
水光分解用のモノリス触媒システム
本発明は、光を用いて水を水素と酸素に分解するためのモノリス触媒システム(1)を提供する。本システムは、単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水から酸素と陽子を生成させることができる第一光活性物質(50)と、単独で、あるいは補助物質の少なくとも一種および/または補助触媒(92)の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水中の陽子を水素に還元することができる第二光活性物質(60)とを含有し、第一光活性物質(50)と第二光活性物質(60)は一種またはそれ以上の電子伝導性物質(30、20、40、60a)を介して電気的、特に直接電気的な接触状態にあることを特徴とする。それと共に、この触媒システムを用いて水から酸素と水素を生成する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は水を光を用いて水素と酸素に分解するための触媒システムおよび本触媒システムを用いて水素および酸素を製造する方法に関する。
【背景技術】
【0002】
水素は未来の物体的なエネルギー担体になると考えられ、環境に優しい方法、即ち二酸化炭素の副生がなく、通常はコストが高く環境に優しくない場合が多い従来の電解を利用しない、水素の製法が注目を集めている。
【0003】
米国特許US 6,936,143 B1号では、グレッツエルらは光を用いて水を水素と酸素に分解するタンデムセルまたは光電化学システムであって、両セルは電気的に接続されているものを開示した。 この電気的接続では、WO3 またはor Fe2O3 等からなる光陽極から色素増感メソ多孔性 TiO2 皮膜からなる光陰極への電子伝導のために、有機酸化還元電解質を使用している。 前記文献には、この有機酸化還元電解質についての記載は一切ないが、電解質における電荷の運搬は通常イオンによるものであるので、有機酸化還元電解質という概念から電子の運搬はイオン性導電によって行なわれることは明らかである。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許US 6,936,143号明細書
【発明の概要】
【課題を解決するための手段】
【0005】
本発明は光を用いて水を水素と酸素に分解するために用いるモノリス触媒システムを提供する。このシステムは、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水から酸素と陽子を生成させることができる第一光活性物質と、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水中の陽子を水素に還元することができる第二光活性物質と
を含有し、
第一光活性物質および第二光活性物質は、一種またはそれ以上の電子伝導性物質を介して電気的、特に直接電気的な接触状態にあることを特徴とする。
【0006】
さらに、本発明は光と触媒を用いて水から水素と酸素を生成する方法を提供する。この方法は、
第一光活性物質を含有するか、あるいは、第一光活性物質とこれと組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第一区域において、本発明による触媒システムを水または水性液体または水性溶液に接触させ、
第二光活性物質を含有するか、あるいは、第一光活性物質とこれに組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第二区域において、前記触媒システムを水または水性液体または水性溶液に接触させ、
前記触媒システムに光を照射し、
陽子が第一区域から第二区域に移動できるように、第一区域に接触している水または水性液体または水性溶液と、第二区域に接触している水または水性液体または水性溶液とが相互に接触状態にあるようにすることを特徴とする。
【0007】
本発明の好ましい実施形態は、特許請求項に記載のとおりである。
【図面の簡単な説明】
【0008】
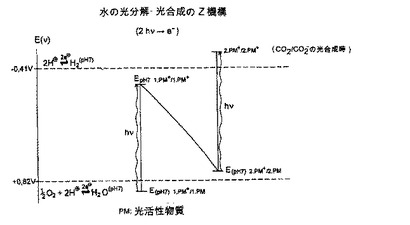
【図1】図1は、本発明による触媒システムにおいて用いる、植物 バクテリアにおける光合成あるいは水の光分解のいわゆるZ機構を示す図である。
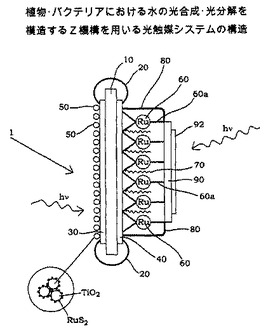
【図2】図2は、本発明による触媒システムの一例を示す概略断面図である。
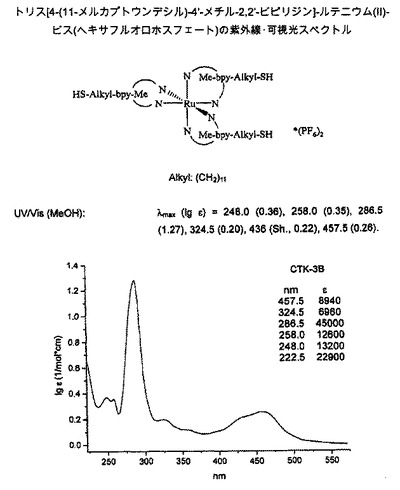
【図3】図3は、トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス(ヘキサフルオロホスフェート)の紫外線/可視光スペクトルを示す図である。
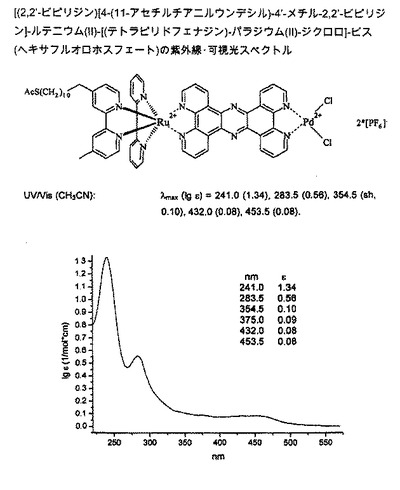
【図4】図4は、[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート)の紫外線/可視光スペクトルを示す図である。
【図5】図5は、Mn4O4(フェニル2PO2)の紫外線/可視光スペクトルを示す図である。
【発明を実施するための形態】
【0009】
[いわゆるZ機構による水素 酸素の発生原理]
本発明の触媒システムでは、水の1/2O2とH2への分解のために4つの陽子を使っている。 この場合における各ステップとそれらのエネルギーレベルによる関係を概略的に図1に示す。
【0010】
下記の反応には触媒システムの酸化側(以下、第一光活性物質とも言う)で2つの陽子が要求される。
H2O + 2 光子 → 1/2 O2 + 2 H+ + 2 e- (EO2/H2O(pH7) = +0.82 V).
【0011】
この反応は植物/バクテリアの光化学系2における反応である。
【0012】
次に示す反応には触媒システムの還元側(以下、第二光活性物質とも言う)で2つの陽子が要求される。
2 H+ + 2 e- + 2 光子 → H2 (EH+/H2(pH7) = -0.41 V)
【0013】
この反応は、水素を発生する酵素ヒドロゲナーゼがかかわり、数種類のバクテリアの光化学系に起こり得る反応である。
【0014】
総合的には、この反応は次のようにまとめられる。
H2O + 4 光子 → H2 + 1/2 O2 (EpH7 = 1.23 V).
【0015】
したがって、この場合には、1 e- が水中の酸素から分離し、それがH+イオンに移動 (2 hν → 1 e-) するように2つの光子 (2 hν) を使用するプロセスが必要とされる。 また、この反応は植物 バクテリアの光合成のZ機構における反応の一つである。
【0016】
なお、本明細書において水素、陽子、H+、H+イオンの概念はジュウテリウム、ジュウテリウムイオン、D+、D+イオン等という意味も含んでいる。 同様に、H2とは「HD」、「D2’’」の意味を含んでいる。しかし、「D2」には「HD」と「H2」は含んでいない。
【0017】
本発明による触媒システムの酸化側(電気化学の表現では陽極である)で放出される電子は、電子伝達性物質の1種または2種以上を介して直接本触媒システムの還元側(電気化学の表現では陰極である)に伝導される。 電子伝導性物質という概念にはイオン伝導体、液体状酸化還元電解質、固体電解質は含んでいない。 pn接合などの接合による電子伝導は本発明のような、第一光活性物質と第二光活性物質の間における電子伝導性物質には無関係であるものとする。
【0018】
本発明は、二種の光活性物質間の電子伝導の面でも植物 バクテリアにおけるZ機構を模造するものである。 実質的に植物 バクテリアにおける光合成、もっと詳しく言えば、バクテリアのある種類における水の光分解を人工的に行なうことを図っている。
【0019】
勿論、本システムを働かせるために、外部から電圧を印加する必要がない。
【0020】
本出願においてモノリス触媒システムとは、コンパクトな構造によるシステムを意味し、本システムから伸びたり、または本システムに一体化されていない、目で見られるような電線、導体、電極など、例えば導電性ワイヤ、バンド、シート等によってシステムに接続された電極のないシステムを指している。 このようなシステムの形状として、プレート状、フィルム状、チューブ状等が挙げられる。 「モノリス(monolithic)」とは必ずしも本システムが一つの部品として製造されたものを指すものではない。
【0021】
また、本願における光活性物質とは、他の光活性物質と組み合わせたとき、光合成 分解におけるZ機構に対応する酸化還元電位パターンを示す物質の意味であって、その総電位差が波長420nm以上、好ましくは430nm以上、もっと好ましくは440nm以上、特に450nm以上の光の照射下、水を水素と酸素に分解するのに十分なものである。 さらに両方の光触媒のうちのいずれかまたは両方は波長700nm以上の電磁波しか吸収しないものではないほうが好ましい。
【0022】
図1に示すZ機構から明らかなように、第一および第二の光活性物質の酸化還元電位は、以下の酸化還元電位および酸化還元電位間の関係を示すものである。
1. 第一光活性物質のイオン化された状態の酸化還元電位および第一光活性物質の正に荷電された価電子帯の酸化還元電位は、両方とも+0.82Vより陽性側にある。
2. 第二光活性物質の励起された状態の酸化還元電位および第二光活性物質の伝導帯の酸化還元電位は、両方とも-0.41Vより陰性側にある。
3. 第一光活性物質の励起された状態の酸化還元電位および第一光活性物質の伝導帯の酸化還元電位は、それぞれ、第二光活性物質のイオン化された状態の酸化還元電位および第二光活性物質の正に荷電された価電子帯の酸化還元電位より陰性である。
【0023】
第一光活性物質の励起されていない状態の酸化還元電位および第一光活性物質の価電子帯の酸化還元電位は、それぞれ、基本的に第二光活性物質の励起されていない状態の酸化還元電位および第二光活性物質の価電子帯の酸化還元電位より陽性である。
【0024】
本触媒システムは、波長420nm以上の可視光の照射下で使用するので、その励起状態および伝導帯が、それぞれ、前記波長の光を用いて誘発されることが求められている。
【0025】
第一光活性物質(電気化学的には陽極またはその一部を形成するもの)と第二光活性物質(電気化学的には陰極またはその一部を形成するもの)という概念には、複数の第一光活性物質と第二光活性物質(或いはそれぞれの混合物)を使用する場合も含んでいる。
【0026】
波長420nm以上の光の照射下、第一光活性物質(酸化を促進させる物質)として様々な非分子性化合物、分子性化合物、ポリマー化合物を使用することができる。 第一光活性物質(酸化を促進させる物質)としては、これに限定することなくドープされてもよい酸化物および/あるいは酸化物と硫化物を含有する物質、特にRuS2、 貴金属または遷移金属を含有する錯体またはクラスタ、光活性を示すポリマー性物質を挙げることができる。 例えば、ドープされてもよいRuS2、貴金属を含有してもよいWO3、他元素の原子でドープされてもよい酸化鉄、Sb/M(ただし、MはCr、Niおよび/またはCu)でドープされてもよいTiO2 、Mn4系かご錯体、Ru4系クラスタ錯体、Ru3+ 系錯体、本願の実施例1~3に記載の光活性物質から選ぶことができるが、それに限定するものではない。
【0027】
水中の酸素から電子が分離される過程については、それぞれの機械的なステップの詳細がいまだにくわしく知られていない場合がある。 しかし、一般的には波長420nm以上の光子により、電子的に励起された状態が生じ、空間的に第一光活性物質から離れた励起された電子が1単位ぐらいあげた酸化状態、または水中の酸素から由来する電子で埋められる孔を残し、即座にO2と陽子が発生するが、その時に、2〜4つの陽子がさらに同時に発生する場合が多い。
【0028】
酸素の発生を促進させる目的で、第一光活性物質を前記意味で光活性を示さない補助物質および/または補助触媒、つまり照射されると酸素を生成するものではなく、その代わりに酸素の発生を促進させるものと組み合わせることができる。 このような補助物質および/または補助触媒は、RuO2、パラジウム、白金などの貴金属、または金属性コバルトを水中においてホスフェートと反応させることによりin situで形成される化合物から選ぶことができるが、但しこれに限定するものではない。
【0029】
本発明の触媒システムを使用する際、第一光活性物質が、またそれと補助物質および/または補助触媒とを組み合わせて用いる場合には、そのいずれかまたは両方が水に接触するようにする。
【0030】
第二光活性物質(還元を促進させる物質)としても、波長420nm以上の光を吸収する様々な物質を使用することができ、この物質は、非分子性固体であっても、固体の分子性化合物およびポリマー化合物であってもよい。 本発明の第二光活性物質は、例えば数多くあるルテニウム(II)系錯体から選択でき、これらには窒素を含有する配位子が結合されたものが多く、PdまたはPtのように電子が供給されると陽子を水素に還元することができる一般的な触媒種と組み合わせられると、波長420nm以上の光で励起されたときに、水中に存在する陽子(より詳細には、本明細書において「H+」と記載されることもあるヒドロニウムイオン)をH2に還元することができることが公知である。 そのほかにも、他の金属含有錯体、例えば貴金属錯体、天然葉緑素(Mgを中心原子とするもの)、Cu-クロリン、Cu-2-a-オキシメソイソクロリン、その他の金属含有フタロシアニン類、金属含有ポルフィリン類、またはH2-クロリンやプロフラビンなどの拡張π電子系を有する純有機化合物も、 必要に応じて電荷分離を起こす励起電子を適当な補助物質および または補助触媒(例えばPd、Pt、Ruまたは亜鉛を含有する触媒種)への伝導性トランスファーと組み合わせた形態で、可視光の照射下、H+を1/2H2に還元するのに十分なエネルギーを有する励起状態を示す。
【0031】
また、数多くの、必要に応じてドープされていてもよい酸化物含有物質および酸化窒化物含有物質、これほどは好ましくないが、リン化物、砒素化物、アンチモン化物、硫化物、またはセレン化物を含有する物質 [例えばCr/SbまたはRhを添加したSrTiO3やZnIn2S4、TaON、NiM2O6 (M = Nb, Ta)]も、場合によりPd、Pt、Ruなどの補助物質および/または補助触媒の存在下、波長420nmの光を照射すれば水からH2を生成させることができる。 さらに、光半導性のポリマーも、必要に応じて他の非ポリマー性の有機物質と組み合わせて、光起電力効果を示し、使用可能である。
【0032】
光活性物質の具体例は本願の実施例3〜5に示す。
【0033】
本発明の触媒システムを使用する際、第二光活性物質が、またそれと補助物質および/または補助触媒とを組み合わせて用いる場合、そのいずれかまたは両方が水に接触するようにする。
【0034】
本発明では、第一光活性物質は、植物 バクテリアが、水を酸素と陽子に分解する光化学系IIまたはその変更されたものを完全に形成しないことが好ましい。 特に、ポリペプチドまたは蛋白質を含有しないことが好ましい。 なぜなら、天然の光化学系IIは非常に不安定であるからである。
【0035】
本発明では、第二光活性物質は、植物・バクテリアが、NADP+をNADPH+H、あるいは特別な場合ではヒドロゲナーゼを用いて陽子を水素に還元する光化学系IIまたはその変更されたものを完全に形成しないことが好ましい。 特に、ポリペプチドまたは蛋白質を含有しないことが好ましい。 その理由は、ヒドロゲナーゼは酸素に対して非常に敏感であって、陽子の還元のための補助触媒によっては完全な光化学系IIに連結できないものがあり得ることにある。
【0036】
第一光活性物質として、単結晶または単結晶からドーピングにより得たものを使用することは原理的には有用であるものの、下記の理由から好ましくない。 単結晶またはエピタキシャル成長などによって得られるドープされた結晶は、製造コストが高い。
【0037】
同じ理由から、第二光活性物質の場合も単結晶または単結晶からドーピングにより得たものを使用することは原理的には有用であるものの、下記の理由から好ましくない。
【0038】
また、そのコストが高いから、ドープされた珪素を光活性物質として使用しないほうが好ましい。
【0039】
また、本発明では、光起電力の分野で使われる従来の半導体あるいは光半導体、例えば遷移金属の1価、2価および/または3価のカチオンと元素の周期表におけるVa族およびVIa族のアニオンを含有してもよいIII-V族半導体、II-IV族半導体、III-VI族半導体、またそれに類似している半導体を両方の光活性物質に用いないことが好ましい。 また、光活性物質の両方ともが、上述の半導体を使用しないことがさらに好ましい。 これらの半導体は原理的には有用ではあるが、コストが高く、公害を起こす金属を含有したり、水の存在下、不安定になり、それに伴って毒性ガスが発生することがしばしばある。
【0040】
