
樹脂組成物、成型体、樹脂組成物の製造方法、及びポリオレフィン系樹脂を光酸化劣化させる方法
【課題】優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供する。また、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供する。
【解決手段】樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシドと、光触媒と、フィラーと、を含有する。ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される。
【解決手段】樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシドと、光触媒と、フィラーと、を含有する。ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、樹脂組成物、成型体、樹脂組成物の製造方法、及びポリオレフィン系樹脂を光酸化劣化させる方法に関する。
【背景技術】
【0002】
地球環境保全の意識の高まりに伴い、環境調和型プラスチックの開発が求められている。現在、環境調和型プラスチックとして研究が進められているものの一つに、生分解性プラスチックが挙げられる。生分解性プラスチックは、微生物等によって代謝・分解される性質を有するため、炭素循環を可能にする材料として注目されている。
【0003】
現在、プラスチックの材料としては、優れた物性を有するポリオレフィンが汎用されている。しかしながら、高分子であるポリオレフィンは微生物の細胞膜を通過できないため、生分解性に乏しい。そこで、近年、ポリオレフィンの生分解手法(Oxo−biodegradation)が開発された。この手法では、ポリオレフィンに酸化促進剤を添加することで、ポリオレフィンを低分子にまで酸化劣化させ(非生物分解)、微生物による代謝(生分解)を可能とする。この手法を利用した生分解性プラスチックがいくつか見出され、報告されている。
【0004】
非特許文献1〜3には、ポリオレフィンの一つであるポリプロピレンに、酸化促進剤(酸化チタン)を含有するポリエチレンオキシド(PEO)マイクロカプセルを導入した生分解性プラスチックが記載されている。この生分解性プラスチックにおいては、酸化チタンの光触媒反応により、PEOが分解されて酸及びアルデヒドが発生し、この酸及びアルデヒドがポリプロピレンの酸化劣化を促進する。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】K.Miyazaki et al,Polymer Degradation and Stability,94(2009)2114−2120
【非特許文献2】K.Miyazaki et al,Polymer Degradation and Stability,95(2010)1557−1567
【非特許文献3】K.Miyazaki et al,Polymer Degradation and Stability,96(2011)1039−1046
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、非特許文献1〜3に記載の生分解性プラスチックは、低弾性率体であるPEOを含有するため、力学物性に難点を有し、また、PEOは水溶性であるため、水などに濡れるとPEOとともに酸化チタンが溶出し、酸化劣化を確実に進めることができない場合があり、実用化という点で課題を残していた。
【0007】
本発明は、上記事情に鑑みてなされたものであり、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することを目的とする。また本発明は、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記目的を達成するため、本発明の第1の観点に係る樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシドと、光触媒と、フィラーと、を含有する。
【0009】
前記ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有されていてもよい。
【0010】
前記フィラーは、前記樹脂組成物中に0.1質量%〜7.0質量%含有されていてもよい。
【0011】
前記光触媒は、前記樹脂組成物中に0.1質量%〜3.0質量%含有されていてもよい。
【0012】
前記フィラーはセルロースであってもよい。
【0013】
前記光触媒は酸化チタンであってもよい。
【0014】
前記ポリオレフィン系樹脂はポリプロピレン類を含有していてもよい。
【0015】
前記ポリプロピレン系樹脂はプロピレンホモポリマーを含有していてもよい。
【0016】
さらに、前記樹脂組成物100質量部に対して、光酸化劣化遅延剤を0.01質量部〜10.0質量部含有していてもよい。
【0017】
前記光酸化劣化遅延剤はアパタイト誘導体であってもよい。
【0018】
本発明の第2の観点に係る成型体は、本発明の第1の観点に係る樹脂組成物からなる。
【0019】
本発明の第3の観点に係る樹脂組成物の製造方法は、ポリエチレンオキシドと、光触媒と、フィラーと、を混練して触媒組成物を得る工程と、前記触媒組成物をポリオレフィン系樹脂と混練する工程と、を含む、ことを特徴とする。
【0020】
本発明の第4の観点に係るポリオレフィン系樹脂を光酸化劣化させる方法は、光触媒とフィラーとを含有するポリオレフィン系樹脂に、ポリエチレンオキシドを添加する、ことを特徴とする。
【発明の効果】
【0021】
本発明によれば、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することができる。また、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することができる。
【図面の簡単な説明】
【0022】
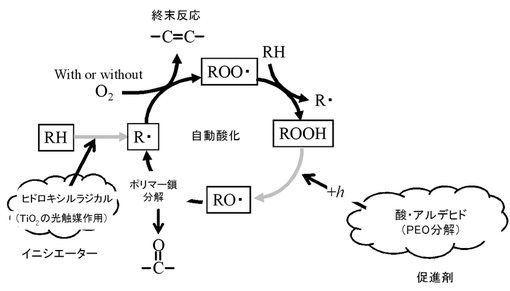
【図1】PPの光酸化劣化メカニズム(自動酸化)を表す概念図である。
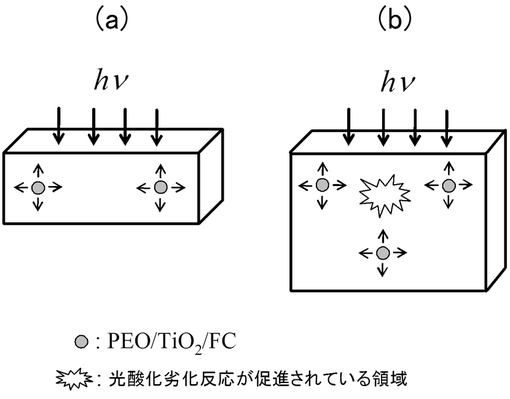
【図2】(a)はサンプル厚が薄い場合、(b)はサンプル厚が厚い場合の厚さ方向への光酸化劣化の進行を表す概念図である。
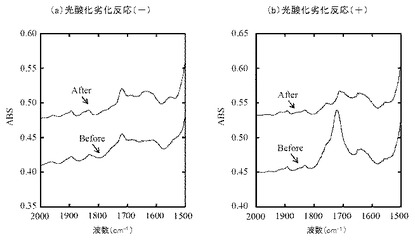
【図3】(a)は樹脂組成物1、(b)は樹脂組成物2の光酸化劣化前後におけるエチレン性不飽和基及びカルボニル基の発生量を表すFT−IRスペクトルの図である。
【図4】(a)は比較組成物1、(b)は比較組成物2、(c)は樹脂組成物1、及び(d)は樹脂組成物2の光酸化劣化前後における水酸基の発生量を表すFT−IRスペクトルの図である。
【図5】(a)は樹脂組成物3、(b)は樹脂組成物4、(c)は樹脂組成物5、及び(d)は樹脂組成物6の光酸化劣化前後におけるエチレン性不飽和基及びカルボニル基の発生量を表すFT−IRスペクトルの図である。
【図6】(a)は樹脂組成物3、(b)は樹脂組成物4、(c)は樹脂組成物5、及び(d)は樹脂組成物6の光酸化劣化前後における水酸基の発生量を表すFT−IRスペクトルの図である。
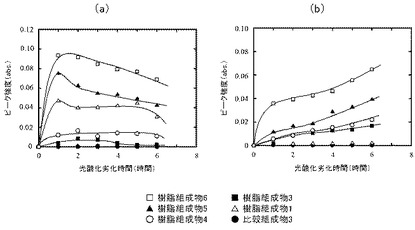
【図7】光酸化劣化開始後の樹脂組成物1及び3〜6、並びに比較組成物3について、(a)はエチレン性不飽和基、(b)はカルボニル基の発生量の経時的変化を表すグラフ図である。
【図8】光酸化劣化開始後の厚さ100μmの樹脂組成物3及び6について、カルボニル基の発生量の経時的変化を表すグラフ図である。
【図9】各々厚さ50μm及び100μmの(a)樹脂組成物3及び(b)樹脂組成物6について、光酸化劣化によるカルボニル基の発生量を表すグラフ図である。
【図10】樹脂組成物1及び3〜6、並びに比較組成物3について、(a)はヤング率、(b)は引張強度、(c)は破断ひずみ(ただし、(c)については、比較組成物3のデータ無し)を表すグラフ図である。
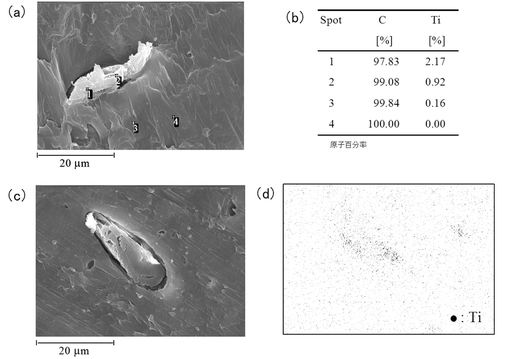
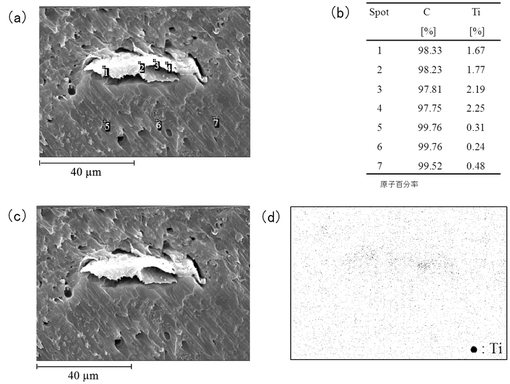
【図11】エッチングをしなかった場合の樹脂組成物6について、(a)及び(c)はSEM像、(b)は(a)の各スポットの原子百分率、(d)は(c)に対応するTi原子の分布を表す図である。
【図12】エッチングをした場合の樹脂組成物6について、(a)及び(c)はSEM像、(b)は(a)の各スポットの原子百分率、(d)は(c)に対応するTi原子の分布を表す図である。
【図13】(a)は光酸化劣化させなかった樹脂組成物3、(b)は光酸化劣化させた樹脂組成物3について、埋土試験後のSEM像を示す図である。
【図14】(a)は光酸化劣化させなかった樹脂組成物3、(b)は光酸化劣化させた樹脂組成物3について、埋土試験前後のFT−IRスペクトルを表す図である。
【図15】(a)〜(d)は、光酸化劣化させた樹脂組成物3の埋土試験後の光学顕微鏡像を示す図である。
【発明を実施するための形態】
【0023】
以下、本発明の実施形態について詳細に説明する。
【0024】
本発明による樹脂組成物は、光酸化劣化されることにより(非生物分解)、微生物による代謝(生分解)が可能となる。
【0025】
本発明による樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシド(PEO)と、光触媒と、フィラーと、を含有する。
【0026】
(1.ポリオレフィン系樹脂)
本発明による樹脂組成物に含まれるポリオレフィン系樹脂は、主としてポリオレフィンを含有する樹脂である。ポリオレフィンとしては、特に限定されるものではなく、プロピレンホモポリマー(PP)(アイソタクチック型及びシンジオタクチック型を含む)、プロピレン−エチレンブロック共重合体、プロピレン−エチレンランダム共重合体等のポリプロピレン類、高密度ポリエチレン、中密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、超低密度ポリエチレン、メタロセン触媒によるポリエチレン等のポリエチレン類、ポリプロレン、ポリブテン、ポリメチルペンテン、ポリスチレン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明において、良好な力学物性を有するという点において、ポリプロピレン類が好適に用いられ、PPがさらに好適に用いられる。PPについては、例えば、平均分子量1.0×104〜1.0×106のものが用いられる。本発明の効果を奏するポリオレフィン系樹脂であれば、適宜選択され得る。
【0027】
(2.PEO)
本発明による樹脂組成物に含まれるPEOとしては、例えば、数平均分子量2.0×103〜1.0×106のものが用いられる。本発明の効果を奏する分子量のPEOであれば、適宜選択され得る。
【0028】
PEOは、本発明による樹脂組成物中、例えば、0.1質量%〜7.0質量%含有される。PEOは、力学物性に影響を与え得る低弾性率体である一方で、後述の光酸化劣化における促進剤でもある。優れた力学物性と確実な光酸化劣化能との両方を実現するために、樹脂組成物中PEOを0.3質量%〜5.0質量%含有した樹脂組成物が好適に用いられる。さらに確実な光酸化劣化能を実現するために、樹脂組成物中PEOを1.0質量%〜5.0質量%含有した樹脂組成物がさらに好適に用いられる。
