
治療用バックミンスターフラーレンのマロン酸/酢酸C60三重アダクトおよびこれに関連する方法
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトをここに開示し、特許請求の範囲に記載する。同物質の調製方法、ならびにニューロン傷害を治療するためおよび寿命延長のための使用もここに開示し、特許請求の範囲に記載する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規C60誘導体、こうした誘導体を調製する方法、および治療方法に関する。さらに詳細には、(a)新規なバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトおよび同物質を調製する方法、(b)治療的有効量のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトでニューロン傷害を治療する組成物および方法、および(c)治療的有効量のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトで後生動物のまたは後生動物細胞における予想される生存期間(または「寿命」と呼ばれる)の長さまたは期間を延長するための組成物および方法を、ここに開示し、特許請求の範囲に記載する。
【背景技術】
【0002】
ヒトおよびペット動物の総合的健康および寿命を増進させる方法は、非常に活発な研究分野になっている。この分野の現在の見解は、カロリー制限が後生動物の生存期間を延ばすために役立つことを提唱している。
【0003】
後生動物全体にわたる細胞または発育方法の保存特性を仮定して、C.エレガンス(C.elegans)およびキイロショウジョウバエ(D.melanogaster)を含む多数のモデル生物が、寿命の研究に利用されてきた。
【0004】
例えば、C.エレガンスの遺伝子分析は、寿命決定に関与する幾つかの遺伝子を明らかにした。Daf−2(インスリン受容体)およびClk−1(「Clock 1」、発生および行動のタイミングに関する多数の側面に影響を与える遺伝子)における突然変異は成人の寿命を延ばすことが証明された。しかし、Clk−1突然変異体は、寿命初期での死滅率が高い。成長後期では、Clk−1突然変異体は寿命の増加を示し、これは、おそらく寿命初期での長寿個体の選別による。Clk−1寿命表現型は、スーパーオキシド/フリーラジカル代謝に関与する、カタラーゼをコードしている遺伝子の突然変異によって、完全に破壊される。加えて、C.エレガンス中で補酵素Qを排除すると、寿命が延びることが証明された。
【0005】
Eat遺伝子に突然変異を有するC.エレガンスも、寿命の増加を示したが、摂食量の低下および代謝の遅速も示した。この突然変異に随伴する寿命の増進は、カロリー制限に帰するものであり、後生動物においても寿命を増加させることが証明された。
【0006】
ショウジョウバエ属では、スーパーオキシドジスムターゼ(SOD)およびカタラーゼの過発現が、ショウジョウバエの寿命を35%増加させた。メトセラ遺伝子(「Mth」)の突然変異も、寿命を20%増加させることが証明された。Mth、G蛋白共役受容体、の機能は不明であるが、突然変異体は、パラコート(スーパーオキシドラジカル傷害誘発物質)毒性に対する耐性増加を示し、これがストレス応答遺伝子であることを示唆している。
【0007】
カロリー制限(CR)は、今日までに研究されたすべての動物(マウス、ラット、数種のサル、イヌ、ヒト、ならびに非後生動物種、例えばクモ、線虫およびショウジョウバエ)において寿命を25から35%増加させることが証明された。(注意:すべての動物は、後生動物である)。しかし、寿命への強い効果を達成するためには、カロリー摂取を30%から40%ほども減少させる必要がある。米国国立老化研究所(National Institute of Aging(NIA))におけるアカゲザルおよびセアカリスザルでの進行中の研究(Rothら,Eur.J.Clin.Nutr.S:15,2000)では、齧歯動物において報告された変化と同様の生化学的変化が、カロリー制限したサルに見出され、このため脊椎動物種全体にわたる生化学的方法に対するカロリー制限の普遍性を支持している。
【0008】
最近、2−デオキシグルコースを使用して、経口摂取量を制限することなくカロリー制限をもたらした。2−デオキシグルコースで治療した動物には、カロリー制限した動物において観察される変化と同様の体温低下および血漿中インスリンレベル低下があった(Rothら,Ann.NY Acad.Sci.,928:305,2001)。寿命に対する2−デオキシグルコースの効果に関する科学的研究は終わっていないが、サイエンス(2002年2月8日)における最近の論説は、これらの研究の主任研究員(Geoge Roth、NIA)が、「2−デオキシグルコースで治療した彼のサルのうちの一匹が、平均生存期間の25ヶ月ではなく、38ヶ月生きた」と述べたことを引用した。しかし、こうした主張は、サンプルサイズが小さいことから、科学的に指示されない。対照集団の中で最も長く生きたサルの年齢に関するコメントもなかった。
【0009】
マウスの予想される生存期間における20%までの増加が、遺伝子操作または成長因子拮抗物質投与のいずれかによる成長因子剥奪によって示された。残念なことに、小人症が、成長因子剥奪の副作用である。ヒトにおいて、小人症または晩年期成長ホルモン欠損症は、寿命を減少させるようであり、このことが、成長因子剥奪が予想される生存の期間を増加させる1つの手段として有効であるか否かの論争をさらに混乱させている。
【0010】
デプレニール(パーキンソン病の治療に使用される選択的モノアミンオキシダーゼ(MAO)B阻害剤)が多くの種の寿命を増加させることを、幾つかの学術論文が示している(例えば、Knoll,Mech Ageing Dev.46:237,1988参照)。ある研究では、週齢96週から死ぬまでラットのデプレニールでの長期治療が、「生存を増進した」。対照ラットは、147+/−1週生き、これに対してデプレニールで治療したラットは、198+/−2週生きた。しかし、この学術論文においてはっきりと述べられているこれらのラットの平均予想寿命は182週であった。このため、この研究における対照群は、早期死亡率を有したようである。これらの研究所からの他の研究では、高能力のラットを選択し、次いで、これらがデプレニール寿命試験に登録された。このために人為的に結果が歪められた可能性がある。
【0011】
第二の研究は、344匹のFisherラット(Kitaniら,Life Sci 52:281,1993)を使用して、18ヶ月齢でデプレニール治療を開始した。対照の平均生存は、28ヶ月であり、治療動物の平均生存は、30ヶ月であった。このことは、7%の寿命の増加を示している。しかし、これらの結果は、統計学的に有意ではないと証明された。
【0012】
対照的に、同じ用量のデプレニールを用いた344匹のFisherラットでの別の研究(Carilloら,Life Sci 67:2539,2000)では、デプレニール治療動物において、より高い死亡率および短縮された寿命が観察された。さらに、NIAからの研究では、18ヶ月齢で長期デプレニール治療を開始したC57B6マウスにおいて何の生存利益も示されなかった(Ingramら,Neurobiol Aging 14:431,1993)。同様に、ショウジョウバエ属におけるデプレニールの対照試験は、寿命の増加を示さなかった(Jordenら,Neurochem Res 24:227,1999)。
【0013】
同様に、ヒトでのデプレニール試験は、寿命に関して相反する結果を示した。パーキンソン病患者における「公開、非対照」試験は、9年で生存増加を示した(Birkmayerら,J.Neural Transm.64:114,1985)が、他の研究は、デプレニールを特にL−ドパと共に摂取しているPD患者の死亡率増加を示唆している(例えば、Ben−Shlomoら,BMJ 316:1191,1998)。
【0014】
全体的に見て、データは、デプレニールが寿命に対して弱い効果を及ぼすことがあり、または及ぼさないこともあることを示唆している。
【0015】
マウスの幾つかの遺伝子は、「寿命」遺伝子として特定されており、これは、これらの遺伝子に突然変異を有するマウスが、対照マウスの予想される生存期間に比して長い平均寿命を有するからである。これらの遺伝子は、エイムズ矮性突然変異およびスネル矮性突然変異を含む。しかし、これらの突然変異によって、飼育が難しい小さく虚弱なマウスが生じることとなる。これらの突然変異によりもたらされる寿命は、本質的にカロリー制限に起因すると考えられる。遺伝子アレイ分析、または蠕虫、ハエおよび齧歯動物における寿命表現型に関連した遺伝子についての他の遺伝子スクリーニングを利用する最近の試みによって、多数の候補遺伝子が見つけ出された。しかし、一般に、これらは多くの場合、「ストレス応答」遺伝子である。
【0016】
イチョウ(Gingko)、ニンジン(Ginseng)、ビタミンCなどの多数の化合物が、生存を改善するために提案されてきたが、これらの化合物に関する利点を報告する管理された、統計学的に有意な生存研究は、知られていない。ビタミンCおよび多数の薬物は、一定の疾病状態、例えば心血管疾患の発生率を低下させ、このためおそらく総合的には寿命を増進するであろう。
【0017】
バックミンスターフラーレン、C60、は、芳香族系溶媒には可溶性であるが、水には可溶性ではない、12の五角形および20の六角形を有する炭素球である。
【0018】
C60(C(COOH)2)n(式中、nは、1から4の整数である)の使用は、ニューロン傷害の治療に関し、2001年7月24日に発行されたChoiらの米国特許第6,265,443号に開示されている。前記特許は、この全文が本明細書に参考として援用されている。
【0019】
この文献において報告されているC3ヘキサカルボン酸(「C3」または「Hexa」)の調製では、細胞培養スクリーニングでの再生能および変異機能が劣る、一部未確認のプロドラッグの混合物が生成される。
【発明の開示】
【0020】
上記にかんがみて、下で説明し、特許請求の範囲に記載する組成物および方法を開発した。
【0021】
第一の実施態様は、後生動物の寿命を増加する結果となる後生動物への組成物の投与を含み、前記組成物は、ペンダント炭素に結合しているx対の隣接した炭素原子を有するC60化合物などのC60フラーレンのカルボキシル化誘導体(「カルボキシフラーレン」)を含み、ここで、前記ペンダント炭素原子は、一般式−COOHおよび−R(Rは、−COOHおよびHから成る群より独立して選択される)の2つの基にさらに結合しており、ならびにxは、少なくとも1である。
【0022】
有用な化合物の別の実施態様は、一般式C60[(CHCOOH)]x[C(COOH)2]y(式中、xは、0から3の整数であり、yは、1から4の整数であり、x+yは、2から4の整数である)と記述され得る。
【0023】
追加の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトである。さらに詳細な実施態様は、Penta Pair、Tetra QuartetおよびC3−ライト バックミンスターフラーレンのマロン酸/酢酸三重アダクト(後でより詳細に説明する)を含む。
【0024】
追加の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは−Hまたは−COOMeである)のバックミンスターフラーレンの三重アダクトである。
【0025】
さらなる実施態様は、Penta Pair、Tetra QuartetおよびC3−ライト バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを含む、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを調製する方法を含む。
【0026】
さらに追加の実施態様は、ニューロン傷害を治療および寿命を延長するための(C3に類似した)新規バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトの使用を含む。
【0027】
バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレンの使用は、カロリー制限の中での固有の難しさ(一般に、摂食量の厳格な制限ならびにヒトでの使用の実行不可能性が挙げられるが、これらに限定されない)からして、後生動物、特にヒトの寿命を実質的に増加させる方法として、カロリー制限を越える実質的な改善をもたらすと考えられる。新規バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトは、C3トリスマロン酸C60化合物に類似した望ましい特性を有するが、動物体内での半減期がより長く、その結果、インビボでの有効期間が延長されることも明らかにする。加えて、Penta−1、Penta−2、Tetra QuartetおよびC3−ライトを含むバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトは、C3より親油性が高いため、脳などの高脂質組織内に集中することができる。
【0028】
明細書に組み込まれており、この一部を形成している添付の図面は、本発明の様々な実施態様を説明するものであり、この記述とともに本発明の原理の説明に役立つ。
【0029】
添付の図面を参照して示すと(同じ参照番号が同じ要素を示す):
図1aは、HPLCによるC3調製物の分析を開示するものであり、(全体の99%を上回る)3つの主成分が同定されている。
【0030】
図1bは、3つのピークすべてが、C60核へのe,e,e(C3)付加に特有の吸収スペクトルを有することを示すものであり、C3のこれら成分ピークが、C60上のシクロプロパン炭素に結合している先端基が異なるe,e,e位置異性体を表すことを示している。
【0031】
図1c(1)から1c(3)は、HPLCによってHexaカルボン酸C3(1、80%)と二つの異性体Pentaカルボン酸(2および3、各10%)に分離し、次いで、質量分析によって判定した化合物1〜3を示す。
【0032】
図1d(1)から1d(3)は、前記Hexa異性体(1)および各Pentaカルボン酸(2)、(3)に対して行った質量分析を示す。
【0033】
図2は、2つのビス異性体、2つのトリス異性体、および1つのテトラ異性体を含む様々なカルボキシフラーレンを表す。
【0034】
図3は、C3対称性を有するe,e,eトリスマロン酸位置異性体(「C3」)を空間が詰まった構造と化学構造の両方として示すものである。
【0035】
図4は、月齢12ヶ月から死ぬまで飲み水に入れた食用着色剤(対照)またはC3(0.5mg/kg/日)のいずれかで治療したC57B6マウスの寿命を示すKaplan−Meier生存曲線である。各マウスについての自然死の日付を記録し、これを使用して寿命を計算した。NIA齧歯動物コロニーから受け取ったマウスには、誕生月の記録はあったが、具体的な誕生日の記録がなかったため、各マウスの寿命は月ごとに計算した。平均生存率を治療ごとに計算し、t検定を用い、有意性をp<0.05(実際にはp=0.033)に設定して平均寿命を比較した。第一群からのデータは、性別(A、B)および混成(C)でグラフにし、各治療グループを同性の対照と比較した。治療および未治療マウスの体重(g)は、(性による)差がなかった。例えば、19ヶ月の体重は、雌については、対照:27±1、C3治療:29±1であり、雄については、対照:35±4、C3治療:35±6(平均±SD)であった。
【0036】
図5は、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト上の官能基の立体配置の詳細図である。
【0037】
図6は、C3−ライト(e,e,eトリス酢酸C60)およびC3の構造を表す。C3−ライトは、3つのシクロプロパン炭素各々に結合しているカルボン酸の対のうちの1つがプロトンで置換されている点で、C3とは異なる。
【0038】
図7は、Penta−1化合物の構造を表す。HOMO−LUMOエネルギー分布(上部)、および球と棒によるモデル(下部)。Penta−1は、C3のマロン酸基のうちの1つが、酢酸によって置換されている。Penta−1では、このプロトンが、二つのマロン酸基の方に向いている。
【0039】
図8は、Penta−2化合物の構造を表す。HOMO−LUMOエネルギー分布(上部)、および球と棒によるモデル(下部)。Penta−2は、C3のマロン酸基のうちの1つが、酢酸によって置換されている。Penta−1では、このプロトンが、二つのマロン酸基とは反対の方に向いている。
【0040】
図9は、大脳皮質細胞培養物におけるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトHexa、Penta−1、Penta−2およびC3−ライトによる、NMDA受容体媒介興奮毒性に対する神経保護を表す。C3誘導体(0.05から100μM)を含有する、または含有しない200μMのNMDAに培養物を10分間暴露した。10分後、すべての薬物を洗浄除去し、細胞を24時間、細胞培養器に戻した。次に、ニューロンを乾燥させることにより乳酸デヒドロゲナーゼ(LDH)の放出を測定することによって、神経細胞死を評価した。細胞死および保護は、死滅したニューロンのヨウ化プロピジウム染色を画像化することにより、および位相差顕微鏡を使用してニューロンの形態を評価することにより、確認した。各化合物についての用量応答曲線を示す。値は、NMDAのみ(C60誘導体を含有しないもの)に暴露した培養物において観察された細胞死%を表す。
【0041】
図10は、大脳皮質細胞培養物におけるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトHexa、Penta−1、Penta−2およびC3−ライトによる、AMPA受容体媒介興奮毒性に対する神経保護を表す。C3誘導体(0.5から100μM)を含有する、または含有しない6μMのAMPAに培養物を24時間暴露した。ニューロンを乾燥させることにより乳酸デヒドロゲナーゼ(LDH)の放出を測定することによって、24時間での神経細胞死を評価した。細胞死および保護は、死滅したニューロンのヨウ化プロピジウム染色を画像化することにより、および位相差顕微鏡を使用してニューロンの形態を評価することにより、確認した。各化合物についての用量応答曲線を示す。値は、AMPAのみ(C60誘導体を含有しないもの)に暴露した培養物において観察された細胞死%を表す。
【0042】
図11は、Hexa、Penta−1およびPenta−2の血漿中薬物動態(AからB)およびHexaC3の組織分布(C)を表す。図11(A)は、80% Hexa、10% Penta−1、10% Penta−2を含有する14C−C3を注射したマウスからの血漿のHPLC分析である。サンプルは、t=0(注射前のC3サンプル)、t=2時間(C3の腹腔内注射から2時間後の血漿)、t=18時間(注射から18時間後の血漿)であった。図11(B)は、C3の静脈内、腹腔内、皮下および経口投与後の血漿中レベルを表す。Hexa、Petnta−1およびPenta−2についての血漿中T1/2は、すべて8.2時間であった。図11(C)は、腹腔内投与後、様々な時点でのC3の組織内レベルを表しており、肝臓および腎臓による蓄積を示している。
【0043】
図12は、C3−ライトの血漿中薬物動態および組織分布を表す。図(A)は、14C−C3−ライトの血漿中薬物動態を示すものであり、15時間のT1/2を実証している。図12(B)は、腹腔内投与後のC3−ライトの組織内分布を表す。
【発明を実施するための最良の形態】
【0044】
全体を通して、以下の用語を使用する:
「寿命」または「予想される生存期間」は、ある特定の環境において後生動物が、カルボキシフラーレンでの治療を受けなかった場合、生きることが予想される(生まれてから死ぬまでの)生涯(すなわち、「属に特有の」予想される生存期間)の予想平均長である。
【0045】
「マロン酸/酢酸C60誘導体」は、C60の酢酸誘導体、C60のマロン酸誘導体、およびC60のマロン酸/酢酸混合型誘導体である。
【0046】
「Hexa」およびC3は、C3(C60(C(COOH)2)3を表し、この場合、マロン酸基は、すべて、e,e,e位にある(化合物1、下記)。
【0047】
「Penta」または「Penta Pair」は、(C60(C(COOH)2)2(C(CHCOOH))を意味し、この場合、R基は、e,e,e位でシクロプロパン炭素に結合している。2つの立体異性体Penta−1およびPenta−2がある(化合物2、下記)。
【0048】
「Tetra」または「Tetra Quartet」は、(C60(C(COOH)2)(C(CHCOOH))2を意味し、この場合、酢酸/マロン酸基は、e,e,e位でシクロプロパン炭素に結合している。4つの立体異性体がある(化合物3、下記)。
【0049】
「C3−ライト」は、(C60(CHCOOH))3を意味し、この場合、酢酸基は、e,e,e位でシクロプロパン炭素に結合している。4つの立体異性体がある(化合物4、下記)。
【0050】
「バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト」または「マロン酸/酢酸C60誘導体」は、マロン酸基(>C(COOH)2)および酢酸基(>CHCOOH)から独立して選択される3つのペンダント基がe,e,e位にあるバックミンスターフラーレン、すなわち、
【0051】
【化1】
を意味する。
【0052】
寿命の予想長または期間を延長するための、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレン
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させる。スーパーオキシド(O2−●)および過酸化水素(H2O2)を含む反応性酸素種は、特定の生理機能に利用される一方で、細胞および組織の生存力および完全性に脅威を与え続けているという問題ももたらしている。これに応じて、細胞および生体は、自らをO2−●およびH2O2から防御する様々なメカニズムを発達させた。後生動物では、O2−●は、二つの金属酵素、Cu,Zn−スーパーオキシドジスムターゼ(SOD1)、およびMnSOD(SOD2)によって除去される。また、H2O2は、カタラーゼ(ヘム鉄含有金属酵素)またはグルタチオンペルオキシダーゼ(セレノシステインとグルタチオンを併用してH2O2をO2とH2Oに変換する蛋白質の一科)によって除去される。しかし、これらの内因性抗酸化物質防御系は、病的状態の下では圧倒されてしまうことがある。これは、細胞の抗酸化物質防御を補足する小分子としてのさらなる抗酸化物質(酸化を抑制する、または酸素もしくは過酸化物によって促進される反応を抑制する、有用な物質)を潜在的治療薬として開発する試みを導いた。
【0053】
多数の水溶性C60誘導体(スーパーオキシドジスムターゼ模擬体)は、これらの親フラーレン分子の抗酸化特性を保有しており、このため生体系においてこのフリーラジカルスカベンジ能力を利用することができ、その結果、細胞の損傷および死滅を減少させる因子としての機能を果たすことができる。
【0054】
C60誘導体、カルボキシフラーレンの1つの基は、H2O2およびO2−●の分解酵素としての機能を果たす。MnTMPypを含むマンガン含有プロトポルフィリン化合物は、O2−●/H2O2の分解酵素としての機能を果たすと報告されているが、これらの化合物は、マンガン原子の酸化−還元に依存して分解を触媒する。C3は、非金属化合物であるが、同様の触媒性状を有することを、今般、本発明者らは発見した。本化合物は、こうした方法で動作する最初の非金属化合物であると考えられる。
【0055】
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させるため、潜在的治療薬として細胞の抗酸化物質防御を補足することができる抗酸化性分子は、治療上有用である。これは、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトなどのカルボキシフラーレンを含む。これらの新規カルボキシフラーレン組成物は、抗酸化特性を有する。本研究を通じて、O2−●およびH2O2とC3の反応性を特性付けした。O2−●に対するC3のKiを計算して、3x106M−1秒−1になった。O2−●およびH2O2との相互作用後のC3の分析は、C60部分においても、マロン酸基においても永続的な化学的または構造的変化が発生しないことを示した。これは、C3が真の触媒であるという本特許請求事項を支持している。マンガン含有プロトポルフィリンおよびサレン化合物も、O2−●/H2O2の分解のための触媒としての機能を果たすと報告されているが、これらの化合物は、金属原子の酸化−還元に依存して分解を触媒し、これに対して本マロン酸フラーレン誘導体は、反応性酸素種の分解を触媒するために金属原子を必要としない。
【0056】
従って、上述の発見にかんがみて、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレンは、反応性酸素種、特に、過酸化水素(H2O2)およびスーパーオキシド(O2−●)などの生理関連の反応性酸素種の排除に有用である。
【0057】
後生動物または後生動物の細胞の寿命を延ばす結果となる、抗酸化物質の治療的有効量を投与することにより後生動物の予想される生存期間を増加させる方法を、ここで説明し、特許請求の範囲に記載する。詳細には、抗酸化性カルボキシフラーレンを含む組成物を治療薬として使用して、後生動物または後生動物細胞の寿命を増加させる。
【0058】
従って、ここで有用な化合物は、このC60フラーレンのx対の隣接炭素原子が、少なくとも1つのペンダント炭素に結合しており、このペンダント炭素原子が、一般式−COOHおよび−Rの2つの基にさらに結合している、カルボキシフラーレン化合物、これらの対応する塩およびエステルであり、この場合、Rは、−COOHおよび−Hから成る群より独立して選択され、xは、少なくとも1である。この一般式の異性体の例を図1から3に示す。一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトも、ここでは有用である。
【0059】
従って、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを後生動物に投与することによる、哺乳動物およびさらに詳細にはヒトを含む後生動物または後生動物細胞の予想される生存期間を延ばす方法を提供する。
