
治療化合物
本発明は、タンパク質結合相互作用/結合化合物及びそれらを同定し、使用する方法に関する。本発明はさらに、GPCR及び/又はRSKが関与する疾患及び障害をはじめとする5−HT2C及び/又はRSK障害を治療するための医薬組成物及び方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
[優先権の主張]
本出願は、2009年5月5日に出願された米国仮出願第61/215,504号、2009年9月10日に出願された米国仮出願第61/241,384号、及び2009年9月24日に出願された米国仮出願第61/277,408号の恩恵を主張するPCT国際出願である。前記米国仮出願のそれぞれの全内容は、本明細書中に組み込まれる。
[連邦政府により支援された研究のもとでなされた発明に対する権利の記載]
【0002】
本研究は、一部、アメリカ合衆国公衆衛生局認可MH068655により支援された。本研究は、一部、国立衛生研究所認可DA023928とMH081193とにより支援された。政府は本発明にある権利を有する。
【背景技術】
【0003】
セロトニン(5−ヒドロキシトリプタミン、5HT)は、5HT1〜5HT7ファミリーに分類される14の哺乳動物の5HT受容体サブタイプにより、様々な中枢及び末梢の心理学的及び生理学的効果に関与する(Sanders−Bush and Mayer, 2006)。5HT2ファミリーは、主にGαqを介してシグナルを送り、ホスホリパーゼ(PL)C並びにイノシトールリン酸(IP)及びジアシルグリセロール(DAG)第2メッセンジャーの形成を活性化する、5HT2A、5HT2B、及び5HT2c膜結合型Gタンパク質共役受容体(GPCR)から構成される(Raymond et al., 2001)。ヒト5HT2c受容体(Saltzman et al., 1991)は、脳のみで見いだされるようであり、脳で、広く発現され、おそらくは、摂食行動(Tecott et al., 1995)、コカイン中毒(Fletcher et al.,2002;Rocha et al., 2002;Muller and Huston, 2006)、睡眠ホメオスタシス(Frank et al., 2002)、不安(Kennett et al., 1994;Sard et al., 2005;Heisler et al., 2007)、鬱病(Tohda et al., 1989;Palvimaki et al., 1996)、てんかん(Heisler et al., 1998)、アルツハイマー病(Arjona et al.,2002;Stein et al., 2004)、運動機能(Heisler and Tecott, 2000;Segman et al., 2000)、精神病(Marquis et al., 2007;Siuciak et al., 2007)及び抗精神病薬に対する反応(Veenstra−VanderWeele et al., 2000;Reynolds et al., 2005)をはじめとするいくつかの(疾患)−生理学的及び心理学的プロセスに関与する。したがって、5HT2C受容体の薬物療法学的標的としての重要性は約10年間にわたり明らかであるが、5HT2c特異的薬剤は開発されていない。
【0004】
5HT2c受容体を標的とする創薬に関する1つの課題は、このGPCRが、5HT2A受容体と約80%の膜通過ドメイン(TMD)配列同一性を共有し、そして5HT2B受容体と約70%の膜通過ドメイン(TMD)配列同一性を共有することである(Julius et al., 1988;1990)。高度に保存されたTMD及び類似の第2メッセンジャーカップリングのために、5HT2C受容体に対して選択的なアゴニストリガンドの開発が特に困難になっている。それにもかかわらず、5HT2c受容体の活性化が食物摂取を減らして、抗肥満効果につながるという動かぬ証拠がある。たとえば、5−HT2cノックアウトマウスは摂食の増大や肥満を示し、d−フェンフルラミンの食欲抑制作用に対して耐性を示す(Tecott et al., 1995;Vickers et al., 1999;2001;Heisler et al., 2002)。フェンフェルラミンは現在、禁止されている。なぜなら、この薬剤を使用している人々は脳5HT2c受容体の活性化のために体重が減少するが、フェンフェルラミンはまた、5HT2A受容体を活性化し、このことは有害な精神医学的(幻覚誘発性)効果をもたらし得(Nichols, 2004)、また5HT2B受容体を活性化し、これにより心臓弁膜症(Connolly et al., 1997;Fitzgerald et al., 2000;Rothman et al., 2000;Roth, 2007)及び肺高血圧症(Pouwels et al., 1990;Launay et al., 2002)が引き起こされ、5HT2Bが関与する効果の結果、死に至ったためである。
【0005】
5HT2c対5HT2A及び/又は5HT2B受容体に関して真に選択的なアゴニストリガンドは本論文までは報告されていないが、ラットコカイン自己投与パラダイム(self−administration paradigm)において非常に(すなわち少なくとも100倍)選択的な5HT2A及び5HT2c拮抗物質を使用して脳5HT2c受容体がコカイン使用及び依存性を軽減する役割を部分的に説明することが可能である。たとえば、選択的な5HT2A拮抗物質M100907(Kehne et al., 1996)はコカイン自己投与に対する反応速度を変えないが、選択的な5HT2c拮抗物質SB242084(Bromidge et al., 1997)は、コカイン自己投与速度を用量に依存して増大させる(Fletcher et al., 2002)。覚醒剤中毒に対する5HT2cアゴニスト薬物療法の多大な可能性は現在、広く認識されている(Bubar and Cunningham, 2006)。
【0006】
肥満及び覚醒剤中毒等の神経精神障害における5HT2c受容体の薬物療法学的関連性により、製薬会社は強い関心を刺激されて、選択的な5HT2Cアゴニストを開発してきたが、報告されている5HT2cアゴニストはすべて、これまでのところ5HT2A及び/又は5HT2B受容体も刺激する(Nilsson, 2006)。それにもかかわらず、5HT2アゴニストロルカセリン(APD356)は、5HT2c受容体の活性化について5HT2A受容体よりも15倍程度の選択性しかないが、最近、肥満治療の第3相臨床試験まで進んだ(Jensen, 2006;Smith et al., 2006)。しかし、本明細書中で報告する結果は、本発明者等の実験室で合成した新規化合物、すなわち、フェニル−3−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレン(PAT)及びシクリル−3−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレン(CAT)化合物に基づき、これらはヒト5HT2c受容体での十分に有効なアゴニストであり、その上、5HT2A及び5HT2B受容体での拮抗物質である。
【0007】
Gタンパク質共役受容体(GPCR)は、2種以上のGタンパク質を活性化することができ、その結果、複数の生理学的/薬理学的効果(薬物療法学的かつ不適切な副作用)が起こる(Moniri et al., Journal of Pharmacology and Experimental Therapeutics, 311:274−281(2004))。単一のGPCRと関連する複数のシグナル伝達経路の現象は、GPCR活性化の3状態モデルのフレームワーク内で記載することができ、この場合、GPCRは、不活性状態と構成的に活性な状態との間で異性化する。GPCR活性化は、ヘテロ三量体(α,β,γ)Gタンパク質サブユニットの解離を引き起こし、−Gαサブユニットは次いでトランスデューサータンパク質(例えば、PLC、AC)を活性化して、第2メッセンジャー濃度を変えることができる。同じGPCRが異なるGαタンパク質とカップリングして、その結果、「多機能シグナル伝達」を得ることができることがわかっている。GPCR多機能シグナル伝達理論の重要な仮定は、活性受容体コンフォメーションの異質性が存在し、アゴニストリガンドは、「刺激輸送」仮説で記載されるように、受容体コンフォメーションを誘発するか、安定化するか、又は選択するそれらの能力が異なる。結合すると、アゴニストリガンド化学構造パラメータは、Gαタンパク質の種類及び活性化されるシグナル伝達経路に影響を及ぼすGPCRコンフォメーションの最も重要な決定因子に含まれる。
【0008】
73の生物学的Gタンパク質共役受容体標的に対する380の拮抗物質の活性に関する105の論文の調査によると、この試料データセットにおいて、322が逆アゴニストであり、58(15%)が中性拮抗物質である。逆アゴニズムの優位性は、中性拮抗物質が薬理学的空間において少数種であることを示す理論的予想と一致する(Kenakin, Mol Pharmacol.(2004);65:2−11)。
【0009】
p90リボソームs6キナーゼ(RSK)は、ヒトkinome中のAGCサブファミリーの構成要素であるセリン/トレオニンキナーゼの一群である(Nguyen, 2008)。4つのRSKイソ型(RSK1〜4)があり、それぞれ別個の遺伝子の産物である。RSKイソ形は、75%〜80%の配列同一性によって特徴付けられる。RSKイソ型はヒト組織において広く分布しているが、様々な組織発現を示し、このことは、それらが異なる機能に関与することを示す。RSKイソ型は、Ras−ERK経路を刺激する細胞外シグナル伝達分子によって活性化される。これらの分子は、様々な成長因子、サイトカイン、ペプチドホルモン及び神経伝達物質を含む。なお、現時点で、RSKイソ型のそれぞれについての生物学的/薬理学的役割は、癌をはじめとするほとんどの疾患について確立されていない。
【0010】
RSKは、ファミリーとしてのRSKを細胞増殖、細胞分化、生存、及び遊走などの多くの生物学的プロセスと関連づける、転写因子、最初期遺伝子産物、翻訳調節因子、酵素、及び構造タンパク質をはじめとする様々なタンパク質をリン酸化する(Anjum and Blenis, 2008a )。
【0011】
RSKイソ形は、ヒト乳癌組織試料の50%で過剰発現され、このことは、RSKの調節が損なわれたことを示し、RSK活性と腫瘍細胞増殖との間の関連により、RSKが新規癌治療薬標的であることが明らかになる(Gioeli et al., 1999;Smith et al., 2005)。
【0012】
RSKとHIV感染との間の関連について動かぬ証拠がある。RSK2はHIV−1 Tatと相互関係を有することが示された。RSK2はHIV−1 Tatにより動員され、活性化され、それ自体も、正常なTat機能に対して重要である(Hetzer et al., 2005)。さらに、カポジ肉腫関連ヘルペスウイルスはRSKと相互作用し、それらのキナーゼ活性を強力に刺激する(Kuang et al., 2009)。
【0013】
構造的に、RSKイソ型は非常に類似している(Nguyen, 2008)。イソ型のそれぞれは、大きな100アミノ酸リンカー領域によって分離されている2つの機能的触媒ドメインを含む。この2つのキナーゼドメインは別個であり、同じでないATP結合部位を含む。NTKDはp70S6キナーゼ(p70 S6K1)と類似し、CTKDはカルシウム/カルモジュリンタンパク質キナーゼと類似している。その分子構造のために、RSは阻害に関して2つの論理的部位:NTKDにおけるATP結合部位及びCTKDにおけるものを提供する。両ATPポケットの部位特異的阻害剤が同定されている。
【0014】
90kDaリボソームS6キナーゼ(RSK)の上流アクチベータの異常な活性化は、癌をはじめとする多くのヒト疾患と関連付けられている。RSK1及びRSK2イソ型は、胸部及び前立腺腫瘍で増幅される。造血変換(hoematopoietic transformation)におけるRSK2活性化の重要性は、最近、線維芽細胞成長因子受容体−3(FGFR3)を発現する複数の骨髄腫細胞で示された。RSK3は、最近、卵巣癌において潜在的な腫瘍サプレッサとして機能することが示された。RSK4はp53依存的細胞増殖停止に関与することが示されたが、RSK4の異常な発現が乳癌で観察された。ヒトRSK2遺伝子における突然変異は、コフィン・ローリー症候群(CLS)(重篤な精神運動遅延、指及び顔の変形、並びに進行性骨格奇形によって特徴付けられるX連鎖障害)と関連する。CLS患者由来の線維芽細胞において、転写因子CREBの誘発されたSerl33リン酸化の大幅な減弱が、上皮成長因子刺激に反応して検出された。CLS患者から誘導された線維芽細胞は、このヒト疾患に関するRSK2の機能を決定する際に有用であったが;これらの細胞とRSK2欠損マウス線維芽細胞との間の相違は、CLSが多変量疾患である可能性があり、マウスはRSK2機能を研究するのに理想的な系でない可能性があることを示唆する。RSK2及びRSK4遺伝子はどちらもX染色体上に位置し、最近のデータも、非特異的X連鎖精神遅滞にRSKを関連づけるが、RSK4に関する決定的証拠はまだ得られていない(Anjum and Blenis, 2008b)。
【発明の概要】
【発明が解決しようとする課題】
【0015】
一態様では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に5−HT2結合相互作用を調節できる治療有効量の化合物(例えば、本明細書中で記載する化合物)を投与することを含む方法を提供する。一実施形態では、化合物は5−HT2cを刺激することができる。別の実施形態では、化合物は、5−HT2a及び/又は5−HT2bを拮抗するが、5−HT2cを刺激することができる。
【0016】
一態様では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に治療有効量の5−HT2c刺激化合物を投与することを含む。
【0017】
別の態様では、本発明は神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、5−HT2cを直接、好ましくは5−HT2a及び/又は5−HT2bと比べて選択的に調節することによって、5−HT2結合相互作用を調節することができる治療有効量の化合物を投与することを含む。
【0018】
別の実施形態では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とすると認定された対象に、治療有効量の5−HT2c刺激化合物又は5−HT2c選択的化合物を投与することを含む。
【0019】
別の態様では、本発明は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安、睡眠ホメオスタシスをはじめとする神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は対象に有効量の5−HT2cを刺激できる投与することを含む。
【0020】
別の態様では、本発明は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安に罹っているか又は罹りやすい対象を治療する方法であって、当該対象に、5−HT2cを刺激する(5−HT2a及び/又は5−HT2bと比べて選択的であることを包含する)ことができる有効量の化合物を投与して、当該対象が治療されるようにすることを含む方法を提供する。
【0021】
一態様では、本発明は、GPCR障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に、治療有効量のGPCR結合相互作用を調節することができる化合物を投与することを含む方法を提供する。一実施形態では、化合物はGPCRを刺激することができる。別の実施形態では、化合物はGPCRを拮抗することができる。
【0022】
別の態様において、本明細書中の化合物は、セロトニンヒスタミンH1、5HT2A、2B、2C、及びアセチルコリンムスカリン性M1〜M5GPCRを標的とする機能的に選択的な化合物である。態様において、本発明は、対象においてセロトニンヒスタミンH1、5HT2A、2B、2C、及びアセチルコリンムスカリン性M1〜M5GPCRを選択的に標的とする方法であって、当該対象に本明細書中の化合物を投与することを含む方法を提供する。
【0023】
別の態様では、本発明は、対象におけるGPCR介在性障害を治療又は予防する方法であって、それらを必要とすると認定された対象に対して、本明細書中で記載される化合物を投与することを含む方法を提供する。ある実施形態では、化合物は、表1{下記)の化合物である。ある実施形態では、化合物は、式(I):
【化1】

(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化2】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり、
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0024】
ある実施形態では、化合物は、各R1が−NMe2であるものである。
【0025】
他の実施形態では、R1は
【化3】
であり、これは場合によって置換されている。
【0026】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでないものである。
【0027】
ある実施形態では、化合物は、R8が1個のR5で置換され、R5が、H、アルキル、又はハロでない置換基であるものである。
【0028】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0029】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0030】
ある実施形態では、化合物は、各R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、ここで各R5は独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0031】
ある実施形態では、本発明は、そのような治療を必要とすると認定された対象において、疾患、障害又はその症状を治療する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。
【0032】
ある実施形態では、障害は、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、又は泌尿生殖路(genitor−urinary tract)障害(例えば、膀胱制御)である。ある実施形態では、障害は覚醒剤(例えば、コカイン、アンフェタミン、メタンフェタミン)薬物中毒である。ある実施形態では、障害は肥満である。ある実施形態では、障害は認知障害である。他の実施形態では、障害はアレルギー又は炎症性障害である。
【0033】
別の態様では、本発明は、そのような治療を必要とすると認定された対象において5−HT2cを阻害する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。
【0034】
別の態様では、本発明は、対象において肥満を治療する方法であって、それを必要とすると認定された対象に、5−HT2a又は5−HT2bと比べて、5−HT2cを選択的に阻害することができる化合物を投与することを含む方法を提供する。ある実施形態では、5−HT2cを阻害するための結合相互作用は、5−HT2a又は5−HT2bのいずれかについてよりも少なくとも5倍(或いは少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い。ある実施形態では、5−HT2cを阻害するための結合相互作用は、5−HT2a又は5−HT2bのいずれかについてよりも少なくとも100倍高い。
【0035】
別の態様では、本発明は、5−HT2c活性を調節することができる化合物を同定するための方法であって;(i)分子若しくは分子複合体の三次元表示(前記分子若しくは分子複合体は、5−HT2cの構造座標によって規定される結合ポケットを含む);又はb)前記分子又は分子複合体の相同体の三次元表示(ここで、前記相同体は、約2.0オングストローム以下の前記アミノ酸の骨格からの二乗平均平方根偏差(root measn square deviation)を有する結合ポケットを含む)を作製し;(ii)試験化合物の三次元表示を作製し;(iii)試験化合物の標的との結合相互作用を評価することを含む方法を提供する。ある実施形態では、当該方法は、試験化合物を5−HT2cと接触させ、化合物の結合活性を測定することを更に含む。
【0036】
一態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に治療有効量のRSK結合相互作用を調節することができる化合物(例えば、本明細書中に記載するいずれかの式の化合物)を投与することを含む方法を提供する。
【0037】
一態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、治療有効量のRSK阻害剤化合物を投与することを含む。
【0038】
別の態様では、本発明は、RSK介在性疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、治療有効量の、本明細書中に記載するいずれかの式の化合物(例えば、RSK調節化合物(直接的若しくは間接的)、RSK阻害化合物)を投与することを含む。
【0039】
別の態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とすると認定された対象に、治療有効量の、1以上の他のキナーゼよりもRSK阻害について選択的な化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、1以上の他のキナーゼよりもRSK阻害について選択的であると認定された化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、RSK−2よりもRSK−1、RSK−3又はRSK−4の阻害について選択的な化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、5−HT2(例えば、5−HT2a、5−HT2b、5−HT2c)の調節(例えば、逆アゴニスト活性)によりRSK−2を調節するための化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、化合物が、5−HT2(例えば、5−HT2a、5−HT2b、5−HT2c)の調節(例えば、逆アゴニスト活性)によりRSK−2を調節する(例えば、本明細書中に記載する化合物)と認定される方法であり得る。
【0040】
他の態様では、当該方法には、化合物が、他のキナーゼよりもRSK−1、RSK−3又はRSK−4について選択的である方法が含まれる。選択性は、1つの標的(すなわちキナーゼ)について、他の標的と比べて例えば2倍、3倍、10倍、100倍、X倍(ここで、Xは数値である)等である。別の実施形態では、本発明は、化合物が標的酵素に対する活性範囲、及び標的以外の酵素に対する異なる活性範囲の選択性を示す、本明細書中で記載する方法を提供する。
【0041】
他の態様では、当該方法には、化合物が、RSK2又は他のキナーゼよりも、RSK1並びに特に3、及び4を優先的に阻害する方法が含まれる。他の態様では、当該方法には、化合物が1.0マイクロモル濃度でRSK1並びに特に3、そして4を優先的に阻害し、RSK2を阻害しない方法が含まれる。このように、(−)−トランス−CAT及び誘導体は、RSK1、3、及び4の物理的及び生物学的特性をRSK2から区別するため、及びRSK1、3、4の生物学を438の他のキナーゼから区別するための分子生物学的手段として非常に有用であり(表4を参照);そしてRSK1、3、及び4の物理的及び生物学的特性をRSK2から区別するため、そしてRSK1、3、4の生物学を438の他のキナーゼから区別するための分子生物学的手段として非常に有用であり;そして癌(特に、造血、胸部及び前立腺)、HIV、及びコフィン・ローリー症候群の生物学及び病理を1つのRSKイソ型の関与に関して、これらの疾患における別のイソ型に対して、及びこれらの疾患における他のキナーゼに対して特徴付けるために有用である。さらに、(−)−トランス−CAT及び本明細書中の化合物は、RSK1、3、及び4とのそれらの結合相互作用により、これらのキナーゼの構造を特徴付けるための有用な生化学的プローブである。
【0042】
ある実施形態では、本発明は、化合物が、式(I):
【化4】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化5】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシである)であり;
R6は独立してH又はアルキルであり;
R7は独立してH、若しくはN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、若しくは複素環(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
により表されるか、又はその塩、水和物若しくは溶媒和物である前記方法を提供する。
【0043】
ある実施形態では、化合物は、各R1が−NMe2である化合物である。
【0044】
ある実施形態では、R1は
【化6】
であり、これは場合によって置換されている。
【0045】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでない化合物である。
【0046】
ある実施形態では、化合物は、R8が1つのR5で置換され、R5が、H、アルキル、又はハロでない置換基である、化合物である。
【0047】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0048】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0049】
ある実施形態では、化合物は、R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、各R5が独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0050】
ある実施形態では、本発明は、そのような治療を必要とすると認定された対象において、疾患、障害、又はその症状を治療する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。一態様では、化合物は(−)−トランス−CATである。
【0051】
ある実施形態では、障害は癌(特に、造血、胸部及び前立腺)、HIV、又はコフィン・ローリー症候群である。ある実施形態では、障害は乳癌である。ある実施形態では、障害は前立腺癌である。ある実施形態では、障害はHIVである。
【0052】
一態様では、本発明は、式(I):
【化7】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化8】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
により表される化合物又はその塩、水和物若しくは溶媒和物を提供する。
【0053】
ある実施形態では、化合物は、各R1が−NMe2である化合物である。
【0054】
ある実施形態では、R1は
【化9】
であり;場合によって置換されている;
【0055】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでない化合物である。
【0056】
ある実施形態では、化合物は、R8が1個のR5で置換され、R5が、H、アルキル、又はハロでない置換基である化合物である。
【0057】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0058】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0059】
ある実施形態では、化合物は、各R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、各R5が独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0060】
ある実施形態では、1位及び3位での化合物置換基は、互いにトランス配位である。
【0061】
ある実施形態では、化合物は式(II):
【化10】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化11】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリール;式中、環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり;
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;
mは1、2又は3である)によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0062】
ある実施形態では、各R’は独立して、メチル、エチル、プロピル、イソプロピル、ブチル、s−ブチル、t−ブチル、アルケニル、ビニル、又はアルキニルである。
【0063】
ある実施形態では、1位及び3位での化合物置換基は互いにトランス配位である。
【0064】
ある実施形態では、化合物R1は−NMe2である。ある実施形態では、PAT化合物は(1R,3S)−(−)トランス−1−フェニル−3−N,N−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレンである。
【0065】
さらなる実施形態では、化合物は式(II−a):
【化12】
(式中、
R5は、H、メチル、エチル、プロピル、イソプロピル、ブチル、s−ブチル、t−ブチル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、フェニル、F、Cl、若しくはBrであり;そして
mは1、2、若しくは3である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0066】
ある実施形態では、化合物は、式(III):
【化13】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化14】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
nは2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
R5は、H、アルキル、又はハロであり;
R6は独立してH又はアルキルであり;
R7はH、又はN(アルキル)2であり;そして
mは1、2、又は3である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0067】
別の態様では、本発明は、式(IV):
【化15】
(式中、
R1は独立してH、又はN(アルキル)2であり;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、NH2、NH(R’)、N(RM)2であるか;
又は
【化16】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は場合によって1、2、3、若しくは4個の独立したR5で置換された複素環式である)
により表される化合物、又はその塩、水和物若しくは溶媒和物を提供する。
【0068】
前記式のいずれかのある実施形態では、1位及び3位の化合物置換基は互いにトランス配位である。
【0069】
ある実施形態では、化合物R1は−NMe2である。ある実施形態では、PAT化合物は(1R,3S)−(−)−トランス−1−フェニル−3−N,N−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレンである。
【0070】
別の態様では、本発明は、式(V):
【化17】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化18】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;R3はH、OH、又はハロであり;
R4はH、OH、アリール、ヘテロアリール、又はハロであり;
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキルであり;
R7はH、又はN(アルキル)2であり;そして
Zはアリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルであり、そのそれぞれは場合によって1、2、3、若しくは4個の独立したR5で置換されている)によって表される化合物、又はその塩、水和物若しくは溶媒和物を提供する。
【0071】
ある実施形態では、R1はN(R’)2であり;そして各R’はアルキルである。他の実施形態では、Zはフェニルであり、これはハロアルキル、アルコキシ、又はハロで置換されていてもよい。他の実施形態では、R4はフェニルであり、これはハロアルキル、アルコキシ、又はハロで置換されていてもよい。
【0072】
別の態様では、本発明は、本明細書中で記載される化合物(例えば、本明細書中のいずれかの式の化合物)及び薬剤的に許容される担体を含む組成物を提供する。
【0073】
別の態様では、本発明は、本明細書中で記載される化合物(例えば、本明細書中のいずれかの式の化合物)及び薬剤的に許容される担体を組み合わせることを含む、組成物の作製法を提供する。
【0074】
構造−活性関係(SAR)研究の結果は、H1対5HT2A対5HT2B対5HT2C対、M1対M2対M3対M4対M5GPCRについての本発明の化合物の親和選択性(affinity selectivity)は、テトラヒドロナフタレン環系の、C1位での置換基(例えば、ペンダントフェニル又は他の芳香族、ヘテロ芳香族、シクリル、シクロアルキル)及びC3位での置換基(アミン)の立体化学並びに、C1及びC3置換基の化学的性質、並びにC6及びC7位での化学置換基に依存することを示す(炭素ナンバリングは式I、表1のとおり)。同様に、H1対5HT2A対5HT2B対5HT2C対、M1対M2対M3対M4対M5GPCRでのアゴニスト対逆アゴニスト対拮抗物質活性は、C1、C3、C6、及びC7での置換基の化学的性質及び立体化学によって決定される。例えば、Bucholtz, E. C, Wyrick, S.D., Owens, C.E., and Booth, R.G. 1−Phenyl−3−dimethylaminotetralins (PATs): Effect of stereochemistry on binding and function at brain histamine receptors. Medicinal Chemistry Research 8:322−332 (1998).; Bucholtz, E.C., Brown., R.L., Tropsha, A., Booth, R.G, and Wyrick, S.D. Synthesis, Evaluation and Comparative Molecular Field Analysis of 1−Phenyl−3−amino−1,2,3,4−tetrahydronaphthalenes as Ligands for Histamine H1 Receptors. Journal of Medicinal Chemsitry.42:3041−3054 (1999);Choksi, N.Y., Nix, William B., Wyrick, S.D., and Booth, R.G. A novel phenylaminotetralin recognizes Histamine H1 receptors and stimulates dopamine synthesis in vivo in rat brain. Brain Research 852:151−160 (2000); Booth RG, Moniri NH, Bakker RA, Choksi NY, Timmerman H, and Leurs R. A novel phenylaminotetralin radioligand reveals a sub−population of Histamine H1 receptors. Journal of Pharmacology and Experimental Therapeutics 302:328−336 (2002); Moniri NH, Covington−Strachan D, Booth RG. Ligand−directed functional heterogeneity of Histamine H1 receptors: Novel dual−function ligands selectively activate and block H1−meditated phospholipas C and adenylyl cyclase signaling. Journal of Pharmacology and Experimental Therapeutics, 311 :274−281 (2004); Booth RG, Moniri NH. Ligand−directed multifunctional signaling of histamine H1 receptors Inflammation Research 54: S44−45 (2005); Ghoneim OM, Legere JA, Glbraikh A, Tropsha A, Booth RG. Novel ligands for the human histamine H1 receptor: Synthesis, pharmacology, and comparative molecular field analysis studies of 2−dimethylamino−5−(6)−phenyl−1,2,3,4−tetrahydronaphthalenes. Bioorganic and Medicinal Chemistry, 14:6640−6658 (2006); Booth RG, Moniri NH. Novel Ligands Stabilize Stereo−Selective Conformations of the Histamine H1 Receptor to Activate Catecholamine Synthesis. Inflammation Research 56:1−12 (2007)を参照。
【0075】
別の実施形態では、本明細書中の化合物は、アデニル酸シクラーゼ(AC)/cAMP対ホスホリパーゼC(PLC)/イノシトールリン酸(IP)細胞内シグナル伝達経路とカップリングして、脳カテコールアミン(ドーパミン、ノルエピネフリン)神経伝達物質合成を調節する脳H1受容体を識別し、選択的に活性化することができる。複数の態様では、本発明は、アデニル酸シクラーゼ(AC)/cAMP対ホスホリパーゼC(PLC)/イノシトールリン酸(IP)細胞内シグナル伝達経路とカップリングして、H1/AC/cAMPシグナル伝達に対して感受性のプロセス、例えば対象における脳カテコールアミン(ドーパミン、ノルエピネフリン)神経伝達物質合成の調節に生理学的に影響を及ぼす脳H1受容体を選択的に活性化する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0076】
別の態様では、本明細書中の化合物は、H1が関与するAC/cAMPシグナル伝達を選択的に増強して、改変されたカテコールアミン神経伝達を含むある神経精神病に罹っている患者を治療する化合物である。複数の態様では、本発明は、H1が関与するAC/cAMPシグナル伝達を選択的に増強して、改変されたカテコールアミン神経伝達を含むある神経精神病に罹っている対象を治療する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0077】
別の実施形態では、本明細書中の化合物は、H1/PLC/IPシグナル伝達によって進行する不適切なH1が関与する効果、例えば、呼吸困難(気管支狭窄)、下痢(GI収縮)、並びに浮腫及び低血圧(増大した血管透過性)、特に末梢アレルギー反応に関連するものの拮抗物質及び逆アゴニストである化合物である。複数の態様では、本発明は、対象においてPLC/IP経路によって進行する不適切なH1が介在する効果を拮抗(例えば遮断)する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0078】
別の態様では、本明細書中の化合物は、セロトニン5HT2A及び5HT2B受容体での拮抗物質並びに逆アゴニストである。
【0079】
別の実施形態では、本明細書中の化合物は、PLC/IPシグナル伝達と関連するヒスタミンH1受容体での拮抗物質並びに逆アゴニストである。
【0080】
別の態様では、本明細書中の化合物は、アセチルコリンムスカリン性M1〜M5受容体での拮抗物質並びに逆アゴニストである。
【0081】
別の態様では、本明細書中の化合物は、アセチルコリンムスカリン性M1〜M5受容体での拮抗物質並びに逆アゴニスト及びアゴニストである。別の態様では、本明細書中の化合物は同時にセロトニン5HT2A及び5HT2B受容体での逆アゴニストであり、かつ5HT2c受容体でのアゴニストである。複数の態様では、本発明は、対象において疾患又は障害(例えば、精神障害;肥満)を治療又は予防する方法であって、対象に、同時にセロトニン5HT2A及び5HT2B受容体での逆アゴニストであり、5HT2c受容体でのアゴニストである化合物を投与することを含む方法を提供する。別の実施形態では、当該方法は、対象が精神障害及び肥満の両方の治療を必要とする方法である。
【0082】
一実施形態では、当該化合物は、対象においてアセチルコリンムスカリン受容体系の障害から起こる疾患又は障害を薬理学的に治療するための方法であって、対象に本明細書中のいずれかの式の化合物を投与することを含む方法を提供する。本明細書中のいずれかの式の化合物は、ムスカリン性M1、M2、M3、M4、及び/又はM5受容体に対して薬理学的に関連する親和性を有し、1以上のムスカリン受容体でアゴニスト、逆アゴニスト、及び/又は拮抗物質として機能的に挙動する。
【0083】
ムスカリン性M1、M2、M3、M4、及び/又はM5受容体の薬理学の調節に反応する典型的な疾患又は障害(Brown and Taylor, 2006, Muscarinic Agonists and Antagonists. In: Brunton L.L., Lazo, J.S., Parker, K.L. (Eds.), Goodman and Gilman’s The Pharmacological Basis of Therapeutics 11th Edition. McGranw−Hill, New York, NY, pp. 183−200)としては、これらに限定されるものではないが:胃腸管の障害(便秘、下痢、胃酸過多、痙縮)、尿路の障害(頻尿、排尿不足、多尿)、緑内障、喘息、パーキンソン病、アルツハイマー病、外分泌腺が関係する様々な障害(発汗、涙液形成、唾液形成、粘液形成に関連する問題)、及びあるキノコ(例えば、天然のムスカリン誘導体を含むもの)による中毒の治療が挙げられる。
【0084】
別の態様では、本発明は、5−HT2cを調節する化合物を同定するための方法を提供し、この方法は、5−HT2cタンパク質を得るか、又は5−HT2cタンパク質の結晶構造に関連する情報を得、試験化合物を5−HT2cタンパク質構造中又は構造上でモデル化して、5−HT2cタンパク質の相互作用を調節するかどうかを判定することを含む。ある実施形態では、モデリングのステップは、化合物が、5−HT2cの1以上の膜通過ドメイン1〜7の構造座標により規定される結合ポケットに結合するか、又はこれに関連する能力をモデル化又は判定することを含む。
【0085】
