
炎症部位集積性化合物、核医学画像診断剤及び標識前駆体
【課題】生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体を提供する。
【解決手段】
式(1)で表される炎症部位集積性化合物。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基;YはMet又はNle;Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;Tはスペーサー、mは0又は1;Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1;HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
【解決手段】
式(1)で表される炎症部位集積性化合物。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基;YはMet又はNle;Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;Tはスペーサー、mは0又は1;Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1;HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、炎症部位集積性化合物、核医学画像診断剤及び標識前駆体に関するものである。更に詳しくは、本発明は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体に関するものである。
【背景技術】
【0002】
ヒトをはじめとする哺乳動物は、有害な外部刺激を受けた場合、防御作用として免疫応答反応を行う。免疫応答反応の代表的なものとして、炎症反応をあげることができる。炎症反応により、個体に侵入した異物の除去、侵された組織の破壊、破壊された組織の修復等が行われる。
【0003】
ところで、糖尿病に伴う重大な病変として、糖尿病足病変がある。糖尿病足病変とは、糖尿病患者の下肢に生じる、主に感染症を起因とした、潰瘍、深部組織の破壊性病変であり、神経障害や種々の程度の末梢血流障害を合併している病変である。糖尿病足病変が進行すると、病変部組織に壊死が発生し、足を切断する必要が生じるという重大な事態に陥る。したがって、糖尿病足病変を早期に発見して治療し、治療の効果を追跡しつつ、効果的な治療を行う必要がある。ところが、かかる要求を十分に満たす方法は見出されていない。本発明はかかる現状においてなされたものである。
【0004】
糖尿病足病変を含む炎症部位には、防御反応として、ホルミル化ペプチド受容体(FPR)を発現する白血球が集積することが知られている。
【0005】
FPRへの親和性を有するペプチドとして、走化性ホルミル化ペプチドであるホルミル−メチオニル−ロイシル−フェニルアラニル(fMLF)含有ペプチドが知られている。非特許文献1には、125Iで放射性核種標識したfMLFが記載されている。非特許文献2には、125Iで放射性核種標識したfMLFが生体内で炎症に集積することが記載されている。特許文献1には、DTPA(ジエチレントリアミン5酢酸)を介した111InfMLFが開示されている。非特許文献3には、メルカプトアセチルグリシルグリシンを介したTc−99mfMLFが記載されている。非特許文献4には、ジアミノジチオール化合物を介したTc−99mfMLFが記載されている。特許文献2には、放射性核種fMLFの光親和性を介して身体外で白血球を放射性核種標識するための使用が記載されている。特許文献3には、放射性核種標識可能なfMLFが記載されている。特許文献4には、白血球の受容体FPRとの結合部位、全白血球中の単球及びリンパ球への結合性を向上させる部位並びに放射性金属で標識可能な部位を含むペプチドが開示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】 特許第2931093号明細書
【特許文献2】 米国特許第4,986,979号明細書
【特許文献3】 米国特許第5,792,444号明細書
【特許文献4】 国際公開第2004/029080号パンフレット
【非特許文献】
【0007】
【非特許文献1】 Day,AR.et al.,FEBS Lett.77,291−294(1977)
【非特許文献2】 Jiang,MS.et al.,Nuklearmedizin,21,110−113(1982)
【非特許文献3】 Verbeke,K.et al,,Nuclear Medicine & Biology,27,769−779(2000)
【非特許文献4】 Baidoo,K.E.et al.,Bioconjugate Chemistry,9,208−217(1998)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、PETをはじめとする核医学診断を行うために最適な構造を持ったペプチドがさらに求められており、またその放射性ペプチドを核医学診断剤として利用するためには、自然界に存在するfMLFより、ターゲットである白血球に発現するFPRへの十分に高い親和性、つまり集積性を有することが望ましい。ところが、従来の技術によると、かかる要求を十分に満たす放射性化合物は見出されていなかった。
【0009】
かかる状況において、本発明が解決しようとする課題は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体を提供する点にある。
【課題を解決するための手段】
【0010】
すなわち、本発明のうち一の発明は、下記式(1)で表される炎症部位集積性化合物に係るものである。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表し;
HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
また、本発明のうち第二の発明は、上記第一の発明の炎症部位集積性化合物を主成分とする核医学画像診断剤に係るものである。
また、本発明のうち第三の発明は、上記第一の発明の化合物の標識前駆体であって、下記式(3)で表される標識前駆体に係るものである。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m)−(U)k(NH2) (3)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【発明の効果】
【0011】
本発明により、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体を提供することができる。
【図面の簡単な説明】
【0012】
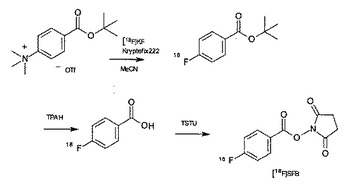
【図1】放射性ハロゲン含有モノマー[18F]SFBの合成スキーム
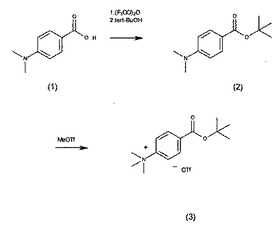
【図2】t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateの合成スキーム
【発明を実施するための形態】
【0013】
以下、本発明を実施するための形態について、詳細に説明する。
【0014】
本明細書で用いるアミノ酸は全て三文字表記で記し、特に断りのないかぎり左側をN末端側、右側をC末端側として表記した。アミノ酸に続くかっこ内は、特に断りのないかぎり側鎖に結合したペプチドならびに有機化合物を表すものである。また、かっこ内のアミノ酸配列は全体構造を把握しやすくするために、右側をN末端側、左側をC末端側として表記した。さらに、本明細書において、D体のアミノ酸は三文字表記ではDアミノ酸で記載した。
【0015】
本発明の炎症部位集積性化合物は、下記式(1)で表される炎症部位集積性化合物である。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
【0016】
式(1)中、Zはアミノ基の保護基を表す。Zの具体例としては、ホルミル基、アセチル基などの炭素数1〜9のアシル基、t−Boc基(ter−ブトキシカルボニル基)などの炭素数2〜9のアシルオキシ基、メチル、エチル、プロピルなどの炭素数1〜6の低級アルキル基、カルバミル基などをあげることができる。これらのうちでは、FPRへの親和性の観点から、ホルミル基が好ましい。
【0017】