【0060】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むすべてのカルボキシフラーレン化合物は、活性化合物および前記化合物と相溶性の医薬適合性担体を含有する組成物として全身投与することができる。こうした組成物の調製では、従来どおりの医薬適合性担体を利用することができる。この薬物を経口投与する際には、一般に、一定の間隔を開けて投与する。
【0061】
治療使用の際、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、薬物が従来投与されているあらゆる経路によって投与することができる。こうした経路には、静脈内経路、筋肉内経路、皮下経路、髄腔内経路、腹腔内経路、局所経路および経口経路が挙げられる。
【0062】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む医薬組成物は、経口投与のための固体形、例えば錠剤、カプセル、ピル、粉末、顆粒などを含む、あらゆる従来型の形態で製造することができる。これらの医薬組成物は、滅菌することができ、ならびに/または保存薬、安定剤、湿潤剤、乳化剤、浸透圧を変化させるための塩、および/もしくは緩衝剤などのアジュバントを含有することができる。
【0063】
静脈内投与のための局所製剤は、水/緩衝溶液を含む滅菌水溶液であろう。静脈内用ビヒクルには、液体、栄養および電解質補充液が挙げられる。保存薬および他の添加剤、例えば抗生物質および抗酸化物質が存在してもよい。
【0064】
ここで説明し、特許請求の範囲に記載するバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、医薬適合性の経口方式で有用である。これらの医薬組成物は、前記化合物と併せて相溶性の医薬適合性担体材料を含有する。あらゆる従来型担体材料を利用することができる。あらゆる従来型経口剤形、例えば錠剤、カプセル、ピル、粉末、顆粒などを使用することができる。担体材料は、経口投与に適する有機または無機不活性担体材料であり得る。適する担体には、水、ゼラチン、アラビアゴム、ラクトース、デンプン、ステアリン酸マグネシウム、タルク、植物油、ポリアルキレン−グリコール、ワセリンなどが挙げられる。さらに、本医薬組成物は、他の薬学的に活性な薬剤を含有することができる。さらなる添加剤、例えば着香剤、保存薬、安定剤、乳化剤、緩衝剤などを、薬剤の調剤の一般に是認されているやり方に従って、添加してもよい。
【0065】
経口剤形は、例えば錠剤、硬質もしくは軟質ゼラチン、メチルセルロースの、または消化管内で容易に溶解する別の適切な材料のカプセルを含む。考えられる経口投薬量は、処方する医師が判定するような個々の患者の必要性に従って変化するだろう。この経口剤形実施態様の一例は、50から500mgのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含有するカプセルまたは錠剤を含む。
【0066】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトでの治療方法は、一般には成人に毎日、好ましくは経口的、筋肉内的、皮下的または静脈内的に投与すことができる。筋肉内的、静脈内的または皮下的に投与する場合、治療薬は、約0.1mg/kgほどの少ない量から3mg/kgほどもの多い量で投与されることとなり、この正確な投薬量は、患者の必要性に依存して変化するであろう。経口投与の場合の日用量は、0.1mg/kgほどの少ない量から15mg/kgほどもの多い量になると予想される。一般に、この療法は、無期限に、予防的に行うことができる。
【0067】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、常習的に(例えば毎日)投与してもよいし、または頻繁に(例えば一週間に一度)投与してもよい。C3 Hexa異性体、Penta Pair、Tetra QuartetおよびC3−ライトは、より有効な薬剤であると予想される。静脈内、筋肉内または皮下送達によって投与する場合、C3異性体の予想日用量は、約0.1mg/kgから約3mg/kgであろう。経口的に投与する場合、この日用量は、0.1mg/kgと15mg/kgの間の範囲であると予想されよう。
【0068】
上の投薬情報は、マウスにおいて行った薬物動態試験、マウスでの毒性試験およびラットでの毒性試験に基づくものである。マウスにおいて、C3、Penta−1およびPenta−2の血漿中半減期は、約8時間であると計算され、一方、C3−ライトの血漿中半減期は、約15時間であった。1回のカルボキシフラーレン注射についての50%致死量(LD50)は、70mg/kgより上であった。C3、Penta−1およびPenta−2は、肝臓および腎臓による排泄によってマウスから一掃され、一方、C3−ライトは、肝臓および脂肪によって一掃された。この薬物動態データに基づく計算を用いると、治療的血漿中レベルは、0.1μg/mLと1μg/mLの間であるようである。本化合物を静脈内、腹膜内または皮下投与によって与えると、同量のカルボキシフラーレンが吸収されるが、(例えば、飲料水の中に入れて)経口投与すると、この用量の約15分の1しか吸収されない。しかし、経口送達のためのカルボキシフラーレンの標準的な医薬調合物(例えば、カルボキシフラーレンを時間放出性錠剤に組み込んだもの)は、経口投与されるカルボキシフラーレンのバイオアベイラビリティを有意に増大させると予想される。加えて、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、C3より親油性が高く、このことが、これらを脳などの脂質豊富な組織に集中させる。
【0069】
本特許請求の範囲に記載するこれらの方法は、脊椎動物、さらに具体的には、ヒトおよびペット動物を含む哺乳類を含む、すべての後生動物に有用である。
【0070】
カルボキシフラーレンによって増加される寿命とは、後生動物が、カルボキシフラーレンで治療を受けなかった場合、生きることが予想される(生まれてから死ぬまでの)時間の予想平均長である。実施例2および図4の結果が示すように、この治療を受けたマウスは、23.5ヶ月の対照マウスの寿命より約20%長い寿命に相当する28.7ヶ月の実際寿命を有した。この実施例で使用した対照マウスの寿命は、一般「予想寿命」を表す。
【0071】
哺乳動物の生存期間を延ばすために有用な化合物の非限定的実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0072】
さらなる非限定的実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを少なくとも1つ含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばす方法である。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pari、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。前記組成物は、少なくとも1つのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体を含み、これらの成分は、前記組成物中に治療的有効量で存在すると考えられる。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、静脈内、筋肉内、皮下または経口投与することができる。この方法は、脊椎動物、哺乳類およびヒトを含む(しかし、これらに限定されない)すべての後生動物で使用することができる。
【0073】
バックミンスターフラーレンのマロン酸/酢酸三重アダクト
C60e,e,eマロン酸/酢酸三重アダクトは、増大した水溶性を含むさらなる望ましい特質を示す。1)C60の酢酸誘導体および2)バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、幾つかの新規C60e,e,eマロン酸/酢酸三重アダクトを合成し、特性付けした。「Penta−1」(図7)および「Penta−2」(図8)と呼ばれる2つの化合物(2)は、C60に付加している2つのマロン酸基と1つの酢酸基を有するC60のe,e,e誘導体である。Penta−1およびPenta−2は、酢酸基のシクロプロパン炭素に結合しているプロトンが、マロン酸基の方に向いているか、マロン酸基とは反対の方に向いているかという点で異なる。第二組の4化合物「Tetra−Quartet」(3)は、e,e,e位に1つのマロン酸基と2つの酢酸基を有する。第三組の化合物は、C60のe,e,eトリス酢酸誘導体(C3−ライト)(4)(図6)である。これらのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトには、Penta−1、Penta−2、4つのTetraおよびC3−ライト化合物、これらの医薬適合性の塩およびエステルが挙げられる。
【0074】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトについての一般式は、C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)を含む。
【0075】
神経保護性のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクト
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させるため、細胞の抗酸化防御を補足することができる抗酸化性分子は、潜在的治療薬として治療上有用である。
【0076】
ここで説明し、特許請求の範囲に記載するバックミンスターフラーレン誘導体のe,e,eマロン酸/酢酸三重アダクトを使用して、フリーラジカル、特にグルタメートの神経毒性(「興奮毒性」)の結果として放出されるときのフリーラジカルによって引き起こされるあらゆる疾病状態の進行を予防、治療または緩和することができる。興奮毒性傷害の治療は、周囲の細胞から放出されたグルタメートによって損傷した中枢神経に対する損傷の程度を低減させること意味する。興奮毒性などの神経毒性事象は、低酸素/虚血などの多数のタイプの急性神経性発作中に発生し得る。例えば、卒中、低血糖、癲癇または外傷中に発生する。神経毒性事象は、神経変性疾患、例えば、ハンチングトン病、アルツハイマー病、筋萎縮性側索硬化症(「ALS」)、およびAIDSの神経変成作用によって引き起こされる慢性神経障害にも関与し得る。従って、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、神経毒性傷害が発生する疾病の治療法にも有用である。
【0077】
アラキドン酸(「AA」)は、グルタメートによるNMDA受容体刺激によって引き起こされる神経細胞への過剰なCa2+の流入に起因してニューロンにおいて放出される(このグルタメートは、神経毒性事象によって損傷したニューロン自体によって放出されたものである)。過剰なCa2+の流入は、ホスホリパーゼA2(AAを遊離する細胞膜を破壊するカルシウム依存酵素)を活性化する。内因性リポキシゲナーゼおよびシクロオキシゲナーゼによるAAの代謝は、ニューロンの損傷または死をもたらす神経脂質膜の過酸化性分解を開始させる酸素フリーラジカルの生成を導く。従って、フリーラジカル・スカベンジ性のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを含む組成物の投与による酸素由来フリーラジカルの低減は、グルタメート誘発神経毒性を抑制する代替メカニズムを提供する。
【0078】
個体の寿命を増加させるための上記方法および組成物と同様に、神経毒性傷害に罹患している患者において神経毒性障害を治療するための組成物および方法は、前記患者に、この個体に対する治療的に有効な量のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを含む組成物を投与することを含む。さらに詳細には、これらの実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを少なくとも1つ投与することを含む。更なる実施態様は、患者へのC3−ライト、Penta−1、Penta−2およびTetra Quartetの投与を含む。これらの新規化合物での治療方法は、静脈内、筋肉内、皮下または経口送達することができる。
【0079】
第一の実施態様は、ニューロン傷害治療用の化合物を含む。こうした化合物の非限定例には、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが挙げられる。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトのさらなる非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0080】
第二の実施態様は、神経毒性傷害に罹患している患者において、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステル、ならびに医薬適合性の担体を含む組成物(この場合、前記化合物は、前記神経毒性障害を治療するために有効な量で前記組成物中に存在する)を前記患者に投与することによる、神経毒性障害の治療方法を含む。ニューロン傷害の治療に有用なバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択されるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが挙げられる。
【0081】
上で示したように、この方法の化合物は、1日に約1.5mg/kgから約1500mg/kg、または1日に約10mg/kgから約60mg/kgの量で投与することができる。
【0082】
第三の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、患者において、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされる神経毒性傷害を抑制する方法である。この方法に有用なバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0083】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、1日に約1.5mg/kgから約1500mg/kgの量で、または1日に約10mg/kgから約60mg/kgの量で投与することができる。
【0084】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの化学合成
文献に報告されているC3の調製では、細胞培養スクリーニングでの再生能および変異機能が劣る、一部正体未確認のプロドラッグの混合物が生産される。従って、特定の成分へのより信頼性できる、直接的な方法が必要である。
【0085】
高濃度のHexaを合成する方法
下記合成方法を用いて、一般に90、94および97%を超える結果で、大量のHexaを生じさせることができる。
1つのこうした方法は、水、メタノールおよび溶媒中のC3メチルエステルの初期溶液の調製を含む。水のエステルに対するモル比は、約100:1から約20:1である。C3メチルエステルを溶解するために有用な溶媒には、芳香族系溶媒、例えばトルエン、トルエン中の酢酸t−ブチルなどが挙げられる。次に、この初期溶液を完全に(約1時間)混合する。これは、最初は濁っているように見えることがある。次に、メタノール中、約1Mまでの水酸化ナトリウムを添加した後、この溶液を、溶液の着色がなくなるまで(約2時間以下)激しく攪拌する。反応の完了を確認するために、TLCを使用してもよい。
【0086】
反応が完了したら、水を添加して非水性層と水性層を形成する。次に、デカント、および分液漏斗または層を分離するために有用な他のあらゆる器具の使用を含む(しかし、これらに限定されない)あらゆる標準法によって、これらの層を分離する。次に、一切の残留溶媒を真空下でストリップする。生成物を含有する水性層を、不活性雰囲気(N2など)中、約60℃で約1から約2時間加熱する。
【0087】
異性体配分は、HPLCプロトコル1および2(下記)を使用して判定することができ、一般には90、94または97%より上である。
【0088】
脱カルボキシル化によるPenta PairまたはTetra Quartetの直接合成
溶解度を最大にするように選択された芳香族系溶媒、例えばトルエンまたはベンゼン中の(Bingel、米国特許第5,739,376号(この全文が本明細書に参考として援用されている)により調製されるような)ジ−t−ブチルマロニルC60に、ブロモマロン酸のエステル(ブロモマロン酸ジメチルを含むが、これに限定されない)を先ず添加し、続いて、(Bingelの‘376、カラム4、44から45行目に開示されているとおり調製した)1,8−ジアザビシクロ(5,4,0)ウンデク−7−エン(「DBU」)または水素化ナトリウムを含む(しかしこれらに限定されない)第三アミンを添加して、反応混合物を作ることにより、Penta PairまたはTetra Quartetを直接合成する。Penta Pairを調製するためには、これらの反応物は、C60対マロネート対塩基比1:2:2の濃度である。Tetra Quartetを調製するためには、比率1:1:1を用いる。適切な時間(約30分)を経過させて、確実にこの反応混合物を完全混合する。反応の終点は、TLCによって判定することができる。次に、この反応混合物を、上で選択した溶媒中のシリカゲルのカラムに注入する。ビス異性体を調製するために、このカラムは、すべてのビス異性体が出てくるまで溶媒で溶離しなければならない。次に、溶媒は、酢酸エチル/溶媒混合物に換えるべきであり、酢酸エチルの漸増濃度、例えば0.5%→1%→約2%で添加すべきである。これらの混合物は、次の4つの成分を溶出するであろう。D3(トランス−3,トランス−3,トランス−3 トリスマロン酸C60);e,トランス−3,トランス−2;e,トランス−4,トランス−3;およびC3エステル。溶出されたC3エステル画分を、次いで、真空下で蒸発させる。
【0089】
次にp−トルエンスルホン酸・一水和物、トリフルオロ酢酸(TFA)またはメタンスルホン酸を含む(しかし、これらに限定されない)上記溶媒に混和可能な酸を、芳香族系溶媒(トルエンを含むが、これに限定されない)中のC3エステル画分の溶液に添加して、前記画分を溶解する。この溶液を約88から89℃に加熱する。より低い温度を用いてもよいが、反応は、より遅い速度で進行することになる。TFAを用いる場合、室温で充分であろう。反応が開始したら(約45分後、またはアッセイにより判定する)、追加の酸を添加すべきである。約10分後に沈殿が形成し始めるだろう。加熱は、反応が完了するまで(約90分、またはTLCもしくはHPLCなどのアッセイによって判定する)継続する。次に、溶媒をこの沈殿から除去する。これらの固体に水および酢酸エチルを添加し、酢酸エチル/水混合物を作る。この酢酸エチル溶液を分離し、次いで、水で洗浄して触媒の酸を除去する。必要な場合には、酢酸エチルを複数回、洗浄しなければならない。次いで、酢酸エチルを真空下で蒸発させて、固体を残す。
【0090】
次に、固体を約1:1:2のアセトニトリル:水:アセトン混合物に溶解する。材料の可溶化を最大にするために、他の比率(アセトン単独使用を含む)を用いてもよい。この混合物を油浴、加熱マントルなどで約50℃、約76.5℃または約100℃に加熱して、反応を完了させる。反応が完了していることを確認するために、アッセイを行ってもよい。次に、沈殿物を充分に溶解するために一定量のアセトンを添加する。加熱は、約5時間から約7.5時間、継続する。次に、一切の揮発性溶媒を(例えば真空下で)除去し、次いで、酢酸エチルを添加して均質な溶液を作る。次いで、この溶液を真空下で蒸発させて、固体を生じさせる。
【0091】
次に、この固体を、メタノールおよび水を含有する芳香族系溶媒(トルエンまたはトルエン中5%の酢酸t−ブチル(1mg/mL))に溶解する。様々な比率(100:4.4:0.2など)を用いることができるが、二相の生成を回避するために、限られた量の水を使用すべきである。次いで、この溶液を、約1時間攪拌しながら充分に混合する。混合後、比率20対1の1M 水酸化ナトリウム/メタノールを添加する。沈殿完了後(色が完全に消えたら(これは、約1時間から約2時間かかることがある))、水を添加する。次いで、溶媒層を完全に分離し、一切の残留溶媒を確実に除去する。水性層を約60℃から約110℃で約2時間+/−30分間加熱する。アッセイを行って、反応の完了を判定する。
【0092】
冷却後、硫酸などの強酸(酢酸エチルに抽出されないであろうもの)を、上からの塩基を中和させるために充分な量でこの溶液に添加する。最終生成物は、上で説明したように酢酸エチルで抽出する。
【0093】
(溶液またはニートでの)C3−ライトの直接合成
C3−ライトは、Hexa、Penta−1およびPenta−2のC3誘導体の乾燥または溶解サンプルを加熱することにより、直接合成することができる。溶解サンプルを使用する場合、前記誘導体は、アセトニトリル:水の混合物に溶解すべきである。加熱マントル、油浴などを含む(しかし、これらに限定されない)あらゆる標準的加熱方法を用いることができる。得られた溶液/サンプルを、C3−ライトを生じさせるために充分な時間(約24時間未満)、約60℃から約70℃から約81度(アセトニトリルの沸点)で加熱する。次いで、溶媒を真空下で除去して、固体生成物を得る。
【0094】
C3誘導体のニートサンプルを用いる場合、このサンプルは、約−30mmHgの真空オーブン内で、約150℃で加熱する。
【0095】
Petna、TetraまたはC3−ライトを生じさせるための溶液中でのC3の熱分解
Petnta,TetraまたはC3−ライトを含む生成物は、Hexa、Penta−1およびPenta−2を含有するC3誘導体のサンプルを、比率1:1のアセトニトリル:水に溶解することによって生成する。得られた溶液を約60℃に加熱する。約1.5時間加熱後、Penta濃度は、ほぼこの最大値である。加熱の適用が長いほど(約3.5時間から約5.5時間)、Penta濃度は低下するであろうが、C3−ライトおよびTetraの濃度は、上昇するであろう。
【0096】
所望の結果が得られたら、溶媒を真空下で除去して、固体生成物を得る。
【0097】
HexaとPentaの混合物を合成するためのNaOMe/MeOHの使用
N2などの不活性ガス下、芳香族系溶媒中のe,e,e−トリスジメチルマロニルフラーレン(「C3エステル」)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作る。芳香族系溶媒は、トルエンおよびトルエン中の酢酸t−ブチルから成る(しかし、これらに限定されない)群より選択することができる。ナトリウムメトキシドとC3エステルは、比率16から20:1である。赤橙色の沈殿が、直ぐに形成し始める。適切な時間(約1から2時間)の後、水を添加して、水性層と非水性層を形成する。あらゆる色が水性層に移るであろう。次に、層を分離する。水性層を氷浴などで0から5℃に冷却し、次いで、過剰な硫酸で約2のpHに酸性化する。次に、この酸性溶液を酢酸エチルで抽出して、すべての色を酢酸エチル抽出物に移す。次に、併せた酢酸エチル抽出物を水で洗浄して、一切の黄色不純物を除去する。次いで、この溶液を、真空下、ほぼ室温で蒸発および乾燥させる。
【0098】
細胞スクリーニングのための溶液は、0.1Nの水酸化ナトリウムにこの固体を溶解することにより調製する。充分な塩基を添加して、すべてのカルボキシルを中和することができる。有効濃度は、トルエン中のこのエステルの前駆体で決定した吸光係数4400を使用して、uvなどのアッセイにより決定することができる。HPLCプロトコル1を用いて、Hexa、Penta−1およびPenta−2の百分率を判定することができる。この時、HPLCによりこれら三成分を分離することができる。
【0099】
ここでのデータは、開示するカルボキシフラーレンが、酸素由来のフリーラジカルを分解するユニークな能力を有する新しいクラスの抗酸化物質であること、およびこれらの化合物が、個体の寿命を延ばす、並外れた広い強力な能力を有することを証明している。
【0100】
上記組成物および方法のさらなる特徴および利点、ならびに様々な実施態様の構造および操作は、添付の図面を参照しながら下で詳細に説明する。
【0101】
上の開示は、幾つかの好ましい実施態様を説明するものであり、いかなる点でも範囲を制限するものではない。熟練した技術者は、これらの方法および組成物の実施の際、本明細書には明白に開示されていない他の実施態様に気づくであろう。下に記載する実施例によって上の実施態様をさらに説明する。これらの実施例は、これらの実施態様を説明する意味を持つものであり、いかなる点でも範囲を制限するものとは解釈すべきでない。
【0102】
本明細書おいて言及するすべての参照および関連技術は、本技術の現状の一部であり、従って、これら全体が本明細書に援用されている。
【実施例1】
【0103】
C3カルボキシフラーレンの調製
材料
シリカゲル(Merck グレード9385、260−400、60A)は、Aldrich Chemials(ミズーリ州、セントルイス)から入手した。他の試薬は、Sigma Chemical Co.(ミズーリ州、セントルイス)および他の標準的な供給業者から購入した。
【0104】
マロン酸C60のC3位置異性体(e,e,eC60[C(COOH)2]3)は、一晩攪拌することによりC6(720mg、1.00mmol)をトルエンに1mg/mLの濃度で溶解することによって合成した。ブロモマロン酸ジメチル(632.4mg、2.69mmol)を添加し、続いて1,8−ジアザビシクロ(5.4.0)ウンデク−7−エン(DBU、493mg、3.24mmol)を添加した。この反応混合物を2時間攪拌し、シリカゲルのパッドに通して濾過し、真空下で濃縮した。残留物を、トルエン中で開始する、シリカゲルの450mLカラム(Merck、280から400メッシュ)でのクロマトグラフィーに付した。漸増量の酢酸エチル(EtOAc)をトルエンに添加することにより、着色成分を分離した。C3画分は、トルエン中5%のEtOAcで溶出した。C60マロン酸エステル画分の純度をTLCおよびHPLCでモニターした。このC3エステル(0.25g、0.23mmol)をトルエン(250mL)に溶解し、窒素でスパージした。ナトリウムメトキシド(2.2Mのものを2.22mL、4.88mmol)の添加により、数分以内に沈殿が生じた。この混合物を窒素下、室温で1時間攪拌した。水(20mL)を添加し、この混合物を一晩攪拌した。すべての着色生成物が、水性層に行った。層を分離し、水性層を冷却して、20%硫酸(1.32mL)で酸性化した。この溶液をEtOAcで3回抽出した結果、すべての色が有機層に行った。この有機層を水で数回洗浄して、黄色の不純物を抽出した。次いで、EtOAc層を蒸発させ、残留物223.2mg(理論量の89%)を凍結乾燥させた。
