
活性成分を含有するカチオンリポソームの投与方法
【課題】パクリタキセルの、これを必要とする被験体への治療有効量での、深刻な副作用のない投与方法の提供。
【解決手段】少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤。
【解決手段】少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明の詳細な説明
本発明は、必要とするヒト患者への投与のための、パクリタキセルを含有する医薬組成物の使用に関する。
【背景技術】
【0002】
増強した有糸分裂に関連する疾病に苦しむヒト患者のための治療薬としての、有糸分裂阻害剤、例えばタキサンの使用は公知技術において公知である。
【0003】
パクリタキセルは、独特の作用機構及び広い範囲の抗増殖性活性を有し、これはパクリタキセルが微小管に結合し、チューブリン重合を促進し、この構築された微小管を安定化するためである。この結果、パクリタキセルは細胞周期を前期でブロックし、G2/M期での細胞の蓄積を生じる。
【0004】
残念ながら、パクリタキセルは水中での極めて低い溶解性を有し、これにより適した剤形を提供することが困難になる。最近では、パクリタキセルはクレモホールEL(Cremophor EL)(ポリエトキシル化されたひまし油)及びエタノールを50:50(vol/vol)の比で含有する媒体中で処方され、投与される。前記溶液を1:10で食塩水中で、ヒトに投与する前に希釈する。しかし、様々な深刻な副作用、例えば過敏反応及び高血圧性反応、腎毒性及び神経毒性などが、クレモホールEL処方のせいで患者で報告されてきた。
【0005】
更に、パクリタキセルは(その他の抗腫瘍剤のうちで)、有効な、確立した標準的な抗腫瘍剤({Rowinsky, 1995#1},{Awada, 2002#2},{Seidman, 2003#3},{Romanini, 2003#4})ではあるが、薬剤不応答性の腫瘍及び転移がしばしば癌患者で観察される({Blom, 1996#5},{Modi, 2002#6},{Ozols, 2003#7})。遺伝的に不安定な、急速に分裂する腫瘍細胞は、この選択された抗癌剤の成長阻害効果を克服する能力を獲得する({Vogelstein,1988#8},{Kerbel,1991 #9})。前記能力は通常は単独の薬剤(二次治療)に限定されずに、この最初の耐性の発生後に使用されるその他の薬剤にまで及ぶ。従って、この現象は多剤耐性(MDR)と呼ばれる。利用可能でかつ承認された抗腫瘍剤の数が多数の癌の種類に関わらず非常に限定されているために、多数の患者は、彼らの癌組織がMDRを示し屈する。この明白な問題は従って、既にそれぞれの薬剤に対して耐性のある、薬剤耐性腫瘍、特に薬剤耐性細胞を致死するための方法及び手段を見出すことである。
【0006】
いくつかのアプローチが、上述の問題を取り扱うために試みられた。従来の戦略は、最大耐量(MTD)にまで用量を上昇させ、かつ全ての腫瘍細胞を可能な限り素早くかつ完全に根絶することを試みることである({Schuenemann, 1999#10},{Heidemann, 1997#11})。前記戦略が深刻な副作用を引き起こし、より長期間へと延長可能でないことは明白である。従って、前記治療スケジュールは、MTDでの1つの短い治療期間(通常1日間〜1週間)と、患者を不可避な副作用から回復させるための数週間(通常3〜4週間)の治療なしの間隔との周期からなる({Schuenemann, 1999#10},{Heidemann, 1997#11},{Romanini, 2003#4})。多数の場合に、腫瘍成長はこの薬剤なしの期間の間に再開してもよい。最も重要なことに、腫瘍細胞が高レベルの耐性を発生し、これにより前記腫瘍細胞がMTDでの薬剤濃度を収容することが可能である多数の患者では前記アプローチは失敗する。この患者は、治療抵抗性になる。
【0007】
最も一般的な解決法は、治療を第2の薬剤で開始することである({Blom, 1996#5}, {Awada, 2002#2}, {Seidman, 2003#3}, {Heinemann, 2003#12}, {Thigpen, 2003#13})。最も良好な場合では、この二次治療は奏功し、かつこの患者は治癒する。一般的な経験ではしかしながら、腫瘍が一定の時間の間応答し、これにより前記腫瘍の一時的な退縮を生じるのみである。この後では腫瘍は前記の第2の薬剤に対しても耐性になってしまう。前記戦略を続行すると、全ての使用可能な抗癌剤に対して抵抗性である、多剤耐性腫瘍の発生を最終的に生じる({Blom, 1996#5},{Seidman, 2003#3},{Thigpen, 2003#13})。その他の可能性は、患者をすぐさま2以上の薬剤の併用により処置することである({Heinemann, 2003#12},{Kuenen, 2002#14},{Sledge, 2003#15},{Ozols, 2003#7},{Reck, 2003#17},{Romanini, 2003#4})。前記戦略はより成功する可能性があり、というのは二重の薬剤耐性の発生の見込みを減少させるからである。しかし前記戦略は、時間及び費用集中的に適した薬剤の組み合わせを調査する必要がある。第2の不利な点は、この副作用は上昇する可能性もあることである({Kuenen, 2002#14},{Ozols, 2003#7})。前記の治療窓口は同時に小さくなり、この毒性作用は前記のイメージした治療上の利点をカバーしてしまう可能性がある。この場合にも、多剤耐性が生じ、前記の治療は無効になる可能性がある({Zimpfer−Rechner,2003#18},{Sledge,2003#15},{Sledge,2003#16},{Ozols,2003 #7})。
【0008】
前記の従来の治療戦略でのネガティブな経験の結果は、上述の治療の選択肢を拡大するためのより多くの新規の薬剤を開発することである。明らかにこれは、結局は多くの場合で治療抵抗性の腫瘍を生じる、より有効な薬剤のための極めて時間及び費用集中的なレースである。近年ではこの認識により、腫瘍の耐性を回避するための新規のアプローチが導かれた。これは前記MDRが、細胞に化学療法剤を排出させる酵素の過剰発現により引き起こされるという仮定に基づく。前記酵素のカテゴリーのうち最も有名なメンバーは、p−糖タンパク質(p−gp)と呼ばれる。これは細胞質膜に存在し、ATPにより駆動された方法({Nobmann, 2001#19},{Thomas, 2003#20})で、パクリタキセル又はドキソルビシンなどの化合物を排出する({Harker, 1985#21},{Fellner, 2002#22},{Kiesewetter, 2003#23})。この概念は、p−gp媒介薬剤耐性を逆転させるためのp−gp阻害剤の開発を導いた。従って、前記の種類の分子に対して化学増感剤(chemosensitizer)との用語が造語される。試験した最初の例の1つは、ベラパミルである。臨床試験はしかしながら、ことによると低い比活性のせいである、不十分な結果を示した({Thomas, 2003#20}, {Kohler, 2003#24})。更なる研究により第2世代の化合物が生じ、これはやはり臨床的に適用可能でないことが見出された({Leonard, 2002#25},{Thomas, 2003#20})。今日、第3世代のいくつかの物質(1つはタリキダール(tariquidar)として公知である)が臨床試験にある({Agrawal, 2003#26},{Callies, 2003#27})。前記化合物の有用性及び広い適用性はしかし、未だ不明である({Leonard, 2002#25}, {Thomas, 2003#20})。第1世代の化学増感剤に比較してより改善してはいても、第3世代化合物もまた副作用を引き起こし、全身に対して予期せぬ結果を有する可能性がある。大規模な臨床試験が必要とされ、これまでのところ将来に前記アプローチが一般的な実施となるかは不明瞭である({Leonard, 2002#25},{Thomas, 2003#20})。
【0009】
様々なデリバリー系がパクリタキセルの効果を促進するため及び/又は毒性を減少させるために使用されてきた。リポソームは、水溶性、かくしてより低い毒性と組み合わさった効率を促進するために開発された多数のキャリヤーのうちの1つである。
【0010】
U.S.Pat.No.5,648,090,U.S.Pat.No.5,424,073及びU.S.Pat.No.6,146,659(Rahman et al.)は、哺乳類の癌の治療方法のために、リポソームによりカプセル化したパクリタキセルを提供する。前記特許はリポソームの治療有効量の医薬組成物をこのホストに投与する方法を開示し、前記医薬組成物はリポソーム形成材料、カルジオリピン、及びパクリタキセルのような薬剤、又はパクリタキセルの抗腫瘍性誘導体、又はこの混合物を、医薬的に許容可能な添加物と共に含む。U.S.Pat.No.6,146,659では、タキサンを一時間より短い時間にわたって75〜300mg/m2の量で投与する(その際、前記タキサンはリポソームによりカプセル化されている)ことによるタキサンの患者への投与方法が提供される。前記特許中で開示されたリポソームは負に帯電している。
【0011】
McDonald etal.,U.S.Pat.No.5,837,283,の開示から、正に帯電したリポソームは血管新生内皮細胞を特異的に標的とすることが公知である。ヒト患者に関する臨床データは提示されていない。
【発明の概要】
【発明が解決しようとする課題】
【0012】
従って、本発明の基礎をなす課題は、パクリタキセルの、これを必要とする被験体への治療有効量での、深刻な副作用のない投与方法を提供することである。特に、公知技術の製剤、例えばクレモホール製剤の適用におけるパクリタキセルの高い初期治療量を用いる必要性によって引き起こされる副作用の発生は、避けることが望ましい。
【課題を解決するための手段】
【0013】
本出願発明において、パクリタキセルを含有するカチオンリポソーム製剤の投与のヒトに関する臨床データが提示される。
【発明の効果】
【0014】
意外にも、活性成分であるパクリタキセルの高含量を有するカチオンリポソーム製剤の投与が、これを必要とするヒト患者の血管新生内皮細胞に選択的に影響を与えることが見出された。従って本発明は前記患者への、パクリタキセル約0.25〜約100mg、特に〜約60mg/前記患者体重kgの一月量での、カチオン脂質約30〜約98モル%、少なくとも約2モル%の量でパクリタキセル及び中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の投与を含む。
【0015】
本発明の更なる観点は、パクリタキセル約0.25〜約60mg/前記患者体重kgの一月量での、少なくとも1つのカチオン脂質約30モル%〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の、これを必要とするヒト患者への投与を含む。
【図面の簡単な説明】
【0016】
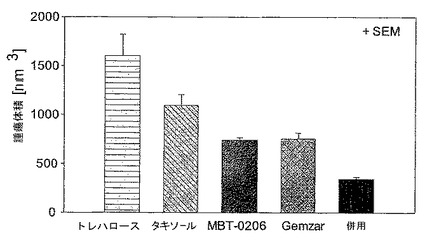
【図1】図1は、治療後の腫瘍体積を示す図である。
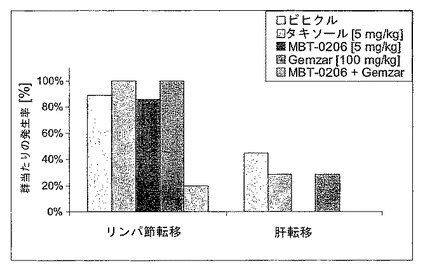
【図2】図2は、治療後の転移を示す図である。
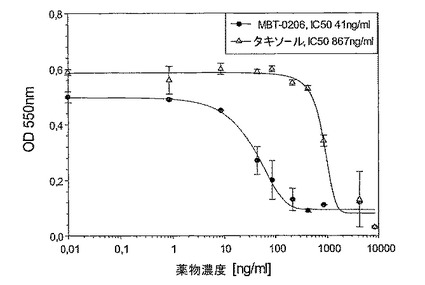
【図3】図3は、極めて薬剤耐性である子宮肉腫系Mes−SA/Dx−5MBTに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図4】図4は、適度に薬剤耐性である子宮肉腫系Mes−SA/Dx−5に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
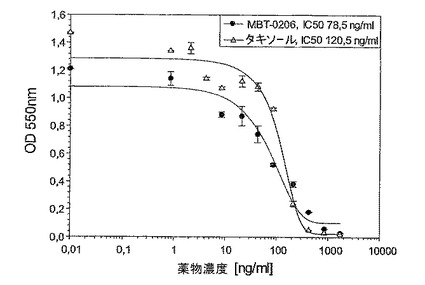
【図5】図5は、薬剤感受性であるヒト子宮肉腫系Mes−SAに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図6】図6は、極めて薬剤耐性であるマウス結腸癌腫系Colon−26MBTに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図7】図7は親系統の薬剤感受性であるマウス結腸癌腫系Colon−26に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図8】図8は、薬剤感受性であるヒト内皮系EA.hy926に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図9】図9は、感受性親系統Mes−SA及び前記耐性誘導体Mes−SA/Dx−5MBTに対するパクリタキセルのin vitro阻害能力を示す図である。
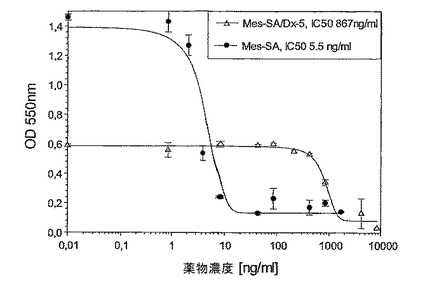
【図10】図10は、薬剤耐性ヒト皮膚メラノーマ系Sk−Mel28に対するパクリタキセルの阻害能力を示す図である。
【発明を実施するための形態】
【0017】
本明細書で使用された用語「約」は、所定の値からの±5%のずれを表す。上述した製剤は、多剤耐性の腫瘍及び/又は腫瘍転移の予防及び/又は治療に、場合により、その他の治療プロトコルと併用した多剤耐性の腫瘍及び/又は腫瘍転移の予防及び/又は治療に特に適する。
【0018】
本発明の利点は以下のようである:
−多量の活性成分
−選択的ターゲティング
−改善した有効性
−多剤耐性の回避(様々な標的)
−耐性細胞を直接的に致死させることにより薬剤耐性に影響を与える
−転移に影響を与える
−従来の化学療法、又は中性又はアニオンリポソームに比較してより低い副作用
−疾病に関連した痛みの減少
−生活の質の改善
−治療の間の体重の安定化
−従来の治療方式との協力効果。
【0019】
本発明の医薬組成物は、約0.25〜約100mg、特に〜約60mgのリポソーム状パクリタキセル/患者体重kg(bw)、有利には約0.5〜約30mgのリポソーム状パクリタキセル/kg bw、より有利には約1.0〜約15mgのリポソーム状パクリタキセル/kg bwの一月量で投与されてよい。平均的にヒト患者は約70kg体重を有し、約172cmの高さである。
【0020】
投与計画は、1日間に複数回から1ヶ月間に複数回の範囲にあってよく、このそれぞれの回は1日間〜3週間の間隔により隔てられている。この全治療期間は有利には少なくとも1ヶ月間である。
【0021】
前記医薬組成物はまた、少なくとも3ヶ月間の、少なくとも4ヶ月間の、少なくとも6ヶ月間の、又は少なくとも12ヶ月間の、及び6ヶ月間までの、12ヶ月間までの、18ヶ月間までの、24ヶ月間までの、またはより長期の長期投与にも適している。
【0022】
長期治療においてさえも、薬剤耐性又は有害な副作用、例えば脱毛症、腎症、又はその他は通常は生じない。更に、通常は前投薬、コルチコステロイド又は抗ヒスタミン剤などの前投薬は必要でない。
【0023】
本発明による医薬組成物は、約0.01〜100mg、特に〜約60mgのリポソーム状パクリタキセル/体重kgの1回量ユニット(single dose unit)方式で投与されてよい。本発明の有利な実施態様において、約0.01〜約10mg、例えば約0.05〜約5mgのリポソーム状パクリタキセル/体重kgが1回量ユニットで投与される。有利には、1回量ユニットにつき0.1〜2.5mgのリポソーム状パクリタキセル/体重kgが投与される。
【0024】
更なる有利な実施態様において、一月量につき約1〜約10mgのリポソーム状パクリタキセル/bw kgが投与される。また更なる有利な実施態様において、一月量につき約20〜約60mgのリポソーム状パクリタキセル/kg bwが投与される。更なる有利な実施態様において、一月量につき約1〜約7.5mgのリポソーム状パクリタキセル/bw kgが投与される。
【0025】
ヒト患者に対する適用のためのリポソーム状パクリタキセルの適した用量は、約0.01〜2.5、有利には0.02〜1.7、より有利には0.05〜0.5mg/kg bwの量で、少なくとも1日1回、例えば1日につき2回、3回、又はそれ以上;隔日で、約0.01〜5.0、有利には0.02〜2.5、より有利には0.05〜1.7mg/kg bw;1週間に1回で、約0.01〜10、有利には0.02〜5.0、より有利には0.05〜2.5mg/kg bwである。
【0026】
一月量は有利には複数回の1回量ユニットで投与される。低用量の複数回の投与は、単回の高用量の投与と少なくとも同じくらい効果的であってよい。前記治療間隔の間に、前記の用量ユニット及び投与間隔は、一定のままであってよい。その一方で、前記用量ユニットは前記治療間隔の間に増加してよく、例えば開始用量で始め、一段階又は複数段階で、前記開始用量の3又は4倍より高くてよい固定用量へと漸増させる。付加的に又は代替的に、前記の単回投与の間の治療間隔を変化させてよく、例えば前記治療期間の間に減少させるか上昇させてよい。
【0027】
単回投与のための有利な用量は約0.25〜約1.75mg/kg bwである。
【0028】
前記カチオンリポソーム製剤の投与を含む更なる有利な治療プロトコルは、
(i)最初の週に少なくとも3回、特に3〜5回、その後に投与なしで1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、その際、この一月量は有利には約1〜約10mg/kg bw、特に約5〜約7.5mg/kg bw、
(ii)最初の週に1回、その後に投与なしで少なくとも1週間、特に1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、その際、この一月量は有利には約20〜約100mg/kg bw、特に約20〜約60mg/kg bwである。
