
炎症性疾患治療剤

【課題】炎症性疾患に対して有効な炎症性疾患治療剤を提供する。
【解決手段】下記の一般式(I)(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。

【解決手段】下記の一般式(I)(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、炎症性疾患治療剤に関する。
【背景技術】
【0002】
炎症性疾患には、種々の炎症性サイトカインの調節機能に異常が生じて過剰分泌することに起因するものがある。炎症性サイトカインとは、炎症を促進するサイトカインの総称であり、TNFα、Fasリガンド(FasL)、IL−1α、IL−1β、IL−6、IFNγ、IL−8、IL−10、IL−12、IL−15、IL−18及びCD40リガンド等が知られている。このため、炎症性疾患の治療方法のひとつとして、炎症性サイトカインの分泌抑制が挙げられ、炎症性サイトカインの分泌抑制に効果がある薬剤がいくつか提案されている(例えば、特許文献1及び2)。
【0003】
また炎症性サイトカインの中には、マトリックスメタロプロテアーゼ(MMP)により膜型から可溶型に変換して活性型になるものが多数あることが知られている。MMPは、このような作用の他に、がんの転移や慢性関節リウマチ、変形性関節症、炎症、糖尿病などの疾患や老化に関与していると考えられている。このため、MMPの作用を阻害又はMMPの活性化を阻害することによって、MMPに関与する種々の疾患の症状を緩和することができると考えられ、多くのMMP阻害剤が開発又は提案されている(例えば、特許文献3及び4)。
【0004】
一方、血栓溶解酵素であるプラスミンを特異的に阻害する物質として、特定構造のアミド化合物YO−2及びその誘導体が知られている(非特許文献1及び2)。また、YO−2には、ヒトのメラノーマ細胞、Bリンパ腫細胞、大腸ガン細胞等の増殖を抑制し、腫瘍細胞の増殖阻害作用とがんの転移抑制作用が認められることについても報告されている(非特許文献3)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−235447号公報
【特許文献2】特開2008−303171号公報
【特許文献3】特開2002−201130号公報
【特許文献4】特表2010−511027号公報
【非特許文献】
【0006】
【非特許文献1】Thrombosis Research, (1996) Vol.82, No.1, pp.79-86
【非特許文献2】Chem. Pharm. Bull., (2001) Vol.49(11), pp.1457-1463
【非特許文献3】In vivo, (2002) Vol.16, pp.281-286
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、炎症性疾患は複数の炎症性サイトカインが関与しているため、ひとつ又は数種の炎症性サイトカインの過剰分泌を阻害したとしても充分な治療効果が得られるとは限らない。また、種々開発されているMMP阻害剤は、その強い効果と引き替えに、肩又は腕の筋肉痛、関節痛、関節滑膜の肥厚、関節包中への炎症細胞の浸潤など、強い副作用を示すものも多く、重篤な副作用に耐えかねて使用を継続でない場合もある。このためMMP阻害剤を使用しても、その作用を充分に発揮することができないことが知られている。
従って本発明の目的は、炎症性疾患に対して有効な新規の炎症性疾患治療剤を提供することである。
【課題を解決するための手段】
【0008】
本発明は以下のとおりである。
[1] 下記の一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。
【0009】
【化1】
【0010】
(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)
[2] 前記アミド誘導体が下記一般式(I−1)で表される[1]に記載の炎症性疾患治療剤。
【0011】
【化2】
【0012】
(式中R及びL2は一般式(I)と同様である。)
[3] 前記L2が、フェニレン基及び炭素数2〜9のアルキレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す[1]又は[2]に記載の炎症性疾患治療剤。
[4] 下記の化合物の少なくとも一方を含む[1]〜[3]のいずれかに記載の炎症性疾患治療剤。
【0013】
【化3】
【0014】
[5] 前記炎症性疾患が、移植片対宿主疾患(GVHD)、全身性炎症反応症候群、炎症性腸疾患、膠原病、及び癌悪液質からなる群より選択された少なくとも1つである[1]〜[4]のいずれかに記載の炎症性疾患治療剤。
【発明の効果】
【0015】
本発明によれば、炎症性疾患に対して有効な炎症性疾患治療剤を提供することができる。
【図面の簡単な説明】
【0016】
【図1】実施例2に係る急性GVHDモデルマウスの生存率を示す図である。
【図2】実施例2に係る急性GVHDモデルマウスの体重の変化を示すグラフである。
【図3】実施例2に係る急性GVHDモデルマウスの各血清サイトカイン濃度(図3(A)はTNF−α、図3(B)は全MMP−9、図3(C)はFasL)の変化を示すグラフである。
【図4】参考例1に係る抗TNFα抗体及びMMP阻害剤を投与した急性GVHDモデルマウスの生存率を示す図である。
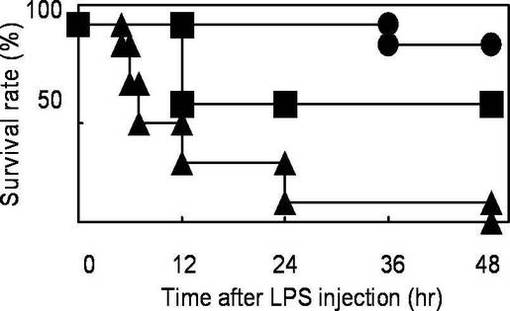
【図5】実施例3に係る敗血症性ショックモデルマウスの生存率を示す図である。
【発明を実施するための最良の形態】
【0017】
本発明の炎症性疾患治療剤は、上記一般式(I)で示されるアミド誘導体又はその塩を有効成分とするものである。
一般式(I)で示されるアミド誘導体は、プラスミン阻害剤として既知の物質であるが、今回、MMP活性阻害作用があることが新たに見出された。本発明は、この知見に基づくものである。
【0018】
これを詳細に説明すれば、MMPは、不活性型のプロMMPで体内に存在しており、活性化因子の存在下で活性化するとMMPとして作用可能となる。線溶作用を有するプラスミンは、フィブリンに作用してフィブリン分解物を生成する作用を有する一方で、プロMMPの活性化にも作用すると推測されている。活性化されたMMPは、種々の炎症性サイトカインの外部ドメインを脱落させ、この結果、炎症性サイトカインの可溶性部分を離脱させて炎症性サイトカインを活性化すると考えられている。本発明に係るアミド誘導体は、プラスミノーゲンの活性化を阻害することによって、MMPの関与する複数の炎症性サイトカインの活性化を抑制し、その結果、MMPに関与する炎症性疾患の治療効果を奏すると推測されるが、この理論に限定されない。また、前記アミド誘導体には、強い副作用も認められない。従って、本発明に係る前記アミド誘導体を有効成分とする医薬組成物を用いることより、MMPが関与する種々の炎症性疾患に対して、高い治療効果が期待できる。
【0019】
本発明において「治療」とは、本発明にかかる組成物の投与前後において炎症性疾患に特有の症状が軽減又は、重篤化が抑制されることを意味する。また、この用語には、本発明にかかる組成物の投与後において炎症性疾患に特有の症状の顕著な発生を抑制する「予防」も包含される。
【0020】
本明細書において「工程」との語は、独立した工程だけでなく、他の工程と明確に区別できない場合であっても本工程の所期の作用が達成されれば、本用語に含まれる。
また、本明細書において「〜」を用いて示された数値範囲は、「〜」の前後に記載される数値をそれぞれ最小値及び最大値として含む範囲を示す。
また、本発明において、組成物中の各成分の量について言及する場合、組成物中に各成分に該当する物質が複数存在する場合には、特に断らない限り、組成物中に存在する当該複数の物質の合計量を意味する。
以下、本発明について説明する。
【0021】
本発明にかかるアミド誘導体は、一般式(I)で示される化合物である。
【0022】
【化4】
【0023】
式(I)中、Arは、炭素数4〜6の芳香族基を表し、溶解性の観点から好ましくは炭素数4または6の芳香族基を表す。当該芳香族基は、置換されていても、未置換であってもよい。また環に、硫黄原子、窒素原子又は酸素原子などのヘテロ原子を1つ又は2つ以上含んでいてよい。Arで表される芳香族基が置換基を有する場合、置換基の例としては、水酸基、アミノ基、カルボキシ基、炭素数1〜4のアルキル基及びハロゲンを挙げることができる。Arで表される芳香族基における置換基は、2つ以上存在していてもよいが、未置換であることが好ましい。
Arで表される芳香族基の例としては、フェニル基、ピリジル基、ピリミジル基及びピリダジル基等を挙げることができ、溶解性の観点からピリジル基であることが好ましい。
【0024】
式(I)中L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す。前記L1としては、炭素数1又は2のアルキレンオキシ基であることが好ましく、メチレンオキシ基であることがより好ましい。
【0025】
式(I)中L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表す。前記脂肪族基は、飽和又は不飽和であってもよく、また直鎖構造のみならず、分岐鎖又は環状構造を有していてもよい。前記脂肪族基の例としては、炭素数2〜9の直鎖又は分岐アルキレン基及びシクロアルキレン基等を挙げることができる。アリーレン基の例としては、置換基を有していてもよいフェニレン基を挙げることができる。前記アリーレン基の置換基の例としては、アルキル基、アミノ基及びカルボキシル基等を挙げることができ、前記アリーレン基における置換基は2つ以上存在していてもよい。
式(I)中L2として表される炭化水素基としては、例えば以下のものを挙げることができる。なお、下記構造中「*」は隣接する原子との連結部分を表す。
【0026】
【化5】
【0027】
前記L2で表される炭化水素基としては、疎水性と嵩高さの観点から、炭素数2〜9のアルキレン基及びフェニレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基であることが好ましい。炭素数2〜9のアルキレン基としては、炭素数2〜9の直鎖又は分岐アルキレン基、シクロヘキシレン基及びフェニレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基であることが好ましい。