【実施例2】
【0105】
マウスでの寿命試験についての実験法
月齢12ヶ月の雄および雌(同数)C57B6NIHマウスを米国国立老化研究所(NIA)齧歯動物老化コロニーから購入した。このコロニーから出荷されたマウスを健康、腫瘍または他の能力障害で一切選別せず、このコロニーから入手したすべてのマウスを、続いて、この研究に登録した。マウスは、同性のものを入れるケージに1ケージあたり2匹で無作為に配置し、識別のために耳にパンチし、計量した。次に、マウスを週2回の3セッションで、回転棒での訓練を行い、次いで、ベースラインの運動能力を測定するために3セッションで回転棒での試験を行った。次に、治療薬Aまたは治療薬Bのいずれかを受けるように、これらの治療が何であるのか知らない観察者が、ケージを割り当てた。
【0106】
治療薬Aは、水中のC3(28.75μM)の溶液であり、治療薬Bは、赤色のC3溶液に合うように添加した市販の食用色素であった。溶液が何かを知らない者が、溶液Aまたは溶液Bを採水瓶に入れ、週に2回、瓶の口のところまで溶液を補充し、週に2回、濾過して一切の微粒子を除去した。週齢19ヶ月でマウスを再び計量し、別のラウンドの回転棒訓練および試験を受けさせた。マウスを自然死させ、動物収容施設のスタッフがこの施設の一般操作手順の一部として死亡日を記録した。施設のスタッフは、動物には抗生物質溶液を用いたと信じており、この研究の目的を知らなかった。動物が死亡したとき、このゲージ番号、動物の由来、および死亡日を死亡通知に記録し、次いで、これを研究所に送り、そこで情報をデータベースに入力した。
【0107】
これらの実験の結果を図4に表示する。これらは、マウスの寿命の顕著な増加(約20%)を示している。加えて、薬物の経口投薬により寿命が増加したので、これは、後生動物の寿命増加を達成する初めての実用的方法である。C3治療マウスの寿命増加は、体重減少を随伴しなかった。
【実施例3】
【0108】
ラットを用いる毒性研究
二系統のラット(Sprague−DawleyおよびLong−Evans)を用いてC3のラット毒性試験も行い、前記ラットに30日間、10mg/kg以下を与えたが、毒性(すなわち、生存率低下、毛づくろいの欠陥、または摂食障害)は一切示さなかった。
【0109】
(実施例4から13)
幾つかの新規バックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを下記で同定する。C3 Hexaの調製からの主成分は、ほぼ等しい存在比での2つの異性ペンタカルボン酸(Penta Pair)を含み、マイナーな生成物は、4つの異性テトラ酸(Tetra Quarted)および4つの異性トリ−脱カルボキシル化生成物(C3−ライト)を含む。これらの生成物は、C3より水溶性が劣り(親油性が高く)、このことが、組織におけるこれら新規化合物の吸収および保持を増大させ得る。
【0110】
以下の方法は、94+%のヘキサカルボン酸濃度をもたらし、他の新規成分への直接的経路となる。Penta Pair、Tetra QuartetおよびC3−ライトの含有率が高い混合物が、脱カルボキシル化により得られた。Penta PairおよびTetra Quartetは、t−ブチル保護を用いる別戦略によっても得られた。
【0111】
誘導体を分析するためのHPLC法
以下のすべてのHPLC法に、四液ポンプ(Quarternary pump)およびダイオードアレイ検出器を具備するHewlett Pakard/Agilent 1100シリーズ HPLCを使用した。分離は、室温で維持した、Zorbax SB−C18 4.6X250mmカラム(パッキング 5μm)(カラムA)またはZorbax SB−C8 4.6X250mmカラム(パッキング 5μm)(カラムB)を用いて行った。すべての方法に、1mL/分の溶媒流量を用いた。
【0112】
プロトコル1 − HexaおよびPenta化合物の分析
溶媒は、水中0.1%のTFA(溶媒A)、およびアセトニトリル95%と水5%中0.1%のTFA(溶媒B)であった。サンプルは、15分間かけてA:B;40:60からA:B;10:90への勾配を用い、さらに15分、A:B;10:90でカラムから溶離した。化合物は、インラインダイオードアレイ検出器を使用してこれらのUV−vis 吸収によりモニターし、同定した。
【0113】
プロトコル2 − HexaおよびPenta化合物を分離するための別法
HPLC溶媒は、水中0.1%のTFA(溶媒A)、および2−プロパノール95%と水5%中0.1%のTFA(溶媒B)であった。化合物は、10分かけてA:B;95:5からA:B;52:48への勾配、10分かけてA:B;51:49にさらに進め、次いで、5分かけてA:B;21:79にさらに進める勾配を用いて、(カラムAまたはB)から溶離した。次に、これをさらに2分間、イソクラティックにしておいた。
【0114】
プロトコル3 − Tetra Quartetを分離するための方法
Tetra Quartetの調製物は、水中0.1%のTFA(溶媒A)、およびアセトニトリル95%と水5%中0.1%のTFA(溶媒B)を使用して、(カラムAまたはB)から溶離した。この溶媒組成を35分間、A:B;30:70で維持し、次いで、2分かけてA:B;5:95への勾配、続いて、さらに10分間、A:B;5:95で維持した。
【0115】
プロトコル4 − C3−ライトの分析
C3−ライトの調製物は、(カラムAまたはB)のいずれかでプロトコル1について説明したとおり溶媒を使用して分析した。勾配は、A:B;95:5で開始、5分間維持し、続いて、30分かけてA:B;5:95に勾配を変化させるように修正した。この溶媒組成をさらに35分間、A:B;5:95のままにしておいた。
【0116】
プロトコル5 − t−ブチル、メチルエステルの分析
溶媒は、アセトニトリル50%とジクロロメタン50%(溶媒A)であった。サンプルは、5分かけて100%(溶媒A)のイソクラティックプログラムで(カラムA)から溶離した。
【0117】
プロトコル6 − 部分メチルエステルの分析
HPLC溶媒は、アセトニトリル95%と水5%中0.1%のTFA(溶媒A)であった。サンプルは、120分かけてイソクラティック100%(溶媒A)溶液で(カラムAまたはB)から溶離した。
【実施例4】
【0118】
Hexa異性体とPenta異性体の混合物を合成すためのMaOMe/MeOH法
ナトリウムメトキシド(2.2Mのもの1.84mL、4.05mmol)をN2下、224mLのトルエン中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)(224.1mg、0.202mmol)の溶液に添加した。赤橙色の固体の沈殿が、直ぐに始まった。1時間後、水を添加し、すべての色が水性層に行った。層を分離して、水性層を冷却し、硫酸(3.7Mのもの1.10mL、4.07mmol)で酸性化し、酢酸エチル(2x40mLおよび1x10mL)で抽出した。併せた酢酸エチル抽出物を3x40mLの水で洗浄し、これによって黄色の不純物を除去した。真空下、室温での蒸発および乾燥によって、199.2mg(Hexaカルボン酸に基づく理論値の96.1%)が生じた。細胞スクリーニング用の溶液は、この固体を0.1Nの水酸化ナトリウムに溶解して重量で約25mMの溶液を得ることによって調製した。次に、この有効濃度を、トルエン中のこのエステル前駆体を基に決定した吸光係数4400を用い、UVによって決定した。UV−vis λmax(nm)は488であり、max/min比(488/414nm)は、2.19であった。HPLCプロトコル1を用いて、この合成により65.1%のHexa異性体、14.3%のPenta−1および20.6%のPenta−2が生成されたと判定した。これら三成分をHPLCにより分離し、質量分析(表3)および1H NMR(表4)によって同定した。
【実施例5】
【0119】
95% Hexaを生成させるためのトルエン、6% MeOH、および等量の水とNaOH
500mLのトルエン中の水(0.216mL、12mmol)、メタノール(17.1mL)およびC3メチルエステルの溶液を1時間攪拌した。この溶液は、最初は濁っているように見えた。塩基を直ちに添加すると、Hexaの収率は低下する。メタノール中の水酸化ナトリウム(0.93Mのもの12.9mL、12.0mmol)を添加した。1.5時間後、TLCおよび溶液の色によって評価すると、溶液中にエステルは残存しなかった。水100mLを添加した。トルエン層を分離し、水性層を濃縮してメタノールおよびトルエンを除去し、次いで、N2下、60°2時間加熱して、加水分解を完了させた。抽出による処理によって、490.8mg(理論収率の79.5%)を得た。UV−vis λmaxは、487nmであり、max/min比(487/411nm)は、3.10であった。HPLCプロトコル1を用いて、異性体分配は、95.2% Hexa、1.7% Penta−1、および3.0% Penta−2であると判定した。
【実施例6】
【0120】
トルエン中5%の酢酸t−ブチルを添加した後に混合した、等量のNaOHと水および2%のMeOHを使用して、88% HexaおよびPenta異性体を生じさせる方法
水酸化ナトリウム(3.679g、0.092mol)を92mLのメタノールおよび1.655mL(0.092mol)の水に溶解して、0.982M溶液を得た。この溶液の一部(12.3mL、12.0mmol)を、N2下、室温で、5%の酢酸t−ブチルを含有する585mLのトルエン(1.0mg/mLと比較して1.14mg/mL)に溶解したC3メチルエステル(670mg、0.604mmol)に添加した。沈殿がゆっくりと形成した。1時間後、この反応混合物に水を添加した。相を分離し、水性層を濃縮してメタノールおよびトルエンを除去し、次いで、60°で2時間加熱した。生成物573.3mg(理論値の92.5%)を、上に記載した酢酸エチル抽出によって水性層から除去した。UV−vis λmaxは、489nmであり、max/min比(489/411nm)は、3.37であった。HPLCプロトコル1を用いて、異性体分配は、88.4% Hexa、5.6% Penta−1、および6.0% Penta−2であると判定した。
【実施例7】
【0121】
酢酸t−ブチルと5個の水分子を使用して96% Hexaを生じさせる方法
950mLの5% 酢酸t−ブチル中に水(1.52mL、84.4mmol)、メタノール(40mL)およびC3メチルエステル(UVにより0.845mmol)を含有する溶液を、N2下で30分間攪拌した。メタノール中の水酸化ナトリウム(1Nのもの16.9mL、16.9mmol)を添加した。2時間後、すべての色が沈殿した。水50mLを添加し、トルエンをデカントして、残留トルエンを真空下でストリップした。水性層をN2下、60°で2時間加熱した。抽出による処理によって、798.1mg(理論値の92.1%)を得た。UV−vis λmaxは、489nmであり、max/min比(489/412nm)は、2.71であった。HPLCプロトコル1を用いて、異性体分配は、96.7% Hexa、1.5%の各Pentaであると判定した。
【0122】
合計メタノール濃度は、6%である。塩基を直ぐに添加すると、またはエステルの添加前に塩基とすべてのメタノールを併せると、異性体分配は、より低いHexa含有率へとシフトする(実施例6参照)。
【実施例8】
【0123】
トルエンのみを使用する97% Hexaの別合成
この手順は、トルエン中5%の酢酸t−ブチルの代わりにトルエンを使用したことを除き、実施例7と同じであった。HPLCプロトコル1を用いて、この生成物は、97.2% Hexa、1.3% Penta−1および1.5% Penta−2の異性体分配を有すると判定した。UV−vis maxは、491nmであり、max/min比(491/412nm)は、2.97であり、収率は、90.5%であった。13C NMR(K2CO3,D2O,600MHz):δ173.84,173.76,152.55,151.33,149.49,149.41,149.05,148.98,148.92,148.78,148.61,148.13,147.96,146.55,146.46,145.89,144.81,143.82,143.07,142.99,80.69,79.88,69.55。
【実施例9】
【0124】
Penta Pair直接合成
段階1 − ブロモマロン酸ジメチル(281.3mg、1.20mmol)を、200mLのトルエン中のジ−t−ブチルマロニルC60(0.592mmol)に添加し、続いて、DBU(202.4mg、1.33mmol)を添加した(Bingelの‘376、カラム4、44から45行目)。30分後、この反応混合物を、トルエン中のシリカゲルの410mL(4x310cm)カラムに注入した。このカラムを、すべてのビス異性体(UV−visにより25%)が出てくるまで、トルエンで溶離した。溶媒を酢酸エチル/トルエン混合物(0.5%対2%)に換えた。以下の成分の含有量が多いエステル調合物が溶出した(HPLCプロトコル5を使用):D3(トランス−3,トランス−3,トランス−3 トリスジメチルマロニルC60);e,トランス−3,トランス−2 トリスジメチルマロニルC60;e,トランス−4,トランス−3 トリスジメチルマロニルC60;およびC3。C3エステル画分(HPLCプロトコル1)を真空下で蒸発させた。質量分析および1H NMRデータは、表3および4にある。
【0125】
段階2 − p−トルエンスルホン酸・一水和物(22.5mg、0.118mmol)をトルエン中のC3画分の溶液(15mL)に添加し、88から89°の油浴に入れた。45分後、追加のp−トルエンスルホン酸・一水和物(15.6mg、0.082mmol)を添加した。生成物は、10分以内に沈殿し始めた。90分間、加熱し続けた。沈殿からトルエンを除去し、これらの固体に水および酢酸エチルを添加した。この酢酸エチルを水で3回洗浄してp−トルエンスルホン酸を除去した。酢酸エチルを真空下で蒸発させることにより、118.2mgの固体が残った。HPLCプロトコル6を用いて化合物を分析した。
【0126】
段階3 − これらの固体を、10mLの1:1:2 アセトニトリル:水:アセトンに溶解した。サンプル0.2mLを分析用にとった。残りは、76.5°の油浴で加熱した。30分後、沈殿が始まって、分離した。アセトン(10mL)を添加し、固体を溶解した。加熱を継続し、1.75時間後、脱カルボキシル化は73%完了した。合計7.5時間加熱した後、真空下で揮発性溶媒を除去し、酢酸エチルを添加して、均質溶液を得た。分析用に一部(1.9mg)をとった。残りは、真空下で蒸発させて、0.13gの固体を得た。化合物をHPLCプロトコル1によって分析した。
【0127】
段階4 − 上記固体(0.125mmol)を、メタノール(4.9mL)および水(0.128mL、7.1mmol)を含有するトルエン(120mL)に溶解し、1時間攪拌した後、メタノール中の水酸化ナトリウム(1.0Mのもの2.3mL、2.3mmol)を添加した。5分以内に固体が沈殿し始めた。1時間後、水50mLを添加した。トルエンを分離し、水性層をメタノールおよびトルエンでストリップして、60°で2時間加熱した。冷却した溶液に硫酸(3.7Mのもの0.621mL、2.3mmol)を添加し、上に記載したように酢酸エチルを用いて生成物を抽出した。この乾燥重量は、91.1mgであった。U−vis λmax(nm)。HPLCプロトコル1を用いて化合物を分析した。質量分析および1H NMRデータは、表3および4にある。13C NMR(K2CO3,D2O,600MHz):δ175.39,175.36,173.94,173.88,173.82,173.80,173.74,173.68,173.65,153.39,152.46,152.32,152.23,152.15,151.37,151.22,149.66,149.46,149.38,149.24,149.14,149.04,148.97,148.86,148.55,148.45,148.42,148.36,148.35,148.13,147.96,147.78,147.04,146.96,146.87,146.70,146.66,146.60,146.42,146.38,146.03,145.94,145.70,145.55,145.22,144.86,144.79,144.67,144.58,144.00,143.92,143.88,143.81,143.76,143.62,143.07,143.02,142.98,142.93,142.11,142.05,80.72,80.69,80.58,79.97,79.75,79.67,77.21,77.12,76.38,76.30,69.52,69.44,69.38,69.72。
【実施例10】
【0128】
Tetra Quartetの合成
段階1 − ブロモマロン酸ジメチル(38mg、0.16mmol)を、30mLのトルエン中のC60のマロン酸ビス−ジ−t−ブチル(UVにより0.167mmol)のサンプルに添加した。DBU(28mg、0.18mmol)を添加し、この反応をTLC(シリカゲルを用いて、トルエン中2%のEtAc)によって追跡した。1時間後に反応が完了した。この反応混合物を、トルエン中のシリカゲルの330x2.5cmカラムに注入した。ビス画分(12%、4成分、HPLCプロトコル5)をトルエン中で回収し、次いで、溶離液をトルエン中0.5%の酢酸エチルに換えた。D3、e,トランス−3,トランス−2、e,トランス−4,トランス−3およびC3のエステルを含む画分を得た。C3画分は、HPLCプロトコル1によると純度88.5%であり、さらに精製せずに使用した。UV−vis(トルエン中0.5%のEtAc)λmaxは、486nmである、max/min比は、3.57であった。
【0129】
段階2 − p−トルエンスルホン酸・水和物(4.9mg、0.0258mmol)を4.5mLのトルエン中のC3エステル(0.0154mmol)に添加し、この混合物を77°の油浴に入れた。この温度を15分かけて85°に上昇させ、さらなるp−トルエンスルホン酸(5.9mg、0.0310mmol)を添加した。5分以内に沈殿が開始した。30分間加熱し続けた。この混合物を冷却し、トルエンをデカントした。水および酢酸エチルの添加によって、すべての色が酢酸エチル層にある二相混合物が生成した。この酢酸エチルを水で3回洗浄し、蒸発させて、18.3mgの固体を得た。HPLCプロトコル1を用いて化合物も分析した。
【0130】
段階3 − この固体を5mLの4:1 アセトニトリル:水に溶解し、0.546mLを分析用にとった。残りを74°の油浴で加熱した。65分後、固体が存在した。3mLのアセトニトリルの添加により、均質溶液を得、5.5時間加熱し続けた。この溶液の一部0.8mLを分析用にとり、残り7.0mLは濃縮して、アセトンおよびアセトニトリルを除去した。HPLC(95:5:0.1,ACN:H2O:TFA,min,%)19.341(26.2),38.180(25.1,43.798(22.2)および80.874(21.6)。HPLCプロトコル1を用いて化合物も分析した。
【0131】
段階4 − 水酸化ナトリウム(0.1Nのもの0.59mL、0.059mmol)を、約3mLの水中の上で得られた物質の懸濁液に添加した。固体が溶解した。水酸化ナトリウム(1Nのもの0.6mL、0.6mmol)を添加し、溶液を62℃で1時間加熱した。HPLCは、加水分解の完了を示した。この溶液を冷却し、硫酸(3.7Mのもの0.162mL)で酸性化した。酢酸エチルでの抽出により、11.4mgの固体が生じた。HPLC(95:5:0.1,ACN:H2O:TFA,min,%)6.076(26.4),10.784(24.5),13.883(26.0),23.589(23.1)。HPLCプロトコル1を用いて化合物を分析した。質量分析および1H NMRデータは、表3および4にある。
【実施例11】
【0132】
溶液中でのC3−ライトの直接合成
C3のサンプル(329.7mg、0.32mmol、71 %Hexa、15% Penta 1および14% Penta 2)を6mLの1:1 アセトニトリル:水に溶解し、70℃の油浴に入れた。この反応の進行をHPLC(プロトコル2)によってモニターした。2.25時間後、反応は、ほぼ完了した。5時間後、固体が沈殿し始めた。24時間加熱し続けた。真空下で溶媒を除去して、199.3mgの生成物を得、これは、HPLCプロトコル1によると以下の組成を有した。C3−ライト ピーク1(14.0%)、ピーク2(39.4%)、ピーク3(37.0%)、ピーク4(8.7%)。LC−MS pos m/eは、各々895であり、neg m/eは、各々1007であった(M+TFA−H)。すべてが、C3(C60へのe,e,e付加)のUV−visスペクトルを有した。1H NMRおよび質量分析データは、表3および4にある。この固体は、水酸化ナトリウムに可溶性であり、アセトンおよび酢酸エチルには、中等度に可溶性であり、水、メタノールおよびアセトニトリルへの溶解性は乏しかった。
【実施例12】
【0133】
Penta、TetraまたはC3−ライトを生じさせるための溶液中でのC3の熱分解
97.4% Hexa、1.2% Penta 1および1.3% Penta 2を含有するC3のサンプル(74.8mg)を10mLの1:1 アセトニトリル:水に溶解し、60℃で加熱した。この組成を、プロトコル2および4を使用してHPLCによりモニターした(表1参照)。
【0134】
【表1】
【実施例13】
【0135】
C3からC3−ライトへのニート脱カルボキシル化
93.8% Hexa、3.5% Penta 1および2.7% Penta 2を含有するC3のサンプルを乾燥粉末として、150℃、30mmHgの真空オーブンに入れた。この組成をHPLCプロトコル2によってモニターした。表2は、C3−ライトを含むC3の脱カルボキシル化生成物の時間依存性生成を示すものである(表2参照)。
【0136】
【表2】
【0137】
【表3】
【0138】
【表4】
【0139】
【表5】
【0140】
【表6】
【0141】
【表7】
【0142】
上記にかんがみて、上の実施態様および実施例の幾つかの利点が達成され、成し遂げられたことがわかるだろう。他の当業者が様々な実施態様において本発明を最良に利用することができ、考えられる特定の使用に適するよう様々な変更を施すことができるように、実施態様を選択し、記載して、本発明の原理およびこの実際の適用を最良に説明した。
【0143】
本明細書に記載し、説明する構造および方法に、本発明の範囲を逸脱することなく、様々な変更を施すことができるので、上記明細書に含まれているまたは添付の図面に示されているすべての事柄は、限定的なものではなく、説明的なものとして解釈されよう。例えば、上に記載したような方法は、ヒトを含む(しかし、これに限定されない)他の後生動物に容易に同じ結果で適用することができた。従って、本発明の広さおよび範囲は、上に記載した具体例としての実施態様のいずれによっても、当然、制限されず、本明細書に続く添付の請求項およびこれらに相当するもののみに基づいて定義されるべきものである。
【図面の簡単な説明】
【0144】
【図1A】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1B】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1C】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1D】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図2】様々な有用なカルボキシフラーレンを示す図である。
【図3】C3トリスマロン酸位置異性体を示す図である。
【図4】対照溶液で治療したC57B6マウスに対する経口C3で治療したC57B6マウスの生存を表す図である。
【図5】バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト上の官能基の立体配置を表す図である。
【図6】C3−ライト(e,e,eトリス酢酸C60)およびC3(e,e,eトリスマロン酸C60)の構造を表す図である。
【図7】Penta−1の構造を表す図である。
【図8】Penta−2の構造を表す図である。
【図9】NMDA毒性に対するHexa、Penta−1、Penta−2およびC3−ライトによる神経保護を表す図である。
【図10】AMPA毒性に対するHexa、Penta−1、Penta−2およびC3−ライトによる神経保護を表す図である。
【図11】Hexa、Penta−1、Penta−2およびC3−ライトの血漿中動態、ならびにC3の組織分布の詳細図である。
【図12】C3−ライトの血漿中薬物動態および組織分布の詳細図である。
【技術分野】
【0001】
本発明は、新規C60誘導体、こうした誘導体を調製する方法、および治療方法に関する。さらに詳細には、(a)新規なバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトおよび同物質を調製する方法、(b)治療的有効量のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトでニューロン傷害を治療する組成物および方法、および(c)治療的有効量のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトで後生動物のまたは後生動物細胞における予想される生存期間(または「寿命」と呼ばれる)の長さまたは期間を延長するための組成物および方法を、ここに開示し、特許請求の範囲に記載する。
【背景技術】
【0002】
ヒトおよびペット動物の総合的健康および寿命を増進させる方法は、非常に活発な研究分野になっている。この分野の現在の見解は、カロリー制限が後生動物の生存期間を延ばすために役立つことを提唱している。
【0003】
後生動物全体にわたる細胞または発育方法の保存特性を仮定して、C.エレガンス(C.elegans)およびキイロショウジョウバエ(D.melanogaster)を含む多数のモデル生物が、寿命の研究に利用されてきた。
【0004】
例えば、C.エレガンスの遺伝子分析は、寿命決定に関与する幾つかの遺伝子を明らかにした。Daf−2(インスリン受容体)およびClk−1(「Clock 1」、発生および行動のタイミングに関する多数の側面に影響を与える遺伝子)における突然変異は成人の寿命を延ばすことが証明された。しかし、Clk−1突然変異体は、寿命初期での死滅率が高い。成長後期では、Clk−1突然変異体は寿命の増加を示し、これは、おそらく寿命初期での長寿個体の選別による。Clk−1寿命表現型は、スーパーオキシド/フリーラジカル代謝に関与する、カタラーゼをコードしている遺伝子の突然変異によって、完全に破壊される。加えて、C.エレガンス中で補酵素Qを排除すると、寿命が延びることが証明された。
