
正浸透膜流動システム及び正浸透膜流動システム用複合半透膜
【課題】高い効率を示す正浸透膜流動システムを提供する。
【解決手段】
正浸透膜流動システム1は、高浸透圧流体が供給される高浸透圧流体流動部2と、高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部3と、高浸透圧流体流動部と低浸透圧流体流動部とを隔てる半透膜4と、を備えている。低浸透圧流体流動部3から高浸透圧流体流動部2に半透膜4を介して流体移動が生じることにより高浸透圧流体流動部2の流量を増加させる。半透膜4が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である。
【解決手段】
正浸透膜流動システム1は、高浸透圧流体が供給される高浸透圧流体流動部2と、高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部3と、高浸透圧流体流動部と低浸透圧流体流動部とを隔てる半透膜4と、を備えている。低浸透圧流体流動部3から高浸透圧流体流動部2に半透膜4を介して流体移動が生じることにより高浸透圧流体流動部2の流量を増加させる。半透膜4が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、正浸透膜流動システム、正浸透膜流動システムに用いられる複合半透膜に関する。
【背景技術】
【0002】
エネルギーに対する世界的な需要の高まりは、再生可能なエネルギー源及び燃料使用効率の改善への関心を高めている。特に、逆浸透膜による海水淡水化の工程においては、海水から人類の営みに必要な淡水を生成する一方で、一般に海水の浸透圧以上のエネルギーをかける必要があるため、多量のエネルギーを必要とする。さらに、海水淡水化の工程において、淡水からこし出された塩分は濃縮海水として存在するため、これを廃棄する必要がある。通常はこのような濃縮海水は海水と混合して塩分濃度を下げた状態で海に排出されている。これに対して、この濃縮海水を希釈するために正浸透技術を利用した方法(例えば、特許文献1参照。)や、海水淡水化とともに前記濃縮海水を用いた浸透圧発電を行うことで、濃縮海水の濃度低下と海水淡水化に必要なエネルギーの一部を賄うことを可能とするシステムが提案されている(例えば、特許文献2参照。)。
【0003】
ところで、支持体上に酢酸セルロース系スキン層を設けた膜、又は、ポリスルホン支持体上にポリアミドスキン層を設けた膜などの従来逆浸透分野で用いられてきた半透膜を正浸透用の半透膜として流用することが考えられる。しかしながら、これらの膜は正浸透において逆浸透時とは異なる性能及び挙動を示すため、これらの膜を正浸透膜として流用したシステムの効率は十分ではなく、正浸透技術を利用したシステムの効率をさらに向上させることが求められていた。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−279540号公報
【特許文献2】特開2003−176775号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、高い効率を示す正浸透膜流動システム、そのような正浸透膜流動システムに用いられる複合半透膜を提供することを目的とする。
【課題を解決するための手段】
【0006】
上記の事情に鑑み、本発明は、高浸透圧流体が供給される高浸透圧流体流動部と、前記高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部と、前記高浸透圧流体流動部と前記低浸透圧流体流動部とを隔てる半透膜と、を備え、前記低浸透圧流体流動部から前記高浸透圧流体流動部に前記半透膜を介して流体移動が生じることにより前記高浸透圧流体流動部の流量を増加させ、前記半透膜が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である、正浸透膜流動システムを提供する。
【0007】
また別の側面から、本発明は、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した、正浸透膜流動システム用複合半透膜を提供する。
【発明の効果】
【0008】
上記の正浸透膜流動システムによれば、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜であるので、正浸透流動時に半透膜中を移動する流体の量(透過流束)が大きい。そのため、正浸透膜流動システムの効率が高まる。
【0009】
また、上記の正浸透膜流動システム用複合半透膜を正浸透膜流動システムに用いることにより、正浸透膜中を移動する流体の量(透過流束)を大きくすることできる。その結果、高い効率を示す正浸透膜流動システムを実現することができる。
【図面の簡単な説明】
【0010】
【図1】本実施形態に係る正浸透膜流動システムを模式的に示す図
【発明を実施するための形態】
【0011】
以下、本発明の実施形態について説明する。なお、以下の説明は本発明の一例に関するものであり、本発明はこれらによって限定されるものではない。
【0012】
本実施形態に係る正浸透膜流動システムは、半透膜の両面にそれぞれ浸透圧の異なる流体を供給し、低浸透圧流体側から高浸透圧流体側に流体移動が生じることで高浸透圧流体側の流量を増加させる正浸透現象を利用したものである。そして、本実施形態に係る正浸透膜流動システムは、三次元網目構造により連通した空孔を有するエポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜を用いる。
【0013】
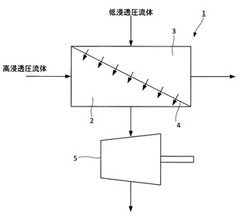
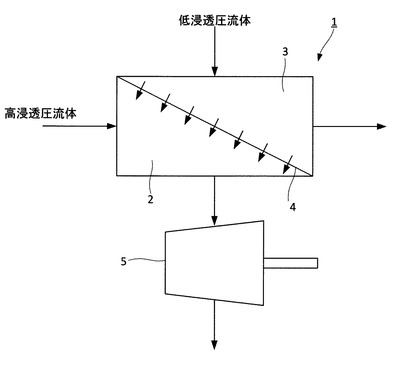
図1は、本実施形態の正浸透膜流動システムを模式的に示した図である。正浸透膜流動システム1は、高浸透圧流体が供給される高浸透圧流体流動部2と、高浸透圧流体の浸透圧よりも浸透圧の低い低浸透圧流体が供給される低浸透圧流体流動部3と、高浸透圧流体流動部2と低浸透圧流体流動部3とを隔てる半透膜4と、を有している。半透膜4としては、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜が用いられる。
【0014】
高浸透圧流体が高浸透圧流体流動部2に供給される。すなわち、半透膜4の一方の面に高浸透圧流体を供給する。また、低浸透圧流体が低浸透圧流体流動部3に供給される。すなわち、半透膜4の他方の面に高浸透圧流体を供給する。これにより、低浸透圧流体側から半透膜4を介して高浸透圧流体側に流体移動が生じる。この流体移動により、高浸透圧流体側の流量が増加し、高浸透圧流体の圧力が増加する。図1に示すように、本実施形態の正浸透膜流動システム1は、タービン5をさらに有している。タービン5は、高浸透圧流体流動部2を流出した高浸透圧流体の圧力によって回転する。タービン5の回転によって発電が行われる。
【0015】
本実施形態に係る正浸透膜流動システム1用の半透膜4は、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である。従来の逆浸透膜の分野では、1970年代頃から透過性能を向上させるために、常識的に膜面の表裏で孔径の異なる非対称性の支持膜を用いていた。本発明者らは、鋭意検討した結果、この支持膜が意外にも正浸透現象の流動の妨げとなっていることを見出した。そこで、従来用いられていた支持膜よりも自立性が高く、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を用いることにより、正浸透膜流動システム用の半透膜の薄型化を可能とし、正浸透膜流動システムの効率向上を実現した。
【0016】
本実施形態の正浸透膜流動システム1は、高浸透圧流体流動部2において、高浸透圧流体に高浸透圧流体の浸透圧と低浸透圧流体の浸透圧との差よりも低い圧力を加える、PRO(Pressure Retarded Osmosis)システムとして構成されていてもよい。この場合に、高浸透圧流体に印加する圧力(供給圧力)は、例えば高浸透圧流体が海水である場合、3MPa以下程度であり、2MPa以下とすることが好ましい。この供給圧力が高すぎると、低浸透圧流体側から高浸透圧流体側に流体移動する浸透水の流量が低下してしまう。また、この供給圧力は0.01MPa以上であればよく、0.1MPa以上とすることが好ましい。この供給圧力が低すぎると半透膜の界面付近で流体が滞留しやすくなる。そのため、濃度分極が生じ易くなり、時間とともに低浸透圧流体側から高浸透圧流体側に流体移動する浸透水の流量が低下していく傾向がある。
【0017】
本実施形態の正浸透膜流動システム1では、複合半透膜のいずれの面に低浸透圧流体及び高浸透圧流体のいずれを供給するか特に限定されない。複合半透膜のスキン層側に高浸透圧流体を供給する方法が一般的である。しかしながら、本実施形態では、複合半透膜のスキン層側に低浸透圧流体を供給する方法も好ましく用いることができる。低浸透圧流体をスキン層側に供給することで、汚染物質を膜面に付着しにくくできるとともに、汚染物質が付着した場合でも膜面の洗浄が容易になる。
【0018】
本実施形態の正浸透膜流動システム1では、エポキシ樹脂多孔質膜を半透膜4の支持体として用いる。このエポキシ樹脂多孔質膜は、従来のポリスルホン支持膜が必要とした不織布等の支持基材を必要とせず、自立性が高い。このような自立性の高い支持膜を用いることで半透膜4の支持体の薄膜化を可能としている。また、このエポキシ樹脂多孔質膜は、好ましくは、孔径が膜の厚さ方向で大きく変化しない対称膜であってもよい。対称膜は、ポリスルホン支持膜のような非対称膜と比べて、汚染物質が付着しにくく、かつ、濃度分極を抑制しやすい。
【0019】
本実施形態で使用できるエポキシ樹脂多孔質膜はエポキシ樹脂、硬化剤及び空孔を形成するためのポロゲンを分散させることで作製することができる。
【0020】
