
炭素担持された白金合金触媒
本発明は、ガス拡散電極のための、及び/又は、触媒がコートされた膜(catalyst−coated membrane)内の、担持白金合金電極触媒に関する。炭素担持された白金合金触媒は、炭素担体上での、インサイチュで形成された(in situ formed)二酸化白金及び少なくとも1つの遷移金属水和酸化物(transition metal hydrous oxide)の同時の化学的還元によって得られる。該遷移金属は、好ましくはニッケル、クロム、コバルト、バナジウム及び鉄より選択される。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
触媒、より詳細には、ガス拡散電極又は触媒被覆膜構造への組み込みに適した炭素担持された白金合金電極触媒。
【背景技術】
【0002】
発明の背景
炭素担持白金(Carbon−supported platinum)は、例えば、燃料電池、電気分解及びセンサー用途において、ガス拡散電極及び触媒被覆膜構造(catalyst−coated membrane structures)への組み込みのためによく知られた触媒である。場合によっては、異なる目的のために白金と他の遷移金属を合金するのが望ましい;例えば他の貴金属(例えばルテニウム)との白金合金の場合は、一酸化炭素耐性アノード触媒及び直接メタノール燃料電池(または他の直接酸化燃料電池)のためのガス拡散アノードの分野ではよく知られている。炭素担持された、非貴の遷移金属(non−noble transition metals)との白金合金は、燃料電池の分野、特にガス拡散カソードのために有用であることも知られている。ニッケル、クロム又はコバルトとの白金合金は、通常酸素還元に対して優れた活性を示す。これらの合金は、直接酸化燃料電池カソードのためにより有用であり得る。なぜなら、それらの高い活性に加えて、それらはまた、アルコール燃料(これらはセパレーターとして使用される半透膜を超えてある程度拡散し、一般的にこれらの電池のカソードコンポーネントを重要なほどに汚染する)によって、より汚染されにくいからである。
【0003】
このタイプの炭素担持白金合金触媒は、例えばJohnson Matthey PLCのUS 5,068,161に開示されており、それには、塩化白金酸及び金属塩を二酸化炭素及び炭素担体の存在下で煮沸することによる、例えばニッケル、クロム、コバルト又はマンガンを含む2種混合又は3種混合の白金合金の調製が記載されている。白金及び関連した共金属(co−metals)の混合酸化物は、最初ホルムアルデヒドを溶液に加えることによって、それから窒素中で930℃の熱処理を行うことで、炭素担体上に沈殿し、その後還元される。ゆえに、白金及び共金属は、次の2つの別個のステップにより還元されると想定され得る:Ptの還元はほとんど水相内で完了するようであり、一方他の酸化物(例えばニッケル又はクロム酸化物)は、次の熱処理中に(おそらく900℃以上で)金属に転化されているようである。
これは、なぜ合金化の程度が、個々の元素の大きなドメインと限定された合金相(limited alloyed phase)の形成を伴って重要な程度まで凝離が起こることを示すXRDスキャンによって証明されたように、相当低いのかを、説明する。
いくつかの適当な白金触媒としての望ましい電気化学的特質が失われるのに加えて、この構造均一性の欠如はまた、不満足な平均粒度及びその分布をもたらす。そのうえ、塩化白金酸の使用は系中に、完全に除去するのが困難で、かつ触媒毒として働き得、その活性を低下させる塩化物イオンを導入する。
【0004】
白金合金触媒を得るための代替方法がChemcat Corp.の米国特許番号 5,876,867に開示されており、この明細書では、炭素担持された白金触媒が2番目の金属の可溶性塩(例えば硝酸コバルト)とともに水溶液中で処理され、乾燥され、合金形成を誘導するために高温で熱せられる。しかしまた、このケースでは、合金の程度は概して不十分である。最初の炭素担持白金触媒(これは再び一般的に塩化白金酸ルートを通じて製造される)上に存在し得る残留塩化物イオンは、毒効果(poisoning effect)に加えて、Ptと2番目の金属間での均質な合金の形成を妨害し得る。
【発明の開示】
【発明が解決しようとする課題】
【0005】
発明の目的
本発明の目的は、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を提供することである。
【0006】
本発明の他の目的は、導電性ウェブに、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を組み込んだ、電気化学的用途の使用のためのガス拡散電極を提供することである。
【0007】
本発明のさらなる目的は、イオン交換膜に、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を組み込んだ、電気化学的用途の使用のための触媒がコートされた膜を提供することである。
【0008】
本発明の目的はまた、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒の形成のための方法を提供することである。
【0009】
本発明の、これら及び他の目的並びに利益は、次の詳細な記載から明らかとなるであろう。
【課題を解決するための手段】
【0010】
本発明
第1の局面において、本発明は、炭素担体上の二酸化白金及び少なくとも1つの遷移金属水和酸化物MOX−YH2O(ここでMは任意の遷移金属であり、より有利にはニッケル、コバルト、クロム、バナジウム及び鉄の中から選択される)の同時の化学的還元により得られる炭素担持白金合金触媒からなる。好ましい実施形態において、二酸化白金は、白金酸としてもまた知られたヘキサヒドロキソ白金(IV)酸(dihydrogen hexahydroxyplatinate:H2Pt(OH)6)から沈殿し、遷移金属水和酸化物は可溶遷移金属塩、好ましくは硝酸塩の転化により得られる。1を超える遷移金属水和酸化物は、例えば炭素担持された3成分系又は4成分系合金の形成のために、二酸化白金と同時に還元され得る。
