
活性炭及びその製造方法
【課題】本発明は、電極の単位体積当たりの比容量が大きく、かつ、セル内部抵抗が極めて低い電気二重層キャパシタの電極用活性炭を提供する。また、本発明は、かかる活性炭を用いて製造される電極、及び該電極を備えた電気二重層キャパシタを提供する。
【解決手段】炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【解決手段】炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、電気二重層キャパシタの電極材料として有用な活性炭及びその製造方法、並びに該活性炭を電極材料として用いる電気二重層キャパシタに関する。
【背景技術】
【0002】
電気二重層キャパシタは、固体と液体の界面に生じる電気二重層を利用したコンデンサーであり、充放電サイクル特性や急速充放電にも優れ、またメンテナンスフリーで、環境汚染を招く恐れがないため、電気自動車用途や太陽光発電の蓄電装置(道路鋲に使用されている)として最近特に注目されている。
【0003】
電気二重層キャパシタにおける上記固体は分極性電極であり、通常粉末活性炭が使用される。該活性炭としては、種々の賦活処理されたものが報告されている。
【0004】
例えば、アルカリ金属水酸化物や塩化亜鉛及び燐酸などの薬品で賦活された活性炭は、製造直後は極めて高い静電容量を示すものの、一般に内部抵抗も大きく、その静電容量が短時間内に低下するという課題を有している。
【0005】
一方、薬品によらないガス賦活などの賦活法で製造された活性炭は、静電容量の経時低下はあまりなく、薬品賦活炭と比較すれば内部抵抗も小さいが、キャパシタの高性能化には更なる低抵抗化が望まれている。
【0006】
例えば、特許文献1には、合成有機高分子又はピッチ等をガス賦活乃至水蒸気賦活して得られる繊維状活性炭が記載されている。しかし、繊維状活性炭を電極として用いた場合には、粉末状活性炭に比べ充填密度が低くなり、内部抵抗が大きくなる傾向にある。
【0007】
また、特許文献2には、水蒸気賦活されたヤシ殻活性炭であり、中位径が6〜10μmかつBET比表面積が1000〜1500m2/gの電気二重層コンデンサーの電極用活性炭が記載されている。しかし、該活性炭は、賦活温度900℃以上かつ賦活時間8時間以上で処理されていないため、セル内部抵抗が増大する傾向にある。
【特許文献1】特許第2502986号
【特許文献2】特開平9−63907号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、電極の単位体積当たりの比容量が大きく、かつ、セル内部抵抗が極めて低い電気二重層キャパシタの電極用活性炭を提供することを目的とする。また、本発明は、かかる活性炭を用いて製造される電極、及び該電極を備えた電気二重層キャパシタを提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者は、上記課題を解決するために鋭意研究を行った結果、炭素質材料(特に、ヤシ殻系炭化物)を、ガス化速度を遅くした所定の反応速度にて、温度900℃以上かつ8時間以上で保持してガス賦活処理することにより得られる活性炭が、極めて低い圧縮時導電率を有することを見出した。さらに、該活性炭を電極として用いた電気二重層キャパシタでは、セル内部抵抗が極めて低くなることを見出した。かかる知見に基づき、さらに研究を重ねることにより本発明を完成するに至った。
【0010】
すなわち、本発明は、下記の活性炭、その製造方法、該活性炭を電極に用いた電気二重層キャパシタを提供する。
【0011】
項1.炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【0012】
項2.炭素質材料を賦活温度900℃未満で1次賦活した後に、項1に記載の条件で2次賦活を施すことを特徴とする活性炭の製造方法。
【0013】
項3.炭素質材料がヤシ殻又はその炭化物である項1又は2に記載の活性炭の製造方法。
【0014】
項4.項1〜3のいずれかに記載の活性炭の製造方法により製造される活性炭。
【0015】
項5.圧縮時導電率が2.0S/cm以上であり、かつ窒素吸着法により測定したBET比表面積が1500〜3000m2/gであることを特徴とする活性炭。
【0016】
項6.項4又は5に記載の活性炭を含む電極。
【0017】
項7.項6に記載の電極を備えた電気二重層キャパシタ。
【発明の効果】
【0018】
本発明の方法により製造される活性炭は、高い圧縮時導電率を有しており、電気二重層キャパシタの電極用材料として好適である。本発明の活性炭を電極に用いた電気二重層キャパシタは、電極の単位体積当たりの比容量が大きく、かつ、セル内部抵抗が極めて低いという優れた特性を有している。
【発明を実施するための最良の形態】
【0019】
以下、本発明について詳細に説明する。
【0020】
本発明において用いられる炭素質材料としては、通常活性炭原料として用いられる炭素源であれば特に限定されるものではなく、たとえば、木材、木粉、ヤシ殻、パルプ製造時の副産物、バカス、廃糖蜜などの植物系原料;泥炭、亜炭、褐炭、瀝青炭、無煙炭、石油蒸留残渣成分、石油ピッチ、コークス、コールタールなどの化石系原料;フェノール樹脂、塩化ビニル樹脂、酢酸ビニル樹脂、メラミン樹脂、尿素樹脂、レゾルシノール樹脂、セルロイド、エポキシ樹脂、ポリウレタン樹脂、ポリエステル樹脂、ポリアミド樹脂などの各種合成樹脂;ポリブチレン、ポリブタジエン、ポリクロロプレンなどの合成ゴム;その他合成木材、合成パルプなど、或いは、それらの炭化物が挙げられる。
【0021】
これらの炭素質材料の中では、単位体積当たり高静電容量の活性炭、即ちセル内部抵抗の小さい活性炭を得るためには、ヤシ殻、木粉などの植物系原料、又はそれらの炭化物が好ましく、ヤシ殻炭が特に好ましい。
【0022】
炭素質材料の炭化、賦活方式としては、たとえば固定床方式、移動床方式、流動床方式、スラリー方式、ロータリーキルン方式などの公知の方式を採用できる。
【0023】
炭素質材料の炭化方法としては、窒素、二酸化炭素、ヘリウム、アルゴン、キセノン、ネオン、一酸化炭素、燃焼排ガスなどの不活性ガス、或いはこれらの不活性ガスを主成分とした他のガスとの混合ガスを使用して、400〜700℃程度で30分〜10時間程度焼成する方法が挙げられる。
【0024】
炭素質材料の賦活方法としては、水蒸気、二酸化炭素、酸素などの賦活ガスを用いて焼成するガス賦活法が用いられる。このうち、賦活ガスとして、水蒸気又は二酸化炭素が好ましく、特に、反応速度が小さい点で二酸化炭素が好ましい。なお、薬品賦活法は内部抵抗が高くなるため、本発明においては実質的に薬品賦活法は採用しない。
【0025】
賦活温度は900℃以上、好ましくは900〜1200℃程度、より好ましくは900〜1100℃程度である。また、賦活時間は通常8時間以上とする。なお、賦活化の効率及び賦活品の生産性等の観点から、賦活時管を8〜50時間程度とするのが好ましい。賦活温度が900℃未満であったり、賦活時間が短かすぎたりすると、得られる活性炭の(圧縮時)導電率が低くなる傾向にあり、電極とした場合に内部抵抗が増大してしまう。
【0026】
さらに、本発明では、炭素質材料の賦活化の反応速度(即ち、炭素質材料のガス化速度)を一定値以下に制御することも重要である。即ち、下記式(1)で算出される値を反応速度(hr−1)とした場合に、この反応速度を0.06hr−1以下、好ましくは0.04hr−1以下の範囲で制御して賦活処理する。なお、賦活化の効率及び賦活品の生産性等の観点から、反応速度を0.005〜0.04hr−1とするのが好ましい。