本発明のさらに別の態様は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安を治療又は予防するために有用な化合物を同定する方法である。当該方法は、5−HT2c複合体を試験化合物と接触させ、そして試験化合物が5−HT2cを調節(例えば、刺激又は拮抗)する能力を評価することを含む。
【0086】
本発明のさらに別の態様は、5−HT2cの活性を調節する化合物を同定する方法であって、この方法は、5−HT2cの1以上の膜通過ドメイン1〜7の原子座標を用いて、結合ポケットを含む分子の三次元構造(例えば、コンピュータによる)を生成させ、そしてこの三次元構造を用いて5−HT2cの1以上の膜通過ドメイン1〜7の活性を調節する化合物を同定することを含む。
【0087】
別の態様では、本発明は、治療有効量の5−HT2cアゴニスト化合物及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。当該組成物は、神経精神障害(例えば、肥満)に罹っているか又は罹りやすい対象を治療するために処方され、神経精神障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0088】
一態様では、本発明は、対象における神経精神障害を治療するためのキットを提供し、このキットは、本明細書中の化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、並びに使用説明書を含む。さらなる態様では、本発明は、5−HT2cを刺激し、対象における抗肥満治療薬の有効性を評価し、5−HT2cアゴニストで治療される対象の経過をモニタリングし、神経精神障害の対象を5−HT2cアゴニストでの治療について選択し、及び/又は神経精神障害(例えば、肥満)に罹っているか又は罹りやすい対象を治療するためのキットを提供する。ある実施形態では、本発明は:対象における神経精神障害を治療するためのキットであって、5−HT2cアゴニスト活性を調節(例えば、刺激)することができる化合物を含むキットを提供する。他の態様では、当該化合物は、5−HT2a及び/又は5−HT2bと比べて5−HT2cを選択的に刺激する。他の態様では、当該化合物は、5−HT2a及び/又は5−HT2bを選択的に拮抗する。
【0089】
別の態様では、本発明は、5−HT2cの1以上の膜通過ドメイン1〜7(それぞれ単独又は組み合わせ)の三次元構造に関する。
【0090】
したがって、本発明は、これらの結合ポケットのいずれか1つ若しくは両方又は類似した三次元形状を有するいずれかの結合ポケットの相同体を含む分子若しくは分子複合体を提供する。
【0091】
本発明は、本明細書中で記載される化合物の医薬組成物であって、5−HT2cを刺激することができる化合物;5−HT2a及び/又は5−HT2bと比べて5−HT2cを選択的に刺激することができる化合物;5−HT2cを刺激し、5−HT2a及び/又は5−HT2bを拮抗することができる化合物;又はその薬剤的に許容されるエステル、塩、若しくはプロドラッグを、薬剤的に許容される担体とともに含む医薬組成物も提供する。
【0092】
別の態様では、本発明は、機械可読記憶媒体であって、5−HT2cの1以上の膜通過ドメイン1〜7を規定する結合ポケットの構造座標を含む記憶媒体を提供する。
【0093】
別の態様では、本発明は、分子又は分子複合体の三次元表示(前記分子又は分子複合体は、5−HT2cの1以上の膜通過ドメイン1〜7の構造座標によって規定される結合ポケットである);又はb)前記分子又は分子複合体の相同体の三次元表示(前記相同体は、前記アミノ酸の骨格原子からの二乗平均平方根偏差が約2.0オングストローム以下である結合ポケットを含む)を作成するコンピュータを提供する。コンピュータは:(i)機械可読データでコード化されたデータ記憶材料を含む、機械可読データ記憶媒体(ここで、前記データは5−HT2cの1以上の膜通過ドメイン1〜7の構造座標を含む);(ii)前記機械可読データを処理するための命令を記憶するためのワーキングメモリ;(iii)前記機械可読データを前記三次元表示に処理するための前記ワーキングメモリ及び前記機械可読データ記憶媒体に接続された中央処理装置;並びに(iv)前記三次元表示を表示するための前記中央処理装置に接続されたディスプレーを含む。
【0094】
別の態様では、本発明は、治療有効量のRSK阻害剤化合物(例えば、選択的なRSK−1、RSK−3、又はRSK−4阻害剤化合物)及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。当該組成物は、疾患又は障害(例えば、癌、HIV、RSK介在性疾患)に罹っているか又は罹りやすい対象を治療するために処方することができ、そして疾患又は障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0095】
一態様では、本発明は、対象における疾患又は障害を治療するためのキットを提供し、このキットは、本明細書中の化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、及び使用説明書を含む。さらなる態様では、本発明は、RSKを阻害するためのキットを提供する。ある実施形態では、本発明は:対象における疾患又は障害(すなわち、RSK介在性疾患又は障害)を治療するためのキットであって、RSK活性を調節(例えば、阻害)することができる化合物を含むキットを提供する。他の態様では、当該化合物は、他のキナーゼと比べてRSKを選択的に阻害する。他の態様では、当該化合物は、他のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害する。
【0096】
本発明はまた、本明細書中で記載される化合物の医薬組成物であって、RSK活性を調節(例えば阻害)することができる化合物又はその薬剤的に許容されるエステル、塩、若しくはプロドラッグを、薬剤的に許容される担体とともに含む医薬組成物も提供する。他の態様では、当該化合物は、他のキナーゼと比べてRSKを選択的に阻害する。他の態様では、当該化合物は、他のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害する。他の態様では、当該化合物は、5−HT2(例えば、1以上の5−HT2a、b、又はc)の調節(例えば、逆アゴニスト活性)によりRSKを調節する。
【0097】
本発明はまた、前記結合ポケットと結合する化合物の設計、評価及び同定方法も提供する。本発明の他の実施形態を以下で開示する。
【図面の簡単な説明】
【0098】
本発明を以下の非限定的な例を参照し、そして以下の図面を参照して、さらに説明する。
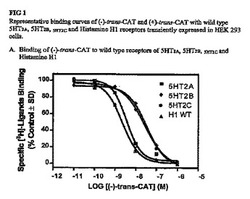
【図1】図1A〜1Eは、HEK293細胞において一時的に発現された野生型5HT2A、5HT2B、5HT2c及びヒスタミンH1受容体に関する(−)−トランス−CAT及び(+)−トランス−CATの代表的な結合曲線である。
【図2】野生型5HT2A、5HT2B、5HT2C受容体を一時的に発現するHEK293細胞におけるPLC活性/IP形成の活性に対する(−)−トランス−CAT効果の代表的な曲線を表す。データは、(−)−トランス−CATが、5HT2A、5HT2B及び5HT2c受容体の逆アゴニストであることを示す。
【図3】PLC活性/[3H]−IP形成のヒスタミンH1受容体が関与する刺激のヒスタミン活性化は、(−)−トランス−CATにより競合的に拮抗されることを表す。
【発明を実施するための形態】
【0099】
本発明者らは、5−HT2c及び/又はRSKを選択的に標的とすることによる選択的な疾患治療及び予防(すなわち有害な副作用が軽減又は最小限に抑えられた)を対象とする治療法を発見した。そのような相互作用は、特に、5−HT2メカニズムが重要な役割を果たすある種の神経精神障害における5−HT2c介在性障害の調節に関連する。
【0100】
本発明は、少なくとも一つには、5−HT2c相互作用が、神経精神障害治療の標的として有用(例えば選択的)であるという発見に関する。
【0101】
1.定義
本発明のさらなる説明の前に、本発明がより容易に理解されるように、便宜のために、いくつかの用語をまず定義し、ここにまとめる。
【0102】
「投与」又は「投与する」という用語は、本発明の化合物(複数可)を対象に対して、それらが目的とする機能を発揮するように導入する経路を包含する。使用できる投与経路の例としては、注射(皮下、静脈内、非経口的、腹腔内、髄腔内)、経口、吸入、直腸及び経皮が挙げられる。医薬製剤は、各投与経路に適した形態で投与することができる。たとえば、これらの製剤は、錠剤又はカプセル形態で、注射、吸入、洗眼液、軟膏、坐薬など、注射、注入又は吸入による投与;ローション又は軟膏による局所;及び坐薬により直腸で投与される。経口投与が好ましい。注射は、ボーラスであってもよいし、又は連続注入であってもよい。投与経路に応じて、本発明の化合物を、その目的とする機能を果たす能力に悪影響を及ぼし得る自然の条件から保護するために選択された材料でコーティングするか又は材料中に入れることができる。本発明の化合物は、単独、或いは前述の他の薬剤若しくは薬剤的に許容される担体又は両者と併用して、投与することができる。本発明の化合物は、他の薬剤の投与前に、薬剤と同時に、又は薬剤の投与後に、投与することができる。さらに、本発明の化合物は、インビボでその活性代謝物、又はさらに活性の高い代謝物に変換されるプロドラッグ形態で投与することもできる。
【0103】
「アルキル」という用語は、直鎖アルキル基、分枝アルキル基、シクロアルキル(脂環式)基、アルキル置換シクロアルキル基、及びシクロアルキル置換アルキル基をはじめとする飽和脂肪族基のラジカルを指す。アルキルという用語は、炭化水素骨格の1以上の炭素を置換する酸素、窒素、硫黄又はリン原子、たとえば酸素、窒素、硫黄又はリン原子をさらに含み得るアルキル基をさらに包含する。好適な実施形態では、直鎖又は分枝鎖アルキルは、その骨格中に、30個以下の炭素原子(例えば、直鎖についてはC1〜C30、分枝鎖についてはC3〜C30)、好ましくは26個以下、そしてさらに好ましくは20個以下、なお一層好ましくは4個以下を有する。同様に、好ましいシクロアルキルは、それらの館構造中に3〜10個の炭素原子、そしてさらに好ましくは3、4、5、6又は7個の炭素を環構造中に有する。
【0104】
さらに、アルキルという用語は、本明細書及び文全体を通して用いられる場合、「非置換アルキル」及び「置換アルキル」の両方を包含することが意図され、後者は、炭化水素骨格の1以上の炭素上の置換基を置換する置換基を有するアルキル部分を指す。そのような置換基としては、たとえば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分を挙げることができる。当業者らには、炭化水素鎖上で置換された部分は、適切ならばそれら自体が置換され得ると理解されるであろう。シクロアルキルを、たとえば前述の置換基でさらに置換することができる。「アルキルアリール」部分は、アリールで置換されたアルキル(例えば、フェニルメチル(ベンジル))である。「アルキル」という用語は、長さ及びアルキルに対する可能な置換は類似しているが、少なくとも1つの二重又は三重結合をそれぞれ含む不飽和脂肪族基も包含する。
【0105】
炭素数について特に指定がない限り、本明細書中で用いられる「低級アルキル」は、前記定義のとおりであるが、1〜10個の炭素、さらに好ましくは1〜6個、なお一層好ましくは1〜4個の炭素原子をその骨格構造中に有し、直鎖又は分枝鎖であってよいアルキル基を意味する。低級アルキル基の例としては、メチル、エチル、n−プロピル、i−プロピル、tert−ブチル、ヘキシル、ヘプチル、オクチルなどが挙げられる。好ましい実施形態において、「低級アルキル」という用語は、その骨格中に、4個以下炭素原子を有する直鎖アルキル、例えば、C1〜C4アルキルを包含する。
【0106】
「アルコキシアルキル」、「ポリアミノアルキル」及び「チオアルコキシアルキル」という用語は、前述のアルキル基であって、炭化水素骨格の1個以上の炭素を置換する酸素、窒素又は硫黄原子、たとえば酸素、窒素又は硫黄原子をさらに含むアルキル基をさす。
【0107】
「アルケニル」及び「アルキニル」という用語は、前述のアルキルと長さ及び可能な置換は類似しているが、それぞれ少なくとも1個の二重又は三重結合をさらに含む不飽和脂肪族基を指す。たとえば、本発明は、シアノ及びプロパルギル基を想定する。
【0108】
「アリール」という用語は、本明細書中で用いられる場合、0〜4個のヘテロ原子を含み得る5員及び6員単環芳香族基をはじめとするアリール基のラジカル、たとえば、ベンゼン、ピロール、フラン、チオフェン、イミダゾール、ベンゾキサゾール、ベンゾチアゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピラジン、ピリダジン及びピリミジンなどを指す。アリール基は、ナフチル、キノリル、インドリルなどの多環式縮合芳香族基も包含する。環構造中にヘテロ原子を有するこれらのアリール基は、「アリールヘテロ環」、「ヘテロアリール」又は「ヘテロ芳香族」とも称する。芳香環は、1以上の環位置で、前述のような置換基、たとえば、ハロゲン、ヒドロキシル、アルコキシ、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分で置換することができる。アリール基は、芳香族でない脂環式又は複素環式環と縮合又は架橋して、多環(例えば、テトラリン)を形成することもできる。
【0109】
「〜と結合している」という用語は、化学成分若しくは化合物、又はその一部と、タンパク質上の結合ポケット若しくは結合部位との間の近接の状態を指す。会合は、非共有的(この場合、近位は、水素結合又はファンデルワールス又は静電的相互作用により支援される)であってもよいし、又は共有的であってもよい。
【0110】
「結合ポケット」という用語は、本明細書で用いられる場合、その形状の結果として、有利に別の化学成分又は化合物と会合する分子又は分子複合体の部分を指す。
【0111】
本発明の化合物の「生物学的活性」という言葉には、反応性の細胞において本発明の化合物により誘発されるすべての活性が含まれる。この言葉は、これらの化合物により誘発されるゲノム及び非ゲノム活性を含む。
【0112】
「生物学的組成物」又は「生物学的試料」とは、細胞若しくはバイオポリマーを含むか、又は細胞若しくはバイオポリマーから誘導される組成物を指す。細胞含有組成物には、たとえば、哺乳動物の血液、赤血球濃縮物、血小板濃縮物、白血球濃縮物、血球タンパク質、血漿、血小板浮遊血漿、血漿濃縮物、血漿の分画から得られる沈殿物、血漿の分画から得られる上清、血漿タンパク質画分、精製若しくは部分的に精製された血液タンパク質又は他の成分、血清、精液、哺乳動物の初乳、乳、唾液、胎盤抽出物、クリオ沈殿物(cryopercipitate)、クリオ上清(cryosupernatant)、細胞可溶化物、哺乳動物の細胞培養若しくは培養培地、発酵産物、腹水、血球中で誘発されるタンパク質、及び正常若しくは形質転換細胞により細胞培養で産生される産物(例えば、組換えDNA若しくはモノクローナル抗体技術による)が含まれる。生物学的組成物は無細胞であり得る。好ましい実施形態では、好適な生物学的組成物又は生物学的試料は、赤血球懸濁液である。いくつかの実施形態では、血球懸濁液は哺乳動物血球を含む。好ましくは、血球は、ヒト、非ヒト霊長類、イヌ、ネコ、ウマ、ウシ、ヤギ、ヒツジ又はブタから得られる。好適な実施形態では、血球懸濁液には、赤血球及び/又は血小板及び/又は白血球及び/又は骨髄細胞が含まれる。
【0113】
「キラル」という用語は、鏡像パートナーの重ね合わせることができない性質を有する分子を指し、一方、「アキラル」という用語は、それらの鏡像パートナー上に重ねることができる分子を指す。
【0114】
「ジアステレオマー」という用語は、2以上の不斉中心を有し、それらの分子が互いに鏡像でない立体異性体を指す。
【0115】
「有効量」という用語には、所望の結果を達成するために必要な投与量及び期間で有効な量、例えば本明細書中で記載される障害を治療するために十分な量が含まれる。本発明の化合物の有効量は、対象の疾患状態、年齢、及び体重、並びに当該対象において所望の反応を誘発する本発明の化合物の能力などの因子によって変化し得る。投与計画は、最適の治療反応を提供するために調整することができる。有効量は、本発明の化合物の任意の毒性又は有害な効果(例えば副作用)よりも治療上有益な効果が上回る量でもある。
【0116】
本発明の化合物の治療有効量(すなわち、有効投与量)は、約0.001〜30mg/kg体重、好ましくは約0.01〜25mg/kg体重、さらに好ましくは約0.1〜20mg/kg体重、なおいっそう好ましくは約1〜10mg/kg、2〜9mg/kg、3〜8mg/kg、4〜7mg/kg、又は5〜6mg/kg体重の範囲であり得る。当業者は、これらに限定されるものではないが、疾患若しくは障害の重篤度、以前の治療、対象の全体的な健康及び/又は年齢、並びに存在する他の疾患をはじめとするある因子が、対象を有効に治療するために必要な投与量に影響を及ぼし得ることを理解するであろう。さらに、治療有効量の本発明の化合物での対象の治療は、単一治療を含み得るか、又は好ましくは一連の治療を含み得る。一例では、対象を、約0.1〜20mg/kg体重の範囲の本発明の化合物で、約1〜10週間、好ましくは2〜8週間、さらに好ましくは約3〜7週間、なおいっそう好ましくは約4、5、又は6週間、1週につき1回で治療する。治療に用いられる本発明の化合物な有効投与量は、特定の治療過程にわたって増大又は減少し得ることも理解されるであろう。
【0117】
「エナンチオマー」という用語は、互いに重ね合わせることができない鏡像である化合物の2つの立体異性体を指す。2つのエナンチオマーの等モル混合物は、「ラセミ混合物」又は「ラセミ体」と呼ばれる。
【0118】
「ハロアルキル」という用語は、ハロゲンで一置換、二置換又は多置換された前記定義のアルキル基、例えばフルオロメチル及びトリフルオロメチルを包含することが意図される。
【0119】
「ハロゲン」及び「ハロ」という用語は、−F、−Cl、−Br又は−Iを表す。
【0120】
「ヘテロアリール」という用語は、典型的には5〜18個の環原子を含む芳香族ヘテロシクリルを意味する。ヘテロアリールは、単環、又は二環以上の縮合環であってよい。5員ヘテロアリールの非限定的な例としては、イミダゾリル;フラニル;チオフェニル(又はチエニル若しくはチオフラニル);ピラゾリル;オキサゾリル;イソキサゾリル;チアゾリル;1,2,3−、1,2,4−、1,2,5−、及び1,3,4−オキサジアゾリル;及びイソチアゾリルが挙げられる。6員ヘテロアリールの非限定的な例としては、ピリジニル;ピラジニル;ピリミジニル;ピリダジニル;並びに1,3,5−、1,2,4−、及び1,2,3−トリアジニルが挙げられる。6/5員縮合環ヘテロアリールの非限定的な例としては、ベンゾチオフラニル、イソベンゾチオフラニル、ベンズイソキサゾリル、ベンゾオキサゾリル、プリニル、及びアントラニリルが挙げられる。6/6員縮合環ヘテロアリールの非限定的な例としては、キノリニル;イソキノリニル;並びにベンゾオキサジニル(シンノリニル及びキナゾリニルを包含する)が挙げられる。
【0121】
「複素環式」又は「ヘテロシクロ」又は「ヘテロシクリル」という用語は、典型的には3〜18個の環原子を含む、飽和(例えば、「ヘテロシクロアルキル」)、部分不飽和(例えば、「ヘテロシクロアルケニル」又は「ヘテロシクロアルキニル」)又は完全不飽和(例えば、「ヘテロアリール」)環系であって、環原子の少なくとも1つがヘテロ原子(すなわち、窒素、酸素若しくは硫黄)であり、残りの環原子が炭素、窒素、酸素及び硫黄からなる群から独立して選択されるものを指す。ヘテロシクリル基は、安定な分子が得られるならば、親分子部分と基中の任意の置換可能な炭素若しくは窒素原子により結合することができる。ヘテロシクリルは、制限なく、典型的には3〜14個の環原子、3〜8個の環原子、3〜6個の環原子、又は5〜6個の環原子を含む単環であってよい。単環ヘテロシクリルの非限定的な例としては、フラニル、ジヒドロフラニル、ピロリル、イソピロリル、ピロリニル、ピロリジニル、イミダゾリル、イソイミダゾリル、イミダゾリニル、イミダゾリジニル、ピラゾリル、ピラゾリニル、ピラゾリジニル、トリアゾリル、テトラゾリル、ジチオリル、オキサチオリル、オキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、チアゾリニル、イソチアゾリニル、チアゾリジニル、イソチアゾリジニル、チオジアゾリル、オキサチアゾリル、オキサジアゾリル、ピラニル、ジヒドロピラニル、ピリジニル、ピペリジニル、ピリダジニル、ピリミジニル、ピラジニル、ピペラジニル、トリアジニル、イソキサジニル、オキサゾリジニル、イソキサゾリジニル、オキサチアジニル、オキサジアジニル、モルホリニル、アゼピニル、オキセピニル、チエピニル、又はジアゼピニルが挙げられる。ヘテロシクリルは、制限なく、互いに縮合した2以上の環を含んでもよく、例えば、ナフチリジニル、チアゾールピリミジニル、チエノピリミジニル、ピリミドピリミジニル、又はピリドピリミジニルである。ヘテロシクリルは、環構成要素として1以上の硫黄原子を含んでもよく;場合によっては、硫黄原子(複数可)はSO又はSO2に酸化される。ヘテロシクリル中の窒素ヘテロ原子(複数可)は四級化されていても、四級化されていなくてもよく、又はNオキシドに酸化されていても、酸化されていなくてもよい。加えて、窒素ヘテロ原子(複数可)はN保護されていても、N保護されていなくてもよい。
【0122】
「ヒドロキシル」という用語は、−OHを意味する。
【0123】
「ヘテロ原子」という用語は、本明細書中で用いられる場合、炭素又は水素以外の任意の元素の原子を意味する。好ましいヘテロ原子は、窒素、酸素、硫黄及びリンである。
【0124】
「ホメオスタシス」という用語は、内部環境における静的、又は一定の状態の維持を意味することが当該技術分野で承認されている。
【0125】
「改善された生物学的特性」という語法は、インビボでのその有効性を増強する本発明の化合物中に内在する活性を指す。好ましい実施形態では、この用語は、本発明の化合物の任意の質的又は量的に改善された治療特性、例えば低下した毒性を指す。
【0126】
「GPCR障害という用語は、Gタンパク質共役受容体(例えば、5−HT2a、5−HT2b、5−HT2c、ムスカリン性M1〜M5)が関与する任意の疾患、障害又はその症状を包含する。そのようなGPCRが関与する疾患及び障害としては、たとえば、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安、鬱病、統合失調症、精神病、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、及び泌尿生殖路障害(例えば、膀胱制御)が挙げられる。
【0127】
「M1〜M5 GPCR」という語法は、コリン作動性ムスカリン性M1〜M5神経伝達物質Gタンパク質共役受容体(本明細書中に記載されるものを包含する)を指す。
【0128】
「5−HT2」という語法は、セロトニン受容体(本明細書中に記載されるものを包含する)、例えば5−HT2a、5−HT2b及び5−HT2cサブタイプを指す。
【0129】
「場合によって置換された」という用語は、非置換である基又は1以上の利用可能な位置で水素以外によって、典型的には1、2、3、4若しくは5位で、1以上の好適な基(同一であっても、異なっていてもよい)により置換されている基を包含することが意図される、そのような任意の置換基としては、たとえば、ヒドロキシ、ハロゲン、シアノ、ニトロ、C1〜C8アルキル、C2〜C8アルケニル、C2〜C8アルキニル、C1〜C8アルコキシ、C2〜C8アルキルエーテル、C3〜C8アルカノン、C1〜C8アルキルチオ、アミノ、モノ−若しくはジ−(C1〜C8アルキル)アミノ、ハロC1〜C8アルキル、ハロC1〜C8アルコキシ、C1〜C8アルカノイル、C2〜C8アルカノイルオキシ、C1〜C8アルコキシカルボニル、−COOH、−CONH2、モノ−若しくはジ−(C1〜C8アルキル)アミノカルボニル、−SO2NH2、及び/又はモノ若しくはジ(C1〜C8アルキル)スルホンアミド、並びに炭素環及び複素環式基が挙げられる。任意の置換は、「0〜X個の置換基で置換された」という慣用句によっても表され、この場合、Xは可能な置換基の最大数である。ある場合によって置換された基は、0〜2、3又は4個の独立して選択された置換基で置換されている(すなわち、非置換であるか、又は記載された最大数までの置換基で置換されている)。
【0130】
「異性体」又は「立体異性体」という用語は、同じ化学構造を有するが、空間における原子又は基の配置に関して異なっている化合物を指す。
【0131】
「調節する」という用語は、所望の最終結果、例えば治療結果が得られるように、例えば、対象(例えば、動物、ヒト)において本発明の化合物への暴露に反応して標的の活性を阻害する化合物の能力の増加又は減少を指す。
【0132】
本明細書中で記載される「標的を調節(刺激、拮抗)できる化合物を得る」においてのような「得る」という用語は、当該化合物を購入、合成又は他の方法で取得することを包含することが意図される。
【0133】
本明細書中で用いられる「非経口投与」及び「非経口的に投与される」という表現は、腸内及び局所投与以外の、通常は注射による投与様式を意味し、静脈内、筋肉内、動脈内、髄腔内、包内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、被膜下、クモ膜下、脊椎内及び胸骨内注射並びに注入を包含するが、これらに限定されない。
【0134】
「ポリシクリル」又は「多環式ラジカル」という用語は、2以上の環(例えば、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/又はヘテロシクリル)のラジカルであって、2個以上の炭素が2つの隣接する環で共通であるもの(例えばこの環は「縮合環」である)を指す。隣接しない原子によって連結された環は、「架橋」環と呼ばれる。多環の環のそれぞれを、前記のような置換基、たとえば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分で置換することができる。
【0135】
「プロドラッグ」又は「プロ−ドラッグ」という用語には、インビボで代謝することができる部分を有する化合物が含まれる。一般に、プロドラッグは、エステラーゼによるか又は他のメカニズムによってインビボで代謝されて、活性薬物になる。プロドラッグの例及びそれらの使用は、当該技術分野で周知である(例えば、Berge et al.(1977)“Pharmaceutical Salts”、J. Pharm. Sci. 66:1−19を参照)。プロドラッグは、化合物の最終単離及び精製中にその場で、又は、その遊離酸形態若しくはヒドロキシルで精製された化合物を好適なエステル化剤と別に反応させることによって調製することができる。ヒドロキシル基は、カルボン酸での処理によってエステルに変換することができる。プロドラッグ部分の例としては、置換及び非置換、分枝又は非分枝低級アルキルエステル部分、(例えば、プロピオン酸(propionoic acid)エステル)、低級アルケニルエステル、ジ低級アルキル−アミノ低級アルキルエステル(例えば、ジメチルアミノエチルエステル)、アシルアミノ低級アルキルエステル(例えば、アセチルオキシメチルエステル)、アシルオキシ低級アルキルエステル(例えば、ピバロイルオキシメチルエステル)、アリールエステル(フェニルエステル)、アリール−低級アルキルエステル(例えば、ベンジルエステル)、(例えば、メチル、ハロ、若しくはメトキシ置換基で)置換されたアリール及びアリール低級アルキルエステル、アミド、低級アルキルアミド、ジ低級アルキルアミド、及びヒドロキシアミドが挙げられる。好ましいプロドラッグ部分は、プロピオン酸(propionoic acid)エステル及びアシルエステルである。インビボで他のメカニズムにより活性形態に変換されるプロドラッグも含まれる。
【0136】
化合物の「予防的有効量」という語法は、患者に1回又は複数回投与すると、本明細書中で記載する障害の予防又は治療に有効である、本明細書中の任意の式又は本明細書中で他の方法で記載された本発明の化合物の量を指す。
【0137】
「低減された毒性」という語法は、インビボで投与された場合に、本発明の化合物によって誘発される任意の望ましくない副作用の軽減を包含することが意図される。
【0138】
「スルフヒドリル」又は「チオール」という用語は、−SHを意味する。
【0139】
「対象」という用語には、本明細書中の障害に罹っている可能性があるか、又は本発明の化合物の投与から恩恵を受け得る生物、例えばヒト及びヒト以外の動物が含まれる。好ましいヒトには、本明細書中で記載されるような、神経精神障害又は関連する状態に罹っているか又は罹りやすいヒト患者が含まれる。本発明の「ヒト以外の動物」という用語には、全ての脊椎動物、例えば哺乳類、例えば齧歯類、例えばマウス、及び非哺乳類、例えば非ヒト霊長類、例えばヒツジ、イヌ、ウシ、ニワトリ、両生類、は虫類等が含まれる。
【0140】
「神経精神障害に罹りやすい」という用語は、例えば本明細書中に記載されるものをはじめとする神経精神障害を発症する危険性がある対象、すなわち、神経精神障害又はその症状に罹っている対象、神経精神障害又はその症状の家族歴又は病歴を有する対象などを包含することを意味する。
【0141】
本明細書中で用いられる「全身投与」、「全身的に投与される」、「末梢投与」及び「末梢的に投与される」という表現は、本発明の化合物(複数可)、薬物又は他の物質が、患者の系中に侵入し、したがって代謝及び他の同様のプロセスを受けるような投与、例えば皮下投与を意味する。
【0142】
本発明の化合物の「治療有効量」という語法は、患者に1回又は複数回投与すると、神経精神障害の神経精神障害及び/又は症状の治療若しくは予防において、或いはそのような神経精神障害に罹っている患者の生存性を、そのような治療が存在しない場合に予想されるよりも延長するのに有効である薬剤の量を指す。
【0143】
キラル中心の命名法に関して、「d」及び「l」構造という用語は、IUPAC Recommendationsによって定義されるとおりである。この用語の使用に関して、ジアステレオマー、ラセミ体、エピマー及びエナンチオマーは、調製物の立体化学を記載するためのそれらの通常の状況で用いられる。
【0144】
2.本発明の化合物
一態様では、本発明は、5−HT及び/又はRSK結合活性を(直接的若しくは間接的に)調節(例えば阻害若しくは刺激)することができる化合物を提供する。
【0145】
一実施形態では、本発明は、5−HT2c及び/又はRSKを刺激することができる化合物;並びにその薬剤的に許容されるエステル、塩、及びプロドラッグを提供する。ある実施形態では、化合物は式(I)の化合物である。ある実施形態では、化合物は、式(II)、(II−a)、(III)、(IV)、又は(V)の化合物である。
【0146】
ある好ましい化合物には、本明細書中に具体的に記載される化合物が含まれる:
表1:化合物:
【化19】
構造は、C1&C3での立体化学である
【表1】
【0147】
ある好ましい化合物には、本明細書中に具体的に記載される化合物が含まれる:
表2:化合物:
【化20】
【表2】
【0148】
【化21】
【0149】
本発明のさらなる化合物には、式(V)の以下の化合物が含まれる:
【化22】
【0150】
前記化合物のいずれかの全てのエナンチオマー、ジアステレオマー、ラセミ体、ラセミ混合物、エナンチオ富化(enantioenriched)混合物、及びジアステレオマー混合物が本発明によって想定される。
【0151】
突然変異誘発及びモデリングデータから、PATプロトン化(pH7.4)アミンは保存5HT2残基D3.32とイオン結合を形成する(Boothら、2009)。Aim 3におけるモデリング結果も、PATアミンがS3.36&Y7.43に近い(〜2Å)配置であることを示し、したがって、さらなるヘテロ原子を有するアミン置換基(R1、チャート1)は、これらの残基と水素結合を形成することが提示されている。さらに、Aim3モデリング結果は、PAT N−アルキル部分が立体的に寛容な受容体空間中で結合し、したがって、提案される立体的に大きなR1複素環式置換基が近くの5HT2残基とファンデルワールス相互作用を形成し得ることを示す。パラ置換PAT類似体は、アンフェタミン挙動を強力に調節し(Aim4)、したがって、パラ−ハロゲン化PATがまず合成され、続いて、先に合成されたメタ置換PATの未完了のインビボ結果であるメタ−ハロゲン化を行う。
【0152】
メタ−ハロゲン化PATは、最高の5HT2親和性及び強力な機能的選択性を示した。分子モデリングは、PATペンダントフェニル環のメタ位での置換基との相互作用に関して立体的に寛容であることを示す(Booth et al., 2009;Aim 3 results)。したがって、種々の類似体は、PAT(C4)ペンダントフェニル部分と相互作用すると仮定され、相互作用することが知られている(突然変異誘発データ)保存された5HT2残基W6.48 F6.51 F6.52及びY7.43とのさらなる水素結合及び芳香族相互作用を促進するように設計された。
【0153】
モデリング研究(Booth, 2009;Aim 3)に基づいて、さらなる類似体が、PATテトラヒドロナフチル環の芳香族部分と相互作用すると仮定され、相互作用することが知られている(突然変異誘発データ)、保存された5HT2残基V3.33、S3.36、S5.43&F6.52とさらなる水素結合を形成するように設計された。
【0154】
本発明はまた、前記化合物の薬剤的に許容される塩及びエステルに関する。
【0155】
天然に存在するか又は合成異性体は、当該技術分野で公知の幾つかの方法で分離することができる。2つのエナンチオマーのラセミ混合物を分離するための方法には、キラル固定相を用いたクロマトグラフィー(例えば、“Chiral Liquid Chromatography”, WJ. Lough, Ed. Chapman and Hall, New York(1989)を参照)が含まれる。エナンチオマーは、古典的な分割技術によって分離することができる。たとえば、ジアステレオマー塩の形成及び分別結晶を用いてエナンチオマーを分離することもできる。カルボン酸のエナンチオマーの分離に関して、エナンチオマー的に純粋なキラル塩基、例えばブルシン、キニーネ、エフェドリン、ストリキニーネなどの添加により、ジアステレオマー塩を形成することができる。或いは、ジアステレオマーエステルを、エナンチオマー的に純粋なキラルアルコール、例えばメントールを用いて形成することができ、続いてジアステレオマーエステルを分離し、加水分解して、遊離したエナンチオマー的に富化されたカルボン酸を得る。アミノ化合物の光学異性体の分離のために、キラルカルボン酸又はスルホン酸、例えばカンファースルホン酸、酒石酸、マンデル酸、又は乳酸の添加によって、ジアステレオマー塩を形成することができる。
【0156】
別の実施形態によれば、本発明は、本明細書中に記載される方法によって産生又は同定される、GPCR又はその結合ポケットと会合又は結合する化合物を提供する。
【0157】
3.本発明の化合物の使用
一実施形態では、本発明は、GPCR介在性障害に関して対象を治療するための方法であって、対象に、GPCR標的を調節(刺激、拮抗)することができる有効量の化合物を投与することによる方法を提供する。GPCR障害には、そのようなGPCRが関与する疾患及び障害が含まれる。本明細書中に記載される化合物、組成物及び方法は、たとえば、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、5−HT2b介在性疾患、高血圧)、胃腸障害(例えば、過敏性腸症候群)、及び泌尿生殖路障害(例えば、膀胱制御)をはじめとする障害の治療又は予防に有用である。ある実施形態では、対象は、哺乳類、例えば霊長類、例えばヒトである。
【0158】
一実施形態では、本発明は、ヒスタミン標的を調節(刺激、拮抗)することができる有効量の化合物を投与することによって、ヒスタミン(例えば、H1、H2、H3、H4)が関与する障害に関して対象を治療するための化合物及び方法を提供する。ヒスタミン障害には、そのようなヒスタミン(例えば、H1)が関与する疾患及び障害が含まれる。本明細書中で記載される化合物、組成物及び方法は、たとえば、呼吸困難(例えば、気管支狭窄)、下痢(GI収縮)、浮腫、及び低血圧(例えば、増大した血管透過性)、アレルギー反応、並びに本明細書中の神経精神医学的、神経変性及び神経障害をはじめとする障害の治療又は予防に有用である。
【0159】
この実施形態では、本発明の化合物は、5−HT2c又はその特定のドメインの活性を直接的又は間接的のいずれかで調節(例えば、刺激、刺激)することができる。細胞は、本発明の化合物と接触して、5−HT2cを刺激し、そして5−HT2c介在性活性を調節することができる。細胞を接触させるか、又は本発明の化合物を対象に投与することは、不必要な若しくは望ましくない5−HT2C介在性活性又は5−HT2c介在性障害に罹っているか或いは罹りやすい細胞又は対象を治療する一方法である。
【0160】
一実施形態では、5−HT2c障害に罹っているか又は罹りやすい対象を治療する方法は、それを必要とする対象に、5−HT2cの活性を直接的若しくは間接的に調節することができる治療有効量の化合物を投与し、それによって対象を治療することを含む。例示的化合物には、本明細書中に記載される化合物又は式(式I、II、II−a、III、IV又はV)が含まれる。
【0161】
したがって、一実施形態では、本発明は、5−HT2C障害に関して対象を治療する方法であって、対象に有効量の5−HT2cを刺激することができる化合物を投与することによる方法を提供する。
【0162】
別の態様では、本発明は、対象において肥満を治療又は予防する方法であって、それを必要とすると認定された対象に対して、本明細書中のいずれかの式の化合物とアンフェタミン化合物との組み合わせを投与することを含む方法を提供する。化合物を、併用して、同時に、又は連続して投与することができる。複数の態様では、アンフェタミン化合物はフェンテルミンである。
【0163】
ある実施形態では、本発明の方法は、治療有効量の本発明の化合物を、別の医薬活性化合物と組み合わせて対象に投与することを含む。使用できる他の医薬活性化合物は、Harrison’s Principles of Internal Medicine, Thirteenth Edition, Eds. T.R. Harrison et al. McGraw−Hill N.Y., NY;及びthe Physicians Desk Reference 50th Edition 1997, Oradell New Jersey, Medical Economics Co.(その全内容は参照することによって明確に本明細書中に組み込まれる)で見出すことができる。本発明の化合物及び医薬活性化合物を、同じ医薬組成物中で、又は異なる医薬組成物中で(同時に又は異なる時間に)対象に投与することができる。
【0164】
一態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に対して、5−HT2C受容体(例えば、本明細書中の化合物)を活性化することができる治療有効量の化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。一実施形態では、化合物は5−HT2c受容体を活性化することができる。別の実施形態では、化合物は、5−HT2A及び/又は5−HT2β受容体を拮抗するが、5−HT2cを活性化することができる。別の実施形態では、化合物は、5−HT2の活性化、及び/又は5−HT2A及び/又は5−HT2β受容体の拮抗が可能である。別の実施形態では、化合物は、5−HT2A及び/又は5−HT2β受容体を拮抗することができる。
【0165】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に、治療有効量の5−HT2c活性化化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0166】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に、好ましくは5−HT2a及び/又は5−HT2bと比べて選択的に5−HT2cを直接調節することにより5−HT2結合相互作用を調節することができる治療有効量の化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0167】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とすると認定された対象(すなわち、肥満対象)に、治療有効量の5−HT2c刺激化合物又は5−HT2c選択的化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0168】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、対象に、5−HT2c刺激化合物をアンフェタミン化合物と組み合わせて投与して、肥満が防止、改善又は治療されるようにする(例えば体重減少が観察され、体重増加が防止される)ことを含む方法を提供する。
【0169】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、5−HT2a及び/又は5−HT2b活性化と比べて選択的に(単独又はアンフェタミンとの組み合わせで)5−HT2cを活性化することができる有効量の化合物を、アンフェタミン化合物と組み合わせて、対象に投与して、対象が治療されるようにすることを含む方法を提供する。
【0170】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、5−HT2a又は5−HT2b受容体を刺激(及び/又は拮抗)しない(又はあまりしない)が、(例えば、5−HT2a及び/又は5−HT2bと比べて選択的に)5−HT2cを刺激することができる有効量の化合物を、アンフェタミン化合物と組み合わせて対象に投与して、対象が治療されるようにすることを含む方法を提供する。
【0171】
別の態様では、本発明は、疾患、障害又はその症状に苦しんでいるか又は罹りやすい対象を治療する方法であって、5−HT2cアゴニストとして同定された有効量の化合物(例えば、本明細書中の式の化合物)を投与することを含む方法を提供する。別の態様では、化合物は、5−HT2cアゴニスト及び5−HT2a逆アゴニスト(例えば、本明細書中の式の化合物)として同定される。別の態様では、化合物は、5−HT2c逆アゴニスト及び5−HT2a逆アゴニスト(例えば、本明細書中の式の化合物)として同定される。別の態様では、化合物は、5−HT2a又は5−HT2b(例えば、本明細書中の式の化合物)を刺激しない(又はあまりしない)、選択的な5−HT2cアゴニストとして同定される。
【0172】
別の態様では、治療方法は、対象(例えば、患者)が、そのような治療を必要としていると認定されることを含む。別の態様では、治療方法は、対象(例えば、患者)が、本明細書中の化合物又は組成物を投与されて、本明細書中に記載される疾患、障害又は症状について治療されることを含む。
【0173】
別の態様では、本発明は、アンフェタミン化合物と組み合わせた本明細書中のいずれかの式の治療有効量の化合物及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。組成物は、肥満に苦しんでいるか又は肥満になりやすい対象を治療するために処方することができ、肥満に苦しんでいるか又は肥満になりやすい対象を治療するための説明書とともにパッケージすることができる。
【0174】
ある実施形態では、本発明の化合物は、通常の抗肥満薬(例えば、脂肪吸収遮断薬)との併用療法で用いることができる。ある5−HT薬物(例えば、5−HTa又は5−HTb拮抗物質)は、それらをある患者にとって最適ではないか又は適しないものとする傾向がある望ましくない副作用を有する。すなわち、それらは心臓血管(例えば、心臓弁膜症、肺高血圧症、新毒性)又は精神医学的な望ましくない及び/又は生命を脅かす副作用特性を示す。
【0175】
