
正極活物質および電極の製造方法、ならびに電極
【課題】リチウム−遷移金属複合酸化物の性能低下を抑制しつつ、リチウム−遷移金属複合酸化物の表面に形成された被膜を除去する手段を提供することを目的とする。
【解決手段】リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法。
【解決手段】リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、正極活物質および電極の製造方法、ならびに電極に関する。より詳しくは、本発明は、リチウム−遷移金属複合酸化物の性能低下を抑制しつつ、リチウム−遷移金属複合酸化物の表面に形成される被膜の少なくとも一部を効果的に除去する手段に関する。
【背景技術】
【0002】
近年、地球温暖化に対処するため、二酸化炭素排出量の低減が切に望まれている。自動車業界では、電気自動車(EV)やハイブリッド電気自動車(HEV)の導入による二酸化炭素排出量の低減に期待が集まっており、これらの実用化の鍵を握るモータ駆動用二次電池の開発が盛んに行われている。
【0003】
モータ駆動用二次電池としては、携帯電話やノートパソコン等に使用される民生用リチウムイオン二次電池と比較して極めて高い出力特性、および高いエネルギー密度を発揮することが求められている。したがって、全ての電池の中で最も高い理論エネルギーを有するリチウムイオン二次電池が注目を集めており、現在急速に開発が進められている。
【0004】
リチウムイオン二次電池は、正極と負極とが、電解質を含む電解質層を介して接続され、電池ケースに収納される構成を有している。上記正極または負極は、一般に、正極活物質または負極活物質とバインダとが溶媒中に分散されてなる活物質スラリーを集電体の表面に塗布することによって形成される。
【0005】
このようなリチウムイオン二次電池の正極活物質として、例えば、リチウム−ニッケル複合酸化物、リチウム−マンガン複合酸化物、リチウム−コバルト複合酸化物、リチウム−鉄複合酸化物などの、リチウム−遷移金属複合酸化物などが用いられうる。このうち、特に、リチウム−ニッケル複合酸化物は、反応性、サイクル特性に優れ、コストが低いことからもモータ駆動用二次電池に好適であり、その実用化が期待されている。
【0006】
しかしながら、リチウム−ニッケル複合酸化物をはじめとしたリチウム−遷移金属複合酸化物からなる正極活物質は、その表面に被膜が形成されることが知られている。しかしながら、このような被膜が形成された正極活物質をそのまま用いて電池を作製した場合、電池性能に悪影響が生じることが以前から問題となっており、その解決方法が模索されていた。
【0007】
例えば、特許文献1では、リチウム−ニッケル複合酸化物などを主成分とする正極活物質の製造方法において、当該被膜が形成された未処理正極活物質をpH10〜13に調整した処理水溶液に接触させる表面処理工程を設けることが提案されている。そして、当該方法により、正極活物質中からリチウムイオンが溶出するのを抑制しつつ、正極活物質の表面を被覆しているリチウム塩(炭酸リチウムなど)の少なくとも一部を除去することができる、としている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2009−99461号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、本発明者らは、上記特許文献1に記載された方法を試みたところ、新たな問題が生じることが判明した。すなわち、当該文献の実施例1の方法にしたがい、リチウム−ニッケル複合酸化物からなる正極活物質を水酸化リチウム溶液に投入し、プロペラ攪拌機を用いて攪拌するという表面処理を行ったところ、被膜の一部は除去されたものの、正極活物質そのものも破砕されてしまったのである。正極活物質粒子が破砕されてしまうと、正極活物質の粒径を所望の範囲に制御することが困難となるばかりでなく、正極活物質の性能低下をも招来する虞がある。また、正極活物質粒子の破砕を防ぐため、正極活物質を水酸化リチウム溶液に投入した後、攪拌を行わない方法(浸漬のみ)や、プロペラ攪拌機の回転数を少なくする方法も試みたが、この場合は表面の被膜はほとんど除去することができなかった。つまり、特許文献1に記載された方法では、リチウム−遷移金属複合酸化物の性能を維持しつつ、表面に形成された被膜を効果的に除去することは不可能であった。
【0010】
そこで、本発明は、リチウム−遷移金属複合酸化物の性能低下を抑制しつつ、リチウム−遷移金属複合酸化物の表面に形成された被膜の少なくとも一部を効果的に除去する手段を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記の課題に鑑み鋭意研究を積み重ねた。その過程で、所定の酸解離定数(pKa)を有する酸を用いた表面処理により、上記課題が解決されうることを見出し、本発明を完成させるに至った。
【0012】
すなわち、本発明の一形態によれば、リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法が提供される。
【発明の効果】
【0013】
本発明によれば、酸の酸解離定数(pKa)を6.35未満とすることにより、リチウム−遷移金属複合酸化物の表面に形成された被膜を構成する化合物が酸と反応し溶解するため、被膜の少なくとも一部を除去することができる。また、酸解離定数(pKa)が4以上の酸を用いることにより、リチウム−遷移金属複合酸化物のリチウムイオンのプロトン置換や、リチウムイオンの脱離を防ぐことができるため、正極活物質としての性能低下を抑制することができる。
【図面の簡単な説明】
【0014】
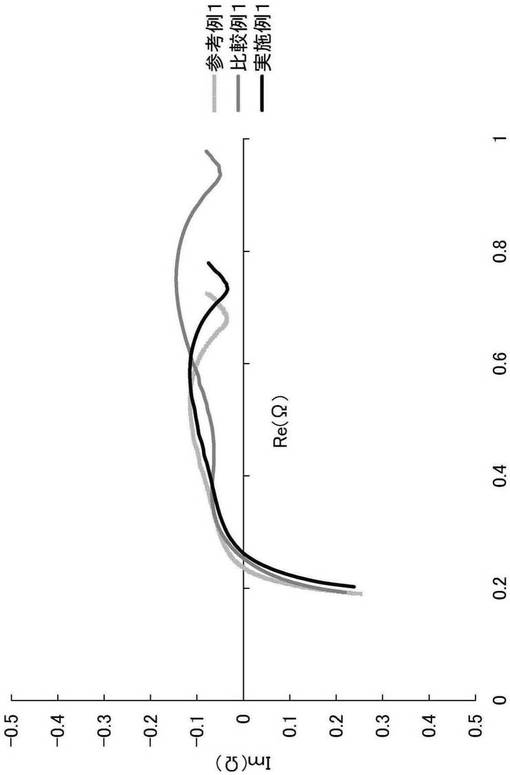
【図1】実施例1および比較例1のEIS試験の結果を示すグラフである。
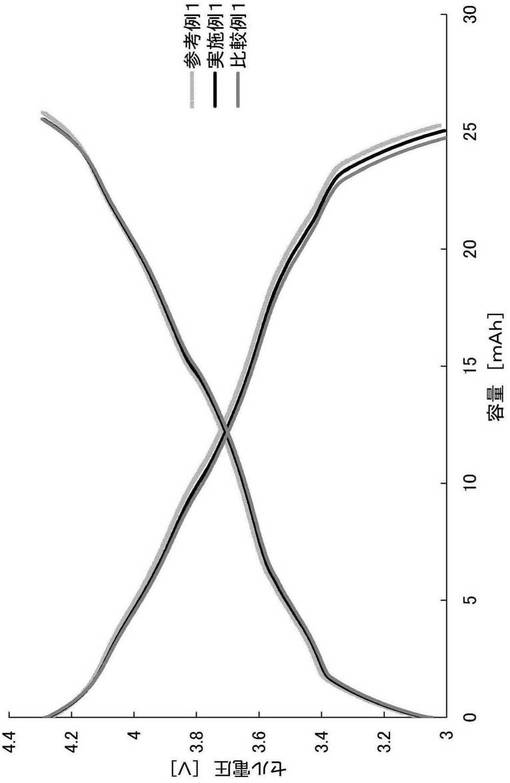
【図2】実施例1および比較例1の充放電容量試験の結果を示すグラフである。
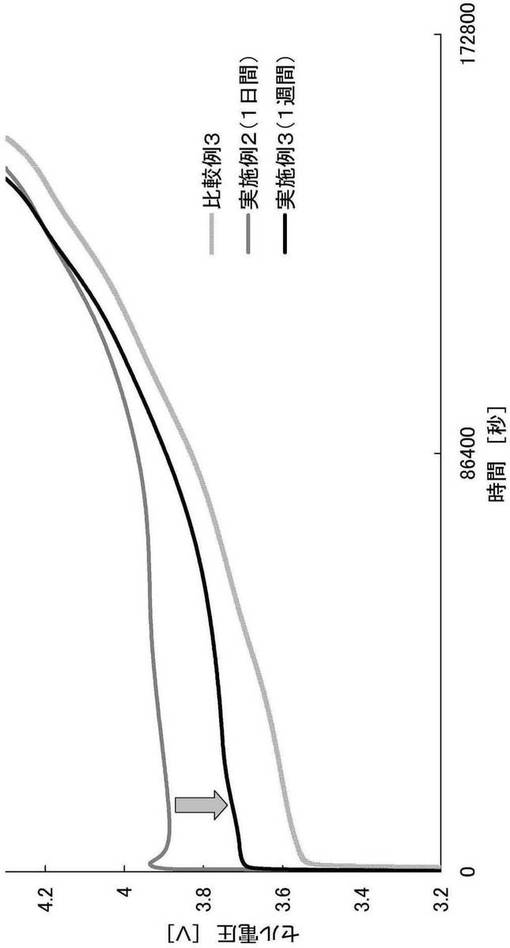
【図3】実施例2および3ならびに比較例3の過電圧試験の結果を示すグラフである。
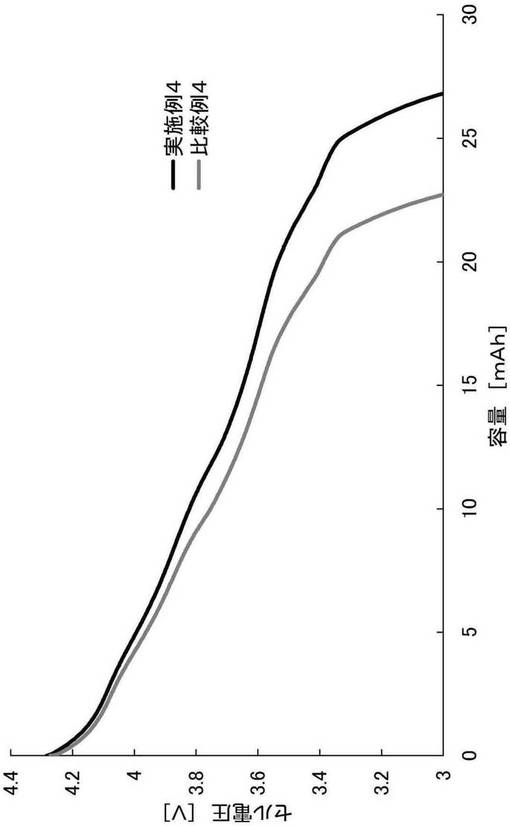
【図4】実施例4および比較例4の放電容量試験の結果を示すグラフである。
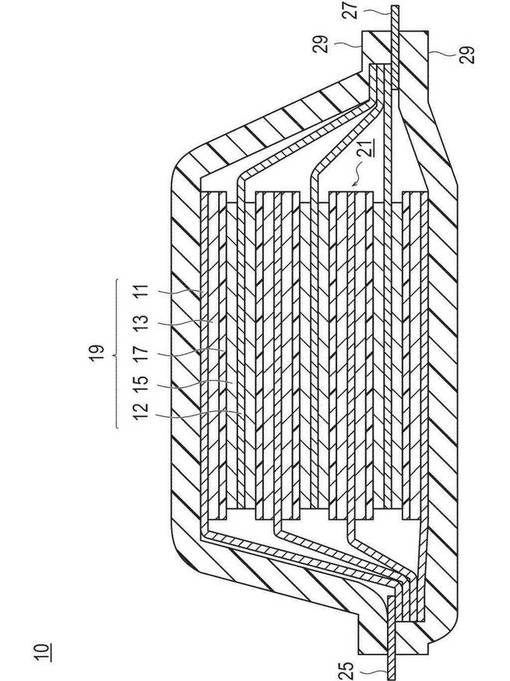
【図5】本発明の一実施形態に係る積層型のリチウムイオン二次電池の全体構造を模式的に表した概略図である。
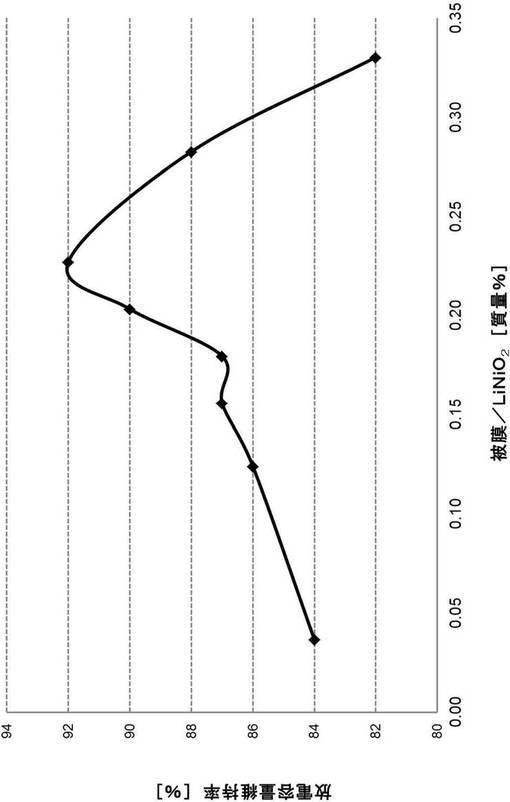
【図6】被膜/LiNiO2の割合に対する放電容量維持率の関係を表すグラフである。
【発明を実施するための形態】
【0015】
<処理済み正極活物質の製造方法>
以下、本発明の好ましい形態を説明する。本発明の一形態は、リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法に関する。なお、以降の本明細書中では、「酸解離定数(pKa)が4以上、6.35未満の酸」を、「酸解離定数(pKa)が4以上、6.35未満の弱酸」または単に「弱酸」とも称する。
【0016】
まず、本形態の理解を容易にするために、本形態の処理工程によってリチウム−遷移金属複合酸化物の表面に形成される被膜が除去されるメカニズムについて、リチウム−ニッケル複合酸化物を例に挙げて説明する。ただし、本発明の技術的範囲は、あくまでも特許請求の範囲の記載に基づいて定められるべきであり、以下のメカニズムにより限定されるものではない。したがって、本発明の効果が以下メカニズム以外のメカニズムによって生じていたとしても、本発明の技術的範囲は限定的に解釈されるものではない。
【0017】
リチウム−ニッケル複合酸化物を通常の空気雰囲気に曝すと、表面に被膜が形成される。この被膜の形成は、下記化学式1に示される2段階の化学反応により起こると考えられていた。すなわち、まず空気中に含まれる水分により、表面のリチウム−ニッケル複合酸化物(例えば、LiNiO2)が加水分解されて水酸化リチウム(LiOH)が生じる。次に当該LiOHが空気中の二酸化炭素(CO2)と反応することによって炭酸リチウム(Li2CO3)が生じ、表面に被膜が形成される。つまり、これまでの技術常識によると、リチウム−ニッケル複合酸化物の表面に形成される被膜の構成成分は、Li2CO3であると考えられていたのである。
【0018】
【化1】

【0019】
しかしながら、今回、本発明者らが被膜の構成成分を解析するために熱分析(不活性ガス雰囲気下で25℃から500℃まで加熱)を行ったところ、被膜は200℃付近で熱分解し、それと同時にCO2とH2Oとを生じることが判明した。Li2CO3の融点は約723℃であるため、少なくとも200℃付近で熱分解することはない。また、Li2CO3がたとえ分解したとしてもH2Oが生じることはない。この事実より、被膜の構成成分は、従来考えられていたLi2CO3ではなく、準安定塩である炭酸水素リチウム(LiHCO3)であることが推測された。この場合、LiHCO3が形成される反応は、以下の化学式2のようであると考えられる。
【0020】
【化2】
【0021】
本発明者らは、上記知見を基に、リチウム−ニッケル複合酸化物の表面の被膜に、炭酸の酸解離定数(pKa1)6.35よりも小さいpKa値を有する酸を接触させたところ、被膜が容易に除去された。これにより被膜の構成成分がLiHCO3であるとの本発明者らの推測はより確からしいことが示された。さらに、酸として強酸を用いた場合は、リチウム−ニッケル複合酸化物の性能が低下してしまうこと;酸の酸解離定数(pKa)を4以上とすることにより、リチウム−ニッケル複合酸化物の性能低下を抑制できることも判明した。加えて、リチウム−ニッケル複合酸化物以外のリチウム−遷移金属複合酸化物においても同様の効果が得られることが確認された。これらの結果より、酸解離定数(pKa)が4以上、6.35未満である弱酸を用いることにより、被膜の除去および性能低下の抑制の両立を可能としたリチウム−遷移金属複合酸化物の処理方法を完成させたのである。以下、本形態の製造方法について詳細に説明する。
【0022】
本形態の製造方法の原料として用いられる正極活物質(以下、「未処理の正極活物質」とも称する)は、リチウム−遷移金属複合酸化物を必須に含む。
【0023】
本形態におけるリチウム−遷移金属複合酸化物は、リチウムイオンを吸蔵・放出可能な正極活物質として使用される材料であれば特に制限はなく、本技術分野で使用されうるものを当業者が適宜採用することができる。このようなリチウム−遷移金属複合酸化物の一例として、リチウム−ニッケル複合酸化物が挙げられる。なお、本明細書において、「リチウム−ニッケル複合酸化物」は、金属元素としてLiおよびNiのみを含む複合酸化物のみならず、Niの一部がCo、Mn、Alなどの元素で置換された複合酸化物をも含む。このようなリチウム−ニッケル複合酸化物としては、具体的には、LiNiO2、LiNi0.5Mn0.5O2、LiNi0.9Co0.1O2、LiNi0.8Co0.2O2、LiNi0.8Co0.18Mn0.02O2、LiNi1/3Co1/3Mn1/3O2、LiNi0.8Co0.15Al0.05O2、LiNi0.6Co0.1Mn0.25Al0.05O2などが例示される。
【0024】
リチウム−ニッケル複合酸化物以外にも、リチウム−遷移金属複合酸化物として、LiCoO2等のリチウム−コバルト複合酸化物、スピネルLiMn2O4等のリチウム−マンガン複合酸化物、LiFeO2等のリチウム−鉄複合酸化物およびこれらの遷移金属(Co、Mn、Fe)の一部を他の元素により置換したもの等が使用できる。
【0025】
これらのうち、本形態の処理工程では、リチウム−ニッケル複合酸化物からなる群から選択される少なくとも1種を含む正極活物質を用いることが好ましい。リチウム−ニッケル複合酸化物は、特に吸湿しやすいため、上述の被膜の問題が他のリチウム−遷移金属複合酸化物よりも顕著に現れる。したがって、本形態の処理工程を、リチウム−ニッケル複合酸化物を含む正極活物質に適用することによって、本発明の効果をより一層享受することができる。
【0026】
なお、これらのリチウム−遷移金属複合酸化物は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。また、リチウム−遷移金属複合酸化物の形状やサイズについても、特に制限はなく、当該分野で使用されうるあらゆる形状やサイズのリチウム−遷移金属複合酸化物に対して有効である。
【0027】
さらに、本形態の正極活物質は、リチウム−遷移金属複合酸化物以外の他の正極活物質材料を含んでも構わない。他の正極活物質材料として、LiFePO4等の遷移金属とリチウムのリン酸化合物や硫酸化合物;V2O5、MnO2、TiS2、MoS2、MoO3等の遷移金属酸化物や硫化物;PbO2、AgO、NiOOH等が使用可能である。これらの他の正極活物質材料も、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0028】
上記リチウム−遷移金属複合酸化物は、その表面に被膜が形成されているものでありうる。上述のように、本発明者らの知見によると、当該被膜は、構成成分としてLiHCO3を含んでいるものと考えられる。このような被膜は、リチウム−遷移金属複合酸化物を、空気中で保管した際に形成される。また、後述のように、リチウム−遷移金属複合酸化物に対する被膜量の程度が、電池性能や電池寿命に大きく影響する。
【0029】
弱酸は、リチウム−遷移金属複合酸化物の表面に形成される被膜を分解し、除去する役割を果たす。したがって、弱酸により被膜の少なくとも一部を除去することにより、リチウム−遷移金属複合酸化物に対する被膜量を制御し、電池性能や電池寿命を改善することが可能となる。上述のように、本発明者らの知見によると、被膜にはその構成成分としてLiHCO3が含まれると考えられる。LiHCO3は、理論上、炭酸の酸解離定数(pKa1)6.35よりも小さい酸と反応するため、本形態で使用される弱酸は、酸解離定数(pKa)が6.35未満であることが必須である。一方、LiHCO3は、酸解離定数(pKa)が6.35未満の酸であれば強酸であっても反応するが、酸の強度が大きくなりすぎると、リチウム−遷移金属複合酸化物の正極活物質としての性能が低下する虞がある。この性能の低下は、リチウム−遷移金属複合酸化物において、リチウムイオンのプロトン置換(例えば、リチウム−ニッケル複合酸化物においては、LiNiO2+H+→HNiO2+Li+)や、リチウムイオンの脱離が起こることが一因である。したがって、このような好ましくない反応を抑えるため、本形態においては、弱酸の酸解離定数(pKa)を4以上とすることが必須である。なお、本明細書において、「酸解離定数(pKa)」は水中、25℃における値であり、具体的には、赤岩英夫編、「分析化学実験」、丸善、1999年発行に記載の方法により測定される。