【0028】
式I中Rは、水素原子、アミノ基又はカルボキシ基を表し、水素原子又はアミノ基であることが好ましい。
【0029】
前記アミド誘導体としては、下記一般式(I−1)で表されるアミド誘導体であることが、MMP阻害活性の観点から好ましい。式(I−1)中、L2及びRは、前記式(I)におけるL2及びRと同一であり、L2及びRの好ましい例も同一である。特に、MMP阻害活性の観点から、下記一般式(II)におけるL2としては、炭素数2〜9の未置換の直鎖アルキレン基、未置換のフェニレン基及びシクロヘキシレン基であることが好ましく、Rとしては水素原子又はアミノ基であることが好ましい。
【0030】
【化6】
【0031】
本発明におけるアミド誘導体としては、MMP阻害活性及び抗炎症効果の観点から、以下のYO−2又はYO−57であることが特に好ましい。
【0032】
【化7】
【0033】
前記アミド誘導体は、公知の方法に従って合成することができ、例えばChem. Pharm. Bull., (2002), 49 (11), 1457-1463に合成方法が開示されている。簡単に説明すれば、まず、Boc-Tyr(O-Pic)-OH(Boc: t-Butoxycarbonyl、Tyr: tyrosine、O-Pic: O-Picolyl)を、トリエチルアミンの存在下、イソブチルクロロホルメイトで無水物に変換し、対応するアミンと反応し、Boc-Tyr(O-Pic)-NHX(酸アミド体)を得る。次に、Boc基をTFA(トリフルオロ酢酸)−anisoleで除去し、生じたアミンをBoc-4-aminomethylcyclo-hexanecarboxylic acid (Tra)の無水物と反応してBoc-Tra-Tyr(O-Pic)-NHX を得る。これをTFA−anisoleで処理すると、目的とするアミド誘導体(I)を得ることができる。
【0034】
前記アミド誘導体は、通常の薬理的に許容される酸と容易に塩を形成し得る。かかる酸としては、例えば硫酸、硝酸、塩酸、臭化水素酸等の無機酸、酢酸、トリフルオロ酢酸、p−トルエンスルホン酸、エタンスルホン酸、シュウ酸、マレイン酸、フマル酸、クエン酸、コハク酸、安息香酸等の有機酸を例示できる。
【0035】
本発明の炎症性疾患治療剤は、前記アミド誘導体を単独で又は2種以上を組み合わせて含んでもよい。
【0036】
本発明の炎症性疾患治療剤は、医薬として許容可能な担体と組み合わせてもよい。このような担体の例としては、医薬製剤として適用される形態に応じた公知の担体を挙げることができる。
例えば、注射剤として調製される場合、液剤、乳剤及び懸濁剤は殺菌され、且つ血液と等張であるのが好ましく、これらの形態に成形するに際しては、希釈剤としてこの分野において慣用されているものをすべて使用できる。例えば水、エチルアルコール、マクロゴール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオキシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等を使用できる。なお、この場合、等張性の溶液を調製するに充分な量の食塩、ブドウ糖あるいはグリセリンを医薬製剤中に含有してもよく、また通常の溶解補助剤、緩衝剤、無痛化剤等を添加してもよい。更に必要に応じて着色剤、保存剤、香料、風味剤、甘味剤等や他の医薬品を組成物中に含んでもよい。
またその他、組成物の形態に応じて、適宜、通常使用される充填剤、増量剤、結合剤、付湿剤、崩壊剤、表面活性剤、滑沢剤等の希釈剤あるいは賦形剤を含んでもよい。
【0037】
本発明の炎症性疾患治療剤に含有されるべき前記アミド誘導体の量としては、特に限定されず広範囲から適宜選択されるが、例えば、組成物の全質量の約1〜70質量%、好ましくは約5〜50質量%とするのがよい。
本発明の炎症性疾患治療剤は、MMPによって細胞外ドメイン分泌されるサイトカインに起因する多くの炎症性疾患に広く適用可能である。適用可能な炎症性疾患としては、移植片対宿主疾患(GVHD)、全身性炎症反応症候群(敗血症性ショック]、潰瘍性大腸炎等の炎症性腸疾患、一部の全身性炎症性疾患(例えば、関節リウマチ等の膠原病)、癌悪液質等を挙げることができ、これらの組み合わせに対して用いてもよい。
【0038】
上記の炎症性疾患はいずれもMMPによりその細胞外ドメイン分泌が制御される複数の炎症性サイトカインとそのシグナルに起因する炎症性疾患である。例えば、GVHD、全身性炎症反応症候群、炎症性腸疾患、関節リウマチ、及び癌悪液質では、TNFαは、病態上、これらの炎症性疾患の発症及び進行において最も重要なサイトカインの一つと考えられている。また、これらの炎症性疾患ではFasリガンドやIL−1、IL−2、IL−6の他CD40リガンド等の他の複数の炎症性サイトカインの関与が示唆されている。MMPはこれらのサイトカインあるいはこれらの受容体の細胞膜表面からの細胞外ドメイン分泌を制御することにより、サイトカイン血中濃度の上昇あるいは可溶型受容体によるサイトカイン作用の抑制等の形でこれらの炎症性疾患の病態形成に関与していると考えられる。従ってこれらの複数の炎症性サイトカインあるいはこれらの受容体の細胞外ドメイン分泌が前記アミド誘導体によって制御されることにより、上述した炎症性疾患はいずれも予防又は治療が可能となる。
【0039】
本発明の炎症性疾患治療剤の投与方法は特に制限はなく、各種製剤形態、患者の年齢、性別その他の条件、疾患の種類又は疾患の程度等に応じた方法を選択することができ、経口投与及び非経口投与のいずれであってもよい。
本発明の炎症性疾患治療剤の投与量は、用法、患者の年齢、性別その他の条件、疾患の程度等により適宜選択されるが、例えば、前記アミド誘導体の量が、1日当り体重1kgあたり約3.0mg〜8.0mg程度とすることができる。
【0040】
本発明はまた、前記一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤を、炎症性疾患の患者又は、炎症性疾患を生じる可能性がある若しくは疑いのある患者に投与することを含む炎症性疾患の予防又は治療方法を含む。
本炎症性疾患の予防又は治療方法によれば、前記アミド誘導体を投与することによって、MMPに関連する炎症性疾患を予防又は治療することができる。
【0041】
本予防又は治療方法における前記炎症性疾患治療剤については、前述した事項をそのまま適用する。
また、前記炎症性疾患については、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用する。
前記投与の方法、経路等についても、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用する。
その他の事項についても、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用すればよい。
【実施例】
【0042】
以下、本発明を実施例にて詳細に説明する。しかしながら、本発明はそれらに何ら限定されるものではない。なお、特に断りのない限り、「%」は質量基準である。
【0043】
[実施例1]
<アミド誘導体の特性評価>
(1) アミド誘導体の合成
下記アミド誘導体YO−2及びYO−57はChem. Pharm. Bull., (2002), 49 (11), 1457-1463に記載された方法に従って以下のように合成した。なお、一般的操作は以下のとおりである。
融点は未補正、微量融点測定器(柳本製作所製)で測定した。
旋光度は自動旋光計 model DPI-360 [日本分光(株)]で測定した。
Matrix assisted laser desorption time-of flight mass spectra (MALDI-TOF-MS) にはKratos MALDI IV 質量分析器(Kratos Analytical)を用いた。
分析または分取HPLCにはWaters model 600Eを使用した。それぞれ、COSMOSIL C18 column AR-II (4.6 x 250 mm, nacalai tesque)、COSMOSIL C18 column AR-II (20 x 250 mm, nacalai tesque)を用いた。
保持時間はtRで示した。移動層には0.05% TFAを含む水−0.05% TFAを含むアセトニトリルを用いた。1分間あたり1%ずつアセトニトリルの濃度が高くなる濃度勾配をかけて溶出した。検出は220 nmで行い、流速は、分析または分取HPLCにおいて、それぞれ1.0 ml/min、10.0 ml/minで行った。
【0044】
薄層クロマトグラフィー(TLC)はKiesel gel 60G(Merck社製)を用いて行った。展開溶媒にはRf1 (クロロホルム:メタノール:酢酸=90:8:2)、Rf2 (クロロホルム:メタノール:水=89:10:1)、Rf3(n-ブタノール:酢酸:水=4:1:5、上層)Rf4 (n-ブタノール:酢酸:ピリジン:水=4:1:1: 2)を用いた。発色試薬としてアミノ基の検出には01%ニンヒドリン−アセトン溶液を用いた。Boc保護体の検出には臭化水素-ニンヒドリン法を用いた。BocおよびFmoc保護体の検出には254 nmのUV照射を用いた。
【0045】
(1−1)YO−2の合成
1) Boc-Tyr(O-Pic)-NH-Octylの合成
Boc-Tyr(O-Pic)-OH (1.48 g , 0.004 mol)をテトラヒドロフラン(THF, 20 ml) に溶解し、トリエチルアミン (TEA) (0.56 ml, 0.004 mol)を加えて-15℃に冷却した。イソブチルクロロホルメート(IBCF) (0.52 ml, 0.004 mol)を加え、-15℃で10分間攪拌した。そこにオクチルアミン(0.66 ml, 0.004 mol)を加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取した。エタノールから再結晶し、白色パウダーとして目的物を得た。
収量 1.32 g (68.3 %)、融点143-148℃、Rf1= 0.46.
