
潜在型TGF−βの活性化抑制剤
本発明の課題は、潜在型TGF−βの活性化抑制剤、TGF−β活性化抑制物質のスクリーニング方法、またはTGF−βが関与する疾患の治療薬を提供することである。本発明は、ビメンチンとLAPの部分断片との結合を阻害する物質を含有する、潜在型TGF−βの活性化抑制剤およびTGF−βが関与する疾患の治療薬、ならびにビメンチンとLAPの部分断片との結合を阻害する物質を評価することを特徴とするTGF−β活性化抑制物質のスクリーニング方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、潜在型トランスフォーミング・グロース・ファクター−β(transforming growth factor−β、以下「TGF−β」という。)の活性化抑制剤、TGF−β活性化抑制物質のスクリーニング方法、およびTGF−βが関与する疾患の治療薬に関する。
【背景技術】
【0002】
TGF−βは、細胞増殖・分化、細胞外マトリックス合成、アポトーシス、免疫機能などの制御等、多彩な生理活性を有するサイトカインとして知られており、その機能不全は、ヒトの病態、例えば癌、組織の線維化、自己免疫疾患等に関連していることが知られている(非特許文献1参照)。
【0003】
TGF−βは不活性の潜在型分子として分泌されるため、生理作用を発揮するためには細胞から分泌された後に活性化を受ける必要がある(非特許文献2参照)。潜在型TGF−βの活性化段階を制御することはTGF−βの活性を制御することにつながることから、その活性化機構が精力的に研究されている。これまでに、プロテアーゼによる潜在型TGF−βの限定分解を介した機構、細胞表面分子への結合に伴う構造変化を介した機構、またはその両者を組み合わせた機構等が報告されている。例えば、潜在型TGF−βを限定分解するプロテアーゼとして、プラスミン(非特許文献3参照)、マトリックスメタロプロテアーゼ(非特許文献4参照)等が報告されている。潜在型TGF−βが結合し、その構造を変化させる細胞表面分子として、トロンボスポンジン−1(非特許文献5参照)、インテグリンαvβ6(非特許文献6参照)等が報告されている。インテグリンαvβ8への結合とそれに伴う構造変化により、マトリックスメタロプロテアーゼによる限定分解が生じるという機構も報告されている(非特許文献7参照)。
【0004】
一方、潜在型TGF−βを構成するタンパク質であるラテンシー・アソシエイテッド・ペプチド(Latency Associated Peptide、以下「LAP」という。)の部分断片が、潜在型TGF−βの活性化を促進すること、該部分断片が血管内皮細胞の表面に結合することが報告されている(非特許文献8参照)。
【0005】
ビメンチンは、多くの種類の細胞に存在する中間径フィラメントのサブユニットタンパク質であり、ビメンチン繊維として細胞質に網目状構造で存在する。ビメンチンに結合する物質としては、抗ビメンチンモノクローナル抗体が知られている。抗ビメンチンモノクローナル抗体VIM3B4は、げっ歯類以外の哺乳動物のビメンチンに結合し、その認識部位はビメンチンの第二ロッド領域であることが報告されている(非特許文献9参照)。また、抗ビメンチンモノクローナル抗体V9(非特許文献9参照)は、ヒトマクロファージの活性化に伴い細胞表面または細胞外に分泌されるビメンチンに結合し得ることが報告されている(非特許文献10参照)。
【0006】
【非特許文献1】「ニュー・イングランド・ジャーナル・オブ・メディシン(New England Journal of Medicine)」、2000年、第342巻、p.1350−1358
【非特許文献2】「ジャーナル・オブ・セル・サイエンス(Journal of Cell Science)」、2003年、第116巻、p.217−224
【非特許文献3】「ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology)」、1989年、第109巻、p.309−315
【非特許文献4】「ジーンズ・アンド・デベロップメント(Genes and Development)」、2000年、第14巻、p.163−176
【非特許文献5】「サイトカイン・アンド・グロース・ファクター・レビュー(Cytokine and Growth Factor Review)11,59−69(2000)」、2000年、第11巻、p.59−69
【非特許文献6】「セル(Cell)」、1999年、第96巻、p.319−328
【非特許文献7】「ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology)」、2002年、第157巻、p.493−507
【非特許文献8】「エンドセリウム(Endothelium)」、2002年、第9巻、p.25−36
【非特許文献9】「エクスペリメンタル・セル・リサーチ(Experimental Cell Research)」、1992年、第201巻、p.1−7
【非特許文献10】「ネイチャー・セル・バイオロジー(Nature Cell Biology)」、2003年、第5巻、p.59−63
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、潜在型TGF−βの活性化抑制剤、TGF−β活性化抑制物質のスクリーニング方法、またはTGF−βが関与する疾患の治療薬を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明は、以下の(1)〜(10)に関する。
(1)ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤。
(2)ビメンチンとLAPの部分断片との結合を阻害する物質が、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体または該抗体断片である、上記(1)のTGF−β活性化抑制剤。
(3)LAPの部分断片が配列番号1〜16のいずれかで表されろアミノ酸配列を有するペプチドである、上記(1)または(2)のいずれか1つのTGF−β活性化抑制剤。
(4)(i)LAPの部分断片を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(ii)LAPの部分断片およびスクリーニングの対象となる物質を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(iii)(i)および(ii)の測定量から、該物質の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価すること、
(iv)該結合阻害活性を有する物質をTGF−β活性化抑制物質として選択すること、
からなるTGF−β活性化抑制物質のスクリーニング方法。
(5)ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する、TGF−βが関与する疾患の治療薬。
(6)TGF−βが関与する疾患が動脈硬化症、癌、炎症、線維症である、上記(5)の治療薬。
(7)ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−β活性化抑制方法。
(8)ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−βが関与する疾患の治療方法。
(9)TGF−β活性化抑制剤の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
(10)TGF−βが関与する疾患の治療薬の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【発明の効果】
【0009】
本発明により、ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤、またはTGF−β活性化抑制物質のスクリーニング方法が提供される。本発明のTGF−βの活性化抑制剤は、TGF−βが関与する疾患の治療薬として利用することができる。
【図面の簡単な説明】
【0010】
【図1】図1は、BAECに対する潜在型TGF−β活性化ペプチドの結合度(RLU/ウェル)を示す図である。「Peptide−25」のカラムは100μg/mlのPeptide−25、「Peptide−25+Ab」のカラムは100μg/mlのPeptide−25および5μg/mlの抗ビメンチン抗体VIM3B4、「Peptide−25N」のカラムは100μg/mlのPeptide−25N、「Peptide−25N+Ab」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのVIM3B4をそれぞれ添加した場合の結合度を示す。**は、スチューデントのt検定において、危険率1%未満で有意差があることを示す。
【図2】図2は、BAECを剃刀で剥離した後、剥離部分に遊走してきた観察視野当たりの細胞数(個/HPF)を示す図である。「Control+NI」のカラムは5μg/mlの非免疫マウスIgG、「Peptide−21+NI」のカラムは100μg/mlのPeptide−21および5μg/mlのIgG、「Peptide−21+Ab」のカラムは100μg/mlのPeptide−21および5μg/mlの抗ビメンチン抗体V9、「Peptide−25+NI」のカラムは100μg/mlのPeptide−25および5μg/mlのIgG、「Peptide−25+Ab」のカラムは100μg/mlのPeptide−25および5μg/mlのV9をそれぞれ添加した場合の細胞数を示す。*および**は、スチューデントのt検定において、それぞれ危険率5%および1%未満で有意差があることを示す。
【図3】図3は、BAECに対する潜在型TGF−β活性化ペプチドの結合度(RLU/well)を示す図である。「Peptide−25+KM511」のカラムは100μg/mlのPeptide−25および5μg/mlの抗G−CSF変異体抗体KM511、「Peptide−25+KM3565」のカラムは100μg/mlのPeptide−25および5μg/mlの抗ビメンチン抗体KM3565、「Peptide−25N+KM511」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのKM511、「Peptide−25N+KM3565」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのKM3565をそれぞれ添加した場合の結合度を示す。**は、スチューデントのt検定において、危険率1%未満で有意差があることを示す。
【図4】図4は、酵素免疫測定法により測定した、モノクローナル抗体KM3565のヒトビメンチン部分ペプチドに対する反応性を示す図である。上段が化合物1を、下段が化合物2を、それぞれ固層化したプレートを用いた測定結果を示す。X軸は添加したKM3565の濃度を、Y軸は抗体の結合度を表す吸光度をそれぞれ示す。
【図5】図5は、酵素免疫測定法により測定した、モノクローナル抗体KM3565のヒトビメンチン蛋白に対する反応性を示す図である。上段がビメンチン蛋白を、下段が大腸菌由来夾雑蛋白を、それぞれ固層化したプレートを用いた測定結果を示す。X軸は添加したKM3565の濃度を、Y軸は抗体の結合度を表す吸光度をそれぞれ示す。
【図6】図6は、酵素免疫測定法により測定した、市販抗ビメンチン抗体(▲;VIM3B4、■;V9)の化合物1およびビメンチン蛋白に対する反応性を示す図である。上段が化合物1を、下段がビメンチン蛋白を、それぞれ固層化したプレートを用いた測定結果を示す。
【発明を実施するための最良の形態】
【0011】
本発明において、ビメンチンとLAPの部分断片との結合を阻害する物質としては、該阻害活性を有する物質であれば、合成化合物でも天然物質でも特に制限なく用いることができるが、好ましくは、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体が用いられる。
抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体のうち、ビメンチンとLAPの部分断片との結合を阻害する活性を有するものの選択方法としては、後述の、本発明のTGF−βの活性化を抑制する物質のスクリーニング方法を用いることができる。
本発明に用いる抗ビメンチン抗体は、ポリクローナル抗体およびモノクローナル抗体を包含する。
【0012】
モノクローナル抗体としては、ハイブリドーマが産生する抗体、ヒト化抗体およびヒト抗体等があげられる。
ハイブリドーマとは、ヒト以外の哺乳動物に抗原を免疫して取得されたB細胞と、マウス等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を産生する細胞である。
ヒト化抗体としては、ヒト型キメラ抗体、ヒト型相同性決定領域(complementarity determining region、以下「CDR」という。)移植抗体等があげられる。
【0013】
ヒト型キメラ抗体は、ヒト以外の動物の抗体重鎖可変領域(以下、重鎖はH鎖として、可変領域はV領域として「HV」または「VH」ともいう。)および抗体軽鎖可変領域(以下、軽鎖はL鎖として「LV」または「VL」ともいう。)とヒト抗体の重鎖定常領域(以下、定常領域はC領域として「CH」ともいう。)およびヒト抗体の軽鎖定常領域(以下「CL」ともいう。)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0014】
本発明に用いるヒト型キメラ抗体は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する動物細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下「hIg」という。)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0015】
ヒト型キメラL鎖とは、ヒト以外の動物のVLとヒト抗体のCLとからなる抗体L鎖を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
本発明に用いるヒト型キメラL鎖は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VLをコードするcDNAを取得し、ヒト抗体CLをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型キメラL鎖発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラL鎖のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0016】
ヒト型キメラH鎖とは、ヒト以外の動物のVHとヒト抗体のCHとからなる抗体H鎖を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0017】
本発明に用いるヒト型キメラH鎖は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VHをコードするcDNAを取得し、ヒト抗体CHをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型キメラH鎖発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラH鎖のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。
【0018】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体を意味する。
本発明に用いるヒト型CDR移植抗体は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する動物細胞用発現ベクターにそれぞれ挿入してヒト型CDR移植抗体発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植抗体を発現させ、製造することができる。
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型CDR移植抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0019】
本発明に用いるヒト型CDR移植キメラL鎖は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVLのCDR配列を任意のヒト抗体のVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCLをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型CDR移植キメラL鎖発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植キメラL鎖を発現させ、製造することができる。
ヒト型CDR移植キメラL鎖のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0020】
本発明に用いるヒト型CDR移植キメラH鎖は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVHのCDR配列を任意のヒト抗体のVHのCDR配列に移植したH領域をコードするcDNAを構築し、ヒト抗体のCHをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型CDR移植キメラH鎖発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植キメラH鎖を発現させ、製造することができる。
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。
【0021】
ヒト抗体は、元来、ヒト体内に天然に存在する抗体を意味するが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーおよびヒト抗体産生トランスジェニック動物から得られる抗体等も含まれる。
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を産生するリンパ球を培養でき、培養物中より該抗体を精製することができる。
【0022】
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab(fragment of antigen bindingの略)、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0023】
ヒト抗体産生トランスジェニック動物は、ヒト抗体遺伝子が細胞内に組込まれた動物を意味する。具体的には、マウスES細胞へヒト抗体遺伝子を導入し、該ES細胞を他のマウスの初期胚へ移植後、発生させることによりヒト抗体産生トランスジェニック動物を作製することができる。ヒト抗体産生トランスジェニック動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体産生ハイブリドーマを得、培養することで培養物中にヒト抗体を産生蓄積させることができる。
【0024】
本発明に用いる抗体または抗体のL鎖もしくはH鎖の部分断片としては、Fab、Fab’、F(ab’)2、一本鎖抗体(single chain Fv、以下「scFv」という。)、ジスルフィド安定化抗体(disulfide stabilized Fv、以下「dsFv」という。)、CDRを含むペプチド等があげられる。
【0025】
Fabは、IgGを蛋白質分解酵素パパインで処理して得られる断片のうち(H鎖の224番目のアミノ酸残基で切断される)、H鎖のN末端側約半分のアミノ酸とL鎖全体がジスルフィド結合で結合した分子量約5万の抗原結合活性を有する抗体断片である。
本発明に用いるFabは、ビメンチンに特異的に反応する抗体を蛋白質分解酵素パパインで処理して得ることができる。または、該抗体のFabをコードするDNAを原核生物用発現ベクターもしくは真核生物用発現ベクターに挿入し、該ベクターを原核生物もしくは真核生物へ導入することにより発現させ、Fabを製造することができる。
【0026】
F(ab’)2は、IgGを蛋白質分解酵素ペプシンで処理して得られる断片のうち(H鎖の234番目のアミノ酸残基で切断される)、Fabがヒンジ領域のジスルフィド結合を介して結合されたものよりやや大きい、分子量約10万の抗原結合活性を有する抗体断片である。
本発明に用いるF(ab’)2は、ビメンチンに特異的に反応する抗体を蛋白質分解酵素ペプシンで処理して得ることができる。または、下記のFab’をチオエーテル結合もしくはジスルフィド結合させ、作製することができる。
Fab’は、上記F(ab’)2のヒンジ領域のジスルフィド結合を切断した分子量約5万の抗原結合活性を有する抗体断片である。
本発明に用いるFab’は、ビメンチンに特異的に反応するF(ab’)2を還元剤ジチオスレイトール処理して得ることができる。または、該抗体のFab’断片をコードするDNAを原核生物用発現ベクターもしくは真核生物用発現ベクターに挿入し、該ベクターを原核生物もしくは真核生物へ導入することによりFab’を発現させ、製造することができる。
【0027】
scFvは、一本のVHと一本のVLとを適当なペプチドリンカー(以下「P」という。)を用いて連結した、VH−P−VLまたはVL−P−VHポリペプチドを示す。本発明に用いるscFvに含まれるVHおよびVLは、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体のいずれをも用いることができる。
本発明に用いるscFvは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得し、scFvをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
【0028】
dsFvは、VHおよびVL中のそれぞれ1アミノ酸残基をシステイン残基に置換したポリペプチドを該システイン残基間のジスルフィド結合を介して結合させたものをいう。システイン残基に置換するアミノ酸残基はReiterらにより示された方法[Protein Engineering,7,697(1994)]に従って、抗体の立体構造予測に基づいて選択することができる。本発明に用いるdsFvに含まれるVHおよびVLは、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体のいずれをも用いることができる。
【0029】
本発明に用いるdsFvは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得し、dsFvをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
【0030】
CDRを含むペプチドは、H鎖またはL鎖CDRの少なくとも1領域以上を含んで構成される。複数のCDRは、直接または適当なペプチドリンカーを介して結合させることができる。
本発明に用いるCDRを含むペプチドは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得した後、CDRをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
また、CDRを含むペプチドは、Fmoc法(フルオレニルメチルオキシカルボニル法)、tBoc法(t−ブチルオキシカルボニル法)等の化学合成法によって製造することもできる。
【0031】
本発明に用いる抗体の誘導体としては、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体またはそれらの抗体断片に放射性同位元素、蛋白質または低分子の化合物等を結合させた抗体の誘導体等があげられる。
本発明に用いる抗体の誘導体は、ビメンチンに特異的に反応する抗体または抗体断片のH鎖もしくはL鎖のN末端側もしくはC末端側、抗体または抗体断片中の適当な置換基もしくは側鎖、さらには抗体または抗体断片中の糖鎖に放射性同位元素、蛋白質もしくは低分子の化合物等を化学的手法[抗体工学入門(金光修著1994年(株)地人書館)]により結合させることにより製造することができる。
【0032】
または、ビメンチンに特異的に反応する抗体もしくは抗体断片をコードするDNAと、結合させたい蛋白質をコードするDNAを連結させて発現ベクターに挿入し、該発現ベクターを宿主細胞へ導入する、遺伝子工学的手法によっても製造することができる。