一実施形態では、本発明の化合物は、RSK又はその特定のドメインの活性を直接的又は間接的にいずれかで調節(例えば阻害)することができる。細胞を本発明の化合物と接触させて、RSKを阻害し、RSK介在性活性を調節することができる。細胞を接触させるか、又は本発明の化合物を対象に投与することは、不必要又は望ましくないRSK介在性活性又はRSKが関与する障害に苦しんでいるか、又は影響を受けやすい細胞又は対象を治療する一方法である。
【0176】
一実施形態では、RSKが関与する障害に罹っているか、又は罹りやすい対象を治療する方法は、それを必要とする対象に、RSKの活性を直接的又は間接的に調節することができる治療有効量の化合物を投与し、それによって対象を治療することを含む。例示的化合物には、本明細書中に記載される化合物又は式(式I、II、II−a、III、IV、又はV)が含まれる。
【0177】
したがって、一実施形態では、本発明は、RSKが関与する障害について対象を治療する方法であって、RSK−1、RSK−3、又はRSK−4を阻害することができる有効量の化合物を対象に投与することによる方法を提供する。
【0178】
別の態様では、本発明は、対象における癌(例えば、胸部及び前立腺)、HIV、又はコフィン・ローリー症候群を治療又は予防する方法であって、それを必要とすると認定された対象に、本明細書中のいずれかの式の化合物及びさらなる治療化合物(例えば、抗がん剤、抗HIV剤、コフィン・ローリー症候群剤)の組み合わせを投与することを含む方法を提供する。化合物を、併用して、同時に、又は連続して投与することができる。
【0179】
ある実施形態では、本発明の方法は、対象に、治療有効量の本発明の化合物を別の医薬活性化合物と組み合わせて投与することを含む。使用できる他の医薬活性化合物は、Harrison’s Principles of Internal Medicine, Thirteenth Edition, Eds. T.R. Harrison et al. McGraw−Hill N.Y., NY;及びthe Physicians Desk Reference 50th Edition 1997, Oradell New Jersey, Medical Economics Co.,(その全内容は、参照することによって明確に本明細書中に組み込まれる)で見いだすことができる。本発明の化合物及び医薬活性化合物は、同じ医薬組成物中又は異なる医薬組成物中で(同時に又は異なる時点で)対象に投与することができる。
【0180】
別の態様では、治療方法は、対象(例えば、患者)がそのような治療を必要であると認定されることを含む。別の態様では、治療方法は、対象(例えば、患者)に本明細書中の化合物又は組成物を投与して、その対象が、本明細書中に記載される疾患、障害又は症状について治療されるようにすることを含む。
【0181】
本発明の別の目的は、RSK介在性障害の治療において使用するための医薬の製造における、本明細書中に記載される(例えば、本明細書中の任意の式の)化合物の使用である。本発明の別の目的は、RSKが関与する障害の治療において使用するための、(例えば、本明細書中の任意の式の)本明細書中に記載される化合物の使用である。
【0182】
本発明の化合物の治療有効量又は予防有効量は、当業者として、医師又は獣医師(「担当臨床医」)が、公知技術を使用することによるか、又は類似の状況下で得られる結果を観察することによって決定することができる。投与量は、担当臨床医の判断で、患者の要求;治療される状態の重篤度及び用いられる特定の化合物に応じて変えることができる。治療有効量又は用量、及び予防有効量又は用量の決定において、これらに限定されるものではないが:関係する特定の5−HT2c及び/又はRSK障害;特定の薬剤の薬力学的特性並びにその投与様式及び経路;治療の所望の時間経過;哺乳類の種;そのサイズ、年齢、及び全体的な健康;関係する特定の疾患;疾患の程度又は疾患の関与又は重篤度;個々の患者の反応;投与される特定の化合物;投与様式;投与される製剤の生体利用性;選択される投与計画;併用療法の種類(すなわち、本発明の化合物と他の同時投与される治療薬との相互作用);及び他の関連する状況をはじめとする多くの因子が担当臨床医によって考慮される。
【0183】
治療は、より少ない用量から始めることができ、この量は、化合物の最適用量よりも少ない。その後、その状況下で最適の効果に達するまで、投与量を少しずつ増やすことができる。便宜上、所望により、総一日量を分割し、一日中で数回に分けて投与することができる。本発明の化合物の治療有効量及び予防有効量は、1日につき体重1キログラムあたり約0.1ミリグラム(mg/kg/日)〜約100mg/kg/日で変化すると予想される。
【0184】
動物、例えば、イヌ、ニワトリ、及び齧歯類において、5−HT2c及び/又はRSK障害の予防又は治療に有効であることが見いだされた化合物は、ヒトにおける5−HT2c及び/又はRSK障害の治療にも有用である可能性がある。ヒトにおける5−HT2c及び/又はRSK疾患の治療をする当業者らは、動物実験で得られたデータに基づいて、ヒトへの当該化合物の投与量及び経路がわかるであろう。一般的に、ヒトにおける投与量及び投与経路は、動物においてと類似していると予想される。
【0185】
5−HT2C及び/又はRSK障害の予防的治療を必要とする患者の認定は、十分に当業者の技術及び知識の範囲内である。対象の方法により治療することができる5−HT2C及び/又はRSK障害を発症する危険性がある患者を特定するためのある方法、たとえば家族歴、及び対象患者における疾患状態の発症に関連する危険因子の存在は、医術で理解される。当該技術に熟達した臨床医は、そのような候補患者を、たとえば、臨床試験、健康診断及び病歴/家族歴の使用により、容易に特定することができる。
【0186】
対象における治療の有効性を評価する方法は、当該技術分野で周知の方法(例えば、5−HT2C及び/又はRSK障害のマーカーのレベルを測定すること)によって5−HT2C及び/又はRSK障害の前処理の程度を測定し、次いで治療有効量の本明細書中に記載される化合物を本発明にしたがって対象に投与することを含む。化合物の投与の適切な期間(例えば、1日、1週、2週、1か月、6か月)の後に、5−HT2C及び/又はRSK障害の程度を再度測定する。5−HT2C及び/又はRSK障害の程度又は侵襲性の調節(たとえば、低減)は、治療の有効性を示す。5−HT2C及び/又はRSK障害の程度又は侵襲性は、治療を通して定期的に測定することができる。たとえば、5−HT2C及び/又はRSK障害の程度又は侵襲性を、数時間ごと、数日ごと又は数週間ごとにチェックして、治療のさらなる有効性を評価することができる。5−HT2C及び/又はRSK障害の程度又は侵襲性の減少は、治療が有効であることを示す。記載した方法を用いて、5−HT2C及び/又はRSK障害の調節化合物を用いた治療から恩恵を得る可能性がある患者をスクリーニング又は選択することができる。
【0187】
対象における治療の有効性を評価する方法は、当該技術分野で周知の方法による肥満(太りすぎ)の前治療の程度を測定し、次いで治療有効量の本明細書中に記載される化合物を単独又はそれぞれとアンフェタミン化合物との組み合わせで本発明にしたがって対象に投与することを含む。化合物の投与後の適切な期間(例えば、1日、1週、2週、1か月、6か月)の後、肥満(太りすぎ)の程度を再度測定する。太りすぎの程度の調節(たとえば減少)は、治療の有効性を示す。肥満の程度又は侵襲性は、治療を通して定期的に測定することができる。たとえば、太りすぎの程度又は侵襲性を、数時間ごと、数日ごと、又は数週間ごとにチェックして、治療の有効性をさらに評価することができる。太りすぎの程度又は侵襲性の減少は、治療が有効であることを示す。記載される方法を用いて、アンフェタミン化合物との組み合わせで選択的に5−HT2c(対5−HT2a、b)受容体を活性化することができる調節化合物での治療から恩恵を受ける可能性がある患者をスクリーニング又は選択することができる。
【0188】
本明細書中で用いられるように、「対象から生物学的試料を得る」には、本明細書中に記載される方法で使用するための試料を得ることが含まれる。生物学的試料は前述されている。
【0189】
さらに別の態様は、5−HT2C及び/又はRSK又はその特定のドメインの相互作用を調節する化合物を同定するための方法を提供する。この方法は、試験化合物の存在下及び/又は非存在下で、5−HT2C及び/又はRSK又はその特定のドメインの結晶構造(場合によってアポ形若しくは複合体化)を得るか、或いは5−HT2C及び/又はRSK又はその特定のドメインの結晶構造(場合によってアポ形若しくは複合体化)に関する情報を得ることを含み得る。化合物を次いで5−HT2C及び/又はRSK構造、又はその特定のドメイン(例えば、結晶構造の結合部位)中又は上でコンピュータでモデル化して、5−HT2C及び/又はRSK又はその特定のドメイン及び試験化合物間の相互作用の安定化を予想することができる。潜在的な調節化合物が同定されたら、細胞アッセイ、たとえば本明細書中で特定されたもの及び当該技術分野で公知の競合アッセイを用いてその化合物をスクリーニングすることができる。このようにして特定された化合物は、治療薬として有用である。
【0190】
別の態様では、本発明の化合物は、治療有効量で、薬剤的に許容される担体又は希釈剤とともにパッケージされる。組成物は、5−HT2C及び/又はRSK障害に罹っているか又は罹りやすい対象を治療するために処方することができ、そして5−HT2C及び/又はRSK障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0191】
別の態様では、本発明は、5−HT2C及び/又はRSK疾患を調節するための方法を提供する。一実施形態では、本発明による5−HT2C(又は5−HT2C障害)を調節する方法は、細胞と、5−HT2C(若しくは5−HT2C障害)、又はその特定のドメインを調節することができる化合物とを接触させることを含む。一実施形態では、本発明によるRSK(又はRSK障害)を調節する方法は、細胞と、RSK(若しくはRSK障害)、又はその特定のドメインを調節することができる化合物とを接触させることを含む。いずれの実施形態においても、接触は、インビトロで、たとえば、細胞を取り巻く流体、たとえば、細胞がその中で生きているか又は存在している成長媒体に化合物を添加することによってであってもよい。接触は、化合物を細胞に直接接触させることによってであってもよい。別法として、接触は、インビボで、たとえば、投与経路に応じて、たとえば投与後に、対象中に化合物を通過させることによってであってもよく、化合物は、消化管又は血流中で運ばれる可能性があるか、又は処理を必要とする細胞に直接適用若しくは投与することができる。
【0192】
別の態様では、対象における5−HT2C及び/又はRSK障害を阻害する方法は、有効量の本発明の化合物(すなわち、本明細書中で記載される化合物)を対象に投与することを含む。投与は、製薬分野で公知の任意の投与経路によってであってよい。対象は、5−HT2C及び/又はRSK障害を有する可能性があるか、5−HT2C及び/又はRSK障害を発症する危険性があるか、又は5−HT2C及び/又はRSK障害に対する感受性を増大させ得る状態に対する予定された暴露又は予期せぬ暴露の前に予防的治療を必要とする可能性がある。
【0193】
一態様では、本明細書中の化合物で治療される対象の経過をモニタリングする方法は、5−HT2C及び/又はRSK障害の治療前状態(例えば、進行、標的特性、マーカー特性)を測定し、治療有効量の本明細書中の化合物を対象に投与し、そして当該化合物での治療の初期期間後の5−HT2C及び/又はRSK障害の状態(例えば、進行、標的特性、マーカー特性)を測定することを含み、この場合、状態の調節は、治療の有効性を示す。
【0194】
対象は、5−HT2C及び/又はRSK障害の危険性の可能性があり、5−HT2C及び/又はRSK障害の症状を示す可能性があり、5−HT2C及び/又はRSK障害に罹りやすい可能性があり、及び/又は5−HT2C及び/又はRSK障害を有すると診断されている可能性がある。
【0195】
状態の調節が、対象が治療に対して有利な臨床反応を有し得ることを示す場合、対象は、当該化合物で治療することができる。たとえば、対象に治療有効量の化合物を投与することができる。
【0196】
別の態様では、試験化合物を評価するための方法は、5−HT2C及び/又はRSK又はその特定のドメインを試験化合物(複合体)と接触させ、そして接触後の結合相互作用を評価することを含み、この場合、参考値に対する複合体の安定性における変化は、複合体の安定性を調節することの表れである。
【0197】
5−HT2C及び/又はRSK又はその特定のドメイン複合体をコンピュータでモデル化することができるか、或いは細胞から単離されるか、細胞若しくは組換え発現系から組み換え発現されるか、精製されるか、若しくは単離されるか、又は細胞若しくは組換え発現系から部分的に精製されるか若しくは単離された細胞内の複合体であり得る。
【0198】
本発明のキットは、対象における5−HT2C及び/又はRSK障害を治療するためのキットを含む。キットは、本発明の化合物,たとえば本明細書中で記載される化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、並びに使用説明書を含み得る。使用説明書は、用量、送達方法、キットの保存などに関する情報を含み得る。キットは、試薬、たとえば、試験化合物、緩衝液、培地(例えば、細胞増殖培地)、細胞なども含み得る。試験化合物は、既知化合物又は新たに発見された化合物、たとえば化合物のコンビナトリアルライブラリを含み得る。1以上の本発明のキットを合わせてパッケージすることができ、たとえば、5−HT2C及び/又はRSK障害についての治療の有効性を評価するためのキットを、本発明による5−HT2C及び/又はRSK障害に関して治療される対象の経過をモニタリングするためのキットとともにパッケージすることができる。
【0199】
本発明の方法は、培養物中、たとえばインビトロ若しくはエクスビボの細胞、又は動物対象中に存在する細胞、たとえばインビボの細胞に関して実施することができる。本発明の化合物は、まず、形質転換細胞などの細胞の一次培養を用いてインビトロで試験することができる。
【0200】
本発明の方法は、培養物中、たとえばインビトロ若しくはエクスビボの細胞、又は動物対象中に存在する細胞、たとえばインビボの細胞に関して実施することができる。本発明の化合物は、まず、齧歯類胎仔(たとえば、米国特許第5,179,109号−胎仔ラット細胞培養を参照)、又は他の哺乳類(たとえば、米国特許第5,089,517号−胎仔マウス組織培養を参照)又は非哺乳動物の動物モデルからの気道由来の細胞を用いてインビトロで試験することができる。
【0201】
或いは、本発明の化合物の効果は、動物モデルを用いてインビボで特性化することができる。
【0202】
4.医薬組成物
本発明は、有効量の化合物及び薬剤的に許容される担体を含む医薬組成物も提供する。さらなる実施形態では、有効量は、前述のように、5−HT2C及び/又はRSK障害を治療するために有効である。
【0203】
一実施形態では、本発明の化合物は、薬剤的に許容される処方、たとえば、薬剤的に許容される処方が対象に投与された後、少なくとも12時間、24時間、36時間、48時間、1週間、2週間、3週間、又は4週間、本発明の化合物を対象に持続送達する薬剤的に許容される処方を用いて対象に投与される。
【0204】
ある実施形態では、これらの医薬組成物は、対象への局所又は経口投与に適している。他の実施形態では、以下で詳細に記載するように、本発明の医薬組成物は、特に:(1)経口投与(たとえば、飲薬(水性若しくは非水性溶液又は懸濁液)、錠剤、ボーラス、粉末、顆粒、ペースト);(2)非経口投与(例えば滅菌溶液若しくは懸濁液として皮下、筋肉内又は静脈内注射による);(3)局所適用(たとえば、皮膚に適用されるクリーム、軟膏又はスプレー);(4)膣内又は直腸内で(例えばペッサリー、クリーム又はフォームとして);或いは(5)エアゾル(例えば化合物を含有する水性エアゾル、リポソーム製剤又は固体粒子として)のために適用されるものをはじめとする、固体又は液体形態での投与用に処方することができる。
【0205】
「薬剤的に許容される」という表現は、健全な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応又は他の問題若しくは合併症がなく、ヒト及び動物の組織と接触して使用するのに適し、妥当な損益比に見合う、本発明の化合物、そのような化合物を含む組成物、及び/又は投与形態を指す。
【0206】
「薬剤的に許容される担体」という表現は、1つの器官、又は身体の部分から、別の器官、若しくは身体の部分への対象の化学物質の運搬若しくは輸送に関与する、薬剤的に許容される物質、組成物又はビヒクル、たとえば液体若しくは固体フィラー、希釈剤、賦形剤、溶媒又は封入材料を包含する。各担体は、処方の他の成分と適合性であり、患者に有害でないという意味で、「許容される」。薬剤的に許容される担体として機能し得る材料の数例としては:(1)糖、たとえばラクトース、グルコース及びスクロース;(2)デンプン、たとえばコーンスターチ及びジャガイモデンプン;(3)セルロース、及びその誘導体、たとえばカルボキシメチルセルロースナトリウム、エチルセルロース及び酢酸セルロース;(4)トラガカント粉末;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤、たとえばカカオバター及び坐薬ワックス;(9)油、たとえばピーナッツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、コーン油及び大豆油;(10)グリコール、たとえばプロピレングリコール;(11)ポリオール、たとえばグリセリン、ソルビトール、マンニトール及びポリエチレングリコール;(12)エステル、たとえばオレイン酸エチル及びラウリン酸エチル;(13)寒天;(14)緩衝剤、たとえば水酸化マグネシウム及び水酸化アルミニウム;(15)アルギン酸;(16)パイロジェンフリー水;(17)等張性生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸塩緩衝液;及び(21)医薬処方で用いられる他の非毒性適合性物質が挙げられる。
【0207】
湿潤剤、乳化剤及び潤滑剤、たとえばラウリル硫酸ナトリウム及びステアリン酸マグネシウム、並びに着色剤、放出剤、コーティング剤、甘味料、矯味矯臭剤及び香料、保存料及び酸化防止剤も組成物中に存在してもよい。
【0208】
薬剤的に許容される酸化防止剤の例としては:(1)水溶性酸化防止剤、たとえばアスコルビン酸、塩酸システイン、硫酸水素ナトリウム、メタ亜硫酸水素ナトリウム、亜硫酸ナトリウムなど;(2)油溶性酸化防止剤、たとえばパルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、アルファ−トコフェロールなど;及び(3)金属キレート剤、たとえばクエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などが挙げられる。
【0209】
本発明の化合物を含む組成物には、経口、鼻、局所(頬側及び舌下を包含する)、直腸、膣、エアゾル及び/又は非経口投与に適したものが含まれる。組成物は、便宜上、単位投与形態で提供することができ、調剤の技術分野で周知の方法によって調製することができる。単一投与形態を得るために担体材料と組み合わせることができる活性成分の量は、治療される宿主、特定の投与様式に応じて変わるであろう。単一投与形態を得るために担体材料と組み合わせることができる活性成分の量は、一般的に、治療効果をもたらす化合物の量であろう。一般的に、100パーセントのうち、この量は、活性成分の約1パーセント〜約99パーセント、好ましくは約5パーセント〜約70パーセント、さらに好ましくは約10パーセント〜約30パーセントの範囲である。
【0210】
これらの組成物の調製法は、本発明の化合物(複数可)を担体、及び場合によって1以上の補助成分と会合させるステップを含む。一般的に、処方は、本発明の化合物を液体担体、又は固体担体微粉末、又は両者と均一かつ密接に会合され、次いで必要ならば生成物を成形することにより調製される。
【0211】
経口投与に適した本発明の組成物は、カプセル、カシェ剤、丸薬、錠剤、ロゼンジ(フレーバー付主成分、通常、スクロース及びアカシア又はトラガカントを使用)、粉末、顆粒の形態で、又は水性若しくは非水性液体中溶液若しくは懸濁液として、又は水中油若しくは油中水液体エマルジョンとして、又はエリキシル若しくはシロップとして、又はパステル(不活性基剤、たとえばゼラチン及びグリセリン、又はスクロース及びアカシアを使用)として及び/又は洗口剤などとしてであってよく、それぞれは、あらかじめ決められた量の本発明の化合物(複数可)を活性成分として含む。化合物は、ボーラス、舐剤又はペーストとして投与することもできる。
【0212】
経口投与用の本発明の固体投与形態(カプセル、錠剤、丸薬、糖衣剤、粉末、顆粒など)において、活性成分を、1以上の薬剤的に許容される担体、たとえば、クエン酸ナトリウム又はリン酸二カルシウム、及び/又は以下のいずれか:(1)フィラー又は増量剤、たとえばデンプン、ラクトース、スクロース、グルコース、マンニトール、及び/又はケイ酸;(2)結合剤、たとえば、カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロース及び/又はアカシア;(3)湿潤剤、たとえばグリセロール;(4)崩壊剤、たとえば寒天−寒天、炭酸カルシウム、ジャガイモ若しくはタピオカデンプン、アルギン酸、ある種のケイ酸塩、及び炭酸ナトリウム;(5)溶解遅延剤、たとえばパラフィン;(6)吸収促進剤、たとえば第四アンモニウム化合物;(7)湿潤剤、たとえば、アセチルアルコール及びグリセロールモノステアレート;(8)吸収剤、たとえばカオリン及びベントナイトクレイ;(9)潤滑剤、たとえばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、及びそれらの混合物;並びに(10)着色剤と混合する。カプセル、錠剤及び丸薬の場合、医薬組成物は緩衝剤も含み得る。類似の種類の固体組成物は、ラクトース又は乳糖などの賦形剤、並びに高分子量ポリエチレングリコールなどを用いて、ソフト及びハード充填ゼラチンカプセル中フィラーとして用いることもできる。
【0213】
場合によって1以上の補助成分とともに圧縮又は成形することによって錠剤を作成することができる。圧縮錠は、結合剤(たとえば、ゼラチン若しくはヒドロキシプロピルメチルセルロース)、潤滑剤、不活性希釈剤、保存料、崩壊剤(たとえば、デンプングリコール酸ナトリウム若しくは架橋カルボキシメチルセルロースナトリウム)、表面活性剤又は分散剤を用いて調製することができる。成形錠剤は、好適な機械中で、不活性液体希釈剤で湿らせた活性成分粉末の混合物を成形することによって作成することができる。
【0214】
錠剤、及び糖衣剤、カプセル、丸薬及び顆粒などの他の固体投与形態の本発明の医薬組成物は、場合によって、刻み目をつけるか、又は腸溶コーティング及び他の医薬処方技術分野で周知の他のコーティングなどのコーティング及びシェルを用いて調製することができる。それらは、たとえば、所望の放出特性を得るために様々な割合のヒドロキシプロピルメチルセルロース、他のポリマーマトリックス、リポソーム及び/又は微小球を用いて、その中の活性成分の遅延又は制御放出をもたらすように処方することもできる。それらは、細菌保持フィルターを通した濾過によるか、又は滅菌水、又は他の滅菌注射可能媒体中に使用直前に溶解させることができる滅菌固体組成物の形態中に滅菌剤を組み入れることによって、滅菌することができる。これらの組成物は、場合によって、不透明化剤を含んでもよく、胃腸管のある部分でのみ、又は胃腸管のある部分で優先的に、場合によって遅延された方法で、活性成分を放出する組成物であってもよい。使用できる包埋組成物の例としては、ポリマー物質及びワックスが挙げられる。活性成分は、適切ならば前記賦形剤とともにミクロカプセル化された形態であってもよい。
【0215】
本発明の化合物の経口投与用液体投与形態には、薬剤的に許容されるエマルジョン、ミクロエマルジョン、溶液、懸濁液、シロップ及びエリキシルが含まれる。活性成分に加えて、液体投与形態は、当該技術分野で通常用いられる不活性希釈剤、たとえば、水又は他の溶媒、可溶化剤及び乳化剤、たとえば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、コーン油、胚芽油、オリーブ油、ヒマシ油及びゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコール及びソルビタンの脂肪酸エステル、並びにそれらの混合物を含んでもよい。
【0216】
不活性希釈剤に加えて、経口組成物は、湿潤剤、乳化剤及び懸濁化剤、甘味料、矯味矯臭剤、着色剤、香料及び保存料などのアジュバントを含むことができる。
【0217】
懸濁液は、本発明の活性化合物に加えて、たとえば、エトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトール及びソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天−寒天及びトラガカント、並びにそれらの混合物などの懸濁化剤を含んでもよい。
【0218】
直腸又は膣投与用の本発明の医薬組成物は、坐薬として提供することができ、坐薬は、1以上の本発明の化合物を、たとえば、カカオバター、ポリエチレングリコール、坐薬ワックス又はサリチレートを含む1以上の好適な非刺激性賦形剤又は担体と混合することによって調製することができ、室温で固体であるが、体温で液体であり、したがって直腸又は膣腔中で融解し、活性剤を放出する。
【0219】
膣投与に適した本発明の組成物としては、当該技術分野で適切であると知られている担体を含むペッサリー、タンポン、クリーム、ゲル、ペースト、フォーム又はスプレー処方が挙げられる。
【0220】
本発明の化合物の局所又は経皮投与用投与形態には、粉末、スプレー、軟膏、ペースト、クリーム、ローション、ゲル、溶液、貼付剤及び吸入剤が含まれる。本発明の活性化合物は、無菌条件下で、薬剤的に許容される担体と、及び任意の保存料、緩衝液、又は必要とされ得るプロペラントと混合することができる。
【0221】
軟膏、ペースト、クリーム及びゲルは、本発明の化合物に加えて、賦形剤、たとえば動物性及び植物性脂、油、ワックス、パラフィン、デンプン、トラガカント、セルロース誘導体、ポリエチレングリコール、シリコーン、ベントナイト、ケイ酸、タルク及び酸化亜鉛、又はそれらの混合物を含んでもよい。
【0222】
粉末及びスプレーは、本発明の化合物に加えて、賦形剤、たとえばラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム及びポリアミド粉末、又はこれらの物質の混合物を含むことができる。スプレーは、慣例的なプロペラント、たとえばクロロフルオロ炭化水素及び揮発性非置換炭化水素、たとえばブタン及びプロパンをさらに含むことができる。
【0223】
本発明の化合物(複数可)は、別法として、エアゾルにより投与することができる。これは、当該化合物を含む水性エアゾル、リポソーム製剤又は固体粒子を調製することによって達成される。非水性(例えば、フルオロカーボンプロペラント)懸濁液を使用することができる。音波ネブライザーが好ましい。なぜなら、これらは化合物が結果として分解される可能性があるせん断に暴露されることを最小限に抑えるからである。
【0224】
通常、水性エアゾルは、薬剤の水性溶液又は懸濁液を通常の薬剤的に許容される担体及び安定剤とともに処方することによって調製される。担体及び安定剤は、特定の化合物の要件によって変わるが、典型的には、非イオン性界面活性剤(Tween、Pluronic、又はポリエチレングリコール)、血清アルブミンなどの無害タンパク質、ソルビタンエステル、オレイン酸、レシチン、グリシンなどのアミノ酸、緩衝液、塩、糖又は糖アルコールを含む。エアゾルは、一般的に、等張性溶液から調製される。
【0225】
経皮貼付剤は、本発明の化合物の身体への制御送達を提供するさらなる利点を有する。そのような投与形態は、適切な媒体中に当該薬剤を溶解又は分散させることによって作成することができる。吸収促進剤を用いて、皮膚を越える活性成分の流動を増大させることもできる。そのような流動速度は、速度制御膜を提供するか、又はポリマーマトリックス若しくはゲル中に活性成分を分散させるかのいずれかによって制御することができる。
【0226】
眼用処方、眼軟膏、粉末、溶液なども本発明の範囲内に含まれることが想定される。
【0227】
非経口投与に適した本発明の医薬組成物は、1以上の本発明の化合物を1以上の薬剤的に許容される滅菌等張性水性若しくは非水性溶液、分散液、懸濁液又はエマルジョン、或いは使用直前に滅菌注射可能な溶液又は分散液中に再構成することができる滅菌粉末と組み合わせて含み、酸化防止剤、緩衝液、静菌剤、処方を意図されるレシピエントの血液と等張性にする溶質、又は懸濁剤若しくは増粘剤を含んでもよい。
【0228】
本発明の医薬組成物において用いることができる好適な水性及び非水性担体の例としては、水、エタノール、ポリオール(たとえばグリセロール、プロピレングリコール、ポリエチレングリコールなど)、及びそれらの好適な混合物、植物性油、たとえばオリーブ油、並びに注射可能な有機エステル、たとえばオレイン酸エチルが挙げられる。適切な流動性は、たとえば、コーティング材料、たとえばレシチンの使用により、分散液の場合は必要とされる粒子サイズの維持により、そして界面活性剤の使用により、維持することができる。
【0229】
これらの組成物は、保存料、湿潤剤、乳化剤及び分散剤などのアジュバントを含んでもよい。微生物の作用の予防は、様々な抗菌剤及び抗真菌剤、たとえば、パラベン、クロロブタノール、フェノールソルビン酸などを含めることによって確実にすることができる。等張剤、たとえば糖、塩化ナトリウムなどを組成物中に含めることも望ましい場合がある。加えて、注射可能な医薬形態の長時間にわたる吸収は、アルミニウムモノステアレート及びゼラチンなどの吸収を遅らせる薬剤を含めることによってもたらされ得る。
【0230】
場合によっては、薬物の効果を延長するために、皮下又は筋肉内注射からの薬物の吸収を遅らせることが望ましい。これは、水溶性が低い結晶性又はアモルファス材料の懸濁液の使用により達成することができる。そして薬物の吸収速度は、その溶解速度に依存し、これは次に結晶サイズ及び結晶形態に依存し得る。或いは、非経口投与された薬物形態の遅延吸収は、薬物を油性ビヒクル中に溶解又は懸濁させることによって達成される。
【0231】
注射可能なデポー形態は、ポリラクチド−ポリグリコリドなどの生分解性ポリマー中本発明の化合物のマイクロカプセル化マトリックスを形成することによって作製される。薬物のポリマーに対する割合に応じて、そして用いられる特定のポリマーの性質に応じて、薬物放出速度を制御することができる。生分解性ポリマーの例としては、ポリ(オルトエステル)及びポリ(無水物)が挙げられる。デポー注射可能な処方は、身体組織と適合性であるリポソーム又はミクロエマルジョン中に薬物をトラップすることによっても調製される。
【0232】
本発明の化合物を医薬としてヒト及び動物に投与する場合、それらはそのまま又はたとえば、0.1〜99.5%(更に好ましくは、0.5〜90%)の活性成分を薬剤的に許容される担体と組み合わせて含む医薬組成物として与えることができる。
【0233】
選択された投与経路に関係なく、好適な水和形態、及び/又は医薬本発明の組成物で使用できる本発明の化合物は、当業者に公知の通常の方法によって薬剤的に許容される投与形態に処方される。
【0234】
本発明の医薬組成物中の活性成分の投与の実際の投与量レベル及び時間経過を変えて、患者に対して毒性でない、特定の患者、組成物、及び投与様式に対する所望の治療反応を達成するために有効である活性成分の量を得ることができる。用量範囲の一例は、1日あたり0.1〜10mgである。
【0235】
本発明に関して本発明の化合物の好ましい用量は、患者が耐えることができ、重篤な副作用を発現しない最大量である。好ましくは、本発明の化合物を体重1キログラムあたり約0.001mg〜約100mg、約0.001〜約10mg/kg又は約0.001mg〜約100mg/kg体重の濃度で投与する。前記値の中間の範囲は本発明の一部であることが意図される。
【0236】
合成スキーム
本発明の化合物を以下の合成スキームにしたがって作成することができる。
1)
【化23】
スキームA:チャート1におけるN−置換PATの合成。スチレン又はハロスチレン7の[2]との反応により、4−置換−テトラレン−2−オールフェニルアセテート8を得る。8の還元により、テトラール−2−オール9を得る。トランス−(2S,4R)−N−置換PAT10は、9のトシル化とそれに続く環状アミン(ピロリジン、ピペリジン等)を用いた環状アミンSΝ2反転及びキラル固定相(CSP)−HPLCによるエナンチオマー分離によって調製される(Vincek & Booth 2009)。
【化24】
スキームB:別の方法。フェニルアセトン11のベンズアルデヒド又はハロベンズアルデヒド12とのKOHの存在下でのアルドール縮合により、エノン誘導体13を得る。テトラロン中間体14は、すでに報告されているように、13のポリリン酸での処理から得られる(Bucholtz 1999)。トランス−(2S,4R)−PAT10は、14の還元的アミノ化(Abdel−Magid 1996)により調製され、続いてCSP−HPLCを行う。
【化25】
スキームC:チャート2におけるメタ置換PATの合成。本発明者らは(Vincek & Booth, 2009)、メタ置換−フェニルPATを得るためのいくつかの方法を公開した。容易に入手可能な試薬1及び[2]により、汎用3−置換−フェニルテトラレン−2−オールフェニルアセテート3を得る。3上の3−置換アリール及びエノールフェニルアセテート官能基は、これらの分子を不斉転換及び様々な有機合成に有用にする。(a)不斉移動水素化、(b)トシル化、及び(c)ジメチルアミンを用いたSN2反転の3−ステップで、エナンチオ富化シス−(2R,4R)−4、シス−(2R−4R)−5;トランス−(2R−4S)及び(2S,4R)生成物がCSP−HPLCにより得られる。
【化26】
スキームD:別の方法。反応(d)における4−(3−ブロモフェニル)−テトラレン−2−オールフェニルアセテート3a(すなわち、スキーム1中の3(式中、R1=Br))の置換ボロン酸とのスズキカップリングによって、4−(3−置換フェニル)−テトラレン−2−オールフェニルアセテート6を得る(Vincek & Booth, 2009)。多くのボロン酸スズキカップリング試薬が利用可能である(Chemfiles, 2007)。
【化27】
スキームE:チャート3におけるテトラヒドロナフチル置換基を有するPATの合成。
容易に調製される1及び市販の2により、無水トリフルオロ酢酸(TFAA)を用いて置換フェニルテトラレン−2−オールフェニルアセテート3を得た(Vincek & Booth, 2009)。化合物3を水素化ホウ素ナトリウムにより還元して、ラセミのシス−オール4を得、これを直接トシル化し、トランス−ジメチルアミン5に変換する。CSP−HPLCを用いて、(+)−トランス−(2R,4S)−及び(−)−トランス−(2S,4R)−5を分離する。これらの新規PAT類似体を三臭化ホウ素により還元して、別のシリーズの新規化合物6にすることができた。ミツノブ反応により4を7に変換し、続いて,トシル化、アミノ化及びCSP−HPLCエナンチオマー分離を行って、(+)−及び(−)−シス−PAT8及び9を得る。
【化28】
スキームF:別の方法:本発明者らは以前にスキーム6を使用し(Bucholtz et al., 1999;Booth et al., 2009)、この場合、ラセミのシス及びラセミのトランス異性体をカラムクロマトグラフィーにより分離し、続いてジアステレオマー再結晶又はCSP−HPLCを行って、1つのエナンチオマーPAT類似体を得る。
【0237】
実施例
本発明を説明するが、本発明の範囲を制限しないことを意図する以下の実施例により、本発明をさらに説明する。
【0238】
実施例1:化学物質
すべての試薬は、商業的供給元から入手し、さらに精製することなく使用した。1H及び13C NMRを対応して共振周波数400及び100MHzで、CDCl3中で集めた。化学シフトをテトラメチルシランからのppmで報告する。フラッシュカラムクロマトグラフィーを、シリカゲル60(230〜400メッシュ)を用いて実施した。融点は、水銀温度計を備えたMel−Temp装置で測定した。HPLCキラル分離は、RegisCell(商標)(5μm、25cm×10mm i.d.)カラムを備えたHPLC装置によって測定した。いくつかの1H及び13C NMRスペクトルの小さなピーク(<5%)は、位置異性体の形成が原因であった。
【0239】
[3H]−ケタンセリン(比活性72.2Ci/ミリモル)及びミオ−[2−3H(N)]−イノシトール(比活性18.5Ci/ミリモル)をPerkin−Elmer Life Science(Boston, MA)から購入し、[N6−メチル−3H]−メスレルギン(比活性72.0Ci/ミリモル)をAmersham Biosciences(GE healthcare, Piscataway, NJ)から購入した。他の化合物は高純度でSigma−Aldrich(St. Louis, MO)から入手した。
【0240】
クローン細胞培養及びトランスフェクション
すべての細胞系をATCCの示唆にしたがって維持した。チャイニーズハムスター卵巣細胞(CHO−Kl、ATCC CCL−61)は10%のウシ胎仔血清、1%の重炭酸ナトリウム(Mediatech 25−035−CI)、10IU/mlのペニシリン及び10ug/mlのストレプトマイシンを追加したHamのF−12培地中、並びにヒト胚腎臓(HEK)293は、1.5g/Lの重炭酸ナトリウム、0.1mMの非必須アミノ酸、及び1.0mMのピルビン酸ナトリウム(90%)と10%ウシ胎仔血清、10IU/mlのペニシリン及び10ug/mlのストレプトマイシンを含むように調節された2mMのL−グルタミンを含む最小必須培地(イーグル)(MEM)中。細胞を、5%のCO2を含む加湿インキュベータ中37℃で増殖させる。ヒトHT2A、5−HT2B、及び5−HT2C(野生型)をコード化するcDNAを、クローン細胞の一過性トランスフェクションのためにUMR(Rolla, MO)から購入する。放射性受容体結合アッセイのために、5−HT2A、5−HT2B、及び5−HT2C受容体膜をトランスフェクトされたCHO−K1細胞から調製する。PLC/IP形成の活性を測定する機能的アッセイのために、5−HT2A及び5−HT2C受容体についてはトランスフェクトされたCHO−K1細胞を使用する。しかし、5HT2B受容体については、トランスフェクトされたHEK細胞を用いて、PLC/IPアッセイに関してさらに強力かつ一貫した結果が得られる(Setola et al., 2005)。トランスフェクションの24時間前に、細胞を、放射性受容体結合アッセイについては100mm皿中、40%コンフルエンシーで、又は機能的アッセイについては12ウェルプレート中、1ウェルあたり105細胞で蒔く。CHO−K1細胞を、放射性受容体結合アッセイについては100mm皿につき12μgのプラスミド及び32μlのリポフェクタミン(Invitrogen)で、又は機能的アッセイについてはウェルあたり0.8μgのプラスミド及び4.0μlのリポフェクタミンで、一時的にトランスフェクトする。HEK細胞を用いた5−HT2B機能的アッセイについて、24μgのプラスミドDNAを60μlのリポフェクタミン2000(Invitrogen)と混合して、10cmプレート中1〜2×106細胞をトランスフェクトする。細胞にトランスフェクトされた受容体をさらに24時間発現させる(Herrick, 1997)。
【0241】
放射性受容体アッセイ
放射性受容体飽和及び競合結合アッセイを、膜ホモジネートを用い、系統学的に密接に関連するヒスタミンH1GPCRについて以前に報告されている本発明者らの方法と同様にして実施する(Booth, 2002;Moniri et al., 2004)。[3H]−ケタンセリンを用いて、5−HT2A受容体を放射標識し、[3H]−メスレルギンを5−HT2B及び5−HT2c受容体について使用する。手短に言うと、CHO細胞トランスフェクションの48時間後、細胞を収集し、0.1%のアスコルビン酸及び4.0mMのCaCl2を含むpH7.4の50mMのTris−HCl(アッセイ緩衝液)中で均質化する。ホモジネートを35,000gで25分間遠心分離し、結果として得られる膜ペレットをアッセイ緩衝液中に再懸濁させる。タンパク質濃度を、Lowryらの方法(Lowry, 1951)により測定する。飽和結合アッセイに関して、100μgのタンパク質を含む膜懸濁液を、0.1〜5.0nMの[3H]−ケタンセリン(5−HT2A受容体)又は0.1〜20nMの[3H]−メスレルギン(5−HT2B及び5−HT2c受容体)とともに250μlの合計アッセイ緩衝液体積中でインキュベートする。非特異的結合を、10μMのメチセルギド(5−HT2A受容体)又は1.0μMのミアンセリン(5−HT2B及び5−HT2c受容体)の存在下で測定する。競合結合アッセイを、1.0nMの[3H]−ケタンセリン又は[3H]−メスレルギンを用いて同様に実施する。放射性受容体結合アッセイ混合物のインキュベーションは、1.0時間37℃で、96ウェルセルハーベスター(Tomtec, Hamden, CT)を用いてWhatman GF/Bフィルターを通した急速ろ過法により終了させる。フィルターディスク上に保持される膜結合[3H]放射性リガンドを、液体シンチレーション分光測定法によって定量化する。データを、Prism 4.03(GraphPad Software Inc., San Diego, CA)のS字形曲線適合アルゴリズムを用いた非線形回帰により分析する。リガンド親和性は、IC50データを、式K0.5=/C50/I+LIKQ(式中、Lは親和性KDを有する放射性リガンドの濃度である)を用いてK0.5値に変換することによって、Ki値の近似値として表す(Cheng, 1973)。
【0242】
PLCの活性化及び[3H]−IP形成についてのアッセイ
PLCの機能的活性化を、すでに報告されているようにして、セロトニン5−HT2C受容体を一時的に発現するCHO細胞又はセロトニン5−HT2A若しくは5−HT2B受容体を一時的に発現するHEK細胞における[3H]−IP形成として測定する(Moniri et al., 2004)。手短に言うと、トランスフェクションの32時間後に、イノシトールを含まないダルベッコの修飾イーグル培地(DMEM)中の細胞を12時間1.0μCi/mlのミオ−[2−3H]−イノシトール(PLC−β基質ホスファチジルイノシトールの放射標識された前駆体)とともにインキュベートする。細胞を次いで洗浄し、10mMの塩化リチウム、10μMのパルギリン(HEK細胞について5%の透析されたウシ胎仔血清を添加)、及び種々の濃度の試験リガンドを含むDMEM中で45〜60分間37℃でインキュベートする。培地を吸引した後、50mMのギ酸とともにインキュベーション(15〜60分)することによって、ウェルを溶解させる。ギ酸を水酸化アンモニウムで中和し、各ウェルからの内容物を個々のAG1−X8 200−400ホルメート樹脂アニオン交換カラムに添加する。ギ酸アンモニウム/ギ酸(1.2M/0.1M)を用いて、液体シンチレーション分光測定法によってトリチウムを計数するために、[3H]−IPをシンチレーションバイアル中に直接溶出させる。結果として得られるデータを、Prism 4.03の非線形回帰アルゴリズムを用いて分析し、対照[3H]−IP形成の平均パーセンテージとして表し、効力は、最大基礎(構成的)[3H]−IP形成を50%±S.E.M.(n≧3)刺激(EC50)又は阻害(IC50)するために必要な濃度として表す。
【0243】
CHO−K1及びHEK細胞における[3H]−IP形成の測定
