
機能グループII莢膜遺伝子クラスターを有しないE.COLIBL21株
本発明は、グループII莢膜遺伝子クラスターの欠失を含む新規非病原性大腸菌(E,coli) B BL21株、及びペプチド製造のためのその使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、大腸菌(E.coli)ゲノムの操作、改善した非病原性E.Coli B株、並びにペプチド及びタンパク質製造のためのその使用に関する。
【背景技術】
【0002】
細菌は、広範な市販の製品の製造のために使用されている。遺伝子操作した細菌、例えば、大腸菌を宿主細胞として生物試薬、例えば、ペプチド及び核酸の生産のために実験室で使用すること及び工業的に使用することは、当該技術分野においてよく知られている。自然な環境における細菌は、標準的な工業的又は実験室における増殖で通常与えられるものではない多数の環境に曝されるため、それらのゲノムは、多数の環境依存的なストレス誘導遺伝子、又はそうでなければ非必須遺伝子を有し、それらは、前記生物の工業的又は実験室における使用には必要とされない可能性があり、又は望ましくない可能性すらある。
【0003】
細菌性病原体は、毒性因子、例えば、細胞表面における莢膜多糖類を含み、それは、細菌とその周囲の環境との間の相互作用を媒介し、細菌の病原性に直接関連する多数の機能を有する。80を超える血清学的及び化学的に異なるタイプの多糖類莢膜が、E.Coliについて開示されており、K抗原と称されている。多糖類莢膜は、4つのグループに分類され、そのうちグループII莢膜多糖類は、インフルエンザ菌及び髄膜炎菌のものと類似している。グループII莢膜遺伝子クラスターは、侵襲性疾患と関連する莢膜のタイプの大半を含み、3つの機能的領域からなる保存された遺伝子構成を有する。領域1及び3は、グループII遺伝子クラスターの間で保存されており、合成部位から細胞表面に多糖類を輸送するために必要なペプチドをコードしており、領域2は、血清型特異的であり、特定の多糖類を含む個々の単糖類の生合成及び重合に関与する酵素をコードしている。領域1は、6つの遺伝子のkpsFEDUCSを含み、それらは1つの転写単位に構成されている。領域2は、血清型特異的であり、グループ2抗原の間で異なる。領域3は、2つの遺伝子のkpsM及びkpsTを含み、それらは1つの転写単位に構成されている[Andreishcheva et al. Gene (2006) 384: 113-119]。
【0004】
E.ColiのK−12株及びB株は、莢膜を有さず、ペプチドの製造に一般的に使用される株である。E.Coli K−12のゲノムは、完全に配列決定されており、莢膜遺伝子を全く含まない。組換えペプチドの高収量発現のための好ましい株であるE.coli B BL21(DE3)は、グループII莢膜多糖類をコードする遺伝子クラスターを含むことが認められており、E.coli Bの祖先が水平伝播によってこれらの莢膜遺伝子を獲得したに違いないと解されている。E.coliの莢膜に位置するK抗原は、遺伝子の機能形態を有する株の病理表現形と関連する場合が多い。E.coli B BL21では、グループII莢膜遺伝子クラスターの領域2が、挿入エレメント(IS1)によって不活性状態になっており、抗原の生産が妨げられているが、野生型の莢膜抗原の排出及び提示を補助する領域1及び3はインタクトなままである[Andreishcheva et al. Gene (2006) 384: 113-119]。しかしながら、機能的なグループII莢膜遺伝子クラスターの再構築の可能性が理論的にはあり、例えば、領域2のIS1が無くなり、それによって、グループIIクラスターが再構築される可能性がある。代替的には、IS1変異領域が外来性の野生型の領域2と交換されると、グループIIクラスターも再構築される可能性がある。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Andreishcheva et al. Gene (2006) 384: 113-119
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、グループII莢膜遺伝子クラスターの永久的なノックアウト(KO)を有するE.coli B BL21(DE3)株を提供し、当該株を他の商業的に重要な株、例えば、E.coli K−12と同じ安全な種類にする。
【課題を解決するための手段】
【0007】
本発明は、不可逆的に不活性化されたグループII莢膜遺伝子クラスターを有するE.coli B BL21株に関する。
【0008】
1つの実施態様では、E.coli B BL21株は、遺伝子II莢膜遺伝子クラスターの全体又は一部の欠失を有することを特徴とする。
【0009】
1つの実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの全体の欠失を有することを特徴とする。
【0010】
1つの実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの一部の欠失を有することを特徴とする。
【0011】
さらなる実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの1又は複数の点変異を有することを特徴とする。
【0012】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも5%が欠失によって永久的に不活性化されていることを特徴とする。
【0013】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも25%が欠失によって永久的に不活性化されていることを特徴とする。
【0014】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも40%が欠失によって永久的に不活性化されていることを特徴とする。
【0015】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも55%が欠失によって永久的に不活性化されていることを特徴とする。
【0016】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの75から100%が欠失によって永久的に不活性化されていることを特徴とする。
【0017】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも85%が欠失によって永久的に不活性化されていることを特徴とする。
【0018】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも95%が欠失によって永久的に不活性化されていることを特徴とする。
【0019】
1つの実施態様では、グループII莢膜遺伝子クラスターの30%が欠失されている。
【0020】
他の実施態様では、グループII莢膜遺伝子クラスターの50%が欠失されている。
【0021】
他の実施態様では、グループII莢膜遺伝子クラスターの70%が欠失されている。
【0022】
他の実施態様では、グループII莢膜遺伝子クラスターの80%が欠失されている。
【0023】
他の実施態様では、グループII莢膜遺伝子クラスターの90%が欠失されている。
【0024】
1つの実施態様では、E.coliの株がE.coli B BL21(DE3)であり、さらなる実施態様では、E.coliの株がE.coli B BL21(DE3)ΔdadX Δalrである。
【0025】
本発明は、
a.選択マーカーによってグループII莢膜遺伝子クラスターを置換する工程、
b.選択マーカーを除去する工程、
c.PCR分析によって欠失を確認する工程
を含む方法によってグループII莢膜遺伝子クラスターが欠失される、E.coli B株の製造方法も企図する。
【0026】
1つの実施態様では、選択マーカーは抗生物質耐性遺伝子である。
【0027】
他の実施態様では、抗生物質耐性遺伝子は、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン、及びエリスロマイシンからなる群から選択される。
【0028】
さらなる他の実施態様では、抗生物質耐性遺伝子はクロラムフェニコールである。
【0029】
本発明は、本発明のE.coli株に、ペプチドをコードするDNA配列を含む発現ベクターを導入することによって、ペプチドを製造するための方法にも関する。
【0030】
1つの実施態様では、本発明は、ペプチドをコードするDNAの発現のための条件で適切な培養培地において、本発明に係るE.coli B株を培養する工程を含む、ペプチドの製造方法も企図する。発現ベクターで発現するペプチドは、E.coliで発現することができる任意のペプチドであってよい。そのようなペプチドの代表例は、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、並びに酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアントである。
【0031】
1つの実施態様では、本発明のE.coli株は、ペプチドをコードするDNA配列を含む発現ベクターを含む。
【0032】
1つの実施態様では、製造するペプチドはhGHである。
【0033】
他の実施態様では、製造するペプチドはFGF−21である。
【0034】
他の実施態様では、製造するペプチドはアミリンである。
【0035】
他の実施態様では、製造するペプチドはレプチンである。
【0036】
他の実施態様では、製造するペプチドはシアリダーゼである。
【図面の簡単な説明】
【0037】
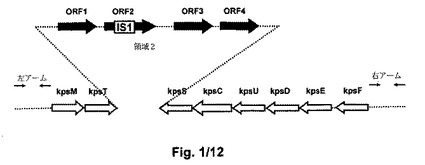
【図1】図1は、非コード隣接領域を有するE.coli B BL21(DE3)莢膜遺伝子クラスターの遺伝子構成を示す。B BL21(DE3)莢膜遺伝子クラスターのオープンリーディングフレーム(ORF)(大きい矢印);転写方向(矢頭);領域1遺伝子(灰色の矢印);領域2のORF(黒色の矢印);領域3の遺伝子(白色の矢印);IS1エレメント(白色ボックス)[Andreishcheva et al. Gene (2006) 384: 113-119]。小さい矢印は、非コード左アーム(LA)及び右アーム(RA)の位置を示す。
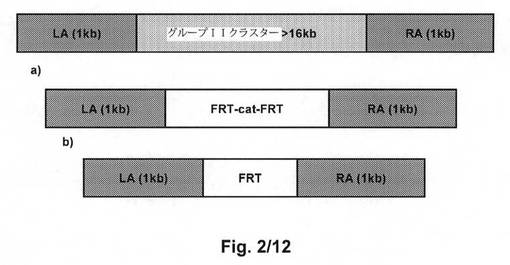
【図2】図2は、グループII莢膜遺伝子クラスターノックアウト方法:a)抗生物質耐性遺伝子によるグループII莢膜遺伝子クラスターの置換、及びb)抗生物質耐性遺伝子の除去のワークフローの略図である。
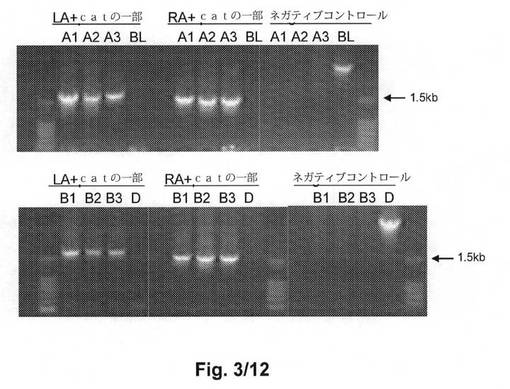
【図3】図3は、ノックアウト候補のPCRスクリーニングを示す。ノックアウト候補(親株ではない)(BL又はD)から増幅した、正しいサイズである1686bp(LA+catの一部、上の図のA1からA3、下の図のB1からB3、及び1578bp(RA+catの一部、上の図のA1からA3、下の図のB1からB3)は、グループII遺伝子クラスターのノックアウトが成功したことを示す。陰性対照については、親株のみであると約3kbのサイズのバンドを有するが、グループIIノックアウト株ではそうではない。BLはBL21(DE3)を示し、DはBL21(DE3)ΔdadXΔalr株を示す。A1からA3は、異なるコロニーナンバーのBL21(DE3)ノックアウト候補であり、B1からB3はBL21(DE3)ΔdadXΔalr株のノックアウト候補である。
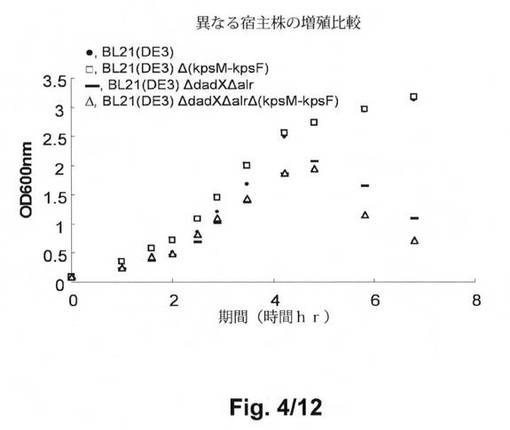
【図4】図4は、振盪したフラスコ中の宿主E.coli B株の増殖の比較である。使用する記号は、以下の株を示す:●:BL21(DE3);□:BL21(DE3)Δ(kpsM−kpsF);−:BL21(DE3)ΔdadXΔalr;△:BL21(DE3)ΔdadXΔalr Δ(kpsM−kpsF)。
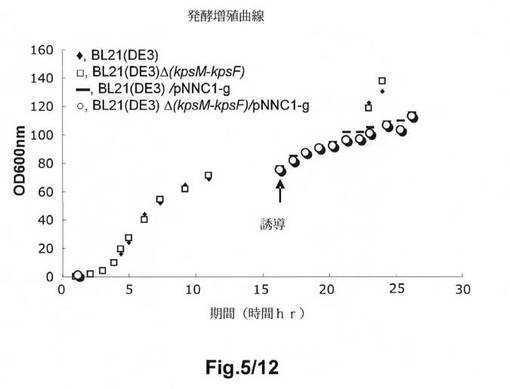
【図5】図5は、異なるE.coli B株の発酵増殖曲線を示す。使用する記号は、以下の株を示す:◆:BL21(DE3);□:BL21(DE3)Δ(kpsM−kpsF);−:BL21(DE3)/pNNC1−g;○:BL21(DE3)Δ(kpsM−kpsF)/pNNC1−g。
【図6】図6は、異なる誘導時点におけるhGH発現のSDSポリアクリルアミドゲル電気誘導(PAGE)を示す:a)E.coli B BL21(DE3)/pNNC−1gにおける発現、及びb)E.coli B BL21(DE3)Δ(kpsM−kpsF)/pNNc1−gにおける発現。a)及びb)の双方において、レーン1から3は、hGHの異なる濃度:5μg、2.5μg、及び1.25μgの各々を示す。レーン4は誘導前のhGHの発現を示し、レーン5から14は、1から10時間の毎時の異なる誘導時点におけるhGHの発現を示す。全てのサンプルは、同じロードODに対して正規化した。
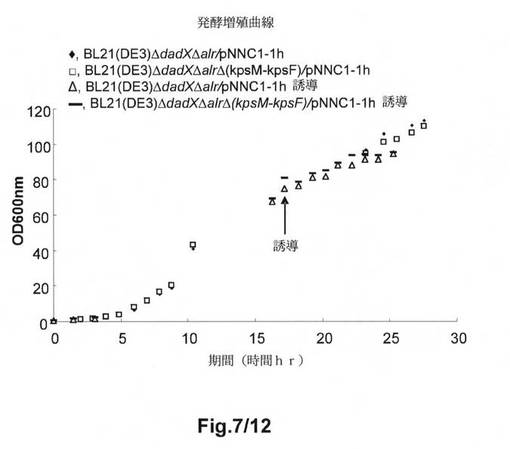
【図7】図7は、hGH発現の誘導の前及び後の異なるE.coli B株の増殖を示す。使用する記号は、以下の株を示す:◆:BL21(DE3)ΔdadXΔalr/pNNC1−1h;□:BL21ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1h;Δ:BL21(DE3)ΔdadXΔalr/pNNC1−1h誘導;−:BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1h誘導。
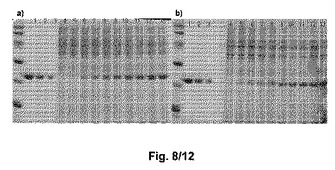
【図8】図8は、異なる誘導時点におけるSDS−PAGEによって分析したhGHの発現を示す:a)E.coli B BL21(DE3)ΔdadΔalr/pNNC1−1h;b)E.coli B BL21(DE3)ΔdadΔalrΔ(kpsM−kpsF)/pNNC1−1h。a)及びb)の双方において、レーン1から3は異なる濃度:5μg、2.5μg、及び1.25μgの各々におけるhGHの発現を示す。レーン4は誘導前のhGHの発現を示し、レーン5から13は1から9時間の毎時の誘導時点におけるhGHの発現を示す。全てのサンプルは、同じロードODに対して正規化した。
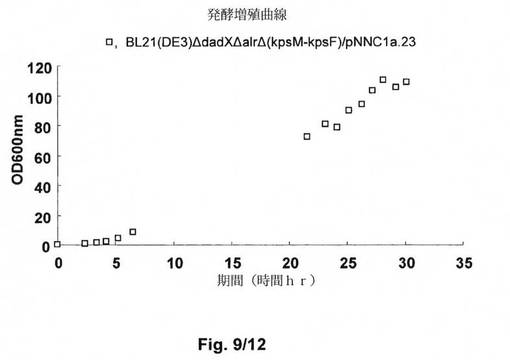
【図9】図9は、FDM1−2培地における高細胞密度発酵におけるE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1a.23の増殖曲線を示す。hGH変異体であるMEAE−hGH(L101C)の発現は、細胞密度が70OD600に達した際に、0.2mMの終濃度までIPTGを添加することによって誘導し、25℃で培養し続けた。最終的な細胞密度は、誘導の8時間後に約100OD600に達した。
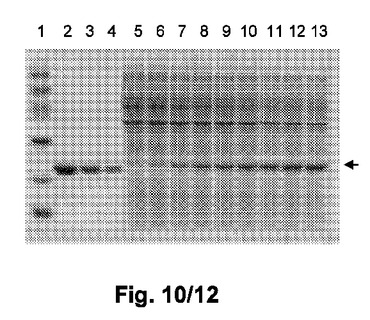
【図10】図10は、高細胞密度発酵の間のMEAE−hGH(L101C)の発現を示す。レーン1:マーカー;レーン2から4:5、2.5、1.25μgの各々の濃度の標準としての精製hGH;レーン5から13:0、1、2、3、4、5、6、7、及び8時間の各々の間にわたって誘導した後の異なる時間間隔で得たサンプル。矢印は標的タンパク質であるMEAE−hGH(L101C)を示す。
【図11】図11は、E.coli B BL21(DE3)Δ(kpsM−kpsF)におけるアースロバクター ウレアファシエンス(Arthrobacter ureafaciens)シアリダーゼの発現のための発酵サンプルのSDS−PAGE分析を示す。レーン1:マーカー;レーン2:誘導前のホモジネート;レーン3:誘導後のホモジネート;レーン4:最終サンプルのホモジネート;レーン5:誘導前の不溶性画分;レーン6:誘導後の不溶性画分:レーン7:最終サンプルの不溶性画分:レーン8:誘導前の可溶性画分;レーン9:誘導語の可溶性画分;レーン10:最終サンプルの可溶性画分;レーン11から12:標準シアリダーゼ(150及び300mg/l)。全てのサンプルは10倍希釈した。
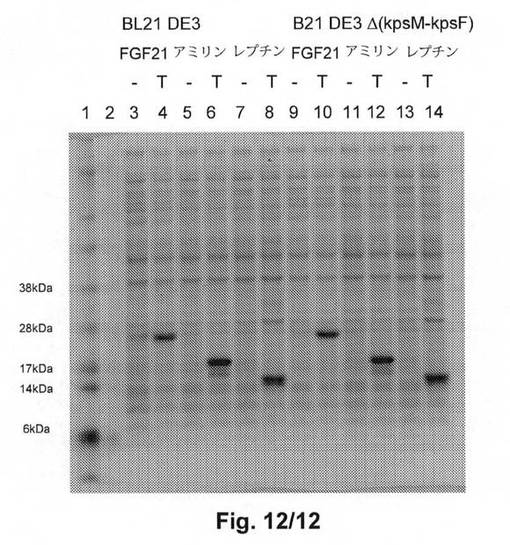
