
液晶性有機半導体ポリマー、その製造方法及び有機ナノポーラス材料
【課題】有機FETなどの有機デバイスとしての用途に有用な有機ナノポーラス材料を作製し得るブロック共重合体を提供する。
【解決手段】一般式(1)で表されるカルボン酸基含有重合体における任意の−COOH基に、特定の水酸基含有オリゴチオフェン誘導体をエステル化反応させてなるブロック共重合体単独、又は該ブロック共重合体と、下記一般式(1)で表されるカルボン酸基含有重合体とを含み、ブロック共重合体におけるオリゴチオフェン含有エステル基と、ブロック共重合体及びカルボン酸基含有重合体の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー。

【解決手段】一般式(1)で表されるカルボン酸基含有重合体における任意の−COOH基に、特定の水酸基含有オリゴチオフェン誘導体をエステル化反応させてなるブロック共重合体単独、又は該ブロック共重合体と、下記一般式(1)で表されるカルボン酸基含有重合体とを含み、ブロック共重合体におけるオリゴチオフェン含有エステル基と、ブロック共重合体及びカルボン酸基含有重合体の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、液晶性有機半導体ポリマー、その製造方法、有機ナノポーラス材料及び有機電界効果トランジスターに関する。さらに詳しくは、本発明は、分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成し得る特定構造のブロック共重合体からなる液晶性有機半導体ポリマー、その効果的な製造方法、前記液晶性有機半導体ポリマーを用いて得られた、有機電界効果トランジスター(以下、有機FETと略記することがある。)などの有機デバイスとしての用途に有用な有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETに関するものである。
【背景技術】
【0002】
近年、ナノメートルオーダーの均一サイズを有する細孔(ポア)が規則的に並んだナノポーラス材料が注目されており、より規則性の高い有機ナノポーラス材料を得ることは、この分野において課題であった。
非特許文献1には、ポリスチレンブロックの連続相と、親水性ブロックの非連続相からなるミクロ相分離構造を有し、光開裂型開始剤を含有する組成物に、紫外線(UV)を照射し、ポリスチレンブロックと親水性ブロックとの間のエステル結合を加水分解し、リンス処理を行うことで親水性ブロックを除去し、有機ナノポーラス材料を得る方法が報告されている。
【0003】
また、非特許文献2及び3には、液晶性のブロックを有するブロック共重合体を用いることで、規則性の高いミクロ相分離構造を形成しうることが示されている。
前記非特許文献2には、水滴で開けた孔(Figure2、ミクロ相分離ではない)と、相分離構造(Figure6、ミクロ相分離)が入り混じった構造が示されており、また、非特許文献3には、液晶性のアゾベンゼンとポリエチレンオキシド(PEO)のミクロ相分離構造を得ることが示されている。しかしながら、これらの文献には、ナノポアの形成については何の示唆もされてない。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】B.Moonら,Macromolecules 2009年,42巻,455ページ
【非特許文献2】:T.Hayakawaら,Angrew.Chem.Int.Ed,2003年,42巻,2285ページ
【非特許文献3】:T.Iyodaら,Adv.Mater.2007年,19巻,1267ページ
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、このような状況下になされたもので、有機FETなどの有機デバイスとしての用途に有用な有機ナノポーラス材料を作製し得るブロック共重合体、その効果的な製造方法、前記ブロック共重合体を用いて得られた有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETを提供することを目的とするものである。
【課題を解決するための手段】
【0006】
本発明者らは、前記目的を達成するために鋭意研究を重ねた結果、下記の知見を得た。
分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有する特定構造のペンダント型ブロック共重合体からなる、液晶性有機半導体ポリマー組成物に、所定の処理を施すことにより、規則性の高い液晶性の配向を有する有機ナノポーラス材料が得られること、そして、前記液晶性有機半導体ポリマー組成物は、特定の工程を施すことにより、効率よく製造し得ることを見出した。本発明は、かかる知見に基づいて完成したものである。
【0007】
すなわち、本発明は、
[1]下記一般式(1)で表されるカルボン酸基含有重合体(B)における任意の−COOH基に、下記一般式(2)で表されるオリゴチオフェン誘導体(C)をエステル化反応させてなるブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含み、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー組成物、
【0008】
【化1】
【0009】
(一般式(1)中、R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基、Xは親水基、Zはハロゲン原子、mは2〜500の整数、pは0又は1を示す。)
【0010】
【化2】
(一般式(2)中、R4及びR5は、それぞれ独立に水素原子又は炭素数1〜3のアルキル基、Xは親水性基、Yは炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基、Aは炭素数2〜16のアルカンジイル基、kは2〜8の整数、nは0又は1を示す。)
【0011】
[2]一般式(1)におけるXで示される親水性基が、ポリエーテル残基である上記[1]に記載の液晶性有機半導体ポリマー組成物、
[3]上記[1]又は[2]に記載の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする有機ナノポーラス材料、
[4]電磁波照射処理が、紫外線照射処理である上記[3]に記載の有機ナノポーラス材料、
[5]紫外線の波長領域が、ブロック共重合体(A)の分子構造中に含まれるo−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェンエステル基の極大吸収波長を実質上含まない上記[4]に記載の有機ナノポーラス材料、
[6]上記[3]〜[5]のいずれかに記載の有機ナノポーラス材料を用いることを特徴とする有機電界効果トランジスター、
[7]以下の工程を有する上記[1]又は[2]に記載の液晶性有機半導体ポリマー組成物の製造方法、
工程(a)一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程。
工程(b)o−ニトロベンジル基の2〜5位のいずれかに親水性基の結合した官能基を有する原子移動ラジカル重合制御剤を用いて、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合法により重合することにより、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体を製造する工程。
工程(c)前記工程(a)で得られたオリゴチオフェン誘導体と、前記工程(b)で得られたカルボン酸基含有重合体とをエステル化反応させる工程。
[8]工程(b)におけるカルボン酸基1個以上を有するビニル化合物単量体が、(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸又はシトラコン酸である上記[7]に記載の液晶性有機半導体ポリマーの製造方法、及び
[9]ビニル化合物単量体が、(メタ)アクリル酸である上記[8]に記載の液晶性有機半導体ポリマーの製造方法、
を提供するものである。
【発明の効果】
【0012】
本発明によれば、分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成し得る特定構造のブロック共重合体からなる液晶性有機半導体ポリマー組成物、その効果的な製造方法、前記液晶性有機半導体ポリマー組成物を用いて得られた、有機FETなどの有機デバイスとしての用途に有用な有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETを提供することができる。
【図面の簡単な説明】
【0013】
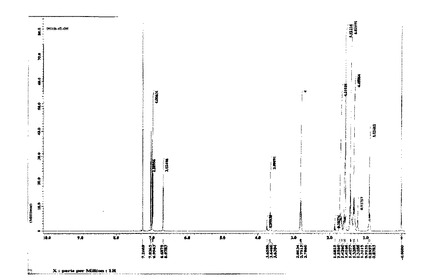
【図1】実施例1で得られた4T−OHの1H−NMRチャートである。
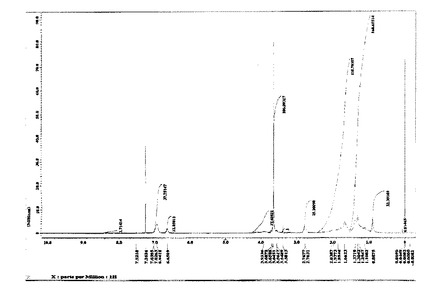
【図2】実施例1で得られた重合体(B’−6)の1H−NMRチャートである。
【図3】実施例1で得られたブロックコポリマー(BCP1)の1H−NMRチャートである。
【図4】実施例4で得られたナノポーラス薄膜のSEM写真である。
【図5】比較例1で得られた薄膜のSEM写真である。
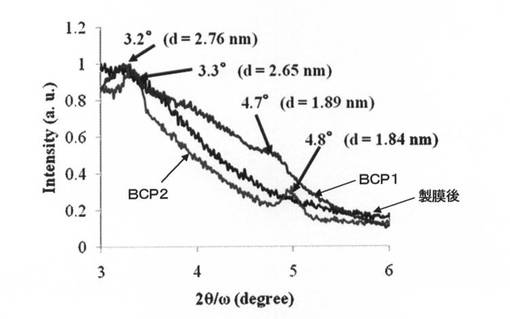
【図6】実施例4及び5で得られた各薄膜のXRDチャートである。
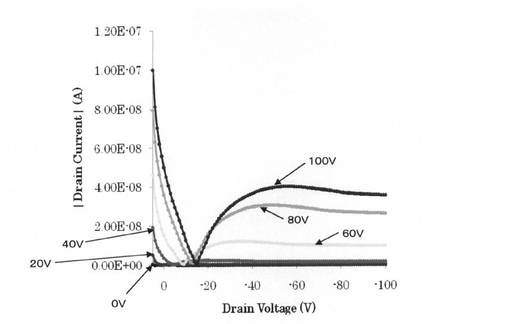
【図7】実施例4で得られた有機FETのId−Vd曲線である。
【発明を実施するための形態】
【0014】
まず、本発明の液晶性有機半導体ポリマー組成物について説明する。
[液晶性有機半導体ポリマー組成物]
本発明の液晶性有機半導体ポリマー組成物は、ブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含むことを特徴とする。
【0015】
(ブロック共重合体(A))
本発明の液晶性有機半導体ポリマー組成物において、成分(A)として用いられるブロック共重合体は、下記一般式(1)
【0016】
【化3】
【0017】
で表される共重合体における任意の−COOH基に、下記一般式(2)
【0018】
【化4】
【0019】
で表されるオリゴチオフェン誘導体(C)がエステル結合している構造を有する。
なお、前記一般式(1)中、
R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基を示す。
本発明において、オリゴチオフェン縮合重合体構造とは、前記一般式(1)中の任意の−COOH基に前記一般式(2)のオリゴチオフェン誘導体(C)がエステル結合したm個の繰り返し単位構造をいう。また、オリゴチオフェン含有エステル基とは、オリゴチオフェン誘導体(C)がエステル結合した−COOH基のことをいう。
【0020】
一般式(1)におけるXは親水性基を示す。なお、本発明における親水性基とは、ブロック共重合体(A)が親水性基とオリゴチオフェン縮合重合体構造との親水性の差により、ミクロ相分離構造を形成しうる程度に、かつ後述する電離放射線照射後の水及び/又は低級アルコールによるリンス処理により除去可能な程度に親水性を有することを指す。当該親水性基は、効果的にオリゴチオフェン縮合重合体構造とミクロ相分離を形成し、また、上記リンス処理により除去されうる。
【0021】
このような親水性基としては、親水性ポリマー残基であることが好ましく、ポリエーテル残基、ポリエーテルポリウレタン残基、ポリ(メタ)アクリル酸(又はその塩)残基、ポリ(メタ)アクリルアミド誘導体残基、ポリビニルアルコール残基、セルロースアルキルエステル残基等の1種または2種以上の組み合わせが挙げられる。とりわけ、後述のリンス処理による除去の容易性から、ポリエーテル残基が好ましい。ここで、ポリエーテル残基は、ポリエチレングリコール残基、ポリプロピレングリコール残基のようなポリアルキレンエーテル残基であってもよく、ポリグリセリン残基のように側鎖に水酸基を有するものであってもよい。
【0022】
当該親水性基は、親水性の発現及び後述の形成される有機ナノポーラス材料のポアサイズの観点から、重合度(単量体単位数)が2〜500程度、好ましくは100〜300の親水性ポリマー残基であることが望ましい。
親水性基Xは、酸素原子を介して、又は介さずに、o−ニトロベンジロキシカルボニル基のベンゼン環に結合しており、上記o−ニトロベンジロキシカルボニル基におけるo−ニトロベンジル基は、後述の電磁波を照射する際に、光開裂型開始機能を発現し、上記の親水性基が結合したo−ニトロベンジロキシカルボニル基のエステル結合部が容易に加水分解される。
【0023】
また、前記一般式(1)において、Zはハロゲン原子を示す。ハロゲン原子としては、フッ素原子、塩素原子、臭素原子及びヨウ素原子を挙げることができるが、これらの中で、特に臭素原子が好適である。
【0024】
ブロック共重合体(A)は、上述のオリゴチオフェン縮合重合体構造のオリゴチオフェン構造の配向性により、親水性基とのミクロ相分離構造をより規則性高く形成でき、また後述のように、オリゴチオフェン縮合重合体構造の連続相と、親水性基の非連続相の間に存在するエステル結合を電磁波照射により光開裂させて、その後のリンス処理により親水性基を除去することによってナノポーラスを形成することができる。
親水性基が親水性ポリマー残基である場合には、ブロック共重合体(A)における親水セポリマー残基の数平均分子量Mnと、オリゴチオフェン縮合重合体構造の比率は、親水性ポリマー残基の数平均分思慮Mn:オリゴチオフェン縮合体構造のMnとして、15:85〜40:60であることが好ましい。親水性ポリマー残基のMnが15:85以上であれば、ミクロ相分離の形成が得やすくなる。一方、40:60以下であると、ラメラ構造を形成しにくくなり、オリゴチオフェン縮合重合体構造の連続相を形成しやすくなる。
【0025】
一般式(1)におけるmはカルボン酸基1個以上を有するビニル化合物単量体の重合度を示し、2〜500の整数である。またpは0又は1を示す。
【0026】
ブロック共重合体(A)の数平均分子量は、5,000〜50,000であることが好ましい。5,000以上であると材料が半固体になることがないので、構造体の維持が容易となり、50,000以下であると材料の溶解性が低下することがない。より好ましくは、10,000〜30,000であり、さらに好ましくは、12,000〜25,000である。
【0027】
一方、前記一般式(2)において、R4及びR5はそれぞれ独立に水素原子又は炭素数1〜3のアルキル基を示す。炭素数1〜3のアルキル基としては、メチル基、エチル基、n−プロピル基及びイソプロピル基が挙げられる。
Yは、炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基を示す。炭素数1〜16のアルキル基は直鎖状、分岐状のいずれであってもよく、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、各種ペンチル基、各種ヘキシル基、各種オクチル基、各種デシル基、各種ドデシル基、各種テトラデシル基、各種ヘキサデシル基などを挙げることができる。
