
機能性核酸を用いた分子センサー
【課題】特殊なプローブや検出機器を用いなくとも、目的分子の存在、濃度を簡便に検出し得る機能性核酸及びこれを用いて目的分子を検出する方法を提供する。
【解決手段】本発明の機能性核酸分子は、(a)レポーター遺伝子配列、(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含む。目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成し、そして目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。
【解決手段】本発明の機能性核酸分子は、(a)レポーター遺伝子配列、(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含む。目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成し、そして目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、機能性核酸を用いた分子センサーに関し、より詳細には、目的分子との結合により自己切断酵素活性をもつ機能性核酸(アプタザイム)とレポーター遺伝子とを含む核酸分子を用いて、目的分子を検出する方法、及びこのような方法に用いる核酸分子等に関する。
【背景技術】
【0002】
近年、インビトロで選択されたRNAアプタマー、又はDNAアプタマーは、種々の分子に対するセンサーとして大きな注目を集めている。アプタマーは、目的とする有機分子、イオン、タンパク質、又は細胞等を特異的に認識することができる。この認識機構は、アプタマー分子内に存在するリン酸基や有機塩基の有する疎水性及び親水性の両方の性質によって形成されている。アプタマーがその目的分子と複合体を形成すると分子量の増加をもたらし、目的分子が大きい場合には、質量検出器によって直接検出することができる。また、いくつかのアプタマーは目的分子との結合により構造変化を起こす。このような構造変化と、蛍光的又は電気化学的方法とを組み合わせることにより、低分子量の目的分子でも検出することが可能である。ただし、その際は、アプタマーを直接的若しくは間接的に蛍光色素又は電気応答性物質で標識しなければならない。
【0003】
「アプタザイム」又は「アロステリックリボザイム」と称される核酸は、特定の分子(コファクター)によってアロステリックに活性化される酵素機能を有し、センサーとして利用可能である。アプタザイムは、アプタマードメイン及びリボザイムドメインの両方を分子内に有し、コファクターとの複合体形成という信号を、例えば自己切断のようなより明確な触媒活性へ容易に伝達することができる。
【0004】
例えば、国際公開公報WO03/014375号パンフレット(特許文献1)には、アプタザイムを蛍光標識することによって、目的分子の結合を光学的に検出するシステムが開示されている。また、国際公開公報WO00/26226号パンフレット(特許文献2)には、アプタザイムの5’末端を放射性同位元素である32Pで標識して切断されたアプタザイムを検出する方法が開示されている。質量変化を測定する方法としては、アプタザイムを金表面に固定化し、特定の分子との結合によって誘起された酵素活性によるアプタザイムの質量変化を測定する方法が開示されている(例えば、特許文献3参照)。
【0005】
一方、塩基配列の相同性に基づいてDNAを検出する方法として、分子ビーコン法(例えば、非特許文献1参照)が知られている。この方法は、目的配列と相補的な配列をループドメインに有するヘアピン構造のオリゴヌクレオチドプローブを用いて、例えば、5’末端及び3’末端に標識されたFRETを検出する方法である。さらに、無細胞タンパク質合成系において、分子ビーコン型リボ核酸と連結したレポーター遺伝子の発現を指標として、目的とするDNAを検出する方法が報告されている(例えば、非特許文献2参照)。しかしながら、これらの方法は核酸配列の相補性に基づくため、DNA等の核酸分子の検出に利用できるに過ぎず、低分子化合物やペプチド、タンパク質等の検出は不可能である。
【0006】
【特許文献1】国際公開公報WO03/014375号パンフレット
【特許文献2】国際公開公報WO00/26226号パンフレット
【特許文献3】米国出願公開公報第20040175693号
【非特許文献1】Tyagi, S., Kramer, F.R., Nat. Biotechnol. 1996, Vol. 14, pp.303-308
【非特許文献2】Sando, S., Narita A., Abe, K., and Aoyama Y., J. Am. Chem. Soc., 2005, Vol.127, pp.5300-5301
【発明の開示】
【発明が解決しようとする課題】
【0007】
上記従来技術によれば、アプタザイムを用いて目的分子を検出するためには、何れも蛍光物質や放射性同位元素等で標識する必要があり、そのための時間やコスト、さらには特殊な検出装置が必要になるという課題がある。また、これらの標識のために不安定なRNAを直接用いなければならないという問題点もある。本発明は、特殊なプローブや検出機器を用いなくとも、目的分子の存在、濃度を簡便に検出し得る機能性核酸及びこれを用いて目的分子を検出する方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明はかかる課題を解決するためになされたものであって、目的分子の存在下において自己切断触媒活性を示すアプタザイムと、タンパク質の翻訳段階におけるレポーター遺伝子の発現制御機構とを組み合わせることによって完成されたものである。
【0009】
すなわち、第一の観点において、目的分子を検出するための本発明の機能性核酸分子は、(a)レポーター遺伝子配列、(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含むことを特徴とする。目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成する。そして目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、それによって前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。翻訳反応系が共存すると、当該核酸分子のリボソーム結合部位にリボソームが結合して翻訳が開始され、前記レポーター遺伝子が発現される。目的分子に対するアプタザイムを選択することによって所望の目的分子を検出することが可能であるが、検出対象としては、例えば、低分子化合物、ペプチド及びタンパク質等が挙げられる。
【0010】
別の観点において、本発明は上記核酸分子をコードする二本鎖DNAであって、当該コード領域の上流に機能的に連結された転写プロモーター配列を含む二本鎖DNAを提供する。転写プロモーター配列としては、用いる翻訳反応系において機能しうるものであれば特に限定されないが、強力なプロモーター活性を有する、ファージ由来のT7プロモーターが好ましい。
【0011】
さらに異なる観点において、本発明は上記核酸分子を用いる目的分子の検出方法を提供する。当該検出方法は、上記核酸分子と、目的分子を含む試料とを、翻訳反応系内において所定の時間接触させる工程、及び前記レポーター遺伝子の発現量を測定する工程、を含むことを特徴とする。前記核酸分子が二本鎖DNAの場合は、転写/翻訳共役反応系を用いることができる。
【0012】
さらになお異なる観点において、本発明は、目的分子の検出のための本発明の核酸分子を含むキット、及び本発明の核酸分子によって形質転換された原核生物の細胞等を提供するものである。
【発明の効果】
【0013】
本発明の方法によれば、従来技術のように蛍光標識や放射性標識を行うことなく、且つ特殊な機器を用いなくとも目的分子を検出することが可能である。また、不安定であるRNAを直接扱わなくても、より安定なDNAをセンサー前駆体として用いることができる。本発明に係るDNAには転写プロモーターが導入されており、用いる翻訳系にRNAポリメラーゼを添加することによってRNAを容易に合成できるからである。機能性RNA自身をセンサーとして用いることも可能であるが、センサー前駆体であるDNAを用いた方がより効率的であり感度も向上するという知見が得られている。
【発明を実施するための最良の形態】
【0014】
(定義)
本明細書中で使用される用語「リボザイム」とは、酵素のような作用を有するRNAのオリゴヌクレオチドであり「RNA酵素」若しくは「核酸酵素」と呼ばれる場合もある。一般的に、これらはRNA分子の切断を触媒するエンドリボヌクレアーゼ作用を示す。切断部位の位置は高度に配列特異的である。リボザイムの中には、ポリメラーゼ又は脱リン酸化酵素のように作用するものもある。
【0015】
用語「アプタマー」とは、標的リガンドに最適に結合するように特異的に選択される核酸をいう。標的リガンドとしては、増殖因子、酵素、受容体、膜タンパク質、ウイルスタンパク質等の種々のタンパク質が挙げられるが、金属イオンや分子量の小さい有機化合物、ペプチド、ウイルス等とも結合することが分かってきた。標的リガンドに特異的なアプタマーを探し出す方法としては、SELEX法(試験管内人工進化法 Systematic Evolution of Ligands by Exponential Enrichment)(Tuerk, C., Gold, L., Science, 1990 Vol. 249, pp.505-510参照)が知られている。この方法は、標的とする目的分子に対し、ランダムな配列をもつ多数のオリゴヌクレオチドの混合物であるオリゴヌクレオチドライブラリを作用させることにより、親和性の高いオリゴヌクレオチドを選び出し、選び出されたオリゴヌクレオチドを増幅生産するという作業を繰り返す。これによって、標的とする目的分子に対し特異的に結合するオリゴヌクレオチドを得る方法である。
【0016】
用語「アプタザイム」とは、エフェクターによって調節されるリボザイム又は核酸酵素を意味する。アプタザイムは、典型的にはエフェクターを認識するアプタマードメインと、触媒作用を示すリボザイムドメインを含む。エフェクターは、アプタマードメインとリボザイムドメインとの特異的な相互作用を通じてアプタザイムの触媒活性を高め、又は低下させることができる。従って、アプタザイムの活性はエフェクターの存在の有無やその量を監視するために用いられうる。この方法は、これまで種々の診断及び検出のためのアプタザイムセンサーを設計するために用いられている。アプタザイムの設計に用いる核酸分子としては、RNA(リボザイム)、DNA(デオキシリボザイム)、DNA/RNAハイブリッド、及びペプチド核酸(PNA)等が挙げられる。一般的には、リボザイムはデオキシリボザイムよりも不安定である。種々のエフェクター分子、例えば、金属イオンや低分子化合物により調節されるアプタザイムの作製方法は公知であり(米国特許第6630306号等参照)、その内容は参照により本願に組み込まれる。
【0017】
(機能性核酸分子)
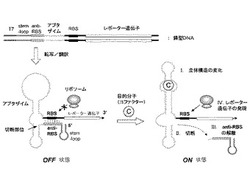
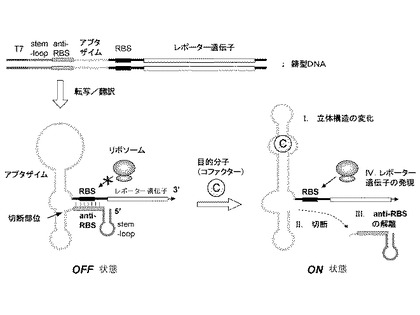
以下に図面を参照しながら本発明の機能性核酸分子について説明する。図1は、本発明の好ましい実施形態における機能性核酸分子を模式的に表したものである。図1上部の二本鎖鋳型DNAは、T7プロモーターの制御下において、レポーター遺伝子をコードする。レポーター遺伝子上流には、5’末端から順に、ステム−ループ構造、アンチ−リボソーム結合部位(アンチ−RBS)、アプタザイム配列、及びリボソーム結合部位(RBS)を含む。T7RNAポリメラーゼを含む無細胞タンパク質合成系においてこの鋳型DNAが転写されると、アンチ−RBSはRBSとハイブリダイズしてリボソームがRBSに結合することを阻害する(左下部のOFF状態)。mRNAの5’末端側(領域)に存在するステム−ループ構造は転写産物を安定化し、同時にアンチ−RBSが正確にRBSと結合して二重鎖を形成するために有用である。このようなステム−ループ構造は、少なくとも10塩基長程度の長さがあればよく、好ましくは10〜200塩基長、より好ましくは13〜100塩基長、さらに好ましくは17〜50塩基長程度である。アプタザイムに対するコファクターの存在下では、(I)アプタザイムが立体構造の変化を起こし、(II)これによって自己切断活性を誘導する。一般的に分子間で形成された二重鎖の融解温度(Tm)は、分子内二重鎖の融解温度よりも低いため、(III)所定の条件下において切断されたアンチ−RBSドメインがRBSから解離する。これによってリボソームがRBSに接触可能となり、下流のレポーター遺伝子の発現が活性化される(右下部のON状態)。