【0011】
インサイチュで形成されたPtO2コロイドからの炭素担持白金触媒の有利な形成は、同様係属中の特許出願シリアル番号60/561 ,207, 9月4日, 2004に出願、に記載されており、ここにその全体が参照として組み込まれる。PtO2コロイド形成についての熱動力学的コントロールは、非常に多くの粒子を同時に沈殿でき、それらがあるサイズを超えて成長する前に炭素担体上へ素早く吸収させる。本発明のケースでは、単一の混合溶液中で分離することなく、PtO2及び水和した遷移金属酸化物MOX−YH2Oが形成される。引用される同時係属中の出願の教示に従ったPtO2の形成後、金属塩溶液、好ましくは金属硝酸塩溶液を加える。そして、水和金属酸化物の形成を誘導するために化学薬剤を加え、該酸化物をPtO2で含浸した炭素担体上へ吸収させる。共に吸収させたPtO2と水和金属酸化物MOX−YH2Oは次いで濾過により回収し、乾燥させ、水素中高温(好ましくは300℃超)で共に還元する。次の高温処理(好ましくは600℃超)は、単にアニーリングおよび合金形成を完了するために実行され、一方任意の炭素質の粒子が炭素担体として使用され得、高表面積(少なくとも50m2/g)のカーボンブラックがやはり好ましい。
【0012】
このようにして形成されたPt合金は原子スケールにおいて均一であり、よくコントロールされた粒度をもたらし、最小限の外来イオンの汚染しかない。この触媒は、電気化学的プロセスの広い領域、例えば直接酸化燃料電池を含む燃料電池のためのガス拡散カソード及びアノードに使用され得る。
【0013】
第2の局面において、本発明は、上に開示した触媒を導電性ウェブ(例えば炭素織布又は不織布あるいは炭素紙)に組み込むことによって得られる、ガス拡散電極からなる。他の局面において、本発明は、上に開示した触媒をイオン交換膜へ組み込むことによって得られる、触媒がコートされた膜からなる。
【0014】
さらに他の局面において、本発明は、同時に還元するインサイチュで形成された二酸化白金及び少なくとも1つの遷移金属水和酸化物を炭素担体上に含んだ炭素担持白金合金触媒の製造のための方法からなる。好ましい実施形態において、二酸化白金のインサイチュでの形成は、必要に応じて炭素担体上にプレ吸着(pre−adsorbed)した、ヘキサヒドロキソ白金(IV)酸前駆体の転化によってなされる。こういった転化は、好ましくはpH及び/又は温度の変動により、必要に応じて、例えばpH2から9の間へ到達するまで、苛性ソーダといったアルカリ又はアンモニアの酸性開始溶液への添加の制御により、及び/又は、温度を室温から30℃から100℃の間を含んだ最終温度、好ましくは70℃へ上げることにより、実行される。
【0015】
高アクティブエリアカーボンブラックは、炭素担体として好ましく用いられ、好ましい実施形態においては、前駆体の吸着より前に、カーボンブラック担体は濃縮硝酸中でスラリーにされ、得られたスラリーは白金酸を容易に溶解するために使用され得る。他の好ましい非錯化強酸(例えばHClO4、H2SO4、CF3COOH、トルエンスルホン酸又はトリフルオロメタン−スルホン酸)が硝酸の代わりに使用され得る。PtO2のインサイチュで形成がなされた後、少なくとも1つの遷移金属酸化物、好ましくは可溶性塩、さらに好ましくは硝酸塩の適した前駆体が溶液に加えられる。それから前駆体は、例えばさらなるアルカリの添加によって、遷移金属水和酸化物へ転化される。濾過及び乾燥後、共に吸収させたPtO2及び水和金属酸化物は、好ましくは高温(300℃以上)において水素により、対応する金属へ還元される。最終段階においては、合金形成を完了するため、600℃あるいはそれより高い温度における高温アニーリング工程が実行される。
【発明を実施するための最良の形態】
【0016】
次の実施例において、本発明を例証するためいくつかの好ましい実施態様が述べられるが、本発明がこの特定の実施態様に限定されることを意味するものではないと理解されるべきである。
実施例1
Vulcan XC−72カーボンブラック上の30重量%Pt−Ni触媒(Pt:Ni 1:1、原子ベース)100gを、つぎの手順に従って調製した:
70gのCabot Corp./アメリカ合衆国 社製のVulcan XC−72を4リットルビーカー中で2.5リットルのイオン化水(ionized water)により懸濁した。5分間の超音波処理により炭素を微細に分散させ、そしてスラリーをマグネチックスターラーにより撹拌し、そして87mlの濃HNO3(〜69%)をそこへ加えた。
【0017】
36.03gの白金酸、PTA(23.06gのPtに相当)を分離フラスコ内の413mlの4.0M HNO3へ加えた。該溶液をPTAが完全に溶解するまで撹拌し、赤みがかった色に着色させた。このPTA溶液をカーボンスラリーへ移し、そして周囲温度で30分撹拌した。そしてビーカーを1℃/分の割合で70℃まで加熱し、撹拌しながらこの温度を1時間維持した。加熱を止め、15.0M NaOH溶液を10ml/分の割合で、pHが3から3.5の間に達するまでスラリーへ加えた(およそ200mlのNaOH溶液を加えた)。該溶液をさらに撹拌しながら、室温まで冷却した。
【0018】
34.37gのNi(NO3)2・6H2O(20.19%Ni、トータルで6.94gのNi)を150mlの脱イオン水に溶解し、そしてスラリーへ加えた。30分後、加熱を再開し、温度を1℃/分の割合で75℃に上昇させた。該溶液は全プロセスの間撹拌し、さらにNaOHを加えてpHを〜8.5に制御した。75℃に達した後、加熱及び撹拌の両方を1時間維持した。スラリーは室温まで冷却し、濾過した。触媒ケーキ(catalyst cake)を1.5リットルの脱イオン水で洗い、300mlアリコートへと細分し、そして水分含有量2%に達するまで125℃で乾燥した。乾燥したケーキは10メッシュの顆粒へと砕き、得られた触媒を30分間500℃で水素蒸気中で還元し、そしてアルゴン中で850℃で1時間焼結し、ボールミルで処理して微細粉末とした。