エポキシ樹脂多孔質膜に用いられるエポキシ樹脂としては、例えば、ビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、スチルベン型エポキシ樹脂、ビフェニル型エポキシ樹脂、ビスフェノールAノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、ジアミノジフェニルメタン型エポキシ樹脂、及びテトラキス(ヒドロキシフェニル)エタンベースなどのポリフェニルベースエポキシ樹脂、フルオレン含有エポキシ樹脂、トリグリシジルイソシアヌレート、複素芳香環(例えば、トリアジン環など)を含有するエポキシ樹脂などの芳香族エポキシ樹脂;脂肪族グリシジルエーテル型エポキシ樹脂、脂肪族グリシジルエステル型エポキシ樹脂、脂環式グリシジルエーテル型エポキシ樹脂、脂環式グリシジルエステル型エポキシ樹脂などの非芳香族エポキシ樹脂が挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0021】
これらのうち、均一な三次元網目構造と均一な空孔を形成するため、また耐薬品性や膜強度を確保するために、ビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、フルオレン含有エポキシ樹脂、及びトリグリシジルイソシアヌレートからなる群より選択される少なくとも1種の芳香族エポキシ樹脂;脂環式グリシジルエーテル型エポキシ樹脂、及び脂環式グリシジルエステル型エポキシ樹脂からなる群より選択される少なくとも1種の脂環式エポキシ樹脂を用いることが好ましい。特に、エポキシ当量が6000以下で、融点が170℃以下であるビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、フルオレン含有エポキシ樹脂、及びトリグリシジルイソシアヌレートからなる群より選択される少なくとも1種の芳香族エポキシ樹脂;エポキシ当量が6000以下で、融点が170℃以下である脂環式グリシジルエーテル型エポキシ樹脂、及び脂環式グリシジルエステル型エポキシ樹脂からなる群より選択される少なくとも1種の脂環式エポキシ樹脂を用いることが好ましい。
【0022】
本実施形態で使用できる硬化剤としては、例えば、芳香族アミン(例えば、メタフェニレンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルホン、ベンジルジメチルアミン、ジメチルアミノメチルベンゼンなど)、芳香族酸無水物(例えば、無水フタル酸、無水トリメリット酸、無水ピロメリット酸など)、フェノール樹脂、フェノールノボラック樹脂、複素芳香環含有アミン(例えば、トリアジン環含有アミンなど)などの芳香族硬化剤;脂肪族アミン類(例えば、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン、1,3,6−トリスアミノメチルヘキサン、ポリメチレンジアミン、トリメチルヘキサメチレンジアミン、ポリエーテルジアミンなど)、脂環式アミン類(イソホロンジアミン、メンタンジアミン、N−アミノエチルピペラジン、3,9−ビス(3−アミノプロピル)2,4,8,10−テトラオキサスピロ(5,5)ウンデカンアダクト、ビス(4−アミノ−3−メチルシクロヘキシル)メタン、ビス(4−アミノシクロヘキシル)メタン、これらの変性品など)、ポリアミン類とダイマー酸からなる脂肪族ポリアミドアミンなどの非芳香族硬化剤が挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0023】
これらのうち、均一な三次元網目構造と均一な空孔を形成するため、また膜強度と弾性率を確保するために、分子内に一級アミンを2つ以上有するメタフェニレンジアミン、ジアミノジフェニルメタン、及びジアミノジフェニルスルホンからなる群より選択される少なくとも1種の芳香族アミン硬化剤;分子内に一級アミンを2つ以上有するビス(4−アミノ−3−メチルシクロヘキシル)メタン、及びビス(4−アミノシクロヘキシル)メタンからなる群より選択される少なくとも1種の脂環式アミン硬化剤を用いることが好ましい。
【0024】
また、エポキシ樹脂と硬化剤の組み合わせとしては、芳香族エポキシ樹脂と脂環式アミン硬化剤の組み合わせ、又は、脂環式エポキシ樹脂と芳香族アミン硬化剤の組み合わせが好ましい。これらの組み合わせにより、得られるエポキシ樹脂多孔質膜の耐熱性が高くなり、複合半透膜の多孔性支持体として好適に用いられる。
【0025】
本実施形態で使用できるポロゲンとは、エポキシ樹脂及び硬化剤を溶かすことができ、かつ、エポキシ樹脂と硬化剤が重合した後、反応誘起相分離を生ぜしめることが可能な溶剤をいい、例えば、メチルセロソルブ、エチルセロソルブなどのセロソルブ類、エチレングリコールモノメチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、及びポリエチレングリコール、ポリプロピレングリコールなどのグリコール類などが挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0026】
これらのうち、均一な三次元網目構造と均一な空孔を形成するために、メチルセロソルブ、エチルセロソルブ、分子量600以下のポリエチレングリコール、エチレングリコールモノメチルエーテルアセテート、ポリプロピレングリコール、ポリオキシエチレンモノメチルエーテル、ポリオキシエチレンジメチルエーテル、及びプロピレングリコールモノメチルエーテルアセテートからなる群より選択される少なくとも1種を用いることが好ましく、特に分子量200以下のポリエチレングリコール、分子量500以下のポリプロピレングリコール、ポリオキシエチレンモノメチルエーテル、及びプロピレングリコールモノメチルエーテルアセテートからなる群より選択される少なくとも1種を用いることが好ましい。
【0027】
また、個々のエポキシ樹脂又は硬化剤と常温で不溶又は難溶である溶剤であっても、エポキシ樹脂と硬化剤との反応物が可溶である溶剤についてはポロゲンとして使用可能である。このようなポロゲンとしては、例えば臭素化ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、「エピコート5058」)などが挙げられる。
【0028】
エポキシ樹脂多孔質膜の空孔率、平均孔径、孔径分布などは、使用するエポキシ樹脂、硬化剤、ポロゲンなどの原料の種類や配合比率、及び反応誘起相分離時における加熱温度や加熱時間などの反応条件により変化するため、目的とする空孔率、平均孔径、孔径分布を得るために系の相図を作成して最適な条件を選択することが好ましい。また、相分離時におけるエポキシ樹脂架橋体の分子量、分子量分布、系の粘度、架橋反応速度などを制御することにより、エポキシ樹脂架橋体とポロゲンとの共連続構造を特定の状態で固定し、安定した多孔構造を得ることができる。
【0029】
また、エポキシ樹脂多孔質膜を構成する全炭素原子に対する芳香環由来の炭素原子比率が0.1〜0.65の範囲になるように、エポキシ樹脂及び硬化剤の種類と配合割合を決定することが好ましい。上記値が0.1未満の場合には、エポキシ樹脂多孔質膜の特性である分離媒体の平面構造の認識性が低下する傾向にある。一方、0.65を超える場合には、均一な三次元網目構造を形成することが困難になる。
【0030】
また、エポキシ樹脂に対する硬化剤の配合割合は、エポキシ基1当量に対して硬化剤当量が0.6〜1.5であることが好ましい。硬化剤当量が0.6未満の場合には、硬化体の架橋密度が低くなり、耐熱性、耐溶剤性などが低下する傾向にある。一方、1.5を超える場合には、未反応の硬化剤が残留し、架橋密度の向上を阻害する傾向にある。なお、本実施形態では、上述した硬化剤の他に、目的とする多孔構造を得るために、溶液中に硬化促進剤を添加してもよい。硬化促進剤としては、公知のものを使用することができ、例えば、トリエチルアミン、トリブチルアミンなどの三級アミン、2−フェノール−4−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェノール−4,5−ジヒドロキシイミダゾールなどのイミダゾール類などが挙げられる。
【0031】
エポキシ樹脂多孔質膜はより良いスキン層を形成するために、水銀圧入法で測定したときの平均孔径を0.01〜0.4μmとすることが好ましい。エポキシ樹脂多孔質膜の平均孔径を0.01〜0.4μmに調整するためには、エポキシ樹脂、硬化剤及びポロゲンの総重量に対してポロゲンを40〜80重量%用いることが好ましい。ポロゲンの量が40重量%未満の場合にはエポキシ樹脂多孔質膜の平均孔径が小さくなりすぎ、空孔が形成されなくなる傾向にある。一方、ポロゲンの量が80重量%を超える場合にはエポキシ樹脂多孔質膜の平均孔径が大きくなりすぎて均一なスキン層を多孔体上に形成することができず、塩阻止率が著しく低下する傾向にある。エポキシ樹脂多孔質膜の平均孔径は、0.05〜0.3μmであることが好ましく、より好ましくは0.05〜0.2μmである。そのためにはポロゲンを60〜70重量%用いることがより好ましく、特に好ましくは60〜65重量%である。
【0032】
また、エポキシ樹脂多孔質膜の平均孔径を0.01〜0.4μmに調整する方法として、エポキシ当量の異なる2種以上のエポキシ樹脂を混合して用いる方法も好適である。その際、エポキシ当量の差は100以上であることが好ましい。
【0033】
また、エポキシ樹脂多孔質膜の平均孔径は、全体のエポキシ当量とポロゲンの割合、硬化温度などの諸条件を適宜設定することにより目的の範囲に調整できる。
【0034】
エポキシ樹脂多孔質膜は、例えば、以下の方法で作製することができる。
【0035】
1)エポキシ樹脂、硬化剤及びポロゲンを含むエポキシ樹脂組成物を基板上に塗布し、その後、塗布したエポキシ樹脂組成物を加熱してエポキシ樹脂を三次元架橋させる。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、得られたエポキシ樹脂シートからポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。使用する基板は特に制限されず、例えば、プラスチック基板、ガラス基板、及び金属板などが挙げられる。