【0029】
(3.光触媒)
本発明による樹脂組成物に含まれる光触媒としては、後述の光酸化劣化の反応を触媒できるものであれば特に限定されるものではなく、酸化チタン(TiO2)(アナターゼ型、ルチル型、及びブルッカイト型を含む)、モリブデン酸化物、酸化亜鉛、酸化錫、チタン酸ストロンチウム、酸化スズ、酸化タングステン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明において、入手が容易であるという点、及び紫外線といった短い波長の光により光触媒活性を示すことができるという点で、TiO2が好適に用いられる。本発明の効果を奏する光触媒であれば、適宜選択され得る。
【0030】
光触媒は、本発明による樹脂組成物中、例えば0.1質量%〜3.0質量%、好ましくは0.1質量%〜1.0質量%含有される。樹脂組成物中0.1質量%以上含有されることで、光触媒としての機能を十分に発揮することができ、後述の光酸化劣化を確実に進めることができる。また、光触媒が樹脂組成物中3.0質量%以下含有されることで、コストを抑えた樹脂組成物が可能となる。
【0031】
(4.フィラー)
本発明による樹脂組成物に含まれるフィラーとしては、樹脂組成物の力学物性を向上させ、光触媒に親和性を有し、かつ、光酸化劣化能及び生分解能に影響を及ぼさないものであれば適宜用いることができ、特に限定されるものではなく、繊維状セルロース(以下、FCという)、タルク、ガラス繊維、カオリン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明においては、FCが好適に用いられる。本発明による樹脂組成物にFCを用いた場合、優れた力学物性(例えば、ヤング率、引張強度、破断ひずみ等で規定される)が得られ、また、後述の通り、FCは光触媒との親和性が高く、光触媒を樹脂組成物内に保持することができるため、確実な光酸化劣化能が得られる。本発明の効果を奏するフィラーであれば、適宜選択され得る。
【0032】
フィラーは、本発明による樹脂組成物中、例えば0.1質量%〜7.0質量%含有される。優れた力学物性を実現するために、樹脂組成物中フィラーを1.0質量%〜7.0質量%含有した樹脂組成物が好適に用いられる。樹脂組成物の透明度を保ち、光の透過性を確保することで、確実な光酸化劣化能を実現するために、樹脂組成物中フィラーを1.0質量%〜5.0質量%含有した樹脂組成物がさらに好適に用いられる。
【0033】
本発明による樹脂組成物は、例えば、ポリオレフィン系樹脂としてPP、光触媒としてTiO2、及びフィラーとしてFCを含有する(以下、このような樹脂組成物を、PP/PEO/TiO2/FCという)。本発明による樹脂組成物中の各成分の含量に関して、PP/PEO/TiO2/FCの場合、例えば、樹脂組成物中PPを97.5質量%、PEOを1.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを96.5質量%、PEOを2.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを95.5質量%、PEOを3.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを93.5質量%、PEOを5.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの等が好適に用いられる。このようなPP/PEO/TiO2/FCは、優れた力学物性及び確実な光酸化劣化能を有する。
【0034】
(5.光酸化劣化遅延剤)
本発明による樹脂組成物は、光酸化劣化遅延剤をさらに含有していてもよい。光酸化劣化遅延剤として、例えば、Ca8(HPO4)2(OOC−R−COO)(PO4)4・mH2O(コハク酸イオン含有オクタカルシウムホスフェート;OCPC)、CaHPO4・2H2O(ジカルシウムホスフェートジハイドレート;DCPC)、Ca8(HPO4)2(PO4)4・5H2O(オクタカルシウムホスフェート;OCP)等といったアパタイト誘導体が挙げられる。これらは、単独で、又は組み合わせて用いられ得る。例えば、光触媒としてTiO2を用いた場合、アパタイト誘導体はTiO2の表面に結晶を形成するため、光酸化劣化を遅延させることが可能となる。また、他の光酸化劣化遅延剤として、例えば、ヒンダードアミン等といったアミン系光安定化剤が挙げられる。ヒンダードアミンは、PEOから生じた酸と中和反応することで、光酸化劣化を遅延させることができる。なお、確実な光酸化劣化遅延効果を得るために、アパタイト誘導体をさらに含有した樹脂組成物が好適に用いられる。本発明の効果を奏する光酸化劣化遅延剤であれば、適宜選択され得る。
【0035】
本発明による樹脂組成物は、前述の光酸化劣化分解遅延剤を含むことで、時限分解性を実現することができる。例えば、樹脂組成物の時限分解性を制御することで、樹脂組成物からなる成型体の材料寿命を自在に設計することが可能となる。樹脂組成物の時限分解性の制御は、例えば、光酸化劣化遅延剤の含有量及び反応時間を調節すること等により可能となる。光酸化劣化遅延剤は、例えば、樹脂組成物100質量部に対して0.01質量部〜10.0質量部含有される。
【0036】
本発明による樹脂組成物は、上記以外に、光酸化劣化能及び生分解能に影響を及ぼさない範囲で、一般的な樹脂組成物に通常用いられる添加剤をさらに含むことができる。例えば、光吸収剤、光安定剤、帯電防止剤、滑剤、着色剤、発泡剤、可塑剤、難燃剤、架橋剤、抗菌剤、耐候劣化防止剤等が挙げられ、これらを単独又は組み合わせで用いることができる。
【0037】
(6.光酸化劣化)
一例としてPP/PEO/TiO2/FCを用いて、光酸化劣化について説明する。PP/PEO/TiO2/FCは、疎水性のPP中に、親水性のPEO、TiO2、及びFCからなる粒子状物が分散される特徴を有する。
【0038】
この樹脂組成物を構成するTiO2に光が照射されると、h+(正孔)とe−(電子)とが発生する。正孔は水と反応して、・OH(ヒドロキシルラジカル)を生成する。電子はO2と反応して、HOO・とOH−とを生成する。生成したOH−は正孔と反応して、ヒドロキシルラジカルを生成する。生成したHOO・が2分子結合して、H2O2とO2とを生成し、このH2O2に光が照射されるとヒドロキシルラジカルが生成される。このように、種々の段階でヒドロキシルラジカルが発生する。
【0039】
【化1】

【0040】
この樹脂組成物を構成するPEOは、前述で発生したヒドロキシルラジカルにより活性化され、−CH2−・CH−O−と水とが発生する。−CH2−・CH−O−はO2と反応して、−CH2−C(−OO・)H−O−が生成される。−CH2−C(−OO・)H−O−が2分子結合して、O2が発生し、それにより−CH2−C(−O・)H−O−が生成される。−CH2−C(−O・)H−O−から−CH2−COH(アルデヒド)と・O−CH2とが生成される。また、−CH2−C(−O・)H−O−から−CH2−COO−(エステル)が生成され、さらにヒドロキシラジカルにより活性化されて−CH2−COO−・CH−CH2−O−が生成される。−CH2−COO−・CH−CH2−O−が分解されて、−CO−O・−とOHC−CH2−(アルデヒド)が生成される。−CO−O・−とOHC−CH2−(アルデヒド)とから、−COOH(酸)が生成される。このように、PEOはTiO2の光触媒作用により分解され、酸とアルデヒドを生成する。また、PEOが活性化されることにより生じた水は、前述のTiO2の光触媒反応に利用される。
【0041】
【化2】
【0042】
この樹脂組成物を構成するPPは、図1に示される自動酸化のサイクルが継続することにより光酸化劣化される。PPの自動酸化は、前述で発生したヒドロキシルラジカルにより開始される。PP(図1において、RH)は、ヒドロキシルラジカルによりR・(アルキルラジカル)となる。ここで、一部のアルキルラジカルは、停止反応へと移行し、エチレン性不飽和基となる。自動酸化のサイクルが停止反応で止まってしまうと、PPはそれ以上光酸化劣化されない。一方、他のアルキルラジカルは、ROO・(ペルオキシラジカル)を経て、ROOH(ヒドロペルオキシド)となる。ヒドロペルオキシドは、光照射によってRO・(アルコシラジカル)に分解される。前述のPEO分解により発生した酸及びアルデヒドは、このヒドロペルオキシドの分解を促進する。PP鎖の末端は、アルコシラジカルを経て、カルボニル基となる。このような自動酸化のサイクルが継続することで、PPは主鎖の一端から順次分解されていく。したがって、光酸化劣化が進むにつれて、カルボニル基の発生量が増加する。また、この自動酸化のサイクルにおいて、TiO2の光触媒作用により発生したヒドロキシルラジカルは「イニシエーター」としての役割を有し、PEO分解により発生した酸及びアルデヒドは「促進剤」としての役割を有する。
【0043】
前述の自動酸化のサイクルで必要とされる光は、例えば紫外線といった短い波長の光である。また、PPの光酸化劣化に要する時間は、例えば2〜48時間、数日、数十日である。生分解が可能となる程度にまでPPが分解されればよく、光酸化劣化に要する時間は適宜選択され得る。
【0044】
本発明による樹脂組成物に含まれる光触媒は、前述の通り、フィラーに親和性を有する。このため、樹脂組成物が水などに濡れて一部のPEOが溶出した場合でも、光触媒はフィラーに保持されることにより樹脂組成物内に留まっている。光触媒は光酸化劣化に必須であるため、光触媒が保持されていることで、樹脂組成物は確実な光酸化劣化能を有する。例えば、PP/PEO/TiO2/FCを屋外で使用した場合、雨などに濡れて一部のPEOが溶出したとしても、FCに親和性を有するTiO2は溶出することなく、FCとともに樹脂内に保持されている。光酸化劣化に必須であるTiO2が保持されるため、PP/PEO/TiO2/FCは確実な光酸化劣化能を有する。なお、雨などに濡れて一部のPEOが溶出してしまったとしても、後からPEOを添加(PEO溶液を樹脂組成物に振り掛ける等)することで、光酸化劣化を促進させることができる。
【0045】
本発明による樹脂組成物は、光酸化劣化により低分子にまで分解されることにより、生分解能を有するようになる。具体的には、樹脂組成物に含まれるポリオレフィンは、光酸化劣化により分子量1,000以下程度にまで分解されることで、微生物による代謝が可能となる。
【0046】
(7.成型体)
本発明による樹脂組成物は、種々の形状に成型することができる。すなわち、本発明は、前述の樹脂組成物からなる成型体を提供するものである。成型体としては、厚さ数十μm〜数十mmのシート状物のほか、一般にポリオレフィン系樹脂として成型される種々の形状に成型することができる。本発明による成型体の厚さは、その用途により適宜選択され、例えば、数十μm〜数十cmとすることができる。
【0047】
本発明による成型体は、表面から内部に向かって光酸化劣化を進行させることができる。例えば、PP/PEO/TiO2/FCからなる成型体の場合(図2)、成型体内部に存在するTiO2が光を受けてヒドロキシルラジカルを発生させ、かつ、PEOが酸及びアルデヒドを生成させることで、光酸化劣化が進行する。