(iii)1週又は連続した複数週にわたって、有利には少なくとも4週にわたって、1週間に1回、その際、この一月量は有利には約1〜約7.5mg/kg bw、特に約3〜約6mg/kg bw、又は
(iv) (i)、(ii)、及び/又は(iii)の組み合わせ
である。
【0029】
ヒト医薬品での適用のために、本発明の医薬組成物は有利には、有利には約9mg〜約3700mg、特に〜約2237mg/ヒト体表(bs)m2、有利には約18〜約1168mg/m2 bs、特に有利には約37〜約584mg/m2 bsの一月量で投与されてよい。平均的に、ヒト患者は約1.84m2の体表を有する。従って、体重70kg及び身長172cmの平均的な人間に対して、一月量、1回量その他のための有利な値は、上記ではmg/体重(bw)kgで示されてきたが、ヒト適用のためには公知の方法により、種特異的な因数でのかけ算によってmg/ヒト体表(bs)m2の相応する値に変換されてよい。
【0030】
更なる観点において、本発明の前記カチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜約99.9モル%、有利には〜約98モル%、少なくとも約0.1モル%、有利には少なくとも約2モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のために便利である。
【0031】
有利な実施態様において、前記リポソーム製剤はパクリタキセルを、約0.1モル%〜、特に約2モル%〜約8モル%の量で、有利には約0.5モル%〜、特に約2モル%〜約5モル%の量で、より有利には約1モル%〜約4モル%の量で、最も有利には約2.5モル%〜約3.5モル%の量で含有する。本発明のカチオンリポソーム製剤は、パクリタキセル結晶をほぼ含有しない。
【0032】
本発明のリポソーム製剤は、約30モル%〜約99.9モル%、特に〜約70モル%、有利には約40モル%〜約60モル%、最も有利には約45モル%〜約55モル%の量でカチオン脂質を含有するカチオンリポソーム製剤である。前記製剤及び前記カチオン脂質は、正のゼータ電位を、約pH7.5の約0.05M KCl溶液中で、室温で有することにより特徴付けられている。
【0033】
前記リポソーム製剤の有利なカチオン脂質は、N−[1−(2,3−ジオレオイロキシ)プロピル]−N,N,N−トリメチルアンモニウム塩、例えば、このメチルスルファート(DOTAP)である。その他の本発明のために有利な脂質は以下を含んでよい:DDAB、ジメチルジオクタデシルアンモニウムブロミド;1,2−ジアシルオキシ−3−トリメチルアンモニウムプロパン、(以下を含むがこれに限定されない:ジオレオイル、ジミリストイル、ジラウロイル、ジパルミトイル及びジステアロイル;2つの異なるアシル鎖も前記グリセロール骨格に結合していてよい);N−[1−(2,3−ジオロイロキシ)プロピル]−N,N−ジメチルアミン(DODAP);1,2−ジアシルオキシ−3−ジメチルアンモニウムプロパン(以下を含むがこれに限定されない:ジオレオイル、ジミリストイル、ジラウロイル、ジパルミトイル、及びジステアロイル;2つの異なるアシル鎖も、前記グリセロール骨格に結合していてよい);N−[1−(2,3−ジオレイロキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド(DOTMA);1,2−ジアルキルオキシ−3−ジメチルアンモニウムプロパン、(以下を含むがこれに限定されない;ジオレイル、ジミリスチル、ジラウリル、ジパルミチル、及びジステアリル;2つの異なるアルキル鎖も、前記グリセロール骨格に結合していてよい);ジオクタデシルアミドグリシルスペルミン(DOGS);3β−[N−(N’,N’−ジメチルアミノエタン)カルバモイル]コレステロール(DC−Chol);2,3−ジオレオイロキシ−N−(2−(スペルミンカルボキサミド)−エチル)−N,N−ジメチル−1−プロパンアミニウムトリフルオロアセタート(DOSPA);β−アラニルコレステロール;セチルトリメチルアンモニウムブロミド(CTAB);ジC14アミジン;N−tert−ブチル−N’−テトラデシル−3−テトラデシルアミノプロピオンアミジン;14Dea2;N−(アルファ−トリメチルアンモニオアセチル)ジドデシル−D−グルタマートクロリド(TMAG);O,O’−ジテトラデカノイル−N−(トリメチルアンモニオ−アセチル)ジエタノールアミンクロリド;1,3−ジオレオイロキシ−2−(6−カルボキシ−スペルミル)−プロピルアミド(DOSPER);N,N,N’,N’−テトラメチル−N,N’−ビス(2−ヒドロキシエチル)−2,3−ジオレオイロキシ−1,4−ブタンジアンモニウムヨージド;1−[2−(アシルオキシ)エチル]2−アルキル(アルケニル)−3−(2−ヒドロキシエチル)−イミダゾリニウムクロリド誘導体(Solodin et al. (1995) Biochem. 43: 13537-13544によって述べられたもの)、例えば1−[2−(9(Z)−オクタデセノイルオキシ)エチル]−2−(8(Z)ーヘプタデセニル−3−(2−ヒドロキシエチル)イミダゾリニウムクロリド(DOTIM)、1−[2−(ヘキサデカノイルオキシ)エチル]−2−ペンタデシル−3−(2−ヒドロキシエチル)イミダゾリニウムクロリド(DPTIM)、第四級アミンにヒドロキシアルキル部を含有する、2,3−ジアルキルオキシプロピル第四級アンモニウム化合物誘導体(例えばFelgner et al[Felgner et al. J. Biol. Chem. 1994,269, 2550-2561]によって説明されたとおり)、例えば:1,2−ジオレオイル−3−ジメチルヒドロキシエチルアンモニウムブロミド(DORI)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DORIE)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシプロピルアンモニウムブロミド(DORIE−HP)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシブチルアンモニウムブロミド(DORIE−HB)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシペンチルアンモニウムブロミド(DORIE−Hpe)、1,2−ジミリスチロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DMRIE)、1,2−ジパルミチロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DPRIE)、1,2−ジステリロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DSRIE);Santaniello et al. [US5498633]によって報告されたアシルカルニチンのカチオンエステル;ホスファチジルコリンのカチオントリエステル、例えば1,2−ジアシル−sn−グリセロール−3−エチルホスホコリン、その際この炭化水素は飽和又は不飽和であってよく、及び分枝又は非分枝であってよく、C12〜C24の鎖長を有し、この2つのアシル鎖は必ずしも同一でない。
【0034】
有利な実施態様において、前記リポソーム製剤は場合により、少なくとも1つの中性及び/又はアニオン脂質を含有する。中性脂質は、中性の正味の電荷を有する脂質である。アニオン脂質又は両親媒性物質は、負の実効電荷を有する分子である。これらは中性又は負の実効電荷を有する、ステロール又は脂質、例えば、コレステロール、リン脂質、リゾ脂質、リゾリン脂質、スフィンゴ脂質又はペグ化(pegylated)脂質から選択されてよい。有利な中性及びアニオン脂質はこの際以下を含む:カルボン酸基を含む、ホスファチジルセリン、ホスファチジルグリセロール、ホスファチジルイノシトール(特定の糖に限定されない)、脂肪酸、ステロール、例えばコレステロール、1,2−ジアシル−sn−グリセロ−3−ホスホエタノールアミンであり、DOPE、1,2−ジアシル−グリセロ−3−ホスホコリン及びスフィンゴミエリンを含むがこれに限定されない。グリセロール骨格に結合した脂肪酸は、特定の長さ又は二重結合の数に限定されない。リン脂質は、2つの異なる脂肪酸をも有してよい。有利には、この更なる脂質は室温で液状の結晶状態にあり、かつ前記脂質は前記の使用したカチオン脂質と、これらが適用される割合で混合可能である(即ち、均一な相が形成されてよく、相分離又は分域形成は生じない)。有利な実施態様において、前記中性脂質はDOPCである。
【0035】
更なる有利な実施態様において、前記リポソーム製剤は、場合により中性及び/又はアニオン脂質を、有利にはDOPCを、約30〜約70モル%、有利には約40〜約60モル%、特に有利には約45〜約55モル%の量で含有する。
【0036】
本発明の更なる対象は、本発明において使用される前記カチオンリポソーム製剤を脱水し、脱水したまま長期間の間貯蔵し、次に、前記脱水、貯蔵及び再水和の工程の間にこの内容物の大部分を失うことなしに、これを使用すべき時に及び場で再水和することである。後者を達成するために、1つ又はそれ以上の保護剤、例えば凍結保護防止剤が存在してよい。このように、本発明によるカチオンリポソーム製剤は有利には凍結防止剤を含有し、その際前記凍結保護防止剤を糖又はアルコール又はこの組み合わせから選択する。有利には、前記凍結保護防止剤を、トレハロース、マルトース、スクロース、グルコース、ラクトース、デキストラン、マンニトール、又はソルビトールから選択する。
【0037】
更なる有利な実施態様において、前記リポソーム製剤はトレハロースを、前記製剤の総体積に対して、約5%(m/v)〜約15%(m/v)の範囲で含有する。
【0038】
本発明の前記カチオンリポソームの処方は変化してよい。特に有利な実施態様において、DOTAP、DOPC、及びパクリタキセルのモル比は、50:47:3モル%である。この処方は、MBT−0206又はEndoTAG−1とも命名される。
【0039】
様々なサイズのリポソームが本発明において使用される。本発明の有利な実施態様において、カチオンリポソームは約25nm〜約500nm、有利には約50nm〜約500nm、より有利には約100nm〜約300nmの平均粒子直径を有する。
【0040】
本発明のリポソーム製剤は、全身に、有利には静脈内に投与されてよい。
【0041】
本発明のカチオンリポソームは、増加した血管新生に関連した全て形態の症状、例えば癌を処置するために使用されてよい。
【0042】
本発明の医薬組成物は、ヒト患者における腫瘍、例えば膀胱癌、乳癌、結腸直腸癌、子宮内膜癌、白血病、肺癌、リンパ腫、メラノーマ、非小細胞肺癌、卵巣癌、前立腺癌を治療するために、及び、小児癌、例えば脳幹部神経膠腫、小脳星状細胞腫、大脳星状細胞腫、上衣細胞腫、ユーイング肉腫/腫瘍ファミリー、胚細胞腫瘍、頭蓋外の、ホジキン病、白血病、急性リンパ芽球性、白血病、急性骨髄性の、肝臓癌、髄芽腫、神経芽腫、ホジキン病でないリンパ腫、骨の骨肉腫/悪性の線維性組織球腫、網膜芽腫、横紋筋肉腫、軟部組織肉腫、テント上性原始性、神経外胚葉性及び松果体部の腫瘍、普通でない小児癌、視覚路及び視床下部膠腫、ウィルムス腫瘍、及びその他の小児の腎臓癌の治療、及びこれらより一般的でない、以下を含む癌、急性リンパ性白血病、成人の急性骨髄性白血病、成人のホジキン病でないリンパ腫、脳腫瘍、子宮頸癌、小児癌、小児肉腫、慢性リンパ性白血病、慢性骨髄性白血病、食道癌、ヘアリーセル白血病、腎臓癌、肝臓癌、多発性骨髄腫、神経芽腫、口頭癌、膵癌、原発性中枢神経系リンパ腫、皮膚癌、小細胞肺癌、頭頸部癌、胆嚢及び胆管癌、胃癌、消化管癌、カポシ肉腫、尿路上皮細胞癌腫、甲状腺癌腫、精巣癌腫、膣癌、血管肉腫、軟部組織肉腫、及び中皮腫を処置するために特に有利である。特に、前記癌は転移性癌及び/又は標準的な(化学)療法耐性癌であってよい。本発明の組成物の投与は、疾病の進行を遅めるか又は停止させ、又は部分的な又は完全な寛解を導いてよい。更なる症状は、創傷治癒、又は炎症性疾患又は慢性的な炎症性疾患、例えば関節リウマチ、皮膚炎、子宮内膜症、又は乾癬であってよい。
【0043】
本発明のカチオンリポソーム製剤は、上述した癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌、及びメラノーマの治療に、単独療法として、又は更なる治療療法、例えば以下に詳細に示した更なる活性成分との、特に化学療法剤、例えばDNA/RNA代謝拮抗産物、例えばゲムシタビンとの併用療法として、特に適する。
【0044】
意外にも、カチオンリポソーム中に負荷した活性成分は、内皮性又は非内皮性薬剤耐性細胞、特に薬剤耐性の内皮性又は非内皮性腫瘍細胞に対して直接的に作用することが見出された。従って、この新しい知見は非内皮性薬剤耐性細胞を更なる標的として定義し、これによりカチオンリポソームの抗腫瘍性の適用を強める。これは本発明の更なる観点である。
【0045】
従って、活性成分を含有するカチオンリポソーム製剤を、内皮性又は非内皮性の薬剤耐性細胞に対する医薬品の製造のために使用することは本発明の更なる観点である。本発明は、活性成分を含有するカチオンリポソーム製剤の、これを必要とする被験体の薬剤耐性細胞へと、疾病、例えば癌に作用するための治療有効量で投与する方法も提供する。
【0046】
前記観点の更なる実施態様は、薬剤耐性細胞の発生に関連しかつ/または付随する疾病の予防又は治療のための、例えば薬剤耐性腫瘍の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用に関し、特に、癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌及びメラノーマのための二次治療又は三次治療としての使用を意味する。
【0047】
初期治療の失敗の主な原因は、薬剤耐性細胞クローンの出現である。二次治療は、この開始投与が失敗した後に投与される。二次化学療法は、様々な作用様式を有する様々な化学療法剤を用いた単独療法又は複数の更なる薬物又は治療の併用であってよい。しかし、二次治療は、多剤耐性細胞クローンの発生により、同様に失敗する可能性もある。従って、このような腫瘍再発のために三次治療が存在してよい。前記カチオンリポソーム製剤は上述したように二次治療又は三次治療として特に適する。
【0048】
本発明の前記観点のカチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜薬99.9モル%、特に〜約98モル%、少なくとも約0.1モル%、特に少なくとも約2モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、薬剤耐性細胞の発生に関連しかつ、または付随する疾病を軽減する(退縮を引き起こして)か、又は最終的に治癒するように、薬剤耐性細胞に作用するための医薬組成物の製造のために有利である。
【0049】
本発明のうちの更なる意外な知見は、活性成分を含有するカチオンリポソームが単独で、又は転移形成に対する少なくとも1つのその他の治療療法との併用で作用することである。
【0050】
従って本発明の更なる対象は、活性成分を含有するカチオンリポソーム製剤を、転移に対する医薬品を準備するために使用することである。本発明はまた、活性成分を含有するカチオンリポソーム製剤を、これを必要とする被験体に、転移形成の発現及び/又は進行に作用するための、例えば転移性疾病を遅延するか及び/又は回避する、治療有効量で投与する方法をも提供する。「作用する」との用語は本発明において一般的に、所望の薬理学的及び/又は生理学的効果、例えば疾病の発現及び/又は進行を、遅延及び/又は回避する効果が得られることを意味する。有利な実施態様において、本発明は肝臓の転移形成の遅延及び/又は回避のために使用される。
【0051】
また更なる観点においても、本発明のカチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜約99.9モル%、特に〜約98モル%、少なくとも約0.1モル%、特に少なくとも約2モル%の量で第1の活性成分、例えばパクリタキセル、及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、転移形成の遅延及び/又は回避のための、少なくとも1つの更なる活性成分の有効量、例えば第2の非リポソーム状活性成分及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のために有利である。
【0052】
前記カチオンリポソーム製剤中に負荷した活性成分、例えば単独療法のための活性成分又は、併用療法のための第1の活性成分は、細胞毒性の又は細胞分裂抑制性の物質、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質から選択されてよい。より有利な実施態様においては、前記活性成分は、タキサン、カンプトテシン、スタチン、デプシペプチド、サリドマイド、微小管と相互作用するその他の薬剤、例えばジスコデルモリド(discodermolide)、ラウリマリド(laulimalide)、イソラウリマリド(isolaulimalide)、エレウレテロビン(eleutherobin)、サルコジクチン(Sarcodictyin)A及びBから選択され、最も有利な実施態様においては、パクリタキセル、ドセタキセル、カンプトテシン又はこれらの全ての誘導体から選択される。
【0053】