放射性同位元素としては、32P、3H、14C、131I、125I等があげられ、例えば、クロラミンT法等により、抗体に結合させることができる。
【0033】
低分子の薬剤としては、ハイドロコーチゾン、プレドニゾン等のステロイド剤、アスピリン、インドメタシン等の非ステロイド剤、金チオマレート、ペニシラミン等の免疫調節剤、アドリアマイシン、ダウノマイシン等の免疫抑制作用を有する抗癌剤、サイクロスポリン、FK506、サイクロフォスファミド、アザチオプリン等の免疫抑制剤、マレイン酸クロルフェニラミン、クレマシチンのような抗ヒスタミン剤等の抗炎症剤[炎症と抗炎症療法昭和57年 医歯薬出版株式会社]等があげられる。
【0034】
例えば、ダウノマイシンと抗体とを結合させる方法としては、グルタールアルデヒドを介してダウノマイシンと抗体のアミノ基間を結合させる方法、水溶性カルボジイミドを介してダウノマイシンのアミノ基と抗体のカルボキシル基を結合させる方法等があげられる。
【0035】
蛋白質としては、β−ガラクトシダーゼ、グルコースオキシダーゼ、ペルオキシダーゼ、アルカリホスファターゼ、グルコース−6−リン酸脱水素酵素等があげられる。
蛋白質と抗体とを結合させる方法としては、抗体または抗体断片をコードするcDNAに蛋白質をコードするcDNAを連結させ、融合抗体をコードするDNAを構築し、該DNAを原核生物または真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより蛋白質と抗体が融合した融合抗体を発現させる方法等があげられる。
【0036】
以下、ビメンチンに反応するポリクローナル抗体の作製方法について記す。
ビメンチンをコードするcDNAを含む発現ベクターを大腸菌、酵母、昆虫細胞、動物細胞等に導入して形質転換株を作製し、該形質転換株を培養・精製することにより、リコンビナントビメンチンを得る。または、ヒト血管内皮細胞、例えば臍帯静脈血管内皮細胞(HUVEC、クラボウ社製)等を培養・精製することによりビメンチンを得る。これらのビメンチン、ビメンチンの部分断片ポリペプチドの精製標品、またはビメンチンの一部のアミノ酸配列を有するペプチドを抗原に用いる。
【0037】
これら抗原はそのまま、またはキーホールリンペットヘモシアニン(KLH)、牛血清アルブミン(BSA)、メチル化牛血清アルブミン(メチル化BSA)、牛チログロブリン(THY)等の分子量の大きいキャリアタンパク質と結合させて投与する。
免疫に用いる動物としては、マウス、ラット、ハムスター、ラビット等ハイブリドーマを作製することが可能であれば、いかなるものでもよい。下記に、マウスおよびラットを用いる例について説明する。
【0038】
3〜20週令のマウスまたはラットに、上記方法で調製した抗原を免疫し、その動物の脾臓、リンパ節、末梢血より抗体産生細胞を採取する。免疫は、動物の皮下、静脈内または腹腔内に、適当なアジュバントとともに抗原を数回投与することにより行う。アジュバンドとしては、フロインドの完全アジュバント(Complete Freund’s Adjuvant)または、水酸化アルミニウムゲルと百日咳菌ワクチン等があげられる。各投与後3〜7日目に免疫動物の眼底静脈叢または尾静脈より採血し、免疫に用いた抗原に対しての反応性について、酵素免疫測定法〔酵素免疫測定法(ELISA法):医学書院刊(1976年)、Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory(1988)〕等で確認する。
【0039】
免疫に用いた抗原に対し、その血清が充分な抗体価を示した非ヒト哺乳動物より血清を取得し、該血清よりポリクローナル抗体を分離、精製する。
ポリクローナル抗体を分離、精製する方法としては、遠心分離、40〜50%飽和硫酸アンモニウムによる塩析、カプリル酸沈殿〔Antibodies,A Laboratory manuml,Cold Spring Harbor Laboratory,(1988)〕、またはDEAE−セファロースカラム、陰イオン交換カラム、プロテインAもしくはG−カラム、ゲル濾過カラム等を用いるクロマトグラフィー等を、単独または組み合わせて処理する方法があげられる。
【0040】
以下、ビメンチンに反応するモノクローナル抗体の作製方法について記す。
1.抗ビメンチンモノクローナル抗体の作製
(1)抗体産生細胞の調製
免疫に用いた抗原に対し、その血清が十分な抗体価を示したマウスまたはラットを抗体産生細胞の供給源とする。抗原物質の最終投与後3〜7日目に、免疫したマウスまたはラットより公知の方法[アンティボディズ・ア・ラボラトリー・マニュアル、コールド・スプリングハーバー・ラボラトリー(Antibodies−A Laboratory Manual Cold Spring Harbor Laboratory,1988)、以下「アンチボディズ・ア・ラボラトリー・マニュアル」という。]に準じて脾臓を摘出し、脾細胞と骨髄腫細胞とを融合させる。
【0041】
(2)骨髄腫細胞の調製
骨髄腫細胞としては、マウスから得られた株化細胞である、8−アザグアニン耐性マウス(BALB/c由来)骨髄腫細胞株P3−X63Ag8−U1(P3−U1)[Euro.J.Immunol.,6,511(1976)]、SP2/0−Ag14(SP−2)[Nature,276,269(1978)]、P3−X63−Ag8653(653)[J.Immunol.,123,1548(1979)]、P3−X63−Ag8(X63)[Nature,256,495(1975)]等、イン・ビトロ(in vitro)で増殖可能な骨髄腫細胞であればいかなるものでもよい。これらの細胞株の培養および継代については公知の方法(アンチボディズ・ア・ラボラトリー・マニュアル)に従い、細胞融合時までに2×107個以上の細胞数を確保する。
【0042】
(3)細胞融合
上記で得られた抗体産生細胞と骨髄腫細胞とを洗浄したのち、ポリエチレングライコール−1000(PEG−1000)などの細胞凝集性媒体を加え、細胞を融合させ、培地中に懸濁させる。細胞の洗浄にはMEM培地またはPBS(リン酸二ナトリウム1.83g、リン酸一カリウム0.21g、塩化ナトリウム7.65g、蒸留水1リットル、pH7.2)等を用いる。また、融合細胞を懸濁させる培地としては、目的の融合細胞のみを選択的に得られるように、HAT培地{正常培地[RPMI−1640培地にグルタミン(1.5mmol/l)、2−メルカプトエタノール(5×10−5mol/l)、ジェンタマイシン(10μg/ml)および牛胎児血清(FCS)(CSL社製、10%)を加えた培地]にヒポキサンチン(10−4mol/l)、チミジン(1.5×10−5mol/l)およびアミノプテリン(4×10−7mol/l)を加えた培地}を用いる。
培養後、培養上清の一部をとり、酵素免疫測定法により、抗原蛋白質に反応し、非抗原蛋白質に反応しないサンプルを選択する。ついで、限界希釈法によりクローニングを行い、酵素免疫測定法により安定して高い抗体価の認められたものをモノクローナル抗体産生ハイブリドーマ株として選択する。
【0043】
(4)抗ビメンチンモノクローナル抗体産生ハイブリドーマの選択
抗ビメンチンモノクローナル抗体を産生するハイブリドーマの選択は、アンチボディズ・ア・ラボラトリー・マニュアルに述べられている方法等に従い、以下に述べる測定法により行う。これらの方法により、後述する抗ビメンチンヒト化抗体、該抗体断片を産生する形質転換株の培養上清中に含まれる抗ビメンチン抗体またはすべての精製抗ビメンチン抗体の結合活性を測定することができる。
【0044】
酵素免疫測定法
抗原または抗原を発現した細胞等を96ウェルプレートにコートし、ハイブリドーマ培養上清または上述の方法で得られる精製抗体を第一抗体として反応させる。
第一抗体反応後、プレートを洗浄して第二抗体を添加する。
第二抗体とは、第一抗体のイムノグロブリンを認識できる抗体を、ビオチン、酵素、化学発光物質または放射線化合物等で標識した抗体である。具体的にはハイブリドーマ作製の際にマウスを用いたのであれば、第二抗体としては、マウスイムノグロブリンを認識できる抗体を用いる。
反応後、第二抗体を標識した物質に応じた反応を行ない、抗原に特異的に反応するモノクローナル抗体を生産するハイブリドーマとして選択する。
【0045】
(5)モノクローナル抗体の精製
プリスタン処理〔2,6,10,14−テトラメチルペンタデカン(Pristane)0.5mlを腹腔内投与し、2週間飼育する〕した8〜10週令のマウスまたはヌードマウスに、1(3)で得られた抗ビメンチンモノクローナル抗体産生ハイブリドーマ細胞2×107〜5×106細胞/匹を腹腔内に注射する。10〜21日間でハイブリドーマは腹水癌化する。該マウスまたはヌードマウスから腹水を採取し、遠心分離、40〜50%飽和硫酸アンモニウムによる塩析、カプリル酸沈殿法、DEAE−セファロースカラム、プロテインA−カラムまたはセルロファインGSL2000(生化学工業社製)のカラムなどを用いて、IgGまたはIgM画分を回収し、精製モノクローナル抗体とする。
精製モノクローナル抗体のサブクラスの決定は、マウスモノクローナル抗体タイピングキットまたはラットモノクローナル抗体タイピングキット等を用いて行うことができる。蛋白質量は、ローリー法または280nmでの吸光度より算出することができる。
抗体のサブクラスとは、クラス内のアイソタイプのことで、マウスでは、IgG1、IgG2a、IgG2b、IgG3、ヒトでは、IgG1、IgG2、IgG3、IgG4があげられる。
【0046】
(6)抗ビメンチンモノクローナル抗体の反応性
抗ビメンチンモノクローナル抗体の反応性を調べる方法として、インヒビションELISAがあげられる。
まず、1(4)に記したように抗原を固相化したプレートを準備し、第一抗体として抗ビメンチンモノクローナル抗体を反応させる。同時に適当に希釈したビメンチンを加えプレートに固相化したビメンチンと競合させる。その後は1(4)に示した方法と同様に検出することができる。抗ビメンチンモノクローナル抗体がビメンチンと反応する場合には、液相系で加えたビメンチン濃度依存的に固相化ビメンチンへの結合が阻害され、OD値が低下する。
【0047】
2.ヒト化抗体の作製方法(I)−抗ビメンチンヒト化抗体の作製方法
(1)ヒト化抗体発現用ベクターの構築
ヒト以外の動物の抗体からヒト化抗体を作製するために必要なヒト化抗体発現用ベクターを構築する。ヒト化抗体発現用ベクターとは、ヒト抗体のC領域であるCHおよびCLをコードする遺伝子が組み込まれた動物細胞用発現ベクターであり、動物細胞用発現ベクターにヒト抗体のCHおよびCLをコードする遺伝子をそれぞれ挿入することにより構築されたものである。
【0048】
ヒト抗体のC領域としては、例えば、ヒト抗体H鎖ではCγ1やCγ4、ヒト抗体L鎖ではCκ等の任意のヒト抗体のC領域を用いることができる。ヒト抗体のC領域をコードする遺伝子としてはエキソンとイントロンより成る染色体DNAを用いることができ、また、cDNAを用いることもできる。動物細胞用発現ベクターとしては、ヒト抗体C領域をコードする遺伝子を組込み発現できるものであればいかなるものでも用いることができる。
【0049】
例えば、pAGE107[Cytotechnology,3,133(1990)]、pAGE103[J.Biochem.,101,1307(1987)]、pHSG274[Gene,27,223(1984)]、pKCR[Proc.Natl.Acad.Sci.USA,78,1527(1981)]、pSG1 β d2−4[Cytotechnology,4,173(1990)]等があげられる。動物細胞用発現ベクターに用いるプロモーターとエンハンサーとしては、SV40の初期プロモーターとエンハンサー[J.Biochem.,101,1307(1987)]、モロニーマウス白血病ウイルスのLTRプロモーターとエンハンサー[Biochem.Biophys.Res.Comun.,149,960(1987)]、および免疫グロブリンH鎖のプロモーター[Cell,41,479(1985)]とエンハンサー[Cell,33,717(1983)]等があげられる。
【0050】
ヒト化抗体発現用ベクターは、抗体H鎖、L鎖が別々のベクター上に存在するタイプまたは同一のベクター上に存在するタイプ(タンデム型)のどちらでも用いることができるが、ヒト化抗体発現ベクターの構築のしやすさ、動物細胞への導入のし易さ、動物細胞内での抗体H鎖およびL鎖の発現量のバランスがとれる等の点でタンデム型のヒト化抗体発現用ベクターの方が好ましい[J.Immunol.Methods,167,271(1994)]。
【0051】
(2)ヒト以外の動物の抗体のVHおよびVLをコードするcDNAの取得
ヒト以外の動物の抗体、例えば、マウス抗ビメンチンモノクローナル抗体のVHおよびVLをコードするcDNAは以下のようにして取得する。
抗ビメンチンモノクローナル抗体を産生する細胞、例えば、マウスビメンチン抗体産生ハイブリドーマ等よりmRNAを抽出し、cDNAを合成する。合成したcDNAを、ファージまたはプラスミドなどのベクターに挿入し、cDNAライブラリーを作製する。該ライブラリーより、ヒト以外の動物の抗体、例えば、マウス抗体のC領域部分またはV領域部分をプローブとして用い、VHをコードするcDNAを有する組換えファージまたは組換えプラスミド、およびVLをコードするcDNAを有する組換えファージまたは組換えプラスミドをそれぞれ単離する。組換えファージまたは組換えプラスミド上の目的とする抗体のVHおよびVLの全塩基配列を決定し、塩基配列よりVHおよびVLの全アミノ酸配列を推定する。
【0052】
(3)ヒト型キメラ抗体発現ベクターの構築
前記2(1)で構築したヒト化抗体発現用ベクターのヒト抗体のCHおよびCLをコードする遺伝子の上流に、ヒト以外の動物の抗体のVHおよびVLをコードするcDNAを挿入し、ヒト型キメラ抗体発現ベクターを構築することができる。例えば、キメラ抗体発現用ベクターのヒト抗体のCHおよびCLをコードする遺伝子の上流にあらかじめヒト以外の動物の抗体のVHおよびVLをコードするcDNAをクローニングするための制限酵素の認識配列を設けておき、このクローニングサイトにヒト以外の動物の抗体のV領域をコードするcDNAを下記に述べる合成DNAを介して挿入することにより、ヒト型キメラ抗体発現ベクターを製造することができる。合成DNAは、ヒト以外の動物の抗体のV領域の3’末端側の塩基配列とヒト抗体のC領域の5’末端側の塩基配列とからなるものであり、両端に適当な制限酵素部位を有するようにDNA合成機を用いて製造する。
【0053】
(4)ヒト以外の動物の抗体のCDR配列の同定
抗体の抗原結合部位を形成するVHおよびVLは、配列の比較的保存された4個のフレームワーク領域(以下、FR領域と称す)とそれらを連結する配列の変化に富んだ3個の相補性決定領域(CDR)から成っている[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,(1991)、以下「シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト」という。]。そして各CDRアミノ酸配列(CDR配列)は、既知の抗体のV領域のアミノ酸配列(シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト)と比較することにより同定することができる。
【0054】
(5)ヒト型CDR移植抗体のV領域をコードするcDNAの構築
ヒト型CDR移植抗体のVHおよびVLをコードするcDNAは以下のようにして取得することができる。
まず、目的のヒト以外の動物の抗体のV領域のCDRを移植するためのヒト抗体のV領域のFRのアミノ酸配列をVH、VLそれぞれについて選択する。ヒト抗体のV領域のFRのアミノ酸配列としては、ヒト抗体由来のV領域のFRのアミノ酸配列であればいかなるものでも用いることができる。
【0055】
例えば、Protein Data Bankに登録されているヒト抗体のV領域のFRのアミノ酸配列、ヒト抗体のV領域のFRの各サブグループの共通アミノ酸配列(シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト)があげられるが、充分な活性を有するヒト型CDR移植抗体を創製するためには、目的のヒト以外の動物の抗体のV領域のアミノ酸配列と高い相同性、好ましくは60%以上の相同性を有することが望ましい。次に、選択したヒト抗体のV領域のFRのアミノ酸配列をコードするDNA配列と目的のヒト以外の動物の抗体のV領域のCDRのアミノ酸配列をコードするDNA配列を連結させて、VH、VLそれぞれのアミノ酸配列をコードするDNA配列を設計する。CDR移植抗体可変領域遺伝子を構築するために設計したDNA配列を得るためには、全DNA配列をカバーするように各鎖について数本の合成DNAを設計し、それらを用いてポリメラーゼ・チェイン・リアクション(Polymerase Chain Reaction、以下「PCR」という。)を行う。PCRでの反応効率および合成可能なDNAの長さから各鎖について、好ましくは、6本の合成DNAを設計する。反応後、増幅断片を適当なベクターにサブクローニングし、その塩基配列を決定し、目的のヒト型CDR移植抗体の各鎖のV領域のアミノ酸配列をコードするcDNAを含むプラスミドを取得する。また、約100塩基よりなる合成DNAを用いてセンス、アンチセンスともに全配列を合成し、それらをアニーリング、連結することで、目的のヒト型CDR移植抗体の各鎖のV領域のアミノ酸配列をコードするcDNAを構築することもできる。
【0056】
(6)ヒト型CDR移植抗体のV領域のアミノ酸配列の改変
ヒト型CDR移植抗体は目的のヒト以外の動物の抗体のV領域のCDRのみをヒト抗体のV領域のFR間に、単純に移植しただけでは、その活性はもとのヒト以外の動物の抗体の活性に比べて低下してしまうことが知られている[BIO/TECHNOLOGY,9,266(1991)]。そこでヒト抗体のV領域のFRのアミノ酸配列のうち、直接抗原との結合に関与しているアミノ酸残基、CDRのアミノ酸残基と相互作用をしているアミノ酸残基、または抗体の立体構造の維持に関与している等の可能性を有するアミノ酸残基をもとのヒト以外の動物の抗体に見出されるアミノ酸残基に改変し、活性を上昇させることが行われている。そして、それらのアミノ酸残基を効率よく同定するため、X線結晶解析あるいはコンピューターモデリング等を用いた抗体の立体構造の構築および解析を行っている。しかし、いかなる抗体にも適応可能なヒト型CDR移植抗体の製造法は未だ確立されておらず、現状では個々の抗体によって種々の試行錯誤が必要である。
【0057】
選択したヒト抗体のV領域のFRのアミノ酸配列の改変は各種の変異導入プライマーを用いて前記2(5)に記載のPCRを行うことにより達成できる。PCR後の増幅断片を適当なベクターにサブクローニング後、その塩基配列を決定し、目的の変異が導入されたcDNAを含むベクター(以下「アミノ酸配列改変ベクター」という。)を取得する。
また、狭い領域のアミノ酸配列の改変であれば、20〜35塩基からなる変異導入プライマーを用いたPCR変異導入法により行うことができる。具体的には、改変後のアミノ酸残基をコードするDNA配列を含む20〜35塩基からなるセンス変異プライマーおよびアンチセンス変異プライマーを合成し、改変すべきV領域のアミノ酸配列をコードするcDNAを含むプラスミドを鋳型として2段階のPCRを行う。最終増幅断片を適当なベクターにサブクローニング後、その塩基配列を決定し、目的の変異が導入されたcDNAを含むアミノ酸配列改変ベクターを取得する。
【0058】
(7)ヒト型CDR移植抗体発現ベクターの構築
前記2(1)のヒト化抗体発現用ベクターのヒト抗体のCH及びCLをコードする遺伝子の上流に、前記2(5)および2(6)で取得したヒト型CDR移植抗体のVHおよびVLをコードするcDNAを挿入し、ヒト型CDR移植抗体発現ベクターを構築することができる。例えば、ヒト型CDR移植抗体のVHおよびVLのアミノ酸配列をコードするcDNAを構築するためのPCRの際に5’末端および3’末端の合成DNAの末端に適当な制限酵素の認識配列を導入することで、所望のヒト抗体のC領域をコードする遺伝子の上流にそれらが適切な形で発現するように挿入することができる。
【0059】
(8)ヒト化抗体の一過性(トランジェント)発現および活性評価
多種類のヒト化抗体の活性を効率的に評価するために、前記2(3)のヒト型キメラ抗体発現ベクター、および前記2(7)のヒト型CDR移植抗体発現ベクターまたはそれらの改変ベクターをCOS−7細胞(ATCC CRL1651)に導入してヒト化抗体の一過性発現[Methods in Nucleic Acids Res.,CRC Press,pp283(1991)]を行い、その活性を測定することができる。
COS−7細胞への発現ベクターの導入法としては、DEAE−デキストラン法[Methods in Nucleic Acids Res.,CRC Press,pp283(1991)]、リポフェクション法[Proc.Natl.Acad.Sci.USA,84,7413(1987)]等があげられる。
ベクターの導入後、培養上清中のヒト化抗体の活性は前記1(5)に記載の酵素免疫測定法(ELISA法)等により測定することができる。
【0060】
(9)ヒト化抗体の安定(ステーブル)発現および活性評価
前記2(3)のヒト型キメラ抗体発現ベクターおよび前記2(7)のヒト型CDR移植抗体発現ベクターを適当な宿主細胞に導入することによりヒト化抗体を安定に生産する形質転換株を得ることができる。
【0061】
宿主細胞への発現ベクターの導入法としては、エレクトロポレーション法[特開平2−257891;Cytotechnology,3,133(1990)]等があげられる。
ヒト化抗体発現ベクターを導入する宿主細胞としては、ヒト化抗体を発現させることができる宿主細胞であれば、いかなる細胞でも用いることができる。例えば、マウスSP2/0−Ag14細胞(ATCC CRL1581)、マウスP3X63−Ag8.653細胞(ATCC CRL1580)、ジヒドロ葉酸還元酵素遺伝子(以下「DHFR遺伝子」という。)が欠損したCHO細胞[Proc.Natl.Acad.Sci.USA,77,4216(1980)]、ラットYB2/3HL.P2.G11.16Ag.20細胞(ATCC CRL1662、以下「YB2/0細胞」という。)等があげられる。
【0062】
ベクターの導入後、ヒト化抗体を安定に生産する形質転換株は、特開平2−257891に開示されている方法に従い、G418およびFCSを含むRPMI1640培地により選択する。得られた形質転換株を培地中で培養することで培養液中にヒト化抗体を生産蓄積させることができる。培養液中のヒト化抗体の活性は前記1(5)に記載の方法等により測定する。また、形質転換株は、特開平2−257891に開示されている方法に従い、DHFR遺伝子増幅系等を利用してヒト化抗体の生産量を上昇させることができる。
【0063】
ヒト化抗体は、形質転換株の培養上清よりプロテインAカラムを用いて精製することができる(アンチボディズ・ア・ラボラトリー・マニュアル第8章)。また、その他に、通常の蛋白質で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィーおよび限外濾過等を組合せて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖または抗体分子全体の分子量は、ポリアクリルアミドゲル電気泳動(SDS−PAGE)[Nature,227,680(1970)]やウエスタンブロッティング法(アンチボディズ・ア・ラボラトリー・マニュアル第12章)等で測定する。
精製したヒト化抗体の反応性、また、ヒト化抗体のビメンチンに対する結合活性の測定は前記1(4)に記載の方法などにより測定することができる。
【0064】
3.抗体断片の作製
抗体断片は、上記2に記載のヒト化抗体を元に遺伝子工学的手法または蛋白質化学的手法により、作製することができる。抗体断片としては、Fab、F(ab’)2、Fab’、scFv、Diabody、dsFv、CDRを含むペプチド等があげられる。
【0065】
(1)Fabの作製