【0018】
ここで、転写プロモーターとしては、反応系において転写されるプロモーターであれば特に限定されないが、強力なプロモーターであるT7プロモーターが好ましい。また、レポーター遺伝子も翻訳系において検出可能なものであれば何れも使用可能できるが、検出が容易で感度が高いものとして、例えば、ルシフェラーゼ遺伝子やβ−ガラクトシダーゼ遺伝子等が好ましい。
【0019】
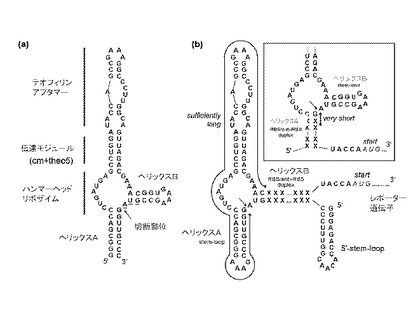
アプタザイム配列としては、目的分子によって調節可能な核酸酵素であれば特に限定されないが、好ましくは、目的分子としてテオフィリンのような低分子によってリボザイム活性が調節されるアプタザイムが用いられる。Breakerらによって試験管内で選択されたテオフィリン依存性アプタザイムが報告されている(Soukup, G. A., Emilsson, G.A.M., Breaker, R.R., J. Mol. Biol., 2000, Vol.298, pp.623-)。このアプタザイムは、図2(a)に示したように、アプタマードメイン、伝達モジュール、及びハンマーヘッドリボザイムの3つのドメインからなっている。コファクターであるテオフィリンがアプタマードメインに結合することによってリボザイムドメインの立体構造が変化し、ヘリックスAとヘリックスBの間の切断部位(矢印で示す)における自己切断活性を誘導する。この自己切断活性の発現に必要な切断部位に隣接するGUAトリプレット配列に下線を付加した。このGUAトリプレット以外の2つのヘリックスA及びヘリックスBの配列は、当該アプタザイムの酵素活性とは関係ないことが知られているため、上記RBS/アンチ−RBS二重鎖をこれら2つのヘリックス構造の何れかと置換した核酸分子を作製した。ヘリックスAの代わりにRBS/アンチ−RBS二重鎖を挿入した場合は、アプタザイム配列とRBSの間が切断されるため5’末端からRBSまでの配列が短くなり(図2(b)右上挿入図参照)効率的な翻訳が起こらなかった。一方、ヘリックスBに上記RBS/アンチ−RBS二重鎖を挿入した核酸分子では、アプタザイム配列とアンチ−RBSの間が切断されるため5’末端からRBSまでの配列が長く(図2(b)参照)効率的な翻訳が起こった。さらにこの場合、切断後のmRNAの5’末端側にステム−ループ構造(ヘリックスA)が存在するため、mRNAが安定であることも利点としてあげられる。
【0020】
リボソーム結合部位(RBS)は、mRNAの翻訳開始コドン近傍にあり、リボソームの16srRNA分子の3’末端近くにある配列と塩基対を形成してリボソーム小サブユニットと結合するために必要な配列である。大腸菌のmRNAのリボソーム結合部位は、シャイン−ダルガルノ配列とも呼ばれる。この配列はプリンを多く含んでいるという傾向はあるものの、16srRNAと完全に相補的な配列となることはあまりなく、また長さも3塩基から9塩基程度である。mRNAのRBSに結合したリボソーム小サブユニットは遺伝子の開始コドン(AUG:メチオニンに該当)までmRNA上を移動し、メチオニン−tRNAが開始コドンに結合して30s開始複合体を形成する。シャイン−ダルガルノ配列と翻訳開始コドンとの距離は、翻訳の効率と関連性のあることが知られ、最適距離は4塩基から9塩基であると推定されている。mRNA上において、このRBSが強固に二重鎖を形成しているとリボソームがRBSに接触できないため翻訳が開始されない。
【0021】
アンチリボソーム結合部位(アンチ−RBS)は、上記RBSと分子内でハイブリダイズし得るものであれば特に限定されないが、目的分子の存在又は非存在下においてのON/OFF転換効率が向上するように最適化されることが好ましい。具体的には、目的分子の非存在下では分子内でRBS/アンチ−RBS二重鎖が安定に形成されるが、目的分子の存在下では自己切断により前記二重鎖が解離しやすいことである。一般的に分子間で形成される核酸配列二重鎖の融解温度(Tm)は、分子内二重鎖の融解温度よりも低いことが知られている。DNAやRNAのTm値の計算は、種々のコンピューターソフトウェアを用いて行うことができる。最も正確なTm値の推定ソフトウェアの一つとして、例えば、HyTher(商標)が知られており、オンライン上で使用可能である(http://ozone3.chem.wayne.edu/)。Tmでは、二本鎖状態の核酸と一本鎖状態の核酸が同率比で存在しており、Tm以上では、ほとんどがハイブリダイズできずに一本鎖状態になっていると考えられる。
【0022】
本発明に係る核酸分子は、一本鎖RNA又は一本鎖DNAの何れでもよいが、20〜40℃の水溶液中において分子内で会合し、RBS/アンチ−RBS二重鎖を含むステム−ループ構造を形成する一本鎖RNAであることが好ましい。さらに好ましくは、上記RBS/アンチ−RBS二重鎖のTmは反応溶液温度程度か、それ以下である。従って、好ましい実施形態において、上記RBS/アンチ−RBS二重鎖は、当該RBSと、アンチ−RBSと、それらの隣接配列とによって形成され、その二重鎖の長さは6〜20塩基対である。より好ましくは8〜15塩基対である。当該二重鎖にはG−Uペア、又は1〜数個のミスマッチを含んでもよい。
【0023】
(レポーター遺伝子)
本発明において用いられるレポーター遺伝子は、翻訳反応系において発現し検出が容易なものであれば特に限定されないが、例えば、ルシフェラーゼ、β−ガラクトシダーゼ、アルカリフォスファターゼ、緑色蛍光タンパク質(GFP)及びエクオリン等の遺伝子を挙げることができる。ホタルルシフェラーゼはATPの存在下でルシフェリンの酸化を触媒し、光子を生成する化学ルミネッセンス反応を触媒する。ルシフェラーゼは単量体のタンパク質で翻訳後の修飾を必要としないから、遺伝子発現のレポーターとして適している。大腸菌由来のβ−ガラクトシダーゼ遺伝子(lacZ)は種々の細胞の形質転換におけるレポーター遺伝子としてよく使用される。この酵素の生体内での基質はラクトースであるが、ほぼあらゆるβ−ガラクトシドを加水分解することができ、例えば、無色の基質であるオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を加水分解して420nmの光を吸収する黄色のオルト−ニトロフェノールを生成する。また、x−galはこの酵素により分解されてインディゴブルーの呈色反応を示す。アルカリフォスファターゼは、分子触媒活性の極めて高い酵素であり、種々の蛍光及び発光基質も開発されているため高感度検出に好適である。GFPはオワンクラゲ(Aequorea Victoria)等から発見された蛍光タンパク質であり、共存する発光タンパク質であるエクオリンの青色化学発光をエネルギー変換して緑色にすることが知られている。このGFPは上記ルシフェラーゼやエクオリンのように特殊な蛍光団を必要とせず、自らアポタンパク質上で自動的に蛍光団を形成する。このため、クローン化されたGFPのcDNAを用いて多くの細菌や動物細胞等で発現され、遺伝子発現の指標として利用されている。また、GFP遺伝子に種々の突然変異を導入して多くのGFP誘導体が作製され、これらの中には野生型GFPよりも強い蛍光強度を示したり、異なる波長の蛍光を発する誘導体もあることが報告されている。
【0024】
(目的分子又はコファクター)
本発明の方法において検出可能な目的分子としては、アプタザイムのコファクターとなりうるもので、かつ翻訳反応を阻害しないものであれば特に限定されないが、例えば、低分子化合物、細菌又はウイルス由来の抗原タンパク質、毒物分子、コカイン、ヒト免疫不全ウイルス(HIV)及びHIV−由来分子などを含む。
【0025】
また、本発明の別の観点において、上記レポーター遺伝子に代えて所望の遺伝子を挿入した核酸分子は、原核生物の翻訳反応系において上記目的分子又はコファクターに相当するリガンド分子による遺伝子発現制御システムとして利用することもできる。すなわち、リガンド分子の非存在下ではアンチ−RBSがRBSに結合して当該所望の遺伝子の発現を抑制し、リガンド分子を添加することによって、前記アプタザイムが自己切断触媒活性を示し、それによって前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。その結果、前記所望の遺伝子の発現を誘導しうるのである。
【0026】
(目的分子の検出方法)
本発明の検出方法は、無細胞タンパク質合成系において、目的分子を含む試料と本発明の機能性核酸分子を所定の時間接触させることにより、目的分子の濃度に応じて発現するレポーター遺伝子の活性を測定する工程を含む。用いる機能性核酸分子としては、RNA及びこれをコードする鋳型DNAの何れでもよいが、検出感度及び取扱いの容易さの観点から二本鎖鋳型DNAを用いることが好ましい。DNAはRNAに比べて酵素的に分解されにくく安定に存在するからである。これらの核酸分子を調製する方法は、化学合成法、PCR、DNAのクローン化と複製、その他の公知技術を用いることができる。鋳型DNAとしてはPCRにより合成した増幅産物をそのまま用いることができるが、好ましくはプラスミドなどにクローン化し、組換え体を用いて大量に調製及び精製したDNAの方が検出感度に優れる。RNAの場合は、無細胞系として翻訳反応系を用いるが、二本鎖鋳型DNAの場合は転写/翻訳共役反応系を用いる必要がある。このような無細胞系としては、大腸菌由来のもの、コムギ胚芽由来のもの、ウサギ網状赤血球由来のものが市販されているが、大腸菌等の原核細胞由来の抽出液が好ましい。大腸菌の抽出液では、転写、翻訳の共役反応によりDNAからタンパク質を合成できることが知られ、例えば、ズベイらによって大腸菌のS30抽出液を用いる方法が系統的に開発されている(例えば、ジェフリー・ズベイ(Geoffrey Zubay)、「アニュアル・レビュー・オブ・ジェネティクス(Annual Review of Genetics)」1973年、第7巻、p.267−287参照)。S30抽出液には、mRNAの翻訳に必要なリボソーム、アミノアシルtRNA合成酵素、ポリペプチド鎖開始因子(IF)、同伸長因子(EF)及び同終結因子が含まれている。合成タンパク質の鋳型としてDNAを用いる場合は、強力なプロモーター(一般的にはT7プロモーター)の下流に目的タンパク質遺伝子を配置したDNAが用いられ、転写と翻訳の両反応を共役させるために、系内にT7RNAポリメラーゼと4種類のリボヌクレオチド(ATP、GTP、CTP及びUTP)を添加する。アミノアシルtRNAの合成、及びmRNAの翻訳反応にはATPのエネルギーが必要であることから、クレアチンキナーゼ−クレアチンリン酸系等のエネルギー再生系を無細胞系に加える。このような構成成分により、細胞内で起こるタンパク質合成反応を試験管内で再構成している。
【0027】
大腸菌S30抽出液は、転写及び翻訳に必要な大腸菌の全ての酵素と因子を含んでいるが、更に補充的な混合液を添加することができる。具体的な調製方法としては、まず最初に大腸菌を培養し、菌体を遠心分離等により回収する。回収された菌体は、洗浄後、緩衝液に再懸濁し、フレンチプレスやガラスビーズ、ワーリングブレンダー等を用いて破砕する。破砕された大腸菌の不溶物質を遠心分離で除去し、プレインキュベーション混合液と混合してインキュベーションする。この操作によって内在性のDNA、RNAが分解されるが、更に、カルシウム塩やマイクロコッカスのヌクレアーゼ等を添加して内在性の核酸を分解させてもよい。続いて、透析により内在性のアミノ酸、核酸、ヌクレオシド等を除き、適量ずつ分注して液体窒素又は−80℃にて保存する。
【0028】
転写、翻訳及びエネルギー再生に必要な全てのタンパク質因子にタグを付けて別々に調製し、これらを再構成した無細胞タンパク質合成系も知られている。これらのタンパク質因子に大腸菌70Sリボソーム、tRNA、アミノ酸、NTP等の転写、翻訳に必要な成分を添加したキットも販売されている。
【0029】
(検出キット)
本発明の方法に用いるために上記方法により調製された核酸分子、及び大腸菌の細胞抽出液等の無細胞翻訳反応試薬は、使用しやすいように一定量ごと分注して、目的分子の検出キットとして配送することができる。これらの製品は凍結又は乾燥状態で保存することができ、保存及び輸送に適した容器に収容してキットとして販売される。キットには取扱説明書や陽性コントロールサンプル等を添付することができる。
【0030】
(バイオセンサー)
本発明はまた、上記核酸分子によって形質転換された原核細胞からなるバイオセンサーに関する。特に、本発明の二本鎖DNAからなる核酸分子は安定であり、プラスミドDNAとして種々の原核細胞で安定に存在しうる。或いは、本発明の二本鎖DNAを支持体に固定化したバイオセンサーを用いてもよい。
