
殺微生物剤としての新規ピラゾール−4−N−アルコキシカルボキサミド
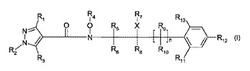
式(I)(ここで置換基は請求項1に記載するとおりである)は、殺微生物剤としての使用に適している。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、殺微生物剤として活性の、特に殺真菌剤として活性の、新規カルボキサミドに関する。更に、本発明は、これらの化合物の調製に使用される中間体、これらの化合物を含有する組成物、及び植物への植物病原性微生物、好ましくは真菌の繁殖を防除又は予防するための、農業又は園芸における、それらの使用に関する。
【背景技術】
【0002】
殺真菌剤として活性のカルボキサミドは、EP 1787981及びEP 1792901に記載されている。
【0003】
特定の置換基のパターンを有する新規のカルボキサミドが、殺微生物活性を有することが見出された。
【発明の概要】
【課題を解決するための手段】
【0004】
故に、本発明は、式I
【化1】
[式中、
R1はC1-C4アルキル又はC1-C4ハロアルキルであり;
R2はC1-C4アルキルであり;
R3は水素又はハロゲンであり;
R4は水素、C1-C4アルキル又はC1-C4ハロゲンアルキルであり;
R5、R6、R8、R9及びR10は、互いに独立して、水素、ハロゲン、C1-C4アルキル又はC1-C4ハロアルキルであり;
R7は水素、ハロゲン、C1-C4アルキル、C2-C6アルケニル又はC3-C6アルキニルであり;
R11は水素、ハロゲン又はC1-C6アルキルであり;
R12は水素、ハロゲン、C1-C6アルキル、C2-C6アルケニル、C3-C6アルキニル、C3-C6シクロアルキル-C3-C6アルキニル、ハロフェノキシ、ハロフェニル-C3-C6アルキニル、C(C1-C4アルキル)=NO-C1-C4アルキル、C1-C6ハロアルキル、C1-C6ハロアルコキシ、C2-C6ハロアルケニル、又はC2-C6ハロアルケニルオキシであり;
R13は水素、ハロゲン、C1-C6アルキルであり;
Xは酸素又は硫黄又は存在せず;但しXが酸素又は硫黄であるときR7はハロゲンではなく;
nは0又は1である]
の化合物、及びその農学的に許容される塩/異性体/構造異性体/立体異性体/ジアステレオ異性体/鏡像異性体/互換異性体、並びにそれらの化合物のN-オキシドに関する。
【0005】
前記置換基の定義にあるアルキル基は、直鎖又は分岐鎖であることができ、例えば、メチル、エチル、n-プロピル、n-ブチル、n-ペンチル、n-ヘキシル、iso-プロピル、n-ブチル、sec-ブチル、iso-ブチル又はtert-ブチルである。前記アルコキシ、アルケニル及びアルキニル基は、上記アルキル基に由来する。前記アルケニル及びアルキニル基は、一不飽和又は二不飽和であることができる。前記置換基の定義にあるシクロアルキル基は、例えば、シクロプロピル、シクロブチル、シクロペンチル又はシクロヘキシルである。ハロゲンは、一般に、フッ素、塩素、臭素又はヨウ素であって、好ましくはフッ素、臭素又は塩素である。これは、他の語との組合せ、例えばハロゲンアルキル又はハロゲンアルコキシ等の中のハロゲンにも当てはまる。ハロアルキル基は、好ましくは、1〜4個の炭素原子の長さの鎖を有する。ハロアルキルは、例えば、フルオロメチル、ジフルオロメチル、トリフルオロメチル、クロロメチル、ジクロロメチル、トリクロロメチル、2,2,2-トリフルオロエチル、2-フルオロエチル、2-クロロエチル、ペンタフルオロエチル、1,1-ジフルオロ-2,2,2-トリクロロエチル、2,2,3,3-テトラフルオロエチル及び2,2,2-トリクロロエチル;好ましくはトリクロロメチル、ジフルオロクロロメチル、ジフルオロメチル、トリフルオロメチル及びジクロロフルオロメチルである。アルコキシは、例えば、メトキシ、エトキシ、プロポキシ、i-プロポキシ、n-ブトキシ、iso-ブトキシ、sec-ブトキシ及びtert-ブトキシ;好ましくはメトキシ及びエトキシである。ハロゲンアルコキシは、例えば、フルオロメトキシ、ジフルオロメトキシトリフルオロメトキシ、2,2,2-トリフルオロエトキシ、1,1,2,2-テトラフルオロエトキシ、2-フルオロエトキシ、2-クロロエトキシ、2,2-ジフルオロエトキシ及び2,2,2-トリクロロエトキシ;好ましくはジフルオロメトキシ、2-クロロエトキシ及びトリフルオロメトキシである。
【0006】
好ましい式Iの化合物において、互いに独立して、
a)R1はジフルオロメチル、トリフルオロメチル又はメチル;
b)R2はメチル;
c)R3は水素又はフルオロ;
d)R4は水素、メチル又はエチル;
e)R4はメチル;
f)R5は水素又はメチル;
g)nは0;
h)Xは酸素;
i)R8、R9及びR10は水素;
j)R11、R12及びR13は水素又はクロロ;
k)R12はクロロ又はC1-C4アルキル;
I)R6は水素;そして
m)R7はメチル
である。
【0007】
特に好ましい式Iの化合物は、
R1はジフルオロメチル又はトリフルオロメチル;
R2はメチル;
R3は水素;
R4はメチル;
R11、R12及びR13は、互いに独立して、水素又はハロゲン、好ましくは水素又は塩素である。
【0008】
更に好ましい式Iの化合物は、
Xが酸素で、同時にR7がC1-C4アルキル、好ましくはメチル;又はXが存在せずR7が水素である。
【0009】
式Iの化合物は、式II
【化2】
で表され、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が式Iで定義したものである化合物を、式III
【化3】
で表され、R1、R2及びR3が式Iで定義したもので、R*がハロゲン、ヒドロキシ又はC1-6アルコキシ、好ましくはクロロである化合物と反応させることにより、調製されてもよい。
【0010】
式Iの化合物を得るための反応は、非プロトン性の有機溶媒中で実施されるのが有利である。そのような溶媒は、ベンゼン、トルエン、キシレン若しくはシクロヘキサン等の炭化水素、ジクロロメタン、トリクロロメタン、テトラクロロメタン又はクロロベンゼン等の塩素化炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、アセトニトリル又はプロピオニトリル等のニトリル、N,N-ジメチルホルムアミド、ジエチルホルムアミド若しくはN-メチルピロリジノン等のアミドである。前記反応の温度は、-20℃〜+120℃の間が有利である。一般に、前記反応は僅かに発熱性であるので、通常、常温で実施されることができる。反応時間を短縮するために、又は反応を開始するために、混合物が沸点近くまで短時間加熱されてもよい。前記反応時間は、反応触媒として数滴の塩基を添加することによっても短縮できる。適切な塩基は、特に、第三級アミン、例えばトリメチルアミン、トリエチルアミン、キヌクリジン(quinuclidine)、1,4−ジアザビシクロ[2.2.2]オクタン、1,5−ジアザビシクロ[4.3.0]ノン-5−エン又は1,5−ジアザビシクロ[5.4.0]ウンデク-7−エンである。しかしながら、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基も、塩基として使用されてもよい。前記塩基は、単独で、あるいは触媒として使用される量の相間移動触媒、例えばクラウンエーテル特に18−クラウン−6、又はテトラアルキルアンモニウム塩等と共に使用できる。
【0011】
R*がヒドロキシのとき、ベンゾトリアゾール-
1−イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート、bis-(2−オキソ−3−オキサゾリジニル)−ホスフィン酸塩化物(BOP-CI)、N,N'-ジシクロヘキシルカルボジイミド(DCC)又は1,1’−ジイミダゾール(CDI)等のカップリング剤が使用されてもよい。
【0012】
式II
【化4】
で表され、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が式Iで定義したものであり、好ましくはR4がC1-C4アルキルである中間体は、新規であって、式Iの化合物の調製のために特別に開発された。故に、式IIの中間体は、本発明の対象の部分を形成する。
【0013】
式Iの化合物の好ましい置換基の設定は、式IIの化合物にも適用される。故に、好ましい式IIの化合物は、互いに独立して、
a)R4は水素、メチル又はエチル;特に好ましくはメチル;
b)R5は水素又はメチル;
c)nは0;
d)Xは酸素;
e)R8、R9及びR10は水素;
f)R11、R12及びR13は水素又はクロロ;
g)R12はクロロ又はC1-C4アルキル;
h)R6は水素;そして
i)R7はメチル
である。
【0014】
式IIA
【化5】
で表され、R4、R5、X、R7、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム1に記載のようにして調製されてもよい。
【0015】
【化6】
式VIで表され、R5、R11、R12及びR13が式IIAで定義したものであるニトロアルケンは、常温と対流温度の間の温度で、酢酸及び酢酸アンモニウムの存在下、式Vで表され、R5が式IIAで定義したものであるニトロアルカンを、式(IV)で表され、R11、R12及びR13が式IIAで定義したものであるカルボニル化合物と、Henry反応(ニトロアルドール反応)させることにより、調製できる。
【0016】
式VIIaで表され、R7及び式Iで定義したものであり、更にMがLi、Na、K又は水素である化合物の、式VIで表されるニトロアルケンへのMichael付加は、対応するアルコール、チオール、トルエン又はエーテル溶媒、例えばジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン中のアルカリ土類アルコラート、好ましくはナトリウム、カリウム、リチウム塩等を使用して達成されてもよく、これにより、式VIIIで表され、R5、R7、R11、R12及びR13が式IIAで定義したものであるニトロアルカンを形成する。
【0017】
式VIで表され、R5、R11、R12及びR13が式IIAで定義したものであるニトロアルケンは、鉄及び塩酸を用いて還元されてもよく、これにより、式IXで表され、R5、R11、R12及びR13が式IIAで定義したものであるオキシムが得られる。前記オキシムは、式Xaで表され、R5、R11、R12及びR13が式IIAで定義したものであるケトンに加水分解できる。これは、例えば、M. Kulka and H. Hibbert J. Am. Chem. Soc. 65, 1180 (1943)及びPrasun K. Pradhan et al. Synthetic Commun., 35, 913-922, 2005に記載されている。前記反応は、40〜100℃の間の温度で、メタノール、エタノール、tert-ブチル、トリフルオロエタノール又はジオキサン等の、手近な有機溶媒中で実施される。
【0018】
式Xaで表されるケトンを、R7−Yで表され、R7が式IIAで定義したものであり、Yが、ハロゲン、メシレート又はトシレート等の離脱基を表す化合物を用いて塩基の存在下でアルキル化することにより、式Xbで表され、R5、R7、R11、R12及びR13が式IIAで定義したものであるα-アルキル化ケトンが得られる。前記アルキル化反応は、非プロトン性の不活性有機溶媒中で実施するのが有利である。そのような溶媒は、ベンゼン、トルエン、キシレン又はシクロヘキサン等の炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、N,N-ジメチルホルムアミド、ジエチルホルムアミド又はN-メチルピロリドン等のアミドである。反応温度は、−20℃〜+120℃の間である。適切な塩基は、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基である。前記塩基は、単独で、あるいは触媒として使用される量の相間移動触媒、例えばクラウンエーテル特に18−クラウン−6、又はテトラアルキルアンモニウム塩等と共に使用できる。
【0019】
式XIIa、XIIbで表されるO-アルコキシオキシム誘導体は、式Xa及びXbで表されるケトンを、式XIで表されるO-アルキルヒドロキシアミン誘導体又はその塩を用いてオキシム化することにより、調製されてもよい。前記オキシム化工程を実行するのに適した溶媒は、ベンゼン、トルエン、キシレン若しくはシクロヘキサン等の炭化水素、ジクロロメタン、トリクロロメタン、テトラクロロメタン又はクロロベンゼン等の塩素化炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、アセトニトリル又はプロピオニトリル等のニトリル、N,N-ジメチルホルムアミド、ジエチルホルムアミド若しくはN-メチルピロリジノン水又は混合物等のアミドである。前記反応温度は、−20℃〜+120℃の間であるのが有利である。一般に、前記反応は、常温で実施できる。適切な塩基は、具体的には、ピリジン、第三級アミン、例えばトリメチルアミン、トリエチルアミン、キヌクリジン、1,4−ジアザビシクロ[2.2.2]オクタン、1,5−ジアザビシクロ[4.3.0]ノン-5−エン又は1,5−ジアザビシクロ[5.4.0]ウンデク-7−エンである。しかしながら、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基も、塩基として使用されてもよい。
【0020】
式XIIIで表されるオキシム誘導体は、式VIで表されるニトロアルケンを、式VIIbで表されるアルコール又はチオール中で、SnCI2.2H2Oを使用するR.S. Varma and G.W. Kabalka Chem. Lett, 243-244 (1985)で報告された手順に従い、選択的に還元することにより調製されてもよい。式XIIcで表されるオキシムエーテル誘導体は、式XIIIで表されるオキシム誘導体を、R4−Yで表され、R4が式IIAで定義したものであり、Yが、ハロゲン、メシレート又はトシレート等の離脱基を表す化合物を用いて、塩基の存在下でO-アルキル化することにより調製されてもよい。あるいは、式XIIIcで表されるオキシムエーテル誘導体は、式XIVで表されるケトンを、式XIで表されるO-アルキルヒドロキシルアミン誘導体、又はその塩でオキシム化することによって調製されてもよい。
【0021】
式XIVで表されるケトンは、式VIIIで表されるニトロアルカンを、J. M Aizpurua and C. Palomo THL, Vol. 28, No.44,pp 5361-5364 (1987)に記載される手順に従い、Nef反応で変換することにより調製されてもよい。
【0022】
式IIAa、IIAb及びIIAcで表されるO-アルキルヒドロキシアミンは、式XIIa、XIIb及びXIIcで表されるO-アルコキシオキシム誘導体の還元により調製されてもよい。
【0023】
式IIA
【化7】
で表され、R4、R5、R7、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム2に記載されるように調製されてもよい。
【0024】
【化8】
式VIで表されるαアルキル化アルキルアリール酢酸塩誘導体は、塩基の存在下で、式XVのアリール酢酸誘導体をR7-Y等のハライドでアルキル化することにより合成でき、ここでR7は式IIAで定義したものであり、Yは、ハロゲン、メシレート、トシレート等の離脱基を表す。式XVIの化合物は、LiOH等の水酸化物により加水分解される。得られた式XVIIの酸を、対応するアシルクロライドに変換することができ、このアシルクロライドを、そのままN,O-ジメチルヒドロキシルアミンと反応させて、式XVIIIで表され、R7、R11、R12及びR13が式IIAで定義したものであるWeinrebアミドを得ることができる。続いての、式R5-MgBrで表され、が式IIAで定義したものであるグリニャール試薬との反応で、式Xbのケトンが得られ、これが、スキーム1に記載の反応により、式IIAbの化合物に変換されることができる。
【0025】
式Xb1のアルデヒド誘導体は、式XVIIIのWeinrebアミドを、LiAlH又はDIBAL-Hを用いて部分的に還元することにより調製されてもよい。
【0026】
式IIA
【化9】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム3に記載されるように調製されてもよい。
【0027】
【化10】
式Xa1のアルデヒド誘導体は、式XIXの酸誘導体を還元して式XXのアルコール誘導体を得て、続いてこれを式XXIの活性化ベンジル誘導体に変換し、続いてこれを式XXIIのニトリル誘導体に変換し、続いてこれを還元して、式Xa1のアルデヒド誘導体を得ることにより、調製されることができる。この過程は、「調製」の項に記載されている。続いての式XXIIのニトリル誘導体と、R5-MgBrで表され、R5が式IIAで定義したものであるグリニャール試薬と反応させて、式Xaのケトンを取得し、これを、スキーム1に記載の反応により、式IIAbの化合物に転換することができる。
【0028】
式IIB
【化11】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム4に記載されるように調製されてもよい。
【0029】
【化12】
式VIのアルデヒドは、Meldrum酸及びトリエチル蟻酸アンモニウムで、式XXIIIの対応する酸に変換することができる。この反応は、例えば、G.Toth et. al. in Synth. Commun. 25 (19), 3067-3074 (1995)に記載されている。この酸を、N,O-ジメチルヒドロキシルアミンとそのまま反応させて、式XXIVのWeinrebアミドが得られる。当該式XXIVのWeinrebアミドを、R5-MgYで表され、R5が式IIAで定義したものであるグリニャール試薬と反応させて、式XXVaのケトンが得られる。
【0030】
式XXVbのアルデヒドは、式XXIVのWeinrebアミドをLiAlH4又はDIBAL-Hで部分的に還元することにより調製されてもよい。あるいは、当該アルデヒドは、式XXVIのアルコールの酸化により調製されてもよい。適切な酸化剤として、ピリジニウムクロロクロメート(PCC)、Swern試薬(オキサリルクロライド/DMSO)、Dess-Martin Periodinane、及びMnO2が挙げられる。適切な溶媒として、ジクロロメタン及びTHFが挙げられる。反応温度は、典型的には、−50℃〜20℃の範囲内である。
【0031】
式XXVa及びXXVbのケトン及びアルデヒド誘導体は、更に、スキーム1に記載の反応により、式IIBa及びIIBbの化合物に変換することができる。
【0032】
式IIB
【化13】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム5に記載されるように調製されてもよい。
【0033】
【化14】
式XXIXのケトン又はアルデヒド誘導体は、式XXVIIIのアリル型アルコールを、水中の式XXVIIのアリールヨーダイドで、パラジウム触媒アリール化することにより、調製されてもよく、この方法は、例えばHong Zhao, Ming-Zhong Cai et. al. in Synth. Commun. 31 (23), 3665-3669 (2001); Alberto Scrivanti, Ugo Matteoli et. al. in Tetrahedron 64, 543- 548 (2008)に記載されている。式XXIXのケトン及びアルデヒド誘導体は、更に、スキーム1に記載の反応により、式IIBcの化合物に変換されることができる。
【0034】
化合物I及び適切な場合その互換異性体は、適切な場合、水和物及び/又は、現在固体状態の化合物の結晶化に使用された他の溶媒を含有する形態で得られることができる。
【0035】
本発明の式Iの化合物は、事実上、真菌、細菌又はウイルス等の植物病原性微生物により引き起こされる病害から有用植物を保護するのに、非常に有利な範囲の活性を有することが見出された。
【0036】
本発明は、有用植物への植物病原性微生物の繁殖を防除又は予防する方法に関し、当該方法において、式Iの化合物を、有効成分として、植物、その部分又はその生育場所に施用する。本発明の式Iの化合物は、低い施用率での優秀な活性、植物に対する良好な寛容性、及び環境安全性を特徴とする。それらは、非常に有用な、治療的、予防的及び浸透性の特徴を有し、多くの有用植物の保護に使用される。式Iの化合物は、様々な有用植物の作物の植物体又は植物の部分(果実、花、葉、茎、塊茎、根)に生じる病害を阻害又は死滅させるのに使用されるが、同時に、後日生育する植物の部分をも、植物病原性微生物から保護される。
【0037】
植物育苗材料、具体的には種子(果実、塊茎、穀物)及び挿し木(plant cutting)(例えばイネ)等を、真菌の繁殖、及び土壌中に発生する植物病原性の真菌から保護するための処理用のドレッシング剤として、式Iの化合物を使用することも可能である。
【0038】
更に、本発明の式Iの化合物は、関連する領域、例えば技術材料、例えば木材及び木材に関連する技術製品の保護、食料の保存、又は衛生管理において、真菌を防除するのに使用されてもよい。
【0039】
式Iの化合物は、例えば、以下のクラスの植物病原性の真菌に対して効果的である:
不完全菌類(例えばボトリティス(Botrytis)、ピリクラリア(Pyricularia)、ヘルミントスポリウム(Helminthosporium)、フサリウム(Fusarium)、セプトリア(Septoria)、ケルコスポラ(Cercospora)及びアルテルナリア(Alternaria)等)並びに担子菌類(例えばリゾクトニア(Rhizoctonia)、ヘミレイア(Hemileia)、プッキニア(Puccinia)。加えて、子嚢菌類のクラス(例えばウェンツリア(Venturia)及びエリシフェ(Erysiphe)、ポドスファエラ(Podosphaera)、モニリニア(Monilinia)、ウンキヌラ(Uncinula))並びに卵菌類(例えばフィトフトラ(Phytophthora)、ピチウム(Pythium)、プラスモパラ(Plasmopara))に対しても効果的である。うどん粉病菌(エリシフェ(Erysiphe spp.))に対して、卓越した活性が観察された。更に、当該式Iの新規化合物は、植物病原性の細菌及びウイルス、例えばキサントモナス(Xanthomonas spp)、シュードモナス(Pseudomonas spp)、エルウィニア・アミロウォラ(Erwinia amylovora)に対して、及びタバコモザイクウイルスに対して効果的である。アジアダイズサビ病(ファコプソラ・パキリジ(Phakopsora pachyrhizi))に対して、良好な活性が観察された。
【0040】
本発明の範囲内で、保護される有用植物は、典型的には、以下の植物種である:穀類(コムギ、オオムギ、ライムギ、オーツムギ、イネ、トウモロコシ、ソルガム及び関連する種);ビート(テンサイ及び飼料用テンサイ);漿果、石果及び軟果(リンゴ、洋ナシ、プラム、桃、アーモンド、桜桃、イチゴ、ラズベリー及びブラックベリー);マメ科植物(マメ、レンティル、エンドウ、ダイズ);油脂植物(アブラナ、カラシナ、ポピー、オリーブ、ヒマワリ、ココヤシ、トウゴマ、カカオ豆、ラッカセイ);ウリ科植物(カボチャ、キュウリ、メロン);線維植物(綿花;亜麻;ヘンプ;ジュート);柑橘類(オレンジ、レモン、グレープフルーツ、マンダリン);野菜(ホウレンソウ、レタス、アスパラガス、キャベツ、ニンジン、タマネギ、トマト、ジャガイモ、パプリカ);クスノキ科植物(アボカド、シナモン、樟脳)又はタバコ、ナッツ、コーヒー、ナス、サトウキビ、茶、コショウ、ブドウ、ホップ、バナナ及び天然ゴム、更に観葉植物。
【0041】
「有用植物」という用語は、公知の育種又は遺伝子組換えの方法の結果として、ブロモキシニル等の除草剤又は除草剤のクラス(例えばHPPD阻害剤、ALS阻害剤、例えばプリミスルフロン、プロスルフロン及びトリフロキシスルフロン、EPSPS(5−エノール−プロビル−シキメート−3−リン酸−シンターゼ)阻害剤、GS(グルタミンシンターゼ)阻害剤又はPPO(プロトポルフィリノーゲン−オキシダーゼ)阻害剤等)に対して耐性を獲得した有用植物をも含むものとして理解されたい。公知の育種方法(突然変異生成)によりイミダゾリノン、例えばイマザモックスに対して耐性を獲得した作物の一例として、Clearfield(登録商標)夏アブラナ(Canola)が挙げられる。遺伝子組換え手法により除草剤又は除草剤のクラスに対して耐性を獲得した作物の例として、RoundupReady(登録商標)、Herculex I(登録商標)及びLibertyLink(登録商標)の商標の下で市販される、グリホサート及びグルフォシネート耐性トウモロコシ変異体が含まれる。
【0042】
また、「有用植物」という用語は、組換えDNA技術を使用して、例えば毒素生産細菌に由来するものとして知られる1つ以上の選択的に作用する毒素、特にバチルス(Bacillus)属等に由来する毒素等を生産するように形質転換した有用植物をも含むものとして理解されたい。
【0043】
そのような植物の例として:YieldGard(登録商標)(CryIA(b)毒素を発現するトウモロコシ変異体);YieldGard Rootworm(登録商標)(CryIIIB(b1)毒素を発現するトウモロコシ変異体);YieldGard Plus(登録商標)(CryIA(b)及びCryIIIB(b1)毒素を発現するトウモロコシ変異体;Starlink(登録商標)(Cry9(c)毒素を発現するトウモロコシ変異体);Herculex I(登録商標)(CryIF(a2)毒素及び酵素ホスフィノツリシンN-アセチルトランスフェラーゼ(PAT)を発現し、グルフォシネートアンモニウムに対する耐性を得たトウモロコシ変異体);NuCOTN 33B(登録商標)(CryIA(c)毒素を発現する綿花変異体);Bollgard I(登録商標)(CryIA(c)毒素を発現する綿花変異体);Bollgard II(登録商標)(CryIA(c)及びCryIIA(b)毒素を発現する綿花変異体);VIPCOT(登録商標)(VIP毒素を発現する綿花変異体);NewLeaf(登録商標)(CryIIIA毒素を発現するジャガイモ変異体);Nature- Gard(登録商標)Agrisure(登録商標)GT Advantage(GA21グリホサート耐性形質), Agrisure(登録商標)CB Advantage (Bt11 アワノメイガ(CB)形質)、Agrisure(登録商標)RW (根きり虫(corn rootworm)形質)及びProtecta(登録商標)が挙げられる。
【0044】
また、「有用植物」という用語は、組換えDNA技術を使用して、いわゆる「病原性関連タンパク質」(PRP、例えばEP−A−0 392 225を参照されたい)等の、選択的活性を有する抗病原性物質を合成するように形質転換した有用植物をも含むものとして理解されたい。そのような抗病原性物質及びそのような抗病原性物質を合成できる組換え植物は公知であり、例えばEP−A−0 392 225、WO 95/33818、及びEP−A−0 353 191に記載されている。そのような遺伝子組換え植物を作製する方法は、当業者に周知であり、また、例えば上で示した文献中等に記載されている。
【0045】
本明細書中で使用されるとき、有用植物の「生育場所」という用語は、有用植物が生育する場所、有用植物の植物育苗材料が播種される場所、又は有用植物の植物育苗材料が土中に埋められる予定の場所を含む。そのような生育場所の一例が、作物植物が生育している土地である。
【0046】
「植物育苗材料」という用語は、子孫を繁殖させるのに使用され得る、種子、及び植物材料、例えば挿し木又は例えばジャガイモの塊茎等の、植物の生産的部分を意味するものとして理解されたい。狭義では種子を指すが、根、果実、塊茎、鱗茎、根茎、及び植物の部分をも指す。発芽後、又は土壌から出芽後に移植されるべき発芽した植物又は幼若植物も該当し得る。これらの幼若植物は、浸漬による全体又は部分処理により、移植前に保護される。好ましくは「植物育苗材料」は、種子を意味するものとして理解されたい。
【0047】
式Iの化合物は、未修飾の形態で使用することができ、又は、好ましくは、製剤の分野で伝統的に採用される担体及び助剤と共に使用することができる。
【0048】
従って、本発明は、式Iの化合物及び不活性の担体を含有する、植物病原性微生物を防除及び防御するための組成物、並びに、不活性の担体及び有効成分として式Iの化合物を含有する組成物が、植物、その部分又はその生育場所に適用される、有用植物への植物病原性微生物の繁殖を防除又は予防する方法にも関する。
【0049】