【図12】図12は、E.coli B BL21 DE3及びE.coli B BL21(DE3)Δ(kpsM−kpsF)のヒト由来FGF21、アミリン、及びレプチンの発現を示す。レーン1:マーカー;レーン3から8:BL21 DE3における発現;レーン9から14:BL21 DE3Δ(kpsM−kpsF)における発現;レーン3及び9:非誘導;レーン4及び10:誘導したFGF21;レーン5及び11:非誘導;レーン6及び12:誘導したアミリン;レーン7及び13:非誘導;レーン8及び14:誘導したレプチン。
【発明を実施するための形態】
【0038】
本発明は、不可逆的に不活性化したグループIIクラスターを有するE.coli B BL21株に関連し、ペプチド製造のための永久的に安全な株を提供する。
【0039】
E.coli B BL21(DE3)の野生型又は任意の他の株のグループII莢膜遺伝子クラスターの損傷又は不活性化は、クラスター全体又はその一部の欠失を含む任意の適切な方法又は遺伝子クラスターの1又は複数の点変異によって達成されてよい。
【0040】
本発明のE.coli株は、一般的に産業的に使用されていた株、例えば、E.coli K−12と比較して、ペプチド製造のためのより着実かつ高収量な方法を提供するという利点を有する。
【0041】
本発明で使用するE.coli B BL21(DE3)は、WO2008/025744に記載のダブルノックアウト(2×KO)であってよい。このE.coli B BL21(DE3)株は、双方がD,L−アラニンラセマーゼをコードするalr及びdadXの2遺伝子を欠失している。このダブルノックアウト株は、D−アラニンの増殖培地に対する添加又は必要なD−アラニンを供給することができるプラスミド上のラセマーゼ遺伝子の1つの存在のいずれかが無ければ増殖することができない。この2×KO D−アラニン栄養要求性E.coli株は、本明細書においてE.coli. B BL21(DE3)ΔalrΔdadXと称され、細胞中のプラスミドの維持のための非抗生物質選択方法を可能にする。
【0042】
本発明の1つの実施態様では、野生型E.coli B BL21(DE3)又はE.coli B BL21(DE3)ΔalrΔdadX株のグループII莢膜遺伝子クラスター内の1又は複数の遺伝子を欠失する。E.coli株B BL21(DE3)はStudier and Moffat[J.Mol.Biol. (1986) 189:113-130]及びStudier et al[Methods in Enzymology (1990) 185:60-89]に開示されており、Stratagene及びNovagenから入手可能である。
【0043】
本発明の1つの実施態様では、野生型E.coli B BL21(DE3)のグループII莢膜遺伝子クラスター内の1又は複数の遺伝子が欠失される。本発明の他の実施態様では、E.coli B BL21(DE3)ΔalrΔdadXのグループII莢膜遺伝子クラスター内の1又は複数の遺伝子が欠失される。
【0044】
他の実施態様では、グループII莢膜遺伝子クラスター内の欠失に選択される1又は複数の遺伝子は、潜在的な病原性に関与するものである。16kbのグループII莢膜遺伝子クラスター全体の欠失は、病原性の莢膜の表現型の潜在的な再構築の予防を提供する。図1は、領域1遺伝子kpsFEDUCS、領域2 ORF1−4、及び領域3 kpsM及びkpsTを含む莢膜を有する株のグループII莢膜遺伝子クラスターに含まれる遺伝子を示す。これらの遺伝子のいずれか1つ又はそれらの組合せが欠失されてよい。
【0045】
グループII莢膜遺伝子クラスター全体の欠失は、3つの領域の欠失、すなわち、Δ(kpsM−kpsF)を意味する。得られるE.coli B株は、E.coli B BL21(DE3)Δ(kpsM−kpsF)及びE.coli B BL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)である。
【0046】
1つの実施態様では、本発明は、挿入エレメント(IS1)は別として、グループII遺伝子クラスターを不活性化するのに十分な任意の修飾を企図する。
【0047】
細菌は1倍体生物であり、すなわち、それらは1つのみの染色体を有するため、組換えはたいてい、環境から新規DNAを獲得した際にのみ、例えば、形質転換を通じて生じる。さらに、例えば、線状DNAを分解する細胞内エキソンクレアーゼのため、細菌は、線状DNAでは容易に形質転換できない。しかしながら、組換え複合体のエキソヌクレアーゼを欠いている、組換え能力がある変異体は、線状DNAで形質転換をすることができる。リコンビナーゼは、天然の組換え反応を触媒する酵素であり、λ Redは、他の組換え系と比較した際に組換え速度の向上を示すことが示されている。本発明の株のグループII莢膜遺伝子クラスターの欠失方法は、Datsenko and Wanner[(2000) PNAS 97(12): 6640-6645]によって開発されたRed媒介組換え技術又は他の従来の方法論に基づいてよい。
【0048】
図2は、本発明の株の16kbのグループII莢膜遺伝子クラスターの欠失方法の略図を示す。1つの実施態様では、前記方法の第一工程は、ヌクレオチド相同性伸長部(nucleotide homolgy extension)又はRA及びLA領域(図1参照)とともにプライマーを使用することによるPCRによって生成される、選択マーカーを使用する、16kbのグループII莢膜遺伝子クラスターの置換である。これは、これらの隣接相同性伸長部におけるRed媒介組換えによって達成される。選択の後の、本方法の第二工程は、選択マーカーに隣接する直接反復FRT(FLP認識標的)部位に作用するFLPリコンビナーゼを発現するヘルパープラスミドを使用することによる選択マーカーの除去である。Red及びFLPヘルパープラスミドは、温度感受性レプリコンであるため、37℃における増殖によって取除く。第三工程は、PCR分析による欠失の確認を含む。
【0049】
細菌の形質転換は、DNAを取込むことによる安定な遺伝子変化と称されてよい。コンピテンスは、外来性DNAを取込むことができる状態を示す。ある細菌は、天然に、実験室条件下でDNAを取込むことができ、そのような細菌は、細胞の1又は複数の膜を越えてDNAを輸送するための機構を特定する遺伝子セットを有するが、他のものは、天然には通常生じない条件を使用して、細胞が受動的にDNA透過性にされる実験手法によって誘導しなければならない。二価陽イオン、例えば、Ca2+(CaCl2中)の存在下で細胞を冷却することは、プラスミドDNAに透過性になるように細胞壁を調製する。細胞をDNAとともに氷上でインキュベートし、次いで、短くヒートショックを与え(例えば、30から120秒間、42℃)、DNAを細胞内に入れる。これは当該技術分野においてよく知られた方法である[Sambrook et al., A Laboratory Manual (1989) CSH]。エレクトロポレーションは、細菌(及び他の)細胞に穴を作製する他の方法であり、それらにkV範囲の電場で短い間でショックを与えることによる。この穴を通って、プラスミドDNAは細胞に入ることができる。この方法は、大きなプラスミドDNAの使用に適している。天然の膜修復機構は、ショックの後に穴を直ぐに閉じるであろう。細胞に維持及び安定に保持するために、プラスミドDNA分子は、染色体からは独立して細胞内で複製することを可能にする複製起点を含む必要がある。
【0050】
本明細書に記載の任意の遺伝子不活性化方法を使用する際に、グループII遺伝子クラスターの一部又は全体の任意の標的とした不活性化が選択マーカーによって検出されて、修飾されたE.coli B BL21株クローンが同定及び単離される。
【0051】
選択マーカーは、任意の適切なマーカー遺伝子、例えば、抗生物質耐性遺伝子又は栄養要求性マーカーであってよい。
【0052】
適切な抗生物質耐性遺伝子は、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン及びエリスロマイシン若しくはβ−ラクタムの任意の他の代表例、アミノグリコシド、グリコペプチド、又はマクロライド抗生物質である。
【0053】
本明細書で使用する用語「クローニング」は、DNAの配列の多数の同一のコピーの作製を伴う。標的DNA配列は、次いで、クローニングベクターに挿入される。このベクターは自己複製ウイルス、プラスミド、又は高等生物細胞に由来するため、適当なサイズのDNAが挿入される際は、「標的及びベクターDNA断片を、次いで、連結し」、組換えDNA分子を作製する。次いで、形質転換後に幾つかの同一のコピーを生産する細菌株(通常はE.coli)に組換えDNA分子を入れる。形質転換は、細菌が有するDNA取込み機構である。しかしながら、所定の複製起点を有する1つの組換えDNA分子のみが、1つの細菌細胞内で確立することができ、そのため、各クローンは1つのDNAインサートのみを有する。
【0054】
本明細書で使用する用語「ゲノム」又は「遺伝的構成物」は、遺伝することが可能であり及び/又はそうでなければ伝達性である、遺伝的物質全体、その構造及び機能の双方を指し、特定の細胞又は微生物内に含まれている。微生物のゲノム(遺伝的構成物)は、かくして、染色体DNA及び染色体外DNA(例えば、プラスミド及びR因子など)を含むと解される。
【0055】
本明細書で使用する用語「約」は、所定の数値の合理的な近傍、例えば、±10%を意味する。
【0056】
本明細書で使用する用語「ノックアウト」は、1つ又は複数の遺伝子が欠失又は不活性化されている(生物から「ノックアウト」されている)、遺伝子操作された細菌株を意味する。ノックアウト生物は、配列決定されているが、未知の機能を有する遺伝子の役割を研究するためのモデルとして通常は使用される。ノックアウト技術を使用して、興味のある生物から望ましくない遺伝子及び/又は潜在的な病原性遺伝子を除去してよい。
【0057】
本明細書で使用する用語「遺伝子クラスター」は、遺伝的及び機能的に連関した2以上の遺伝子のセットを意味する。共通の祖先に由来する集団は同じ遺伝子クラスターの種を有する傾向があり、細菌の進化の過程を遡るのに有用である。
【0058】
用語「親株」は、縮小したゲノムの株の由来である任意の細菌株であってよい。本発明に係る親株の代表例は、E.coli株、例えば、B若しくはC、又はそれらと実質的に同一のゲノム配列を有する株を含むが、それらに限らない。
【0059】
所定の細菌宿主細胞で製造されるペプチドの「レベル」又は「量」なる本明細書で使用される用語は、選択した製造方法に従って、細胞培養物中に存在する又は検出可能なペプチドの定量的尺度であってよい。例えば、選択したペプチド製造方法が細胞自体の内部におけるペプチドの蓄積をもたらす場合には、製造されたペプチドのレベル又は量は、細胞自体の内部に存在するペプチドの定量的尺度である。ペプチド製造方法がペプチドの分泌を伴う場合には、ペプチド製造レベルは、増殖培地に存在するペプチドの定量的尺度であろう。
【0060】
ペプチドの「収量」なる本明細書で使用する用語は、本質的に純粋なペプチドの定量値であり、ここでいうペプチドは、宿主株と天然に関連を有するペプチドから本質的に遊離して回収及び/又は精製されている。細菌宿主細胞、例えば、E.coli B BL21株によって製造されたペプチドの定量的な「収量」又は「量」を確認する際に、従来のペプチド特異的な方法、例えば、イムノアッセイ、高速液体クロマトグラフィー(HPLC)、酵素活性、分光光度法、及びSDS−PAGEなどを使用することができる。
【0061】
本明細書で使用する用語「発現調節配列」は、作動可能に連結した核酸の転写を管理する転写因子結合部位の配列又はプロモーターを意味する。
【0062】
二本鎖核酸に関連して使用する用語「遊離端」は、平滑遊離端若しくは付着遊離端又はそれらの組合せを有する線状核酸を意味してよい。
【0063】
本明細書で使用する用語「遺伝子」は、ポリペプチド又はその前駆体をコードする核酸配列を含む核酸(例えば、DNA又はRNA)である。ポリペプチドは、全長又は断片の所望の活性又は機能的性質(例えば、酵素活性、リガンド結合、シグナル伝達、抗原性など)が維持されている限りにおいて、コード配列の全長又はコード配列の任意の一部によってコードされ得る。前記用語は、全長mRNAに転写される遺伝子に寄与する、5’及び3’末端の双方のコード領域に隣接して位置する配列も包含する。用語「遺伝子」は、遺伝子のcDNA及びゲノム形態(genomic form)を含む。遺伝子のゲノム形態又はクローンは、例えばイントロンと称される非コード配列で中断されるコード領域を含んでよい。
【0064】
用語「導入する」又は「導入された」は、株への核酸の添加と関連して使用される際には、核酸が、株の染色体に組み込まれるか又はベクター、例えば、プラスミド上で株に含まれることを意味する。
【0065】
本明細書で使用する用語「オープンリーディングフレーム」又は「ORF」は、終止コドンを含まない遺伝子配列である。
【0066】
用語「ペプチド」、「ポリペプチド」、又は「タンパク質」は、本明細書において互換的に使用され、天然又は組換えのいずれかの、ペプチド、ポリペプチド、及びタンパク質、並びにその断片、誘導体、ホモログ、バリアント、及び融合体を意味する。
【0067】
グループIIノックアウト株の増殖は、振盪したフラスコ中及び高密度発酵槽中で、それらの親株と比較して評価した。実験によって、グループIIノックアウトは、いずれの株の背景においても細胞増殖に影響を与えないことが示された。高密度発酵槽において、発現試験をするために、複数のペプチドを有する発現ベクターでグループII欠失株を形質転換した。
【0068】
1つの実施態様では、本発明のE.coli B株において製造するためのペプチドは、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアント、並びに任意の他のペプチド又はE.coliで発現系が作動可能であるペプチドからなるが、それらに限らない群から選択されてよい。
【0069】
本発明は、上述のヒトペプチドの製造方法にも関する。一般的には、クローン化した野生型ペプチドの核酸配列は、所望のタンパク質をコードするように修飾される。この修飾配列は、次いで、発現ベクターに挿入され、次に宿主細胞に形質転換又は形質移入される。作動可能な発現系は、典型的には、Studier and Moffat[J.Mol.Biol. (1986) 189:113-130]及びStudier et al.[Methods in Enzymology (1990) 185:60-89]に記載の従来の遺伝子発現成分を含む。
【0070】
宿主細胞中で製造するペプチドは、融合ペプチドとして合成されるか、培地中又は細胞膜周辺腔に分泌されるか、細胞質中に遊離の形態で製造されるか、又は細胞内構造体、例えば、封入体に蓄積される。同様に、ペプチド精製は、興味のあるペプチドに対して従来技術によって適合される、上述の従来の一般的に利用可能な方法の使用を伴う。
【0071】
本発明は、任意のタイプのペプチドの製造に適している。
【0072】
1つの実施態様では、グループIIノックアウトE.coli B BL21宿主細胞中における製造に選択されるペプチドは、ヒト成長ホルモン(hGH)であり、1つの実施態様では、ヒト成長ホルモン(hGH)は、hGH前駆体からhGHを作製する方法を開示するEP217814に記載の前駆体ペプチドから製造される。
【0073】
さらなる実施態様では、成長ホルモンは、hGHアナログの前駆体であるMEAE(SEQ ID 16)−hGH(L101C)であり、hGH分子の101位のロイシンがシステインによって置換されている。
【0074】
他の実施態様では、グルカゴン様ペプチドは、GLP−1、GLP−1、及び/又はその誘導体である。
【0075】
本明細書で使用する用語「グルカゴン様ペプチド」は、グルカゴンファミリーのペプチド、エキセンジン、及びそのアナログを意味する。グルカゴンファミリーのペプチドは、プレグルカゴン遺伝子によってコードされ、高い相同性を有する3つの小さなペプチド、すなわち、グルカゴン(1−29)、GLP−1(1−37)、及びGLP−2(1−33)を含む。エキセンジンは、トカゲで発現しているペプチドであり、かつ、GLP−1のようにインスリン分泌を促進する。エキセンジンの例は、エキセンジン−3及びエキセンジン−4である。