【0028】
炭素数1〜16のアルコキシ基を構成するアルキル基は直鎖状、分岐状のいずれであってもよく、炭素数1〜16のアルコキシ基としては、例えばメトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、各種ペントキシ基、各種ヘキソキシ基、各種オクトキシ基、各種デシロキシ基、各種ドデシロキシ基、各種テトラデシロキシ基、各種ヘキサデシロキシ基などを挙げることができる。
【0029】
また、炭素数2〜17のアルコキシカルボニル基を構成するアルキル基は直鎖状、分岐状のいずれであってもよく、炭素数2〜17のアルコキシカルボニル基としては、例えばメトキシカルボニル基、エトキシカルボニル基、n−プロポキシカルボニル基、イソプロポキシカルボニル基、n−ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基、各種ペントキシカルボニル基、各種ヘキソキシカルボニル基、各種オクトキシカルボニル基、各種デシロキシカルボニル基、各種ドデシロキシカルボニル基、各種テトラデシロキシカルボニル基、各種ヘキサデシロキシカルボニル基などを挙げることができる。
【0030】
前記一般式(2)におけるAは、炭素数2〜16のアルカンジイル基を示し、直鎖状、分岐状のいずれであってもよく、例えば各種エタンジイル基、各種プロパンジイル基、各種ブタンジイル基、各種ペンタンジイル基、各種ヘキサンジイル基、各種オクタンジイル基、各種デカンジイル基、各種ドデカンジイル基、各種テトラデカンジイル基、各種ヘキサデカンジイル基などを挙げることができる。
kはオリゴチオフェンの重合数を示し、2〜8の整数、nは0又は1を示す。
【0031】
ブロック共重合体(A)においては、チオフェン構造の繰り返しにより、隣接する単量体単位と結合したオリゴチオフェン構造同士が、π-電子の相互作用によるいわゆるπ-πスタッキングを生じるために配向を生じやすく、効果的に、規則性の高いミクロ相分離構造を形成することができる。
オリゴチオフェンを形成するチオフェン構造単位の繰り返し数は、2単位〜8単位であることを要する。1単位である場合には、上述のスタッキングを生じることが難しく、9単位以上では、結晶性が高くなり取り扱い性が悪くなる。チオフェン構造の繰り返しは、2単位〜6単位であることがより好ましく、3単位〜5単位であることがさらに好ましい。
カルボン酸基含有重合体の単量体単位の2以上の単位にオリゴチオフェン化合物を縮合させることで、オリゴチオフェン縮合重合体構造を得ることができる。この縮合反応は、ジシクロヘキシルカルボジイミド等の公知の縮合剤を用いて行うことができる。
なお、ブロック共重合体(A)は、後述する本発明の液晶性有機半導体ポリマー組成物の製造方法によって、効率よく製造することができる。
【0032】
(カルボン酸基含有重合体(B))
本発明の液晶性有機半導体ポリマー組成物において、成分(B)として用いられるカルボン酸基含有重合体は、下記一般式(1)
【0033】
【化5】
【0034】
で表される構造を有している。
前記一般式(1)において、X、Z、R1〜R3、m及びpは、前述の通りである。
本発明の液晶性有機半導体ポリマー組成物は、ブロック共重合体(A)単独、又は該ブロック共重合体(A)と、前記一般式(1)で表されるカルボン酸基含有重合体(B)とを含むものであって、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸基との割合は、100:0〜35超:65未満(モル比)であることが好ましく、100:0〜45:55(モル比)であることがさらに好ましい。前記割合が上記範囲内であれば、本発明の液晶製有機半導体ポリマー組成物は液晶性が発現されやすくなる。
【0035】
次に、本発明の液晶性有機半導体ポリマー組成物の製造方法について説明する。
[液晶性有機半導体ポリマー組成物の製造方法]
本発明の液晶性有機半導体ポリマー組成物の製造方法(以下、単に製造方法と略記することがある。)は、下記に示す工程(a)、工程(b)及び工程(c)を有することを特徴とする。
【0036】
(工程(a))
本発明の製造方法における工程(a)は、一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程である。
すなわち、下記一般式(2)
【0037】
【化6】
【0038】
(一般式(2)中、A、R4、R5、Y、n及びkは、前記と同じである。)
で表されるオリゴチオフェン誘導体(C)を製造する工程である。
このオリゴチオフェン誘導体(C)は、例えば、下記の反応式(a)によって製造することができる。
【0039】
【化7】
【0040】
上記反応式(a)は、得られるオリゴチオフェンが4量体であって、R4及びR5がいずれも水素原子、n=0、−A−OHが−(CH2)6−OH、Yが−C6H13、kが4の例である。
まず、2,2’−ビチオフェン(C−1)に、n−ブチルリチウムの存在下に、水酸基がテトラヒドロ−2H−ピラン(THP)で保護された6−ブロモヘキシルアルコール(C−2)を反応させて、水酸基がTHPで保護された6−(2,2’−ビチオフェン−5−イル)へキシルアルコール(C−3)を合成する。
次いで、上記の水酸基がTHPで保護された6−(2,2’−ビチオフェン−5−イル)へキシルアルコール(C−3)に、ブロム化剤のNBS(N−ブロモスクシンイミド)を反応させて、水酸基がTHPで保護された6−(5’−ブロモ−2,2’−ビチオフェン−5−イル)ヘキシルアルコール(C−4)を合成する。
次に、この水酸基がTHPで保護された6−(5’−ブロモ−2,2’−ビチオフェン−5−イル)ヘキシルアルコール(C−4)に、(5’−ヘキシル−2,2’−ビチオフェン−5−イル)ボロン酸ピナコールエステル(C−5)を、テトラキス(トリフェニルホスフィン)パラジウム[Pd(PPh3)4]の存在下反応させることにより、水酸基がTHPで保護された6−(5’−ヘキシル−2,2’:5,2’’:5’’,2’’’−クォータチオフェン−5’’’−イル)ヘキシルアルコール(C−6)を合成する。
最後に、該化合物(C−6)にp−トルエンスルホン酸を作用させることにより、チオフェン環が4個結合したオリゴチオフェンの片末端のチオフェン環の5位にヘキサノールを結合すると共に、他方の末端のチオフェン環の5位にヘキシル基が結合してなる、前記一般式(2)で表されるオリゴチオフェン誘導体の一例である(4T−OH)が得られる。
当該工程(a)においては、前記の反応式(a)で示すような方法により、片末端のチオフェン環の5位にヒドロキシアルキル基を有する一般式(2)で表されるオリゴチオフェン誘導体(C)が得られる。
【0041】
(工程(b))
本発明の製造方法における工程(b)は、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合により、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)を製造する工程である。
<原子移動ラジカル重合>
原子移動ラジカル重合(ATRP)は、成長しているポリマー鎖と重合制御剤との間の不安定なラジカルが容易に移動することを媒介して、リビングラジカル重合を行う触媒可逆的レドックス法である。
この原子移動ラジカル重合制御剤としては、例えば有機ハロゲン化物、又は有機ハロゲン化合物と遷移金属錯体との組合わせを用いることができる。上記有機ハロゲン化物としては、特に反応性の高い炭素−ハロゲン結合を有する化合物(例えばα位にハロゲンを有するカルボニル化合物や、ベンジル位にハロゲンを有する化合物など)、あるいはハロゲン化スルホニル化合物などが好適である。
一方、遷移金属錯体としては、特に制限はないが、好ましくは周期表第7〜11族に属する元素を中心金属とする金属錯体であり、より好ましくは0価又は1価の銅、2価のルテニウム、2価の鉄、2価のニッケルの錯体であり、特に好ましくは銅の錯体である。
このATRP法によれば、得られるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)は、分子量の制御が容易であると共に、重量平均分子量/数平均分子量(Mw/Mn)比が極めて小さい、例えば1.3以下程度となる。
前記一般式(2)で表されるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)は、例えば以下に示す反応式(b)によって製造することができる。
【0042】
【化8】
【0043】
(反応式(b)中、Rは炭素数1〜5の低級アルキル基であり、R1〜R3、X、Z、m及びpは前記と同じである。)
まず、親水性基を有するo−ニトロベンジルアルコール(B−1)に、α−ハロゲノイソ酪酸ハライド(B−2)を、メチレンジクロリドなどの溶媒中において、三級アミンなどの存在下に反応させて、α−ハロゲノイソ酪酸エステル化合物(B−3)を合成する。次いで、このα−ハロゲノイソ酪酸エステル化合物(B−3)の存在下、カルボン酸基1個以上を有するビニル化合物単量体(B−4)をATRP法により重合させて、ポリマーの側鎖にエステル結合を有する重合体(B−5)を合成する。このATRP法による重合においては、溶媒としてアニソールを用いることが好ましく、また、リガンドとして、N,N,N’,N’’,N’’−ペンタメチルジエチレントリアミン(PMDTA)を用いることが好ましい。反応温度は、通常60〜120℃程度である。
最後にこの重合体(B−5)を加水分解(脱保護)することにより、一般式(1)で表されるポリマーの側鎖にカルボン酸基含有重合体(B)(この場合は、B−6とする。)が得られる。
【0044】
前記のカルボン酸基1個以上を有するビニル化合物単量体としては、例えば(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸、シトラコン酸などを用いることができるが、これらの中で(メタ)アクリル酸が好適である。ここで、(メタ)アクリル酸は、アクリル酸又はメタクリル酸を示す。
前記ビニル化合物単量体がアクリル酸である場合には、一般式(1)の重合体(B)におけるR1〜R3は全て水素原子であり、メタクリル酸である場合には、R1はメチル基、R2及びR3は水素原子である。マレイン酸やフマル酸の場合には、R1及びR2は水素原子であり、R3は−COOHである。イタコン酸である場合には、R1は−CH2COOHであり、R2及びR3は水素原子である。シトラコン酸である場合には、R1は水素原子、R2はメチル基及びR3は−COOHである。
なお、ATRP法による重合反応時には、各カルボン酸基は低級アルキルエステルの形態であることが好ましく、このエステルは、最終工程での加水分解反応(脱保護反応)により、遊離のカルボン酸基となる。本発明におけるカルボン酸基は、カルボン酸低級アルキルエステル基及び遊離のカルボン酸基(−COOH基)のいずれをも含む広い意味である。
【0045】
(工程(c))
本発明の製造方法における工程(c)は、前記工程(a)で得られた一般式(2)で表されるオリゴチオフェン誘導体(C)と、前記工程(b)で得られた一般式(1)で表されるカルボン酸基含有重合体(B)とをエステル化反応させる工程である。
この工程(c)における反応を、下記の反応式(c)で示す。
【0046】
【化9】
【0047】
(反応式(c)中、R1〜R5、A、X、Y、Z、k、m、n及びpは、前記と同じである。)
ジメチルホルムアミドなどの溶媒中において、ジシクロヘキシルカルボジイミドや4−ジメチルアミノピリジンなどの縮合剤の存在下に、一般式(1)で表されるカルボン酸基含有重合体(B)と、一般式(2)で表されるオリゴチオフェン誘導体(C)とを、60〜120℃程度の温度で縮合させることにより、ブロック共重合体(A)が得られる。
この場合、任意のカルボン酸基含有重合体(B)の単量体単位に、オリゴチオフェン誘導体(C)を縮合させることにより、ペンダント型ブロック重合体を得ることができる。
【0048】
また、カルボン酸基含有重合体のカルボン酸基のうち、35%(官能基の数の割合)よりも大きい比率で、オリゴチオフェン誘導体と縮合されることを要する。35%以下である場合には、液晶性を呈しにくい。カルボン酸基は、50%よりも大きい比率でオリゴチオフェン誘導体と縮合されることがより好ましく、70%よりも大きい比率で縮合されていることがさらに好ましい。
ペンダント型ブロック共重合体の数平均分子量は、5,000〜50,000であることが好ましい。5,000未満であると材料が半固体になってしまい、構造体の維持が困難になってしまう、50,000を超えると材料の溶解性が低下してしまう、より好ましくは、10,000〜30,000であり、さらに好ましくは、12,000〜25,000である。
なお、上記数平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)法で測定される標準ポリスチレン換算の値である。
【0049】
次に、本発明の有機ナノポーラス材料について説明する。
[有機ナノポーラス材料]
本発明の有機ナノポーラス材料は、前述した本発明の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理、ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする。
前述した本発明の液晶性有機半導体ポリマー組成物は、分子内に有機半導体部位と、親水性基(ポリマー型)を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成するペンダント型ブロック共重合体である。
このような性状を有する液晶性有機半導体ポリマー組成物にアニール処理を施すことにより、オリゴチオフェン縮合重合体構造の連続相と、親水性基(ポリマー型)の非連続相からなるミクロ相分離構造を誘起させることができ、これに電磁波を照射することにより、親水性基(ポリマー型)が結合してなるo−ニトロベンジル基の光開裂型開始機能により、該o−ニトロベンジル基に隣接するエステル結合が切断される。したがって、これに水及び/又は低級アルコールによるリンス処理を施すことによって、親水性基を有する部位は除去されるので、有機ナノポーラス材料が形成される。
【0050】
(アニール処理)
アニール処理は、前述したように、オリゴチオフェン縮合重合体構造の連続相と、親水性基(ポリマー型)の非連続相からなるミクロ相分離構造を誘起させるための処理である。
アニール処理は、高温雰囲気下に一定時間留置する熱アニール処理や、溶媒濃度の高い雰囲気下に一定時間留置するソルベントアニール処理が挙げられる。
【0051】
熱アニール処理は、液晶性有機半導体ポリマー組成物の液晶温度範囲内の環境に液晶性有機半導体ポリマー組成物を留置することにより行うことができる。なお、上記の液晶温度範囲とは、液晶性物質の、結晶相と液晶相の転移温度を下限とし、液晶相と等方性液体相の転移温度を上限とする範囲を意味する。本発明の有機半導体ポリマー組成物では、結晶相と液晶相の転移温度は通常40〜250℃であり、液晶相と等方性液体相の転移温度は通常100〜400℃である。これらの転移温度は示差熱分析測定により決定できる。熱アニール処理は、5分〜96時間の間で行うことが好ましい。