成型体が薄い場合(図2(a))、TiO2の光触媒作用により成型体内部のPEOが消費され、自動酸化が促進される。前述の通り、PEO分解により生じた酸及びアルデヒドは、自動酸化において促進剤としての役割を有するため、光酸化劣化の進行はPEO含量に依存するといえる。
一方、成型体が厚い場合(図2(b))、TiO2の光触媒作用により、まず成型体の表面近くに存在するPEOが消費される。成型体表面近くに存在するPEOが消費し尽くされたら、次に成型体表面から厚さ方向に少し離れた場所に存在するPEOが順次消費され、光酸化劣化が進行する。このように、PEOが成型体内部に分散して存在しており、成型体の厚さ方向に光酸化劣化を進めることができるため、厚い成型体を調製することが可能である。本発明による成型体は、数十cm程度にまで厚くしても、確実な光酸化劣化能を有する。
【0048】
(8.樹脂組成物の製造方法)
本発明による樹脂組成物の製造方法には限定はない。しかしながら、例えば、樹脂組成物100質量部に対して、PEO0.1質量部〜7.0質量部と、光触媒0.1質量部〜3.0質量部と、及びフィラー0.1質量部〜7.0質量部と、を混練して触媒組成物を得、次いで該触媒組成物をポリオレフィン系樹脂質83.0質量部〜99.7質量部と混練することで好適に製造することができる。
【0049】
本発明で使用する光触媒とフィラーとをPEOとともに混練し、これを乾燥させると、フィラーに光触媒が付着したものがPEO中に均一に分散した触媒組成物が得られる。この触媒組成物をポリオレフィン系樹脂に混練させると、PEOが乳化剤のように作用して、疎水性のポリオレフィン系樹脂中で該触媒組成物が均一に混練される。この際、ポリオレフィン系樹脂中で該触媒組成物は粒子状に形成され、光触媒及びフィラーは該触媒組成物の粒子中に存在する。なお、光触媒及びフィラーをPEOとともに混練する際、水を添加してもよい。水の存在によって、均一に混練することができる。この際、水を残したままポリオレフィン系樹脂と混練してもよい。PPとの混練中に水が蒸発するからである。また、ポリオレフィン系樹脂との混練時に、亜リン酸エステル系酸化防止剤といった酸化防止剤を添加してもよい。
【0050】
(9.成型体の製造方法)
本発明による成型体は、前述の樹脂組成物を従来公知の方法で成型して製造することができる。例えば、圧縮成型、押出成型、射出成型等により、用途に合わせて所望の形状、厚さ、大きさ等に成型することができる。
【0051】
(10.成型体の用途)
本発明による成型体は、プラスチック材を使用するあらゆる用途に使用可能である。例えば、土木工事資材、家具材、家電材、建材、使い捨て容器等の構造物に用いることができる。本発明による成型体は、生分解能を有するため、山間部等の回収困難な場所に設置される構造物、コスト及び技術等の問題からリサイクルが容易ではない構造物等に好適に用いることができる。また、前述の通り、本発明による成型体が光酸化劣化遅延剤を含有する場合、時限分解が可能となるため、例えば、構造物を廃材としたい時期に光酸化劣化が開始されるように、材料寿命を設計することができる。また、本発明による成型体は、熱を加えることなく光酸化劣化を進めることができる。光を照射すれば光酸化劣化を進めることができるため、0℃以下の低温環境で用いられる構造物にも用いることができる。
【0052】
(11.ポリオレフィン系樹脂を光酸化劣化させる方法)
本発明は、樹脂組成物を構成するTiO2への光照射によってヒドロキシルラジカルが発生し、このヒドロキシルラジカルによってPEOが活性化されてアルデヒド及び酸が発生し、図1に示すようにこれらが相互に作用してポリオレフィン系樹脂を継続的に光酸化劣化させるものである。PEOは、光酸化劣化の際に光触媒、フィラー、及びポリオレフィン系樹脂と共存していればよく、光触媒とフィラーとを含有するポリオレフィン系樹脂に後からPEOを添加しても、ポリオレフィン系樹脂を光酸化劣化させることができる。すなわち、本発明は、光触媒とフィラーとを含有するポリオレフィン系樹脂に、PEOを添加する、ことを特徴とするポリオレフィン系樹脂を光酸化劣化させる方法を提供するものである。この方法において使用可能な光触媒、フィラー、ポリオレフィン系樹脂、及びPEOの種類等については、前述の通りである。また、この場合の「PEOを添加する」とは、例えば、PEO水溶液を該ポリオレフィン系樹脂に振り掛ける(例えば、スプレーする)ことを意味する。
【実施例1】
【0053】
以下、実施例を挙げて本発明を具体的に説明する。ただし、本発明はこれらの実施例に限定されるものではない。
【0054】
(PP/PEO/TiO2/FCの調製)
PP(日本ポリプロ社製、メソペンタッド分率=98%、平均分子量(Mn):4.6×104、多分散性(Mw/Mn):5.7)、PEO(和光純薬工業社製、平均分子量(Mn):5.0×105)、TiO2(和光純薬工業社製、アナターゼ型(98.5%以上)、表面積:約6m2/g)、及びFC(日本製紙ケミカル社製、商品名「W−100GK」、90質量%が100メッシュの篩(150μm以下)を通過、平均長:37μm)を用いて、表1に示す組成の樹脂組成物を調製した。なお、FCは、デシケーターで7日間乾燥させ、FCの水分量が0.7質量%以下となったものを調製に用いた。
0.1Lのガラスビーカーに純水50mL、PEO0.3g、TiO20.5g、FC1.0gを入れ、24時間、23℃で、スターラーで撹拌しながら混合させた。その後、ロータリーエバポレーターを用いて、水を蒸発させた。その後、このPEO/TiO2/FC混合物を真空オーブンで24時間、50℃で乾燥させた。その後、このPEO/TiO2/FC混合物と、PP98.2g及び亜リン酸エステル系酸化防止剤(ADEKA社製、商品名「ADKSTAB PEP−36」)0.1gをミキサー(井元製作所製、商品名「IMC−1884」)に仕込み、180℃に加温し、5分間、100rpmで混練した。このようにして、100gの樹脂組成物1を調製した。
上記と同様にして、樹脂組成物2〜6を調製した。また、比較例として、上記と同様にして、比較組成物1〜3を調製した。
【0055】
【表1】
【実施例2】
【0056】
(光酸化劣化試験)
樹脂組成物1及び2を、5MPa、190℃で5分間圧縮成型し、厚さ50μmのフィルム状に成型した。各々を20mm×20mmの大きさに切断し、バイアル(パイレックス社製)に入れた。リコーロータリー光化学反応装置(理工科学産業社製、商品名「RH400−10W」)を用いて、空気中、30℃で光酸化劣化試験を行った。この光化学反応装置には、400Wの高圧水銀灯(理工科学産業社製、UVL−400HA型超高圧水銀灯(5mW/cm2))が備えられている。光酸化劣化試験前後におけるエチレン性不飽和基、カルボニル基、及び水酸基の発生量を、フーリエ変換型赤外分光(FT−IR)分析により測定した。FT−IRスペクトル測定は、FT−IRスペクトロメータ(パーキンエルマー社製、商品名「Spectrum One」)を用いて(16スキャン、分解能:2cm−1)、中赤外領域(400−4000cm−1)で行った。光酸化劣化時間は、6時間とした。なお、比較組成物1,2及び5についても、同様に水酸基の発生量を測定した。結果を図3及び図4に示す。
【0057】
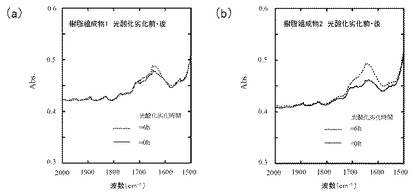
図3(a)は樹脂組成物1、及び図3(b)は樹脂組成物2のFT−IRスペクトルを示す。
樹脂組成物1及び2において、光酸化劣化時間6時間では、カルボニル基のピーク(1720cm−1付近)はほとんど見られなかったものの、光酸化劣化後においてエチレン性不飽和基のピーク(1642cm−1付近)の増大がみられた。このことから、PPの自動酸化が進みつつあることが示唆された。
【0058】
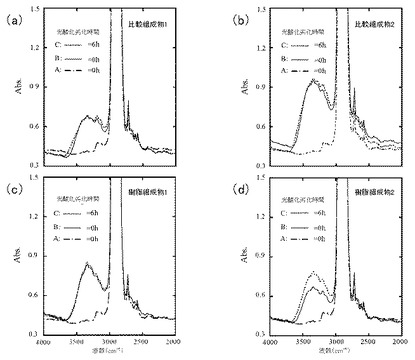
図4(a)は比較組成物1、図4(b)は比較組成物2、図4(c)は樹脂組成物1、及び図4(d)は樹脂組成物2のFT−IRスペクトルを示す。図4(a)〜(d)のAは、比較組成物3のFT−IRスペクトルを示す。
比較組成物3のAでは水酸基のピーク(3350cm−1付近)が見られなかった一方で、いずれの比較組成物においても、水酸基のピークが見られた。
比較組成物1に比して、比較組成物2では水酸基のピークが高いものの、光酸化劣化前後でピーク強度に変化はみられなかった。このことから、これらのピークには自動酸化により生じるヒドロペルオキシド由来の水酸基は含まれておらず、FC由来の水酸基が含まれていることが示唆された。つまり、PEOが含まれない比較組成物1及び2では、PPの自動酸化が進行しないことが示唆された。
一方、PEOが0.3質量%含まれる樹脂組成物1では、比較組成物1よりもFC含量が少ないにもかかわらず、比較組成物1に比して水酸基のピークが増大しており、またそのピークは鋭い形状であった。このことから、このピークにはヒドロペルオキシド由来の水酸基が含まれていることが示唆された。
さらに、樹脂組成物2においても、水酸基のピークがみられ、光酸化劣化後にそのピークが増大していた。このことから、この水酸基のピークにはヒドロペルオキシド由来の水酸基が含まれていること、また、自動酸化のサイクルにおいてヒドロペルオキシドが分解されずに若干蓄積しているものの、PPの自動酸化が進みつつあることが示唆された。
以上のことから、PEOが0.3質量%含まれる樹脂組成物1及び2において、光酸化劣化開始後6時間の時点では、自動酸化が継続する程度には至っていないものの、自動酸化が進みつつあることが示唆された。
【0059】
前述において、PEO含量は0.3質量%であったが、さらにPEO含量を増やして光酸化劣化の進行を検討した。樹脂組成物1及び3〜6、並びに比較組成物3を前述と同様に、厚さ50μmに成型し、前述と同様に光酸化劣化試験を行った。光酸化劣化前後におけるエチレン性不飽和基、カルボニル基、及び水酸基の発生量を測定した。光酸化劣化時間は、6時間とした。結果を図5〜図7に示す。
【0060】
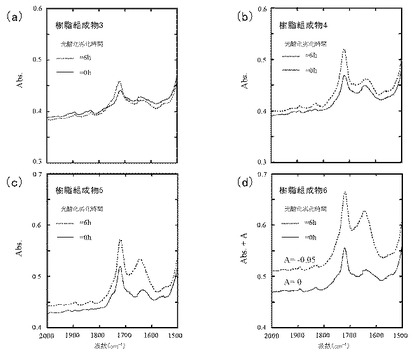
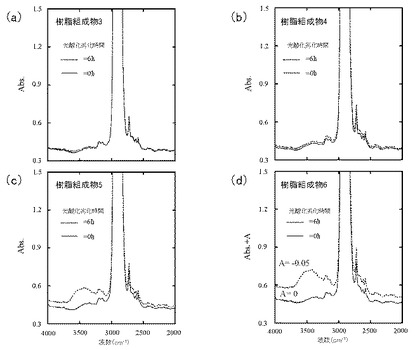
図5(a)は樹脂組成物3、図5(b)は樹脂組成物4、図5(c)は樹脂組成物5、及び図5(d)は樹脂組成物6のFT−IRスペクトルを示す。
PEO含量依存的に、エチレン性不飽和基及びカルボニル基のピーク強度が増大した。いずれにおいても、エチレン性不飽和基のピーク強度よりも、カルボニル基のピーク強度の方が大きかった。また、PEO含量依存的に、光酸化劣化前後におけるピーク強度増大の程度が大きくなった。このことから、PEO含量を増やすことで、自動酸化のサイクルにおいて、停止反応のみならず、ヒドロペルオキシド分解の方向にも反応が進み、自動酸化のサイクルが継続する状態になっていることが示唆された。
【0061】
図6(a)は樹脂組成物3、図6(b)は樹脂組成物4、図6(c)は樹脂組成物5、及び図6(d)は樹脂組成物6のFT−IRスペクトルを示す。
樹脂組成物3及び4では、水酸基のピーク強度はほぼ0であり、光酸化劣化前後でピーク強度に変化はみられなかった。このこと、並びに図5(a)及び図5(b)においてカルボニル基の発生が確認されたことから、樹脂組成物1及び2ではヒドロペルオキシドがほとんど分解され、PPの自動酸化が進行していることが示唆された。
また、樹脂組成物5及び6では、光酸化劣化後に水酸基のピーク強度増大がみられた。このことから、樹脂組成物5及び6では、自動酸化がサンプルの厚さ方向に進み、サンプル内部における自動酸化により新たに発生したヒドロペルオキシドが蓄積しつつあることが示唆された。
