
炭素材料およびこれを利用した金属担持炭素材料、ならびにその製造方法
【課題】固体表面上に金属を高分散で担持させるための炭素材料を提供する。
【解決手段】細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【解決手段】細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、炭素材料、およびこれを利用した金属担持炭素材料に関する。より詳細には、燃料電池用の水素吸蔵材料や電極触媒に適用して好適な炭素材料、およびこれを利用した金属担持炭素材料、ならびにその製造方法に関する。
【背景技術】
【0002】
多量の水素の貯蔵は燃料電池自動車の実用化に必要不可欠な技術であり、これまでに水素吸蔵合金、化学水素化物、吸着系材料などの水素吸蔵材料の研究が活発に行なわれてきた。水素吸蔵合金および化学水素化物は、吸着系材料と比較して貯蔵量が大きく、水素吸蔵能が5重量%を超えるものが得られているものの、水素の放出に加熱が必要であること、寿命が短いことなどの問題がある。一方、吸着系材料としては、例えば活性炭やカーボンナノチューブをはじめとする炭素材料が挙げられる。吸着系材料では、物理吸着を利用するために水素の吸蔵・放出過程で加熱は不要であるが、水素吸蔵合金や化学水素化物と比較して吸着量が少ないという問題がある。そこで近年、物理吸着に加えてスピルオーバーを利用した水素貯蔵方式が注目されている。
【0003】
スピルオーバーとは、固体表面上に白金などの金属を担持すると、気相中の水素分子が金属の作用により金属表面上で水素原子に解離し、固体表面上に流出する現象である。流出した水素原子が固体表面に貯蔵されるため、水素を分子のまま吸着させる物理吸着と組み合わせて水素吸蔵能を向上させることができると考えられる。例えば、非特許文献1では、高表面積の活性炭に白金ナノ粒子を担持させると、常温における水素吸蔵能が担持前に比べて大幅に増大することが報告されており、これは、物理吸着に加えてスピルオーバーの効果によるものとされている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】J.Phys.Chem.C,111(2007)11086.
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、燃料電池自動車の実用化のためには、より多量の水素を貯蔵できる水素吸蔵材料が求められている。また、固体表面に担持させる金属は、主に白金などの高価な金属であり、スピルオーバーに寄与するのは金属粒子の表面部分だけであるため、同じ量の金属を担持するならば、粒子径を小さくして粒子の表面積をできるだけ大きくすることが不可欠である。しかしながら、金属粒子の微粒子化には限界がある。
【0006】
そこで本発明は、固体表面上に金属を高分散で担持させるための炭素材料、およびこれに金属を担持させた金属担持材料を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記の課題に鑑み、鋭意研究を積み重ねた。その過程で、金属を粒子としてではなく、金属錯体として炭素材料の表面に担持させることで、金属を酸化させることなく原子レベルで担持させることを検討した。そして、炭素材料の表面に配位子を導入することで、金属を錯体として容易に導入できることを見出し、本発明を完成させた。
【0008】
すなわち本発明は、細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【発明の効果】
【0009】
本発明によれば、炭素材料に導入されたリン配位子部分に金属錯体を導入することができ、したがって金属を原子レベルで炭素材料に分散させて担持させることが可能になる。そのため、水素のスピルオーバーの効率が高まり、水素貯蔵効率が向上した水素吸蔵材料が得られうる。
【図面の簡単な説明】
【0010】
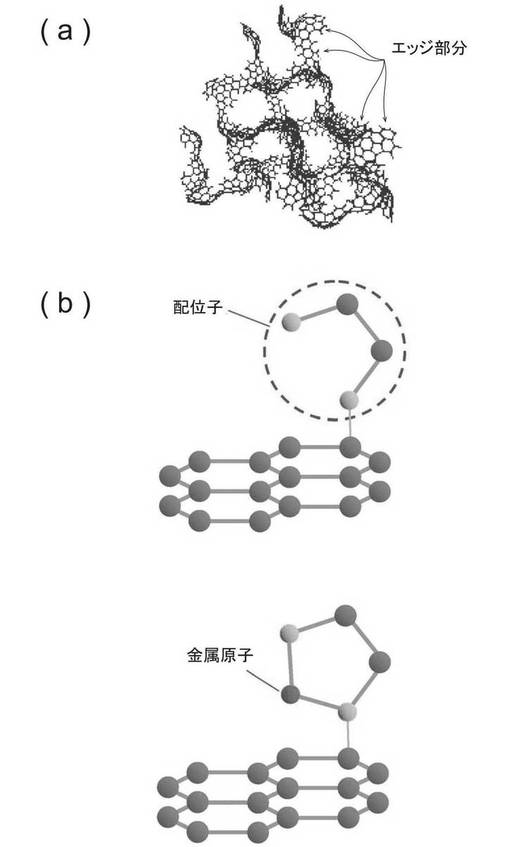
【図1】(a)炭素材料のエッジ部分、(b)炭素材料に配位子が導入された炭素材料、(c)金属原子が配位子を介して導入された金属担持炭素材料をそれぞれ表す模式図である。
【図2】ミクロポーラス炭素材料を表す模式図である。
【図3】合成例1〜5で製造した試料のXRD測定の結果を表す図である。
【図4】合成例3で製造した試料の水蒸気吸脱着等温線を表す図である。
【図5】実施例6で製造した試料のXRD測定の結果を表す図である。
【図6】実施例6で製造した試料のXPS測定の結果を表す図である。
【図7】実施例6で製造した試料の固体31P−NMR測定の結果を表す図である。
【図8】実施例7で製造した試料の固体31P−NMR測定の結果を表す図である。
【図9】実施例6で製造した試料の水素吸脱着等温線を表す図である。
【図10】実施例7で製造した試料の水素吸脱着等温線を表す図である。
【図11】実施例6で製造した試料の水素吸脱着等温線を表す図である。
【図12】実施例8で製造した試料のXRD測定の結果を表す図である。
【図13】実施例8で製造した試料のXPS測定の結果を表す図である。
【図14】実施例8で製造した試料の水素吸脱着等温線を表す図である。
【発明を実施するための形態】
【0011】
以下、添付した図面を参照しながら、本発明の実施形態を説明する。なお、図面の説明において同一の要素には同一の符号を付し、重複する説明を省略する。また、図面の寸法比率は、説明の都合上誇張されており、実際の比率とは異なる場合がある。
【0012】
<リン配位子を有する炭素材料>
本発明の一実施形態は、細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【0013】
本発明の一実施形態によるリン配位子を有する炭素材料は、図1(b)に示すように、炭素材料の表面、好ましくは前記炭素材料の表面のエッジ部分に、リン配位子が結合してなる。ここで炭素材料のエッジは、例えば図1(a)に示すようにグラフェンシートの構造における炭素縮合環の端部が配列した面である。
【0014】
本実施形態による炭素材料は、炭素材料の表面の少なくとも一部にリン配位子が導入されてなる。好ましくは、前記炭素材料の表面のエッジ部分の少なくとも一部にリン配位子が導入されてなる。リン配位子を導入することによって、金属錯体をリン配位子に安定に配位させることができる。そのため、金属を原子レベルで炭素材料に担持させることが可能になる。
【0015】
この際、用いられるリン配位子としては、特に制限されないが、好ましくはPPh1Ph2(Ph1およびPh1は同一であっても異なっていてもよく、置換または非置換のフェニル基である)で表されるリン配位子が好ましく用いられうる。Ph1またはPh2の置換基としては、特に制限されないが、例えば炭素数1〜8のアルキル基、アルコキシ基、ハロゲン化アルキル基である。
【0016】
Pはd軌道が大きいため、8〜10属の金属と配位しやすい。またPh1およびPh2が置換または非置換のフェニル基であればZTCの細孔に入る大きさであるため、反応に有利である。
【0017】
前記リン配位子の例としては、特に制限されないが、ジフェニルホスフィン、ビス(3,5−ジメチルフェニル)ホスフィン、ビス(4−メトキシフェニル)ホスフィン、ビス[4−(トリフルオロメチル)フェニル]ホスフィン、ジ(p−トリル)ホスフィン、ビス(3,5−ジ−tert−ブチル−4−メトキシフェニル)ホスフィン、ビス[3,5−ビス(トリフルオロメチル)フェニル]ホスフィン、などが挙げられる。
【0018】
炭素材料としては、上記のように細孔を有し、エッジを有するものであれば特に制限はなく、カーボンブラック、活性炭、コークス、天然黒鉛、人造黒鉛などからなるカーボン粒子などが用いられうるが、特にゼオライト鋳型カーボン(ZTC)のようなミクロポーラス炭素材料が好ましく用いられうる。ZTCは、球面状の細いグラフェンシートが3次元状に規則的につながっており、エッジの炭素原子の数が相対的に多い。さらに、超高表面積であるため、水素吸着特性に優れる。
【0019】
図2に、ミクロポーラス炭素材料としてのゼオライト鋳型カーボンの一例を模式的に示す。ゼオライト鋳型カーボン2は、ゼオライト1を鋳型として得られたゼオライト炭素2である。より詳細には、ゼオライト鋳型カーボン2の作製には、まず、図2(a)に示すゼオライト1のミクロ孔1aに炭素源である有機化合物を導入した後に加熱処理して図2(b)に示すゼオライト1とゼオライト炭素2との複合体3を調製する。その後にゼオライト1のみを除去することによって、ゼオライト鋳型カーボン2が得られる(図2(c))。ゼオライト鋳型カーボン2は、鋳型として用いたゼオライト1の構造的特徴が反映された、3次元の長周期規則構造と内部にミクロ細孔2aとを有する。
【0020】
ゼオライト鋳型カーボン2は、その製造にあたり、使用する鋳型材である特定の3次元規則構造を有するゼオライト1が備える構造的特徴を反映した多孔性炭素材料である。ゼオライト鋳型カーボン2は、直径が0.1〜2nmの範囲内にある細孔(ミクロ細孔2a)が網目状に連結した構造を有する。具体的には、ゼオライト鋳型カーボン2は、0.5〜100nmの範囲内の3次元長周期規則構造を有すると共に、ミクロ細孔2aを有する。より具体的には、ゼオライト鋳型カーボン2は、3次元長周期規則構造を構成する炭素鎖と炭素鎖の間の距離が、好ましくは0.5〜100nmであり、より好ましくは0.7〜50nmであり、さらに好ましくは0.7〜2nmである。このように、ゼオライト鋳型カーボン2は、炭素鎖と炭素鎖の間の距離が任意の間隔で3次元的に長周期にわたって規則的に繰り返した構造を有する炭素材料である。なお、IUPAC(国際純正及び応用化学連合)では、直径2nm以下の細孔をミクロ細孔(micropore)、直径2〜50nmの細孔をメソ細孔(mesopore)、直径50nm以上の細孔をマクロ細孔(macropore)と定義している。ミクロ細孔を有する物質を総称してミクロ(マイクロ)ポーラス材料と称している。
【0021】
本実施形態において、前記炭素材料は、エッジ部分の少なくとも一部に官能基が導入されてなるエッジ修飾炭素材料でありうる。好ましくは、前記炭素材料のエッジ部分の少なくとも一部にアミジン構造を有する官能基が導入されてなる炭素材料である。この際、前記アミジン構造を有する官能基に、リン配位子が結合される。
【0022】
本明細書中、アミジン構造は、1つの炭素原子に二重結合を介して1つの窒素原子が結合し、単結合を介して1つの窒素原子が結合した構造である。アミジン構造は強塩基で求核性を示すため、後述するようにアミジン構造部分にさらにリン配位子を導入し、金属錯体を導入することができる。本発明に用いられるアミジン構造を有する官能基としては、例えば、下記化学式1で表される官能基が挙げられる。
【0023】
【化1】

【0024】
式中、R1、R2およびR3は独立して、水素原子、置換もしくは非置換の炭素数1〜10のアルキル基または置換もしくは非置換の炭素数3〜6のシクロアルキル基であり、R1とR2とが結合して環を形成してもよく、R2とR3とが結合して環を形成してもよい。
【0025】
アルキル基は、直鎖であっても、分岐であってもよい。アルキル基またはシクロアルキル基は、置換であっても非置換であってもよい。アルキル基の有する炭素数は、特に限定されないが、好ましくは1〜10個、より好ましくは1〜5個である。シクロアルキル基の有する炭素数は、特に限定されないが、好ましくは3〜6個である。アルキル基またはシクロアルキル基の置換基も特に制限されないが、例えば、水酸基、カルボキシル基、アミノ基、シアノ基、ハロゲン原子などが挙げられる。アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、などが挙げられる。シクロアルキル基としては、シクロプロピル基、シクロペンチル基、シクロヘキシル基などが挙げられる。これら以外のアルキル基またはシクロアルキル基が用いられてもよい。R1とR2とが結合して環を形成する場合、または、R2とR3とが結合して環を形成する場合、当該環は好ましくは5〜6員環であり、より好ましくは5員環である。
【0026】
中でも、下記化学式で表される官能基が好適に用いられうる。
【0027】
【化2】
【0028】
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料、リン配位子を有する炭素材料、または後述する金属担持炭素材料は、そのBET表面積が1500m2/g以上であることが好ましい。例えばゼオライト鋳型カーボンを用いた炭素材料、または金属担持炭素材料は、3次元長周期規則構造とミクロ細孔とを有することによりBET表面積が大きい。一般に吸着材への適用に関しては、BET表面積が大きいことが好ましい。また、吸着する分子サイズにも影響されるが、ミクロ孔が存在することも重要であると考えられる。これに対して、メソ孔は前述した用途への適用に際してはあまり効果がないと考えられる。このため、所望の高い機能を発現させるためには、相対的にミクロ孔が多く存在することが重要であり、なるべくメソ孔は少ない方が良いと考えられる。この目安として、BET表面積は1500m2/g以上であることが好ましい。また、2000m2/g以上であることがより好ましく、さらには2500m2/g以上であることが好ましい。
【0029】
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料、リン配位子を有する炭素材料、または後述する金属担持炭素材料は、その構造的特徴として2次元積層規則性が少ないほど、吸着力が高くなる。例えば、粉末X線回折測定を行った場合には、得られるX線回折パターンは、2次元積層規則性を示す通常26°付近に現れる回折ピークはできるだけ少ないほうが好ましい。この26°付近に現れる回折ピークの存在は、無孔質の炭素層の増加を意味し、BET表面積の低下を意味する。
【0030】
また、上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料は、25℃において、水蒸気吸着等温線の傾きの極大値を示す相対圧(P/P0)が0.70以下であることが好ましい。上記範囲であれば、炭素材料の表面の親水性が高く、炭素材料に機能性を付与することができる。
【0031】
上記アミジン構造を有する官能基が導入されてなる炭素材料は親水性を有するため、水蒸気をはじめとする極性を有するガス、酸性ガス、含酸素炭化水素蒸気等の吸着剤として有用である。また、電気二重層キャパシタ(EDLC)の電極として応用した場合にも性能の向上が期待できる。一般に、EDLCの電解液は極性が大きく、電極表面との親和性(濡れ性)を考慮すると親水性を示す電極が望まれる。活性炭、ミクロポーラス炭素材料も含めて炭素材料は疎水性が強く、キャパシタ等へ応用する場合に親和性(濡れ性)を改変する。このため、EDLCの直接的な性能に関与しない二次的な表面改質処理を行うことがある。これに対し、上記アミジン構造を有する官能基が導入されてなる炭素材料は二次的な表面改質処理を行う必要がなく、そのまま用いることが可能となる。このため、よりすぐれた効果が期待できる。その他、水蒸気の吸脱着を利用する吸着式ヒートポンプにおいて、吸着剤として現在使用されているゼオライトと比べて大幅な性能向上が可能なため、装置の小型化等、性能向上が期待できる。
【0032】
(エッジ修飾炭素材料の製造方法)
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料は、エッジを有する炭素材料(炭素材料前駆体)を準備する段階と、前記炭素材料を、アミジン構造を含むアゾ化合物と反応させて、前記炭素材料のエッジにアミジン構造を有する官能基を導入する段階と、を有する方法によって製造することができる。
【0033】
原料となる炭素材料(炭素材料前駆体)の入手経路については特に制限はない。商業的に入手可能な商品を用いてもよいし、自ら調製してもよい。以下、ゼオライト担持カーボンなどのミクロポーラス炭素材料を用いる場合を説明する。
【0034】
まず、上記した構造的な特徴を有するミクロポーラス炭素材料を得るためには、構造内部に空孔を有し、この空孔が網目状に連結した構造を有する多孔質材料を鋳型として用いる。そして、この多孔質材料の表面及びミクロ細孔内部に加熱条件下で有機化合物を導入し、加熱することによって有機化合物を炭化し、多孔質材料に炭素を堆積させる。有機化合物の炭化・炭素の堆積は、例えば化学気相成長(Chemical Vapor Deposition:CVD)法により行う。次に、鋳型である多孔質材料を除去する。この方法により、ミクロ細孔を有するミクロポーラス炭素材料を容易に製造することができる。ただし、後述するように、鋳型の除去はアミジン構造を有する官能基を導入する反応と同時に行うことができるので、多孔質材料に炭素を堆積させた複合体を原料として用いてアミジン構造を有するアゾ化合物と反応させ、エッジ修飾炭素材料を製造してもよい。