また、第一光活性物質と第二光活性物質との組み合わせは、第一光活性物質により吸収されなかった黄色光、赤色光、近赤外光が第二の光活性物質材に伝達されるように、両物質が列に配置されている限り、太陽光の青色部分と緑色部分を吸収する半導性酸化物と陽子の還元に太陽光の黄色部分と赤色部分と近赤外部分を使用するメソ多孔性皮膜からなる組み合わせではないことが好ましい。 この組み合わせでは、水の分解のために本発明において好ましくない特殊のタンデムセルを必要とする。
【0041】
第一光活性物質と第二光活性物質とは、互いに異なる化学種からなっていることが好ましく、同じ元素から構成されていないことがさらに好ましい。
【0042】
第一光活性物質と第二光活性物質とは、上述のZ機構に従って組み合わせることができる。
【0043】
第二光活性物質(還元を促進させる物質)が光で照射されるとき、そこの電子は励起状態となり、そのエネルギーが十分な場合、水中の陽子に伝達され(ただし、それにはPt、Ruなどの補助物質または補助触媒を用いる場合が多い)、その結果、水素が発生すると共に、光活性の還元促進物質または第二光活性物質に正孔が生じるか、その酸化状態が1つの単位上がる。
【0044】
このサイクルは第一光活性物質から励起電子を与えられ、それにより酸化された第二光活性物質が還元を受けることで閉じられる。
【0045】
具体例を一つ挙げれば、例えば適当なRu(II)錯体が励起されたとき、その電子が、必要に応じて補助物質および/または補助触媒を介して陽子側に伝達され、すると、前記錯体はRu(III)錯体に酸化されるが、この酸化された錯体は電子伝導性物質を介してRuS2のように励起された適当な酸化促進物質から電子を分離させる作用を発揮することで、水を分解する電気回路が閉じられるのである。 陽子が連続的に還元されるので、水の pH値は一定である。
【0046】
第一光活性物質および第二光活性物質の放射、あるいは照射なしの失活または電子正孔の組み換えを避ける目的で、隣接する区域においてできるだけ不可逆的に電荷を分離する作用を有し、必要に応じて電荷(電子)を中継できる電子受容体を設けることが望まれる場合があり得る。 このような電子受容体は、例えば、ナノ結晶性二酸化チタン、錫でドープされたIn2O3 、キノンやメチルビオロゲンなどの有機受容化合物、金または例えば分子性ワイヤを介して電子を中継可能なその他の錯体などから選択することができるが、それに限定するものではない。
【0047】
本発明において補助物質とは、場合によって伝導体または半導体を介して酸素から第一光活性物質への電子伝達を促進する物質(例えばRuO2、Pt、またはコバルトを水中においてホスフェートと反応させることによりin situで形成される化合物)または場合によって伝導体または半導体(例えばAu、PtまたはRu)を介して第二光活性物質からの電子伝達を促進する物質を意味する。 しかし、このような補助物質自体は光活性を示さないものであって、そのほかに光活性物質が存在しなければ、水は分解されない。
【0048】
本発明の触媒システムにおける電子伝導のためには、任意の公知の電子伝導性物質を使用できる。 このような電子伝導性物質としては、例えば金属、合金、半導体、導電性酸化物、導電性ポリマー、更にいわゆる分子性ワイヤ(様々な構造における炭素鎖、炭化水素鎖、または一般的には共有結合された直鎖もしくは分岐鎖であって、化合物の官能基として、又はそれとは別な形で存在しており、電子を伝導可能な物)、またはいわゆるナノワイヤ〔ナノメートルスケール(10-9メートル)の直径を有し、例えばNi、Pt、Au等のような金属性のもの、Si、InP、GaN等の半導体、SiO2やTiO2のようにマクロレベルでは絶縁体を形成するもの、有機(DNA等) 無機(e.g. Mo6S9-xlx等)を問わず繰り返し単位からなるナノワイヤ〕を挙げることができる。 また、物質の組み合わせにより、電子は分子から分子へ飛ぶ(hop)場合もあり得る。
【0049】
有機化合物や錯体の配位子において、それらの官能基の1個以上は、保護されていてもよいチオール基であってよく、また保護されていてもよいチオール基が結合する電子伝導性物質は、金を含んでいてもよい。
【0050】
例えば、第一光活性物質から第二光活性物質への電子伝導は、ナノ結晶性に酸化チタン/インジウムすず酸化物(ITO)/銅/金/分子性ワイヤの順番で起こり得る。 ただし、その他の順番も考えられる。 同様に、第二(還元を促進する)光活性物質由来の電子も、いくつか異なる経路を通じて陽子またはヒドロニウムイオンに伝達されうる。 電子を陽子へ伝導する際、補助触媒を使用することができ、このような場合には第二光活性物質と補助触媒の間に1種または1種以上の電子伝導性物質が配置されていてもよい。
【0051】
この伝導性物質としては、天然の光合成系の場合とは異なり、有機分子または錯体だけからなっていないものが好ましい。
【0052】
本発明において、「電気的接触」あるいは「直接電気的接触」とは、それぞれの物質間の電気的接触が、電子を伝導 中継する1種または1種以上の固体物質によるものであって、その他の物質、例えば流体や固体の中でイオンを伝導するイオン伝導性物質や液状酸化還元電解質などは使用しないことを意味する。
【0053】
第一(酸化促進)光活性物質および第二(還元促進)光活性物質のいずれかが、有機分子または有機配位子を有する錯体である場合、通常、両光活性物質間の伝導は、有機物質から無機物質への、またはその反対方向への電子伝達が行なわれるが、その中には配位子を介して錯体の中心原子から伝導物質への、または配位子を介して伝導物質から錯体の中心原子への伝達という特殊な場合がある。
【0054】
これは、電子の中心原子から配位子への伝達の場合には別に問題を起こさないので、配位子の置換基は分子性ワイヤになるようになるよう選択される。 しかし、配位子またはその置換基からの、例えば無機伝導体への伝達は直接起こるものではない。 このような場合、配位子またはその末端に区域する置換基に、電子の伝導が可能になるほど無機材と強い相互作用を発揮する官能基を導入することにより、よい結果が得られる。 その代表的な例として、チオール基類を金表面と結合させることを挙げることができるが、上述相互作用は、その他にも、例えばホスホン酸類、カルボン酸の無水物、シラン類との無機酸化物との相互作用など、数多く実現可能であり、その概要は、例えばElena Galoppini 著 "Linkers for anchoring sensitizers to semiconductor nanoparticles" [Coordination Chemistry Reviews 2004 248, 1283-1297]に記載されている。
【0055】
第二光活性物質を構成する錯体として、少なくとも2種の官能基を含有し、そのうち少なくとも一方は電子伝導性物質に結合され、他方は電子が供給されると陽子を水に還元することができる化学種を含有するその他の電子伝導性物質に結合されるものが挙げられる。
【0056】
いくつかの光活性物質、特に還元を促進する錯体は、光と水と酸素が同時に存在すると不安定になる。 本発明では、このような不安定性を避けるために、上述のような物質を水と酸素を除去した「カスケット」(棺)、例えば側面が遮断性の壁からなり、薄い透明な「金製カスケット」に入れておいて水と酸素に対して保護する方法を採用することを薦める。 この場合、還元促進性の錯体はチオール基など、少なくとも二つの官能基を含有し、そのうちの一方により上述のような「カスケット」の底面を介して酸化促進性の物質への電子伝導が確保され、その他方は例えば金/白金系合金など、前記「カスケット」のふたを形成する合金材(これは錯体の中心原子から放出され分子性ワイヤを通してこの合金に中継された励起電子を用いて行われる水素生成の触媒である)への伝導を可能にするものである。 勿論、その他のカスケット型、またはサンドウィッチ型の構造も採用可能である。
【0057】
第一(酸化促進性)光活性物質と第二(還元促進生)光活性物質は、物理蒸着、ケミカル ボンディングなどを採用して、1つ以上の基板に固定されていても、他の方法により前記基板と接続されてもよい。 第一(酸化促進性)光活性物質と第二(還元促進生)光活性物質は、物理蒸着、化学蒸着などを採用して、1つ以上の基板に固定されていても、他の方法により前記基板と接続されていてもよい。 基板は、電気的 光化学的に不活性であってもそうでないものであってもよく、また直接照射された光活性物質により吸収されなかった光が浸透できるように、例えばガラスのような透明 半透明なものでも、そうでないものでもよい。 基板に使用する材料としては、これに限定することなく、コーヒング処理を施していてもよいガラス、セラミックス、金属または金属合金、半金属、カーボンまたはそれに誘導された材料、および各種の無機および有機ポリマー材料を挙げることができる。
【0058】
このような基板を用いて、板状など平面状、または管状やその他に適切な形状を有する触媒システム、例えばその一方の側に酸化促進性の光活性物質を、その他方側に還元促進性の光活性物質を配置したもの、または適切な構造を採用した場合、両物質を同じ側に配置したもの等を、製造することができる。 基板として透明 半透明な材料が選択されたとき、場合によって光をその一方の側だけに照射することにより、光を他方側まで供給できる。
【0059】
光活性物質をそれぞれ対抗する側に配置した平面状、例えば板状の触媒システムを水性液体に浸漬するとき、一方の側では水素が発生され、他方側では酸素が発生する。 このような構成によれば、水素と酸素が空間的に離れて発生するので、酸素 水素間の反応のリスクが大幅に低減する。 両方の光活性物質を空間的に離れた区域に配置した構成、例えば陽子と水だけが浸透可能な材料(例えばナフィオン(登録商標)製の膜)を用いて反応器を二つの区画に区切り、または反応器システムを2区画システムにすることで、水素と酸素を完全に隔離することが可能である。 電荷の補償を確保する目的で、陽子が両区画間で往復できるようにする必要がある。
【0060】
板状などの平面状の触媒システムを浸漬する水性液体としては、通常水を使用し、場合によって塩類、酸類、塩基類を各種添加することができるが、必然的ではない。 また、水の光分解に悪影響を与えない限り、溶剤、界面活性剤など水溶性成分との混合物、また必要に応じて、光分解反応に参加しない水性乳化液なども使用可能な溶媒である。
【0061】
本発明の製法において、触媒を照射するために使用される光としては、太陽光が好ましい。
【0062】
また、照射される第一区域と第二区域は、陽子と水だけが浸透可能な膜、例えばナフィオン(登録商標)製の膜を用いて、互いに隔離して設けることが好ましい。
【0063】
本装置が十分に透明であるかあるいは部分的に透明である場合、触媒システムの第一区域だけに光を照射してもよい。 また、逆に触媒システムの第二区域だけに光を照射してもよい。 しかし、前記両区域に光を照射する場合が多い。
【0064】
本触媒システムと光を用いて水から発生する酸素および/または水素は、連続的に捕集することができる。
【0065】
本発明の触媒システムは多くの効果がある。 水素と酸素は、酸素/水素ガスを生成することなく、別々に生成可能である。 本発明の触媒システムは粉体状ではなく、モノリス型の触媒システム、例えば単に水性溶媒に浸漬される板状のものであり、多くの場合では、製法のコストや環境負担を増加させる、塩類・酸類・塩基類などの添加を必要とせず、(ただし、これを除外するものではない)、また圧力の印加が必要な特殊セルや、溶剤に対して密閉になるようにカプセル化処理の必要な酸化還元電解質を使用する必要もない。 本発明のシステムは水酸化触媒(第一光活性物質)と水(陽子)還元触媒(第二光活性物質)を幅広い範囲から選択できることから、非常にフレキシブルであるので、各種用途などに最適な組み合わせの設計することができる。
【0066】
[代表的な触媒システムの構成]
図2は、Z機構と同様に作用する光触媒システム1の構成を示す概略断面図であり、板型の不活性基板10aの一方の側は酸化インジウムスズ(ITO)からなる透明な伝導性皮膜30が、他方側には金からなる透明皮膜40がそれぞれ設けられている。 ITO皮膜30および金皮膜40とは銅バンド20により電気的に接続されている。
【0067】
ITO皮膜30の上部はRuS2でコーティングされたナノ結晶性TiO2 50が焼結されている。 金皮膜40にはメルカプトアルキル置換基60a(実施例III.2.b参照)を3つ有するルテニウム系錯体60とアルキルチオール70とからなる単分子層(monolayer)(極端に拡大して表示)が設けられている。 同単分子層のそれぞれの端部は、すべての辺上がレジスト80で覆われ、これにより同単分子層はレジストの幅の狭いバンドによって囲まれている。 ルテニウム錯体60およびアルキルチオール70の単分子層上には、わずか2〜3層の金の層からなる透明な金皮膜90が、真空蒸着によりレジスト80にわたって薄く設けられている。 また、金皮膜90の上には白金皮膜92が設けられており、同皮膜を形成する原子の数は前記の単分子層を形成する原子の数よりもさらに少ない。
【0068】
図2に示す触媒システムは、水に浸漬され、かつ、波長420nmの光で照射されると1/2 O2 + 2H+ に酸化された H2O の酸素原子から由来する電子が、二硫化ルテニウムでコートされているTiO2 50からITO皮膜30と銅バンド20を介して金皮膜40に移動する。 照射を受けることでルテニウム錯体60が一つの電子をH+に与えるため、一つの電子が金皮膜40からチオール基およびルテニウム錯体60のメルカプトアルキル置換基60aのアルキル鎖を介して、Ru中心原子に移動する。 照射によりルテニウム錯体60は励起状態となり、これによってアルキル鎖およびもう一つのメルカプトアルキル置換基60aを介して白金92でコートされた金皮膜90に、一つの電子が与えられると共にそこからは電子が白金92を介して水中の陽子(H+) に与えられることによって前記陽子は1/2 H2に還元される。
【実施例】
【0069】
以下、本発明は実施例を挙げて更に説明するが、本発明をそれらの例に限定するものではない。
【0070】
[実施例1]
A. TiO2/RuS2の5%懸濁液(TiO2 に対しRuS2を2重量%含有)としてのTiO2上での酸化促進性の第一光活性物質の調製
水15mlで希釈したTiO2の懸濁液(10%, Aldrich社製)5gに、ルテニウム(III)-クロライド23mg(0.11mmol)を添加し、超音波槽で処理して、乾燥するまで減圧下で濃縮した。
【0071】
TiO2粉末に析出したRuCl3を、まず不活性雰囲気下、水素ガス(H2)流中で、金属に還元した。 即ち試料を300℃に加熱し、3時間流量 50 ml/min の水素流中で処理し、温度を400℃にし、流量10 ml/min の硫化水素を添加することにより、Ruの黒色硫化ルテニウム (RuS2) への硫化を誘発し、これを4時間続けた [A. Ishiguro, T. Nakajima, T. Iwata, M. Fujita, T. Minato, F. Kiyotaki, Y.Kiyotaki, Y. Izumi, K.-i. Aika, M. Uchida, K.Uchida, K. Kimoto, Y. Matsui, Y. Wakatsuki, Chem. Eur. J. 2002, 8 (14), 3260-3268. / K. Hara, K. Sayama, H. Arakawa, Appl. Catal. A.: Gen. 1999, 189 (1), 127-137.] この結果得られる灰色の粉末から50mgを取り、水1mlと混合した後、長音波槽によく懸濁させることにより、薄い灰色を呈するTiO2上のRuS2(2重量%)の懸濁液が得られる。
【0072】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
一方の側の酸化インジウムスズ(ITO)で被覆された市販のガラススライド(納入元: PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)のITO側に、TiO2の10%水性懸濁液(粒子径40nmより小さいもの、Aldrich社製)を薄く塗布し、450℃で4時間の焼結処理を施した後で、TiO2に対してRuS2(上述のように調製したもの)2重量%の割合でTiO2/RuS2を含有する5%水性懸濁液を塗布した後、スライドを不活性ガス雰囲気下で再度450℃で60分焼結した。
【0073】
以下、この結果得られるスライドを、Ox-Iと呼ぶ。
【0074】
[実施例2]
A. 第一光活性物質としての光活性WO3ナノ粒子と、それに白金処理を施したものの調製
光活性のWO3 ナノ粒子を、下記の引用文献に記載の方法に従って調製した (J. Polleux, M. Antonietti, M. Niederberger, J. Mater. / T.Chem. 2006, 16 (40), 3969-3975. / M. Niederberger, J. H. Bartl, G. D. Stucky, J. / T.Chem. Soc. 2002, 124 (46), 13642-13643. J. Polleux, M. Pinna, M. Antonietti, M. Niederberger, J. / T.Chem. Soc. 2005, 127 (44), 15595-15601.)