【0062】
光酸化劣化の間のエチレン性不飽和基(図7(a))及びカルボニル基(図7(b))の発生量を経時的に測定した。いずれにおいても、PEO含量依存的にピーク強度の増大がみられた。エチレン性不飽和基については、樹脂組成物1及び3〜6のいずれにおいても光酸化劣化開始1時間後に最大ピークがみられ、その後ピーク強度は減少した。一方で、カルボニル基については、時間とともにピーク強度が増大した。このことから、自動酸化のサイクルにおいて、停止反応への進行は光酸化劣化開始後ある程度の時間経過後の段階で止まり、その後の段階では自動酸化のサイクルが継続する状態に移行することが示唆された。
【実施例3】
【0063】
(サンプルの厚さとPEO含量との関係)
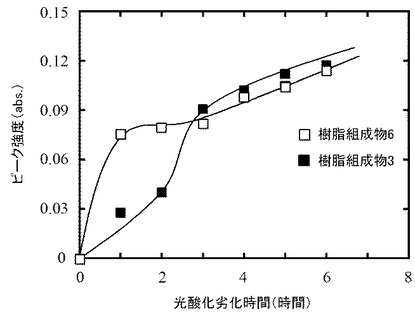
自動酸化の進行におけるサンプルの厚さとPEO含量との関係について検討した。樹脂組成物3及び6を、前述と同様の方法で厚さ100μmに成型し、前述と同様に光酸化劣化試験を行った。光酸化劣化後におけるカルボニル基のピーク強度を、樹脂組成物3と樹脂組成物6との間で比較した。光酸化劣化時間は、6時間とした。結果を図8に示す。
【0064】
光酸化劣化開始2時間後までは、樹脂組成物3に比して樹脂組成物6のピーク強度が高かった。しかしながら、光酸化劣化開始3時間後以降、樹脂組成物3のピーク強度は、樹脂組成物6のピーク強度よりも高くなった。このように、サンプルの厚さが50μmの場合(図7(b))とは異なる傾向がみられた。
このことから、サンプル厚が厚い場合、光酸化劣化開始直後では、PEO含量の多い方でより良好に自動酸化が進行するが、それ以降の段階では、PEO含量が少なくても良好に自動酸化が進むことが示唆された。PEOの分解によって酸及びアルデヒドが発生し、これらによってヒドロペルオキシドの分解が促進され、サンプルの厚さ方向に自動酸化が進行したものと考えられる。
【0065】
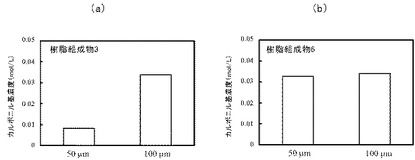
自動酸化の進行におけるサンプルの厚さとPEO含量との関係についてさらに詳細に検討した。樹脂組成物3及び樹脂組成物6について、各々厚さ50μm及び100μmのサンプルを前述と同様の方法で調製し、光酸化劣化反応前後におけるカルボニル基の増加量を測定した。カルボニル基濃度は、PP平均モル吸光係数(330mol l−1 cm−1)及びPP密度(0.900g/mL)を用いて、ランベルト・ベールの法則により推定した。光酸化劣化時間は、5時間とした。結果を図9に示す。
【0066】
図9(a)は樹脂組成物3、図9(b)は樹脂組成物6の光酸化劣化前後におけるカルボニル基の増加量を示す。
樹脂組成物3において、100μmでは50μmに比して約4倍のピーク強度がみられた。一方、樹脂組成物6において、50μmと100μmとの間で差はみられなかった。このことから、樹脂組成物3の厚さ100μmのサンプルでは、PEOによる自動酸化促進作用により、サンプルの厚さ方向に光酸化劣化が進行したことが示唆された。
【実施例4】
【0067】
(力学物性試験)
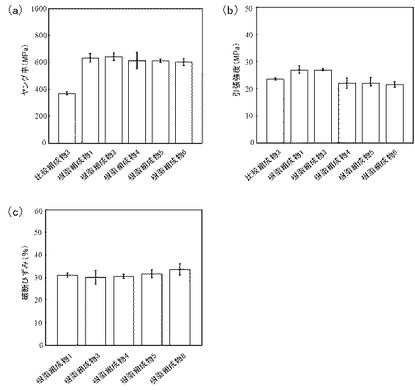
低弾性率体であるPEOを含有するサンプルの力学物性について検討した。樹脂組成物1及び3〜6、並びに比較組成物3について、力学試験機(島津製作所社製、商品名「SHIMAZU EZ−S」)を用いて、ヤング率、引張強度、及び破断ひずみを測定した。各サンプルを、前述と同様の方法で、30mm×5mm×0.05mmの大きさに調製した。クロスヘッド速度3mm/分、20℃で試験を行った。ヤング率は、応力−ひずみ曲線の傾き(ひずみ値約1%まで)より得た。各々10回測定による平均値を測定値とした。なお、PPのみで構成された比較組成物3は伸びが大きいため、破断ひずみについては測定不可能であった。結果を図10に示す。
【0068】
ヤング率(図10(a))については、比較組成物3に比して、FCを含有する樹脂組成物1及び3〜6では1.8倍程度増大した。引張強度(図10(b))については、比較組成物3と樹脂組成物1及び3〜6との間で大きな差はみられなかった。破断ひずみ(図10(c))については、PEO含量を多くしてもほぼ一定に保たれることが示された。このことから、本発明による樹脂組成物は、FCを含むことにより、低弾性率体であるPEOの含量を多くしても、優れた力学物性を有することが示された。
【実施例5】
【0069】
(エッチング試験)
サンプルのTiO2保持性を検証するためにエッチング試験を行った。サンプルとして、樹脂組成物6を前述と同様に約10mm×約10mm×約0.5mmの大きさに調製したものを用いた。エッチング試験は、テトラヒドロフラン(THF)15mLを入れた30mLのサンプル管において、該サンプルをTHFに室温で24時間浸すことで行った。エッチング後のサンプルについて、走査型電子顕微鏡(日本電子社製、商品名「JOEL JSM−5800」、20kV)を用いて、走査型電子顕微鏡(SEM)分析及び走査型電子顕微鏡/エネルギー分散型X線分析(SEM/EDS)によりTiO2保持性を検証した。Ti原子分布については、EDS(オックスフォード・インストゥルメンツ社製、INCA微量分析)を用いて調べた。SEMイメージングにおいては、30分間、27℃で乾燥させ、金でスパッタコーディングしたサンプルを用いた。結果を図11及び図12に示す。
【0070】
図11は、エッチングをしなかった場合の樹脂組成物6のサンプル表面のSEM像(図11(a),図11(c))、図11(a)の各スポットの原子百分率(図11(b))、及び図11(c)に対応するTi原子の分布像(図11(d))を示す。スポット1及び2(図11(a))において、Ti原子が分布していることが示された(図11(b))。また、図11(c)に表された略楕円形の形状(セルロースが存在する)に沿って、Ti原子が分布していることが示された(図11(d))。このことから、TiO2がFCに付着することにより保持されていることが示された。
【0071】
図12は、エッチングをした場合の樹脂組成物6のサンプル表面のSEM像(図12(a),図12(c))、図12(a)の各スポットの原子百分率(図12(b))、及び図12(c)に対応するTi原子の分布像(図12(d))を示す。スポット1,2,3及び4(図12(a))において、Ti原子が分布していることが示された(図12(b))。また、図12(c)に表された略楕円形の形状(セルロースが存在する)に沿って、Ti原子が分布していることが示された(図12(d))。このことから、エッチングした場合においても、図11と同様にTiO2が保持されていることが示された。このように、樹脂組成物6は、TiO2と親和性を有するFCを含有することにより、エッチングされて一部のPEOが溶出した場合でも、TiO2が保持されることが明らかとなった。
【実施例6】
【0072】
(生分解性試験)
光酸化劣化されたサンプルが実際に生分解されるかについて検証した。前述と同様の方法で20mm×20mm×0.05mmの大きさに調製した樹脂組成物3を用いて、埋土試験を行った。埋土試験の前に、30℃で24時間、前述と同様の方法で、サンプルを光酸化劣化させた。北見工業大学の敷地内の花壇の土が入った容器内にサンプルを置き、20℃で28日間、埋土試験を行った。該容器内の土が乾燥するのを防ぐため、埋土試験中は毎日、水をスプレーした。また、光酸化劣化させなかったサンプルについても、同様に埋土試験を行った。
埋土試験終了後、回収したサンプルを4℃で1〜2時間、0.1Mリン酸緩衝液(pH7.4)中の3%グルタルアルデヒドで固定し、0.1Mリン酸緩衝液で3回洗浄した。得られたサンプルを1時間、0.1Mリン酸緩衝液(pH7.4)中の1%オスミウム酸で固定し、エタノールシリーズ(50,70,95,及び100%、15分ごとに3回)で脱水した。サンプルを30分間、27℃で乾燥させ、金でスパッタコーディングし、前述と同様にSEMイメージングを行った。
また、埋土試験前後のサンプルについて、前述と同様にFT−IRスペクトルを測定した。
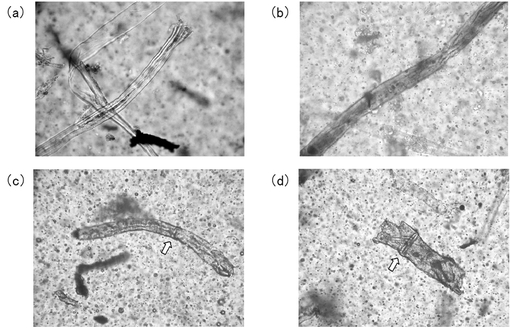
さらに、埋土試験後のサンプルを、ラクトフェノールコットン青(関東化学社製)で染色し、光学顕微鏡(ニコン社製、商品名「Nikon ECLIPSE 50/POL」)で観察した。結果を図13〜15に示す。
【0073】
図13に埋土試験後のサンプルのSEM像を示す。光酸化劣化させなかったサンプル(図13(a))では、微生物は見られなかった。一方、光酸化劣化させたサンプル(図13(b))では、微生物が見られた。
【0074】
図14に埋土試験前後のサンプルのFT−IRスペクトルを示す。光酸化劣化させなかったサンプル(図14(a))では、カルボニル基のピーク強度は低く、埋土試験前後でほとんど変化はみられなかった。一方、光酸化劣化させたサンプル(図14(b))では、埋土試験前でカルボニル基のピーク強度が増大しており、埋土試験後にこのピークが消失した。このことから、光酸化劣化させたサンプルは、埋土試験中、微生物により代謝されたことが示された。
【0075】
図15に光酸化劣化させたサンプルの埋土試験後の光学顕微鏡像を示す。埋土試験後のサンプルにカーブラリア属の菌が見られた(図15(a)〜(d))。図15(c)及び図15(d)においては、カーブラリア属に特徴的である隔壁構造が観察された(矢印)。このように、光酸化劣化させたサンプルを代謝する微生物が実際に確認された。
【0076】
以上説明したように、本発明によれば、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することができる。また、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することができる。
【技術分野】
【0001】
本発明は、樹脂組成物、成型体、樹脂組成物の製造方法、及びポリオレフィン系樹脂を光酸化劣化させる方法に関する。
【背景技術】
【0002】
地球環境保全の意識の高まりに伴い、環境調和型プラスチックの開発が求められている。現在、環境調和型プラスチックとして研究が進められているものの一つに、生分解性プラスチックが挙げられる。生分解性プラスチックは、微生物等によって代謝・分解される性質を有するため、炭素循環を可能にする材料として注目されている。
【0003】
現在、プラスチックの材料としては、優れた物性を有するポリオレフィンが汎用されている。しかしながら、高分子であるポリオレフィンは微生物の細胞膜を通過できないため、生分解性に乏しい。そこで、近年、ポリオレフィンの生分解手法(Oxo−biodegradation)が開発された。この手法では、ポリオレフィンに酸化促進剤を添加することで、ポリオレフィンを低分子にまで酸化劣化させ(非生物分解)、微生物による代謝(生分解)を可能とする。この手法を利用した生分解性プラスチックがいくつか見出され、報告されている。
【0004】
非特許文献1〜3には、ポリオレフィンの一つであるポリプロピレンに、酸化促進剤(酸化チタン)を含有するポリエチレンオキシド(PEO)マイクロカプセルを導入した生分解性プラスチックが記載されている。この生分解性プラスチックにおいては、酸化チタンの光触媒反応により、PEOが分解されて酸及びアルデヒドが発生し、この酸及びアルデヒドがポリプロピレンの酸化劣化を促進する。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】K.Miyazaki et al,Polymer Degradation and Stability,94(2009)2114−2120
