
炭素系固体酸及びその製造方法
【課題】安価に入手でき、しかも量産に適した原料から製造可能な炭素固体酸を提供するとともに、当該炭素系固体酸の製造に好適な調製条件を満たす製造方法を提供する。併せて、濃硫酸や発煙硫酸を用いた反応よりも効率を高めた製造方法も提案する。
【解決手段】スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であり、三酸化硫黄または三酸化硫黄を含有したスルホ化剤をセルロース含有原料または植物系原料に接触させてスルホ化する。または、植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理する。
【解決手段】スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であり、三酸化硫黄または三酸化硫黄を含有したスルホ化剤をセルロース含有原料または植物系原料に接触させてスルホ化する。または、植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、炭素系固体酸及びその製造方法に関し、炭素材料の表面にスルホン酸基を導入して得た炭素系固体酸、並びに当該炭素系固体酸の製造方法に関する。
【背景技術】
【0002】
硫酸は高い活性を有し、炭化水素化合物を反応させる際の触媒としても広く利用される。例えば、遊離高級脂肪酸とアルコールとを反応させて、高級脂肪酸エステルを得るエステル化反応の促進、セルロース等の糖鎖から単糖への加水分解反応の促進、その他、炭化水素燃料を合成するアルキル化反応の促進等の用途である。
【0003】
硫酸は触媒として各種の反応促進に寄与した後、中和、洗浄され、その都度消費されていた。硫酸は液体であるため回収が容易ではない。回収処理と新規投入との経費差から、現状は使い捨てが主流である。しかし、使用済みの硫酸の中和、洗浄に加え、環境基準に準拠した排水処理までを考慮すると、この負担は大きい。このことから、触媒として連続使用に耐えうるとともに、反応後の分離、回収に容易なより利便性の高い触媒が求められるようになってきた。
【0004】
そのような触媒として固体酸が挙げられる。例えば、硫酸処理を施したジルコニア、PTFEにスルホン酸基を導入したフッ素樹脂である。前記のジルコニアの場合、単位重量あたりのスルホン酸基濃度が低いため、触媒活性が低い欠点がある。また、前記のフッ素樹脂に関しては、熱に弱く、適用できる反応種が限られている問題がある。
【0005】
そこで、十分な触媒活性と耐熱性も併せ持つ固体酸として、炭素系の固体酸が提案された(特許文献1、特許文献2参照)。特許文献1の固体酸は、多環式芳香族炭化水素を濃硫酸中で加熱処理して得ることができる。
【0006】
さらに、固体酸がその内部に細孔構造による適度な表面積(比表面積)を有していればより吸着が増す。このため、吸着界面における濃度がバルク相における濃度よりも高くなる。このことから、固体酸内部の吸着界面では溶媒中の溶質濃度が固体酸表面と比較して高くなり、細孔構造を有する固体酸のほうが反応を加速することができる。
【0007】
その後、発明者らは、炭素系の固体酸の性能向上を鋭意研究するうちに、安価に入手でき、しかも量産に適した原料、並びにその調製条件を見出すに至った。
【0008】
これと併せて発明者らは、濃硫酸、発煙硫酸中で加熱処理する方法に対する改良も試みた。すなわち、濃硫酸と多環式芳香族炭化水素類の反応により芳香族スルホン酸及び水が生成する。しかし、この反応は平衡反応であるため反応系内の水の増加により逆反応が進み、固体酸に導入されるスルホン酸基の量が低下する点を内包していた。この方法では、多量の濃硫酸、発煙硫酸を用いる必要があり、また、反応系内の水を除去しなければならないため高い反応温度で長時間維持する必要がある。さらに、加熱処理後の固体酸から濃硫酸、発煙硫酸を除去するために減圧蒸留としていたため製造コストが高くなる傾向にあった。
【0009】
また、固体状の芳香族加工物をスルホ化する方法としては、塩素系有機溶媒に溶解した状態でスルホ化剤(クロルスルホン酸、発煙硫酸、濃硫酸等)を反応させる方法が一般的に適用されている。例えば、ポリスチレンをスルホ化する際にエチレンジクロライドを溶媒として使用する方法が知られている。この場合、ポリスチレンの分子間や分子内でSO2により架橋が生成してしまう。このため、ルイス塩基や水、アルコール化合物、陰イオン系化合物、非イオン系化合物を反応性に添加したり、あるいは高剪断型反応機を用いたりして架橋の生成を抑える必要があった(例えば、特許文献3、4、5等参照)。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特許第4041409号公報
【特許文献2】WO2005/029508
【特許文献3】特公昭50−33838号公報
【特許文献4】特公昭51−37226号公報
【特許文献5】特公昭51−37227号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、上記状況に鑑み提案されたものであり、安価に入手でき、しかも量産に適した原料から製造可能な炭素固体酸を提供するとともに、当該炭素系固体酸の製造に好適な調製条件を満たす製造方法を提供する。併せて、濃硫酸や発煙硫酸を用いた反応よりも効率を高めた製造方法も提案する。
【課題を解決するための手段】
【0012】
すなわち、請求項1の発明は、スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であることを特徴とする炭素系固体酸に係る。
【0013】
BET比表面積に関して、試料を200℃の窒素雰囲気下において3時間乾燥した後、77K(−195℃)における窒素吸着等温線を日本ベル株式会社製BELSORP MINIにより測定し、BET法により比表面積(m2/g)を求めた。
【0014】
請求項2の発明は、前記多孔質炭素がセルロース含有原料に由来する請求項1に記載の炭素系固体酸に係る。
【0015】
請求項3の発明は、前記セルロース含有原料が植物系原料である請求項2に記載の炭素系固体酸に係る。
【0016】
請求項4の発明は、前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものである請求項1ないし3のいずれか1項に記載の炭素系固体酸に係る。
【0017】
請求項5の発明は、前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものである請求項3に記載の炭素系固体酸に係る。
【0018】
請求項6の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものである請求項5に記載の炭素系固体酸に係る。
【0019】
請求項7の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0020】
請求項8の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0021】
請求項9の発明は、前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0022】
請求項10の発明は、前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種である請求項3ないし9のいずれか1項に記載の炭素系固体酸に係る。
【0023】
請求項11の発明は、請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化することを特徴とする炭素系固体酸の製造方法に係る。
【0024】
請求項12の発明は、前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われる請求項11に記載の炭素系固体酸の製造方法に係る。
【0025】
請求項13の発明は、請求項4に記載の炭素系固体酸を製造するに際し、前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化することを特徴とする炭素系固体酸の製造方法に係る。
【0026】
請求項14の発明は、前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状である請求項13に記載の炭素系固体酸の製造方法に係る。
【0027】
請求項15の発明は、前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液である請求項14に記載の炭素系固体酸の製造方法に係る。
【発明の効果】
【0028】
請求項1の発明に係る炭素系固体酸によると、スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であるため、良好な触媒活性を得ることができる。特にスルホン酸基が寄与する良好な触媒活性を得ることができる。
【0029】
請求項2の発明に係る炭素系固体酸によると、請求項1の発明において前記多孔質炭素がセルロース含有原料に由来するため、焼成後に炭素の環構造を保持しやすい。
【0030】
請求項3の発明に係る炭素系固体酸によると、請求項2の発明において、前記セルロース含有原料が植物系原料であるため、比較的容易に、大量に調達できる
【0031】
請求項4の発明に係る炭素系固体酸によると、請求項1ないし3のいずれかの発明において、前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものであるため、必ずしも硫酸や発煙硫酸を用いることなく、低温度域で短時間の反応で得ることができた。
【0032】
請求項5の発明に係る炭素系固体酸によると、請求項3の発明において、前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものであるため、量産に適した原料から製造可能な触媒用多孔質炭素を得るに至った。
【0033】
請求項6の発明に係る炭素系固体酸によると、請求項5の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものであるため、量産に適した原料を用いて各種の反応に有効な炭素系固体酸を得ることができた。
【0034】
請求項7の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものであるため、加水分解反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0035】
請求項8の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものであるため、エステル化反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0036】
請求項9の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものであるため、アルキル化反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0037】
請求項10の発明に係る炭素系固体酸によると、請求項3ないし9のいずれかの発明において、前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種であるため、原料を安価に入手でき、製造コストを圧縮することができる。
【0038】
請求項11の発明に係る炭素系固体酸の製造方法によると、請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化するため、賦活により多孔質化した炭化物表面へのスルホン酸基の導入を促進できる。
【0039】
請求項12の発明に係る炭素系固体酸の製造方法によると、請求項11の発明において、前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われるため、スルホン酸基と水分との接触を抑制して炭素表面に導入するスルホン酸基濃度の希釈を防ぐことができる。
【0040】
請求項13の発明に係る炭素系固体酸の製造方法によると、請求項4に記載の炭素系固体酸を製造するに際し、前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化するため、硫酸や発煙硫酸を用いることなく、より低温度域で短時間の反応によるスルホ化の製造方法を構築することができた。
【0041】
請求項14の発明に係る炭素系固体酸の製造方法によると、請求項13の発明において、前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状であるため、硫酸や発煙硫酸を用いないスルホ化が可能となった。
【0042】
請求項15の発明に係る炭素系固体酸の製造方法によると、請求項14の発明において、前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液であるため、三酸化硫黄と塩素系有機溶媒との反応を抑制し、炭化物に対するスルホン酸基の保持量の低下を防ぐことができる。
【図面の簡単な説明】
【0043】
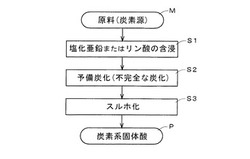
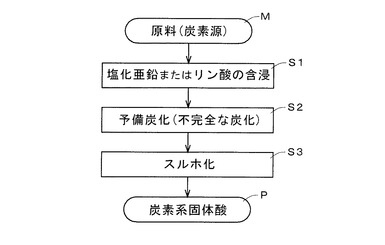
【図1】本発明の製造方法に係る概略工程図である。
【発明を実施するための形態】
【0044】
本発明の炭素系固体酸を形成する主原料は、純粋セルロース等のセルロース含有原料をはじめ、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種を炭素源(出発原料)とし、これらの植物系原料が炭素系固体酸を構成する構造骨格となる。植物系原料として、例えば、木材、間伐材、建築廃木材、オガ屑(オガコ)、椰子殻、コーヒーの出し殻、クルミの殻、桃などの果実の種子、パルプ製造時の副生成物、リグニン廃液、製糖廃棄物、廃糖蜜、海藻、レーヨン、セロハン等を列記することができる。植物系原料は未焼成物であっても焼成物(ただし不完全な焼成物である)であってもよい。これらの原料の特徴としては、いずれもセルロースを構成成分として有しており、比較的容易かつ、大量に調達できる材料に由来する。セルロースは焼成後に炭素の環構造を保持しやすいため好ましく用いられる。
【0045】
前掲の炭素系固体酸を形成する他の原料としては、例えば、ベンゼン、アントラセン、ペリレン、コロネン、またはそのスルホ化合物より選択される、少なくとも1種の多環式芳香族炭化水素類を使用することができ、好ましくはナフタレンなどを使用することができる。また、芳香族炭化水素類を含む重油、ピッチ、タール、アスファルト等も使用することができる。以上の有機物は、単独で使用してもよいし、2種類以上の複数種の混合物であってもよい。
【0046】
不完全(中途半端)な炭化とは、10〜20個の芳香族6員環からなる多環式芳香族炭化水素で構成されたアモルファスカーボンであり、一例としてはベンゼン環が10〜20個並んだ状態のものである。粉末X線回析パターンにおいては、半値幅(2θ)が5〜30°の炭素(002)面の回析ピークが検出されるような状態のものである。これは、有機物を完全に炭化すると、炭素だけになってしまい、後記するスルホン酸基が結合できないことから必須となる。すなわち、この要件は、有機物を不完全に炭化することで、炭素と水素との結合を残存させて、そこにスルホン酸基を結合させるようにする。