【0076】
本明細書で使用する用語「グルカゴン様ペプチドアナログ」は、修飾されたグルカゴン様ペプチドであり、グルカゴン様ペプチドの1又は複数のアミノ酸残基が他のアミノ酸残基によって置換されており、及び/又は1又は複数のアミノ酸残基がグルカゴン様ペプチドから欠失されており、及び/又は1又は複数のアミノ酸残基がグルカゴン様ペプチドに付加されている。そのようなアミノ酸残基の付加又は欠失はペプチドの任意の位置で起こり得る。
【0077】
実験
株のグリセロールストックを調製するために、適当な抗生物質を含む2mlのLB培地を含む12ml試験管に1つのコロニー移して、220rpmで振盪しながら2時間にわたって37℃で増殖させた。
【0078】
発酵種菌を調製するために、グリセロールストックの細胞を、アンピシリンを含むか又は含まないLBプレートにストリークする。一晩の後に細胞をLB培地で洗浄して、各々適当な抗生物質濃度を有する100mlの発酵複合培地を含む500ml振盪フラスコに植菌して、37℃において220rpmで浸透しながら3〜4時間にわたって増殖させた。次いで、約0.5 OD600の濃度に細胞をリアクターに植菌した。
【0079】
適当な抗生物質を含む2.5Lの初期容量の発酵複合培地を含む5Lバイオリアクターにおいて、好気条件下で発酵を実施した。0時点において、リアクターは約0.5のOD600に植菌した。5N NH3及びH2SO4を使用してpHを7.0に維持した。6ml/分の一定空気流速を培養期間に培養物に拡散させた。温度は37℃に維持した。20%の飽和O2まで溶存酸素レベルを維持することによって、攪拌をカスケード制御した。
【0080】
フェドバッチ実験を実施して、高密度発酵槽において、グループII欠失株の増殖及びペプチド発現を親株と比較した。27g/lのグリセロールを添加した複合培地を発酵槽に入れた。0時点において、発酵槽は0.5のOD600に植菌した。溶存酸素の増加によって示された、基礎のグリセロールの摂取に伴って、工程を増加する際にグリセロール及び酵母抽出物を連続的に添加した。OD600及びタンパク質発現を測定するためのサンプルは、発酵槽から回収して、分光光度計及びSDS−PAGEによって分析した。
【0081】
細菌中のタンパク質発現の誘導は、当該技術分野においてよく知られている。本発明では、タンパク質発現の誘導はイソプロピル−β−D−チオガラクトピラノシド(IPTG)の添加によって実施する。
【0082】
本発明のある実施態様で使用するプラスミド及び株を、表1及び2の各々に記載する。プラスミドの作製は、Sambrook et al.[A Laboratory Manual (1989) CSH]に記載された標準的な分子生物学的技術を使用して実施した。
【0083】
【表1】

【0084】
【表2】
【0085】
【表3】
【実施例】
【0086】
実施例1:置換カセットの構築
PCR増幅のためのDNAの起源として、E.coli B BL21(DE3)株を使用した。オリゴヌクレオチドプライマー(LA_BL_GII_F/LA_BL_GII_R)(表3)を合成して、SacII/EcoRIクローニング部位を含むKpsMの5’末端の上流の1001bp領域を標的とした。左アーム(LA)として、ゲノムDNAから当該領域を増幅した。右アーム(RA)に関しては、オリゴヌクレオチドプライマー(RA1_BL_GII_F/RA1_BL_GII_R)(表3)を合成して、BamHI/SacIクローニング部位を含むKpsFの5’末端の上流の1108bp領域を標的とした。左アーム及び右アームのPCR産物を産生した。以下のパラメータ:94℃で50秒間の変性、57℃で60秒間のアニーリング、及び72℃で60秒間のプライマー伸長を全部で30サイクル含むプログラムを使用して、サーマルサイクラーにおいてDNA増幅反応を実施した。正確なバンドサイズのPCR増幅産物を1%アガロースゲル上でゲル電気泳動によって観察して、50μlの増幅産物をゲルから抽出して、置換断片を構築するために使用した。オリゴヌクレオチド配列のリストを表3に示す。
【0087】
増幅した左アームは、ゲルから精製して、プラスミドpEZ−Tに連結し、プラスミド−T−LAを製造した。制限酵素EcoRI/BamHIによって、FRT−cat−FRT断片を消化して、pEZ−T−LAのEcoRI/BamHI部位に連結し、pEZ−T−LA−FRT−cat−FRTをもたらした。増幅した右アームをプラスミドpGEM−Tに連結して、BamHI/SacIで消化した。アンピシリン耐性遺伝子を有するBamHI/SacI消化したDNA断片を、プラスミドpEZ−LA−FRT−cat−FRTに連結した。クロラムフェニコール耐性遺伝子は、置換断片を同定するための選択マーカーを提供する。得られたプラスミドpEZ−LA−FRT−cat−FRT−RAを使用して、ノックアウトBL21(DE3)株を製造した。
【0088】
実施例2:ノックアウトBL21(DE3)株の製造
LB培地で一晩にわたって37℃においてE.coli BL21(DE3)を増殖させ、BL21(DE3)のヒートショックコンピテント細胞を標準的な方法によって調製した。プラスミドpKD46でBL21(DE3)コンピテント細胞を形質転換して、形質転換した細胞を0.4OD600nmまで100μg/mlのアンピシリンを添加したLB培地中で増殖させた。Redリコンビナーゼの発現は、L−アラビノース(終濃度1mmol/l)によって1時間にわたって誘導した。
【0089】
グループII遺伝子クラスターノックアウトについては、pEZ−T−LA−FRT−cat−FRT−RAから増幅した置換断片の500ngを、BL21(DE3)コンピテント細胞に、エレクトロポレーションによって移した。エレクトロポレーションコンピテント細胞は標準的な方法で調製した。細胞の回収は、30℃で1.5時間にわたって実施した。8μg/mlのクロラムフェニコールを添加したLB寒天に細胞をプレーティングした。BL21(DE3)ΔkpsM−kpsF)::catのクロラムフェニコール耐性コロニーが、30℃における一晩のインキュベーション後に形成された。
【0090】
染色体の正しい位置におけるグループII遺伝子クラスターの完全なノックアウトを確実なものとするために、グループII遺伝子クラスターの左及び右アームの外側に相同性を有するオリゴヌクレオチドプライマーを設計した。近いフランキング領域の高い相同性に基づいて、2つのゲノムのグループIIクラスターの類似している位置を推定して、E.coli 536(Accession no.CP000247.1)の18kbの公開配列の外側のE.coli B BL21(DE3)のフランキング配列の欠失をPCRスクリーニングプライマーの設計のために使用した。3つのクロラムフェニコール耐性コロニーを遺伝子置換について試験した。
【0091】
クロラムフェニコール耐性コロニーの表現型試験を、少なくとも2世代にわたってLB+Cm(25μg/ml)における増殖によって実施した。クロラムフェニコール耐性コロニーの全てが、LB+Cm(25μg/ml)培地において非常に良好に増殖することができるであろう。クロラムフェニコール耐性D−アラニン栄養要求性株も、D−アラニンを添加して同様の方法において試験した。
【0092】
置換の成功が、1686bp及び1578bpのバンドによって確認された。
【0093】
オリゴヌクレオチドプライマー(LA_SEQ_F1/Cat_ID_R)を使用して、左アーム及びcat遺伝子の一部を含むバンド(1686bpのサイズ)を増幅し、オリゴヌクレオチドプライマー(RA_SEQ_R1/Cat_SEQ_F)を使用して、右アーム及びcat遺伝子の一部を含むバンド(1578bpのサイズ)を増幅した。3つのクロラムフェニコール耐性コロニーに関しては、バンドの正確なサイズによって、16kbのグループII遺伝子クラスターがcat遺伝子によって置換されたことが示された(図3)。kpsF及びkpsEの一部を含む右アームを含む3000bpのバンドのネガティブコントロールを作製した。これは、コントロール株(置換前)にのみ現れて、クロラムフェニコール耐性株には現れないべきである。オリゴヌクレオチドプライマー(RA6_BL_GII_F/RA6_BL_GII_R)(表3)を使用して、kpsF及びkpsEを増幅した。PCRの結果は、対照株のみ当該バンドを有し、クロラムフェニコール耐性株が有しないことを示した。
【0094】
LB培地における増殖の間にD−アラニンの添加を必要とする、D−アラニン栄養要求株であるBL21(DE3)ΔdadxΔalrに同じ消去法を適用した。
【0095】
クロラムフェニコール耐性株E.coli B BL21(DE3)Δ(kpsM−kpsF)::cat及びBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)::catを、それらの表現型及びPCRスクリーニングによって確認し、グループII莢膜遺伝子クラスターがcat遺伝子によって置換されたことを確認した。
【0096】
実施例3:薬剤耐性マーカーの除去及びグループIIノックアウト株の特性決定
凍結ストックからのE.coli BL21(DE3)Δ(kpsM−kpsF)::cat及びBL21(DE3)ΔdadXΔalr(kpsM−kpsF)::cat細胞を、LB培地において、37℃で、25μg/mlのクロラムフェニコール又は第二の株には25μg/mlのクロラムフェニコールに100mMのD−アラニンとともに増殖させた。2つのノックアウト株のエレクトロポレーションコンピテント細胞を標準的な方法によって調製した。プラスミドpCP20(CmR、AmpR)で2つのノックアウトコンピテント株を形質転換した。薬剤耐性マーカーの除去は、穏やかに振盪しながら1時間にわたって30℃においてSOCブロスで形質転換した細胞を増殖させることによって実施した。プレート状のアンピシリン耐性候補クローンの複数のシングルコロニーを、30℃で一晩培養した後に選択した。候補クローンを、LB培地のみ又はD−アラニンを添加したLB培地で37℃において3時間にわたって増殖させ、プラスミドpCP20を除去した。LB培地のみ又はD−アラニンを添加したLB培地で現れた、5つのシングルコロニーをPCRスクリーニングによってさらに特性決定した。
【0097】
LB、LB+Cm(8μg/ml)、及びLB+Ampの複数のシングルコロニーを増殖させることによって、cat除去株の表現型試験を実施した。全てのcatを除去したコロニーは、LB培地でのみ増殖できるであろう。catを除去したD−アラニン栄養要求性株については、5つのコロニーの全てがD−アラニンを添加したLB培地のみで増殖できるであろう。かくして、cat遺伝子が除去された。
【0098】
cat遺伝子の除去の成功は、2381bpバンドによって確認されるべきである。オリゴヌクレオチドプライマー(LA_SEQ_F2/RA_SEQ_R1)を使用して、左アーム及び右アームを含むバンドを増幅した。全てのシングルコロニーについて、2381bpのバンドの出現によって、cat遺伝子が除去されたことが示された。
【0099】
ノックアウト株E.coli B BL21(DE3)Δ(kpsM−kpsF)及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)は、それらの表現型及びPCRスクリーニングによって確認した。双方の株のグリセロールストックを保存のために調製した。
【0100】
実施例4:振盪フラスコ中におけるノックアウト宿主株の増殖
凍結ストックからのE.coli B BL21(DE3)及びB BL21(DE3)Δ(kpsM−kpsF)細胞を、LBプレートで一晩にわたって37℃で増殖させた。一晩たったプレートから回収した適当量の細胞を、25mlのLB培地を含むバッフル付き250ml振盪フラスコ(baffled shake flask)に約0.05 OD600の濃度まで植菌した。220rpmで振盪しながら8時間にわたって、細胞を37℃で増殖させた。各株のサンプルを1/2時間毎に回収し、OD600で細胞の増殖を比較した。
【0101】
振盪フラスコにおける増殖の比較のために、D−アラニンを添加して、同一の作業順序をE.coli B BL21(DE3)ΔdadXΔalr及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)に対して実施した。
【0102】
図4の結果は、BL21Δ(kpsM−kpsF)の一晩の増殖は、LB培地を用いた振盪フラスコにおいて、親株BL21(DE3)と同一であったことを示す。BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)をBL21(DE3)ΔdadXΔalrと比較した際も同じ結果が得られるであろう。
【0103】
実施例5:高密度発酵における宿主株BL21(DE3)及びBL21(DE3)Δ(kpsM−kpsF)の間の増殖及びMEAE−hGH発現の比較
凍結ストックからのE.coli B BL21(DE3)Δ(kpsM−kpsF)細胞をLBプレート上で増殖させた。ノックアウト株のヒートショックコンピテント細胞を標準的な方法によって調製した。hGHの発現カセットを含むプラスミドpNNC1−g(pET11d−MEAE−hGH(AmpR))で、B BL21(DE3)Δ(kpsM−kpsF)コンピテント細胞を形質転換し、そこから形質転換した細胞のグリセロールストックを調製した。
【0104】
高密度発酵は、上述のとおり好気条件下で実施した。
【0105】
図5は、BL21Δ(kpsM−kpsF)の経時的な増殖が、複合培地中で高密度発酵槽において、親株BL21(DE3)と同一であったことを示す。同じ結果が、BL21(DE3)Δ(kpsM−kpsF)/pNNC1−gをBL21(DE3)/pNNC1−gと比較した際にも得られるであろう。非誘導状態において、細胞密度が約140のOG600に達した。hGHの発現は、細胞の増殖が80のOD600に達した際に、1mMの濃度までIPTGを添加することによって実施した。30℃におけるIPTG誘導の10時間後に、細胞密度が約100のOD600に達した。
【0106】
E.coli B BL21(DE3)/pNNC1−g及びB BL21(DE3)Δ(kpsM−kpsF)/pNNC1−gは、SDS−PAGEで認められるとおり、hGHの製造に有効である。hGH発現レベルにおける増大は、SDS−PAGEゲルで示される1から10時間の誘導時間の増大にしたがう(図6)。hGHを製造する2つの株の能力は同一である。すなわち、遺伝子IIクラスターを有するB BL21(DE3)と比較した際の、宿主株B BL21(DE3)Δ(kpsM−kpsF)によるhGH発現における顕著な差異は認められなかった。
【0107】
実施例6:高密度発酵における宿主株BL21(DE3)ΔalrΔdadX及びBL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)の間の増殖及びMEAE−hGH発現の比較
凍結ストックからのE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)細胞を、D−アラニンを添加したLB培地で増殖させた。ノックアウト株のヒートショックコンピテント細胞を標準的な方法によって調製した。D−アラニン補足遺伝子を含むpNNC1−1h(pET11d−MEAE−hGH及びalrP−alrCD(AmpR))をAatII/PsiIを使用して消化し、その後の平滑末端及びプラスミドのセルフライゲーションによって、プラスミド上のalr遺伝子の選択のためのbla−プラスミドを得た。プラスミドpNNC1−1h(pET11d−MEAE−hGH,alrP−alrCD,bla−)で、BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)コンピテント細胞を形質転換した。D−アラニンを添加していないLBプレート上に一晩であらわれたコロニーを、グリセロールストックの調製のために単離した。
【0108】
高密度発酵を図5に記載のとおりに実施した。
【0109】