実施例2
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 2:1触媒を得るため、改変した。この目的のために、PTAの量を40.75g(トータル26.08gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は19.43g(20.19%Ni、トータルで3.92gのNi)に減らした。
実施例3
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 3:1触媒を得るため、改変した。この目的のために、PTAの量を42.60g(トータル27.27gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は13.54g(20.19%Ni、トータルで2.73gのNi)に減らした。
実施例4
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 4:1触媒を得るため、改変した。この目的のために、PTAの量を43.60g(トータル27.90gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は10.39g(20.19%Ni、トータルで2.10gのNi)に減らした。
実施例5
実施例3の手順を、Vulcan XC−72上の30重量%のPt:Co 3:1触媒を得るため、改変した。この目的のために、硝酸ニッケルをモル当量の硝酸コバルトへと置き換えた。
実施例6
Vulcan XC−72カーボンブラック上の30重量%Pt−Cr触媒(Pt:Cr 3:1)100gを、つぎの手順に従って調製した:
4リットルビーカー中で2.5リットルの脱イオン化水(deionized water)に70gのCabot Corp./アメリカ合衆国 社製のVulcan XC−72を懸濁し、そして15分間の超音波処理により該カーボンを微細に分散させた。さらにスラリーをマグネチックスターラーにより撹拌し、そして87mlの濃HNO3(〜69%)をそこへ加えた。
【0019】
43.05gの白金酸、PTA(27.55gのPtに相当)を分離フラスコ内の413mlの4.0M HNO3へ加えた。該溶液をPTAが完全に溶解するまで撹拌し、赤みがかった色に着色させた。このPTA溶液を次いでカーボンスラリーへ移し、そして周囲温度で30分撹拌した。そしてビーカーを1℃/分の割合で70℃まで加熱し、撹拌しながらこの温度を1時間維持した。次いで加熱を止め、濃アンモニア(〜30%)を10ml/分の割合で、pHが3から3.5の間に達するまでスラリーへ加えた(およそ200mlのアンモニアを加えた)。該溶液をさらに撹拌しながら、室温まで冷却した。
【0020】
18.88gのCr(NO3)・9H2O(12.98%Cr、トータルで2.45gのCr)を150mlの脱イオン水に溶解し、そしてスラリーへ加えた。30分後、0.5M NH4OHによりスラリーのpHを〜4.5に調整し、さらにその30分後加熱を再開し、温度を75℃まで1℃/分の割合で上昇させた。該溶液は全プロセスの間撹拌し、さらにアンモニアを加えてpHを〜5.5に制御した。75℃に達した後、加熱及び撹拌の両方とも1時間維持し、それからスラリーを室温まで冷却し、濾過した。触媒ケーキを1.5リットルの脱イオン水で洗い、300mlアリコートへと細分し、そして水分含有量2%に達するまで125℃で乾燥した。乾燥ケーキは10メッシュの顆粒へと砕き、得られた触媒を30分間500℃で水素蒸気中で還元し、そしてアルゴン中で850℃で1時間焼結し、ボールミルで処理して微細粉末とした。
実施例7
グラビア/ローラーコーティング機を用いてテキストロン(Textron)カーボンクロス上のインク溶液からShawiniganアセチレンブラック(SAB)/PTFE層(60/40wt)を第1層に適用し、そしてVulcan XC−72/PTFEを第2層に適用することによって、ガス拡散電極を調製した。被覆カーボンクロスは340℃で焼結した。このようにして得られた焼結させたガス拡散層を重量2:1の触媒/アイオノマー懸濁インクに対し支持体として使用し、一方で、触媒は実施例6のPtCr/Cであり、フルオロカーボンポリマーアイオノマー懸濁液はアルコール中で9%の市販フルオロカーボン材料から調製した。約0.4―0.5 mg/cm2でロードされたPtが幾つかの被膜中に得られた。所望の白金充填に達した後、100−130℃で最終アニーリングを行った。
比較例1
ガス拡散電極を、使用した触媒が、実施例1の手順(但し硝酸ニッケルの添加及びその後の転化を省略)に従って白金酸を用いて調製した30%Pt/Cであることを除いては、実施例7に記載の手順に従って調製した。
実施例8
膜−電極接合体(MEA)を、実施例7で調製したガス拡散電極をカソードとして、そして標準的な機械製品の30%PT/Cガス拡散電極(公知技術として知られたフルオロカーボンポリマーアイオノマーで含浸され、標準的な手順に従って市販膜の反対側にホットプレスされたもの)を、アノードとして組み込むことにより作製した。他のMEAを同じ手順(但し比較例1のガス拡散電極をカソードとして使用)で作製した。どちらのMEAも実験燃料電池へ導入し、70℃、100%に調湿の反応ガス(空気/純粋(pure)H2)で作動させた。圧力は、カソード側に4バール絶対圧、アノード側3.5バール絶対圧の一定のフローレート(flow−rates)とし、1.2A/cm2の空気2、水素1.5の化学量論の比率に相当する。
【0021】
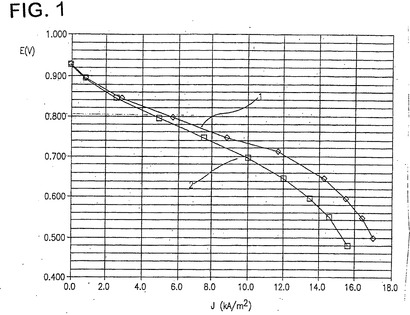