式(1)中、Yはアミノ酸であるMet又はNleを表す。
【0018】
式(1)中、(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサーであり、nは0又は1である。なお、Xが(−Nle−Tyr−)であり、nが1であることが好ましい。
【0019】
式(1)中、Tはスペーサーであり、mは0又は1を表す。
【0020】
本発明の炎症部位集積性化合物の好ましい具体例として、Tが下記式(2)で表され、mが1であるものをあげることができる。
−HN−(C2H4O)pCH2COO− (2)
(式(2)おいて、pは1〜6の整数を表す。)
【0021】
式(1)中、HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。HalBのハロゲンとしては、SPECT用として121I、123I、125I及び131Iがあげられ、PET用としては124I及び18Fをあげることができる。なお、PET用としては、汎用性の観点から、18F(この場合のHalBを「[18F]FB」と表す。)が好ましい。
【0022】
式(1)中、Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【0023】
本発明の炎症部位集積性化合物の、より具体的で好ましい例として、下記のものをあげることができる。
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−NH2;(配列番号1)
ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([18F]FB)−NH2;(配列番号2)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−NH2;(配列番号3)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;(配列番号4)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[18F]FB)−NH2;(配列番号5)
【0024】
本発明の炎症部位集積性化合物は、たとえば以下に示すステップ−1〜ステップ−3を用いる方法により合成することができる。
【0025】
[ステップ−1]標識前駆体の合成
標識前駆体であるペプチドは、「固相法」又は「液相法」として知られるペプチド合成法により、調製することができる。例えば、社団法人日本生化学会編集『生化学実験講座』、第1巻、「タンパク質IV」、第207〜495頁、1977年、東京化学同人発行及び社団法人日本生化学会編集『新生化学実験講座』、第1巻、「タンパク質VI」、第3〜74頁、1992年、東京化学同人発行などにはペプチド合成の詳細が記載されている。また、Fmoc(9−フルオレニルメトオキシカルボニル)固相合成法を用いてペプチド合成機にて合成することができる。すなわち、合成する各ペプチドのC末端に相当するアミノ酸が導入されているFmocアミノ酸を樹脂に結合させ、(I)Fmoc基の脱保護と洗浄、(II)Fmocアミノ酸の縮合と洗浄、の操作を繰り返してペプチド鎖を延長し、最後に最終脱保護反応させて、目的とするペプチドを合成することができる。
【0026】
ペプチドを単離精製するためには、公知の分離操作を組み合わせて行うことができる。例えば、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、逆相クロマトグラフィー、高速液体クロマトグラフィーなどのペプチド又は蛋白質を精製するための方法が用いられ、必要に応じて、これら方法を適宜組合せてもよい。そして、最終使用形態に応じて、精製したペプチドを濃縮、また必要に応じてさらに凍結乾燥して単離すればよい。
【0027】
[ステップ−2]放射性ハロゲン含有モノマー[18F]SFBの合成
下記に記載した手順に従い、放射性ハロゲン含有モノマー[18F]SFBを得ることができる(図1)。
【0028】
1.相間移動触媒を遮光バイアル中で脱水アセトニトリルに溶かし、18F−のK2CO3水溶液を必要な放射能分を加えて攪拌する。相間移動触媒としては、18Fイオンとの間で包摂体を形成する性質を有する種々の化合物を用いることができる。具体的には、放射性フッ素標識有機化合物の製造に用いられている種々の化合物を用いることができ、18−クラウン−6−エーテル及びその他の種々のアミノポリエーテルを用いることができる。最も好ましい態様としては、クリプトフィックス2.2.2.(商品名、メルク社製)を用いることができる。
2.窒素ガスを吹き付けながら加熱して溶媒を飛ばす。更に脱水アセトニトリルを加えて溶媒を飛ばし、完全に水を飛ばす。
3.t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateを脱水アセトニトリルに溶かし、反応バイアルに加えて強く攪拌し、反応させる。なお、t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateと脱水アセトニトリルの量比については、t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateが完全に溶解する限りにおいて限定されないが、好ましくはt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate 0.3〜0.5mgの場合、脱水アセトニトリルは100〜200μLとすることができる。
4.反応後、tetrapropylammonium hydroxideを加えて攪拌し、反応させる。
5.反応後、TSTUを(脱水)アセトニトリルに溶かして反応バイアルに加えて攪拌し、反応させる。
6.反応液を5%酢酸水溶液で希釈し、アセトニトリルと水で活性化したSep−Pak(登録商標、日本ウォーターズ株式会社製)plus PS−2 に通し、水/アセトニトリルでカラムを洗浄し、アセトニトリルで放射性ハロゲン含有モノマーである[18F]SFBを溶出する。
【0029】
なお、上記の方法による出発原料であるt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateの製造方法は公知であり(たとえば、Applied Radiation and Isotopes 59(2003) 43−48)次の手順に従って合成することができる。
4−N,N−dimethylamino benzoic acid (1)を含む冷却された乾燥THF中にtrifluoroacetic anhydrideを加える。しばらく後(たとえば30分後)にtert−BuOHを加え、室温に保つ(たとえば2時間)。その後、飽和NaHCO3水溶液に注ぎ、CH2Cl2で抽出する。抽出物をショートシリカゲルカラムに通し、減圧下に溶媒を除去することにより、tert−butyl ester (2)が得られる。
(2)をnitromethaneに溶解させ、冷却する。methyl triflateを加え、攪拌する(たとえば1時間)。反応物をdiethyl etherに注ぎ、真空乾燥することによりt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate (3)が得られる。
【0030】
[ステップ−3]ペプチドの標識
標識前駆体であるペプチドをアセトニトリル(以下、MeCNと表記する)とBorate Bufferの混合液に溶かし、70℃ Ar気流下で濃縮した[18F]SFBに加える。MeCN/トリエチルアミン(以下、Et3Nと表記する)=98/2でpHを8.5〜9.0にし、反応させる。HPLCを用いて分取、精製し、純度確認を行う。
【0031】
本発明の炎症部位集積性化合物は、炎症部位への高い集積性を有し、糖尿病足病変に伴う炎症部位の診断をはじめ、核医学画像診断剤の主成分として最適に用いることができる。なお、炎症を伴う疾患としては、糖尿病足病変、炎症性腸疾患等を例示することができる。
【0032】
本発明の核医学画像診断剤は、本発明に係る放射性ヨウ素標識ペプチドを溶解した液として調製することができる。放射性ヨウ素標識ペプチドを溶解する液は、水、生理食塩水やリンゲル液等を用いることができる。放射性ヨウ素標識ペプチドの水溶性が低い場合には、必要に応じて可溶化剤を添加するか、当該ペプチドを溶解させることができる液に溶解後、生体認容性ある液と混合する。たとえば、放射性ヨウ素標識ペプチドとして、配列番号1〜5に記載のペプチドを用いた場合には、当該ペプチドをDMSOに溶解し、10%DMSOになるよう、binding bufferあるいは生理食塩液を加えていき、水溶液を作製するといった方法を用いればよい。また、必要に応じて、安定化剤を配合してもよい。