【非特許文献2】K.Miyazaki et al,Polymer Degradation and Stability,95(2010)1557−1567
【非特許文献3】K.Miyazaki et al,Polymer Degradation and Stability,96(2011)1039−1046
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、非特許文献1〜3に記載の生分解性プラスチックは、低弾性率体であるPEOを含有するため、力学物性に難点を有し、また、PEOは水溶性であるため、水などに濡れるとPEOとともに酸化チタンが溶出し、酸化劣化を確実に進めることができない場合があり、実用化という点で課題を残していた。
【0007】
本発明は、上記事情に鑑みてなされたものであり、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することを目的とする。また本発明は、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
上記目的を達成するため、本発明の第1の観点に係る樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシドと、光触媒と、フィラーと、を含有する。
【0009】
前記ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有されていてもよい。
【0010】
前記フィラーは、前記樹脂組成物中に0.1質量%〜7.0質量%含有されていてもよい。
【0011】
前記光触媒は、前記樹脂組成物中に0.1質量%〜3.0質量%含有されていてもよい。
【0012】
前記フィラーはセルロースであってもよい。
【0013】
前記光触媒は酸化チタンであってもよい。
【0014】
前記ポリオレフィン系樹脂はポリプロピレン類を含有していてもよい。
【0015】
前記ポリプロピレン系樹脂はプロピレンホモポリマーを含有していてもよい。
【0016】
さらに、前記樹脂組成物100質量部に対して、光酸化劣化遅延剤を0.01質量部〜10.0質量部含有していてもよい。
【0017】
前記光酸化劣化遅延剤はアパタイト誘導体であってもよい。
【0018】
本発明の第2の観点に係る成型体は、本発明の第1の観点に係る樹脂組成物からなる。
【0019】
本発明の第3の観点に係る樹脂組成物の製造方法は、ポリエチレンオキシドと、光触媒と、フィラーと、を混練して触媒組成物を得る工程と、前記触媒組成物をポリオレフィン系樹脂と混練する工程と、を含む、ことを特徴とする。
【0020】
本発明の第4の観点に係るポリオレフィン系樹脂を光酸化劣化させる方法は、光触媒とフィラーとを含有するポリオレフィン系樹脂に、ポリエチレンオキシドを添加する、ことを特徴とする。
【発明の効果】
【0021】
本発明によれば、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することができる。また、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することができる。
【図面の簡単な説明】
【0022】
【図1】PPの光酸化劣化メカニズム(自動酸化)を表す概念図である。
【図2】(a)はサンプル厚が薄い場合、(b)はサンプル厚が厚い場合の厚さ方向への光酸化劣化の進行を表す概念図である。
【図3】(a)は樹脂組成物1、(b)は樹脂組成物2の光酸化劣化前後におけるエチレン性不飽和基及びカルボニル基の発生量を表すFT−IRスペクトルの図である。
【図4】(a)は比較組成物1、(b)は比較組成物2、(c)は樹脂組成物1、及び(d)は樹脂組成物2の光酸化劣化前後における水酸基の発生量を表すFT−IRスペクトルの図である。
【図5】(a)は樹脂組成物3、(b)は樹脂組成物4、(c)は樹脂組成物5、及び(d)は樹脂組成物6の光酸化劣化前後におけるエチレン性不飽和基及びカルボニル基の発生量を表すFT−IRスペクトルの図である。
【図6】(a)は樹脂組成物3、(b)は樹脂組成物4、(c)は樹脂組成物5、及び(d)は樹脂組成物6の光酸化劣化前後における水酸基の発生量を表すFT−IRスペクトルの図である。
【図7】光酸化劣化開始後の樹脂組成物1及び3〜6、並びに比較組成物3について、(a)はエチレン性不飽和基、(b)はカルボニル基の発生量の経時的変化を表すグラフ図である。
【図8】光酸化劣化開始後の厚さ100μmの樹脂組成物3及び6について、カルボニル基の発生量の経時的変化を表すグラフ図である。
【図9】各々厚さ50μm及び100μmの(a)樹脂組成物3及び(b)樹脂組成物6について、光酸化劣化によるカルボニル基の発生量を表すグラフ図である。
【図10】樹脂組成物1及び3〜6、並びに比較組成物3について、(a)はヤング率、(b)は引張強度、(c)は破断ひずみ(ただし、(c)については、比較組成物3のデータ無し)を表すグラフ図である。
【図11】エッチングをしなかった場合の樹脂組成物6について、(a)及び(c)はSEM像、(b)は(a)の各スポットの原子百分率、(d)は(c)に対応するTi原子の分布を表す図である。
【図12】エッチングをした場合の樹脂組成物6について、(a)及び(c)はSEM像、(b)は(a)の各スポットの原子百分率、(d)は(c)に対応するTi原子の分布を表す図である。
【図13】(a)は光酸化劣化させなかった樹脂組成物3、(b)は光酸化劣化させた樹脂組成物3について、埋土試験後のSEM像を示す図である。
【図14】(a)は光酸化劣化させなかった樹脂組成物3、(b)は光酸化劣化させた樹脂組成物3について、埋土試験前後のFT−IRスペクトルを表す図である。
【図15】(a)〜(d)は、光酸化劣化させた樹脂組成物3の埋土試験後の光学顕微鏡像を示す図である。
【発明を実施するための形態】
【0023】
以下、本発明の実施形態について詳細に説明する。
【0024】
本発明による樹脂組成物は、光酸化劣化されることにより(非生物分解)、微生物による代謝(生分解)が可能となる。
【0025】
本発明による樹脂組成物は、ポリオレフィン系樹脂と、ポリエチレンオキシド(PEO)と、光触媒と、フィラーと、を含有する。
【0026】
(1.ポリオレフィン系樹脂)
本発明による樹脂組成物に含まれるポリオレフィン系樹脂は、主としてポリオレフィンを含有する樹脂である。ポリオレフィンとしては、特に限定されるものではなく、プロピレンホモポリマー(PP)(アイソタクチック型及びシンジオタクチック型を含む)、プロピレン−エチレンブロック共重合体、プロピレン−エチレンランダム共重合体等のポリプロピレン類、高密度ポリエチレン、中密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、超低密度ポリエチレン、メタロセン触媒によるポリエチレン等のポリエチレン類、ポリプロレン、ポリブテン、ポリメチルペンテン、ポリスチレン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明において、良好な力学物性を有するという点において、ポリプロピレン類が好適に用いられ、PPがさらに好適に用いられる。PPについては、例えば、平均分子量1.0×104〜1.0×106のものが用いられる。本発明の効果を奏するポリオレフィン系樹脂であれば、適宜選択され得る。
【0027】
(2.PEO)
本発明による樹脂組成物に含まれるPEOとしては、例えば、数平均分子量2.0×103〜1.0×106のものが用いられる。本発明の効果を奏する分子量のPEOであれば、適宜選択され得る。
【0028】
PEOは、本発明による樹脂組成物中、例えば、0.1質量%〜7.0質量%含有される。PEOは、力学物性に影響を与え得る低弾性率体である一方で、後述の光酸化劣化における促進剤でもある。優れた力学物性と確実な光酸化劣化能との両方を実現するために、樹脂組成物中PEOを0.3質量%〜5.0質量%含有した樹脂組成物が好適に用いられる。さらに確実な光酸化劣化能を実現するために、樹脂組成物中PEOを1.0質量%〜5.0質量%含有した樹脂組成物がさらに好適に用いられる。
【0029】
(3.光触媒)
本発明による樹脂組成物に含まれる光触媒としては、後述の光酸化劣化の反応を触媒できるものであれば特に限定されるものではなく、酸化チタン(TiO2)(アナターゼ型、ルチル型、及びブルッカイト型を含む)、モリブデン酸化物、酸化亜鉛、酸化錫、チタン酸ストロンチウム、酸化スズ、酸化タングステン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明において、入手が容易であるという点、及び紫外線といった短い波長の光により光触媒活性を示すことができるという点で、TiO2が好適に用いられる。本発明の効果を奏する光触媒であれば、適宜選択され得る。
【0030】
光触媒は、本発明による樹脂組成物中、例えば0.1質量%〜3.0質量%、好ましくは0.1質量%〜1.0質量%含有される。樹脂組成物中0.1質量%以上含有されることで、光触媒としての機能を十分に発揮することができ、後述の光酸化劣化を確実に進めることができる。また、光触媒が樹脂組成物中3.0質量%以下含有されることで、コストを抑えた樹脂組成物が可能となる。
【0031】
(4.フィラー)
本発明による樹脂組成物に含まれるフィラーとしては、樹脂組成物の力学物性を向上させ、光触媒に親和性を有し、かつ、光酸化劣化能及び生分解能に影響を及ぼさないものであれば適宜用いることができ、特に限定されるものではなく、繊維状セルロース(以下、FCという)、タルク、ガラス繊維、カオリン等が挙げられ、これらを単独で又は組み合わせて用いることができる。本発明においては、FCが好適に用いられる。本発明による樹脂組成物にFCを用いた場合、優れた力学物性(例えば、ヤング率、引張強度、破断ひずみ等で規定される)が得られ、また、後述の通り、FCは光触媒との親和性が高く、光触媒を樹脂組成物内に保持することができるため、確実な光酸化劣化能が得られる。本発明の効果を奏するフィラーであれば、適宜選択され得る。
【0032】
フィラーは、本発明による樹脂組成物中、例えば0.1質量%〜7.0質量%含有される。優れた力学物性を実現するために、樹脂組成物中フィラーを1.0質量%〜7.0質量%含有した樹脂組成物が好適に用いられる。樹脂組成物の透明度を保ち、光の透過性を確保することで、確実な光酸化劣化能を実現するために、樹脂組成物中フィラーを1.0質量%〜5.0質量%含有した樹脂組成物がさらに好適に用いられる。
【0033】