【0036】
2)エポキシ樹脂、硬化剤、及びポロゲンを含むエポキシ樹脂組成物を基板上に塗布し、その後、塗布したエポキシ樹脂組成物上に別の基板を被せてサンドイッチ構造体を作製する。なお、基板間に一定の厚さを設けるために、基板の四隅にスペーサー(例えば、両面テープ)を設けておくことが好ましい。そして、該サンドイッチ構造体を加熱してエポキシ樹脂を三次元架橋させる。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、得られたエポキシ樹脂シートを取り出し、ポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。使用する基板は特に制限されず、例えば、プラスチック基板、ガラス基板、及び金属板などが挙げられるが、特にガラス基板を用いることが好ましい。
【0037】
3)エポキシ樹脂、硬化剤、及びポロゲンを含むエポキシ樹脂組成物を所定形状のモールド内に充填し、その後、反応を促進させてエポキシ樹脂を三次元架橋させて、円筒状又は円柱状樹脂ブロックを作製する。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、その円柱状樹脂ブロックを円筒軸又は円柱軸を中心に回転させながら該ブロックの表面を所定厚さで切削して長尺状のエポキシ樹脂シートを作製する。そして、エポキシ樹脂シート中のポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。
【0038】
エポキシ樹脂組成物を硬化する際の条件(加熱硬化又は室温硬化など)は特に制限されないが、均一な孔を有する多孔シートを形成するためには、エポキシ樹脂組成物を室温で硬化させることが好ましい。エポキシ樹脂組成物を室温で硬化させる場合、20〜40℃程度で硬化を開始させ、硬化時間は3〜100時間程度であり、好ましくは20〜50時間程度である。エポキシ樹脂組成物を加熱硬化させる場合、温度は40〜120℃程度であり、好ましくは60〜100℃程度であり、硬化時間は10〜300分程度であり、好ましくは30〜180分程度である。
【0039】
得られたエポキシ樹脂シートからポロゲンを除去するために用いられる溶剤としては、例えば、水、DMF、DMSO、THF及びこれらの混合溶剤などが挙げられ、ポロゲンの種類に応じて適宜選択する。
【0040】
ポロゲンを除去したエポキシ樹脂多孔質膜の乾燥条件は特に制限されないが、温度は40〜120℃程度であり、乾燥時間は3分〜3時間程度である。
【0041】
エポキシ樹脂多孔質膜の厚さは、複合半透膜の製造又は使用に必要な強度及び実用性を満たす限り、特に制限されない。ところで、複合半透膜のスキン層は溶質を透過させないため、スキン層付近では溶質濃度の高い流体が溜まりやすく、複合半透膜の厚さ方向に溶質濃度の偏りが生じる。濃度分極と呼ばれるこのような溶質濃度の偏りは、一般に複合半透膜の透過流量を減らし、正浸透膜流動システムの効率を低下させてしまう。本発明者らは、このエポキシ樹脂多孔質膜の厚さが薄い方が、濃度分極が抑制されやすいことを見出した。一方で、エポキシ樹脂多孔質膜の厚さが薄すぎると、エポキシ樹脂多孔質膜上にスキン層を形成する際に、エポキシ樹脂多孔質膜の取り扱いが困難となる。また、エポキシ樹脂多孔質膜の厚さが薄すぎると、正浸透処理、特に、PRO処理行う場合に、エポキシ樹脂多孔質膜が破れる場合がある。このような観点から、エポキシ樹脂多孔質膜の厚さは、例えば10μm以上であり、20μm以上が好ましく、25μm以上がより好ましい。また、エポキシ樹脂多孔質膜の厚さは、例えば150μm以下であり、100μm以下が好ましく、60μm以下がより好ましい。また、エポキシ樹脂多孔質膜の裏面を織布、不織布などで補強してもよい。
【0042】
一方、本実施形態におけるエポキシ樹脂多孔質膜上に形成するスキン層はポリアミド系樹脂からなるものであり、多官能アミン成分と多官能酸ハライド成分とを重合してなるポリアミド系樹脂を含むスキン層であることが好ましい。
【0043】
多官能アミン成分とは、2以上の反応性アミノ基を有する多官能アミンであり、芳香族、脂肪族、及び脂環式の多官能アミンが挙げられる。
【0044】
芳香族多官能アミンとしては、例えば、m−フェニレンジアミン、p−フェニレンジアミン、o−フェニレンジアミン、1,3,5−トリアミノベンゼン、1,2,4−トリアミノベンゼン、3,5−ジアミノ安息香酸、2,4−ジアミノトルエン、2,6−ジアミノトルエン、N,N’−ジメチル−m−フェニレンジアミン、2,4−ジアミノアニソール、アミドール、キシリレンジアミン等が挙げられる。
【0045】
脂肪族多官能アミンとしては、例えば、エチレンジアミン、プロピレンジアミン、トリス(2−アミノエチル)アミン、n−フェニル−エチレンジアミン等が挙げられる。
【0046】
脂環式多官能アミンとしては、例えば、1,3−ジアミノシクロヘキサン、1,2−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、ピペラジン、2,5−ジメチルピペラジン、4−アミノメチルピペラジン等が挙げられる。
【0047】
これらの多官能アミンは1種で用いてもよく、2種以上を併用してもよい。高塩阻止性能のスキン層を得るためには、芳香族多官能アミンを用いることが好ましい。
【0048】
多官能酸ハライド成分とは、反応性カルボニル基を2個以上有する多官能酸ハライドである。
【0049】
多官能酸ハライドとしては、芳香族、脂肪族、及び脂環式の多官能酸ハライドが挙げられる。
【0050】
芳香族多官能酸ハライドとしては、例えば、トリメシン酸トリクロライド、テレフタル酸ジクロライド、イソフタル酸ジクロライド、ビフェニルジカルボン酸ジクロライド、ナフタレンジカルボン酸ジクロライド、ベンゼントリスルホン酸トリクロライド、ベンゼンジスルホン酸ジクロライド、クロロスルホニルベンゼンジカルボン酸ジクロライド等が挙げられる。
【0051】
脂肪族多官能酸ハライドとしては、例えば、プロパンジカルボン酸ジクロライド、ブタンジカルボン酸ジクロライド、ペンタンジカルボン酸ジクロライド、プロパントリカルボン酸トリクロライド、ブタントリカルボン酸トリクロライド、ペンタントリカルボン酸トリクロライド、グルタリルハライド、アジポイルハライド等が挙げられる。
【0052】
脂環式多官能酸ハライドとしては、例えば、シクロプロパントリカルボン酸トリクロライド、シクロブタンテトラカルボン酸テトラクロライド、シクロペンタントリカルボン酸トリクロライド、シクロペンタンテトラカルボン酸テトラクロライド、シクロヘキサントリカルボン酸トリクロライド、テトラハイドロフランテトラカルボン酸テトラクロライド、シクロペンタンジカルボン酸ジクロライド、シクロブタンジカルボン酸ジクロライド、シクロヘキサンジカルボン酸ジクロライド、テトラハイドロフランジカルボン酸ジクロライド等が挙げられる。
【0053】
これら多官能酸ハライドは1種で用いてもよく、2種以上を併用してもよい。高い塩阻止性能のスキン層を得るためには、芳香族多官能酸ハライドを用いることが好ましい。また、多官能酸ハライド成分の少なくとも一部に3価以上の多官能酸ハライドを用いて、架橋構造を形成するのが好ましい。
【0054】
また、ポリアミド系樹脂を含むスキン層の性能を向上させるために、ポリビニルアルコール、ポリビニルピロリドン、ポリアクリル酸などのポリマー、ソルビトール、グリセリンなどの多価アルコールなどを共重合させてもよい。
【0055】
ポリアミド系樹脂を含むスキン層をエポキシ樹脂多孔質膜の表面に形成する方法は特に制限されず、あらゆる公知の手法を用いることができる。例えば、界面縮合法、相分離法、薄膜塗布法などが挙げられる。界面縮合法とは、具体的には、多官能アミン成分を含有するアミン水溶液と、多官能酸ハライド成分を含有する有機溶液とを接触させて界面重合させることによりスキン層を形成し、そのスキン層をエポキシ樹脂多孔質膜上に載置する方法、又は、エポキシ樹脂多孔質膜上での界面重合によりポリアミド系樹脂のスキン層をエポキシ樹脂多孔質膜上に直接形成する方法などである。かかる界面縮合法の条件等の詳細は、特開昭58−24303号公報、特開平1−180208号公報等に記載されており、それらの公知技術を適宜採用することができる。
【0056】
本実施形態において、多官能アミン成分を含むアミン水溶液からなる水溶液被覆層をエポキシ樹脂多孔質膜上に形成し、次いで多官能酸ハライド成分を含有する有機溶液と水溶液被覆層とを接触させて界面重合させることによりスキン層を形成する方法が好ましい。
【0057】
界面重合法において、アミン水溶液中の多官能アミン成分の濃度は特に制限されないが、0.1〜5重量%であることが好ましく、さらに好ましくは1〜4重量%である。多官能アミン成分の濃度が低すぎる場合にはスキン層にピンホール等の欠陥が生じやすくなり、また塩阻止性能が低下する傾向にある。一方、多官能アミン成分の濃度が高すぎる場合には、膜厚が厚くなりすぎて透過抵抗が大きくなって透過流束が低下する傾向にある。
【0058】
有機溶液中の多官能酸ハライド成分の濃度は特に制限されないが、0.01〜5重量%であることが好ましく、さらに好ましくは0.05〜3重量%である。多官能酸ハライド成分の濃度が低すぎる場合には、未反応多官能アミン成分が残留しやすくなり、スキン層にピンホール等の欠陥が生じやすくなって塩阻止性能が低下する傾向にある。一方、多官能酸ハライド成分の濃度が高すぎる場合には、未反応多官能酸ハライド成分が残留しやすくなり、膜厚が厚くなりすぎて透過抵抗が大きくなり、透過流束が低下する傾向にある。
【0059】
上述の有機溶液に用いられる有機溶媒としては、水に対する溶解度が低く、エポキシ樹脂多孔質膜を劣化させず、多官能酸ハライド成分を溶解するものであれば特に限定されず、例えば、シクロヘキサン、ヘプタン、オクタン、及びノナン等の飽和炭化水素、1,1,2−トリクロロトリフルオロエタン等のハロゲン置換炭化水素などを挙げることができる。好ましくは沸点が300℃以下、さらに好ましくは沸点が200℃以下の飽和炭化水素である。