従って、本発明の有利な実施態様において、前記リポソーム製剤は、タキサン、有利にはパクリタキセル又はドセタキセル、又はこれらの誘導体を、約0.1モル%〜約20モル%の量で、有利には約0,5モル%〜約10モル%の量で、より有利には約1モル%〜約5モル%の量で、最も有利には約2モル%〜約4モル%の量で含有する。
【0054】
前記の少なくとも1つの更なる活性成分は、上述したように細胞毒性の又は細胞分裂抑制性の、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質、過敏反応又は化学増感剤を減少させるか又は消去する化合物であってよい。有利には、前記の少なくとも1つの更なる活性成分は、非リポソーム製剤中に存在する。更に、前記活性成分及び前記の更なる活性成分が異なることが有利である。
【0055】
有利な実施態様において、前記の更なる活性成分は、抗腫瘍剤、特に有糸分裂阻害剤、例えばパクリタキセル、アルキル化剤、特に白金含有化合物、特にシスプラチン、カルボプラチン、DNAトポイソメラーゼ阻害剤、例えばカンプトテシン、又はドキソルビシン、RNA/DNA代謝拮抗物質、特に5−フルオロウラシル又はゲムシタビン、及び/又はその他の抗腫瘍活性を有する化合物から選択される。特に有利なのは、シスプラチン又はカルボプラチンとの、又は5−フルオロウラシル又はゲムシタビンとの併用療法である。
【0056】
更なる有利な実施態様において、過敏反応を減少させるか又は消去する化合物は、致命的なアナフィラキシー反応を妨げるために十分な量にある、ステロイド、抗ヒスタミン剤、H2レセプターアンタゴニスト、及びこれらの組み合わせを含む(しかしこれらに限定されない)グループから選択される。より更に有利な実施態様において、前記化合物は、ラニチジン、デキサメタゾン、ジフェンヒドラミン、ファモチジン、ヒドロコルチゾン、クレマスチン、シメチジン、プレドニゾロン、プレドニゾン、クロルフェニラミン、クロルフェナミン、マレイン酸ジメチンデン、インドメタジン、及びプロメタジン又はこれらの全ての誘導体を含むグループから選択される。
【0057】
有利な実施態様において、前記化学増感剤は、細胞周期調節剤、薬剤耐性を戻す物質、例えばベラパミル、血管作動性物質、例えば抗高血圧剤、カチオンリポソームと血液成分との電荷に関連した相互作用を変更する物質、例えばプロタミンを含む(しかしこれらに限定されない)グループから選択される。
【0058】
1つ又は複数の本発明の観点に対して論じられた全ての有利な実施態様は、その他全ての観点にも関することに留意されたい。これは特に、カチオン脂質の量及び種類、中性及び/又はアニオン脂質の量及び種類、活性成分の量及び種類、併用療法のための更なる活性成分の量及び種類、及び処置すべき疾病の種類を指す。
【0059】
図の説明
図1:治療後の腫瘍体積
治療開始後19日目のL3.6pl膵臓腫瘍の腫瘍サイズ。10%トレハロース、パクリタキセル、MBT−0206、Gemzar(ゲムシタビン)及びMBT−0206とGemzarとの両方の併用での治療を腫瘍細胞接種後8日目に開始した。Gemzarをi.p.で100mg/kgの用量で、1週間に2回(月、木)適用した。パクリタキセル及びMBT−0206は、i.v.で、月、水、金スケジュールで、5mg/kg bwのパクリタキセル用量で適用した。前記併用グループは、それぞれのスケジュールでMBT−0206及びGemzar両方を得た。腫瘍をキャリパーを用いた触診によって、23日目及び27日目に測定した。平均±SEM;グループにつきn=9。
【0060】
図2:治療後の転移
治療開始後19日目の転移。10%トレハロース、パクリタキセル、MBT−0206、Gemzar(ゲムシタビン)及びMBT−0206とGemzarとの両方の併用での治療を腫瘍細胞接種後8日目に開始した。Gemzarをi.p.で100mg/kg bwの用量で、1週間に2回(月、木)適用した。パクリタキセル及びMBT−0206は、i.v.で、月、水、金スケジュールで、5mg/kg bwのパクリタキセル用量で適用した。前記併用グループは、それぞれのスケジュールでMBT−0206及びGemzar両方を得た(グループにつきn=9)。
【0061】
図3〜5:成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
図3:極めて薬剤耐性である子宮肉腫系Mes−SA/Dx−5MBTに対するMBT−0206及びパクリタキセルの阻害能力
図4:適度に薬剤耐性である子宮肉腫系Mes−SA/Dx−5に対するMBT−0206及びパクリタキセルの阻害能力。
図5:薬剤感受性であるヒト子宮肉腫系Mes−SAに対するMBT−0206及びパクリタキセルの阻害能力。
【0062】
図6〜7:成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
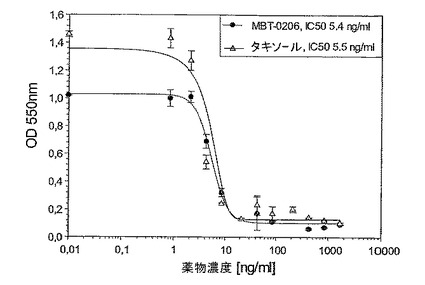
図6:極めて薬剤耐性であるマウス結腸癌腫系Colon−26MBTに対するMBT−0206及びパクリタキセルの阻害能力
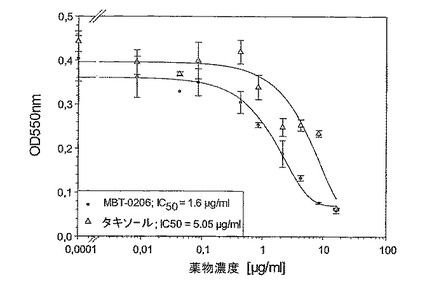
図7:親系統の薬剤感受性であるマウス結腸癌腫系Colon−26に対するMBT−0206及びパクリタキセルの阻害能力。
【0063】
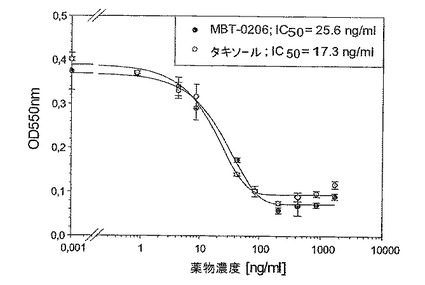
図8:薬剤感受性であるヒト内皮系統EA.hy926に対するMBT−0206及びパクリタキセルの阻害能力
成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【0064】
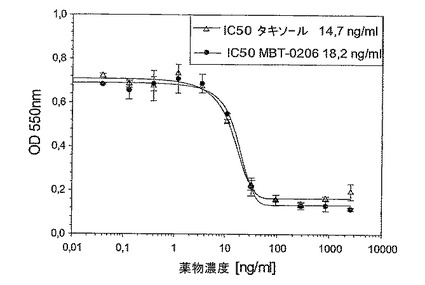
図9:前記感受性親系統Mes−SA及び前記耐性誘導体Mes−SA/Dx−5MBTに対するパクリタキセルのin vitro阻害能力。成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に試験した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【0065】
図10:薬剤耐性ヒト皮膚メラノーマ系統Sk−Mel28に対するパクリタキセルの阻害能力。成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に試験した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【実施例】
【0066】
以下の実施例は、本発明の範囲を単に説明するものであり制限する意図はないものとする。その他の一般的な及び特定の構成は当業者に公知である。
【0067】
実施例
1.ヒト治療法プロトコル
この実施例は、開示した製剤を使用するヒト治療プロトコルに関する。治療は、増強した血管新生活性に関連する様々なヒト疾病及び障害を予防及び/又は処置するために使用される。これは抗腫瘍療法において、例えば充実性腫瘍及び血液の悪性腫瘍を有する患者の処置において、様々な慢性的な炎症性疾患、例えば関節リウマチ又は乾癬に対する治療において、特に有利であると考えられる。
【0068】
本発明の特徴は、様々な分類の疾病及び/又は異常が、前記異常に関係する組織又は細胞を直接的にターゲッティングすることなしに直接的に血管新生内皮細胞を標的とすることで治療されてよいことであり、例えば血管新生を阻害することにより腫瘍へのこの血液供給は遮断され、全く前記腫瘍細胞を直接的にターゲッティングすることなしに前記腫瘍は致死する。疾病及び/又は異常のその他の分類は、血管新生内皮細胞を直接的にターゲッティングすることにより、かつ前記異常に関係する組織又は細胞を直接的に標的とすることにより治療されてよい。
【0069】
その他の適用において、薬剤耐性細胞、例えば薬剤耐性癌細胞、又は関節リウマチにおけるきわめて増殖性の滑膜細胞に、直接的に作用してよい。
【0070】
臨床試験の実施(患者の治療及び観察を含む)の様々な要素は、本発明の開示の観点において、当業者に公知である。
【0071】
規定の承認目的のために、試験のために選択された患者が、従来の治療法の少なくとも1クールに応答しないであろうこと、かつ理学的検査、実験室技術、又はX線撮影方法により決定される客観的に測定可能な疾病を有することが検討された。前記患者は心臓又は腎疾患の履歴を有さず、かついかなる化学療法も前記試験の開始少なくとも2週間前に停止すべきものである。
【0072】
適用の前に、前記製剤が凍結乾燥されている場合には、これを水溶液中に再構成してよい。以上に概略したように、必要な適用体積は、患者の体重及び投与スケジュールから計算した。
【0073】
開示した製剤を、短い注入時間の間に投与してよい。いかなる用量レベルでも、投与される注入は、それぞれの注入後に達成される毒性に依存すべきものである。従って、いかなる単独注入後にも、又は一定の割合の注入のための特定の期間にも、IIグレードの毒性に達する場合には、更なる用量を控えるか又は前記の一定の割合の注入を、毒性が改善しない限り停止すべきものである。患者の約60%が全てのカテゴリーにおいて許容不可能なIII又はIVグレードの毒性を示すまで、増加用量を患者グループ投与する。この値の2/3の用量を、安全用量として定義する。
【0074】
理学的検査、腫瘍測定及び実験室試験を無論、治療以前に、及び約3〜4週間後の間隔の際に実施するものである。実験室試験は、全血球計算、血清クレアチニン、クレアチンキナーゼ、電解質、尿素、窒素、SGOT、ビリルビン、アルブミン及び総血清タンパク質を含むものである。
【0075】
臨床反応は、許容可能な測定又は実験室値、例えば腫瘍マーカーの変化によって定義される。例えば、完全寛解は、全ての測定可能な疾病の少なくとも一ヶ月間の消滅により定義されてよく、その一方で部分寛解は50%又はそれ以上の減少によって定義されてよい。
【0076】
本発明において開示及び請求された全ての組成物及び方法は、本発明の開示の観点において過度の実験なしに製造され、かつ実施されてよい。本発明の組成物及び方法は、有利な実施態様の観点において説明されているが、本発明の概念、趣旨及び範囲から逸脱することなく、本発明において説明された組成物に、方法に及び方法の工程において、又は一連の方法の工程において、変形を適用してよいことが当業者には明白である。より詳細には、同等の又は類似の結果が達成される一方で、化学的にも生理学的にも関連するある種の薬剤が、本発明において説明された薬剤の代わりに代用されてよいことが明白である。当業者に公知であるこのような類似の代用物及び修正の全ては、添付の特許請求の範囲によって定義される本発明の趣旨、範囲及び概念のうちにあると考えられる。
【0077】
用量におけるいくつかの変形は、処置される被験体の状態に依存して必然的に生じる。投与の責任者は、いかなる場合においても、この個々の被験体にとって適した用量を決定する。更に、ヒトへの適用に対しては、製剤は、the FDA Office of Biologics standardsが要求する無菌性、発熱原性、一般的な安全性及び純度水準を満たすものである。
【0078】
本発明は、活性成分の本発明の製剤の医薬有効量を、これを必要とする被験体の標的部位、例えば血管新生性の血管標的部位へと、投与する方法も含む。「これを必要とする被験体」とは、哺乳類、例えばヒトを指す。
【0079】
投与経路は有利には、腹膜又は非経口適用を含む。
【0080】
本発明での使用のために、これを必要とする被験体へ投与される化合物の「薬理学的有効量」は、様々な範囲の因子に依存して変化する。前記化合物の量は、この患者の大きさ、年齢、性別、重量及び状態にも、投与される前記物質の効力にも依存する。投与に関して様々な変形が存在することを示してきたが、当業者は、本発明の開示を用いて容易に、極めて少量を最初に投与し、所望の結果が得られるまで前記用量を増分して上昇させることによる、適切な投与を決定することが可能であると考えられている。前記投与量は上述した因子によって非常に変化するが、一般的には、病的組織を標的とするに過ぎない、例えばこの腫瘍細胞自体を標的とするに過ぎないデリバリー系に比較して、大幅に少ない量でいかなる物質をも投与することが本発明により可能になる。
【0081】
2.単独療法プロトコル
【0082】
【表1】

【0083】
【表2】
有効性
応答を、WHO又はRECIST判定基準に従って評価した。
【0084】
3.併用療法プロトコル
【0085】
【表3】
【0086】
更に計画された試験は、リポソーム状パクリタキセル、例えば0.5又は1.0又は1.5mg/kg及びゲムシタビン、例えば1000mg/m2を、週に1回、3週間の間、その後に治療なしで1週間、有利には少なくとも1年間の間隔の投与を含む。
【0087】
リポソーム状パクリタキセルの前記治療スケジュールは上述したように進行中の試験のためである。
【0088】
有効性
応答を、WHO又はRECIST判定基準に従って評価した。
【0089】
4.症例報告#1
患者:
−喉頭の粘膜表皮癌の、大きな治療耐性再発を有する49歳の患者
−頸部、鎖骨上、腋窩、縦隔及び肺の転移
−最初の腫瘍切除の5年後、頸部腫瘍切開及び補助放射線療法
−複数回の切除、形成外科手術、放射線療法及び化学療法を用いた再発の繰り返した治療後。
投与スケジュール:
−MBT−0206:50/47/3(DOTAP/DOPC/パクリタキセル)
−リポソーム状パクリタキセル 0.06、0.25、0.5及び1.0mg/kg bwの投与、i.v.
−1週間に3回の1サイクル(1、3、及び5日目)
結果:
−注入時及び後の心臓血管、肺、及び血清のパラメーター観察の間の良好な認容性
−急性又は慢性毒性の徴候なし
−腫瘍の血液循環の減少
−3ヶ月の間、腫瘍成長の強力に減少した進行。
【0090】
5.症例報告#2
多重化学療法後に疾病進行を有した、肝臓細胞癌腫を有する一患者をMBT−0206で治療した。
【0091】
凍結乾燥したMBT−0206を、注射のために水により再構成し、総注入体積300〜400mlのMBT−0206(リポソーム状パクリタキセル1.0mg/体重kgの用量に相当)を、中心又は末梢静脈注入により2〜4hの期間にわたり投与した。この注入率を、最大速度2.5ml/minにまでゆっくりと上昇させた。前投薬は、前記患者の性別、年齢又は条件に依存した。上述した患者の特定の場合には、デキサメタゾン及び抗ヒスタミン剤が投与されている。
【0092】
MBT−0206を、1週間に1回、2回のリポソーム状パクリタキセル0.25mg/kg bw 1回のリポソーム状パクリタキセル0.5mg/kg bw)で開始し、次に19回のリポソーム状パクリタキセル1.0mg/kg bwの強化用量にする、用量漸増スケジュールでもって投与した。前記治療は22週間の投与後にいまだ進行中であり、かつ今現在まで、薬物有害反応は報告されていない。好ましい安全特性の他に、肝臓のCT−スキャンによって実施された腫瘍サイズの最後の評価では、安定した疾病を示した。
【0093】
6.症例報告#3
その他の症例において、ホルモン療法に治療抵抗性になった前立腺癌患者を、リポソーム状パクリタキセル1.0mg/kg bwで、週に3回、3日毎に、上述した製剤及び投与と同様の条件下で処置した。この前投薬は、デキサメタゾン及び抗ヒスタミン剤を含有した。前記患者に対するリポソーム状パクリタキセルの蓄積用量は、リポソーム状パクリタキセル3.0mg/kg bwであった。
【0094】
7.リポソーム状パクリタキセルでの低用量スケジュール
不死化内皮細胞(EA.hy926)を24ウェルプレート(4×104細胞/ウェル)中に播き、一晩育てた。翌日、9つのウェルを1hの間、MBT−0206として処方した51.2ng/ml(60nM)のリポソーム状パクリタキセルの低用量で処理した。更に、処方につき3ウェルを、MBT−0206として処方した153.7ng/mlの高用量(180nM)で1h処理し、3ウェルは未処理のままにした。約24h後に、9つの低用量処理ウェルのうち6つを、再度同様の低用量MBT−0206で1h処置した(即ち:2×処理群)。再度24h後、前記6つの2回処理したウェルのうち3つを、MBT−0206として処方した51.2ng/mlのパクリタキセルで3度目に、1hの間処置した(3×処置群)。前記の3度目の処理の約96h後に、全てのウェルの細胞生存率を定量化した。この目的のために、テトラゾリウム塩の3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)を用いたミトコンドリア脱水素酵素の活性を測定するアッセイを、標準的なプロトコルに従って適用した(例えば、わずかに改変した{Lindl, 1994#28})。
【0095】
この結果は、3回の低用量MBT−0206で処理した細胞の生存率は、高用量で1回のみ処理した細胞の生存率と少なくとも同じくらい強力に減少していたことを示す。1回又は2回低用量MBTで処理した細胞は、幾分上昇した生存率を示したが、これは未処理細胞に比較すると減少している。
【0096】
結論
高用量のMBT−0206での処理を、より高い頻度での低用量を用いて置き換えてよい。治療密度(1週間における治療の数)と治療有効性との間には相関がある。低用量での1週間に3回の治療は、1週間に1又は2回の治療よりも優れていている。この最適化した投与治療方式は、高用量治療により引き起こされる毒性副作用を潜在的に減少させる。
【0097】
8.L3.6pl膵臓腫瘍中での、ゲムシタビン(50mg/kg)と併用したMBT−0206の抗腫瘍有効性
動物モデル
種:雄Balb/c nu/nuマウス
腫瘍モデル:正所性L3.6pl膵臓充実性腫瘍(極めて転移性:Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999;1(1) : 50-62.)