対応する分極曲線を図1に示す。これは、カーボン上の30%Pt:Cr(1)が、カーボン上の標準的な30%Pt(2)よりも、より活性なカソード触媒であることを明確に示している。
実施例9
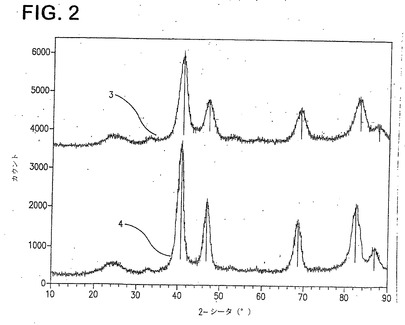
図2は、実施例6の3:1 PtCr触媒(3)及び米国特許番号5876867の教示に従って調製した3:1 PtCr触媒(4)のXRDスペクトルを示す。Pt220ピーク(2θ=68−69)は実施例6のより高い値を示すものであり、合金化の優れた程度を示すものである。その上、2θ=40から48の間に、より顕著な実施例6の触媒の“超格子ピーク”がみられる。これらのピークは、良好なO2還元活性と関連する。実施例6の触媒はまた、公知技術の触媒のXRDサイズ(53Å)と比較して、より小さいXRDサイズ(37Å)を有する。これは、実施例6の触媒が、より良好な性能と関連するより高い表面積を有することを示す。
【0022】
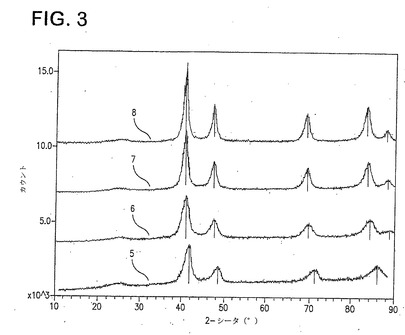
図3は、実施例1(5)、2(6)、3(7)及び4(8)の触媒のXRDスペクトルを示し、ピーク位置のシフトがあるものの、そのパターンはPt/Cのものと同じである。これは、Ni金属単相は検出できないものであり、PtとNi間の合金化の非常に高い程度を示す。ニッケル含有量が、Pt4Ni(8)からPtNi(5)へと増すにつれ、各二次ピークは対応するPtのピークからより遠くなる。より多くのNiがPt格子内へと組み込まれると、d−間隔(d−spacing)はより小さくなる。例えば、Pt{220}ピーク(2θ=72)では、Pt4Ni、Pt3Ni、Pt2Ni及びPtNiのd−間隔はそれぞれ、1.3649、1.3569、1.3498及び1.3270である。30%Pt/Cのd−間隔は1.3877である。
【0023】
上記触媒は、本発明の精神又は範囲から逸脱しない限り、変更され得ること、並びに、本発明は添付されたクレームの範囲にのみ限定されるものではないことを理解することができる。
【図面の簡単な説明】
【0024】
【図1】図1は、本発明の触媒及び公知技術の触媒に関する燃料電池分極曲線の群である。
【図2】図2及び図3は、本発明の触媒および公知技術に関するXRDスペクトルである。
【図3】図2及び図3は、本発明の触媒および公知技術に関するXRDスペクトルである。
【技術分野】
【0001】
発明の分野
触媒、より詳細には、ガス拡散電極又は触媒被覆膜構造への組み込みに適した炭素担持された白金合金電極触媒。
【背景技術】
【0002】
発明の背景
炭素担持白金(Carbon−supported platinum)は、例えば、燃料電池、電気分解及びセンサー用途において、ガス拡散電極及び触媒被覆膜構造(catalyst−coated membrane structures)への組み込みのためによく知られた触媒である。場合によっては、異なる目的のために白金と他の遷移金属を合金するのが望ましい;例えば他の貴金属(例えばルテニウム)との白金合金の場合は、一酸化炭素耐性アノード触媒及び直接メタノール燃料電池(または他の直接酸化燃料電池)のためのガス拡散アノードの分野ではよく知られている。炭素担持された、非貴の遷移金属(non−noble transition metals)との白金合金は、燃料電池の分野、特にガス拡散カソードのために有用であることも知られている。ニッケル、クロム又はコバルトとの白金合金は、通常酸素還元に対して優れた活性を示す。これらの合金は、直接酸化燃料電池カソードのためにより有用であり得る。なぜなら、それらの高い活性に加えて、それらはまた、アルコール燃料(これらはセパレーターとして使用される半透膜を超えてある程度拡散し、一般的にこれらの電池のカソードコンポーネントを重要なほどに汚染する)によって、より汚染されにくいからである。
【0003】
このタイプの炭素担持白金合金触媒は、例えばJohnson Matthey PLCのUS 5,068,161に開示されており、それには、塩化白金酸及び金属塩を二酸化炭素及び炭素担体の存在下で煮沸することによる、例えばニッケル、クロム、コバルト又はマンガンを含む2種混合又は3種混合の白金合金の調製が記載されている。白金及び関連した共金属(co−metals)の混合酸化物は、最初ホルムアルデヒドを溶液に加えることによって、それから窒素中で930℃の熱処理を行うことで、炭素担体上に沈殿し、その後還元される。ゆえに、白金及び共金属は、次の2つの別個のステップにより還元されると想定され得る:Ptの還元はほとんど水相内で完了するようであり、一方他の酸化物(例えばニッケル又はクロム酸化物)は、次の熱処理中に(おそらく900℃以上で)金属に転化されているようである。
これは、なぜ合金化の程度が、個々の元素の大きなドメインと限定された合金相(limited alloyed phase)の形成を伴って重要な程度まで凝離が起こることを示すXRDスキャンによって証明されたように、相当低いのかを、説明する。
いくつかの適当な白金触媒としての望ましい電気化学的特質が失われるのに加えて、この構造均一性の欠如はまた、不満足な平均粒度及びその分布をもたらす。そのうえ、塩化白金酸の使用は系中に、完全に除去するのが困難で、かつ触媒毒として働き得、その活性を低下させる塩化物イオンを導入する。
【0004】
白金合金触媒を得るための代替方法がChemcat Corp.の米国特許番号 5,876,867に開示されており、この明細書では、炭素担持された白金触媒が2番目の金属の可溶性塩(例えば硝酸コバルト)とともに水溶液中で処理され、乾燥され、合金形成を誘導するために高温で熱せられる。しかしまた、このケースでは、合金の程度は概して不十分である。最初の炭素担持白金触媒(これは再び一般的に塩化白金酸ルートを通じて製造される)上に存在し得る残留塩化物イオンは、毒効果(poisoning effect)に加えて、Ptと2番目の金属間での均質な合金の形成を妨害し得る。