【0060】
上述のアミン水溶液や有機溶液には、製膜を容易にし、得られる複合半透膜の性能を向上させるための目的で各種の添加剤を加えることができる。添加剤としては、例えば、ドデシルベンゼンスルホン酸ナトリウム、ドデシル硫酸ナトリウム及びラウリル硫酸ナトリウム等の界面活性剤、重合により生成するハロゲン化水素を除去する水酸化ナトリウム、リン酸三ナトリウム及びトリエチルアミン等の塩基性化合物、アシル化触媒、特開平8−224452号公報記載の溶解度パラメータが8〜14(cal/cm3)1/2の化合物などが挙げられる。
【0061】
エポキシ樹脂多孔質膜上にアミン水溶液を塗布してから有機溶液を塗布するまでの時間は、アミン水溶液の組成、粘度及びエポキシ樹脂多孔質膜の表面の孔径にもよるが、1〜180秒程度であり、好ましくは2〜120秒であり、より好ましくは2〜40秒であり、特に好ましくは2〜10秒である。溶液の塗布間隔が長すぎる場合には、アミン水溶液がエポキシ樹脂多孔質膜の内部深くまで浸透・拡散し、未反応多官能アミン成分がエポキシ樹脂多孔質膜中に大量に残存する可能性がある。また、エポキシ樹脂多孔質膜の内部深くまで浸透した未反応多官能アミン成分は、その後の膜洗浄処理でも除去し難い傾向にある。溶液の塗布間隔が短すぎる場合には、余分なアミン水溶液が残存しすぎてしまい、膜性能が低下する傾向にある。
【0062】
本実施形態においては、アミン水溶液からなる水溶液被覆層と有機溶液との接触後、エポキシ樹脂多孔質膜上の過剰な有機溶液を除去し、エポキシ樹脂多孔質膜上の形成膜を70℃以上で加熱乾燥してスキン層を形成することが好ましい。形成膜を加熱処理することによりその機械的強度や耐熱性等を高めることができる。加熱温度は70〜200℃であることがより好ましく、特に好ましくは80〜130℃である。加熱時間は30秒〜10分程度が好ましく、さらに好ましくは40秒〜7分程度である。
【0063】
エポキシ樹脂多孔質膜上に形成したスキン層の厚さは特に制限されないが、通常0.05〜2μm程度であり、好ましくは、0.1〜1μmである。
【0064】
複合半透膜の厚さは特に制限されない。強度、実用的な透水性及び塩阻止性の観点から、表面処理が行われてもよく、補強材が積層されてもよい。複合半透膜の厚さは、例えば10〜250μmであり、好ましくは10〜150μmである。
【0065】
また、この複合半透膜の形状は特に制限されない。すなわち、平膜状、スパイラルエレメント状など、公知の形態に加工して用いればよい。
【0066】
本実施形態の正浸透膜流動システムは、例えば、特開2003−176775に記載の浸透圧発電システムや、特開2005−279540に記載の海水淡水化における濃縮水の希釈システムとして使用することができる。このとき、上述の浸透圧発電システムにおける発電量と比例する出力密度[W/m2]は、正浸透時の透過流束によって簡易的に求めることができる。
【実施例】
【0067】
以下に本発明の効果について具体例を用いて詳細に説明する。なお、本発明はこれらの実施例に限定されるものではない。
【0068】
<実施例1>
(エポキシ樹脂多孔質基材の作製)
ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート828)139重量部、ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート1010)93.2重量部、ビス(4−アミノシクロヘキシル)メタン52重量部、及びポリエチレングリコール200(三洋化成工業社製)500重量部を調製したエポキシ樹脂組成物を、円筒状モールド(外径35cm、内径10.5cm)内に高さ30cmまで充填して25℃で12時間室温硬化し、さらに130℃で18時間反応硬化させて円筒状樹脂ブロックを作製した。この樹脂ブロックを、円筒軸を中心に回転させながら切削装置(東芝機械社製)を用いて、その表面から厚さ145μmで連続的にスライスし、長尺状のエポキシ樹脂シート(長さ:100m)を得た。このエポキシ樹脂シートを純水中に12時間浸漬してポリエチレングリコールを除去し、50℃の乾燥機内で約4時間乾燥することでエポキシ樹脂多孔質膜(厚さ130μm、空孔率45%、平均孔径0.04μm)を得た。
【0069】
(複合半透膜の作製)
m-フェニレンジアミン3.0g、ラウリル硫酸ナトリウム0.15g、ベンゼンスルホン酸6.0g、トリエチルアミン3.0g、及び水87.85gを混合して水溶液(B)を調製した。水溶液(B)を大気圧プラズマ処理したエポキシ樹脂多孔質膜上に塗布して余分なアミン水溶液を除去した。次に、トリメシン酸クロライド0.2%を含むイソオクタン溶液をエポキシ樹脂多孔質膜上に塗布した。その後、余分なイソオクタン溶液を除去して100℃の乾燥器内で2分間保持することで、エポキシ樹脂多孔質膜上にポリアミドからなるスキン層(厚さ約200nm)を形成して、実施例1の複合半透膜を得た。最終的に得られた実施例1に係る複合半透膜の厚さは130μmであった。
【0070】
<実施例2>
エポキシ樹脂多孔質膜の作製において、樹脂ブロックの切削厚さを90μmに変更した点以外は、実施例1と同様にして実施例2の複合半透膜を得た。最終的に得られた実施例2に係る複合半透膜の厚さは80μmであった。
【0071】
<実施例3>
エポキシ樹脂多孔質膜の作製において、樹脂ブロックの切削厚さを55μmに変更した点以外は、実施例1と同様にして実施例2の複合半透膜を得た。最終的に得られた実施例3に係る複合半透膜の厚さは50μmであった。
【0072】
<比較例1>
ポリエステル製不織布(70g/m2、厚さ90μm)上にポリスルホン(ソルベイアドバンストポリマーズ社製、P‐3500)18.3重量%とジメチルホルムアルデヒド81、7重量%の混合液を塗布した。その後、その混合液を塗布したポリエステル製不織布を20℃の純水に浸漬し、さらに、45℃の純水に浸漬した。このようにして、約130μmの厚さのポリスルホン支持膜を得た。実施例1の複合半透膜の作成の工程で用いられた水溶液(B)をポリスルホン支持膜上に塗布し、実施例1と同様に処理して、比較例1の複合半透膜を得た。最終的に得られた比較例1に係る複合半透膜の厚さは130μmであった。
【0073】
(逆浸透方式によるNaCl阻止率の測定)
作製した実施例1〜3及び比較例1の複合半透膜を逆浸透膜として用いて、1500mg/LのNaCl水溶液を原水として、25℃、pH6.5、操作圧力1.5MPaの条件で逆浸透方式による透過試験を行った。その結果、NaCl阻止率99%であった。なお、NaCl阻止率は下記式で求めた。
NaCl阻止率(%)=(1−(逆浸透膜透過液中のNaCl濃度/原水中のNaCl濃度))×100
【0074】
(正浸透方式による透過流束の測定)
作製した平膜状の実施例1〜3及び比較例1の複合半透膜を所定の形状、サイズに切断し、平膜評価用のセル(日東電工株式会社製、C10−T)にセットした。NaCl135gを溶解させた5LのNaCl水溶液が入ったタンクを準備し、ポリアミドからなるスキン層側に220ml/minの流量でNaCl水溶液を供給した。また、5Lの純水が入ったタンクを準備し、エポキシ樹脂多孔質膜側又はポリスルホン支持膜の不織布側に純水を60ml/minの流量で循環供給した。この操作によって純水タンクの減少重量を計測し、減少重量を透過流束とした。
【0075】
実施例1〜3及び比較例1に係る複合半透膜の上記の測定の測定結果を表1に示す。
【表1】

【0076】
上記の通り、実施例1〜3及び比較例1に係る複合半透膜の逆浸透方式の透過流束は同程度であった。これに対し、実施例1〜3に係る複合半透膜の正浸透方式の透過流束は、比較例1に係る複合半透膜の正浸透方式の透過流束の2倍以上であった。また、実施例1〜3において、複合半透膜の厚さが薄いほど正浸透方式の透過流束は大きくなる傾向があった。なお、エポキシ樹脂多孔質膜の厚さが複合半透膜の厚さの大部分を占めるので、エポキシ樹脂多孔質膜の厚さが薄いほど正浸透方式の透過流束が大きくなる傾向があるということもできる。従って、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜を用いて正浸透膜流動システムを構成することにより、正浸透膜流動システムの効率が向上することが示唆された。
【産業上の利用可能性】
【0077】
本発明に係る正浸透膜流動システムは、例えば、タービンを回転させて発電するシステム、又は、高浸透圧流体を希釈するシステム等に使用することができる。
【符号の説明】
【0078】
1 正浸透膜流動システム
2 高浸透圧流体流動部
3 低浸透圧流体流動部
4 半透膜
5 タービン
【技術分野】
【0001】
本発明は、正浸透膜流動システム、正浸透膜流動システムに用いられる複合半透膜に関する。
【背景技術】
【0002】
エネルギーに対する世界的な需要の高まりは、再生可能なエネルギー源及び燃料使用効率の改善への関心を高めている。特に、逆浸透膜による海水淡水化の工程においては、海水から人類の営みに必要な淡水を生成する一方で、一般に海水の浸透圧以上のエネルギーをかける必要があるため、多量のエネルギーを必要とする。さらに、海水淡水化の工程において、淡水からこし出された塩分は濃縮海水として存在するため、これを廃棄する必要がある。通常はこのような濃縮海水は海水と混合して塩分濃度を下げた状態で海に排出されている。これに対して、この濃縮海水を希釈するために正浸透技術を利用した方法(例えば、特許文献1参照。)や、海水淡水化とともに前記濃縮海水を用いた浸透圧発電を行うことで、濃縮海水の濃度低下と海水淡水化に必要なエネルギーの一部を賄うことを可能とするシステムが提案されている(例えば、特許文献2参照。)。
【0003】
