
海洋プランクトン由来発光タンパク質

【課題】新規な発光タンパク質を提供する。
【解決手段】海洋プランクトン由来の新規な発光タンパク質(ルシフェラーゼ)とその遺伝子群、並びにそれらより推測された発光タンパク質活性を有する祖先発光タンパク質及びその祖先発光タンパク質をコードする遺伝子に関する。海洋プランクトンがCalanoida目、Augaptiloidea上科、Metridinidae科、Lucicutiidae科、Heterorhabdidae科に属する動物プランクトンのアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質の完全長アミノ酸配列、塩基配列も記載する。
【解決手段】海洋プランクトン由来の新規な発光タンパク質(ルシフェラーゼ)とその遺伝子群、並びにそれらより推測された発光タンパク質活性を有する祖先発光タンパク質及びその祖先発光タンパク質をコードする遺伝子に関する。海洋プランクトンがCalanoida目、Augaptiloidea上科、Metridinidae科、Lucicutiidae科、Heterorhabdidae科に属する動物プランクトンのアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質の完全長アミノ酸配列、塩基配列も記載する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、海洋プランクトン由来の新規な発光タンパク質(ルシフェラーゼ)とその遺伝子群、並びにそれらより推測された発光タンパク質活性を有する祖先発光タンパク質及びその祖先発光タンパク質をコードする遺伝子に関する。
【背景技術】
【0002】
北米ホタルのフォチヌス・ピラリス(Photinus pyralis)由来のルシフェラーゼ(FLuc:Firefly luciferase)、あるいはその改変体タンパク質は、異種細胞内、例えば各種の哺乳動物細胞内において、組換え発現可能であり、得られる組換えタンパク質は、宿主細胞内において、発光基質のD-ルシフェリンを分解して発光特性を示す。またウミシイタケ(Renilla reniformis)由来のルシフェラーゼ(RLuc:Renilla luciferase)も同様に動物細胞を含む各種生物において発現が可能であり、発光基質のセレンテラジン(coelenterazine)を分解する発光活性を有する。これらルシフェラーゼならびにその変異体により得られる発光量は定量性に優れており、生化学、細胞生物学、医学分野において、主に動物細胞のプロモーター(遺伝子発現制御領域)のRNA転写活性を計測する、プロモータアッセイのレポータータンパク質として広く利用が図られている(非特許文献1)。
【0003】
一方、P. pyralisやR. reniformis由来のルシフェラーゼ以外に、ウミホタル(Vargula hilgendorfii)から分泌型ルシフェラーゼがクローニングされており(非特許文献2)、更には、同じ節足動物門(Arthropoda)カイアシ亜綱(Copepoda)のガウシア・プリンセップス(Gaussia princeps)からも、分泌型ルシフェラーゼがクローニングされている(非特許文献3)。これらカイアシ亜綱(Copepoda)のガウシア・プリンセップス(G. princeps)由来ルシフェラーゼはウミホタル(V. hilgendorfii)や北米ホタル(P. pyralis)とは構造的に異なる発光基質を特異的に分解することから、生物進化的には、異なる起源を有するであろうことが報告されている。その発光基質(ルシフェリン)は、R. reniformis由来のルシフェラーゼと同じくセレンテラジンであることが同定されている。
【0004】
【化1】

【0005】
他にも同じカイアシ亜綱(Copepoda)メトリディア・ロンガ(Metridia longa)由来のルシフェラーゼ(非特許文献4)、我々が以前クローニングしたメトリディア・パシフィカ(Metridia pacifica)由来のルシフェラーゼ(非特許文献5)、オワンクラゲ(Aequorea victoria)由来のイクオリン(非特許文献1)、十脚類(Decapoda)に属するヒオドシエビ(Oplophorus gracilirostris)由来のルシフェラーゼ(非特許文献6)等が、上記セレンテラジンをその発光基質(ルシフェリン)として、利用可能であることが報告されている。
【0006】
北米ホタル(P. pyralis)由来のルシフェラーゼ(FLuc)およびウミシイタケ(R. reniformis)のルシフェラーゼ(RLuc)に関しては、動物細胞内で安定して発現可能な、in vivo発光レポータータンパク質として広く利用されているが、非分泌型のルシフェラーゼである。そのためプロモータアッセイを行うためには、ホストの動物細胞等を破砕してルシフェラーゼ活性を測定する必要がある。一方、海洋生物、特に動物性プランクトン類のガウシア・プリンセップス(G. princeps)、メトリディア・ロンガ(Metridia longa)、メトリディア・パシフィカ(Metridia pacifica)由来ルシフェラーゼはウミシイタケ(R. reniformis)と同じ発光基質(セレンテラジン)を酸化分解して発光するが、R. reniformisとは違って分泌型のルシフェラーゼである。そのためレポーターであるルシフェラーゼタンパク質はホストの細胞外へ速やかに分泌され、培地中のルシフェラーゼ活性をモニタリングすればよく、細胞破砕の必要性がない。その結果、同一細胞を用いた連続モニタリングやタイムコース実験を簡単に行うことができる。またウミホタル(V. hilgendorfii)ルシフェラーゼも同じく分泌型であるが、その発光基質であるウミホタル・ルシフェリンはセレンテラジンとは全く異なる構造を有する。またガウシア・プリンセップス(G. princeps)やメトリディア・パシフィカ(M. pacifica)由来ルシフェラーゼは顕著な耐熱性を示すことが報告されているが、有機溶媒等に対しては耐性が低い性質を有し、さらなる改善の余地がある(非特許文献5、非特許文献7)。
【0007】
そこで宿主動物細胞内で発現可能で、より高輝度、高安定性な、新規なルシフェラーゼの探索・開発が待望されていた。
【0008】
ルシフェラーゼタンパク質を、宿主細胞内で発現可能な、in vivo発光性レポータータンパク質として利用する際、特定のプロモーターを用いて宿主細胞内でルシフェラーゼタンパク質を翻訳する。翻訳されたルシフェラーゼタンパク質は適切な構造に折りたたまれてその発光活性を発揮する。一般的に特定のタンパク質の機能や局在を観察する場合これまで蛍光性マーカータンパク質が利用されてきた。すなわち特定のタンパク質のN末端やC末端に蛍光性マーカータンパク質のアミノ酸配列を融合した形で発現させ、宿主細胞に励起光を照射して蛍光性マーカータンパク質から得られる蛍光を検出する。例えば下村脩博士が研究し、ノーベル賞を授与されたことで特に注目を浴びたGFPなどがよく利用されてきた。しかし最近、ルシフェラーゼタンパク質を機能性ペプチドやタンパク質に融合させて、細胞内のシグナルを検出する系の開発も進んでいる(非特許文献8)。このような検出系において、融合させるルシフェラーゼの分子量は融合タンパク質の機能に大きな影響をおよぼす。これまでルシフェラーゼタンパク質としてよく利用されてきた北米ホタル(P. pyralis)由来のルシフェラーゼ(FLuc)は61 kDa、ウミシイタケ(R. reniformis)由来のルシフェラーゼ(RLuc)は36 kDa、ウミホタル(V. hilgendorfii)由来のルシフェラーゼは62 kDaと融合タンパク質としては比較的分子量の大きいタンパク質であり、より分子量が小さく高い輝度をもつ、新規な発光性レポータータンパク質の提供が望まれていた。動物性プランクトン類のガウシア・プリンセップス(G. princeps)、メトリディア・ロンガ(M. longa)、メトリディア・パシフィカ(M. pacifica)由来ルシフェラーゼはそれぞれ19.9 kDa、23.8 kDa、22.7 kDaであることから、その分子量が30 kDa以下であった。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Bioluminescence, chemical principles and methods, Osamu Shimomura, World Scientific Publishing Co. Ltd, 2006
【非特許文献2】Thompson, EM, Nagata, S, and Tsuji, FI. Cloning and expression of cDNA for the luciferase from the marine ostracod Vargula hilgendorfii. Proc. Natl. Acad. Sci. USA, 86, 6567-6571, 1989
【非特許文献3】Verhaegen, M, and Christopoulos, TK. Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal. Chem., 74, 4378-4385, 2002
【非特許文献4】Markova, SV, Golz, S., Frank, LA, Kalthof, B. and Vysotski, ES. Cloning and expression of cDNA for a luciferase from the marine copepod Metridia longa. J. Biol. Chem., 279, 3212-3217, 2004
【非特許文献5】Takenaka, Y, Masuda, H, Yamaguchi, A, Nishikawa, S, Shigeri, Y, Yoshida, Y, and Mizuno, H. Two forms of secreted and thermostable luciferase from the marine copepod crustacean, Metridia pacifica. Gene, 425, 28-35, 2008
【非特許文献6】Inouye, S, and Sasaki, S. Overexpression, purification and characterization of the catalytic component of Oplophorus luciferase in the deep-sea shrimp, Oplophorus gracilirostris. Protein Expr. Purif., 56, 261-268, 2007
【非特許文献7】Rathnayaka, T, Tawa, M, Sohya, S, Yohda, M, and Kuroda, Y. Biophysical characterization of highly active recombinant Gaussia luciferase expressed in Escherichia coli. Biochim. Biophys. Acta, 1804, 1902-1907, 2010
【非特許文献8】Kim, S, Sato, M, and Tao, H. Genetically encoded bioluminescent indicators for stress hormones. Anal. Chem., 81, 3760-3768, 2009
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、新規な発光タンパク質及び該発光タンパク質をコードする遺伝子を提供することを目的とする。
【課題を解決するための手段】
【0011】
そこで上記課題を解決するために、発光能を有する海洋プランクトンを日本近海、具体的には、函館湾沖、親潮域で採取し、発光タンパク質(ルシフェラーゼ)を網羅的にクローニングし、これらの配列を元にバイオインフォマティクスを駆使し、ルシフェラーゼの祖先遺伝子の予測を行い、この配列からルシフェラーゼ活性の発揮に必須な最小機能領域を特定して、発光機能を保持した分子量十数kDaのさらに小さいルシフェラーゼを開発した。