【0027】
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
式(1)中、Vは炭素質材料に含まれる揮発分の含有量(重量%)を示し、Yは賦活前の炭素質材料に対する賦活後の活性炭の重量%、即ち収率(重量%)を示し、Tは賦活時間(hr)を示す。
【0028】
この反応速度が0.06hr−1を越える場合には、得られる活性炭の(圧縮時)導電率が低くなる傾向にあり、電極にした場合に内部抵抗が増大してしまう。
【0029】
このように、本発明では、上記の賦活温度及び賦活時間に加えて、上記の式(1)での反応速度を満たすことが重要となる。上記の式(1)で示される反応速度は、ガス賦活により炭素質材料からガス化する炭素質の重量%を、賦活処理時間(hr)で除したものである。これによれば、短い賦活時間でガス化量が大きい場合には、素材抵抗が大きくなるため、活性炭に所望の導電性が付与されなくなる。一方、賦活時間が8時間以上と時間をかけてガス化をゆっくりと進行させる場合には、素材抵抗が小さくなるため、所望の導電性が付与された活性炭を得ることができる。
【0030】
一般に、賦活温度が高くなるとガス化速度が増大する傾向にあるが、本発明では、賦活温度を高くしても積極的に炭素質材料のガス化速度を低減させることで、所望の高い(圧縮時)導電率を有する活性炭を得ることができるのである。
【0031】
この反応速度(ガス化速度)を0.06hr−1以下に制御する方法としては、例えば、原料仕込量に対する賦活ガスの供給量を少なくすることや反応性の乏しい賦活ガスを選択する等の方法が挙げられる。例えば、上記した水蒸気、二酸化炭素などの賦活ガスを、炭素質材料の仕込量1kg当たり、供給速度0.5kg/hr以下、好ましくは0.3kg/hr以下で反応炉内へ供給するなどの方法が例示される。かかる方法は、いずれも本願明細書の実施例の記載から容易に理解することができる。
【0032】
さらに、上記の炭素質材料の賦活処理に先立ち、あらかじめ炭素質材料を1次賦活してもよい。通常、炭素質材料を水蒸気、二酸化炭素、酸素などの賦活ガスを用いて、900℃未満の温度で焼成してガス賦活すればよい。その後、上記したように賦活温度は900℃以上、賦活時間は8時間以上、反応速度を0.06hr−1以下の条件で2次賦活すればよい。このように1次賦活した炭素質材料を用いて2次賦活すると、2次賦活における収率は見掛け上高くなり(炭化品からの収率は同程度)、2次賦活では反応速度を小さくできるメリットがある。また、同じ反応速度で同程度の比表面積の活性炭を得るためには賦活時間を短くできるというメリットがあり、工業的には有効な手段となる。いずれも式(1)から容易に理解できる。
【0033】
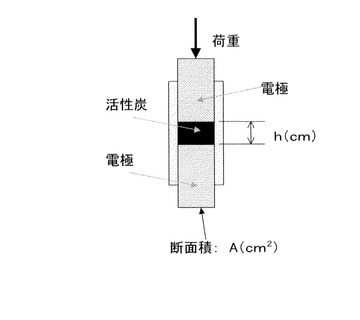
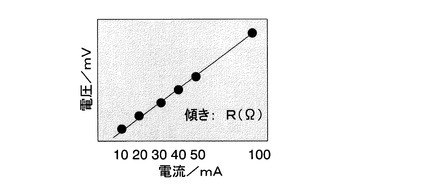
かくして得られる本発明の活性炭は、従来にはない高い導電性を有している。得られる活性炭の導電性を評価する方法として、次の圧縮時導電率が採用される。ここで、圧縮時導電率とは、活性炭を断面積A(cm2)の電極間に挟んだ後、これに一定荷重をかけて圧縮して保持した時の厚さをh(cm)とし(図1を参照)、その後電極の両端に電圧をかけて電流を測定して圧縮された活性炭の抵抗R(Ω)を求めて(図2を参照)、次の計算式(2)を用いて算出した値である。
【0034】
圧縮時導電率(S/cm)=h/(A×R) (2)
式(2)中、Aは電極の断面積(cm2)を示し、hは活性炭を電極間に挟みこれに一定荷重をかけて体積が変化しなくなるまで圧縮して保持した時の厚さ(cm)を示し、Rは圧縮された活性炭の抵抗(Ω)を示す。具体的には実施例1、図1及び図2を参照。
【0035】
なお、測定に用いる活性炭の重量は、圧縮されて電極間に保持される量であればよく、また、圧縮時の荷重は、活性炭の形状破壊が起こらない程度でかつ活性炭の体積変化がない程度にまで圧縮できる荷重であればよい。
【0036】
なお、上記の式(2)で表される圧縮時導電率を採用したのは、粉末状の活性炭の場合には、ひとつの粒子の導電率を測定することは不可能であるが、粉体にある以上の荷重をかけて体積が変化しなくなるまで圧縮した状態(ひとつの塊)で測定することにより活性炭の固有の導電率を評価できるからである。
【0037】
一般的に、電気二重層キャパシタはメンテナンスフリー(劣化しない)で長期間使用できることが要求される。そのため、活性炭に要求される項目としては静電容量が高いことは重要であるが、セル内部抵抗を小さくできることが極めて重要となる。つまり、静電容量が高くてもセル内部抵抗が大きければ、サイクル特性(充放電の繰り返し試験)において初期の静電容量から充放電回数が増加するについて徐々に変化(低下)するため電極の設計が非常に難しくなる。また、サイクル特性を調べるためにはかなりの労力と時間等を必要とすることから、セル内部抵抗が大きくなる材料は実用的でない。そこで、本発明では、電極セル内部抵抗と相関関係があるとされている材料(活性炭)の圧縮時導電性を採用したのである。
【0038】
本発明の活性炭は、この式(2)で算出される圧縮時導電率が2.0S/cm以上、好ましくは3.5〜10S/cmと高い値となる。
【0039】
また、本発明の活性炭は、高い比表面積を有しており、窒素吸着法により測定したBET比表面積が1500〜3000m2/g、好ましくは1800〜2800m2/g、より好ましくは2000〜2500m2/gとなる。
【0040】
また、本発明の活性炭の単位重量当たりの比容量は、25F/g以上、好ましくは30F/g以上程度である。なお、活性炭の単位重量当たりの比容量は、実施例1に記載の方法に従って測定される。
【0041】
本発明の活性炭は、その優れた特徴から電気二重層キャパシタにおける電極材料として有用である。本発明の活性炭を用いて電気二重層キャパシタの電極を製造するには、公知の方法を採用することができる。
【0042】
例えば、本発明の粉末状活性炭を、カーボンブラック及びバインダーと混合した後、その混合物を成形することにより電極を製造することができる。
【0043】
カーボンブラックの使用量は、例えば、活性炭100重量部に対し0.5〜30重量部程度、好ましくは1〜20重量部程度でよい。バインダーとしては、例えば、ポリテトラフルオロエチレン樹脂、スチレンブタジエンゴム(SBR)、ポリ弗化ビニリデン(PVDF)等が挙げられ、ポリテトラフルオロエチレン樹脂が好ましい。バインダーは、成形性を容易にするため、粉末状のものが好ましい。バインダーの使用量は、例えば、活性炭100重量部に対し1〜30重量部程度でよい。
【0044】
活性炭、カーボンブラック及びバインダーを混合方法は、特に限定はなく公知の混合方法を用いればよいが、例えば、通常のミキサー、ニーダー等を用いる方法が挙げられる。
【0045】
得られる混合物の成形方法は、例えば、プレス成形、押し出し成形等が挙げられる。特に、プレス成形が好ましい。電極の形状は、使用目的に応じ適宜選択することができるが、シート状のものが好ましい。その厚さは、0.2〜0.8mm程度である。電極の乾燥は、十分に水分を除去できればよく、通常70〜280℃程度で、10時間程度乾燥すればよい。
【0046】
次いで、上記で得られる電極を正極及び負極とした後、セパレータ、電解液を加えて電気二重層キャパシタを製造することができる。
【0047】