図7は、BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hの経時的な増殖が、FMC1培地中で高密度発酵槽において、親株BL21(DE3)ΔdadXΔalr/pNNC1−1hと同一であったことを示す。同じ結果が、B BL21(DE3)ΔdadXΔalr/pNNC1−1hをBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hと比較した際にもhGHの誘導下で得られるであろう。非誘導状態において、細胞密度が約120のOG600に達した。hGHの発現は、細胞の増殖が80のOD600に達した際に、1mMの濃度までIPTGを添加することによって実施した。30℃におけるIPTG誘導の9時間後に、細胞密度が約100のOD600に達した。
【0110】
E.coli B BL21(DE3)ΔdadXΔalr/pNNC1−1h及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hは、SDS−PAGEによる結果で認められるとおり、hGHの製造に有効である(図8)。hGH発現レベルにおける増大は、誘導時間の増大にしたがう。hGHを製造する2つの株の能力は同一である。
【0111】
実施例7:高細胞密度発酵における宿主株E.coli B BL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)を使用した細胞増殖及びMEAE−hGH(L101C)の発現の評価
凍結ストックからのE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)細胞を、D−アラニンを添加したLB培地で増殖させた。ヒートショック方法を使用する形質転換のために、このトリプルノックアウト株のコンピテント細胞を標準的な方法によって調製した。
【0112】
アンピシリン耐性をコードするβ−ラクタマーゼ遺伝子を欠失するために、MEAE−hGH(L101C)をコードし、かつ、宿主細胞のD−アラニン欠乏を補足するalrP−alrCDを含むプラスミドpNNC1a.23(pET11d−MEAE−hGH(L101C))をAatII/PsiIで消化し、その後に末端平滑化及びβラクタマーゼ遺伝子の除去のためのセルフライゲーションを実施した。得られたプラスミドであるpNNC1a.23(pET11d−MEAE−hGH,alr,bla−)で宿主細胞であるBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)をヒートショック法によって形質転換した。D−アラニンを添加していないLBプレートで増殖したシングルコロニーを選択して、−80℃における保存のためのグリセロールストックを調製した。
【0113】
2つのバッチの高細胞密度発酵を実施例5に記載のとおりに実施した。プロセスの間の細胞増殖を観察した。結果は、図9に示す。
【0114】
プラスミドpNNC1a.23を有する株E.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)によるMEAE−hGH(L101C)の発現を高細胞密度発酵において評価した。結果は、回収時の細胞密度が、OD600nmで測定した際に約100に達し得ることを示し、標的タンパク質であるMEAE−hGH(L101C)の発現レベルが、図10に示すとおり、8時間の誘導の後に約7g/lに達した。
【0115】
実施例8:E.coli BL21(DE3)Δ(kpsM−kpsF)におけるアースロバクター ウレアファシエンス(Arthrobacter ureafaciens)シアリダーゼの発現
アースロバクター ウレアファシエンスのシアリダーゼ(AUS,53.4kDa理論的質量)を、C末端のHisタグとともに大腸菌BL21(DE3)Δ(kpsM−kpsF)で製造した。C末端にHis6タグを有する、アースロバクター ウレアファシエンスのシアリダーゼの遺伝子は、T7プロモーターの制御の下に発現ベクターpET−24a(Novagen)にクローニングした。アースロバクター ウレアファシエンス由来のシアリダーゼ酵素を製造するために、以下のタンパク質配列をコードする合成コドン最適化遺伝子を産生した(SEQ ID NO:12):
MAPTPPNSPTLPPGSFSETNLAADRTAANFFYRIPALTYLGNDVVLAAWDGRPGSAADAPNPNSIVQRRSTDGGKTWGPVQVIAAGHVADASGPRYGYSDPSYIYDAEANKVFAFFVYSKDQGFGGSQFGNDDADRNVISSAVIESSDAGVTWSQPRLITSVTKPGTSKTNPAAGDVRSNFASSGEGIQLKYGPHKGRLIQQYAGDVRQADGSNKIQAYSVYSDDHGVTWHKGANVGDRMDENKTVELSDGRVLLNSRDNANRGYRKVAVSTDGGATYGPVSQDTELPDPANNGAIARMFPNAAQGSADAKKLIFTNANSKTGRENVSARVSCDDGETWPGVRTIRSGFSAYSTVTRLADGKFGVLYEGNYTDNMPFATFDDAWLNYVCAPLAVPAVNIAPSATQEVPVTVTNQEATTLSGATATVYTPSGWSATTVPVPDVAPGASVTVTVALTAPADASGPRSLNAAFTTADGRVSQFTFTATTPVAPQVGLTITGSALEHHHHHH。
【0116】
合成遺伝子は、pET−24a発現ベクターにXbaI−XhoI断片として導入し、AUS発現ベクターpLLC028を製造した。得られたベクターで、コンピテントBL21(DE3)Δ(kpsM−kpsF)を形質転換し、形質転換体を−80℃でグリセロール中で保存した。
【0117】
凍結した最初の細胞クローン(ICC)の1つの凍結チューブ(cryotube)を使用して、2つの酵母抽出物媒体(酵母抽出物5g/l、グルコース10g/l、寒天20g/l)寒天フラスコに植菌した。寒天媒体には選択のためにカナマイシンを含めた。インキュベーションを30℃で25時間にわたって実施した。インキュベーションの後に、滅菌0.9%NaCl溶液を使用して細胞を寒天表面から洗って回収した。10L発酵は、2つの寒天フラスコからの前培養物を使用して植菌した。得られた植菌材料の容量は75mで、OD600nmは4.3であり、発酵槽の始めのOD600nmは約0.03であった。発酵は、10Lの開始作業容量で20Lステンレススチール容器で実施した。発酵は、40g/l酵母抽出物、15g/lグリセロール(87%)、塩、及び微量金属を含む複合培地に対して8時間のバッチフェーズで開始し、続いて、所定のプロファイルでグリセロールを加えた。最初の17時間は、発酵温度は37であった。その後、温度を30℃まで下げた。発酵時間が18時間経過した時点で、誘導因子であるIPTGを0.1mMで添加して、タンパク質発現を誘導した。誘導の期間は22時間までであり得る。回収プロセスは、遠心分離、高圧ホモジナイゼーション、0.2μmの孔径を有するSartocon Slice Cassetteを使用したクロスフロー精密濾過からなる。
【0118】
AUS発酵は、SDS−PAGE及び酵素活性アッセイを使用して分析した。SDS−PAGE分析によって、発現したシアリダーゼが、ビーズミリングの後に、可溶性及びペレット画分の双方にあらわれることが明らかになった。図11は、30℃の誘導温度及び0.1mMのIPTGで得られた10Lバッチに由来する典型的な発酵の結果を示す。
【0119】
実施例9:BL21 DE3及びBL21 DE3 Δ(kpsM−kpsF)におけるヒト線維芽細胞増殖因子21(FGF21)の発現
ヒトFGF21のDNA及びアミノ酸配列は、例えば、Biochim. Biophys. Acta 1492(1):203−206 (2000)のNishimura et alに開示されている。前記配列は、公開データベースからも得られる(各々、Accession nos. EMBL:AB021975及びUNIPROT:Q9NSA1)。
【0120】
天然のポリペプチドは、分泌のための28アミノ酸のシグナルペプチド(以下に斜体で記載)とともに合成されるが、成熟FGFポリペプチドは、残部の181アミノ酸からなる(SEQ ID NO:13):
MDSDETGFEHSGLWVSVLAGLLLGACQAHPIPDSSPLLQFGGQVRQRYLYTDDAQQTEAHLEIREDGTVGGAADQSPESLLQLKALKPGVIQILGVKTSRFLCQRPDGALYGSLHFDPEACSFRELLLEDGYNVYQSEAHGLPLHLPGNKSPHRDPAPRGPARFLPLPGLPPA-LPEPPGILAPQPPDVGSSDPLSMVGPS QGRSPSYAS。
【0121】
シグナルペプチドはないが、N末端のメチオニンを添加して、成熟FGF21ポリペプチドをクローニングして、E.coliで細胞内タンパク質として発現させた。特に、3’末端にメチオニンのATGコドン並びにNde1及びBamH1制限酵素部位を3’末端及び5’末端の各々に含む550bpのコード領域を、ファージT7プロモーターの制御下のNde1−BamH1において発現ベクターpET 11c(Novagen)に挿入して、E.coli B BL21(DE3)及びE.coli B BL21 DE3 Δ(kpsM−kpsF)を形質転換した。細胞をLB amp 100μg/ml中においてOD450が0.5になるまで増殖させ、0.3mM IPTGで4時間にわたって37℃で発現を誘導した。FGF21発現分析のために細胞の粗抽出物を超音波処理によって作製した。
【0122】
図12のクーマシー染色したSDS−PAGEゲルは、E.coli B BL21 DE3およびE.coli B BL21 DE3Δ(kpsM−kpsF)におけるFGF21の発現が等しく成功したことを示す。発現したFGF21(Met−FGF21)の計算分子量(Mw)は19.5kDであるが、ゲル上では25kDタンパク質として移動し、これはタンパク質の移動を遅らせるプロリンが多く含まれているためであろう。
【0123】
本実施例は、FGF21の産生量は宿主細胞の遺伝子操作によって影響を受けないことを示す。
【0124】
実施例10:BL21 DE3及びBL21 DE3Δ(kpsM−kpsF)におけるヒトアミリンの発現
37アミノ酸のアミロイドペプチドであるアミリンの配列は、Proc. Natl. Acad. Sci. (84) : 8628−8632, (1987)のCooper et al.によって公開された。遺伝子をクローニングして、サーモトガ・マリティマ(thermotoga maritima)由来のリボソームタンパク質RL23にN末端に融合した。146アミノ酸の融合タンパク質は、以下の配列(SEQ ID NO:14)を有する:
KQEKLSLHDVLIRPIITEKALILREQRKYVFEVNPLANKNLVKEAVEKLFNVKVEKVNILNMKPKPKRRGIFEGKTRSWKKAVVTLKEGYTIKELEGEHSSSSDDDDKKCNTATCATQRLA-NFLVHSSNNFGAILSSTNVGSNTY。
【0125】
前記遺伝子を、実施例9に記載のとおり、Nde1−BamH1断片として、T7プロモーターの制御下においてpET11cにクローニングして、プラスミドを使用してBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)を形質転換した。
【0126】
発現は実施例9に記載のとおり実施し、結果は図12に示す。図12は、2種のE.coli B宿主であるBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)における等しく高い産生量の発現を示す。
【0127】
実施例11:BL21 DE3及びBL21 DE3Δ(kpsM−kpsF)におけるヒトレプチンの発現
146アミノ酸長のヒトレプチンの配列は、Diabetes (44): 855−858 (1995)のMasuzaki et alに公開されている。前記タンパク質は、21アミノ酸のシグナルペプチドが添加されてヒトにおいて分泌される。E.coliで細胞内に発現させた成熟レプチンは、以下の配列(SEQ ID NO:15)を有する:
MVPIQKVQDDTKTLIKTIVTRINDISHTQSVSSKQKVTGLDFIPGLHPILTLSKMDQTLAVYQQILTSMPSRNVIQISNDLENLRDLLHVLAFSKSCHLPWASGLETLDSLGGVLEASGYSTEVVALSRLQGSLQDMLWQLDLSPGC。
【0128】
レプチン遺伝子は、実施例9に記載のとおり、450bpのNde1−BamH1断片として、T7プロモーターの制御下においてpET 11cにクローニングし、このプラスミドを使用してBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)を形質転換した。
【0129】
発現は実施例9に記載のとおりに実施した。結果は図12に示す。図12は、2種のE.coli宿主であるBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)における等しく高い産生量の発現を示す。
【0130】
グループII莢膜遺伝子クラスターの除去は、いずれの増殖条件における細胞の増殖にも影響を与えることがないと結論付けられる。グループII莢膜遺伝子クラスターの除去がタンパク質発現に影響を与えないとも結論付けられる。したがって、治療用タンパク質製造のための市販の宿主株としてE.coli B BL21(DE3)を使用するための、グループII莢膜遺伝子クラスターを完全に取り除いたより安全な株がこれにより製造される。E.coli B BL21(DE3)のグループII莢膜遺伝子を欠失する遺伝子操作によって、E.coli K12と同じ安全カテゴリーになる。グループII遺伝子クラスターが、これら2種の株の唯一の顕著な違いだからである。
【技術分野】
【0001】
本発明は、大腸菌(E.coli)ゲノムの操作、改善した非病原性E.Coli B株、並びにペプチド及びタンパク質製造のためのその使用に関する。
【背景技術】
【0002】
細菌は、広範な市販の製品の製造のために使用されている。遺伝子操作した細菌、例えば、大腸菌を宿主細胞として生物試薬、例えば、ペプチド及び核酸の生産のために実験室で使用すること及び工業的に使用することは、当該技術分野においてよく知られている。自然な環境における細菌は、標準的な工業的又は実験室における増殖で通常与えられるものではない多数の環境に曝されるため、それらのゲノムは、多数の環境依存的なストレス誘導遺伝子、又はそうでなければ非必須遺伝子を有し、それらは、前記生物の工業的又は実験室における使用には必要とされない可能性があり、又は望ましくない可能性すらある。
【0003】
細菌性病原体は、毒性因子、例えば、細胞表面における莢膜多糖類を含み、それは、細菌とその周囲の環境との間の相互作用を媒介し、細菌の病原性に直接関連する多数の機能を有する。80を超える血清学的及び化学的に異なるタイプの多糖類莢膜が、E.Coliについて開示されており、K抗原と称されている。多糖類莢膜は、4つのグループに分類され、そのうちグループII莢膜多糖類は、インフルエンザ菌及び髄膜炎菌のものと類似している。グループII莢膜遺伝子クラスターは、侵襲性疾患と関連する莢膜のタイプの大半を含み、3つの機能的領域からなる保存された遺伝子構成を有する。領域1及び3は、グループII遺伝子クラスターの間で保存されており、合成部位から細胞表面に多糖類を輸送するために必要なペプチドをコードしており、領域2は、血清型特異的であり、特定の多糖類を含む個々の単糖類の生合成及び重合に関与する酵素をコードしている。領域1は、6つの遺伝子のkpsFEDUCSを含み、それらは1つの転写単位に構成されている。領域2は、血清型特異的であり、グループ2抗原の間で異なる。領域3は、2つの遺伝子のkpsM及びkpsTを含み、それらは1つの転写単位に構成されている[Andreishcheva et al. Gene (2006) 384: 113-119]。
【0004】
