
無機粉体混合物及びフィラー含有組成物
【課題】取り扱い性に優れ、高い流動性をもつ無機粉体混合物を提供すること。
【解決手段】シリカ粒子をシランカップリング剤とオルガノシラザンとで表面処理して体積平均粒径が5nm〜100nmであるシリカ粒子材料を得る。シランカップリング剤とオルガノシラザンとのモル比を特定の範囲にすることで、シリカ粒子材料の表面に存在するX1(フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基)とR(炭素数1〜3のアルキル基)との存在数比を特定の範囲にし、樹脂に対する親和性と凝集抑制とを両立させる。このシリカ粒子材料をそれよりも粒径が大きい無機粉末と混合した無機粉体混合物は高い流動性をもつ。
【解決手段】シリカ粒子をシランカップリング剤とオルガノシラザンとで表面処理して体積平均粒径が5nm〜100nmであるシリカ粒子材料を得る。シランカップリング剤とオルガノシラザンとのモル比を特定の範囲にすることで、シリカ粒子材料の表面に存在するX1(フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基)とR(炭素数1〜3のアルキル基)との存在数比を特定の範囲にし、樹脂に対する親和性と凝集抑制とを両立させる。このシリカ粒子材料をそれよりも粒径が大きい無機粉末と混合した無機粉体混合物は高い流動性をもつ。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、流動性に優れた無機粉体混合物及びその無機粉体混合物をフィラーとして含むフィラー含有樹脂組成物に関する。
【背景技術】
【0002】
シリカ粒子の一種であるコロイダルシリカは、水ガラスを原料として製造する方法、金属ケイ素粉末を原料として製造する方法、気相合成法等により製造される。水ガラスを原料として製造する方法は、水ガラスを中和したり、イオン交換することで水ガラスを沈殿させることによって微小な粒子を生成する(例えば特許文献1参照)。
【0003】
このようなシリカ粒子の粒径は、例えばコロイダルシリカでは数nm〜数百nm程度と非常に小さい。粒径の小さなシリカ粒子は種々の用途に供される。その一方で粒径の小さなシリカ粒子の比表面積は非常に大きい。このため粒径の小さなシリカ粒子は凝集し易い特性を持つ。一旦凝集したシリカ粒子は再分散し難い。このため、粒径の小さなシリカ粒子は取り扱い性に劣る問題がある。
【0004】
すなわち、シリカ粒子は、液状媒体中における分散性には比較的優れても、乾粉状態になると(すなわち、シリカ粒子が単体で存在する場合には)凝集する。凝集したシリカ粒子は、例えば、高性能の分散機等を用いたとしても解砕し難い。
【0005】
従って、シリカ粒子を樹脂用のフィラーとして用いる場合、シリカ粒子は乾燥した状態で凝集するために、そのまま樹脂中に混合することは困難である。そのため、シリカ粒子は液状媒体中に分散された状態で液状媒体と共に樹脂材料中に配合することになる。この場合、液状媒体と樹脂材料との相互作用から適用できる組み合わせに制限が生じたり、本来混合が望まれていない液状媒体が樹脂材料中に導入される問題がある。このため、乾燥状態でも取り扱い性に優れるシリカ粒子が望まれている事情がある。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2006−45039号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
ところで、コロイダルシリカのような粒径が小さな材料をそれよりも大きな粒径をもつ粒子に混合すると、コロイダルシリカがころのような作用を発揮して混合物全体としても高い流動性を示すことが知られている。
【0008】
しかしながら、コロイダルシリカは前述したように凝集しやすいため他の粒子と混合することは困難であった。従って、他の粒子に混合することで高い流動性が期待できるとしても、その実現は困難であった。
【0009】
本発明は上記事情に鑑みてなされたものであり、取り扱い性に優れ、高い流動性をもつ無機粉体混合物、及び、その無機粉体混合物をフィラーとして採用したフィラー含有組成物を提供することを解決すべき課題とする。
【課題を解決するための手段】
【0010】
(a)上記課題を解決する本発明の無機粉体混合物は、式(1):−OSiX1X2X3で表される官能基及び式(2):−OSiY1Y2Y3で表される官能基と、両官能基が表面に結合するシリカ粒子とからなり、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%である。なお、上記式(1)、(2)中;X1はフェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基であり;X2、X3は−OSiR3及び−OSiY4Y5Y6よりそれぞれ独立して選択され;Y1はRであり;Y2、Y3はR及び−OSiY4Y5Y6よりそれぞれ独立して選択される。Y4はRであり;Y5及びY6は、R及び−OSiR3からそれぞれ独立して選択され;Rは炭素数1〜3のアルキル基から独立して選択される。X2、X3、Y2、Y3、Y5、及びY6の何れかは、近接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。
【0011】
本発明の無機粉体混合物は、下記の(i)〜(iii)の何れかを備えることが好ましく、(i)〜(iii)の複数を備えることがより好ましい。(i)前記式(1)で表される官能基と前記式(2)で表される官能基との存在数比が1:12〜1:60である。(ii)前記X1は前記シリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個である。(iii)前記Rは前記シリカ粒子材料の単位表面積(nm2)あたり1〜10個である。
(b)上記課題を解決する無機粉体混合物は、水を含む液状媒体中でシランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する表面処理工程をもつ表面処理方法により処理され、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%であり、前記シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基と、を持ち、前記シランカップリング剤と前記オルガノシラザンとのモル比は、前記シランカップリング剤:前記オルガノシラザン=1:2〜1:10である。
【0012】
本発明の無機粉体混合物は、下記の(iv)〜(viii)の何れかを備えることが好ましく、(iv)〜(viii)の複数を備えることがより好ましい。(iv)前記表面処理工程は、前記シリカ粒子を前記シランカップリング剤で処理する第1の処理工程と、前記シリカ粒子を前記オルガノシラザンで処理する第2の処理工程と、を持ち、前記第2の処理工程は、前記第1の処理工程後に行う。(v)前記第2の処理工程において、3つのアルコキシ基と炭素数1〜3のアルキル基とを持つ第2のシランカップリング剤で前記オルガノシラザンの一部を置き換え、前記第2の処理工程後に、さらに前記シリカ粒子を前記オルガノシラザンで処理する第3の処理工程を持つ。(vi)前記表面処理工程後に、前記シリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る固形化工程を備える。(vii)前記シランカップリング剤は、フェニルトリメトキシシラン、ビニルトリメトキシシラン、エポキシトリメトキシシラン、メタクリルトリメトキシシラン、アミノトリメトキシシラン、ウレイドトリメトキシシラン、メルカプトトリメトキシシラン、イソシアネート、又はアクリルトリメトキシシランから選ばれる少なくとも一種である。(viii)前記オルガノシラザンは、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザンから選ばれる少なくとも一種である。
(c)上述した(a)及び(b)に係る無機粉体混合物は前記無機粉体の体積平均粒径が0.5μm〜50μmであることが好ましい。
(d)上記課題を解決する本発明のフィラー含有組成物は、上述した(a)〜(c)の何れかに開示の本発明の無機粉体混合物からなるフィラーと、樹脂材料及び/又は樹脂材料前駆体と、を含むことを特徴とする。
【発明の効果】
【0013】
(a)本発明の無機粉体混合物がもつシリカ粒子材料は、X1(フェニル基、ビニル基、エポキシ基、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基)と、R(炭素数1〜3のアルキル基)とを表面に持つ。Rすなわち炭素数1〜3のアルキル基は疎水性が高いために、互いに反発し合う。従って、本発明の無機粉体混合物がもつシリカ粒子材料は炭素数1〜3のアルキル基同士の反発力により凝集し難く乾燥状態で流動性が高い粒子が得られるため、無機粉体に混合したときに流動性の高い無機粉体混合物が得られる。
【0014】
