
炎症性疾患治療用候補薬剤をスクリーニングするためのアッセイ

【課題】サイトカイン産生に起因する炎症性疾患の治療用候補薬剤を選択するためのアッセイの提供。
【解決手段】a)前記候補薬剤を腸細胞に接触させることと、b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、NF−κBファミリーの転写因子の転写活性の破壊、p65(RelA)の特異的ヒストンアセチル化、腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び1以上のサイトカインのレベルの変化から成る群から選択される、前記候補薬剤の作用を解析する。
【解決手段】a)前記候補薬剤を腸細胞に接触させることと、b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、NF−κBファミリーの転写因子の転写活性の破壊、p65(RelA)の特異的ヒストンアセチル化、腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び1以上のサイトカインのレベルの変化から成る群から選択される、前記候補薬剤の作用を解析する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、感染に対する腸の反応の調節、特にバクテロイデス・テタイオタオミクロン(Bacteroides thetaiotaomicron)を用いた調節に関する。
【背景技術】
【0002】
哺乳動物の腸においては、数百種類の細菌がコロニーを形成し、この細菌の数は結腸、即ち、感染や炎症性疾患、癌を起こしやすい解剖学的部位において劇的に増加する。しかし、成長した動物においては、消化管内に常在する細菌叢によって感染に対する高い防御作用(コロニー形成耐性)(colonisation resistance)が存在する(ファン・デル・ワーイ(van der Waaij)、1984年;サルミネン(Salminen)ら、1998年)。その結果、この環境に遭遇する日和見病原菌の多くは足掛かりを築くことができなくなり、急速に排除される(ファン・デル・ワーイ(van der Waaij)、1984年;サルミネン(Salminen)ら、1998年)。しかし、共生細菌叢が損なわれている場合、日和見病原菌が腸内に存在し続けることがある。例えば、緑膿菌は通常ネズミの腸に存在し続けることはないが、抗生物質によって共生細菌叢が破壊されると、緑膿菌はコロニーを形成し、重篤な感染を引き起こす(ピア(Pier)ら、1992年)。また、共生細菌もサルモネラ菌等の悪性病原菌に対してある程度の防御作用をもたらす。しかし、このような悪性病原菌は、その存在する数が非常に多い場合、共生細菌叢の防御作用を克服したりその作用から免れたりして、重篤な感染を引き起こすことがある。
【0003】
コロニー形成耐性(Colonisation resistance)は有害細菌の競合排除に一部起因するものであるが、この排除は、栄養素や基質を優先的に用いたり、或いは有害細菌が付着する可能性のある腸の部位を共生細菌叢によってブロックすることによって行われる。しかし、共生細菌が宿主の腸細胞及び免疫系を調節することもある(ブライ(Bry)ら、1996年;へリアス(Herias)ら、1998年、1999年;フーパー(Hooper)ら、2000年、2001年;セブラ(Cebra)、1999年;スネル(Snel)ら、1998年;タルハム(Talham)ら、1999年;ロペス−ボアド(Lopez-Boado)ら、2000年;シュー(Shu)ら、2000年;キャンベル(Campbell)ら、2001年)。また、共生細菌叢は消化管を変化させ、この細菌叢にはよく適合するが他の細菌には適合しないニッチミクロ環境(niche microenvironments)を形成することがある。或いは、共生細菌叢は有害細菌に対する上皮細胞の反応を変化させ(キャンベル(Campbell)ら、2001年)、これによってコロニー形成や浸潤を促進する変化を抑制することもある。
【0004】
健康な腸は様々な細菌の負荷に対して低反応状態(hyporesponsive tone)を維持するが、閾値レベルの病原性細菌が存在すると、腸組織内での炎症誘発性遺伝子の発現を急速にアップレギュレートする転写系が十分に活性化される。こうして得られた転写物によって、一連の反応(例えば、腸の感染部位の粘膜固有層への多形核(PMN)細胞の遊走)が引き起こされる。これらのイベントは細菌の排除には必須であるが、病状を悪化させ得る組織異変の原因にもなる。管腔内細菌が最初に接触する場所は、腸免疫系と接続する或いは遮断される上皮細胞の連続層である。腸上皮細胞が病原性細菌と非病原性細菌とを区別する能力は、コロニー形成細菌に対する有害で不適切な反応を防ぎ、腸を健康な状態に維持する上で極めて重要である。この区別機能は細菌認識系及び細胞シグナル伝達系に刷り込まれている。細菌細胞の表面構造の認識は、上皮細胞の先端側表面及び基底外側表面に発現するToll様受容体の機能の1つであり、この受容体は核因子κB(NF−κB)仲介による免疫活性化を引き起こす(ガーウィルツ(Gerwirtz)ら、2001年)。従来、免疫抑制剤の活性に関連した受容体系については確認されていない。
【0005】
ヒト共生細菌B.テタイオタオミクロンを無菌マウスに投与することによって、腸の成熟やバリア機能の発達に関連する重要な遺伝子の発現が引き起こされた(フーパー(Hooper)ら、2001年)。この投与によって、Fucα1,2Galβ−グリカンの腸内レベルも上昇し(ブライ(Bry)ら、1996年;フーパー(Hooper)ら、2001年)、また、マトリリシンの腸内レベルも上昇した(ロペス−ボアド(Lopez-Boado)ら、2000年)。また、B.テタイオタオミクロンは、サルモネラ菌を用いたインビトロチャレンジに対する上皮細胞の反応を変化させることも見出された(キャンベル(Campbell)ら、2001年)。特に、炎症誘発経路の一部が抑制された(キャンベル(Campbell)ら、2001年)。B.テタイオタオミクロンはこのような潜在的防御特性を有するが、ウェルシュ菌血清型Aの感染に対する完全無菌マウスの耐性を増加させることはなかった(ユルドゥセフ(Yurdusev)ら、1989年)。しかし、このマウスをフソバクテリウム・ネクロジーン(ユルドゥセフ(Yurdusev)ら、1989年)及び非病原性クロストリジウム菌株CI(ユルドゥセフ(Yurdusev)ら、1986年)と併用してB.テタイオタオミクロンで処理すると、病原菌が腸から排除された。このことから、B.テタイオタオミクロン単独でも腸内の潜在的防御変化を誘発することが可能であるが、感染に対する全耐性を大幅に高めるためには他の共生菌株と協力して作用させる必要のあることが示唆された。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明においては、非病原性細菌並びにPPARγの核−細胞質内分布及び(NF−κB)の転写活性の双方による新規なメカニズムを明らかにする。
【0007】
更に本発明は、炎症反応の調節のための非病原性細菌の使用に関する。更に本発明は、IκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化についての実証データと、ペルオキシソーム増殖因子によって活性化された受容体(PPARγ)の新規な作用モデルについての実証データとを提供する。特に本発明は、非病原性細菌を用いてPPARγの核−細胞質内分布とNF−κBの転写活性とを変化させることによる上皮炎症性遺伝子発現の抑制について記載する。更に本発明は、炎症を軽減して疾患や炎症性障害を治療及び予防する手段を提供する。
【0008】
本発明に記載の新規な経路を用いて炎症性サイトカインの産生を調節する新規な方法及び新規な製品をスクリーニングすることができる。更に、本発明は、炎症性サイトカインの産生を抑制し、免疫系をホメオスタシスへ回復させることにおける非病原性細菌の使用について記載する。
【0009】
本明細書に記載の炎症性サイトカイン産生の調節は、公知の刊行物に記載のものとは多くの面で異なる。
【0010】
米国特許第5,925,657号は、PPARγのアゴニストについて記載している。このアゴニストはチアゾリジンジオン、即ち、チオゾリジンジオン核に置換アリール部位が結合した化学的化合物である。本発明においては、非病原性細菌を用い、PPARγを直接活性化することにより炎症反応を軽減する。
【0011】
ネイシュら(2000年)は、IκBαユビキチン化の阻害による上皮反応の調節について記載している。本発明とネイシュら(2000年)の記載のものとでは、用いる作用モードが異なる。
【0012】
IκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化についての実証データと、ペルオキシソーム増殖因子によって活性化された受容体(PPARγ)の新規な作用モデルについての実証データとを提供する。この新規なデータによって、炎症性サイトカインの産生を調節するための新規な手段及び新規な製品を考案する可能性が提供される。また、得られた結果のインビボでの検証についても記載する。
【課題を解決するための手段】
【0013】
本発明は、サイトカイン産生に起因する炎症性疾患の治療用候補薬剤を選択するためのアッセイを提供する。本発明のアッセイは、
a)候補薬剤を腸細胞に接触させることと、
b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び
PPARγの核質内破壊から成る群から選択される、前記候補薬剤の作用を解析することを含む。
【0014】
本発明のアッセイは、
NF−κBファミリーの転写因子の核外移行の促進或いは核内移行の抑制、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の増加、及び
PPARγの核質内破壊から成る群から選択される少なくとも1つの作用を示す候補薬剤を選択する工程を更に含んでもよい。
【0015】
好ましい実施形態において、解析される候補薬剤の作用は、NF−κBファミリーの転写因子の核外移行又は腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化である。
【0016】
他の好ましい実施形態において、本発明のアッセイは、TNF−α、IL−8、MIP−2α及びCox−2から成る群から選択される1以上のサイトカイン(好ましくは、IL−8及び/又はMIP−2α)のレベルの変化を解析する工程を更に含む。
【0017】
好適には、本発明のアッセイに用いる腸細胞は、細胞培養で維持される腸細胞株の形態を有する。適切な細胞株としてはCaco−2細胞株が挙げられる。或いは、本発明のアッセイは、哺乳動物(例えば、マウス)等の適切な動物を用いてインビボで行うことができる。
【0018】
任意的には、本発明のアッセイは、サルモネラ菌等の公知の病原性細菌の存在下で行ってもよい。
【0019】
更に本発明は、炎症性サイトカイン産生に伴う疾患の治療方法であって、
NF−κBファミリーの転写因子の核外移行或いは核内移行、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内のPPARγ/RelA複合体量、又は
PPARγの核質内破壊を調節することができる剤の治療有効量を投与する工程を含む方法を提供する。
【0020】
好ましくは、前記剤を炎症性疾患、特に腸細胞の炎症反応によって少なくとも一部引き起こされる疾患の治療に用いる。一般に、このような疾患は炎症性サイトカインの産生を伴う。好適には、前記剤はバクテロイデス・テタイオタオミクロン又はその一構成成分である。本明細書において「その一構成成分」とは、通常B.テタイオタオミクロンの一部を形成し、免疫反応を軽減させることのできるあらゆる部分(タンパク質やポリペプチド、糖、核酸等)を意味する。
【0021】
更に本発明は、B.テタイオタオミクロン又はその一構成成分の薬剤としての使用を提供する。好ましくは、B.テタイオタオミクロン又はその一構成成分を炎症性疾患、特に腸細胞の炎症反応によって少なくとも一部引き起こされる炎症性疾患の治療に用いる。従って、本発明は、炎症性サイトカイン産生に伴う疾患や病態を軽減、予防又は治療するためのB.テタイオタオミクロンの使用を提供する。このような疾患や病態として、炎症性腸疾患(特にクローン病や過敏性腸症候群について言及する場合もある)や、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイト増殖阻害、皮膚の過剰増殖及び炎症性障害の阻害(例えば、乾癬や尋常性ざ瘡が挙げられるが、これらに限定されない)等が挙げられるが、これらに限定されるものではない。
【0022】
更に本発明は、炎症性疾患治療用薬剤の製造におけるB.テタイオタオミクロンの使用を提供する。
【0023】
更に本発明は、B.テタイオタオミクロン又はその一構成成分の治療有効量を投与することを含む、腸細胞の炎症反応の処置方法、或いは該反応を軽減する方法を提供する。
【0024】
より詳細には、B.テタイオタオミクロン又はその一構成成分を用いて、NF−κB経路を破壊し、及び/又はp65(RelA)反応を阻害し、及び/又はPPARγの刺激剤或いは阻害剤として作用させることができる。従って、B.テタイオタオミクロン又はその一構成成分を炎症性サイトカイン産生に伴う疾患の治療に用いることができる。
【0025】
B.テタイオタオミクロンは食品や座薬を用いて生菌の状態で患者に投与することができる。
【0026】
従って、本発明は、B.テタイオタオミクロン又はその一構成成分の治療有効量を投与する工程を含む、腸細胞の炎症反応を治癒させる方法について記載する。
【0027】
ここに記載された病態を予防或いは治療するためには、適当な製剤やキャリア、賦形剤、希釈剤、安定剤を用いてB.テタイオタオミクロンを経口投与し、消化管の作用部位に優先的に供給する。このような供給手段としては、錠剤やカプセル剤等の固形製剤、ヨーグルトや飲料、懸濁剤等の液状製剤等の製剤が挙げられるが、これらに限定されない。優先的な供給手段の1つとして、B.テタイオタオミクロンを経口的に、好ましくは不都合なく胃の酸性環境を経由させて腸内の作用部位に供給する手段が挙げられる。B.テタイオタオミクロンはプレバイオティックと共に投与してもよい。
【0028】
更に本発明は、本明細書に記載の新規な経路を介して炎症性サイトカイン産生をアゴニスト的或いはアンタゴニスト的に調節する可能性のある特定のリガンドをスクリーニングするための、PPARγの作用とIκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化との新規モードを提供する。
【0029】
更に本発明は、炎症性サイトカイン産生に伴う疾患の治療方法であって、NF−κBの転写活性を変化させること或いは腸細胞のサイトゾル内のPPARγ/RelA複合体量を増加させることが可能な化合物の治療有効量を投与する工程を含む方法を提供する。
【0030】
更に本発明は、哺乳動物の免疫系をホメオスタシスに回復させ維持させる方法であって、NF−κBの転写活性を変化させること或いは腸細胞のサイトゾル内のPPARγ/RelA複合体量を増加させることが可能な化合物の治療有効量を投与する工程を含む方法を提供する。
【0031】
本発明者らは、B.テタイオタオミクロンの抗炎症活性が、NF−κBの核外移行の促進とサイトゾル内でのPPARγ仲介による隔離とによって特徴づけられる全く新規な作用モードを伴うことを見出した。実験的検討(詳細は実施例に記載)は次の3段階で行った。
1)病原性/非病原性細菌への接触後のCaco−2細胞内での炎症性サイトカイン遺伝子の発現。データはcDNAマクロアレイ、リアルタイムPCR及びノーザンハイブリダイゼーション解析を用いて得た。
2)B.テタイオタオミクロンの抗炎症作用の生理学的妥当性のインビトロ(Caco−2トランスウェル培養)及びインビボ(最少量細菌叢ラット)での検証。
3)NF−κB及びAP−1シグナル変換経路の解析、並びにサイトゾル内でのRelAとPPARγとの関係の解析。
【0032】
実験的検討を行った結果、次の事項が示された。
1)B.テタイオタオミクロンによる調節の主なターゲットはNF−κBであり、AP−1ではない。
2)B.テタイオタオミクロン及び腸炎菌(S. enteritidis)で処理した細胞内にはp65(RelA)が蓄積する(最長30分間検出した)。
3)B.テタイオタオミクロン存在下でのp65の転写作用の低下は、p65の核外排除(移行)の促進に起因し、この核外移行はLMB感受性である(即ち、crm−1仲介による)。
4)データは全て、B.テタイオタオミクロンに起因する、炎症性サイトカイン/ケモカインの発現の低下、IκBαの発現、PMNの補充、及びラットにおいてインビボで示された生理的低レベルの炎症に関連付けられる。
5)B.テタイオタオミクロンの存在下でPPARγはサイトゾルに局在する。
6)PPARγとp65がサイトゾル内に共存する場合、両者は物理的に結合する。
【図面の簡単な説明】
【0033】
【図1】(a)1)非感染Caco−2細胞、2)108個の腸炎菌を感染させたCaco−2細胞、3)108個の腸炎菌と109個のB.テタイオタオミクロンを感染させたCaco−2細胞、及び4)109個のB.テタイオタオミクロンを感染させたCaco−2細胞のTNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたサイトカインmRNAのノーザンブロットハイブリダイゼーション。 (b)IL8及びG3PDHの半定量的PCR。(1)培地のみでインキュベートしたCaco−2細胞;(2)108個の腸炎菌、108個の大腸菌0157H7、PMA(300ng/ml)或いはIL−1α/β(20ng/ml)(図示の通り)と共にインキュベートしたCaco−2細胞;(3)109個のB.テタイオタオミクロンの存在下で上記(2)のようにインキュベートしたCaco−2細胞;(4)109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞。 (c)1)非感染Caco−2細胞、2)108個の腸炎菌を感染させたCaco−2細胞、3)108個の腸炎菌と109個のB.ブルガータスを感染させたCaco−2細胞、及び4)109個のB.ブルガータスを感染させたCaco−2細胞のTNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたmRNAのノーザンブロットハイブリダイゼーション。 (d)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にCaco−2細胞を2時間或いは4時間(図示の通り)インキュベートした。 (e)上記(a)の処理群(1)〜(4)のMPOアッセイにより測定した、Caco−2細胞単層を経由するPMN細胞の経上皮遊走。 (f)(1)対照、(2)108個の腸炎菌、(3)B.テタイオタオミクロンに次いで108個の腸炎菌、及び(4)B.テタイオタオミクロンのみでチャレンジしたラットの処理後6日目の回腸粘膜のMPOアッセイ。データは平均±SD(n=3)。
【図2】1)非感染Caco−2細胞、2)108個の腸炎菌と共にインキュベートしたCaco−2細胞、3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞、及び4)109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞。次の一次抗体を用いて試験した。抗RelA(A〜D)、抗PPARγ(E〜H)、抗IκBα(I〜L)及び抗pIκβα(M〜P)。スケールバーは全て25μm。O及びP中の挿入図は点状核標識の詳細を示す。
【図3】(A)図1Aで上述した群(1)〜(4)に関して、コンセンサス32P標識NF−κB結合配列オリゴヌクレオチドを用い、抗RelA抗体と共にインキュベートしたCaco−2核抽出物について行ったスーパーシフトEMSA。ここで、上記B.テタイオタオミクロンは70℃、15分で熱失活させた。 (B)上記実験処理群(1)〜(4)のmRNAのノーザンハイブリダイゼーション。ブロットは特異的32P標識IκBα及びG3PDHプローブを用いて試験した。 (C)Caco−2細胞由来の核抽出物のウェスタンブロット。処理群は上述の通りである(1〜4)。免疫ブロットは、抗p38及び抗pp38(ニューイングリッシュバイオラボ)(New English Biolabs)に特異的な抗体を用いて試験した。 (D)c−Fos及びc−Junの超誘発は、Caco−2細胞から得た全RNAについてのノーザンハイブリダイゼーションによって測定した。Caco−2細胞の処理はシクロヘキサミドの非存在下(−C)或いはシクロヘキサミド(10μg/ml)の存在下で上述のように(1〜4)行った。ブロットは、c−Fos、c−Jun及びG3PDHに特異的な32P標識プローブでハイブリダイズした。 (E、F)Caco−2細胞由来の核抽出物のウェスタンブロット。処理群は上述の通りである(1〜4)。c−Fos(サンタクルズ)、ATF−2及びpATF−2(ニューイングランドバイオラボ)(New England Biolabs)に特異的な抗体を用いた。 (G)IκBα及びpIκBαを用いた免疫沈降(IP)。Caco−2細胞は標準培養プロトコルに従い調製した。処理群(1〜4)。IPはウェスタンブロッティングで解析した。NSは非特異的であることを示し、Blは細胞抽出物を含まない対照である。
【図4】(A)特異的32P標識PPARγ及びPPARαプローブで解析した、(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートした後のCaco−2細胞から得たmRNAのノーザンハイブリダイゼーション。細菌の添加は2時間行った。 (B)処理(1)及び(2)は上述の通りであり、更に(3a)30μMの15PG−J2の存在下で108個の腸炎菌と共にインキュベートしたCaco−2細胞、及び(3b)30μMのフェノフィブラートの存在下で108個の腸炎菌と共にインキュベートしたCaco−2細胞を用いた(図示の通り)。細菌及びこれら薬剤の添加は2時間行った。32P標識PPARγ特異的プローブを用いてノーザンハイブリダイゼーションによりmRNAを解析した。 (C)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)(4)(5)10、20或いは30μMの15PG−J2の存在下で108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(6)30μMの15PG−J2のみと共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (D)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(3)(4)10或いは30μMのシグリタゾンの存在下で108個の腸炎菌と共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (E)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(3)108個の腸炎菌及び30μMのフェノフィブラートと共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (F)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートした後のCaco−2細胞から得た核抽出物及び細胞質抽出物を抗PPARγ(サンタクルズ)を用いたウェスタンブロッティングで解析した。細菌は2時間適用した。Caco−2細胞は核画分、細胞質画分、洗浄剤可溶画分(D.可溶)或いは洗浄剤不溶画分(D.不溶)として調製した。 (G)108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間共培養したCaco−2細胞を1%ノニデットP40溶液含有PBSに溶解し、ポリクローナル抗RelAセファロース(サンタクルズ)と共に一晩インキュベートした。免疫沈降物をSDS−PAGEを用いて分離した。モノクローナル抗RelA[NF−κB p65]、抗PPARγ抗体及び抗HDAC3(サンタクルズ)を用いてウェスタンブロットを展開した。 (H)PPARγ(f〜m、o)及びRelA(n)の免疫蛍光顕微鏡検査。プレートn及びo(p)からPPARγとRelAとの共存を確認。培地のみで(t、j)、108個の腸炎菌と共に(g、k)、108個の腸炎菌及び109個のB.テタイオタオミクロンと共に(h、l、n〜p)、或いは109個のB.テタイオタオミクロンと共に(i、m)Caco−2細胞を増殖させた。4μMのTSAの非存在下(t〜i、n〜p)及び存在下(j〜m)で処理を行った。スケールバーは全て25μm。
【図5】PPARγの変異はB.テタイオタオミクロン仲介によるNF−κBの抑制及びRelAのサイトゾル内隔離を妨害する。 (A)RelAの免疫検出。Caco−2細胞は標準培養プロトコルに従い調製した。処理群(1〜4)。免疫沈降物は抗PPARγセファロース複合体を用いて調製した。 (B)RelA/PPARγ結合を示すインビトロ翻訳 (C)PPARγDNとNF−κBルシフェラーゼレポーター構造体をトランスフェクトしたCaco−2細胞(DN)と、NF−κBルシフェラーゼレポーターのみをトランスフェクトした対照細胞(MOCK)とを比較した。(1)非感染Caco−2細胞、(2)108個の腸炎菌と共に更に6時間インキュベートした後のCaco−2細胞、及び(3)109個のB.テタイオタオミクロンの存在下で108個の腸炎菌と共に更に6時間インキュベートした後のCaco−2細胞において、ルシフェラーゼ活性(%刺激で示す)を測定した。 (D)構成的に発現する緑色蛍光タンパク質(GFP)構造体をトランスフェクトしたCaco−2細胞(B、C)及びPPARγDNとGFPをトランスフェクトしたCaco−2細胞(D、E)。細胞は全て108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。次いで、細胞を抗RelA(p65)特異的抗体(サンタクルズ)で免疫染色し、LSCMで試験した。(B)及び(D)はGFP(緑)とRelA(赤)の複合画像を示す2チャンネルキャプチャである。(B)及び(D)はそれぞれ(B)及び(D)と面積が同じであり、RelAのみを示す1チャンネルキャプチャである。スケールバー=25μM。 (E)カルボキシ末端に黄色蛍光タンパク質(YFP)を有するRelAキメラ構造体とPPARγ(d〜f)或いはPPARγDN(g〜i)とをCaco−2細胞にトランスフェクトした。PPARγ及びPPARγDNはいずれもカルボキシ末端にシアン蛍光タンパク質(CFP)を有するキメラ構造体である。トランスフェクション後2日目に、細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。CFP蛍光(d、g)及びYFP蛍光(e、h)。CFP−PPARγとYFP−RelAとの共存をプレートd及びe(f)から確認し、CFP−PPARγ(DN)とYFP−RelAとの共存をプレートg及びh(i)から確認した。スケールバーは全て25μm。(j、k)CFP−PPARγをHela細胞にトランスフェクトし、トランスフェクション後2日目に、細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。Hela細胞を固定し、透過処理し、免疫染色してSC35を確認した(immuno-stained for SC35)。CFP−PPARγ(j)及びSC35の間接免疫蛍光(k)。スケールバー=10μm。
【図6】図6は、対照ラット、バクテロイデス・テタイオタオミクロンを経口投与したラット(BT)、腸炎菌を経口投与したラット(SE)、及び腸炎菌とB.テタイオタオミクロンとを経口投与したラット(SE+BT)での骨格筋(□:湿重量、■:乾燥重量)の成長(mg/ラット/日)を示す。*は、乾燥重量及び湿重量それぞれについて対照、BT及びSE+BTと有意差があることを示す(p≦0.05)。骨格筋を腓腹筋で代表させたが、若いHooded−Listerラットの全骨格筋重量は腓腹筋重量の約47倍である(バルドクス(Bardocz)ら、1996年)。腓腹筋の初期重量は、初期新鮮体重100g当り湿重量で804±10mg[乾燥重量で194±7mg]。
【図7】図7は、ケモカイン/サイトカインmRNA発現への作用を示す定量的データ(ノーザンブロット及びリアルタイムPCR)を示す。S.e.=腸炎菌。B.t.=B.テタイオタオミクロン。
【図8】図8は、Caco−2細胞単層と共に2時間インキュベートした後の細菌の数を示す。 a)B.tの存在下及び非存在下でCaco−2細胞に付随するB.t(細胞単層洗浄後に残ったB.t)。 b)B.tの存在下及び非存在下でCaco−2細胞に付随するS.e。 c)B.tの存在下及び非存在下でCaco−2細胞に浸潤した腸炎菌(細胞洗浄及びゲンタマイシン処理(100μg/ml、4時間)後に残った腸炎菌)。n=6、+/−標準偏差。
【図9A】:EMSAを用いて測定した、Caco−2細胞における腸炎菌に呼応するNF−κB活性化を示す。
【図9B】:ノーザンハイブリダイゼーションによるサイトカイン反応の解析 a)108及び1010cfuのS.eに2時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。 b)109個のS.eに各時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。2時間或いは4時間まで接触させた際のデータも示す(図9D)。 c)107〜1010個のB.t.に2時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。 d)Caco−2細胞から得たmRNAを解析して炎症性サイトカインの発現(COX−2及びIL−8)を確認した。2時間及び4時間でのB.t.の抑制作用を示す。
【図9C】:免疫細胞化学的検出法を用いて得た、腸炎菌及びB.テタイオタオミクロンにCaco−2細胞を接触させた後のRel(p65)活性化の経時データ。S.e.+/−B.t.に2時間接触させたCaco−2細胞を4%パラホルムアルデヒドに固定し、Triton X−100で透過処理し、抗p65(RelA)(サンタクルズ)で免疫標識した。二次検出はAlexa Fluor488抗ウサギIgG(モレキュラープローブ)を用いて行った。Axiovert2000顕微鏡に搭載したZeiss Axiocamを用いて画像をデジタル記録した。
【図9D】:免疫細胞化学的検出法を用いて得た、腸炎菌とB.テタイオタオミクロンにCaco−2細胞を接触させた後のPPARγ作用の経時データ。 S.e.+/−B.t.に2時間接触させたCaco−2細胞を4%パラホルムアルデヒドに固定し、のTriton X−100で透過処理し、抗PPARγ(サンタクルズ)で免疫標識した。二次検出はAlexa Fluor568抗ヤギIgG(モレキュラープローブ)を用いて行った。Axiovert200顕微鏡に搭載したZeiss Axiocamを用いて画像をデジタル記録した。
【図9E】:B.テタイオタオミクロンと腸炎菌に2時間接触させた後のCaco−2細胞におけるNF−κB p65とPPARγとの共標識化(co-labelling)を示す。
【図10】対照ラット、B.テタイオタオミクロンを経口投与したラット[B.t]、腸炎菌を経口投与したラット[S.e]、及び腸炎菌とB.テタイオタオミクロンとを経口投与したラット[S.e+B.t]での骨格筋(腓腹筋)(□:湿重量、■:乾燥重量)の増量(mg/ラット/日)。n=6、+/−標準誤差。
【図11】SDS−PAGEを用いて分離し、エレクトロブロットし、アセチル化リジンモノクローナル抗体で免疫染色したp65免疫沈降物を示す。
【発明を実施するための形態】
【0034】
以下、非限定的な実施例と図面とを参照し本発明について更に説明する。
【0035】
実施例1:B.テタイオタオミクロンとB.ブルガ−タス(B. vulgatus)の存在下及び非存在下で腸炎菌に接触したCaco−2細胞の炎症反応
【0036】
細菌による宿主炎症反応の調節についての実証データは、バクテロイデス・テタイオタオミクロンの存在下及び非存在下で共培養した腸炎菌にCaco−2細胞を短時間接触させた後に炎症性遺伝子の発現を検討した結果から得た。cDNAマクロアレイ技法(クロンテック、Atlasヒトサイトカイン/受容体アレイシステム)を用いて、TNF−α、IL−8、MIP−2α及びCOX−2を含む数種類の遺伝子を同定した。腸炎菌との接触後のこれら遺伝子の誘発はB.テタイオタオミクロンの存在により低下した。この結果はノーザンハイブリダイゼーション及びリアルタイムPCRを用いて確認された(図1a、f)。また、IL−1α、IL1−β、TNF−α、PMA、LPS及び腸出血性大腸菌0157H7(図1F)を含む他の炎症性メディエータに対するB.テタイオタオミクロンの抗炎症活性についても検討した。Caco−2細胞内でIL−8発現を誘発したこれらリガンドの内、PMA、腸炎菌及び大腸菌0157H7の作用のみがB.テタイオタオミクロンによって低下した。
【0037】