Fabは、IgGを蛋白質分解酵素パパインで処理することにより、作製することができる。パパインの処理後は、元の抗体がプロテインA結合性を有するIgGサブクラスであれば、プロテインAカラムに通すことで、IgG分子やFc断片と分離し、均一なFabとして回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,1995)。プロテインA結合性を持たないIgGサブクラスの抗体の場合は、イオン交換クロマトグラフィーにより、Fabは低塩濃度で溶出される画分中に回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,1995)。また、Fabは、大腸菌を用いて遺伝子工学的に作製することもできる。例えば、上記2の(2)および(3)に記載の抗体のV領域をコードするDNAを、Fab発現用ベクターにクローニングし、Fab発現ベクターを作製することができる。Fab発現用ベクターとしては、Fab用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pIT106[Science,240,1041,(1988)]等があげられる。Fab発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にFabを生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるFabとすることができ、また、ペリプラズマ層に発現させた場合は、培養上清中に活性を持ったFabが漏出する。リフォールディング後または培養上清からは、抗原を結合させたカラムを用いることにより、均一なFabを精製することができる(Antibody Engineering,A Practical Guide,W.H.Freeman and Company,1992)。
【0066】
(2)F(ab’)2の作製
F(ab’)2は、IgGを蛋白質分解酵素ペプシンで処理することにより、作製することができる。ペプシンの処理後は、Fabと同様の精製操作により、均一なF(ab’)2として回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,Academic Press,1995)。また、上記2(3)に記載のFab’をo−PDMやビスマレイミドヘキサン等のようなマレイミドで処理し、チオエーテル結合させる方法や、DTNBで処理し、S−S結合させる方法によっても作製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0067】
(3)Fab’の作製
Fab’は、大腸菌を用いて遺伝子工学的に作製することができる。例えば、上記2(2)および(3)に記載の抗体のV領域をコードするDNAを、Fab’発現用ベクターにクローニングし、Fab’発現ベクターを作製することができる。Fab’発現用ベクターとしては、Fab’用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pAK19[Bio/Technology,10,163,(1992)]等があげられる。Fab’発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にFab’を生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるFab’とすることができ、また、ペリプラズマ層に発現させた場合は、リゾチームによる部分消化、浸透圧ショック、ソニケーション等の処理により菌を破砕し、菌体外へ回収させることができる。リフォールディング後または菌の破砕液からは、プロテインGカラム等を用いることにより、均一なFab’を精製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0068】
(4)scFvの作製
scFvは遺伝子工学的には、ファージまたは大腸菌を用いて作製することができる。例えば、上記2(2)および(3)に記載の抗体のVHおよびVLをコードするDNAを、12残基以上のアミノ酸配列からなるポリペプチドリンカーをコードするDNAを介して連結し、scFvをコードするDNAを作製する。作製したDNAをscFv発現用ベクターにクローニングし、scFv発現ベクターを作製することができる。scFv発現用ベクターとしては、scFvのDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pCANTAB5E(Pharmacia社製)、Phfa[Hum.Antibody Hybridoma,5,48(1994)]等があげられる。scFv発現ベクターを適当な大腸菌に導入し、ヘルパーファージを感染させることで、ファージ表面にscFvがファージ表面蛋白質と融合した形で発現するファージを得ることができる。また、scFv発現ベクターを導入した大腸菌の封入体またはペリプラズマ層にscFvを生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるscFvとすることができ、また、ペリプラズマ層に発現させた場合は、リゾチームによる部分消化、浸透圧ショック、ソニケーション等の処理により菌を破砕し、菌体外へ回収することができる。リフォールディング後または菌の破砕液からは、陽イオン交換クロマトグラフィー等を用いることにより、均一なscFvを精製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0069】
(5)Diabodyの作製
Diabodyは、上記のscFvを作製する際のポリペプチドリンカーを3〜10残基程度にすることで、作製することができる。1種類の抗体のVHおよびVLを用いた場合には、2価のDiabodyを、2種類の抗体のVHおよびVLを用いた場合は、2特異性を有するDiabodyを作製することができる[FEBS Letters,453,164(1999)、Int.J.Cancer,77,763(1998)]。
【0070】
(6)dsFvの作製
dsFvは、大腸菌を用いて遺伝子工学的に作製することができる。まず、上記2(2)および(3)に記載の抗体のVHおよびVLをコードするDNAの適当な位置に変異を導入し、コードするアミノ酸残基がシステインに置換されたDNAを作製する。作製した各DNAをdsFv発現用ベクターにクローニングし、VHおよびVLの発現ベクターを作製することができる。dsFv発現用ベクターとしては、dsFv用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pULI9[Protein Engineering,7,697(1994)]等があげられる。VHおよびVLの発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にVHおよびVLを生成蓄積させることができる。封入体またはペリプラズマ層からVHおよびVLを得、混合し、通常蛋白質で用いられるリフォールディング法により、活性のあるdsFvとすることができる。リフォールディング後は、イオン交換クロマトグラフィーおよびゲル濾過等により、さらに精製することができる[Protein Engineering,7,697(1994)]。
【0071】
(7)CDRペプチドの作製
CDRを含むペプチドは、Fmoc法またはtBoc法等の化学合成法によって作製することができる。また、CDRを含むペプチドをコードするDNAを作製し、作製したDNAを適当な発現用ベクターにクローニングし、CDRペプチド発現ベクターを作製することができる。発現用ベクターとしては、CDRを含むペプチドをコードするDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pLEX(Invitrogen社製)、pAX4a+(Invitrogen社製)等があげられる。発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にCDRを含むペプチドを生成蓄積させることができる。封入体またはペリプラズマ層からCDRを含むペプチドを得、イオン交換クロマトグラフィーおよびゲル濾過等により、精製することができる[Protein Engineering,7,697(1994)]。
【0072】
4.融合抗体および融合ペプチドの作製方法
本発明で使用される抗体またはペプチドに、放射性同位元素、毒素、サイトカインまたは酵素等の蛋白質、低分子の薬剤等を、化学的または遺伝子工学的に結合させた融合抗体または融合ペプチドも抗体の誘導体として使用することができる。
【0073】
抗体またはペプチドと毒素蛋白質とを化学的に結合させた融合抗体または融合ペプチドは、文献[Anticancer Research,11,2003(1991);Nature Medicine,3,350(1996)]記載の方法等に従って作製することができる。
抗体またはペプチドと、毒素、サイトカインまたは酵素等の蛋白質とを遺伝子工学的に結合させた融合抗体または融合ペプチドは、文献[Proc.Natl.Acad.Sci.USA,93,974(1996);Proc.Natl.Acad.Sci.USA,93,7826(1996)]記載の方法等に従って作製することができる。
【0074】
抗体またはペプチドと低分子抗癌剤を化学的に結合させた融合抗体または融合ペプチドは、文献[Science,261,212(1993)]記載の方法等に従って作製することができる。
抗体またはペプチドと放射性同位元素を化学的に結合させた融合抗体または融合ペプチドは、文献[Antibody Immunoconjugates and Radiopharmaceuticals,3,60(1990);Anticancer Research,11,2003(1991)]記載の方法等に従って作製することができる。
【0075】
これらの誘導体は、抗体分子の特異性に従って放射性同位元素、毒素、サイトカインまたは酵素等の蛋白質、低分子の薬剤等を標的組織周辺に集積させることで、より効果的で副作用の少ない診断または治療を可能にすることが期待されている。
【0076】
5.ヒト化抗体または抗体断片の活性評価
精製したヒト化抗体の抗原との結合性、ビメンチンに対する結合活性はELISA法および蛍光抗体法[Cancer Immunol.Immunother.,36,373(1993)]、BIAcoreTM等を用いた表面プラズモン共鳴等により測定できる。
【0077】
6.本発明に用いる抗体を用いたビメンチンの定量および検出方法
本発明に用いる抗ビメンチン抗体またはその抗体断片を用いる、ビメンチンを免疫学的に定量または検出する方法としては、蛍光抗体法、免疫酵素抗体法(ELISA)、放射性物質標識免疫抗体法(RIA)、免疫組織染色法、免疫細胞染色法などの免疫組織化学染色法(ABC法、CSA法等)、ウェスタンブロッティング法、免疫沈降法、上記に記した酵素免疫測定法、サンドイッチELISA法[単クローン抗体実験マニュアル(講談社サイエンティフィック、1987年)、続生化学実験講座5免疫生化学研究法(東京化学同人、1986年)]等を用いることができる。
【0078】
蛍光抗体法は、文献[Monoclonal Antibodies:Principles and practice,Third edition(Academic Press,1996),単クローン抗体実験マニュアル(講談社サイエンティフィック、1987)]等に記載された方法を用いて行うことができる。具体的には、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いられるモノクローナル抗体またはその抗体断片を反応させ、さらにフルオレシン・イソチオシアネート(FITC)またはフィコエリスリンなどの蛍光物質でラベルした抗イムノグロブリン抗体または結合断片を反応させた後、蛍光色素をフローサイトメーターで測定する方法である。
【0079】
免疫細胞染色法、免疫組織染色法等の免疫組織化学染色法(ABC法、CSA法等)は、文献[Monoclonal Antibodies:Principles and practice,Third edition(Academic Press,1996),単クローン抗体実験マニュアル(講談社サイエンティフィック,1987)]等に記載された方法を用いて行うことができる。
免疫酵素抗体法(ELISA)は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにペルオキシダーゼ、ビオチン等の酵素標識等を施した抗イムノグロブリン抗体または結合断片を反応させた後、発色色素を吸光光度計で測定する方法である。
【0080】
放射性物質標識免疫抗体法(RIA)は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらに放射線標識を施した抗イムノグロブリン抗体または結合断片を反応させた後、シンチレーションカウンター等で測定する方法である。
免疫細胞染色法、免疫組織染色法は、生体内から分離された細胞または組織等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにフルオレシン・イソチオシアネート(FITC)等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素標識を施した抗イムノグロブリン抗体または結合断片を反応させた後、顕微鏡を用いて観察する方法である。
【0081】
ウェスタンブロッティング法は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等をSDS−ポリアクリルアミドゲル電気泳動[Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988]で分画した後、該ゲルをPVDF膜またはニトロセルロース膜にブロッティングし、該膜に本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにFITC等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素標識を施した抗マウスIgG抗体または結合断片を反応させた後、確認する。
【0082】
免疫沈降法とは、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等を本発明に用いるモノクローナル抗体またはその抗体断片と反応させた後、プロテインG−セファロース等のイムノグロブリンに特異的な結合能を有する担体を加えて抗原抗体複合体を沈降させるものである。
サンドイッチELISA法とは、本発明に用いるモノクローナル抗体またはその抗体断片で、抗原認識部位の異なる2種類のモノクローナル抗体のうち、あらかじめ一方のモノクローナル抗体または抗体断片はプレートに吸着させ、もう一方のモノクローナル抗体または抗体断片はFITC等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素で標識しておく。抗体吸着プレートに、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等を反応後、標識したモノクローナル抗体またはその抗体断片を反応させ、標識物質に応じた反応を行う方法である。
【0083】
本発明でいうLAPの部分断片としては、細胞表面または細胞外に分泌されるビメンチンに結合し得るLAPの部分断片であればいずれのものも用いることができ、例えば、国際公開第98/51704号の第2表に記載された化合物1〜16等を使用することができるが、化合物15が好ましく使用される。化合物1〜16のアミノ酸配列を配列番号1〜16に示す。
【0084】
以下、本発明のTGF−βの活性化抑制物質のスクリーニング方法について記す。
本発明のスクリーニング方法の対象となる物質は、合成化合物でも天然物質でも特に制限なく用いることができる。
本発明の方法に使用するLAPの部分断片としては、上記のLAPの部分断片を用いることができる。
【0085】
本発明の方法に使用するビメンチンおよびTGF−βを発現する細胞としては、例えば、血管内皮細胞、マクロファージ等があげられる。細胞は、ヒト、ウシ、ブタ、ネズミ等の動物から、単離・精製したもの、またはそれら細胞由来の培養細胞を用いることができる。単離・精製方法としては、例えば、オカダ(Okada)らの方法[ジャーナル・オブ・バイオケミストリー(Journal of Biochemistry),106,304(1989)]等があげられる。
【0086】
細胞へのLAPの部分断片の結合は、例えば細胞を培地で培養し該部分断片を添加しインキュベートした後、細胞を洗浄し、細胞に結合している該部分断片を測定することにより行うことができる。
LAPの部分断片は、例えばクロラミンT法[モレキュラー・アンド・セルラー・バイオロジー(Molecular & Cellular Biology),2,599(1982)]により125I標識したもの、フルオレセインイソチオシアネート、AMCA、ローダミン6G、ローダミンレッド−X、Cy3、テキサスレッド等の蛍光色素で標識をしたもの等を用いるのが好ましい。これにより、細胞に結合したLAPの部分断片は、それぞれ、放射活性、発光強度等を測定することにより検出および定量することができる。
【0087】
また、細胞表面上に発現するビメンチンにLAPの部分断片が結合することにより潜在型TGF−βが活性化されるので、細胞に結合するLAPの部分断片量の測定は、活性型TGF−β量を測定することにより行うこともできる。活性型TGF−βの定量法は特に限定されないが、直接抗TGF−β抗体を用いた酵素免疫測定方法[メソッヅ・イン・エンザイモロジー(Methods in Enzymology),198,303(1991)]または安部らのルシフェラーゼ・アッセイ・システム[アナリティカル・バイオケミストリー(Analytical Biochemistry),216,276(1994)]等で測定することができる。また、血管内皮細胞の遊走度[ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology),123,1249(1993)]、血管平滑筋細胞の増殖[トウホク・ジャーナル・オブ・エクスペリメンタル・メディスン(Tohoku Journal of Experimental Medicine),179,23(1996)]、各種癌細胞の増殖阻害[ジャーナル・オブ・クリニカル・インベスティゲーション(Journal of Clinical Investigation),87,277(1991),エンドクリノロジー(Endocrinology),128,1981(1991)]、ミンク肺上皮細胞の増殖阻害[メソッヅ・イン・エンザイモロジー(Methods in Enzymology),198,317(1991)]等の方法で測定することができる。
【0088】
スクリーニングの対象となる物質を添加しないときの、細胞に結合するLAPの部分断片量または活性型TGF−β量と、該物質を添加したときの、細胞に結合するLAPの部分断片量または活性型TGF−β量とを比較し、その差異から、該化合物の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価することができる。該結合阻害活性を有する物質をTGF−βの活性化を抑制する目的物質として選択する。目的物質の選択は、例えば、添加濃度1mmol/lで、対象となる化合物を添加しない場合の活性型TGF−β量に対し、添加した場合の活性型TGF−β量が10%以上減少の認められる物質を選択することにより行うことができる。
【0089】
本発明のビメンチンとLAPの部分断片との結合を阻害する物質は潜在型TGF−βの活性化を抑制することから、潜在型TGF−βの活性化抑制剤として用いることができ、TGF−βが関与する各種疾患の治療薬として用いることができる。
TGF−βが関与する疾患としては、例えば、癌、IgA腎症、巣状および分節性糸球体腎炎、ループス腎炎、糖尿病性腎症、HIV腎症、C型肝炎、アルコール性および自己免疫性肝線維症、ブレオマイシン誘発性および突発性肺線維症、全身性硬化症、骨髄線維症、増殖性硝子体網膜症、クローン病、好酸球増多筋肉痛症候群、肝硬変、梗塞後心線維症、手術後腹内癒着、血行再建術後再狭窄、高血圧性血管障害、移植腎拒絶、ケロイド、肥厚性火傷瘢痕、眼内線維症、リウマチ性関節炎、鼻ポリープ等があげられる。本発明のビメンチンとLAPの部分断片との結合を阻害する物質は、上記のTGF−βが関与するいずれの疾患の治療にも用いることができるが、好ましくは、動脈硬化症、癌、炎症、線維症の治療に用いることができる。
【0090】
本発明のビメンチンとLAPの部分断片との結合を阻害する物質は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つまたはそれ以上の担体と一緒に混合し、製剤学の技術分野においてよく知られる任意の方法により製造した医薬製剤として提供するのが望ましい。
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体またはペプチド製剤の場合、望ましくは静脈内投与をあげることができる。
【0091】
投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。
乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。
【0092】
カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0093】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。
注射剤は、塩溶液、ブドウ糖溶液、または両者の混合物からなる担体等を用いて調製される。
座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。
また、噴霧剤は本発明のビメンチンとLAPの部分断片との結合を阻害する物質そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該物質を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0094】
担体として具体的には乳糖、グリセリン等が例示される。該物質および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
投与量または投与回数は、目的とする治療の効果、投与方法、治療期間、年齢、体重等により異なるが、ビメンチンとLAPの部分断片との結合を阻害する物質が抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体である場合、通常成人1日当たり10μg/kg〜8mg/kgである。
以下に本発明の実施例を示す。
【実施例1】
【0095】
抗ビメンチン抗体によるビメンチンとLAPの部分断片との結合阻害(1)
本試験は安部(Abe)らの方法[エンドセリウム(Endothelium)9,25−36(2002)]に従って行った。また、本実験で使用したペプチドのビオチン化は国際公開第98/51704号に記載の方法に従った。
すなわち、10%FBS、4mmol/lのL−グルタミン、10μg/mlのカナマイシン(明治製菓社製)を含むDMEM培地に懸濁したウシ血管内皮細胞(以下、BAECという。)を96穴黒色プレート(コーニング・コースター社製)に、1穴あたり2.0×104個ずつ播種し、5%CO2条件下37℃で1日間培養した後に、以下の実験に供した。
【0096】
細胞をPBSで3回洗浄した後、5μg/mlの抗ビメンチン抗体(VIM3B4、PROGEN社製)で37℃30分間インキュベートし、その後、配列番号15で表されるペプチド(Peptide−25)または配列番号17で表されるビオチン化ペプチド(Peptide−25N)および0.1%BSAを含むDMEM培地を、各ペプチドの最終濃度が100μg/mlとなるように添加し、37℃で60分間インキュベートした。次にPBSで1回洗浄し、PBSで500倍希釈したHRP−conjugated streptavidin(アマシャム社製)を1穴あたり100μl加えて、37℃で30分間インキュベートした。その後PBSで3回洗浄し、100μlのECL western blotting detection reagent(アマシャム社製)を加えて、ルミネセンサーJNR(アトー社製)により各ウェルの発光強度を測定した。
【0097】
結果を図1に示す。図1から明らかなように、Peptide−25Nは細胞表面に結合し、その結合は抗ビメンチン抗体により抑制された。
【実施例2】
【0098】
抗ビメンチン抗体によるTGF−β活性化抑制
本試験は安部(Abe)らの方法[エンドセリウム(Endothelium)9,25−36(2002)]に従って行った。
すなわち、BAECを35mmディッシュに播種し、コンフルエントになるまで培養した。配列番号15で表されるペプチド(Peptide−25)または配列番号18で表されるペプチド(Peptide−21)を0.1%BSA含有DMEM培地中に最終濃度100μg/mlとなるように調製し、細胞に添加して37℃で6時間プレインキュベートした。その後、剃刀で一部の細胞を剥ぎ取り、PBSで洗浄してからプレインキュベート時と同組成の培地で24時間インキュベートした。プレインキュベート時および剥ぎ取り後のインキュベート時には、各ペプチドに加えて5μg/mlの抗ビメンチン抗体(V9、Neo Markers社製)またはマウスIgG1抗体(Ancell社製)をそれぞれ添加し、剥ぎ取った領域に遊走してくる細胞数に対する各抗体の影響を調べた。該細胞数は、剥ぎ取り後24時間に位相差顕微鏡下で計数した。