【0052】
ソルベントアニール処理の方法は、密閉された系内に溶媒と液晶性有機半導体ポリマー組成物を留置することにより行うことができる。
このソルベントアニール処理に用いる溶媒は、オリゴチオフェン縮合重合体構造、親水性基(ポリマー型)のいずれかの良溶媒又はそれらの混合溶媒を用いるのがよい。具体的にはアセトン、アセトニトリル、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ジクロロメタン、クロロホルム、四塩化炭素、THF、ジオキサン、二硫化炭素、シクロヘキサノン、シクロヘキサン、酢酸エチル、ジメチルスルホキシド、ジメチルホルムアミドなどが好適に用いられ、中でも、ベンゼン、ジオキサン、アセトンが特に好ましい。
【0053】
処理方法として室温あるいは使用溶媒の沸点付近まで加熱を行い、液晶性有機半導体ポリマー組成物の処理雰囲気の溶媒蒸気濃度を高めることが望ましい。ソルベントアニール処理を行う時間は、5分〜96時間であることが好ましい。
また、上記した熱アニール処理、ソルベントアニール処理のうち、より規則正しいミクロ相分離構造が得られやすいため、ソルベントアニール処理がより好ましい。
【0054】
(電磁波照射処理)
本発明においては、前述したようなアニール処理により、ミクロ相分離構造を誘起させた液晶性有機半導体ポリマー組成物に、電磁波照射処理を施す。
当該液晶性有機半導体ポリマー組成物は、分子内に光開裂開始機能をもつo−ニトロベンジル基を有することから、電磁波照射により、該o−ニトロベンジル基が開裂し、発生したラジカルによって、下記の反応式(d)で示すように、該o−ニトロベンジル基に隣接するエステル結合が効果的に光開裂され、親水性化合物(D−1)とオリゴチオフェン縮合重合体(D−2)が生成する。
【0055】
【化10】
【0056】
(反応式(d)中、R1〜R5、A、X、Y、Z、k、m、n及びpは、前記と同じである。)
なお、光開裂型開始機能をもつ基としては、前記のo−ニトロベンジル基以外に、ジアリルメチルエステル残基などがある。
電磁波照射としては、可視光線、紫外線(UV)、X線、ガンマ線等の照射が挙げられるが、当該エステル結合に与えるエネルギーの大きさの妥当性の面から、UV照射であることが好ましい。
【0057】
一方で、UVの照射により、オリゴチオフェン縮合重合体構造の分解が引き起こされる可能性がある。オリゴチオフェン縮合重合体構造の分解が起こると、分解物であるオリゴチオフェン化合物が、リンス処理において除去されてしまうために、ポーラスのサイズが大きくなり、有機ナノポーラス材料のポアのサイズを制御する上で好ましくない。
オリゴチオフェン縮合重合体構造の不本意な分解を防ぐためには、UVにフィルタリングを行うことが好ましい。より好ましくは、UVは、o−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない波長選択UVであることが好ましい。このようなUVの照射は、一定の波長領域のみを透過するバンドパスフィルターを用いた選択的照射により可能であり、オリゴチオフェン縮合重合体構造の分解が効果的に防止できる。
【0058】
(リンス処理)
本発明においては、前述のようにして、アニール処理及び電磁波照射処理が施された液晶性有機半導体ポリマー組成物に、水及び/又は低級アルコール(メタノール、エタノール、プロパノール、イソプロパノールなど)によるリンス処理を施し、前記反応式(d)における親水性化合物(D−1)を除去することによって、有機ナノポーラス材料を形成することができる。
【0059】
(有機ナノポーラス材料の作製)
本発明の有機ナノポーラス材料の具体的な作製方法ついて説明する。
まず、ガラス基板やSiO2基板などの適当な基板上に、クロロホルムなどの溶媒中に、本発明の液晶性有機半導体ポリマー組成物を含む塗工液をスピンコート法などにより塗工し、乾燥して、厚さ20〜400nm程度の薄膜を形成させる。次いで、密封可能なガラス容器内で、適当な溶媒を用いてソルベントアニール処理を施し、ミクロ相分離構造を誘起させる。ソルベントアニール処理の条件は、使用する溶媒の種類に応じて、適宜選択する。
次に、一定の波長領域のみを透過するバンドパスフィルターを用いて、紫外線をソルベントアニール処理された薄膜に照射する。なお、上記一定の波長領域とは、当該液晶性有機半導体ポリマー組成物における、o−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない波長領域を指す。
最後に、UV照射された薄膜に、水及び/又は低級アルコール(メタノール、エタノール、プロパノール、イソプロパノールなど)によるリンス処理を施すことにより、本発明の有機ナノポーラス材料を作製することができる。
このようにして得た有機ナノポーラス材料は、液晶性に起因して、オリゴチオフェン縮合重合体のオリゴチオフェン構造が配向している。
配向を有する有機ナノポーラス材料は、有機FETや、有機薄膜太陽電池の材料などとしての利用が期待される。
【0060】
次に、本発明の有機電界効果トランジスター(FET)について説明する。
[有機電界効果トランジスター(FET)]
本発明の有機FETは、前述した本発明の有機ナノポーラス材料を用いることを特徴とする。
薄膜トランジスター(TFT)は、エレクトロニクスにおけるスイッチ素子として広く使用されており、とりわけアクティブ・マトリックス型液晶表示装置やスマート・カードなど、広範囲にわたる適用分野で使用されている。薄膜トランジスター(TFT)の殆どは、電界効果トランジスター(FET)である。
現在、殆どのTFTデバイスは、半導体材料としてアモルファスシリコンを使用して作製されている。しかしながら、アモルファスシリコンTFTの製造にはプラズマ強化化学気相成長法などの高コストの装置が必要であり、そのプロセスも真空中、高温(約360℃)で行わねばならず、高コストであるのに加え、フレキシブルプラスチック基板を用いることが難しいという問題がある。
したがって、TFTやFETの半導体材料として、有機半導体材料が注目されている。
【0061】
本発明の有機ナノポーラス材料を有機FETとして用いる場合の利点は以下の通りである。すなわち、本来ポーラス構造を有する故にポーラス構造を有しない場合に比べて移動度の大幅な低下が懸念されるにも関らず、配向を生じていることにより、ポーラス構造体が明確に形成されており、且つ導電性部位であるオリゴチオフェン部位が移動度向上に有利な基板材料に対して垂直方向に高く配向しているため、有機FETとして利用可能な程度の移動度を呈する。さらに、ポアのサイズを変えることによって、配向の程度が変わり、移動度を変えることができるので、移動度を精密に制御できる可能性がある。また、単にミクロ相分離したブロック共重合体を用いる場合には、非導電性部位となる非液晶性ブロックに起因する問題、例えば吸湿性等、を生じるおそれがあるが、有機ナノポーラス材料においては、非導電性部位は空隙であるため、そのような問題を避けることができる。
【実施例】
【0062】
次に、本発明を実施例により、さらに詳細に説明するが、本発明はこれらの例によってなんら限定されるものではない。
実施例1 ブロックコポリマー(BCP1)の製造
(1)4T−OHの合成
反応式(a)に従って、4T−OH[6−(5’−ヘキシル−2,2’:5,2’’:5’’,2’’’−クォーターチオフェン−5’’’−イル)ヘキシルアルコール]を合成する。
(イ)化合物(C−2)の合成
500mL三口フラスコに6−ブロモ−1−ヘキサノール20g(東京化成製、0.11mol)とパラトルエンスルホン酸一水和物2.8g(東京化成製、0.017mol)、テトラヒドロフラン(THF)200mLを加えた。そこに3,4−ジヒドロ−2H−ピラン13.9g(東京化成製、0.165mol)を室温で滴下し、室温で20時間撹拌を行った。反応終了後、反応液を濃縮し、展開溶媒としてクロロホルム-ヘキサン(質量比2:8)を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い、化合物(C−2)が合成できていることを確認した。収率は80%であった。
なお、1H−NMR装置として、日本電子社製「JNM ECP400」を用いた。以下同様である。
【0063】
(ロ)化合物(C−3)の合成
300mL三口フラスコに2,2’−ビチオフェン(C−1)7g(東京化成製、42.2mmol)を加えた。系内を封止し乾燥窒素チャージと脱気を3回繰り返すことで系内の酸素を除去した。この状態で脱水THF40mLを加え、反応液の温度を-40℃以下に保った状態でn−ブチルリチウム(関東化学製、1.6mol/L、29mL、46.42mmol)加えた。反応液の温度を保持しながら、1時間撹拌しリチオ化を行った。そこに化合物(C−2)をTHF15mLに溶解した溶液を滴下した。滴下終了後、室温に戻して20時間反応を行った。反応を終了させるために、水10mLを加え処理し、酢酸エチル−水で抽出を行った。反応液を濃縮し展開溶媒として酢酸エチル−ヘキサン(質量比1:12)を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い化合物(C−3)が合成できていることを確認した。収率は51%であった。
【0064】
(ハ)化合物(C−4)の合成
化合物(C−3)(2.62g、9.85mmol)を、ジメチルホルムアミド(DMF)70mL溶解し、N−ブロモスクシンイミド(NBS)2.1g (和光純薬製、11.8mmol)を加え24時間遮光下で反応を行った。反応終了後、クロロホルムで抽出を行い、有機層を濃縮し、クロロホルム:ヘキサン(質量比4:6)の展開溶媒を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い、チオフェン基のプロトン積分比の和が4となっていることから、目的の化合物(C−4)が合成できていることを確認した。収率は73%であった。
【0065】
(ニ)化合物(C−6)の合成
化合物(C−4)(2.04g、5.9mmol)および5’−ヘキシル−2,2’−ビチオフェン−5−ボロン酸ピナコールエステル化合物(C−5)(アルドリッチ製、2.78g、7.4mmol)をトルエン25mLに溶解し、エタノール10mL、24質量%炭酸ナトリウム水溶液を加えた。凍結脱気(反応液を液体窒素で凍結し、ポンプにより脱気を行い、脱気後常温に戻す)を2回行った。そこにテトラキス(トリフェニルホスフィン)パラジウム(東京化成製、0.48g、0.41mmol)を加え、窒素雰囲気下で24時間加熱還流を行った。反応終了後、クロロホルムで抽出を行い、有機層を濃縮し、クロロホルム−メタノールで再結晶を行い精製した。1H−NMR測定を行い、チオフェン基のプロトン積分比の総和が8となっていることから、目的の化合物(C−6)ができていることを確認した。収率は51%であった。
【0066】
(ホ)4T−OHの合成
500mL三口フラスコに化合物(C−6)を5.11g(8.53mmol)とメタノール200mLを加えた。そこにp−トルエンスルホン酸一水和物0.294g(1.71mmol)を加え、加熱還流を20時間行った。反応終了後、反応液をろ過し、ろ物をメタノールで洗浄することにより精製を行った。1H−NMR測定により目的の4T−OHができていることを確認した。収率は94%であった。
図1に、4T−OHの1H−NMRチャートを示す。
【0067】
(2)一般式(2)のカルボン酸基含有重合体(B)の合成
反応式(b)に従って、一般式(2)のカルボン酸基含有重合体(B−6)を合成した。
なお、化合物(B−1)として、5位に親水性基であるH3C−(O−CH2CH2)n−O−が結合したo−ニトロベンジルアルコール(化合物(B’−1)を、化合物(B−2)として、α−ブロモイソ酪酸ブロミドを、化合物(B−4)として、tert−ブチルメタクリレートを用いた。すなわち、X:数平均分子量Mnが5,000のポリエチレングリコールメチルエーテル残基、Z:臭素原子、p:1である。
【0068】
(イ)化合物(B’−1)の合成
THF5mLに水素化ナトリウム77mg(3.2mmol)を懸濁させた液に、THF5mL中に5−ヒドロキシ−2−ニトロベンジルアルコール541mg(3.2mmol)を溶解させた溶液を0℃で徐々に加え、0℃にて30分間撹拌した。次いで、これに、THF10mL中にα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド4g(0.8mmol、Mn=5000、分子量5000のモノメチルポリエチレングリコール(Aldrich社製))を用いて、非特許文献1に開示の方法により合成したものである。)を含む溶液を0℃にて徐々に添加した。この溶液を70℃にて一晩撹拌したのち、蒸留水1mLを加えて反応を終了させた。
反応混合物を塩基性アルミナ層を通してろ過し、ろ液をジエチルエーテル中に加え、生成した沈殿物をろ取し、24時間真空乾燥することにより、化合物(B’−1)を得た。収率は88%であった。
【0069】
(ロ)化合物(B’−3)の合成
撹拌機を備えた250mLフラスコに、上記(イ)で得た化合物(B’−1)800mg(0.16mmol)と触媒量の4−ジメチルアミノピリジンの混合物を仕込み、次いで、脱気と窒素ガスチャージを3回繰り返すことで酸素を除去した。このフラスコにシリンジを用いてメチレンジクロリド10mLを加え、前記混合物を溶解させたのち、トリエチルアミン(TEA)0.4mLと、α−ブロモイソ酪酸ブロミド80μL(0.64mmol)を順に加えた。この溶液を室温で20時間撹拌後、メタノール1mLを加えて反応を終了させ、さらにTHFを加えて希釈した。
次いで、反応混合物を塩基性アルミナ層を通してHBr・TEAをろ別したのち、ろ液をジエチルエーテル中に加え、生成した沈殿物をろ取し、一晩真空乾燥することにより、下記式(3)で表される白色固体650mgを得た。収率は81%であった。
この白色固体は、1H−NMR分析により、下記式(3)で表される化合物(B’−3)であることが確認された。
【0070】
【化11】
【0071】
(式(3)中、nは、ポリエチレングリコールメチルエーテル残基のMnが5000となる整数を示す。)
【0072】
(ハ)重合体(B’−5)の合成
30mLのシュレンク管内部から、脱気と窒素チャージを3回繰り返すことで、酸素を除去した。ここに、N,N,N’,N’’,N’’−ペンタメチルジエチレントリアミン(PMDTA)(Aldrich社製、56μL、0.268mmol)、脱水アニソール(Aldrich社製、2.5mL)、tert−ブチルメタクリレート(アルドリッチ製、0.76g、5.36mmol)を、この順序でシリンジを用いて反応容器に注入した。その後、反応混合物内の酸素を除去するために、凍結脱気を2回行った。窒素の流量を強くして臭化銅(I)粉末(アルドリッチ製、19mg、0.134mmol)を加えた。最後に、上記(ロ)で得た化合物(B’−3)0.2g(0.067mmol)を加え容器を封じて70℃で24時間重合を行った。重合終了後、THF:5mLを加え、展開溶媒にTHFを用いたアルミナカラムに通すことで不溶物を取り除いた。溶媒を減圧留去し、ヘキサン溶液で2回再沈澱を行い精製することにより、重合体(B’−5)を得た。
