
炭酸エステル化合物の製造法
【課題】触媒の存在下に脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造する。
【解決手段】一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【解決手段】一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を製造する方法に関するものである。
【背景技術】
【0002】
脂肪族アルコールと一酸化炭素及び酸素から酸化カルボニル化反応によって炭酸エステル化合物を製造する方法は広く知られており、多くの公知文献がある。
その殆どの文献では、触媒成分として銅及び/またはパラジウムのハロゲン化物や、これらに他の金属元素のハロゲン化物を加えたものが用いられている(特許文献1〜3参照)。
この方法では、酸化反応中に腐食性の強いハロゲン化水素酸が副生するために、製造設備には高価な耐食性金属材料やグラスライニング等の耐食保護材料を用いなければならない。更に酸化カルボニル化反応では、加圧条件下で有利に反応が進行するために、殆どの文献において数気圧〜数十気圧程度の加圧条件が採用されている。そのため、より高価な耐腐食性耐圧反応装置が必要となり、炭酸エステルを安価に製造する観点から著しく不利である。
【0003】
ハロゲン化合物を含まない触媒系を用いた酸化カルボニル化反応による炭酸エステル化合物の製造法としては、酸化銅,水酸化銅,酢酸銅のようなハロゲンを含まない弱酸の銅塩から選ばれた少なくとも一種類の銅化合物を触媒として用いる方法(特許文献4参照)、酸化銅と周期律表第13〜16族元素の酸化物からなる触媒を用いる方法(特許文献5参照)、水酸化銅,酸化銅,銅の弱酸塩から選ばれる少なくとも一種類の銅化合物と硝酸銅からなる触媒を用いる方法(特許文献6参照)、及び銅アルコラート化合物を用いる方法(特許文献7参照)などが開示されている。
これらの文献例では炭酸エステルの生産性は高くなく、改善の余地は大きい。また、酸化銅、水酸化銅、銅アルコラート等は脂肪族アルコールのような還元性物質、反応で副生する水、熱などに対して非常に不安定であり、容易に分解して触媒として失活しやすいという欠点があった。
【0004】
また、酸化カルボニル化による炭酸エステル合成反応において層状化合物を用いる方法が知られている。例えば、層状粘土化合物の層間あるいは表面に一価または二価の銅を担持した触媒を用いる方法(特許文献8参照)、粘土化合物または黒鉛などの層状物質の層間に銅の塩、カチオン、錯体化合物等の触媒成分が挿入された固体触媒を用いる方法(特許文献9参照)が公知である。これらは無機層状化合物の層間に従来公知な触媒活性成分をインターカレートまたはイオン交換させることによって触媒性能の向上を図ったものと考えられる。
【0005】
さらに、酸化カルボニル化による炭酸エステル合成反応を二段反応で行う方法も公知である。前段反応として脂肪族アルコールと一酸化窒素及び酸素から亜硝酸エステルと水を作り、水を分離してから、後段反応として亜硝酸エステルと一酸化炭素を触媒によって炭酸エステルと一酸化窒素へ転化させ、再生した一酸化窒素を前段反応へリサイクルする方法である(特許文献10及び11参照)。この方法は比較的温和な反応条件で反応が進行し、且つ腐食性の問題が少ない有効な方法ではあるが、後段反応の触媒として高価な白金族元素が必須であること、二段反応であるために一段合成法に比較して付帯設備が多く、反応系が複雑になるなどの不利益があった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平2−169549号公報
【特許文献2】特開平5−208137号公報
【特許文献3】特開平9−10591号公報
【特許文献4】特開平6−218284号公報
【特許文献5】特開平7−313879号公報
【特許文献6】特開平6−92908号公報
【特許文献7】特開平10−114720号公報
【特許文献8】特開平5−255201号公報
【特許文献9】特開平6−116212号公報
【特許文献10】特開昭60−11443号公報
【特許文献11】特開平3−141243号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、従来技術における上記のような課題を解決し、触媒の存在下に脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造する方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は触媒の存在下に脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を一段反応によって製造する方法について鋭意研究を重ねた結果、層状水酸化銅カルボン酸塩の存在下で反応を行うことによって効率よく、炭酸エステル化合物を製造できることを見いだし本発明に到達した。
【0009】
すなわち、本発明は、一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法に関するものである。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【発明の効果】
【0010】
本発明の方法により、ハロゲン元素を含まない固体触媒存在下に一段反応で脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造することができる。
【図面の簡単な説明】
【0011】
【図1】参考調製例1で得られたA1試料のX線回折チャート。
【図2】参考調製例2で得られたA2試料のX線回折チャート。
【図3】参考調製例3で得られたA3試料のX線回折チャート。
【発明を実施するための形態】
【0012】
以下に本発明の各構成要件について説明する。
本発明に用いる脂肪族アルコール化合物(以下、「原料アルコール」ともいう。)は、上記一般式(2)で表される化合物であれば特に制限はない。具体的にはメタノール,エタノール,1-プロパノール,1-ブタノール,2-ブタノール,2-メチル-1-プロパノール,2-メチル-2-プロパノール等が挙げられる。
これらのアルコールは、単独で又は2種以上を組み合わせて用いることができる。本発明では、これらから選ばれる複数の脂肪族アルコールを混合して用いることも可能ではあるが、その場合には対称、非対称の複数の炭酸エステル化合物が同時に生成するため分離操作などが複雑になる傾向がある。そのため特段の理由がない限り、単独の脂肪族アルコールを用いることがプロセスを簡便にする観点からは好ましい。
【0013】
本発明に用いる一酸化炭素ガスに特に制限はなく、従来公知な酸化カルボニル化反応に用いられているものを採用することができる。その製造方法などにも制限はないが、圧力スイング吸着法(Pressure Swing Adsorption method)などによってより高純度に精製したものがより好ましい。
本発明に用いる酸素は分子状酸素ガスであれば特に制限はなく、従来公知な酸化反応に用いられているものを採用することができる。
一酸化炭素ガス、酸素ガスのいずれも反応に不活性なガスによって任意の割合で希釈された混合ガスを用いることが出来る。反応に不活性なガスの例としては窒素ガス、ヘリウムガス、アルゴンガス、二酸化炭素ガス等であり、例えば酸素ガス源として空気を用いたり、反応器出口ガス中の不活性ガスを原料ガスとしてリサイクルすることなどが可能である。
【0014】
これらの原料を用いて製造される炭酸エステル化合物は、下記一般式(3)で表される化合物である。また、酸化カルボニル化による炭酸エステル化合物の生成反応は下記化学式(4)で表される。
R2O−C(=O)−OR3 (3)
2R1OH + CO + 1/2O2 → R2O−C(=O)−OR3 + H2O (4)
(ただし、R1は上記一般式(2)と同じである。R2,R3は、R1と同じであるが、R2とR3はそれぞれ同一でも異なってもよい。)