【0031】
以下に具体的な実施例を示して本発明をさらに詳細に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
【実施例】
【0032】
[実験材料及び方法]
1 鋳型DNAの調製
ルシフェラーゼ遺伝子をコードするプラスミドpBESTluc、及びβ−ガラクトシダーゼ遺伝子をコードするプラスミドpSV-β-Galactosidaseは、プロメガ社から購入した。本実施例で用いた全ての鋳型DNAは、以下に説明するポリメラーゼ連鎖反応(PCR)及びβ−ガラクトシダーゼ用の鋳型の調製については、これに続いて連結反応により調製した。PCRは、20μLの反応混合液中、4pmolの正方向プライマー、4pmolの逆方向プライマー、0.5ユニットのPrimeSTAR HS DNAポリメラーゼ(商品名、タカラバイオ社)、それぞれ4nmolのdNTPs(タカラバイオ社)、及び4μLの5倍濃度PrimeSTAR反応用緩衝液を含み、第一次PCR用には200pgのpBESTluc又はpSV-β-Galactosidaseを、第二次以降のPCR用には約20fmolの精製したPCR産物を鋳型として用いた。PCR産物は、アガロースゲル電気泳動により、GFX PCR DNA and Gel Band Purification Kit(商品名、GEヘルスケアバイオサイエンス株式会社)を用いて精製し、260nmの吸光度を測定して定量した。
【0033】
2 鋳型DNAのインビトロにおける転写/翻訳共役反応
インビトロにおける転写/翻訳反応は、T7RNAポリメラーゼを含む再構成された無細胞翻訳システム(ピュアシステム、クラシックII、ポストゲノム研究所)を用いて行った。約0.1μgの鋳型DNAを含む10μLの反応溶液を、種々の濃度のテオフィリン、カフェイン又はcGMPの存在下又は非存在下において、37℃で1時間インキュベートした。反応終了後の翻訳溶液はさらに精製することなく次のアッセイに用いた。
【0034】
3 mRNA鋳型の調製
鋳型DNAのインビトロ転写反応は、MEGAscript T7 Kit(商品名、アンビオン)を用いて行った。0.3μgの鋳型DNAを含む10μlの反応溶液を37℃で4.5時間インキュベートした。当該キットに添付されているTURBO DNaseを1ユニット添加した後、さらに15分間37℃でインキュベートした。転写産物は、RNeasy MinElute Cleanup Kit(商品名、キアゲン)を用いて精製し、上述と同様の方法で定量した。
【0035】
4 mRNA鋳型のインビトロ翻訳反応
インビトロ翻訳反応は、ピュアシステム、クラシックIIを用いて行った。約1μgのmRNAを含む10μlの反応溶液を、種々の濃度のコファクター(テオフィリン)の存在又は非存在下で37℃、1時間インキュベートした。生成した翻訳産物を含む溶液は精製せずに次のアッセイに用いた。
【0036】
5 ルシフェラーゼアッセイ
翻訳溶液と水を等量含む5μlの混合物を100μlのルシフェラーゼアッセイ試薬(プロメガ)と混合した。化学発光強度の測定は、ワラック社の1420ARVOsx装置により、黒色96穴プレート(コーニングコースター3915)を用いて行った。化学発光の映像はクールセイバーAE−6955(アトー)により取得した。
【0037】
6 β−ガラクトシダーゼアッセイ
10μlの翻訳溶液を、40μlの1×レポーターリシスバッファー(プロメガ)、及び50μlのオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を含む2×アッセイバッファー(プロメガ社のβ−ガラクトシダーゼアッセイシステム)と混合した。混合溶液は37℃にて3時間インキュベートした。この反応溶液の405nmにおける吸光度をワラック社の1420ARVOsx装置により、透明の96穴プレート(コーニングコースター3635)を用いて測定した。β−ガラクトシダーゼを含まない(鋳型なしの翻訳溶液)バックグラウンド強度をそれぞれの測定データから差し引いた。
【0038】
[実施例1]テオフィリン誘導アプタザイムとルシフェラーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたテオフィリンの検出
ルシフェラーゼ遺伝子をコードするプラスミドpBESTlucと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号1)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0039】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、第三次、及び第四次PCRを行った。
第二次PCR用
正方向プライマー:5'-ACCCTGATGAGCCTGGATACCAGCCGAAAGGCCCTTGGCAGTTAGACGAAACAAGAAGGAGATATACCAATG-3'(配列番号3)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第三次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCCTGG-3'(配列番号4)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第四次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTATCACCTTTTTGTAGGTTG-3'(配列番号5)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0040】
第三次PCRによって増幅された二本鎖DNAは、5’末端のT7プロモーターから転写されるテオフィリン依存性アプタザイムとルシフェラーゼをコードしており、RBS/アンチ−RBSによって形成される二重鎖は、表1の二重鎖2の配列を有する。また、第四次PCRによって増幅された二本鎖DNAは、同様に5’末端のT7プロモーターから転写されるテオフィリン依存性アプタザイムとルシフェラーゼをコードしており、RBS/アンチ−RBS二重鎖は、表1の二重鎖5の配列を有する(全体配列を配列番号24に示す)。
【0041】
対照実験に用いるための通常の5’非翻訳領域(T7ファージのg10リーダー配列)を有する鋳型DNAは、以下のプライマー対を用いて増幅した。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号6)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第二次PCR用
正方向プライマー:5'-GAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCA-3'(配列番号7)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0042】
このようにして、種々の塩基配列及び長さのRBS/アンチ−RBS二重鎖を有する核酸分子を作製し、ルシフェラーゼ遺伝子をレポーター遺伝子として翻訳のON/OFF転換効率を調べた。ON/OFF転換効率とは、ここではテオフィリンの非存在下(OFF)に対する存在下(ON)における翻訳されたルシフェラーゼ活性として規定される。その結果を下記の表1に示す。
【0043】
【表1】

【0044】
ハンマーヘッド型リボザイムの触媒活性を維持する上で、切断部位の上流に隣接するGUAのトリプレット塩基が不可欠であることが分かっているため、このGUAトリプレットに隣接して直接RBSを結合した核酸分子を作製した(表1の二重鎖1)。この核酸分子1のテオフィリン1mMの場合のON/OFF転換効率は、表1に示したように約2.2倍であった。テオフィリン存在下(ON状態)でのルシフェラーゼ活性は5708cpsであり、タンパク質大量合成のために一般的に用いられる5’−非翻訳領域(T7ファージのg10リーダー配列)を有する鋳型から転写される場合(100000cps以上)と比べて極めて小さいことが分かった。そこで、RBS配列の5’側に、T7ファージのg10リーダー配列と同じ3つのプリン塩基AAGを挿入した一連の核酸分子2〜5を作製した。このようにして作製した核酸分子2は、ON状態において約6倍のルシフェラーゼ活性を示したが、ON/OFF転換効率は、わずかに1.7倍上昇したに過ぎなかった。これはOFF状態においても活性が上昇したことに起因する。テオフィリン非存在下(OFF状態)における発現を抑えるために、次に、アンチ−RBS部位の相補鎖を延長した核酸分子3及び4を作製した。
【0045】
より長いアンチ−RBS部位及びその隣接塩基からなる配列:5'-UUUCCUUUUU-3'(配列番号8)及び5'-AUAUUUCCUUUUU-3'(配列番号9)を含む核酸は、OFF状態における発現を効率的に抑えたが、同時にON状態における発現量も低下し、ON/OFF転換効率はそれぞれ4.4及び4.6と中程度に留まった。そこで、アンチ−RBS二重鎖の中にミスマッチを導入した配列5:5'-UAUCACCUUUUU-3'(配列番号10)を含む核酸を作製したところ、予想外にも極めて高いON/OFF転換効率(7.2)を有することが分かった。
【0046】
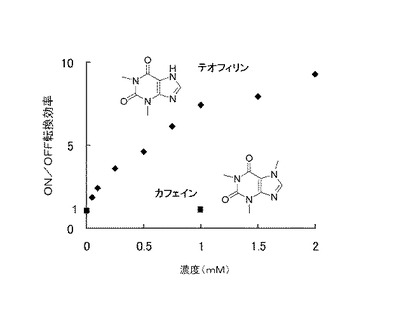
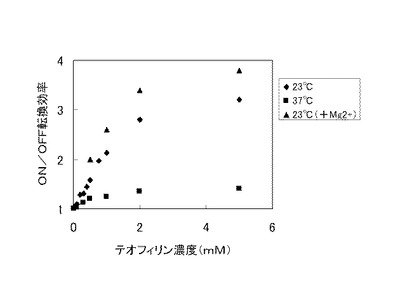
この最適化された二重鎖5を含む核酸分子を用いて、種々の濃度のテオフィリン又はカフェイン存在下でON/OFF転換効率を測定した結果を図3に示した。カフェインはテオフィリンと比べてN7位にメチル基を有する以外は類似する構造を有するにもかかわらず、1mMの濃度でON/OFF転換効率がほぼ1であり、当該核酸分子が高い特異性(テオフィリン依存性及びテオフィリン直交性)を有することが示された。
【0047】
鋳型DNAの代わりに鋳型mRNAを用いて同様の実験を行った結果を図4に示す。これらの結果より、本核酸分子の活性は転写レベルではなくて翻訳レベルにおいて制御されていることが示唆される。また、mRNAを用いた場合よりも鋳型DNAを用いた場合の方が効率が良い。
【0048】
[実施例2]テオフィリン誘導アプタザイムとβ−ガラクトシダーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたテオフィリンの検出
β−ガラクトシダーゼ遺伝子は、約3000bpと非常に長いため、最初に前方部分と後方部分とに分けてPCRを行い、続いてそれぞれの増幅断片を制限酵素AatIIで切断し、両方のDNA断片をライゲーションハイ(商品名、東洋紡)により連結した。下記プライマー配列中のAatIIサイトを下線で示した。
【0049】
前方部分
β−ガラクトシダーゼ遺伝子をコードするプラスミドpSV-β-Galactosidaseと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGCGCTGTATGGCGAGATC-3'(配列番号11)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号12)
【0050】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、第三次、第四次、及び第五次PCRを行った。
第二次PCR用
正方向プライマー:5'-AGGAGATATACCAATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTC-3'(配列番号13)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号12)
第三次PCR用
正方向プライマー:5'-ACCCTGATGAGCCTGGATACCAGCCGAAAGGCCCTTGGCAGTTAGACGAAACAAGAAGGAGATATACCAATG-3'(配列番号14)
逆方向プライマー:5'-GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
第四次PCR用
正方向プライマー:5'- CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCC TCCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCCTGG-3'(配列番号16)