【0012】
本発明は、以下の新規な発光タンパク質およびそれをコードする遺伝子を提供するものである。
項1. 配列番号22若しくは配列番号24のアミノ酸配列、または、1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列を有することを特徴とする発光タンパク質。
項2. 配列番号1〜21のいずれかアミノ酸配列を有する項1に記載の発光タンパク質。
項3. 項1または2に記載の発光タンパク質をコードするDNAまたはその相補鎖。
【発明の効果】
【0013】
本発明によれば、発光タンパク質の祖先遺伝子とアミノ酸配列を同定することができた。
【0014】
このような祖先遺伝子が明らかになったことで、より優れた性質を有する改変型発光タンパク質が開発されることが期待される。
【図面の簡単な説明】
【0015】
【図1】Metridinidae科およびLucicutiidae科由来ルシフェラーゼとそこから推測される改変型祖先ルシフェラーゼ aCopLuc43mut(配列番号22)及び改変前の祖先発光タンパク質 aCopLuc43(配列番号23)のアミノ酸配列のアラインメントを示す。
【図2】Heterorhabdidae科由来ルシフェラーゼとそこから推測される祖先発光タンパク質aCopLuc48(配列番号24)のアミノ酸配列のアラインメントを示す。
【図3】発光プランクトンMetridia okhotensisの(a)紫外線照射下での蛍光顕微鏡写真及び(b)白色光下での顕微鏡写真を示す。
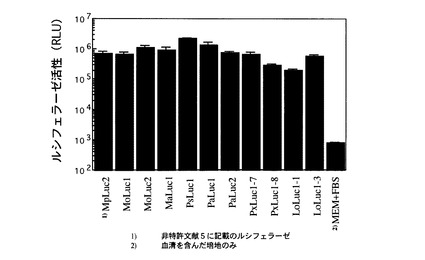
【図4】ヒト培養細胞(HEK293)により培地中に分泌発現されたカイアシ類由来ルシフェラーゼの活性を示す。
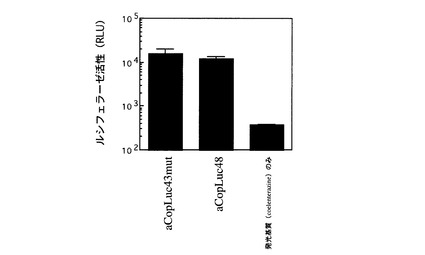
【図5】実施例2において、大腸菌により発現された祖先遺伝子(aCopLuc43mutとaCopLuc48)のルシフェラーゼ活性を示す。
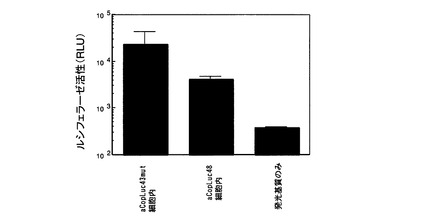
【図6】実施例3において、ヒト培養細胞(HEK293)により発現された祖先遺伝子(aCopLuc43mutとaCopLuc48)のルシフェラーゼ活性を示す。
【発明を実施するための形態】
【0016】
本発明の1つの特徴は、海洋プランクトン由来の発光タンパク質において、Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼの遺伝子配列およびアミノ酸配列を同定し、それにより祖先遺伝子とアミノ酸配列を決定したことにある。Metridinidae科プランクトン及びLucicutiidae科プランクトンの祖先発光タンパク質のアミノ酸配列は、配列番号22に示され、該アミノ酸配列をコードする塩基配列は、配列番号46に示される。また、Heterorhabdidae科プランクトンの祖先発光タンパク質のアミノ酸配列は、配列番号24に示され、該アミノ酸配列をコードする塩基配列は、配列番号48に示される。
【0017】
配列番号22に記載のアミノ酸配列は、配列番号23に記載のアミノ酸配列において、91位のCys→Aspに、92位のSerがCysに置換されたものである。
【0018】
本発明者らが見出したMetridinidae科とLucicutiidae科の海洋プランクトン由来の発光タンパク質(配列番号1〜15)を用いて得られた配列番号23の祖先発光タンパク質は発光活性を有しなかったが、91位と92位のアミノ酸を上記のように置換した配列番号22に記載のアミノ酸配列を有する改変型の祖先発光タンパク質は発光活性を有することを確認した。
【0019】
Heterorhabdidae科プランクトンの場合には、本発明者らが見出した配列番号16〜21のアミノ酸配列を用いて得られた祖先発光タンパク質(配列番号24)は発光活性を有することを確認した。
【0020】
配列番号22,24に示される祖先発光タンパク質は、本発明により初めて明らかにされたものである。
【0021】
本発明は、配列番号22,24の配列において、図1(配列番号23のaCopLuc43を除く),図2に示される範囲内でアミノ酸置換が可能である。
【0022】
例えば図1において、N末端のアミノ酸は、配列番号22のaCopLuc43mutではMであるが、このアミノ酸は、図1の他の発光タンパク質で確認できるH,L,Gに置換可能である。同様に、N末端から4番目の配列番号22のアミノ酸Kは、Q、Lに置換可能である。N末端から10番目の配列番号22のアミノ酸Vは、他の発光タンパク質が全てVであるため、置換できない。同様に、11番目のアミノ酸Lも置換不可能である。図2のアミノ酸配列に関しても、配列番号24のアミノ酸配列において同様なアミノ酸置換は可能である。
【0023】
さらに配列番号22、24のアミノ酸配列のN末端又はC末端側には、図1、図2で示される他のアミノ酸が付加可能である。
【0024】
本発明における、配列番号22、24のアミノ酸配列において1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列とは、上記のアミノ酸置換ないし付加が包含される。ここで置換、付加、欠失又は挿入される1又は複数のアミノ酸とは、150個以下、145個以下、140個以下、135個以下、130個以下、125個以下、120個以下、115個以下、110個以下、105個以下、100個以下、95個以下、90個以下、85個以下、80個以下、75個以下、70個以下、65個以下、60個以下、55個以下、50個以下、45個以下、40個以下、35個以下、30個以下、25個以下、20個以下(例えば1、2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19又は20個)のアミノ酸の置換、付加、欠失又は挿入を意味する。例えば図1に示されるMoLuc1は図1に示す番号で1番〜48番,57番〜85番、146番、148番、226番の合計80個のアミノ酸が配列番号22の改変型祖先発光タンパク質に対して付加され、さらに87番(P/G)、94番(A/E)、97番(I/K)、104番(F/K)、118番(K/H)、127番(E/K)、128番(Y/F)、135番(D/S)、137番(G/E)、141番(K/D)、143番(G/A)、145番(A/G)、149番(V/I)、151番(A/P)、159番(S/P)、163番(E/D)、165番(G/A)、178番(A/V)、183番(G/R)、192番(K/Q)、195番(A/D)、196番(L/Q)、203番(D/T)、206番(A/T)、207番(S/T)、210番(D/S)、214番(R/K)、217番(H/D)、218番(N/T)、224番(G/D)の合計30個のアミノ酸が置換されている。従って、MoLuc1の場合、配列番号22のアミノ酸配列に対し110個のアミノ酸が置換、付加、欠失又は挿入されることになり、このようなアミノ酸の置換、付加、欠失又は挿入は、本発明の範囲内である。また、置換、付加、欠失、挿入されるアミノ酸は、図1,図2に示される範囲内であることが好ましく、MoLuc1と配列番号22の祖先発光タンパク質(aCopLuc43Mut)を比較して最大110個のアミノ酸が置換、付加、欠失又は挿入可能であるが、1〜109個のアミノ酸を適宜選択して置換、付加、欠失又は挿入してもよい。上記の記載は、MoLuc1を例に取り説明したが、MoLuc2、McLuc1、McLuc2、MaLuc1、MaLuc2、PsLuc1、PsLuc2、PaLuc1、PaLuc2、PxLuc1-7、PxLuc1-8、PxLuc2、LoLuc1-1、LoLuc1-3についても同様に、図1に示される範囲でアミノ酸の置換、付加、欠失又は挿入が可能である。さらに、HtLuc1-1-2、HtLuc1-1-3、HtLuc1-2-2、HtLuc2、HmLuc1、HmLuc2についても同様に、図2に示される範囲でアミノ酸の置換、付加、欠失又は挿入が可能である。
【実施例】
【0025】
以下、本発明を実施例に従いより詳細に説明するが、本発明がこれら実施例に限定されないことは言うまでもない。
【0026】
実施例1:発光プランクトン由来の新規ルシフェラーゼ
1.アミノ酸配列及び塩基配列
海洋に棲息する動物性プランクトンより、発光プランクトンを探索した。具体的には、北海道大学大学院水産科学研究院の山口篤准教授(産総研客員研究員)と共同で、附属練習船おしょろ丸に乗船し、函館湾沖、親潮域で採取された海洋中に存在する、動物性プランクトンの分類を進める過程で、数多くの発光プランクトンを採取した。更に、これら発光プランクトンのうち、分類学的に節足動物門(Arthropoda)カイアシ亜綱(Copepoda)に属する動物性プランクトンであって、体外に分泌発現型の発光タンパク質を発現しているものを選別した。この動物プランクトン採集で見出された、分泌型の発光タンパク質を発現しているカイアシ亜綱(Copepoda)の分類を試み、Calanoida目、Augaptiloidea上科に属するMetridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトンの同定を行った。さらにMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質について、それをコードする遺伝子の塩基配列の特定と、推定アミノ酸配列の決定を試みた。先ず、Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトンから、全RNAを抽出し、含まれるmRNAを精製し、逆転写酵素を用い、定法に従って、対応するcDNAを合成した。これまでに報告されているMetridinidae科プランクトン由来のルシフェラーゼ遺伝子の配列を参考にPCR法によりcDNAより部分配列の増幅を行った。その結果、約250塩基のルシフェラーゼ遺伝子の部分配列を得た。次にその塩基配列をもとに、cDNA全長の増幅と塩基配列解析を行い、目的とするMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質の配列を解明した。このMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質の完全長アミノ酸配列をアライメントして、コンピュータプログラム(MEGA5)を用いて分子量15 kDaおよび13.8 kDaの祖先ルシフェラーゼタンパク質配列を予測し、ルシフェラーゼタンパク質をコードする遺伝子配列を人工遺伝子合成して、大腸菌およびほ乳類培養細胞において発現させ、そのルシフェラーゼ機能を確認するに至った。
【0027】
本発明にかかるCalanoida目、Augaptiloidea上科、Metridinidae科、Lucicutiidae科、Heterorhabdidae科に属する動物プランクトン由来の発光タンパク質、カイアシ類Metridinidae科およびLucicutiidae科に属する動物プランクトン由来の発光タンパク質のアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質(aCopLuc43)とその改変体(aCopLuc43mut)、カイアシ類Heterorhabdidae科に属する動物プランクトン由来の発光タンパク質のアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質(aCopLuc48)の名称と完全長アミノ酸配列、塩基配列の配列番号の対応関係を以下に示す。
【0028】
【表1】
【0029】
2.実験方法
カイアシ類Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン体内でのタンパク質の発現に伴い、残留している多種のmRNAのうち、当該ルシフェラーゼの遺伝子を選別することを試みた。具体的には、mRNAよりcDNAライブラリーを作製し、このcDNAライブラリーからPCR法によりルシフェラーゼ遺伝子の部分配列および全長配列の取得を行った。
