
活性薬剤デリバリーの、システム、医学用装置、及び方法
本発明は、医学用装置における使用のための活性薬剤デリバリーシステムを提供し、当該活性薬剤デリバリーシステムは、活性薬剤と混和性ポリマーブレンドを含む。
【発明の詳細な説明】
【発明の開示】
【0001】
関連出願のクロスリファレンス
本出願は、2002年8月13日に出願された合衆国仮特許出願第60/403,352号に対する優先権を主張し、その全体を引用により本明細書に編入する。
【0002】
発明の背景
医学用装置上へのポリマーのコーティングは活性薬剤(例えば、治療剤)の被験者へのデリバリーのための貯蔵所(repository)として機能するかもしれない。多数のそのような応用のために、ポリマーコーティングは可能な限り薄くなければならない。活性薬剤をデリバリーすることにおける使用のためのポリマー材料も、様々な三次元の形状をとり得る。
【0003】
慣用の活性薬剤のデリバリーシステムは、装置表面からのクラッキング及び層間剥離による構造上の破損を含む限界をこうむる。さらに、それらは、使用可能な活性薬剤の範囲、デリバリーシステム内に含まれ得る活性薬剤の量の範囲、及びそれからデリバリーされる含有活性薬剤の比率の範囲により、限定される傾向にある。これは、しばしば、多数の慣用のシステムが単一のポリマーを含むからである。
【0004】
即ち、多大な融通性及び整調性を伴う活性薬剤デリバリーシステムに関しての継続する要求が存在する。
発明の概要
本発明は、活性薬剤のデリバリーを制御することにおいて広く良好な融通性及び整調性を有する活性薬剤デリバリーシステムを提供する。一般に、そのような利点は、2つ又はそれより多い混和性のポリマーのブレンドの使用によりもたらされる。これらのデリバリーシステムは医学用装置、例えばステント、ステントグラフト、吻合術用コネクターに、所望ならば取込ませることができる。
【0005】
本発明の活性薬剤デリバリーシステムは少なくとも2つの混和性ポリマーのブレンドを含み、少なくとも1つのポリマー(好ましくは、混和性ポリマーのうちのひとつ)は、活性薬剤のデリバリーが好ましくは浸透性制御下で支配的に生じるように、活性薬剤の溶解性に適合される。このコンテクストにおいて、浸透性制御に関して「支配的に」とは、全活性薬剤の負荷の、少なくとも50%、好ましくは少なくとも75%、そしてより好ましくは少なくとも90%が、浸透性制御によりデリバリーされることを意味する。
【0006】
浸透性制御は、一般に、ミクロンスケールレベル上に「臨界の(critical)」寸法を有する混和性ポリマーのブレンドを活性薬剤が通過するようなシステムから活性薬剤をデリバリーすることにおいて重要である(即ち、拡散ネットの通路が約1000マイクロメートルより大きくないが、成型された対象物に関して、約10,000ミクロンまでであり得る)。さらに、活性薬剤の不均一な取込みにより不利益に影響されることなく所望の機械特性を提供する特定の活性薬剤のためにポリマーを選択することが一般に望まれる。
【0007】
第1の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は疎水性であり、約1200g/molを超えない(即ち、未満又は等しい)分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、各々が少なくとも一つの可溶性パラメーターを有し、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えず(好ましくは、25J1/2/cm3/2を超えない)、そして分子の膨張率が10体積%を超えない。
【0008】
第2の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は親水性であり、約1200g/molを超えない分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超え(好ましくは、25J1/2/cm3/2を超える)、そして分子の膨張率が10体積%を超えない。
【0009】
第3の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は疎水性であり、約1200g/molを超える分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えず(好ましくは、25J1/2/cm3/2を超えない)、そして分子の膨張率が10体積%を超える。
【0010】
第4の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は親水性であり、約1200g/molを超える分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超え(好ましくは、25J1/2/cm3/2を超える)、そして分子の膨張率が10体積%を超える。
【0011】
本発明は、活性薬剤デリバリーシステムを含む医学用装置も提供する。
本発明は、被験者に活性薬剤をデリバリーする方法も提供する。一つの態様において、デリバリー方法は、上記のとおりに活性薬剤デリバリーシステムを用意し、そして活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【0012】
本発明は、予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする(及び作成する)方法も提供する。
【0013】
一つの態様において、上記方法は、約1200g/mlを超えない分子量を有する活性薬剤を用意し;少なくとも2つのポリマーを選択するが、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして少なくとも二つのポリマーの各々の少なくとも一つのTgが目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;そして混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【0014】
別の態様において、上記方法は、約1200g/mlを超えない分子量を有する活性薬剤を用意し;少なくとも2つのポリマーを選択するが、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして上記少なくとも2つのポリマーの膨張性の間の差が目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;そして混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【0015】
本発明の上記の概要は、本発明の各開示された態様又は各手段(implementation)を記載することを意図しない。続く記載は例示の態様を、より特定して例示する。出願を通して幾つかの場所において、実施例のリストを通してガイダンスが提供されるが、当該実例は様々な組み合わせにおいて使用可能である。各例において、記載されたリストは代表群としてのみ提供され、そして排他的リストとして解釈されるべきではない。
【0016】
例示的態様の詳細な説明
本発明は、被験者にデリバリーされる活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムを提供する。当該デリバリーシステムは、少なくとも2つが本明細書にて定義されるとおりに混和性である限り、様々なポリマーを含み得る。活性薬剤は、ブレンドからそれが溶解するように、混和性ポリマーブレンド内に取込まれてよいか、あるいは、活性薬剤が通過する環境に対してバリヤーとしてブレンドが機能し得る。
【0017】
混和性ポリマーブレンドは、非混和混合物又は例えば単一のポリマーを含む慣用のシステムができるよりも広い範囲の活性薬剤に関して多大な融通性及び整調性を提供し得るため、有利である。即ち、2つまたはそれより多いポリマーを使用すると、そのうちの少なくとも2つが混和し、ポリマーの一方のみを用いたデリバリーシステムよりも、より融通のきく活性薬剤デリバリーシステムを一般的には提供できる。広い範囲の種類の活性薬剤が一般的には使用できる。広い範囲の量の活性薬剤が一般的には本発明のデリバリーシステムに取込まれて、デリバリーされ得る(好ましくは浸透性制御下で支配的に)。活性薬剤に関して広い範囲のデリバリー速度が本発明のデリバリーシステムにより一般的には提供できる。少なくとも一部において、これは、少なくとも2つの混和性ポリマーを含む混和性ブレンドの使用のためである。理解されるべきことは、本明細書の記載は2つのポリマーに言及しているが、発明は混和性ポリマーブレンドが少なくとも2つの混和性ポリマーを含んで形成される限り、2つより多いポリマーを含むシステムも包含することである。
【0018】
本発明の混和性ポリマーブレンドは連続部分を形成するのに十分な量の少なくとも2つの混和性ポリマーを有し、活性薬剤の放出の速度を整調させる助けとなる。そのような連続部分(即ち、連続相)は、顕微鏡によるか、又は選択された溶剤エッチングにより、同定することができる。好ましくは、少なくとも2つの混和性ポリマーが混和性ポリマーブレンドの少なくとも50体積%を形成する。
【0019】
混和性ポリマーブレンドは、任意に、分散した(即ち、不連続な)非混和性部分を含むことできる。連続部分と分散部分の両方が存在するなら、活性薬剤は何れかの部分に取込むことができる。好ましくは、活性薬剤は、連続部分に負荷されて、支配的な浸透性制御下において活性薬剤のデリバリーを提供する。活性薬剤を負荷するためには、活性薬剤の可溶性パラメーターと、可溶性薬剤の大部分が負荷される混和性ポリマーブレンドの部分を適合させる(典型的には、約10J1/2/cm3/2を超えない範囲まで、好ましくは5J1/2/cm3/2を超えない範囲まで、そしてより好ましくは約3J1/2/cm3/2を超えない範囲まで)。連続相は、活性薬剤が負荷される場所に拘わらず、活性薬剤の放出を制御する。
【0020】
本明細書にて使用される、混和性ポリマーブレンドは、2つ又はそれより多いポリマーの完全に混和性の多数のブレンド、並びに2つ又はそれより多いポリマーの部分的に混和性の多数のブレンドを包含する。完全に混和性のポリマーのブレンドは、理想的には、単一のガラス遷移温度(Tg)を、好ましくは全濃度範囲にわたり分子レベルにて混合するために、セグメント化されたポリマーに関して各相内(典型的には、ハード相とソフト相)に一つのTgを有する。部分的に混和性のポリマーのブレンドは複数のTg’sを有してよく、セグメント化されたポリマーに関してハード相及びソフト相の一方又は両方の中に存在することができるが、分子レベルでの混合が全濃度範囲のほんの一部にのみ限定されるからである。これらの部分的に混和性のポリマーのブレンドは、ブレンド内の少なくとも2つのポリマーの各々に関しての少なくとも一つのTg内の差(Tgポリマー1−Tgポリマー2)の絶対値がブレンディングの作用により減少する限り、用語「混和性のポリマーのブレンド」の範囲内に含まれる。Tg’sは、機械特性、熱特性、電気特性等を測定することにより、温度の函数として決定できる。
【0021】
混和性ポリマーブレンドは、その光学特性に基づいて決定することもできる。完全に混和性のポリマーブレンドは、安定且つ均質な透明のドメインを形成し、不混和性ポリマーブレンドは、成分が同一の屈折率を有さないと、光を散乱して視覚上濁って見える不均質なドメインを形成する。さらに、不混和性ブレンドの相−分離構造は顕微鏡により直接観察することができる。混和性をチェックするために本発明において使用される単純な方法は、ポリマーを混合し、そして約10マイクロメートルから約50マイクロメートルの薄いフィルムを形成することを含む。そのようなフィルムが、ブレンド前の個々のポリマーのクリアーさ及び透明性の劣る同じ厚さのフィルムほど広く(generally)クリアー及び透明性であるなら、ポリマーは完全に混和性である。
【0022】
ポリマー間の混和性は、それらの間の相互作用及びそれらの分子構造及び分子量に依存する。ポリマー間の相互作用は、いわゆるフローリー−ハギンスのパラメーター(χ)により特性決定できる。χがゼロ(0)に近いか又は負の場合、ポリマーは極めておそらく(very likely)混和性である。理論上、χはポリマーの可溶性パラメーターから見積もることができ、即ち、χはそれらの間の差の二乗に比例する。よって、ポリマーの混和性は、ほぼ予測することができる。例えば、2つのポリマーのχパラメーターが近ければ近いほど、2つのポリマーが混和性である可能性は高くなる。ポリマー間の混和性はそれらの分子量が増加するにつれて低下する傾向にある。
【0023】
即ち、実験による決定に加え、ポリマーの混和性はフローリー−ハギンス相互作用パラメーターに基づくか、又はより単純には、成分の可溶性パラメーターに基づいて、単純に予測することができる。しかしながら、分子量効果のため、接近した可溶性パラメーターは必ずしも混和性を保証しない。
【0024】
ポリマーの混合物は、混和性であるとして本明細書において提供された定義の一つを満すことのみを要求されることを理解するべきである。さらに、ポリマーの混合物は、活性薬剤の取込みに際して混和性ブレンドになってよい。本発明の特定の態様は、セグメント化されたポリマーを含む。本明細書にて使用される「セグメント化されたポリマー」は、複数のブロックから構成され、それらの各々は主にそれ自体から構成される相に分離し得る。本明細書にて使用される、ポリマーの「ハード」セグメント又は「ハード」相は、使用温度において結晶質であるか又は使用温度を超えるガラス遷移温度の非晶質(即ち、ガラス質)であるかの何れかであり、そしてポリマーの「ソフト」セグメント又は「ソフト」相は、使用温度未満のガラス遷移温度の非晶質である(即ち、ゴム質)。本明細書にて、「セグメント」は化学的フォーミュレーションを意味し、そして「相」は形態を意味し、主に、対応するセグメント(例えば、ハードセグメントはハード相を形成する)を含むが、他のセグメントの幾つかを含むことができる(例えば、ハード相中のソフトセグメント)。
【0025】
本明細書にて使用されるブレンドの「ハード」相は、主に、セグメント化されたポリマーのハードセグメントを含み、そして任意に、その中にブレンドされた第2のポリマーの少なくとも一部を含む。同様に、ブレンドの「ソフト」相は、セグメント化されたポリマーのソフトセグメントを支配的に含み、そして任意に、その中にブレンドされた第2のポリマーの少なくとも一部を含む。好ましくは、本発明のポリマーの混和性ブレンドは、セグメント化されたポリマーのソフトセグメントのブレンドを含む。
【0026】
セグメント化されたポリマーの可溶性パラメーターに言及する際は「セグメント」が用いられ、そしてセグメント化されたポリマーのTgに言及する際は「相」が用いられる。即ち、可溶性パラメーターは、一般にセグメント化されたポリマーに関しての計算値であり、個々のポリマー分子のハード及び/又はソフトセグメントを意味し、Tgは、一般に測定値であり、バルクのポリマーのハード及び/又はソフト相を意味する。
【0027】
ポリマー及び活性薬剤の種類及び量は、一般に、予め選択された溶解時間を有するシステムを、混和性ポリマーブレンドの予め選択された臨界寸法を通して形成させるために選択される。ポリマーのガラス遷移温度、膨張性、及び可溶性パラメーターは、活性薬剤が混和性ポリマーブレンドに取込まれていようがいまいが、活性薬剤デリバリーシステム中の成分の適切な組み合わせを選択するように当業者を導く際に使用することができる。可溶性パラメーターは、一般に、ポリマーの混和性を決定し、そして活性薬剤の可溶性を混和性ポリマーブレンドの可溶性に適合させるのに有用である。ガラス遷移温度及び/又は膨張性は、一般に、活性薬剤の溶解時間(又は速度)を整調するのに有用である。これらのコンセプトは、以下において詳細に論じられる。
【0028】
混和性ポリマーブレンドは、当該混和性ポリマーブレンドが活性薬剤のデリバリーを制御する限り、様々なフォーマットにおいて本発明のデリバリーシステム内で活性薬剤と化合して使用することができる。
【0029】
一つの態様において、混和性ポリマーブレンドは、その中に活性薬剤を取込んで有する。好ましくは、そのような活性薬剤は浸透性制御下において支配的に溶解される、大多数の活性薬剤が連続部分に負荷されていようがいまいが、ポリマーブレンドの連続する部分(即ち、混和性部分)内で活性薬剤の少なくとも幾つかの可溶性を必要とする。活性薬剤の溶解の間に孔性チャネリングがほとんど又は全く生じず、そして分散したドメインのサイズがブレンドの臨界寸法よりはるかに小さい限り、分散は容認され、そして物理的特性は、所望の機械的性能のための組成を通して、一般に均一である。この態様は、しばしば「マトリックス」システムと呼ばれる。
【0030】
別の態様において、混和性ポリマーブレンドは、最初は、活性薬剤の浸透に対してバリヤーを提供する。この態様は、しばしば、「リザーバー」システムと呼ばれる。リザーバーシステムは、2つ又はそれよい多い層を有する多数のフォーマットを採り得る。例えば、混和性ポリマーブレンドは、別の物質の内部層にわたり外部層を形成することができる(本明細書においては内部マトリックス物質と呼ぶ)。別の例において、リザーバーシステムはコア−シェルの形態を採ることができ、混和性ポリマーブレンドがコアマトリックスの周囲にシェルを形成する(即ち、内部マトリックス物質)。形成の少なくとも初期には、シェル又は外部層の中の混和性ポリマーブレンドが、活性薬剤を実質上含まないことが可能である。のちに、活性薬剤は、内部マトリックスから、並びに被験者へのデリバリーのための混和性ポリマーブレンドを通して、浸透する。内部マトリックス物質は、活性薬剤のデリバリーにおいて使用される様々な慣用の物質を含むことができる。これらは、例えば、有機ポリマー、例えば、混和性ポリマーブレンド中での使用のための本明細書にて記載された有機ポリマー、又はワックス、又は別の混和性ポリマーブレンドを含む。あるいは、内部マトリックス物質は活性薬剤自身であり得る。
【0031】
リザーバーシステムに関しては、活性薬剤の放出速度を、外部層の物質の選択により整調することができる。内部マトリックスはポリマーの非混和性の混合物を含み得るか、又は外部層がポリマーの混和性ブレンドならば、内部マトリックスはホモポリマーであり得る。
【0032】
マトリックスシステムを用いた場合のように、リザーバーシステム中の活性薬剤は、好ましくは、支配的に、浸透性制御下で、バリヤー層の混和性ポリマーブレンド(即ち、バリヤーポリマーブレンド)を通して溶解され、当該バリヤーポリマーブレンド中で活性薬剤の少なくともいくらかの溶解性を必要とする。再び、活性薬剤の溶解の間に孔性チャネリングがほとんど又は全く生じず、そして分散したドメインのサイズがブレンドの臨界寸法よりはるかに小さい限り、分散は容認され、そして物理特性は所望の機械的性能のための組成を通して、一般に均一である。これらの考察は内部マトリックスのためにも望まれるかもしれないが、それらは必ずしも必要ではない要求である。
【0033】
典型的には、本発明の活性薬剤デリバリーシステム内の活性薬剤の量は、デリバリーされる量及びデリバリーされる時間により決定される。他の因子も存在する活性薬剤のレベルに貢献することができ、例えば、組成物が基質上で均一なフィルムを形成する能力を含む。
【0034】
好ましくは、マトリックスシステムに関して、活性薬剤は、混和性ポリマーブレンドと活性薬剤の全重量の、少なくとも約0.1重量パーセント(wt%)、より好ましくは少なくとも約1wt%、そしてさらにより好ましくは少なくとも約5wt%の量にて混和性ポリマーブレンド内に存在する(即ち、内に取込まれる)。好ましくは、マトリックスシステムに関して、活性薬剤は、混和性ポリマーブレンドと活性薬剤の全重量の、約80wt%を超えず、より好ましくは約50wt%を超えず、そしてもっとも好ましくは約30wt%を超えない量にて、混和性ポリマーブレンド内に存在する。典型的に且つ好ましくは、活性薬剤の量は、混和性ポリマーブレンド内のその可溶性限界になるか又はそれ未満になる。
【0035】
好ましくは、リザーバーシステムに関して、活性薬剤は、内部マトリックス(活性薬剤を含む)の全重量の、少なくとも約0.1wt%、より好ましくは約10wt%、そしてさらにより好ましくは約25wt%の量にて、内部マトリックス内に存在する。好ましくは、リザーバーシステムに関して、活性薬剤は、内部マトリックス(活性薬剤を含む)の全重量の、100wt%まで、そしてより好ましくは約80wt%を超えない量にて、内部マトリックス内に存在する。
【0036】
本発明の活性薬剤デリバリーシステムにおいて、活性薬剤は混和性ポリマーブレンドを通して溶解可能である。溶解は、好ましくは、活性薬剤を混和性ポリマーブレンドを通して支配的に浸透させることにより制御される。即ち、活性薬剤は最初に混和性ポリマーブレンド内に溶解して、次に、浸透性制御下において支配的に混和性ポリマーブレンドを通して拡散する。即ち、上記のとおり、特定の好ましい態様に関して、活性薬剤は混和性ポリマーブレンドの可溶性限界であるか又はそれ未満である。理論に拘束されることを意図しないが、この機構のために、本発明の活性薬剤デリバリーシステムは顕著なレベルの整調可能性を有すると信じられる。
【0037】
活性薬剤が混和性ポリマーブレンドの可溶性を超え、そして不溶の活性薬剤の量が濾過限界を超えたなら、そのときは、活性薬剤が孔性機構を通して支配的に溶解され得る。さらに、活性薬剤の不溶性の相(例えば、粒子又は粒子の凝集物)の最大寸法が混和性ポリマーブレンドの臨界寸法と同じ桁ならば、そのときは、活性薬剤が孔性機構を通して支配的に溶解され得る。孔度の制御による溶解は、典型的には、不所望であるが、何故ならば、有効な予測性及び制御可能性を提供しないからである。
【0038】
本発明の活性薬剤デリバリーシステムは、好ましくはミクロンスケールのレベルの臨界寸法を有するため、十分な量の活性薬剤を含むこと及び孔性機構によるデリバリーを回避することが難しくなり得る。即ち、活性薬剤と混和性ポリマーブレンドの少なくとも一つのポリマーの可溶性パラメーターを適合させることにより、孔性機構によるデリバリーに関する傾向を低下させながら負荷のレベルを最大にする。
【0039】
放出される活性薬剤の量対時間(t)の溶解プロフィールを検査することにより、浸透制御放出機構が存在するか否かを決定することができる。マトリックスシステムからの浸透制御放出に関しては、当該プロフィールがt1/2に正比例する。リザーバーシステムからの浸透制御放出に関しては、当該プロフィールがtに正比例する。あるいは、シンク条件(即ち、ポリマーブレンドと、活性薬剤が溶解される溶媒との間に速度制限バリヤーがない条件)下では、孔度制御溶解がバースト効果(即ち、活性薬剤の初期の極めて素早い放出)をもたらすことができた。
【0040】
本発明の活性薬剤デリバリーシステムは、マトリックスシステムの形態かリザーバーシステムの形態かを問わず、限定ではないが、基材のコーティング(例えば、開いたか又は閉じたセルの泡、織り込まれたか又は織り込まれていない材料)、フィルム(例えば、パッチ中のようにフリースタンドであり得る)、成形されたオブジェクト(微小球、ビーズ、ロッド、ファイバー、又は他の成形されたオブジェクト)、曲がりくねったパッキング剤等の形態であり得る。
【0041】
本明細書にて使用される「活性薬剤」は、被験者(例えば、動物)において局所性又は全身性の効果を生じる薬剤である。一般には、それは、薬学上活性な薬剤である。当該用語は、疾患の診断、治療(cure)、軽減、処置(treatment)、又は予防(prevention)においてか、又は被験者における所望の外科的又は精神的発達又は症状の増強においての使用のために意図されたあらゆる物資を包含するように使用される。本明細書にて使用される用語「被験者」は、ヒト、ヒツジ、ウマ、畜牛、ブタ、イヌ、ネコ、ラット、マウス、鳥、爬虫類、魚、昆虫、蛛形類、原生動物(例えば、ポロトゾア)、及び原核細菌を含むものと見なされる。好ましくは、被験者はヒト又は他の哺乳類である。
【0042】
活性薬剤(active agent)は、合成又は天然であることができ、そして限定ではないが、有機又は無機化学薬剤、ポリペプチド(ペプチド、オリゴペプチド、蛋白質、酵素、ホルモン等を含むあらゆる長さのL−又はD−アミノ酸のポリマーを包含するように本明細書では使用される)、ポリヌクレオチド(オリゴヌクレオチド、一本鎖及び二本鎖のDNA、一本及び二本鎖のRNA,DNA/RNAキメラ等を含むあらゆる長さの核酸のポリマーを包含するように本明細書では使用される)、サッカライド(例えば、モノ−、ジ−、ポリ−サッカライド及びムコポリサッカライド)、ビタミン、ウイルス因子(agent)、及び他の生物材料、放射性核種等を含む。例としては、抗血栓性及び抗凝血性薬剤、例えばヘパリン、クマジン、クマリン、プロタミン、及びヒルジン;抗細菌剤、例えば抗生物質;抗新生物剤及び抗増殖剤、例えばエトポシド、ポドフィロトキシン;アスピリン及びジピリダモルを含む抗血小板剤;抗有糸分裂剤(細胞障害性薬剤)及び抗代謝剤、例えばメトトレキセート、コルチシン、アザチオプリン、ビンクリスチン、ブンブラスチン、フルオロウラシル、アダリアマイシン、及びムタミシン核酸;抗糖尿病薬剤、例えばロシグリタゾンマレート;及び抗炎症剤を含む。本発明における使用のための抗炎症剤は、グルココルチコイド、それらの塩、及びそれらの誘導体、例えばコルチゾール、コルチゾン、フルドロコルチゾン、プレドニソン、プレドニソロン、6α−メチルプレドニソロン、トリアムシノロン、ベータメタゾン、デキサメタゾン、ベクロメタゾン、アクロメタゾン、アムシノニド、クレベタゾール及びクロコルトコーンを含む。好ましくは、活性薬剤はヘパリンではない。
【0043】
本発明の好ましい活性薬剤デリバリーシステムに関して、活性薬剤はポリマーブレンドの混和性部分の可溶性に適合されるのが典型的である。即ち、活性薬剤が親水性である本発明の態様に関しては、好ましくは、混和性ポリマーブレンドの少なくとも一つの混和性ポリマーが親水性である。活性薬剤が疎水性である本発明の態様に関しては、好ましくは、混和性ポリマーブレンドの少なくとも一つの混和性ポリマーが疎水性である。しかしながら、これは必ずしも必要ではなく、そして水によるポリマーの膨張の可能性のため及び活性薬剤の制御されたデリバリーの損失のため、低分子量の親水性活性薬剤のためのデリバリーシステムを有することは望まれないかもしれない。本明細書にて使用される、このコンテクストにおける(ブレンドのポリマーのコンテクストにおける)用語「親水性」は、最初に来るにせよ、体温(即ち、約37℃)において水で膨張したときに、10体積%を超えるか又は重量で少なくとも10%まで増加する物質を意味する。本明細書にて使用される、このコンテクストにおける(ブレンドのポリマーのコンテクストにおける)用語「疎水性」は、最初のものはどれも(whichever comes first)、体温(即ち、約37℃)において水で膨張したときに、10体積%を超えるか又は重量で10%を超えるまで増加しない物質を意味する。
【0044】
本明細書にて使用される、このコンテクストにおける(活性薬剤のコンテクストにおける)用語「親水性」は、ミリリットルあたり200マイクログラムを超える水に可溶性を有する活性薬剤を意味する。本明細書にて使用される、このコンテクストにおける(活性薬剤のコンテクストにおける)用語「疎水性」は、ミリリットルあたり200マイクログラムを超えない水に可溶性を有する活性薬剤を意味する。
【0045】
活性薬剤のサイズが十分に大きくなるにつれ、ポリマーを通じての拡散が影響される。即ち、活性薬剤は分子量に基づいて分類することができ、そしてポリマーは活性薬剤の分子量の範囲に依存して選択することができる。
【0046】
本発明の特定の好ましい活性薬剤デリバリーシステムに関して、活性薬剤は約1200g/molより大きい分子量を有する。本発明の特定の他の好ましい活性薬剤デリバリーシステムに関して、活性薬剤は約1200g/molを超えない(即ち、未満又は等しい)分子量を有する。さらにより好ましい態様に関して、約800g/molを超えない分子量の活性薬剤が望まれる。
【0047】
活性薬剤及びデリバリーのためのフォーマット(例えば、時間/速度及び臨界寸法)が選択されたら、当業者は本発明の教示を利用することにより、活性薬剤デリバリーシステムを提供するための少なくとも2つのポリマーの適切な組み合わせを選択することができる。
【0048】
上記のとおり、ポリマー及び活性薬剤の種類及び量は、予め選択された時間(t)を有するシステムを、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して形成するように選択されるのが典型的である。これは目標の拡散性を提供するように少なくとも2つのポリマーを選択することを含み、所定の活性薬剤に関して、臨界寸法の二乗割る時間(x2/t)に正比例する。
【0049】
所望の活性薬剤、所望の溶解時間(又は速度)、及び所望の臨界寸法のためのポリマーの選択を正確にすることにおいて、所望の活性薬剤のためにポリマーを選択する際に考え得るパラメーターは、ポリマーのガラス遷移温度、ポリマーの膨張性、ポリマーの可溶性パラメーター、及び活性薬剤の活性パラメーターを含む。これらは、活性薬剤を混和性ポリマーブレンドに取込もうが取込むまいが、当業者が活性薬剤デリバリーシステム内の成分の適切な組み合わせを選択する手引きにおいて使用することができる。
【0050】
浸透性制御デリバリーシステムの融通性を増強するためには、例えば、好ましくは、以下の関係の少なくとも一つが真実であるようにポリマーを選択する:(1)活性薬剤の可溶性パラメーターと少なくとも一つのポリマーの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えない(好ましくは、約5J1/2/cm3/2を超えず、約3J1/2/cm3/2を超えない);及び(2)少なくとも2つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない(好ましくは、約3J1/2/cm3/2を超えない)。より好ましくは、両方の関係が真実である。もっとも好ましくは、両方の関係がブレンドの全部のポリマーに関して真実である。
【0051】