そのために、式Iの化合物及び不活性の担体は、公知の方法で、乳剤、被覆ペースト、直接スプレー可能な、又は希釈可能な溶液、希釈乳濁物、水和剤、可溶性の粉末、粉塵、顆粒、及びポリマー物質のカプセル等に、容易に製剤化される。前記組成物の種類と同様に、スプレー、散布(atomising)、散粉(dusting)、散乱(scattering)、コーティング又は注入等の施用方法が、本来の目的及び周囲の状況に従い選択される。また、前記組成物は、安定化剤、消泡剤、粘度調整剤、結合剤又は粘着付与剤(tackifier)等の更なる助剤、及び肥料、微量栄養素供与剤又は他の特別な機能を付与する製剤を含んでもよい。
【0050】
適切な担体及び助剤は個体又は液体であり、製剤技術において使用される、例えば天然又は再生された鉱物性物質、溶媒、分散剤、水和剤、粘着付与剤、増粘剤、結合剤又は肥料等の物質である。そのような担体は、例えばWO 97/33890に記載されている。
【0051】
式Iの化合物又は不活性の担体及び有効成分として式Iを含有する組成物は、処理される植物の生育場所又は植物体に、更なる化合物と同時に、又は連続して適用することができる。これらの更なる化合物は、例えば、肥料若しくは微量栄養素供与剤、又は植物の生育に影響する他の調製物等であることができる。また、それらは、選択的な除草剤、及び殺虫剤、殺真菌剤、殺細菌剤、殺線虫剤、殺軟体動物剤、又はこれらの調製物の幾つかの混合物であることができ、必要に応じて、更なる担体、界面活性剤、又は製剤分野で日常的に採用される施用を促進する助剤を加えてもよい。
【0052】
式Iの化合物又は不活性の担体及び有効成分として式Iを含有する組成物を施用する好ましい方法は、葉上施用である。施用の頻度及び施用率は、関連する病原体の繁殖のリスクに依存する。しかしながら、式Iの化合物は、液体製剤で植物の生育場所を濡らすことにより、又は顆粒等の固体形態の前記化合物を土壌に施用することにより(土壌施用)、土壌越しに根を通じて植物体に浸透させることもできる(全身作用)。水稲の作物において、そのような顆粒を、水田に適用することができる。また、式Iの化合物は、前記殺真菌剤の液体製剤で種子又は塊茎を浸含し、あるいは固体製剤でそれらをコーティングすることにより種子に施用されてもよい。
【0053】
製剤、即ち式Iの化合物及び必要に応じて固体又は液体の助剤を含有する組成物は、公知の方法、典型的には、化合物を、増量剤、例えば溶媒、固体担体、任意で界面活性化合物(界面活性剤)等と共に、念入りに混合及び/又は挽き潰すことにより調製される。
【0054】
前記農業化学製剤は、通常、0.1〜99重量%、好ましくは0.1〜95重量%の式Iの化合物、99.0〜1重量%、好ましくは99.8〜5重量%の固体又は液体助剤、及び0〜25重量%、好ましくは0.1〜25重量%の界面活性剤を含む。
【0055】
市販の製品としては濃縮品として製剤するのが好ましいが、最終消費者は、通常は希釈製剤を使用する。
【0056】
有利な施用率は、通常5g〜2kg有効成分(a.i.)/ヘクタール(ha)、好ましくは10g〜1kg a.i./ha、最も好ましくは20g〜600g a.i./haである。種子浸漬剤として使用されるとき、簡便な施用率は、種子1kgあたり有効成分10mg〜1gである。所望の効果を奏する施用率は、実験により決定できる。当該施用率は、例えば、作用の種類、有用植物の発達段階、及び施用(場所、時期、施用方法)に依存し、これらのパラメーターにより、広い範囲で変化する。
【0057】
驚くべきことに、式Iの化合物は、有用植物の作物への植物病原性微生物の攻撃を防御する方法に加え、植物病原性微生物が繁殖した有用植物の作物の処理方法にも使用することができ、当該処理方法は、グリホサート及び1つ以上の式Iの化合物の組合せ剤を、グリホサートに耐性又は感受性の植物の植物体又はその生育場所に投与することを含む。
【0058】
前記方法は、グリホサート無しで式Iの化合物を使用する場合と比較して、病害の防除作用の予期せぬ改善をもたらし得る。当該方法は、式Iの化合物による病害の防除の促進において、効果的であってもよい。グリホサートと1つ以上の式Iの化合物との混合物は、少なくとも部分的には、式Iの化合物によりコントロールされる病害の範囲を増やす場合もあるが、式Iの化合物によりある程度まで防除されることが既に知られている病害の種類に対する式Iの化合物の活性の増大も、観察される効果であることができる。
【0059】
当該方法は、菌界、担子菌門、サビ菌(Uredinomycetes)綱、サビ菌(urediniomycetidae)亜綱、及びサビ菌(Uredinales)目の植物病原性生物(一般的に、サビ菌類と呼ばれる)に対して特に有効である。農業において特に重大な影響を与えるサビ菌の種は、ファコプソラセア(Phakopsoraceae)科、特にファコプソラ属の種、例えばファコプソラ・パシリジ(Phakopsora pachyrhizi)(当該種は、アジアダイズサビと呼ばれている)、及びプッシニアセア(Pucciniaceae)科、特にプッシニア(Puccinia)属の種、例えばプッシニア・グラミニス(Puccinia graminis)(茎サビ又は黒色サビとして知られており、当該サビ病は穀草作物において問題となる病害である)、及び茶色サビとして知られているプッシニア・レコンジタ(Puccinia recondita)を含む。
【0060】
本方法の態様は、植物病原性生物による攻撃に対して有用植物である作物を保護する方法、及び/又は植物病原性生物に感染した有用植物の作物を治療する方法であって、当該方法は、その塩又はエステルを含むグリホサート、及び式Iの少なくとも1つの化合物を、植物、植物の部分、及び植物の生育場所からなる群から選択される少なくとも1のメンバーに同時に施用することを含む方法である。
【0061】
上記式Iの化合物、又はその医薬上の塩は、動物における微生物感染の治療及び/又は予防のための活性の有利な適用範囲をも有してもよい。「動物」は、任意の動物、例えば昆虫、哺乳動物、は虫類、魚、両生類であってもよく、好ましくは哺乳動物、最も好ましくはヒトである。「治療」は、感染の増加又は広がりを減少又は緩和又は停止するため、又は感染を低下又は感染を治癒するための微生物感染を有する動物における使用を意味する。「予防」は、微生物感染の明らかな兆候を有さない動物に対して、将来の感染を予防するため、又は将来の感染の増加又は広がりを低減又は緩和するために、使用することを意味する。
【0062】
本発明に従うと、動物において微生物感染の治療及び/又は予防において使用するための医薬の製造における式Iの化合物の使用も提供される。式Iの化合物を医薬として使用することも提供される。動物の治療において抗菌剤として式Iの化合物を使用することも提供される。本発明に従うと、活性成分として式Iの化合物、又はその医薬として許容される塩を含む医薬組成物が提供される。当該組成物は、動物において抗菌感染の治療及び/又は予防のために使用することができる。当該医薬組成物は、経口投与に適した形態、例えば錠剤、ロゼンジ、ハードカプセル、水性懸濁液、油状懸濁液、乳濁物、エマルジョン分散粉末、粉末可能顆粒、シロップ及びエリクシルの形態であってもよい。或いは、当該医薬組成物は、局所適用に適した形態、例えばスプレー、クリーム又はローションの形態であってもよい。或いは、この医薬組成物は、非経口投与に適した形態、例えば注射であってもよい。或いは、この医薬組成物は、吸入可能形態、例えばエアロゾルスプレーであってもよい。
【0063】
式Iの化合物は、動物において微生物感染を引き起こすことができる様々な微生物種に対して有効である。かかる微生物種の例は、アスペルギルス症を引き起こす微生物種、例えば、アスペルギルス・フミガツス(Aspergillus fumigatus)、A.フラブス(A.flavus)、A.テルス(A.terrus)、A.ニジュランス(A.nidulans)及びA.ニガー(A. niger)、ブラストミセス症を引き起こす種、例えば、ブラストマイシス・デルマチチジス(Blastomyces dermatitidis);カンジダ症を引き起こす種、例えばカンジタ・アルビカンス(Candida albicans)、C.グラブラタ(C. glabrata)、C.トロピカリス(C. tropicalis)、C.パラプシロシス(C. parapsilosis)、C.クルゼイ(C. krusei)及びC.ルシタニア(C. lusitaniae); コクシジオイデス症を引き起こす微生物種、例えばコクシジオイデス・イミチス(Coccidioides immitis);クリプトコッカス症を引き起こす種、例えばクリプトコッカス・ネオフォルマンス(Cryptococcus neoformans);ヒストプラスマ症を引き起こす種、例えばヒストプラスマ・カプスラツム(Histoplasma capsulatum)及び接合菌症を引き起こす種、例えばアブシジア・コリムビフェラ(Absidia corymbifera)、リゾムコール・プシルス(Rhizomucor pusillus)及びリゾプス・アリズス(Rhizopus arrhizus)である。さらなる例は、フサリウム種、例えばフサリウム・オキシスポルム(Fusarium oxysporum)及びフサリウム・ソラニ(Fusarium solani)及びセンドスポリウム種、例えばセンドスポリウム・アピオスペルマム(Scedosporium apiospermum)及びセドスポリウム・プロリフィカンス(Scedosporium prolificans)である。さらなる例は、マイクロスポルム種(Microsporum spp.)、トリコフィトン種(Trichophyton Spp)、エピデルモフィトン種(Epidermophyton Spp)、ムコール種(Mucor Spp)、スポロトリックス種(Sporothorix Spp)、フィアロフォア種(Phialophora Spp)、クラドスポリウム種(Cladosporium Spp)、ペトリエリジウム種(Petriellidium spp)、パラコッシジオイデス種(Paracoccidioides Spp)及びヒストプラスマ種(Histoplasma Spp.)である。
【0064】
以下の非限定的な実施例は、上記発明を、限定すること無く、より詳細に例示する。
【実施例】
【0065】
調製の例:
実施例P1:3-ジフルオロメチル-1-メチル-1H-pyrazole−4-カルボン酸[2-(4-クロロ-フェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.001 )の調製
【化15】
ジクロロメタン(5ml)中の3−ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(564mg;2.9mmol)を、実施例P13に記載のように調製されたN-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン(600mg;2.9mmol)、トリエチルアミン(0.80ml;5.8mmol)の0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を6時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c-ヘキサン/酢酸エチル1:1)。
【0066】
0.99g(理論上93.4%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(4-クロロ-フェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.001)が、樹脂状の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.33-1.37(d,3H); 2.77-2.82(dd,1H); 3.07-3.13(dd,1H); 3.64(s,3H); 3.94(s,3H); 4.63-4.68(m,1H); 6.98-7.28(m, 5H); 7.61 (s,1H)。
MS [M+H]+ 358/360。
【0067】
実施例P2:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.002)の調製
【化16】
3ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.91 g; 4.7 mmol)のジクロロメタン(5ml)溶液を、実施例P14に記載のように調製されたN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン(1.0 g; 4.27 mmol)、トリエチルアミン(0.90 ml; 6.4 mmol)の0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を1.5時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0068】
1.35g(理論上80.3%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.002)は、白色の固体の形態で取得された(m.p. 98-1020C)。
1H NMR: (CDCI3, 400MHz):
1.41-1.46(d,3H); 2.99-3.04(dd,1 H); 3.17-3.23(dd,1H); 3.60(s,3H); 3.95(s,3H); 4.68-4.70(m,1H); 7.10-7.62(m,5H)。
MS [M+H]+ 392/394/396。
【0069】
実施例P3:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-2-(2,4,6-triクロロフェニル)-エチル]-アミド(化合物1.003)の調製:
【化17】
実施例P15dに記載のように調製されたO-メチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.65 g, 2.4 mmol)のジクロロメタン(5 ml)溶液にトリエチルアミン(0.844 ml, 6.0 mmol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.519 g, 2.67 mmol)の0℃のジクロロメタン溶液を滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブライン溶液で洗浄し、これを硫酸ナトリウムで脱水し、そして溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-アミド(0.51 g, 49%)を、m.p:110〜112℃の白色の固体として取得した。
【0070】
1H NMR(400MHz, CDCI3):δ 1.38-1.39(d,3H), 3.20-3.26(dd,1H), 3.32-3.37(dd,1H), 3.70(s,3H), 3.97(s,3H), 4.88-4.93(m,1H), 7.02-7.29(t,1H), 7.27(s,2H), 7.81(s,1H)
MS [M+H]+ 426/428/430
【0071】
実施例P4:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸{2-[4-(4-クロロ-phenoxy)-phenyl]-1-メチル-エチル)-メトキシ-アミド(化合物1.015)の調製:
【化18】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(797 mg; 4.1 mmol)のジクロロメタン(5ml)溶液を、実施例P16のように調製したN-{2-[4-(4-クロロ-フェノキシ)-フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミン(1.2 g; 4.1 mmol)、トリエチルアミン(1.10 ml; 8.2 mmol)の、0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を常温で一昼夜攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル1:1)。
【0072】
1.2g(理論上66%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸{2-[4-(4- クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-メトキシ-アミド(化合物1.015)は、油状の形態で得られた。
1H NMR: (CDCI3,400MHz):
1.36-1.39(d,3H); 2.78-2.84(dd,1H); 3.05-3.12(dd,1H); 3.65(s,3H); 3.94(s,3H); 4.64-4.68(m,1H); 6.80-6.90(m, 4H);6.95-7.23(t,1H,CHF2);7.17-7.26(m,4H);7.67(s,1 H)。
MS [M+H]+ 450/452。
【0073】
実施例P5:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシ-アミド(化合物1.028)の調製:
【化19】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(2.10 g; 11.0 mmol)のジクロロメタン(5ml)溶液を、P17に記載のように調製したN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミン (2.0 g; 9.10 mmol)、トリエチルアミン(3.10 ml; 22 mmol)の、0℃の攪拌したジクロロメタン(15ml)溶液に滴下した。当該反応混合物を室温で一昼夜攪拌した。当該反応混合物を1M HCl(50ml)に注ぎ、ジクロロメタン(3x20ml)で抽出し、Na2SO4で脱水した。溶媒の除去後、残留物(5.08g)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル7:3)。
【0074】
1.51g(理論上43.8 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシ-アミド(化合物1.028)は、白色の固体の形態で取得された(m.p. 162-167°C)。
1H NMR:(CDCI3, 400MHz):
1.40-1.41(d,3H);2.89-2.94(dd,1H);3.03-3.14(dd,1H);3.86(s,3H);4.3-4.5(mbr,1H);6.5-7.0(mbr,2H); 7.19-7.21(m, 2H); 7.36(m,1H); 7.8-8.6(mbr,1H)。
MS [M+H]+ 378/380/382。
【0075】
実施例P6:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-エトキシ-アミド(化合物1.031)の調製:
【化20】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.86 g; 4.4 mmol)のジクロロメタン(5ml)溶液を、実施例P18に記載のように調製したN-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミン(1.0 g; 4.0 mmol)、トリエチルアミン(0.82 ml;6.0 mmol)の、0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を1.5時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0076】
0.75g(理論上45.7%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-エトキシ-アミド(化合物1.031 )は、白色の固体の形態で取得された(m.p. 116-118℃)。
1H NMR: (CDCI3, 400MHz):
1.14-1.20(t,3H);1.36-1.45(2d,3H);2.98-3.03(dd,1H);3.19-3.25(dd,1H);3.74-3.82(q,3H);3.94(s,3H); 4.64-4.70(m,1H); 6.93-7.65(m, 5H)。
MS [M+H]+ 406/408/410。
【0077】
実施例P7:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸エトキシ-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-アミド(化合物1.032)の調製:
【化21】
O-エチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.5g, 1.63 mmol)のジクロロメタン(5 ml)溶液に、トリエチルアミン(0.566 ml, 4.07 mmlol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.34g, 1.79mmol)のジクロロメタン溶液を0℃で滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブラインで洗浄し、これを硫酸ナトリウムで脱水し、そして減圧下で溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸エトキシ-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-アミド(0.390g, 50%)を、m.p:111〜114℃のオフホワイトの固体として取得した。
【0078】
1H NMR (400MHz, CDCI3): δ 1.20-1.24(t,3H), 1.35-1.37(d,3H), 3.23-3.28(dd,1H), 3.33-3.38(dd,1H), 3.84-3.88(q,2H), 3.96(s,3H), 4.86-4.91(m,1H), 7.01-7.25(t,1H), 7.28(s,2H), 7.83(s,1H)
MS [M+H]+ 439.9/441.83
【0079】
実施例P8:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-isoプロポキシ-アミド(化合物1.033)の調製:
【化22】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(1.1 g; 5.7 mmol)のジクロロメタン(5ml)溶液を実施例P20に記載のように調製したN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミン (1.5 g; 5.7 mmol)、トリエチルアミン(1.6 ml;11.4 mmol)の、0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を常温で一昼夜攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0080】
1.30g(理論上54.0 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-isoプロポキシ-アミド(化合物1.033)は、黄色の油の形態で取得された。
1H NMR: (CDCI3, 400MHz):
0.91-0.93+1.11-1.13(2d,6H);1.42-1.48(2d,3H);3.00-3.07(dd,1H);3.29-3.36(dd,1H);3.92(s,3H); 3.97-4.36(m,1H);4.36-4.45(m,1H); 6.97-7.59(m,5H)。
MS [M+H]+ 420/422/424。
【0081】
実施例P9:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-ペンチル]-メトキシ-アミド(化合物1.059)の調製:
【化23】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(2.20g; 1 1.0mmol)のジクロロメタン(10ml)溶液を、実施例P22に記載のように調製したN-[2-(2,4- ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミン (3.0 g; 11.0 mmol)、トリエチルアミン(3.0 ml; 22 mmol)の、0℃の攪拌したジクロロメタン(20ml)溶液に滴下した。当該反応混合物を常温で6時間攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル1:1)。
【0082】
3.85gm(理論上83 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-ペンチル]-メトキシ-アミド(化合物1.059)は、黄色の油の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.84-0.87(t,3H);1.14-1.25(m,2H);1.61-1.69(m,2H);3.55(s,3H);3.71-3.80(m,1H);3.80-3.84(dd,1H); 3.96(s,3H); 3.99-4.05(dd,1H); 7.07-7.36(m,4H);7.79(s,1H)。
MS [M+H]+ 420/422/424。
【0083】
実施例P10:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(化合物2.003)の調製:
【化24】
実施例P23dに記載のように調製したO-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.6g, 2.1 mmol)のジクロロメタン(6 ml)溶液に、トリエチルアミン(0.73 ml, 5.2 mmol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.415 g, 2.1 mmol)のジクロロメタン溶液を0℃で滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブライン溶液で洗浄し、これを硫酸ナトリウムで脱水し、そして減圧下で溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(0.53 g, 57%)を、ゴム状生産物として取得した。
【0084】
物理データ:
1H NMR: (CDCI3, 400MHz):
1.41 (d,3H,CH3),1.71-1.80(m,1H,CH2),1.94-2.04(m,1H,CH2),1.87-1.92(m,2H,CH2),3.78(s,3H,CH3),3.98(s,3H,CH3),4.66-4.71(m,1H,CH),7.11(t,1H,CHF2),7.28(s,2H,Ar-H),
7.88(s,1 H,ピラゾール-H)
MS [M+H]+ 440/442/444/446。
【0085】
実施例P11:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(化合物2.040)の調製:
【化25】
実施例P24cに記載のように調製されたO-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.35 g, 1.3 mmol)のジクロロメタン(5 ml)溶液に、トリエチルアミン(0.55 ml, 3.9 mmol)及び3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド酸のジクロロメタン(2ml)溶液を、0℃でゆっくり添加した。酸塩化物を完全に添加した後、混合物を常温で3時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブラインで洗浄した。当該有機相を無水硫酸ナトリウムで脱水し、そして減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュシリカゲル、ヘキサン中26%酢酸エチル)にかけて精製し、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(0.5g, 91 %)を、m.p- 95-96℃の白色の結晶固体として取得した。
【0086】
物理データ:
1H NMR: (CDCI3, 400MHz):
1.89-1.96(m,2H, CH2), 2.91-2.95(m,2H,CH2), 3.66(s,3H,CH3), 3.83-3.86(t,2H,CH2), 3.97(s,3H,CH3), 7.31 (t,1H,CHF2), 7.28(s,2H,Ar-H), 7.89(s,1H,ピラゾール-H)
MS [M+H]+ 426/428/430/432
【0087】