【0075】
代表的な実験では、まず無水ベンジルアルコール(またはそれと4-tert.-ブチルベンジルアルコール)20mlに、六塩化ウォルフラム (WCl6)を430mg溶解した。 密閉反応器を100℃に加熱し48時間攪拌を続けた。 生成物を、沈澱とデカンテーションを交互に行うことによって捕集し、EtOH15ミリリットルを用いて3回洗浄した。 得られた物質を数時間60℃で風乾して、WO3の黄色の粉末を得た。
【0076】
白金添加の最適化を目的に、粉末50mgを50%のエタノールと50%の水からなる混合物に懸濁した。 Pt助触媒(WO3の量に対して2重量%)の添加は光堆積法によりH2PtCl6・6H2Oの中性にした水溶液から行なった (K. Yamaguti, S. Sato, J. / T.Chem. Soc. Faraday Trans 1 1985, 81 (5), 1237-1246. / T. Sakata, T. Kawai, K. Hashimoto, Chem. Phys. Lett. 1982, 88 (1), 50-54.)
【0077】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
白金添加触媒(灰色)または白金無添加の触媒(黄色)の粉末をそれぞれ20mgずつ超音波処理により無水イソプロパノール0.4mlおよび超純水0.2mlからなる混合物に再度懸濁した。 それぞれの懸濁液の分割量を、適切なITO被覆ガラスのスライドに塗布した。 触媒を塗布したスライドは15分風乾後2時間450℃で焼結した。
【0078】
以下、この結果得られる、純WO3で被覆されたスライドをOx-IIaと呼び、白金添加のWO3で被覆されたスライドをOx-IIbと呼ぶ。
【0079】
[実施例3]
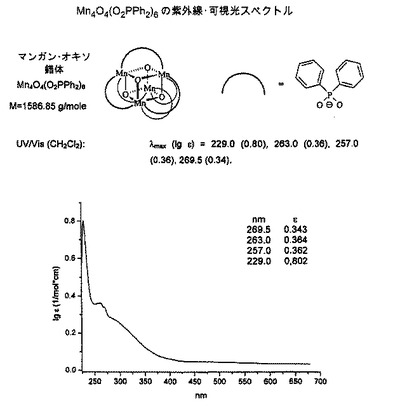
A. 酸化促進性の第一光活性物質に使用するMn4O4オキソクバン錯体Mn4O4 (フェニル2PO2)の調製
[下記の引用文献記載の方法に準拠した: R. Brimblecombe, G. F. Swiegers, G. C. Dismukes, L. Spiccia, Angew. / T.Chem. Int. Ed. 2008, 47 (38), 7335-7338. / T. G.C. Carrell, S. Cohen, G. C. Dismukes, L. Mol. Cat. A 2002, 187 (1), 3-15.]
【0080】
【化1】
【0081】
NaOH60mg(1.5mmol)を、不活性ガス(N2)雰囲気下、DMF20mlに溶解してNaOHのDMF溶液を調製した。 これに、ジフェニルホスフィン酸330mg(1.5 mmol)および過塩素酸マンガン(II)255mg(0.7mmol)をDMF8mlに溶解したものを、激しく攪拌しながら添加した。 反応時間として15分経過後、KMnO4 50mg(0.3mmol)をDMF18mlに溶解したものを、添加漏斗を通して徐々に滴下した。 これにより、褐色のかかった赤色の懸濁液が形成され、これを室温で16時間攪拌した。 懸濁液を濾過して得られた残渣を、それぞれ40mlのエタノールとエーテルを用いて洗浄した後、乾燥した。 目的の錯体は、褐色がかった赤色を呈する粉末として得られる。
【0082】
【化2】
【0083】
UV/Vis (CH2Cl2): λmax (lg ε) = 229.0 (0.80), 263.0 (0.36), 257.0 (0.36), 269.5 (0.34).
紫外線/可視光のスペクトルは、図5を参照されたい。
【0084】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
前記のMn4O4(フェニル2PO2)錯体を、下記の引用文献記載の方法に従ってITO被覆ガラスのスライドに塗布した(M. Yagi, K. Nagai, A. Kira, M./ T.Chem. Kaneko, J. Electroanal. / T.Chem. 1995, 394 (1-2), 169-175.)
【0085】
片側が酸化インジウムスズ(ITO)で被覆された市販のガラススライド(納入元: PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)のITO側に、Mn4O4(フェニル2PO2)錯体の 1mM 溶液(この錯体をナフィオン(登録商標)117と無水エタノールの1/1の混合物に溶解したもの)を塗布した後、12時間風乾した。
【0086】
以下、この結果得られるスライドを、Ox-IIIと呼ぶ。
【0087】
[実施例4]
A. 還元促進性の第二光活性物質に用いるトリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート)
【0088】
A.1 保護されたビピリジン配位子である4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
[D. K. Ellison, R. T. Iwamato, Tet. Lett. 1983, 24 (1), 31-32. / P. K. Gosh, T. G.C. Spiro, J. / T.Chem. Soc. 1980, 102 (17), 5543-5549に準拠して調製した。]
【0089】
【化3】
【0090】
a) 4-(11-ブロモウンデシル)-4’-メチル-2,2’-ビピリジン (A)
不活性ガス雰囲気下で、無水テトラヒドロフレン(THF)100mlを0℃まで冷却し、4,4’-ジメチル-2,2’-ビピリジン2.00g(10.9 mmol)と混合した。 0℃で15分攪拌を続けた後、リチウムジイソプロピルアミド(LDA)の2M溶液5.45ml(10.9mmol)を、前記のTHFに添加した後、冷却しながら2時間反応させた。 反応液を、30分かけて、0℃にて、3.30g(11.0mmol)ジブロモデカンを無水THFに溶解して得た溶液に滴下した。褐色がかった透明感のある溶液を、0℃で2時間攪拌してから更に室温で16時間攪拌を続けた後、H2Oを用いて冷却した。 この懸濁液を、減圧下で殆んど乾いた状態になるまで濃縮した。 水性残渣を、25mlの水に移して食塩水25mlを添加した後、混合物を、CHCl3 75mlを用いて3回抽出した。 各有機相を、Na2SO4で乾燥した後、乾いた状態になるまで減圧下で濃縮した。 生成物の精製は、エチルアセテートを用いて、シリカゲル上で行なった。 生成物Aは、ベージュ色を呈する結晶性粉末の形態で得られる。
【0091】
b) 4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
[生成法は、D. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, Tetrahedron 2006, 62 (9), 1955-1966参照.]
【0092】
前記Aと、1.20g(2.97mmol)無水エタノール(EtOH)とを、THF(50ml、1/1、V/V)に溶解して得られた溶液を、不活性ガス(N2)の雰囲気下、 チオアセテート2.04g(6 equ.、17.9mmol)と混合した後、2時間還流した。 反応が完了した後、赤褐色を呈する懸濁液を濾過し、赤桃色を呈する透明感のある濾液を、回転蒸発器を用いて減圧下で濃縮した。 それから残渣をクロロホルム50mlに移し、水40mlずつで2回、食塩水40mlで1回洗浄した後、Na2SO4上での乾燥後、減圧下で溶剤を除去した。 残渣の精製は、EtOAc/n-ヘキサン (1 : 1)を用いてシリカゲル上で行なった。 生成物Bは黄色を呈する固体として得られる。
【0093】
4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
【0094】
【化4】
【0095】
1H-NMR (300 MHz, CDCl3): δ = 8.53 (dd, 2H, J5/6 = J5’/6’= 4.2 Hz, J3/6 = J3’/6’= 0.6 Hz, 6-H, 6’-H), 8.21 (br s, 2H, 3-H, 3’-H), 7.14 (dd, 2H, J5/6 = J5’/6’= 4.8 Hz, J3/5 = J3’/5’= 1.5 Hz, 5-H, 5’-H), 2.84 (t, J = 7.5 Hz, 2H, 17-CH2, S-CH2), 2.67 (t, J = 7.8 Hz, 2H, 7-CH2, Aryl-CH2), 2.42 (s, 3H, 7’-CH3, Aryl-CH3), 2.30 (s, 3H, 20-CH3, CO-CH3), 1.72 - 1.62 (m, 2H, 8-CH2), 1.59 - 1.49 (m, 2H, 16-CH2), 1.38 - 1.24 (m, 14H, 9/10/11/12/13/ 14/15-CH2).
13C-NMR (75.5 MHz, CDCl3): δ = 196.0 (C-19), 156.1 (C-2), 156.0 (C-2’), 152.9 (C-4), 148.9 (C-6), 148.9 (C-6’), 148.1 (C-4’), 124.6 (C-3’), 123.9 (C-3), 122.0 (C-5’), 121.2 (C-5), 35.5 (C-7), 30.6 (C-17), 30.4 (C-8), 29.4 (C-11/12), 29.4 (C-10), 29.4 (C-13), 29.3 (C-15), 29.3 (C-9), 29.1 (C-16), 29.0 (C-20), 28.8 (C-14), 21.2 (C-7’).
EI-MS (70 eV): m/z (%) = 398 (4) [M+], 357 (18), 356 (68), 355 (90) [M+-Me, -CO], 324 (9), 323 (39), 309 (18), 295 (12), 281 (10), 267 (6), 253 (6), 239 (6), 211 (13), 209 (5), 198 (21), 197 (100), 185 (12), 184 (95), 183 (10), 170 (5), 43 (30), 41 (5).
紫外線/可視光 (CH2Cl2): lmax (lg e) = 229.0 (0.80), 263.0 (0.36), 257.0 (0.36), 269.5 (0.34).
【0096】
A.2: トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C) の調製
a)トリス[4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート]ルテニウム(II)ビス(ヘキサフルオロホスフェート) (保護されたC)
[R. A. Palmer, T. S. Piper, Inorg. / T.Chem. 1966, 5 (5), 864-878. および Y-R. Hong, C. B. Gorman, J. Org. / T.Chem. 2003, 68 (23), 9019-9025に準拠。]
【0097】
【化5】
【0098】
不活性ガス(窒素ガス)の雰囲気下で窒素ガスを流し込んだ無水エタノール30mlを、実施例4.A.bで調製したB180mg(452μmol/RuCl3に対して25%過剰)と混合した。 この反応液に、RuCl3・H2O 25mg(120.5μmol)を、無水エタノール5mlに溶解したものを添加し、暗所で65時間還流した。 反応完了後、赤褐色を呈する懸濁液を濾過し、透明の赤桃色の濾液を、減圧下で回転蒸発器にて濃縮した。 赤色を呈する残渣をCH2Cl2 20mlに移し、2回にわたって水を20mlずつ用いて洗浄した。 各有機相は、Na2SO4で乾燥した後、乾いた状態になるまで回転蒸発器を用いて減圧下で濃縮した。 この結果として得られる赤褐色を呈する生成物の混合物をCHCl3/methanol (15/1)を用いてシリカゲル上で精製することにより、目的の化合物が得られる。
【0099】
b) トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C) の調製
[H. Hotta, K.Imahori, A. Yoshida, T.Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, Tetrahedron 2006, 62 (9), 1955-1966. F. Ono, S. Kanemasa, J. Tanaka, Tetrahedron Lett. 2005, 46 (44), 7623-7626. / T. Suzuki, A. Matsuura, A. Kouketsu, S. Hisakawa, H. Nakagawa, N. Miyata, Bioorg. Med.Med. / T.Chem. 2005, 13 (13), 4332-4342に準拠]
【0100】
100ml容量の三つ口丸底フラスコに無水エタノール10mlを入れ、30分窒素ガスを流し込む。 無水THF10mlと共にA.2.a号に記載の通り調製した保護されたルテニウム錯体 44mg((32.2μmol)を室温にてフラスコに移し、100 mg KOH (55 eq.)と混合する。. 反応液を室温で60分攪拌した後、食塩水15mlの上に注いでから、25mlのCH2Cl2で抽出する。 有機相は、Na2SO4の上で短時間乾燥した後、減圧下で濃縮する。 赤色の錯体は、NH4PF6 (0.6 mmol,、約20 eq.)を105mgをメタノール5mlに溶解したものと組み合わせてから、暗所で120分室温で攪拌する。 赤色を呈する溶液は2回水洗した後、Na2SO4の上で少々乾燥した後、回転蒸発器を用いて減圧下で溶剤を除去する。 残渣は少量のCH2Cl2に溶解して、それから石油エーテルを用いて粒状化させる。 目的のルテニウム錯体Cは赤桃色を呈する粉末として沈降する。
【0101】
トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C)
【0102】
【化6】
【0103】
1H-NMR (300 MHz, CDCl3): δ = 8.17 (d, 2H*3, J = 7.8 Hz, 6-H*3, 6’-H*3), 7.58-7.45 (m, 2H*3, 3-H*3, 3’-H*3), 7.23 (dd, 2H*3 J = 5 Hz, J = 2 Hz, 5-H*3, 5’-H*3), 2.79 (t, 2H*3, J = 8 Hz, 7-CH2, Aryl-CH2), 2.53 (s, 3H*3, 7’-CH3, Aryl-Me), 2.50 (t, 2H*3, J = 7.2 Hz, 17-CH2, S-CH2), 1.72 - 1.62 (m, 2H*3, 8-CH2), 1.61- 1.54 (m, 2H*3, 16-CH2), 1.32 (t, 3*1H, J = 7.5 Hz, -SH) 1.38 - 1.23 (m, 14H*3, 9/10/11/12/13/14/15-CH2).
13C-NMR (75.5 MHz, CDCl3): δ = 156.4 (C-2), 156.3 (C-2’), 154.5 (C-4), 150.8 (C-6), 150.6 (C-6’), 150.0 (C-4’), 129.0 (C-3’), 128.0 (C-3), 124.6 (C-5’), 123.8 (C-5), 35.4 (C-7), 34.0 (C-16), 30.0 (C-8), 29.5 (C-10), 29.4 (C-11/12), 29.4 (C-13), 29.3 (C-15), 29.0 (C-9), 28.3 (C-14), 24.6 (C-17), 21.3 (C-7’).
ESI-MS: m/z = 1315.6 ([M-(PF6)]+), 585.3 ([M-(2*PF6)]2+).
UV/Vis (MeOH): λmax (lg ε) = 286.5 (1.27), 248.0 (0.36), 258.0 (0.35), 457.5 (0.26), 436 (sh, 0.22), 324.5 (0.20).