【0047】
これより、図1の概略工程図を踏まえ炭素系固体酸の製造方法から説明する。まず、前出の炭素源となるセルロース含有原料、あるいは植物系原料が準備される(M)。続いて原料(炭素源:セルロース含有原料、植物系原料)に塩化亜鉛、またはリン酸が含浸される(S1)。含浸と併せて原料(炭素源)は焼成(予備炭化、不完全な炭化)され(S2)、予備炭化物が得られる(多孔質炭素となる)。予備炭化物(多孔質炭素)に濃硫酸または発煙硫酸中あるいは三酸化硫黄ガスが添加されて80〜350℃の温度域でスルホ化され(S3)、予備炭化物にスルホン酸基(−SO2(OH))が導入されたスルホ化物が得られる。こうして炭素系固体酸(P)が出来上がる。なお、スルホン酸基はスルホ基とも称される。
【0048】
原料(炭素源)の準備(M)、特には植物系原料においては、その原料の水分含量、粒径等を可能な限り均一化するべく乾燥、篩い分け等の処理が行われる。水分含量、粒径にばらつきが多くなると、後述の焼成にむらが生じ品質の安定が得られないためである。乾燥時間や粒径は、植物系原料の種別、最終的な形態に応じて適切に設定される。
【0049】
S1,S2の塩化亜鉛、リン酸の含浸及び焼成は、前出の原料(炭素源)に対する賦活処理及び予備炭化に相当する。この時点で多孔質が発達する。当該S1,S2の工程は、原料(炭素源:セルロース含有原料、植物系原料)に対する不完全な炭化ということができる。すなわち、完全に炭化されて炭素のみの構造骨格となっているわけではなく、水素をはじめ、一部に他の官能基等を残存させている状態である。特に、植物系原料の場合、複雑な成分を有する天然物であるため、顕著である。なお、前記S1の塩化亜鉛やリン酸による賦活処理が省略され、原料(炭素源)の予備炭化(不完全な炭化)の後、スルホ化される場合もある(後記実施例における試作例21ないし24参照)。
【0050】
多孔質炭素としては、一般に活性炭が知られている。通常、活性炭は水蒸気または炭酸ガス等を用いたガス賦活により製造されることが多い。ガス賦活は800℃〜1000℃の温度条件下で行われる。このような温度下で得られた活性炭は、加熱によって黒鉛微結晶(以下、グラフェンシート様炭素という。)が発達する。これに対して、塩化亜鉛やリン酸による薬品賦活の場合、ガス賦活よりも低温度である200〜700℃の温度域において予備炭化温度を制御することができる。ガス賦活と比較してより微細なグラフェンシート様炭素の状態が残存すると予想され、炭素表面または炭素端面の反応部位にスルホ化によるスルホン酸基の付与量の増加が期待できる。
【0051】
原料(炭素源)に対する塩化亜鉛(ZnCl2)の含浸に際し、塩化亜鉛は水または希塩酸等に溶解され、濃度50〜70%(w/w)に調製される。植物系原料にあっては、その100重量部に対し、塩化亜鉛の重量換算として、塩化亜鉛はおよそ200〜400重量部(2〜4重量倍)、好ましく300〜350重量部(3〜3.5重量倍)含浸される。植物系原料に含浸させる塩化亜鉛が2重量倍を下回る場合、賦活後に得られる炭素質の細孔の発達が悪く十分な比表面積を得ることができない。植物系原料に含浸させる塩化亜鉛が4重量倍を上回る場合、反応が進行しすぎて炭素質の多孔質構造が脆弱化する。そこで、触媒としての作用や実際の取り扱いの便宜を考慮して、前記の含浸量が好適となる。
【0052】
塩化亜鉛の含浸後の焼成(予備炭化)は、200〜600℃、好ましくは400〜550℃の温度域で塩酸を含んだ状態で行われる。予備炭化は設備、規模、植物系原料の種類やその形状により40〜120分間行われる。予備炭化は、大気中で行うこともできるものの、過剰な炭化を抑制するため、窒素ガス、炭酸ガス、ヘリウムガスの通気、あるいは燃焼時の排気ガスを循環させること等の不活性ガス雰囲気下における加熱として行われる。嫌気性条件とすることにより、過剰な炭化を抑制できる。予備炭化は、後述の実施例のように塩化亜鉛賦活の段階と塩酸含浸後の加熱の2段階として行うことも可能である。
【0053】
低めの温度とする場合、細孔の発達を加味して炭化時間は長く設定さる。高めの温度とする場合、賦活が進みすぎないように炭化時間は短く設定される。得られた予備炭化物は、濃度0.2〜35%の十分量の塩酸により洗浄され、乾燥される。洗浄には塩酸の代わりに硝酸、硫酸を用いることもできる。
【0054】
セルロース含有原料もしくは前出の植物系原料にリン酸(H3PO4)を含浸させるときには、リン酸は、濃リン酸ないし濃度50%(w/w)のリン酸溶液が用いられる。例えば、植物系原料100重量部に対し、リン酸の重量換算として、リン酸はおよそ100〜200重量部(1〜2重量倍)含浸される。植物系原料に含浸させるリン酸が1重量倍を下回る場合、賦活後に得られる炭素質の細孔の発達が悪く十分な比表面積を得ることができない。植物系原料に含浸させるリン酸が4重量倍を上回る場合、反応が進行しすぎて炭素質の多孔質構造が脆弱化する。そこで、触媒としての作用や実際の取り扱いの便宜を考慮して、前記の含浸量が好適となる。
【0055】
リン酸の含浸後の焼成(予備炭化)は、リン酸を含んだ状態のまま200〜600℃の温度域で行われる。予備炭化は設備、規模、植物系原料の種類やその形状によるものの40〜120分間行われる。低めの温度とする場合、細孔の発達を加味して炭化時間は長めとなる。高めの温度とする場合、賦活が進みすぎないように炭化時間は短めとなる。得られた予備炭化物は、十分な量の水、温水により洗浄され、乾燥される。なお、使用できるリン酸には、H3PO4の他にピロリン酸等も含められる。
【0056】
原料(炭素源)、特には植物系原料に対する予備炭化の温度と触媒となる炭素系固体酸の用途の間には、後記の実施例から把握されるように、関連性が確認できる。そこで、植物系原料の予備炭化の加熱処理温度が200〜600℃であるとするならば、当該炭素系固体酸は縮合、分解反応の触媒として効果的である。より詳しく説明すると、植物系原料の予備炭化の加熱処理温度が200〜450℃であるとするならば、当該炭素系固体酸は加水分解反応の触媒として効果的である。次に、植物系原料の予備炭化の加熱処理温度が200〜500℃とするならば、当該炭素系固体酸はエステル化反応の触媒として効果的である。さらに、植物系原料の予備炭化の加熱処理温度が400〜500℃であるとするならば、当該炭素系固体酸はアルキル化反応の触媒として効果的である。好適な触媒反応種と予備炭化の加熱温度との関連性は現在のところ十分に解明されていない。おそらく、細孔の発達、後述するスルホン酸基の修飾量の差異が影響していることが推察される。
【0057】
スルホ化の工程(S3)にあっては、予備炭化物をそのまま、もしくは適当に粉砕して粒径を調整した後、植物系原料に由来する予備炭化物は濃硫酸中または発煙硫酸中に浸漬される。あるいは、予備炭化物は三酸化硫黄ガスが充満したチャンバ内に搬送される。こうして、80〜350℃の温度条件下、好ましくは80〜150℃の温度条件下で加熱(スルホ化)され、植物系原料に由来する予備炭化物のスルホ化が促進し、賦活により多孔質化した炭化物表面にスルホン酸基が導入されスルホ化物が得られる。
【0058】
また、前記のスルホ化の工程(S3)は、不活性ガス、または乾燥空気流中において行われる。硫酸は非常に水分を吸収しやすい。そこで、水分をできる限り抑制することにより、炭素表面に導入するスルホン酸基濃度の低下を防ぐ必要からの措置である。
【0059】
スルホ化の加熱温度が低すぎるとスルホン酸基導入の反応が遅くなる。温度を高くしすぎる場合、炭化が進みすぎ、収量が減少する。また、温度、設備、処理量等の影響を受けるものの、スルホ化の加熱時間は、30分〜20時間、好ましくは1〜10時間行われる。加熱時間が短ければスルホン酸基の導入は不十分となる。加熱時間を長くしすぎても反応が頭打ちになった後の加熱はエネルギーの無駄である。なお、スルホ化に際しても、予備炭化と同様に窒素ガス、炭酸ガス、ヘリウムガスの通気、あるいは燃焼時の排気ガスを循環させること等の不活性ガス雰囲気下において加熱が行われる。スルホ化の後、当該スルホ化物は水洗され硫酸等の成分が除去され、乾燥される。
【0060】
各処理を経ることにより、本発明に好適な炭素系固体酸(P)が得られる。そこで、当該炭素系固体酸が、好適な触媒活性等をはじめとする作用を発揮する上で具備すべき物理的条件は、スルホン酸基を導入した後、つまりスルホ化後における多孔質炭素のBET比表面積が3〜1600m2/gを満たすことである。当該比表面積が3m2/gを下回る場合、細孔の発達が不十分であり、反応効率を求めることが難しくなる。また、比表面積1600m2/gは現状の製造方法における上限と考えられる。BET比表面積の測定の詳細は、後記の実施例にて述べる。簡単には77K(およそ−195℃)における窒素吸着等温線からBET法により求めた比表面積(m2/g)である。比表面積の最適範囲は、炭素系固体酸を適用する反応種によっても変動する。
【0061】
さらに、既述の各処理を経て得られた炭素系固体酸(P)のスルホン酸基の量は、0.2mmol/g以上、より好ましくは0.5mmol/g以上を満たすことである。スルホン酸基の測定の詳細も後記の実施例にて述べる。簡単には酸−アルカリの中和反応の滴定や元素分析による算出により可能である。後記の実施例から把握できるように、出来上がった炭素系固体酸の分析結果を勘案して0.2mmol/g以上となる。そのうち、炭素系固体酸のスルホ基の量は多くなるほど触媒反応は促進する。概ね0.5mmol/g以上の試料からは良好な触媒活性が確認できたためである。
【0062】
炭素系固体酸を粉末状の触媒として用いる場合、通常、多孔質炭素は塊状物で得られるため、粉砕された上で用いられる。粉砕後の粒径(メジアン径)は、1〜100μm、好ましくは2〜50μm、より好ましくは5〜20μmである。粒径の分布が均一であるほど反応後の分離、回収が容易となるためである。粉砕に際し、ボールミル、ハンマーミル、ジェットミル等の公知の粉砕装置が用いられる。また、炭素系固体酸を固定床に敷設する触媒として用いる場合、多孔質炭素塊状物は適宜の篩別により、0.1〜4mm、好ましくは0.2〜3mm、より好ましくは0.4〜2.5mmの粒状物に分けられる。むろん、粉末、粒状のいずれにおいても、当該炭素系固体酸を適用する反応、設備、耐久性等の諸要因を勘案して粒径は規定される。
【0063】
これまでの説明は主に原料(炭素源)としてセルロース含有原料や植物系原料を用い、これに濃硫酸、発煙硫酸を反応させてスルホ化する処理である。続いて、原料(炭素源)となるセルロース含有原料や植物系原料の不完全な炭化物、つまりは、前述の塩化亜鉛やリン酸による賦活を終えて予備炭化を経た予備炭化物等に対する三酸化硫黄によるスルホ化を説明する。
【0064】
すなわち、原料(炭素源)となるセルロース含有原料や植物系原料は不完全に炭化され(図1のS1,S2の工程参照)、これに三酸化硫黄が接触されることにより、炭化不完全な原料の縮合、スルホ化が行われる。ここで、炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に三酸化硫黄を接触させる場合、窒素、アルゴン等の不活性ガス気流下、あるいは乾燥空気気流下で行うことがスルホ基密度の高い炭素系固体酸を製造する上で重要となる。
【0065】
炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に三酸化硫黄を接触させる方法としては、液体状または気体状の三酸化硫黄を直接接触(含浸)させてもよいし、三酸化硫黄を含有する液状物またはガス状物として接触(含浸)させるようにしてもよい。
【0066】
液体状または気体状の三酸化硫黄を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に直接接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、液体状または気体状の三酸化硫黄を徐々に加えることが望ましい。ただし、それに限られず、液体状または気体状の三酸化硫黄に対し炭化不完全な有機物を徐々に加えるようにしてもよい。
【0067】
三酸化硫黄を含有するガス状物を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、三酸化硫黄を含有するガス状物を徐々に加えることが望ましい。ただし、この場合も、三酸化硫黄を含有するガス状物に炭化不完全な有機物を徐々に加えてもよい。ガス状物に含まれる三酸化硫黄以外のガス成分としては、スルホ化反応に不活性な気体であれば特に制約はなく、例えば乾燥空気、窒素やアルゴン等が挙げられる。
【0068】
三酸化硫黄を含有する液状物を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、三酸化硫黄を含有する液状物を徐々に加えることができる。この場合も、三酸化硫黄を含有する液状物に炭化不完全な有機物を徐々に加えてもよい。また、炭化不完全な有機物を予め三酸化硫黄を含まない液状物に分散させておき、三酸化硫黄を含有する液状物または三酸化硫黄を徐々に加えてもよいし、三酸化硫黄を含有する液状物または三酸化硫黄に、三酸化硫黄を含まない液状物に分散させた炭化不完全な有機物を徐々に加えてもよい。液状物に含まれる三酸化硫黄以外の液体成分としては、スルホ化反応に不活性な液体であれば特に制約はなく、例えばジクロロメタン、エチレンジクロライド等の塩素系有機溶媒等が挙げられる。
【0069】
三酸化硫黄による処理(縮合、スルホ化)温度としては、スルホ化反応を進行させる温度であれば特に限定されないが、通常は0℃から100℃であり、好ましくは20℃から80℃である。
【0070】
三酸化硫黄による処理(縮合、スルホ化)時間は、反応温度や目標とするスルホ基の導入率にも依存するが、通常は1分から24時間、好ましくは30分から10時間である。また、三酸化硫黄による処理(縮合、スルホ化)圧力は、スルホ化反応の進行させる圧力であれば特に限定されない。
【0071】
炭化不完全な有機物(予備炭化物)の縮合、スルホ化に使用するスルホ化剤は、通常の三酸化硫黄であるが、塩素系有機溶媒等を用いる場合には、安定剤を含まない三酸化硫黄が好適である。
【0072】
ここで、安定剤を含まない三酸化硫黄とは、三酸化ホウ素、ジメチル硫酸等の安定化剤を敢えて含まない三酸化硫黄である。これらの安定剤は、ジクロロメタンにおける「C−Cl」の結合内にSO3を挿入する反応を触媒してしまうことが知られているためである。反応系に安定剤入りの三酸化硫黄を添加すると、炭化物にスルホ基として結合する前にジクロロメタン等の塩素系有機溶媒と反応してしまい、SO2による架橋生成の原因となる。このため、炭化物に対するスルホ基の保持量の低下を防ぐためにも、安定剤を含まない三酸化硫黄の使用が良好な結果をもたらす。
【0073】