また、シリカ粒子材料が表面にRのみならずX1をも持つ場合には、極性の大きな官能基を持つ材料(例えば樹脂材料等)に対する親和性が向上する。このようなシリカ粒子材料は、樹脂材料中に均一分散し易くなる。シリカ粒子材料はX1を表面に持つため、樹脂材料との親和性に優れ、樹脂材料中に均一分散できる。特に、無機粉末の間に入ってころとしての作用を発揮できるために無機粉体混合物全体としての流動性も高い。換言すると、本発明の無機粉体混合物は、樹脂に対する親和性に優れるとともに、凝集抑制効果にも優れている。このため本発明の無機粉体混合物は、樹脂材料用のフィラーとして好適に使用できる。さらに、本発明の無機粉体混合物は凝集し難いため、液状媒体に分散しなくても良い。このことによっても、本発明の無機粉体混合物は、樹脂材料用のフィラーとして好適に使用できる。
【0015】
また、シリカ粒子材料の表面にRが多く存在するほど凝集抑制効果が向上するが、その一方で、樹脂に対する親和性が低下する。シリカ粒子材料の表面にX1が多く存在するほど樹脂に対する親和性が向上するが、その一方で、Rの数が少なくなり凝集抑制効果が低減する。従って、RとX1との存在数比には、好ましい範囲が存在する。式(1)で表される官能基と式(2)で表される官能基との存在数比が1:12〜1:60の範囲内であれば、樹脂に対する優れた親和性と優れた凝集抑制効果とを両立することができる。また、X1がシリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個であれば、樹脂に対する優れた親和性と優れた凝集抑制効果とを両立することができる。
(b)本発明の無機粉体混合物は上述の表面処理方法にてシリカ粒子を表面処理することにより流動性が高いシリカ粒子材料を乾燥状態で得ることができる。この表面処理方法にて処理して得られたシリカ粒子材料の表面には、上式(1)で表されるシランカップリング剤由来の官能基と、上式(2)で表されるオルガノシラザン由来の官能基とをもつことが予測される。そして、シランカップリング剤とオルガノシラザンとのモル比が1:2〜1:10の範囲内であれば、上式(1)で表される官能基と上式(2)で表される官能基とを、シリカ粒子材料の表面にバランス良く存在させ得るものと思われる。このため、樹脂に対する親和性と凝集抑制効果とが両立したシリカ粒子材料を得ることができる。
(c)本発明のフィラー含有組成物は、無機粉体混合物を乾燥状態で樹脂組成物に混合するため樹脂組成物中に分散媒が混入されない。特に熱可塑性樹脂にフィラーを混合する場合には一度混合された分散媒は除去しがたく最初から混合されない本発明のフィラー含有組成物は有利である。
【図面の簡単な説明】
【0016】
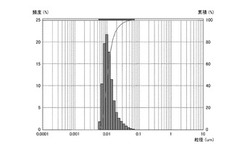
【図1】実施例1のシリカ粒子材料の粒度分布を表すグラフである。
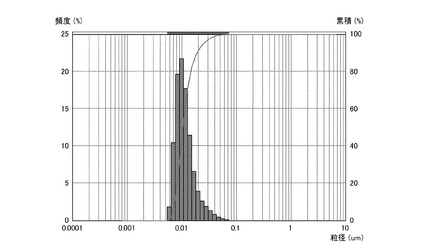
【図2】実施例2のシリカ粒子材料の粒度分布を表すグラフである。
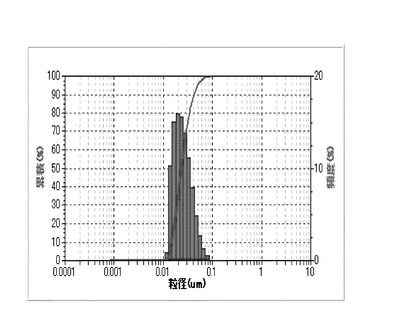
【図3】実施例3のシリカ粒子材料の粒度分布を表すグラフである。
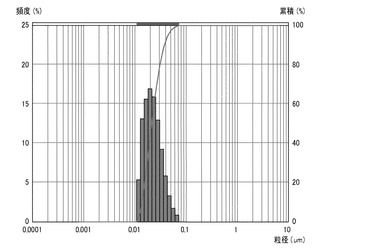
【図4】実施例4のシリカ粒子材料の粒度分布を表すグラフである。
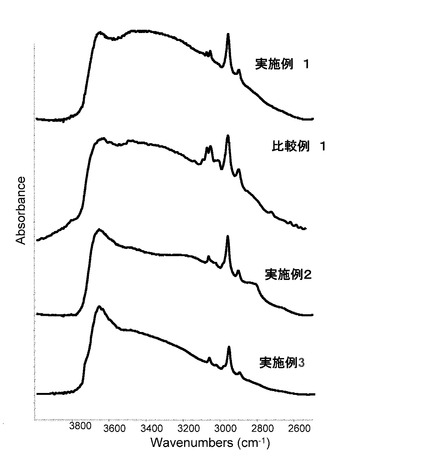
【図5】実施例1〜3及び比較例1のシリカ粒子材料の赤外吸収スペクトルを表すグラフである。
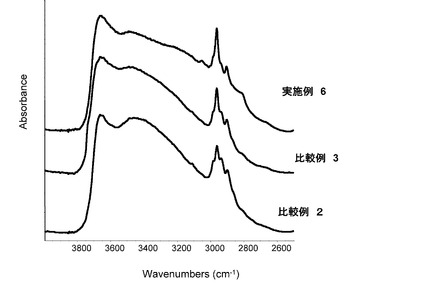
【図6】実施例4及び比較例2、3のシリカ粒子材料の赤外吸収スペクトルを表すグラフである。
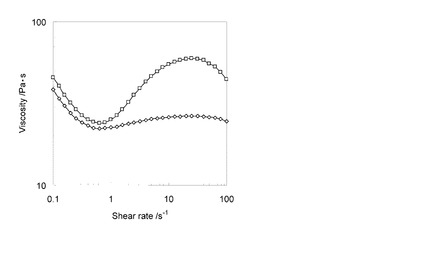
【図7】実施例6のフィラー含有樹脂組成物及び比較例4のフィラー含有樹脂組成物の粘度を表すグラフである。
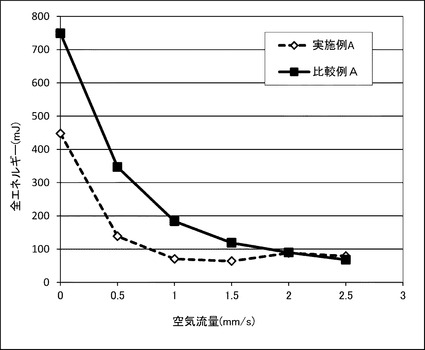
【図8】実施例A及び比較例Aの無機粉体混合物の粉体流動特性を表すグラフである。
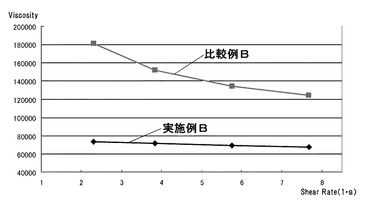
【図9】実施例B及び比較例Bのフィラー含有組成物について25℃におけるシェアレート−粘度の関係を示すグラフである。
【発明を実施するための形態】
【0017】
本発明の無機粉体混合物及びフィラー含有組成物について実施形態に基づき以下詳細に説明を行う。本実施形態の無機粉体混合物は乾燥した状態で熱硬化性樹脂(硬化前のもの)、光硬化性樹脂(硬化前のもの)、熱可塑性樹脂(加熱溶融した状態のもの)などに簡単に分散可能である。この無機粉体混合物は流動性に優れているため、これらの樹脂に混合しても高い流動性をもつことができる。特に乾燥状態で高い流動性が得られるため、熱可塑性樹脂への混合も容易に行うことができる。また、熱硬化性樹脂や光硬化性樹脂においても乾燥状態で混合できるので添加が望まれない分散媒が混入することなくそれらの樹脂に添加することができる。
(無機粉体混合物)
本実施形態の無機粉体混合物は無機粉末とシリカ粒子材料とを乾燥状態で含む混合物である。両者の混合方法は特に限定されず公知の方法が採用できる。本実施形態の無機粉体混合物の構成要素であるシリカ粒子材料は粒径が小さいにもかかわらず流動性・分散性に優れており、他の粉体と容易に混合可能である。
【0018】
無機粉末とシリカ粒子材料との混合割合としては無機粉体混合物の質量を基準としてシリカ粒子材料の量が0.05%〜10%であること以外は特に限定されない。シリカ粒子材料の量が1%以上であることが望ましく、5%以下であることが望ましい。
【0019】
本明細書中における「乾燥状態」との語は粒子の表面に吸着していない溶媒が存在していないこととの意味である。具体的に乾燥状態にあるか否かの判断は、無機粉体混合物を取り扱うときに、スラリー状になっていない状態で流動性をもっているかどうかで簡易的に判定できる。スラリーになっていない状態で流動性をもっていれば乾燥状態にあるものと判断できる。
・無機粉末
無機粉末は無機物から形成され、適用される目的に応じてその組成が決定される。本実施形態の無機粉体混合物の高い流動性は、より粒径が小さいシリカ粒子材料がころとして作用することに起因するため、無機粉末の組成には殆ど影響されないものと考えられる。例えばシリカ、アルミナ、ジルコニア、チタニア、酸化亜鉛、ベーマイト、窒化ホウ素、それらの混合物、複合酸化物が例示できる。
【0020】
無機粉末は後述するシリカ粒子材料よりも体積平均粒径が大きい材料である。例えば、0.5μm以上にすることが望ましく、1μm以上にすることが更に望ましい。また、50μm以下にすることが望ましく、40μm以下にすることが更に望ましい。
【0021】
無機粉末は真球度が高い方が流動性が向上するため望ましい。真球度としては0.8以上にすることが望ましく、0.9以上にすることが更に望ましい。真球度の測定は、SEMで写真を撮り、その観察される粒子の面積と周囲長から、(真球度)={4π×(面積)÷(周囲長)2}で算出される値として算出する。1に近づくほど真球に近い。具体的には画像処理装置(シスメックス株式会社:FPIA−3000)を用い、無作為に抽出した100個の粒子について測定した平均値を採用する。
・シリカ粒子材料(その1)
シリカ粒子材料は体積平均粒径が5nm〜100nmのシリカからなる粒子である。特に望ましくは10nm以上である。また、50nm以下が望ましい。そしてその表面には後述する官能基をもつか、後述する表面処理が行われているかの何れかである。
【0022】
シリカ粒子材料は、式(1):−OSiX1X2X3で表される官能基と、式(2):−OSiY1Y2Y3で表される官能基とが表面に結合したシリカ粒子材料である。以下、式(1)で表される官能基を第1の官能基と呼び、式(2)で表される官能基を第2の官能基と呼ぶ。
【0023】
第1の官能基におけるX1は、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基である。X2、X3は、それぞれ、−OSiR3又は−OSiY4Y5Y6である。Y4はRである。Y5、Y6は、それぞれ、R又は−OSiR3である。
【0024】
第2の官能基におけるY1はRである。Y2、Y3は、それぞれ、−OSiR3又は−OSiY4Y5Y6である。
【0025】
第1の官能基及び第2の官能基に含まれる−OSiR3が多い程、シリカ粒子材料の表面にRを多く持つ。第1の官能基及び第2の官能基に含まれるR(炭素数1〜3のアルキル基)が多い程、シリカ粒子材料は凝集し難い。
【0026】
第1の官能基に関していえば、X2、X3がそれぞれ−OSiR3である場合に、Rの数が最小となる。また、X2及びX3がそれぞれ−OSiY4Y5Y6であり、かつ、Y5、Y6がそれぞれ−OSiR3である場合に、Rの数が最大となる。
【0027】
第2の官能基に関していえば、Y2、Y3がそれぞれ−OSiR3である場合に、Rの数が最小となる。また、Y2及びY3がそれぞれ−OSiY4Y5Y6であり、かつ、Y5、Y6がぞれぞれ−OSiR3である場合に、Rの数が最大となる。
【0028】
第1の官能基に含まれるX1の数、第1の官能基に含まれるRの数、第2の官能基に含まれるRの数は、RとX1との存在数比や、シリカ粒子材料の粒径や用途に応じて適宜設定すれば良い。
【0029】