実施例P12:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-メトキシ-アミド(化合物2.057)の調製:
【化26】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.89 g; 4.6 mmol)のジクロロメタン(5ml)溶液を、実施例P25に記載のように調製したN-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミン(1.0 g; 4.2 mmol)、トリエチルアミン(0.86 ml; 6.3 mmol)の、0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を、常温で1.5時間攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0088】
0.59g(理論上35.3%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-メトキシ-アミド(化合物2.057)は、樹脂状の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.90-0.95(d,3H);1.30(s,9H);2.24-2.36(m,1H);2.39-2.45(dd,1H);2.67-2.72(dd,1H);3.58(s,3H); 3.68-3.70(d,2H);3.98(s,3H); 7.09-7.11 (d, 2H); 7.15-7.42(m,3H);7.90(s,1H)。
MS [M+H]+ 394。
【0089】
実施例P13:N-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシルアミンの調製:
a) 1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化27】
1-(4-クロロ-フェニル)-プロパン-2−オン(8.5 g, 50.4 mmole)のメタノール(100ml)溶液をピリジン(5.2 ml, 62 mmol)、続いてO-メチルヒドロキシアミン塩酸塩(5.20 g, 62 mmol)で処理した。得られた混合物を、23℃で一昼夜16時間攪拌した。当該反応混合物を水(200ml)に注ぎ、そしてジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル9:1)。
【0090】
7.38g(理論上74%)の1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシムは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.71 (s,3H);3.48+3.52(2s,2H);3.87-3.89(2s,3H); 7.13-7.22(m,4H). MS [M+H]+ 198/200。
【0091】
b)N-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンの調製:
【化28】
1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.0 g, 5.1mmol)のacetic acid (7.6 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(641 mg, 10.2 mmol)を10分間にわたり添加して処理し、得られた溶液を24℃で5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、1M NaOHでpHを9に調整した。水相をジクロロメタン(2x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(1OOmbar; 45℃)、残留物(910mg)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル9:1)。
【0092】
830mg(理論上82.0%)のN-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.02-1.06(d,3H);2.54-2.59(dd,1H);2.79-2.84(dd,1H);3.14-3.24(m,1H),3.52(s,3H); 5.3-5.5(sbr,1H); 7.12-7.16(m,2H); 7.25-7.28(m,2H)。
MS[M+H]+ 200/202。
【0093】
実施例P14:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化29】
O-メチルヒドロキシアミン塩酸塩(7.04g, 0.0843mol)の(130ml)とTHF(50ml)の溶液を、酢酸ナトリウム(5.9g, 0.0720mol)、続いて1-(2,4-ジクロロフェニル)-プロパン-2−オン(10g, 0.0496mole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、そして無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0094】
13.5g(理論上100%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシムは、更なる精製を行わずに次の工程に使用される透明な液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
1.76(s,3H);3.60+3.78(2s,2H);3.87(s,3H); 7.12-7.19(m,2H); 7.39(d,1H)。
MS [M+H]+ 232/234/236。
【0095】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンの調製:
【化30】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシム(10.0g, 0.0431mol)の酢酸(100 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(5.41g, 0.0862mol)を15分間にわたり少量ずつ添加して処理し、得られた溶液を24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を減圧下で除去した(1OOmbar; 45℃)。
【0096】
8.13g (理論上81.3% of theory)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンは、更なる精製を行わずに次の工程に使用される透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.04-1.09(d,3H);2.66-2.71 (dd,1H);2.93-2.99(dd,1H);3.25-3.33(m,1H),3.52(s,3H); 5.2-5.4(sbr,1H); 7.17-7.18(m,2H); 7.35(d, 1H)。
MS [M+H]+ 234/236/238。
【0097】
実施例P15:O-メチル-N-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-ヒドロキシアミンの調製:
a)1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼンの調製:
【化31】
2,4,6-トリクロロ-ベンズアルデヒド(19g, 90.69mmol)及び酢酸アンモニウム(16.75g, 217mmol)の攪拌した酢酸(76ml, 4Vol.)溶液に、0℃でニトロエタン(45.1ml, 625mmol)を滴下した。当該反応生産物を、100℃で90分間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を室温にし、続いて冷水(700ml)で希釈し、これを更に酢酸エチル(3 x 50 ml)で抽出した。一まとめにした酢酸エチルの相を飽和炭酸水素ナトリウム溶液で洗浄してpHを中性にし、続いて水で洗浄し、続いてブラインで洗浄し、これをNa2SO4で脱水し、その後溶媒を完全に蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中1%酢酸エチルを溶出系として回収して、1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼン(15g, 61%)を得た。
1H NMR (400MHz, CDCI3): δ2.10(s,3H,CH3), 7.24(s,2H,Ar-H),7.77(s,1H,CH)
【0098】
b)1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンの調製:
【化32】
上記で取得した攪拌されたニトロスチレン(5g, 18.72mmol)の水(20 ml)及びメタノール(60 ml)溶液に、鉄粉末(2.355gms , 42.12mmol)、続いて濃HCl(11.5ml, 112 mmol)を、窒素大気中、常温で添加した。当該反応混合物を70℃で1時間攪拌し、更に鉄粉(2.355 gms, 42.12 mmol)及び濃HCl(11.5 ml, 112 mmol)をこの反応生産物に添加し、70℃で2時間攪拌を続行した。前記反応の完遂がTLCで確認されたら、前記反応物を室温に冷し、続いてロータベイパー(rotavapor)を用いてエタノールを除去した。得られた残留物を水で希釈し、続いて酢酸エチル(3 x 80 ml)で抽出した。一まとめにした酢酸エチルの相を最後に水で洗浄し、続いてブラインで洗浄し、これを無水硫酸ナトリウムで脱水し、その後溶媒を完全に蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中5%酢酸エチルを溶出系として回収して、1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オン(3.7 g, 83.3%)を得た。
1H NMR (400MHz, CDCI3): δ 2.26(s,3H,CH3), 4.05(s,2H,CH2), 7.34(s,2H,Ar-H)
【0099】
c)1-(2,4,6-トリクロロフェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化33】
O-メチル-ヒドロキシアミン塩酸塩(0.79 g, 9.4 mmol)のメタノール(20 ml)溶液を、トリエチルアミン(1.32 ml, 9.4 mmol)、続いて1-(2,4,6-トリクロロフェニル)-プロパン-2−オン(1.5 g, 6.31 mmole)で処理した。得られた混合物を、60℃で3時間攪拌した。当該反応混合物を水で浸漬し、続いてメタノールを蒸発させた。得られた水性媒体を、酢酸エチル(3x50ml)で抽出した。一まとめにした有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100 mbar; 45℃)。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中2%酢酸エチルを溶出系として回収して、1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.2 g, 76%)を得て、これを更に還元した。
【0100】
d)O-メチル-N-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-ヒドロキシアミンの調製:
【化34】
上記で取得した1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.2g, 4.51 mmol)の酢酸12mlの溶液に、シアノ水素化ホウ素ナトリウム(0.568, 9 mmol)を添加した。当該反応混合物を、常温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、溶媒を減圧下で蒸発させた(トルエンとの共蒸発2回)。得られた残留物を水で希釈し、続いて2Nの水酸化ナトリウムでpHを9に調製して、それから酢酸エチル(3 x 50 ml)で抽出した。一まとめにした有機相をブラインで洗浄し、これを無水硫酸ナトリウムで脱水して、溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中5%酢酸エチルを溶出系として回収して、O-メチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.91 g, 75%)を得た。
1H NMR(400MHz, CDCI3):δ 0.91-0.93(d,3H), 2.72-2.77(dd,1H), 2.98-3.03(dd,1H), 3.25-3.30(m,1H), 3.93(s,3H), 7.15(s,2H)
【0101】
実施例P16:N-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンの調製:
a)1-[4-(4-クロロフェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシムの調製:
【化35】
1-[4-(4-クロロ-フェノキシ)−フェニル]-プロパン-2−オン(10.0 g, 38 mmole)のメタノール(65 ml)溶液を、O-メチルヒドロキシアミン塩酸塩(3.90 g, 47 mmol)、続いてピリジン(4.0 ml, 47 mmol)で処理した。得られた混合物を、室温で一昼夜攪拌した。当該反応混合物を水(150ml)に注ぎ、そしてジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。
【0102】
11.0g(理論上100%)の粗1-[4-(4-クロロ-フェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシムは、更なる精製を行わずに次の工程に使用される黄色の液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
1.73+1.81(2s,3H);3.44+3.65(2s,2H);3.88+3.89(2s,3H); 6.89-6.94(m,4H);7.15-7.28(m,4H)。
MS [M+H]+ 290/292。
【0103】
b)N-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンの調製:
【化36】
粗1-[4-(4-クロロフェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシム(6.0 g, 20.7 mmol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.6 g, 41.4 mmol)を少量ずつ10分間かけて添加して処理し、そして得られた溶液を、23℃で3.5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを11に調整した。水相をジクロロメタン(2x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(1OOmbar; 45℃)、残留物(7.17g)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン)。
【0104】
3.91 g(理論上65.0%)のN-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.07-1.09(d,3H);2.53-2.62(dd,1H);2.72-2.81(dd,1H);3.16-3.26(m,1H),3.54(s,3H); 5.3- 5.5(Sbr,1H); 6.86-6.94(m,4H);7.14-7.18(m,2H);7.24-7.29(m,2H)。
MS [M+H]+ 292/294。
【0105】
実施例P17:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシムの調製:
【化37】
1-(2,4-ジクロロフェニル)-プロパン-2−オン(3.46 g, 16.7 mmole)のメタノール(40 ml)溶液を、ヒドロキシアミン塩酸塩(1.40 g, 20.6 mmol)、続いてピリジン(1.7 ml, 20.6 mmol)で処理した。得られた混合物を常温で一昼夜16時間攪拌した。当該反応混合物を水(200ml)に注いだ。固体の産物を濾過により回収し、水で洗浄し、乾燥させた(40℃、100mbar)。
【0106】
3.63g(理論上100%)の1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシムは、白色の固体の形態で取得された(m.p. 120-1240C)。
1H NMR: (CDCI3, 400MHz):
1.86(s,3H);3.61 (s,2H); 7.12-7.21 (m,2H); 7.39-7.41 (d,1H); 8.2(sbr,1H)。
MS [M+H]+ 218/220/222。
【0107】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンの調製:
【化38】
1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシム(3.3 g, 15.1 mmol)の酢酸(30 ml)溶液を、シアノ水素化ホウ素ナトリウム(1.90 g, 30.3 mmol)を少量ずつ10分間添加して処理し、そして得られた溶液を、23℃で5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを10に調整した。水相をジクロロメタン(3x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後3.18g (理論上96.0 %)のN-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンは、白色の固体の形態で取得された(m.p. 83-86℃)。
1H NMR: (CDCI3, 400MHz):
1.08-1.11(d,3H);2.69-2.74(dd,1H);2.98-3.03(dd,1H);3.24-3.32(m,1H); 5.3-5.6(sbr,1H);7.18-7.19(m,2H);7.39(m,1H);7.5-8.2(sbr,1H)。
MS[M+H]+ 220/222/224。
【0108】
実施例P18:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-エチル-オキシムの調製:
【化39】
O-エチルヒドロキシアミン塩酸塩(4.10 g, 42.1 mmol)の水(130 ml)及びTHF(50 ml)の混合物の溶液を、酢酸ナトリウム(2.95 g, 72 mmol)、続いて1-(2,4-ジクロロフェニル)-プロパン-2−オン(5 g, 24.8 mmole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、そして無水Na2SO4で脱水した。溶媒を減圧下で蒸発させた(90mbar; 45℃)。
【0109】
6.0g(理論上99%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-エチル-オキシムは、更なる精製をせずに次の工程に使用されるE/Z−混合物として、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.23-1.28(2t,3H);1.74+1.76(2s,3H);3.60+3.78(2s,3H);4.09-4.16(2q,2H);7.12-7.19(m,2H);7.38(d,1H)。
MS [M+H]+ 246/248/250。
【0110】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミンの調製:
【化40】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシム(10.0g, 0.0406mol)の酢酸(100 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(5.41 g, 0.0862 mol)を少量ずつ10分間にわたり添加して処理し、得られた溶液を、24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0111】
5.09g(理論上51.0 %)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル- ヒドロキシアミンは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.98-1.02+1.08-1.12(2d,3H);1.13-1.18(2t,3H);2.63-2.68+2.70-2.78(2dd,1H);2.94-3.10+3.15-3.21(m,1H),3.25-3.31+3.71-3.78(2q,2H);5.0-5.5(sbr,1H);7.16-7.19(m,2H);7.34-7.36(m,1H)。
MS [M+H]+ 248/250/252。
【0112】
実施例P20:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシムの調製:
【化41】
1-(2,4-ジクロロフェニル)-プロパン-2−オン(5.1 g, 25.0 mmole)のメタノール(45ml)溶液を、O-isoプロピル-ヒドロキシアミン塩酸塩(3.50 g, 31 mmol)、続いてピリジン(2.6 ml, 31 mmole)で処理し、そして得られた混合物を、23℃で一昼夜攪拌した。当該反応混合物を水(100ml)に注ぎ、ジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒を減圧下で蒸発させた(90mbar; 45℃)。
【0113】
6.48g (理論上99%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシムは、更なる精製をせずに次の工程に使用されるE/Z−混合物として、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.23-1.26(d,6H);1.74(s,3H);3.60+3.78(2s,3H);4.25-4.38(2m,1H);7.14-7.21(m,2H);7.39(d,1H)。
MS [M+H]+ 260/262/264。
【0114】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミンの調製:
【化42】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシム(6.0g, 23 mmol)の酢酸(40 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.90 g, 46 mmol)を少量ずつ15分間にわたり添加して処理し、得られた溶液を、24℃で4時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを10に調整した。水相をジクロロメタン(2x80ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0115】
5.90g (理論上98.0%)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル- ヒドロキシアミンは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
1H NMR: (CDCI3,400MHz):
1.03-1.07(d,3H);1.13-1.19(m,6H);2.61-2.68+2.94-3.03(2dd,2H);3.21-3.31(m,1H),3.78-3.84(m,1H);5.2(sbr,1H);7.14-7.18(m,2H);7.35-7.36(m,1H)。
MS [M+H]+ 262/264/266。
【0116】
実施例P21:O-メチル-N-[2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミンの調製:
a)1,3,5-トリクロロ-2-クロロメチル-ベンゼンの調製:
【化43】
窒素大気中に置いて攪拌した2,4,6-トリクロロベンジルアルコール(10.0g; 47.3 mmoles)のクロロホルム(100ml)溶液に、塩化チオニル(6.07ml, 85.1 mmole)を、0℃で、15分間にわたりゆっくり添加し、続いて触媒として使用される量のDMFを添加した。当該反応混合物を、常温で3時間攪拌した。当該反応混合物を50mlの水で滴定し;水相をDCM(3x100ml)で抽出した。一まとめにした有機相を5%炭酸水素ナトリウム(2x50ml)、続いてブライン(50ml)で洗浄し、そして無水硫酸ナトリウムで脱水した。溶媒を減圧下で蒸発させた。10.9g(理論上100.0%)の1,3,5-トリクロロ-2-クロロメチル-ベンゼンは、白色の固体の形態で取得された。
1HNMR (CDCI3, 400MHz): δ=7.37(2H,s);4.82(2H,s)
Mass: M = 229.8
【0117】
b)(2,4,6-トリクロロ-フェニル)-アセトニトリルの調製:
【化44】
攪拌した1,3,5-トリクロロ-2-クロロメチル-ベンゼン (12.85 g, 58.8mmole)のエタノール(45ml)溶液に、NaCN(3.20, 67.0mmole)の(15ml)溶液を、常温で添加した。当該反応混合物を4時間再還流して、反応を完遂した。エタノールを蒸発させ、反応混合物を水(50ml)で希釈した。水相をEtOAc(3x100ml)で抽出した。一まとめにした有機相をブライン(30ml)で洗浄し、続いて硫酸ナトリウムで脱水した。有機相を真空中で濃縮して、白色の固体を得た。粗生産物をシリカゲルカラムのクロマトグラフィーで精製して、9.Og(理論上75.0%)の(2,4,6-トリクロロ-フェニル)-アセトニトリルを得た。
1HNMR (CDCI3,400MHz): 7.41(s,2H); 3.97(s,2H)。
Mass:M = 218.9
【0118】
c)(2,4,6-トリクロロ-フェニル)-アセトアルデヒドの調製:
【化45】
N2大気中に置いて、攪拌して冷却(-70℃)した(2,4,6-トリクロロ-フェニル)-アセトニトリル(5.7g, 26.0mmole)のトルエン(120ml)溶液に、DIBAL-H(1MのTHF溶液28.1ml、28.6mmole)を、30分間以上滴下により添加した。反応は、-70℃で3時間攪拌して行われた。反応終了後、-40℃でHCl(60ml、2N)の滴下により滴定し、そして常温に30分間置いた。トルエン相を分離し、水相をブライン(30ml)で洗浄し、硫酸ナトリウムで脱水した。有機相を、真空下で濃縮した。4.5g(理論上78.0%)の(2,4,6-トリクロロ-フェニル)-アセトアルデヒドは、白色の固体の形態で取得された。
1HNMR (CDCI3, 400MHz: 9.70 (s, 1 H); 7.38 (s, 2H); 4.08 (s, 2H)
Mass: M = 246.9
【0119】
実施例P22:N-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンの調製:
a)2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシムの調製:
【化46】
2-(4-クロロ-フェニル)-ペンタナル(10.0 g, 43.0 mmole)のメタノール(75 ml)溶液を、O-メチルヒドロキシアミン塩酸塩(4.40 g, 53 mmol)、続いてピリジン(4.5 ml, 53 mmol)で処理した。得られた混合物を、23℃で一昼夜攪拌した。当該反応混合物を水(200ml)に注ぎ、ジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。減圧下で溶媒を蒸発させた(100mbar;45℃)。
【0120】
11.2Og(理論上100.0%)の粗2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシムは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 260/262/264.