【発明の開示】
【発明が解決しようとする課題】
【0005】
発明の目的
本発明の目的は、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を提供することである。
【0006】
本発明の他の目的は、導電性ウェブに、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を組み込んだ、電気化学的用途の使用のためのガス拡散電極を提供することである。
【0007】
本発明のさらなる目的は、イオン交換膜に、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒を組み込んだ、電気化学的用途の使用のための触媒がコートされた膜を提供することである。
【0008】
本発明の目的はまた、高度の合金化及び微少で均質な粒度により特徴付けられる炭素担持白金合金触媒の形成のための方法を提供することである。
【0009】
本発明の、これら及び他の目的並びに利益は、次の詳細な記載から明らかとなるであろう。
【課題を解決するための手段】
【0010】
本発明
第1の局面において、本発明は、炭素担体上の二酸化白金及び少なくとも1つの遷移金属水和酸化物MOX−YH2O(ここでMは任意の遷移金属であり、より有利にはニッケル、コバルト、クロム、バナジウム及び鉄の中から選択される)の同時の化学的還元により得られる炭素担持白金合金触媒からなる。好ましい実施形態において、二酸化白金は、白金酸としてもまた知られたヘキサヒドロキソ白金(IV)酸(dihydrogen hexahydroxyplatinate:H2Pt(OH)6)から沈殿し、遷移金属水和酸化物は可溶遷移金属塩、好ましくは硝酸塩の転化により得られる。1を超える遷移金属水和酸化物は、例えば炭素担持された3成分系又は4成分系合金の形成のために、二酸化白金と同時に還元され得る。
【0011】
インサイチュで形成されたPtO2コロイドからの炭素担持白金触媒の有利な形成は、同様係属中の特許出願シリアル番号60/561 ,207, 9月4日, 2004に出願、に記載されており、ここにその全体が参照として組み込まれる。PtO2コロイド形成についての熱動力学的コントロールは、非常に多くの粒子を同時に沈殿でき、それらがあるサイズを超えて成長する前に炭素担体上へ素早く吸収させる。本発明のケースでは、単一の混合溶液中で分離することなく、PtO2及び水和した遷移金属酸化物MOX−YH2Oが形成される。引用される同時係属中の出願の教示に従ったPtO2の形成後、金属塩溶液、好ましくは金属硝酸塩溶液を加える。そして、水和金属酸化物の形成を誘導するために化学薬剤を加え、該酸化物をPtO2で含浸した炭素担体上へ吸収させる。共に吸収させたPtO2と水和金属酸化物MOX−YH2Oは次いで濾過により回収し、乾燥させ、水素中高温(好ましくは300℃超)で共に還元する。次の高温処理(好ましくは600℃超)は、単にアニーリングおよび合金形成を完了するために実行され、一方任意の炭素質の粒子が炭素担体として使用され得、高表面積(少なくとも50m2/g)のカーボンブラックがやはり好ましい。
【0012】
このようにして形成されたPt合金は原子スケールにおいて均一であり、よくコントロールされた粒度をもたらし、最小限の外来イオンの汚染しかない。この触媒は、電気化学的プロセスの広い領域、例えば直接酸化燃料電池を含む燃料電池のためのガス拡散カソード及びアノードに使用され得る。
【0013】
第2の局面において、本発明は、上に開示した触媒を導電性ウェブ(例えば炭素織布又は不織布あるいは炭素紙)に組み込むことによって得られる、ガス拡散電極からなる。他の局面において、本発明は、上に開示した触媒をイオン交換膜へ組み込むことによって得られる、触媒がコートされた膜からなる。
【0014】
さらに他の局面において、本発明は、同時に還元するインサイチュで形成された二酸化白金及び少なくとも1つの遷移金属水和酸化物を炭素担体上に含んだ炭素担持白金合金触媒の製造のための方法からなる。好ましい実施形態において、二酸化白金のインサイチュでの形成は、必要に応じて炭素担体上にプレ吸着(pre−adsorbed)した、ヘキサヒドロキソ白金(IV)酸前駆体の転化によってなされる。こういった転化は、好ましくはpH及び/又は温度の変動により、必要に応じて、例えばpH2から9の間へ到達するまで、苛性ソーダといったアルカリ又はアンモニアの酸性開始溶液への添加の制御により、及び/又は、温度を室温から30℃から100℃の間を含んだ最終温度、好ましくは70℃へ上げることにより、実行される。
【0015】
高アクティブエリアカーボンブラックは、炭素担体として好ましく用いられ、好ましい実施形態においては、前駆体の吸着より前に、カーボンブラック担体は濃縮硝酸中でスラリーにされ、得られたスラリーは白金酸を容易に溶解するために使用され得る。他の好ましい非錯化強酸(例えばHClO4、H2SO4、CF3COOH、トルエンスルホン酸又はトリフルオロメタン−スルホン酸)が硝酸の代わりに使用され得る。PtO2のインサイチュで形成がなされた後、少なくとも1つの遷移金属酸化物、好ましくは可溶性塩、さらに好ましくは硝酸塩の適した前駆体が溶液に加えられる。それから前駆体は、例えばさらなるアルカリの添加によって、遷移金属水和酸化物へ転化される。濾過及び乾燥後、共に吸収させたPtO2及び水和金属酸化物は、好ましくは高温(300℃以上)において水素により、対応する金属へ還元される。最終段階においては、合金形成を完了するため、600℃あるいはそれより高い温度における高温アニーリング工程が実行される。
【発明を実施するための最良の形態】
【0016】
次の実施例において、本発明を例証するためいくつかの好ましい実施態様が述べられるが、本発明がこの特定の実施態様に限定されることを意味するものではないと理解されるべきである。