この重合体(B’−5)のGPC法で測定された標準ポリスチレン換算の数平均分子量Mnは10,000であり、分子量分布Mw/Mnは1.21であった。なお、GPC装置として、東ソー株式会社製、機種名「GPC−8020」を用いた。以下、同様である。
【0073】
(ニ)重合体(B’−6)の合成
100mL三口フラスコに上記(ハ)で得られた重合体(B’−5)0.5g(4mmol)とジクロロメタン10.9g(128mmol)を加えた。ここにトリフルオロ酢酸(和光純薬製、5.9g、52mmol)を加え室温で20時間反応させた。反応終了後エーテルに再沈澱させることでポリマーの精製を行うことにより、下記式(4)で表される重合体(B’−6)を得た。
【0074】
【化12】
【0075】
(式(4)中、m’はポリメタクリル酸部位の重合度を示し、nは前記式(3)と同じである。)
この重合体(B’−6)の1H−NMR、GPC測定により、
GPC;Mn=4380、Mw/Mn=1.12
1H−NMR;Mn=4462、末端カルボン酸の個数=17(反応点17個)
であった。
図2に、重合体(B’−6)の1H−NMRチャートを示す。
【0076】
(3)ブロックコポリマー(BCP1)の合成
反応式(c)に従って、ブロックコポリマー(BCP1)を合成した。
なお、一般式(1)で表されるカルボン酸基含有重合体(B)として、式(4)で表される重合体(B’−6)を用い、一般式(2)で表されるオリゴチオフェン誘導体(C)として4T−OHを用いた。
30mLシュレンク管に重合体(B’−6)0.13g(0.01mmol)、4T−OH0.36g(0.69mmol)、N,N−ジメチルアミノピリジン(東京化成製、28mg、0.23mmol)を加え、系内を窒素置換した。そこに脱水DMF4mLを加え、50 ℃に加温し4T−OHを溶解させた。さらにジシクロヘキシルカルボジイミド(東京化成製、0.54g、2.64mmol)を加え80℃に昇温し72時間加熱撹拌を行った。ポリマーの精製は展開溶媒としてTHFを用いBio−Beads S−X1(Bio−Rad製)を用いて行った。
このようにして、下記式(5)で表されるブロックコポリマー(BCP1、ポリエチレングリコールメチルエーテル残基の分子量Mn=5000)を得た。
図3に、このBCP1の1H−NMRチャートを示す。
【0077】
【化13】
【0078】
実施例2 ブロックコポリマー(BCP2)の製造
実施例1(2)−(イ)において、ポリエチレングリコールメチルエーテル残基のMnが5000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシドの代わりに、ポリエチレングリコールメチルエーテル残基のMnが2000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド(分子量2000のモノメチルポリエチレングリコール(Aldrich社製)を用いて合成したものである。)を用いた以外は、実施例1と同様な操作を行い、ブロックコポリマー(BCP2、ポリエチレングリコール鎖のMn=2000)を製造した。
【0079】
実施例3 ブロックコポリマー(BCP3)の製造
実施例1(2)−(イ)において、ポリエチレングリコールメチルエーテル残基のMnが5000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシドの代わりに、ポリエチレングリコールメチルエーテル残基のMnが750のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド(分子量750のモノメチルポリエチレングリコール(Aldrich社製)を用いて合成したものである。)を用いた以外は、実施例1と同様な操作を行い、ブロックコポリマー(BCP3、ポリエチレングリコール鎖のMn=750)を製造した。
【0080】
前記の実施例1で得られたブロックコポリマー(BCP1)、実施例2で得られたブロックコポリマー(BCP2)及び比較例1で得られたブロックコポリマー(BCP3)の分析結果及び熱特性を表1に示す。
【0081】
【表1】
【0082】
[注]
1)X:重合体中におけるポリエチレングリコールユニットの分子量割合
2)Y:重合体中におけるペンダント型チオフェンユニットの分子量割合
3)PDI:分子量分布(Mw/Mn)
4)層転移温度:示差熱分析装置[SII社製「DSC220C」]を用いた示差熱分析測定と、偏光顕微鏡[OLYMPUS社製「BX51」及び加熱ステージ[METTLER社製「FP80HT」]を用いた観察により決定した。C=結晶相、Sm=スメクチック液晶相、I=等方性液体相を表し、表1中の「C XXX Sm YYYI」のごとき表記は、XXX℃で結晶相からスメクチック液晶層に転移し、YYY℃でスメクチック液晶相から等方性液体相に転移することを示す。
【0083】
実施例4
予め、過酸化水素・硫酸混合液で表面処理を行っておいたシリカ基板上に、数平均分子量Mn22,000、分子量分布1.22のBCP1薄膜(厚さ:40nm)を、実施例1で得られたBCP1のクロロホルム溶液を塗液として、スピンコート法により製膜した。その後、密封可能なガラス容器内にベンゼンを満たしたサンプル管および上記BCP1薄膜を入れて密封し、室温で48時間ソルベントアニール処理を行うことで、ミクロ相分離を誘起した。
さらにこのフィルムに307nm付近のUVのみを透過する307nmバンドパスフィルターを用いて、365nmのUV光を照射強度4.5mW/cm2で照射し、メタノール溶媒でリンス処理を行うことでポーラスフィルムを作製した。このフィルムに真空蒸着法を用いて酸化モリブデン(MoO3)を3nm、さらに金を50nm積層することでトップコンタクト型電界効果トランジスター(FET)素子を得た。
【0084】
実施例5
ブロックコポリマーとして、数平均分子量Mn14600、分子量分布1.18の実施例2で得られたBCP2を用いた以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0085】
実施例6
実施例4において、UV照射時に307nmバンドパスフィルターを用いる代わりに、紫外線透過可視吸収フィルターを用いた以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0086】
実施例7
実施例4において、307nmバンドパスフィルターを用いなかったこと以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0087】
比較例1
実施例4で用いたBCP1の製造における、(3)ブロックコポリマー(BCP1)の合成において、系内にジシクロヘキシルカルボジイミドを加え80℃に昇温した後の加熱攪拌時間を24時間とした以外は、実施例4と同様にして薄膜を得た。合成により得られたブロックコポリマーにおける、カルボン酸のオリゴチオフェン化合物との縮合率は30%であった。
【0088】
比較例2
実施例1(3)のブロックコポリマー(BCP1)の合成において、オリゴチオフェン誘導体として、4T−OHの代わりに、チオフェン単位の繰り返しを9単位としたものを用いて、実施例1(3)と同様にしてペンダント型ブロック重合体の合成を試みたが、オリゴチオフェン誘導体が溶媒に溶解せず、合成できなかった。
【0089】
試験1 (電子顕微鏡観察)
実施例4〜7、及び比較例1の薄膜表面にJEOL製「JFC−1600」イオンプレーティング装置を用いて白金でイオンコーティング処理を行い、走査型電子顕微鏡(日本電子製「Carry scope JCM−5700」)により観察し、ナノポーラス形成の有無を確認した。実施例4〜7ではナノポーラスの形成が確認されたが比較例1ではミクロ相分離由来のポーラス構造体は形成されていなかった。
ナノポーラスが形成された場合の走査型電子顕微鏡(SEM)写真を、代表として実施例4について図4に示す。また、比較例1についてのSEM写真を図5に示す。
【0090】
試験2 (原子間力顕微鏡観察)
実施例4〜7の薄膜表面を、原子間力顕微鏡(JEOL製「JSPM−5400」)により観察し、ナノポーラスの平均直径、ナノポーラスの平均深さ、及び空孔密度を測定した。
なお、平均直径は5μm角の範囲で原子間力顕微鏡のAC−AFMモードにより測定し、断面解析処理において測定範囲内のポーラス構造体の平均を取った。
また、平均深さは、5μm角の範囲で原子間力顕微鏡のAC−AFMモードにより測定し、断面解析処理において測定範囲内のポーラス構造体の平均を取った。
そして、ナノポア密度は、任意の5点において、1μm2中のナノポアの個数を数えることにより測定した。
結果を表2に示す。
【0091】
【表2】
【0092】
実施例4と5を比較すると、より分子量の低い実施例5では、ポアのサイズが小さくなるが、ポアの密度も小さくなってしまう。実施例4と比較して実施例6はナノポア直径がより大きく、実施例7はさらに直径が大きくなっている。バンドパスフィルターを用いた場合、紫外線透過可視吸収フィルターを用いた場合、無処理の場合の順でナノポア直径が大きくなっているのは、波長選択性の低いUVを照射するほどオリゴチオフェン縮合重合体構造中の結合の分解が進行し、発生した分子の断片がリンスにより洗い流されることに起因していると考えられる。このことから、バンドパスフィルターを用いることで、より精密にポーラスサイズを制御できることがわかる。
【0093】
実施例4及び5の薄膜について、RIGAKU社製「Ultima IV」を用いてXRD測定を行った。図6に、実施例4及び5の薄膜(図中、「BCP1」、「BCP2」と記載。)、及び実施例4のソルベントアニール処理を行う前のBCP1薄膜(図中、「製膜後」と記載。)のXRDパターンを示す。実施例4及び5のいずれのパターンにも、チオフェン四量体1単位の分子長に起因する回折ピークが得られ、チオフェン四量体構造が基板に対して垂直に配向していることが示された。なお、図中には掲載されていないが、実施例5のソルベントアニール処理を行う前のBCP2薄膜のXRD測定においても、実施例4のソルベントアニール処理を行う前のBCP1薄膜と同様に、回折ピークは検出されなかった。
【0094】
試験4 (移動度測定)
実施例4及び5の有機FETの電気特性評価は、1.3×10-3Pa以下の真空・遮光下で行った。Id−Vg特性を測定するために(ドレイン電流が飽和領域をとるゲート電圧値で、ゲート電圧を一定に保ち、ドレイン電圧を走査する)アジレント社製の半導体パラメーターアナライザー「AgilentB1500A」を用いて作製した有機FETにゲート電圧;−100V、ドレイン電圧;+20V→−100V、−1Vstepの条件でゲート電圧を印加して、室温(25℃)での特性を測定した。測定したドレイン電流−ソース・ドレイン電圧曲線のチャネルコンダクタンスから移動度(μ)を見積もった。実施例4及び5の有機EFTの移動度及び閾値電圧を表3に示す。
【0095】
【表3】
【0096】
さらにId−Vd特性の測定を行うために(ゲート電圧をある一定値にしておき、ドレイン電圧を走査しその時のドレイン電流を測定する。順次、ゲート電圧を変化させ何回も測定する。)半導体パラメーターアナライザー「AgilentB1500A」を用いて作製した有機電界効果トランジスターゲート電圧;20V→−100V、−10Vステップ、ドレイン電圧;+20V→−100V、−1Vstepの条件で測定を行ったところ図7のように、ゲート電圧の増加と共に電流値が増加していく、典型的なp型のトランジスター特性を示した。
【産業上の利用可能性】
【0097】
本発明により得られる有機ナノポーラス材料は、有機FET及びその他の有機デバイスとしての利用について有望であり、それらを用いた電子機器等の開発の発展に寄与することが期待される。
【技術分野】
【0001】
本発明は、液晶性有機半導体ポリマー、その製造方法、有機ナノポーラス材料及び有機電界効果トランジスターに関する。さらに詳しくは、本発明は、分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成し得る特定構造のブロック共重合体からなる液晶性有機半導体ポリマー、その効果的な製造方法、前記液晶性有機半導体ポリマーを用いて得られた、有機電界効果トランジスター(以下、有機FETと略記することがある。)などの有機デバイスとしての用途に有用な有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETに関するものである。
【背景技術】
【0002】
近年、ナノメートルオーダーの均一サイズを有する細孔(ポア)が規則的に並んだナノポーラス材料が注目されており、より規則性の高い有機ナノポーラス材料を得ることは、この分野において課題であった。
非特許文献1には、ポリスチレンブロックの連続相と、親水性ブロックの非連続相からなるミクロ相分離構造を有し、光開裂型開始剤を含有する組成物に、紫外線(UV)を照射し、ポリスチレンブロックと親水性ブロックとの間のエステル結合を加水分解し、リンス処理を行うことで親水性ブロックを除去し、有機ナノポーラス材料を得る方法が報告されている。
【0003】
また、非特許文献2及び3には、液晶性のブロックを有するブロック共重合体を用いることで、規則性の高いミクロ相分離構造を形成しうることが示されている。
前記非特許文献2には、水滴で開けた孔(Figure2、ミクロ相分離ではない)と、相分離構造(Figure6、ミクロ相分離)が入り混じった構造が示されており、また、非特許文献3には、液晶性のアゾベンゼンとポリエチレンオキシド(PEO)のミクロ相分離構造を得ることが示されている。しかしながら、これらの文献には、ナノポアの形成については何の示唆もされてない。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】B.Moonら,Macromolecules 2009年,42巻,455ページ
【非特許文献2】:T.Hayakawaら,Angrew.Chem.Int.Ed,2003年,42巻,2285ページ
【非特許文献3】:T.Iyodaら,Adv.Mater.2007年,19巻,1267ページ
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、このような状況下になされたもので、有機FETなどの有機デバイスとしての用途に有用な有機ナノポーラス材料を作製し得るブロック共重合体、その効果的な製造方法、前記ブロック共重合体を用いて得られた有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETを提供することを目的とするものである。
【課題を解決するための手段】
【0006】
本発明者らは、前記目的を達成するために鋭意研究を重ねた結果、下記の知見を得た。
分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有する特定構造のペンダント型ブロック共重合体からなる、液晶性有機半導体ポリマー組成物に、所定の処理を施すことにより、規則性の高い液晶性の配向を有する有機ナノポーラス材料が得られること、そして、前記液晶性有機半導体ポリマー組成物は、特定の工程を施すことにより、効率よく製造し得ることを見出した。本発明は、かかる知見に基づいて完成したものである。