【0030】
本形態において、弱酸は、酸解離定数(pKa)が4以上、6.35未満であれば、特に制限はなく、無機酸および有機酸のいずれも使用可能である。また、酸の物質状態も特に制限はなく、常温常圧(25℃、1気圧)下において、固体、液体、および気体のいずれであってもよい。このような弱酸としては、例えば、アジ化水素(pKa=4.65)、アクリル酸(pKa=4.26)、アジピン酸(pKa1=4.26、pKa2=5.03)、アゼライン酸(pKa1=4.39、pKa2=5.12)、o−アニス酸(pKa=4.09)、m−アニス酸(pKa=4.09)、p−アニス酸(pKa=4.48)、アニリン(pKa=4.65)、安息香酸(pKa=4.20)、イソ吉草酸(pKa=4.58)、イソ酪酸(pKa=4.63)、オクタン酸(pKa=4.89)、吉草酸(pKa=4.64)、キナルジン酸(pKa=4.75)、グルタル酸(pKa1=4.13、pKa2=5.03)、2−ナフトエ酸(pKa=4.16)、ビニル酢酸(pKa=4.12)、ピメリン酸(pKa1=4.31、pKa2=5.08)、フェニル酢酸(pKa=4.10)、p−フルオロ安息香酸(pKa=4.14)、p−ブロモ安息香酸(pKa=4.00)、ヘキサン酸(pKa=4.63)、ヘプタン酸(pKa=4.66)、p−ヨード安息香酸(pKa=4.00)、酪酸(pKa=4.63)、レブリン酸(pKa=4.44)などが挙げられる。これらの酸は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0031】
上記弱酸が常温常圧(25℃、1気圧)下において固体である場合、弱酸を適当な溶媒に溶解した弱酸溶液をリチウム−遷移金属複合酸化物と接触させてもよい。この際に使用される溶媒は特に制限はなく、例えば、水、メタノール、エタノール、n−プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、エチレングリコール、プロピレングリコールなどのプロトン性の極性溶媒;ジエチルエーテル、酢酸エチル、ギ酸メチル、プロピオン酸メチル、テトラヒドロフラン、ジメチルスルホキシド、アセトニトリル、シクロヘキサノン、アセトン、メチルエチルケトン、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、メチルエチルカーボネート、1,2−ジメトキシエタン、1,2−ジエトキシエタン、1,4−ジオキサン、1,3−ジオキソラン、エチレングリコールジメチルエーテル、γ−ブチロラクトン、γ−バレロラクトンなどの非プロトン性の極性溶媒などを用いることができる。これらの溶媒は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。なお、弱酸溶液中の弱酸の濃度は、弱酸の酸強度等に応じて当業者が適宜設定することができる。
【0032】
未処理の正極活物質と弱酸(または弱酸溶液)とを接触させる処理工程において、弱酸(または弱酸溶液)に含まれる水分は少ないほど好ましい。具体的には、弱酸(または弱酸溶液)の含水率は、50体積ppm以下であることが好ましく、30体積ppm以下であることがより好ましい。一方、下限値は特に制限はないが、通常0.01体積ppm程度である。このように、弱酸(または弱酸溶液)に含まれる水分を少なくすることにより、未処理の正極活物質中に含まれるリチウム−遷移金属複合酸化物が水と接触して加水分解され、LiOHが生成するのを防ぐことができる。これにより、リチウム−遷移金属複合酸化物の正極活物質としての性能低下をより一層防ぐことができる。なお、ここで述べた、弱酸(または弱酸溶液)に含まれる水分を少なくするという形態は、あくまでも発明の好ましい形態に過ぎず、上述の弱酸溶液に使用される溶媒としての水の使用を妨げるものではない。
【0033】
本形態で使用される好ましい弱酸として、酢酸が挙げられる。酢酸は、常温常圧(25℃、1気圧)下において液体であり、そのまま未処理の正極活物質との接触に用いることができる点で本形態に好適である。また、酢酸は、含水率の低いもの(氷酢酸)が比較的容易に入手可能な点でも有利である。
【0034】
本形態において、未処理の正極活物質と弱酸とを接触させる方法は、特に制限はない。一例を挙げると、正極活物質を弱酸(または弱酸溶液)中に浸漬させる方法;正極活物質の表面に弱酸(または弱酸溶液)を塗布または噴霧する方法などが挙げられる。このうち、十分に正極活物質と弱酸を接触させることができるという観点から、正極活物質を弱酸(または弱酸溶液)中に浸漬させる方法であることが好ましい。なお、浸漬の際には、正極活物質の性能を著しく低下させない限りにおいて、正極活物質が入った弱酸(または弱酸溶液)を攪拌しても構わない。また、正極活物質と弱酸とを接触させる際の、弱酸(または弱酸溶液)の温度も特に制限はないが、好ましくは20〜40℃である。
【0035】
本形態において、正極活物質を弱酸に接触させる際の接触時間は特に制限はない。しかしながら、被膜が完全に除去されたリチウム−遷移金属複合酸化物を弱酸に接触させておくと、リチウムイオンのプロトン置換や、リチウムイオンの脱離によって正極活物質としての性能が低下する虞があることから、接触時間を必要以上に長くしないことが好ましい。接触時間は、処理するリチウム−遷移金属複合酸化物の量や、被膜の厚さ、使用する弱酸、接触させる方法によるため、一概に規定することは困難である。よって、本形態では、正極活物質を、弱酸に接触させる工程の終点を、被膜と弱酸との反応により発生するCO2の増加率によって決定することが好ましい。本明細書において、「CO2増加率」は、下記数式1により定義される。
【0036】
【数1】
【0037】
具体的には、正極活物質を弱酸に接触させる際に、単位時間(例えば1時間)当たりに発生するCO2量を随時モニタリングする。発生するCO2量は、反応する被膜量に相当するため、通常、接触開始直後が最大となり、接触時間が長くなるにつれ低下する。すなわち、接触開始直後は、CO2増加率は1であるが、次第にその値は小さくなり、最終的には0となる。よって、予め設定した所定の数値範囲内にCO2増加率が到達した時点で接触を終了させることにより、様々な条件下においても、接触工程の終点を容易に判断することができるのである。より具体的には、上記数値範囲が、0.05〜0.1であることが好ましい。下限値を0.05とすることにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離による正極活物質としての性能低下をより一層防ぐことができる。一方、上限値を0.1とすることにより、リチウム−遷移金属複合酸化物の表面に残存する被膜の量を低減させることができる。
【0038】
本形態では、正極活物質を弱酸に接触させる工程の後、正極活物質の表面に付着した弱酸を取り除くために、洗浄用の溶媒等で洗浄する工程をさらに含んでもよい。使用される洗浄用溶媒は、特に制限はなく、例えば、上述の弱酸溶液に使用される溶媒と同様のものを適宜しようすることができる。また、当該洗浄工程の後、正極活物質の表面に付着した洗浄用溶媒を、真空乾燥機などを用いて乾燥させる、乾燥工程をさらに設けてもよい。以上の工程によって、リチウム−遷移金属複合酸化物の表面の被膜の少なくとも一部が取り除かれた、処理済み正極活物質が得られる。
【0039】
本形態の処理工程によると、リチウム−遷移金属複合酸化物の表面の被膜の少なくとも一部が弱酸によって除去される。当該被膜は、上述のように、LiHCO3を主成分としていると考えられる。したがって、リチウム−遷移金属複合酸化物と弱酸との反応により、リチウムと弱酸との塩が生じる。例えば、弱酸として酢酸を使用した場合には、反応の結果、酢酸リチウム(CH3COOLi)が生じる(下記化学式3参照)。
【0040】
【化3】
【0041】
反応により生じたCH3COOLi等のリチウム塩は、洗浄工程を行う場合などは、その大部分が表面から除去される。しかしながら、その一部は、リチウム−遷移金属複合酸化物の表面に物理的に付着したままとなりうる。リチウム−遷移金属複合酸化物表面に付着したCH3COOLi等のリチウム塩は、リチウム−遷移金属複合酸化物の表面をX線光電子分光(XPS)分析することにより検出されうる。よって、リチウム−遷移金属複合酸化物の表面にCH3COOLi等のリチウム塩が存在するか否かによって、本形態の処理工程が行われたか否かを判定することが可能である。換言すると、本形態の処理工程を経た正極活物質は、リチウム−遷移金属複合酸化物と、弱酸のリチウム塩とを含む。
【0042】
<処理済み電極の製造方法>
以上、本形態の処理済み正極活物質の製造方法について説明したが、本発明は、リチウム−遷移金属複合酸化物を含む電極に対しても適用することが可能である。すなわち、本発明の他の一形態によると、集電体と、集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を有する正極活物質層と、を含む電極の少なくとも正極活物質層を、酸解離定数(pKa)が4以上、6.35未満の弱酸に接触させる処理工程を含む、処理済み電極の製造方法が提供される。
【0043】
上述のように、リチウム−遷移金属複合酸化物は、通常の空気雰囲気に曝されただけでも、空気中に含まれるH2OやCO2によって反応し、表面に被膜が形成されうる。従来、被膜が形成された状態のリチウム−遷移金属複合酸化物を正極活物質として用いて電池を作製すると、電池の充放電時に被膜が分解されてCO2ガスを発生させ、電池内圧の上昇による電池特性の低下や電池の膨脹を引き起こす、といった問題が生じていた。したがって、これまでは被膜の形成を防ぐために、電極の製造工程をドライボックス中で行う、などといった対応が必要であった。しかしながら、車両用などの大型の電池をドライボックス中で製造することは、量産化の足枷となるばかりか、設備コストが膨大となるため結果的に電池コストを抑えられず、電気自動車等の普及の足枷にもなりかねない。そこで、特に電池に組み込む前の電極に対して適用できるような処理方法が切に求められていた。本形態の処理工程は、このような要請にも応えるものであり、電極の性能を低下させずに被膜の少なくとも一部を除去することを可能とするものである。また、本形態の処理工程を行った後の処理済み電極を用いて電池の組み立てを行えば、電池内部に持ち込まれる被膜量を抑えることができるため、充放電反応時に被膜が分解されることによるガスの発生を抑制することができる。以下、本形態の製造方法について説明する。
【0044】
本形態において、電極は、集電体の一方の面にリチウム−遷移金属複合酸化物を含む正極活物質層を有する限りにおいては、特に制限はない。例えば、集電体の一方の面にのみ、リチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなる正極;集電体の両方の面にリチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなる正極;集電体の一方の面にリチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなり、集電体の他方の面に負極活物質を含む負極活物質層が形成されてなる双極型電極;等の形態が挙げられるが、本形態の処理工程は、これらのいずれの電極に対しても適用可能である。以下、本形態の電極について説明する。
【0045】
(集電体)
本形態において、集電体を構成する材料に特に制限はない。例えば、金属や、導電性高分子材料または非導電性高分子材料に導電性フィラーが添加された樹脂が採用されうる。具体的には、金属としては、アルミニウム、ニッケル、鉄、ステンレス鋼、チタン、銅等が挙げられる。これらのほか、ニッケルとアルミニウムとのクラッド材、銅とアルミニウムとのクラッド材、あるいはこれらの金属を組み合わせためっき材等が好ましく用いられうる。また、金属表面にアルミニウムが被覆されてなる箔であってもよい。なかでも、電子伝導性や電池作動電位の観点からは、アルミニウム、ステンレス鋼、銅が好ましい。
【0046】
また、導電性高分子材料としては、例えば、ポリアニリン、ポリピロール、ポリチオフェン、ポリアセチレン、ポリパラフェニレン、ポリフェニレンビニレン、ポリアクリロニトリル、およびポリオキサジアゾール等が挙げられる。かような導電性高分子材料は、導電性フィラーを添加しなくても十分な導電性を有するため、製造工程の容易化または集電体の軽量化の点において有利である。
【0047】
非導電性高分子材料としては、例えば、ポリエチレン(PE;高密度ポリエチレン(HDPE)、低密度ポリエチレン(LDPE))、ポリプロピレン(PP)、ポリエチレンテレフタレート(PET)、ポリエーテルニトリル(PEN)、ポリイミド(PI)、ポリアミドイミド(PAI)、ポリアミド(PA)、ポリテトラフルオロエチレン(PTFE)、スチレン−ブタジエンゴム(SBR)、ポリアクリロニトリル(PAN)、ポリメチルアクリレート(PMA)、ポリメチルメタクリレート(PMMA)、ポリ塩化ビニル(PVC)、ポリフッ化ビニリデン(PVdF)、およびポリスチレン(PS)等が挙げられる。かような非導電性高分子材料は、優れた耐電位性または耐溶媒性を有しうる。
【0048】
上記の導電性高分子材料または非導電性高分子材料には、必要に応じて導電性フィラーが添加されうる。特に、集電体の基材となる樹脂が非導電性高分子のみからなる場合は、樹脂に導電性を付与するために必然的に導電性フィラーが必須となる。導電性フィラーは、導電性を有する物質であれば特に制限なく用いることができる。例えば、導電性、耐電位性、またはリチウムイオン遮断性に優れた材料として、金属および導電性カーボン等が挙げられる。金属としては、特に制限はないが、Ni、Ti、Al、Cu、Pt、Fe、Cr、Sn、Zn、In、Sb、およびKからなる群から選択される少なくとも1種の金属もしくはこれらの金属を含む合金または金属酸化物を含むことが好ましい。また、導電性カーボンとしては、特に制限はないが、アセチレンブラック、バルカン、ブラックパール、カーボンナノファイバー、ケッチェンブラック、カーボンナノチューブ、カーボンナノホーン、カーボンナノバルーン、およびフラーレンからなる群から選択される少なくとも1種を含むことが好ましい。導電性フィラーの添加量は、集電体に十分な導電性を付与できる量であれば特に制限はなく、一般的には、5〜35質量%程度である。
【0049】
集電体の大きさは、電池の使用用途に応じて決定される。例えば、高エネルギー密度が要求される大型の電池に用いられるのであれば、面積の大きな集電体が用いられる。集電体の厚さについても特に制限はないが、通常は1〜100μm程度である。
【0050】
(正極活物質層)