【0033】
本発明に係る核医学画像診断剤の投与量は、投与された薬剤の分布を画像化するために十分な濃度であれば特に限定する必要はない。たとえば、18F標識ペプチドの場合は、体重60kgの成人一人当り50〜600MBq程度、静脈投与又は局所投与して使用することができる。投与された薬剤の分布は、PET装置やSPECT装置を用いて公知の方法により画像化することができる。
【実施例】
【0034】
次に、本発明を実施例により説明する。
実施例に用いた化合物は次のとおりである。なお、本実施例においては、炎症部位への集積性を評価するという目的に鑑み、非標識化合物を用いるコールドテストとした。
【0035】
本発明による化合物の非標識化合物
化合物1:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−NH2;
化合物2:ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([19F]FB)−NH2;
化合物3:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−NH2;
化合物4:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;
化合物5:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[19F]FB)−NH2;
【0036】
以下の方法により、上記化合物1〜5を得た。
ここで、アミノ酸の表示については、アラニン(Ala、A)、システイン(Cys、C)、アスパラギン酸(Asp、D)、グルタミン酸(Glu、E)、フェニルアラニン(Phe、F)、グリシン(Gly、G)、ヒスチジン(His、H)、イソロイシン(Ile、I)、リジン(Lys、K)、ロイシン(Leu、L)、メチオニン(Met、M)、アスパラギン(Asn、N)、プロリン(Pro、P)、グルタミン(Gln、Q)、アルギニン(Arg、R)、セリン(Ser、S)、スレオニン(Thr、T)、バリン(Val、V)、トリプトファン(Trp、W)、ノルロイシン(Nle)と1文字あるいは3文字表示とする。
また、ペプチドは、ペプチド自動合成機(433A型:Applied Biosystems社製)を用い、樹脂に固定したアミノ酸誘導体に、1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。
【0037】
なお、化合物1〜5(非標識化合物)の合成においては、公知の方法に準じてリジンの側鎖に非放射性FBを導入したアミノ酸を予め調製し、自動合成機を用いた合成の原料として用いた。
リジンの側鎖に非放射性FBを導入したアミノ酸は次のとおり調製した。すなわち、Fmoc−Lys(5g,13.6mmol)を水:THF(1:9,30ml)に溶解し、DIEA(4.7ml,27.1mmol、diisopropylethylamine)を加えた後、氷冷下に攪拌しながらFB−Cl(1.53ml,12.9mmol,4−F−benzoic acid chloride)を加えた。その後反応液を一昼夜攪拌の後、0.5mol/L塩酸水溶液(300ml)に反応液を加えた後、酢酸エチル300mlで目的とする表題誘導体を抽出した。得られた酢酸エチル層を硫酸ナトリウムで乾燥し、減圧濃縮により白色固体粉末を得た。このものを該当するペプチド合成のアミノ酸誘導体原料として使用した。
【0038】
実施例1 化合物1(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin (0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H20−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
HPLC分析条件:
カラム YMC A−302(ODS,150×4.6mm I.D.)
カラム温度 40℃
溶離液 MeCN/Water/0.1%TFA(25min linear)
流速 1.0mL/min
検出器 220nm
注入量 1μL
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 20.3min,純度 92.3%
m/z 945.5([M+H]+ 946.1),m/z 473.4([M+2H]2+ 473.6) 分子量 945.1
【0039】
実施例2 ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−[18F]FBの合成
[ステップ−1]標識前駆体の合成
標識前駆体:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。
使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(Boc) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SALResin保護ペプチド樹脂を得た。その後ホルミル−MetをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
【0040】
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別後、TFAを留去し、残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
【0041】
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
HPLC分析条件:
カラム YMC A−302(ODS,150×4.6mm I.D.)
カラム温度 40℃
溶離液 MeCN/Water/0.1%TFA(25min linear)
流速 1.0mL/min
検出器 220nm
注入量 1μL
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 14.5min,純度 93.9%
m/z 823.5([M+H]+ 824.0),m/z 412.4([M+2H]2+ 412.5) 分子量 823.0
【0042】
[ステップ−2]放射性ハロゲン含有モノマーの合成
1.Kryptofix(商品名、メルク社製)2.2.2.(商品名、メルク社製)(10mg)を遮光バイアル中で脱水アセトニトリル(500 μL)に溶かし、18F−のK2CO3水溶液(100〜500 μL)を加えて攪拌した。
2.窒素ガスを吹き付けながら110℃の油浴で加熱して溶媒を飛ばした(目安:10 min)。更に脱水アセトニトリル(400 μL×3, 目安:各3 min)を加えて溶媒を飛ばし、完全に水を飛ばした。
3.t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate(0.5mg)を脱水アセトニトリル(1 mL)に溶かし、反応バイアルに加えて強く攪拌し、90℃で10 min反応させた。
4.反応後、tetrapropylammonium hydroxide(1 mol/L in H2O,20 μL)を加えて攪拌し、120℃で5 min反応させた。
5.反応後、TSTU(15 mg)を(脱水)アセトニトリル(100 μL)に溶かして反応バイアルに加えて攪拌し、90℃で2 min反応させた。
6.反応液を5%酢酸水溶液(10 mL)で希釈し、アセトニトリルと水(各5 mL)で活性化したSep−Pak(登録商標、日本ウォーターズ株式会社製) plus PS−2に通し、水/アセトニトリル(80/20, 20 mL)でカラムを洗浄し、アセトニトリル(2.5mL)で[18F]SFBを溶出した。
【0043】
[ステップ−3]ペプチドの放射性フッ素標識
標識前駆体であるペプチド0.3mgをMeCN 40μL, Borate Buffer 40μLに溶かし、70℃ Ar気流下で濃縮した[18F]SFBに加えた。MeCN/Et3N=98/2でpHを8.5−9.0にし、1時間30分反応させた。HPLCを用いて分取、純度確認を行った。
HPLC分析条件:
カラム Cosmosil(5C18−ARII,250×10mm I.D.)