元素分析 C28H41N3O4
計算値: C, 69.5: H, 8.55: N, 8.69
実測値: C, 69.4: H, 8.55: N, 8.49
【0046】
2) Boc-Tra-Tyr(O-Pic)-NH-Octyl
Boc-Tra-OH (Tra: trans-アミノメチルシクロヘキサンカルボン酸、トラネキサム酸) (0.69 g, 0.0027 mol) をTHF(20 ml) に溶解し、TEA (0.37 ml, 0.0027 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、TFA. H-Tyr(O-Pic)-NH-Octyl [Boc-Tyr(O-Pic)-NH-Octyl (1.32 g, 0.0027 mol)とTFA-アニソール(5.0 ml-0.50 ml)から予め調整した]とTEA (0.75 ml, 0.0054 mol)を含んだジメチルホルムアミド(DMF)溶液10 mlを加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取、白色パウダーとして目的物を得た。
収量 1.17 g (70.1 %)、融点188-191℃、[α]D=-11.0°(c=1, DMF)、 Rf1 = 0.28.
元素分析 C36H54N4O5
計算値: C, 69.4: H, 8.74: N, 9.00
実測値: C, 69.1: H, 8.86: N, 8.90
【0047】
3) H-Tra-Tyr(O-Pic)-NH-Octyl. 2HCl (YO-2)
Boc-Tra-Tyr(O-Pic)-NH-Octyl (0.79 g, 0.0012 mol )をTFA-アニソール(1.8 ml-0.18 ml)で室温、90分間処理して後、エーテルを加えて生じた沈殿物をろ取した。粗生成物をHPLCで精製し、1 mol 塩酸水溶液を加えて凍結乾燥を繰り返し、白色の非晶質として目的物を得た。
収量 0.50 g (69.9 %)、[α]D=+2.5°(c=1, MeOH)、Rf3 = 0.18、tR = 30.7 min.
TOF-MS m/Z: 523.8(M+H)+ (calc. for C31H46N4O3 :522.7).
元素分析 C31H46N4O3.2HCl. 2.5H2O
計算値: C, 68.7: H, 8.33: N, 8.74
実測値: C, 68.4: H, 8.50: N, 8.66
【0048】
(1−2)YO−57の合成
1) [Fmoc-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide
Fmoc-Tyr(O-Pic)−OH (0.50 g, 0.001 mol) をDMF(20 ml) に溶解し、N-メチルモルホリン (NMM) (0.11 ml, 0.001 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、4-(Boc-aminomethyl)aniline (0.45 g, 0.001 mol)を含んだDMF-THF (1:1)溶液20 mlを加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、10%クエン酸水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取し粗結晶を得た。粗結晶をクロロホルム (5 ml)に溶解し、シリカゲルカラム (BW-127ZH, 3 x 18 cm)にアプライした。クロロホルム (400 ml)で溶出した後、クロロホルム (800 ml) を集め留去した。残査に石油エーテルを加え、生じた沈殿物をろ過し、白色パウダーとして目的物を得た。
収量 0.25 g (36.0 %)、融点140-142℃、[α]D=+11.2°(c=0.67, DMF)、Rf1 = 0.58、Rf2 = 0.54.
元素分析 C42H42N4O6
計算値: C, 72.2: H, 6.06: N, 8.20
実測値: C, 71.9: H, 6.18: N, 7.91
【0049】
2) [Boc-Tra-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide
Boc-Tra-OH (0.074 g, 0.00029 mol) をTHF(10 ml) に溶解し、NMM (0.032 ml, 0.00029 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、H-Tyr(O-Pic)-4-(Boc-aminomethyl)anilide {[Fmoc-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide (0.20 g, 0.00027 mol)を20%ピペリジン/DMFで処理し、予め調整した}を含んだDMF溶液 (10 ml)を加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸水素ナトリウム水溶液、10%クエン酸水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取した。粗結晶を酢酸エチルから再結晶して、白色パウダーとして目的物を得た。
収量 0.13 g (63 %)、融点186-188℃、 [α]D=-11.0°(c=1, DMF)、 Rf1 = 0.56、Rf2 = 0.37.
元素分析 C40H53N5O7.0.25H20
計算値: C, 66.7: H, 7.48: N, 9.72
実測値: C, 66.6: H, 7.36: N, 9.71
【0050】
3) [H-Tra-Tyr(O-Pic)]-4-(aminomethyl)anilide. 2TFA
[Boc-Tra-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide (0.10 g, 0.0014 mol )をTFA-アニソール(0.50 ml-0.050 ml)で室温、90分間処理し、エーテルを加えて生じた沈殿物をろ取した。粗生成物を3%酢酸に溶解し、Sephadex G-15 カラム(2.2 x 49 cm)にアプライした。3%酢酸で溶出して13 gずつ集め、試験管番号 (4〜10) を集め凍結乾燥して、白色の非晶質として目的物を得た。
収量 0.10 g (83 %)、[α]D=+22.9°(c=0.86, 10%酢酸)、Rf3= 0.10、Rf4 = 0.62、tR = 20.8 min.
TOF-MS m/Z: 516.4 (M+H)+ (calc. for C30H37N5O3 :515.6).