【0121】
b)N-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンの調製:
【化47】
2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシム(6.0g, 23mmol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.90 g, 46 mmol)を少量ずつ10分間にわたり添加して処理し、得られた溶液を、24℃で4時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを11に調整した。水相をジクロロメタン(2x50ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(100mbar; 45℃)、残留物(6.2g)を、シリカゲルのフラッシュクロマトグラフィーで精製した(25Og;溶出液:c-ヘキサン)。
【0122】
5.15g(理論上79.0%)のN-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
0.82-0.89(t,3H);1.12-1.27(m,2H);1.49-1.58(m,1H);1.63-1.72(m,1H);3.05-3.10(dd,1H);3.12-3.18(dd,1H);3.47(s,3H),3.50-3.51 (m,1H); 5.0-5.50(sbr,1H); 7.14-7.16(m,1H); 7.21-7.25(m,1H);7.37-7.39(m,1H)。
MS [M+H]+ 262/264/266。
【0123】
実施例P23:O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
a)3-(2,4,6-トリクロロ-フェニル)-プロピオン酸の調製:
【化48】
常温で攪拌しながらMeldrum酸(2.06 g, 14.32 mmoles)をTEAF (7.05 ml)に加え、当該本能混合物を窒素大気中に置いた。10分後、当該反応混合物に、2,4,6-トリクロロ-ベンズアルデヒド(3g, 14.32mmoles)を添加した。続いて、これを、3時間、120℃で再還流した。この反応の収量後、当該混合物を常温で冷却し、そして氷水(50ml)に注いだ。それから、水相を酢酸エチル(3x80ml)で抽出し、一まとめにした有機相をブライン(40ml)で洗浄し、硫酸ナトリウムで洗浄した。有機相を、真空下で濃縮した。310g(理論上86.0%)の3-(2,4,6-トリクロロ-フェニル)-プロピオン酸は、白色の固体の形態で取得された。
1H NMR (400 MHz, CDCI3):
7.32(s,2H); 3.26-3.22 (m,2H); 2.63-2.58(m,2H)。
MS [M+H]+ 253/255/257
【0124】
TEAF(トリエチル蟻酸アンモニウム)の調製:窒素大気中においた蟻酸(1.12g 〜0.93 ml,24.5mmoles)溶液に、0℃で、トリエチルアミン(1g 〜1.37ml,9.8mmoles)をゆっくり添加し、混合物を、常温で1時間半攪拌した。
【0125】
b)N-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミドの調製:
【化49】
酸塩化物のクロロホルム溶液[酸塩化物は、対応する3-(2,4,6-トリクロロ-フェニル)-プロピオン酸(5g ,19.7mmoles)から、塩化チオニル(5.87g ,49.4mmole)を使用して、110℃で3時間の還流条件下で調製された]に、0℃で、N,O-ジメチルヒドロキシアミン塩酸塩(2.3g, 23.9mmoles)、続いてピリジン(3.77ml,47.7mmole)を添加した。その後、当該反応混合物を、70mlのDCM及び40mlの水で希釈した。有機相を分離し、そして水相をDCM(50mlx2)で2回抽出した。一まとめにした有機相をブライン(40ml)で洗浄し、続いて硫酸ナトリウムで脱水した。溶媒の除去後、残留物を、シリカゲルのクロマトグラフィーで洗浄した。3.Og(理論上52.0%)のN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミドは、固体の形態で取得された。
1H NMR(CDCI3, 400MHz):
7.31(s,2H); 3.67(s,3H);3.24-3.19(m,2H);3.19(s,3H);2.67-2.63(m,2H)
MS [M+H]+ 296/298/300
【0126】
c) 4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンの調製:
【化50】
攪拌し冷却(0℃)したN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミド(1g, 3.37mmol)のTHF(15ml)溶液を窒素大気中に置き、MeMgI(1.3mlの3Mエーテル溶液、3.9 mmole)を添加し、攪拌を0℃で2時間継続した。反応混合物を、0℃で5%HCl(10ml)で滴定し、そして水相をEtOAc(3x30ml)で抽出した。一まとめにした有機相を20mlのブラインで洗浄し、硫酸ナトリウムで脱水し;真空中で濃縮して、オフホワイトの固体を得た。当該残留物を、シリカゲルのクロマトグラフィーにより精製した。0.5Og(理論上60.0%)の4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンは、固体の形態で取得された。
1H NMR(CDCI3, 400MHz):
2.14(s,3H,CH3), 2.72-2.76(t,2H,CH2), 2.93-2.97(t,2H,CH2), 7.13-7.16+7.16-7.19(2d,2H,Ar-H), 7.34-7.35(d,1H,Ar-H)。
MS [M+H]+ 252/254/256
【0127】
d)O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
【化51】
攪拌した4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オン(0.9 g, 3.57 mmol)のメタノール(5 ml)溶液に、トリエチルアミン(0.73 ml, 5.3 mmol)及びO-メチル-ヒドロキシアミン塩酸塩(0.447 g, 5.3mmol)を添加した。当該反応混合物を、60℃で3時間加熱した。前記反応の完遂がTLCで確認されたら、前記反応物を常温にした。当該反応混合物を水で希釈した後、メタノールを蒸発させ、水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチル相を、水で、続いてブライン溶液で洗浄し、硫酸ナトリウムで脱水した後、溶媒を完全に蒸発させて、粗4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンO-メチル-オキシム(0.98Og)を得た。これは、更なる精製を行わずに、還元に付された。
【0128】
4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンO-メチル-オキシム(0.650g, 2.33 mmol)の酢酸溶液に、シアノ水素化ホウ素ナトリウム(0.293 g, 4.66 mmol)を添加した。当該反応物を、室温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、当該反応物中の酢酸を、共沸蒸留により除去した。その後10%NaOHで残留物を塩基性化し、酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチルの相を水で、続いてブライン溶液で洗浄し、硫酸ナトリウムで脱水し、その後濃縮した。得られた粗生産物を、60〜120μメッシュのシリカゲルを使用するカラムクロマトグラフィーにより精製し、ヘキサン中3%酢酸エチルを溶出液として生産物を回収して、O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.6 g, 90%)を得た。
1H NMR (400MHz, CDCI3): δ 1.21(d,3H), 1.52-1.59(m,1H), 1.68-1.76(m,1H), 2.80-2.83(q,2H), 3.06-3.13(m,1H), 3.55(s,3H), 5.45(s,1H), 7.29(s,2H)
物理データ:
1H NMR (CDCI3, 400MHz):
1.21(d,3H,CH3), 1.52-1.59(m,1H,CH2), 1.68-1.76(m,1H,CH2), 2.80-2.83(m,2H,CH2), 3.06-3.13(m,1H,CH), 1.45(s,1H,NH), 7.29(s,2H,Ar-H)
【0129】
実施例P24:O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
a)3-(2,4,6-トリクロロ-フェニル)-プロパン-1-オールの調製:
【化52】
物理データ:
1H NMR (CDCI3, 400MHz):
1.79-1.86(m,2H, CH2), 2.95-2.99(m,2H,CH2), 3.71-3.749(t,2H,CH2), 7.30(s,2H,Ar-H)
【0130】
b)3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドの調製:
【化53】
攪拌したN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミド(0.9 g, 3.04mmol)のトルエン(18mL)溶液に、-60℃で、DIBAL-H(15.2ml, 15.2mmol)を20分かけてゆっくり滴下し、当該反応物を、-60℃で3時間攪拌した。更に、反応物を、希塩酸(20ml)を滴下して滴定し、続いて水(10ml)で希釈し、15分間攪拌した。トルエン相を分離し、得られた水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチルの相を水で、続いてブライン溶液で洗浄した。当該有機相を無水硫酸ナトリウムで脱水し、その後減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中2%酢酸エチル)で精製して、3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒド(0.45g,62.5%)の白色の結晶固体を得た。
1H NMR (400MHz, CDCI3): δ 2.68(t,2H), 3.19-3.23(t,2H), 7.29(s,2H), 9.86(s,1H) MS [M+H]+: 235.9 /239.9
物理データ:
1H NMR (CDCI3, 400MHz):
2.68-2.72(m,2H,CH2), 3.19-3.23(m,2H,CH2), 7.29(s,2H,Ar-H), 9.86(s,1H,CHO)
【0131】
c)O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
【化54】
3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒド(0.45 g, 1.8 mmol)のメタノール(5 ml)溶液に、トリエチルアミン(0.4 ml, 2.8 mmol)、続いてO-メチルヒドロキシアミン-塩酸塩(0.24 g, 2.8mmol)を添加し、当該混合物を60℃で2時間加熱した。前記反応の完遂がTLCで確認されたら、前記反応物を常温にし、水で滴定し、ロータベイパーでメタノールを蒸発させた。得られた水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチル相を、水で洗浄し、硫酸ナトリウムで脱水した後、溶媒を完全に蒸発させて、粗生産物を得た。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中1%酢酸エチル)で精製して、3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドO-メチル-オキシム(0.5 g, 80%)を得た。
【0132】
攪拌した3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドO-メチル-オキシムの酢酸溶液に、NaCNBH3を、0℃〜10℃で添加した。当該反応混合物を、室温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、当該反応物中の酢酸を、共沸蒸留により除去した。その後残留物を10%NaOH溶液で残留物を塩基性化し、水溶液を、酢酸エチル(3x30ml)で抽出した。一まとめにした有機相を水で、続いてブライン溶液で洗浄した。当該有機相を、続いて、無水硫酸ナトリウムで脱水し、その後減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中3%酢酸エチル)で精製して、O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン (0.4 g, 88%)を得た。
1H NMR (400MHz, DMSO): δ 1.74-1.82(m,2H), 2.92-3.01 (m,4H), 3.55(s,3H), 5.60(s,1H), 7.29(s,2H)
MS [M+H]+ : 268.13 /272.17
物理データ:
1H NMR (CDCI3, 400MHz):
1.74-1.82(m,2H, CH2), 2.92-3.01 (m,4H,CH2-CH2), 3.55(s,3H,CH3), 5.60(s,1H,NH), 7.29(s,2H,Ar-H)。
【0133】
実施例P25:N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンの調製:
a)3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシムの調製:
【化55】
O-メチル ヒドロキシアミン塩酸塩(3.30 g, 0.041 mol)のwater(65 ml)及びTHF(25 ml)の混合物の溶液を、酢酸ナトリウム(2.85 g, 0.0348 mol)、続いて3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒド(5 g, 0.024 mole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、更に無水硫酸ナトリウムで脱水した。溶媒を減圧下で蒸発させた(100mbar; 45℃)。
【0134】
6.01g(理論上100 %)の粗3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシムは、更なる精製を要さずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 234。
【0135】
b)N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンの調製:
【化56】
3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシム(5.0 g, 0.0213 mol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.63g, 0.042mol)を15分間にわたり添加して処理し、得られた溶液を24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(1OOmbar; 45℃)。
【0136】
4.52g(理論上89.5 %)の粗N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンは、更なる精製を要さずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 236。
【0137】
実施例P26:2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンの調製:
a)2,4-ジクロロ-1-((E)-2-ニトロ-プロペニル)-ベンゼンの調製:
【化57】
スルホン化フラスコ中で、2,4-ジクロロ-ベンズアルデヒド(77g, 0.44mol)、-ニトロエタン(216ml, 3.04mol)及び酢酸アンモニウム(81.4g, 1.06mol)を、氷酢酸(600ml)に添加した。固体の生産物を濾過により回収し、水で洗浄し、そしてエタノールから再結晶化した。55.9g(理論上55%)の2,4-ジクロロ-1-((E)-2--ニトロ-プロペニル)-ベンゼンは、黄色の固体の形態で取得された(m.p. 79-81℃)。
1H NMR (400MHz, CDCI3): δ 8.11(s,1H), 7.51(d,1H), 7.34(dd,1H), 7.27(d,1H), 2.33(s,3H,CH3)。
【0138】
a)2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンの調製:
【化58】
攪拌した黄色の2,4-ジクロロ-1-((E)-2-ニトロ-プロペニル)-ベンゼン(4 mmol, 0.93 g)の無水トルエン(20ml)溶液に、N2大気中、0℃で、メタノール(16.2mmol, 3ml)中の5.4M CH3ONaとメタノール(2ml)の混合物を2分間かけて滴下した。1.5時間の攪拌後、氷酢酸(3ml)、続いて水(20ml)を添加した。当該水溶液をジクロロメタン(2x30ml)で抽出し、有機相を一まとめにし、脱水し(MgSO4)、濾過し、そして減圧下で蒸発させて、0.78gの粗1-アリール-1-メトキシ-2-ニトロプロパンの黄色の油を得た。この生材料をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル8:2)で精製して、0.45g(理論上43%)の2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンを、液体の形態で、ジアステレオ異性体の混合物として得た。
1H NMR (400MHz, CDCI3): δ 1.35-1.37+1.39-1.40(2d,3H, CH3), 3.18+3.21(2s,3H,CH3), 3.88+3.92(2s, 3H,CH3),4.69-4.75(m,1H,CH), 5.16-5.18+5.39-5.40(2d,1H,CH), 7.15-7.47(m,3H,Ar-H)。
MS [M+H]+ 264/266/268。
【0139】
実施例P27:2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンの調製:
a)1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールの調製:
【化59】
攪拌したニトロエタン(8.3g, 0.11 mol)のアセトニトリル(150 ml)溶液に、無水リン酸カリウム(1.Og, 4.6mmol)、続いて2,4-ジクロロ-ベンズアルデヒド(17.5g, O.10mol)を添加した。当該反応混合物を4時間攪拌した。水(300ml)を添加し、更に当該反応混合物をジエチルエーテル(200ml)で抽出した。有機抽出物を水で洗浄し、無水Na2SO4で脱水し、溶媒を除去し、そして得られた残留物を、シリカゲルのフラッシュクロマトグラフィー(溶出液:シクロヘキサン/酢酸エチル9:1)で精製した。20.7g (理論上82.5%)の1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールのトレオ/エリトロ混合物が得られた。シクロヘキサンから結晶化して、純粋なエリトロ1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールが得られた。
(エリトロ型)1H NMR(400MHz, CDCI3):δ 1.43(d,3H,CH3), 2.92(d,1H,OH), 4.84(m,1H,CH), 5.79(t,1H,CH), 7.34(d,1H,Ar-H), 7.40(d,1H,Ar-H), 7.59(d,1H,Ar-H)。
【0140】
b)2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンの調製:
【化60】
攪拌したエリトロ1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オール(2.5g, 10.0mmol)の無水ジクロロメタン(20ml)混合物に、窒素大気中で、ジクロロメタン(5ml)中のDAST(1.3ml, 10.0mmol)を、5℃に冷却しながら滴下した。当該溶液を、室温で1時間攪拌した。ジクロロメタン(80ml)を添加し、有機相を、飽和NaHCO3(50ml)、1M HC1(30ml)及び単独(sole)(30ml)で連続的に洗浄した。当該有機相をNaSO4で洗浄し、濾過し、そして濃縮した。2.5gの2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンは、褐色の油の形態で取得された。
【0141】
実施例P28:1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンの調製:
【化61】
攪拌した1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼン (5g, 18.72 mmol)のH2O (20 ml)及びMeOH(60 ml)溶液に常温で、窒素環境下で、鉄粉(2.355g, 42.12mmoles)及び濃塩酸(11.5 ml,112mmol)を添加した。当該反応混合物を、70℃で1時間攪拌した。1時間後、更に鉄粉(2.355g, 42.12mmoles)及び濃塩酸(11.5ml, 112mmol)を添加し、70℃で2時間攪拌を継続した。当該反応の完遂後(TLCにより確認)、反応物を室温に冷却し、MeOHを蒸発させた。得られた残留物を水で希釈し、酢酸エチル(3x80ml)で抽出した。一まとめにした酢酸エチルの相を水及びブラインで洗浄し、無水Na2SO4で脱水した。当該有機相を、真空下で濃縮した。粗材料をカラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン/酢酸エチル95:5)で精製して、3.7g(理論上83.3%)の1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンを得た。
【0142】
表1〜6:式Iaの化合物:
本発明は、下記表1〜6に列挙される、式(Ia)で表される好ましい個々の化合物により、更に例示された。特性のデータは、表9に示される。
【化62】
【0143】
式Iaの化合物において、Aは、A1
【化63】
A2
【化64】
及びA3
【化65】
殻なる群から選択され、そしてnは0又は1である。
【0144】
下記表Yの後の表1〜6のそれぞれは、100個の式(Ia)の化合物を含む。ここで、式(Ia)中、R4、R5、R7、X、R11、R12及びR13は表Yに記載の数値をとり、そしてAは関連する表1〜6の定義に記載のものが選択され、そしてnは関連する表1〜6の定義に記載の数値をとる。故に、表1は、表YのYが1で、且つAが表1の定義に記載のものであるときの表Yに対応し、表1は、表YのYが2で、且つAが表2の定義に記載のものであるときの表Yに対応し、そして以下表3〜6も同様である。
【0145】
表1〜6において「Me」はメチル、「Et」はエチル、「i-Pr」はイソプロピル、「c−Pr」はシクロプロピル、及び「t−Bu」は第三級ブチルを意味する。
【0146】
【表1】
【表2】
【表3】
【表4】
【0147】
表1は、100個の式(Ia)の化合物を提供し、ここでAはA1
【化66】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物1.001は、以下の構造を有する:
【化67】
【0148】
表2は、100個の式(Ia)の化合物を提供し、ここでAはA1
【化68】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物2.057は、以下の構造を有する:
【化69】
【0149】
表3は、100個の式(Ia)の化合物を提供し、ここでAはA2
【化70】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物3.002は、以下の構造を有する:
【化71】
【0150】
表4は、100個の式(Ia)の化合物を提供し、ここでAはA2
【化72】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物4.003は、以下の構造を有する:
【化73】
【0151】
表5は、100個の式(Ia)の化合物を提供し、ここでAはA3
【化74】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物5.002は、以下の構造を有する:
【化75】
【0152】
表6は、100個の式(Ia)の化合物を提供し、ここでAはA3
【化76】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物6.003は、以下の構造を有する:
【化77】
【0153】
表7及び8:
上記表Yの後の表7及び8のそれぞれは、100個の式(IIb)の化合物を含む。ここで、式(IIb)中、R4、R5、R7、X、R11、R12及びR13は表Yに記載の数値をとり、そしてnは関連する表7及び8の定義に記載の数値をとる。故に、表7は、表YのYが7であるときの表Yに対応し、表8は、表YのYが8であるときの表Yに対応する。
【0154】
表7は、100個の式(IIb)
【化78】
の化合物を提供し、ここでnは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物7.001は、以下の構造を有する:
【化79】
【0155】
表8は、100個の式(IIb)
【化80】
の化合物を提供し、ここでnは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。
【0156】
表9:特性データ:
表9は、表1〜6の化合物の選択された融点及び選択されたNMRデータを示す。特に言及が無い限り、NMR測定の溶媒として、CDCI3が使用される。溶媒の混合物が存在する場合、例えば:)と表される。いかなる場合も、全ての特性データを列記することを試みていない。表9及び以下の記載全体において、温度はセルシウス度で示され;「NMR」は核磁気共鳴スペクトルを意味し;MSは質量スペクトルを意味し;「%」は、対応する濃度が他の単位で示されていない限り、重量パーセントである。以下の略語は、本願全体で使用されている:
【表5】
【0157】
【表6】
【表7】
【0158】
式Iの化合物の製剤の例:
実施例F-1.1〜F-1.2:乳剤
【表8】
これらの濃縮物を水で希釈することにより、任意の所望の濃度の乳濁物を調製することができる。
【0159】
実施例F-2:乳剤
【表9】
これらの濃縮物を水で希釈することにより、任意の所望の濃度の乳濁物を調製することができる。
【0160】
実施例F-3.1〜F-3.4:溶液
【表10】
これらの溶液は、微液滴の形態での使用に適している。
【0161】
実施例F-4.1〜F-4.4:顆粒
【表11】
本発明の新規化合物をジクロロメタン中に溶解し、当該溶液を担体上に噴霧し、そして溶媒を真空下で蒸発により除去する。
【0162】
実施例F-5.1及びF-5.2:粉塵
【表12】
全ての材料を念入りに混合して、使用可能な粉塵が得られる。
【0163】
実施例F-6.1〜F-6.3:水和剤
【表13】
全ての材料を混合し、当該混合物を適切なミル中で激しく挽き潰すことにより、水和物が得られる。当該水和物を水で希釈することにより、任意の所望の濃度の懸濁物を得ることができる。
【0164】
実施例F7:種子処理用流動性濃縮物
【表14】
細かく挽き潰した有効成分を助剤と念入りに混合することにより、懸濁濃縮物が得られる。当該懸濁濃縮物を水で希釈することにより、任意の所望の希釈率の懸濁物を取得することができる。そのような希釈物を、噴霧、注入又は浸漬により使用して、生きた植物の他に植物育苗材料の、微生物の繁殖を処理及び防御できる。
【0165】
生物学的実施例:殺真菌作用:
実施例B-1:ボトリティス・キネレア(Botrytis cinerea)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、3〜4日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0166】
マイコスファエレラ・アラキディス(Mycosphaerella arachidis)(ラッカセイの早期斑点病(early leaf spot);ケルコスポラ・アラキディコラ(Cercospora arachidicola)[アナモルフ])に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、6〜7日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059、2.003及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0167】
実施例B-3:セプトリア・トリティキ(Septoria tritici)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、72時間後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059、2.003及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0168】
実施例B-4:タペシア・ワルンダエ(Tapesia vallundae)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、6〜7日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0169】
実施例B-5:モノグラフェラ・ニワリス(Monographella nivalis)(アナモルフ:フサリウム・ニワレ(Fusarium nivale)、ミクロドキウム・ニワレ(Microdochium nivale);雪腐れ病)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、72時間後に、増殖の阻害を光学的に測定した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.057は、この試験で良好な活性を示している(≦50%阻害率)。
【0170】
実施例B-6:リゾクトニア・ソラニ(Rhizoctonia solani)に対する作用-真菌増殖アッセイ
前記真菌の新しく増殖させた液体培養物の菌糸断片を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、3〜4日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率で表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.003は、この試験で良好な活性を示している(≦50%阻害率)。
【0171】
実施例B-7:エリシフェ・グラミニスf.sp.トリティキ(Erysiphe graminis f.sp. tritici)(うどんこ病)に対する作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の7日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.033、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0172】
実施例B-8:コムギのプッキニア・レコンディタ(Puccinia recondita)(茶さび(brown rust))に対する作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の8日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.032及び1.057は、この試験で良好な活性を示している(≦50%阻害率)。