カラム温度 40℃
溶離液 MeOH/Water/0.1%TFA
流速 1.0mL/min
検出器 220nm
注入量 25μL
分析結果:MeOH 70−90%,
retention time 22.8min,放射化学的収率 31%、放射化学的純度 99%以上
【0044】
実施例3 化合物2(ホルミル−Met−Leu−Phe−Met−Tyr−Lys(NH2)−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin (0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v)脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 18.8min,純度 96.1%
m/z 963.8([M+H]+ 964.2),m/z 482.5([M+2H]2+ 482.6) 分子量 963.2
【0045】
実施例4 化合物3(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−NH2)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 18.6min,純度 91.5%
m/z 1290.8([M+H]+ 1291.4),m/z 646.0([M+2H]2+ 646.2) 分子量 1290.4
【0046】
実施例5 化合物4(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−DArg−Ser−NH2)の合成
(1)保護ペプチド樹脂の合成
Applied Blosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB)そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/400μL 75%MeCN/H2O
注入量 2μL,retention time 16.7min,純度 97.5%
m/z 767.6([M+2H]2+ 767.8) 分子量1533.7
【0047】
実施例6 化合物5(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−PEG3−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB)そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 30−80%,溶液 1mg/200μL DMSO
retention time 14.3min, 純度 98.0%
m/z 1134.9([M+H]+ 1135.3),m/z 568.3([M+2H]2+ 568.2) 分子量 1134.3
【0048】
実施例7 Binding Assay/Inhibition Assay:ホルミル化ペプチド受容体(FPR)に対する結合親和性の評価
得られた化合物1〜5について、次の方法によりFPRに対する結合親和性を評価した。
binding buffer(170μL)中に様々な濃度のペプチド(DMSO溶液,10μL)と、2nmol/Lの[125I]WKYMVm(10μL)と、FPR(10μL)を加え、25℃で1時間インキュベート後、ポリリジンbufferによりコーティングしたGF/Cフィルターを用いて濾取(セルハーベスタ)し、wash後、フィルター上に残った放射能をγカウンターで測定した。
※binding buffer:50mmol/L Hepes,pH 7.4,5mmol/L MgCl2,1mmol/L CaCl2,0.2% BSA
wash buffer:50mmol/L Hepes,pH 7.4,500mmol/L NaCl,0.1% BSA
ポリリジンbuffer:ポリL−リジン臭化水素酸塩100mg/wash buffer 100mL
評価結果を表1に示した。
【0049】
【表1】

【0050】
表1の結果から、本発明による化合物はFMLPに比べ、低いKiを示し、よって高いFPRへの親和性、すなわち高い炎症集積性を有することがわかる。
【産業上の利用可能性】
【0051】
本発明は、炎症部位集積性化合物及び核医学画像診断剤に関するものである。更に詳しくは、本発明は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体に利用することができる。
【技術分野】
【0001】
本発明は、炎症部位集積性化合物、核医学画像診断剤及び標識前駆体に関するものである。更に詳しくは、本発明は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体に関するものである。
【背景技術】
【0002】
ヒトをはじめとする哺乳動物は、有害な外部刺激を受けた場合、防御作用として免疫応答反応を行う。免疫応答反応の代表的なものとして、炎症反応をあげることができる。炎症反応により、個体に侵入した異物の除去、侵された組織の破壊、破壊された組織の修復等が行われる。
【0003】
ところで、糖尿病に伴う重大な病変として、糖尿病足病変がある。糖尿病足病変とは、糖尿病患者の下肢に生じる、主に感染症を起因とした、潰瘍、深部組織の破壊性病変であり、神経障害や種々の程度の末梢血流障害を合併している病変である。糖尿病足病変が進行すると、病変部組織に壊死が発生し、足を切断する必要が生じるという重大な事態に陥る。したがって、糖尿病足病変を早期に発見して治療し、治療の効果を追跡しつつ、効果的な治療を行う必要がある。ところが、かかる要求を十分に満たす方法は見出されていない。本発明はかかる現状においてなされたものである。
【0004】
糖尿病足病変を含む炎症部位には、防御反応として、ホルミル化ペプチド受容体(FPR)を発現する白血球が集積することが知られている。
【0005】
FPRへの親和性を有するペプチドとして、走化性ホルミル化ペプチドであるホルミル−メチオニル−ロイシル−フェニルアラニル(fMLF)含有ペプチドが知られている。非特許文献1には、125Iで放射性核種標識したfMLFが記載されている。非特許文献2には、125Iで放射性核種標識したfMLFが生体内で炎症に集積することが記載されている。特許文献1には、DTPA(ジエチレントリアミン5酢酸)を介した111InfMLFが開示されている。非特許文献3には、メルカプトアセチルグリシルグリシンを介したTc−99mfMLFが記載されている。非特許文献4には、ジアミノジチオール化合物を介したTc−99mfMLFが記載されている。特許文献2には、放射性核種fMLFの光親和性を介して身体外で白血球を放射性核種標識するための使用が記載されている。特許文献3には、放射性核種標識可能なfMLFが記載されている。特許文献4には、白血球の受容体FPRとの結合部位、全白血球中の単球及びリンパ球への結合性を向上させる部位並びに放射性金属で標識可能な部位を含むペプチドが開示されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】 特許第2931093号明細書
【特許文献2】 米国特許第4,986,979号明細書
【特許文献3】 米国特許第5,792,444号明細書
【特許文献4】 国際公開第2004/029080号パンフレット
【非特許文献】
【0007】
【非特許文献1】 Day,AR.et al.,FEBS Lett.77,291−294(1977)
【非特許文献2】 Jiang,MS.et al.,Nuklearmedizin,21,110−113(1982)
【非特許文献3】 Verbeke,K.et al,,Nuclear Medicine & Biology,27,769−779(2000)
【非特許文献4】 Baidoo,K.E.et al.,Bioconjugate Chemistry,9,208−217(1998)
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながら、PETをはじめとする核医学診断を行うために最適な構造を持ったペプチドがさらに求められており、またその放射性ペプチドを核医学診断剤として利用するためには、自然界に存在するfMLFより、ターゲットである白血球に発現するFPRへの十分に高い親和性、つまり集積性を有することが望ましい。ところが、従来の技術によると、かかる要求を十分に満たす放射性化合物は見出されていなかった。
【0009】
かかる状況において、本発明が解決しようとする課題は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体を提供する点にある。