元素分析 C30H37N5O3.3TFA. 0.75H2O
計算値: C, 49.6: H, 4.80: N, 8.04
実測値: C, 49.5: H, 4.60: N, 8.14
【0051】
【化8】
【0052】
[実施例2]
<GVHD予防活性>
(1) 急性GVHDモデルマウスの作製
6週齢のメスの(BALB/c × C57BL/6)F1(CBF1;H−2b/d)(SLC、日本)マウスに、6週齢のメスのC57BL/6:H−2Kbマウス(SLC、日本)の脾臓細胞1×108個を、尾静脈から0日目と7日目にそれぞれ投与することにより、急性GVHDマウスを作製した。
【0053】
(2)YO−2及びYO−57の急性GVHD予防活性評価
上記(1)で得られた急性GVHDモデルマウスを、それぞれ7〜10匹ずつ、YO−2投与群、YO−57投与群及び対照群(PBS投与群)に分けた。YO−2投与群及びYO−57投与群については、上記(1)に記載のGHVDモデルマウスの作製手順と同様に、マウスあたり3.75mg/kgの投与量で各薬剤を、0日目以降毎日それぞれ腹腔内投与した。なお比較用として、それぞれの薬剤をGHVDモデルマウスではなく正常マウスにも投与した。
【0054】
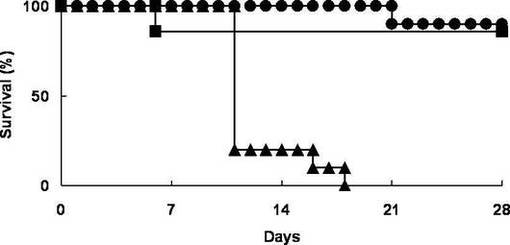
生存率の結果を図1に示す。また、比較対象としてPBSのみを用いた以外は各薬剤投与群と同様にして、GHVDモデルマウスに投与し、対照群とした。対照群の結果を、YO−2投与群及びYO−57投与群と同様に図1に示す。なお、図1において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。(Mann−Whitney Utest P<0.01)
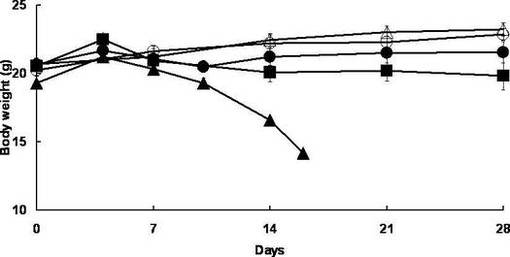
また、体重の変化については図2に示す。図2において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。また白丸はYO−2を投与した正常マウス、白三角はPBSを投与した正常マウスをそれぞれ表す。
また、YO−2投与群、YO−57投与群及びPBSのみの対照群について、10日目に、肝臓、皮膚、小腸、脾臓、骨髄及び胸腺の組織を採取して、常法に従って各組織の病理切片を作製した。
【0055】
(3)サイトカインのレベル評価
上記(2)のYO−2投与群及び対照群の各マウスの静脈血を内眼角より採取し、常法により試料を調製した後、TNF−α、Fasリガンド(FasL)、及び全MMP−9の血清中の濃度を確認した。各種サイトカイン及びMMP−9レベル(濃度)についてはELISA法(酵素免疫吸着法)により測定した。TNF−αはマウス血清中、その他は血漿中の値を測定した。
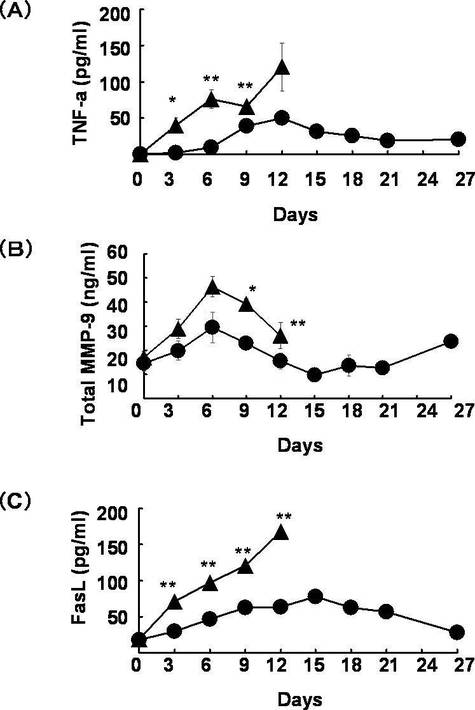
結果を図3に示す。図3において黒三角は対照群、黒丸はYO−2投与群を表す。図3において「*」は有意差p値が0.05以下であることを示し、「**」は有意差p値が0.01以下を示す。
【0056】
(4)結果
図1に示されるように、急性GVHDモデルマウスについて、YO−2投与群及びYO−57投与群の両群では共に、20日以上にわたって80%以上の生存率を示し、特にYO−2投与群では21日目まで100%の生存率であった。これに対して対照群では18日目で生存率0%となった。
また、図2に示されるように、体重の変化についても、YO−2投与群及びYO−57投与群は対照群に対して有意な体重減少抑制を示した。
【0057】
病理組織所見については、対照群ではGVHD特有の変化である肝臓のグリソン鞘付近の著明な細胞浸潤、消化管では消化管絨毛の平低下及び開大、並びに腺窩の増高等が認められ、皮膚でも炎症性細胞浸潤や脱毛、浮腫、及び潰瘍形成が認められた。また、脾臓、胸腺及び骨髄では、いずれも著明な細胞密度の減少が認められ、リンパ系細胞の減少、及びリンパ組織の萎縮、さらにこれに伴う組織構造の破壊が認められた。
これに対し、YO−2投与群ではこれらのいずれの臓器の病理組織においてもこれらのGVHDに基づくと考えられる組織変化が抑制されていた。
【0058】
また、対照群ではMMP−9の血中濃度の増大を認め(図3(B)参照)、これに伴って炎症性サイトカインTNF−α(図3(A)参照)、及びFasL(図3(C)参照)の血中濃度の上昇が認められた。これに対しYO−2投与群では、対照群と比較して有意にMMP−9濃度が減少しており(図3(B)参照)、さらにGVHDの組織傷害の主たる原因となるこれら炎症性サイトカインの増加も抑制されていた(図3(A)及び(C)参照)。
【0059】
特にTNF−αとFasLは細胞死(アポトーシス)の主たる誘導因子と考えられているため、YO−2投与群におけるこれらの血中濃度の低下は、骨髄、脾臓、胸腺の病理組織所見におけるGVHDに特有なリンパ系細胞の減少、及びリンパ組織の萎縮といった病変の改善にYO−2が関与していることを示唆するものである。またTNF−αの上昇は、敗血症性ショックの直接な誘導因子と考えられており、YO−2の投与がこうした病態に対しても奏効することは、これらのデータからも容易に推察される。
【0060】
このように、YO−2及びYO−57にはGVHDに対する予防効果が認められた。
また、これらの病理組織所見、マウス重量のデータに示されるように、各薬剤投与に伴う食欲低下、行動能の低下等の症状は認められず、各種臓器に病理組織上の変化等も認められないことから、副作用が少ないことも明らかであった。
【0061】
YO−2及びYO−57などの本発明にかかるアミド誘導体は、プラスミン阻害活性を有することは既に知られている。同様に線溶阻害活性を有する薬剤として、フィブリンの分解抑制作用を有するトラネキサム酸が知られているが、トラネキサム酸には、GVHD予防活性があるとの報告はない。
上記結果から、本発明にかかるアミド誘導体は、トラネキサム酸とは異なる機構により線溶阻害活性を示すものであることが示唆される。
【0062】
[参考例1]
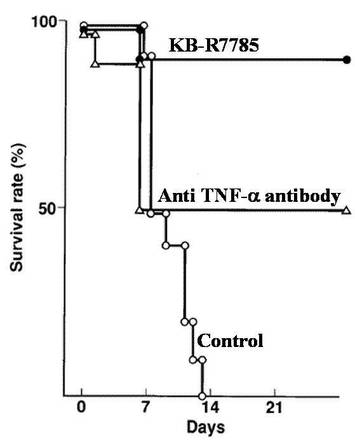
急性GVHDモデルマウスは、それぞれ10匹ずつ、抗TNFα抗体投与群、MMP阻害剤投与群及び対照群(PBS投与群)に分けた。抗TNFα抗体投与群では、0.5mgの抗TNFα抗体(PharMingen社)を含有するPBS溶液を投与に使用し、2mgのMMP阻害剤(KB−R7785:カネボウ社-製薬)を含有するPBS溶液を投与に使用して、実施例1(3)と同様に各マウスに、0日目以降毎日それぞれ腹腔内投与した。結果を図4に示す。図4において黒丸がMMP阻害剤投与群、白三角が抗TNFα抗体投与群、白丸が対照群をそれぞれ表す。
図4に示されるように、抗TNFα抗体投与群の生存率は7日目以降で50%であり、GVHDの予防としては、TNF−αのみを抑えても充分な効果が得られないことは明らかである。また、KB−R7785投与群の生存率は7日目以降で90%であり、本発明のアミド誘導体を超えるものではなかった。
【0063】
[実施例3]
<全身性炎症反応症候群予防活性>
(1)敗血症性ショックモデルマウスの作製
10週齢のメスのC57BL/6マウス(SLC、日本)の腹腔内にリポ多糖(LPS)0.5μg/kgとD−(+)−塩酸ガラクトサミン400mg/kgを0日目に投与することにより、敗血症性ショックを誘導するモデルマウスを作製した。
【0064】
(2)YO−2及びYO−57の敗血症性ショック予防活性評価
上記(1)で得られた敗血症性ショックモデルマウスを、YO−2投与群(n=10)、YO−57投与群(n=5)及び対照群(リン酸緩衝生理食塩水(PBS)投与群、n=10)に分けた。YO−2投与群及びYO−57投与群については、各薬剤を、マウスあたり3.75mg/kgの投与量で、上記のLPS投与5日前から0日目まで連日腹腔内投与した。
生存率の結果を図5に示す。なお、図5において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。
【0065】
図5に示されるように、YO−2投与群では、48時間以降も80%以上の生存率であった。特に、36時間までは100%の生存率であった。また、YO−57投与群では、48時間で50%以上の生存率であった。これに対して対照群では48時間で生存率0%となった。
【0066】
このように、実施例2及び実施例3の結果から、本発明にかかるアミド誘導体は、副作用が少なく、GVHD予防作用及び/又は全身性炎症反応症候群予防作用の効果が高いことがわかる。
従って、本発明によれば、炎症性疾患に対して有効な炎症性疾患治療剤を提供することができるのは明らかである。
【技術分野】
【0001】
本発明は、炎症性疾患治療剤に関する。
【背景技術】
【0002】
炎症性疾患には、種々の炎症性サイトカインの調節機能に異常が生じて過剰分泌することに起因するものがある。炎症性サイトカインとは、炎症を促進するサイトカインの総称であり、TNFα、Fasリガンド(FasL)、IL−1α、IL−1β、IL−6、IFNγ、IL−8、IL−10、IL−12、IL−15、IL−18及びCD40リガンド等が知られている。このため、炎症性疾患の治療方法のひとつとして、炎症性サイトカインの分泌抑制が挙げられ、炎症性サイトカインの分泌抑制に効果がある薬剤がいくつか提案されている(例えば、特許文献1及び2)。
【0003】