集電体としては、例えば、ステンレスメッシュ、アルミニウム等が挙げられるが、中でもステンレスメッシュのものが好ましい。集電体の厚さは、例えば、0.02〜0.5mm程度であればよい。
【0048】
セパレータの構成は、特に限定されるものではないが、単層又は複層のセパレータを用いることができする。また、セパレータの材質も、特に限定されるものではないが、例えば、電解コンデンサー紙、ポリエチレン、ポリプロピレンなどのポリオレフィン、ポリアミド、クラフト紙、ガラス、セルロース系材料等が挙げられ、電池の耐熱性、安全性設計に応じ適宜決定される。中でも、電解コンデンサー紙が好ましい。また、セパレータは十分に乾燥したものが好ましい。
【0049】
電解液としては、例えば、公知のアンモニウム塩を含む非水系電解質を使用することができる。具体的には、トリエチルメチルアンモニウム・テトラフルオロボレート(Et3MeNBF4)、テトラエチルアンモニウム・テトラフルオロボレート(Et4NBF4)等のアンモニウム塩を、プロピレンカーボネート、エチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、メチルエチルカーボネート、ジメトキシエタン、γ−ブチロラクトン、酢酸メチル、蟻酸メチル、或いはこれら2種以上の混合溶媒等の有機溶媒に溶解したもの等が例示される。また、電解液の濃度は特に限定されるものではないが、一般的に0.5〜2mol/Lが実用的である。該電解液は当然のことながら、水分が100ppm以下のものを用いることが好ましい。
【0050】
上記の電極、セパレータ、電解液を、例えば、ドライボックス中で組み立てることにより電気二重層キャパシタを得ることができる。
【0051】
かくして得られる本発明の電気二重層キャパシタは、電極単位体積当たりの比容量が大きく、内部抵抗(セル内部電気抵抗)が極めて小さいという特徴を有している。
【0052】
本発明の電気二重層キャパシタにおける電極のセル内部抵抗は、10Ω以下程度、好ましくは5Ω以下程度である。本発明の電気二重層キャパシタにおける電極の単位体積当たりの比容量は、13F/ml以上、好ましくは14F/ml以上程度である。また、電極の単位重量当たりの比容量は、例えば、23F/g以上、好ましくは28F/g以上程度である。
【0053】
なお、電極のセル内部抵抗、電極の単位体積当たりの比容量及び電極単位重量当たりの比容量の測定方法は、実施例1に記載の方法に従う。
【実施例】
【0054】
次に、本発明を、実施例を用いてより詳細説明するが、本発明はこれに限定されるものではない。
[実施例1]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化された活性炭を得た(表1)。
【0055】
得られた活性炭を空間速度4hr-1に保持された工水で10時間通水洗浄を行った後に水切りした。その後、115℃に保持された電気乾燥機内で10時間乾燥した後に、ボールミルで1時間粉砕を行い、本発明の活性炭を得た。
【0056】
炭素質材料の賦活化の反応速度(即ち、炭素質材料のガス化速度)は、式(1)のようにして求めた。
【0057】
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
式(1)中、Vは炭素質材料に含まれる揮発分の含有量(重量%)を示し、Yは賦活前の炭素質材料に対する賦活後の活性炭の重量%、即ち収率(重量%)を示し、Tは賦活時間(hr)を示す。
【0058】
得られた活性炭について、窒素を吸着質とし等温線の測定を行ない(測定装置:マイクロメリテクス社製“ASAP−2400”)、得られた等温線からBET法により比表面積値を求めた。その結果を表2に示す。
【0059】
また、活性炭約0.05gを断面積A(cm2)(=0.09cm2)の電極間に挟んだ後に荷重7000gで圧縮した状態を保持した(図1参照)。圧縮された活性炭の厚さはh(cm)であった。その後、電極の両端に電圧をかけて電流を測定して図2の関係を得て、圧縮された活性炭の抵抗値R(Ω)を求めた。式(2)を用いて圧縮時導電性(S/cm)を算出した。なお、1S(ジーメンス)=1/Ωを意味する。
【0060】
圧縮時導電率(S/cm)=h/(A×R) (2)
上記測定および計算による結果を表2に示す。
【0061】
次いで、上記の炭素質材料を粉砕し、この粉末100重量部に対し、カーボンブラック10重量部と、バインダーとしてのポリテトラフルオロエチレン樹脂粉末8重量部を混合した後、プレス成形することにより、厚さ0.5mmの電極を得た。
【0062】
上記で得られたシート状電極を1.5cm×1.5cmにカットし、150℃で2時間乾燥した。得られた電極を正極および負極とし、集電体として厚さ0.2mmのステンレスメッシュを用い、セパレータとして充分に乾燥した電解コンデンサー紙を用い、電解液として、濃度1.5mol/Lのトリエチルメチルアンモニウム・テトラフルオロボレート(Et3MeNBF4)/プロピレンカーボネート(PC)溶液を用いて、ドライボックス中でキャパシタを組み立てた。
【0063】
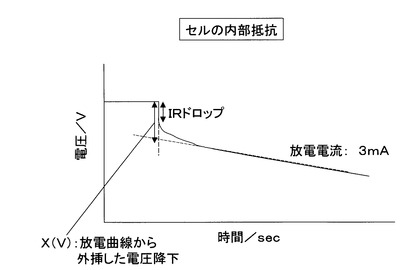
次いで、得られたキャパシタを用いて単位体積当たりのイオン吸着量(比容量)を求めた。比容量は、キャパシタの単位体積当りの電気容量(F/ml)として測定した。すなわち、キャパシタの最大充電電流を50mAに規制し、2.5Vで1時間充電した後、3mAの定電流にてキャパシタ電圧が0Vになるまで放電した。その時に得られた放電曲線を図3に示した。その放電曲線から2.0V及び1.5Vになる時間t2.0及びt1. 5を読み取り、式(3)より電気容量(F)を求めた。
【0064】
電気容量(F)=(t1.5−t2.0)/(0.5/0.003) (3)
式(2)より得られた値を正極及び負極の全体積で除せば電極の体積当たりの比容量(F/ml)が得られ、正極及び負極の全重量で除せば電極の重量当たりの比容量(F/g)が得られた。
【0065】
また、カーボンブラック及びバインダーの比容量をそれぞれ24F/g及び0F/gとして、それぞれの配合割合から活性炭重量当たりの比容量(F/g)を式(4)より算出する。つまり、前述したように電極は、活性炭100部、カーボンブラック10部とバインダー8部の混合物であるので、電極全体では118部となり、各物質の電気容量の合計が電極の電気容量である。
【0066】
比容量(活性炭重量)=(118×F−10×24−8×0)/100 (4)
なお、式(4)中、Fは先に求めた電極の重量当たりの比容量(F/g)である。
【0067】
また、図3より放電曲線を外挿して読み取った電圧降下量(IRドロップ)X(V)からセル内部抵抗を式(5)により算出した。
【0068】
セル内部抵抗(Ω)=X/0.003 (5)
なお、キャパシタ電極を構成する活性炭や電解質等それぞれに抵抗があるため、充電電圧(外部印加電圧)と電極に蓄電された電圧は実際等しくない。つまり、電極等に由来する抵抗に消費される電圧はキャパシタの充電には全く寄与していないため、放電初期に急激な電圧降下を生じる現象が図3の「IRドロップ」である。このIRドロップは計算式からも明白なようにセル内部抵抗と関係があることから、セル内部抵抗を出来るだけ小さくすれば有効充電電圧(=充電電圧−電圧降下)が大きくなりキャパシタ性能も向上することになる。
【0069】
上記測定および計算による結果を表3に示す。