【0007】
すなわち、本発明は、
[1]下記一般式(1)で表されるカルボン酸基含有重合体(B)における任意の−COOH基に、下記一般式(2)で表されるオリゴチオフェン誘導体(C)をエステル化反応させてなるブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含み、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー組成物、
【0008】
【化1】
【0009】
(一般式(1)中、R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基、Xは親水基、Zはハロゲン原子、mは2〜500の整数、pは0又は1を示す。)
【0010】
【化2】
(一般式(2)中、R4及びR5は、それぞれ独立に水素原子又は炭素数1〜3のアルキル基、Xは親水性基、Yは炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基、Aは炭素数2〜16のアルカンジイル基、kは2〜8の整数、nは0又は1を示す。)
【0011】
[2]一般式(1)におけるXで示される親水性基が、ポリエーテル残基である上記[1]に記載の液晶性有機半導体ポリマー組成物、
[3]上記[1]又は[2]に記載の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする有機ナノポーラス材料、
[4]電磁波照射処理が、紫外線照射処理である上記[3]に記載の有機ナノポーラス材料、
[5]紫外線の波長領域が、ブロック共重合体(A)の分子構造中に含まれるo−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェンエステル基の極大吸収波長を実質上含まない上記[4]に記載の有機ナノポーラス材料、
[6]上記[3]〜[5]のいずれかに記載の有機ナノポーラス材料を用いることを特徴とする有機電界効果トランジスター、
[7]以下の工程を有する上記[1]又は[2]に記載の液晶性有機半導体ポリマー組成物の製造方法、
工程(a)一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程。
工程(b)o−ニトロベンジル基の2〜5位のいずれかに親水性基の結合した官能基を有する原子移動ラジカル重合制御剤を用いて、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合法により重合することにより、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体を製造する工程。
工程(c)前記工程(a)で得られたオリゴチオフェン誘導体と、前記工程(b)で得られたカルボン酸基含有重合体とをエステル化反応させる工程。
[8]工程(b)におけるカルボン酸基1個以上を有するビニル化合物単量体が、(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸又はシトラコン酸である上記[7]に記載の液晶性有機半導体ポリマーの製造方法、及び
[9]ビニル化合物単量体が、(メタ)アクリル酸である上記[8]に記載の液晶性有機半導体ポリマーの製造方法、
を提供するものである。
【発明の効果】
【0012】
本発明によれば、分子内に有機半導体部位と、親水性基を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成し得る特定構造のブロック共重合体からなる液晶性有機半導体ポリマー組成物、その効果的な製造方法、前記液晶性有機半導体ポリマー組成物を用いて得られた、有機FETなどの有機デバイスとしての用途に有用な有機ナノポーラス材料、及びこの有機ナノポーラス材料を用いてなる有機FETを提供することができる。
【図面の簡単な説明】
【0013】
【図1】実施例1で得られた4T−OHの1H−NMRチャートである。
【図2】実施例1で得られた重合体(B’−6)の1H−NMRチャートである。
【図3】実施例1で得られたブロックコポリマー(BCP1)の1H−NMRチャートである。
【図4】実施例4で得られたナノポーラス薄膜のSEM写真である。
【図5】比較例1で得られた薄膜のSEM写真である。
【図6】実施例4及び5で得られた各薄膜のXRDチャートである。
【図7】実施例4で得られた有機FETのId−Vd曲線である。
【発明を実施するための形態】
【0014】
まず、本発明の液晶性有機半導体ポリマー組成物について説明する。
[液晶性有機半導体ポリマー組成物]
本発明の液晶性有機半導体ポリマー組成物は、ブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含むことを特徴とする。
【0015】
(ブロック共重合体(A))
本発明の液晶性有機半導体ポリマー組成物において、成分(A)として用いられるブロック共重合体は、下記一般式(1)
【0016】
【化3】
【0017】
で表される共重合体における任意の−COOH基に、下記一般式(2)
【0018】
【化4】
【0019】
で表されるオリゴチオフェン誘導体(C)がエステル結合している構造を有する。
なお、前記一般式(1)中、
R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基を示す。
本発明において、オリゴチオフェン縮合重合体構造とは、前記一般式(1)中の任意の−COOH基に前記一般式(2)のオリゴチオフェン誘導体(C)がエステル結合したm個の繰り返し単位構造をいう。また、オリゴチオフェン含有エステル基とは、オリゴチオフェン誘導体(C)がエステル結合した−COOH基のことをいう。
【0020】
一般式(1)におけるXは親水性基を示す。なお、本発明における親水性基とは、ブロック共重合体(A)が親水性基とオリゴチオフェン縮合重合体構造との親水性の差により、ミクロ相分離構造を形成しうる程度に、かつ後述する電離放射線照射後の水及び/又は低級アルコールによるリンス処理により除去可能な程度に親水性を有することを指す。当該親水性基は、効果的にオリゴチオフェン縮合重合体構造とミクロ相分離を形成し、また、上記リンス処理により除去されうる。
【0021】
このような親水性基としては、親水性ポリマー残基であることが好ましく、ポリエーテル残基、ポリエーテルポリウレタン残基、ポリ(メタ)アクリル酸(又はその塩)残基、ポリ(メタ)アクリルアミド誘導体残基、ポリビニルアルコール残基、セルロースアルキルエステル残基等の1種または2種以上の組み合わせが挙げられる。とりわけ、後述のリンス処理による除去の容易性から、ポリエーテル残基が好ましい。ここで、ポリエーテル残基は、ポリエチレングリコール残基、ポリプロピレングリコール残基のようなポリアルキレンエーテル残基であってもよく、ポリグリセリン残基のように側鎖に水酸基を有するものであってもよい。
【0022】
当該親水性基は、親水性の発現及び後述の形成される有機ナノポーラス材料のポアサイズの観点から、重合度(単量体単位数)が2〜500程度、好ましくは100〜300の親水性ポリマー残基であることが望ましい。
親水性基Xは、酸素原子を介して、又は介さずに、o−ニトロベンジロキシカルボニル基のベンゼン環に結合しており、上記o−ニトロベンジロキシカルボニル基におけるo−ニトロベンジル基は、後述の電磁波を照射する際に、光開裂型開始機能を発現し、上記の親水性基が結合したo−ニトロベンジロキシカルボニル基のエステル結合部が容易に加水分解される。
【0023】
また、前記一般式(1)において、Zはハロゲン原子を示す。ハロゲン原子としては、フッ素原子、塩素原子、臭素原子及びヨウ素原子を挙げることができるが、これらの中で、特に臭素原子が好適である。
【0024】
ブロック共重合体(A)は、上述のオリゴチオフェン縮合重合体構造のオリゴチオフェン構造の配向性により、親水性基とのミクロ相分離構造をより規則性高く形成でき、また後述のように、オリゴチオフェン縮合重合体構造の連続相と、親水性基の非連続相の間に存在するエステル結合を電磁波照射により光開裂させて、その後のリンス処理により親水性基を除去することによってナノポーラスを形成することができる。
親水性基が親水性ポリマー残基である場合には、ブロック共重合体(A)における親水セポリマー残基の数平均分子量Mnと、オリゴチオフェン縮合重合体構造の比率は、親水性ポリマー残基の数平均分思慮Mn:オリゴチオフェン縮合体構造のMnとして、15:85〜40:60であることが好ましい。親水性ポリマー残基のMnが15:85以上であれば、ミクロ相分離の形成が得やすくなる。一方、40:60以下であると、ラメラ構造を形成しにくくなり、オリゴチオフェン縮合重合体構造の連続相を形成しやすくなる。
【0025】
一般式(1)におけるmはカルボン酸基1個以上を有するビニル化合物単量体の重合度を示し、2〜500の整数である。またpは0又は1を示す。
【0026】
ブロック共重合体(A)の数平均分子量は、5,000〜50,000であることが好ましい。5,000以上であると材料が半固体になることがないので、構造体の維持が容易となり、50,000以下であると材料の溶解性が低下することがない。より好ましくは、10,000〜30,000であり、さらに好ましくは、12,000〜25,000である。
【0027】
一方、前記一般式(2)において、R4及びR5はそれぞれ独立に水素原子又は炭素数1〜3のアルキル基を示す。炭素数1〜3のアルキル基としては、メチル基、エチル基、n−プロピル基及びイソプロピル基が挙げられる。
Yは、炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基を示す。炭素数1〜16のアルキル基は直鎖状、分岐状のいずれであってもよく、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、各種ペンチル基、各種ヘキシル基、各種オクチル基、各種デシル基、各種ドデシル基、各種テトラデシル基、各種ヘキサデシル基などを挙げることができる。
【0028】
炭素数1〜16のアルコキシ基を構成するアルキル基は直鎖状、分岐状のいずれであってもよく、炭素数1〜16のアルコキシ基としては、例えばメトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、各種ペントキシ基、各種ヘキソキシ基、各種オクトキシ基、各種デシロキシ基、各種ドデシロキシ基、各種テトラデシロキシ基、各種ヘキサデシロキシ基などを挙げることができる。
【0029】
また、炭素数2〜17のアルコキシカルボニル基を構成するアルキル基は直鎖状、分岐状のいずれであってもよく、炭素数2〜17のアルコキシカルボニル基としては、例えばメトキシカルボニル基、エトキシカルボニル基、n−プロポキシカルボニル基、イソプロポキシカルボニル基、n−ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基、各種ペントキシカルボニル基、各種ヘキソキシカルボニル基、各種オクトキシカルボニル基、各種デシロキシカルボニル基、各種ドデシロキシカルボニル基、各種テトラデシロキシカルボニル基、各種ヘキサデシロキシカルボニル基などを挙げることができる。
【0030】
前記一般式(2)におけるAは、炭素数2〜16のアルカンジイル基を示し、直鎖状、分岐状のいずれであってもよく、例えば各種エタンジイル基、各種プロパンジイル基、各種ブタンジイル基、各種ペンタンジイル基、各種ヘキサンジイル基、各種オクタンジイル基、各種デカンジイル基、各種ドデカンジイル基、各種テトラデカンジイル基、各種ヘキサデカンジイル基などを挙げることができる。
kはオリゴチオフェンの重合数を示し、2〜8の整数、nは0又は1を示す。
【0031】
ブロック共重合体(A)においては、チオフェン構造の繰り返しにより、隣接する単量体単位と結合したオリゴチオフェン構造同士が、π-電子の相互作用によるいわゆるπ-πスタッキングを生じるために配向を生じやすく、効果的に、規則性の高いミクロ相分離構造を形成することができる。
オリゴチオフェンを形成するチオフェン構造単位の繰り返し数は、2単位〜8単位であることを要する。1単位である場合には、上述のスタッキングを生じることが難しく、9単位以上では、結晶性が高くなり取り扱い性が悪くなる。チオフェン構造の繰り返しは、2単位〜6単位であることがより好ましく、3単位〜5単位であることがさらに好ましい。
カルボン酸基含有重合体の単量体単位の2以上の単位にオリゴチオフェン化合物を縮合させることで、オリゴチオフェン縮合重合体構造を得ることができる。この縮合反応は、ジシクロヘキシルカルボジイミド等の公知の縮合剤を用いて行うことができる。
なお、ブロック共重合体(A)は、後述する本発明の液晶性有機半導体ポリマー組成物の製造方法によって、効率よく製造することができる。
【0032】
(カルボン酸基含有重合体(B))
本発明の液晶性有機半導体ポリマー組成物において、成分(B)として用いられるカルボン酸基含有重合体は、下記一般式(1)
【0033】
【化5】
【0034】
で表される構造を有している。
前記一般式(1)において、X、Z、R1〜R3、m及びpは、前述の通りである。
本発明の液晶性有機半導体ポリマー組成物は、ブロック共重合体(A)単独、又は該ブロック共重合体(A)と、前記一般式(1)で表されるカルボン酸基含有重合体(B)とを含むものであって、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸基との割合は、100:0〜35超:65未満(モル比)であることが好ましく、100:0〜45:55(モル比)であることがさらに好ましい。前記割合が上記範囲内であれば、本発明の液晶製有機半導体ポリマー組成物は液晶性が発現されやすくなる。
【0035】
次に、本発明の液晶性有機半導体ポリマー組成物の製造方法について説明する。
[液晶性有機半導体ポリマー組成物の製造方法]
本発明の液晶性有機半導体ポリマー組成物の製造方法(以下、単に製造方法と略記することがある。)は、下記に示す工程(a)、工程(b)及び工程(c)を有することを特徴とする。
【0036】
(工程(a))
本発明の製造方法における工程(a)は、一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程である。
すなわち、下記一般式(2)
【0037】
【化6】
【0038】
(一般式(2)中、A、R4、R5、Y、n及びkは、前記と同じである。)
で表されるオリゴチオフェン誘導体(C)を製造する工程である。
このオリゴチオフェン誘導体(C)は、例えば、下記の反応式(a)によって製造することができる。
【0039】
【化7】
【0040】
上記反応式(a)は、得られるオリゴチオフェンが4量体であって、R4及びR5がいずれも水素原子、n=0、−A−OHが−(CH2)6−OH、Yが−C6H13、kが4の例である。