ところで、支持体上に酢酸セルロース系スキン層を設けた膜、又は、ポリスルホン支持体上にポリアミドスキン層を設けた膜などの従来逆浸透分野で用いられてきた半透膜を正浸透用の半透膜として流用することが考えられる。しかしながら、これらの膜は正浸透において逆浸透時とは異なる性能及び挙動を示すため、これらの膜を正浸透膜として流用したシステムの効率は十分ではなく、正浸透技術を利用したシステムの効率をさらに向上させることが求められていた。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−279540号公報
【特許文献2】特開2003−176775号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、高い効率を示す正浸透膜流動システム、そのような正浸透膜流動システムに用いられる複合半透膜を提供することを目的とする。
【課題を解決するための手段】
【0006】
上記の事情に鑑み、本発明は、高浸透圧流体が供給される高浸透圧流体流動部と、前記高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部と、前記高浸透圧流体流動部と前記低浸透圧流体流動部とを隔てる半透膜と、を備え、前記低浸透圧流体流動部から前記高浸透圧流体流動部に前記半透膜を介して流体移動が生じることにより前記高浸透圧流体流動部の流量を増加させ、前記半透膜が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である、正浸透膜流動システムを提供する。
【0007】
また別の側面から、本発明は、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した、正浸透膜流動システム用複合半透膜を提供する。
【発明の効果】
【0008】
上記の正浸透膜流動システムによれば、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜であるので、正浸透流動時に半透膜中を移動する流体の量(透過流束)が大きい。そのため、正浸透膜流動システムの効率が高まる。
【0009】
また、上記の正浸透膜流動システム用複合半透膜を正浸透膜流動システムに用いることにより、正浸透膜中を移動する流体の量(透過流束)を大きくすることできる。その結果、高い効率を示す正浸透膜流動システムを実現することができる。
【図面の簡単な説明】
【0010】
【図1】本実施形態に係る正浸透膜流動システムを模式的に示す図
【発明を実施するための形態】
【0011】
以下、本発明の実施形態について説明する。なお、以下の説明は本発明の一例に関するものであり、本発明はこれらによって限定されるものではない。
【0012】
本実施形態に係る正浸透膜流動システムは、半透膜の両面にそれぞれ浸透圧の異なる流体を供給し、低浸透圧流体側から高浸透圧流体側に流体移動が生じることで高浸透圧流体側の流量を増加させる正浸透現象を利用したものである。そして、本実施形態に係る正浸透膜流動システムは、三次元網目構造により連通した空孔を有するエポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜を用いる。
【0013】
図1は、本実施形態の正浸透膜流動システムを模式的に示した図である。正浸透膜流動システム1は、高浸透圧流体が供給される高浸透圧流体流動部2と、高浸透圧流体の浸透圧よりも浸透圧の低い低浸透圧流体が供給される低浸透圧流体流動部3と、高浸透圧流体流動部2と低浸透圧流体流動部3とを隔てる半透膜4と、を有している。半透膜4としては、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜が用いられる。
【0014】
高浸透圧流体が高浸透圧流体流動部2に供給される。すなわち、半透膜4の一方の面に高浸透圧流体を供給する。また、低浸透圧流体が低浸透圧流体流動部3に供給される。すなわち、半透膜4の他方の面に高浸透圧流体を供給する。これにより、低浸透圧流体側から半透膜4を介して高浸透圧流体側に流体移動が生じる。この流体移動により、高浸透圧流体側の流量が増加し、高浸透圧流体の圧力が増加する。図1に示すように、本実施形態の正浸透膜流動システム1は、タービン5をさらに有している。タービン5は、高浸透圧流体流動部2を流出した高浸透圧流体の圧力によって回転する。タービン5の回転によって発電が行われる。
【0015】
本実施形態に係る正浸透膜流動システム1用の半透膜4は、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である。従来の逆浸透膜の分野では、1970年代頃から透過性能を向上させるために、常識的に膜面の表裏で孔径の異なる非対称性の支持膜を用いていた。本発明者らは、鋭意検討した結果、この支持膜が意外にも正浸透現象の流動の妨げとなっていることを見出した。そこで、従来用いられていた支持膜よりも自立性が高く、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を用いることにより、正浸透膜流動システム用の半透膜の薄型化を可能とし、正浸透膜流動システムの効率向上を実現した。
【0016】
本実施形態の正浸透膜流動システム1は、高浸透圧流体流動部2において、高浸透圧流体に高浸透圧流体の浸透圧と低浸透圧流体の浸透圧との差よりも低い圧力を加える、PRO(Pressure Retarded Osmosis)システムとして構成されていてもよい。この場合に、高浸透圧流体に印加する圧力(供給圧力)は、例えば高浸透圧流体が海水である場合、3MPa以下程度であり、2MPa以下とすることが好ましい。この供給圧力が高すぎると、低浸透圧流体側から高浸透圧流体側に流体移動する浸透水の流量が低下してしまう。また、この供給圧力は0.01MPa以上であればよく、0.1MPa以上とすることが好ましい。この供給圧力が低すぎると半透膜の界面付近で流体が滞留しやすくなる。そのため、濃度分極が生じ易くなり、時間とともに低浸透圧流体側から高浸透圧流体側に流体移動する浸透水の流量が低下していく傾向がある。
【0017】
本実施形態の正浸透膜流動システム1では、複合半透膜のいずれの面に低浸透圧流体及び高浸透圧流体のいずれを供給するか特に限定されない。複合半透膜のスキン層側に高浸透圧流体を供給する方法が一般的である。しかしながら、本実施形態では、複合半透膜のスキン層側に低浸透圧流体を供給する方法も好ましく用いることができる。低浸透圧流体をスキン層側に供給することで、汚染物質を膜面に付着しにくくできるとともに、汚染物質が付着した場合でも膜面の洗浄が容易になる。
【0018】
本実施形態の正浸透膜流動システム1では、エポキシ樹脂多孔質膜を半透膜4の支持体として用いる。このエポキシ樹脂多孔質膜は、従来のポリスルホン支持膜が必要とした不織布等の支持基材を必要とせず、自立性が高い。このような自立性の高い支持膜を用いることで半透膜4の支持体の薄膜化を可能としている。また、このエポキシ樹脂多孔質膜は、好ましくは、孔径が膜の厚さ方向で大きく変化しない対称膜であってもよい。対称膜は、ポリスルホン支持膜のような非対称膜と比べて、汚染物質が付着しにくく、かつ、濃度分極を抑制しやすい。
【0019】
本実施形態で使用できるエポキシ樹脂多孔質膜はエポキシ樹脂、硬化剤及び空孔を形成するためのポロゲンを分散させることで作製することができる。
【0020】
エポキシ樹脂多孔質膜に用いられるエポキシ樹脂としては、例えば、ビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、スチルベン型エポキシ樹脂、ビフェニル型エポキシ樹脂、ビスフェノールAノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、ジアミノジフェニルメタン型エポキシ樹脂、及びテトラキス(ヒドロキシフェニル)エタンベースなどのポリフェニルベースエポキシ樹脂、フルオレン含有エポキシ樹脂、トリグリシジルイソシアヌレート、複素芳香環(例えば、トリアジン環など)を含有するエポキシ樹脂などの芳香族エポキシ樹脂;脂肪族グリシジルエーテル型エポキシ樹脂、脂肪族グリシジルエステル型エポキシ樹脂、脂環式グリシジルエーテル型エポキシ樹脂、脂環式グリシジルエステル型エポキシ樹脂などの非芳香族エポキシ樹脂が挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0021】
これらのうち、均一な三次元網目構造と均一な空孔を形成するため、また耐薬品性や膜強度を確保するために、ビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、フルオレン含有エポキシ樹脂、及びトリグリシジルイソシアヌレートからなる群より選択される少なくとも1種の芳香族エポキシ樹脂;脂環式グリシジルエーテル型エポキシ樹脂、及び脂環式グリシジルエステル型エポキシ樹脂からなる群より選択される少なくとも1種の脂環式エポキシ樹脂を用いることが好ましい。特に、エポキシ当量が6000以下で、融点が170℃以下であるビスフェノールA型エポキシ樹脂、臭素化ビスフェノールA型エポキシ樹脂、ビスフェノールAD型エポキシ樹脂、フルオレン含有エポキシ樹脂、及びトリグリシジルイソシアヌレートからなる群より選択される少なくとも1種の芳香族エポキシ樹脂;エポキシ当量が6000以下で、融点が170℃以下である脂環式グリシジルエーテル型エポキシ樹脂、及び脂環式グリシジルエステル型エポキシ樹脂からなる群より選択される少なくとも1種の脂環式エポキシ樹脂を用いることが好ましい。
【0022】