紫外線/可視光のスペクトルは、図3を参照されたい。
【0104】
B. 前記還元促進性光活性物質Cの不活性基板への被覆処理
一方の側に膜厚50nmの金皮膜と、膜厚5〜10nmのクロムとのプレコートを有し、寸法が第一光活性物質を塗布するときに使用されたものと同様であるガラススライドを、トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (C)をエタノールに溶解して得られた溶液で処理する。 本発明の一つの実施形態によれば、溶解した錯体を塗布するとき、Sと同様に共吸着物である、鎖長がC8、C10、C12、C14、C16またはC18のアルキルチオールの存在下で行なう( J. Summer, S. E. Creager, J. / T.Chem. Soc. 2000, 122 (48), 11914-11920に準拠。)錯体の溶解度を向上する目的で、必要に応じてジクロロメタンを少量その溶液に添加する。 本発明の他の実施形態によれば、Y. S. Obeng, A. J. Bard, Langmuir 1991, 7 (1), 195-201に記載のように、共吸着物を添加しないで、塗布処理を施す。塗布した後、スライドを乾燥する。
【0105】
以下、この結果得られるスライドを、Red-Iと呼ぶ。
【0106】
[実施例5]
A. 還元を促進する第二光活性物質としての[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート)の調製
(以下、配位子を略して表示するが、bpyは2,2'―ビピリジン、Me-bpyは4'―メチル―2,2'―ビピリジン、またtppzはテトラピリドフェナジンを指す。)
【0107】
以下の調製方法は、P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway, A. H. White, Inorg. / T.Chem. 1995, 34 (24), 6145-6157に準拠する。
【0108】
A.1: cis-ジカルボニル-(2.2'ービピリジン([4-(11-アセチルスルファニルウンデシル)-4'-メチル-2,2'-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E)の調製
【0109】
【化7】
【0110】
窒素ガスによる不活性雰囲気下で、実施例4の4.1.b号で合成した4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B) 195mg(0.49mmol)を、無水エタノール30mlに溶解する。 この溶液に、cis,cis-[Ru(bpy)(CO)2(CF3SO3)2] (D) (0.25 mmol; P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway, A. H. White, Inorg. / T.Chem. 1995, 34 (24), 6145-6157に準拠して調製したもの) 150mgを添加した後、還流しながら2時間過熱する。透明感のある赤色を呈する溶液を、乾いた状態になるまで減圧下で濃縮する。残渣を、水25mlに溶解してから濾過する。濾液は、室温まで冷却し、冷たい状態で飽和するNH4PF6 の水溶液5mlを添加すると、即座に無色の沈殿物が発生する。この沈殿物を濾過分離してから冷水30mlで洗浄する。目的の生成物(E)は少量のエタノール・アセトンで再結晶されるピンク色を呈する個体として得られる。
【0111】
cis-ジカルボニル(2,2’-ビpリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E)
【0112】
【化8】
【0113】
化合物Eには、二つの幾何学的異性体A/B が存在する[T. J. Rutherford, F. R. Keene, Inorg. / T.Chem. 1997, 36 (13), 2872-2878]。
【0114】
1H-NMR (300 MHz, (CD3)2CO): δ = 9.50 (m, H, Aryl-H), 9.31 (dd, H, J = 6.0 Hz, J = 8.0 Hz, Aryl-H), 8.94 (d, H, J = 8.4 Hz, Aryl-H), 8.85 (m, H, Aryl-H), 8.82 (d, H, J = 8.4 Hz, Aryl-H), 8.73 (m, H, Aryl-H), 8.63 (m, H, Aryl-H), 8.39 (m, H, Aryl-H), 8.11 (m, H, Aryl-H), 7.96 (m, H, Aryl-H), 7.82 (m, H, Aryl-H), 7.69 (m, H, Aryl-H), 7.65 (t, H, J = 6.0 Hz, Aryl-H), 7.52 (m, H, Aryl-H), 3.04 / 2.82 (t, 2H, J = 8 Hz, 7-CH2 A/B), 2.85 / 2.83 (t, 2H, J = 7.2 Hz, 17-CH2 A/B), 2.75 / 2.53 (s, 3H, Aryl-CH3 A/B), 2.29 / 2.28 (s, 3H, CO-CH3 A/B), 1.86 / 1.65 (m, 2H, 8-CH2 A/B), 1.57-1.45 (m, (m, 2H, 16-CH2 A/B), 1.31 - 1.25 (m, 14H, 9/10/11/12/13/ 14/15-CH2 A/B).
EI-MS (70 eV): m/z (%) = 713 (8) [M-H+-2*PF6], 616 (12), 506 (9), 449 (14), 353 (22), 322 (35), 197 (100), 184 (88), 156 (28), 65 (17).
【0115】
A.2 (2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート)の調製 (G)
【0116】
【化9】
【0117】
前記A.1号に記載のように合成したcis-ジカルボニル (2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E) 145 mg (145μmol)を無水2-メトキシエタノール40mlに溶解し、これに、テトラピリドフェナジン (F) (tppz; 0.43 mmol, 3eq.; W. Paw, R. Eisenberg, Inorg. / T.Chem. 1997, 36 (11), 2287-2293. J. Bolger, A. Gourdon, E. Ishow, J.-P. Launay, J. / T.Chem. Soc., Chem. Commun. 1995, 1799-1800に準拠して合成したもの)165 mgを無水2-メトキシエタノール10mlに溶解したものを添加する。黄色を呈する懸濁液を、窒素ガスで連続的にゆっくり置換する。100 °Cに加熱し、約10分窒素ガスで置換してからトリメチルアミン-N-オキサイド(ZMMO)56mg(0.5 mmol、約3.5 eq.) を 無水2-メトキシエタノール5mlに溶解したものを添加する。得られる懸濁液は連続的に窒素ガスを流し込みながら暗所で24時間還流させる。反応終了後、赤褐色を呈する懸濁液を容量が半分に減るまで濃縮し、30分暗所で放置する。沈降した未反応のtppzを濾過分離する。ろ過器に残った残渣を、エタノール15mlで洗浄し、濃い赤色を呈する濾液を、減圧下で濃縮する。これにより、ほとんど黒い濃い赤色を呈するオイル状の物質が得られる。CHCl3/メタノール(5:1)を用いるシリカゲル上のクロマトグラフィーによる分離・精製後、生成物(G)を、赤桃色のエネメル質状の固体として得る。
【0118】
(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート) (G)
【0119】
【化10】
【0120】
ESI-MS (CH3CN): m/z = 566.2 ([M-(2*PF6)+HCOOH]2+).
UV/Vis (CH3CN): λmax (lg ε) = 282.0 (1.02), 244.0 (0.60), 379.5 (0.19), 359.0 (0.16), 453.5 (0.16), 434.0 (sh, 0.15).
【0121】
A.3 [(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]
ビス (ヘキサフルオロホスフェート) (H)の調製
以下の反応は、S. Rau, B. Schafer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Gorls, W. Henry, J. G.C. Vos, Angew. / T.Chem. Int. Ed. 2006, 45, 6215-6218の方法に準拠する。
【0122】
【化11】
【0123】
不活性ガス(窒素ガス)の雰囲気下、6 mg of ビス(アセトニトリル)-パラジウム(II)-ジクロライド6mg (23μmol、15 %過剰、Aldrich社)を無水ジクロロメタン(DCM)20mlと混合した後、(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート)26mgを無水DCM 2mlに懸濁したものを添加する。反応混合物を加熱し、窒素ガス雰囲気下で6時間還流する。反応終了後、懸濁液を室温まで冷却した後、濾過することにより得られる赤褐色を呈する残渣を、少量のDCMで洗浄する。乾燥することにより、目的の生成物を赤褐色を呈する個体として得る。
【0124】
[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート) (H)
【0125】
【化12】
【0126】
ESI-MS (CH3CN): m/z = 673.2 ([M-(2*PF6)]2++HCOOH+H2O]).
FAB-MS (-): m/z = 1526.2 (MH -+H2O).
UV/Vis (CH3CN): λmax (lg ε) = 241.0 (1.34), 283.5 (0.56), 354.5 (sh, 0.10), 432.0 (0.08), 453.5 (0.08).
紫外線/可視光スペクトルは、図4参照
【0127】
B. 不活性な基板への還元促進性の第二光活性物質Hの適用
錯体Hの被覆処理は S. J. Summer, S. E. Creager, J. / T.Chem. Soc. 2000, 122 (48), 11914-11920 / S. E. Craeger, G. K. Rowe, J. Electroanal. / T.Chem. 1994, 370 (1-2), 203-211 (coating in CH3CN) / J. M. Tour, L. Jones II, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh, S. V. Atre, J. / T.Chem. Soc. 1995, 117 (37), 9529-9534. / L. Cai, Y. Yao, J. Yang, D. W. Price jr., J. M. Tour, Chem. Mater. 2002, 14 (7), 2905-2909 (アセチルスルファニル基でのコーティング)に準拠して行なう。
【0128】
第一光活性物質の皮膜処理のときと同様の寸法を有し、片側に膜厚50nmの金皮膜および膜厚5〜10nmのクロム・プレコートを設けたガラススライドに、錯体HのCH3CN溶液を塗布する。塗布時には、炭素数12または16のアルキルチオールなどの共吸着物が存在していてもよいが、存在していなくてもよい。塗布の後のスライドを、洗浄してから乾燥する。
【0129】
この結果得られるスライドを、以下、Red-IIと呼ぶ。
【0130】
[実施例6]
A. 還元促進性の第二光活性物質として2.5%アンチモンと2.0%クロムでドープした白金添加SrTiO3 の調製
(共ドープしたSrTiO3 の調製はH. Kato, A. Kudo, J. Phys. / T.Chem. B 2002, 106 (19), 5029-5034に準拠して行なった。)
【0131】
化学式SrTi1-X-YSbXCrYO3 に従って出発原料であるSrCO3、TiO2、Sb2O3、Cr2O3 のそれぞれの適量を充分に混合した。混合物を、アルミナ製ルツボに移し、大気雰囲気中で1050℃で少なくとも30時間焼成した。ボールミル(GERMATECH GmbH, Osterfeldstr. 3, D-56235 Ransbach-Baumbach, ドイツ)を用いて微細粉末を得るまで粉砕した。
【0132】
白金添加のためにエタノール20%および水80%からなる混合液に懸濁した。Pt助触媒(WO3の量に対して2重量%)を、光堆積法によって、H2PtCl6・6H2Oの中性に調製した水溶液から堆積させた (K. Yamaguti, S. Sato, J. / T.Chem. Soc. Faraday Trans 1 1985, 81 (5), 1237-1246. / T. Sakata, T. Kawai, K. Hashimoto, Chem. Phys. Lett. 1982, 88 (1), 50-54参照。)
【0133】
B. 前記の還元促進性光活性物質でのITO 基板の被覆処理
2.5%アンチモンおよび2.0%クロムでドープした白金添加SrTiO3 の乾燥粉末100mgを、無水イソプロパノール2mlおよび超純水1mlからなる混合液に、超音波処理によって再懸濁させた。懸濁液の少量の分割量を、片側が酸化インジウムスズ(ITO)でコーティングされた、市販のガラススライド (PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, [ドイツ]から入手)に塗布した。15分風乾燥後、スライドを450℃で2時間焼結した。
【0134】
この結果得られるスライドを、以下、Red-IIIと呼ぶ。
【0135】
[実施例7]
第一および第二の光活性物質を含有する触媒ユニットの触媒システムへの組立およびその照射
7.1.a Ox-I とRed-Iの組み合わせ
酸化促進性第一光活性物質RuS4と、還元促進性第二光活性物質トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート)とをそれぞれ含む、実施例1および4で作製した触媒ユニット(スライドOx-IおよびスライドRed-I)の両方を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。その後、Red-Iの金の側面をレジストでコーティングして、銅バンドと、金皮膜の端部と、金皮膜の上で前記錯体を含有するスライドのそれぞれの端とに沿って、幅の狭い帯状の区域が被覆されるようにした。このように組み立てられて電動可能に接続した触媒ユニットを乾燥した後、錯体でコーティングされた金側に再度非常に薄い金皮膜(5 nm)を蒸着し、隣接するレジスト層が被覆されるようにした。最終的には、この金皮膜を更に白金の0.5〜0.7単分子層(ML)を設けて、触媒システムを得た。
【0136】
7.1.b (Ox-I)-(Red-I)による触媒システムの照射
7.1.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含むこの膜を、光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を満たし、上部をオイル層でカバーした。Ox-Iに隣接する区画に、黄色のロイコインジゴカルミンを導入した。その後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。約50℃に加熱した後で、黄色のロイコインジゴカルミンが変色して青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が発生した。
D2OおよびH218O、またフィルタとして亜硝酸塩を使用した実験で、マススペクトロメトリーで分析した結果、D2 、また自然量より大きい量の16O18Oが発生したことが確認されたことからも、水の分解が実証された。
【0137】
7.2.a Ox-IとRed-IIの組み合わせ
酸化促進性第一光活性物質RuS4 と還元促進性第二光活性物質[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]ビス-(ヘキサフルオロホスフェート)とをそれぞれ含む、実施例1および5で作製した触媒ユニット(スライドOx-IおよびスライドRed-II)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0138】
7.2.b (Ox-I)-(Red-II)による触媒システムの照射
7.2.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-Iに隣接する区画に、黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。室温にて、Red-IIに隣接する区画のpHを、硫酸の添加により約5の酸性側に調製した後で、黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が発生した。
【0139】
7.3.a Ox-IとRed-IIIの組み合わせ
酸化促進性第一光活性物質RuS4 と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3をそれぞれ含む実施例1および6で作製した触媒ユニット(スライドOx-IおよびスライドRed-III)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0140】
7.3.b (Ox-I)-(Red-III)による触媒システムの照射
7.3.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-Iに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0141】
7.4.a Ox-IIとRed-IIIの組み合わせ
酸化促進性第一光活性物質WO3 (白金添加のものと白金無添加のもの) と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3とをそれぞれ含む、実施例2および6で作製した触媒ユニット(スライドOx-IIおよびスライドRed-III)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0142】
7.4.b (Ox-II)-(Red-III)による触媒システムの照射
7.4.a号で説明したように作製した触媒システムをナフィオン(登録商標)膜に接着し、また触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-IIに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0143】
7.5.a Ox-IIIとRed-IIIの組み合わせ
酸化促進性第一光活性物質Mn4O4(フェニル2PO2)と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3をそれぞれ含む実施例3と6で作製した触媒ユニット(スライドOx-IIとスライドRed-III)をそれぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0144】
7.5.b (Ox-III)-(Red-III)による触媒システムの照射
7.5.a号で説明したように作製した触媒システムをナフィオン(登録商標)膜に接着し、また触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-IIIに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0145】
本願に引用されている文献、例えばジャーナルの記事・論文、本、特許、特願などに開示されている内容はすべて参照により参照により、本明細書中に組み込まれているものとする。
【技術分野】
【0001】
本発明は水を光を用いて水素と酸素に分解するための触媒システムおよび本触媒システムを用いて水素および酸素を製造する方法に関する。
【背景技術】
【0002】
水素は未来の物体的なエネルギー担体になると考えられ、環境に優しい方法、即ち二酸化炭素の副生がなく、通常はコストが高く環境に優しくない場合が多い従来の電解を利用しない、水素の製法が注目を集めている。
【0003】
米国特許US 6,936,143 B1号では、グレッツエルらは光を用いて水を水素と酸素に分解するタンデムセルまたは光電化学システムであって、両セルは電気的に接続されているものを開示した。 この電気的接続では、WO3 またはor Fe2O3 等からなる光陽極から色素増感メソ多孔性 TiO2 皮膜からなる光陰極への電子伝導のために、有機酸化還元電解質を使用している。 前記文献には、この有機酸化還元電解質についての記載は一切ないが、電解質における電荷の運搬は通常イオンによるものであるので、有機酸化還元電解質という概念から電子の運搬はイオン性導電によって行なわれることは明らかである。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許US 6,936,143号明細書
【発明の概要】
【課題を解決するための手段】
【0005】
本発明は光を用いて水を水素と酸素に分解するために用いるモノリス触媒システムを提供する。このシステムは、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水から酸素と陽子を生成させることができる第一光活性物質と、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水中の陽子を水素に還元することができる第二光活性物質と
を含有し、
第一光活性物質および第二光活性物質は、一種またはそれ以上の電子伝導性物質を介して電気的、特に直接電気的な接触状態にあることを特徴とする。
【0006】
さらに、本発明は光と触媒を用いて水から水素と酸素を生成する方法を提供する。この方法は、
第一光活性物質を含有するか、あるいは、第一光活性物質とこれと組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第一区域において、本発明による触媒システムを水または水性液体または水性溶液に接触させ、
第二光活性物質を含有するか、あるいは、第一光活性物質とこれに組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第二区域において、前記触媒システムを水または水性液体または水性溶液に接触させ、
前記触媒システムに光を照射し、
陽子が第一区域から第二区域に移動できるように、第一区域に接触している水または水性液体または水性溶液と、第二区域に接触している水または水性液体または水性溶液とが相互に接触状態にあるようにすることを特徴とする。
【0007】
本発明の好ましい実施形態は、特許請求項に記載のとおりである。
【図面の簡単な説明】
【0008】
【図1】図1は、本発明による触媒システムにおいて用いる、植物 バクテリアにおける光合成あるいは水の光分解のいわゆるZ機構を示す図である。
【図2】図2は、本発明による触媒システムの一例を示す概略断面図である。
【図3】図3は、トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス(ヘキサフルオロホスフェート)の紫外線/可視光スペクトルを示す図である。
【図4】図4は、[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート)の紫外線/可視光スペクトルを示す図である。
【図5】図5は、Mn4O4(フェニル2PO2)の紫外線/可視光スペクトルを示す図である。
【発明を実施するための形態】
【0009】
[いわゆるZ機構による水素 酸素の発生原理]
本発明の触媒システムでは、水の1/2O2とH2への分解のために4つの陽子を使っている。 この場合における各ステップとそれらのエネルギーレベルによる関係を概略的に図1に示す。
【0010】
下記の反応には触媒システムの酸化側(以下、第一光活性物質とも言う)で2つの陽子が要求される。
H2O + 2 光子 → 1/2 O2 + 2 H+ + 2 e- (EO2/H2O(pH7) = +0.82 V).