三酸化硫黄の添加量は、炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に導入したいスルホ基の総量によるが、通常は当該炭化不完全な有機物の重量(予備炭化物の重量)に対して、三酸化硫黄重量として、1〜500重量%、好ましくは5〜300重量%、さらには10〜100重量%が好ましい。これは、三酸化硫黄の添加量が少なすぎると、炭化不完全な有機物(予備炭化物)へのスルホ基導入率が低くなり、多すぎると余剰の三酸化硫黄の除去に多大な時間や経費が必要となるので実用的ではないからである。一方、炭化不完全な有機物(予備炭化物)を縮合、スルホ化した後、反応液である三酸化硫黄または三酸化硫黄を含有したスルホ化剤より固体酸を採取する方法は、実施例で行ったような通常の方法に従って行うことができる。
【0074】
以上の炭素系固体酸の製造方法によれば、従来のごとく濃硫酸または発煙硫酸を使用せず、塩素系有機溶媒を用いる場合にも、スルホ架橋用の抑制剤等を使用せずに、高収率で再現性にも優れ、硫酸に匹敵するような炭素系固体酸が得られる。
【0075】
一連の説明から理解されるように、製造される炭素系固体酸は、プロトン伝導性材料や固体酸触媒として好適となるとともに、耐久性つまり耐熱性、耐酸性あるいは化学的安定性にも優れ、コスト低減も図られることから、イオン交換体、プロトン伝導性材料、電解質膜、反応触媒等として非常に有用である。むろん、本発明の炭素系固体酸を利用して固体電解質膜を作製し、それを用いて膜電極接合体や燃料電池を作製することが可能である。
【実施例】
【0076】
〔炭素系固体酸の試作(発煙硫酸によるスルホ化)〕
炭素源となる植物系原料として米松(ベイマツ)の大鋸粉(オガコ)を篩により篩別し、篩別したオガコの木粉を用い、以下に記載の試作例の手順に基づいて塩化亜鉛の含浸、予備炭化、発煙硫酸によるスルホ化を行い、炭素系固体酸を試作した。スルホ化物が各試作例の炭素系固体酸となる。
【0077】
〈試作例1〉
オガコを105±5℃に保った乾燥機内で8時間乾燥後、4.7〜83meshの篩(粒径180〜4000μmに相当)により篩別し、木粉20gを取り分けた。木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。350℃まで60分間かけて昇温し、その後350℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。
【0078】
予備炭化物10gを300mLの15%発煙硫酸に加えて80℃で10時間加熱しスルホ化した。その後、過剰な濃硫酸を100℃の蒸留水で繰り返し洗浄し、洗浄後の蒸留水中の硫酸が元素分析の検出限界以下になるまで洗浄を繰り返した。水洗後、105±5℃に保った乾燥機内で8時間乾燥してスルホ化物を得た(比表面積:15m2/g,スルホン酸基量:1.27mmol/g)。試作例1の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0079】
〈試作例2〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。400℃まで70分かけて昇温し、その後400℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例2の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:155m2/g,スルホン酸基量:1.34mmol/g)。試作例2の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0080】
〈試作例3〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。450℃まで80分間かけて昇温し、その後450℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、ボールミルにより粉砕後した。粉砕品を83meshの篩(粒径180μmに相当)により篩別し、篩下物を予備炭化物とした。試作例3の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:818m2/g,スルホン酸基量:1.27mmol/g)。試作例3の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0081】
〈試作例4〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。250℃まで40分間かけて昇温し、その後250℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例4の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:3m2/g,スルホン酸基量:1.33mmol/g)。試作例4の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0082】
〈試作例5〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。300℃まで50分間かけて昇温し、その後300℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例5の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:4m2/g,スルホン酸基量:1.27mmol/g)。試作例5の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0083】
〈試作例6〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。500℃まで90分間かけて昇温し、その後500℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例6の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1345m2/g,スルホン酸基量:1.04mmol/g)。試作例6の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0084】
〈試作例7〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。550℃まで100分間かけて昇温し、その後550℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例7の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1492m2/g,スルホン酸基量:0.70mmol/g)。試作例7の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0085】
〈試作例8〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。600℃まで110分間かけて昇温し、その後600℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例8の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1453m2/g,スルホン酸基量:0.47mmol/g)。試作例8の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0086】
〈試作例9〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。200℃まで30分間かけて昇温し、その後200℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例9の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:3m2/g,スルホン酸基量:1.34mmol/g)。試作例9の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0087】
〈試作例10〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。200℃まで30分間かけて昇温し、その後200℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を10〜22meshの篩により篩別し、予備炭化物を得た。試作例10の予備炭化物に対するスルホ化は、試作例1と同一の条件により行った。スルホ化時に試作例10の予備炭化物は形状維持ができず砕けてしまい、回収することができなかった。このため、以降の測定は実施できなかった。
【0088】
〈試作例11〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。300℃まで50分間かけて昇温し、その後300℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を10〜22meshの篩により篩別し、予備炭化物を得た。試作例11の予備炭化物に対するスルホ化は、試作例1と同一の条件により行った。スルホ化時に試作例11の予備炭化物も形状維持ができず砕けてしまい、回収することができなかった。このため、以降の測定は実施できなかった。
【0089】
〈試作例12〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。400℃まで70分間かけて昇温し、その後400℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例12の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:5m2/g,スルホン酸基量:0.84mmol/g)。
【0090】
〈試作例13〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。500℃まで90分間かけて昇温し、その後500℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例13の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:20m2/g,スルホン酸基量:0.29mmol/g)。
【0091】
〈試作例14〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。600℃まで110分間かけて昇温し、その後600℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例14の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:13m2/g,スルホン酸基量:0.10mmol/g)。
【0092】
〔物性の測定方法〕
・比表面積
試作例の炭素系固体酸の比表面積(m2/g)はBET法に基づいて測定した。実施例及び比較例の試料を200℃の窒素雰囲気下において3時間乾燥した後、77Kにおける窒素吸着等温線を日本ベル株式会社製:「BELSORP MINI」により測定した。
【0093】
・スルホン酸基量の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、2mgを分取し、エレメンタール社製:CHNS元素分析計vario Micro cubeを用いてS(硫黄分)の量(mmol/g)を測定し、等価としてスルホン基酸量(mmol/g)を求めた。
【0094】
〔触媒活性の測定〕
・加水分解反応の測定
各試作例の炭素系固体酸より0.3gを分取し、これにセロビオース0.025g(73.04μmol)、水0.7gを添加し、90℃を維持しながら1時間反応させた。反応液をHPLCに装填し、グルコース等の単糖類のピーク面積比よりセロビオースから分解されて生成した糖類量を求めた。
【0095】
・エステル化反応の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、0.2g分取し、これに酢酸0.1mol、エタノール1.0molを添加し、70℃を維持しながら3時間反応させた。反応液中に含まれる酢酸エチルの生成量を株式会社島津製作所製のFID−ガスクロマトグラフィー:GC−17Aにより求めた。
【0096】
・アルキル化反応の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、0.2g分取し、これにトルエン100mmol、塩化ベンジル(クロロメチルベンゼン)10mmolを添加し、100℃を維持しながら4時間反応させた。反応液中に含まれるベンジルトルエンの生成量をガスクロマトグラフィーにより求めた。
【0097】
各試作例について、予備炭化時の植物系原料(木粉)に対する塩化亜鉛換算の添加重量比、予備炭化条件(焼成温度(℃)、昇温時間(分)、維持時間(分))、スルホン酸基量(スルホ基量)(mmol/g)、加水分解量(μmol)、エステル化反応量(mmol/g・min)、アルキル化反応量(μmol/g・min)、及び比表面積(m2/g)を表1にまとめた。表では、賦活剤を用いた試作例と賦活剤を用いなかった試作例の双方をそれぞれ予備炭化時の焼成温度の低い方から順に並べた。
【0098】
【表1】

【0099】
〔結果,考察〕
・加水分解反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下、200〜450℃の焼成温度域で予備炭化して得た試作例については、加水分解反応において良好な触媒作用の結果を示した。予備炭化時の温度が高くなるほど性能が低下することから、前記の焼成温度域にすることが望ましい。
【0100】
・エステル化反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下において、200〜500℃の焼成温度域で予備炭化して得た試作例については、エステル化反応において良好な触媒作用の結果を示した。予備炭化時の温度が高くなるほど性能が低下することから、前記の焼成温度域にすることが望ましい。
【0101】
・アルキル化反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下において、400〜500℃の焼成温度域で予備炭化して得た試作例については、アルキル化反応において良好な触媒作用の結果を示した。同反応の場合、焼成温度が400℃を下回る場合及び500℃を上回る場合に極端に効果が低下する。
【0102】
一般に焼成温度が高くなるほど比表面積は大きくなるため、反応性は向上していることが予想できる。しかしながら、スルホン酸基(スルホ基)の量は、焼成温度500℃までは概ね安定している。このため、炭素表面に発達した細孔と、触媒反応の対象となる分子の大きさ、構造、性質との親和性や相互作用等により、予備炭化時の焼成温度の違いから触媒活性の差異が生じたものと予想できる。また、植物系原料を賦活、炭化して炭素系固体酸を得ているため、炭素表面に残存する官能基量も影響していると考えられる。なお、賦活剤を用いず低温度域で予備炭化して、スルホ化を行った試作例では形状維持が困難となったことから、本発明の硫酸や発煙硫酸によるスルホ化を伴うスルホン酸基を有する炭素系固体酸には薬品賦活が必須である。
【0103】