上記検討に用いる実験条件の設定に当り、多くの最適化実験(時間経過、細菌量/増殖相、Caco−2細胞継代/コンフルエンス検討を含む)を行った。重要な点は、細菌の増殖、付着及び浸潤が培養/共培養条件に影響を受けないことを立証し、これによってデータが特異的付着/浸潤に起因する可能性を排除したことである(図8参照)。
【0038】
(1)非感染Caco−2細胞、(2)108個の腸炎菌のみと共にインキュベートしたCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートしたCaco−2細胞において炎症性遺伝子の発現を測定した。細菌の添加は2時間行った。細胞を洗浄し回収してmRNAを単離した。mRNA(5μg)を、TNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたノーザンハイブリダイゼーションによって解析した。
【0039】
B.テタイオタオミクロンの作用を示す結果の一部は、フーパー(Hooper)ら(2001年)によって報告された最近の分子解析と一致する。本発明者らによる結果は、ノーザンハイブリダイゼーション及びリアルタイムPCRを用いて確認された。TNF−α、IL−8、MIP−2α及びCOX−2のレベルは、腸炎菌チャレンジに呼応して有意に上昇した(図1a)。しかし、B.テタイオタオミクロンの存在下では、TFG−βの場合を除き、これら遺伝子の全てにおいて発現レベルが低下した。TFG−βの発現レベルは腸炎菌によって低下したが、B.テタイオタオミクロンの存在により対照値に維持された。これら両方の細菌の生残性及び増殖速度、並びに上皮細胞に付着し浸潤した腸炎菌の数については上述の処理の間で差がなく、培養手順による影響を受けなかった。
【0040】
上記結果を立証する定量的データを図7に示すが、このデータは、ビヒクル(対照)、5×108個の腸炎菌(S.e)、5×108個のS.e及び1.5×109個のB.テタイオタオミクロン(B.t)、或いはB.tのみと共に2時間インキュベートした後のCaco−2細胞から精製したmRNAを用いて得た。ノーザンブロットをデンシトメトリーによって定量化し、G3PDHレベルに対して規準化した。また、ABI Taqmanを用いてリアルタイムPCRを行い、18SrRNAレベルに対して規準化した(n=4 +/−標準偏差)。
【0041】
炎症性サイトカイン発現の低下がB.テタイオタオミクロンに特異的なものかどうかを調べるため、関連する耐気性菌株であるB.ブルガ−タスについても検討した。
【0042】
(1)と(2)については上述の様に処理を行い、更に、(3a)108個の腸炎菌及び109個のB.ブルガ−タスと共にインキュベートしたCaco−2細胞、及び(4a)109個のB.ブルガ−タスのみと共にインキュベートしたCaco−2細胞を用いた。細菌の添加は2時間行った。
【0043】
B.ブルガ−タスは抗炎症活性について陰性であった(図1b参照)。
【0044】
得られた結果において、上記共生菌株の両方によって誘発される炎症性遺伝子の発現が低かったことは、対数期後期の細菌を試験した事実に関連するかもしれないが、注目に値する。細菌細胞片を有しない対数期初期の細菌を用いれば、炎症反応を完全に抑制することができるであろう。従って、Caco−2細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間或いは4時間インキュベートする実験を更に行った。IL−8発現に対するB.テタイオタオミクロンの阻害作用は長時間持続した(図1d)が、TNF−αの場合はこれとは対照的であった。この結果から、上記遺伝子の発現を調節するメカニズムはB.テタイオタオミクロンによって特異的に影響を受けることが示唆される。
【0045】
B.テタイオタオミクロンによる炎症性サイトカインの抑制を示すデータの生理学的妥当性を、PMN補充の機能性インビトロモデル及びインビボ腸炎菌ラット感染モデルを用いて確認した。インビトロ及びインビボでのPMN補充はミエロペルオキシダーゼ(MPO)活性を用いてモニターした。
【0046】
Caco−2細胞をインバーテッドトランスウェル(コーニング)に播種し、様々な組合せの細菌を該細胞の先端表面に添加した。新たに単離したヒトPMNを該細胞の側底区画に添加し、該PMNの経上皮遊走をMPOアッセイ(ネイシュ(Neish)ら、2000年)で測定した。処理群(1〜4)については上述の通りである。細胞を2時間インキュベートした後、細菌を除去し新しい培地を添加した。細胞を更に2時間インキュベートした後、HBSSで洗浄した。細胞と先端区画から得た培地とを1%のTriton C−100に可溶化し、MPOを定量した。
【0047】
多形核白血球(PMN)補充アッセイのインビトロモデルから得た結果から、B.テタイオタオミクロンの抗炎症活性が確認された(図1e)。
【0048】
ラットの腸の腸炎菌感染が主に回腸、即ち、B.テタイオタオミクロンの棲息部位で起こることは注目に値する。
【0049】
離乳したばかりの(21日齢)最少量細菌叢ラット(通常の実験飼料で飼育)を2つの群に分け、一方のラット群を更に108cfuのB.テタイオタオミクロンを含む嫌気的に調製したゼリー(0.5g/日)で19日間飼育した。各群のラットの半数に対して108個の腸炎菌で経口チャレンジを行った。全てのラットにおける炎症反応の重症度を、腸炎菌感染後6日目に回腸粘膜内のMPOレベルを測定することによって評価した。ラットに対して行った処理は次の通りである。(1)B.テタイオタオミクロン投与無し、腸炎菌投与無し、(2)B.テタイオタオミクロン投与無し、108個の腸炎菌投与、(3)B.テタイオタオミクロン投与に次いで108個の腸炎菌投与、及び(4)B.テタイオタオミクロンのみ投与。実験は少なくとも3回行ったが、いずれも同様の結果が得られた。
【0050】
腸炎菌でチャレンジしたラットの回腸粘膜内のMPOレベルは上昇した(p<0.005)が、B.テタイオタオミクロンの先行経口接種及び細菌叢内での安定化によりMPOレベルは有意に低下した(p<0.001)(図1f)。また、PMN補充実験から、腸炎菌によって400pg/mlのIL−8タンパク質が培養上清内に4時間に亘り誘導されるが、B.テタイオタオミクロンの存在下で共培養した場合、IL−8タンパク質濃度は230pg/mlに有意に低下することを見出した。B.テタイオタオミクロンによるコロニー形成は、腸組織の特異的プライマー増幅によって確認した。IL−8およびMIP−2αはPMN経上皮遊走に対する必須ケモカインである(マコーミック(McCormick)ら、1993年;ハング(Hang)ら、1999年)ため、B.テタイオタオミクロンの作用は、IL−8及びMIP−2αの転写の減少に起因し、この転写減少は次いで活性タンパク質の翻訳及び分泌に影響を及ぼす。ネイシュ(Neish)ら(2000年)の記載と一致するが、本発明者らは、遺伝子発現を調節する宿主系を破壊する(subverting)ことにより非病原性細菌が免疫抑制作用を発揮し得ることを新規なモードシステムを用いて立証した。重要なのは、本発明者らによる知見がインビボでも検証されたことである。
【0051】
上述したMPOデータに加えて、図10は、腸炎菌によって誘発される炎症反応時の筋細胞(muscle biology)に対するB.テタイオタオミクロンの防御作用の実証データを示す。
【0052】
実施例2:B.テタイオタオミクロンは、NF−κB及びPPARγタンパク質の細胞内分布と活性化状態を変化させることによって炎症を軽減し、また、NF−κBシグナル変換経路を標的破壊する。
【0053】
転写因子のNF−κBファミリーは炎症反応の調節において中心的役割を果たす。これらのタンパク質は、Rel相同ドメインと称される高いNH2末端保存配列を共有するが、この配列は、タンパク質のサブユニット二量体化、DNA結合、及び阻害性IκBタンパク質との相互作用に必要である。NF−κBを誘発するシグナルは阻害性IκBタンパク質(Ser−32及びSer−36ではIκBα)のリン酸化を引き起こし、次いでユビキチン化とプロテオソーム仲介による分解とをターゲットとする。これによって、RelA(p65)等のRelタンパク質は遊離し、核に転位しDNAを結合する。
【0054】
非感染Caco−2細胞(1)を108個の腸炎菌のみ(2)、108個の腸炎菌及び109個のB.テタイオタオミクロン(3)、及び109個のB.テタイオタオミクロンのみ(4)と共にそれぞれ2時間インキュベートした。細胞を4%パラホルムアルデヒドに室温で30分間固定し、0.2%のTriton−X1000を用いて4℃で透過処理し、間接免疫蛍光顕微鏡検査法にて試験した。一次抗体(サンタクルズ):抗RelA[NF−κB p65](A〜D)、抗PPARγ(E〜H)、抗IκBα(I〜L)及び抗pκBα(M〜P)。二次抗体:Alexa Fluor種特異的抗IgG(モレキュラープローブ)。
【0055】
Caco−2細胞を腸炎菌に接触させることによって一連のイベントが引き起こされ、その結果、RelAの核への転位が促進されることが判明した(図2B)。
【0056】
特筆べき点として、B.テタイオタオミクロンによってRelAの核への転位が無くなることが見出されたが、B.テタイオタオミクロンへ2時間接触させた後、ほぼ全てのRelAはサイトゾルに局在した(図2C、D)。
【0057】
IκBαのリン酸化と、重要なこととしてのIκBαの分解は、B.テタイオタオミクロンの存在下及び非存在下での腸炎菌への接触後に観察された(図2G)。2時間の接触後、特に腸炎菌のみに接触させた細胞と腸炎菌及びB.テタイオタオミクロンに接触させた細胞において、IκBαのmRNA(図3B)及びIκBαタンパク質(図2J、K)のレベルが上昇したが、このことは転写活性NF−κBの存在を示している(チェン(Cheng)ら、1994年;チャオ(Chiao)ら、1994年)。
【0058】
生残B.テタイオタオミクロンは、NF−κBタンパク質の転写活性を選択的に妨害するが、AP−1タンパク質の合成やリン酸化には影響を及ぼさない。
【0059】
(1)非感染Caco−2細胞、(2)108個の腸炎菌と共に2時間インキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間インキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共に2時間インキュベートしたCaco−2細胞から得た核抽出物を、RelA[NF−κB p65]特異的抗体(サンタクルズ)と共にインキュベートし、この抽出物に対してスーパーシフトEMSAを行った。上記B.テタイオタオミクロンはCaco−2細胞へ添加する前に70℃、15分で熱失活させた。
【0060】
電気泳動移動度シフトアッセイ(EMSA)によって、RelAが腸炎菌により活性化される主要なサブユニットであることが確認された(図3A)が、p52の活性化レベルが低いことも明らかになった(データは図示せず)。次に、本発明者らは、B.テタイオタオミクロンがNF−κBシグナル変換経路を破壊することによって、腸炎菌により引き起こされる免疫反応メディエータの産生を低下させるという仮説を立てた。
【0061】
同様の実験を行った結果を図9Aに示すが、Caco−2細胞内で腸炎菌に接触後2時間でp65の活性化はピークに達することがEMSAスーパーシフトにより示された。レーン1:タンパク質無し、レーン2、5、8、11:対照細胞から得た核抽出物、レーン3、6、9、12:S.e.を感染させた細胞から得た核抽出物、レーン4、7、10、13:S.e.を感染させp65(RelA)抗体でスーパーシフトさせた細胞から得た核抽出物。
【0062】
更に、熱失活していない生残B.テタイオタオミクロンによってRelA反応が阻害されることが示された(図3A)。また、培養上清及び調製培地(B.テタイオタオミクロンに接触させたCaco−2細胞から得た培地)のいずれも生物活性を有しないことも示された(データは図示せず)。このことから、上皮細胞への接触がB.テタイオタオミクロンの抗炎症活性には必須であることが示唆される。腸炎菌に接触させてから20分以内に、B.テタイオタオミクロンの存在下及び非存在下でIκBαのリン酸化及び分解を観察した(データは図示せず)。また、検討対象細菌株の生残性、増殖、付着及び浸潤は培養処理による影響を受けなかった(データは図示せず)。従って、上記観察された作用は受容体認識及び活性化の差には起因しないと考えられる。
【0063】
上記と同じ実験群(1〜4)を用いて、細菌の添加を2時間行った後、mRNAをノーザンハイブリダイゼーションで解析し、ブロットを特異的32P標識IκBα及びG3PDHプローブを用いて試験した。IκBαのmRNA(図3B)及びIκBαタンパク質(図2J、K)のレベルはいずれも上昇した。このレベルの上昇は、IκBαタンパク質の新生合成を誘発するRelAの転写活性に起因するものとされる(チェン(Cheng)ら、1994年)。IκBαは次いで核に侵入しRelAと結合し、RelAをDNAプロモーター部位から除去する(アレンザナ−セイスデドス(Arenzana- Seisdedos)ら、1995年)。興味深いことに、PPARα仲介によるIκBα合成の刺激についても報告されている(デレライブ(Delerive)ら、2000年)。一過性のRelA転位がB.テタイオタオミクロンで処理した細胞内で起こるかどうかを更に調べるため、CRM−1依存型核外移行の特異的阻害剤であるレプトマイシンB(LMB)を用いて実験を行った。Relタンパク質(例えば、p65)の細胞質内での位置はRelA/IκBα複合体のCRM−1依存型核外移行によって維持される(ファン(Huang)ら、2000年;タム(Tam)ら、2000年)というのが理論的根拠である。LMBによってRelAが腸炎菌及びB.テタイオタオミクロンの両方と共培養した細胞の核内に蓄積することを見出したが(結果は図示せず)、このことは、B.テタイオタオミクロンの存在に関係なく、核への一過性RelA転位が起こることを示している。この結果は、腸細胞を細菌へ初期接触させた後のサイトゾル内のIκBα/RelA複合体のリン酸化及びユビキチン化、並びにIκBα遺伝子発現の活性化と一致する。
【0064】
実施例3:B.テタイオタオミクロンはPPARγの核−細胞質間輸送を誘発させ、RelAを隔離する。
【0065】
非病原性細菌による免疫抑制についての最近の報告によると、その抑制メカニズムはIκBαのユビキチン化及び分解の阻害に起因するものとされている(ネイシュ(Neish)ら、2000年)が、このことは本発明者らによるモデル系には明らかに適用されない。腸炎菌及びB.テタイオタオミクロンの両方と共培養した細胞におけるNF−κB複合体のサイトゾル内での主な位置は、IκBα仲介による核外移行が核内局在化よりも効率よく起こることから説明されるであろう。腸炎菌及びB.テタイオタオミクロンの両方で処理した細胞の核内でのIκBαタンパク質のレベルは、腸炎菌のみで処理した場合に比べて若干高いが、このことはNF−κBの不活化を促進し、B.テタイオタオミクロンの全抗炎症特性に寄与する可能性がある。しかし、RelAの特異的脱アセチル化がより適切な説明となる。核内RelA活性化の継続時間が可逆的アセチル化によって測定されることが最近報告されている(チェン(Chen)ら、2001年)。アセチル化されたRelAはIκBαに対する親和性が低いが、ヒストンデアセチラーゼ3(HDAC3)でRelAを脱アセチル化することにより、IκBαへの結合が促進され、CRM−1仲介によるRelAの核外移行が促進される。本発明者らは、B.テタイオタオミクロンがHDAC3のRelAとの結合を促進することを示した(図4G)。ヒストンデアセチラーゼの特異的阻害剤であるトリコスタチンA(TSA)を用いた実験を行った。その結果、TSA処理(800nM、4時間)に呼応してHDACが阻害され、腸炎菌のみで処理した細胞においてp65が部分的に蓄積し、重要なこととしてPPARγのサイトゾルへの移行が促進されることが示された。得られたデータから、B.テタイオタオミクロン処理細胞内でのp65及びPPARγの核外移行を促進する上でアセチル化/脱アセチル化が重要な役割を果たしていることが裏付けられた(図11参照)。
【0066】
更に、RelAとペルオキシソーム増殖因子によって活性化された受容体γ(PPARγ)タンパク質とが同じ細胞区画に共存することが分かった(図2C、G)ため、本発明者らは、B.テタイオタオミクロン処理細胞内での更なるRelA核内転位を制限する重要なメカニズムはPPARγによるRelAのサイトゾル内隔離を伴うという仮説を立てた。この仮説を裏付ける実験的証拠を以下に示す。
【0067】
AP−1複合体は炎症性遺伝子発現の調節において重要な役割を果たし、増殖因子やサイトカイン、細菌等の様々な細胞外刺激剤によって急速に活性化される(マイヤー−テア−フェーン(Meyer-ter-Vehn)ら、2000年)。本発明者らは、B.テタイオタオミクロンの阻害作用がこのシグナル伝達経路まで及ぶかどうかを調べた。AP−1活性は2つの主要なメカニズム、即ち、プロトオンコジーンファミリーのホモ及びヘテロ二量体から成るAP−1サブユニットの発現の上昇と該サブユニットのリン酸化とによって調節される。プロトオンコジーンファミリーとしては、Fos(c−Fos、FosB、Fra−1及びFra−2)、Jun(JunB、c−Jun及びJunD)及びATF(ATF2、ATF3/LRF2及びB−ATF)が挙げられ、メンバーは全てDNA結合タンパク質のロイシンジッパーファミリーである。これらのタンパク質は、マイトジェン活性化プロテイン(MAP)キナーゼと総称される3種類の関連キナーゼによって主に制御される。p42及びp44(細胞外シグナル調節キナーゼ;ERK)キナーゼ、c−JunN末端キナーゼ(JNK)/ストレス活性化プロテイン(SAP)キナーゼ及びp38キナーゼのタンパク質リン酸化のレベルが腸炎菌及びB.テタイオタオミクロンに呼応して変化するかどうかを調べた。腸炎菌によってp38MAPキナーゼは活性化されるが(図3C)ERKやJNKは試験時に活性化されない(結果は図示せず)ことを見出した。この知見は、p38が炎症性刺激剤によって急速に活性化され(レインギュード(Raingeaud)ら、1996年)、一方、JNKの活性化はもっと遅い時点で起こる(クジメ(Kujime)ら、2000年)という事実と一致する。p38の活性化によってelk−1のリン酸化が引き起こされ、elk−1は血清反応因子と共にc−fosプロモーター内の血清反応成分に結合し、c−fosの転写及び翻訳を促進するが、これは遺伝子及びタンパク質レベルの変化によって示される(図3D、E)。腸炎菌によって誘発されるp38のリン酸化はB.テタイオタオミクロンを用いた共培養によって変化しないことが見出した。メッセージレベル及びタンパク質レベルはいずれも腸炎菌及びB.テタイオタオミクロンに呼応して上昇し、c−fos遺伝子発現への作用は相加的なものであることが分かった。デノボc−Fox合成によって、10倍のDNA結合親和性を有するJun−Fosへテロ二量体が形成され、AP−1活性が増加する(ムスティ(Musti)ら、1997年;スミール(Smeal)ら、1991年)。ATF−2もp38MAPキナーゼ及びJNKシグナル変換経路の標的である。転写因子ATF−2はp38MAPキナーゼによりThr−69及びThr−71上でリン酸化される。一方、JNKはATF−2及びc−Junの両方をリン酸化し活性化する。従って、ATF−2とc−Junは違った形でp38及びJNKシグナル変換経路によって調節される(レインギュード(Raingeaud)ら、1996年)。本発明者らの検討においては、c−Junタンパク質レベルの上昇とc−Junリン酸化状態は腸炎菌及びB.テタイオタオミクロンのいずれにも呼応しなかったが、このことは、腸炎菌に呼応するATF−2のリン酸化(図3F)を誘発しやすいのはp38MAPキナーゼであってJNKではないことを示している。以上のデータはp38MAPキナーゼ活性に対する腸炎菌の直接の作用と一致する。腸炎菌に呼応してc−jun遺伝子転写を刺激するc−Jun/ATF−2ヘテロ二量体の形成はATF−2の活性化によって引き起こされやすい(図3D)。B.テタイオタオミクロンの抗炎症作用はNF−κB経路を選択的にターゲットとすることが分かるが、これにより、NF−κBに対して絶対条件を有する遺伝子(例えば、IL−8)(ムカイダ(Mukaida)ら、1994年;エリオット(Elliott)ら、2001年)がB.テタイオタオミクロンによる阻害に対し特に敏感である理由が説明される。
【0068】
実施例4
非病原性細菌による免疫抑制のメカニズムを更に調べるため、本発明者らは、先ず、抗炎症性サイトカインIL−10及びTGF−βについて検討した。IL−10遺伝子発現はB.テタイオタオミクロンを用いた処理には影響を受けなかった(データは図示せず)が、このことは構成タンパク質が関与する可能性を排除するわけではない。抗炎症性サイトカインTGF−βが関与する可能性を示すデータから多少の示唆が得られた。しかし、この短時間の検討の時間経過においてはデノボサイトカイン合成は恐らく十分に行われてないので、TGF−βが炎症反応をダウンレギュレートするように作用しているとすれば、TGF−βは長期の抗炎症作用に関与している可能性が高い。
【0069】
PPARが炎症プロセスの重要なモジュレータとして浮上してきた(ナカジマ(Nakajima)ら、2001年)。PPARはリガンド活性転写因子であり、ヘテロ二量体のパートナーとしてのレチノイドX受容体(RXR)と共に、PPAR反応成分(PPRE)と称される特異的DNA配列成分に結合し、遺伝子発現を調節する。しかし、最近の研究から、AP−1及びNF−κB仲介による遺伝子発現を調節する上でPPARγのリガンド活性化が重要であることが示唆された(スー(Su)ら、1999年)。本発明者らは、B.テタイオタオミクロンによるNF−κBの調節におけるPPARの役割について調べた。腸炎菌とB.テタイオタオミクロンの両方に接触させた細胞内でPPARα及びPPARγのmRNAが減少すること(図4A)、そして、特異的PPARγリガンドである15−デオキシΔ12,14−プロスタグランジンJ2(15d−PGJ2)に接触させた細胞内でもmRNAが減少する一方で、特異的PPARαリガンドであるフェノフィブラートに接触させた細胞内ではmRNAが減少しないこと(図5B)を示すことができたが、この知見は受容体活性化と一致する。従来、PPARγの活性化は、3T3−L1脂肪細胞におけるPPARγのmRNA及びタンパク質のダウンレギュレーションを伴うものとされていた(キャンプ(Camp)ら、1999年)。本発明者らは更に、15d−PGJ2及びシグリタゾンを用いて、生理学的と考えられる濃度範囲に亘って腸炎菌による炎症性サイトカイン発現の活性化が低下することを示した(図4C、D)。15d−PGJ2がIκBキナーゼを直接阻害できる(シュトラウス(Straus)ら、2000年)ものとして、PPARγの2種類のリガンドについて試験した。15d−PGJ2及びシグリタゾンのいずれもヒト腸上皮細胞におけるAP−1及びCOX−2誘発を阻害することが報告されており(サバラマイア(Subbaramaiah)ら、2001年)、また、これらは結腸炎の処置においても治療上の利点を有する(スー(Su)ら、1999年)。本発明者らは、PPARγ及びPPARαのリガンドが腸炎菌仲介によるサイトカイン誘発を低下させることができることを見出した(図4C、D、E)。この結果は、最近示されたPPARγアゴニストに関する報告、即ち、H.ピロリ(H. pylori)で処理した胃上皮細胞におけるNK−κB及びIL−8の発現がPPARγアゴニストによって低下すること(グプタ(Gupta)ら、2001年)、また、虚血再灌流誘発型腸障害におけるPMNの浸潤がPPARγアゴニストによって阻害されること(ナカジマ(Nakajima)ら、2001年)と一致する。
【0070】
PPARγ及びPPARαアゴニストの作用について述べてきたが、結腸粘膜ではPPARγの発現レベルがPPARαに比べて遥かに高いことが示されている(マンセン(Mansen)ら、1996年;ファジャス(Fajas)ら、1997年)ため、下の実験においてはPPARγに注目した。
【0071】
PPREを介して遺伝子転写を調節することに加え、最近では、PPARがDNA結合とは独立したメカニズムによって他の転写因子経路を妨害することにより遺伝子転写を阻害することが示された(デレライブ(Delerive)ら、1999年)。PPARγ2はサイトゾル区画及び核区画で見出される(スイリアー(Thuillier)ら、1998年)が、サイトゾルPPARγの生理学的妥当性については現在知られていない。本発明者らは、固定したCaco−2細胞に対して免疫細胞化学的局在化技法を用い、腸炎菌がPPARγの核内蓄積を誘発することを見出した(図2F)。PPARγのNH2末端ドメイン(Ser−122)のMAPキナーゼによるリン酸化によって、リガンド結合親和性が低下し、PPARγの転写及び生物学的機能が負に調節される(シャオ(Shao)ら、1998年)ことについて言及することは重要である。これによって、腸炎菌に対する炎症反応の初期段階で、PPARγの核内蓄積が炎症性遺伝子転写の抑制に対し効果的でない理由が説明される。腸炎菌をB.テタイオタオミクロンと共培養した後、PPARγはサイトゾルへ再分布された(図2G、H)。B.テタイオタオミクロンの存在下で腸炎菌に接触させたCaco−2細胞内のPPARγの特異的分布についても、核抽出物及び細胞質抽出物を用いたウェスタンブロッティングによって示された(図4F)。
【0072】
PPARγに関して公表されたデータは全て、PPARγの核内作用部位に注目している。本発明者らは、免疫細胞化学的技法を用いて、腸炎菌がCaco−2細胞におけるPPARγの核内蓄積を誘発することを見出した(図4H(g))。一方、腸炎菌とB.テタイオタオミクロンを用いて共培養した後、PPARγはサイトゾルへ再分布された(図4H(h、i))。経時的検討によって、この再分布がこれらの細菌への接触後60分で明らかになり、2時間経過するまでにはほぼ完了することが示された(結果は図示せず)。腸炎菌とB.テタイオタオミクロンとに接触させたCaco−2細胞内のPPARγの特異的分布についても、核抽出物及び細胞質抽出物を用いたウェスタンブロッティングによって示された(図4F)。
【0073】
腸炎菌で誘発されたPPARγタンパク質はRXRαとヘテロ二量化複合体を形成し(IPで実証済み、結果は図示せず)、洗浄剤不溶性細胞画分へと分配された。一方、B.テタイオタオミクロンで誘発されたPPARγタンパク質は洗浄剤可溶性であった(図4F)。腸炎菌とB.テタイオタオミクロンの共培養によって誘発されたPPARγの核−細胞質間輸送は、レプトマイシンB(LMB)処理ではブロックされず(結果は図示せず)、従って、PPARγの核−細胞質間輸送は、核外移行受容体crm−1(この受容体は他の核内受容体と類似する)によって促進されなかった(ブン(Bunn)ら、2001年)。しかし、冷却を行うこと及び代謝阻害剤を用いることによって、核外移行は有意に減少した(結果は図示せず)。 TSA(ヒストンデアセチラーゼ阻害剤)(図4H(j〜m)やSB(p38MAPキナーゼ阻害剤)(データは図示せず)等の他の生物学的阻害剤を腸炎菌で処理したCaco−2細胞に添加した。これらの阻害剤が点状サイトゾル標識化及びPPARγの核外移行を誘発することが示された(図4H(k))が、この作用はB.テタイオタオミクロンの作用と類似する。p38を含むAP−1シグナル伝達経路はB.テタイオタオミクロンによって阻害されなかった(結果は図示せず)が、このことはアセチル化/脱アセチル化反応がPPARγ核外移行メカニズムに潜在的に関連していることを示す。
【0074】
また、PPARγ及びPPARαリガンドは腸炎菌仲介によるサイトカイン誘発を抑制する(結果は図示せず)が、上記実験で試験したPPARリガンドの作用はB.テタイオタオミクロンのPPARγの細胞内再局在化に対する作用とは異なる。このことは、PPARγを調節する新規な内因性リガンドやメカニズムが存在することを示唆する。
【0075】
本発明者らは、二重標識免疫細胞化学的技法を用いて、PPARγタンパク質の多くがRelAと共存することを見出した(図4H(n〜p))。PPARγとRelAとの物理的結合が、腸炎菌及びB.テタイオタオミクロンに接触させた腸細胞における細胞質内局在化を促進する重要な因子であるという仮説を立てた。
【0076】
核内PPARγとNF−κBが不活性な複合体を形成し得ることが示唆されている(リコテ(Ricote)ら、1999年)。更に、グルタチオンSトランスフェラーゼプルダウン実験により、PPARαがc−Jun及びRelA p65と物理的に相互作用することが示されている(デレライブ(Delerive)ら、1999年)。本発明者らの検討においては、免疫精製(IP)プロトコルを用い、B.テタイオタオミクロン存在下、腸炎菌で処理した細胞からPPARγを単離することによってRelAが共精製される(図4G)ことが示されたが、この結果は、PPARγとRelAが物理的に結合し、層状多タンパク質複合体を構成している可能性を示唆する。PPARγとRelAとの直接相互作用についてはインビトロ翻訳及びIPによって確認した(図4b)。
【0077】
実施例5
RelAの調節におけるPPARγの重要性を更に調べるため、PPARγのドミナントネガティブ(DN)型(英国ケンブリッジ大学チャタージー(Chatterjee)教授の寄贈)(ガーネル(Gurnell)ら、2000年)を用いた。PPAR受容体内ではロイシンとグルタミン酸の保存性が高い。これらのアミノ酸残基はリガンド結合及び核コアクチベーターの補充において必須である。これらのアミノ酸残基の変異によって上記受容体のDN型が産生し、この受容体が持つコアクチベーターを補充する能力と2種類のコリプレッサー、即ち、レチノイド甲状腺受容体のサイレンシングメディエータ(SMRT)と核コリプレッサー(NcoR)を遊離する能力が低下する(ガーネル(Gurnell)ら、2000年)。DN PPARγを用いた免疫共沈降実験によって、SMRTがPPARγとインビボで相互作用すること、そして、変異したPPARγは強力な転写リプレッサーとなることが示された。ヒトPPARγのキメラ蛍光タンパク構造体と、シアン蛍光タンパク質(CFP)をカルボキシ末端ドメインに有するPPARγのドミナントネガティブ型とを先に報告されたクローン(ガーネル(Gurnell)ら、2000年)からPCR増幅によって調製した。YFPと結合したキメラRelAは、J シュミット博士(Dr. J Schmid)から寄贈された(シュミット(Schmid)ら、2000年)。発現の成功は、抗ヒトPPARγ及び抗ヒトRelA抗体を用いた一過性トランスフェクトHela細胞のウェスタンブロット解析によって確認した。
【0078】
PPARγ DN及びNF−κBルシフェラーゼレポーター(パスツール研究所(フランス、パリ)イスラエル博士(Dr. Israel)の寄贈)を用いた二重トランスフェクションについて検討した。本発明者らは、病原性腸炎菌に長時間接触させた細胞によってルシフェラーゼ活性が失活することを見出したが、この知見は最近の報告(サフコヴィック(Savkovic)ら、2000年)と一致する。この現象を回避するため、上記細胞を2時間刺激し、洗浄によって腸炎菌を除去し、8時間後にルシフェラーゼタンパク質の産生を測定した。対照細胞及びPPARγ DNをトランスフェクトした細胞において、腸炎菌によりNF−κB活性化とルシフェラーゼタンパク質合成が誘発されることを見出した(P<0.001)(図5C)。PPARγ野生型(NT)をトランスフェクトした細胞(ルシフェラーゼのみ)においては、上記反応はB.テタイオタオミクロンによって低下した(P<0.028)が、PPARγドミナントネガティブ型(DN)をトランスフェクトした細胞においてはこのような反応の低下は見られなかった(図5C)。同様に、PPARγ DNの存在下及び非存在下で緑色蛍光タンパク質(GFP)をトランスフェクトした細胞においては、免疫細胞化学的技法及びレーザー走査型共焦点顕微鏡検査法(LSCM)で測定されたように、RelAの細胞内分布が変化した。対照細胞(GFPのみ)においては、トランスフェクトしていない細胞について上述した様に、RelAは主にサイトゾルに分布していた(図5D(挿入図B、C))。PPARγ DNをトランスフェクトした細胞においては、RelA分布は一定ではなく、RelAはサイトゾル区画及び核区画の両方に局在化していた(図5D(挿入図D、E))。以上のデータから、RelAを隔離し核内転位を防ぐ上でPPARγが必須であることが示された。
【0079】