【0005】
Eat遺伝子に突然変異を有するC.エレガンスも、寿命の増加を示したが、摂食量の低下および代謝の遅速も示した。この突然変異に随伴する寿命の増進は、カロリー制限に帰するものであり、後生動物においても寿命を増加させることが証明された。
【0006】
ショウジョウバエ属では、スーパーオキシドジスムターゼ(SOD)およびカタラーゼの過発現が、ショウジョウバエの寿命を35%増加させた。メトセラ遺伝子(「Mth」)の突然変異も、寿命を20%増加させることが証明された。Mth、G蛋白共役受容体、の機能は不明であるが、突然変異体は、パラコート(スーパーオキシドラジカル傷害誘発物質)毒性に対する耐性増加を示し、これがストレス応答遺伝子であることを示唆している。
【0007】
カロリー制限(CR)は、今日までに研究されたすべての動物(マウス、ラット、数種のサル、イヌ、ヒト、ならびに非後生動物種、例えばクモ、線虫およびショウジョウバエ)において寿命を25から35%増加させることが証明された。(注意:すべての動物は、後生動物である)。しかし、寿命への強い効果を達成するためには、カロリー摂取を30%から40%ほども減少させる必要がある。米国国立老化研究所(National Institute of Aging(NIA))におけるアカゲザルおよびセアカリスザルでの進行中の研究(Rothら,Eur.J.Clin.Nutr.S:15,2000)では、齧歯動物において報告された変化と同様の生化学的変化が、カロリー制限したサルに見出され、このため脊椎動物種全体にわたる生化学的方法に対するカロリー制限の普遍性を支持している。
【0008】
最近、2−デオキシグルコースを使用して、経口摂取量を制限することなくカロリー制限をもたらした。2−デオキシグルコースで治療した動物には、カロリー制限した動物において観察される変化と同様の体温低下および血漿中インスリンレベル低下があった(Rothら,Ann.NY Acad.Sci.,928:305,2001)。寿命に対する2−デオキシグルコースの効果に関する科学的研究は終わっていないが、サイエンス(2002年2月8日)における最近の論説は、これらの研究の主任研究員(Geoge Roth、NIA)が、「2−デオキシグルコースで治療した彼のサルのうちの一匹が、平均生存期間の25ヶ月ではなく、38ヶ月生きた」と述べたことを引用した。しかし、こうした主張は、サンプルサイズが小さいことから、科学的に指示されない。対照集団の中で最も長く生きたサルの年齢に関するコメントもなかった。
【0009】
マウスの予想される生存期間における20%までの増加が、遺伝子操作または成長因子拮抗物質投与のいずれかによる成長因子剥奪によって示された。残念なことに、小人症が、成長因子剥奪の副作用である。ヒトにおいて、小人症または晩年期成長ホルモン欠損症は、寿命を減少させるようであり、このことが、成長因子剥奪が予想される生存の期間を増加させる1つの手段として有効であるか否かの論争をさらに混乱させている。
【0010】
デプレニール(パーキンソン病の治療に使用される選択的モノアミンオキシダーゼ(MAO)B阻害剤)が多くの種の寿命を増加させることを、幾つかの学術論文が示している(例えば、Knoll,Mech Ageing Dev.46:237,1988参照)。ある研究では、週齢96週から死ぬまでラットのデプレニールでの長期治療が、「生存を増進した」。対照ラットは、147+/−1週生き、これに対してデプレニールで治療したラットは、198+/−2週生きた。しかし、この学術論文においてはっきりと述べられているこれらのラットの平均予想寿命は182週であった。このため、この研究における対照群は、早期死亡率を有したようである。これらの研究所からの他の研究では、高能力のラットを選択し、次いで、これらがデプレニール寿命試験に登録された。このために人為的に結果が歪められた可能性がある。
【0011】
第二の研究は、344匹のFisherラット(Kitaniら,Life Sci 52:281,1993)を使用して、18ヶ月齢でデプレニール治療を開始した。対照の平均生存は、28ヶ月であり、治療動物の平均生存は、30ヶ月であった。このことは、7%の寿命の増加を示している。しかし、これらの結果は、統計学的に有意ではないと証明された。
【0012】
対照的に、同じ用量のデプレニールを用いた344匹のFisherラットでの別の研究(Carilloら,Life Sci 67:2539,2000)では、デプレニール治療動物において、より高い死亡率および短縮された寿命が観察された。さらに、NIAからの研究では、18ヶ月齢で長期デプレニール治療を開始したC57B6マウスにおいて何の生存利益も示されなかった(Ingramら,Neurobiol Aging 14:431,1993)。同様に、ショウジョウバエ属におけるデプレニールの対照試験は、寿命の増加を示さなかった(Jordenら,Neurochem Res 24:227,1999)。
【0013】
同様に、ヒトでのデプレニール試験は、寿命に関して相反する結果を示した。パーキンソン病患者における「公開、非対照」試験は、9年で生存増加を示した(Birkmayerら,J.Neural Transm.64:114,1985)が、他の研究は、デプレニールを特にL−ドパと共に摂取しているPD患者の死亡率増加を示唆している(例えば、Ben−Shlomoら,BMJ 316:1191,1998)。
【0014】
全体的に見て、データは、デプレニールが寿命に対して弱い効果を及ぼすことがあり、または及ぼさないこともあることを示唆している。
【0015】
マウスの幾つかの遺伝子は、「寿命」遺伝子として特定されており、これは、これらの遺伝子に突然変異を有するマウスが、対照マウスの予想される生存期間に比して長い平均寿命を有するからである。これらの遺伝子は、エイムズ矮性突然変異およびスネル矮性突然変異を含む。しかし、これらの突然変異によって、飼育が難しい小さく虚弱なマウスが生じることとなる。これらの突然変異によりもたらされる寿命は、本質的にカロリー制限に起因すると考えられる。遺伝子アレイ分析、または蠕虫、ハエおよび齧歯動物における寿命表現型に関連した遺伝子についての他の遺伝子スクリーニングを利用する最近の試みによって、多数の候補遺伝子が見つけ出された。しかし、一般に、これらは多くの場合、「ストレス応答」遺伝子である。
【0016】
イチョウ(Gingko)、ニンジン(Ginseng)、ビタミンCなどの多数の化合物が、生存を改善するために提案されてきたが、これらの化合物に関する利点を報告する管理された、統計学的に有意な生存研究は、知られていない。ビタミンCおよび多数の薬物は、一定の疾病状態、例えば心血管疾患の発生率を低下させ、このためおそらく総合的には寿命を増進するであろう。
【0017】
バックミンスターフラーレン、C60、は、芳香族系溶媒には可溶性であるが、水には可溶性ではない、12の五角形および20の六角形を有する炭素球である。
【0018】
C60(C(COOH)2)n(式中、nは、1から4の整数である)の使用は、ニューロン傷害の治療に関し、2001年7月24日に発行されたChoiらの米国特許第6,265,443号に開示されている。前記特許は、この全文が本明細書に参考として援用されている。
【0019】
この文献において報告されているC3ヘキサカルボン酸(「C3」または「Hexa」)の調製では、細胞培養スクリーニングでの再生能および変異機能が劣る、一部未確認のプロドラッグの混合物が生成される。
【発明の開示】
【0020】
上記にかんがみて、下で説明し、特許請求の範囲に記載する組成物および方法を開発した。
【0021】
第一の実施態様は、後生動物の寿命を増加する結果となる後生動物への組成物の投与を含み、前記組成物は、ペンダント炭素に結合しているx対の隣接した炭素原子を有するC60化合物などのC60フラーレンのカルボキシル化誘導体(「カルボキシフラーレン」)を含み、ここで、前記ペンダント炭素原子は、一般式−COOHおよび−R(Rは、−COOHおよびHから成る群より独立して選択される)の2つの基にさらに結合しており、ならびにxは、少なくとも1である。
【0022】
有用な化合物の別の実施態様は、一般式C60[(CHCOOH)]x[C(COOH)2]y(式中、xは、0から3の整数であり、yは、1から4の整数であり、x+yは、2から4の整数である)と記述され得る。
【0023】
追加の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトである。さらに詳細な実施態様は、Penta Pair、Tetra QuartetおよびC3−ライト バックミンスターフラーレンのマロン酸/酢酸三重アダクト(後でより詳細に説明する)を含む。
【0024】
追加の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは−Hまたは−COOMeである)のバックミンスターフラーレンの三重アダクトである。
【0025】
さらなる実施態様は、Penta Pair、Tetra QuartetおよびC3−ライト バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを含む、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを調製する方法を含む。
【0026】
さらに追加の実施態様は、ニューロン傷害を治療および寿命を延長するための(C3に類似した)新規バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトの使用を含む。
【0027】
バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレンの使用は、カロリー制限の中での固有の難しさ(一般に、摂食量の厳格な制限ならびにヒトでの使用の実行不可能性が挙げられるが、これらに限定されない)からして、後生動物、特にヒトの寿命を実質的に増加させる方法として、カロリー制限を越える実質的な改善をもたらすと考えられる。新規バックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトは、C3トリスマロン酸C60化合物に類似した望ましい特性を有するが、動物体内での半減期がより長く、その結果、インビボでの有効期間が延長されることも明らかにする。加えて、Penta−1、Penta−2、Tetra QuartetおよびC3−ライトを含むバックミンスターフラーレンのe,e,eマロン酸/酢酸三重アダクトは、C3より親油性が高いため、脳などの高脂質組織内に集中することができる。
【0028】
明細書に組み込まれており、この一部を形成している添付の図面は、本発明の様々な実施態様を説明するものであり、この記述とともに本発明の原理の説明に役立つ。
【0029】
添付の図面を参照して示すと(同じ参照番号が同じ要素を示す):
図1aは、HPLCによるC3調製物の分析を開示するものであり、(全体の99%を上回る)3つの主成分が同定されている。
【0030】
図1bは、3つのピークすべてが、C60核へのe,e,e(C3)付加に特有の吸収スペクトルを有することを示すものであり、C3のこれら成分ピークが、C60上のシクロプロパン炭素に結合している先端基が異なるe,e,e位置異性体を表すことを示している。
【0031】
図1c(1)から1c(3)は、HPLCによってHexaカルボン酸C3(1、80%)と二つの異性体Pentaカルボン酸(2および3、各10%)に分離し、次いで、質量分析によって判定した化合物1〜3を示す。
【0032】
図1d(1)から1d(3)は、前記Hexa異性体(1)および各Pentaカルボン酸(2)、(3)に対して行った質量分析を示す。
【0033】
図2は、2つのビス異性体、2つのトリス異性体、および1つのテトラ異性体を含む様々なカルボキシフラーレンを表す。
【0034】
図3は、C3対称性を有するe,e,eトリスマロン酸位置異性体(「C3」)を空間が詰まった構造と化学構造の両方として示すものである。
【0035】
図4は、月齢12ヶ月から死ぬまで飲み水に入れた食用着色剤(対照)またはC3(0.5mg/kg/日)のいずれかで治療したC57B6マウスの寿命を示すKaplan−Meier生存曲線である。各マウスについての自然死の日付を記録し、これを使用して寿命を計算した。NIA齧歯動物コロニーから受け取ったマウスには、誕生月の記録はあったが、具体的な誕生日の記録がなかったため、各マウスの寿命は月ごとに計算した。平均生存率を治療ごとに計算し、t検定を用い、有意性をp<0.05(実際にはp=0.033)に設定して平均寿命を比較した。第一群からのデータは、性別(A、B)および混成(C)でグラフにし、各治療グループを同性の対照と比較した。治療および未治療マウスの体重(g)は、(性による)差がなかった。例えば、19ヶ月の体重は、雌については、対照:27±1、C3治療:29±1であり、雄については、対照:35±4、C3治療:35±6(平均±SD)であった。
【0036】
図5は、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト上の官能基の立体配置の詳細図である。
【0037】
図6は、C3−ライト(e,e,eトリス酢酸C60)およびC3の構造を表す。C3−ライトは、3つのシクロプロパン炭素各々に結合しているカルボン酸の対のうちの1つがプロトンで置換されている点で、C3とは異なる。
【0038】
図7は、Penta−1化合物の構造を表す。HOMO−LUMOエネルギー分布(上部)、および球と棒によるモデル(下部)。Penta−1は、C3のマロン酸基のうちの1つが、酢酸によって置換されている。Penta−1では、このプロトンが、二つのマロン酸基の方に向いている。
【0039】
図8は、Penta−2化合物の構造を表す。HOMO−LUMOエネルギー分布(上部)、および球と棒によるモデル(下部)。Penta−2は、C3のマロン酸基のうちの1つが、酢酸によって置換されている。Penta−1では、このプロトンが、二つのマロン酸基とは反対の方に向いている。
【0040】
図9は、大脳皮質細胞培養物におけるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトHexa、Penta−1、Penta−2およびC3−ライトによる、NMDA受容体媒介興奮毒性に対する神経保護を表す。C3誘導体(0.05から100μM)を含有する、または含有しない200μMのNMDAに培養物を10分間暴露した。10分後、すべての薬物を洗浄除去し、細胞を24時間、細胞培養器に戻した。次に、ニューロンを乾燥させることにより乳酸デヒドロゲナーゼ(LDH)の放出を測定することによって、神経細胞死を評価した。細胞死および保護は、死滅したニューロンのヨウ化プロピジウム染色を画像化することにより、および位相差顕微鏡を使用してニューロンの形態を評価することにより、確認した。各化合物についての用量応答曲線を示す。値は、NMDAのみ(C60誘導体を含有しないもの)に暴露した培養物において観察された細胞死%を表す。
【0041】
図10は、大脳皮質細胞培養物におけるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトHexa、Penta−1、Penta−2およびC3−ライトによる、AMPA受容体媒介興奮毒性に対する神経保護を表す。C3誘導体(0.5から100μM)を含有する、または含有しない6μMのAMPAに培養物を24時間暴露した。ニューロンを乾燥させることにより乳酸デヒドロゲナーゼ(LDH)の放出を測定することによって、24時間での神経細胞死を評価した。細胞死および保護は、死滅したニューロンのヨウ化プロピジウム染色を画像化することにより、および位相差顕微鏡を使用してニューロンの形態を評価することにより、確認した。各化合物についての用量応答曲線を示す。値は、AMPAのみ(C60誘導体を含有しないもの)に暴露した培養物において観察された細胞死%を表す。
【0042】
図11は、Hexa、Penta−1およびPenta−2の血漿中薬物動態(AからB)およびHexaC3の組織分布(C)を表す。図11(A)は、80% Hexa、10% Penta−1、10% Penta−2を含有する14C−C3を注射したマウスからの血漿のHPLC分析である。サンプルは、t=0(注射前のC3サンプル)、t=2時間(C3の腹腔内注射から2時間後の血漿)、t=18時間(注射から18時間後の血漿)であった。図11(B)は、C3の静脈内、腹腔内、皮下および経口投与後の血漿中レベルを表す。Hexa、Petnta−1およびPenta−2についての血漿中T1/2は、すべて8.2時間であった。図11(C)は、腹腔内投与後、様々な時点でのC3の組織内レベルを表しており、肝臓および腎臓による蓄積を示している。
【0043】
図12は、C3−ライトの血漿中薬物動態および組織分布を表す。図(A)は、14C−C3−ライトの血漿中薬物動態を示すものであり、15時間のT1/2を実証している。図12(B)は、腹腔内投与後のC3−ライトの組織内分布を表す。
【発明を実施するための最良の形態】
【0044】
全体を通して、以下の用語を使用する:
「寿命」または「予想される生存期間」は、ある特定の環境において後生動物が、カルボキシフラーレンでの治療を受けなかった場合、生きることが予想される(生まれてから死ぬまでの)生涯(すなわち、「属に特有の」予想される生存期間)の予想平均長である。
【0045】
「マロン酸/酢酸C60誘導体」は、C60の酢酸誘導体、C60のマロン酸誘導体、およびC60のマロン酸/酢酸混合型誘導体である。
【0046】
「Hexa」およびC3は、C3(C60(C(COOH)2)3を表し、この場合、マロン酸基は、すべて、e,e,e位にある(化合物1、下記)。
【0047】
「Penta」または「Penta Pair」は、(C60(C(COOH)2)2(C(CHCOOH))を意味し、この場合、R基は、e,e,e位でシクロプロパン炭素に結合している。2つの立体異性体Penta−1およびPenta−2がある(化合物2、下記)。
【0048】
「Tetra」または「Tetra Quartet」は、(C60(C(COOH)2)(C(CHCOOH))2を意味し、この場合、酢酸/マロン酸基は、e,e,e位でシクロプロパン炭素に結合している。4つの立体異性体がある(化合物3、下記)。
【0049】
「C3−ライト」は、(C60(CHCOOH))3を意味し、この場合、酢酸基は、e,e,e位でシクロプロパン炭素に結合している。4つの立体異性体がある(化合物4、下記)。
【0050】
「バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト」または「マロン酸/酢酸C60誘導体」は、マロン酸基(>C(COOH)2)および酢酸基(>CHCOOH)から独立して選択される3つのペンダント基がe,e,e位にあるバックミンスターフラーレン、すなわち、
【0051】
【化1】
を意味する。
【0052】
寿命の予想長または期間を延長するための、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレン
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させる。スーパーオキシド(O2−●)および過酸化水素(H2O2)を含む反応性酸素種は、特定の生理機能に利用される一方で、細胞および組織の生存力および完全性に脅威を与え続けているという問題ももたらしている。これに応じて、細胞および生体は、自らをO2−●およびH2O2から防御する様々なメカニズムを発達させた。後生動物では、O2−●は、二つの金属酵素、Cu,Zn−スーパーオキシドジスムターゼ(SOD1)、およびMnSOD(SOD2)によって除去される。また、H2O2は、カタラーゼ(ヘム鉄含有金属酵素)またはグルタチオンペルオキシダーゼ(セレノシステインとグルタチオンを併用してH2O2をO2とH2Oに変換する蛋白質の一科)によって除去される。しかし、これらの内因性抗酸化物質防御系は、病的状態の下では圧倒されてしまうことがある。これは、細胞の抗酸化物質防御を補足する小分子としてのさらなる抗酸化物質(酸化を抑制する、または酸素もしくは過酸化物によって促進される反応を抑制する、有用な物質)を潜在的治療薬として開発する試みを導いた。
【0053】
多数の水溶性C60誘導体(スーパーオキシドジスムターゼ模擬体)は、これらの親フラーレン分子の抗酸化特性を保有しており、このため生体系においてこのフリーラジカルスカベンジ能力を利用することができ、その結果、細胞の損傷および死滅を減少させる因子としての機能を果たすことができる。
【0054】
C60誘導体、カルボキシフラーレンの1つの基は、H2O2およびO2−●の分解酵素としての機能を果たす。MnTMPypを含むマンガン含有プロトポルフィリン化合物は、O2−●/H2O2の分解酵素としての機能を果たすと報告されているが、これらの化合物は、マンガン原子の酸化−還元に依存して分解を触媒する。C3は、非金属化合物であるが、同様の触媒性状を有することを、今般、本発明者らは発見した。本化合物は、こうした方法で動作する最初の非金属化合物であると考えられる。
【0055】
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させるため、潜在的治療薬として細胞の抗酸化物質防御を補足することができる抗酸化性分子は、治療上有用である。これは、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトなどのカルボキシフラーレンを含む。これらの新規カルボキシフラーレン組成物は、抗酸化特性を有する。本研究を通じて、O2−●およびH2O2とC3の反応性を特性付けした。O2−●に対するC3のKiを計算して、3x106M−1秒−1になった。O2−●およびH2O2との相互作用後のC3の分析は、C60部分においても、マロン酸基においても永続的な化学的または構造的変化が発生しないことを示した。これは、C3が真の触媒であるという本特許請求事項を支持している。マンガン含有プロトポルフィリンおよびサレン化合物も、O2−●/H2O2の分解のための触媒としての機能を果たすと報告されているが、これらの化合物は、金属原子の酸化−還元に依存して分解を触媒し、これに対して本マロン酸フラーレン誘導体は、反応性酸素種の分解を触媒するために金属原子を必要としない。
【0056】
従って、上述の発見にかんがみて、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むカルボキシフラーレンは、反応性酸素種、特に、過酸化水素(H2O2)およびスーパーオキシド(O2−●)などの生理関連の反応性酸素種の排除に有用である。
【0057】
後生動物または後生動物の細胞の寿命を延ばす結果となる、抗酸化物質の治療的有効量を投与することにより後生動物の予想される生存期間を増加させる方法を、ここで説明し、特許請求の範囲に記載する。詳細には、抗酸化性カルボキシフラーレンを含む組成物を治療薬として使用して、後生動物または後生動物細胞の寿命を増加させる。
【0058】
従って、ここで有用な化合物は、このC60フラーレンのx対の隣接炭素原子が、少なくとも1つのペンダント炭素に結合しており、このペンダント炭素原子が、一般式−COOHおよび−Rの2つの基にさらに結合している、カルボキシフラーレン化合物、これらの対応する塩およびエステルであり、この場合、Rは、−COOHおよび−Hから成る群より独立して選択され、xは、少なくとも1である。この一般式の異性体の例を図1から3に示す。一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトも、ここでは有用である。
【0059】
従って、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを後生動物に投与することによる、哺乳動物およびさらに詳細にはヒトを含む後生動物または後生動物細胞の予想される生存期間を延ばす方法を提供する。
【0060】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含むすべてのカルボキシフラーレン化合物は、活性化合物および前記化合物と相溶性の医薬適合性担体を含有する組成物として全身投与することができる。こうした組成物の調製では、従来どおりの医薬適合性担体を利用することができる。この薬物を経口投与する際には、一般に、一定の間隔を開けて投与する。
【0061】
治療使用の際、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、薬物が従来投与されているあらゆる経路によって投与することができる。こうした経路には、静脈内経路、筋肉内経路、皮下経路、髄腔内経路、腹腔内経路、局所経路および経口経路が挙げられる。
【0062】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む医薬組成物は、経口投与のための固体形、例えば錠剤、カプセル、ピル、粉末、顆粒などを含む、あらゆる従来型の形態で製造することができる。これらの医薬組成物は、滅菌することができ、ならびに/または保存薬、安定剤、湿潤剤、乳化剤、浸透圧を変化させるための塩、および/もしくは緩衝剤などのアジュバントを含有することができる。
【0063】
静脈内投与のための局所製剤は、水/緩衝溶液を含む滅菌水溶液であろう。静脈内用ビヒクルには、液体、栄養および電解質補充液が挙げられる。保存薬および他の添加剤、例えば抗生物質および抗酸化物質が存在してもよい。
【0064】
ここで説明し、特許請求の範囲に記載するバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、医薬適合性の経口方式で有用である。これらの医薬組成物は、前記化合物と併せて相溶性の医薬適合性担体材料を含有する。あらゆる従来型担体材料を利用することができる。あらゆる従来型経口剤形、例えば錠剤、カプセル、ピル、粉末、顆粒などを使用することができる。担体材料は、経口投与に適する有機または無機不活性担体材料であり得る。適する担体には、水、ゼラチン、アラビアゴム、ラクトース、デンプン、ステアリン酸マグネシウム、タルク、植物油、ポリアルキレン−グリコール、ワセリンなどが挙げられる。さらに、本医薬組成物は、他の薬学的に活性な薬剤を含有することができる。さらなる添加剤、例えば着香剤、保存薬、安定剤、乳化剤、緩衝剤などを、薬剤の調剤の一般に是認されているやり方に従って、添加してもよい。
【0065】