本実施形態で使用できる硬化剤としては、例えば、芳香族アミン(例えば、メタフェニレンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルホン、ベンジルジメチルアミン、ジメチルアミノメチルベンゼンなど)、芳香族酸無水物(例えば、無水フタル酸、無水トリメリット酸、無水ピロメリット酸など)、フェノール樹脂、フェノールノボラック樹脂、複素芳香環含有アミン(例えば、トリアジン環含有アミンなど)などの芳香族硬化剤;脂肪族アミン類(例えば、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン、1,3,6−トリスアミノメチルヘキサン、ポリメチレンジアミン、トリメチルヘキサメチレンジアミン、ポリエーテルジアミンなど)、脂環式アミン類(イソホロンジアミン、メンタンジアミン、N−アミノエチルピペラジン、3,9−ビス(3−アミノプロピル)2,4,8,10−テトラオキサスピロ(5,5)ウンデカンアダクト、ビス(4−アミノ−3−メチルシクロヘキシル)メタン、ビス(4−アミノシクロヘキシル)メタン、これらの変性品など)、ポリアミン類とダイマー酸からなる脂肪族ポリアミドアミンなどの非芳香族硬化剤が挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0023】
これらのうち、均一な三次元網目構造と均一な空孔を形成するため、また膜強度と弾性率を確保するために、分子内に一級アミンを2つ以上有するメタフェニレンジアミン、ジアミノジフェニルメタン、及びジアミノジフェニルスルホンからなる群より選択される少なくとも1種の芳香族アミン硬化剤;分子内に一級アミンを2つ以上有するビス(4−アミノ−3−メチルシクロヘキシル)メタン、及びビス(4−アミノシクロヘキシル)メタンからなる群より選択される少なくとも1種の脂環式アミン硬化剤を用いることが好ましい。
【0024】
また、エポキシ樹脂と硬化剤の組み合わせとしては、芳香族エポキシ樹脂と脂環式アミン硬化剤の組み合わせ、又は、脂環式エポキシ樹脂と芳香族アミン硬化剤の組み合わせが好ましい。これらの組み合わせにより、得られるエポキシ樹脂多孔質膜の耐熱性が高くなり、複合半透膜の多孔性支持体として好適に用いられる。
【0025】
本実施形態で使用できるポロゲンとは、エポキシ樹脂及び硬化剤を溶かすことができ、かつ、エポキシ樹脂と硬化剤が重合した後、反応誘起相分離を生ぜしめることが可能な溶剤をいい、例えば、メチルセロソルブ、エチルセロソルブなどのセロソルブ類、エチレングリコールモノメチルエーテルアセテート、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、及びポリエチレングリコール、ポリプロピレングリコールなどのグリコール類などが挙げられる。これらは1種単独で用いてもよく、2種以上を併用してもよい。
【0026】
これらのうち、均一な三次元網目構造と均一な空孔を形成するために、メチルセロソルブ、エチルセロソルブ、分子量600以下のポリエチレングリコール、エチレングリコールモノメチルエーテルアセテート、ポリプロピレングリコール、ポリオキシエチレンモノメチルエーテル、ポリオキシエチレンジメチルエーテル、及びプロピレングリコールモノメチルエーテルアセテートからなる群より選択される少なくとも1種を用いることが好ましく、特に分子量200以下のポリエチレングリコール、分子量500以下のポリプロピレングリコール、ポリオキシエチレンモノメチルエーテル、及びプロピレングリコールモノメチルエーテルアセテートからなる群より選択される少なくとも1種を用いることが好ましい。
【0027】
また、個々のエポキシ樹脂又は硬化剤と常温で不溶又は難溶である溶剤であっても、エポキシ樹脂と硬化剤との反応物が可溶である溶剤についてはポロゲンとして使用可能である。このようなポロゲンとしては、例えば臭素化ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、「エピコート5058」)などが挙げられる。
【0028】
エポキシ樹脂多孔質膜の空孔率、平均孔径、孔径分布などは、使用するエポキシ樹脂、硬化剤、ポロゲンなどの原料の種類や配合比率、及び反応誘起相分離時における加熱温度や加熱時間などの反応条件により変化するため、目的とする空孔率、平均孔径、孔径分布を得るために系の相図を作成して最適な条件を選択することが好ましい。また、相分離時におけるエポキシ樹脂架橋体の分子量、分子量分布、系の粘度、架橋反応速度などを制御することにより、エポキシ樹脂架橋体とポロゲンとの共連続構造を特定の状態で固定し、安定した多孔構造を得ることができる。
【0029】
また、エポキシ樹脂多孔質膜を構成する全炭素原子に対する芳香環由来の炭素原子比率が0.1〜0.65の範囲になるように、エポキシ樹脂及び硬化剤の種類と配合割合を決定することが好ましい。上記値が0.1未満の場合には、エポキシ樹脂多孔質膜の特性である分離媒体の平面構造の認識性が低下する傾向にある。一方、0.65を超える場合には、均一な三次元網目構造を形成することが困難になる。
【0030】
また、エポキシ樹脂に対する硬化剤の配合割合は、エポキシ基1当量に対して硬化剤当量が0.6〜1.5であることが好ましい。硬化剤当量が0.6未満の場合には、硬化体の架橋密度が低くなり、耐熱性、耐溶剤性などが低下する傾向にある。一方、1.5を超える場合には、未反応の硬化剤が残留し、架橋密度の向上を阻害する傾向にある。なお、本実施形態では、上述した硬化剤の他に、目的とする多孔構造を得るために、溶液中に硬化促進剤を添加してもよい。硬化促進剤としては、公知のものを使用することができ、例えば、トリエチルアミン、トリブチルアミンなどの三級アミン、2−フェノール−4−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェノール−4,5−ジヒドロキシイミダゾールなどのイミダゾール類などが挙げられる。
【0031】
エポキシ樹脂多孔質膜はより良いスキン層を形成するために、水銀圧入法で測定したときの平均孔径を0.01〜0.4μmとすることが好ましい。エポキシ樹脂多孔質膜の平均孔径を0.01〜0.4μmに調整するためには、エポキシ樹脂、硬化剤及びポロゲンの総重量に対してポロゲンを40〜80重量%用いることが好ましい。ポロゲンの量が40重量%未満の場合にはエポキシ樹脂多孔質膜の平均孔径が小さくなりすぎ、空孔が形成されなくなる傾向にある。一方、ポロゲンの量が80重量%を超える場合にはエポキシ樹脂多孔質膜の平均孔径が大きくなりすぎて均一なスキン層を多孔体上に形成することができず、塩阻止率が著しく低下する傾向にある。エポキシ樹脂多孔質膜の平均孔径は、0.05〜0.3μmであることが好ましく、より好ましくは0.05〜0.2μmである。そのためにはポロゲンを60〜70重量%用いることがより好ましく、特に好ましくは60〜65重量%である。
【0032】
また、エポキシ樹脂多孔質膜の平均孔径を0.01〜0.4μmに調整する方法として、エポキシ当量の異なる2種以上のエポキシ樹脂を混合して用いる方法も好適である。その際、エポキシ当量の差は100以上であることが好ましい。
【0033】
また、エポキシ樹脂多孔質膜の平均孔径は、全体のエポキシ当量とポロゲンの割合、硬化温度などの諸条件を適宜設定することにより目的の範囲に調整できる。
【0034】
エポキシ樹脂多孔質膜は、例えば、以下の方法で作製することができる。
【0035】
1)エポキシ樹脂、硬化剤及びポロゲンを含むエポキシ樹脂組成物を基板上に塗布し、その後、塗布したエポキシ樹脂組成物を加熱してエポキシ樹脂を三次元架橋させる。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、得られたエポキシ樹脂シートからポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。使用する基板は特に制限されず、例えば、プラスチック基板、ガラス基板、及び金属板などが挙げられる。
【0036】
2)エポキシ樹脂、硬化剤、及びポロゲンを含むエポキシ樹脂組成物を基板上に塗布し、その後、塗布したエポキシ樹脂組成物上に別の基板を被せてサンドイッチ構造体を作製する。なお、基板間に一定の厚さを設けるために、基板の四隅にスペーサー(例えば、両面テープ)を設けておくことが好ましい。そして、該サンドイッチ構造体を加熱してエポキシ樹脂を三次元架橋させる。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、得られたエポキシ樹脂シートを取り出し、ポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。使用する基板は特に制限されず、例えば、プラスチック基板、ガラス基板、及び金属板などが挙げられるが、特にガラス基板を用いることが好ましい。
【0037】
3)エポキシ樹脂、硬化剤、及びポロゲンを含むエポキシ樹脂組成物を所定形状のモールド内に充填し、その後、反応を促進させてエポキシ樹脂を三次元架橋させて、円筒状又は円柱状樹脂ブロックを作製する。その際に、エポキシ樹脂架橋体とポロゲンとの相分離により共連続構造が形成される。その後、その円柱状樹脂ブロックを円筒軸又は円柱軸を中心に回転させながら該ブロックの表面を所定厚さで切削して長尺状のエポキシ樹脂シートを作製する。そして、エポキシ樹脂シート中のポロゲンを洗浄除去し、乾燥することにより、三次元網目構造及び連通する空孔を有するエポキシ樹脂多孔質膜を作製する。
【0038】
エポキシ樹脂組成物を硬化する際の条件(加熱硬化又は室温硬化など)は特に制限されないが、均一な孔を有する多孔シートを形成するためには、エポキシ樹脂組成物を室温で硬化させることが好ましい。エポキシ樹脂組成物を室温で硬化させる場合、20〜40℃程度で硬化を開始させ、硬化時間は3〜100時間程度であり、好ましくは20〜50時間程度である。エポキシ樹脂組成物を加熱硬化させる場合、温度は40〜120℃程度であり、好ましくは60〜100℃程度であり、硬化時間は10〜300分程度であり、好ましくは30〜180分程度である。
【0039】
得られたエポキシ樹脂シートからポロゲンを除去するために用いられる溶剤としては、例えば、水、DMF、DMSO、THF及びこれらの混合溶剤などが挙げられ、ポロゲンの種類に応じて適宜選択する。
【0040】
ポロゲンを除去したエポキシ樹脂多孔質膜の乾燥条件は特に制限されないが、温度は40〜120℃程度であり、乾燥時間は3分〜3時間程度である。
【0041】