【課題を解決するための手段】
【0010】
すなわち、本発明のうち一の発明は、下記式(1)で表される炎症部位集積性化合物に係るものである。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表し;
HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
また、本発明のうち第二の発明は、上記第一の発明の炎症部位集積性化合物を主成分とする核医学画像診断剤に係るものである。
また、本発明のうち第三の発明は、上記第一の発明の化合物の標識前駆体であって、下記式(3)で表される標識前駆体に係るものである。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m)−(U)k(NH2) (3)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【発明の効果】
【0011】
本発明により、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体を提供することができる。
【図面の簡単な説明】
【0012】
【図1】放射性ハロゲン含有モノマー[18F]SFBの合成スキーム
【図2】t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateの合成スキーム
【発明を実施するための形態】
【0013】
以下、本発明を実施するための形態について、詳細に説明する。
【0014】
本明細書で用いるアミノ酸は全て三文字表記で記し、特に断りのないかぎり左側をN末端側、右側をC末端側として表記した。アミノ酸に続くかっこ内は、特に断りのないかぎり側鎖に結合したペプチドならびに有機化合物を表すものである。また、かっこ内のアミノ酸配列は全体構造を把握しやすくするために、右側をN末端側、左側をC末端側として表記した。さらに、本明細書において、D体のアミノ酸は三文字表記ではDアミノ酸で記載した。
【0015】
本発明の炎症部位集積性化合物は、下記式(1)で表される炎症部位集積性化合物である。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
【0016】
式(1)中、Zはアミノ基の保護基を表す。Zの具体例としては、ホルミル基、アセチル基などの炭素数1〜9のアシル基、t−Boc基(ter−ブトキシカルボニル基)などの炭素数2〜9のアシルオキシ基、メチル、エチル、プロピルなどの炭素数1〜6の低級アルキル基、カルバミル基などをあげることができる。これらのうちでは、FPRへの親和性の観点から、ホルミル基が好ましい。
【0017】
式(1)中、Yはアミノ酸であるMet又はNleを表す。
【0018】
式(1)中、(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサーであり、nは0又は1である。なお、Xが(−Nle−Tyr−)であり、nが1であることが好ましい。
【0019】
式(1)中、Tはスペーサーであり、mは0又は1を表す。
【0020】
本発明の炎症部位集積性化合物の好ましい具体例として、Tが下記式(2)で表され、mが1であるものをあげることができる。
−HN−(C2H4O)pCH2COO− (2)
(式(2)おいて、pは1〜6の整数を表す。)
【0021】
式(1)中、HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。HalBのハロゲンとしては、SPECT用として121I、123I、125I及び131Iがあげられ、PET用としては124I及び18Fをあげることができる。なお、PET用としては、汎用性の観点から、18F(この場合のHalBを「[18F]FB」と表す。)が好ましい。
【0022】
式(1)中、Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【0023】
本発明の炎症部位集積性化合物の、より具体的で好ましい例として、下記のものをあげることができる。
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−NH2;(配列番号1)
ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([18F]FB)−NH2;(配列番号2)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−NH2;(配列番号3)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;(配列番号4)
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[18F]FB)−NH2;(配列番号5)
【0024】
本発明の炎症部位集積性化合物は、たとえば以下に示すステップ−1〜ステップ−3を用いる方法により合成することができる。
【0025】
[ステップ−1]標識前駆体の合成
標識前駆体であるペプチドは、「固相法」又は「液相法」として知られるペプチド合成法により、調製することができる。例えば、社団法人日本生化学会編集『生化学実験講座』、第1巻、「タンパク質IV」、第207〜495頁、1977年、東京化学同人発行及び社団法人日本生化学会編集『新生化学実験講座』、第1巻、「タンパク質VI」、第3〜74頁、1992年、東京化学同人発行などにはペプチド合成の詳細が記載されている。また、Fmoc(9−フルオレニルメトオキシカルボニル)固相合成法を用いてペプチド合成機にて合成することができる。すなわち、合成する各ペプチドのC末端に相当するアミノ酸が導入されているFmocアミノ酸を樹脂に結合させ、(I)Fmoc基の脱保護と洗浄、(II)Fmocアミノ酸の縮合と洗浄、の操作を繰り返してペプチド鎖を延長し、最後に最終脱保護反応させて、目的とするペプチドを合成することができる。
【0026】
ペプチドを単離精製するためには、公知の分離操作を組み合わせて行うことができる。例えば、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、逆相クロマトグラフィー、高速液体クロマトグラフィーなどのペプチド又は蛋白質を精製するための方法が用いられ、必要に応じて、これら方法を適宜組合せてもよい。そして、最終使用形態に応じて、精製したペプチドを濃縮、また必要に応じてさらに凍結乾燥して単離すればよい。
【0027】
[ステップ−2]放射性ハロゲン含有モノマー[18F]SFBの合成
下記に記載した手順に従い、放射性ハロゲン含有モノマー[18F]SFBを得ることができる(図1)。
【0028】
1.相間移動触媒を遮光バイアル中で脱水アセトニトリルに溶かし、18F−のK2CO3水溶液を必要な放射能分を加えて攪拌する。相間移動触媒としては、18Fイオンとの間で包摂体を形成する性質を有する種々の化合物を用いることができる。具体的には、放射性フッ素標識有機化合物の製造に用いられている種々の化合物を用いることができ、18−クラウン−6−エーテル及びその他の種々のアミノポリエーテルを用いることができる。最も好ましい態様としては、クリプトフィックス2.2.2.(商品名、メルク社製)を用いることができる。
2.窒素ガスを吹き付けながら加熱して溶媒を飛ばす。更に脱水アセトニトリルを加えて溶媒を飛ばし、完全に水を飛ばす。
3.t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateを脱水アセトニトリルに溶かし、反応バイアルに加えて強く攪拌し、反応させる。なお、t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateと脱水アセトニトリルの量比については、t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateが完全に溶解する限りにおいて限定されないが、好ましくはt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate 0.3〜0.5mgの場合、脱水アセトニトリルは100〜200μLとすることができる。
4.反応後、tetrapropylammonium hydroxideを加えて攪拌し、反応させる。
5.反応後、TSTUを(脱水)アセトニトリルに溶かして反応バイアルに加えて攪拌し、反応させる。