【0030】
以下に、本発明のカイアシ類由来のルシフェラーゼに関して、より詳しく説明する。
【0031】
カイアシ類発光プランクトンの採集、同定
まず、本発明のルシフェラーゼの起源である動物性発光プランクトンは、函館湾沖、水深50〜1000mから採取された親潮域深層水中に見出された、Calanoida目、Augaptiloidea上科に属するプランクトンである。代表的な発光プランクトンMetridia okhotensisの形態は、図3(a)に示すように、紫外線照射下において、蛍光顕微鏡により観察すると発光基質を含む分泌腺が青緑色に見える。また、白色光下で顕微鏡観察すると、図3(b)に示すように、Metridia okhotensisのプランクトンの体内は透明から若干白色がかって観察される。本発明でルシフェラーゼ遺伝子のクローニングを行ったプランクトンはMetridinidae(メトリディニダエ科)に属するものとして、Metridia okhotensis、Metridia curticauda、Metridia asymmetrica、Pleuromamma abdominalis、Pleuromamma scutullata、Pleuromamma xiphias、Lucicutiidae(ルチクチイダエ科)に属するものとしてLucicutia ovaliformis、またHeterorhabdidae(ヘテロハブディダエ科)に属するものとして、Heterorhabdus tanneri、Heterostylites majorの計5属9種である。これらの動物プランクトンの詳細な分類学上の同定では、Metazoa(動物界)、Arthropoda(節足動物門)、Crustacea(甲殻亜門)、Maxillopoda(アゴアシ綱)、Copepoda(カイアシ亜綱)、Neocopepoda(カイアシ下網)、Gymnoplea(前脚類)、Calanoida(カラヌス目)、Metridinidae(メトリディニダエ科)もしくはLucicutiidae(ルチクチイダエ科)もしくはHeterorhabdidae科(ヘテロハブディダエ科)となる。
【0032】
本発明者らは、これらのMetridinidae科、Lucicutiidae科またはHeterorhabdidae科に属するプランクトンが物理的刺激を与えることにより発光を示し、またその粗抽出液を発光基質セレンテラジンと混合した際に発光が観測されることを詳細に調べた結果、ルシフェラーゼを産生するバクテリアの寄生、付着に起因するものではなく、このMetridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン自体に由来するルシフェラーゼに因ると結論した。そこで、該プランクトンを集め、ルシフェラーゼタンパク質の単離を進めることを検討したが、入手されるプランクトン量が十分でなく、そのアミノ酸配列解析に十分なタンパク質量を回収することは困難であると判断された。従って、近縁のMetridia属のMetridia longa由来のルシフェラーゼ(GenBank accession number, AY364164; 文献4)とGaussia属に属する、Gaussia princeps由来のルシフェラーゼ(GenBank accession number, AY015993; 文献3)のアミノ酸配列に基づき、相同性の高いアミノ酸配列の一部をコードする、縮重プライマーを作製し、cDNAから、当該ルシフェラーゼの遺伝子をPCR法により、クローニングする手法を適用した。
【0033】
カイアシ類発光プランクトンRNAの精製およびcDNAの合成
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトンより、市販のRNA抽出試薬;RNeasy Mini kit(Qiagen社製)を用いて全RNAを抽出し、次いで、市販の精製キット;Oligotex-dT30 <SUPER> mRNA Purification kit(Takara Bio社製)を用いてPoly(A)+mRNAの精製を行った。更に、精製済みのmRNAから、市販のcDNA調製キット:SMART RACE cDNA増幅キット(Takara Bio社製)を利用して、それぞれ5’末端側増幅用および3’末端側増幅用cDNAの合成をおこなった。
【0034】
PCR反応による、カイアシ類ルシフェラーゼcDNA断片の取得
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン由来のルシフェラーゼをコードする遺伝子クローニング工程においては、mRNAから調製されるcDNAを鋳型として、前記上流側混合プライマー二種と下流側混合プライマー二種とを組み合わせた、計4種のPCR用プライマー対についてPCR反応を行った。
【0035】
具体的には、以下に示す、上流側混合プライマー2種と下流側混合プライマー2種の塩基配列を選択した。
【0036】
上流側混合プライマー
White luc UP1 (26 mer:16種の混合プライマー)
5’-GGC TGC ACY AGG GGA TGY CTK ATM TC-3’
(Y=T, C;K=G, T;M=A, C)
White luc UP2 (26 mer:16種の混合プライマー)
5’-GCT ATT GTT GAY ATY CCY GAR AT-3’
(Y=T, C;R=G, A)
【0037】
下流側混合プライマー
White luc LP2 (26 mer:16種の混合プライマー)
5’-TC AAG TTG WTC AAT RAA YTG YTC CAT-3’
(W=A, T;R=G, A;Y=T, C)
White luc LP1 (23 mer:12種の混合プライマー)
5’-AC ATT GGC AAG ACC YTT VAG RCA-3’
(Y=T, C;V=A, G, C;R=G, A)
【0038】
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン由来のmRNAを鋳型として、調製される3’-Ready cDNAから、上記に示す4組のプライマー対を利用して、常法に従いPCR増幅産物を調製する。
【0039】
回収されたDNA溶液(PCR増幅産物)に、10xLoading dye液 2μLを添加した後、1.6% TAEアガロースゲルにアプライし、各レーンDNA溶液 15μLを泳動した。目的分子量のバンドをゲルから切り出し、Montageゲル抽出キット(Millipore社製)に含まれるマイクロチューブカラムにゲル片を入れて、7,000 rpmで10分間遠心する。メンブレンを通して抽出されてきたDNA溶液をさらにMinElute PCR purification kit(Qiagen社製)を用いて精製を行う。DNA溶液1容量当たり、バッファーPBを5容量を加え、ボルテックスにかけた後、MinEluteカラムに移した。30秒間遠心し、DNAをカラムに吸着させる。ついでカラムを、バッファーPE 0.7 mLで洗浄し、さらに1分間、遠心(15,000 rpm)することによりバッファーPEをカラムから完全に除く。その後、溶出バッファーEB 10μLを加えて、室温で、1分間静置する。最後に、1分間、遠心(15,000 rpm)し、カラムから溶出してきたDNAを1.5 mL用マイクロチューブ中に回収する。
【0040】
上記精製済みPCR反応産物を、クローニング・ベクターpCR2.1-TOPO(Invitrogen社製)中に挿入する。PCR反応産物2μLにベクターに添付のSalt solution 0.5μLおよびベクター溶液0.5μLを加えて、室温で5〜15分間インキュベートしてライゲーションを行う。この反応液に含まれるDNAをChemical competent cell DH5alpha株へ導入し、形質転換株を選択する。プラスミドベクターの宿主大腸菌へ導入は、以下の手順で実施した。宿主大腸菌に利用する、DH5alpha株は、凍結保存されているCompetent cellを、氷温で解凍する。PCR産物挿入プラスミドベクター溶液3μLを、解凍された宿主大腸菌DH5alpha株懸濁液 50μLに加える。そのまま氷上で10分間保持した後、42℃、30秒加温し、氷上に戻した。ついで100μLのSOC培地を加えて、37℃、10分間振盪培養する。その後、培養液に含まれる形質転換株を、クローニング・ベクターpCR2.1-TOPOに由来する選択マーカーを利用して薬剤耐性株を選択する。50μg/mL抗生剤カルベニシリンを含んだLBプレート上に出現するコロニーを解析して、PCR産物を保持するプラスミドベクターをもつ大腸菌を選択する。
【0041】
PCR産物を保持するプラスミド・ベクターの複製、精製
選別されたクローンから、以下の手順で、導入されているプラスミドベクターの精製を行う。
【0042】
PCR産物が挿入されたプラスミドベクターを保持する宿主大腸菌 DH5alpha株のコロニー(クローン)を、それぞれ2 mLのLB/カルベニシリン液体培地中に懸濁し、37℃、16時間培養を行った。培養液を、10分間、遠心(5,000 x g)して、細胞を分取した。
【0043】
分取した細胞から、市販のプラスミド精製キット:Qiagen Plasmid purification kit(Qiagen社製)を利用して、プラスミドを分離、精製する。分取した細胞に、同精製キットに添付のP1液 0.25mLを添加し、ボルテックスにかけて、よく分散させる。この細胞分散液に、添付のP2液 0.25mLを添加、混和して室温(20℃)にて5分間静置する。溶菌処理後、添付のN3液 0.35mLを添加し、混和する。次いで15分間、4℃で遠心(15,000 rpm)し、プラスミドDNAを含む上清を分離、回収する。上清を、同精製キットのQIAprep miniカラムにかける。4℃で遠心(15,000 rpm)し、液層を除去する。バッファーPB 0.5 mLを加えて、カラムを洗浄し、引き続き、バッファーPE 0.75 mLを加えて、再度洗浄する。最終的に、1分間、4℃で遠心(15,000 rpm)し、洗浄液を完全に除去する。精製キットのQIAprep miniカラムに吸着されているプラスミドDNAは、バッファーEB 30μLにより溶出、回収する。この精製済みプラスミドを含む液 30μLのうち、1μLをとり、蒸留水 99μLを加えて、100倍希釈液とし260nmの吸光度を測定してDNA濃度の定量を行った。このプラスミドベクターのDNA濃度を250ng/μLとなるように、濃度調整を行った。
【0044】
選別クローン中のcDNA断片(PCR産物)の塩基配列解析
プラスミドベクター中に挿入されている、cDNA断片(PCR産物)を、以下の手順で塩基配列解析用の核酸鎖伸長反応産物を調製する。
【0045】
選択クローンから回収、次いで、市販のシークエンス用DNA試料調製キット:BigDye Terminator Cycle Sequencing Ready Reaction Kit ver. 1.1(Life Technologies Japan社製)により、この精製済みプラスミドベクターを鋳型として、塩基配列解析用の試料を、2種のプライマー;M13 sense M4およびM13 reverseをシークエンス・プライマーとして利用し、プラスミドベクター中に挿入されているcDNA断片を含む領域のシーケンス反応生成物を調製する。 調製された塩基配列解析用の試料の精製は、以下の手順で行う。
【0046】
各反応用チューブから、調製済み試料溶液を別の0.5 mL容チューブに移す。試料溶液 5 μL当たり、3M 酢酸ナトリウム水溶液 0.5 μL、95%エタノール12.5μLの比率で混合した液を、別途、1.5 mL容チューブに予め用意する。この1.5 mL容チューブ中に、予め集めた試料溶液を投入する。均一に混合した上で、氷冷下に、10分間静置し、含まれるDNA断片をエタノール沈澱(析出)させる。20分間、遠心(15,000 rpm)し、析出したDNA断片を沈積させ、上清を除去する。次いで、125μLの70%エタノールを加え、沈殿DNAをリンスする。再び、5分間、遠心(15,000 rpm)し上清を吸引除去する。残った沈殿DNA断片のペレットを乾燥する。