経口剤形は、例えば錠剤、硬質もしくは軟質ゼラチン、メチルセルロースの、または消化管内で容易に溶解する別の適切な材料のカプセルを含む。考えられる経口投薬量は、処方する医師が判定するような個々の患者の必要性に従って変化するだろう。この経口剤形実施態様の一例は、50から500mgのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含有するカプセルまたは錠剤を含む。
【0066】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトでの治療方法は、一般には成人に毎日、好ましくは経口的、筋肉内的、皮下的または静脈内的に投与すことができる。筋肉内的、静脈内的または皮下的に投与する場合、治療薬は、約0.1mg/kgほどの少ない量から3mg/kgほどもの多い量で投与されることとなり、この正確な投薬量は、患者の必要性に依存して変化するであろう。経口投与の場合の日用量は、0.1mg/kgほどの少ない量から15mg/kgほどもの多い量になると予想される。一般に、この療法は、無期限に、予防的に行うことができる。
【0067】
本発明のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、常習的に(例えば毎日)投与してもよいし、または頻繁に(例えば一週間に一度)投与してもよい。C3 Hexa異性体、Penta Pair、Tetra QuartetおよびC3−ライトは、より有効な薬剤であると予想される。静脈内、筋肉内または皮下送達によって投与する場合、C3異性体の予想日用量は、約0.1mg/kgから約3mg/kgであろう。経口的に投与する場合、この日用量は、0.1mg/kgと15mg/kgの間の範囲であると予想されよう。
【0068】
上の投薬情報は、マウスにおいて行った薬物動態試験、マウスでの毒性試験およびラットでの毒性試験に基づくものである。マウスにおいて、C3、Penta−1およびPenta−2の血漿中半減期は、約8時間であると計算され、一方、C3−ライトの血漿中半減期は、約15時間であった。1回のカルボキシフラーレン注射についての50%致死量(LD50)は、70mg/kgより上であった。C3、Penta−1およびPenta−2は、肝臓および腎臓による排泄によってマウスから一掃され、一方、C3−ライトは、肝臓および脂肪によって一掃された。この薬物動態データに基づく計算を用いると、治療的血漿中レベルは、0.1μg/mLと1μg/mLの間であるようである。本化合物を静脈内、腹膜内または皮下投与によって与えると、同量のカルボキシフラーレンが吸収されるが、(例えば、飲料水の中に入れて)経口投与すると、この用量の約15分の1しか吸収されない。しかし、経口送達のためのカルボキシフラーレンの標準的な医薬調合物(例えば、カルボキシフラーレンを時間放出性錠剤に組み込んだもの)は、経口投与されるカルボキシフラーレンのバイオアベイラビリティを有意に増大させると予想される。加えて、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、C3より親油性が高く、このことが、これらを脳などの脂質豊富な組織に集中させる。
【0069】
本特許請求の範囲に記載するこれらの方法は、脊椎動物、さらに具体的には、ヒトおよびペット動物を含む哺乳類を含む、すべての後生動物に有用である。
【0070】
カルボキシフラーレンによって増加される寿命とは、後生動物が、カルボキシフラーレンで治療を受けなかった場合、生きることが予想される(生まれてから死ぬまでの)時間の予想平均長である。実施例2および図4の結果が示すように、この治療を受けたマウスは、23.5ヶ月の対照マウスの寿命より約20%長い寿命に相当する28.7ヶ月の実際寿命を有した。この実施例で使用した対照マウスの寿命は、一般「予想寿命」を表す。
【0071】
哺乳動物の生存期間を延ばすために有用な化合物の非限定的実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0072】
さらなる非限定的実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを少なくとも1つ含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばす方法である。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pari、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。前記組成物は、少なくとも1つのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体を含み、これらの成分は、前記組成物中に治療的有効量で存在すると考えられる。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、静脈内、筋肉内、皮下または経口投与することができる。この方法は、脊椎動物、哺乳類およびヒトを含む(しかし、これらに限定されない)すべての後生動物で使用することができる。
【0073】
バックミンスターフラーレンのマロン酸/酢酸三重アダクト
C60e,e,eマロン酸/酢酸三重アダクトは、増大した水溶性を含むさらなる望ましい特質を示す。1)C60の酢酸誘導体および2)バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、幾つかの新規C60e,e,eマロン酸/酢酸三重アダクトを合成し、特性付けした。「Penta−1」(図7)および「Penta−2」(図8)と呼ばれる2つの化合物(2)は、C60に付加している2つのマロン酸基と1つの酢酸基を有するC60のe,e,e誘導体である。Penta−1およびPenta−2は、酢酸基のシクロプロパン炭素に結合しているプロトンが、マロン酸基の方に向いているか、マロン酸基とは反対の方に向いているかという点で異なる。第二組の4化合物「Tetra−Quartet」(3)は、e,e,e位に1つのマロン酸基と2つの酢酸基を有する。第三組の化合物は、C60のe,e,eトリス酢酸誘導体(C3−ライト)(4)(図6)である。これらのバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトには、Penta−1、Penta−2、4つのTetraおよびC3−ライト化合物、これらの医薬適合性の塩およびエステルが挙げられる。
【0074】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトについての一般式は、C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)を含む。
【0075】
神経保護性のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクト
多数の重要な生物学的反応が、意図的に、または望ましくない毒性副生成物として、反応性酸素種を発生させるため、細胞の抗酸化防御を補足することができる抗酸化性分子は、潜在的治療薬として治療上有用である。
【0076】
ここで説明し、特許請求の範囲に記載するバックミンスターフラーレン誘導体のe,e,eマロン酸/酢酸三重アダクトを使用して、フリーラジカル、特にグルタメートの神経毒性(「興奮毒性」)の結果として放出されるときのフリーラジカルによって引き起こされるあらゆる疾病状態の進行を予防、治療または緩和することができる。興奮毒性傷害の治療は、周囲の細胞から放出されたグルタメートによって損傷した中枢神経に対する損傷の程度を低減させること意味する。興奮毒性などの神経毒性事象は、低酸素/虚血などの多数のタイプの急性神経性発作中に発生し得る。例えば、卒中、低血糖、癲癇または外傷中に発生する。神経毒性事象は、神経変性疾患、例えば、ハンチングトン病、アルツハイマー病、筋萎縮性側索硬化症(「ALS」)、およびAIDSの神経変成作用によって引き起こされる慢性神経障害にも関与し得る。従って、バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、神経毒性傷害が発生する疾病の治療法にも有用である。
【0077】
アラキドン酸(「AA」)は、グルタメートによるNMDA受容体刺激によって引き起こされる神経細胞への過剰なCa2+の流入に起因してニューロンにおいて放出される(このグルタメートは、神経毒性事象によって損傷したニューロン自体によって放出されたものである)。過剰なCa2+の流入は、ホスホリパーゼA2(AAを遊離する細胞膜を破壊するカルシウム依存酵素)を活性化する。内因性リポキシゲナーゼおよびシクロオキシゲナーゼによるAAの代謝は、ニューロンの損傷または死をもたらす神経脂質膜の過酸化性分解を開始させる酸素フリーラジカルの生成を導く。従って、フリーラジカル・スカベンジ性のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを含む組成物の投与による酸素由来フリーラジカルの低減は、グルタメート誘発神経毒性を抑制する代替メカニズムを提供する。
【0078】
個体の寿命を増加させるための上記方法および組成物と同様に、神経毒性傷害に罹患している患者において神経毒性障害を治療するための組成物および方法は、前記患者に、この個体に対する治療的に有効な量のバックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを含む組成物を投与することを含む。さらに詳細には、これらの実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを少なくとも1つ投与することを含む。更なる実施態様は、患者へのC3−ライト、Penta−1、Penta−2およびTetra Quartetの投与を含む。これらの新規化合物での治療方法は、静脈内、筋肉内、皮下または経口送達することができる。
【0079】
第一の実施態様は、ニューロン傷害治療用の化合物を含む。こうした化合物の非限定例には、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが挙げられる。バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトのさらなる非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0080】
第二の実施態様は、神経毒性傷害に罹患している患者において、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステル、ならびに医薬適合性の担体を含む組成物(この場合、前記化合物は、前記神経毒性障害を治療するために有効な量で前記組成物中に存在する)を前記患者に投与することによる、神経毒性障害の治療方法を含む。ニューロン傷害の治療に有用なバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択されるバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが挙げられる。
【0081】
上で示したように、この方法の化合物は、1日に約1.5mg/kgから約1500mg/kg、または1日に約10mg/kgから約60mg/kgの量で投与することができる。
【0082】
第三の実施態様は、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、患者において、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされる神経毒性傷害を抑制する方法である。この方法に有用なバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの非限定例には、Penta Pair、Tetra Quartet、C3−ライト、これらの立体異性体、これらの混合物などから成る群より選択される化合物が挙げられる。
【0083】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトは、1日に約1.5mg/kgから約1500mg/kgの量で、または1日に約10mg/kgから約60mg/kgの量で投与することができる。
【0084】
バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトの化学合成
文献に報告されているC3の調製では、細胞培養スクリーニングでの再生能および変異機能が劣る、一部正体未確認のプロドラッグの混合物が生産される。従って、特定の成分へのより信頼性できる、直接的な方法が必要である。
【0085】
高濃度のHexaを合成する方法
下記合成方法を用いて、一般に90、94および97%を超える結果で、大量のHexaを生じさせることができる。
1つのこうした方法は、水、メタノールおよび溶媒中のC3メチルエステルの初期溶液の調製を含む。水のエステルに対するモル比は、約100:1から約20:1である。C3メチルエステルを溶解するために有用な溶媒には、芳香族系溶媒、例えばトルエン、トルエン中の酢酸t−ブチルなどが挙げられる。次に、この初期溶液を完全に(約1時間)混合する。これは、最初は濁っているように見えることがある。次に、メタノール中、約1Mまでの水酸化ナトリウムを添加した後、この溶液を、溶液の着色がなくなるまで(約2時間以下)激しく攪拌する。反応の完了を確認するために、TLCを使用してもよい。
【0086】
反応が完了したら、水を添加して非水性層と水性層を形成する。次に、デカント、および分液漏斗または層を分離するために有用な他のあらゆる器具の使用を含む(しかし、これらに限定されない)あらゆる標準法によって、これらの層を分離する。次に、一切の残留溶媒を真空下でストリップする。生成物を含有する水性層を、不活性雰囲気(N2など)中、約60℃で約1から約2時間加熱する。
【0087】
異性体配分は、HPLCプロトコル1および2(下記)を使用して判定することができ、一般には90、94または97%より上である。
【0088】
脱カルボキシル化によるPenta PairまたはTetra Quartetの直接合成
溶解度を最大にするように選択された芳香族系溶媒、例えばトルエンまたはベンゼン中の(Bingel、米国特許第5,739,376号(この全文が本明細書に参考として援用されている)により調製されるような)ジ−t−ブチルマロニルC60に、ブロモマロン酸のエステル(ブロモマロン酸ジメチルを含むが、これに限定されない)を先ず添加し、続いて、(Bingelの‘376、カラム4、44から45行目に開示されているとおり調製した)1,8−ジアザビシクロ(5,4,0)ウンデク−7−エン(「DBU」)または水素化ナトリウムを含む(しかしこれらに限定されない)第三アミンを添加して、反応混合物を作ることにより、Penta PairまたはTetra Quartetを直接合成する。Penta Pairを調製するためには、これらの反応物は、C60対マロネート対塩基比1:2:2の濃度である。Tetra Quartetを調製するためには、比率1:1:1を用いる。適切な時間(約30分)を経過させて、確実にこの反応混合物を完全混合する。反応の終点は、TLCによって判定することができる。次に、この反応混合物を、上で選択した溶媒中のシリカゲルのカラムに注入する。ビス異性体を調製するために、このカラムは、すべてのビス異性体が出てくるまで溶媒で溶離しなければならない。次に、溶媒は、酢酸エチル/溶媒混合物に換えるべきであり、酢酸エチルの漸増濃度、例えば0.5%→1%→約2%で添加すべきである。これらの混合物は、次の4つの成分を溶出するであろう。D3(トランス−3,トランス−3,トランス−3 トリスマロン酸C60);e,トランス−3,トランス−2;e,トランス−4,トランス−3;およびC3エステル。溶出されたC3エステル画分を、次いで、真空下で蒸発させる。
【0089】
次にp−トルエンスルホン酸・一水和物、トリフルオロ酢酸(TFA)またはメタンスルホン酸を含む(しかし、これらに限定されない)上記溶媒に混和可能な酸を、芳香族系溶媒(トルエンを含むが、これに限定されない)中のC3エステル画分の溶液に添加して、前記画分を溶解する。この溶液を約88から89℃に加熱する。より低い温度を用いてもよいが、反応は、より遅い速度で進行することになる。TFAを用いる場合、室温で充分であろう。反応が開始したら(約45分後、またはアッセイにより判定する)、追加の酸を添加すべきである。約10分後に沈殿が形成し始めるだろう。加熱は、反応が完了するまで(約90分、またはTLCもしくはHPLCなどのアッセイによって判定する)継続する。次に、溶媒をこの沈殿から除去する。これらの固体に水および酢酸エチルを添加し、酢酸エチル/水混合物を作る。この酢酸エチル溶液を分離し、次いで、水で洗浄して触媒の酸を除去する。必要な場合には、酢酸エチルを複数回、洗浄しなければならない。次いで、酢酸エチルを真空下で蒸発させて、固体を残す。
【0090】
次に、固体を約1:1:2のアセトニトリル:水:アセトン混合物に溶解する。材料の可溶化を最大にするために、他の比率(アセトン単独使用を含む)を用いてもよい。この混合物を油浴、加熱マントルなどで約50℃、約76.5℃または約100℃に加熱して、反応を完了させる。反応が完了していることを確認するために、アッセイを行ってもよい。次に、沈殿物を充分に溶解するために一定量のアセトンを添加する。加熱は、約5時間から約7.5時間、継続する。次に、一切の揮発性溶媒を(例えば真空下で)除去し、次いで、酢酸エチルを添加して均質な溶液を作る。次いで、この溶液を真空下で蒸発させて、固体を生じさせる。
【0091】
次に、この固体を、メタノールおよび水を含有する芳香族系溶媒(トルエンまたはトルエン中5%の酢酸t−ブチル(1mg/mL))に溶解する。様々な比率(100:4.4:0.2など)を用いることができるが、二相の生成を回避するために、限られた量の水を使用すべきである。次いで、この溶液を、約1時間攪拌しながら充分に混合する。混合後、比率20対1の1M 水酸化ナトリウム/メタノールを添加する。沈殿完了後(色が完全に消えたら(これは、約1時間から約2時間かかることがある))、水を添加する。次いで、溶媒層を完全に分離し、一切の残留溶媒を確実に除去する。水性層を約60℃から約110℃で約2時間+/−30分間加熱する。アッセイを行って、反応の完了を判定する。
【0092】
冷却後、硫酸などの強酸(酢酸エチルに抽出されないであろうもの)を、上からの塩基を中和させるために充分な量でこの溶液に添加する。最終生成物は、上で説明したように酢酸エチルで抽出する。
【0093】
(溶液またはニートでの)C3−ライトの直接合成
C3−ライトは、Hexa、Penta−1およびPenta−2のC3誘導体の乾燥または溶解サンプルを加熱することにより、直接合成することができる。溶解サンプルを使用する場合、前記誘導体は、アセトニトリル:水の混合物に溶解すべきである。加熱マントル、油浴などを含む(しかし、これらに限定されない)あらゆる標準的加熱方法を用いることができる。得られた溶液/サンプルを、C3−ライトを生じさせるために充分な時間(約24時間未満)、約60℃から約70℃から約81度(アセトニトリルの沸点)で加熱する。次いで、溶媒を真空下で除去して、固体生成物を得る。
【0094】
C3誘導体のニートサンプルを用いる場合、このサンプルは、約−30mmHgの真空オーブン内で、約150℃で加熱する。
【0095】
Petna、TetraまたはC3−ライトを生じさせるための溶液中でのC3の熱分解
Petnta,TetraまたはC3−ライトを含む生成物は、Hexa、Penta−1およびPenta−2を含有するC3誘導体のサンプルを、比率1:1のアセトニトリル:水に溶解することによって生成する。得られた溶液を約60℃に加熱する。約1.5時間加熱後、Penta濃度は、ほぼこの最大値である。加熱の適用が長いほど(約3.5時間から約5.5時間)、Penta濃度は低下するであろうが、C3−ライトおよびTetraの濃度は、上昇するであろう。
【0096】
所望の結果が得られたら、溶媒を真空下で除去して、固体生成物を得る。
【0097】
HexaとPentaの混合物を合成するためのNaOMe/MeOHの使用
N2などの不活性ガス下、芳香族系溶媒中のe,e,e−トリスジメチルマロニルフラーレン(「C3エステル」)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作る。芳香族系溶媒は、トルエンおよびトルエン中の酢酸t−ブチルから成る(しかし、これらに限定されない)群より選択することができる。ナトリウムメトキシドとC3エステルは、比率16から20:1である。赤橙色の沈殿が、直ぐに形成し始める。適切な時間(約1から2時間)の後、水を添加して、水性層と非水性層を形成する。あらゆる色が水性層に移るであろう。次に、層を分離する。水性層を氷浴などで0から5℃に冷却し、次いで、過剰な硫酸で約2のpHに酸性化する。次に、この酸性溶液を酢酸エチルで抽出して、すべての色を酢酸エチル抽出物に移す。次に、併せた酢酸エチル抽出物を水で洗浄して、一切の黄色不純物を除去する。次いで、この溶液を、真空下、ほぼ室温で蒸発および乾燥させる。
【0098】
細胞スクリーニングのための溶液は、0.1Nの水酸化ナトリウムにこの固体を溶解することにより調製する。充分な塩基を添加して、すべてのカルボキシルを中和することができる。有効濃度は、トルエン中のこのエステルの前駆体で決定した吸光係数4400を使用して、uvなどのアッセイにより決定することができる。HPLCプロトコル1を用いて、Hexa、Penta−1およびPenta−2の百分率を判定することができる。この時、HPLCによりこれら三成分を分離することができる。
【0099】
ここでのデータは、開示するカルボキシフラーレンが、酸素由来のフリーラジカルを分解するユニークな能力を有する新しいクラスの抗酸化物質であること、およびこれらの化合物が、個体の寿命を延ばす、並外れた広い強力な能力を有することを証明している。
【0100】
上記組成物および方法のさらなる特徴および利点、ならびに様々な実施態様の構造および操作は、添付の図面を参照しながら下で詳細に説明する。
【0101】
上の開示は、幾つかの好ましい実施態様を説明するものであり、いかなる点でも範囲を制限するものではない。熟練した技術者は、これらの方法および組成物の実施の際、本明細書には明白に開示されていない他の実施態様に気づくであろう。下に記載する実施例によって上の実施態様をさらに説明する。これらの実施例は、これらの実施態様を説明する意味を持つものであり、いかなる点でも範囲を制限するものとは解釈すべきでない。
【0102】
本明細書おいて言及するすべての参照および関連技術は、本技術の現状の一部であり、従って、これら全体が本明細書に援用されている。
【実施例1】
【0103】
C3カルボキシフラーレンの調製
材料
シリカゲル(Merck グレード9385、260−400、60A)は、Aldrich Chemials(ミズーリ州、セントルイス)から入手した。他の試薬は、Sigma Chemical Co.(ミズーリ州、セントルイス)および他の標準的な供給業者から購入した。
【0104】
マロン酸C60のC3位置異性体(e,e,eC60[C(COOH)2]3)は、一晩攪拌することによりC6(720mg、1.00mmol)をトルエンに1mg/mLの濃度で溶解することによって合成した。ブロモマロン酸ジメチル(632.4mg、2.69mmol)を添加し、続いて1,8−ジアザビシクロ(5.4.0)ウンデク−7−エン(DBU、493mg、3.24mmol)を添加した。この反応混合物を2時間攪拌し、シリカゲルのパッドに通して濾過し、真空下で濃縮した。残留物を、トルエン中で開始する、シリカゲルの450mLカラム(Merck、280から400メッシュ)でのクロマトグラフィーに付した。漸増量の酢酸エチル(EtOAc)をトルエンに添加することにより、着色成分を分離した。C3画分は、トルエン中5%のEtOAcで溶出した。C60マロン酸エステル画分の純度をTLCおよびHPLCでモニターした。このC3エステル(0.25g、0.23mmol)をトルエン(250mL)に溶解し、窒素でスパージした。ナトリウムメトキシド(2.2Mのものを2.22mL、4.88mmol)の添加により、数分以内に沈殿が生じた。この混合物を窒素下、室温で1時間攪拌した。水(20mL)を添加し、この混合物を一晩攪拌した。すべての着色生成物が、水性層に行った。層を分離し、水性層を冷却して、20%硫酸(1.32mL)で酸性化した。この溶液をEtOAcで3回抽出した結果、すべての色が有機層に行った。この有機層を水で数回洗浄して、黄色の不純物を抽出した。次いで、EtOAc層を蒸発させ、残留物223.2mg(理論量の89%)を凍結乾燥させた。
【実施例2】
【0105】
マウスでの寿命試験についての実験法
月齢12ヶ月の雄および雌(同数)C57B6NIHマウスを米国国立老化研究所(NIA)齧歯動物老化コロニーから購入した。このコロニーから出荷されたマウスを健康、腫瘍または他の能力障害で一切選別せず、このコロニーから入手したすべてのマウスを、続いて、この研究に登録した。マウスは、同性のものを入れるケージに1ケージあたり2匹で無作為に配置し、識別のために耳にパンチし、計量した。次に、マウスを週2回の3セッションで、回転棒での訓練を行い、次いで、ベースラインの運動能力を測定するために3セッションで回転棒での試験を行った。次に、治療薬Aまたは治療薬Bのいずれかを受けるように、これらの治療が何であるのか知らない観察者が、ケージを割り当てた。
【0106】
治療薬Aは、水中のC3(28.75μM)の溶液であり、治療薬Bは、赤色のC3溶液に合うように添加した市販の食用色素であった。溶液が何かを知らない者が、溶液Aまたは溶液Bを採水瓶に入れ、週に2回、瓶の口のところまで溶液を補充し、週に2回、濾過して一切の微粒子を除去した。週齢19ヶ月でマウスを再び計量し、別のラウンドの回転棒訓練および試験を受けさせた。マウスを自然死させ、動物収容施設のスタッフがこの施設の一般操作手順の一部として死亡日を記録した。施設のスタッフは、動物には抗生物質溶液を用いたと信じており、この研究の目的を知らなかった。動物が死亡したとき、このゲージ番号、動物の由来、および死亡日を死亡通知に記録し、次いで、これを研究所に送り、そこで情報をデータベースに入力した。
【0107】
これらの実験の結果を図4に表示する。これらは、マウスの寿命の顕著な増加(約20%)を示している。加えて、薬物の経口投薬により寿命が増加したので、これは、後生動物の寿命増加を達成する初めての実用的方法である。C3治療マウスの寿命増加は、体重減少を随伴しなかった。
【実施例3】
【0108】
ラットを用いる毒性研究
二系統のラット(Sprague−DawleyおよびLong−Evans)を用いてC3のラット毒性試験も行い、前記ラットに30日間、10mg/kg以下を与えたが、毒性(すなわち、生存率低下、毛づくろいの欠陥、または摂食障害)は一切示さなかった。