【0035】
鋳型として用いる多孔質材料は、ミクロ細孔内部に有機化合物が導入できること、CVD法の際に元の構造を安定に保つこと、生成したミクロポーラス炭素材料と分離できることが必要である。このため、例えば多孔質酸化物等の耐熱性に優れ、且つ、酸やアルカリで溶解する材料が望ましい。また、既に述べたように、ミクロポーラス炭素材料は鋳型の形態を転写した状態で合成される。このため、鋳型として用いる多孔質材料は、結晶(構造)が十分に発達し、粒子径の揃った構造及び組成が均一な材料であることが望ましい。多孔質材料の備えるべき材料物性と、得られるミクロポーラス炭素材料の物性を考慮すると、多孔質材料としてゼオライトを用いることが好ましい。ゼオライトは、シリカ構造のケイ素(Si)の一部がアルミニウム(Al)で置換されたアルミノケイ酸塩であり、骨格自体が負電荷を持つことから構造内にカチオンが分布した構造を有する。また、ゼオライトは、Si/Alモル比、カチオンの種類や量、及びカチオンに水和した水分子の数によって多様な結晶構造を有し、例えば細孔が2次元的に連結した構造や3次元的に連結した構造等の、多様な大きさの細孔を有する多孔質材料である。代表的なゼオライトとしては、ケージ又はスーパーケージといった空隙構造を有するものが挙げられ、ゼオライトの中でもFAU型ゼオライト、FAU型ゼオライトの中でもY型ゼオライトを用いることが望ましい。多孔質材料の除去は、生成したミクロポーラス炭素材料を分離できる方法であれば如何なる方法を用いても良い。例えば、ゼオライトは酸で溶解可能であり、例えば、塩酸やフッ化水素酸を用いることで容易に溶解できる。
【0036】
有機化合物を炭化して炭素を堆積するために用いるCVD法は、鋳型等の基板上に特定の元素又は元素組成からなる薄膜(例えば炭素からなる薄膜)を作る工業的手法である。通常、原料物質を含むガスに熱や光によってエネルギーを与えたり、高周波でプラズマ化することにより、化学反応や熱分解によって原料物質がラジカル化して反応性に富むようになり、基板上に原料物質が吸着して堆積することを利用する技術である。温度を上げて原料物質を堆積させるものを熱CVD法、化学反応や熱分解を促進させるために光を照射するものを光CVD法、ガスをプラズマ状態に励起する方法をプラズマCVD法と区別することもある。
【0037】
CVD法で用いる有機化合物は、常温で気体であるか、又は気化できるものが好ましい。気化の方法は、沸点以上に熱する方法や雰囲気を減圧にする方法等がある。用いる有機化合物は、当業者に知られた炭素源物質の中から適宜選択して使用できる。特に、加熱により熱分解する化合物が好ましい。例えば、CVD法で鋳型として用いる多孔質材料の骨格上(例えばシリカゲル骨格上)に炭素を堆積することができる化合物が好ましい。
【0038】
また、用いる有機化合物は、水素を含む有機化合物でも良い。この有機化合物は、不飽和又は飽和の有機化合物でも良く、これらの混合物でも良い。用いる有機化合物は、二重結合及び/又は三重結合を有する不飽和直鎖又は分枝鎖の炭化水素、飽和直鎖又は分枝鎖の炭化水素等が含まれて良く、飽和環式炭化水素や芳香族炭化水素等を含んでいても良い。有機化合物は、例えば、アセチレン、メチルアセチレン、エチレン、プロピレン、イソプレン、シクロプロパン、メタン、エタン、プロパン、ベンゼン、ビニル化合物、エチレンオキサイド等があげられる。中でも、用いる有機化合物は、多孔質のミクロ細孔内に入り込むことが可能なもの、例えばアセチレン、エチレン、メタン、エタン等を用いることが望ましい。有機化合物は、より高温でのCVDに用いるものと、より低温でCVDに用いるものとでは互いに同一のものであっても異なっていても良い。例えば、低温でのCVDではアセチレン、エチレン等を使用し、高温でのCVDにはプロピレン、イソプレン、ベンゼン等を使用しても良い。
【0039】
多孔質材料のミクロ細孔内部に有機化合物を導入する際は、多孔質材料を予め減圧にしても良く、系自体を減圧下にしても良い。多孔質材料は安定であるので、CVDにより炭素が堆積する方法であれば如何なる方法を用いても良い。通常は、多孔質材料の骨格上に有機化合物の化学反応又は熱分解で生成した炭素を堆積(又は吸着)させ、多孔質材料と炭素を含むミクロポーラス炭素材料からなる複合体を得る。CVDを行う際は、加熱温度は、使用する有機化合物によって適宜適切な温度を選択できる。通常は、400〜1500℃であることが好ましい。加熱温度は、450〜1100℃であることがより好ましく、500〜900℃が更に好ましい。また、550〜800℃であることがより好ましく、575〜750℃更には約600〜700℃の範囲内にすることが望ましい。加熱温度はCVD処理時間及び/又は反応系内の圧力に応じて適宜適切な温度を選択することもできる。CVDの処理時間は、十分に炭素堆積が得られる時間とすることが好ましく、使用する有機化合物や温度によって適宜適切な時間を選択できる。
【0040】
CVDは、減圧又は真空下、加圧下、若しくは不活性ガス雰囲気下で行うことができる。不活性ガス雰囲気下で行う場合には、不活性ガスとしては例えばN2ガス、ヘリウム、ネオン、アルゴン等があげられる。CVD法では、通常、気体状の有機化合物をキャリアガスと共に多孔質材料に接触させるように流通させながら加熱し、容易に気相で多孔質材料上に炭素を堆積させることができる。キャリアガスの種類、流速、流量及び加熱温度は使用する有機化合物や多孔質材料の種類によって適宜調節する。キャリアガスは、例えば上記の不活性ガス等があげられる。爆発限界を考慮して、酸素ガス又は水素ガスとの混合物等であっても良い。
【0041】
CVD法により多孔質材料のミクロ細孔内部に炭素を堆積させる条件として、ミクロ細孔中の炭素の充填量は10〜40wt%の範囲内であることが好ましい。また、炭素の充填量は多孔質材料の重量を基準として15〜30wt%の範囲内に制御することがより好ましい。炭素の充填量が10wt%以上であれば、炭素骨格形成に必要な量の炭素が導入されるため、安定な規則性構造が得られうる。炭素の充填量が40wt%以下であれば、必要以上の炭素が付着することなく、ミクロ細孔容積及びBET表面積が維持されうる。
【0042】
CVDによる炭素の堆積(吸着)後、多孔質材料とミクロポーラス炭素系材料の複合体を、CVD温度より高い温度で更に加熱しても良い。この加熱温度は、使用する有機化合物によって適宜選択できるが、通常は700〜1500℃である。加熱温度は、750〜1200℃であることが好ましく、800〜1100℃であることがより好ましい。また、825〜1000℃であることが好ましく、850〜950℃、更には約875〜925℃の範囲内にすることが好ましい。また、加熱温度は、加熱時間及び/又は反応系内の圧力に応じて適宜選択することもできる。また、加熱時間は生成物を分析し、その結果に基づいて十分な炭素堆積に要求される時間を設定することができる。
【0043】
また、多孔質材料とミクロポーラス炭素材料の複合体に更に有機化合物を導入して加熱し、更に炭素を堆積させても良い。この場合には、CVD法により得られたミクロポーラス炭素材料の構造がより安定する。炭化は、CVD法によって行っても良く、他の加熱方法で行っても良い。また、加熱温度はCVD温度より高温であっても良く、低温であっても良い。また、導入する有機化合物は、CVD法で導入した有機化合物と同じであっても良く、異なっていても良い。この操作は、複数回行っても構わない。
【0044】
多孔質材料の表面及びミクロ細孔内に有機化合物を導入してCVDを行う前に、有機化合物を含浸して炭化しても良い。含浸する有機化合物は、多孔質材料のミクロ細孔径より小さな分子サイズを有する有機化合物であれば使用できる。具体的には、有機化合物は、炭化歩留まりの高いフルフリルアルコール等の熱重合性モノマーを用いることが好ましい。有機化合物の含浸方法は、モノマーが液体であればそのまま、固体であれば溶媒に溶解して多孔質材料と接触させる等、公知の手段を採用することができる。なお、多孔質材料の表面に残った過剰なモノマーは、予め洗浄等で除去することが好ましい。例えば、多孔質材料を室温減圧下でフルフリルアルコールと接触させた後、混合物を大気圧に戻すことにより、多孔質材料のミクロ細孔内にフルフリルアルコールを導入することができる。また、多孔質材料の表面に付着した余分なアルコールは、有機溶剤による洗浄で除去できる。
【0045】
用いる有機化合物は、多孔質材料のミクロ細孔内に挿入可能な大きさを有し、且つ、炭化時に炭素としてミクロ細孔内に残留するものであれば特に制限は無く用いることができる。例えば、有機化合物として、酢酸ビニル・アクリロニトリル・塩化ビニル等のビニル化合物、塩化ビニリデン・メタクリル酸メチル等のビニリデン化合物、無水マレイン酸等のビニレン化合物、エチレンオキサイド等のエポキシ誘導体があげられる。また、グルコース・サッカロース等の糖類、脂肪族多価アルコール類、レゾルシノール・カテコール等の芳香族多価アルコール(ジオール)類、チオフェン等の含窒素複素環化合物、ピリジン・ピリミジン等の含窒素複素環化合物も利用することができる。
【0046】
次いで、このようにして得られた炭素材料のエッジにアミジン構造を有する官能基を導入する。炭素材料の表面におけるエッジは活性面であり、ラジカルを生成しうる重合開始剤などを用いて各種のアミジン構造を導入することができる。炭素材料のエッジにアミジン構造を有する官能基を導入する方法は特に制限されない。例えば、エッジ部分を有する炭素材料をアミジン構造を有するアゾ化合物(例えば、水溶性アゾ重合開始剤)と反応させて、前記炭素材料のエッジにアミジン構造を有する官能基を導入する工程を有する方法が好ましく用いられうる。具体的には、水溶性アゾ重合開始剤を用いてラジカルを生成させ、炭素材料のエッジと反応させる方法が好ましく用いられうる。このような方法によれば、例えば炭素材料としてZTCなどを用いた場合、ZTCの構造規則性を低下させることなくエッジ部分に目的の親水性官能基を導入することができる。
【0047】
また、通常、ZTCを調製する際に、ZTCの前駆体としてゼオライトと炭素との複合体を調製した後、ゼオライトをフッ化水素酸などで処理して取り除く工程を必要とするが、親水性官能基を導入する工程は、上記のゼオライトを取り除く工程で同時に行うことができる。そのため、親水性官能基を導入するために反応ステップを増やす必要がなく、複雑な実験操作も必要としない。さらに、多くの水溶性アゾ重合開始剤は工業的にも利用されているため比較的安価で使用できる。
【0048】
ここで、アミジン構造を導入するために用いられうる水溶性アゾ重合開始剤としては、特に制限されないが、例えば、下記式に示される化合物が好ましく用いられうる。
【0049】
【化3】
【0050】
炭素材料とアミジン構造を有するアゾ化合物とを反応させる条件は特に制限されない。好ましくは、反応容器に窒素置換しながらアミジン構造を有するアゾ化合物および炭素材料を加え、加熱、攪拌しながら反応させる。反応温度は用いられる水溶性アゾ重合開始剤の10時間半減期温度に応じて、ラジカルが十分発生するように選択されうる。例えば、水溶性アゾ重合開始剤である、水中の10時間半減期温度が44℃であるVA−044を用いる場合、反応温度は、例えば0〜85℃であり、好ましくは30〜70℃であり、より好ましくは40〜65℃である。反応時間は、例えば、0.5〜10時間であり、好ましくは1〜6時間である。
【0051】
アミジン構造を有するアゾ化合物の使用量は、特に制限されないが、炭素材料の質量に対して、例えば10〜10000質量%であり、好ましくは100〜5000質量%であり、より好ましくは100〜500質量%である。上記範囲であれば、炭素材料のBET表面積の大きな減少がなく、炭素材料の構造を維持しながら、アミジン構造を有する官能基を効率的に炭素材料のエッジに導入することができる。
【0052】
なお、エッジ修飾ZTCを製造する場合、ZTCを原料として用いて水溶性アゾ重合開始剤と反応させてもよいが、ゼオライトと炭素との複合体を原料として用い、フッ化水素酸などの酸溶液中で水溶性アゾ重合開始剤と反応させ、アミジン構造の導入とゼオライトの除去を同時に行ってもよい。ZTCを原料として用いる場合は、例えば、一般的なガラスフラスコ中で蒸留水とZTCとをよく混合し、不活性ガスをバブリングさせてから水溶性アゾ重合開始剤を添加して反応させることでエッジ修飾ZTCを製造することができる。
【0053】
炭素材料のエッジ部分にアミジン官能基を導入すると、この官能基の部分に多様な化学構造をさらに導入することができるが、この際、アミジン官能基の部分にリン配位子を導入することによって、後述する金属錯体を安定に配位させることができる。
【0054】
アミジン構造を有する官能基は強塩基で求核性を示すため、このアミジン構造を基点として、炭素材料にリン配位子を導入することができる。その後、後述するようにこのリン配位子に触媒となる金属を含む金属錯体を導入する。この反応では、金属錯体の配位子が、配位力の強いリン配位子と置き換わる配位子交換反応によって進行しうる。その結果、リン配位子と強塩基性のアミジン構造からなる二座配位子が形成される。したがって金属原子にリン原子と窒素原子が同時にキレート配位し、金属を安定な錯体として担持させることができる。このように金属錯体を担持させることができれば、金属を粒子ではなく原子レベルで分散させることが可能となる。
【0055】
この際、用いられるリン配位子を導入するためのリン化合物としては、特に制限されないが、好ましくは下記化学式で表されるリン化合物が好ましく用いられうる。
【0056】
【化4】
【0057】
式中、Ph1およびPh2は同一であっても異なっていてもよく、置換または非置換のフェニル基であり、Xは、ハロゲン原子である。ハロゲン原子としては、Cl、Brが好ましく、Clがより好ましい。Ph1またはPh2の置換基としては、特に制限されないが、例えば炭素数1〜8のアルキル基、アルコキシ基、ハロゲン化アルキル基である。
【0058】
前記リン化合物の例としては、クロロジフェニルホスフィン、クロロビス(3,5−ジメチルフェニル)ホスフィン、クロロビス(4−メトキシフェニル)ホスフィン、クロロビス[4−(トリフルオロメチル)フェニル]ホスフィン、クロロジ(p−トリル)ホスフィン、ビス(3,5−ジ−tert−ブチル−4−メトキシフェニル)クロロホスフィン、クロロビス[3,5−ビス(トリフルオロメチル)フェニル]ホスフィンなどが挙げられる。
【0059】
前記エッジ修飾炭素材料とリン化合物とを反応させる条件についても特に制限されない。好ましくは、あらかじめ真空加熱乾燥させて放冷した前記エッジ修飾炭素材料と、リン化合物とを、窒素置換しながら、好ましくはアミン存在下で反応させる。反応温度、反応時間については特に制限されない。好ましくは、はじめに前記エッジ修飾炭素材料に溶媒に溶解させたアミンを含浸させ、これにリン化合物を、例えば−100〜10℃、好ましくは−100〜5℃で少しずつ添加して混合する。その後、反応溶液の温度を、例えば0〜100℃、好ましくは10〜30℃に上昇させ、攪拌しながら反応させる。反応温度を上昇させた後の反応時間は、好ましくは0.5〜72時間であり、より好ましくは2〜24時間であり、さらに好ましくは12〜24時間である。
【0060】
この際、溶媒としては、特に限定されないが、非プロトン性極性溶媒が好ましく用いられうる。非プロトン性極性溶媒としては、例えば、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)、ヘキサメチルリン酸トリアミド(HMPT)、ジメチルホルムアミド(DMF)、アセトン、アセトニトリルなどが挙げられる。中でも、THFが好ましい。
【0061】
アミンとしては、3級アミンを用いることが好ましい。3級アミンとしては、特に制限されないが、例えば、トリメチルアミン、トリエチルアミン、トリ−n−プロピルアミン、トリイソプロピルアミン、トリ−n−ブチルアミン、ジエチルメチルアミン(N,N−ジエチルメチルアミン)、ジメチルエチルアミン(N,N−ジメチルエチルアミン)などが挙げられる。中でも、トリエチルアミンが好ましい。
【0062】
リン化合物の使用量も特に制限されないが、好ましくは、炭素材料に結合したアミジン構造を有する官能基1モルに対して0.5〜100モルであり、より好ましくは1〜10モルである。上記範囲であれば、反応が効率的に進行しうる。
【0063】