以上の知見から明らかなように、塩化亜鉛の含浸に伴う薬品賦活に際し、その焼成時の温度域を調整することによって、目的とする反応に対応した活性を発現する触媒を得ることができた。よって、植物系原料を用いたものであっても、反応種や用途に応じた触媒の作り分けが簡便かつ容易となり、実需要に極めて柔軟に応えることができる炭素系固体酸を安価に提供することができる。
【0104】
〔炭素系固体酸の試作(三酸化硫黄によるスルホ化)〕
続いて、発明者らは三酸化硫黄によるスルホ化を試みた。炭素源として、セルロース含有原料に微結晶セルロース(メルク社製)、植物系原料(大鋸粉(オガコ))由来の顆粒活性炭(フタムラ化学株式会社製「SG」,7.5〜42mesh(粒径355〜400μmに相当))、粉末活性炭(フタムラ化学株式会社製「S」,メジアン径42.5μm(株式会社島津製作所製:SALD−200Vによる測定))を用い、以下に記載の各試作例の手順に基づいて三酸化硫黄によるスルホ化を行い、炭素系固体酸を試作した。スルホ化物が各試作例の炭素系固体酸となる。試作例に用いた2種類の植物系原料由来の活性炭は、いずれも塩化亜鉛により賦活し、450℃で大気下において予備炭化、焼成した活性炭である。
【0105】
炭化不完全な有機物の縮合、スルホ化するために使用したロータリーエバポレーターは、ビュッヒ・ラボテクニック社(BUCHI Labortechnik AG(スイス))製、ROTAVAPOR RE120である。使用フラスコはナス型フラスコ:1L容積を使用した。また、評価は、固体酸中の硫黄分測定を成分ガス分析・計測装置として、浜田理科株式会社製:燃焼フラスコ(FHO−A型)装置により測定した。
【0106】
〔セルロース含有原料の不完全な炭化〕
前出の微結晶セルロース(メルク社製)20gを、500mL容積の三つ口フラスコに入れ、窒素ガス気流下、450℃で5時間加熱し、9gの不完全な炭化物を得た(以下、これを「不完全炭化物」という)。この操作を繰り返すことで所定重量の不完全炭化物を確保し、必要量を計量して使用した。
【0107】
〈試作例21〉
前記の不完全炭化物の所定重量を110℃で1時間乾燥した。前記ロータリーエバポレーターは、コンデンサー上部に注入コックを有し、その注入コックに三酸化硫黄ガス吹込みラインを接続し、エバポレーター内を窒素ガスで置換した。不完全炭化物20.2gを1L容積のナス型フラスコに入れた後、該ナス型フラスコをエバポレーターに取り付け、窒素ガスで再度置換した。そして、該ナス型フラスコを60℃に加温して回転(約80rpm)させるとともに、真空ポンプによりエバポレーター内を脱気(0.5kPa)し、密閉した。
【0108】
三酸化硫黄(日曹金属化学株式会社製:製品名「日曹サルファン」)6.1gをガス化用三つ口フラスコに量り取った。エバポレーターのコンデンサー上部にある注入コックを開けて、三酸化硫黄を徐々にエバポレーター内に導入した。そして、三酸化硫黄を導入した後、不完全炭化物と三酸化硫黄の反応を、ナス型フラスコを回転(約80rpm)させながら60℃で2時間行った。反応後は、三酸化硫黄ガス吹込みラインを切り離し、エバポレーター内の三酸化硫黄ガスを窒素ガスで置換した。
【0109】
ナス型フラスコをエバポレーターから外し、該ナス型フラスコ内に約500mLの蒸留水を加えて10分間攪拌した。温度は30℃以下に保持した。その後は、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。その後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸20.9gを得た。同炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、0.94wt%であった(試作例21の炭素系固体酸)。
【0110】
〈試作例22〉
前記の不完全炭化物の所定重量を110℃で1時間乾燥した。500mL容積の三つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、内部を窒素ガスで置換した。そして、三つ口フラスコに乾燥した不完全炭化物20.0gとジクロロメタン271.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0111】
三酸化硫黄として、安定剤を添加しない液体状の三酸化硫黄30.1gを滴下ロートにて量り取った後、前記の三つ口フラスコ内にゆっくりと滴下した。滴下後、(不完全炭化物+ジクロロメタン)と液体状の三酸化硫黄とを25〜30℃で、2時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン300mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約500mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0112】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。乾燥後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸24.2gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、5.26wt%であった(試作例22の炭素系固体酸)。
【0113】
〈試作例23〉
前出の微結晶セルロース(メルク社製)500gを、5000mL容積のセパラブルフラスコに入れ、窒素ガス気流下、400℃で1時間加熱し、150gの不完全炭化物を得るとともに、80℃で1日間乾燥した。この不完全炭化物20.2gを1L容積のナス型フラスコに入れた後、該ナス型フラスコを試作例21と同様のロータリーエバポレーターに取り付け、窒素ガスで再度置換した。そして、該ナス型フラスコを60℃に加温して回転(約80rpm)させるとともに、真空ポンプによりエバポレーター内を脱気(0.5kPa)し、密閉した。
【0114】
三酸化硫黄5.8gをガス化用三つ口フラスコに量り取った。エバポレーターのコンデンサー上部にある注入コックを開けて、気化した三酸化硫黄を徐々にエバポレーター内に導入した。そして、三酸化硫黄を導入した後、不完全炭化物と三酸化硫黄の反応を、ナス型フラスコを回転(約80rpm)させながら60℃で2時間行った。反応後は、三酸化硫黄ガス吹込みラインを切り離し、エバポレーター内の三酸化硫黄ガスを窒素ガスで置換した。
【0115】
ナス型フラスコをエバポレーターから外し、該ナス型フラスコ内に約500mLの蒸留水を加えて10分間攪拌した。温度は30℃以下に保持した。その後は、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。その後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸19.4gを得た。同炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、0.85wt%であった(試作例23の炭素系固体酸)。
【0116】
〈試作例24〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、内部を窒素ガスで置換した。そして、四つ口フラスコに試作例23にて調製し乾燥した不完全炭化物20.0gとジクロロメタン432.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0117】
日曹金属化学株式会社製:製品名「日曹サルファン」より安定剤を含まない三酸化硫黄(液状)を別途調製した。安定剤を含まない三酸化硫黄60.2gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0118】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸26.7gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、7.32wt%であった(試作例24の炭素系固体酸)。
【0119】
〈試作例25〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、四つ口フラスコに植物系原料(オガコ)由来の顆粒活性炭(フタムラ化学株式会社製「SG」)20.0gとジクロロメタン240.9gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0120】
試作例24にて用いた安定剤を含まない三酸化硫黄60.3gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0121】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸23.9gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、3.82wt%であった(試作例25の炭素系固体酸)。
【0122】
〈試作例26〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、四つ口フラスコに植物系原料(オガコ)由来の粉末活性炭(フタムラ化学株式会社製「S」)20.0gとジクロロメタン240.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0123】
試作例24にて用いた安定剤を含まない三酸化硫黄60.2gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0124】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸23.8gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、3.88wt%であった(試作例26の炭素系固体酸)。
【0125】
試作例23ないし26の結果について、原料、焼成雰囲気、賦活剤、スルホ化剤、スルホ化剤の状態(ガス状または液体)、スルホ基量(mmol/g)、比表面積(m2/g)を表2のとおりまとめた。表中のスルホ基量(スルホン酸基量)、比表面積は、前掲の物性の測定方法と同様である。
【0126】
【表2】
【0127】
〔三酸化硫黄によるスルホ化の結果と考察〕
スルホ化剤の反応においては、液体状の三酸化硫黄を使用する方が炭素系固体酸の単位重量あたりのスルホ基量の多くすることができる。なお、試作例24,25,26にあっては、発煙硫酸によるスルホ化(表1参照)と比較しても遜色ない。また、オガコ由来の活性炭製品をスルホ化しても十分なスルホ基量を得ることができることから、調達容易な原料を用いての量産も有望である。さらに、三酸化硫黄によるスルホ化では、硫酸等を用いた際のスルホ化よりも反応条件が比較的穏和であり、短時間となることから生産性の向上やエネルギー消費量の抑制等に有効であるといえる。なお、三酸化硫黄によるスルホ化により生じた炭素系固体酸のスルホ基量及び表1の結果を勘案すると、同様に触媒効果を示すものと予想できる。
【技術分野】
【0001】
本発明は、炭素系固体酸及びその製造方法に関し、炭素材料の表面にスルホン酸基を導入して得た炭素系固体酸、並びに当該炭素系固体酸の製造方法に関する。
【背景技術】
【0002】
硫酸は高い活性を有し、炭化水素化合物を反応させる際の触媒としても広く利用される。例えば、遊離高級脂肪酸とアルコールとを反応させて、高級脂肪酸エステルを得るエステル化反応の促進、セルロース等の糖鎖から単糖への加水分解反応の促進、その他、炭化水素燃料を合成するアルキル化反応の促進等の用途である。
【0003】
硫酸は触媒として各種の反応促進に寄与した後、中和、洗浄され、その都度消費されていた。硫酸は液体であるため回収が容易ではない。回収処理と新規投入との経費差から、現状は使い捨てが主流である。しかし、使用済みの硫酸の中和、洗浄に加え、環境基準に準拠した排水処理までを考慮すると、この負担は大きい。このことから、触媒として連続使用に耐えうるとともに、反応後の分離、回収に容易なより利便性の高い触媒が求められるようになってきた。
【0004】
そのような触媒として固体酸が挙げられる。例えば、硫酸処理を施したジルコニア、PTFEにスルホン酸基を導入したフッ素樹脂である。前記のジルコニアの場合、単位重量あたりのスルホン酸基濃度が低いため、触媒活性が低い欠点がある。また、前記のフッ素樹脂に関しては、熱に弱く、適用できる反応種が限られている問題がある。
【0005】
そこで、十分な触媒活性と耐熱性も併せ持つ固体酸として、炭素系の固体酸が提案された(特許文献1、特許文献2参照)。特許文献1の固体酸は、多環式芳香族炭化水素を濃硫酸中で加熱処理して得ることができる。
【0006】
さらに、固体酸がその内部に細孔構造による適度な表面積(比表面積)を有していればより吸着が増す。このため、吸着界面における濃度がバルク相における濃度よりも高くなる。このことから、固体酸内部の吸着界面では溶媒中の溶質濃度が固体酸表面と比較して高くなり、細孔構造を有する固体酸のほうが反応を加速することができる。
【0007】
その後、発明者らは、炭素系の固体酸の性能向上を鋭意研究するうちに、安価に入手でき、しかも量産に適した原料、並びにその調製条件を見出すに至った。
【0008】
これと併せて発明者らは、濃硫酸、発煙硫酸中で加熱処理する方法に対する改良も試みた。すなわち、濃硫酸と多環式芳香族炭化水素類の反応により芳香族スルホン酸及び水が生成する。しかし、この反応は平衡反応であるため反応系内の水の増加により逆反応が進み、固体酸に導入されるスルホン酸基の量が低下する点を内包していた。この方法では、多量の濃硫酸、発煙硫酸を用いる必要があり、また、反応系内の水を除去しなければならないため高い反応温度で長時間維持する必要がある。さらに、加熱処理後の固体酸から濃硫酸、発煙硫酸を除去するために減圧蒸留としていたため製造コストが高くなる傾向にあった。
【0009】
また、固体状の芳香族加工物をスルホ化する方法としては、塩素系有機溶媒に溶解した状態でスルホ化剤(クロルスルホン酸、発煙硫酸、濃硫酸等)を反応させる方法が一般的に適用されている。例えば、ポリスチレンをスルホ化する際にエチレンジクロライドを溶媒として使用する方法が知られている。この場合、ポリスチレンの分子間や分子内でSO2により架橋が生成してしまう。このため、ルイス塩基や水、アルコール化合物、陰イオン系化合物、非イオン系化合物を反応性に添加したり、あるいは高剪断型反応機を用いたりして架橋の生成を抑える必要があった(例えば、特許文献3、4、5等参照)。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特許第4041409号公報
【特許文献2】WO2005/029508
【特許文献3】特公昭50−33838号公報
【特許文献4】特公昭51−37226号公報
【特許文献5】特公昭51−37227号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、上記状況に鑑み提案されたものであり、安価に入手でき、しかも量産に適した原料から製造可能な炭素固体酸を提供するとともに、当該炭素系固体酸の製造に好適な調製条件を満たす製造方法を提供する。併せて、濃硫酸や発煙硫酸を用いた反応よりも効率を高めた製造方法も提案する。