【0109】
(実施例4から13)
幾つかの新規バックミンスターフラーレンe,e,eマロン酸/酢酸三重アダクトを下記で同定する。C3 Hexaの調製からの主成分は、ほぼ等しい存在比での2つの異性ペンタカルボン酸(Penta Pair)を含み、マイナーな生成物は、4つの異性テトラ酸(Tetra Quarted)および4つの異性トリ−脱カルボキシル化生成物(C3−ライト)を含む。これらの生成物は、C3より水溶性が劣り(親油性が高く)、このことが、組織におけるこれら新規化合物の吸収および保持を増大させ得る。
【0110】
以下の方法は、94+%のヘキサカルボン酸濃度をもたらし、他の新規成分への直接的経路となる。Penta Pair、Tetra QuartetおよびC3−ライトの含有率が高い混合物が、脱カルボキシル化により得られた。Penta PairおよびTetra Quartetは、t−ブチル保護を用いる別戦略によっても得られた。
【0111】
誘導体を分析するためのHPLC法
以下のすべてのHPLC法に、四液ポンプ(Quarternary pump)およびダイオードアレイ検出器を具備するHewlett Pakard/Agilent 1100シリーズ HPLCを使用した。分離は、室温で維持した、Zorbax SB−C18 4.6X250mmカラム(パッキング 5μm)(カラムA)またはZorbax SB−C8 4.6X250mmカラム(パッキング 5μm)(カラムB)を用いて行った。すべての方法に、1mL/分の溶媒流量を用いた。
【0112】
プロトコル1 − HexaおよびPenta化合物の分析
溶媒は、水中0.1%のTFA(溶媒A)、およびアセトニトリル95%と水5%中0.1%のTFA(溶媒B)であった。サンプルは、15分間かけてA:B;40:60からA:B;10:90への勾配を用い、さらに15分、A:B;10:90でカラムから溶離した。化合物は、インラインダイオードアレイ検出器を使用してこれらのUV−vis 吸収によりモニターし、同定した。
【0113】
プロトコル2 − HexaおよびPenta化合物を分離するための別法
HPLC溶媒は、水中0.1%のTFA(溶媒A)、および2−プロパノール95%と水5%中0.1%のTFA(溶媒B)であった。化合物は、10分かけてA:B;95:5からA:B;52:48への勾配、10分かけてA:B;51:49にさらに進め、次いで、5分かけてA:B;21:79にさらに進める勾配を用いて、(カラムAまたはB)から溶離した。次に、これをさらに2分間、イソクラティックにしておいた。
【0114】
プロトコル3 − Tetra Quartetを分離するための方法
Tetra Quartetの調製物は、水中0.1%のTFA(溶媒A)、およびアセトニトリル95%と水5%中0.1%のTFA(溶媒B)を使用して、(カラムAまたはB)から溶離した。この溶媒組成を35分間、A:B;30:70で維持し、次いで、2分かけてA:B;5:95への勾配、続いて、さらに10分間、A:B;5:95で維持した。
【0115】
プロトコル4 − C3−ライトの分析
C3−ライトの調製物は、(カラムAまたはB)のいずれかでプロトコル1について説明したとおり溶媒を使用して分析した。勾配は、A:B;95:5で開始、5分間維持し、続いて、30分かけてA:B;5:95に勾配を変化させるように修正した。この溶媒組成をさらに35分間、A:B;5:95のままにしておいた。
【0116】
プロトコル5 − t−ブチル、メチルエステルの分析
溶媒は、アセトニトリル50%とジクロロメタン50%(溶媒A)であった。サンプルは、5分かけて100%(溶媒A)のイソクラティックプログラムで(カラムA)から溶離した。
【0117】
プロトコル6 − 部分メチルエステルの分析
HPLC溶媒は、アセトニトリル95%と水5%中0.1%のTFA(溶媒A)であった。サンプルは、120分かけてイソクラティック100%(溶媒A)溶液で(カラムAまたはB)から溶離した。
【実施例4】
【0118】
Hexa異性体とPenta異性体の混合物を合成すためのMaOMe/MeOH法
ナトリウムメトキシド(2.2Mのもの1.84mL、4.05mmol)をN2下、224mLのトルエン中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)(224.1mg、0.202mmol)の溶液に添加した。赤橙色の固体の沈殿が、直ぐに始まった。1時間後、水を添加し、すべての色が水性層に行った。層を分離して、水性層を冷却し、硫酸(3.7Mのもの1.10mL、4.07mmol)で酸性化し、酢酸エチル(2x40mLおよび1x10mL)で抽出した。併せた酢酸エチル抽出物を3x40mLの水で洗浄し、これによって黄色の不純物を除去した。真空下、室温での蒸発および乾燥によって、199.2mg(Hexaカルボン酸に基づく理論値の96.1%)が生じた。細胞スクリーニング用の溶液は、この固体を0.1Nの水酸化ナトリウムに溶解して重量で約25mMの溶液を得ることによって調製した。次に、この有効濃度を、トルエン中のこのエステル前駆体を基に決定した吸光係数4400を用い、UVによって決定した。UV−vis λmax(nm)は488であり、max/min比(488/414nm)は、2.19であった。HPLCプロトコル1を用いて、この合成により65.1%のHexa異性体、14.3%のPenta−1および20.6%のPenta−2が生成されたと判定した。これら三成分をHPLCにより分離し、質量分析(表3)および1H NMR(表4)によって同定した。
【実施例5】
【0119】
95% Hexaを生成させるためのトルエン、6% MeOH、および等量の水とNaOH
500mLのトルエン中の水(0.216mL、12mmol)、メタノール(17.1mL)およびC3メチルエステルの溶液を1時間攪拌した。この溶液は、最初は濁っているように見えた。塩基を直ちに添加すると、Hexaの収率は低下する。メタノール中の水酸化ナトリウム(0.93Mのもの12.9mL、12.0mmol)を添加した。1.5時間後、TLCおよび溶液の色によって評価すると、溶液中にエステルは残存しなかった。水100mLを添加した。トルエン層を分離し、水性層を濃縮してメタノールおよびトルエンを除去し、次いで、N2下、60°2時間加熱して、加水分解を完了させた。抽出による処理によって、490.8mg(理論収率の79.5%)を得た。UV−vis λmaxは、487nmであり、max/min比(487/411nm)は、3.10であった。HPLCプロトコル1を用いて、異性体分配は、95.2% Hexa、1.7% Penta−1、および3.0% Penta−2であると判定した。
【実施例6】
【0120】
トルエン中5%の酢酸t−ブチルを添加した後に混合した、等量のNaOHと水および2%のMeOHを使用して、88% HexaおよびPenta異性体を生じさせる方法
水酸化ナトリウム(3.679g、0.092mol)を92mLのメタノールおよび1.655mL(0.092mol)の水に溶解して、0.982M溶液を得た。この溶液の一部(12.3mL、12.0mmol)を、N2下、室温で、5%の酢酸t−ブチルを含有する585mLのトルエン(1.0mg/mLと比較して1.14mg/mL)に溶解したC3メチルエステル(670mg、0.604mmol)に添加した。沈殿がゆっくりと形成した。1時間後、この反応混合物に水を添加した。相を分離し、水性層を濃縮してメタノールおよびトルエンを除去し、次いで、60°で2時間加熱した。生成物573.3mg(理論値の92.5%)を、上に記載した酢酸エチル抽出によって水性層から除去した。UV−vis λmaxは、489nmであり、max/min比(489/411nm)は、3.37であった。HPLCプロトコル1を用いて、異性体分配は、88.4% Hexa、5.6% Penta−1、および6.0% Penta−2であると判定した。
【実施例7】
【0121】
酢酸t−ブチルと5個の水分子を使用して96% Hexaを生じさせる方法
950mLの5% 酢酸t−ブチル中に水(1.52mL、84.4mmol)、メタノール(40mL)およびC3メチルエステル(UVにより0.845mmol)を含有する溶液を、N2下で30分間攪拌した。メタノール中の水酸化ナトリウム(1Nのもの16.9mL、16.9mmol)を添加した。2時間後、すべての色が沈殿した。水50mLを添加し、トルエンをデカントして、残留トルエンを真空下でストリップした。水性層をN2下、60°で2時間加熱した。抽出による処理によって、798.1mg(理論値の92.1%)を得た。UV−vis λmaxは、489nmであり、max/min比(489/412nm)は、2.71であった。HPLCプロトコル1を用いて、異性体分配は、96.7% Hexa、1.5%の各Pentaであると判定した。
【0122】
合計メタノール濃度は、6%である。塩基を直ぐに添加すると、またはエステルの添加前に塩基とすべてのメタノールを併せると、異性体分配は、より低いHexa含有率へとシフトする(実施例6参照)。
【実施例8】
【0123】
トルエンのみを使用する97% Hexaの別合成
この手順は、トルエン中5%の酢酸t−ブチルの代わりにトルエンを使用したことを除き、実施例7と同じであった。HPLCプロトコル1を用いて、この生成物は、97.2% Hexa、1.3% Penta−1および1.5% Penta−2の異性体分配を有すると判定した。UV−vis maxは、491nmであり、max/min比(491/412nm)は、2.97であり、収率は、90.5%であった。13C NMR(K2CO3,D2O,600MHz):δ173.84,173.76,152.55,151.33,149.49,149.41,149.05,148.98,148.92,148.78,148.61,148.13,147.96,146.55,146.46,145.89,144.81,143.82,143.07,142.99,80.69,79.88,69.55。
【実施例9】
【0124】
Penta Pair直接合成
段階1 − ブロモマロン酸ジメチル(281.3mg、1.20mmol)を、200mLのトルエン中のジ−t−ブチルマロニルC60(0.592mmol)に添加し、続いて、DBU(202.4mg、1.33mmol)を添加した(Bingelの‘376、カラム4、44から45行目)。30分後、この反応混合物を、トルエン中のシリカゲルの410mL(4x310cm)カラムに注入した。このカラムを、すべてのビス異性体(UV−visにより25%)が出てくるまで、トルエンで溶離した。溶媒を酢酸エチル/トルエン混合物(0.5%対2%)に換えた。以下の成分の含有量が多いエステル調合物が溶出した(HPLCプロトコル5を使用):D3(トランス−3,トランス−3,トランス−3 トリスジメチルマロニルC60);e,トランス−3,トランス−2 トリスジメチルマロニルC60;e,トランス−4,トランス−3 トリスジメチルマロニルC60;およびC3。C3エステル画分(HPLCプロトコル1)を真空下で蒸発させた。質量分析および1H NMRデータは、表3および4にある。
【0125】
段階2 − p−トルエンスルホン酸・一水和物(22.5mg、0.118mmol)をトルエン中のC3画分の溶液(15mL)に添加し、88から89°の油浴に入れた。45分後、追加のp−トルエンスルホン酸・一水和物(15.6mg、0.082mmol)を添加した。生成物は、10分以内に沈殿し始めた。90分間、加熱し続けた。沈殿からトルエンを除去し、これらの固体に水および酢酸エチルを添加した。この酢酸エチルを水で3回洗浄してp−トルエンスルホン酸を除去した。酢酸エチルを真空下で蒸発させることにより、118.2mgの固体が残った。HPLCプロトコル6を用いて化合物を分析した。
【0126】
段階3 − これらの固体を、10mLの1:1:2 アセトニトリル:水:アセトンに溶解した。サンプル0.2mLを分析用にとった。残りは、76.5°の油浴で加熱した。30分後、沈殿が始まって、分離した。アセトン(10mL)を添加し、固体を溶解した。加熱を継続し、1.75時間後、脱カルボキシル化は73%完了した。合計7.5時間加熱した後、真空下で揮発性溶媒を除去し、酢酸エチルを添加して、均質溶液を得た。分析用に一部(1.9mg)をとった。残りは、真空下で蒸発させて、0.13gの固体を得た。化合物をHPLCプロトコル1によって分析した。
【0127】
段階4 − 上記固体(0.125mmol)を、メタノール(4.9mL)および水(0.128mL、7.1mmol)を含有するトルエン(120mL)に溶解し、1時間攪拌した後、メタノール中の水酸化ナトリウム(1.0Mのもの2.3mL、2.3mmol)を添加した。5分以内に固体が沈殿し始めた。1時間後、水50mLを添加した。トルエンを分離し、水性層をメタノールおよびトルエンでストリップして、60°で2時間加熱した。冷却した溶液に硫酸(3.7Mのもの0.621mL、2.3mmol)を添加し、上に記載したように酢酸エチルを用いて生成物を抽出した。この乾燥重量は、91.1mgであった。U−vis λmax(nm)。HPLCプロトコル1を用いて化合物を分析した。質量分析および1H NMRデータは、表3および4にある。13C NMR(K2CO3,D2O,600MHz):δ175.39,175.36,173.94,173.88,173.82,173.80,173.74,173.68,173.65,153.39,152.46,152.32,152.23,152.15,151.37,151.22,149.66,149.46,149.38,149.24,149.14,149.04,148.97,148.86,148.55,148.45,148.42,148.36,148.35,148.13,147.96,147.78,147.04,146.96,146.87,146.70,146.66,146.60,146.42,146.38,146.03,145.94,145.70,145.55,145.22,144.86,144.79,144.67,144.58,144.00,143.92,143.88,143.81,143.76,143.62,143.07,143.02,142.98,142.93,142.11,142.05,80.72,80.69,80.58,79.97,79.75,79.67,77.21,77.12,76.38,76.30,69.52,69.44,69.38,69.72。
【実施例10】
【0128】
Tetra Quartetの合成
段階1 − ブロモマロン酸ジメチル(38mg、0.16mmol)を、30mLのトルエン中のC60のマロン酸ビス−ジ−t−ブチル(UVにより0.167mmol)のサンプルに添加した。DBU(28mg、0.18mmol)を添加し、この反応をTLC(シリカゲルを用いて、トルエン中2%のEtAc)によって追跡した。1時間後に反応が完了した。この反応混合物を、トルエン中のシリカゲルの330x2.5cmカラムに注入した。ビス画分(12%、4成分、HPLCプロトコル5)をトルエン中で回収し、次いで、溶離液をトルエン中0.5%の酢酸エチルに換えた。D3、e,トランス−3,トランス−2、e,トランス−4,トランス−3およびC3のエステルを含む画分を得た。C3画分は、HPLCプロトコル1によると純度88.5%であり、さらに精製せずに使用した。UV−vis(トルエン中0.5%のEtAc)λmaxは、486nmである、max/min比は、3.57であった。
【0129】
段階2 − p−トルエンスルホン酸・水和物(4.9mg、0.0258mmol)を4.5mLのトルエン中のC3エステル(0.0154mmol)に添加し、この混合物を77°の油浴に入れた。この温度を15分かけて85°に上昇させ、さらなるp−トルエンスルホン酸(5.9mg、0.0310mmol)を添加した。5分以内に沈殿が開始した。30分間加熱し続けた。この混合物を冷却し、トルエンをデカントした。水および酢酸エチルの添加によって、すべての色が酢酸エチル層にある二相混合物が生成した。この酢酸エチルを水で3回洗浄し、蒸発させて、18.3mgの固体を得た。HPLCプロトコル1を用いて化合物も分析した。
【0130】
段階3 − この固体を5mLの4:1 アセトニトリル:水に溶解し、0.546mLを分析用にとった。残りを74°の油浴で加熱した。65分後、固体が存在した。3mLのアセトニトリルの添加により、均質溶液を得、5.5時間加熱し続けた。この溶液の一部0.8mLを分析用にとり、残り7.0mLは濃縮して、アセトンおよびアセトニトリルを除去した。HPLC(95:5:0.1,ACN:H2O:TFA,min,%)19.341(26.2),38.180(25.1,43.798(22.2)および80.874(21.6)。HPLCプロトコル1を用いて化合物も分析した。
【0131】
段階4 − 水酸化ナトリウム(0.1Nのもの0.59mL、0.059mmol)を、約3mLの水中の上で得られた物質の懸濁液に添加した。固体が溶解した。水酸化ナトリウム(1Nのもの0.6mL、0.6mmol)を添加し、溶液を62℃で1時間加熱した。HPLCは、加水分解の完了を示した。この溶液を冷却し、硫酸(3.7Mのもの0.162mL)で酸性化した。酢酸エチルでの抽出により、11.4mgの固体が生じた。HPLC(95:5:0.1,ACN:H2O:TFA,min,%)6.076(26.4),10.784(24.5),13.883(26.0),23.589(23.1)。HPLCプロトコル1を用いて化合物を分析した。質量分析および1H NMRデータは、表3および4にある。
【実施例11】
【0132】
溶液中でのC3−ライトの直接合成
C3のサンプル(329.7mg、0.32mmol、71 %Hexa、15% Penta 1および14% Penta 2)を6mLの1:1 アセトニトリル:水に溶解し、70℃の油浴に入れた。この反応の進行をHPLC(プロトコル2)によってモニターした。2.25時間後、反応は、ほぼ完了した。5時間後、固体が沈殿し始めた。24時間加熱し続けた。真空下で溶媒を除去して、199.3mgの生成物を得、これは、HPLCプロトコル1によると以下の組成を有した。C3−ライト ピーク1(14.0%)、ピーク2(39.4%)、ピーク3(37.0%)、ピーク4(8.7%)。LC−MS pos m/eは、各々895であり、neg m/eは、各々1007であった(M+TFA−H)。すべてが、C3(C60へのe,e,e付加)のUV−visスペクトルを有した。1H NMRおよび質量分析データは、表3および4にある。この固体は、水酸化ナトリウムに可溶性であり、アセトンおよび酢酸エチルには、中等度に可溶性であり、水、メタノールおよびアセトニトリルへの溶解性は乏しかった。
【実施例12】
【0133】
Penta、TetraまたはC3−ライトを生じさせるための溶液中でのC3の熱分解
97.4% Hexa、1.2% Penta 1および1.3% Penta 2を含有するC3のサンプル(74.8mg)を10mLの1:1 アセトニトリル:水に溶解し、60℃で加熱した。この組成を、プロトコル2および4を使用してHPLCによりモニターした(表1参照)。
【0134】
【表1】
【実施例13】
【0135】
C3からC3−ライトへのニート脱カルボキシル化
93.8% Hexa、3.5% Penta 1および2.7% Penta 2を含有するC3のサンプルを乾燥粉末として、150℃、30mmHgの真空オーブンに入れた。この組成をHPLCプロトコル2によってモニターした。表2は、C3−ライトを含むC3の脱カルボキシル化生成物の時間依存性生成を示すものである(表2参照)。
【0136】
【表2】
【0137】
【表3】
【0138】
【表4】
【0139】
【表5】
【0140】
【表6】
【0141】
【表7】
【0142】
上記にかんがみて、上の実施態様および実施例の幾つかの利点が達成され、成し遂げられたことがわかるだろう。他の当業者が様々な実施態様において本発明を最良に利用することができ、考えられる特定の使用に適するよう様々な変更を施すことができるように、実施態様を選択し、記載して、本発明の原理およびこの実際の適用を最良に説明した。
【0143】
本明細書に記載し、説明する構造および方法に、本発明の範囲を逸脱することなく、様々な変更を施すことができるので、上記明細書に含まれているまたは添付の図面に示されているすべての事柄は、限定的なものではなく、説明的なものとして解釈されよう。例えば、上に記載したような方法は、ヒトを含む(しかし、これに限定されない)他の後生動物に容易に同じ結果で適用することができた。従って、本発明の広さおよび範囲は、上に記載した具体例としての実施態様のいずれによっても、当然、制限されず、本明細書に続く添付の請求項およびこれらに相当するもののみに基づいて定義されるべきものである。
【図面の簡単な説明】
【0144】
【図1A】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1B】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1C】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図1D】3つの主カルボキシフラーレン成分を同定するHPLCによるC3調製物の分析を開示する図である。
【図2】様々な有用なカルボキシフラーレンを示す図である。
【図3】C3トリスマロン酸位置異性体を示す図である。
【図4】対照溶液で治療したC57B6マウスに対する経口C3で治療したC57B6マウスの生存を表す図である。
【図5】バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト上の官能基の立体配置を表す図である。
【図6】C3−ライト(e,e,eトリス酢酸C60)およびC3(e,e,eトリスマロン酸C60)の構造を表す図である。
【図7】Penta−1の構造を表す図である。
【図8】Penta−2の構造を表す図である。
【図9】NMDA毒性に対するHexa、Penta−1、Penta−2およびC3−ライトによる神経保護を表す図である。
【図10】AMPA毒性に対するHexa、Penta−1、Penta−2およびC3−ライトによる神経保護を表す図である。
【図11】Hexa、Penta−1、Penta−2およびC3−ライトの血漿中動態、ならびにC3の組織分布の詳細図である。
【図12】C3−ライトの血漿中薬物動態および組織分布の詳細図である。
【特許請求の範囲】
【請求項1】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト。
【請求項2】
前記化合物が、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項1に記載のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト。
【請求項3】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは−Hおよび−COOMeから成る群より選択されることを条件とする)のバックミンスターフラーレンの三重アダクト。
【請求項4】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、ニューロン傷害の治療用化合物。
【請求項5】
Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項4に記載の化合物。
【請求項6】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)の化合物、この医薬適合性の塩および医薬適合性のエステルを含むバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトならびに医薬適合性担体を患者に投与することにより(この場合、前記化合物は、前記組成物中に神経毒性傷害を治療するために有効な量で存在する)、神経毒性傷害を前記傷害に罹患している患者において治療する方法。
【請求項7】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項6に記載の方法。
【請求項8】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項7に記載の方法。
【請求項9】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項8に記載の方法。
【請求項10】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項8に記載の方法。
【請求項11】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項8に記載の方法。
【請求項12】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項8に記載の方法。
【請求項13】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、患者において、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされる神経毒性傷害を抑制する方法。
【請求項14】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項13に記載の方法。
【請求項15】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項13に記載の方法。
【請求項16】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項15に記載の方法。
【請求項17】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項15に記載の方法。
【請求項18】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項15に記載の方法。
【請求項19】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項15に記載の方法。
【請求項20】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、哺乳類の生存期間を延ばすための化合物。
【請求項21】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項20に記載の化合物。
【請求項22】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばすための方法。