また炎症性サイトカインの中には、マトリックスメタロプロテアーゼ(MMP)により膜型から可溶型に変換して活性型になるものが多数あることが知られている。MMPは、このような作用の他に、がんの転移や慢性関節リウマチ、変形性関節症、炎症、糖尿病などの疾患や老化に関与していると考えられている。このため、MMPの作用を阻害又はMMPの活性化を阻害することによって、MMPに関与する種々の疾患の症状を緩和することができると考えられ、多くのMMP阻害剤が開発又は提案されている(例えば、特許文献3及び4)。
【0004】
一方、血栓溶解酵素であるプラスミンを特異的に阻害する物質として、特定構造のアミド化合物YO−2及びその誘導体が知られている(非特許文献1及び2)。また、YO−2には、ヒトのメラノーマ細胞、Bリンパ腫細胞、大腸ガン細胞等の増殖を抑制し、腫瘍細胞の増殖阻害作用とがんの転移抑制作用が認められることについても報告されている(非特許文献3)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−235447号公報
【特許文献2】特開2008−303171号公報
【特許文献3】特開2002−201130号公報
【特許文献4】特表2010−511027号公報
【非特許文献】
【0006】
【非特許文献1】Thrombosis Research, (1996) Vol.82, No.1, pp.79-86
【非特許文献2】Chem. Pharm. Bull., (2001) Vol.49(11), pp.1457-1463
【非特許文献3】In vivo, (2002) Vol.16, pp.281-286
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、炎症性疾患は複数の炎症性サイトカインが関与しているため、ひとつ又は数種の炎症性サイトカインの過剰分泌を阻害したとしても充分な治療効果が得られるとは限らない。また、種々開発されているMMP阻害剤は、その強い効果と引き替えに、肩又は腕の筋肉痛、関節痛、関節滑膜の肥厚、関節包中への炎症細胞の浸潤など、強い副作用を示すものも多く、重篤な副作用に耐えかねて使用を継続でない場合もある。このためMMP阻害剤を使用しても、その作用を充分に発揮することができないことが知られている。
従って本発明の目的は、炎症性疾患に対して有効な新規の炎症性疾患治療剤を提供することである。
【課題を解決するための手段】
【0008】
本発明は以下のとおりである。
[1] 下記の一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。
【0009】
【化1】
【0010】
(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)
[2] 前記アミド誘導体が下記一般式(I−1)で表される[1]に記載の炎症性疾患治療剤。
【0011】
【化2】
【0012】
(式中R及びL2は一般式(I)と同様である。)
[3] 前記L2が、フェニレン基及び炭素数2〜9のアルキレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す[1]又は[2]に記載の炎症性疾患治療剤。
[4] 下記の化合物の少なくとも一方を含む[1]〜[3]のいずれかに記載の炎症性疾患治療剤。
【0013】
【化3】
【0014】
[5] 前記炎症性疾患が、移植片対宿主疾患(GVHD)、全身性炎症反応症候群、炎症性腸疾患、膠原病、及び癌悪液質からなる群より選択された少なくとも1つである[1]〜[4]のいずれかに記載の炎症性疾患治療剤。
【発明の効果】
【0015】
本発明によれば、炎症性疾患に対して有効な炎症性疾患治療剤を提供することができる。
【図面の簡単な説明】
【0016】
【図1】実施例2に係る急性GVHDモデルマウスの生存率を示す図である。
【図2】実施例2に係る急性GVHDモデルマウスの体重の変化を示すグラフである。
【図3】実施例2に係る急性GVHDモデルマウスの各血清サイトカイン濃度(図3(A)はTNF−α、図3(B)は全MMP−9、図3(C)はFasL)の変化を示すグラフである。
【図4】参考例1に係る抗TNFα抗体及びMMP阻害剤を投与した急性GVHDモデルマウスの生存率を示す図である。
【図5】実施例3に係る敗血症性ショックモデルマウスの生存率を示す図である。
【発明を実施するための最良の形態】
【0017】
本発明の炎症性疾患治療剤は、上記一般式(I)で示されるアミド誘導体又はその塩を有効成分とするものである。
一般式(I)で示されるアミド誘導体は、プラスミン阻害剤として既知の物質であるが、今回、MMP活性阻害作用があることが新たに見出された。本発明は、この知見に基づくものである。
【0018】
これを詳細に説明すれば、MMPは、不活性型のプロMMPで体内に存在しており、活性化因子の存在下で活性化するとMMPとして作用可能となる。線溶作用を有するプラスミンは、フィブリンに作用してフィブリン分解物を生成する作用を有する一方で、プロMMPの活性化にも作用すると推測されている。活性化されたMMPは、種々の炎症性サイトカインの外部ドメインを脱落させ、この結果、炎症性サイトカインの可溶性部分を離脱させて炎症性サイトカインを活性化すると考えられている。本発明に係るアミド誘導体は、プラスミノーゲンの活性化を阻害することによって、MMPの関与する複数の炎症性サイトカインの活性化を抑制し、その結果、MMPに関与する炎症性疾患の治療効果を奏すると推測されるが、この理論に限定されない。また、前記アミド誘導体には、強い副作用も認められない。従って、本発明に係る前記アミド誘導体を有効成分とする医薬組成物を用いることより、MMPが関与する種々の炎症性疾患に対して、高い治療効果が期待できる。
【0019】
本発明において「治療」とは、本発明にかかる組成物の投与前後において炎症性疾患に特有の症状が軽減又は、重篤化が抑制されることを意味する。また、この用語には、本発明にかかる組成物の投与後において炎症性疾患に特有の症状の顕著な発生を抑制する「予防」も包含される。
【0020】
本明細書において「工程」との語は、独立した工程だけでなく、他の工程と明確に区別できない場合であっても本工程の所期の作用が達成されれば、本用語に含まれる。
また、本明細書において「〜」を用いて示された数値範囲は、「〜」の前後に記載される数値をそれぞれ最小値及び最大値として含む範囲を示す。
また、本発明において、組成物中の各成分の量について言及する場合、組成物中に各成分に該当する物質が複数存在する場合には、特に断らない限り、組成物中に存在する当該複数の物質の合計量を意味する。
以下、本発明について説明する。
【0021】
本発明にかかるアミド誘導体は、一般式(I)で示される化合物である。
【0022】
【化4】
【0023】
式(I)中、Arは、炭素数4〜6の芳香族基を表し、溶解性の観点から好ましくは炭素数4または6の芳香族基を表す。当該芳香族基は、置換されていても、未置換であってもよい。また環に、硫黄原子、窒素原子又は酸素原子などのヘテロ原子を1つ又は2つ以上含んでいてよい。Arで表される芳香族基が置換基を有する場合、置換基の例としては、水酸基、アミノ基、カルボキシ基、炭素数1〜4のアルキル基及びハロゲンを挙げることができる。Arで表される芳香族基における置換基は、2つ以上存在していてもよいが、未置換であることが好ましい。
Arで表される芳香族基の例としては、フェニル基、ピリジル基、ピリミジル基及びピリダジル基等を挙げることができ、溶解性の観点からピリジル基であることが好ましい。
【0024】
式(I)中L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す。前記L1としては、炭素数1又は2のアルキレンオキシ基であることが好ましく、メチレンオキシ基であることがより好ましい。
【0025】
式(I)中L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表す。前記脂肪族基は、飽和又は不飽和であってもよく、また直鎖構造のみならず、分岐鎖又は環状構造を有していてもよい。前記脂肪族基の例としては、炭素数2〜9の直鎖又は分岐アルキレン基及びシクロアルキレン基等を挙げることができる。アリーレン基の例としては、置換基を有していてもよいフェニレン基を挙げることができる。前記アリーレン基の置換基の例としては、アルキル基、アミノ基及びカルボキシル基等を挙げることができ、前記アリーレン基における置換基は2つ以上存在していてもよい。
式(I)中L2として表される炭化水素基としては、例えば以下のものを挙げることができる。なお、下記構造中「*」は隣接する原子との連結部分を表す。
【0026】
【化5】
【0027】
前記L2で表される炭化水素基としては、疎水性と嵩高さの観点から、炭素数2〜9のアルキレン基及びフェニレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基であることが好ましい。炭素数2〜9のアルキレン基としては、炭素数2〜9の直鎖又は分岐アルキレン基、シクロヘキシレン基及びフェニレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基であることが好ましい。
【0028】
式I中Rは、水素原子、アミノ基又はカルボキシ基を表し、水素原子又はアミノ基であることが好ましい。
【0029】
前記アミド誘導体としては、下記一般式(I−1)で表されるアミド誘導体であることが、MMP阻害活性の観点から好ましい。式(I−1)中、L2及びRは、前記式(I)におけるL2及びRと同一であり、L2及びRの好ましい例も同一である。特に、MMP阻害活性の観点から、下記一般式(II)におけるL2としては、炭素数2〜9の未置換の直鎖アルキレン基、未置換のフェニレン基及びシクロヘキシレン基であることが好ましく、Rとしては水素原子又はアミノ基であることが好ましい。