【0099】
結果を図2に示す。図2から明らかなように、Peptide−25を添加することにより遊走細胞数が減少した。これはPeptide−25が潜在型TGF−βの活性化を促進したことにより、TGF−βによる細胞遊走阻害作用が増強されたためである。一方、Peptide−25に加えて抗ビメンチン抗体を添加した場合、遊走細胞数が有意に増加したことから、抗ビメンチン抗体がPeptide−25の潜在型TGF−βの活性化を抑制することが明らかとなった。なお、潜在型TGF−β活性化能を有さないPeptide−21を添加した場合は、細胞遊走数に変化は見られず、そこに抗ビメンチン抗体を添加しても何ら変化は見られなかった。
【実施例3】
【0100】
抗ビメンチン抗体によるビメンチンとLAPの部分断片との結合阻害(2)
実施例1と同様の方法で、参考例で作製した抗ヒトビメンチンモノクローナル抗体KM3565のビメンチンとLAPの部分断片との結合阻害活性を評価した。本実施例においては、実施例1におけるVIM3B4の代りに、5μg/mlのKM3565または陰性対照として同濃度の抗G−CSF変異体抗体KM511(Agric.Biol.Chem.,53(4),1095−1101,1989)のいずれかを添加した。
結果を図3に示す。図3から明らかなように、KM3565はPeptide−25Nの細胞表面への結合を抑制した。
【0101】
参考例
抗ヒトビメンチンモノクローナル抗体の作製
(1)抗原ペプチドの調製
(1−1)化合物1(ヒトビメンチン部分ペプチド:配列番号19)の合成
本発明において使用したアミノ酸およびその保護基に関する略号は、生化学命名に関するIUPAC−IUB委員会(IUPAC−IUB Joint Commission on Biochemical Nomenclature)の勧告〔ヨーロピアン・ジャーナル・オブ・バイオケミストリー(European Journal of Biochemistry),138巻,9頁(1984年)〕に従った。
【0102】
以下の略号は、特に断わらない限り対応する下記のアミノ酸を表す。
Ala:L−アラニン
Arg:L−アルギニン
Asn:L−アスパラギン
Asp:L−アスパラギン酸
Asx:L−アスパラギン酸またはL−アスパラギン
Cys:L−システイン
Gln:L−グルタミン
Glu:L−グルタミン酸
Glx:L−グルタミン酸またはL−グルタミン
Gly:グリシン
Ile:L−イソロイシン
Leu:L−ロイシン
Lys:L−リジン
Met:L−メチオニン
Pro:L−プロリン
Ser:L−セリン
Thr:L−スレオニン
Tyr:L−チロシン
【0103】
以下の略号は、対応する下記のアミノ酸の保護基または側鎖保護アミノ酸を表す。
Fmoc:9−フルオレニルメチルオキシカルボニル
t−Bu:t−ブチル
Trt:トリチル
Pmc:2,2,5,7,8−ペンタメチルクロマン−6−スルホニル
Boc:t−ブチルオキシカルボニル
Fmoc−Ser(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−セリン
Fmoc−Thr(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−スレオニン
Fmoc−Lys(Boc)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nε−t−ブチルオキシカルボニル−L−リジン
Fmoc−Asn(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nγ−トリチル−L−アスパラギン
Fmoc−Asp(O−t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−L−アスパラギン酸−β−t−ブチルエステル
Fmoc−Glu(O−t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−L−グルタミン酸−γ−t−ブチルエステル
Fmoc−Gln(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nγ−トリチル−L−グルタミン
Fmoc−Arg(Pmc)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Ng−2,2,5,7,8−ペンタメチルクロマン−6−スルホニル−L−アルギニン
Fmoc−Tyr(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−チロシン
Fmoc−Cys(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−S−トリチル−L−システイン
【0104】
以下の略号は、対応する下記の反応溶媒、反応試薬等を表す。
HBTU:2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム・ヘキサフルオロホスフェート
HOBt:N−ヒドロキシベンゾトリアゾール
DMF:N,N−ジメチルホルムアミド
TFA:トリフルオロ酢酸
DIEA:ジイソプロピルエチルアミン
【0105】
アミドリンカー16.5μmolが結合した担体樹脂(Rink−Amide MBHA樹脂、Nova Biochem社製)30mgを自動合成機(島津製作所PSSM−8)の反応容器に入れ、島津製作所の合成プログラムに従い次の操作を行った。
(a)担体樹脂を500μlのDMFで1分間洗浄し、該溶液を排出した
(b)30%ピペリジン−DMF溶液858μlを加えて混合物を4分間攪拌し、該溶液を排出し、この操作をもう1回繰り返した。
(c)担体樹脂を500μlのDMFで1分間洗浄し、該溶液を排出し、この操作を5回繰り返した。
(d)Fmoc−Glu(O−t−Bu)−OH(165μmol)、HBTU(165μmol)、HOBt 1水和物(165μmol)およびDIEA(330μmol)をDMF(858μl)中で3分間攪拌し、得られた溶液を樹脂に加えて混合物を30分間攪拌し、溶液を排出した。
(e)担体樹脂を858μlのDMFで1分間洗浄し、これを5回繰り返した。こうして、Fmoc−Glu(O−t−Bu)−アミドを担体上に合成した。
【0106】
次に(b)、(c)のFmoc基脱保護工程および洗浄工程を行い、Fmoc基を除去したH−Glu(O−t−Bu)−アミドの結合した担体樹脂を得た。
次に、(d)の工程でFmoc−Glu(O−t−Bu)−OHを用いて縮合反応を行い、(e)の洗浄工程、次いで(b)、(c)の脱保護、洗浄工程を経て、H−Glu(O−t−Bu)−Glu(O−t−Bu)−アミドを担体上に合成した。以下、工程(d)において、Fmoc−Lys(Boc)−OH、
Fmoc−Met−OH、Fmoc−Asn(Trt)−OH、Fmoc−Gln(Trt)−OH、Fmoc−Ile−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Asp(O−t−Bu)−OH、Fmoc−Gln(Trt)−OH、
Fmoc−Leu−OH、Fmoc−Arg(Pmc)−OH、Fmoc−Gly−OH、Fmoc−Ile−OH、
Fmoc−Thr(t−Bu)−OH、Fmoc−Asp(O−t−Bu)−OH、Fmoc−Gln(Trt)−OH、
Fmoc−Tyr(t−Bu)−OH、Fmoc−Asn(Trt)−OH、Fmoc−Ala−OHを順次用いて、(d)、(e)、(b)、(c)の順に工程を繰り返した。最後にFmoc−Cys(Trt)−OHを用いて(d)、(e)の操作を2度繰り返した後に、(a)、(b)の操作を行った。
【0107】
得られた樹脂をメタノール、ブチルエーテルで順次洗浄し、減圧下12時間乾燥して、側鎖保護ペプチドの結合した担体樹脂を得た。これに、TFA(82.5%)、チオアニソール(5%)、水(5%)、エチルメチルスルフィド(3%)、1,2−エタンジチオール(2.5%)およびチオフェノール(2%)からなる混合溶液600μlを加えて室温で8時間放置し、側鎖保護基を除去するとともに樹脂よりペプチドを切り出した。樹脂を濾別後、得られた溶液にエーテル約10mlを加え、生成した沈澱を遠心分離およびデカンテーションにより回収し、粗ペプチド42.7mgを取得した。この粗ペプチドを酢酸水溶液に溶解後、逆相カラム(資生堂製、CAPCELL PAK C18 30mmI.D.X25mm)を用いたHPLCで精製した。0.1%TFA水溶液に、TFA0.1%を含む90%アセトニトリル水溶液を加えていく直線濃度勾配法で溶出し、220nmで検出し、化合物1を含む画分を得た。この画分を凍結乾燥して、化合物1を8.4mg得た。化合物1の物理化学的性質は以下の通りである。
質量分析[TOF−MS];m/z=2497.25(M+H+)
アミノ酸分析;Glx 6.0(6),Asx 3.7(4),Arg 1.1(1),Thr 1.0(1),Ala 1.0(1),Ile 1.9(2),Leu 1.1(1),Lys 0.9(1),Tyr 0.9(1),Met 0.9(1),Gly 1.3(1),Cys 1.4(1)
【0108】
(1−2)化合物2(対照ペプチド:配列番号20)の合成
アミドリンカー16.5μmolが結合した担体樹脂(Rink−Amide MBHA樹脂、Nova Biochem社製)30mgを出発物質とし、(1−1)と同様にして、Fmoc−Pro−OH、
Fmoc−Leu−OH、Fmoc−Pro−OH、Fmoc−Leu−OH、Fmoc−Ser(t−Bu)−OH、
Fmoc−Ile−OH、Fmoc−Arg(Pmc)−OH、Fmoc−Ser(t−Bu)−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Glu(O−t−Bu)−OH、Fmoc−Gly−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Leu−OH、Fmoc−Leu−OH、Fmoc−Lys(Boc)−OH、
Fmoc−Arg(Pmc)−OH、Fmoc−Tyr(t−Bu)−OH、Fmoc−Thr(t−Bu)−OH、
Fmoc−Cys(Trt)−OHを順次縮合した後に、洗浄、乾燥を経て、側鎖保護ペプチドの結合した担体樹脂を得た。(1−1)と同様にして側鎖保護基の切断および樹脂からの切り出しを行い、粗ペプチド38.7mgを取得した。これを(1−1)と同様に精製し、化合物2を4.7mg得た。化合物2の物理化学的性質は以下の通りである。
質量分析(TOF−MS);m/z=2203.19(M+H+)
アミノ酸分析:アミノ酸分析;Glx 3.1(3),Arg 1.9(2),Thr 0.9(1),Ser 1.9(2),Ile 1.0(1),Leu 4.1(4),Lys 0.9(1),Tyr 0.9(1),Gly 1.1(1),Pro 2.2(2),Cys 1.1(1)
【0109】
(2)免疫原の調製
上記(1)で得られたヒトビメンチン部分ペプチドは、免疫原性を高める目的で以下の方法でKLH(カルビオケム社)とのコンジュゲートを作製し、免疫原とした。すなわち、KLHをPBSに溶解して10mg/mlに調整し、1/10容量の25mg/ml MBS(ナカライテスク社)を滴下して30分間撹拌反応させた。あらかじめPBSで平衡化したセファデックスG−25カラムでフリーのMBSを除いて得られたKLH−MBS 2.5mgを0.1Mりん酸ナトリウムバッファー(pH7.0)に溶解したペプチド1mgと混合し、室温で3時間、攪拌反応させた。反応後、反応物をPBSで透析し、ヒトビメンチン部分ペプチド−KLHコンジュゲートを得た。
【0110】
(3)動物の免疫と抗体産生細胞の調製
上記(2)で調製したヒトビメンチン部分ペプチド−KLHコンジュゲート100μgをアルミニウムゲル2mgおよび百日咳ワクチン(千葉県血清研究所製)1×109細胞とともに5週令雌マウス(Balb/c)に投与し、2週間後より100μgの該コンジュゲートを1週間に1回、計4回投与した。眼底静脈叢より採血し、血清抗体価を下記(4)に示す酵素免疫測定法で調べ、十分な抗体価を示したマウスから最終免疫3日後に脾臓を摘出した。
脾臓をMEM培地(日水製薬社製)中で細断し、ピンセットでほぐし、1200rpmで5分間遠心分離した。上清を捨て、トリス−塩化アンモニウム緩衝液(pH7.65)で1〜2分間処理し赤血球を除去し、MEM培地で3回洗浄したものを細胞融合に用いた。
【0111】
(4)酵素免疫測定法
アッセイ用の抗原には上記(1)で得られたヒトビメンチン部分ペプチドとサイログロブリン(以下、THYと略す)とのコンジュゲートを用いた。該コンジュゲートは、架橋剤としてMBSの代わりにSMCC(シグマ社)を用いた以外、上記(2)と同様の方法により作製した。
96穴のEIA用プレート(グライナー社)に、上記コンジュゲートを10μg/ml、50μl/穴で分注し、4℃で一晩放置して吸着させた。洗浄後、1%BSA(ウシ血清アルブミン)−PBSを100μl/穴で加え、室温1時間反応させて残っている活性基をブロックした。
1%BSA−PBSを捨て、被免疫マウス抗血清、抗ビメンチンモノクローナル抗体の培養上清または精製モノクローナル抗体を50μl/穴で分注し2時間反応させた。
0.05%tween−PBSで洗浄後、ペルオキシダーゼ標識ウサギ抗マウスイムノグロブリン(ダコ社)を50μl/穴で加えて室温、1時間反応させ、0.05%tween−PBSで洗浄後ABTS基質液[2.2−アジノビス(3−エチルベンゾチアゾール−6−スルホン酸)アンモニウム]を用いて発色させ、OD415nmの吸光度をプレートリーダー(Emax;Molecular Devices社)にて測定した。
【0112】
(5)マウス骨髄腫細胞の調製
8−アザグアニン耐性マウス骨髄腫細胞株P3−U1を正常培地で培養し、細胞融合時に2×107以上の細胞を確保し、細胞融合に親株として供した。
(6)ハイブリドーマの作製
上記(3)で得られたマウス脾細胞と上記(5)で得られた骨髄腫細胞とを10:1になるよう混合し、1,200rpmで5分間遠心分離した。上清を捨て、沈澱した細胞群をよくほぐした後、ポリエチレングライコール−1,000(PEG−1,000)2g、MEM培地2mlおよびジメチルスルホキシド0.7mlの混液を0.2〜1ml/108マウス脾細胞となるように加えて37℃で攪拌した。1〜2分間毎にMEM培地1〜2mlを数回加えた後、MEM培地を加えて全量が50mlになるようにした。900rpmで5分遠心分離した後、上清を捨て、ゆるやかに細胞をほぐした後、メスピペットによる吸込み・吸出しを繰り返し、細胞をHAT培地100ml中に懸濁した。
【0113】
この懸濁液を96穴培養用プレートに100μl/穴ずつ分注し、5%CO2インキュベーター中、37℃で10〜14日間、CO25%条件下で培養した。この培養上清を上記(4)に記載した酵素免疫測定法で調べ、ヒトビメンチン部分ペプチド(化合物1)に反応して対照ペプチド(化合物2)に反応しない穴を選び、HAT培地を正常培地に換えて2回クローニングを繰り返し、抗ヒトビメンチンモノクローナル抗体産生ハイブリドーマを確立した。
以上の結果、化合物1に特異的な反応性を示すモノクローナル抗体KM3565を産生するハイブリドーマ株KM3565を取得した。なお、ハイブリドーマ株KM3565はKM3565の株名で、平成17年4月19日付けで独立行政法人産業技術総合研究所特許生物寄託センター(日本国茨城県つくば市東1丁目1番地1中央第6)にFERM ABP−10324として寄託されている。
また、モノクローナル抗体KM3565の抗体クラスは抗体クラス特異的な標識化抗体を用いた酵素免疫測定法によりIgG3と決定された。
【0114】
(7)モノクローナル抗体の精製
プリスタン処理した8週令ヌード雌マウス(Balb/c)に上記(6)で得られたハイブリドーマ株を5〜20×106細胞/匹で腹腔内注射した。10〜21日後に、ハイブリドーマは腹水癌化した。腹水のたまったマウスから、腹水を採取(1〜8ml/匹)し、3,000rpmで5分遠心分離して固形分を除去した。得られた上清をカプリル酸沈殿法〔Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988〕により精製し、精製モノクローナル抗体を得た。
【0115】
(8)モノクローナル抗体KM3565のヒトビメンチン部分ペプチドとの反応性(酵素免疫測定法)
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン部分ペプチドとの反応性を、上記(4)の酵素免疫測定法にて調べた。図4に示すように、モノクローナル抗体KM3565は化合物1特異的に抗体濃度依存的な反応性を示した。
(9)モノクローナル抗体KM3565のヒトビメンチン蛋白との反応性(酵素免疫測定法)
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン蛋白との反応性を上記(4)の酵素免疫測定法にて調べた。大腸菌で発現させたヒトビメンチン蛋白(PROGEN社)を添付のデータシート記載の方法に従って蒸留水にて溶解後、PBSで2μg/mlに希釈してプレートに固層化した。陰性対照抗原として大腸菌由来夾雑蛋白をPBSで10μg/mlに希釈してプレートに固層化したものを用いた。図5に示すように、モノクローナル抗体KM3565はビメンチン特異的に抗体濃度依存的な反応性を示した。
【0116】
(10)ウェスタンブロッティング
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン蛋白との反応性をウエスタンブロッティングにて調べた。
大腸菌で発現させたヒトビメンチン蛋白、または大腸菌由来夾雑蛋白を0.1μg/レーンでSDS−ポリアクリルアミド電気泳動〔Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988〕にて分画後、PVDF膜にブロッティングした1%BSA−PBSでブロッキング後、1次抗体としてKM3565のハイブリドーマ培養上清原液、市販の抗ビメンチン抗体2種(PROGEN社製VIM3B4およびSanta Cruz Biotechnology社製V9、いずれも精製抗体5μg/ml)、および陰性対照抗体としてKM511(抗G−CSF変異体抗体、Agric.Biol.Chem.,53(4),1095−1101,1989、5μg/ml)を室温で2時間反応させた。0.05%tween−PBSでよく洗浄した後、第二抗体としてペルオキシダーゼ標識抗マウスイムノグロブリン抗体(DAKO社製)を室温で1時間反応させた。
0.05%tween−PBSでよく洗浄した後、ECL−detection kit(アマシャム社)による反応を行った。
その結果、モノクローナル抗体KM3565は、市販抗体と同様、ビメンチン蛋白に特異的な反応性を示した。
【0117】
(11)モノクローナル抗体KM3565と市販抗ビメンチン抗体との認識エピトープの異同
市販抗ビメンチン抗体2種(PROGEN社製VIM3B4およびSanta Cruz Biotechnology社製V9)とモノクローナル抗体KM3565の認識エピトープの異同を検討する目的で、該市販抗体の化合物1に対する反応性を上記(4)の酵素免疫測定法にて調べた。その結果、図6に示すように該市販抗体はいずれもビメンチン蛋白に反応性を有するが、化合物1に対する反応性を示さなかったことから、これらの抗体はビメンチン蛋白上のモノクローナル抗体KM3565とは異なる別のエピトープを認識していることが明らかとなった。
【産業上の利用可能性】
【0118】
本発明により、ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤、またはTGF−β活性化抑制物質のスクリーニング方法が提供される。本発明のTGF−βの活性化抑制剤は、TGF−βが関与する疾患の治療薬として利用することができる。
【配列表フリーテキスト】
【0119】
配列番号1−人工配列の説明:合成ペプチド
配列番号2−人工配列の説明:合成ペプチド
配列番号3−人工配列の説明:合成ペプチド
配列番号4−人工配列の説明:合成ペプチド
配列番号5−人工配列の説明:合成ペプチド
配列番号6−人工配列の説明:合成ペプチド
配列番号7−人工配列の説明:合成ペプチド
配列番号8−人工配列の説明:合成ペプチド
配列番号9−人工配列の説明:合成ペプチド
配列番号10−人工配列の説明:合成ペプチド
配列番号11−人工配列の説明:合成ペプチド
配列番号12−人工配列の説明:合成ペプチド
配列番号13−人工配列の説明:合成ペプチド
配列番号14−人工配列の説明:合成ペプチド
配列番号15−人工配列の説明:合成ペプチド
配列番号16−人工配列の説明:合成ペプチド
配列番号17−人工配列の説明:合成ペプチド
配列番号18−人工配列の説明:合成ペプチド
配列番号19−人工配列の説明:合成ペプチド
配列番号20−人工配列の説明:合成ペプチド
【技術分野】
【0001】
本発明は、潜在型トランスフォーミング・グロース・ファクター−β(transforming growth factor−β、以下「TGF−β」という。)の活性化抑制剤、TGF−β活性化抑制物質のスクリーニング方法、およびTGF−βが関与する疾患の治療薬に関する。
【背景技術】
【0002】
TGF−βは、細胞増殖・分化、細胞外マトリックス合成、アポトーシス、免疫機能などの制御等、多彩な生理活性を有するサイトカインとして知られており、その機能不全は、ヒトの病態、例えば癌、組織の線維化、自己免疫疾患等に関連していることが知られている(非特許文献1参照)。
【0003】
TGF−βは不活性の潜在型分子として分泌されるため、生理作用を発揮するためには細胞から分泌された後に活性化を受ける必要がある(非特許文献2参照)。潜在型TGF−βの活性化段階を制御することはTGF−βの活性を制御することにつながることから、その活性化機構が精力的に研究されている。これまでに、プロテアーゼによる潜在型TGF−βの限定分解を介した機構、細胞表面分子への結合に伴う構造変化を介した機構、またはその両者を組み合わせた機構等が報告されている。例えば、潜在型TGF−βを限定分解するプロテアーゼとして、プラスミン(非特許文献3参照)、マトリックスメタロプロテアーゼ(非特許文献4参照)等が報告されている。潜在型TGF−βが結合し、その構造を変化させる細胞表面分子として、トロンボスポンジン−1(非特許文献5参照)、インテグリンαvβ6(非特許文献6参照)等が報告されている。インテグリンαvβ8への結合とそれに伴う構造変化により、マトリックスメタロプロテアーゼによる限定分解が生じるという機構も報告されている(非特許文献7参照)。
【0004】
一方、潜在型TGF−βを構成するタンパク質であるラテンシー・アソシエイテッド・ペプチド(Latency Associated Peptide、以下「LAP」という。)の部分断片が、潜在型TGF−βの活性化を促進すること、該部分断片が血管内皮細胞の表面に結合することが報告されている(非特許文献8参照)。
【0005】
ビメンチンは、多くの種類の細胞に存在する中間径フィラメントのサブユニットタンパク質であり、ビメンチン繊維として細胞質に網目状構造で存在する。ビメンチンに結合する物質としては、抗ビメンチンモノクローナル抗体が知られている。抗ビメンチンモノクローナル抗体VIM3B4は、げっ歯類以外の哺乳動物のビメンチンに結合し、その認識部位はビメンチンの第二ロッド領域であることが報告されている(非特許文献9参照)。また、抗ビメンチンモノクローナル抗体V9(非特許文献9参照)は、ヒトマクロファージの活性化に伴い細胞表面または細胞外に分泌されるビメンチンに結合し得ることが報告されている(非特許文献10参照)。
【0006】
【非特許文献1】「ニュー・イングランド・ジャーナル・オブ・メディシン(New England Journal of Medicine)」、2000年、第342巻、p.1350−1358
【非特許文献2】「ジャーナル・オブ・セル・サイエンス(Journal of Cell Science)」、2003年、第116巻、p.217−224
【非特許文献3】「ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology)」、1989年、第109巻、p.309−315
【非特許文献4】「ジーンズ・アンド・デベロップメント(Genes and Development)」、2000年、第14巻、p.163−176