【0173】
実施例B-9:コムギのプッキニア・レコンディタ(Puccinia recondita)(茶さび(brown rust))に対する治療的作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、前記真菌の胞子懸濁物を接種した。接種の翌日、当該葉の断片に試験溶液(0.02%有効成分)を噴霧した。適切なインキュベーションの後、化合物の活性を、前記添加の8日後の殺真菌活性の防止の程度として評価した。化合物1.002及び1.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.015は、この試験で良好な活性を示している(≦50%阻害率)。
【0174】
実施例B-10:オオムギのピレノフォラ・テラス(Pyrenophora teres)(網斑病)に対する作用
オオムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の4日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.033、1.059及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【技術分野】
【0001】
本発明は、殺微生物剤として活性の、特に殺真菌剤として活性の、新規カルボキサミドに関する。更に、本発明は、これらの化合物の調製に使用される中間体、これらの化合物を含有する組成物、及び植物への植物病原性微生物、好ましくは真菌の繁殖を防除又は予防するための、農業又は園芸における、それらの使用に関する。
【背景技術】
【0002】
殺真菌剤として活性のカルボキサミドは、EP 1787981及びEP 1792901に記載されている。
【0003】
特定の置換基のパターンを有する新規のカルボキサミドが、殺微生物活性を有することが見出された。
【発明の概要】
【課題を解決するための手段】
【0004】
故に、本発明は、式I
【化1】
[式中、
R1はC1-C4アルキル又はC1-C4ハロアルキルであり;
R2はC1-C4アルキルであり;
R3は水素又はハロゲンであり;
R4は水素、C1-C4アルキル又はC1-C4ハロゲンアルキルであり;
R5、R6、R8、R9及びR10は、互いに独立して、水素、ハロゲン、C1-C4アルキル又はC1-C4ハロアルキルであり;
R7は水素、ハロゲン、C1-C4アルキル、C2-C6アルケニル又はC3-C6アルキニルであり;
R11は水素、ハロゲン又はC1-C6アルキルであり;
R12は水素、ハロゲン、C1-C6アルキル、C2-C6アルケニル、C3-C6アルキニル、C3-C6シクロアルキル-C3-C6アルキニル、ハロフェノキシ、ハロフェニル-C3-C6アルキニル、C(C1-C4アルキル)=NO-C1-C4アルキル、C1-C6ハロアルキル、C1-C6ハロアルコキシ、C2-C6ハロアルケニル、又はC2-C6ハロアルケニルオキシであり;
R13は水素、ハロゲン、C1-C6アルキルであり;
Xは酸素又は硫黄又は存在せず;但しXが酸素又は硫黄であるときR7はハロゲンではなく;
nは0又は1である]
の化合物、及びその農学的に許容される塩/異性体/構造異性体/立体異性体/ジアステレオ異性体/鏡像異性体/互換異性体、並びにそれらの化合物のN-オキシドに関する。
【0005】
前記置換基の定義にあるアルキル基は、直鎖又は分岐鎖であることができ、例えば、メチル、エチル、n-プロピル、n-ブチル、n-ペンチル、n-ヘキシル、iso-プロピル、n-ブチル、sec-ブチル、iso-ブチル又はtert-ブチルである。前記アルコキシ、アルケニル及びアルキニル基は、上記アルキル基に由来する。前記アルケニル及びアルキニル基は、一不飽和又は二不飽和であることができる。前記置換基の定義にあるシクロアルキル基は、例えば、シクロプロピル、シクロブチル、シクロペンチル又はシクロヘキシルである。ハロゲンは、一般に、フッ素、塩素、臭素又はヨウ素であって、好ましくはフッ素、臭素又は塩素である。これは、他の語との組合せ、例えばハロゲンアルキル又はハロゲンアルコキシ等の中のハロゲンにも当てはまる。ハロアルキル基は、好ましくは、1〜4個の炭素原子の長さの鎖を有する。ハロアルキルは、例えば、フルオロメチル、ジフルオロメチル、トリフルオロメチル、クロロメチル、ジクロロメチル、トリクロロメチル、2,2,2-トリフルオロエチル、2-フルオロエチル、2-クロロエチル、ペンタフルオロエチル、1,1-ジフルオロ-2,2,2-トリクロロエチル、2,2,3,3-テトラフルオロエチル及び2,2,2-トリクロロエチル;好ましくはトリクロロメチル、ジフルオロクロロメチル、ジフルオロメチル、トリフルオロメチル及びジクロロフルオロメチルである。アルコキシは、例えば、メトキシ、エトキシ、プロポキシ、i-プロポキシ、n-ブトキシ、iso-ブトキシ、sec-ブトキシ及びtert-ブトキシ;好ましくはメトキシ及びエトキシである。ハロゲンアルコキシは、例えば、フルオロメトキシ、ジフルオロメトキシトリフルオロメトキシ、2,2,2-トリフルオロエトキシ、1,1,2,2-テトラフルオロエトキシ、2-フルオロエトキシ、2-クロロエトキシ、2,2-ジフルオロエトキシ及び2,2,2-トリクロロエトキシ;好ましくはジフルオロメトキシ、2-クロロエトキシ及びトリフルオロメトキシである。
【0006】
好ましい式Iの化合物において、互いに独立して、
a)R1はジフルオロメチル、トリフルオロメチル又はメチル;
b)R2はメチル;
c)R3は水素又はフルオロ;
d)R4は水素、メチル又はエチル;
e)R4はメチル;
f)R5は水素又はメチル;
g)nは0;
h)Xは酸素;
i)R8、R9及びR10は水素;
j)R11、R12及びR13は水素又はクロロ;
k)R12はクロロ又はC1-C4アルキル;
I)R6は水素;そして
m)R7はメチル
である。
【0007】
特に好ましい式Iの化合物は、
R1はジフルオロメチル又はトリフルオロメチル;
R2はメチル;
R3は水素;
R4はメチル;
R11、R12及びR13は、互いに独立して、水素又はハロゲン、好ましくは水素又は塩素である。
【0008】
更に好ましい式Iの化合物は、
Xが酸素で、同時にR7がC1-C4アルキル、好ましくはメチル;又はXが存在せずR7が水素である。
【0009】
式Iの化合物は、式II
【化2】
で表され、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が式Iで定義したものである化合物を、式III
【化3】
で表され、R1、R2及びR3が式Iで定義したもので、R*がハロゲン、ヒドロキシ又はC1-6アルコキシ、好ましくはクロロである化合物と反応させることにより、調製されてもよい。
【0010】
式Iの化合物を得るための反応は、非プロトン性の有機溶媒中で実施されるのが有利である。そのような溶媒は、ベンゼン、トルエン、キシレン若しくはシクロヘキサン等の炭化水素、ジクロロメタン、トリクロロメタン、テトラクロロメタン又はクロロベンゼン等の塩素化炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、アセトニトリル又はプロピオニトリル等のニトリル、N,N-ジメチルホルムアミド、ジエチルホルムアミド若しくはN-メチルピロリジノン等のアミドである。前記反応の温度は、-20℃〜+120℃の間が有利である。一般に、前記反応は僅かに発熱性であるので、通常、常温で実施されることができる。反応時間を短縮するために、又は反応を開始するために、混合物が沸点近くまで短時間加熱されてもよい。前記反応時間は、反応触媒として数滴の塩基を添加することによっても短縮できる。適切な塩基は、特に、第三級アミン、例えばトリメチルアミン、トリエチルアミン、キヌクリジン(quinuclidine)、1,4−ジアザビシクロ[2.2.2]オクタン、1,5−ジアザビシクロ[4.3.0]ノン-5−エン又は1,5−ジアザビシクロ[5.4.0]ウンデク-7−エンである。しかしながら、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基も、塩基として使用されてもよい。前記塩基は、単独で、あるいは触媒として使用される量の相間移動触媒、例えばクラウンエーテル特に18−クラウン−6、又はテトラアルキルアンモニウム塩等と共に使用できる。
【0011】
R*がヒドロキシのとき、ベンゾトリアゾール-
1−イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート、bis-(2−オキソ−3−オキサゾリジニル)−ホスフィン酸塩化物(BOP-CI)、N,N'-ジシクロヘキシルカルボジイミド(DCC)又は1,1’−ジイミダゾール(CDI)等のカップリング剤が使用されてもよい。
【0012】
式II
【化4】
で表され、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が式Iで定義したものであり、好ましくはR4がC1-C4アルキルである中間体は、新規であって、式Iの化合物の調製のために特別に開発された。故に、式IIの中間体は、本発明の対象の部分を形成する。
【0013】
式Iの化合物の好ましい置換基の設定は、式IIの化合物にも適用される。故に、好ましい式IIの化合物は、互いに独立して、
a)R4は水素、メチル又はエチル;特に好ましくはメチル;
b)R5は水素又はメチル;
c)nは0;
d)Xは酸素;
e)R8、R9及びR10は水素;
f)R11、R12及びR13は水素又はクロロ;
g)R12はクロロ又はC1-C4アルキル;
h)R6は水素;そして
i)R7はメチル
である。
【0014】
式IIA
【化5】
で表され、R4、R5、X、R7、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム1に記載のようにして調製されてもよい。
【0015】
【化6】
式VIで表され、R5、R11、R12及びR13が式IIAで定義したものであるニトロアルケンは、常温と対流温度の間の温度で、酢酸及び酢酸アンモニウムの存在下、式Vで表され、R5が式IIAで定義したものであるニトロアルカンを、式(IV)で表され、R11、R12及びR13が式IIAで定義したものであるカルボニル化合物と、Henry反応(ニトロアルドール反応)させることにより、調製できる。
【0016】
式VIIaで表され、R7及び式Iで定義したものであり、更にMがLi、Na、K又は水素である化合物の、式VIで表されるニトロアルケンへのMichael付加は、対応するアルコール、チオール、トルエン又はエーテル溶媒、例えばジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン中のアルカリ土類アルコラート、好ましくはナトリウム、カリウム、リチウム塩等を使用して達成されてもよく、これにより、式VIIIで表され、R5、R7、R11、R12及びR13が式IIAで定義したものであるニトロアルカンを形成する。
【0017】
式VIで表され、R5、R11、R12及びR13が式IIAで定義したものであるニトロアルケンは、鉄及び塩酸を用いて還元されてもよく、これにより、式IXで表され、R5、R11、R12及びR13が式IIAで定義したものであるオキシムが得られる。前記オキシムは、式Xaで表され、R5、R11、R12及びR13が式IIAで定義したものであるケトンに加水分解できる。これは、例えば、M. Kulka and H. Hibbert J. Am. Chem. Soc. 65, 1180 (1943)及びPrasun K. Pradhan et al. Synthetic Commun., 35, 913-922, 2005に記載されている。前記反応は、40〜100℃の間の温度で、メタノール、エタノール、tert-ブチル、トリフルオロエタノール又はジオキサン等の、手近な有機溶媒中で実施される。
【0018】
式Xaで表されるケトンを、R7−Yで表され、R7が式IIAで定義したものであり、Yが、ハロゲン、メシレート又はトシレート等の離脱基を表す化合物を用いて塩基の存在下でアルキル化することにより、式Xbで表され、R5、R7、R11、R12及びR13が式IIAで定義したものであるα-アルキル化ケトンが得られる。前記アルキル化反応は、非プロトン性の不活性有機溶媒中で実施するのが有利である。そのような溶媒は、ベンゼン、トルエン、キシレン又はシクロヘキサン等の炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、N,N-ジメチルホルムアミド、ジエチルホルムアミド又はN-メチルピロリドン等のアミドである。反応温度は、−20℃〜+120℃の間である。適切な塩基は、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基である。前記塩基は、単独で、あるいは触媒として使用される量の相間移動触媒、例えばクラウンエーテル特に18−クラウン−6、又はテトラアルキルアンモニウム塩等と共に使用できる。
【0019】
式XIIa、XIIbで表されるO-アルコキシオキシム誘導体は、式Xa及びXbで表されるケトンを、式XIで表されるO-アルキルヒドロキシアミン誘導体又はその塩を用いてオキシム化することにより、調製されてもよい。前記オキシム化工程を実行するのに適した溶媒は、ベンゼン、トルエン、キシレン若しくはシクロヘキサン等の炭化水素、ジクロロメタン、トリクロロメタン、テトラクロロメタン又はクロロベンゼン等の塩素化炭化水素、ジエチルエーテル、エチレングリコールジメチルエーテル、ジエチレングリコールジメチルエーテル、テトラヒドロフラン又はジオキサン等のエーテル、アセトニトリル又はプロピオニトリル等のニトリル、N,N-ジメチルホルムアミド、ジエチルホルムアミド若しくはN-メチルピロリジノン水又は混合物等のアミドである。前記反応温度は、−20℃〜+120℃の間であるのが有利である。一般に、前記反応は、常温で実施できる。適切な塩基は、具体的には、ピリジン、第三級アミン、例えばトリメチルアミン、トリエチルアミン、キヌクリジン、1,4−ジアザビシクロ[2.2.2]オクタン、1,5−ジアザビシクロ[4.3.0]ノン-5−エン又は1,5−ジアザビシクロ[5.4.0]ウンデク-7−エンである。しかしながら、水素化物、例えば水素化ナトリウム若しくは水素化カルシウム、水酸化物、例えば水酸化ナトリウム又は水酸化カリウム、炭酸塩、例えば炭酸ナトリウム及び炭酸カリウム、又は炭酸水素塩、例えば炭酸水素カリウム及び炭酸水素ナトリウム等の無機塩基も、塩基として使用されてもよい。
【0020】
式XIIIで表されるオキシム誘導体は、式VIで表されるニトロアルケンを、式VIIbで表されるアルコール又はチオール中で、SnCI2.2H2Oを使用するR.S. Varma and G.W. Kabalka Chem. Lett, 243-244 (1985)で報告された手順に従い、選択的に還元することにより調製されてもよい。式XIIcで表されるオキシムエーテル誘導体は、式XIIIで表されるオキシム誘導体を、R4−Yで表され、R4が式IIAで定義したものであり、Yが、ハロゲン、メシレート又はトシレート等の離脱基を表す化合物を用いて、塩基の存在下でO-アルキル化することにより調製されてもよい。あるいは、式XIIIcで表されるオキシムエーテル誘導体は、式XIVで表されるケトンを、式XIで表されるO-アルキルヒドロキシルアミン誘導体、又はその塩でオキシム化することによって調製されてもよい。
【0021】
式XIVで表されるケトンは、式VIIIで表されるニトロアルカンを、J. M Aizpurua and C. Palomo THL, Vol. 28, No.44,pp 5361-5364 (1987)に記載される手順に従い、Nef反応で変換することにより調製されてもよい。
【0022】
式IIAa、IIAb及びIIAcで表されるO-アルキルヒドロキシアミンは、式XIIa、XIIb及びXIIcで表されるO-アルコキシオキシム誘導体の還元により調製されてもよい。
【0023】
式IIA
【化7】
で表され、R4、R5、R7、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム2に記載されるように調製されてもよい。
【0024】
【化8】
式VIで表されるαアルキル化アルキルアリール酢酸塩誘導体は、塩基の存在下で、式XVのアリール酢酸誘導体をR7-Y等のハライドでアルキル化することにより合成でき、ここでR7は式IIAで定義したものであり、Yは、ハロゲン、メシレート、トシレート等の離脱基を表す。式XVIの化合物は、LiOH等の水酸化物により加水分解される。得られた式XVIIの酸を、対応するアシルクロライドに変換することができ、このアシルクロライドを、そのままN,O-ジメチルヒドロキシルアミンと反応させて、式XVIIIで表され、R7、R11、R12及びR13が式IIAで定義したものであるWeinrebアミドを得ることができる。続いての、式R5-MgBrで表され、が式IIAで定義したものであるグリニャール試薬との反応で、式Xbのケトンが得られ、これが、スキーム1に記載の反応により、式IIAbの化合物に変換されることができる。
【0025】
式Xb1のアルデヒド誘導体は、式XVIIIのWeinrebアミドを、LiAlH又はDIBAL-Hを用いて部分的に還元することにより調製されてもよい。
【0026】
式IIA
【化9】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム3に記載されるように調製されてもよい。
【0027】
【化10】
式Xa1のアルデヒド誘導体は、式XIXの酸誘導体を還元して式XXのアルコール誘導体を得て、続いてこれを式XXIの活性化ベンジル誘導体に変換し、続いてこれを式XXIIのニトリル誘導体に変換し、続いてこれを還元して、式Xa1のアルデヒド誘導体を得ることにより、調製されることができる。この過程は、「調製」の項に記載されている。続いての式XXIIのニトリル誘導体と、R5-MgBrで表され、R5が式IIAで定義したものであるグリニャール試薬と反応させて、式Xaのケトンを取得し、これを、スキーム1に記載の反応により、式IIAbの化合物に転換することができる。
【0028】
式IIB
【化11】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム4に記載されるように調製されてもよい。
【0029】
【化12】
式VIのアルデヒドは、Meldrum酸及びトリエチル蟻酸アンモニウムで、式XXIIIの対応する酸に変換することができる。この反応は、例えば、G.Toth et. al. in Synth. Commun. 25 (19), 3067-3074 (1995)に記載されている。この酸を、N,O-ジメチルヒドロキシルアミンとそのまま反応させて、式XXIVのWeinrebアミドが得られる。当該式XXIVのWeinrebアミドを、R5-MgYで表され、R5が式IIAで定義したものであるグリニャール試薬と反応させて、式XXVaのケトンが得られる。
【0030】
式XXVbのアルデヒドは、式XXIVのWeinrebアミドをLiAlH4又はDIBAL-Hで部分的に還元することにより調製されてもよい。あるいは、当該アルデヒドは、式XXVIのアルコールの酸化により調製されてもよい。適切な酸化剤として、ピリジニウムクロロクロメート(PCC)、Swern試薬(オキサリルクロライド/DMSO)、Dess-Martin Periodinane、及びMnO2が挙げられる。適切な溶媒として、ジクロロメタン及びTHFが挙げられる。反応温度は、典型的には、−50℃〜20℃の範囲内である。
【0031】
式XXVa及びXXVbのケトン及びアルデヒド誘導体は、更に、スキーム1に記載の反応により、式IIBa及びIIBbの化合物に変換することができる。
【0032】
式IIB
【化13】
で表され、R4、R5、R11、R12及びR13が式Iで定義したものである中間体は、反応スキーム5に記載されるように調製されてもよい。
【0033】
【化14】
式XXIXのケトン又はアルデヒド誘導体は、式XXVIIIのアリル型アルコールを、水中の式XXVIIのアリールヨーダイドで、パラジウム触媒アリール化することにより、調製されてもよく、この方法は、例えばHong Zhao, Ming-Zhong Cai et. al. in Synth. Commun. 31 (23), 3665-3669 (2001); Alberto Scrivanti, Ugo Matteoli et. al. in Tetrahedron 64, 543- 548 (2008)に記載されている。式XXIXのケトン及びアルデヒド誘導体は、更に、スキーム1に記載の反応により、式IIBcの化合物に変換されることができる。
【0034】
化合物I及び適切な場合その互換異性体は、適切な場合、水和物及び/又は、現在固体状態の化合物の結晶化に使用された他の溶媒を含有する形態で得られることができる。
【0035】
本発明の式Iの化合物は、事実上、真菌、細菌又はウイルス等の植物病原性微生物により引き起こされる病害から有用植物を保護するのに、非常に有利な範囲の活性を有することが見出された。
【0036】
本発明は、有用植物への植物病原性微生物の繁殖を防除又は予防する方法に関し、当該方法において、式Iの化合物を、有効成分として、植物、その部分又はその生育場所に施用する。本発明の式Iの化合物は、低い施用率での優秀な活性、植物に対する良好な寛容性、及び環境安全性を特徴とする。それらは、非常に有用な、治療的、予防的及び浸透性の特徴を有し、多くの有用植物の保護に使用される。式Iの化合物は、様々な有用植物の作物の植物体又は植物の部分(果実、花、葉、茎、塊茎、根)に生じる病害を阻害又は死滅させるのに使用されるが、同時に、後日生育する植物の部分をも、植物病原性微生物から保護される。
【0037】
植物育苗材料、具体的には種子(果実、塊茎、穀物)及び挿し木(plant cutting)(例えばイネ)等を、真菌の繁殖、及び土壌中に発生する植物病原性の真菌から保護するための処理用のドレッシング剤として、式Iの化合物を使用することも可能である。
【0038】
更に、本発明の式Iの化合物は、関連する領域、例えば技術材料、例えば木材及び木材に関連する技術製品の保護、食料の保存、又は衛生管理において、真菌を防除するのに使用されてもよい。
【0039】
式Iの化合物は、例えば、以下のクラスの植物病原性の真菌に対して効果的である:
不完全菌類(例えばボトリティス(Botrytis)、ピリクラリア(Pyricularia)、ヘルミントスポリウム(Helminthosporium)、フサリウム(Fusarium)、セプトリア(Septoria)、ケルコスポラ(Cercospora)及びアルテルナリア(Alternaria)等)並びに担子菌類(例えばリゾクトニア(Rhizoctonia)、ヘミレイア(Hemileia)、プッキニア(Puccinia)。加えて、子嚢菌類のクラス(例えばウェンツリア(Venturia)及びエリシフェ(Erysiphe)、ポドスファエラ(Podosphaera)、モニリニア(Monilinia)、ウンキヌラ(Uncinula))並びに卵菌類(例えばフィトフトラ(Phytophthora)、ピチウム(Pythium)、プラスモパラ(Plasmopara))に対しても効果的である。うどん粉病菌(エリシフェ(Erysiphe spp.))に対して、卓越した活性が観察された。更に、当該式Iの新規化合物は、植物病原性の細菌及びウイルス、例えばキサントモナス(Xanthomonas spp)、シュードモナス(Pseudomonas spp)、エルウィニア・アミロウォラ(Erwinia amylovora)に対して、及びタバコモザイクウイルスに対して効果的である。アジアダイズサビ病(ファコプソラ・パキリジ(Phakopsora pachyrhizi))に対して、良好な活性が観察された。
【0040】
本発明の範囲内で、保護される有用植物は、典型的には、以下の植物種である:穀類(コムギ、オオムギ、ライムギ、オーツムギ、イネ、トウモロコシ、ソルガム及び関連する種);ビート(テンサイ及び飼料用テンサイ);漿果、石果及び軟果(リンゴ、洋ナシ、プラム、桃、アーモンド、桜桃、イチゴ、ラズベリー及びブラックベリー);マメ科植物(マメ、レンティル、エンドウ、ダイズ);油脂植物(アブラナ、カラシナ、ポピー、オリーブ、ヒマワリ、ココヤシ、トウゴマ、カカオ豆、ラッカセイ);ウリ科植物(カボチャ、キュウリ、メロン);線維植物(綿花;亜麻;ヘンプ;ジュート);柑橘類(オレンジ、レモン、グレープフルーツ、マンダリン);野菜(ホウレンソウ、レタス、アスパラガス、キャベツ、ニンジン、タマネギ、トマト、ジャガイモ、パプリカ);クスノキ科植物(アボカド、シナモン、樟脳)又はタバコ、ナッツ、コーヒー、ナス、サトウキビ、茶、コショウ、ブドウ、ホップ、バナナ及び天然ゴム、更に観葉植物。
【0041】
「有用植物」という用語は、公知の育種又は遺伝子組換えの方法の結果として、ブロモキシニル等の除草剤又は除草剤のクラス(例えばHPPD阻害剤、ALS阻害剤、例えばプリミスルフロン、プロスルフロン及びトリフロキシスルフロン、EPSPS(5−エノール−プロビル−シキメート−3−リン酸−シンターゼ)阻害剤、GS(グルタミンシンターゼ)阻害剤又はPPO(プロトポルフィリノーゲン−オキシダーゼ)阻害剤等)に対して耐性を獲得した有用植物をも含むものとして理解されたい。公知の育種方法(突然変異生成)によりイミダゾリノン、例えばイマザモックスに対して耐性を獲得した作物の一例として、Clearfield(登録商標)夏アブラナ(Canola)が挙げられる。遺伝子組換え手法により除草剤又は除草剤のクラスに対して耐性を獲得した作物の例として、RoundupReady(登録商標)、Herculex I(登録商標)及びLibertyLink(登録商標)の商標の下で市販される、グリホサート及びグルフォシネート耐性トウモロコシ変異体が含まれる。
【0042】
また、「有用植物」という用語は、組換えDNA技術を使用して、例えば毒素生産細菌に由来するものとして知られる1つ以上の選択的に作用する毒素、特にバチルス(Bacillus)属等に由来する毒素等を生産するように形質転換した有用植物をも含むものとして理解されたい。
【0043】
そのような植物の例として:YieldGard(登録商標)(CryIA(b)毒素を発現するトウモロコシ変異体);YieldGard Rootworm(登録商標)(CryIIIB(b1)毒素を発現するトウモロコシ変異体);YieldGard Plus(登録商標)(CryIA(b)及びCryIIIB(b1)毒素を発現するトウモロコシ変異体;Starlink(登録商標)(Cry9(c)毒素を発現するトウモロコシ変異体);Herculex I(登録商標)(CryIF(a2)毒素及び酵素ホスフィノツリシンN-アセチルトランスフェラーゼ(PAT)を発現し、グルフォシネートアンモニウムに対する耐性を得たトウモロコシ変異体);NuCOTN 33B(登録商標)(CryIA(c)毒素を発現する綿花変異体);Bollgard I(登録商標)(CryIA(c)毒素を発現する綿花変異体);Bollgard II(登録商標)(CryIA(c)及びCryIIA(b)毒素を発現する綿花変異体);VIPCOT(登録商標)(VIP毒素を発現する綿花変異体);NewLeaf(登録商標)(CryIIIA毒素を発現するジャガイモ変異体);Nature- Gard(登録商標)Agrisure(登録商標)GT Advantage(GA21グリホサート耐性形質), Agrisure(登録商標)CB Advantage (Bt11 アワノメイガ(CB)形質)、Agrisure(登録商標)RW (根きり虫(corn rootworm)形質)及びProtecta(登録商標)が挙げられる。
【0044】
また、「有用植物」という用語は、組換えDNA技術を使用して、いわゆる「病原性関連タンパク質」(PRP、例えばEP−A−0 392 225を参照されたい)等の、選択的活性を有する抗病原性物質を合成するように形質転換した有用植物をも含むものとして理解されたい。そのような抗病原性物質及びそのような抗病原性物質を合成できる組換え植物は公知であり、例えばEP−A−0 392 225、WO 95/33818、及びEP−A−0 353 191に記載されている。そのような遺伝子組換え植物を作製する方法は、当業者に周知であり、また、例えば上で示した文献中等に記載されている。
【0045】
本明細書中で使用されるとき、有用植物の「生育場所」という用語は、有用植物が生育する場所、有用植物の植物育苗材料が播種される場所、又は有用植物の植物育苗材料が土中に埋められる予定の場所を含む。そのような生育場所の一例が、作物植物が生育している土地である。
【0046】
「植物育苗材料」という用語は、子孫を繁殖させるのに使用され得る、種子、及び植物材料、例えば挿し木又は例えばジャガイモの塊茎等の、植物の生産的部分を意味するものとして理解されたい。狭義では種子を指すが、根、果実、塊茎、鱗茎、根茎、及び植物の部分をも指す。発芽後、又は土壌から出芽後に移植されるべき発芽した植物又は幼若植物も該当し得る。これらの幼若植物は、浸漬による全体又は部分処理により、移植前に保護される。好ましくは「植物育苗材料」は、種子を意味するものとして理解されたい。
【0047】
式Iの化合物は、未修飾の形態で使用することができ、又は、好ましくは、製剤の分野で伝統的に採用される担体及び助剤と共に使用することができる。
【0048】
従って、本発明は、式Iの化合物及び不活性の担体を含有する、植物病原性微生物を防除及び防御するための組成物、並びに、不活性の担体及び有効成分として式Iの化合物を含有する組成物が、植物、その部分又はその生育場所に適用される、有用植物への植物病原性微生物の繁殖を防除又は予防する方法にも関する。
【0049】
そのために、式Iの化合物及び不活性の担体は、公知の方法で、乳剤、被覆ペースト、直接スプレー可能な、又は希釈可能な溶液、希釈乳濁物、水和剤、可溶性の粉末、粉塵、顆粒、及びポリマー物質のカプセル等に、容易に製剤化される。前記組成物の種類と同様に、スプレー、散布(atomising)、散粉(dusting)、散乱(scattering)、コーティング又は注入等の施用方法が、本来の目的及び周囲の状況に従い選択される。また、前記組成物は、安定化剤、消泡剤、粘度調整剤、結合剤又は粘着付与剤(tackifier)等の更なる助剤、及び肥料、微量栄養素供与剤又は他の特別な機能を付与する製剤を含んでもよい。
【0050】
適切な担体及び助剤は個体又は液体であり、製剤技術において使用される、例えば天然又は再生された鉱物性物質、溶媒、分散剤、水和剤、粘着付与剤、増粘剤、結合剤又は肥料等の物質である。そのような担体は、例えばWO 97/33890に記載されている。
【0051】
式Iの化合物又は不活性の担体及び有効成分として式Iを含有する組成物は、処理される植物の生育場所又は植物体に、更なる化合物と同時に、又は連続して適用することができる。これらの更なる化合物は、例えば、肥料若しくは微量栄養素供与剤、又は植物の生育に影響する他の調製物等であることができる。また、それらは、選択的な除草剤、及び殺虫剤、殺真菌剤、殺細菌剤、殺線虫剤、殺軟体動物剤、又はこれらの調製物の幾つかの混合物であることができ、必要に応じて、更なる担体、界面活性剤、又は製剤分野で日常的に採用される施用を促進する助剤を加えてもよい。
【0052】
式Iの化合物又は不活性の担体及び有効成分として式Iを含有する組成物を施用する好ましい方法は、葉上施用である。施用の頻度及び施用率は、関連する病原体の繁殖のリスクに依存する。しかしながら、式Iの化合物は、液体製剤で植物の生育場所を濡らすことにより、又は顆粒等の固体形態の前記化合物を土壌に施用することにより(土壌施用)、土壌越しに根を通じて植物体に浸透させることもできる(全身作用)。水稲の作物において、そのような顆粒を、水田に適用することができる。また、式Iの化合物は、前記殺真菌剤の液体製剤で種子又は塊茎を浸含し、あるいは固体製剤でそれらをコーティングすることにより種子に施用されてもよい。
【0053】
製剤、即ち式Iの化合物及び必要に応じて固体又は液体の助剤を含有する組成物は、公知の方法、典型的には、化合物を、増量剤、例えば溶媒、固体担体、任意で界面活性化合物(界面活性剤)等と共に、念入りに混合及び/又は挽き潰すことにより調製される。
【0054】
前記農業化学製剤は、通常、0.1〜99重量%、好ましくは0.1〜95重量%の式Iの化合物、99.0〜1重量%、好ましくは99.8〜5重量%の固体又は液体助剤、及び0〜25重量%、好ましくは0.1〜25重量%の界面活性剤を含む。