本発明者らは更に、蛍光顕微鏡検査法を用いて、WT及びDN PPARγのキメラ構造体、CFP、キメラYFP、及びRelAをトランスフェクトしたCaco−2細胞とHela細胞とにおけるPPARγ及びRelAの細胞内分布について調べた。発現したPPARγ/RelAタンパク質の多くは、既に報告されているように(シュミット(Schmid)ら、2000年)、トランスフェクトした細胞の核内に局在した。同様に、PPARγ WTとRelAとをトランスフェクトした細胞においても、発現は主に核内で起こったが、B.テタイオタオミクロンを用いたインキュベーションの後、点状サイトゾル標識化及びPPARγとRelAとの共存が非常に明確に示された(図5E(f))。しかし、PPARγ DNをトランスフェクトした細胞において、RelAはサイトゾル区画には存在しなかった(図5E(i))。以上の結果から、DN PPARγの核外移行(即ちRelAとの共存)が阻害されたことが示され、よって、腸細胞内でB.テタイオタオミクロンによって誘発されるRelAの核外移行及びサイトゾル内分布に対してはPPARγが不可欠であることが結論づけられる。Caco−2細胞トランスフェクション実験及びHela細胞トランスフェクション実験のいずれにおいても、発現したPPARγ及びRelAタンパク質の核内貯蔵が顕著であった。この場合、WTやDNの状態とは関係無く、腸炎菌及びB.テタイオタオミクロンの存在下で培養した細胞の核内にPPARγとRelAとの凝集物がはっきりと見られた(IPによっても確認、結果は図示せず)が、この結果から、PPARγとRelAとの相互作用が核区画内で起こることが証明された。本発明者らは、PPAR及びRelAの核内での分布が、核斑(nuclear speckles)やスプライセオソーム内で起こるスプライシング因子の分布と類似していることに注目した。共存についての検討において、 本発明者らはPPARγタンパク質がSC35、即ち、特異的プレmRNAスプライシング因子と共存することを見出した(図5E(k))が、この知見から、PPARγ/RelA複合体が細胞のmRNA機構と相互作用し得ることが示される。核内受容体とスプライシング区画との相互作用についての実証データが、本明細書作成中に公表された(ゾア(Zhoa)ら、2002年)。以上から明らかなように、Caco−2細胞では、B.テタイオタオミクロンの存在によりPPARγの核外移行が促進され、また、これに便乗する形で転写活性RelAの核外移行も促進されることにより、炎症持続中の更なる核内移行及びRelA仲介による転写が防止される。このメカニズムによって、B.テタイオタオミクロンは強力な抗炎症作用を発揮する。
【0080】
炎症を抑制する細菌が消化管内に存在することは確実と思われる。炎症を抑制する腸内微生物と炎症反応を持続させる腸内微生物とのバランスが変化すると、それが消化管の炎症性疾患の原因に直接関係する場合がある。従って、炎症性腸疾患が免疫ホメオスタシスに積極的に寄与する細菌株の減少に関係することは妥当と思われ、これによって、プロバイオティック療法に反応する患者もいることの理由が説明される(マッドセン(Madsen)ら、2001年)。また同様に、受容体タンパク質のレベルが不十分なこと、或いは炎症病態への感受性を高める特定の対立遺伝子変異体の腸内での発現に起因するPPARの機能障害が大いに関係する場合もある。この見解は、虚血再灌流誘発型腸炎症及び障害に対するヘテロ接合型PPARγ欠乏性マウスの感受性が有意に高いことを示す最近公表されたデータ(ナカジマ(Nakajima)ら、2001年)によって更に支持される。
【0081】
方法
【0082】
試薬
【0083】
組織培養試薬はシグマ及びインビトロゲンより入手した。抗体はサンタクルズ、モレキュラープローブ、ニューイングランドバイオラボ(New England Biolabs)より入手した。また、分子試薬はプロメガ、インビトロゲン及びアマシャムより入手した。
【0084】
S.エンテリティディス/B.テタイオタオミクロン共培養モデル
【0085】
Caco−2細胞及びHela細胞は35mm培養皿内で常法によって培養した。通常の実験では、次の4種類の処理でインキュベートした細胞を要した。(1)培地のみでインキュベートした細胞、(2)108個の腸炎菌(S.エンテリティディス)と共にインキュベートした細胞、(3)108個の腸炎菌(S.エンテリティディス)及び109個のバクテロイデス・テタイオタオミクロンと共にインキュベートした細胞、及び(4)109個のB.テタイオタオミクロンと共にインキュベートした細胞。上記細菌は2時間或いは4時間添加した後、十分に洗浄して除去した。試験に用いた他の細菌及びリガンドとしては、大腸菌0157H7、B.ブルガータス、PMA、IL−1α/β及びTNFαが挙げられる。実験は全て最適化し、プロトコルは詳細な用量反応及び時間経過に基づくものであった。Caco−2細胞単層を経由するPMN細胞の経上皮遊走はMPOアッセイ(パーコス(Parkos)ら、1991年)によって測定した。Caco−2細胞を2時間インキュベートした後、上記細菌を除去し新しい培地に取り替えた。その後、細胞を更に2時間インキュベートした。次いで、先端区画から得た培地及び細胞をいずれも1%のTriton X−100に可溶化し、MPOを定量した。
【0086】
サイトカイン解析
【0087】
サイトカイン解析はクロンテックマクロアレイ、ノーザンハイブリダイゼーション、及びCaco−2細胞から単離したRNAのリアルタイムPCRを用いて行った。全RNA及びmRNAを単離し、cDNAを産生し、PCRを標準条件下で行った。IL−8タンパク質濃度はELISAによって測定した。
【0088】
NF−κB及びAP−1のEMSA解析
【0089】
NF−κBのコンセンサス結合配列(5’−AGT TGA GGG GAC TTT CCC AGG C−3’)或いはAP−1のコンセンサス結合配列(5’−CGC TTG ATG AGT CAG CCG GAA−3’)を含む一本鎖32P標識オリゴヌクレオチドプローブと共に核抽出物をインキュベートし、電気泳動によって分離し、オートラジオグラフィーによって可視化した。特異的NF−κBサブユニット抗体を用いてEMSAスーパーシフトを行った。EMSA、ウェスタンブロッティング(タンパク質及びリンタンパク質)、プロモーター特異的レポーター解析及び標的遺伝子発現(例えば、fos及びjun)によって、NFκB及びAP−1のシグナル伝達に対するB.テタイオタオミクロンの作用を測定した。
【0090】
免疫蛍光解析
【0091】
実験的処理後、35mm培養皿で増殖したCaco2細胞及びHela細胞を4%パラホルムアルデヒドに固定し、0.2%のTriton−X100/PBSで透過処理した。二次抗体を産生させた種由来の血清1%を含むPBSに希釈した一次抗体(1μg/ml)と共に細胞を室温で1時間インキュベートした。二次抗体(1μ/ml)としては、Alexa Fluor594ロバ抗ヤギIgG或いはAlexa Fluor488ヤギ抗ウサギIgG(モレキュラープローブ)を必要に応じて用いた。標識した細胞をVectorshield(ベクター)でスライドに載せ(mounted)、Zeiss Axioskop50広視野蛍光顕微鏡或いはBio−Rad Radiance2100レーザー走査型顕微鏡にて検査した。代表的デジタル画像をAdobe Photoshop6.0に取り込み最終調整を行った。
【0092】
蛍光標識PPAR・の作成
【0093】
PPARγのコード配列及びPPARγのドミナントネガティブ変異体(ガーネル(Gurnell)ら、2000年)をPCR増幅時に修飾し、生成物の5’末端にXhoI認識配列とSacII認識配列を、3’末端にはSacII配列を付加した。PCR増幅はPfuDNAポリメラーゼを用いて行った。生成物を制限しpECFP−C1及びpEYFP−C1(プロメガ)にクローン化した。クローン化の成功はDNAシークエンシングによって確認した。pEYFP−C1へクローン化したヒトp65については既に報告されている(シュミット(Schmid)ら、2000年)。
【0094】
Caco2細胞及びHela細胞を35mm培養皿に播種し、90%コンフルエントまで増殖させ、標準リポフェクタミン仲介法(インビトロゲン)を用いてトランスフェクトした。48時間後、細菌インキュベーションを上述の様に行い、細胞を室温で30分間4%パラホルムアルデヒド0.1Mリン酸ナトリウム緩衝液(pH7.4)に固定し、PBSで洗浄し、CFP及びYFP用カスタムフィルター(オメガオプティカル)を備えたZeiss Axioskop50広視野蛍光顕微鏡にて検査した。代表的画像をZeiss AxioCamデジタルカメラで記録し、Zeiss AxioVision3.0ソフトウェアで処理し、Adobe Photoshop6.0に取り込み最終調整を行った。一部の実験では、トランスフェクトした細胞を上述の免疫蛍光解析に付した。
【0095】
実施例6:インビボラット研究
本実施例では、B.テタイオタオミクロンによって、特定病原不在ラットの腸機能が変化し、該ラットの病原菌チャレンジに対する耐性が増加するどうかを立証した。一般に、バクテロイデスは哺乳期から離乳期への移行時に小腸に現われる(チャン(Chang)ら、1994年;フーパー(Hooper)ら、2001年)。従って、特定病原不在Hooded Listerラットに離乳期(19日齢)から成熟期(34〜40日齢)までこの細菌を毎日投与し、腸や免疫の発達に重要なこの時期にこの菌が確実に多数存在するようにした。34日齢でラットに対し腸炎菌S1400の経口チャレンジを行った。
【0096】
方法
【0097】
細菌の培養:
【0098】
ドイチュ・サムルング・フォン・ミクロオーガニズメン・ウント・GmBH(Deutsche Sammlung von Microorganismen und GmBH)(ドイツ、ブラウンシュヴァイク(Braunschweig))から入手したB.テタイオタオミクロンをWilkins−Chalgren嫌気性寒天で凍結保持した。B.テタイオタオミクロンをWilkins−Chalgren嫌気性ブロス或いはM10培地[Hungateチューブ内10ml(ブライアント(Bryant)、1972年)]に継代培養し、37℃で48時間増殖させた。サンプル(0.5ml)を新しい培地[Hungateチューブ内10ml]に移し、37℃で24時間増殖させた。得られた培養物には約108〜109CFU/mlのB.テタイオタオミクロンが含まれていた。
【0099】
B.テタイオタオミクロン培養物(10ml)を遠沈し(2000g、12分間)、得られたペレットを0.05Mリン酸緩衝生理食塩水(pH7.2)で洗浄した後、市販のゼリー[10ml、37℃、嫌気条件下で調製済]に再懸濁した。このゼリーをCO2下で滅菌ペトリ皿に注入し4℃で放置した。約1時間後、ゼリーを一定分量(0.5g、B.テタイオタオミクロン(〜108CFU))に分割し上記ラットに与えた。B.テタイオタオミクロンはこの条件下で少なくとも6時間は生存していたが、通常、ゼリーはケージに入れて数分以内には全て食べられた。
【0100】
腸炎菌S1400は当初家禽感染症から単離されたものであり、特徴付けが行われてる(アレン−ヴェルコー(Allen-Vercoe)及びウッドワード(Woodward)、1999年)。Dorset卵スロープ(Dorset egg slopes)で4℃で保持した菌株をLuria−Bertani寒天プレートに継代培養し37℃で一晩増殖させた。該プレートから得た5〜10個のコロニーをLuria−Bertaniブロス(10ml)に接種し、37℃で一晩攪拌しながらインキュベートし、約1×109CFU/mlの腸炎菌S1400を得た。
【0101】
動物実験:ローウェット研究所は英国動物(科学的処置)法1986の下で認可を受けている。同研究所の倫理審査委員会及び動物保護部門、更には適切な政府視察団(appropriate governmental inspectorate)によって動物実験は全て視察、監視されている。管理及び実験手続については倫理委員会によって承認され、その手続は実施許可を得た研究所員が上記動物法の厳しい条件に従って行った。
【0102】
ローウェット研究所の小動物部門で飼育した、24匹の特定病原不在Hooded Lister雄性ラット(40g)を19日目に離乳させ、すぐにClassII施設へ移した。実験継続期間中(21日間)、ラットを個々にフレキシフィルムアイソレータ(モードゥン、アニマルヘルス、英国、ペニクイク(Penicuik))内の代謝ケージ(テクニプラスト、英国、ケッターリング)に収容した。入れ子状トンネルを各代謝ケージに設け、ラットが互いを視認できても接触できないように該アイソレータ内に代謝ケージを配置した。滅菌蒸留水をいつでも摂取できるようにした。ラットを毎日計量し、糞便を実験期間中採取した。実験は少なくとも3回行ったが、いずれも同様の結果が得られた。データは平均ISDである(n=3)。先ず、ラットに良質の半合成(100gタンパク質/kg)ラクトアルブミンベースの食餌(グラント(Grant)ら、2000年)を自由に与えた。ラットが約80gに達した時に(約30日齢)、食餌摂取量を3〜4日間かけて7g/ラット/日(1日2食)まで徐々に減らし、実験の残りの期間中この摂取量を維持した。この摂取量は、腸炎菌を経口感染させた後の同一日齢ラットの1日当りの自由摂取量の平均であり、この量は非感染ラットによる上記食餌の自由摂取量の約70%であった。ラットを離乳時からこのように収容、管理し、細菌への環境曝露や離乳後の相互汚染を減少させた。(グラント(Grant)、1996年)。
【0103】
離乳時(19日目)から実験終了時まで、12匹のラットにB.テタイオタオミクロン(約108CFU)を1日1回投与した。34日齢時に、B.テタイオタオミクロンで前処理した6匹のラットと6匹の対照ラットに、胃管栄養法によって単一用量0.8mlの腸炎菌S1400培養物(約109CFU)を投与した。残りのラットには相当量の培地を投与した。ラットを全て代謝ケージに戻し、ラクトアルブミンベースの食餌(7g/ラット/日)を更に6日間与えた。最終日にラットに2gを食餌を与え、その丁度2時間後、ハロタン(ローヌメリュー、英国、エセックス)の過剰摂取及び失血によりラットを殺した。開腹して組織を無菌的に除去した。胃及び小腸をリン酸緩衝生理食塩水(PBS、pH7.2)で流して内容物や非付着細菌を除去した。空腸10cm(幽門から5〜15cm)と回腸10cm(回盲連結部から5〜15cm)を除去した。こうして除去した空腸と回腸、胃組織、盲腸及びその内容物、結腸及びその内容物、腸間膜リンパ節、肝臓と脾臓の代表的部分(200〜400mg)及び腎臓1個を処理し生細胞数を測定した。
【0104】
更に空腸片(幽門から15〜25cm)及び回腸片(回盲連結部から15〜25cm)を採取し液体窒素で凍結し、生化学的解析を行った。同様の処理を残りの小腸組織(〜40cm)、肝臓、脾臓、腎臓、胸腺、肺、心臓及腓腹後肢筋についても行った。これらを計量し、凍結乾燥した後再計量した。
【0105】
ラット組織中の生残細菌
【0106】
組織サンプルを計量した後、Janke−Kunkel Ultra−TurraxT25組織ホモジナイザー(20,000rev/分、30秒)を用いてMaximum Recovery Diluent(MRD、フィッシャーサイエンティフィック、英国)にホモジナイズした。最大6回の希釈工程を続けて行い、一次ホモジネート(1:10v/v)をMRDに形成した。各希釈工程におけるサンプルを十分に乾燥したXLD寒天(フィッシャーサイエンティフィック、英国)及びMacConkey寒天No.3(フィッシャーサイエンティフィック、英国)プレートの表面に載せ37℃で一晩インキュベートした。生細胞数をマイルズ(Miles)及びミスラ(Misra)(1938年)の方法或いはスプレッドプレート法(コリンズ(Collins)及びライム(Lyme)、1989年)によって評価した。
【0107】
PCR
【0108】
QIAamp DNA Stool Mini Kit(キアゲン社、英国、クローリー)を用いてDNAを抽出、精製した。PCRはテング(Teng)らの方法(2000年)に基づいた。テング(Teng)ら(2000年)によって同定されたプライマー対、即ち、5’−TGGAGTTTTACTTTGAATGGAC−3’(BTH−F)及び5’−CTGCCCTTTTACAATGGG−3’(BTH−R)はシグマジェノシス社(英国、ケンブリッジ)から購入した。TaqPCR Core Kit(キアゲン社、英国、クローリー)から入手した試薬に基づく反応混合物(50μl)には、上記2種類のプライマー(50pmol)、dNTP混合物(10nmol)、10X Qiagen PCRバッファー(5μl)、5X Q溶液(10μl)、サンプル(10μl)及びTaqDNAポリメラーゼ(1.25U)が含まれていた。ホットスタートPCRプログラムを用いた。TaqDNAポリメラーゼを含まない反応混合物を94℃で15秒間加熱した後、(94℃×10秒、55℃×30秒、74℃×1分)のサイクルを35回行って加熱し、更にその後(74℃×2分、45℃×2秒)のサイクルを1回行って加熱した。アンプリコンを臭化エチジウム(1μg/mlゲル)を含むアガロースゲル(10g/l)で分離した。糞便及びB.テタイオタオミクロンからDNAと共に単一アンプリコン(721bp)を得た。これらのアンプリコンの配列は互いに一致しており、また公表された配列(テング(Teng)ら、2000年)とも一致することが分かった。
【0109】
MPO
【0110】
Janke−Kunkel Ultra−TurraxT25組織ホモジナイザー(20,000rev/分、30秒)を用いて、組織サンプルを氷冷5mMリン酸カリウム緩衝液(pH6.0)にホモジナイズし(1:80w/v)、遠沈した(3000g×30分、4℃)。臭化ヘキサデシルトリメチルアンモニウム(HETAB、5g/l)及びエチレンジアミン四酢酸(EDTA、3.72g/l)を含む氷冷0.5Mリン酸カリウム緩衝液(pH6.0)で、得られたペレット(1:20w/v)を超音波処理し(3×5秒)、30分間氷上に放置した後、遠沈した(3000g×30分、4℃)[ストゥッキ(Stucchi)ら、2000年]。得られた上清をアッセイを行うまで凍結した。50mMリン酸カリウム緩衝液(pH6.0)での3,3’,5,5’−テトラメチルベンジジン(TMB、ダイネックステクノロジーズ、英国、アッシュフォード)のH2O2依存型酸化をモニターすることによってミエロペルオキシダーゼ(MPO)活性を測定した(ツィマーマン(Zimmerman)及びグランガー(Granger)、1990年)。450nmにおける吸光度は0.18MH2SO4との反応終了後に測定した。ヒトミエロペルオキシダーゼ(カルバイオケム、英国)を標準品として用い、数値はMPO当量として示した。
【0111】
腸内容物や糞便内の免疫反応性MPOを競合ELISA法で測定した。10mMリン酸緩衝生理食塩水(pH7.4)[PBS]に溶解したヒトミエロペルオキシダーゼでマイクロタイタープレート(Immulon4、ダイネックステクノロジーズ、、アッシュフォード、英国)を4℃で一晩コートした[10ng/ウェル]。Tween−20(1ml/l)を含むPBSで洗浄後、プレートをウシ血清アルブミン(BSA、10g/l)含有PBSで室温下1時間ブロックした。プレートを洗浄し、サンプル及び標準品を添加し[50μl/ウェル]、BSA(1g/l)、Tween−20(1ml/l)及びロイペプチン(1mg/l)を含むPBSで連続的に希釈した。更に、ウサギ抗ヒトミエロペルオキシダーゼ抗体[カルバイオケム、英国](1:4000希釈品、50μl/ウェル)も添加した。プレートを1時間インキュベートし洗浄した後、ビオチン化抗ウサギIgGと1時間反応させ、次いでExtravidin/ペルオキシダーゼ(EXTRA−3キット、シグマアルドリッチ、英国、プール)と反応させた。洗浄後、TMB試薬を添加し(7μl/ウェル)、暗所で最大2時間インキュベートした。0.18MH2SO4の添加(50μl/ウェル)によって反応を終了させ、吸光度を450nmで測定した。数値は免疫反応性MPO当量として示した。
【0112】
IgG
【0113】
IgGは競合ELISAによって測定した。PBSに溶解したラットIgG(シグマアルドリッチ、英国、プール)でマイクロタイタープレートを4℃で一晩コートした[1μg/ウェル]。プレートをブロック、洗浄した後、サンプル及びラットIgG標準品を添加し[50μl/ウェル]、BSA(1g/l)及びTween−20(1ml/l)を含むPBSで連続的に希釈した。更に、ビオチン化抗ラットIgG抗体(シグマアルドリッチ、英国、プール;1:1000希釈品)も添加し(50μl/ウェル)、プレートを1時間インキュベートした。次いで、MPOイムノアッセイによりプレートをExtravidin/ペルオキシダーゼ及びTMB試薬と反応させた。数値はIgG当量として示した。
【0114】
IgA
【0115】
IgAは捕捉ELISAによって測定した。ヤギ抗ラットIgA(シグマアルドリッチ、英国、プール;1:100PBS希釈品)でマイクロタイタープレートを4℃で一晩コートした(100μl/ウェル)。プレートをブロック、洗浄した後、サンプル及び標準品を添加し[100μl/ウェル]、BSA(1g/l)、Tween−20(1ml/l)及びロイペプチン(1mg/l)を含むPBSで希釈し、1時間インキュベートした。洗浄後、マウス抗ラットIgA抗体(シグマアルドリッチ、英国、プール;1:500希釈品)と共にプレートを1時間インキュベートし、再洗浄後、ビオチン化抗マウスIgG抗体(1:1000希釈品)と共に更に1時間インキュベートした。次いで、MPOイムノアッセイによりプレートを処理した。数値はIgA当量として示した。
LPS特異的IgG及びIgA
【0116】
腸炎菌(シグマアルドリッチ、ドーセット、プール)由来のリポ多糖類(LPS)でマイクロタイタープレートをコートした(10μg/ウェル)。プレートをブロック、洗浄後、サンプル及び標準品を添加した[100μl/ウェル]。1時間後プレートを洗浄し、ビオチン化抗ラットIgG及び抗ラットIgAを添加した後、プレートを上述の様に処理した。ELISAにおいて吸光度が少なくとも0.2となる容量(μl)に含まれるLPS特異的抗体の量を該抗体の1ユニットとした。腸炎菌で感染後22日目のラットから採取した血清(LPS−IgA:3.2×104ユニット/ml、LPS−IgG:1.7×105ユニット/ml)を陽性対照とした。
【0117】
エラスターゼ
【0118】
凍結乾燥した糞便及び腸内容物を、HETAB、EDTA及びNaN3(1g/l)を含む氷冷0.5Mリン酸カリウム緩衝液(pH6.0)に抽出し(1:10w/v)、氷上で30分間放置した後、遠沈し(3000g、30分、4℃)、得られた上清を凍結した。
【0119】
マイクロタイタープレート上で、サンプル及び白血球エラスターゼ(シグマアルドリッチ、英国、プール)を1MNaCl及びロイペプチン(1mg/l)を含む0.2MトリスHCl(pH8.0)で連続的に希釈した。基質(N−スクシニル−ala−ala−val−p−ニトロアニリド、0.2g/l、シグマアルドリッチ、英国、プール)を添加し、その直後、405nmにおける吸光度をモニターした。更に、37℃でのインキュベーション時に最大20時間の間隔で吸光度をモニターした。数値は白血球エラスターゼ当量として示した。
【0120】
DNA、RNA、タンパク質
【0121】
DNA、RNA、タンパク質は、従来通り、ジフェニルアミン法、オルシノール法及び改良ローリー法によって測定した(グラント(Grant)ら、2000年)。サケ精巣DNA、酵母DNA及びウシ血清アルブミンを標準品として用いた。
【0122】
統計解析
【0123】
Instat Statistical Package(グラフパッドソフトウェア社、米国、サンディエゴ)を用い、一元配置分散分析(ANOVA)とチューキーの多重比較検定とを組み合わせてデータを評価した。
【0124】
結果
【0125】
バクテロイデス・テタイオタオミクロン(BT)はBT処理ラットから採取した糞便サンプルで検出された。また、BTは対照ラットの糞便でも検出された。しかし、対照ラットの糞便で検出されたBTのレベル(約104CFU/g糞便に相当)は、毎日BTで処理したラットの糞便で検出されたBTのレベル(約106CFU/g糞便に相当)よりも低かった。
【0126】
体重増加、主要臓器重量、小腸成分及び肝臓成分については、BT処理ラットと対照ラットとの間では差が無かった(表1〜4)。BT処理ラットの場合、乳糖発酵菌及び非乳糖発酵菌の消化管全体への分布は影響を受けなかったと思われ、また、対照ラットの場合と同様に、腸間膜リンパ節、肝臓、脾臓及び腎臓サンプルでは乳糖発酵菌及び非乳糖発酵菌のいずれも検出されなかった。更に、腸内容物や糞便中の非特異的IgA、エラスターゼ及び免疫反応性MPOのレベル、腸組織中のMPOのレベル、及び血清中の非特異的IgG及びIgAのレベルについても、対照ラットから採取したサンプルの場合と同程度であった。
【0127】
【表1】

【0128】
腸炎菌[SE]は経口チャレンジ後6日目に消化管全体で検出された(表1)。更に、腸炎菌は腸間膜リンパ節、肝臓及び脾臓でも検出された。上記ラットにおいて非乳糖発酵菌は全て同様の組織内分布を示した(表1)。腸炎菌感染の腸内乳糖発酵菌数に対する影響については明らかではなかった。しかし、かなりの数の乳糖発酵菌がSE感染ラットの腸間膜リンパ節、肝臓及び脾臓に存在した(表1)。
【0129】
腸炎菌チャレンジ後に肝臓及び脾臓で検出された生残腸炎菌の数は、ラットをB.テタイオタオミクロンでも処理した場合[SE+BT]には大幅に減少した(表1)。肝臓及び脾臓内では全非乳糖発酵菌の数も減少したが、乳糖発酵菌数は変化しなかった。消化管全体及び腸間膜リンパ節内の腸炎菌、全非乳糖発酵菌及び乳糖発酵菌のレベルは、SE+BTとSEとの間では差が無かった(表1)。また、6日間の実験中、腸炎菌の糞便への分泌が腸炎菌感染ラットでは同程度で起こった(SE+BT:7.0±0.8Log10CFU/g/日、SE:6.2±0.6Log10CFU/g/日)。
【0130】
【表2】
【0131】
【表3】
【0132】
腸炎菌感染の結果、小腸の重量が増加した(表2、3)。重量の増加は回腸組織で最も顕著であり、これは水分の蓄積と組織内のタンパク質量、RNA量及びDNA量の増加とによるものであると分かる(表3)。BTでの処理により感染に対する反応がわずかに変化した。即ち、SE+BTラットから採取した小腸の水分量[748±22mg/g]はSEラットから採取した小腸の水分量[799±17mg/g]に比べてかなり少ない。実際には、SE+BTラットの小腸の水分量は非感染ラットから採取した小腸の水分量[BT:745±24mg/g、対照:760±10mg/g]と同等である。一方、小腸乾燥重量、DNA量、RNA量及びタンパク質量についてはSEラットとSE+BTラットとの間に有意差は無かった。
【0133】
【表4】
【0134】
SEラットから採取した回腸組織においてミエロペルオキシダーゼ(MPO)活性は有意に上昇した(表4)。また、腸内容物及び糞便における免疫反応性MPO及びエラスターゼの活性も大幅に上昇した(表4)。一方、SE+BTラットから採取した腸内容物及び糞便におけるこれらの酵素のレベルは対照ラットの場合と同等か或いはごくわずかに高かった。
【0135】
腸炎菌感染の結果、腸内容物、糞便及び血清中の免疫反応性非特異的IgAは増加した(表4)。しかし、IgA反応についてはSE+BTラットとSEラットとの間では差が無かった。血清及び腸内容物では少量のLPS特異的IgAも検出された。この場合も、SE+BTラットとSEラットとの間では有意差が無かったが、SE+BTラットの方が腸内でのLPS特異的IgAレベルがより高くなる傾向にあった。
【0136】
腓腹後肢筋重量については対照ラットよりSEラットの方が有意に小さかった(表2)。この傾向はSE+BTラットでは明らかでなく、よって、この重量減少は筋肉成長の低下によるものと分かった。SEラットの場合、腓腹筋は1日当り湿重量で約6.6±5.7mg[乾燥重量で1.4±1.2mg]成長し、一方、SE+BTラットの場合、腓腹筋の成長率は1日当り湿重量で17.1±4.1mg[乾燥重量で4.2±1.0mg]であった。また、対照ラットの場合、腓腹筋の成長は1日当り湿重量で15.5±2.9mg[乾燥重量で3.8±0.7mg]であり、BTラットの場合、1日当り湿重量で17.0±4.2mg[乾燥重量で4.2±1.0mg]であった。この結果から、1日当りの全骨格筋の成長については、SEラットの場合、平均0.3g(湿重量)であり、SE+BT、BT及び対照ラットの場合、約0.7〜0.8gであることが示唆される(図6)。
【0137】
腸炎菌感染ラットにおいては、肝臓、脾臓及び腸間膜リンパ節の湿重量は有意に増加した(表3)。これは主に、肝臓及び脾臓での水分の蓄積と腸間膜リンパ節での水分及び乾燥物の増加によるものである(表4)。腸炎菌感染ラットの腸間膜リンパ節でのペルオキシダーゼ活性も上昇した[SE:31.0±25.0μg、SE+BT:37.7±23.8μg、BT:0.9±0.9μg、対照:0.4±0.3μg]。しかし、腸間膜リンパ節でのペルオキシダーゼ活性については、SE群とSE+BT群との間では有意差が無かった。
【0138】
考察
【0139】
サルモネラ症:腸炎菌S1400(Salmonera enterica var Enteritidis S1400)はラットの消化管全体でコロニーを形成し、腸間膜リンパ節、肝臓及び脾臓に転位した。また、腸炎菌感染により小腸の湿重量及び乾燥重量は大幅に増加した。この重量増加は回腸において最も顕著であり、回腸内での水分の蓄積とタンパク質、RNA及びDNAのレベルの上昇とに関連していた。従って、腸炎菌感染の基本的な特徴は、ラットモデルで他のエンテリティディス菌株やネズミチフス菌(S.ティフィムリウム)株感染において見られる特徴(ノートン(Naughton)ら、1996年、2001年;イーウェン(Ewen)ら、1997年;ボヴィー−オーデンホーヴェン(Bovee-Oudenhoven)ら、1999年;イスラム(Islam)ら、2000年;ハヴェラール(Havelaar)ら、2001年)と類似していた。
【0140】
エンテリティディスやティフィムリウムによってラットで自己制限感染が引き起こされ、この細菌は主に消化管に局在し回腸を経由して浸潤するが、全身への伝播は制限されることが分かっている(ノートン(Naughton)ら、1996年;ボヴィー−オーデンホーヴェン(Bovee-Oudenhoven)ら、1997年、1999年;イスラム(Islam)ら、2000年)。また、ラットの健康状態や腸の完全性が感染以前に他の因子によって損なわれていなければ、重篤な菌血症や死亡が起こることはまれである。これは、エンテリティディスやティフィムリウムによって死亡率の高い重篤な腸チフス様疾患が引き起こされた、マウスモデルの一部におけるサルモネラ症とは全く対照的である(ルー(Lu)ら、1999年;キングスレー(Kingsley)及びバウムラー(Baumler)、2000年;シェヒター(Schechter)及びリー(Lee)、2000年)。このように、ラットにおけるサルモネラ症は、エンテリティディスやティフィムリウムに感染したヒトや家畜に共通する自己制限胃腸炎型感染症と非常に類似している。
【0141】
本発明の検討においては、腸炎菌感染による強い炎症反応が小腸の基部ではなく末端部で引き起こされることを見出した。従って、回腸組織でのミエロペルオキシダーゼ(MPO、好中球マーカー)の活性は有意に上昇した。また、腸内容物や糞便での免疫反応性MPO及び白血球エラスターゼの活性レベルも上昇した。この結果は、先にサルモネラ症の動物モデルで観察されたように(ノートン(Naughton)ら、1995年、1996年;ヴァシロヤナコポウロス(Vassiloyanakopoulos)ら、1998年;ダーウィン(Darwin)及びミラー(Miller)、1999年;ヘンダーソン(Henderson)ら、1999年)、多形核白血球や他の炎症細胞の回腸組織への浸潤及び炎症細胞の腸管内腔への逃避(exvasion)と一致する。また、この結果は、回腸がサルモネラ菌によるコロニー形成及び浸潤の主要部位であるという知見(カーター(Carter)及びコリンズ(Collins)、1974;ノートン(Naughton)ら、1996年)とも一致する。
【0142】
腸炎菌感染によって血清、腸内容物及び糞便中の非特異的IgAは有意に増加した。また、感染後6日目までにはLPS特異的IgA反応の発生が示された。更に、腸内容物中のタンパク質レベルと同様に、糞便乾燥物、水分及びタンパク質の排出量(outputs)も増加した。腸炎菌感染によって肝臓及び脾臓の重量も増加した。
【0143】
サルモネラ症によってラットの骨格筋代謝が損なわれることも見出された。腸炎菌感染ラットと対照ラットは共に同じ乾燥物(7g/ラット/日)及びタンパク質(0.7g/ラット/日)を摂取したが、感染ラットの場合、骨格筋の成長は続いたものの一日当りの成長率は対照ラットの場合の約40〜50%であった。これは、感染に対する防御反応を維持するための栄養素の転用や利用(クラシング(Klasing)及びカルバート(Calvert)、1999年)及び/又は内毒素や他の細菌性因子の筋肉タンパク質合成への作用(フライマン(Friman)ら、1984年;ラング(Lang)ら、2000年)に起因するものであろう。
【0144】
B.テタイオタオミクロン:本発明の検討においては、腸が急速に成長、成熟し且つ免疫系が発達する時期を通して、ラットを高レベルの外因性B.テタイオタオミクロンに接触させた。バクテロイデス菌株の一部は日和見病原菌であり、この発達時期に有害な作用を示す可能性がある。しかし、B.テタイオタオミクロンのみを感染させた成熟完全無菌動物を用いた研究(フーパー(Hooper)ら、1999年、2001年;ノアック(Noack)ら、2000年)の場合と同様に、B.テタイオタオミクロンが特定病原不在ラットの腸や全身代謝に悪影響を及ぼさないことが示された。実際、B.テタイオタオミクロンのみで処理したラットでモニターした全パラメータ(炎症マーカーや免疫グロブリンのレベルを含む)は対照ラットについて得たものと同等であった。