【0047】
精製した解析用試料DNA断片は、15 μLホルムアミド中に再溶解する。ボルテックスにかけて、混合した後、遠心して液を集める。95℃、2分間加熱し、一本鎖DNAに分離し、氷冷する。その後、解析用試料DNA断片は、市販のシークエンス装置:ABI PRISM 3100 Genetic Analyzer(Life Technologies Japan社製)にかけ、塩基配列解析を行う。
【0048】
センス鎖の配列解析結果と、アンチセンス鎖の配列解析結果とを総合し、挿入されているcDNA断片(PCR反応産物)の塩基配列を決定する。
【0049】
得られたカイアシ類由来の各ルシフェラーゼの塩基配列及びアミノ酸配列を表1及び配列表に記載する。
【0050】
カイアシ類由来ルシフェラーゼタンパク質のヒト細胞発現用発現ベクターのHEK293細胞への導入
ほ乳類培養細胞発現用ベクターpcDNA3.2 V5-His TOPO(Invitrogen社製)中へ上記で得られたカイアシ類由来ルシフェラーゼ遺伝子のコード領域を挿入する。プラスミドpcDNA3.2 V5-His TOPOのクローニング・サイト中に各遺伝子が挿入されていることをシーケンスにより確認する。作製したほ乳類培養細胞発現ベクターのうち、MoLuc1, MoLuc2, MaLuc1, PsLuc1, PaLuc1, PaLuc2, PxLuc1-7, PxLuc1-8, LoLuc1-1, LoLuc1-3を代表として培養細胞で遺伝子発現させた。精製済みのヒト細胞発現ベクターを、HEK293細胞中に、Lipofectamine 2000 (Invitrogen社製)を用いて遺伝子導入した。宿主のHEK293細胞は、10 mL血清添加培地(MEM+10% FBS+Penicillin/streptomycin)を用いて、100 mm dish上で、95%コンフルエントな状態まで培養されたものを利用する。まずプラスミド溶液(DNA濃度:1 μg/μL)24 μLを1.5 mLの無血清Opti-MEM培地に懸濁したものと、Lipofectamine 2000 60 μLを同様に1.5 mLの無血清Opti-MEM培地に懸濁したものを用意する。この2液を混合して室温にて20分間インキュベートする。この混合液全量を100 mm dish上の細胞培地中に添加して、均一になるように軽く撹拌する。トランスフェクション処理後、HEK293細胞は、5%CO2下、37℃にて48時間以上インキュベーションし、導入遺伝子の発現を行う。
【0051】
培養上清 10 mLをすべて新しい15 mLの遠心管に移し、1000 rpmで2分間遠心して、遠心上清を培地画分とした。一方、 dish 上に残った細胞に新しい培地10 mLを加えて、セルスクレーパーを使って細胞をはがした後すべて15 mLの遠心管に回収した。この懸濁液を氷上で10秒間超音波破砕を行い、15,000 rpm で5分間遠心した。上清を細胞内画分として回収し、培地画分と同時に発光活性を測定した。そのタンパク質は、細胞外に分泌され、培地中に蓄積されることが観測された(図4)。 またその時の分泌効率について表2に記した。ルシフェラーゼ活性の測定法は、後述する「カイアシ類由来ルシフェラーゼ活性の測定」に記載した。以上より、ヒト由来のHEK293細胞内においても、カイアシ類由来ルシフェラーゼタンパク質のコード遺伝子に基づき、完全長のアミノ酸配列を有するプレ・タンパク質型組換え体へと翻訳がなされる。その後、そのN末端に存在するシグナルペプチド部を利用して、細胞外へ成熟型活性タンパク質として、分泌されることを明確に示している。
【0052】
【表2】
【0053】
実施例2:祖先遺伝子の推測と大腸菌による発現、活性の確認
カイアシ類由来ルシフェラーゼの祖先遺伝子の推測
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列を、それぞれの科ごとにタンパク質配列データの分子進化・系統学的解析を行うためのソフトウェアMEGA5にて解析を行った。まずアミノ酸配列をもとに多重配列アライメントをClustalWを用いて行った。この多重配列アライメントデータをもとにMaximum likelihood法により系統樹を再構成し、MEGA5の祖先配列推測機能を用いて、祖先遺伝子の配列推定を行った。具体的にはMetridinidae科およびLucicutiidae科のプランクトンに由来する15種のルシフェラーゼのアミノ酸配列から推定したその祖先遺伝子のアミノ酸配列として137アミノ酸からなるaCopLuc43と、Heterorhabdidae科プランクトンに由来する6種のルシフェラーゼのアミノ酸配列から推定したその祖先遺伝子のアミノ酸配列として132アミノ酸からなるaCopLuc48を決定した。Metridinidae科およびLucicutiidae科の共通祖先遺伝子を推測したのは、この2つの科から見いだされたルシフェラーゼのアミノ酸配列が、Heterorhabdidae科由来のものと比較して相同性が高かったからである。このaCopLuc43のアミノ酸配列を、クローニングしたMetridinidae科およびLucicutiidae科のプランクトンに由来する15種のルシフェラーゼとアライメントしたところ(図1)、15種のルシフェラーゼのアミノ酸配列中に強く保存されているアミノ酸残基Cys-X(3)-Cys-Leu-X(2)-Leu-X(4)-Cys-X(8)-Pro-X-Arg-Cys (X, アミノ酸)の並び方に合致しない領域を発見したため、aCopLuc43のアミノ酸配列をコードする塩基配列を人工合成した後、当該部位(配列番号23の91番目と92番目)に変異を導入して、この法則に合致したクローン、aCopLuc43mut (配列番号22)を作成した。
【0054】
カイアシ類由来ルシフェラーゼの祖先遺伝子の大腸菌による発現
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列の解析により得られたカイアシ類ルシフェラーゼの祖先遺伝子aCopLuc43, aCopLuc43mutおよびaCopLuc48のアミノ酸配列をコードする塩基配列を、大腸菌で使用されるコドン頻度に最適化して、オペロンバイオテクノロジー社の人工遺伝子合成サービスを利用して合成した。このプラスミドベクターをNdeIおよびBamHIにて制限酵素処理し、そのインサートDNA断片を1.6%アガロースゲル上で泳動し、目的分子量の制限酵素断片を確認し、ゲル切片からMontageゲル抽出キット(Millipore社製)とMinElute PCR purification kit(Qiagen社製)を用いた前述の方法でDNAを回収した。
【0055】
精製済みインサートDNA(コード配列部分)を、同様にNdeIおよびBamHIにて制限酵素処理した大腸菌発現用プラスミドpET-16b(Novagen社製)中にライゲーションする。ライゲーションにはLigation High (東洋紡社製)を用いた。反応後ライゲーション液を、Chemical competent cell DH5alpha株へ導入し、LB/カルベニシリンプレート上で形質転換株を選択する。上記プレート上にはえてきたコロニーからcolony PCR法を利用するスクリーングによって、祖先ルシフェラーゼ遺伝子のコード領域を有する発現ベクターpET-16bを保持している大腸菌クローンを選択する。選択されたコロニー(クローン)を培養し、Qiagen Plasmid purification kit(Qiagen社製)を利用して、プラスミドベクターを回収・精製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit ver. 1.1(Life Technologies Japan社製)により、pET-16bに挿入された遺伝子のコード配列部分およびその連結部分の塩基配列を前述した方法でシーケンスし、遺伝子の配列および向きを確認した。シーケンスが確認されたプラスミドベクターを大腸菌発現株BL21 CodonPlus RIPL (Stratagene社製)へ導入する。形質転換体をLB/カルベニシリンプレート上で37℃で一晩培養し、得られたコロニーを10 mLのZYM5052培地(50μg/mLカルベニシリン含)に植菌し、16℃、180 rpmで6日間培養した。ZYM5052培地の組成は以下の通りである。
(1% N-Z-amine, 0.5% Yeast extract, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.5% glycerol, 0.05% glucose, 0.2% lactose)
培養後7,000 rpm、10分間の遠心を行って大腸菌を沈殿させ、上清培地を除いた後、1 mLの溶解バッファー(20 mM Tris-HCl (pH8.0), 10 mM MgCl2)に再懸濁した。この懸濁液を氷上で10秒間超音波破砕を行い、15,000 rpm で5分間遠心した。上清を可溶性組換えタンパク質を含む画分として回収し、その活性を測定したところ発光活性が認められた(図5)。
【0056】
実施例3:祖先遺伝子のヒト細胞(HEK293)における発現(図6)
カイアシ類由来ルシフェラーゼの祖先遺伝子のヒト細胞による発現
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列の解析により得られたカイアシ類ルシフェラーゼの祖先遺伝子aCopLuc43mutおよびaCopLuc48のアミノ酸配列をコードする塩基配列を、ほ乳類培養細胞発現用ベクターpcDNA3.2 V5-His TOPO中へ挿入する。精製済みのヒト細胞発現ベクターを、ヒト培養細胞HEK293に遺伝子導入・発現させ、祖先遺伝子の発光活性を測定した。100 mm dishにてHEK293細胞を培養した。カイアシ類由来ルシフェラーゼの祖先遺伝子の発現ベクターのLipofectamine 2000 (Invitrogen社製)によるトランスフェクション処理後、細胞は5%CO2下、37℃にて72時間インキュベーションし、導入遺伝子の発現を行った。この結果、細胞内画分において発光活性が認められた(図6)。ルシフェラーゼ活性の測定法は、後述する「カイアシ類由来ルシフェラーゼ活性の測定」に記載した。
【0057】
カイアシ類由来ルシフェラーゼ活性の測定
カイアシ類ルシフェラーゼの活性測定には以下の方法を用いた。1μg/μLに調製した発光基質セレンテラジン(coelenterazine)のメタノール溶液より1 μLをとって、1 mLの基質希釈用バッファー(20 mM Tris-HCl, pH8.0, 50 mM MgCl2)で希釈した。希釈後の発光基質液は氷上に保持した。カイアシ類ルシフェラーゼを発現するプラスミドベクターを導入されたHEK293細胞からの培養上清または大腸菌破砕液5 μLをポリスチレン製テストチューブに分取して、ルミノメーターLB9506(ベルトールド社製)の測定部にセットし、10 μLの上記希釈発光基質を添加した後、直ちに測定を開始した。発光の測定は10秒間行った。発光が強すぎて測定限界を超えた場合は、試料希釈バッファー(20 mM Tris-HCl, pH8.0, 10 mM MgCl2)にて10倍希釈を行って、同様に測定した。得られた発光値をルミノメーターに附属のプリンターに出力した後、次の試料の測定に移った。
【産業上の利用可能性】
【0058】
本発明にかかるカイアシ類由来のルシフェラーゼとその祖先ルシフェラーゼのタンパク質をコードする遺伝子とは、いずれも、哺乳動物細胞を利用する培養系において、該宿主細胞内で発現可能であり、かつ、かかる遺伝子から発現された組換え発現型ルシフェラーゼタンパク質は、宿主細胞外へと分泌され、または祖先ルシフェラーゼは細胞内に発現するレポータータンパク質として利用することが可能である。