逆方向プライマー:5'- GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
第五次PCR用
正方向プライマー:5'- CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCC TATCACCTTTTTGTAGGTTG-3'(配列番号17)
逆方向プライマー:5'- GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
【0051】
後方部分
第一次PCR用
正方向プライマー:5'-CATTTTCCGTGACGTCTCGTTG-3'(配列番号18)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号19)
【0052】
上記PCRにより増幅した前方部分と後方部分とを連結後、全長DNAを鋳型として、以下の最終プライマー対を用いてPCRを行い、5’末端のT7プロモーターから転写されるテオフィリン依存性リボスイッチをコードする二本鎖DNA断片を取得した。
【0053】
最終的なPCR
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGAC-3'(配列番号20)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号19)
【0054】
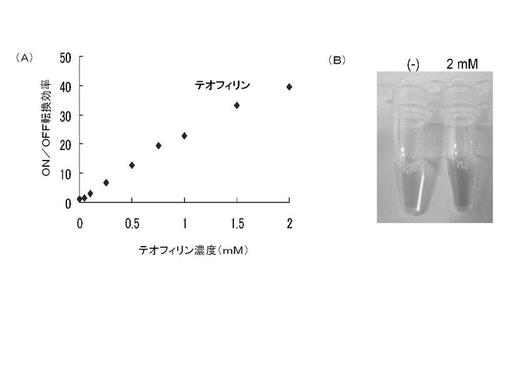
このようにして作製したテオフィリン誘導アプタザイムとβ−ガラクトシダーゼ遺伝子とを含む二本鎖DNA(最適化されたRBS/アンチ−RBS二重鎖を含む)を用いて、種々の濃度のテオフィリン存在下でON/OFF転換効率を測定した結果を図5(A)に示す。本測定方法においては、翻訳産物であるβ−ガラクトシダーゼが無色のオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を黄色のオルト−ニトロフェノールに加水分解するため酵素活性の発現を可視化することができる。実際に、インビトロにおける転写/翻訳共役反応(37℃、1時間)の後、反応溶液にONPGを添加してさらに数時間37℃でインキュベートするだけで特殊な検出器を用いずにテオフィリンを可視的に検出することができた(図5(B))。1mM又は2mMのテオフィリンに対するON/OFF転換効率は、それぞれ20及び40であり、ルシフェラーゼ遺伝子を用いた場合(約7及び9)に比べてより高い値を示した。その理由の一つとしては、大腸菌由来の酵素であるβ−ガラクトシダーゼ遺伝子に比べて、ルシフェラーゼ遺伝子がレアコドンを多く有していることが挙げられる。レアコドンとは、それに対応するtRNA量が翻訳反応系において乏しいコドンをいう。このことは、翻訳反応系の条件を最適化することによって、さらに検出感度が向上する可能性を示唆する。
【0055】
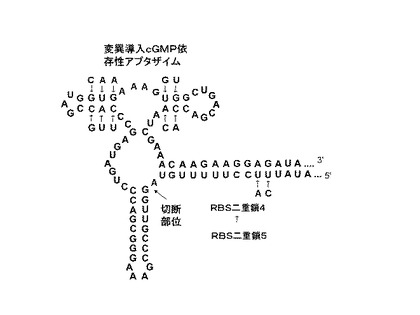
[実施例3]cGMP依存性アプタザイムとルシフェラーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたcGMPの検出
ルシフェラーゼ遺伝子をコードするプラスミドpBESTlucと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号21)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0056】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、及び第三次PCRを行った。
第二次PCR用
正方向プライマー:5'-CCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCC CACCGATGGTGAAAGTGGCTGACGACCAATCGAAACAAGAAGGAGATATACCAATG-3'(配列番号22)
第三次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCATATTTCCTTTTTGTAGGTTG-3'(配列番号23)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0057】
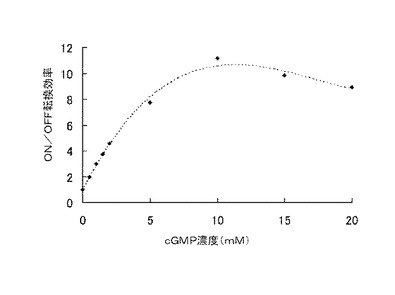
このようにして作製したcGMP依存性アプタザイムの塩基配列の一部及びその推定二次構造を図6に示した。Breaker et al.によって報告されたcGMP依存性アプタザイムは、実施例1で最適化されたアンチ−RBS配列5と相補的な配列を含むため、まず最初に、当該リボザイムの活性を損なわないように塩基置換を行った変異体アプタザイムを合成した。しかしながら、この変異体アプタザイムもなお、正確なRBS/アンチ−RBS二重鎖を形成するには不十分であったため、上述した表1のアンチ−RBS配列4を用いたところ、アンチ−RBS配列5を用いた場合よりもON/OFF転換効率が高くなることが分かった。種々の濃度のcGMP存在下においてルシフェラーゼアッセイを行い、ON/OFF転換効率を調べた結果を図7に示す。10mMのcGMP存在下において、約10の高いON/OFF転換効率を示した。この結果は、異なるアプタザイムを用いる場合においても、それぞれの目的分子に合わせて容易に本発明の核酸分子をデザインできることを示す。
【0058】
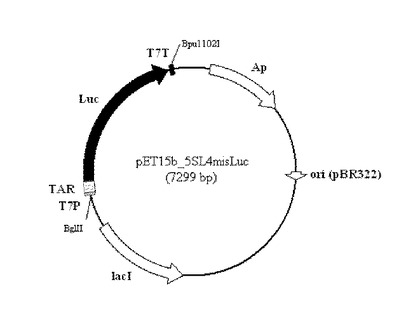
[実施例4]インビボアッセイ
実施例1で作製したテオフィリン依存性アプタザイムを含む核酸分子を図8に示すプラスミドpET15b_5SL4misLucのTAR(Theophyline-Aptazyme-Riboswitch)領域にクローン化した。図8に示したように、本発明の機能性核酸分子はT7プロモーターの制御下において発現が調節される。このプラスミドを大腸菌BL21(DE3)pLysS株(ノバジェン、ラクトースオペロンの制御下のT7RNAポリメラーゼ遺伝子をゲノムに含む)に形質転換した。この形質転換大腸菌を一晩培養後、培養液で1/20に希釈後、さらに37℃で振とう培養し、培養液の600nmでの吸光度が約0.5に達したときに、最終濃度が1mMとなるようにIPTGを添加した。また、最終濃度が0〜5mMの種々の濃度となるようにテオフィリンを添加した。引き続き23℃又は37℃で1.5〜2.5時間培養を続け、菌体を回収して溶解し、ルシフェラーゼ活性を測定した。その結果を図9に示した。図9に示したように、23℃で培養したときにテオフィリンの濃度依存的にON/OFF転換効率が増加することが認められた。37℃に比べて23℃の方が効率が良い理由としては、アプタザイム活性の至適温度が23℃付近であるから、及び低温の方が転写/翻訳反応の速度が遅くなり、細胞内で合成されたmRNAのRBS/アンチ−RBS二重鎖が形成し易くなるから、並びにアプタザイムによって切断されたmRNAの分解速度が遅くなるから等の理由が考えられる。また、培養液に5mMのマグネシウムを添加したときの方が若干活性が上昇した。これらの結果より、本発明のリボスイッチを用いる目的分子の検出方法は、インビボにおいても適用可能であることが示された。
【図面の簡単な説明】
【0059】
【図1】本発明に係る核酸分子及びこれを用いる目的分子(コファクター)の検出方法の一実施形態を示した模式図である。
【図2】本発明の一実施形態に係るテオフィリン依存性アプタザイムの構造を示した模式図である。
【図3】実施例1において、RBS二重鎖5を用いて調製した本発明の核酸分子について、種々の濃度のテオフィリン又はカフェイン存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図4】実施例1において、RBS二重鎖5を用いて調製した本発明の核酸分子について、直接mRNAを用いて反応した場合のルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図5】実施例2において、RBS二重鎖5を用いて調製した本発明の核酸分子について、種々の濃度のテオフィリン存在下におけるβ−ガラクトシダーゼアッセイのON/OFF転換効率を測定した結果(A)、及びテオフィリンの非存在(−)及び存在(2mM)下におけるアッセイ溶液の写真を示す。
【図6】実施例3において調製したcGMP依存性アプタザイムを含む本発明の核酸分子の塩基配列と推定二次構造を示した模式図である。
【図7】実施例3において、種々の濃度のcGMP存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図8】本発明に係る核酸分子をTAR領域に挿入したプラスミド構造を示した模式図である。
【図9】実施例4で調製した形質転換大腸菌を用いて、種々の濃度のテオフィリン存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【技術分野】
【0001】
本発明は、機能性核酸を用いた分子センサーに関し、より詳細には、目的分子との結合により自己切断酵素活性をもつ機能性核酸(アプタザイム)とレポーター遺伝子とを含む核酸分子を用いて、目的分子を検出する方法、及びこのような方法に用いる核酸分子等に関する。
【背景技術】
【0002】
近年、インビトロで選択されたRNAアプタマー、又はDNAアプタマーは、種々の分子に対するセンサーとして大きな注目を集めている。アプタマーは、目的とする有機分子、イオン、タンパク質、又は細胞等を特異的に認識することができる。この認識機構は、アプタマー分子内に存在するリン酸基や有機塩基の有する疎水性及び親水性の両方の性質によって形成されている。アプタマーがその目的分子と複合体を形成すると分子量の増加をもたらし、目的分子が大きい場合には、質量検出器によって直接検出することができる。また、いくつかのアプタマーは目的分子との結合により構造変化を起こす。このような構造変化と、蛍光的又は電気化学的方法とを組み合わせることにより、低分子量の目的分子でも検出することが可能である。ただし、その際は、アプタマーを直接的若しくは間接的に蛍光色素又は電気応答性物質で標識しなければならない。
【0003】
「アプタザイム」又は「アロステリックリボザイム」と称される核酸は、特定の分子(コファクター)によってアロステリックに活性化される酵素機能を有し、センサーとして利用可能である。アプタザイムは、アプタマードメイン及びリボザイムドメインの両方を分子内に有し、コファクターとの複合体形成という信号を、例えば自己切断のようなより明確な触媒活性へ容易に伝達することができる。
【0004】
例えば、国際公開公報WO03/014375号パンフレット(特許文献1)には、アプタザイムを蛍光標識することによって、目的分子の結合を光学的に検出するシステムが開示されている。また、国際公開公報WO00/26226号パンフレット(特許文献2)には、アプタザイムの5’末端を放射性同位元素である32Pで標識して切断されたアプタザイムを検出する方法が開示されている。質量変化を測定する方法としては、アプタザイムを金表面に固定化し、特定の分子との結合によって誘起された酵素活性によるアプタザイムの質量変化を測定する方法が開示されている(例えば、特許文献3参照)。
【0005】
一方、塩基配列の相同性に基づいてDNAを検出する方法として、分子ビーコン法(例えば、非特許文献1参照)が知られている。この方法は、目的配列と相補的な配列をループドメインに有するヘアピン構造のオリゴヌクレオチドプローブを用いて、例えば、5’末端及び3’末端に標識されたFRETを検出する方法である。さらに、無細胞タンパク質合成系において、分子ビーコン型リボ核酸と連結したレポーター遺伝子の発現を指標として、目的とするDNAを検出する方法が報告されている(例えば、非特許文献2参照)。しかしながら、これらの方法は核酸配列の相補性に基づくため、DNA等の核酸分子の検出に利用できるに過ぎず、低分子化合物やペプチド、タンパク質等の検出は不可能である。
【0006】
【特許文献1】国際公開公報WO03/014375号パンフレット
【特許文献2】国際公開公報WO00/26226号パンフレット
【特許文献3】米国出願公開公報第20040175693号
【非特許文献1】Tyagi, S., Kramer, F.R., Nat. Biotechnol. 1996, Vol. 14, pp.303-308
【非特許文献2】Sando, S., Narita A., Abe, K., and Aoyama Y., J. Am. Chem. Soc., 2005, Vol.127, pp.5300-5301