【0011】
この反応は植物/バクテリアの光化学系2における反応である。
【0012】
次に示す反応には触媒システムの還元側(以下、第二光活性物質とも言う)で2つの陽子が要求される。
2 H+ + 2 e- + 2 光子 → H2 (EH+/H2(pH7) = -0.41 V)
【0013】
この反応は、水素を発生する酵素ヒドロゲナーゼがかかわり、数種類のバクテリアの光化学系に起こり得る反応である。
【0014】
総合的には、この反応は次のようにまとめられる。
H2O + 4 光子 → H2 + 1/2 O2 (EpH7 = 1.23 V).
【0015】
したがって、この場合には、1 e- が水中の酸素から分離し、それがH+イオンに移動 (2 hν → 1 e-) するように2つの光子 (2 hν) を使用するプロセスが必要とされる。 また、この反応は植物 バクテリアの光合成のZ機構における反応の一つである。
【0016】
なお、本明細書において水素、陽子、H+、H+イオンの概念はジュウテリウム、ジュウテリウムイオン、D+、D+イオン等という意味も含んでいる。 同様に、H2とは「HD」、「D2’’」の意味を含んでいる。しかし、「D2」には「HD」と「H2」は含んでいない。
【0017】
本発明による触媒システムの酸化側(電気化学の表現では陽極である)で放出される電子は、電子伝達性物質の1種または2種以上を介して直接本触媒システムの還元側(電気化学の表現では陰極である)に伝導される。 電子伝導性物質という概念にはイオン伝導体、液体状酸化還元電解質、固体電解質は含んでいない。 pn接合などの接合による電子伝導は本発明のような、第一光活性物質と第二光活性物質の間における電子伝導性物質には無関係であるものとする。
【0018】
本発明は、二種の光活性物質間の電子伝導の面でも植物 バクテリアにおけるZ機構を模造するものである。 実質的に植物 バクテリアにおける光合成、もっと詳しく言えば、バクテリアのある種類における水の光分解を人工的に行なうことを図っている。
【0019】
勿論、本システムを働かせるために、外部から電圧を印加する必要がない。
【0020】
本出願においてモノリス触媒システムとは、コンパクトな構造によるシステムを意味し、本システムから伸びたり、または本システムに一体化されていない、目で見られるような電線、導体、電極など、例えば導電性ワイヤ、バンド、シート等によってシステムに接続された電極のないシステムを指している。 このようなシステムの形状として、プレート状、フィルム状、チューブ状等が挙げられる。 「モノリス(monolithic)」とは必ずしも本システムが一つの部品として製造されたものを指すものではない。
【0021】
また、本願における光活性物質とは、他の光活性物質と組み合わせたとき、光合成 分解におけるZ機構に対応する酸化還元電位パターンを示す物質の意味であって、その総電位差が波長420nm以上、好ましくは430nm以上、もっと好ましくは440nm以上、特に450nm以上の光の照射下、水を水素と酸素に分解するのに十分なものである。 さらに両方の光触媒のうちのいずれかまたは両方は波長700nm以上の電磁波しか吸収しないものではないほうが好ましい。
【0022】
図1に示すZ機構から明らかなように、第一および第二の光活性物質の酸化還元電位は、以下の酸化還元電位および酸化還元電位間の関係を示すものである。
1. 第一光活性物質のイオン化された状態の酸化還元電位および第一光活性物質の正に荷電された価電子帯の酸化還元電位は、両方とも+0.82Vより陽性側にある。
2. 第二光活性物質の励起された状態の酸化還元電位および第二光活性物質の伝導帯の酸化還元電位は、両方とも-0.41Vより陰性側にある。
3. 第一光活性物質の励起された状態の酸化還元電位および第一光活性物質の伝導帯の酸化還元電位は、それぞれ、第二光活性物質のイオン化された状態の酸化還元電位および第二光活性物質の正に荷電された価電子帯の酸化還元電位より陰性である。
【0023】
第一光活性物質の励起されていない状態の酸化還元電位および第一光活性物質の価電子帯の酸化還元電位は、それぞれ、基本的に第二光活性物質の励起されていない状態の酸化還元電位および第二光活性物質の価電子帯の酸化還元電位より陽性である。
【0024】
本触媒システムは、波長420nm以上の可視光の照射下で使用するので、その励起状態および伝導帯が、それぞれ、前記波長の光を用いて誘発されることが求められている。
【0025】
第一光活性物質(電気化学的には陽極またはその一部を形成するもの)と第二光活性物質(電気化学的には陰極またはその一部を形成するもの)という概念には、複数の第一光活性物質と第二光活性物質(或いはそれぞれの混合物)を使用する場合も含んでいる。
【0026】
波長420nm以上の光の照射下、第一光活性物質(酸化を促進させる物質)として様々な非分子性化合物、分子性化合物、ポリマー化合物を使用することができる。 第一光活性物質(酸化を促進させる物質)としては、これに限定することなくドープされてもよい酸化物および/あるいは酸化物と硫化物を含有する物質、特にRuS2、 貴金属または遷移金属を含有する錯体またはクラスタ、光活性を示すポリマー性物質を挙げることができる。 例えば、ドープされてもよいRuS2、貴金属を含有してもよいWO3、他元素の原子でドープされてもよい酸化鉄、Sb/M(ただし、MはCr、Niおよび/またはCu)でドープされてもよいTiO2 、Mn4系かご錯体、Ru4系クラスタ錯体、Ru3+ 系錯体、本願の実施例1~3に記載の光活性物質から選ぶことができるが、それに限定するものではない。
【0027】
水中の酸素から電子が分離される過程については、それぞれの機械的なステップの詳細がいまだにくわしく知られていない場合がある。 しかし、一般的には波長420nm以上の光子により、電子的に励起された状態が生じ、空間的に第一光活性物質から離れた励起された電子が1単位ぐらいあげた酸化状態、または水中の酸素から由来する電子で埋められる孔を残し、即座にO2と陽子が発生するが、その時に、2〜4つの陽子がさらに同時に発生する場合が多い。
【0028】
酸素の発生を促進させる目的で、第一光活性物質を前記意味で光活性を示さない補助物質および/または補助触媒、つまり照射されると酸素を生成するものではなく、その代わりに酸素の発生を促進させるものと組み合わせることができる。 このような補助物質および/または補助触媒は、RuO2、パラジウム、白金などの貴金属、または金属性コバルトを水中においてホスフェートと反応させることによりin situで形成される化合物から選ぶことができるが、但しこれに限定するものではない。
【0029】
本発明の触媒システムを使用する際、第一光活性物質が、またそれと補助物質および/または補助触媒とを組み合わせて用いる場合には、そのいずれかまたは両方が水に接触するようにする。
【0030】
第二光活性物質(還元を促進させる物質)としても、波長420nm以上の光を吸収する様々な物質を使用することができ、この物質は、非分子性固体であっても、固体の分子性化合物およびポリマー化合物であってもよい。 本発明の第二光活性物質は、例えば数多くあるルテニウム(II)系錯体から選択でき、これらには窒素を含有する配位子が結合されたものが多く、PdまたはPtのように電子が供給されると陽子を水素に還元することができる一般的な触媒種と組み合わせられると、波長420nm以上の光で励起されたときに、水中に存在する陽子(より詳細には、本明細書において「H+」と記載されることもあるヒドロニウムイオン)をH2に還元することができることが公知である。 そのほかにも、他の金属含有錯体、例えば貴金属錯体、天然葉緑素(Mgを中心原子とするもの)、Cu-クロリン、Cu-2-a-オキシメソイソクロリン、その他の金属含有フタロシアニン類、金属含有ポルフィリン類、またはH2-クロリンやプロフラビンなどの拡張π電子系を有する純有機化合物も、 必要に応じて電荷分離を起こす励起電子を適当な補助物質および または補助触媒(例えばPd、Pt、Ruまたは亜鉛を含有する触媒種)への伝導性トランスファーと組み合わせた形態で、可視光の照射下、H+を1/2H2に還元するのに十分なエネルギーを有する励起状態を示す。
【0031】
また、数多くの、必要に応じてドープされていてもよい酸化物含有物質および酸化窒化物含有物質、これほどは好ましくないが、リン化物、砒素化物、アンチモン化物、硫化物、またはセレン化物を含有する物質 [例えばCr/SbまたはRhを添加したSrTiO3やZnIn2S4、TaON、NiM2O6 (M = Nb, Ta)]も、場合によりPd、Pt、Ruなどの補助物質および/または補助触媒の存在下、波長420nmの光を照射すれば水からH2を生成させることができる。 さらに、光半導性のポリマーも、必要に応じて他の非ポリマー性の有機物質と組み合わせて、光起電力効果を示し、使用可能である。
【0032】
光活性物質の具体例は本願の実施例3〜5に示す。
【0033】
本発明の触媒システムを使用する際、第二光活性物質が、またそれと補助物質および/または補助触媒とを組み合わせて用いる場合、そのいずれかまたは両方が水に接触するようにする。
【0034】
本発明では、第一光活性物質は、植物 バクテリアが、水を酸素と陽子に分解する光化学系IIまたはその変更されたものを完全に形成しないことが好ましい。 特に、ポリペプチドまたは蛋白質を含有しないことが好ましい。 なぜなら、天然の光化学系IIは非常に不安定であるからである。
【0035】
本発明では、第二光活性物質は、植物・バクテリアが、NADP+をNADPH+H、あるいは特別な場合ではヒドロゲナーゼを用いて陽子を水素に還元する光化学系IIまたはその変更されたものを完全に形成しないことが好ましい。 特に、ポリペプチドまたは蛋白質を含有しないことが好ましい。 その理由は、ヒドロゲナーゼは酸素に対して非常に敏感であって、陽子の還元のための補助触媒によっては完全な光化学系IIに連結できないものがあり得ることにある。
【0036】
第一光活性物質として、単結晶または単結晶からドーピングにより得たものを使用することは原理的には有用であるものの、下記の理由から好ましくない。 単結晶またはエピタキシャル成長などによって得られるドープされた結晶は、製造コストが高い。
【0037】
同じ理由から、第二光活性物質の場合も単結晶または単結晶からドーピングにより得たものを使用することは原理的には有用であるものの、下記の理由から好ましくない。
【0038】
また、そのコストが高いから、ドープされた珪素を光活性物質として使用しないほうが好ましい。
【0039】
また、本発明では、光起電力の分野で使われる従来の半導体あるいは光半導体、例えば遷移金属の1価、2価および/または3価のカチオンと元素の周期表におけるVa族およびVIa族のアニオンを含有してもよいIII-V族半導体、II-IV族半導体、III-VI族半導体、またそれに類似している半導体を両方の光活性物質に用いないことが好ましい。 また、光活性物質の両方ともが、上述の半導体を使用しないことがさらに好ましい。 これらの半導体は原理的には有用ではあるが、コストが高く、公害を起こす金属を含有したり、水の存在下、不安定になり、それに伴って毒性ガスが発生することがしばしばある。
【0040】
また、第一光活性物質と第二光活性物質との組み合わせは、第一光活性物質により吸収されなかった黄色光、赤色光、近赤外光が第二の光活性物質材に伝達されるように、両物質が列に配置されている限り、太陽光の青色部分と緑色部分を吸収する半導性酸化物と陽子の還元に太陽光の黄色部分と赤色部分と近赤外部分を使用するメソ多孔性皮膜からなる組み合わせではないことが好ましい。 この組み合わせでは、水の分解のために本発明において好ましくない特殊のタンデムセルを必要とする。
【0041】
第一光活性物質と第二光活性物質とは、互いに異なる化学種からなっていることが好ましく、同じ元素から構成されていないことがさらに好ましい。
【0042】
第一光活性物質と第二光活性物質とは、上述のZ機構に従って組み合わせることができる。
【0043】
第二光活性物質(還元を促進させる物質)が光で照射されるとき、そこの電子は励起状態となり、そのエネルギーが十分な場合、水中の陽子に伝達され(ただし、それにはPt、Ruなどの補助物質または補助触媒を用いる場合が多い)、その結果、水素が発生すると共に、光活性の還元促進物質または第二光活性物質に正孔が生じるか、その酸化状態が1つの単位上がる。
【0044】
このサイクルは第一光活性物質から励起電子を与えられ、それにより酸化された第二光活性物質が還元を受けることで閉じられる。
【0045】
具体例を一つ挙げれば、例えば適当なRu(II)錯体が励起されたとき、その電子が、必要に応じて補助物質および/または補助触媒を介して陽子側に伝達され、すると、前記錯体はRu(III)錯体に酸化されるが、この酸化された錯体は電子伝導性物質を介してRuS2のように励起された適当な酸化促進物質から電子を分離させる作用を発揮することで、水を分解する電気回路が閉じられるのである。 陽子が連続的に還元されるので、水の pH値は一定である。
【0046】
第一光活性物質および第二光活性物質の放射、あるいは照射なしの失活または電子正孔の組み換えを避ける目的で、隣接する区域においてできるだけ不可逆的に電荷を分離する作用を有し、必要に応じて電荷(電子)を中継できる電子受容体を設けることが望まれる場合があり得る。 このような電子受容体は、例えば、ナノ結晶性二酸化チタン、錫でドープされたIn2O3 、キノンやメチルビオロゲンなどの有機受容化合物、金または例えば分子性ワイヤを介して電子を中継可能なその他の錯体などから選択することができるが、それに限定するものではない。
【0047】
本発明において補助物質とは、場合によって伝導体または半導体を介して酸素から第一光活性物質への電子伝達を促進する物質(例えばRuO2、Pt、またはコバルトを水中においてホスフェートと反応させることによりin situで形成される化合物)または場合によって伝導体または半導体(例えばAu、PtまたはRu)を介して第二光活性物質からの電子伝達を促進する物質を意味する。 しかし、このような補助物質自体は光活性を示さないものであって、そのほかに光活性物質が存在しなければ、水は分解されない。
【0048】
本発明の触媒システムにおける電子伝導のためには、任意の公知の電子伝導性物質を使用できる。 このような電子伝導性物質としては、例えば金属、合金、半導体、導電性酸化物、導電性ポリマー、更にいわゆる分子性ワイヤ(様々な構造における炭素鎖、炭化水素鎖、または一般的には共有結合された直鎖もしくは分岐鎖であって、化合物の官能基として、又はそれとは別な形で存在しており、電子を伝導可能な物)、またはいわゆるナノワイヤ〔ナノメートルスケール(10-9メートル)の直径を有し、例えばNi、Pt、Au等のような金属性のもの、Si、InP、GaN等の半導体、SiO2やTiO2のようにマクロレベルでは絶縁体を形成するもの、有機(DNA等) 無機(e.g. Mo6S9-xlx等)を問わず繰り返し単位からなるナノワイヤ〕を挙げることができる。 また、物質の組み合わせにより、電子は分子から分子へ飛ぶ(hop)場合もあり得る。
【0049】
有機化合物や錯体の配位子において、それらの官能基の1個以上は、保護されていてもよいチオール基であってよく、また保護されていてもよいチオール基が結合する電子伝導性物質は、金を含んでいてもよい。
【0050】
例えば、第一光活性物質から第二光活性物質への電子伝導は、ナノ結晶性に酸化チタン/インジウムすず酸化物(ITO)/銅/金/分子性ワイヤの順番で起こり得る。 ただし、その他の順番も考えられる。 同様に、第二(還元を促進する)光活性物質由来の電子も、いくつか異なる経路を通じて陽子またはヒドロニウムイオンに伝達されうる。 電子を陽子へ伝導する際、補助触媒を使用することができ、このような場合には第二光活性物質と補助触媒の間に1種または1種以上の電子伝導性物質が配置されていてもよい。
【0051】
この伝導性物質としては、天然の光合成系の場合とは異なり、有機分子または錯体だけからなっていないものが好ましい。
【0052】
本発明において、「電気的接触」あるいは「直接電気的接触」とは、それぞれの物質間の電気的接触が、電子を伝導 中継する1種または1種以上の固体物質によるものであって、その他の物質、例えば流体や固体の中でイオンを伝導するイオン伝導性物質や液状酸化還元電解質などは使用しないことを意味する。
【0053】
第一(酸化促進)光活性物質および第二(還元促進)光活性物質のいずれかが、有機分子または有機配位子を有する錯体である場合、通常、両光活性物質間の伝導は、有機物質から無機物質への、またはその反対方向への電子伝達が行なわれるが、その中には配位子を介して錯体の中心原子から伝導物質への、または配位子を介して伝導物質から錯体の中心原子への伝達という特殊な場合がある。
【0054】
これは、電子の中心原子から配位子への伝達の場合には別に問題を起こさないので、配位子の置換基は分子性ワイヤになるようになるよう選択される。 しかし、配位子またはその置換基からの、例えば無機伝導体への伝達は直接起こるものではない。 このような場合、配位子またはその末端に区域する置換基に、電子の伝導が可能になるほど無機材と強い相互作用を発揮する官能基を導入することにより、よい結果が得られる。 その代表的な例として、チオール基類を金表面と結合させることを挙げることができるが、上述相互作用は、その他にも、例えばホスホン酸類、カルボン酸の無水物、シラン類との無機酸化物との相互作用など、数多く実現可能であり、その概要は、例えばElena Galoppini 著 "Linkers for anchoring sensitizers to semiconductor nanoparticles" [Coordination Chemistry Reviews 2004 248, 1283-1297]に記載されている。