【技術分野】
【0001】
本発明は、海洋プランクトン由来の新規な発光タンパク質(ルシフェラーゼ)とその遺伝子群、並びにそれらより推測された発光タンパク質活性を有する祖先発光タンパク質及びその祖先発光タンパク質をコードする遺伝子に関する。
【背景技術】
【0002】
北米ホタルのフォチヌス・ピラリス(Photinus pyralis)由来のルシフェラーゼ(FLuc:Firefly luciferase)、あるいはその改変体タンパク質は、異種細胞内、例えば各種の哺乳動物細胞内において、組換え発現可能であり、得られる組換えタンパク質は、宿主細胞内において、発光基質のD-ルシフェリンを分解して発光特性を示す。またウミシイタケ(Renilla reniformis)由来のルシフェラーゼ(RLuc:Renilla luciferase)も同様に動物細胞を含む各種生物において発現が可能であり、発光基質のセレンテラジン(coelenterazine)を分解する発光活性を有する。これらルシフェラーゼならびにその変異体により得られる発光量は定量性に優れており、生化学、細胞生物学、医学分野において、主に動物細胞のプロモーター(遺伝子発現制御領域)のRNA転写活性を計測する、プロモータアッセイのレポータータンパク質として広く利用が図られている(非特許文献1)。
【0003】
一方、P. pyralisやR. reniformis由来のルシフェラーゼ以外に、ウミホタル(Vargula hilgendorfii)から分泌型ルシフェラーゼがクローニングされており(非特許文献2)、更には、同じ節足動物門(Arthropoda)カイアシ亜綱(Copepoda)のガウシア・プリンセップス(Gaussia princeps)からも、分泌型ルシフェラーゼがクローニングされている(非特許文献3)。これらカイアシ亜綱(Copepoda)のガウシア・プリンセップス(G. princeps)由来ルシフェラーゼはウミホタル(V. hilgendorfii)や北米ホタル(P. pyralis)とは構造的に異なる発光基質を特異的に分解することから、生物進化的には、異なる起源を有するであろうことが報告されている。その発光基質(ルシフェリン)は、R. reniformis由来のルシフェラーゼと同じくセレンテラジンであることが同定されている。
【0004】
【化1】
【0005】
他にも同じカイアシ亜綱(Copepoda)メトリディア・ロンガ(Metridia longa)由来のルシフェラーゼ(非特許文献4)、我々が以前クローニングしたメトリディア・パシフィカ(Metridia pacifica)由来のルシフェラーゼ(非特許文献5)、オワンクラゲ(Aequorea victoria)由来のイクオリン(非特許文献1)、十脚類(Decapoda)に属するヒオドシエビ(Oplophorus gracilirostris)由来のルシフェラーゼ(非特許文献6)等が、上記セレンテラジンをその発光基質(ルシフェリン)として、利用可能であることが報告されている。
【0006】
北米ホタル(P. pyralis)由来のルシフェラーゼ(FLuc)およびウミシイタケ(R. reniformis)のルシフェラーゼ(RLuc)に関しては、動物細胞内で安定して発現可能な、in vivo発光レポータータンパク質として広く利用されているが、非分泌型のルシフェラーゼである。そのためプロモータアッセイを行うためには、ホストの動物細胞等を破砕してルシフェラーゼ活性を測定する必要がある。一方、海洋生物、特に動物性プランクトン類のガウシア・プリンセップス(G. princeps)、メトリディア・ロンガ(Metridia longa)、メトリディア・パシフィカ(Metridia pacifica)由来ルシフェラーゼはウミシイタケ(R. reniformis)と同じ発光基質(セレンテラジン)を酸化分解して発光するが、R. reniformisとは違って分泌型のルシフェラーゼである。そのためレポーターであるルシフェラーゼタンパク質はホストの細胞外へ速やかに分泌され、培地中のルシフェラーゼ活性をモニタリングすればよく、細胞破砕の必要性がない。その結果、同一細胞を用いた連続モニタリングやタイムコース実験を簡単に行うことができる。またウミホタル(V. hilgendorfii)ルシフェラーゼも同じく分泌型であるが、その発光基質であるウミホタル・ルシフェリンはセレンテラジンとは全く異なる構造を有する。またガウシア・プリンセップス(G. princeps)やメトリディア・パシフィカ(M. pacifica)由来ルシフェラーゼは顕著な耐熱性を示すことが報告されているが、有機溶媒等に対しては耐性が低い性質を有し、さらなる改善の余地がある(非特許文献5、非特許文献7)。
【0007】
そこで宿主動物細胞内で発現可能で、より高輝度、高安定性な、新規なルシフェラーゼの探索・開発が待望されていた。
【0008】
ルシフェラーゼタンパク質を、宿主細胞内で発現可能な、in vivo発光性レポータータンパク質として利用する際、特定のプロモーターを用いて宿主細胞内でルシフェラーゼタンパク質を翻訳する。翻訳されたルシフェラーゼタンパク質は適切な構造に折りたたまれてその発光活性を発揮する。一般的に特定のタンパク質の機能や局在を観察する場合これまで蛍光性マーカータンパク質が利用されてきた。すなわち特定のタンパク質のN末端やC末端に蛍光性マーカータンパク質のアミノ酸配列を融合した形で発現させ、宿主細胞に励起光を照射して蛍光性マーカータンパク質から得られる蛍光を検出する。例えば下村脩博士が研究し、ノーベル賞を授与されたことで特に注目を浴びたGFPなどがよく利用されてきた。しかし最近、ルシフェラーゼタンパク質を機能性ペプチドやタンパク質に融合させて、細胞内のシグナルを検出する系の開発も進んでいる(非特許文献8)。このような検出系において、融合させるルシフェラーゼの分子量は融合タンパク質の機能に大きな影響をおよぼす。これまでルシフェラーゼタンパク質としてよく利用されてきた北米ホタル(P. pyralis)由来のルシフェラーゼ(FLuc)は61 kDa、ウミシイタケ(R. reniformis)由来のルシフェラーゼ(RLuc)は36 kDa、ウミホタル(V. hilgendorfii)由来のルシフェラーゼは62 kDaと融合タンパク質としては比較的分子量の大きいタンパク質であり、より分子量が小さく高い輝度をもつ、新規な発光性レポータータンパク質の提供が望まれていた。動物性プランクトン類のガウシア・プリンセップス(G. princeps)、メトリディア・ロンガ(M. longa)、メトリディア・パシフィカ(M. pacifica)由来ルシフェラーゼはそれぞれ19.9 kDa、23.8 kDa、22.7 kDaであることから、その分子量が30 kDa以下であった。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Bioluminescence, chemical principles and methods, Osamu Shimomura, World Scientific Publishing Co. Ltd, 2006
【非特許文献2】Thompson, EM, Nagata, S, and Tsuji, FI. Cloning and expression of cDNA for the luciferase from the marine ostracod Vargula hilgendorfii. Proc. Natl. Acad. Sci. USA, 86, 6567-6571, 1989
【非特許文献3】Verhaegen, M, and Christopoulos, TK. Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal. Chem., 74, 4378-4385, 2002
【非特許文献4】Markova, SV, Golz, S., Frank, LA, Kalthof, B. and Vysotski, ES. Cloning and expression of cDNA for a luciferase from the marine copepod Metridia longa. J. Biol. Chem., 279, 3212-3217, 2004
【非特許文献5】Takenaka, Y, Masuda, H, Yamaguchi, A, Nishikawa, S, Shigeri, Y, Yoshida, Y, and Mizuno, H. Two forms of secreted and thermostable luciferase from the marine copepod crustacean, Metridia pacifica. Gene, 425, 28-35, 2008
【非特許文献6】Inouye, S, and Sasaki, S. Overexpression, purification and characterization of the catalytic component of Oplophorus luciferase in the deep-sea shrimp, Oplophorus gracilirostris. Protein Expr. Purif., 56, 261-268, 2007
【非特許文献7】Rathnayaka, T, Tawa, M, Sohya, S, Yohda, M, and Kuroda, Y. Biophysical characterization of highly active recombinant Gaussia luciferase expressed in Escherichia coli. Biochim. Biophys. Acta, 1804, 1902-1907, 2010
【非特許文献8】Kim, S, Sato, M, and Tao, H. Genetically encoded bioluminescent indicators for stress hormones. Anal. Chem., 81, 3760-3768, 2009
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、新規な発光タンパク質及び該発光タンパク質をコードする遺伝子を提供することを目的とする。
【課題を解決するための手段】
【0011】
そこで上記課題を解決するために、発光能を有する海洋プランクトンを日本近海、具体的には、函館湾沖、親潮域で採取し、発光タンパク質(ルシフェラーゼ)を網羅的にクローニングし、これらの配列を元にバイオインフォマティクスを駆使し、ルシフェラーゼの祖先遺伝子の予測を行い、この配列からルシフェラーゼ活性の発揮に必須な最小機能領域を特定して、発光機能を保持した分子量十数kDaのさらに小さいルシフェラーゼを開発した。
【0012】
本発明は、以下の新規な発光タンパク質およびそれをコードする遺伝子を提供するものである。
項1. 配列番号22若しくは配列番号24のアミノ酸配列、または、1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列を有することを特徴とする発光タンパク質。
項2. 配列番号1〜21のいずれかアミノ酸配列を有する項1に記載の発光タンパク質。
項3. 項1または2に記載の発光タンパク質をコードするDNAまたはその相補鎖。
【発明の効果】
【0013】
本発明によれば、発光タンパク質の祖先遺伝子とアミノ酸配列を同定することができた。
【0014】
このような祖先遺伝子が明らかになったことで、より優れた性質を有する改変型発光タンパク質が開発されることが期待される。
【図面の簡単な説明】
【0015】
【図1】Metridinidae科およびLucicutiidae科由来ルシフェラーゼとそこから推測される改変型祖先ルシフェラーゼ aCopLuc43mut(配列番号22)及び改変前の祖先発光タンパク質 aCopLuc43(配列番号23)のアミノ酸配列のアラインメントを示す。