6.反応液を5%酢酸水溶液で希釈し、アセトニトリルと水で活性化したSep−Pak(登録商標、日本ウォーターズ株式会社製)plus PS−2 に通し、水/アセトニトリルでカラムを洗浄し、アセトニトリルで放射性ハロゲン含有モノマーである[18F]SFBを溶出する。
【0029】
なお、上記の方法による出発原料であるt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflateの製造方法は公知であり(たとえば、Applied Radiation and Isotopes 59(2003) 43−48)次の手順に従って合成することができる。
4−N,N−dimethylamino benzoic acid (1)を含む冷却された乾燥THF中にtrifluoroacetic anhydrideを加える。しばらく後(たとえば30分後)にtert−BuOHを加え、室温に保つ(たとえば2時間)。その後、飽和NaHCO3水溶液に注ぎ、CH2Cl2で抽出する。抽出物をショートシリカゲルカラムに通し、減圧下に溶媒を除去することにより、tert−butyl ester (2)が得られる。
(2)をnitromethaneに溶解させ、冷却する。methyl triflateを加え、攪拌する(たとえば1時間)。反応物をdiethyl etherに注ぎ、真空乾燥することによりt−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate (3)が得られる。
【0030】
[ステップ−3]ペプチドの標識
標識前駆体であるペプチドをアセトニトリル(以下、MeCNと表記する)とBorate Bufferの混合液に溶かし、70℃ Ar気流下で濃縮した[18F]SFBに加える。MeCN/トリエチルアミン(以下、Et3Nと表記する)=98/2でpHを8.5〜9.0にし、反応させる。HPLCを用いて分取、精製し、純度確認を行う。
【0031】
本発明の炎症部位集積性化合物は、炎症部位への高い集積性を有し、糖尿病足病変に伴う炎症部位の診断をはじめ、核医学画像診断剤の主成分として最適に用いることができる。なお、炎症を伴う疾患としては、糖尿病足病変、炎症性腸疾患等を例示することができる。
【0032】
本発明の核医学画像診断剤は、本発明に係る放射性ヨウ素標識ペプチドを溶解した液として調製することができる。放射性ヨウ素標識ペプチドを溶解する液は、水、生理食塩水やリンゲル液等を用いることができる。放射性ヨウ素標識ペプチドの水溶性が低い場合には、必要に応じて可溶化剤を添加するか、当該ペプチドを溶解させることができる液に溶解後、生体認容性ある液と混合する。たとえば、放射性ヨウ素標識ペプチドとして、配列番号1〜5に記載のペプチドを用いた場合には、当該ペプチドをDMSOに溶解し、10%DMSOになるよう、binding bufferあるいは生理食塩液を加えていき、水溶液を作製するといった方法を用いればよい。また、必要に応じて、安定化剤を配合してもよい。
【0033】
本発明に係る核医学画像診断剤の投与量は、投与された薬剤の分布を画像化するために十分な濃度であれば特に限定する必要はない。たとえば、18F標識ペプチドの場合は、体重60kgの成人一人当り50〜600MBq程度、静脈投与又は局所投与して使用することができる。投与された薬剤の分布は、PET装置やSPECT装置を用いて公知の方法により画像化することができる。
【実施例】
【0034】
次に、本発明を実施例により説明する。
実施例に用いた化合物は次のとおりである。なお、本実施例においては、炎症部位への集積性を評価するという目的に鑑み、非標識化合物を用いるコールドテストとした。
【0035】
本発明による化合物の非標識化合物
化合物1:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−NH2;
化合物2:ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([19F]FB)−NH2;
化合物3:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−NH2;
化合物4:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;
化合物5:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[19F]FB)−NH2;
【0036】
以下の方法により、上記化合物1〜5を得た。
ここで、アミノ酸の表示については、アラニン(Ala、A)、システイン(Cys、C)、アスパラギン酸(Asp、D)、グルタミン酸(Glu、E)、フェニルアラニン(Phe、F)、グリシン(Gly、G)、ヒスチジン(His、H)、イソロイシン(Ile、I)、リジン(Lys、K)、ロイシン(Leu、L)、メチオニン(Met、M)、アスパラギン(Asn、N)、プロリン(Pro、P)、グルタミン(Gln、Q)、アルギニン(Arg、R)、セリン(Ser、S)、スレオニン(Thr、T)、バリン(Val、V)、トリプトファン(Trp、W)、ノルロイシン(Nle)と1文字あるいは3文字表示とする。
また、ペプチドは、ペプチド自動合成機(433A型:Applied Biosystems社製)を用い、樹脂に固定したアミノ酸誘導体に、1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。
【0037】
なお、化合物1〜5(非標識化合物)の合成においては、公知の方法に準じてリジンの側鎖に非放射性FBを導入したアミノ酸を予め調製し、自動合成機を用いた合成の原料として用いた。
リジンの側鎖に非放射性FBを導入したアミノ酸は次のとおり調製した。すなわち、Fmoc−Lys(5g,13.6mmol)を水:THF(1:9,30ml)に溶解し、DIEA(4.7ml,27.1mmol、diisopropylethylamine)を加えた後、氷冷下に攪拌しながらFB−Cl(1.53ml,12.9mmol,4−F−benzoic acid chloride)を加えた。その後反応液を一昼夜攪拌の後、0.5mol/L塩酸水溶液(300ml)に反応液を加えた後、酢酸エチル300mlで目的とする表題誘導体を抽出した。得られた酢酸エチル層を硫酸ナトリウムで乾燥し、減圧濃縮により白色固体粉末を得た。このものを該当するペプチド合成のアミノ酸誘導体原料として使用した。
【0038】
実施例1 化合物1(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin (0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H20−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
HPLC分析条件:
カラム YMC A−302(ODS,150×4.6mm I.D.)
カラム温度 40℃
溶離液 MeCN/Water/0.1%TFA(25min linear)
流速 1.0mL/min
検出器 220nm
注入量 1μL
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 20.3min,純度 92.3%
m/z 945.5([M+H]+ 946.1),m/z 473.4([M+2H]2+ 473.6) 分子量 945.1
【0039】
実施例2 ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−[18F]FBの合成
[ステップ−1]標識前駆体の合成
標識前駆体:ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。
使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(Boc) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SALResin保護ペプチド樹脂を得た。その後ホルミル−MetをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
【0040】
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別後、TFAを留去し、残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
【0041】
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
HPLC分析条件:
カラム YMC A−302(ODS,150×4.6mm I.D.)