一方、エッジ修飾されていない炭素材料に、直接リン配位子を導入することもできる。リン配位子を導入するためのリン化合物や、リン化合物を炭素材料と反応させる際の反応条件は、上述のエッジ修飾炭素材料の場合と同様のものが採用されうる。エッジ修飾されていない炭素材料を用いた場合、リン化合物は炭素材料の表面の含酸素官能基と反応することによって炭素材料に導入されるものと考えられる。このため、エッジ部分の割合が高いZTCだけではなく、エッジ部分の割合が比較的少ない炭素材料であっても効率よくリン配位子を導入することが可能であると考えられる。また、エッジ修飾の工程を省略することができるため、コスト低減が可能である。
【0064】
<金属担持炭素材料>
本発明の一実施形態は、上記のリン配位子を有する炭素材料において、リン配位子に8〜10族の金属が配位してなる、金属担持炭素材料である。
【0065】
本実施形態による金属担持炭素材料は、上記図1(c)のように、炭素材料の表面に8〜10属の金属が錯体として配位子を介して配位されることにより、金属が担持されている。すなわち、前記金属は、粒子としてではなく、錯体として担持されている。このようにすることで、金属が酸化されることなく、金属を原子レベルで分散させることができるので、より高効率なスピルオーバーが可能となる。
【0066】
好ましくは、前記金属は、炭素材料において、0.01〜10wt%の濃度範囲内で担持することが好ましい。担持されている金属が0.01wt%以上であれば、金属の機能を十分に得ることができる。一方、担持されている金属が10wt%以下である場合には、細孔機能を維持し、高いBET表面積が得られうる。
【0067】
担持する金属は、機能性を付与するという観点で8〜10族の金属であることが好ましい。また、前記金属は単体だけではなく、2種類以上の金属が担持されていてもよい。中でも、水素吸蔵材料として用いる場合には、Pt、Pd、Ir、Rh、Co、Ni、Ru、Feを用いることが好ましく、Pt、Pd、Niを用いることがさらに好ましく、Ptが特に好ましい。
【0068】
本実施形態による金属担持炭素材料は、金属を担持した状態で、ミクロ細孔2aの占める容積が1.0cm3/g以上であることが好ましい。ミクロ細孔2aの占める容積は、1.2cm3/g以上であることがより好ましく、1.5cm3/g以上であることが更に好ましい。また、金属を担持した状態で、BET表面積が1500m2/g以上であることが好ましい。BET表面積は、例えば2000m2/g以上、好ましくは2500m2/g以上であり、3000m2/g以上であることがより好ましく、BET表面積が3500m2/g以上であることが更に好ましい。ミクロ細孔の占める容積が1.0cm3/g以上、又はBET表面積が1500m3/g以上である場合には、十分な水素貯蔵性能が得られうる。
【0069】
また、前記金属担持炭素材料は、粉末X線回折測定を行った場合に得られるX線回折パターンは、2次元積層規則性を示す通常26°付近に現れる回折ピークはできるだけ少ないほうが好ましい。
【0070】
本実施形態に係る金属担持炭素材料は、−40℃から150℃の範囲で水素を吸蔵放出させることができる。従来、金属担持されていない炭素材料は、温度上昇と共に水素吸蔵量が低下する場合があったが、本実施形態に係る金属担持炭素材料は温度上昇と共に吸蔵能が向上する。また、粒子状の金属を担持させた場合に比較して、水素分子の解離吸着が促進され、水素吸蔵能が向上しうる。そのため、前記金属担持炭素材料を水素吸蔵材料として用いると、高効率の水素の吸蔵及び放出が可能となる。
【0071】
(金属担持炭素材料の製造方法)
本実施形態による金属担持炭素材料は、細孔を有する炭素材料を準備する段階と、前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階と、前記リン配位子を有する炭素材料を、8〜10族金属化合物と反応させて、前記リン配位子に8〜10族金属を配位させる段階と、を有する方法によって調製されうる。
【0072】
ここで、細孔を有する炭素材料を準備する段階および前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階については上述と同様であるため、説明を省略する。リン配位子を有する炭素材料を得た後、好ましくはこれを洗浄して真空加熱乾燥した後、金属錯体と反応させる。用いられる金属錯体についても特に制限されない。好ましい白金錯体としては、例えば、(1,5−シクロオクタジエン)ジメチル白金(II)、(2,2’−ビピリジン)ジクロロ白金(II)、(エチレンジアミン)ヨード白金ダイマー、ジクロロ(1,10−フェナントロリン)白金(II)、ジクロロ(エチレンジアミン)白金(II)、ジクロロビス(ジメチルスルフィド)白金(II)、ジクロロビス(エチレンジアミン)白金(II)、トリクロロ(エチレン)白金(II)酸塩(例えばトリクロロ(エチレン)白金(II)酸カリウム)、ヘキサヒドロキシ白金(IV)酸塩(例えばヘキサヒドロキシ白金(IV)酸ナトリウム)、cis−ビス(アセトニトリル)ジクロロ白金(II)、cis−ジアンミンテトラクロロ白金(IV)、cis−ジクロロビス(ピリジン)白金(II)、cis−ジアンミン白金(II)ジクロリド、cis−ジクロロビス(ジエチルスルフィド)白金(II)、cis−ジクロロビス(トリエチルホスフィン)白金(II)、cis−ジクロロビス(トリフェニルホスフィン)白金(II)、cis−ビス(ベンゾニトリル)ジクロロ白金(II)、trans−ジクロロビス(トリフェニルホスフィン)白金(II)、trans−ジクロロビス(トリエチルホスフィン)白金(II)、エチレンビス(トリフェニルホスフィン)白金(0)、ジアミンジニトリト白金(II)、白金(0)−1,3−ジビニル−1,1,3,3−テトラメチルジシロキサン錯体、白金(0)−2,4,6,8−テトラメチル−2,4,6,8−テトラビニルシクロテトラシロキサン錯体、白金(II)アセチルアセトナート、硝酸テトラアンミン白金(II)、ジクロロ(1,2−ジアミノシクロヘキサン)白金(II)、ジクロロ(1,5−シクロオクタジエン)白金(II)、ジクロロ(ジシクロペンタジエニル)白金(II)、テトラアンミン白金(II)クロリド、テトラキス(トリフェニルホスフィン)白金(0)、トリメチル(メチルシクロペンタジエニル)白金(IV)、ビス(トリ−tert−ブチルホスフィン)白金(0)、ヘキサクロロ白金(IV)酸塩(例えばヘキサクロロ白金(IV)酸カリウム、ヘキサクロロ白金(IV)酸テトラブチルアンモニウム)、ヨウ化白金、塩化白金、などが挙げられる。
【0073】
好ましいパラジウム錯体としては、例えば、塩化パラジウム、クロロ(1,5−シクロオクタジエン)メチルパラジウム(II)、ジクロロ(1,5−シクロオクタジエン)パラジウム(II)、ビシクロ([2.2.1]ヘプタ−2,5−ジエン)ジクロロパラジウム(II)、(エチレンジアミン)パラジウム(II)クロリドなどが挙げられる。
【0074】
前記リン配位子を有する炭素材料と金属錯体とを反応させる条件についても特に制限されない。好ましくは、前記リン配位子を有する炭素材料と金属錯体とを窒素雰囲気中で還流しながら反応させる。反応温度は、例えば0〜150℃であり、好ましくは20〜100℃である。反応時間は、例えば、0.5〜72時間であり、好ましくは1〜12時間である。
【0075】
金属錯体の使用量も特に制限されないが、好ましくは、リン配位子1モルに対して0.001〜10モルであり、より好ましくは0.01〜5モルである。上記範囲であれば、反応が効率的に進行しうる。反応後、濾過、洗浄し、真空加熱乾燥して、金属担持炭素材料を得ることができる。導入されたリン配位子に対する金属錯体の導入率は、誘導結合プラズマ(ICP)発光分光分析と31P−NMR測定から見積もることができる。
【0076】
<水素吸蔵材料>
上述の金属担持炭素材料は、高い水素吸蔵能を有し、100℃以下の温度で水素の吸蔵、放出が可能である。また、水素の吸蔵、放出に化学反応を伴わないため、耐久性に優れる。そのため、特に燃料電池自動車用の水素吸蔵材料に好適に用いられうる。
【実施例】
【0077】
本発明の作用効果を以下の実施例を用いて説明する。ただし、本発明の技術的範囲が以下の実施例のみに制限されるわけではない。
【0078】
1.ミクロポーラス炭素材料へのアミジン構造の導入
1−1.試料の調製
<参考例1:NaY−PFA−P7(1)−H9(3)>
乾燥したゼオライト(NaY5.5)にフルフリルアルコール(FA)を含浸した。これを、150℃で8時間熱処理してFAを重合させ、PFA/ゼオライト複合体とした。これをN2雰囲気下5℃/minで850℃まで昇温し、次いで700℃で1時間プロピレンCVDを行った。その後N2雰囲気下5℃/minで900℃まで昇温して3時間保持し、炭素/ゼオライト複合体を調製した。最後に、この複合体を47wt%のフッ素水素酸100mlに投入後、5時間攪拌してフッ化水素酸処理し、鋳型であるゼオライトを溶解除去してミクロポーラス炭素材料(MPC)を取り出した。こうして得た試料をNaY−PFA−P7(1)−H9(3)とした。
【0079】
<合成例1>
下記反応式(1)、(2)にしたがってエッジ修飾ミクロポーラス炭素材料を調製した。
【0080】
【化5】
【0081】
具体的には、PTFE製4口フラスコ(500ml)に46wt%フッ化水素酸(200g)を加え、溶解している酸素ガスを取り除くために事前に窒素ガスを100cc/分で30分間以上バブリングさせた。続いて水溶性アゾ重合開始剤であるVA−044を反応溶液の全重量に対して0.15wt%になるように加え、オイルバスの設定温度を70℃にして加熱を開始させた。フラスコ内のフッ化水素酸の温度が50±2℃となった時点で、上記参考例1で調製した炭素/ゼオライト複合体(0.47g)を加え、一定時間攪拌した。炭素/ゼオライト複合体を加えた後約1時間で、フラスコ内のフッ化水素酸の温度は60±2℃になった。反応時間は、フッ化水素酸の温度が60℃に達してから6時間とした。
【0082】
反応後、反応溶液を濾過して生成物を500mlの蒸留水で良く洗い、灰分が残らないようにするためにさらに蒸留水(500g)で30分間攪拌した。その後、濾過して蒸留水で洗浄して、エッジ修飾ミクロポーラス炭素材料を得た。収量は0.11gであった。
【0083】
<合成例2>
VA−044を反応溶液の全重量に対して0.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0084】
<合成例3>
VA−044を反応溶液の全重量に対して2.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0085】
<合成例4>
VA−044を反応溶液の全重量に対して2.50wt%になるように加え、フッ化水素酸の温度が60℃に達してから1時間反応させたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0086】
<合成例5>
15か月前に調製した炭素/ゼオライト複合体を用いたこと、およびVA−044を反応溶液の全重量に対して2.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0087】
1−2.評価
合成例1〜5及び参考例1で合成したミクロポーラス炭素材料の粉末X線回折測定は、島津製作所製XRD−6100を用いて行い、線源はCu−Kα、電圧30kV、電流20mAで行った。BET表面積の測定は、日本ベル製BELSORP miniを用いて行い、−196℃の温度で、多点法で行った。水蒸気吸着等温線は、日本ベル製BELSORP MAXを用いて25℃の温度で測定した。
【0088】
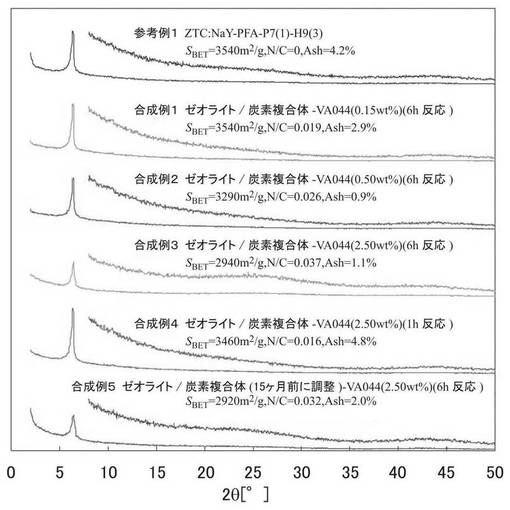
合成例1〜5及び参考例1で合成したミクロポーラス炭素材料のBET表面積SBET、窒素含有量(N/C)、窒素吸脱着測定結果から算出した試料の細孔構造を表1、図3に示す。ここで、VTotal、Vmicro、およびVmesoは、それぞれ、全細孔の占める容積、ミクロ細孔の占める容積、およびメソ細孔の占める容積を表す。長周期規則構造の有無を示す粉末X線回折装置(XRD)を用いて測定した結果を図3にまとめた。
【0089】
【表1】
【0090】
表1、図3の結果から、VA−044の仕込み濃度を増加させるほど、また反応時間を1時間から6時間に長くすると、N/C比が増加し、アミジン構造を有する官能基が多く導入されていることがわかる。また、アミジン構造を有する官能基が多く導入されるほど、BET表面積が低下する傾向にあり、合成例3では参考例1に比べて17%減少している。しかしながらアミジン構造を有する官能基を導入した場合であっても2000m2/g以上の十分大きいBET表面積を維持することが分かった。
【0091】
図3に示すように、合成例1〜5の試料のX線回折パターンは2θ=6°付近の長周期規則構造を示すピークが明確に観察され、また2θ=20〜30°に炭素網面の積層に由来するピークの強度が小さかった。このことから、得られた試料では、ミクロポーラス炭素材料(MPC)の規則構造が保持されていることが確認された。
【0092】
ZTCの質量、H/C比からZTCのエッジの割合を求め、エッジがすべてラジカルであると仮定しても、エッジのラジカルを全て反応させるためにはVA−044の濃度が0.15wt%で充分であると考えられるが、合成例1〜3を比較すると、VA−044の濃度が高くなるにつれてN/Cが増加している。さらに、同じ濃度で反応時間の異なる合成例3と合成例4を比較すると、反応時間が長いほどN/Cが増加していることがわかる。これは、VA−044が過剰である場合や反応時間が長い場合、VA−044由来のラジカルとZTCのラジカルサイトとの反応だけではなく、ZTCのエッジの水素がVA−044由来のラジカルに引き抜かれ、その結果ZTCに生成したラジカルサイトが、VA−044由来のラジカルと再結合する機構も関与しているものと考えられる。
【0093】
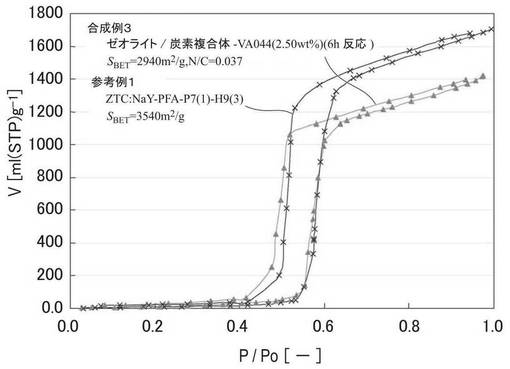
図4に、参考例1および合成例3の水蒸気吸脱着等温線を示す。細孔径が同等の場合、吸着量が急激に増加する相対圧が小さいほど材料表面は親水性である。参考例1および合成例3の水蒸気吸着等温線を比較すると、立ち上がりはいずれも相対圧0.5付近であり、ほぼ同じである。これは、親水性官能基を導入した効果に加えて、表1の元素分析結果から示されるように、試料に一定量含まれる含酸素官能基の影響があるためと考えられる。
【0094】
2.エッジ修飾ミクロポーラス炭素材料への原子オーダーの白金の導入
2−1.試料の調製
<実施例6>
(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)
下記式にしたがって反応を行った。
【0095】
【化6】
【0096】
合成例3と同様の手順で、アミジン構造を導入したZTCを調製した。
【0097】
アミジン構造を導入したZTC(ZTC−VA044)780mgとスターラーバーとを外径35mmの直線状のフラスコに加え、150℃で6時間真空加熱乾燥後、室温まで放冷した。続いてスターラーを攪拌させながら2.62wt%トリエチルアミン/THF溶液をシリンジを用いてフラスコ内に50ml(トリエチルアミン1.13g、11.20mmol)注入して真空含浸させた。その後、恒温槽を用いて0℃のウォーターバス(エチレングリコールを約15重量%加える)でフラスコを冷却した。30分後、大気圧までフラスコ内に窒素をパージし、フラスコ内に空気が入らないように窒素を流しながらクロロジフェニルホスフィン(アルドリッチ社製)2.26g(10.25mmol)をシリンジを用いてフラスコ内に加えて10分間攪拌した。その後、ウォーターバスの温度を25℃に設定し、25℃に達してから15時間反応させた。その後、反応溶液にTHF80mlを加えて数分間攪拌してからメンブレンフィルター(0.1μm、ADVANTEC社製T010A047A)で濾過し、さらに500mlのTHFでよく洗浄した。次いで試料を2500mlの蒸留水中で30分間攪拌して良く洗浄し、メンブレンフィルター(0.1μm、ADVANTEC社製H010A047A)で濾過した後、試料を150℃で真空乾燥させた。収量は0.87gであった。この試料をZTC−VA044−PPh2と表す。
【0098】
この試料の元素分析結果を下記表2に示す。ZTC−VA044からZTC−VA044−PPh2への反応前後の試料の重量変化から計算したZTC−VA044−PPh2のN/Cは、元素分析によるN/Cの値とよく一致した。
【0099】
また、誘導結合プラズマ(ICP)発光分光分析法により測定したリンの含有量は、2.53wt%であった。窒素とリンの含有量から算出した結果、82.76%のアミジン構造を有する官能基にリン配位子が導入されたことがわかった。ここで、リンの含有量は、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて測定した値を用いた。
【0100】
【表2】
【0101】