PLCの機能的活性化は、本発明者らの研究室により以前に報告されているように、5−HT2A又は5−HT2C受容体を一時的に発現するCHO細胞及び5HT2B受容体を一時的に発現するHEK細胞において[3H]−IP形成として測定される(Booth, 2002;Moniri et al., 2004)。手短に言うと、トランスフェクションの32時間後、イノシトールを含まないダルベッコの修飾イーグル培地(DMEM)中の細胞を、1μCi/mlのミオ−[2−3H]−イノシトール(PLC−β基質ホスファチジルイノシトールの前駆体)で標識する。細胞を次いで洗浄し、25mMのHepes(pH7.4)、10mMのLiCl、10μMのパルギリン(HEK細胞について、5%の透析されたFBSを添加)、並びに様々な濃度の試験リガンドを含むDMEM中、45〜60分間37℃でインキュベートする。培地を吸引した後、ウェルを氷上に置き、50mMのギ酸(15〜60分)とともにインキュベーションすることによって溶解させる。ギ酸を水酸化アンモニウムで中和し、各ウェルからのすべての内容物を個々のAG1−X8 200−400ホルメート樹脂アニオン交換カラムに添加する。ギ酸アンモニウム/ギ酸(1.2M/0.1M)を使用して、液体シンチレーション分光測定法によってトリチウムを計数するために、[3H]−IPをシンチレーションバイアル中に直接溶出させる。結果として得られるデータを、Prism 4.03の非線形回帰アルゴリズムを用いて分析し、対照[3H]−IP形成の平均パーセンテージとして表し、効力を50%最大[3H]−IP形成(EC50)±S.E.M(n≧3)を得るために必要な濃度として表す。
【0244】
実施例2
5HT−サブタイプ受容体の放射性リガンド飽和結合分析:ヌルトランスフェクト(null−transfected)CHO及びHEK細胞から調製された膜を用いて測定可能な特異的放射性リガンド結合はない。しかし、5−HT2A、5−HT2B、又は5−HT2CcDNAで一時的にトランスフェクトされたCHO細胞から調製された膜を用いて、飽和可能な特異的放射性リガンド結合が起こる。[3H]−ケタンセリンは、5HT2A受容体の見かけ上1つの集団(Bmax=1.73±0.11pmol/mgタンパク質)と高親和性(KD=0.80±0.03nM)で結合する。同様に、[3H]−メスレルギンは、Bmax=1.13±0.39pmol/mgタンパク質及びKD=5.19±0.36nMで5HT2B受容体の1つの集団を標識する。[3H]−メスレルギンはまた、高親和性(KD=0.88±0.03nM)で5HT2c受容体の見かけ上1つの集団(Bmax=8.37±0.15pmol/mgprot)も標識する。
【0245】
実施例3
RSK及び他のキナーゼのキナーゼ阻害活性は、当該技術分野で公知である。RSK活性に関するアッセイは、基本的に、Jeffrey A. Smith, Celeste E. Poteet−Smith, Yaming Xu, Timothy M. Errington,, Sidney M. Hecht, and Deborah A. Lannigan Cancer Res 2005;65:(3), pp. 1027−1034. February 1, 2005で記載されているようにして実施される。
【0246】
実施例4
メタ置換化合物の合成及びエナンチオマーの分離
一般的合成法:本発明の化合物を合成するのに有用な関連する方法は当該技術分野で公知であり、本発明者等の合成医薬品化学刊行物(例えば、Ghoneim, O. M.;Legere, J. A.;Golbraikh, A.;Tropsha, A.;Booth, R. G. Bioorg. Med. Chem. 2006, 14, 6640;Bucholtz, E. C;Brown, R. L.;Tropsha, A.;Booth, R. G.;Wyrick, S. D. J. .Med. Chem. 1999, 42, 3041;Wyrick SD, Booth RG, Myers AM, Owens CE, KuIa NS, Baldessarini RJ, McPhail AT, and Mailman RB(1993)Synthesis and pharmacological evaluation of 1−phenyl−3−amino 1,2,3,4−tetrahydronaphthalenes as ligands for a novel receptor with sigma−like neuromedulatory activity. J Med Chem 36:2542−2551を包含する)で記載されている。インビトロの薬理学的研究は、まず、ラセミのシス及びトランス生成物を使用する。ラセミ化合物は、ジアステレオマー塩に誘導体化し、続いて分画晶出(differential crystallization)を行うことにより(+)−及び(−)−エナンチオマーに分割することができるか、又はキラル還元ステップを用いて新たに合成することができる。すでに合成された純粋なエナンチオマーと比較することにより単結晶X線結晶学又は分光測光法(NMR、旋光度)によって絶対配置を帰属させる。NMR、元素分析、質量分析、融点及び薄層クロマトグラフィーを用いて生成物を純度について特性化する(HCl塩として)。
【0247】
スキーム1:メタ置換化合物:方法は、前記方法及び当該技術分野で公知の他の方法から変更した。メタ置換アルデヒド2を用いたクライゼン・シュミット反応によって、α,β−不飽和ケトン3を得、これを環化してケトン4にし、そしてNaBH4を用いて還元した。(±)−シス及び(±)−トランス遊離塩基7を(1R)−(−)−又は(1S)−(+)−カンファー−10−スルホン酸ジアステレオマー塩に変換し、これを分画再結晶して、(+)−又は(−)−エナンチオマーを得、これをアルキル化して、生成物8を得る。
【化29】
【0248】
スキーム2:PAT類似体立体異性体を得るためのキラル還元剤の使用:
ジイソピノカンフェニル−ボラン(DIP)類似体は、最近、スキーム2におけるケトン4と類似した構造を有するケトンについて立体選択的な還元剤として報告された(例えば、Cha et al., 2005を参照)。
【0249】
C(1)ペンダントフェニル置換基への変更を加えた新規PAT類似体の合成
結合、機能、3D QSAR、及び分子モデリング結果に基づいて、(−)−トランス−置換フェニルアミノテトラリン(PAT)C(1)ペンダントフェニル部分は、5HT2A及び5HT2B受容体の活性化なしに完全に有効な5HT2cアゴニスト活性を提供するのに重要であることが示された。
【化30】
【0250】
スキーム3 ステップAでは、β−テトラロン(1)をベンゼンルテニウム(II)クロリド二量体及びキラルリガンド(R,R)−N−(2−アミノ−1,2−ジフェニルエチル)−p−トルエンスルホンアミド((R,R)−ΝAPTS)とともに還流して、(R)−β−テトラロール(2)を得た(Mogi et al., 2004)。ステップBでは、(R)−β−テトラロール(2)をtert−ブチルジメチルシリル(TBDMS)誘導体(3)(隣接するベンジル位でのブロム化を防止するためのTBDMS保護基)に変換した。ステップCでは、TBDMS保護化合物を無水CCl4中還流下、N−ブロモスクシンイミド(Agarwal et al., 1990)でブロム化して、ブロム化された一般的中間体(4)を得、フラッシュカラムクロマトグラフィーによりシス及びトランスブロモ化合物に分離した。ステップDでは、各(シス及びトランス)ブロム化中間体(4)を市販のボロン酸(R2B(OH)2、R2=アルキル、アルケニル、又はアリール基;例えば、表7のボロン酸化合物で例示される基を参照)と、ニッケル触媒作用を受けたスズキ反応下(Gonzalez−Bobes et al)、NiI2/トランス−2−アミノシクロヘキサノールとナトリウムビス(トリメチルシリル)アミドを還流下で用いて反応させて、種々のシス及びトランスPAT類似体を作成した。ステップEでは、TBDMS保護PAT類似体を、テトラヒドロフラン中テトラブチルアンモニウムフルオリド(TBAF)を用いて脱保護した。ステップFでは、シス及びトランスヒドロキシルPAT類似体をワンポットで、アジ化亜鉛/ビスピリジン複合体、ジイソプロピルアゾジカルボキシレート(DIAD)及びトリフェニルホスフェン(phosphene)とのミツノブ反応を用いて、対応するトランス及びシスアジドPAT中間体(7)に変換した(Vorogushin et al.、2003)。ステップgでは、アジドPAT誘導体を対応するPATアミン(8)に還元した。ステップHでは、これらのエナンチオマーシス及びトランスアミンを、還流下、ギ酸/ホルムアルデヒドでのEschweiler−Clarkeメチル化を用いて、ジメチル化PAT類似体(9)に変換する。
【化31】
化合物を、非メチル化遊離アミンをジアステレオマー塩に誘導体化し、続いて分画晶出することにより(+)−及び(−)−エナンチオマーに分割するか、又はキラル還元ステップを用いて新たに合成した。すでに合成された純粋なエナンチオマーと比較することにより単結晶X線結晶学又は分光測光法(NMR、旋光度)によって絶対配置を帰属させた。生成物(HCl塩として)は、NMR、元素分析、質量分析、融点及び薄層クロマトグラフィーを用いて純度について特性化した。本明細書中で具体的に記載される化合物には、R2が、表7のボロン酸化合物中のホウ素原子に結合した化学基又は部分(例えば、置換若しくは非置換アルキル、アルケニル、又はアリール基)であるものが含まれる。本明細書中の化合物を調製するための別の手順は、PCT国際公開番号WO2008/156707(出願番号PCT/US2008/007458)にも記載されている。
【0251】
類似体の合成
トリフルオロアセチル混合無水物及び3−ハロスチレン(フルオロ、クロロ、及びブロモ)を、溶媒を追加することなく、4−(3−ハロフェニル)−3,4−ジヒドロナフタレン−2−イルフェニルアセテート(ブロモ、50%)に関するカスケードフリーデルクラフツ環状−アシルアルキル化、エノール化、及びO−アシル化に付した。マスクされた4−フェニルテトラール−2−オンの塩基アルコール分解により、インサイチュ不斉移動水素化のためのケト基が明らかになった。ブロモ−誘導体は、4−(ビフェン−3−イル)−3,4−ジヒドロナフタレン−2−イルフェニルアセテートを得るためにフェニルボロン酸とのスズキカップリングを受け、トランス−4−フェニル−2−アミノテトラリンへの短い有効な経路を提供した。
【0252】
カスケードFC−CAA、エノール化、及びO−アシル化を、TFAA活性化フェニル酢酸、スチレン又は3−ハロスチレンを用いて調査した。反応性スチレン12aを[9]とともに穏やかに加熱すると、本質的に遅いエノール化が加速され(a)Nevy, J. B.;Hawkinson, D.. C;Blotny, G.;Yao, X.;Pollack, R. M. J. Am. Chem. Soc. 1997, 119, 12722. b)Yao, X.;Gold, M. A.;Pollack, R. M. J. Am. Chem. Soc. 1999, 121, 6220を参照)、4a(15%)を得た。加熱の非存在下で、複合体混合物は、結果として反応性3aの損失をもたらした。中程度に反応性である3−ハロスチレンと、等モル濃度又は3当量までの[9]により、ハロゲン化3及び4を得た。フルオロスチレン12bと等モル濃度の[9]により、多量の3b(42%)及び少量の4b(8%)を得た。クロロスチレン12cと3当量の[9]により3c(70%)を得た。3cを等モル濃度の[9]でさらに処理して、4c(38%)を得た。24時間にわたって室温まで温めて、ブロモスチレン12cと3当量の[9]により、4d(50%)を得、非ハロゲン化4aからの収率で3倍増加した。
【0253】
【表3】
スキーム4:4dの(例えば、表5からの)フェニルボロン酸でのスズキカップリング(a)Wolfe, J. P.;Singer, R, A.;Yang, B. H.;Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9550. b)Wolfe, J. P.;Buchwald, S. L. Angew. Chem. Int. Ed. 1999, 38, 2413を参照)によりスムーズに4−(ビフェニル−3−イル)−3,4−ジヒドロ−ナフタレン−2−イルフェニル−アセテート14を得、これをスキーム3におけるようなプロトコルにしたがって所望の置換フェニルアミノテトラリンに変換した。
【0254】
スキーム4:
【化32】
【0255】
実施例5:
CAT化合物の合成
スキーム5
【化33】
【0256】
4−シクロヘキシル−3,4−ジヒドロナフタレン−2−イルフェニルアセテート(1a)。(Gray AD, and Smyth TP(2001) Clean−chemistry synthesis of 2−tetralones in a single−stage acylation−cycloalkylation process. J Org Chem 66:7113−7117)フェニル酢酸(10.9g、0.08モル)を無水トリフルオロ酢酸TFAA(11mL、0.08モル)中に室温で溶解させて、その場で混合無水物を生成させた。窒素ガスを使用して、両頭針を通して、ビニルシクロヘキサン(0.04モル)を含み、60℃で撹拌したフラスコ中に混合無水物を押し込んだ。30分後、反応を水(100mL)でクエンチし、酢酸エチル(100mL、3×)で抽出した。有機層を硫酸ナトリウム上で乾燥し、ろ過し、そして濃縮した。粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、ラセミ1a(30%、油状物)及び1b(〜30%、4−シクロヘキシル−3,4−ジヒドロナフタレン−2(1H)−オン)を得た。1H NMR(CDCl3); 0.92−1.26(m, 5H), 1.40−1.44(m, 1H), 1.57−1.76(m, 5H), 2.36(dd J=3.0, 17.1 Hz, 1H), 2.61(dt, J=2.9, 7.0 Hz, 1H), 2.76(ddd, J=2.5, 7.6, 17.0 Hz, 1H), 3.73(s, 2H), 6.15(d, J=2.3 Hz, 1H), 6.95(dd, J=2.2, 7.2 Hz, 1H), 7.02−7.19(m, 3H), 7.26−7.36(m, 5H). 13C NMR(D6−DMSO); 25.9, 26.0, 26.1, 28.2, 29.2, 30.5, 40.2, 40.6, 43.5, 113.8, 126.1, 126.2, 126.5, 127.0, 128.4, 129.4, 132.5, 133.9, 135.3, 149.9, 169.6. 元素分析 C24H26O2についての計算値C=83.20、H=7.56。実測値:C=83.02、H=7.91。HRMS m/z C24H26O2についての計算値 346.1933 [M]+、実測値346.1921。
【0257】
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−オール(2a).(Peach, P, Cross DJ, Kenny JA, Mann I, Houson I, Campbell L, Walsgrove T, and Wills M(2006)Asymmetric transfer hydrogenation of a,3−unsaturated, a−tosyloxy and a−substituted ketones. Tetrahedron 62:1864−1876),(Alcock NJ, Mann I, Peach P, and Wills M(2002)Dynamic kinetic resolution−asymmetric transfer hydrogenation of 1−aryl−substituted cyclic ketones. Tetrahedron: Asymmetry 13:2485−2490),(Mogi M, Fuji K, and Node M(2004)Asymmetric reduction of methoxy substituted 3−tetralnes using transfer hydrogenation. Tetrahedron: Asymmetry 15:3715−3717)
ベンゼンルテニウム(II)クロリド二量体(55mg、0.11ミリモル)及び(1R,2R)−(−)−N−(4−トルエンスルホニル)−1,2−ジフェニルエチレンジアミン(80mg、11ミリモル)をイソプロパノール(25mL)中で混合し、80℃で30分間加熱すると、触媒混合物が生成した。別に、4−シクロヘキシル−3,4−ジヒドロナフタレン−2−イルフェニルアセテート1a(950mg、2.8ミリモル)をイソプロパノール(25mL)中で50℃まで加熱した。窒素ガスを用いて、両頭針を通してIaを含むフラスコに触媒混合物を押し込み、直後にKOH(0.5g)のイソプロパノール(50mL)中混合物を添加した。3時間後、セライト上シリカゲルのパッドを通して反応物を濾過し、次いで減圧下で濃縮した。粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、2aを40%の収率で得た。
【0258】
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−イル4−メチルベンゼンスルホネート(3a)。(Wyrick SD, Booth RG, Myers AM, Owens CE, Kula NS, Baldessarini RJ, McPhail AT, and Mailman RB(1993) Synthesis and pharmacological evaluation of 1−phenyl−3−amino−1,2,3,4−tetrahydronaphthalenes as ligands for a novel receptor with sigma−like neuromodulatory activity. J Med Chem 36:2542−2551)
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−オール2a(0.23g、0.1モル)、p−トルエンスルホニルクロリド(0.20g、0.11モル)、及びピリジン(1.5mL)を溶液中で24時間、室温、不活性雰囲気(N2)下で撹拌した。溶媒を除去し、粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、3aを62%の収率で得た。HRMS m/z C23H28O3Sについての計算値 407.1651 [M+Na]+、実測値 407.1631。
【0259】
(2S,4R)−4−シクロヘキシル−N,N−ジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(4a).(Bosse K, Marineau J, Nason DM, Fliri AJ, Segelstein BE, Desai K, and Volkmann RA(2006)Expanding the medicinal chemistry toolbox: stereospecific generation of methyl group−containing propylene linkers. Tetrahedron Letters 47:7285−7287)
ジメチルアミン(H2O中40%、6mL)及び(2R,4R)−4−シクロヘキシル1,2,3,4−テトラヒドロナフタレン−2−イル4−メチルベンゼンスルホネート3a(0.19g、0.5ミリモル)を試験管中に入れ、密封し、83℃で24時間加熱した。溶媒を除去し、粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、4a(収率70%)及び5a(30%、(R)−1−シクロヘキシル−1,2−ジヒドロナフタレン)を得た。ベンチクロマトグラフィーの後、4aを、キラル(RegisCell(商標)、EtOH(15):ヘキサン(85))HPLC技術を用いて調べて、UV波形によりeeが98%を超えることが示された。1H NMR(CDCl3);1.00−1.31(m, 5H), 1.39−1.47(m, 1H), 1.57−1.85(m, 6H), 2.48−2.52(m, 1H), 2.72−2.77(m, 1H), 2.80(s, 6H), 3.03(dd, J=9.0, 16.6 Hz, 1H), 3.34(dd, J=6.8, 16.0 Hz, 1H), 3.72−3.75(m, 1H), 7.09−7.29(m, 4H). 13CNMR(CDCl3);26.5, 26.8, 27.3, 30.2, 31.8, 32.3, 41.6, 41.9, 43.5, 56.8, 124.9, 125.7, 129.1, 129.2, 136.0, 139.7. HRMS m/z C18H27Nについての計算値258.2216 [M+H]+、実測値258.2220。薬理学的研究のために、遊離塩基をHCl塩に変換した;MP:174〜176℃。C18H27N+HCl+H2Oの元素分析計算値:C=69.32、H=9.70、N=4.49。実測値:C=69.48、H=9.98、N=4.34。
【0260】
実施例6:
セロトニン5HT2A、5HT2B、5HT2C、及びヒスタミンH1Gタンパク質共役受容体でのCATの薬理学的活性
(−)−トランス−CAT及び(+)−トランス−CATの活性を、基本的に当該技術分野で公知のとおりであり、本明細書中に記載されるプロトコルを用いて評価した。結果を以下の表及び本明細書中の図面で示す(例えば、図1〜3)。
【0261】
(+)−(2R,4S)−及び(−)−(2S,4R)−4−シクロヘキシル−N,Nジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(或いは、4−シクロヘキシル−2−ジメチルアミノテトラリン、CAT)の分子構造
【化34】
【0262】
【表4】
【表5】
【0263】
442の異なるキナーゼについての親和性に関する(−)−トランス−CATの薬理学的データのまとめ
(−)−(2S,4R)−4−シクロヘキシル−N,N−ジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(或いは、4−シクロヘキシル−2−ジメチルアミノテトラリン、CAT)の分子構造
【化35】
【0264】
(−)−トランス−CATはRSK3ときわめて強力に結合する(1.0uMのCATで標識タグ結合の80%阻害)
【0265】
(−)−トランス−CATはRSK4に非常に強力に結合する(1.0uMのCATで結合する標識されたタグの80%阻害)
【0266】
(−)−トランス−CATはRSK1と中程度の親和性を有する(1.0uMのCATで結合するタグの20%阻害)。
【0267】
1.0uMまでの濃度で、(−)−トランス−CATは、本明細書中に記載されるキナーゼに対してRSK2に同様に有意な結合を示さず、有意な選択性を示した。
【0268】
1.0uMまでの濃度で、(−)−トランス−CATは、本明細書中に記載されるキナーゼに対して試験した438の他のキナーゼ(表4を参照)と有意な結合として示さず、有意な選択性を示した。
【0269】
RSK1(わずかな結合)及び2(結合なし)よりもRSK3及び4の阻害についての(−)−トランス−CATの非常に高い特異性を有する分子は現在のところ報告されていない。RSK2及び438の他のキナーゼからRSK(1、3及び4)の阻害について(−)−トランス−CATの非常に高い特異性を有する分子は現在のところ報告されていない。
【0270】
実施例7
化合物の5−HT2機能的活性は、PCT国際公開番号第WO2008/156707号(出願番号PCT/US2008/007458)で記載されているものをはじめとする当該技術分野で公知の方法によって評価することができる。
【0271】
【表6】
【表7】
参考文献:
Arjona AA, Pooler AM, Lee RK, Wurtman RJ. Effect of a
5-HT2C serotonin agonist, dexnorfenfluramine, on amyloid precursor
protein metabolism in guinea pigs. Brain Res. 2002 951 :135-140.
Baldessarini RJ, Tarazi FI. Pharmacotherapy of Psychosis
and mania. In: Brunton LL, Laxo JS, Parker KL, eds. The Pharmacological Basis
of Therapeutics. 11th ed. New York:
McGraw-Hill, 2006:461-500.
Bubar MJ, Cunningham KA. Distribution of serotonin
5-HT(2C) receptors in the ventral tegmental area. Neuroscience. 2007 (doi:
10.1016/j.neuroscience.2006.12.071).
Bubar MJ, Cunningham KA. Serotonin 5-HT2A and 5-HT2C
receptors as potential targets for modulation of psychostimulant use and
dependence. Current Topics and Medicinal Chemistry 2006; 6:1971-1985.
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS,
Edwards WD, Schaff HV. Valvular heart disease associated with
fenfiuramine-phentermine.N Engl J Med. 1997;337:581-5888. Erratum in: N Engl J
Med 1997;337:1783.
Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay
MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, et al. Possible role
of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with
fenfluramine. MoI Pharmacol 2000 57: 75-81.
Fletcher PJ, Grottick AJ, Higgins GA. Differential
effects of the 5-HT(2A) receptor antagonist M 100907 and the 5-HT(2C) receptor
antagonist SB242084 on cocaine-induced locomotor activity, cocaine
self-administration and cocaine-induced reinstatement of responding. Neuropsychopharmacology
2002 27:576-586.
Frank MG, Stryker MP, Tecott LH. Sleep and sleep
homeostasis in mice lacking the 5-HT2c receptor. Neuropsychopharmacology. 2002
27:869-873.
Ghoneim et al., Bioorg. Med. Chem., 14, 6640-6658 (2006).
Giorgetti M, Tecott LH. Contributions of 5-HT(2C)
receptors to multiple actions of central serotonin systems. Eur J Pharmacol.
2004 488: 1-9.
Heisler LK, Chu HM,
Tecott LH. Epilepsy and obesity in serotonin 5-HT2C receptor mutant mice.Ann N
Y Acad Sci. 1998 861 :74-78.
Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart
JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, et al. Activation of central
melanocortin pathways by fenfluramine. Science (Wash DC) 2002 297: 609-611.
Heisler LK, Tecott LH. A paradoxical locomotor response
in serotonin 5-HT(2C) receptor mutant mice. J Neurosci. 200020:RC71.
Heisler LK, Zhou L, Bajwa P, Hsu J, Tecott LH Serotonin
5-HT(2C) receptors regulate anxiety-like behavior.Genes Brain Behav. 2007 (DOI
10.1 1 1 l/j.l601-183X.2007.00316.x)
Jensen MD. Potential role of new therapies in modifying
cardiovascular risk in overweight patients with metabolic risk factors.
Obesity. 2006 14:143S-149S.
Julius D, Huang KN, Livelli TJ, Axel R, Jessel TM. The
5HT2 receptor defines a family of structurally distinct but functionally
conserved serotonin receptors. Proc. Natl. Acad. Sci. 1990 87:928-932.
Julius D, MacDermott
AB, Axel R, Jessell^
TM. Molecular Characterization of a functional cDNA encoding the serotonin Ic
receptor. Science 1988 241 :558-564.
Kennett GA, Pittaway K, Blackburn TP: Evidence that 5-HT2C receptor
antagonists are anxiolytic in the rat Geller-Seifter model of anxiety.
Psychopharmacology (Bed.) (1994) 114:90-96.
Launay JM, Herve P, Peoc'h K, Tournois C, Callebert J,
Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, et al. Function of the
serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med
2002 8: 1129-1135.
Marquis KL, Sabb AL, Logue SF, Brennan JA, Piesla MJ,
Comery TA, Grauer SM, Ashby CR Jr, Nguyen HQ, Dawson LA, Barrett JE, Stack G,
Meltzer HY, Harrison BL, Rosenzweig-Lipson S . W A Y- 163909 [(7bR, 1 OaR)-
1,2,3,4,8,9,10,1 Oa-octahydro-7bH-cyclopenta-[b][1,4]diazepino[6,7,lhi]indole]:
A novel 5-hydroxytryptamine 2C receptor-selective agonist with preclinical
antipsychotic-like activity. J Pharmacol Exp Ther. 2007 320:486-496.
Muller CP, Huston JP. Determining the region-specific
contributions of 5-HT receptors to the psychostimulant effects of cocaine.
Trends Pharmacol Sci. 2006 27:105-112.
Nichols DE. Hallucinogens, Pharmacol. Ther. 2004 101:131-181.
Nilsson BM. 5-Hydroxytryptamine 2C (5-HT2C) receptor
agonists as potential antiobesity agents. J Med Chem. 2006 49:4023-4034.
Palvimaki EP, Roth BL, Majasuo H, Laakso A, Kuoppamaki M,
Syvalahti E, Hietala J. Interactions of selective serotonin reuptake inhibitors
with the serotonin 5-HT2c receptor, sychopharmacology (Berl). 1996 126:234-240.
Pouwels HM, Smeets JL, Cheriex EC, Wouters EF. Pulmonary
hypertension and fenfluramine. Eur Respir J. 1990 May;3(5):606-7.
Raymond JR. Mukhin YV. Gelasco A. Turner J. Collinsworth
G. Gettys TW. Grewal JS. Garnovskaya
MN. Multiplicity of mechanisms of
serotonin receptor signal transduction. Pharmacol Ther. 2001 92: 179-212.
Reynolds GP, Yao Z, Zhang X, Sun J, Zhang Z. Pharmacogenetics
of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor
polymorphisms separately associate with positive and negative symptom response.
Eur Neuropsychopharmacol. 2005 Mar; 15(2): 143-51.
Rocha BA, Goulding EH, O1DeIl LE, Mead AN, Coufal
NG, Parsons LH, Tecott LH. Enhanced locomotor, reinforcing, and neurochemical
effects of cocaine in serotonin 5-hydroxytryptamine 2C receptor mutant mice. J
Neurosci. 2002 ;22: 10039-10045.
Rosenzweig-Lipson S, Sabb A, Stack G, Mitchell P, Lucki
I, Malberg JE, Grauer S, Brennan J, Cryan JF, Sukoff Rizzo SJ, Dunlop J,
Barrett JE, Marquis KL. Antidepressant-like effects of the novel, selective,
5-HT(2C) receptor agonist WAY-163909 in rodents. Psychopharmacology (Berl).
2007 192:159-170.
Roth BL. Drugs and valvular heart disease. N Engl J Med.
2007; 356:6-9.
Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A,
Hufeisen SJ, and Roth BL. Evidence for possible involvement of 5-HT(2B)
receptors in the cardiac valvulopathy associated with fenfluramine and other
serotonergic medications. Circulation 2000 102: 2836-2841.
Saltzman AG, Morse B, Whitman MM, Ivanshchenko Y, Jaye M,
Felder S. Cloning of the human serotonin 5-HT2 and 5-HTlC receptor subtypes.
Biochem Biophys Res Commun. 1991 181 :1469-7148.
Sanders-Bush E, Mayer SE. Serotonin Receptor Agonists and
Antagonists. Chapter 11, in Goodman and Gilman's The Pharmacological Basis of
Therapeutics 11th Edition. Brunton LL, Lazo JS, Parker KL, Editors, McGranw-Hill, New
York, 297-315, 2006.
Sard H, Kumaran G, Morency C, Roth BL, Toth BA, He P,
Shuster L. SAR of psilocybin analogs: discovery of a selective 5-HT 2C agonist.
Bioorg Med Chem Lett. 2005 15:4555-4559.
Segman RH, Heresco-levy U, Finkel B, Inbar R, Neeman T,
Schlafman M, Dorevitch A, Yakir A, Lerner A, Goltser T, Shelevoy A, Lerer B.
Association between the serotonin 2C receptor gene and tardive dyskinesia in
chronic schizophrenia: additive contribution of 5-HT2CSer and DRD3Gly alleles
to susceptibility, Psychopharmacology 2000 152:408-413.
Setola V, Dukat M, Glennon RA, Roth BL. Molecular
determinants for the interaction of the valvulopathic anorexigen
norfenfluramine with the 5-HT2B receptor. Mol Pharmacol 2005
68:20-33.
Simansky KJ. N1H symposium series: ingestive mechanisms
in obesity, substance abuse and mental disorders. Physiology & Behavior
2005; 86: 1-4.
Siuciak JA, Chapin DS, McCarthy SA, Guanowsky V, Brown J,
Chiang P, Marala R, Patterson T, Seymour PA, Swick A, Iredale PA. CP-809,101, a
selective 5-HT2C agonist, shows activity in animal models of antipsychotic
activity. Neuropharmacology. 2007 52:279-290.
Smith SR, Prosser W, Donahue D, Anderson C, Shanahan W. Lorcaserin Phase 2b
Clinical Study. American Diabetes Association, 2006.
Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP,
Johnson JA. Neutralization of transthyretin reverses the neuroprotective
effects of secreted amyloid precursor protein (APP) in APPSw mice
resulting in tau phosphorylation and loss of hippocampal neurons: support for
the amyloid hypothesis. J. Neurosci. 2004 24:7707-7717.
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH,
Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c
serotonin receptors. Nature. 1995 374:542-546
Tohda M, Takasu T, Nomura Y. Effects of antidepressants
on serotonin-evoked current in Xenopus oocytes injected with rat brain mRNA.
Eur J Pharmacol. 1989166:57-63.
Veenstra-VanderWeele J, G.M. Anderson GM, Cook EH.
Pharmacogenetics and the serotonin system: initial studies and furure
directions. Eur. J. Pharmacol. 2000 410:65-181.
Vickers SP, Dourish
CT, and Kennett GA. Evidence that hypophagia
induced by D-fenfluramine and D-norfenfluramine in the rat is mediated by
5-HT2C receptors. Neuropharmacology 2001 41: 200-209.
Vickers, S.P., Clifton,
P.G., Dourish, CT. and Tecott, L.H., 1999. Reduced satiating effect of
d-fenfluramine in serotonin 5-HT2C receptor mutant mice. Psychopharmacology
1999 143:309-314.
Agarwal R, Boyd
DR, McMordie RAS, O'Kane GA, Porter P, Sharma ND, Dalton H, Gray DJ. J.
Chiral arene hydrates of naphthalene: Enzymatic and chemical syntheses. Chem
Soc Chem Commun. 1990 171 1-1713.
Mogi M, Fugi K, Node M. Asymmetric reduction of methoxy
substituted β-tetralones using
hydrogenation. Tetrahedron: Asymmetry. 2004 15:3715-3717.
Vorogushin AV, Predeus A, Wuff WD, Hansen H. Diels- Alder
reaction-aromatization approach toward functionalized ring C allocolchicinoids.
Enantioselective total synthesis of (-)-7S-Allocolchicine. J Org Chem.