【0145】
B.テタイオタオミクロンとサルモネラ症:B.テタイオタオミクロンによって、ラットでの腸炎菌S1400による感染の特徴が変化し、感染全体の重症度が低下した。特に、小腸での炎症反応が制限され、肝臓及び脾臓で検出された生残腸炎菌の数が大幅に減少し、骨格筋成長率はほぼ対照レベルへ回復した。このように、B.テタイオタオミクロンは腸内で局所的に作用を示すと同時に、全身組織に対しても遠隔的に作用を示した。
【0146】
しかし、サルモネラ症の特徴の一部はB.テタイオタオミクロンによっては影響されなかった。消化管及び腸間膜リンパ節内の腸炎菌数は変化せず、また、腸炎菌の糞便排泄についても変化しなかった。また、腸炎菌感染に伴う小腸の拡張(即ち、乾燥重量の増加とDNA、RNA及びタンパク質量の増加)もB.テタイオタオミクロンと腸炎菌を投与したラットにおいて明らかであった。この結果から、B.テタイオタオミクロンが腸炎菌自身を直接妨害したり、宿主の代謝に対する腸炎菌の一般的な作用をブロックするのではないことが示唆される。むしろ、B.テタイオタオミクロンは宿主の感染に対する反応を選択的に調節し、腸の完全性を損なう可能性のある反応を標的にするのであろう。
【0147】
最近では数多くの細菌株がエンテリティディス感染やティフィムリウム感染を部分的に防御することが示されている。このような細菌株の一部は腸内の付着部位や栄養素における病原菌を打ち負かしたり、殺菌性化合物を産生したりする。その結果、腸内で検出され、腸間膜リンパ節、肝臓及び脾臓に到達するサルモネラ菌の数が減少する(バーネット−カマード(Bernet-Camard)ら、1997年;フドールト(Hudault)ら、1997年、2001年;ヘンドリクソン(Hendriksson)及びコンウェイ(Conway)、2001年)。一方、上記細菌株の多くは腸内のサルモネラ菌の数に対し殆ど或いは全く影響を及ぼさない。しかし、それでもB.テタイオタオミクロンの場合と同様に、サルモネラ菌感染を有意に改善する(シルヴァ(Silva)ら、1999年;フィルホ−リマ(Filho-Lima)ら、2000年;シュー(Schu)ら、2000年;ヘンドリクソン(Hendriksson)及びコンウェイ(Conway)、2001年;マイア(Maia)ら、2001年)。従って、後者の細菌株が示す防御作用は競合排除や殺菌性化合物分泌以外のメカニズムによるものである。
【0148】
サルモネラ菌は、細胞代謝、細胞間相互作用及び宿主反応メカニズムを妨害することによって腸上皮を突破する(ダーウィン(Darwin)及びミラー(Miller)、1999年:ネテア(Netea)ら、2000年;イーヴス−ピレス(Eaves-Pyles)、2001年;ゲウィルツ(Gewirtz)ら、2001年;ルー(Lu)及びウォーカー(Walker)、2001年;オール(Ohl)及びミラー(Miller)、2001年)。特に、サルモネラ菌は多形核白血球の腸上皮への急速な浸潤を誘発し、腸の急性炎症や重篤な破壊を引き起こす(マダラ(Madara)、1997年;オール(Ohl)及びミラー(Miller)、2001年)。腸への好中球の浸潤は、サルモネラ菌感染に呼応して上皮細胞により分泌される化学誘引物質ケモカイン(例えばIL−8)によって仲介される(マコーミック(McCormick)ら、1995年;マダラ(Madara)、1997年;ダーウィン(Darwin)及びミラー(Miller)、1999年;フレックスタイン(Fleckstein)及びコペッコ(Kopecko)、2001年)。上皮細胞をインビトロでサルモネラ菌と共に培養した場合、この化学誘引物質の遊離が促進された(マコーミック(McCormick)ら、1995年;キャンベル(Campbell)ら、2001年)。一方、上皮細胞をサルモネラ菌及びB.テタイオタオミクロンと共に培養した場合、この化学誘引物質の産生量は大幅に低下した(キャンベル(Campbell)ら、2001年)。
【0149】
B.テタイオタオミクロンと腸炎菌とを投与したラットの回腸組織及び腸内容物中のミエロペルオキシダーゼのレベルは、腸炎菌のみを投与したラットから採取した比較サンプルに比べて遥かに低かった。このように、ラットをB.テタイオタオミクロンでも処理した場合、腸炎菌により通常腸内で誘発される炎症反応は明らかに抑制された。インビトロで見出されたように(キャンベル(Campbell)ら、2001年)、B.テタイオタオミクロンは、サルモネラ菌が関係する腸上皮細胞による化学誘引物質ケモカインの産生をブロックし、腸上皮への好中球の補充を防ぐと思われる。このインビボでの宿主反応をB.テタイオタオミクロンが調節することにより、感染の結果起こる腸の炎症や障害の程度を抑制し、腸の完全性を維持するであろう。これによって、炎症の回復に腸が必要とする栄養素の量が減少し(クラシング(Klasing)及びカルバート(Calvert)、1999年)、骨格筋等の他の組織へより多くの栄養素を供給することができる。
【0150】
サルモネラ菌は一旦腸上皮を通過すると腸間膜リンパ節へ流入する(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年)。次いで、サルモネラ菌はマクロファージによって腸間膜リンパ節から排除されるか、或いは腸間膜リンパ節から抜け出て血液、肝臓及び脾臓へと伝播し得る(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年;ルー(Lu)及びウォーカー(Walker)、2001年)。腸炎菌感染ラットの腸間膜リンパ節、肝臓及び脾臓ではかなりの数の腸炎菌が検出された。B.テタイオタオミクロンでラットを処理することにより、肝臓及び脾臓で検出される生残腸炎菌のレベルは大幅に減少した。しかし、腸間膜リンパ節での腸炎菌数はこの処理の影響を受けなかった。従って、B.テタイオタオミクロンは腸代謝に大きく作用するが、腸炎菌の腸間膜リンパ節への浸潤や流入を制限しないことが分かった。腸炎菌のレベルが全身組織で減少したことから、B.テタイオタオミクロンの主な防御作用は全身代謝の変化によって生じることが示されるであろう。
【0151】
共生細菌は強力な免疫調節物質となる(へリアス(Herias)ら、1999年;タルハム(Talham)ら、1999年;シャレック(Scharek)ら、2000年;イソラウリ(Isolauri)ら、2001年;ルー(Lu)及びウォーカー(Walker)、2001年)。特に、ビフィドバクテリウム・ラクティス(Bifidobacterium lactis)でマウスを処理すると、マウスの免疫機能やサルモネラ菌に対する反応性が向上することが分かった(シュー(Schu)ら、2000年)。血液や腹膜細胞での貪食活性、リンパ球分裂促進反応性及びティフィムリウム特異的抗体の分泌が向上した。B.テタイオタオミクロンも全身代謝に対して同様の作用を示すと思われる。従って、B.テタイオタオミクロンで処理したラットの肝臓及び脾臓で腸炎菌数が減少したことは、血液や内臓からの腸炎菌の排除が高まった結果によるものであろう。
【0152】
乳糖発酵菌(大腸菌)も、数こそ少ないが、腸炎菌感染ラットの肝臓及び脾臓で検出された。しかし、腸炎菌の場合と異なり、乳糖発酵菌のレベルはB.テタイオタオミクロンの影響を受けなかった。このことから、B.テタイオタオミクロンによって促進される細菌の排除には選択性があると思われる。或いは、大腸菌はラットの常在細菌叢に由来するため、不適切な組織部位に存在している場合でも、潜在的有害物質として容易に認識されないのであろう。
【0153】
B.テタイオタオミクロンは腸に局所的に作用することによって、サルモネラ菌の全身への伝播を抑制すると思われる。サルモネラ菌が腸上皮に付着し、腸上皮内に浸潤し、腸間膜リンパ節へ流入し、肝臓や脾臓へ伝播することは一般に認められている(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年)。しかし、最近の研究では、サルモネラ菌の全身への伝播が別の経路で起こり得ることが示唆されている。樹状細胞により管腔に摂取されるか、或いはCD18を発現する貪食細胞により上皮下組織へ摂取されたサルモネラ菌は、リンパ系へ流入することなく、肝臓や脾臓に直接伝播し得る(ヴァルケス−トレス(Varquez-Torres)ら、1999年;アイスバーグ(Isberg)及びバーンズ(Barnes)、2000年;レスチグノ(Rescigno)ら、2001年)。その結果、肝臓や脾臓で検出されるサルモネラ菌は2種類の摂取経路に由来する可能性がある。上記貪食細胞の補充は、上皮細胞により産生する化学誘引物質ケモカインによって仲介される可能性がある(イザドパナー(Izadpanah)ら、2001年;ケレンナン(Kellennan)及びマッケヴォイ(McEvoy)、2001年)。B.テタイオタオミクロンは腸内での好中球補充や炎症を引き起こすのに必要な化学誘引物質ケモカインの分泌を防ぐことによって、これらの補充や炎症を抑制することが分かっているため(キャンベル(Campbell)ら、2001年)、樹状細胞やCD18を発現する貪食細胞の補充に必要とされるケモカインも抑制し、その結果、このような系によるサルモネラ菌の摂取を防止或いは抑制するであろう。
【0154】
概要
【0155】
経口投与したB.テタイオタオミクロンによって、ラットにおける腸炎菌による感染の重症度が低下した。肝臓及び脾臓内の生残腸炎菌数は大幅に減少し、骨格筋成長率は正常値に回復し、また、小腸内の炎症反応は軽減された。これは、免疫反応性が向上したことや遠隔部位(例えば、肝臓や脾臓)で腸炎菌が急速に排除されたことによるものと思われる。しかし、少なくとも一部は、B.テタイオタオミクロンの腸への局所作用に起因するものでもある。腸内での好中球補充や炎症はB.テタイオタオミクロンの作用によって防止或いは遅延された。また、B.テタイオタオミクロンは、腸炎菌の貪食細胞への直接摂取と肝臓や脾臓への伝播もブロックしたと思われる。
【0156】
〔参考文献〕
1. E. Cario et al. J Immunol. 164, 966 (2000).
2. A. T. Gewirtz, T. A. Navas, S. Lyons, P. J. Godowski, J. L. Madara, J. Immunol. 167, 1882 (2001).
3. B. A. McCormick, S. P. Colgan, C. Delp-Archer, S. I. Miller, J. L. Madara, J. Cell Biol. 123, 895 (1993).
4. L. Hang et AL., J. Immunol. 162, 3037 (1999).
5. S. Ghosh, M. J. May, E. B. Kopp Annu. Rev. Immunol. 16, 225-260 (1998).
6. M. J. May, S. Ghosh. Immunol. Today (1998).
7. Q. Cheng et al., J. Biol. Chem. 269, 13551 (1994).
8. P. J. Chiao, S. Miyamoto, I. M. Verma. Proc. Natl. Acad. Sci. USA 91, 28-32. (1994).
9. P. Renard et al. J. Biol. Chem. 275, 15193 (2000).
10. A. S. Neish et al., Science 289, 1560 (2000).
11. T. Meyer-ter-Vehn, A. Covacci, M. Kist, H. L. Pahl, J. Biol. Chem. 275, 16064 (2000).
12. J. Raingeaud et al., J. Biol. Chem. 270, 7420 (1995).
13. Nakajima et al. Gastroenterology 120, 460 2001.
14. E. D. Rosen, B. M. Spiegelman. J. Biol. Chem. 276, 37731-37734 (2001).
15. C. G. Su et al., J. Clin. Invest. 104, 383 (1999).
16. C. F. Bunn et al., Mol. Endocrinol. 15, 512 (2001).
17. M. Ricote, J. T. Huang, J. S. Welch, C. K. Glass, J. Leukoc. Biol. 66, 733 (1999).
18. P. Delerive et al., J. Biol. Chem. 274, 32048 (1999).
19. M. Gurnell et al., J. Biol. Chem. 275, 5754 (2000).
20. S. D. Savkovic, A. Koutsouris, G. Wu, G. Hecht, Biotechniques 29, 514 (2000).
21. A. Birbach et al. J. Biol. Chem. 277, 10842 (2002).
22. J. Schmid et al. J. Biol. Chem. 275, 17035 (2002).
23. Y. Zhoa et al., J. Biol. Chem. 277, 30031-30039 (2002).
24. K. Madsen et al., Gastroenterology 121, 580 (2001).
25. C. A. Parkos et al., J. Clin. Invest. 88, 1605 (1991).
26. Allen-Vercoe et al., J Med Microbiol 48, 771-780 (1999).
27. Bardocz, S. et al., Br J Nutr 76, 613-626 (1996).
28. Bernet-Camard et al., Appl Environ Microbial 63, 2747-2753 (1997).
29. Bovee-Oudenhoven et al., Gastroenterology 113, 550-557 (1997).
30. Bovee-Oudenhoven et al., J Nutr 129,607-612 (1999).
31. Bry, L et al., Science 273, 1380-1383 (1996).
32. Bryant, M. P. Am J Clin Nutr 25, 1324-1328 (1972).
33. Carter, J Exp Med 139, 1189-1203 (1974).
34. Cebra, J. J., Am J CLIN Nutr 69, 1046S-1051S (1999).
35. Chang, J. et al., J Appl Bacteriol 77, 709-718 (1994).
36. Collins, C. H. L. P. M. (1989) Microbiological Methods Butterworths, London.
37. Darwin, K. H. et al., Clin Microbiol Rev 12, 405-428 (1999).
38. Eaves-Pyles, T. et al., J Immunol 166, 1248-1260 (2001).
39. Ewen, S. W. et al., FEMS Immunol Med Microbiol 18, 185-192 (1997).
40. Filho-Lima, J. V. et al., J Appl Microbiol 88, 365-370 (2000).
41. Fleckenstein, J. M. et al., J Clin Invest 107, 27-30 (2001).
42. Friman, G. et al., Scand J Infect Dis 16, 111-119 (1984).
43. Gewirtz, A. T. et al., J Immunol 167, 1882-1885 (2001).
44. Grant, G. (1996) Management of Animal Experiments. COST 98. Effects of antinutrients on the nutritional value of legume diets (Bardocz, S; Pusztai. A., ed) pp. 44-51, European Commision, Brussels.
45. Grant, G. et al., Pancreas 20, 305-312 (2000).
46. Havelaar, A. H. et al., J Appl Microbiol 91,442-452 (2001).
47. Henderson, S. C. et al., Infect Immun 67, 3580-3586 (1999).
48. Henriksson et al., J Appl Microbiol 90, 223-228 (2001).
49. Herias et al., Scand J Immunol 48, 277-282 (1998).
50. Herias, M. V. et al., Clin Exp Immunol 116, 283-290 (1999).
51. Hooper, L. V. et al., Proc Natl Acad Sci U S A 96, 9833-9838 (1999).52. Hooper, L. V. et al., Curr Opin Microbiol 3, 79-85 (2000).
53. Hooper, L. V. et al., Glycobiology 11, 1R-10R (2001).
54. Hooper, L. V. et al., Science 291, 881-884 (2001).
55. Hudault, S. et al., Appl Environ Microbiol 63, 513-518 (1997).
56. Hudault, S. et al., Gut 49,47-55 (2001).
57. Isberg, R. R. et al., Trends Microbiol 8, 291-293 (2000).
58. Islam, A. F. et al., Infect Immun 68, 1-5 (2000).
59. Isolauri, E. et al., Am J Clin Nutr 73, 444S-450S (2001).
60. Izadpanah, A. et al., Am J Physiol Gastrointest Liver Physiol 280, G710-G719 (2001).
61. Kellermann, S. A. et al., J Immunol 167, 682-690 (2001).
62. Kingsley, R. A et al., Bacterial Invasion into Eukaryotic Cells (Oelschlaeger, T. A. and Hacker, J., eds) pp. 321-342, Kluwer Academic/Plenum Publishers, New York (2000).
63. Klasing, K. C. C. C. C. Protein Metabolism and Nutrition (Lobely, G. E; Wnite, . A. and MacRae, J. C., eds) pp. 253-264, Wageningen Pers, Wageningen (1999).
64. Lang, C. H. et al., Am J Physiol Endocrinol Metab 278, E1133-E1143 (2000).
65. Lopez-Boado, Y. S. et al., J Cell Biol 148, 1305-1315 (2000).
66. Lu, L. et al., Am J Clin Nutr 73, 1124S-1130S (2001).
67. Lu, S. et al., Infect Immun 67, 5651-5657 (1999).
68. Madara, J. L. Aliment Pharmacol Ther 11 Suppl 3, 57-62 (1997).
69. Maia, O. B. et al., Vet Microbiol 79, 183-189 (2001).
70. McCormick, B. A. et al., J Cell Biol 131, 1599-1608 (1995).
71. Miles, A. A. et al., J Hyg 38, 749 (1993). 72. Naughton, P. J. et al., FEMS Immunol Med Microbiol 12, 251-258 (1995).
73. Naughton, P. J. et al., J Appl Bacteriol 81, 651-656 (1996).
74. Naughton, P. J. et al., J Med Microbiol 50, 191-197 (2001).
75. Netea, M. G. et al., J Immunol 164, 2644-2649 (2000).
76. Noack, J. et al., J Nutr 130, 1225-1231 (2000).
77. Pier, G. B. et al., Infect Immun 60, 4768-4776 (1992).
78. Rescigno, M. et al., Nat Immunol 2, 361-367 (2001).
79. Salminen, S. et al., Br J Nutr 80 Suppl 1, S147-S171 (1998).
80. Scharek, L. et al., Immunobiology 202, 429-441 (2000).
81. Schechter, L. M. et al., Bacterial Invasion into Eukaryotic Cells. (Oelschlaeger, T. A. H. J., ed) pp. 289-320, Kluwer Academic/Plenum Publishers, New York (2000).
82. Shu, Q. et al., Med Microbiol Immunol (BERL) 189, 147-152 (2001).
83. Silva, A. M. et al., J Appl Microbiol 86, 331-336 (1999).
84. Snel, J. et al., Can J Microbiol 44, 1177-1182 (1998).
85. Stucchi, A. F. et al., Am J Physiol Gastrointest Liver Physiol 279, G1298-G1306 (2000).
86. Talham, G. L. et al., Infect Immun 67, 1992-2000 (1999).
87. Teng, L. J. et al., J Clin Microbiol 38, 1672-1675 (2000).
88. Van Der Waaij, D. The Germ-free Animal in Biomedical Research (Coates, M. E. Gustafsson,. B. E., ed) pp. 155-165, Laboratory Animals Ltd, London (1984).
89. Vassiloyanakopoulos, A. P. et al., Proc Natl Acad Sci U S A 95, 7676-7681 (1998).
90. Vazquez-Torres, A. et al., Nature 401, 804-808 (1999).
91. Yurdusev, N. et al., Can J Microbiol 33, 226-231 (1987).
92. Yurdusev, N. et al., Infect Immun 57, 724-731 (1989).
93. Zimmerman, B. J. et al., Am J Physiol 259, H390-H394 (1990).
【技術分野】
【0001】
本発明は、感染に対する腸の反応の調節、特にバクテロイデス・テタイオタオミクロン(Bacteroides thetaiotaomicron)を用いた調節に関する。
【背景技術】
【0002】
哺乳動物の腸においては、数百種類の細菌がコロニーを形成し、この細菌の数は結腸、即ち、感染や炎症性疾患、癌を起こしやすい解剖学的部位において劇的に増加する。しかし、成長した動物においては、消化管内に常在する細菌叢によって感染に対する高い防御作用(コロニー形成耐性)(colonisation resistance)が存在する(ファン・デル・ワーイ(van der Waaij)、1984年;サルミネン(Salminen)ら、1998年)。その結果、この環境に遭遇する日和見病原菌の多くは足掛かりを築くことができなくなり、急速に排除される(ファン・デル・ワーイ(van der Waaij)、1984年;サルミネン(Salminen)ら、1998年)。しかし、共生細菌叢が損なわれている場合、日和見病原菌が腸内に存在し続けることがある。例えば、緑膿菌は通常ネズミの腸に存在し続けることはないが、抗生物質によって共生細菌叢が破壊されると、緑膿菌はコロニーを形成し、重篤な感染を引き起こす(ピア(Pier)ら、1992年)。また、共生細菌もサルモネラ菌等の悪性病原菌に対してある程度の防御作用をもたらす。しかし、このような悪性病原菌は、その存在する数が非常に多い場合、共生細菌叢の防御作用を克服したりその作用から免れたりして、重篤な感染を引き起こすことがある。
【0003】
コロニー形成耐性(Colonisation resistance)は有害細菌の競合排除に一部起因するものであるが、この排除は、栄養素や基質を優先的に用いたり、或いは有害細菌が付着する可能性のある腸の部位を共生細菌叢によってブロックすることによって行われる。しかし、共生細菌が宿主の腸細胞及び免疫系を調節することもある(ブライ(Bry)ら、1996年;へリアス(Herias)ら、1998年、1999年;フーパー(Hooper)ら、2000年、2001年;セブラ(Cebra)、1999年;スネル(Snel)ら、1998年;タルハム(Talham)ら、1999年;ロペス−ボアド(Lopez-Boado)ら、2000年;シュー(Shu)ら、2000年;キャンベル(Campbell)ら、2001年)。また、共生細菌叢は消化管を変化させ、この細菌叢にはよく適合するが他の細菌には適合しないニッチミクロ環境(niche microenvironments)を形成することがある。或いは、共生細菌叢は有害細菌に対する上皮細胞の反応を変化させ(キャンベル(Campbell)ら、2001年)、これによってコロニー形成や浸潤を促進する変化を抑制することもある。
【0004】
健康な腸は様々な細菌の負荷に対して低反応状態(hyporesponsive tone)を維持するが、閾値レベルの病原性細菌が存在すると、腸組織内での炎症誘発性遺伝子の発現を急速にアップレギュレートする転写系が十分に活性化される。こうして得られた転写物によって、一連の反応(例えば、腸の感染部位の粘膜固有層への多形核(PMN)細胞の遊走)が引き起こされる。これらのイベントは細菌の排除には必須であるが、病状を悪化させ得る組織異変の原因にもなる。管腔内細菌が最初に接触する場所は、腸免疫系と接続する或いは遮断される上皮細胞の連続層である。腸上皮細胞が病原性細菌と非病原性細菌とを区別する能力は、コロニー形成細菌に対する有害で不適切な反応を防ぎ、腸を健康な状態に維持する上で極めて重要である。この区別機能は細菌認識系及び細胞シグナル伝達系に刷り込まれている。細菌細胞の表面構造の認識は、上皮細胞の先端側表面及び基底外側表面に発現するToll様受容体の機能の1つであり、この受容体は核因子κB(NF−κB)仲介による免疫活性化を引き起こす(ガーウィルツ(Gerwirtz)ら、2001年)。従来、免疫抑制剤の活性に関連した受容体系については確認されていない。
【0005】
ヒト共生細菌B.テタイオタオミクロンを無菌マウスに投与することによって、腸の成熟やバリア機能の発達に関連する重要な遺伝子の発現が引き起こされた(フーパー(Hooper)ら、2001年)。この投与によって、Fucα1,2Galβ−グリカンの腸内レベルも上昇し(ブライ(Bry)ら、1996年;フーパー(Hooper)ら、2001年)、また、マトリリシンの腸内レベルも上昇した(ロペス−ボアド(Lopez-Boado)ら、2000年)。また、B.テタイオタオミクロンは、サルモネラ菌を用いたインビトロチャレンジに対する上皮細胞の反応を変化させることも見出された(キャンベル(Campbell)ら、2001年)。特に、炎症誘発経路の一部が抑制された(キャンベル(Campbell)ら、2001年)。B.テタイオタオミクロンはこのような潜在的防御特性を有するが、ウェルシュ菌血清型Aの感染に対する完全無菌マウスの耐性を増加させることはなかった(ユルドゥセフ(Yurdusev)ら、1989年)。しかし、このマウスをフソバクテリウム・ネクロジーン(ユルドゥセフ(Yurdusev)ら、1989年)及び非病原性クロストリジウム菌株CI(ユルドゥセフ(Yurdusev)ら、1986年)と併用してB.テタイオタオミクロンで処理すると、病原菌が腸から排除された。このことから、B.テタイオタオミクロン単独でも腸内の潜在的防御変化を誘発することが可能であるが、感染に対する全耐性を大幅に高めるためには他の共生菌株と協力して作用させる必要のあることが示唆された。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明においては、非病原性細菌並びにPPARγの核−細胞質内分布及び(NF−κB)の転写活性の双方による新規なメカニズムを明らかにする。
【0007】
更に本発明は、炎症反応の調節のための非病原性細菌の使用に関する。更に本発明は、IκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化についての実証データと、ペルオキシソーム増殖因子によって活性化された受容体(PPARγ)の新規な作用モデルについての実証データとを提供する。特に本発明は、非病原性細菌を用いてPPARγの核−細胞質内分布とNF−κBの転写活性とを変化させることによる上皮炎症性遺伝子発現の抑制について記載する。更に本発明は、炎症を軽減して疾患や炎症性障害を治療及び予防する手段を提供する。
【0008】
本発明に記載の新規な経路を用いて炎症性サイトカインの産生を調節する新規な方法及び新規な製品をスクリーニングすることができる。更に、本発明は、炎症性サイトカインの産生を抑制し、免疫系をホメオスタシスへ回復させることにおける非病原性細菌の使用について記載する。
【0009】
本明細書に記載の炎症性サイトカイン産生の調節は、公知の刊行物に記載のものとは多くの面で異なる。
【0010】
米国特許第5,925,657号は、PPARγのアゴニストについて記載している。このアゴニストはチアゾリジンジオン、即ち、チオゾリジンジオン核に置換アリール部位が結合した化学的化合物である。本発明においては、非病原性細菌を用い、PPARγを直接活性化することにより炎症反応を軽減する。
【0011】
ネイシュら(2000年)は、IκBαユビキチン化の阻害による上皮反応の調節について記載している。本発明とネイシュら(2000年)の記載のものとでは、用いる作用モードが異なる。
【0012】
IκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化についての実証データと、ペルオキシソーム増殖因子によって活性化された受容体(PPARγ)の新規な作用モデルについての実証データとを提供する。この新規なデータによって、炎症性サイトカインの産生を調節するための新規な手段及び新規な製品を考案する可能性が提供される。また、得られた結果のインビボでの検証についても記載する。
【課題を解決するための手段】
【0013】
本発明は、サイトカイン産生に起因する炎症性疾患の治療用候補薬剤を選択するためのアッセイを提供する。本発明のアッセイは、
a)候補薬剤を腸細胞に接触させることと、
b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び
PPARγの核質内破壊から成る群から選択される、前記候補薬剤の作用を解析することを含む。
【0014】
本発明のアッセイは、
NF−κBファミリーの転写因子の核外移行の促進或いは核内移行の抑制、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の増加、及び
PPARγの核質内破壊から成る群から選択される少なくとも1つの作用を示す候補薬剤を選択する工程を更に含んでもよい。
【0015】
好ましい実施形態において、解析される候補薬剤の作用は、NF−κBファミリーの転写因子の核外移行又は腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化である。