実施例1
Vulcan XC−72カーボンブラック上の30重量%Pt−Ni触媒(Pt:Ni 1:1、原子ベース)100gを、つぎの手順に従って調製した:
70gのCabot Corp./アメリカ合衆国 社製のVulcan XC−72を4リットルビーカー中で2.5リットルのイオン化水(ionized water)により懸濁した。5分間の超音波処理により炭素を微細に分散させ、そしてスラリーをマグネチックスターラーにより撹拌し、そして87mlの濃HNO3(〜69%)をそこへ加えた。
【0017】
36.03gの白金酸、PTA(23.06gのPtに相当)を分離フラスコ内の413mlの4.0M HNO3へ加えた。該溶液をPTAが完全に溶解するまで撹拌し、赤みがかった色に着色させた。このPTA溶液をカーボンスラリーへ移し、そして周囲温度で30分撹拌した。そしてビーカーを1℃/分の割合で70℃まで加熱し、撹拌しながらこの温度を1時間維持した。加熱を止め、15.0M NaOH溶液を10ml/分の割合で、pHが3から3.5の間に達するまでスラリーへ加えた(およそ200mlのNaOH溶液を加えた)。該溶液をさらに撹拌しながら、室温まで冷却した。
【0018】
34.37gのNi(NO3)2・6H2O(20.19%Ni、トータルで6.94gのNi)を150mlの脱イオン水に溶解し、そしてスラリーへ加えた。30分後、加熱を再開し、温度を1℃/分の割合で75℃に上昇させた。該溶液は全プロセスの間撹拌し、さらにNaOHを加えてpHを〜8.5に制御した。75℃に達した後、加熱及び撹拌の両方を1時間維持した。スラリーは室温まで冷却し、濾過した。触媒ケーキ(catalyst cake)を1.5リットルの脱イオン水で洗い、300mlアリコートへと細分し、そして水分含有量2%に達するまで125℃で乾燥した。乾燥したケーキは10メッシュの顆粒へと砕き、得られた触媒を30分間500℃で水素蒸気中で還元し、そしてアルゴン中で850℃で1時間焼結し、ボールミルで処理して微細粉末とした。
実施例2
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 2:1触媒を得るため、改変した。この目的のために、PTAの量を40.75g(トータル26.08gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は19.43g(20.19%Ni、トータルで3.92gのNi)に減らした。
実施例3
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 3:1触媒を得るため、改変した。この目的のために、PTAの量を42.60g(トータル27.27gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は13.54g(20.19%Ni、トータルで2.73gのNi)に減らした。
実施例4
実施例1の手順を、Vulcan XC−72上の30重量%のPt:Ni 4:1触媒を得るため、改変した。この目的のために、PTAの量を43.60g(トータル27.90gのPt)に増やし、一方スラリーに加えるNi(NO3)2・6H2Oの量は10.39g(20.19%Ni、トータルで2.10gのNi)に減らした。
実施例5
実施例3の手順を、Vulcan XC−72上の30重量%のPt:Co 3:1触媒を得るため、改変した。この目的のために、硝酸ニッケルをモル当量の硝酸コバルトへと置き換えた。
実施例6
Vulcan XC−72カーボンブラック上の30重量%Pt−Cr触媒(Pt:Cr 3:1)100gを、つぎの手順に従って調製した:
4リットルビーカー中で2.5リットルの脱イオン化水(deionized water)に70gのCabot Corp./アメリカ合衆国 社製のVulcan XC−72を懸濁し、そして15分間の超音波処理により該カーボンを微細に分散させた。さらにスラリーをマグネチックスターラーにより撹拌し、そして87mlの濃HNO3(〜69%)をそこへ加えた。
【0019】
43.05gの白金酸、PTA(27.55gのPtに相当)を分離フラスコ内の413mlの4.0M HNO3へ加えた。該溶液をPTAが完全に溶解するまで撹拌し、赤みがかった色に着色させた。このPTA溶液を次いでカーボンスラリーへ移し、そして周囲温度で30分撹拌した。そしてビーカーを1℃/分の割合で70℃まで加熱し、撹拌しながらこの温度を1時間維持した。次いで加熱を止め、濃アンモニア(〜30%)を10ml/分の割合で、pHが3から3.5の間に達するまでスラリーへ加えた(およそ200mlのアンモニアを加えた)。該溶液をさらに撹拌しながら、室温まで冷却した。
【0020】
18.88gのCr(NO3)・9H2O(12.98%Cr、トータルで2.45gのCr)を150mlの脱イオン水に溶解し、そしてスラリーへ加えた。30分後、0.5M NH4OHによりスラリーのpHを〜4.5に調整し、さらにその30分後加熱を再開し、温度を75℃まで1℃/分の割合で上昇させた。該溶液は全プロセスの間撹拌し、さらにアンモニアを加えてpHを〜5.5に制御した。75℃に達した後、加熱及び撹拌の両方とも1時間維持し、それからスラリーを室温まで冷却し、濾過した。触媒ケーキを1.5リットルの脱イオン水で洗い、300mlアリコートへと細分し、そして水分含有量2%に達するまで125℃で乾燥した。乾燥ケーキは10メッシュの顆粒へと砕き、得られた触媒を30分間500℃で水素蒸気中で還元し、そしてアルゴン中で850℃で1時間焼結し、ボールミルで処理して微細粉末とした。
実施例7