エポキシ樹脂多孔質膜の厚さは、複合半透膜の製造又は使用に必要な強度及び実用性を満たす限り、特に制限されない。ところで、複合半透膜のスキン層は溶質を透過させないため、スキン層付近では溶質濃度の高い流体が溜まりやすく、複合半透膜の厚さ方向に溶質濃度の偏りが生じる。濃度分極と呼ばれるこのような溶質濃度の偏りは、一般に複合半透膜の透過流量を減らし、正浸透膜流動システムの効率を低下させてしまう。本発明者らは、このエポキシ樹脂多孔質膜の厚さが薄い方が、濃度分極が抑制されやすいことを見出した。一方で、エポキシ樹脂多孔質膜の厚さが薄すぎると、エポキシ樹脂多孔質膜上にスキン層を形成する際に、エポキシ樹脂多孔質膜の取り扱いが困難となる。また、エポキシ樹脂多孔質膜の厚さが薄すぎると、正浸透処理、特に、PRO処理行う場合に、エポキシ樹脂多孔質膜が破れる場合がある。このような観点から、エポキシ樹脂多孔質膜の厚さは、例えば10μm以上であり、20μm以上が好ましく、25μm以上がより好ましい。また、エポキシ樹脂多孔質膜の厚さは、例えば150μm以下であり、100μm以下が好ましく、60μm以下がより好ましい。また、エポキシ樹脂多孔質膜の裏面を織布、不織布などで補強してもよい。
【0042】
一方、本実施形態におけるエポキシ樹脂多孔質膜上に形成するスキン層はポリアミド系樹脂からなるものであり、多官能アミン成分と多官能酸ハライド成分とを重合してなるポリアミド系樹脂を含むスキン層であることが好ましい。
【0043】
多官能アミン成分とは、2以上の反応性アミノ基を有する多官能アミンであり、芳香族、脂肪族、及び脂環式の多官能アミンが挙げられる。
【0044】
芳香族多官能アミンとしては、例えば、m−フェニレンジアミン、p−フェニレンジアミン、o−フェニレンジアミン、1,3,5−トリアミノベンゼン、1,2,4−トリアミノベンゼン、3,5−ジアミノ安息香酸、2,4−ジアミノトルエン、2,6−ジアミノトルエン、N,N’−ジメチル−m−フェニレンジアミン、2,4−ジアミノアニソール、アミドール、キシリレンジアミン等が挙げられる。
【0045】
脂肪族多官能アミンとしては、例えば、エチレンジアミン、プロピレンジアミン、トリス(2−アミノエチル)アミン、n−フェニル−エチレンジアミン等が挙げられる。
【0046】
脂環式多官能アミンとしては、例えば、1,3−ジアミノシクロヘキサン、1,2−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、ピペラジン、2,5−ジメチルピペラジン、4−アミノメチルピペラジン等が挙げられる。
【0047】
これらの多官能アミンは1種で用いてもよく、2種以上を併用してもよい。高塩阻止性能のスキン層を得るためには、芳香族多官能アミンを用いることが好ましい。
【0048】
多官能酸ハライド成分とは、反応性カルボニル基を2個以上有する多官能酸ハライドである。
【0049】
多官能酸ハライドとしては、芳香族、脂肪族、及び脂環式の多官能酸ハライドが挙げられる。
【0050】
芳香族多官能酸ハライドとしては、例えば、トリメシン酸トリクロライド、テレフタル酸ジクロライド、イソフタル酸ジクロライド、ビフェニルジカルボン酸ジクロライド、ナフタレンジカルボン酸ジクロライド、ベンゼントリスルホン酸トリクロライド、ベンゼンジスルホン酸ジクロライド、クロロスルホニルベンゼンジカルボン酸ジクロライド等が挙げられる。
【0051】
脂肪族多官能酸ハライドとしては、例えば、プロパンジカルボン酸ジクロライド、ブタンジカルボン酸ジクロライド、ペンタンジカルボン酸ジクロライド、プロパントリカルボン酸トリクロライド、ブタントリカルボン酸トリクロライド、ペンタントリカルボン酸トリクロライド、グルタリルハライド、アジポイルハライド等が挙げられる。
【0052】
脂環式多官能酸ハライドとしては、例えば、シクロプロパントリカルボン酸トリクロライド、シクロブタンテトラカルボン酸テトラクロライド、シクロペンタントリカルボン酸トリクロライド、シクロペンタンテトラカルボン酸テトラクロライド、シクロヘキサントリカルボン酸トリクロライド、テトラハイドロフランテトラカルボン酸テトラクロライド、シクロペンタンジカルボン酸ジクロライド、シクロブタンジカルボン酸ジクロライド、シクロヘキサンジカルボン酸ジクロライド、テトラハイドロフランジカルボン酸ジクロライド等が挙げられる。
【0053】
これら多官能酸ハライドは1種で用いてもよく、2種以上を併用してもよい。高い塩阻止性能のスキン層を得るためには、芳香族多官能酸ハライドを用いることが好ましい。また、多官能酸ハライド成分の少なくとも一部に3価以上の多官能酸ハライドを用いて、架橋構造を形成するのが好ましい。
【0054】
また、ポリアミド系樹脂を含むスキン層の性能を向上させるために、ポリビニルアルコール、ポリビニルピロリドン、ポリアクリル酸などのポリマー、ソルビトール、グリセリンなどの多価アルコールなどを共重合させてもよい。
【0055】
ポリアミド系樹脂を含むスキン層をエポキシ樹脂多孔質膜の表面に形成する方法は特に制限されず、あらゆる公知の手法を用いることができる。例えば、界面縮合法、相分離法、薄膜塗布法などが挙げられる。界面縮合法とは、具体的には、多官能アミン成分を含有するアミン水溶液と、多官能酸ハライド成分を含有する有機溶液とを接触させて界面重合させることによりスキン層を形成し、そのスキン層をエポキシ樹脂多孔質膜上に載置する方法、又は、エポキシ樹脂多孔質膜上での界面重合によりポリアミド系樹脂のスキン層をエポキシ樹脂多孔質膜上に直接形成する方法などである。かかる界面縮合法の条件等の詳細は、特開昭58−24303号公報、特開平1−180208号公報等に記載されており、それらの公知技術を適宜採用することができる。
【0056】
本実施形態において、多官能アミン成分を含むアミン水溶液からなる水溶液被覆層をエポキシ樹脂多孔質膜上に形成し、次いで多官能酸ハライド成分を含有する有機溶液と水溶液被覆層とを接触させて界面重合させることによりスキン層を形成する方法が好ましい。
【0057】
界面重合法において、アミン水溶液中の多官能アミン成分の濃度は特に制限されないが、0.1〜5重量%であることが好ましく、さらに好ましくは1〜4重量%である。多官能アミン成分の濃度が低すぎる場合にはスキン層にピンホール等の欠陥が生じやすくなり、また塩阻止性能が低下する傾向にある。一方、多官能アミン成分の濃度が高すぎる場合には、膜厚が厚くなりすぎて透過抵抗が大きくなって透過流束が低下する傾向にある。
【0058】
有機溶液中の多官能酸ハライド成分の濃度は特に制限されないが、0.01〜5重量%であることが好ましく、さらに好ましくは0.05〜3重量%である。多官能酸ハライド成分の濃度が低すぎる場合には、未反応多官能アミン成分が残留しやすくなり、スキン層にピンホール等の欠陥が生じやすくなって塩阻止性能が低下する傾向にある。一方、多官能酸ハライド成分の濃度が高すぎる場合には、未反応多官能酸ハライド成分が残留しやすくなり、膜厚が厚くなりすぎて透過抵抗が大きくなり、透過流束が低下する傾向にある。
【0059】
上述の有機溶液に用いられる有機溶媒としては、水に対する溶解度が低く、エポキシ樹脂多孔質膜を劣化させず、多官能酸ハライド成分を溶解するものであれば特に限定されず、例えば、シクロヘキサン、ヘプタン、オクタン、及びノナン等の飽和炭化水素、1,1,2−トリクロロトリフルオロエタン等のハロゲン置換炭化水素などを挙げることができる。好ましくは沸点が300℃以下、さらに好ましくは沸点が200℃以下の飽和炭化水素である。
【0060】
上述のアミン水溶液や有機溶液には、製膜を容易にし、得られる複合半透膜の性能を向上させるための目的で各種の添加剤を加えることができる。添加剤としては、例えば、ドデシルベンゼンスルホン酸ナトリウム、ドデシル硫酸ナトリウム及びラウリル硫酸ナトリウム等の界面活性剤、重合により生成するハロゲン化水素を除去する水酸化ナトリウム、リン酸三ナトリウム及びトリエチルアミン等の塩基性化合物、アシル化触媒、特開平8−224452号公報記載の溶解度パラメータが8〜14(cal/cm3)1/2の化合物などが挙げられる。
【0061】
エポキシ樹脂多孔質膜上にアミン水溶液を塗布してから有機溶液を塗布するまでの時間は、アミン水溶液の組成、粘度及びエポキシ樹脂多孔質膜の表面の孔径にもよるが、1〜180秒程度であり、好ましくは2〜120秒であり、より好ましくは2〜40秒であり、特に好ましくは2〜10秒である。溶液の塗布間隔が長すぎる場合には、アミン水溶液がエポキシ樹脂多孔質膜の内部深くまで浸透・拡散し、未反応多官能アミン成分がエポキシ樹脂多孔質膜中に大量に残存する可能性がある。また、エポキシ樹脂多孔質膜の内部深くまで浸透した未反応多官能アミン成分は、その後の膜洗浄処理でも除去し難い傾向にある。溶液の塗布間隔が短すぎる場合には、余分なアミン水溶液が残存しすぎてしまい、膜性能が低下する傾向にある。
【0062】
本実施形態においては、アミン水溶液からなる水溶液被覆層と有機溶液との接触後、エポキシ樹脂多孔質膜上の過剰な有機溶液を除去し、エポキシ樹脂多孔質膜上の形成膜を70℃以上で加熱乾燥してスキン層を形成することが好ましい。形成膜を加熱処理することによりその機械的強度や耐熱性等を高めることができる。加熱温度は70〜200℃であることがより好ましく、特に好ましくは80〜130℃である。加熱時間は30秒〜10分程度が好ましく、さらに好ましくは40秒〜7分程度である。
【0063】
エポキシ樹脂多孔質膜上に形成したスキン層の厚さは特に制限されないが、通常0.05〜2μm程度であり、好ましくは、0.1〜1μmである。
【0064】
複合半透膜の厚さは特に制限されない。強度、実用的な透水性及び塩阻止性の観点から、表面処理が行われてもよく、補強材が積層されてもよい。複合半透膜の厚さは、例えば10〜250μmであり、好ましくは10〜150μmである。
【0065】
また、この複合半透膜の形状は特に制限されない。すなわち、平膜状、スパイラルエレメント状など、公知の形態に加工して用いればよい。
【0066】