【請求項23】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項22に記載の方法。
【請求項24】
前記組成物が、前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体(これらは、前記組成物中に治療的有効量で存在する)を含む、請求項22に記載の方法。
【請求項25】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内、筋肉内、皮下または経口投与される、請求項24に記載の方法。
【請求項26】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で静脈内、筋肉内または皮下投与される、請求項25に記載の方法。
【請求項27】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約3mg/kgの量で静脈内、筋肉内または皮下投与される、請求項26に記載の方法。
【請求項28】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項29】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約15mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項30】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、毎日投与される、請求項25に記載の方法。
【請求項31】
前記後生動物が、脊椎動物である、請求項25に記載の方法。
【請求項32】
前記後生動物が、哺乳類である、請求項31に記載の方法。
【請求項33】
前記後生動物が、ヒトである、請求項32に記載の方法。
【請求項34】
a)水とメタノールと溶媒中のC3メチルエステルとを混合して、初期溶液を作ること;
b)メタノール中の水酸化ナトリウムを前記初期溶液に添加すること;
c)水を添加して、水性層と非水性層を形成すること;
d)前記水性層を加熱すること;および
e)前記加熱水性層からHexa生成物を抽出すること
を含む、Hexaを合成するための方法。
【請求項35】
前記初期溶液を攪拌する、請求項34に記載の方法。
【請求項36】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項34に記載の方法。
【請求項37】
前記C3メチルエステルがもはや溶液状態でなくなるまで、前記初期溶液を水酸化ナトリウムとともに放置する、請求項34に記載の方法。
【請求項38】
前記水性層を加熱して、加水分解を完了させる、請求項34に記載の方法。
【請求項39】
前記水性層をN2下、約60℃で約2時間加熱して、加水分解を完了させる、請求項38に記載の方法。
【請求項40】
前記Hexaが、約94%より多い量で得られる、請求項34に記載の方法。
【請求項41】
前記非水性層をデカントして、一切の溶媒を除去する、請求項34に記載の方法。
【請求項42】
一切の残留溶媒を真空下でストリップする、請求項41に記載の方法。
【請求項43】
a)溶媒中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作ること;
b)水を添加して、水性層と非水性層を形成すること;
c)前記水性層と非水性層を分離すること;
d)前記水性層を冷却して、硫酸で酸性化し、酢酸エチルで抽出して、酢酸エチル抽出物を作ること;
d)前記酢酸エチル抽出物から一切の不純物を除去すること;および
e)洗浄した抽出物を蒸発および乾燥させて、Hexa、Penta−1およびPenta−2を含む固体生成物を作ること
を含む、HexaおよびPentaを合成するための方法。
【請求項44】
前記初期溶液をN2下で混合する、請求項43に記載の方法。
【請求項45】
前記酢酸エチル抽出物を水で洗浄することにより前記不純物を除去する、請求項43に記載の方法。
【請求項46】
洗浄した抽出物を真空下、室温で蒸発および乾燥させる、請求項43に記載の方法。
【請求項47】
前記初期溶液を約1時間混合する、請求項43に記載の方法。
【請求項48】
a)C60をマロン酸ジ−t−ブチルと反応させ、続いて、カラムクロマトグラフィーによりジ−t−ブチルマロニルC60とビス−t−ブチルマロニルC60を分離すること;
b)溶媒中のジ−t−ブチルマロニルC60またはビス−t−ブチルマロニルC60にブロモマロン酸エステルを添加し、続いて、第三アミンまたは水酸化ナトリウムを添加して、反応混合物を作ること;
c)溶媒中のシリカゲルのカラムに前記反応混合物を注入し、このカラムを溶媒で溶離すること;
d)このカラムを酢酸エチル/溶媒混合物で溶離して、C3画分を作ること;
e)C3画分を蒸発させ、次いで、溶媒を添加すること;
f)溶媒中のC3画分に酸を添加し、次いで、加熱すること;
g)追加の酸を添加すること;
h)熱を加えながら、生成物を放置して沈殿させること;
i)この沈殿から溶媒を除去すること;
j)水および酢酸エチルをこの沈殿に添加して、酢酸エチル溶液を作ること;
k)この酢酸エチル溶液を洗浄すること;
l)真空下でこの酢酸エチル溶液を蒸発させて、固体を得ること;
m)前記固体をアセトニトリル、水およびアセトンの混合物に溶解すること;
n)溶解した固体を加熱すること;
o)このアセトニトリル、水およびアセトン混合物からの沈殿を分離させること;
p)アセトンを添加して、このアセトニトリル、水およびアセトン混合物からの沈殿を溶解すること;
q)溶解したこのアセトニトリル、水およびアセトン混合物からの沈殿を加熱すること;
r)一切の揮発性溶媒を真空下で除去すること;
s)酢酸エチルを添加して、均質溶液を生じさせること;
t)前記均質溶液を真空下で蒸発させて、酢酸エチルから固体を得ること;
u)メタノールおよび水を含有する溶媒にこの酢酸エチルからの固体を溶解し、次いで、攪拌すること;
v)メタノール中の水酸化ナトリウムを添加すること;
w)水を添加して、水性層を形成すること;
x)加熱しながらこの水性層を分離し、次いで、この分離した水性層を冷却すること;
y)冷却した水性層に硫酸を添加すること;および
z)生成物を酢酸エチルで抽出すること
を含む、Penta PairまたはTetra Quartetを合成するための方法。
【請求項49】
前記ブロモマロン酸エステルが、ブロモマロン酸ジメチルである、請求項48に記載の方法。
【請求項50】
a)において第三アミンを利用する、請求項48に記載の方法。
【請求項51】
前記第三アミンが、DBUである、請求項50に記載の方法。
【請求項52】
前記酸が、トルエン混和性の酸である、請求項48に記載の方法。
【請求項53】
前記酸が、p−トルエンスルホン酸・一水和物、TFAおよびメタン硫酸から成る群より選択される、請求項48に記載の方法。
【請求項54】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項48に記載の方法。
【請求項55】
約0.5%、約1.0%および約2.0%の酢酸エチル/溶媒混合物で前記カラムを溶離する、請求項48に記載の方法。
【請求項56】
前記生成物が、Tetra Quartetを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:1:1である、請求項48に記載の方法。
【請求項57】
前記生成物が、Penta Pairを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:2:2である、請求項48に記載の方法。
【請求項58】
前記酢酸エチル溶液を洗浄して、前記酸を除去する、請求項48に記載の方法。
【請求項59】
前記酢酸エチル溶液を水で洗浄する、請求項58に記載の方法。
【請求項60】
前記水性層を約60℃から約110℃に加熱する、請求項48に記載の方法。
【請求項61】
添加した酸と共に前記C3画分を約88℃に加熱する、請求項48に記載の方法。
【請求項62】
前記アセトニトリル、水およびアセトンが、比率1:1:2である、請求項48に記載の方法。
【請求項63】
溶解した固体を約76.5℃で加熱する、請求項48に記載の方法。
【請求項64】
C3誘導体のサンプルを加熱してC3−ライトを作ることを含む、C3−ライトを生産するための定方向方法。
【請求項65】
前記C3誘導体のサンプルが、ニートであり、および約150℃の真空オーブンで加熱される、請求項64に記載の方法。
【請求項66】
前記サンプルを約1.5時間未満加熱する、請求項65に記載の方法。
【請求項67】
前記サンプルを約1.5時間より長く加熱する、請求項65に記載の方法。
【請求項68】
−30mmHgでの、請求項65に記載の方法。
【請求項69】
前記C3誘導体が、溶液状態である、請求項64に記載の方法。
【特許請求の範囲】
【請求項1】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体。
【請求項2】
前記化合物が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項1に記載のバックミンスターフラーレンの誘導体。
【請求項3】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeからなる群から独立して選択されるが、但し、全R1およびR2のうちの少なくとも1つは−Hおよび−COOMeから成る群より選択される)の化合物を含むバックミンスターフラーレンの三重アダクト。
【請求項4】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体を含む組成物のニューロン傷害を治療するための使用。
【請求項5】
前記誘導体が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項4に記載の使用。
【請求項6】
患者における神経毒性障害を治療する方法であって、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を患者に投与する(この場合、前記化合物は、組成物中に神経毒性傷害を治療するために有効な量で存在する)ことによる、方法。
【請求項7】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項6に記載の方法。
【請求項8】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項7に記載の方法。
【請求項9】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項8に記載の方法。
【請求項10】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項8に記載の方法。
【請求項11】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項8に記載の方法。
【請求項12】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項8に記載の方法。
【請求項13】
患者における神経毒性障害を抑制する方法であって、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、ここで前記障害は、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされるものである、方法。
【請求項14】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項13に記載の方法。
【請求項15】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項13に記載の方法。
【請求項16】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項15に記載の方法。
【請求項17】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項15に記載の方法。
【請求項18】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項15に記載の方法。
【請求項19】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項15に記載の方法。
【請求項20】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体を含む組成物の、哺乳類の生存期間を延ばすための使用。
【請求項21】
前記バックミンスターフラーレン誘導体が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項20に記載の使用。
【請求項22】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばすための方法。
【請求項23】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項22に記載の方法。
【請求項24】
前記組成物が、前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体(これらは、前記組成物中に治療的有効量で存在する)を含む、請求項22に記載の方法。
【請求項25】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内、筋肉内、皮下または経口投与される、請求項24に記載の方法。
【請求項26】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で静脈内、筋肉内または皮下投与される、請求項25に記載の方法。
【請求項27】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約3mg/kgの量で静脈内、筋肉内または皮下投与される、請求項26に記載の方法。
【請求項28】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項29】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約15mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項30】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、毎日投与される、請求項25に記載の方法。
【請求項31】
前記後生動物が、脊椎動物である、請求項25に記載の方法。
【請求項32】
前記後生動物が、哺乳類である、請求項31に記載の方法。
【請求項33】
前記後生動物が、ヒトである、請求項32に記載の方法。
【請求項34】
a)水とメタノールと溶媒中のC3メチルエステルとを混合して、初期溶液を作ること;
b)メタノール中の水酸化ナトリウムを前記初期溶液に添加すること;
c)水を添加して、水性層と非水性層を形成すること;
d)前記水性層を加熱すること;および
e)前記加熱水性層からC3(C60(C(COOH)2)3)生成物を抽出すること
を含む、C3(C60(C(COOH)2)3)を合成するための方法。
【請求項35】
前記初期溶液を攪拌する、請求項34に記載の方法。
【請求項36】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項34に記載の方法。
【請求項37】
前記C3メチルエステルが溶液中になくなるまで、水酸化ナトリウムを含む初期溶液を放置する、請求項34に記載の方法。
【請求項38】
前記水性層を加熱して、加水分解を完了させる、請求項34に記載の方法。
【請求項39】
前記水性層をN2下、約60℃で約2時間加熱して、加水分解を完了させる、請求項38に記載の方法。
【請求項40】
前記Hexaが、約94%より多い量で得られる、請求項34に記載の方法。
【請求項41】
前記非水性層をデカントして、一切の溶媒を除去する、請求項34に記載の方法。
【請求項42】
一切の残留溶媒を真空下でストリップする、請求項41に記載の方法。
【請求項43】
a)溶媒中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作ること;
b)水を添加して、水性層と非水性層を形成すること;
c)前記水性層と非水性層を分離すること;
d)前記水性層を冷却して、硫酸で酸性化し、酢酸エチルで抽出して、酢酸エチル抽出物を作ること;
d)前記酢酸エチル抽出物から一切の不純物を除去すること;および
e)洗浄した抽出物を蒸発および乾燥させて、C3(C60(C(COOH)2)3)、C60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を含む固体生成物を作ること
を含む、C3(C60(C(COOH)2)3)およびC60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を合成するための方法。
【請求項44】
前記初期溶液をN2下で混合する、請求項43に記載の方法。
【請求項45】
前記酢酸エチル抽出物を水で洗浄することにより前記不純物を除去する、請求項43に記載の方法。
【請求項46】
洗浄した抽出物を真空下、室温で蒸発および乾燥させる、請求項43に記載の方法。
【請求項47】
前記初期溶液を約1時間混合する、請求項43に記載の方法。
【請求項48】
a)C60をマロン酸ジ−t−ブチルと反応させ、続いて、カラムクロマトグラフィーによりジ−t−ブチルマロニルC60とビス−t−ブチルマロニルC60を分離すること;
b)溶媒中のジ−t−ブチルマロニルC60またはビス−t−ブチルマロニルC60にブロモマロン酸エステルを添加し、続いて、第三級アミンまたは水酸化ナトリウムを添加して、反応混合物を作ること;
c)溶媒中のシリカゲルのカラムに前記反応混合物を注入し、このカラムを溶媒で溶離すること;
d)このカラムを酢酸エチル/溶媒混合物で溶離して、C3画分を作ること;
e)C3画分を蒸発させ、次いで、溶媒を添加すること;
f)溶媒中のC3画分に酸を添加し、次いで、加熱すること;
g)追加の酸を添加すること;
h)熱を加えながら、生成物を沈殿させること;
i)この沈殿物から溶媒を除去すること;
j)水および酢酸エチルをこの沈殿物に添加して、酢酸エチル溶液を作ること;
k)この酢酸エチル溶液を洗浄すること;
l)真空下でこの酢酸エチル溶液を蒸発させて、固体を得ること;
m)前記固体をアセトニトリル、水およびアセトンの混合物に溶解すること;
n)溶解した固体を加熱すること;
o)このアセトニトリル、水およびアセトン混合物から沈殿物を分離させること;
p)アセトンを添加して、このアセトニトリル、水およびアセトン混合物からの沈殿物を溶解すること;
q)溶解したこのアセトニトリル、水およびアセトン混合物からの沈殿物を加熱すること;
r)一切の揮発性溶媒を真空下で除去すること;
s)酢酸エチルを添加して、均質溶液を生じさせること;
t)前記均質溶液を真空下で蒸発させて、酢酸エチルから固体を得ること;
u)メタノールおよび水を含有する溶媒にこの酢酸エチルからの固体を溶解し、次いで、攪拌すること;
v)メタノール中の水酸化ナトリウムを添加すること;
w)水を添加して、水性層を形成すること;
x)加熱しながらこの水性層を分離し、次いで、この分離した水性層を冷却すること;
y)冷却した水性層に硫酸を添加すること;および
z)生成物を酢酸エチルで抽出すること
を含む、C60(C(COOH)2)(C(CHCOOH))2又はC60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を合成するための方法。
【請求項49】
前記ブロモマロン酸エステルが、ブロモマロン酸ジメチルである、請求項48に記載の方法。
【請求項50】
a)において第三級アミンを利用する、請求項48に記載の方法。
【請求項51】
前記第三級アミンが、DBUである、請求項50に記載の方法。
【請求項52】
前記酸が、トルエン混和性の酸である、請求項48に記載の方法。
【請求項53】
前記酸が、p−トルエンスルホン酸・一水和物、TFAおよびメタン硫酸から成る群より選択される、請求項48に記載の方法。
【請求項54】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項48に記載の方法。
【請求項55】
約0.5%、約1.0%および約2.0%の酢酸エチル/溶媒混合物で前記カラムを溶離する、請求項48に記載の方法。
【請求項56】
前記生成物が、Tetra Quartetを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:1:1である、請求項48に記載の方法。
【請求項57】
前記生成物が、Penta Pairを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:2:2である、請求項48に記載の方法。
【請求項58】
前記酢酸エチル溶液を洗浄して、前記酸を除去する、請求項48に記載の方法。
【請求項59】
前記酢酸エチル溶液を水で洗浄する、請求項58に記載の方法。
【請求項60】
前記水性層を約60℃から約110℃に加熱する、請求項48に記載の方法。
【請求項61】
添加した酸と共に前記C3画分を約88℃に加熱する、請求項48に記載の方法。
【請求項62】
前記アセトニトリル、水およびアセトンが、比率1:1:2である、請求項48に記載の方法。
【請求項63】
溶解した固体を約76.5℃で加熱する、請求項48に記載の方法。
【請求項64】
C3誘導体のサンプルを加熱してC3−ライトを作ることを含む、C3−ライトを生産するための示された方法。
【請求項65】
前記C3誘導体のサンプルが、ニートであり、および約150℃の真空オーブンで加熱される、請求項64に記載の方法。
【請求項66】
前記サンプルを約1.5時間未満加熱する、請求項65に記載の方法。
【請求項67】
前記サンプルを約1.5時間より長く加熱する、請求項65に記載の方法。
【請求項68】
−30mmHgで行う、請求項65に記載の方法。
【請求項69】
前記C3誘導体が、溶液中にある、請求項64に記載の方法。
【請求項1】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト。
【請求項2】
前記化合物が、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項1に記載のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト。
【請求項3】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは−Hおよび−COOMeから成る群より選択されることを条件とする)のバックミンスターフラーレンの三重アダクト。
【請求項4】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、ニューロン傷害の治療用化合物。
【請求項5】
Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項4に記載の化合物。
【請求項6】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)の化合物、この医薬適合性の塩および医薬適合性のエステルを含むバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトならびに医薬適合性担体を患者に投与することにより(この場合、前記化合物は、前記組成物中に神経毒性傷害を治療するために有効な量で存在する)、神経毒性傷害を前記傷害に罹患している患者において治療する方法。
【請求項7】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項6に記載の方法。
【請求項8】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項7に記載の方法。
【請求項9】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項8に記載の方法。
【請求項10】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項8に記載の方法。
【請求項11】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項8に記載の方法。
【請求項12】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項8に記載の方法。
【請求項13】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、患者において、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされる神経毒性傷害を抑制する方法。
【請求項14】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項13に記載の方法。
【請求項15】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項13に記載の方法。
【請求項16】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項15に記載の方法。
【請求項17】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項15に記載の方法。
【請求項18】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項15に記載の方法。
【請求項19】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項15に記載の方法。
【請求項20】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む、哺乳類の生存期間を延ばすための化合物。
【請求項21】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項20に記載の化合物。
【請求項22】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、R1(複数)およびR2(複数)のうちの少なくとも1つは水素であることを条件とする)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばすための方法。