本発明による樹脂組成物は、例えば、ポリオレフィン系樹脂としてPP、光触媒としてTiO2、及びフィラーとしてFCを含有する(以下、このような樹脂組成物を、PP/PEO/TiO2/FCという)。本発明による樹脂組成物中の各成分の含量に関して、PP/PEO/TiO2/FCの場合、例えば、樹脂組成物中PPを97.5質量%、PEOを1.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを96.5質量%、PEOを2.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを95.5質量%、PEOを3.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの、樹脂組成物中PPを93.5質量%、PEOを5.0質量%、TiO2を0.5質量%、FCを1.0質量%含有したもの等が好適に用いられる。このようなPP/PEO/TiO2/FCは、優れた力学物性及び確実な光酸化劣化能を有する。
【0034】
(5.光酸化劣化遅延剤)
本発明による樹脂組成物は、光酸化劣化遅延剤をさらに含有していてもよい。光酸化劣化遅延剤として、例えば、Ca8(HPO4)2(OOC−R−COO)(PO4)4・mH2O(コハク酸イオン含有オクタカルシウムホスフェート;OCPC)、CaHPO4・2H2O(ジカルシウムホスフェートジハイドレート;DCPC)、Ca8(HPO4)2(PO4)4・5H2O(オクタカルシウムホスフェート;OCP)等といったアパタイト誘導体が挙げられる。これらは、単独で、又は組み合わせて用いられ得る。例えば、光触媒としてTiO2を用いた場合、アパタイト誘導体はTiO2の表面に結晶を形成するため、光酸化劣化を遅延させることが可能となる。また、他の光酸化劣化遅延剤として、例えば、ヒンダードアミン等といったアミン系光安定化剤が挙げられる。ヒンダードアミンは、PEOから生じた酸と中和反応することで、光酸化劣化を遅延させることができる。なお、確実な光酸化劣化遅延効果を得るために、アパタイト誘導体をさらに含有した樹脂組成物が好適に用いられる。本発明の効果を奏する光酸化劣化遅延剤であれば、適宜選択され得る。
【0035】
本発明による樹脂組成物は、前述の光酸化劣化分解遅延剤を含むことで、時限分解性を実現することができる。例えば、樹脂組成物の時限分解性を制御することで、樹脂組成物からなる成型体の材料寿命を自在に設計することが可能となる。樹脂組成物の時限分解性の制御は、例えば、光酸化劣化遅延剤の含有量及び反応時間を調節すること等により可能となる。光酸化劣化遅延剤は、例えば、樹脂組成物100質量部に対して0.01質量部〜10.0質量部含有される。
【0036】
本発明による樹脂組成物は、上記以外に、光酸化劣化能及び生分解能に影響を及ぼさない範囲で、一般的な樹脂組成物に通常用いられる添加剤をさらに含むことができる。例えば、光吸収剤、光安定剤、帯電防止剤、滑剤、着色剤、発泡剤、可塑剤、難燃剤、架橋剤、抗菌剤、耐候劣化防止剤等が挙げられ、これらを単独又は組み合わせで用いることができる。
【0037】
(6.光酸化劣化)
一例としてPP/PEO/TiO2/FCを用いて、光酸化劣化について説明する。PP/PEO/TiO2/FCは、疎水性のPP中に、親水性のPEO、TiO2、及びFCからなる粒子状物が分散される特徴を有する。
【0038】
この樹脂組成物を構成するTiO2に光が照射されると、h+(正孔)とe−(電子)とが発生する。正孔は水と反応して、・OH(ヒドロキシルラジカル)を生成する。電子はO2と反応して、HOO・とOH−とを生成する。生成したOH−は正孔と反応して、ヒドロキシルラジカルを生成する。生成したHOO・が2分子結合して、H2O2とO2とを生成し、このH2O2に光が照射されるとヒドロキシルラジカルが生成される。このように、種々の段階でヒドロキシルラジカルが発生する。
【0039】
【化1】
【0040】
この樹脂組成物を構成するPEOは、前述で発生したヒドロキシルラジカルにより活性化され、−CH2−・CH−O−と水とが発生する。−CH2−・CH−O−はO2と反応して、−CH2−C(−OO・)H−O−が生成される。−CH2−C(−OO・)H−O−が2分子結合して、O2が発生し、それにより−CH2−C(−O・)H−O−が生成される。−CH2−C(−O・)H−O−から−CH2−COH(アルデヒド)と・O−CH2とが生成される。また、−CH2−C(−O・)H−O−から−CH2−COO−(エステル)が生成され、さらにヒドロキシラジカルにより活性化されて−CH2−COO−・CH−CH2−O−が生成される。−CH2−COO−・CH−CH2−O−が分解されて、−CO−O・−とOHC−CH2−(アルデヒド)が生成される。−CO−O・−とOHC−CH2−(アルデヒド)とから、−COOH(酸)が生成される。このように、PEOはTiO2の光触媒作用により分解され、酸とアルデヒドを生成する。また、PEOが活性化されることにより生じた水は、前述のTiO2の光触媒反応に利用される。
【0041】
【化2】
【0042】
この樹脂組成物を構成するPPは、図1に示される自動酸化のサイクルが継続することにより光酸化劣化される。PPの自動酸化は、前述で発生したヒドロキシルラジカルにより開始される。PP(図1において、RH)は、ヒドロキシルラジカルによりR・(アルキルラジカル)となる。ここで、一部のアルキルラジカルは、停止反応へと移行し、エチレン性不飽和基となる。自動酸化のサイクルが停止反応で止まってしまうと、PPはそれ以上光酸化劣化されない。一方、他のアルキルラジカルは、ROO・(ペルオキシラジカル)を経て、ROOH(ヒドロペルオキシド)となる。ヒドロペルオキシドは、光照射によってRO・(アルコシラジカル)に分解される。前述のPEO分解により発生した酸及びアルデヒドは、このヒドロペルオキシドの分解を促進する。PP鎖の末端は、アルコシラジカルを経て、カルボニル基となる。このような自動酸化のサイクルが継続することで、PPは主鎖の一端から順次分解されていく。したがって、光酸化劣化が進むにつれて、カルボニル基の発生量が増加する。また、この自動酸化のサイクルにおいて、TiO2の光触媒作用により発生したヒドロキシルラジカルは「イニシエーター」としての役割を有し、PEO分解により発生した酸及びアルデヒドは「促進剤」としての役割を有する。
【0043】
前述の自動酸化のサイクルで必要とされる光は、例えば紫外線といった短い波長の光である。また、PPの光酸化劣化に要する時間は、例えば2〜48時間、数日、数十日である。生分解が可能となる程度にまでPPが分解されればよく、光酸化劣化に要する時間は適宜選択され得る。
【0044】
本発明による樹脂組成物に含まれる光触媒は、前述の通り、フィラーに親和性を有する。このため、樹脂組成物が水などに濡れて一部のPEOが溶出した場合でも、光触媒はフィラーに保持されることにより樹脂組成物内に留まっている。光触媒は光酸化劣化に必須であるため、光触媒が保持されていることで、樹脂組成物は確実な光酸化劣化能を有する。例えば、PP/PEO/TiO2/FCを屋外で使用した場合、雨などに濡れて一部のPEOが溶出したとしても、FCに親和性を有するTiO2は溶出することなく、FCとともに樹脂内に保持されている。光酸化劣化に必須であるTiO2が保持されるため、PP/PEO/TiO2/FCは確実な光酸化劣化能を有する。なお、雨などに濡れて一部のPEOが溶出してしまったとしても、後からPEOを添加(PEO溶液を樹脂組成物に振り掛ける等)することで、光酸化劣化を促進させることができる。
【0045】
本発明による樹脂組成物は、光酸化劣化により低分子にまで分解されることにより、生分解能を有するようになる。具体的には、樹脂組成物に含まれるポリオレフィンは、光酸化劣化により分子量1,000以下程度にまで分解されることで、微生物による代謝が可能となる。
【0046】
(7.成型体)
本発明による樹脂組成物は、種々の形状に成型することができる。すなわち、本発明は、前述の樹脂組成物からなる成型体を提供するものである。成型体としては、厚さ数十μm〜数十mmのシート状物のほか、一般にポリオレフィン系樹脂として成型される種々の形状に成型することができる。本発明による成型体の厚さは、その用途により適宜選択され、例えば、数十μm〜数十cmとすることができる。
【0047】
本発明による成型体は、表面から内部に向かって光酸化劣化を進行させることができる。例えば、PP/PEO/TiO2/FCからなる成型体の場合(図2)、成型体内部に存在するTiO2が光を受けてヒドロキシルラジカルを発生させ、かつ、PEOが酸及びアルデヒドを生成させることで、光酸化劣化が進行する。
成型体が薄い場合(図2(a))、TiO2の光触媒作用により成型体内部のPEOが消費され、自動酸化が促進される。前述の通り、PEO分解により生じた酸及びアルデヒドは、自動酸化において促進剤としての役割を有するため、光酸化劣化の進行はPEO含量に依存するといえる。
一方、成型体が厚い場合(図2(b))、TiO2の光触媒作用により、まず成型体の表面近くに存在するPEOが消費される。成型体表面近くに存在するPEOが消費し尽くされたら、次に成型体表面から厚さ方向に少し離れた場所に存在するPEOが順次消費され、光酸化劣化が進行する。このように、PEOが成型体内部に分散して存在しており、成型体の厚さ方向に光酸化劣化を進めることができるため、厚い成型体を調製することが可能である。本発明による成型体は、数十cm程度にまで厚くしても、確実な光酸化劣化能を有する。
【0048】
(8.樹脂組成物の製造方法)
本発明による樹脂組成物の製造方法には限定はない。しかしながら、例えば、樹脂組成物100質量部に対して、PEO0.1質量部〜7.0質量部と、光触媒0.1質量部〜3.0質量部と、及びフィラー0.1質量部〜7.0質量部と、を混練して触媒組成物を得、次いで該触媒組成物をポリオレフィン系樹脂質83.0質量部〜99.7質量部と混練することで好適に製造することができる。
【0049】
本発明で使用する光触媒とフィラーとをPEOとともに混練し、これを乾燥させると、フィラーに光触媒が付着したものがPEO中に均一に分散した触媒組成物が得られる。この触媒組成物をポリオレフィン系樹脂に混練させると、PEOが乳化剤のように作用して、疎水性のポリオレフィン系樹脂中で該触媒組成物が均一に混練される。この際、ポリオレフィン系樹脂中で該触媒組成物は粒子状に形成され、光触媒及びフィラーは該触媒組成物の粒子中に存在する。なお、光触媒及びフィラーをPEOとともに混練する際、水を添加してもよい。水の存在によって、均一に混練することができる。この際、水を残したままポリオレフィン系樹脂と混練してもよい。PPとの混練中に水が蒸発するからである。また、ポリオレフィン系樹脂との混練時に、亜リン酸エステル系酸化防止剤といった酸化防止剤を添加してもよい。
【0050】
(9.成型体の製造方法)
本発明による成型体は、前述の樹脂組成物を従来公知の方法で成型して製造することができる。例えば、圧縮成型、押出成型、射出成型等により、用途に合わせて所望の形状、厚さ、大きさ等に成型することができる。
【0051】
(10.成型体の用途)
本発明による成型体は、プラスチック材を使用するあらゆる用途に使用可能である。例えば、土木工事資材、家具材、家電材、建材、使い捨て容器等の構造物に用いることができる。本発明による成型体は、生分解能を有するため、山間部等の回収困難な場所に設置される構造物、コスト及び技術等の問題からリサイクルが容易ではない構造物等に好適に用いることができる。また、前述の通り、本発明による成型体が光酸化劣化遅延剤を含有する場合、時限分解が可能となるため、例えば、構造物を廃材としたい時期に光酸化劣化が開始されるように、材料寿命を設計することができる。また、本発明による成型体は、熱を加えることなく光酸化劣化を進めることができる。光を照射すれば光酸化劣化を進めることができるため、0℃以下の低温環境で用いられる構造物にも用いることができる。