なお、X2、X3、Y2、Y3、Y5、及びY6の何れかは、隣接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。例えば、第1の官能基のX2、X3、Y5、及びY6の何れかが、この第1の官能基に隣接する第1の官能基のX2、X3、Y5、及びY6の何れかと−O−にて結合していても良い。同様に、第2の官能基のY2、Y3、Y5、及びY6の何れかが、この第2の官能基に隣接する第2の官能基のY2、Y3、Y5、及びY6の何れかと−O−にて結合していても良い。さらには、第1の官能基のX2、X3、Y5、及びY6の何れかが、この第1の官能基に隣接する第2の官能基のY2、Y3、Y5、及びY6の何れかと−O−にて結合していても良い。
【0030】
第1の官能基と第2の官能基との存在数比が1:12〜1:60であれば、シリカ粒子材料の表面にX1とRとがバランス良く存在する。このため、第1の官能基と第2の官能基との存在数比が1:12〜1:60であるシリカ粒子材料は、樹脂に対する親和性及び凝集抑制効果に特に優れる。また、X1がシリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個であれば、シリカ粒子材料の表面に充分な数の第1の官能基が結合し、第1の官能基及び第2の官能基に由来するRもまた充分な数存在する。従ってこの場合にも、樹脂に対する親和性及びシリカ粒子材料の凝集抑制効果が充分に発揮される。
【0031】
何れの場合にも、シリカ粒子材料の単位表面積(nm2)あたりのRは、1個〜10個であるのが好ましい。この場合には、シリカ粒子材料の表面に存在するX1の数とRの数とのバランスが良くなり、樹脂に対する親和性及びシリカ粒子材料の凝集抑制効果との両方がバランス良く発揮される。
【0032】
シリカ粒子の表面に存在していた水酸基の全部が第1の官能基又は第2の官能基で置換されていることが好ましい。第1の官能基と第2の官能基との和が、シリカ粒子材料の単位表面積(nm2)あたり2.0個以上であれば、シリカ粒子材料において、シリカ粒子の表面に存在していた水酸基のほぼ全部が第1の官能基又は第2の官能基で置換されているといえる。
【0033】
シリカ粒子材料は、表面にRを持つ。これは、赤外線吸収スペクトルによって確認できる。詳しくは、シリカ粒子材料の赤外線吸収スペクトルを固体拡散反射法で測定すると、2962±2cm−1にC−H伸縮振動の極大吸収がある。このため、本実施形態の無機粉体混合物がもつシリカ粒子材料であるか否かは、赤外線吸収スペクトルによって確認できる。
【0034】
また、上述したように本発明の無機粉体混合物がもつシリカ粒子材料は凝集し難い。従って、シリカ粒子材料としては粒径の小さなものに採用できる。例えば、シリカ粒子材料は、平均粒径3nm〜5000nm程度にできる。平均粒径3〜200nmのシリカ粒子材料に適用するのが好ましい。
【0035】
なお、シリカ粒子材料は、例え僅かに凝集した場合にも、超音波処理することによって再度分散可能である。詳しくは、シリカ粒子材料をメチルエチルケトンに分散させたものに、発振周波数39kHz、出力500Wの超音波を照射することで、シリカ粒子材料を実質的に一次粒子にまで分散できる。このときの超音波照射時間は10分間以下で良い。シリカ粒子材料が一次粒子にまで分散したか否かは、粒度分布を測定することで確認できる。詳しくは、このシリカ粒子材料のメチルエチルケトン分散材料をマイクロトラック装置等の粒度分布測定装置で測定し、シリカ粒子材料の粒度分布があれば、シリカ粒子材料が一次粒子にまで分散したといえる。
【0036】
このシリカ粒子材料は、凝集し難いため、水やアルコール等の液状媒体に分散されていないシリカ粒子材料として提供できる。また、シリカ粒子材料は凝集し難いために、水で容易に洗浄できる。
・シリカ粒子材料(その2)
その1に示すシリカ粒子材料に代えて、以下に示す表面処理を行ったシリカ粒子材料を採用することもできる。なお、以下の方法によりシリカ粒子材料(その1)を得ることもできるため、その1とその2とは排他的なものではない。
【0037】
シリカ粒子材料の表面処理方法は、水を含む液状媒体中で、シランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する工程(表面処理工程)を持つ。シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基(すなわち上記のX1)とを持つ。
【0038】
シランカップリング剤で表面処理することで、シリカ粒子の表面に存在する水酸基がシランカップリング剤に由来する官能基で置換される。シランカップリング剤に由来する官能基は式(3);−OSiX1X4X5で表される。式(3)で表される官能基を第3の官能基と呼ぶ。第3の官能基におけるX1は式(1)で表される官能基におけるX1と同じである。X4、X5は、それぞれ、アルキコキシ基である。オルガノシラザンで表面処理することで、第3の官能基のX4、X5がオルガノシラザンに由来する−OSiY1Y2Y3(式(2)で表される官能基、第2の官能基)で置換される。シリカ粒子の表面に存在する水酸基の全てが第3の官能基で置換されていない場合には、シリカ粒子の表面に残存する水酸基が第2の官能基で置換される。このため、表面処理されたシリカ粒子材料の表面には、式(1):−OSiX1X2X3で表される官能基(すなわち第1の官能基)と、式(2):−OSiY1Y2Y3で表される官能基と(すなわち第2の官能基)が結合する。なお、シランカップリング剤とオルガノシラザンとのモル比は、シランカップリング剤:オルガノシラザン=1:2〜1:10であるため、得られたシリカ粒子材料における第1の官能基と第2の官能基との存在数比は理論上1:12〜1:60となる。
【0039】
表面処理工程においては、シリカ粒子をシランカップリング剤及びオルガノシラザンで同時に表面処理しても良い。又は、先ずシリカ粒子をシランカップリング剤で表面処理し、次いでオルガノシラザンで表面処理しても良い。又は、先ずシリカ粒子をオルガノシラザンで表面処理し、次いでシランカップリング剤で表面処理し、さらにその後にオルガノシラザンで表面処理しても良い。何れの場合にも、シリカ粒子の表面に存在する水酸基全てが第2の官能基で置換されないように、オルガノシラザンの量を調整すれば良い。なお、シリカ粒子の表面に存在する水酸基は、全てが第3の官能基で置換されても良いし、一部のみが第3の官能基で置換され、他の部分が第2の官能基で置換されても良い。第3の官能基に含まれるX4、X5は、全て第2の官能基で置換されるのが良い。
【0040】
なお、オルガノシラザンの一部を、第2のシランカップリング剤で置き換えても良い。第2のシランカップリング剤としては、3つのアルコキシ基と、1つのアルキル基とを持つものを用いることができる。この場合には、第3の官能基に含まれるX4、X5が、第2のシランカップリング剤に由来する第4の官能基で置換される。第4の官能基は式(4);−OSiY1X6X7で表される。Y1は第2の官能基におけるY1と同じRであり、X6、X7はそれぞれアルコキシ基又は水酸基である。第4の官能基に含まれるX6、X7は、オルガノシラザンに由来する第2の官能基で置換されるか、又は、別の第4の官能基で置換される。この場合には、シリカ粒子材料の表面に存在するRの量をさらに多くする事ができる。なお、オルガノシラザンの一部を、第2のシランカップリング剤に置き換える場合、第2のシランカップリング剤で表面処理した後に、再度オルガノシラザンで表面処理する必要がある。第4の官能基に含まれるX6、X7を、最終的にはオルガノシラザンに由来する第2の官能基で置換するためである。
【0041】
オルガノシラザンの一部を第2のシランカップリング剤で置き換える場合、上述した第1の官能基に含まれるX4、X5は、オルガノシラザンに由来する第2の官能基で置換されるか、第2のシランカップリング剤に由来する第4の官能基で置換される。X4、X5が第4の官能基で置換された場合、第4の官能基に含まれるX6、X7は、第2の官能基で置換されるか、別の第4の官能基によって置換される。第4の官能基に含まれるX6、X7が別の第4の官能基によって置換された場合、第4の官能基に含まれるX6、X7は、第2の官能基で置換される。このため第2のシランカップリング剤は、第1のカップリング剤及びオルガノシラザンのみで表面処理する場合(オルガノシラザンを第2のシランカップリング剤で置き換えなかった場合)に設定されるオルガノシラザンの量(a)molに対して、最大限5a/3mol置き換えることができる。この場合に必要になるオルガノシラザンの量は、8a/3molである。
【0042】
シランカップリング剤及び第2のシランカップリング剤のアルコキシ基は特に限定しないが、比較的炭素数の小さなものが好ましく、炭素数1〜12であることが好ましい。アルコキシ基の加水分解性を考慮すると、アルコキシ基はメトキシ基、エトキシ基、プロポキシ基、ブトキシ基の何れかであることがより好ましい。
【0043】
シランカップリング剤として、具体的には、フェニルトリメトキシシラン、ビニルトリメトキシシラン、ビニルトリエトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、3−グリシドキシプロピルトリメトキシシラン、3−グリシドキシプロピルトリエトキシシラン、p−スチリルトリメトキシシラン、3−メタクリロキシプロピルトリメトキシシラン、3−メタクリロキシプロピルトリエトキシシラン、3−アクリロキシプロピルトリメトキシシラン、N−フェニル−3−アミノプロピルトリメトキシシランが挙げられる。