供給者:Charles River France。
【0098】
治療
治療開始:腫瘍接種後7日目
最後の治療:腫瘍接種後26日目
用量:適用につきパクリタキセル5mg/kg bwを有するMBTー0206
:適用につきパクリタキセル5mg/kg bwのパクリタキセル
:適用につき50mg/kgのゲムシタビン(Gemzar)
スケジュール:
MBT−0206、パクリタキセル、トレハロース:d7、10、12、14、17、19、21、24、26
ゲムシタビン:d7、11、14、18、21、25
併用:併用した単独治療
適用経路:MBT−0206、パクリタキセル、トレハロース:尾静脈中へのi.v.ボーラス
ゲムシタビン:i.p.ボーラス。
【0099】
【表4】
【0100】
前記死亡率は21日目前では低かった:対照動物1匹及びMBT−0206動物1匹が死んだのみであった。24日目には、不明の理由により死亡率が上昇した:3匹のMBT−0206、1匹のGemzar、及び4匹の対照動物が24日目に死んだ。
【0101】
前記体重はいずれの治療によっても影響を受けなかったが、対照腫瘍の重量がこの最後の週の間に18%だけ減少した。
【0102】
観察パラメーター
−接種後10、12、14、17、19、21、24日目に触診した、及び回収後26、28日目の腫瘍体積
−26、及び28日目に回収後に剖検。
【0103】
結果
いかなる治療の効果も、治療の開始後3日間、触診によっては観察されなかった。強力な抗腫瘍効果が、触診により1週間後及び24日目に、以下の有効性の順位で観察された。Gemzar−50=パクリタキセル<MBT−0206<MBT−0206+Gemzar。しかし、26日目の回収後、全ての群のこの測定した腫瘍体積は、24日目に比較して明らかに小さかった。この大きさの差は、回収前の不正確な触診のためのようである。24日目に、この対照群(n=2)に比較して単独治療により腫瘍サイズは〜30%に減少した。MBT−0206+Gemzarの併用は、13%へとこの腫瘍サイズの最も強力な減少を生じ、これはパクリタキセル、Gemzar及びMBT−0206単独に比較して、有意に(p<0.05)より有効であった。28日目の前記対照群(n=2)を示さないのは、この2つの腫瘍のうち1つが、その他の腫瘍(24、26、28日目)に比較して極めて小さく、従って代表とは見なされないからである。前記単独療法後の腫瘍は、26日目(パクリタキセル:536mm3;MBT−0206:392mm3;Gemzar:398mm3)に比較して腫瘍サイズに微弱な増加を示したが、その一方で前記併用療法は、26日目と28日目の間に88mm3とわずかな腫瘍の退縮を生じた。
【0104】
前記データは前記モデル中でのパクリタキセル、Gemzar及びMBT−0206の強力な抗腫瘍性効能を示す。MBT−0206の抗腫瘍作用は、パクリタキセルよりわずかに強力であるが、Gemzarと類似している。MBT−0206とGemzatとの併用は、顕著な抗腫瘍性効能を示した。
【0105】
9.L3.6pl膵臓腫瘍中のゲムシタビン(100mg/kg)と併用したMBT−0206の抗腫瘍性効能
動物モデル
種:雄Bald/c nu/nuマウス
腫瘍モデル:正所性L3.6pl膵臓充実性腫瘍(極めて転移性)
供給者:Charles River France。
【0106】
治療
治療開始:腫瘍接種後8日目(01.05.03)
最後の治療:腫瘍接種後26日目(19.05.03)
スケジュール:
MBT−0206、パクリタキセル、トレハロース:d9、12、14、16、19、21、23、26
ゲムシタビン:d8、12、15、19、22、26
併用:併用した単独治療。
【0107】
【表5】
観察パラメーター
−接種後23、26日目に触診した及び回収後の腫瘍体積
−1、7、12、16、19、21、23、27日目の体重
−27日目に回収後の剖検。
【0108】
結果(図1及び2参照)
全ての治療的療法の明らかな抗腫瘍効果が、併用療法の顕著な効能と共に、腫瘍接種後23日目に観察された(図1)。腫瘍阻害の順位:パクリタキセル<MBT−0206=Gemzar−50<MBT−0206+Gemzar。この対照群と比較して、前記の最終的な腫瘍体積は、MBT−0206によって46%(p<0.05)にまで、Gemzatによって47%(p<0.01)にまで、及び併用療法によって22%(p<0.01)にまで、27日目には有意に減少した。パクリタキセル治療は、最終的な腫瘍体積を68%にまで減少させたが、これは有意でなかった。興味深いことに、MBT−0206及び併用療法の効能は、23日目でより顕著であった。この原因は、23日目と27日目との週末の間に延長した治療間隔である可能性がある。前記データは、パクリタキセル、Gemzar、及びMBT−0206の前記動物モデル中での明らかな抗腫瘍性効能を明らかにした。MBT−0206の抗腫瘍性作用は、パクリタキセルよりわずかに強力であったが、Gemzarに類似していた。MBT−0206及びGemzarとの併用の両方共に、転移形成を阻害した(図2)。肝転移は、前記の2つの群でのみ存在しなかった。更に、併用群においてのみ、このリンパ節転移がほとんどなかった。前記データは、MBT−0206とGemzarとの併用が、いずれかの単独療法の抗腫瘍性効能を促進することを明らかにした。
【0109】
この死亡率は、前記治療群で、特に併用治療の間にわずかに増加した(4匹のマウスが死亡)。回収前にはどの対照動物も死亡しなかった。
【0110】
対照マウスの体重は、この最後の11日間の間に18%だけ減少した。この期間の間に、一過的な減少もまた前記処置マウスに関して観察され、これはパクリタキセルに関しては2%の、MBT−0206に関しては12%の、併用に関しては19%の、及びGemzarに関しては22%の重量損失を生じた。
【0111】
10.パクリタキセル耐性細胞(例えば、腫瘍細胞系)の致死
(多)薬剤耐性を発現している腫瘍を直接的に致死させるためのMBT−0206の能力を実証するために、2つの極めてパクリタキセルに耐性である哺乳類腫瘍細胞系をin vitroで調査した。前記細胞系を、段階的に上昇するパクリタキセル濃度により培地中で選択した。両方の細胞系は、高い耐性レベルを発現し、これはパクリタキセル約1〜5μM(867又は5000ng/ml)に対する50%の成長阻害(IC50値)のための濃度により反映されている。両方の場合において、MBT−0206はパクリタキセルよりも、薬剤耐性腫瘍細胞の致死において明らかに優れていた。対照的に、薬剤耐性の又は耐性の低い細胞系において、MBT−0206はパクリタキセルと幾分同様の致死能力を有した。
【0112】
MBT−0206とヒト子宮肉腫由来の細胞系Mes−SAとその誘導体の系統
極めてパクリタキセル耐性である誘導体細胞系Mes−SA/Dx−5MBTを、パクリタキセル濃度の増加により、市販の系Mes−SA/Dx−5(ATCC,{Harker,1986 #29})から選択した。図3に示したように、前記誘導体細胞系はIC50値867ng/mlによって示されるように、パクリタキセルに極めて耐性であった。意外にも、MBT−0206が前記細胞系を、約20倍低いIC50値によって反映されるように、より効果的に致死させることが見出された。前記の市販の系Mes−SA/Dx−5は、親系統であるMes−SAからドキソルビシンにより選択される({Harker, 1986#29})が、パクリタキセルに対して低レベルの交差耐性を発現した(図3〜5を比較のこと)。パクリタキセルに対するIC50値は、Mes−SA/Dx−5MBTにおける値よりも約7倍低かった。これに伴い、前記細胞系において、パクリタキセルに比較して、MBT−0206のより高い致死能力のわずかな傾向が存在するにすぎない(図4)。前記親系統Mes−SAは、5.5ng/mlの低いIC50値によって示されるように、極めてパクリタキセル感受性であった(図5)。前記薬剤感受性系統に対して、MBT−0206はパクリタキセルと同様の致死能力を有した。これは、これまで調査した、その他全てのパクリタキセル感受性細胞系にも当てはまった。前記知見に対する例として、MBT−0206及びパクリタキセルでの、不死化内皮系統、EA.hy926の治療の結果を図8に示した。
【0113】
MBT−0206と細胞系マウス結腸癌腫由来のColon−26
Mes−SA/Dx−5MBTと同様に、マウス結腸癌腫系統Colon−26の極めてパクリタキセル耐性である誘導体系統(Cell lines Service,Heidelberg)を樹立し、Colon−26MBTと呼んだ。パクリタキセルに対するIC50値は約5μg/mlであった(図6)。再度、Mes−SA/Dx−5MBTと同様に、MBT−0206はこの細胞系の成長を阻害するための明らかにより高い能力を有した。前記細胞系において、前記IC50値は、3倍だけ違っていた。Mes−SA及びEA.hy926細胞に対して示した前記結果に沿って、この親系統の薬剤感受性Colon−26は、MBT−0206及びパクリタキセルに対して同じ様に感受性であった(図7)。
【0114】
結論
極めてパクリタキセル耐性である細胞系において、MBT−0206はパクリタキセルの致死能力よりも有意により高い致死能力を有した。パクリタキセル感受性系においては、両方のパクリタキセル製剤は匹敵する効能を有した。MBT−0206は従って、(多)薬剤耐性腫瘍をも直接的にin vitro及びin vivoで致死させることができる可能性がある。従ってこれは、パクリタキセルに対して不応答性になったヒト腫瘍(又はその他の疾病)を治療する新規のアプローチである可能性がある。
【0115】
11.再発性頭頸部扁平上皮癌の治療
パクリタキセル負荷したカチオンリポソーム(MBT−0206)の安全性評価を、再発性の治療抵抗性の頭頸部扁平上皮癌を有する患者において実施した。
【0116】
試験設計
−再発性の治療抵抗性の頭頸部扁平上皮癌を有する患者
−少なくともスパイラルCT/MRIスキャンでの次元において、正確に測定されえる、少なくとも1つの障害として定義された測定可能な疾病
−カルノフスキー動作指数>60%
−余命>4ヶ月。
【0117】
薬剤投与
−カチオンリポソーム中のカプセル化パクリタキセルを2つの用量で投与した:パクリタキセル0.55mg/kg及びパクリタキセル1.10mg/kg
−スクリーニング期間の後、1、3、及び5日目に前記薬剤を静脈内注射した。
【0118】
結論
注射の間及び後には、急性又は慢性の毒性の徴候は観察されなかった;バイタル及び実験室安全パラメーラーはほぼ一定のままであった。
【0119】
前記結果は、前臨床毒性学試験から提案された前記用量及びスケジュールが、前記患者により良好に認容されたことを示す。
【0120】
カチオンリポソームはヒト頭頸部扁平上皮癌の内皮を選択的に標的とした。レーザードップラーフローメトリーは、前記療法の抗血管新生作用機構を確認した。
【0121】
12.MBT−0206(EndoTag 1)は、パクリタキセル耐性腫瘍に対してin vivoで効果がある。
パクリタキセルでのin vitroでの72hの処理で高い生存率を有する2つの腫瘍細胞系を、マウス腫瘍モデルとして、MBT−0206の前記能力をin vivoで調査するために使用した。前記腫瘍モデルでは、MBTー0206が腫瘍成長を、パクリタキセルよりも、有意により効果的に阻害することが実証されてよい。
【0122】
1.極めてパクリタキセル耐性である誘導体(Mes−SA/Dx−5MBT、IC50、0.87μg/ml)を、前記の市販のヒト子宮肉腫由来の細胞Mes−SA/Dx−5(ECACC)から選択した。前記の適度に耐性である系統は、極めて感受性である親系統のMes−SAから生じる。図9に示したとおり、前記系統Mes−SA/Dx−5MBTは前記親系統のMesSAと比較すると、このそれぞれのIC50値により評価して、〜150倍まで増加したパクリタキセル認容性を示した。in vivo実験のために、Mes−SA/Dx−5MBT細胞をNMRIヌードマウス中に皮下注射(s.c.)した。MBT−0206又はパクリタキセルでの治療を12日目に開始し、21日目まで、1週間に3回の用量で延長した(5mg/kg b.w.のパクリタキセル)。MBT−0206処置動物の平均腫瘍サイズを、パクリタキセル処置動物の腫瘍サイズと、この最後の治療後2日目(治療開始後23日目)に比較した。MBT−0206が誘導した減少を、パクリタキセルと比較して%で示した(下の表参照)。下記表及び図9中に示したように、MBT−0206は、パクリタキセルよりも、前記腫瘍モデル中で、前記腫瘍成長を〜1/3だけ減少させることにおいて明らかにより効果があった。
【0123】
2.ヒトの皮膚メラノーマ系統Sk−Mel28に関して、市販の系統は既に高いパクリタキセル耐性(IC50、5.8μg/ml、図10)を示し、かつ感受性の誘導体の系統は比較のためには使用可能でなかった。in vivoにおいて、前記細胞を、ヒトの皮膚の移植とマウスのアクセプター皮膚との間でのヒトの皮膚の移植を有するSCIDマウス中に注射した。治療を腫瘍細胞注射後17日目に開始し、2日毎に用量(12.5mg/kg b.w.)で28日目まで延長した。MBT−0206及びパクリタキセルの治療能力を、治療開始後7日目に比較した(下の表参照)。前記腫瘍モデル中で、MBT−0206は腫瘍発達をほぼ完全にブロックし、その一方でパクリタキセルは完全に効果がなかった。MBT−0206で処理すると、腫瘍はパクリタキセル(パクリタキセル)−処理腫瘍の大きさの約1/10へとほんのわずかになった。
【0124】
表
【0125】
【表6】
Mes−SA/Dx−5MBT腫瘍モデル:治療開始後23日目の腫瘍体積(9つの治療、即ち腫瘍細胞注射後週に3回、12日目から31日目まで)
Sk−Mel腫瘍モデル:治療開始後7日目の腫瘍体積(一日おきの6つの治療、即ち、腫瘍細胞注入後17日目から28日目まで)。
【0126】
【表7】
【0127】
【表8】
【0128】
【表9】
【0129】
【表10】
【技術分野】
【0001】
本発明の詳細な説明
本発明は、必要とするヒト患者への投与のための、パクリタキセルを含有する医薬組成物の使用に関する。
【背景技術】
【0002】
増強した有糸分裂に関連する疾病に苦しむヒト患者のための治療薬としての、有糸分裂阻害剤、例えばタキサンの使用は公知技術において公知である。
【0003】
パクリタキセルは、独特の作用機構及び広い範囲の抗増殖性活性を有し、これはパクリタキセルが微小管に結合し、チューブリン重合を促進し、この構築された微小管を安定化するためである。この結果、パクリタキセルは細胞周期を前期でブロックし、G2/M期での細胞の蓄積を生じる。
【0004】
残念ながら、パクリタキセルは水中での極めて低い溶解性を有し、これにより適した剤形を提供することが困難になる。最近では、パクリタキセルはクレモホールEL(Cremophor EL)(ポリエトキシル化されたひまし油)及びエタノールを50:50(vol/vol)の比で含有する媒体中で処方され、投与される。前記溶液を1:10で食塩水中で、ヒトに投与する前に希釈する。しかし、様々な深刻な副作用、例えば過敏反応及び高血圧性反応、腎毒性及び神経毒性などが、クレモホールEL処方のせいで患者で報告されてきた。
【0005】
更に、パクリタキセルは(その他の抗腫瘍剤のうちで)、有効な、確立した標準的な抗腫瘍剤({Rowinsky, 1995#1},{Awada, 2002#2},{Seidman, 2003#3},{Romanini, 2003#4})ではあるが、薬剤不応答性の腫瘍及び転移がしばしば癌患者で観察される({Blom, 1996#5},{Modi, 2002#6},{Ozols, 2003#7})。遺伝的に不安定な、急速に分裂する腫瘍細胞は、この選択された抗癌剤の成長阻害効果を克服する能力を獲得する({Vogelstein,1988#8},{Kerbel,1991 #9})。前記能力は通常は単独の薬剤(二次治療)に限定されずに、この最初の耐性の発生後に使用されるその他の薬剤にまで及ぶ。従って、この現象は多剤耐性(MDR)と呼ばれる。利用可能でかつ承認された抗腫瘍剤の数が多数の癌の種類に関わらず非常に限定されているために、多数の患者は、彼らの癌組織がMDRを示し屈する。この明白な問題は従って、既にそれぞれの薬剤に対して耐性のある、薬剤耐性腫瘍、特に薬剤耐性細胞を致死するための方法及び手段を見出すことである。
【0006】
いくつかのアプローチが、上述の問題を取り扱うために試みられた。従来の戦略は、最大耐量(MTD)にまで用量を上昇させ、かつ全ての腫瘍細胞を可能な限り素早くかつ完全に根絶することを試みることである({Schuenemann, 1999#10},{Heidemann, 1997#11})。前記戦略が深刻な副作用を引き起こし、より長期間へと延長可能でないことは明白である。従って、前記治療スケジュールは、MTDでの1つの短い治療期間(通常1日間〜1週間)と、患者を不可避な副作用から回復させるための数週間(通常3〜4週間)の治療なしの間隔との周期からなる({Schuenemann, 1999#10},{Heidemann, 1997#11},{Romanini, 2003#4})。多数の場合に、腫瘍成長はこの薬剤なしの期間の間に再開してもよい。最も重要なことに、腫瘍細胞が高レベルの耐性を発生し、これにより前記腫瘍細胞がMTDでの薬剤濃度を収容することが可能である多数の患者では前記アプローチは失敗する。この患者は、治療抵抗性になる。
【0007】
最も一般的な解決法は、治療を第2の薬剤で開始することである({Blom, 1996#5}, {Awada, 2002#2}, {Seidman, 2003#3}, {Heinemann, 2003#12}, {Thigpen, 2003#13})。最も良好な場合では、この二次治療は奏功し、かつこの患者は治癒する。一般的な経験ではしかしながら、腫瘍が一定の時間の間応答し、これにより前記腫瘍の一時的な退縮を生じるのみである。この後では腫瘍は前記の第2の薬剤に対しても耐性になってしまう。前記戦略を続行すると、全ての使用可能な抗癌剤に対して抵抗性である、多剤耐性腫瘍の発生を最終的に生じる({Blom, 1996#5},{Seidman, 2003#3},{Thigpen, 2003#13})。その他の可能性は、患者をすぐさま2以上の薬剤の併用により処置することである({Heinemann, 2003#12},{Kuenen, 2002#14},{Sledge, 2003#15},{Ozols, 2003#7},{Reck, 2003#17},{Romanini, 2003#4})。前記戦略はより成功する可能性があり、というのは二重の薬剤耐性の発生の見込みを減少させるからである。しかし前記戦略は、時間及び費用集中的に適した薬剤の組み合わせを調査する必要がある。第2の不利な点は、この副作用は上昇する可能性もあることである({Kuenen, 2002#14},{Ozols, 2003#7})。前記の治療窓口は同時に小さくなり、この毒性作用は前記のイメージした治療上の利点をカバーしてしまう可能性がある。この場合にも、多剤耐性が生じ、前記の治療は無効になる可能性がある({Zimpfer−Rechner,2003#18},{Sledge,2003#15},{Sledge,2003#16},{Ozols,2003 #7})。
【0008】
前記の従来の治療戦略でのネガティブな経験の結果は、上述の治療の選択肢を拡大するためのより多くの新規の薬剤を開発することである。明らかにこれは、結局は多くの場合で治療抵抗性の腫瘍を生じる、より有効な薬剤のための極めて時間及び費用集中的なレースである。近年ではこの認識により、腫瘍の耐性を回避するための新規のアプローチが導かれた。これは前記MDRが、細胞に化学療法剤を排出させる酵素の過剰発現により引き起こされるという仮定に基づく。前記酵素のカテゴリーのうち最も有名なメンバーは、p−糖タンパク質(p−gp)と呼ばれる。これは細胞質膜に存在し、ATPにより駆動された方法({Nobmann, 2001#19},{Thomas, 2003#20})で、パクリタキセル又はドキソルビシンなどの化合物を排出する({Harker, 1985#21},{Fellner, 2002#22},{Kiesewetter, 2003#23})。この概念は、p−gp媒介薬剤耐性を逆転させるためのp−gp阻害剤の開発を導いた。従って、前記の種類の分子に対して化学増感剤(chemosensitizer)との用語が造語される。試験した最初の例の1つは、ベラパミルである。臨床試験はしかしながら、ことによると低い比活性のせいである、不十分な結果を示した({Thomas, 2003#20}, {Kohler, 2003#24})。更なる研究により第2世代の化合物が生じ、これはやはり臨床的に適用可能でないことが見出された({Leonard, 2002#25},{Thomas, 2003#20})。今日、第3世代のいくつかの物質(1つはタリキダール(tariquidar)として公知である)が臨床試験にある({Agrawal, 2003#26},{Callies, 2003#27})。前記化合物の有用性及び広い適用性はしかし、未だ不明である({Leonard, 2002#25}, {Thomas, 2003#20})。第1世代の化学増感剤に比較してより改善してはいても、第3世代化合物もまた副作用を引き起こし、全身に対して予期せぬ結果を有する可能性がある。大規模な臨床試験が必要とされ、これまでのところ将来に前記アプローチが一般的な実施となるかは不明瞭である({Leonard, 2002#25},{Thomas, 2003#20})。