(リン配位子を有するエッジ修飾ミクロポーラス炭素材料への白金錯体の導入)
下記式にしたがって反応を行った。
【0102】
【化7】
【0103】
上記で調製したZTC−VA044−PPh20.30gを、150℃で6時間真空加熱乾燥した後、室温まで放冷してスターラーを攪拌させながらシリンジを用いてフラスコ内に1.56重量%(1,5−シクロオクタジエン)ジメチル白金(II)錯体/アセトニトリル溶液12mlを注入して真空含浸させ、10分間ほど攪拌した後に大気圧に達するまでフラスコ内に窒素をパージさせた。続いてフラスコに冷却器を取り付けて100℃に加熱したオイルバスで12時間還流させた。還流後、反応溶液を室温まで冷却してから反応溶液をメンブレンフィルター(0.1μm、ADVANTEC社製H010A047A)で濾過し、100mlのアセトニトリル、続いて100mlのジエチルエーテルで洗浄してから真空加熱乾燥した(100℃、6時間)。この試料をZTC−VA044−PPh2−PtMe2と表す。収量は0.37gであった。
【0104】
実施例6で調製した試料であるZTC−VA044−PPh2−PtMe2に含まれるリンおよび白金の定量分析を、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて行なった。試料中に含まれるリンは1.62wt%であり、白金は11.1wt%であった。
【0105】
また、窒素吸脱着測定によって算出したZTC−VA044−PPh2およびZTC−VA044−PPh2−PtMe2のBET表面積は、それぞれ1910m2/gおよび1550g/m2であった。
【0106】
<実施例7>
(ミクロポーラス炭素材料へのリン配位子の導入反応)
アミジン構造を導入したZTCに代えて、参考例1と同様の手順で調製したミクロポーラス炭素材料を用いたことを除いては、実施例6(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)と同様の手順でリン配位子を有する炭素材料を得た。この試料をZTC−PPh2と表す。
【0107】
さらに、ZTC−VA044−PPh2に代えて、上記で調製したZTC−PPh2を用いたことを除いては、実施例6(リン配位子を有するエッジ修飾ミクロポーラス炭素材料への白金錯体の導入)と同様の手順でリン配位子を有する炭素材料を得た。この試料をZTC−PPh2−PtMe2と表す。
【0108】
実施例7で調製した試料であるZTC−PPh2−PtMe2に含まれるリンおよび白金の定量分析を、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて行なった。試料中に含まれるリンは1.48wt%であり、白金は10.3wt%であった。
【0109】
また、窒素吸脱着測定によって算出したZTC−PPh2およびZTC−PPh2−PtMe2のBET表面積は、それぞれ1930m2/gおよび1860g/m2であった。
【0110】
<比較例1>
参考例1と同様の手順で、ミクロポーラス炭素材料(MPC)を得た。
【0111】
次に、Pt水溶液として、Ptを含む化合物であるジアンミンジニトロ白金[Pt(NO2)2(NH3)2]の、0.096wt%の水溶液A6.7mlと、還元剤水溶液である水素化ホウ素ナトリウムの水溶液B66.7mlとを調製し、溶液A、Bを0℃に冷却した。溶液A,Bの濃度は白金の担持量が得られたMPCに対し3.8wt%になるよう計算した。次に、100mgのMPCを0℃の溶液Aに投入し、減圧雰囲気下0℃で30分攪拌した。次に、これを遠心分離して0℃の溶液Bと混合し、0℃で10分間攪拌することによりPtを還元して白金ナノ粒子を生成させた。最後に、白金ナノ粒子を担持させたMPCを濾過して純水で洗浄する操作を数回繰り返した後、150℃で6時間真空乾燥させることにより、白金を担持させたMPCを得た。この試料を、Pt/ZTCと表す。
【0112】
<比較例2>
参考例1で調製した炭素材料を真空加熱乾燥した後、(1,5−シクロオクタジエン)ジメチル白金(II)錯体を注入して真空含浸させ、白金錯体を含浸させ、炭素材料ZTC−PtMe2CODを得た。この試料に含まれる白金の量は、13.3wt%であった。
【0113】
2−2.評価
実施例6で調製した試料について、XRD測定、水素吸脱着測定を行なった。XRD測定は上記1−2と同様の方法で行なった。
【0114】
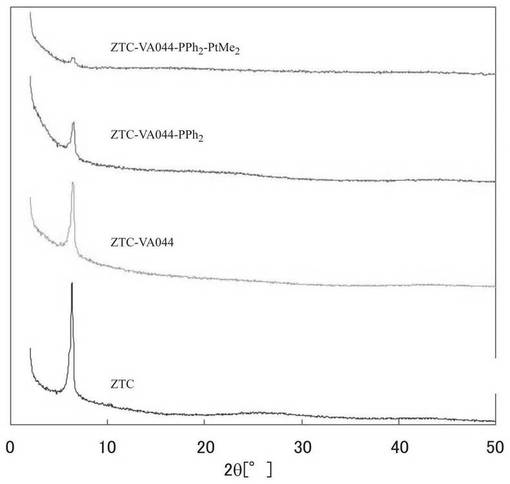
図5に、実施例6で調製した試料であるZTC−VA044−PPh2およびZTC−VA044−PPh2−PtMe2のX線回折パターンを示す。比較のために、アミジン構造を導入していないZTCおよびアミジン構造を導入した段階の試料のX線回折パターンを示した。図5に示すように、ZTC−VA044−PPh2、ZTC−VA044−PPh2−PtMe2のX線回折パターンは、参考例1のMPCのX線回折パターンと比較すると弱いものの、2Xで示す2θ=6°付近にピークを示し、また2θ=20〜30°のピーク強度は小さい。このことから、実施例6で得られた試料では、MPCの規則構造が保持されていることがわかる。また、0価の白金に由来するピークは確認されなかった。
【0115】
図6に、実施例6で得られたZTC−VA044−PPh2−PtMe2のXPSスペクトルを示す。72.3eV付近および75.9eV付近に2価の白金に由来するピークが観察された。図5、図6の結果から、2価の白金がZTCに担持されていることが確認された。なお、XPSスペクトルの測定は、KRATOS社製ESCA−3400を用い、線源はMg−Kα、電圧10kV、電流10mAで行なった。
【0116】
図7、図8に、実施例6で得られた試料の固体31P−NMRスペクトルを示す。Pt錯体を担持していないZTC−VA044−PPh2の固体31P−NMRスペクトル(図8)は21.08ppmに1本のピークを示すのに対して、ZTC−VA044−PPh2−PtMe2ではピークが2つに分裂し、試料に含まれるリン配位子の一部にPtが錯体として担持されていることがわかる。2つのピークの積分比は、3.1(48.79ppm):84.7(21.39ppm)であった。この積分比と、試料に含まれる白金の量(11.1wt%)から、リン配位子に配位している白金の割合は、試料に含まれる白金の量を基準として、3.2%と見積もることができる。
【0117】
一方、図8に示すように、実施例7で得られた試料の固体31P−NMRスペクトルを実施例6の試料と比較すると、ZTC−PPh2は、ZTC−VA044−PPh2と異なる位置にピークトップを示すことがわかる。これは、ZTC−PPh2では、導入したリン配位子のリン原子が結合した原子が両者において異なることを示唆している。ZTC−VA044−PPh2では、ZTC合成の最後のフッ化水素酸処理によるゼオライト除去の際にラジカルを反応させることにより、ZTC表面において含酸素官能基の生成が抑制される。そのため、ZTC−VA044には含酸素官能基の量が少なく、アミジンはクロロジフェニルホスフィンと反応したと考えられる。しかしながら、ZTC−PPh2とクロロジフェニルホスフィンとの反応では、ZTCの表面の含酸素官能基とクロロジフェニルホスフィンとが反応したものと考えられる。白金錯体を担持後のZTC−PPh2−PtMe2ではピークがわずかに高磁場側にシフトした。
【0118】
得られた試料の水素吸蔵能を評価するために、実施例6の試料について、ジーベルツ法(容量法、JIS H 7201)に従って圧力−組成等温線(PCT線)を得た。水素吸蔵能は、国立標準規格技術研究所(National Institute of Standards and Technology:NIST)で定められている圧縮係数を用いて測定した。測定精度はサンプルの充填量に依存する。最低でも1g以上を充填し、必要に応じて上記合成スキームを繰り返し行い、所要量を準備した。試料を秤量して測定用耐圧試料管に入れ、100℃で4時間真空引きして試料管内に残留しているガスを放出させて、水素が吸蔵されていない原点を得た後測定した。測定温度は25℃、70℃、100℃とした。測定順序は、新しい試料で25℃で測定後に引き続き70℃で測定した。その後、新しい試料で100℃で測定した。水素吸蔵能の測定後、大気圧まで減圧して水素放出量の確認を行った。測定結果を図9に示す。図9中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。
【0119】
図9の結果から、実施例6で調製したZTC−VA044−PPh2−PtMe2の水素吸着量は、温度の上昇に伴って水素吸着量が大幅に増加することがわかった。図9中、H2/Ptは、各測定点における吸着した水素分子と担持された白金とのモル比である。図9に示すように、実施例6の試料ではH2/Ptの値は最大で4.82と大きな値であり、水素分子が白金錯体に解離吸着しただけではなく、解離した水素原子が流れ出ている(スピルオーバー)ことが考えられる。
【0120】
実施例7の試料について、上記と同様に水素吸蔵能を用いて測定した。試料を100℃で6時間真空引きして試料管内に残留しているガスを放出させて、水素が吸蔵されていない原点を得た後測定した。測定温度は25℃、70℃、100℃とした。測定順序は、新しい試料で25℃で測定後に引き続き70℃で、その後100℃で測定した(図10上)。その後、新しい試料で100℃で測定した(図10下、図10中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである)。図10に示すように、実施例7の試料ではH2/Ptの値は最大で4.65の値が得られた。したがって、エッジ修飾していない炭素材料を用いた場合も、アミジンでエッジ修飾した炭素材料を用いた場合と同様にリン配位子に金属錯体を担持させることができ、優れた水素吸着特性が得られることが確認された。
【0121】
比較例2の試料について、上記実施例6の試料と同様の手法で25℃、70℃、100℃で水素吸蔵能の測定を行なった。上記実施例6の試料と同様、水素吸着量は温度の上昇に伴って増加した(図示せず)。吸着量の最大値におけるH2/Ptの値は1.67であり、白金錯体1分子に対して水素1.7分子が吸着し、解離しているものと考えられる。
【0122】
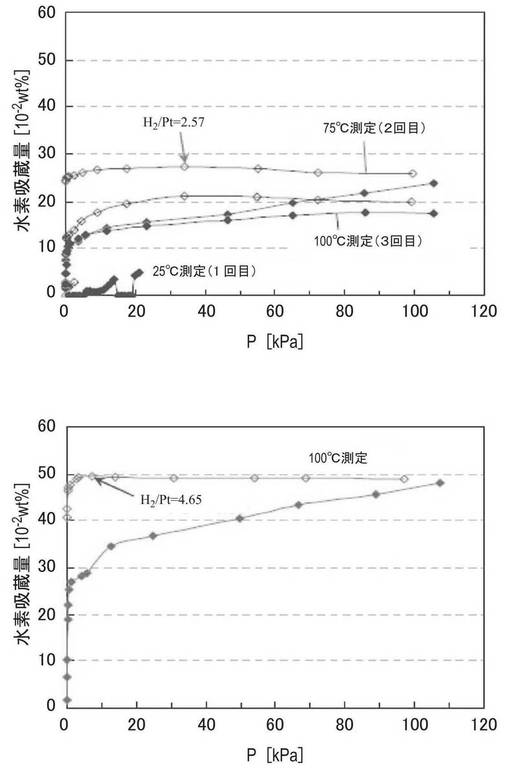
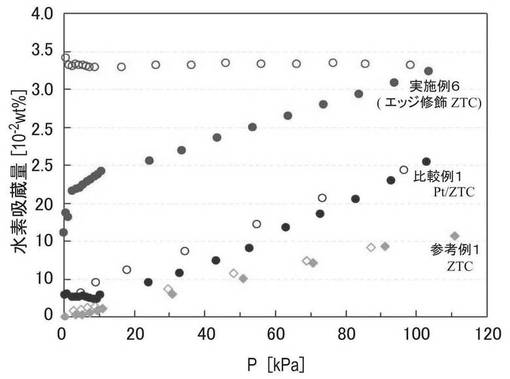
図11は、実施例6で調製したZTC−VA044−PPh2−PtMe2、参考例1のZTC、比較例1のPt/ZTCについて、25℃で測定した、試料の重量あたりの水素吸着量を示す図である。図11中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。測定条件は、温度条件以外は実施例6と同様である。比較のために、温度25℃で測定した実施例6の試料の結果を併せて示した。図11の結果から、実施例6の試料は、参考例1、比較例1の試料と比較して、試料の重量当たりの水素吸着量が改善されていることがわかる。
【0123】
さらに、実施例6、実施例7の試料では温度の上昇に伴って水素吸着量が増加した(図9、図10)が、参考例1、比較例1の試料では、高温では水素吸着量が減少する傾向にあった(図示せず)。
【0124】
このように、炭素材料に金属錯体を担持させた場合、金属を含まない場合や、粒子状の金属を担持させた場合に比べて水素吸蔵能が大幅に改善されることが明らかになった。
【0125】
3.エッジ修飾ミクロポーラス炭素材料への原子オーダーのパラジウムの導入
3−1.試料の調製
<実施例8>
(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)
実施例6と同様の手順で、アミジン構造を導入したZTC(ZTC−VA044)にリン配位子であるクロロジフェニルホスフィンを導入し、ZTC−VA044−PPh2を調製した。
【0126】
(リン配位子を有するエッジ修飾ミクロポーラス炭素材料へのパラジウム錯体の導入)
下記式にしたがって反応を行った。
【0127】
【化8】
【0128】
上記で調製したZTC−VA044−PPh219.8mgを、150℃で一晩真空加熱乾燥した後、塩化パラジウム/アセトニトリル溶液15mlを真空含浸させ、80℃で3時間攪拌した。ここで、塩化パラジウム/アセトニトリル溶液は、窒素雰囲気下で塩化パラジウム25.3mgにアセトニトリル20mlを加え、約80℃に加熱して塩化パラジウムを溶解させて調製したものを用いた。その後、メンブレンフィルターで反応溶液を濾過し、200mlのアセトニトリルで洗浄後、さらに200mlのTHFで洗浄してから150℃で6時間真空加熱乾燥させた。この試料をZTC−VA044−PPh2−PdCl2と表す。収量は17.4mgであった。
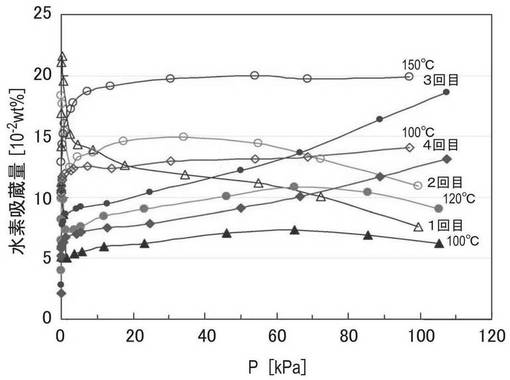
【0129】
3−2.評価
図12に、上記で作製したZTC−VA044−PPh2−PdCl2のXRD測定結果を示す。装置、測定条件は上述のものと同様である。図12に示されるように、2θ=6°付近の長周期規則構造を示すピークが観察され、MPCの規則構造が保持されていることがわかる。また、2θ=40°付近に0価のPd粒子に由来するピークが観察された。
【0130】
図13に、上記で作製したZTC−VA044−PPh2−PdCl2のXPS測定結果を示す。装置、測定条件は上述のものと同様である。340.4eVおよび335.2eVに0価のパラジウムに由来するピークが観察される。一方、337.5eV付近に2価のパラジウムに由来するピークが観察され、これは、リン配位子に配位した2価のPd錯体のほかに、配位していないPdCl2によるものと考えられる。
【0131】
図14に、上記で作製したZTC−VA044−PPh2−PdCl2の水素吸脱着測定結果を示す。測定温度は、100℃、120℃、140℃であり、測定順序は、1回目:100℃、2回目:120℃、3回目:150℃、4回目:100℃の順で行なった。図14中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。
温度の上昇に伴って吸着量が増加し、吸着量の最大値は約0.2wt%であることがわかる。したがって、Pd錯体を担持したZTCにおいても、Pt錯体を担持した場合と同様に高い水素吸蔵能が得られることが明らかになった。
【0132】
また、温度の上昇に伴って吸着量が増加することや、特に3回目(150℃)や4回目(100℃)の測定で水素の脱着も確認できることから、水素の吸着に主に寄与しているのは0価のPd粒子ではなく、2価のPd錯体であると考えられる。
【0133】
さらに、4回測定しても試料の劣化が生じにくく、これはPd錯体が熱安定性や寿命が良好であるためと考えられる。
【符号の説明】
【0134】
1 ゼオライト、
1a、2a ミクロ孔、
2 ゼオライト炭素(ゼオライト鋳型カーボン)、
3 複合体。
【技術分野】
【0001】
本発明は、炭素材料、およびこれを利用した金属担持炭素材料に関する。より詳細には、燃料電池用の水素吸蔵材料や電極触媒に適用して好適な炭素材料、およびこれを利用した金属担持炭素材料、ならびにその製造方法に関する。
【背景技術】
【0002】
多量の水素の貯蔵は燃料電池自動車の実用化に必要不可欠な技術であり、これまでに水素吸蔵合金、化学水素化物、吸着系材料などの水素吸蔵材料の研究が活発に行なわれてきた。水素吸蔵合金および化学水素化物は、吸着系材料と比較して貯蔵量が大きく、水素吸蔵能が5重量%を超えるものが得られているものの、水素の放出に加熱が必要であること、寿命が短いことなどの問題がある。一方、吸着系材料としては、例えば活性炭やカーボンナノチューブをはじめとする炭素材料が挙げられる。吸着系材料では、物理吸着を利用するために水素の吸蔵・放出過程で加熱は不要であるが、水素吸蔵合金や化学水素化物と比較して吸着量が少ないという問題がある。そこで近年、物理吸着に加えてスピルオーバーを利用した水素貯蔵方式が注目されている。
【0003】
スピルオーバーとは、固体表面上に白金などの金属を担持すると、気相中の水素分子が金属の作用により金属表面上で水素原子に解離し、固体表面上に流出する現象である。流出した水素原子が固体表面に貯蔵されるため、水素を分子のまま吸着させる物理吸着と組み合わせて水素吸蔵能を向上させることができると考えられる。例えば、非特許文献1では、高表面積の活性炭に白金ナノ粒子を担持させると、常温における水素吸蔵能が担持前に比べて大幅に増大することが報告されており、これは、物理吸着に加えてスピルオーバーの効果によるものとされている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】J.Phys.Chem.C,111(2007)11086.