【0044】
オルガノシラザンとしては、シリカ粒子の表面に存在する水酸基及びシランカップリング剤に由来するアルコキシ基を、上述した第2の官能基で置換できるものであれば良いが、分子量の小さなものを用いるのが好ましい。具体的には、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザン等が挙げられる。
【0045】
第2のシランカップリング剤としては、メチルトリメトキシシラン、メチルトリエトキシシラン、n−プロピルトリメトキシシラン、n−プロピルトリエトキシシラン、ヘキシルトリメトキシシラン、ヘキシルトリエトキシシラン等が挙げられる。
【0046】
なお、表面処理工程において、シランカップリング剤の重合や第2のシランカップリング剤の重合を抑制するため、重合禁止剤を加えても良い。重合禁止剤としては、3,5−ジブチル−4−ヒドロキシトルエン(BHT)、p−メトキシフェノール(メトキノン)等の一般的なものを用いることができる。
【0047】
シリカ粒子材料を得るための表面処理について説明する。本表面処理方法は、表面処理工程後に固形化工程を備えても良い。固形化工程は、表面処理後のシリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る工程である。上述したように、一般的なシリカ粒子材料は非常に凝集し易いため、一旦固形化したシリカ粒子材料を再度分散するのは非常に困難である。しかし、シリカ粒子材料は凝集し難いため、固形化しても凝集し難く、また、例え凝集しても再分散し易い。なお、上述したように、シリカ粒子材料を水で洗浄することで、電子部品等の用途に用いられるシリカ粒子材料を容易に製造できる。なお、洗浄工程においては、シリカ粒子材料の抽出水(詳しくは、シリカ粒子材料を121℃で24時間浸漬した水)の電気伝導度が50μS/cm以下となるまで、洗浄を繰り返すのが好ましい。
【0048】
固形化工程で用いる鉱酸としては塩酸、硝酸、硫酸、リン酸などが例示でき、特に塩酸が望ましい。鉱酸はそのまま用いても良いが、鉱酸水溶液として用いるのが好ましい。鉱酸水溶液における鉱酸の濃度は0.1質量%以上が望ましく、0.5質量%以上が更に望ましい。鉱酸水溶液の量は、洗浄対象であるシリカ粒子材料の質量を基準として6〜12倍程度にすることができる。
【0049】
鉱酸水溶液による洗浄は複数回数行うことも可能である。鉱酸水溶液による洗浄はシリカ粒子材料を鉱酸水溶液に浸漬後、撹拌することが望ましい。また、浸漬した状態で1時間から24時間、更には72時間程度放置することができる。放置する際には撹拌を継続することもできるし、撹拌しないこともできる。鉱酸含有液中にて洗浄する際には常温以上に加熱することもできる。
【0050】
その後、洗浄して懸濁させたシリカ粒子材料をろ取した後、水にて洗浄する。使用する水はアルカリ金属などのイオンを含まない(例えば質量基準で1ppm以下)ことが望ましい。例えば、イオン交換水、蒸留水、純水などである。水による洗浄は鉱酸水溶液による洗浄と同じく、シリカ粒子材料を分散、懸濁させた後、ろ過することもできるし、ろ取したシリカ粒子材料に対して水を継続的に通過させることによっても可能である。水による洗浄の終了時期は、上述した抽出水の電気伝導度で判断しても良いし、シリカ粒子材料を洗浄した後の排水中のアルカリ金属濃度が1ppm以下になった時点としても良いし、抽出水のアルカリ金属濃度が5ppm以下になった時点としても良い。なお、水で洗浄する際には常温以上に加熱することもできる。
【0051】
シリカ粒子材料の乾燥は、常法により行うことができる。例えば、加熱や、減圧(真空)下に放置する等である。
・フィラー含有組成物
本実施形態のフィラー含有組成物は、上述した無機粉体混合物と、樹脂材料及び/又は樹脂材料前駆体とを有する。無機粉体混合物は乾燥状態のまま樹脂材料及び/又は樹脂材料前駆体に混合される。フィラーの含有量は特に限定されず必要な量が添加される。上述した無機粉体混合物は流動性が高いため、大量に樹脂材料(又は樹脂材料前駆体)中に混合することができる。例えば、フィラー含有組成物全体の質量を基準として80%以上含有させることができる。また、90%以上含有させることもできる。このように大量にフィラーを含有させても流動性を示す組成物が得られる。
【0052】
樹脂材料は高分子材料であって、樹脂材料前駆体は高分子又は低分子の材料であり、更に反応が進行することにより分子量が増大したり、架橋が進行したりして樹脂材料を形成できる材料である。樹脂材料、並びに、樹脂材料前駆体により形成される樹脂材料は熱可塑性樹脂、熱硬化性樹脂、光硬化性樹脂などの一般的な樹脂材料が例示できる。樹脂材料前駆体は、エポキシ基、オキセタン基、水酸基、ブロックされたイソシアネート基、アミノ基、ハーフエステル基、アミック基、カルボキシ基、及び炭素-炭素二重結合基を化学構造中に有することが望ましい。これらの官能基は好適な反応条件を設定することで互いに結合可能な官能基(重合性官能基)であり、混合材料の分子量を向上できる。好適な反応条件としては単純に加熱や光照射を行ったり、熱や光照射によりラジカルやイオン(アニオン、カチオン)などの反応性種を生成したり、それらの官能基間を結合する反応開始剤(重合開始剤)を添加して加熱や光照射を行うことなどである。重合反応に際して必要な化合物を硬化剤として添加したり、その反応に対する触媒を添加することもできる。
【0053】
樹脂材料前駆体としては重合により高分子材料を形成する単量体や、上述したような重合性官能基により修飾した高分子材料が好ましいものとして挙げられる。例えば、硬化前の、エポキシ樹脂、アクリル樹脂、ウレタン樹脂などのプレポリマーが好適である。
【0054】
樹脂材料前駆体に対して無機粉体混合物を混合する場合にはそのまま混合することもできる。樹脂材料として熱可塑性樹脂を採用する場合に、その樹脂材料に混合するときには熱可塑性樹脂を加熱により溶融させた後に混合することができる。
【実施例】
【0055】
以下、本発明の無機粉体混合物について実施例に基づき詳細に説明する。
(本発明の無機粉体混合物が含有するシリカ粒子材料について)
(実施例1)
(材料)
シリカ粒子として、コロイダルシリカの一種であるスノーテックスOS(日産化学工業株式会社製、平均粒径10nm、水中に分散されており固形分濃度20%)を準備した。
【0056】
アルコールとして、イソプロパノールを準備した。
【0057】
シランカップリング剤として、フェニルトリメトキシシラン(信越化学工業株式会社製、KBM−103)を準備した。
【0058】
オルガノシラザンとして、ヘキサメチルジシラザン(HMDS、信越化学工業株式会社製、HDMS−1)を準備した。
【0059】
(表面処理工程)
(1)準備工程
シリカ粒子が20質量%の濃度で水に分散したスラリー100質量部にイソプロパノール60質量部を加え、室温(約25℃)で混合することで、シリカ粒子が液状媒体に分散されてなる分散液を得た。
【0060】
(2)第1工程
この分散液にフェニルトリメトキシシラン1.8質量部を加え、40℃で72時間混合した。この工程により、シリカ粒子の表面に存在する水酸基をシランカップリング剤で表面処理した。なお、このときフェニルトリメトキシシランは、必要な量の水酸基(一部)が表面処理されず残存するように計算して加えた。
【0061】
(3)第2工程
次いで、この混合物に、ヘキサメチルジシラザン3.7質量部を加え、40℃で72時間放置した。この工程によって、シリカ粒子が表面処理され、シリカ粒子材料が得られた。表面処理の進行に伴い、疎水性になったシリカ粒子が水及びイソプロパノールの中で安定に存在できなくなり、凝集・沈殿した。なお、フェニルトリメトキシシランとヘキサメチルジシラザンとのモル比は2:5であった。
【0062】
(固形化工程)
表面処理工程で得られた混合物全量に35%塩酸水溶液を5質量部を加え、シリカ粒子材料を沈殿させた。沈殿物をろ紙(アドバンテック社製 5A)で濾過した。濾過残渣(固形分)を純水で洗浄した後に100℃で真空乾燥して、シリカ粒子材料の固形物を得た。
【0063】
(実施例2)
実施例2のシリカ粒子の表面処理方法は、フェニルトリメトキシシランにかえてビニルトリメトキシシランを用い、ビニルトリメトキシシランとヘキサメチルジシラザンとのモル比が2:5であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、ビニルトリメトキシシラン1.36質量部を加え、第2工程においてはヘキサメチルジシラザン3.7質量部を加えた。
【0064】
なおビニルトリメトキシシランとしては、信越化学工業株式会社製 KBM−1003を用いた。
【0065】
(実施例3)
実施例3のシリカ粒子の表面処理方法は、フェニルトリメトキシシランにかえてビニルトリメトキシシランを用い、ビニルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:5であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、ビニルトリメトキシシラン1.36質量部を加え、第2工程においてはヘキサメチルジシラザン7.41質量部を加えた。
【0066】
(実施例4)