【0016】
他の好ましい実施形態において、本発明のアッセイは、TNF−α、IL−8、MIP−2α及びCox−2から成る群から選択される1以上のサイトカイン(好ましくは、IL−8及び/又はMIP−2α)のレベルの変化を解析する工程を更に含む。
【0017】
好適には、本発明のアッセイに用いる腸細胞は、細胞培養で維持される腸細胞株の形態を有する。適切な細胞株としてはCaco−2細胞株が挙げられる。或いは、本発明のアッセイは、哺乳動物(例えば、マウス)等の適切な動物を用いてインビボで行うことができる。
【0018】
任意的には、本発明のアッセイは、サルモネラ菌等の公知の病原性細菌の存在下で行ってもよい。
【0019】
更に本発明は、炎症性サイトカイン産生に伴う疾患の治療方法であって、
NF−κBファミリーの転写因子の核外移行或いは核内移行、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内のPPARγ/RelA複合体量、又は
PPARγの核質内破壊を調節することができる剤の治療有効量を投与する工程を含む方法を提供する。
【0020】
好ましくは、前記剤を炎症性疾患、特に腸細胞の炎症反応によって少なくとも一部引き起こされる疾患の治療に用いる。一般に、このような疾患は炎症性サイトカインの産生を伴う。好適には、前記剤はバクテロイデス・テタイオタオミクロン又はその一構成成分である。本明細書において「その一構成成分」とは、通常B.テタイオタオミクロンの一部を形成し、免疫反応を軽減させることのできるあらゆる部分(タンパク質やポリペプチド、糖、核酸等)を意味する。
【0021】
更に本発明は、B.テタイオタオミクロン又はその一構成成分の薬剤としての使用を提供する。好ましくは、B.テタイオタオミクロン又はその一構成成分を炎症性疾患、特に腸細胞の炎症反応によって少なくとも一部引き起こされる炎症性疾患の治療に用いる。従って、本発明は、炎症性サイトカイン産生に伴う疾患や病態を軽減、予防又は治療するためのB.テタイオタオミクロンの使用を提供する。このような疾患や病態として、炎症性腸疾患(特にクローン病や過敏性腸症候群について言及する場合もある)や、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイト増殖阻害、皮膚の過剰増殖及び炎症性障害の阻害(例えば、乾癬や尋常性ざ瘡が挙げられるが、これらに限定されない)等が挙げられるが、これらに限定されるものではない。
【0022】
更に本発明は、炎症性疾患治療用薬剤の製造におけるB.テタイオタオミクロンの使用を提供する。
【0023】
更に本発明は、B.テタイオタオミクロン又はその一構成成分の治療有効量を投与することを含む、腸細胞の炎症反応の処置方法、或いは該反応を軽減する方法を提供する。
【0024】
より詳細には、B.テタイオタオミクロン又はその一構成成分を用いて、NF−κB経路を破壊し、及び/又はp65(RelA)反応を阻害し、及び/又はPPARγの刺激剤或いは阻害剤として作用させることができる。従って、B.テタイオタオミクロン又はその一構成成分を炎症性サイトカイン産生に伴う疾患の治療に用いることができる。
【0025】
B.テタイオタオミクロンは食品や座薬を用いて生菌の状態で患者に投与することができる。
【0026】
従って、本発明は、B.テタイオタオミクロン又はその一構成成分の治療有効量を投与する工程を含む、腸細胞の炎症反応を治癒させる方法について記載する。
【0027】
ここに記載された病態を予防或いは治療するためには、適当な製剤やキャリア、賦形剤、希釈剤、安定剤を用いてB.テタイオタオミクロンを経口投与し、消化管の作用部位に優先的に供給する。このような供給手段としては、錠剤やカプセル剤等の固形製剤、ヨーグルトや飲料、懸濁剤等の液状製剤等の製剤が挙げられるが、これらに限定されない。優先的な供給手段の1つとして、B.テタイオタオミクロンを経口的に、好ましくは不都合なく胃の酸性環境を経由させて腸内の作用部位に供給する手段が挙げられる。B.テタイオタオミクロンはプレバイオティックと共に投与してもよい。
【0028】
更に本発明は、本明細書に記載の新規な経路を介して炎症性サイトカイン産生をアゴニスト的或いはアンタゴニスト的に調節する可能性のある特定のリガンドをスクリーニングするための、PPARγの作用とIκBα仲介によるRelAの搬出に影響を及ぼすp65(RelA)の特異的ヒストンアセチル化との新規モードを提供する。
【0029】
更に本発明は、炎症性サイトカイン産生に伴う疾患の治療方法であって、NF−κBの転写活性を変化させること或いは腸細胞のサイトゾル内のPPARγ/RelA複合体量を増加させることが可能な化合物の治療有効量を投与する工程を含む方法を提供する。
【0030】
更に本発明は、哺乳動物の免疫系をホメオスタシスに回復させ維持させる方法であって、NF−κBの転写活性を変化させること或いは腸細胞のサイトゾル内のPPARγ/RelA複合体量を増加させることが可能な化合物の治療有効量を投与する工程を含む方法を提供する。
【0031】
本発明者らは、B.テタイオタオミクロンの抗炎症活性が、NF−κBの核外移行の促進とサイトゾル内でのPPARγ仲介による隔離とによって特徴づけられる全く新規な作用モードを伴うことを見出した。実験的検討(詳細は実施例に記載)は次の3段階で行った。
1)病原性/非病原性細菌への接触後のCaco−2細胞内での炎症性サイトカイン遺伝子の発現。データはcDNAマクロアレイ、リアルタイムPCR及びノーザンハイブリダイゼーション解析を用いて得た。
2)B.テタイオタオミクロンの抗炎症作用の生理学的妥当性のインビトロ(Caco−2トランスウェル培養)及びインビボ(最少量細菌叢ラット)での検証。
3)NF−κB及びAP−1シグナル変換経路の解析、並びにサイトゾル内でのRelAとPPARγとの関係の解析。
【0032】
実験的検討を行った結果、次の事項が示された。
1)B.テタイオタオミクロンによる調節の主なターゲットはNF−κBであり、AP−1ではない。
2)B.テタイオタオミクロン及び腸炎菌(S. enteritidis)で処理した細胞内にはp65(RelA)が蓄積する(最長30分間検出した)。
3)B.テタイオタオミクロン存在下でのp65の転写作用の低下は、p65の核外排除(移行)の促進に起因し、この核外移行はLMB感受性である(即ち、crm−1仲介による)。
4)データは全て、B.テタイオタオミクロンに起因する、炎症性サイトカイン/ケモカインの発現の低下、IκBαの発現、PMNの補充、及びラットにおいてインビボで示された生理的低レベルの炎症に関連付けられる。
5)B.テタイオタオミクロンの存在下でPPARγはサイトゾルに局在する。
6)PPARγとp65がサイトゾル内に共存する場合、両者は物理的に結合する。
【図面の簡単な説明】
【0033】
【図1】(a)1)非感染Caco−2細胞、2)108個の腸炎菌を感染させたCaco−2細胞、3)108個の腸炎菌と109個のB.テタイオタオミクロンを感染させたCaco−2細胞、及び4)109個のB.テタイオタオミクロンを感染させたCaco−2細胞のTNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたサイトカインmRNAのノーザンブロットハイブリダイゼーション。 (b)IL8及びG3PDHの半定量的PCR。(1)培地のみでインキュベートしたCaco−2細胞;(2)108個の腸炎菌、108個の大腸菌0157H7、PMA(300ng/ml)或いはIL−1α/β(20ng/ml)(図示の通り)と共にインキュベートしたCaco−2細胞;(3)109個のB.テタイオタオミクロンの存在下で上記(2)のようにインキュベートしたCaco−2細胞;(4)109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞。 (c)1)非感染Caco−2細胞、2)108個の腸炎菌を感染させたCaco−2細胞、3)108個の腸炎菌と109個のB.ブルガータスを感染させたCaco−2細胞、及び4)109個のB.ブルガータスを感染させたCaco−2細胞のTNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたmRNAのノーザンブロットハイブリダイゼーション。 (d)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にCaco−2細胞を2時間或いは4時間(図示の通り)インキュベートした。 (e)上記(a)の処理群(1)〜(4)のMPOアッセイにより測定した、Caco−2細胞単層を経由するPMN細胞の経上皮遊走。 (f)(1)対照、(2)108個の腸炎菌、(3)B.テタイオタオミクロンに次いで108個の腸炎菌、及び(4)B.テタイオタオミクロンのみでチャレンジしたラットの処理後6日目の回腸粘膜のMPOアッセイ。データは平均±SD(n=3)。
【図2】1)非感染Caco−2細胞、2)108個の腸炎菌と共にインキュベートしたCaco−2細胞、3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞、及び4)109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞。次の一次抗体を用いて試験した。抗RelA(A〜D)、抗PPARγ(E〜H)、抗IκBα(I〜L)及び抗pIκβα(M〜P)。スケールバーは全て25μm。O及びP中の挿入図は点状核標識の詳細を示す。
【図3】(A)図1Aで上述した群(1)〜(4)に関して、コンセンサス32P標識NF−κB結合配列オリゴヌクレオチドを用い、抗RelA抗体と共にインキュベートしたCaco−2核抽出物について行ったスーパーシフトEMSA。ここで、上記B.テタイオタオミクロンは70℃、15分で熱失活させた。 (B)上記実験処理群(1)〜(4)のmRNAのノーザンハイブリダイゼーション。ブロットは特異的32P標識IκBα及びG3PDHプローブを用いて試験した。 (C)Caco−2細胞由来の核抽出物のウェスタンブロット。処理群は上述の通りである(1〜4)。免疫ブロットは、抗p38及び抗pp38(ニューイングリッシュバイオラボ)(New English Biolabs)に特異的な抗体を用いて試験した。 (D)c−Fos及びc−Junの超誘発は、Caco−2細胞から得た全RNAについてのノーザンハイブリダイゼーションによって測定した。Caco−2細胞の処理はシクロヘキサミドの非存在下(−C)或いはシクロヘキサミド(10μg/ml)の存在下で上述のように(1〜4)行った。ブロットは、c−Fos、c−Jun及びG3PDHに特異的な32P標識プローブでハイブリダイズした。 (E、F)Caco−2細胞由来の核抽出物のウェスタンブロット。処理群は上述の通りである(1〜4)。c−Fos(サンタクルズ)、ATF−2及びpATF−2(ニューイングランドバイオラボ)(New England Biolabs)に特異的な抗体を用いた。 (G)IκBα及びpIκBαを用いた免疫沈降(IP)。Caco−2細胞は標準培養プロトコルに従い調製した。処理群(1〜4)。IPはウェスタンブロッティングで解析した。NSは非特異的であることを示し、Blは細胞抽出物を含まない対照である。
【図4】(A)特異的32P標識PPARγ及びPPARαプローブで解析した、(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートした後のCaco−2細胞から得たmRNAのノーザンハイブリダイゼーション。細菌の添加は2時間行った。 (B)処理(1)及び(2)は上述の通りであり、更に(3a)30μMの15PG−J2の存在下で108個の腸炎菌と共にインキュベートしたCaco−2細胞、及び(3b)30μMのフェノフィブラートの存在下で108個の腸炎菌と共にインキュベートしたCaco−2細胞を用いた(図示の通り)。細菌及びこれら薬剤の添加は2時間行った。32P標識PPARγ特異的プローブを用いてノーザンハイブリダイゼーションによりmRNAを解析した。 (C)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)(4)(5)10、20或いは30μMの15PG−J2の存在下で108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(6)30μMの15PG−J2のみと共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (D)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(3)(4)10或いは30μMのシグリタゾンの存在下で108個の腸炎菌と共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (E)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、及び(3)108個の腸炎菌及び30μMのフェノフィブラートと共にインキュベートした後のCaco−2細胞から得たmRNAを、32P標識TNF−α、IL−8、COX−2及びG3PDHを用いたノーザンハイブリダイゼーションで解析した。細菌及び薬剤の添加は2時間行った。 (F)(1)非感染Caco−2細胞、(2)108個の腸炎菌と共にインキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートした後のCaco−2細胞から得た核抽出物及び細胞質抽出物を抗PPARγ(サンタクルズ)を用いたウェスタンブロッティングで解析した。細菌は2時間適用した。Caco−2細胞は核画分、細胞質画分、洗浄剤可溶画分(D.可溶)或いは洗浄剤不溶画分(D.不溶)として調製した。 (G)108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間共培養したCaco−2細胞を1%ノニデットP40溶液含有PBSに溶解し、ポリクローナル抗RelAセファロース(サンタクルズ)と共に一晩インキュベートした。免疫沈降物をSDS−PAGEを用いて分離した。モノクローナル抗RelA[NF−κB p65]、抗PPARγ抗体及び抗HDAC3(サンタクルズ)を用いてウェスタンブロットを展開した。 (H)PPARγ(f〜m、o)及びRelA(n)の免疫蛍光顕微鏡検査。プレートn及びo(p)からPPARγとRelAとの共存を確認。培地のみで(t、j)、108個の腸炎菌と共に(g、k)、108個の腸炎菌及び109個のB.テタイオタオミクロンと共に(h、l、n〜p)、或いは109個のB.テタイオタオミクロンと共に(i、m)Caco−2細胞を増殖させた。4μMのTSAの非存在下(t〜i、n〜p)及び存在下(j〜m)で処理を行った。スケールバーは全て25μm。
【図5】PPARγの変異はB.テタイオタオミクロン仲介によるNF−κBの抑制及びRelAのサイトゾル内隔離を妨害する。 (A)RelAの免疫検出。Caco−2細胞は標準培養プロトコルに従い調製した。処理群(1〜4)。免疫沈降物は抗PPARγセファロース複合体を用いて調製した。 (B)RelA/PPARγ結合を示すインビトロ翻訳 (C)PPARγDNとNF−κBルシフェラーゼレポーター構造体をトランスフェクトしたCaco−2細胞(DN)と、NF−κBルシフェラーゼレポーターのみをトランスフェクトした対照細胞(MOCK)とを比較した。(1)非感染Caco−2細胞、(2)108個の腸炎菌と共に更に6時間インキュベートした後のCaco−2細胞、及び(3)109個のB.テタイオタオミクロンの存在下で108個の腸炎菌と共に更に6時間インキュベートした後のCaco−2細胞において、ルシフェラーゼ活性(%刺激で示す)を測定した。 (D)構成的に発現する緑色蛍光タンパク質(GFP)構造体をトランスフェクトしたCaco−2細胞(B、C)及びPPARγDNとGFPをトランスフェクトしたCaco−2細胞(D、E)。細胞は全て108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。次いで、細胞を抗RelA(p65)特異的抗体(サンタクルズ)で免疫染色し、LSCMで試験した。(B)及び(D)はGFP(緑)とRelA(赤)の複合画像を示す2チャンネルキャプチャである。(B)及び(D)はそれぞれ(B)及び(D)と面積が同じであり、RelAのみを示す1チャンネルキャプチャである。スケールバー=25μM。 (E)カルボキシ末端に黄色蛍光タンパク質(YFP)を有するRelAキメラ構造体とPPARγ(d〜f)或いはPPARγDN(g〜i)とをCaco−2細胞にトランスフェクトした。PPARγ及びPPARγDNはいずれもカルボキシ末端にシアン蛍光タンパク質(CFP)を有するキメラ構造体である。トランスフェクション後2日目に、細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。CFP蛍光(d、g)及びYFP蛍光(e、h)。CFP−PPARγとYFP−RelAとの共存をプレートd及びe(f)から確認し、CFP−PPARγ(DN)とYFP−RelAとの共存をプレートg及びh(i)から確認した。スケールバーは全て25μm。(j、k)CFP−PPARγをHela細胞にトランスフェクトし、トランスフェクション後2日目に、細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートした。Hela細胞を固定し、透過処理し、免疫染色してSC35を確認した(immuno-stained for SC35)。CFP−PPARγ(j)及びSC35の間接免疫蛍光(k)。スケールバー=10μm。
【図6】図6は、対照ラット、バクテロイデス・テタイオタオミクロンを経口投与したラット(BT)、腸炎菌を経口投与したラット(SE)、及び腸炎菌とB.テタイオタオミクロンとを経口投与したラット(SE+BT)での骨格筋(□:湿重量、■:乾燥重量)の成長(mg/ラット/日)を示す。*は、乾燥重量及び湿重量それぞれについて対照、BT及びSE+BTと有意差があることを示す(p≦0.05)。骨格筋を腓腹筋で代表させたが、若いHooded−Listerラットの全骨格筋重量は腓腹筋重量の約47倍である(バルドクス(Bardocz)ら、1996年)。腓腹筋の初期重量は、初期新鮮体重100g当り湿重量で804±10mg[乾燥重量で194±7mg]。
【図7】図7は、ケモカイン/サイトカインmRNA発現への作用を示す定量的データ(ノーザンブロット及びリアルタイムPCR)を示す。S.e.=腸炎菌。B.t.=B.テタイオタオミクロン。
【図8】図8は、Caco−2細胞単層と共に2時間インキュベートした後の細菌の数を示す。 a)B.tの存在下及び非存在下でCaco−2細胞に付随するB.t(細胞単層洗浄後に残ったB.t)。 b)B.tの存在下及び非存在下でCaco−2細胞に付随するS.e。 c)B.tの存在下及び非存在下でCaco−2細胞に浸潤した腸炎菌(細胞洗浄及びゲンタマイシン処理(100μg/ml、4時間)後に残った腸炎菌)。n=6、+/−標準偏差。
【図9A】:EMSAを用いて測定した、Caco−2細胞における腸炎菌に呼応するNF−κB活性化を示す。
【図9B】:ノーザンハイブリダイゼーションによるサイトカイン反応の解析 a)108及び1010cfuのS.eに2時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。 b)109個のS.eに各時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。2時間或いは4時間まで接触させた際のデータも示す(図9D)。 c)107〜1010個のB.t.に2時間接触させた後のCaco−2細胞から得たmRNAを解析して炎症誘発性サイトカインの誘発を確認し、G3PDHに対して規準化した。 d)Caco−2細胞から得たmRNAを解析して炎症性サイトカインの発現(COX−2及びIL−8)を確認した。2時間及び4時間でのB.t.の抑制作用を示す。
【図9C】:免疫細胞化学的検出法を用いて得た、腸炎菌及びB.テタイオタオミクロンにCaco−2細胞を接触させた後のRel(p65)活性化の経時データ。S.e.+/−B.t.に2時間接触させたCaco−2細胞を4%パラホルムアルデヒドに固定し、Triton X−100で透過処理し、抗p65(RelA)(サンタクルズ)で免疫標識した。二次検出はAlexa Fluor488抗ウサギIgG(モレキュラープローブ)を用いて行った。Axiovert2000顕微鏡に搭載したZeiss Axiocamを用いて画像をデジタル記録した。
【図9D】:免疫細胞化学的検出法を用いて得た、腸炎菌とB.テタイオタオミクロンにCaco−2細胞を接触させた後のPPARγ作用の経時データ。 S.e.+/−B.t.に2時間接触させたCaco−2細胞を4%パラホルムアルデヒドに固定し、のTriton X−100で透過処理し、抗PPARγ(サンタクルズ)で免疫標識した。二次検出はAlexa Fluor568抗ヤギIgG(モレキュラープローブ)を用いて行った。Axiovert200顕微鏡に搭載したZeiss Axiocamを用いて画像をデジタル記録した。
【図9E】:B.テタイオタオミクロンと腸炎菌に2時間接触させた後のCaco−2細胞におけるNF−κB p65とPPARγとの共標識化(co-labelling)を示す。
【図10】対照ラット、B.テタイオタオミクロンを経口投与したラット[B.t]、腸炎菌を経口投与したラット[S.e]、及び腸炎菌とB.テタイオタオミクロンとを経口投与したラット[S.e+B.t]での骨格筋(腓腹筋)(□:湿重量、■:乾燥重量)の増量(mg/ラット/日)。n=6、+/−標準誤差。
【図11】SDS−PAGEを用いて分離し、エレクトロブロットし、アセチル化リジンモノクローナル抗体で免疫染色したp65免疫沈降物を示す。
【発明を実施するための形態】
【0034】
以下、非限定的な実施例と図面とを参照し本発明について更に説明する。
【0035】
実施例1:B.テタイオタオミクロンとB.ブルガ−タス(B. vulgatus)の存在下及び非存在下で腸炎菌に接触したCaco−2細胞の炎症反応
【0036】
細菌による宿主炎症反応の調節についての実証データは、バクテロイデス・テタイオタオミクロンの存在下及び非存在下で共培養した腸炎菌にCaco−2細胞を短時間接触させた後に炎症性遺伝子の発現を検討した結果から得た。cDNAマクロアレイ技法(クロンテック、Atlasヒトサイトカイン/受容体アレイシステム)を用いて、TNF−α、IL−8、MIP−2α及びCOX−2を含む数種類の遺伝子を同定した。腸炎菌との接触後のこれら遺伝子の誘発はB.テタイオタオミクロンの存在により低下した。この結果はノーザンハイブリダイゼーション及びリアルタイムPCRを用いて確認された(図1a、f)。また、IL−1α、IL1−β、TNF−α、PMA、LPS及び腸出血性大腸菌0157H7(図1F)を含む他の炎症性メディエータに対するB.テタイオタオミクロンの抗炎症活性についても検討した。Caco−2細胞内でIL−8発現を誘発したこれらリガンドの内、PMA、腸炎菌及び大腸菌0157H7の作用のみがB.テタイオタオミクロンによって低下した。
【0037】
上記検討に用いる実験条件の設定に当り、多くの最適化実験(時間経過、細菌量/増殖相、Caco−2細胞継代/コンフルエンス検討を含む)を行った。重要な点は、細菌の増殖、付着及び浸潤が培養/共培養条件に影響を受けないことを立証し、これによってデータが特異的付着/浸潤に起因する可能性を排除したことである(図8参照)。
【0038】
(1)非感染Caco−2細胞、(2)108個の腸炎菌のみと共にインキュベートしたCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共にインキュベートしたCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共にインキュベートしたCaco−2細胞において炎症性遺伝子の発現を測定した。細菌の添加は2時間行った。細胞を洗浄し回収してmRNAを単離した。mRNA(5μg)を、TNF−α、IL−8、MIP−2α、TGF−β、COX−2及びG3PDHに特異的な32P標識プローブを用いたノーザンハイブリダイゼーションによって解析した。
【0039】
B.テタイオタオミクロンの作用を示す結果の一部は、フーパー(Hooper)ら(2001年)によって報告された最近の分子解析と一致する。本発明者らによる結果は、ノーザンハイブリダイゼーション及びリアルタイムPCRを用いて確認された。TNF−α、IL−8、MIP−2α及びCOX−2のレベルは、腸炎菌チャレンジに呼応して有意に上昇した(図1a)。しかし、B.テタイオタオミクロンの存在下では、TFG−βの場合を除き、これら遺伝子の全てにおいて発現レベルが低下した。TFG−βの発現レベルは腸炎菌によって低下したが、B.テタイオタオミクロンの存在により対照値に維持された。これら両方の細菌の生残性及び増殖速度、並びに上皮細胞に付着し浸潤した腸炎菌の数については上述の処理の間で差がなく、培養手順による影響を受けなかった。
【0040】
上記結果を立証する定量的データを図7に示すが、このデータは、ビヒクル(対照)、5×108個の腸炎菌(S.e)、5×108個のS.e及び1.5×109個のB.テタイオタオミクロン(B.t)、或いはB.tのみと共に2時間インキュベートした後のCaco−2細胞から精製したmRNAを用いて得た。ノーザンブロットをデンシトメトリーによって定量化し、G3PDHレベルに対して規準化した。また、ABI Taqmanを用いてリアルタイムPCRを行い、18SrRNAレベルに対して規準化した(n=4 +/−標準偏差)。
【0041】
炎症性サイトカイン発現の低下がB.テタイオタオミクロンに特異的なものかどうかを調べるため、関連する耐気性菌株であるB.ブルガ−タスについても検討した。
【0042】
(1)と(2)については上述の様に処理を行い、更に、(3a)108個の腸炎菌及び109個のB.ブルガ−タスと共にインキュベートしたCaco−2細胞、及び(4a)109個のB.ブルガ−タスのみと共にインキュベートしたCaco−2細胞を用いた。細菌の添加は2時間行った。
【0043】
B.ブルガ−タスは抗炎症活性について陰性であった(図1b参照)。
【0044】
得られた結果において、上記共生菌株の両方によって誘発される炎症性遺伝子の発現が低かったことは、対数期後期の細菌を試験した事実に関連するかもしれないが、注目に値する。細菌細胞片を有しない対数期初期の細菌を用いれば、炎症反応を完全に抑制することができるであろう。従って、Caco−2細胞を108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間或いは4時間インキュベートする実験を更に行った。IL−8発現に対するB.テタイオタオミクロンの阻害作用は長時間持続した(図1d)が、TNF−αの場合はこれとは対照的であった。この結果から、上記遺伝子の発現を調節するメカニズムはB.テタイオタオミクロンによって特異的に影響を受けることが示唆される。
【0045】
B.テタイオタオミクロンによる炎症性サイトカインの抑制を示すデータの生理学的妥当性を、PMN補充の機能性インビトロモデル及びインビボ腸炎菌ラット感染モデルを用いて確認した。インビトロ及びインビボでのPMN補充はミエロペルオキシダーゼ(MPO)活性を用いてモニターした。
【0046】
Caco−2細胞をインバーテッドトランスウェル(コーニング)に播種し、様々な組合せの細菌を該細胞の先端表面に添加した。新たに単離したヒトPMNを該細胞の側底区画に添加し、該PMNの経上皮遊走をMPOアッセイ(ネイシュ(Neish)ら、2000年)で測定した。処理群(1〜4)については上述の通りである。細胞を2時間インキュベートした後、細菌を除去し新しい培地を添加した。細胞を更に2時間インキュベートした後、HBSSで洗浄した。細胞と先端区画から得た培地とを1%のTriton C−100に可溶化し、MPOを定量した。
【0047】
多形核白血球(PMN)補充アッセイのインビトロモデルから得た結果から、B.テタイオタオミクロンの抗炎症活性が確認された(図1e)。
【0048】
ラットの腸の腸炎菌感染が主に回腸、即ち、B.テタイオタオミクロンの棲息部位で起こることは注目に値する。
【0049】
離乳したばかりの(21日齢)最少量細菌叢ラット(通常の実験飼料で飼育)を2つの群に分け、一方のラット群を更に108cfuのB.テタイオタオミクロンを含む嫌気的に調製したゼリー(0.5g/日)で19日間飼育した。各群のラットの半数に対して108個の腸炎菌で経口チャレンジを行った。全てのラットにおける炎症反応の重症度を、腸炎菌感染後6日目に回腸粘膜内のMPOレベルを測定することによって評価した。ラットに対して行った処理は次の通りである。(1)B.テタイオタオミクロン投与無し、腸炎菌投与無し、(2)B.テタイオタオミクロン投与無し、108個の腸炎菌投与、(3)B.テタイオタオミクロン投与に次いで108個の腸炎菌投与、及び(4)B.テタイオタオミクロンのみ投与。実験は少なくとも3回行ったが、いずれも同様の結果が得られた。
【0050】
腸炎菌でチャレンジしたラットの回腸粘膜内のMPOレベルは上昇した(p<0.005)が、B.テタイオタオミクロンの先行経口接種及び細菌叢内での安定化によりMPOレベルは有意に低下した(p<0.001)(図1f)。また、PMN補充実験から、腸炎菌によって400pg/mlのIL−8タンパク質が培養上清内に4時間に亘り誘導されるが、B.テタイオタオミクロンの存在下で共培養した場合、IL−8タンパク質濃度は230pg/mlに有意に低下することを見出した。B.テタイオタオミクロンによるコロニー形成は、腸組織の特異的プライマー増幅によって確認した。IL−8およびMIP−2αはPMN経上皮遊走に対する必須ケモカインである(マコーミック(McCormick)ら、1993年;ハング(Hang)ら、1999年)ため、B.テタイオタオミクロンの作用は、IL−8及びMIP−2αの転写の減少に起因し、この転写減少は次いで活性タンパク質の翻訳及び分泌に影響を及ぼす。ネイシュ(Neish)ら(2000年)の記載と一致するが、本発明者らは、遺伝子発現を調節する宿主系を破壊する(subverting)ことにより非病原性細菌が免疫抑制作用を発揮し得ることを新規なモードシステムを用いて立証した。重要なのは、本発明者らによる知見がインビボでも検証されたことである。
【0051】
上述したMPOデータに加えて、図10は、腸炎菌によって誘発される炎症反応時の筋細胞(muscle biology)に対するB.テタイオタオミクロンの防御作用の実証データを示す。
【0052】
実施例2:B.テタイオタオミクロンは、NF−κB及びPPARγタンパク質の細胞内分布と活性化状態を変化させることによって炎症を軽減し、また、NF−κBシグナル変換経路を標的破壊する。
【0053】
転写因子のNF−κBファミリーは炎症反応の調節において中心的役割を果たす。これらのタンパク質は、Rel相同ドメインと称される高いNH2末端保存配列を共有するが、この配列は、タンパク質のサブユニット二量体化、DNA結合、及び阻害性IκBタンパク質との相互作用に必要である。NF−κBを誘発するシグナルは阻害性IκBタンパク質(Ser−32及びSer−36ではIκBα)のリン酸化を引き起こし、次いでユビキチン化とプロテオソーム仲介による分解とをターゲットとする。これによって、RelA(p65)等のRelタンパク質は遊離し、核に転位しDNAを結合する。
【0054】
非感染Caco−2細胞(1)を108個の腸炎菌のみ(2)、108個の腸炎菌及び109個のB.テタイオタオミクロン(3)、及び109個のB.テタイオタオミクロンのみ(4)と共にそれぞれ2時間インキュベートした。細胞を4%パラホルムアルデヒドに室温で30分間固定し、0.2%のTriton−X1000を用いて4℃で透過処理し、間接免疫蛍光顕微鏡検査法にて試験した。一次抗体(サンタクルズ):抗RelA[NF−κB p65](A〜D)、抗PPARγ(E〜H)、抗IκBα(I〜L)及び抗pκBα(M〜P)。二次抗体:Alexa Fluor種特異的抗IgG(モレキュラープローブ)。
【0055】
Caco−2細胞を腸炎菌に接触させることによって一連のイベントが引き起こされ、その結果、RelAの核への転位が促進されることが判明した(図2B)。
【0056】
特筆べき点として、B.テタイオタオミクロンによってRelAの核への転位が無くなることが見出されたが、B.テタイオタオミクロンへ2時間接触させた後、ほぼ全てのRelAはサイトゾルに局在した(図2C、D)。
【0057】
IκBαのリン酸化と、重要なこととしてのIκBαの分解は、B.テタイオタオミクロンの存在下及び非存在下での腸炎菌への接触後に観察された(図2G)。2時間の接触後、特に腸炎菌のみに接触させた細胞と腸炎菌及びB.テタイオタオミクロンに接触させた細胞において、IκBαのmRNA(図3B)及びIκBαタンパク質(図2J、K)のレベルが上昇したが、このことは転写活性NF−κBの存在を示している(チェン(Cheng)ら、1994年;チャオ(Chiao)ら、1994年)。
【0058】