[実施例2]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0070】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0071】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例3]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを設定温度1000℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0072】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0073】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例4]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で16時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0074】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0075】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例5]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で9時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0076】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0077】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例6]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度1050℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで二酸化炭素を炉内へ投入した。その状態で24時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0078】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0079】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例7]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで二酸化炭素を炉内へ投入した。その状態で18時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0080】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0081】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例1]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度8kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0082】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0083】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例2]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で23時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0084】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0085】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例3]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度1000℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0086】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0087】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例4]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度850℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で23時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0088】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0089】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例5]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで二酸化炭素を炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0090】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0091】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
【0092】
【表1】

【0093】
【表2】
【0094】
【表3】
【図面の簡単な説明】
【0095】
【図1】活性炭の圧縮時導電率を測定する装置の概略図である。
【図2】圧縮された活性炭に流れる電流と電圧の関係を示す模式図である。
【図3】キャパシタの放電曲線を示す模式図である。
【技術分野】
【0001】
本発明は、電気二重層キャパシタの電極材料として有用な活性炭及びその製造方法、並びに該活性炭を電極材料として用いる電気二重層キャパシタに関する。
【背景技術】
【0002】
電気二重層キャパシタは、固体と液体の界面に生じる電気二重層を利用したコンデンサーであり、充放電サイクル特性や急速充放電にも優れ、またメンテナンスフリーで、環境汚染を招く恐れがないため、電気自動車用途や太陽光発電の蓄電装置(道路鋲に使用されている)として最近特に注目されている。
【0003】
電気二重層キャパシタにおける上記固体は分極性電極であり、通常粉末活性炭が使用される。該活性炭としては、種々の賦活処理されたものが報告されている。
【0004】
例えば、アルカリ金属水酸化物や塩化亜鉛及び燐酸などの薬品で賦活された活性炭は、製造直後は極めて高い静電容量を示すものの、一般に内部抵抗も大きく、その静電容量が短時間内に低下するという課題を有している。
【0005】
一方、薬品によらないガス賦活などの賦活法で製造された活性炭は、静電容量の経時低下はあまりなく、薬品賦活炭と比較すれば内部抵抗も小さいが、キャパシタの高性能化には更なる低抵抗化が望まれている。
【0006】
例えば、特許文献1には、合成有機高分子又はピッチ等をガス賦活乃至水蒸気賦活して得られる繊維状活性炭が記載されている。しかし、繊維状活性炭を電極として用いた場合には、粉末状活性炭に比べ充填密度が低くなり、内部抵抗が大きくなる傾向にある。