E.ColiのK−12株及びB株は、莢膜を有さず、ペプチドの製造に一般的に使用される株である。E.Coli K−12のゲノムは、完全に配列決定されており、莢膜遺伝子を全く含まない。組換えペプチドの高収量発現のための好ましい株であるE.coli B BL21(DE3)は、グループII莢膜多糖類をコードする遺伝子クラスターを含むことが認められており、E.coli Bの祖先が水平伝播によってこれらの莢膜遺伝子を獲得したに違いないと解されている。E.coliの莢膜に位置するK抗原は、遺伝子の機能形態を有する株の病理表現形と関連する場合が多い。E.coli B BL21では、グループII莢膜遺伝子クラスターの領域2が、挿入エレメント(IS1)によって不活性状態になっており、抗原の生産が妨げられているが、野生型の莢膜抗原の排出及び提示を補助する領域1及び3はインタクトなままである[Andreishcheva et al. Gene (2006) 384: 113-119]。しかしながら、機能的なグループII莢膜遺伝子クラスターの再構築の可能性が理論的にはあり、例えば、領域2のIS1が無くなり、それによって、グループIIクラスターが再構築される可能性がある。代替的には、IS1変異領域が外来性の野生型の領域2と交換されると、グループIIクラスターも再構築される可能性がある。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Andreishcheva et al. Gene (2006) 384: 113-119
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、グループII莢膜遺伝子クラスターの永久的なノックアウト(KO)を有するE.coli B BL21(DE3)株を提供し、当該株を他の商業的に重要な株、例えば、E.coli K−12と同じ安全な種類にする。
【課題を解決するための手段】
【0007】
本発明は、不可逆的に不活性化されたグループII莢膜遺伝子クラスターを有するE.coli B BL21株に関する。
【0008】
1つの実施態様では、E.coli B BL21株は、遺伝子II莢膜遺伝子クラスターの全体又は一部の欠失を有することを特徴とする。
【0009】
1つの実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの全体の欠失を有することを特徴とする。
【0010】
1つの実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの一部の欠失を有することを特徴とする。
【0011】
さらなる実施態様では、E.coli B BL21株は、グループII莢膜遺伝子クラスターの1又は複数の点変異を有することを特徴とする。
【0012】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも5%が欠失によって永久的に不活性化されていることを特徴とする。
【0013】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも25%が欠失によって永久的に不活性化されていることを特徴とする。
【0014】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも40%が欠失によって永久的に不活性化されていることを特徴とする。
【0015】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも55%が欠失によって永久的に不活性化されていることを特徴とする。
【0016】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの75から100%が欠失によって永久的に不活性化されていることを特徴とする。
【0017】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも85%が欠失によって永久的に不活性化されていることを特徴とする。
【0018】
他の実施態様では、E.coli株は、グループII莢膜遺伝子クラスターの少なくとも95%が欠失によって永久的に不活性化されていることを特徴とする。
【0019】
1つの実施態様では、グループII莢膜遺伝子クラスターの30%が欠失されている。
【0020】
他の実施態様では、グループII莢膜遺伝子クラスターの50%が欠失されている。
【0021】
他の実施態様では、グループII莢膜遺伝子クラスターの70%が欠失されている。
【0022】
他の実施態様では、グループII莢膜遺伝子クラスターの80%が欠失されている。
【0023】
他の実施態様では、グループII莢膜遺伝子クラスターの90%が欠失されている。
【0024】
1つの実施態様では、E.coliの株がE.coli B BL21(DE3)であり、さらなる実施態様では、E.coliの株がE.coli B BL21(DE3)ΔdadX Δalrである。
【0025】
本発明は、
a.選択マーカーによってグループII莢膜遺伝子クラスターを置換する工程、
b.選択マーカーを除去する工程、
c.PCR分析によって欠失を確認する工程
を含む方法によってグループII莢膜遺伝子クラスターが欠失される、E.coli B株の製造方法も企図する。
【0026】
1つの実施態様では、選択マーカーは抗生物質耐性遺伝子である。
【0027】
他の実施態様では、抗生物質耐性遺伝子は、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン、及びエリスロマイシンからなる群から選択される。
【0028】
さらなる他の実施態様では、抗生物質耐性遺伝子はクロラムフェニコールである。
【0029】
本発明は、本発明のE.coli株に、ペプチドをコードするDNA配列を含む発現ベクターを導入することによって、ペプチドを製造するための方法にも関する。
【0030】
1つの実施態様では、本発明は、ペプチドをコードするDNAの発現のための条件で適切な培養培地において、本発明に係るE.coli B株を培養する工程を含む、ペプチドの製造方法も企図する。発現ベクターで発現するペプチドは、E.coliで発現することができる任意のペプチドであってよい。そのようなペプチドの代表例は、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、並びに酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアントである。
【0031】
1つの実施態様では、本発明のE.coli株は、ペプチドをコードするDNA配列を含む発現ベクターを含む。
【0032】
1つの実施態様では、製造するペプチドはhGHである。
【0033】
他の実施態様では、製造するペプチドはFGF−21である。
【0034】
他の実施態様では、製造するペプチドはアミリンである。
【0035】
他の実施態様では、製造するペプチドはレプチンである。
【0036】
他の実施態様では、製造するペプチドはシアリダーゼである。
【図面の簡単な説明】
【0037】
【図1】図1は、非コード隣接領域を有するE.coli B BL21(DE3)莢膜遺伝子クラスターの遺伝子構成を示す。B BL21(DE3)莢膜遺伝子クラスターのオープンリーディングフレーム(ORF)(大きい矢印);転写方向(矢頭);領域1遺伝子(灰色の矢印);領域2のORF(黒色の矢印);領域3の遺伝子(白色の矢印);IS1エレメント(白色ボックス)[Andreishcheva et al. Gene (2006) 384: 113-119]。小さい矢印は、非コード左アーム(LA)及び右アーム(RA)の位置を示す。
【図2】図2は、グループII莢膜遺伝子クラスターノックアウト方法:a)抗生物質耐性遺伝子によるグループII莢膜遺伝子クラスターの置換、及びb)抗生物質耐性遺伝子の除去のワークフローの略図である。
【図3】図3は、ノックアウト候補のPCRスクリーニングを示す。ノックアウト候補(親株ではない)(BL又はD)から増幅した、正しいサイズである1686bp(LA+catの一部、上の図のA1からA3、下の図のB1からB3、及び1578bp(RA+catの一部、上の図のA1からA3、下の図のB1からB3)は、グループII遺伝子クラスターのノックアウトが成功したことを示す。陰性対照については、親株のみであると約3kbのサイズのバンドを有するが、グループIIノックアウト株ではそうではない。BLはBL21(DE3)を示し、DはBL21(DE3)ΔdadXΔalr株を示す。A1からA3は、異なるコロニーナンバーのBL21(DE3)ノックアウト候補であり、B1からB3はBL21(DE3)ΔdadXΔalr株のノックアウト候補である。
【図4】図4は、振盪したフラスコ中の宿主E.coli B株の増殖の比較である。使用する記号は、以下の株を示す:●:BL21(DE3);□:BL21(DE3)Δ(kpsM−kpsF);−:BL21(DE3)ΔdadXΔalr;△:BL21(DE3)ΔdadXΔalr Δ(kpsM−kpsF)。
【図5】図5は、異なるE.coli B株の発酵増殖曲線を示す。使用する記号は、以下の株を示す:◆:BL21(DE3);□:BL21(DE3)Δ(kpsM−kpsF);−:BL21(DE3)/pNNC1−g;○:BL21(DE3)Δ(kpsM−kpsF)/pNNC1−g。
【図6】図6は、異なる誘導時点におけるhGH発現のSDSポリアクリルアミドゲル電気誘導(PAGE)を示す:a)E.coli B BL21(DE3)/pNNC−1gにおける発現、及びb)E.coli B BL21(DE3)Δ(kpsM−kpsF)/pNNc1−gにおける発現。a)及びb)の双方において、レーン1から3は、hGHの異なる濃度:5μg、2.5μg、及び1.25μgの各々を示す。レーン4は誘導前のhGHの発現を示し、レーン5から14は、1から10時間の毎時の異なる誘導時点におけるhGHの発現を示す。全てのサンプルは、同じロードODに対して正規化した。
【図7】図7は、hGH発現の誘導の前及び後の異なるE.coli B株の増殖を示す。使用する記号は、以下の株を示す:◆:BL21(DE3)ΔdadXΔalr/pNNC1−1h;□:BL21ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1h;Δ:BL21(DE3)ΔdadXΔalr/pNNC1−1h誘導;−:BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1h誘導。
【図8】図8は、異なる誘導時点におけるSDS−PAGEによって分析したhGHの発現を示す:a)E.coli B BL21(DE3)ΔdadΔalr/pNNC1−1h;b)E.coli B BL21(DE3)ΔdadΔalrΔ(kpsM−kpsF)/pNNC1−1h。a)及びb)の双方において、レーン1から3は異なる濃度:5μg、2.5μg、及び1.25μgの各々におけるhGHの発現を示す。レーン4は誘導前のhGHの発現を示し、レーン5から13は1から9時間の毎時の誘導時点におけるhGHの発現を示す。全てのサンプルは、同じロードODに対して正規化した。
【図9】図9は、FDM1−2培地における高細胞密度発酵におけるE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1a.23の増殖曲線を示す。hGH変異体であるMEAE−hGH(L101C)の発現は、細胞密度が70OD600に達した際に、0.2mMの終濃度までIPTGを添加することによって誘導し、25℃で培養し続けた。最終的な細胞密度は、誘導の8時間後に約100OD600に達した。
【図10】図10は、高細胞密度発酵の間のMEAE−hGH(L101C)の発現を示す。レーン1:マーカー;レーン2から4:5、2.5、1.25μgの各々の濃度の標準としての精製hGH;レーン5から13:0、1、2、3、4、5、6、7、及び8時間の各々の間にわたって誘導した後の異なる時間間隔で得たサンプル。矢印は標的タンパク質であるMEAE−hGH(L101C)を示す。
【図11】図11は、E.coli B BL21(DE3)Δ(kpsM−kpsF)におけるアースロバクター ウレアファシエンス(Arthrobacter ureafaciens)シアリダーゼの発現のための発酵サンプルのSDS−PAGE分析を示す。レーン1:マーカー;レーン2:誘導前のホモジネート;レーン3:誘導後のホモジネート;レーン4:最終サンプルのホモジネート;レーン5:誘導前の不溶性画分;レーン6:誘導後の不溶性画分:レーン7:最終サンプルの不溶性画分:レーン8:誘導前の可溶性画分;レーン9:誘導語の可溶性画分;レーン10:最終サンプルの可溶性画分;レーン11から12:標準シアリダーゼ(150及び300mg/l)。全てのサンプルは10倍希釈した。
【図12】図12は、E.coli B BL21 DE3及びE.coli B BL21(DE3)Δ(kpsM−kpsF)のヒト由来FGF21、アミリン、及びレプチンの発現を示す。レーン1:マーカー;レーン3から8:BL21 DE3における発現;レーン9から14:BL21 DE3Δ(kpsM−kpsF)における発現;レーン3及び9:非誘導;レーン4及び10:誘導したFGF21;レーン5及び11:非誘導;レーン6及び12:誘導したアミリン;レーン7及び13:非誘導;レーン8及び14:誘導したレプチン。
【発明を実施するための形態】
【0038】
本発明は、不可逆的に不活性化したグループIIクラスターを有するE.coli B BL21株に関連し、ペプチド製造のための永久的に安全な株を提供する。
【0039】
E.coli B BL21(DE3)の野生型又は任意の他の株のグループII莢膜遺伝子クラスターの損傷又は不活性化は、クラスター全体又はその一部の欠失を含む任意の適切な方法又は遺伝子クラスターの1又は複数の点変異によって達成されてよい。
【0040】
本発明のE.coli株は、一般的に産業的に使用されていた株、例えば、E.coli K−12と比較して、ペプチド製造のためのより着実かつ高収量な方法を提供するという利点を有する。
【0041】
本発明で使用するE.coli B BL21(DE3)は、WO2008/025744に記載のダブルノックアウト(2×KO)であってよい。このE.coli B BL21(DE3)株は、双方がD,L−アラニンラセマーゼをコードするalr及びdadXの2遺伝子を欠失している。このダブルノックアウト株は、D−アラニンの増殖培地に対する添加又は必要なD−アラニンを供給することができるプラスミド上のラセマーゼ遺伝子の1つの存在のいずれかが無ければ増殖することができない。この2×KO D−アラニン栄養要求性E.coli株は、本明細書においてE.coli. B BL21(DE3)ΔalrΔdadXと称され、細胞中のプラスミドの維持のための非抗生物質選択方法を可能にする。
【0042】
本発明の1つの実施態様では、野生型E.coli B BL21(DE3)又はE.coli B BL21(DE3)ΔalrΔdadX株のグループII莢膜遺伝子クラスター内の1又は複数の遺伝子を欠失する。E.coli株B BL21(DE3)はStudier and Moffat[J.Mol.Biol. (1986) 189:113-130]及びStudier et al[Methods in Enzymology (1990) 185:60-89]に開示されており、Stratagene及びNovagenから入手可能である。
【0043】
本発明の1つの実施態様では、野生型E.coli B BL21(DE3)のグループII莢膜遺伝子クラスター内の1又は複数の遺伝子が欠失される。本発明の他の実施態様では、E.coli B BL21(DE3)ΔalrΔdadXのグループII莢膜遺伝子クラスター内の1又は複数の遺伝子が欠失される。
【0044】
他の実施態様では、グループII莢膜遺伝子クラスター内の欠失に選択される1又は複数の遺伝子は、潜在的な病原性に関与するものである。16kbのグループII莢膜遺伝子クラスター全体の欠失は、病原性の莢膜の表現型の潜在的な再構築の予防を提供する。図1は、領域1遺伝子kpsFEDUCS、領域2 ORF1−4、及び領域3 kpsM及びkpsTを含む莢膜を有する株のグループII莢膜遺伝子クラスターに含まれる遺伝子を示す。これらの遺伝子のいずれか1つ又はそれらの組合せが欠失されてよい。
【0045】
グループII莢膜遺伝子クラスター全体の欠失は、3つの領域の欠失、すなわち、Δ(kpsM−kpsF)を意味する。得られるE.coli B株は、E.coli B BL21(DE3)Δ(kpsM−kpsF)及びE.coli B BL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)である。
【0046】