生残B.テタイオタオミクロンは、NF−κBタンパク質の転写活性を選択的に妨害するが、AP−1タンパク質の合成やリン酸化には影響を及ぼさない。
【0059】
(1)非感染Caco−2細胞、(2)108個の腸炎菌と共に2時間インキュベートした後のCaco−2細胞、(3)108個の腸炎菌及び109個のB.テタイオタオミクロンと共に2時間インキュベートした後のCaco−2細胞、及び(4)109個のB.テタイオタオミクロンのみと共に2時間インキュベートしたCaco−2細胞から得た核抽出物を、RelA[NF−κB p65]特異的抗体(サンタクルズ)と共にインキュベートし、この抽出物に対してスーパーシフトEMSAを行った。上記B.テタイオタオミクロンはCaco−2細胞へ添加する前に70℃、15分で熱失活させた。
【0060】
電気泳動移動度シフトアッセイ(EMSA)によって、RelAが腸炎菌により活性化される主要なサブユニットであることが確認された(図3A)が、p52の活性化レベルが低いことも明らかになった(データは図示せず)。次に、本発明者らは、B.テタイオタオミクロンがNF−κBシグナル変換経路を破壊することによって、腸炎菌により引き起こされる免疫反応メディエータの産生を低下させるという仮説を立てた。
【0061】
同様の実験を行った結果を図9Aに示すが、Caco−2細胞内で腸炎菌に接触後2時間でp65の活性化はピークに達することがEMSAスーパーシフトにより示された。レーン1:タンパク質無し、レーン2、5、8、11:対照細胞から得た核抽出物、レーン3、6、9、12:S.e.を感染させた細胞から得た核抽出物、レーン4、7、10、13:S.e.を感染させp65(RelA)抗体でスーパーシフトさせた細胞から得た核抽出物。
【0062】
更に、熱失活していない生残B.テタイオタオミクロンによってRelA反応が阻害されることが示された(図3A)。また、培養上清及び調製培地(B.テタイオタオミクロンに接触させたCaco−2細胞から得た培地)のいずれも生物活性を有しないことも示された(データは図示せず)。このことから、上皮細胞への接触がB.テタイオタオミクロンの抗炎症活性には必須であることが示唆される。腸炎菌に接触させてから20分以内に、B.テタイオタオミクロンの存在下及び非存在下でIκBαのリン酸化及び分解を観察した(データは図示せず)。また、検討対象細菌株の生残性、増殖、付着及び浸潤は培養処理による影響を受けなかった(データは図示せず)。従って、上記観察された作用は受容体認識及び活性化の差には起因しないと考えられる。
【0063】
上記と同じ実験群(1〜4)を用いて、細菌の添加を2時間行った後、mRNAをノーザンハイブリダイゼーションで解析し、ブロットを特異的32P標識IκBα及びG3PDHプローブを用いて試験した。IκBαのmRNA(図3B)及びIκBαタンパク質(図2J、K)のレベルはいずれも上昇した。このレベルの上昇は、IκBαタンパク質の新生合成を誘発するRelAの転写活性に起因するものとされる(チェン(Cheng)ら、1994年)。IκBαは次いで核に侵入しRelAと結合し、RelAをDNAプロモーター部位から除去する(アレンザナ−セイスデドス(Arenzana- Seisdedos)ら、1995年)。興味深いことに、PPARα仲介によるIκBα合成の刺激についても報告されている(デレライブ(Delerive)ら、2000年)。一過性のRelA転位がB.テタイオタオミクロンで処理した細胞内で起こるかどうかを更に調べるため、CRM−1依存型核外移行の特異的阻害剤であるレプトマイシンB(LMB)を用いて実験を行った。Relタンパク質(例えば、p65)の細胞質内での位置はRelA/IκBα複合体のCRM−1依存型核外移行によって維持される(ファン(Huang)ら、2000年;タム(Tam)ら、2000年)というのが理論的根拠である。LMBによってRelAが腸炎菌及びB.テタイオタオミクロンの両方と共培養した細胞の核内に蓄積することを見出したが(結果は図示せず)、このことは、B.テタイオタオミクロンの存在に関係なく、核への一過性RelA転位が起こることを示している。この結果は、腸細胞を細菌へ初期接触させた後のサイトゾル内のIκBα/RelA複合体のリン酸化及びユビキチン化、並びにIκBα遺伝子発現の活性化と一致する。
【0064】
実施例3:B.テタイオタオミクロンはPPARγの核−細胞質間輸送を誘発させ、RelAを隔離する。
【0065】
非病原性細菌による免疫抑制についての最近の報告によると、その抑制メカニズムはIκBαのユビキチン化及び分解の阻害に起因するものとされている(ネイシュ(Neish)ら、2000年)が、このことは本発明者らによるモデル系には明らかに適用されない。腸炎菌及びB.テタイオタオミクロンの両方と共培養した細胞におけるNF−κB複合体のサイトゾル内での主な位置は、IκBα仲介による核外移行が核内局在化よりも効率よく起こることから説明されるであろう。腸炎菌及びB.テタイオタオミクロンの両方で処理した細胞の核内でのIκBαタンパク質のレベルは、腸炎菌のみで処理した場合に比べて若干高いが、このことはNF−κBの不活化を促進し、B.テタイオタオミクロンの全抗炎症特性に寄与する可能性がある。しかし、RelAの特異的脱アセチル化がより適切な説明となる。核内RelA活性化の継続時間が可逆的アセチル化によって測定されることが最近報告されている(チェン(Chen)ら、2001年)。アセチル化されたRelAはIκBαに対する親和性が低いが、ヒストンデアセチラーゼ3(HDAC3)でRelAを脱アセチル化することにより、IκBαへの結合が促進され、CRM−1仲介によるRelAの核外移行が促進される。本発明者らは、B.テタイオタオミクロンがHDAC3のRelAとの結合を促進することを示した(図4G)。ヒストンデアセチラーゼの特異的阻害剤であるトリコスタチンA(TSA)を用いた実験を行った。その結果、TSA処理(800nM、4時間)に呼応してHDACが阻害され、腸炎菌のみで処理した細胞においてp65が部分的に蓄積し、重要なこととしてPPARγのサイトゾルへの移行が促進されることが示された。得られたデータから、B.テタイオタオミクロン処理細胞内でのp65及びPPARγの核外移行を促進する上でアセチル化/脱アセチル化が重要な役割を果たしていることが裏付けられた(図11参照)。
【0066】
更に、RelAとペルオキシソーム増殖因子によって活性化された受容体γ(PPARγ)タンパク質とが同じ細胞区画に共存することが分かった(図2C、G)ため、本発明者らは、B.テタイオタオミクロン処理細胞内での更なるRelA核内転位を制限する重要なメカニズムはPPARγによるRelAのサイトゾル内隔離を伴うという仮説を立てた。この仮説を裏付ける実験的証拠を以下に示す。
【0067】
AP−1複合体は炎症性遺伝子発現の調節において重要な役割を果たし、増殖因子やサイトカイン、細菌等の様々な細胞外刺激剤によって急速に活性化される(マイヤー−テア−フェーン(Meyer-ter-Vehn)ら、2000年)。本発明者らは、B.テタイオタオミクロンの阻害作用がこのシグナル伝達経路まで及ぶかどうかを調べた。AP−1活性は2つの主要なメカニズム、即ち、プロトオンコジーンファミリーのホモ及びヘテロ二量体から成るAP−1サブユニットの発現の上昇と該サブユニットのリン酸化とによって調節される。プロトオンコジーンファミリーとしては、Fos(c−Fos、FosB、Fra−1及びFra−2)、Jun(JunB、c−Jun及びJunD)及びATF(ATF2、ATF3/LRF2及びB−ATF)が挙げられ、メンバーは全てDNA結合タンパク質のロイシンジッパーファミリーである。これらのタンパク質は、マイトジェン活性化プロテイン(MAP)キナーゼと総称される3種類の関連キナーゼによって主に制御される。p42及びp44(細胞外シグナル調節キナーゼ;ERK)キナーゼ、c−JunN末端キナーゼ(JNK)/ストレス活性化プロテイン(SAP)キナーゼ及びp38キナーゼのタンパク質リン酸化のレベルが腸炎菌及びB.テタイオタオミクロンに呼応して変化するかどうかを調べた。腸炎菌によってp38MAPキナーゼは活性化されるが(図3C)ERKやJNKは試験時に活性化されない(結果は図示せず)ことを見出した。この知見は、p38が炎症性刺激剤によって急速に活性化され(レインギュード(Raingeaud)ら、1996年)、一方、JNKの活性化はもっと遅い時点で起こる(クジメ(Kujime)ら、2000年)という事実と一致する。p38の活性化によってelk−1のリン酸化が引き起こされ、elk−1は血清反応因子と共にc−fosプロモーター内の血清反応成分に結合し、c−fosの転写及び翻訳を促進するが、これは遺伝子及びタンパク質レベルの変化によって示される(図3D、E)。腸炎菌によって誘発されるp38のリン酸化はB.テタイオタオミクロンを用いた共培養によって変化しないことが見出した。メッセージレベル及びタンパク質レベルはいずれも腸炎菌及びB.テタイオタオミクロンに呼応して上昇し、c−fos遺伝子発現への作用は相加的なものであることが分かった。デノボc−Fox合成によって、10倍のDNA結合親和性を有するJun−Fosへテロ二量体が形成され、AP−1活性が増加する(ムスティ(Musti)ら、1997年;スミール(Smeal)ら、1991年)。ATF−2もp38MAPキナーゼ及びJNKシグナル変換経路の標的である。転写因子ATF−2はp38MAPキナーゼによりThr−69及びThr−71上でリン酸化される。一方、JNKはATF−2及びc−Junの両方をリン酸化し活性化する。従って、ATF−2とc−Junは違った形でp38及びJNKシグナル変換経路によって調節される(レインギュード(Raingeaud)ら、1996年)。本発明者らの検討においては、c−Junタンパク質レベルの上昇とc−Junリン酸化状態は腸炎菌及びB.テタイオタオミクロンのいずれにも呼応しなかったが、このことは、腸炎菌に呼応するATF−2のリン酸化(図3F)を誘発しやすいのはp38MAPキナーゼであってJNKではないことを示している。以上のデータはp38MAPキナーゼ活性に対する腸炎菌の直接の作用と一致する。腸炎菌に呼応してc−jun遺伝子転写を刺激するc−Jun/ATF−2ヘテロ二量体の形成はATF−2の活性化によって引き起こされやすい(図3D)。B.テタイオタオミクロンの抗炎症作用はNF−κB経路を選択的にターゲットとすることが分かるが、これにより、NF−κBに対して絶対条件を有する遺伝子(例えば、IL−8)(ムカイダ(Mukaida)ら、1994年;エリオット(Elliott)ら、2001年)がB.テタイオタオミクロンによる阻害に対し特に敏感である理由が説明される。
【0068】
実施例4
非病原性細菌による免疫抑制のメカニズムを更に調べるため、本発明者らは、先ず、抗炎症性サイトカインIL−10及びTGF−βについて検討した。IL−10遺伝子発現はB.テタイオタオミクロンを用いた処理には影響を受けなかった(データは図示せず)が、このことは構成タンパク質が関与する可能性を排除するわけではない。抗炎症性サイトカインTGF−βが関与する可能性を示すデータから多少の示唆が得られた。しかし、この短時間の検討の時間経過においてはデノボサイトカイン合成は恐らく十分に行われてないので、TGF−βが炎症反応をダウンレギュレートするように作用しているとすれば、TGF−βは長期の抗炎症作用に関与している可能性が高い。
【0069】
PPARが炎症プロセスの重要なモジュレータとして浮上してきた(ナカジマ(Nakajima)ら、2001年)。PPARはリガンド活性転写因子であり、ヘテロ二量体のパートナーとしてのレチノイドX受容体(RXR)と共に、PPAR反応成分(PPRE)と称される特異的DNA配列成分に結合し、遺伝子発現を調節する。しかし、最近の研究から、AP−1及びNF−κB仲介による遺伝子発現を調節する上でPPARγのリガンド活性化が重要であることが示唆された(スー(Su)ら、1999年)。本発明者らは、B.テタイオタオミクロンによるNF−κBの調節におけるPPARの役割について調べた。腸炎菌とB.テタイオタオミクロンの両方に接触させた細胞内でPPARα及びPPARγのmRNAが減少すること(図4A)、そして、特異的PPARγリガンドである15−デオキシΔ12,14−プロスタグランジンJ2(15d−PGJ2)に接触させた細胞内でもmRNAが減少する一方で、特異的PPARαリガンドであるフェノフィブラートに接触させた細胞内ではmRNAが減少しないこと(図5B)を示すことができたが、この知見は受容体活性化と一致する。従来、PPARγの活性化は、3T3−L1脂肪細胞におけるPPARγのmRNA及びタンパク質のダウンレギュレーションを伴うものとされていた(キャンプ(Camp)ら、1999年)。本発明者らは更に、15d−PGJ2及びシグリタゾンを用いて、生理学的と考えられる濃度範囲に亘って腸炎菌による炎症性サイトカイン発現の活性化が低下することを示した(図4C、D)。15d−PGJ2がIκBキナーゼを直接阻害できる(シュトラウス(Straus)ら、2000年)ものとして、PPARγの2種類のリガンドについて試験した。15d−PGJ2及びシグリタゾンのいずれもヒト腸上皮細胞におけるAP−1及びCOX−2誘発を阻害することが報告されており(サバラマイア(Subbaramaiah)ら、2001年)、また、これらは結腸炎の処置においても治療上の利点を有する(スー(Su)ら、1999年)。本発明者らは、PPARγ及びPPARαのリガンドが腸炎菌仲介によるサイトカイン誘発を低下させることができることを見出した(図4C、D、E)。この結果は、最近示されたPPARγアゴニストに関する報告、即ち、H.ピロリ(H. pylori)で処理した胃上皮細胞におけるNK−κB及びIL−8の発現がPPARγアゴニストによって低下すること(グプタ(Gupta)ら、2001年)、また、虚血再灌流誘発型腸障害におけるPMNの浸潤がPPARγアゴニストによって阻害されること(ナカジマ(Nakajima)ら、2001年)と一致する。
【0070】
PPARγ及びPPARαアゴニストの作用について述べてきたが、結腸粘膜ではPPARγの発現レベルがPPARαに比べて遥かに高いことが示されている(マンセン(Mansen)ら、1996年;ファジャス(Fajas)ら、1997年)ため、下の実験においてはPPARγに注目した。
【0071】
PPREを介して遺伝子転写を調節することに加え、最近では、PPARがDNA結合とは独立したメカニズムによって他の転写因子経路を妨害することにより遺伝子転写を阻害することが示された(デレライブ(Delerive)ら、1999年)。PPARγ2はサイトゾル区画及び核区画で見出される(スイリアー(Thuillier)ら、1998年)が、サイトゾルPPARγの生理学的妥当性については現在知られていない。本発明者らは、固定したCaco−2細胞に対して免疫細胞化学的局在化技法を用い、腸炎菌がPPARγの核内蓄積を誘発することを見出した(図2F)。PPARγのNH2末端ドメイン(Ser−122)のMAPキナーゼによるリン酸化によって、リガンド結合親和性が低下し、PPARγの転写及び生物学的機能が負に調節される(シャオ(Shao)ら、1998年)ことについて言及することは重要である。これによって、腸炎菌に対する炎症反応の初期段階で、PPARγの核内蓄積が炎症性遺伝子転写の抑制に対し効果的でない理由が説明される。腸炎菌をB.テタイオタオミクロンと共培養した後、PPARγはサイトゾルへ再分布された(図2G、H)。B.テタイオタオミクロンの存在下で腸炎菌に接触させたCaco−2細胞内のPPARγの特異的分布についても、核抽出物及び細胞質抽出物を用いたウェスタンブロッティングによって示された(図4F)。
【0072】
PPARγに関して公表されたデータは全て、PPARγの核内作用部位に注目している。本発明者らは、免疫細胞化学的技法を用いて、腸炎菌がCaco−2細胞におけるPPARγの核内蓄積を誘発することを見出した(図4H(g))。一方、腸炎菌とB.テタイオタオミクロンを用いて共培養した後、PPARγはサイトゾルへ再分布された(図4H(h、i))。経時的検討によって、この再分布がこれらの細菌への接触後60分で明らかになり、2時間経過するまでにはほぼ完了することが示された(結果は図示せず)。腸炎菌とB.テタイオタオミクロンとに接触させたCaco−2細胞内のPPARγの特異的分布についても、核抽出物及び細胞質抽出物を用いたウェスタンブロッティングによって示された(図4F)。
【0073】
腸炎菌で誘発されたPPARγタンパク質はRXRαとヘテロ二量化複合体を形成し(IPで実証済み、結果は図示せず)、洗浄剤不溶性細胞画分へと分配された。一方、B.テタイオタオミクロンで誘発されたPPARγタンパク質は洗浄剤可溶性であった(図4F)。腸炎菌とB.テタイオタオミクロンの共培養によって誘発されたPPARγの核−細胞質間輸送は、レプトマイシンB(LMB)処理ではブロックされず(結果は図示せず)、従って、PPARγの核−細胞質間輸送は、核外移行受容体crm−1(この受容体は他の核内受容体と類似する)によって促進されなかった(ブン(Bunn)ら、2001年)。しかし、冷却を行うこと及び代謝阻害剤を用いることによって、核外移行は有意に減少した(結果は図示せず)。 TSA(ヒストンデアセチラーゼ阻害剤)(図4H(j〜m)やSB(p38MAPキナーゼ阻害剤)(データは図示せず)等の他の生物学的阻害剤を腸炎菌で処理したCaco−2細胞に添加した。これらの阻害剤が点状サイトゾル標識化及びPPARγの核外移行を誘発することが示された(図4H(k))が、この作用はB.テタイオタオミクロンの作用と類似する。p38を含むAP−1シグナル伝達経路はB.テタイオタオミクロンによって阻害されなかった(結果は図示せず)が、このことはアセチル化/脱アセチル化反応がPPARγ核外移行メカニズムに潜在的に関連していることを示す。
【0074】
また、PPARγ及びPPARαリガンドは腸炎菌仲介によるサイトカイン誘発を抑制する(結果は図示せず)が、上記実験で試験したPPARリガンドの作用はB.テタイオタオミクロンのPPARγの細胞内再局在化に対する作用とは異なる。このことは、PPARγを調節する新規な内因性リガンドやメカニズムが存在することを示唆する。
【0075】
本発明者らは、二重標識免疫細胞化学的技法を用いて、PPARγタンパク質の多くがRelAと共存することを見出した(図4H(n〜p))。PPARγとRelAとの物理的結合が、腸炎菌及びB.テタイオタオミクロンに接触させた腸細胞における細胞質内局在化を促進する重要な因子であるという仮説を立てた。
【0076】
核内PPARγとNF−κBが不活性な複合体を形成し得ることが示唆されている(リコテ(Ricote)ら、1999年)。更に、グルタチオンSトランスフェラーゼプルダウン実験により、PPARαがc−Jun及びRelA p65と物理的に相互作用することが示されている(デレライブ(Delerive)ら、1999年)。本発明者らの検討においては、免疫精製(IP)プロトコルを用い、B.テタイオタオミクロン存在下、腸炎菌で処理した細胞からPPARγを単離することによってRelAが共精製される(図4G)ことが示されたが、この結果は、PPARγとRelAが物理的に結合し、層状多タンパク質複合体を構成している可能性を示唆する。PPARγとRelAとの直接相互作用についてはインビトロ翻訳及びIPによって確認した(図4b)。
【0077】
実施例5
RelAの調節におけるPPARγの重要性を更に調べるため、PPARγのドミナントネガティブ(DN)型(英国ケンブリッジ大学チャタージー(Chatterjee)教授の寄贈)(ガーネル(Gurnell)ら、2000年)を用いた。PPAR受容体内ではロイシンとグルタミン酸の保存性が高い。これらのアミノ酸残基はリガンド結合及び核コアクチベーターの補充において必須である。これらのアミノ酸残基の変異によって上記受容体のDN型が産生し、この受容体が持つコアクチベーターを補充する能力と2種類のコリプレッサー、即ち、レチノイド甲状腺受容体のサイレンシングメディエータ(SMRT)と核コリプレッサー(NcoR)を遊離する能力が低下する(ガーネル(Gurnell)ら、2000年)。DN PPARγを用いた免疫共沈降実験によって、SMRTがPPARγとインビボで相互作用すること、そして、変異したPPARγは強力な転写リプレッサーとなることが示された。ヒトPPARγのキメラ蛍光タンパク構造体と、シアン蛍光タンパク質(CFP)をカルボキシ末端ドメインに有するPPARγのドミナントネガティブ型とを先に報告されたクローン(ガーネル(Gurnell)ら、2000年)からPCR増幅によって調製した。YFPと結合したキメラRelAは、J シュミット博士(Dr. J Schmid)から寄贈された(シュミット(Schmid)ら、2000年)。発現の成功は、抗ヒトPPARγ及び抗ヒトRelA抗体を用いた一過性トランスフェクトHela細胞のウェスタンブロット解析によって確認した。
【0078】
PPARγ DN及びNF−κBルシフェラーゼレポーター(パスツール研究所(フランス、パリ)イスラエル博士(Dr. Israel)の寄贈)を用いた二重トランスフェクションについて検討した。本発明者らは、病原性腸炎菌に長時間接触させた細胞によってルシフェラーゼ活性が失活することを見出したが、この知見は最近の報告(サフコヴィック(Savkovic)ら、2000年)と一致する。この現象を回避するため、上記細胞を2時間刺激し、洗浄によって腸炎菌を除去し、8時間後にルシフェラーゼタンパク質の産生を測定した。対照細胞及びPPARγ DNをトランスフェクトした細胞において、腸炎菌によりNF−κB活性化とルシフェラーゼタンパク質合成が誘発されることを見出した(P<0.001)(図5C)。PPARγ野生型(NT)をトランスフェクトした細胞(ルシフェラーゼのみ)においては、上記反応はB.テタイオタオミクロンによって低下した(P<0.028)が、PPARγドミナントネガティブ型(DN)をトランスフェクトした細胞においてはこのような反応の低下は見られなかった(図5C)。同様に、PPARγ DNの存在下及び非存在下で緑色蛍光タンパク質(GFP)をトランスフェクトした細胞においては、免疫細胞化学的技法及びレーザー走査型共焦点顕微鏡検査法(LSCM)で測定されたように、RelAの細胞内分布が変化した。対照細胞(GFPのみ)においては、トランスフェクトしていない細胞について上述した様に、RelAは主にサイトゾルに分布していた(図5D(挿入図B、C))。PPARγ DNをトランスフェクトした細胞においては、RelA分布は一定ではなく、RelAはサイトゾル区画及び核区画の両方に局在化していた(図5D(挿入図D、E))。以上のデータから、RelAを隔離し核内転位を防ぐ上でPPARγが必須であることが示された。
【0079】
本発明者らは更に、蛍光顕微鏡検査法を用いて、WT及びDN PPARγのキメラ構造体、CFP、キメラYFP、及びRelAをトランスフェクトしたCaco−2細胞とHela細胞とにおけるPPARγ及びRelAの細胞内分布について調べた。発現したPPARγ/RelAタンパク質の多くは、既に報告されているように(シュミット(Schmid)ら、2000年)、トランスフェクトした細胞の核内に局在した。同様に、PPARγ WTとRelAとをトランスフェクトした細胞においても、発現は主に核内で起こったが、B.テタイオタオミクロンを用いたインキュベーションの後、点状サイトゾル標識化及びPPARγとRelAとの共存が非常に明確に示された(図5E(f))。しかし、PPARγ DNをトランスフェクトした細胞において、RelAはサイトゾル区画には存在しなかった(図5E(i))。以上の結果から、DN PPARγの核外移行(即ちRelAとの共存)が阻害されたことが示され、よって、腸細胞内でB.テタイオタオミクロンによって誘発されるRelAの核外移行及びサイトゾル内分布に対してはPPARγが不可欠であることが結論づけられる。Caco−2細胞トランスフェクション実験及びHela細胞トランスフェクション実験のいずれにおいても、発現したPPARγ及びRelAタンパク質の核内貯蔵が顕著であった。この場合、WTやDNの状態とは関係無く、腸炎菌及びB.テタイオタオミクロンの存在下で培養した細胞の核内にPPARγとRelAとの凝集物がはっきりと見られた(IPによっても確認、結果は図示せず)が、この結果から、PPARγとRelAとの相互作用が核区画内で起こることが証明された。本発明者らは、PPAR及びRelAの核内での分布が、核斑(nuclear speckles)やスプライセオソーム内で起こるスプライシング因子の分布と類似していることに注目した。共存についての検討において、 本発明者らはPPARγタンパク質がSC35、即ち、特異的プレmRNAスプライシング因子と共存することを見出した(図5E(k))が、この知見から、PPARγ/RelA複合体が細胞のmRNA機構と相互作用し得ることが示される。核内受容体とスプライシング区画との相互作用についての実証データが、本明細書作成中に公表された(ゾア(Zhoa)ら、2002年)。以上から明らかなように、Caco−2細胞では、B.テタイオタオミクロンの存在によりPPARγの核外移行が促進され、また、これに便乗する形で転写活性RelAの核外移行も促進されることにより、炎症持続中の更なる核内移行及びRelA仲介による転写が防止される。このメカニズムによって、B.テタイオタオミクロンは強力な抗炎症作用を発揮する。
【0080】
炎症を抑制する細菌が消化管内に存在することは確実と思われる。炎症を抑制する腸内微生物と炎症反応を持続させる腸内微生物とのバランスが変化すると、それが消化管の炎症性疾患の原因に直接関係する場合がある。従って、炎症性腸疾患が免疫ホメオスタシスに積極的に寄与する細菌株の減少に関係することは妥当と思われ、これによって、プロバイオティック療法に反応する患者もいることの理由が説明される(マッドセン(Madsen)ら、2001年)。また同様に、受容体タンパク質のレベルが不十分なこと、或いは炎症病態への感受性を高める特定の対立遺伝子変異体の腸内での発現に起因するPPARの機能障害が大いに関係する場合もある。この見解は、虚血再灌流誘発型腸炎症及び障害に対するヘテロ接合型PPARγ欠乏性マウスの感受性が有意に高いことを示す最近公表されたデータ(ナカジマ(Nakajima)ら、2001年)によって更に支持される。
【0081】
方法
【0082】
試薬
【0083】
組織培養試薬はシグマ及びインビトロゲンより入手した。抗体はサンタクルズ、モレキュラープローブ、ニューイングランドバイオラボ(New England Biolabs)より入手した。また、分子試薬はプロメガ、インビトロゲン及びアマシャムより入手した。
【0084】
S.エンテリティディス/B.テタイオタオミクロン共培養モデル
【0085】
Caco−2細胞及びHela細胞は35mm培養皿内で常法によって培養した。通常の実験では、次の4種類の処理でインキュベートした細胞を要した。(1)培地のみでインキュベートした細胞、(2)108個の腸炎菌(S.エンテリティディス)と共にインキュベートした細胞、(3)108個の腸炎菌(S.エンテリティディス)及び109個のバクテロイデス・テタイオタオミクロンと共にインキュベートした細胞、及び(4)109個のB.テタイオタオミクロンと共にインキュベートした細胞。上記細菌は2時間或いは4時間添加した後、十分に洗浄して除去した。試験に用いた他の細菌及びリガンドとしては、大腸菌0157H7、B.ブルガータス、PMA、IL−1α/β及びTNFαが挙げられる。実験は全て最適化し、プロトコルは詳細な用量反応及び時間経過に基づくものであった。Caco−2細胞単層を経由するPMN細胞の経上皮遊走はMPOアッセイ(パーコス(Parkos)ら、1991年)によって測定した。Caco−2細胞を2時間インキュベートした後、上記細菌を除去し新しい培地に取り替えた。その後、細胞を更に2時間インキュベートした。次いで、先端区画から得た培地及び細胞をいずれも1%のTriton X−100に可溶化し、MPOを定量した。
【0086】
サイトカイン解析
【0087】
サイトカイン解析はクロンテックマクロアレイ、ノーザンハイブリダイゼーション、及びCaco−2細胞から単離したRNAのリアルタイムPCRを用いて行った。全RNA及びmRNAを単離し、cDNAを産生し、PCRを標準条件下で行った。IL−8タンパク質濃度はELISAによって測定した。
【0088】
NF−κB及びAP−1のEMSA解析
【0089】
NF−κBのコンセンサス結合配列(5’−AGT TGA GGG GAC TTT CCC AGG C−3’)或いはAP−1のコンセンサス結合配列(5’−CGC TTG ATG AGT CAG CCG GAA−3’)を含む一本鎖32P標識オリゴヌクレオチドプローブと共に核抽出物をインキュベートし、電気泳動によって分離し、オートラジオグラフィーによって可視化した。特異的NF−κBサブユニット抗体を用いてEMSAスーパーシフトを行った。EMSA、ウェスタンブロッティング(タンパク質及びリンタンパク質)、プロモーター特異的レポーター解析及び標的遺伝子発現(例えば、fos及びjun)によって、NFκB及びAP−1のシグナル伝達に対するB.テタイオタオミクロンの作用を測定した。
【0090】
免疫蛍光解析
【0091】
実験的処理後、35mm培養皿で増殖したCaco2細胞及びHela細胞を4%パラホルムアルデヒドに固定し、0.2%のTriton−X100/PBSで透過処理した。二次抗体を産生させた種由来の血清1%を含むPBSに希釈した一次抗体(1μg/ml)と共に細胞を室温で1時間インキュベートした。二次抗体(1μ/ml)としては、Alexa Fluor594ロバ抗ヤギIgG或いはAlexa Fluor488ヤギ抗ウサギIgG(モレキュラープローブ)を必要に応じて用いた。標識した細胞をVectorshield(ベクター)でスライドに載せ(mounted)、Zeiss Axioskop50広視野蛍光顕微鏡或いはBio−Rad Radiance2100レーザー走査型顕微鏡にて検査した。代表的デジタル画像をAdobe Photoshop6.0に取り込み最終調整を行った。
【0092】
蛍光標識PPAR・の作成
【0093】
PPARγのコード配列及びPPARγのドミナントネガティブ変異体(ガーネル(Gurnell)ら、2000年)をPCR増幅時に修飾し、生成物の5’末端にXhoI認識配列とSacII認識配列を、3’末端にはSacII配列を付加した。PCR増幅はPfuDNAポリメラーゼを用いて行った。生成物を制限しpECFP−C1及びpEYFP−C1(プロメガ)にクローン化した。クローン化の成功はDNAシークエンシングによって確認した。pEYFP−C1へクローン化したヒトp65については既に報告されている(シュミット(Schmid)ら、2000年)。
【0094】
Caco2細胞及びHela細胞を35mm培養皿に播種し、90%コンフルエントまで増殖させ、標準リポフェクタミン仲介法(インビトロゲン)を用いてトランスフェクトした。48時間後、細菌インキュベーションを上述の様に行い、細胞を室温で30分間4%パラホルムアルデヒド0.1Mリン酸ナトリウム緩衝液(pH7.4)に固定し、PBSで洗浄し、CFP及びYFP用カスタムフィルター(オメガオプティカル)を備えたZeiss Axioskop50広視野蛍光顕微鏡にて検査した。代表的画像をZeiss AxioCamデジタルカメラで記録し、Zeiss AxioVision3.0ソフトウェアで処理し、Adobe Photoshop6.0に取り込み最終調整を行った。一部の実験では、トランスフェクトした細胞を上述の免疫蛍光解析に付した。
【0095】
実施例6:インビボラット研究
本実施例では、B.テタイオタオミクロンによって、特定病原不在ラットの腸機能が変化し、該ラットの病原菌チャレンジに対する耐性が増加するどうかを立証した。一般に、バクテロイデスは哺乳期から離乳期への移行時に小腸に現われる(チャン(Chang)ら、1994年;フーパー(Hooper)ら、2001年)。従って、特定病原不在Hooded Listerラットに離乳期(19日齢)から成熟期(34〜40日齢)までこの細菌を毎日投与し、腸や免疫の発達に重要なこの時期にこの菌が確実に多数存在するようにした。34日齢でラットに対し腸炎菌S1400の経口チャレンジを行った。
【0096】
方法
【0097】
細菌の培養:
【0098】
ドイチュ・サムルング・フォン・ミクロオーガニズメン・ウント・GmBH(Deutsche Sammlung von Microorganismen und GmBH)(ドイツ、ブラウンシュヴァイク(Braunschweig))から入手したB.テタイオタオミクロンをWilkins−Chalgren嫌気性寒天で凍結保持した。B.テタイオタオミクロンをWilkins−Chalgren嫌気性ブロス或いはM10培地[Hungateチューブ内10ml(ブライアント(Bryant)、1972年)]に継代培養し、37℃で48時間増殖させた。サンプル(0.5ml)を新しい培地[Hungateチューブ内10ml]に移し、37℃で24時間増殖させた。得られた培養物には約108〜109CFU/mlのB.テタイオタオミクロンが含まれていた。
【0099】