【0015】
上記の酸化カルボニル化反応によって得られる炭酸エステル化合物の例としては、ジメチルカーボネート、ジエチルカーボネート、ジプロピルカーボネート、ジブチルカーボネート、メチルエチルカーボネート、メチルプロピルカーボネート、メチルブチルカーボネート、エチルプロピルカーボネート、エチルブチルカーボネート等が挙げられる。
【0016】
本発明では、下記一般式(1)で表される層状水酸化銅カルボン酸塩を触媒として用いる。
Cu2(OH)3Xと表記される水酸化銅塩類については特に塩化物において結晶形が詳細に検討されており、アタカマイト(atacamite),パラアタカマイト(paratacamite),クリノアタカマイト(clinoatacamite),ボタラカイト(botallackite)の結晶構造が知られている。本発明で用いる層状水酸化銅カルボン酸塩はボタラカイトと同様の層状結晶構造を持ち、下記一般式(1)で表される化合物であれば、特に制限はない。
Cu2(OH)4-n(RCOO)n (1)
上記一般式(1)において、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価のカルボン酸アニオンを表す。nは0.9〜1.1である。
【0017】
従来公知な層状水酸化銅カルボン酸塩の製造法としては、特定の濃度の銅カルボン酸塩溶液を塩基性物質で部分的に中和する方法(Solid.State.Ionics,53−56(1992)527−533等)、銅カルボン酸塩の水溶液を特定の温度条件下で熱処理する方法(J.Solid.State.Chem.,131(1997)252)、層状水酸化銅無機酸塩を別途合成し、カルボン酸塩を用いてイオン交換する方法(J.Mater.Chem.,9,169−174(1999)等)等が知られている。いずれの方法によって製造されたものであっても本発明に用いることができ、その出発原料の種類、組成比や添加方法等について何ら制限はない。
【0018】
工業的利用の見地からは、これらの方法によって水酸化銅カルボン酸塩を廉価で且つ安定に製造できることが好ましい。そのためにはカルボン酸源として最も入手が容易で廉価な酢酸塩を用いることが好ましい。
【0019】
層状水酸化銅カルボン酸塩のカルボン酸基と水酸基との量論比では、上記一般式(1)のnは1となるが、これらの層状水酸化銅カルボン酸塩の製造方法や条件によっては、カルボン酸基や水酸基の含有率に若干の変動があることが知られている。そのために、本発明に用いる層状水酸化銅カルボン酸塩は、その組成によっては上記一般式(1)のnの値が1を中心に若干の変動幅を有していると考えられるが、上記一般式(1)のnの範囲を満たすものであれば問題はない。また通常、水溶液から製造した層状水酸化銅カルボン酸塩は結晶水を含むが、結晶水の有無は本発明の効果に影響しない。
【0020】
本発明に用いる層状水酸化銅カルボン酸塩の粒子径について特に制限はなく、その製造条件や利用目的に適合した粒子径分布のものを用いることができる。
また、製造条件によっては粒子形状が直方体(Solid.State.Ionics,53−56(1992)527−533)や、円盤状(J.Mater.Res.,20,(10),2997−3003(2005))になることが知られているが、いずれの粒子形状であっても本発明に用いるに何ら問題はない。
【0021】
本発明に用いる水酸化銅カルボン酸塩は層状構造を有する。その構造を確認するにはSolid.State.Ionics,178(2007)p1148または粘土鉱物学(白水晴雄著、朝倉書店1988年)p57に記載があるようにX線回折による方法が簡便で有効である。
層状水酸化銅カルボン酸塩の層間距離(d)、X線波長(λ)と入射(反射)角(θ)が、2d×sinθ=kλ(kは整数)を満たすときに回折現象が起こり、層間距離に相当する入射角度で強い回折ピークが得られる(k=1)。次に、2次反射、3次反射といった特定の回折ピークが観察される。
またChemistry Letters,p.1869−1872(1989)に記載があるように層状水酸化銅カルボン酸塩はカルボン酸アニオン種の種類によってほぼ決まった層間距離を持つため、その物質のX線回折が特定の回折パターンと層間距離を示すことにより容易に確認することができる。
【0022】
本発明の触媒は層状水酸化銅カルボン酸塩を含むものであれば加工方法、形状、含有量などに特に制限はない。例えば、層状水酸化銅カルボン酸塩のみを粉末状,粒状,成型品などの任意の形状で用いる方法、層状水酸化銅カルボン酸塩に任意の割合で他の化合物類を混合,混練などした混合物を粉末状,粒状,押出成型品,打錠成形品などの任意の形状で用いる方法、層状水酸化銅カルボン酸塩を従来公知な任意の形状の触媒担体上に析出−沈殿,含浸したものを用いる方法などから最適な方法を選んで用いることができる。
【0023】
前述の層状水酸化銅カルボン酸塩の製法では層状構造が積層した数〜十数ミクロン程度の比較的大きな結晶物が得られることが一般的である。
さらに、本発明では、溶媒中で水酸化銅カルボン酸塩の積層構造を剥離、分散させた状態となった化合物についても用いることができる。このような分散液を用いると簡便であり、かつ高分散な状態で原料アルコールと混合したり、触媒調製のために他の無機化合物と混合、あるいは担体物質へ担持することが可能である。
【0024】
本発明に用いる層状水酸化銅カルボン酸塩は、触媒担体に担持して用いることができる。
触媒担体としては、特に制限はなく従来公知なものを用いることができる。例えばシリカ、アルミナ、チタニア、ジルコニア、マグネシア、活性炭、シリコンカーバイドなどを用いることができる。中でも、担持し易さの観点から、活性炭、シリカが好ましく、活性炭がより好ましい。
担持量としては、特に制限はないが、担持触媒全体に対して0.1〜50質量%が好ましく、0.5〜30質量%がより好ましい。
また、担体の形状、粒度、表面積、気孔率などの物性値や、触媒に担持する方法ついても特に制限はなく、反応方式や条件に好適な担体、担持方法を選択して用いることができる。
【0025】
本発明における反応条件としては、反応温度は、通常50〜300℃、好ましくは80〜250℃、さらに好ましくは100〜200℃で、反応圧力は、通常ゲージ圧で0(常圧)〜5MPa、好ましくは0.05〜2MPa、さらに好ましくは0.1〜1MPaであることが望ましい。
【0026】
本発明における反応は気相、気液混相(トリクル相を含む)のいずれの状態でも行うことができる。本発明の反応における原料の供給方法は流通方式、回分方式のいずれであっても良い。
本発明の反応における触媒の使用方法は固定床、流動床、懸濁床など従来公知な方式を採用することができ、特に制限はない。これらの中では生成物と触媒の分離が容易であること、接触時間を制御し易いことなどから気相条件で流通方式の反応方式がより好適である。
【0027】
本発明における反応を流通方式で行う場合、供給原料の一酸化炭素の使用量は、原料アルコールに対する一酸化炭素のモル比〔一酸化炭素/原料アルコール〕で、好ましくは0.1〜100、より好ましくは0.2〜50、さらに好ましくは0.5〜20の範囲である。
供給原料の酸素の使用量は、原料アルコールに対する酸素のモル比〔酸素/原料アルコール〕で、好ましくは0.01〜2.0、より好ましくは0.02〜1.0、さらに好ましくは0.05〜0.5である。
また、供給原料ガスの空時速度は好ましくは100〜20000h-1程度である。
【実施例】
【0028】
以下に、本発明の方法について実施例および比較例をあげて更に具体的に説明するが、本発明は要旨を超えない限り、これらの実施例に限定されるものではない。
【0029】
下記調製例で得られた各試料のX線回折の分析条件は以下のとおりである。
機器 マックサイエンス社製 MXP-18
線源 Cu Kα線(λ=0.154056nm)
管電圧/管電流 100kV/40mA
走査速度 2°(2θ)/min.