【0009】
様々なデリバリー系がパクリタキセルの効果を促進するため及び/又は毒性を減少させるために使用されてきた。リポソームは、水溶性、かくしてより低い毒性と組み合わさった効率を促進するために開発された多数のキャリヤーのうちの1つである。
【0010】
U.S.Pat.No.5,648,090,U.S.Pat.No.5,424,073及びU.S.Pat.No.6,146,659(Rahman et al.)は、哺乳類の癌の治療方法のために、リポソームによりカプセル化したパクリタキセルを提供する。前記特許はリポソームの治療有効量の医薬組成物をこのホストに投与する方法を開示し、前記医薬組成物はリポソーム形成材料、カルジオリピン、及びパクリタキセルのような薬剤、又はパクリタキセルの抗腫瘍性誘導体、又はこの混合物を、医薬的に許容可能な添加物と共に含む。U.S.Pat.No.6,146,659では、タキサンを一時間より短い時間にわたって75〜300mg/m2の量で投与する(その際、前記タキサンはリポソームによりカプセル化されている)ことによるタキサンの患者への投与方法が提供される。前記特許中で開示されたリポソームは負に帯電している。
【0011】
McDonald etal.,U.S.Pat.No.5,837,283,の開示から、正に帯電したリポソームは血管新生内皮細胞を特異的に標的とすることが公知である。ヒト患者に関する臨床データは提示されていない。
【発明の概要】
【発明が解決しようとする課題】
【0012】
従って、本発明の基礎をなす課題は、パクリタキセルの、これを必要とする被験体への治療有効量での、深刻な副作用のない投与方法を提供することである。特に、公知技術の製剤、例えばクレモホール製剤の適用におけるパクリタキセルの高い初期治療量を用いる必要性によって引き起こされる副作用の発生は、避けることが望ましい。
【課題を解決するための手段】
【0013】
本出願発明において、パクリタキセルを含有するカチオンリポソーム製剤の投与のヒトに関する臨床データが提示される。
【発明の効果】
【0014】
意外にも、活性成分であるパクリタキセルの高含量を有するカチオンリポソーム製剤の投与が、これを必要とするヒト患者の血管新生内皮細胞に選択的に影響を与えることが見出された。従って本発明は前記患者への、パクリタキセル約0.25〜約100mg、特に〜約60mg/前記患者体重kgの一月量での、カチオン脂質約30〜約98モル%、少なくとも約2モル%の量でパクリタキセル及び中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の投与を含む。
【0015】
本発明の更なる観点は、パクリタキセル約0.25〜約60mg/前記患者体重kgの一月量での、少なくとも1つのカチオン脂質約30モル%〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の、これを必要とするヒト患者への投与を含む。
【図面の簡単な説明】
【0016】
【図1】図1は、治療後の腫瘍体積を示す図である。
【図2】図2は、治療後の転移を示す図である。
【図3】図3は、極めて薬剤耐性である子宮肉腫系Mes−SA/Dx−5MBTに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図4】図4は、適度に薬剤耐性である子宮肉腫系Mes−SA/Dx−5に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図5】図5は、薬剤感受性であるヒト子宮肉腫系Mes−SAに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図6】図6は、極めて薬剤耐性であるマウス結腸癌腫系Colon−26MBTに対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図7】図7は親系統の薬剤感受性であるマウス結腸癌腫系Colon−26に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図8】図8は、薬剤感受性であるヒト内皮系EA.hy926に対するMBT−0206及びパクリタキセルの阻害能力を示す図である。
【図9】図9は、感受性親系統Mes−SA及び前記耐性誘導体Mes−SA/Dx−5MBTに対するパクリタキセルのin vitro阻害能力を示す図である。
【図10】図10は、薬剤耐性ヒト皮膚メラノーマ系Sk−Mel28に対するパクリタキセルの阻害能力を示す図である。
【発明を実施するための形態】
【0017】
本明細書で使用された用語「約」は、所定の値からの±5%のずれを表す。上述した製剤は、多剤耐性の腫瘍及び/又は腫瘍転移の予防及び/又は治療に、場合により、その他の治療プロトコルと併用した多剤耐性の腫瘍及び/又は腫瘍転移の予防及び/又は治療に特に適する。
【0018】
本発明の利点は以下のようである:
−多量の活性成分
−選択的ターゲティング
−改善した有効性
−多剤耐性の回避(様々な標的)
−耐性細胞を直接的に致死させることにより薬剤耐性に影響を与える
−転移に影響を与える
−従来の化学療法、又は中性又はアニオンリポソームに比較してより低い副作用
−疾病に関連した痛みの減少
−生活の質の改善
−治療の間の体重の安定化
−従来の治療方式との協力効果。
【0019】
本発明の医薬組成物は、約0.25〜約100mg、特に〜約60mgのリポソーム状パクリタキセル/患者体重kg(bw)、有利には約0.5〜約30mgのリポソーム状パクリタキセル/kg bw、より有利には約1.0〜約15mgのリポソーム状パクリタキセル/kg bwの一月量で投与されてよい。平均的にヒト患者は約70kg体重を有し、約172cmの高さである。
【0020】
投与計画は、1日間に複数回から1ヶ月間に複数回の範囲にあってよく、このそれぞれの回は1日間〜3週間の間隔により隔てられている。この全治療期間は有利には少なくとも1ヶ月間である。
【0021】
前記医薬組成物はまた、少なくとも3ヶ月間の、少なくとも4ヶ月間の、少なくとも6ヶ月間の、又は少なくとも12ヶ月間の、及び6ヶ月間までの、12ヶ月間までの、18ヶ月間までの、24ヶ月間までの、またはより長期の長期投与にも適している。
【0022】
長期治療においてさえも、薬剤耐性又は有害な副作用、例えば脱毛症、腎症、又はその他は通常は生じない。更に、通常は前投薬、コルチコステロイド又は抗ヒスタミン剤などの前投薬は必要でない。
【0023】
本発明による医薬組成物は、約0.01〜100mg、特に〜約60mgのリポソーム状パクリタキセル/体重kgの1回量ユニット(single dose unit)方式で投与されてよい。本発明の有利な実施態様において、約0.01〜約10mg、例えば約0.05〜約5mgのリポソーム状パクリタキセル/体重kgが1回量ユニットで投与される。有利には、1回量ユニットにつき0.1〜2.5mgのリポソーム状パクリタキセル/体重kgが投与される。
【0024】
更なる有利な実施態様において、一月量につき約1〜約10mgのリポソーム状パクリタキセル/bw kgが投与される。また更なる有利な実施態様において、一月量につき約20〜約60mgのリポソーム状パクリタキセル/kg bwが投与される。更なる有利な実施態様において、一月量につき約1〜約7.5mgのリポソーム状パクリタキセル/bw kgが投与される。
【0025】
ヒト患者に対する適用のためのリポソーム状パクリタキセルの適した用量は、約0.01〜2.5、有利には0.02〜1.7、より有利には0.05〜0.5mg/kg bwの量で、少なくとも1日1回、例えば1日につき2回、3回、又はそれ以上;隔日で、約0.01〜5.0、有利には0.02〜2.5、より有利には0.05〜1.7mg/kg bw;1週間に1回で、約0.01〜10、有利には0.02〜5.0、より有利には0.05〜2.5mg/kg bwである。
【0026】
一月量は有利には複数回の1回量ユニットで投与される。低用量の複数回の投与は、単回の高用量の投与と少なくとも同じくらい効果的であってよい。前記治療間隔の間に、前記の用量ユニット及び投与間隔は、一定のままであってよい。その一方で、前記用量ユニットは前記治療間隔の間に増加してよく、例えば開始用量で始め、一段階又は複数段階で、前記開始用量の3又は4倍より高くてよい固定用量へと漸増させる。付加的に又は代替的に、前記の単回投与の間の治療間隔を変化させてよく、例えば前記治療期間の間に減少させるか上昇させてよい。
【0027】
単回投与のための有利な用量は約0.25〜約1.75mg/kg bwである。
【0028】
前記カチオンリポソーム製剤の投与を含む更なる有利な治療プロトコルは、
(i)最初の週に少なくとも3回、特に3〜5回、その後に投与なしで1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、その際、この一月量は有利には約1〜約10mg/kg bw、特に約5〜約7.5mg/kg bw、
(ii)最初の週に1回、その後に投与なしで少なくとも1週間、特に1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、その際、この一月量は有利には約20〜約100mg/kg bw、特に約20〜約60mg/kg bwである。
(iii)1週又は連続した複数週にわたって、有利には少なくとも4週にわたって、1週間に1回、その際、この一月量は有利には約1〜約7.5mg/kg bw、特に約3〜約6mg/kg bw、又は
(iv) (i)、(ii)、及び/又は(iii)の組み合わせ
である。
【0029】
ヒト医薬品での適用のために、本発明の医薬組成物は有利には、有利には約9mg〜約3700mg、特に〜約2237mg/ヒト体表(bs)m2、有利には約18〜約1168mg/m2 bs、特に有利には約37〜約584mg/m2 bsの一月量で投与されてよい。平均的に、ヒト患者は約1.84m2の体表を有する。従って、体重70kg及び身長172cmの平均的な人間に対して、一月量、1回量その他のための有利な値は、上記ではmg/体重(bw)kgで示されてきたが、ヒト適用のためには公知の方法により、種特異的な因数でのかけ算によってmg/ヒト体表(bs)m2の相応する値に変換されてよい。
【0030】
更なる観点において、本発明の前記カチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜約99.9モル%、有利には〜約98モル%、少なくとも約0.1モル%、有利には少なくとも約2モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のために便利である。
【0031】
有利な実施態様において、前記リポソーム製剤はパクリタキセルを、約0.1モル%〜、特に約2モル%〜約8モル%の量で、有利には約0.5モル%〜、特に約2モル%〜約5モル%の量で、より有利には約1モル%〜約4モル%の量で、最も有利には約2.5モル%〜約3.5モル%の量で含有する。本発明のカチオンリポソーム製剤は、パクリタキセル結晶をほぼ含有しない。
【0032】
本発明のリポソーム製剤は、約30モル%〜約99.9モル%、特に〜約70モル%、有利には約40モル%〜約60モル%、最も有利には約45モル%〜約55モル%の量でカチオン脂質を含有するカチオンリポソーム製剤である。前記製剤及び前記カチオン脂質は、正のゼータ電位を、約pH7.5の約0.05M KCl溶液中で、室温で有することにより特徴付けられている。
【0033】
前記リポソーム製剤の有利なカチオン脂質は、N−[1−(2,3−ジオレオイロキシ)プロピル]−N,N,N−トリメチルアンモニウム塩、例えば、このメチルスルファート(DOTAP)である。その他の本発明のために有利な脂質は以下を含んでよい:DDAB、ジメチルジオクタデシルアンモニウムブロミド;1,2−ジアシルオキシ−3−トリメチルアンモニウムプロパン、(以下を含むがこれに限定されない:ジオレオイル、ジミリストイル、ジラウロイル、ジパルミトイル及びジステアロイル;2つの異なるアシル鎖も前記グリセロール骨格に結合していてよい);N−[1−(2,3−ジオロイロキシ)プロピル]−N,N−ジメチルアミン(DODAP);1,2−ジアシルオキシ−3−ジメチルアンモニウムプロパン(以下を含むがこれに限定されない:ジオレオイル、ジミリストイル、ジラウロイル、ジパルミトイル、及びジステアロイル;2つの異なるアシル鎖も、前記グリセロール骨格に結合していてよい);N−[1−(2,3−ジオレイロキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド(DOTMA);1,2−ジアルキルオキシ−3−ジメチルアンモニウムプロパン、(以下を含むがこれに限定されない;ジオレイル、ジミリスチル、ジラウリル、ジパルミチル、及びジステアリル;2つの異なるアルキル鎖も、前記グリセロール骨格に結合していてよい);ジオクタデシルアミドグリシルスペルミン(DOGS);3β−[N−(N’,N’−ジメチルアミノエタン)カルバモイル]コレステロール(DC−Chol);2,3−ジオレオイロキシ−N−(2−(スペルミンカルボキサミド)−エチル)−N,N−ジメチル−1−プロパンアミニウムトリフルオロアセタート(DOSPA);β−アラニルコレステロール;セチルトリメチルアンモニウムブロミド(CTAB);ジC14アミジン;N−tert−ブチル−N’−テトラデシル−3−テトラデシルアミノプロピオンアミジン;14Dea2;N−(アルファ−トリメチルアンモニオアセチル)ジドデシル−D−グルタマートクロリド(TMAG);O,O’−ジテトラデカノイル−N−(トリメチルアンモニオ−アセチル)ジエタノールアミンクロリド;1,3−ジオレオイロキシ−2−(6−カルボキシ−スペルミル)−プロピルアミド(DOSPER);N,N,N’,N’−テトラメチル−N,N’−ビス(2−ヒドロキシエチル)−2,3−ジオレオイロキシ−1,4−ブタンジアンモニウムヨージド;1−[2−(アシルオキシ)エチル]2−アルキル(アルケニル)−3−(2−ヒドロキシエチル)−イミダゾリニウムクロリド誘導体(Solodin et al. (1995) Biochem. 43: 13537-13544によって述べられたもの)、例えば1−[2−(9(Z)−オクタデセノイルオキシ)エチル]−2−(8(Z)ーヘプタデセニル−3−(2−ヒドロキシエチル)イミダゾリニウムクロリド(DOTIM)、1−[2−(ヘキサデカノイルオキシ)エチル]−2−ペンタデシル−3−(2−ヒドロキシエチル)イミダゾリニウムクロリド(DPTIM)、第四級アミンにヒドロキシアルキル部を含有する、2,3−ジアルキルオキシプロピル第四級アンモニウム化合物誘導体(例えばFelgner et al[Felgner et al. J. Biol. Chem. 1994,269, 2550-2561]によって説明されたとおり)、例えば:1,2−ジオレオイル−3−ジメチルヒドロキシエチルアンモニウムブロミド(DORI)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DORIE)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシプロピルアンモニウムブロミド(DORIE−HP)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシブチルアンモニウムブロミド(DORIE−HB)、1,2−ジオレイロキシプロピル−3−ジメチル−ヒドロキシペンチルアンモニウムブロミド(DORIE−Hpe)、1,2−ジミリスチロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DMRIE)、1,2−ジパルミチロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DPRIE)、1,2−ジステリロキシプロピル−3−ジメチル−ヒドロキシエチルアンモニウムブロミド(DSRIE);Santaniello et al. [US5498633]によって報告されたアシルカルニチンのカチオンエステル;ホスファチジルコリンのカチオントリエステル、例えば1,2−ジアシル−sn−グリセロール−3−エチルホスホコリン、その際この炭化水素は飽和又は不飽和であってよく、及び分枝又は非分枝であってよく、C12〜C24の鎖長を有し、この2つのアシル鎖は必ずしも同一でない。
【0034】
有利な実施態様において、前記リポソーム製剤は場合により、少なくとも1つの中性及び/又はアニオン脂質を含有する。中性脂質は、中性の正味の電荷を有する脂質である。アニオン脂質又は両親媒性物質は、負の実効電荷を有する分子である。これらは中性又は負の実効電荷を有する、ステロール又は脂質、例えば、コレステロール、リン脂質、リゾ脂質、リゾリン脂質、スフィンゴ脂質又はペグ化(pegylated)脂質から選択されてよい。有利な中性及びアニオン脂質はこの際以下を含む:カルボン酸基を含む、ホスファチジルセリン、ホスファチジルグリセロール、ホスファチジルイノシトール(特定の糖に限定されない)、脂肪酸、ステロール、例えばコレステロール、1,2−ジアシル−sn−グリセロ−3−ホスホエタノールアミンであり、DOPE、1,2−ジアシル−グリセロ−3−ホスホコリン及びスフィンゴミエリンを含むがこれに限定されない。グリセロール骨格に結合した脂肪酸は、特定の長さ又は二重結合の数に限定されない。リン脂質は、2つの異なる脂肪酸をも有してよい。有利には、この更なる脂質は室温で液状の結晶状態にあり、かつ前記脂質は前記の使用したカチオン脂質と、これらが適用される割合で混合可能である(即ち、均一な相が形成されてよく、相分離又は分域形成は生じない)。有利な実施態様において、前記中性脂質はDOPCである。
【0035】
更なる有利な実施態様において、前記リポソーム製剤は、場合により中性及び/又はアニオン脂質を、有利にはDOPCを、約30〜約70モル%、有利には約40〜約60モル%、特に有利には約45〜約55モル%の量で含有する。
【0036】
本発明の更なる対象は、本発明において使用される前記カチオンリポソーム製剤を脱水し、脱水したまま長期間の間貯蔵し、次に、前記脱水、貯蔵及び再水和の工程の間にこの内容物の大部分を失うことなしに、これを使用すべき時に及び場で再水和することである。後者を達成するために、1つ又はそれ以上の保護剤、例えば凍結保護防止剤が存在してよい。このように、本発明によるカチオンリポソーム製剤は有利には凍結防止剤を含有し、その際前記凍結保護防止剤を糖又はアルコール又はこの組み合わせから選択する。有利には、前記凍結保護防止剤を、トレハロース、マルトース、スクロース、グルコース、ラクトース、デキストラン、マンニトール、又はソルビトールから選択する。
【0037】
更なる有利な実施態様において、前記リポソーム製剤はトレハロースを、前記製剤の総体積に対して、約5%(m/v)〜約15%(m/v)の範囲で含有する。
【0038】
本発明の前記カチオンリポソームの処方は変化してよい。特に有利な実施態様において、DOTAP、DOPC、及びパクリタキセルのモル比は、50:47:3モル%である。この処方は、MBT−0206又はEndoTAG−1とも命名される。
【0039】
様々なサイズのリポソームが本発明において使用される。本発明の有利な実施態様において、カチオンリポソームは約25nm〜約500nm、有利には約50nm〜約500nm、より有利には約100nm〜約300nmの平均粒子直径を有する。
【0040】
本発明のリポソーム製剤は、全身に、有利には静脈内に投与されてよい。
【0041】