1つの実施態様では、本発明は、挿入エレメント(IS1)は別として、グループII遺伝子クラスターを不活性化するのに十分な任意の修飾を企図する。
【0047】
細菌は1倍体生物であり、すなわち、それらは1つのみの染色体を有するため、組換えはたいてい、環境から新規DNAを獲得した際にのみ、例えば、形質転換を通じて生じる。さらに、例えば、線状DNAを分解する細胞内エキソンクレアーゼのため、細菌は、線状DNAでは容易に形質転換できない。しかしながら、組換え複合体のエキソヌクレアーゼを欠いている、組換え能力がある変異体は、線状DNAで形質転換をすることができる。リコンビナーゼは、天然の組換え反応を触媒する酵素であり、λ Redは、他の組換え系と比較した際に組換え速度の向上を示すことが示されている。本発明の株のグループII莢膜遺伝子クラスターの欠失方法は、Datsenko and Wanner[(2000) PNAS 97(12): 6640-6645]によって開発されたRed媒介組換え技術又は他の従来の方法論に基づいてよい。
【0048】
図2は、本発明の株の16kbのグループII莢膜遺伝子クラスターの欠失方法の略図を示す。1つの実施態様では、前記方法の第一工程は、ヌクレオチド相同性伸長部(nucleotide homolgy extension)又はRA及びLA領域(図1参照)とともにプライマーを使用することによるPCRによって生成される、選択マーカーを使用する、16kbのグループII莢膜遺伝子クラスターの置換である。これは、これらの隣接相同性伸長部におけるRed媒介組換えによって達成される。選択の後の、本方法の第二工程は、選択マーカーに隣接する直接反復FRT(FLP認識標的)部位に作用するFLPリコンビナーゼを発現するヘルパープラスミドを使用することによる選択マーカーの除去である。Red及びFLPヘルパープラスミドは、温度感受性レプリコンであるため、37℃における増殖によって取除く。第三工程は、PCR分析による欠失の確認を含む。
【0049】
細菌の形質転換は、DNAを取込むことによる安定な遺伝子変化と称されてよい。コンピテンスは、外来性DNAを取込むことができる状態を示す。ある細菌は、天然に、実験室条件下でDNAを取込むことができ、そのような細菌は、細胞の1又は複数の膜を越えてDNAを輸送するための機構を特定する遺伝子セットを有するが、他のものは、天然には通常生じない条件を使用して、細胞が受動的にDNA透過性にされる実験手法によって誘導しなければならない。二価陽イオン、例えば、Ca2+(CaCl2中)の存在下で細胞を冷却することは、プラスミドDNAに透過性になるように細胞壁を調製する。細胞をDNAとともに氷上でインキュベートし、次いで、短くヒートショックを与え(例えば、30から120秒間、42℃)、DNAを細胞内に入れる。これは当該技術分野においてよく知られた方法である[Sambrook et al., A Laboratory Manual (1989) CSH]。エレクトロポレーションは、細菌(及び他の)細胞に穴を作製する他の方法であり、それらにkV範囲の電場で短い間でショックを与えることによる。この穴を通って、プラスミドDNAは細胞に入ることができる。この方法は、大きなプラスミドDNAの使用に適している。天然の膜修復機構は、ショックの後に穴を直ぐに閉じるであろう。細胞に維持及び安定に保持するために、プラスミドDNA分子は、染色体からは独立して細胞内で複製することを可能にする複製起点を含む必要がある。
【0050】
本明細書に記載の任意の遺伝子不活性化方法を使用する際に、グループII遺伝子クラスターの一部又は全体の任意の標的とした不活性化が選択マーカーによって検出されて、修飾されたE.coli B BL21株クローンが同定及び単離される。
【0051】
選択マーカーは、任意の適切なマーカー遺伝子、例えば、抗生物質耐性遺伝子又は栄養要求性マーカーであってよい。
【0052】
適切な抗生物質耐性遺伝子は、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン及びエリスロマイシン若しくはβ−ラクタムの任意の他の代表例、アミノグリコシド、グリコペプチド、又はマクロライド抗生物質である。
【0053】
本明細書で使用する用語「クローニング」は、DNAの配列の多数の同一のコピーの作製を伴う。標的DNA配列は、次いで、クローニングベクターに挿入される。このベクターは自己複製ウイルス、プラスミド、又は高等生物細胞に由来するため、適当なサイズのDNAが挿入される際は、「標的及びベクターDNA断片を、次いで、連結し」、組換えDNA分子を作製する。次いで、形質転換後に幾つかの同一のコピーを生産する細菌株(通常はE.coli)に組換えDNA分子を入れる。形質転換は、細菌が有するDNA取込み機構である。しかしながら、所定の複製起点を有する1つの組換えDNA分子のみが、1つの細菌細胞内で確立することができ、そのため、各クローンは1つのDNAインサートのみを有する。
【0054】
本明細書で使用する用語「ゲノム」又は「遺伝的構成物」は、遺伝することが可能であり及び/又はそうでなければ伝達性である、遺伝的物質全体、その構造及び機能の双方を指し、特定の細胞又は微生物内に含まれている。微生物のゲノム(遺伝的構成物)は、かくして、染色体DNA及び染色体外DNA(例えば、プラスミド及びR因子など)を含むと解される。
【0055】
本明細書で使用する用語「約」は、所定の数値の合理的な近傍、例えば、±10%を意味する。
【0056】
本明細書で使用する用語「ノックアウト」は、1つ又は複数の遺伝子が欠失又は不活性化されている(生物から「ノックアウト」されている)、遺伝子操作された細菌株を意味する。ノックアウト生物は、配列決定されているが、未知の機能を有する遺伝子の役割を研究するためのモデルとして通常は使用される。ノックアウト技術を使用して、興味のある生物から望ましくない遺伝子及び/又は潜在的な病原性遺伝子を除去してよい。
【0057】
本明細書で使用する用語「遺伝子クラスター」は、遺伝的及び機能的に連関した2以上の遺伝子のセットを意味する。共通の祖先に由来する集団は同じ遺伝子クラスターの種を有する傾向があり、細菌の進化の過程を遡るのに有用である。
【0058】
用語「親株」は、縮小したゲノムの株の由来である任意の細菌株であってよい。本発明に係る親株の代表例は、E.coli株、例えば、B若しくはC、又はそれらと実質的に同一のゲノム配列を有する株を含むが、それらに限らない。
【0059】
所定の細菌宿主細胞で製造されるペプチドの「レベル」又は「量」なる本明細書で使用される用語は、選択した製造方法に従って、細胞培養物中に存在する又は検出可能なペプチドの定量的尺度であってよい。例えば、選択したペプチド製造方法が細胞自体の内部におけるペプチドの蓄積をもたらす場合には、製造されたペプチドのレベル又は量は、細胞自体の内部に存在するペプチドの定量的尺度である。ペプチド製造方法がペプチドの分泌を伴う場合には、ペプチド製造レベルは、増殖培地に存在するペプチドの定量的尺度であろう。
【0060】
ペプチドの「収量」なる本明細書で使用する用語は、本質的に純粋なペプチドの定量値であり、ここでいうペプチドは、宿主株と天然に関連を有するペプチドから本質的に遊離して回収及び/又は精製されている。細菌宿主細胞、例えば、E.coli B BL21株によって製造されたペプチドの定量的な「収量」又は「量」を確認する際に、従来のペプチド特異的な方法、例えば、イムノアッセイ、高速液体クロマトグラフィー(HPLC)、酵素活性、分光光度法、及びSDS−PAGEなどを使用することができる。
【0061】
本明細書で使用する用語「発現調節配列」は、作動可能に連結した核酸の転写を管理する転写因子結合部位の配列又はプロモーターを意味する。
【0062】
二本鎖核酸に関連して使用する用語「遊離端」は、平滑遊離端若しくは付着遊離端又はそれらの組合せを有する線状核酸を意味してよい。
【0063】
本明細書で使用する用語「遺伝子」は、ポリペプチド又はその前駆体をコードする核酸配列を含む核酸(例えば、DNA又はRNA)である。ポリペプチドは、全長又は断片の所望の活性又は機能的性質(例えば、酵素活性、リガンド結合、シグナル伝達、抗原性など)が維持されている限りにおいて、コード配列の全長又はコード配列の任意の一部によってコードされ得る。前記用語は、全長mRNAに転写される遺伝子に寄与する、5’及び3’末端の双方のコード領域に隣接して位置する配列も包含する。用語「遺伝子」は、遺伝子のcDNA及びゲノム形態(genomic form)を含む。遺伝子のゲノム形態又はクローンは、例えばイントロンと称される非コード配列で中断されるコード領域を含んでよい。
【0064】
用語「導入する」又は「導入された」は、株への核酸の添加と関連して使用される際には、核酸が、株の染色体に組み込まれるか又はベクター、例えば、プラスミド上で株に含まれることを意味する。
【0065】
本明細書で使用する用語「オープンリーディングフレーム」又は「ORF」は、終止コドンを含まない遺伝子配列である。
【0066】
用語「ペプチド」、「ポリペプチド」、又は「タンパク質」は、本明細書において互換的に使用され、天然又は組換えのいずれかの、ペプチド、ポリペプチド、及びタンパク質、並びにその断片、誘導体、ホモログ、バリアント、及び融合体を意味する。
【0067】
グループIIノックアウト株の増殖は、振盪したフラスコ中及び高密度発酵槽中で、それらの親株と比較して評価した。実験によって、グループIIノックアウトは、いずれの株の背景においても細胞増殖に影響を与えないことが示された。高密度発酵槽において、発現試験をするために、複数のペプチドを有する発現ベクターでグループII欠失株を形質転換した。
【0068】
1つの実施態様では、本発明のE.coli B株において製造するためのペプチドは、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアント、並びに任意の他のペプチド又はE.coliで発現系が作動可能であるペプチドからなるが、それらに限らない群から選択されてよい。
【0069】
本発明は、上述のヒトペプチドの製造方法にも関する。一般的には、クローン化した野生型ペプチドの核酸配列は、所望のタンパク質をコードするように修飾される。この修飾配列は、次いで、発現ベクターに挿入され、次に宿主細胞に形質転換又は形質移入される。作動可能な発現系は、典型的には、Studier and Moffat[J.Mol.Biol. (1986) 189:113-130]及びStudier et al.[Methods in Enzymology (1990) 185:60-89]に記載の従来の遺伝子発現成分を含む。
【0070】
宿主細胞中で製造するペプチドは、融合ペプチドとして合成されるか、培地中又は細胞膜周辺腔に分泌されるか、細胞質中に遊離の形態で製造されるか、又は細胞内構造体、例えば、封入体に蓄積される。同様に、ペプチド精製は、興味のあるペプチドに対して従来技術によって適合される、上述の従来の一般的に利用可能な方法の使用を伴う。
【0071】
本発明は、任意のタイプのペプチドの製造に適している。
【0072】
1つの実施態様では、グループIIノックアウトE.coli B BL21宿主細胞中における製造に選択されるペプチドは、ヒト成長ホルモン(hGH)であり、1つの実施態様では、ヒト成長ホルモン(hGH)は、hGH前駆体からhGHを作製する方法を開示するEP217814に記載の前駆体ペプチドから製造される。
【0073】
さらなる実施態様では、成長ホルモンは、hGHアナログの前駆体であるMEAE(SEQ ID 16)−hGH(L101C)であり、hGH分子の101位のロイシンがシステインによって置換されている。
【0074】
他の実施態様では、グルカゴン様ペプチドは、GLP−1、GLP−1、及び/又はその誘導体である。
【0075】
本明細書で使用する用語「グルカゴン様ペプチド」は、グルカゴンファミリーのペプチド、エキセンジン、及びそのアナログを意味する。グルカゴンファミリーのペプチドは、プレグルカゴン遺伝子によってコードされ、高い相同性を有する3つの小さなペプチド、すなわち、グルカゴン(1−29)、GLP−1(1−37)、及びGLP−2(1−33)を含む。エキセンジンは、トカゲで発現しているペプチドであり、かつ、GLP−1のようにインスリン分泌を促進する。エキセンジンの例は、エキセンジン−3及びエキセンジン−4である。
【0076】
本明細書で使用する用語「グルカゴン様ペプチドアナログ」は、修飾されたグルカゴン様ペプチドであり、グルカゴン様ペプチドの1又は複数のアミノ酸残基が他のアミノ酸残基によって置換されており、及び/又は1又は複数のアミノ酸残基がグルカゴン様ペプチドから欠失されており、及び/又は1又は複数のアミノ酸残基がグルカゴン様ペプチドに付加されている。そのようなアミノ酸残基の付加又は欠失はペプチドの任意の位置で起こり得る。
【0077】
実験
株のグリセロールストックを調製するために、適当な抗生物質を含む2mlのLB培地を含む12ml試験管に1つのコロニー移して、220rpmで振盪しながら2時間にわたって37℃で増殖させた。
【0078】
発酵種菌を調製するために、グリセロールストックの細胞を、アンピシリンを含むか又は含まないLBプレートにストリークする。一晩の後に細胞をLB培地で洗浄して、各々適当な抗生物質濃度を有する100mlの発酵複合培地を含む500ml振盪フラスコに植菌して、37℃において220rpmで浸透しながら3〜4時間にわたって増殖させた。次いで、約0.5 OD600の濃度に細胞をリアクターに植菌した。
【0079】
適当な抗生物質を含む2.5Lの初期容量の発酵複合培地を含む5Lバイオリアクターにおいて、好気条件下で発酵を実施した。0時点において、リアクターは約0.5のOD600に植菌した。5N NH3及びH2SO4を使用してpHを7.0に維持した。6ml/分の一定空気流速を培養期間に培養物に拡散させた。温度は37℃に維持した。20%の飽和O2まで溶存酸素レベルを維持することによって、攪拌をカスケード制御した。
【0080】
フェドバッチ実験を実施して、高密度発酵槽において、グループII欠失株の増殖及びペプチド発現を親株と比較した。27g/lのグリセロールを添加した複合培地を発酵槽に入れた。0時点において、発酵槽は0.5のOD600に植菌した。溶存酸素の増加によって示された、基礎のグリセロールの摂取に伴って、工程を増加する際にグリセロール及び酵母抽出物を連続的に添加した。OD600及びタンパク質発現を測定するためのサンプルは、発酵槽から回収して、分光光度計及びSDS−PAGEによって分析した。
【0081】
細菌中のタンパク質発現の誘導は、当該技術分野においてよく知られている。本発明では、タンパク質発現の誘導はイソプロピル−β−D−チオガラクトピラノシド(IPTG)の添加によって実施する。
【0082】
本発明のある実施態様で使用するプラスミド及び株を、表1及び2の各々に記載する。プラスミドの作製は、Sambrook et al.[A Laboratory Manual (1989) CSH]に記載された標準的な分子生物学的技術を使用して実施した。
【0083】
【表1】
【0084】
【表2】
【0085】
【表3】
【実施例】
【0086】
実施例1:置換カセットの構築
PCR増幅のためのDNAの起源として、E.coli B BL21(DE3)株を使用した。オリゴヌクレオチドプライマー(LA_BL_GII_F/LA_BL_GII_R)(表3)を合成して、SacII/EcoRIクローニング部位を含むKpsMの5’末端の上流の1001bp領域を標的とした。左アーム(LA)として、ゲノムDNAから当該領域を増幅した。右アーム(RA)に関しては、オリゴヌクレオチドプライマー(RA1_BL_GII_F/RA1_BL_GII_R)(表3)を合成して、BamHI/SacIクローニング部位を含むKpsFの5’末端の上流の1108bp領域を標的とした。左アーム及び右アームのPCR産物を産生した。以下のパラメータ:94℃で50秒間の変性、57℃で60秒間のアニーリング、及び72℃で60秒間のプライマー伸長を全部で30サイクル含むプログラムを使用して、サーマルサイクラーにおいてDNA増幅反応を実施した。正確なバンドサイズのPCR増幅産物を1%アガロースゲル上でゲル電気泳動によって観察して、50μlの増幅産物をゲルから抽出して、置換断片を構築するために使用した。オリゴヌクレオチド配列のリストを表3に示す。