【0030】
参考調製例1
<触媒の調製>
水酸化銅酢酸塩 Cu2(OH)3(CH3COO)をSolid.State.Ionics,53−56(1992)527−533に記載の方法に準じて以下の方法で合成した。
酢酸銅(II)一水和物(和光純薬工業株式会社製、特級試薬、以下同じ)19.95gをイオン交換水999.6gに溶解させて酢酸銅水溶液を調製した。水酸化ナトリウム(和光純薬工業株式会社製、特級試薬、以下同じ)3.92gをイオン交換水1005.2gに溶解させて水酸化ナトリウム水溶液を調製した。
次いで、酢酸銅水溶液を撹拌しているところへ、水酸化ナトリウム水溶液を常温で全量添加し、添加終了後も約2時間撹拌して、青緑色の沈降性結晶を得た。静置して沈降させた後に上澄み液を除去し、アセトン400mlでの洗浄とデカンテーションを数回繰り返した後に、0.2μm径ポリテトラフルオロエチレン(PTFE)製フィルターで濾過し、これを真空ポンプによって常温下で減圧乾燥した後に回収した(以下、A1試料とする)。このA1試料のX線回折のチャートを図1に示した。
これによりLangmuir,24(19),11164−11168(2008)に記載の水酸化銅酢酸塩と同様のXRDパターンが得られていること、層間距離が9.32Åと上記の文献値(9.27〜9.4Å)に一致することを確認した。
また、J.Mater.Res.,20,(11),2997−3003(2005)に記載の示差熱分析を参考にして重量法により以下の方法でA1試料の組成を求めた。粉末状A1試料を105℃で加熱処理して無水化した。得られた試料0.929gを空気中で400℃、3時間加熱処理することによって酸化銅(CuO)へ転化させた。ここで得られた酸化銅は0.626gであった。この重量減少率(67.4%)から求められるA1試料の組成は、Cu2(OH)4-n(CH3COO)nの表記に従うとn=0.97であった。
【0031】
参考調製例2
<触媒の調製>
水酸化銅ギ酸塩 Cu2(OH)3(HCOO)を参考調製例1に準じて以下の方法で合成した。
ギ酸銅(II)四水和物(和光純薬工業株式会社製、Practical.Grade.)11.28gをイオン交換水500.2gに溶解させてギ酸銅水溶液を調製した。水酸化ナトリウム2.00gをイオン交換水500.0gに溶解させて水酸化ナトリウム水溶液を調製した。ギ酸銅水溶液を撹拌しているところへ、水酸化ナトリウム水溶液を常温で全量添加し、添加終了後も約2時間撹拌して、沈降性結晶を得た。以降、参考調製例1と同様にして粉末試料(以下、A2試料とする)を得た。
このA2試料のX線回折のチャートを図2に示した。回折ピークから求められた層間距離は6.66Åであった。
【0032】
参考調製例3
<触媒の調製>
水酸化銅塩化物 Cu2(OH)3ClをSolid.State.Ionics,53−56(1992)527−533に記載の方法に準じて以下の方法で合成した。
塩化ナトリウム(和光純薬工業株式会社製、特級試薬)11.71gをイオン交換水199.96gに溶かした。この塩化ナトリウム水溶液に参考調製例1で得たA1試料粉末〔Cu2(OH)3(CH3COO)〕2.00gを加えて一晩攪拌した。次いで0.2μm径PTFE製フィルターで濾過し、イオン交換水で洗浄した後に、真空ポンプによって常温下で減圧乾燥して、粉末試料(以下、A3試料とする)を得た。このA3試料のX線回折のチャートを図3に示した。
これによりJ.Mater.Res.,20,(11),2997−3003(2005)に記載の水酸化銅塩化物と同様のXRDパターンが得られていることを確認した。(ただし、該文献ではCo Kα線,λ=0.178897nmによる測定であるため、本測定のCu Kα線,λ=0.154056nmとの比を補正して比較した)
また、回折ピークから求められた層間距離は5.71Åであり、文献値(5.71Å)と一致することを確認した。
【0033】
調製例1
<担持触媒の調製>
粒状シリカ担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物1.99g及びイオン交換水60.4gからなる溶液に、水酸化ナトリウム0.40g及びイオン交換水60.1gからなる溶液を、攪拌しながら加えた。水酸化ナトリウム水溶液の添加終了後、粒状シリカ(富士シリシア化学株式会社製、「キャリアクトQ−10」(商品名)の破砕品、粒径0.5〜1.0mm)6.23gを加え、更に一晩攪拌した。攪拌終了後、溶液中には層状水酸化銅酢酸塩の結晶が生成し、シリカ粒子は、析出した結晶により青く着色した。イオン交換水での洗浄とデカンテーションを数回繰り返してシリカ粒子のみを回収し、60℃,1.33kPa(10torr)の条件で加熱減圧乾燥させて触媒(以下、B1触媒とする)を得た。誘導結合プラズマ発光分析(ICP分析)からB1触媒の銅担持量は2.2質量%であった。
【0034】
調製例2
<担持触媒の調製>
粒状シリカ担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物1.86g及びイオン交換水31.4gからなる溶液に、調製例1で用いた粒状シリカ6.23gを加えた。その後に水酸化ナトリウム0.37g及びイオン交換水40.0gからなる溶液を、攪拌しながら更に加えた。水酸化ナトリウム水溶液の添加終了後、更に一晩攪拌した。攪拌終了後、溶液中には層状水酸化銅酢酸塩の結晶が生成し、シリカ粒子は析出した結晶により青く着色した。イオン交換水での洗浄とデカンテーションを数回繰り返してシリカ粒子のみ回収し、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、B2触媒とする)を得た。B2触媒の銅担持量は3.2質量%であった。
【0035】
比較調製例1
<担持触媒の調製>
酢酸銅(II)一水和物1.43gをイオン交換水26.2gに溶解させた溶液に、調製例1で用いたシリカ粒子10.04gを加え、一晩攪拌した。次いで調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C1触媒とする)を得た。C1触媒の銅担持量は3.9質量%であった。
【0036】
比較調製例2
<担持触媒の調製>
水酸化銅(II)(和光純薬工業株式会社製、特級試薬)0.31gを28%アンモニア水約30gに溶解させた。この溶液に、調製例1で用いたシリカ粒子6.20gを加え、一晩攪拌した。その後に調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C2触媒とする)を得た。C2触媒の銅担持量は2.8質量%であった。
【0037】
実施例1
調製例1で得られたB1触媒2.47gをパイレックス(登録商標)製ガラス反応管(内径12mm)に充填した。常圧下で一酸化炭素ガス毎分50ml、空気毎分10ml、メタノール毎時3.0gの組成の混合ガスを触媒層へ流通しながら165℃で反応を行った。反応器出口のドライアイス−メタノールトラップによる凝縮回収液をガスクロマトグラフ装置で分析したところ、炭酸ジメチルの空時収量は8.5g−炭酸ジメチル/kg−触媒・時間であった。
【0038】
実施例2
B1触媒に代えて調製例2で得られたB2触媒2.51gを用いた以外は実施例1と同様の反応操作を行うことにより炭酸ジメチルが空時収量10.6g−炭酸ジメチル/kg−触媒・時間で得られた。
【0039】
比較例1
B1触媒に代えて比較調製例1で得られたC1触媒3.32gを用い、反応温度160℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量3.9g−炭酸ジメチル/kg−触媒・時間で得られた。