【非特許文献5】「サイトカイン・アンド・グロース・ファクター・レビュー(Cytokine and Growth Factor Review)11,59−69(2000)」、2000年、第11巻、p.59−69
【非特許文献6】「セル(Cell)」、1999年、第96巻、p.319−328
【非特許文献7】「ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology)」、2002年、第157巻、p.493−507
【非特許文献8】「エンドセリウム(Endothelium)」、2002年、第9巻、p.25−36
【非特許文献9】「エクスペリメンタル・セル・リサーチ(Experimental Cell Research)」、1992年、第201巻、p.1−7
【非特許文献10】「ネイチャー・セル・バイオロジー(Nature Cell Biology)」、2003年、第5巻、p.59−63
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、潜在型TGF−βの活性化抑制剤、TGF−β活性化抑制物質のスクリーニング方法、またはTGF−βが関与する疾患の治療薬を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明は、以下の(1)〜(10)に関する。
(1)ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤。
(2)ビメンチンとLAPの部分断片との結合を阻害する物質が、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体または該抗体断片である、上記(1)のTGF−β活性化抑制剤。
(3)LAPの部分断片が配列番号1〜16のいずれかで表されろアミノ酸配列を有するペプチドである、上記(1)または(2)のいずれか1つのTGF−β活性化抑制剤。
(4)(i)LAPの部分断片を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(ii)LAPの部分断片およびスクリーニングの対象となる物質を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(iii)(i)および(ii)の測定量から、該物質の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価すること、
(iv)該結合阻害活性を有する物質をTGF−β活性化抑制物質として選択すること、
からなるTGF−β活性化抑制物質のスクリーニング方法。
(5)ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する、TGF−βが関与する疾患の治療薬。
(6)TGF−βが関与する疾患が動脈硬化症、癌、炎症、線維症である、上記(5)の治療薬。
(7)ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−β活性化抑制方法。
(8)ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−βが関与する疾患の治療方法。
(9)TGF−β活性化抑制剤の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
(10)TGF−βが関与する疾患の治療薬の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【発明の効果】
【0009】
本発明により、ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤、またはTGF−β活性化抑制物質のスクリーニング方法が提供される。本発明のTGF−βの活性化抑制剤は、TGF−βが関与する疾患の治療薬として利用することができる。
【図面の簡単な説明】
【0010】
【図1】図1は、BAECに対する潜在型TGF−β活性化ペプチドの結合度(RLU/ウェル)を示す図である。「Peptide−25」のカラムは100μg/mlのPeptide−25、「Peptide−25+Ab」のカラムは100μg/mlのPeptide−25および5μg/mlの抗ビメンチン抗体VIM3B4、「Peptide−25N」のカラムは100μg/mlのPeptide−25N、「Peptide−25N+Ab」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのVIM3B4をそれぞれ添加した場合の結合度を示す。**は、スチューデントのt検定において、危険率1%未満で有意差があることを示す。
【図2】図2は、BAECを剃刀で剥離した後、剥離部分に遊走してきた観察視野当たりの細胞数(個/HPF)を示す図である。「Control+NI」のカラムは5μg/mlの非免疫マウスIgG、「Peptide−21+NI」のカラムは100μg/mlのPeptide−21および5μg/mlのIgG、「Peptide−21+Ab」のカラムは100μg/mlのPeptide−21および5μg/mlの抗ビメンチン抗体V9、「Peptide−25+NI」のカラムは100μg/mlのPeptide−25および5μg/mlのIgG、「Peptide−25+Ab」のカラムは100μg/mlのPeptide−25および5μg/mlのV9をそれぞれ添加した場合の細胞数を示す。*および**は、スチューデントのt検定において、それぞれ危険率5%および1%未満で有意差があることを示す。
【図3】図3は、BAECに対する潜在型TGF−β活性化ペプチドの結合度(RLU/well)を示す図である。「Peptide−25+KM511」のカラムは100μg/mlのPeptide−25および5μg/mlの抗G−CSF変異体抗体KM511、「Peptide−25+KM3565」のカラムは100μg/mlのPeptide−25および5μg/mlの抗ビメンチン抗体KM3565、「Peptide−25N+KM511」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのKM511、「Peptide−25N+KM3565」のカラムは100μg/mlのPeptide−25Nおよび5μg/mlのKM3565をそれぞれ添加した場合の結合度を示す。**は、スチューデントのt検定において、危険率1%未満で有意差があることを示す。
【図4】図4は、酵素免疫測定法により測定した、モノクローナル抗体KM3565のヒトビメンチン部分ペプチドに対する反応性を示す図である。上段が化合物1を、下段が化合物2を、それぞれ固層化したプレートを用いた測定結果を示す。X軸は添加したKM3565の濃度を、Y軸は抗体の結合度を表す吸光度をそれぞれ示す。
【図5】図5は、酵素免疫測定法により測定した、モノクローナル抗体KM3565のヒトビメンチン蛋白に対する反応性を示す図である。上段がビメンチン蛋白を、下段が大腸菌由来夾雑蛋白を、それぞれ固層化したプレートを用いた測定結果を示す。X軸は添加したKM3565の濃度を、Y軸は抗体の結合度を表す吸光度をそれぞれ示す。
【図6】図6は、酵素免疫測定法により測定した、市販抗ビメンチン抗体(▲;VIM3B4、■;V9)の化合物1およびビメンチン蛋白に対する反応性を示す図である。上段が化合物1を、下段がビメンチン蛋白を、それぞれ固層化したプレートを用いた測定結果を示す。
【発明を実施するための最良の形態】
【0011】
本発明において、ビメンチンとLAPの部分断片との結合を阻害する物質としては、該阻害活性を有する物質であれば、合成化合物でも天然物質でも特に制限なく用いることができるが、好ましくは、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体が用いられる。
抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体のうち、ビメンチンとLAPの部分断片との結合を阻害する活性を有するものの選択方法としては、後述の、本発明のTGF−βの活性化を抑制する物質のスクリーニング方法を用いることができる。
本発明に用いる抗ビメンチン抗体は、ポリクローナル抗体およびモノクローナル抗体を包含する。
【0012】
モノクローナル抗体としては、ハイブリドーマが産生する抗体、ヒト化抗体およびヒト抗体等があげられる。
ハイブリドーマとは、ヒト以外の哺乳動物に抗原を免疫して取得されたB細胞と、マウス等に由来するミエローマ細胞とを細胞融合させて得られる、所望の抗原特異性を有したモノクローナル抗体を産生する細胞である。
ヒト化抗体としては、ヒト型キメラ抗体、ヒト型相同性決定領域(complementarity determining region、以下「CDR」という。)移植抗体等があげられる。
【0013】
ヒト型キメラ抗体は、ヒト以外の動物の抗体重鎖可変領域(以下、重鎖はH鎖として、可変領域はV領域として「HV」または「VH」ともいう。)および抗体軽鎖可変領域(以下、軽鎖はL鎖として「LV」または「VL」ともいう。)とヒト抗体の重鎖定常領域(以下、定常領域はC領域として「CH」ともいう。)およびヒト抗体の軽鎖定常領域(以下「CL」ともいう。)とからなる抗体を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0014】
本発明に用いるヒト型キメラ抗体は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VHおよびVLをコードするcDNAを取得し、ヒト抗体CHおよびヒト抗体CLをコードする遺伝子を有する動物細胞用発現ベクターにそれぞれ挿入してヒト型キメラ抗体発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラ抗体のCHとしては、ヒトイムノグロブリン(以下「hIg」という。)に属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型キメラ抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0015】
ヒト型キメラL鎖とは、ヒト以外の動物のVLとヒト抗体のCLとからなる抗体L鎖を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
本発明に用いるヒト型キメラL鎖は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VLをコードするcDNAを取得し、ヒト抗体CLをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型キメラL鎖発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラL鎖のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0016】
ヒト型キメラH鎖とは、ヒト以外の動物のVHとヒト抗体のCHとからなる抗体H鎖を意味する。ヒト以外の動物としては、マウス、ラット、ハムスター、ラビット等、ハイブリドーマを作製することが可能であれば、いかなるものも用いることができる。
【0017】
本発明に用いるヒト型キメラH鎖は、ビメンチンに特異的に反応するモノクローナル抗体を生産するハイブリドーマより、VHをコードするcDNAを取得し、ヒト抗体CHをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型キメラH鎖発現ベクターを構築し、動物細胞へ導入することにより発現させ、製造することができる。
ヒト型キメラH鎖のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。
【0018】
ヒト型CDR移植抗体は、ヒト以外の動物の抗体のVHおよびVLのCDRのアミノ酸配列をヒト抗体のVHおよびVLの適切な位置に移植した抗体を意味する。
本発明に用いるヒト型CDR移植抗体は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVHおよびVLのCDR配列を任意のヒト抗体のVHおよびVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCHおよびヒト抗体のCLをコードする遺伝子を有する動物細胞用発現ベクターにそれぞれ挿入してヒト型CDR移植抗体発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植抗体を発現させ、製造することができる。
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。また、ヒト型CDR移植抗体のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0019】
本発明に用いるヒト型CDR移植キメラL鎖は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVLのCDR配列を任意のヒト抗体のVLのCDR配列に移植したV領域をコードするcDNAを構築し、ヒト抗体のCLをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型CDR移植キメラL鎖発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植キメラL鎖を発現させ、製造することができる。
ヒト型CDR移植キメラL鎖のCLとしては、hIgに属すればいかなるものでもよく、κクラスまたはλクラスのものを用いることができる。
【0020】
本発明に用いるヒト型CDR移植キメラH鎖は、ビメンチンに特異的に反応するヒト以外の動物の抗体のVHのCDR配列を任意のヒト抗体のVHのCDR配列に移植したH領域をコードするcDNAを構築し、ヒト抗体のCHをコードする遺伝子を有する動物細胞用発現ベクターに挿入してヒト型CDR移植キメラH鎖発現ベクターを構築し、該発現ベクターを動物細胞へ導入することによりヒト型CDR移植キメラH鎖を発現させ、製造することができる。
ヒト型CDR移植抗体のCHとしては、hIgに属すればいかなるものでもよいが、hIgGクラスのものが好適であり、更にhIgGクラスに属するhIgG1、hIgG2、hIgG3、hIgG4といったサブクラスのいずれも用いることができる。
【0021】
ヒト抗体は、元来、ヒト体内に天然に存在する抗体を意味するが、最近の遺伝子工学的、細胞工学的、発生工学的な技術の進歩により作製されたヒト抗体ファージライブラリーおよびヒト抗体産生トランスジェニック動物から得られる抗体等も含まれる。
ヒト体内に存在する抗体は、例えば、ヒト末梢血リンパ球を単離し、EBウイルス等を感染させ不死化、クローニングすることにより、該抗体を産生するリンパ球を培養でき、培養物中より該抗体を精製することができる。
【0022】
ヒト抗体ファージライブラリーは、ヒトB細胞から調製した抗体遺伝子をファージ遺伝子に挿入することによりFab(fragment of antigen bindingの略)、一本鎖抗体等の抗体断片をファージ表面に発現させたライブラリーである。該ライブラリーより、抗原を固定化した基質に対する結合活性を指標として所望の抗原結合活性を有する抗体断片を発現しているファージを回収することができる。該抗体断片は、更に遺伝子工学的手法により、2本の完全なH鎖および2本の完全なL鎖からなるヒト抗体分子へも変換することができる。
【0023】
ヒト抗体産生トランスジェニック動物は、ヒト抗体遺伝子が細胞内に組込まれた動物を意味する。具体的には、マウスES細胞へヒト抗体遺伝子を導入し、該ES細胞を他のマウスの初期胚へ移植後、発生させることによりヒト抗体産生トランスジェニック動物を作製することができる。ヒト抗体産生トランスジェニック動物からのヒト抗体の作製方法は、通常のヒト以外の哺乳動物で行われているハイブリドーマ作製方法によりヒト抗体産生ハイブリドーマを得、培養することで培養物中にヒト抗体を産生蓄積させることができる。
【0024】
本発明に用いる抗体または抗体のL鎖もしくはH鎖の部分断片としては、Fab、Fab’、F(ab’)2、一本鎖抗体(single chain Fv、以下「scFv」という。)、ジスルフィド安定化抗体(disulfide stabilized Fv、以下「dsFv」という。)、CDRを含むペプチド等があげられる。
【0025】
Fabは、IgGを蛋白質分解酵素パパインで処理して得られる断片のうち(H鎖の224番目のアミノ酸残基で切断される)、H鎖のN末端側約半分のアミノ酸とL鎖全体がジスルフィド結合で結合した分子量約5万の抗原結合活性を有する抗体断片である。
本発明に用いるFabは、ビメンチンに特異的に反応する抗体を蛋白質分解酵素パパインで処理して得ることができる。または、該抗体のFabをコードするDNAを原核生物用発現ベクターもしくは真核生物用発現ベクターに挿入し、該ベクターを原核生物もしくは真核生物へ導入することにより発現させ、Fabを製造することができる。
【0026】
F(ab’)2は、IgGを蛋白質分解酵素ペプシンで処理して得られる断片のうち(H鎖の234番目のアミノ酸残基で切断される)、Fabがヒンジ領域のジスルフィド結合を介して結合されたものよりやや大きい、分子量約10万の抗原結合活性を有する抗体断片である。
本発明に用いるF(ab’)2は、ビメンチンに特異的に反応する抗体を蛋白質分解酵素ペプシンで処理して得ることができる。または、下記のFab’をチオエーテル結合もしくはジスルフィド結合させ、作製することができる。
Fab’は、上記F(ab’)2のヒンジ領域のジスルフィド結合を切断した分子量約5万の抗原結合活性を有する抗体断片である。
本発明に用いるFab’は、ビメンチンに特異的に反応するF(ab’)2を還元剤ジチオスレイトール処理して得ることができる。または、該抗体のFab’断片をコードするDNAを原核生物用発現ベクターもしくは真核生物用発現ベクターに挿入し、該ベクターを原核生物もしくは真核生物へ導入することによりFab’を発現させ、製造することができる。
【0027】
scFvは、一本のVHと一本のVLとを適当なペプチドリンカー(以下「P」という。)を用いて連結した、VH−P−VLまたはVL−P−VHポリペプチドを示す。本発明に用いるscFvに含まれるVHおよびVLは、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体のいずれをも用いることができる。
本発明に用いるscFvは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得し、scFvをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
【0028】
dsFvは、VHおよびVL中のそれぞれ1アミノ酸残基をシステイン残基に置換したポリペプチドを該システイン残基間のジスルフィド結合を介して結合させたものをいう。システイン残基に置換するアミノ酸残基はReiterらにより示された方法[Protein Engineering,7,697(1994)]に従って、抗体の立体構造予測に基づいて選択することができる。本発明に用いるdsFvに含まれるVHおよびVLは、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体のいずれをも用いることができる。
【0029】
本発明に用いるdsFvは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得し、dsFvをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
【0030】
CDRを含むペプチドは、H鎖またはL鎖CDRの少なくとも1領域以上を含んで構成される。複数のCDRは、直接または適当なペプチドリンカーを介して結合させることができる。
本発明に用いるCDRを含むペプチドは、ビメンチンに特異的に反応する抗体のVHおよびVLをコードするcDNAを取得した後、CDRをコードするDNAを構築し、該DNAを原核生物用発現ベクターまたは真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより発現させ、製造することができる。
また、CDRを含むペプチドは、Fmoc法(フルオレニルメチルオキシカルボニル法)、tBoc法(t−ブチルオキシカルボニル法)等の化学合成法によって製造することもできる。
【0031】
本発明に用いる抗体の誘導体としては、ハイブリドーマが産生する抗体、ヒト化抗体、ヒト抗体またはそれらの抗体断片に放射性同位元素、蛋白質または低分子の化合物等を結合させた抗体の誘導体等があげられる。
本発明に用いる抗体の誘導体は、ビメンチンに特異的に反応する抗体または抗体断片のH鎖もしくはL鎖のN末端側もしくはC末端側、抗体または抗体断片中の適当な置換基もしくは側鎖、さらには抗体または抗体断片中の糖鎖に放射性同位元素、蛋白質もしくは低分子の化合物等を化学的手法[抗体工学入門(金光修著1994年(株)地人書館)]により結合させることにより製造することができる。
【0032】
または、ビメンチンに特異的に反応する抗体もしくは抗体断片をコードするDNAと、結合させたい蛋白質をコードするDNAを連結させて発現ベクターに挿入し、該発現ベクターを宿主細胞へ導入する、遺伝子工学的手法によっても製造することができる。
放射性同位元素としては、32P、3H、14C、131I、125I等があげられ、例えば、クロラミンT法等により、抗体に結合させることができる。
【0033】
低分子の薬剤としては、ハイドロコーチゾン、プレドニゾン等のステロイド剤、アスピリン、インドメタシン等の非ステロイド剤、金チオマレート、ペニシラミン等の免疫調節剤、アドリアマイシン、ダウノマイシン等の免疫抑制作用を有する抗癌剤、サイクロスポリン、FK506、サイクロフォスファミド、アザチオプリン等の免疫抑制剤、マレイン酸クロルフェニラミン、クレマシチンのような抗ヒスタミン剤等の抗炎症剤[炎症と抗炎症療法昭和57年 医歯薬出版株式会社]等があげられる。
【0034】
例えば、ダウノマイシンと抗体とを結合させる方法としては、グルタールアルデヒドを介してダウノマイシンと抗体のアミノ基間を結合させる方法、水溶性カルボジイミドを介してダウノマイシンのアミノ基と抗体のカルボキシル基を結合させる方法等があげられる。
【0035】
蛋白質としては、β−ガラクトシダーゼ、グルコースオキシダーゼ、ペルオキシダーゼ、アルカリホスファターゼ、グルコース−6−リン酸脱水素酵素等があげられる。
蛋白質と抗体とを結合させる方法としては、抗体または抗体断片をコードするcDNAに蛋白質をコードするcDNAを連結させ、融合抗体をコードするDNAを構築し、該DNAを原核生物または真核生物用発現ベクターに挿入し、該発現ベクターを原核生物または真核生物へ導入することにより蛋白質と抗体が融合した融合抗体を発現させる方法等があげられる。
【0036】
以下、ビメンチンに反応するポリクローナル抗体の作製方法について記す。
ビメンチンをコードするcDNAを含む発現ベクターを大腸菌、酵母、昆虫細胞、動物細胞等に導入して形質転換株を作製し、該形質転換株を培養・精製することにより、リコンビナントビメンチンを得る。または、ヒト血管内皮細胞、例えば臍帯静脈血管内皮細胞(HUVEC、クラボウ社製)等を培養・精製することによりビメンチンを得る。これらのビメンチン、ビメンチンの部分断片ポリペプチドの精製標品、またはビメンチンの一部のアミノ酸配列を有するペプチドを抗原に用いる。
【0037】
これら抗原はそのまま、またはキーホールリンペットヘモシアニン(KLH)、牛血清アルブミン(BSA)、メチル化牛血清アルブミン(メチル化BSA)、牛チログロブリン(THY)等の分子量の大きいキャリアタンパク質と結合させて投与する。
免疫に用いる動物としては、マウス、ラット、ハムスター、ラビット等ハイブリドーマを作製することが可能であれば、いかなるものでもよい。下記に、マウスおよびラットを用いる例について説明する。
【0038】
3〜20週令のマウスまたはラットに、上記方法で調製した抗原を免疫し、その動物の脾臓、リンパ節、末梢血より抗体産生細胞を採取する。免疫は、動物の皮下、静脈内または腹腔内に、適当なアジュバントとともに抗原を数回投与することにより行う。アジュバンドとしては、フロインドの完全アジュバント(Complete Freund’s Adjuvant)または、水酸化アルミニウムゲルと百日咳菌ワクチン等があげられる。各投与後3〜7日目に免疫動物の眼底静脈叢または尾静脈より採血し、免疫に用いた抗原に対しての反応性について、酵素免疫測定法〔酵素免疫測定法(ELISA法):医学書院刊(1976年)、Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory(1988)〕等で確認する。