本発明のカチオンリポソームは、増加した血管新生に関連した全て形態の症状、例えば癌を処置するために使用されてよい。
【0042】
本発明の医薬組成物は、ヒト患者における腫瘍、例えば膀胱癌、乳癌、結腸直腸癌、子宮内膜癌、白血病、肺癌、リンパ腫、メラノーマ、非小細胞肺癌、卵巣癌、前立腺癌を治療するために、及び、小児癌、例えば脳幹部神経膠腫、小脳星状細胞腫、大脳星状細胞腫、上衣細胞腫、ユーイング肉腫/腫瘍ファミリー、胚細胞腫瘍、頭蓋外の、ホジキン病、白血病、急性リンパ芽球性、白血病、急性骨髄性の、肝臓癌、髄芽腫、神経芽腫、ホジキン病でないリンパ腫、骨の骨肉腫/悪性の線維性組織球腫、網膜芽腫、横紋筋肉腫、軟部組織肉腫、テント上性原始性、神経外胚葉性及び松果体部の腫瘍、普通でない小児癌、視覚路及び視床下部膠腫、ウィルムス腫瘍、及びその他の小児の腎臓癌の治療、及びこれらより一般的でない、以下を含む癌、急性リンパ性白血病、成人の急性骨髄性白血病、成人のホジキン病でないリンパ腫、脳腫瘍、子宮頸癌、小児癌、小児肉腫、慢性リンパ性白血病、慢性骨髄性白血病、食道癌、ヘアリーセル白血病、腎臓癌、肝臓癌、多発性骨髄腫、神経芽腫、口頭癌、膵癌、原発性中枢神経系リンパ腫、皮膚癌、小細胞肺癌、頭頸部癌、胆嚢及び胆管癌、胃癌、消化管癌、カポシ肉腫、尿路上皮細胞癌腫、甲状腺癌腫、精巣癌腫、膣癌、血管肉腫、軟部組織肉腫、及び中皮腫を処置するために特に有利である。特に、前記癌は転移性癌及び/又は標準的な(化学)療法耐性癌であってよい。本発明の組成物の投与は、疾病の進行を遅めるか又は停止させ、又は部分的な又は完全な寛解を導いてよい。更なる症状は、創傷治癒、又は炎症性疾患又は慢性的な炎症性疾患、例えば関節リウマチ、皮膚炎、子宮内膜症、又は乾癬であってよい。
【0043】
本発明のカチオンリポソーム製剤は、上述した癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌、及びメラノーマの治療に、単独療法として、又は更なる治療療法、例えば以下に詳細に示した更なる活性成分との、特に化学療法剤、例えばDNA/RNA代謝拮抗産物、例えばゲムシタビンとの併用療法として、特に適する。
【0044】
意外にも、カチオンリポソーム中に負荷した活性成分は、内皮性又は非内皮性薬剤耐性細胞、特に薬剤耐性の内皮性又は非内皮性腫瘍細胞に対して直接的に作用することが見出された。従って、この新しい知見は非内皮性薬剤耐性細胞を更なる標的として定義し、これによりカチオンリポソームの抗腫瘍性の適用を強める。これは本発明の更なる観点である。
【0045】
従って、活性成分を含有するカチオンリポソーム製剤を、内皮性又は非内皮性の薬剤耐性細胞に対する医薬品の製造のために使用することは本発明の更なる観点である。本発明は、活性成分を含有するカチオンリポソーム製剤の、これを必要とする被験体の薬剤耐性細胞へと、疾病、例えば癌に作用するための治療有効量で投与する方法も提供する。
【0046】
前記観点の更なる実施態様は、薬剤耐性細胞の発生に関連しかつ/または付随する疾病の予防又は治療のための、例えば薬剤耐性腫瘍の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用に関し、特に、癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌及びメラノーマのための二次治療又は三次治療としての使用を意味する。
【0047】
初期治療の失敗の主な原因は、薬剤耐性細胞クローンの出現である。二次治療は、この開始投与が失敗した後に投与される。二次化学療法は、様々な作用様式を有する様々な化学療法剤を用いた単独療法又は複数の更なる薬物又は治療の併用であってよい。しかし、二次治療は、多剤耐性細胞クローンの発生により、同様に失敗する可能性もある。従って、このような腫瘍再発のために三次治療が存在してよい。前記カチオンリポソーム製剤は上述したように二次治療又は三次治療として特に適する。
【0048】
本発明の前記観点のカチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜薬99.9モル%、特に〜約98モル%、少なくとも約0.1モル%、特に少なくとも約2モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、薬剤耐性細胞の発生に関連しかつ、または付随する疾病を軽減する(退縮を引き起こして)か、又は最終的に治癒するように、薬剤耐性細胞に作用するための医薬組成物の製造のために有利である。
【0049】
本発明のうちの更なる意外な知見は、活性成分を含有するカチオンリポソームが単独で、又は転移形成に対する少なくとも1つのその他の治療療法との併用で作用することである。
【0050】
従って本発明の更なる対象は、活性成分を含有するカチオンリポソーム製剤を、転移に対する医薬品を準備するために使用することである。本発明はまた、活性成分を含有するカチオンリポソーム製剤を、これを必要とする被験体に、転移形成の発現及び/又は進行に作用するための、例えば転移性疾病を遅延するか及び/又は回避する、治療有効量で投与する方法をも提供する。「作用する」との用語は本発明において一般的に、所望の薬理学的及び/又は生理学的効果、例えば疾病の発現及び/又は進行を、遅延及び/又は回避する効果が得られることを意味する。有利な実施態様において、本発明は肝臓の転移形成の遅延及び/又は回避のために使用される。
【0051】
また更なる観点においても、本発明のカチオンリポソーム製剤は、少なくとも1つのカチオン脂質約30〜約99.9モル%、特に〜約98モル%、少なくとも約0.1モル%、特に少なくとも約2モル%の量で第1の活性成分、例えばパクリタキセル、及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有し、転移形成の遅延及び/又は回避のための、少なくとも1つの更なる活性成分の有効量、例えば第2の非リポソーム状活性成分及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のために有利である。
【0052】
前記カチオンリポソーム製剤中に負荷した活性成分、例えば単独療法のための活性成分又は、併用療法のための第1の活性成分は、細胞毒性の又は細胞分裂抑制性の物質、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質から選択されてよい。より有利な実施態様においては、前記活性成分は、タキサン、カンプトテシン、スタチン、デプシペプチド、サリドマイド、微小管と相互作用するその他の薬剤、例えばジスコデルモリド(discodermolide)、ラウリマリド(laulimalide)、イソラウリマリド(isolaulimalide)、エレウレテロビン(eleutherobin)、サルコジクチン(Sarcodictyin)A及びBから選択され、最も有利な実施態様においては、パクリタキセル、ドセタキセル、カンプトテシン又はこれらの全ての誘導体から選択される。
【0053】
従って、本発明の有利な実施態様において、前記リポソーム製剤は、タキサン、有利にはパクリタキセル又はドセタキセル、又はこれらの誘導体を、約0.1モル%〜約20モル%の量で、有利には約0,5モル%〜約10モル%の量で、より有利には約1モル%〜約5モル%の量で、最も有利には約2モル%〜約4モル%の量で含有する。
【0054】
前記の少なくとも1つの更なる活性成分は、上述したように細胞毒性の又は細胞分裂抑制性の、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質、過敏反応又は化学増感剤を減少させるか又は消去する化合物であってよい。有利には、前記の少なくとも1つの更なる活性成分は、非リポソーム製剤中に存在する。更に、前記活性成分及び前記の更なる活性成分が異なることが有利である。
【0055】
有利な実施態様において、前記の更なる活性成分は、抗腫瘍剤、特に有糸分裂阻害剤、例えばパクリタキセル、アルキル化剤、特に白金含有化合物、特にシスプラチン、カルボプラチン、DNAトポイソメラーゼ阻害剤、例えばカンプトテシン、又はドキソルビシン、RNA/DNA代謝拮抗物質、特に5−フルオロウラシル又はゲムシタビン、及び/又はその他の抗腫瘍活性を有する化合物から選択される。特に有利なのは、シスプラチン又はカルボプラチンとの、又は5−フルオロウラシル又はゲムシタビンとの併用療法である。
【0056】
更なる有利な実施態様において、過敏反応を減少させるか又は消去する化合物は、致命的なアナフィラキシー反応を妨げるために十分な量にある、ステロイド、抗ヒスタミン剤、H2レセプターアンタゴニスト、及びこれらの組み合わせを含む(しかしこれらに限定されない)グループから選択される。より更に有利な実施態様において、前記化合物は、ラニチジン、デキサメタゾン、ジフェンヒドラミン、ファモチジン、ヒドロコルチゾン、クレマスチン、シメチジン、プレドニゾロン、プレドニゾン、クロルフェニラミン、クロルフェナミン、マレイン酸ジメチンデン、インドメタジン、及びプロメタジン又はこれらの全ての誘導体を含むグループから選択される。
【0057】
有利な実施態様において、前記化学増感剤は、細胞周期調節剤、薬剤耐性を戻す物質、例えばベラパミル、血管作動性物質、例えば抗高血圧剤、カチオンリポソームと血液成分との電荷に関連した相互作用を変更する物質、例えばプロタミンを含む(しかしこれらに限定されない)グループから選択される。
【0058】
1つ又は複数の本発明の観点に対して論じられた全ての有利な実施態様は、その他全ての観点にも関することに留意されたい。これは特に、カチオン脂質の量及び種類、中性及び/又はアニオン脂質の量及び種類、活性成分の量及び種類、併用療法のための更なる活性成分の量及び種類、及び処置すべき疾病の種類を指す。
【0059】
図の説明
図1:治療後の腫瘍体積
治療開始後19日目のL3.6pl膵臓腫瘍の腫瘍サイズ。10%トレハロース、パクリタキセル、MBT−0206、Gemzar(ゲムシタビン)及びMBT−0206とGemzarとの両方の併用での治療を腫瘍細胞接種後8日目に開始した。Gemzarをi.p.で100mg/kgの用量で、1週間に2回(月、木)適用した。パクリタキセル及びMBT−0206は、i.v.で、月、水、金スケジュールで、5mg/kg bwのパクリタキセル用量で適用した。前記併用グループは、それぞれのスケジュールでMBT−0206及びGemzar両方を得た。腫瘍をキャリパーを用いた触診によって、23日目及び27日目に測定した。平均±SEM;グループにつきn=9。
【0060】
図2:治療後の転移
治療開始後19日目の転移。10%トレハロース、パクリタキセル、MBT−0206、Gemzar(ゲムシタビン)及びMBT−0206とGemzarとの両方の併用での治療を腫瘍細胞接種後8日目に開始した。Gemzarをi.p.で100mg/kg bwの用量で、1週間に2回(月、木)適用した。パクリタキセル及びMBT−0206は、i.v.で、月、水、金スケジュールで、5mg/kg bwのパクリタキセル用量で適用した。前記併用グループは、それぞれのスケジュールでMBT−0206及びGemzar両方を得た(グループにつきn=9)。
【0061】
図3〜5:成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
図3:極めて薬剤耐性である子宮肉腫系Mes−SA/Dx−5MBTに対するMBT−0206及びパクリタキセルの阻害能力
図4:適度に薬剤耐性である子宮肉腫系Mes−SA/Dx−5に対するMBT−0206及びパクリタキセルの阻害能力。
図5:薬剤感受性であるヒト子宮肉腫系Mes−SAに対するMBT−0206及びパクリタキセルの阻害能力。
【0062】
図6〜7:成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
図6:極めて薬剤耐性であるマウス結腸癌腫系Colon−26MBTに対するMBT−0206及びパクリタキセルの阻害能力
図7:親系統の薬剤感受性であるマウス結腸癌腫系Colon−26に対するMBT−0206及びパクリタキセルの阻害能力。
【0063】
図8:薬剤感受性であるヒト内皮系統EA.hy926に対するMBT−0206及びパクリタキセルの阻害能力
成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に実施した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【0064】
図9:前記感受性親系統Mes−SA及び前記耐性誘導体Mes−SA/Dx−5MBTに対するパクリタキセルのin vitro阻害能力。成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に試験した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の10〜11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【0065】
図10:薬剤耐性ヒト皮膚メラノーマ系統Sk−Mel28に対するパクリタキセルの阻害能力。成長阻害アッセイを24ウェルプレート中で、各薬剤濃度でもって二重に試験した(n=2ウェル)。4×104細胞/ウェルを24ウェルプレート中に播き、一晩インキュベートした。翌日、それぞれの製剤の11つの濃度を72hの間加え、それぞれのグラフに記した範囲を包含した。最後に、前記細胞の生存率をミトコンドリアの脱水素酵素活性を測定する標準的なMTTアッセイにより決定した。
【実施例】
【0066】
以下の実施例は、本発明の範囲を単に説明するものであり制限する意図はないものとする。その他の一般的な及び特定の構成は当業者に公知である。
【0067】
実施例
1.ヒト治療法プロトコル
この実施例は、開示した製剤を使用するヒト治療プロトコルに関する。治療は、増強した血管新生活性に関連する様々なヒト疾病及び障害を予防及び/又は処置するために使用される。これは抗腫瘍療法において、例えば充実性腫瘍及び血液の悪性腫瘍を有する患者の処置において、様々な慢性的な炎症性疾患、例えば関節リウマチ又は乾癬に対する治療において、特に有利であると考えられる。
【0068】
本発明の特徴は、様々な分類の疾病及び/又は異常が、前記異常に関係する組織又は細胞を直接的にターゲッティングすることなしに直接的に血管新生内皮細胞を標的とすることで治療されてよいことであり、例えば血管新生を阻害することにより腫瘍へのこの血液供給は遮断され、全く前記腫瘍細胞を直接的にターゲッティングすることなしに前記腫瘍は致死する。疾病及び/又は異常のその他の分類は、血管新生内皮細胞を直接的にターゲッティングすることにより、かつ前記異常に関係する組織又は細胞を直接的に標的とすることにより治療されてよい。
【0069】
その他の適用において、薬剤耐性細胞、例えば薬剤耐性癌細胞、又は関節リウマチにおけるきわめて増殖性の滑膜細胞に、直接的に作用してよい。
【0070】
臨床試験の実施(患者の治療及び観察を含む)の様々な要素は、本発明の開示の観点において、当業者に公知である。
【0071】
規定の承認目的のために、試験のために選択された患者が、従来の治療法の少なくとも1クールに応答しないであろうこと、かつ理学的検査、実験室技術、又はX線撮影方法により決定される客観的に測定可能な疾病を有することが検討された。前記患者は心臓又は腎疾患の履歴を有さず、かついかなる化学療法も前記試験の開始少なくとも2週間前に停止すべきものである。
【0072】
適用の前に、前記製剤が凍結乾燥されている場合には、これを水溶液中に再構成してよい。以上に概略したように、必要な適用体積は、患者の体重及び投与スケジュールから計算した。
【0073】
開示した製剤を、短い注入時間の間に投与してよい。いかなる用量レベルでも、投与される注入は、それぞれの注入後に達成される毒性に依存すべきものである。従って、いかなる単独注入後にも、又は一定の割合の注入のための特定の期間にも、IIグレードの毒性に達する場合には、更なる用量を控えるか又は前記の一定の割合の注入を、毒性が改善しない限り停止すべきものである。患者の約60%が全てのカテゴリーにおいて許容不可能なIII又はIVグレードの毒性を示すまで、増加用量を患者グループ投与する。この値の2/3の用量を、安全用量として定義する。
【0074】
理学的検査、腫瘍測定及び実験室試験を無論、治療以前に、及び約3〜4週間後の間隔の際に実施するものである。実験室試験は、全血球計算、血清クレアチニン、クレアチンキナーゼ、電解質、尿素、窒素、SGOT、ビリルビン、アルブミン及び総血清タンパク質を含むものである。
【0075】
臨床反応は、許容可能な測定又は実験室値、例えば腫瘍マーカーの変化によって定義される。例えば、完全寛解は、全ての測定可能な疾病の少なくとも一ヶ月間の消滅により定義されてよく、その一方で部分寛解は50%又はそれ以上の減少によって定義されてよい。
【0076】
本発明において開示及び請求された全ての組成物及び方法は、本発明の開示の観点において過度の実験なしに製造され、かつ実施されてよい。本発明の組成物及び方法は、有利な実施態様の観点において説明されているが、本発明の概念、趣旨及び範囲から逸脱することなく、本発明において説明された組成物に、方法に及び方法の工程において、又は一連の方法の工程において、変形を適用してよいことが当業者には明白である。より詳細には、同等の又は類似の結果が達成される一方で、化学的にも生理学的にも関連するある種の薬剤が、本発明において説明された薬剤の代わりに代用されてよいことが明白である。当業者に公知であるこのような類似の代用物及び修正の全ては、添付の特許請求の範囲によって定義される本発明の趣旨、範囲及び概念のうちにあると考えられる。
【0077】
用量におけるいくつかの変形は、処置される被験体の状態に依存して必然的に生じる。投与の責任者は、いかなる場合においても、この個々の被験体にとって適した用量を決定する。更に、ヒトへの適用に対しては、製剤は、the FDA Office of Biologics standardsが要求する無菌性、発熱原性、一般的な安全性及び純度水準を満たすものである。
【0078】
本発明は、活性成分の本発明の製剤の医薬有効量を、これを必要とする被験体の標的部位、例えば血管新生性の血管標的部位へと、投与する方法も含む。「これを必要とする被験体」とは、哺乳類、例えばヒトを指す。
【0079】
投与経路は有利には、腹膜又は非経口適用を含む。
【0080】
本発明での使用のために、これを必要とする被験体へ投与される化合物の「薬理学的有効量」は、様々な範囲の因子に依存して変化する。前記化合物の量は、この患者の大きさ、年齢、性別、重量及び状態にも、投与される前記物質の効力にも依存する。投与に関して様々な変形が存在することを示してきたが、当業者は、本発明の開示を用いて容易に、極めて少量を最初に投与し、所望の結果が得られるまで前記用量を増分して上昇させることによる、適切な投与を決定することが可能であると考えられている。前記投与量は上述した因子によって非常に変化するが、一般的には、病的組織を標的とするに過ぎない、例えばこの腫瘍細胞自体を標的とするに過ぎないデリバリー系に比較して、大幅に少ない量でいかなる物質をも投与することが本発明により可能になる。
【0081】
2.単独療法プロトコル
【0082】
【表1】
【0083】
【表2】
有効性
応答を、WHO又はRECIST判定基準に従って評価した。
【0084】
3.併用療法プロトコル
【0085】
【表3】
【0086】
更に計画された試験は、リポソーム状パクリタキセル、例えば0.5又は1.0又は1.5mg/kg及びゲムシタビン、例えば1000mg/m2を、週に1回、3週間の間、その後に治療なしで1週間、有利には少なくとも1年間の間隔の投与を含む。
【0087】
リポソーム状パクリタキセルの前記治療スケジュールは上述したように進行中の試験のためである。
【0088】
有効性
応答を、WHO又はRECIST判定基準に従って評価した。
【0089】
4.症例報告#1
患者:
−喉頭の粘膜表皮癌の、大きな治療耐性再発を有する49歳の患者
−頸部、鎖骨上、腋窩、縦隔及び肺の転移
−最初の腫瘍切除の5年後、頸部腫瘍切開及び補助放射線療法
−複数回の切除、形成外科手術、放射線療法及び化学療法を用いた再発の繰り返した治療後。
投与スケジュール:
−MBT−0206:50/47/3(DOTAP/DOPC/パクリタキセル)
−リポソーム状パクリタキセル 0.06、0.25、0.5及び1.0mg/kg bwの投与、i.v.