一般に、化合物は可溶性パラメーターをたった一つしか持たないが、特定のポリマー、例えばセグメント化されたコポリマー及びブロックコポリマーは、例えば一つより多い可溶性パラメーターを有することができる。可溶性パラメーターは測定することができるか、又はそれらはホイ(Hoy)法及びホフティザー−ファンクレヴェレン(Hoftyzer-van Krevelen)法(化学基寄与法)を用いて計算された値の平均値を用いて計算され、D.W.van Krevelen,Properties of Polymers,第3版、Elsevier,アムステルダムに開示されるとおりである。これらの値を計算するためには、各化学物質の体積が必要であり、同じ文献に開示されたフェドーズ(Fedors)法を用いて計算することができる。
【0052】
可溶性パラメーターは、コンピューターシミュレーション、例えば、モリキュラーダイナミックスシミュレーション及びモンテカルロシミュレーションによっても計算することができる。特定すれば、モリキュラーダイナミックスシミュレーションは、アクセルリスマテリアルズスチュディオ(Accelrys Materials Studio)、アクセルリス、サンディエゴ、カリフォルニアにより実施され得る。当該コンピューターシミュレーションはフローリー−ハギンスのパラメーターを直接計算するのに使用することができる。
【0053】
様々なポリマー及び活性薬剤に関する可溶性パラメーターの例を表1に示す。
【0054】
【表1】

【0055】
【表2】
【0056】
【表3】
【0057】
【表4】
【0058】
【表5】
【0059】
【表6】
活性薬剤が疎水性であるデリバリーシステムに関しては、分子量に拘わらず、混和性ポリマーブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように、ポリマーを選択するのが一般的である。本明細書における「モル平均可溶性パラメーター」は、互いに混和可能であって且つ混和性ポリマーブレンドの連続部分を形成するブレンド成分の可溶性パラメーターの平均を意味する。これらは、ポリマーブレンドへ取込まれる活性薬剤無しに、ブレンド中のそれらのモルパーセンテージにより秤量される。
【0060】
例えば、約1200g/mlを超えない疎水性活性薬剤、例えばグループコントリビューション法によると27J1/2/cm3/2の可溶性パラメーター又はモリキュラーダイナミックスシミュレーションによると21J1/2/cm3/2の可溶性パラメーターを有するデキサメタゾンに関しては、例示のポリマーブレンドは、セルロースアセテートブチレート(CAB)とポリビニルアセテート(PVAC)を含む。これらは、それぞれ、22J1/2/cm3/2の可溶性パラメーター及び21J1/2/cm3/2の可溶性パラメーターを有する。これらのポリマーの適当なブレンド(1:1モル比はCABのPVACに対する)は、21.5J1/2/cm3/2のモル平均可溶性パラメーターを有する。この値は、本明細書にては22*0.5+21*0.5=21.5(J1/2/cm3/2)として計算された。CABの繰り返しユニットの分子量は、アセチル基、ブチリル基、及びOH基の全数が繰り返しユニットあたり3であるという事実に基づいて、303g/molであると見積もられる。PVACの繰り返しユニットの分子量は86g/mlである。次に、PVACに対するCABの質量比は、この1:1モル比のブレンドに関して0.78/0.22である。
【0061】
活性薬剤が親水性であるデリバリーシステムに関しては、分子量に拘わらず、混和性ポリマーブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように、ポリマーを選択するのが一般的である。
【0062】
低分子量の活性薬剤に関しての浸透性制御溶解時間(速度)の整調性を増強するためには、好ましくは、少なくとも2つのポリマーの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当するように、ポリマーを選択することができる。
【0063】
あるいは、高分子量の活性薬剤に関しての浸透性制御溶解時間(速度)の整調性を増強するためには、好ましくは、少なくとも2つのポリマーの膨張性の差が目標の拡散性を含む拡散性の範囲に相当するように、ポリマーを選択することができる。目標の拡散性は、デリバリーに関しての予め選択された時間(t)及びポリマー組成の予め選択された臨界寸法(x)により決定され、そしてx2/tに正比例する。
【0064】
目標の拡散性は、以下の等式を用いて溶解分析により容易に測定することができる(例えば、Kinam Park編纂、制御されたドラッグデリバリー:チャレンジと戦略、アメリカンケミカルソサイエティー、ワシントン、DC,1997):
【0065】
【数1】
式中、D=拡散定数;Mt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。この等式は、活性薬剤の初期負荷の60重量%までの溶解の間、有効である。また、ブレンドサンプルはフィルムの形態であるべきである。
【0066】
一般に、少なくとも一つのポリマーが目標の拡散性より高い活性薬剤拡散性を有し、そして少なくとも一つのポリマーが目標の拡散性より低い活性薬剤拡散性を有する。ポリマーシステムの拡散性は溶解分析により容易に測定することができ、実施例のセクションにおいて詳細に記載される。個々のポリマー各々からの活性薬剤の拡散性は、溶解分析により測定できるが、各ポリマーの主要な相の相対的なTg’s又は膨張性により見積もることができる。
【0067】
拡散性は、疎水性又は親水性ポリマーのガラス遷移温度に相関することができ、低分子量の活性薬剤(例えば、約1200g/mlを超えない分子量を有するもの)のためのデリバリーシステムをデザインするのに使用することができる。あるいは、拡散性は、疎水性又は親水性ポリマーの膨張性に相関することができ、高分子量の活性薬剤(例えば、約1200g/mlを超える分子量を有するもの)のためのデリバリーシステムをデザインするのに使用することができる。これは、類似の可溶性の活性薬剤に関して極めて異なる溶解速度を包含するために混和性ブレンドの範囲を使用できるため、有利である。
【0068】
ポリマーのガラス遷移温度は、よく知られたパラメーターであり、一般には測定された値である。例示の値を表1に掲載する。セグメント化されたポリマー(例えば、セグメント化されたポリウレタン)に関して、上記Tgはバルクポリマーの特定の相を意味する。一般には、低分子量の活性薬剤に関しては、混和性の相対的に低いTgのポリマーと相対的に高いTgのポリマーを選択することにより、システムの溶解キネティクスを整調させることができる。これは、小分子量の薬剤(例えば、約1200g/mlを超えない)がTg’sに直接相関する経路を通して拡散し、即ちポリマーブレンドの自由体積が温度の直接の函数であり、温度がTgを超えたときにスロープが大きくなるからである。
【0069】
好ましくは、少なくとも一つの相対的に高いTgを有するポリマーを、少なくとも一つの相対的に低いTgを有するポリマーと組み合わせる。
例えば、1200g/mlを超えない分子量を有する活性薬剤のための混和性ポリマーブレンドは、100−120℃のTgを有するセルロースアセテートブチレート、及び20−30℃のTgを有するポリビニルアセテートを含む。1200g/mlを超えない分子量を有する活性薬剤のための混和性ポリマーブレンドの別の例は、約10−80℃のハード相Tgのポリウレタンと、約140℃のTgのポリカーボネートを含む。そのような高いTgのポリマーと低いTgのポリマーを組み合わせることにより、活性薬剤デリバリーシステムが、活性薬剤の所望の溶解時間に関して整調され得る。
【0070】
図1は、低分子量の疎水性活性薬剤、ロシグリタゾンマレートをデリバリーさせるための混和性ポリマーブレンドのための適切なポリマー候補を示す。これは、選択されたポリマーのTg対可溶性パラメーターのチャートである。ロシグリタゾンマレートの可溶性パラメーターを中心とするボックスは、この活性薬剤のための候補を囲む。
【0071】
水中のポリマーの膨張性は、容易に測定することができる。しかしながら、膨張性が水の取り込みによりもたらされるのであって、温度の上昇によりもたらされるのではないことを認識するべきである。一般に、高分子量の活性薬剤に関しては、混和性の相対的に低い膨張性のポリマーと相対的に高い膨張性のポリマーを選択することにより、システムの溶解キネティクスを整調させることができる。これらのシステムをデザインするのにポリマーの膨張性を使用できるが、何故ならば、相対的に高い分子量(例えば、約1200g/mlを超える)の活性薬剤のためのフリー体積を増加させることによりポリマーブレンド外に拡散するためには、水がポリマーの中に拡散することを必要とするからである。
【0072】
好ましくは、相対的に高い膨張性を有するポリマーを、相対的に低い膨張性を有するポリマーと組み合わせる。例えば、1200g/mlを超える分子量を有する混和性ポリマーブレンドは、100%を超える膨張性を有する(即ち、水溶性)ポリビニルピロリドン−ビニルアセテート−コポリマー、及び60%の膨張性を有するポリ(エーテルウレタン)を含む。そのような高い膨張性のポリマーと低い膨張性のポリマーを組み合わせることにより、活性薬剤デリバリーシステムを活性薬剤の所望の溶解時間に関して整調させることができる。
【0073】
混和性ポリマーブレンドの膨張性は、特定の活性薬剤のためにポリマーの組み合わせを決定することにおけるファクターとしても使用される。活性薬剤が1200g/mlを超える分子量を有するデリバリーシステムに関しては、それが疎水性であるか親水性であるかに拘わらず、ポリマーは、ブレンドの膨張性が10体積%を超えるように選択される。ブレンドの膨張性は、そこに取り込まれる活性薬剤無しに評価される。
【0074】
疎水性であり且つ1200g/mlを超えない分子量を有する活性薬剤の第1群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0075】
活性薬剤の第1群のためのポリマーブレンドの適切な組み合わせの例は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,477及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A HYDROPHOBIC CELLULOSE DERIVATIVE,MEDICAL DEVICE,AND METHOD」と題する出願;2002年8月13日に出願された、合衆国仮出願連続番号60/403,478及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLYURETHANE,MEDICAL DEVICE,AND METHOD」と題する出願;及び2002年8月13日に出願された、合衆国仮出願連続番号60/403,413及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLY(ETHYLENE−CO−(METH)ACRYLATE),MEDICAL DEVICE,AND METHOD」と題する出願において詳述されている。そのようなブレンドの特定の例は、実施例のセクションに例示される。好ましくは、活性薬剤の第1の群を用いた使用のために適した混和性ポリマーブレンドは、以下を含まない:疎水性セルロース誘導体とポリウレタン及びポリビニルピロリドンのブレンド;及び/又はポリアルキルメタクリレートとポリエチレン−コ−ビニルアセテートのブレンド。
【0076】
親水性であり且つ1200g/mlを超えない分子量を有する活性薬剤の第2群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0077】
この第2の群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、ポリアクリロニトリル、シアノアクリレート、メタクリロニトリル、親水性セルロース誘導体、等、及びそれらの組み合わせを含む。このコンテクストにおいて、「組み合わせ(combination)」は、混合物及びそれらのコポリマーを意味する。混合物及びコポリマーは、一つ又はそれより多いメンバーの群及び/又は他のモノマー/ポリマーを含み得る。好ましくは、活性薬剤の第2群による使用のために適した混和性ポリマーブレンドは、疎水性セルロース誘導体とポリビニルピロリドンの両方を含まない。
【0078】
活性薬剤の第1群のためのポリマーブレンドの適切な組み合わせの例は、出願人の譲渡人の係属中の、同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLYURETHANE,MEDICAL DEVICE,AND METHOD」と題する出願において詳述されている。
【0079】
疎水性であり且つ1200g/mlを超える分子量を有する活性薬剤の第3群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0080】
この第3群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、少なくとも一つの疎水性ポリマーを含み、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースニトレート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、メチルエチルセルロース、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースプロピオネートブチレート、セルロースアセテートプロピオネートブチレート、及びそれらの組み合わせを含む。これらのシステムのためのポリマーブレンドは、疎水性又は親水性の何れかの第2のポリマーを含み得る。例えば、親水性ポリマーは浸水性ポリウレタンであり得る。好ましい親水性ポリウレタンは、その中にポリエチレンオキシドユニットを有するソフトセグメント含む。適切な親水性ポリウレタンの例は、サーメディックス社(ウオーバーン、マサチューセッツ)によりTECOPHILICの商標名で市販されているポリ(エーテルウレタン)である。好ましくは、活性薬剤の第3群による使用のために適した混和性ポリマーブレンドは、以下を含まない:疎水性セルロース誘導体とポリウレタン又はポリビニルピロリドンのブレンド;及び/又はポリアルキルメタクリレートとポリエチレン−コ−ビニルアセテートのブレンド。
【0081】
親水性であり且つ1200g/mlを超える分子量を有する活性薬剤の第4群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0082】
この第4群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,392及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A HYDROPHILIC POLYMER,MEDICAL DEVICE,AND METHOD」と題する出願(アトーニードケット番号P−10858.00)において詳述されている。そのようなブレンドの特定の例は、実施例のセクションに例示される。好ましくは、活性薬剤の第4の群を用いた使用のために適した混和性ポリマーブレンドは、疎水性セルロース誘導体とポリビニルピロリドンの両方を含まない。
【0083】
混和性ポリマーブレンド中のポリマーを架橋結合させることもさせないこともできる。同様に、ブレンドされたポリマーを架橋結合させることもさせないこともできる。そのような架橋結合は、標準の技術を用いてブレンドした後に、当業者により実施され得る。
【0084】
本発明の活性薬剤のシステムにおいては、活性薬剤が「臨界」の寸法を有する混和性ポリマーブレンドを通って通過する。この臨界寸法は、活性薬剤のネットの拡散経路であり、そして好ましくは1000マイクロメートル(即ち、ミクロン)を超えないが、成形されたオブジェクトに関しては、約10,000ミクロンまでであり得る。
【0085】
混和性ポリマーブレンドがコーティング又はフリースタンディングフィルム(共に一般的には「フィルム」と呼ばれる)を形成する態様に関しては、臨界寸法はフィルムの厚さであり、そして好ましくは約1000ミクロンを超えず、より好ましくは約500ミクロンを超えず、そしてもっとも好ましくは約100ミクロンを超えない。フィルムは望みどおり薄くすることができるが(例えば、1ナノメートル)、好ましくは約10ナノメートルより薄くなく、より好ましくは約100ナノメートルより薄くない。一般に、最少のフィルム厚は、活性薬剤の必要な用量を保持するのに要求される体積により決定され、一般的には上記物質を形成するのに使用されるプロセスによってのみ限定される。本明細書中の全ての態様に関して、フィルムの厚さが一定又は均一である必要はない。さらに、フィルムの厚さは、活性薬剤が放出されるのにかかる時間を整調するのに使用することができる。
【0086】
混和性ポリマーブレンドが成形されたオブジェクトを形成する態様に関しては(例えば、微小球、ビーズ、ロッド、ファイバー、又は他の成形されたオブジェクト)、オブジェクトの臨界寸法(例えば、微小球又はロッドの直径)は、好ましくは、約10,000ミクロンを超えず、より好ましくは約1000ミクロンを超えず、さらにより好ましくは約500ミクロンを超えず、そしてもっとも好ましくは約100ミクロンを超えない。オブジェクトは望みどおり小さくすることができる(例えば、臨界寸法に関して10ナノメートル)。好ましくは、臨界寸法は、約100ミクロンを下回らず、そしてより好ましくは約500ナノメートルを下回らない。
【0087】
一つの態様において、本発明は、混和性ポリマーブレンドを含むポリマーのトップコート層、好ましくはポリマーのアンダーコート(プライマー)層を重層された基材表面により特徴付けされる医学用装置を提供する。当該装置を使用するとき、混和性ポリマーブレンドを、被験者の体液、器官、又は組織に接触させる。
【0088】
発明は医学用装置の性質により限定されない;むしろ、何れの医学用装置も混和性ポリマーブレンドを含むポリマーコーティング層を含み得る。即ち、本明細書にて使用される用語「医学用装置」は、それらの使用及び操作の通常の過程において、体組織、器官又は体液、例えば血液と接触させることができる表面を有するあらゆる装置を意味する。医学用装置の例は、限定ではないが、ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、ポンプ、血管グラフト、弁、ペースメーカー等を含む。医学用装置は体外装置、例えば外科手術のときに使用される装置であり得、例えば、のちに被験者に戻される血液と接触する、血液酸素発生機、血液ポンプ、血液センサー、又は血液を運ぶのに使用されるチューブ等を含む。医学用装置は同様に移植用装置、例えば、血管グラフト、ステント、ステントグラフト、吻合コネクター、電気刺激用リード、心臓弁、整形外科用装置、カテーテル、シャント、センサー、髄核(nucleus pulposus)のための置換装置、蝸牛又は中耳の移植物、眼球内レンズ、等であり得る。移植可能な装置は、経皮装置、例えば薬剤注射ポート(ports)等を含む。
【0089】
一般に、発明の医学用装置を製作するのに使用されるのに好ましい材料はバイオマテリアルである。「バイオマテリアル」は、ヒト体内への移植及び/又は体液、組織、器官等への接触のために意図された材料であり、且つ、意図された目的のために機能するのに要求される機械的強度、例えば耐久性(strength)、弾性、浸透性及び柔軟性を有する材料である。移植用装置に関しては特に、使用される材料が生物適合性材料、即ち細胞又は組織に対して過度に毒性ではなく、そしてからだに対して不適当な害をもたらさない材料であることが好ましい。
【0090】
発明は、混和性ポリマーブレンドがポリマーコーティングを形成する態様に関しての基材表面の性質により制限されない。例えば、基材表面は、セラミック、ガラス、金属、ポリマー、又はそれらの組み合わせから構成され得る。金属基材表面を有する態様においては、金属は、典型的には、鉄、ニッケル、金、コバルト、銅、クロム、モリブデン、チタン、タンタル、アルミニウム、銀、プラチナ、炭素、及びそれらの合金である。好ましい金属は、ステンレス鋼、ニッケルチタン合金、例えばNITINOL、又はコバルトクロム合金、例えばNP35Nである。
【0091】
混和性ポリマーブレンドを含むポリマーコーティングを、共有又は非共有相互作用の何れかにより基材表面に接着することができる。非共有相互作用は、イオン性相互作用、水素結合、双極子相互作用、疎水性相互作用及びファンデルワース相互作用を例えば含む。
【0092】
好ましくは、混和性ポリマーブレンドコーティングの塗布の前に、基材表面は活性化又は官能基化されないが、幾つかの態様においては、接着を促進するために、基材表面の前処理が望まれるかもしれない。例えば、ポリマーのアンダーコート層(即ち、プライマー)を使用することにより、基材表面へのポリマーコーティングの接着を増強することができる。適切なポリマーアンダーコート層は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,479及び同日出願された合衆国特許出願番号 を有する、MEDICAL DEVICE EXHIBITING IMPROVED ADHESION BETWEEN POLYMERIC COATING AND SUBSTRATEと題する出願に開示されている。本明細書に開示された特定の好ましいアンダーコート層は、本質的にポリウレタンからなる。そのような好ましいアンダーコート層は、ポリウレタン以外のポリマーを含むポリマーブレンドを含むが、独占的にポリウレタンのアンダーコート層に比較して、アンダーコート層のデュロメーター、耐久性(durability)、接着特性、構造上の完全性及び弾性に相当に影響しないほど少量においてである。
【0093】
ステント又は他の血管補綴器具(vascular prosthesis)を被験者に移植する場合、損傷直後から始まって約1から6カ月後までの期間は、血管の再狭症がしばしば観察される。即ち、ステントを含む発明の態様に関しては、意図される一般的な溶解速度は、補填器具がルーメンの壁に固まって細胞増殖を減少させた直後に活性薬剤が理想的には放出されるべきであるような速度である。活性薬剤は、次に、全部で約1から6カ月まで溶解し続けるべきである。
【0094】
発明は、基材表面にポリマーブレンドを塗布することによりコーティングを形成させるために使用されるプロセスにより制限されない。適切なコーティングプロセスの例は、溶液プロセス、粉末コーティング、熔融押出、又は蒸気蒸着(vapor deposition)を含む。
【0095】
好ましい方法は溶液コーティングである。溶液コーティングプロセスに関して、溶液プロセスの例は、スプレーコーティング、ディップコーティング、及びスピンコーティングを含む。溶液プロセスでの使用のための典型的な溶剤は、テトラヒドロフラン(THF)、メタノール、エタノール、エチルアセテート、ジメチルフォルムアミド(DMF)、ジメチアセトアミド(DMA)、ジメチルスルフォキシド(DMSO)、ジオキサン、N−メチルピロリドン、クロロホルム、ヘキサン、ヘプタン、シクロヘキサン、トルエン、ギ酸、酢酸、及び/又はジクロロメタンを含む。単一のコート又は複数の薄層を塗布することができる。
【0096】
同様に、発明は、成形されたオブジェクト内に混和性ポリマーブレンドを形成させるのに用いられるプロセスにより制限されない。そのような方法は、成形されたオブジェクトの種類に依存するはずである。適切なプロセスの例は、押出、鋳造(molding)、マイクロマシニング、押出重合法、電気スプレー法等を含む。
【0097】
活性薬剤デリバリーシステムが基材表面に塗布される一つ又はそれより多いコーティング層を含む好ましい態様に関しては、好ましい態様はプライマーの使用を含み、好ましくは「逆流(reflow)法」を用いて塗布され、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,479及び同日出願された合衆国特許出願番号 を有する、共にMEDICAL DEVICE EXHIBITING IMPROVED ADHESION BETWEEN POLYMERIC COATING AND SUBSTRATEと題する出願に開示されている。
【0098】
好ましくは、この「逆流法」において、装置作成プロセスは、最初にアンダーコートポリマーを基材表面に塗布することによりポリマーアンダーコート層を形成させ、次に当該ポリマーアンダーコート層を処理することによりアンダーコートポリマーを逆流させ、次に混和性ポリマーブレンドを、好ましくはその中に取り込まれた活性薬剤と共に、逆流されたアンダーコート層に塗布することにより、ポリマーのトップコート層を形成させることを含む。アンダーコートポリマーの逆流は、あらゆる便利な様式、例えば熱処理、紫外線処理、マイクロ波処理、RF処理、機械的圧縮、又は溶剤処理において達成することができる。アンダーコートポリマーを逆流させるためには、アンダーコート層を、少なくともアンダーコートポリマーの「熔融流出(melt flow)温度」ほどの高温まで、ポリマーを逆流させるのに十分な時間、加熱する。ポリマーが液体流状態に入る温度(即ち、「熔融流出温度」)は、発明に従いポリマーを逆流させるのに使用される好ましい最少温度である。典型的には、1から10分が発明に従い熱処理を用いてポリマーを逆流させるのに使用される時間である。ポリマーに関する熔融流出温度は、一般的には、ポリマーのTg(ガラスの熔融温度)及びTm(結晶の熔融温度)を上回る。
【0099】
実施例
本発明は以下の実施例により例示される。特定の実施例、材料、量、及び手法は本明細書にて以前に記載された発明の範囲及び精神に従い広く解釈されるできことが認識されるべきである。
【0100】
実施例1−5及び7−9は、相対的に低い(即ち、約1200g/mlを超えない)分子量を有する疎水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
実施例6は、相対的に高い(即ち、約1200g/mlを超える)分子量を有する親水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
【0101】
実施例10は、相対的に低い(即ち、約1200g/mlを超えない)分子量を有する親水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
実施例1
デキサメタゾン(疎水性活性薬剤)とのポリ(カーボネートウレタン)/
ポリ(ビス−フェノールAカーボネート)ブレンド
ブレンドの製造と混和性試験
ポリ(カーボネートウレタン)75D(PCU 75D)をポリマーテクノロジーグループ社、バークレー、カリフォルニアから購入した。それはヒドロキシル末端化ポリカーボネート、芳香族ジイソシアネート、及び低分子量のグリコールのコポリマーである。7グラム/10分のメルトインデックス(300℃/1.2kg,ASTM D 1238)を有するポリ(ビス−フェノールAカーボネート)(PC)を、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシン、から購入した。ブレンドする前に、上記のポリマーを60℃から70℃において減圧下で乾燥させた。2つの乾燥させたポリマーを様々な比率で乾燥混合して、次に、2つのローラーブレードを備えたバッチミキサー(サーモハーケー、カールスルー、バーデンビュッテムベルグ、ドイツ)を用いて約200−225℃において熔融ブレンドした。ブレンドは1分あたり50回転(rpm)にて実施した。トルクをレベルオフしたときに(2から3分以内)、rpmを100に上昇させた。トルクを再びレベルオフした後に(2から3分以内)、rpmを50rpmに設定し直した。ブレンドをさらに1分間続けた。混合を完全にした後に、サンプルを回収して、空気中で室温に冷やした。ブレンドの際の酸化を防止するため、0.1−0.2wt%のIRGANOX 1010抗酸化剤(チバスペシャリティーズケミカル社、テリータウン、ニューヨーク)を熔融混合の前にブレンドに添加した。
【0102】
ブレンドの機械的特性からブレンドの熱繊維温度を測定することにより、PCU 75DとPCの間の混和性を測定した。ブレンドサンプルを2つのホットプレートの間で約230℃において約5分間プレスすることにより、フィルムサンプルを用意した。典型的には、フィルムは約0.1ミリメートル(mm)から0.5mmの厚さ、5mmから7mmの幅、そして2センチメートル(cm)の長さであった。これらのフィルムを、レエトリックソリッドアナライザー(Rheometric Solids Analyzer)III(RSAIII)(レオメトリックサイエンティフィック社、ピスカタウエイ、ニュージャージー)のフィルム/ファイバー固定物の中にマウントした。最初のギャップを約5mmに設定した。試験はダイナミックモードにて1Hzの周波数にて行った。機械特性は、5℃/分の速度において−80℃から200℃にサンプルを加熱する間に記録した。命令された圧力(commanded strain)は、−80℃から200℃で0.1%、0℃から150℃で0.5%、そして150℃から200℃で1%に設定した。
【0103】
図2は、貯蔵弾性率対温度を示す。純粋なPCの率は約140℃において下落し始めた。よって、PCのTgは約140℃であった。純粋なPCUは、約10℃にて開始し約180℃までの類似の遷移を有した。PCUとPCの両方を含むブレンドに関しては、2つのガラス遷移が存在した。PCの含有量が増加すればするほど、両方のTg’sは増加して、共に接近した。これは、PCUとPCが混和性であったことを示唆した。
デキサメタゾンを用いたサンプル調製
溶解サンプルを溶剤ブレンド化により用意した。PCU 75Dポリ(カーボネートウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥させ、次に、熔融して、2つのホットプレートの間で約230℃において約5−10分間プレスした。次に、フィルムを冷やして、無水テトラヒドロフラン(THF)中に約60℃において入れた。混合物をポリマーが溶解するまで磁気バーを用いて撹拌した。少量のゲルが場合により溶液中に検出されたが、溶液を0.45ミクロン(μm)のフィルターを用いて濾過することにより除去した。PCUの濃度は1.16wt%であった。PCを最初に室温においてクロロホルム中に溶解することにより5wt%溶液を作成した。次に、当該溶液を無水THFにより1wt%まで希釈した。無水THF中のデキサメタゾン(シグマ−アルドリッチ)の1wt%溶液も室温において作成した。次に、上記の3つの溶液を比率を変えながら混合することにより、表2に示す組成の異なるサンプルを作成した。