【請求項23】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項22に記載の方法。
【請求項24】
前記組成物が、前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体(これらは、前記組成物中に治療的有効量で存在する)を含む、請求項22に記載の方法。
【請求項25】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内、筋肉内、皮下または経口投与される、請求項24に記載の方法。
【請求項26】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で静脈内、筋肉内または皮下投与される、請求項25に記載の方法。
【請求項27】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約3mg/kgの量で静脈内、筋肉内または皮下投与される、請求項26に記載の方法。
【請求項28】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項29】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約15mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項30】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、毎日投与される、請求項25に記載の方法。
【請求項31】
前記後生動物が、脊椎動物である、請求項25に記載の方法。
【請求項32】
前記後生動物が、哺乳類である、請求項31に記載の方法。
【請求項33】
前記後生動物が、ヒトである、請求項32に記載の方法。
【請求項34】
a)水とメタノールと溶媒中のC3メチルエステルとを混合して、初期溶液を作ること;
b)メタノール中の水酸化ナトリウムを前記初期溶液に添加すること;
c)水を添加して、水性層と非水性層を形成すること;
d)前記水性層を加熱すること;および
e)前記加熱水性層からHexa生成物を抽出すること
を含む、Hexaを合成するための方法。
【請求項35】
前記初期溶液を攪拌する、請求項34に記載の方法。
【請求項36】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項34に記載の方法。
【請求項37】
前記C3メチルエステルがもはや溶液状態でなくなるまで、前記初期溶液を水酸化ナトリウムとともに放置する、請求項34に記載の方法。
【請求項38】
前記水性層を加熱して、加水分解を完了させる、請求項34に記載の方法。
【請求項39】
前記水性層をN2下、約60℃で約2時間加熱して、加水分解を完了させる、請求項38に記載の方法。
【請求項40】
前記Hexaが、約94%より多い量で得られる、請求項34に記載の方法。
【請求項41】
前記非水性層をデカントして、一切の溶媒を除去する、請求項34に記載の方法。
【請求項42】
一切の残留溶媒を真空下でストリップする、請求項41に記載の方法。
【請求項43】
a)溶媒中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作ること;
b)水を添加して、水性層と非水性層を形成すること;
c)前記水性層と非水性層を分離すること;
d)前記水性層を冷却して、硫酸で酸性化し、酢酸エチルで抽出して、酢酸エチル抽出物を作ること;
d)前記酢酸エチル抽出物から一切の不純物を除去すること;および
e)洗浄した抽出物を蒸発および乾燥させて、Hexa、Penta−1およびPenta−2を含む固体生成物を作ること
を含む、HexaおよびPentaを合成するための方法。
【請求項44】
前記初期溶液をN2下で混合する、請求項43に記載の方法。
【請求項45】
前記酢酸エチル抽出物を水で洗浄することにより前記不純物を除去する、請求項43に記載の方法。
【請求項46】
洗浄した抽出物を真空下、室温で蒸発および乾燥させる、請求項43に記載の方法。
【請求項47】
前記初期溶液を約1時間混合する、請求項43に記載の方法。
【請求項48】
a)C60をマロン酸ジ−t−ブチルと反応させ、続いて、カラムクロマトグラフィーによりジ−t−ブチルマロニルC60とビス−t−ブチルマロニルC60を分離すること;
b)溶媒中のジ−t−ブチルマロニルC60またはビス−t−ブチルマロニルC60にブロモマロン酸エステルを添加し、続いて、第三アミンまたは水酸化ナトリウムを添加して、反応混合物を作ること;
c)溶媒中のシリカゲルのカラムに前記反応混合物を注入し、このカラムを溶媒で溶離すること;
d)このカラムを酢酸エチル/溶媒混合物で溶離して、C3画分を作ること;
e)C3画分を蒸発させ、次いで、溶媒を添加すること;
f)溶媒中のC3画分に酸を添加し、次いで、加熱すること;
g)追加の酸を添加すること;
h)熱を加えながら、生成物を放置して沈殿させること;
i)この沈殿から溶媒を除去すること;
j)水および酢酸エチルをこの沈殿に添加して、酢酸エチル溶液を作ること;
k)この酢酸エチル溶液を洗浄すること;
l)真空下でこの酢酸エチル溶液を蒸発させて、固体を得ること;
m)前記固体をアセトニトリル、水およびアセトンの混合物に溶解すること;
n)溶解した固体を加熱すること;
o)このアセトニトリル、水およびアセトン混合物からの沈殿を分離させること;
p)アセトンを添加して、このアセトニトリル、水およびアセトン混合物からの沈殿を溶解すること;
q)溶解したこのアセトニトリル、水およびアセトン混合物からの沈殿を加熱すること;
r)一切の揮発性溶媒を真空下で除去すること;
s)酢酸エチルを添加して、均質溶液を生じさせること;
t)前記均質溶液を真空下で蒸発させて、酢酸エチルから固体を得ること;
u)メタノールおよび水を含有する溶媒にこの酢酸エチルからの固体を溶解し、次いで、攪拌すること;
v)メタノール中の水酸化ナトリウムを添加すること;
w)水を添加して、水性層を形成すること;
x)加熱しながらこの水性層を分離し、次いで、この分離した水性層を冷却すること;
y)冷却した水性層に硫酸を添加すること;および
z)生成物を酢酸エチルで抽出すること
を含む、Penta PairまたはTetra Quartetを合成するための方法。
【請求項49】
前記ブロモマロン酸エステルが、ブロモマロン酸ジメチルである、請求項48に記載の方法。
【請求項50】
a)において第三アミンを利用する、請求項48に記載の方法。
【請求項51】
前記第三アミンが、DBUである、請求項50に記載の方法。
【請求項52】
前記酸が、トルエン混和性の酸である、請求項48に記載の方法。
【請求項53】
前記酸が、p−トルエンスルホン酸・一水和物、TFAおよびメタン硫酸から成る群より選択される、請求項48に記載の方法。
【請求項54】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項48に記載の方法。
【請求項55】
約0.5%、約1.0%および約2.0%の酢酸エチル/溶媒混合物で前記カラムを溶離する、請求項48に記載の方法。
【請求項56】
前記生成物が、Tetra Quartetを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:1:1である、請求項48に記載の方法。
【請求項57】
前記生成物が、Penta Pairを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:2:2である、請求項48に記載の方法。
【請求項58】
前記酢酸エチル溶液を洗浄して、前記酸を除去する、請求項48に記載の方法。
【請求項59】
前記酢酸エチル溶液を水で洗浄する、請求項58に記載の方法。
【請求項60】
前記水性層を約60℃から約110℃に加熱する、請求項48に記載の方法。
【請求項61】
添加した酸と共に前記C3画分を約88℃に加熱する、請求項48に記載の方法。
【請求項62】
前記アセトニトリル、水およびアセトンが、比率1:1:2である、請求項48に記載の方法。
【請求項63】
溶解した固体を約76.5℃で加熱する、請求項48に記載の方法。
【請求項64】
C3誘導体のサンプルを加熱してC3−ライトを作ることを含む、C3−ライトを生産するための定方向方法。
【請求項65】
前記C3誘導体のサンプルが、ニートであり、および約150℃の真空オーブンで加熱される、請求項64に記載の方法。
【請求項66】
前記サンプルを約1.5時間未満加熱する、請求項65に記載の方法。
【請求項67】
前記サンプルを約1.5時間より長く加熱する、請求項65に記載の方法。
【請求項68】
−30mmHgでの、請求項65に記載の方法。
【請求項69】
前記C3誘導体が、溶液状態である、請求項64に記載の方法。
【特許請求の範囲】
【請求項1】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体。
【請求項2】
前記化合物が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項1に記載のバックミンスターフラーレンの誘導体。
【請求項3】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−H、−COOHおよび−COOMeからなる群から独立して選択されるが、但し、全R1およびR2のうちの少なくとも1つは−Hおよび−COOMeから成る群より選択される)の化合物を含むバックミンスターフラーレンの三重アダクト。
【請求項4】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体を含む組成物のニューロン傷害を治療するための使用。
【請求項5】
前記誘導体が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項4に記載の使用。
【請求項6】
患者における神経毒性障害を治療する方法であって、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を患者に投与する(この場合、前記化合物は、組成物中に神経毒性傷害を治療するために有効な量で存在する)ことによる、方法。
【請求項7】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項6に記載の方法。
【請求項8】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項7に記載の方法。
【請求項9】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項8に記載の方法。
【請求項10】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項8に記載の方法。
【請求項11】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項8に記載の方法。
【請求項12】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項8に記載の方法。
【請求項13】
患者における神経毒性障害を抑制する方法であって、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHから独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルを含む化合物ならびに医薬適合性担体を含む組成物(この場合、前記化合物は、前記組成物中に神経毒性傷害を抑制するために充分な量で存在する)を患者に投与することにより、ここで前記障害は、ニューロンのNMDA受容体のグルタメートによる刺激に起因してニューロンが放出するフリーラジカル酸素種によって引き起こされるものである、方法。
【請求項14】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項13に記載の方法。
【請求項15】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約1.5mg/kgから約1500mg/kgの量で投与される、請求項13に記載の方法。
【請求項16】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、1日に約10mg/kgから約60mg/kgの量で投与される、請求項15に記載の方法。
【請求項17】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、経口投与される、請求項15に記載の方法。
【請求項18】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内投与される、請求項15に記載の方法。
【請求項19】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、C3−ライトである、請求項15に記載の方法。
【請求項20】
バックミンスターフラーレン・e,e,eマロン酸三重アダクト、バックミンスターフラーレン・e,e,e酢酸三重アダクト、またはバックミンスターフラーレン・e,e,eマロン酸三重アダクトとバックミンスターフラーレン・e,e,e酢酸三重アダクトの混合物である、一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、全R1とR2のうちの少なくとも1つは水素であり、全R1とR2のうちの少なくとも1つは、COOHまたはこのエステルである)のバックミンスターフラーレンの誘導体を含む組成物の、哺乳類の生存期間を延ばすための使用。
【請求項21】
前記バックミンスターフラーレン誘導体が、C60(C(COOH)2)2(C(CHCOOH))、C60(C(COOH)2)(C(CHCOOH))2、およびC60(CHCOOH)3から成る群より選択される、請求項20に記載の使用。
【請求項22】
一般式C60R3(式中、各Rは、式−CR1R2の基から独立して選択され、この場合の各R1およびR2は、−Hおよび−COOHからなる群から独立して選択されるが、但し、R1(複数)およびR2(複数)のうちの少なくとも1つは水素である)のバックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトを含む組成物を後生動物に投与することを含む、後生動物または後生動物細胞の寿命を延ばすための方法。
【請求項23】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、Penta Pair、Tetra QuartetおよびC3−ライトから成る群より選択される、請求項22に記載の方法。
【請求項24】
前記組成物が、前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクト、この医薬適合性の塩および医薬適合性のエステルならびに医薬適合性担体(これらは、前記組成物中に治療的有効量で存在する)を含む、請求項22に記載の方法。
【請求項25】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、静脈内、筋肉内、皮下または経口投与される、請求項24に記載の方法。
【請求項26】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で静脈内、筋肉内または皮下投与される、請求項25に記載の方法。
【請求項27】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約3mg/kgの量で静脈内、筋肉内または皮下投与される、請求項26に記載の方法。
【請求項28】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、少なくとも0.1mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項29】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、約15mg/kgの量で経口投与される、請求項25に記載の方法。
【請求項30】
前記バックミンスターフラーレン・e,e,eマロン酸/酢酸三重アダクトが、毎日投与される、請求項25に記載の方法。
【請求項31】
前記後生動物が、脊椎動物である、請求項25に記載の方法。
【請求項32】
前記後生動物が、哺乳類である、請求項31に記載の方法。
【請求項33】
前記後生動物が、ヒトである、請求項32に記載の方法。
【請求項34】
a)水とメタノールと溶媒中のC3メチルエステルとを混合して、初期溶液を作ること;
b)メタノール中の水酸化ナトリウムを前記初期溶液に添加すること;
c)水を添加して、水性層と非水性層を形成すること;
d)前記水性層を加熱すること;および
e)前記加熱水性層からC3(C60(C(COOH)2)3)生成物を抽出すること
を含む、C3(C60(C(COOH)2)3)を合成するための方法。
【請求項35】
前記初期溶液を攪拌する、請求項34に記載の方法。
【請求項36】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項34に記載の方法。
【請求項37】
前記C3メチルエステルが溶液中になくなるまで、水酸化ナトリウムを含む初期溶液を放置する、請求項34に記載の方法。
【請求項38】
前記水性層を加熱して、加水分解を完了させる、請求項34に記載の方法。
【請求項39】
前記水性層をN2下、約60℃で約2時間加熱して、加水分解を完了させる、請求項38に記載の方法。
【請求項40】
前記Hexaが、約94%より多い量で得られる、請求項34に記載の方法。
【請求項41】
前記非水性層をデカントして、一切の溶媒を除去する、請求項34に記載の方法。
【請求項42】
一切の残留溶媒を真空下でストリップする、請求項41に記載の方法。
【請求項43】
a)溶媒中のe,e,e−トリスジメチルマロニルフラーレン(C3エステル)の溶液にナトリウムメトキシドまたは水酸化ナトリウムを添加して、初期溶液を作ること;
b)水を添加して、水性層と非水性層を形成すること;
c)前記水性層と非水性層を分離すること;
d)前記水性層を冷却して、硫酸で酸性化し、酢酸エチルで抽出して、酢酸エチル抽出物を作ること;
d)前記酢酸エチル抽出物から一切の不純物を除去すること;および
e)洗浄した抽出物を蒸発および乾燥させて、C3(C60(C(COOH)2)3)、C60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を含む固体生成物を作ること
を含む、C3(C60(C(COOH)2)3)およびC60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を合成するための方法。
【請求項44】
前記初期溶液をN2下で混合する、請求項43に記載の方法。
【請求項45】
前記酢酸エチル抽出物を水で洗浄することにより前記不純物を除去する、請求項43に記載の方法。
【請求項46】
洗浄した抽出物を真空下、室温で蒸発および乾燥させる、請求項43に記載の方法。
【請求項47】
前記初期溶液を約1時間混合する、請求項43に記載の方法。
【請求項48】
a)C60をマロン酸ジ−t−ブチルと反応させ、続いて、カラムクロマトグラフィーによりジ−t−ブチルマロニルC60とビス−t−ブチルマロニルC60を分離すること;
b)溶媒中のジ−t−ブチルマロニルC60またはビス−t−ブチルマロニルC60にブロモマロン酸エステルを添加し、続いて、第三級アミンまたは水酸化ナトリウムを添加して、反応混合物を作ること;
c)溶媒中のシリカゲルのカラムに前記反応混合物を注入し、このカラムを溶媒で溶離すること;
d)このカラムを酢酸エチル/溶媒混合物で溶離して、C3画分を作ること;
e)C3画分を蒸発させ、次いで、溶媒を添加すること;
f)溶媒中のC3画分に酸を添加し、次いで、加熱すること;
g)追加の酸を添加すること;
h)熱を加えながら、生成物を沈殿させること;
i)この沈殿物から溶媒を除去すること;
j)水および酢酸エチルをこの沈殿物に添加して、酢酸エチル溶液を作ること;
k)この酢酸エチル溶液を洗浄すること;
l)真空下でこの酢酸エチル溶液を蒸発させて、固体を得ること;
m)前記固体をアセトニトリル、水およびアセトンの混合物に溶解すること;
n)溶解した固体を加熱すること;
o)このアセトニトリル、水およびアセトン混合物から沈殿物を分離させること;
p)アセトンを添加して、このアセトニトリル、水およびアセトン混合物からの沈殿物を溶解すること;
q)溶解したこのアセトニトリル、水およびアセトン混合物からの沈殿物を加熱すること;
r)一切の揮発性溶媒を真空下で除去すること;
s)酢酸エチルを添加して、均質溶液を生じさせること;
t)前記均質溶液を真空下で蒸発させて、酢酸エチルから固体を得ること;
u)メタノールおよび水を含有する溶媒にこの酢酸エチルからの固体を溶解し、次いで、攪拌すること;
v)メタノール中の水酸化ナトリウムを添加すること;
w)水を添加して、水性層を形成すること;
x)加熱しながらこの水性層を分離し、次いで、この分離した水性層を冷却すること;
y)冷却した水性層に硫酸を添加すること;および
z)生成物を酢酸エチルで抽出すること
を含む、C60(C(COOH)2)(C(CHCOOH))2又はC60(C(COOH)2)2(C(CHCOOH))またはこの立体異性体を合成するための方法。
【請求項49】
前記ブロモマロン酸エステルが、ブロモマロン酸ジメチルである、請求項48に記載の方法。
【請求項50】
a)において第三級アミンを利用する、請求項48に記載の方法。
【請求項51】
前記第三級アミンが、DBUである、請求項50に記載の方法。
【請求項52】
前記酸が、トルエン混和性の酸である、請求項48に記載の方法。
【請求項53】
前記酸が、p−トルエンスルホン酸・一水和物、TFAおよびメタン硫酸から成る群より選択される、請求項48に記載の方法。
【請求項54】
前記溶媒が、トルエンおよびトルエン中の酢酸t−ブチルから成る群より選択される、請求項48に記載の方法。
【請求項55】
約0.5%、約1.0%および約2.0%の酢酸エチル/溶媒混合物で前記カラムを溶離する、請求項48に記載の方法。
【請求項56】
前記生成物が、Tetra Quartetを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:1:1である、請求項48に記載の方法。
【請求項57】
前記生成物が、Penta Pairを含み、前記ブロモマロン酸エステル、ジ−t−ブチルマロニルC60および溶媒が、比率1:2:2である、請求項48に記載の方法。
【請求項58】
前記酢酸エチル溶液を洗浄して、前記酸を除去する、請求項48に記載の方法。
【請求項59】
前記酢酸エチル溶液を水で洗浄する、請求項58に記載の方法。
【請求項60】
前記水性層を約60℃から約110℃に加熱する、請求項48に記載の方法。
【請求項61】
添加した酸と共に前記C3画分を約88℃に加熱する、請求項48に記載の方法。
【請求項62】
前記アセトニトリル、水およびアセトンが、比率1:1:2である、請求項48に記載の方法。
【請求項63】
溶解した固体を約76.5℃で加熱する、請求項48に記載の方法。
【請求項64】
C3誘導体のサンプルを加熱してC3−ライトを作ることを含む、C3−ライトを生産するための示された方法。
【請求項65】
前記C3誘導体のサンプルが、ニートであり、および約150℃の真空オーブンで加熱される、請求項64に記載の方法。
【請求項66】
前記サンプルを約1.5時間未満加熱する、請求項65に記載の方法。
【請求項67】
前記サンプルを約1.5時間より長く加熱する、請求項65に記載の方法。
【請求項68】
−30mmHgで行う、請求項65に記載の方法。
【請求項69】
前記C3誘導体が、溶液中にある、請求項64に記載の方法。
【図1A】

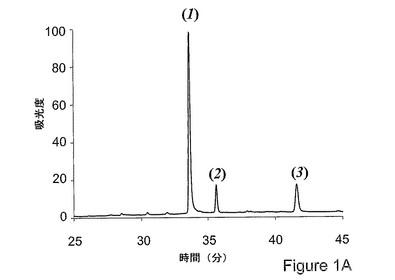
【図1B】
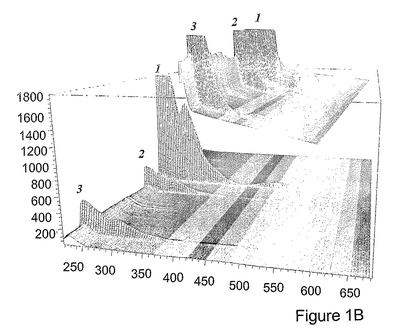
【図1C】
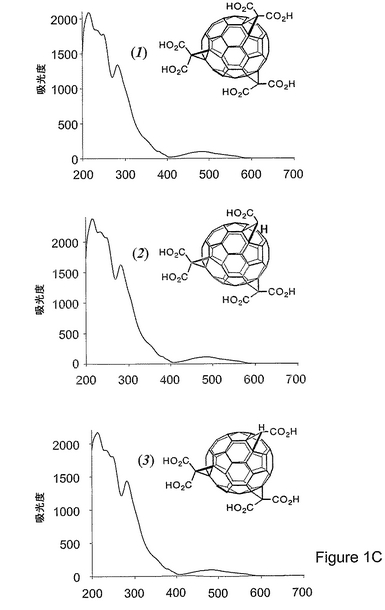
【図1D】
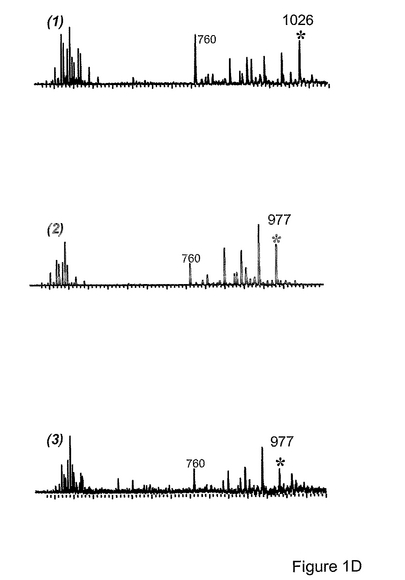
【図2】
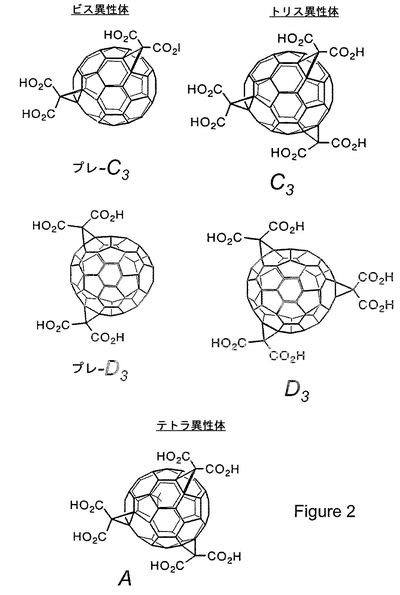
【図3】
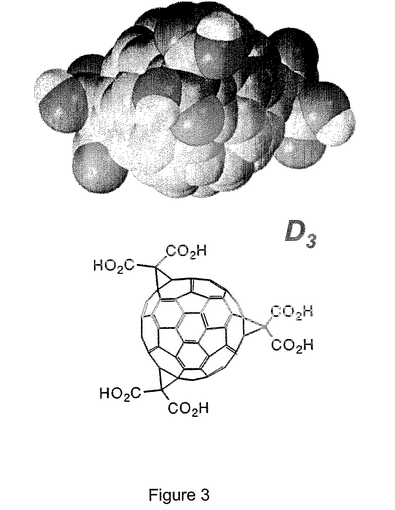
【図4】
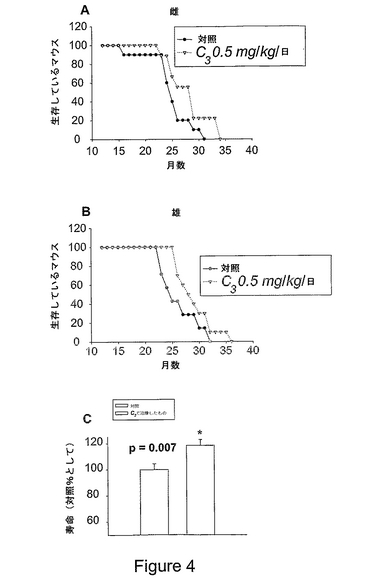
【図5】
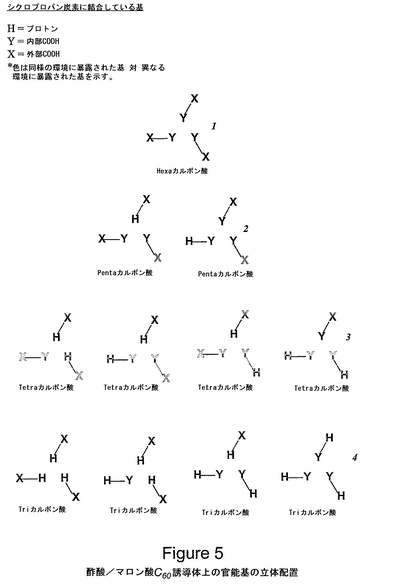
【図6】
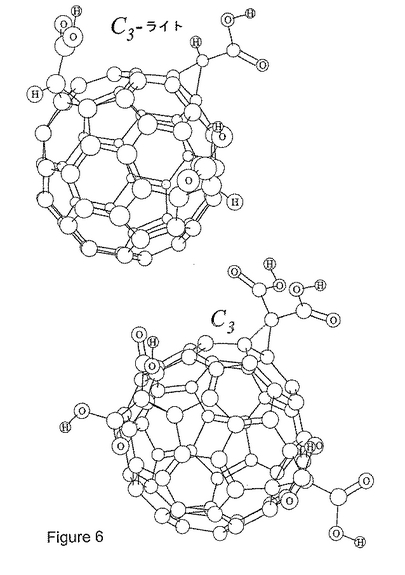
【図7】
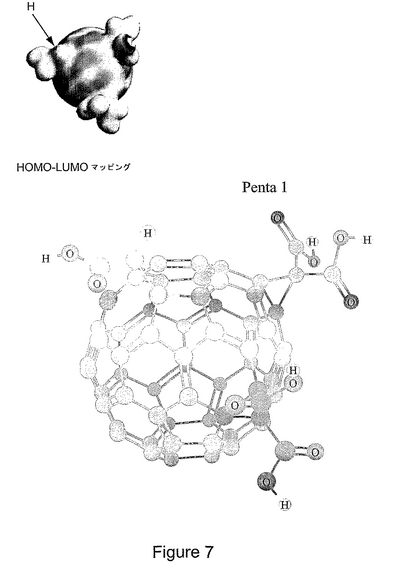
【図8】
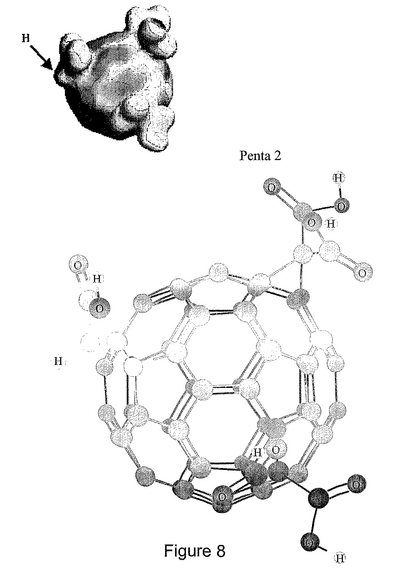
【図9】
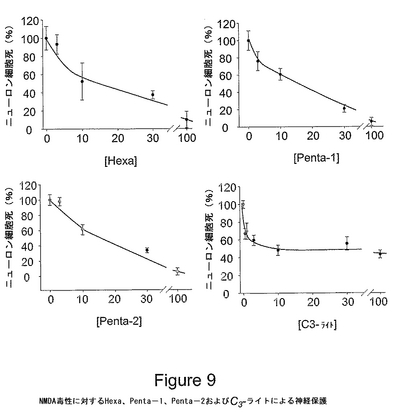
【図10】
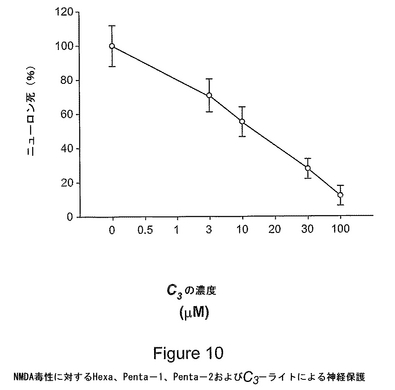
【図11】
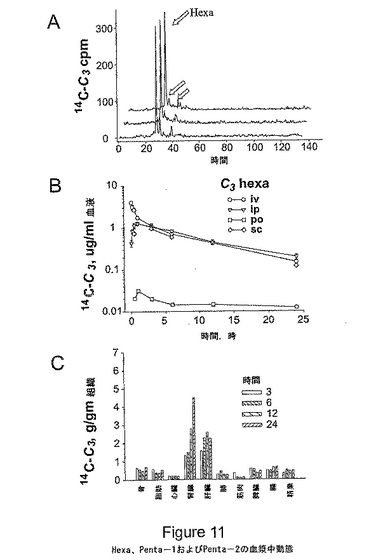
【図12】
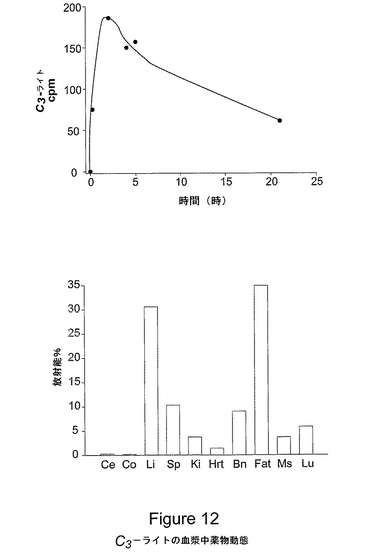
【図1B】
【図1C】
【図1D】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2006−518760(P2006−518760A)
【公表日】平成18年8月17日(2006.8.17)
【国際特許分類】
【出願番号】特願2006−503837(P2006−503837)
【出願日】平成16年2月24日(2004.2.24)
【国際出願番号】PCT/US2004/005442
【国際公開番号】WO2004/076349
【国際公開日】平成16年9月10日(2004.9.10)
【出願人】(500204278)ワシントン ユニヴァーシティー (14)
【Fターム(参考)】
【公表日】平成18年8月17日(2006.8.17)
【国際特許分類】
【出願日】平成16年2月24日(2004.2.24)
【国際出願番号】PCT/US2004/005442
【国際公開番号】WO2004/076349
【国際公開日】平成16年9月10日(2004.9.10)
【出願人】(500204278)ワシントン ユニヴァーシティー (14)
【Fターム(参考)】
[ Back to top ]