【図2】Heterorhabdidae科由来ルシフェラーゼとそこから推測される祖先発光タンパク質aCopLuc48(配列番号24)のアミノ酸配列のアラインメントを示す。
【図3】発光プランクトンMetridia okhotensisの(a)紫外線照射下での蛍光顕微鏡写真及び(b)白色光下での顕微鏡写真を示す。
【図4】ヒト培養細胞(HEK293)により培地中に分泌発現されたカイアシ類由来ルシフェラーゼの活性を示す。
【図5】実施例2において、大腸菌により発現された祖先遺伝子(aCopLuc43mutとaCopLuc48)のルシフェラーゼ活性を示す。
【図6】実施例3において、ヒト培養細胞(HEK293)により発現された祖先遺伝子(aCopLuc43mutとaCopLuc48)のルシフェラーゼ活性を示す。
【発明を実施するための形態】
【0016】
本発明の1つの特徴は、海洋プランクトン由来の発光タンパク質において、Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼの遺伝子配列およびアミノ酸配列を同定し、それにより祖先遺伝子とアミノ酸配列を決定したことにある。Metridinidae科プランクトン及びLucicutiidae科プランクトンの祖先発光タンパク質のアミノ酸配列は、配列番号22に示され、該アミノ酸配列をコードする塩基配列は、配列番号46に示される。また、Heterorhabdidae科プランクトンの祖先発光タンパク質のアミノ酸配列は、配列番号24に示され、該アミノ酸配列をコードする塩基配列は、配列番号48に示される。
【0017】
配列番号22に記載のアミノ酸配列は、配列番号23に記載のアミノ酸配列において、91位のCys→Aspに、92位のSerがCysに置換されたものである。
【0018】
本発明者らが見出したMetridinidae科とLucicutiidae科の海洋プランクトン由来の発光タンパク質(配列番号1〜15)を用いて得られた配列番号23の祖先発光タンパク質は発光活性を有しなかったが、91位と92位のアミノ酸を上記のように置換した配列番号22に記載のアミノ酸配列を有する改変型の祖先発光タンパク質は発光活性を有することを確認した。
【0019】
Heterorhabdidae科プランクトンの場合には、本発明者らが見出した配列番号16〜21のアミノ酸配列を用いて得られた祖先発光タンパク質(配列番号24)は発光活性を有することを確認した。
【0020】
配列番号22,24に示される祖先発光タンパク質は、本発明により初めて明らかにされたものである。
【0021】
本発明は、配列番号22,24の配列において、図1(配列番号23のaCopLuc43を除く),図2に示される範囲内でアミノ酸置換が可能である。
【0022】
例えば図1において、N末端のアミノ酸は、配列番号22のaCopLuc43mutではMであるが、このアミノ酸は、図1の他の発光タンパク質で確認できるH,L,Gに置換可能である。同様に、N末端から4番目の配列番号22のアミノ酸Kは、Q、Lに置換可能である。N末端から10番目の配列番号22のアミノ酸Vは、他の発光タンパク質が全てVであるため、置換できない。同様に、11番目のアミノ酸Lも置換不可能である。図2のアミノ酸配列に関しても、配列番号24のアミノ酸配列において同様なアミノ酸置換は可能である。
【0023】
さらに配列番号22、24のアミノ酸配列のN末端又はC末端側には、図1、図2で示される他のアミノ酸が付加可能である。
【0024】
本発明における、配列番号22、24のアミノ酸配列において1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列とは、上記のアミノ酸置換ないし付加が包含される。ここで置換、付加、欠失又は挿入される1又は複数のアミノ酸とは、150個以下、145個以下、140個以下、135個以下、130個以下、125個以下、120個以下、115個以下、110個以下、105個以下、100個以下、95個以下、90個以下、85個以下、80個以下、75個以下、70個以下、65個以下、60個以下、55個以下、50個以下、45個以下、40個以下、35個以下、30個以下、25個以下、20個以下(例えば1、2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19又は20個)のアミノ酸の置換、付加、欠失又は挿入を意味する。例えば図1に示されるMoLuc1は図1に示す番号で1番〜48番,57番〜85番、146番、148番、226番の合計80個のアミノ酸が配列番号22の改変型祖先発光タンパク質に対して付加され、さらに87番(P/G)、94番(A/E)、97番(I/K)、104番(F/K)、118番(K/H)、127番(E/K)、128番(Y/F)、135番(D/S)、137番(G/E)、141番(K/D)、143番(G/A)、145番(A/G)、149番(V/I)、151番(A/P)、159番(S/P)、163番(E/D)、165番(G/A)、178番(A/V)、183番(G/R)、192番(K/Q)、195番(A/D)、196番(L/Q)、203番(D/T)、206番(A/T)、207番(S/T)、210番(D/S)、214番(R/K)、217番(H/D)、218番(N/T)、224番(G/D)の合計30個のアミノ酸が置換されている。従って、MoLuc1の場合、配列番号22のアミノ酸配列に対し110個のアミノ酸が置換、付加、欠失又は挿入されることになり、このようなアミノ酸の置換、付加、欠失又は挿入は、本発明の範囲内である。また、置換、付加、欠失、挿入されるアミノ酸は、図1,図2に示される範囲内であることが好ましく、MoLuc1と配列番号22の祖先発光タンパク質(aCopLuc43Mut)を比較して最大110個のアミノ酸が置換、付加、欠失又は挿入可能であるが、1〜109個のアミノ酸を適宜選択して置換、付加、欠失又は挿入してもよい。上記の記載は、MoLuc1を例に取り説明したが、MoLuc2、McLuc1、McLuc2、MaLuc1、MaLuc2、PsLuc1、PsLuc2、PaLuc1、PaLuc2、PxLuc1-7、PxLuc1-8、PxLuc2、LoLuc1-1、LoLuc1-3についても同様に、図1に示される範囲でアミノ酸の置換、付加、欠失又は挿入が可能である。さらに、HtLuc1-1-2、HtLuc1-1-3、HtLuc1-2-2、HtLuc2、HmLuc1、HmLuc2についても同様に、図2に示される範囲でアミノ酸の置換、付加、欠失又は挿入が可能である。
【実施例】
【0025】
以下、本発明を実施例に従いより詳細に説明するが、本発明がこれら実施例に限定されないことは言うまでもない。
【0026】
実施例1:発光プランクトン由来の新規ルシフェラーゼ
1.アミノ酸配列及び塩基配列
海洋に棲息する動物性プランクトンより、発光プランクトンを探索した。具体的には、北海道大学大学院水産科学研究院の山口篤准教授(産総研客員研究員)と共同で、附属練習船おしょろ丸に乗船し、函館湾沖、親潮域で採取された海洋中に存在する、動物性プランクトンの分類を進める過程で、数多くの発光プランクトンを採取した。更に、これら発光プランクトンのうち、分類学的に節足動物門(Arthropoda)カイアシ亜綱(Copepoda)に属する動物性プランクトンであって、体外に分泌発現型の発光タンパク質を発現しているものを選別した。この動物プランクトン採集で見出された、分泌型の発光タンパク質を発現しているカイアシ亜綱(Copepoda)の分類を試み、Calanoida目、Augaptiloidea上科に属するMetridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトンの同定を行った。さらにMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質について、それをコードする遺伝子の塩基配列の特定と、推定アミノ酸配列の決定を試みた。先ず、Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトンから、全RNAを抽出し、含まれるmRNAを精製し、逆転写酵素を用い、定法に従って、対応するcDNAを合成した。これまでに報告されているMetridinidae科プランクトン由来のルシフェラーゼ遺伝子の配列を参考にPCR法によりcDNAより部分配列の増幅を行った。その結果、約250塩基のルシフェラーゼ遺伝子の部分配列を得た。次にその塩基配列をもとに、cDNA全長の増幅と塩基配列解析を行い、目的とするMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質の配列を解明した。このMetridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトン由来のルシフェラーゼタンパク質の完全長アミノ酸配列をアライメントして、コンピュータプログラム(MEGA5)を用いて分子量15 kDaおよび13.8 kDaの祖先ルシフェラーゼタンパク質配列を予測し、ルシフェラーゼタンパク質をコードする遺伝子配列を人工遺伝子合成して、大腸菌およびほ乳類培養細胞において発現させ、そのルシフェラーゼ機能を確認するに至った。
【0027】
本発明にかかるCalanoida目、Augaptiloidea上科、Metridinidae科、Lucicutiidae科、Heterorhabdidae科に属する動物プランクトン由来の発光タンパク質、カイアシ類Metridinidae科およびLucicutiidae科に属する動物プランクトン由来の発光タンパク質のアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質(aCopLuc43)とその改変体(aCopLuc43mut)、カイアシ類Heterorhabdidae科に属する動物プランクトン由来の発光タンパク質のアミノ酸配列から推測されるその祖先ルシフェラーゼタンパク質(aCopLuc48)の名称と完全長アミノ酸配列、塩基配列の配列番号の対応関係を以下に示す。
【0028】
【表1】
【0029】
2.実験方法
カイアシ類Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン体内でのタンパク質の発現に伴い、残留している多種のmRNAのうち、当該ルシフェラーゼの遺伝子を選別することを試みた。具体的には、mRNAよりcDNAライブラリーを作製し、このcDNAライブラリーからPCR法によりルシフェラーゼ遺伝子の部分配列および全長配列の取得を行った。
【0030】
以下に、本発明のカイアシ類由来のルシフェラーゼに関して、より詳しく説明する。
【0031】
カイアシ類発光プランクトンの採集、同定
まず、本発明のルシフェラーゼの起源である動物性発光プランクトンは、函館湾沖、水深50〜1000mから採取された親潮域深層水中に見出された、Calanoida目、Augaptiloidea上科に属するプランクトンである。代表的な発光プランクトンMetridia okhotensisの形態は、図3(a)に示すように、紫外線照射下において、蛍光顕微鏡により観察すると発光基質を含む分泌腺が青緑色に見える。また、白色光下で顕微鏡観察すると、図3(b)に示すように、Metridia okhotensisのプランクトンの体内は透明から若干白色がかって観察される。本発明でルシフェラーゼ遺伝子のクローニングを行ったプランクトンはMetridinidae(メトリディニダエ科)に属するものとして、Metridia okhotensis、Metridia curticauda、Metridia asymmetrica、Pleuromamma abdominalis、Pleuromamma scutullata、Pleuromamma xiphias、Lucicutiidae(ルチクチイダエ科)に属するものとしてLucicutia ovaliformis、またHeterorhabdidae(ヘテロハブディダエ科)に属するものとして、Heterorhabdus tanneri、Heterostylites majorの計5属9種である。これらの動物プランクトンの詳細な分類学上の同定では、Metazoa(動物界)、Arthropoda(節足動物門)、Crustacea(甲殻亜門)、Maxillopoda(アゴアシ綱)、Copepoda(カイアシ亜綱)、Neocopepoda(カイアシ下網)、Gymnoplea(前脚類)、Calanoida(カラヌス目)、Metridinidae(メトリディニダエ科)もしくはLucicutiidae(ルチクチイダエ科)もしくはHeterorhabdidae科(ヘテロハブディダエ科)となる。