【0104】
【表7】
溶解サンプルを、THFでリンスすることにより清潔にしたステンレス鋼(316L)シーム(shims)により調製した。清潔にされたシームに、THFに溶解した1wt%のポリ(エーテルウレタン)(PELLETHANE 75D,ダウケミカル社、ミッドランド、ミシシッピ)の溶液でコートした。PELLETHANE 75Dポリ(エーテルウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥し、次に、熔融して、2つのホットプレートの間で約230℃において約5−10分間プレスした。次に、フィルムを冷やして、磁気バーで一晩撹拌することにより、無水テトラヒドロフラン(THF)内で約25℃において溶解した。
【0105】
コートされたシームを窒素下で一晩乾燥させた。続いて、それらを215−220℃において5−10分間、熱処理した。この前処理により、ポリマー/活性薬剤層によるそれらの接着を促進したシームの表面上にプライマーの形成を導く。プライマー処理されたシームを上に掲載された溶液でコートし、そして一晩窒素下で乾燥させた。シームを各工程の後に秤量した。重量の違いに基づき、コーティングの厚さのときのように、ポリマー/活性薬剤コーティングの全量を測定した。典型的にコーティングの厚さは約10ミクロンであった。
デキサメタゾンの溶解
リン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン(HyClone)、ローガン、ユタから購入)を含んだガラスバイアル内にコートされたシームを入れることにより、PCU 75D/PCポリマーマトリックスからのデキサメタゾンの溶解を実施した。各シームは約2ミリグラム(mg)のコーティング(約0.2mgのデキサメタゾン)を有し、そして各バイアルは3ミリリットル(mL)のPBSを含んだ。バイアルはインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて撹拌した。PBSをバイアルから回収して、新鮮なPBSと置換した。公知濃度のデキサメタゾン溶液のシリーズを用いて目盛を定められたUV−Vis分光光度計(HP 4152A)によりデキサメタゾンの濃度を測定した。
溶解データ分析
図3は、ブレンド中のPCUの増量により増加したデキサメタゾンの累積放出を示す。これらの放出曲線は、明らかに、デキサメタゾンの放出速度がブレンド中のPCUの含有量を変えることにより調節し得たことを示す。当該曲線に基づいて、これらのブレンドからのデキサメタゾンの拡散係数が以下の式を用いて計算され、そして図4のブレンド組成の函数としてプロットされた。
【0106】
【数2】
式中、D=拡散係数;Mt=累積放出;MMt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。
【0107】
図4は、ブレンド組成のほぼ直線函数であった拡散係数のlogを示し、混和性ポリマーブレンドを用いることにより活性薬剤放出速度が整調され得たことを証明した。さらに、図3に提示されたデータはバーストを示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0108】
実施例2
デキサメタゾン(疎水性活性薬剤)との
ポリ(エーテルウレタン)/フェノキシブレンド
ポリ(エーテルウレタン)(PELLETHANE 75D)とデキサメタゾンは実施例1において使用されたのと同じであった。フェノキシ樹脂(PX)、直鎖ポリ(ビス−フェノールAエポキシド)をフェノキシスペシャリティーズ社、ロックヒル、カリフォルニア)から得た。本実施例において使用されたグレードは、モルあたり約10−16キログラムの平均分子量及び95℃のTgを有するPKHJであった。この物質は、PCが実施例1において示したようにデキサメタゾンの放出速度をスローダウンさせることを示すことが予測された。
【0109】
PELLETHANE 75Dとデキサメタゾンを実施例1において記載されたとおりにTHFに溶解した(以下の手法の全ては、特に示さない限り実施例1において使用されたのと同じであった)。PXを無水THF中へ、室温において、溶液中の1wt%のポリマーと共に溶解した。これらの3つの溶液を様々な比率で混合し、そして実施例に記載されたのと同じ手法によりプライマー処理されたステンレス鋼ステント上にコートした。コーティングを乾燥させた後に、溶解及びUV−Vis分析を実施した。
【0110】
PELLETHANE 75D/PXブレンドからのデキサメタゾンの累積放出を図5においてプロットした。デキサメタゾンの放出速度はブレンド中のPELLETHANE 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、PELLETHANE 75DとPXの含有量を変化させることにより、デキサメタゾンの放出速度が調節され得たことを示す。さらに、図5に提示したデータはバースト示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0111】
PELLETHANE 75D/PXブレンドのTg遷移をPYRIS 1示差走査型熱量計(DSC)、パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ、により測定することにより、PELLETHANE 75DとPXの間の混和性を試験した。約5wt%のPELLETHANE 75DとPXのTHF溶液を上で記載されたのと同じ手法により別々に作成した。各約10mgをブレンドサンプルをDSCに負荷し、そして−100から230℃において40℃/分にてスキャンした。各サンプルを2回スキャンした。第2のスキャンはノイズが少ないので使用した。PYRISソフトウエアバージョン5.0を用いることにより、Tg遷移の開始を決定した。図6に示すとおり、純粋なPELLETHANE 75Dは約22℃においてガラス遷移を有し、熔融様の遷移を173℃に有した。このTgは樹脂のハードなドメインに関連していると考えられた。検出できるのなら、ポリ(エーテルウレタン)のソフトドメインのTgは、通常は0℃を下回る。純粋なPXは高温に(77℃)Tg遷移を有する。PELLETHANE 75DとPXをブレンドしたとき、2つの変化が存在した。第1に、純粋なPELLETHANE 75DとPXのTg遷移はブレンドサンプルからはもはや明確に同定できなかった。純粋なPELLETHANE 75DのTgに比較して、高い開始温度の広いTg遷移範囲が存在した。これは、PELLETHANE 75DとPXが少なくとも部分的に混和していることを示唆する(本明細書にて定義されたものとして)。第2に、3つ全てのブレンドにおいてTg選の直後に、結晶性成分を表す新規な遷移が存在した。これは、PXがPELLETHANE 75Dの中でより早い結晶化遷移を引き起こしたことを示唆することから、PXとPELLETHANE 75Dハードドメインの間の相互作用の存在が示される。これは、さらに、2つの物質の間の混和性を示唆する。
【0112】
実施例3
デキサメタゾン(疎水性活性薬剤)とのポリ(カーボネートウレタン)
75D/ポリ(カーボネートウレタン)55Dブレンド
ポリ(カーボネートウレタン)75D(PCU 75D)とデキサメタゾンの溶液は、実施例1において使用したものと同じであった。PCU 55Dはポリマーテクノロジーグループにより作成されたポリ(カーボネートウレタン)ファミリーの別のメンバーに関する商業上の名称であるが、PCU 75Dよりもソフトである。それを実施例1に記載されたのと同じ手法にてTHFに溶解したが、PCU 75Dに関しては溶解が室温どころか60℃において(at room temperature rather at 60 oc)起きた。これらの3つの溶液を様々な比率で混合し、ステンレス鋼の上にコートし、そして実施例1において記載されたのと同じ手法を用いて乾燥させた。溶解試験は実施例1に記載されたとおりに実施した。
【0113】
PCU75D/PCU 55Dブレンドからのデキサメタゾンの累積放出を図7に示す。デキサメタゾンの放出速度はブレンド中のPCU 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、よりソフトな(即ち、低いデュロメーター)PCUをよりハードなPCUにブレンドすることにより、活性薬剤の放出速度が調節され得たことを示す。
【0114】
PCU 75D 100とPCU 75D 70の間のクロスオーバーが2つの厚さの違いによったことも指摘するべきである。放出速度は、曲線が最初は直線領域であるが、のちに平坦な部分により、決定した。デキサメタゾンは、PCU 75D 100からは、PCU 75D 70よりも早く放出された。
【0115】
実施例4
ロシグリタゾンマレート(疎水性活性薬剤)との
ポリ(エーテルウレタン)/フェノキシ樹脂
スミスクラインビーチャム、大英帝国から市販のロシグリタゾンマレートを、実施例2に記載されたとおりに、PELLETHANE 75D/PXブレンドから放出させた。当該ブレンド組成及び全てのサンプル調製及び試験手法は実施例2に記載されたのと同じであった。
【0116】
この活性薬剤の累積放出を図8にプロットした。放出速度はブレンド中のPELLETHANE 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、ロシグリタゾンマレートの放出速度が混和性ポリマーブレンドを用いることにより整調されたことを示す。
【0117】
実施例5
デキサメタゾンを含むポリビニルアセテート(PVAC)/
セルロースアセテートブチレート(CAB)
熱刺激電流試験法
熱刺激電流(TherMold Partner,L.P.,スタムフォード、コネチカット)を用いることにより、PVAC/CABブレンド中の熱遷移を測定した。約1センチメーター(cm)掛ける1cmの1片のフィルムをポリテトラフルオロエチレン(PTFE)フィルム(約50ミクロン厚)の表面の上に置いた。2つのフィルムをTSCのプレート−ピボット電極の間に置いた。別にHeガス(超高度純粋、トールガスアンドウエルディング社、プリマス、マサチューセッツ)上に及び真空で3回回転させることにより、試験用チャンバーをパージした。Heの圧力は約0.08メガパスカル(MPa)から0.12MPaであった。パージ後に、チャンバーを同じ圧力のHeガスで満した。サンプルを200℃に加熱し、そして200ボルト/ミリリットルの電圧をサンプルとPTFEフィルムの厚さを横切るように適用した。2分後、サンプルを約10分間50℃にてクエンチしたが、200V/分の電圧は保持した。電場を次に消して、サンプルを2℃/分から200℃に加熱した。フィルムを横切る電流を、この熱プロセスの間記録した。記録された電流−温度曲線を用いることにより、熱遷移を決定した。PTFEフィルムをプレート電極とサンプルフィルムの間に用いた場合のように、15−25℃からのその熱遷移ピークのうちの一方が全てのサンプルのTSC曲線において出現した。熱遷移温度を比較するために、各サンプルのもっとも高いピークを1に低下させるように、当該曲線をスケールした。よって、図の中の曲線の値は任意のユニットであった。
デキサメタゾンによるサンプル調製
ポリビニルアセテート(PVAC,Mw(重量平均分子量)=167から500キログラム/モル(kg/mol)及びセルロースアセテートブチレート(CAB、29.5wt%アセチル及び17wt%ブチリル、Mn(数平均分子量)=65kg/mol)、共にシグマ−アルドリッチカンパニー、ミルウオーキー、ウイスコンシンを真空オーブン中で乾燥させ、そして別々に無水テトラヒドロフラン(THF)に溶解した。両溶液のポリマー濃度は約1wt%であった。1wt%のデキサメタゾン(シグマ−アルドリッチ)を含むTHF溶液も類似の様式にて作成した。3つの溶液を様々な比率で混合して、表3に示す組成の5つの異なるサンプルを作成した。
【0118】
【表8】
溶解サンプルをTHFでリンスして乾燥させることにより洗浄されたステンレス鋼(316L)シームにより調製した。洗浄されたシームを、THFに溶解した1wt%のポリ(エーテルウレタン)(PELLETHANE 75D、ダウケミカル社、ミッドランド、ミシガン)の溶液でコートした。PELLETHANE 75Dポリ(エーテルウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥させ、次に、熔融し、そして2つのホットプレートの間で230℃において5−10分間プレスした。次に、フィルムを冷やし、そして磁気バーを用いて一晩撹拌することにより無水テトラヒドロフラン(THF)中に約25℃において溶解した。
【0119】
コートされたシームを一晩窒素下で乾燥させ、そして215−220℃において5−10分間熱処理した。この前処理はシームの表面上にプライマーを形成させることにより、ポリマー/活性薬剤の層による接着を促進させた。プライムされたシームは上に掲載された溶液でコートし、そして窒素下で一晩乾燥させた。当該シームを各工程後に秤量した。重量の違いを基にして、ポリマー/活性薬剤コーティングの全量を、コーティングの厚さのときのように、測定した。典型的な溶解サンプルは4−5ミリグラム(mg)の乾燥コーティングを約10ミクロン厚のシームあたり有した。
【0120】
プライマーコーティングが無い以外は、類似の様式にて混和性試験のためのサンプルを調製した。典型的なサンプルの厚さは約100ミクロンであり、そしてその中に活性薬剤は含まなかった。
混和性
様々なブレンドの熱遷移温度を測定することにより、PVACとCABの混和性を測定した。示差走査型熱量計(DSC)、動的機械分析(DMA)、及び熱刺激電流(TCS)を用いることにより、ガラス遷移温度(Tg)と他の遷移を測定した。TSCはもっとも強いシグナルを有した。図9A−Dに示されるとおりに一致した結果が提供された。純粋なPVACに関しては(図9A)、TSCが2つの遷移ピークを示し、それぞれ34℃及び62℃を中央とした。純粋なCAB(図9A)はそのTSC曲線中の約110℃を中央とする一つのピークを有した。30wt%のCABをPVAC中にブレンドした場合、PVACの遷移ピークのいずれもが変化しなかった(図9B)。しかしながら、純粋なCABのガラス遷移は消失し、このブレンドが混和性であったことを示した。CABの量を50wt%に増加させた場合、PVACの2つの遷移ピークは高い温度にシフトしたが、純粋なCABに関するTgピークは何も観察されなかった(図9C)。これは、PVACとCABが50/50ブレンドにおいても混和性であったことを示唆する。70wt%のCABを含むブレンドにおいては、遷移ピークの温度がいっそう高く、再び混和性ブレンドを示唆する(図9D)。全てのフィルムはクリアー(clear)で透明(transparent)であり、これらが混和性であったとの我々の結論を支持する。
【0121】
DSC分析はPYRIS 1DSC(パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ)を用いて実施した。−50℃から220℃まで、40℃/分にて、スキャニングをプログラムした。サンプルサイズは約10ミリグラム(mg)であった。図10に示すとおり、純粋なPVACは約39℃にTg遷移を有し、そして純粋なCABは約167℃にTg遷移を有した。PVACとCABを70/30の重量比でブレンドした場合、PVACに相当するTgが55℃に上昇した。これは、PVACとCABがこの比率では部分的に混和性であることを示す。CABをもっと加えると、PVACに相当するTgがさらに上昇したが、遅い速度においてであった。CABに相当するTgはPVACとの混合に際して低下した。これらの全ての結果は、PVACとCABが混合の全ての範囲において部分的に混和性であることを示す。この結果は、上で記載されたTSC試験に基づく結果とわずかに異なる。しかしながら、本発明の混和性の定義を用いた結論は同じであり、即ち、PVACとCABは混和性である。
デキサメタゾンの溶解
PVAC/CABポリマーマトリックスからのデキサメタゾンの溶解を、上記のとおりに調製されたポリマー/活性薬剤コートシームを用いて実施した。コートされたシームを小片に切断し、測定し、そして標準化のために面積を計測した。各小片を3ミリリットル(mL)のリン酸緩衝塩溶液(PBS、リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含むバイアルに浸した。サンプルとPBS溶液の量は、活性薬剤の濃度がUV−Vis分光光度計により検出可能であり、しかもサンプル中の活性薬剤の濃度は実験の間にPBS中の活性薬剤の可溶性の5%を超えない(シンク(sink)条件)ように選択した。約200ミリグラムの活性薬剤を含む約2ミリグラム(mg)のコーティング及び予め37℃に温めた3ミリリットル(mL)のPBSを用いた。溶解試験は37℃において実施し、そしてサンプルをシェーカー中で1分あたり約10サイクルにて撹拌した。PBSをサンプルバイアルから取り出し、そして様々な時間で分析することにより、各サンプル中の活性薬剤の濃度を測定した。PBS中の活性薬剤の濃度はUV−Vis分光光度計(HP 4152A)を用いて243ナノメートル(nm)の波長において測定した。各サンプル中の活性薬剤の濃度は、連続希釈法により創製された標準曲線に比較することにより計算した。各測定後にキュベットを注意深く洗浄することにより、キュベット表面上への疎水性活性薬剤の蓄積を最小にした。ベースラインが、測定された活性薬剤のシグナルよりも少なくとも一桁低い場合に、キュベットは清浄であったと考えた。PBSを各分析時間点において新鮮にした。
溶解データ分析
図11は、ブレンド中のPVACの量を増加させて増加させたデキサメタゾンの累積放出を示す。これらの累積曲線は、明らかに、PVACとCABをブレンドすることにより、2つのホモポリマーの相対的な量を変化させて放出速度を変えることが可能であったことを示す。当該曲線に基づき、これらのブレンドからのデキサメタゾンの拡散係数を以下の指揮を用いて計算し、そして図12のブレンド組成の函数としてプロットした。
【0122】
【数3】
式中、D=拡散係数;Mt=累積放出;MMt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。
【0123】
拡散係数のlogがブレンド組成のほぼ直線函数であったことから活性薬剤の放出速度は混和性ポリマーブレンドを用いることにより整調され得ることを証明した。さらに、図11に提示されたデータはバーストを示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0124】
実施例6
Resten NGを含む疎水性ポリウレタン及び
ポリ(ビニルアセテート−コ−ビニルピロリドン)
TECOPHILIC HP−60D−60ポリウレタン、サーメディックス社、ウオーバーン、マサチューセッツ、及びポリ(ビニルアセテート−コ−ピロリドン)(PVP−VA),シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシが、この実施例で使用されたマトリックスポリマーであった。RESTEN NG,7000の分子量の水溶性アンチセンスオリゴヌクレオチド、AVIバイオファーマ、コーヴァリス、オレゴン、がこの実施例で使用された活性薬剤であった。TECOPHILICポリウレタンのソフトセグメントは、ポリ(エチレンオキシド)(PEO)及びポリ(テトラメチレンオキシド)(PTMO)の混合物を含む。このソフトセグメントの可溶性パラメーターは、ホフティザーとファンクレヴェレンの(Hoftyzer and van Kevelen's)(H−vK)方法(化学薬品の体積はフェドーズ(Fedor's)法に基づいて計算した)(第7章、D.W.van Krevelen,Properties of Polymers,第3版、Elsevier,1990,表7.8がHoftyzer and van Kevelen’s法に関し、表7.3がFedor’s法に関した)に基づくと、19J1/3/cm3/2(PTMO)から23J1/3/cm3/2(PEO)であると見積もられた。PVP−VAの可溶性パラメーターは、同じ方法に基づいて23J1/3/cm3/2であると見積もられた(ポリマー中のそれらの質量比に基づいたPVPとVAモノマーを超えた(over)モル平均)。
【0125】
TECOPHILICポリウレタンを、無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシに、1wt%ポリウレタンの濃度にて溶解した。ポリウレタンと溶剤をガラスバイアル中で混合し、シールし、そしてポリウレタンが完全に溶解するまで(視覚観察による)撹拌した。メタノール中での超音波により以前に洗浄して空気乾燥されていた、メドトロニックモデルS−670冠動脈ステント(3.0mm x 18mm)を、上で製造された50から100マイクログラムのポリウレタンコーティングでスプレーコートした。専有の(proprietary)スプレーユニットを用いることにより、この実施例においてステントをコートしたが、上記ポリマーの微細に霧状にされた噴霧の上記ステントへの塗布が可能な如何なるスプレーユニットも十分であるべきである。ポリウレタンの50から100マイクログラムのスプレーコーティングの後に、ステントを実験室環境条件にて、25℃及び15%相対湿度(RH)において、4時間乾燥させた。ステントが乾燥した後に、それらをオーブン中に220℃において20分間置くことにより、プライマーコーティングを逆流させた(reflow)。逆流の後で、ステントをオーブンから取り出して、室温において冷やした。
【0126】
TECOPHILICポリウレタンを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。TECOPHILICの溶液中の濃度は1wt%であった。この溶液をAと呼んだ。
【0127】
RESTEN NGオリゴヌクレオチドを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。RESTEN NGの溶液中の濃度は1wt%であった。この溶液をBと呼んだ。
【0128】
PVP−VAを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。RESTEN NGの溶液中の濃度は1wt%であった。この溶液をCと呼んだ。
【0129】
溶液A,B及びCを以下の表4に示すとおりに混合することにより、1%の全(overall)「固形物」濃度の溶液を作成した。各溶液中の「固形物」は、10wt%のRESTEN NG、そして残りがTECOPHILICポリウレタンとPVP−VAのブレンドであり、表4に示されるとおりである。
【0130】
【表9】
溶液1−3を0.45ミクロン(マイクログラム)のフィルターで濾過し、そして上で製造されたプライムされたステントの上にスプレーした。ステントをプライムするのに使用した同じ持主のスプレーユニットとプロセスを用いてトップコートを塗布したが、ポリマーと薬剤の溶液の微細に霧状にされた噴霧をステントに塗布することができるらゆるスプレーユニットを使用することができた。コートされたステントは45℃において真空オーブン中で12時間乾燥した。約2000マイクログラム(μg)のコーティングを各ステントに塗布し、そして実際のコーティング重量を用いることにより、コーティング溶液製剤に基づいて各ステント上の活性薬剤の理論上の量を計算した。
【0131】
溶解試験を上でコートされたステント上で実施した。各ステントを、予め37℃に温められた3.0ミリリットル(mL)のリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含むバイアル内に入れた。バイアルをインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて撹拌した。示された時間(この研究においては1分、1時間、3時間、1日、2日、3日、及び4日)において、PVACの全容量をサンプルバイアルから取り出し(バイアルに素早く予め温められた3.0mLの新鮮なPBSを満した)、そしてUV−Vis分光光度計(HP 4152A)により260ナノメートル(nm)にて分析した。各サンプル中のRESTEN NGの濃度を標準曲線との比較により測定した。放出されたRESTEN NGの累積量を、各ステントに関する理論上のRESTEN NG負荷で割り、そして時間の平方根に対してプロットした。結果を図13に提示する。
【0132】
最初の時間にかけて放出されたRESTEN NGの初期バーストが存在したが、放出曲線の残りは時間の平方根に比例したことから、RESTEN NGが透過制御下で放出されたことを示唆する。デリバリーの速度は上記マトリックスポリマーブレンド中のTECOPHILICのPVP−VAに対する比に相関した。より多いPVP−VAによるコーティングはRESTEN NGをより早くデリバリーした。
【0133】
TECOPHILICポリウレタンとPVP−VAの間の混和性を、PYRIS 1示差走査型熱量計(DSC)、パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ、により試験した。TECOPHILICポリウレタンとPVP−VAを同じ溶剤の中に同じ様式にて溶解することにより、約5wt%の溶液を作成した。2つの溶液を様々な比率で混合することにより、PVP−VAの重量パーセンテージを0から100%の範囲で含むサンプルを作成した。ブレンドサンプルを窒素ガス保護の下で乾燥した。試験を実施する前に、サンプルをさらに減圧下で室温にて乾燥した。DSCスキャンを−100℃から230℃に40℃/分にてプログラムした。サンプルを2回スキャンした。第2のスキャンはノイズが少ないので使用した。サンプルサイズは約10ミリグラム(mg)であった。同じサンプルをこの実施例においては全てのTg遷移のために使用した。
【0134】
図14に示すとおり、純粋なTECOPHILICポリウレタンは約−53℃においてガラス遷移を有した(PYRISバージョン5.0ソフトウエアにより測定された開始温度)。このTgはソフトなドメインに関連していると考えられた。ハードドメインのTgは室温よりも高かったが、何故ならばこの樹脂が室温においては全く剛直だからである(デュロメーター41D)。純粋なPVP−VAは高温にTg遷移を有した(76℃)。TECOPHILICポリウレタンを20wt%のPVP−VAと混合した場合、そのDSCは本質的には変わらなかった;が、PVP−VAのTgが消失した。2つのポリマーを50/50の重量比で混合したら、TECOPHILICポリウレタンのTg遷移が消失した。PVP−VAのTg付近の温度に極めて弱い遷移ピークが存在した。Tg遷移の消失は、2つのポリマーが少なくとも一部混和性であったことを示した。
【0135】
DSC試験に関するのと同じサンプルを用いて膨張性試験を実施した。完全に乾燥したサンプル(ウエイト1=50から100mg)を、5mLのリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内に入れた。バイアルをインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて1日撹拌した。サンプルをバイアルから取り出した。一片の組織を使用して、サンプル表面から遊離のPBSを吸い取った。サンプルを再び秤量した(ウエイト2)。次に、サンプルを減圧下で室温において乾燥した。サンプルを3回秤量した(ウエイト3)。膨張性のパーセンテージは、ウエイト2からウエイト3を引いて、ウエイト3で割ることにより計算した。純粋なTECOPHILICポリウレタンは約56%膨張した。純粋なPVP−VAはPBSに完全に溶解した。膨張性のパーセンテージを、20wt%までのPVP−VAを含むサンプルに関して図15中のPVP−VA含有量の函数としてプロットした。PVP−VAのPBSへの浸出による重量の損失(ウエイト1−ウエイト3)は、10wt%を超えないPVP−VAを含むサンプルに関しては1wt%に満たなかった。
【0136】
実施例7
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアセテート)
(PEcMA)/ポリ(ビニルフォーマル)(PVM)
PEcMAとPVMをこの実施例では使用して、デキサメタゾン(DX)の放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、商人の情報を表5に掲載する。
【0137】
【表10】
2つのポリマーの可溶性パラメーターの差が約3.5J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。当該2つのポリマーを室温において減圧下で一晩乾燥し、そして次に個々に無水テトラヒドロフラン(THF)(シグマ−アルドリッチ社)に溶解することにより、4wt%及び5wt%の溶液を作成した。DXを同じTHFを用いて溶解することにより、約0.141wt%の溶液を作成した。上記3つの溶液を異なる量にて混合することにより、固形物の全重量に基づいて約0wt%、40wt%及び100wt%のPEcMAを含んだ3つのブレンド溶液を作成した。
【0138】
各溶液は固形物の全重量に基づいて約10wt%のDXを含んだ。ブレンド溶液を約1.27cmかける3.81cmのステンレス鋼(316L)シームの表面にコートしたが、それらは前もってTHFで洗浄して乾燥させてあった。コートされたシームを窒素ガス下で室温において一晩保存することにより、溶剤を除去した。シームを実験の各工程の後に秤量した。コーティングの厚さのときのように、重量差に基づいて、薬剤/ポリマーコーティングの全量を各シームに関して測定した。この実施例では、乾燥されたコーティングの典型的な重量は約4ミリグラム(mg)から10mg/シームであり、そして厚さは約10マイクロメーター(ミクロン)から20ミクロンであった。
【0139】
PEcMA/PVMポリマーマトリックスの溶解を、上で記載したポリマー/薬剤をコートされたシームを用いて実施した。コートされたシームを、約2mgのコーティングを含んだ小片に切断した。各小片を、3ミリリットル(mL)の予め37℃に温めたリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内に入れた。溶解試験を37℃において実施し、そしてサンプルをシェーカー上で約10回転/分(rpm)にて撹拌した。サンプルを様々な時間において分析することにより、PBSを回収することにより、サンプル中の薬剤の濃度を測定した。各回収の後に、PBSを新鮮にした。PBS中のDXの濃度を、UV−Vis分光光度計(HP 4152A)を用いて243ナノメートル(nm)の波長において測定した。各サンプル中のDXの濃度は、公知の濃度の一連の溶液を用いて創製された標準曲線と比較することにより、計算した。