【0030】
【化6】
【0031】
本発明におけるアミド誘導体としては、MMP阻害活性及び抗炎症効果の観点から、以下のYO−2又はYO−57であることが特に好ましい。
【0032】
【化7】
【0033】
前記アミド誘導体は、公知の方法に従って合成することができ、例えばChem. Pharm. Bull., (2002), 49 (11), 1457-1463に合成方法が開示されている。簡単に説明すれば、まず、Boc-Tyr(O-Pic)-OH(Boc: t-Butoxycarbonyl、Tyr: tyrosine、O-Pic: O-Picolyl)を、トリエチルアミンの存在下、イソブチルクロロホルメイトで無水物に変換し、対応するアミンと反応し、Boc-Tyr(O-Pic)-NHX(酸アミド体)を得る。次に、Boc基をTFA(トリフルオロ酢酸)−anisoleで除去し、生じたアミンをBoc-4-aminomethylcyclo-hexanecarboxylic acid (Tra)の無水物と反応してBoc-Tra-Tyr(O-Pic)-NHX を得る。これをTFA−anisoleで処理すると、目的とするアミド誘導体(I)を得ることができる。
【0034】
前記アミド誘導体は、通常の薬理的に許容される酸と容易に塩を形成し得る。かかる酸としては、例えば硫酸、硝酸、塩酸、臭化水素酸等の無機酸、酢酸、トリフルオロ酢酸、p−トルエンスルホン酸、エタンスルホン酸、シュウ酸、マレイン酸、フマル酸、クエン酸、コハク酸、安息香酸等の有機酸を例示できる。
【0035】
本発明の炎症性疾患治療剤は、前記アミド誘導体を単独で又は2種以上を組み合わせて含んでもよい。
【0036】
本発明の炎症性疾患治療剤は、医薬として許容可能な担体と組み合わせてもよい。このような担体の例としては、医薬製剤として適用される形態に応じた公知の担体を挙げることができる。
例えば、注射剤として調製される場合、液剤、乳剤及び懸濁剤は殺菌され、且つ血液と等張であるのが好ましく、これらの形態に成形するに際しては、希釈剤としてこの分野において慣用されているものをすべて使用できる。例えば水、エチルアルコール、マクロゴール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオキシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等を使用できる。なお、この場合、等張性の溶液を調製するに充分な量の食塩、ブドウ糖あるいはグリセリンを医薬製剤中に含有してもよく、また通常の溶解補助剤、緩衝剤、無痛化剤等を添加してもよい。更に必要に応じて着色剤、保存剤、香料、風味剤、甘味剤等や他の医薬品を組成物中に含んでもよい。
またその他、組成物の形態に応じて、適宜、通常使用される充填剤、増量剤、結合剤、付湿剤、崩壊剤、表面活性剤、滑沢剤等の希釈剤あるいは賦形剤を含んでもよい。
【0037】
本発明の炎症性疾患治療剤に含有されるべき前記アミド誘導体の量としては、特に限定されず広範囲から適宜選択されるが、例えば、組成物の全質量の約1〜70質量%、好ましくは約5〜50質量%とするのがよい。
本発明の炎症性疾患治療剤は、MMPによって細胞外ドメイン分泌されるサイトカインに起因する多くの炎症性疾患に広く適用可能である。適用可能な炎症性疾患としては、移植片対宿主疾患(GVHD)、全身性炎症反応症候群(敗血症性ショック]、潰瘍性大腸炎等の炎症性腸疾患、一部の全身性炎症性疾患(例えば、関節リウマチ等の膠原病)、癌悪液質等を挙げることができ、これらの組み合わせに対して用いてもよい。
【0038】
上記の炎症性疾患はいずれもMMPによりその細胞外ドメイン分泌が制御される複数の炎症性サイトカインとそのシグナルに起因する炎症性疾患である。例えば、GVHD、全身性炎症反応症候群、炎症性腸疾患、関節リウマチ、及び癌悪液質では、TNFαは、病態上、これらの炎症性疾患の発症及び進行において最も重要なサイトカインの一つと考えられている。また、これらの炎症性疾患ではFasリガンドやIL−1、IL−2、IL−6の他CD40リガンド等の他の複数の炎症性サイトカインの関与が示唆されている。MMPはこれらのサイトカインあるいはこれらの受容体の細胞膜表面からの細胞外ドメイン分泌を制御することにより、サイトカイン血中濃度の上昇あるいは可溶型受容体によるサイトカイン作用の抑制等の形でこれらの炎症性疾患の病態形成に関与していると考えられる。従ってこれらの複数の炎症性サイトカインあるいはこれらの受容体の細胞外ドメイン分泌が前記アミド誘導体によって制御されることにより、上述した炎症性疾患はいずれも予防又は治療が可能となる。
【0039】
本発明の炎症性疾患治療剤の投与方法は特に制限はなく、各種製剤形態、患者の年齢、性別その他の条件、疾患の種類又は疾患の程度等に応じた方法を選択することができ、経口投与及び非経口投与のいずれであってもよい。
本発明の炎症性疾患治療剤の投与量は、用法、患者の年齢、性別その他の条件、疾患の程度等により適宜選択されるが、例えば、前記アミド誘導体の量が、1日当り体重1kgあたり約3.0mg〜8.0mg程度とすることができる。
【0040】
本発明はまた、前記一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤を、炎症性疾患の患者又は、炎症性疾患を生じる可能性がある若しくは疑いのある患者に投与することを含む炎症性疾患の予防又は治療方法を含む。
本炎症性疾患の予防又は治療方法によれば、前記アミド誘導体を投与することによって、MMPに関連する炎症性疾患を予防又は治療することができる。
【0041】
本予防又は治療方法における前記炎症性疾患治療剤については、前述した事項をそのまま適用する。
また、前記炎症性疾患については、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用する。
前記投与の方法、経路等についても、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用する。
その他の事項についても、前記炎症性疾患治療剤に関連して前述した事項をそのまま適用すればよい。
【実施例】
【0042】
以下、本発明を実施例にて詳細に説明する。しかしながら、本発明はそれらに何ら限定されるものではない。なお、特に断りのない限り、「%」は質量基準である。
【0043】
[実施例1]
<アミド誘導体の特性評価>
(1) アミド誘導体の合成
下記アミド誘導体YO−2及びYO−57はChem. Pharm. Bull., (2002), 49 (11), 1457-1463に記載された方法に従って以下のように合成した。なお、一般的操作は以下のとおりである。
融点は未補正、微量融点測定器(柳本製作所製)で測定した。
旋光度は自動旋光計 model DPI-360 [日本分光(株)]で測定した。
Matrix assisted laser desorption time-of flight mass spectra (MALDI-TOF-MS) にはKratos MALDI IV 質量分析器(Kratos Analytical)を用いた。
分析または分取HPLCにはWaters model 600Eを使用した。それぞれ、COSMOSIL C18 column AR-II (4.6 x 250 mm, nacalai tesque)、COSMOSIL C18 column AR-II (20 x 250 mm, nacalai tesque)を用いた。
保持時間はtRで示した。移動層には0.05% TFAを含む水−0.05% TFAを含むアセトニトリルを用いた。1分間あたり1%ずつアセトニトリルの濃度が高くなる濃度勾配をかけて溶出した。検出は220 nmで行い、流速は、分析または分取HPLCにおいて、それぞれ1.0 ml/min、10.0 ml/minで行った。
【0044】
薄層クロマトグラフィー(TLC)はKiesel gel 60G(Merck社製)を用いて行った。展開溶媒にはRf1 (クロロホルム:メタノール:酢酸=90:8:2)、Rf2 (クロロホルム:メタノール:水=89:10:1)、Rf3(n-ブタノール:酢酸:水=4:1:5、上層)Rf4 (n-ブタノール:酢酸:ピリジン:水=4:1:1: 2)を用いた。発色試薬としてアミノ基の検出には01%ニンヒドリン−アセトン溶液を用いた。Boc保護体の検出には臭化水素-ニンヒドリン法を用いた。BocおよびFmoc保護体の検出には254 nmのUV照射を用いた。
【0045】
(1−1)YO−2の合成
1) Boc-Tyr(O-Pic)-NH-Octylの合成
Boc-Tyr(O-Pic)-OH (1.48 g , 0.004 mol)をテトラヒドロフラン(THF, 20 ml) に溶解し、トリエチルアミン (TEA) (0.56 ml, 0.004 mol)を加えて-15℃に冷却した。イソブチルクロロホルメート(IBCF) (0.52 ml, 0.004 mol)を加え、-15℃で10分間攪拌した。そこにオクチルアミン(0.66 ml, 0.004 mol)を加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取した。エタノールから再結晶し、白色パウダーとして目的物を得た。
収量 1.32 g (68.3 %)、融点143-148℃、Rf1= 0.46.