【0039】
免疫に用いた抗原に対し、その血清が充分な抗体価を示した非ヒト哺乳動物より血清を取得し、該血清よりポリクローナル抗体を分離、精製する。
ポリクローナル抗体を分離、精製する方法としては、遠心分離、40〜50%飽和硫酸アンモニウムによる塩析、カプリル酸沈殿〔Antibodies,A Laboratory manuml,Cold Spring Harbor Laboratory,(1988)〕、またはDEAE−セファロースカラム、陰イオン交換カラム、プロテインAもしくはG−カラム、ゲル濾過カラム等を用いるクロマトグラフィー等を、単独または組み合わせて処理する方法があげられる。
【0040】
以下、ビメンチンに反応するモノクローナル抗体の作製方法について記す。
1.抗ビメンチンモノクローナル抗体の作製
(1)抗体産生細胞の調製
免疫に用いた抗原に対し、その血清が十分な抗体価を示したマウスまたはラットを抗体産生細胞の供給源とする。抗原物質の最終投与後3〜7日目に、免疫したマウスまたはラットより公知の方法[アンティボディズ・ア・ラボラトリー・マニュアル、コールド・スプリングハーバー・ラボラトリー(Antibodies−A Laboratory Manual Cold Spring Harbor Laboratory,1988)、以下「アンチボディズ・ア・ラボラトリー・マニュアル」という。]に準じて脾臓を摘出し、脾細胞と骨髄腫細胞とを融合させる。
【0041】
(2)骨髄腫細胞の調製
骨髄腫細胞としては、マウスから得られた株化細胞である、8−アザグアニン耐性マウス(BALB/c由来)骨髄腫細胞株P3−X63Ag8−U1(P3−U1)[Euro.J.Immunol.,6,511(1976)]、SP2/0−Ag14(SP−2)[Nature,276,269(1978)]、P3−X63−Ag8653(653)[J.Immunol.,123,1548(1979)]、P3−X63−Ag8(X63)[Nature,256,495(1975)]等、イン・ビトロ(in vitro)で増殖可能な骨髄腫細胞であればいかなるものでもよい。これらの細胞株の培養および継代については公知の方法(アンチボディズ・ア・ラボラトリー・マニュアル)に従い、細胞融合時までに2×107個以上の細胞数を確保する。
【0042】
(3)細胞融合
上記で得られた抗体産生細胞と骨髄腫細胞とを洗浄したのち、ポリエチレングライコール−1000(PEG−1000)などの細胞凝集性媒体を加え、細胞を融合させ、培地中に懸濁させる。細胞の洗浄にはMEM培地またはPBS(リン酸二ナトリウム1.83g、リン酸一カリウム0.21g、塩化ナトリウム7.65g、蒸留水1リットル、pH7.2)等を用いる。また、融合細胞を懸濁させる培地としては、目的の融合細胞のみを選択的に得られるように、HAT培地{正常培地[RPMI−1640培地にグルタミン(1.5mmol/l)、2−メルカプトエタノール(5×10−5mol/l)、ジェンタマイシン(10μg/ml)および牛胎児血清(FCS)(CSL社製、10%)を加えた培地]にヒポキサンチン(10−4mol/l)、チミジン(1.5×10−5mol/l)およびアミノプテリン(4×10−7mol/l)を加えた培地}を用いる。
培養後、培養上清の一部をとり、酵素免疫測定法により、抗原蛋白質に反応し、非抗原蛋白質に反応しないサンプルを選択する。ついで、限界希釈法によりクローニングを行い、酵素免疫測定法により安定して高い抗体価の認められたものをモノクローナル抗体産生ハイブリドーマ株として選択する。
【0043】
(4)抗ビメンチンモノクローナル抗体産生ハイブリドーマの選択
抗ビメンチンモノクローナル抗体を産生するハイブリドーマの選択は、アンチボディズ・ア・ラボラトリー・マニュアルに述べられている方法等に従い、以下に述べる測定法により行う。これらの方法により、後述する抗ビメンチンヒト化抗体、該抗体断片を産生する形質転換株の培養上清中に含まれる抗ビメンチン抗体またはすべての精製抗ビメンチン抗体の結合活性を測定することができる。
【0044】
酵素免疫測定法
抗原または抗原を発現した細胞等を96ウェルプレートにコートし、ハイブリドーマ培養上清または上述の方法で得られる精製抗体を第一抗体として反応させる。
第一抗体反応後、プレートを洗浄して第二抗体を添加する。
第二抗体とは、第一抗体のイムノグロブリンを認識できる抗体を、ビオチン、酵素、化学発光物質または放射線化合物等で標識した抗体である。具体的にはハイブリドーマ作製の際にマウスを用いたのであれば、第二抗体としては、マウスイムノグロブリンを認識できる抗体を用いる。
反応後、第二抗体を標識した物質に応じた反応を行ない、抗原に特異的に反応するモノクローナル抗体を生産するハイブリドーマとして選択する。
【0045】
(5)モノクローナル抗体の精製
プリスタン処理〔2,6,10,14−テトラメチルペンタデカン(Pristane)0.5mlを腹腔内投与し、2週間飼育する〕した8〜10週令のマウスまたはヌードマウスに、1(3)で得られた抗ビメンチンモノクローナル抗体産生ハイブリドーマ細胞2×107〜5×106細胞/匹を腹腔内に注射する。10〜21日間でハイブリドーマは腹水癌化する。該マウスまたはヌードマウスから腹水を採取し、遠心分離、40〜50%飽和硫酸アンモニウムによる塩析、カプリル酸沈殿法、DEAE−セファロースカラム、プロテインA−カラムまたはセルロファインGSL2000(生化学工業社製)のカラムなどを用いて、IgGまたはIgM画分を回収し、精製モノクローナル抗体とする。
精製モノクローナル抗体のサブクラスの決定は、マウスモノクローナル抗体タイピングキットまたはラットモノクローナル抗体タイピングキット等を用いて行うことができる。蛋白質量は、ローリー法または280nmでの吸光度より算出することができる。
抗体のサブクラスとは、クラス内のアイソタイプのことで、マウスでは、IgG1、IgG2a、IgG2b、IgG3、ヒトでは、IgG1、IgG2、IgG3、IgG4があげられる。
【0046】
(6)抗ビメンチンモノクローナル抗体の反応性
抗ビメンチンモノクローナル抗体の反応性を調べる方法として、インヒビションELISAがあげられる。
まず、1(4)に記したように抗原を固相化したプレートを準備し、第一抗体として抗ビメンチンモノクローナル抗体を反応させる。同時に適当に希釈したビメンチンを加えプレートに固相化したビメンチンと競合させる。その後は1(4)に示した方法と同様に検出することができる。抗ビメンチンモノクローナル抗体がビメンチンと反応する場合には、液相系で加えたビメンチン濃度依存的に固相化ビメンチンへの結合が阻害され、OD値が低下する。
【0047】
2.ヒト化抗体の作製方法(I)−抗ビメンチンヒト化抗体の作製方法
(1)ヒト化抗体発現用ベクターの構築
ヒト以外の動物の抗体からヒト化抗体を作製するために必要なヒト化抗体発現用ベクターを構築する。ヒト化抗体発現用ベクターとは、ヒト抗体のC領域であるCHおよびCLをコードする遺伝子が組み込まれた動物細胞用発現ベクターであり、動物細胞用発現ベクターにヒト抗体のCHおよびCLをコードする遺伝子をそれぞれ挿入することにより構築されたものである。
【0048】
ヒト抗体のC領域としては、例えば、ヒト抗体H鎖ではCγ1やCγ4、ヒト抗体L鎖ではCκ等の任意のヒト抗体のC領域を用いることができる。ヒト抗体のC領域をコードする遺伝子としてはエキソンとイントロンより成る染色体DNAを用いることができ、また、cDNAを用いることもできる。動物細胞用発現ベクターとしては、ヒト抗体C領域をコードする遺伝子を組込み発現できるものであればいかなるものでも用いることができる。
【0049】
例えば、pAGE107[Cytotechnology,3,133(1990)]、pAGE103[J.Biochem.,101,1307(1987)]、pHSG274[Gene,27,223(1984)]、pKCR[Proc.Natl.Acad.Sci.USA,78,1527(1981)]、pSG1 β d2−4[Cytotechnology,4,173(1990)]等があげられる。動物細胞用発現ベクターに用いるプロモーターとエンハンサーとしては、SV40の初期プロモーターとエンハンサー[J.Biochem.,101,1307(1987)]、モロニーマウス白血病ウイルスのLTRプロモーターとエンハンサー[Biochem.Biophys.Res.Comun.,149,960(1987)]、および免疫グロブリンH鎖のプロモーター[Cell,41,479(1985)]とエンハンサー[Cell,33,717(1983)]等があげられる。
【0050】
ヒト化抗体発現用ベクターは、抗体H鎖、L鎖が別々のベクター上に存在するタイプまたは同一のベクター上に存在するタイプ(タンデム型)のどちらでも用いることができるが、ヒト化抗体発現ベクターの構築のしやすさ、動物細胞への導入のし易さ、動物細胞内での抗体H鎖およびL鎖の発現量のバランスがとれる等の点でタンデム型のヒト化抗体発現用ベクターの方が好ましい[J.Immunol.Methods,167,271(1994)]。
【0051】
(2)ヒト以外の動物の抗体のVHおよびVLをコードするcDNAの取得
ヒト以外の動物の抗体、例えば、マウス抗ビメンチンモノクローナル抗体のVHおよびVLをコードするcDNAは以下のようにして取得する。
抗ビメンチンモノクローナル抗体を産生する細胞、例えば、マウスビメンチン抗体産生ハイブリドーマ等よりmRNAを抽出し、cDNAを合成する。合成したcDNAを、ファージまたはプラスミドなどのベクターに挿入し、cDNAライブラリーを作製する。該ライブラリーより、ヒト以外の動物の抗体、例えば、マウス抗体のC領域部分またはV領域部分をプローブとして用い、VHをコードするcDNAを有する組換えファージまたは組換えプラスミド、およびVLをコードするcDNAを有する組換えファージまたは組換えプラスミドをそれぞれ単離する。組換えファージまたは組換えプラスミド上の目的とする抗体のVHおよびVLの全塩基配列を決定し、塩基配列よりVHおよびVLの全アミノ酸配列を推定する。
【0052】
(3)ヒト型キメラ抗体発現ベクターの構築
前記2(1)で構築したヒト化抗体発現用ベクターのヒト抗体のCHおよびCLをコードする遺伝子の上流に、ヒト以外の動物の抗体のVHおよびVLをコードするcDNAを挿入し、ヒト型キメラ抗体発現ベクターを構築することができる。例えば、キメラ抗体発現用ベクターのヒト抗体のCHおよびCLをコードする遺伝子の上流にあらかじめヒト以外の動物の抗体のVHおよびVLをコードするcDNAをクローニングするための制限酵素の認識配列を設けておき、このクローニングサイトにヒト以外の動物の抗体のV領域をコードするcDNAを下記に述べる合成DNAを介して挿入することにより、ヒト型キメラ抗体発現ベクターを製造することができる。合成DNAは、ヒト以外の動物の抗体のV領域の3’末端側の塩基配列とヒト抗体のC領域の5’末端側の塩基配列とからなるものであり、両端に適当な制限酵素部位を有するようにDNA合成機を用いて製造する。
【0053】
(4)ヒト以外の動物の抗体のCDR配列の同定
抗体の抗原結合部位を形成するVHおよびVLは、配列の比較的保存された4個のフレームワーク領域(以下、FR領域と称す)とそれらを連結する配列の変化に富んだ3個の相補性決定領域(CDR)から成っている[シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト(Sequences of Proteins of Immunological Interest),US Dept.Health and Human Services,(1991)、以下「シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト」という。]。そして各CDRアミノ酸配列(CDR配列)は、既知の抗体のV領域のアミノ酸配列(シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト)と比較することにより同定することができる。
【0054】
(5)ヒト型CDR移植抗体のV領域をコードするcDNAの構築
ヒト型CDR移植抗体のVHおよびVLをコードするcDNAは以下のようにして取得することができる。
まず、目的のヒト以外の動物の抗体のV領域のCDRを移植するためのヒト抗体のV領域のFRのアミノ酸配列をVH、VLそれぞれについて選択する。ヒト抗体のV領域のFRのアミノ酸配列としては、ヒト抗体由来のV領域のFRのアミノ酸配列であればいかなるものでも用いることができる。
【0055】
例えば、Protein Data Bankに登録されているヒト抗体のV領域のFRのアミノ酸配列、ヒト抗体のV領域のFRの各サブグループの共通アミノ酸配列(シーケンシズ・オブ・プロテインズ・オブ・イムノロジカル・インタレスト)があげられるが、充分な活性を有するヒト型CDR移植抗体を創製するためには、目的のヒト以外の動物の抗体のV領域のアミノ酸配列と高い相同性、好ましくは60%以上の相同性を有することが望ましい。次に、選択したヒト抗体のV領域のFRのアミノ酸配列をコードするDNA配列と目的のヒト以外の動物の抗体のV領域のCDRのアミノ酸配列をコードするDNA配列を連結させて、VH、VLそれぞれのアミノ酸配列をコードするDNA配列を設計する。CDR移植抗体可変領域遺伝子を構築するために設計したDNA配列を得るためには、全DNA配列をカバーするように各鎖について数本の合成DNAを設計し、それらを用いてポリメラーゼ・チェイン・リアクション(Polymerase Chain Reaction、以下「PCR」という。)を行う。PCRでの反応効率および合成可能なDNAの長さから各鎖について、好ましくは、6本の合成DNAを設計する。反応後、増幅断片を適当なベクターにサブクローニングし、その塩基配列を決定し、目的のヒト型CDR移植抗体の各鎖のV領域のアミノ酸配列をコードするcDNAを含むプラスミドを取得する。また、約100塩基よりなる合成DNAを用いてセンス、アンチセンスともに全配列を合成し、それらをアニーリング、連結することで、目的のヒト型CDR移植抗体の各鎖のV領域のアミノ酸配列をコードするcDNAを構築することもできる。
【0056】
(6)ヒト型CDR移植抗体のV領域のアミノ酸配列の改変
ヒト型CDR移植抗体は目的のヒト以外の動物の抗体のV領域のCDRのみをヒト抗体のV領域のFR間に、単純に移植しただけでは、その活性はもとのヒト以外の動物の抗体の活性に比べて低下してしまうことが知られている[BIO/TECHNOLOGY,9,266(1991)]。そこでヒト抗体のV領域のFRのアミノ酸配列のうち、直接抗原との結合に関与しているアミノ酸残基、CDRのアミノ酸残基と相互作用をしているアミノ酸残基、または抗体の立体構造の維持に関与している等の可能性を有するアミノ酸残基をもとのヒト以外の動物の抗体に見出されるアミノ酸残基に改変し、活性を上昇させることが行われている。そして、それらのアミノ酸残基を効率よく同定するため、X線結晶解析あるいはコンピューターモデリング等を用いた抗体の立体構造の構築および解析を行っている。しかし、いかなる抗体にも適応可能なヒト型CDR移植抗体の製造法は未だ確立されておらず、現状では個々の抗体によって種々の試行錯誤が必要である。
【0057】
選択したヒト抗体のV領域のFRのアミノ酸配列の改変は各種の変異導入プライマーを用いて前記2(5)に記載のPCRを行うことにより達成できる。PCR後の増幅断片を適当なベクターにサブクローニング後、その塩基配列を決定し、目的の変異が導入されたcDNAを含むベクター(以下「アミノ酸配列改変ベクター」という。)を取得する。
また、狭い領域のアミノ酸配列の改変であれば、20〜35塩基からなる変異導入プライマーを用いたPCR変異導入法により行うことができる。具体的には、改変後のアミノ酸残基をコードするDNA配列を含む20〜35塩基からなるセンス変異プライマーおよびアンチセンス変異プライマーを合成し、改変すべきV領域のアミノ酸配列をコードするcDNAを含むプラスミドを鋳型として2段階のPCRを行う。最終増幅断片を適当なベクターにサブクローニング後、その塩基配列を決定し、目的の変異が導入されたcDNAを含むアミノ酸配列改変ベクターを取得する。
【0058】
(7)ヒト型CDR移植抗体発現ベクターの構築
前記2(1)のヒト化抗体発現用ベクターのヒト抗体のCH及びCLをコードする遺伝子の上流に、前記2(5)および2(6)で取得したヒト型CDR移植抗体のVHおよびVLをコードするcDNAを挿入し、ヒト型CDR移植抗体発現ベクターを構築することができる。例えば、ヒト型CDR移植抗体のVHおよびVLのアミノ酸配列をコードするcDNAを構築するためのPCRの際に5’末端および3’末端の合成DNAの末端に適当な制限酵素の認識配列を導入することで、所望のヒト抗体のC領域をコードする遺伝子の上流にそれらが適切な形で発現するように挿入することができる。
【0059】
(8)ヒト化抗体の一過性(トランジェント)発現および活性評価
多種類のヒト化抗体の活性を効率的に評価するために、前記2(3)のヒト型キメラ抗体発現ベクター、および前記2(7)のヒト型CDR移植抗体発現ベクターまたはそれらの改変ベクターをCOS−7細胞(ATCC CRL1651)に導入してヒト化抗体の一過性発現[Methods in Nucleic Acids Res.,CRC Press,pp283(1991)]を行い、その活性を測定することができる。
COS−7細胞への発現ベクターの導入法としては、DEAE−デキストラン法[Methods in Nucleic Acids Res.,CRC Press,pp283(1991)]、リポフェクション法[Proc.Natl.Acad.Sci.USA,84,7413(1987)]等があげられる。
ベクターの導入後、培養上清中のヒト化抗体の活性は前記1(5)に記載の酵素免疫測定法(ELISA法)等により測定することができる。
【0060】
(9)ヒト化抗体の安定(ステーブル)発現および活性評価
前記2(3)のヒト型キメラ抗体発現ベクターおよび前記2(7)のヒト型CDR移植抗体発現ベクターを適当な宿主細胞に導入することによりヒト化抗体を安定に生産する形質転換株を得ることができる。
【0061】
宿主細胞への発現ベクターの導入法としては、エレクトロポレーション法[特開平2−257891;Cytotechnology,3,133(1990)]等があげられる。
ヒト化抗体発現ベクターを導入する宿主細胞としては、ヒト化抗体を発現させることができる宿主細胞であれば、いかなる細胞でも用いることができる。例えば、マウスSP2/0−Ag14細胞(ATCC CRL1581)、マウスP3X63−Ag8.653細胞(ATCC CRL1580)、ジヒドロ葉酸還元酵素遺伝子(以下「DHFR遺伝子」という。)が欠損したCHO細胞[Proc.Natl.Acad.Sci.USA,77,4216(1980)]、ラットYB2/3HL.P2.G11.16Ag.20細胞(ATCC CRL1662、以下「YB2/0細胞」という。)等があげられる。
【0062】
ベクターの導入後、ヒト化抗体を安定に生産する形質転換株は、特開平2−257891に開示されている方法に従い、G418およびFCSを含むRPMI1640培地により選択する。得られた形質転換株を培地中で培養することで培養液中にヒト化抗体を生産蓄積させることができる。培養液中のヒト化抗体の活性は前記1(5)に記載の方法等により測定する。また、形質転換株は、特開平2−257891に開示されている方法に従い、DHFR遺伝子増幅系等を利用してヒト化抗体の生産量を上昇させることができる。
【0063】
ヒト化抗体は、形質転換株の培養上清よりプロテインAカラムを用いて精製することができる(アンチボディズ・ア・ラボラトリー・マニュアル第8章)。また、その他に、通常の蛋白質で用いられる精製方法を使用することができる。例えば、ゲル濾過、イオン交換クロマトグラフィーおよび限外濾過等を組合せて行い、精製することができる。精製したヒト化抗体のH鎖、L鎖または抗体分子全体の分子量は、ポリアクリルアミドゲル電気泳動(SDS−PAGE)[Nature,227,680(1970)]やウエスタンブロッティング法(アンチボディズ・ア・ラボラトリー・マニュアル第12章)等で測定する。
精製したヒト化抗体の反応性、また、ヒト化抗体のビメンチンに対する結合活性の測定は前記1(4)に記載の方法などにより測定することができる。
【0064】
3.抗体断片の作製
抗体断片は、上記2に記載のヒト化抗体を元に遺伝子工学的手法または蛋白質化学的手法により、作製することができる。抗体断片としては、Fab、F(ab’)2、Fab’、scFv、Diabody、dsFv、CDRを含むペプチド等があげられる。
【0065】
(1)Fabの作製
Fabは、IgGを蛋白質分解酵素パパインで処理することにより、作製することができる。パパインの処理後は、元の抗体がプロテインA結合性を有するIgGサブクラスであれば、プロテインAカラムに通すことで、IgG分子やFc断片と分離し、均一なFabとして回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,1995)。プロテインA結合性を持たないIgGサブクラスの抗体の場合は、イオン交換クロマトグラフィーにより、Fabは低塩濃度で溶出される画分中に回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,1995)。また、Fabは、大腸菌を用いて遺伝子工学的に作製することもできる。例えば、上記2の(2)および(3)に記載の抗体のV領域をコードするDNAを、Fab発現用ベクターにクローニングし、Fab発現ベクターを作製することができる。Fab発現用ベクターとしては、Fab用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pIT106[Science,240,1041,(1988)]等があげられる。Fab発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にFabを生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるFabとすることができ、また、ペリプラズマ層に発現させた場合は、培養上清中に活性を持ったFabが漏出する。リフォールディング後または培養上清からは、抗原を結合させたカラムを用いることにより、均一なFabを精製することができる(Antibody Engineering,A Practical Guide,W.H.Freeman and Company,1992)。
【0066】
(2)F(ab’)2の作製
F(ab’)2は、IgGを蛋白質分解酵素ペプシンで処理することにより、作製することができる。ペプシンの処理後は、Fabと同様の精製操作により、均一なF(ab’)2として回収することができる(Monoclonal Antibodies:Principles and Practice,third edition,Academic Press,1995)。また、上記2(3)に記載のFab’をo−PDMやビスマレイミドヘキサン等のようなマレイミドで処理し、チオエーテル結合させる方法や、DTNBで処理し、S−S結合させる方法によっても作製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0067】
(3)Fab’の作製
Fab’は、大腸菌を用いて遺伝子工学的に作製することができる。例えば、上記2(2)および(3)に記載の抗体のV領域をコードするDNAを、Fab’発現用ベクターにクローニングし、Fab’発現ベクターを作製することができる。Fab’発現用ベクターとしては、Fab’用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pAK19[Bio/Technology,10,163,(1992)]等があげられる。Fab’発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にFab’を生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるFab’とすることができ、また、ペリプラズマ層に発現させた場合は、リゾチームによる部分消化、浸透圧ショック、ソニケーション等の処理により菌を破砕し、菌体外へ回収させることができる。リフォールディング後または菌の破砕液からは、プロテインGカラム等を用いることにより、均一なFab’を精製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0068】