正極活物質層は正極活物質として、リチウム−遷移金属複合酸化物を必須に含み、必要に応じて、リチウム−遷移金属複合酸化物以外の他の正極活物質材料を含みうる。リチウム−遷移金属複合酸化物や、他の正極活物質材料の詳細については、上述の処理済み正極活物質の製造方法において述べた内容と同様であるので、ここでは詳細な説明を省略する。
【0051】
(負極活物質層)
本形態の電極は、負極活物質層を含みうる。負極活物質層に含まれる負極活物質は、リチウムを可逆的に吸蔵および放出できるものであれば特に制限されない。負極活物質の例としては、SiやSn等の金属、あるいはTiO、Ti2O3、TiO2、もしくはSiO2、SiO、SnO2等の金属酸化物、Li4/3Ti5/3O4もしくはLi7MnN等のリチウムと遷移金属との複合酸化物、Li−Pb系合金、Li−Al系合金、Li、または天然黒鉛、人造黒鉛、カーボンブラック、活性炭、カーボンファイバー、コークス、ソフトカーボン、もしくはハードカーボン等の炭素材料等が好ましく挙げられる。また、負極活物質は、リチウムと合金化する元素を含むことが好ましい。リチウムと合金化する元素を用いることにより、従来の炭素系材料に比べて高いエネルギー密度を有する高容量および優れた出力特性の電池を得ることが可能となる。上記負極活物質は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0052】
上記のリチウムと合金化する元素としては、以下に制限されることはないが、具体的には、Si、Ge、Sn、Pb、Al、In、Zn、H、Ca、Sr、Ba、Ru、Rh、Ir、Pd、Pt、Ag、Au、Cd、Hg、Ga、Tl、C、N、Sb、Bi、O、S、Se、Te、Cl等が挙げられる。これらの中でも、容量およびエネルギー密度に優れた電池を構成できる観点から、炭素材料、ならびに/またはSi、Ge、Sn、Pb、Al、In、およびZnからなる群より選択される少なくとも1種以上の元素を含むことが好ましく、炭素材料、Si、またはSnの元素を含むことが特に好ましい。これらは1種単独で使用しても良いし、2種以上を併用してもよい。なお、負極活物質の形状やサイズは、特に制限はなく、当業者により適宜設定されうる。
【0053】
正極活物質層または負極活物質層(以下、単に「活物質層」とも称する)には、必要であれば、その他の物質が含まれてもよい。例えば、導電助剤、バインダ等が含まれうる。また、イオン伝導性ポリマーが含まれる場合には、前記ポリマーを重合させるための重合開始剤が含まれてもよい。
【0054】
導電助剤とは、活物質層の導電性を向上させるために配合される添加物をいう。導電助剤としては、アセチレンブラック、カーボンブラック、ケッチェンブラック、グラファイト等のカーボン粉末や、気相成長炭素繊維(VGCF;登録商標)等の種々の炭素繊維、膨張黒鉛等が挙げられる。しかし、導電助剤がこれらに限定されないことはいうまでもない。
【0055】
バインダとしては、ポリフッ化ビニリデン(PVdF)、ポリイミド、PTFE、SBR、合成ゴム系バインダ等が挙げられる。しかし、バインダがこれらに限定されないことはいうまでもない。また、バインダとゲル電解質として用いるマトリックスポリマーとが同じ場合には、バインダを使用する必要はない。
【0056】
活物質層に含まれる成分の配合比および活物質層の厚さについては特に限定されず、リチウムイオン二次電池用の電極についての公知の知見を参照し、当業者が適宜設定することができる。
【0057】
本形態の製造方法に用いられる電極(以下、「未処理の電極」)は、特に制限されず、従来公知の知見を適宜参照することにより製造されうる。例えば、活物質、バインダ、および粘度調整用溶媒を含む活物質スラリーを調製し、当該活物質スラリーを集電体上に塗布し、乾燥させた後、プレスすることで未処理の電極が作製されうる。
【0058】
上記未処理の電極に含まれるリチウム−遷移金属複合酸化物は、その表面に被膜が形成されているものでありうる。上述のように、本発明者らの知見によると、当該被膜は、構成成分としてLiHCO3を含んでいるものと考えられる。このような被膜は、リチウム−遷移金属複合酸化物や電極を、空気中で保管した際に形成されうる。
【0059】
本形態の製造方法は、上述の未処理の電極の少なくとも正極活物質層を酸解離定数(pKa)が4以上、6.35未満の弱酸に接触させる処理工程を含む点に特徴を有する。なお、弱酸との接触は、電極の正極活物質層のみならず、集電体や負極活物質層等を含む電極全体に対して行ってもよい。使用される弱酸や、接触方法、および処理工程の終点の決定方法等の諸条件は、上述の処理済み正極活物質の製造方法と同様であるので、ここでは詳細な説明を省略する。
【0060】
本形態の処理工程によると、電極の少なくとも正極活物質層を酸解離定数(pKa)6.35未満の弱酸に接触させることにより、電極の正極活物質中に含まれるリチウム−遷移金属複合酸化物の表面に形成される被膜の少なくとも一部を除去することが可能である。また、弱酸の酸解離定数(pKa)を4以上とすることにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離によるリチウム−遷移金属複合酸化物の正極活物質としての性能の低下を抑制することができる。また、弱酸の酸解離定数(pKa)を4以上とすることにより、集電体や活物質層中に含まれる他の部材との反応を抑えることができるため、電極の性能の低下を抑制することができる。さらに、本形態の処理工程を行った後の処理済み電極を用いて電池の組み立てを行うことにより、電池内部に持ち込まれる被膜量を抑えることができるので、充放電反応時に被膜が分解されることによるガスの発生を抑制することも可能である。
【0061】
<電極>
以上、処理済み電極の製造方法について説明してきたが、当該製造方法によると、電極における正極活物質層中に含まれるリチウム−遷移金属複合酸化物の表面に存在する被膜の量を制御することができる。本発明者らの検討によると、後述の実施例で示すように、上述の処理工程により被膜の少なくとも一部を除去した場合、全く除去しない場合と比較して、電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されることが分かった。さらに、被膜を完全に除去した場合よりも、被膜を所定量残すように処理工程を行う場合の方が、電池のサイクル特性(放電容量維持率)の点で特に優れることが分かった。
【0062】
したがって、本発明の他の一形態によると、上述の処理済み電極の製造方法により得られうる電極が提供される。本形態の電極は、リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、当該被膜の量は、リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%であることが好ましい。
【0063】
また、本発明の他の一形態によると、集電体と、集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層とを含む電極であって、リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、当該被膜の量は、リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である電極が提供される。
【0064】
上記被膜は、リチウム−遷移金属複合酸化物の表面に存在する、リチウム−遷移金属複合酸化物以外のリチウム塩から構成される。本発明者らの知見によると、当該被膜は、上述で説明したように、LiHCO3を含みうる。しかしながら、被膜の全ての構成成分がLiHCO3であるとは限らず、被膜の形成条件等によりLi2CO3や、弱酸処理により生じる弱酸のリチウム塩等が含まれる場合もある。
【0065】
本形態では、上記被膜の量はリチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%であることが好ましく、より好ましくは0.15〜0.29質量%であり、さらに好ましくは0.2〜0.26質量%であり、特に好ましくは0.20〜0.23質量%である。被膜量を上記範囲内とすることにより、後述の実施例で示すように、サイクル特性(放電容量維持率)を有意に向上させることができる。
【0066】
電極における被膜量の測定方法は特に制限はないが、空気中で電極を作製する際は、経時的に被膜量が増加するため、直接的に被膜量を測定することが難しい。この場合は、後述の実施例で示すように、所定の温度・湿度下に曝した際の時間に対する被膜量についての検量線を予め作成する。そして、内挿によりある時間(電極を作製するのに掛かる時間)における被膜量を求める。その後、(被膜量)÷(被膜を有しないリチウム−遷移金属複合酸化物量)×100を算出することにより、被膜のリチウム−遷移金属複合酸化物に対する質量割合[質量%]を算出することが可能である。
【0067】
なお、本形態の電極に含まれる、集電体、活物質(正極活物質、負極活物質)、および、任意に含まれる導電助剤、バインダ等は、上述の処理済み電極の製造方法において述べた内容と同様であるので、ここでは詳細な説明を省略する。
【0068】
本形態の電極において電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されるメカニズムは定かではないが、本発明者らは以下のように推測している。すなわち、サイクル特性に関しては、リチウム−遷移金属複合酸化物の表面の所定量の被膜が、リチウム−遷移金属複合酸化物の結晶構造の変化(例えば、充電時の高電位環境や、電解液との接点増加に起因する結晶構造の変化)を防ぐ働きをしていると考えている。その結果、リチウム−遷移金属複合酸化物の耐久性が向上し、サイクル特性(放電容量維持率)が向上するものと推測される。一方、被膜量が0.31質量%を超えると、電池の充放電時に被膜が分解されてCO2ガスが発生することに起因する電池特性の低下の影響が大きくなるため、サイクル特性が低下すると考えられる。また、放電容量に関しては、リチウム−遷移金属複合酸化物へのリチウムイオンの吸蔵・放出が被膜により妨げられていることが原因であると考えられる。また、インピーダンス抵抗に関しては、被膜が抵抗増大の要因となっていると考えられる。
【0069】
<電気デバイス>
上述の本形態の電極は、サイクル特性に優れるため、様々な電気デバイスに好適に使用されうる。すなわち、本発明の一形態によると上記電極を含む電気デバイスが提供される。電気デバイスは、上述の電極を要するものであれば特に制限はなく、リチウムイオン二次電池などの二次電池および電気二重層キャパシタなどが挙げられる。以下、電気デバイスの一例として、本形態の電極を具備したリチウムイオン二次電池について説明する。
【0070】
図5に、本発明の一実施形態に係る積層型のリチウムイオン二次電池の全体構造を模式的に表した概略図を示す。本実施形態のリチウムイオン二次電池10は、実際に充放電反応が進行する略矩形の発電要素17が、電池外装材であるラミネートフィルム22の内部に封止された構造を有する。詳しくは、高分子−金属複合ラミネートフィルムを電池外装材として用いて、その周辺部の全部を熱融着にて接合することにより、発電要素17を収納し密封した構成を有している。
【0071】
発電要素17は、負極集電体11の両面(発電要素の最下層用および最上層用は片面のみ)に負極活物質層12が配置された負極と、電解質層13と、正極集電体14の両面に正極活物質層15が配置された正極とを積層した構成を有している。具体的には、1つの負極活物質層12とこれに隣接する正極活物質層15とが、電解質層13を介して対向するようにして、負極、電解質層13、正極がこの順に積層されている。
【0072】
これにより、隣接する負極、電解質層13および正極は、1つの単電池層16を構成する。したがって、本実施形態のリチウムイオン二次電池10は、単電池層16が複数積層されることで、電気的に並列接続されてなる構成を有するともいえる。また、単電池層16の外周には、隣接する負極集電体11と正極集電体14との間を絶縁するためのシール部(絶縁層)(図示せず)が設けられていてもよい。発電要素17の両最外層に位置する最外層負極集電体11aには、いずれも片面のみに負極活物質層12が配置されている。なお、負極および正極の配置を逆にすることで、発電要素17の両最外層に最外層正極集電体が位置するようにし、該最外層正極集電体の片面のみに正極活物質層が配置されているようにしてもよい。
【0073】
負極集電体11および正極集電体14には、各電極(負極および正極)と導通される負極集電板(負極タブ)18および正極集電板(正極タブ)19がそれぞれ取り付けられ、ラミネートフィルム22の端部に挟まれるようにラミネートフィルム22の外部に導出される構造を有している。負極集電板(負極タブ)18および正極集電板(正極タブ)19は、必要に応じて負極端子リード20および正極端子リード21を介して、各電極の負極集電体11および正極集電体14に超音波溶接や抵抗溶接などにより取り付けられていてもよい。ただし、負極集電体11が延長されて負極集電板(負極タブ)18とされ、ラミネートフィルム22から導出されていてもよい。同様に、正極集電体14が延長されて正極集電板(正極タブ)19とされ、同様に電池外装材22から導出される構造としてもよい。
【0074】
本実施形態のリチウムイオン二次電池10では、正極として上述の本形態の電極が使用される。これにより、リチウムイオン二次電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されうる。
【実施例】
【0075】
以下、本発明の作用効果を実施例および比較例を用いて説明する。ただし、本発明の技術的効果は以下の実施例に制限されるものではない。
【0076】
[実施例1]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0077】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0078】
(3)弱酸処理
保管後の正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1週間浸漬させ、その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0079】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0080】
[参考例1]
上記(2)正極の保管、および上記(3)弱酸処理を行わなかった(つまり被膜が形成されていない)ことを除いては、実施例1と同様の方法で、評価用コインセル(ブランク)を作製した。
【0081】
[比較例1]
上記(3)弱酸処理を行わなかったことを除いては、実施例1と同様の方法で、評価用コインセルを作製した。
【0082】