グラビア/ローラーコーティング機を用いてテキストロン(Textron)カーボンクロス上のインク溶液からShawiniganアセチレンブラック(SAB)/PTFE層(60/40wt)を第1層に適用し、そしてVulcan XC−72/PTFEを第2層に適用することによって、ガス拡散電極を調製した。被覆カーボンクロスは340℃で焼結した。このようにして得られた焼結させたガス拡散層を重量2:1の触媒/アイオノマー懸濁インクに対し支持体として使用し、一方で、触媒は実施例6のPtCr/Cであり、フルオロカーボンポリマーアイオノマー懸濁液はアルコール中で9%の市販フルオロカーボン材料から調製した。約0.4―0.5 mg/cm2でロードされたPtが幾つかの被膜中に得られた。所望の白金充填に達した後、100−130℃で最終アニーリングを行った。
比較例1
ガス拡散電極を、使用した触媒が、実施例1の手順(但し硝酸ニッケルの添加及びその後の転化を省略)に従って白金酸を用いて調製した30%Pt/Cであることを除いては、実施例7に記載の手順に従って調製した。
実施例8
膜−電極接合体(MEA)を、実施例7で調製したガス拡散電極をカソードとして、そして標準的な機械製品の30%PT/Cガス拡散電極(公知技術として知られたフルオロカーボンポリマーアイオノマーで含浸され、標準的な手順に従って市販膜の反対側にホットプレスされたもの)を、アノードとして組み込むことにより作製した。他のMEAを同じ手順(但し比較例1のガス拡散電極をカソードとして使用)で作製した。どちらのMEAも実験燃料電池へ導入し、70℃、100%に調湿の反応ガス(空気/純粋(pure)H2)で作動させた。圧力は、カソード側に4バール絶対圧、アノード側3.5バール絶対圧の一定のフローレート(flow−rates)とし、1.2A/cm2の空気2、水素1.5の化学量論の比率に相当する。
【0021】
対応する分極曲線を図1に示す。これは、カーボン上の30%Pt:Cr(1)が、カーボン上の標準的な30%Pt(2)よりも、より活性なカソード触媒であることを明確に示している。
実施例9
図2は、実施例6の3:1 PtCr触媒(3)及び米国特許番号5876867の教示に従って調製した3:1 PtCr触媒(4)のXRDスペクトルを示す。Pt220ピーク(2θ=68−69)は実施例6のより高い値を示すものであり、合金化の優れた程度を示すものである。その上、2θ=40から48の間に、より顕著な実施例6の触媒の“超格子ピーク”がみられる。これらのピークは、良好なO2還元活性と関連する。実施例6の触媒はまた、公知技術の触媒のXRDサイズ(53Å)と比較して、より小さいXRDサイズ(37Å)を有する。これは、実施例6の触媒が、より良好な性能と関連するより高い表面積を有することを示す。
【0022】
図3は、実施例1(5)、2(6)、3(7)及び4(8)の触媒のXRDスペクトルを示し、ピーク位置のシフトがあるものの、そのパターンはPt/Cのものと同じである。これは、Ni金属単相は検出できないものであり、PtとNi間の合金化の非常に高い程度を示す。ニッケル含有量が、Pt4Ni(8)からPtNi(5)へと増すにつれ、各二次ピークは対応するPtのピークからより遠くなる。より多くのNiがPt格子内へと組み込まれると、d−間隔(d−spacing)はより小さくなる。例えば、Pt{220}ピーク(2θ=72)では、Pt4Ni、Pt3Ni、Pt2Ni及びPtNiのd−間隔はそれぞれ、1.3649、1.3569、1.3498及び1.3270である。30%Pt/Cのd−間隔は1.3877である。
【0023】
上記触媒は、本発明の精神又は範囲から逸脱しない限り、変更され得ること、並びに、本発明は添付されたクレームの範囲にのみ限定されるものではないことを理解することができる。
【図面の簡単な説明】
【0024】
【図1】図1は、本発明の触媒及び公知技術の触媒に関する燃料電池分極曲線の群である。
【図2】図2及び図3は、本発明の触媒および公知技術に関するXRDスペクトルである。
【図3】図2及び図3は、本発明の触媒および公知技術に関するXRDスペクトルである。
【特許請求の範囲】
【請求項1】
インサイチュで形成された(in situ―formed)二酸化白金及び炭素担体上の少なくとも1つの遷移金属水和酸化物(transition metal hydrous oxide)の同時の化学的還元により得られ得る、炭素担持された白金合金触媒。
【請求項2】
前記炭素担体が、少なくとも50m2/gのアクティブエリア(active area)を有するカーボンブラックである、請求項1の触媒。
【請求項3】
前記インサイチュで形成された二酸化白金が、前記炭素担体上でのヘキサヒドロキソ白金(IV)酸(dihydrogen hexahydroxyplatinate)の転化により得られる、請求項1の触媒。
【請求項4】
前記少なくとも1つの遷移金属水和酸化物が、前記炭素担体上での可溶性塩の転化によって得られる、請求項1の触媒。
【請求項5】
前記可溶性塩が硝酸塩である、請求項4の触媒。
【請求項6】
前記遷移金属が、Ni、Cr、Co、V及びFeからなる群より選択される、請求項1の触媒。
【請求項7】
前記化学的還元が、少なくとも300℃の温度において水素ガスにより行われる、請求項1の触媒。
【請求項8】
さらに、少なくとも600℃の温度に制御された雰囲気中でアニーリング処理に供される、請求項1の触媒。
【請求項9】
前記制御された雰囲気が、不活性なアルゴン又は窒素雰囲気である、請求項8の触媒。
【請求項10】
導電性ウェブ、及びその中に組み込まれた請求項1の触媒を含む、ガス拡散電極。
【請求項11】
イオン交換膜及び少なくとも1つのその中に組み込まれた請求項10のガス拡散電極を含む、膜−電極接合体。
【請求項12】
インサイチュで形成された二酸化白金及び少なくとも1つの遷移金属水和酸化物を炭素担体上で同時に還元すること含む、炭素担持された白金合金触媒の製造方法。
【請求項13】
前記インサイチュで形成された二酸化白金が、pH及び/又は温度の変化により、前記炭素担体上のヘキサヒドロキソ白金(IV)酸前駆体を転化することで得られる、請求項12の方法。
【請求項14】