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、燃料電池自動車の実用化のためには、より多量の水素を貯蔵できる水素吸蔵材料が求められている。また、固体表面に担持させる金属は、主に白金などの高価な金属であり、スピルオーバーに寄与するのは金属粒子の表面部分だけであるため、同じ量の金属を担持するならば、粒子径を小さくして粒子の表面積をできるだけ大きくすることが不可欠である。しかしながら、金属粒子の微粒子化には限界がある。
【0006】
そこで本発明は、固体表面上に金属を高分散で担持させるための炭素材料、およびこれに金属を担持させた金属担持材料を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記の課題に鑑み、鋭意研究を積み重ねた。その過程で、金属を粒子としてではなく、金属錯体として炭素材料の表面に担持させることで、金属を酸化させることなく原子レベルで担持させることを検討した。そして、炭素材料の表面に配位子を導入することで、金属を錯体として容易に導入できることを見出し、本発明を完成させた。
【0008】
すなわち本発明は、細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【発明の効果】
【0009】
本発明によれば、炭素材料に導入されたリン配位子部分に金属錯体を導入することができ、したがって金属を原子レベルで炭素材料に分散させて担持させることが可能になる。そのため、水素のスピルオーバーの効率が高まり、水素貯蔵効率が向上した水素吸蔵材料が得られうる。
【図面の簡単な説明】
【0010】
【図1】(a)炭素材料のエッジ部分、(b)炭素材料に配位子が導入された炭素材料、(c)金属原子が配位子を介して導入された金属担持炭素材料をそれぞれ表す模式図である。
【図2】ミクロポーラス炭素材料を表す模式図である。
【図3】合成例1〜5で製造した試料のXRD測定の結果を表す図である。
【図4】合成例3で製造した試料の水蒸気吸脱着等温線を表す図である。
【図5】実施例6で製造した試料のXRD測定の結果を表す図である。
【図6】実施例6で製造した試料のXPS測定の結果を表す図である。
【図7】実施例6で製造した試料の固体31P−NMR測定の結果を表す図である。
【図8】実施例7で製造した試料の固体31P−NMR測定の結果を表す図である。
【図9】実施例6で製造した試料の水素吸脱着等温線を表す図である。
【図10】実施例7で製造した試料の水素吸脱着等温線を表す図である。
【図11】実施例6で製造した試料の水素吸脱着等温線を表す図である。
【図12】実施例8で製造した試料のXRD測定の結果を表す図である。
【図13】実施例8で製造した試料のXPS測定の結果を表す図である。
【図14】実施例8で製造した試料の水素吸脱着等温線を表す図である。
【発明を実施するための形態】
【0011】
以下、添付した図面を参照しながら、本発明の実施形態を説明する。なお、図面の説明において同一の要素には同一の符号を付し、重複する説明を省略する。また、図面の寸法比率は、説明の都合上誇張されており、実際の比率とは異なる場合がある。
【0012】
<リン配位子を有する炭素材料>
本発明の一実施形態は、細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料である。
【0013】
本発明の一実施形態によるリン配位子を有する炭素材料は、図1(b)に示すように、炭素材料の表面、好ましくは前記炭素材料の表面のエッジ部分に、リン配位子が結合してなる。ここで炭素材料のエッジは、例えば図1(a)に示すようにグラフェンシートの構造における炭素縮合環の端部が配列した面である。
【0014】
本実施形態による炭素材料は、炭素材料の表面の少なくとも一部にリン配位子が導入されてなる。好ましくは、前記炭素材料の表面のエッジ部分の少なくとも一部にリン配位子が導入されてなる。リン配位子を導入することによって、金属錯体をリン配位子に安定に配位させることができる。そのため、金属を原子レベルで炭素材料に担持させることが可能になる。
【0015】
この際、用いられるリン配位子としては、特に制限されないが、好ましくはPPh1Ph2(Ph1およびPh1は同一であっても異なっていてもよく、置換または非置換のフェニル基である)で表されるリン配位子が好ましく用いられうる。Ph1またはPh2の置換基としては、特に制限されないが、例えば炭素数1〜8のアルキル基、アルコキシ基、ハロゲン化アルキル基である。
【0016】
Pはd軌道が大きいため、8〜10属の金属と配位しやすい。またPh1およびPh2が置換または非置換のフェニル基であればZTCの細孔に入る大きさであるため、反応に有利である。
【0017】
前記リン配位子の例としては、特に制限されないが、ジフェニルホスフィン、ビス(3,5−ジメチルフェニル)ホスフィン、ビス(4−メトキシフェニル)ホスフィン、ビス[4−(トリフルオロメチル)フェニル]ホスフィン、ジ(p−トリル)ホスフィン、ビス(3,5−ジ−tert−ブチル−4−メトキシフェニル)ホスフィン、ビス[3,5−ビス(トリフルオロメチル)フェニル]ホスフィン、などが挙げられる。
【0018】
炭素材料としては、上記のように細孔を有し、エッジを有するものであれば特に制限はなく、カーボンブラック、活性炭、コークス、天然黒鉛、人造黒鉛などからなるカーボン粒子などが用いられうるが、特にゼオライト鋳型カーボン(ZTC)のようなミクロポーラス炭素材料が好ましく用いられうる。ZTCは、球面状の細いグラフェンシートが3次元状に規則的につながっており、エッジの炭素原子の数が相対的に多い。さらに、超高表面積であるため、水素吸着特性に優れる。
【0019】
図2に、ミクロポーラス炭素材料としてのゼオライト鋳型カーボンの一例を模式的に示す。ゼオライト鋳型カーボン2は、ゼオライト1を鋳型として得られたゼオライト炭素2である。より詳細には、ゼオライト鋳型カーボン2の作製には、まず、図2(a)に示すゼオライト1のミクロ孔1aに炭素源である有機化合物を導入した後に加熱処理して図2(b)に示すゼオライト1とゼオライト炭素2との複合体3を調製する。その後にゼオライト1のみを除去することによって、ゼオライト鋳型カーボン2が得られる(図2(c))。ゼオライト鋳型カーボン2は、鋳型として用いたゼオライト1の構造的特徴が反映された、3次元の長周期規則構造と内部にミクロ細孔2aとを有する。
【0020】
ゼオライト鋳型カーボン2は、その製造にあたり、使用する鋳型材である特定の3次元規則構造を有するゼオライト1が備える構造的特徴を反映した多孔性炭素材料である。ゼオライト鋳型カーボン2は、直径が0.1〜2nmの範囲内にある細孔(ミクロ細孔2a)が網目状に連結した構造を有する。具体的には、ゼオライト鋳型カーボン2は、0.5〜100nmの範囲内の3次元長周期規則構造を有すると共に、ミクロ細孔2aを有する。より具体的には、ゼオライト鋳型カーボン2は、3次元長周期規則構造を構成する炭素鎖と炭素鎖の間の距離が、好ましくは0.5〜100nmであり、より好ましくは0.7〜50nmであり、さらに好ましくは0.7〜2nmである。このように、ゼオライト鋳型カーボン2は、炭素鎖と炭素鎖の間の距離が任意の間隔で3次元的に長周期にわたって規則的に繰り返した構造を有する炭素材料である。なお、IUPAC(国際純正及び応用化学連合)では、直径2nm以下の細孔をミクロ細孔(micropore)、直径2〜50nmの細孔をメソ細孔(mesopore)、直径50nm以上の細孔をマクロ細孔(macropore)と定義している。ミクロ細孔を有する物質を総称してミクロ(マイクロ)ポーラス材料と称している。
【0021】
本実施形態において、前記炭素材料は、エッジ部分の少なくとも一部に官能基が導入されてなるエッジ修飾炭素材料でありうる。好ましくは、前記炭素材料のエッジ部分の少なくとも一部にアミジン構造を有する官能基が導入されてなる炭素材料である。この際、前記アミジン構造を有する官能基に、リン配位子が結合される。
【0022】
本明細書中、アミジン構造は、1つの炭素原子に二重結合を介して1つの窒素原子が結合し、単結合を介して1つの窒素原子が結合した構造である。アミジン構造は強塩基で求核性を示すため、後述するようにアミジン構造部分にさらにリン配位子を導入し、金属錯体を導入することができる。本発明に用いられるアミジン構造を有する官能基としては、例えば、下記化学式1で表される官能基が挙げられる。
【0023】
【化1】
【0024】
式中、R1、R2およびR3は独立して、水素原子、置換もしくは非置換の炭素数1〜10のアルキル基または置換もしくは非置換の炭素数3〜6のシクロアルキル基であり、R1とR2とが結合して環を形成してもよく、R2とR3とが結合して環を形成してもよい。
【0025】
アルキル基は、直鎖であっても、分岐であってもよい。アルキル基またはシクロアルキル基は、置換であっても非置換であってもよい。アルキル基の有する炭素数は、特に限定されないが、好ましくは1〜10個、より好ましくは1〜5個である。シクロアルキル基の有する炭素数は、特に限定されないが、好ましくは3〜6個である。アルキル基またはシクロアルキル基の置換基も特に制限されないが、例えば、水酸基、カルボキシル基、アミノ基、シアノ基、ハロゲン原子などが挙げられる。アルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、などが挙げられる。シクロアルキル基としては、シクロプロピル基、シクロペンチル基、シクロヘキシル基などが挙げられる。これら以外のアルキル基またはシクロアルキル基が用いられてもよい。R1とR2とが結合して環を形成する場合、または、R2とR3とが結合して環を形成する場合、当該環は好ましくは5〜6員環であり、より好ましくは5員環である。
【0026】
中でも、下記化学式で表される官能基が好適に用いられうる。
【0027】
【化2】
【0028】
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料、リン配位子を有する炭素材料、または後述する金属担持炭素材料は、そのBET表面積が1500m2/g以上であることが好ましい。例えばゼオライト鋳型カーボンを用いた炭素材料、または金属担持炭素材料は、3次元長周期規則構造とミクロ細孔とを有することによりBET表面積が大きい。一般に吸着材への適用に関しては、BET表面積が大きいことが好ましい。また、吸着する分子サイズにも影響されるが、ミクロ孔が存在することも重要であると考えられる。これに対して、メソ孔は前述した用途への適用に際してはあまり効果がないと考えられる。このため、所望の高い機能を発現させるためには、相対的にミクロ孔が多く存在することが重要であり、なるべくメソ孔は少ない方が良いと考えられる。この目安として、BET表面積は1500m2/g以上であることが好ましい。また、2000m2/g以上であることがより好ましく、さらには2500m2/g以上であることが好ましい。
【0029】
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料、リン配位子を有する炭素材料、または後述する金属担持炭素材料は、その構造的特徴として2次元積層規則性が少ないほど、吸着力が高くなる。例えば、粉末X線回折測定を行った場合には、得られるX線回折パターンは、2次元積層規則性を示す通常26°付近に現れる回折ピークはできるだけ少ないほうが好ましい。この26°付近に現れる回折ピークの存在は、無孔質の炭素層の増加を意味し、BET表面積の低下を意味する。
【0030】
また、上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料は、25℃において、水蒸気吸着等温線の傾きの極大値を示す相対圧(P/P0)が0.70以下であることが好ましい。上記範囲であれば、炭素材料の表面の親水性が高く、炭素材料に機能性を付与することができる。
【0031】
上記アミジン構造を有する官能基が導入されてなる炭素材料は親水性を有するため、水蒸気をはじめとする極性を有するガス、酸性ガス、含酸素炭化水素蒸気等の吸着剤として有用である。また、電気二重層キャパシタ(EDLC)の電極として応用した場合にも性能の向上が期待できる。一般に、EDLCの電解液は極性が大きく、電極表面との親和性(濡れ性)を考慮すると親水性を示す電極が望まれる。活性炭、ミクロポーラス炭素材料も含めて炭素材料は疎水性が強く、キャパシタ等へ応用する場合に親和性(濡れ性)を改変する。このため、EDLCの直接的な性能に関与しない二次的な表面改質処理を行うことがある。これに対し、上記アミジン構造を有する官能基が導入されてなる炭素材料は二次的な表面改質処理を行う必要がなく、そのまま用いることが可能となる。このため、よりすぐれた効果が期待できる。その他、水蒸気の吸脱着を利用する吸着式ヒートポンプにおいて、吸着剤として現在使用されているゼオライトと比べて大幅な性能向上が可能なため、装置の小型化等、性能向上が期待できる。
【0032】
(エッジ修飾炭素材料の製造方法)
上記アミジン構造を有する官能基が導入されてなるエッジ修飾炭素材料は、エッジを有する炭素材料(炭素材料前駆体)を準備する段階と、前記炭素材料を、アミジン構造を含むアゾ化合物と反応させて、前記炭素材料のエッジにアミジン構造を有する官能基を導入する段階と、を有する方法によって製造することができる。
【0033】
原料となる炭素材料(炭素材料前駆体)の入手経路については特に制限はない。商業的に入手可能な商品を用いてもよいし、自ら調製してもよい。以下、ゼオライト担持カーボンなどのミクロポーラス炭素材料を用いる場合を説明する。
【0034】
まず、上記した構造的な特徴を有するミクロポーラス炭素材料を得るためには、構造内部に空孔を有し、この空孔が網目状に連結した構造を有する多孔質材料を鋳型として用いる。そして、この多孔質材料の表面及びミクロ細孔内部に加熱条件下で有機化合物を導入し、加熱することによって有機化合物を炭化し、多孔質材料に炭素を堆積させる。有機化合物の炭化・炭素の堆積は、例えば化学気相成長(Chemical Vapor Deposition:CVD)法により行う。次に、鋳型である多孔質材料を除去する。この方法により、ミクロ細孔を有するミクロポーラス炭素材料を容易に製造することができる。ただし、後述するように、鋳型の除去はアミジン構造を有する官能基を導入する反応と同時に行うことができるので、多孔質材料に炭素を堆積させた複合体を原料として用いてアミジン構造を有するアゾ化合物と反応させ、エッジ修飾炭素材料を製造してもよい。
【0035】
鋳型として用いる多孔質材料は、ミクロ細孔内部に有機化合物が導入できること、CVD法の際に元の構造を安定に保つこと、生成したミクロポーラス炭素材料と分離できることが必要である。このため、例えば多孔質酸化物等の耐熱性に優れ、且つ、酸やアルカリで溶解する材料が望ましい。また、既に述べたように、ミクロポーラス炭素材料は鋳型の形態を転写した状態で合成される。このため、鋳型として用いる多孔質材料は、結晶(構造)が十分に発達し、粒子径の揃った構造及び組成が均一な材料であることが望ましい。多孔質材料の備えるべき材料物性と、得られるミクロポーラス炭素材料の物性を考慮すると、多孔質材料としてゼオライトを用いることが好ましい。ゼオライトは、シリカ構造のケイ素(Si)の一部がアルミニウム(Al)で置換されたアルミノケイ酸塩であり、骨格自体が負電荷を持つことから構造内にカチオンが分布した構造を有する。また、ゼオライトは、Si/Alモル比、カチオンの種類や量、及びカチオンに水和した水分子の数によって多様な結晶構造を有し、例えば細孔が2次元的に連結した構造や3次元的に連結した構造等の、多様な大きさの細孔を有する多孔質材料である。代表的なゼオライトとしては、ケージ又はスーパーケージといった空隙構造を有するものが挙げられ、ゼオライトの中でもFAU型ゼオライト、FAU型ゼオライトの中でもY型ゼオライトを用いることが望ましい。多孔質材料の除去は、生成したミクロポーラス炭素材料を分離できる方法であれば如何なる方法を用いても良い。例えば、ゼオライトは酸で溶解可能であり、例えば、塩酸やフッ化水素酸を用いることで容易に溶解できる。
【0036】
有機化合物を炭化して炭素を堆積するために用いるCVD法は、鋳型等の基板上に特定の元素又は元素組成からなる薄膜(例えば炭素からなる薄膜)を作る工業的手法である。通常、原料物質を含むガスに熱や光によってエネルギーを与えたり、高周波でプラズマ化することにより、化学反応や熱分解によって原料物質がラジカル化して反応性に富むようになり、基板上に原料物質が吸着して堆積することを利用する技術である。温度を上げて原料物質を堆積させるものを熱CVD法、化学反応や熱分解を促進させるために光を照射するものを光CVD法、ガスをプラズマ状態に励起する方法をプラズマCVD法と区別することもある。
【0037】
CVD法で用いる有機化合物は、常温で気体であるか、又は気化できるものが好ましい。気化の方法は、沸点以上に熱する方法や雰囲気を減圧にする方法等がある。用いる有機化合物は、当業者に知られた炭素源物質の中から適宜選択して使用できる。特に、加熱により熱分解する化合物が好ましい。例えば、CVD法で鋳型として用いる多孔質材料の骨格上(例えばシリカゲル骨格上)に炭素を堆積することができる化合物が好ましい。