実施例4のシリカ粒子の表面処理方法においては、シリカ粒子として、コロイダルシリカの一種であるスノーテックスOL(日産化学工業株式会社製、平均粒径50nm、水中に分散されており固形分濃度20%)を用いた。また、第1工程においてシランカップリング剤として3−メタクリロキシプロピルトリメトキシシラン(信越化学工業株式会社製、KBM−503)0.48質量部を加えた。さらに、このシランカップリング剤に加えて重合禁止剤(3,5−ジブチル−4−ヒドロキシトルエン(BHT)、関東化学株式会社製)を0.01質量部加えた。また、第2工程において、ヘキサメチルジシラザン0.78質量部を加えた。さらに、固形化工程においては、表面処理工程で得られた混合物全量に35%塩酸水溶液2.6質量部を加えてシリカ粒子材料を沈殿させた。これ以外は、実施例4のシリカ粒子の表面処理方法は、実施例1のシリカ粒子の表面処理方法と同じであった。なお、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比は2:5であった。
【0067】
(比較例1)
比較例1のシリカ粒子の表面処理方法は、フェニルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:1であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、フェニルトリメトキシシラン4.5質量部を加え、第2工程においてはヘキサメチルジシラザン3.7質量部を加えた。
【0068】
(比較例2)
比較例2のシリカ粒子の表面処理方法は、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比が2:1であったこと以外は、実施例4のシリカ粒子の表面処理方法と同じである。第1工程においては、3−メタクリロキシプロピルトリメトキシシラン0.48質量部を加え、第2工程においてはヘキサメチルジシラザン0.16質量部を加えた。
【0069】
(比較例3)
比較例3のシリカ粒子の表面処理方法は、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:1であったこと以外は、実施例4のシリカ粒子の表面処理方法と同じである。第1工程においては、3−メタクリロキシプロピルトリメトキシシラン0.48質量部を加え、第2工程においてはヘキサメチルジシラザン0.31質量部を加えた。
【0070】
(凝集性評価試験)
実施例1〜4及び比較例1〜3のシリカ粒子材料について、液状媒体中における凝集性を測定した。
【0071】
詳しくは、実施例1〜3及び比較例1については、シリカ粒子材料10gとメチルエチルケトン40gとの混合物を攪拌し、シリカ粒子材料の分散試料を得た。
【0072】
実施例4及び比較例2、3については、シリカ粒子材料10gとメチルエチルケトン10gとの混合物を攪拌し、シリカ粒子材料の分散試料を得た。得られた各分散試料に含まれるシリカ粒子材料の粒度分布を、粒祖分布測定装置(日機装株式会社製、マイクロトラック)により測定した。凝集性評価試験の結果を図1〜4に示す。なお、図1は実施例1のシリカ粒子材料の粒度分布を表す。図2は実施例2のシリカ粒子材料の粒度分布を表す。図3は実施例3のシリカ粒子材料の粒度分布を表す。図4は実施例4のシリカ粒子材料の粒度分布を表す。
【0073】
図1〜4に示すように、実施例1〜4のシリカ粒子材料は、凝集のない一次粒子の状態で分散している。これは、図1〜4のグラフにおいて、各シリカ粒子材料の粒度分布(ピーク)が、粒子径10nm程度の位置に一つのみ現れていることから裏付けられる。シリカ粒子材料が二次粒子であれば(すなわち、少しでも凝集があれば)、粒子径100nm以上の位置に少なくとも一つのピークが現れる。このため、実施例1〜4のシリカ粒子材料は、一旦固形化したにもかかわらず、その殆どが一次粒子であり、殆ど凝集していないことがわかる。これに対して、比較例1〜3のシリカ粒子材料は、攪拌するだけでは分散せず、攪拌後に発振周波数39kHz、出力500Wで1時間以上超音波照射しても、肉眼で凝集が確認でき、一次粒子にまで分散しなかった。この結果から、シランカップリング剤とオルガノシラザンとのモル比を1:2〜1:10の範囲にすることで、固形化しても凝集し難いシリカ粒子材料を製造できることがわかる。なお、実施例1のシリカ粒子材料の平均粒径は10nm、実施例2のシリカ粒子材料の平均粒径は10nm、実施例3のシリカ粒子材料の平均粒径は10nm、実施例4のシリカ粒子材料の平均粒径は50nmであった。この結果から、凝集抑制のためには、シランカップリング剤とオルガノシラザンとのモル比を1:5〜2:5の範囲にするのが好ましいことがわかる。
【0074】
(極大吸収測定試験)
実施例1〜4及び比較例1〜3のシリカ粒子材料を準備し、この試料の赤外線吸収スペクトルを、サーモニコレット社製 FT−IR Avatorを用いた粉体拡散反射法で測定した。このときの測定条件は、分解能4、スキャン回数64であった。極大吸収測定試験の結果を表すグラフを図5〜図6に示す。図5〜図6に示すように、実施例1〜4及び比較例1〜3のシリカ粒子材料の赤外吸収スペクトルは、何れも、2962cm−1にC-H伸縮振動の極大吸収(ピーク)を持つ。このため、実施例1〜4及び比較例1〜3のシリカ粒子材料は、アルキル基を持つこと(すなわち、アルキル基を持つオルガノシラザンで表面処理されていること)がわかる。なお、比較例1〜3のシリカ粒子材料のピーク高さは実施例1〜4のシリカ粒子材料のピーク高さに比べて低かった。この結果は、比較例1〜3のシリカ粒子材料においては、充分な量のアルキル基を持たないことを示唆している。詳しくは、実施例1〜4のシリカ粒子材料の赤外線吸収スペクトルにおいては、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが3倍以上であった。比較例1〜3のシリカ粒子材料の赤外線吸収スペクトルにおいては、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが2倍以下であった。上述したように、実施例1〜4のシリカ粒子材料は凝集し難く、比較例1〜3のシリカ粒子材料は凝集し易かった。これらの結果から、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが3倍以上であるシリカ粒子材料は凝集し難いといえる。
【0075】
(実施例5)
実1のシリカ材料100質量部とメチルイソブチルケトン100質量部とを混合し、シリカ粒子材料とメチルイソブチルケトンとの混合物を得た。この混合物4質量部にアクリル樹脂(根上工業株式会社製 アートレジン UK904)6質量部を加え混合した。この混合物100質量部に対して5質量部の重合開始剤(チバ・スペシャルティ・ケミカルズ株式会社製、ダロキュアTPO)を加え、さらに、固形分濃度が30質量%となるようにメチルイソブチルケトンを加えた。この混合物をさらに攪拌して、フィラー含有樹脂組成物(前駆体)を得た。この前駆体は、均一溶液状であった。実施例5のフィラー含有樹脂組成物を、基板に塗布し、実施例5のフィラー含有樹脂組成物(フィルム)を成膜した。
【0076】
(樹脂に対する親和性評価I)
実施例5のフィラー含有樹脂組成物(フィルム)を目視にて観察したところ、このフィルムは硬く透明であった。実施例5のフィルムが硬いのは、シリカ粒子材料が樹脂材料中に略均一に分散しているためだと考えられる。また、実施例5のフィルムが透明なのは、樹脂材料に対するシリカ粒子材料の親和性に優れるためだと考えられる。すなわち、実施例1のシリカ粒子材料は、凝集し難く、かつ、樹脂に対する親和性に優れていた。また、実施例1のシリカ粒子材料を用いた実施例5のフィラー含有樹脂組成物(前駆体)は硬く透明なフィルムを形成できた。さらに、実施例1のシリカ粒子材料を用いた実施例5のフィラー含有樹脂組成物(フィルム)は、硬く透明であった。
【0077】
(実施例6)
実施例4のシリカ粒子材料100質量部とイソプロパノール100質量部とを混合し、シリカ粒子材料とイソプロパノールとの混合物を得た。この混合物に、発振周波数39kHz、出力500Wで15分間超音波照射し、シリカ粒子材料含有スラリーを得た。
【0078】
アドマファイン SE2200(株式会社アドマテックス製、平均粒径約500nmのシリカ粒子材料)をイソプロパノールに等量配合し、シリカ粒子材料を50質量%含むスラリーを準備した。このスラリーをアドマファインスラリーと呼ぶ。
【0079】
上述したシリカ粒子材料含有スラリーとアドマファインスラリーとをシリカ粒子材料含有スラリー:アドマファインスラリー=1:10(シリカ質量比)となるように混合した。その後、減圧加熱することでこの混合物からイソプロパノールを揮発させ、乾燥させた。この乾燥混合物をエポキシ樹脂(東都化成株式会社製、ZX1059)に加えた。このとき、乾燥混合物とエポキシ樹脂との総量を100質量%として、シリカ濃度が70質量%になるようにした。この混合物を三本ロール機(EXAKT社製、80s)にかけて、実施例4のシリカ粒子材料及びアドマファインをエポキシ樹脂中に略均一に分散させた。以上の工程で、実施例6のフィラー含有樹脂組成物を得た。
【0080】
(比較例4)