<EIS(電気化学インピーダンススペクトル法)試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流定電圧方式(CCCV、電流:1C)で4.3Vまで充電し、5分間休止させた。その後、印加電圧10mV、周波数100kHz〜1MHzにおいてインピーダンスを測定した。インピーダンス測定の結果をコール−コールプロットしたものを図1に示す。
【0083】
図1の結果より、温度30℃、相対湿度60%中で保管し、弱酸処理を行わなかった比較例1は、参考例1と比較してインピーダンスの上昇が確認された(図1において、横軸上のレジスタンスが約0.7[Ω]から約0.9[Ω]に上昇)。これは保管中に、リチウム−ニッケル複合酸化物の表面に被膜が形成されたことによるものである。一方、保管後に、弱酸処理を行った実施例1では、インピーダンスが参考例1に近いところまで減少していることが示された。これは、本発明の弱酸処理により、被膜が除去されたことを示す。
【0084】
<充放電容量試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:1C)でセル電圧3.0Vまで放電させた。当該充放電の際の充放電容量[mAh]を図2に示す。
【0085】
図2の結果より、保管後に弱酸処理を行った実施例1、および保管後に弱酸処理を行わなかった比較例1のいずれにおいても、参考例1と比較して充放電容量の大幅な減少は見られなかった。これは、本発明の弱酸処理を行っても、充放電容量の低下が起こりにくいことを示す。
【0086】
[実施例2]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0087】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0088】
(3)弱酸処理およびCO2濃度測定
保管後の正極を、ガス採取口の付いた密閉容器に入れた酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1日間浸漬させた。次いで、ガス採取口から注射器で密閉容器内のガスを採取し、採取したガス内のCO2量をガスクロマトグラフィーで測定し、1日後の密閉容器内のCO2濃度を求めた。その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0089】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0090】
[実施例3]
「(3)弱酸処理およびCO2濃度測定」において、酢酸中に浸漬させた時間を1週間としたことを除いては、実施例2と同様の方法で評価用コインセルを作製した。
【0091】
[比較例2]
「(2)正極の保管」を行わなかったことを除いては、実施例2と同様の方法で「(1)電極の作製」および「(3)弱酸処理およびCO2濃度測定」を行った。
【0092】
[比較例3]
「(2)正極の保管」を行わなかったことを除いては、実施例3と同様の方法で評価用コインセルを作製した。
【0093】
<CO2濃度および過電圧の関係>
上記実施例2および3、ならびに比較例2および3で測定したCO2濃度の結果を表1にまとめる。
【0094】
【表1】
【0095】
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C、電圧:4.3V)で充電した。充電の際のセル電圧を図3に示す。
【0096】
表1より、比較例2および3よりも、実施例2および3の方が二酸化炭素濃度が著しく高いことが示された。このことから、当該二酸化炭素濃度の差は、被膜と酢酸との反応により発生する二酸化炭素量の差に起因していることがわかる。また、実施例2よりも実施例3の方が二酸化炭素濃度が高いが、これは、1日間の浸漬後も、なお未反応の被膜が残存していることを意味する。
【0097】
図3より、実施例2は、実施例3よりも過電圧(電流印加前の電圧と電流印加20秒後の電圧の差(ΔV))が大きいことが示された。これは、上述のように、1日間の浸漬後も、なおリチウム−ニッケル複合酸化物の表面に被膜が残存し、電極の抵抗が上昇したためであると考えられた。
【0098】
[実施例4]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0099】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0100】
(3)弱酸処理
保管後の正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1週間浸漬させ、その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0101】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0102】
[比較例4]
「(3)弱酸処理」において、酢酸に代えて、フッ化水素(HF)溶液を用いたこと以外は、実施例4と同様の方法で評価用コインセルを作製した。なお、HFの酸解離定数(pKa)は3.17である。
【0103】
当該HF溶液は、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液(電解液)に、水を2000体積ppm混合することにより調製した。下記化学式4に示すように、電解液中のLiPF6は水と反応し、HFを生じる。
【0104】
【化4】
【0105】
<充放電容量試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:1C)でセル電圧3.0Vまで放電させた。当該放電の際の放電容量[mAh]を図4に示す。
【0106】
図4の結果より、HF溶液で処理した比較例4は、酢酸で処理した実施例4と比較して、放電容量の低下が確認された。これは、酸解離定数(pKa)が3.17のHFで処理することにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離が起こったためであると考えられた。
【0107】
[実施例5]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm、被膜量0.22質量%)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:20μm)の一方の面側に塗布し乾燥させた(正極活物質層の目付量:14mg/cm2)。その後、プレス処理を行い、積層方向から見た投影面積が100cm2(10cm×10cmの正方形)となるように切り出し、正極活物質層を片面に有する正極を作製した。以上の操作は、30℃、相対湿度(RH)50%の雰囲気下で行った。また、当該電極中に含まれるLiNiO2は1260mgであり、被膜は4.16mgであった。
【0108】
なお、当該被膜量は、予め作成した検量線に基づく内挿により算出した。検量線は、30℃、相対湿度(RH)50%の雰囲気下での、下記表2に示す各時間におけるLiNiO2量に対する被膜量の割合(質量%)をプロットすることにより作成した。
【0109】
【表2】
【0110】
(2)弱酸処理および被膜量測定
得られた正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1分間浸漬させ、次いで、温度80℃の真空乾燥機で1分間乾燥させた。弱酸処理前後の電極の質量を測定し、下記数式2に基づき弱酸処理後の被膜量を算出したところ、3.56[mg]であった。その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0111】
【数2】
【0112】
(3)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比3:7)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0113】
[比較例5]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させなかったことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0114】
[実施例6]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を3分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0115】
[実施例7]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を5分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0116】
[実施例8]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を10分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0117】
[実施例9]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を15分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0118】
[実施例10]
上記(2)弱酸処理および質量測定において、酢酸中に浸漬させた時間を20分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0119】
[実施例11]
上記(2)弱酸処理および質量測定において、酢酸中に浸漬させた時間を60分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0120】
<EIS(電気化学インピーダンススペクトル法)試験>
上記実施例5〜8および比較例5で作製した評価用コインセルを、25℃の雰囲気下、定電流―定電圧方式(CCCV、電流:0.1C)で電圧4.3Vまで充電し、5分間休止させた。その後、印加電圧10mV、周波数100kHz〜1MHzにおいてインピーダンスを測定し、コール−コールプロットを求めた。次に、コール−コールプロットから得たX軸との2交点である半円部分をインピーダンス抵抗として算出した。結果を表3に示す。
【0121】
<充放電容量試験>
上記実施例5〜11および比較例5で作製した各評価用コインセルを、25℃の雰囲気下、定電流―定電圧方式(CCCV、電流:0.1C)で電圧4.3Vまで充電し、その後、電流が50マイクロAになるまで4.3Vで保持した。その後、定電流(CC、電流:0.1C)でセル電圧3.0Vまで放電させ、コインセル容量を測定した。結果を表3に示す。
【0122】
<充放電サイクル試験>
上記実施例5〜11および比較例5で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:0.1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:0.1C)でセル電圧3.0Vまで放電し、5分間休止させた。この繰返しを1サイクルとし、300サイクル実施した。300サイクル後の放電容量と1サイクル目の放電容量からの維持率を求めた。結果を表3および図6に示す。
【0123】
【表3】
【0124】
表3および図6の結果より、被膜量がLiNiO2量に対して0.036〜0.31質量%である実施例5〜11は、比較例5と比較して、優れたサイクル特性を示すことが示された。さらに、図6より、LiNiO2に対する被膜の割合が0.15〜0.29質量%、0.2〜0.26質量%、0.20〜0.23質量%の順で、サイクル特性はより高くなることが分かった。以上の結果より、LiNiO2の表面の所定量の被膜が、LiNiO2の結晶構造の変化を防ぐ働きをし、その結果、リチウム−遷移金属複合酸化物の耐久性が向上し、サイクル特性が向上するものと推測された。
【0125】
また、表3より、LiNiO2の表面の被膜量が減少するにつれ、初期の放電容量が増加する傾向にあることが示された。これにより、LiNiO2へのチウムイオンの吸蔵・放出が被膜により妨げられていることが示唆された。
【0126】
また、表3より、LiNiO2の表面の被膜量が減少するにつれ、インピーダンス抵抗が減少する傾向にあることが示された。これにより、被膜の存在により抵抗が増大することが示唆された。
【技術分野】
【0001】
本発明は、正極活物質および電極の製造方法、ならびに電極に関する。より詳しくは、本発明は、リチウム−遷移金属複合酸化物の性能低下を抑制しつつ、リチウム−遷移金属複合酸化物の表面に形成される被膜の少なくとも一部を効果的に除去する手段に関する。
【背景技術】
【0002】
近年、地球温暖化に対処するため、二酸化炭素排出量の低減が切に望まれている。自動車業界では、電気自動車(EV)やハイブリッド電気自動車(HEV)の導入による二酸化炭素排出量の低減に期待が集まっており、これらの実用化の鍵を握るモータ駆動用二次電池の開発が盛んに行われている。
【0003】
モータ駆動用二次電池としては、携帯電話やノートパソコン等に使用される民生用リチウムイオン二次電池と比較して極めて高い出力特性、および高いエネルギー密度を発揮することが求められている。したがって、全ての電池の中で最も高い理論エネルギーを有するリチウムイオン二次電池が注目を集めており、現在急速に開発が進められている。
【0004】
リチウムイオン二次電池は、正極と負極とが、電解質を含む電解質層を介して接続され、電池ケースに収納される構成を有している。上記正極または負極は、一般に、正極活物質または負極活物質とバインダとが溶媒中に分散されてなる活物質スラリーを集電体の表面に塗布することによって形成される。
【0005】
このようなリチウムイオン二次電池の正極活物質として、例えば、リチウム−ニッケル複合酸化物、リチウム−マンガン複合酸化物、リチウム−コバルト複合酸化物、リチウム−鉄複合酸化物などの、リチウム−遷移金属複合酸化物などが用いられうる。このうち、特に、リチウム−ニッケル複合酸化物は、反応性、サイクル特性に優れ、コストが低いことからもモータ駆動用二次電池に好適であり、その実用化が期待されている。
【0006】
しかしながら、リチウム−ニッケル複合酸化物をはじめとしたリチウム−遷移金属複合酸化物からなる正極活物質は、その表面に被膜が形成されることが知られている。しかしながら、このような被膜が形成された正極活物質をそのまま用いて電池を作製した場合、電池性能に悪影響が生じることが以前から問題となっており、その解決方法が模索されていた。
【0007】