【0272】
本明細書中の変数の定義における要素のリストの説明は、いずれか1つの要素又は記載された要素の組み合わせとしての変数の定義を包含する。本明細書中の要素、又は実施形態の説明は、いずれか1つの要素若しくは実施形態又は任意の他の要素、実施形態若しくはそれらの一部との組み合わせを包含する。
【0273】
これらに限定されるものではないが、抄録、論文、雑誌、刊行物、テキスト、専門書、技術的データシート、インターネットウェブサイト、データベース及び特許、特許出願、及び特許公報をはじめとする本明細書中の全ての引用文献は、印刷物、電子的、コンピュータ可読記憶媒体又は他の形態であるかに関わらず、参照することによってそれらの全体として本明細書中に明らかに組み込まれる。
【0274】
本発明を具体的な実施例を参照して開示したが、本発明の真の精神及び範囲から逸脱することなく本発明の他の実施形態及び変形を他の当業者らが考案できることは明らかである。特許請求の範囲は、そのような実施形態及び等価な変形を包含すると解釈されるものである。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図2A】
【図2B】
【図2C】
【技術分野】
【0001】
[優先権の主張]
本出願は、2009年5月5日に出願された米国仮出願第61/215,504号、2009年9月10日に出願された米国仮出願第61/241,384号、及び2009年9月24日に出願された米国仮出願第61/277,408号の恩恵を主張するPCT国際出願である。前記米国仮出願のそれぞれの全内容は、本明細書中に組み込まれる。
[連邦政府により支援された研究のもとでなされた発明に対する権利の記載]
【0002】
本研究は、一部、アメリカ合衆国公衆衛生局認可MH068655により支援された。本研究は、一部、国立衛生研究所認可DA023928とMH081193とにより支援された。政府は本発明にある権利を有する。
【背景技術】
【0003】
セロトニン(5−ヒドロキシトリプタミン、5HT)は、5HT1〜5HT7ファミリーに分類される14の哺乳動物の5HT受容体サブタイプにより、様々な中枢及び末梢の心理学的及び生理学的効果に関与する(Sanders−Bush and Mayer, 2006)。5HT2ファミリーは、主にGαqを介してシグナルを送り、ホスホリパーゼ(PL)C並びにイノシトールリン酸(IP)及びジアシルグリセロール(DAG)第2メッセンジャーの形成を活性化する、5HT2A、5HT2B、及び5HT2c膜結合型Gタンパク質共役受容体(GPCR)から構成される(Raymond et al., 2001)。ヒト5HT2c受容体(Saltzman et al., 1991)は、脳のみで見いだされるようであり、脳で、広く発現され、おそらくは、摂食行動(Tecott et al., 1995)、コカイン中毒(Fletcher et al.,2002;Rocha et al., 2002;Muller and Huston, 2006)、睡眠ホメオスタシス(Frank et al., 2002)、不安(Kennett et al., 1994;Sard et al., 2005;Heisler et al., 2007)、鬱病(Tohda et al., 1989;Palvimaki et al., 1996)、てんかん(Heisler et al., 1998)、アルツハイマー病(Arjona et al.,2002;Stein et al., 2004)、運動機能(Heisler and Tecott, 2000;Segman et al., 2000)、精神病(Marquis et al., 2007;Siuciak et al., 2007)及び抗精神病薬に対する反応(Veenstra−VanderWeele et al., 2000;Reynolds et al., 2005)をはじめとするいくつかの(疾患)−生理学的及び心理学的プロセスに関与する。したがって、5HT2C受容体の薬物療法学的標的としての重要性は約10年間にわたり明らかであるが、5HT2c特異的薬剤は開発されていない。
【0004】
5HT2c受容体を標的とする創薬に関する1つの課題は、このGPCRが、5HT2A受容体と約80%の膜通過ドメイン(TMD)配列同一性を共有し、そして5HT2B受容体と約70%の膜通過ドメイン(TMD)配列同一性を共有することである(Julius et al., 1988;1990)。高度に保存されたTMD及び類似の第2メッセンジャーカップリングのために、5HT2C受容体に対して選択的なアゴニストリガンドの開発が特に困難になっている。それにもかかわらず、5HT2c受容体の活性化が食物摂取を減らして、抗肥満効果につながるという動かぬ証拠がある。たとえば、5−HT2cノックアウトマウスは摂食の増大や肥満を示し、d−フェンフルラミンの食欲抑制作用に対して耐性を示す(Tecott et al., 1995;Vickers et al., 1999;2001;Heisler et al., 2002)。フェンフェルラミンは現在、禁止されている。なぜなら、この薬剤を使用している人々は脳5HT2c受容体の活性化のために体重が減少するが、フェンフェルラミンはまた、5HT2A受容体を活性化し、このことは有害な精神医学的(幻覚誘発性)効果をもたらし得(Nichols, 2004)、また5HT2B受容体を活性化し、これにより心臓弁膜症(Connolly et al., 1997;Fitzgerald et al., 2000;Rothman et al., 2000;Roth, 2007)及び肺高血圧症(Pouwels et al., 1990;Launay et al., 2002)が引き起こされ、5HT2Bが関与する効果の結果、死に至ったためである。
【0005】
5HT2c対5HT2A及び/又は5HT2B受容体に関して真に選択的なアゴニストリガンドは本論文までは報告されていないが、ラットコカイン自己投与パラダイム(self−administration paradigm)において非常に(すなわち少なくとも100倍)選択的な5HT2A及び5HT2c拮抗物質を使用して脳5HT2c受容体がコカイン使用及び依存性を軽減する役割を部分的に説明することが可能である。たとえば、選択的な5HT2A拮抗物質M100907(Kehne et al., 1996)はコカイン自己投与に対する反応速度を変えないが、選択的な5HT2c拮抗物質SB242084(Bromidge et al., 1997)は、コカイン自己投与速度を用量に依存して増大させる(Fletcher et al., 2002)。覚醒剤中毒に対する5HT2cアゴニスト薬物療法の多大な可能性は現在、広く認識されている(Bubar and Cunningham, 2006)。
【0006】
肥満及び覚醒剤中毒等の神経精神障害における5HT2c受容体の薬物療法学的関連性により、製薬会社は強い関心を刺激されて、選択的な5HT2Cアゴニストを開発してきたが、報告されている5HT2cアゴニストはすべて、これまでのところ5HT2A及び/又は5HT2B受容体も刺激する(Nilsson, 2006)。それにもかかわらず、5HT2アゴニストロルカセリン(APD356)は、5HT2c受容体の活性化について5HT2A受容体よりも15倍程度の選択性しかないが、最近、肥満治療の第3相臨床試験まで進んだ(Jensen, 2006;Smith et al., 2006)。しかし、本明細書中で報告する結果は、本発明者等の実験室で合成した新規化合物、すなわち、フェニル−3−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレン(PAT)及びシクリル−3−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレン(CAT)化合物に基づき、これらはヒト5HT2c受容体での十分に有効なアゴニストであり、その上、5HT2A及び5HT2B受容体での拮抗物質である。
【0007】
Gタンパク質共役受容体(GPCR)は、2種以上のGタンパク質を活性化することができ、その結果、複数の生理学的/薬理学的効果(薬物療法学的かつ不適切な副作用)が起こる(Moniri et al., Journal of Pharmacology and Experimental Therapeutics, 311:274−281(2004))。単一のGPCRと関連する複数のシグナル伝達経路の現象は、GPCR活性化の3状態モデルのフレームワーク内で記載することができ、この場合、GPCRは、不活性状態と構成的に活性な状態との間で異性化する。GPCR活性化は、ヘテロ三量体(α,β,γ)Gタンパク質サブユニットの解離を引き起こし、−Gαサブユニットは次いでトランスデューサータンパク質(例えば、PLC、AC)を活性化して、第2メッセンジャー濃度を変えることができる。同じGPCRが異なるGαタンパク質とカップリングして、その結果、「多機能シグナル伝達」を得ることができることがわかっている。GPCR多機能シグナル伝達理論の重要な仮定は、活性受容体コンフォメーションの異質性が存在し、アゴニストリガンドは、「刺激輸送」仮説で記載されるように、受容体コンフォメーションを誘発するか、安定化するか、又は選択するそれらの能力が異なる。結合すると、アゴニストリガンド化学構造パラメータは、Gαタンパク質の種類及び活性化されるシグナル伝達経路に影響を及ぼすGPCRコンフォメーションの最も重要な決定因子に含まれる。
【0008】
73の生物学的Gタンパク質共役受容体標的に対する380の拮抗物質の活性に関する105の論文の調査によると、この試料データセットにおいて、322が逆アゴニストであり、58(15%)が中性拮抗物質である。逆アゴニズムの優位性は、中性拮抗物質が薬理学的空間において少数種であることを示す理論的予想と一致する(Kenakin, Mol Pharmacol.(2004);65:2−11)。
【0009】
p90リボソームs6キナーゼ(RSK)は、ヒトkinome中のAGCサブファミリーの構成要素であるセリン/トレオニンキナーゼの一群である(Nguyen, 2008)。4つのRSKイソ型(RSK1〜4)があり、それぞれ別個の遺伝子の産物である。RSKイソ形は、75%〜80%の配列同一性によって特徴付けられる。RSKイソ型はヒト組織において広く分布しているが、様々な組織発現を示し、このことは、それらが異なる機能に関与することを示す。RSKイソ型は、Ras−ERK経路を刺激する細胞外シグナル伝達分子によって活性化される。これらの分子は、様々な成長因子、サイトカイン、ペプチドホルモン及び神経伝達物質を含む。なお、現時点で、RSKイソ型のそれぞれについての生物学的/薬理学的役割は、癌をはじめとするほとんどの疾患について確立されていない。
【0010】
RSKは、ファミリーとしてのRSKを細胞増殖、細胞分化、生存、及び遊走などの多くの生物学的プロセスと関連づける、転写因子、最初期遺伝子産物、翻訳調節因子、酵素、及び構造タンパク質をはじめとする様々なタンパク質をリン酸化する(Anjum and Blenis, 2008a )。
【0011】
RSKイソ形は、ヒト乳癌組織試料の50%で過剰発現され、このことは、RSKの調節が損なわれたことを示し、RSK活性と腫瘍細胞増殖との間の関連により、RSKが新規癌治療薬標的であることが明らかになる(Gioeli et al., 1999;Smith et al., 2005)。
【0012】
RSKとHIV感染との間の関連について動かぬ証拠がある。RSK2はHIV−1 Tatと相互関係を有することが示された。RSK2はHIV−1 Tatにより動員され、活性化され、それ自体も、正常なTat機能に対して重要である(Hetzer et al., 2005)。さらに、カポジ肉腫関連ヘルペスウイルスはRSKと相互作用し、それらのキナーゼ活性を強力に刺激する(Kuang et al., 2009)。
【0013】
構造的に、RSKイソ型は非常に類似している(Nguyen, 2008)。イソ型のそれぞれは、大きな100アミノ酸リンカー領域によって分離されている2つの機能的触媒ドメインを含む。この2つのキナーゼドメインは別個であり、同じでないATP結合部位を含む。NTKDはp70S6キナーゼ(p70 S6K1)と類似し、CTKDはカルシウム/カルモジュリンタンパク質キナーゼと類似している。その分子構造のために、RSは阻害に関して2つの論理的部位:NTKDにおけるATP結合部位及びCTKDにおけるものを提供する。両ATPポケットの部位特異的阻害剤が同定されている。
【0014】
90kDaリボソームS6キナーゼ(RSK)の上流アクチベータの異常な活性化は、癌をはじめとする多くのヒト疾患と関連付けられている。RSK1及びRSK2イソ型は、胸部及び前立腺腫瘍で増幅される。造血変換(hoematopoietic transformation)におけるRSK2活性化の重要性は、最近、線維芽細胞成長因子受容体−3(FGFR3)を発現する複数の骨髄腫細胞で示された。RSK3は、最近、卵巣癌において潜在的な腫瘍サプレッサとして機能することが示された。RSK4はp53依存的細胞増殖停止に関与することが示されたが、RSK4の異常な発現が乳癌で観察された。ヒトRSK2遺伝子における突然変異は、コフィン・ローリー症候群(CLS)(重篤な精神運動遅延、指及び顔の変形、並びに進行性骨格奇形によって特徴付けられるX連鎖障害)と関連する。CLS患者由来の線維芽細胞において、転写因子CREBの誘発されたSerl33リン酸化の大幅な減弱が、上皮成長因子刺激に反応して検出された。CLS患者から誘導された線維芽細胞は、このヒト疾患に関するRSK2の機能を決定する際に有用であったが;これらの細胞とRSK2欠損マウス線維芽細胞との間の相違は、CLSが多変量疾患である可能性があり、マウスはRSK2機能を研究するのに理想的な系でない可能性があることを示唆する。RSK2及びRSK4遺伝子はどちらもX染色体上に位置し、最近のデータも、非特異的X連鎖精神遅滞にRSKを関連づけるが、RSK4に関する決定的証拠はまだ得られていない(Anjum and Blenis, 2008b)。
【発明の概要】
【発明が解決しようとする課題】
【0015】
一態様では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に5−HT2結合相互作用を調節できる治療有効量の化合物(例えば、本明細書中で記載する化合物)を投与することを含む方法を提供する。一実施形態では、化合物は5−HT2cを刺激することができる。別の実施形態では、化合物は、5−HT2a及び/又は5−HT2bを拮抗するが、5−HT2cを刺激することができる。
【0016】
一態様では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に治療有効量の5−HT2c刺激化合物を投与することを含む。
【0017】
別の態様では、本発明は神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、5−HT2cを直接、好ましくは5−HT2a及び/又は5−HT2bと比べて選択的に調節することによって、5−HT2結合相互作用を調節することができる治療有効量の化合物を投与することを含む。
【0018】
別の実施形態では、本発明は、神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とすると認定された対象に、治療有効量の5−HT2c刺激化合物又は5−HT2c選択的化合物を投与することを含む。
【0019】
別の態様では、本発明は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安、睡眠ホメオスタシスをはじめとする神経精神障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は対象に有効量の5−HT2cを刺激できる投与することを含む。
【0020】
別の態様では、本発明は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安に罹っているか又は罹りやすい対象を治療する方法であって、当該対象に、5−HT2cを刺激する(5−HT2a及び/又は5−HT2bと比べて選択的であることを包含する)ことができる有効量の化合物を投与して、当該対象が治療されるようにすることを含む方法を提供する。
【0021】
一態様では、本発明は、GPCR障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に、治療有効量のGPCR結合相互作用を調節することができる化合物を投与することを含む方法を提供する。一実施形態では、化合物はGPCRを刺激することができる。別の実施形態では、化合物はGPCRを拮抗することができる。
【0022】
別の態様において、本明細書中の化合物は、セロトニンヒスタミンH1、5HT2A、2B、2C、及びアセチルコリンムスカリン性M1〜M5GPCRを標的とする機能的に選択的な化合物である。態様において、本発明は、対象においてセロトニンヒスタミンH1、5HT2A、2B、2C、及びアセチルコリンムスカリン性M1〜M5GPCRを選択的に標的とする方法であって、当該対象に本明細書中の化合物を投与することを含む方法を提供する。
【0023】
別の態様では、本発明は、対象におけるGPCR介在性障害を治療又は予防する方法であって、それらを必要とすると認定された対象に対して、本明細書中で記載される化合物を投与することを含む方法を提供する。ある実施形態では、化合物は、表1{下記)の化合物である。ある実施形態では、化合物は、式(I):
【化1】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化2】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり、
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0024】
ある実施形態では、化合物は、各R1が−NMe2であるものである。
【0025】
他の実施形態では、R1は
【化3】
であり、これは場合によって置換されている。
【0026】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでないものである。
【0027】
ある実施形態では、化合物は、R8が1個のR5で置換され、R5が、H、アルキル、又はハロでない置換基であるものである。
【0028】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0029】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0030】
ある実施形態では、化合物は、各R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、ここで各R5は独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0031】
ある実施形態では、本発明は、そのような治療を必要とすると認定された対象において、疾患、障害又はその症状を治療する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。
【0032】
ある実施形態では、障害は、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、又は泌尿生殖路(genitor−urinary tract)障害(例えば、膀胱制御)である。ある実施形態では、障害は覚醒剤(例えば、コカイン、アンフェタミン、メタンフェタミン)薬物中毒である。ある実施形態では、障害は肥満である。ある実施形態では、障害は認知障害である。他の実施形態では、障害はアレルギー又は炎症性障害である。
【0033】
別の態様では、本発明は、そのような治療を必要とすると認定された対象において5−HT2cを阻害する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。
【0034】
別の態様では、本発明は、対象において肥満を治療する方法であって、それを必要とすると認定された対象に、5−HT2a又は5−HT2bと比べて、5−HT2cを選択的に阻害することができる化合物を投与することを含む方法を提供する。ある実施形態では、5−HT2cを阻害するための結合相互作用は、5−HT2a又は5−HT2bのいずれかについてよりも少なくとも5倍(或いは少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い。ある実施形態では、5−HT2cを阻害するための結合相互作用は、5−HT2a又は5−HT2bのいずれかについてよりも少なくとも100倍高い。
【0035】
別の態様では、本発明は、5−HT2c活性を調節することができる化合物を同定するための方法であって;(i)分子若しくは分子複合体の三次元表示(前記分子若しくは分子複合体は、5−HT2cの構造座標によって規定される結合ポケットを含む);又はb)前記分子又は分子複合体の相同体の三次元表示(ここで、前記相同体は、約2.0オングストローム以下の前記アミノ酸の骨格からの二乗平均平方根偏差(root measn square deviation)を有する結合ポケットを含む)を作製し;(ii)試験化合物の三次元表示を作製し;(iii)試験化合物の標的との結合相互作用を評価することを含む方法を提供する。ある実施形態では、当該方法は、試験化合物を5−HT2cと接触させ、化合物の結合活性を測定することを更に含む。
【0036】
一態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法であって、それを必要とする対象に治療有効量のRSK結合相互作用を調節することができる化合物(例えば、本明細書中に記載するいずれかの式の化合物)を投与することを含む方法を提供する。
【0037】
一態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、治療有効量のRSK阻害剤化合物を投与することを含む。
【0038】
別の態様では、本発明は、RSK介在性疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とする対象に、治療有効量の、本明細書中に記載するいずれかの式の化合物(例えば、RSK調節化合物(直接的若しくは間接的)、RSK阻害化合物)を投与することを含む。
【0039】
別の態様では、本発明は、疾患又は障害に罹っているか又は罹りやすい対象を治療する方法を提供する。当該方法は、それを必要とすると認定された対象に、治療有効量の、1以上の他のキナーゼよりもRSK阻害について選択的な化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、1以上の他のキナーゼよりもRSK阻害について選択的であると認定された化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、RSK−2よりもRSK−1、RSK−3又はRSK−4の阻害について選択的な化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、それを必要とすると認定された対象に、治療有効量の、5−HT2(例えば、5−HT2a、5−HT2b、5−HT2c)の調節(例えば、逆アゴニスト活性)によりRSK−2を調節するための化合物(例えば、本明細書中に記載する化合物)を投与することを含む。当該方法は、化合物が、5−HT2(例えば、5−HT2a、5−HT2b、5−HT2c)の調節(例えば、逆アゴニスト活性)によりRSK−2を調節する(例えば、本明細書中に記載する化合物)と認定される方法であり得る。
【0040】
他の態様では、当該方法には、化合物が、他のキナーゼよりもRSK−1、RSK−3又はRSK−4について選択的である方法が含まれる。選択性は、1つの標的(すなわちキナーゼ)について、他の標的と比べて例えば2倍、3倍、10倍、100倍、X倍(ここで、Xは数値である)等である。別の実施形態では、本発明は、化合物が標的酵素に対する活性範囲、及び標的以外の酵素に対する異なる活性範囲の選択性を示す、本明細書中で記載する方法を提供する。
【0041】
他の態様では、当該方法には、化合物が、RSK2又は他のキナーゼよりも、RSK1並びに特に3、及び4を優先的に阻害する方法が含まれる。他の態様では、当該方法には、化合物が1.0マイクロモル濃度でRSK1並びに特に3、そして4を優先的に阻害し、RSK2を阻害しない方法が含まれる。このように、(−)−トランス−CAT及び誘導体は、RSK1、3、及び4の物理的及び生物学的特性をRSK2から区別するため、及びRSK1、3、4の生物学を438の他のキナーゼから区別するための分子生物学的手段として非常に有用であり(表4を参照);そしてRSK1、3、及び4の物理的及び生物学的特性をRSK2から区別するため、そしてRSK1、3、4の生物学を438の他のキナーゼから区別するための分子生物学的手段として非常に有用であり;そして癌(特に、造血、胸部及び前立腺)、HIV、及びコフィン・ローリー症候群の生物学及び病理を1つのRSKイソ型の関与に関して、これらの疾患における別のイソ型に対して、及びこれらの疾患における他のキナーゼに対して特徴付けるために有用である。さらに、(−)−トランス−CAT及び本明細書中の化合物は、RSK1、3、及び4とのそれらの結合相互作用により、これらのキナーゼの構造を特徴付けるための有用な生化学的プローブである。
【0042】
ある実施形態では、本発明は、化合物が、式(I):
【化4】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化5】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシである)であり;
R6は独立してH又はアルキルであり;
R7は独立してH、若しくはN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、若しくは複素環(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
により表されるか、又はその塩、水和物若しくは溶媒和物である前記方法を提供する。
【0043】
ある実施形態では、化合物は、各R1が−NMe2である化合物である。
【0044】
ある実施形態では、R1は
【化6】
であり、これは場合によって置換されている。
【0045】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでない化合物である。
【0046】
ある実施形態では、化合物は、R8が1つのR5で置換され、R5が、H、アルキル、又はハロでない置換基である、化合物である。
【0047】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0048】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0049】
ある実施形態では、化合物は、R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、各R5が独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0050】
ある実施形態では、本発明は、そのような治療を必要とすると認定された対象において、疾患、障害、又はその症状を治療する方法であって、本明細書中に記載する化合物を投与することを含む方法を提供する。一態様では、化合物は(−)−トランス−CATである。
【0051】
ある実施形態では、障害は癌(特に、造血、胸部及び前立腺)、HIV、又はコフィン・ローリー症候群である。ある実施形態では、障害は乳癌である。ある実施形態では、障害は前立腺癌である。ある実施形態では、障害はHIVである。
【0052】
一態様では、本発明は、式(I):
【化7】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化8】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)
により表される化合物又はその塩、水和物若しくは溶媒和物を提供する。
【0053】
ある実施形態では、化合物は、各R1が−NMe2である化合物である。
【0054】
ある実施形態では、R1は
【化9】
であり;場合によって置換されている;
【0055】
ある実施形態では、化合物は、各R5がH、アルキル、又はハロでない化合物である。
【0056】
ある実施形態では、化合物は、R8が1個のR5で置換され、R5が、H、アルキル、又はハロでない置換基である化合物である。
【0057】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)シクロアルキルである化合物である。
【0058】
ある実施形態では、化合物は、R8が場合によって置換された(例えば、置換、非置換)アリールである化合物である。
【0059】
ある実施形態では、化合物は、各R8が独立してアリール又はシクロアルキルであり、各R8が2、3又は4個の独立したR5で置換され、各R5が独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである化合物である。
【0060】
ある実施形態では、1位及び3位での化合物置換基は、互いにトランス配位である。
【0061】
ある実施形態では、化合物は式(II):
【化10】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化11】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリール;式中、環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり;
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;
mは1、2又は3である)によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0062】
ある実施形態では、各R’は独立して、メチル、エチル、プロピル、イソプロピル、ブチル、s−ブチル、t−ブチル、アルケニル、ビニル、又はアルキニルである。
【0063】
ある実施形態では、1位及び3位での化合物置換基は互いにトランス配位である。
【0064】
ある実施形態では、化合物R1は−NMe2である。ある実施形態では、PAT化合物は(1R,3S)−(−)トランス−1−フェニル−3−N,N−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレンである。
【0065】
さらなる実施形態では、化合物は式(II−a):
【化12】
(式中、
R5は、H、メチル、エチル、プロピル、イソプロピル、ブチル、s−ブチル、t−ブチル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、フェニル、F、Cl、若しくはBrであり;そして
mは1、2、若しくは3である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0066】
ある実施形態では、化合物は、式(III):
【化13】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化14】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
nは2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
R5は、H、アルキル、又はハロであり;
R6は独立してH又はアルキルであり;
R7はH、又はN(アルキル)2であり;そして
mは1、2、又は3である)
によって表されるか、又はその塩、水和物若しくは溶媒和物である。
【0067】
別の態様では、本発明は、式(IV):
【化15】
(式中、
R1は独立してH、又はN(アルキル)2であり;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、NH2、NH(R’)、N(RM)2であるか;
又は
【化16】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は場合によって1、2、3、若しくは4個の独立したR5で置換された複素環式である)
により表される化合物、又はその塩、水和物若しくは溶媒和物を提供する。
【0068】
前記式のいずれかのある実施形態では、1位及び3位の化合物置換基は互いにトランス配位である。
【0069】
ある実施形態では、化合物R1は−NMe2である。ある実施形態では、PAT化合物は(1R,3S)−(−)−トランス−1−フェニル−3−N,N−ジメチルアミノ−1,2,3,4−テトラヒドロナフタレンである。
【0070】
別の態様では、本発明は、式(V):
【化17】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化18】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;R3はH、OH、又はハロであり;
R4はH、OH、アリール、ヘテロアリール、又はハロであり;
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキルであり;
R7はH、又はN(アルキル)2であり;そして
Zはアリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルであり、そのそれぞれは場合によって1、2、3、若しくは4個の独立したR5で置換されている)によって表される化合物、又はその塩、水和物若しくは溶媒和物を提供する。
【0071】
ある実施形態では、R1はN(R’)2であり;そして各R’はアルキルである。他の実施形態では、Zはフェニルであり、これはハロアルキル、アルコキシ、又はハロで置換されていてもよい。他の実施形態では、R4はフェニルであり、これはハロアルキル、アルコキシ、又はハロで置換されていてもよい。
【0072】
別の態様では、本発明は、本明細書中で記載される化合物(例えば、本明細書中のいずれかの式の化合物)及び薬剤的に許容される担体を含む組成物を提供する。
【0073】
別の態様では、本発明は、本明細書中で記載される化合物(例えば、本明細書中のいずれかの式の化合物)及び薬剤的に許容される担体を組み合わせることを含む、組成物の作製法を提供する。
【0074】
構造−活性関係(SAR)研究の結果は、H1対5HT2A対5HT2B対5HT2C対、M1対M2対M3対M4対M5GPCRについての本発明の化合物の親和選択性(affinity selectivity)は、テトラヒドロナフタレン環系の、C1位での置換基(例えば、ペンダントフェニル又は他の芳香族、ヘテロ芳香族、シクリル、シクロアルキル)及びC3位での置換基(アミン)の立体化学並びに、C1及びC3置換基の化学的性質、並びにC6及びC7位での化学置換基に依存することを示す(炭素ナンバリングは式I、表1のとおり)。同様に、H1対5HT2A対5HT2B対5HT2C対、M1対M2対M3対M4対M5GPCRでのアゴニスト対逆アゴニスト対拮抗物質活性は、C1、C3、C6、及びC7での置換基の化学的性質及び立体化学によって決定される。例えば、Bucholtz, E. C, Wyrick, S.D., Owens, C.E., and Booth, R.G. 1−Phenyl−3−dimethylaminotetralins (PATs): Effect of stereochemistry on binding and function at brain histamine receptors. Medicinal Chemistry Research 8:322−332 (1998).; Bucholtz, E.C., Brown., R.L., Tropsha, A., Booth, R.G, and Wyrick, S.D. Synthesis, Evaluation and Comparative Molecular Field Analysis of 1−Phenyl−3−amino−1,2,3,4−tetrahydronaphthalenes as Ligands for Histamine H1 Receptors. Journal of Medicinal Chemsitry.42:3041−3054 (1999);Choksi, N.Y., Nix, William B., Wyrick, S.D., and Booth, R.G. A novel phenylaminotetralin recognizes Histamine H1 receptors and stimulates dopamine synthesis in vivo in rat brain. Brain Research 852:151−160 (2000); Booth RG, Moniri NH, Bakker RA, Choksi NY, Timmerman H, and Leurs R. A novel phenylaminotetralin radioligand reveals a sub−population of Histamine H1 receptors. Journal of Pharmacology and Experimental Therapeutics 302:328−336 (2002); Moniri NH, Covington−Strachan D, Booth RG. Ligand−directed functional heterogeneity of Histamine H1 receptors: Novel dual−function ligands selectively activate and block H1−meditated phospholipas C and adenylyl cyclase signaling. Journal of Pharmacology and Experimental Therapeutics, 311 :274−281 (2004); Booth RG, Moniri NH. Ligand−directed multifunctional signaling of histamine H1 receptors Inflammation Research 54: S44−45 (2005); Ghoneim OM, Legere JA, Glbraikh A, Tropsha A, Booth RG. Novel ligands for the human histamine H1 receptor: Synthesis, pharmacology, and comparative molecular field analysis studies of 2−dimethylamino−5−(6)−phenyl−1,2,3,4−tetrahydronaphthalenes. Bioorganic and Medicinal Chemistry, 14:6640−6658 (2006); Booth RG, Moniri NH. Novel Ligands Stabilize Stereo−Selective Conformations of the Histamine H1 Receptor to Activate Catecholamine Synthesis. Inflammation Research 56:1−12 (2007)を参照。
【0075】
別の実施形態では、本明細書中の化合物は、アデニル酸シクラーゼ(AC)/cAMP対ホスホリパーゼC(PLC)/イノシトールリン酸(IP)細胞内シグナル伝達経路とカップリングして、脳カテコールアミン(ドーパミン、ノルエピネフリン)神経伝達物質合成を調節する脳H1受容体を識別し、選択的に活性化することができる。複数の態様では、本発明は、アデニル酸シクラーゼ(AC)/cAMP対ホスホリパーゼC(PLC)/イノシトールリン酸(IP)細胞内シグナル伝達経路とカップリングして、H1/AC/cAMPシグナル伝達に対して感受性のプロセス、例えば対象における脳カテコールアミン(ドーパミン、ノルエピネフリン)神経伝達物質合成の調節に生理学的に影響を及ぼす脳H1受容体を選択的に活性化する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0076】
別の態様では、本明細書中の化合物は、H1が関与するAC/cAMPシグナル伝達を選択的に増強して、改変されたカテコールアミン神経伝達を含むある神経精神病に罹っている患者を治療する化合物である。複数の態様では、本発明は、H1が関与するAC/cAMPシグナル伝達を選択的に増強して、改変されたカテコールアミン神経伝達を含むある神経精神病に罹っている対象を治療する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0077】
別の実施形態では、本明細書中の化合物は、H1/PLC/IPシグナル伝達によって進行する不適切なH1が関与する効果、例えば、呼吸困難(気管支狭窄)、下痢(GI収縮)、並びに浮腫及び低血圧(増大した血管透過性)、特に末梢アレルギー反応に関連するものの拮抗物質及び逆アゴニストである化合物である。複数の態様では、本発明は、対象においてPLC/IP経路によって進行する不適切なH1が介在する効果を拮抗(例えば遮断)する方法であって、対象に本明細書中の化合物を投与することを含む方法を提供する。
【0078】
別の態様では、本明細書中の化合物は、セロトニン5HT2A及び5HT2B受容体での拮抗物質並びに逆アゴニストである。
【0079】
別の実施形態では、本明細書中の化合物は、PLC/IPシグナル伝達と関連するヒスタミンH1受容体での拮抗物質並びに逆アゴニストである。
【0080】
別の態様では、本明細書中の化合物は、アセチルコリンムスカリン性M1〜M5受容体での拮抗物質並びに逆アゴニストである。
【0081】
別の態様では、本明細書中の化合物は、アセチルコリンムスカリン性M1〜M5受容体での拮抗物質並びに逆アゴニスト及びアゴニストである。別の態様では、本明細書中の化合物は同時にセロトニン5HT2A及び5HT2B受容体での逆アゴニストであり、かつ5HT2c受容体でのアゴニストである。複数の態様では、本発明は、対象において疾患又は障害(例えば、精神障害;肥満)を治療又は予防する方法であって、対象に、同時にセロトニン5HT2A及び5HT2B受容体での逆アゴニストであり、5HT2c受容体でのアゴニストである化合物を投与することを含む方法を提供する。別の実施形態では、当該方法は、対象が精神障害及び肥満の両方の治療を必要とする方法である。
【0082】
一実施形態では、当該化合物は、対象においてアセチルコリンムスカリン受容体系の障害から起こる疾患又は障害を薬理学的に治療するための方法であって、対象に本明細書中のいずれかの式の化合物を投与することを含む方法を提供する。本明細書中のいずれかの式の化合物は、ムスカリン性M1、M2、M3、M4、及び/又はM5受容体に対して薬理学的に関連する親和性を有し、1以上のムスカリン受容体でアゴニスト、逆アゴニスト、及び/又は拮抗物質として機能的に挙動する。
【0083】
ムスカリン性M1、M2、M3、M4、及び/又はM5受容体の薬理学の調節に反応する典型的な疾患又は障害(Brown and Taylor, 2006, Muscarinic Agonists and Antagonists. In: Brunton L.L., Lazo, J.S., Parker, K.L. (Eds.), Goodman and Gilman’s The Pharmacological Basis of Therapeutics 11th Edition. McGranw−Hill, New York, NY, pp. 183−200)としては、これらに限定されるものではないが:胃腸管の障害(便秘、下痢、胃酸過多、痙縮)、尿路の障害(頻尿、排尿不足、多尿)、緑内障、喘息、パーキンソン病、アルツハイマー病、外分泌腺が関係する様々な障害(発汗、涙液形成、唾液形成、粘液形成に関連する問題)、及びあるキノコ(例えば、天然のムスカリン誘導体を含むもの)による中毒の治療が挙げられる。
【0084】
別の態様では、本発明は、5−HT2cを調節する化合物を同定するための方法を提供し、この方法は、5−HT2cタンパク質を得るか、又は5−HT2cタンパク質の結晶構造に関連する情報を得、試験化合物を5−HT2cタンパク質構造中又は構造上でモデル化して、5−HT2cタンパク質の相互作用を調節するかどうかを判定することを含む。ある実施形態では、モデリングのステップは、化合物が、5−HT2cの1以上の膜通過ドメイン1〜7の構造座標により規定される結合ポケットに結合するか、又はこれに関連する能力をモデル化又は判定することを含む。
【0085】
本発明のさらに別の態様は、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安を治療又は予防するために有用な化合物を同定する方法である。当該方法は、5−HT2c複合体を試験化合物と接触させ、そして試験化合物が5−HT2cを調節(例えば、刺激又は拮抗)する能力を評価することを含む。
【0086】
本発明のさらに別の態様は、5−HT2cの活性を調節する化合物を同定する方法であって、この方法は、5−HT2cの1以上の膜通過ドメイン1〜7の原子座標を用いて、結合ポケットを含む分子の三次元構造(例えば、コンピュータによる)を生成させ、そしてこの三次元構造を用いて5−HT2cの1以上の膜通過ドメイン1〜7の活性を調節する化合物を同定することを含む。
【0087】
別の態様では、本発明は、治療有効量の5−HT2cアゴニスト化合物及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。当該組成物は、神経精神障害(例えば、肥満)に罹っているか又は罹りやすい対象を治療するために処方され、神経精神障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0088】
一態様では、本発明は、対象における神経精神障害を治療するためのキットを提供し、このキットは、本明細書中の化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、並びに使用説明書を含む。さらなる態様では、本発明は、5−HT2cを刺激し、対象における抗肥満治療薬の有効性を評価し、5−HT2cアゴニストで治療される対象の経過をモニタリングし、神経精神障害の対象を5−HT2cアゴニストでの治療について選択し、及び/又は神経精神障害(例えば、肥満)に罹っているか又は罹りやすい対象を治療するためのキットを提供する。ある実施形態では、本発明は:対象における神経精神障害を治療するためのキットであって、5−HT2cアゴニスト活性を調節(例えば、刺激)することができる化合物を含むキットを提供する。他の態様では、当該化合物は、5−HT2a及び/又は5−HT2bと比べて5−HT2cを選択的に刺激する。他の態様では、当該化合物は、5−HT2a及び/又は5−HT2bを選択的に拮抗する。
【0089】
別の態様では、本発明は、5−HT2cの1以上の膜通過ドメイン1〜7(それぞれ単独又は組み合わせ)の三次元構造に関する。
【0090】
したがって、本発明は、これらの結合ポケットのいずれか1つ若しくは両方又は類似した三次元形状を有するいずれかの結合ポケットの相同体を含む分子若しくは分子複合体を提供する。
【0091】
本発明は、本明細書中で記載される化合物の医薬組成物であって、5−HT2cを刺激することができる化合物;5−HT2a及び/又は5−HT2bと比べて5−HT2cを選択的に刺激することができる化合物;5−HT2cを刺激し、5−HT2a及び/又は5−HT2bを拮抗することができる化合物;又はその薬剤的に許容されるエステル、塩、若しくはプロドラッグを、薬剤的に許容される担体とともに含む医薬組成物も提供する。
【0092】
別の態様では、本発明は、機械可読記憶媒体であって、5−HT2cの1以上の膜通過ドメイン1〜7を規定する結合ポケットの構造座標を含む記憶媒体を提供する。
【0093】