【0007】
また、特許文献2には、水蒸気賦活されたヤシ殻活性炭であり、中位径が6〜10μmかつBET比表面積が1000〜1500m2/gの電気二重層コンデンサーの電極用活性炭が記載されている。しかし、該活性炭は、賦活温度900℃以上かつ賦活時間8時間以上で処理されていないため、セル内部抵抗が増大する傾向にある。
【特許文献1】特許第2502986号
【特許文献2】特開平9−63907号公報
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、電極の単位体積当たりの比容量が大きく、かつ、セル内部抵抗が極めて低い電気二重層キャパシタの電極用活性炭を提供することを目的とする。また、本発明は、かかる活性炭を用いて製造される電極、及び該電極を備えた電気二重層キャパシタを提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者は、上記課題を解決するために鋭意研究を行った結果、炭素質材料(特に、ヤシ殻系炭化物)を、ガス化速度を遅くした所定の反応速度にて、温度900℃以上かつ8時間以上で保持してガス賦活処理することにより得られる活性炭が、極めて低い圧縮時導電率を有することを見出した。さらに、該活性炭を電極として用いた電気二重層キャパシタでは、セル内部抵抗が極めて低くなることを見出した。かかる知見に基づき、さらに研究を重ねることにより本発明を完成するに至った。
【0010】
すなわち、本発明は、下記の活性炭、その製造方法、該活性炭を電極に用いた電気二重層キャパシタを提供する。
【0011】
項1.炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【0012】
項2.炭素質材料を賦活温度900℃未満で1次賦活した後に、項1に記載の条件で2次賦活を施すことを特徴とする活性炭の製造方法。
【0013】
項3.炭素質材料がヤシ殻又はその炭化物である項1又は2に記載の活性炭の製造方法。
【0014】
項4.項1〜3のいずれかに記載の活性炭の製造方法により製造される活性炭。
【0015】
項5.圧縮時導電率が2.0S/cm以上であり、かつ窒素吸着法により測定したBET比表面積が1500〜3000m2/gであることを特徴とする活性炭。
【0016】
項6.項4又は5に記載の活性炭を含む電極。
【0017】
項7.項6に記載の電極を備えた電気二重層キャパシタ。
【発明の効果】
【0018】
本発明の方法により製造される活性炭は、高い圧縮時導電率を有しており、電気二重層キャパシタの電極用材料として好適である。本発明の活性炭を電極に用いた電気二重層キャパシタは、電極の単位体積当たりの比容量が大きく、かつ、セル内部抵抗が極めて低いという優れた特性を有している。
【発明を実施するための最良の形態】
【0019】
以下、本発明について詳細に説明する。
【0020】
本発明において用いられる炭素質材料としては、通常活性炭原料として用いられる炭素源であれば特に限定されるものではなく、たとえば、木材、木粉、ヤシ殻、パルプ製造時の副産物、バカス、廃糖蜜などの植物系原料;泥炭、亜炭、褐炭、瀝青炭、無煙炭、石油蒸留残渣成分、石油ピッチ、コークス、コールタールなどの化石系原料;フェノール樹脂、塩化ビニル樹脂、酢酸ビニル樹脂、メラミン樹脂、尿素樹脂、レゾルシノール樹脂、セルロイド、エポキシ樹脂、ポリウレタン樹脂、ポリエステル樹脂、ポリアミド樹脂などの各種合成樹脂;ポリブチレン、ポリブタジエン、ポリクロロプレンなどの合成ゴム;その他合成木材、合成パルプなど、或いは、それらの炭化物が挙げられる。
【0021】
これらの炭素質材料の中では、単位体積当たり高静電容量の活性炭、即ちセル内部抵抗の小さい活性炭を得るためには、ヤシ殻、木粉などの植物系原料、又はそれらの炭化物が好ましく、ヤシ殻炭が特に好ましい。
【0022】
炭素質材料の炭化、賦活方式としては、たとえば固定床方式、移動床方式、流動床方式、スラリー方式、ロータリーキルン方式などの公知の方式を採用できる。
【0023】
炭素質材料の炭化方法としては、窒素、二酸化炭素、ヘリウム、アルゴン、キセノン、ネオン、一酸化炭素、燃焼排ガスなどの不活性ガス、或いはこれらの不活性ガスを主成分とした他のガスとの混合ガスを使用して、400〜700℃程度で30分〜10時間程度焼成する方法が挙げられる。
【0024】
炭素質材料の賦活方法としては、水蒸気、二酸化炭素、酸素などの賦活ガスを用いて焼成するガス賦活法が用いられる。このうち、賦活ガスとして、水蒸気又は二酸化炭素が好ましく、特に、反応速度が小さい点で二酸化炭素が好ましい。なお、薬品賦活法は内部抵抗が高くなるため、本発明においては実質的に薬品賦活法は採用しない。
【0025】
賦活温度は900℃以上、好ましくは900〜1200℃程度、より好ましくは900〜1100℃程度である。また、賦活時間は通常8時間以上とする。なお、賦活化の効率及び賦活品の生産性等の観点から、賦活時管を8〜50時間程度とするのが好ましい。賦活温度が900℃未満であったり、賦活時間が短かすぎたりすると、得られる活性炭の(圧縮時)導電率が低くなる傾向にあり、電極とした場合に内部抵抗が増大してしまう。
【0026】
さらに、本発明では、炭素質材料の賦活化の反応速度(即ち、炭素質材料のガス化速度)を一定値以下に制御することも重要である。即ち、下記式(1)で算出される値を反応速度(hr−1)とした場合に、この反応速度を0.06hr−1以下、好ましくは0.04hr−1以下の範囲で制御して賦活処理する。なお、賦活化の効率及び賦活品の生産性等の観点から、反応速度を0.005〜0.04hr−1とするのが好ましい。
【0027】
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
式(1)中、Vは炭素質材料に含まれる揮発分の含有量(重量%)を示し、Yは賦活前の炭素質材料に対する賦活後の活性炭の重量%、即ち収率(重量%)を示し、Tは賦活時間(hr)を示す。
【0028】
この反応速度が0.06hr−1を越える場合には、得られる活性炭の(圧縮時)導電率が低くなる傾向にあり、電極にした場合に内部抵抗が増大してしまう。
【0029】
このように、本発明では、上記の賦活温度及び賦活時間に加えて、上記の式(1)での反応速度を満たすことが重要となる。上記の式(1)で示される反応速度は、ガス賦活により炭素質材料からガス化する炭素質の重量%を、賦活処理時間(hr)で除したものである。これによれば、短い賦活時間でガス化量が大きい場合には、素材抵抗が大きくなるため、活性炭に所望の導電性が付与されなくなる。一方、賦活時間が8時間以上と時間をかけてガス化をゆっくりと進行させる場合には、素材抵抗が小さくなるため、所望の導電性が付与された活性炭を得ることができる。
【0030】
一般に、賦活温度が高くなるとガス化速度が増大する傾向にあるが、本発明では、賦活温度を高くしても積極的に炭素質材料のガス化速度を低減させることで、所望の高い(圧縮時)導電率を有する活性炭を得ることができるのである。
【0031】
この反応速度(ガス化速度)を0.06hr−1以下に制御する方法としては、例えば、原料仕込量に対する賦活ガスの供給量を少なくすることや反応性の乏しい賦活ガスを選択する等の方法が挙げられる。例えば、上記した水蒸気、二酸化炭素などの賦活ガスを、炭素質材料の仕込量1kg当たり、供給速度0.5kg/hr以下、好ましくは0.3kg/hr以下で反応炉内へ供給するなどの方法が例示される。かかる方法は、いずれも本願明細書の実施例の記載から容易に理解することができる。