前記少なくとも1つの遷移金属水和酸化物が、pH及び/又は温度の変化により、前記二酸化白金を含む炭素担体上の可溶性塩を転化することで得られる、請求項12の方法。
【請求項15】
前記pHの変化が、アルカリ、必要に応じて苛性ソーダ、又はアンモニアの添加により得られる、請求項13の方法。
【請求項16】
前記アルカリ又はアンモニアの添加により、pHを2と9の間にする、請求項15の方法。
【請求項17】
前記温度の変化が、前記水溶液を、室温から、30から100℃の終温度にすることからなる、請求項13の方法。
【請求項18】
前記炭素担体が、50m2/g以上のアクティブエリアを有するカーボンブラックである、請求項12の方法。
【請求項19】
前記カーボンブラックが、強酸中のスラリーである、請求項18の方法。
【請求項20】
前記遷移金属が、Ni、Cr、Co、V及びFeからなる群より選択される、請求項12の方法。
【請求項21】
前記遷移金属の可溶性塩が硝酸塩である、請求項14の方法。
【請求項22】
前記化学的還元が、少なくとも300℃の温度において水素ガスで行われる、請求項12の方法。
【請求項23】
さらに、少なくとも600℃の温度において制御された雰囲気中でのアニーリング処理を含む、請求項22の方法。
【請求項24】
前記制御された雰囲気が不活性雰囲気である、請求項23の方法。
【請求項1】
インサイチュで形成された(in situ―formed)二酸化白金及び炭素担体上の少なくとも1つの遷移金属水和酸化物(transition metal hydrous oxide)の同時の化学的還元により得られ得る、炭素担持された白金合金触媒。
【請求項2】
前記炭素担体が、少なくとも50m2/gのアクティブエリア(active area)を有するカーボンブラックである、請求項1の触媒。
【請求項3】
前記インサイチュで形成された二酸化白金が、前記炭素担体上でのヘキサヒドロキソ白金(IV)酸(dihydrogen hexahydroxyplatinate)の転化により得られる、請求項1の触媒。
【請求項4】
前記少なくとも1つの遷移金属水和酸化物が、前記炭素担体上での可溶性塩の転化によって得られる、請求項1の触媒。
【請求項5】
前記可溶性塩が硝酸塩である、請求項4の触媒。
【請求項6】
前記遷移金属が、Ni、Cr、Co、V及びFeからなる群より選択される、請求項1の触媒。
【請求項7】
前記化学的還元が、少なくとも300℃の温度において水素ガスにより行われる、請求項1の触媒。
【請求項8】
さらに、少なくとも600℃の温度に制御された雰囲気中でアニーリング処理に供される、請求項1の触媒。
【請求項9】
前記制御された雰囲気が、不活性なアルゴン又は窒素雰囲気である、請求項8の触媒。
【請求項10】
導電性ウェブ、及びその中に組み込まれた請求項1の触媒を含む、ガス拡散電極。
【請求項11】
イオン交換膜及び少なくとも1つのその中に組み込まれた請求項10のガス拡散電極を含む、膜−電極接合体。
【請求項12】
インサイチュで形成された二酸化白金及び少なくとも1つの遷移金属水和酸化物を炭素担体上で同時に還元すること含む、炭素担持された白金合金触媒の製造方法。
【請求項13】
前記インサイチュで形成された二酸化白金が、pH及び/又は温度の変化により、前記炭素担体上のヘキサヒドロキソ白金(IV)酸前駆体を転化することで得られる、請求項12の方法。
【請求項14】
前記少なくとも1つの遷移金属水和酸化物が、pH及び/又は温度の変化により、前記二酸化白金を含む炭素担体上の可溶性塩を転化することで得られる、請求項12の方法。
【請求項15】
前記pHの変化が、アルカリ、必要に応じて苛性ソーダ、又はアンモニアの添加により得られる、請求項13の方法。
【請求項16】
前記アルカリ又はアンモニアの添加により、pHを2と9の間にする、請求項15の方法。
【請求項17】
前記温度の変化が、前記水溶液を、室温から、30から100℃の終温度にすることからなる、請求項13の方法。
【請求項18】
前記炭素担体が、50m2/g以上のアクティブエリアを有するカーボンブラックである、請求項12の方法。
【請求項19】
前記カーボンブラックが、強酸中のスラリーである、請求項18の方法。
【請求項20】
前記遷移金属が、Ni、Cr、Co、V及びFeからなる群より選択される、請求項12の方法。
【請求項21】
前記遷移金属の可溶性塩が硝酸塩である、請求項14の方法。
【請求項22】
前記化学的還元が、少なくとも300℃の温度において水素ガスで行われる、請求項12の方法。
【請求項23】
さらに、少なくとも600℃の温度において制御された雰囲気中でのアニーリング処理を含む、請求項22の方法。
【請求項24】
前記制御された雰囲気が不活性雰囲気である、請求項23の方法。
【図1】


【図2】
【図3】
【図2】
【図3】
【公表番号】特表2008−532732(P2008−532732A)
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願番号】特願2007−541859(P2007−541859)
【出願日】平成17年11月28日(2005.11.28)
【国際出願番号】PCT/EP2005/012676
【国際公開番号】WO2006/056470
【国際公開日】平成18年6月1日(2006.6.1)
【出願人】(505118475)ベーアーエスエフ フューエル セル ゲーエムベーハー (24)
【Fターム(参考)】
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願日】平成17年11月28日(2005.11.28)
【国際出願番号】PCT/EP2005/012676
【国際公開番号】WO2006/056470
【国際公開日】平成18年6月1日(2006.6.1)
【出願人】(505118475)ベーアーエスエフ フューエル セル ゲーエムベーハー (24)
【Fターム(参考)】
[ Back to top ]