例えば、特許文献1では、リチウム−ニッケル複合酸化物などを主成分とする正極活物質の製造方法において、当該被膜が形成された未処理正極活物質をpH10〜13に調整した処理水溶液に接触させる表面処理工程を設けることが提案されている。そして、当該方法により、正極活物質中からリチウムイオンが溶出するのを抑制しつつ、正極活物質の表面を被覆しているリチウム塩(炭酸リチウムなど)の少なくとも一部を除去することができる、としている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2009−99461号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
しかしながら、本発明者らは、上記特許文献1に記載された方法を試みたところ、新たな問題が生じることが判明した。すなわち、当該文献の実施例1の方法にしたがい、リチウム−ニッケル複合酸化物からなる正極活物質を水酸化リチウム溶液に投入し、プロペラ攪拌機を用いて攪拌するという表面処理を行ったところ、被膜の一部は除去されたものの、正極活物質そのものも破砕されてしまったのである。正極活物質粒子が破砕されてしまうと、正極活物質の粒径を所望の範囲に制御することが困難となるばかりでなく、正極活物質の性能低下をも招来する虞がある。また、正極活物質粒子の破砕を防ぐため、正極活物質を水酸化リチウム溶液に投入した後、攪拌を行わない方法(浸漬のみ)や、プロペラ攪拌機の回転数を少なくする方法も試みたが、この場合は表面の被膜はほとんど除去することができなかった。つまり、特許文献1に記載された方法では、リチウム−遷移金属複合酸化物の性能を維持しつつ、表面に形成された被膜を効果的に除去することは不可能であった。
【0010】
そこで、本発明は、リチウム−遷移金属複合酸化物の性能低下を抑制しつつ、リチウム−遷移金属複合酸化物の表面に形成された被膜の少なくとも一部を効果的に除去する手段を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記の課題に鑑み鋭意研究を積み重ねた。その過程で、所定の酸解離定数(pKa)を有する酸を用いた表面処理により、上記課題が解決されうることを見出し、本発明を完成させるに至った。
【0012】
すなわち、本発明の一形態によれば、リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法が提供される。
【発明の効果】
【0013】
本発明によれば、酸の酸解離定数(pKa)を6.35未満とすることにより、リチウム−遷移金属複合酸化物の表面に形成された被膜を構成する化合物が酸と反応し溶解するため、被膜の少なくとも一部を除去することができる。また、酸解離定数(pKa)が4以上の酸を用いることにより、リチウム−遷移金属複合酸化物のリチウムイオンのプロトン置換や、リチウムイオンの脱離を防ぐことができるため、正極活物質としての性能低下を抑制することができる。
【図面の簡単な説明】
【0014】
【図1】実施例1および比較例1のEIS試験の結果を示すグラフである。
【図2】実施例1および比較例1の充放電容量試験の結果を示すグラフである。
【図3】実施例2および3ならびに比較例3の過電圧試験の結果を示すグラフである。
【図4】実施例4および比較例4の放電容量試験の結果を示すグラフである。
【図5】本発明の一実施形態に係る積層型のリチウムイオン二次電池の全体構造を模式的に表した概略図である。
【図6】被膜/LiNiO2の割合に対する放電容量維持率の関係を表すグラフである。
【発明を実施するための形態】
【0015】
<処理済み正極活物質の製造方法>
以下、本発明の好ましい形態を説明する。本発明の一形態は、リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法に関する。なお、以降の本明細書中では、「酸解離定数(pKa)が4以上、6.35未満の酸」を、「酸解離定数(pKa)が4以上、6.35未満の弱酸」または単に「弱酸」とも称する。
【0016】
まず、本形態の理解を容易にするために、本形態の処理工程によってリチウム−遷移金属複合酸化物の表面に形成される被膜が除去されるメカニズムについて、リチウム−ニッケル複合酸化物を例に挙げて説明する。ただし、本発明の技術的範囲は、あくまでも特許請求の範囲の記載に基づいて定められるべきであり、以下のメカニズムにより限定されるものではない。したがって、本発明の効果が以下メカニズム以外のメカニズムによって生じていたとしても、本発明の技術的範囲は限定的に解釈されるものではない。
【0017】
リチウム−ニッケル複合酸化物を通常の空気雰囲気に曝すと、表面に被膜が形成される。この被膜の形成は、下記化学式1に示される2段階の化学反応により起こると考えられていた。すなわち、まず空気中に含まれる水分により、表面のリチウム−ニッケル複合酸化物(例えば、LiNiO2)が加水分解されて水酸化リチウム(LiOH)が生じる。次に当該LiOHが空気中の二酸化炭素(CO2)と反応することによって炭酸リチウム(Li2CO3)が生じ、表面に被膜が形成される。つまり、これまでの技術常識によると、リチウム−ニッケル複合酸化物の表面に形成される被膜の構成成分は、Li2CO3であると考えられていたのである。
【0018】
【化1】
【0019】
しかしながら、今回、本発明者らが被膜の構成成分を解析するために熱分析(不活性ガス雰囲気下で25℃から500℃まで加熱)を行ったところ、被膜は200℃付近で熱分解し、それと同時にCO2とH2Oとを生じることが判明した。Li2CO3の融点は約723℃であるため、少なくとも200℃付近で熱分解することはない。また、Li2CO3がたとえ分解したとしてもH2Oが生じることはない。この事実より、被膜の構成成分は、従来考えられていたLi2CO3ではなく、準安定塩である炭酸水素リチウム(LiHCO3)であることが推測された。この場合、LiHCO3が形成される反応は、以下の化学式2のようであると考えられる。
【0020】
【化2】
【0021】
本発明者らは、上記知見を基に、リチウム−ニッケル複合酸化物の表面の被膜に、炭酸の酸解離定数(pKa1)6.35よりも小さいpKa値を有する酸を接触させたところ、被膜が容易に除去された。これにより被膜の構成成分がLiHCO3であるとの本発明者らの推測はより確からしいことが示された。さらに、酸として強酸を用いた場合は、リチウム−ニッケル複合酸化物の性能が低下してしまうこと;酸の酸解離定数(pKa)を4以上とすることにより、リチウム−ニッケル複合酸化物の性能低下を抑制できることも判明した。加えて、リチウム−ニッケル複合酸化物以外のリチウム−遷移金属複合酸化物においても同様の効果が得られることが確認された。これらの結果より、酸解離定数(pKa)が4以上、6.35未満である弱酸を用いることにより、被膜の除去および性能低下の抑制の両立を可能としたリチウム−遷移金属複合酸化物の処理方法を完成させたのである。以下、本形態の製造方法について詳細に説明する。
【0022】
本形態の製造方法の原料として用いられる正極活物質(以下、「未処理の正極活物質」とも称する)は、リチウム−遷移金属複合酸化物を必須に含む。
【0023】
本形態におけるリチウム−遷移金属複合酸化物は、リチウムイオンを吸蔵・放出可能な正極活物質として使用される材料であれば特に制限はなく、本技術分野で使用されうるものを当業者が適宜採用することができる。このようなリチウム−遷移金属複合酸化物の一例として、リチウム−ニッケル複合酸化物が挙げられる。なお、本明細書において、「リチウム−ニッケル複合酸化物」は、金属元素としてLiおよびNiのみを含む複合酸化物のみならず、Niの一部がCo、Mn、Alなどの元素で置換された複合酸化物をも含む。このようなリチウム−ニッケル複合酸化物としては、具体的には、LiNiO2、LiNi0.5Mn0.5O2、LiNi0.9Co0.1O2、LiNi0.8Co0.2O2、LiNi0.8Co0.18Mn0.02O2、LiNi1/3Co1/3Mn1/3O2、LiNi0.8Co0.15Al0.05O2、LiNi0.6Co0.1Mn0.25Al0.05O2などが例示される。
【0024】
リチウム−ニッケル複合酸化物以外にも、リチウム−遷移金属複合酸化物として、LiCoO2等のリチウム−コバルト複合酸化物、スピネルLiMn2O4等のリチウム−マンガン複合酸化物、LiFeO2等のリチウム−鉄複合酸化物およびこれらの遷移金属(Co、Mn、Fe)の一部を他の元素により置換したもの等が使用できる。
【0025】
これらのうち、本形態の処理工程では、リチウム−ニッケル複合酸化物からなる群から選択される少なくとも1種を含む正極活物質を用いることが好ましい。リチウム−ニッケル複合酸化物は、特に吸湿しやすいため、上述の被膜の問題が他のリチウム−遷移金属複合酸化物よりも顕著に現れる。したがって、本形態の処理工程を、リチウム−ニッケル複合酸化物を含む正極活物質に適用することによって、本発明の効果をより一層享受することができる。
【0026】
なお、これらのリチウム−遷移金属複合酸化物は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。また、リチウム−遷移金属複合酸化物の形状やサイズについても、特に制限はなく、当該分野で使用されうるあらゆる形状やサイズのリチウム−遷移金属複合酸化物に対して有効である。
【0027】
さらに、本形態の正極活物質は、リチウム−遷移金属複合酸化物以外の他の正極活物質材料を含んでも構わない。他の正極活物質材料として、LiFePO4等の遷移金属とリチウムのリン酸化合物や硫酸化合物;V2O5、MnO2、TiS2、MoS2、MoO3等の遷移金属酸化物や硫化物;PbO2、AgO、NiOOH等が使用可能である。これらの他の正極活物質材料も、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0028】
上記リチウム−遷移金属複合酸化物は、その表面に被膜が形成されているものでありうる。上述のように、本発明者らの知見によると、当該被膜は、構成成分としてLiHCO3を含んでいるものと考えられる。このような被膜は、リチウム−遷移金属複合酸化物を、空気中で保管した際に形成される。また、後述のように、リチウム−遷移金属複合酸化物に対する被膜量の程度が、電池性能や電池寿命に大きく影響する。
【0029】
弱酸は、リチウム−遷移金属複合酸化物の表面に形成される被膜を分解し、除去する役割を果たす。したがって、弱酸により被膜の少なくとも一部を除去することにより、リチウム−遷移金属複合酸化物に対する被膜量を制御し、電池性能や電池寿命を改善することが可能となる。上述のように、本発明者らの知見によると、被膜にはその構成成分としてLiHCO3が含まれると考えられる。LiHCO3は、理論上、炭酸の酸解離定数(pKa1)6.35よりも小さい酸と反応するため、本形態で使用される弱酸は、酸解離定数(pKa)が6.35未満であることが必須である。一方、LiHCO3は、酸解離定数(pKa)が6.35未満の酸であれば強酸であっても反応するが、酸の強度が大きくなりすぎると、リチウム−遷移金属複合酸化物の正極活物質としての性能が低下する虞がある。この性能の低下は、リチウム−遷移金属複合酸化物において、リチウムイオンのプロトン置換(例えば、リチウム−ニッケル複合酸化物においては、LiNiO2+H+→HNiO2+Li+)や、リチウムイオンの脱離が起こることが一因である。したがって、このような好ましくない反応を抑えるため、本形態においては、弱酸の酸解離定数(pKa)を4以上とすることが必須である。なお、本明細書において、「酸解離定数(pKa)」は水中、25℃における値であり、具体的には、赤岩英夫編、「分析化学実験」、丸善、1999年発行に記載の方法により測定される。
【0030】
本形態において、弱酸は、酸解離定数(pKa)が4以上、6.35未満であれば、特に制限はなく、無機酸および有機酸のいずれも使用可能である。また、酸の物質状態も特に制限はなく、常温常圧(25℃、1気圧)下において、固体、液体、および気体のいずれであってもよい。このような弱酸としては、例えば、アジ化水素(pKa=4.65)、アクリル酸(pKa=4.26)、アジピン酸(pKa1=4.26、pKa2=5.03)、アゼライン酸(pKa1=4.39、pKa2=5.12)、o−アニス酸(pKa=4.09)、m−アニス酸(pKa=4.09)、p−アニス酸(pKa=4.48)、アニリン(pKa=4.65)、安息香酸(pKa=4.20)、イソ吉草酸(pKa=4.58)、イソ酪酸(pKa=4.63)、オクタン酸(pKa=4.89)、吉草酸(pKa=4.64)、キナルジン酸(pKa=4.75)、グルタル酸(pKa1=4.13、pKa2=5.03)、2−ナフトエ酸(pKa=4.16)、ビニル酢酸(pKa=4.12)、ピメリン酸(pKa1=4.31、pKa2=5.08)、フェニル酢酸(pKa=4.10)、p−フルオロ安息香酸(pKa=4.14)、p−ブロモ安息香酸(pKa=4.00)、ヘキサン酸(pKa=4.63)、ヘプタン酸(pKa=4.66)、p−ヨード安息香酸(pKa=4.00)、酪酸(pKa=4.63)、レブリン酸(pKa=4.44)などが挙げられる。これらの酸は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0031】
上記弱酸が常温常圧(25℃、1気圧)下において固体である場合、弱酸を適当な溶媒に溶解した弱酸溶液をリチウム−遷移金属複合酸化物と接触させてもよい。この際に使用される溶媒は特に制限はなく、例えば、水、メタノール、エタノール、n−プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、エチレングリコール、プロピレングリコールなどのプロトン性の極性溶媒;ジエチルエーテル、酢酸エチル、ギ酸メチル、プロピオン酸メチル、テトラヒドロフラン、ジメチルスルホキシド、アセトニトリル、シクロヘキサノン、アセトン、メチルエチルケトン、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、メチルエチルカーボネート、1,2−ジメトキシエタン、1,2−ジエトキシエタン、1,4−ジオキサン、1,3−ジオキソラン、エチレングリコールジメチルエーテル、γ−ブチロラクトン、γ−バレロラクトンなどの非プロトン性の極性溶媒などを用いることができる。これらの溶媒は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。なお、弱酸溶液中の弱酸の濃度は、弱酸の酸強度等に応じて当業者が適宜設定することができる。
【0032】
未処理の正極活物質と弱酸(または弱酸溶液)とを接触させる処理工程において、弱酸(または弱酸溶液)に含まれる水分は少ないほど好ましい。具体的には、弱酸(または弱酸溶液)の含水率は、50体積ppm以下であることが好ましく、30体積ppm以下であることがより好ましい。一方、下限値は特に制限はないが、通常0.01体積ppm程度である。このように、弱酸(または弱酸溶液)に含まれる水分を少なくすることにより、未処理の正極活物質中に含まれるリチウム−遷移金属複合酸化物が水と接触して加水分解され、LiOHが生成するのを防ぐことができる。これにより、リチウム−遷移金属複合酸化物の正極活物質としての性能低下をより一層防ぐことができる。なお、ここで述べた、弱酸(または弱酸溶液)に含まれる水分を少なくするという形態は、あくまでも発明の好ましい形態に過ぎず、上述の弱酸溶液に使用される溶媒としての水の使用を妨げるものではない。