【発明の開示】
【発明が解決しようとする課題】
【0007】
上記従来技術によれば、アプタザイムを用いて目的分子を検出するためには、何れも蛍光物質や放射性同位元素等で標識する必要があり、そのための時間やコスト、さらには特殊な検出装置が必要になるという課題がある。また、これらの標識のために不安定なRNAを直接用いなければならないという問題点もある。本発明は、特殊なプローブや検出機器を用いなくとも、目的分子の存在、濃度を簡便に検出し得る機能性核酸及びこれを用いて目的分子を検出する方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明はかかる課題を解決するためになされたものであって、目的分子の存在下において自己切断触媒活性を示すアプタザイムと、タンパク質の翻訳段階におけるレポーター遺伝子の発現制御機構とを組み合わせることによって完成されたものである。
【0009】
すなわち、第一の観点において、目的分子を検出するための本発明の機能性核酸分子は、(a)レポーター遺伝子配列、(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含むことを特徴とする。目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成する。そして目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、それによって前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。翻訳反応系が共存すると、当該核酸分子のリボソーム結合部位にリボソームが結合して翻訳が開始され、前記レポーター遺伝子が発現される。目的分子に対するアプタザイムを選択することによって所望の目的分子を検出することが可能であるが、検出対象としては、例えば、低分子化合物、ペプチド及びタンパク質等が挙げられる。
【0010】
別の観点において、本発明は上記核酸分子をコードする二本鎖DNAであって、当該コード領域の上流に機能的に連結された転写プロモーター配列を含む二本鎖DNAを提供する。転写プロモーター配列としては、用いる翻訳反応系において機能しうるものであれば特に限定されないが、強力なプロモーター活性を有する、ファージ由来のT7プロモーターが好ましい。
【0011】
さらに異なる観点において、本発明は上記核酸分子を用いる目的分子の検出方法を提供する。当該検出方法は、上記核酸分子と、目的分子を含む試料とを、翻訳反応系内において所定の時間接触させる工程、及び前記レポーター遺伝子の発現量を測定する工程、を含むことを特徴とする。前記核酸分子が二本鎖DNAの場合は、転写/翻訳共役反応系を用いることができる。
【0012】
さらになお異なる観点において、本発明は、目的分子の検出のための本発明の核酸分子を含むキット、及び本発明の核酸分子によって形質転換された原核生物の細胞等を提供するものである。
【発明の効果】
【0013】
本発明の方法によれば、従来技術のように蛍光標識や放射性標識を行うことなく、且つ特殊な機器を用いなくとも目的分子を検出することが可能である。また、不安定であるRNAを直接扱わなくても、より安定なDNAをセンサー前駆体として用いることができる。本発明に係るDNAには転写プロモーターが導入されており、用いる翻訳系にRNAポリメラーゼを添加することによってRNAを容易に合成できるからである。機能性RNA自身をセンサーとして用いることも可能であるが、センサー前駆体であるDNAを用いた方がより効率的であり感度も向上するという知見が得られている。
【発明を実施するための最良の形態】
【0014】
(定義)
本明細書中で使用される用語「リボザイム」とは、酵素のような作用を有するRNAのオリゴヌクレオチドであり「RNA酵素」若しくは「核酸酵素」と呼ばれる場合もある。一般的に、これらはRNA分子の切断を触媒するエンドリボヌクレアーゼ作用を示す。切断部位の位置は高度に配列特異的である。リボザイムの中には、ポリメラーゼ又は脱リン酸化酵素のように作用するものもある。
【0015】
用語「アプタマー」とは、標的リガンドに最適に結合するように特異的に選択される核酸をいう。標的リガンドとしては、増殖因子、酵素、受容体、膜タンパク質、ウイルスタンパク質等の種々のタンパク質が挙げられるが、金属イオンや分子量の小さい有機化合物、ペプチド、ウイルス等とも結合することが分かってきた。標的リガンドに特異的なアプタマーを探し出す方法としては、SELEX法(試験管内人工進化法 Systematic Evolution of Ligands by Exponential Enrichment)(Tuerk, C., Gold, L., Science, 1990 Vol. 249, pp.505-510参照)が知られている。この方法は、標的とする目的分子に対し、ランダムな配列をもつ多数のオリゴヌクレオチドの混合物であるオリゴヌクレオチドライブラリを作用させることにより、親和性の高いオリゴヌクレオチドを選び出し、選び出されたオリゴヌクレオチドを増幅生産するという作業を繰り返す。これによって、標的とする目的分子に対し特異的に結合するオリゴヌクレオチドを得る方法である。
【0016】
用語「アプタザイム」とは、エフェクターによって調節されるリボザイム又は核酸酵素を意味する。アプタザイムは、典型的にはエフェクターを認識するアプタマードメインと、触媒作用を示すリボザイムドメインを含む。エフェクターは、アプタマードメインとリボザイムドメインとの特異的な相互作用を通じてアプタザイムの触媒活性を高め、又は低下させることができる。従って、アプタザイムの活性はエフェクターの存在の有無やその量を監視するために用いられうる。この方法は、これまで種々の診断及び検出のためのアプタザイムセンサーを設計するために用いられている。アプタザイムの設計に用いる核酸分子としては、RNA(リボザイム)、DNA(デオキシリボザイム)、DNA/RNAハイブリッド、及びペプチド核酸(PNA)等が挙げられる。一般的には、リボザイムはデオキシリボザイムよりも不安定である。種々のエフェクター分子、例えば、金属イオンや低分子化合物により調節されるアプタザイムの作製方法は公知であり(米国特許第6630306号等参照)、その内容は参照により本願に組み込まれる。
【0017】
(機能性核酸分子)
以下に図面を参照しながら本発明の機能性核酸分子について説明する。図1は、本発明の好ましい実施形態における機能性核酸分子を模式的に表したものである。図1上部の二本鎖鋳型DNAは、T7プロモーターの制御下において、レポーター遺伝子をコードする。レポーター遺伝子上流には、5’末端から順に、ステム−ループ構造、アンチ−リボソーム結合部位(アンチ−RBS)、アプタザイム配列、及びリボソーム結合部位(RBS)を含む。T7RNAポリメラーゼを含む無細胞タンパク質合成系においてこの鋳型DNAが転写されると、アンチ−RBSはRBSとハイブリダイズしてリボソームがRBSに結合することを阻害する(左下部のOFF状態)。mRNAの5’末端側(領域)に存在するステム−ループ構造は転写産物を安定化し、同時にアンチ−RBSが正確にRBSと結合して二重鎖を形成するために有用である。このようなステム−ループ構造は、少なくとも10塩基長程度の長さがあればよく、好ましくは10〜200塩基長、より好ましくは13〜100塩基長、さらに好ましくは17〜50塩基長程度である。アプタザイムに対するコファクターの存在下では、(I)アプタザイムが立体構造の変化を起こし、(II)これによって自己切断活性を誘導する。一般的に分子間で形成された二重鎖の融解温度(Tm)は、分子内二重鎖の融解温度よりも低いため、(III)所定の条件下において切断されたアンチ−RBSドメインがRBSから解離する。これによってリボソームがRBSに接触可能となり、下流のレポーター遺伝子の発現が活性化される(右下部のON状態)。
【0018】
ここで、転写プロモーターとしては、反応系において転写されるプロモーターであれば特に限定されないが、強力なプロモーターであるT7プロモーターが好ましい。また、レポーター遺伝子も翻訳系において検出可能なものであれば何れも使用可能できるが、検出が容易で感度が高いものとして、例えば、ルシフェラーゼ遺伝子やβ−ガラクトシダーゼ遺伝子等が好ましい。
【0019】
アプタザイム配列としては、目的分子によって調節可能な核酸酵素であれば特に限定されないが、好ましくは、目的分子としてテオフィリンのような低分子によってリボザイム活性が調節されるアプタザイムが用いられる。Breakerらによって試験管内で選択されたテオフィリン依存性アプタザイムが報告されている(Soukup, G. A., Emilsson, G.A.M., Breaker, R.R., J. Mol. Biol., 2000, Vol.298, pp.623-)。このアプタザイムは、図2(a)に示したように、アプタマードメイン、伝達モジュール、及びハンマーヘッドリボザイムの3つのドメインからなっている。コファクターであるテオフィリンがアプタマードメインに結合することによってリボザイムドメインの立体構造が変化し、ヘリックスAとヘリックスBの間の切断部位(矢印で示す)における自己切断活性を誘導する。この自己切断活性の発現に必要な切断部位に隣接するGUAトリプレット配列に下線を付加した。このGUAトリプレット以外の2つのヘリックスA及びヘリックスBの配列は、当該アプタザイムの酵素活性とは関係ないことが知られているため、上記RBS/アンチ−RBS二重鎖をこれら2つのヘリックス構造の何れかと置換した核酸分子を作製した。ヘリックスAの代わりにRBS/アンチ−RBS二重鎖を挿入した場合は、アプタザイム配列とRBSの間が切断されるため5’末端からRBSまでの配列が短くなり(図2(b)右上挿入図参照)効率的な翻訳が起こらなかった。一方、ヘリックスBに上記RBS/アンチ−RBS二重鎖を挿入した核酸分子では、アプタザイム配列とアンチ−RBSの間が切断されるため5’末端からRBSまでの配列が長く(図2(b)参照)効率的な翻訳が起こった。さらにこの場合、切断後のmRNAの5’末端側にステム−ループ構造(ヘリックスA)が存在するため、mRNAが安定であることも利点としてあげられる。
【0020】
リボソーム結合部位(RBS)は、mRNAの翻訳開始コドン近傍にあり、リボソームの16srRNA分子の3’末端近くにある配列と塩基対を形成してリボソーム小サブユニットと結合するために必要な配列である。大腸菌のmRNAのリボソーム結合部位は、シャイン−ダルガルノ配列とも呼ばれる。この配列はプリンを多く含んでいるという傾向はあるものの、16srRNAと完全に相補的な配列となることはあまりなく、また長さも3塩基から9塩基程度である。mRNAのRBSに結合したリボソーム小サブユニットは遺伝子の開始コドン(AUG:メチオニンに該当)までmRNA上を移動し、メチオニン−tRNAが開始コドンに結合して30s開始複合体を形成する。シャイン−ダルガルノ配列と翻訳開始コドンとの距離は、翻訳の効率と関連性のあることが知られ、最適距離は4塩基から9塩基であると推定されている。mRNA上において、このRBSが強固に二重鎖を形成しているとリボソームがRBSに接触できないため翻訳が開始されない。
【0021】
アンチリボソーム結合部位(アンチ−RBS)は、上記RBSと分子内でハイブリダイズし得るものであれば特に限定されないが、目的分子の存在又は非存在下においてのON/OFF転換効率が向上するように最適化されることが好ましい。具体的には、目的分子の非存在下では分子内でRBS/アンチ−RBS二重鎖が安定に形成されるが、目的分子の存在下では自己切断により前記二重鎖が解離しやすいことである。一般的に分子間で形成される核酸配列二重鎖の融解温度(Tm)は、分子内二重鎖の融解温度よりも低いことが知られている。DNAやRNAのTm値の計算は、種々のコンピューターソフトウェアを用いて行うことができる。最も正確なTm値の推定ソフトウェアの一つとして、例えば、HyTher(商標)が知られており、オンライン上で使用可能である(http://ozone3.chem.wayne.edu/)。Tmでは、二本鎖状態の核酸と一本鎖状態の核酸が同率比で存在しており、Tm以上では、ほとんどがハイブリダイズできずに一本鎖状態になっていると考えられる。
【0022】
本発明に係る核酸分子は、一本鎖RNA又は一本鎖DNAの何れでもよいが、20〜40℃の水溶液中において分子内で会合し、RBS/アンチ−RBS二重鎖を含むステム−ループ構造を形成する一本鎖RNAであることが好ましい。さらに好ましくは、上記RBS/アンチ−RBS二重鎖のTmは反応溶液温度程度か、それ以下である。従って、好ましい実施形態において、上記RBS/アンチ−RBS二重鎖は、当該RBSと、アンチ−RBSと、それらの隣接配列とによって形成され、その二重鎖の長さは6〜20塩基対である。より好ましくは8〜15塩基対である。当該二重鎖にはG−Uペア、又は1〜数個のミスマッチを含んでもよい。