【0055】
第二光活性物質を構成する錯体として、少なくとも2種の官能基を含有し、そのうち少なくとも一方は電子伝導性物質に結合され、他方は電子が供給されると陽子を水に還元することができる化学種を含有するその他の電子伝導性物質に結合されるものが挙げられる。
【0056】
いくつかの光活性物質、特に還元を促進する錯体は、光と水と酸素が同時に存在すると不安定になる。 本発明では、このような不安定性を避けるために、上述のような物質を水と酸素を除去した「カスケット」(棺)、例えば側面が遮断性の壁からなり、薄い透明な「金製カスケット」に入れておいて水と酸素に対して保護する方法を採用することを薦める。 この場合、還元促進性の錯体はチオール基など、少なくとも二つの官能基を含有し、そのうちの一方により上述のような「カスケット」の底面を介して酸化促進性の物質への電子伝導が確保され、その他方は例えば金/白金系合金など、前記「カスケット」のふたを形成する合金材(これは錯体の中心原子から放出され分子性ワイヤを通してこの合金に中継された励起電子を用いて行われる水素生成の触媒である)への伝導を可能にするものである。 勿論、その他のカスケット型、またはサンドウィッチ型の構造も採用可能である。
【0057】
第一(酸化促進性)光活性物質と第二(還元促進生)光活性物質は、物理蒸着、ケミカル ボンディングなどを採用して、1つ以上の基板に固定されていても、他の方法により前記基板と接続されてもよい。 第一(酸化促進性)光活性物質と第二(還元促進生)光活性物質は、物理蒸着、化学蒸着などを採用して、1つ以上の基板に固定されていても、他の方法により前記基板と接続されていてもよい。 基板は、電気的 光化学的に不活性であってもそうでないものであってもよく、また直接照射された光活性物質により吸収されなかった光が浸透できるように、例えばガラスのような透明 半透明なものでも、そうでないものでもよい。 基板に使用する材料としては、これに限定することなく、コーヒング処理を施していてもよいガラス、セラミックス、金属または金属合金、半金属、カーボンまたはそれに誘導された材料、および各種の無機および有機ポリマー材料を挙げることができる。
【0058】
このような基板を用いて、板状など平面状、または管状やその他に適切な形状を有する触媒システム、例えばその一方の側に酸化促進性の光活性物質を、その他方側に還元促進性の光活性物質を配置したもの、または適切な構造を採用した場合、両物質を同じ側に配置したもの等を、製造することができる。 基板として透明 半透明な材料が選択されたとき、場合によって光をその一方の側だけに照射することにより、光を他方側まで供給できる。
【0059】
光活性物質をそれぞれ対抗する側に配置した平面状、例えば板状の触媒システムを水性液体に浸漬するとき、一方の側では水素が発生され、他方側では酸素が発生する。 このような構成によれば、水素と酸素が空間的に離れて発生するので、酸素 水素間の反応のリスクが大幅に低減する。 両方の光活性物質を空間的に離れた区域に配置した構成、例えば陽子と水だけが浸透可能な材料(例えばナフィオン(登録商標)製の膜)を用いて反応器を二つの区画に区切り、または反応器システムを2区画システムにすることで、水素と酸素を完全に隔離することが可能である。 電荷の補償を確保する目的で、陽子が両区画間で往復できるようにする必要がある。
【0060】
板状などの平面状の触媒システムを浸漬する水性液体としては、通常水を使用し、場合によって塩類、酸類、塩基類を各種添加することができるが、必然的ではない。 また、水の光分解に悪影響を与えない限り、溶剤、界面活性剤など水溶性成分との混合物、また必要に応じて、光分解反応に参加しない水性乳化液なども使用可能な溶媒である。
【0061】
本発明の製法において、触媒を照射するために使用される光としては、太陽光が好ましい。
【0062】
また、照射される第一区域と第二区域は、陽子と水だけが浸透可能な膜、例えばナフィオン(登録商標)製の膜を用いて、互いに隔離して設けることが好ましい。
【0063】
本装置が十分に透明であるかあるいは部分的に透明である場合、触媒システムの第一区域だけに光を照射してもよい。 また、逆に触媒システムの第二区域だけに光を照射してもよい。 しかし、前記両区域に光を照射する場合が多い。
【0064】
本触媒システムと光を用いて水から発生する酸素および/または水素は、連続的に捕集することができる。
【0065】
本発明の触媒システムは多くの効果がある。 水素と酸素は、酸素/水素ガスを生成することなく、別々に生成可能である。 本発明の触媒システムは粉体状ではなく、モノリス型の触媒システム、例えば単に水性溶媒に浸漬される板状のものであり、多くの場合では、製法のコストや環境負担を増加させる、塩類・酸類・塩基類などの添加を必要とせず、(ただし、これを除外するものではない)、また圧力の印加が必要な特殊セルや、溶剤に対して密閉になるようにカプセル化処理の必要な酸化還元電解質を使用する必要もない。 本発明のシステムは水酸化触媒(第一光活性物質)と水(陽子)還元触媒(第二光活性物質)を幅広い範囲から選択できることから、非常にフレキシブルであるので、各種用途などに最適な組み合わせの設計することができる。
【0066】
[代表的な触媒システムの構成]
図2は、Z機構と同様に作用する光触媒システム1の構成を示す概略断面図であり、板型の不活性基板10aの一方の側は酸化インジウムスズ(ITO)からなる透明な伝導性皮膜30が、他方側には金からなる透明皮膜40がそれぞれ設けられている。 ITO皮膜30および金皮膜40とは銅バンド20により電気的に接続されている。
【0067】
ITO皮膜30の上部はRuS2でコーティングされたナノ結晶性TiO2 50が焼結されている。 金皮膜40にはメルカプトアルキル置換基60a(実施例III.2.b参照)を3つ有するルテニウム系錯体60とアルキルチオール70とからなる単分子層(monolayer)(極端に拡大して表示)が設けられている。 同単分子層のそれぞれの端部は、すべての辺上がレジスト80で覆われ、これにより同単分子層はレジストの幅の狭いバンドによって囲まれている。 ルテニウム錯体60およびアルキルチオール70の単分子層上には、わずか2〜3層の金の層からなる透明な金皮膜90が、真空蒸着によりレジスト80にわたって薄く設けられている。 また、金皮膜90の上には白金皮膜92が設けられており、同皮膜を形成する原子の数は前記の単分子層を形成する原子の数よりもさらに少ない。
【0068】
図2に示す触媒システムは、水に浸漬され、かつ、波長420nmの光で照射されると1/2 O2 + 2H+ に酸化された H2O の酸素原子から由来する電子が、二硫化ルテニウムでコートされているTiO2 50からITO皮膜30と銅バンド20を介して金皮膜40に移動する。 照射を受けることでルテニウム錯体60が一つの電子をH+に与えるため、一つの電子が金皮膜40からチオール基およびルテニウム錯体60のメルカプトアルキル置換基60aのアルキル鎖を介して、Ru中心原子に移動する。 照射によりルテニウム錯体60は励起状態となり、これによってアルキル鎖およびもう一つのメルカプトアルキル置換基60aを介して白金92でコートされた金皮膜90に、一つの電子が与えられると共にそこからは電子が白金92を介して水中の陽子(H+) に与えられることによって前記陽子は1/2 H2に還元される。
【実施例】
【0069】
以下、本発明は実施例を挙げて更に説明するが、本発明をそれらの例に限定するものではない。
【0070】
[実施例1]
A. TiO2/RuS2の5%懸濁液(TiO2 に対しRuS2を2重量%含有)としてのTiO2上での酸化促進性の第一光活性物質の調製
水15mlで希釈したTiO2の懸濁液(10%, Aldrich社製)5gに、ルテニウム(III)-クロライド23mg(0.11mmol)を添加し、超音波槽で処理して、乾燥するまで減圧下で濃縮した。
【0071】
TiO2粉末に析出したRuCl3を、まず不活性雰囲気下、水素ガス(H2)流中で、金属に還元した。 即ち試料を300℃に加熱し、3時間流量 50 ml/min の水素流中で処理し、温度を400℃にし、流量10 ml/min の硫化水素を添加することにより、Ruの黒色硫化ルテニウム (RuS2) への硫化を誘発し、これを4時間続けた [A. Ishiguro, T. Nakajima, T. Iwata, M. Fujita, T. Minato, F. Kiyotaki, Y.Kiyotaki, Y. Izumi, K.-i. Aika, M. Uchida, K.Uchida, K. Kimoto, Y. Matsui, Y. Wakatsuki, Chem. Eur. J. 2002, 8 (14), 3260-3268. / K. Hara, K. Sayama, H. Arakawa, Appl. Catal. A.: Gen. 1999, 189 (1), 127-137.] この結果得られる灰色の粉末から50mgを取り、水1mlと混合した後、長音波槽によく懸濁させることにより、薄い灰色を呈するTiO2上のRuS2(2重量%)の懸濁液が得られる。
【0072】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
一方の側の酸化インジウムスズ(ITO)で被覆された市販のガラススライド(納入元: PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)のITO側に、TiO2の10%水性懸濁液(粒子径40nmより小さいもの、Aldrich社製)を薄く塗布し、450℃で4時間の焼結処理を施した後で、TiO2に対してRuS2(上述のように調製したもの)2重量%の割合でTiO2/RuS2を含有する5%水性懸濁液を塗布した後、スライドを不活性ガス雰囲気下で再度450℃で60分焼結した。
【0073】
以下、この結果得られるスライドを、Ox-Iと呼ぶ。
【0074】
[実施例2]
A. 第一光活性物質としての光活性WO3ナノ粒子と、それに白金処理を施したものの調製
光活性のWO3 ナノ粒子を、下記の引用文献に記載の方法に従って調製した (J. Polleux, M. Antonietti, M. Niederberger, J. Mater. / T.Chem. 2006, 16 (40), 3969-3975. / M. Niederberger, J. H. Bartl, G. D. Stucky, J. / T.Chem. Soc. 2002, 124 (46), 13642-13643. J. Polleux, M. Pinna, M. Antonietti, M. Niederberger, J. / T.Chem. Soc. 2005, 127 (44), 15595-15601.)
【0075】
代表的な実験では、まず無水ベンジルアルコール(またはそれと4-tert.-ブチルベンジルアルコール)20mlに、六塩化ウォルフラム (WCl6)を430mg溶解した。 密閉反応器を100℃に加熱し48時間攪拌を続けた。 生成物を、沈澱とデカンテーションを交互に行うことによって捕集し、EtOH15ミリリットルを用いて3回洗浄した。 得られた物質を数時間60℃で風乾して、WO3の黄色の粉末を得た。
【0076】
白金添加の最適化を目的に、粉末50mgを50%のエタノールと50%の水からなる混合物に懸濁した。 Pt助触媒(WO3の量に対して2重量%)の添加は光堆積法によりH2PtCl6・6H2Oの中性にした水溶液から行なった (K. Yamaguti, S. Sato, J. / T.Chem. Soc. Faraday Trans 1 1985, 81 (5), 1237-1246. / T. Sakata, T. Kawai, K. Hashimoto, Chem. Phys. Lett. 1982, 88 (1), 50-54.)
【0077】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
白金添加触媒(灰色)または白金無添加の触媒(黄色)の粉末をそれぞれ20mgずつ超音波処理により無水イソプロパノール0.4mlおよび超純水0.2mlからなる混合物に再度懸濁した。 それぞれの懸濁液の分割量を、適切なITO被覆ガラスのスライドに塗布した。 触媒を塗布したスライドは15分風乾後2時間450℃で焼結した。
【0078】
以下、この結果得られる、純WO3で被覆されたスライドをOx-IIaと呼び、白金添加のWO3で被覆されたスライドをOx-IIbと呼ぶ。
【0079】
[実施例3]
A. 酸化促進性の第一光活性物質に使用するMn4O4オキソクバン錯体Mn4O4 (フェニル2PO2)の調製
[下記の引用文献記載の方法に準拠した: R. Brimblecombe, G. F. Swiegers, G. C. Dismukes, L. Spiccia, Angew. / T.Chem. Int. Ed. 2008, 47 (38), 7335-7338. / T. G.C. Carrell, S. Cohen, G. C. Dismukes, L. Mol. Cat. A 2002, 187 (1), 3-15.]
【0080】
【化1】
【0081】
NaOH60mg(1.5mmol)を、不活性ガス(N2)雰囲気下、DMF20mlに溶解してNaOHのDMF溶液を調製した。 これに、ジフェニルホスフィン酸330mg(1.5 mmol)および過塩素酸マンガン(II)255mg(0.7mmol)をDMF8mlに溶解したものを、激しく攪拌しながら添加した。 反応時間として15分経過後、KMnO4 50mg(0.3mmol)をDMF18mlに溶解したものを、添加漏斗を通して徐々に滴下した。 これにより、褐色のかかった赤色の懸濁液が形成され、これを室温で16時間攪拌した。 懸濁液を濾過して得られた残渣を、それぞれ40mlのエタノールとエーテルを用いて洗浄した後、乾燥した。 目的の錯体は、褐色がかった赤色を呈する粉末として得られる。
【0082】
【化2】
【0083】
UV/Vis (CH2Cl2): λmax (lg ε) = 229.0 (0.80), 263.0 (0.36), 257.0 (0.36), 269.5 (0.34).
紫外線/可視光のスペクトルは、図5を参照されたい。
【0084】
B. 前記酸化促進性光活性物質のITO基板への被覆処理
前記のMn4O4(フェニル2PO2)錯体を、下記の引用文献記載の方法に従ってITO被覆ガラスのスライドに塗布した(M. Yagi, K. Nagai, A. Kira, M./ T.Chem. Kaneko, J. Electroanal. / T.Chem. 1995, 394 (1-2), 169-175.)
【0085】
片側が酸化インジウムスズ(ITO)で被覆された市販のガラススライド(納入元: PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)のITO側に、Mn4O4(フェニル2PO2)錯体の 1mM 溶液(この錯体をナフィオン(登録商標)117と無水エタノールの1/1の混合物に溶解したもの)を塗布した後、12時間風乾した。
【0086】
以下、この結果得られるスライドを、Ox-IIIと呼ぶ。
【0087】
[実施例4]
A. 還元促進性の第二光活性物質に用いるトリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート)
【0088】
A.1 保護されたビピリジン配位子である4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
[D. K. Ellison, R. T. Iwamato, Tet. Lett. 1983, 24 (1), 31-32. / P. K. Gosh, T. G.C. Spiro, J. / T.Chem. Soc. 1980, 102 (17), 5543-5549に準拠して調製した。]
【0089】
【化3】
【0090】
a) 4-(11-ブロモウンデシル)-4’-メチル-2,2’-ビピリジン (A)
不活性ガス雰囲気下で、無水テトラヒドロフレン(THF)100mlを0℃まで冷却し、4,4’-ジメチル-2,2’-ビピリジン2.00g(10.9 mmol)と混合した。 0℃で15分攪拌を続けた後、リチウムジイソプロピルアミド(LDA)の2M溶液5.45ml(10.9mmol)を、前記のTHFに添加した後、冷却しながら2時間反応させた。 反応液を、30分かけて、0℃にて、3.30g(11.0mmol)ジブロモデカンを無水THFに溶解して得た溶液に滴下した。褐色がかった透明感のある溶液を、0℃で2時間攪拌してから更に室温で16時間攪拌を続けた後、H2Oを用いて冷却した。 この懸濁液を、減圧下で殆んど乾いた状態になるまで濃縮した。 水性残渣を、25mlの水に移して食塩水25mlを添加した後、混合物を、CHCl3 75mlを用いて3回抽出した。 各有機相を、Na2SO4で乾燥した後、乾いた状態になるまで減圧下で濃縮した。 生成物の精製は、エチルアセテートを用いて、シリカゲル上で行なった。 生成物Aは、ベージュ色を呈する結晶性粉末の形態で得られる。
【0091】
b) 4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
[生成法は、D. Imahori, A. Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, Tetrahedron 2006, 62 (9), 1955-1966参照.]