溶解データの分析
PEcMA/PVMブレンドマトリックスからのデキサメタゾンの累積放出を図16にプロットした。PEcMAからのデキサメタゾンの放出速度は、PVMからよりもはるかに早かった。混和性ブレンドポリマーに関する放出速度は、ブレンドしていないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いてブレンド中のポリマー濃度の比を調節することにより、放出速度が整調できることを示す。3つのマトリックス全部からの累積放出は時間の平方根に対してほぼ直線であったことから、バーストがなく、そしてDXのデリバリーが透過制御下であったことを示す。
【0140】
実施例8
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアクリレート)
(PEcMA)/ポリスチレン(PS)
PEcMAとPSをこの実施例において用いることにより、DXの放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、売り主の情報を表5に掲載する。2つのポリマーの可溶性パラメーターの差が約1.3J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、実施例7で使用されたのと同じであった。サンプルの調製、溶解、及びデータ分析は実施例7におけるのと同じであった。放出曲線を図17に示す。デキサメタゾンの放出速度は、PEcMAからよりもPSからの方が遅かった。PSとPEcMAの混和性ブレンドからのDXの放出速度はブレンドしないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いることにより放出速度は整調できることを示す。DXの累積放出は時間の平方根に比例したことから(バーストが観察されなかった)、PEcMA/PSブレンドからのDXのデリバリーが透過制御下であったことを示唆する。
【0141】
実施例9
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアクリレート)
(PEcMA)/ポリ(メチルメタクリレート)(PMMA)
PEcMAとPMMAをこの実施例において用いることにより、DXの放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、売り主の情報を表5に掲載する。2つのポリマーの可溶性パラメーターの差が約2.1J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、実施例7で使用されたのと同じであった。サンプルの調製、溶解、及びデータ分析は、実施例7におけるのと同じであった。図18に示すとおり、PEcMAからのDXの放出速度は、PMMAからよりも相当早かった。PMMAとPEcMAの混和性ブレンドからのDXの放出速度は、ブレンドしないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いることにより放出速度が整調できることを示す。DXの累積放出も時間の平方根に比例したことから(バーストが観察されなかった)、PEcMA/PMMAブレンドからのDXのデリバリーが透過制御下であったことを示唆する。
【0142】
実施例10
クマリン(疎水性活性薬剤)を含むポリ(エーテルウレタン)ブレンド
PELLETHANE 75D(PL75D)、ポリ(エーテルウレタン)をダウケミカルカンパニー、ミッドランド、ミシガンから購入した。TECOPLAST(TP)(TP−470)及びTECOPHILIC(TL)60D60,他の2つのポリ(エーテルウレタン)を、サーメディックス社、ウオーバーン、マサチューセッツから購入した。TPは82Dのショア硬度を有する。クマリンはシグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。メルクインデックス(13版、メルクアンドカンパニー社、ホワイトハウスステーション、ニュージャージー)に基づいて、1グラムのクマリンを400mLの氷水に溶解した。この実施例で使用した無水テトラヒドロフラン(THF)、無水メタノール、及びアセトニトリル(HPLC)も、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。
【0143】
PL75Dを70℃にて減圧下で一晩乾燥した。乾燥したペレットを、予め230℃に温めた2つのプレートの間で圧縮し、そして約5分間維持した。サンプルを室温に冷やした後、THFを持たしたバイアルの中にそれを入れて、溶解するまで撹拌した(視覚観察による)。TPとTLを、室温にて混合物を撹拌することにより直接THFに溶解した。クマリンもTHFに溶解した。全ての溶液の濃度は約1wt%であった。TL溶液及びクマリン溶液を1:1の重量比にて混合した。この混合物がリザーバーシステムのベースコーティング溶液である。TP溶液とPL75D溶液を様々な重量比で混合することによ、TPのPL75Dに対する重量比が100:0、75:25、50:50、25:75、及び0:100の5つの異なる混合物を作成した。これらの溶液を本明細書ではリザーバーシステムのキャップコーティング溶液と呼ぶ。
【0144】
溶解サンプルをTHFによりリンスして洗浄されたステンレス鋼(316L)シーム(12.1 X 38.1mm2)により調製した。洗浄されたシームをPL75D/THF溶液でコートした。コートされたシームを一晩窒素下で乾燥させた。次に、それらを215−220℃にて5−10分間熱処理した。この熱処理は、ポリマー/活性薬剤層によるそれらの接着を促進したシームの表面へのプライマーの形成を導く。プライマーコーティングの厚さは約1マイクロメーター(ミクロン)であった。5つのプライマー処理されたシームを、次に、ベースコーティング溶液でコートして、一晩乾燥した。次に、これらのシームを以下の様式にて異なるキャップコーティング溶液によりディップコートした:シームを2から3秒間キャップコーティング溶液の一つの中に浸し、次に窒素ガス中で乾燥した(約1分間)。そのようなディッピング及び乾燥のプロセスを8回各シームに繰り返した。全プロセスを窒素を満したグローブボックス中で終えた。各シーム中のキャップコーティングの厚さは約1.7から3.4ミクロンであった。全てのコーティングがクリアー(clear)で、透明(transparent)であった。
クマリンの溶解
リン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内にコートされたシームを入れることにより、キャップコートされたシームからのクマリンの溶解を実施した。各バイアルは4ミリリットル(mL)のPBSを含んだ。バイアルはインキュベーター−シェーカー内で37℃に保存し、そして約50rpmにて撹拌した。予め決定された時間において、PBSをバイアルから回収して、新鮮なPBSと置換した。1週間後に、溶解試験を停止し、そして残りのコーティングを4mLのアセトニトリルに溶解した。これらの溶液全ての中のクマリンの濃度を、UV検出器を備えた液体クロマトグラフィー(HP 1090)により測定した。可動相は50wt%の酢酸ナトリウム水溶液と50wt%のアセトニトリル(HPLC)の混合物であった。Zorbax Eclipse(5ミクロン)カラムを使用した。UV検出は277nmの波長にて実施した。標準曲線は公知濃度のクマリン溶液のシリーズにより得た。これらの標準クマリン溶液は、メタノールにクマリンを溶解することにより濃縮溶液(約1wt%)を作成してこの濃縮溶液をPBSで希釈することで作成した。
溶解データ分析
クマリンの累積パーセンテージ放出対キャップコートされたシーム中のPL75D含有量を図19においてプロットした。クマリンのシーム中の全量は、溶解した溶液中の全クマリンと残りのコーティング中に残ったクマリンを一緒に加えることにより測定した。プロット中に示されるように、クマリンは100%TPをコートされたシームよりも100%PL75Dをコートされたシームからの方が相当素早く放出された。PL75D/TPブレンドからの放出速度は2つの純粋なポリマーからの速度の間であった。より興味を引くこととして、速度がブレンド中のPL75D含有量に対して平行であった。これらの結果は、明らかに、ブレンドの組成を変更することによりクマリンの放出速度が調節できたことを示す。
【0145】
PL75D/TPブレンドをTL/クマリン層のトップへのキャップコーティングとしてコートされたため、我々は、放出曲線にタイムラグが存在したはずであると予測した。しかしながら、図19に示した結果はこれを示さなかった。我々は、これがTL/クマリンがディップコーティングプロセスの間に再溶解したことによったと推測した。
混和性試験
溶解サンプルが有したのと同じTP/PL75D比を含むように、混和性試験のためのサンプルを作成した。これらのサンプル中にクマリンは存在しなかった。サンプルをPYRIS 1示差走査型熱量計(DSC)(パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ)により試験した。スキャニングは−100から220℃まで、40℃/分にプログラムした。サンプルのサイズは約10ミリグラム(mg)から16mgであった。図20に示すとおり、純粋なPL75Dは約22℃にTg遷移を有し、熔融様遷移を約173℃に有した。このTgは樹脂のハードドメインに関連すると考えられた。純粋なTPは約72℃にガラス遷移を有した。PL75DとTPを50/50の比でブレンドした場合、たった一つのTg遷移しか有さず、それは約50℃であった。これは、PL75DとTPがこの比率で混和性であることを示唆する。
【0146】
特許、特許出願、仮特許出願及び公開を含む、及び電子上で利用可能な本明細書にて引用された資料(materials)は、引用により本明細書に編入される。前記の詳細な説明及び実施例は理解の明瞭化のために提供された。発明は示され且つ記載された厳密な詳述にのみ限定されない;多くの変更が当業者には明らかになり、そして請求の範囲に定義された発明の範囲に含まれることが意図される。
【図面の簡単な説明】
【0147】
【図1】選択されたポリマーのTg対可溶性パラメーターのチャート。ロシグリタゾンマレートの可溶性パラメーターにて一点に集められたボックスは、ロシグリタゾンマレートに関する候補を囲む。
【図2】様々なポリ(カーボネートウレタン)及ポリ(ビス−フェノールAカーボネート)ブレンド(PCU75D/PCブレンド)の率対温度のグラフ。PCの含有量が増加するにつれて、ブレンドの個々のポリマーのTgが共により接近してシフトすることから、PCU75D/PCブレンドは混和性であったことが示される。
【図3】様々なPCU75D/PCブレンドからのデキサメタゾンの累積放出対時間の平方根のグラフ。ブレンドのPCU75Dの量を変化させることにより放出速度は整調された(tuned)。
【図4】PCU75D/PC中のデキサメタゾンの拡散係数対ブレンドの組成のグラフ。拡散係数はブレンドのPCU75D含有量の函数として増加した。
【図5】様々なPELLETHANE 75D/PXブレンド(PX=直鎖状ポリ(ビス−フェノールAエポキシド樹脂、レジェンド内のPLのあとの数はPELLETHANE 75Dの重量パーセント(wt%)を示す)の累積放出対時間の平方根のグラフ。ブレンドのPELLETHANE 75Dの量を変化させることにより放出速度は整調された。
【図6】PELLETHANE 75D/PHENOXYブレンドのDSC曲線。
【図7】様々なPCU75D/PCU55Dブレンドからのデキサメタゾンの累積放出のグラフ(2つの異なるポリ(カーボネートウレタン)のブレンド、レジェンド内のPCU75Dのあとの数はPCU75Dのwt%を示す)。ブレンドのPCU55Dの量を変化させることにより放出速度は整調された。
【図8】様々なPELLETHANE 75D/PXブレンドからのロシグリタゾンマレートの累積放出(レジェンド内のPLのあとの数はPELLETHANE 75Dのwt%を示す)。ブレンドのPELLETHANE 75Dの量を変化させることにより放出速度は整調された。
【図9A】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9B】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9C】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9D】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図10】PVAC/CABブレンドのDSCスキャン。ブレンドのガラス遷移は、ブレンドのPVAC含有量の函数として変化した。
【図11】様々なPVAC/CABブレンドからのデキサメタゾンの累積放出対時間の平方根のグラフ。ブレンドのPVACの量を変化させることにより放出速度は整調された。
【図12】PVAC/CABブレンド中のデキサメタゾンの拡散係数対ブレンドの組成のグラフ。拡散係数はブレンドのPVACの含有量の函数として増加した。
【図13】親水性ポリウレタンとポリ(ビニルアセテート−コ−ビニルピロリドン)のブレンドからのResten NGのデリバリーのグラフ。
【図14】TECOPHILIC HP−60D−60/PVP−VAブレンドのDSC曲線のグラフ。
【図15】PVP−VA含有量の函数としてのTECOPHILIC HP−60D−60/PVP−VAブレンドの膨張パーセンテージのグラフ。
【図16】42wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と58%のポリ(ビニルフォーマル)(PVM)のブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図17】45wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と55%のポリスチレンのブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図18】45wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と55%のポリ(メチルメタクリレート)のブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図19】PL75D/TPブレンドキャップコートされたシームからのクマリンの累積パーセンテージ放出。
【図20】PL75D/TPブレンドのDSC曲線であり、これらの2つのポリマーの間の混和性を示した。
【発明の開示】
【0001】
関連出願のクロスリファレンス
本出願は、2002年8月13日に出願された合衆国仮特許出願第60/403,352号に対する優先権を主張し、その全体を引用により本明細書に編入する。
【0002】
発明の背景
医学用装置上へのポリマーのコーティングは活性薬剤(例えば、治療剤)の被験者へのデリバリーのための貯蔵所(repository)として機能するかもしれない。多数のそのような応用のために、ポリマーコーティングは可能な限り薄くなければならない。活性薬剤をデリバリーすることにおける使用のためのポリマー材料も、様々な三次元の形状をとり得る。
【0003】
慣用の活性薬剤のデリバリーシステムは、装置表面からのクラッキング及び層間剥離による構造上の破損を含む限界をこうむる。さらに、それらは、使用可能な活性薬剤の範囲、デリバリーシステム内に含まれ得る活性薬剤の量の範囲、及びそれからデリバリーされる含有活性薬剤の比率の範囲により、限定される傾向にある。これは、しばしば、多数の慣用のシステムが単一のポリマーを含むからである。
【0004】
即ち、多大な融通性及び整調性を伴う活性薬剤デリバリーシステムに関しての継続する要求が存在する。
発明の概要
本発明は、活性薬剤のデリバリーを制御することにおいて広く良好な融通性及び整調性を有する活性薬剤デリバリーシステムを提供する。一般に、そのような利点は、2つ又はそれより多い混和性のポリマーのブレンドの使用によりもたらされる。これらのデリバリーシステムは医学用装置、例えばステント、ステントグラフト、吻合術用コネクターに、所望ならば取込ませることができる。
【0005】
本発明の活性薬剤デリバリーシステムは少なくとも2つの混和性ポリマーのブレンドを含み、少なくとも1つのポリマー(好ましくは、混和性ポリマーのうちのひとつ)は、活性薬剤のデリバリーが好ましくは浸透性制御下で支配的に生じるように、活性薬剤の溶解性に適合される。このコンテクストにおいて、浸透性制御に関して「支配的に」とは、全活性薬剤の負荷の、少なくとも50%、好ましくは少なくとも75%、そしてより好ましくは少なくとも90%が、浸透性制御によりデリバリーされることを意味する。
【0006】
浸透性制御は、一般に、ミクロンスケールレベル上に「臨界の(critical)」寸法を有する混和性ポリマーのブレンドを活性薬剤が通過するようなシステムから活性薬剤をデリバリーすることにおいて重要である(即ち、拡散ネットの通路が約1000マイクロメートルより大きくないが、成型された対象物に関して、約10,000ミクロンまでであり得る)。さらに、活性薬剤の不均一な取込みにより不利益に影響されることなく所望の機械特性を提供する特定の活性薬剤のためにポリマーを選択することが一般に望まれる。
【0007】
第1の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は疎水性であり、約1200g/molを超えない(即ち、未満又は等しい)分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、各々が少なくとも一つの可溶性パラメーターを有し、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えず(好ましくは、25J1/2/cm3/2を超えない)、そして分子の膨張率が10体積%を超えない。
【0008】
第2の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は親水性であり、約1200g/molを超えない分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超え(好ましくは、25J1/2/cm3/2を超える)、そして分子の膨張率が10体積%を超えない。
【0009】
第3の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は疎水性であり、約1200g/molを超える分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えず(好ましくは、25J1/2/cm3/2を超えない)、そして分子の膨張率が10体積%を超える。
【0010】
第4の好ましい態様において、本発明は、活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステム(目標の拡散率を有する)を提供するが、但し:活性薬剤は親水性であり、約1200g/molを超える分子量を有し;そして混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;ブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超え(好ましくは、25J1/2/cm3/2を超える)、そして分子の膨張率が10体積%を超える。
【0011】
本発明は、活性薬剤デリバリーシステムを含む医学用装置も提供する。
本発明は、被験者に活性薬剤をデリバリーする方法も提供する。一つの態様において、デリバリー方法は、上記のとおりに活性薬剤デリバリーシステムを用意し、そして活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【0012】
本発明は、予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする(及び作成する)方法も提供する。
【0013】
一つの態様において、上記方法は、約1200g/mlを超えない分子量を有する活性薬剤を用意し;少なくとも2つのポリマーを選択するが、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして少なくとも二つのポリマーの各々の少なくとも一つのTgが目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;そして混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【0014】
別の態様において、上記方法は、約1200g/mlを超えない分子量を有する活性薬剤を用意し;少なくとも2つのポリマーを選択するが、但し:活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして上記少なくとも2つのポリマーの膨張性の間の差が目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;そして混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【0015】
本発明の上記の概要は、本発明の各開示された態様又は各手段(implementation)を記載することを意図しない。続く記載は例示の態様を、より特定して例示する。出願を通して幾つかの場所において、実施例のリストを通してガイダンスが提供されるが、当該実例は様々な組み合わせにおいて使用可能である。各例において、記載されたリストは代表群としてのみ提供され、そして排他的リストとして解釈されるべきではない。
【0016】
例示的態様の詳細な説明
本発明は、被験者にデリバリーされる活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムを提供する。当該デリバリーシステムは、少なくとも2つが本明細書にて定義されるとおりに混和性である限り、様々なポリマーを含み得る。活性薬剤は、ブレンドからそれが溶解するように、混和性ポリマーブレンド内に取込まれてよいか、あるいは、活性薬剤が通過する環境に対してバリヤーとしてブレンドが機能し得る。
【0017】
混和性ポリマーブレンドは、非混和混合物又は例えば単一のポリマーを含む慣用のシステムができるよりも広い範囲の活性薬剤に関して多大な融通性及び整調性を提供し得るため、有利である。即ち、2つまたはそれより多いポリマーを使用すると、そのうちの少なくとも2つが混和し、ポリマーの一方のみを用いたデリバリーシステムよりも、より融通のきく活性薬剤デリバリーシステムを一般的には提供できる。広い範囲の種類の活性薬剤が一般的には使用できる。広い範囲の量の活性薬剤が一般的には本発明のデリバリーシステムに取込まれて、デリバリーされ得る(好ましくは浸透性制御下で支配的に)。活性薬剤に関して広い範囲のデリバリー速度が本発明のデリバリーシステムにより一般的には提供できる。少なくとも一部において、これは、少なくとも2つの混和性ポリマーを含む混和性ブレンドの使用のためである。理解されるべきことは、本明細書の記載は2つのポリマーに言及しているが、発明は混和性ポリマーブレンドが少なくとも2つの混和性ポリマーを含んで形成される限り、2つより多いポリマーを含むシステムも包含することである。
【0018】
本発明の混和性ポリマーブレンドは連続部分を形成するのに十分な量の少なくとも2つの混和性ポリマーを有し、活性薬剤の放出の速度を整調させる助けとなる。そのような連続部分(即ち、連続相)は、顕微鏡によるか、又は選択された溶剤エッチングにより、同定することができる。好ましくは、少なくとも2つの混和性ポリマーが混和性ポリマーブレンドの少なくとも50体積%を形成する。
【0019】
混和性ポリマーブレンドは、任意に、分散した(即ち、不連続な)非混和性部分を含むことできる。連続部分と分散部分の両方が存在するなら、活性薬剤は何れかの部分に取込むことができる。好ましくは、活性薬剤は、連続部分に負荷されて、支配的な浸透性制御下において活性薬剤のデリバリーを提供する。活性薬剤を負荷するためには、活性薬剤の可溶性パラメーターと、可溶性薬剤の大部分が負荷される混和性ポリマーブレンドの部分を適合させる(典型的には、約10J1/2/cm3/2を超えない範囲まで、好ましくは5J1/2/cm3/2を超えない範囲まで、そしてより好ましくは約3J1/2/cm3/2を超えない範囲まで)。連続相は、活性薬剤が負荷される場所に拘わらず、活性薬剤の放出を制御する。
【0020】
本明細書にて使用される、混和性ポリマーブレンドは、2つ又はそれより多いポリマーの完全に混和性の多数のブレンド、並びに2つ又はそれより多いポリマーの部分的に混和性の多数のブレンドを包含する。完全に混和性のポリマーのブレンドは、理想的には、単一のガラス遷移温度(Tg)を、好ましくは全濃度範囲にわたり分子レベルにて混合するために、セグメント化されたポリマーに関して各相内(典型的には、ハード相とソフト相)に一つのTgを有する。部分的に混和性のポリマーのブレンドは複数のTg’sを有してよく、セグメント化されたポリマーに関してハード相及びソフト相の一方又は両方の中に存在することができるが、分子レベルでの混合が全濃度範囲のほんの一部にのみ限定されるからである。これらの部分的に混和性のポリマーのブレンドは、ブレンド内の少なくとも2つのポリマーの各々に関しての少なくとも一つのTg内の差(Tgポリマー1−Tgポリマー2)の絶対値がブレンディングの作用により減少する限り、用語「混和性のポリマーのブレンド」の範囲内に含まれる。Tg’sは、機械特性、熱特性、電気特性等を測定することにより、温度の函数として決定できる。
【0021】
混和性ポリマーブレンドは、その光学特性に基づいて決定することもできる。完全に混和性のポリマーブレンドは、安定且つ均質な透明のドメインを形成し、不混和性ポリマーブレンドは、成分が同一の屈折率を有さないと、光を散乱して視覚上濁って見える不均質なドメインを形成する。さらに、不混和性ブレンドの相−分離構造は顕微鏡により直接観察することができる。混和性をチェックするために本発明において使用される単純な方法は、ポリマーを混合し、そして約10マイクロメートルから約50マイクロメートルの薄いフィルムを形成することを含む。そのようなフィルムが、ブレンド前の個々のポリマーのクリアーさ及び透明性の劣る同じ厚さのフィルムほど広く(generally)クリアー及び透明性であるなら、ポリマーは完全に混和性である。
【0022】
ポリマー間の混和性は、それらの間の相互作用及びそれらの分子構造及び分子量に依存する。ポリマー間の相互作用は、いわゆるフローリー−ハギンスのパラメーター(χ)により特性決定できる。χがゼロ(0)に近いか又は負の場合、ポリマーは極めておそらく(very likely)混和性である。理論上、χはポリマーの可溶性パラメーターから見積もることができ、即ち、χはそれらの間の差の二乗に比例する。よって、ポリマーの混和性は、ほぼ予測することができる。例えば、2つのポリマーのχパラメーターが近ければ近いほど、2つのポリマーが混和性である可能性は高くなる。ポリマー間の混和性はそれらの分子量が増加するにつれて低下する傾向にある。
【0023】
即ち、実験による決定に加え、ポリマーの混和性はフローリー−ハギンス相互作用パラメーターに基づくか、又はより単純には、成分の可溶性パラメーターに基づいて、単純に予測することができる。しかしながら、分子量効果のため、接近した可溶性パラメーターは必ずしも混和性を保証しない。
【0024】
ポリマーの混合物は、混和性であるとして本明細書において提供された定義の一つを満すことのみを要求されることを理解するべきである。さらに、ポリマーの混合物は、活性薬剤の取込みに際して混和性ブレンドになってよい。本発明の特定の態様は、セグメント化されたポリマーを含む。本明細書にて使用される「セグメント化されたポリマー」は、複数のブロックから構成され、それらの各々は主にそれ自体から構成される相に分離し得る。本明細書にて使用される、ポリマーの「ハード」セグメント又は「ハード」相は、使用温度において結晶質であるか又は使用温度を超えるガラス遷移温度の非晶質(即ち、ガラス質)であるかの何れかであり、そしてポリマーの「ソフト」セグメント又は「ソフト」相は、使用温度未満のガラス遷移温度の非晶質である(即ち、ゴム質)。本明細書にて、「セグメント」は化学的フォーミュレーションを意味し、そして「相」は形態を意味し、主に、対応するセグメント(例えば、ハードセグメントはハード相を形成する)を含むが、他のセグメントの幾つかを含むことができる(例えば、ハード相中のソフトセグメント)。
【0025】
本明細書にて使用されるブレンドの「ハード」相は、主に、セグメント化されたポリマーのハードセグメントを含み、そして任意に、その中にブレンドされた第2のポリマーの少なくとも一部を含む。同様に、ブレンドの「ソフト」相は、セグメント化されたポリマーのソフトセグメントを支配的に含み、そして任意に、その中にブレンドされた第2のポリマーの少なくとも一部を含む。好ましくは、本発明のポリマーの混和性ブレンドは、セグメント化されたポリマーのソフトセグメントのブレンドを含む。
【0026】
セグメント化されたポリマーの可溶性パラメーターに言及する際は「セグメント」が用いられ、そしてセグメント化されたポリマーのTgに言及する際は「相」が用いられる。即ち、可溶性パラメーターは、一般にセグメント化されたポリマーに関しての計算値であり、個々のポリマー分子のハード及び/又はソフトセグメントを意味し、Tgは、一般に測定値であり、バルクのポリマーのハード及び/又はソフト相を意味する。
【0027】
ポリマー及び活性薬剤の種類及び量は、一般に、予め選択された溶解時間を有するシステムを、混和性ポリマーブレンドの予め選択された臨界寸法を通して形成させるために選択される。ポリマーのガラス遷移温度、膨張性、及び可溶性パラメーターは、活性薬剤が混和性ポリマーブレンドに取込まれていようがいまいが、活性薬剤デリバリーシステム中の成分の適切な組み合わせを選択するように当業者を導く際に使用することができる。可溶性パラメーターは、一般に、ポリマーの混和性を決定し、そして活性薬剤の可溶性を混和性ポリマーブレンドの可溶性に適合させるのに有用である。ガラス遷移温度及び/又は膨張性は、一般に、活性薬剤の溶解時間(又は速度)を整調するのに有用である。これらのコンセプトは、以下において詳細に論じられる。