【0023】
(レポーター遺伝子)
本発明において用いられるレポーター遺伝子は、翻訳反応系において発現し検出が容易なものであれば特に限定されないが、例えば、ルシフェラーゼ、β−ガラクトシダーゼ、アルカリフォスファターゼ、緑色蛍光タンパク質(GFP)及びエクオリン等の遺伝子を挙げることができる。ホタルルシフェラーゼはATPの存在下でルシフェリンの酸化を触媒し、光子を生成する化学ルミネッセンス反応を触媒する。ルシフェラーゼは単量体のタンパク質で翻訳後の修飾を必要としないから、遺伝子発現のレポーターとして適している。大腸菌由来のβ−ガラクトシダーゼ遺伝子(lacZ)は種々の細胞の形質転換におけるレポーター遺伝子としてよく使用される。この酵素の生体内での基質はラクトースであるが、ほぼあらゆるβ−ガラクトシドを加水分解することができ、例えば、無色の基質であるオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を加水分解して420nmの光を吸収する黄色のオルト−ニトロフェノールを生成する。また、x−galはこの酵素により分解されてインディゴブルーの呈色反応を示す。アルカリフォスファターゼは、分子触媒活性の極めて高い酵素であり、種々の蛍光及び発光基質も開発されているため高感度検出に好適である。GFPはオワンクラゲ(Aequorea Victoria)等から発見された蛍光タンパク質であり、共存する発光タンパク質であるエクオリンの青色化学発光をエネルギー変換して緑色にすることが知られている。このGFPは上記ルシフェラーゼやエクオリンのように特殊な蛍光団を必要とせず、自らアポタンパク質上で自動的に蛍光団を形成する。このため、クローン化されたGFPのcDNAを用いて多くの細菌や動物細胞等で発現され、遺伝子発現の指標として利用されている。また、GFP遺伝子に種々の突然変異を導入して多くのGFP誘導体が作製され、これらの中には野生型GFPよりも強い蛍光強度を示したり、異なる波長の蛍光を発する誘導体もあることが報告されている。
【0024】
(目的分子又はコファクター)
本発明の方法において検出可能な目的分子としては、アプタザイムのコファクターとなりうるもので、かつ翻訳反応を阻害しないものであれば特に限定されないが、例えば、低分子化合物、細菌又はウイルス由来の抗原タンパク質、毒物分子、コカイン、ヒト免疫不全ウイルス(HIV)及びHIV−由来分子などを含む。
【0025】
また、本発明の別の観点において、上記レポーター遺伝子に代えて所望の遺伝子を挿入した核酸分子は、原核生物の翻訳反応系において上記目的分子又はコファクターに相当するリガンド分子による遺伝子発現制御システムとして利用することもできる。すなわち、リガンド分子の非存在下ではアンチ−RBSがRBSに結合して当該所望の遺伝子の発現を抑制し、リガンド分子を添加することによって、前記アプタザイムが自己切断触媒活性を示し、それによって前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離する。その結果、前記所望の遺伝子の発現を誘導しうるのである。
【0026】
(目的分子の検出方法)
本発明の検出方法は、無細胞タンパク質合成系において、目的分子を含む試料と本発明の機能性核酸分子を所定の時間接触させることにより、目的分子の濃度に応じて発現するレポーター遺伝子の活性を測定する工程を含む。用いる機能性核酸分子としては、RNA及びこれをコードする鋳型DNAの何れでもよいが、検出感度及び取扱いの容易さの観点から二本鎖鋳型DNAを用いることが好ましい。DNAはRNAに比べて酵素的に分解されにくく安定に存在するからである。これらの核酸分子を調製する方法は、化学合成法、PCR、DNAのクローン化と複製、その他の公知技術を用いることができる。鋳型DNAとしてはPCRにより合成した増幅産物をそのまま用いることができるが、好ましくはプラスミドなどにクローン化し、組換え体を用いて大量に調製及び精製したDNAの方が検出感度に優れる。RNAの場合は、無細胞系として翻訳反応系を用いるが、二本鎖鋳型DNAの場合は転写/翻訳共役反応系を用いる必要がある。このような無細胞系としては、大腸菌由来のもの、コムギ胚芽由来のもの、ウサギ網状赤血球由来のものが市販されているが、大腸菌等の原核細胞由来の抽出液が好ましい。大腸菌の抽出液では、転写、翻訳の共役反応によりDNAからタンパク質を合成できることが知られ、例えば、ズベイらによって大腸菌のS30抽出液を用いる方法が系統的に開発されている(例えば、ジェフリー・ズベイ(Geoffrey Zubay)、「アニュアル・レビュー・オブ・ジェネティクス(Annual Review of Genetics)」1973年、第7巻、p.267−287参照)。S30抽出液には、mRNAの翻訳に必要なリボソーム、アミノアシルtRNA合成酵素、ポリペプチド鎖開始因子(IF)、同伸長因子(EF)及び同終結因子が含まれている。合成タンパク質の鋳型としてDNAを用いる場合は、強力なプロモーター(一般的にはT7プロモーター)の下流に目的タンパク質遺伝子を配置したDNAが用いられ、転写と翻訳の両反応を共役させるために、系内にT7RNAポリメラーゼと4種類のリボヌクレオチド(ATP、GTP、CTP及びUTP)を添加する。アミノアシルtRNAの合成、及びmRNAの翻訳反応にはATPのエネルギーが必要であることから、クレアチンキナーゼ−クレアチンリン酸系等のエネルギー再生系を無細胞系に加える。このような構成成分により、細胞内で起こるタンパク質合成反応を試験管内で再構成している。
【0027】
大腸菌S30抽出液は、転写及び翻訳に必要な大腸菌の全ての酵素と因子を含んでいるが、更に補充的な混合液を添加することができる。具体的な調製方法としては、まず最初に大腸菌を培養し、菌体を遠心分離等により回収する。回収された菌体は、洗浄後、緩衝液に再懸濁し、フレンチプレスやガラスビーズ、ワーリングブレンダー等を用いて破砕する。破砕された大腸菌の不溶物質を遠心分離で除去し、プレインキュベーション混合液と混合してインキュベーションする。この操作によって内在性のDNA、RNAが分解されるが、更に、カルシウム塩やマイクロコッカスのヌクレアーゼ等を添加して内在性の核酸を分解させてもよい。続いて、透析により内在性のアミノ酸、核酸、ヌクレオシド等を除き、適量ずつ分注して液体窒素又は−80℃にて保存する。
【0028】
転写、翻訳及びエネルギー再生に必要な全てのタンパク質因子にタグを付けて別々に調製し、これらを再構成した無細胞タンパク質合成系も知られている。これらのタンパク質因子に大腸菌70Sリボソーム、tRNA、アミノ酸、NTP等の転写、翻訳に必要な成分を添加したキットも販売されている。
【0029】
(検出キット)
本発明の方法に用いるために上記方法により調製された核酸分子、及び大腸菌の細胞抽出液等の無細胞翻訳反応試薬は、使用しやすいように一定量ごと分注して、目的分子の検出キットとして配送することができる。これらの製品は凍結又は乾燥状態で保存することができ、保存及び輸送に適した容器に収容してキットとして販売される。キットには取扱説明書や陽性コントロールサンプル等を添付することができる。
【0030】
(バイオセンサー)
本発明はまた、上記核酸分子によって形質転換された原核細胞からなるバイオセンサーに関する。特に、本発明の二本鎖DNAからなる核酸分子は安定であり、プラスミドDNAとして種々の原核細胞で安定に存在しうる。或いは、本発明の二本鎖DNAを支持体に固定化したバイオセンサーを用いてもよい。
【0031】
以下に具体的な実施例を示して本発明をさらに詳細に説明するが、本発明の範囲はこれらの実施例に限定されるものではない。
【実施例】
【0032】
[実験材料及び方法]
1 鋳型DNAの調製
ルシフェラーゼ遺伝子をコードするプラスミドpBESTluc、及びβ−ガラクトシダーゼ遺伝子をコードするプラスミドpSV-β-Galactosidaseは、プロメガ社から購入した。本実施例で用いた全ての鋳型DNAは、以下に説明するポリメラーゼ連鎖反応(PCR)及びβ−ガラクトシダーゼ用の鋳型の調製については、これに続いて連結反応により調製した。PCRは、20μLの反応混合液中、4pmolの正方向プライマー、4pmolの逆方向プライマー、0.5ユニットのPrimeSTAR HS DNAポリメラーゼ(商品名、タカラバイオ社)、それぞれ4nmolのdNTPs(タカラバイオ社)、及び4μLの5倍濃度PrimeSTAR反応用緩衝液を含み、第一次PCR用には200pgのpBESTluc又はpSV-β-Galactosidaseを、第二次以降のPCR用には約20fmolの精製したPCR産物を鋳型として用いた。PCR産物は、アガロースゲル電気泳動により、GFX PCR DNA and Gel Band Purification Kit(商品名、GEヘルスケアバイオサイエンス株式会社)を用いて精製し、260nmの吸光度を測定して定量した。
【0033】
2 鋳型DNAのインビトロにおける転写/翻訳共役反応
インビトロにおける転写/翻訳反応は、T7RNAポリメラーゼを含む再構成された無細胞翻訳システム(ピュアシステム、クラシックII、ポストゲノム研究所)を用いて行った。約0.1μgの鋳型DNAを含む10μLの反応溶液を、種々の濃度のテオフィリン、カフェイン又はcGMPの存在下又は非存在下において、37℃で1時間インキュベートした。反応終了後の翻訳溶液はさらに精製することなく次のアッセイに用いた。
【0034】
3 mRNA鋳型の調製
鋳型DNAのインビトロ転写反応は、MEGAscript T7 Kit(商品名、アンビオン)を用いて行った。0.3μgの鋳型DNAを含む10μlの反応溶液を37℃で4.5時間インキュベートした。当該キットに添付されているTURBO DNaseを1ユニット添加した後、さらに15分間37℃でインキュベートした。転写産物は、RNeasy MinElute Cleanup Kit(商品名、キアゲン)を用いて精製し、上述と同様の方法で定量した。
【0035】
4 mRNA鋳型のインビトロ翻訳反応
インビトロ翻訳反応は、ピュアシステム、クラシックIIを用いて行った。約1μgのmRNAを含む10μlの反応溶液を、種々の濃度のコファクター(テオフィリン)の存在又は非存在下で37℃、1時間インキュベートした。生成した翻訳産物を含む溶液は精製せずに次のアッセイに用いた。
【0036】
5 ルシフェラーゼアッセイ
翻訳溶液と水を等量含む5μlの混合物を100μlのルシフェラーゼアッセイ試薬(プロメガ)と混合した。化学発光強度の測定は、ワラック社の1420ARVOsx装置により、黒色96穴プレート(コーニングコースター3915)を用いて行った。化学発光の映像はクールセイバーAE−6955(アトー)により取得した。
【0037】
6 β−ガラクトシダーゼアッセイ
10μlの翻訳溶液を、40μlの1×レポーターリシスバッファー(プロメガ)、及び50μlのオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を含む2×アッセイバッファー(プロメガ社のβ−ガラクトシダーゼアッセイシステム)と混合した。混合溶液は37℃にて3時間インキュベートした。この反応溶液の405nmにおける吸光度をワラック社の1420ARVOsx装置により、透明の96穴プレート(コーニングコースター3635)を用いて測定した。β−ガラクトシダーゼを含まない(鋳型なしの翻訳溶液)バックグラウンド強度をそれぞれの測定データから差し引いた。
【0038】
[実施例1]テオフィリン誘導アプタザイムとルシフェラーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたテオフィリンの検出
ルシフェラーゼ遺伝子をコードするプラスミドpBESTlucと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号1)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0039】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、第三次、及び第四次PCRを行った。
第二次PCR用