まず、2,2’−ビチオフェン(C−1)に、n−ブチルリチウムの存在下に、水酸基がテトラヒドロ−2H−ピラン(THP)で保護された6−ブロモヘキシルアルコール(C−2)を反応させて、水酸基がTHPで保護された6−(2,2’−ビチオフェン−5−イル)へキシルアルコール(C−3)を合成する。
次いで、上記の水酸基がTHPで保護された6−(2,2’−ビチオフェン−5−イル)へキシルアルコール(C−3)に、ブロム化剤のNBS(N−ブロモスクシンイミド)を反応させて、水酸基がTHPで保護された6−(5’−ブロモ−2,2’−ビチオフェン−5−イル)ヘキシルアルコール(C−4)を合成する。
次に、この水酸基がTHPで保護された6−(5’−ブロモ−2,2’−ビチオフェン−5−イル)ヘキシルアルコール(C−4)に、(5’−ヘキシル−2,2’−ビチオフェン−5−イル)ボロン酸ピナコールエステル(C−5)を、テトラキス(トリフェニルホスフィン)パラジウム[Pd(PPh3)4]の存在下反応させることにより、水酸基がTHPで保護された6−(5’−ヘキシル−2,2’:5,2’’:5’’,2’’’−クォータチオフェン−5’’’−イル)ヘキシルアルコール(C−6)を合成する。
最後に、該化合物(C−6)にp−トルエンスルホン酸を作用させることにより、チオフェン環が4個結合したオリゴチオフェンの片末端のチオフェン環の5位にヘキサノールを結合すると共に、他方の末端のチオフェン環の5位にヘキシル基が結合してなる、前記一般式(2)で表されるオリゴチオフェン誘導体の一例である(4T−OH)が得られる。
当該工程(a)においては、前記の反応式(a)で示すような方法により、片末端のチオフェン環の5位にヒドロキシアルキル基を有する一般式(2)で表されるオリゴチオフェン誘導体(C)が得られる。
【0041】
(工程(b))
本発明の製造方法における工程(b)は、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合により、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)を製造する工程である。
<原子移動ラジカル重合>
原子移動ラジカル重合(ATRP)は、成長しているポリマー鎖と重合制御剤との間の不安定なラジカルが容易に移動することを媒介して、リビングラジカル重合を行う触媒可逆的レドックス法である。
この原子移動ラジカル重合制御剤としては、例えば有機ハロゲン化物、又は有機ハロゲン化合物と遷移金属錯体との組合わせを用いることができる。上記有機ハロゲン化物としては、特に反応性の高い炭素−ハロゲン結合を有する化合物(例えばα位にハロゲンを有するカルボニル化合物や、ベンジル位にハロゲンを有する化合物など)、あるいはハロゲン化スルホニル化合物などが好適である。
一方、遷移金属錯体としては、特に制限はないが、好ましくは周期表第7〜11族に属する元素を中心金属とする金属錯体であり、より好ましくは0価又は1価の銅、2価のルテニウム、2価の鉄、2価のニッケルの錯体であり、特に好ましくは銅の錯体である。
このATRP法によれば、得られるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)は、分子量の制御が容易であると共に、重量平均分子量/数平均分子量(Mw/Mn)比が極めて小さい、例えば1.3以下程度となる。
前記一般式(2)で表されるo−ニトロベンジル基を有するカルボン酸基含有重合体(B)は、例えば以下に示す反応式(b)によって製造することができる。
【0042】
【化8】
【0043】
(反応式(b)中、Rは炭素数1〜5の低級アルキル基であり、R1〜R3、X、Z、m及びpは前記と同じである。)
まず、親水性基を有するo−ニトロベンジルアルコール(B−1)に、α−ハロゲノイソ酪酸ハライド(B−2)を、メチレンジクロリドなどの溶媒中において、三級アミンなどの存在下に反応させて、α−ハロゲノイソ酪酸エステル化合物(B−3)を合成する。次いで、このα−ハロゲノイソ酪酸エステル化合物(B−3)の存在下、カルボン酸基1個以上を有するビニル化合物単量体(B−4)をATRP法により重合させて、ポリマーの側鎖にエステル結合を有する重合体(B−5)を合成する。このATRP法による重合においては、溶媒としてアニソールを用いることが好ましく、また、リガンドとして、N,N,N’,N’’,N’’−ペンタメチルジエチレントリアミン(PMDTA)を用いることが好ましい。反応温度は、通常60〜120℃程度である。
最後にこの重合体(B−5)を加水分解(脱保護)することにより、一般式(1)で表されるポリマーの側鎖にカルボン酸基含有重合体(B)(この場合は、B−6とする。)が得られる。
【0044】
前記のカルボン酸基1個以上を有するビニル化合物単量体としては、例えば(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸、シトラコン酸などを用いることができるが、これらの中で(メタ)アクリル酸が好適である。ここで、(メタ)アクリル酸は、アクリル酸又はメタクリル酸を示す。
前記ビニル化合物単量体がアクリル酸である場合には、一般式(1)の重合体(B)におけるR1〜R3は全て水素原子であり、メタクリル酸である場合には、R1はメチル基、R2及びR3は水素原子である。マレイン酸やフマル酸の場合には、R1及びR2は水素原子であり、R3は−COOHである。イタコン酸である場合には、R1は−CH2COOHであり、R2及びR3は水素原子である。シトラコン酸である場合には、R1は水素原子、R2はメチル基及びR3は−COOHである。
なお、ATRP法による重合反応時には、各カルボン酸基は低級アルキルエステルの形態であることが好ましく、このエステルは、最終工程での加水分解反応(脱保護反応)により、遊離のカルボン酸基となる。本発明におけるカルボン酸基は、カルボン酸低級アルキルエステル基及び遊離のカルボン酸基(−COOH基)のいずれをも含む広い意味である。
【0045】
(工程(c))
本発明の製造方法における工程(c)は、前記工程(a)で得られた一般式(2)で表されるオリゴチオフェン誘導体(C)と、前記工程(b)で得られた一般式(1)で表されるカルボン酸基含有重合体(B)とをエステル化反応させる工程である。
この工程(c)における反応を、下記の反応式(c)で示す。
【0046】
【化9】
【0047】
(反応式(c)中、R1〜R5、A、X、Y、Z、k、m、n及びpは、前記と同じである。)
ジメチルホルムアミドなどの溶媒中において、ジシクロヘキシルカルボジイミドや4−ジメチルアミノピリジンなどの縮合剤の存在下に、一般式(1)で表されるカルボン酸基含有重合体(B)と、一般式(2)で表されるオリゴチオフェン誘導体(C)とを、60〜120℃程度の温度で縮合させることにより、ブロック共重合体(A)が得られる。
この場合、任意のカルボン酸基含有重合体(B)の単量体単位に、オリゴチオフェン誘導体(C)を縮合させることにより、ペンダント型ブロック重合体を得ることができる。
【0048】
また、カルボン酸基含有重合体のカルボン酸基のうち、35%(官能基の数の割合)よりも大きい比率で、オリゴチオフェン誘導体と縮合されることを要する。35%以下である場合には、液晶性を呈しにくい。カルボン酸基は、50%よりも大きい比率でオリゴチオフェン誘導体と縮合されることがより好ましく、70%よりも大きい比率で縮合されていることがさらに好ましい。
ペンダント型ブロック共重合体の数平均分子量は、5,000〜50,000であることが好ましい。5,000未満であると材料が半固体になってしまい、構造体の維持が困難になってしまう、50,000を超えると材料の溶解性が低下してしまう、より好ましくは、10,000〜30,000であり、さらに好ましくは、12,000〜25,000である。
なお、上記数平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)法で測定される標準ポリスチレン換算の値である。
【0049】
次に、本発明の有機ナノポーラス材料について説明する。
[有機ナノポーラス材料]
本発明の有機ナノポーラス材料は、前述した本発明の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理、ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする。
前述した本発明の液晶性有機半導体ポリマー組成物は、分子内に有機半導体部位と、親水性基(ポリマー型)を有すると共に、電磁波照射時に光開裂型開始機能をもつ基を含むポリマー部位とを有し、液晶性ミクロ相分離構造を形成するペンダント型ブロック共重合体である。
このような性状を有する液晶性有機半導体ポリマー組成物にアニール処理を施すことにより、オリゴチオフェン縮合重合体構造の連続相と、親水性基(ポリマー型)の非連続相からなるミクロ相分離構造を誘起させることができ、これに電磁波を照射することにより、親水性基(ポリマー型)が結合してなるo−ニトロベンジル基の光開裂型開始機能により、該o−ニトロベンジル基に隣接するエステル結合が切断される。したがって、これに水及び/又は低級アルコールによるリンス処理を施すことによって、親水性基を有する部位は除去されるので、有機ナノポーラス材料が形成される。
【0050】
(アニール処理)
アニール処理は、前述したように、オリゴチオフェン縮合重合体構造の連続相と、親水性基(ポリマー型)の非連続相からなるミクロ相分離構造を誘起させるための処理である。
アニール処理は、高温雰囲気下に一定時間留置する熱アニール処理や、溶媒濃度の高い雰囲気下に一定時間留置するソルベントアニール処理が挙げられる。
【0051】
熱アニール処理は、液晶性有機半導体ポリマー組成物の液晶温度範囲内の環境に液晶性有機半導体ポリマー組成物を留置することにより行うことができる。なお、上記の液晶温度範囲とは、液晶性物質の、結晶相と液晶相の転移温度を下限とし、液晶相と等方性液体相の転移温度を上限とする範囲を意味する。本発明の有機半導体ポリマー組成物では、結晶相と液晶相の転移温度は通常40〜250℃であり、液晶相と等方性液体相の転移温度は通常100〜400℃である。これらの転移温度は示差熱分析測定により決定できる。熱アニール処理は、5分〜96時間の間で行うことが好ましい。
【0052】
ソルベントアニール処理の方法は、密閉された系内に溶媒と液晶性有機半導体ポリマー組成物を留置することにより行うことができる。
このソルベントアニール処理に用いる溶媒は、オリゴチオフェン縮合重合体構造、親水性基(ポリマー型)のいずれかの良溶媒又はそれらの混合溶媒を用いるのがよい。具体的にはアセトン、アセトニトリル、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、ジクロロメタン、クロロホルム、四塩化炭素、THF、ジオキサン、二硫化炭素、シクロヘキサノン、シクロヘキサン、酢酸エチル、ジメチルスルホキシド、ジメチルホルムアミドなどが好適に用いられ、中でも、ベンゼン、ジオキサン、アセトンが特に好ましい。
【0053】
処理方法として室温あるいは使用溶媒の沸点付近まで加熱を行い、液晶性有機半導体ポリマー組成物の処理雰囲気の溶媒蒸気濃度を高めることが望ましい。ソルベントアニール処理を行う時間は、5分〜96時間であることが好ましい。
また、上記した熱アニール処理、ソルベントアニール処理のうち、より規則正しいミクロ相分離構造が得られやすいため、ソルベントアニール処理がより好ましい。
【0054】
(電磁波照射処理)
本発明においては、前述したようなアニール処理により、ミクロ相分離構造を誘起させた液晶性有機半導体ポリマー組成物に、電磁波照射処理を施す。
当該液晶性有機半導体ポリマー組成物は、分子内に光開裂開始機能をもつo−ニトロベンジル基を有することから、電磁波照射により、該o−ニトロベンジル基が開裂し、発生したラジカルによって、下記の反応式(d)で示すように、該o−ニトロベンジル基に隣接するエステル結合が効果的に光開裂され、親水性化合物(D−1)とオリゴチオフェン縮合重合体(D−2)が生成する。
【0055】
【化10】
【0056】
(反応式(d)中、R1〜R5、A、X、Y、Z、k、m、n及びpは、前記と同じである。)
なお、光開裂型開始機能をもつ基としては、前記のo−ニトロベンジル基以外に、ジアリルメチルエステル残基などがある。
電磁波照射としては、可視光線、紫外線(UV)、X線、ガンマ線等の照射が挙げられるが、当該エステル結合に与えるエネルギーの大きさの妥当性の面から、UV照射であることが好ましい。
【0057】
一方で、UVの照射により、オリゴチオフェン縮合重合体構造の分解が引き起こされる可能性がある。オリゴチオフェン縮合重合体構造の分解が起こると、分解物であるオリゴチオフェン化合物が、リンス処理において除去されてしまうために、ポーラスのサイズが大きくなり、有機ナノポーラス材料のポアのサイズを制御する上で好ましくない。
オリゴチオフェン縮合重合体構造の不本意な分解を防ぐためには、UVにフィルタリングを行うことが好ましい。より好ましくは、UVは、o−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない波長選択UVであることが好ましい。このようなUVの照射は、一定の波長領域のみを透過するバンドパスフィルターを用いた選択的照射により可能であり、オリゴチオフェン縮合重合体構造の分解が効果的に防止できる。
【0058】
(リンス処理)
本発明においては、前述のようにして、アニール処理及び電磁波照射処理が施された液晶性有機半導体ポリマー組成物に、水及び/又は低級アルコール(メタノール、エタノール、プロパノール、イソプロパノールなど)によるリンス処理を施し、前記反応式(d)における親水性化合物(D−1)を除去することによって、有機ナノポーラス材料を形成することができる。
【0059】
(有機ナノポーラス材料の作製)
本発明の有機ナノポーラス材料の具体的な作製方法ついて説明する。
まず、ガラス基板やSiO2基板などの適当な基板上に、クロロホルムなどの溶媒中に、本発明の液晶性有機半導体ポリマー組成物を含む塗工液をスピンコート法などにより塗工し、乾燥して、厚さ20〜400nm程度の薄膜を形成させる。次いで、密封可能なガラス容器内で、適当な溶媒を用いてソルベントアニール処理を施し、ミクロ相分離構造を誘起させる。ソルベントアニール処理の条件は、使用する溶媒の種類に応じて、適宜選択する。
次に、一定の波長領域のみを透過するバンドパスフィルターを用いて、紫外線をソルベントアニール処理された薄膜に照射する。なお、上記一定の波長領域とは、当該液晶性有機半導体ポリマー組成物における、o−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない波長領域を指す。
最後に、UV照射された薄膜に、水及び/又は低級アルコール(メタノール、エタノール、プロパノール、イソプロパノールなど)によるリンス処理を施すことにより、本発明の有機ナノポーラス材料を作製することができる。
このようにして得た有機ナノポーラス材料は、液晶性に起因して、オリゴチオフェン縮合重合体のオリゴチオフェン構造が配向している。
配向を有する有機ナノポーラス材料は、有機FETや、有機薄膜太陽電池の材料などとしての利用が期待される。
【0060】