アドマファインスラリーを減圧加熱することで、アドマファインスラリーからイソプロパノールを揮発させ、乾燥させた。この乾燥物をエポキシ樹脂(東都化成株式会社製、ZX1059)に加えた。このとき、乾燥物とエポキシ樹脂との総量を100質量%として、シリカ濃度が70質量%になるようにした。この混合物を三本ロール機(EXAKT社製、80s)にかけて、アドマファインをエポキシ樹脂中に略均一に分散させた。以上の工程で、比較例4のフィラー含有樹脂組成物を得た。
【0081】
(樹脂に対する親和性評価II)
実施例6のフィラー含有樹脂組成物及び比較例4のフィラー含有樹脂組成物の粘度を、レオメータ(TA Instruments社製、ARES G−2)により測定した。その結果を表すグラフを図7に示す。なお、図7中縦軸は粘度を表し、横軸は剪断速度を表す。
【0082】
図7に示すように、実施例6のフィラー含有樹脂組成物の粘度は、比較例4のフィラー含有樹脂組成物の粘度に比べて低かった。この結果から、実施例4のシリカ粒子材料を樹脂材料用のフィラーとして用いる場合には、従来のシリカ粒子材料を樹脂材料用のフィラーとして用いる場合に比べて、フィラー含有樹脂組成物の粘度を大幅に低下させ得る事がわかる。これは、実施例4のシリカ粒子材料が凝集なく、かつ、樹脂に対する親和性に優れるためだと考えられる。なお、粘度の低いフィラー含有樹脂組成物は、成形性に優れる。
【0083】
(炭素量測定試験)
実施例1〜4のシリカ粒子材料及び比較例1〜3のシリカ粒子材料について、シリカ粒子材料の質量あたりに存在する炭素の量(質量%)を測定した。測定には、有機炭素測定装置(HORIBA社製、EMIA−320V)を用いた。
【0084】
その結果、実施例1のシリカ粒子材料の炭素量は3.5質量%であり、実施例2のシリカ粒子材料の炭素量は2.6質量%であり、実施例3のシリカ粒子材料の炭素量は2.8質量%であり、実施例4のシリカ粒子材料の炭素量は0.96質量%であった。比較例1のシリカ粒子材料の炭素量は4.0質量%であり、比較例2のシリカ粒子材料の炭素量は1.8質量%であり、比較例3のシリカ粒子材料の炭素量は1.0質量%であった。
【0085】
(X1数測定試験)
実施例1〜4のシリカ粒子材料及び比較例1〜3のシリカ粒子材料について、シリカ粒子材料の単位表面積(nm2)あたりのX1の存在数を測定した。実施例1及び比較例1のシリカ粒子材料におけるX1はフェニル基であり、実施例2、3のシリカ粒子材料におけるX1はビニル基であり、実施例4及び比較例2、3のシリカ粒子材料におけるX1はメタクリロキシ基であった。シリカ粒子材料の表面積(比表面積)は窒素を用いたBET法で測定した。X1の存在数はシリカ粒子材料の炭素量を基に算出した。詳しくは、第1工程後のシリカ粒子を、水で洗浄し遠心分離した後に乾燥して、シランカップリング剤処理後のシリカ粒子試料を得た。この試料の炭素量を、有機炭素測定装置を用いて測定し、測定値を基にX1数を算出した。
【0086】
その結果、実施例1のシリカ粒子材料におけるX1数は、約1.2個/nm2であった。実施例2のシリカ粒子材料におけるX1数は、約1.1個/nm2であった。実施例3のシリカ粒子材料におけるX1数は、約1.1個/nm2であった。実施例4のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。比較例1のシリカ粒子材料におけるX1数は、約1.7個/nm2であった。比較例2のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。比較例3のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。参考までに、シランカップリング剤処理後のシリカ粒子試料の炭素量は、実施例1のシリカ粒子材料では3.6質量%、実施例2のシリカ粒子材料では1.1質量%、実施例3のシリカ粒子材料では1.1質量%、実施例4のシリカ粒子材料では1.5質量%であった。また、比較例1のシリカ粒子材料では5.0質量%、比較例2のシリカ粒子材料では1.5質量%、比較例3のシリカ粒子材料では1.5質量%であった。
【0087】
上述したように、シリカ粒子材料の樹脂材料に対する親和性はX1の数及び種類によって異なり、実施例1のシリカ粒子材料及び実施例4のシリカ粒子材料は、樹脂材料に対する親和性に優れていた。この結果から、樹脂材料に対して優れた親和性を発揮するためには、シリカ粒子材料の単位表面積(nm2)あたりのX1は0.5個〜2.5個であるのが好ましく、1.0個〜2.0個であるのがより好ましいといえる。
(高流動性粉体への応用)
以下に本発明の無機粉体混合物について説明する。
(実施例A)
無機粉末としてのシリカ粉末(アドマヒューズFEB24C:体積平均粒径12μm、24μm以上の粒子をカット:株式会社アドマテックス製)を63質量部、無機粉末としてのシリカ粉末(アドマヒューズFEA24A:体積平均粒径6μm、24μm以上の粒子をカット:株式会社アドマテックス製)を10質量部、シリカ粒子材料(アドマナノスラリー(シリカ濃度30質量%):体積平均粒径10nm:株式会社アドマテックス製:ビニルシランで表面処理)を粉末固形分換算で3質量部加え、80℃で乾燥させた後、ミキサーで解砕した。
【0088】
ここに、無機粉末としてのシリカ粉末(アドマファインSE5200−SQV:体積平均粒径1.5μm:24μm以上の粒子をカット:株式会社アドマテックス製)を8質量部、無機粉末としてのシリカ粉末(アドマファインSE2200−SQV体積平均粒径0.5μm:24μm以上の粒子をカット:株式会社アドマテックス製)を16質量部を加え、ミキサーで混合し、実施例Aの無機粉体混合物を得た。この粉体流動特性をパウダーテスター(篩通過性)、Sysmex製パウダーレオメータFT−4(粉体流動性)で測定した。
(実施例B)
実施例Aの無機粉体混合物を、エポキシ樹脂ZX1059に全体の質量を基準として75質量%分散させ、25℃での粘度を測定した。
(実施例C)
無機粉末としてのシリカ粉末(アドマファインSE2050−SEJ:体積平均粒径0.5μm:5μm以上の粒子をカット:エポキシシランで表面処理:株式会社アドマテックス製)を100質量部、シリカ粒子材料(アドマナノYA010C−SM1:体積平均粒径10nm:メタクリルシランにて表面処理)を3質量部混合後、エポキシ樹脂ZX1059に分散させた。シリカ濃度は全体の質量を基準として70%にした。更に、硬化剤を添加し、実施例Cのフィラー含有組成物とした。得られた本実施例のフィラー含有組成物について、110℃での浸透性を測定した。
(比較例A〜C)
それぞれ実施例A〜Cにおけるシリカ粒子材料を添加しない以外は同様の組成及び方法で組成物を調製し、同様の試験を行った。
(比較例D)
シリカ粒子材料に代えて表面処理を行っていないシリカ粒子(IPA−ST:日産化学製)を用いた以外は実施例Aと同様の組成及び方法で組成物を調製し、同様の試験を行った。
(結果)
・篩透過性
測定装置としてホソカワミクロンのTYPE−Eを用い、バイブレーション設定をRheostat7.5として、実施例A、比較例A,Dのそれぞれ100gについて粉体流動特性を測定した。結果を表1に示す。
【0089】
【表1】

【0090】
表1より明らかなように、実施例Aの無機粉体混合物は篩目が250μmであっても残渣もなく全て篩を通過させることができた。それに対して、シリカ粒子材料を含有させていない比較例Aの無機粉体混合物は目詰まりを起こして篩目250μmを通過することができず、篩目355μmでは実施例Aの4倍以上(更には残渣もあった)、篩目710μmでは実施例Aの2倍以上の時間がかかっており、実施例Aの無機粉体混合物の篩通過性は優れたものであることが分かった。
【0091】
また、表面処理が為されていないシリカ粒子を混ぜた比較例Dの無機粉体混合物では乾燥によって凝集が生じてどの篩目においても目詰まりが生じて通過できなかった。
・粉体流動特性
Sysmex製パウダーレオメータFT−4を用い、実施例A及び比較例Aの無機粉体混合物について粉体流動特性を測定した。具体的には円筒状の測定容器内に測定対象の無機粉体混合物を充填し、下部より空気を導入しながら所定の大きさの回転式ブレード(回転翼)をらせん状に回転させたときの回転トルクから粉体流動特性情報を得る方法である。
【0092】
結果を図8に示す。図8より明らかなように、実施例Aの無機粉体混合物の方が比較例Aの無機粉体混合物よりも小さなトルクで回転できることが分かり、流動性に優れていることが明らかになった。なお、空気流速が2〜2.5mm/s程度でトルクが逆転しているが、目視による観察によると、実施例Aの無機粉体混合物では流動性が良すぎるために空気によって吹きこぼれが生じたために、かえってトルクが上昇したものと考えられる。
・粘度測定
実施例B及び比較例Bのフィラー含有組成物について25℃における粘度を測定した。結果を図9に示す。図9より明らかなように、シェアレート2(s−1)〜8(s−1)程度の間で顕著な差が認められた。具体的には実施例Bのフィラー含有組成物の方が比較例Bのフィラー含有組成物よりも常に小さな粘度であった。
・浸透性評価