【0028】
混和性ポリマーブレンドは、当該混和性ポリマーブレンドが活性薬剤のデリバリーを制御する限り、様々なフォーマットにおいて本発明のデリバリーシステム内で活性薬剤と化合して使用することができる。
【0029】
一つの態様において、混和性ポリマーブレンドは、その中に活性薬剤を取込んで有する。好ましくは、そのような活性薬剤は浸透性制御下において支配的に溶解される、大多数の活性薬剤が連続部分に負荷されていようがいまいが、ポリマーブレンドの連続する部分(即ち、混和性部分)内で活性薬剤の少なくとも幾つかの可溶性を必要とする。活性薬剤の溶解の間に孔性チャネリングがほとんど又は全く生じず、そして分散したドメインのサイズがブレンドの臨界寸法よりはるかに小さい限り、分散は容認され、そして物理的特性は、所望の機械的性能のための組成を通して、一般に均一である。この態様は、しばしば「マトリックス」システムと呼ばれる。
【0030】
別の態様において、混和性ポリマーブレンドは、最初は、活性薬剤の浸透に対してバリヤーを提供する。この態様は、しばしば、「リザーバー」システムと呼ばれる。リザーバーシステムは、2つ又はそれよい多い層を有する多数のフォーマットを採り得る。例えば、混和性ポリマーブレンドは、別の物質の内部層にわたり外部層を形成することができる(本明細書においては内部マトリックス物質と呼ぶ)。別の例において、リザーバーシステムはコア−シェルの形態を採ることができ、混和性ポリマーブレンドがコアマトリックスの周囲にシェルを形成する(即ち、内部マトリックス物質)。形成の少なくとも初期には、シェル又は外部層の中の混和性ポリマーブレンドが、活性薬剤を実質上含まないことが可能である。のちに、活性薬剤は、内部マトリックスから、並びに被験者へのデリバリーのための混和性ポリマーブレンドを通して、浸透する。内部マトリックス物質は、活性薬剤のデリバリーにおいて使用される様々な慣用の物質を含むことができる。これらは、例えば、有機ポリマー、例えば、混和性ポリマーブレンド中での使用のための本明細書にて記載された有機ポリマー、又はワックス、又は別の混和性ポリマーブレンドを含む。あるいは、内部マトリックス物質は活性薬剤自身であり得る。
【0031】
リザーバーシステムに関しては、活性薬剤の放出速度を、外部層の物質の選択により整調することができる。内部マトリックスはポリマーの非混和性の混合物を含み得るか、又は外部層がポリマーの混和性ブレンドならば、内部マトリックスはホモポリマーであり得る。
【0032】
マトリックスシステムを用いた場合のように、リザーバーシステム中の活性薬剤は、好ましくは、支配的に、浸透性制御下で、バリヤー層の混和性ポリマーブレンド(即ち、バリヤーポリマーブレンド)を通して溶解され、当該バリヤーポリマーブレンド中で活性薬剤の少なくともいくらかの溶解性を必要とする。再び、活性薬剤の溶解の間に孔性チャネリングがほとんど又は全く生じず、そして分散したドメインのサイズがブレンドの臨界寸法よりはるかに小さい限り、分散は容認され、そして物理特性は所望の機械的性能のための組成を通して、一般に均一である。これらの考察は内部マトリックスのためにも望まれるかもしれないが、それらは必ずしも必要ではない要求である。
【0033】
典型的には、本発明の活性薬剤デリバリーシステム内の活性薬剤の量は、デリバリーされる量及びデリバリーされる時間により決定される。他の因子も存在する活性薬剤のレベルに貢献することができ、例えば、組成物が基質上で均一なフィルムを形成する能力を含む。
【0034】
好ましくは、マトリックスシステムに関して、活性薬剤は、混和性ポリマーブレンドと活性薬剤の全重量の、少なくとも約0.1重量パーセント(wt%)、より好ましくは少なくとも約1wt%、そしてさらにより好ましくは少なくとも約5wt%の量にて混和性ポリマーブレンド内に存在する(即ち、内に取込まれる)。好ましくは、マトリックスシステムに関して、活性薬剤は、混和性ポリマーブレンドと活性薬剤の全重量の、約80wt%を超えず、より好ましくは約50wt%を超えず、そしてもっとも好ましくは約30wt%を超えない量にて、混和性ポリマーブレンド内に存在する。典型的に且つ好ましくは、活性薬剤の量は、混和性ポリマーブレンド内のその可溶性限界になるか又はそれ未満になる。
【0035】
好ましくは、リザーバーシステムに関して、活性薬剤は、内部マトリックス(活性薬剤を含む)の全重量の、少なくとも約0.1wt%、より好ましくは約10wt%、そしてさらにより好ましくは約25wt%の量にて、内部マトリックス内に存在する。好ましくは、リザーバーシステムに関して、活性薬剤は、内部マトリックス(活性薬剤を含む)の全重量の、100wt%まで、そしてより好ましくは約80wt%を超えない量にて、内部マトリックス内に存在する。
【0036】
本発明の活性薬剤デリバリーシステムにおいて、活性薬剤は混和性ポリマーブレンドを通して溶解可能である。溶解は、好ましくは、活性薬剤を混和性ポリマーブレンドを通して支配的に浸透させることにより制御される。即ち、活性薬剤は最初に混和性ポリマーブレンド内に溶解して、次に、浸透性制御下において支配的に混和性ポリマーブレンドを通して拡散する。即ち、上記のとおり、特定の好ましい態様に関して、活性薬剤は混和性ポリマーブレンドの可溶性限界であるか又はそれ未満である。理論に拘束されることを意図しないが、この機構のために、本発明の活性薬剤デリバリーシステムは顕著なレベルの整調可能性を有すると信じられる。
【0037】
活性薬剤が混和性ポリマーブレンドの可溶性を超え、そして不溶の活性薬剤の量が濾過限界を超えたなら、そのときは、活性薬剤が孔性機構を通して支配的に溶解され得る。さらに、活性薬剤の不溶性の相(例えば、粒子又は粒子の凝集物)の最大寸法が混和性ポリマーブレンドの臨界寸法と同じ桁ならば、そのときは、活性薬剤が孔性機構を通して支配的に溶解され得る。孔度の制御による溶解は、典型的には、不所望であるが、何故ならば、有効な予測性及び制御可能性を提供しないからである。
【0038】
本発明の活性薬剤デリバリーシステムは、好ましくはミクロンスケールのレベルの臨界寸法を有するため、十分な量の活性薬剤を含むこと及び孔性機構によるデリバリーを回避することが難しくなり得る。即ち、活性薬剤と混和性ポリマーブレンドの少なくとも一つのポリマーの可溶性パラメーターを適合させることにより、孔性機構によるデリバリーに関する傾向を低下させながら負荷のレベルを最大にする。
【0039】
放出される活性薬剤の量対時間(t)の溶解プロフィールを検査することにより、浸透制御放出機構が存在するか否かを決定することができる。マトリックスシステムからの浸透制御放出に関しては、当該プロフィールがt1/2に正比例する。リザーバーシステムからの浸透制御放出に関しては、当該プロフィールがtに正比例する。あるいは、シンク条件(即ち、ポリマーブレンドと、活性薬剤が溶解される溶媒との間に速度制限バリヤーがない条件)下では、孔度制御溶解がバースト効果(即ち、活性薬剤の初期の極めて素早い放出)をもたらすことができた。
【0040】
本発明の活性薬剤デリバリーシステムは、マトリックスシステムの形態かリザーバーシステムの形態かを問わず、限定ではないが、基材のコーティング(例えば、開いたか又は閉じたセルの泡、織り込まれたか又は織り込まれていない材料)、フィルム(例えば、パッチ中のようにフリースタンドであり得る)、成形されたオブジェクト(微小球、ビーズ、ロッド、ファイバー、又は他の成形されたオブジェクト)、曲がりくねったパッキング剤等の形態であり得る。
【0041】
本明細書にて使用される「活性薬剤」は、被験者(例えば、動物)において局所性又は全身性の効果を生じる薬剤である。一般には、それは、薬学上活性な薬剤である。当該用語は、疾患の診断、治療(cure)、軽減、処置(treatment)、又は予防(prevention)においてか、又は被験者における所望の外科的又は精神的発達又は症状の増強においての使用のために意図されたあらゆる物資を包含するように使用される。本明細書にて使用される用語「被験者」は、ヒト、ヒツジ、ウマ、畜牛、ブタ、イヌ、ネコ、ラット、マウス、鳥、爬虫類、魚、昆虫、蛛形類、原生動物(例えば、ポロトゾア)、及び原核細菌を含むものと見なされる。好ましくは、被験者はヒト又は他の哺乳類である。
【0042】
活性薬剤(active agent)は、合成又は天然であることができ、そして限定ではないが、有機又は無機化学薬剤、ポリペプチド(ペプチド、オリゴペプチド、蛋白質、酵素、ホルモン等を含むあらゆる長さのL−又はD−アミノ酸のポリマーを包含するように本明細書では使用される)、ポリヌクレオチド(オリゴヌクレオチド、一本鎖及び二本鎖のDNA、一本及び二本鎖のRNA,DNA/RNAキメラ等を含むあらゆる長さの核酸のポリマーを包含するように本明細書では使用される)、サッカライド(例えば、モノ−、ジ−、ポリ−サッカライド及びムコポリサッカライド)、ビタミン、ウイルス因子(agent)、及び他の生物材料、放射性核種等を含む。例としては、抗血栓性及び抗凝血性薬剤、例えばヘパリン、クマジン、クマリン、プロタミン、及びヒルジン;抗細菌剤、例えば抗生物質;抗新生物剤及び抗増殖剤、例えばエトポシド、ポドフィロトキシン;アスピリン及びジピリダモルを含む抗血小板剤;抗有糸分裂剤(細胞障害性薬剤)及び抗代謝剤、例えばメトトレキセート、コルチシン、アザチオプリン、ビンクリスチン、ブンブラスチン、フルオロウラシル、アダリアマイシン、及びムタミシン核酸;抗糖尿病薬剤、例えばロシグリタゾンマレート;及び抗炎症剤を含む。本発明における使用のための抗炎症剤は、グルココルチコイド、それらの塩、及びそれらの誘導体、例えばコルチゾール、コルチゾン、フルドロコルチゾン、プレドニソン、プレドニソロン、6α−メチルプレドニソロン、トリアムシノロン、ベータメタゾン、デキサメタゾン、ベクロメタゾン、アクロメタゾン、アムシノニド、クレベタゾール及びクロコルトコーンを含む。好ましくは、活性薬剤はヘパリンではない。
【0043】
本発明の好ましい活性薬剤デリバリーシステムに関して、活性薬剤はポリマーブレンドの混和性部分の可溶性に適合されるのが典型的である。即ち、活性薬剤が親水性である本発明の態様に関しては、好ましくは、混和性ポリマーブレンドの少なくとも一つの混和性ポリマーが親水性である。活性薬剤が疎水性である本発明の態様に関しては、好ましくは、混和性ポリマーブレンドの少なくとも一つの混和性ポリマーが疎水性である。しかしながら、これは必ずしも必要ではなく、そして水によるポリマーの膨張の可能性のため及び活性薬剤の制御されたデリバリーの損失のため、低分子量の親水性活性薬剤のためのデリバリーシステムを有することは望まれないかもしれない。本明細書にて使用される、このコンテクストにおける(ブレンドのポリマーのコンテクストにおける)用語「親水性」は、最初に来るにせよ、体温(即ち、約37℃)において水で膨張したときに、10体積%を超えるか又は重量で少なくとも10%まで増加する物質を意味する。本明細書にて使用される、このコンテクストにおける(ブレンドのポリマーのコンテクストにおける)用語「疎水性」は、最初のものはどれも(whichever comes first)、体温(即ち、約37℃)において水で膨張したときに、10体積%を超えるか又は重量で10%を超えるまで増加しない物質を意味する。
【0044】
本明細書にて使用される、このコンテクストにおける(活性薬剤のコンテクストにおける)用語「親水性」は、ミリリットルあたり200マイクログラムを超える水に可溶性を有する活性薬剤を意味する。本明細書にて使用される、このコンテクストにおける(活性薬剤のコンテクストにおける)用語「疎水性」は、ミリリットルあたり200マイクログラムを超えない水に可溶性を有する活性薬剤を意味する。
【0045】
活性薬剤のサイズが十分に大きくなるにつれ、ポリマーを通じての拡散が影響される。即ち、活性薬剤は分子量に基づいて分類することができ、そしてポリマーは活性薬剤の分子量の範囲に依存して選択することができる。
【0046】
本発明の特定の好ましい活性薬剤デリバリーシステムに関して、活性薬剤は約1200g/molより大きい分子量を有する。本発明の特定の他の好ましい活性薬剤デリバリーシステムに関して、活性薬剤は約1200g/molを超えない(即ち、未満又は等しい)分子量を有する。さらにより好ましい態様に関して、約800g/molを超えない分子量の活性薬剤が望まれる。
【0047】
活性薬剤及びデリバリーのためのフォーマット(例えば、時間/速度及び臨界寸法)が選択されたら、当業者は本発明の教示を利用することにより、活性薬剤デリバリーシステムを提供するための少なくとも2つのポリマーの適切な組み合わせを選択することができる。
【0048】
上記のとおり、ポリマー及び活性薬剤の種類及び量は、予め選択された時間(t)を有するシステムを、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して形成するように選択されるのが典型的である。これは目標の拡散性を提供するように少なくとも2つのポリマーを選択することを含み、所定の活性薬剤に関して、臨界寸法の二乗割る時間(x2/t)に正比例する。
【0049】
所望の活性薬剤、所望の溶解時間(又は速度)、及び所望の臨界寸法のためのポリマーの選択を正確にすることにおいて、所望の活性薬剤のためにポリマーを選択する際に考え得るパラメーターは、ポリマーのガラス遷移温度、ポリマーの膨張性、ポリマーの可溶性パラメーター、及び活性薬剤の活性パラメーターを含む。これらは、活性薬剤を混和性ポリマーブレンドに取込もうが取込むまいが、当業者が活性薬剤デリバリーシステム内の成分の適切な組み合わせを選択する手引きにおいて使用することができる。
【0050】
浸透性制御デリバリーシステムの融通性を増強するためには、例えば、好ましくは、以下の関係の少なくとも一つが真実であるようにポリマーを選択する:(1)活性薬剤の可溶性パラメーターと少なくとも一つのポリマーの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えない(好ましくは、約5J1/2/cm3/2を超えず、約3J1/2/cm3/2を超えない);及び(2)少なくとも2つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない(好ましくは、約3J1/2/cm3/2を超えない)。より好ましくは、両方の関係が真実である。もっとも好ましくは、両方の関係がブレンドの全部のポリマーに関して真実である。
【0051】
一般に、化合物は可溶性パラメーターをたった一つしか持たないが、特定のポリマー、例えばセグメント化されたコポリマー及びブロックコポリマーは、例えば一つより多い可溶性パラメーターを有することができる。可溶性パラメーターは測定することができるか、又はそれらはホイ(Hoy)法及びホフティザー−ファンクレヴェレン(Hoftyzer-van Krevelen)法(化学基寄与法)を用いて計算された値の平均値を用いて計算され、D.W.van Krevelen,Properties of Polymers,第3版、Elsevier,アムステルダムに開示されるとおりである。これらの値を計算するためには、各化学物質の体積が必要であり、同じ文献に開示されたフェドーズ(Fedors)法を用いて計算することができる。
【0052】
可溶性パラメーターは、コンピューターシミュレーション、例えば、モリキュラーダイナミックスシミュレーション及びモンテカルロシミュレーションによっても計算することができる。特定すれば、モリキュラーダイナミックスシミュレーションは、アクセルリスマテリアルズスチュディオ(Accelrys Materials Studio)、アクセルリス、サンディエゴ、カリフォルニアにより実施され得る。当該コンピューターシミュレーションはフローリー−ハギンスのパラメーターを直接計算するのに使用することができる。
【0053】
様々なポリマー及び活性薬剤に関する可溶性パラメーターの例を表1に示す。
【0054】
【表1】
【0055】
【表2】
【0056】
【表3】
【0057】
【表4】
【0058】
【表5】
【0059】
【表6】
活性薬剤が疎水性であるデリバリーシステムに関しては、分子量に拘わらず、混和性ポリマーブレンドのモル平均可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように、ポリマーを選択するのが一般的である。本明細書における「モル平均可溶性パラメーター」は、互いに混和可能であって且つ混和性ポリマーブレンドの連続部分を形成するブレンド成分の可溶性パラメーターの平均を意味する。これらは、ポリマーブレンドへ取込まれる活性薬剤無しに、ブレンド中のそれらのモルパーセンテージにより秤量される。
【0060】
例えば、約1200g/mlを超えない疎水性活性薬剤、例えばグループコントリビューション法によると27J1/2/cm3/2の可溶性パラメーター又はモリキュラーダイナミックスシミュレーションによると21J1/2/cm3/2の可溶性パラメーターを有するデキサメタゾンに関しては、例示のポリマーブレンドは、セルロースアセテートブチレート(CAB)とポリビニルアセテート(PVAC)を含む。これらは、それぞれ、22J1/2/cm3/2の可溶性パラメーター及び21J1/2/cm3/2の可溶性パラメーターを有する。これらのポリマーの適当なブレンド(1:1モル比はCABのPVACに対する)は、21.5J1/2/cm3/2のモル平均可溶性パラメーターを有する。この値は、本明細書にては22*0.5+21*0.5=21.5(J1/2/cm3/2)として計算された。CABの繰り返しユニットの分子量は、アセチル基、ブチリル基、及びOH基の全数が繰り返しユニットあたり3であるという事実に基づいて、303g/molであると見積もられる。PVACの繰り返しユニットの分子量は86g/mlである。次に、PVACに対するCABの質量比は、この1:1モル比のブレンドに関して0.78/0.22である。
【0061】
活性薬剤が親水性であるデリバリーシステムに関しては、分子量に拘わらず、混和性ポリマーブレンドのモル平均可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように、ポリマーを選択するのが一般的である。
【0062】
低分子量の活性薬剤に関しての浸透性制御溶解時間(速度)の整調性を増強するためには、好ましくは、少なくとも2つのポリマーの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当するように、ポリマーを選択することができる。
【0063】
あるいは、高分子量の活性薬剤に関しての浸透性制御溶解時間(速度)の整調性を増強するためには、好ましくは、少なくとも2つのポリマーの膨張性の差が目標の拡散性を含む拡散性の範囲に相当するように、ポリマーを選択することができる。目標の拡散性は、デリバリーに関しての予め選択された時間(t)及びポリマー組成の予め選択された臨界寸法(x)により決定され、そしてx2/tに正比例する。
【0064】
目標の拡散性は、以下の等式を用いて溶解分析により容易に測定することができる(例えば、Kinam Park編纂、制御されたドラッグデリバリー:チャレンジと戦略、アメリカンケミカルソサイエティー、ワシントン、DC,1997):
【0065】
【数1】
式中、D=拡散定数;Mt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。この等式は、活性薬剤の初期負荷の60重量%までの溶解の間、有効である。また、ブレンドサンプルはフィルムの形態であるべきである。
【0066】
一般に、少なくとも一つのポリマーが目標の拡散性より高い活性薬剤拡散性を有し、そして少なくとも一つのポリマーが目標の拡散性より低い活性薬剤拡散性を有する。ポリマーシステムの拡散性は溶解分析により容易に測定することができ、実施例のセクションにおいて詳細に記載される。個々のポリマー各々からの活性薬剤の拡散性は、溶解分析により測定できるが、各ポリマーの主要な相の相対的なTg’s又は膨張性により見積もることができる。
【0067】
拡散性は、疎水性又は親水性ポリマーのガラス遷移温度に相関することができ、低分子量の活性薬剤(例えば、約1200g/mlを超えない分子量を有するもの)のためのデリバリーシステムをデザインするのに使用することができる。あるいは、拡散性は、疎水性又は親水性ポリマーの膨張性に相関することができ、高分子量の活性薬剤(例えば、約1200g/mlを超える分子量を有するもの)のためのデリバリーシステムをデザインするのに使用することができる。これは、類似の可溶性の活性薬剤に関して極めて異なる溶解速度を包含するために混和性ブレンドの範囲を使用できるため、有利である。
【0068】
ポリマーのガラス遷移温度は、よく知られたパラメーターであり、一般には測定された値である。例示の値を表1に掲載する。セグメント化されたポリマー(例えば、セグメント化されたポリウレタン)に関して、上記Tgはバルクポリマーの特定の相を意味する。一般には、低分子量の活性薬剤に関しては、混和性の相対的に低いTgのポリマーと相対的に高いTgのポリマーを選択することにより、システムの溶解キネティクスを整調させることができる。これは、小分子量の薬剤(例えば、約1200g/mlを超えない)がTg’sに直接相関する経路を通して拡散し、即ちポリマーブレンドの自由体積が温度の直接の函数であり、温度がTgを超えたときにスロープが大きくなるからである。
【0069】
好ましくは、少なくとも一つの相対的に高いTgを有するポリマーを、少なくとも一つの相対的に低いTgを有するポリマーと組み合わせる。
例えば、1200g/mlを超えない分子量を有する活性薬剤のための混和性ポリマーブレンドは、100−120℃のTgを有するセルロースアセテートブチレート、及び20−30℃のTgを有するポリビニルアセテートを含む。1200g/mlを超えない分子量を有する活性薬剤のための混和性ポリマーブレンドの別の例は、約10−80℃のハード相Tgのポリウレタンと、約140℃のTgのポリカーボネートを含む。そのような高いTgのポリマーと低いTgのポリマーを組み合わせることにより、活性薬剤デリバリーシステムが、活性薬剤の所望の溶解時間に関して整調され得る。
【0070】
図1は、低分子量の疎水性活性薬剤、ロシグリタゾンマレートをデリバリーさせるための混和性ポリマーブレンドのための適切なポリマー候補を示す。これは、選択されたポリマーのTg対可溶性パラメーターのチャートである。ロシグリタゾンマレートの可溶性パラメーターを中心とするボックスは、この活性薬剤のための候補を囲む。
【0071】
水中のポリマーの膨張性は、容易に測定することができる。しかしながら、膨張性が水の取り込みによりもたらされるのであって、温度の上昇によりもたらされるのではないことを認識するべきである。一般に、高分子量の活性薬剤に関しては、混和性の相対的に低い膨張性のポリマーと相対的に高い膨張性のポリマーを選択することにより、システムの溶解キネティクスを整調させることができる。これらのシステムをデザインするのにポリマーの膨張性を使用できるが、何故ならば、相対的に高い分子量(例えば、約1200g/mlを超える)の活性薬剤のためのフリー体積を増加させることによりポリマーブレンド外に拡散するためには、水がポリマーの中に拡散することを必要とするからである。
【0072】
好ましくは、相対的に高い膨張性を有するポリマーを、相対的に低い膨張性を有するポリマーと組み合わせる。例えば、1200g/mlを超える分子量を有する混和性ポリマーブレンドは、100%を超える膨張性を有する(即ち、水溶性)ポリビニルピロリドン−ビニルアセテート−コポリマー、及び60%の膨張性を有するポリ(エーテルウレタン)を含む。そのような高い膨張性のポリマーと低い膨張性のポリマーを組み合わせることにより、活性薬剤デリバリーシステムを活性薬剤の所望の溶解時間に関して整調させることができる。
【0073】
混和性ポリマーブレンドの膨張性は、特定の活性薬剤のためにポリマーの組み合わせを決定することにおけるファクターとしても使用される。活性薬剤が1200g/mlを超える分子量を有するデリバリーシステムに関しては、それが疎水性であるか親水性であるかに拘わらず、ポリマーは、ブレンドの膨張性が10体積%を超えるように選択される。ブレンドの膨張性は、そこに取り込まれる活性薬剤無しに評価される。
【0074】
疎水性であり且つ1200g/mlを超えない分子量を有する活性薬剤の第1群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0075】
活性薬剤の第1群のためのポリマーブレンドの適切な組み合わせの例は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,477及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A HYDROPHOBIC CELLULOSE DERIVATIVE,MEDICAL DEVICE,AND METHOD」と題する出願;2002年8月13日に出願された、合衆国仮出願連続番号60/403,478及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLYURETHANE,MEDICAL DEVICE,AND METHOD」と題する出願;及び2002年8月13日に出願された、合衆国仮出願連続番号60/403,413及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLY(ETHYLENE−CO−(METH)ACRYLATE),MEDICAL DEVICE,AND METHOD」と題する出願において詳述されている。そのようなブレンドの特定の例は、実施例のセクションに例示される。好ましくは、活性薬剤の第1の群を用いた使用のために適した混和性ポリマーブレンドは、以下を含まない:疎水性セルロース誘導体とポリウレタン及びポリビニルピロリドンのブレンド;及び/又はポリアルキルメタクリレートとポリエチレン−コ−ビニルアセテートのブレンド。
【0076】
親水性であり且つ1200g/mlを超えない分子量を有する活性薬剤の第2群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0077】
この第2の群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、ポリアクリロニトリル、シアノアクリレート、メタクリロニトリル、親水性セルロース誘導体、等、及びそれらの組み合わせを含む。このコンテクストにおいて、「組み合わせ(combination)」は、混合物及びそれらのコポリマーを意味する。混合物及びコポリマーは、一つ又はそれより多いメンバーの群及び/又は他のモノマー/ポリマーを含み得る。好ましくは、活性薬剤の第2群による使用のために適した混和性ポリマーブレンドは、疎水性セルロース誘導体とポリビニルピロリドンの両方を含まない。
【0078】
活性薬剤の第1群のためのポリマーブレンドの適切な組み合わせの例は、出願人の譲渡人の係属中の、同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A POLYURETHANE,MEDICAL DEVICE,AND METHOD」と題する出願において詳述されている。
【0079】
疎水性であり且つ1200g/mlを超える分子量を有する活性薬剤の第3群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが28J1/2/cm3/2を超えない(好ましくは、25J1/2/cm3/2を超えない)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0080】
この第3群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、少なくとも一つの疎水性ポリマーを含み、メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースニトレート、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルエチルセルロース、メチルエチルセルロース、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースプロピオネートブチレート、セルロースアセテートプロピオネートブチレート、及びそれらの組み合わせを含む。これらのシステムのためのポリマーブレンドは、疎水性又は親水性の何れかの第2のポリマーを含み得る。例えば、親水性ポリマーは浸水性ポリウレタンであり得る。好ましい親水性ポリウレタンは、その中にポリエチレンオキシドユニットを有するソフトセグメント含む。適切な親水性ポリウレタンの例は、サーメディックス社(ウオーバーン、マサチューセッツ)によりTECOPHILICの商標名で市販されているポリ(エーテルウレタン)である。好ましくは、活性薬剤の第3群による使用のために適した混和性ポリマーブレンドは、以下を含まない:疎水性セルロース誘導体とポリウレタン又はポリビニルピロリドンのブレンド;及び/又はポリアルキルメタクリレートとポリエチレン−コ−ビニルアセテートのブレンド。
【0081】
親水性であり且つ1200g/mlを超える分子量を有する活性薬剤の第4群に関して、混和性ポリマーブレンドのためのポリマーは、ブレンドの混和性ポリマーの可溶性パラメーターが21J1/2/cm3/2を超える(好ましくは、25J1/2/cm3/2を超える)ように;そしてブレンドの膨張性が10体積%を超えないように選択される。
【0082】
この第4群から活性薬剤をデリバリーするシステムのための適したポリマーの例は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,392及び同日出願された合衆国特許出願番号 を有する「ACTIVE AGENT DELIVERY SYSTEM INCLUDING A HYDROPHILIC POLYMER,MEDICAL DEVICE,AND METHOD」と題する出願(アトーニードケット番号P−10858.00)において詳述されている。そのようなブレンドの特定の例は、実施例のセクションに例示される。好ましくは、活性薬剤の第4の群を用いた使用のために適した混和性ポリマーブレンドは、疎水性セルロース誘導体とポリビニルピロリドンの両方を含まない。