【0032】
本発明者らは、これらのMetridinidae科、Lucicutiidae科またはHeterorhabdidae科に属するプランクトンが物理的刺激を与えることにより発光を示し、またその粗抽出液を発光基質セレンテラジンと混合した際に発光が観測されることを詳細に調べた結果、ルシフェラーゼを産生するバクテリアの寄生、付着に起因するものではなく、このMetridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン自体に由来するルシフェラーゼに因ると結論した。そこで、該プランクトンを集め、ルシフェラーゼタンパク質の単離を進めることを検討したが、入手されるプランクトン量が十分でなく、そのアミノ酸配列解析に十分なタンパク質量を回収することは困難であると判断された。従って、近縁のMetridia属のMetridia longa由来のルシフェラーゼ(GenBank accession number, AY364164; 文献4)とGaussia属に属する、Gaussia princeps由来のルシフェラーゼ(GenBank accession number, AY015993; 文献3)のアミノ酸配列に基づき、相同性の高いアミノ酸配列の一部をコードする、縮重プライマーを作製し、cDNAから、当該ルシフェラーゼの遺伝子をPCR法により、クローニングする手法を適用した。
【0033】
カイアシ類発光プランクトンRNAの精製およびcDNAの合成
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科プランクトンより、市販のRNA抽出試薬;RNeasy Mini kit(Qiagen社製)を用いて全RNAを抽出し、次いで、市販の精製キット;Oligotex-dT30 <SUPER> mRNA Purification kit(Takara Bio社製)を用いてPoly(A)+mRNAの精製を行った。更に、精製済みのmRNAから、市販のcDNA調製キット:SMART RACE cDNA増幅キット(Takara Bio社製)を利用して、それぞれ5’末端側増幅用および3’末端側増幅用cDNAの合成をおこなった。
【0034】
PCR反応による、カイアシ類ルシフェラーゼcDNA断片の取得
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン由来のルシフェラーゼをコードする遺伝子クローニング工程においては、mRNAから調製されるcDNAを鋳型として、前記上流側混合プライマー二種と下流側混合プライマー二種とを組み合わせた、計4種のPCR用プライマー対についてPCR反応を行った。
【0035】
具体的には、以下に示す、上流側混合プライマー2種と下流側混合プライマー2種の塩基配列を選択した。
【0036】
上流側混合プライマー
White luc UP1 (26 mer:16種の混合プライマー)
5’-GGC TGC ACY AGG GGA TGY CTK ATM TC-3’
(Y=T, C;K=G, T;M=A, C)
White luc UP2 (26 mer:16種の混合プライマー)
5’-GCT ATT GTT GAY ATY CCY GAR AT-3’
(Y=T, C;R=G, A)
【0037】
下流側混合プライマー
White luc LP2 (26 mer:16種の混合プライマー)
5’-TC AAG TTG WTC AAT RAA YTG YTC CAT-3’
(W=A, T;R=G, A;Y=T, C)
White luc LP1 (23 mer:12種の混合プライマー)
5’-AC ATT GGC AAG ACC YTT VAG RCA-3’
(Y=T, C;V=A, G, C;R=G, A)
【0038】
Metridinidae科、Lucicutiidae科またはHeterorhabdidae科のプランクトン由来のmRNAを鋳型として、調製される3’-Ready cDNAから、上記に示す4組のプライマー対を利用して、常法に従いPCR増幅産物を調製する。
【0039】
回収されたDNA溶液(PCR増幅産物)に、10xLoading dye液 2μLを添加した後、1.6% TAEアガロースゲルにアプライし、各レーンDNA溶液 15μLを泳動した。目的分子量のバンドをゲルから切り出し、Montageゲル抽出キット(Millipore社製)に含まれるマイクロチューブカラムにゲル片を入れて、7,000 rpmで10分間遠心する。メンブレンを通して抽出されてきたDNA溶液をさらにMinElute PCR purification kit(Qiagen社製)を用いて精製を行う。DNA溶液1容量当たり、バッファーPBを5容量を加え、ボルテックスにかけた後、MinEluteカラムに移した。30秒間遠心し、DNAをカラムに吸着させる。ついでカラムを、バッファーPE 0.7 mLで洗浄し、さらに1分間、遠心(15,000 rpm)することによりバッファーPEをカラムから完全に除く。その後、溶出バッファーEB 10μLを加えて、室温で、1分間静置する。最後に、1分間、遠心(15,000 rpm)し、カラムから溶出してきたDNAを1.5 mL用マイクロチューブ中に回収する。
【0040】
上記精製済みPCR反応産物を、クローニング・ベクターpCR2.1-TOPO(Invitrogen社製)中に挿入する。PCR反応産物2μLにベクターに添付のSalt solution 0.5μLおよびベクター溶液0.5μLを加えて、室温で5〜15分間インキュベートしてライゲーションを行う。この反応液に含まれるDNAをChemical competent cell DH5alpha株へ導入し、形質転換株を選択する。プラスミドベクターの宿主大腸菌へ導入は、以下の手順で実施した。宿主大腸菌に利用する、DH5alpha株は、凍結保存されているCompetent cellを、氷温で解凍する。PCR産物挿入プラスミドベクター溶液3μLを、解凍された宿主大腸菌DH5alpha株懸濁液 50μLに加える。そのまま氷上で10分間保持した後、42℃、30秒加温し、氷上に戻した。ついで100μLのSOC培地を加えて、37℃、10分間振盪培養する。その後、培養液に含まれる形質転換株を、クローニング・ベクターpCR2.1-TOPOに由来する選択マーカーを利用して薬剤耐性株を選択する。50μg/mL抗生剤カルベニシリンを含んだLBプレート上に出現するコロニーを解析して、PCR産物を保持するプラスミドベクターをもつ大腸菌を選択する。
【0041】
PCR産物を保持するプラスミド・ベクターの複製、精製
選別されたクローンから、以下の手順で、導入されているプラスミドベクターの精製を行う。
【0042】
PCR産物が挿入されたプラスミドベクターを保持する宿主大腸菌 DH5alpha株のコロニー(クローン)を、それぞれ2 mLのLB/カルベニシリン液体培地中に懸濁し、37℃、16時間培養を行った。培養液を、10分間、遠心(5,000 x g)して、細胞を分取した。
【0043】
分取した細胞から、市販のプラスミド精製キット:Qiagen Plasmid purification kit(Qiagen社製)を利用して、プラスミドを分離、精製する。分取した細胞に、同精製キットに添付のP1液 0.25mLを添加し、ボルテックスにかけて、よく分散させる。この細胞分散液に、添付のP2液 0.25mLを添加、混和して室温(20℃)にて5分間静置する。溶菌処理後、添付のN3液 0.35mLを添加し、混和する。次いで15分間、4℃で遠心(15,000 rpm)し、プラスミドDNAを含む上清を分離、回収する。上清を、同精製キットのQIAprep miniカラムにかける。4℃で遠心(15,000 rpm)し、液層を除去する。バッファーPB 0.5 mLを加えて、カラムを洗浄し、引き続き、バッファーPE 0.75 mLを加えて、再度洗浄する。最終的に、1分間、4℃で遠心(15,000 rpm)し、洗浄液を完全に除去する。精製キットのQIAprep miniカラムに吸着されているプラスミドDNAは、バッファーEB 30μLにより溶出、回収する。この精製済みプラスミドを含む液 30μLのうち、1μLをとり、蒸留水 99μLを加えて、100倍希釈液とし260nmの吸光度を測定してDNA濃度の定量を行った。このプラスミドベクターのDNA濃度を250ng/μLとなるように、濃度調整を行った。
【0044】
選別クローン中のcDNA断片(PCR産物)の塩基配列解析
プラスミドベクター中に挿入されている、cDNA断片(PCR産物)を、以下の手順で塩基配列解析用の核酸鎖伸長反応産物を調製する。
【0045】
選択クローンから回収、次いで、市販のシークエンス用DNA試料調製キット:BigDye Terminator Cycle Sequencing Ready Reaction Kit ver. 1.1(Life Technologies Japan社製)により、この精製済みプラスミドベクターを鋳型として、塩基配列解析用の試料を、2種のプライマー;M13 sense M4およびM13 reverseをシークエンス・プライマーとして利用し、プラスミドベクター中に挿入されているcDNA断片を含む領域のシーケンス反応生成物を調製する。 調製された塩基配列解析用の試料の精製は、以下の手順で行う。
【0046】
各反応用チューブから、調製済み試料溶液を別の0.5 mL容チューブに移す。試料溶液 5 μL当たり、3M 酢酸ナトリウム水溶液 0.5 μL、95%エタノール12.5μLの比率で混合した液を、別途、1.5 mL容チューブに予め用意する。この1.5 mL容チューブ中に、予め集めた試料溶液を投入する。均一に混合した上で、氷冷下に、10分間静置し、含まれるDNA断片をエタノール沈澱(析出)させる。20分間、遠心(15,000 rpm)し、析出したDNA断片を沈積させ、上清を除去する。次いで、125μLの70%エタノールを加え、沈殿DNAをリンスする。再び、5分間、遠心(15,000 rpm)し上清を吸引除去する。残った沈殿DNA断片のペレットを乾燥する。
【0047】
精製した解析用試料DNA断片は、15 μLホルムアミド中に再溶解する。ボルテックスにかけて、混合した後、遠心して液を集める。95℃、2分間加熱し、一本鎖DNAに分離し、氷冷する。その後、解析用試料DNA断片は、市販のシークエンス装置:ABI PRISM 3100 Genetic Analyzer(Life Technologies Japan社製)にかけ、塩基配列解析を行う。
【0048】
センス鎖の配列解析結果と、アンチセンス鎖の配列解析結果とを総合し、挿入されているcDNA断片(PCR反応産物)の塩基配列を決定する。
【0049】
得られたカイアシ類由来の各ルシフェラーゼの塩基配列及びアミノ酸配列を表1及び配列表に記載する。
【0050】
カイアシ類由来ルシフェラーゼタンパク質のヒト細胞発現用発現ベクターのHEK293細胞への導入