次に、本発明の有機電界効果トランジスター(FET)について説明する。
[有機電界効果トランジスター(FET)]
本発明の有機FETは、前述した本発明の有機ナノポーラス材料を用いることを特徴とする。
薄膜トランジスター(TFT)は、エレクトロニクスにおけるスイッチ素子として広く使用されており、とりわけアクティブ・マトリックス型液晶表示装置やスマート・カードなど、広範囲にわたる適用分野で使用されている。薄膜トランジスター(TFT)の殆どは、電界効果トランジスター(FET)である。
現在、殆どのTFTデバイスは、半導体材料としてアモルファスシリコンを使用して作製されている。しかしながら、アモルファスシリコンTFTの製造にはプラズマ強化化学気相成長法などの高コストの装置が必要であり、そのプロセスも真空中、高温(約360℃)で行わねばならず、高コストであるのに加え、フレキシブルプラスチック基板を用いることが難しいという問題がある。
したがって、TFTやFETの半導体材料として、有機半導体材料が注目されている。
【0061】
本発明の有機ナノポーラス材料を有機FETとして用いる場合の利点は以下の通りである。すなわち、本来ポーラス構造を有する故にポーラス構造を有しない場合に比べて移動度の大幅な低下が懸念されるにも関らず、配向を生じていることにより、ポーラス構造体が明確に形成されており、且つ導電性部位であるオリゴチオフェン部位が移動度向上に有利な基板材料に対して垂直方向に高く配向しているため、有機FETとして利用可能な程度の移動度を呈する。さらに、ポアのサイズを変えることによって、配向の程度が変わり、移動度を変えることができるので、移動度を精密に制御できる可能性がある。また、単にミクロ相分離したブロック共重合体を用いる場合には、非導電性部位となる非液晶性ブロックに起因する問題、例えば吸湿性等、を生じるおそれがあるが、有機ナノポーラス材料においては、非導電性部位は空隙であるため、そのような問題を避けることができる。
【実施例】
【0062】
次に、本発明を実施例により、さらに詳細に説明するが、本発明はこれらの例によってなんら限定されるものではない。
実施例1 ブロックコポリマー(BCP1)の製造
(1)4T−OHの合成
反応式(a)に従って、4T−OH[6−(5’−ヘキシル−2,2’:5,2’’:5’’,2’’’−クォーターチオフェン−5’’’−イル)ヘキシルアルコール]を合成する。
(イ)化合物(C−2)の合成
500mL三口フラスコに6−ブロモ−1−ヘキサノール20g(東京化成製、0.11mol)とパラトルエンスルホン酸一水和物2.8g(東京化成製、0.017mol)、テトラヒドロフラン(THF)200mLを加えた。そこに3,4−ジヒドロ−2H−ピラン13.9g(東京化成製、0.165mol)を室温で滴下し、室温で20時間撹拌を行った。反応終了後、反応液を濃縮し、展開溶媒としてクロロホルム-ヘキサン(質量比2:8)を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い、化合物(C−2)が合成できていることを確認した。収率は80%であった。
なお、1H−NMR装置として、日本電子社製「JNM ECP400」を用いた。以下同様である。
【0063】
(ロ)化合物(C−3)の合成
300mL三口フラスコに2,2’−ビチオフェン(C−1)7g(東京化成製、42.2mmol)を加えた。系内を封止し乾燥窒素チャージと脱気を3回繰り返すことで系内の酸素を除去した。この状態で脱水THF40mLを加え、反応液の温度を-40℃以下に保った状態でn−ブチルリチウム(関東化学製、1.6mol/L、29mL、46.42mmol)加えた。反応液の温度を保持しながら、1時間撹拌しリチオ化を行った。そこに化合物(C−2)をTHF15mLに溶解した溶液を滴下した。滴下終了後、室温に戻して20時間反応を行った。反応を終了させるために、水10mLを加え処理し、酢酸エチル−水で抽出を行った。反応液を濃縮し展開溶媒として酢酸エチル−ヘキサン(質量比1:12)を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い化合物(C−3)が合成できていることを確認した。収率は51%であった。
【0064】
(ハ)化合物(C−4)の合成
化合物(C−3)(2.62g、9.85mmol)を、ジメチルホルムアミド(DMF)70mL溶解し、N−ブロモスクシンイミド(NBS)2.1g (和光純薬製、11.8mmol)を加え24時間遮光下で反応を行った。反応終了後、クロロホルムで抽出を行い、有機層を濃縮し、クロロホルム:ヘキサン(質量比4:6)の展開溶媒を用いたシリカゲルカラムクロマトグラフィーにより精製を行った。1H−NMR測定を行い、チオフェン基のプロトン積分比の和が4となっていることから、目的の化合物(C−4)が合成できていることを確認した。収率は73%であった。
【0065】
(ニ)化合物(C−6)の合成
化合物(C−4)(2.04g、5.9mmol)および5’−ヘキシル−2,2’−ビチオフェン−5−ボロン酸ピナコールエステル化合物(C−5)(アルドリッチ製、2.78g、7.4mmol)をトルエン25mLに溶解し、エタノール10mL、24質量%炭酸ナトリウム水溶液を加えた。凍結脱気(反応液を液体窒素で凍結し、ポンプにより脱気を行い、脱気後常温に戻す)を2回行った。そこにテトラキス(トリフェニルホスフィン)パラジウム(東京化成製、0.48g、0.41mmol)を加え、窒素雰囲気下で24時間加熱還流を行った。反応終了後、クロロホルムで抽出を行い、有機層を濃縮し、クロロホルム−メタノールで再結晶を行い精製した。1H−NMR測定を行い、チオフェン基のプロトン積分比の総和が8となっていることから、目的の化合物(C−6)ができていることを確認した。収率は51%であった。
【0066】
(ホ)4T−OHの合成
500mL三口フラスコに化合物(C−6)を5.11g(8.53mmol)とメタノール200mLを加えた。そこにp−トルエンスルホン酸一水和物0.294g(1.71mmol)を加え、加熱還流を20時間行った。反応終了後、反応液をろ過し、ろ物をメタノールで洗浄することにより精製を行った。1H−NMR測定により目的の4T−OHができていることを確認した。収率は94%であった。
図1に、4T−OHの1H−NMRチャートを示す。
【0067】
(2)一般式(2)のカルボン酸基含有重合体(B)の合成
反応式(b)に従って、一般式(2)のカルボン酸基含有重合体(B−6)を合成した。
なお、化合物(B−1)として、5位に親水性基であるH3C−(O−CH2CH2)n−O−が結合したo−ニトロベンジルアルコール(化合物(B’−1)を、化合物(B−2)として、α−ブロモイソ酪酸ブロミドを、化合物(B−4)として、tert−ブチルメタクリレートを用いた。すなわち、X:数平均分子量Mnが5,000のポリエチレングリコールメチルエーテル残基、Z:臭素原子、p:1である。
【0068】
(イ)化合物(B’−1)の合成
THF5mLに水素化ナトリウム77mg(3.2mmol)を懸濁させた液に、THF5mL中に5−ヒドロキシ−2−ニトロベンジルアルコール541mg(3.2mmol)を溶解させた溶液を0℃で徐々に加え、0℃にて30分間撹拌した。次いで、これに、THF10mL中にα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド4g(0.8mmol、Mn=5000、分子量5000のモノメチルポリエチレングリコール(Aldrich社製))を用いて、非特許文献1に開示の方法により合成したものである。)を含む溶液を0℃にて徐々に添加した。この溶液を70℃にて一晩撹拌したのち、蒸留水1mLを加えて反応を終了させた。
反応混合物を塩基性アルミナ層を通してろ過し、ろ液をジエチルエーテル中に加え、生成した沈殿物をろ取し、24時間真空乾燥することにより、化合物(B’−1)を得た。収率は88%であった。
【0069】
(ロ)化合物(B’−3)の合成
撹拌機を備えた250mLフラスコに、上記(イ)で得た化合物(B’−1)800mg(0.16mmol)と触媒量の4−ジメチルアミノピリジンの混合物を仕込み、次いで、脱気と窒素ガスチャージを3回繰り返すことで酸素を除去した。このフラスコにシリンジを用いてメチレンジクロリド10mLを加え、前記混合物を溶解させたのち、トリエチルアミン(TEA)0.4mLと、α−ブロモイソ酪酸ブロミド80μL(0.64mmol)を順に加えた。この溶液を室温で20時間撹拌後、メタノール1mLを加えて反応を終了させ、さらにTHFを加えて希釈した。
次いで、反応混合物を塩基性アルミナ層を通してHBr・TEAをろ別したのち、ろ液をジエチルエーテル中に加え、生成した沈殿物をろ取し、一晩真空乾燥することにより、下記式(3)で表される白色固体650mgを得た。収率は81%であった。
この白色固体は、1H−NMR分析により、下記式(3)で表される化合物(B’−3)であることが確認された。
【0070】
【化11】
【0071】
(式(3)中、nは、ポリエチレングリコールメチルエーテル残基のMnが5000となる整数を示す。)
【0072】
(ハ)重合体(B’−5)の合成
30mLのシュレンク管内部から、脱気と窒素チャージを3回繰り返すことで、酸素を除去した。ここに、N,N,N’,N’’,N’’−ペンタメチルジエチレントリアミン(PMDTA)(Aldrich社製、56μL、0.268mmol)、脱水アニソール(Aldrich社製、2.5mL)、tert−ブチルメタクリレート(アルドリッチ製、0.76g、5.36mmol)を、この順序でシリンジを用いて反応容器に注入した。その後、反応混合物内の酸素を除去するために、凍結脱気を2回行った。窒素の流量を強くして臭化銅(I)粉末(アルドリッチ製、19mg、0.134mmol)を加えた。最後に、上記(ロ)で得た化合物(B’−3)0.2g(0.067mmol)を加え容器を封じて70℃で24時間重合を行った。重合終了後、THF:5mLを加え、展開溶媒にTHFを用いたアルミナカラムに通すことで不溶物を取り除いた。溶媒を減圧留去し、ヘキサン溶液で2回再沈澱を行い精製することにより、重合体(B’−5)を得た。
この重合体(B’−5)のGPC法で測定された標準ポリスチレン換算の数平均分子量Mnは10,000であり、分子量分布Mw/Mnは1.21であった。なお、GPC装置として、東ソー株式会社製、機種名「GPC−8020」を用いた。以下、同様である。
【0073】
(ニ)重合体(B’−6)の合成
100mL三口フラスコに上記(ハ)で得られた重合体(B’−5)0.5g(4mmol)とジクロロメタン10.9g(128mmol)を加えた。ここにトリフルオロ酢酸(和光純薬製、5.9g、52mmol)を加え室温で20時間反応させた。反応終了後エーテルに再沈澱させることでポリマーの精製を行うことにより、下記式(4)で表される重合体(B’−6)を得た。
【0074】
【化12】
【0075】
(式(4)中、m’はポリメタクリル酸部位の重合度を示し、nは前記式(3)と同じである。)
この重合体(B’−6)の1H−NMR、GPC測定により、
GPC;Mn=4380、Mw/Mn=1.12
1H−NMR;Mn=4462、末端カルボン酸の個数=17(反応点17個)
であった。
図2に、重合体(B’−6)の1H−NMRチャートを示す。
【0076】
(3)ブロックコポリマー(BCP1)の合成
反応式(c)に従って、ブロックコポリマー(BCP1)を合成した。
なお、一般式(1)で表されるカルボン酸基含有重合体(B)として、式(4)で表される重合体(B’−6)を用い、一般式(2)で表されるオリゴチオフェン誘導体(C)として4T−OHを用いた。
30mLシュレンク管に重合体(B’−6)0.13g(0.01mmol)、4T−OH0.36g(0.69mmol)、N,N−ジメチルアミノピリジン(東京化成製、28mg、0.23mmol)を加え、系内を窒素置換した。そこに脱水DMF4mLを加え、50 ℃に加温し4T−OHを溶解させた。さらにジシクロヘキシルカルボジイミド(東京化成製、0.54g、2.64mmol)を加え80℃に昇温し72時間加熱撹拌を行った。ポリマーの精製は展開溶媒としてTHFを用いBio−Beads S−X1(Bio−Rad製)を用いて行った。
このようにして、下記式(5)で表されるブロックコポリマー(BCP1、ポリエチレングリコールメチルエーテル残基の分子量Mn=5000)を得た。
図3に、このBCP1の1H−NMRチャートを示す。
【0077】
【化13】
【0078】
実施例2 ブロックコポリマー(BCP2)の製造
実施例1(2)−(イ)において、ポリエチレングリコールメチルエーテル残基のMnが5000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシドの代わりに、ポリエチレングリコールメチルエーテル残基のMnが2000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド(分子量2000のモノメチルポリエチレングリコール(Aldrich社製)を用いて合成したものである。)を用いた以外は、実施例1と同様な操作を行い、ブロックコポリマー(BCP2、ポリエチレングリコール鎖のMn=2000)を製造した。
【0079】
実施例3 ブロックコポリマー(BCP3)の製造
実施例1(2)−(イ)において、ポリエチレングリコールメチルエーテル残基のMnが5000のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシドの代わりに、ポリエチレングリコールメチルエーテル残基のMnが750のα−メトキシ−ω−トルエンスルホニル−ポリエチレンオキシド(分子量750のモノメチルポリエチレングリコール(Aldrich社製)を用いて合成したものである。)を用いた以外は、実施例1と同様な操作を行い、ブロックコポリマー(BCP3、ポリエチレングリコール鎖のMn=750)を製造した。
【0080】
前記の実施例1で得られたブロックコポリマー(BCP1)、実施例2で得られたブロックコポリマー(BCP2)及び比較例1で得られたブロックコポリマー(BCP3)の分析結果及び熱特性を表1に示す。
【0081】
【表1】
【0082】
[注]
1)X:重合体中におけるポリエチレングリコールユニットの分子量割合
2)Y:重合体中におけるペンダント型チオフェンユニットの分子量割合
3)PDI:分子量分布(Mw/Mn)
4)層転移温度:示差熱分析装置[SII社製「DSC220C」]を用いた示差熱分析測定と、偏光顕微鏡[OLYMPUS社製「BX51」及び加熱ステージ[METTLER社製「FP80HT」]を用いた観察により決定した。C=結晶相、Sm=スメクチック液晶相、I=等方性液体相を表し、表1中の「C XXX Sm YYYI」のごとき表記は、XXX℃で結晶相からスメクチック液晶層に転移し、YYY℃でスメクチック液晶相から等方性液体相に転移することを示す。
【0083】
実施例4
予め、過酸化水素・硫酸混合液で表面処理を行っておいたシリカ基板上に、数平均分子量Mn22,000、分子量分布1.22のBCP1薄膜(厚さ:40nm)を、実施例1で得られたBCP1のクロロホルム溶液を塗液として、スピンコート法により製膜した。その後、密封可能なガラス容器内にベンゼンを満たしたサンプル管および上記BCP1薄膜を入れて密封し、室温で48時間ソルベントアニール処理を行うことで、ミクロ相分離を誘起した。