正方向プライマー:5'-ACCCTGATGAGCCTGGATACCAGCCGAAAGGCCCTTGGCAGTTAGACGAAACAAGAAGGAGATATACCAATG-3'(配列番号3)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第三次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCCTGG-3'(配列番号4)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第四次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTATCACCTTTTTGTAGGTTG-3'(配列番号5)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0040】
第三次PCRによって増幅された二本鎖DNAは、5’末端のT7プロモーターから転写されるテオフィリン依存性アプタザイムとルシフェラーゼをコードしており、RBS/アンチ−RBSによって形成される二重鎖は、表1の二重鎖2の配列を有する。また、第四次PCRによって増幅された二本鎖DNAは、同様に5’末端のT7プロモーターから転写されるテオフィリン依存性アプタザイムとルシフェラーゼをコードしており、RBS/アンチ−RBS二重鎖は、表1の二重鎖5の配列を有する(全体配列を配列番号24に示す)。
【0041】
対照実験に用いるための通常の5’非翻訳領域(T7ファージのg10リーダー配列)を有する鋳型DNAは、以下のプライマー対を用いて増幅した。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号6)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
第二次PCR用
正方向プライマー:5'-GAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCA-3'(配列番号7)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0042】
このようにして、種々の塩基配列及び長さのRBS/アンチ−RBS二重鎖を有する核酸分子を作製し、ルシフェラーゼ遺伝子をレポーター遺伝子として翻訳のON/OFF転換効率を調べた。ON/OFF転換効率とは、ここではテオフィリンの非存在下(OFF)に対する存在下(ON)における翻訳されたルシフェラーゼ活性として規定される。その結果を下記の表1に示す。
【0043】
【表1】
【0044】
ハンマーヘッド型リボザイムの触媒活性を維持する上で、切断部位の上流に隣接するGUAのトリプレット塩基が不可欠であることが分かっているため、このGUAトリプレットに隣接して直接RBSを結合した核酸分子を作製した(表1の二重鎖1)。この核酸分子1のテオフィリン1mMの場合のON/OFF転換効率は、表1に示したように約2.2倍であった。テオフィリン存在下(ON状態)でのルシフェラーゼ活性は5708cpsであり、タンパク質大量合成のために一般的に用いられる5’−非翻訳領域(T7ファージのg10リーダー配列)を有する鋳型から転写される場合(100000cps以上)と比べて極めて小さいことが分かった。そこで、RBS配列の5’側に、T7ファージのg10リーダー配列と同じ3つのプリン塩基AAGを挿入した一連の核酸分子2〜5を作製した。このようにして作製した核酸分子2は、ON状態において約6倍のルシフェラーゼ活性を示したが、ON/OFF転換効率は、わずかに1.7倍上昇したに過ぎなかった。これはOFF状態においても活性が上昇したことに起因する。テオフィリン非存在下(OFF状態)における発現を抑えるために、次に、アンチ−RBS部位の相補鎖を延長した核酸分子3及び4を作製した。
【0045】
より長いアンチ−RBS部位及びその隣接塩基からなる配列:5'-UUUCCUUUUU-3'(配列番号8)及び5'-AUAUUUCCUUUUU-3'(配列番号9)を含む核酸は、OFF状態における発現を効率的に抑えたが、同時にON状態における発現量も低下し、ON/OFF転換効率はそれぞれ4.4及び4.6と中程度に留まった。そこで、アンチ−RBS二重鎖の中にミスマッチを導入した配列5:5'-UAUCACCUUUUU-3'(配列番号10)を含む核酸を作製したところ、予想外にも極めて高いON/OFF転換効率(7.2)を有することが分かった。
【0046】
この最適化された二重鎖5を含む核酸分子を用いて、種々の濃度のテオフィリン又はカフェイン存在下でON/OFF転換効率を測定した結果を図3に示した。カフェインはテオフィリンと比べてN7位にメチル基を有する以外は類似する構造を有するにもかかわらず、1mMの濃度でON/OFF転換効率がほぼ1であり、当該核酸分子が高い特異性(テオフィリン依存性及びテオフィリン直交性)を有することが示された。
【0047】
鋳型DNAの代わりに鋳型mRNAを用いて同様の実験を行った結果を図4に示す。これらの結果より、本核酸分子の活性は転写レベルではなくて翻訳レベルにおいて制御されていることが示唆される。また、mRNAを用いた場合よりも鋳型DNAを用いた場合の方が効率が良い。
【0048】
[実施例2]テオフィリン誘導アプタザイムとβ−ガラクトシダーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたテオフィリンの検出
β−ガラクトシダーゼ遺伝子は、約3000bpと非常に長いため、最初に前方部分と後方部分とに分けてPCRを行い、続いてそれぞれの増幅断片を制限酵素AatIIで切断し、両方のDNA断片をライゲーションハイ(商品名、東洋紡)により連結した。下記プライマー配列中のAatIIサイトを下線で示した。
【0049】
前方部分
β−ガラクトシダーゼ遺伝子をコードするプラスミドpSV-β-Galactosidaseと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGCGCTGTATGGCGAGATC-3'(配列番号11)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号12)
【0050】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、第三次、第四次、及び第五次PCRを行った。
第二次PCR用
正方向プライマー:5'-AGGAGATATACCAATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTC-3'(配列番号13)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号12)
第三次PCR用
正方向プライマー:5'-ACCCTGATGAGCCTGGATACCAGCCGAAAGGCCCTTGGCAGTTAGACGAAACAAGAAGGAGATATACCAATG-3'(配列番号14)
逆方向プライマー:5'-GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
第四次PCR用
正方向プライマー:5'- CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCC TCCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCCTGG-3'(配列番号16)
逆方向プライマー:5'- GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
第五次PCR用
正方向プライマー:5'- CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCC TATCACCTTTTTGTAGGTTG-3'(配列番号17)
逆方向プライマー:5'- GTTTATGCAGCAACGAGACGTCAC-3'(配列番号15)
【0051】
後方部分
第一次PCR用
正方向プライマー:5'-CATTTTCCGTGACGTCTCGTTG-3'(配列番号18)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号19)
【0052】
上記PCRにより増幅した前方部分と後方部分とを連結後、全長DNAを鋳型として、以下の最終プライマー対を用いてPCRを行い、5’末端のT7プロモーターから転写されるテオフィリン依存性リボスイッチをコードする二本鎖DNA断片を取得した。
【0053】
最終的なPCR
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGAC-3'(配列番号20)
逆方向プライマー:5'-GGATTAGTTATTCATTATTTTTGACACCAGACCAAC-3'(配列番号19)
【0054】
このようにして作製したテオフィリン誘導アプタザイムとβ−ガラクトシダーゼ遺伝子とを含む二本鎖DNA(最適化されたRBS/アンチ−RBS二重鎖を含む)を用いて、種々の濃度のテオフィリン存在下でON/OFF転換効率を測定した結果を図5(A)に示す。本測定方法においては、翻訳産物であるβ−ガラクトシダーゼが無色のオルト−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)を黄色のオルト−ニトロフェノールに加水分解するため酵素活性の発現を可視化することができる。実際に、インビトロにおける転写/翻訳共役反応(37℃、1時間)の後、反応溶液にONPGを添加してさらに数時間37℃でインキュベートするだけで特殊な検出器を用いずにテオフィリンを可視的に検出することができた(図5(B))。1mM又は2mMのテオフィリンに対するON/OFF転換効率は、それぞれ20及び40であり、ルシフェラーゼ遺伝子を用いた場合(約7及び9)に比べてより高い値を示した。その理由の一つとしては、大腸菌由来の酵素であるβ−ガラクトシダーゼ遺伝子に比べて、ルシフェラーゼ遺伝子がレアコドンを多く有していることが挙げられる。レアコドンとは、それに対応するtRNA量が翻訳反応系において乏しいコドンをいう。このことは、翻訳反応系の条件を最適化することによって、さらに検出感度が向上する可能性を示唆する。
【0055】
[実施例3]cGMP依存性アプタザイムとルシフェラーゼ遺伝子とを用いた核酸分子(リボスイッチ)の作製と、これを用いたcGMPの検出
ルシフェラーゼ遺伝子をコードするプラスミドpBESTlucと、以下のプライマー対とを用いて第一次PCRを行った。
第一次PCR用
正方向プライマー:5'-AGGAGATATACCAATGGAAGACGCCAAAAACATAAAGAAAG-3'(配列番号21)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0056】
引き続き、以下のそれぞれのプライマー対と、直前のPCR増幅産物とを用いて順に第二次、及び第三次PCRを行った。
第二次PCR用
正方向プライマー:5'-CCTTTTTGTAGGTTGCCCGAAAGGGCGACCCTGATGAGCC CACCGATGGTGAAAGTGGCTGACGACCAATCGAAACAAGAAGGAGATATACCAATG-3'(配列番号22)
第三次PCR用
正方向プライマー:5'-CTTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCATATTTCCTTTTTGTAGGTTG-3'(配列番号23)
逆方向プライマー:5'-TATTCATTACAATTTGGACTTTCCGCCCTTCTTGG-3'(配列番号2)
【0057】
このようにして作製したcGMP依存性アプタザイムの塩基配列の一部及びその推定二次構造を図6に示した。Breaker et al.によって報告されたcGMP依存性アプタザイムは、実施例1で最適化されたアンチ−RBS配列5と相補的な配列を含むため、まず最初に、当該リボザイムの活性を損なわないように塩基置換を行った変異体アプタザイムを合成した。しかしながら、この変異体アプタザイムもなお、正確なRBS/アンチ−RBS二重鎖を形成するには不十分であったため、上述した表1のアンチ−RBS配列4を用いたところ、アンチ−RBS配列5を用いた場合よりもON/OFF転換効率が高くなることが分かった。種々の濃度のcGMP存在下においてルシフェラーゼアッセイを行い、ON/OFF転換効率を調べた結果を図7に示す。10mMのcGMP存在下において、約10の高いON/OFF転換効率を示した。この結果は、異なるアプタザイムを用いる場合においても、それぞれの目的分子に合わせて容易に本発明の核酸分子をデザインできることを示す。
【0058】
[実施例4]インビボアッセイ