ほ乳類培養細胞発現用ベクターpcDNA3.2 V5-His TOPO(Invitrogen社製)中へ上記で得られたカイアシ類由来ルシフェラーゼ遺伝子のコード領域を挿入する。プラスミドpcDNA3.2 V5-His TOPOのクローニング・サイト中に各遺伝子が挿入されていることをシーケンスにより確認する。作製したほ乳類培養細胞発現ベクターのうち、MoLuc1, MoLuc2, MaLuc1, PsLuc1, PaLuc1, PaLuc2, PxLuc1-7, PxLuc1-8, LoLuc1-1, LoLuc1-3を代表として培養細胞で遺伝子発現させた。精製済みのヒト細胞発現ベクターを、HEK293細胞中に、Lipofectamine 2000 (Invitrogen社製)を用いて遺伝子導入した。宿主のHEK293細胞は、10 mL血清添加培地(MEM+10% FBS+Penicillin/streptomycin)を用いて、100 mm dish上で、95%コンフルエントな状態まで培養されたものを利用する。まずプラスミド溶液(DNA濃度:1 μg/μL)24 μLを1.5 mLの無血清Opti-MEM培地に懸濁したものと、Lipofectamine 2000 60 μLを同様に1.5 mLの無血清Opti-MEM培地に懸濁したものを用意する。この2液を混合して室温にて20分間インキュベートする。この混合液全量を100 mm dish上の細胞培地中に添加して、均一になるように軽く撹拌する。トランスフェクション処理後、HEK293細胞は、5%CO2下、37℃にて48時間以上インキュベーションし、導入遺伝子の発現を行う。
【0051】
培養上清 10 mLをすべて新しい15 mLの遠心管に移し、1000 rpmで2分間遠心して、遠心上清を培地画分とした。一方、 dish 上に残った細胞に新しい培地10 mLを加えて、セルスクレーパーを使って細胞をはがした後すべて15 mLの遠心管に回収した。この懸濁液を氷上で10秒間超音波破砕を行い、15,000 rpm で5分間遠心した。上清を細胞内画分として回収し、培地画分と同時に発光活性を測定した。そのタンパク質は、細胞外に分泌され、培地中に蓄積されることが観測された(図4)。 またその時の分泌効率について表2に記した。ルシフェラーゼ活性の測定法は、後述する「カイアシ類由来ルシフェラーゼ活性の測定」に記載した。以上より、ヒト由来のHEK293細胞内においても、カイアシ類由来ルシフェラーゼタンパク質のコード遺伝子に基づき、完全長のアミノ酸配列を有するプレ・タンパク質型組換え体へと翻訳がなされる。その後、そのN末端に存在するシグナルペプチド部を利用して、細胞外へ成熟型活性タンパク質として、分泌されることを明確に示している。
【0052】
【表2】
【0053】
実施例2:祖先遺伝子の推測と大腸菌による発現、活性の確認
カイアシ類由来ルシフェラーゼの祖先遺伝子の推測
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列を、それぞれの科ごとにタンパク質配列データの分子進化・系統学的解析を行うためのソフトウェアMEGA5にて解析を行った。まずアミノ酸配列をもとに多重配列アライメントをClustalWを用いて行った。この多重配列アライメントデータをもとにMaximum likelihood法により系統樹を再構成し、MEGA5の祖先配列推測機能を用いて、祖先遺伝子の配列推定を行った。具体的にはMetridinidae科およびLucicutiidae科のプランクトンに由来する15種のルシフェラーゼのアミノ酸配列から推定したその祖先遺伝子のアミノ酸配列として137アミノ酸からなるaCopLuc43と、Heterorhabdidae科プランクトンに由来する6種のルシフェラーゼのアミノ酸配列から推定したその祖先遺伝子のアミノ酸配列として132アミノ酸からなるaCopLuc48を決定した。Metridinidae科およびLucicutiidae科の共通祖先遺伝子を推測したのは、この2つの科から見いだされたルシフェラーゼのアミノ酸配列が、Heterorhabdidae科由来のものと比較して相同性が高かったからである。このaCopLuc43のアミノ酸配列を、クローニングしたMetridinidae科およびLucicutiidae科のプランクトンに由来する15種のルシフェラーゼとアライメントしたところ(図1)、15種のルシフェラーゼのアミノ酸配列中に強く保存されているアミノ酸残基Cys-X(3)-Cys-Leu-X(2)-Leu-X(4)-Cys-X(8)-Pro-X-Arg-Cys (X, アミノ酸)の並び方に合致しない領域を発見したため、aCopLuc43のアミノ酸配列をコードする塩基配列を人工合成した後、当該部位(配列番号23の91番目と92番目)に変異を導入して、この法則に合致したクローン、aCopLuc43mut (配列番号22)を作成した。
【0054】
カイアシ類由来ルシフェラーゼの祖先遺伝子の大腸菌による発現
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列の解析により得られたカイアシ類ルシフェラーゼの祖先遺伝子aCopLuc43, aCopLuc43mutおよびaCopLuc48のアミノ酸配列をコードする塩基配列を、大腸菌で使用されるコドン頻度に最適化して、オペロンバイオテクノロジー社の人工遺伝子合成サービスを利用して合成した。このプラスミドベクターをNdeIおよびBamHIにて制限酵素処理し、そのインサートDNA断片を1.6%アガロースゲル上で泳動し、目的分子量の制限酵素断片を確認し、ゲル切片からMontageゲル抽出キット(Millipore社製)とMinElute PCR purification kit(Qiagen社製)を用いた前述の方法でDNAを回収した。
【0055】
精製済みインサートDNA(コード配列部分)を、同様にNdeIおよびBamHIにて制限酵素処理した大腸菌発現用プラスミドpET-16b(Novagen社製)中にライゲーションする。ライゲーションにはLigation High (東洋紡社製)を用いた。反応後ライゲーション液を、Chemical competent cell DH5alpha株へ導入し、LB/カルベニシリンプレート上で形質転換株を選択する。上記プレート上にはえてきたコロニーからcolony PCR法を利用するスクリーングによって、祖先ルシフェラーゼ遺伝子のコード領域を有する発現ベクターpET-16bを保持している大腸菌クローンを選択する。選択されたコロニー(クローン)を培養し、Qiagen Plasmid purification kit(Qiagen社製)を利用して、プラスミドベクターを回収・精製し、BigDye Terminator Cycle Sequencing Ready Reaction Kit ver. 1.1(Life Technologies Japan社製)により、pET-16bに挿入された遺伝子のコード配列部分およびその連結部分の塩基配列を前述した方法でシーケンスし、遺伝子の配列および向きを確認した。シーケンスが確認されたプラスミドベクターを大腸菌発現株BL21 CodonPlus RIPL (Stratagene社製)へ導入する。形質転換体をLB/カルベニシリンプレート上で37℃で一晩培養し、得られたコロニーを10 mLのZYM5052培地(50μg/mLカルベニシリン含)に植菌し、16℃、180 rpmで6日間培養した。ZYM5052培地の組成は以下の通りである。
(1% N-Z-amine, 0.5% Yeast extract, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.5% glycerol, 0.05% glucose, 0.2% lactose)
培養後7,000 rpm、10分間の遠心を行って大腸菌を沈殿させ、上清培地を除いた後、1 mLの溶解バッファー(20 mM Tris-HCl (pH8.0), 10 mM MgCl2)に再懸濁した。この懸濁液を氷上で10秒間超音波破砕を行い、15,000 rpm で5分間遠心した。上清を可溶性組換えタンパク質を含む画分として回収し、その活性を測定したところ発光活性が認められた(図5)。
【0056】
実施例3:祖先遺伝子のヒト細胞(HEK293)における発現(図6)
カイアシ類由来ルシフェラーゼの祖先遺伝子のヒト細胞による発現
Metridinidae科、Lucicutiidae科およびHeterorhabdidae科プランクトン由来ルシフェラーゼのアミノ酸配列の解析により得られたカイアシ類ルシフェラーゼの祖先遺伝子aCopLuc43mutおよびaCopLuc48のアミノ酸配列をコードする塩基配列を、ほ乳類培養細胞発現用ベクターpcDNA3.2 V5-His TOPO中へ挿入する。精製済みのヒト細胞発現ベクターを、ヒト培養細胞HEK293に遺伝子導入・発現させ、祖先遺伝子の発光活性を測定した。100 mm dishにてHEK293細胞を培養した。カイアシ類由来ルシフェラーゼの祖先遺伝子の発現ベクターのLipofectamine 2000 (Invitrogen社製)によるトランスフェクション処理後、細胞は5%CO2下、37℃にて72時間インキュベーションし、導入遺伝子の発現を行った。この結果、細胞内画分において発光活性が認められた(図6)。ルシフェラーゼ活性の測定法は、後述する「カイアシ類由来ルシフェラーゼ活性の測定」に記載した。
【0057】
カイアシ類由来ルシフェラーゼ活性の測定
カイアシ類ルシフェラーゼの活性測定には以下の方法を用いた。1μg/μLに調製した発光基質セレンテラジン(coelenterazine)のメタノール溶液より1 μLをとって、1 mLの基質希釈用バッファー(20 mM Tris-HCl, pH8.0, 50 mM MgCl2)で希釈した。希釈後の発光基質液は氷上に保持した。カイアシ類ルシフェラーゼを発現するプラスミドベクターを導入されたHEK293細胞からの培養上清または大腸菌破砕液5 μLをポリスチレン製テストチューブに分取して、ルミノメーターLB9506(ベルトールド社製)の測定部にセットし、10 μLの上記希釈発光基質を添加した後、直ちに測定を開始した。発光の測定は10秒間行った。発光が強すぎて測定限界を超えた場合は、試料希釈バッファー(20 mM Tris-HCl, pH8.0, 10 mM MgCl2)にて10倍希釈を行って、同様に測定した。得られた発光値をルミノメーターに附属のプリンターに出力した後、次の試料の測定に移った。
【産業上の利用可能性】
【0058】
本発明にかかるカイアシ類由来のルシフェラーゼとその祖先ルシフェラーゼのタンパク質をコードする遺伝子とは、いずれも、哺乳動物細胞を利用する培養系において、該宿主細胞内で発現可能であり、かつ、かかる遺伝子から発現された組換え発現型ルシフェラーゼタンパク質は、宿主細胞外へと分泌され、または祖先ルシフェラーゼは細胞内に発現するレポータータンパク質として利用することが可能である。
【特許請求の範囲】
【請求項1】
配列番号22若しくは配列番号24のアミノ酸配列、または、1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列を有することを特徴とする発光タンパク質。
【請求項2】
配列番号1〜21のいずれかアミノ酸配列を有する請求項1に記載の発光タンパク質。
【請求項3】
請求項1または2に記載の発光タンパク質をコードするDNAまたはその相補鎖。
【請求項1】
配列番号22若しくは配列番号24のアミノ酸配列、または、1又は複数のアミノ酸が置換、付加、欠失又は挿入された改変アミノ酸配列を有することを特徴とする発光タンパク質。
【請求項2】
配列番号1〜21のいずれかアミノ酸配列を有する請求項1に記載の発光タンパク質。
【請求項3】
請求項1または2に記載の発光タンパク質をコードするDNAまたはその相補鎖。
【図1】


【図2】

【図3】

【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−249619(P2012−249619A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2011−126900(P2011−126900)
【出願日】平成23年6月7日(2011.6.7)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成23年6月7日(2011.6.7)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]