【0087】
増幅した左アームは、ゲルから精製して、プラスミドpEZ−Tに連結し、プラスミド−T−LAを製造した。制限酵素EcoRI/BamHIによって、FRT−cat−FRT断片を消化して、pEZ−T−LAのEcoRI/BamHI部位に連結し、pEZ−T−LA−FRT−cat−FRTをもたらした。増幅した右アームをプラスミドpGEM−Tに連結して、BamHI/SacIで消化した。アンピシリン耐性遺伝子を有するBamHI/SacI消化したDNA断片を、プラスミドpEZ−LA−FRT−cat−FRTに連結した。クロラムフェニコール耐性遺伝子は、置換断片を同定するための選択マーカーを提供する。得られたプラスミドpEZ−LA−FRT−cat−FRT−RAを使用して、ノックアウトBL21(DE3)株を製造した。
【0088】
実施例2:ノックアウトBL21(DE3)株の製造
LB培地で一晩にわたって37℃においてE.coli BL21(DE3)を増殖させ、BL21(DE3)のヒートショックコンピテント細胞を標準的な方法によって調製した。プラスミドpKD46でBL21(DE3)コンピテント細胞を形質転換して、形質転換した細胞を0.4OD600nmまで100μg/mlのアンピシリンを添加したLB培地中で増殖させた。Redリコンビナーゼの発現は、L−アラビノース(終濃度1mmol/l)によって1時間にわたって誘導した。
【0089】
グループII遺伝子クラスターノックアウトについては、pEZ−T−LA−FRT−cat−FRT−RAから増幅した置換断片の500ngを、BL21(DE3)コンピテント細胞に、エレクトロポレーションによって移した。エレクトロポレーションコンピテント細胞は標準的な方法で調製した。細胞の回収は、30℃で1.5時間にわたって実施した。8μg/mlのクロラムフェニコールを添加したLB寒天に細胞をプレーティングした。BL21(DE3)ΔkpsM−kpsF)::catのクロラムフェニコール耐性コロニーが、30℃における一晩のインキュベーション後に形成された。
【0090】
染色体の正しい位置におけるグループII遺伝子クラスターの完全なノックアウトを確実なものとするために、グループII遺伝子クラスターの左及び右アームの外側に相同性を有するオリゴヌクレオチドプライマーを設計した。近いフランキング領域の高い相同性に基づいて、2つのゲノムのグループIIクラスターの類似している位置を推定して、E.coli 536(Accession no.CP000247.1)の18kbの公開配列の外側のE.coli B BL21(DE3)のフランキング配列の欠失をPCRスクリーニングプライマーの設計のために使用した。3つのクロラムフェニコール耐性コロニーを遺伝子置換について試験した。
【0091】
クロラムフェニコール耐性コロニーの表現型試験を、少なくとも2世代にわたってLB+Cm(25μg/ml)における増殖によって実施した。クロラムフェニコール耐性コロニーの全てが、LB+Cm(25μg/ml)培地において非常に良好に増殖することができるであろう。クロラムフェニコール耐性D−アラニン栄養要求性株も、D−アラニンを添加して同様の方法において試験した。
【0092】
置換の成功が、1686bp及び1578bpのバンドによって確認された。
【0093】
オリゴヌクレオチドプライマー(LA_SEQ_F1/Cat_ID_R)を使用して、左アーム及びcat遺伝子の一部を含むバンド(1686bpのサイズ)を増幅し、オリゴヌクレオチドプライマー(RA_SEQ_R1/Cat_SEQ_F)を使用して、右アーム及びcat遺伝子の一部を含むバンド(1578bpのサイズ)を増幅した。3つのクロラムフェニコール耐性コロニーに関しては、バンドの正確なサイズによって、16kbのグループII遺伝子クラスターがcat遺伝子によって置換されたことが示された(図3)。kpsF及びkpsEの一部を含む右アームを含む3000bpのバンドのネガティブコントロールを作製した。これは、コントロール株(置換前)にのみ現れて、クロラムフェニコール耐性株には現れないべきである。オリゴヌクレオチドプライマー(RA6_BL_GII_F/RA6_BL_GII_R)(表3)を使用して、kpsF及びkpsEを増幅した。PCRの結果は、対照株のみ当該バンドを有し、クロラムフェニコール耐性株が有しないことを示した。
【0094】
LB培地における増殖の間にD−アラニンの添加を必要とする、D−アラニン栄養要求株であるBL21(DE3)ΔdadxΔalrに同じ消去法を適用した。
【0095】
クロラムフェニコール耐性株E.coli B BL21(DE3)Δ(kpsM−kpsF)::cat及びBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)::catを、それらの表現型及びPCRスクリーニングによって確認し、グループII莢膜遺伝子クラスターがcat遺伝子によって置換されたことを確認した。
【0096】
実施例3:薬剤耐性マーカーの除去及びグループIIノックアウト株の特性決定
凍結ストックからのE.coli BL21(DE3)Δ(kpsM−kpsF)::cat及びBL21(DE3)ΔdadXΔalr(kpsM−kpsF)::cat細胞を、LB培地において、37℃で、25μg/mlのクロラムフェニコール又は第二の株には25μg/mlのクロラムフェニコールに100mMのD−アラニンとともに増殖させた。2つのノックアウト株のエレクトロポレーションコンピテント細胞を標準的な方法によって調製した。プラスミドpCP20(CmR、AmpR)で2つのノックアウトコンピテント株を形質転換した。薬剤耐性マーカーの除去は、穏やかに振盪しながら1時間にわたって30℃においてSOCブロスで形質転換した細胞を増殖させることによって実施した。プレート状のアンピシリン耐性候補クローンの複数のシングルコロニーを、30℃で一晩培養した後に選択した。候補クローンを、LB培地のみ又はD−アラニンを添加したLB培地で37℃において3時間にわたって増殖させ、プラスミドpCP20を除去した。LB培地のみ又はD−アラニンを添加したLB培地で現れた、5つのシングルコロニーをPCRスクリーニングによってさらに特性決定した。
【0097】
LB、LB+Cm(8μg/ml)、及びLB+Ampの複数のシングルコロニーを増殖させることによって、cat除去株の表現型試験を実施した。全てのcatを除去したコロニーは、LB培地でのみ増殖できるであろう。catを除去したD−アラニン栄養要求性株については、5つのコロニーの全てがD−アラニンを添加したLB培地のみで増殖できるであろう。かくして、cat遺伝子が除去された。
【0098】
cat遺伝子の除去の成功は、2381bpバンドによって確認されるべきである。オリゴヌクレオチドプライマー(LA_SEQ_F2/RA_SEQ_R1)を使用して、左アーム及び右アームを含むバンドを増幅した。全てのシングルコロニーについて、2381bpのバンドの出現によって、cat遺伝子が除去されたことが示された。
【0099】
ノックアウト株E.coli B BL21(DE3)Δ(kpsM−kpsF)及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)は、それらの表現型及びPCRスクリーニングによって確認した。双方の株のグリセロールストックを保存のために調製した。
【0100】
実施例4:振盪フラスコ中におけるノックアウト宿主株の増殖
凍結ストックからのE.coli B BL21(DE3)及びB BL21(DE3)Δ(kpsM−kpsF)細胞を、LBプレートで一晩にわたって37℃で増殖させた。一晩たったプレートから回収した適当量の細胞を、25mlのLB培地を含むバッフル付き250ml振盪フラスコ(baffled shake flask)に約0.05 OD600の濃度まで植菌した。220rpmで振盪しながら8時間にわたって、細胞を37℃で増殖させた。各株のサンプルを1/2時間毎に回収し、OD600で細胞の増殖を比較した。
【0101】
振盪フラスコにおける増殖の比較のために、D−アラニンを添加して、同一の作業順序をE.coli B BL21(DE3)ΔdadXΔalr及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)に対して実施した。
【0102】
図4の結果は、BL21Δ(kpsM−kpsF)の一晩の増殖は、LB培地を用いた振盪フラスコにおいて、親株BL21(DE3)と同一であったことを示す。BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)をBL21(DE3)ΔdadXΔalrと比較した際も同じ結果が得られるであろう。
【0103】
実施例5:高密度発酵における宿主株BL21(DE3)及びBL21(DE3)Δ(kpsM−kpsF)の間の増殖及びMEAE−hGH発現の比較
凍結ストックからのE.coli B BL21(DE3)Δ(kpsM−kpsF)細胞をLBプレート上で増殖させた。ノックアウト株のヒートショックコンピテント細胞を標準的な方法によって調製した。hGHの発現カセットを含むプラスミドpNNC1−g(pET11d−MEAE−hGH(AmpR))で、B BL21(DE3)Δ(kpsM−kpsF)コンピテント細胞を形質転換し、そこから形質転換した細胞のグリセロールストックを調製した。
【0104】
高密度発酵は、上述のとおり好気条件下で実施した。
【0105】
図5は、BL21Δ(kpsM−kpsF)の経時的な増殖が、複合培地中で高密度発酵槽において、親株BL21(DE3)と同一であったことを示す。同じ結果が、BL21(DE3)Δ(kpsM−kpsF)/pNNC1−gをBL21(DE3)/pNNC1−gと比較した際にも得られるであろう。非誘導状態において、細胞密度が約140のOG600に達した。hGHの発現は、細胞の増殖が80のOD600に達した際に、1mMの濃度までIPTGを添加することによって実施した。30℃におけるIPTG誘導の10時間後に、細胞密度が約100のOD600に達した。
【0106】
E.coli B BL21(DE3)/pNNC1−g及びB BL21(DE3)Δ(kpsM−kpsF)/pNNC1−gは、SDS−PAGEで認められるとおり、hGHの製造に有効である。hGH発現レベルにおける増大は、SDS−PAGEゲルで示される1から10時間の誘導時間の増大にしたがう(図6)。hGHを製造する2つの株の能力は同一である。すなわち、遺伝子IIクラスターを有するB BL21(DE3)と比較した際の、宿主株B BL21(DE3)Δ(kpsM−kpsF)によるhGH発現における顕著な差異は認められなかった。
【0107】
実施例6:高密度発酵における宿主株BL21(DE3)ΔalrΔdadX及びBL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)の間の増殖及びMEAE−hGH発現の比較
凍結ストックからのE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)細胞を、D−アラニンを添加したLB培地で増殖させた。ノックアウト株のヒートショックコンピテント細胞を標準的な方法によって調製した。D−アラニン補足遺伝子を含むpNNC1−1h(pET11d−MEAE−hGH及びalrP−alrCD(AmpR))をAatII/PsiIを使用して消化し、その後の平滑末端及びプラスミドのセルフライゲーションによって、プラスミド上のalr遺伝子の選択のためのbla−プラスミドを得た。プラスミドpNNC1−1h(pET11d−MEAE−hGH,alrP−alrCD,bla−)で、BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)コンピテント細胞を形質転換した。D−アラニンを添加していないLBプレート上に一晩であらわれたコロニーを、グリセロールストックの調製のために単離した。
【0108】
高密度発酵を図5に記載のとおりに実施した。
【0109】
図7は、BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hの経時的な増殖が、FMC1培地中で高密度発酵槽において、親株BL21(DE3)ΔdadXΔalr/pNNC1−1hと同一であったことを示す。同じ結果が、B BL21(DE3)ΔdadXΔalr/pNNC1−1hをBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hと比較した際にもhGHの誘導下で得られるであろう。非誘導状態において、細胞密度が約120のOG600に達した。hGHの発現は、細胞の増殖が80のOD600に達した際に、1mMの濃度までIPTGを添加することによって実施した。30℃におけるIPTG誘導の9時間後に、細胞密度が約100のOD600に達した。
【0110】
E.coli B BL21(DE3)ΔdadXΔalr/pNNC1−1h及びB BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)/pNNC1−1hは、SDS−PAGEによる結果で認められるとおり、hGHの製造に有効である(図8)。hGH発現レベルにおける増大は、誘導時間の増大にしたがう。hGHを製造する2つの株の能力は同一である。
【0111】
実施例7:高細胞密度発酵における宿主株E.coli B BL21(DE3)ΔalrΔdadXΔ(kpsM−kpsF)を使用した細胞増殖及びMEAE−hGH(L101C)の発現の評価
凍結ストックからのE.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)細胞を、D−アラニンを添加したLB培地で増殖させた。ヒートショック方法を使用する形質転換のために、このトリプルノックアウト株のコンピテント細胞を標準的な方法によって調製した。
【0112】
アンピシリン耐性をコードするβ−ラクタマーゼ遺伝子を欠失するために、MEAE−hGH(L101C)をコードし、かつ、宿主細胞のD−アラニン欠乏を補足するalrP−alrCDを含むプラスミドpNNC1a.23(pET11d−MEAE−hGH(L101C))をAatII/PsiIで消化し、その後に末端平滑化及びβラクタマーゼ遺伝子の除去のためのセルフライゲーションを実施した。得られたプラスミドであるpNNC1a.23(pET11d−MEAE−hGH,alr,bla−)で宿主細胞であるBL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)をヒートショック法によって形質転換した。D−アラニンを添加していないLBプレートで増殖したシングルコロニーを選択して、−80℃における保存のためのグリセロールストックを調製した。
【0113】
2つのバッチの高細胞密度発酵を実施例5に記載のとおりに実施した。プロセスの間の細胞増殖を観察した。結果は、図9に示す。
【0114】
プラスミドpNNC1a.23を有する株E.coli B BL21(DE3)ΔdadXΔalrΔ(kpsM−kpsF)によるMEAE−hGH(L101C)の発現を高細胞密度発酵において評価した。結果は、回収時の細胞密度が、OD600nmで測定した際に約100に達し得ることを示し、標的タンパク質であるMEAE−hGH(L101C)の発現レベルが、図10に示すとおり、8時間の誘導の後に約7g/lに達した。
【0115】
実施例8:E.coli BL21(DE3)Δ(kpsM−kpsF)におけるアースロバクター ウレアファシエンス(Arthrobacter ureafaciens)シアリダーゼの発現
アースロバクター ウレアファシエンスのシアリダーゼ(AUS,53.4kDa理論的質量)を、C末端のHisタグとともに大腸菌BL21(DE3)Δ(kpsM−kpsF)で製造した。C末端にHis6タグを有する、アースロバクター ウレアファシエンスのシアリダーゼの遺伝子は、T7プロモーターの制御の下に発現ベクターpET−24a(Novagen)にクローニングした。アースロバクター ウレアファシエンス由来のシアリダーゼ酵素を製造するために、以下のタンパク質配列をコードする合成コドン最適化遺伝子を産生した(SEQ ID NO:12):