【0092】
前記Aと、1.20g(2.97mmol)無水エタノール(EtOH)とを、THF(50ml、1/1、V/V)に溶解して得られた溶液を、不活性ガス(N2)の雰囲気下、 チオアセテート2.04g(6 equ.、17.9mmol)と混合した後、2時間還流した。 反応が完了した後、赤褐色を呈する懸濁液を濾過し、赤桃色を呈する透明感のある濾液を、回転蒸発器を用いて減圧下で濃縮した。 それから残渣をクロロホルム50mlに移し、水40mlずつで2回、食塩水40mlで1回洗浄した後、Na2SO4上での乾燥後、減圧下で溶剤を除去した。 残渣の精製は、EtOAc/n-ヘキサン (1 : 1)を用いてシリカゲル上で行なった。 生成物Bは黄色を呈する固体として得られる。
【0093】
4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B)
【0094】
【化4】
【0095】
1H-NMR (300 MHz, CDCl3): δ = 8.53 (dd, 2H, J5/6 = J5’/6’= 4.2 Hz, J3/6 = J3’/6’= 0.6 Hz, 6-H, 6’-H), 8.21 (br s, 2H, 3-H, 3’-H), 7.14 (dd, 2H, J5/6 = J5’/6’= 4.8 Hz, J3/5 = J3’/5’= 1.5 Hz, 5-H, 5’-H), 2.84 (t, J = 7.5 Hz, 2H, 17-CH2, S-CH2), 2.67 (t, J = 7.8 Hz, 2H, 7-CH2, Aryl-CH2), 2.42 (s, 3H, 7’-CH3, Aryl-CH3), 2.30 (s, 3H, 20-CH3, CO-CH3), 1.72 - 1.62 (m, 2H, 8-CH2), 1.59 - 1.49 (m, 2H, 16-CH2), 1.38 - 1.24 (m, 14H, 9/10/11/12/13/ 14/15-CH2).
13C-NMR (75.5 MHz, CDCl3): δ = 196.0 (C-19), 156.1 (C-2), 156.0 (C-2’), 152.9 (C-4), 148.9 (C-6), 148.9 (C-6’), 148.1 (C-4’), 124.6 (C-3’), 123.9 (C-3), 122.0 (C-5’), 121.2 (C-5), 35.5 (C-7), 30.6 (C-17), 30.4 (C-8), 29.4 (C-11/12), 29.4 (C-10), 29.4 (C-13), 29.3 (C-15), 29.3 (C-9), 29.1 (C-16), 29.0 (C-20), 28.8 (C-14), 21.2 (C-7’).
EI-MS (70 eV): m/z (%) = 398 (4) [M+], 357 (18), 356 (68), 355 (90) [M+-Me, -CO], 324 (9), 323 (39), 309 (18), 295 (12), 281 (10), 267 (6), 253 (6), 239 (6), 211 (13), 209 (5), 198 (21), 197 (100), 185 (12), 184 (95), 183 (10), 170 (5), 43 (30), 41 (5).
紫外線/可視光 (CH2Cl2): lmax (lg e) = 229.0 (0.80), 263.0 (0.36), 257.0 (0.36), 269.5 (0.34).
【0096】
A.2: トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C) の調製
a)トリス[4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート]ルテニウム(II)ビス(ヘキサフルオロホスフェート) (保護されたC)
[R. A. Palmer, T. S. Piper, Inorg. / T.Chem. 1966, 5 (5), 864-878. および Y-R. Hong, C. B. Gorman, J. Org. / T.Chem. 2003, 68 (23), 9019-9025に準拠。]
【0097】
【化5】
【0098】
不活性ガス(窒素ガス)の雰囲気下で窒素ガスを流し込んだ無水エタノール30mlを、実施例4.A.bで調製したB180mg(452μmol/RuCl3に対して25%過剰)と混合した。 この反応液に、RuCl3・H2O 25mg(120.5μmol)を、無水エタノール5mlに溶解したものを添加し、暗所で65時間還流した。 反応完了後、赤褐色を呈する懸濁液を濾過し、透明の赤桃色の濾液を、減圧下で回転蒸発器にて濃縮した。 赤色を呈する残渣をCH2Cl2 20mlに移し、2回にわたって水を20mlずつ用いて洗浄した。 各有機相は、Na2SO4で乾燥した後、乾いた状態になるまで回転蒸発器を用いて減圧下で濃縮した。 この結果として得られる赤褐色を呈する生成物の混合物をCHCl3/methanol (15/1)を用いてシリカゲル上で精製することにより、目的の化合物が得られる。
【0099】
b) トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C) の調製
[H. Hotta, K.Imahori, A. Yoshida, T.Fujimoto, S. Kang, H. Hotta, K. Yoshida, T. Umeyama, Y. Matano, S. Isoda, Tetrahedron 2006, 62 (9), 1955-1966. F. Ono, S. Kanemasa, J. Tanaka, Tetrahedron Lett. 2005, 46 (44), 7623-7626. / T. Suzuki, A. Matsuura, A. Kouketsu, S. Hisakawa, H. Nakagawa, N. Miyata, Bioorg. Med.Med. / T.Chem. 2005, 13 (13), 4332-4342に準拠]
【0100】
100ml容量の三つ口丸底フラスコに無水エタノール10mlを入れ、30分窒素ガスを流し込む。 無水THF10mlと共にA.2.a号に記載の通り調製した保護されたルテニウム錯体 44mg((32.2μmol)を室温にてフラスコに移し、100 mg KOH (55 eq.)と混合する。. 反応液を室温で60分攪拌した後、食塩水15mlの上に注いでから、25mlのCH2Cl2で抽出する。 有機相は、Na2SO4の上で短時間乾燥した後、減圧下で濃縮する。 赤色の錯体は、NH4PF6 (0.6 mmol,、約20 eq.)を105mgをメタノール5mlに溶解したものと組み合わせてから、暗所で120分室温で攪拌する。 赤色を呈する溶液は2回水洗した後、Na2SO4の上で少々乾燥した後、回転蒸発器を用いて減圧下で溶剤を除去する。 残渣は少量のCH2Cl2に溶解して、それから石油エーテルを用いて粒状化させる。 目的のルテニウム錯体Cは赤桃色を呈する粉末として沈降する。
【0101】
トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート) (C)
【0102】
【化6】
【0103】
1H-NMR (300 MHz, CDCl3): δ = 8.17 (d, 2H*3, J = 7.8 Hz, 6-H*3, 6’-H*3), 7.58-7.45 (m, 2H*3, 3-H*3, 3’-H*3), 7.23 (dd, 2H*3 J = 5 Hz, J = 2 Hz, 5-H*3, 5’-H*3), 2.79 (t, 2H*3, J = 8 Hz, 7-CH2, Aryl-CH2), 2.53 (s, 3H*3, 7’-CH3, Aryl-Me), 2.50 (t, 2H*3, J = 7.2 Hz, 17-CH2, S-CH2), 1.72 - 1.62 (m, 2H*3, 8-CH2), 1.61- 1.54 (m, 2H*3, 16-CH2), 1.32 (t, 3*1H, J = 7.5 Hz, -SH) 1.38 - 1.23 (m, 14H*3, 9/10/11/12/13/14/15-CH2).
13C-NMR (75.5 MHz, CDCl3): δ = 156.4 (C-2), 156.3 (C-2’), 154.5 (C-4), 150.8 (C-6), 150.6 (C-6’), 150.0 (C-4’), 129.0 (C-3’), 128.0 (C-3), 124.6 (C-5’), 123.8 (C-5), 35.4 (C-7), 34.0 (C-16), 30.0 (C-8), 29.5 (C-10), 29.4 (C-11/12), 29.4 (C-13), 29.3 (C-15), 29.0 (C-9), 28.3 (C-14), 24.6 (C-17), 21.3 (C-7’).
ESI-MS: m/z = 1315.6 ([M-(PF6)]+), 585.3 ([M-(2*PF6)]2+).
UV/Vis (MeOH): λmax (lg ε) = 286.5 (1.27), 248.0 (0.36), 258.0 (0.35), 457.5 (0.26), 436 (sh, 0.22), 324.5 (0.20).
紫外線/可視光のスペクトルは、図3を参照されたい。
【0104】
B. 前記還元促進性光活性物質Cの不活性基板への被覆処理
一方の側に膜厚50nmの金皮膜と、膜厚5〜10nmのクロムとのプレコートを有し、寸法が第一光活性物質を塗布するときに使用されたものと同様であるガラススライドを、トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (C)をエタノールに溶解して得られた溶液で処理する。 本発明の一つの実施形態によれば、溶解した錯体を塗布するとき、Sと同様に共吸着物である、鎖長がC8、C10、C12、C14、C16またはC18のアルキルチオールの存在下で行なう( J. Summer, S. E. Creager, J. / T.Chem. Soc. 2000, 122 (48), 11914-11920に準拠。)錯体の溶解度を向上する目的で、必要に応じてジクロロメタンを少量その溶液に添加する。 本発明の他の実施形態によれば、Y. S. Obeng, A. J. Bard, Langmuir 1991, 7 (1), 195-201に記載のように、共吸着物を添加しないで、塗布処理を施す。塗布した後、スライドを乾燥する。
【0105】
以下、この結果得られるスライドを、Red-Iと呼ぶ。
【0106】
[実施例5]
A. 還元を促進する第二光活性物質としての[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート)の調製
(以下、配位子を略して表示するが、bpyは2,2'―ビピリジン、Me-bpyは4'―メチル―2,2'―ビピリジン、またtppzはテトラピリドフェナジンを指す。)
【0107】
以下の調製方法は、P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway, A. H. White, Inorg. / T.Chem. 1995, 34 (24), 6145-6157に準拠する。
【0108】
A.1: cis-ジカルボニル-(2.2'ービピリジン([4-(11-アセチルスルファニルウンデシル)-4'-メチル-2,2'-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E)の調製
【0109】
【化7】
【0110】
窒素ガスによる不活性雰囲気下で、実施例4の4.1.b号で合成した4-(4’-メチル-2,2’-ビピリジン)-ウンデシルチオ-S-アセテート (B) 195mg(0.49mmol)を、無水エタノール30mlに溶解する。 この溶液に、cis,cis-[Ru(bpy)(CO)2(CF3SO3)2] (D) (0.25 mmol; P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway, A. H. White, Inorg. / T.Chem. 1995, 34 (24), 6145-6157に準拠して調製したもの) 150mgを添加した後、還流しながら2時間過熱する。透明感のある赤色を呈する溶液を、乾いた状態になるまで減圧下で濃縮する。残渣を、水25mlに溶解してから濾過する。濾液は、室温まで冷却し、冷たい状態で飽和するNH4PF6 の水溶液5mlを添加すると、即座に無色の沈殿物が発生する。この沈殿物を濾過分離してから冷水30mlで洗浄する。目的の生成物(E)は少量のエタノール・アセトンで再結晶されるピンク色を呈する個体として得られる。
【0111】
cis-ジカルボニル(2,2’-ビpリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E)
【0112】
【化8】
【0113】
化合物Eには、二つの幾何学的異性体A/B が存在する[T. J. Rutherford, F. R. Keene, Inorg. / T.Chem. 1997, 36 (13), 2872-2878]。
【0114】
1H-NMR (300 MHz, (CD3)2CO): δ = 9.50 (m, H, Aryl-H), 9.31 (dd, H, J = 6.0 Hz, J = 8.0 Hz, Aryl-H), 8.94 (d, H, J = 8.4 Hz, Aryl-H), 8.85 (m, H, Aryl-H), 8.82 (d, H, J = 8.4 Hz, Aryl-H), 8.73 (m, H, Aryl-H), 8.63 (m, H, Aryl-H), 8.39 (m, H, Aryl-H), 8.11 (m, H, Aryl-H), 7.96 (m, H, Aryl-H), 7.82 (m, H, Aryl-H), 7.69 (m, H, Aryl-H), 7.65 (t, H, J = 6.0 Hz, Aryl-H), 7.52 (m, H, Aryl-H), 3.04 / 2.82 (t, 2H, J = 8 Hz, 7-CH2 A/B), 2.85 / 2.83 (t, 2H, J = 7.2 Hz, 17-CH2 A/B), 2.75 / 2.53 (s, 3H, Aryl-CH3 A/B), 2.29 / 2.28 (s, 3H, CO-CH3 A/B), 1.86 / 1.65 (m, 2H, 8-CH2 A/B), 1.57-1.45 (m, (m, 2H, 16-CH2 A/B), 1.31 - 1.25 (m, 14H, 9/10/11/12/13/ 14/15-CH2 A/B).
EI-MS (70 eV): m/z (%) = 713 (8) [M-H+-2*PF6], 616 (12), 506 (9), 449 (14), 353 (22), 322 (35), 197 (100), 184 (88), 156 (28), 65 (17).
【0115】
A.2 (2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート)の調製 (G)
【0116】
【化9】
【0117】
前記A.1号に記載のように合成したcis-ジカルボニル (2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-ビス(ヘキサフルオロホスフェート) (E) 145 mg (145μmol)を無水2-メトキシエタノール40mlに溶解し、これに、テトラピリドフェナジン (F) (tppz; 0.43 mmol, 3eq.; W. Paw, R. Eisenberg, Inorg. / T.Chem. 1997, 36 (11), 2287-2293. J. Bolger, A. Gourdon, E. Ishow, J.-P. Launay, J. / T.Chem. Soc., Chem. Commun. 1995, 1799-1800に準拠して合成したもの)165 mgを無水2-メトキシエタノール10mlに溶解したものを添加する。黄色を呈する懸濁液を、窒素ガスで連続的にゆっくり置換する。100 °Cに加熱し、約10分窒素ガスで置換してからトリメチルアミン-N-オキサイド(ZMMO)56mg(0.5 mmol、約3.5 eq.) を 無水2-メトキシエタノール5mlに溶解したものを添加する。得られる懸濁液は連続的に窒素ガスを流し込みながら暗所で24時間還流させる。反応終了後、赤褐色を呈する懸濁液を容量が半分に減るまで濃縮し、30分暗所で放置する。沈降した未反応のtppzを濾過分離する。ろ過器に残った残渣を、エタノール15mlで洗浄し、濃い赤色を呈する濾液を、減圧下で濃縮する。これにより、ほとんど黒い濃い赤色を呈するオイル状の物質が得られる。CHCl3/メタノール(5:1)を用いるシリカゲル上のクロマトグラフィーによる分離・精製後、生成物(G)を、赤桃色のエネメル質状の固体として得る。
【0118】
(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート) (G)
【0119】
【化10】
【0120】
ESI-MS (CH3CN): m/z = 566.2 ([M-(2*PF6)+HCOOH]2+).
UV/Vis (CH3CN): λmax (lg ε) = 282.0 (1.02), 244.0 (0.60), 379.5 (0.19), 359.0 (0.16), 453.5 (0.16), 434.0 (sh, 0.15).
【0121】
A.3 [(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]
ビス (ヘキサフルオロホスフェート) (H)の調製
以下の反応は、S. Rau, B. Schafer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Gorls, W. Henry, J. G.C. Vos, Angew. / T.Chem. Int. Ed. 2006, 45, 6215-6218の方法に準拠する。
【0122】
【化11】
【0123】
不活性ガス(窒素ガス)の雰囲気下、6 mg of ビス(アセトニトリル)-パラジウム(II)-ジクロライド6mg (23μmol、15 %過剰、Aldrich社)を無水ジクロロメタン(DCM)20mlと混合した後、(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-(テトラピリドフェナジン)-ビス(ヘキサフルオロホスフェート)26mgを無水DCM 2mlに懸濁したものを添加する。反応混合物を加熱し、窒素ガス雰囲気下で6時間還流する。反応終了後、懸濁液を室温まで冷却した後、濾過することにより得られる赤褐色を呈する残渣を、少量のDCMで洗浄する。乾燥することにより、目的の生成物を赤褐色を呈する個体として得る。
【0124】
[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]-ビス(ヘキサフルオロホスフェート) (H)
【0125】
【化12】
【0126】
ESI-MS (CH3CN): m/z = 673.2 ([M-(2*PF6)]2++HCOOH+H2O]).
FAB-MS (-): m/z = 1526.2 (MH -+H2O).
UV/Vis (CH3CN): λmax (lg ε) = 241.0 (1.34), 283.5 (0.56), 354.5 (sh, 0.10), 432.0 (0.08), 453.5 (0.08).