(4)scFvの作製
scFvは遺伝子工学的には、ファージまたは大腸菌を用いて作製することができる。例えば、上記2(2)および(3)に記載の抗体のVHおよびVLをコードするDNAを、12残基以上のアミノ酸配列からなるポリペプチドリンカーをコードするDNAを介して連結し、scFvをコードするDNAを作製する。作製したDNAをscFv発現用ベクターにクローニングし、scFv発現ベクターを作製することができる。scFv発現用ベクターとしては、scFvのDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pCANTAB5E(Pharmacia社製)、Phfa[Hum.Antibody Hybridoma,5,48(1994)]等があげられる。scFv発現ベクターを適当な大腸菌に導入し、ヘルパーファージを感染させることで、ファージ表面にscFvがファージ表面蛋白質と融合した形で発現するファージを得ることができる。また、scFv発現ベクターを導入した大腸菌の封入体またはペリプラズマ層にscFvを生成蓄積させることができる。封入体からは、通常蛋白質で用いられるリフォールディング法により、活性のあるscFvとすることができ、また、ペリプラズマ層に発現させた場合は、リゾチームによる部分消化、浸透圧ショック、ソニケーション等の処理により菌を破砕し、菌体外へ回収することができる。リフォールディング後または菌の破砕液からは、陽イオン交換クロマトグラフィー等を用いることにより、均一なscFvを精製することができる(Antibody Engineering,A Practical Approach,IRL PRESS,1996)。
【0069】
(5)Diabodyの作製
Diabodyは、上記のscFvを作製する際のポリペプチドリンカーを3〜10残基程度にすることで、作製することができる。1種類の抗体のVHおよびVLを用いた場合には、2価のDiabodyを、2種類の抗体のVHおよびVLを用いた場合は、2特異性を有するDiabodyを作製することができる[FEBS Letters,453,164(1999)、Int.J.Cancer,77,763(1998)]。
【0070】
(6)dsFvの作製
dsFvは、大腸菌を用いて遺伝子工学的に作製することができる。まず、上記2(2)および(3)に記載の抗体のVHおよびVLをコードするDNAの適当な位置に変異を導入し、コードするアミノ酸残基がシステインに置換されたDNAを作製する。作製した各DNAをdsFv発現用ベクターにクローニングし、VHおよびVLの発現ベクターを作製することができる。dsFv発現用ベクターとしては、dsFv用のDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pULI9[Protein Engineering,7,697(1994)]等があげられる。VHおよびVLの発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にVHおよびVLを生成蓄積させることができる。封入体またはペリプラズマ層からVHおよびVLを得、混合し、通常蛋白質で用いられるリフォールディング法により、活性のあるdsFvとすることができる。リフォールディング後は、イオン交換クロマトグラフィーおよびゲル濾過等により、さらに精製することができる[Protein Engineering,7,697(1994)]。
【0071】
(7)CDRペプチドの作製
CDRを含むペプチドは、Fmoc法またはtBoc法等の化学合成法によって作製することができる。また、CDRを含むペプチドをコードするDNAを作製し、作製したDNAを適当な発現用ベクターにクローニングし、CDRペプチド発現ベクターを作製することができる。発現用ベクターとしては、CDRを含むペプチドをコードするDNAを組み込み発現できるものであればいかなるものも用いることができる。例えば、pLEX(Invitrogen社製)、pAX4a+(Invitrogen社製)等があげられる。発現ベクターを適当な大腸菌に導入し、封入体またはペリプラズマ層にCDRを含むペプチドを生成蓄積させることができる。封入体またはペリプラズマ層からCDRを含むペプチドを得、イオン交換クロマトグラフィーおよびゲル濾過等により、精製することができる[Protein Engineering,7,697(1994)]。
【0072】
4.融合抗体および融合ペプチドの作製方法
本発明で使用される抗体またはペプチドに、放射性同位元素、毒素、サイトカインまたは酵素等の蛋白質、低分子の薬剤等を、化学的または遺伝子工学的に結合させた融合抗体または融合ペプチドも抗体の誘導体として使用することができる。
【0073】
抗体またはペプチドと毒素蛋白質とを化学的に結合させた融合抗体または融合ペプチドは、文献[Anticancer Research,11,2003(1991);Nature Medicine,3,350(1996)]記載の方法等に従って作製することができる。
抗体またはペプチドと、毒素、サイトカインまたは酵素等の蛋白質とを遺伝子工学的に結合させた融合抗体または融合ペプチドは、文献[Proc.Natl.Acad.Sci.USA,93,974(1996);Proc.Natl.Acad.Sci.USA,93,7826(1996)]記載の方法等に従って作製することができる。
【0074】
抗体またはペプチドと低分子抗癌剤を化学的に結合させた融合抗体または融合ペプチドは、文献[Science,261,212(1993)]記載の方法等に従って作製することができる。
抗体またはペプチドと放射性同位元素を化学的に結合させた融合抗体または融合ペプチドは、文献[Antibody Immunoconjugates and Radiopharmaceuticals,3,60(1990);Anticancer Research,11,2003(1991)]記載の方法等に従って作製することができる。
【0075】
これらの誘導体は、抗体分子の特異性に従って放射性同位元素、毒素、サイトカインまたは酵素等の蛋白質、低分子の薬剤等を標的組織周辺に集積させることで、より効果的で副作用の少ない診断または治療を可能にすることが期待されている。
【0076】
5.ヒト化抗体または抗体断片の活性評価
精製したヒト化抗体の抗原との結合性、ビメンチンに対する結合活性はELISA法および蛍光抗体法[Cancer Immunol.Immunother.,36,373(1993)]、BIAcoreTM等を用いた表面プラズモン共鳴等により測定できる。
【0077】
6.本発明に用いる抗体を用いたビメンチンの定量および検出方法
本発明に用いる抗ビメンチン抗体またはその抗体断片を用いる、ビメンチンを免疫学的に定量または検出する方法としては、蛍光抗体法、免疫酵素抗体法(ELISA)、放射性物質標識免疫抗体法(RIA)、免疫組織染色法、免疫細胞染色法などの免疫組織化学染色法(ABC法、CSA法等)、ウェスタンブロッティング法、免疫沈降法、上記に記した酵素免疫測定法、サンドイッチELISA法[単クローン抗体実験マニュアル(講談社サイエンティフィック、1987年)、続生化学実験講座5免疫生化学研究法(東京化学同人、1986年)]等を用いることができる。
【0078】
蛍光抗体法は、文献[Monoclonal Antibodies:Principles and practice,Third edition(Academic Press,1996),単クローン抗体実験マニュアル(講談社サイエンティフィック、1987)]等に記載された方法を用いて行うことができる。具体的には、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いられるモノクローナル抗体またはその抗体断片を反応させ、さらにフルオレシン・イソチオシアネート(FITC)またはフィコエリスリンなどの蛍光物質でラベルした抗イムノグロブリン抗体または結合断片を反応させた後、蛍光色素をフローサイトメーターで測定する方法である。
【0079】
免疫細胞染色法、免疫組織染色法等の免疫組織化学染色法(ABC法、CSA法等)は、文献[Monoclonal Antibodies:Principles and practice,Third edition(Academic Press,1996),単クローン抗体実験マニュアル(講談社サイエンティフィック,1987)]等に記載された方法を用いて行うことができる。
免疫酵素抗体法(ELISA)は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにペルオキシダーゼ、ビオチン等の酵素標識等を施した抗イムノグロブリン抗体または結合断片を反応させた後、発色色素を吸光光度計で測定する方法である。
【0080】
放射性物質標識免疫抗体法(RIA)は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらに放射線標識を施した抗イムノグロブリン抗体または結合断片を反応させた後、シンチレーションカウンター等で測定する方法である。
免疫細胞染色法、免疫組織染色法は、生体内から分離された細胞または組織等に、本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにフルオレシン・イソチオシアネート(FITC)等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素標識を施した抗イムノグロブリン抗体または結合断片を反応させた後、顕微鏡を用いて観察する方法である。
【0081】
ウェスタンブロッティング法は、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等をSDS−ポリアクリルアミドゲル電気泳動[Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988]で分画した後、該ゲルをPVDF膜またはニトロセルロース膜にブロッティングし、該膜に本発明に用いるモノクローナル抗体またはその抗体断片を反応させ、さらにFITC等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素標識を施した抗マウスIgG抗体または結合断片を反応させた後、確認する。
【0082】
免疫沈降法とは、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等を本発明に用いるモノクローナル抗体またはその抗体断片と反応させた後、プロテインG−セファロース等のイムノグロブリンに特異的な結合能を有する担体を加えて抗原抗体複合体を沈降させるものである。
サンドイッチELISA法とは、本発明に用いるモノクローナル抗体またはその抗体断片で、抗原認識部位の異なる2種類のモノクローナル抗体のうち、あらかじめ一方のモノクローナル抗体または抗体断片はプレートに吸着させ、もう一方のモノクローナル抗体または抗体断片はFITC等の蛍光物質、ペルオキシダーゼ、ビオチン等の酵素で標識しておく。抗体吸着プレートに、生体内から分離された細胞またはその破砕液、組織またはその破砕液、細胞培養上清、血清等を反応後、標識したモノクローナル抗体またはその抗体断片を反応させ、標識物質に応じた反応を行う方法である。
【0083】
本発明でいうLAPの部分断片としては、細胞表面または細胞外に分泌されるビメンチンに結合し得るLAPの部分断片であればいずれのものも用いることができ、例えば、国際公開第98/51704号の第2表に記載された化合物1〜16等を使用することができるが、化合物15が好ましく使用される。化合物1〜16のアミノ酸配列を配列番号1〜16に示す。
【0084】
以下、本発明のTGF−βの活性化抑制物質のスクリーニング方法について記す。
本発明のスクリーニング方法の対象となる物質は、合成化合物でも天然物質でも特に制限なく用いることができる。
本発明の方法に使用するLAPの部分断片としては、上記のLAPの部分断片を用いることができる。
【0085】
本発明の方法に使用するビメンチンおよびTGF−βを発現する細胞としては、例えば、血管内皮細胞、マクロファージ等があげられる。細胞は、ヒト、ウシ、ブタ、ネズミ等の動物から、単離・精製したもの、またはそれら細胞由来の培養細胞を用いることができる。単離・精製方法としては、例えば、オカダ(Okada)らの方法[ジャーナル・オブ・バイオケミストリー(Journal of Biochemistry),106,304(1989)]等があげられる。
【0086】
細胞へのLAPの部分断片の結合は、例えば細胞を培地で培養し該部分断片を添加しインキュベートした後、細胞を洗浄し、細胞に結合している該部分断片を測定することにより行うことができる。
LAPの部分断片は、例えばクロラミンT法[モレキュラー・アンド・セルラー・バイオロジー(Molecular & Cellular Biology),2,599(1982)]により125I標識したもの、フルオレセインイソチオシアネート、AMCA、ローダミン6G、ローダミンレッド−X、Cy3、テキサスレッド等の蛍光色素で標識をしたもの等を用いるのが好ましい。これにより、細胞に結合したLAPの部分断片は、それぞれ、放射活性、発光強度等を測定することにより検出および定量することができる。
【0087】
また、細胞表面上に発現するビメンチンにLAPの部分断片が結合することにより潜在型TGF−βが活性化されるので、細胞に結合するLAPの部分断片量の測定は、活性型TGF−β量を測定することにより行うこともできる。活性型TGF−βの定量法は特に限定されないが、直接抗TGF−β抗体を用いた酵素免疫測定方法[メソッヅ・イン・エンザイモロジー(Methods in Enzymology),198,303(1991)]または安部らのルシフェラーゼ・アッセイ・システム[アナリティカル・バイオケミストリー(Analytical Biochemistry),216,276(1994)]等で測定することができる。また、血管内皮細胞の遊走度[ジャーナル・オブ・セル・バイオロジー(Journal of Cell Biology),123,1249(1993)]、血管平滑筋細胞の増殖[トウホク・ジャーナル・オブ・エクスペリメンタル・メディスン(Tohoku Journal of Experimental Medicine),179,23(1996)]、各種癌細胞の増殖阻害[ジャーナル・オブ・クリニカル・インベスティゲーション(Journal of Clinical Investigation),87,277(1991),エンドクリノロジー(Endocrinology),128,1981(1991)]、ミンク肺上皮細胞の増殖阻害[メソッヅ・イン・エンザイモロジー(Methods in Enzymology),198,317(1991)]等の方法で測定することができる。
【0088】
スクリーニングの対象となる物質を添加しないときの、細胞に結合するLAPの部分断片量または活性型TGF−β量と、該物質を添加したときの、細胞に結合するLAPの部分断片量または活性型TGF−β量とを比較し、その差異から、該化合物の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価することができる。該結合阻害活性を有する物質をTGF−βの活性化を抑制する目的物質として選択する。目的物質の選択は、例えば、添加濃度1mmol/lで、対象となる化合物を添加しない場合の活性型TGF−β量に対し、添加した場合の活性型TGF−β量が10%以上減少の認められる物質を選択することにより行うことができる。
【0089】
本発明のビメンチンとLAPの部分断片との結合を阻害する物質は潜在型TGF−βの活性化を抑制することから、潜在型TGF−βの活性化抑制剤として用いることができ、TGF−βが関与する各種疾患の治療薬として用いることができる。
TGF−βが関与する疾患としては、例えば、癌、IgA腎症、巣状および分節性糸球体腎炎、ループス腎炎、糖尿病性腎症、HIV腎症、C型肝炎、アルコール性および自己免疫性肝線維症、ブレオマイシン誘発性および突発性肺線維症、全身性硬化症、骨髄線維症、増殖性硝子体網膜症、クローン病、好酸球増多筋肉痛症候群、肝硬変、梗塞後心線維症、手術後腹内癒着、血行再建術後再狭窄、高血圧性血管障害、移植腎拒絶、ケロイド、肥厚性火傷瘢痕、眼内線維症、リウマチ性関節炎、鼻ポリープ等があげられる。本発明のビメンチンとLAPの部分断片との結合を阻害する物質は、上記のTGF−βが関与するいずれの疾患の治療にも用いることができるが、好ましくは、動脈硬化症、癌、炎症、線維症の治療に用いることができる。
【0090】
本発明のビメンチンとLAPの部分断片との結合を阻害する物質は、治療薬として単独で投与することも可能ではあるが、通常は薬理学的に許容される一つまたはそれ以上の担体と一緒に混合し、製剤学の技術分野においてよく知られる任意の方法により製造した医薬製剤として提供するのが望ましい。
投与経路は、治療に際して最も効果的なものを使用するのが望ましく、経口投与、または口腔内、気道内、直腸内、皮下、筋肉内および静脈内等の非経口投与をあげることができ、抗体またはペプチド製剤の場合、望ましくは静脈内投与をあげることができる。
【0091】
投与形態としては、噴霧剤、カプセル剤、錠剤、顆粒剤、シロップ剤、乳剤、座剤、注射剤、軟膏、テープ剤等があげられる。
経口投与に適当な製剤としては、乳剤、シロップ剤、カプセル剤、錠剤、散剤、顆粒剤等があげられる。
乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビトール、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ごま油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を添加剤として用いて製造できる。
【0092】
カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニトール等の賦形剤、デンプン、アルギン酸ナトリウム等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を添加剤として用いて製造できる。
【0093】
非経口投与に適当な製剤としては、注射剤、座剤、噴霧剤等があげられる。
注射剤は、塩溶液、ブドウ糖溶液、または両者の混合物からなる担体等を用いて調製される。
座剤はカカオ脂、水素化脂肪またはカルボン酸等の担体を用いて調製される。
また、噴霧剤は本発明のビメンチンとLAPの部分断片との結合を阻害する物質そのもの、ないしは受容者の口腔および気道粘膜を刺激せず、かつ該物質を微細な粒子として分散させ吸収を容易にさせる担体等を用いて調製される。
【0094】
担体として具体的には乳糖、グリセリン等が例示される。該物質および用いる担体の性質により、エアロゾル、ドライパウダー等の製剤が可能である。また、これらの非経口剤においても経口剤で添加剤として例示した成分を添加することもできる。
投与量または投与回数は、目的とする治療の効果、投与方法、治療期間、年齢、体重等により異なるが、ビメンチンとLAPの部分断片との結合を阻害する物質が抗ビメンチン抗体もしくは該抗体断片またはそれらの誘導体である場合、通常成人1日当たり10μg/kg〜8mg/kgである。
以下に本発明の実施例を示す。
【実施例1】
【0095】
抗ビメンチン抗体によるビメンチンとLAPの部分断片との結合阻害(1)
本試験は安部(Abe)らの方法[エンドセリウム(Endothelium)9,25−36(2002)]に従って行った。また、本実験で使用したペプチドのビオチン化は国際公開第98/51704号に記載の方法に従った。
すなわち、10%FBS、4mmol/lのL−グルタミン、10μg/mlのカナマイシン(明治製菓社製)を含むDMEM培地に懸濁したウシ血管内皮細胞(以下、BAECという。)を96穴黒色プレート(コーニング・コースター社製)に、1穴あたり2.0×104個ずつ播種し、5%CO2条件下37℃で1日間培養した後に、以下の実験に供した。
【0096】
細胞をPBSで3回洗浄した後、5μg/mlの抗ビメンチン抗体(VIM3B4、PROGEN社製)で37℃30分間インキュベートし、その後、配列番号15で表されるペプチド(Peptide−25)または配列番号17で表されるビオチン化ペプチド(Peptide−25N)および0.1%BSAを含むDMEM培地を、各ペプチドの最終濃度が100μg/mlとなるように添加し、37℃で60分間インキュベートした。次にPBSで1回洗浄し、PBSで500倍希釈したHRP−conjugated streptavidin(アマシャム社製)を1穴あたり100μl加えて、37℃で30分間インキュベートした。その後PBSで3回洗浄し、100μlのECL western blotting detection reagent(アマシャム社製)を加えて、ルミネセンサーJNR(アトー社製)により各ウェルの発光強度を測定した。
【0097】
結果を図1に示す。図1から明らかなように、Peptide−25Nは細胞表面に結合し、その結合は抗ビメンチン抗体により抑制された。
【実施例2】
【0098】
抗ビメンチン抗体によるTGF−β活性化抑制
本試験は安部(Abe)らの方法[エンドセリウム(Endothelium)9,25−36(2002)]に従って行った。
すなわち、BAECを35mmディッシュに播種し、コンフルエントになるまで培養した。配列番号15で表されるペプチド(Peptide−25)または配列番号18で表されるペプチド(Peptide−21)を0.1%BSA含有DMEM培地中に最終濃度100μg/mlとなるように調製し、細胞に添加して37℃で6時間プレインキュベートした。その後、剃刀で一部の細胞を剥ぎ取り、PBSで洗浄してからプレインキュベート時と同組成の培地で24時間インキュベートした。プレインキュベート時および剥ぎ取り後のインキュベート時には、各ペプチドに加えて5μg/mlの抗ビメンチン抗体(V9、Neo Markers社製)またはマウスIgG1抗体(Ancell社製)をそれぞれ添加し、剥ぎ取った領域に遊走してくる細胞数に対する各抗体の影響を調べた。該細胞数は、剥ぎ取り後24時間に位相差顕微鏡下で計数した。
【0099】
結果を図2に示す。図2から明らかなように、Peptide−25を添加することにより遊走細胞数が減少した。これはPeptide−25が潜在型TGF−βの活性化を促進したことにより、TGF−βによる細胞遊走阻害作用が増強されたためである。一方、Peptide−25に加えて抗ビメンチン抗体を添加した場合、遊走細胞数が有意に増加したことから、抗ビメンチン抗体がPeptide−25の潜在型TGF−βの活性化を抑制することが明らかとなった。なお、潜在型TGF−β活性化能を有さないPeptide−21を添加した場合は、細胞遊走数に変化は見られず、そこに抗ビメンチン抗体を添加しても何ら変化は見られなかった。
【実施例3】
【0100】
抗ビメンチン抗体によるビメンチンとLAPの部分断片との結合阻害(2)
実施例1と同様の方法で、参考例で作製した抗ヒトビメンチンモノクローナル抗体KM3565のビメンチンとLAPの部分断片との結合阻害活性を評価した。本実施例においては、実施例1におけるVIM3B4の代りに、5μg/mlのKM3565または陰性対照として同濃度の抗G−CSF変異体抗体KM511(Agric.Biol.Chem.,53(4),1095−1101,1989)のいずれかを添加した。
結果を図3に示す。図3から明らかなように、KM3565はPeptide−25Nの細胞表面への結合を抑制した。
【0101】
参考例
抗ヒトビメンチンモノクローナル抗体の作製
(1)抗原ペプチドの調製
(1−1)化合物1(ヒトビメンチン部分ペプチド:配列番号19)の合成
本発明において使用したアミノ酸およびその保護基に関する略号は、生化学命名に関するIUPAC−IUB委員会(IUPAC−IUB Joint Commission on Biochemical Nomenclature)の勧告〔ヨーロピアン・ジャーナル・オブ・バイオケミストリー(European Journal of Biochemistry),138巻,9頁(1984年)〕に従った。
【0102】
以下の略号は、特に断わらない限り対応する下記のアミノ酸を表す。
Ala:L−アラニン
Arg:L−アルギニン
Asn:L−アスパラギン
Asp:L−アスパラギン酸
Asx:L−アスパラギン酸またはL−アスパラギン
Cys:L−システイン
Gln:L−グルタミン
Glu:L−グルタミン酸
Glx:L−グルタミン酸またはL−グルタミン
Gly:グリシン
Ile:L−イソロイシン
Leu:L−ロイシン
Lys:L−リジン
Met:L−メチオニン
Pro:L−プロリン