カラム温度 40℃
溶離液 MeCN/Water/0.1%TFA(25min linear)
流速 1.0mL/min
検出器 220nm
注入量 1μL
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 14.5min,純度 93.9%
m/z 823.5([M+H]+ 824.0),m/z 412.4([M+2H]2+ 412.5) 分子量 823.0
【0042】
[ステップ−2]放射性ハロゲン含有モノマーの合成
1.Kryptofix(商品名、メルク社製)2.2.2.(商品名、メルク社製)(10mg)を遮光バイアル中で脱水アセトニトリル(500 μL)に溶かし、18F−のK2CO3水溶液(100〜500 μL)を加えて攪拌した。
2.窒素ガスを吹き付けながら110℃の油浴で加熱して溶媒を飛ばした(目安:10 min)。更に脱水アセトニトリル(400 μL×3, 目安:各3 min)を加えて溶媒を飛ばし、完全に水を飛ばした。
3.t−butyl 4−N,N,N−trimethyl−ammoniumbenzoate triflate(0.5mg)を脱水アセトニトリル(1 mL)に溶かし、反応バイアルに加えて強く攪拌し、90℃で10 min反応させた。
4.反応後、tetrapropylammonium hydroxide(1 mol/L in H2O,20 μL)を加えて攪拌し、120℃で5 min反応させた。
5.反応後、TSTU(15 mg)を(脱水)アセトニトリル(100 μL)に溶かして反応バイアルに加えて攪拌し、90℃で2 min反応させた。
6.反応液を5%酢酸水溶液(10 mL)で希釈し、アセトニトリルと水(各5 mL)で活性化したSep−Pak(登録商標、日本ウォーターズ株式会社製) plus PS−2に通し、水/アセトニトリル(80/20, 20 mL)でカラムを洗浄し、アセトニトリル(2.5mL)で[18F]SFBを溶出した。
【0043】
[ステップ−3]ペプチドの放射性フッ素標識
標識前駆体であるペプチド0.3mgをMeCN 40μL, Borate Buffer 40μLに溶かし、70℃ Ar気流下で濃縮した[18F]SFBに加えた。MeCN/Et3N=98/2でpHを8.5−9.0にし、1時間30分反応させた。HPLCを用いて分取、純度確認を行った。
HPLC分析条件:
カラム Cosmosil(5C18−ARII,250×10mm I.D.)
カラム温度 40℃
溶離液 MeOH/Water/0.1%TFA
流速 1.0mL/min
検出器 220nm
注入量 25μL
分析結果:MeOH 70−90%,
retention time 22.8min,放射化学的収率 31%、放射化学的純度 99%以上
【0044】
実施例3 化合物2(ホルミル−Met−Leu−Phe−Met−Tyr−Lys(NH2)−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin (0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v)脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 18.8min,純度 96.1%
m/z 963.8([M+H]+ 964.2),m/z 482.5([M+2H]2+ 482.6) 分子量 963.2
【0045】
実施例4 化合物3(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−NH2)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB) そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/200μL DMSO
retention time 18.6min,純度 91.5%
m/z 1290.8([M+H]+ 1291.4),m/z 646.0([M+2H]2+ 646.2) 分子量 1290.4
【0046】
実施例5 化合物4(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([19F]FB)−Asp−Asp−Asp−DArg−Ser−NH2)の合成
(1)保護ペプチド樹脂の合成
Applied Blosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB)そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 20−70%,溶液 1mg/400μL 75%MeCN/H2O
注入量 2μL,retention time 16.7min,純度 97.5%
m/z 767.6([M+2H]2+ 767.8) 分子量1533.7
【0047】
実施例6 化合物5(ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys(NH2)−PEG3−[19F]FB)の合成
(1)保護ペプチド樹脂の合成
Applied Biosystems社製のペプチド自動合成機(433A)を用いて添付のソフトに従って1個ずつアミノ酸をカルボキシル末端側から結合させていく方法(固相合成法)によりペプチドを合成した。保護ペプチド樹脂の合成を行った。
Fmoc−SAL Resin(0.65mol/g、0.32mmol scal)を出発樹脂担体として使用し、通常のFmoc−ペプチド合成法に使われる各Fmoc−アミノ酸誘導体を原料として、配列にしたがって逐次ペプチド鎖の延長を行った。Fmoc−アミノ酸誘導体を上記ペプチド合成機の反応容器にセットし、合成機に添付されているソフトウエアーに従って、活性化剤として、1−[ビスジメチルアミノメチレン]−1H−ベンゾトリアゾリウムー3−オキシドーヘキサフルオロホスフェイト(HBTu),1−ヒドロキシベンゾトリアゾール(HOBt)とジメチルホルムアミド(DMF)に溶解して反応槽に加えて反応させた。得られた樹脂をピペリジン含有N−メチルピロリドン中で緩やかに攪拌してFmoc基を除いて次のアミノ酸誘導体の縮合に進めた。使用したFmocアミノ酸誘導体は側鎖に官能基のあるアミノ酸はそれぞれTyr(OBu),Lys(FB)そしてFmoc−Nleを用いた。配列に従って逐次アミノ酸を延長してH−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−SAL Resin保護ペプチド樹脂を得た。その後ホルミル−NleをDIC−HOOBtで縮合して目的とする配列の保護ペプチド樹脂の構築を行った。
(2)脱保護と樹脂からの切り出し
得られた保護ペプチド樹脂をトリフルオロ酢酸を用いる定法のTFA−TIS−H2O−(95/2.5/2.5,v/v) 脱保護条件で室温、2時間処理し脱保護と樹脂からのペプチドの切り離しを同時に行った。反応液から担体樹脂をろ別の後、TFAを留去。残渣にエーテルを加えて得られる粗生成物の沈殿をろ取した。
(3)ペプチドの単離精製
得られた粗生成ペプチドを島津製LC−8A−1のHPLC分取装置(カラム:ODS30×250mm)を用いて0.1%トリフルオロ酢酸を含む水−アセトニトリルの系で分取精製し、目的のペプチドの分画を得、アセトニトリルを留去した後、凍結乾燥粉末とし、目的物をトリフルオロ酢酸塩として得た。
得られたペプチドが目的のものであることを確認するために、EMI−MS及びHPLCの分析を行った。
分析結果:
溶離液MeCN 30−80%,溶液 1mg/200μL DMSO
retention time 14.3min, 純度 98.0%
m/z 1134.9([M+H]+ 1135.3),m/z 568.3([M+2H]2+ 568.2) 分子量 1134.3
【0048】
実施例7 Binding Assay/Inhibition Assay:ホルミル化ペプチド受容体(FPR)に対する結合親和性の評価