MAPTPPNSPTLPPGSFSETNLAADRTAANFFYRIPALTYLGNDVVLAAWDGRPGSAADAPNPNSIVQRRSTDGGKTWGPVQVIAAGHVADASGPRYGYSDPSYIYDAEANKVFAFFVYSKDQGFGGSQFGNDDADRNVISSAVIESSDAGVTWSQPRLITSVTKPGTSKTNPAAGDVRSNFASSGEGIQLKYGPHKGRLIQQYAGDVRQADGSNKIQAYSVYSDDHGVTWHKGANVGDRMDENKTVELSDGRVLLNSRDNANRGYRKVAVSTDGGATYGPVSQDTELPDPANNGAIARMFPNAAQGSADAKKLIFTNANSKTGRENVSARVSCDDGETWPGVRTIRSGFSAYSTVTRLADGKFGVLYEGNYTDNMPFATFDDAWLNYVCAPLAVPAVNIAPSATQEVPVTVTNQEATTLSGATATVYTPSGWSATTVPVPDVAPGASVTVTVALTAPADASGPRSLNAAFTTADGRVSQFTFTATTPVAPQVGLTITGSALEHHHHHH。
【0116】
合成遺伝子は、pET−24a発現ベクターにXbaI−XhoI断片として導入し、AUS発現ベクターpLLC028を製造した。得られたベクターで、コンピテントBL21(DE3)Δ(kpsM−kpsF)を形質転換し、形質転換体を−80℃でグリセロール中で保存した。
【0117】
凍結した最初の細胞クローン(ICC)の1つの凍結チューブ(cryotube)を使用して、2つの酵母抽出物媒体(酵母抽出物5g/l、グルコース10g/l、寒天20g/l)寒天フラスコに植菌した。寒天媒体には選択のためにカナマイシンを含めた。インキュベーションを30℃で25時間にわたって実施した。インキュベーションの後に、滅菌0.9%NaCl溶液を使用して細胞を寒天表面から洗って回収した。10L発酵は、2つの寒天フラスコからの前培養物を使用して植菌した。得られた植菌材料の容量は75mで、OD600nmは4.3であり、発酵槽の始めのOD600nmは約0.03であった。発酵は、10Lの開始作業容量で20Lステンレススチール容器で実施した。発酵は、40g/l酵母抽出物、15g/lグリセロール(87%)、塩、及び微量金属を含む複合培地に対して8時間のバッチフェーズで開始し、続いて、所定のプロファイルでグリセロールを加えた。最初の17時間は、発酵温度は37であった。その後、温度を30℃まで下げた。発酵時間が18時間経過した時点で、誘導因子であるIPTGを0.1mMで添加して、タンパク質発現を誘導した。誘導の期間は22時間までであり得る。回収プロセスは、遠心分離、高圧ホモジナイゼーション、0.2μmの孔径を有するSartocon Slice Cassetteを使用したクロスフロー精密濾過からなる。
【0118】
AUS発酵は、SDS−PAGE及び酵素活性アッセイを使用して分析した。SDS−PAGE分析によって、発現したシアリダーゼが、ビーズミリングの後に、可溶性及びペレット画分の双方にあらわれることが明らかになった。図11は、30℃の誘導温度及び0.1mMのIPTGで得られた10Lバッチに由来する典型的な発酵の結果を示す。
【0119】
実施例9:BL21 DE3及びBL21 DE3 Δ(kpsM−kpsF)におけるヒト線維芽細胞増殖因子21(FGF21)の発現
ヒトFGF21のDNA及びアミノ酸配列は、例えば、Biochim. Biophys. Acta 1492(1):203−206 (2000)のNishimura et alに開示されている。前記配列は、公開データベースからも得られる(各々、Accession nos. EMBL:AB021975及びUNIPROT:Q9NSA1)。
【0120】
天然のポリペプチドは、分泌のための28アミノ酸のシグナルペプチド(以下に斜体で記載)とともに合成されるが、成熟FGFポリペプチドは、残部の181アミノ酸からなる(SEQ ID NO:13):
MDSDETGFEHSGLWVSVLAGLLLGACQAHPIPDSSPLLQFGGQVRQRYLYTDDAQQTEAHLEIREDGTVGGAADQSPESLLQLKALKPGVIQILGVKTSRFLCQRPDGALYGSLHFDPEACSFRELLLEDGYNVYQSEAHGLPLHLPGNKSPHRDPAPRGPARFLPLPGLPPA-LPEPPGILAPQPPDVGSSDPLSMVGPS QGRSPSYAS。
【0121】
シグナルペプチドはないが、N末端のメチオニンを添加して、成熟FGF21ポリペプチドをクローニングして、E.coliで細胞内タンパク質として発現させた。特に、3’末端にメチオニンのATGコドン並びにNde1及びBamH1制限酵素部位を3’末端及び5’末端の各々に含む550bpのコード領域を、ファージT7プロモーターの制御下のNde1−BamH1において発現ベクターpET 11c(Novagen)に挿入して、E.coli B BL21(DE3)及びE.coli B BL21 DE3 Δ(kpsM−kpsF)を形質転換した。細胞をLB amp 100μg/ml中においてOD450が0.5になるまで増殖させ、0.3mM IPTGで4時間にわたって37℃で発現を誘導した。FGF21発現分析のために細胞の粗抽出物を超音波処理によって作製した。
【0122】
図12のクーマシー染色したSDS−PAGEゲルは、E.coli B BL21 DE3およびE.coli B BL21 DE3Δ(kpsM−kpsF)におけるFGF21の発現が等しく成功したことを示す。発現したFGF21(Met−FGF21)の計算分子量(Mw)は19.5kDであるが、ゲル上では25kDタンパク質として移動し、これはタンパク質の移動を遅らせるプロリンが多く含まれているためであろう。
【0123】
本実施例は、FGF21の産生量は宿主細胞の遺伝子操作によって影響を受けないことを示す。
【0124】
実施例10:BL21 DE3及びBL21 DE3Δ(kpsM−kpsF)におけるヒトアミリンの発現
37アミノ酸のアミロイドペプチドであるアミリンの配列は、Proc. Natl. Acad. Sci. (84) : 8628−8632, (1987)のCooper et al.によって公開された。遺伝子をクローニングして、サーモトガ・マリティマ(thermotoga maritima)由来のリボソームタンパク質RL23にN末端に融合した。146アミノ酸の融合タンパク質は、以下の配列(SEQ ID NO:14)を有する:
KQEKLSLHDVLIRPIITEKALILREQRKYVFEVNPLANKNLVKEAVEKLFNVKVEKVNILNMKPKPKRRGIFEGKTRSWKKAVVTLKEGYTIKELEGEHSSSSDDDDKKCNTATCATQRLA-NFLVHSSNNFGAILSSTNVGSNTY。
【0125】
前記遺伝子を、実施例9に記載のとおり、Nde1−BamH1断片として、T7プロモーターの制御下においてpET11cにクローニングして、プラスミドを使用してBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)を形質転換した。
【0126】
発現は実施例9に記載のとおり実施し、結果は図12に示す。図12は、2種のE.coli B宿主であるBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)における等しく高い産生量の発現を示す。
【0127】
実施例11:BL21 DE3及びBL21 DE3Δ(kpsM−kpsF)におけるヒトレプチンの発現
146アミノ酸長のヒトレプチンの配列は、Diabetes (44): 855−858 (1995)のMasuzaki et alに公開されている。前記タンパク質は、21アミノ酸のシグナルペプチドが添加されてヒトにおいて分泌される。E.coliで細胞内に発現させた成熟レプチンは、以下の配列(SEQ ID NO:15)を有する:
MVPIQKVQDDTKTLIKTIVTRINDISHTQSVSSKQKVTGLDFIPGLHPILTLSKMDQTLAVYQQILTSMPSRNVIQISNDLENLRDLLHVLAFSKSCHLPWASGLETLDSLGGVLEASGYSTEVVALSRLQGSLQDMLWQLDLSPGC。
【0128】
レプチン遺伝子は、実施例9に記載のとおり、450bpのNde1−BamH1断片として、T7プロモーターの制御下においてpET 11cにクローニングし、このプラスミドを使用してBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)を形質転換した。
【0129】
発現は実施例9に記載のとおりに実施した。結果は図12に示す。図12は、2種のE.coli宿主であるBL21 DE3及びBL21 DE3Δ(kpsM−kpsF)における等しく高い産生量の発現を示す。
【0130】
グループII莢膜遺伝子クラスターの除去は、いずれの増殖条件における細胞の増殖にも影響を与えることがないと結論付けられる。グループII莢膜遺伝子クラスターの除去がタンパク質発現に影響を与えないとも結論付けられる。したがって、治療用タンパク質製造のための市販の宿主株としてE.coli B BL21(DE3)を使用するための、グループII莢膜遺伝子クラスターを完全に取り除いたより安全な株がこれにより製造される。E.coli B BL21(DE3)のグループII莢膜遺伝子を欠失する遺伝子操作によって、E.coli K12と同じ安全カテゴリーになる。グループII遺伝子クラスターが、これら2種の株の唯一の顕著な違いだからである。
【特許請求の範囲】
【請求項1】
不可逆的に不活性化されたグループII莢膜遺伝子クラスターを有することを特徴とする、E.coli B BL21株。
【請求項2】
グループII莢膜遺伝子クラスターの全体又は一部の欠失を有することを特徴とする、請求項1に記載のE.coli B BL21株。
【請求項3】
グループII莢膜遺伝子クラスターの全体の欠失を有することを特徴とする、請求項1に記載のE.coli B BL21株。
【請求項4】
前記E.coliがE.coli B BL21(DE3)である、請求項1から3のいずれか一項に記載のE.coli B BL21株。
【請求項5】
前記E.coliがE.coli B BL21(DE3)ΔdadXΔalrである、請求項1から3のいずれか一項に記載のE.coli株。
【請求項6】
ペプチドをコードする挿入DNA配列を有する発現ベクターも含む、請求項1から5のいずれか一項に記載のE.coli B BL21株。
【請求項7】
前記ペプチドが、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、並びに酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアントからなる群から選択される、請求項1から6のいずれか一項に記載のE.coli B BL21株。
【請求項8】
前記グループII莢膜遺伝子クラスターが、
a.選択マーカーによってグループII莢膜遺伝子クラスターを置換する工程、
b.前記選択マーカーを除去する工程、
c.PCR分析によって欠失を確認する工程
を含む方法によって欠失される、請求項1から7のいずれか一項に記載のE.coli B株の製造方法。
【請求項9】
前記選択マーカーが抗生物質耐性遺伝子である、請求項8に記載のE.coli B株の製造方法。
【請求項10】
前記抗生物質耐性遺伝子が、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン、及びエリスロマイシンからなる群から選択される、請求項9に記載のE.coli B株の製造方法。
【請求項11】
前記抗生物質がクロラムフェニコールである、請求項9に記載のE.coli B株の製造方法。
【請求項12】
請求項1から7のいずれか一項に記載のE.coli B株を適切な培養培地で培養して、その後に、よく知られた技術によって発現したペプチドを単離及び精製することを含む、ペプチドの製造方法。
【請求項13】
前記ペプチドがhGHである、請求項12に記載の方法。
【請求項14】
前記ペプチドがFGF−21である、請求項12に記載の方法。
【請求項15】
前記ペプチドがアミリンである、請求項12に記載の方法。
【請求項1】
不可逆的に不活性化されたグループII莢膜遺伝子クラスターを有することを特徴とする、E.coli B BL21株。
【請求項2】
グループII莢膜遺伝子クラスターの全体又は一部の欠失を有することを特徴とする、請求項1に記載のE.coli B BL21株。
【請求項3】
グループII莢膜遺伝子クラスターの全体の欠失を有することを特徴とする、請求項1に記載のE.coli B BL21株。
【請求項4】
前記E.coliがE.coli B BL21(DE3)である、請求項1から3のいずれか一項に記載のE.coli B BL21株。
【請求項5】
前記E.coliがE.coli B BL21(DE3)ΔdadXΔalrである、請求項1から3のいずれか一項に記載のE.coli株。
【請求項6】
ペプチドをコードする挿入DNA配列を有する発現ベクターも含む、請求項1から5のいずれか一項に記載のE.coli B BL21株。
【請求項7】
前記ペプチドが、hGH、グルカゴン様ペプチド、インターロイキン、インスリンアナログ、野生型アジポネクチン、FGF−21、トリプシン、アプロチニン、アミリン及びレプチン並びにそれらのアナログ、並びに酵素、例えば、シアリダーゼ、トランスグルタミナーゼ(tGase)、HRV(ヒトライノウイルス)3Cプロテアーゼ、タバコエッチウイルス(TEV)プロテアーゼ、及びそれらのバリアントからなる群から選択される、請求項1から6のいずれか一項に記載のE.coli B BL21株。
【請求項8】
前記グループII莢膜遺伝子クラスターが、
a.選択マーカーによってグループII莢膜遺伝子クラスターを置換する工程、
b.前記選択マーカーを除去する工程、
c.PCR分析によって欠失を確認する工程
を含む方法によって欠失される、請求項1から7のいずれか一項に記載のE.coli B株の製造方法。
【請求項9】
前記選択マーカーが抗生物質耐性遺伝子である、請求項8に記載のE.coli B株の製造方法。
【請求項10】
前記抗生物質耐性遺伝子が、クロラムフェニコール、テトラサイクリン、アンピシリン、カナマイシン、バンコマイシン、及びエリスロマイシンからなる群から選択される、請求項9に記載のE.coli B株の製造方法。
【請求項11】
前記抗生物質がクロラムフェニコールである、請求項9に記載のE.coli B株の製造方法。
【請求項12】
請求項1から7のいずれか一項に記載のE.coli B株を適切な培養培地で培養して、その後に、よく知られた技術によって発現したペプチドを単離及び精製することを含む、ペプチドの製造方法。
【請求項13】
前記ペプチドがhGHである、請求項12に記載の方法。
【請求項14】
前記ペプチドがFGF−21である、請求項12に記載の方法。
【請求項15】
前記ペプチドがアミリンである、請求項12に記載の方法。
【図1】

【図6】

【図8】
【図10】
【図11】

【図2】
【図3】
【図4】
【図5】
【図7】
【図9】
【図12】
【図6】
【図8】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図7】
【図9】
【図12】
【公表番号】特表2012−508001(P2012−508001A)
【公表日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願番号】特願2011−535134(P2011−535134)
【出願日】平成21年11月10日(2009.11.10)
【国際出願番号】PCT/EP2009/064903
【国際公開番号】WO2010/052335
【国際公開日】平成22年5月14日(2010.5.14)
【出願人】(511099685)
【Fターム(参考)】
【公表日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願日】平成21年11月10日(2009.11.10)
【国際出願番号】PCT/EP2009/064903
【国際公開番号】WO2010/052335
【国際公開日】平成22年5月14日(2010.5.14)
【出願人】(511099685)
【Fターム(参考)】
[ Back to top ]