【課題を解決するための手段】
【0012】
すなわち、請求項1の発明は、スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であることを特徴とする炭素系固体酸に係る。
【0013】
BET比表面積に関して、試料を200℃の窒素雰囲気下において3時間乾燥した後、77K(−195℃)における窒素吸着等温線を日本ベル株式会社製BELSORP MINIにより測定し、BET法により比表面積(m2/g)を求めた。
【0014】
請求項2の発明は、前記多孔質炭素がセルロース含有原料に由来する請求項1に記載の炭素系固体酸に係る。
【0015】
請求項3の発明は、前記セルロース含有原料が植物系原料である請求項2に記載の炭素系固体酸に係る。
【0016】
請求項4の発明は、前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものである請求項1ないし3のいずれか1項に記載の炭素系固体酸に係る。
【0017】
請求項5の発明は、前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものである請求項3に記載の炭素系固体酸に係る。
【0018】
請求項6の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものである請求項5に記載の炭素系固体酸に係る。
【0019】
請求項7の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0020】
請求項8の発明は、前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0021】
請求項9の発明は、前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸に係る。
【0022】
請求項10の発明は、前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種である請求項3ないし9のいずれか1項に記載の炭素系固体酸に係る。
【0023】
請求項11の発明は、請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化することを特徴とする炭素系固体酸の製造方法に係る。
【0024】
請求項12の発明は、前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われる請求項11に記載の炭素系固体酸の製造方法に係る。
【0025】
請求項13の発明は、請求項4に記載の炭素系固体酸を製造するに際し、前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化することを特徴とする炭素系固体酸の製造方法に係る。
【0026】
請求項14の発明は、前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状である請求項13に記載の炭素系固体酸の製造方法に係る。
【0027】
請求項15の発明は、前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液である請求項14に記載の炭素系固体酸の製造方法に係る。
【発明の効果】
【0028】
請求項1の発明に係る炭素系固体酸によると、スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であるため、良好な触媒活性を得ることができる。特にスルホン酸基が寄与する良好な触媒活性を得ることができる。
【0029】
請求項2の発明に係る炭素系固体酸によると、請求項1の発明において前記多孔質炭素がセルロース含有原料に由来するため、焼成後に炭素の環構造を保持しやすい。
【0030】
請求項3の発明に係る炭素系固体酸によると、請求項2の発明において、前記セルロース含有原料が植物系原料であるため、比較的容易に、大量に調達できる
【0031】
請求項4の発明に係る炭素系固体酸によると、請求項1ないし3のいずれかの発明において、前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものであるため、必ずしも硫酸や発煙硫酸を用いることなく、低温度域で短時間の反応で得ることができた。
【0032】
請求項5の発明に係る炭素系固体酸によると、請求項3の発明において、前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものであるため、量産に適した原料から製造可能な触媒用多孔質炭素を得るに至った。
【0033】
請求項6の発明に係る炭素系固体酸によると、請求項5の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものであるため、量産に適した原料を用いて各種の反応に有効な炭素系固体酸を得ることができた。
【0034】
請求項7の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものであるため、加水分解反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0035】
請求項8の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものであるため、エステル化反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0036】
請求項9の発明に係る炭素系固体酸によると、請求項6の発明において、前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものであるため、アルキル化反応の触媒として有効に作用する温度域を見出して炭素系固体酸の触媒を得ることができた。
【0037】
請求項10の発明に係る炭素系固体酸によると、請求項3ないし9のいずれかの発明において、前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種であるため、原料を安価に入手でき、製造コストを圧縮することができる。
【0038】
請求項11の発明に係る炭素系固体酸の製造方法によると、請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化するため、賦活により多孔質化した炭化物表面へのスルホン酸基の導入を促進できる。
【0039】
請求項12の発明に係る炭素系固体酸の製造方法によると、請求項11の発明において、前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われるため、スルホン酸基と水分との接触を抑制して炭素表面に導入するスルホン酸基濃度の希釈を防ぐことができる。
【0040】
請求項13の発明に係る炭素系固体酸の製造方法によると、請求項4に記載の炭素系固体酸を製造するに際し、前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化するため、硫酸や発煙硫酸を用いることなく、より低温度域で短時間の反応によるスルホ化の製造方法を構築することができた。
【0041】
請求項14の発明に係る炭素系固体酸の製造方法によると、請求項13の発明において、前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状であるため、硫酸や発煙硫酸を用いないスルホ化が可能となった。
【0042】
請求項15の発明に係る炭素系固体酸の製造方法によると、請求項14の発明において、前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液であるため、三酸化硫黄と塩素系有機溶媒との反応を抑制し、炭化物に対するスルホン酸基の保持量の低下を防ぐことができる。
【図面の簡単な説明】
【0043】
【図1】本発明の製造方法に係る概略工程図である。
【発明を実施するための形態】
【0044】
本発明の炭素系固体酸を形成する主原料は、純粋セルロース等のセルロース含有原料をはじめ、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種を炭素源(出発原料)とし、これらの植物系原料が炭素系固体酸を構成する構造骨格となる。植物系原料として、例えば、木材、間伐材、建築廃木材、オガ屑(オガコ)、椰子殻、コーヒーの出し殻、クルミの殻、桃などの果実の種子、パルプ製造時の副生成物、リグニン廃液、製糖廃棄物、廃糖蜜、海藻、レーヨン、セロハン等を列記することができる。植物系原料は未焼成物であっても焼成物(ただし不完全な焼成物である)であってもよい。これらの原料の特徴としては、いずれもセルロースを構成成分として有しており、比較的容易かつ、大量に調達できる材料に由来する。セルロースは焼成後に炭素の環構造を保持しやすいため好ましく用いられる。
【0045】
前掲の炭素系固体酸を形成する他の原料としては、例えば、ベンゼン、アントラセン、ペリレン、コロネン、またはそのスルホ化合物より選択される、少なくとも1種の多環式芳香族炭化水素類を使用することができ、好ましくはナフタレンなどを使用することができる。また、芳香族炭化水素類を含む重油、ピッチ、タール、アスファルト等も使用することができる。以上の有機物は、単独で使用してもよいし、2種類以上の複数種の混合物であってもよい。
【0046】
不完全(中途半端)な炭化とは、10〜20個の芳香族6員環からなる多環式芳香族炭化水素で構成されたアモルファスカーボンであり、一例としてはベンゼン環が10〜20個並んだ状態のものである。粉末X線回析パターンにおいては、半値幅(2θ)が5〜30°の炭素(002)面の回析ピークが検出されるような状態のものである。これは、有機物を完全に炭化すると、炭素だけになってしまい、後記するスルホン酸基が結合できないことから必須となる。すなわち、この要件は、有機物を不完全に炭化することで、炭素と水素との結合を残存させて、そこにスルホン酸基を結合させるようにする。
【0047】
これより、図1の概略工程図を踏まえ炭素系固体酸の製造方法から説明する。まず、前出の炭素源となるセルロース含有原料、あるいは植物系原料が準備される(M)。続いて原料(炭素源:セルロース含有原料、植物系原料)に塩化亜鉛、またはリン酸が含浸される(S1)。含浸と併せて原料(炭素源)は焼成(予備炭化、不完全な炭化)され(S2)、予備炭化物が得られる(多孔質炭素となる)。予備炭化物(多孔質炭素)に濃硫酸または発煙硫酸中あるいは三酸化硫黄ガスが添加されて80〜350℃の温度域でスルホ化され(S3)、予備炭化物にスルホン酸基(−SO2(OH))が導入されたスルホ化物が得られる。こうして炭素系固体酸(P)が出来上がる。なお、スルホン酸基はスルホ基とも称される。
【0048】
原料(炭素源)の準備(M)、特には植物系原料においては、その原料の水分含量、粒径等を可能な限り均一化するべく乾燥、篩い分け等の処理が行われる。水分含量、粒径にばらつきが多くなると、後述の焼成にむらが生じ品質の安定が得られないためである。乾燥時間や粒径は、植物系原料の種別、最終的な形態に応じて適切に設定される。
【0049】
S1,S2の塩化亜鉛、リン酸の含浸及び焼成は、前出の原料(炭素源)に対する賦活処理及び予備炭化に相当する。この時点で多孔質が発達する。当該S1,S2の工程は、原料(炭素源:セルロース含有原料、植物系原料)に対する不完全な炭化ということができる。すなわち、完全に炭化されて炭素のみの構造骨格となっているわけではなく、水素をはじめ、一部に他の官能基等を残存させている状態である。特に、植物系原料の場合、複雑な成分を有する天然物であるため、顕著である。なお、前記S1の塩化亜鉛やリン酸による賦活処理が省略され、原料(炭素源)の予備炭化(不完全な炭化)の後、スルホ化される場合もある(後記実施例における試作例21ないし24参照)。
【0050】
多孔質炭素としては、一般に活性炭が知られている。通常、活性炭は水蒸気または炭酸ガス等を用いたガス賦活により製造されることが多い。ガス賦活は800℃〜1000℃の温度条件下で行われる。このような温度下で得られた活性炭は、加熱によって黒鉛微結晶(以下、グラフェンシート様炭素という。)が発達する。これに対して、塩化亜鉛やリン酸による薬品賦活の場合、ガス賦活よりも低温度である200〜700℃の温度域において予備炭化温度を制御することができる。ガス賦活と比較してより微細なグラフェンシート様炭素の状態が残存すると予想され、炭素表面または炭素端面の反応部位にスルホ化によるスルホン酸基の付与量の増加が期待できる。
【0051】
原料(炭素源)に対する塩化亜鉛(ZnCl2)の含浸に際し、塩化亜鉛は水または希塩酸等に溶解され、濃度50〜70%(w/w)に調製される。植物系原料にあっては、その100重量部に対し、塩化亜鉛の重量換算として、塩化亜鉛はおよそ200〜400重量部(2〜4重量倍)、好ましく300〜350重量部(3〜3.5重量倍)含浸される。植物系原料に含浸させる塩化亜鉛が2重量倍を下回る場合、賦活後に得られる炭素質の細孔の発達が悪く十分な比表面積を得ることができない。植物系原料に含浸させる塩化亜鉛が4重量倍を上回る場合、反応が進行しすぎて炭素質の多孔質構造が脆弱化する。そこで、触媒としての作用や実際の取り扱いの便宜を考慮して、前記の含浸量が好適となる。
【0052】
塩化亜鉛の含浸後の焼成(予備炭化)は、200〜600℃、好ましくは400〜550℃の温度域で塩酸を含んだ状態で行われる。予備炭化は設備、規模、植物系原料の種類やその形状により40〜120分間行われる。予備炭化は、大気中で行うこともできるものの、過剰な炭化を抑制するため、窒素ガス、炭酸ガス、ヘリウムガスの通気、あるいは燃焼時の排気ガスを循環させること等の不活性ガス雰囲気下における加熱として行われる。嫌気性条件とすることにより、過剰な炭化を抑制できる。予備炭化は、後述の実施例のように塩化亜鉛賦活の段階と塩酸含浸後の加熱の2段階として行うことも可能である。
【0053】
低めの温度とする場合、細孔の発達を加味して炭化時間は長く設定さる。高めの温度とする場合、賦活が進みすぎないように炭化時間は短く設定される。得られた予備炭化物は、濃度0.2〜35%の十分量の塩酸により洗浄され、乾燥される。洗浄には塩酸の代わりに硝酸、硫酸を用いることもできる。
【0054】
セルロース含有原料もしくは前出の植物系原料にリン酸(H3PO4)を含浸させるときには、リン酸は、濃リン酸ないし濃度50%(w/w)のリン酸溶液が用いられる。例えば、植物系原料100重量部に対し、リン酸の重量換算として、リン酸はおよそ100〜200重量部(1〜2重量倍)含浸される。植物系原料に含浸させるリン酸が1重量倍を下回る場合、賦活後に得られる炭素質の細孔の発達が悪く十分な比表面積を得ることができない。植物系原料に含浸させるリン酸が4重量倍を上回る場合、反応が進行しすぎて炭素質の多孔質構造が脆弱化する。そこで、触媒としての作用や実際の取り扱いの便宜を考慮して、前記の含浸量が好適となる。
【0055】
リン酸の含浸後の焼成(予備炭化)は、リン酸を含んだ状態のまま200〜600℃の温度域で行われる。予備炭化は設備、規模、植物系原料の種類やその形状によるものの40〜120分間行われる。低めの温度とする場合、細孔の発達を加味して炭化時間は長めとなる。高めの温度とする場合、賦活が進みすぎないように炭化時間は短めとなる。得られた予備炭化物は、十分な量の水、温水により洗浄され、乾燥される。なお、使用できるリン酸には、H3PO4の他にピロリン酸等も含められる。