【0033】
本形態で使用される好ましい弱酸として、酢酸が挙げられる。酢酸は、常温常圧(25℃、1気圧)下において液体であり、そのまま未処理の正極活物質との接触に用いることができる点で本形態に好適である。また、酢酸は、含水率の低いもの(氷酢酸)が比較的容易に入手可能な点でも有利である。
【0034】
本形態において、未処理の正極活物質と弱酸とを接触させる方法は、特に制限はない。一例を挙げると、正極活物質を弱酸(または弱酸溶液)中に浸漬させる方法;正極活物質の表面に弱酸(または弱酸溶液)を塗布または噴霧する方法などが挙げられる。このうち、十分に正極活物質と弱酸を接触させることができるという観点から、正極活物質を弱酸(または弱酸溶液)中に浸漬させる方法であることが好ましい。なお、浸漬の際には、正極活物質の性能を著しく低下させない限りにおいて、正極活物質が入った弱酸(または弱酸溶液)を攪拌しても構わない。また、正極活物質と弱酸とを接触させる際の、弱酸(または弱酸溶液)の温度も特に制限はないが、好ましくは20〜40℃である。
【0035】
本形態において、正極活物質を弱酸に接触させる際の接触時間は特に制限はない。しかしながら、被膜が完全に除去されたリチウム−遷移金属複合酸化物を弱酸に接触させておくと、リチウムイオンのプロトン置換や、リチウムイオンの脱離によって正極活物質としての性能が低下する虞があることから、接触時間を必要以上に長くしないことが好ましい。接触時間は、処理するリチウム−遷移金属複合酸化物の量や、被膜の厚さ、使用する弱酸、接触させる方法によるため、一概に規定することは困難である。よって、本形態では、正極活物質を、弱酸に接触させる工程の終点を、被膜と弱酸との反応により発生するCO2の増加率によって決定することが好ましい。本明細書において、「CO2増加率」は、下記数式1により定義される。
【0036】
【数1】
【0037】
具体的には、正極活物質を弱酸に接触させる際に、単位時間(例えば1時間)当たりに発生するCO2量を随時モニタリングする。発生するCO2量は、反応する被膜量に相当するため、通常、接触開始直後が最大となり、接触時間が長くなるにつれ低下する。すなわち、接触開始直後は、CO2増加率は1であるが、次第にその値は小さくなり、最終的には0となる。よって、予め設定した所定の数値範囲内にCO2増加率が到達した時点で接触を終了させることにより、様々な条件下においても、接触工程の終点を容易に判断することができるのである。より具体的には、上記数値範囲が、0.05〜0.1であることが好ましい。下限値を0.05とすることにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離による正極活物質としての性能低下をより一層防ぐことができる。一方、上限値を0.1とすることにより、リチウム−遷移金属複合酸化物の表面に残存する被膜の量を低減させることができる。
【0038】
本形態では、正極活物質を弱酸に接触させる工程の後、正極活物質の表面に付着した弱酸を取り除くために、洗浄用の溶媒等で洗浄する工程をさらに含んでもよい。使用される洗浄用溶媒は、特に制限はなく、例えば、上述の弱酸溶液に使用される溶媒と同様のものを適宜しようすることができる。また、当該洗浄工程の後、正極活物質の表面に付着した洗浄用溶媒を、真空乾燥機などを用いて乾燥させる、乾燥工程をさらに設けてもよい。以上の工程によって、リチウム−遷移金属複合酸化物の表面の被膜の少なくとも一部が取り除かれた、処理済み正極活物質が得られる。
【0039】
本形態の処理工程によると、リチウム−遷移金属複合酸化物の表面の被膜の少なくとも一部が弱酸によって除去される。当該被膜は、上述のように、LiHCO3を主成分としていると考えられる。したがって、リチウム−遷移金属複合酸化物と弱酸との反応により、リチウムと弱酸との塩が生じる。例えば、弱酸として酢酸を使用した場合には、反応の結果、酢酸リチウム(CH3COOLi)が生じる(下記化学式3参照)。
【0040】
【化3】
【0041】
反応により生じたCH3COOLi等のリチウム塩は、洗浄工程を行う場合などは、その大部分が表面から除去される。しかしながら、その一部は、リチウム−遷移金属複合酸化物の表面に物理的に付着したままとなりうる。リチウム−遷移金属複合酸化物表面に付着したCH3COOLi等のリチウム塩は、リチウム−遷移金属複合酸化物の表面をX線光電子分光(XPS)分析することにより検出されうる。よって、リチウム−遷移金属複合酸化物の表面にCH3COOLi等のリチウム塩が存在するか否かによって、本形態の処理工程が行われたか否かを判定することが可能である。換言すると、本形態の処理工程を経た正極活物質は、リチウム−遷移金属複合酸化物と、弱酸のリチウム塩とを含む。
【0042】
<処理済み電極の製造方法>
以上、本形態の処理済み正極活物質の製造方法について説明したが、本発明は、リチウム−遷移金属複合酸化物を含む電極に対しても適用することが可能である。すなわち、本発明の他の一形態によると、集電体と、集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を有する正極活物質層と、を含む電極の少なくとも正極活物質層を、酸解離定数(pKa)が4以上、6.35未満の弱酸に接触させる処理工程を含む、処理済み電極の製造方法が提供される。
【0043】
上述のように、リチウム−遷移金属複合酸化物は、通常の空気雰囲気に曝されただけでも、空気中に含まれるH2OやCO2によって反応し、表面に被膜が形成されうる。従来、被膜が形成された状態のリチウム−遷移金属複合酸化物を正極活物質として用いて電池を作製すると、電池の充放電時に被膜が分解されてCO2ガスを発生させ、電池内圧の上昇による電池特性の低下や電池の膨脹を引き起こす、といった問題が生じていた。したがって、これまでは被膜の形成を防ぐために、電極の製造工程をドライボックス中で行う、などといった対応が必要であった。しかしながら、車両用などの大型の電池をドライボックス中で製造することは、量産化の足枷となるばかりか、設備コストが膨大となるため結果的に電池コストを抑えられず、電気自動車等の普及の足枷にもなりかねない。そこで、特に電池に組み込む前の電極に対して適用できるような処理方法が切に求められていた。本形態の処理工程は、このような要請にも応えるものであり、電極の性能を低下させずに被膜の少なくとも一部を除去することを可能とするものである。また、本形態の処理工程を行った後の処理済み電極を用いて電池の組み立てを行えば、電池内部に持ち込まれる被膜量を抑えることができるため、充放電反応時に被膜が分解されることによるガスの発生を抑制することができる。以下、本形態の製造方法について説明する。
【0044】
本形態において、電極は、集電体の一方の面にリチウム−遷移金属複合酸化物を含む正極活物質層を有する限りにおいては、特に制限はない。例えば、集電体の一方の面にのみ、リチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなる正極;集電体の両方の面にリチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなる正極;集電体の一方の面にリチウム−遷移金属複合酸化物を含む正極活物質層が形成されてなり、集電体の他方の面に負極活物質を含む負極活物質層が形成されてなる双極型電極;等の形態が挙げられるが、本形態の処理工程は、これらのいずれの電極に対しても適用可能である。以下、本形態の電極について説明する。
【0045】
(集電体)
本形態において、集電体を構成する材料に特に制限はない。例えば、金属や、導電性高分子材料または非導電性高分子材料に導電性フィラーが添加された樹脂が採用されうる。具体的には、金属としては、アルミニウム、ニッケル、鉄、ステンレス鋼、チタン、銅等が挙げられる。これらのほか、ニッケルとアルミニウムとのクラッド材、銅とアルミニウムとのクラッド材、あるいはこれらの金属を組み合わせためっき材等が好ましく用いられうる。また、金属表面にアルミニウムが被覆されてなる箔であってもよい。なかでも、電子伝導性や電池作動電位の観点からは、アルミニウム、ステンレス鋼、銅が好ましい。
【0046】
また、導電性高分子材料としては、例えば、ポリアニリン、ポリピロール、ポリチオフェン、ポリアセチレン、ポリパラフェニレン、ポリフェニレンビニレン、ポリアクリロニトリル、およびポリオキサジアゾール等が挙げられる。かような導電性高分子材料は、導電性フィラーを添加しなくても十分な導電性を有するため、製造工程の容易化または集電体の軽量化の点において有利である。
【0047】
非導電性高分子材料としては、例えば、ポリエチレン(PE;高密度ポリエチレン(HDPE)、低密度ポリエチレン(LDPE))、ポリプロピレン(PP)、ポリエチレンテレフタレート(PET)、ポリエーテルニトリル(PEN)、ポリイミド(PI)、ポリアミドイミド(PAI)、ポリアミド(PA)、ポリテトラフルオロエチレン(PTFE)、スチレン−ブタジエンゴム(SBR)、ポリアクリロニトリル(PAN)、ポリメチルアクリレート(PMA)、ポリメチルメタクリレート(PMMA)、ポリ塩化ビニル(PVC)、ポリフッ化ビニリデン(PVdF)、およびポリスチレン(PS)等が挙げられる。かような非導電性高分子材料は、優れた耐電位性または耐溶媒性を有しうる。
【0048】
上記の導電性高分子材料または非導電性高分子材料には、必要に応じて導電性フィラーが添加されうる。特に、集電体の基材となる樹脂が非導電性高分子のみからなる場合は、樹脂に導電性を付与するために必然的に導電性フィラーが必須となる。導電性フィラーは、導電性を有する物質であれば特に制限なく用いることができる。例えば、導電性、耐電位性、またはリチウムイオン遮断性に優れた材料として、金属および導電性カーボン等が挙げられる。金属としては、特に制限はないが、Ni、Ti、Al、Cu、Pt、Fe、Cr、Sn、Zn、In、Sb、およびKからなる群から選択される少なくとも1種の金属もしくはこれらの金属を含む合金または金属酸化物を含むことが好ましい。また、導電性カーボンとしては、特に制限はないが、アセチレンブラック、バルカン、ブラックパール、カーボンナノファイバー、ケッチェンブラック、カーボンナノチューブ、カーボンナノホーン、カーボンナノバルーン、およびフラーレンからなる群から選択される少なくとも1種を含むことが好ましい。導電性フィラーの添加量は、集電体に十分な導電性を付与できる量であれば特に制限はなく、一般的には、5〜35質量%程度である。
【0049】
集電体の大きさは、電池の使用用途に応じて決定される。例えば、高エネルギー密度が要求される大型の電池に用いられるのであれば、面積の大きな集電体が用いられる。集電体の厚さについても特に制限はないが、通常は1〜100μm程度である。
【0050】
(正極活物質層)
正極活物質層は正極活物質として、リチウム−遷移金属複合酸化物を必須に含み、必要に応じて、リチウム−遷移金属複合酸化物以外の他の正極活物質材料を含みうる。リチウム−遷移金属複合酸化物や、他の正極活物質材料の詳細については、上述の処理済み正極活物質の製造方法において述べた内容と同様であるので、ここでは詳細な説明を省略する。
【0051】
(負極活物質層)
本形態の電極は、負極活物質層を含みうる。負極活物質層に含まれる負極活物質は、リチウムを可逆的に吸蔵および放出できるものであれば特に制限されない。負極活物質の例としては、SiやSn等の金属、あるいはTiO、Ti2O3、TiO2、もしくはSiO2、SiO、SnO2等の金属酸化物、Li4/3Ti5/3O4もしくはLi7MnN等のリチウムと遷移金属との複合酸化物、Li−Pb系合金、Li−Al系合金、Li、または天然黒鉛、人造黒鉛、カーボンブラック、活性炭、カーボンファイバー、コークス、ソフトカーボン、もしくはハードカーボン等の炭素材料等が好ましく挙げられる。また、負極活物質は、リチウムと合金化する元素を含むことが好ましい。リチウムと合金化する元素を用いることにより、従来の炭素系材料に比べて高いエネルギー密度を有する高容量および優れた出力特性の電池を得ることが可能となる。上記負極活物質は、1種のみが単独で使用されてもよいし、2種以上が組み合わされて使用されてもよい。
【0052】
上記のリチウムと合金化する元素としては、以下に制限されることはないが、具体的には、Si、Ge、Sn、Pb、Al、In、Zn、H、Ca、Sr、Ba、Ru、Rh、Ir、Pd、Pt、Ag、Au、Cd、Hg、Ga、Tl、C、N、Sb、Bi、O、S、Se、Te、Cl等が挙げられる。これらの中でも、容量およびエネルギー密度に優れた電池を構成できる観点から、炭素材料、ならびに/またはSi、Ge、Sn、Pb、Al、In、およびZnからなる群より選択される少なくとも1種以上の元素を含むことが好ましく、炭素材料、Si、またはSnの元素を含むことが特に好ましい。これらは1種単独で使用しても良いし、2種以上を併用してもよい。なお、負極活物質の形状やサイズは、特に制限はなく、当業者により適宜設定されうる。
【0053】
正極活物質層または負極活物質層(以下、単に「活物質層」とも称する)には、必要であれば、その他の物質が含まれてもよい。例えば、導電助剤、バインダ等が含まれうる。また、イオン伝導性ポリマーが含まれる場合には、前記ポリマーを重合させるための重合開始剤が含まれてもよい。
【0054】
導電助剤とは、活物質層の導電性を向上させるために配合される添加物をいう。導電助剤としては、アセチレンブラック、カーボンブラック、ケッチェンブラック、グラファイト等のカーボン粉末や、気相成長炭素繊維(VGCF;登録商標)等の種々の炭素繊維、膨張黒鉛等が挙げられる。しかし、導電助剤がこれらに限定されないことはいうまでもない。
【0055】
バインダとしては、ポリフッ化ビニリデン(PVdF)、ポリイミド、PTFE、SBR、合成ゴム系バインダ等が挙げられる。しかし、バインダがこれらに限定されないことはいうまでもない。また、バインダとゲル電解質として用いるマトリックスポリマーとが同じ場合には、バインダを使用する必要はない。
【0056】
活物質層に含まれる成分の配合比および活物質層の厚さについては特に限定されず、リチウムイオン二次電池用の電極についての公知の知見を参照し、当業者が適宜設定することができる。
【0057】
本形態の製造方法に用いられる電極(以下、「未処理の電極」)は、特に制限されず、従来公知の知見を適宜参照することにより製造されうる。例えば、活物質、バインダ、および粘度調整用溶媒を含む活物質スラリーを調製し、当該活物質スラリーを集電体上に塗布し、乾燥させた後、プレスすることで未処理の電極が作製されうる。