【0038】
また、用いる有機化合物は、水素を含む有機化合物でも良い。この有機化合物は、不飽和又は飽和の有機化合物でも良く、これらの混合物でも良い。用いる有機化合物は、二重結合及び/又は三重結合を有する不飽和直鎖又は分枝鎖の炭化水素、飽和直鎖又は分枝鎖の炭化水素等が含まれて良く、飽和環式炭化水素や芳香族炭化水素等を含んでいても良い。有機化合物は、例えば、アセチレン、メチルアセチレン、エチレン、プロピレン、イソプレン、シクロプロパン、メタン、エタン、プロパン、ベンゼン、ビニル化合物、エチレンオキサイド等があげられる。中でも、用いる有機化合物は、多孔質のミクロ細孔内に入り込むことが可能なもの、例えばアセチレン、エチレン、メタン、エタン等を用いることが望ましい。有機化合物は、より高温でのCVDに用いるものと、より低温でCVDに用いるものとでは互いに同一のものであっても異なっていても良い。例えば、低温でのCVDではアセチレン、エチレン等を使用し、高温でのCVDにはプロピレン、イソプレン、ベンゼン等を使用しても良い。
【0039】
多孔質材料のミクロ細孔内部に有機化合物を導入する際は、多孔質材料を予め減圧にしても良く、系自体を減圧下にしても良い。多孔質材料は安定であるので、CVDにより炭素が堆積する方法であれば如何なる方法を用いても良い。通常は、多孔質材料の骨格上に有機化合物の化学反応又は熱分解で生成した炭素を堆積(又は吸着)させ、多孔質材料と炭素を含むミクロポーラス炭素材料からなる複合体を得る。CVDを行う際は、加熱温度は、使用する有機化合物によって適宜適切な温度を選択できる。通常は、400〜1500℃であることが好ましい。加熱温度は、450〜1100℃であることがより好ましく、500〜900℃が更に好ましい。また、550〜800℃であることがより好ましく、575〜750℃更には約600〜700℃の範囲内にすることが望ましい。加熱温度はCVD処理時間及び/又は反応系内の圧力に応じて適宜適切な温度を選択することもできる。CVDの処理時間は、十分に炭素堆積が得られる時間とすることが好ましく、使用する有機化合物や温度によって適宜適切な時間を選択できる。
【0040】
CVDは、減圧又は真空下、加圧下、若しくは不活性ガス雰囲気下で行うことができる。不活性ガス雰囲気下で行う場合には、不活性ガスとしては例えばN2ガス、ヘリウム、ネオン、アルゴン等があげられる。CVD法では、通常、気体状の有機化合物をキャリアガスと共に多孔質材料に接触させるように流通させながら加熱し、容易に気相で多孔質材料上に炭素を堆積させることができる。キャリアガスの種類、流速、流量及び加熱温度は使用する有機化合物や多孔質材料の種類によって適宜調節する。キャリアガスは、例えば上記の不活性ガス等があげられる。爆発限界を考慮して、酸素ガス又は水素ガスとの混合物等であっても良い。
【0041】
CVD法により多孔質材料のミクロ細孔内部に炭素を堆積させる条件として、ミクロ細孔中の炭素の充填量は10〜40wt%の範囲内であることが好ましい。また、炭素の充填量は多孔質材料の重量を基準として15〜30wt%の範囲内に制御することがより好ましい。炭素の充填量が10wt%以上であれば、炭素骨格形成に必要な量の炭素が導入されるため、安定な規則性構造が得られうる。炭素の充填量が40wt%以下であれば、必要以上の炭素が付着することなく、ミクロ細孔容積及びBET表面積が維持されうる。
【0042】
CVDによる炭素の堆積(吸着)後、多孔質材料とミクロポーラス炭素系材料の複合体を、CVD温度より高い温度で更に加熱しても良い。この加熱温度は、使用する有機化合物によって適宜選択できるが、通常は700〜1500℃である。加熱温度は、750〜1200℃であることが好ましく、800〜1100℃であることがより好ましい。また、825〜1000℃であることが好ましく、850〜950℃、更には約875〜925℃の範囲内にすることが好ましい。また、加熱温度は、加熱時間及び/又は反応系内の圧力に応じて適宜選択することもできる。また、加熱時間は生成物を分析し、その結果に基づいて十分な炭素堆積に要求される時間を設定することができる。
【0043】
また、多孔質材料とミクロポーラス炭素材料の複合体に更に有機化合物を導入して加熱し、更に炭素を堆積させても良い。この場合には、CVD法により得られたミクロポーラス炭素材料の構造がより安定する。炭化は、CVD法によって行っても良く、他の加熱方法で行っても良い。また、加熱温度はCVD温度より高温であっても良く、低温であっても良い。また、導入する有機化合物は、CVD法で導入した有機化合物と同じであっても良く、異なっていても良い。この操作は、複数回行っても構わない。
【0044】
多孔質材料の表面及びミクロ細孔内に有機化合物を導入してCVDを行う前に、有機化合物を含浸して炭化しても良い。含浸する有機化合物は、多孔質材料のミクロ細孔径より小さな分子サイズを有する有機化合物であれば使用できる。具体的には、有機化合物は、炭化歩留まりの高いフルフリルアルコール等の熱重合性モノマーを用いることが好ましい。有機化合物の含浸方法は、モノマーが液体であればそのまま、固体であれば溶媒に溶解して多孔質材料と接触させる等、公知の手段を採用することができる。なお、多孔質材料の表面に残った過剰なモノマーは、予め洗浄等で除去することが好ましい。例えば、多孔質材料を室温減圧下でフルフリルアルコールと接触させた後、混合物を大気圧に戻すことにより、多孔質材料のミクロ細孔内にフルフリルアルコールを導入することができる。また、多孔質材料の表面に付着した余分なアルコールは、有機溶剤による洗浄で除去できる。
【0045】
用いる有機化合物は、多孔質材料のミクロ細孔内に挿入可能な大きさを有し、且つ、炭化時に炭素としてミクロ細孔内に残留するものであれば特に制限は無く用いることができる。例えば、有機化合物として、酢酸ビニル・アクリロニトリル・塩化ビニル等のビニル化合物、塩化ビニリデン・メタクリル酸メチル等のビニリデン化合物、無水マレイン酸等のビニレン化合物、エチレンオキサイド等のエポキシ誘導体があげられる。また、グルコース・サッカロース等の糖類、脂肪族多価アルコール類、レゾルシノール・カテコール等の芳香族多価アルコール(ジオール)類、チオフェン等の含窒素複素環化合物、ピリジン・ピリミジン等の含窒素複素環化合物も利用することができる。
【0046】
次いで、このようにして得られた炭素材料のエッジにアミジン構造を有する官能基を導入する。炭素材料の表面におけるエッジは活性面であり、ラジカルを生成しうる重合開始剤などを用いて各種のアミジン構造を導入することができる。炭素材料のエッジにアミジン構造を有する官能基を導入する方法は特に制限されない。例えば、エッジ部分を有する炭素材料をアミジン構造を有するアゾ化合物(例えば、水溶性アゾ重合開始剤)と反応させて、前記炭素材料のエッジにアミジン構造を有する官能基を導入する工程を有する方法が好ましく用いられうる。具体的には、水溶性アゾ重合開始剤を用いてラジカルを生成させ、炭素材料のエッジと反応させる方法が好ましく用いられうる。このような方法によれば、例えば炭素材料としてZTCなどを用いた場合、ZTCの構造規則性を低下させることなくエッジ部分に目的の親水性官能基を導入することができる。
【0047】
また、通常、ZTCを調製する際に、ZTCの前駆体としてゼオライトと炭素との複合体を調製した後、ゼオライトをフッ化水素酸などで処理して取り除く工程を必要とするが、親水性官能基を導入する工程は、上記のゼオライトを取り除く工程で同時に行うことができる。そのため、親水性官能基を導入するために反応ステップを増やす必要がなく、複雑な実験操作も必要としない。さらに、多くの水溶性アゾ重合開始剤は工業的にも利用されているため比較的安価で使用できる。
【0048】
ここで、アミジン構造を導入するために用いられうる水溶性アゾ重合開始剤としては、特に制限されないが、例えば、下記式に示される化合物が好ましく用いられうる。
【0049】
【化3】
【0050】
炭素材料とアミジン構造を有するアゾ化合物とを反応させる条件は特に制限されない。好ましくは、反応容器に窒素置換しながらアミジン構造を有するアゾ化合物および炭素材料を加え、加熱、攪拌しながら反応させる。反応温度は用いられる水溶性アゾ重合開始剤の10時間半減期温度に応じて、ラジカルが十分発生するように選択されうる。例えば、水溶性アゾ重合開始剤である、水中の10時間半減期温度が44℃であるVA−044を用いる場合、反応温度は、例えば0〜85℃であり、好ましくは30〜70℃であり、より好ましくは40〜65℃である。反応時間は、例えば、0.5〜10時間であり、好ましくは1〜6時間である。
【0051】
アミジン構造を有するアゾ化合物の使用量は、特に制限されないが、炭素材料の質量に対して、例えば10〜10000質量%であり、好ましくは100〜5000質量%であり、より好ましくは100〜500質量%である。上記範囲であれば、炭素材料のBET表面積の大きな減少がなく、炭素材料の構造を維持しながら、アミジン構造を有する官能基を効率的に炭素材料のエッジに導入することができる。
【0052】
なお、エッジ修飾ZTCを製造する場合、ZTCを原料として用いて水溶性アゾ重合開始剤と反応させてもよいが、ゼオライトと炭素との複合体を原料として用い、フッ化水素酸などの酸溶液中で水溶性アゾ重合開始剤と反応させ、アミジン構造の導入とゼオライトの除去を同時に行ってもよい。ZTCを原料として用いる場合は、例えば、一般的なガラスフラスコ中で蒸留水とZTCとをよく混合し、不活性ガスをバブリングさせてから水溶性アゾ重合開始剤を添加して反応させることでエッジ修飾ZTCを製造することができる。
【0053】
炭素材料のエッジ部分にアミジン官能基を導入すると、この官能基の部分に多様な化学構造をさらに導入することができるが、この際、アミジン官能基の部分にリン配位子を導入することによって、後述する金属錯体を安定に配位させることができる。
【0054】
アミジン構造を有する官能基は強塩基で求核性を示すため、このアミジン構造を基点として、炭素材料にリン配位子を導入することができる。その後、後述するようにこのリン配位子に触媒となる金属を含む金属錯体を導入する。この反応では、金属錯体の配位子が、配位力の強いリン配位子と置き換わる配位子交換反応によって進行しうる。その結果、リン配位子と強塩基性のアミジン構造からなる二座配位子が形成される。したがって金属原子にリン原子と窒素原子が同時にキレート配位し、金属を安定な錯体として担持させることができる。このように金属錯体を担持させることができれば、金属を粒子ではなく原子レベルで分散させることが可能となる。
【0055】
この際、用いられるリン配位子を導入するためのリン化合物としては、特に制限されないが、好ましくは下記化学式で表されるリン化合物が好ましく用いられうる。
【0056】
【化4】
【0057】
式中、Ph1およびPh2は同一であっても異なっていてもよく、置換または非置換のフェニル基であり、Xは、ハロゲン原子である。ハロゲン原子としては、Cl、Brが好ましく、Clがより好ましい。Ph1またはPh2の置換基としては、特に制限されないが、例えば炭素数1〜8のアルキル基、アルコキシ基、ハロゲン化アルキル基である。
【0058】
前記リン化合物の例としては、クロロジフェニルホスフィン、クロロビス(3,5−ジメチルフェニル)ホスフィン、クロロビス(4−メトキシフェニル)ホスフィン、クロロビス[4−(トリフルオロメチル)フェニル]ホスフィン、クロロジ(p−トリル)ホスフィン、ビス(3,5−ジ−tert−ブチル−4−メトキシフェニル)クロロホスフィン、クロロビス[3,5−ビス(トリフルオロメチル)フェニル]ホスフィンなどが挙げられる。
【0059】
前記エッジ修飾炭素材料とリン化合物とを反応させる条件についても特に制限されない。好ましくは、あらかじめ真空加熱乾燥させて放冷した前記エッジ修飾炭素材料と、リン化合物とを、窒素置換しながら、好ましくはアミン存在下で反応させる。反応温度、反応時間については特に制限されない。好ましくは、はじめに前記エッジ修飾炭素材料に溶媒に溶解させたアミンを含浸させ、これにリン化合物を、例えば−100〜10℃、好ましくは−100〜5℃で少しずつ添加して混合する。その後、反応溶液の温度を、例えば0〜100℃、好ましくは10〜30℃に上昇させ、攪拌しながら反応させる。反応温度を上昇させた後の反応時間は、好ましくは0.5〜72時間であり、より好ましくは2〜24時間であり、さらに好ましくは12〜24時間である。
【0060】
この際、溶媒としては、特に限定されないが、非プロトン性極性溶媒が好ましく用いられうる。非プロトン性極性溶媒としては、例えば、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)、ヘキサメチルリン酸トリアミド(HMPT)、ジメチルホルムアミド(DMF)、アセトン、アセトニトリルなどが挙げられる。中でも、THFが好ましい。
【0061】
アミンとしては、3級アミンを用いることが好ましい。3級アミンとしては、特に制限されないが、例えば、トリメチルアミン、トリエチルアミン、トリ−n−プロピルアミン、トリイソプロピルアミン、トリ−n−ブチルアミン、ジエチルメチルアミン(N,N−ジエチルメチルアミン)、ジメチルエチルアミン(N,N−ジメチルエチルアミン)などが挙げられる。中でも、トリエチルアミンが好ましい。
【0062】
リン化合物の使用量も特に制限されないが、好ましくは、炭素材料に結合したアミジン構造を有する官能基1モルに対して0.5〜100モルであり、より好ましくは1〜10モルである。上記範囲であれば、反応が効率的に進行しうる。
【0063】
一方、エッジ修飾されていない炭素材料に、直接リン配位子を導入することもできる。リン配位子を導入するためのリン化合物や、リン化合物を炭素材料と反応させる際の反応条件は、上述のエッジ修飾炭素材料の場合と同様のものが採用されうる。エッジ修飾されていない炭素材料を用いた場合、リン化合物は炭素材料の表面の含酸素官能基と反応することによって炭素材料に導入されるものと考えられる。このため、エッジ部分の割合が高いZTCだけではなく、エッジ部分の割合が比較的少ない炭素材料であっても効率よくリン配位子を導入することが可能であると考えられる。また、エッジ修飾の工程を省略することができるため、コスト低減が可能である。
【0064】
<金属担持炭素材料>
本発明の一実施形態は、上記のリン配位子を有する炭素材料において、リン配位子に8〜10族の金属が配位してなる、金属担持炭素材料である。
【0065】
本実施形態による金属担持炭素材料は、上記図1(c)のように、炭素材料の表面に8〜10属の金属が錯体として配位子を介して配位されることにより、金属が担持されている。すなわち、前記金属は、粒子としてではなく、錯体として担持されている。このようにすることで、金属が酸化されることなく、金属を原子レベルで分散させることができるので、より高効率なスピルオーバーが可能となる。
【0066】
好ましくは、前記金属は、炭素材料において、0.01〜10wt%の濃度範囲内で担持することが好ましい。担持されている金属が0.01wt%以上であれば、金属の機能を十分に得ることができる。一方、担持されている金属が10wt%以下である場合には、細孔機能を維持し、高いBET表面積が得られうる。
【0067】
担持する金属は、機能性を付与するという観点で8〜10族の金属であることが好ましい。また、前記金属は単体だけではなく、2種類以上の金属が担持されていてもよい。中でも、水素吸蔵材料として用いる場合には、Pt、Pd、Ir、Rh、Co、Ni、Ru、Feを用いることが好ましく、Pt、Pd、Niを用いることがさらに好ましく、Ptが特に好ましい。
【0068】
本実施形態による金属担持炭素材料は、金属を担持した状態で、ミクロ細孔2aの占める容積が1.0cm3/g以上であることが好ましい。ミクロ細孔2aの占める容積は、1.2cm3/g以上であることがより好ましく、1.5cm3/g以上であることが更に好ましい。また、金属を担持した状態で、BET表面積が1500m2/g以上であることが好ましい。BET表面積は、例えば2000m2/g以上、好ましくは2500m2/g以上であり、3000m2/g以上であることがより好ましく、BET表面積が3500m2/g以上であることが更に好ましい。ミクロ細孔の占める容積が1.0cm3/g以上、又はBET表面積が1500m3/g以上である場合には、十分な水素貯蔵性能が得られうる。
【0069】
また、前記金属担持炭素材料は、粉末X線回折測定を行った場合に得られるX線回折パターンは、2次元積層規則性を示す通常26°付近に現れる回折ピークはできるだけ少ないほうが好ましい。
【0070】
本実施形態に係る金属担持炭素材料は、−40℃から150℃の範囲で水素を吸蔵放出させることができる。従来、金属担持されていない炭素材料は、温度上昇と共に水素吸蔵量が低下する場合があったが、本実施形態に係る金属担持炭素材料は温度上昇と共に吸蔵能が向上する。また、粒子状の金属を担持させた場合に比較して、水素分子の解離吸着が促進され、水素吸蔵能が向上しうる。そのため、前記金属担持炭素材料を水素吸蔵材料として用いると、高効率の水素の吸蔵及び放出が可能となる。
【0071】
(金属担持炭素材料の製造方法)