元素分析 C28H41N3O4
計算値: C, 69.5: H, 8.55: N, 8.69
実測値: C, 69.4: H, 8.55: N, 8.49
【0046】
2) Boc-Tra-Tyr(O-Pic)-NH-Octyl
Boc-Tra-OH (Tra: trans-アミノメチルシクロヘキサンカルボン酸、トラネキサム酸) (0.69 g, 0.0027 mol) をTHF(20 ml) に溶解し、TEA (0.37 ml, 0.0027 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、TFA. H-Tyr(O-Pic)-NH-Octyl [Boc-Tyr(O-Pic)-NH-Octyl (1.32 g, 0.0027 mol)とTFA-アニソール(5.0 ml-0.50 ml)から予め調整した]とTEA (0.75 ml, 0.0054 mol)を含んだジメチルホルムアミド(DMF)溶液10 mlを加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取、白色パウダーとして目的物を得た。
収量 1.17 g (70.1 %)、融点188-191℃、[α]D=-11.0°(c=1, DMF)、 Rf1 = 0.28.
元素分析 C36H54N4O5
計算値: C, 69.4: H, 8.74: N, 9.00
実測値: C, 69.1: H, 8.86: N, 8.90
【0047】
3) H-Tra-Tyr(O-Pic)-NH-Octyl. 2HCl (YO-2)
Boc-Tra-Tyr(O-Pic)-NH-Octyl (0.79 g, 0.0012 mol )をTFA-アニソール(1.8 ml-0.18 ml)で室温、90分間処理して後、エーテルを加えて生じた沈殿物をろ取した。粗生成物をHPLCで精製し、1 mol 塩酸水溶液を加えて凍結乾燥を繰り返し、白色の非晶質として目的物を得た。
収量 0.50 g (69.9 %)、[α]D=+2.5°(c=1, MeOH)、Rf3 = 0.18、tR = 30.7 min.
TOF-MS m/Z: 523.8(M+H)+ (calc. for C31H46N4O3 :522.7).
元素分析 C31H46N4O3.2HCl. 2.5H2O
計算値: C, 68.7: H, 8.33: N, 8.74
実測値: C, 68.4: H, 8.50: N, 8.66
【0048】
(1−2)YO−57の合成
1) [Fmoc-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide
Fmoc-Tyr(O-Pic)−OH (0.50 g, 0.001 mol) をDMF(20 ml) に溶解し、N-メチルモルホリン (NMM) (0.11 ml, 0.001 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、4-(Boc-aminomethyl)aniline (0.45 g, 0.001 mol)を含んだDMF-THF (1:1)溶液20 mlを加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸ナトリウム水溶液、10%クエン酸水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取し粗結晶を得た。粗結晶をクロロホルム (5 ml)に溶解し、シリカゲルカラム (BW-127ZH, 3 x 18 cm)にアプライした。クロロホルム (400 ml)で溶出した後、クロロホルム (800 ml) を集め留去した。残査に石油エーテルを加え、生じた沈殿物をろ過し、白色パウダーとして目的物を得た。
収量 0.25 g (36.0 %)、融点140-142℃、[α]D=+11.2°(c=0.67, DMF)、Rf1 = 0.58、Rf2 = 0.54.
元素分析 C42H42N4O6
計算値: C, 72.2: H, 6.06: N, 8.20
実測値: C, 71.9: H, 6.18: N, 7.91
【0049】
2) [Boc-Tra-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide
Boc-Tra-OH (0.074 g, 0.00029 mol) をTHF(10 ml) に溶解し、NMM (0.032 ml, 0.00029 mol)を加えて-15℃に冷却した。IBCF (0.33 ml, 0.0027 mol)を加え、-15℃で10分間攪拌した。そこに、H-Tyr(O-Pic)-4-(Boc-aminomethyl)anilide {[Fmoc-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide (0.20 g, 0.00027 mol)を20%ピペリジン/DMFで処理し、予め調整した}を含んだDMF溶液 (10 ml)を加え、0℃で30分間、さらに室温で15時間攪拌した。反応溶媒を留去した後、残査を酢酸エチルに溶解し、有機層を5%炭酸水素ナトリウム水溶液、10%クエン酸水溶液、水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去後、残査に石油エーテルを加え、生じた沈殿をろ取した。粗結晶を酢酸エチルから再結晶して、白色パウダーとして目的物を得た。
収量 0.13 g (63 %)、融点186-188℃、 [α]D=-11.0°(c=1, DMF)、 Rf1 = 0.56、Rf2 = 0.37.
元素分析 C40H53N5O7.0.25H20
計算値: C, 66.7: H, 7.48: N, 9.72
実測値: C, 66.6: H, 7.36: N, 9.71
【0050】
3) [H-Tra-Tyr(O-Pic)]-4-(aminomethyl)anilide. 2TFA
[Boc-Tra-Tyr(O-Pic)]-4-(Boc-aminomethyl)anilide (0.10 g, 0.0014 mol )をTFA-アニソール(0.50 ml-0.050 ml)で室温、90分間処理し、エーテルを加えて生じた沈殿物をろ取した。粗生成物を3%酢酸に溶解し、Sephadex G-15 カラム(2.2 x 49 cm)にアプライした。3%酢酸で溶出して13 gずつ集め、試験管番号 (4〜10) を集め凍結乾燥して、白色の非晶質として目的物を得た。
収量 0.10 g (83 %)、[α]D=+22.9°(c=0.86, 10%酢酸)、Rf3= 0.10、Rf4 = 0.62、tR = 20.8 min.
TOF-MS m/Z: 516.4 (M+H)+ (calc. for C30H37N5O3 :515.6).