実施例C及び比較例Cのフィラー含有組成物について110℃で浸透性の測定を行った。浸透性の評価は、スライドガラスの上にギャップ70μmでスライドガラスを載せた松波硝子社製MUR−300(セル幅18mm)を用い、試料滴下用の一辺(短辺)にシリンジを用いて満遍なく各実施例及び比較例のフィラー含有組成物を載置した。載置してから対辺まで進んで(17.8mm)到達した時間を計測した。その結果、実施例Cのフィラー含有組成物では2分12秒60であり、比較例Cでは10分15秒39となった。つまり、70μmという狭い隙間のセル内を非常に早く充填することができるため、実施例Cの方が浸透性に優れていることが分かった。
(熱可塑性樹脂への無機粉体混合物の添加)
実施例Aの無機粉体混合物と比較例Aの無機粉体混合物との双方について熱可塑性樹脂(ポリエチレン)に混練した。混練の条件は200℃で行い、全体の質量を基準として40%の量を加えて混練した。
【0093】
その結果、実施例Aの無機粉体混合物では樹脂中に均一に分散されており、得られたフィラー含有組成物についても元の熱可塑性樹脂よりも機械的強度に優れたものになった。それに対して比較例Aの無機粉体混合物では樹脂中において無機粉体混合物が凝集していることが肉眼でも確認可能であり、得られたフィラー含有組成物の強度も元になった熱可塑性樹脂よりも低下することが分かった。
【産業上の利用可能性】
【0094】
本発明の無機粉体混合物は、EMC、ダイアタッチ、フィルム、プリプレーグ、TIM、UF、穴埋め材、ダム材、レジスト、リッドシーツ接着剤、放熱材接着剤、LCD接着剤、LCD封止材、光学接着剤、導波路材、電子ペーパ・有機ELにおける隔壁材、MEMS、インプリンティング転写材、ハードコート、プラスチック塗料、自動車用塗料などの組成物に添加して利用可能である。また、透明性が要求されるディスプレー、発光素子等のハードコート、カラーフィルタ等のオーバーコート、レジストインキ、MEMS等の光ナノインプリンティング材料に用いられる組成物の構成要素としてのシリカ粒子材料としても好適である。これらの組成物はエポキシ樹脂、アクリル樹脂などの熱硬化性樹脂、熱可塑性樹脂などを含む本発明のフィラー含有組成物として用いることもできる。
【技術分野】
【0001】
本発明は、流動性に優れた無機粉体混合物及びその無機粉体混合物をフィラーとして含むフィラー含有樹脂組成物に関する。
【背景技術】
【0002】
シリカ粒子の一種であるコロイダルシリカは、水ガラスを原料として製造する方法、金属ケイ素粉末を原料として製造する方法、気相合成法等により製造される。水ガラスを原料として製造する方法は、水ガラスを中和したり、イオン交換することで水ガラスを沈殿させることによって微小な粒子を生成する(例えば特許文献1参照)。
【0003】
このようなシリカ粒子の粒径は、例えばコロイダルシリカでは数nm〜数百nm程度と非常に小さい。粒径の小さなシリカ粒子は種々の用途に供される。その一方で粒径の小さなシリカ粒子の比表面積は非常に大きい。このため粒径の小さなシリカ粒子は凝集し易い特性を持つ。一旦凝集したシリカ粒子は再分散し難い。このため、粒径の小さなシリカ粒子は取り扱い性に劣る問題がある。
【0004】
すなわち、シリカ粒子は、液状媒体中における分散性には比較的優れても、乾粉状態になると(すなわち、シリカ粒子が単体で存在する場合には)凝集する。凝集したシリカ粒子は、例えば、高性能の分散機等を用いたとしても解砕し難い。
【0005】
従って、シリカ粒子を樹脂用のフィラーとして用いる場合、シリカ粒子は乾燥した状態で凝集するために、そのまま樹脂中に混合することは困難である。そのため、シリカ粒子は液状媒体中に分散された状態で液状媒体と共に樹脂材料中に配合することになる。この場合、液状媒体と樹脂材料との相互作用から適用できる組み合わせに制限が生じたり、本来混合が望まれていない液状媒体が樹脂材料中に導入される問題がある。このため、乾燥状態でも取り扱い性に優れるシリカ粒子が望まれている事情がある。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2006−45039号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
ところで、コロイダルシリカのような粒径が小さな材料をそれよりも大きな粒径をもつ粒子に混合すると、コロイダルシリカがころのような作用を発揮して混合物全体としても高い流動性を示すことが知られている。
【0008】
しかしながら、コロイダルシリカは前述したように凝集しやすいため他の粒子と混合することは困難であった。従って、他の粒子に混合することで高い流動性が期待できるとしても、その実現は困難であった。
【0009】
本発明は上記事情に鑑みてなされたものであり、取り扱い性に優れ、高い流動性をもつ無機粉体混合物、及び、その無機粉体混合物をフィラーとして採用したフィラー含有組成物を提供することを解決すべき課題とする。
【課題を解決するための手段】
【0010】
(a)上記課題を解決する本発明の無機粉体混合物は、式(1):−OSiX1X2X3で表される官能基及び式(2):−OSiY1Y2Y3で表される官能基と、両官能基が表面に結合するシリカ粒子とからなり、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%である。なお、上記式(1)、(2)中;X1はフェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基であり;X2、X3は−OSiR3及び−OSiY4Y5Y6よりそれぞれ独立して選択され;Y1はRであり;Y2、Y3はR及び−OSiY4Y5Y6よりそれぞれ独立して選択される。Y4はRであり;Y5及びY6は、R及び−OSiR3からそれぞれ独立して選択され;Rは炭素数1〜3のアルキル基から独立して選択される。X2、X3、Y2、Y3、Y5、及びY6の何れかは、近接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。
【0011】
本発明の無機粉体混合物は、下記の(i)〜(iii)の何れかを備えることが好ましく、(i)〜(iii)の複数を備えることがより好ましい。(i)前記式(1)で表される官能基と前記式(2)で表される官能基との存在数比が1:12〜1:60である。(ii)前記X1は前記シリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個である。(iii)前記Rは前記シリカ粒子材料の単位表面積(nm2)あたり1〜10個である。
(b)上記課題を解決する無機粉体混合物は、水を含む液状媒体中でシランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する表面処理工程をもつ表面処理方法により処理され、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%であり、前記シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基と、を持ち、前記シランカップリング剤と前記オルガノシラザンとのモル比は、前記シランカップリング剤:前記オルガノシラザン=1:2〜1:10である。
【0012】
本発明の無機粉体混合物は、下記の(iv)〜(viii)の何れかを備えることが好ましく、(iv)〜(viii)の複数を備えることがより好ましい。(iv)前記表面処理工程は、前記シリカ粒子を前記シランカップリング剤で処理する第1の処理工程と、前記シリカ粒子を前記オルガノシラザンで処理する第2の処理工程と、を持ち、前記第2の処理工程は、前記第1の処理工程後に行う。(v)前記第2の処理工程において、3つのアルコキシ基と炭素数1〜3のアルキル基とを持つ第2のシランカップリング剤で前記オルガノシラザンの一部を置き換え、前記第2の処理工程後に、さらに前記シリカ粒子を前記オルガノシラザンで処理する第3の処理工程を持つ。(vi)前記表面処理工程後に、前記シリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る固形化工程を備える。(vii)前記シランカップリング剤は、フェニルトリメトキシシラン、ビニルトリメトキシシラン、エポキシトリメトキシシラン、メタクリルトリメトキシシラン、アミノトリメトキシシラン、ウレイドトリメトキシシラン、メルカプトトリメトキシシラン、イソシアネート、又はアクリルトリメトキシシランから選ばれる少なくとも一種である。(viii)前記オルガノシラザンは、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザンから選ばれる少なくとも一種である。
(c)上述した(a)及び(b)に係る無機粉体混合物は前記無機粉体の体積平均粒径が0.5μm〜50μmであることが好ましい。
(d)上記課題を解決する本発明のフィラー含有組成物は、上述した(a)〜(c)の何れかに開示の本発明の無機粉体混合物からなるフィラーと、樹脂材料及び/又は樹脂材料前駆体と、を含むことを特徴とする。
【発明の効果】
【0013】
(a)本発明の無機粉体混合物がもつシリカ粒子材料は、X1(フェニル基、ビニル基、エポキシ基、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基)と、R(炭素数1〜3のアルキル基)とを表面に持つ。Rすなわち炭素数1〜3のアルキル基は疎水性が高いために、互いに反発し合う。従って、本発明の無機粉体混合物がもつシリカ粒子材料は炭素数1〜3のアルキル基同士の反発力により凝集し難く乾燥状態で流動性が高い粒子が得られるため、無機粉体に混合したときに流動性の高い無機粉体混合物が得られる。