B.テタイオタオミクロン培養物(10ml)を遠沈し(2000g、12分間)、得られたペレットを0.05Mリン酸緩衝生理食塩水(pH7.2)で洗浄した後、市販のゼリー[10ml、37℃、嫌気条件下で調製済]に再懸濁した。このゼリーをCO2下で滅菌ペトリ皿に注入し4℃で放置した。約1時間後、ゼリーを一定分量(0.5g、B.テタイオタオミクロン(〜108CFU))に分割し上記ラットに与えた。B.テタイオタオミクロンはこの条件下で少なくとも6時間は生存していたが、通常、ゼリーはケージに入れて数分以内には全て食べられた。
【0100】
腸炎菌S1400は当初家禽感染症から単離されたものであり、特徴付けが行われてる(アレン−ヴェルコー(Allen-Vercoe)及びウッドワード(Woodward)、1999年)。Dorset卵スロープ(Dorset egg slopes)で4℃で保持した菌株をLuria−Bertani寒天プレートに継代培養し37℃で一晩増殖させた。該プレートから得た5〜10個のコロニーをLuria−Bertaniブロス(10ml)に接種し、37℃で一晩攪拌しながらインキュベートし、約1×109CFU/mlの腸炎菌S1400を得た。
【0101】
動物実験:ローウェット研究所は英国動物(科学的処置)法1986の下で認可を受けている。同研究所の倫理審査委員会及び動物保護部門、更には適切な政府視察団(appropriate governmental inspectorate)によって動物実験は全て視察、監視されている。管理及び実験手続については倫理委員会によって承認され、その手続は実施許可を得た研究所員が上記動物法の厳しい条件に従って行った。
【0102】
ローウェット研究所の小動物部門で飼育した、24匹の特定病原不在Hooded Lister雄性ラット(40g)を19日目に離乳させ、すぐにClassII施設へ移した。実験継続期間中(21日間)、ラットを個々にフレキシフィルムアイソレータ(モードゥン、アニマルヘルス、英国、ペニクイク(Penicuik))内の代謝ケージ(テクニプラスト、英国、ケッターリング)に収容した。入れ子状トンネルを各代謝ケージに設け、ラットが互いを視認できても接触できないように該アイソレータ内に代謝ケージを配置した。滅菌蒸留水をいつでも摂取できるようにした。ラットを毎日計量し、糞便を実験期間中採取した。実験は少なくとも3回行ったが、いずれも同様の結果が得られた。データは平均ISDである(n=3)。先ず、ラットに良質の半合成(100gタンパク質/kg)ラクトアルブミンベースの食餌(グラント(Grant)ら、2000年)を自由に与えた。ラットが約80gに達した時に(約30日齢)、食餌摂取量を3〜4日間かけて7g/ラット/日(1日2食)まで徐々に減らし、実験の残りの期間中この摂取量を維持した。この摂取量は、腸炎菌を経口感染させた後の同一日齢ラットの1日当りの自由摂取量の平均であり、この量は非感染ラットによる上記食餌の自由摂取量の約70%であった。ラットを離乳時からこのように収容、管理し、細菌への環境曝露や離乳後の相互汚染を減少させた。(グラント(Grant)、1996年)。
【0103】
離乳時(19日目)から実験終了時まで、12匹のラットにB.テタイオタオミクロン(約108CFU)を1日1回投与した。34日齢時に、B.テタイオタオミクロンで前処理した6匹のラットと6匹の対照ラットに、胃管栄養法によって単一用量0.8mlの腸炎菌S1400培養物(約109CFU)を投与した。残りのラットには相当量の培地を投与した。ラットを全て代謝ケージに戻し、ラクトアルブミンベースの食餌(7g/ラット/日)を更に6日間与えた。最終日にラットに2gを食餌を与え、その丁度2時間後、ハロタン(ローヌメリュー、英国、エセックス)の過剰摂取及び失血によりラットを殺した。開腹して組織を無菌的に除去した。胃及び小腸をリン酸緩衝生理食塩水(PBS、pH7.2)で流して内容物や非付着細菌を除去した。空腸10cm(幽門から5〜15cm)と回腸10cm(回盲連結部から5〜15cm)を除去した。こうして除去した空腸と回腸、胃組織、盲腸及びその内容物、結腸及びその内容物、腸間膜リンパ節、肝臓と脾臓の代表的部分(200〜400mg)及び腎臓1個を処理し生細胞数を測定した。
【0104】
更に空腸片(幽門から15〜25cm)及び回腸片(回盲連結部から15〜25cm)を採取し液体窒素で凍結し、生化学的解析を行った。同様の処理を残りの小腸組織(〜40cm)、肝臓、脾臓、腎臓、胸腺、肺、心臓及腓腹後肢筋についても行った。これらを計量し、凍結乾燥した後再計量した。
【0105】
ラット組織中の生残細菌
【0106】
組織サンプルを計量した後、Janke−Kunkel Ultra−TurraxT25組織ホモジナイザー(20,000rev/分、30秒)を用いてMaximum Recovery Diluent(MRD、フィッシャーサイエンティフィック、英国)にホモジナイズした。最大6回の希釈工程を続けて行い、一次ホモジネート(1:10v/v)をMRDに形成した。各希釈工程におけるサンプルを十分に乾燥したXLD寒天(フィッシャーサイエンティフィック、英国)及びMacConkey寒天No.3(フィッシャーサイエンティフィック、英国)プレートの表面に載せ37℃で一晩インキュベートした。生細胞数をマイルズ(Miles)及びミスラ(Misra)(1938年)の方法或いはスプレッドプレート法(コリンズ(Collins)及びライム(Lyme)、1989年)によって評価した。
【0107】
PCR
【0108】
QIAamp DNA Stool Mini Kit(キアゲン社、英国、クローリー)を用いてDNAを抽出、精製した。PCRはテング(Teng)らの方法(2000年)に基づいた。テング(Teng)ら(2000年)によって同定されたプライマー対、即ち、5’−TGGAGTTTTACTTTGAATGGAC−3’(BTH−F)及び5’−CTGCCCTTTTACAATGGG−3’(BTH−R)はシグマジェノシス社(英国、ケンブリッジ)から購入した。TaqPCR Core Kit(キアゲン社、英国、クローリー)から入手した試薬に基づく反応混合物(50μl)には、上記2種類のプライマー(50pmol)、dNTP混合物(10nmol)、10X Qiagen PCRバッファー(5μl)、5X Q溶液(10μl)、サンプル(10μl)及びTaqDNAポリメラーゼ(1.25U)が含まれていた。ホットスタートPCRプログラムを用いた。TaqDNAポリメラーゼを含まない反応混合物を94℃で15秒間加熱した後、(94℃×10秒、55℃×30秒、74℃×1分)のサイクルを35回行って加熱し、更にその後(74℃×2分、45℃×2秒)のサイクルを1回行って加熱した。アンプリコンを臭化エチジウム(1μg/mlゲル)を含むアガロースゲル(10g/l)で分離した。糞便及びB.テタイオタオミクロンからDNAと共に単一アンプリコン(721bp)を得た。これらのアンプリコンの配列は互いに一致しており、また公表された配列(テング(Teng)ら、2000年)とも一致することが分かった。
【0109】
MPO
【0110】
Janke−Kunkel Ultra−TurraxT25組織ホモジナイザー(20,000rev/分、30秒)を用いて、組織サンプルを氷冷5mMリン酸カリウム緩衝液(pH6.0)にホモジナイズし(1:80w/v)、遠沈した(3000g×30分、4℃)。臭化ヘキサデシルトリメチルアンモニウム(HETAB、5g/l)及びエチレンジアミン四酢酸(EDTA、3.72g/l)を含む氷冷0.5Mリン酸カリウム緩衝液(pH6.0)で、得られたペレット(1:20w/v)を超音波処理し(3×5秒)、30分間氷上に放置した後、遠沈した(3000g×30分、4℃)[ストゥッキ(Stucchi)ら、2000年]。得られた上清をアッセイを行うまで凍結した。50mMリン酸カリウム緩衝液(pH6.0)での3,3’,5,5’−テトラメチルベンジジン(TMB、ダイネックステクノロジーズ、英国、アッシュフォード)のH2O2依存型酸化をモニターすることによってミエロペルオキシダーゼ(MPO)活性を測定した(ツィマーマン(Zimmerman)及びグランガー(Granger)、1990年)。450nmにおける吸光度は0.18MH2SO4との反応終了後に測定した。ヒトミエロペルオキシダーゼ(カルバイオケム、英国)を標準品として用い、数値はMPO当量として示した。
【0111】
腸内容物や糞便内の免疫反応性MPOを競合ELISA法で測定した。10mMリン酸緩衝生理食塩水(pH7.4)[PBS]に溶解したヒトミエロペルオキシダーゼでマイクロタイタープレート(Immulon4、ダイネックステクノロジーズ、、アッシュフォード、英国)を4℃で一晩コートした[10ng/ウェル]。Tween−20(1ml/l)を含むPBSで洗浄後、プレートをウシ血清アルブミン(BSA、10g/l)含有PBSで室温下1時間ブロックした。プレートを洗浄し、サンプル及び標準品を添加し[50μl/ウェル]、BSA(1g/l)、Tween−20(1ml/l)及びロイペプチン(1mg/l)を含むPBSで連続的に希釈した。更に、ウサギ抗ヒトミエロペルオキシダーゼ抗体[カルバイオケム、英国](1:4000希釈品、50μl/ウェル)も添加した。プレートを1時間インキュベートし洗浄した後、ビオチン化抗ウサギIgGと1時間反応させ、次いでExtravidin/ペルオキシダーゼ(EXTRA−3キット、シグマアルドリッチ、英国、プール)と反応させた。洗浄後、TMB試薬を添加し(7μl/ウェル)、暗所で最大2時間インキュベートした。0.18MH2SO4の添加(50μl/ウェル)によって反応を終了させ、吸光度を450nmで測定した。数値は免疫反応性MPO当量として示した。
【0112】
IgG
【0113】
IgGは競合ELISAによって測定した。PBSに溶解したラットIgG(シグマアルドリッチ、英国、プール)でマイクロタイタープレートを4℃で一晩コートした[1μg/ウェル]。プレートをブロック、洗浄した後、サンプル及びラットIgG標準品を添加し[50μl/ウェル]、BSA(1g/l)及びTween−20(1ml/l)を含むPBSで連続的に希釈した。更に、ビオチン化抗ラットIgG抗体(シグマアルドリッチ、英国、プール;1:1000希釈品)も添加し(50μl/ウェル)、プレートを1時間インキュベートした。次いで、MPOイムノアッセイによりプレートをExtravidin/ペルオキシダーゼ及びTMB試薬と反応させた。数値はIgG当量として示した。
【0114】
IgA
【0115】
IgAは捕捉ELISAによって測定した。ヤギ抗ラットIgA(シグマアルドリッチ、英国、プール;1:100PBS希釈品)でマイクロタイタープレートを4℃で一晩コートした(100μl/ウェル)。プレートをブロック、洗浄した後、サンプル及び標準品を添加し[100μl/ウェル]、BSA(1g/l)、Tween−20(1ml/l)及びロイペプチン(1mg/l)を含むPBSで希釈し、1時間インキュベートした。洗浄後、マウス抗ラットIgA抗体(シグマアルドリッチ、英国、プール;1:500希釈品)と共にプレートを1時間インキュベートし、再洗浄後、ビオチン化抗マウスIgG抗体(1:1000希釈品)と共に更に1時間インキュベートした。次いで、MPOイムノアッセイによりプレートを処理した。数値はIgA当量として示した。
LPS特異的IgG及びIgA
【0116】
腸炎菌(シグマアルドリッチ、ドーセット、プール)由来のリポ多糖類(LPS)でマイクロタイタープレートをコートした(10μg/ウェル)。プレートをブロック、洗浄後、サンプル及び標準品を添加した[100μl/ウェル]。1時間後プレートを洗浄し、ビオチン化抗ラットIgG及び抗ラットIgAを添加した後、プレートを上述の様に処理した。ELISAにおいて吸光度が少なくとも0.2となる容量(μl)に含まれるLPS特異的抗体の量を該抗体の1ユニットとした。腸炎菌で感染後22日目のラットから採取した血清(LPS−IgA:3.2×104ユニット/ml、LPS−IgG:1.7×105ユニット/ml)を陽性対照とした。
【0117】
エラスターゼ
【0118】
凍結乾燥した糞便及び腸内容物を、HETAB、EDTA及びNaN3(1g/l)を含む氷冷0.5Mリン酸カリウム緩衝液(pH6.0)に抽出し(1:10w/v)、氷上で30分間放置した後、遠沈し(3000g、30分、4℃)、得られた上清を凍結した。
【0119】
マイクロタイタープレート上で、サンプル及び白血球エラスターゼ(シグマアルドリッチ、英国、プール)を1MNaCl及びロイペプチン(1mg/l)を含む0.2MトリスHCl(pH8.0)で連続的に希釈した。基質(N−スクシニル−ala−ala−val−p−ニトロアニリド、0.2g/l、シグマアルドリッチ、英国、プール)を添加し、その直後、405nmにおける吸光度をモニターした。更に、37℃でのインキュベーション時に最大20時間の間隔で吸光度をモニターした。数値は白血球エラスターゼ当量として示した。
【0120】
DNA、RNA、タンパク質
【0121】
DNA、RNA、タンパク質は、従来通り、ジフェニルアミン法、オルシノール法及び改良ローリー法によって測定した(グラント(Grant)ら、2000年)。サケ精巣DNA、酵母DNA及びウシ血清アルブミンを標準品として用いた。
【0122】
統計解析
【0123】
Instat Statistical Package(グラフパッドソフトウェア社、米国、サンディエゴ)を用い、一元配置分散分析(ANOVA)とチューキーの多重比較検定とを組み合わせてデータを評価した。
【0124】
結果
【0125】
バクテロイデス・テタイオタオミクロン(BT)はBT処理ラットから採取した糞便サンプルで検出された。また、BTは対照ラットの糞便でも検出された。しかし、対照ラットの糞便で検出されたBTのレベル(約104CFU/g糞便に相当)は、毎日BTで処理したラットの糞便で検出されたBTのレベル(約106CFU/g糞便に相当)よりも低かった。
【0126】
体重増加、主要臓器重量、小腸成分及び肝臓成分については、BT処理ラットと対照ラットとの間では差が無かった(表1〜4)。BT処理ラットの場合、乳糖発酵菌及び非乳糖発酵菌の消化管全体への分布は影響を受けなかったと思われ、また、対照ラットの場合と同様に、腸間膜リンパ節、肝臓、脾臓及び腎臓サンプルでは乳糖発酵菌及び非乳糖発酵菌のいずれも検出されなかった。更に、腸内容物や糞便中の非特異的IgA、エラスターゼ及び免疫反応性MPOのレベル、腸組織中のMPOのレベル、及び血清中の非特異的IgG及びIgAのレベルについても、対照ラットから採取したサンプルの場合と同程度であった。
【0127】
【表1】
【0128】
腸炎菌[SE]は経口チャレンジ後6日目に消化管全体で検出された(表1)。更に、腸炎菌は腸間膜リンパ節、肝臓及び脾臓でも検出された。上記ラットにおいて非乳糖発酵菌は全て同様の組織内分布を示した(表1)。腸炎菌感染の腸内乳糖発酵菌数に対する影響については明らかではなかった。しかし、かなりの数の乳糖発酵菌がSE感染ラットの腸間膜リンパ節、肝臓及び脾臓に存在した(表1)。
【0129】
腸炎菌チャレンジ後に肝臓及び脾臓で検出された生残腸炎菌の数は、ラットをB.テタイオタオミクロンでも処理した場合[SE+BT]には大幅に減少した(表1)。肝臓及び脾臓内では全非乳糖発酵菌の数も減少したが、乳糖発酵菌数は変化しなかった。消化管全体及び腸間膜リンパ節内の腸炎菌、全非乳糖発酵菌及び乳糖発酵菌のレベルは、SE+BTとSEとの間では差が無かった(表1)。また、6日間の実験中、腸炎菌の糞便への分泌が腸炎菌感染ラットでは同程度で起こった(SE+BT:7.0±0.8Log10CFU/g/日、SE:6.2±0.6Log10CFU/g/日)。
【0130】
【表2】
【0131】
【表3】
【0132】
腸炎菌感染の結果、小腸の重量が増加した(表2、3)。重量の増加は回腸組織で最も顕著であり、これは水分の蓄積と組織内のタンパク質量、RNA量及びDNA量の増加とによるものであると分かる(表3)。BTでの処理により感染に対する反応がわずかに変化した。即ち、SE+BTラットから採取した小腸の水分量[748±22mg/g]はSEラットから採取した小腸の水分量[799±17mg/g]に比べてかなり少ない。実際には、SE+BTラットの小腸の水分量は非感染ラットから採取した小腸の水分量[BT:745±24mg/g、対照:760±10mg/g]と同等である。一方、小腸乾燥重量、DNA量、RNA量及びタンパク質量についてはSEラットとSE+BTラットとの間に有意差は無かった。
【0133】
【表4】
【0134】
SEラットから採取した回腸組織においてミエロペルオキシダーゼ(MPO)活性は有意に上昇した(表4)。また、腸内容物及び糞便における免疫反応性MPO及びエラスターゼの活性も大幅に上昇した(表4)。一方、SE+BTラットから採取した腸内容物及び糞便におけるこれらの酵素のレベルは対照ラットの場合と同等か或いはごくわずかに高かった。
【0135】
腸炎菌感染の結果、腸内容物、糞便及び血清中の免疫反応性非特異的IgAは増加した(表4)。しかし、IgA反応についてはSE+BTラットとSEラットとの間では差が無かった。血清及び腸内容物では少量のLPS特異的IgAも検出された。この場合も、SE+BTラットとSEラットとの間では有意差が無かったが、SE+BTラットの方が腸内でのLPS特異的IgAレベルがより高くなる傾向にあった。
【0136】
腓腹後肢筋重量については対照ラットよりSEラットの方が有意に小さかった(表2)。この傾向はSE+BTラットでは明らかでなく、よって、この重量減少は筋肉成長の低下によるものと分かった。SEラットの場合、腓腹筋は1日当り湿重量で約6.6±5.7mg[乾燥重量で1.4±1.2mg]成長し、一方、SE+BTラットの場合、腓腹筋の成長率は1日当り湿重量で17.1±4.1mg[乾燥重量で4.2±1.0mg]であった。また、対照ラットの場合、腓腹筋の成長は1日当り湿重量で15.5±2.9mg[乾燥重量で3.8±0.7mg]であり、BTラットの場合、1日当り湿重量で17.0±4.2mg[乾燥重量で4.2±1.0mg]であった。この結果から、1日当りの全骨格筋の成長については、SEラットの場合、平均0.3g(湿重量)であり、SE+BT、BT及び対照ラットの場合、約0.7〜0.8gであることが示唆される(図6)。
【0137】
腸炎菌感染ラットにおいては、肝臓、脾臓及び腸間膜リンパ節の湿重量は有意に増加した(表3)。これは主に、肝臓及び脾臓での水分の蓄積と腸間膜リンパ節での水分及び乾燥物の増加によるものである(表4)。腸炎菌感染ラットの腸間膜リンパ節でのペルオキシダーゼ活性も上昇した[SE:31.0±25.0μg、SE+BT:37.7±23.8μg、BT:0.9±0.9μg、対照:0.4±0.3μg]。しかし、腸間膜リンパ節でのペルオキシダーゼ活性については、SE群とSE+BT群との間では有意差が無かった。
【0138】
考察
【0139】
サルモネラ症:腸炎菌S1400(Salmonera enterica var Enteritidis S1400)はラットの消化管全体でコロニーを形成し、腸間膜リンパ節、肝臓及び脾臓に転位した。また、腸炎菌感染により小腸の湿重量及び乾燥重量は大幅に増加した。この重量増加は回腸において最も顕著であり、回腸内での水分の蓄積とタンパク質、RNA及びDNAのレベルの上昇とに関連していた。従って、腸炎菌感染の基本的な特徴は、ラットモデルで他のエンテリティディス菌株やネズミチフス菌(S.ティフィムリウム)株感染において見られる特徴(ノートン(Naughton)ら、1996年、2001年;イーウェン(Ewen)ら、1997年;ボヴィー−オーデンホーヴェン(Bovee-Oudenhoven)ら、1999年;イスラム(Islam)ら、2000年;ハヴェラール(Havelaar)ら、2001年)と類似していた。
【0140】
エンテリティディスやティフィムリウムによってラットで自己制限感染が引き起こされ、この細菌は主に消化管に局在し回腸を経由して浸潤するが、全身への伝播は制限されることが分かっている(ノートン(Naughton)ら、1996年;ボヴィー−オーデンホーヴェン(Bovee-Oudenhoven)ら、1997年、1999年;イスラム(Islam)ら、2000年)。また、ラットの健康状態や腸の完全性が感染以前に他の因子によって損なわれていなければ、重篤な菌血症や死亡が起こることはまれである。これは、エンテリティディスやティフィムリウムによって死亡率の高い重篤な腸チフス様疾患が引き起こされた、マウスモデルの一部におけるサルモネラ症とは全く対照的である(ルー(Lu)ら、1999年;キングスレー(Kingsley)及びバウムラー(Baumler)、2000年;シェヒター(Schechter)及びリー(Lee)、2000年)。このように、ラットにおけるサルモネラ症は、エンテリティディスやティフィムリウムに感染したヒトや家畜に共通する自己制限胃腸炎型感染症と非常に類似している。
【0141】
本発明の検討においては、腸炎菌感染による強い炎症反応が小腸の基部ではなく末端部で引き起こされることを見出した。従って、回腸組織でのミエロペルオキシダーゼ(MPO、好中球マーカー)の活性は有意に上昇した。また、腸内容物や糞便での免疫反応性MPO及び白血球エラスターゼの活性レベルも上昇した。この結果は、先にサルモネラ症の動物モデルで観察されたように(ノートン(Naughton)ら、1995年、1996年;ヴァシロヤナコポウロス(Vassiloyanakopoulos)ら、1998年;ダーウィン(Darwin)及びミラー(Miller)、1999年;ヘンダーソン(Henderson)ら、1999年)、多形核白血球や他の炎症細胞の回腸組織への浸潤及び炎症細胞の腸管内腔への逃避(exvasion)と一致する。また、この結果は、回腸がサルモネラ菌によるコロニー形成及び浸潤の主要部位であるという知見(カーター(Carter)及びコリンズ(Collins)、1974;ノートン(Naughton)ら、1996年)とも一致する。
【0142】
腸炎菌感染によって血清、腸内容物及び糞便中の非特異的IgAは有意に増加した。また、感染後6日目までにはLPS特異的IgA反応の発生が示された。更に、腸内容物中のタンパク質レベルと同様に、糞便乾燥物、水分及びタンパク質の排出量(outputs)も増加した。腸炎菌感染によって肝臓及び脾臓の重量も増加した。
【0143】
サルモネラ症によってラットの骨格筋代謝が損なわれることも見出された。腸炎菌感染ラットと対照ラットは共に同じ乾燥物(7g/ラット/日)及びタンパク質(0.7g/ラット/日)を摂取したが、感染ラットの場合、骨格筋の成長は続いたものの一日当りの成長率は対照ラットの場合の約40〜50%であった。これは、感染に対する防御反応を維持するための栄養素の転用や利用(クラシング(Klasing)及びカルバート(Calvert)、1999年)及び/又は内毒素や他の細菌性因子の筋肉タンパク質合成への作用(フライマン(Friman)ら、1984年;ラング(Lang)ら、2000年)に起因するものであろう。
【0144】
B.テタイオタオミクロン:本発明の検討においては、腸が急速に成長、成熟し且つ免疫系が発達する時期を通して、ラットを高レベルの外因性B.テタイオタオミクロンに接触させた。バクテロイデス菌株の一部は日和見病原菌であり、この発達時期に有害な作用を示す可能性がある。しかし、B.テタイオタオミクロンのみを感染させた成熟完全無菌動物を用いた研究(フーパー(Hooper)ら、1999年、2001年;ノアック(Noack)ら、2000年)の場合と同様に、B.テタイオタオミクロンが特定病原不在ラットの腸や全身代謝に悪影響を及ぼさないことが示された。実際、B.テタイオタオミクロンのみで処理したラットでモニターした全パラメータ(炎症マーカーや免疫グロブリンのレベルを含む)は対照ラットについて得たものと同等であった。
【0145】
B.テタイオタオミクロンとサルモネラ症:B.テタイオタオミクロンによって、ラットでの腸炎菌S1400による感染の特徴が変化し、感染全体の重症度が低下した。特に、小腸での炎症反応が制限され、肝臓及び脾臓で検出された生残腸炎菌の数が大幅に減少し、骨格筋成長率はほぼ対照レベルへ回復した。このように、B.テタイオタオミクロンは腸内で局所的に作用を示すと同時に、全身組織に対しても遠隔的に作用を示した。
【0146】
しかし、サルモネラ症の特徴の一部はB.テタイオタオミクロンによっては影響されなかった。消化管及び腸間膜リンパ節内の腸炎菌数は変化せず、また、腸炎菌の糞便排泄についても変化しなかった。また、腸炎菌感染に伴う小腸の拡張(即ち、乾燥重量の増加とDNA、RNA及びタンパク質量の増加)もB.テタイオタオミクロンと腸炎菌を投与したラットにおいて明らかであった。この結果から、B.テタイオタオミクロンが腸炎菌自身を直接妨害したり、宿主の代謝に対する腸炎菌の一般的な作用をブロックするのではないことが示唆される。むしろ、B.テタイオタオミクロンは宿主の感染に対する反応を選択的に調節し、腸の完全性を損なう可能性のある反応を標的にするのであろう。
【0147】
最近では数多くの細菌株がエンテリティディス感染やティフィムリウム感染を部分的に防御することが示されている。このような細菌株の一部は腸内の付着部位や栄養素における病原菌を打ち負かしたり、殺菌性化合物を産生したりする。その結果、腸内で検出され、腸間膜リンパ節、肝臓及び脾臓に到達するサルモネラ菌の数が減少する(バーネット−カマード(Bernet-Camard)ら、1997年;フドールト(Hudault)ら、1997年、2001年;ヘンドリクソン(Hendriksson)及びコンウェイ(Conway)、2001年)。一方、上記細菌株の多くは腸内のサルモネラ菌の数に対し殆ど或いは全く影響を及ぼさない。しかし、それでもB.テタイオタオミクロンの場合と同様に、サルモネラ菌感染を有意に改善する(シルヴァ(Silva)ら、1999年;フィルホ−リマ(Filho-Lima)ら、2000年;シュー(Schu)ら、2000年;ヘンドリクソン(Hendriksson)及びコンウェイ(Conway)、2001年;マイア(Maia)ら、2001年)。従って、後者の細菌株が示す防御作用は競合排除や殺菌性化合物分泌以外のメカニズムによるものである。
【0148】
サルモネラ菌は、細胞代謝、細胞間相互作用及び宿主反応メカニズムを妨害することによって腸上皮を突破する(ダーウィン(Darwin)及びミラー(Miller)、1999年:ネテア(Netea)ら、2000年;イーヴス−ピレス(Eaves-Pyles)、2001年;ゲウィルツ(Gewirtz)ら、2001年;ルー(Lu)及びウォーカー(Walker)、2001年;オール(Ohl)及びミラー(Miller)、2001年)。特に、サルモネラ菌は多形核白血球の腸上皮への急速な浸潤を誘発し、腸の急性炎症や重篤な破壊を引き起こす(マダラ(Madara)、1997年;オール(Ohl)及びミラー(Miller)、2001年)。腸への好中球の浸潤は、サルモネラ菌感染に呼応して上皮細胞により分泌される化学誘引物質ケモカイン(例えばIL−8)によって仲介される(マコーミック(McCormick)ら、1995年;マダラ(Madara)、1997年;ダーウィン(Darwin)及びミラー(Miller)、1999年;フレックスタイン(Fleckstein)及びコペッコ(Kopecko)、2001年)。上皮細胞をインビトロでサルモネラ菌と共に培養した場合、この化学誘引物質の遊離が促進された(マコーミック(McCormick)ら、1995年;キャンベル(Campbell)ら、2001年)。一方、上皮細胞をサルモネラ菌及びB.テタイオタオミクロンと共に培養した場合、この化学誘引物質の産生量は大幅に低下した(キャンベル(Campbell)ら、2001年)。
【0149】
B.テタイオタオミクロンと腸炎菌とを投与したラットの回腸組織及び腸内容物中のミエロペルオキシダーゼのレベルは、腸炎菌のみを投与したラットから採取した比較サンプルに比べて遥かに低かった。このように、ラットをB.テタイオタオミクロンでも処理した場合、腸炎菌により通常腸内で誘発される炎症反応は明らかに抑制された。インビトロで見出されたように(キャンベル(Campbell)ら、2001年)、B.テタイオタオミクロンは、サルモネラ菌が関係する腸上皮細胞による化学誘引物質ケモカインの産生をブロックし、腸上皮への好中球の補充を防ぐと思われる。このインビボでの宿主反応をB.テタイオタオミクロンが調節することにより、感染の結果起こる腸の炎症や障害の程度を抑制し、腸の完全性を維持するであろう。これによって、炎症の回復に腸が必要とする栄養素の量が減少し(クラシング(Klasing)及びカルバート(Calvert)、1999年)、骨格筋等の他の組織へより多くの栄養素を供給することができる。
【0150】
サルモネラ菌は一旦腸上皮を通過すると腸間膜リンパ節へ流入する(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年)。次いで、サルモネラ菌はマクロファージによって腸間膜リンパ節から排除されるか、或いは腸間膜リンパ節から抜け出て血液、肝臓及び脾臓へと伝播し得る(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年;ルー(Lu)及びウォーカー(Walker)、2001年)。腸炎菌感染ラットの腸間膜リンパ節、肝臓及び脾臓ではかなりの数の腸炎菌が検出された。B.テタイオタオミクロンでラットを処理することにより、肝臓及び脾臓で検出される生残腸炎菌のレベルは大幅に減少した。しかし、腸間膜リンパ節での腸炎菌数はこの処理の影響を受けなかった。従って、B.テタイオタオミクロンは腸代謝に大きく作用するが、腸炎菌の腸間膜リンパ節への浸潤や流入を制限しないことが分かった。腸炎菌のレベルが全身組織で減少したことから、B.テタイオタオミクロンの主な防御作用は全身代謝の変化によって生じることが示されるであろう。
【0151】
共生細菌は強力な免疫調節物質となる(へリアス(Herias)ら、1999年;タルハム(Talham)ら、1999年;シャレック(Scharek)ら、2000年;イソラウリ(Isolauri)ら、2001年;ルー(Lu)及びウォーカー(Walker)、2001年)。特に、ビフィドバクテリウム・ラクティス(Bifidobacterium lactis)でマウスを処理すると、マウスの免疫機能やサルモネラ菌に対する反応性が向上することが分かった(シュー(Schu)ら、2000年)。血液や腹膜細胞での貪食活性、リンパ球分裂促進反応性及びティフィムリウム特異的抗体の分泌が向上した。B.テタイオタオミクロンも全身代謝に対して同様の作用を示すと思われる。従って、B.テタイオタオミクロンで処理したラットの肝臓及び脾臓で腸炎菌数が減少したことは、血液や内臓からの腸炎菌の排除が高まった結果によるものであろう。
【0152】
乳糖発酵菌(大腸菌)も、数こそ少ないが、腸炎菌感染ラットの肝臓及び脾臓で検出された。しかし、腸炎菌の場合と異なり、乳糖発酵菌のレベルはB.テタイオタオミクロンの影響を受けなかった。このことから、B.テタイオタオミクロンによって促進される細菌の排除には選択性があると思われる。或いは、大腸菌はラットの常在細菌叢に由来するため、不適切な組織部位に存在している場合でも、潜在的有害物質として容易に認識されないのであろう。
【0153】
B.テタイオタオミクロンは腸に局所的に作用することによって、サルモネラ菌の全身への伝播を抑制すると思われる。サルモネラ菌が腸上皮に付着し、腸上皮内に浸潤し、腸間膜リンパ節へ流入し、肝臓や脾臓へ伝播することは一般に認められている(キングスレー(Kingsley)及びボールマー(Baulmer)、2000年)。しかし、最近の研究では、サルモネラ菌の全身への伝播が別の経路で起こり得ることが示唆されている。樹状細胞により管腔に摂取されるか、或いはCD18を発現する貪食細胞により上皮下組織へ摂取されたサルモネラ菌は、リンパ系へ流入することなく、肝臓や脾臓に直接伝播し得る(ヴァルケス−トレス(Varquez-Torres)ら、1999年;アイスバーグ(Isberg)及びバーンズ(Barnes)、2000年;レスチグノ(Rescigno)ら、2001年)。その結果、肝臓や脾臓で検出されるサルモネラ菌は2種類の摂取経路に由来する可能性がある。上記貪食細胞の補充は、上皮細胞により産生する化学誘引物質ケモカインによって仲介される可能性がある(イザドパナー(Izadpanah)ら、2001年;ケレンナン(Kellennan)及びマッケヴォイ(McEvoy)、2001年)。B.テタイオタオミクロンは腸内での好中球補充や炎症を引き起こすのに必要な化学誘引物質ケモカインの分泌を防ぐことによって、これらの補充や炎症を抑制することが分かっているため(キャンベル(Campbell)ら、2001年)、樹状細胞やCD18を発現する貪食細胞の補充に必要とされるケモカインも抑制し、その結果、このような系によるサルモネラ菌の摂取を防止或いは抑制するであろう。
【0154】
概要
【0155】
経口投与したB.テタイオタオミクロンによって、ラットにおける腸炎菌による感染の重症度が低下した。肝臓及び脾臓内の生残腸炎菌数は大幅に減少し、骨格筋成長率は正常値に回復し、また、小腸内の炎症反応は軽減された。これは、免疫反応性が向上したことや遠隔部位(例えば、肝臓や脾臓)で腸炎菌が急速に排除されたことによるものと思われる。しかし、少なくとも一部は、B.テタイオタオミクロンの腸への局所作用に起因するものでもある。腸内での好中球補充や炎症はB.テタイオタオミクロンの作用によって防止或いは遅延された。また、B.テタイオタオミクロンは、腸炎菌の貪食細胞への直接摂取と肝臓や脾臓への伝播もブロックしたと思われる。