得られた化合物1〜5について、次の方法によりFPRに対する結合親和性を評価した。
binding buffer(170μL)中に様々な濃度のペプチド(DMSO溶液,10μL)と、2nmol/Lの[125I]WKYMVm(10μL)と、FPR(10μL)を加え、25℃で1時間インキュベート後、ポリリジンbufferによりコーティングしたGF/Cフィルターを用いて濾取(セルハーベスタ)し、wash後、フィルター上に残った放射能をγカウンターで測定した。
※binding buffer:50mmol/L Hepes,pH 7.4,5mmol/L MgCl2,1mmol/L CaCl2,0.2% BSA
wash buffer:50mmol/L Hepes,pH 7.4,500mmol/L NaCl,0.1% BSA
ポリリジンbuffer:ポリL−リジン臭化水素酸塩100mg/wash buffer 100mL
評価結果を表1に示した。
【0049】
【表1】
【0050】
表1の結果から、本発明による化合物はFMLPに比べ、低いKiを示し、よって高いFPRへの親和性、すなわち高い炎症集積性を有することがわかる。
【産業上の利用可能性】
【0051】
本発明は、炎症部位集積性化合物及び核医学画像診断剤に関するものである。更に詳しくは、本発明は、放射性ハロゲンを含有し、かつ糖尿病足病変をはじめとする種々の病変に伴って発生する生体中の炎症部位への選択的集積性に優れる炎症部位集積性化合物、該化合物を主成分とする核医学画像診断剤及び該化合物の標識前駆体に利用することができる。
【特許請求の範囲】
【請求項1】
下記式(1)で表される炎症部位集積性化合物。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表し;
HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
【請求項2】
Zがホルミル基である請求項1記載の炎症部位集積性化合物。
【請求項3】
Xが(−Nle−Tyr−)であり、nが1である請求項1記載の炎症部位集積性化合物。
【請求項4】
Uが、(−Asp−Asp−Asp−)又は(−Asp−Asp−Asp−DArg−Ser)である請求項1記載の炎症部位集積性化合物。
【請求項5】
mが0である請求項1記載の炎症部位集積性化合物。
【請求項6】
Tが下記式(2)で表され、mが1である請求項1記載の炎症部位集積性化合物。
−HN−(C2H4O)pCH2COO− (2)
(式(2)おいて、pは1〜6の整数を表す。)
【請求項7】
pが3である(この場合の式(2)を「PEG3」と表す。)請求項6記載の炎症部位集積性化合物。
【請求項8】
HalBのハロゲンが、121I、123I、125I、131I、124I又は18Fである請求項1記載の炎症部位集積性化合物。
【請求項9】
HalBのハロゲンが18Fである(この場合のHalBを「[18F]FB」と表す。)請求項1記載の炎症部位集積性化合物。
【請求項10】
式(1)で表される炎症部位集積性化合物が、
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−NH2;
ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([18F]FB)−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[18F]FB)−NH2;
よりなる群から選ばれる一である請求項1記載の炎症部位集積性化合物。
【請求項11】
請求項1〜10のいずれかに記載の炎症部位集積性化合物を主成分とする核医学画像診断剤。
【請求項12】
糖尿病足病変に伴う炎症部位を診断する請求項11記載の核医学画像診断剤。
【請求項13】
請求項1記載の化合物の標識前駆体であって、下記式(3)で表される標識前駆体。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m)−(U)k(NH2)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【請求項1】
下記式(1)で表される炎症部位集積性化合物。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m−HalB)−(U)k(NH2) (1)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表し;
HalBはベンゼン核に放射性ハロゲンを有する置換安息香酸の残基を表す。
【請求項2】
Zがホルミル基である請求項1記載の炎症部位集積性化合物。
【請求項3】
Xが(−Nle−Tyr−)であり、nが1である請求項1記載の炎症部位集積性化合物。
【請求項4】
Uが、(−Asp−Asp−Asp−)又は(−Asp−Asp−Asp−DArg−Ser)である請求項1記載の炎症部位集積性化合物。
【請求項5】
mが0である請求項1記載の炎症部位集積性化合物。
【請求項6】
Tが下記式(2)で表され、mが1である請求項1記載の炎症部位集積性化合物。
−HN−(C2H4O)pCH2COO− (2)
(式(2)おいて、pは1〜6の整数を表す。)
【請求項7】
pが3である(この場合の式(2)を「PEG3」と表す。)請求項6記載の炎症部位集積性化合物。
【請求項8】
HalBのハロゲンが、121I、123I、125I、131I、124I又は18Fである請求項1記載の炎症部位集積性化合物。
【請求項9】
HalBのハロゲンが18Fである(この場合のHalBを「[18F]FB」と表す。)請求項1記載の炎症部位集積性化合物。
【請求項10】
式(1)で表される炎症部位集積性化合物が、
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−NH2;
ホルミル−Met−Leu−Phe−Met−Tyr−Lys−ε([18F]FB)−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε([18F]FB)−Asp−Asp−Asp−DArg−Ser−NH2;
ホルミル−Nle−Leu−Phe−Nle−Tyr−Lys−ε(−PEG3−[18F]FB)−NH2;
よりなる群から選ばれる一である請求項1記載の炎症部位集積性化合物。
【請求項11】
請求項1〜10のいずれかに記載の炎症部位集積性化合物を主成分とする核医学画像診断剤。
【請求項12】
糖尿病足病変に伴う炎症部位を診断する請求項11記載の核医学画像診断剤。
【請求項13】
請求項1記載の化合物の標識前駆体であって、下記式(3)で表される標識前駆体。
Z−Y−Leu−Phe−(X)n−Lys(−(T)m)−(U)k(NH2)
(式(1)中、Zはアミノ基の保護基を表し;
YはMet又はNleを表し;
(X)nにおいて、Xは1個もしくはそれ以上のアミノ酸及び/又は有機合成可能な化合物よりなるスペーサー、nは0又は1を表し;
Tはスペーサー、mは0又は1を表し;
Uは少なくとも一種類の酸性アミノ酸を含むペプチドよりなるスペーサー、kは0又は1を表す。
【図1】

【図2】
【図2】
【公開番号】特開2011−246427(P2011−246427A)
【公開日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願番号】特願2010−133386(P2010−133386)
【出願日】平成22年5月26日(2010.5.26)
【出願人】(000230250)日本メジフィジックス株式会社 (75)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公開日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願日】平成22年5月26日(2010.5.26)
【出願人】(000230250)日本メジフィジックス株式会社 (75)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]