実施例1で作製したテオフィリン依存性アプタザイムを含む核酸分子を図8に示すプラスミドpET15b_5SL4misLucのTAR(Theophyline-Aptazyme-Riboswitch)領域にクローン化した。図8に示したように、本発明の機能性核酸分子はT7プロモーターの制御下において発現が調節される。このプラスミドを大腸菌BL21(DE3)pLysS株(ノバジェン、ラクトースオペロンの制御下のT7RNAポリメラーゼ遺伝子をゲノムに含む)に形質転換した。この形質転換大腸菌を一晩培養後、培養液で1/20に希釈後、さらに37℃で振とう培養し、培養液の600nmでの吸光度が約0.5に達したときに、最終濃度が1mMとなるようにIPTGを添加した。また、最終濃度が0〜5mMの種々の濃度となるようにテオフィリンを添加した。引き続き23℃又は37℃で1.5〜2.5時間培養を続け、菌体を回収して溶解し、ルシフェラーゼ活性を測定した。その結果を図9に示した。図9に示したように、23℃で培養したときにテオフィリンの濃度依存的にON/OFF転換効率が増加することが認められた。37℃に比べて23℃の方が効率が良い理由としては、アプタザイム活性の至適温度が23℃付近であるから、及び低温の方が転写/翻訳反応の速度が遅くなり、細胞内で合成されたmRNAのRBS/アンチ−RBS二重鎖が形成し易くなるから、並びにアプタザイムによって切断されたmRNAの分解速度が遅くなるから等の理由が考えられる。また、培養液に5mMのマグネシウムを添加したときの方が若干活性が上昇した。これらの結果より、本発明のリボスイッチを用いる目的分子の検出方法は、インビボにおいても適用可能であることが示された。
【図面の簡単な説明】
【0059】
【図1】本発明に係る核酸分子及びこれを用いる目的分子(コファクター)の検出方法の一実施形態を示した模式図である。
【図2】本発明の一実施形態に係るテオフィリン依存性アプタザイムの構造を示した模式図である。
【図3】実施例1において、RBS二重鎖5を用いて調製した本発明の核酸分子について、種々の濃度のテオフィリン又はカフェイン存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図4】実施例1において、RBS二重鎖5を用いて調製した本発明の核酸分子について、直接mRNAを用いて反応した場合のルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図5】実施例2において、RBS二重鎖5を用いて調製した本発明の核酸分子について、種々の濃度のテオフィリン存在下におけるβ−ガラクトシダーゼアッセイのON/OFF転換効率を測定した結果(A)、及びテオフィリンの非存在(−)及び存在(2mM)下におけるアッセイ溶液の写真を示す。
【図6】実施例3において調製したcGMP依存性アプタザイムを含む本発明の核酸分子の塩基配列と推定二次構造を示した模式図である。
【図7】実施例3において、種々の濃度のcGMP存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【図8】本発明に係る核酸分子をTAR領域に挿入したプラスミド構造を示した模式図である。
【図9】実施例4で調製した形質転換大腸菌を用いて、種々の濃度のテオフィリン存在下におけるルシフェラーゼアッセイのON/OFF転換効率を測定した結果を示す。
【特許請求の範囲】
【請求項1】
目的分子を検出するための核酸分子であって、
(a)レポーター遺伝子配列、
(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、
(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び
(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含み、
目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成し、そして
目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離することを特徴とする核酸分子。
【請求項2】
前記核酸分子が一本鎖RNAからなり、前記二重鎖を含むステム−ループ構造を形成することを特徴とする請求項1に記載の核酸分子。
【請求項3】
前記ステム−ループ構造は、20〜40℃の温度の水溶液中において前記一本鎖RNAが分子内で会合することにより形成されることを特徴とする請求項2に記載の核酸分子。
【請求項4】
前記アプタザイム配列が、目的分子と結合するアプタマー領域と、触媒活性を有するリボザイム領域と、を含み、前記目的分子がアプタマー領域と結合したとき、リボザイム領域の構造変化を引き起こして前記自己切断触媒活性を示すことを特徴とする請求項1〜3何れか記載の核酸分子。
【請求項5】
前記自己切断部位が、アプタザイム配列とアンチリボソーム結合部位との間に位置することを特徴とする請求項1〜4何れか記載の核酸分子。
【請求項6】
前記リボソーム結合部位と、アンチリボソーム結合部位と、それらの隣接配列とによって分子内で形成される二重鎖が、6〜20塩基対の長さを有することを特徴とする請求項1〜5何れか記載の核酸分子。
【請求項7】
前記アンチリボソーム結合部位の5’側に、少なくとも10塩基長のステム−ループ構造を形成しうる配列を含むことを特徴とする請求項1〜6何れか記載の核酸分子。
【請求項8】
前記アンチリボソーム結合部位及びその隣接塩基が、配列番号9又は10で表される塩基配列からなることを特徴とする請求項1〜7何れか記載の核酸分子。
【請求項9】
前記レポーター遺伝子が、ルシフェラーゼ遺伝子又はβ−ガラクトシダーゼ遺伝子である請求項1〜8何れか記載の核酸分子。
【請求項10】
請求項1〜9何れか記載の核酸分子をコードする二本鎖DNAであって、当該コード領域の上流に機能的に連結された転写プロモーター配列を含むことを特徴とする二本鎖DNA。
【請求項11】
(a)請求項1〜9何れか記載の核酸分子と、目的分子を含む試料とを、翻訳反応系内において所定の時間接触させる工程、及び
(b)前記レポーター遺伝子の発現量を測定する工程、
を含むことを特徴とする目的分子の検出方法。
【請求項12】
(a)請求項10に記載の二本鎖DNAと、目的分子を含む試料とを、転写/翻訳共役反応系内において所定の時間接触させる工程、及び
(b)前記レポーター遺伝子の発現量を測定する工程、
を含むことを特徴とする目的分子の検出方法。
【請求項13】
請求項1〜9何れか記載の核酸分子と、無細胞翻訳反応試薬とを含むことを特徴とする目的分子の検出キット。
【請求項14】
請求項10に記載の二本鎖DNAと、無細胞転写/翻訳共役反応試薬とを含むことを特徴とする目的分子の検出キット。
【請求項15】
請求項10に記載の二本鎖DNAによって形質転換された組換え原核細胞。
【請求項16】
原核生物の翻訳反応系においてリガンド分子の添加によって遺伝子発現を誘導し得る核酸分子であって、
(a)所望の遺伝子配列、
(b)前記遺伝子配列と機能的に結合したリボソーム結合部位、
(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び
(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含み、
前記アプタザイムがリガンドの存在下において自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離することを特徴とする核酸分子。
【請求項1】
目的分子を検出するための核酸分子であって、
(a)レポーター遺伝子配列、
(b)前記レポーター遺伝子配列と機能的に結合したリボソーム結合部位、
(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び
(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含み、
目的分子の非存在下において、前記アンチリボソーム結合部位は、前記リボソーム結合部位とハイブリダイズして二重鎖を形成し、そして
目的分子の存在下において、前記アプタザイムは自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離することを特徴とする核酸分子。
【請求項2】
前記核酸分子が一本鎖RNAからなり、前記二重鎖を含むステム−ループ構造を形成することを特徴とする請求項1に記載の核酸分子。
【請求項3】
前記ステム−ループ構造は、20〜40℃の温度の水溶液中において前記一本鎖RNAが分子内で会合することにより形成されることを特徴とする請求項2に記載の核酸分子。
【請求項4】
前記アプタザイム配列が、目的分子と結合するアプタマー領域と、触媒活性を有するリボザイム領域と、を含み、前記目的分子がアプタマー領域と結合したとき、リボザイム領域の構造変化を引き起こして前記自己切断触媒活性を示すことを特徴とする請求項1〜3何れか記載の核酸分子。
【請求項5】
前記自己切断部位が、アプタザイム配列とアンチリボソーム結合部位との間に位置することを特徴とする請求項1〜4何れか記載の核酸分子。
【請求項6】
前記リボソーム結合部位と、アンチリボソーム結合部位と、それらの隣接配列とによって分子内で形成される二重鎖が、6〜20塩基対の長さを有することを特徴とする請求項1〜5何れか記載の核酸分子。
【請求項7】
前記アンチリボソーム結合部位の5’側に、少なくとも10塩基長のステム−ループ構造を形成しうる配列を含むことを特徴とする請求項1〜6何れか記載の核酸分子。
【請求項8】
前記アンチリボソーム結合部位及びその隣接塩基が、配列番号9又は10で表される塩基配列からなることを特徴とする請求項1〜7何れか記載の核酸分子。
【請求項9】
前記レポーター遺伝子が、ルシフェラーゼ遺伝子又はβ−ガラクトシダーゼ遺伝子である請求項1〜8何れか記載の核酸分子。
【請求項10】
請求項1〜9何れか記載の核酸分子をコードする二本鎖DNAであって、当該コード領域の上流に機能的に連結された転写プロモーター配列を含むことを特徴とする二本鎖DNA。
【請求項11】
(a)請求項1〜9何れか記載の核酸分子と、目的分子を含む試料とを、翻訳反応系内において所定の時間接触させる工程、及び
(b)前記レポーター遺伝子の発現量を測定する工程、
を含むことを特徴とする目的分子の検出方法。
【請求項12】
(a)請求項10に記載の二本鎖DNAと、目的分子を含む試料とを、転写/翻訳共役反応系内において所定の時間接触させる工程、及び
(b)前記レポーター遺伝子の発現量を測定する工程、
を含むことを特徴とする目的分子の検出方法。
【請求項13】
請求項1〜9何れか記載の核酸分子と、無細胞翻訳反応試薬とを含むことを特徴とする目的分子の検出キット。
【請求項14】
請求項10に記載の二本鎖DNAと、無細胞転写/翻訳共役反応試薬とを含むことを特徴とする目的分子の検出キット。
【請求項15】
請求項10に記載の二本鎖DNAによって形質転換された組換え原核細胞。
【請求項16】
原核生物の翻訳反応系においてリガンド分子の添加によって遺伝子発現を誘導し得る核酸分子であって、
(a)所望の遺伝子配列、
(b)前記遺伝子配列と機能的に結合したリボソーム結合部位、
(c)前記リボソーム結合部位と分子内でハイブリダイズし得るアンチリボソーム結合部位、及び
(d)前記リボソーム結合部位とアンチリボソーム結合部位との間に連結されたアプタザイム配列、を含み、
前記アプタザイムがリガンドの存在下において自己切断触媒活性を示し、前記アンチリボソーム結合部位を含む当該切断部位から5’側の核酸断片が前記リボソーム結合部位から解離することを特徴とする核酸分子。
【図1】

【図2】
【図3】
【図4】

【図6】
【図7】
【図8】
【図9】
【図5】
【図2】
【図3】
【図4】
【図6】
【図7】
【図8】
【図9】
【図5】
【公開番号】特開2008−220191(P2008−220191A)
【公開日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願番号】特願2007−58760(P2007−58760)
【出願日】平成19年3月8日(2007.3.8)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
【公開日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願日】平成19年3月8日(2007.3.8)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
[ Back to top ]