【0040】
比較例2
B1触媒に代えて比較調製例2で得られたC2触媒3.18gを用い、反応温度155℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量4.2g−炭酸ジメチル/kg−触媒・時間で得られた。
【0041】
調製例3
<担持触媒の調製>
粒状活性炭担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物0.99g及びイオン交換水43.2gからなる溶液に、粒状活性炭(クラレケミカル株式会社製、商品名「クラレコールGW」、10〜32mesh)6.25gを加えた。その後に水酸化ナトリウム0.17g及びイオン交換水43.0gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B3触媒とする)を得た。B3触媒の銅担持量は3.5質量%であった。
【0042】
調製例4
<担持触媒の調製>
繊維状活性炭担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物3.99g及びイオン交換水200.2gからなる溶液に、繊維状活性炭(クラレケミカル株式会社製、商品名「クラレコールFR−20」、数cm長に裁断したものを使用)5.36gを加えた。その後に水酸化ナトリウム0.79g及びイオン交換水100.3gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B4触媒とする)を得た。B4触媒の銅担持量は4.8質量%であった。
【0043】
調製例5
<担持触媒の調製>
粒状活性炭担体の存在下で参考調製例2と同様の操作を行って担持触媒を調製した。
ギ酸銅(II)四水和物1.12g及びイオン交換水44.4gからなる溶液に、調製例3で使用した粒状活性炭6.25gを加えた。その後に水酸化ナトリウム0.18g及びイオン交換水45.0gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B5触媒とする)を得た。B5触媒の銅担持量は2.9質量%であった。
【0044】
調製例6
<担持触媒の調製>
参考調製例1で得られたA1試料粉末0.45gにアセト酢酸メチル(和光純薬工業株式会社製、一級試薬)0.55g、1−プロパノール(和光純薬工業株式会社製、特級試薬)100.2gを加えて攪拌することにより、分散コロイド液を調製した。この溶液43.7gに調製例3で用いた粒状活性炭4.53gを加えた後、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、B6触媒とする)を得た。B6触媒の銅担持量は2.0質量%であった。
【0045】
比較調製例3
<担持触媒の調製>
酢酸銅(II)一水和物0.81gをイオン交換水30.2gに溶解させた溶液に、調製例3で用いた粒状活性炭6.31gを加え、一晩攪拌した。その後に調製例1と同様の条件で熱減圧乾燥させて触媒(以下、C3触媒とする)を得た。C3触媒の銅担持量は3.6質量%であった。
【0046】
比較調製例4
<担持触媒の調製>
水酸化銅(II)(和光純薬特級)0.42gを28%アンモニア水約28gに溶解させた溶液に、調製例3で用いた粒状活性炭6.28gを加え、一晩攪拌した。その後に調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C4触媒とする)を得た。C4触媒の銅担持量は2.9質量%であった。
【0047】
比較調製例5
<担持触媒の調製>
酢酸銅(II)一水和物0.99g及びイオン交換水43.2gからなる溶液に、調製例3で用いた粒状活性炭6.25gを加えた。その後に水酸化ナトリウム0.17g及びイオン交換水43.0gからなる溶液を、攪拌しながら更に加えた。水酸化ナトリウム水溶液の添加終了後、更に一晩攪拌することにより層状水酸化銅酢酸塩を担持した。イオン交換水での洗浄とデカンテーションを数回繰り返した触媒の半分量に、1mol/Lの塩化ナトリウム水溶液20mlを加え、室温で一晩攪拌し、参考調製例3と同様の層状水酸化銅塩化物へのイオン交換処理を行った。その後、イオン交換水で洗浄−デカンテーションを繰り返して、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C5触媒とする)を得た。C5触媒の銅担持量は3.0質量%であった。
【0048】
実施例3
B1触媒に代えて調製例3で得られたB3触媒2.54gを用い、反応温度を160℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量28.8g−炭酸ジメチル/kg−触媒・時間で得られた。
【0049】
実施例4
B1触媒に代えて調製例4で得られたB4触媒1.40gを用い、反応温度を160℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量33.7g−炭酸ジメチル/kg−触媒・時間で得られた。
【0050】
実施例5
B1触媒に代えて調製例5で得られたB5触媒2.53gを用い、反応温度を150℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量22.0g−炭酸ジメチル/kg−触媒・時間で得られた。
【0051】
実施例6
B1触媒に代えて調製例6で得られたB6触媒2.97gを用い、反応温度を170℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量22.1g−炭酸ジメチル/kg−触媒・時間で得られた。
【0052】
比較例3
B1触媒に代えて比較調製例3で得られたC3触媒2.57gを用い、反応温度160℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量12.2g−炭酸ジメチル/kg−触媒・時間で得られた。
【0053】
比較例4
B1触媒に代えて比較調製例4で得られたC4触媒2.57gを用い、反応温度155℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量15.5g−炭酸ジメチル/kg−触媒・時間で得られた。
【0054】
比較例5
B1触媒に代えて比較調製例5で得られたC5触媒2.53gを用い、反応温度150〜165℃で行った以外は実施例1と同様の反応操作を行ったところ生成した炭酸ジメチルは痕跡量であった。
【産業上の利用可能性】
【0055】
本発明の方法により、ハロゲン元素を含まない固体触媒存在下に一段反応で脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造することができる。
【技術分野】
【0001】
本発明は、脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を製造する方法に関するものである。
【背景技術】
【0002】
脂肪族アルコールと一酸化炭素及び酸素から酸化カルボニル化反応によって炭酸エステル化合物を製造する方法は広く知られており、多くの公知文献がある。
その殆どの文献では、触媒成分として銅及び/またはパラジウムのハロゲン化物や、これらに他の金属元素のハロゲン化物を加えたものが用いられている(特許文献1〜3参照)。