【0056】
原料(炭素源)、特には植物系原料に対する予備炭化の温度と触媒となる炭素系固体酸の用途の間には、後記の実施例から把握されるように、関連性が確認できる。そこで、植物系原料の予備炭化の加熱処理温度が200〜600℃であるとするならば、当該炭素系固体酸は縮合、分解反応の触媒として効果的である。より詳しく説明すると、植物系原料の予備炭化の加熱処理温度が200〜450℃であるとするならば、当該炭素系固体酸は加水分解反応の触媒として効果的である。次に、植物系原料の予備炭化の加熱処理温度が200〜500℃とするならば、当該炭素系固体酸はエステル化反応の触媒として効果的である。さらに、植物系原料の予備炭化の加熱処理温度が400〜500℃であるとするならば、当該炭素系固体酸はアルキル化反応の触媒として効果的である。好適な触媒反応種と予備炭化の加熱温度との関連性は現在のところ十分に解明されていない。おそらく、細孔の発達、後述するスルホン酸基の修飾量の差異が影響していることが推察される。
【0057】
スルホ化の工程(S3)にあっては、予備炭化物をそのまま、もしくは適当に粉砕して粒径を調整した後、植物系原料に由来する予備炭化物は濃硫酸中または発煙硫酸中に浸漬される。あるいは、予備炭化物は三酸化硫黄ガスが充満したチャンバ内に搬送される。こうして、80〜350℃の温度条件下、好ましくは80〜150℃の温度条件下で加熱(スルホ化)され、植物系原料に由来する予備炭化物のスルホ化が促進し、賦活により多孔質化した炭化物表面にスルホン酸基が導入されスルホ化物が得られる。
【0058】
また、前記のスルホ化の工程(S3)は、不活性ガス、または乾燥空気流中において行われる。硫酸は非常に水分を吸収しやすい。そこで、水分をできる限り抑制することにより、炭素表面に導入するスルホン酸基濃度の低下を防ぐ必要からの措置である。
【0059】
スルホ化の加熱温度が低すぎるとスルホン酸基導入の反応が遅くなる。温度を高くしすぎる場合、炭化が進みすぎ、収量が減少する。また、温度、設備、処理量等の影響を受けるものの、スルホ化の加熱時間は、30分〜20時間、好ましくは1〜10時間行われる。加熱時間が短ければスルホン酸基の導入は不十分となる。加熱時間を長くしすぎても反応が頭打ちになった後の加熱はエネルギーの無駄である。なお、スルホ化に際しても、予備炭化と同様に窒素ガス、炭酸ガス、ヘリウムガスの通気、あるいは燃焼時の排気ガスを循環させること等の不活性ガス雰囲気下において加熱が行われる。スルホ化の後、当該スルホ化物は水洗され硫酸等の成分が除去され、乾燥される。
【0060】
各処理を経ることにより、本発明に好適な炭素系固体酸(P)が得られる。そこで、当該炭素系固体酸が、好適な触媒活性等をはじめとする作用を発揮する上で具備すべき物理的条件は、スルホン酸基を導入した後、つまりスルホ化後における多孔質炭素のBET比表面積が3〜1600m2/gを満たすことである。当該比表面積が3m2/gを下回る場合、細孔の発達が不十分であり、反応効率を求めることが難しくなる。また、比表面積1600m2/gは現状の製造方法における上限と考えられる。BET比表面積の測定の詳細は、後記の実施例にて述べる。簡単には77K(およそ−195℃)における窒素吸着等温線からBET法により求めた比表面積(m2/g)である。比表面積の最適範囲は、炭素系固体酸を適用する反応種によっても変動する。
【0061】
さらに、既述の各処理を経て得られた炭素系固体酸(P)のスルホン酸基の量は、0.2mmol/g以上、より好ましくは0.5mmol/g以上を満たすことである。スルホン酸基の測定の詳細も後記の実施例にて述べる。簡単には酸−アルカリの中和反応の滴定や元素分析による算出により可能である。後記の実施例から把握できるように、出来上がった炭素系固体酸の分析結果を勘案して0.2mmol/g以上となる。そのうち、炭素系固体酸のスルホ基の量は多くなるほど触媒反応は促進する。概ね0.5mmol/g以上の試料からは良好な触媒活性が確認できたためである。
【0062】
炭素系固体酸を粉末状の触媒として用いる場合、通常、多孔質炭素は塊状物で得られるため、粉砕された上で用いられる。粉砕後の粒径(メジアン径)は、1〜100μm、好ましくは2〜50μm、より好ましくは5〜20μmである。粒径の分布が均一であるほど反応後の分離、回収が容易となるためである。粉砕に際し、ボールミル、ハンマーミル、ジェットミル等の公知の粉砕装置が用いられる。また、炭素系固体酸を固定床に敷設する触媒として用いる場合、多孔質炭素塊状物は適宜の篩別により、0.1〜4mm、好ましくは0.2〜3mm、より好ましくは0.4〜2.5mmの粒状物に分けられる。むろん、粉末、粒状のいずれにおいても、当該炭素系固体酸を適用する反応、設備、耐久性等の諸要因を勘案して粒径は規定される。
【0063】
これまでの説明は主に原料(炭素源)としてセルロース含有原料や植物系原料を用い、これに濃硫酸、発煙硫酸を反応させてスルホ化する処理である。続いて、原料(炭素源)となるセルロース含有原料や植物系原料の不完全な炭化物、つまりは、前述の塩化亜鉛やリン酸による賦活を終えて予備炭化を経た予備炭化物等に対する三酸化硫黄によるスルホ化を説明する。
【0064】
すなわち、原料(炭素源)となるセルロース含有原料や植物系原料は不完全に炭化され(図1のS1,S2の工程参照)、これに三酸化硫黄が接触されることにより、炭化不完全な原料の縮合、スルホ化が行われる。ここで、炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に三酸化硫黄を接触させる場合、窒素、アルゴン等の不活性ガス気流下、あるいは乾燥空気気流下で行うことがスルホ基密度の高い炭素系固体酸を製造する上で重要となる。
【0065】
炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に三酸化硫黄を接触させる方法としては、液体状または気体状の三酸化硫黄を直接接触(含浸)させてもよいし、三酸化硫黄を含有する液状物またはガス状物として接触(含浸)させるようにしてもよい。
【0066】
液体状または気体状の三酸化硫黄を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に直接接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、液体状または気体状の三酸化硫黄を徐々に加えることが望ましい。ただし、それに限られず、液体状または気体状の三酸化硫黄に対し炭化不完全な有機物を徐々に加えるようにしてもよい。
【0067】
三酸化硫黄を含有するガス状物を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、三酸化硫黄を含有するガス状物を徐々に加えることが望ましい。ただし、この場合も、三酸化硫黄を含有するガス状物に炭化不完全な有機物を徐々に加えてもよい。ガス状物に含まれる三酸化硫黄以外のガス成分としては、スルホ化反応に不活性な気体であれば特に制約はなく、例えば乾燥空気、窒素やアルゴン等が挙げられる。
【0068】
三酸化硫黄を含有する液状物を炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に接触させる場合は、炭化不完全な有機物を攪拌し流動化させながら、三酸化硫黄を含有する液状物を徐々に加えることができる。この場合も、三酸化硫黄を含有する液状物に炭化不完全な有機物を徐々に加えてもよい。また、炭化不完全な有機物を予め三酸化硫黄を含まない液状物に分散させておき、三酸化硫黄を含有する液状物または三酸化硫黄を徐々に加えてもよいし、三酸化硫黄を含有する液状物または三酸化硫黄に、三酸化硫黄を含まない液状物に分散させた炭化不完全な有機物を徐々に加えてもよい。液状物に含まれる三酸化硫黄以外の液体成分としては、スルホ化反応に不活性な液体であれば特に制約はなく、例えばジクロロメタン、エチレンジクロライド等の塩素系有機溶媒等が挙げられる。
【0069】
三酸化硫黄による処理(縮合、スルホ化)温度としては、スルホ化反応を進行させる温度であれば特に限定されないが、通常は0℃から100℃であり、好ましくは20℃から80℃である。
【0070】
三酸化硫黄による処理(縮合、スルホ化)時間は、反応温度や目標とするスルホ基の導入率にも依存するが、通常は1分から24時間、好ましくは30分から10時間である。また、三酸化硫黄による処理(縮合、スルホ化)圧力は、スルホ化反応の進行させる圧力であれば特に限定されない。
【0071】
炭化不完全な有機物(予備炭化物)の縮合、スルホ化に使用するスルホ化剤は、通常の三酸化硫黄であるが、塩素系有機溶媒等を用いる場合には、安定剤を含まない三酸化硫黄が好適である。
【0072】
ここで、安定剤を含まない三酸化硫黄とは、三酸化ホウ素、ジメチル硫酸等の安定化剤を敢えて含まない三酸化硫黄である。これらの安定剤は、ジクロロメタンにおける「C−Cl」の結合内にSO3を挿入する反応を触媒してしまうことが知られているためである。反応系に安定剤入りの三酸化硫黄を添加すると、炭化物にスルホ基として結合する前にジクロロメタン等の塩素系有機溶媒と反応してしまい、SO2による架橋生成の原因となる。このため、炭化物に対するスルホ基の保持量の低下を防ぐためにも、安定剤を含まない三酸化硫黄の使用が良好な結果をもたらす。
【0073】
三酸化硫黄の添加量は、炭化不完全な有機物(セルロース含有原料や植物系原料の予備炭化物)に導入したいスルホ基の総量によるが、通常は当該炭化不完全な有機物の重量(予備炭化物の重量)に対して、三酸化硫黄重量として、1〜500重量%、好ましくは5〜300重量%、さらには10〜100重量%が好ましい。これは、三酸化硫黄の添加量が少なすぎると、炭化不完全な有機物(予備炭化物)へのスルホ基導入率が低くなり、多すぎると余剰の三酸化硫黄の除去に多大な時間や経費が必要となるので実用的ではないからである。一方、炭化不完全な有機物(予備炭化物)を縮合、スルホ化した後、反応液である三酸化硫黄または三酸化硫黄を含有したスルホ化剤より固体酸を採取する方法は、実施例で行ったような通常の方法に従って行うことができる。
【0074】
以上の炭素系固体酸の製造方法によれば、従来のごとく濃硫酸または発煙硫酸を使用せず、塩素系有機溶媒を用いる場合にも、スルホ架橋用の抑制剤等を使用せずに、高収率で再現性にも優れ、硫酸に匹敵するような炭素系固体酸が得られる。
【0075】
一連の説明から理解されるように、製造される炭素系固体酸は、プロトン伝導性材料や固体酸触媒として好適となるとともに、耐久性つまり耐熱性、耐酸性あるいは化学的安定性にも優れ、コスト低減も図られることから、イオン交換体、プロトン伝導性材料、電解質膜、反応触媒等として非常に有用である。むろん、本発明の炭素系固体酸を利用して固体電解質膜を作製し、それを用いて膜電極接合体や燃料電池を作製することが可能である。
【実施例】
【0076】
〔炭素系固体酸の試作(発煙硫酸によるスルホ化)〕
炭素源となる植物系原料として米松(ベイマツ)の大鋸粉(オガコ)を篩により篩別し、篩別したオガコの木粉を用い、以下に記載の試作例の手順に基づいて塩化亜鉛の含浸、予備炭化、発煙硫酸によるスルホ化を行い、炭素系固体酸を試作した。スルホ化物が各試作例の炭素系固体酸となる。
【0077】
〈試作例1〉
オガコを105±5℃に保った乾燥機内で8時間乾燥後、4.7〜83meshの篩(粒径180〜4000μmに相当)により篩別し、木粉20gを取り分けた。木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。350℃まで60分間かけて昇温し、その後350℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。
【0078】
予備炭化物10gを300mLの15%発煙硫酸に加えて80℃で10時間加熱しスルホ化した。その後、過剰な濃硫酸を100℃の蒸留水で繰り返し洗浄し、洗浄後の蒸留水中の硫酸が元素分析の検出限界以下になるまで洗浄を繰り返した。水洗後、105±5℃に保った乾燥機内で8時間乾燥してスルホ化物を得た(比表面積:15m2/g,スルホン酸基量:1.27mmol/g)。試作例1の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0079】
〈試作例2〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。400℃まで70分かけて昇温し、その後400℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例2の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:155m2/g,スルホン酸基量:1.34mmol/g)。試作例2の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0080】
〈試作例3〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。450℃まで80分間かけて昇温し、その後450℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間煮沸しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、ボールミルにより粉砕後した。粉砕品を83meshの篩(粒径180μmに相当)により篩別し、篩下物を予備炭化物とした。試作例3の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:818m2/g,スルホン酸基量:1.27mmol/g)。試作例3の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0081】
〈試作例4〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。250℃まで40分間かけて昇温し、その後250℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例4の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:3m2/g,スルホン酸基量:1.33mmol/g)。試作例4の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0082】
〈試作例5〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。300℃まで50分間かけて昇温し、その後300℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例5の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:4m2/g,スルホン酸基量:1.27mmol/g)。試作例5の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0083】