【0156】
〔参考文献〕
1. E. Cario et al. J Immunol. 164, 966 (2000).
2. A. T. Gewirtz, T. A. Navas, S. Lyons, P. J. Godowski, J. L. Madara, J. Immunol. 167, 1882 (2001).
3. B. A. McCormick, S. P. Colgan, C. Delp-Archer, S. I. Miller, J. L. Madara, J. Cell Biol. 123, 895 (1993).
4. L. Hang et AL., J. Immunol. 162, 3037 (1999).
5. S. Ghosh, M. J. May, E. B. Kopp Annu. Rev. Immunol. 16, 225-260 (1998).
6. M. J. May, S. Ghosh. Immunol. Today (1998).
7. Q. Cheng et al., J. Biol. Chem. 269, 13551 (1994).
8. P. J. Chiao, S. Miyamoto, I. M. Verma. Proc. Natl. Acad. Sci. USA 91, 28-32. (1994).
9. P. Renard et al. J. Biol. Chem. 275, 15193 (2000).
10. A. S. Neish et al., Science 289, 1560 (2000).
11. T. Meyer-ter-Vehn, A. Covacci, M. Kist, H. L. Pahl, J. Biol. Chem. 275, 16064 (2000).
12. J. Raingeaud et al., J. Biol. Chem. 270, 7420 (1995).
13. Nakajima et al. Gastroenterology 120, 460 2001.
14. E. D. Rosen, B. M. Spiegelman. J. Biol. Chem. 276, 37731-37734 (2001).
15. C. G. Su et al., J. Clin. Invest. 104, 383 (1999).
16. C. F. Bunn et al., Mol. Endocrinol. 15, 512 (2001).
17. M. Ricote, J. T. Huang, J. S. Welch, C. K. Glass, J. Leukoc. Biol. 66, 733 (1999).
18. P. Delerive et al., J. Biol. Chem. 274, 32048 (1999).
19. M. Gurnell et al., J. Biol. Chem. 275, 5754 (2000).
20. S. D. Savkovic, A. Koutsouris, G. Wu, G. Hecht, Biotechniques 29, 514 (2000).
21. A. Birbach et al. J. Biol. Chem. 277, 10842 (2002).
22. J. Schmid et al. J. Biol. Chem. 275, 17035 (2002).
23. Y. Zhoa et al., J. Biol. Chem. 277, 30031-30039 (2002).
24. K. Madsen et al., Gastroenterology 121, 580 (2001).
25. C. A. Parkos et al., J. Clin. Invest. 88, 1605 (1991).
26. Allen-Vercoe et al., J Med Microbiol 48, 771-780 (1999).
27. Bardocz, S. et al., Br J Nutr 76, 613-626 (1996).
28. Bernet-Camard et al., Appl Environ Microbial 63, 2747-2753 (1997).
29. Bovee-Oudenhoven et al., Gastroenterology 113, 550-557 (1997).
30. Bovee-Oudenhoven et al., J Nutr 129,607-612 (1999).
31. Bry, L et al., Science 273, 1380-1383 (1996).
32. Bryant, M. P. Am J Clin Nutr 25, 1324-1328 (1972).
33. Carter, J Exp Med 139, 1189-1203 (1974).
34. Cebra, J. J., Am J CLIN Nutr 69, 1046S-1051S (1999).
35. Chang, J. et al., J Appl Bacteriol 77, 709-718 (1994).
36. Collins, C. H. L. P. M. (1989) Microbiological Methods Butterworths, London.
37. Darwin, K. H. et al., Clin Microbiol Rev 12, 405-428 (1999).
38. Eaves-Pyles, T. et al., J Immunol 166, 1248-1260 (2001).
39. Ewen, S. W. et al., FEMS Immunol Med Microbiol 18, 185-192 (1997).
40. Filho-Lima, J. V. et al., J Appl Microbiol 88, 365-370 (2000).
41. Fleckenstein, J. M. et al., J Clin Invest 107, 27-30 (2001).
42. Friman, G. et al., Scand J Infect Dis 16, 111-119 (1984).
43. Gewirtz, A. T. et al., J Immunol 167, 1882-1885 (2001).
44. Grant, G. (1996) Management of Animal Experiments. COST 98. Effects of antinutrients on the nutritional value of legume diets (Bardocz, S; Pusztai. A., ed) pp. 44-51, European Commision, Brussels.
45. Grant, G. et al., Pancreas 20, 305-312 (2000).
46. Havelaar, A. H. et al., J Appl Microbiol 91,442-452 (2001).
47. Henderson, S. C. et al., Infect Immun 67, 3580-3586 (1999).
48. Henriksson et al., J Appl Microbiol 90, 223-228 (2001).
49. Herias et al., Scand J Immunol 48, 277-282 (1998).
50. Herias, M. V. et al., Clin Exp Immunol 116, 283-290 (1999).
51. Hooper, L. V. et al., Proc Natl Acad Sci U S A 96, 9833-9838 (1999).52. Hooper, L. V. et al., Curr Opin Microbiol 3, 79-85 (2000).
53. Hooper, L. V. et al., Glycobiology 11, 1R-10R (2001).
54. Hooper, L. V. et al., Science 291, 881-884 (2001).
55. Hudault, S. et al., Appl Environ Microbiol 63, 513-518 (1997).
56. Hudault, S. et al., Gut 49,47-55 (2001).
57. Isberg, R. R. et al., Trends Microbiol 8, 291-293 (2000).
58. Islam, A. F. et al., Infect Immun 68, 1-5 (2000).
59. Isolauri, E. et al., Am J Clin Nutr 73, 444S-450S (2001).
60. Izadpanah, A. et al., Am J Physiol Gastrointest Liver Physiol 280, G710-G719 (2001).
61. Kellermann, S. A. et al., J Immunol 167, 682-690 (2001).
62. Kingsley, R. A et al., Bacterial Invasion into Eukaryotic Cells (Oelschlaeger, T. A. and Hacker, J., eds) pp. 321-342, Kluwer Academic/Plenum Publishers, New York (2000).
63. Klasing, K. C. C. C. C. Protein Metabolism and Nutrition (Lobely, G. E; Wnite, . A. and MacRae, J. C., eds) pp. 253-264, Wageningen Pers, Wageningen (1999).
64. Lang, C. H. et al., Am J Physiol Endocrinol Metab 278, E1133-E1143 (2000).
65. Lopez-Boado, Y. S. et al., J Cell Biol 148, 1305-1315 (2000).
66. Lu, L. et al., Am J Clin Nutr 73, 1124S-1130S (2001).
67. Lu, S. et al., Infect Immun 67, 5651-5657 (1999).
68. Madara, J. L. Aliment Pharmacol Ther 11 Suppl 3, 57-62 (1997).
69. Maia, O. B. et al., Vet Microbiol 79, 183-189 (2001).
70. McCormick, B. A. et al., J Cell Biol 131, 1599-1608 (1995).
71. Miles, A. A. et al., J Hyg 38, 749 (1993). 72. Naughton, P. J. et al., FEMS Immunol Med Microbiol 12, 251-258 (1995).
73. Naughton, P. J. et al., J Appl Bacteriol 81, 651-656 (1996).
74. Naughton, P. J. et al., J Med Microbiol 50, 191-197 (2001).
75. Netea, M. G. et al., J Immunol 164, 2644-2649 (2000).
76. Noack, J. et al., J Nutr 130, 1225-1231 (2000).
77. Pier, G. B. et al., Infect Immun 60, 4768-4776 (1992).
78. Rescigno, M. et al., Nat Immunol 2, 361-367 (2001).
79. Salminen, S. et al., Br J Nutr 80 Suppl 1, S147-S171 (1998).
80. Scharek, L. et al., Immunobiology 202, 429-441 (2000).
81. Schechter, L. M. et al., Bacterial Invasion into Eukaryotic Cells. (Oelschlaeger, T. A. H. J., ed) pp. 289-320, Kluwer Academic/Plenum Publishers, New York (2000).
82. Shu, Q. et al., Med Microbiol Immunol (BERL) 189, 147-152 (2001).
83. Silva, A. M. et al., J Appl Microbiol 86, 331-336 (1999).
84. Snel, J. et al., Can J Microbiol 44, 1177-1182 (1998).
85. Stucchi, A. F. et al., Am J Physiol Gastrointest Liver Physiol 279, G1298-G1306 (2000).
86. Talham, G. L. et al., Infect Immun 67, 1992-2000 (1999).
87. Teng, L. J. et al., J Clin Microbiol 38, 1672-1675 (2000).
88. Van Der Waaij, D. The Germ-free Animal in Biomedical Research (Coates, M. E. Gustafsson,. B. E., ed) pp. 155-165, Laboratory Animals Ltd, London (1984).
89. Vassiloyanakopoulos, A. P. et al., Proc Natl Acad Sci U S A 95, 7676-7681 (1998).
90. Vazquez-Torres, A. et al., Nature 401, 804-808 (1999).
91. Yurdusev, N. et al., Can J Microbiol 33, 226-231 (1987).
92. Yurdusev, N. et al., Infect Immun 57, 724-731 (1989).
93. Zimmerman, B. J. et al., Am J Physiol 259, H390-H394 (1990).
【特許請求の範囲】
【請求項1】
サイトカイン産生に起因する炎症性疾患の治療用候補薬剤を選択するためのアッセイであって、
a)前記候補薬剤を腸細胞に接触させることと、
b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び
PPARγの核質内破壊から成る群から選択される、前記候補薬剤の作用を解析することを含むアッセイ。
【請求項2】
NF−κBファミリーの転写因子の核外移行の促進或いは核内移行の抑制、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の増加、及び
PPARγの核質内破壊から成る群から選択される少なくとも1つの作用を示す候補薬剤を選択する工程を更に含む、請求項1に記載のアッセイ。
【請求項3】
解析される候補薬剤の作用がNF−κBファミリーの転写因子の核外移行である、請求項1又は2に記載のアッセイ。
【請求項4】
解析される候補薬剤の作用が腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化である、請求項1又は2に記載のアッセイ。
【請求項5】
TNF−α、IL−8、MIP−2α及びCox−2から成る群から選択される1以上のサイトカインのレベルに対する候補薬剤の作用を解析する工程を更に含む、請求項1又は2に記載のアッセイ。
【請求項6】
解析される候補薬剤の作用がIL−8及び/又はMIP−2αのレベルの変化である、請求項5に記載のアッセイ。
【請求項7】
腸細胞が細胞培養で維持される細胞株から得られるものである、請求項1〜6のいずれか1項に記載のアッセイ。
【請求項8】
前記細胞株がCaco−2細胞株である、請求項1〜7のいずれか1項に記載のアッセイ。
【請求項9】
動物に対してインビボで行う、請求項1〜6のいずれか1項に記載のアッセイ。
【請求項10】
前記動物が哺乳動物である、請求項9に記載のアッセイ。
【請求項11】
公知の病原性細菌の存在下で行う、請求項1〜10のいずれか1項に記載のアッセイ。
【請求項12】
前記公知の病原性細菌がサルモネラ菌である、請求項11に記載のアッセイ。
【請求項13】
炎症性サイトカイン産生に伴う疾患の治療方法であって、
NF−κBファミリーの転写因子の核外移行或いは核内移行、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内のPPARγ/RelA複合体量、又は
PPARγの核質内破壊を調節することが可能な化合物の治療有効量を投与する工程を含む方法。
【請求項14】
前記化合物がNF−κBの転写活性を変化させることが可能な化合物である、請求項13に記載の方法。
【請求項15】
前記化合物が腸細胞のサイトゾル内でPPARγ/RelA複合体量を増加させることが可能な化合物である、請求項13に記載の方法。
【請求項16】
哺乳動物の免疫系をホメオスタシスへ回復させ且つ維持させる、請求項13〜15のいずれか1項に記載の方法。
【請求項17】
炎症性疾患の治療のための請求項1〜12のいずれか1項に記載のアッセイによって選択された剤の使用。
【請求項18】
炎症性疾患が腸細胞の炎症反応によって少なくとも一部引き起こされる疾患である、請求項17に記載の剤の使用。
【請求項19】
炎症性疾患が炎症性サイトカインの産生に伴う疾患である、請求項17又は18に記載の剤の使用。
【請求項20】
炎症性疾患が、炎症性腸疾患、クローン病、過敏性腸症候群、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイトの増殖、皮膚の過剰増殖及び炎症性障害、乾癬及び尋常性ざ瘡から成る群から選択される疾患である、請求項17〜19のいずれか1項に記載の剤の使用。
【請求項21】
バクテロイデス・テタイオタオミクロン又はその一構成成分の治療有効量を投与する工程を含む、腸細胞の炎症反応の処置方法。
【請求項22】
食品や座薬を用いてバクテロイデス・テタイオタオミクロンを生菌の状態で患者に投与する、請求項21に記載の方法。
【請求項23】
炎症性サイトカイン産生に伴う疾患の治療のための請求項21又は22に記載の方法。
【請求項24】
炎症性腸疾患、クローン病、過敏性腸症候群、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイトの増殖、皮膚の過剰増殖及び炎症性障害、乾癬及び尋常性ざ瘡から成る群から選択される疾患の治療のための請求項21〜23のいずれか1項に記載の方法。
【請求項25】
バクテロイデス・テタイオタオミクロン又はその一構成成分の治療有効量を経口投与する、請求項21〜24のいずれか1項に記載の方法。
【請求項1】
サイトカイン産生に起因する炎症性疾患の治療用候補薬剤を選択するためのアッセイであって、
a)前記候補薬剤を腸細胞に接触させることと、
b)NF−κBファミリーの転写因子の核外移行或いは核内移行の変化、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化、及び
PPARγの核質内破壊から成る群から選択される、前記候補薬剤の作用を解析することを含むアッセイ。
【請求項2】
NF−κBファミリーの転写因子の核外移行の促進或いは核内移行の抑制、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内でのPPARγ/RelA複合体量の増加、及び
PPARγの核質内破壊から成る群から選択される少なくとも1つの作用を示す候補薬剤を選択する工程を更に含む、請求項1に記載のアッセイ。
【請求項3】
解析される候補薬剤の作用がNF−κBファミリーの転写因子の核外移行である、請求項1又は2に記載のアッセイ。
【請求項4】
解析される候補薬剤の作用が腸細胞のサイトゾル内でのPPARγ/RelA複合体量の変化である、請求項1又は2に記載のアッセイ。
【請求項5】
TNF−α、IL−8、MIP−2α及びCox−2から成る群から選択される1以上のサイトカインのレベルに対する候補薬剤の作用を解析する工程を更に含む、請求項1又は2に記載のアッセイ。
【請求項6】
解析される候補薬剤の作用がIL−8及び/又はMIP−2αのレベルの変化である、請求項5に記載のアッセイ。
【請求項7】
腸細胞が細胞培養で維持される細胞株から得られるものである、請求項1〜6のいずれか1項に記載のアッセイ。
【請求項8】
前記細胞株がCaco−2細胞株である、請求項1〜7のいずれか1項に記載のアッセイ。
【請求項9】
動物に対してインビボで行う、請求項1〜6のいずれか1項に記載のアッセイ。
【請求項10】
前記動物が哺乳動物である、請求項9に記載のアッセイ。
【請求項11】
公知の病原性細菌の存在下で行う、請求項1〜10のいずれか1項に記載のアッセイ。
【請求項12】
前記公知の病原性細菌がサルモネラ菌である、請求項11に記載のアッセイ。
【請求項13】
炎症性サイトカイン産生に伴う疾患の治療方法であって、
NF−κBファミリーの転写因子の核外移行或いは核内移行、
NF−κBファミリーの転写因子の転写活性の破壊、
p65(RelA)の特異的ヒストンアセチル化、
腸細胞のサイトゾル内のPPARγ/RelA複合体量、又は
PPARγの核質内破壊を調節することが可能な化合物の治療有効量を投与する工程を含む方法。
【請求項14】
前記化合物がNF−κBの転写活性を変化させることが可能な化合物である、請求項13に記載の方法。
【請求項15】
前記化合物が腸細胞のサイトゾル内でPPARγ/RelA複合体量を増加させることが可能な化合物である、請求項13に記載の方法。
【請求項16】
哺乳動物の免疫系をホメオスタシスへ回復させ且つ維持させる、請求項13〜15のいずれか1項に記載の方法。
【請求項17】
炎症性疾患の治療のための請求項1〜12のいずれか1項に記載のアッセイによって選択された剤の使用。
【請求項18】
炎症性疾患が腸細胞の炎症反応によって少なくとも一部引き起こされる疾患である、請求項17に記載の剤の使用。
【請求項19】
炎症性疾患が炎症性サイトカインの産生に伴う疾患である、請求項17又は18に記載の剤の使用。
【請求項20】
炎症性疾患が、炎症性腸疾患、クローン病、過敏性腸症候群、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイトの増殖、皮膚の過剰増殖及び炎症性障害、乾癬及び尋常性ざ瘡から成る群から選択される疾患である、請求項17〜19のいずれか1項に記載の剤の使用。
【請求項21】
バクテロイデス・テタイオタオミクロン又はその一構成成分の治療有効量を投与する工程を含む、腸細胞の炎症反応の処置方法。
【請求項22】
食品や座薬を用いてバクテロイデス・テタイオタオミクロンを生菌の状態で患者に投与する、請求項21に記載の方法。
【請求項23】
炎症性サイトカイン産生に伴う疾患の治療のための請求項21又は22に記載の方法。
【請求項24】
炎症性腸疾患、クローン病、過敏性腸症候群、関節リウマチ、免疫不全症候群、悪液質、多発性硬化症、ケラチノサイトの増殖、皮膚の過剰増殖及び炎症性障害、乾癬及び尋常性ざ瘡から成る群から選択される疾患の治療のための請求項21〜23のいずれか1項に記載の方法。
【請求項25】
バクテロイデス・テタイオタオミクロン又はその一構成成分の治療有効量を経口投与する、請求項21〜24のいずれか1項に記載の方法。
【図1】

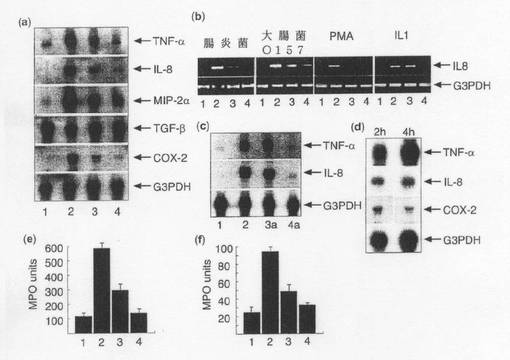
【図2】

【図3】
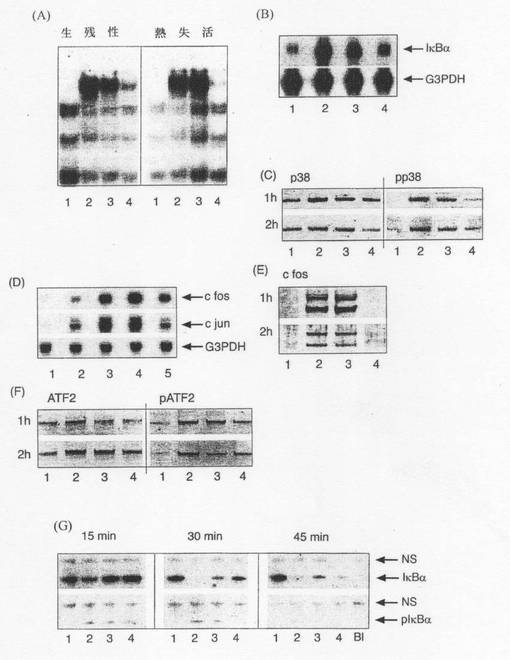
【図4】
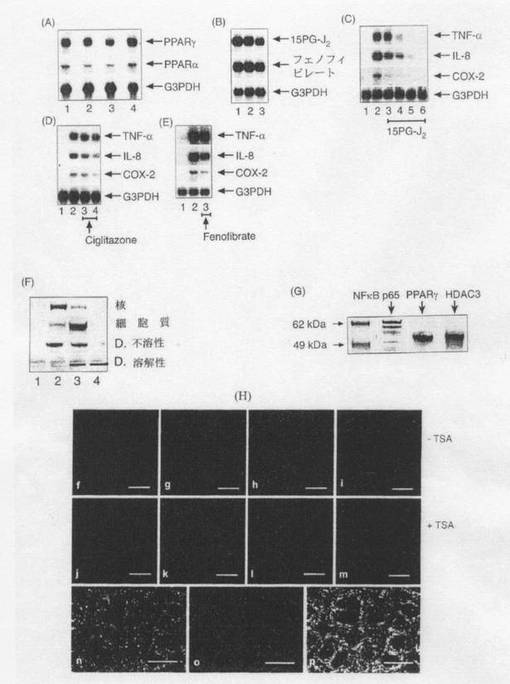
【図5】
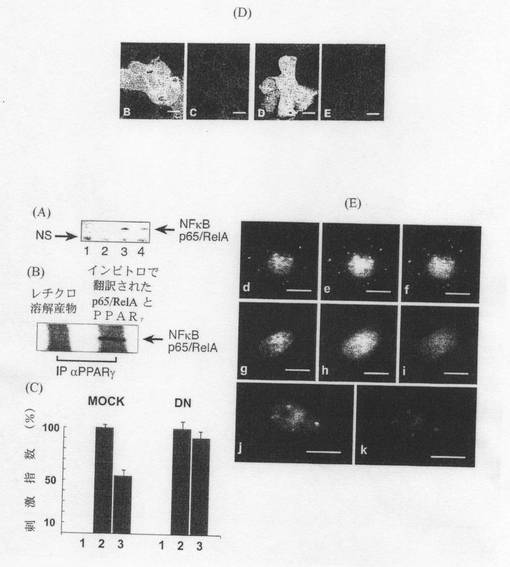
【図6】
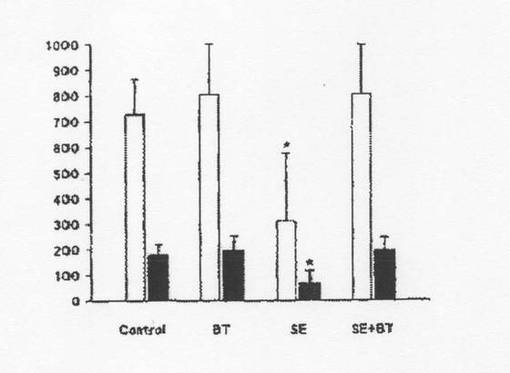
【図7】
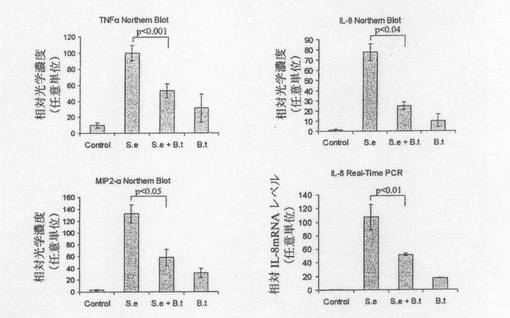
【図8】

【図9A】
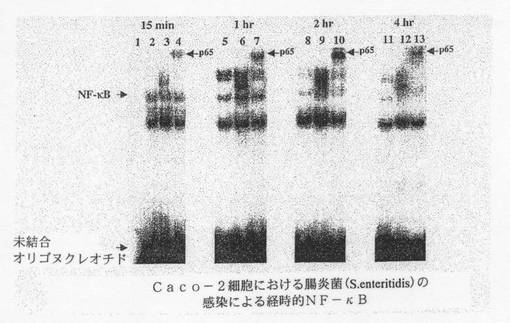
【図9B】
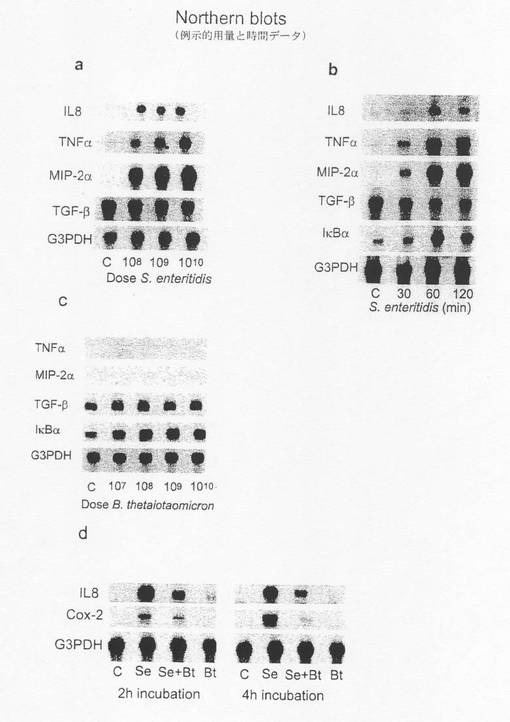
【図9C】
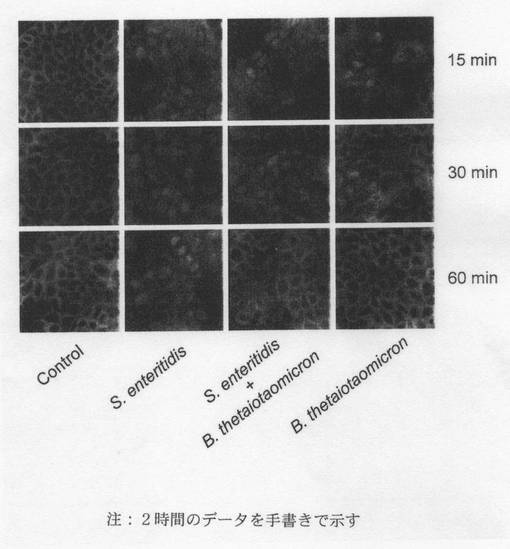
【図9D】

【図9E】
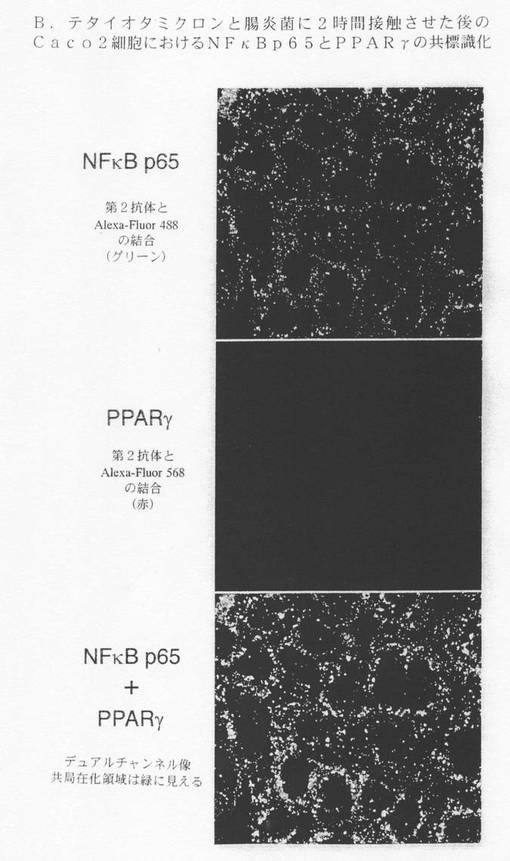
【図10】
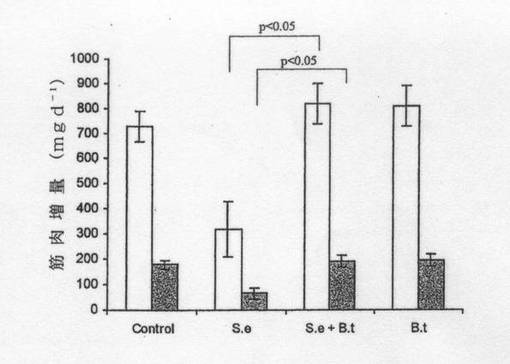
【図11】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図10】
【図11】
【公開番号】特開2013−46615(P2013−46615A)
【公開日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−196103(P2012−196103)
【出願日】平成24年9月6日(2012.9.6)
【分割の表示】特願2003−547968(P2003−547968)の分割
【原出願日】平成14年11月20日(2002.11.20)
【出願人】(503124676)ロウェット、リサーチ、インスティテュート (1)
【氏名又は名称原語表記】ROWETT RESEARCH INSTITUTE
【住所又は居所原語表記】Greenburn Road, Bucksburn, Aberdeen AB21 9SB, GB
【Fターム(参考)】
【公開日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成24年9月6日(2012.9.6)
【分割の表示】特願2003−547968(P2003−547968)の分割
【原出願日】平成14年11月20日(2002.11.20)
【出願人】(503124676)ロウェット、リサーチ、インスティテュート (1)
【氏名又は名称原語表記】ROWETT RESEARCH INSTITUTE
【住所又は居所原語表記】Greenburn Road, Bucksburn, Aberdeen AB21 9SB, GB
【Fターム(参考)】
[ Back to top ]