この方法では、酸化反応中に腐食性の強いハロゲン化水素酸が副生するために、製造設備には高価な耐食性金属材料やグラスライニング等の耐食保護材料を用いなければならない。更に酸化カルボニル化反応では、加圧条件下で有利に反応が進行するために、殆どの文献において数気圧〜数十気圧程度の加圧条件が採用されている。そのため、より高価な耐腐食性耐圧反応装置が必要となり、炭酸エステルを安価に製造する観点から著しく不利である。
【0003】
ハロゲン化合物を含まない触媒系を用いた酸化カルボニル化反応による炭酸エステル化合物の製造法としては、酸化銅,水酸化銅,酢酸銅のようなハロゲンを含まない弱酸の銅塩から選ばれた少なくとも一種類の銅化合物を触媒として用いる方法(特許文献4参照)、酸化銅と周期律表第13〜16族元素の酸化物からなる触媒を用いる方法(特許文献5参照)、水酸化銅,酸化銅,銅の弱酸塩から選ばれる少なくとも一種類の銅化合物と硝酸銅からなる触媒を用いる方法(特許文献6参照)、及び銅アルコラート化合物を用いる方法(特許文献7参照)などが開示されている。
これらの文献例では炭酸エステルの生産性は高くなく、改善の余地は大きい。また、酸化銅、水酸化銅、銅アルコラート等は脂肪族アルコールのような還元性物質、反応で副生する水、熱などに対して非常に不安定であり、容易に分解して触媒として失活しやすいという欠点があった。
【0004】
また、酸化カルボニル化による炭酸エステル合成反応において層状化合物を用いる方法が知られている。例えば、層状粘土化合物の層間あるいは表面に一価または二価の銅を担持した触媒を用いる方法(特許文献8参照)、粘土化合物または黒鉛などの層状物質の層間に銅の塩、カチオン、錯体化合物等の触媒成分が挿入された固体触媒を用いる方法(特許文献9参照)が公知である。これらは無機層状化合物の層間に従来公知な触媒活性成分をインターカレートまたはイオン交換させることによって触媒性能の向上を図ったものと考えられる。
【0005】
さらに、酸化カルボニル化による炭酸エステル合成反応を二段反応で行う方法も公知である。前段反応として脂肪族アルコールと一酸化窒素及び酸素から亜硝酸エステルと水を作り、水を分離してから、後段反応として亜硝酸エステルと一酸化炭素を触媒によって炭酸エステルと一酸化窒素へ転化させ、再生した一酸化窒素を前段反応へリサイクルする方法である(特許文献10及び11参照)。この方法は比較的温和な反応条件で反応が進行し、且つ腐食性の問題が少ない有効な方法ではあるが、後段反応の触媒として高価な白金族元素が必須であること、二段反応であるために一段合成法に比較して付帯設備が多く、反応系が複雑になるなどの不利益があった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平2−169549号公報
【特許文献2】特開平5−208137号公報
【特許文献3】特開平9−10591号公報
【特許文献4】特開平6−218284号公報
【特許文献5】特開平7−313879号公報
【特許文献6】特開平6−92908号公報
【特許文献7】特開平10−114720号公報
【特許文献8】特開平5−255201号公報
【特許文献9】特開平6−116212号公報
【特許文献10】特開昭60−11443号公報
【特許文献11】特開平3−141243号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、従来技術における上記のような課題を解決し、触媒の存在下に脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造する方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者は触媒の存在下に脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を一段反応によって製造する方法について鋭意研究を重ねた結果、層状水酸化銅カルボン酸塩の存在下で反応を行うことによって効率よく、炭酸エステル化合物を製造できることを見いだし本発明に到達した。
【0009】
すなわち、本発明は、一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法に関するものである。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【発明の効果】
【0010】
本発明の方法により、ハロゲン元素を含まない固体触媒存在下に一段反応で脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造することができる。
【図面の簡単な説明】
【0011】
【図1】参考調製例1で得られたA1試料のX線回折チャート。
【図2】参考調製例2で得られたA2試料のX線回折チャート。
【図3】参考調製例3で得られたA3試料のX線回折チャート。
【発明を実施するための形態】
【0012】
以下に本発明の各構成要件について説明する。
本発明に用いる脂肪族アルコール化合物(以下、「原料アルコール」ともいう。)は、上記一般式(2)で表される化合物であれば特に制限はない。具体的にはメタノール,エタノール,1-プロパノール,1-ブタノール,2-ブタノール,2-メチル-1-プロパノール,2-メチル-2-プロパノール等が挙げられる。
これらのアルコールは、単独で又は2種以上を組み合わせて用いることができる。本発明では、これらから選ばれる複数の脂肪族アルコールを混合して用いることも可能ではあるが、その場合には対称、非対称の複数の炭酸エステル化合物が同時に生成するため分離操作などが複雑になる傾向がある。そのため特段の理由がない限り、単独の脂肪族アルコールを用いることがプロセスを簡便にする観点からは好ましい。
【0013】
本発明に用いる一酸化炭素ガスに特に制限はなく、従来公知な酸化カルボニル化反応に用いられているものを採用することができる。その製造方法などにも制限はないが、圧力スイング吸着法(Pressure Swing Adsorption method)などによってより高純度に精製したものがより好ましい。
本発明に用いる酸素は分子状酸素ガスであれば特に制限はなく、従来公知な酸化反応に用いられているものを採用することができる。
一酸化炭素ガス、酸素ガスのいずれも反応に不活性なガスによって任意の割合で希釈された混合ガスを用いることが出来る。反応に不活性なガスの例としては窒素ガス、ヘリウムガス、アルゴンガス、二酸化炭素ガス等であり、例えば酸素ガス源として空気を用いたり、反応器出口ガス中の不活性ガスを原料ガスとしてリサイクルすることなどが可能である。
【0014】
これらの原料を用いて製造される炭酸エステル化合物は、下記一般式(3)で表される化合物である。また、酸化カルボニル化による炭酸エステル化合物の生成反応は下記化学式(4)で表される。
R2O−C(=O)−OR3 (3)
2R1OH + CO + 1/2O2 → R2O−C(=O)−OR3 + H2O (4)
(ただし、R1は上記一般式(2)と同じである。R2,R3は、R1と同じであるが、R2とR3はそれぞれ同一でも異なってもよい。)
【0015】
上記の酸化カルボニル化反応によって得られる炭酸エステル化合物の例としては、ジメチルカーボネート、ジエチルカーボネート、ジプロピルカーボネート、ジブチルカーボネート、メチルエチルカーボネート、メチルプロピルカーボネート、メチルブチルカーボネート、エチルプロピルカーボネート、エチルブチルカーボネート等が挙げられる。
【0016】