〈試作例6〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。500℃まで90分間かけて昇温し、その後500℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例6の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1345m2/g,スルホン酸基量:1.04mmol/g)。試作例6の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0084】
〈試作例7〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。550℃まで100分間かけて昇温し、その後550℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例7の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1492m2/g,スルホン酸基量:0.70mmol/g)。試作例7の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0085】
〈試作例8〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。600℃まで110分間かけて昇温し、その後600℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例8の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:1453m2/g,スルホン酸基量:0.47mmol/g)。試作例8の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0086】
〈試作例9〉
前記の乾燥、篩別条件を経た木粉20gに1Nの塩酸に溶解した濃度65%(w/w)の塩化亜鉛溶液108gを加え混合し、この混合物をるつぼに入れて電気炉内に置いた。200℃まで30分間かけて昇温し、その後200℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を濾布の付いた洗浄層に移した。ここに、20%に希釈した塩酸200mLを添加し、1時間攪拌しながら洗浄した。炭化物の水切りをした後、50〜60℃の水で水洗した。水洗後の炭化物を105±5℃に保った乾燥機内で8時間乾燥し、10〜22meshの篩により篩別し、予備炭化物を得た。試作例9の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:3m2/g,スルホン酸基量:1.34mmol/g)。試作例9の炭素系固体酸の予備炭化時における塩化亜鉛の添加量は、植物系原料(木粉)に対し塩化亜鉛換算の重量比で3.5重量倍である。
【0087】
〈試作例10〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。200℃まで30分間かけて昇温し、その後200℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を10〜22meshの篩により篩別し、予備炭化物を得た。試作例10の予備炭化物に対するスルホ化は、試作例1と同一の条件により行った。スルホ化時に試作例10の予備炭化物は形状維持ができず砕けてしまい、回収することができなかった。このため、以降の測定は実施できなかった。
【0088】
〈試作例11〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。300℃まで50分間かけて昇温し、その後300℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して炭化物を10〜22meshの篩により篩別し、予備炭化物を得た。試作例11の予備炭化物に対するスルホ化は、試作例1と同一の条件により行った。スルホ化時に試作例11の予備炭化物も形状維持ができず砕けてしまい、回収することができなかった。このため、以降の測定は実施できなかった。
【0089】
〈試作例12〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。400℃まで70分間かけて昇温し、その後400℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例12の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:5m2/g,スルホン酸基量:0.84mmol/g)。
【0090】
〈試作例13〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。500℃まで90分間かけて昇温し、その後500℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例13の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:20m2/g,スルホン酸基量:0.29mmol/g)。
【0091】
〈試作例14〉
前記の乾燥、篩別条件を経た木粉20gをるつぼに入れて電気炉内に置いた。600℃まで110分間かけて昇温し、その後600℃を60分間維持し焼成した(予備炭化)。冷却後電気炉からるつぼを取り出して、10〜22meshの篩により篩別し、予備炭化物を得た。試作例14の予備炭化物に対するスルホ化は、試作例1と同一の条件により行い、スルホ化物を得た(比表面積:13m2/g,スルホン酸基量:0.10mmol/g)。
【0092】
〔物性の測定方法〕
・比表面積
試作例の炭素系固体酸の比表面積(m2/g)はBET法に基づいて測定した。実施例及び比較例の試料を200℃の窒素雰囲気下において3時間乾燥した後、77Kにおける窒素吸着等温線を日本ベル株式会社製:「BELSORP MINI」により測定した。
【0093】
・スルホン酸基量の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、2mgを分取し、エレメンタール社製:CHNS元素分析計vario Micro cubeを用いてS(硫黄分)の量(mmol/g)を測定し、等価としてスルホン基酸量(mmol/g)を求めた。
【0094】
〔触媒活性の測定〕
・加水分解反応の測定
各試作例の炭素系固体酸より0.3gを分取し、これにセロビオース0.025g(73.04μmol)、水0.7gを添加し、90℃を維持しながら1時間反応させた。反応液をHPLCに装填し、グルコース等の単糖類のピーク面積比よりセロビオースから分解されて生成した糖類量を求めた。
【0095】
・エステル化反応の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、0.2g分取し、これに酢酸0.1mol、エタノール1.0molを添加し、70℃を維持しながら3時間反応させた。反応液中に含まれる酢酸エチルの生成量を株式会社島津製作所製のFID−ガスクロマトグラフィー:GC−17Aにより求めた。
【0096】
・アルキル化反応の測定
各試作例の炭素系固体酸を150℃で1時間真空排気した後、0.2g分取し、これにトルエン100mmol、塩化ベンジル(クロロメチルベンゼン)10mmolを添加し、100℃を維持しながら4時間反応させた。反応液中に含まれるベンジルトルエンの生成量をガスクロマトグラフィーにより求めた。
【0097】
各試作例について、予備炭化時の植物系原料(木粉)に対する塩化亜鉛換算の添加重量比、予備炭化条件(焼成温度(℃)、昇温時間(分)、維持時間(分))、スルホン酸基量(スルホ基量)(mmol/g)、加水分解量(μmol)、エステル化反応量(mmol/g・min)、アルキル化反応量(μmol/g・min)、及び比表面積(m2/g)を表1にまとめた。表では、賦活剤を用いた試作例と賦活剤を用いなかった試作例の双方をそれぞれ予備炭化時の焼成温度の低い方から順に並べた。
【0098】
【表1】
【0099】
〔結果,考察〕
・加水分解反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下、200〜450℃の焼成温度域で予備炭化して得た試作例については、加水分解反応において良好な触媒作用の結果を示した。予備炭化時の温度が高くなるほど性能が低下することから、前記の焼成温度域にすることが望ましい。
【0100】
・エステル化反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下において、200〜500℃の焼成温度域で予備炭化して得た試作例については、エステル化反応において良好な触媒作用の結果を示した。予備炭化時の温度が高くなるほど性能が低下することから、前記の焼成温度域にすることが望ましい。
【0101】
・アルキル化反応の評価
試作例の炭素系固体酸において、塩化亜鉛の存在下において、400〜500℃の焼成温度域で予備炭化して得た試作例については、アルキル化反応において良好な触媒作用の結果を示した。同反応の場合、焼成温度が400℃を下回る場合及び500℃を上回る場合に極端に効果が低下する。
【0102】
一般に焼成温度が高くなるほど比表面積は大きくなるため、反応性は向上していることが予想できる。しかしながら、スルホン酸基(スルホ基)の量は、焼成温度500℃までは概ね安定している。このため、炭素表面に発達した細孔と、触媒反応の対象となる分子の大きさ、構造、性質との親和性や相互作用等により、予備炭化時の焼成温度の違いから触媒活性の差異が生じたものと予想できる。また、植物系原料を賦活、炭化して炭素系固体酸を得ているため、炭素表面に残存する官能基量も影響していると考えられる。なお、賦活剤を用いず低温度域で予備炭化して、スルホ化を行った試作例では形状維持が困難となったことから、本発明の硫酸や発煙硫酸によるスルホ化を伴うスルホン酸基を有する炭素系固体酸には薬品賦活が必須である。
【0103】
以上の知見から明らかなように、塩化亜鉛の含浸に伴う薬品賦活に際し、その焼成時の温度域を調整することによって、目的とする反応に対応した活性を発現する触媒を得ることができた。よって、植物系原料を用いたものであっても、反応種や用途に応じた触媒の作り分けが簡便かつ容易となり、実需要に極めて柔軟に応えることができる炭素系固体酸を安価に提供することができる。
【0104】
〔炭素系固体酸の試作(三酸化硫黄によるスルホ化)〕
続いて、発明者らは三酸化硫黄によるスルホ化を試みた。炭素源として、セルロース含有原料に微結晶セルロース(メルク社製)、植物系原料(大鋸粉(オガコ))由来の顆粒活性炭(フタムラ化学株式会社製「SG」,7.5〜42mesh(粒径355〜400μmに相当))、粉末活性炭(フタムラ化学株式会社製「S」,メジアン径42.5μm(株式会社島津製作所製:SALD−200Vによる測定))を用い、以下に記載の各試作例の手順に基づいて三酸化硫黄によるスルホ化を行い、炭素系固体酸を試作した。スルホ化物が各試作例の炭素系固体酸となる。試作例に用いた2種類の植物系原料由来の活性炭は、いずれも塩化亜鉛により賦活し、450℃で大気下において予備炭化、焼成した活性炭である。
【0105】
炭化不完全な有機物の縮合、スルホ化するために使用したロータリーエバポレーターは、ビュッヒ・ラボテクニック社(BUCHI Labortechnik AG(スイス))製、ROTAVAPOR RE120である。使用フラスコはナス型フラスコ:1L容積を使用した。また、評価は、固体酸中の硫黄分測定を成分ガス分析・計測装置として、浜田理科株式会社製:燃焼フラスコ(FHO−A型)装置により測定した。
【0106】
〔セルロース含有原料の不完全な炭化〕
前出の微結晶セルロース(メルク社製)20gを、500mL容積の三つ口フラスコに入れ、窒素ガス気流下、450℃で5時間加熱し、9gの不完全な炭化物を得た(以下、これを「不完全炭化物」という)。この操作を繰り返すことで所定重量の不完全炭化物を確保し、必要量を計量して使用した。
【0107】
〈試作例21〉
前記の不完全炭化物の所定重量を110℃で1時間乾燥した。前記ロータリーエバポレーターは、コンデンサー上部に注入コックを有し、その注入コックに三酸化硫黄ガス吹込みラインを接続し、エバポレーター内を窒素ガスで置換した。不完全炭化物20.2gを1L容積のナス型フラスコに入れた後、該ナス型フラスコをエバポレーターに取り付け、窒素ガスで再度置換した。そして、該ナス型フラスコを60℃に加温して回転(約80rpm)させるとともに、真空ポンプによりエバポレーター内を脱気(0.5kPa)し、密閉した。
【0108】
三酸化硫黄(日曹金属化学株式会社製:製品名「日曹サルファン」)6.1gをガス化用三つ口フラスコに量り取った。エバポレーターのコンデンサー上部にある注入コックを開けて、三酸化硫黄を徐々にエバポレーター内に導入した。そして、三酸化硫黄を導入した後、不完全炭化物と三酸化硫黄の反応を、ナス型フラスコを回転(約80rpm)させながら60℃で2時間行った。反応後は、三酸化硫黄ガス吹込みラインを切り離し、エバポレーター内の三酸化硫黄ガスを窒素ガスで置換した。
【0109】
ナス型フラスコをエバポレーターから外し、該ナス型フラスコ内に約500mLの蒸留水を加えて10分間攪拌した。温度は30℃以下に保持した。その後は、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。その後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸20.9gを得た。同炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、0.94wt%であった(試作例21の炭素系固体酸)。
【0110】
〈試作例22〉
前記の不完全炭化物の所定重量を110℃で1時間乾燥した。500mL容積の三つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、内部を窒素ガスで置換した。そして、三つ口フラスコに乾燥した不完全炭化物20.0gとジクロロメタン271.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0111】
三酸化硫黄として、安定剤を添加しない液体状の三酸化硫黄30.1gを滴下ロートにて量り取った後、前記の三つ口フラスコ内にゆっくりと滴下した。滴下後、(不完全炭化物+ジクロロメタン)と液体状の三酸化硫黄とを25〜30℃で、2時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン300mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約500mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0112】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。乾燥後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸24.2gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、5.26wt%であった(試作例22の炭素系固体酸)。