【0055】
市販の製品としては濃縮品として製剤するのが好ましいが、最終消費者は、通常は希釈製剤を使用する。
【0056】
有利な施用率は、通常5g〜2kg有効成分(a.i.)/ヘクタール(ha)、好ましくは10g〜1kg a.i./ha、最も好ましくは20g〜600g a.i./haである。種子浸漬剤として使用されるとき、簡便な施用率は、種子1kgあたり有効成分10mg〜1gである。所望の効果を奏する施用率は、実験により決定できる。当該施用率は、例えば、作用の種類、有用植物の発達段階、及び施用(場所、時期、施用方法)に依存し、これらのパラメーターにより、広い範囲で変化する。
【0057】
驚くべきことに、式Iの化合物は、有用植物の作物への植物病原性微生物の攻撃を防御する方法に加え、植物病原性微生物が繁殖した有用植物の作物の処理方法にも使用することができ、当該処理方法は、グリホサート及び1つ以上の式Iの化合物の組合せ剤を、グリホサートに耐性又は感受性の植物の植物体又はその生育場所に投与することを含む。
【0058】
前記方法は、グリホサート無しで式Iの化合物を使用する場合と比較して、病害の防除作用の予期せぬ改善をもたらし得る。当該方法は、式Iの化合物による病害の防除の促進において、効果的であってもよい。グリホサートと1つ以上の式Iの化合物との混合物は、少なくとも部分的には、式Iの化合物によりコントロールされる病害の範囲を増やす場合もあるが、式Iの化合物によりある程度まで防除されることが既に知られている病害の種類に対する式Iの化合物の活性の増大も、観察される効果であることができる。
【0059】
当該方法は、菌界、担子菌門、サビ菌(Uredinomycetes)綱、サビ菌(urediniomycetidae)亜綱、及びサビ菌(Uredinales)目の植物病原性生物(一般的に、サビ菌類と呼ばれる)に対して特に有効である。農業において特に重大な影響を与えるサビ菌の種は、ファコプソラセア(Phakopsoraceae)科、特にファコプソラ属の種、例えばファコプソラ・パシリジ(Phakopsora pachyrhizi)(当該種は、アジアダイズサビと呼ばれている)、及びプッシニアセア(Pucciniaceae)科、特にプッシニア(Puccinia)属の種、例えばプッシニア・グラミニス(Puccinia graminis)(茎サビ又は黒色サビとして知られており、当該サビ病は穀草作物において問題となる病害である)、及び茶色サビとして知られているプッシニア・レコンジタ(Puccinia recondita)を含む。
【0060】
本方法の態様は、植物病原性生物による攻撃に対して有用植物である作物を保護する方法、及び/又は植物病原性生物に感染した有用植物の作物を治療する方法であって、当該方法は、その塩又はエステルを含むグリホサート、及び式Iの少なくとも1つの化合物を、植物、植物の部分、及び植物の生育場所からなる群から選択される少なくとも1のメンバーに同時に施用することを含む方法である。
【0061】
上記式Iの化合物、又はその医薬上の塩は、動物における微生物感染の治療及び/又は予防のための活性の有利な適用範囲をも有してもよい。「動物」は、任意の動物、例えば昆虫、哺乳動物、は虫類、魚、両生類であってもよく、好ましくは哺乳動物、最も好ましくはヒトである。「治療」は、感染の増加又は広がりを減少又は緩和又は停止するため、又は感染を低下又は感染を治癒するための微生物感染を有する動物における使用を意味する。「予防」は、微生物感染の明らかな兆候を有さない動物に対して、将来の感染を予防するため、又は将来の感染の増加又は広がりを低減又は緩和するために、使用することを意味する。
【0062】
本発明に従うと、動物において微生物感染の治療及び/又は予防において使用するための医薬の製造における式Iの化合物の使用も提供される。式Iの化合物を医薬として使用することも提供される。動物の治療において抗菌剤として式Iの化合物を使用することも提供される。本発明に従うと、活性成分として式Iの化合物、又はその医薬として許容される塩を含む医薬組成物が提供される。当該組成物は、動物において抗菌感染の治療及び/又は予防のために使用することができる。当該医薬組成物は、経口投与に適した形態、例えば錠剤、ロゼンジ、ハードカプセル、水性懸濁液、油状懸濁液、乳濁物、エマルジョン分散粉末、粉末可能顆粒、シロップ及びエリクシルの形態であってもよい。或いは、当該医薬組成物は、局所適用に適した形態、例えばスプレー、クリーム又はローションの形態であってもよい。或いは、この医薬組成物は、非経口投与に適した形態、例えば注射であってもよい。或いは、この医薬組成物は、吸入可能形態、例えばエアロゾルスプレーであってもよい。
【0063】
式Iの化合物は、動物において微生物感染を引き起こすことができる様々な微生物種に対して有効である。かかる微生物種の例は、アスペルギルス症を引き起こす微生物種、例えば、アスペルギルス・フミガツス(Aspergillus fumigatus)、A.フラブス(A.flavus)、A.テルス(A.terrus)、A.ニジュランス(A.nidulans)及びA.ニガー(A. niger)、ブラストミセス症を引き起こす種、例えば、ブラストマイシス・デルマチチジス(Blastomyces dermatitidis);カンジダ症を引き起こす種、例えばカンジタ・アルビカンス(Candida albicans)、C.グラブラタ(C. glabrata)、C.トロピカリス(C. tropicalis)、C.パラプシロシス(C. parapsilosis)、C.クルゼイ(C. krusei)及びC.ルシタニア(C. lusitaniae); コクシジオイデス症を引き起こす微生物種、例えばコクシジオイデス・イミチス(Coccidioides immitis);クリプトコッカス症を引き起こす種、例えばクリプトコッカス・ネオフォルマンス(Cryptococcus neoformans);ヒストプラスマ症を引き起こす種、例えばヒストプラスマ・カプスラツム(Histoplasma capsulatum)及び接合菌症を引き起こす種、例えばアブシジア・コリムビフェラ(Absidia corymbifera)、リゾムコール・プシルス(Rhizomucor pusillus)及びリゾプス・アリズス(Rhizopus arrhizus)である。さらなる例は、フサリウム種、例えばフサリウム・オキシスポルム(Fusarium oxysporum)及びフサリウム・ソラニ(Fusarium solani)及びセンドスポリウム種、例えばセンドスポリウム・アピオスペルマム(Scedosporium apiospermum)及びセドスポリウム・プロリフィカンス(Scedosporium prolificans)である。さらなる例は、マイクロスポルム種(Microsporum spp.)、トリコフィトン種(Trichophyton Spp)、エピデルモフィトン種(Epidermophyton Spp)、ムコール種(Mucor Spp)、スポロトリックス種(Sporothorix Spp)、フィアロフォア種(Phialophora Spp)、クラドスポリウム種(Cladosporium Spp)、ペトリエリジウム種(Petriellidium spp)、パラコッシジオイデス種(Paracoccidioides Spp)及びヒストプラスマ種(Histoplasma Spp.)である。
【0064】
以下の非限定的な実施例は、上記発明を、限定すること無く、より詳細に例示する。
【実施例】
【0065】
調製の例:
実施例P1:3-ジフルオロメチル-1-メチル-1H-pyrazole−4-カルボン酸[2-(4-クロロ-フェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.001 )の調製
【化15】
ジクロロメタン(5ml)中の3−ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(564mg;2.9mmol)を、実施例P13に記載のように調製されたN-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン(600mg;2.9mmol)、トリエチルアミン(0.80ml;5.8mmol)の0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を6時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c-ヘキサン/酢酸エチル1:1)。
【0066】
0.99g(理論上93.4%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(4-クロロ-フェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.001)が、樹脂状の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.33-1.37(d,3H); 2.77-2.82(dd,1H); 3.07-3.13(dd,1H); 3.64(s,3H); 3.94(s,3H); 4.63-4.68(m,1H); 6.98-7.28(m, 5H); 7.61 (s,1H)。
MS [M+H]+ 358/360。
【0067】
実施例P2:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.002)の調製
【化16】
3ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.91 g; 4.7 mmol)のジクロロメタン(5ml)溶液を、実施例P14に記載のように調製されたN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン(1.0 g; 4.27 mmol)、トリエチルアミン(0.90 ml; 6.4 mmol)の0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を1.5時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0068】
1.35g(理論上80.3%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-メトキシ-アミド(化合物1.002)は、白色の固体の形態で取得された(m.p. 98-1020C)。
1H NMR: (CDCI3, 400MHz):
1.41-1.46(d,3H); 2.99-3.04(dd,1 H); 3.17-3.23(dd,1H); 3.60(s,3H); 3.95(s,3H); 4.68-4.70(m,1H); 7.10-7.62(m,5H)。
MS [M+H]+ 392/394/396。
【0069】
実施例P3:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-2-(2,4,6-triクロロフェニル)-エチル]-アミド(化合物1.003)の調製:
【化17】
実施例P15dに記載のように調製されたO-メチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.65 g, 2.4 mmol)のジクロロメタン(5 ml)溶液にトリエチルアミン(0.844 ml, 6.0 mmol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.519 g, 2.67 mmol)の0℃のジクロロメタン溶液を滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブライン溶液で洗浄し、これを硫酸ナトリウムで脱水し、そして溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-アミド(0.51 g, 49%)を、m.p:110〜112℃の白色の固体として取得した。
【0070】
1H NMR(400MHz, CDCI3):δ 1.38-1.39(d,3H), 3.20-3.26(dd,1H), 3.32-3.37(dd,1H), 3.70(s,3H), 3.97(s,3H), 4.88-4.93(m,1H), 7.02-7.29(t,1H), 7.27(s,2H), 7.81(s,1H)
MS [M+H]+ 426/428/430
【0071】
実施例P4:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸{2-[4-(4-クロロ-phenoxy)-phenyl]-1-メチル-エチル)-メトキシ-アミド(化合物1.015)の調製:
【化18】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(797 mg; 4.1 mmol)のジクロロメタン(5ml)溶液を、実施例P16のように調製したN-{2-[4-(4-クロロ-フェノキシ)-フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミン(1.2 g; 4.1 mmol)、トリエチルアミン(1.10 ml; 8.2 mmol)の、0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を常温で一昼夜攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル1:1)。
【0072】
1.2g(理論上66%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸{2-[4-(4- クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-メトキシ-アミド(化合物1.015)は、油状の形態で得られた。
1H NMR: (CDCI3,400MHz):
1.36-1.39(d,3H); 2.78-2.84(dd,1H); 3.05-3.12(dd,1H); 3.65(s,3H); 3.94(s,3H); 4.64-4.68(m,1H); 6.80-6.90(m, 4H);6.95-7.23(t,1H,CHF2);7.17-7.26(m,4H);7.67(s,1 H)。
MS [M+H]+ 450/452。
【0073】
実施例P5:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシ-アミド(化合物1.028)の調製:
【化19】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(2.10 g; 11.0 mmol)のジクロロメタン(5ml)溶液を、P17に記載のように調製したN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミン (2.0 g; 9.10 mmol)、トリエチルアミン(3.10 ml; 22 mmol)の、0℃の攪拌したジクロロメタン(15ml)溶液に滴下した。当該反応混合物を室温で一昼夜攪拌した。当該反応混合物を1M HCl(50ml)に注ぎ、ジクロロメタン(3x20ml)で抽出し、Na2SO4で脱水した。溶媒の除去後、残留物(5.08g)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル7:3)。
【0074】
1.51g(理論上43.8 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシ-アミド(化合物1.028)は、白色の固体の形態で取得された(m.p. 162-167°C)。
1H NMR:(CDCI3, 400MHz):
1.40-1.41(d,3H);2.89-2.94(dd,1H);3.03-3.14(dd,1H);3.86(s,3H);4.3-4.5(mbr,1H);6.5-7.0(mbr,2H); 7.19-7.21(m, 2H); 7.36(m,1H); 7.8-8.6(mbr,1H)。
MS [M+H]+ 378/380/382。
【0075】
実施例P6:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-エトキシ-アミド(化合物1.031)の調製:
【化20】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.86 g; 4.4 mmol)のジクロロメタン(5ml)溶液を、実施例P18に記載のように調製したN-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミン(1.0 g; 4.0 mmol)、トリエチルアミン(0.82 ml;6.0 mmol)の、0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を1.5時間常温で攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0076】
0.75g(理論上45.7%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-エトキシ-アミド(化合物1.031 )は、白色の固体の形態で取得された(m.p. 116-118℃)。
1H NMR: (CDCI3, 400MHz):
1.14-1.20(t,3H);1.36-1.45(2d,3H);2.98-3.03(dd,1H);3.19-3.25(dd,1H);3.74-3.82(q,3H);3.94(s,3H); 4.64-4.70(m,1H); 6.93-7.65(m, 5H)。
MS [M+H]+ 406/408/410。
【0077】
実施例P7:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸エトキシ-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-アミド(化合物1.032)の調製:
【化21】
O-エチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.5g, 1.63 mmol)のジクロロメタン(5 ml)溶液に、トリエチルアミン(0.566 ml, 4.07 mmlol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.34g, 1.79mmol)のジクロロメタン溶液を0℃で滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブラインで洗浄し、これを硫酸ナトリウムで脱水し、そして減圧下で溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸エトキシ-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-アミド(0.390g, 50%)を、m.p:111〜114℃のオフホワイトの固体として取得した。
【0078】
1H NMR (400MHz, CDCI3): δ 1.20-1.24(t,3H), 1.35-1.37(d,3H), 3.23-3.28(dd,1H), 3.33-3.38(dd,1H), 3.84-3.88(q,2H), 3.96(s,3H), 4.86-4.91(m,1H), 7.01-7.25(t,1H), 7.28(s,2H), 7.83(s,1H)
MS [M+H]+ 439.9/441.83
【0079】
実施例P8:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-isoプロポキシ-アミド(化合物1.033)の調製:
【化22】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(1.1 g; 5.7 mmol)のジクロロメタン(5ml)溶液を実施例P20に記載のように調製したN-[2-(2,4- ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミン (1.5 g; 5.7 mmol)、トリエチルアミン(1.6 ml;11.4 mmol)の、0℃の攪拌したジクロロメタン(10ml)溶液に滴下した。当該反応混合物を常温で一昼夜攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0080】
1.30g(理論上54.0 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-isoプロポキシ-アミド(化合物1.033)は、黄色の油の形態で取得された。
1H NMR: (CDCI3, 400MHz):
0.91-0.93+1.11-1.13(2d,6H);1.42-1.48(2d,3H);3.00-3.07(dd,1H);3.29-3.36(dd,1H);3.92(s,3H); 3.97-4.36(m,1H);4.36-4.45(m,1H); 6.97-7.59(m,5H)。
MS [M+H]+ 420/422/424。
【0081】
実施例P9:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-ペンチル]-メトキシ-アミド(化合物1.059)の調製:
【化23】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(2.20g; 1 1.0mmol)のジクロロメタン(10ml)溶液を、実施例P22に記載のように調製したN-[2-(2,4- ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミン (3.0 g; 11.0 mmol)、トリエチルアミン(3.0 ml; 22 mmol)の、0℃の攪拌したジクロロメタン(20ml)溶液に滴下した。当該反応混合物を常温で6時間攪拌した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル1:1)。
【0082】
3.85gm(理論上83 %)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[2-(2,4-ジクロロフェニル)-ペンチル]-メトキシ-アミド(化合物1.059)は、黄色の油の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.84-0.87(t,3H);1.14-1.25(m,2H);1.61-1.69(m,2H);3.55(s,3H);3.71-3.80(m,1H);3.80-3.84(dd,1H); 3.96(s,3H); 3.99-4.05(dd,1H); 7.07-7.36(m,4H);7.79(s,1H)。
MS [M+H]+ 420/422/424。
【0083】
実施例P10:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(化合物2.003)の調製:
【化24】
実施例P23dに記載のように調製したO-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.6g, 2.1 mmol)のジクロロメタン(6 ml)溶液に、トリエチルアミン(0.73 ml, 5.2 mmol)を添加し、続いて3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.415 g, 2.1 mmol)のジクロロメタン溶液を0℃で滴下した。酸塩化物を完全に添加した後、混合物を常温で18時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブライン溶液で洗浄し、これを硫酸ナトリウムで脱水し、そして減圧下で溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中30%酢酸エチルを溶出液として、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(0.53 g, 57%)を、ゴム状生産物として取得した。
【0084】
物理データ:
1H NMR: (CDCI3, 400MHz):
1.41 (d,3H,CH3),1.71-1.80(m,1H,CH2),1.94-2.04(m,1H,CH2),1.87-1.92(m,2H,CH2),3.78(s,3H,CH3),3.98(s,3H,CH3),4.66-4.71(m,1H,CH),7.11(t,1H,CHF2),7.28(s,2H,Ar-H),
7.88(s,1 H,ピラゾール-H)
MS [M+H]+ 440/442/444/446。
【0085】
実施例P11:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(化合物2.040)の調製:
【化25】
実施例P24cに記載のように調製されたO-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.35 g, 1.3 mmol)のジクロロメタン(5 ml)溶液に、トリエチルアミン(0.55 ml, 3.9 mmol)及び3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド酸のジクロロメタン(2ml)溶液を、0℃でゆっくり添加した。酸塩化物を完全に添加した後、混合物を常温で3時間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を水で希釈し、ジクロロメタン(3 x 60 ml)で抽出した。ジクロロメタンの相を一まとめにし、2N HCl、続いて飽和NaHCO3、そして水、最後にブラインで洗浄した。当該有機相を無水硫酸ナトリウムで脱水し、そして減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュシリカゲル、ヘキサン中26%酢酸エチル)にかけて精製し、3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸メトキシ-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-アミド(0.5g, 91 %)を、m.p- 95-96℃の白色の結晶固体として取得した。
【0086】
物理データ:
1H NMR: (CDCI3, 400MHz):
1.89-1.96(m,2H, CH2), 2.91-2.95(m,2H,CH2), 3.66(s,3H,CH3), 3.83-3.86(t,2H,CH2), 3.97(s,3H,CH3), 7.31 (t,1H,CHF2), 7.28(s,2H,Ar-H), 7.89(s,1H,ピラゾール-H)
MS [M+H]+ 426/428/430/432
【0087】
実施例P12:3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-メトキシ-アミド(化合物2.057)の調製:
【化26】
3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボニルクロライド(0.89 g; 4.6 mmol)のジクロロメタン(5ml)溶液を、実施例P25に記載のように調製したN-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミン(1.0 g; 4.2 mmol)、トリエチルアミン(0.86 ml; 6.3 mmol)の、0℃の攪拌したジクロロメタン(7ml)溶液に滴下した。当該反応混合物を、常温で1.5時間攪拌した。当該反応混合物を1M NaOH(20ml)、1M HCl(20ml)ブライン(20ml)で洗浄し、Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:ヘキサン/酢酸エチル7:3)。
【0088】
0.59g(理論上35.3%)の3-ジフルオロメチル-1-メチル-1H-ピラゾール-4-カルボン酸[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-メトキシ-アミド(化合物2.057)は、樹脂状の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.90-0.95(d,3H);1.30(s,9H);2.24-2.36(m,1H);2.39-2.45(dd,1H);2.67-2.72(dd,1H);3.58(s,3H); 3.68-3.70(d,2H);3.98(s,3H); 7.09-7.11 (d, 2H); 7.15-7.42(m,3H);7.90(s,1H)。
MS [M+H]+ 394。
【0089】
実施例P13:N-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシルアミンの調製:
a) 1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化27】
1-(4-クロロ-フェニル)-プロパン-2−オン(8.5 g, 50.4 mmole)のメタノール(100ml)溶液をピリジン(5.2 ml, 62 mmol)、続いてO-メチルヒドロキシアミン塩酸塩(5.20 g, 62 mmol)で処理した。得られた混合物を、23℃で一昼夜16時間攪拌した。当該反応混合物を水(200ml)に注ぎ、そしてジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒の除去後、残留物をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル9:1)。
【0090】
7.38g(理論上74%)の1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシムは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.71 (s,3H);3.48+3.52(2s,2H);3.87-3.89(2s,3H); 7.13-7.22(m,4H). MS [M+H]+ 198/200。
【0091】
b)N-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンの調製:
【化28】
1-(4-クロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.0 g, 5.1mmol)のacetic acid (7.6 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(641 mg, 10.2 mmol)を10分間にわたり添加して処理し、得られた溶液を24℃で5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、1M NaOHでpHを9に調整した。水相をジクロロメタン(2x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(1OOmbar; 45℃)、残留物(910mg)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン/酢酸エチル9:1)。
【0092】
830mg(理論上82.0%)のN-[2-(4-クロロ-フェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.02-1.06(d,3H);2.54-2.59(dd,1H);2.79-2.84(dd,1H);3.14-3.24(m,1H),3.52(s,3H); 5.3-5.5(sbr,1H); 7.12-7.16(m,2H); 7.25-7.28(m,2H)。
MS[M+H]+ 200/202。
【0093】
実施例P14:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミン:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化29】
O-メチルヒドロキシアミン塩酸塩(7.04g, 0.0843mol)の(130ml)とTHF(50ml)の溶液を、酢酸ナトリウム(5.9g, 0.0720mol)、続いて1-(2,4-ジクロロフェニル)-プロパン-2−オン(10g, 0.0496mole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、そして無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0094】