【0083】
混和性ポリマーブレンド中のポリマーを架橋結合させることもさせないこともできる。同様に、ブレンドされたポリマーを架橋結合させることもさせないこともできる。そのような架橋結合は、標準の技術を用いてブレンドした後に、当業者により実施され得る。
【0084】
本発明の活性薬剤のシステムにおいては、活性薬剤が「臨界」の寸法を有する混和性ポリマーブレンドを通って通過する。この臨界寸法は、活性薬剤のネットの拡散経路であり、そして好ましくは1000マイクロメートル(即ち、ミクロン)を超えないが、成形されたオブジェクトに関しては、約10,000ミクロンまでであり得る。
【0085】
混和性ポリマーブレンドがコーティング又はフリースタンディングフィルム(共に一般的には「フィルム」と呼ばれる)を形成する態様に関しては、臨界寸法はフィルムの厚さであり、そして好ましくは約1000ミクロンを超えず、より好ましくは約500ミクロンを超えず、そしてもっとも好ましくは約100ミクロンを超えない。フィルムは望みどおり薄くすることができるが(例えば、1ナノメートル)、好ましくは約10ナノメートルより薄くなく、より好ましくは約100ナノメートルより薄くない。一般に、最少のフィルム厚は、活性薬剤の必要な用量を保持するのに要求される体積により決定され、一般的には上記物質を形成するのに使用されるプロセスによってのみ限定される。本明細書中の全ての態様に関して、フィルムの厚さが一定又は均一である必要はない。さらに、フィルムの厚さは、活性薬剤が放出されるのにかかる時間を整調するのに使用することができる。
【0086】
混和性ポリマーブレンドが成形されたオブジェクトを形成する態様に関しては(例えば、微小球、ビーズ、ロッド、ファイバー、又は他の成形されたオブジェクト)、オブジェクトの臨界寸法(例えば、微小球又はロッドの直径)は、好ましくは、約10,000ミクロンを超えず、より好ましくは約1000ミクロンを超えず、さらにより好ましくは約500ミクロンを超えず、そしてもっとも好ましくは約100ミクロンを超えない。オブジェクトは望みどおり小さくすることができる(例えば、臨界寸法に関して10ナノメートル)。好ましくは、臨界寸法は、約100ミクロンを下回らず、そしてより好ましくは約500ナノメートルを下回らない。
【0087】
一つの態様において、本発明は、混和性ポリマーブレンドを含むポリマーのトップコート層、好ましくはポリマーのアンダーコート(プライマー)層を重層された基材表面により特徴付けされる医学用装置を提供する。当該装置を使用するとき、混和性ポリマーブレンドを、被験者の体液、器官、又は組織に接触させる。
【0088】
発明は医学用装置の性質により限定されない;むしろ、何れの医学用装置も混和性ポリマーブレンドを含むポリマーコーティング層を含み得る。即ち、本明細書にて使用される用語「医学用装置」は、それらの使用及び操作の通常の過程において、体組織、器官又は体液、例えば血液と接触させることができる表面を有するあらゆる装置を意味する。医学用装置の例は、限定ではないが、ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、ポンプ、血管グラフト、弁、ペースメーカー等を含む。医学用装置は体外装置、例えば外科手術のときに使用される装置であり得、例えば、のちに被験者に戻される血液と接触する、血液酸素発生機、血液ポンプ、血液センサー、又は血液を運ぶのに使用されるチューブ等を含む。医学用装置は同様に移植用装置、例えば、血管グラフト、ステント、ステントグラフト、吻合コネクター、電気刺激用リード、心臓弁、整形外科用装置、カテーテル、シャント、センサー、髄核(nucleus pulposus)のための置換装置、蝸牛又は中耳の移植物、眼球内レンズ、等であり得る。移植可能な装置は、経皮装置、例えば薬剤注射ポート(ports)等を含む。
【0089】
一般に、発明の医学用装置を製作するのに使用されるのに好ましい材料はバイオマテリアルである。「バイオマテリアル」は、ヒト体内への移植及び/又は体液、組織、器官等への接触のために意図された材料であり、且つ、意図された目的のために機能するのに要求される機械的強度、例えば耐久性(strength)、弾性、浸透性及び柔軟性を有する材料である。移植用装置に関しては特に、使用される材料が生物適合性材料、即ち細胞又は組織に対して過度に毒性ではなく、そしてからだに対して不適当な害をもたらさない材料であることが好ましい。
【0090】
発明は、混和性ポリマーブレンドがポリマーコーティングを形成する態様に関しての基材表面の性質により制限されない。例えば、基材表面は、セラミック、ガラス、金属、ポリマー、又はそれらの組み合わせから構成され得る。金属基材表面を有する態様においては、金属は、典型的には、鉄、ニッケル、金、コバルト、銅、クロム、モリブデン、チタン、タンタル、アルミニウム、銀、プラチナ、炭素、及びそれらの合金である。好ましい金属は、ステンレス鋼、ニッケルチタン合金、例えばNITINOL、又はコバルトクロム合金、例えばNP35Nである。
【0091】
混和性ポリマーブレンドを含むポリマーコーティングを、共有又は非共有相互作用の何れかにより基材表面に接着することができる。非共有相互作用は、イオン性相互作用、水素結合、双極子相互作用、疎水性相互作用及びファンデルワース相互作用を例えば含む。
【0092】
好ましくは、混和性ポリマーブレンドコーティングの塗布の前に、基材表面は活性化又は官能基化されないが、幾つかの態様においては、接着を促進するために、基材表面の前処理が望まれるかもしれない。例えば、ポリマーのアンダーコート層(即ち、プライマー)を使用することにより、基材表面へのポリマーコーティングの接着を増強することができる。適切なポリマーアンダーコート層は、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,479及び同日出願された合衆国特許出願番号 を有する、MEDICAL DEVICE EXHIBITING IMPROVED ADHESION BETWEEN POLYMERIC COATING AND SUBSTRATEと題する出願に開示されている。本明細書に開示された特定の好ましいアンダーコート層は、本質的にポリウレタンからなる。そのような好ましいアンダーコート層は、ポリウレタン以外のポリマーを含むポリマーブレンドを含むが、独占的にポリウレタンのアンダーコート層に比較して、アンダーコート層のデュロメーター、耐久性(durability)、接着特性、構造上の完全性及び弾性に相当に影響しないほど少量においてである。
【0093】
ステント又は他の血管補綴器具(vascular prosthesis)を被験者に移植する場合、損傷直後から始まって約1から6カ月後までの期間は、血管の再狭症がしばしば観察される。即ち、ステントを含む発明の態様に関しては、意図される一般的な溶解速度は、補填器具がルーメンの壁に固まって細胞増殖を減少させた直後に活性薬剤が理想的には放出されるべきであるような速度である。活性薬剤は、次に、全部で約1から6カ月まで溶解し続けるべきである。
【0094】
発明は、基材表面にポリマーブレンドを塗布することによりコーティングを形成させるために使用されるプロセスにより制限されない。適切なコーティングプロセスの例は、溶液プロセス、粉末コーティング、熔融押出、又は蒸気蒸着(vapor deposition)を含む。
【0095】
好ましい方法は溶液コーティングである。溶液コーティングプロセスに関して、溶液プロセスの例は、スプレーコーティング、ディップコーティング、及びスピンコーティングを含む。溶液プロセスでの使用のための典型的な溶剤は、テトラヒドロフラン(THF)、メタノール、エタノール、エチルアセテート、ジメチルフォルムアミド(DMF)、ジメチアセトアミド(DMA)、ジメチルスルフォキシド(DMSO)、ジオキサン、N−メチルピロリドン、クロロホルム、ヘキサン、ヘプタン、シクロヘキサン、トルエン、ギ酸、酢酸、及び/又はジクロロメタンを含む。単一のコート又は複数の薄層を塗布することができる。
【0096】
同様に、発明は、成形されたオブジェクト内に混和性ポリマーブレンドを形成させるのに用いられるプロセスにより制限されない。そのような方法は、成形されたオブジェクトの種類に依存するはずである。適切なプロセスの例は、押出、鋳造(molding)、マイクロマシニング、押出重合法、電気スプレー法等を含む。
【0097】
活性薬剤デリバリーシステムが基材表面に塗布される一つ又はそれより多いコーティング層を含む好ましい態様に関しては、好ましい態様はプライマーの使用を含み、好ましくは「逆流(reflow)法」を用いて塗布され、出願人の譲渡人の係属中の、2002年8月13日に出願された、合衆国仮出願連続番号60/403,479及び同日出願された合衆国特許出願番号 を有する、共にMEDICAL DEVICE EXHIBITING IMPROVED ADHESION BETWEEN POLYMERIC COATING AND SUBSTRATEと題する出願に開示されている。
【0098】
好ましくは、この「逆流法」において、装置作成プロセスは、最初にアンダーコートポリマーを基材表面に塗布することによりポリマーアンダーコート層を形成させ、次に当該ポリマーアンダーコート層を処理することによりアンダーコートポリマーを逆流させ、次に混和性ポリマーブレンドを、好ましくはその中に取り込まれた活性薬剤と共に、逆流されたアンダーコート層に塗布することにより、ポリマーのトップコート層を形成させることを含む。アンダーコートポリマーの逆流は、あらゆる便利な様式、例えば熱処理、紫外線処理、マイクロ波処理、RF処理、機械的圧縮、又は溶剤処理において達成することができる。アンダーコートポリマーを逆流させるためには、アンダーコート層を、少なくともアンダーコートポリマーの「熔融流出(melt flow)温度」ほどの高温まで、ポリマーを逆流させるのに十分な時間、加熱する。ポリマーが液体流状態に入る温度(即ち、「熔融流出温度」)は、発明に従いポリマーを逆流させるのに使用される好ましい最少温度である。典型的には、1から10分が発明に従い熱処理を用いてポリマーを逆流させるのに使用される時間である。ポリマーに関する熔融流出温度は、一般的には、ポリマーのTg(ガラスの熔融温度)及びTm(結晶の熔融温度)を上回る。
【0099】
実施例
本発明は以下の実施例により例示される。特定の実施例、材料、量、及び手法は本明細書にて以前に記載された発明の範囲及び精神に従い広く解釈されるできことが認識されるべきである。
【0100】
実施例1−5及び7−9は、相対的に低い(即ち、約1200g/mlを超えない)分子量を有する疎水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
実施例6は、相対的に高い(即ち、約1200g/mlを超える)分子量を有する親水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
【0101】
実施例10は、相対的に低い(即ち、約1200g/mlを超えない)分子量を有する親水性の活性薬剤を含む活性薬剤デリバリーシステムを示す。
実施例1
デキサメタゾン(疎水性活性薬剤)とのポリ(カーボネートウレタン)/
ポリ(ビス−フェノールAカーボネート)ブレンド
ブレンドの製造と混和性試験
ポリ(カーボネートウレタン)75D(PCU 75D)をポリマーテクノロジーグループ社、バークレー、カリフォルニアから購入した。それはヒドロキシル末端化ポリカーボネート、芳香族ジイソシアネート、及び低分子量のグリコールのコポリマーである。7グラム/10分のメルトインデックス(300℃/1.2kg,ASTM D 1238)を有するポリ(ビス−フェノールAカーボネート)(PC)を、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシン、から購入した。ブレンドする前に、上記のポリマーを60℃から70℃において減圧下で乾燥させた。2つの乾燥させたポリマーを様々な比率で乾燥混合して、次に、2つのローラーブレードを備えたバッチミキサー(サーモハーケー、カールスルー、バーデンビュッテムベルグ、ドイツ)を用いて約200−225℃において熔融ブレンドした。ブレンドは1分あたり50回転(rpm)にて実施した。トルクをレベルオフしたときに(2から3分以内)、rpmを100に上昇させた。トルクを再びレベルオフした後に(2から3分以内)、rpmを50rpmに設定し直した。ブレンドをさらに1分間続けた。混合を完全にした後に、サンプルを回収して、空気中で室温に冷やした。ブレンドの際の酸化を防止するため、0.1−0.2wt%のIRGANOX 1010抗酸化剤(チバスペシャリティーズケミカル社、テリータウン、ニューヨーク)を熔融混合の前にブレンドに添加した。
【0102】
ブレンドの機械的特性からブレンドの熱繊維温度を測定することにより、PCU 75DとPCの間の混和性を測定した。ブレンドサンプルを2つのホットプレートの間で約230℃において約5分間プレスすることにより、フィルムサンプルを用意した。典型的には、フィルムは約0.1ミリメートル(mm)から0.5mmの厚さ、5mmから7mmの幅、そして2センチメートル(cm)の長さであった。これらのフィルムを、レエトリックソリッドアナライザー(Rheometric Solids Analyzer)III(RSAIII)(レオメトリックサイエンティフィック社、ピスカタウエイ、ニュージャージー)のフィルム/ファイバー固定物の中にマウントした。最初のギャップを約5mmに設定した。試験はダイナミックモードにて1Hzの周波数にて行った。機械特性は、5℃/分の速度において−80℃から200℃にサンプルを加熱する間に記録した。命令された圧力(commanded strain)は、−80℃から200℃で0.1%、0℃から150℃で0.5%、そして150℃から200℃で1%に設定した。
【0103】
図2は、貯蔵弾性率対温度を示す。純粋なPCの率は約140℃において下落し始めた。よって、PCのTgは約140℃であった。純粋なPCUは、約10℃にて開始し約180℃までの類似の遷移を有した。PCUとPCの両方を含むブレンドに関しては、2つのガラス遷移が存在した。PCの含有量が増加すればするほど、両方のTg’sは増加して、共に接近した。これは、PCUとPCが混和性であったことを示唆した。
デキサメタゾンを用いたサンプル調製
溶解サンプルを溶剤ブレンド化により用意した。PCU 75Dポリ(カーボネートウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥させ、次に、熔融して、2つのホットプレートの間で約230℃において約5−10分間プレスした。次に、フィルムを冷やして、無水テトラヒドロフラン(THF)中に約60℃において入れた。混合物をポリマーが溶解するまで磁気バーを用いて撹拌した。少量のゲルが場合により溶液中に検出されたが、溶液を0.45ミクロン(μm)のフィルターを用いて濾過することにより除去した。PCUの濃度は1.16wt%であった。PCを最初に室温においてクロロホルム中に溶解することにより5wt%溶液を作成した。次に、当該溶液を無水THFにより1wt%まで希釈した。無水THF中のデキサメタゾン(シグマ−アルドリッチ)の1wt%溶液も室温において作成した。次に、上記の3つの溶液を比率を変えながら混合することにより、表2に示す組成の異なるサンプルを作成した。
【0104】
【表7】
溶解サンプルを、THFでリンスすることにより清潔にしたステンレス鋼(316L)シーム(shims)により調製した。清潔にされたシームに、THFに溶解した1wt%のポリ(エーテルウレタン)(PELLETHANE 75D,ダウケミカル社、ミッドランド、ミシシッピ)の溶液でコートした。PELLETHANE 75Dポリ(エーテルウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥し、次に、熔融して、2つのホットプレートの間で約230℃において約5−10分間プレスした。次に、フィルムを冷やして、磁気バーで一晩撹拌することにより、無水テトラヒドロフラン(THF)内で約25℃において溶解した。
【0105】
コートされたシームを窒素下で一晩乾燥させた。続いて、それらを215−220℃において5−10分間、熱処理した。この前処理により、ポリマー/活性薬剤層によるそれらの接着を促進したシームの表面上にプライマーの形成を導く。プライマー処理されたシームを上に掲載された溶液でコートし、そして一晩窒素下で乾燥させた。シームを各工程の後に秤量した。重量の違いに基づき、コーティングの厚さのときのように、ポリマー/活性薬剤コーティングの全量を測定した。典型的にコーティングの厚さは約10ミクロンであった。
デキサメタゾンの溶解
リン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン(HyClone)、ローガン、ユタから購入)を含んだガラスバイアル内にコートされたシームを入れることにより、PCU 75D/PCポリマーマトリックスからのデキサメタゾンの溶解を実施した。各シームは約2ミリグラム(mg)のコーティング(約0.2mgのデキサメタゾン)を有し、そして各バイアルは3ミリリットル(mL)のPBSを含んだ。バイアルはインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて撹拌した。PBSをバイアルから回収して、新鮮なPBSと置換した。公知濃度のデキサメタゾン溶液のシリーズを用いて目盛を定められたUV−Vis分光光度計(HP 4152A)によりデキサメタゾンの濃度を測定した。
溶解データ分析
図3は、ブレンド中のPCUの増量により増加したデキサメタゾンの累積放出を示す。これらの放出曲線は、明らかに、デキサメタゾンの放出速度がブレンド中のPCUの含有量を変えることにより調節し得たことを示す。当該曲線に基づいて、これらのブレンドからのデキサメタゾンの拡散係数が以下の式を用いて計算され、そして図4のブレンド組成の函数としてプロットされた。
【0106】
【数2】
式中、D=拡散係数;Mt=累積放出;MMt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。
【0107】
図4は、ブレンド組成のほぼ直線函数であった拡散係数のlogを示し、混和性ポリマーブレンドを用いることにより活性薬剤放出速度が整調され得たことを証明した。さらに、図3に提示されたデータはバーストを示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0108】
実施例2
デキサメタゾン(疎水性活性薬剤)との
ポリ(エーテルウレタン)/フェノキシブレンド
ポリ(エーテルウレタン)(PELLETHANE 75D)とデキサメタゾンは実施例1において使用されたのと同じであった。フェノキシ樹脂(PX)、直鎖ポリ(ビス−フェノールAエポキシド)をフェノキシスペシャリティーズ社、ロックヒル、カリフォルニア)から得た。本実施例において使用されたグレードは、モルあたり約10−16キログラムの平均分子量及び95℃のTgを有するPKHJであった。この物質は、PCが実施例1において示したようにデキサメタゾンの放出速度をスローダウンさせることを示すことが予測された。
【0109】
PELLETHANE 75Dとデキサメタゾンを実施例1において記載されたとおりにTHFに溶解した(以下の手法の全ては、特に示さない限り実施例1において使用されたのと同じであった)。PXを無水THF中へ、室温において、溶液中の1wt%のポリマーと共に溶解した。これらの3つの溶液を様々な比率で混合し、そして実施例に記載されたのと同じ手法によりプライマー処理されたステンレス鋼ステント上にコートした。コーティングを乾燥させた後に、溶解及びUV−Vis分析を実施した。
【0110】
PELLETHANE 75D/PXブレンドからのデキサメタゾンの累積放出を図5においてプロットした。デキサメタゾンの放出速度はブレンド中のPELLETHANE 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、PELLETHANE 75DとPXの含有量を変化させることにより、デキサメタゾンの放出速度が調節され得たことを示す。さらに、図5に提示したデータはバースト示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0111】
PELLETHANE 75D/PXブレンドのTg遷移をPYRIS 1示差走査型熱量計(DSC)、パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ、により測定することにより、PELLETHANE 75DとPXの間の混和性を試験した。約5wt%のPELLETHANE 75DとPXのTHF溶液を上で記載されたのと同じ手法により別々に作成した。各約10mgをブレンドサンプルをDSCに負荷し、そして−100から230℃において40℃/分にてスキャンした。各サンプルを2回スキャンした。第2のスキャンはノイズが少ないので使用した。PYRISソフトウエアバージョン5.0を用いることにより、Tg遷移の開始を決定した。図6に示すとおり、純粋なPELLETHANE 75Dは約22℃においてガラス遷移を有し、熔融様の遷移を173℃に有した。このTgは樹脂のハードなドメインに関連していると考えられた。検出できるのなら、ポリ(エーテルウレタン)のソフトドメインのTgは、通常は0℃を下回る。純粋なPXは高温に(77℃)Tg遷移を有する。PELLETHANE 75DとPXをブレンドしたとき、2つの変化が存在した。第1に、純粋なPELLETHANE 75DとPXのTg遷移はブレンドサンプルからはもはや明確に同定できなかった。純粋なPELLETHANE 75DのTgに比較して、高い開始温度の広いTg遷移範囲が存在した。これは、PELLETHANE 75DとPXが少なくとも部分的に混和していることを示唆する(本明細書にて定義されたものとして)。第2に、3つ全てのブレンドにおいてTg選の直後に、結晶性成分を表す新規な遷移が存在した。これは、PXがPELLETHANE 75Dの中でより早い結晶化遷移を引き起こしたことを示唆することから、PXとPELLETHANE 75Dハードドメインの間の相互作用の存在が示される。これは、さらに、2つの物質の間の混和性を示唆する。
【0112】
実施例3
デキサメタゾン(疎水性活性薬剤)とのポリ(カーボネートウレタン)
75D/ポリ(カーボネートウレタン)55Dブレンド
ポリ(カーボネートウレタン)75D(PCU 75D)とデキサメタゾンの溶液は、実施例1において使用したものと同じであった。PCU 55Dはポリマーテクノロジーグループにより作成されたポリ(カーボネートウレタン)ファミリーの別のメンバーに関する商業上の名称であるが、PCU 75Dよりもソフトである。それを実施例1に記載されたのと同じ手法にてTHFに溶解したが、PCU 75Dに関しては溶解が室温どころか60℃において(at room temperature rather at 60 oc)起きた。これらの3つの溶液を様々な比率で混合し、ステンレス鋼の上にコートし、そして実施例1において記載されたのと同じ手法を用いて乾燥させた。溶解試験は実施例1に記載されたとおりに実施した。
【0113】
PCU75D/PCU 55Dブレンドからのデキサメタゾンの累積放出を図7に示す。デキサメタゾンの放出速度はブレンド中のPCU 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、よりソフトな(即ち、低いデュロメーター)PCUをよりハードなPCUにブレンドすることにより、活性薬剤の放出速度が調節され得たことを示す。
【0114】
PCU 75D 100とPCU 75D 70の間のクロスオーバーが2つの厚さの違いによったことも指摘するべきである。放出速度は、曲線が最初は直線領域であるが、のちに平坦な部分により、決定した。デキサメタゾンは、PCU 75D 100からは、PCU 75D 70よりも早く放出された。
【0115】
実施例4
ロシグリタゾンマレート(疎水性活性薬剤)との
ポリ(エーテルウレタン)/フェノキシ樹脂
スミスクラインビーチャム、大英帝国から市販のロシグリタゾンマレートを、実施例2に記載されたとおりに、PELLETHANE 75D/PXブレンドから放出させた。当該ブレンド組成及び全てのサンプル調製及び試験手法は実施例2に記載されたのと同じであった。
【0116】
この活性薬剤の累積放出を図8にプロットした。放出速度はブレンド中のPELLETHANE 75Dの量を増加させると共に増加した。これらの放出曲線は、明らかに、ロシグリタゾンマレートの放出速度が混和性ポリマーブレンドを用いることにより整調されたことを示す。
【0117】
実施例5
デキサメタゾンを含むポリビニルアセテート(PVAC)/
セルロースアセテートブチレート(CAB)
熱刺激電流試験法
熱刺激電流(TherMold Partner,L.P.,スタムフォード、コネチカット)を用いることにより、PVAC/CABブレンド中の熱遷移を測定した。約1センチメーター(cm)掛ける1cmの1片のフィルムをポリテトラフルオロエチレン(PTFE)フィルム(約50ミクロン厚)の表面の上に置いた。2つのフィルムをTSCのプレート−ピボット電極の間に置いた。別にHeガス(超高度純粋、トールガスアンドウエルディング社、プリマス、マサチューセッツ)上に及び真空で3回回転させることにより、試験用チャンバーをパージした。Heの圧力は約0.08メガパスカル(MPa)から0.12MPaであった。パージ後に、チャンバーを同じ圧力のHeガスで満した。サンプルを200℃に加熱し、そして200ボルト/ミリリットルの電圧をサンプルとPTFEフィルムの厚さを横切るように適用した。2分後、サンプルを約10分間50℃にてクエンチしたが、200V/分の電圧は保持した。電場を次に消して、サンプルを2℃/分から200℃に加熱した。フィルムを横切る電流を、この熱プロセスの間記録した。記録された電流−温度曲線を用いることにより、熱遷移を決定した。PTFEフィルムをプレート電極とサンプルフィルムの間に用いた場合のように、15−25℃からのその熱遷移ピークのうちの一方が全てのサンプルのTSC曲線において出現した。熱遷移温度を比較するために、各サンプルのもっとも高いピークを1に低下させるように、当該曲線をスケールした。よって、図の中の曲線の値は任意のユニットであった。
デキサメタゾンによるサンプル調製
ポリビニルアセテート(PVAC,Mw(重量平均分子量)=167から500キログラム/モル(kg/mol)及びセルロースアセテートブチレート(CAB、29.5wt%アセチル及び17wt%ブチリル、Mn(数平均分子量)=65kg/mol)、共にシグマ−アルドリッチカンパニー、ミルウオーキー、ウイスコンシンを真空オーブン中で乾燥させ、そして別々に無水テトラヒドロフラン(THF)に溶解した。両溶液のポリマー濃度は約1wt%であった。1wt%のデキサメタゾン(シグマ−アルドリッチ)を含むTHF溶液も類似の様式にて作成した。3つの溶液を様々な比率で混合して、表3に示す組成の5つの異なるサンプルを作成した。
【0118】
【表8】
溶解サンプルをTHFでリンスして乾燥させることにより洗浄されたステンレス鋼(316L)シームにより調製した。洗浄されたシームを、THFに溶解した1wt%のポリ(エーテルウレタン)(PELLETHANE 75D、ダウケミカル社、ミッドランド、ミシガン)の溶液でコートした。PELLETHANE 75Dポリ(エーテルウレタン)をTHFに溶解する前に、一晩70℃において減圧下で乾燥させ、次に、熔融し、そして2つのホットプレートの間で230℃において5−10分間プレスした。次に、フィルムを冷やし、そして磁気バーを用いて一晩撹拌することにより無水テトラヒドロフラン(THF)中に約25℃において溶解した。
【0119】
コートされたシームを一晩窒素下で乾燥させ、そして215−220℃において5−10分間熱処理した。この前処理はシームの表面上にプライマーを形成させることにより、ポリマー/活性薬剤の層による接着を促進させた。プライムされたシームは上に掲載された溶液でコートし、そして窒素下で一晩乾燥させた。当該シームを各工程後に秤量した。重量の違いを基にして、ポリマー/活性薬剤コーティングの全量を、コーティングの厚さのときのように、測定した。典型的な溶解サンプルは4−5ミリグラム(mg)の乾燥コーティングを約10ミクロン厚のシームあたり有した。
【0120】
プライマーコーティングが無い以外は、類似の様式にて混和性試験のためのサンプルを調製した。典型的なサンプルの厚さは約100ミクロンであり、そしてその中に活性薬剤は含まなかった。
混和性
様々なブレンドの熱遷移温度を測定することにより、PVACとCABの混和性を測定した。示差走査型熱量計(DSC)、動的機械分析(DMA)、及び熱刺激電流(TCS)を用いることにより、ガラス遷移温度(Tg)と他の遷移を測定した。TSCはもっとも強いシグナルを有した。図9A−Dに示されるとおりに一致した結果が提供された。純粋なPVACに関しては(図9A)、TSCが2つの遷移ピークを示し、それぞれ34℃及び62℃を中央とした。純粋なCAB(図9A)はそのTSC曲線中の約110℃を中央とする一つのピークを有した。30wt%のCABをPVAC中にブレンドした場合、PVACの遷移ピークのいずれもが変化しなかった(図9B)。しかしながら、純粋なCABのガラス遷移は消失し、このブレンドが混和性であったことを示した。CABの量を50wt%に増加させた場合、PVACの2つの遷移ピークは高い温度にシフトしたが、純粋なCABに関するTgピークは何も観察されなかった(図9C)。これは、PVACとCABが50/50ブレンドにおいても混和性であったことを示唆する。70wt%のCABを含むブレンドにおいては、遷移ピークの温度がいっそう高く、再び混和性ブレンドを示唆する(図9D)。全てのフィルムはクリアー(clear)で透明(transparent)であり、これらが混和性であったとの我々の結論を支持する。
【0121】
