項7に記載の電子装置の製造方法において、
前記樹脂組成物を硬化する前記工程は、加圧流体を用いて前記樹脂組成物を加圧しながら硬化することを特徴とする電子装置の製造方法。
【請求項9】
請求項7または8に記載の電子装置の製造方法において、
前記加圧流体は、ガス及び/又は空気であることを特徴とする電子装置の製造方法。
【請求項10】
請求項7乃至9いずれか一項に記載の電子装置の製造方法において、
前記硬化後の樹脂組成物の反応率(B)に対する、前記溶融接合直後の樹脂組成物の反応率(A)が、0.01≦A/B≦0.7 であることを特徴とする電子装置の製造方法。
【請求項11】
請求項7乃至10いずれか一項に記載の電子装置の製造方法において、
前記加圧流体による加圧が、0.1MPa以上10MPa以下であることを特徴とする電子装置の製造方法。
【請求項12】
請求項7乃至11いずれか一項に記載の電子装置の製造方法において、
前記樹脂組成物を硬化する前記工程は、圧力容器内で行われ、前記圧力容器を前記加圧流体により加圧することを特徴とする電子装置の製造方法。
【請求項13】
請求項7乃至12いずれか一項に記載の電子装置の製造方法において、
前記樹脂組成物の硬化は、100℃以上250℃以下で行われることを特徴とする電子装置の製造方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2012−89571(P2012−89571A)
【公開日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願番号】特願2010−232747(P2010−232747)
【出願日】平成22年10月15日(2010.10.15)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
【公開日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願日】平成22年10月15日(2010.10.15)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
[ Back to top ]