本実施形態による金属担持炭素材料は、細孔を有する炭素材料を準備する段階と、前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階と、前記リン配位子を有する炭素材料を、8〜10族金属化合物と反応させて、前記リン配位子に8〜10族金属を配位させる段階と、を有する方法によって調製されうる。
【0072】
ここで、細孔を有する炭素材料を準備する段階および前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階については上述と同様であるため、説明を省略する。リン配位子を有する炭素材料を得た後、好ましくはこれを洗浄して真空加熱乾燥した後、金属錯体と反応させる。用いられる金属錯体についても特に制限されない。好ましい白金錯体としては、例えば、(1,5−シクロオクタジエン)ジメチル白金(II)、(2,2’−ビピリジン)ジクロロ白金(II)、(エチレンジアミン)ヨード白金ダイマー、ジクロロ(1,10−フェナントロリン)白金(II)、ジクロロ(エチレンジアミン)白金(II)、ジクロロビス(ジメチルスルフィド)白金(II)、ジクロロビス(エチレンジアミン)白金(II)、トリクロロ(エチレン)白金(II)酸塩(例えばトリクロロ(エチレン)白金(II)酸カリウム)、ヘキサヒドロキシ白金(IV)酸塩(例えばヘキサヒドロキシ白金(IV)酸ナトリウム)、cis−ビス(アセトニトリル)ジクロロ白金(II)、cis−ジアンミンテトラクロロ白金(IV)、cis−ジクロロビス(ピリジン)白金(II)、cis−ジアンミン白金(II)ジクロリド、cis−ジクロロビス(ジエチルスルフィド)白金(II)、cis−ジクロロビス(トリエチルホスフィン)白金(II)、cis−ジクロロビス(トリフェニルホスフィン)白金(II)、cis−ビス(ベンゾニトリル)ジクロロ白金(II)、trans−ジクロロビス(トリフェニルホスフィン)白金(II)、trans−ジクロロビス(トリエチルホスフィン)白金(II)、エチレンビス(トリフェニルホスフィン)白金(0)、ジアミンジニトリト白金(II)、白金(0)−1,3−ジビニル−1,1,3,3−テトラメチルジシロキサン錯体、白金(0)−2,4,6,8−テトラメチル−2,4,6,8−テトラビニルシクロテトラシロキサン錯体、白金(II)アセチルアセトナート、硝酸テトラアンミン白金(II)、ジクロロ(1,2−ジアミノシクロヘキサン)白金(II)、ジクロロ(1,5−シクロオクタジエン)白金(II)、ジクロロ(ジシクロペンタジエニル)白金(II)、テトラアンミン白金(II)クロリド、テトラキス(トリフェニルホスフィン)白金(0)、トリメチル(メチルシクロペンタジエニル)白金(IV)、ビス(トリ−tert−ブチルホスフィン)白金(0)、ヘキサクロロ白金(IV)酸塩(例えばヘキサクロロ白金(IV)酸カリウム、ヘキサクロロ白金(IV)酸テトラブチルアンモニウム)、ヨウ化白金、塩化白金、などが挙げられる。
【0073】
好ましいパラジウム錯体としては、例えば、塩化パラジウム、クロロ(1,5−シクロオクタジエン)メチルパラジウム(II)、ジクロロ(1,5−シクロオクタジエン)パラジウム(II)、ビシクロ([2.2.1]ヘプタ−2,5−ジエン)ジクロロパラジウム(II)、(エチレンジアミン)パラジウム(II)クロリドなどが挙げられる。
【0074】
前記リン配位子を有する炭素材料と金属錯体とを反応させる条件についても特に制限されない。好ましくは、前記リン配位子を有する炭素材料と金属錯体とを窒素雰囲気中で還流しながら反応させる。反応温度は、例えば0〜150℃であり、好ましくは20〜100℃である。反応時間は、例えば、0.5〜72時間であり、好ましくは1〜12時間である。
【0075】
金属錯体の使用量も特に制限されないが、好ましくは、リン配位子1モルに対して0.001〜10モルであり、より好ましくは0.01〜5モルである。上記範囲であれば、反応が効率的に進行しうる。反応後、濾過、洗浄し、真空加熱乾燥して、金属担持炭素材料を得ることができる。導入されたリン配位子に対する金属錯体の導入率は、誘導結合プラズマ(ICP)発光分光分析と31P−NMR測定から見積もることができる。
【0076】
<水素吸蔵材料>
上述の金属担持炭素材料は、高い水素吸蔵能を有し、100℃以下の温度で水素の吸蔵、放出が可能である。また、水素の吸蔵、放出に化学反応を伴わないため、耐久性に優れる。そのため、特に燃料電池自動車用の水素吸蔵材料に好適に用いられうる。
【実施例】
【0077】
本発明の作用効果を以下の実施例を用いて説明する。ただし、本発明の技術的範囲が以下の実施例のみに制限されるわけではない。
【0078】
1.ミクロポーラス炭素材料へのアミジン構造の導入
1−1.試料の調製
<参考例1:NaY−PFA−P7(1)−H9(3)>
乾燥したゼオライト(NaY5.5)にフルフリルアルコール(FA)を含浸した。これを、150℃で8時間熱処理してFAを重合させ、PFA/ゼオライト複合体とした。これをN2雰囲気下5℃/minで850℃まで昇温し、次いで700℃で1時間プロピレンCVDを行った。その後N2雰囲気下5℃/minで900℃まで昇温して3時間保持し、炭素/ゼオライト複合体を調製した。最後に、この複合体を47wt%のフッ素水素酸100mlに投入後、5時間攪拌してフッ化水素酸処理し、鋳型であるゼオライトを溶解除去してミクロポーラス炭素材料(MPC)を取り出した。こうして得た試料をNaY−PFA−P7(1)−H9(3)とした。
【0079】
<合成例1>
下記反応式(1)、(2)にしたがってエッジ修飾ミクロポーラス炭素材料を調製した。
【0080】
【化5】
【0081】
具体的には、PTFE製4口フラスコ(500ml)に46wt%フッ化水素酸(200g)を加え、溶解している酸素ガスを取り除くために事前に窒素ガスを100cc/分で30分間以上バブリングさせた。続いて水溶性アゾ重合開始剤であるVA−044を反応溶液の全重量に対して0.15wt%になるように加え、オイルバスの設定温度を70℃にして加熱を開始させた。フラスコ内のフッ化水素酸の温度が50±2℃となった時点で、上記参考例1で調製した炭素/ゼオライト複合体(0.47g)を加え、一定時間攪拌した。炭素/ゼオライト複合体を加えた後約1時間で、フラスコ内のフッ化水素酸の温度は60±2℃になった。反応時間は、フッ化水素酸の温度が60℃に達してから6時間とした。
【0082】
反応後、反応溶液を濾過して生成物を500mlの蒸留水で良く洗い、灰分が残らないようにするためにさらに蒸留水(500g)で30分間攪拌した。その後、濾過して蒸留水で洗浄して、エッジ修飾ミクロポーラス炭素材料を得た。収量は0.11gであった。
【0083】
<合成例2>
VA−044を反応溶液の全重量に対して0.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0084】
<合成例3>
VA−044を反応溶液の全重量に対して2.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0085】
<合成例4>
VA−044を反応溶液の全重量に対して2.50wt%になるように加え、フッ化水素酸の温度が60℃に達してから1時間反応させたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0086】
<合成例5>
15か月前に調製した炭素/ゼオライト複合体を用いたこと、およびVA−044を反応溶液の全重量に対して2.50wt%になるように加えたことを除いては、合成例1と同様にして、エッジ修飾ミクロポーラス炭素材料を得た。
【0087】
1−2.評価
合成例1〜5及び参考例1で合成したミクロポーラス炭素材料の粉末X線回折測定は、島津製作所製XRD−6100を用いて行い、線源はCu−Kα、電圧30kV、電流20mAで行った。BET表面積の測定は、日本ベル製BELSORP miniを用いて行い、−196℃の温度で、多点法で行った。水蒸気吸着等温線は、日本ベル製BELSORP MAXを用いて25℃の温度で測定した。
【0088】
合成例1〜5及び参考例1で合成したミクロポーラス炭素材料のBET表面積SBET、窒素含有量(N/C)、窒素吸脱着測定結果から算出した試料の細孔構造を表1、図3に示す。ここで、VTotal、Vmicro、およびVmesoは、それぞれ、全細孔の占める容積、ミクロ細孔の占める容積、およびメソ細孔の占める容積を表す。長周期規則構造の有無を示す粉末X線回折装置(XRD)を用いて測定した結果を図3にまとめた。
【0089】
【表1】
【0090】
表1、図3の結果から、VA−044の仕込み濃度を増加させるほど、また反応時間を1時間から6時間に長くすると、N/C比が増加し、アミジン構造を有する官能基が多く導入されていることがわかる。また、アミジン構造を有する官能基が多く導入されるほど、BET表面積が低下する傾向にあり、合成例3では参考例1に比べて17%減少している。しかしながらアミジン構造を有する官能基を導入した場合であっても2000m2/g以上の十分大きいBET表面積を維持することが分かった。
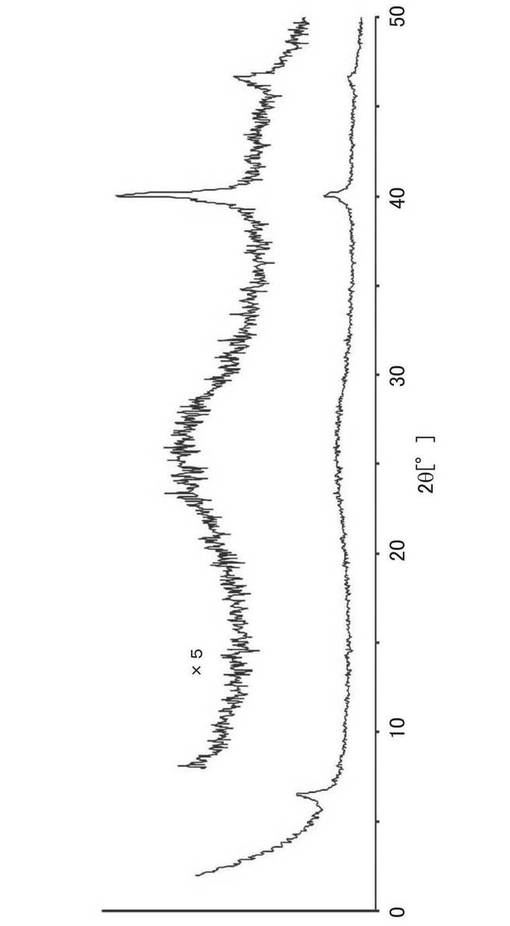
【0091】
図3に示すように、合成例1〜5の試料のX線回折パターンは2θ=6°付近の長周期規則構造を示すピークが明確に観察され、また2θ=20〜30°に炭素網面の積層に由来するピークの強度が小さかった。このことから、得られた試料では、ミクロポーラス炭素材料(MPC)の規則構造が保持されていることが確認された。
【0092】
ZTCの質量、H/C比からZTCのエッジの割合を求め、エッジがすべてラジカルであると仮定しても、エッジのラジカルを全て反応させるためにはVA−044の濃度が0.15wt%で充分であると考えられるが、合成例1〜3を比較すると、VA−044の濃度が高くなるにつれてN/Cが増加している。さらに、同じ濃度で反応時間の異なる合成例3と合成例4を比較すると、反応時間が長いほどN/Cが増加していることがわかる。これは、VA−044が過剰である場合や反応時間が長い場合、VA−044由来のラジカルとZTCのラジカルサイトとの反応だけではなく、ZTCのエッジの水素がVA−044由来のラジカルに引き抜かれ、その結果ZTCに生成したラジカルサイトが、VA−044由来のラジカルと再結合する機構も関与しているものと考えられる。
【0093】
図4に、参考例1および合成例3の水蒸気吸脱着等温線を示す。細孔径が同等の場合、吸着量が急激に増加する相対圧が小さいほど材料表面は親水性である。参考例1および合成例3の水蒸気吸着等温線を比較すると、立ち上がりはいずれも相対圧0.5付近であり、ほぼ同じである。これは、親水性官能基を導入した効果に加えて、表1の元素分析結果から示されるように、試料に一定量含まれる含酸素官能基の影響があるためと考えられる。
【0094】
2.エッジ修飾ミクロポーラス炭素材料への原子オーダーの白金の導入
2−1.試料の調製
<実施例6>
(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)
下記式にしたがって反応を行った。
【0095】
【化6】
【0096】
合成例3と同様の手順で、アミジン構造を導入したZTCを調製した。
【0097】
アミジン構造を導入したZTC(ZTC−VA044)780mgとスターラーバーとを外径35mmの直線状のフラスコに加え、150℃で6時間真空加熱乾燥後、室温まで放冷した。続いてスターラーを攪拌させながら2.62wt%トリエチルアミン/THF溶液をシリンジを用いてフラスコ内に50ml(トリエチルアミン1.13g、11.20mmol)注入して真空含浸させた。その後、恒温槽を用いて0℃のウォーターバス(エチレングリコールを約15重量%加える)でフラスコを冷却した。30分後、大気圧までフラスコ内に窒素をパージし、フラスコ内に空気が入らないように窒素を流しながらクロロジフェニルホスフィン(アルドリッチ社製)2.26g(10.25mmol)をシリンジを用いてフラスコ内に加えて10分間攪拌した。その後、ウォーターバスの温度を25℃に設定し、25℃に達してから15時間反応させた。その後、反応溶液にTHF80mlを加えて数分間攪拌してからメンブレンフィルター(0.1μm、ADVANTEC社製T010A047A)で濾過し、さらに500mlのTHFでよく洗浄した。次いで試料を2500mlの蒸留水中で30分間攪拌して良く洗浄し、メンブレンフィルター(0.1μm、ADVANTEC社製H010A047A)で濾過した後、試料を150℃で真空乾燥させた。収量は0.87gであった。この試料をZTC−VA044−PPh2と表す。
【0098】
この試料の元素分析結果を下記表2に示す。ZTC−VA044からZTC−VA044−PPh2への反応前後の試料の重量変化から計算したZTC−VA044−PPh2のN/Cは、元素分析によるN/Cの値とよく一致した。
【0099】
また、誘導結合プラズマ(ICP)発光分光分析法により測定したリンの含有量は、2.53wt%であった。窒素とリンの含有量から算出した結果、82.76%のアミジン構造を有する官能基にリン配位子が導入されたことがわかった。ここで、リンの含有量は、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて測定した値を用いた。
【0100】
【表2】
【0101】
(リン配位子を有するエッジ修飾ミクロポーラス炭素材料への白金錯体の導入)
下記式にしたがって反応を行った。
【0102】
【化7】
【0103】
上記で調製したZTC−VA044−PPh20.30gを、150℃で6時間真空加熱乾燥した後、室温まで放冷してスターラーを攪拌させながらシリンジを用いてフラスコ内に1.56重量%(1,5−シクロオクタジエン)ジメチル白金(II)錯体/アセトニトリル溶液12mlを注入して真空含浸させ、10分間ほど攪拌した後に大気圧に達するまでフラスコ内に窒素をパージさせた。続いてフラスコに冷却器を取り付けて100℃に加熱したオイルバスで12時間還流させた。還流後、反応溶液を室温まで冷却してから反応溶液をメンブレンフィルター(0.1μm、ADVANTEC社製H010A047A)で濾過し、100mlのアセトニトリル、続いて100mlのジエチルエーテルで洗浄してから真空加熱乾燥した(100℃、6時間)。この試料をZTC−VA044−PPh2−PtMe2と表す。収量は0.37gであった。
【0104】
実施例6で調製した試料であるZTC−VA044−PPh2−PtMe2に含まれるリンおよび白金の定量分析を、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて行なった。試料中に含まれるリンは1.62wt%であり、白金は11.1wt%であった。
【0105】
また、窒素吸脱着測定によって算出したZTC−VA044−PPh2およびZTC−VA044−PPh2−PtMe2のBET表面積は、それぞれ1910m2/gおよび1550g/m2であった。
【0106】
<実施例7>
(ミクロポーラス炭素材料へのリン配位子の導入反応)
アミジン構造を導入したZTCに代えて、参考例1と同様の手順で調製したミクロポーラス炭素材料を用いたことを除いては、実施例6(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)と同様の手順でリン配位子を有する炭素材料を得た。この試料をZTC−PPh2と表す。
【0107】
さらに、ZTC−VA044−PPh2に代えて、上記で調製したZTC−PPh2を用いたことを除いては、実施例6(リン配位子を有するエッジ修飾ミクロポーラス炭素材料への白金錯体の導入)と同様の手順でリン配位子を有する炭素材料を得た。この試料をZTC−PPh2−PtMe2と表す。
【0108】
実施例7で調製した試料であるZTC−PPh2−PtMe2に含まれるリンおよび白金の定量分析を、試料をアルカリ融解により分解し溶液化した後、エスアイアイナノテクノロジー社製誘導結合プラズマ(ICP)発光分光分析装置を用いて行なった。試料中に含まれるリンは1.48wt%であり、白金は10.3wt%であった。
【0109】
また、窒素吸脱着測定によって算出したZTC−PPh2およびZTC−PPh2−PtMe2のBET表面積は、それぞれ1930m2/gおよび1860g/m2であった。
【0110】
<比較例1>
参考例1と同様の手順で、ミクロポーラス炭素材料(MPC)を得た。
【0111】
次に、Pt水溶液として、Ptを含む化合物であるジアンミンジニトロ白金[Pt(NO2)2(NH3)2]の、0.096wt%の水溶液A6.7mlと、還元剤水溶液である水素化ホウ素ナトリウムの水溶液B66.7mlとを調製し、溶液A、Bを0℃に冷却した。溶液A,Bの濃度は白金の担持量が得られたMPCに対し3.8wt%になるよう計算した。次に、100mgのMPCを0℃の溶液Aに投入し、減圧雰囲気下0℃で30分攪拌した。次に、これを遠心分離して0℃の溶液Bと混合し、0℃で10分間攪拌することによりPtを還元して白金ナノ粒子を生成させた。最後に、白金ナノ粒子を担持させたMPCを濾過して純水で洗浄する操作を数回繰り返した後、150℃で6時間真空乾燥させることにより、白金を担持させたMPCを得た。この試料を、Pt/ZTCと表す。