本発明では、下記一般式(1)で表される層状水酸化銅カルボン酸塩を触媒として用いる。
Cu2(OH)3Xと表記される水酸化銅塩類については特に塩化物において結晶形が詳細に検討されており、アタカマイト(atacamite),パラアタカマイト(paratacamite),クリノアタカマイト(clinoatacamite),ボタラカイト(botallackite)の結晶構造が知られている。本発明で用いる層状水酸化銅カルボン酸塩はボタラカイトと同様の層状結晶構造を持ち、下記一般式(1)で表される化合物であれば、特に制限はない。
Cu2(OH)4-n(RCOO)n (1)
上記一般式(1)において、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価のカルボン酸アニオンを表す。nは0.9〜1.1である。
【0017】
従来公知な層状水酸化銅カルボン酸塩の製造法としては、特定の濃度の銅カルボン酸塩溶液を塩基性物質で部分的に中和する方法(Solid.State.Ionics,53−56(1992)527−533等)、銅カルボン酸塩の水溶液を特定の温度条件下で熱処理する方法(J.Solid.State.Chem.,131(1997)252)、層状水酸化銅無機酸塩を別途合成し、カルボン酸塩を用いてイオン交換する方法(J.Mater.Chem.,9,169−174(1999)等)等が知られている。いずれの方法によって製造されたものであっても本発明に用いることができ、その出発原料の種類、組成比や添加方法等について何ら制限はない。
【0018】
工業的利用の見地からは、これらの方法によって水酸化銅カルボン酸塩を廉価で且つ安定に製造できることが好ましい。そのためにはカルボン酸源として最も入手が容易で廉価な酢酸塩を用いることが好ましい。
【0019】
層状水酸化銅カルボン酸塩のカルボン酸基と水酸基との量論比では、上記一般式(1)のnは1となるが、これらの層状水酸化銅カルボン酸塩の製造方法や条件によっては、カルボン酸基や水酸基の含有率に若干の変動があることが知られている。そのために、本発明に用いる層状水酸化銅カルボン酸塩は、その組成によっては上記一般式(1)のnの値が1を中心に若干の変動幅を有していると考えられるが、上記一般式(1)のnの範囲を満たすものであれば問題はない。また通常、水溶液から製造した層状水酸化銅カルボン酸塩は結晶水を含むが、結晶水の有無は本発明の効果に影響しない。
【0020】
本発明に用いる層状水酸化銅カルボン酸塩の粒子径について特に制限はなく、その製造条件や利用目的に適合した粒子径分布のものを用いることができる。
また、製造条件によっては粒子形状が直方体(Solid.State.Ionics,53−56(1992)527−533)や、円盤状(J.Mater.Res.,20,(10),2997−3003(2005))になることが知られているが、いずれの粒子形状であっても本発明に用いるに何ら問題はない。
【0021】
本発明に用いる水酸化銅カルボン酸塩は層状構造を有する。その構造を確認するにはSolid.State.Ionics,178(2007)p1148または粘土鉱物学(白水晴雄著、朝倉書店1988年)p57に記載があるようにX線回折による方法が簡便で有効である。
層状水酸化銅カルボン酸塩の層間距離(d)、X線波長(λ)と入射(反射)角(θ)が、2d×sinθ=kλ(kは整数)を満たすときに回折現象が起こり、層間距離に相当する入射角度で強い回折ピークが得られる(k=1)。次に、2次反射、3次反射といった特定の回折ピークが観察される。
またChemistry Letters,p.1869−1872(1989)に記載があるように層状水酸化銅カルボン酸塩はカルボン酸アニオン種の種類によってほぼ決まった層間距離を持つため、その物質のX線回折が特定の回折パターンと層間距離を示すことにより容易に確認することができる。
【0022】
本発明の触媒は層状水酸化銅カルボン酸塩を含むものであれば加工方法、形状、含有量などに特に制限はない。例えば、層状水酸化銅カルボン酸塩のみを粉末状,粒状,成型品などの任意の形状で用いる方法、層状水酸化銅カルボン酸塩に任意の割合で他の化合物類を混合,混練などした混合物を粉末状,粒状,押出成型品,打錠成形品などの任意の形状で用いる方法、層状水酸化銅カルボン酸塩を従来公知な任意の形状の触媒担体上に析出−沈殿,含浸したものを用いる方法などから最適な方法を選んで用いることができる。
【0023】
前述の層状水酸化銅カルボン酸塩の製法では層状構造が積層した数〜十数ミクロン程度の比較的大きな結晶物が得られることが一般的である。
さらに、本発明では、溶媒中で水酸化銅カルボン酸塩の積層構造を剥離、分散させた状態となった化合物についても用いることができる。このような分散液を用いると簡便であり、かつ高分散な状態で原料アルコールと混合したり、触媒調製のために他の無機化合物と混合、あるいは担体物質へ担持することが可能である。
【0024】
本発明に用いる層状水酸化銅カルボン酸塩は、触媒担体に担持して用いることができる。
触媒担体としては、特に制限はなく従来公知なものを用いることができる。例えばシリカ、アルミナ、チタニア、ジルコニア、マグネシア、活性炭、シリコンカーバイドなどを用いることができる。中でも、担持し易さの観点から、活性炭、シリカが好ましく、活性炭がより好ましい。
担持量としては、特に制限はないが、担持触媒全体に対して0.1〜50質量%が好ましく、0.5〜30質量%がより好ましい。
また、担体の形状、粒度、表面積、気孔率などの物性値や、触媒に担持する方法ついても特に制限はなく、反応方式や条件に好適な担体、担持方法を選択して用いることができる。
【0025】
本発明における反応条件としては、反応温度は、通常50〜300℃、好ましくは80〜250℃、さらに好ましくは100〜200℃で、反応圧力は、通常ゲージ圧で0(常圧)〜5MPa、好ましくは0.05〜2MPa、さらに好ましくは0.1〜1MPaであることが望ましい。
【0026】
本発明における反応は気相、気液混相(トリクル相を含む)のいずれの状態でも行うことができる。本発明の反応における原料の供給方法は流通方式、回分方式のいずれであっても良い。
本発明の反応における触媒の使用方法は固定床、流動床、懸濁床など従来公知な方式を採用することができ、特に制限はない。これらの中では生成物と触媒の分離が容易であること、接触時間を制御し易いことなどから気相条件で流通方式の反応方式がより好適である。
【0027】
本発明における反応を流通方式で行う場合、供給原料の一酸化炭素の使用量は、原料アルコールに対する一酸化炭素のモル比〔一酸化炭素/原料アルコール〕で、好ましくは0.1〜100、より好ましくは0.2〜50、さらに好ましくは0.5〜20の範囲である。
供給原料の酸素の使用量は、原料アルコールに対する酸素のモル比〔酸素/原料アルコール〕で、好ましくは0.01〜2.0、より好ましくは0.02〜1.0、さらに好ましくは0.05〜0.5である。
また、供給原料ガスの空時速度は好ましくは100〜20000h-1程度である。
【実施例】
【0028】
以下に、本発明の方法について実施例および比較例をあげて更に具体的に説明するが、本発明は要旨を超えない限り、これらの実施例に限定されるものではない。
【0029】
下記調製例で得られた各試料のX線回折の分析条件は以下のとおりである。
機器 マックサイエンス社製 MXP-18
線源 Cu Kα線(λ=0.154056nm)
管電圧/管電流 100kV/40mA
走査速度 2°(2θ)/min.