元素分析 C30H37N5O3.3TFA. 0.75H2O
計算値: C, 49.6: H, 4.80: N, 8.04
実測値: C, 49.5: H, 4.60: N, 8.14
【0051】
【化8】
【0052】
[実施例2]
<GVHD予防活性>
(1) 急性GVHDモデルマウスの作製
6週齢のメスの(BALB/c × C57BL/6)F1(CBF1;H−2b/d)(SLC、日本)マウスに、6週齢のメスのC57BL/6:H−2Kbマウス(SLC、日本)の脾臓細胞1×108個を、尾静脈から0日目と7日目にそれぞれ投与することにより、急性GVHDマウスを作製した。
【0053】
(2)YO−2及びYO−57の急性GVHD予防活性評価
上記(1)で得られた急性GVHDモデルマウスを、それぞれ7〜10匹ずつ、YO−2投与群、YO−57投与群及び対照群(PBS投与群)に分けた。YO−2投与群及びYO−57投与群については、上記(1)に記載のGHVDモデルマウスの作製手順と同様に、マウスあたり3.75mg/kgの投与量で各薬剤を、0日目以降毎日それぞれ腹腔内投与した。なお比較用として、それぞれの薬剤をGHVDモデルマウスではなく正常マウスにも投与した。
【0054】
生存率の結果を図1に示す。また、比較対象としてPBSのみを用いた以外は各薬剤投与群と同様にして、GHVDモデルマウスに投与し、対照群とした。対照群の結果を、YO−2投与群及びYO−57投与群と同様に図1に示す。なお、図1において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。(Mann−Whitney Utest P<0.01)
また、体重の変化については図2に示す。図2において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。また白丸はYO−2を投与した正常マウス、白三角はPBSを投与した正常マウスをそれぞれ表す。
また、YO−2投与群、YO−57投与群及びPBSのみの対照群について、10日目に、肝臓、皮膚、小腸、脾臓、骨髄及び胸腺の組織を採取して、常法に従って各組織の病理切片を作製した。
【0055】
(3)サイトカインのレベル評価
上記(2)のYO−2投与群及び対照群の各マウスの静脈血を内眼角より採取し、常法により試料を調製した後、TNF−α、Fasリガンド(FasL)、及び全MMP−9の血清中の濃度を確認した。各種サイトカイン及びMMP−9レベル(濃度)についてはELISA法(酵素免疫吸着法)により測定した。TNF−αはマウス血清中、その他は血漿中の値を測定した。
結果を図3に示す。図3において黒三角は対照群、黒丸はYO−2投与群を表す。図3において「*」は有意差p値が0.05以下であることを示し、「**」は有意差p値が0.01以下を示す。
【0056】
(4)結果
図1に示されるように、急性GVHDモデルマウスについて、YO−2投与群及びYO−57投与群の両群では共に、20日以上にわたって80%以上の生存率を示し、特にYO−2投与群では21日目まで100%の生存率であった。これに対して対照群では18日目で生存率0%となった。
また、図2に示されるように、体重の変化についても、YO−2投与群及びYO−57投与群は対照群に対して有意な体重減少抑制を示した。
【0057】
病理組織所見については、対照群ではGVHD特有の変化である肝臓のグリソン鞘付近の著明な細胞浸潤、消化管では消化管絨毛の平低下及び開大、並びに腺窩の増高等が認められ、皮膚でも炎症性細胞浸潤や脱毛、浮腫、及び潰瘍形成が認められた。また、脾臓、胸腺及び骨髄では、いずれも著明な細胞密度の減少が認められ、リンパ系細胞の減少、及びリンパ組織の萎縮、さらにこれに伴う組織構造の破壊が認められた。
これに対し、YO−2投与群ではこれらのいずれの臓器の病理組織においてもこれらのGVHDに基づくと考えられる組織変化が抑制されていた。
【0058】
また、対照群ではMMP−9の血中濃度の増大を認め(図3(B)参照)、これに伴って炎症性サイトカインTNF−α(図3(A)参照)、及びFasL(図3(C)参照)の血中濃度の上昇が認められた。これに対しYO−2投与群では、対照群と比較して有意にMMP−9濃度が減少しており(図3(B)参照)、さらにGVHDの組織傷害の主たる原因となるこれら炎症性サイトカインの増加も抑制されていた(図3(A)及び(C)参照)。
【0059】
特にTNF−αとFasLは細胞死(アポトーシス)の主たる誘導因子と考えられているため、YO−2投与群におけるこれらの血中濃度の低下は、骨髄、脾臓、胸腺の病理組織所見におけるGVHDに特有なリンパ系細胞の減少、及びリンパ組織の萎縮といった病変の改善にYO−2が関与していることを示唆するものである。またTNF−αの上昇は、敗血症性ショックの直接な誘導因子と考えられており、YO−2の投与がこうした病態に対しても奏効することは、これらのデータからも容易に推察される。
【0060】
このように、YO−2及びYO−57にはGVHDに対する予防効果が認められた。
また、これらの病理組織所見、マウス重量のデータに示されるように、各薬剤投与に伴う食欲低下、行動能の低下等の症状は認められず、各種臓器に病理組織上の変化等も認められないことから、副作用が少ないことも明らかであった。
【0061】
YO−2及びYO−57などの本発明にかかるアミド誘導体は、プラスミン阻害活性を有することは既に知られている。同様に線溶阻害活性を有する薬剤として、フィブリンの分解抑制作用を有するトラネキサム酸が知られているが、トラネキサム酸には、GVHD予防活性があるとの報告はない。
上記結果から、本発明にかかるアミド誘導体は、トラネキサム酸とは異なる機構により線溶阻害活性を示すものであることが示唆される。
【0062】
[参考例1]
急性GVHDモデルマウスは、それぞれ10匹ずつ、抗TNFα抗体投与群、MMP阻害剤投与群及び対照群(PBS投与群)に分けた。抗TNFα抗体投与群では、0.5mgの抗TNFα抗体(PharMingen社)を含有するPBS溶液を投与に使用し、2mgのMMP阻害剤(KB−R7785:カネボウ社-製薬)を含有するPBS溶液を投与に使用して、実施例1(3)と同様に各マウスに、0日目以降毎日それぞれ腹腔内投与した。結果を図4に示す。図4において黒丸がMMP阻害剤投与群、白三角が抗TNFα抗体投与群、白丸が対照群をそれぞれ表す。
図4に示されるように、抗TNFα抗体投与群の生存率は7日目以降で50%であり、GVHDの予防としては、TNF−αのみを抑えても充分な効果が得られないことは明らかである。また、KB−R7785投与群の生存率は7日目以降で90%であり、本発明のアミド誘導体を超えるものではなかった。
【0063】
[実施例3]
<全身性炎症反応症候群予防活性>
(1)敗血症性ショックモデルマウスの作製
10週齢のメスのC57BL/6マウス(SLC、日本)の腹腔内にリポ多糖(LPS)0.5μg/kgとD−(+)−塩酸ガラクトサミン400mg/kgを0日目に投与することにより、敗血症性ショックを誘導するモデルマウスを作製した。
【0064】
(2)YO−2及びYO−57の敗血症性ショック予防活性評価
上記(1)で得られた敗血症性ショックモデルマウスを、YO−2投与群(n=10)、YO−57投与群(n=5)及び対照群(リン酸緩衝生理食塩水(PBS)投与群、n=10)に分けた。YO−2投与群及びYO−57投与群については、各薬剤を、マウスあたり3.75mg/kgの投与量で、上記のLPS投与5日前から0日目まで連日腹腔内投与した。
生存率の結果を図5に示す。なお、図5において黒丸がYO−2投与群、黒四角がYO−57投与群、黒三角が対照群の生存率をそれぞれ表す。
【0065】
図5に示されるように、YO−2投与群では、48時間以降も80%以上の生存率であった。特に、36時間までは100%の生存率であった。また、YO−57投与群では、48時間で50%以上の生存率であった。これに対して対照群では48時間で生存率0%となった。
【0066】
このように、実施例2及び実施例3の結果から、本発明にかかるアミド誘導体は、副作用が少なく、GVHD予防作用及び/又は全身性炎症反応症候群予防作用の効果が高いことがわかる。
従って、本発明によれば、炎症性疾患に対して有効な炎症性疾患治療剤を提供することができるのは明らかである。
【特許請求の範囲】
【請求項1】
下記の一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。
【化1】
(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)
【請求項2】
前記アミド誘導体が下記一般式(I−1)で表される請求項1記載の炎症性疾患治療剤。
【化2】
(式中R及びL2は一般式(I)と同様である。)
【請求項3】
前記L2が、フェニレン基及び炭素数2〜9のアルキレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す請求項1又は請求項2記載の炎症性疾患治療剤。
【請求項4】
下記の化合物の少なくとも一方を含む請求項1〜請求項3のいずれか1項記載の炎症性疾患治療剤。
【化3】
【請求項5】
前記炎症性疾患が、移植片対宿主疾患(GVHD)、全身性炎症反応症候群、炎症性腸疾患、膠原病、及び癌悪液質からなる群より選択された少なくとも1つである請求項1〜請求項4のいずれか1項記載の炎症性疾患治療剤。
【請求項1】
下記の一般式(I)で示されるアミド誘導体又はその塩を有効成分とする炎症性疾患治療剤。
【化1】
(式中Arは炭素数4〜6の芳香族基を表し、L1は、−O−、−O−CO−、炭素数1又は2のアルキレン基及び炭素数2〜4のアルケニレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表し、L2は、炭素数2〜9の脂肪族基及びアリーレン基からなる群より選択された少なくとも1つで構成される二価の炭化水素基を表し、Rは、水素原子、アミノ基又はカルボキシル基を表す。)
【請求項2】
前記アミド誘導体が下記一般式(I−1)で表される請求項1記載の炎症性疾患治療剤。
【化2】
(式中R及びL2は一般式(I)と同様である。)
【請求項3】
前記L2が、フェニレン基及び炭素数2〜9のアルキレン基からなる群より選択された少なくとも1つで構成される二価の連結基を表す請求項1又は請求項2記載の炎症性疾患治療剤。
【請求項4】
下記の化合物の少なくとも一方を含む請求項1〜請求項3のいずれか1項記載の炎症性疾患治療剤。
【化3】
【請求項5】
前記炎症性疾患が、移植片対宿主疾患(GVHD)、全身性炎症反応症候群、炎症性腸疾患、膠原病、及び癌悪液質からなる群より選択された少なくとも1つである請求項1〜請求項4のいずれか1項記載の炎症性疾患治療剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−71905(P2013−71905A)
【公開日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願番号】特願2011−211450(P2011−211450)
【出願日】平成23年9月27日(2011.9.27)
【出願人】(507307374)学校法人神戸学院 (9)
【Fターム(参考)】
【公開日】平成25年4月22日(2013.4.22)
【国際特許分類】
【出願日】平成23年9月27日(2011.9.27)
【出願人】(507307374)学校法人神戸学院 (9)
【Fターム(参考)】
[ Back to top ]