【0113】
〈試作例23〉
前出の微結晶セルロース(メルク社製)500gを、5000mL容積のセパラブルフラスコに入れ、窒素ガス気流下、400℃で1時間加熱し、150gの不完全炭化物を得るとともに、80℃で1日間乾燥した。この不完全炭化物20.2gを1L容積のナス型フラスコに入れた後、該ナス型フラスコを試作例21と同様のロータリーエバポレーターに取り付け、窒素ガスで再度置換した。そして、該ナス型フラスコを60℃に加温して回転(約80rpm)させるとともに、真空ポンプによりエバポレーター内を脱気(0.5kPa)し、密閉した。
【0114】
三酸化硫黄5.8gをガス化用三つ口フラスコに量り取った。エバポレーターのコンデンサー上部にある注入コックを開けて、気化した三酸化硫黄を徐々にエバポレーター内に導入した。そして、三酸化硫黄を導入した後、不完全炭化物と三酸化硫黄の反応を、ナス型フラスコを回転(約80rpm)させながら60℃で2時間行った。反応後は、三酸化硫黄ガス吹込みラインを切り離し、エバポレーター内の三酸化硫黄ガスを窒素ガスで置換した。
【0115】
ナス型フラスコをエバポレーターから外し、該ナス型フラスコ内に約500mLの蒸留水を加えて10分間攪拌した。温度は30℃以下に保持した。その後は、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。また、水洗いとして、固形分を回収して約500mLの蒸留水に再懸濁し、10分間攪拌した後、再び固形分を濾別した。この操作を、濾液のpHがほぼ一定になるまで繰り返した後、固形分を80℃で1日乾燥した。その後は、熱水洗いとして、固形分を約100℃の蒸留水500mLで、熱水洗いした。この操作を、濾液のpHがほぼ一定になるまで繰り返した。熱水洗後は、固形分を80℃で1日乾燥し、炭素系固体酸19.4gを得た。同炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、0.85wt%であった(試作例23の炭素系固体酸)。
【0116】
〈試作例24〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、内部を窒素ガスで置換した。そして、四つ口フラスコに試作例23にて調製し乾燥した不完全炭化物20.0gとジクロロメタン432.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0117】
日曹金属化学株式会社製:製品名「日曹サルファン」より安定剤を含まない三酸化硫黄(液状)を別途調製した。安定剤を含まない三酸化硫黄60.2gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0118】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸26.7gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、7.32wt%であった(試作例24の炭素系固体酸)。
【0119】
〈試作例25〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、四つ口フラスコに植物系原料(オガコ)由来の顆粒活性炭(フタムラ化学株式会社製「SG」)20.0gとジクロロメタン240.9gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0120】
試作例24にて用いた安定剤を含まない三酸化硫黄60.3gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0121】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸23.9gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、3.82wt%であった(試作例25の炭素系固体酸)。
【0122】
〈試作例26〉
500mL容積の四つ口フラスコに、滴下ロート、冷却管、温度計をそれぞれセットした後、四つ口フラスコに植物系原料(オガコ)由来の粉末活性炭(フタムラ化学株式会社製「S」)20.0gとジクロロメタン240.0gとを入れ、それらをマグネチックスターラーで攪拌した。温度は約25℃に保持した。
【0123】
試作例24にて用いた安定剤を含まない三酸化硫黄60.2gを滴下ロートにて量り取った後、前記の四つ口フラスコ内にゆっくりと滴下した。滴下後、「不完全炭化物+ジクロロメタン」と液体状の三酸化硫黄とを25〜30℃で、1時間反応処理した。その後は、加圧濾過(POLYFLON FILTER 東洋PF050(保留粒子5.0μm))を行い、固形分を濾別した。そして、その固形分をジクロロメタン400mLで、2回洗浄した。また、洗浄後の固形分をビーカーに移し、約400mLの蒸留水を加えて、10分間攪拌した。温度は30℃以下に保持した。
【0124】
その後、親水性PTFE性フィルター(ミリポア社製、オムニポア、孔径10μm)を用いて、固形分を吸引濾過した。当該固形分を2NのNaOH水溶液により濾液中より硫酸イオンが検出されなくなるまで洗浄を続けた。洗浄後、固形分を2NのHCl水溶液で活性化(再生)し、余分なHClをイオン交換水により洗い流した。水洗後、固形分を80℃で1日乾燥し、炭素系固体酸23.8gを得た。そして、以上により得られた炭素系固体酸の硫黄含有率を前出の燃焼フラスコ装置で分析したところ、3.88wt%であった(試作例26の炭素系固体酸)。
【0125】
試作例23ないし26の結果について、原料、焼成雰囲気、賦活剤、スルホ化剤、スルホ化剤の状態(ガス状または液体)、スルホ基量(mmol/g)、比表面積(m2/g)を表2のとおりまとめた。表中のスルホ基量(スルホン酸基量)、比表面積は、前掲の物性の測定方法と同様である。
【0126】
【表2】
【0127】
〔三酸化硫黄によるスルホ化の結果と考察〕
スルホ化剤の反応においては、液体状の三酸化硫黄を使用する方が炭素系固体酸の単位重量あたりのスルホ基量の多くすることができる。なお、試作例24,25,26にあっては、発煙硫酸によるスルホ化(表1参照)と比較しても遜色ない。また、オガコ由来の活性炭製品をスルホ化しても十分なスルホ基量を得ることができることから、調達容易な原料を用いての量産も有望である。さらに、三酸化硫黄によるスルホ化では、硫酸等を用いた際のスルホ化よりも反応条件が比較的穏和であり、短時間となることから生産性の向上やエネルギー消費量の抑制等に有効であるといえる。なお、三酸化硫黄によるスルホ化により生じた炭素系固体酸のスルホ基量及び表1の結果を勘案すると、同様に触媒効果を示すものと予想できる。
【特許請求の範囲】
【請求項1】
スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であることを特徴とする炭素系固体酸。
BET比表面積:試料を200℃の窒素雰囲気下において3時間乾燥した後、77K(−195℃)における窒素吸着等温線を日本ベル株式会社製BELSORP MINIにより測定し、BET法により比表面積(m2/g)を求めた。
【請求項2】
前記多孔質炭素がセルロース含有原料に由来する請求項1に記載の炭素系固体酸。
【請求項3】
前記セルロース含有原料が植物系原料である請求項2に記載の炭素系固体酸。
【請求項4】
前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものである請求項1ないし3のいずれか1項に記載の炭素系固体酸。
【請求項5】
前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものである請求項3に記載の炭素系固体酸。
【請求項6】
前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものである請求項5に記載の炭素系固体酸。
【請求項7】
前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項8】
前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項9】
前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項10】
前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種である請求項3ないし9のいずれか1項に記載の炭素系固体酸。
【請求項11】
請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、
前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化することを特徴とする炭素系固体酸の製造方法。
【請求項12】
前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われる請求項11に記載の炭素系固体酸の製造方法。
【請求項13】
請求項4に記載の炭素系固体酸を製造するに際し、
前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化することを特徴とする炭素系固体酸の製造方法。
【請求項14】
前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状である請求項13に記載の炭素系固体酸の製造方法。
【請求項15】
前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液である請求項14に記載の炭素系固体酸の製造方法。
【請求項1】
スルホン酸基が導入された以下に定義されるBET比表面積が3〜1600m2/gである多孔質炭素からなり、前記多孔質炭素のスルホン酸基量が、0.2mmol/g以上であることを特徴とする炭素系固体酸。
BET比表面積:試料を200℃の窒素雰囲気下において3時間乾燥した後、77K(−195℃)における窒素吸着等温線を日本ベル株式会社製BELSORP MINIにより測定し、BET法により比表面積(m2/g)を求めた。
【請求項2】
前記多孔質炭素がセルロース含有原料に由来する請求項1に記載の炭素系固体酸。
【請求項3】
前記セルロース含有原料が植物系原料である請求項2に記載の炭素系固体酸。
【請求項4】
前記多孔質炭素が三酸化硫黄または三酸化硫黄を含有したスルホ化剤を前記セルロース含有原料または前記植物系原料に接触させてスルホ化して得られたものである請求項1ないし3のいずれか1項に記載の炭素系固体酸。
【請求項5】
前記多孔質炭素が、前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱処理して得られたものを、さらに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中でスルホ化として加熱処理して得られたものである請求項3に記載の炭素系固体酸。
【請求項6】
前記植物系原料の予備炭化の加熱処理温度が200℃〜600℃であり、触媒反応に使用されるものである請求項5に記載の炭素系固体酸。
【請求項7】
前記植物系原料の予備炭化の加熱処理温度が200℃〜450℃であり、加水分解反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項8】
前記植物系原料の予備炭化の加熱処理温度が200℃〜500℃であり、エステル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項9】
前記植物系原料の予備炭化の加熱処理温度が400℃〜500℃であり、アルキル化反応触媒に使用されるものである請求項6に記載の炭素系固体酸。
【請求項10】
前記植物系原料が、樹木、草木、果実、種子等または再生セルロースから選ばれる少なくとも1種である請求項3ないし9のいずれか1項に記載の炭素系固体酸。
【請求項11】
請求項5ないし10のいずれか1項に記載の炭素系固体酸を製造するに際し、
前記植物系原料に塩化亜鉛またはリン酸を含浸した後、予備炭化として加熱して多孔質炭素を得て、これに濃硫酸または発煙硫酸中、あるいは三酸化硫黄のガス中または液中で加熱処理を伴ってスルホ化することを特徴とする炭素系固体酸の製造方法。
【請求項12】
前記発煙硫酸中で加熱処理する工程が不活性ガスまたは乾燥空気中で行われる請求項11に記載の炭素系固体酸の製造方法。
【請求項13】
請求項4に記載の炭素系固体酸を製造するに際し、
前記セルロース含有原料または前記植物系原料を不完全に炭化することにより当該炭化物中に炭素と水素の結合を残存させて得た不完全炭化物に、三酸化硫黄または三酸化硫黄を含有したスルホ化剤を接触させて、前記不完全炭化物をスルホ化することを特徴とする炭素系固体酸の製造方法。
【請求項14】
前記三酸化硫黄または前記三酸化硫黄を含有したスルホ化剤が、液体状または気体状である請求項13に記載の炭素系固体酸の製造方法。
【請求項15】
前記三酸化硫黄を含有したスルホ化剤が、安定剤を含まない三酸化硫黄を含有した塩素系有機溶媒溶液である請求項14に記載の炭素系固体酸の製造方法。
【図1】

【公開番号】特開2011−11201(P2011−11201A)
【公開日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願番号】特願2009−210531(P2009−210531)
【出願日】平成21年9月11日(2009.9.11)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年6月23日 インターネットアドレス「http://science24.com/event/emrs2008fall/journal/?item=5」に発表
【出願人】(591243103)財団法人神奈川科学技術アカデミー (271)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(592184876)フタムラ化学株式会社 (60)
【出願人】(000227087)日曹エンジニアリング株式会社 (33)
【Fターム(参考)】
【公開日】平成23年1月20日(2011.1.20)
【国際特許分類】
【出願日】平成21年9月11日(2009.9.11)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年6月23日 インターネットアドレス「http://science24.com/event/emrs2008fall/journal/?item=5」に発表
【出願人】(591243103)財団法人神奈川科学技術アカデミー (271)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(592184876)フタムラ化学株式会社 (60)
【出願人】(000227087)日曹エンジニアリング株式会社 (33)
【Fターム(参考)】
[ Back to top ]