Ser:L−セリン
Thr:L−スレオニン
Tyr:L−チロシン
【0103】
以下の略号は、対応する下記のアミノ酸の保護基または側鎖保護アミノ酸を表す。
Fmoc:9−フルオレニルメチルオキシカルボニル
t−Bu:t−ブチル
Trt:トリチル
Pmc:2,2,5,7,8−ペンタメチルクロマン−6−スルホニル
Boc:t−ブチルオキシカルボニル
Fmoc−Ser(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−セリン
Fmoc−Thr(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−スレオニン
Fmoc−Lys(Boc)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nε−t−ブチルオキシカルボニル−L−リジン
Fmoc−Asn(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nγ−トリチル−L−アスパラギン
Fmoc−Asp(O−t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−L−アスパラギン酸−β−t−ブチルエステル
Fmoc−Glu(O−t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−L−グルタミン酸−γ−t−ブチルエステル
Fmoc−Gln(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Nγ−トリチル−L−グルタミン
Fmoc−Arg(Pmc)−OH:Nα−9−フルオレニルメチルオキシカルボニル−Ng−2,2,5,7,8−ペンタメチルクロマン−6−スルホニル−L−アルギニン
Fmoc−Tyr(t−Bu)−OH:Nα−9−フルオレニルメチルオキシカルボニル−O−t−ブチル−L−チロシン
Fmoc−Cys(Trt)−OH:Nα−9−フルオレニルメチルオキシカルボニル−S−トリチル−L−システイン
【0104】
以下の略号は、対応する下記の反応溶媒、反応試薬等を表す。
HBTU:2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム・ヘキサフルオロホスフェート
HOBt:N−ヒドロキシベンゾトリアゾール
DMF:N,N−ジメチルホルムアミド
TFA:トリフルオロ酢酸
DIEA:ジイソプロピルエチルアミン
【0105】
アミドリンカー16.5μmolが結合した担体樹脂(Rink−Amide MBHA樹脂、Nova Biochem社製)30mgを自動合成機(島津製作所PSSM−8)の反応容器に入れ、島津製作所の合成プログラムに従い次の操作を行った。
(a)担体樹脂を500μlのDMFで1分間洗浄し、該溶液を排出した
(b)30%ピペリジン−DMF溶液858μlを加えて混合物を4分間攪拌し、該溶液を排出し、この操作をもう1回繰り返した。
(c)担体樹脂を500μlのDMFで1分間洗浄し、該溶液を排出し、この操作を5回繰り返した。
(d)Fmoc−Glu(O−t−Bu)−OH(165μmol)、HBTU(165μmol)、HOBt 1水和物(165μmol)およびDIEA(330μmol)をDMF(858μl)中で3分間攪拌し、得られた溶液を樹脂に加えて混合物を30分間攪拌し、溶液を排出した。
(e)担体樹脂を858μlのDMFで1分間洗浄し、これを5回繰り返した。こうして、Fmoc−Glu(O−t−Bu)−アミドを担体上に合成した。
【0106】
次に(b)、(c)のFmoc基脱保護工程および洗浄工程を行い、Fmoc基を除去したH−Glu(O−t−Bu)−アミドの結合した担体樹脂を得た。
次に、(d)の工程でFmoc−Glu(O−t−Bu)−OHを用いて縮合反応を行い、(e)の洗浄工程、次いで(b)、(c)の脱保護、洗浄工程を経て、H−Glu(O−t−Bu)−Glu(O−t−Bu)−アミドを担体上に合成した。以下、工程(d)において、Fmoc−Lys(Boc)−OH、
Fmoc−Met−OH、Fmoc−Asn(Trt)−OH、Fmoc−Gln(Trt)−OH、Fmoc−Ile−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Asp(O−t−Bu)−OH、Fmoc−Gln(Trt)−OH、
Fmoc−Leu−OH、Fmoc−Arg(Pmc)−OH、Fmoc−Gly−OH、Fmoc−Ile−OH、
Fmoc−Thr(t−Bu)−OH、Fmoc−Asp(O−t−Bu)−OH、Fmoc−Gln(Trt)−OH、
Fmoc−Tyr(t−Bu)−OH、Fmoc−Asn(Trt)−OH、Fmoc−Ala−OHを順次用いて、(d)、(e)、(b)、(c)の順に工程を繰り返した。最後にFmoc−Cys(Trt)−OHを用いて(d)、(e)の操作を2度繰り返した後に、(a)、(b)の操作を行った。
【0107】
得られた樹脂をメタノール、ブチルエーテルで順次洗浄し、減圧下12時間乾燥して、側鎖保護ペプチドの結合した担体樹脂を得た。これに、TFA(82.5%)、チオアニソール(5%)、水(5%)、エチルメチルスルフィド(3%)、1,2−エタンジチオール(2.5%)およびチオフェノール(2%)からなる混合溶液600μlを加えて室温で8時間放置し、側鎖保護基を除去するとともに樹脂よりペプチドを切り出した。樹脂を濾別後、得られた溶液にエーテル約10mlを加え、生成した沈澱を遠心分離およびデカンテーションにより回収し、粗ペプチド42.7mgを取得した。この粗ペプチドを酢酸水溶液に溶解後、逆相カラム(資生堂製、CAPCELL PAK C18 30mmI.D.X25mm)を用いたHPLCで精製した。0.1%TFA水溶液に、TFA0.1%を含む90%アセトニトリル水溶液を加えていく直線濃度勾配法で溶出し、220nmで検出し、化合物1を含む画分を得た。この画分を凍結乾燥して、化合物1を8.4mg得た。化合物1の物理化学的性質は以下の通りである。
質量分析[TOF−MS];m/z=2497.25(M+H+)
アミノ酸分析;Glx 6.0(6),Asx 3.7(4),Arg 1.1(1),Thr 1.0(1),Ala 1.0(1),Ile 1.9(2),Leu 1.1(1),Lys 0.9(1),Tyr 0.9(1),Met 0.9(1),Gly 1.3(1),Cys 1.4(1)
【0108】
(1−2)化合物2(対照ペプチド:配列番号20)の合成
アミドリンカー16.5μmolが結合した担体樹脂(Rink−Amide MBHA樹脂、Nova Biochem社製)30mgを出発物質とし、(1−1)と同様にして、Fmoc−Pro−OH、
Fmoc−Leu−OH、Fmoc−Pro−OH、Fmoc−Leu−OH、Fmoc−Ser(t−Bu)−OH、
Fmoc−Ile−OH、Fmoc−Arg(Pmc)−OH、Fmoc−Ser(t−Bu)−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Glu(O−t−Bu)−OH、Fmoc−Gly−OH、
Fmoc−Glu(O−t−Bu)−OH、Fmoc−Leu−OH、Fmoc−Leu−OH、Fmoc−Lys(Boc)−OH、
Fmoc−Arg(Pmc)−OH、Fmoc−Tyr(t−Bu)−OH、Fmoc−Thr(t−Bu)−OH、
Fmoc−Cys(Trt)−OHを順次縮合した後に、洗浄、乾燥を経て、側鎖保護ペプチドの結合した担体樹脂を得た。(1−1)と同様にして側鎖保護基の切断および樹脂からの切り出しを行い、粗ペプチド38.7mgを取得した。これを(1−1)と同様に精製し、化合物2を4.7mg得た。化合物2の物理化学的性質は以下の通りである。
質量分析(TOF−MS);m/z=2203.19(M+H+)
アミノ酸分析:アミノ酸分析;Glx 3.1(3),Arg 1.9(2),Thr 0.9(1),Ser 1.9(2),Ile 1.0(1),Leu 4.1(4),Lys 0.9(1),Tyr 0.9(1),Gly 1.1(1),Pro 2.2(2),Cys 1.1(1)
【0109】
(2)免疫原の調製
上記(1)で得られたヒトビメンチン部分ペプチドは、免疫原性を高める目的で以下の方法でKLH(カルビオケム社)とのコンジュゲートを作製し、免疫原とした。すなわち、KLHをPBSに溶解して10mg/mlに調整し、1/10容量の25mg/ml MBS(ナカライテスク社)を滴下して30分間撹拌反応させた。あらかじめPBSで平衡化したセファデックスG−25カラムでフリーのMBSを除いて得られたKLH−MBS 2.5mgを0.1Mりん酸ナトリウムバッファー(pH7.0)に溶解したペプチド1mgと混合し、室温で3時間、攪拌反応させた。反応後、反応物をPBSで透析し、ヒトビメンチン部分ペプチド−KLHコンジュゲートを得た。
【0110】
(3)動物の免疫と抗体産生細胞の調製
上記(2)で調製したヒトビメンチン部分ペプチド−KLHコンジュゲート100μgをアルミニウムゲル2mgおよび百日咳ワクチン(千葉県血清研究所製)1×109細胞とともに5週令雌マウス(Balb/c)に投与し、2週間後より100μgの該コンジュゲートを1週間に1回、計4回投与した。眼底静脈叢より採血し、血清抗体価を下記(4)に示す酵素免疫測定法で調べ、十分な抗体価を示したマウスから最終免疫3日後に脾臓を摘出した。
脾臓をMEM培地(日水製薬社製)中で細断し、ピンセットでほぐし、1200rpmで5分間遠心分離した。上清を捨て、トリス−塩化アンモニウム緩衝液(pH7.65)で1〜2分間処理し赤血球を除去し、MEM培地で3回洗浄したものを細胞融合に用いた。
【0111】
(4)酵素免疫測定法
アッセイ用の抗原には上記(1)で得られたヒトビメンチン部分ペプチドとサイログロブリン(以下、THYと略す)とのコンジュゲートを用いた。該コンジュゲートは、架橋剤としてMBSの代わりにSMCC(シグマ社)を用いた以外、上記(2)と同様の方法により作製した。
96穴のEIA用プレート(グライナー社)に、上記コンジュゲートを10μg/ml、50μl/穴で分注し、4℃で一晩放置して吸着させた。洗浄後、1%BSA(ウシ血清アルブミン)−PBSを100μl/穴で加え、室温1時間反応させて残っている活性基をブロックした。
1%BSA−PBSを捨て、被免疫マウス抗血清、抗ビメンチンモノクローナル抗体の培養上清または精製モノクローナル抗体を50μl/穴で分注し2時間反応させた。
0.05%tween−PBSで洗浄後、ペルオキシダーゼ標識ウサギ抗マウスイムノグロブリン(ダコ社)を50μl/穴で加えて室温、1時間反応させ、0.05%tween−PBSで洗浄後ABTS基質液[2.2−アジノビス(3−エチルベンゾチアゾール−6−スルホン酸)アンモニウム]を用いて発色させ、OD415nmの吸光度をプレートリーダー(Emax;Molecular Devices社)にて測定した。
【0112】
(5)マウス骨髄腫細胞の調製
8−アザグアニン耐性マウス骨髄腫細胞株P3−U1を正常培地で培養し、細胞融合時に2×107以上の細胞を確保し、細胞融合に親株として供した。
(6)ハイブリドーマの作製
上記(3)で得られたマウス脾細胞と上記(5)で得られた骨髄腫細胞とを10:1になるよう混合し、1,200rpmで5分間遠心分離した。上清を捨て、沈澱した細胞群をよくほぐした後、ポリエチレングライコール−1,000(PEG−1,000)2g、MEM培地2mlおよびジメチルスルホキシド0.7mlの混液を0.2〜1ml/108マウス脾細胞となるように加えて37℃で攪拌した。1〜2分間毎にMEM培地1〜2mlを数回加えた後、MEM培地を加えて全量が50mlになるようにした。900rpmで5分遠心分離した後、上清を捨て、ゆるやかに細胞をほぐした後、メスピペットによる吸込み・吸出しを繰り返し、細胞をHAT培地100ml中に懸濁した。
【0113】
この懸濁液を96穴培養用プレートに100μl/穴ずつ分注し、5%CO2インキュベーター中、37℃で10〜14日間、CO25%条件下で培養した。この培養上清を上記(4)に記載した酵素免疫測定法で調べ、ヒトビメンチン部分ペプチド(化合物1)に反応して対照ペプチド(化合物2)に反応しない穴を選び、HAT培地を正常培地に換えて2回クローニングを繰り返し、抗ヒトビメンチンモノクローナル抗体産生ハイブリドーマを確立した。
以上の結果、化合物1に特異的な反応性を示すモノクローナル抗体KM3565を産生するハイブリドーマ株KM3565を取得した。なお、ハイブリドーマ株KM3565はKM3565の株名で、平成17年4月19日付けで独立行政法人産業技術総合研究所特許生物寄託センター(日本国茨城県つくば市東1丁目1番地1中央第6)にFERM ABP−10324として寄託されている。
また、モノクローナル抗体KM3565の抗体クラスは抗体クラス特異的な標識化抗体を用いた酵素免疫測定法によりIgG3と決定された。
【0114】
(7)モノクローナル抗体の精製
プリスタン処理した8週令ヌード雌マウス(Balb/c)に上記(6)で得られたハイブリドーマ株を5〜20×106細胞/匹で腹腔内注射した。10〜21日後に、ハイブリドーマは腹水癌化した。腹水のたまったマウスから、腹水を採取(1〜8ml/匹)し、3,000rpmで5分遠心分離して固形分を除去した。得られた上清をカプリル酸沈殿法〔Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988〕により精製し、精製モノクローナル抗体を得た。
【0115】
(8)モノクローナル抗体KM3565のヒトビメンチン部分ペプチドとの反応性(酵素免疫測定法)
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン部分ペプチドとの反応性を、上記(4)の酵素免疫測定法にて調べた。図4に示すように、モノクローナル抗体KM3565は化合物1特異的に抗体濃度依存的な反応性を示した。
(9)モノクローナル抗体KM3565のヒトビメンチン蛋白との反応性(酵素免疫測定法)
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン蛋白との反応性を上記(4)の酵素免疫測定法にて調べた。大腸菌で発現させたヒトビメンチン蛋白(PROGEN社)を添付のデータシート記載の方法に従って蒸留水にて溶解後、PBSで2μg/mlに希釈してプレートに固層化した。陰性対照抗原として大腸菌由来夾雑蛋白をPBSで10μg/mlに希釈してプレートに固層化したものを用いた。図5に示すように、モノクローナル抗体KM3565はビメンチン特異的に抗体濃度依存的な反応性を示した。
【0116】
(10)ウェスタンブロッティング
上記(6)で選択された抗ヒトビメンチンモノクローナル抗体KM3565のヒトビメンチン蛋白との反応性をウエスタンブロッティングにて調べた。
大腸菌で発現させたヒトビメンチン蛋白、または大腸菌由来夾雑蛋白を0.1μg/レーンでSDS−ポリアクリルアミド電気泳動〔Antibodies−A Laboratory Manual,Cold Spring Harbor Laboratory,1988〕にて分画後、PVDF膜にブロッティングした1%BSA−PBSでブロッキング後、1次抗体としてKM3565のハイブリドーマ培養上清原液、市販の抗ビメンチン抗体2種(PROGEN社製VIM3B4およびSanta Cruz Biotechnology社製V9、いずれも精製抗体5μg/ml)、および陰性対照抗体としてKM511(抗G−CSF変異体抗体、Agric.Biol.Chem.,53(4),1095−1101,1989、5μg/ml)を室温で2時間反応させた。0.05%tween−PBSでよく洗浄した後、第二抗体としてペルオキシダーゼ標識抗マウスイムノグロブリン抗体(DAKO社製)を室温で1時間反応させた。
0.05%tween−PBSでよく洗浄した後、ECL−detection kit(アマシャム社)による反応を行った。
その結果、モノクローナル抗体KM3565は、市販抗体と同様、ビメンチン蛋白に特異的な反応性を示した。
【0117】
(11)モノクローナル抗体KM3565と市販抗ビメンチン抗体との認識エピトープの異同
市販抗ビメンチン抗体2種(PROGEN社製VIM3B4およびSanta Cruz Biotechnology社製V9)とモノクローナル抗体KM3565の認識エピトープの異同を検討する目的で、該市販抗体の化合物1に対する反応性を上記(4)の酵素免疫測定法にて調べた。その結果、図6に示すように該市販抗体はいずれもビメンチン蛋白に反応性を有するが、化合物1に対する反応性を示さなかったことから、これらの抗体はビメンチン蛋白上のモノクローナル抗体KM3565とは異なる別のエピトープを認識していることが明らかとなった。
【産業上の利用可能性】
【0118】
本発明により、ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する潜在型TGF−βの活性化抑制剤、またはTGF−β活性化抑制物質のスクリーニング方法が提供される。本発明のTGF−βの活性化抑制剤は、TGF−βが関与する疾患の治療薬として利用することができる。
【配列表フリーテキスト】
【0119】
配列番号1−人工配列の説明:合成ペプチド
配列番号2−人工配列の説明:合成ペプチド
配列番号3−人工配列の説明:合成ペプチド
配列番号4−人工配列の説明:合成ペプチド
配列番号5−人工配列の説明:合成ペプチド
配列番号6−人工配列の説明:合成ペプチド
配列番号7−人工配列の説明:合成ペプチド
配列番号8−人工配列の説明:合成ペプチド
配列番号9−人工配列の説明:合成ペプチド
配列番号10−人工配列の説明:合成ペプチド
配列番号11−人工配列の説明:合成ペプチド
配列番号12−人工配列の説明:合成ペプチド
配列番号13−人工配列の説明:合成ペプチド
配列番号14−人工配列の説明:合成ペプチド
配列番号15−人工配列の説明:合成ペプチド
配列番号16−人工配列の説明:合成ペプチド
配列番号17−人工配列の説明:合成ペプチド
配列番号18−人工配列の説明:合成ペプチド
配列番号19−人工配列の説明:合成ペプチド
配列番号20−人工配列の説明:合成ペプチド
【特許請求の範囲】
【請求項1】
ビメンチン(Vimentin)とラテンシー・アソシエイテッド・ペプチド(Latency Associated Peptide、以下「LAP」という。)の部分断片との結合を阻害する物質を有効成分として含有する潜在型トランスフォーミング・グロース・ファクター−β(transforming growth factor−β、以下「TGF−β」という。)の活性化抑制剤。
【請求項2】
ビメンチンとLAPの部分断片との結合を阻害する物質が、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体または該抗体断片である、請求項1に記載のTGF−β活性化抑制剤。
【請求項3】
LAPの部分断片が配列番号1〜16のいずれかで表されるアミノ酸配列を有するペプチドである、請求項1または2のいずれか1項に記載のTGF−β活性化抑制剤。
【請求項4】
(i)LAPの部分断片を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(ii)LAPの部分断片およびスクリーニングの対象となる物質を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(iii)(i)および(i)の測定量から、該物質の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価すること、
(iv)該結合阻害活性を有する物質をTGF−β活性化抑制物質として選択すること、
からなるTGF−β活性化抑制物質のスクリーニング方法。
【請求項5】
ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する、TGF−βが関与する疾患の治療薬。
【請求項6】
TGF−βが関与する疾患が動脈硬化症、癌、炎症、線維症である、請求項5記載の治療薬。
【請求項7】
ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−β活性化抑制方法。
【請求項8】
ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−βが関与する疾患の治療方法。
【請求項9】
TGF−β活性化抑制剤の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【請求項10】
TGF−βが関与する疾患の治療薬の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【請求項1】
ビメンチン(Vimentin)とラテンシー・アソシエイテッド・ペプチド(Latency Associated Peptide、以下「LAP」という。)の部分断片との結合を阻害する物質を有効成分として含有する潜在型トランスフォーミング・グロース・ファクター−β(transforming growth factor−β、以下「TGF−β」という。)の活性化抑制剤。
【請求項2】
ビメンチンとLAPの部分断片との結合を阻害する物質が、ビメンチンとLAPの部分断片との結合を阻害する活性を有する抗ビメンチン抗体または該抗体断片である、請求項1に記載のTGF−β活性化抑制剤。
【請求項3】
LAPの部分断片が配列番号1〜16のいずれかで表されるアミノ酸配列を有するペプチドである、請求項1または2のいずれか1項に記載のTGF−β活性化抑制剤。
【請求項4】
(i)LAPの部分断片を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(ii)LAPの部分断片およびスクリーニングの対象となる物質を、ビメンチンおよびTGF−βを発現する細胞に添加して、該細胞に結合する該部分断片量を測定すること、
(iii)(i)および(i)の測定量から、該物質の、LAPの部分断片とTGF−βを発現する細胞との結合阻害活性を評価すること、
(iv)該結合阻害活性を有する物質をTGF−β活性化抑制物質として選択すること、
からなるTGF−β活性化抑制物質のスクリーニング方法。
【請求項5】
ビメンチンとLAPの部分断片との結合を阻害する物質を有効成分として含有する、TGF−βが関与する疾患の治療薬。
【請求項6】
TGF−βが関与する疾患が動脈硬化症、癌、炎症、線維症である、請求項5記載の治療薬。
【請求項7】
ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−β活性化抑制方法。
【請求項8】
ビメンチンとLAPの部分断片との結合を阻害する物質を被験体に投与する工程を包含するTGF−βが関与する疾患の治療方法。
【請求項9】
TGF−β活性化抑制剤の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【請求項10】
TGF−βが関与する疾患の治療薬の製造のための、ビメンチンとLAPの部分断片との結合を阻害する物質の使用。
【図1】


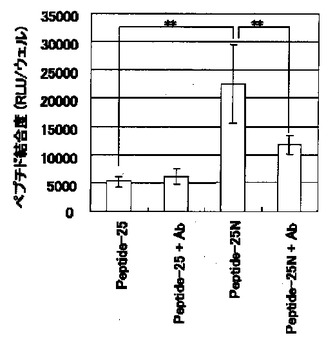
【図2】
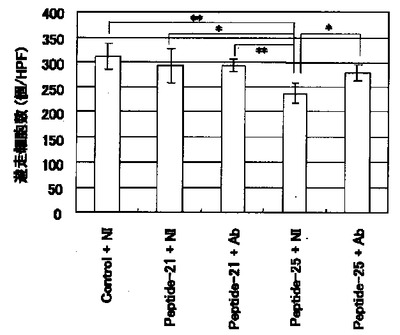
【図3】
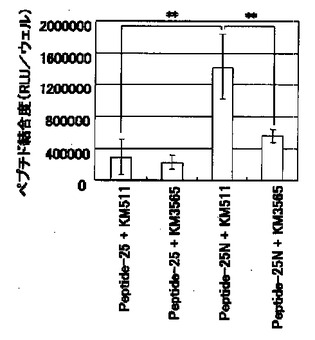
【図4】
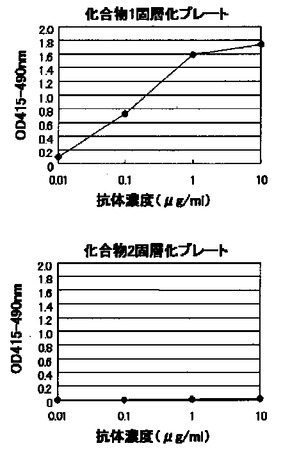
【図5】
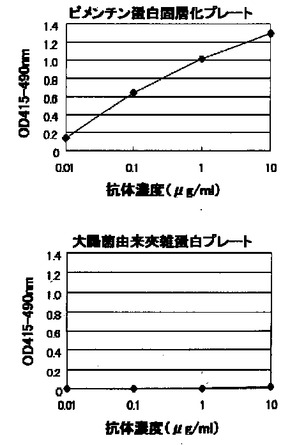
【図6】
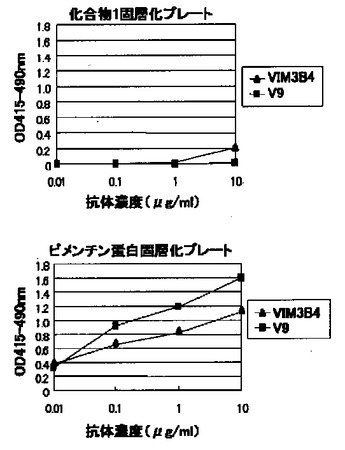
【図2】
【図3】
【図4】
【図5】
【図6】
【国際公開番号】WO2005/105144
【国際公開日】平成17年11月10日(2005.11.10)
【発行日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願番号】特願2006−512831(P2006−512831)
【国際出願番号】PCT/JP2005/008125
【国際出願日】平成17年4月28日(2005.4.28)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
【国際公開日】平成17年11月10日(2005.11.10)
【発行日】平成19年9月13日(2007.9.13)
【国際特許分類】
【国際出願番号】PCT/JP2005/008125
【国際出願日】平成17年4月28日(2005.4.28)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
[ Back to top ]