【0052】
(11.ポリオレフィン系樹脂を光酸化劣化させる方法)
本発明は、樹脂組成物を構成するTiO2への光照射によってヒドロキシルラジカルが発生し、このヒドロキシルラジカルによってPEOが活性化されてアルデヒド及び酸が発生し、図1に示すようにこれらが相互に作用してポリオレフィン系樹脂を継続的に光酸化劣化させるものである。PEOは、光酸化劣化の際に光触媒、フィラー、及びポリオレフィン系樹脂と共存していればよく、光触媒とフィラーとを含有するポリオレフィン系樹脂に後からPEOを添加しても、ポリオレフィン系樹脂を光酸化劣化させることができる。すなわち、本発明は、光触媒とフィラーとを含有するポリオレフィン系樹脂に、PEOを添加する、ことを特徴とするポリオレフィン系樹脂を光酸化劣化させる方法を提供するものである。この方法において使用可能な光触媒、フィラー、ポリオレフィン系樹脂、及びPEOの種類等については、前述の通りである。また、この場合の「PEOを添加する」とは、例えば、PEO水溶液を該ポリオレフィン系樹脂に振り掛ける(例えば、スプレーする)ことを意味する。
【実施例1】
【0053】
以下、実施例を挙げて本発明を具体的に説明する。ただし、本発明はこれらの実施例に限定されるものではない。
【0054】
(PP/PEO/TiO2/FCの調製)
PP(日本ポリプロ社製、メソペンタッド分率=98%、平均分子量(Mn):4.6×104、多分散性(Mw/Mn):5.7)、PEO(和光純薬工業社製、平均分子量(Mn):5.0×105)、TiO2(和光純薬工業社製、アナターゼ型(98.5%以上)、表面積:約6m2/g)、及びFC(日本製紙ケミカル社製、商品名「W−100GK」、90質量%が100メッシュの篩(150μm以下)を通過、平均長:37μm)を用いて、表1に示す組成の樹脂組成物を調製した。なお、FCは、デシケーターで7日間乾燥させ、FCの水分量が0.7質量%以下となったものを調製に用いた。
0.1Lのガラスビーカーに純水50mL、PEO0.3g、TiO20.5g、FC1.0gを入れ、24時間、23℃で、スターラーで撹拌しながら混合させた。その後、ロータリーエバポレーターを用いて、水を蒸発させた。その後、このPEO/TiO2/FC混合物を真空オーブンで24時間、50℃で乾燥させた。その後、このPEO/TiO2/FC混合物と、PP98.2g及び亜リン酸エステル系酸化防止剤(ADEKA社製、商品名「ADKSTAB PEP−36」)0.1gをミキサー(井元製作所製、商品名「IMC−1884」)に仕込み、180℃に加温し、5分間、100rpmで混練した。このようにして、100gの樹脂組成物1を調製した。
上記と同様にして、樹脂組成物2〜6を調製した。また、比較例として、上記と同様にして、比較組成物1〜3を調製した。
【0055】
【表1】
【実施例2】
【0056】
(光酸化劣化試験)
樹脂組成物1及び2を、5MPa、190℃で5分間圧縮成型し、厚さ50μmのフィルム状に成型した。各々を20mm×20mmの大きさに切断し、バイアル(パイレックス社製)に入れた。リコーロータリー光化学反応装置(理工科学産業社製、商品名「RH400−10W」)を用いて、空気中、30℃で光酸化劣化試験を行った。この光化学反応装置には、400Wの高圧水銀灯(理工科学産業社製、UVL−400HA型超高圧水銀灯(5mW/cm2))が備えられている。光酸化劣化試験前後におけるエチレン性不飽和基、カルボニル基、及び水酸基の発生量を、フーリエ変換型赤外分光(FT−IR)分析により測定した。FT−IRスペクトル測定は、FT−IRスペクトロメータ(パーキンエルマー社製、商品名「Spectrum One」)を用いて(16スキャン、分解能:2cm−1)、中赤外領域(400−4000cm−1)で行った。光酸化劣化時間は、6時間とした。なお、比較組成物1,2及び5についても、同様に水酸基の発生量を測定した。結果を図3及び図4に示す。
【0057】
図3(a)は樹脂組成物1、及び図3(b)は樹脂組成物2のFT−IRスペクトルを示す。
樹脂組成物1及び2において、光酸化劣化時間6時間では、カルボニル基のピーク(1720cm−1付近)はほとんど見られなかったものの、光酸化劣化後においてエチレン性不飽和基のピーク(1642cm−1付近)の増大がみられた。このことから、PPの自動酸化が進みつつあることが示唆された。
【0058】
図4(a)は比較組成物1、図4(b)は比較組成物2、図4(c)は樹脂組成物1、及び図4(d)は樹脂組成物2のFT−IRスペクトルを示す。図4(a)〜(d)のAは、比較組成物3のFT−IRスペクトルを示す。
比較組成物3のAでは水酸基のピーク(3350cm−1付近)が見られなかった一方で、いずれの比較組成物においても、水酸基のピークが見られた。
比較組成物1に比して、比較組成物2では水酸基のピークが高いものの、光酸化劣化前後でピーク強度に変化はみられなかった。このことから、これらのピークには自動酸化により生じるヒドロペルオキシド由来の水酸基は含まれておらず、FC由来の水酸基が含まれていることが示唆された。つまり、PEOが含まれない比較組成物1及び2では、PPの自動酸化が進行しないことが示唆された。
一方、PEOが0.3質量%含まれる樹脂組成物1では、比較組成物1よりもFC含量が少ないにもかかわらず、比較組成物1に比して水酸基のピークが増大しており、またそのピークは鋭い形状であった。このことから、このピークにはヒドロペルオキシド由来の水酸基が含まれていることが示唆された。
さらに、樹脂組成物2においても、水酸基のピークがみられ、光酸化劣化後にそのピークが増大していた。このことから、この水酸基のピークにはヒドロペルオキシド由来の水酸基が含まれていること、また、自動酸化のサイクルにおいてヒドロペルオキシドが分解されずに若干蓄積しているものの、PPの自動酸化が進みつつあることが示唆された。
以上のことから、PEOが0.3質量%含まれる樹脂組成物1及び2において、光酸化劣化開始後6時間の時点では、自動酸化が継続する程度には至っていないものの、自動酸化が進みつつあることが示唆された。
【0059】
前述において、PEO含量は0.3質量%であったが、さらにPEO含量を増やして光酸化劣化の進行を検討した。樹脂組成物1及び3〜6、並びに比較組成物3を前述と同様に、厚さ50μmに成型し、前述と同様に光酸化劣化試験を行った。光酸化劣化前後におけるエチレン性不飽和基、カルボニル基、及び水酸基の発生量を測定した。光酸化劣化時間は、6時間とした。結果を図5〜図7に示す。
【0060】
図5(a)は樹脂組成物3、図5(b)は樹脂組成物4、図5(c)は樹脂組成物5、及び図5(d)は樹脂組成物6のFT−IRスペクトルを示す。
PEO含量依存的に、エチレン性不飽和基及びカルボニル基のピーク強度が増大した。いずれにおいても、エチレン性不飽和基のピーク強度よりも、カルボニル基のピーク強度の方が大きかった。また、PEO含量依存的に、光酸化劣化前後におけるピーク強度増大の程度が大きくなった。このことから、PEO含量を増やすことで、自動酸化のサイクルにおいて、停止反応のみならず、ヒドロペルオキシド分解の方向にも反応が進み、自動酸化のサイクルが継続する状態になっていることが示唆された。
【0061】
図6(a)は樹脂組成物3、図6(b)は樹脂組成物4、図6(c)は樹脂組成物5、及び図6(d)は樹脂組成物6のFT−IRスペクトルを示す。
樹脂組成物3及び4では、水酸基のピーク強度はほぼ0であり、光酸化劣化前後でピーク強度に変化はみられなかった。このこと、並びに図5(a)及び図5(b)においてカルボニル基の発生が確認されたことから、樹脂組成物1及び2ではヒドロペルオキシドがほとんど分解され、PPの自動酸化が進行していることが示唆された。
また、樹脂組成物5及び6では、光酸化劣化後に水酸基のピーク強度増大がみられた。このことから、樹脂組成物5及び6では、自動酸化がサンプルの厚さ方向に進み、サンプル内部における自動酸化により新たに発生したヒドロペルオキシドが蓄積しつつあることが示唆された。
【0062】
光酸化劣化の間のエチレン性不飽和基(図7(a))及びカルボニル基(図7(b))の発生量を経時的に測定した。いずれにおいても、PEO含量依存的にピーク強度の増大がみられた。エチレン性不飽和基については、樹脂組成物1及び3〜6のいずれにおいても光酸化劣化開始1時間後に最大ピークがみられ、その後ピーク強度は減少した。一方で、カルボニル基については、時間とともにピーク強度が増大した。このことから、自動酸化のサイクルにおいて、停止反応への進行は光酸化劣化開始後ある程度の時間経過後の段階で止まり、その後の段階では自動酸化のサイクルが継続する状態に移行することが示唆された。
【実施例3】
【0063】
(サンプルの厚さとPEO含量との関係)
自動酸化の進行におけるサンプルの厚さとPEO含量との関係について検討した。樹脂組成物3及び6を、前述と同様の方法で厚さ100μmに成型し、前述と同様に光酸化劣化試験を行った。光酸化劣化後におけるカルボニル基のピーク強度を、樹脂組成物3と樹脂組成物6との間で比較した。光酸化劣化時間は、6時間とした。結果を図8に示す。
【0064】
光酸化劣化開始2時間後までは、樹脂組成物3に比して樹脂組成物6のピーク強度が高かった。しかしながら、光酸化劣化開始3時間後以降、樹脂組成物3のピーク強度は、樹脂組成物6のピーク強度よりも高くなった。このように、サンプルの厚さが50μmの場合(図7(b))とは異なる傾向がみられた。
このことから、サンプル厚が厚い場合、光酸化劣化開始直後では、PEO含量の多い方でより良好に自動酸化が進行するが、それ以降の段階では、PEO含量が少なくても良好に自動酸化が進むことが示唆された。PEOの分解によって酸及びアルデヒドが発生し、これらによってヒドロペルオキシドの分解が促進され、サンプルの厚さ方向に自動酸化が進行したものと考えられる。
【0065】
自動酸化の進行におけるサンプルの厚さとPEO含量との関係についてさらに詳細に検討した。樹脂組成物3及び樹脂組成物6について、各々厚さ50μm及び100μmのサンプルを前述と同様の方法で調製し、光酸化劣化反応前後におけるカルボニル基の増加量を測定した。カルボニル基濃度は、PP平均モル吸光係数(330mol l−1 cm−1)及びPP密度(0.900g/mL)を用いて、ランベルト・ベールの法則により推定した。光酸化劣化時間は、5時間とした。結果を図9に示す。
【0066】
図9(a)は樹脂組成物3、図9(b)は樹脂組成物6の光酸化劣化前後におけるカルボニル基の増加量を示す。
樹脂組成物3において、100μmでは50μmに比して約4倍のピーク強度がみられた。一方、樹脂組成物6において、50μmと100μmとの間で差はみられなかった。このことから、樹脂組成物3の厚さ100μmのサンプルでは、PEOによる自動酸化促進作用により、サンプルの厚さ方向に光酸化劣化が進行したことが示唆された。
【実施例4】
【0067】
(力学物性試験)
低弾性率体であるPEOを含有するサンプルの力学物性について検討した。樹脂組成物1及び3〜6、並びに比較組成物3について、力学試験機(島津製作所社製、商品名「SHIMAZU EZ−S」)を用いて、ヤング率、引張強度、及び破断ひずみを測定した。各サンプルを、前述と同様の方法で、30mm×5mm×0.05mmの大きさに調製した。クロスヘッド速度3mm/分、20℃で試験を行った。ヤング率は、応力−ひずみ曲線の傾き(ひずみ値約1%まで)より得た。各々10回測定による平均値を測定値とした。なお、PPのみで構成された比較組成物3は伸びが大きいため、破断ひずみについては測定不可能であった。結果を図10に示す。