別の態様では、本発明は、分子又は分子複合体の三次元表示(前記分子又は分子複合体は、5−HT2cの1以上の膜通過ドメイン1〜7の構造座標によって規定される結合ポケットである);又はb)前記分子又は分子複合体の相同体の三次元表示(前記相同体は、前記アミノ酸の骨格原子からの二乗平均平方根偏差が約2.0オングストローム以下である結合ポケットを含む)を作成するコンピュータを提供する。コンピュータは:(i)機械可読データでコード化されたデータ記憶材料を含む、機械可読データ記憶媒体(ここで、前記データは5−HT2cの1以上の膜通過ドメイン1〜7の構造座標を含む);(ii)前記機械可読データを処理するための命令を記憶するためのワーキングメモリ;(iii)前記機械可読データを前記三次元表示に処理するための前記ワーキングメモリ及び前記機械可読データ記憶媒体に接続された中央処理装置;並びに(iv)前記三次元表示を表示するための前記中央処理装置に接続されたディスプレーを含む。
【0094】
別の態様では、本発明は、治療有効量のRSK阻害剤化合物(例えば、選択的なRSK−1、RSK−3、又はRSK−4阻害剤化合物)及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。当該組成物は、疾患又は障害(例えば、癌、HIV、RSK介在性疾患)に罹っているか又は罹りやすい対象を治療するために処方することができ、そして疾患又は障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0095】
一態様では、本発明は、対象における疾患又は障害を治療するためのキットを提供し、このキットは、本明細書中の化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、及び使用説明書を含む。さらなる態様では、本発明は、RSKを阻害するためのキットを提供する。ある実施形態では、本発明は:対象における疾患又は障害(すなわち、RSK介在性疾患又は障害)を治療するためのキットであって、RSK活性を調節(例えば、阻害)することができる化合物を含むキットを提供する。他の態様では、当該化合物は、他のキナーゼと比べてRSKを選択的に阻害する。他の態様では、当該化合物は、他のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害する。
【0096】
本発明はまた、本明細書中で記載される化合物の医薬組成物であって、RSK活性を調節(例えば阻害)することができる化合物又はその薬剤的に許容されるエステル、塩、若しくはプロドラッグを、薬剤的に許容される担体とともに含む医薬組成物も提供する。他の態様では、当該化合物は、他のキナーゼと比べてRSKを選択的に阻害する。他の態様では、当該化合物は、他のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害する。他の態様では、当該化合物は、5−HT2(例えば、1以上の5−HT2a、b、又はc)の調節(例えば、逆アゴニスト活性)によりRSKを調節する。
【0097】
本発明はまた、前記結合ポケットと結合する化合物の設計、評価及び同定方法も提供する。本発明の他の実施形態を以下で開示する。
【図面の簡単な説明】
【0098】
本発明を以下の非限定的な例を参照し、そして以下の図面を参照して、さらに説明する。
【図1】図1A〜1Eは、HEK293細胞において一時的に発現された野生型5HT2A、5HT2B、5HT2c及びヒスタミンH1受容体に関する(−)−トランス−CAT及び(+)−トランス−CATの代表的な結合曲線である。
【図2】野生型5HT2A、5HT2B、5HT2C受容体を一時的に発現するHEK293細胞におけるPLC活性/IP形成の活性に対する(−)−トランス−CAT効果の代表的な曲線を表す。データは、(−)−トランス−CATが、5HT2A、5HT2B及び5HT2c受容体の逆アゴニストであることを示す。
【図3】PLC活性/[3H]−IP形成のヒスタミンH1受容体が関与する刺激のヒスタミン活性化は、(−)−トランス−CATにより競合的に拮抗されることを表す。
【発明を実施するための形態】
【0099】
本発明者らは、5−HT2c及び/又はRSKを選択的に標的とすることによる選択的な疾患治療及び予防(すなわち有害な副作用が軽減又は最小限に抑えられた)を対象とする治療法を発見した。そのような相互作用は、特に、5−HT2メカニズムが重要な役割を果たすある種の神経精神障害における5−HT2c介在性障害の調節に関連する。
【0100】
本発明は、少なくとも一つには、5−HT2c相互作用が、神経精神障害治療の標的として有用(例えば選択的)であるという発見に関する。
【0101】
1.定義
本発明のさらなる説明の前に、本発明がより容易に理解されるように、便宜のために、いくつかの用語をまず定義し、ここにまとめる。
【0102】
「投与」又は「投与する」という用語は、本発明の化合物(複数可)を対象に対して、それらが目的とする機能を発揮するように導入する経路を包含する。使用できる投与経路の例としては、注射(皮下、静脈内、非経口的、腹腔内、髄腔内)、経口、吸入、直腸及び経皮が挙げられる。医薬製剤は、各投与経路に適した形態で投与することができる。たとえば、これらの製剤は、錠剤又はカプセル形態で、注射、吸入、洗眼液、軟膏、坐薬など、注射、注入又は吸入による投与;ローション又は軟膏による局所;及び坐薬により直腸で投与される。経口投与が好ましい。注射は、ボーラスであってもよいし、又は連続注入であってもよい。投与経路に応じて、本発明の化合物を、その目的とする機能を果たす能力に悪影響を及ぼし得る自然の条件から保護するために選択された材料でコーティングするか又は材料中に入れることができる。本発明の化合物は、単独、或いは前述の他の薬剤若しくは薬剤的に許容される担体又は両者と併用して、投与することができる。本発明の化合物は、他の薬剤の投与前に、薬剤と同時に、又は薬剤の投与後に、投与することができる。さらに、本発明の化合物は、インビボでその活性代謝物、又はさらに活性の高い代謝物に変換されるプロドラッグ形態で投与することもできる。
【0103】
「アルキル」という用語は、直鎖アルキル基、分枝アルキル基、シクロアルキル(脂環式)基、アルキル置換シクロアルキル基、及びシクロアルキル置換アルキル基をはじめとする飽和脂肪族基のラジカルを指す。アルキルという用語は、炭化水素骨格の1以上の炭素を置換する酸素、窒素、硫黄又はリン原子、たとえば酸素、窒素、硫黄又はリン原子をさらに含み得るアルキル基をさらに包含する。好適な実施形態では、直鎖又は分枝鎖アルキルは、その骨格中に、30個以下の炭素原子(例えば、直鎖についてはC1〜C30、分枝鎖についてはC3〜C30)、好ましくは26個以下、そしてさらに好ましくは20個以下、なお一層好ましくは4個以下を有する。同様に、好ましいシクロアルキルは、それらの館構造中に3〜10個の炭素原子、そしてさらに好ましくは3、4、5、6又は7個の炭素を環構造中に有する。
【0104】
さらに、アルキルという用語は、本明細書及び文全体を通して用いられる場合、「非置換アルキル」及び「置換アルキル」の両方を包含することが意図され、後者は、炭化水素骨格の1以上の炭素上の置換基を置換する置換基を有するアルキル部分を指す。そのような置換基としては、たとえば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分を挙げることができる。当業者らには、炭化水素鎖上で置換された部分は、適切ならばそれら自体が置換され得ると理解されるであろう。シクロアルキルを、たとえば前述の置換基でさらに置換することができる。「アルキルアリール」部分は、アリールで置換されたアルキル(例えば、フェニルメチル(ベンジル))である。「アルキル」という用語は、長さ及びアルキルに対する可能な置換は類似しているが、少なくとも1つの二重又は三重結合をそれぞれ含む不飽和脂肪族基も包含する。
【0105】
炭素数について特に指定がない限り、本明細書中で用いられる「低級アルキル」は、前記定義のとおりであるが、1〜10個の炭素、さらに好ましくは1〜6個、なお一層好ましくは1〜4個の炭素原子をその骨格構造中に有し、直鎖又は分枝鎖であってよいアルキル基を意味する。低級アルキル基の例としては、メチル、エチル、n−プロピル、i−プロピル、tert−ブチル、ヘキシル、ヘプチル、オクチルなどが挙げられる。好ましい実施形態において、「低級アルキル」という用語は、その骨格中に、4個以下炭素原子を有する直鎖アルキル、例えば、C1〜C4アルキルを包含する。
【0106】
「アルコキシアルキル」、「ポリアミノアルキル」及び「チオアルコキシアルキル」という用語は、前述のアルキル基であって、炭化水素骨格の1個以上の炭素を置換する酸素、窒素又は硫黄原子、たとえば酸素、窒素又は硫黄原子をさらに含むアルキル基をさす。
【0107】
「アルケニル」及び「アルキニル」という用語は、前述のアルキルと長さ及び可能な置換は類似しているが、それぞれ少なくとも1個の二重又は三重結合をさらに含む不飽和脂肪族基を指す。たとえば、本発明は、シアノ及びプロパルギル基を想定する。
【0108】
「アリール」という用語は、本明細書中で用いられる場合、0〜4個のヘテロ原子を含み得る5員及び6員単環芳香族基をはじめとするアリール基のラジカル、たとえば、ベンゼン、ピロール、フラン、チオフェン、イミダゾール、ベンゾキサゾール、ベンゾチアゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピラジン、ピリダジン及びピリミジンなどを指す。アリール基は、ナフチル、キノリル、インドリルなどの多環式縮合芳香族基も包含する。環構造中にヘテロ原子を有するこれらのアリール基は、「アリールヘテロ環」、「ヘテロアリール」又は「ヘテロ芳香族」とも称する。芳香環は、1以上の環位置で、前述のような置換基、たとえば、ハロゲン、ヒドロキシル、アルコキシ、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分で置換することができる。アリール基は、芳香族でない脂環式又は複素環式環と縮合又は架橋して、多環(例えば、テトラリン)を形成することもできる。
【0109】
「〜と結合している」という用語は、化学成分若しくは化合物、又はその一部と、タンパク質上の結合ポケット若しくは結合部位との間の近接の状態を指す。会合は、非共有的(この場合、近位は、水素結合又はファンデルワールス又は静電的相互作用により支援される)であってもよいし、又は共有的であってもよい。
【0110】
「結合ポケット」という用語は、本明細書で用いられる場合、その形状の結果として、有利に別の化学成分又は化合物と会合する分子又は分子複合体の部分を指す。
【0111】
本発明の化合物の「生物学的活性」という言葉には、反応性の細胞において本発明の化合物により誘発されるすべての活性が含まれる。この言葉は、これらの化合物により誘発されるゲノム及び非ゲノム活性を含む。
【0112】
「生物学的組成物」又は「生物学的試料」とは、細胞若しくはバイオポリマーを含むか、又は細胞若しくはバイオポリマーから誘導される組成物を指す。細胞含有組成物には、たとえば、哺乳動物の血液、赤血球濃縮物、血小板濃縮物、白血球濃縮物、血球タンパク質、血漿、血小板浮遊血漿、血漿濃縮物、血漿の分画から得られる沈殿物、血漿の分画から得られる上清、血漿タンパク質画分、精製若しくは部分的に精製された血液タンパク質又は他の成分、血清、精液、哺乳動物の初乳、乳、唾液、胎盤抽出物、クリオ沈殿物(cryopercipitate)、クリオ上清(cryosupernatant)、細胞可溶化物、哺乳動物の細胞培養若しくは培養培地、発酵産物、腹水、血球中で誘発されるタンパク質、及び正常若しくは形質転換細胞により細胞培養で産生される産物(例えば、組換えDNA若しくはモノクローナル抗体技術による)が含まれる。生物学的組成物は無細胞であり得る。好ましい実施形態では、好適な生物学的組成物又は生物学的試料は、赤血球懸濁液である。いくつかの実施形態では、血球懸濁液は哺乳動物血球を含む。好ましくは、血球は、ヒト、非ヒト霊長類、イヌ、ネコ、ウマ、ウシ、ヤギ、ヒツジ又はブタから得られる。好適な実施形態では、血球懸濁液には、赤血球及び/又は血小板及び/又は白血球及び/又は骨髄細胞が含まれる。
【0113】
「キラル」という用語は、鏡像パートナーの重ね合わせることができない性質を有する分子を指し、一方、「アキラル」という用語は、それらの鏡像パートナー上に重ねることができる分子を指す。
【0114】
「ジアステレオマー」という用語は、2以上の不斉中心を有し、それらの分子が互いに鏡像でない立体異性体を指す。
【0115】
「有効量」という用語には、所望の結果を達成するために必要な投与量及び期間で有効な量、例えば本明細書中で記載される障害を治療するために十分な量が含まれる。本発明の化合物の有効量は、対象の疾患状態、年齢、及び体重、並びに当該対象において所望の反応を誘発する本発明の化合物の能力などの因子によって変化し得る。投与計画は、最適の治療反応を提供するために調整することができる。有効量は、本発明の化合物の任意の毒性又は有害な効果(例えば副作用)よりも治療上有益な効果が上回る量でもある。
【0116】
本発明の化合物の治療有効量(すなわち、有効投与量)は、約0.001〜30mg/kg体重、好ましくは約0.01〜25mg/kg体重、さらに好ましくは約0.1〜20mg/kg体重、なおいっそう好ましくは約1〜10mg/kg、2〜9mg/kg、3〜8mg/kg、4〜7mg/kg、又は5〜6mg/kg体重の範囲であり得る。当業者は、これらに限定されるものではないが、疾患若しくは障害の重篤度、以前の治療、対象の全体的な健康及び/又は年齢、並びに存在する他の疾患をはじめとするある因子が、対象を有効に治療するために必要な投与量に影響を及ぼし得ることを理解するであろう。さらに、治療有効量の本発明の化合物での対象の治療は、単一治療を含み得るか、又は好ましくは一連の治療を含み得る。一例では、対象を、約0.1〜20mg/kg体重の範囲の本発明の化合物で、約1〜10週間、好ましくは2〜8週間、さらに好ましくは約3〜7週間、なおいっそう好ましくは約4、5、又は6週間、1週につき1回で治療する。治療に用いられる本発明の化合物な有効投与量は、特定の治療過程にわたって増大又は減少し得ることも理解されるであろう。
【0117】
「エナンチオマー」という用語は、互いに重ね合わせることができない鏡像である化合物の2つの立体異性体を指す。2つのエナンチオマーの等モル混合物は、「ラセミ混合物」又は「ラセミ体」と呼ばれる。
【0118】
「ハロアルキル」という用語は、ハロゲンで一置換、二置換又は多置換された前記定義のアルキル基、例えばフルオロメチル及びトリフルオロメチルを包含することが意図される。
【0119】
「ハロゲン」及び「ハロ」という用語は、−F、−Cl、−Br又は−Iを表す。
【0120】
「ヘテロアリール」という用語は、典型的には5〜18個の環原子を含む芳香族ヘテロシクリルを意味する。ヘテロアリールは、単環、又は二環以上の縮合環であってよい。5員ヘテロアリールの非限定的な例としては、イミダゾリル;フラニル;チオフェニル(又はチエニル若しくはチオフラニル);ピラゾリル;オキサゾリル;イソキサゾリル;チアゾリル;1,2,3−、1,2,4−、1,2,5−、及び1,3,4−オキサジアゾリル;及びイソチアゾリルが挙げられる。6員ヘテロアリールの非限定的な例としては、ピリジニル;ピラジニル;ピリミジニル;ピリダジニル;並びに1,3,5−、1,2,4−、及び1,2,3−トリアジニルが挙げられる。6/5員縮合環ヘテロアリールの非限定的な例としては、ベンゾチオフラニル、イソベンゾチオフラニル、ベンズイソキサゾリル、ベンゾオキサゾリル、プリニル、及びアントラニリルが挙げられる。6/6員縮合環ヘテロアリールの非限定的な例としては、キノリニル;イソキノリニル;並びにベンゾオキサジニル(シンノリニル及びキナゾリニルを包含する)が挙げられる。
【0121】
「複素環式」又は「ヘテロシクロ」又は「ヘテロシクリル」という用語は、典型的には3〜18個の環原子を含む、飽和(例えば、「ヘテロシクロアルキル」)、部分不飽和(例えば、「ヘテロシクロアルケニル」又は「ヘテロシクロアルキニル」)又は完全不飽和(例えば、「ヘテロアリール」)環系であって、環原子の少なくとも1つがヘテロ原子(すなわち、窒素、酸素若しくは硫黄)であり、残りの環原子が炭素、窒素、酸素及び硫黄からなる群から独立して選択されるものを指す。ヘテロシクリル基は、安定な分子が得られるならば、親分子部分と基中の任意の置換可能な炭素若しくは窒素原子により結合することができる。ヘテロシクリルは、制限なく、典型的には3〜14個の環原子、3〜8個の環原子、3〜6個の環原子、又は5〜6個の環原子を含む単環であってよい。単環ヘテロシクリルの非限定的な例としては、フラニル、ジヒドロフラニル、ピロリル、イソピロリル、ピロリニル、ピロリジニル、イミダゾリル、イソイミダゾリル、イミダゾリニル、イミダゾリジニル、ピラゾリル、ピラゾリニル、ピラゾリジニル、トリアゾリル、テトラゾリル、ジチオリル、オキサチオリル、オキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、チアゾリニル、イソチアゾリニル、チアゾリジニル、イソチアゾリジニル、チオジアゾリル、オキサチアゾリル、オキサジアゾリル、ピラニル、ジヒドロピラニル、ピリジニル、ピペリジニル、ピリダジニル、ピリミジニル、ピラジニル、ピペラジニル、トリアジニル、イソキサジニル、オキサゾリジニル、イソキサゾリジニル、オキサチアジニル、オキサジアジニル、モルホリニル、アゼピニル、オキセピニル、チエピニル、又はジアゼピニルが挙げられる。ヘテロシクリルは、制限なく、互いに縮合した2以上の環を含んでもよく、例えば、ナフチリジニル、チアゾールピリミジニル、チエノピリミジニル、ピリミドピリミジニル、又はピリドピリミジニルである。ヘテロシクリルは、環構成要素として1以上の硫黄原子を含んでもよく;場合によっては、硫黄原子(複数可)はSO又はSO2に酸化される。ヘテロシクリル中の窒素ヘテロ原子(複数可)は四級化されていても、四級化されていなくてもよく、又はNオキシドに酸化されていても、酸化されていなくてもよい。加えて、窒素ヘテロ原子(複数可)はN保護されていても、N保護されていなくてもよい。
【0122】
「ヒドロキシル」という用語は、−OHを意味する。
【0123】
「ヘテロ原子」という用語は、本明細書中で用いられる場合、炭素又は水素以外の任意の元素の原子を意味する。好ましいヘテロ原子は、窒素、酸素、硫黄及びリンである。
【0124】
「ホメオスタシス」という用語は、内部環境における静的、又は一定の状態の維持を意味することが当該技術分野で承認されている。
【0125】
「改善された生物学的特性」という語法は、インビボでのその有効性を増強する本発明の化合物中に内在する活性を指す。好ましい実施形態では、この用語は、本発明の化合物の任意の質的又は量的に改善された治療特性、例えば低下した毒性を指す。
【0126】
「GPCR障害という用語は、Gタンパク質共役受容体(例えば、5−HT2a、5−HT2b、5−HT2c、ムスカリン性M1〜M5)が関与する任意の疾患、障害又はその症状を包含する。そのようなGPCRが関与する疾患及び障害としては、たとえば、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、精神病、不安、鬱病、統合失調症、精神病、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、及び泌尿生殖路障害(例えば、膀胱制御)が挙げられる。
【0127】
「M1〜M5 GPCR」という語法は、コリン作動性ムスカリン性M1〜M5神経伝達物質Gタンパク質共役受容体(本明細書中に記載されるものを包含する)を指す。
【0128】
「5−HT2」という語法は、セロトニン受容体(本明細書中に記載されるものを包含する)、例えば5−HT2a、5−HT2b及び5−HT2cサブタイプを指す。
【0129】
「場合によって置換された」という用語は、非置換である基又は1以上の利用可能な位置で水素以外によって、典型的には1、2、3、4若しくは5位で、1以上の好適な基(同一であっても、異なっていてもよい)により置換されている基を包含することが意図される、そのような任意の置換基としては、たとえば、ヒドロキシ、ハロゲン、シアノ、ニトロ、C1〜C8アルキル、C2〜C8アルケニル、C2〜C8アルキニル、C1〜C8アルコキシ、C2〜C8アルキルエーテル、C3〜C8アルカノン、C1〜C8アルキルチオ、アミノ、モノ−若しくはジ−(C1〜C8アルキル)アミノ、ハロC1〜C8アルキル、ハロC1〜C8アルコキシ、C1〜C8アルカノイル、C2〜C8アルカノイルオキシ、C1〜C8アルコキシカルボニル、−COOH、−CONH2、モノ−若しくはジ−(C1〜C8アルキル)アミノカルボニル、−SO2NH2、及び/又はモノ若しくはジ(C1〜C8アルキル)スルホンアミド、並びに炭素環及び複素環式基が挙げられる。任意の置換は、「0〜X個の置換基で置換された」という慣用句によっても表され、この場合、Xは可能な置換基の最大数である。ある場合によって置換された基は、0〜2、3又は4個の独立して選択された置換基で置換されている(すなわち、非置換であるか、又は記載された最大数までの置換基で置換されている)。
【0130】
「異性体」又は「立体異性体」という用語は、同じ化学構造を有するが、空間における原子又は基の配置に関して異なっている化合物を指す。
【0131】
「調節する」という用語は、所望の最終結果、例えば治療結果が得られるように、例えば、対象(例えば、動物、ヒト)において本発明の化合物への暴露に反応して標的の活性を阻害する化合物の能力の増加又は減少を指す。
【0132】
本明細書中で記載される「標的を調節(刺激、拮抗)できる化合物を得る」においてのような「得る」という用語は、当該化合物を購入、合成又は他の方法で取得することを包含することが意図される。
【0133】
本明細書中で用いられる「非経口投与」及び「非経口的に投与される」という表現は、腸内及び局所投与以外の、通常は注射による投与様式を意味し、静脈内、筋肉内、動脈内、髄腔内、包内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、被膜下、クモ膜下、脊椎内及び胸骨内注射並びに注入を包含するが、これらに限定されない。
【0134】
「ポリシクリル」又は「多環式ラジカル」という用語は、2以上の環(例えば、シクロアルキル、シクロアルケニル、シクロアルキニル、アリール及び/又はヘテロシクリル)のラジカルであって、2個以上の炭素が2つの隣接する環で共通であるもの(例えばこの環は「縮合環」である)を指す。隣接しない原子によって連結された環は、「架橋」環と呼ばれる。多環の環のそれぞれを、前記のような置換基、たとえば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、及びアルキルアリールアミノを包含する)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル及びウレイドを包含する)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、スルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロシクリル、アルキル、アルキルアリール、又は芳香族若しくはヘテロ芳香族部分で置換することができる。
【0135】
「プロドラッグ」又は「プロ−ドラッグ」という用語には、インビボで代謝することができる部分を有する化合物が含まれる。一般に、プロドラッグは、エステラーゼによるか又は他のメカニズムによってインビボで代謝されて、活性薬物になる。プロドラッグの例及びそれらの使用は、当該技術分野で周知である(例えば、Berge et al.(1977)“Pharmaceutical Salts”、J. Pharm. Sci. 66:1−19を参照)。プロドラッグは、化合物の最終単離及び精製中にその場で、又は、その遊離酸形態若しくはヒドロキシルで精製された化合物を好適なエステル化剤と別に反応させることによって調製することができる。ヒドロキシル基は、カルボン酸での処理によってエステルに変換することができる。プロドラッグ部分の例としては、置換及び非置換、分枝又は非分枝低級アルキルエステル部分、(例えば、プロピオン酸(propionoic acid)エステル)、低級アルケニルエステル、ジ低級アルキル−アミノ低級アルキルエステル(例えば、ジメチルアミノエチルエステル)、アシルアミノ低級アルキルエステル(例えば、アセチルオキシメチルエステル)、アシルオキシ低級アルキルエステル(例えば、ピバロイルオキシメチルエステル)、アリールエステル(フェニルエステル)、アリール−低級アルキルエステル(例えば、ベンジルエステル)、(例えば、メチル、ハロ、若しくはメトキシ置換基で)置換されたアリール及びアリール低級アルキルエステル、アミド、低級アルキルアミド、ジ低級アルキルアミド、及びヒドロキシアミドが挙げられる。好ましいプロドラッグ部分は、プロピオン酸(propionoic acid)エステル及びアシルエステルである。インビボで他のメカニズムにより活性形態に変換されるプロドラッグも含まれる。
【0136】
化合物の「予防的有効量」という語法は、患者に1回又は複数回投与すると、本明細書中で記載する障害の予防又は治療に有効である、本明細書中の任意の式又は本明細書中で他の方法で記載された本発明の化合物の量を指す。
【0137】
「低減された毒性」という語法は、インビボで投与された場合に、本発明の化合物によって誘発される任意の望ましくない副作用の軽減を包含することが意図される。
【0138】
「スルフヒドリル」又は「チオール」という用語は、−SHを意味する。
【0139】
「対象」という用語には、本明細書中の障害に罹っている可能性があるか、又は本発明の化合物の投与から恩恵を受け得る生物、例えばヒト及びヒト以外の動物が含まれる。好ましいヒトには、本明細書中で記載されるような、神経精神障害又は関連する状態に罹っているか又は罹りやすいヒト患者が含まれる。本発明の「ヒト以外の動物」という用語には、全ての脊椎動物、例えば哺乳類、例えば齧歯類、例えばマウス、及び非哺乳類、例えば非ヒト霊長類、例えばヒツジ、イヌ、ウシ、ニワトリ、両生類、は虫類等が含まれる。
【0140】
「神経精神障害に罹りやすい」という用語は、例えば本明細書中に記載されるものをはじめとする神経精神障害を発症する危険性がある対象、すなわち、神経精神障害又はその症状に罹っている対象、神経精神障害又はその症状の家族歴又は病歴を有する対象などを包含することを意味する。
【0141】
本明細書中で用いられる「全身投与」、「全身的に投与される」、「末梢投与」及び「末梢的に投与される」という表現は、本発明の化合物(複数可)、薬物又は他の物質が、患者の系中に侵入し、したがって代謝及び他の同様のプロセスを受けるような投与、例えば皮下投与を意味する。
【0142】
本発明の化合物の「治療有効量」という語法は、患者に1回又は複数回投与すると、神経精神障害の神経精神障害及び/又は症状の治療若しくは予防において、或いはそのような神経精神障害に罹っている患者の生存性を、そのような治療が存在しない場合に予想されるよりも延長するのに有効である薬剤の量を指す。
【0143】
キラル中心の命名法に関して、「d」及び「l」構造という用語は、IUPAC Recommendationsによって定義されるとおりである。この用語の使用に関して、ジアステレオマー、ラセミ体、エピマー及びエナンチオマーは、調製物の立体化学を記載するためのそれらの通常の状況で用いられる。
【0144】
2.本発明の化合物
一態様では、本発明は、5−HT及び/又はRSK結合活性を(直接的若しくは間接的に)調節(例えば阻害若しくは刺激)することができる化合物を提供する。
【0145】
一実施形態では、本発明は、5−HT2c及び/又はRSKを刺激することができる化合物;並びにその薬剤的に許容されるエステル、塩、及びプロドラッグを提供する。ある実施形態では、化合物は式(I)の化合物である。ある実施形態では、化合物は、式(II)、(II−a)、(III)、(IV)、又は(V)の化合物である。
【0146】
ある好ましい化合物には、本明細書中に具体的に記載される化合物が含まれる:
表1:化合物:
【化19】
構造は、C1&C3での立体化学である
【表1】
【0147】
ある好ましい化合物には、本明細書中に具体的に記載される化合物が含まれる:
表2:化合物:
【化20】
【表2】
【0148】
【化21】
【0149】
本発明のさらなる化合物には、式(V)の以下の化合物が含まれる:
【化22】
【0150】
前記化合物のいずれかの全てのエナンチオマー、ジアステレオマー、ラセミ体、ラセミ混合物、エナンチオ富化(enantioenriched)混合物、及びジアステレオマー混合物が本発明によって想定される。
【0151】
突然変異誘発及びモデリングデータから、PATプロトン化(pH7.4)アミンは保存5HT2残基D3.32とイオン結合を形成する(Boothら、2009)。Aim 3におけるモデリング結果も、PATアミンがS3.36&Y7.43に近い(〜2Å)配置であることを示し、したがって、さらなるヘテロ原子を有するアミン置換基(R1、チャート1)は、これらの残基と水素結合を形成することが提示されている。さらに、Aim3モデリング結果は、PAT N−アルキル部分が立体的に寛容な受容体空間中で結合し、したがって、提案される立体的に大きなR1複素環式置換基が近くの5HT2残基とファンデルワールス相互作用を形成し得ることを示す。パラ置換PAT類似体は、アンフェタミン挙動を強力に調節し(Aim4)、したがって、パラ−ハロゲン化PATがまず合成され、続いて、先に合成されたメタ置換PATの未完了のインビボ結果であるメタ−ハロゲン化を行う。
【0152】
メタ−ハロゲン化PATは、最高の5HT2親和性及び強力な機能的選択性を示した。分子モデリングは、PATペンダントフェニル環のメタ位での置換基との相互作用に関して立体的に寛容であることを示す(Booth et al., 2009;Aim 3 results)。したがって、種々の類似体は、PAT(C4)ペンダントフェニル部分と相互作用すると仮定され、相互作用することが知られている(突然変異誘発データ)保存された5HT2残基W6.48 F6.51 F6.52及びY7.43とのさらなる水素結合及び芳香族相互作用を促進するように設計された。
【0153】
モデリング研究(Booth, 2009;Aim 3)に基づいて、さらなる類似体が、PATテトラヒドロナフチル環の芳香族部分と相互作用すると仮定され、相互作用することが知られている(突然変異誘発データ)、保存された5HT2残基V3.33、S3.36、S5.43&F6.52とさらなる水素結合を形成するように設計された。
【0154】
本発明はまた、前記化合物の薬剤的に許容される塩及びエステルに関する。
【0155】
天然に存在するか又は合成異性体は、当該技術分野で公知の幾つかの方法で分離することができる。2つのエナンチオマーのラセミ混合物を分離するための方法には、キラル固定相を用いたクロマトグラフィー(例えば、“Chiral Liquid Chromatography”, WJ. Lough, Ed. Chapman and Hall, New York(1989)を参照)が含まれる。エナンチオマーは、古典的な分割技術によって分離することができる。たとえば、ジアステレオマー塩の形成及び分別結晶を用いてエナンチオマーを分離することもできる。カルボン酸のエナンチオマーの分離に関して、エナンチオマー的に純粋なキラル塩基、例えばブルシン、キニーネ、エフェドリン、ストリキニーネなどの添加により、ジアステレオマー塩を形成することができる。或いは、ジアステレオマーエステルを、エナンチオマー的に純粋なキラルアルコール、例えばメントールを用いて形成することができ、続いてジアステレオマーエステルを分離し、加水分解して、遊離したエナンチオマー的に富化されたカルボン酸を得る。アミノ化合物の光学異性体の分離のために、キラルカルボン酸又はスルホン酸、例えばカンファースルホン酸、酒石酸、マンデル酸、又は乳酸の添加によって、ジアステレオマー塩を形成することができる。
【0156】
別の実施形態によれば、本発明は、本明細書中に記載される方法によって産生又は同定される、GPCR又はその結合ポケットと会合又は結合する化合物を提供する。
【0157】
3.本発明の化合物の使用
一実施形態では、本発明は、GPCR介在性障害に関して対象を治療するための方法であって、対象に、GPCR標的を調節(刺激、拮抗)することができる有効量の化合物を投与することによる方法を提供する。GPCR障害には、そのようなGPCRが関与する疾患及び障害が含まれる。本明細書中に記載される化合物、組成物及び方法は、たとえば、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、5−HT2b介在性疾患、高血圧)、胃腸障害(例えば、過敏性腸症候群)、及び泌尿生殖路障害(例えば、膀胱制御)をはじめとする障害の治療又は予防に有用である。ある実施形態では、対象は、哺乳類、例えば霊長類、例えばヒトである。
【0158】
一実施形態では、本発明は、ヒスタミン標的を調節(刺激、拮抗)することができる有効量の化合物を投与することによって、ヒスタミン(例えば、H1、H2、H3、H4)が関与する障害に関して対象を治療するための化合物及び方法を提供する。ヒスタミン障害には、そのようなヒスタミン(例えば、H1)が関与する疾患及び障害が含まれる。本明細書中で記載される化合物、組成物及び方法は、たとえば、呼吸困難(例えば、気管支狭窄)、下痢(GI収縮)、浮腫、及び低血圧(例えば、増大した血管透過性)、アレルギー反応、並びに本明細書中の神経精神医学的、神経変性及び神経障害をはじめとする障害の治療又は予防に有用である。
【0159】
この実施形態では、本発明の化合物は、5−HT2c又はその特定のドメインの活性を直接的又は間接的のいずれかで調節(例えば、刺激、刺激)することができる。細胞は、本発明の化合物と接触して、5−HT2cを刺激し、そして5−HT2c介在性活性を調節することができる。細胞を接触させるか、又は本発明の化合物を対象に投与することは、不必要な若しくは望ましくない5−HT2C介在性活性又は5−HT2c介在性障害に罹っているか或いは罹りやすい細胞又は対象を治療する一方法である。
【0160】
一実施形態では、5−HT2c障害に罹っているか又は罹りやすい対象を治療する方法は、それを必要とする対象に、5−HT2cの活性を直接的若しくは間接的に調節することができる治療有効量の化合物を投与し、それによって対象を治療することを含む。例示的化合物には、本明細書中に記載される化合物又は式(式I、II、II−a、III、IV又はV)が含まれる。
【0161】
したがって、一実施形態では、本発明は、5−HT2C障害に関して対象を治療する方法であって、対象に有効量の5−HT2cを刺激することができる化合物を投与することによる方法を提供する。
【0162】
別の態様では、本発明は、対象において肥満を治療又は予防する方法であって、それを必要とすると認定された対象に対して、本明細書中のいずれかの式の化合物とアンフェタミン化合物との組み合わせを投与することを含む方法を提供する。化合物を、併用して、同時に、又は連続して投与することができる。複数の態様では、アンフェタミン化合物はフェンテルミンである。
【0163】
ある実施形態では、本発明の方法は、治療有効量の本発明の化合物を、別の医薬活性化合物と組み合わせて対象に投与することを含む。使用できる他の医薬活性化合物は、Harrison’s Principles of Internal Medicine, Thirteenth Edition, Eds. T.R. Harrison et al. McGraw−Hill N.Y., NY;及びthe Physicians Desk Reference 50th Edition 1997, Oradell New Jersey, Medical Economics Co.(その全内容は参照することによって明確に本明細書中に組み込まれる)で見出すことができる。本発明の化合物及び医薬活性化合物を、同じ医薬組成物中で、又は異なる医薬組成物中で(同時に又は異なる時間に)対象に投与することができる。
【0164】
一態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に対して、5−HT2C受容体(例えば、本明細書中の化合物)を活性化することができる治療有効量の化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。一実施形態では、化合物は5−HT2c受容体を活性化することができる。別の実施形態では、化合物は、5−HT2A及び/又は5−HT2β受容体を拮抗するが、5−HT2cを活性化することができる。別の実施形態では、化合物は、5−HT2の活性化、及び/又は5−HT2A及び/又は5−HT2β受容体の拮抗が可能である。別の実施形態では、化合物は、5−HT2A及び/又は5−HT2β受容体を拮抗することができる。
【0165】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に、治療有効量の5−HT2c活性化化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0166】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とする対象に、好ましくは5−HT2a及び/又は5−HT2bと比べて選択的に5−HT2cを直接調節することにより5−HT2結合相互作用を調節することができる治療有効量の化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0167】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、それを必要とすると認定された対象(すなわち、肥満対象)に、治療有効量の5−HT2c刺激化合物又は5−HT2c選択的化合物を、アンフェタミン化合物と組み合わせて投与することを含む方法を提供する。
【0168】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、対象に、5−HT2c刺激化合物をアンフェタミン化合物と組み合わせて投与して、肥満が防止、改善又は治療されるようにする(例えば体重減少が観察され、体重増加が防止される)ことを含む方法を提供する。
【0169】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、5−HT2a及び/又は5−HT2b活性化と比べて選択的に(単独又はアンフェタミンとの組み合わせで)5−HT2cを活性化することができる有効量の化合物を、アンフェタミン化合物と組み合わせて、対象に投与して、対象が治療されるようにすることを含む方法を提供する。
【0170】
別の態様では、本発明は、肥満に苦しんでいるか又は肥満になりやすい対象を治療する方法であって、5−HT2a又は5−HT2b受容体を刺激(及び/又は拮抗)しない(又はあまりしない)が、(例えば、5−HT2a及び/又は5−HT2bと比べて選択的に)5−HT2cを刺激することができる有効量の化合物を、アンフェタミン化合物と組み合わせて対象に投与して、対象が治療されるようにすることを含む方法を提供する。
【0171】
別の態様では、本発明は、疾患、障害又はその症状に苦しんでいるか又は罹りやすい対象を治療する方法であって、5−HT2cアゴニストとして同定された有効量の化合物(例えば、本明細書中の式の化合物)を投与することを含む方法を提供する。別の態様では、化合物は、5−HT2cアゴニスト及び5−HT2a逆アゴニスト(例えば、本明細書中の式の化合物)として同定される。別の態様では、化合物は、5−HT2c逆アゴニスト及び5−HT2a逆アゴニスト(例えば、本明細書中の式の化合物)として同定される。別の態様では、化合物は、5−HT2a又は5−HT2b(例えば、本明細書中の式の化合物)を刺激しない(又はあまりしない)、選択的な5−HT2cアゴニストとして同定される。
【0172】
別の態様では、治療方法は、対象(例えば、患者)が、そのような治療を必要としていると認定されることを含む。別の態様では、治療方法は、対象(例えば、患者)が、本明細書中の化合物又は組成物を投与されて、本明細書中に記載される疾患、障害又は症状について治療されることを含む。
【0173】
別の態様では、本発明は、アンフェタミン化合物と組み合わせた本明細書中のいずれかの式の治療有効量の化合物及び薬剤的に許容される担体又は希釈剤を含むパッケージされた組成物を提供する。組成物は、肥満に苦しんでいるか又は肥満になりやすい対象を治療するために処方することができ、肥満に苦しんでいるか又は肥満になりやすい対象を治療するための説明書とともにパッケージすることができる。
【0174】
ある実施形態では、本発明の化合物は、通常の抗肥満薬(例えば、脂肪吸収遮断薬)との併用療法で用いることができる。ある5−HT薬物(例えば、5−HTa又は5−HTb拮抗物質)は、それらをある患者にとって最適ではないか又は適しないものとする傾向がある望ましくない副作用を有する。すなわち、それらは心臓血管(例えば、心臓弁膜症、肺高血圧症、新毒性)又は精神医学的な望ましくない及び/又は生命を脅かす副作用特性を示す。
【0175】
一実施形態では、本発明の化合物は、RSK又はその特定のドメインの活性を直接的又は間接的にいずれかで調節(例えば阻害)することができる。細胞を本発明の化合物と接触させて、RSKを阻害し、RSK介在性活性を調節することができる。細胞を接触させるか、又は本発明の化合物を対象に投与することは、不必要又は望ましくないRSK介在性活性又はRSKが関与する障害に苦しんでいるか、又は影響を受けやすい細胞又は対象を治療する一方法である。
【0176】
一実施形態では、RSKが関与する障害に罹っているか、又は罹りやすい対象を治療する方法は、それを必要とする対象に、RSKの活性を直接的又は間接的に調節することができる治療有効量の化合物を投与し、それによって対象を治療することを含む。例示的化合物には、本明細書中に記載される化合物又は式(式I、II、II−a、III、IV、又はV)が含まれる。
【0177】
したがって、一実施形態では、本発明は、RSKが関与する障害について対象を治療する方法であって、RSK−1、RSK−3、又はRSK−4を阻害することができる有効量の化合物を対象に投与することによる方法を提供する。
【0178】
別の態様では、本発明は、対象における癌(例えば、胸部及び前立腺)、HIV、又はコフィン・ローリー症候群を治療又は予防する方法であって、それを必要とすると認定された対象に、本明細書中のいずれかの式の化合物及びさらなる治療化合物(例えば、抗がん剤、抗HIV剤、コフィン・ローリー症候群剤)の組み合わせを投与することを含む方法を提供する。化合物を、併用して、同時に、又は連続して投与することができる。
【0179】
ある実施形態では、本発明の方法は、対象に、治療有効量の本発明の化合物を別の医薬活性化合物と組み合わせて投与することを含む。使用できる他の医薬活性化合物は、Harrison’s Principles of Internal Medicine, Thirteenth Edition, Eds. T.R. Harrison et al. McGraw−Hill N.Y., NY;及びthe Physicians Desk Reference 50th Edition 1997, Oradell New Jersey, Medical Economics Co.,(その全内容は、参照することによって明確に本明細書中に組み込まれる)で見いだすことができる。本発明の化合物及び医薬活性化合物は、同じ医薬組成物中又は異なる医薬組成物中で(同時に又は異なる時点で)対象に投与することができる。
【0180】
別の態様では、治療方法は、対象(例えば、患者)がそのような治療を必要であると認定されることを含む。別の態様では、治療方法は、対象(例えば、患者)に本明細書中の化合物又は組成物を投与して、その対象が、本明細書中に記載される疾患、障害又は症状について治療されるようにすることを含む。
【0181】
本発明の別の目的は、RSK介在性障害の治療において使用するための医薬の製造における、本明細書中に記載される(例えば、本明細書中の任意の式の)化合物の使用である。本発明の別の目的は、RSKが関与する障害の治療において使用するための、(例えば、本明細書中の任意の式の)本明細書中に記載される化合物の使用である。
【0182】
本発明の化合物の治療有効量又は予防有効量は、当業者として、医師又は獣医師(「担当臨床医」)が、公知技術を使用することによるか、又は類似の状況下で得られる結果を観察することによって決定することができる。投与量は、担当臨床医の判断で、患者の要求;治療される状態の重篤度及び用いられる特定の化合物に応じて変えることができる。治療有効量又は用量、及び予防有効量又は用量の決定において、これらに限定されるものではないが:関係する特定の5−HT2c及び/又はRSK障害;特定の薬剤の薬力学的特性並びにその投与様式及び経路;治療の所望の時間経過;哺乳類の種;そのサイズ、年齢、及び全体的な健康;関係する特定の疾患;疾患の程度又は疾患の関与又は重篤度;個々の患者の反応;投与される特定の化合物;投与様式;投与される製剤の生体利用性;選択される投与計画;併用療法の種類(すなわち、本発明の化合物と他の同時投与される治療薬との相互作用);及び他の関連する状況をはじめとする多くの因子が担当臨床医によって考慮される。
【0183】
治療は、より少ない用量から始めることができ、この量は、化合物の最適用量よりも少ない。その後、その状況下で最適の効果に達するまで、投与量を少しずつ増やすことができる。便宜上、所望により、総一日量を分割し、一日中で数回に分けて投与することができる。本発明の化合物の治療有効量及び予防有効量は、1日につき体重1キログラムあたり約0.1ミリグラム(mg/kg/日)〜約100mg/kg/日で変化すると予想される。
【0184】
動物、例えば、イヌ、ニワトリ、及び齧歯類において、5−HT2c及び/又はRSK障害の予防又は治療に有効であることが見いだされた化合物は、ヒトにおける5−HT2c及び/又はRSK障害の治療にも有用である可能性がある。ヒトにおける5−HT2c及び/又はRSK疾患の治療をする当業者らは、動物実験で得られたデータに基づいて、ヒトへの当該化合物の投与量及び経路がわかるであろう。一般的に、ヒトにおける投与量及び投与経路は、動物においてと類似していると予想される。
【0185】
5−HT2C及び/又はRSK障害の予防的治療を必要とする患者の認定は、十分に当業者の技術及び知識の範囲内である。対象の方法により治療することができる5−HT2C及び/又はRSK障害を発症する危険性がある患者を特定するためのある方法、たとえば家族歴、及び対象患者における疾患状態の発症に関連する危険因子の存在は、医術で理解される。当該技術に熟達した臨床医は、そのような候補患者を、たとえば、臨床試験、健康診断及び病歴/家族歴の使用により、容易に特定することができる。
【0186】
対象における治療の有効性を評価する方法は、当該技術分野で周知の方法(例えば、5−HT2C及び/又はRSK障害のマーカーのレベルを測定すること)によって5−HT2C及び/又はRSK障害の前処理の程度を測定し、次いで治療有効量の本明細書中に記載される化合物を本発明にしたがって対象に投与することを含む。化合物の投与の適切な期間(例えば、1日、1週、2週、1か月、6か月)の後に、5−HT2C及び/又はRSK障害の程度を再度測定する。5−HT2C及び/又はRSK障害の程度又は侵襲性の調節(たとえば、低減)は、治療の有効性を示す。5−HT2C及び/又はRSK障害の程度又は侵襲性は、治療を通して定期的に測定することができる。たとえば、5−HT2C及び/又はRSK障害の程度又は侵襲性を、数時間ごと、数日ごと又は数週間ごとにチェックして、治療のさらなる有効性を評価することができる。5−HT2C及び/又はRSK障害の程度又は侵襲性の減少は、治療が有効であることを示す。記載した方法を用いて、5−HT2C及び/又はRSK障害の調節化合物を用いた治療から恩恵を得る可能性がある患者をスクリーニング又は選択することができる。
【0187】
対象における治療の有効性を評価する方法は、当該技術分野で周知の方法による肥満(太りすぎ)の前治療の程度を測定し、次いで治療有効量の本明細書中に記載される化合物を単独又はそれぞれとアンフェタミン化合物との組み合わせで本発明にしたがって対象に投与することを含む。化合物の投与後の適切な期間(例えば、1日、1週、2週、1か月、6か月)の後、肥満(太りすぎ)の程度を再度測定する。太りすぎの程度の調節(たとえば減少)は、治療の有効性を示す。肥満の程度又は侵襲性は、治療を通して定期的に測定することができる。たとえば、太りすぎの程度又は侵襲性を、数時間ごと、数日ごと、又は数週間ごとにチェックして、治療の有効性をさらに評価することができる。太りすぎの程度又は侵襲性の減少は、治療が有効であることを示す。記載される方法を用いて、アンフェタミン化合物との組み合わせで選択的に5−HT2c(対5−HT2a、b)受容体を活性化することができる調節化合物での治療から恩恵を受ける可能性がある患者をスクリーニング又は選択することができる。