【0030】
参考調製例1
<触媒の調製>
水酸化銅酢酸塩 Cu2(OH)3(CH3COO)をSolid.State.Ionics,53−56(1992)527−533に記載の方法に準じて以下の方法で合成した。
酢酸銅(II)一水和物(和光純薬工業株式会社製、特級試薬、以下同じ)19.95gをイオン交換水999.6gに溶解させて酢酸銅水溶液を調製した。水酸化ナトリウム(和光純薬工業株式会社製、特級試薬、以下同じ)3.92gをイオン交換水1005.2gに溶解させて水酸化ナトリウム水溶液を調製した。
次いで、酢酸銅水溶液を撹拌しているところへ、水酸化ナトリウム水溶液を常温で全量添加し、添加終了後も約2時間撹拌して、青緑色の沈降性結晶を得た。静置して沈降させた後に上澄み液を除去し、アセトン400mlでの洗浄とデカンテーションを数回繰り返した後に、0.2μm径ポリテトラフルオロエチレン(PTFE)製フィルターで濾過し、これを真空ポンプによって常温下で減圧乾燥した後に回収した(以下、A1試料とする)。このA1試料のX線回折のチャートを図1に示した。
これによりLangmuir,24(19),11164−11168(2008)に記載の水酸化銅酢酸塩と同様のXRDパターンが得られていること、層間距離が9.32Åと上記の文献値(9.27〜9.4Å)に一致することを確認した。
また、J.Mater.Res.,20,(11),2997−3003(2005)に記載の示差熱分析を参考にして重量法により以下の方法でA1試料の組成を求めた。粉末状A1試料を105℃で加熱処理して無水化した。得られた試料0.929gを空気中で400℃、3時間加熱処理することによって酸化銅(CuO)へ転化させた。ここで得られた酸化銅は0.626gであった。この重量減少率(67.4%)から求められるA1試料の組成は、Cu2(OH)4-n(CH3COO)nの表記に従うとn=0.97であった。
【0031】
参考調製例2
<触媒の調製>
水酸化銅ギ酸塩 Cu2(OH)3(HCOO)を参考調製例1に準じて以下の方法で合成した。
ギ酸銅(II)四水和物(和光純薬工業株式会社製、Practical.Grade.)11.28gをイオン交換水500.2gに溶解させてギ酸銅水溶液を調製した。水酸化ナトリウム2.00gをイオン交換水500.0gに溶解させて水酸化ナトリウム水溶液を調製した。ギ酸銅水溶液を撹拌しているところへ、水酸化ナトリウム水溶液を常温で全量添加し、添加終了後も約2時間撹拌して、沈降性結晶を得た。以降、参考調製例1と同様にして粉末試料(以下、A2試料とする)を得た。
このA2試料のX線回折のチャートを図2に示した。回折ピークから求められた層間距離は6.66Åであった。
【0032】
参考調製例3
<触媒の調製>
水酸化銅塩化物 Cu2(OH)3ClをSolid.State.Ionics,53−56(1992)527−533に記載の方法に準じて以下の方法で合成した。
塩化ナトリウム(和光純薬工業株式会社製、特級試薬)11.71gをイオン交換水199.96gに溶かした。この塩化ナトリウム水溶液に参考調製例1で得たA1試料粉末〔Cu2(OH)3(CH3COO)〕2.00gを加えて一晩攪拌した。次いで0.2μm径PTFE製フィルターで濾過し、イオン交換水で洗浄した後に、真空ポンプによって常温下で減圧乾燥して、粉末試料(以下、A3試料とする)を得た。このA3試料のX線回折のチャートを図3に示した。
これによりJ.Mater.Res.,20,(11),2997−3003(2005)に記載の水酸化銅塩化物と同様のXRDパターンが得られていることを確認した。(ただし、該文献ではCo Kα線,λ=0.178897nmによる測定であるため、本測定のCu Kα線,λ=0.154056nmとの比を補正して比較した)
また、回折ピークから求められた層間距離は5.71Åであり、文献値(5.71Å)と一致することを確認した。
【0033】
調製例1
<担持触媒の調製>
粒状シリカ担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物1.99g及びイオン交換水60.4gからなる溶液に、水酸化ナトリウム0.40g及びイオン交換水60.1gからなる溶液を、攪拌しながら加えた。水酸化ナトリウム水溶液の添加終了後、粒状シリカ(富士シリシア化学株式会社製、「キャリアクトQ−10」(商品名)の破砕品、粒径0.5〜1.0mm)6.23gを加え、更に一晩攪拌した。攪拌終了後、溶液中には層状水酸化銅酢酸塩の結晶が生成し、シリカ粒子は、析出した結晶により青く着色した。イオン交換水での洗浄とデカンテーションを数回繰り返してシリカ粒子のみを回収し、60℃,1.33kPa(10torr)の条件で加熱減圧乾燥させて触媒(以下、B1触媒とする)を得た。誘導結合プラズマ発光分析(ICP分析)からB1触媒の銅担持量は2.2質量%であった。
【0034】
調製例2
<担持触媒の調製>
粒状シリカ担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物1.86g及びイオン交換水31.4gからなる溶液に、調製例1で用いた粒状シリカ6.23gを加えた。その後に水酸化ナトリウム0.37g及びイオン交換水40.0gからなる溶液を、攪拌しながら更に加えた。水酸化ナトリウム水溶液の添加終了後、更に一晩攪拌した。攪拌終了後、溶液中には層状水酸化銅酢酸塩の結晶が生成し、シリカ粒子は析出した結晶により青く着色した。イオン交換水での洗浄とデカンテーションを数回繰り返してシリカ粒子のみ回収し、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、B2触媒とする)を得た。B2触媒の銅担持量は3.2質量%であった。
【0035】
比較調製例1
<担持触媒の調製>
酢酸銅(II)一水和物1.43gをイオン交換水26.2gに溶解させた溶液に、調製例1で用いたシリカ粒子10.04gを加え、一晩攪拌した。次いで調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C1触媒とする)を得た。C1触媒の銅担持量は3.9質量%であった。
【0036】
比較調製例2
<担持触媒の調製>
水酸化銅(II)(和光純薬工業株式会社製、特級試薬)0.31gを28%アンモニア水約30gに溶解させた。この溶液に、調製例1で用いたシリカ粒子6.20gを加え、一晩攪拌した。その後に調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C2触媒とする)を得た。C2触媒の銅担持量は2.8質量%であった。
【0037】
実施例1
調製例1で得られたB1触媒2.47gをパイレックス(登録商標)製ガラス反応管(内径12mm)に充填した。常圧下で一酸化炭素ガス毎分50ml、空気毎分10ml、メタノール毎時3.0gの組成の混合ガスを触媒層へ流通しながら165℃で反応を行った。反応器出口のドライアイス−メタノールトラップによる凝縮回収液をガスクロマトグラフ装置で分析したところ、炭酸ジメチルの空時収量は8.5g−炭酸ジメチル/kg−触媒・時間であった。
【0038】
実施例2
B1触媒に代えて調製例2で得られたB2触媒2.51gを用いた以外は実施例1と同様の反応操作を行うことにより炭酸ジメチルが空時収量10.6g−炭酸ジメチル/kg−触媒・時間で得られた。
【0039】
比較例1
B1触媒に代えて比較調製例1で得られたC1触媒3.32gを用い、反応温度160℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量3.9g−炭酸ジメチル/kg−触媒・時間で得られた。
【0040】
比較例2
B1触媒に代えて比較調製例2で得られたC2触媒3.18gを用い、反応温度155℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量4.2g−炭酸ジメチル/kg−触媒・時間で得られた。
【0041】
調製例3
<担持触媒の調製>
粒状活性炭担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物0.99g及びイオン交換水43.2gからなる溶液に、粒状活性炭(クラレケミカル株式会社製、商品名「クラレコールGW」、10〜32mesh)6.25gを加えた。その後に水酸化ナトリウム0.17g及びイオン交換水43.0gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B3触媒とする)を得た。B3触媒の銅担持量は3.5質量%であった。