13.5g(理論上100%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシムは、更なる精製を行わずに次の工程に使用される透明な液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
1.76(s,3H);3.60+3.78(2s,2H);3.87(s,3H); 7.12-7.19(m,2H); 7.39(d,1H)。
MS [M+H]+ 232/234/236。
【0095】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンの調製:
【化30】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシム(10.0g, 0.0431mol)の酢酸(100 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(5.41g, 0.0862mol)を15分間にわたり少量ずつ添加して処理し、得られた溶液を24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を減圧下で除去した(1OOmbar; 45℃)。
【0096】
8.13g (理論上81.3% of theory)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-メチル-ヒドロキシアミンは、更なる精製を行わずに次の工程に使用される透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.04-1.09(d,3H);2.66-2.71 (dd,1H);2.93-2.99(dd,1H);3.25-3.33(m,1H),3.52(s,3H); 5.2-5.4(sbr,1H); 7.17-7.18(m,2H); 7.35(d, 1H)。
MS [M+H]+ 234/236/238。
【0097】
実施例P15:O-メチル-N-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-ヒドロキシアミンの調製:
a)1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼンの調製:
【化31】
2,4,6-トリクロロ-ベンズアルデヒド(19g, 90.69mmol)及び酢酸アンモニウム(16.75g, 217mmol)の攪拌した酢酸(76ml, 4Vol.)溶液に、0℃でニトロエタン(45.1ml, 625mmol)を滴下した。当該反応生産物を、100℃で90分間攪拌した。前記反応の完遂がTLCで確認されたら、前記反応物を室温にし、続いて冷水(700ml)で希釈し、これを更に酢酸エチル(3 x 50 ml)で抽出した。一まとめにした酢酸エチルの相を飽和炭酸水素ナトリウム溶液で洗浄してpHを中性にし、続いて水で洗浄し、続いてブラインで洗浄し、これをNa2SO4で脱水し、その後溶媒を完全に蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中1%酢酸エチルを溶出系として回収して、1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼン(15g, 61%)を得た。
1H NMR (400MHz, CDCI3): δ2.10(s,3H,CH3), 7.24(s,2H,Ar-H),7.77(s,1H,CH)
【0098】
b)1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンの調製:
【化32】
上記で取得した攪拌されたニトロスチレン(5g, 18.72mmol)の水(20 ml)及びメタノール(60 ml)溶液に、鉄粉末(2.355gms , 42.12mmol)、続いて濃HCl(11.5ml, 112 mmol)を、窒素大気中、常温で添加した。当該反応混合物を70℃で1時間攪拌し、更に鉄粉(2.355 gms, 42.12 mmol)及び濃HCl(11.5 ml, 112 mmol)をこの反応生産物に添加し、70℃で2時間攪拌を続行した。前記反応の完遂がTLCで確認されたら、前記反応物を室温に冷し、続いてロータベイパー(rotavapor)を用いてエタノールを除去した。得られた残留物を水で希釈し、続いて酢酸エチル(3 x 80 ml)で抽出した。一まとめにした酢酸エチルの相を最後に水で洗浄し、続いてブラインで洗浄し、これを無水硫酸ナトリウムで脱水し、その後溶媒を完全に蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中5%酢酸エチルを溶出系として回収して、1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オン(3.7 g, 83.3%)を得た。
1H NMR (400MHz, CDCI3): δ 2.26(s,3H,CH3), 4.05(s,2H,CH2), 7.34(s,2H,Ar-H)
【0099】
c)1-(2,4,6-トリクロロフェニル)-プロパン-2−オンO-メチル-オキシムの調製:
【化33】
O-メチル-ヒドロキシアミン塩酸塩(0.79 g, 9.4 mmol)のメタノール(20 ml)溶液を、トリエチルアミン(1.32 ml, 9.4 mmol)、続いて1-(2,4,6-トリクロロフェニル)-プロパン-2−オン(1.5 g, 6.31 mmole)で処理した。得られた混合物を、60℃で3時間攪拌した。当該反応混合物を水で浸漬し、続いてメタノールを蒸発させた。得られた水性媒体を、酢酸エチル(3x50ml)で抽出した。一まとめにした有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100 mbar; 45℃)。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中2%酢酸エチルを溶出系として回収して、1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.2 g, 76%)を得て、これを更に還元した。
【0100】
d)O-メチル-N-[1-メチル-2-(2,4,6-トリクロロフェニル)-エチル]-ヒドロキシアミンの調製:
【化34】
上記で取得した1-(2,4,6-トリクロロ-フェニル)-プロパン-2−オンO-メチル-オキシム(1.2g, 4.51 mmol)の酢酸12mlの溶液に、シアノ水素化ホウ素ナトリウム(0.568, 9 mmol)を添加した。当該反応混合物を、常温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、溶媒を減圧下で蒸発させた(トルエンとの共蒸発2回)。得られた残留物を水で希釈し、続いて2Nの水酸化ナトリウムでpHを9に調製して、それから酢酸エチル(3 x 50 ml)で抽出した。一まとめにした有機相をブラインで洗浄し、これを無水硫酸ナトリウムで脱水して、溶媒を蒸発させた。得られた粗生産物を、60〜120μメッシュシリカゲルを用いたカラムクロマトグラフィーにかけて精製し、ヘキサン中5%酢酸エチルを溶出系として回収して、O-メチル-N-[1-メチル-2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミン(0.91 g, 75%)を得た。
1H NMR(400MHz, CDCI3):δ 0.91-0.93(d,3H), 2.72-2.77(dd,1H), 2.98-3.03(dd,1H), 3.25-3.30(m,1H), 3.93(s,3H), 7.15(s,2H)
【0101】
実施例P16:N-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンの調製:
a)1-[4-(4-クロロフェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシムの調製:
【化35】
1-[4-(4-クロロ-フェノキシ)−フェニル]-プロパン-2−オン(10.0 g, 38 mmole)のメタノール(65 ml)溶液を、O-メチルヒドロキシアミン塩酸塩(3.90 g, 47 mmol)、続いてピリジン(4.0 ml, 47 mmol)で処理した。得られた混合物を、室温で一昼夜攪拌した。当該反応混合物を水(150ml)に注ぎ、そしてジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。
【0102】
11.0g(理論上100%)の粗1-[4-(4-クロロ-フェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシムは、更なる精製を行わずに次の工程に使用される黄色の液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
1.73+1.81(2s,3H);3.44+3.65(2s,2H);3.88+3.89(2s,3H); 6.89-6.94(m,4H);7.15-7.28(m,4H)。
MS [M+H]+ 290/292。
【0103】
b)N-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンの調製:
【化36】
粗1-[4-(4-クロロフェノキシ)−フェニル]-プロパン-2−オンO-メチル-オキシム(6.0 g, 20.7 mmol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.6 g, 41.4 mmol)を少量ずつ10分間かけて添加して処理し、そして得られた溶液を、23℃で3.5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを11に調整した。水相をジクロロメタン(2x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(1OOmbar; 45℃)、残留物(7.17g)をシリカゲルのフラッシュクロマトグラフィーで精製した(溶出:c−ヘキサン)。
【0104】
3.91 g(理論上65.0%)のN-{2-[4-(4-クロロ-フェノキシ)−フェニル]-1-メチル-エチル}-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.07-1.09(d,3H);2.53-2.62(dd,1H);2.72-2.81(dd,1H);3.16-3.26(m,1H),3.54(s,3H); 5.3- 5.5(Sbr,1H); 6.86-6.94(m,4H);7.14-7.18(m,2H);7.24-7.29(m,2H)。
MS [M+H]+ 292/294。
【0105】
実施例P17:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシムの調製:
【化37】
1-(2,4-ジクロロフェニル)-プロパン-2−オン(3.46 g, 16.7 mmole)のメタノール(40 ml)溶液を、ヒドロキシアミン塩酸塩(1.40 g, 20.6 mmol)、続いてピリジン(1.7 ml, 20.6 mmol)で処理した。得られた混合物を常温で一昼夜16時間攪拌した。当該反応混合物を水(200ml)に注いだ。固体の産物を濾過により回収し、水で洗浄し、乾燥させた(40℃、100mbar)。
【0106】
3.63g(理論上100%)の1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシムは、白色の固体の形態で取得された(m.p. 120-1240C)。
1H NMR: (CDCI3, 400MHz):
1.86(s,3H);3.61 (s,2H); 7.12-7.21 (m,2H); 7.39-7.41 (d,1H); 8.2(sbr,1H)。
MS [M+H]+ 218/220/222。
【0107】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンの調製:
【化38】
1-(2,4-ジクロロフェニル)-プロパン-2−オンオキシム(3.3 g, 15.1 mmol)の酢酸(30 ml)溶液を、シアノ水素化ホウ素ナトリウム(1.90 g, 30.3 mmol)を少量ずつ10分間添加して処理し、そして得られた溶液を、23℃で5時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを10に調整した。水相をジクロロメタン(3x20ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後3.18g (理論上96.0 %)のN-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-ヒドロキシアミンは、白色の固体の形態で取得された(m.p. 83-86℃)。
1H NMR: (CDCI3, 400MHz):
1.08-1.11(d,3H);2.69-2.74(dd,1H);2.98-3.03(dd,1H);3.24-3.32(m,1H); 5.3-5.6(sbr,1H);7.18-7.19(m,2H);7.39(m,1H);7.5-8.2(sbr,1H)。
MS[M+H]+ 220/222/224。
【0108】
実施例P18:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-エチル-オキシムの調製:
【化39】
O-エチルヒドロキシアミン塩酸塩(4.10 g, 42.1 mmol)の水(130 ml)及びTHF(50 ml)の混合物の溶液を、酢酸ナトリウム(2.95 g, 72 mmol)、続いて1-(2,4-ジクロロフェニル)-プロパン-2−オン(5 g, 24.8 mmole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、そして無水Na2SO4で脱水した。溶媒を減圧下で蒸発させた(90mbar; 45℃)。
【0109】
6.0g(理論上99%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-エチル-オキシムは、更なる精製をせずに次の工程に使用されるE/Z−混合物として、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.23-1.28(2t,3H);1.74+1.76(2s,3H);3.60+3.78(2s,3H);4.09-4.16(2q,2H);7.12-7.19(m,2H);7.38(d,1H)。
MS [M+H]+ 246/248/250。
【0110】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル-ヒドロキシアミンの調製:
【化40】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-メチル-オキシム(10.0g, 0.0406mol)の酢酸(100 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(5.41 g, 0.0862 mol)を少量ずつ10分間にわたり添加して処理し、得られた溶液を、24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0111】
5.09g(理論上51.0 %)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-エチル- ヒドロキシアミンは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
1H NMR:(CDCI3, 400MHz):
0.98-1.02+1.08-1.12(2d,3H);1.13-1.18(2t,3H);2.63-2.68+2.70-2.78(2dd,1H);2.94-3.10+3.15-3.21(m,1H),3.25-3.31+3.71-3.78(2q,2H);5.0-5.5(sbr,1H);7.16-7.19(m,2H);7.34-7.36(m,1H)。
MS [M+H]+ 248/250/252。
【0112】
実施例P20:N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミンの調製:
a)1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシムの調製:
【化41】
1-(2,4-ジクロロフェニル)-プロパン-2−オン(5.1 g, 25.0 mmole)のメタノール(45ml)溶液を、O-isoプロピル-ヒドロキシアミン塩酸塩(3.50 g, 31 mmol)、続いてピリジン(2.6 ml, 31 mmole)で処理し、そして得られた混合物を、23℃で一昼夜攪拌した。当該反応混合物を水(100ml)に注ぎ、ジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。溶媒を減圧下で蒸発させた(90mbar; 45℃)。
【0113】
6.48g (理論上99%)の粗1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシムは、更なる精製をせずに次の工程に使用されるE/Z−混合物として、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
1.23-1.26(d,6H);1.74(s,3H);3.60+3.78(2s,3H);4.25-4.38(2m,1H);7.14-7.21(m,2H);7.39(d,1H)。
MS [M+H]+ 260/262/264。
【0114】
b)N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル-ヒドロキシアミンの調製:
【化42】
1-(2,4-ジクロロフェニル)-プロパン-2−オンO-isoプロピル-オキシム(6.0g, 23 mmol)の酢酸(40 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.90 g, 46 mmol)を少量ずつ15分間にわたり添加して処理し、得られた溶液を、24℃で4時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを10に調整した。水相をジクロロメタン(2x80ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(100mbar; 45℃)。
【0115】
5.90g (理論上98.0%)の粗N-[2-(2,4-ジクロロフェニル)-1-メチル-エチル]-O-isoプロピル- ヒドロキシアミンは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
1H NMR: (CDCI3,400MHz):
1.03-1.07(d,3H);1.13-1.19(m,6H);2.61-2.68+2.94-3.03(2dd,2H);3.21-3.31(m,1H),3.78-3.84(m,1H);5.2(sbr,1H);7.14-7.18(m,2H);7.35-7.36(m,1H)。
MS [M+H]+ 262/264/266。
【0116】
実施例P21:O-メチル-N-[2-(2,4,6-トリクロロ-フェニル)-エチル]-ヒドロキシアミンの調製:
a)1,3,5-トリクロロ-2-クロロメチル-ベンゼンの調製:
【化43】
窒素大気中に置いて攪拌した2,4,6-トリクロロベンジルアルコール(10.0g; 47.3 mmoles)のクロロホルム(100ml)溶液に、塩化チオニル(6.07ml, 85.1 mmole)を、0℃で、15分間にわたりゆっくり添加し、続いて触媒として使用される量のDMFを添加した。当該反応混合物を、常温で3時間攪拌した。当該反応混合物を50mlの水で滴定し;水相をDCM(3x100ml)で抽出した。一まとめにした有機相を5%炭酸水素ナトリウム(2x50ml)、続いてブライン(50ml)で洗浄し、そして無水硫酸ナトリウムで脱水した。溶媒を減圧下で蒸発させた。10.9g(理論上100.0%)の1,3,5-トリクロロ-2-クロロメチル-ベンゼンは、白色の固体の形態で取得された。
1HNMR (CDCI3, 400MHz): δ=7.37(2H,s);4.82(2H,s)
Mass: M = 229.8
【0117】
b)(2,4,6-トリクロロ-フェニル)-アセトニトリルの調製:
【化44】
攪拌した1,3,5-トリクロロ-2-クロロメチル-ベンゼン (12.85 g, 58.8mmole)のエタノール(45ml)溶液に、NaCN(3.20, 67.0mmole)の(15ml)溶液を、常温で添加した。当該反応混合物を4時間再還流して、反応を完遂した。エタノールを蒸発させ、反応混合物を水(50ml)で希釈した。水相をEtOAc(3x100ml)で抽出した。一まとめにした有機相をブライン(30ml)で洗浄し、続いて硫酸ナトリウムで脱水した。有機相を真空中で濃縮して、白色の固体を得た。粗生産物をシリカゲルカラムのクロマトグラフィーで精製して、9.Og(理論上75.0%)の(2,4,6-トリクロロ-フェニル)-アセトニトリルを得た。
1HNMR (CDCI3,400MHz): 7.41(s,2H); 3.97(s,2H)。
Mass:M = 218.9
【0118】
c)(2,4,6-トリクロロ-フェニル)-アセトアルデヒドの調製:
【化45】
N2大気中に置いて、攪拌して冷却(-70℃)した(2,4,6-トリクロロ-フェニル)-アセトニトリル(5.7g, 26.0mmole)のトルエン(120ml)溶液に、DIBAL-H(1MのTHF溶液28.1ml、28.6mmole)を、30分間以上滴下により添加した。反応は、-70℃で3時間攪拌して行われた。反応終了後、-40℃でHCl(60ml、2N)の滴下により滴定し、そして常温に30分間置いた。トルエン相を分離し、水相をブライン(30ml)で洗浄し、硫酸ナトリウムで脱水した。有機相を、真空下で濃縮した。4.5g(理論上78.0%)の(2,4,6-トリクロロ-フェニル)-アセトアルデヒドは、白色の固体の形態で取得された。
1HNMR (CDCI3, 400MHz: 9.70 (s, 1 H); 7.38 (s, 2H); 4.08 (s, 2H)
Mass: M = 246.9
【0119】
実施例P22:N-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンの調製:
a)2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシムの調製:
【化46】
2-(4-クロロ-フェニル)-ペンタナル(10.0 g, 43.0 mmole)のメタノール(75 ml)溶液を、O-メチルヒドロキシアミン塩酸塩(4.40 g, 53 mmol)、続いてピリジン(4.5 ml, 53 mmol)で処理した。得られた混合物を、23℃で一昼夜攪拌した。当該反応混合物を水(200ml)に注ぎ、ジクロロメタン(3x50ml)で抽出した。有機相をブラインで洗浄し、無水Na2SO4で脱水した。減圧下で溶媒を蒸発させた(100mbar;45℃)。
【0120】
11.2Og(理論上100.0%)の粗2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシムは、更なる精製をせずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 260/262/264.
【0121】
b)N-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンの調製:
【化47】
2-(2,4-ジクロロフェニル)-ペンタナルO-メチル-オキシム(6.0g, 23mmol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.90 g, 46 mmol)を少量ずつ10分間にわたり添加して処理し、得られた溶液を、24℃で4時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、残留物を水でスラリー化し、1M NaOHでpHを11に調整した。水相をジクロロメタン(2x50ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒の除去後(100mbar; 45℃)、残留物(6.2g)を、シリカゲルのフラッシュクロマトグラフィーで精製した(25Og;溶出液:c-ヘキサン)。
【0122】
5.15g(理論上79.0%)のN-[2-(2,4-ジクロロフェニル)-ペンチル]-O-メチル-ヒドロキシアミンは、透明な液体の形態で取得された。
1H NMR: (CDCI3, 400MHz):
0.82-0.89(t,3H);1.12-1.27(m,2H);1.49-1.58(m,1H);1.63-1.72(m,1H);3.05-3.10(dd,1H);3.12-3.18(dd,1H);3.47(s,3H),3.50-3.51 (m,1H); 5.0-5.50(sbr,1H); 7.14-7.16(m,1H); 7.21-7.25(m,1H);7.37-7.39(m,1H)。
MS [M+H]+ 262/264/266。
【0123】
実施例P23:O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
a)3-(2,4,6-トリクロロ-フェニル)-プロピオン酸の調製:
【化48】
常温で攪拌しながらMeldrum酸(2.06 g, 14.32 mmoles)をTEAF (7.05 ml)に加え、当該本能混合物を窒素大気中に置いた。10分後、当該反応混合物に、2,4,6-トリクロロ-ベンズアルデヒド(3g, 14.32mmoles)を添加した。続いて、これを、3時間、120℃で再還流した。この反応の収量後、当該混合物を常温で冷却し、そして氷水(50ml)に注いだ。それから、水相を酢酸エチル(3x80ml)で抽出し、一まとめにした有機相をブライン(40ml)で洗浄し、硫酸ナトリウムで洗浄した。有機相を、真空下で濃縮した。310g(理論上86.0%)の3-(2,4,6-トリクロロ-フェニル)-プロピオン酸は、白色の固体の形態で取得された。
1H NMR (400 MHz, CDCI3):
7.32(s,2H); 3.26-3.22 (m,2H); 2.63-2.58(m,2H)。
MS [M+H]+ 253/255/257
【0124】
TEAF(トリエチル蟻酸アンモニウム)の調製:窒素大気中においた蟻酸(1.12g 〜0.93 ml,24.5mmoles)溶液に、0℃で、トリエチルアミン(1g 〜1.37ml,9.8mmoles)をゆっくり添加し、混合物を、常温で1時間半攪拌した。
【0125】
b)N-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミドの調製:
【化49】
酸塩化物のクロロホルム溶液[酸塩化物は、対応する3-(2,4,6-トリクロロ-フェニル)-プロピオン酸(5g ,19.7mmoles)から、塩化チオニル(5.87g ,49.4mmole)を使用して、110℃で3時間の還流条件下で調製された]に、0℃で、N,O-ジメチルヒドロキシアミン塩酸塩(2.3g, 23.9mmoles)、続いてピリジン(3.77ml,47.7mmole)を添加した。その後、当該反応混合物を、70mlのDCM及び40mlの水で希釈した。有機相を分離し、そして水相をDCM(50mlx2)で2回抽出した。一まとめにした有機相をブライン(40ml)で洗浄し、続いて硫酸ナトリウムで脱水した。溶媒の除去後、残留物を、シリカゲルのクロマトグラフィーで洗浄した。3.Og(理論上52.0%)のN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミドは、固体の形態で取得された。
1H NMR(CDCI3, 400MHz):
7.31(s,2H); 3.67(s,3H);3.24-3.19(m,2H);3.19(s,3H);2.67-2.63(m,2H)
MS [M+H]+ 296/298/300
【0126】
c) 4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンの調製:
【化50】
攪拌し冷却(0℃)したN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミド(1g, 3.37mmol)のTHF(15ml)溶液を窒素大気中に置き、MeMgI(1.3mlの3Mエーテル溶液、3.9 mmole)を添加し、攪拌を0℃で2時間継続した。反応混合物を、0℃で5%HCl(10ml)で滴定し、そして水相をEtOAc(3x30ml)で抽出した。一まとめにした有機相を20mlのブラインで洗浄し、硫酸ナトリウムで脱水し;真空中で濃縮して、オフホワイトの固体を得た。当該残留物を、シリカゲルのクロマトグラフィーにより精製した。0.5Og(理論上60.0%)の4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンは、固体の形態で取得された。
1H NMR(CDCI3, 400MHz):
2.14(s,3H,CH3), 2.72-2.76(t,2H,CH2), 2.93-2.97(t,2H,CH2), 7.13-7.16+7.16-7.19(2d,2H,Ar-H), 7.34-7.35(d,1H,Ar-H)。
MS [M+H]+ 252/254/256
【0127】
d)O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
【化51】
攪拌した4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オン(0.9 g, 3.57 mmol)のメタノール(5 ml)溶液に、トリエチルアミン(0.73 ml, 5.3 mmol)及びO-メチル-ヒドロキシアミン塩酸塩(0.447 g, 5.3mmol)を添加した。当該反応混合物を、60℃で3時間加熱した。前記反応の完遂がTLCで確認されたら、前記反応物を常温にした。当該反応混合物を水で希釈した後、メタノールを蒸発させ、水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチル相を、水で、続いてブライン溶液で洗浄し、硫酸ナトリウムで脱水した後、溶媒を完全に蒸発させて、粗4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンO-メチル-オキシム(0.98Og)を得た。これは、更なる精製を行わずに、還元に付された。
【0128】
4-(2,4,6-トリクロロ-フェニル)-ブタン-2-オンO-メチル-オキシム(0.650g, 2.33 mmol)の酢酸溶液に、シアノ水素化ホウ素ナトリウム(0.293 g, 4.66 mmol)を添加した。当該反応物を、室温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、当該反応物中の酢酸を、共沸蒸留により除去した。その後10%NaOHで残留物を塩基性化し、酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチルの相を水で、続いてブライン溶液で洗浄し、硫酸ナトリウムで脱水し、その後濃縮した。得られた粗生産物を、60〜120μメッシュのシリカゲルを使用するカラムクロマトグラフィーにより精製し、ヘキサン中3%酢酸エチルを溶出液として生産物を回収して、O-メチル-N-[1-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン(0.6 g, 90%)を得た。
1H NMR (400MHz, CDCI3): δ 1.21(d,3H), 1.52-1.59(m,1H), 1.68-1.76(m,1H), 2.80-2.83(q,2H), 3.06-3.13(m,1H), 3.55(s,3H), 5.45(s,1H), 7.29(s,2H)
物理データ:
1H NMR (CDCI3, 400MHz):
1.21(d,3H,CH3), 1.52-1.59(m,1H,CH2), 1.68-1.76(m,1H,CH2), 2.80-2.83(m,2H,CH2), 3.06-3.13(m,1H,CH), 1.45(s,1H,NH), 7.29(s,2H,Ar-H)