【0058】
上記未処理の電極に含まれるリチウム−遷移金属複合酸化物は、その表面に被膜が形成されているものでありうる。上述のように、本発明者らの知見によると、当該被膜は、構成成分としてLiHCO3を含んでいるものと考えられる。このような被膜は、リチウム−遷移金属複合酸化物や電極を、空気中で保管した際に形成されうる。
【0059】
本形態の製造方法は、上述の未処理の電極の少なくとも正極活物質層を酸解離定数(pKa)が4以上、6.35未満の弱酸に接触させる処理工程を含む点に特徴を有する。なお、弱酸との接触は、電極の正極活物質層のみならず、集電体や負極活物質層等を含む電極全体に対して行ってもよい。使用される弱酸や、接触方法、および処理工程の終点の決定方法等の諸条件は、上述の処理済み正極活物質の製造方法と同様であるので、ここでは詳細な説明を省略する。
【0060】
本形態の処理工程によると、電極の少なくとも正極活物質層を酸解離定数(pKa)6.35未満の弱酸に接触させることにより、電極の正極活物質中に含まれるリチウム−遷移金属複合酸化物の表面に形成される被膜の少なくとも一部を除去することが可能である。また、弱酸の酸解離定数(pKa)を4以上とすることにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離によるリチウム−遷移金属複合酸化物の正極活物質としての性能の低下を抑制することができる。また、弱酸の酸解離定数(pKa)を4以上とすることにより、集電体や活物質層中に含まれる他の部材との反応を抑えることができるため、電極の性能の低下を抑制することができる。さらに、本形態の処理工程を行った後の処理済み電極を用いて電池の組み立てを行うことにより、電池内部に持ち込まれる被膜量を抑えることができるので、充放電反応時に被膜が分解されることによるガスの発生を抑制することも可能である。
【0061】
<電極>
以上、処理済み電極の製造方法について説明してきたが、当該製造方法によると、電極における正極活物質層中に含まれるリチウム−遷移金属複合酸化物の表面に存在する被膜の量を制御することができる。本発明者らの検討によると、後述の実施例で示すように、上述の処理工程により被膜の少なくとも一部を除去した場合、全く除去しない場合と比較して、電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されることが分かった。さらに、被膜を完全に除去した場合よりも、被膜を所定量残すように処理工程を行う場合の方が、電池のサイクル特性(放電容量維持率)の点で特に優れることが分かった。
【0062】
したがって、本発明の他の一形態によると、上述の処理済み電極の製造方法により得られうる電極が提供される。本形態の電極は、リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、当該被膜の量は、リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%であることが好ましい。
【0063】
また、本発明の他の一形態によると、集電体と、集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層とを含む電極であって、リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、当該被膜の量は、リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である電極が提供される。
【0064】
上記被膜は、リチウム−遷移金属複合酸化物の表面に存在する、リチウム−遷移金属複合酸化物以外のリチウム塩から構成される。本発明者らの知見によると、当該被膜は、上述で説明したように、LiHCO3を含みうる。しかしながら、被膜の全ての構成成分がLiHCO3であるとは限らず、被膜の形成条件等によりLi2CO3や、弱酸処理により生じる弱酸のリチウム塩等が含まれる場合もある。
【0065】
本形態では、上記被膜の量はリチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%であることが好ましく、より好ましくは0.15〜0.29質量%であり、さらに好ましくは0.2〜0.26質量%であり、特に好ましくは0.20〜0.23質量%である。被膜量を上記範囲内とすることにより、後述の実施例で示すように、サイクル特性(放電容量維持率)を有意に向上させることができる。
【0066】
電極における被膜量の測定方法は特に制限はないが、空気中で電極を作製する際は、経時的に被膜量が増加するため、直接的に被膜量を測定することが難しい。この場合は、後述の実施例で示すように、所定の温度・湿度下に曝した際の時間に対する被膜量についての検量線を予め作成する。そして、内挿によりある時間(電極を作製するのに掛かる時間)における被膜量を求める。その後、(被膜量)÷(被膜を有しないリチウム−遷移金属複合酸化物量)×100を算出することにより、被膜のリチウム−遷移金属複合酸化物に対する質量割合[質量%]を算出することが可能である。
【0067】
なお、本形態の電極に含まれる、集電体、活物質(正極活物質、負極活物質)、および、任意に含まれる導電助剤、バインダ等は、上述の処理済み電極の製造方法において述べた内容と同様であるので、ここでは詳細な説明を省略する。
【0068】
本形態の電極において電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されるメカニズムは定かではないが、本発明者らは以下のように推測している。すなわち、サイクル特性に関しては、リチウム−遷移金属複合酸化物の表面の所定量の被膜が、リチウム−遷移金属複合酸化物の結晶構造の変化(例えば、充電時の高電位環境や、電解液との接点増加に起因する結晶構造の変化)を防ぐ働きをしていると考えている。その結果、リチウム−遷移金属複合酸化物の耐久性が向上し、サイクル特性(放電容量維持率)が向上するものと推測される。一方、被膜量が0.31質量%を超えると、電池の充放電時に被膜が分解されてCO2ガスが発生することに起因する電池特性の低下の影響が大きくなるため、サイクル特性が低下すると考えられる。また、放電容量に関しては、リチウム−遷移金属複合酸化物へのリチウムイオンの吸蔵・放出が被膜により妨げられていることが原因であると考えられる。また、インピーダンス抵抗に関しては、被膜が抵抗増大の要因となっていると考えられる。
【0069】
<電気デバイス>
上述の本形態の電極は、サイクル特性に優れるため、様々な電気デバイスに好適に使用されうる。すなわち、本発明の一形態によると上記電極を含む電気デバイスが提供される。電気デバイスは、上述の電極を要するものであれば特に制限はなく、リチウムイオン二次電池などの二次電池および電気二重層キャパシタなどが挙げられる。以下、電気デバイスの一例として、本形態の電極を具備したリチウムイオン二次電池について説明する。
【0070】
図5に、本発明の一実施形態に係る積層型のリチウムイオン二次電池の全体構造を模式的に表した概略図を示す。本実施形態のリチウムイオン二次電池10は、実際に充放電反応が進行する略矩形の発電要素17が、電池外装材であるラミネートフィルム22の内部に封止された構造を有する。詳しくは、高分子−金属複合ラミネートフィルムを電池外装材として用いて、その周辺部の全部を熱融着にて接合することにより、発電要素17を収納し密封した構成を有している。
【0071】
発電要素17は、負極集電体11の両面(発電要素の最下層用および最上層用は片面のみ)に負極活物質層12が配置された負極と、電解質層13と、正極集電体14の両面に正極活物質層15が配置された正極とを積層した構成を有している。具体的には、1つの負極活物質層12とこれに隣接する正極活物質層15とが、電解質層13を介して対向するようにして、負極、電解質層13、正極がこの順に積層されている。
【0072】
これにより、隣接する負極、電解質層13および正極は、1つの単電池層16を構成する。したがって、本実施形態のリチウムイオン二次電池10は、単電池層16が複数積層されることで、電気的に並列接続されてなる構成を有するともいえる。また、単電池層16の外周には、隣接する負極集電体11と正極集電体14との間を絶縁するためのシール部(絶縁層)(図示せず)が設けられていてもよい。発電要素17の両最外層に位置する最外層負極集電体11aには、いずれも片面のみに負極活物質層12が配置されている。なお、負極および正極の配置を逆にすることで、発電要素17の両最外層に最外層正極集電体が位置するようにし、該最外層正極集電体の片面のみに正極活物質層が配置されているようにしてもよい。
【0073】
負極集電体11および正極集電体14には、各電極(負極および正極)と導通される負極集電板(負極タブ)18および正極集電板(正極タブ)19がそれぞれ取り付けられ、ラミネートフィルム22の端部に挟まれるようにラミネートフィルム22の外部に導出される構造を有している。負極集電板(負極タブ)18および正極集電板(正極タブ)19は、必要に応じて負極端子リード20および正極端子リード21を介して、各電極の負極集電体11および正極集電体14に超音波溶接や抵抗溶接などにより取り付けられていてもよい。ただし、負極集電体11が延長されて負極集電板(負極タブ)18とされ、ラミネートフィルム22から導出されていてもよい。同様に、正極集電体14が延長されて正極集電板(正極タブ)19とされ、同様に電池外装材22から導出される構造としてもよい。
【0074】
本実施形態のリチウムイオン二次電池10では、正極として上述の本形態の電極が使用される。これにより、リチウムイオン二次電池の諸特性(サイクル特性、放電容量、インピーダンス抵抗)が改善されうる。
【実施例】
【0075】
以下、本発明の作用効果を実施例および比較例を用いて説明する。ただし、本発明の技術的効果は以下の実施例に制限されるものではない。
【0076】
[実施例1]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0077】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0078】
(3)弱酸処理
保管後の正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1週間浸漬させ、その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0079】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0080】
[参考例1]
上記(2)正極の保管、および上記(3)弱酸処理を行わなかった(つまり被膜が形成されていない)ことを除いては、実施例1と同様の方法で、評価用コインセル(ブランク)を作製した。
【0081】
[比較例1]
上記(3)弱酸処理を行わなかったことを除いては、実施例1と同様の方法で、評価用コインセルを作製した。
【0082】
<EIS(電気化学インピーダンススペクトル法)試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流定電圧方式(CCCV、電流:1C)で4.3Vまで充電し、5分間休止させた。その後、印加電圧10mV、周波数100kHz〜1MHzにおいてインピーダンスを測定した。インピーダンス測定の結果をコール−コールプロットしたものを図1に示す。
【0083】
図1の結果より、温度30℃、相対湿度60%中で保管し、弱酸処理を行わなかった比較例1は、参考例1と比較してインピーダンスの上昇が確認された(図1において、横軸上のレジスタンスが約0.7[Ω]から約0.9[Ω]に上昇)。これは保管中に、リチウム−ニッケル複合酸化物の表面に被膜が形成されたことによるものである。一方、保管後に、弱酸処理を行った実施例1では、インピーダンスが参考例1に近いところまで減少していることが示された。これは、本発明の弱酸処理により、被膜が除去されたことを示す。
【0084】
<充放電容量試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:1C)でセル電圧3.0Vまで放電させた。当該充放電の際の充放電容量[mAh]を図2に示す。
【0085】
図2の結果より、保管後に弱酸処理を行った実施例1、および保管後に弱酸処理を行わなかった比較例1のいずれにおいても、参考例1と比較して充放電容量の大幅な減少は見られなかった。これは、本発明の弱酸処理を行っても、充放電容量の低下が起こりにくいことを示す。
【0086】
[実施例2]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0087】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0088】
(3)弱酸処理およびCO2濃度測定
保管後の正極を、ガス採取口の付いた密閉容器に入れた酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1日間浸漬させた。次いで、ガス採取口から注射器で密閉容器内のガスを採取し、採取したガス内のCO2量をガスクロマトグラフィーで測定し、1日後の密閉容器内のCO2濃度を求めた。その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0089】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0090】
[実施例3]
「(3)弱酸処理およびCO2濃度測定」において、酢酸中に浸漬させた時間を1週間としたことを除いては、実施例2と同様の方法で評価用コインセルを作製した。
【0091】
[比較例2]
「(2)正極の保管」を行わなかったことを除いては、実施例2と同様の方法で「(1)電極の作製」および「(3)弱酸処理およびCO2濃度測定」を行った。
【0092】
[比較例3]
「(2)正極の保管」を行わなかったことを除いては、実施例3と同様の方法で評価用コインセルを作製した。
【0093】
<CO2濃度および過電圧の関係>
上記実施例2および3、ならびに比較例2および3で測定したCO2濃度の結果を表1にまとめる。
【0094】
【表1】
【0095】
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C、電圧:4.3V)で充電した。充電の際のセル電圧を図3に示す。
【0096】
表1より、比較例2および3よりも、実施例2および3の方が二酸化炭素濃度が著しく高いことが示された。このことから、当該二酸化炭素濃度の差は、被膜と酢酸との反応により発生する二酸化炭素量の差に起因していることがわかる。また、実施例2よりも実施例3の方が二酸化炭素濃度が高いが、これは、1日間の浸漬後も、なお未反応の被膜が残存していることを意味する。