【0042】
調製例4
<担持触媒の調製>
繊維状活性炭担体の存在下で参考調製例1と同様の操作を行って担持触媒を調製した。
酢酸銅(II)一水和物3.99g及びイオン交換水200.2gからなる溶液に、繊維状活性炭(クラレケミカル株式会社製、商品名「クラレコールFR−20」、数cm長に裁断したものを使用)5.36gを加えた。その後に水酸化ナトリウム0.79g及びイオン交換水100.3gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B4触媒とする)を得た。B4触媒の銅担持量は4.8質量%であった。
【0043】
調製例5
<担持触媒の調製>
粒状活性炭担体の存在下で参考調製例2と同様の操作を行って担持触媒を調製した。
ギ酸銅(II)四水和物1.12g及びイオン交換水44.4gからなる溶液に、調製例3で使用した粒状活性炭6.25gを加えた。その後に水酸化ナトリウム0.18g及びイオン交換水45.0gからなる溶液を、攪拌しながら更に加えた。以降、調製例2と同様の操作で触媒(以下、B5触媒とする)を得た。B5触媒の銅担持量は2.9質量%であった。
【0044】
調製例6
<担持触媒の調製>
参考調製例1で得られたA1試料粉末0.45gにアセト酢酸メチル(和光純薬工業株式会社製、一級試薬)0.55g、1−プロパノール(和光純薬工業株式会社製、特級試薬)100.2gを加えて攪拌することにより、分散コロイド液を調製した。この溶液43.7gに調製例3で用いた粒状活性炭4.53gを加えた後、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、B6触媒とする)を得た。B6触媒の銅担持量は2.0質量%であった。
【0045】
比較調製例3
<担持触媒の調製>
酢酸銅(II)一水和物0.81gをイオン交換水30.2gに溶解させた溶液に、調製例3で用いた粒状活性炭6.31gを加え、一晩攪拌した。その後に調製例1と同様の条件で熱減圧乾燥させて触媒(以下、C3触媒とする)を得た。C3触媒の銅担持量は3.6質量%であった。
【0046】
比較調製例4
<担持触媒の調製>
水酸化銅(II)(和光純薬特級)0.42gを28%アンモニア水約28gに溶解させた溶液に、調製例3で用いた粒状活性炭6.28gを加え、一晩攪拌した。その後に調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C4触媒とする)を得た。C4触媒の銅担持量は2.9質量%であった。
【0047】
比較調製例5
<担持触媒の調製>
酢酸銅(II)一水和物0.99g及びイオン交換水43.2gからなる溶液に、調製例3で用いた粒状活性炭6.25gを加えた。その後に水酸化ナトリウム0.17g及びイオン交換水43.0gからなる溶液を、攪拌しながら更に加えた。水酸化ナトリウム水溶液の添加終了後、更に一晩攪拌することにより層状水酸化銅酢酸塩を担持した。イオン交換水での洗浄とデカンテーションを数回繰り返した触媒の半分量に、1mol/Lの塩化ナトリウム水溶液20mlを加え、室温で一晩攪拌し、参考調製例3と同様の層状水酸化銅塩化物へのイオン交換処理を行った。その後、イオン交換水で洗浄−デカンテーションを繰り返して、調製例1と同様の条件で加熱減圧乾燥させて触媒(以下、C5触媒とする)を得た。C5触媒の銅担持量は3.0質量%であった。
【0048】
実施例3
B1触媒に代えて調製例3で得られたB3触媒2.54gを用い、反応温度を160℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量28.8g−炭酸ジメチル/kg−触媒・時間で得られた。
【0049】
実施例4
B1触媒に代えて調製例4で得られたB4触媒1.40gを用い、反応温度を160℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量33.7g−炭酸ジメチル/kg−触媒・時間で得られた。
【0050】
実施例5
B1触媒に代えて調製例5で得られたB5触媒2.53gを用い、反応温度を150℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量22.0g−炭酸ジメチル/kg−触媒・時間で得られた。
【0051】
実施例6
B1触媒に代えて調製例6で得られたB6触媒2.97gを用い、反応温度を170℃としたこと以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量22.1g−炭酸ジメチル/kg−触媒・時間で得られた。
【0052】
比較例3
B1触媒に代えて比較調製例3で得られたC3触媒2.57gを用い、反応温度160℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量12.2g−炭酸ジメチル/kg−触媒・時間で得られた。
【0053】
比較例4
B1触媒に代えて比較調製例4で得られたC4触媒2.57gを用い、反応温度155℃で行った以外は実施例1と同様の反応操作をすることにより炭酸ジメチルが空時収量15.5g−炭酸ジメチル/kg−触媒・時間で得られた。
【0054】
比較例5
B1触媒に代えて比較調製例5で得られたC5触媒2.53gを用い、反応温度150〜165℃で行った以外は実施例1と同様の反応操作を行ったところ生成した炭酸ジメチルは痕跡量であった。
【産業上の利用可能性】
【0055】
本発明の方法により、ハロゲン元素を含まない固体触媒存在下に一段反応で脂肪族アルコール化合物と一酸化炭素及び酸素から炭酸エステル化合物を効率良く製造することができる。
【特許請求の範囲】
【請求項1】
一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【請求項2】
Rがメチル基である請求項1に記載の炭酸エステル化合物の製造方法。
【請求項3】
一般式(1)で表される層状水酸化銅カルボン酸塩が担体に担持された触媒である請求項1または2に記載の炭酸エステル化合物の製造方法。
【請求項4】
担体が活性炭である請求項3に記載の炭酸エステル化合物の製造方法。
【請求項5】
気相流通条件下で反応させる請求項1〜4のいずれかに記載の炭酸エステル化合物の製造方法。
【請求項1】
一般式(1)で表される層状水酸化銅カルボン酸塩の存在下に、一般式(2)で表される脂肪族アルコール化合物と一酸化炭素と酸素を反応させる炭酸エステル化合物の製造方法。
Cu2(OH)4-n(RCOO)n (1)
R1OH (2)
(ただし、Rは水素原子または炭素数1〜3の直鎖若しくは分岐したアルキル基であり、RCOOは一価の脂肪族カルボン酸アニオンを表す。nは0.9〜1.1である。R1は炭素数1〜4の直鎖または分岐したアルキル基である。)
【請求項2】
Rがメチル基である請求項1に記載の炭酸エステル化合物の製造方法。
【請求項3】
一般式(1)で表される層状水酸化銅カルボン酸塩が担体に担持された触媒である請求項1または2に記載の炭酸エステル化合物の製造方法。
【請求項4】
担体が活性炭である請求項3に記載の炭酸エステル化合物の製造方法。
【請求項5】
気相流通条件下で反応させる請求項1〜4のいずれかに記載の炭酸エステル化合物の製造方法。
【図1】



【図2】
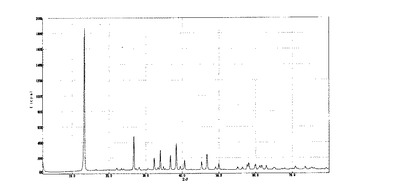
【図3】
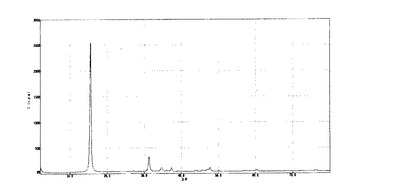
【図2】
【図3】
【公開番号】特開2012−236806(P2012−236806A)
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2011−108149(P2011−108149)
【出願日】平成23年5月13日(2011.5.13)
【出願人】(000004466)三菱瓦斯化学株式会社 (1,281)
【Fターム(参考)】
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成23年5月13日(2011.5.13)
【出願人】(000004466)三菱瓦斯化学株式会社 (1,281)
【Fターム(参考)】
[ Back to top ]