【0112】
<比較例2>
参考例1で調製した炭素材料を真空加熱乾燥した後、(1,5−シクロオクタジエン)ジメチル白金(II)錯体を注入して真空含浸させ、白金錯体を含浸させ、炭素材料ZTC−PtMe2CODを得た。この試料に含まれる白金の量は、13.3wt%であった。
【0113】
2−2.評価
実施例6で調製した試料について、XRD測定、水素吸脱着測定を行なった。XRD測定は上記1−2と同様の方法で行なった。
【0114】
図5に、実施例6で調製した試料であるZTC−VA044−PPh2およびZTC−VA044−PPh2−PtMe2のX線回折パターンを示す。比較のために、アミジン構造を導入していないZTCおよびアミジン構造を導入した段階の試料のX線回折パターンを示した。図5に示すように、ZTC−VA044−PPh2、ZTC−VA044−PPh2−PtMe2のX線回折パターンは、参考例1のMPCのX線回折パターンと比較すると弱いものの、2Xで示す2θ=6°付近にピークを示し、また2θ=20〜30°のピーク強度は小さい。このことから、実施例6で得られた試料では、MPCの規則構造が保持されていることがわかる。また、0価の白金に由来するピークは確認されなかった。
【0115】
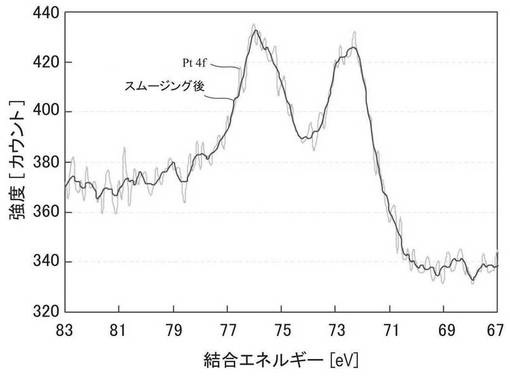
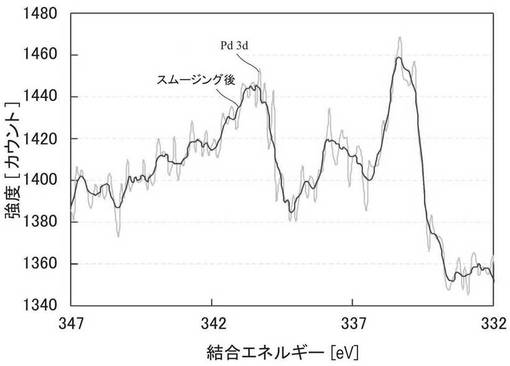
図6に、実施例6で得られたZTC−VA044−PPh2−PtMe2のXPSスペクトルを示す。72.3eV付近および75.9eV付近に2価の白金に由来するピークが観察された。図5、図6の結果から、2価の白金がZTCに担持されていることが確認された。なお、XPSスペクトルの測定は、KRATOS社製ESCA−3400を用い、線源はMg−Kα、電圧10kV、電流10mAで行なった。
【0116】
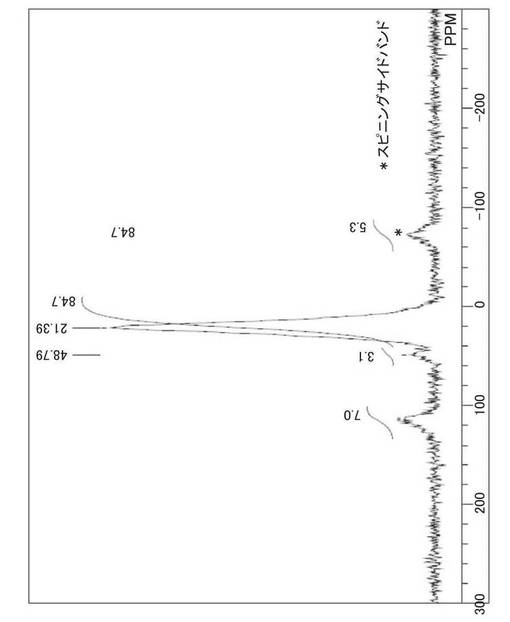
図7、図8に、実施例6で得られた試料の固体31P−NMRスペクトルを示す。Pt錯体を担持していないZTC−VA044−PPh2の固体31P−NMRスペクトル(図8)は21.08ppmに1本のピークを示すのに対して、ZTC−VA044−PPh2−PtMe2ではピークが2つに分裂し、試料に含まれるリン配位子の一部にPtが錯体として担持されていることがわかる。2つのピークの積分比は、3.1(48.79ppm):84.7(21.39ppm)であった。この積分比と、試料に含まれる白金の量(11.1wt%)から、リン配位子に配位している白金の割合は、試料に含まれる白金の量を基準として、3.2%と見積もることができる。
【0117】
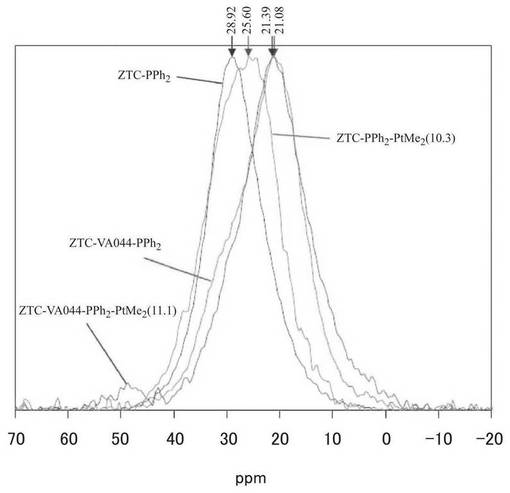
一方、図8に示すように、実施例7で得られた試料の固体31P−NMRスペクトルを実施例6の試料と比較すると、ZTC−PPh2は、ZTC−VA044−PPh2と異なる位置にピークトップを示すことがわかる。これは、ZTC−PPh2では、導入したリン配位子のリン原子が結合した原子が両者において異なることを示唆している。ZTC−VA044−PPh2では、ZTC合成の最後のフッ化水素酸処理によるゼオライト除去の際にラジカルを反応させることにより、ZTC表面において含酸素官能基の生成が抑制される。そのため、ZTC−VA044には含酸素官能基の量が少なく、アミジンはクロロジフェニルホスフィンと反応したと考えられる。しかしながら、ZTC−PPh2とクロロジフェニルホスフィンとの反応では、ZTCの表面の含酸素官能基とクロロジフェニルホスフィンとが反応したものと考えられる。白金錯体を担持後のZTC−PPh2−PtMe2ではピークがわずかに高磁場側にシフトした。
【0118】
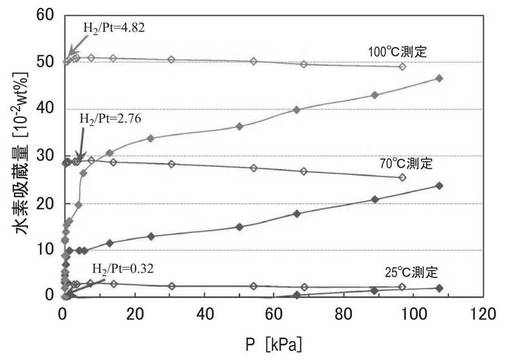
得られた試料の水素吸蔵能を評価するために、実施例6の試料について、ジーベルツ法(容量法、JIS H 7201)に従って圧力−組成等温線(PCT線)を得た。水素吸蔵能は、国立標準規格技術研究所(National Institute of Standards and Technology:NIST)で定められている圧縮係数を用いて測定した。測定精度はサンプルの充填量に依存する。最低でも1g以上を充填し、必要に応じて上記合成スキームを繰り返し行い、所要量を準備した。試料を秤量して測定用耐圧試料管に入れ、100℃で4時間真空引きして試料管内に残留しているガスを放出させて、水素が吸蔵されていない原点を得た後測定した。測定温度は25℃、70℃、100℃とした。測定順序は、新しい試料で25℃で測定後に引き続き70℃で測定した。その後、新しい試料で100℃で測定した。水素吸蔵能の測定後、大気圧まで減圧して水素放出量の確認を行った。測定結果を図9に示す。図9中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。
【0119】
図9の結果から、実施例6で調製したZTC−VA044−PPh2−PtMe2の水素吸着量は、温度の上昇に伴って水素吸着量が大幅に増加することがわかった。図9中、H2/Ptは、各測定点における吸着した水素分子と担持された白金とのモル比である。図9に示すように、実施例6の試料ではH2/Ptの値は最大で4.82と大きな値であり、水素分子が白金錯体に解離吸着しただけではなく、解離した水素原子が流れ出ている(スピルオーバー)ことが考えられる。
【0120】
実施例7の試料について、上記と同様に水素吸蔵能を用いて測定した。試料を100℃で6時間真空引きして試料管内に残留しているガスを放出させて、水素が吸蔵されていない原点を得た後測定した。測定温度は25℃、70℃、100℃とした。測定順序は、新しい試料で25℃で測定後に引き続き70℃で、その後100℃で測定した(図10上)。その後、新しい試料で100℃で測定した(図10下、図10中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである)。図10に示すように、実施例7の試料ではH2/Ptの値は最大で4.65の値が得られた。したがって、エッジ修飾していない炭素材料を用いた場合も、アミジンでエッジ修飾した炭素材料を用いた場合と同様にリン配位子に金属錯体を担持させることができ、優れた水素吸着特性が得られることが確認された。
【0121】
比較例2の試料について、上記実施例6の試料と同様の手法で25℃、70℃、100℃で水素吸蔵能の測定を行なった。上記実施例6の試料と同様、水素吸着量は温度の上昇に伴って増加した(図示せず)。吸着量の最大値におけるH2/Ptの値は1.67であり、白金錯体1分子に対して水素1.7分子が吸着し、解離しているものと考えられる。
【0122】
図11は、実施例6で調製したZTC−VA044−PPh2−PtMe2、参考例1のZTC、比較例1のPt/ZTCについて、25℃で測定した、試料の重量あたりの水素吸着量を示す図である。図11中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。測定条件は、温度条件以外は実施例6と同様である。比較のために、温度25℃で測定した実施例6の試料の結果を併せて示した。図11の結果から、実施例6の試料は、参考例1、比較例1の試料と比較して、試料の重量当たりの水素吸着量が改善されていることがわかる。
【0123】
さらに、実施例6、実施例7の試料では温度の上昇に伴って水素吸着量が増加した(図9、図10)が、参考例1、比較例1の試料では、高温では水素吸着量が減少する傾向にあった(図示せず)。
【0124】
このように、炭素材料に金属錯体を担持させた場合、金属を含まない場合や、粒子状の金属を担持させた場合に比べて水素吸蔵能が大幅に改善されることが明らかになった。
【0125】
3.エッジ修飾ミクロポーラス炭素材料への原子オーダーのパラジウムの導入
3−1.試料の調製
<実施例8>
(エッジ修飾ミクロポーラス炭素材料へのリン配位子の導入反応)
実施例6と同様の手順で、アミジン構造を導入したZTC(ZTC−VA044)にリン配位子であるクロロジフェニルホスフィンを導入し、ZTC−VA044−PPh2を調製した。
【0126】
(リン配位子を有するエッジ修飾ミクロポーラス炭素材料へのパラジウム錯体の導入)
下記式にしたがって反応を行った。
【0127】
【化8】
【0128】
上記で調製したZTC−VA044−PPh219.8mgを、150℃で一晩真空加熱乾燥した後、塩化パラジウム/アセトニトリル溶液15mlを真空含浸させ、80℃で3時間攪拌した。ここで、塩化パラジウム/アセトニトリル溶液は、窒素雰囲気下で塩化パラジウム25.3mgにアセトニトリル20mlを加え、約80℃に加熱して塩化パラジウムを溶解させて調製したものを用いた。その後、メンブレンフィルターで反応溶液を濾過し、200mlのアセトニトリルで洗浄後、さらに200mlのTHFで洗浄してから150℃で6時間真空加熱乾燥させた。この試料をZTC−VA044−PPh2−PdCl2と表す。収量は17.4mgであった。
【0129】
3−2.評価
図12に、上記で作製したZTC−VA044−PPh2−PdCl2のXRD測定結果を示す。装置、測定条件は上述のものと同様である。図12に示されるように、2θ=6°付近の長周期規則構造を示すピークが観察され、MPCの規則構造が保持されていることがわかる。また、2θ=40°付近に0価のPd粒子に由来するピークが観察された。
【0130】
図13に、上記で作製したZTC−VA044−PPh2−PdCl2のXPS測定結果を示す。装置、測定条件は上述のものと同様である。340.4eVおよび335.2eVに0価のパラジウムに由来するピークが観察される。一方、337.5eV付近に2価のパラジウムに由来するピークが観察され、これは、リン配位子に配位した2価のPd錯体のほかに、配位していないPdCl2によるものと考えられる。
【0131】
図14に、上記で作製したZTC−VA044−PPh2−PdCl2の水素吸脱着測定結果を示す。測定温度は、100℃、120℃、140℃であり、測定順序は、1回目:100℃、2回目:120℃、3回目:150℃、4回目:100℃の順で行なった。図14中、塗りつぶしのプロットは吸着側、白抜きのプロットは放出側のデータである。
温度の上昇に伴って吸着量が増加し、吸着量の最大値は約0.2wt%であることがわかる。したがって、Pd錯体を担持したZTCにおいても、Pt錯体を担持した場合と同様に高い水素吸蔵能が得られることが明らかになった。
【0132】
また、温度の上昇に伴って吸着量が増加することや、特に3回目(150℃)や4回目(100℃)の測定で水素の脱着も確認できることから、水素の吸着に主に寄与しているのは0価のPd粒子ではなく、2価のPd錯体であると考えられる。
【0133】
さらに、4回測定しても試料の劣化が生じにくく、これはPd錯体が熱安定性や寿命が良好であるためと考えられる。
【符号の説明】
【0134】
1 ゼオライト、
1a、2a ミクロ孔、
2 ゼオライト炭素(ゼオライト鋳型カーボン)、
3 複合体。
【特許請求の範囲】
【請求項1】
細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料。
【請求項2】
前記リン配位子が、PPh1Ph2(Ph1およびPh2は同一であっても異なっていてもよく、置換または非置換のフェニル基である)である、請求項1に記載のリン配位子を有する炭素材料。
【請求項3】
前記炭素材料が、ゼオライト鋳型カーボン(ZTC)である、請求項1または2に記載のリン配位子を有する炭素材料。
【請求項4】
細孔を有する炭素材料のエッジの少なくとも一部にアミジン構造を有する官能基が導入され、前記アミジン構造を有する官能基に、リン配位子が結合されてなる、請求項1〜3のいずれか1項に記載のリン配位子を有する炭素材料。
【請求項5】
前記アミジン構造を有する官能基が、下記化学式1で表される官能基から選択される少なくとも1種である、請求項4に記載のリン配位子を有する炭素材料:
【化1】
式中、R1、R2およびR3は独立して、水素原子、置換もしくは非置換の炭素数1〜10のアルキル基または置換もしくは非置換の炭素数3〜6のシクロアルキル基であり、R1とR2とが結合して環を形成してもよく、R2とR3とが結合して環を形成してもよい。
【請求項6】
請求項1〜5のいずれか1項に記載のリン配位子を有する炭素材料のリン配位子に、8〜10族の金属が配位されてなる金属担持炭素材料。
【請求項7】
細孔を有する炭素材料を準備する段階と、
前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階と、
前記リン配位子を有する炭素材料を、8〜10族金属化合物と反応させて、前記リン配位子に8〜10族金属を配位させる段階と、
を有する、請求項7に記載の金属担持炭素材料の製造方法。
【請求項8】
前記細孔を有する炭素材料を準備する段階が、
炭素材料前駆体をアミジン構造を有するアゾ化合物と反応させて、前記炭素材料前駆体のエッジにアミジン構造を有する官能基を導入してエッジ修飾炭素材料を得る段階を含む、請求項7に記載の金属担持炭素材料の製造方法。
【請求項9】
請求項6に記載の金属担持炭素材料、または請求項7もしくは8に記載の方法によって製造された金属担持炭素材料を含む、水素吸蔵材料。
【請求項1】
細孔を有する炭素材料にリン配位子が導入されてなる、リン配位子を有する炭素材料。
【請求項2】
前記リン配位子が、PPh1Ph2(Ph1およびPh2は同一であっても異なっていてもよく、置換または非置換のフェニル基である)である、請求項1に記載のリン配位子を有する炭素材料。
【請求項3】
前記炭素材料が、ゼオライト鋳型カーボン(ZTC)である、請求項1または2に記載のリン配位子を有する炭素材料。
【請求項4】
細孔を有する炭素材料のエッジの少なくとも一部にアミジン構造を有する官能基が導入され、前記アミジン構造を有する官能基に、リン配位子が結合されてなる、請求項1〜3のいずれか1項に記載のリン配位子を有する炭素材料。
【請求項5】
前記アミジン構造を有する官能基が、下記化学式1で表される官能基から選択される少なくとも1種である、請求項4に記載のリン配位子を有する炭素材料:
【化1】
式中、R1、R2およびR3は独立して、水素原子、置換もしくは非置換の炭素数1〜10のアルキル基または置換もしくは非置換の炭素数3〜6のシクロアルキル基であり、R1とR2とが結合して環を形成してもよく、R2とR3とが結合して環を形成してもよい。
【請求項6】
請求項1〜5のいずれか1項に記載のリン配位子を有する炭素材料のリン配位子に、8〜10族の金属が配位されてなる金属担持炭素材料。
【請求項7】
細孔を有する炭素材料を準備する段階と、
前記炭素材料を、リン化合物と反応させてリン配位子を有する炭素材料を得る段階と、
前記リン配位子を有する炭素材料を、8〜10族金属化合物と反応させて、前記リン配位子に8〜10族金属を配位させる段階と、
を有する、請求項7に記載の金属担持炭素材料の製造方法。
【請求項8】
前記細孔を有する炭素材料を準備する段階が、
炭素材料前駆体をアミジン構造を有するアゾ化合物と反応させて、前記炭素材料前駆体のエッジにアミジン構造を有する官能基を導入してエッジ修飾炭素材料を得る段階を含む、請求項7に記載の金属担持炭素材料の製造方法。
【請求項9】
請求項6に記載の金属担持炭素材料、または請求項7もしくは8に記載の方法によって製造された金属担持炭素材料を含む、水素吸蔵材料。
【図1】

【図2】

【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2013−14481(P2013−14481A)
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願番号】特願2011−149590(P2011−149590)
【出願日】平成23年7月5日(2011.7.5)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度、独立行政法人新エネルギー・産業技術総合開発機構、水素貯蔵材料先端基盤研究事業/計算科学的手法に基づく水素吸蔵材料の特性評価とメカニズム解明に関する研究委託事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000003997)日産自動車株式会社 (16,386)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願日】平成23年7月5日(2011.7.5)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度、独立行政法人新エネルギー・産業技術総合開発機構、水素貯蔵材料先端基盤研究事業/計算科学的手法に基づく水素吸蔵材料の特性評価とメカニズム解明に関する研究委託事業、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000003997)日産自動車株式会社 (16,386)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
[ Back to top ]