−1週間に3回の1サイクル(1、3、及び5日目)
結果:
−注入時及び後の心臓血管、肺、及び血清のパラメーター観察の間の良好な認容性
−急性又は慢性毒性の徴候なし
−腫瘍の血液循環の減少
−3ヶ月の間、腫瘍成長の強力に減少した進行。
【0090】
5.症例報告#2
多重化学療法後に疾病進行を有した、肝臓細胞癌腫を有する一患者をMBT−0206で治療した。
【0091】
凍結乾燥したMBT−0206を、注射のために水により再構成し、総注入体積300〜400mlのMBT−0206(リポソーム状パクリタキセル1.0mg/体重kgの用量に相当)を、中心又は末梢静脈注入により2〜4hの期間にわたり投与した。この注入率を、最大速度2.5ml/minにまでゆっくりと上昇させた。前投薬は、前記患者の性別、年齢又は条件に依存した。上述した患者の特定の場合には、デキサメタゾン及び抗ヒスタミン剤が投与されている。
【0092】
MBT−0206を、1週間に1回、2回のリポソーム状パクリタキセル0.25mg/kg bw 1回のリポソーム状パクリタキセル0.5mg/kg bw)で開始し、次に19回のリポソーム状パクリタキセル1.0mg/kg bwの強化用量にする、用量漸増スケジュールでもって投与した。前記治療は22週間の投与後にいまだ進行中であり、かつ今現在まで、薬物有害反応は報告されていない。好ましい安全特性の他に、肝臓のCT−スキャンによって実施された腫瘍サイズの最後の評価では、安定した疾病を示した。
【0093】
6.症例報告#3
その他の症例において、ホルモン療法に治療抵抗性になった前立腺癌患者を、リポソーム状パクリタキセル1.0mg/kg bwで、週に3回、3日毎に、上述した製剤及び投与と同様の条件下で処置した。この前投薬は、デキサメタゾン及び抗ヒスタミン剤を含有した。前記患者に対するリポソーム状パクリタキセルの蓄積用量は、リポソーム状パクリタキセル3.0mg/kg bwであった。
【0094】
7.リポソーム状パクリタキセルでの低用量スケジュール
不死化内皮細胞(EA.hy926)を24ウェルプレート(4×104細胞/ウェル)中に播き、一晩育てた。翌日、9つのウェルを1hの間、MBT−0206として処方した51.2ng/ml(60nM)のリポソーム状パクリタキセルの低用量で処理した。更に、処方につき3ウェルを、MBT−0206として処方した153.7ng/mlの高用量(180nM)で1h処理し、3ウェルは未処理のままにした。約24h後に、9つの低用量処理ウェルのうち6つを、再度同様の低用量MBT−0206で1h処置した(即ち:2×処理群)。再度24h後、前記6つの2回処理したウェルのうち3つを、MBT−0206として処方した51.2ng/mlのパクリタキセルで3度目に、1hの間処置した(3×処置群)。前記の3度目の処理の約96h後に、全てのウェルの細胞生存率を定量化した。この目的のために、テトラゾリウム塩の3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)を用いたミトコンドリア脱水素酵素の活性を測定するアッセイを、標準的なプロトコルに従って適用した(例えば、わずかに改変した{Lindl, 1994#28})。
【0095】
この結果は、3回の低用量MBT−0206で処理した細胞の生存率は、高用量で1回のみ処理した細胞の生存率と少なくとも同じくらい強力に減少していたことを示す。1回又は2回低用量MBTで処理した細胞は、幾分上昇した生存率を示したが、これは未処理細胞に比較すると減少している。
【0096】
結論
高用量のMBT−0206での処理を、より高い頻度での低用量を用いて置き換えてよい。治療密度(1週間における治療の数)と治療有効性との間には相関がある。低用量での1週間に3回の治療は、1週間に1又は2回の治療よりも優れていている。この最適化した投与治療方式は、高用量治療により引き起こされる毒性副作用を潜在的に減少させる。
【0097】
8.L3.6pl膵臓腫瘍中での、ゲムシタビン(50mg/kg)と併用したMBT−0206の抗腫瘍有効性
動物モデル
種:雄Balb/c nu/nuマウス
腫瘍モデル:正所性L3.6pl膵臓充実性腫瘍(極めて転移性:Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999;1(1) : 50-62.)
供給者:Charles River France。
【0098】
治療
治療開始:腫瘍接種後7日目
最後の治療:腫瘍接種後26日目
用量:適用につきパクリタキセル5mg/kg bwを有するMBTー0206
:適用につきパクリタキセル5mg/kg bwのパクリタキセル
:適用につき50mg/kgのゲムシタビン(Gemzar)
スケジュール:
MBT−0206、パクリタキセル、トレハロース:d7、10、12、14、17、19、21、24、26
ゲムシタビン:d7、11、14、18、21、25
併用:併用した単独治療
適用経路:MBT−0206、パクリタキセル、トレハロース:尾静脈中へのi.v.ボーラス
ゲムシタビン:i.p.ボーラス。
【0099】
【表4】
【0100】
前記死亡率は21日目前では低かった:対照動物1匹及びMBT−0206動物1匹が死んだのみであった。24日目には、不明の理由により死亡率が上昇した:3匹のMBT−0206、1匹のGemzar、及び4匹の対照動物が24日目に死んだ。
【0101】
前記体重はいずれの治療によっても影響を受けなかったが、対照腫瘍の重量がこの最後の週の間に18%だけ減少した。
【0102】
観察パラメーター
−接種後10、12、14、17、19、21、24日目に触診した、及び回収後26、28日目の腫瘍体積
−26、及び28日目に回収後に剖検。
【0103】
結果
いかなる治療の効果も、治療の開始後3日間、触診によっては観察されなかった。強力な抗腫瘍効果が、触診により1週間後及び24日目に、以下の有効性の順位で観察された。Gemzar−50=パクリタキセル<MBT−0206<MBT−0206+Gemzar。しかし、26日目の回収後、全ての群のこの測定した腫瘍体積は、24日目に比較して明らかに小さかった。この大きさの差は、回収前の不正確な触診のためのようである。24日目に、この対照群(n=2)に比較して単独治療により腫瘍サイズは〜30%に減少した。MBT−0206+Gemzarの併用は、13%へとこの腫瘍サイズの最も強力な減少を生じ、これはパクリタキセル、Gemzar及びMBT−0206単独に比較して、有意に(p<0.05)より有効であった。28日目の前記対照群(n=2)を示さないのは、この2つの腫瘍のうち1つが、その他の腫瘍(24、26、28日目)に比較して極めて小さく、従って代表とは見なされないからである。前記単独療法後の腫瘍は、26日目(パクリタキセル:536mm3;MBT−0206:392mm3;Gemzar:398mm3)に比較して腫瘍サイズに微弱な増加を示したが、その一方で前記併用療法は、26日目と28日目の間に88mm3とわずかな腫瘍の退縮を生じた。
【0104】
前記データは前記モデル中でのパクリタキセル、Gemzar及びMBT−0206の強力な抗腫瘍性効能を示す。MBT−0206の抗腫瘍作用は、パクリタキセルよりわずかに強力であるが、Gemzarと類似している。MBT−0206とGemzatとの併用は、顕著な抗腫瘍性効能を示した。
【0105】
9.L3.6pl膵臓腫瘍中のゲムシタビン(100mg/kg)と併用したMBT−0206の抗腫瘍性効能
動物モデル
種:雄Bald/c nu/nuマウス
腫瘍モデル:正所性L3.6pl膵臓充実性腫瘍(極めて転移性)
供給者:Charles River France。
【0106】
治療
治療開始:腫瘍接種後8日目(01.05.03)
最後の治療:腫瘍接種後26日目(19.05.03)
スケジュール:
MBT−0206、パクリタキセル、トレハロース:d9、12、14、16、19、21、23、26
ゲムシタビン:d8、12、15、19、22、26
併用:併用した単独治療。
【0107】
【表5】
観察パラメーター
−接種後23、26日目に触診した及び回収後の腫瘍体積
−1、7、12、16、19、21、23、27日目の体重
−27日目に回収後の剖検。
【0108】
結果(図1及び2参照)
全ての治療的療法の明らかな抗腫瘍効果が、併用療法の顕著な効能と共に、腫瘍接種後23日目に観察された(図1)。腫瘍阻害の順位:パクリタキセル<MBT−0206=Gemzar−50<MBT−0206+Gemzar。この対照群と比較して、前記の最終的な腫瘍体積は、MBT−0206によって46%(p<0.05)にまで、Gemzatによって47%(p<0.01)にまで、及び併用療法によって22%(p<0.01)にまで、27日目には有意に減少した。パクリタキセル治療は、最終的な腫瘍体積を68%にまで減少させたが、これは有意でなかった。興味深いことに、MBT−0206及び併用療法の効能は、23日目でより顕著であった。この原因は、23日目と27日目との週末の間に延長した治療間隔である可能性がある。前記データは、パクリタキセル、Gemzar、及びMBT−0206の前記動物モデル中での明らかな抗腫瘍性効能を明らかにした。MBT−0206の抗腫瘍性作用は、パクリタキセルよりわずかに強力であったが、Gemzarに類似していた。MBT−0206及びGemzarとの併用の両方共に、転移形成を阻害した(図2)。肝転移は、前記の2つの群でのみ存在しなかった。更に、併用群においてのみ、このリンパ節転移がほとんどなかった。前記データは、MBT−0206とGemzarとの併用が、いずれかの単独療法の抗腫瘍性効能を促進することを明らかにした。
【0109】
この死亡率は、前記治療群で、特に併用治療の間にわずかに増加した(4匹のマウスが死亡)。回収前にはどの対照動物も死亡しなかった。
【0110】
対照マウスの体重は、この最後の11日間の間に18%だけ減少した。この期間の間に、一過的な減少もまた前記処置マウスに関して観察され、これはパクリタキセルに関しては2%の、MBT−0206に関しては12%の、併用に関しては19%の、及びGemzarに関しては22%の重量損失を生じた。
【0111】
10.パクリタキセル耐性細胞(例えば、腫瘍細胞系)の致死
(多)薬剤耐性を発現している腫瘍を直接的に致死させるためのMBT−0206の能力を実証するために、2つの極めてパクリタキセルに耐性である哺乳類腫瘍細胞系をin vitroで調査した。前記細胞系を、段階的に上昇するパクリタキセル濃度により培地中で選択した。両方の細胞系は、高い耐性レベルを発現し、これはパクリタキセル約1〜5μM(867又は5000ng/ml)に対する50%の成長阻害(IC50値)のための濃度により反映されている。両方の場合において、MBT−0206はパクリタキセルよりも、薬剤耐性腫瘍細胞の致死において明らかに優れていた。対照的に、薬剤耐性の又は耐性の低い細胞系において、MBT−0206はパクリタキセルと幾分同様の致死能力を有した。
【0112】
MBT−0206とヒト子宮肉腫由来の細胞系Mes−SAとその誘導体の系統
極めてパクリタキセル耐性である誘導体細胞系Mes−SA/Dx−5MBTを、パクリタキセル濃度の増加により、市販の系Mes−SA/Dx−5(ATCC,{Harker,1986 #29})から選択した。図3に示したように、前記誘導体細胞系はIC50値867ng/mlによって示されるように、パクリタキセルに極めて耐性であった。意外にも、MBT−0206が前記細胞系を、約20倍低いIC50値によって反映されるように、より効果的に致死させることが見出された。前記の市販の系Mes−SA/Dx−5は、親系統であるMes−SAからドキソルビシンにより選択される({Harker, 1986#29})が、パクリタキセルに対して低レベルの交差耐性を発現した(図3〜5を比較のこと)。パクリタキセルに対するIC50値は、Mes−SA/Dx−5MBTにおける値よりも約7倍低かった。これに伴い、前記細胞系において、パクリタキセルに比較して、MBT−0206のより高い致死能力のわずかな傾向が存在するにすぎない(図4)。前記親系統Mes−SAは、5.5ng/mlの低いIC50値によって示されるように、極めてパクリタキセル感受性であった(図5)。前記薬剤感受性系統に対して、MBT−0206はパクリタキセルと同様の致死能力を有した。これは、これまで調査した、その他全てのパクリタキセル感受性細胞系にも当てはまった。前記知見に対する例として、MBT−0206及びパクリタキセルでの、不死化内皮系統、EA.hy926の治療の結果を図8に示した。
【0113】
MBT−0206と細胞系マウス結腸癌腫由来のColon−26
Mes−SA/Dx−5MBTと同様に、マウス結腸癌腫系統Colon−26の極めてパクリタキセル耐性である誘導体系統(Cell lines Service,Heidelberg)を樹立し、Colon−26MBTと呼んだ。パクリタキセルに対するIC50値は約5μg/mlであった(図6)。再度、Mes−SA/Dx−5MBTと同様に、MBT−0206はこの細胞系の成長を阻害するための明らかにより高い能力を有した。前記細胞系において、前記IC50値は、3倍だけ違っていた。Mes−SA及びEA.hy926細胞に対して示した前記結果に沿って、この親系統の薬剤感受性Colon−26は、MBT−0206及びパクリタキセルに対して同じ様に感受性であった(図7)。
【0114】
結論
極めてパクリタキセル耐性である細胞系において、MBT−0206はパクリタキセルの致死能力よりも有意により高い致死能力を有した。パクリタキセル感受性系においては、両方のパクリタキセル製剤は匹敵する効能を有した。MBT−0206は従って、(多)薬剤耐性腫瘍をも直接的にin vitro及びin vivoで致死させることができる可能性がある。従ってこれは、パクリタキセルに対して不応答性になったヒト腫瘍(又はその他の疾病)を治療する新規のアプローチである可能性がある。
【0115】
11.再発性頭頸部扁平上皮癌の治療
パクリタキセル負荷したカチオンリポソーム(MBT−0206)の安全性評価を、再発性の治療抵抗性の頭頸部扁平上皮癌を有する患者において実施した。
【0116】
試験設計
−再発性の治療抵抗性の頭頸部扁平上皮癌を有する患者
−少なくともスパイラルCT/MRIスキャンでの次元において、正確に測定されえる、少なくとも1つの障害として定義された測定可能な疾病
−カルノフスキー動作指数>60%
−余命>4ヶ月。
【0117】
薬剤投与
−カチオンリポソーム中のカプセル化パクリタキセルを2つの用量で投与した:パクリタキセル0.55mg/kg及びパクリタキセル1.10mg/kg
−スクリーニング期間の後、1、3、及び5日目に前記薬剤を静脈内注射した。
【0118】
結論
注射の間及び後には、急性又は慢性の毒性の徴候は観察されなかった;バイタル及び実験室安全パラメーラーはほぼ一定のままであった。
【0119】
前記結果は、前臨床毒性学試験から提案された前記用量及びスケジュールが、前記患者により良好に認容されたことを示す。
【0120】
カチオンリポソームはヒト頭頸部扁平上皮癌の内皮を選択的に標的とした。レーザードップラーフローメトリーは、前記療法の抗血管新生作用機構を確認した。
【0121】
12.MBT−0206(EndoTag 1)は、パクリタキセル耐性腫瘍に対してin vivoで効果がある。
パクリタキセルでのin vitroでの72hの処理で高い生存率を有する2つの腫瘍細胞系を、マウス腫瘍モデルとして、MBT−0206の前記能力をin vivoで調査するために使用した。前記腫瘍モデルでは、MBTー0206が腫瘍成長を、パクリタキセルよりも、有意により効果的に阻害することが実証されてよい。
【0122】
1.極めてパクリタキセル耐性である誘導体(Mes−SA/Dx−5MBT、IC50、0.87μg/ml)を、前記の市販のヒト子宮肉腫由来の細胞Mes−SA/Dx−5(ECACC)から選択した。前記の適度に耐性である系統は、極めて感受性である親系統のMes−SAから生じる。図9に示したとおり、前記系統Mes−SA/Dx−5MBTは前記親系統のMesSAと比較すると、このそれぞれのIC50値により評価して、〜150倍まで増加したパクリタキセル認容性を示した。in vivo実験のために、Mes−SA/Dx−5MBT細胞をNMRIヌードマウス中に皮下注射(s.c.)した。MBT−0206又はパクリタキセルでの治療を12日目に開始し、21日目まで、1週間に3回の用量で延長した(5mg/kg b.w.のパクリタキセル)。MBT−0206処置動物の平均腫瘍サイズを、パクリタキセル処置動物の腫瘍サイズと、この最後の治療後2日目(治療開始後23日目)に比較した。MBT−0206が誘導した減少を、パクリタキセルと比較して%で示した(下の表参照)。下記表及び図9中に示したように、MBT−0206は、パクリタキセルよりも、前記腫瘍モデル中で、前記腫瘍成長を〜1/3だけ減少させることにおいて明らかにより効果があった。
【0123】
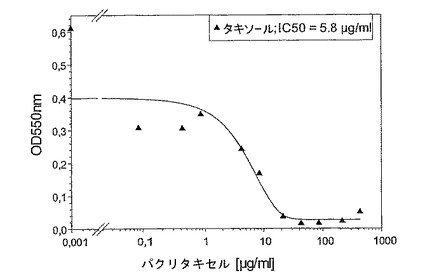
2.ヒトの皮膚メラノーマ系統Sk−Mel28に関して、市販の系統は既に高いパクリタキセル耐性(IC50、5.8μg/ml、図10)を示し、かつ感受性の誘導体の系統は比較のためには使用可能でなかった。in vivoにおいて、前記細胞を、ヒトの皮膚の移植とマウスのアクセプター皮膚との間でのヒトの皮膚の移植を有するSCIDマウス中に注射した。治療を腫瘍細胞注射後17日目に開始し、2日毎に用量(12.5mg/kg b.w.)で28日目まで延長した。MBT−0206及びパクリタキセルの治療能力を、治療開始後7日目に比較した(下の表参照)。前記腫瘍モデル中で、MBT−0206は腫瘍発達をほぼ完全にブロックし、その一方でパクリタキセルは完全に効果がなかった。MBT−0206で処理すると、腫瘍はパクリタキセル(パクリタキセル)−処理腫瘍の大きさの約1/10へとほんのわずかになった。