紫外線/可視光スペクトルは、図4参照
【0127】
B. 不活性な基板への還元促進性の第二光活性物質Hの適用
錯体Hの被覆処理は S. J. Summer, S. E. Creager, J. / T.Chem. Soc. 2000, 122 (48), 11914-11920 / S. E. Craeger, G. K. Rowe, J. Electroanal. / T.Chem. 1994, 370 (1-2), 203-211 (coating in CH3CN) / J. M. Tour, L. Jones II, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides, D. L. Allara, A. N. Parikh, S. V. Atre, J. / T.Chem. Soc. 1995, 117 (37), 9529-9534. / L. Cai, Y. Yao, J. Yang, D. W. Price jr., J. M. Tour, Chem. Mater. 2002, 14 (7), 2905-2909 (アセチルスルファニル基でのコーティング)に準拠して行なう。
【0128】
第一光活性物質の皮膜処理のときと同様の寸法を有し、片側に膜厚50nmの金皮膜および膜厚5〜10nmのクロム・プレコートを設けたガラススライドに、錯体HのCH3CN溶液を塗布する。塗布時には、炭素数12または16のアルキルチオールなどの共吸着物が存在していてもよいが、存在していなくてもよい。塗布の後のスライドを、洗浄してから乾燥する。
【0129】
この結果得られるスライドを、以下、Red-IIと呼ぶ。
【0130】
[実施例6]
A. 還元促進性の第二光活性物質として2.5%アンチモンと2.0%クロムでドープした白金添加SrTiO3 の調製
(共ドープしたSrTiO3 の調製はH. Kato, A. Kudo, J. Phys. / T.Chem. B 2002, 106 (19), 5029-5034に準拠して行なった。)
【0131】
化学式SrTi1-X-YSbXCrYO3 に従って出発原料であるSrCO3、TiO2、Sb2O3、Cr2O3 のそれぞれの適量を充分に混合した。混合物を、アルミナ製ルツボに移し、大気雰囲気中で1050℃で少なくとも30時間焼成した。ボールミル(GERMATECH GmbH, Osterfeldstr. 3, D-56235 Ransbach-Baumbach, ドイツ)を用いて微細粉末を得るまで粉砕した。
【0132】
白金添加のためにエタノール20%および水80%からなる混合液に懸濁した。Pt助触媒(WO3の量に対して2重量%)を、光堆積法によって、H2PtCl6・6H2Oの中性に調製した水溶液から堆積させた (K. Yamaguti, S. Sato, J. / T.Chem. Soc. Faraday Trans 1 1985, 81 (5), 1237-1246. / T. Sakata, T. Kawai, K. Hashimoto, Chem. Phys. Lett. 1982, 88 (1), 50-54参照。)
【0133】
B. 前記の還元促進性光活性物質でのITO 基板の被覆処理
2.5%アンチモンおよび2.0%クロムでドープした白金添加SrTiO3 の乾燥粉末100mgを、無水イソプロパノール2mlおよび超純水1mlからなる混合液に、超音波処理によって再懸濁させた。懸濁液の少量の分割量を、片側が酸化インジウムスズ(ITO)でコーティングされた、市販のガラススライド (PGO Prazisions Glas & Optik GmbH, Im Langen Busch 14, D-58640 Iserlohn, [ドイツ]から入手)に塗布した。15分風乾燥後、スライドを450℃で2時間焼結した。
【0134】
この結果得られるスライドを、以下、Red-IIIと呼ぶ。
【0135】
[実施例7]
第一および第二の光活性物質を含有する触媒ユニットの触媒システムへの組立およびその照射
7.1.a Ox-I とRed-Iの組み合わせ
酸化促進性第一光活性物質RuS4と、還元促進性第二光活性物質トリス[4-(11-メルカプトウンデシル)-4’-メチル-2,2’-ビピリジン]ルテニウム(II)-ビス-(ヘキサフルオロホスフェート)とをそれぞれ含む、実施例1および4で作製した触媒ユニット(スライドOx-IおよびスライドRed-I)の両方を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。その後、Red-Iの金の側面をレジストでコーティングして、銅バンドと、金皮膜の端部と、金皮膜の上で前記錯体を含有するスライドのそれぞれの端とに沿って、幅の狭い帯状の区域が被覆されるようにした。このように組み立てられて電動可能に接続した触媒ユニットを乾燥した後、錯体でコーティングされた金側に再度非常に薄い金皮膜(5 nm)を蒸着し、隣接するレジスト層が被覆されるようにした。最終的には、この金皮膜を更に白金の0.5〜0.7単分子層(ML)を設けて、触媒システムを得た。
【0136】
7.1.b (Ox-I)-(Red-I)による触媒システムの照射
7.1.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含むこの膜を、光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を満たし、上部をオイル層でカバーした。Ox-Iに隣接する区画に、黄色のロイコインジゴカルミンを導入した。その後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。約50℃に加熱した後で、黄色のロイコインジゴカルミンが変色して青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が発生した。
D2OおよびH218O、またフィルタとして亜硝酸塩を使用した実験で、マススペクトロメトリーで分析した結果、D2 、また自然量より大きい量の16O18Oが発生したことが確認されたことからも、水の分解が実証された。
【0137】
7.2.a Ox-IとRed-IIの組み合わせ
酸化促進性第一光活性物質RuS4 と還元促進性第二光活性物質[(2,2’-ビピリジン)[4-(11-アセチルスルファニルウンデシル)-4’-メチル-2,2’-ビピリジン]-ルテニウム(II)-[(テトラピリドフェナジン)-パラジウム(II)-ジクロロ]ビス-(ヘキサフルオロホスフェート)とをそれぞれ含む、実施例1および5で作製した触媒ユニット(スライドOx-IおよびスライドRed-II)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0138】
7.2.b (Ox-I)-(Red-II)による触媒システムの照射
7.2.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-Iに隣接する区画に、黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。室温にて、Red-IIに隣接する区画のpHを、硫酸の添加により約5の酸性側に調製した後で、黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が発生した。
【0139】
7.3.a Ox-IとRed-IIIの組み合わせ
酸化促進性第一光活性物質RuS4 と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3をそれぞれ含む実施例1および6で作製した触媒ユニット(スライドOx-IおよびスライドRed-III)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0140】
7.3.b (Ox-I)-(Red-III)による触媒システムの照射
7.3.a号で説明したように作製した触媒システムを、ナフィオン(登録商標)膜に接着し、触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-Iに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0141】
7.4.a Ox-IIとRed-IIIの組み合わせ
酸化促進性第一光活性物質WO3 (白金添加のものと白金無添加のもの) と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3とをそれぞれ含む、実施例2および6で作製した触媒ユニット(スライドOx-IIおよびスライドRed-III)を、それぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0142】
7.4.b (Ox-II)-(Red-III)による触媒システムの照射
7.4.a号で説明したように作製した触媒システムをナフィオン(登録商標)膜に接着し、また触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-IIに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0143】
7.5.a Ox-IIIとRed-IIIの組み合わせ
酸化促進性第一光活性物質Mn4O4(フェニル2PO2)と還元促進性第二光活性物質2.5%アンチモンと2.0%クロムで共ドープした白金添加のSrTiO3をそれぞれ含む実施例3と6で作製した触媒ユニット(スライドOx-IIとスライドRed-III)をそれぞれのコーティングを施していない面で接着し、電導性銅接着テープ(PGO Prazisions Glas & Optik GmbH社製, Im Langen Busch 14, D-58640 Iserlohn, ドイツ)を用いてコーティングを施した面を電動可能に接続した。
【0144】
7.5.b (Ox-III)-(Red-III)による触媒システムの照射
7.5.a号で説明したように作製した触媒システムをナフィオン(登録商標)膜に接着し、また触媒システムの両側が露出するように同膜に窓を切り抜いた。触媒システムを含む膜を光反応器に挿入し、二つの区画が形成されるようにした。両区画に、窒素ガスが飽和状態にある、酸素を除去した水を入れ、上部をオイル層でカバーした。Ox-IIIに隣接する区画に黄色のロイコインジゴカルミンを導入した後、420nm遮断フィルタを通した500ワットのハロゲンランプを用いて両側から照射した。黄色のロイコインジゴカルミンが変色し青色のインジゴカルミン(酸素用の敏感な指示薬)に変化したことから明らかなように、水素と酸素が室温で発生した。
【0145】
本願に引用されている文献、例えばジャーナルの記事・論文、本、特許、特願などに開示されている内容はすべて参照により参照により、本明細書中に組み込まれているものとする。
【特許請求の範囲】
【請求項1】
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水から酸素と陽子を生成させることができる第一光活性物質と、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水中の陽子を水素に還元することができる第二光活性物質と
を含有し、
第一光活性物質および第二光活性物質は、一種またはそれ以上の電子伝導性物質を介して電気的、特に直接電気的な接触状態にあることを特徴とする、光を用いて水を水素と酸素に分解するためのモノリス触媒システム。
【請求項2】
前記波長が、430nm以上、好ましくは440nm以上、さらに好ましくは450nm以上であることを特徴とする、請求項1に記載のモノリス触媒システム。
【請求項3】
前記酸素を発生する物質と前記水素を発生する物質とのいずれかまたはその両方と組み合わせて、1種以上の補助物質および/または補助触媒を含有することを特徴とする、請求項1または2に記載のモノリス触媒システム。
【請求項4】
前記電子伝導性物質または前記電子伝導性物質の少なくとも1種が、金属、金属合金、または酸化物系の電子伝導性物質を含むことを特徴とする、請求項1〜3のいずれか一項に記載のモノリス触媒システム。
【請求項5】
基板を含有することを特徴とする、請求項1〜4のいずれか一項に記載のモノリス触媒システム。
【請求項6】
第一光活性物質が、ドープされてもよい酸化物および/または硫化物含有物質、特にRuS2、貴金属または遷移金属を含有する錯体またはクラスター、および光活性のポリマー物質から選択されることを特徴とする、請求項1〜5のいずれか一項に記載のモノリス触媒システム。
【請求項7】
第二光活性物質が、金属含有錯体、例えば貴金属を含有する錯体、例えばRu錯体など、拡張π電子系を有する有機化合物、酸化物を含有する物質、酸化窒化物を含有する物質、リン化物を含有する物質、ヒ素化物を含有する物質、アンチモン化物を含有する物質、硫化物を含有する物質、セレン化物を含有する物質、および光半導体型ポリマーからなる群から選択された1種以上であることを特徴とする、請求項1〜6のいずれか一項に記載のモノリス触媒システム。
【請求項8】
第一光活性物質および第二光活性物質のいずれかまたはその両方が、官能基により1種以上の電子伝導性物質に結合されていることを特徴とする、請求項1〜7のいずれか一項に記載のモノリス触媒システム。
【請求項9】
本システムの構造が平面状の多層構造であり、前記構造の一方の側が第一光活性物質含有し且つ他方側が第二光活性物質を含有するか、あるいは前記構造の片側が第一光活性物質および第二光活性物質を含有することを特徴とする、請求項1〜8のいずれか一項に記載のモノリス触媒システム。
【請求項10】
光および触媒システムを用いて水から酸素および水素を製造する方法であって、
第一光活性物質を含有するか、あるいは、第一光活性物質とこれと組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第一区域において、本発明による触媒システムを水または水性液体または水性溶液に接触させ、
第二光活性物質を含有するか、あるいは、第一光活性物質とこれに組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第二区域において、前記触媒システムを水または水性液体または水性溶液に接触させ、
前記触媒システムに光を照射し、かつ、
陽子が第一区域から第二区域に移動できるように、第一区域に接触している前記水または水性液体または水性溶液と、第二区域に接触している前記水または水性液体または水性溶液とを相互に接触した状態にする
ことを特徴とする、方法。
【請求項11】
光が太陽光であることを特徴とする、請求項10に記載の方法。
【請求項12】
請求項9に記載のモノリス触媒システムを使用し、第一区域と第二区域を、陽子と水だけが浸透可能な膜により隔離することを特徴とする、請求項10または11に記載の方法。
【請求項13】
第一区域を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項14】
第二区域を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項15】
第一区域および第二区域の両方を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項16】
酸素および/または水素を、間欠的にまたは連続的に捕集することを特徴とする、請求項10〜15のいずれか一項に記載の方法。
【請求項1】
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水から酸素と陽子を生成させることができる第一光活性物質と、
単独で、あるいは補助物質の少なくとも一種および/または補助触媒の少なくとも一種との組み合わせで、波長420nm以上の光で照射されると水中の陽子を水素に還元することができる第二光活性物質と
を含有し、
第一光活性物質および第二光活性物質は、一種またはそれ以上の電子伝導性物質を介して電気的、特に直接電気的な接触状態にあることを特徴とする、光を用いて水を水素と酸素に分解するためのモノリス触媒システム。
【請求項2】
前記波長が、430nm以上、好ましくは440nm以上、さらに好ましくは450nm以上であることを特徴とする、請求項1に記載のモノリス触媒システム。
【請求項3】
前記酸素を発生する物質と前記水素を発生する物質とのいずれかまたはその両方と組み合わせて、1種以上の補助物質および/または補助触媒を含有することを特徴とする、請求項1または2に記載のモノリス触媒システム。
【請求項4】
前記電子伝導性物質または前記電子伝導性物質の少なくとも1種が、金属、金属合金、または酸化物系の電子伝導性物質を含むことを特徴とする、請求項1〜3のいずれか一項に記載のモノリス触媒システム。
【請求項5】
基板を含有することを特徴とする、請求項1〜4のいずれか一項に記載のモノリス触媒システム。
【請求項6】
第一光活性物質が、ドープされてもよい酸化物および/または硫化物含有物質、特にRuS2、貴金属または遷移金属を含有する錯体またはクラスター、および光活性のポリマー物質から選択されることを特徴とする、請求項1〜5のいずれか一項に記載のモノリス触媒システム。
【請求項7】
第二光活性物質が、金属含有錯体、例えば貴金属を含有する錯体、例えばRu錯体など、拡張π電子系を有する有機化合物、酸化物を含有する物質、酸化窒化物を含有する物質、リン化物を含有する物質、ヒ素化物を含有する物質、アンチモン化物を含有する物質、硫化物を含有する物質、セレン化物を含有する物質、および光半導体型ポリマーからなる群から選択された1種以上であることを特徴とする、請求項1〜6のいずれか一項に記載のモノリス触媒システム。
【請求項8】
第一光活性物質および第二光活性物質のいずれかまたはその両方が、官能基により1種以上の電子伝導性物質に結合されていることを特徴とする、請求項1〜7のいずれか一項に記載のモノリス触媒システム。
【請求項9】
本システムの構造が平面状の多層構造であり、前記構造の一方の側が第一光活性物質含有し且つ他方側が第二光活性物質を含有するか、あるいは前記構造の片側が第一光活性物質および第二光活性物質を含有することを特徴とする、請求項1〜8のいずれか一項に記載のモノリス触媒システム。
【請求項10】
光および触媒システムを用いて水から酸素および水素を製造する方法であって、
第一光活性物質を含有するか、あるいは、第一光活性物質とこれと組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第一区域において、本発明による触媒システムを水または水性液体または水性溶液に接触させ、
第二光活性物質を含有するか、あるいは、第一光活性物質とこれに組み合わせてさらに少なくとも一種の補助物質および/または補助触媒を含有する第二区域において、前記触媒システムを水または水性液体または水性溶液に接触させ、
前記触媒システムに光を照射し、かつ、
陽子が第一区域から第二区域に移動できるように、第一区域に接触している前記水または水性液体または水性溶液と、第二区域に接触している前記水または水性液体または水性溶液とを相互に接触した状態にする
ことを特徴とする、方法。
【請求項11】
光が太陽光であることを特徴とする、請求項10に記載の方法。
【請求項12】
請求項9に記載のモノリス触媒システムを使用し、第一区域と第二区域を、陽子と水だけが浸透可能な膜により隔離することを特徴とする、請求項10または11に記載の方法。
【請求項13】
第一区域を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項14】
第二区域を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項15】
第一区域および第二区域の両方を、直接光で照射することを特徴とする、請求項10〜12のいずれか一項に記載の方法。
【請求項16】
酸素および/または水素を、間欠的にまたは連続的に捕集することを特徴とする、請求項10〜15のいずれか一項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2011−500966(P2011−500966A)
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願番号】特願2010−530362(P2010−530362)
【出願日】平成20年10月31日(2008.10.31)
【国際出願番号】PCT/EP2008/009229
【国際公開番号】WO2009/056348
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(510113313)ツェーエフエスオー・ゲーエムベーハー (1)
【Fターム(参考)】
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願日】平成20年10月31日(2008.10.31)
【国際出願番号】PCT/EP2008/009229
【国際公開番号】WO2009/056348
【国際公開日】平成21年5月7日(2009.5.7)
【出願人】(510113313)ツェーエフエスオー・ゲーエムベーハー (1)
【Fターム(参考)】
[ Back to top ]