【0032】
さらに、上記の炭素質材料の賦活処理に先立ち、あらかじめ炭素質材料を1次賦活してもよい。通常、炭素質材料を水蒸気、二酸化炭素、酸素などの賦活ガスを用いて、900℃未満の温度で焼成してガス賦活すればよい。その後、上記したように賦活温度は900℃以上、賦活時間は8時間以上、反応速度を0.06hr−1以下の条件で2次賦活すればよい。このように1次賦活した炭素質材料を用いて2次賦活すると、2次賦活における収率は見掛け上高くなり(炭化品からの収率は同程度)、2次賦活では反応速度を小さくできるメリットがある。また、同じ反応速度で同程度の比表面積の活性炭を得るためには賦活時間を短くできるというメリットがあり、工業的には有効な手段となる。いずれも式(1)から容易に理解できる。
【0033】
かくして得られる本発明の活性炭は、従来にはない高い導電性を有している。得られる活性炭の導電性を評価する方法として、次の圧縮時導電率が採用される。ここで、圧縮時導電率とは、活性炭を断面積A(cm2)の電極間に挟んだ後、これに一定荷重をかけて圧縮して保持した時の厚さをh(cm)とし(図1を参照)、その後電極の両端に電圧をかけて電流を測定して圧縮された活性炭の抵抗R(Ω)を求めて(図2を参照)、次の計算式(2)を用いて算出した値である。
【0034】
圧縮時導電率(S/cm)=h/(A×R) (2)
式(2)中、Aは電極の断面積(cm2)を示し、hは活性炭を電極間に挟みこれに一定荷重をかけて体積が変化しなくなるまで圧縮して保持した時の厚さ(cm)を示し、Rは圧縮された活性炭の抵抗(Ω)を示す。具体的には実施例1、図1及び図2を参照。
【0035】
なお、測定に用いる活性炭の重量は、圧縮されて電極間に保持される量であればよく、また、圧縮時の荷重は、活性炭の形状破壊が起こらない程度でかつ活性炭の体積変化がない程度にまで圧縮できる荷重であればよい。
【0036】
なお、上記の式(2)で表される圧縮時導電率を採用したのは、粉末状の活性炭の場合には、ひとつの粒子の導電率を測定することは不可能であるが、粉体にある以上の荷重をかけて体積が変化しなくなるまで圧縮した状態(ひとつの塊)で測定することにより活性炭の固有の導電率を評価できるからである。
【0037】
一般的に、電気二重層キャパシタはメンテナンスフリー(劣化しない)で長期間使用できることが要求される。そのため、活性炭に要求される項目としては静電容量が高いことは重要であるが、セル内部抵抗を小さくできることが極めて重要となる。つまり、静電容量が高くてもセル内部抵抗が大きければ、サイクル特性(充放電の繰り返し試験)において初期の静電容量から充放電回数が増加するについて徐々に変化(低下)するため電極の設計が非常に難しくなる。また、サイクル特性を調べるためにはかなりの労力と時間等を必要とすることから、セル内部抵抗が大きくなる材料は実用的でない。そこで、本発明では、電極セル内部抵抗と相関関係があるとされている材料(活性炭)の圧縮時導電性を採用したのである。
【0038】
本発明の活性炭は、この式(2)で算出される圧縮時導電率が2.0S/cm以上、好ましくは3.5〜10S/cmと高い値となる。
【0039】
また、本発明の活性炭は、高い比表面積を有しており、窒素吸着法により測定したBET比表面積が1500〜3000m2/g、好ましくは1800〜2800m2/g、より好ましくは2000〜2500m2/gとなる。
【0040】
また、本発明の活性炭の単位重量当たりの比容量は、25F/g以上、好ましくは30F/g以上程度である。なお、活性炭の単位重量当たりの比容量は、実施例1に記載の方法に従って測定される。
【0041】
本発明の活性炭は、その優れた特徴から電気二重層キャパシタにおける電極材料として有用である。本発明の活性炭を用いて電気二重層キャパシタの電極を製造するには、公知の方法を採用することができる。
【0042】
例えば、本発明の粉末状活性炭を、カーボンブラック及びバインダーと混合した後、その混合物を成形することにより電極を製造することができる。
【0043】
カーボンブラックの使用量は、例えば、活性炭100重量部に対し0.5〜30重量部程度、好ましくは1〜20重量部程度でよい。バインダーとしては、例えば、ポリテトラフルオロエチレン樹脂、スチレンブタジエンゴム(SBR)、ポリ弗化ビニリデン(PVDF)等が挙げられ、ポリテトラフルオロエチレン樹脂が好ましい。バインダーは、成形性を容易にするため、粉末状のものが好ましい。バインダーの使用量は、例えば、活性炭100重量部に対し1〜30重量部程度でよい。
【0044】
活性炭、カーボンブラック及びバインダーを混合方法は、特に限定はなく公知の混合方法を用いればよいが、例えば、通常のミキサー、ニーダー等を用いる方法が挙げられる。
【0045】
得られる混合物の成形方法は、例えば、プレス成形、押し出し成形等が挙げられる。特に、プレス成形が好ましい。電極の形状は、使用目的に応じ適宜選択することができるが、シート状のものが好ましい。その厚さは、0.2〜0.8mm程度である。電極の乾燥は、十分に水分を除去できればよく、通常70〜280℃程度で、10時間程度乾燥すればよい。
【0046】
次いで、上記で得られる電極を正極及び負極とした後、セパレータ、電解液を加えて電気二重層キャパシタを製造することができる。
【0047】
集電体としては、例えば、ステンレスメッシュ、アルミニウム等が挙げられるが、中でもステンレスメッシュのものが好ましい。集電体の厚さは、例えば、0.02〜0.5mm程度であればよい。
【0048】
セパレータの構成は、特に限定されるものではないが、単層又は複層のセパレータを用いることができする。また、セパレータの材質も、特に限定されるものではないが、例えば、電解コンデンサー紙、ポリエチレン、ポリプロピレンなどのポリオレフィン、ポリアミド、クラフト紙、ガラス、セルロース系材料等が挙げられ、電池の耐熱性、安全性設計に応じ適宜決定される。中でも、電解コンデンサー紙が好ましい。また、セパレータは十分に乾燥したものが好ましい。
【0049】
電解液としては、例えば、公知のアンモニウム塩を含む非水系電解質を使用することができる。具体的には、トリエチルメチルアンモニウム・テトラフルオロボレート(Et3MeNBF4)、テトラエチルアンモニウム・テトラフルオロボレート(Et4NBF4)等のアンモニウム塩を、プロピレンカーボネート、エチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、メチルエチルカーボネート、ジメトキシエタン、γ−ブチロラクトン、酢酸メチル、蟻酸メチル、或いはこれら2種以上の混合溶媒等の有機溶媒に溶解したもの等が例示される。また、電解液の濃度は特に限定されるものではないが、一般的に0.5〜2mol/Lが実用的である。該電解液は当然のことながら、水分が100ppm以下のものを用いることが好ましい。
【0050】
上記の電極、セパレータ、電解液を、例えば、ドライボックス中で組み立てることにより電気二重層キャパシタを得ることができる。
【0051】
かくして得られる本発明の電気二重層キャパシタは、電極単位体積当たりの比容量が大きく、内部抵抗(セル内部電気抵抗)が極めて小さいという特徴を有している。
【0052】
本発明の電気二重層キャパシタにおける電極のセル内部抵抗は、10Ω以下程度、好ましくは5Ω以下程度である。本発明の電気二重層キャパシタにおける電極の単位体積当たりの比容量は、13F/ml以上、好ましくは14F/ml以上程度である。また、電極の単位重量当たりの比容量は、例えば、23F/g以上、好ましくは28F/g以上程度である。