DSC分析はPYRIS 1DSC(パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ)を用いて実施した。−50℃から220℃まで、40℃/分にて、スキャニングをプログラムした。サンプルサイズは約10ミリグラム(mg)であった。図10に示すとおり、純粋なPVACは約39℃にTg遷移を有し、そして純粋なCABは約167℃にTg遷移を有した。PVACとCABを70/30の重量比でブレンドした場合、PVACに相当するTgが55℃に上昇した。これは、PVACとCABがこの比率では部分的に混和性であることを示す。CABをもっと加えると、PVACに相当するTgがさらに上昇したが、遅い速度においてであった。CABに相当するTgはPVACとの混合に際して低下した。これらの全ての結果は、PVACとCABが混合の全ての範囲において部分的に混和性であることを示す。この結果は、上で記載されたTSC試験に基づく結果とわずかに異なる。しかしながら、本発明の混和性の定義を用いた結論は同じであり、即ち、PVACとCABは混和性である。
デキサメタゾンの溶解
PVAC/CABポリマーマトリックスからのデキサメタゾンの溶解を、上記のとおりに調製されたポリマー/活性薬剤コートシームを用いて実施した。コートされたシームを小片に切断し、測定し、そして標準化のために面積を計測した。各小片を3ミリリットル(mL)のリン酸緩衝塩溶液(PBS、リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含むバイアルに浸した。サンプルとPBS溶液の量は、活性薬剤の濃度がUV−Vis分光光度計により検出可能であり、しかもサンプル中の活性薬剤の濃度は実験の間にPBS中の活性薬剤の可溶性の5%を超えない(シンク(sink)条件)ように選択した。約200ミリグラムの活性薬剤を含む約2ミリグラム(mg)のコーティング及び予め37℃に温めた3ミリリットル(mL)のPBSを用いた。溶解試験は37℃において実施し、そしてサンプルをシェーカー中で1分あたり約10サイクルにて撹拌した。PBSをサンプルバイアルから取り出し、そして様々な時間で分析することにより、各サンプル中の活性薬剤の濃度を測定した。PBS中の活性薬剤の濃度はUV−Vis分光光度計(HP 4152A)を用いて243ナノメートル(nm)の波長において測定した。各サンプル中の活性薬剤の濃度は、連続希釈法により創製された標準曲線に比較することにより計算した。各測定後にキュベットを注意深く洗浄することにより、キュベット表面上への疎水性活性薬剤の蓄積を最小にした。ベースラインが、測定された活性薬剤のシグナルよりも少なくとも一桁低い場合に、キュベットは清浄であったと考えた。PBSを各分析時間点において新鮮にした。
溶解データ分析
図11は、ブレンド中のPVACの量を増加させて増加させたデキサメタゾンの累積放出を示す。これらの累積曲線は、明らかに、PVACとCABをブレンドすることにより、2つのホモポリマーの相対的な量を変化させて放出速度を変えることが可能であったことを示す。当該曲線に基づき、これらのブレンドからのデキサメタゾンの拡散係数を以下の指揮を用いて計算し、そして図12のブレンド組成の函数としてプロットした。
【0122】
【数3】
式中、D=拡散係数;Mt=累積放出;MMt=累積放出;M∞=活性薬剤の全負荷;x=臨界寸法(例えば、フィルムの厚さ);及びt=溶解時間。
【0123】
拡散係数のlogがブレンド組成のほぼ直線函数であったことから活性薬剤の放出速度は混和性ポリマーブレンドを用いることにより整調され得ることを証明した。さらに、図11に提示されたデータはバーストを示さないことから、活性薬剤の放出が支配的に浸透性制御下にあったことを示す。
【0124】
実施例6
Resten NGを含む疎水性ポリウレタン及び
ポリ(ビニルアセテート−コ−ビニルピロリドン)
TECOPHILIC HP−60D−60ポリウレタン、サーメディックス社、ウオーバーン、マサチューセッツ、及びポリ(ビニルアセテート−コ−ピロリドン)(PVP−VA),シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシが、この実施例で使用されたマトリックスポリマーであった。RESTEN NG,7000の分子量の水溶性アンチセンスオリゴヌクレオチド、AVIバイオファーマ、コーヴァリス、オレゴン、がこの実施例で使用された活性薬剤であった。TECOPHILICポリウレタンのソフトセグメントは、ポリ(エチレンオキシド)(PEO)及びポリ(テトラメチレンオキシド)(PTMO)の混合物を含む。このソフトセグメントの可溶性パラメーターは、ホフティザーとファンクレヴェレンの(Hoftyzer and van Kevelen's)(H−vK)方法(化学薬品の体積はフェドーズ(Fedor's)法に基づいて計算した)(第7章、D.W.van Krevelen,Properties of Polymers,第3版、Elsevier,1990,表7.8がHoftyzer and van Kevelen’s法に関し、表7.3がFedor’s法に関した)に基づくと、19J1/3/cm3/2(PTMO)から23J1/3/cm3/2(PEO)であると見積もられた。PVP−VAの可溶性パラメーターは、同じ方法に基づいて23J1/3/cm3/2であると見積もられた(ポリマー中のそれらの質量比に基づいたPVPとVAモノマーを超えた(over)モル平均)。
【0125】
TECOPHILICポリウレタンを、無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシに、1wt%ポリウレタンの濃度にて溶解した。ポリウレタンと溶剤をガラスバイアル中で混合し、シールし、そしてポリウレタンが完全に溶解するまで(視覚観察による)撹拌した。メタノール中での超音波により以前に洗浄して空気乾燥されていた、メドトロニックモデルS−670冠動脈ステント(3.0mm x 18mm)を、上で製造された50から100マイクログラムのポリウレタンコーティングでスプレーコートした。専有の(proprietary)スプレーユニットを用いることにより、この実施例においてステントをコートしたが、上記ポリマーの微細に霧状にされた噴霧の上記ステントへの塗布が可能な如何なるスプレーユニットも十分であるべきである。ポリウレタンの50から100マイクログラムのスプレーコーティングの後に、ステントを実験室環境条件にて、25℃及び15%相対湿度(RH)において、4時間乾燥させた。ステントが乾燥した後に、それらをオーブン中に220℃において20分間置くことにより、プライマーコーティングを逆流させた(reflow)。逆流の後で、ステントをオーブンから取り出して、室温において冷やした。
【0126】
TECOPHILICポリウレタンを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。TECOPHILICの溶液中の濃度は1wt%であった。この溶液をAと呼んだ。
【0127】
RESTEN NGオリゴヌクレオチドを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。RESTEN NGの溶液中の濃度は1wt%であった。この溶液をBと呼んだ。
【0128】
PVP−VAを、80wt%の無水クロロホルム、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、及び20wt%の無水メタノール、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシ、を含む溶剤に溶解した。ポリマーが完全に溶解するまで(視覚観察による)、混合物を撹拌した。RESTEN NGの溶液中の濃度は1wt%であった。この溶液をCと呼んだ。
【0129】
溶液A,B及びCを以下の表4に示すとおりに混合することにより、1%の全(overall)「固形物」濃度の溶液を作成した。各溶液中の「固形物」は、10wt%のRESTEN NG、そして残りがTECOPHILICポリウレタンとPVP−VAのブレンドであり、表4に示されるとおりである。
【0130】
【表9】
溶液1−3を0.45ミクロン(マイクログラム)のフィルターで濾過し、そして上で製造されたプライムされたステントの上にスプレーした。ステントをプライムするのに使用した同じ持主のスプレーユニットとプロセスを用いてトップコートを塗布したが、ポリマーと薬剤の溶液の微細に霧状にされた噴霧をステントに塗布することができるらゆるスプレーユニットを使用することができた。コートされたステントは45℃において真空オーブン中で12時間乾燥した。約2000マイクログラム(μg)のコーティングを各ステントに塗布し、そして実際のコーティング重量を用いることにより、コーティング溶液製剤に基づいて各ステント上の活性薬剤の理論上の量を計算した。
【0131】
溶解試験を上でコートされたステント上で実施した。各ステントを、予め37℃に温められた3.0ミリリットル(mL)のリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含むバイアル内に入れた。バイアルをインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて撹拌した。示された時間(この研究においては1分、1時間、3時間、1日、2日、3日、及び4日)において、PVACの全容量をサンプルバイアルから取り出し(バイアルに素早く予め温められた3.0mLの新鮮なPBSを満した)、そしてUV−Vis分光光度計(HP 4152A)により260ナノメートル(nm)にて分析した。各サンプル中のRESTEN NGの濃度を標準曲線との比較により測定した。放出されたRESTEN NGの累積量を、各ステントに関する理論上のRESTEN NG負荷で割り、そして時間の平方根に対してプロットした。結果を図13に提示する。
【0132】
最初の時間にかけて放出されたRESTEN NGの初期バーストが存在したが、放出曲線の残りは時間の平方根に比例したことから、RESTEN NGが透過制御下で放出されたことを示唆する。デリバリーの速度は上記マトリックスポリマーブレンド中のTECOPHILICのPVP−VAに対する比に相関した。より多いPVP−VAによるコーティングはRESTEN NGをより早くデリバリーした。
【0133】
TECOPHILICポリウレタンとPVP−VAの間の混和性を、PYRIS 1示差走査型熱量計(DSC)、パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ、により試験した。TECOPHILICポリウレタンとPVP−VAを同じ溶剤の中に同じ様式にて溶解することにより、約5wt%の溶液を作成した。2つの溶液を様々な比率で混合することにより、PVP−VAの重量パーセンテージを0から100%の範囲で含むサンプルを作成した。ブレンドサンプルを窒素ガス保護の下で乾燥した。試験を実施する前に、サンプルをさらに減圧下で室温にて乾燥した。DSCスキャンを−100℃から230℃に40℃/分にてプログラムした。サンプルを2回スキャンした。第2のスキャンはノイズが少ないので使用した。サンプルサイズは約10ミリグラム(mg)であった。同じサンプルをこの実施例においては全てのTg遷移のために使用した。
【0134】
図14に示すとおり、純粋なTECOPHILICポリウレタンは約−53℃においてガラス遷移を有した(PYRISバージョン5.0ソフトウエアにより測定された開始温度)。このTgはソフトなドメインに関連していると考えられた。ハードドメインのTgは室温よりも高かったが、何故ならばこの樹脂が室温においては全く剛直だからである(デュロメーター41D)。純粋なPVP−VAは高温にTg遷移を有した(76℃)。TECOPHILICポリウレタンを20wt%のPVP−VAと混合した場合、そのDSCは本質的には変わらなかった;が、PVP−VAのTgが消失した。2つのポリマーを50/50の重量比で混合したら、TECOPHILICポリウレタンのTg遷移が消失した。PVP−VAのTg付近の温度に極めて弱い遷移ピークが存在した。Tg遷移の消失は、2つのポリマーが少なくとも一部混和性であったことを示した。
【0135】
DSC試験に関するのと同じサンプルを用いて膨張性試験を実施した。完全に乾燥したサンプル(ウエイト1=50から100mg)を、5mLのリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内に入れた。バイアルをインキュベーター−シェーカー内で37℃に保存し、そして約50回転/分(rpm)にて1日撹拌した。サンプルをバイアルから取り出した。一片の組織を使用して、サンプル表面から遊離のPBSを吸い取った。サンプルを再び秤量した(ウエイト2)。次に、サンプルを減圧下で室温において乾燥した。サンプルを3回秤量した(ウエイト3)。膨張性のパーセンテージは、ウエイト2からウエイト3を引いて、ウエイト3で割ることにより計算した。純粋なTECOPHILICポリウレタンは約56%膨張した。純粋なPVP−VAはPBSに完全に溶解した。膨張性のパーセンテージを、20wt%までのPVP−VAを含むサンプルに関して図15中のPVP−VA含有量の函数としてプロットした。PVP−VAのPBSへの浸出による重量の損失(ウエイト1−ウエイト3)は、10wt%を超えないPVP−VAを含むサンプルに関しては1wt%に満たなかった。
【0136】
実施例7
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアセテート)
(PEcMA)/ポリ(ビニルフォーマル)(PVM)
PEcMAとPVMをこの実施例では使用して、デキサメタゾン(DX)の放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、商人の情報を表5に掲載する。
【0137】
【表10】
2つのポリマーの可溶性パラメーターの差が約3.5J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。当該2つのポリマーを室温において減圧下で一晩乾燥し、そして次に個々に無水テトラヒドロフラン(THF)(シグマ−アルドリッチ社)に溶解することにより、4wt%及び5wt%の溶液を作成した。DXを同じTHFを用いて溶解することにより、約0.141wt%の溶液を作成した。上記3つの溶液を異なる量にて混合することにより、固形物の全重量に基づいて約0wt%、40wt%及び100wt%のPEcMAを含んだ3つのブレンド溶液を作成した。
【0138】
各溶液は固形物の全重量に基づいて約10wt%のDXを含んだ。ブレンド溶液を約1.27cmかける3.81cmのステンレス鋼(316L)シームの表面にコートしたが、それらは前もってTHFで洗浄して乾燥させてあった。コートされたシームを窒素ガス下で室温において一晩保存することにより、溶剤を除去した。シームを実験の各工程の後に秤量した。コーティングの厚さのときのように、重量差に基づいて、薬剤/ポリマーコーティングの全量を各シームに関して測定した。この実施例では、乾燥されたコーティングの典型的な重量は約4ミリグラム(mg)から10mg/シームであり、そして厚さは約10マイクロメーター(ミクロン)から20ミクロンであった。
【0139】
PEcMA/PVMポリマーマトリックスの溶解を、上で記載したポリマー/薬剤をコートされたシームを用いて実施した。コートされたシームを、約2mgのコーティングを含んだ小片に切断した。各小片を、3ミリリットル(mL)の予め37℃に温めたリン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内に入れた。溶解試験を37℃において実施し、そしてサンプルをシェーカー上で約10回転/分(rpm)にて撹拌した。サンプルを様々な時間において分析することにより、PBSを回収することにより、サンプル中の薬剤の濃度を測定した。各回収の後に、PBSを新鮮にした。PBS中のDXの濃度を、UV−Vis分光光度計(HP 4152A)を用いて243ナノメートル(nm)の波長において測定した。各サンプル中のDXの濃度は、公知の濃度の一連の溶液を用いて創製された標準曲線と比較することにより、計算した。
溶解データの分析
PEcMA/PVMブレンドマトリックスからのデキサメタゾンの累積放出を図16にプロットした。PEcMAからのデキサメタゾンの放出速度は、PVMからよりもはるかに早かった。混和性ブレンドポリマーに関する放出速度は、ブレンドしていないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いてブレンド中のポリマー濃度の比を調節することにより、放出速度が整調できることを示す。3つのマトリックス全部からの累積放出は時間の平方根に対してほぼ直線であったことから、バーストがなく、そしてDXのデリバリーが透過制御下であったことを示す。
【0140】
実施例8
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアクリレート)
(PEcMA)/ポリスチレン(PS)
PEcMAとPSをこの実施例において用いることにより、DXの放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、売り主の情報を表5に掲載する。2つのポリマーの可溶性パラメーターの差が約1.3J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、実施例7で使用されたのと同じであった。サンプルの調製、溶解、及びデータ分析は実施例7におけるのと同じであった。放出曲線を図17に示す。デキサメタゾンの放出速度は、PEcMAからよりもPSからの方が遅かった。PSとPEcMAの混和性ブレンドからのDXの放出速度はブレンドしないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いることにより放出速度は整調できることを示す。DXの累積放出は時間の平方根に比例したことから(バーストが観察されなかった)、PEcMA/PSブレンドからのDXのデリバリーが透過制御下であったことを示唆する。
【0141】
実施例9
デキサメタゾン(DX)を含むポリ(エチレン−コ−メチルアクリレート)
(PEcMA)/ポリ(メチルメタクリレート)(PMMA)
PEcMAとPMMAをこの実施例において用いることにより、DXの放出を制御した。ポリマー各々に関してのガラス遷移温度、可溶性パラメーター、分子量、売り主の情報を表5に掲載する。2つのポリマーの可溶性パラメーターの差が約2.1J1/2/cm3/2であったように、これら2つのポリマーは本明細書の定義によると混和性ポリマーであると考えられた。デキサメタゾンは、実施例7で使用されたのと同じであった。サンプルの調製、溶解、及びデータ分析は、実施例7におけるのと同じであった。図18に示すとおり、PEcMAからのDXの放出速度は、PMMAからよりも相当早かった。PMMAとPEcMAの混和性ブレンドからのDXの放出速度は、ブレンドしないポリマーの速度の間であった。これらの放出曲線は、明らかに、混和性ポリマーブレンドを用いることにより放出速度が整調できることを示す。DXの累積放出も時間の平方根に比例したことから(バーストが観察されなかった)、PEcMA/PMMAブレンドからのDXのデリバリーが透過制御下であったことを示唆する。
【0142】
実施例10
クマリン(疎水性活性薬剤)を含むポリ(エーテルウレタン)ブレンド
PELLETHANE 75D(PL75D)、ポリ(エーテルウレタン)をダウケミカルカンパニー、ミッドランド、ミシガンから購入した。TECOPLAST(TP)(TP−470)及びTECOPHILIC(TL)60D60,他の2つのポリ(エーテルウレタン)を、サーメディックス社、ウオーバーン、マサチューセッツから購入した。TPは82Dのショア硬度を有する。クマリンはシグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。メルクインデックス(13版、メルクアンドカンパニー社、ホワイトハウスステーション、ニュージャージー)に基づいて、1グラムのクマリンを400mLの氷水に溶解した。この実施例で使用した無水テトラヒドロフラン(THF)、無水メタノール、及びアセトニトリル(HPLC)も、シグマ−アルドリッチ社、ミルウオーキー、ウイスコンシから購入した。
【0143】
PL75Dを70℃にて減圧下で一晩乾燥した。乾燥したペレットを、予め230℃に温めた2つのプレートの間で圧縮し、そして約5分間維持した。サンプルを室温に冷やした後、THFを持たしたバイアルの中にそれを入れて、溶解するまで撹拌した(視覚観察による)。TPとTLを、室温にて混合物を撹拌することにより直接THFに溶解した。クマリンもTHFに溶解した。全ての溶液の濃度は約1wt%であった。TL溶液及びクマリン溶液を1:1の重量比にて混合した。この混合物がリザーバーシステムのベースコーティング溶液である。TP溶液とPL75D溶液を様々な重量比で混合することによ、TPのPL75Dに対する重量比が100:0、75:25、50:50、25:75、及び0:100の5つの異なる混合物を作成した。これらの溶液を本明細書ではリザーバーシステムのキャップコーティング溶液と呼ぶ。
【0144】
溶解サンプルをTHFによりリンスして洗浄されたステンレス鋼(316L)シーム(12.1 X 38.1mm2)により調製した。洗浄されたシームをPL75D/THF溶液でコートした。コートされたシームを一晩窒素下で乾燥させた。次に、それらを215−220℃にて5−10分間熱処理した。この熱処理は、ポリマー/活性薬剤層によるそれらの接着を促進したシームの表面へのプライマーの形成を導く。プライマーコーティングの厚さは約1マイクロメーター(ミクロン)であった。5つのプライマー処理されたシームを、次に、ベースコーティング溶液でコートして、一晩乾燥した。次に、これらのシームを以下の様式にて異なるキャップコーティング溶液によりディップコートした:シームを2から3秒間キャップコーティング溶液の一つの中に浸し、次に窒素ガス中で乾燥した(約1分間)。そのようなディッピング及び乾燥のプロセスを8回各シームに繰り返した。全プロセスを窒素を満したグローブボックス中で終えた。各シーム中のキャップコーティングの厚さは約1.7から3.4ミクロンであった。全てのコーティングがクリアー(clear)で、透明(transparent)であった。
クマリンの溶解
リン酸緩衝化塩溶液(PBS,リン酸二水素カリウム(NF試験された)、0.144グラム/リットル(g/L)、塩化ナトリウム(USP試験された)、9g/L、及びリン酸水素二ナトリウム(USP試験された)0.795g/L,37℃のpH=7.0から7.2、ハイクローン、ローガン、ユタから購入)を含んだガラスバイアル内にコートされたシームを入れることにより、キャップコートされたシームからのクマリンの溶解を実施した。各バイアルは4ミリリットル(mL)のPBSを含んだ。バイアルはインキュベーター−シェーカー内で37℃に保存し、そして約50rpmにて撹拌した。予め決定された時間において、PBSをバイアルから回収して、新鮮なPBSと置換した。1週間後に、溶解試験を停止し、そして残りのコーティングを4mLのアセトニトリルに溶解した。これらの溶液全ての中のクマリンの濃度を、UV検出器を備えた液体クロマトグラフィー(HP 1090)により測定した。可動相は50wt%の酢酸ナトリウム水溶液と50wt%のアセトニトリル(HPLC)の混合物であった。Zorbax Eclipse(5ミクロン)カラムを使用した。UV検出は277nmの波長にて実施した。標準曲線は公知濃度のクマリン溶液のシリーズにより得た。これらの標準クマリン溶液は、メタノールにクマリンを溶解することにより濃縮溶液(約1wt%)を作成してこの濃縮溶液をPBSで希釈することで作成した。
溶解データ分析
クマリンの累積パーセンテージ放出対キャップコートされたシーム中のPL75D含有量を図19においてプロットした。クマリンのシーム中の全量は、溶解した溶液中の全クマリンと残りのコーティング中に残ったクマリンを一緒に加えることにより測定した。プロット中に示されるように、クマリンは100%TPをコートされたシームよりも100%PL75Dをコートされたシームからの方が相当素早く放出された。PL75D/TPブレンドからの放出速度は2つの純粋なポリマーからの速度の間であった。より興味を引くこととして、速度がブレンド中のPL75D含有量に対して平行であった。これらの結果は、明らかに、ブレンドの組成を変更することによりクマリンの放出速度が調節できたことを示す。
【0145】
PL75D/TPブレンドをTL/クマリン層のトップへのキャップコーティングとしてコートされたため、我々は、放出曲線にタイムラグが存在したはずであると予測した。しかしながら、図19に示した結果はこれを示さなかった。我々は、これがTL/クマリンがディップコーティングプロセスの間に再溶解したことによったと推測した。
混和性試験
溶解サンプルが有したのと同じTP/PL75D比を含むように、混和性試験のためのサンプルを作成した。これらのサンプル中にクマリンは存在しなかった。サンプルをPYRIS 1示差走査型熱量計(DSC)(パーキンエルマーカンパニー、ウエルズレイ、マサチューセッツ)により試験した。スキャニングは−100から220℃まで、40℃/分にプログラムした。サンプルのサイズは約10ミリグラム(mg)から16mgであった。図20に示すとおり、純粋なPL75Dは約22℃にTg遷移を有し、熔融様遷移を約173℃に有した。このTgは樹脂のハードドメインに関連すると考えられた。純粋なTPは約72℃にガラス遷移を有した。PL75DとTPを50/50の比でブレンドした場合、たった一つのTg遷移しか有さず、それは約50℃であった。これは、PL75DとTPがこの比率で混和性であることを示唆する。
【0146】
特許、特許出願、仮特許出願及び公開を含む、及び電子上で利用可能な本明細書にて引用された資料(materials)は、引用により本明細書に編入される。前記の詳細な説明及び実施例は理解の明瞭化のために提供された。発明は示され且つ記載された厳密な詳述にのみ限定されない;多くの変更が当業者には明らかになり、そして請求の範囲に定義された発明の範囲に含まれることが意図される。
【図面の簡単な説明】
【0147】
【図1】選択されたポリマーのTg対可溶性パラメーターのチャート。ロシグリタゾンマレートの可溶性パラメーターにて一点に集められたボックスは、ロシグリタゾンマレートに関する候補を囲む。
【図2】様々なポリ(カーボネートウレタン)及ポリ(ビス−フェノールAカーボネート)ブレンド(PCU75D/PCブレンド)の率対温度のグラフ。PCの含有量が増加するにつれて、ブレンドの個々のポリマーのTgが共により接近してシフトすることから、PCU75D/PCブレンドは混和性であったことが示される。
【図3】様々なPCU75D/PCブレンドからのデキサメタゾンの累積放出対時間の平方根のグラフ。ブレンドのPCU75Dの量を変化させることにより放出速度は整調された(tuned)。
【図4】PCU75D/PC中のデキサメタゾンの拡散係数対ブレンドの組成のグラフ。拡散係数はブレンドのPCU75D含有量の函数として増加した。
【図5】様々なPELLETHANE 75D/PXブレンド(PX=直鎖状ポリ(ビス−フェノールAエポキシド樹脂、レジェンド内のPLのあとの数はPELLETHANE 75Dの重量パーセント(wt%)を示す)の累積放出対時間の平方根のグラフ。ブレンドのPELLETHANE 75Dの量を変化させることにより放出速度は整調された。
【図6】PELLETHANE 75D/PHENOXYブレンドのDSC曲線。
【図7】様々なPCU75D/PCU55Dブレンドからのデキサメタゾンの累積放出のグラフ(2つの異なるポリ(カーボネートウレタン)のブレンド、レジェンド内のPCU75Dのあとの数はPCU75Dのwt%を示す)。ブレンドのPCU55Dの量を変化させることにより放出速度は整調された。
【図8】様々なPELLETHANE 75D/PXブレンドからのロシグリタゾンマレートの累積放出(レジェンド内のPLのあとの数はPELLETHANE 75Dのwt%を示す)。ブレンドのPELLETHANE 75Dの量を変化させることにより放出速度は整調された。
【図9A】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9B】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9C】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図9D】ポリビニルアセテート及びセルロースアセテートブチレートブレンド(PVAC/CAB)のTSCスキャン。遷移のピークはブレンドの組成に依存してシフトした。
【図10】PVAC/CABブレンドのDSCスキャン。ブレンドのガラス遷移は、ブレンドのPVAC含有量の函数として変化した。
【図11】様々なPVAC/CABブレンドからのデキサメタゾンの累積放出対時間の平方根のグラフ。ブレンドのPVACの量を変化させることにより放出速度は整調された。
【図12】PVAC/CABブレンド中のデキサメタゾンの拡散係数対ブレンドの組成のグラフ。拡散係数はブレンドのPVACの含有量の函数として増加した。
【図13】親水性ポリウレタンとポリ(ビニルアセテート−コ−ビニルピロリドン)のブレンドからのResten NGのデリバリーのグラフ。
【図14】TECOPHILIC HP−60D−60/PVP−VAブレンドのDSC曲線のグラフ。
【図15】PVP−VA含有量の函数としてのTECOPHILIC HP−60D−60/PVP−VAブレンドの膨張パーセンテージのグラフ。
【図16】42wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と58%のポリ(ビニルフォーマル)(PVM)のブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図17】45wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と55%のポリスチレンのブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図18】45wt%のポリ(エチレン−コ−メチルアクリル)(PEcMA)と55%のポリ(メチルメタクリレート)のブレンドからのデキサメタゾンの放出プロフィールを示すグラフ。ポリマーブレンドからのデキサメタゾンの放出速度は、ブレンドしないポリマー各々の速度の間であったことから、ブレンドシステムの整調性が証明された。累積放出は時間の平方根と比例したことから、浸透性制御によるデリバリーが証明された。