【0188】
本明細書中で用いられるように、「対象から生物学的試料を得る」には、本明細書中に記載される方法で使用するための試料を得ることが含まれる。生物学的試料は前述されている。
【0189】
さらに別の態様は、5−HT2C及び/又はRSK又はその特定のドメインの相互作用を調節する化合物を同定するための方法を提供する。この方法は、試験化合物の存在下及び/又は非存在下で、5−HT2C及び/又はRSK又はその特定のドメインの結晶構造(場合によってアポ形若しくは複合体化)を得るか、或いは5−HT2C及び/又はRSK又はその特定のドメインの結晶構造(場合によってアポ形若しくは複合体化)に関する情報を得ることを含み得る。化合物を次いで5−HT2C及び/又はRSK構造、又はその特定のドメイン(例えば、結晶構造の結合部位)中又は上でコンピュータでモデル化して、5−HT2C及び/又はRSK又はその特定のドメイン及び試験化合物間の相互作用の安定化を予想することができる。潜在的な調節化合物が同定されたら、細胞アッセイ、たとえば本明細書中で特定されたもの及び当該技術分野で公知の競合アッセイを用いてその化合物をスクリーニングすることができる。このようにして特定された化合物は、治療薬として有用である。
【0190】
別の態様では、本発明の化合物は、治療有効量で、薬剤的に許容される担体又は希釈剤とともにパッケージされる。組成物は、5−HT2C及び/又はRSK障害に罹っているか又は罹りやすい対象を治療するために処方することができ、そして5−HT2C及び/又はRSK障害に罹っているか又は罹りやすい対象を治療するための説明書とともにパッケージすることができる。
【0191】
別の態様では、本発明は、5−HT2C及び/又はRSK疾患を調節するための方法を提供する。一実施形態では、本発明による5−HT2C(又は5−HT2C障害)を調節する方法は、細胞と、5−HT2C(若しくは5−HT2C障害)、又はその特定のドメインを調節することができる化合物とを接触させることを含む。一実施形態では、本発明によるRSK(又はRSK障害)を調節する方法は、細胞と、RSK(若しくはRSK障害)、又はその特定のドメインを調節することができる化合物とを接触させることを含む。いずれの実施形態においても、接触は、インビトロで、たとえば、細胞を取り巻く流体、たとえば、細胞がその中で生きているか又は存在している成長媒体に化合物を添加することによってであってもよい。接触は、化合物を細胞に直接接触させることによってであってもよい。別法として、接触は、インビボで、たとえば、投与経路に応じて、たとえば投与後に、対象中に化合物を通過させることによってであってもよく、化合物は、消化管又は血流中で運ばれる可能性があるか、又は処理を必要とする細胞に直接適用若しくは投与することができる。
【0192】
別の態様では、対象における5−HT2C及び/又はRSK障害を阻害する方法は、有効量の本発明の化合物(すなわち、本明細書中で記載される化合物)を対象に投与することを含む。投与は、製薬分野で公知の任意の投与経路によってであってよい。対象は、5−HT2C及び/又はRSK障害を有する可能性があるか、5−HT2C及び/又はRSK障害を発症する危険性があるか、又は5−HT2C及び/又はRSK障害に対する感受性を増大させ得る状態に対する予定された暴露又は予期せぬ暴露の前に予防的治療を必要とする可能性がある。
【0193】
一態様では、本明細書中の化合物で治療される対象の経過をモニタリングする方法は、5−HT2C及び/又はRSK障害の治療前状態(例えば、進行、標的特性、マーカー特性)を測定し、治療有効量の本明細書中の化合物を対象に投与し、そして当該化合物での治療の初期期間後の5−HT2C及び/又はRSK障害の状態(例えば、進行、標的特性、マーカー特性)を測定することを含み、この場合、状態の調節は、治療の有効性を示す。
【0194】
対象は、5−HT2C及び/又はRSK障害の危険性の可能性があり、5−HT2C及び/又はRSK障害の症状を示す可能性があり、5−HT2C及び/又はRSK障害に罹りやすい可能性があり、及び/又は5−HT2C及び/又はRSK障害を有すると診断されている可能性がある。
【0195】
状態の調節が、対象が治療に対して有利な臨床反応を有し得ることを示す場合、対象は、当該化合物で治療することができる。たとえば、対象に治療有効量の化合物を投与することができる。
【0196】
別の態様では、試験化合物を評価するための方法は、5−HT2C及び/又はRSK又はその特定のドメインを試験化合物(複合体)と接触させ、そして接触後の結合相互作用を評価することを含み、この場合、参考値に対する複合体の安定性における変化は、複合体の安定性を調節することの表れである。
【0197】
5−HT2C及び/又はRSK又はその特定のドメイン複合体をコンピュータでモデル化することができるか、或いは細胞から単離されるか、細胞若しくは組換え発現系から組み換え発現されるか、精製されるか、若しくは単離されるか、又は細胞若しくは組換え発現系から部分的に精製されるか若しくは単離された細胞内の複合体であり得る。
【0198】
本発明のキットは、対象における5−HT2C及び/又はRSK障害を治療するためのキットを含む。キットは、本発明の化合物,たとえば本明細書中で記載される化合物、その薬剤的に許容されるエステル、塩、及びプロドラッグ、並びに使用説明書を含み得る。使用説明書は、用量、送達方法、キットの保存などに関する情報を含み得る。キットは、試薬、たとえば、試験化合物、緩衝液、培地(例えば、細胞増殖培地)、細胞なども含み得る。試験化合物は、既知化合物又は新たに発見された化合物、たとえば化合物のコンビナトリアルライブラリを含み得る。1以上の本発明のキットを合わせてパッケージすることができ、たとえば、5−HT2C及び/又はRSK障害についての治療の有効性を評価するためのキットを、本発明による5−HT2C及び/又はRSK障害に関して治療される対象の経過をモニタリングするためのキットとともにパッケージすることができる。
【0199】
本発明の方法は、培養物中、たとえばインビトロ若しくはエクスビボの細胞、又は動物対象中に存在する細胞、たとえばインビボの細胞に関して実施することができる。本発明の化合物は、まず、形質転換細胞などの細胞の一次培養を用いてインビトロで試験することができる。
【0200】
本発明の方法は、培養物中、たとえばインビトロ若しくはエクスビボの細胞、又は動物対象中に存在する細胞、たとえばインビボの細胞に関して実施することができる。本発明の化合物は、まず、齧歯類胎仔(たとえば、米国特許第5,179,109号−胎仔ラット細胞培養を参照)、又は他の哺乳類(たとえば、米国特許第5,089,517号−胎仔マウス組織培養を参照)又は非哺乳動物の動物モデルからの気道由来の細胞を用いてインビトロで試験することができる。
【0201】
或いは、本発明の化合物の効果は、動物モデルを用いてインビボで特性化することができる。
【0202】
4.医薬組成物
本発明は、有効量の化合物及び薬剤的に許容される担体を含む医薬組成物も提供する。さらなる実施形態では、有効量は、前述のように、5−HT2C及び/又はRSK障害を治療するために有効である。
【0203】
一実施形態では、本発明の化合物は、薬剤的に許容される処方、たとえば、薬剤的に許容される処方が対象に投与された後、少なくとも12時間、24時間、36時間、48時間、1週間、2週間、3週間、又は4週間、本発明の化合物を対象に持続送達する薬剤的に許容される処方を用いて対象に投与される。
【0204】
ある実施形態では、これらの医薬組成物は、対象への局所又は経口投与に適している。他の実施形態では、以下で詳細に記載するように、本発明の医薬組成物は、特に:(1)経口投与(たとえば、飲薬(水性若しくは非水性溶液又は懸濁液)、錠剤、ボーラス、粉末、顆粒、ペースト);(2)非経口投与(例えば滅菌溶液若しくは懸濁液として皮下、筋肉内又は静脈内注射による);(3)局所適用(たとえば、皮膚に適用されるクリーム、軟膏又はスプレー);(4)膣内又は直腸内で(例えばペッサリー、クリーム又はフォームとして);或いは(5)エアゾル(例えば化合物を含有する水性エアゾル、リポソーム製剤又は固体粒子として)のために適用されるものをはじめとする、固体又は液体形態での投与用に処方することができる。
【0205】
「薬剤的に許容される」という表現は、健全な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応又は他の問題若しくは合併症がなく、ヒト及び動物の組織と接触して使用するのに適し、妥当な損益比に見合う、本発明の化合物、そのような化合物を含む組成物、及び/又は投与形態を指す。
【0206】
「薬剤的に許容される担体」という表現は、1つの器官、又は身体の部分から、別の器官、若しくは身体の部分への対象の化学物質の運搬若しくは輸送に関与する、薬剤的に許容される物質、組成物又はビヒクル、たとえば液体若しくは固体フィラー、希釈剤、賦形剤、溶媒又は封入材料を包含する。各担体は、処方の他の成分と適合性であり、患者に有害でないという意味で、「許容される」。薬剤的に許容される担体として機能し得る材料の数例としては:(1)糖、たとえばラクトース、グルコース及びスクロース;(2)デンプン、たとえばコーンスターチ及びジャガイモデンプン;(3)セルロース、及びその誘導体、たとえばカルボキシメチルセルロースナトリウム、エチルセルロース及び酢酸セルロース;(4)トラガカント粉末;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤、たとえばカカオバター及び坐薬ワックス;(9)油、たとえばピーナッツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、コーン油及び大豆油;(10)グリコール、たとえばプロピレングリコール;(11)ポリオール、たとえばグリセリン、ソルビトール、マンニトール及びポリエチレングリコール;(12)エステル、たとえばオレイン酸エチル及びラウリン酸エチル;(13)寒天;(14)緩衝剤、たとえば水酸化マグネシウム及び水酸化アルミニウム;(15)アルギン酸;(16)パイロジェンフリー水;(17)等張性生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸塩緩衝液;及び(21)医薬処方で用いられる他の非毒性適合性物質が挙げられる。
【0207】
湿潤剤、乳化剤及び潤滑剤、たとえばラウリル硫酸ナトリウム及びステアリン酸マグネシウム、並びに着色剤、放出剤、コーティング剤、甘味料、矯味矯臭剤及び香料、保存料及び酸化防止剤も組成物中に存在してもよい。
【0208】
薬剤的に許容される酸化防止剤の例としては:(1)水溶性酸化防止剤、たとえばアスコルビン酸、塩酸システイン、硫酸水素ナトリウム、メタ亜硫酸水素ナトリウム、亜硫酸ナトリウムなど;(2)油溶性酸化防止剤、たとえばパルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、アルファ−トコフェロールなど;及び(3)金属キレート剤、たとえばクエン酸、エチレンジアミンテトラ酢酸(EDTA)、ソルビトール、酒石酸、リン酸などが挙げられる。
【0209】
本発明の化合物を含む組成物には、経口、鼻、局所(頬側及び舌下を包含する)、直腸、膣、エアゾル及び/又は非経口投与に適したものが含まれる。組成物は、便宜上、単位投与形態で提供することができ、調剤の技術分野で周知の方法によって調製することができる。単一投与形態を得るために担体材料と組み合わせることができる活性成分の量は、治療される宿主、特定の投与様式に応じて変わるであろう。単一投与形態を得るために担体材料と組み合わせることができる活性成分の量は、一般的に、治療効果をもたらす化合物の量であろう。一般的に、100パーセントのうち、この量は、活性成分の約1パーセント〜約99パーセント、好ましくは約5パーセント〜約70パーセント、さらに好ましくは約10パーセント〜約30パーセントの範囲である。
【0210】
これらの組成物の調製法は、本発明の化合物(複数可)を担体、及び場合によって1以上の補助成分と会合させるステップを含む。一般的に、処方は、本発明の化合物を液体担体、又は固体担体微粉末、又は両者と均一かつ密接に会合され、次いで必要ならば生成物を成形することにより調製される。
【0211】
経口投与に適した本発明の組成物は、カプセル、カシェ剤、丸薬、錠剤、ロゼンジ(フレーバー付主成分、通常、スクロース及びアカシア又はトラガカントを使用)、粉末、顆粒の形態で、又は水性若しくは非水性液体中溶液若しくは懸濁液として、又は水中油若しくは油中水液体エマルジョンとして、又はエリキシル若しくはシロップとして、又はパステル(不活性基剤、たとえばゼラチン及びグリセリン、又はスクロース及びアカシアを使用)として及び/又は洗口剤などとしてであってよく、それぞれは、あらかじめ決められた量の本発明の化合物(複数可)を活性成分として含む。化合物は、ボーラス、舐剤又はペーストとして投与することもできる。
【0212】
経口投与用の本発明の固体投与形態(カプセル、錠剤、丸薬、糖衣剤、粉末、顆粒など)において、活性成分を、1以上の薬剤的に許容される担体、たとえば、クエン酸ナトリウム又はリン酸二カルシウム、及び/又は以下のいずれか:(1)フィラー又は増量剤、たとえばデンプン、ラクトース、スクロース、グルコース、マンニトール、及び/又はケイ酸;(2)結合剤、たとえば、カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロース及び/又はアカシア;(3)湿潤剤、たとえばグリセロール;(4)崩壊剤、たとえば寒天−寒天、炭酸カルシウム、ジャガイモ若しくはタピオカデンプン、アルギン酸、ある種のケイ酸塩、及び炭酸ナトリウム;(5)溶解遅延剤、たとえばパラフィン;(6)吸収促進剤、たとえば第四アンモニウム化合物;(7)湿潤剤、たとえば、アセチルアルコール及びグリセロールモノステアレート;(8)吸収剤、たとえばカオリン及びベントナイトクレイ;(9)潤滑剤、たとえばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、及びそれらの混合物;並びに(10)着色剤と混合する。カプセル、錠剤及び丸薬の場合、医薬組成物は緩衝剤も含み得る。類似の種類の固体組成物は、ラクトース又は乳糖などの賦形剤、並びに高分子量ポリエチレングリコールなどを用いて、ソフト及びハード充填ゼラチンカプセル中フィラーとして用いることもできる。
【0213】
場合によって1以上の補助成分とともに圧縮又は成形することによって錠剤を作成することができる。圧縮錠は、結合剤(たとえば、ゼラチン若しくはヒドロキシプロピルメチルセルロース)、潤滑剤、不活性希釈剤、保存料、崩壊剤(たとえば、デンプングリコール酸ナトリウム若しくは架橋カルボキシメチルセルロースナトリウム)、表面活性剤又は分散剤を用いて調製することができる。成形錠剤は、好適な機械中で、不活性液体希釈剤で湿らせた活性成分粉末の混合物を成形することによって作成することができる。
【0214】
錠剤、及び糖衣剤、カプセル、丸薬及び顆粒などの他の固体投与形態の本発明の医薬組成物は、場合によって、刻み目をつけるか、又は腸溶コーティング及び他の医薬処方技術分野で周知の他のコーティングなどのコーティング及びシェルを用いて調製することができる。それらは、たとえば、所望の放出特性を得るために様々な割合のヒドロキシプロピルメチルセルロース、他のポリマーマトリックス、リポソーム及び/又は微小球を用いて、その中の活性成分の遅延又は制御放出をもたらすように処方することもできる。それらは、細菌保持フィルターを通した濾過によるか、又は滅菌水、又は他の滅菌注射可能媒体中に使用直前に溶解させることができる滅菌固体組成物の形態中に滅菌剤を組み入れることによって、滅菌することができる。これらの組成物は、場合によって、不透明化剤を含んでもよく、胃腸管のある部分でのみ、又は胃腸管のある部分で優先的に、場合によって遅延された方法で、活性成分を放出する組成物であってもよい。使用できる包埋組成物の例としては、ポリマー物質及びワックスが挙げられる。活性成分は、適切ならば前記賦形剤とともにミクロカプセル化された形態であってもよい。
【0215】
本発明の化合物の経口投与用液体投与形態には、薬剤的に許容されるエマルジョン、ミクロエマルジョン、溶液、懸濁液、シロップ及びエリキシルが含まれる。活性成分に加えて、液体投与形態は、当該技術分野で通常用いられる不活性希釈剤、たとえば、水又は他の溶媒、可溶化剤及び乳化剤、たとえば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、コーン油、胚芽油、オリーブ油、ヒマシ油及びゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコール及びソルビタンの脂肪酸エステル、並びにそれらの混合物を含んでもよい。
【0216】
不活性希釈剤に加えて、経口組成物は、湿潤剤、乳化剤及び懸濁化剤、甘味料、矯味矯臭剤、着色剤、香料及び保存料などのアジュバントを含むことができる。
【0217】
懸濁液は、本発明の活性化合物に加えて、たとえば、エトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトール及びソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天−寒天及びトラガカント、並びにそれらの混合物などの懸濁化剤を含んでもよい。
【0218】
直腸又は膣投与用の本発明の医薬組成物は、坐薬として提供することができ、坐薬は、1以上の本発明の化合物を、たとえば、カカオバター、ポリエチレングリコール、坐薬ワックス又はサリチレートを含む1以上の好適な非刺激性賦形剤又は担体と混合することによって調製することができ、室温で固体であるが、体温で液体であり、したがって直腸又は膣腔中で融解し、活性剤を放出する。
【0219】
膣投与に適した本発明の組成物としては、当該技術分野で適切であると知られている担体を含むペッサリー、タンポン、クリーム、ゲル、ペースト、フォーム又はスプレー処方が挙げられる。
【0220】
本発明の化合物の局所又は経皮投与用投与形態には、粉末、スプレー、軟膏、ペースト、クリーム、ローション、ゲル、溶液、貼付剤及び吸入剤が含まれる。本発明の活性化合物は、無菌条件下で、薬剤的に許容される担体と、及び任意の保存料、緩衝液、又は必要とされ得るプロペラントと混合することができる。
【0221】
軟膏、ペースト、クリーム及びゲルは、本発明の化合物に加えて、賦形剤、たとえば動物性及び植物性脂、油、ワックス、パラフィン、デンプン、トラガカント、セルロース誘導体、ポリエチレングリコール、シリコーン、ベントナイト、ケイ酸、タルク及び酸化亜鉛、又はそれらの混合物を含んでもよい。
【0222】
粉末及びスプレーは、本発明の化合物に加えて、賦形剤、たとえばラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム及びポリアミド粉末、又はこれらの物質の混合物を含むことができる。スプレーは、慣例的なプロペラント、たとえばクロロフルオロ炭化水素及び揮発性非置換炭化水素、たとえばブタン及びプロパンをさらに含むことができる。
【0223】
本発明の化合物(複数可)は、別法として、エアゾルにより投与することができる。これは、当該化合物を含む水性エアゾル、リポソーム製剤又は固体粒子を調製することによって達成される。非水性(例えば、フルオロカーボンプロペラント)懸濁液を使用することができる。音波ネブライザーが好ましい。なぜなら、これらは化合物が結果として分解される可能性があるせん断に暴露されることを最小限に抑えるからである。
【0224】
通常、水性エアゾルは、薬剤の水性溶液又は懸濁液を通常の薬剤的に許容される担体及び安定剤とともに処方することによって調製される。担体及び安定剤は、特定の化合物の要件によって変わるが、典型的には、非イオン性界面活性剤(Tween、Pluronic、又はポリエチレングリコール)、血清アルブミンなどの無害タンパク質、ソルビタンエステル、オレイン酸、レシチン、グリシンなどのアミノ酸、緩衝液、塩、糖又は糖アルコールを含む。エアゾルは、一般的に、等張性溶液から調製される。
【0225】
経皮貼付剤は、本発明の化合物の身体への制御送達を提供するさらなる利点を有する。そのような投与形態は、適切な媒体中に当該薬剤を溶解又は分散させることによって作成することができる。吸収促進剤を用いて、皮膚を越える活性成分の流動を増大させることもできる。そのような流動速度は、速度制御膜を提供するか、又はポリマーマトリックス若しくはゲル中に活性成分を分散させるかのいずれかによって制御することができる。
【0226】
眼用処方、眼軟膏、粉末、溶液なども本発明の範囲内に含まれることが想定される。
【0227】
非経口投与に適した本発明の医薬組成物は、1以上の本発明の化合物を1以上の薬剤的に許容される滅菌等張性水性若しくは非水性溶液、分散液、懸濁液又はエマルジョン、或いは使用直前に滅菌注射可能な溶液又は分散液中に再構成することができる滅菌粉末と組み合わせて含み、酸化防止剤、緩衝液、静菌剤、処方を意図されるレシピエントの血液と等張性にする溶質、又は懸濁剤若しくは増粘剤を含んでもよい。
【0228】
本発明の医薬組成物において用いることができる好適な水性及び非水性担体の例としては、水、エタノール、ポリオール(たとえばグリセロール、プロピレングリコール、ポリエチレングリコールなど)、及びそれらの好適な混合物、植物性油、たとえばオリーブ油、並びに注射可能な有機エステル、たとえばオレイン酸エチルが挙げられる。適切な流動性は、たとえば、コーティング材料、たとえばレシチンの使用により、分散液の場合は必要とされる粒子サイズの維持により、そして界面活性剤の使用により、維持することができる。
【0229】
これらの組成物は、保存料、湿潤剤、乳化剤及び分散剤などのアジュバントを含んでもよい。微生物の作用の予防は、様々な抗菌剤及び抗真菌剤、たとえば、パラベン、クロロブタノール、フェノールソルビン酸などを含めることによって確実にすることができる。等張剤、たとえば糖、塩化ナトリウムなどを組成物中に含めることも望ましい場合がある。加えて、注射可能な医薬形態の長時間にわたる吸収は、アルミニウムモノステアレート及びゼラチンなどの吸収を遅らせる薬剤を含めることによってもたらされ得る。
【0230】
場合によっては、薬物の効果を延長するために、皮下又は筋肉内注射からの薬物の吸収を遅らせることが望ましい。これは、水溶性が低い結晶性又はアモルファス材料の懸濁液の使用により達成することができる。そして薬物の吸収速度は、その溶解速度に依存し、これは次に結晶サイズ及び結晶形態に依存し得る。或いは、非経口投与された薬物形態の遅延吸収は、薬物を油性ビヒクル中に溶解又は懸濁させることによって達成される。
【0231】
注射可能なデポー形態は、ポリラクチド−ポリグリコリドなどの生分解性ポリマー中本発明の化合物のマイクロカプセル化マトリックスを形成することによって作製される。薬物のポリマーに対する割合に応じて、そして用いられる特定のポリマーの性質に応じて、薬物放出速度を制御することができる。生分解性ポリマーの例としては、ポリ(オルトエステル)及びポリ(無水物)が挙げられる。デポー注射可能な処方は、身体組織と適合性であるリポソーム又はミクロエマルジョン中に薬物をトラップすることによっても調製される。
【0232】
本発明の化合物を医薬としてヒト及び動物に投与する場合、それらはそのまま又はたとえば、0.1〜99.5%(更に好ましくは、0.5〜90%)の活性成分を薬剤的に許容される担体と組み合わせて含む医薬組成物として与えることができる。
【0233】
選択された投与経路に関係なく、好適な水和形態、及び/又は医薬本発明の組成物で使用できる本発明の化合物は、当業者に公知の通常の方法によって薬剤的に許容される投与形態に処方される。
【0234】
本発明の医薬組成物中の活性成分の投与の実際の投与量レベル及び時間経過を変えて、患者に対して毒性でない、特定の患者、組成物、及び投与様式に対する所望の治療反応を達成するために有効である活性成分の量を得ることができる。用量範囲の一例は、1日あたり0.1〜10mgである。
【0235】
本発明に関して本発明の化合物の好ましい用量は、患者が耐えることができ、重篤な副作用を発現しない最大量である。好ましくは、本発明の化合物を体重1キログラムあたり約0.001mg〜約100mg、約0.001〜約10mg/kg又は約0.001mg〜約100mg/kg体重の濃度で投与する。前記値の中間の範囲は本発明の一部であることが意図される。
【0236】
合成スキーム
本発明の化合物を以下の合成スキームにしたがって作成することができる。
1)
【化23】
スキームA:チャート1におけるN−置換PATの合成。スチレン又はハロスチレン7の[2]との反応により、4−置換−テトラレン−2−オールフェニルアセテート8を得る。8の還元により、テトラール−2−オール9を得る。トランス−(2S,4R)−N−置換PAT10は、9のトシル化とそれに続く環状アミン(ピロリジン、ピペリジン等)を用いた環状アミンSΝ2反転及びキラル固定相(CSP)−HPLCによるエナンチオマー分離によって調製される(Vincek & Booth 2009)。
【化24】
スキームB:別の方法。フェニルアセトン11のベンズアルデヒド又はハロベンズアルデヒド12とのKOHの存在下でのアルドール縮合により、エノン誘導体13を得る。テトラロン中間体14は、すでに報告されているように、13のポリリン酸での処理から得られる(Bucholtz 1999)。トランス−(2S,4R)−PAT10は、14の還元的アミノ化(Abdel−Magid 1996)により調製され、続いてCSP−HPLCを行う。
【化25】
スキームC:チャート2におけるメタ置換PATの合成。本発明者らは(Vincek & Booth, 2009)、メタ置換−フェニルPATを得るためのいくつかの方法を公開した。容易に入手可能な試薬1及び[2]により、汎用3−置換−フェニルテトラレン−2−オールフェニルアセテート3を得る。3上の3−置換アリール及びエノールフェニルアセテート官能基は、これらの分子を不斉転換及び様々な有機合成に有用にする。(a)不斉移動水素化、(b)トシル化、及び(c)ジメチルアミンを用いたSN2反転の3−ステップで、エナンチオ富化シス−(2R,4R)−4、シス−(2R−4R)−5;トランス−(2R−4S)及び(2S,4R)生成物がCSP−HPLCにより得られる。
【化26】
スキームD:別の方法。反応(d)における4−(3−ブロモフェニル)−テトラレン−2−オールフェニルアセテート3a(すなわち、スキーム1中の3(式中、R1=Br))の置換ボロン酸とのスズキカップリングによって、4−(3−置換フェニル)−テトラレン−2−オールフェニルアセテート6を得る(Vincek & Booth, 2009)。多くのボロン酸スズキカップリング試薬が利用可能である(Chemfiles, 2007)。
【化27】
スキームE:チャート3におけるテトラヒドロナフチル置換基を有するPATの合成。
容易に調製される1及び市販の2により、無水トリフルオロ酢酸(TFAA)を用いて置換フェニルテトラレン−2−オールフェニルアセテート3を得た(Vincek & Booth, 2009)。化合物3を水素化ホウ素ナトリウムにより還元して、ラセミのシス−オール4を得、これを直接トシル化し、トランス−ジメチルアミン5に変換する。CSP−HPLCを用いて、(+)−トランス−(2R,4S)−及び(−)−トランス−(2S,4R)−5を分離する。これらの新規PAT類似体を三臭化ホウ素により還元して、別のシリーズの新規化合物6にすることができた。ミツノブ反応により4を7に変換し、続いて,トシル化、アミノ化及びCSP−HPLCエナンチオマー分離を行って、(+)−及び(−)−シス−PAT8及び9を得る。
【化28】
スキームF:別の方法:本発明者らは以前にスキーム6を使用し(Bucholtz et al., 1999;Booth et al., 2009)、この場合、ラセミのシス及びラセミのトランス異性体をカラムクロマトグラフィーにより分離し、続いてジアステレオマー再結晶又はCSP−HPLCを行って、1つのエナンチオマーPAT類似体を得る。
【0237】
実施例
本発明を説明するが、本発明の範囲を制限しないことを意図する以下の実施例により、本発明をさらに説明する。
【0238】
実施例1:化学物質
すべての試薬は、商業的供給元から入手し、さらに精製することなく使用した。1H及び13C NMRを対応して共振周波数400及び100MHzで、CDCl3中で集めた。化学シフトをテトラメチルシランからのppmで報告する。フラッシュカラムクロマトグラフィーを、シリカゲル60(230〜400メッシュ)を用いて実施した。融点は、水銀温度計を備えたMel−Temp装置で測定した。HPLCキラル分離は、RegisCell(商標)(5μm、25cm×10mm i.d.)カラムを備えたHPLC装置によって測定した。いくつかの1H及び13C NMRスペクトルの小さなピーク(<5%)は、位置異性体の形成が原因であった。
【0239】
[3H]−ケタンセリン(比活性72.2Ci/ミリモル)及びミオ−[2−3H(N)]−イノシトール(比活性18.5Ci/ミリモル)をPerkin−Elmer Life Science(Boston, MA)から購入し、[N6−メチル−3H]−メスレルギン(比活性72.0Ci/ミリモル)をAmersham Biosciences(GE healthcare, Piscataway, NJ)から購入した。他の化合物は高純度でSigma−Aldrich(St. Louis, MO)から入手した。
【0240】
クローン細胞培養及びトランスフェクション
すべての細胞系をATCCの示唆にしたがって維持した。チャイニーズハムスター卵巣細胞(CHO−Kl、ATCC CCL−61)は10%のウシ胎仔血清、1%の重炭酸ナトリウム(Mediatech 25−035−CI)、10IU/mlのペニシリン及び10ug/mlのストレプトマイシンを追加したHamのF−12培地中、並びにヒト胚腎臓(HEK)293は、1.5g/Lの重炭酸ナトリウム、0.1mMの非必須アミノ酸、及び1.0mMのピルビン酸ナトリウム(90%)と10%ウシ胎仔血清、10IU/mlのペニシリン及び10ug/mlのストレプトマイシンを含むように調節された2mMのL−グルタミンを含む最小必須培地(イーグル)(MEM)中。細胞を、5%のCO2を含む加湿インキュベータ中37℃で増殖させる。ヒトHT2A、5−HT2B、及び5−HT2C(野生型)をコード化するcDNAを、クローン細胞の一過性トランスフェクションのためにUMR(Rolla, MO)から購入する。放射性受容体結合アッセイのために、5−HT2A、5−HT2B、及び5−HT2C受容体膜をトランスフェクトされたCHO−K1細胞から調製する。PLC/IP形成の活性を測定する機能的アッセイのために、5−HT2A及び5−HT2C受容体についてはトランスフェクトされたCHO−K1細胞を使用する。しかし、5HT2B受容体については、トランスフェクトされたHEK細胞を用いて、PLC/IPアッセイに関してさらに強力かつ一貫した結果が得られる(Setola et al., 2005)。トランスフェクションの24時間前に、細胞を、放射性受容体結合アッセイについては100mm皿中、40%コンフルエンシーで、又は機能的アッセイについては12ウェルプレート中、1ウェルあたり105細胞で蒔く。CHO−K1細胞を、放射性受容体結合アッセイについては100mm皿につき12μgのプラスミド及び32μlのリポフェクタミン(Invitrogen)で、又は機能的アッセイについてはウェルあたり0.8μgのプラスミド及び4.0μlのリポフェクタミンで、一時的にトランスフェクトする。HEK細胞を用いた5−HT2B機能的アッセイについて、24μgのプラスミドDNAを60μlのリポフェクタミン2000(Invitrogen)と混合して、10cmプレート中1〜2×106細胞をトランスフェクトする。細胞にトランスフェクトされた受容体をさらに24時間発現させる(Herrick, 1997)。
【0241】
放射性受容体アッセイ
放射性受容体飽和及び競合結合アッセイを、膜ホモジネートを用い、系統学的に密接に関連するヒスタミンH1GPCRについて以前に報告されている本発明者らの方法と同様にして実施する(Booth, 2002;Moniri et al., 2004)。[3H]−ケタンセリンを用いて、5−HT2A受容体を放射標識し、[3H]−メスレルギンを5−HT2B及び5−HT2c受容体について使用する。手短に言うと、CHO細胞トランスフェクションの48時間後、細胞を収集し、0.1%のアスコルビン酸及び4.0mMのCaCl2を含むpH7.4の50mMのTris−HCl(アッセイ緩衝液)中で均質化する。ホモジネートを35,000gで25分間遠心分離し、結果として得られる膜ペレットをアッセイ緩衝液中に再懸濁させる。タンパク質濃度を、Lowryらの方法(Lowry, 1951)により測定する。飽和結合アッセイに関して、100μgのタンパク質を含む膜懸濁液を、0.1〜5.0nMの[3H]−ケタンセリン(5−HT2A受容体)又は0.1〜20nMの[3H]−メスレルギン(5−HT2B及び5−HT2c受容体)とともに250μlの合計アッセイ緩衝液体積中でインキュベートする。非特異的結合を、10μMのメチセルギド(5−HT2A受容体)又は1.0μMのミアンセリン(5−HT2B及び5−HT2c受容体)の存在下で測定する。競合結合アッセイを、1.0nMの[3H]−ケタンセリン又は[3H]−メスレルギンを用いて同様に実施する。放射性受容体結合アッセイ混合物のインキュベーションは、1.0時間37℃で、96ウェルセルハーベスター(Tomtec, Hamden, CT)を用いてWhatman GF/Bフィルターを通した急速ろ過法により終了させる。フィルターディスク上に保持される膜結合[3H]放射性リガンドを、液体シンチレーション分光測定法によって定量化する。データを、Prism 4.03(GraphPad Software Inc., San Diego, CA)のS字形曲線適合アルゴリズムを用いた非線形回帰により分析する。リガンド親和性は、IC50データを、式K0.5=/C50/I+LIKQ(式中、Lは親和性KDを有する放射性リガンドの濃度である)を用いてK0.5値に変換することによって、Ki値の近似値として表す(Cheng, 1973)。
【0242】
PLCの活性化及び[3H]−IP形成についてのアッセイ
PLCの機能的活性化を、すでに報告されているようにして、セロトニン5−HT2C受容体を一時的に発現するCHO細胞又はセロトニン5−HT2A若しくは5−HT2B受容体を一時的に発現するHEK細胞における[3H]−IP形成として測定する(Moniri et al., 2004)。手短に言うと、トランスフェクションの32時間後に、イノシトールを含まないダルベッコの修飾イーグル培地(DMEM)中の細胞を12時間1.0μCi/mlのミオ−[2−3H]−イノシトール(PLC−β基質ホスファチジルイノシトールの放射標識された前駆体)とともにインキュベートする。細胞を次いで洗浄し、10mMの塩化リチウム、10μMのパルギリン(HEK細胞について5%の透析されたウシ胎仔血清を添加)、及び種々の濃度の試験リガンドを含むDMEM中で45〜60分間37℃でインキュベートする。培地を吸引した後、50mMのギ酸とともにインキュベーション(15〜60分)することによって、ウェルを溶解させる。ギ酸を水酸化アンモニウムで中和し、各ウェルからの内容物を個々のAG1−X8 200−400ホルメート樹脂アニオン交換カラムに添加する。ギ酸アンモニウム/ギ酸(1.2M/0.1M)を用いて、液体シンチレーション分光測定法によってトリチウムを計数するために、[3H]−IPをシンチレーションバイアル中に直接溶出させる。結果として得られるデータを、Prism 4.03の非線形回帰アルゴリズムを用いて分析し、対照[3H]−IP形成の平均パーセンテージとして表し、効力は、最大基礎(構成的)[3H]−IP形成を50%±S.E.M.(n≧3)刺激(EC50)又は阻害(IC50)するために必要な濃度として表す。
【0243】
CHO−K1及びHEK細胞における[3H]−IP形成の測定
PLCの機能的活性化は、本発明者らの研究室により以前に報告されているように、5−HT2A又は5−HT2C受容体を一時的に発現するCHO細胞及び5HT2B受容体を一時的に発現するHEK細胞において[3H]−IP形成として測定される(Booth, 2002;Moniri et al., 2004)。手短に言うと、トランスフェクションの32時間後、イノシトールを含まないダルベッコの修飾イーグル培地(DMEM)中の細胞を、1μCi/mlのミオ−[2−3H]−イノシトール(PLC−β基質ホスファチジルイノシトールの前駆体)で標識する。細胞を次いで洗浄し、25mMのHepes(pH7.4)、10mMのLiCl、10μMのパルギリン(HEK細胞について、5%の透析されたFBSを添加)、並びに様々な濃度の試験リガンドを含むDMEM中、45〜60分間37℃でインキュベートする。培地を吸引した後、ウェルを氷上に置き、50mMのギ酸(15〜60分)とともにインキュベーションすることによって溶解させる。ギ酸を水酸化アンモニウムで中和し、各ウェルからのすべての内容物を個々のAG1−X8 200−400ホルメート樹脂アニオン交換カラムに添加する。ギ酸アンモニウム/ギ酸(1.2M/0.1M)を使用して、液体シンチレーション分光測定法によってトリチウムを計数するために、[3H]−IPをシンチレーションバイアル中に直接溶出させる。結果として得られるデータを、Prism 4.03の非線形回帰アルゴリズムを用いて分析し、対照[3H]−IP形成の平均パーセンテージとして表し、効力を50%最大[3H]−IP形成(EC50)±S.E.M(n≧3)を得るために必要な濃度として表す。
【0244】
実施例2
5HT−サブタイプ受容体の放射性リガンド飽和結合分析:ヌルトランスフェクト(null−transfected)CHO及びHEK細胞から調製された膜を用いて測定可能な特異的放射性リガンド結合はない。しかし、5−HT2A、5−HT2B、又は5−HT2CcDNAで一時的にトランスフェクトされたCHO細胞から調製された膜を用いて、飽和可能な特異的放射性リガンド結合が起こる。[3H]−ケタンセリンは、5HT2A受容体の見かけ上1つの集団(Bmax=1.73±0.11pmol/mgタンパク質)と高親和性(KD=0.80±0.03nM)で結合する。同様に、[3H]−メスレルギンは、Bmax=1.13±0.39pmol/mgタンパク質及びKD=5.19±0.36nMで5HT2B受容体の1つの集団を標識する。[3H]−メスレルギンはまた、高親和性(KD=0.88±0.03nM)で5HT2c受容体の見かけ上1つの集団(Bmax=8.37±0.15pmol/mgprot)も標識する。
【0245】
実施例3
RSK及び他のキナーゼのキナーゼ阻害活性は、当該技術分野で公知である。RSK活性に関するアッセイは、基本的に、Jeffrey A. Smith, Celeste E. Poteet−Smith, Yaming Xu, Timothy M. Errington,, Sidney M. Hecht, and Deborah A. Lannigan Cancer Res 2005;65:(3), pp. 1027−1034. February 1, 2005で記載されているようにして実施される。
【0246】
実施例4
メタ置換化合物の合成及びエナンチオマーの分離
一般的合成法:本発明の化合物を合成するのに有用な関連する方法は当該技術分野で公知であり、本発明者等の合成医薬品化学刊行物(例えば、Ghoneim, O. M.;Legere, J. A.;Golbraikh, A.;Tropsha, A.;Booth, R. G. Bioorg. Med. Chem. 2006, 14, 6640;Bucholtz, E. C;Brown, R. L.;Tropsha, A.;Booth, R. G.;Wyrick, S. D. J. .Med. Chem. 1999, 42, 3041;Wyrick SD, Booth RG, Myers AM, Owens CE, KuIa NS, Baldessarini RJ, McPhail AT, and Mailman RB(1993)Synthesis and pharmacological evaluation of 1−phenyl−3−amino 1,2,3,4−tetrahydronaphthalenes as ligands for a novel receptor with sigma−like neuromedulatory activity. J Med Chem 36:2542−2551を包含する)で記載されている。インビトロの薬理学的研究は、まず、ラセミのシス及びトランス生成物を使用する。ラセミ化合物は、ジアステレオマー塩に誘導体化し、続いて分画晶出(differential crystallization)を行うことにより(+)−及び(−)−エナンチオマーに分割することができるか、又はキラル還元ステップを用いて新たに合成することができる。すでに合成された純粋なエナンチオマーと比較することにより単結晶X線結晶学又は分光測光法(NMR、旋光度)によって絶対配置を帰属させる。NMR、元素分析、質量分析、融点及び薄層クロマトグラフィーを用いて生成物を純度について特性化する(HCl塩として)。
【0247】
スキーム1:メタ置換化合物:方法は、前記方法及び当該技術分野で公知の他の方法から変更した。メタ置換アルデヒド2を用いたクライゼン・シュミット反応によって、α,β−不飽和ケトン3を得、これを環化してケトン4にし、そしてNaBH4を用いて還元した。(±)−シス及び(±)−トランス遊離塩基7を(1R)−(−)−又は(1S)−(+)−カンファー−10−スルホン酸ジアステレオマー塩に変換し、これを分画再結晶して、(+)−又は(−)−エナンチオマーを得、これをアルキル化して、生成物8を得る。
【化29】
【0248】
スキーム2:PAT類似体立体異性体を得るためのキラル還元剤の使用:
ジイソピノカンフェニル−ボラン(DIP)類似体は、最近、スキーム2におけるケトン4と類似した構造を有するケトンについて立体選択的な還元剤として報告された(例えば、Cha et al., 2005を参照)。
【0249】
C(1)ペンダントフェニル置換基への変更を加えた新規PAT類似体の合成
結合、機能、3D QSAR、及び分子モデリング結果に基づいて、(−)−トランス−置換フェニルアミノテトラリン(PAT)C(1)ペンダントフェニル部分は、5HT2A及び5HT2B受容体の活性化なしに完全に有効な5HT2cアゴニスト活性を提供するのに重要であることが示された。
【化30】
【0250】
スキーム3 ステップAでは、β−テトラロン(1)をベンゼンルテニウム(II)クロリド二量体及びキラルリガンド(R,R)−N−(2−アミノ−1,2−ジフェニルエチル)−p−トルエンスルホンアミド((R,R)−ΝAPTS)とともに還流して、(R)−β−テトラロール(2)を得た(Mogi et al., 2004)。ステップBでは、(R)−β−テトラロール(2)をtert−ブチルジメチルシリル(TBDMS)誘導体(3)(隣接するベンジル位でのブロム化を防止するためのTBDMS保護基)に変換した。ステップCでは、TBDMS保護化合物を無水CCl4中還流下、N−ブロモスクシンイミド(Agarwal et al., 1990)でブロム化して、ブロム化された一般的中間体(4)を得、フラッシュカラムクロマトグラフィーによりシス及びトランスブロモ化合物に分離した。ステップDでは、各(シス及びトランス)ブロム化中間体(4)を市販のボロン酸(R2B(OH)2、R2=アルキル、アルケニル、又はアリール基;例えば、表7のボロン酸化合物で例示される基を参照)と、ニッケル触媒作用を受けたスズキ反応下(Gonzalez−Bobes et al)、NiI2/トランス−2−アミノシクロヘキサノールとナトリウムビス(トリメチルシリル)アミドを還流下で用いて反応させて、種々のシス及びトランスPAT類似体を作成した。ステップEでは、TBDMS保護PAT類似体を、テトラヒドロフラン中テトラブチルアンモニウムフルオリド(TBAF)を用いて脱保護した。ステップFでは、シス及びトランスヒドロキシルPAT類似体をワンポットで、アジ化亜鉛/ビスピリジン複合体、ジイソプロピルアゾジカルボキシレート(DIAD)及びトリフェニルホスフェン(phosphene)とのミツノブ反応を用いて、対応するトランス及びシスアジドPAT中間体(7)に変換した(Vorogushin et al.、2003)。ステップgでは、アジドPAT誘導体を対応するPATアミン(8)に還元した。ステップHでは、これらのエナンチオマーシス及びトランスアミンを、還流下、ギ酸/ホルムアルデヒドでのEschweiler−Clarkeメチル化を用いて、ジメチル化PAT類似体(9)に変換する。
【化31】
化合物を、非メチル化遊離アミンをジアステレオマー塩に誘導体化し、続いて分画晶出することにより(+)−及び(−)−エナンチオマーに分割するか、又はキラル還元ステップを用いて新たに合成した。すでに合成された純粋なエナンチオマーと比較することにより単結晶X線結晶学又は分光測光法(NMR、旋光度)によって絶対配置を帰属させた。生成物(HCl塩として)は、NMR、元素分析、質量分析、融点及び薄層クロマトグラフィーを用いて純度について特性化した。本明細書中で具体的に記載される化合物には、R2が、表7のボロン酸化合物中のホウ素原子に結合した化学基又は部分(例えば、置換若しくは非置換アルキル、アルケニル、又はアリール基)であるものが含まれる。本明細書中の化合物を調製するための別の手順は、PCT国際公開番号WO2008/156707(出願番号PCT/US2008/007458)にも記載されている。
【0251】
類似体の合成
トリフルオロアセチル混合無水物及び3−ハロスチレン(フルオロ、クロロ、及びブロモ)を、溶媒を追加することなく、4−(3−ハロフェニル)−3,4−ジヒドロナフタレン−2−イルフェニルアセテート(ブロモ、50%)に関するカスケードフリーデルクラフツ環状−アシルアルキル化、エノール化、及びO−アシル化に付した。マスクされた4−フェニルテトラール−2−オンの塩基アルコール分解により、インサイチュ不斉移動水素化のためのケト基が明らかになった。ブロモ−誘導体は、4−(ビフェン−3−イル)−3,4−ジヒドロナフタレン−2−イルフェニルアセテートを得るためにフェニルボロン酸とのスズキカップリングを受け、トランス−4−フェニル−2−アミノテトラリンへの短い有効な経路を提供した。
【0252】
カスケードFC−CAA、エノール化、及びO−アシル化を、TFAA活性化フェニル酢酸、スチレン又は3−ハロスチレンを用いて調査した。反応性スチレン12aを[9]とともに穏やかに加熱すると、本質的に遅いエノール化が加速され(a)Nevy, J. B.;Hawkinson, D.. C;Blotny, G.;Yao, X.;Pollack, R. M. J. Am. Chem. Soc. 1997, 119, 12722. b)Yao, X.;Gold, M. A.;Pollack, R. M. J. Am. Chem. Soc. 1999, 121, 6220を参照)、4a(15%)を得た。加熱の非存在下で、複合体混合物は、結果として反応性3aの損失をもたらした。中程度に反応性である3−ハロスチレンと、等モル濃度又は3当量までの[9]により、ハロゲン化3及び4を得た。フルオロスチレン12bと等モル濃度の[9]により、多量の3b(42%)及び少量の4b(8%)を得た。クロロスチレン12cと3当量の[9]により3c(70%)を得た。3cを等モル濃度の[9]でさらに処理して、4c(38%)を得た。24時間にわたって室温まで温めて、ブロモスチレン12cと3当量の[9]により、4d(50%)を得、非ハロゲン化4aからの収率で3倍増加した。
【0253】
【表3】