【0097】
図3より、実施例2は、実施例3よりも過電圧(電流印加前の電圧と電流印加20秒後の電圧の差(ΔV))が大きいことが示された。これは、上述のように、1日間の浸漬後も、なおリチウム−ニッケル複合酸化物の表面に被膜が残存し、電極の抵抗が上昇したためであると考えられた。
【0098】
[実施例4]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:10μm)の一方の面側に塗布し乾燥させた。その後、プレス処理を行い正極活物質層を片面に有する正極を作製した。
【0099】
(2)正極の保管
得られた正極を、温度30℃、相対湿度60%の恒温恒湿槽中で3日間保管した。
【0100】
(3)弱酸処理
保管後の正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1週間浸漬させ、その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0101】
(4)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0102】
[比較例4]
「(3)弱酸処理」において、酢酸に代えて、フッ化水素(HF)溶液を用いたこと以外は、実施例4と同様の方法で評価用コインセルを作製した。なお、HFの酸解離定数(pKa)は3.17である。
【0103】
当該HF溶液は、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比1:1)に溶解した溶液(電解液)に、水を2000体積ppm混合することにより調製した。下記化学式4に示すように、電解液中のLiPF6は水と反応し、HFを生じる。
【0104】
【化4】
【0105】
<充放電容量試験>
上記で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:1C)でセル電圧3.0Vまで放電させた。当該放電の際の放電容量[mAh]を図4に示す。
【0106】
図4の結果より、HF溶液で処理した比較例4は、酢酸で処理した実施例4と比較して、放電容量の低下が確認された。これは、酸解離定数(pKa)が3.17のHFで処理することにより、リチウムイオンのプロトン置換や、リチウムイオンの脱離が起こったためであると考えられた。
【0107】
[実施例5]
(1)電極の作製
正極活物質としてLiNiO2(平均粒子径:5μm、被膜量0.22質量%)90質量%、導電助剤としてケッチェンブラック(平均粒子径:300nm)5質量%、バインダとしてポリフッ化ビニリデン(PVdF)5質量%、およびスラリー粘度調整溶媒であるN−メチル−2−ピロリドン(NMP)適量を混合して、正極活物質スラリーを作製した。得られた正極活物質スラリーを、集電体であるアルミニウム箔(厚さ:20μm)の一方の面側に塗布し乾燥させた(正極活物質層の目付量:14mg/cm2)。その後、プレス処理を行い、積層方向から見た投影面積が100cm2(10cm×10cmの正方形)となるように切り出し、正極活物質層を片面に有する正極を作製した。以上の操作は、30℃、相対湿度(RH)50%の雰囲気下で行った。また、当該電極中に含まれるLiNiO2は1260mgであり、被膜は4.16mgであった。
【0108】
なお、当該被膜量は、予め作成した検量線に基づく内挿により算出した。検量線は、30℃、相対湿度(RH)50%の雰囲気下での、下記表2に示す各時間におけるLiNiO2量に対する被膜量の割合(質量%)をプロットすることにより作成した。
【0109】
【表2】
【0110】
(2)弱酸処理および被膜量測定
得られた正極を、酢酸(酸解離定数(pKa)4.76、含水率30体積ppm)中に1分間浸漬させ、次いで、温度80℃の真空乾燥機で1分間乾燥させた。弱酸処理前後の電極の質量を測定し、下記数式2に基づき弱酸処理後の被膜量を算出したところ、3.56[mg]であった。その後、ジメチルカーボネート(DMC)で洗浄した。洗浄後の正極を、温度120℃の真空乾燥機中で10時間乾燥させた。
【0111】
【数2】
【0112】
(3)評価用コインセルの作製
アルゴン雰囲気下のグローブボックス内で、上記正極を直径14mmの円盤形状に打ち抜き、コインセル用の正極とした。負極として、金属リチウムを直径15mmの円盤形状に打ち抜いたものを用いた。また、電解液として、1.0M LiPF6をエチレンカーボネート(EC)とジメチルカーボネート(DMC)との混合溶媒(体積比3:7)に溶解した溶液を準備した。正極と負極とを、セパレータ(材質:ポリプロピレン、厚さ:25μm)を介して積層し、コインセル容器内に入れ、電解液を注入し、上蓋をすることにより評価用コインセルを作製した。
【0113】
[比較例5]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させなかったことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0114】
[実施例6]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を3分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0115】
[実施例7]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を5分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0116】
[実施例8]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を10分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0117】
[実施例9]
「(2)弱酸処理および質量測定」において、酢酸中に浸漬させた時間を15分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0118】
[実施例10]
上記(2)弱酸処理および質量測定において、酢酸中に浸漬させた時間を20分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0119】
[実施例11]
上記(2)弱酸処理および質量測定において、酢酸中に浸漬させた時間を60分間としたことを除いては、実施例5と同様の方法で評価用コインセルを作製した。
【0120】
<EIS(電気化学インピーダンススペクトル法)試験>
上記実施例5〜8および比較例5で作製した評価用コインセルを、25℃の雰囲気下、定電流―定電圧方式(CCCV、電流:0.1C)で電圧4.3Vまで充電し、5分間休止させた。その後、印加電圧10mV、周波数100kHz〜1MHzにおいてインピーダンスを測定し、コール−コールプロットを求めた。次に、コール−コールプロットから得たX軸との2交点である半円部分をインピーダンス抵抗として算出した。結果を表3に示す。
【0121】
<充放電容量試験>
上記実施例5〜11および比較例5で作製した各評価用コインセルを、25℃の雰囲気下、定電流―定電圧方式(CCCV、電流:0.1C)で電圧4.3Vまで充電し、その後、電流が50マイクロAになるまで4.3Vで保持した。その後、定電流(CC、電流:0.1C)でセル電圧3.0Vまで放電させ、コインセル容量を測定した。結果を表3に示す。
【0122】
<充放電サイクル試験>
上記実施例5〜11および比較例5で作製した各評価用コインセルを、25℃の雰囲気下、定電流方式(CC、電流:0.1C)で電圧4.3Vまで充電し、5分間休止させた。その後、定電流(CC、電流:0.1C)でセル電圧3.0Vまで放電し、5分間休止させた。この繰返しを1サイクルとし、300サイクル実施した。300サイクル後の放電容量と1サイクル目の放電容量からの維持率を求めた。結果を表3および図6に示す。
【0123】
【表3】
【0124】
表3および図6の結果より、被膜量がLiNiO2量に対して0.036〜0.31質量%である実施例5〜11は、比較例5と比較して、優れたサイクル特性を示すことが示された。さらに、図6より、LiNiO2に対する被膜の割合が0.15〜0.29質量%、0.2〜0.26質量%、0.20〜0.23質量%の順で、サイクル特性はより高くなることが分かった。以上の結果より、LiNiO2の表面の所定量の被膜が、LiNiO2の結晶構造の変化を防ぐ働きをし、その結果、リチウム−遷移金属複合酸化物の耐久性が向上し、サイクル特性が向上するものと推測された。
【0125】
また、表3より、LiNiO2の表面の被膜量が減少するにつれ、初期の放電容量が増加する傾向にあることが示された。これにより、LiNiO2へのチウムイオンの吸蔵・放出が被膜により妨げられていることが示唆された。
【0126】
また、表3より、LiNiO2の表面の被膜量が減少するにつれ、インピーダンス抵抗が減少する傾向にあることが示された。これにより、被膜の存在により抵抗が増大することが示唆された。
【特許請求の範囲】
【請求項1】
リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法。
【請求項2】
前記処理工程において、前記正極活物質と前記酸との接触を、下記数式1で表されるCO2増加率が所定の数値範囲内に到達した時点で終了させる、請求項1に記載の処理済み正極活物質の製造方法。
【数1】
【請求項3】
前記数値範囲は、0.05〜0.1である、請求項2に記載の処理済み正極活物質の製造方法。
【請求項4】
集電体と、
集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層と、
を有する電極の少なくとも正極活物質層を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み電極の製造方法。
【請求項5】
前記処理工程において、前記正極活物質層と前記酸との接触を、下記数式1で表されるCO2増加率が所定の数値範囲内に到達した時点で終了させる、請求項4に記載の処理済み電極の製造方法。
【数2】
【請求項6】
前記数値範囲は、0.05〜0.1である、請求項5に記載の処理済み電極の製造方法。
【請求項7】
請求項4〜6のいずれか1項に記載の製造方法により得られうる、電極。
【請求項8】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である、請求項7に記載の電極。
【請求項9】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.15〜0.29質量%である、請求項8に記載の電極。
【請求項10】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.2〜0.26質量%である、請求項9に記載の電極。
【請求項11】
前記被膜は、LiHCO3を含む、請求項8〜10のいずれか1項に記載の電極。
【請求項12】
請求項7〜11のいずれか1項に記載の電極を有する、電池デバイス。
【請求項13】
集電体と、
集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層と、
を含む電極であって、
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である電極。
【請求項1】
リチウム−遷移金属複合酸化物を含む正極活物質を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み正極活物質の製造方法。
【請求項2】
前記処理工程において、前記正極活物質と前記酸との接触を、下記数式1で表されるCO2増加率が所定の数値範囲内に到達した時点で終了させる、請求項1に記載の処理済み正極活物質の製造方法。
【数1】
【請求項3】
前記数値範囲は、0.05〜0.1である、請求項2に記載の処理済み正極活物質の製造方法。
【請求項4】
集電体と、
集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層と、
を有する電極の少なくとも正極活物質層を、酸解離定数(pKa)が4以上、6.35未満の酸に接触させる処理工程を含む、処理済み電極の製造方法。
【請求項5】
前記処理工程において、前記正極活物質層と前記酸との接触を、下記数式1で表されるCO2増加率が所定の数値範囲内に到達した時点で終了させる、請求項4に記載の処理済み電極の製造方法。
【数2】
【請求項6】
前記数値範囲は、0.05〜0.1である、請求項5に記載の処理済み電極の製造方法。
【請求項7】
請求項4〜6のいずれか1項に記載の製造方法により得られうる、電極。
【請求項8】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である、請求項7に記載の電極。
【請求項9】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.15〜0.29質量%である、請求項8に記載の電極。
【請求項10】
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.2〜0.26質量%である、請求項9に記載の電極。
【請求項11】
前記被膜は、LiHCO3を含む、請求項8〜10のいずれか1項に記載の電極。
【請求項12】
請求項7〜11のいずれか1項に記載の電極を有する、電池デバイス。
【請求項13】
集電体と、
集電体の一方の面に形成された、リチウム−遷移金属複合酸化物を含む正極活物質層と、
を含む電極であって、
前記リチウム−遷移金属複合酸化物の表面の少なくとも一部に被膜が存在し、
前記被膜の量は、前記リチウム−遷移金属複合酸化物の量100質量%に対して、0.036〜0.31質量%である電極。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−12461(P2013−12461A)
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−72061(P2012−72061)
【出願日】平成24年3月27日(2012.3.27)
【出願人】(000003997)日産自動車株式会社 (16,386)
【Fターム(参考)】
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願日】平成24年3月27日(2012.3.27)
【出願人】(000003997)日産自動車株式会社 (16,386)
【Fターム(参考)】
[ Back to top ]