【図19】PL75D/TPブレンドキャップコートされたシームからのクマリンの累積パーセンテージ放出。
【図20】PL75D/TPブレンドのDSC曲線であり、これらの2つのポリマーの間の混和性を示した。
【特許請求の範囲】
【請求項1】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は疎水性であり、約1200g/molを超えない分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、各々が少なくとも一つの可溶性パラメーターを有し、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超えず;そして
分子の膨張率が10体積%を超えない
システム。
【請求項2】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項1記載のシステム。
【請求項3】
ポリマーの少なくとも2つのうちの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当する、請求項1記載のシステム。
【請求項4】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項1記載のシステム。
【請求項5】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項1記載のシステム。
【請求項6】
活性薬剤が内部マトリックスに取り込まれている、請求項6記載のシステム。
【請求項7】
混和性ポリマーブレンドが少なくとも一つの疎水性ポリマーを含む、請求項1記載のシステム。
【請求項8】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項1記載のシステム。
【請求項9】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項1記載のシステム。
【請求項10】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は親水性であり、約1200g/molを超えない分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超え;そして
分子の膨張率が10体積%を超えない
システム。
【請求項11】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリビニルピロリドンの両方を含まない、請求項10記載のシステム。
【請求項12】
ポリマーの少なくとも2つのうちの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当する、請求項10記載のシステム。
【請求項13】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項10記載のシステム。
【請求項14】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項10記載のシステム。
【請求項15】
活性薬剤が内部マトリックスに取り込まれている、請求項14記載のシステム。
【請求項16】
混和性ポリマーブレンドが少なくとも一つの親水性ポリマーを含む、請求項10記載のシステム。
【請求項17】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項10記載のシステム。
【請求項18】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項10記載のシステム。
【請求項19】
混和性ポリマーブレンドが、ポリアクリロニトリル、シアノアクリレート、メタクリロニトリル、親水性セルロース誘導体、及びそれらの組み合わせからなる群から選択される一つ又はそれより多いポリマーを含む、請求項10記載のシステム。
【請求項20】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は疎水性であり、約1200g/molを超える分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超えず;そして
分子の膨張率が10体積%を超えない
システム。
【請求項21】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない、
請求項20記載のシステム。
【請求項22】
ポリマーの少なくとも2つのうちの膨張性の差が目標の拡散性を含む拡散性の範囲に相当する、請求項20記載のシステム。
【請求項23】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項20記載のシステム。
【請求項24】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項20記載のシステム。
【請求項25】
活性薬剤が内部マトリックスに取り込まれている、請求項24記載のシステム。
【請求項26】
混和性ポリマーブレンドが少なくとも一つの疎水性ポリマーを含む、請求項20記載のシステム。
【請求項27】
混和性ポリマーブレンドが親水性である第2のポリマーを含む、請求項20記載のシステム。
【請求項28】
親水性ポリマーが親水性ポリウレタンである、請求項27記載のシステム。
【請求項29】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項20記載のシステム。
【請求項30】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項20記載のシステム。
【請求項31】
活性薬剤がヘパリンではない、請求項20記載のシステム。
【請求項32】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は親水性であり、約1200g/molを超える分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超え;そして
分子の膨張率が10体積%を超えない
システム。
【請求項33】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリビニルピロリドンの両方を含まない、請求項32記載のシステム。
【請求項34】
ポリマーの少なくとも2つのうちの膨張性の差が目標の拡散性を含む拡散性の範囲に相当する、請求項32記載のシステム。
【請求項35】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項32記載のシステム。
【請求項36】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項32記載のシステム。
【請求項37】
活性薬剤が内部マトリックスに取り込まれている、請求項36記載のシステム。
【請求項38】
混和性ポリマーブレンドが少なくとも一つの親水性ポリマーを含む、請求項32記載のシステム。
【請求項39】
一つのポリマーが親水性ポリウレタンである、請求項38記載のシステム。
【請求項40】
混和性ポリマーブレンドが疎水性である第2のポリマーを含む、請求項38記載のシステム。
【請求項41】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項32記載のシステム。
【請求項42】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項32記載のシステム。
【請求項43】
活性薬剤がヘパリンではない、請求項32記載のシステム。
【請求項44】
請求項1記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項45】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項44記載の医学用装置。
【請求項46】
請求項10記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項47】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項46記載の医学用装置。
【請求項48】
請求項20記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項49】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項48記載の医学用装置。
【請求項50】
請求項32記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項51】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項50記載の医学用装置。
【請求項52】
請求項1記載の活性薬剤デリバリーシステムを含むステント。
【請求項53】
請求項10記載の活性薬剤デリバリーシステムを含むステント。
【請求項54】
請求項20記載の活性薬剤デリバリーシステムを含むステント。
【請求項55】
請求項32記載の活性薬剤デリバリーシステムを含むステント。
【請求項56】
予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする方法であって、当該方法は、
約1200g/mlを超えない分子量を有する活性薬剤を用意し;
少なくとも2つのポリマーを選択するが、但し、
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして
少なくとも二つのポリマーの各々の少なくとも一つのTgが目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;
そして
混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【請求項57】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項56記載の方法。
【請求項58】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項56記載の方法。
【請求項59】
活性薬剤が内部マトリックスに取り込まれている、請求項56記載の方法。
【請求項60】
活性薬剤が疎水性である、請求項56記載の方法。
【請求項61】
活性薬剤が親水性である、請求項5648記載の方法。
【請求項62】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項48記載の方法。
【請求項63】
予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする方法であって、当該方法は、
約1200g/mlを超える分子量を有する活性薬剤を用意し;
少なくとも2つのポリマーを選択するが、但し、
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして
少なくとも二つのポリマーの膨張性の差が目標の拡散率を含むのに十分であり;
少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;
そして
混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【請求項64】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項63記載の方法。
【請求項65】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項63記載の方法。
【請求項66】
活性薬剤が内部マトリックスに取り込まれている、請求項63記載の方法。
【請求項67】
活性薬剤が疎水性である、請求項63記載の方法。
【請求項68】
活性薬剤が親水性である、請求項63記載の方法。
【請求項69】
活性薬剤がヘパリンではない、請求項63記載の方法。
【請求項70】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項63記載の方法。
【請求項71】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項1記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項72】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項10記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項73】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項20記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項74】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項32記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項1】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は疎水性であり、約1200g/molを超えない分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、各々が少なくとも一つの可溶性パラメーターを有し、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超えず;そして
分子の膨張率が10体積%を超えない
システム。
【請求項2】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項1記載のシステム。
【請求項3】
ポリマーの少なくとも2つのうちの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当する、請求項1記載のシステム。
【請求項4】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項1記載のシステム。
【請求項5】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項1記載のシステム。
【請求項6】
活性薬剤が内部マトリックスに取り込まれている、請求項6記載のシステム。
【請求項7】
混和性ポリマーブレンドが少なくとも一つの疎水性ポリマーを含む、請求項1記載のシステム。
【請求項8】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項1記載のシステム。
【請求項9】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項1記載のシステム。
【請求項10】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は親水性であり、約1200g/molを超えない分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超え;そして
分子の膨張率が10体積%を超えない
システム。
【請求項11】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリビニルピロリドンの両方を含まない、請求項10記載のシステム。
【請求項12】
ポリマーの少なくとも2つのうちの少なくとも一つのTgの差が目標の拡散性を含む拡散性の範囲に相当する、請求項10記載のシステム。
【請求項13】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項10記載のシステム。
【請求項14】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項10記載のシステム。
【請求項15】
活性薬剤が内部マトリックスに取り込まれている、請求項14記載のシステム。
【請求項16】
混和性ポリマーブレンドが少なくとも一つの親水性ポリマーを含む、請求項10記載のシステム。
【請求項17】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項10記載のシステム。
【請求項18】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項10記載のシステム。
【請求項19】
混和性ポリマーブレンドが、ポリアクリロニトリル、シアノアクリレート、メタクリロニトリル、親水性セルロース誘導体、及びそれらの組み合わせからなる群から選択される一つ又はそれより多いポリマーを含む、請求項10記載のシステム。
【請求項20】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は疎水性であり、約1200g/molを超える分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超えず;そして
分子の膨張率が10体積%を超えない
システム。
【請求項21】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない、
請求項20記載のシステム。
【請求項22】
ポリマーの少なくとも2つのうちの膨張性の差が目標の拡散性を含む拡散性の範囲に相当する、請求項20記載のシステム。
【請求項23】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項20記載のシステム。
【請求項24】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項20記載のシステム。
【請求項25】
活性薬剤が内部マトリックスに取り込まれている、請求項24記載のシステム。
【請求項26】
混和性ポリマーブレンドが少なくとも一つの疎水性ポリマーを含む、請求項20記載のシステム。
【請求項27】
混和性ポリマーブレンドが親水性である第2のポリマーを含む、請求項20記載のシステム。
【請求項28】
親水性ポリマーが親水性ポリウレタンである、請求項27記載のシステム。
【請求項29】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項20記載のシステム。
【請求項30】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項20記載のシステム。
【請求項31】
活性薬剤がヘパリンではない、請求項20記載のシステム。
【請求項32】
活性薬剤及び混和性ポリマーブレンドを含む活性薬剤デリバリーシステムであって、但し、
活性薬剤は親水性であり、約1200g/molを超える分子量を有し;そして
混和性ポリマーブレンドは少なくとも2つのポリマーを含み、但し、
活性薬剤の可溶性パラメーターとポリマーの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;
少なくとも一つのポリマーが目標の拡散率より高い活性薬剤拡散率を有し、そして少なくとも一つのポリマーが目標の拡散率より低い活性薬剤拡散率を有し;
ブレンドのモル平均可溶性パラメーターが25J1/2/cm3/2を超え;そして
分子の膨張率が10体積%を超えない
システム。
【請求項33】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリビニルピロリドンの両方を含まない、請求項32記載のシステム。
【請求項34】
ポリマーの少なくとも2つのうちの膨張性の差が目標の拡散性を含む拡散性の範囲に相当する、請求項32記載のシステム。
【請求項35】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項32記載のシステム。
【請求項36】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項32記載のシステム。
【請求項37】
活性薬剤が内部マトリックスに取り込まれている、請求項36記載のシステム。
【請求項38】
混和性ポリマーブレンドが少なくとも一つの親水性ポリマーを含む、請求項32記載のシステム。
【請求項39】
一つのポリマーが親水性ポリウレタンである、請求項38記載のシステム。
【請求項40】
混和性ポリマーブレンドが疎水性である第2のポリマーを含む、請求項38記載のシステム。
【請求項41】
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えない、請求項32記載のシステム。
【請求項42】
ポリマーのうちの少なくとも2つの各々の少なくとも一つの可溶性パラメーターの間の差が約3J1/2/cm3/2を超えない、請求項32記載のシステム。
【請求項43】
活性薬剤がヘパリンではない、請求項32記載のシステム。
【請求項44】
請求項1記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項45】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項44記載の医学用装置。
【請求項46】
請求項10記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項47】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項46記載の医学用装置。
【請求項48】
請求項20記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項49】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項48記載の医学用装置。
【請求項50】
請求項32記載の活性薬剤デリバリーシステムを含む医学用装置。
【請求項51】
ステント、ステントグラフト、吻合コネクター、リード、針、ガイドワイヤー、カテーテル、センサー、外科手術用装置、血管形成術用のバルーン、創傷廃液管、シャント、チューブ、尿道挿入物、ペレット、移植物、血液酸素発生機、ポンプ、血管グラフト、弁、ペースメーカー、整形外科用装置、髄核(nucleus pulposus)のための置換装置、及び眼球内レンズからなる群から選択される、請求項50記載の医学用装置。
【請求項52】
請求項1記載の活性薬剤デリバリーシステムを含むステント。
【請求項53】
請求項10記載の活性薬剤デリバリーシステムを含むステント。
【請求項54】
請求項20記載の活性薬剤デリバリーシステムを含むステント。
【請求項55】
請求項32記載の活性薬剤デリバリーシステムを含むステント。
【請求項56】
予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする方法であって、当該方法は、
約1200g/mlを超えない分子量を有する活性薬剤を用意し;
少なくとも2つのポリマーを選択するが、但し、
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして
少なくとも二つのポリマーの各々の少なくとも一つのTgが目標の拡散率を含むのに十分であり;少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;
そして
混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【請求項57】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項56記載の方法。
【請求項58】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項56記載の方法。
【請求項59】
活性薬剤が内部マトリックスに取り込まれている、請求項56記載の方法。
【請求項60】
活性薬剤が疎水性である、請求項56記載の方法。
【請求項61】
活性薬剤が親水性である、請求項5648記載の方法。
【請求項62】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項48記載の方法。
【請求項63】
予め選択された時間(t)をかけて、混和性ポリマーブレンドの予め選択された臨界寸法(x)を通して活性薬剤をデリバリーするための活性薬剤をデリバリーシステムをデザインする方法であって、当該方法は、
約1200g/mlを超える分子量を有する活性薬剤を用意し;
少なくとも2つのポリマーを選択するが、但し、
活性薬剤の可溶性パラメーターとポリマーのうちの少なくとも一つの少なくとも一つの可溶性パラメーターの差が約10J1/2/cm3/2を超えず、そして少なくとも二つのポリマーの各々の少なくとも一つの可溶性パラメーターの差が約5J1/2/cm3/2を超えず;そして
少なくとも二つのポリマーの膨張性の差が目標の拡散率を含むのに十分であり;
少なくとも2つのポリマーを混合することにより混和性ポリマーブレンドを形成させ;
そして
混和性ポリマーブレンドを活性薬剤と混合することにより、予め選択された溶解時間を有する活性薬剤デリバリーシステムを混和性ポリマーブレンドの予め選択された臨界寸法を通して形成することを含む。
【請求項64】
活性薬剤が混和性ポリマーブレンドに取り込まれている、請求項63記載の方法。
【請求項65】
混和性ポリマーブレンドが活性薬剤の浸透のためのバリヤーを最初に提供する、請求項63記載の方法。
【請求項66】
活性薬剤が内部マトリックスに取り込まれている、請求項63記載の方法。
【請求項67】
活性薬剤が疎水性である、請求項63記載の方法。
【請求項68】
活性薬剤が親水性である、請求項63記載の方法。
【請求項69】
活性薬剤がヘパリンではない、請求項63記載の方法。
【請求項70】
混和性ポリマーブレンドが疎水性セルロース誘導体及びポリウレタン又はポリビニルピロリドンのブレンドを含まず;及び/又は
混和性ポリマーブレンドがポリアルキルメタクリレート及びポリエチレン−コ−ポリマーのブレンドを含まない
請求項63記載の方法。
【請求項71】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項1記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項72】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項10記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項73】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項20記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【請求項74】
被験者に活性薬剤をデリバリーする方法であって、当該方法は、
請求項32記載の活性薬剤デリバリーシステムを用意し、そして
活性薬剤デリバリーシステムを被験者の体液、器官、又は組織と接触させることを含む。
【図1】


【図2】

【図3】

【図4】

【図5】
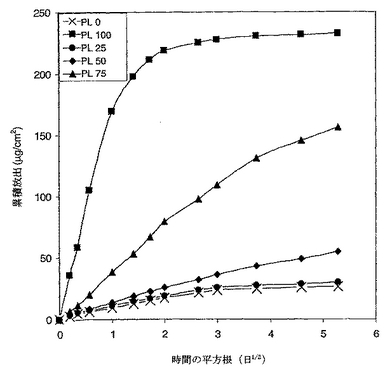
【図6】

【図7】
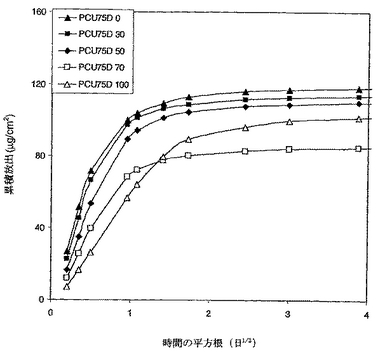
【図8】
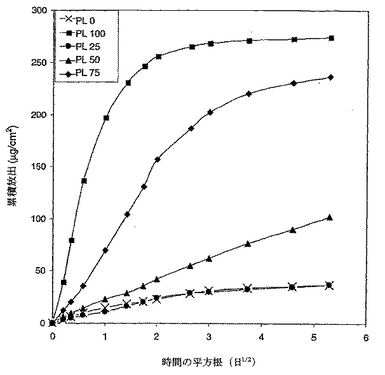
【図9A】

【図9B】
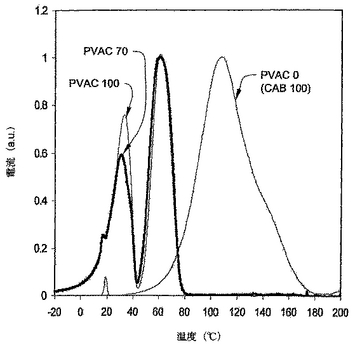
【図9C】
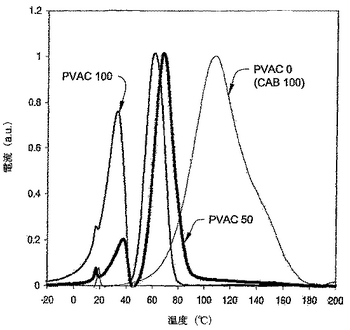
【図9D】

【図10】

【図11】
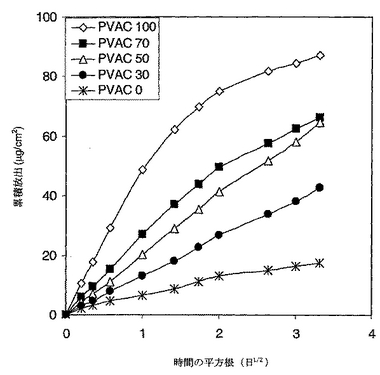
【図12】

【図13】
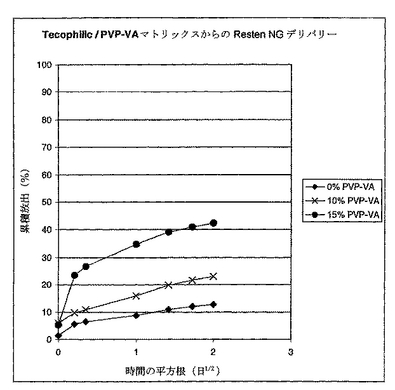
【図14】

【図15】
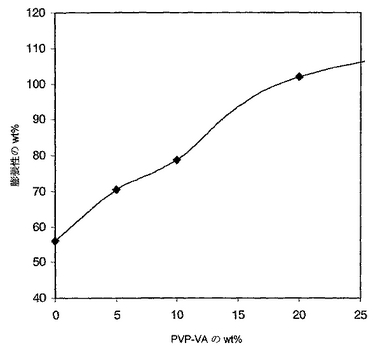
【図16】
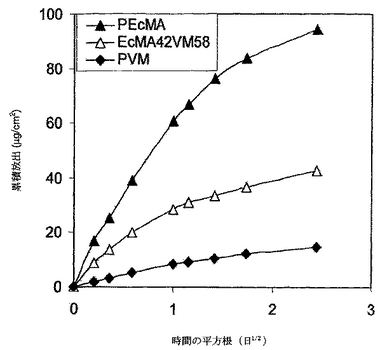
【図17】
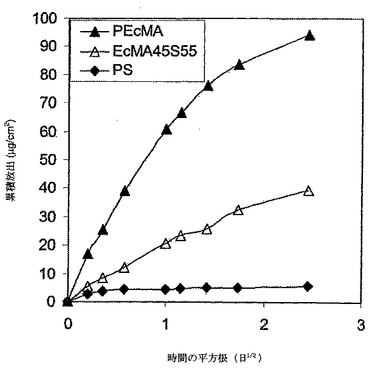
【図18】
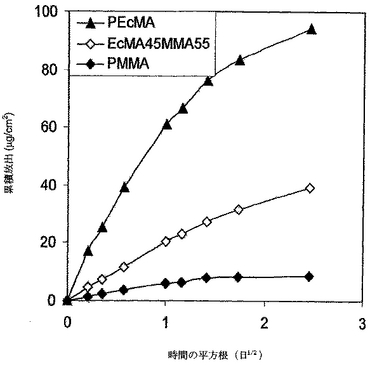
【図19】
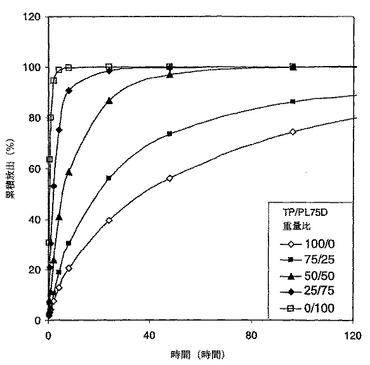
【図20】
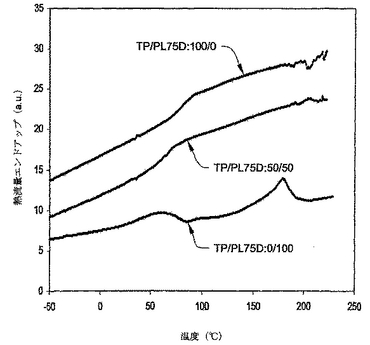
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9A】
【図9B】
【図9C】
【図9D】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公表番号】特表2006−502135(P2006−502135A)
【公表日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願番号】特願2004−528116(P2004−528116)
【出願日】平成15年8月13日(2003.8.13)
【国際出願番号】PCT/US2003/025368
【国際公開番号】WO2004/014451
【国際公開日】平成16年2月19日(2004.2.19)
【出願人】(591007804)メドトロニック・インコーポレーテッド (243)
【Fターム(参考)】
【公表日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願日】平成15年8月13日(2003.8.13)
【国際出願番号】PCT/US2003/025368
【国際公開番号】WO2004/014451
【国際公開日】平成16年2月19日(2004.2.19)
【出願人】(591007804)メドトロニック・インコーポレーテッド (243)
【Fターム(参考)】
[ Back to top ]