スキーム4:4dの(例えば、表5からの)フェニルボロン酸でのスズキカップリング(a)Wolfe, J. P.;Singer, R, A.;Yang, B. H.;Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9550. b)Wolfe, J. P.;Buchwald, S. L. Angew. Chem. Int. Ed. 1999, 38, 2413を参照)によりスムーズに4−(ビフェニル−3−イル)−3,4−ジヒドロ−ナフタレン−2−イルフェニル−アセテート14を得、これをスキーム3におけるようなプロトコルにしたがって所望の置換フェニルアミノテトラリンに変換した。
【0254】
スキーム4:
【化32】
【0255】
実施例5:
CAT化合物の合成
スキーム5
【化33】
【0256】
4−シクロヘキシル−3,4−ジヒドロナフタレン−2−イルフェニルアセテート(1a)。(Gray AD, and Smyth TP(2001) Clean−chemistry synthesis of 2−tetralones in a single−stage acylation−cycloalkylation process. J Org Chem 66:7113−7117)フェニル酢酸(10.9g、0.08モル)を無水トリフルオロ酢酸TFAA(11mL、0.08モル)中に室温で溶解させて、その場で混合無水物を生成させた。窒素ガスを使用して、両頭針を通して、ビニルシクロヘキサン(0.04モル)を含み、60℃で撹拌したフラスコ中に混合無水物を押し込んだ。30分後、反応を水(100mL)でクエンチし、酢酸エチル(100mL、3×)で抽出した。有機層を硫酸ナトリウム上で乾燥し、ろ過し、そして濃縮した。粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、ラセミ1a(30%、油状物)及び1b(〜30%、4−シクロヘキシル−3,4−ジヒドロナフタレン−2(1H)−オン)を得た。1H NMR(CDCl3); 0.92−1.26(m, 5H), 1.40−1.44(m, 1H), 1.57−1.76(m, 5H), 2.36(dd J=3.0, 17.1 Hz, 1H), 2.61(dt, J=2.9, 7.0 Hz, 1H), 2.76(ddd, J=2.5, 7.6, 17.0 Hz, 1H), 3.73(s, 2H), 6.15(d, J=2.3 Hz, 1H), 6.95(dd, J=2.2, 7.2 Hz, 1H), 7.02−7.19(m, 3H), 7.26−7.36(m, 5H). 13C NMR(D6−DMSO); 25.9, 26.0, 26.1, 28.2, 29.2, 30.5, 40.2, 40.6, 43.5, 113.8, 126.1, 126.2, 126.5, 127.0, 128.4, 129.4, 132.5, 133.9, 135.3, 149.9, 169.6. 元素分析 C24H26O2についての計算値C=83.20、H=7.56。実測値:C=83.02、H=7.91。HRMS m/z C24H26O2についての計算値 346.1933 [M]+、実測値346.1921。
【0257】
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−オール(2a).(Peach, P, Cross DJ, Kenny JA, Mann I, Houson I, Campbell L, Walsgrove T, and Wills M(2006)Asymmetric transfer hydrogenation of a,3−unsaturated, a−tosyloxy and a−substituted ketones. Tetrahedron 62:1864−1876),(Alcock NJ, Mann I, Peach P, and Wills M(2002)Dynamic kinetic resolution−asymmetric transfer hydrogenation of 1−aryl−substituted cyclic ketones. Tetrahedron: Asymmetry 13:2485−2490),(Mogi M, Fuji K, and Node M(2004)Asymmetric reduction of methoxy substituted 3−tetralnes using transfer hydrogenation. Tetrahedron: Asymmetry 15:3715−3717)
ベンゼンルテニウム(II)クロリド二量体(55mg、0.11ミリモル)及び(1R,2R)−(−)−N−(4−トルエンスルホニル)−1,2−ジフェニルエチレンジアミン(80mg、11ミリモル)をイソプロパノール(25mL)中で混合し、80℃で30分間加熱すると、触媒混合物が生成した。別に、4−シクロヘキシル−3,4−ジヒドロナフタレン−2−イルフェニルアセテート1a(950mg、2.8ミリモル)をイソプロパノール(25mL)中で50℃まで加熱した。窒素ガスを用いて、両頭針を通してIaを含むフラスコに触媒混合物を押し込み、直後にKOH(0.5g)のイソプロパノール(50mL)中混合物を添加した。3時間後、セライト上シリカゲルのパッドを通して反応物を濾過し、次いで減圧下で濃縮した。粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、2aを40%の収率で得た。
【0258】
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−イル4−メチルベンゼンスルホネート(3a)。(Wyrick SD, Booth RG, Myers AM, Owens CE, Kula NS, Baldessarini RJ, McPhail AT, and Mailman RB(1993) Synthesis and pharmacological evaluation of 1−phenyl−3−amino−1,2,3,4−tetrahydronaphthalenes as ligands for a novel receptor with sigma−like neuromodulatory activity. J Med Chem 36:2542−2551)
(2R,4R)−4−シクロヘキシル−1,2,3,4−テトラヒドロナフタレン−2−オール2a(0.23g、0.1モル)、p−トルエンスルホニルクロリド(0.20g、0.11モル)、及びピリジン(1.5mL)を溶液中で24時間、室温、不活性雰囲気(N2)下で撹拌した。溶媒を除去し、粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、3aを62%の収率で得た。HRMS m/z C23H28O3Sについての計算値 407.1651 [M+Na]+、実測値 407.1631。
【0259】
(2S,4R)−4−シクロヘキシル−N,N−ジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(4a).(Bosse K, Marineau J, Nason DM, Fliri AJ, Segelstein BE, Desai K, and Volkmann RA(2006)Expanding the medicinal chemistry toolbox: stereospecific generation of methyl group−containing propylene linkers. Tetrahedron Letters 47:7285−7287)
ジメチルアミン(H2O中40%、6mL)及び(2R,4R)−4−シクロヘキシル1,2,3,4−テトラヒドロナフタレン−2−イル4−メチルベンゼンスルホネート3a(0.19g、0.5ミリモル)を試験管中に入れ、密封し、83℃で24時間加熱した。溶媒を除去し、粗物質をカラムクロマトグラフィー(Si−ゲル)により精製して、4a(収率70%)及び5a(30%、(R)−1−シクロヘキシル−1,2−ジヒドロナフタレン)を得た。ベンチクロマトグラフィーの後、4aを、キラル(RegisCell(商標)、EtOH(15):ヘキサン(85))HPLC技術を用いて調べて、UV波形によりeeが98%を超えることが示された。1H NMR(CDCl3);1.00−1.31(m, 5H), 1.39−1.47(m, 1H), 1.57−1.85(m, 6H), 2.48−2.52(m, 1H), 2.72−2.77(m, 1H), 2.80(s, 6H), 3.03(dd, J=9.0, 16.6 Hz, 1H), 3.34(dd, J=6.8, 16.0 Hz, 1H), 3.72−3.75(m, 1H), 7.09−7.29(m, 4H). 13CNMR(CDCl3);26.5, 26.8, 27.3, 30.2, 31.8, 32.3, 41.6, 41.9, 43.5, 56.8, 124.9, 125.7, 129.1, 129.2, 136.0, 139.7. HRMS m/z C18H27Nについての計算値258.2216 [M+H]+、実測値258.2220。薬理学的研究のために、遊離塩基をHCl塩に変換した;MP:174〜176℃。C18H27N+HCl+H2Oの元素分析計算値:C=69.32、H=9.70、N=4.49。実測値:C=69.48、H=9.98、N=4.34。
【0260】
実施例6:
セロトニン5HT2A、5HT2B、5HT2C、及びヒスタミンH1Gタンパク質共役受容体でのCATの薬理学的活性
(−)−トランス−CAT及び(+)−トランス−CATの活性を、基本的に当該技術分野で公知のとおりであり、本明細書中に記載されるプロトコルを用いて評価した。結果を以下の表及び本明細書中の図面で示す(例えば、図1〜3)。
【0261】
(+)−(2R,4S)−及び(−)−(2S,4R)−4−シクロヘキシル−N,Nジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(或いは、4−シクロヘキシル−2−ジメチルアミノテトラリン、CAT)の分子構造
【化34】
【0262】
【表4】
【表5】
【0263】
442の異なるキナーゼについての親和性に関する(−)−トランス−CATの薬理学的データのまとめ
(−)−(2S,4R)−4−シクロヘキシル−N,N−ジメチル−1,2,3,4−テトラヒドロナフタレン−2−アミン(或いは、4−シクロヘキシル−2−ジメチルアミノテトラリン、CAT)の分子構造
【化35】
【0264】
(−)−トランス−CATはRSK3ときわめて強力に結合する(1.0uMのCATで標識タグ結合の80%阻害)
【0265】
(−)−トランス−CATはRSK4に非常に強力に結合する(1.0uMのCATで結合する標識されたタグの80%阻害)
【0266】
(−)−トランス−CATはRSK1と中程度の親和性を有する(1.0uMのCATで結合するタグの20%阻害)。
【0267】
1.0uMまでの濃度で、(−)−トランス−CATは、本明細書中に記載されるキナーゼに対してRSK2に同様に有意な結合を示さず、有意な選択性を示した。
【0268】
1.0uMまでの濃度で、(−)−トランス−CATは、本明細書中に記載されるキナーゼに対して試験した438の他のキナーゼ(表4を参照)と有意な結合として示さず、有意な選択性を示した。
【0269】
RSK1(わずかな結合)及び2(結合なし)よりもRSK3及び4の阻害についての(−)−トランス−CATの非常に高い特異性を有する分子は現在のところ報告されていない。RSK2及び438の他のキナーゼからRSK(1、3及び4)の阻害について(−)−トランス−CATの非常に高い特異性を有する分子は現在のところ報告されていない。
【0270】
実施例7
化合物の5−HT2機能的活性は、PCT国際公開番号第WO2008/156707号(出願番号PCT/US2008/007458)で記載されているものをはじめとする当該技術分野で公知の方法によって評価することができる。
【0271】
【表6】
【表7】
参考文献:
Arjona AA, Pooler AM, Lee RK, Wurtman RJ. Effect of a
5-HT2C serotonin agonist, dexnorfenfluramine, on amyloid precursor
protein metabolism in guinea pigs. Brain Res. 2002 951 :135-140.
Baldessarini RJ, Tarazi FI. Pharmacotherapy of Psychosis
and mania. In: Brunton LL, Laxo JS, Parker KL, eds. The Pharmacological Basis
of Therapeutics. 11th ed. New York:
McGraw-Hill, 2006:461-500.
Bubar MJ, Cunningham KA. Distribution of serotonin
5-HT(2C) receptors in the ventral tegmental area. Neuroscience. 2007 (doi:
10.1016/j.neuroscience.2006.12.071).
Bubar MJ, Cunningham KA. Serotonin 5-HT2A and 5-HT2C
receptors as potential targets for modulation of psychostimulant use and
dependence. Current Topics and Medicinal Chemistry 2006; 6:1971-1985.
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS,
Edwards WD, Schaff HV. Valvular heart disease associated with
fenfiuramine-phentermine.N Engl J Med. 1997;337:581-5888. Erratum in: N Engl J
Med 1997;337:1783.
Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay
MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, et al. Possible role
of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with
fenfluramine. MoI Pharmacol 2000 57: 75-81.
Fletcher PJ, Grottick AJ, Higgins GA. Differential
effects of the 5-HT(2A) receptor antagonist M 100907 and the 5-HT(2C) receptor
antagonist SB242084 on cocaine-induced locomotor activity, cocaine
self-administration and cocaine-induced reinstatement of responding. Neuropsychopharmacology
2002 27:576-586.
Frank MG, Stryker MP, Tecott LH. Sleep and sleep
homeostasis in mice lacking the 5-HT2c receptor. Neuropsychopharmacology. 2002
27:869-873.
Ghoneim et al., Bioorg. Med. Chem., 14, 6640-6658 (2006).
Giorgetti M, Tecott LH. Contributions of 5-HT(2C)
receptors to multiple actions of central serotonin systems. Eur J Pharmacol.
2004 488: 1-9.
Heisler LK, Chu HM,
Tecott LH. Epilepsy and obesity in serotonin 5-HT2C receptor mutant mice.Ann N
Y Acad Sci. 1998 861 :74-78.
Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart
JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, et al. Activation of central
melanocortin pathways by fenfluramine. Science (Wash DC) 2002 297: 609-611.
Heisler LK, Tecott LH. A paradoxical locomotor response
in serotonin 5-HT(2C) receptor mutant mice. J Neurosci. 200020:RC71.
Heisler LK, Zhou L, Bajwa P, Hsu J, Tecott LH Serotonin
5-HT(2C) receptors regulate anxiety-like behavior.Genes Brain Behav. 2007 (DOI
10.1 1 1 l/j.l601-183X.2007.00316.x)
Jensen MD. Potential role of new therapies in modifying
cardiovascular risk in overweight patients with metabolic risk factors.
Obesity. 2006 14:143S-149S.
Julius D, Huang KN, Livelli TJ, Axel R, Jessel TM. The
5HT2 receptor defines a family of structurally distinct but functionally
conserved serotonin receptors. Proc. Natl. Acad. Sci. 1990 87:928-932.
Julius D, MacDermott
AB, Axel R, Jessell^
TM. Molecular Characterization of a functional cDNA encoding the serotonin Ic
receptor. Science 1988 241 :558-564.
Kennett GA, Pittaway K, Blackburn TP: Evidence that 5-HT2C receptor
antagonists are anxiolytic in the rat Geller-Seifter model of anxiety.
Psychopharmacology (Bed.) (1994) 114:90-96.
Launay JM, Herve P, Peoc'h K, Tournois C, Callebert J,
Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, et al. Function of the
serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med
2002 8: 1129-1135.
Marquis KL, Sabb AL, Logue SF, Brennan JA, Piesla MJ,
Comery TA, Grauer SM, Ashby CR Jr, Nguyen HQ, Dawson LA, Barrett JE, Stack G,
Meltzer HY, Harrison BL, Rosenzweig-Lipson S . W A Y- 163909 [(7bR, 1 OaR)-
1,2,3,4,8,9,10,1 Oa-octahydro-7bH-cyclopenta-[b][1,4]diazepino[6,7,lhi]indole]:
A novel 5-hydroxytryptamine 2C receptor-selective agonist with preclinical
antipsychotic-like activity. J Pharmacol Exp Ther. 2007 320:486-496.
Muller CP, Huston JP. Determining the region-specific
contributions of 5-HT receptors to the psychostimulant effects of cocaine.
Trends Pharmacol Sci. 2006 27:105-112.
Nichols DE. Hallucinogens, Pharmacol. Ther. 2004 101:131-181.
Nilsson BM. 5-Hydroxytryptamine 2C (5-HT2C) receptor
agonists as potential antiobesity agents. J Med Chem. 2006 49:4023-4034.
Palvimaki EP, Roth BL, Majasuo H, Laakso A, Kuoppamaki M,
Syvalahti E, Hietala J. Interactions of selective serotonin reuptake inhibitors
with the serotonin 5-HT2c receptor, sychopharmacology (Berl). 1996 126:234-240.
Pouwels HM, Smeets JL, Cheriex EC, Wouters EF. Pulmonary
hypertension and fenfluramine. Eur Respir J. 1990 May;3(5):606-7.
Raymond JR. Mukhin YV. Gelasco A. Turner J. Collinsworth
G. Gettys TW. Grewal JS. Garnovskaya
MN. Multiplicity of mechanisms of
serotonin receptor signal transduction. Pharmacol Ther. 2001 92: 179-212.
Reynolds GP, Yao Z, Zhang X, Sun J, Zhang Z. Pharmacogenetics
of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor
polymorphisms separately associate with positive and negative symptom response.
Eur Neuropsychopharmacol. 2005 Mar; 15(2): 143-51.
Rocha BA, Goulding EH, O1DeIl LE, Mead AN, Coufal
NG, Parsons LH, Tecott LH. Enhanced locomotor, reinforcing, and neurochemical
effects of cocaine in serotonin 5-hydroxytryptamine 2C receptor mutant mice. J
Neurosci. 2002 ;22: 10039-10045.
Rosenzweig-Lipson S, Sabb A, Stack G, Mitchell P, Lucki
I, Malberg JE, Grauer S, Brennan J, Cryan JF, Sukoff Rizzo SJ, Dunlop J,
Barrett JE, Marquis KL. Antidepressant-like effects of the novel, selective,
5-HT(2C) receptor agonist WAY-163909 in rodents. Psychopharmacology (Berl).
2007 192:159-170.
Roth BL. Drugs and valvular heart disease. N Engl J Med.
2007; 356:6-9.
Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A,
Hufeisen SJ, and Roth BL. Evidence for possible involvement of 5-HT(2B)
receptors in the cardiac valvulopathy associated with fenfluramine and other
serotonergic medications. Circulation 2000 102: 2836-2841.
Saltzman AG, Morse B, Whitman MM, Ivanshchenko Y, Jaye M,
Felder S. Cloning of the human serotonin 5-HT2 and 5-HTlC receptor subtypes.
Biochem Biophys Res Commun. 1991 181 :1469-7148.
Sanders-Bush E, Mayer SE. Serotonin Receptor Agonists and
Antagonists. Chapter 11, in Goodman and Gilman's The Pharmacological Basis of
Therapeutics 11th Edition. Brunton LL, Lazo JS, Parker KL, Editors, McGranw-Hill, New
York, 297-315, 2006.
Sard H, Kumaran G, Morency C, Roth BL, Toth BA, He P,
Shuster L. SAR of psilocybin analogs: discovery of a selective 5-HT 2C agonist.
Bioorg Med Chem Lett. 2005 15:4555-4559.
Segman RH, Heresco-levy U, Finkel B, Inbar R, Neeman T,
Schlafman M, Dorevitch A, Yakir A, Lerner A, Goltser T, Shelevoy A, Lerer B.
Association between the serotonin 2C receptor gene and tardive dyskinesia in
chronic schizophrenia: additive contribution of 5-HT2CSer and DRD3Gly alleles
to susceptibility, Psychopharmacology 2000 152:408-413.
Setola V, Dukat M, Glennon RA, Roth BL. Molecular
determinants for the interaction of the valvulopathic anorexigen
norfenfluramine with the 5-HT2B receptor. Mol Pharmacol 2005
68:20-33.
Simansky KJ. N1H symposium series: ingestive mechanisms
in obesity, substance abuse and mental disorders. Physiology & Behavior
2005; 86: 1-4.
Siuciak JA, Chapin DS, McCarthy SA, Guanowsky V, Brown J,
Chiang P, Marala R, Patterson T, Seymour PA, Swick A, Iredale PA. CP-809,101, a
selective 5-HT2C agonist, shows activity in animal models of antipsychotic
activity. Neuropharmacology. 2007 52:279-290.
Smith SR, Prosser W, Donahue D, Anderson C, Shanahan W. Lorcaserin Phase 2b
Clinical Study. American Diabetes Association, 2006.
Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP,
Johnson JA. Neutralization of transthyretin reverses the neuroprotective
effects of secreted amyloid precursor protein (APP) in APPSw mice
resulting in tau phosphorylation and loss of hippocampal neurons: support for
the amyloid hypothesis. J. Neurosci. 2004 24:7707-7717.
Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH,
Dallman MF, Julius D. Eating disorder and epilepsy in mice lacking 5-HT2c
serotonin receptors. Nature. 1995 374:542-546
Tohda M, Takasu T, Nomura Y. Effects of antidepressants
on serotonin-evoked current in Xenopus oocytes injected with rat brain mRNA.
Eur J Pharmacol. 1989166:57-63.
Veenstra-VanderWeele J, G.M. Anderson GM, Cook EH.
Pharmacogenetics and the serotonin system: initial studies and furure
directions. Eur. J. Pharmacol. 2000 410:65-181.
Vickers SP, Dourish
CT, and Kennett GA. Evidence that hypophagia
induced by D-fenfluramine and D-norfenfluramine in the rat is mediated by
5-HT2C receptors. Neuropharmacology 2001 41: 200-209.
Vickers, S.P., Clifton,
P.G., Dourish, CT. and Tecott, L.H., 1999. Reduced satiating effect of
d-fenfluramine in serotonin 5-HT2C receptor mutant mice. Psychopharmacology
1999 143:309-314.
Agarwal R, Boyd
DR, McMordie RAS, O'Kane GA, Porter P, Sharma ND, Dalton H, Gray DJ. J.
Chiral arene hydrates of naphthalene: Enzymatic and chemical syntheses. Chem
Soc Chem Commun. 1990 171 1-1713.
Mogi M, Fugi K, Node M. Asymmetric reduction of methoxy
substituted β-tetralones using
hydrogenation. Tetrahedron: Asymmetry. 2004 15:3715-3717.
Vorogushin AV, Predeus A, Wuff WD, Hansen H. Diels- Alder
reaction-aromatization approach toward functionalized ring C allocolchicinoids.
Enantioselective total synthesis of (-)-7S-Allocolchicine. J Org Chem.
【0272】
本明細書中の変数の定義における要素のリストの説明は、いずれか1つの要素又は記載された要素の組み合わせとしての変数の定義を包含する。本明細書中の要素、又は実施形態の説明は、いずれか1つの要素若しくは実施形態又は任意の他の要素、実施形態若しくはそれらの一部との組み合わせを包含する。
【0273】
これらに限定されるものではないが、抄録、論文、雑誌、刊行物、テキスト、専門書、技術的データシート、インターネットウェブサイト、データベース及び特許、特許出願、及び特許公報をはじめとする本明細書中の全ての引用文献は、印刷物、電子的、コンピュータ可読記憶媒体又は他の形態であるかに関わらず、参照することによってそれらの全体として本明細書中に明らかに組み込まれる。
【0274】
本発明を具体的な実施例を参照して開示したが、本発明の真の精神及び範囲から逸脱することなく本発明の他の実施形態及び変形を他の当業者らが考案できることは明らかである。特許請求の範囲は、そのような実施形態及び等価な変形を包含すると解釈されるものである。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図2A】
【図2B】
【図2C】
【特許請求の範囲】
【請求項1】
対象におけるGPCR介在性障害を治療又は予防する方法であって、それを必要とすると認定された対象に式(I):
【化1】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化2】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式又はヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物を投与することを含む、方法。
【請求項2】
対象においてRSK介在性疾患又は障害を治療又は予防する方法であって、それを必要とすると認定された対象に式(I):
【化3】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化4】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物を投与することを含む、方法。
【請求項3】
式(I)の化合物が表1の化合物である、請求項1又は請求項2記載の方法。
【請求項4】
式(I)の化合物において、R1が−NMe2、又は
【化5】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは、場合によって置換されている)である、請求項1又は請求項2記載の方法。
【請求項5】
式(I)の化合物において、R8が場合によって置換されたアリールである、請求項1又は請求項2記載の方法。
【請求項6】
式(I)の化合物において、R8が場合によって置換されたシクロアルキルである、請求項1又は請求項2記載の方法。
【請求項7】
前記障害が、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、又は泌尿生殖路障害(例えば、膀胱制御)である、請求項1記載の方法。
【請求項8】
前記障害がコカイン中毒又はアンフェタミン若しくはメタンフェタミン中毒である、請求項1記載の方法。
【請求項9】
障害が肥満である、請求項1記載の方法。
【請求項10】
前記障害が癌、HIV、又はコフィン・ローリー症候群である、請求項2記載の方法。
【請求項11】
前記障害が乳癌又は前立腺癌である、請求項2記載の方法。
【請求項12】
前記障害がHIVである、請求項2記載の方法。
【請求項13】
そのような治療を必要とすると認定された対象において5−HT2cを阻害する方法であって、請求項1記載の式(I)の化合物を投与することを含む、方法。
【請求項14】
対象において肥満を治療する方法であって、それを必要とすると認定された対象に5−HT2a又は5−HT2bと比べて5−HT2cを選択的に阻害することができる化合物を投与することを含む、方法。
【請求項15】
5−HT2cを阻害するための前記結合相互作用が、5−HT2a又は5−HT2bのいずれかよりも少なくとも5倍(或いは、少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い、請求項14記載の方法。
【請求項16】
5−HT2cを阻害するための前記結合相互作用が、5−HT2a又は5−HT2bのいずれかよりも少なくとも100倍高い、請求項14記載の方法。
【請求項17】
そのような治療を必要とすると認定された対象においてRSKを阻害する方法であって、前記対象に請求項2記載の式(I)の化合物を投与することを含む、方法。
【請求項18】
対象において癌を治療する方法であって、それを必要とすると認定された対象に、別のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害することができる化合物を投与することを含む、方法。
【請求項19】
RSK−1、RSK−3、又はRSK−4の阻害のための結合相互作用が別のキナーゼのいずれかについてよりも少なくとも5倍(或いは少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い、請求項17記載の方法。
【請求項20】
RSK−1、RSK−3、又はRSK−4を阻害するための結合相互作用が、いずれかのRSK−2よりも少なくとも100倍高い、請求項17記載の方法。
【請求項21】
式(I):
【化6】
(式中、
R1は、独立して、H、NH2、NH(R’)、N(R’)2であるか;
又は
【化7】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで、環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよい)であり;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH又はハロであり、各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、若しくは複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物。
【請求項22】
式(I):
【化8】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化9】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで、環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり、
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式であり、ここで各R8は、2、3又は4個の独立したR5で置換され、ここで、各R5は独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである)の化合物又はその塩、水和物若しくは溶媒和物。
【請求項23】
Ri及びR8が互いにトランス配位である、請求項21又は22記載の化合物。
【請求項24】
請求項21又は22記載の化合物及び薬剤的に許容される担体を含む組成物。
【請求項25】
請求項21又は22記載の化合物及び薬剤的に許容される担体を組み合わせることを含む、請求項24記載の組成物の調製法。
【請求項26】
対象におけるGPCR介在性障害を治療又は予防する方法であって、それを必要とすると認定された前記対象に対して、請求項22記載の式(I)の化合物を投与することを含む、方法。
【請求項1】
対象におけるGPCR介在性障害を治療又は予防する方法であって、それを必要とすると認定された対象に式(I):
【化1】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化2】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式又はヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物を投与することを含む、方法。
【請求項2】
対象においてRSK介在性疾患又は障害を治療又は予防する方法であって、それを必要とすると認定された対象に式(I):
【化3】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化4】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは場合によって置換されていてもよい)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物を投与することを含む、方法。
【請求項3】
式(I)の化合物が表1の化合物である、請求項1又は請求項2記載の方法。
【請求項4】
式(I)の化合物において、R1が−NMe2、又は
【化5】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;環Aは、場合によって置換されている)である、請求項1又は請求項2記載の方法。
【請求項5】
式(I)の化合物において、R8が場合によって置換されたアリールである、請求項1又は請求項2記載の方法。
【請求項6】
式(I)の化合物において、R8が場合によって置換されたシクロアルキルである、請求項1又は請求項2記載の方法。
【請求項7】
前記障害が、神経精神障害(例えば、肥満、中毒、コカイン中毒、アンフェタミン/メタンフェタミン中毒、不安、鬱病、統合失調症、及び睡眠障害)、神経変性障害(例えば、パーキンソン病、アルツハイマー病)、神経障害(例えば、てんかん)、心臓血管障害(例えば、高血圧)、胃腸障害(例えば、過敏性腸症候群)、又は泌尿生殖路障害(例えば、膀胱制御)である、請求項1記載の方法。
【請求項8】
前記障害がコカイン中毒又はアンフェタミン若しくはメタンフェタミン中毒である、請求項1記載の方法。
【請求項9】
障害が肥満である、請求項1記載の方法。
【請求項10】
前記障害が癌、HIV、又はコフィン・ローリー症候群である、請求項2記載の方法。
【請求項11】
前記障害が乳癌又は前立腺癌である、請求項2記載の方法。
【請求項12】
前記障害がHIVである、請求項2記載の方法。
【請求項13】
そのような治療を必要とすると認定された対象において5−HT2cを阻害する方法であって、請求項1記載の式(I)の化合物を投与することを含む、方法。
【請求項14】
対象において肥満を治療する方法であって、それを必要とすると認定された対象に5−HT2a又は5−HT2bと比べて5−HT2cを選択的に阻害することができる化合物を投与することを含む、方法。
【請求項15】
5−HT2cを阻害するための前記結合相互作用が、5−HT2a又は5−HT2bのいずれかよりも少なくとも5倍(或いは、少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い、請求項14記載の方法。
【請求項16】
5−HT2cを阻害するための前記結合相互作用が、5−HT2a又は5−HT2bのいずれかよりも少なくとも100倍高い、請求項14記載の方法。
【請求項17】
そのような治療を必要とすると認定された対象においてRSKを阻害する方法であって、前記対象に請求項2記載の式(I)の化合物を投与することを含む、方法。
【請求項18】
対象において癌を治療する方法であって、それを必要とすると認定された対象に、別のキナーゼと比べてRSK−1、RSK−3、又はRSK−4を選択的に阻害することができる化合物を投与することを含む、方法。
【請求項19】
RSK−1、RSK−3、又はRSK−4の阻害のための結合相互作用が別のキナーゼのいずれかについてよりも少なくとも5倍(或いは少なくとも10倍、15倍、20倍、50倍、100倍、500倍)高い、請求項17記載の方法。
【請求項20】
RSK−1、RSK−3、又はRSK−4を阻害するための結合相互作用が、いずれかのRSK−2よりも少なくとも100倍高い、請求項17記載の方法。
【請求項21】
式(I):
【化6】
(式中、
R1は、独立して、H、NH2、NH(R’)、N(R’)2であるか;
又は
【化7】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで、環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよい)であり;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH又はハロであり、各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、若しくは複素環式(場合によって1、2、3、若しくは4個の独立したR5で置換されている)である)の化合物又はその塩、水和物若しくは溶媒和物。
【請求項22】
式(I):
【化8】
(式中、
R1は独立してH、NH2、NH(R’)、N(R’)2であるか;
又は
【化9】
(式中、環Aは、O、N及びSから選択される0〜3個のさらなるヘテロ原子を含む3〜8員複素環式若しくはヘテロアリールであり;ここで、環Aは場合によって置換されている)であり;
各R’は独立してアルキル、アルケニル、又はアルキニルであり、そのそれぞれは場合によって置換されていてもよく;
R2は独立して−(CH2)n−であり;
各nは独立して1又は2であり;
R3は独立してH、OH、又はハロであり;
R4は独立してH、OH、又はハロであり、
各R5は独立してアルキル、アリール、ハロ、ニトロ、アミノ、ヘテロアリール、シクロアルキル、複素環式、又はアルコキシであり;
R6は独立してH又はアルキルであり;
R7は独立してH、又はN(アルキル)2であり;そして
各R8は独立してアリール、シクロアルキル、ヘテロアリール、又は複素環式であり、ここで各R8は、2、3又は4個の独立したR5で置換され、ここで、各R5は独立してH、アルキル、ハロ、アリール、ニトロ、アミノ、ヘテロアリール、シクロアルキルである)の化合物又はその塩、水和物若しくは溶媒和物。
【請求項23】
Ri及びR8が互いにトランス配位である、請求項21又は22記載の化合物。
【請求項24】
請求項21又は22記載の化合物及び薬剤的に許容される担体を含む組成物。
【請求項25】
請求項21又は22記載の化合物及び薬剤的に許容される担体を組み合わせることを含む、請求項24記載の組成物の調製法。
【請求項26】
対象におけるGPCR介在性障害を治療又は予防する方法であって、それを必要とすると認定された前記対象に対して、請求項22記載の式(I)の化合物を投与することを含む、方法。
【図3】

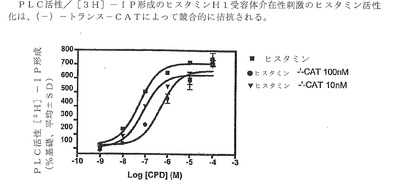
【公表番号】特表2012−526112(P2012−526112A)
【公表日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願番号】特願2012−509789(P2012−509789)
【出願日】平成22年5月5日(2010.5.5)
【国際出願番号】PCT/US2010/001333
【国際公開番号】WO2010/129048
【国際公開日】平成22年11月11日(2010.11.11)
【出願人】(509017000)ユニバーシティ オブ フロリダ リサーチ ファウンデーション,インコーポレイテッド (8)
【Fターム(参考)】
【公表日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願日】平成22年5月5日(2010.5.5)
【国際出願番号】PCT/US2010/001333
【国際公開番号】WO2010/129048
【国際公開日】平成22年11月11日(2010.11.11)
【出願人】(509017000)ユニバーシティ オブ フロリダ リサーチ ファウンデーション,インコーポレイテッド (8)
【Fターム(参考)】
[ Back to top ]