【0124】
表
【0125】
【表6】
Mes−SA/Dx−5MBT腫瘍モデル:治療開始後23日目の腫瘍体積(9つの治療、即ち腫瘍細胞注射後週に3回、12日目から31日目まで)
Sk−Mel腫瘍モデル:治療開始後7日目の腫瘍体積(一日おきの6つの治療、即ち、腫瘍細胞注入後17日目から28日目まで)。
【0126】
【表7】
【0127】
【表8】
【0128】
【表9】
【0129】
【表10】
【特許請求の範囲】
【請求項1】
必要とするヒト患者に、パクリタキセル約0.25mg〜約60mg/前期患者体重kgの一月量で投与するための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項2】
前記一月量が、パクリタキセル約0.5〜約30mg/体重kgである、請求項1記載の使用。
【請求項3】
前記一月量が、パクリタキセル約1.0〜約15mg/体重kgである、請求項1又は2記載の使用。
【請求項4】
前記一月量が、パクリタキセル約1〜約7.5mg/体重kgである、請求項1又は2記載の使用。
【請求項5】
前記一月量がパクリタキセル約20〜約60mg/体重kgである、請求項1又は2記載の使用。
【請求項6】
前記カチオンリポソーム製剤を、1日に少なくとも1回投与する、請求項1から5までのいずれか1項記載の使用。
【請求項7】
前記カチオンリポソーム製剤を1ヶ月間のうち複数回投与し、このそれぞれの回は、1日間〜3週間の間隔により隔てられている、請求項1から6までのいずれか1項記載の使用。
【請求項8】
前記カチオンリポソーム製剤の投与が、
(i)最初の週に少なくとも3回、特に3〜5回、その後に投与なしで1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、
(ii)最初の週に1回、その後に投与なしで少なくとも1週間、特に1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、
(iii)1週又は連続した複数週にわたって1週間に1回、又は
(iv)(i)、(ii)、及び/又は(iii)の組み合わせ
である、請求項1から7までのいずれか1項記載の使用。
【請求項9】
少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項10】
前記組成物が、少なくとも1つの更なる活性成分の有効量との共同での、同時の併用療法のためである、請求項9記載の使用。
【請求項11】
前記カチオンリポソーム製剤が、パクリタキセルを少なくとも約2〜約8モル%の量で含有する、請求項1から10までのいずれか1項記載の使用。
【請求項12】
前記カチオンリポソーム製剤が、パクリタキセルを約2.5〜約3.5モル%の量で含有する、請求項1から11までのいずれか1項記載の使用。
【請求項13】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項1から12までのいずれか1項記載の使用。
【請求項14】
前記カチオンリポソーム製剤が、ほぼパクリタキセル結晶を含有しない、請求項1から13までのいずれか1項記載の使用。
【請求項15】
血管新生に関連した症状の治療のための、請求項1から14までのいずれか1項記載の使用。
【請求項16】
創傷治癒、癌、炎症性疾患又は慢性的な炎症性疾患、例えば関節リウマチ、皮膚炎、乾癬、又は子宮内膜症を治療するための、請求項15記載の使用。
【請求項17】
薬剤耐性細胞の発生に関連しかつ/または付随する疾病の予防又は治療のための、例えば薬剤耐性腫瘍の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項18】
二次治療又は三次治療としての、特に癌のための二次治療又は三次治療としての請求項17記載の使用。
【請求項19】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項17又は18記載の使用。
【請求項20】
転移形成、例えば発現及び/又は進行、特に腫瘍の疾病に関連しかつ/または付随する転移形成の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項21】
肝転移形成の予防又は治療のための医薬組成物の製造のための、請求項20記載の使用。
【請求項22】
転移の発現及び/又は進行、例えば腫瘍に関連しかつ/または付随する転移の発現及び/又は進行に抗する、少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項23】
前記組成物が、少なくとも1つの更なる活性成分の有効量との共同での、同時の併用療法のためである、請求項22記載の使用。
【請求項24】
前記活性成分は、細胞毒性の又は細胞分裂抑制性の物質、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質から選択される、請求項17から23までのいずれか1項記載の使用。
【請求項25】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項20から24までのいずれか1項記載の使用。
【請求項26】
前記活性成分が、タキサン、カンプトテシン、スタチン、デプシペプチド、サリドマイド、微小管と相互作用するその他の薬剤、例えばジスコデルモリド、ラウリマリド、イソラウリマリド、エレウテロビン、サルコジクチンA及びBから選択され、最も有利な実施態様においては、パクリタキセル、ドセタキセル、カンプトテシン又はこれらの全ての誘導体から選択される、請求項20から25までのいずれか1項記載の使用。
【請求項27】
前記の更なる活性成分は、抗内皮細胞性の活性物質、抗腫瘍性の活性物質、化学療法剤、免疫学的活性物質、過敏反応又は化学増感剤を減少させるか又は消去する化合物である、請求項9又は22記載の使用。
【請求項28】
前記の更なる活性成分は、抗腫瘍剤、特に有糸分裂阻害剤、例えばパクリタキセル、アルキル化剤、特に白金含有化合物、例えばシスプラチン、カルボプラチン、DNAトポイソメラーゼ阻害剤、例えばカンプトテシン、又はドキソルビシン、RNA/DNA代謝拮抗物質、特に5−フルオロウラシル又はゲムシタビン、及び抗腫瘍活性を有するその他の化合物から選択される、請求項9、22、又は27記載の使用。
【請求項29】
過敏反応を減少させるか又は消去する前記化合物は、致命的なアナフィラキシー反応を妨げるために十分な量にある、ステロイド、抗ヒスタミン剤、H2レセプターアンタゴニスト、及びこれらの組み合わせを含むグループから選択される、請求項27記載の使用。
【請求項30】
前記化合物が、ラニチジン、デキサメタゾン、ジフェンヒドラミン、ファモチジン、ヒドロコルチゾン、クレマスチン、シメチジン、プレドニゾロン、クロルフェニラミン、クロルフェナミン、マレイン酸ジメチンデン、及びプロメタジンを含むグループから選択される、請求項28記載の使用。
【請求項31】
前記化学増感剤は、細胞周期調節剤、薬剤耐性を戻す物質、例えばベラパミル、血管作動性物質、例えば抗高血圧剤、カチオンリポソームと血液成分との相互作用を変更する物質、例えばプロタミンを含むグループから選択される、請求項27記載の使用。
【請求項32】
癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌、及びメラノーマの治療のための、請求項1から31までのいずれか1項記載の使用。
【請求項33】
前記カチオンリポソーム製剤は、平均粒子直径約25nm〜約500nm、有利には約100nm〜約300nmを有するリポソームを含有する、請求項1から32までのいずれか1項記載の使用。
【請求項34】
前記カチオンリポソーム製剤は、全身に、有利には静脈内に投与される、請求項1から30までのいずれか1項記載の使用。
【請求項35】
必要とするヒト患者に、パクリタキセル約9mg〜約2337mg/前記ヒト患者体表m2の一月量で投与するための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項1】
必要とするヒト患者に、パクリタキセル約0.25mg〜約60mg/前期患者体重kgの一月量で投与するための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項2】
前記一月量が、パクリタキセル約0.5〜約30mg/体重kgである、請求項1記載の使用。
【請求項3】
前記一月量が、パクリタキセル約1.0〜約15mg/体重kgである、請求項1又は2記載の使用。
【請求項4】
前記一月量が、パクリタキセル約1〜約7.5mg/体重kgである、請求項1又は2記載の使用。
【請求項5】
前記一月量がパクリタキセル約20〜約60mg/体重kgである、請求項1又は2記載の使用。
【請求項6】
前記カチオンリポソーム製剤を、1日に少なくとも1回投与する、請求項1から5までのいずれか1項記載の使用。
【請求項7】
前記カチオンリポソーム製剤を1ヶ月間のうち複数回投与し、このそれぞれの回は、1日間〜3週間の間隔により隔てられている、請求項1から6までのいずれか1項記載の使用。
【請求項8】
前記カチオンリポソーム製剤の投与が、
(i)最初の週に少なくとも3回、特に3〜5回、その後に投与なしで1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、
(ii)最初の週に1回、その後に投与なしで少なくとも1週間、特に1〜3週間の間隔、及び場合によりこのプロトコルの1回又は複数回の繰り返し、
(iii)1週又は連続した複数週にわたって1週間に1回、又は
(iv)(i)、(ii)、及び/又は(iii)の組み合わせ
である、請求項1から7までのいずれか1項記載の使用。
【請求項9】
少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項10】
前記組成物が、少なくとも1つの更なる活性成分の有効量との共同での、同時の併用療法のためである、請求項9記載の使用。
【請求項11】
前記カチオンリポソーム製剤が、パクリタキセルを少なくとも約2〜約8モル%の量で含有する、請求項1から10までのいずれか1項記載の使用。
【請求項12】
前記カチオンリポソーム製剤が、パクリタキセルを約2.5〜約3.5モル%の量で含有する、請求項1から11までのいずれか1項記載の使用。
【請求項13】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項1から12までのいずれか1項記載の使用。
【請求項14】
前記カチオンリポソーム製剤が、ほぼパクリタキセル結晶を含有しない、請求項1から13までのいずれか1項記載の使用。
【請求項15】
血管新生に関連した症状の治療のための、請求項1から14までのいずれか1項記載の使用。
【請求項16】
創傷治癒、癌、炎症性疾患又は慢性的な炎症性疾患、例えば関節リウマチ、皮膚炎、乾癬、又は子宮内膜症を治療するための、請求項15記載の使用。
【請求項17】
薬剤耐性細胞の発生に関連しかつ/または付随する疾病の予防又は治療のための、例えば薬剤耐性腫瘍の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項18】
二次治療又は三次治療としての、特に癌のための二次治療又は三次治療としての請求項17記載の使用。
【請求項19】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項17又は18記載の使用。
【請求項20】
転移形成、例えば発現及び/又は進行、特に腫瘍の疾病に関連しかつ/または付随する転移形成の予防又は治療のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項21】
肝転移形成の予防又は治療のための医薬組成物の製造のための、請求項20記載の使用。
【請求項22】
転移の発現及び/又は進行、例えば腫瘍に関連しかつ/または付随する転移の発現及び/又は進行に抗する、少なくとも1つの更なる活性成分の有効量及び/又は温熱療法及び/又は放射線療法及び/又は寒冷療法との共同での、同時の、別々の又は一連の併用療法のための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量で活性成分及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【請求項23】
前記組成物が、少なくとも1つの更なる活性成分の有効量との共同での、同時の併用療法のためである、請求項22記載の使用。
【請求項24】
前記活性成分は、細胞毒性の又は細胞分裂抑制性の物質、例えば抗腫瘍性の又は抗内皮細胞性の活性物質、化学療法剤又は免疫学的活性物質から選択される、請求項17から23までのいずれか1項記載の使用。
【請求項25】
前記カチオンリポソーム製剤が、DOTAP、DOPC、及びパクリタキセル、50:47:3モル%を含有する、請求項20から24までのいずれか1項記載の使用。
【請求項26】
前記活性成分が、タキサン、カンプトテシン、スタチン、デプシペプチド、サリドマイド、微小管と相互作用するその他の薬剤、例えばジスコデルモリド、ラウリマリド、イソラウリマリド、エレウテロビン、サルコジクチンA及びBから選択され、最も有利な実施態様においては、パクリタキセル、ドセタキセル、カンプトテシン又はこれらの全ての誘導体から選択される、請求項20から25までのいずれか1項記載の使用。
【請求項27】
前記の更なる活性成分は、抗内皮細胞性の活性物質、抗腫瘍性の活性物質、化学療法剤、免疫学的活性物質、過敏反応又は化学増感剤を減少させるか又は消去する化合物である、請求項9又は22記載の使用。
【請求項28】
前記の更なる活性成分は、抗腫瘍剤、特に有糸分裂阻害剤、例えばパクリタキセル、アルキル化剤、特に白金含有化合物、例えばシスプラチン、カルボプラチン、DNAトポイソメラーゼ阻害剤、例えばカンプトテシン、又はドキソルビシン、RNA/DNA代謝拮抗物質、特に5−フルオロウラシル又はゲムシタビン、及び抗腫瘍活性を有するその他の化合物から選択される、請求項9、22、又は27記載の使用。
【請求項29】
過敏反応を減少させるか又は消去する前記化合物は、致命的なアナフィラキシー反応を妨げるために十分な量にある、ステロイド、抗ヒスタミン剤、H2レセプターアンタゴニスト、及びこれらの組み合わせを含むグループから選択される、請求項27記載の使用。
【請求項30】
前記化合物が、ラニチジン、デキサメタゾン、ジフェンヒドラミン、ファモチジン、ヒドロコルチゾン、クレマスチン、シメチジン、プレドニゾロン、クロルフェニラミン、クロルフェナミン、マレイン酸ジメチンデン、及びプロメタジンを含むグループから選択される、請求項28記載の使用。
【請求項31】
前記化学増感剤は、細胞周期調節剤、薬剤耐性を戻す物質、例えばベラパミル、血管作動性物質、例えば抗高血圧剤、カチオンリポソームと血液成分との相互作用を変更する物質、例えばプロタミンを含むグループから選択される、請求項27記載の使用。
【請求項32】
癌、特に膵癌、手術不可能な膵癌、消化管癌、肺癌、結腸直腸癌又は胃癌、乳癌、前立腺癌、及びメラノーマの治療のための、請求項1から31までのいずれか1項記載の使用。
【請求項33】
前記カチオンリポソーム製剤は、平均粒子直径約25nm〜約500nm、有利には約100nm〜約300nmを有するリポソームを含有する、請求項1から32までのいずれか1項記載の使用。
【請求項34】
前記カチオンリポソーム製剤は、全身に、有利には静脈内に投与される、請求項1から30までのいずれか1項記載の使用。
【請求項35】
必要とするヒト患者に、パクリタキセル約9mg〜約2337mg/前記ヒト患者体表m2の一月量で投与するための医薬組成物の製造のための、少なくとも1つのカチオン脂質約30〜約99.9モル%、少なくとも約0.1モル%の量でパクリタキセル及び少なくとも1つの中性及び/又はアニオン脂質約0〜約70モル%を含有するカチオンリポソーム製剤の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2012−229255(P2012−229255A)
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願番号】特願2012−159071(P2012−159071)
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2006−534713(P2006−534713)の分割
【原出願日】平成16年10月15日(2004.10.15)
【出願人】(506131019)メディゲーネ アクチエンゲゼルシャフト (7)
【氏名又は名称原語表記】MediGene AG
【住所又は居所原語表記】Lochhamer Strasse 11, D−82152 Planegg/Martinsried, Germany
【Fターム(参考)】
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2006−534713(P2006−534713)の分割
【原出願日】平成16年10月15日(2004.10.15)
【出願人】(506131019)メディゲーネ アクチエンゲゼルシャフト (7)
【氏名又は名称原語表記】MediGene AG
【住所又は居所原語表記】Lochhamer Strasse 11, D−82152 Planegg/Martinsried, Germany
【Fターム(参考)】
[ Back to top ]