さらにこのフィルムに307nm付近のUVのみを透過する307nmバンドパスフィルターを用いて、365nmのUV光を照射強度4.5mW/cm2で照射し、メタノール溶媒でリンス処理を行うことでポーラスフィルムを作製した。このフィルムに真空蒸着法を用いて酸化モリブデン(MoO3)を3nm、さらに金を50nm積層することでトップコンタクト型電界効果トランジスター(FET)素子を得た。
【0084】
実施例5
ブロックコポリマーとして、数平均分子量Mn14600、分子量分布1.18の実施例2で得られたBCP2を用いた以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0085】
実施例6
実施例4において、UV照射時に307nmバンドパスフィルターを用いる代わりに、紫外線透過可視吸収フィルターを用いた以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0086】
実施例7
実施例4において、307nmバンドパスフィルターを用いなかったこと以外は、実施例4と同様にしてポーラスフィルムを作製し、さらに薄膜FET素子を得た。
【0087】
比較例1
実施例4で用いたBCP1の製造における、(3)ブロックコポリマー(BCP1)の合成において、系内にジシクロヘキシルカルボジイミドを加え80℃に昇温した後の加熱攪拌時間を24時間とした以外は、実施例4と同様にして薄膜を得た。合成により得られたブロックコポリマーにおける、カルボン酸のオリゴチオフェン化合物との縮合率は30%であった。
【0088】
比較例2
実施例1(3)のブロックコポリマー(BCP1)の合成において、オリゴチオフェン誘導体として、4T−OHの代わりに、チオフェン単位の繰り返しを9単位としたものを用いて、実施例1(3)と同様にしてペンダント型ブロック重合体の合成を試みたが、オリゴチオフェン誘導体が溶媒に溶解せず、合成できなかった。
【0089】
試験1 (電子顕微鏡観察)
実施例4〜7、及び比較例1の薄膜表面にJEOL製「JFC−1600」イオンプレーティング装置を用いて白金でイオンコーティング処理を行い、走査型電子顕微鏡(日本電子製「Carry scope JCM−5700」)により観察し、ナノポーラス形成の有無を確認した。実施例4〜7ではナノポーラスの形成が確認されたが比較例1ではミクロ相分離由来のポーラス構造体は形成されていなかった。
ナノポーラスが形成された場合の走査型電子顕微鏡(SEM)写真を、代表として実施例4について図4に示す。また、比較例1についてのSEM写真を図5に示す。
【0090】
試験2 (原子間力顕微鏡観察)
実施例4〜7の薄膜表面を、原子間力顕微鏡(JEOL製「JSPM−5400」)により観察し、ナノポーラスの平均直径、ナノポーラスの平均深さ、及び空孔密度を測定した。
なお、平均直径は5μm角の範囲で原子間力顕微鏡のAC−AFMモードにより測定し、断面解析処理において測定範囲内のポーラス構造体の平均を取った。
また、平均深さは、5μm角の範囲で原子間力顕微鏡のAC−AFMモードにより測定し、断面解析処理において測定範囲内のポーラス構造体の平均を取った。
そして、ナノポア密度は、任意の5点において、1μm2中のナノポアの個数を数えることにより測定した。
結果を表2に示す。
【0091】
【表2】
【0092】
実施例4と5を比較すると、より分子量の低い実施例5では、ポアのサイズが小さくなるが、ポアの密度も小さくなってしまう。実施例4と比較して実施例6はナノポア直径がより大きく、実施例7はさらに直径が大きくなっている。バンドパスフィルターを用いた場合、紫外線透過可視吸収フィルターを用いた場合、無処理の場合の順でナノポア直径が大きくなっているのは、波長選択性の低いUVを照射するほどオリゴチオフェン縮合重合体構造中の結合の分解が進行し、発生した分子の断片がリンスにより洗い流されることに起因していると考えられる。このことから、バンドパスフィルターを用いることで、より精密にポーラスサイズを制御できることがわかる。
【0093】
実施例4及び5の薄膜について、RIGAKU社製「Ultima IV」を用いてXRD測定を行った。図6に、実施例4及び5の薄膜(図中、「BCP1」、「BCP2」と記載。)、及び実施例4のソルベントアニール処理を行う前のBCP1薄膜(図中、「製膜後」と記載。)のXRDパターンを示す。実施例4及び5のいずれのパターンにも、チオフェン四量体1単位の分子長に起因する回折ピークが得られ、チオフェン四量体構造が基板に対して垂直に配向していることが示された。なお、図中には掲載されていないが、実施例5のソルベントアニール処理を行う前のBCP2薄膜のXRD測定においても、実施例4のソルベントアニール処理を行う前のBCP1薄膜と同様に、回折ピークは検出されなかった。
【0094】
試験4 (移動度測定)
実施例4及び5の有機FETの電気特性評価は、1.3×10-3Pa以下の真空・遮光下で行った。Id−Vg特性を測定するために(ドレイン電流が飽和領域をとるゲート電圧値で、ゲート電圧を一定に保ち、ドレイン電圧を走査する)アジレント社製の半導体パラメーターアナライザー「AgilentB1500A」を用いて作製した有機FETにゲート電圧;−100V、ドレイン電圧;+20V→−100V、−1Vstepの条件でゲート電圧を印加して、室温(25℃)での特性を測定した。測定したドレイン電流−ソース・ドレイン電圧曲線のチャネルコンダクタンスから移動度(μ)を見積もった。実施例4及び5の有機EFTの移動度及び閾値電圧を表3に示す。
【0095】
【表3】
【0096】
さらにId−Vd特性の測定を行うために(ゲート電圧をある一定値にしておき、ドレイン電圧を走査しその時のドレイン電流を測定する。順次、ゲート電圧を変化させ何回も測定する。)半導体パラメーターアナライザー「AgilentB1500A」を用いて作製した有機電界効果トランジスターゲート電圧;20V→−100V、−10Vステップ、ドレイン電圧;+20V→−100V、−1Vstepの条件で測定を行ったところ図7のように、ゲート電圧の増加と共に電流値が増加していく、典型的なp型のトランジスター特性を示した。
【産業上の利用可能性】
【0097】
本発明により得られる有機ナノポーラス材料は、有機FET及びその他の有機デバイスとしての利用について有望であり、それらを用いた電子機器等の開発の発展に寄与することが期待される。
【特許請求の範囲】
【請求項1】
下記一般式(1)で表されるカルボン酸基含有重合体(B)における任意の−COOH基に、下記一般式(2)で表されるオリゴチオフェン誘導体(C)をエステル化反応させてなるブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含み、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー。
【化1】
(一般式(1)中、R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基、Xは親水基、Zはハロゲン原子、mは2〜500の整数、pは0又は1を示す。)
【化2】
(一般式(2)中、R4及びR5は、それぞれ独立に水素原子又は炭素数1〜3のアルキル基、Xは親水性基、Yは炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基、Aは炭素数2〜16のアルカンジイル基、kは2〜8の整数、nは0又は1を示す。)
【請求項2】
一般式(1)におけるXで示される親水性基が、ポリエーテル残基である請求項1に記載の液晶性有機半導体ポリマー組成物。
【請求項3】
請求項1又は2に記載の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする有機ナノポーラス材料。
【請求項4】
電磁波照射処理が、紫外線照射処理である請求項3に記載の有機ナノポーラス材料。
【請求項5】
紫外線の波長領域が、ブロック共重合体(A)の分子構造中に含まれるo−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない請求項4に記載の有機ナノポーラス材料。
【請求項6】
請求項3〜5のいずれかに記載の有機ナノポーラス材料を用いることを特徴とする有機電界効果トランジスター。
【請求項7】
以下の工程を有する請求項1又は2に記載の液晶性有機半導体ポリマー組成物の製造方法。
工程(a)一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程。
工程(b)o−ニトロベンジル基の2〜5位のいずれかに親水性基の結合した官能基を有する原子移動ラジカル重合制御剤を用いて、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合法により重合することにより、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体を製造する工程。
工程(c)前記工程(a)で得られたオリゴチオフェン誘導体と、前記工程(b)で得られたカルボン酸基含有重合体とをエステル化反応させる工程。
【請求項8】
工程(b)におけるカルボン酸基1個以上を有するビニル化合物単量体が、(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸又はシトラコン酸である請求項7に記載の液晶性有機半導体ポリマーの製造方法。
【請求項9】
ビニル化合物単量体が、(メタ)アクリル酸である請求項8に記載の液晶性有機半導体ポリマーの製造方法。
【請求項1】
下記一般式(1)で表されるカルボン酸基含有重合体(B)における任意の−COOH基に、下記一般式(2)で表されるオリゴチオフェン誘導体(C)をエステル化反応させてなるブロック共重合体(A)単独、又は該ブロック共重合体(A)と、下記一般式(1)で表されるカルボン酸基含有重合体(B)とを含み、ブロック共重合体(A)におけるオリゴチオフェン含有エステル基と、ブロック共重合体(A)及びカルボン酸基含有重合体(B)の合計中における全カルボン酸との割合が100:0〜35超:65未満(モル比)であることを特徴とする液晶性有機半導体ポリマー。
【化1】
(一般式(1)中、R1は水素原子、メチル基又は−CH2COOH基、R2は水素原子又はメチル基、R3は水素原子又は−COOH基、Xは親水基、Zはハロゲン原子、mは2〜500の整数、pは0又は1を示す。)
【化2】
(一般式(2)中、R4及びR5は、それぞれ独立に水素原子又は炭素数1〜3のアルキル基、Xは親水性基、Yは炭素数1〜16のアルキル基若しくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基、Aは炭素数2〜16のアルカンジイル基、kは2〜8の整数、nは0又は1を示す。)
【請求項2】
一般式(1)におけるXで示される親水性基が、ポリエーテル残基である請求項1に記載の液晶性有機半導体ポリマー組成物。
【請求項3】
請求項1又は2に記載の液晶性有機半導体ポリマー組成物に、アニール処理、電磁波照射処理ならびに、水及び/又は低級アルコールによるリンス処理を順次施すことにより得られることを特徴とする有機ナノポーラス材料。
【請求項4】
電磁波照射処理が、紫外線照射処理である請求項3に記載の有機ナノポーラス材料。
【請求項5】
紫外線の波長領域が、ブロック共重合体(A)の分子構造中に含まれるo−ニトロベンジル誘導体基の極大吸収波長を含み、かつオリゴチオフェン含有エステル基の極大吸収波長を実質上含まない請求項4に記載の有機ナノポーラス材料。
【請求項6】
請求項3〜5のいずれかに記載の有機ナノポーラス材料を用いることを特徴とする有機電界効果トランジスター。
【請求項7】
以下の工程を有する請求項1又は2に記載の液晶性有機半導体ポリマー組成物の製造方法。
工程(a)一方の末端のチオフェン環の5位に、酸素原子を介して、もしくは介さずにヒドロキシアルキル基が導入され、他方の末端のチオフェン環の5位に、炭素数1〜16のアルキル基もしくはアルコキシ基又は炭素数2〜17のアルコキシカルボニル基が導入されてなる、チオフェン環数2〜8のオリゴチオフェン誘導体を製造する工程。
工程(b)o−ニトロベンジル基の2〜5位のいずれかに親水性基の結合した官能基を有する原子移動ラジカル重合制御剤を用いて、カルボン酸基1個以上を有するビニル化合物単量体を、原子移動ラジカルリビング重合法により重合することにより、前記一般式(1)で表される親水性基が結合してなるo−ニトロベンジル基を有するカルボン酸基含有重合体を製造する工程。
工程(c)前記工程(a)で得られたオリゴチオフェン誘導体と、前記工程(b)で得られたカルボン酸基含有重合体とをエステル化反応させる工程。
【請求項8】
工程(b)におけるカルボン酸基1個以上を有するビニル化合物単量体が、(メタ)アクリル酸、マレイン酸、フマル酸、イタコン酸又はシトラコン酸である請求項7に記載の液晶性有機半導体ポリマーの製造方法。
【請求項9】
ビニル化合物単量体が、(メタ)アクリル酸である請求項8に記載の液晶性有機半導体ポリマーの製造方法。
【図1】

【図2】

【図3】
【図4】

【図5】

【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−236299(P2011−236299A)
【公開日】平成23年11月24日(2011.11.24)
【国際特許分類】
【出願番号】特願2010−107644(P2010−107644)
【出願日】平成22年5月7日(2010.5.7)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構(ロボット・新技術イノベーションプログラム)、「異分野融合型次世代デバイス製造技術開発・プロジェクト」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504145342)国立大学法人九州大学 (960)
【出願人】(509130000)技術研究組合BEANS研究所 (13)
【出願人】(000102980)リンテック株式会社 (1,750)
【Fターム(参考)】
【公開日】平成23年11月24日(2011.11.24)
【国際特許分類】
【出願日】平成22年5月7日(2010.5.7)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人新エネルギー・産業技術総合開発機構(ロボット・新技術イノベーションプログラム)、「異分野融合型次世代デバイス製造技術開発・プロジェクト」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504145342)国立大学法人九州大学 (960)
【出願人】(509130000)技術研究組合BEANS研究所 (13)
【出願人】(000102980)リンテック株式会社 (1,750)
【Fターム(参考)】
[ Back to top ]