本実施形態の正浸透膜流動システムは、例えば、特開2003−176775に記載の浸透圧発電システムや、特開2005−279540に記載の海水淡水化における濃縮水の希釈システムとして使用することができる。このとき、上述の浸透圧発電システムにおける発電量と比例する出力密度[W/m2]は、正浸透時の透過流束によって簡易的に求めることができる。
【実施例】
【0067】
以下に本発明の効果について具体例を用いて詳細に説明する。なお、本発明はこれらの実施例に限定されるものではない。
【0068】
<実施例1>
(エポキシ樹脂多孔質基材の作製)
ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート828)139重量部、ビスフェノールA型エポキシ樹脂(ジャパンエポキシレジン社製、エピコート1010)93.2重量部、ビス(4−アミノシクロヘキシル)メタン52重量部、及びポリエチレングリコール200(三洋化成工業社製)500重量部を調製したエポキシ樹脂組成物を、円筒状モールド(外径35cm、内径10.5cm)内に高さ30cmまで充填して25℃で12時間室温硬化し、さらに130℃で18時間反応硬化させて円筒状樹脂ブロックを作製した。この樹脂ブロックを、円筒軸を中心に回転させながら切削装置(東芝機械社製)を用いて、その表面から厚さ145μmで連続的にスライスし、長尺状のエポキシ樹脂シート(長さ:100m)を得た。このエポキシ樹脂シートを純水中に12時間浸漬してポリエチレングリコールを除去し、50℃の乾燥機内で約4時間乾燥することでエポキシ樹脂多孔質膜(厚さ130μm、空孔率45%、平均孔径0.04μm)を得た。
【0069】
(複合半透膜の作製)
m-フェニレンジアミン3.0g、ラウリル硫酸ナトリウム0.15g、ベンゼンスルホン酸6.0g、トリエチルアミン3.0g、及び水87.85gを混合して水溶液(B)を調製した。水溶液(B)を大気圧プラズマ処理したエポキシ樹脂多孔質膜上に塗布して余分なアミン水溶液を除去した。次に、トリメシン酸クロライド0.2%を含むイソオクタン溶液をエポキシ樹脂多孔質膜上に塗布した。その後、余分なイソオクタン溶液を除去して100℃の乾燥器内で2分間保持することで、エポキシ樹脂多孔質膜上にポリアミドからなるスキン層(厚さ約200nm)を形成して、実施例1の複合半透膜を得た。最終的に得られた実施例1に係る複合半透膜の厚さは130μmであった。
【0070】
<実施例2>
エポキシ樹脂多孔質膜の作製において、樹脂ブロックの切削厚さを90μmに変更した点以外は、実施例1と同様にして実施例2の複合半透膜を得た。最終的に得られた実施例2に係る複合半透膜の厚さは80μmであった。
【0071】
<実施例3>
エポキシ樹脂多孔質膜の作製において、樹脂ブロックの切削厚さを55μmに変更した点以外は、実施例1と同様にして実施例2の複合半透膜を得た。最終的に得られた実施例3に係る複合半透膜の厚さは50μmであった。
【0072】
<比較例1>
ポリエステル製不織布(70g/m2、厚さ90μm)上にポリスルホン(ソルベイアドバンストポリマーズ社製、P‐3500)18.3重量%とジメチルホルムアルデヒド81、7重量%の混合液を塗布した。その後、その混合液を塗布したポリエステル製不織布を20℃の純水に浸漬し、さらに、45℃の純水に浸漬した。このようにして、約130μmの厚さのポリスルホン支持膜を得た。実施例1の複合半透膜の作成の工程で用いられた水溶液(B)をポリスルホン支持膜上に塗布し、実施例1と同様に処理して、比較例1の複合半透膜を得た。最終的に得られた比較例1に係る複合半透膜の厚さは130μmであった。
【0073】
(逆浸透方式によるNaCl阻止率の測定)
作製した実施例1〜3及び比較例1の複合半透膜を逆浸透膜として用いて、1500mg/LのNaCl水溶液を原水として、25℃、pH6.5、操作圧力1.5MPaの条件で逆浸透方式による透過試験を行った。その結果、NaCl阻止率99%であった。なお、NaCl阻止率は下記式で求めた。
NaCl阻止率(%)=(1−(逆浸透膜透過液中のNaCl濃度/原水中のNaCl濃度))×100
【0074】
(正浸透方式による透過流束の測定)
作製した平膜状の実施例1〜3及び比較例1の複合半透膜を所定の形状、サイズに切断し、平膜評価用のセル(日東電工株式会社製、C10−T)にセットした。NaCl135gを溶解させた5LのNaCl水溶液が入ったタンクを準備し、ポリアミドからなるスキン層側に220ml/minの流量でNaCl水溶液を供給した。また、5Lの純水が入ったタンクを準備し、エポキシ樹脂多孔質膜側又はポリスルホン支持膜の不織布側に純水を60ml/minの流量で循環供給した。この操作によって純水タンクの減少重量を計測し、減少重量を透過流束とした。
【0075】
実施例1〜3及び比較例1に係る複合半透膜の上記の測定の測定結果を表1に示す。
【表1】
【0076】
上記の通り、実施例1〜3及び比較例1に係る複合半透膜の逆浸透方式の透過流束は同程度であった。これに対し、実施例1〜3に係る複合半透膜の正浸透方式の透過流束は、比較例1に係る複合半透膜の正浸透方式の透過流束の2倍以上であった。また、実施例1〜3において、複合半透膜の厚さが薄いほど正浸透方式の透過流束は大きくなる傾向があった。なお、エポキシ樹脂多孔質膜の厚さが複合半透膜の厚さの大部分を占めるので、エポキシ樹脂多孔質膜の厚さが薄いほど正浸透方式の透過流束が大きくなる傾向があるということもできる。従って、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜を用いて正浸透膜流動システムを構成することにより、正浸透膜流動システムの効率が向上することが示唆された。
【産業上の利用可能性】
【0077】
本発明に係る正浸透膜流動システムは、例えば、タービンを回転させて発電するシステム、又は、高浸透圧流体を希釈するシステム等に使用することができる。
【符号の説明】
【0078】
1 正浸透膜流動システム
2 高浸透圧流体流動部
3 低浸透圧流体流動部
4 半透膜
5 タービン
【特許請求の範囲】
【請求項1】
高浸透圧流体が供給される高浸透圧流体流動部と、
前記高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部と、
前記高浸透圧流体流動部と前記低浸透圧流体流動部とを隔てる半透膜と、を備え、
前記低浸透圧流体流動部から前記高浸透圧流体流動部に前記半透膜を介して流体移動が生じることにより前記高浸透圧流体流動部の流量を増加させ、
前記半透膜が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である、正浸透膜流動システム。
【請求項2】
前記エポキシ樹脂多孔質膜の厚さが10〜150μmである、請求項1に記載の正浸透膜流動システム。
【請求項3】
タービンをさらに備え、
前記高浸透圧流体流動部から流出した前記高浸透圧流体の圧力により前記タービンを回転させて発電する、請求項1又は2に記載の正浸透膜流動システム。
【請求項4】
前記高浸透圧流体流動部において、前記高浸透圧流体は、前記高浸透圧流体の浸透圧と前記低浸透圧流体の浸透圧との差よりも低い圧力が加えられている、請求項1〜3のいずれか1項に記載の正浸透膜流動システム。
【請求項5】
前記高浸透圧流体に加えられる圧力が、0.1MPa以上3.0MPa以下である、請求項4記載の正浸透膜流動システム。
【請求項6】
前記複合半透膜の前記ポリアミド系スキン層側に前記低浸透圧流体を供給する、請求項1〜5のいずれか1項に記載の正浸透膜流動システム。
【請求項7】
エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した、正浸透膜流動システム用複合半透膜。
【請求項1】
高浸透圧流体が供給される高浸透圧流体流動部と、
前記高浸透圧流体の浸透圧よりも浸透圧が低い低浸透圧流体が供給される低浸透圧流体流動部と、
前記高浸透圧流体流動部と前記低浸透圧流体流動部とを隔てる半透膜と、を備え、
前記低浸透圧流体流動部から前記高浸透圧流体流動部に前記半透膜を介して流体移動が生じることにより前記高浸透圧流体流動部の流量を増加させ、
前記半透膜が、エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した複合半透膜である、正浸透膜流動システム。
【請求項2】
前記エポキシ樹脂多孔質膜の厚さが10〜150μmである、請求項1に記載の正浸透膜流動システム。
【請求項3】
タービンをさらに備え、
前記高浸透圧流体流動部から流出した前記高浸透圧流体の圧力により前記タービンを回転させて発電する、請求項1又は2に記載の正浸透膜流動システム。
【請求項4】
前記高浸透圧流体流動部において、前記高浸透圧流体は、前記高浸透圧流体の浸透圧と前記低浸透圧流体の浸透圧との差よりも低い圧力が加えられている、請求項1〜3のいずれか1項に記載の正浸透膜流動システム。
【請求項5】
前記高浸透圧流体に加えられる圧力が、0.1MPa以上3.0MPa以下である、請求項4記載の正浸透膜流動システム。
【請求項6】
前記複合半透膜の前記ポリアミド系スキン層側に前記低浸透圧流体を供給する、請求項1〜5のいずれか1項に記載の正浸透膜流動システム。
【請求項7】
エポキシ樹脂多孔質膜上にポリアミド系スキン層を形成した、正浸透膜流動システム用複合半透膜。
【図1】

【公開番号】特開2013−13888(P2013−13888A)
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願番号】特願2012−130622(P2012−130622)
【出願日】平成24年6月8日(2012.6.8)
【出願人】(000003964)日東電工株式会社 (5,557)
【Fターム(参考)】
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願日】平成24年6月8日(2012.6.8)
【出願人】(000003964)日東電工株式会社 (5,557)
【Fターム(参考)】
[ Back to top ]