【0068】
ヤング率(図10(a))については、比較組成物3に比して、FCを含有する樹脂組成物1及び3〜6では1.8倍程度増大した。引張強度(図10(b))については、比較組成物3と樹脂組成物1及び3〜6との間で大きな差はみられなかった。破断ひずみ(図10(c))については、PEO含量を多くしてもほぼ一定に保たれることが示された。このことから、本発明による樹脂組成物は、FCを含むことにより、低弾性率体であるPEOの含量を多くしても、優れた力学物性を有することが示された。
【実施例5】
【0069】
(エッチング試験)
サンプルのTiO2保持性を検証するためにエッチング試験を行った。サンプルとして、樹脂組成物6を前述と同様に約10mm×約10mm×約0.5mmの大きさに調製したものを用いた。エッチング試験は、テトラヒドロフラン(THF)15mLを入れた30mLのサンプル管において、該サンプルをTHFに室温で24時間浸すことで行った。エッチング後のサンプルについて、走査型電子顕微鏡(日本電子社製、商品名「JOEL JSM−5800」、20kV)を用いて、走査型電子顕微鏡(SEM)分析及び走査型電子顕微鏡/エネルギー分散型X線分析(SEM/EDS)によりTiO2保持性を検証した。Ti原子分布については、EDS(オックスフォード・インストゥルメンツ社製、INCA微量分析)を用いて調べた。SEMイメージングにおいては、30分間、27℃で乾燥させ、金でスパッタコーディングしたサンプルを用いた。結果を図11及び図12に示す。
【0070】
図11は、エッチングをしなかった場合の樹脂組成物6のサンプル表面のSEM像(図11(a),図11(c))、図11(a)の各スポットの原子百分率(図11(b))、及び図11(c)に対応するTi原子の分布像(図11(d))を示す。スポット1及び2(図11(a))において、Ti原子が分布していることが示された(図11(b))。また、図11(c)に表された略楕円形の形状(セルロースが存在する)に沿って、Ti原子が分布していることが示された(図11(d))。このことから、TiO2がFCに付着することにより保持されていることが示された。
【0071】
図12は、エッチングをした場合の樹脂組成物6のサンプル表面のSEM像(図12(a),図12(c))、図12(a)の各スポットの原子百分率(図12(b))、及び図12(c)に対応するTi原子の分布像(図12(d))を示す。スポット1,2,3及び4(図12(a))において、Ti原子が分布していることが示された(図12(b))。また、図12(c)に表された略楕円形の形状(セルロースが存在する)に沿って、Ti原子が分布していることが示された(図12(d))。このことから、エッチングした場合においても、図11と同様にTiO2が保持されていることが示された。このように、樹脂組成物6は、TiO2と親和性を有するFCを含有することにより、エッチングされて一部のPEOが溶出した場合でも、TiO2が保持されることが明らかとなった。
【実施例6】
【0072】
(生分解性試験)
光酸化劣化されたサンプルが実際に生分解されるかについて検証した。前述と同様の方法で20mm×20mm×0.05mmの大きさに調製した樹脂組成物3を用いて、埋土試験を行った。埋土試験の前に、30℃で24時間、前述と同様の方法で、サンプルを光酸化劣化させた。北見工業大学の敷地内の花壇の土が入った容器内にサンプルを置き、20℃で28日間、埋土試験を行った。該容器内の土が乾燥するのを防ぐため、埋土試験中は毎日、水をスプレーした。また、光酸化劣化させなかったサンプルについても、同様に埋土試験を行った。
埋土試験終了後、回収したサンプルを4℃で1〜2時間、0.1Mリン酸緩衝液(pH7.4)中の3%グルタルアルデヒドで固定し、0.1Mリン酸緩衝液で3回洗浄した。得られたサンプルを1時間、0.1Mリン酸緩衝液(pH7.4)中の1%オスミウム酸で固定し、エタノールシリーズ(50,70,95,及び100%、15分ごとに3回)で脱水した。サンプルを30分間、27℃で乾燥させ、金でスパッタコーディングし、前述と同様にSEMイメージングを行った。
また、埋土試験前後のサンプルについて、前述と同様にFT−IRスペクトルを測定した。
さらに、埋土試験後のサンプルを、ラクトフェノールコットン青(関東化学社製)で染色し、光学顕微鏡(ニコン社製、商品名「Nikon ECLIPSE 50/POL」)で観察した。結果を図13〜15に示す。
【0073】
図13に埋土試験後のサンプルのSEM像を示す。光酸化劣化させなかったサンプル(図13(a))では、微生物は見られなかった。一方、光酸化劣化させたサンプル(図13(b))では、微生物が見られた。
【0074】
図14に埋土試験前後のサンプルのFT−IRスペクトルを示す。光酸化劣化させなかったサンプル(図14(a))では、カルボニル基のピーク強度は低く、埋土試験前後でほとんど変化はみられなかった。一方、光酸化劣化させたサンプル(図14(b))では、埋土試験前でカルボニル基のピーク強度が増大しており、埋土試験後にこのピークが消失した。このことから、光酸化劣化させたサンプルは、埋土試験中、微生物により代謝されたことが示された。
【0075】
図15に光酸化劣化させたサンプルの埋土試験後の光学顕微鏡像を示す。埋土試験後のサンプルにカーブラリア属の菌が見られた(図15(a)〜(d))。図15(c)及び図15(d)においては、カーブラリア属に特徴的である隔壁構造が観察された(矢印)。このように、光酸化劣化させたサンプルを代謝する微生物が実際に確認された。
【0076】
以上説明したように、本発明によれば、優れた力学物性及び確実な光酸化劣化能を有する樹脂組成物及び成型体、並びにその樹脂組成物の製造方法を提供することができる。また、ポリオレフィン系樹脂を確実に光酸化劣化させる方法を提供することができる。
【特許請求の範囲】
【請求項1】
ポリオレフィン系樹脂と、
ポリエチレンオキシドと、
光触媒と、
フィラーと、
を含有する樹脂組成物。
【請求項2】
前記ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される、
ことを特徴とする請求項1に記載の樹脂組成物。
【請求項3】
前記フィラーは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される、
ことを特徴とする請求項1又は2に記載の樹脂組成物。
【請求項4】
前記光触媒は、前記樹脂組成物中に0.1質量%〜3.0質量%含有される、
ことを特徴とする請求項1乃至3のいずれか1項に記載の樹脂組成物。
【請求項5】
前記フィラーはセルロースである、
ことを特徴とする請求項1乃至4のいずれか1項に記載の樹脂組成物。
【請求項6】
前記光触媒は酸化チタンである、
ことを特徴とする請求項1乃至5のいずれか1項に記載の樹脂組成物。
【請求項7】
前記ポリオレフィン系樹脂はポリプロピレン類を含有する、
ことを特徴とする請求項1乃至6のいずれか1項に記載の樹脂組成物。
【請求項8】
前記ポリプロピレン系樹脂はプロピレンホモポリマーを含有する、
ことを特徴とする請求項7に記載の樹脂組成物。
【請求項9】
さらに、前記樹脂組成物100質量部に対して、光酸化劣化遅延剤を0.01質量部〜10.0質量部含有する、
ことを特徴とする請求項1乃至8のいずれか1項に記載の樹脂組成物。
【請求項10】
前記光酸化劣化遅延剤はアパタイト誘導体である、
ことを特徴とする請求項9項に記載の樹脂組成物。
【請求項11】
請求項1乃至10のいずれか1項に記載の樹脂組成物からなる成型体。
【請求項12】
ポリエチレンオキシドと、光触媒と、フィラーと、を混練して触媒組成物を得る工程と、
前記触媒組成物をポリオレフィン系樹脂と混練する工程と、
を含む、
ことを特徴とする請求項1乃至10のいずれか1項に記載の樹脂組成物の製造方法。
【請求項13】
光触媒とフィラーとを含有するポリオレフィン系樹脂に、ポリエチレンオキシドを添加する、
ことを特徴とするポリオレフィン系樹脂を光酸化劣化させる方法。
【請求項1】
ポリオレフィン系樹脂と、
ポリエチレンオキシドと、
光触媒と、
フィラーと、
を含有する樹脂組成物。
【請求項2】
前記ポリエチレンオキシドは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される、
ことを特徴とする請求項1に記載の樹脂組成物。
【請求項3】
前記フィラーは、前記樹脂組成物中に0.1質量%〜7.0質量%含有される、
ことを特徴とする請求項1又は2に記載の樹脂組成物。
【請求項4】
前記光触媒は、前記樹脂組成物中に0.1質量%〜3.0質量%含有される、
ことを特徴とする請求項1乃至3のいずれか1項に記載の樹脂組成物。
【請求項5】
前記フィラーはセルロースである、
ことを特徴とする請求項1乃至4のいずれか1項に記載の樹脂組成物。
【請求項6】
前記光触媒は酸化チタンである、
ことを特徴とする請求項1乃至5のいずれか1項に記載の樹脂組成物。
【請求項7】
前記ポリオレフィン系樹脂はポリプロピレン類を含有する、
ことを特徴とする請求項1乃至6のいずれか1項に記載の樹脂組成物。
【請求項8】
前記ポリプロピレン系樹脂はプロピレンホモポリマーを含有する、
ことを特徴とする請求項7に記載の樹脂組成物。
【請求項9】
さらに、前記樹脂組成物100質量部に対して、光酸化劣化遅延剤を0.01質量部〜10.0質量部含有する、
ことを特徴とする請求項1乃至8のいずれか1項に記載の樹脂組成物。
【請求項10】
前記光酸化劣化遅延剤はアパタイト誘導体である、
ことを特徴とする請求項9項に記載の樹脂組成物。
【請求項11】
請求項1乃至10のいずれか1項に記載の樹脂組成物からなる成型体。
【請求項12】
ポリエチレンオキシドと、光触媒と、フィラーと、を混練して触媒組成物を得る工程と、
前記触媒組成物をポリオレフィン系樹脂と混練する工程と、
を含む、
ことを特徴とする請求項1乃至10のいずれか1項に記載の樹脂組成物の製造方法。
【請求項13】
光触媒とフィラーとを含有するポリオレフィン系樹脂に、ポリエチレンオキシドを添加する、
ことを特徴とするポリオレフィン系樹脂を光酸化劣化させる方法。
【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図14】
【図1】
【図2】
【図11】
【図12】
【図13】

【図15】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図14】
【図1】
【図2】
【図11】
【図12】
【図13】
【図15】
【公開番号】特開2013−18894(P2013−18894A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2011−154275(P2011−154275)
【出願日】平成23年7月12日(2011.7.12)
【出願人】(504238806)国立大学法人北見工業大学 (80)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成23年7月12日(2011.7.12)
【出願人】(504238806)国立大学法人北見工業大学 (80)
【Fターム(参考)】
[ Back to top ]