【0129】
実施例P24:O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
a)3-(2,4,6-トリクロロ-フェニル)-プロパン-1-オールの調製:
【化52】
物理データ:
1H NMR (CDCI3, 400MHz):
1.79-1.86(m,2H, CH2), 2.95-2.99(m,2H,CH2), 3.71-3.749(t,2H,CH2), 7.30(s,2H,Ar-H)
【0130】
b)3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドの調製:
【化53】
攪拌したN-メトキシ-N-メチル-3-(2,4,6-トリクロロ-フェニル)-プロピオンアミド(0.9 g, 3.04mmol)のトルエン(18mL)溶液に、-60℃で、DIBAL-H(15.2ml, 15.2mmol)を20分かけてゆっくり滴下し、当該反応物を、-60℃で3時間攪拌した。更に、反応物を、希塩酸(20ml)を滴下して滴定し、続いて水(10ml)で希釈し、15分間攪拌した。トルエン相を分離し、得られた水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチルの相を水で、続いてブライン溶液で洗浄した。当該有機相を無水硫酸ナトリウムで脱水し、その後減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中2%酢酸エチル)で精製して、3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒド(0.45g,62.5%)の白色の結晶固体を得た。
1H NMR (400MHz, CDCI3): δ 2.68(t,2H), 3.19-3.23(t,2H), 7.29(s,2H), 9.86(s,1H) MS [M+H]+: 235.9 /239.9
物理データ:
1H NMR (CDCI3, 400MHz):
2.68-2.72(m,2H,CH2), 3.19-3.23(m,2H,CH2), 7.29(s,2H,Ar-H), 9.86(s,1H,CHO)
【0131】
c)O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミンの調製:
【化54】
3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒド(0.45 g, 1.8 mmol)のメタノール(5 ml)溶液に、トリエチルアミン(0.4 ml, 2.8 mmol)、続いてO-メチルヒドロキシアミン-塩酸塩(0.24 g, 2.8mmol)を添加し、当該混合物を60℃で2時間加熱した。前記反応の完遂がTLCで確認されたら、前記反応物を常温にし、水で滴定し、ロータベイパーでメタノールを蒸発させた。得られた水相を酢酸エチル(3x30ml)で抽出した。一まとめにした酢酸エチル相を、水で洗浄し、硫酸ナトリウムで脱水した後、溶媒を完全に蒸発させて、粗生産物を得た。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中1%酢酸エチル)で精製して、3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドO-メチル-オキシム(0.5 g, 80%)を得た。
【0132】
攪拌した3-(2,4,6-トリクロロ-フェニル)-プロピオンアルデヒドO-メチル-オキシムの酢酸溶液に、NaCNBH3を、0℃〜10℃で添加した。当該反応混合物を、室温で6時間攪拌した。前記反応の完遂がTLCで確認されたら、当該反応物中の酢酸を、共沸蒸留により除去した。その後残留物を10%NaOH溶液で残留物を塩基性化し、水溶液を、酢酸エチル(3x30ml)で抽出した。一まとめにした有機相を水で、続いてブライン溶液で洗浄した。当該有機相を、続いて、無水硫酸ナトリウムで脱水し、その後減圧下で濃縮した。得られた粗生産物を、カラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン中3%酢酸エチル)で精製して、O-メチル-N-[3-(2,4,6-トリクロロ-フェニル)-プロピル]-ヒドロキシアミン (0.4 g, 88%)を得た。
1H NMR (400MHz, DMSO): δ 1.74-1.82(m,2H), 2.92-3.01 (m,4H), 3.55(s,3H), 5.60(s,1H), 7.29(s,2H)
MS [M+H]+ : 268.13 /272.17
物理データ:
1H NMR (CDCI3, 400MHz):
1.74-1.82(m,2H, CH2), 2.92-3.01 (m,4H,CH2-CH2), 3.55(s,3H,CH3), 5.60(s,1H,NH), 7.29(s,2H,Ar-H)。
【0133】
実施例P25:N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンの調製:
a)3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシムの調製:
【化55】
O-メチル ヒドロキシアミン塩酸塩(3.30 g, 0.041 mol)のwater(65 ml)及びTHF(25 ml)の混合物の溶液を、酢酸ナトリウム(2.85 g, 0.0348 mol)、続いて3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒド(5 g, 0.024 mole)で処理し、得られた混合物を、23℃で4時間攪拌した。当該反応混合物を酢酸エチルで希釈し、ブラインで洗浄し、更に無水硫酸ナトリウムで脱水した。溶媒を減圧下で蒸発させた(100mbar; 45℃)。
【0134】
6.01g(理論上100 %)の粗3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシムは、更なる精製を要さずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 234。
【0135】
b)N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンの調製:
【化56】
3-(4-tert-ブチル-フェニル)-メチル-プロピオンアルデヒドO-メチル-オキシム(5.0 g, 0.0213 mol)の酢酸(50 ml)溶液を、10℃で、シアノ水素化ホウ素ナトリウム(2.63g, 0.042mol)を15分間にわたり添加して処理し、得られた溶液を24℃で6時間攪拌した。溶媒を減圧下で蒸発させ(トルエンとの共蒸発2回)、そして残留物を水でスラリーにして、2M NaOHでpHを9に調整した。水相をジクロロメタン(2x100ml)で抽出し、ブラインで洗浄し、続いて無水Na2SO4で脱水した。溶媒を、減圧下で蒸発させた(1OOmbar; 45℃)。
【0136】
4.52g(理論上89.5 %)の粗N-[3-(4-tert-ブチル-フェニル)-2-メチル-プロピル]-O-メチル-ヒドロキシアミンは、更なる精製を要さずに次の工程に使用される透明な液体の形態で取得された。
MS [M+H]+ 236。
【0137】
実施例P26:2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンの調製:
a)2,4-ジクロロ-1-((E)-2-ニトロ-プロペニル)-ベンゼンの調製:
【化57】
スルホン化フラスコ中で、2,4-ジクロロ-ベンズアルデヒド(77g, 0.44mol)、-ニトロエタン(216ml, 3.04mol)及び酢酸アンモニウム(81.4g, 1.06mol)を、氷酢酸(600ml)に添加した。固体の生産物を濾過により回収し、水で洗浄し、そしてエタノールから再結晶化した。55.9g(理論上55%)の2,4-ジクロロ-1-((E)-2--ニトロ-プロペニル)-ベンゼンは、黄色の固体の形態で取得された(m.p. 79-81℃)。
1H NMR (400MHz, CDCI3): δ 8.11(s,1H), 7.51(d,1H), 7.34(dd,1H), 7.27(d,1H), 2.33(s,3H,CH3)。
【0138】
a)2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンの調製:
【化58】
攪拌した黄色の2,4-ジクロロ-1-((E)-2-ニトロ-プロペニル)-ベンゼン(4 mmol, 0.93 g)の無水トルエン(20ml)溶液に、N2大気中、0℃で、メタノール(16.2mmol, 3ml)中の5.4M CH3ONaとメタノール(2ml)の混合物を2分間かけて滴下した。1.5時間の攪拌後、氷酢酸(3ml)、続いて水(20ml)を添加した。当該水溶液をジクロロメタン(2x30ml)で抽出し、有機相を一まとめにし、脱水し(MgSO4)、濾過し、そして減圧下で蒸発させて、0.78gの粗1-アリール-1-メトキシ-2-ニトロプロパンの黄色の油を得た。この生材料をカラムクロマトグラフィー(シリカゲル、ヘキサン/酢酸エチル8:2)で精製して、0.45g(理論上43%)の2,4-ジクロロ-1-(1-メトキシ-2-ニトロ-プロピル)-ベンゼンを、液体の形態で、ジアステレオ異性体の混合物として得た。
1H NMR (400MHz, CDCI3): δ 1.35-1.37+1.39-1.40(2d,3H, CH3), 3.18+3.21(2s,3H,CH3), 3.88+3.92(2s, 3H,CH3),4.69-4.75(m,1H,CH), 5.16-5.18+5.39-5.40(2d,1H,CH), 7.15-7.47(m,3H,Ar-H)。
MS [M+H]+ 264/266/268。
【0139】
実施例P27:2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンの調製:
a)1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールの調製:
【化59】
攪拌したニトロエタン(8.3g, 0.11 mol)のアセトニトリル(150 ml)溶液に、無水リン酸カリウム(1.Og, 4.6mmol)、続いて2,4-ジクロロ-ベンズアルデヒド(17.5g, O.10mol)を添加した。当該反応混合物を4時間攪拌した。水(300ml)を添加し、更に当該反応混合物をジエチルエーテル(200ml)で抽出した。有機抽出物を水で洗浄し、無水Na2SO4で脱水し、溶媒を除去し、そして得られた残留物を、シリカゲルのフラッシュクロマトグラフィー(溶出液:シクロヘキサン/酢酸エチル9:1)で精製した。20.7g (理論上82.5%)の1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールのトレオ/エリトロ混合物が得られた。シクロヘキサンから結晶化して、純粋なエリトロ1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オールが得られた。
(エリトロ型)1H NMR(400MHz, CDCI3):δ 1.43(d,3H,CH3), 2.92(d,1H,OH), 4.84(m,1H,CH), 5.79(t,1H,CH), 7.34(d,1H,Ar-H), 7.40(d,1H,Ar-H), 7.59(d,1H,Ar-H)。
【0140】
b)2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンの調製:
【化60】
攪拌したエリトロ1-(2,4-ジクロロフェニル)-2-ニトロ-プロパン-1-オール(2.5g, 10.0mmol)の無水ジクロロメタン(20ml)混合物に、窒素大気中で、ジクロロメタン(5ml)中のDAST(1.3ml, 10.0mmol)を、5℃に冷却しながら滴下した。当該溶液を、室温で1時間攪拌した。ジクロロメタン(80ml)を添加し、有機相を、飽和NaHCO3(50ml)、1M HC1(30ml)及び単独(sole)(30ml)で連続的に洗浄した。当該有機相をNaSO4で洗浄し、濾過し、そして濃縮した。2.5gの2,4-ジクロロ-1-(1-フルオロ-2-ニトロ-プロピル)-ベンゼンは、褐色の油の形態で取得された。
【0141】
実施例P28:1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンの調製:
【化61】
攪拌した1,3,5-トリクロロ-2-((E)-2-ニトロ-プロペニル)-ベンゼン (5g, 18.72 mmol)のH2O (20 ml)及びMeOH(60 ml)溶液に常温で、窒素環境下で、鉄粉(2.355g, 42.12mmoles)及び濃塩酸(11.5 ml,112mmol)を添加した。当該反応混合物を、70℃で1時間攪拌した。1時間後、更に鉄粉(2.355g, 42.12mmoles)及び濃塩酸(11.5ml, 112mmol)を添加し、70℃で2時間攪拌を継続した。当該反応の完遂後(TLCにより確認)、反応物を室温に冷却し、MeOHを蒸発させた。得られた残留物を水で希釈し、酢酸エチル(3x80ml)で抽出した。一まとめにした酢酸エチルの相を水及びブラインで洗浄し、無水Na2SO4で脱水した。当該有機相を、真空下で濃縮した。粗材料をカラムクロマトグラフィー(60〜120μメッシュのシリカゲル、ヘキサン/酢酸エチル95:5)で精製して、3.7g(理論上83.3%)の1-(2,4,6-トリクロロ-フェニル)-プロパン-2-オンを得た。
【0142】
表1〜6:式Iaの化合物:
本発明は、下記表1〜6に列挙される、式(Ia)で表される好ましい個々の化合物により、更に例示された。特性のデータは、表9に示される。
【化62】
【0143】
式Iaの化合物において、Aは、A1
【化63】
A2
【化64】
及びA3
【化65】
殻なる群から選択され、そしてnは0又は1である。
【0144】
下記表Yの後の表1〜6のそれぞれは、100個の式(Ia)の化合物を含む。ここで、式(Ia)中、R4、R5、R7、X、R11、R12及びR13は表Yに記載の数値をとり、そしてAは関連する表1〜6の定義に記載のものが選択され、そしてnは関連する表1〜6の定義に記載の数値をとる。故に、表1は、表YのYが1で、且つAが表1の定義に記載のものであるときの表Yに対応し、表1は、表YのYが2で、且つAが表2の定義に記載のものであるときの表Yに対応し、そして以下表3〜6も同様である。
【0145】
表1〜6において「Me」はメチル、「Et」はエチル、「i-Pr」はイソプロピル、「c−Pr」はシクロプロピル、及び「t−Bu」は第三級ブチルを意味する。
【0146】
【表1】
【表2】
【表3】
【表4】
【0147】
表1は、100個の式(Ia)の化合物を提供し、ここでAはA1
【化66】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物1.001は、以下の構造を有する:
【化67】
【0148】
表2は、100個の式(Ia)の化合物を提供し、ここでAはA1
【化68】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物2.057は、以下の構造を有する:
【化69】
【0149】
表3は、100個の式(Ia)の化合物を提供し、ここでAはA2
【化70】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物3.002は、以下の構造を有する:
【化71】
【0150】
表4は、100個の式(Ia)の化合物を提供し、ここでAはA2
【化72】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物4.003は、以下の構造を有する:
【化73】
【0151】
表5は、100個の式(Ia)の化合物を提供し、ここでAはA3
【化74】
、nは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物5.002は、以下の構造を有する:
【化75】
【0152】
表6は、100個の式(Ia)の化合物を提供し、ここでAはA3
【化76】
、nは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物6.003は、以下の構造を有する:
【化77】
【0153】
表7及び8:
上記表Yの後の表7及び8のそれぞれは、100個の式(IIb)の化合物を含む。ここで、式(IIb)中、R4、R5、R7、X、R11、R12及びR13は表Yに記載の数値をとり、そしてnは関連する表7及び8の定義に記載の数値をとる。故に、表7は、表YのYが7であるときの表Yに対応し、表8は、表YのYが8であるときの表Yに対応する。
【0154】
表7は、100個の式(IIb)
【化78】
の化合物を提供し、ここでnは0、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。例えば、化合物7.001は、以下の構造を有する:
【化79】
【0155】
表8は、100個の式(IIb)
【化80】
の化合物を提供し、ここでnは1、かつR4、R5、R7、X、R11、R12及びR13は表Yに定義したものである。
【0156】
表9:特性データ:
表9は、表1〜6の化合物の選択された融点及び選択されたNMRデータを示す。特に言及が無い限り、NMR測定の溶媒として、CDCI3が使用される。溶媒の混合物が存在する場合、例えば:)と表される。いかなる場合も、全ての特性データを列記することを試みていない。表9及び以下の記載全体において、温度はセルシウス度で示され;「NMR」は核磁気共鳴スペクトルを意味し;MSは質量スペクトルを意味し;「%」は、対応する濃度が他の単位で示されていない限り、重量パーセントである。以下の略語は、本願全体で使用されている:
【表5】
【0157】
【表6】
【表7】
【0158】
式Iの化合物の製剤の例:
実施例F-1.1〜F-1.2:乳剤
【表8】
これらの濃縮物を水で希釈することにより、任意の所望の濃度の乳濁物を調製することができる。
【0159】
実施例F-2:乳剤
【表9】
これらの濃縮物を水で希釈することにより、任意の所望の濃度の乳濁物を調製することができる。
【0160】
実施例F-3.1〜F-3.4:溶液
【表10】
これらの溶液は、微液滴の形態での使用に適している。
【0161】
実施例F-4.1〜F-4.4:顆粒
【表11】
本発明の新規化合物をジクロロメタン中に溶解し、当該溶液を担体上に噴霧し、そして溶媒を真空下で蒸発により除去する。
【0162】
実施例F-5.1及びF-5.2:粉塵
【表12】
全ての材料を念入りに混合して、使用可能な粉塵が得られる。
【0163】
実施例F-6.1〜F-6.3:水和剤
【表13】
全ての材料を混合し、当該混合物を適切なミル中で激しく挽き潰すことにより、水和物が得られる。当該水和物を水で希釈することにより、任意の所望の濃度の懸濁物を得ることができる。
【0164】
実施例F7:種子処理用流動性濃縮物
【表14】
細かく挽き潰した有効成分を助剤と念入りに混合することにより、懸濁濃縮物が得られる。当該懸濁濃縮物を水で希釈することにより、任意の所望の希釈率の懸濁物を取得することができる。そのような希釈物を、噴霧、注入又は浸漬により使用して、生きた植物の他に植物育苗材料の、微生物の繁殖を処理及び防御できる。
【0165】
生物学的実施例:殺真菌作用:
実施例B-1:ボトリティス・キネレア(Botrytis cinerea)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、3〜4日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0166】
マイコスファエレラ・アラキディス(Mycosphaerella arachidis)(ラッカセイの早期斑点病(early leaf spot);ケルコスポラ・アラキディコラ(Cercospora arachidicola)[アナモルフ])に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、6〜7日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059、2.003及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0167】
実施例B-3:セプトリア・トリティキ(Septoria tritici)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、72時間後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059、2.003及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0168】
実施例B-4:タペシア・ワルンダエ(Tapesia vallundae)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、6〜7日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率として表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0169】
実施例B-5:モノグラフェラ・ニワリス(Monographella nivalis)(アナモルフ:フサリウム・ニワレ(Fusarium nivale)、ミクロドキウム・ニワレ(Microdochium nivale);雪腐れ病)に対する作用-真菌増殖アッセイ
前記真菌の凍結保存された分生子を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、72時間後に、増殖の阻害を光学的に測定した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.057は、この試験で良好な活性を示している(≦50%阻害率)。
【0170】
実施例B-6:リゾクトニア・ソラニ(Rhizoctonia solani)に対する作用-真菌増殖アッセイ
前記真菌の新しく増殖させた液体培養物の菌糸断片を栄養ブロス(PDBジャガイモデキストロースブロス)中に直接混入させた。試験化合物(有効成分0.002%)の(DMSO)溶液を滴下したマイクロタイタープレート(96ウェルフォーマット)に、前記真菌胞子を含むブロスを添加した。当該試験プレートを24℃でインキュベーションして、3〜4日後に、増殖の阻害を光学的に測定した。化合物の活性は、真菌増殖の阻害率で表現した(0%は増殖阻害無しを、80%〜99%は良好〜非常に良好な阻害を、そして100%は完全な阻害を意味する)。化合物2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.003は、この試験で良好な活性を示している(≦50%阻害率)。
【0171】
実施例B-7:エリシフェ・グラミニスf.sp.トリティキ(Erysiphe graminis f.sp. tritici)(うどんこ病)に対する作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の7日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.033、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【0172】
実施例B-8:コムギのプッキニア・レコンディタ(Puccinia recondita)(茶さび(brown rust))に対する作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の8日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.059及び2.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.032及び1.057は、この試験で良好な活性を示している(≦50%阻害率)。
【0173】
実施例B-9:コムギのプッキニア・レコンディタ(Puccinia recondita)(茶さび(brown rust))に対する治療的作用
コムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、前記真菌の胞子懸濁物を接種した。接種の翌日、当該葉の断片に試験溶液(0.02%有効成分)を噴霧した。適切なインキュベーションの後、化合物の活性を、前記添加の8日後の殺真菌活性の防止の程度として評価した。化合物1.002及び1.003は、この試験で非常に良好な活性を示している(≦80%阻害率)。化合物1.015は、この試験で良好な活性を示している(≦50%阻害率)。
【0174】
実施例B-10:オオムギのピレノフォラ・テラス(Pyrenophora teres)(網斑病)に対する作用
オオムギの葉の断片をマルチウェルプレート(24ウェルフォーマット)中の寒天上に置き、試験溶液(0.02%有効成分)を噴霧した。乾燥後、前記葉の円盤に、前記真菌の胞子懸濁物を接種した。適切なインキュベーションの後、化合物の活性を、前記添加の4日後の殺真菌活性の防止の程度として評価した。化合物1.001、1.002、1.003、1.015、1.031、1.032、1.033、1.059及び1.057は、この試験で非常に良好な活性を示している(≦80%阻害率)。
【特許請求の範囲】
【請求項1】
式I
【化1】
[式中、
R1はC1-C4アルキル又はC1-C4ハロアルキルであり;
R2はC1-C4アルキルであり;
R3は水素又はハロゲンであり;
R4は水素、C1-C4アルキル又はC1-C4ハロゲンアルキルであり;
R5、R6、R8、R9及びR10は、互いに独立して、水素、ハロゲン、C1-C4アルキル又はC1-C4ハロアルキルであり;
R7は水素、ハロゲン、C1-C4アルキル、C2-C6アルケニル又はC3-C6アルキニルであり;
R11は水素、ハロゲン又はC1-C6アルキルであり;
R12は水素、ハロゲン、C1-C6アルキル、C2-C6アルケニル、C3-C6アルキニル、C3-C6シクロアルキル-C3-C6アルキニル、ハロフェノキシ、ハロフェニル-C3-C6アルキニル、C(C1-C4アルキル)=NO-C1-C4アルキル、C1-C6ハロアルキル、C1-C6ハロアルコキシ、C2-C6ハロアルケニル、又はC2-C6ハロアルケニルオキシであり;
R13は水素、ハロゲン、C1-C6アルキルであり;
Xは酸素又は硫黄であるか、又は存在せず;但しXが酸素又は硫黄であるときR7はハロゲンではなく;
nは0又は1である]
の化合物、及びその農学的に許容される塩/異性体/構造異性体/立体異性体/ジアステレオ異性体/鏡像異性体/互換異性体、並びにそれらの化合物のN-オキシド。
【請求項2】
R1がジフルオロメチル、トリフルオロメチル又はメチルである、請求項1に記載の化合物。
【請求項3】
R2がメチルである、請求項1に記載の化合物。
【請求項4】
R3が水素又はフルオロである、請求項1に記載の化合物。
【請求項5】
R4が水素、メチル又はエチルである、請求項1に記載の化合物。
【請求項6】
R4がメチルである、請求項1に記載の化合物。
【請求項7】
R5が水素又はメチルである、請求項1に記載の化合物。
【請求項8】
nが0である、請求項1に記載の化合物。
【請求項9】
Xが酸素である、請求項1に記載の化合物。
【請求項10】
R8、R9及びR10が水素である、請求項1に記載の化合物。
【請求項11】
R11、R12及びR13が水素又はクロロである、請求項1に記載の化合物。
【請求項12】
R12がクロロ又はC1-C4アルキルである、請求項1に記載の化合物。
【請求項13】
式II
【化2】
の化合物であって、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が、請求項1に記載の式Iで定義したものである、前記化合物。(19条補正で削除)
【請求項14】
有用植物への植物病原性微生物の繁殖を防除又は予防する方法であって、請求項1に記載の式Iの化合物、又は当該化合物を有効成分として含有する組成物を、前記植物、その部分、又はその生育場所に施用する、前記方法。
【請求項15】
請求項1に記載の式Iの化合物及び不活性の担体を含有する、植物病原性微生物に対する防除及び防御のための組成物。
【請求項1】
式I
【化1】
[式中、
R1はC1-C4アルキル又はC1-C4ハロアルキルであり;
R2はC1-C4アルキルであり;
R3は水素又はハロゲンであり;
R4は水素、C1-C4アルキル又はC1-C4ハロゲンアルキルであり;
R5、R6、R8、R9及びR10は、互いに独立して、水素、ハロゲン、C1-C4アルキル又はC1-C4ハロアルキルであり;
R7は水素、ハロゲン、C1-C4アルキル、C2-C6アルケニル又はC3-C6アルキニルであり;
R11は水素、ハロゲン又はC1-C6アルキルであり;
R12は水素、ハロゲン、C1-C6アルキル、C2-C6アルケニル、C3-C6アルキニル、C3-C6シクロアルキル-C3-C6アルキニル、ハロフェノキシ、ハロフェニル-C3-C6アルキニル、C(C1-C4アルキル)=NO-C1-C4アルキル、C1-C6ハロアルキル、C1-C6ハロアルコキシ、C2-C6ハロアルケニル、又はC2-C6ハロアルケニルオキシであり;
R13は水素、ハロゲン、C1-C6アルキルであり;
Xは酸素又は硫黄であるか、又は存在せず;但しXが酸素又は硫黄であるときR7はハロゲンではなく;
nは0又は1である]
の化合物、及びその農学的に許容される塩/異性体/構造異性体/立体異性体/ジアステレオ異性体/鏡像異性体/互換異性体、並びにそれらの化合物のN-オキシド。
【請求項2】
R1がジフルオロメチル、トリフルオロメチル又はメチルである、請求項1に記載の化合物。
【請求項3】
R2がメチルである、請求項1に記載の化合物。
【請求項4】
R3が水素又はフルオロである、請求項1に記載の化合物。
【請求項5】
R4が水素、メチル又はエチルである、請求項1に記載の化合物。
【請求項6】
R4がメチルである、請求項1に記載の化合物。
【請求項7】
R5が水素又はメチルである、請求項1に記載の化合物。
【請求項8】
nが0である、請求項1に記載の化合物。
【請求項9】
Xが酸素である、請求項1に記載の化合物。
【請求項10】
R8、R9及びR10が水素である、請求項1に記載の化合物。
【請求項11】
R11、R12及びR13が水素又はクロロである、請求項1に記載の化合物。
【請求項12】
R12がクロロ又はC1-C4アルキルである、請求項1に記載の化合物。
【請求項13】
式II
【化2】
の化合物であって、R4、R5、R6、R7、X、R8、R9、R10、n、R11、R12及びR13が、請求項1に記載の式Iで定義したものである、前記化合物。(19条補正で削除)
【請求項14】
有用植物への植物病原性微生物の繁殖を防除又は予防する方法であって、請求項1に記載の式Iの化合物、又は当該化合物を有効成分として含有する組成物を、前記植物、その部分、又はその生育場所に施用する、前記方法。
【請求項15】
請求項1に記載の式Iの化合物及び不活性の担体を含有する、植物病原性微生物に対する防除及び防御のための組成物。
【公表番号】特表2012−510974(P2012−510974A)
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2011−538990(P2011−538990)
【出願日】平成21年12月1日(2009.12.1)
【国際出願番号】PCT/EP2009/066119
【国際公開番号】WO2010/063700
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(500584309)シンジェンタ パーティシペーションズ アクチェンゲゼルシャフト (352)
【Fターム(参考)】
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願日】平成21年12月1日(2009.12.1)
【国際出願番号】PCT/EP2009/066119
【国際公開番号】WO2010/063700
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(500584309)シンジェンタ パーティシペーションズ アクチェンゲゼルシャフト (352)
【Fターム(参考)】
[ Back to top ]