【0053】
なお、電極のセル内部抵抗、電極の単位体積当たりの比容量及び電極単位重量当たりの比容量の測定方法は、実施例1に記載の方法に従う。
【実施例】
【0054】
次に、本発明を、実施例を用いてより詳細説明するが、本発明はこれに限定されるものではない。
[実施例1]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化された活性炭を得た(表1)。
【0055】
得られた活性炭を空間速度4hr-1に保持された工水で10時間通水洗浄を行った後に水切りした。その後、115℃に保持された電気乾燥機内で10時間乾燥した後に、ボールミルで1時間粉砕を行い、本発明の活性炭を得た。
【0056】
炭素質材料の賦活化の反応速度(即ち、炭素質材料のガス化速度)は、式(1)のようにして求めた。
【0057】
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
式(1)中、Vは炭素質材料に含まれる揮発分の含有量(重量%)を示し、Yは賦活前の炭素質材料に対する賦活後の活性炭の重量%、即ち収率(重量%)を示し、Tは賦活時間(hr)を示す。
【0058】
得られた活性炭について、窒素を吸着質とし等温線の測定を行ない(測定装置:マイクロメリテクス社製“ASAP−2400”)、得られた等温線からBET法により比表面積値を求めた。その結果を表2に示す。
【0059】
また、活性炭約0.05gを断面積A(cm2)(=0.09cm2)の電極間に挟んだ後に荷重7000gで圧縮した状態を保持した(図1参照)。圧縮された活性炭の厚さはh(cm)であった。その後、電極の両端に電圧をかけて電流を測定して図2の関係を得て、圧縮された活性炭の抵抗値R(Ω)を求めた。式(2)を用いて圧縮時導電性(S/cm)を算出した。なお、1S(ジーメンス)=1/Ωを意味する。
【0060】
圧縮時導電率(S/cm)=h/(A×R) (2)
上記測定および計算による結果を表2に示す。
【0061】
次いで、上記の炭素質材料を粉砕し、この粉末100重量部に対し、カーボンブラック10重量部と、バインダーとしてのポリテトラフルオロエチレン樹脂粉末8重量部を混合した後、プレス成形することにより、厚さ0.5mmの電極を得た。
【0062】
上記で得られたシート状電極を1.5cm×1.5cmにカットし、150℃で2時間乾燥した。得られた電極を正極および負極とし、集電体として厚さ0.2mmのステンレスメッシュを用い、セパレータとして充分に乾燥した電解コンデンサー紙を用い、電解液として、濃度1.5mol/Lのトリエチルメチルアンモニウム・テトラフルオロボレート(Et3MeNBF4)/プロピレンカーボネート(PC)溶液を用いて、ドライボックス中でキャパシタを組み立てた。
【0063】
次いで、得られたキャパシタを用いて単位体積当たりのイオン吸着量(比容量)を求めた。比容量は、キャパシタの単位体積当りの電気容量(F/ml)として測定した。すなわち、キャパシタの最大充電電流を50mAに規制し、2.5Vで1時間充電した後、3mAの定電流にてキャパシタ電圧が0Vになるまで放電した。その時に得られた放電曲線を図3に示した。その放電曲線から2.0V及び1.5Vになる時間t2.0及びt1. 5を読み取り、式(3)より電気容量(F)を求めた。
【0064】
電気容量(F)=(t1.5−t2.0)/(0.5/0.003) (3)
式(2)より得られた値を正極及び負極の全体積で除せば電極の体積当たりの比容量(F/ml)が得られ、正極及び負極の全重量で除せば電極の重量当たりの比容量(F/g)が得られた。
【0065】
また、カーボンブラック及びバインダーの比容量をそれぞれ24F/g及び0F/gとして、それぞれの配合割合から活性炭重量当たりの比容量(F/g)を式(4)より算出する。つまり、前述したように電極は、活性炭100部、カーボンブラック10部とバインダー8部の混合物であるので、電極全体では118部となり、各物質の電気容量の合計が電極の電気容量である。
【0066】
比容量(活性炭重量)=(118×F−10×24−8×0)/100 (4)
なお、式(4)中、Fは先に求めた電極の重量当たりの比容量(F/g)である。
【0067】
また、図3より放電曲線を外挿して読み取った電圧降下量(IRドロップ)X(V)からセル内部抵抗を式(5)により算出した。
【0068】
セル内部抵抗(Ω)=X/0.003 (5)
なお、キャパシタ電極を構成する活性炭や電解質等それぞれに抵抗があるため、充電電圧(外部印加電圧)と電極に蓄電された電圧は実際等しくない。つまり、電極等に由来する抵抗に消費される電圧はキャパシタの充電には全く寄与していないため、放電初期に急激な電圧降下を生じる現象が図3の「IRドロップ」である。このIRドロップは計算式からも明白なようにセル内部抵抗と関係があることから、セル内部抵抗を出来るだけ小さくすれば有効充電電圧(=充電電圧−電圧降下)が大きくなりキャパシタ性能も向上することになる。
【0069】
上記測定および計算による結果を表3に示す。
[実施例2]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0070】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0071】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例3]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを設定温度1000℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で8時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0072】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0073】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例4]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で16時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0074】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0075】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例5]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で9時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0076】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0077】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例6]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度1050℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで二酸化炭素を炉内へ投入した。その状態で24時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0078】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0079】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[実施例7]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで二酸化炭素を炉内へ投入した。その状態で18時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0080】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0081】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例1]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度900℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度8kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0082】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0083】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例2]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度4kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で23時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0084】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0085】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例3]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度1000℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0086】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0087】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例4]
平均粒径約0.5cmの破砕状で揮発分16%のヤシ殻炭化品10kgを、設定温度850℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度2kg/hrで水蒸気を予熱炉で600℃に加温した状態で炉内へ投入した。その状態で23時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0088】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、活性炭を得た。その諸物性を表2に示す。
【0089】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
[比較例5]
平均粒径約0.5cmの破砕状で比表面積1000m2/g程度まで予め賦活された揮発分2%のヤシ殻1次賦活品10kgを、設定温度950℃に保持された窒素気流下の小型回転炉に投入して30分間保持した。その後、設定温度は変更せずに窒素の供給を停止した後に、供給速度1kg/hrで二酸化炭素を炉内へ投入した。その状態で6時間保持した後に容器へ取り出し、窒素雰囲気下で冷却して賦活化した活性炭を得た(表1)。
【0090】
得られた活性炭を実施例1と同様の手法で洗浄・乾燥及び粉砕を行い、本発明の活性炭を得た。その諸物性を表2に示す。
【0091】
得られた活性炭を用いて、実施例1と同様の手法により、電極を作製し、キャパシタを組み立て、充放電を行なった。得られた結果を表3に示す。
【0092】
【表1】
【0093】
【表2】
【0094】
【表3】
【図面の簡単な説明】
【0095】
【図1】活性炭の圧縮時導電率を測定する装置の概略図である。
【図2】圧縮された活性炭に流れる電流と電圧の関係を示す模式図である。
【図3】キャパシタの放電曲線を示す模式図である。
【特許請求の範囲】
【請求項1】
炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【請求項2】
炭素質材料を賦活温度900℃未満で1次賦活した後に、請求項1に記載の条件で2次賦活を施すことを特徴とする活性炭の製造方法。
【請求項3】
炭素質材料がヤシ殻又はその炭化物である請求項1又は2に記載の活性炭の製造方法。
【請求項4】
請求項1〜3のいずれかに記載の活性炭の製造方法により製造される活性炭。
【請求項5】
圧縮時導電率が2.0S/cm以上であり、かつ窒素吸着法により測定したBET比表面積が1500〜3000m2/gであることを特徴とする活性炭。
【請求項6】
請求項4又は5に記載の活性炭を含む電極。
【請求項7】
請求項6に記載の電極を備えた電気二重層キャパシタ。
【請求項1】
炭素質材料を、温度900℃以上かつ8時間以上で保持して、かつ、反応速度0.06hr−1以下でガス賦活処理を施すことを特徴とする活性炭の製造方法であり、該反応速度が、式(1):
反応速度(hr−1)=(100−V−Y)/(100×T) (1)
(式中、Vは炭素質材料に含まれる揮発分の含有量(重量%)、Yは収率(重量%)、Tは賦活時間(hr)を示す。)
で算出される値である。
【請求項2】
炭素質材料を賦活温度900℃未満で1次賦活した後に、請求項1に記載の条件で2次賦活を施すことを特徴とする活性炭の製造方法。
【請求項3】
炭素質材料がヤシ殻又はその炭化物である請求項1又は2に記載の活性炭の製造方法。
【請求項4】
請求項1〜3のいずれかに記載の活性炭の製造方法により製造される活性炭。
【請求項5】
圧縮時導電率が2.0S/cm以上であり、かつ窒素吸着法により測定したBET比表面積が1500〜3000m2/gであることを特徴とする活性炭。
【請求項6】
請求項4又は5に記載の活性炭を含む電極。
【請求項7】
請求項6に記載の電極を備えた電気二重層キャパシタ。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2007−221108(P2007−221108A)
【公開日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願番号】特願2007−6849(P2007−6849)
【出願日】平成19年1月16日(2007.1.16)
【出願人】(503140056)日本エンバイロケミカルズ株式会社 (95)
【Fターム(参考)】
【公開日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願日】平成19年1月16日(2007.1.16)
【出願人】(503140056)日本エンバイロケミカルズ株式会社 (95)
【Fターム(参考)】
[ Back to top ]