【0014】
また、シリカ粒子材料が表面にRのみならずX1をも持つ場合には、極性の大きな官能基を持つ材料(例えば樹脂材料等)に対する親和性が向上する。このようなシリカ粒子材料は、樹脂材料中に均一分散し易くなる。シリカ粒子材料はX1を表面に持つため、樹脂材料との親和性に優れ、樹脂材料中に均一分散できる。特に、無機粉末の間に入ってころとしての作用を発揮できるために無機粉体混合物全体としての流動性も高い。換言すると、本発明の無機粉体混合物は、樹脂に対する親和性に優れるとともに、凝集抑制効果にも優れている。このため本発明の無機粉体混合物は、樹脂材料用のフィラーとして好適に使用できる。さらに、本発明の無機粉体混合物は凝集し難いため、液状媒体に分散しなくても良い。このことによっても、本発明の無機粉体混合物は、樹脂材料用のフィラーとして好適に使用できる。
【0015】
また、シリカ粒子材料の表面にRが多く存在するほど凝集抑制効果が向上するが、その一方で、樹脂に対する親和性が低下する。シリカ粒子材料の表面にX1が多く存在するほど樹脂に対する親和性が向上するが、その一方で、Rの数が少なくなり凝集抑制効果が低減する。従って、RとX1との存在数比には、好ましい範囲が存在する。式(1)で表される官能基と式(2)で表される官能基との存在数比が1:12〜1:60の範囲内であれば、樹脂に対する優れた親和性と優れた凝集抑制効果とを両立することができる。また、X1がシリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個であれば、樹脂に対する優れた親和性と優れた凝集抑制効果とを両立することができる。
(b)本発明の無機粉体混合物は上述の表面処理方法にてシリカ粒子を表面処理することにより流動性が高いシリカ粒子材料を乾燥状態で得ることができる。この表面処理方法にて処理して得られたシリカ粒子材料の表面には、上式(1)で表されるシランカップリング剤由来の官能基と、上式(2)で表されるオルガノシラザン由来の官能基とをもつことが予測される。そして、シランカップリング剤とオルガノシラザンとのモル比が1:2〜1:10の範囲内であれば、上式(1)で表される官能基と上式(2)で表される官能基とを、シリカ粒子材料の表面にバランス良く存在させ得るものと思われる。このため、樹脂に対する親和性と凝集抑制効果とが両立したシリカ粒子材料を得ることができる。
(c)本発明のフィラー含有組成物は、無機粉体混合物を乾燥状態で樹脂組成物に混合するため樹脂組成物中に分散媒が混入されない。特に熱可塑性樹脂にフィラーを混合する場合には一度混合された分散媒は除去しがたく最初から混合されない本発明のフィラー含有組成物は有利である。
【図面の簡単な説明】
【0016】
【図1】実施例1のシリカ粒子材料の粒度分布を表すグラフである。
【図2】実施例2のシリカ粒子材料の粒度分布を表すグラフである。
【図3】実施例3のシリカ粒子材料の粒度分布を表すグラフである。
【図4】実施例4のシリカ粒子材料の粒度分布を表すグラフである。
【図5】実施例1〜3及び比較例1のシリカ粒子材料の赤外吸収スペクトルを表すグラフである。
【図6】実施例4及び比較例2、3のシリカ粒子材料の赤外吸収スペクトルを表すグラフである。
【図7】実施例6のフィラー含有樹脂組成物及び比較例4のフィラー含有樹脂組成物の粘度を表すグラフである。
【図8】実施例A及び比較例Aの無機粉体混合物の粉体流動特性を表すグラフである。
【図9】実施例B及び比較例Bのフィラー含有組成物について25℃におけるシェアレート−粘度の関係を示すグラフである。
【発明を実施するための形態】
【0017】
本発明の無機粉体混合物及びフィラー含有組成物について実施形態に基づき以下詳細に説明を行う。本実施形態の無機粉体混合物は乾燥した状態で熱硬化性樹脂(硬化前のもの)、光硬化性樹脂(硬化前のもの)、熱可塑性樹脂(加熱溶融した状態のもの)などに簡単に分散可能である。この無機粉体混合物は流動性に優れているため、これらの樹脂に混合しても高い流動性をもつことができる。特に乾燥状態で高い流動性が得られるため、熱可塑性樹脂への混合も容易に行うことができる。また、熱硬化性樹脂や光硬化性樹脂においても乾燥状態で混合できるので添加が望まれない分散媒が混入することなくそれらの樹脂に添加することができる。
(無機粉体混合物)
本実施形態の無機粉体混合物は無機粉末とシリカ粒子材料とを乾燥状態で含む混合物である。両者の混合方法は特に限定されず公知の方法が採用できる。本実施形態の無機粉体混合物の構成要素であるシリカ粒子材料は粒径が小さいにもかかわらず流動性・分散性に優れており、他の粉体と容易に混合可能である。
【0018】
無機粉末とシリカ粒子材料との混合割合としては無機粉体混合物の質量を基準としてシリカ粒子材料の量が0.05%〜10%であること以外は特に限定されない。シリカ粒子材料の量が1%以上であることが望ましく、5%以下であることが望ましい。
【0019】
本明細書中における「乾燥状態」との語は粒子の表面に吸着していない溶媒が存在していないこととの意味である。具体的に乾燥状態にあるか否かの判断は、無機粉体混合物を取り扱うときに、スラリー状になっていない状態で流動性をもっているかどうかで簡易的に判定できる。スラリーになっていない状態で流動性をもっていれば乾燥状態にあるものと判断できる。
・無機粉末
無機粉末は無機物から形成され、適用される目的に応じてその組成が決定される。本実施形態の無機粉体混合物の高い流動性は、より粒径が小さいシリカ粒子材料がころとして作用することに起因するため、無機粉末の組成には殆ど影響されないものと考えられる。例えばシリカ、アルミナ、ジルコニア、チタニア、酸化亜鉛、ベーマイト、窒化ホウ素、それらの混合物、複合酸化物が例示できる。
【0020】
無機粉末は後述するシリカ粒子材料よりも体積平均粒径が大きい材料である。例えば、0.5μm以上にすることが望ましく、1μm以上にすることが更に望ましい。また、50μm以下にすることが望ましく、40μm以下にすることが更に望ましい。
【0021】
無機粉末は真球度が高い方が流動性が向上するため望ましい。真球度としては0.8以上にすることが望ましく、0.9以上にすることが更に望ましい。真球度の測定は、SEMで写真を撮り、その観察される粒子の面積と周囲長から、(真球度)={4π×(面積)÷(周囲長)2}で算出される値として算出する。1に近づくほど真球に近い。具体的には画像処理装置(シスメックス株式会社:FPIA−3000)を用い、無作為に抽出した100個の粒子について測定した平均値を採用する。
・シリカ粒子材料(その1)
シリカ粒子材料は体積平均粒径が5nm〜100nmのシリカからなる粒子である。特に望ましくは10nm以上である。また、50nm以下が望ましい。そしてその表面には後述する官能基をもつか、後述する表面処理が行われているかの何れかである。
【0022】
シリカ粒子材料は、式(1):−OSiX1X2X3で表される官能基と、式(2):−OSiY1Y2Y3で表される官能基とが表面に結合したシリカ粒子材料である。以下、式(1)で表される官能基を第1の官能基と呼び、式(2)で表される官能基を第2の官能基と呼ぶ。
【0023】
第1の官能基におけるX1は、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基である。X2、X3は、それぞれ、−OSiR3又は−OSiY4Y5Y6である。Y4はRである。Y5、Y6は、それぞれ、R又は−OSiR3である。
【0024】
第2の官能基におけるY1はRである。Y2、Y3は、それぞれ、−OSiR3又は−OSiY4Y5Y6である。
【0025】
第1の官能基及び第2の官能基に含まれる−OSiR3が多い程、シリカ粒子材料の表面にRを多く持つ。第1の官能基及び第2の官能基に含まれるR(炭素数1〜3のアルキル基)が多い程、シリカ粒子材料は凝集し難い。
【0026】
第1の官能基に関していえば、X2、X3がそれぞれ−OSiR3である場合に、Rの数が最小となる。また、X2及びX3がそれぞれ−OSiY4Y5Y6であり、かつ、Y5、Y6がそれぞれ−OSiR3である場合に、Rの数が最大となる。
【0027】
第2の官能基に関していえば、Y2、Y3がそれぞれ−OSiR3である場合に、Rの数が最小となる。また、Y2及びY3がそれぞれ−OSiY4Y5Y6であり、かつ、Y5、Y6がぞれぞれ−OSiR3である場合に、Rの数が最大となる。
【0028】
第1の官能基に含まれるX1の数、第1の官能基に含まれるRの数、第2の官能基に含まれるRの数は、RとX1との存在数比や、シリカ粒子材料の粒径や用途に応じて適宜設定すれば良い。
【0029】
なお、X2、X3、Y2、Y3、Y5、及びY6の何れかは、隣接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。例えば、第1の官能基のX2、X3、Y5、及びY6の何れかが、この第1の官能基に隣接する第1の官能基のX2、X3、Y5、及びY6の何れかと−O−にて結合していても良い。同様に、第2の官能基のY2、Y3、Y5、及びY6の何れかが、この第2の官能基に隣接する第2の官能基のY2、Y3、Y5、及びY6の何れかと−O−にて結合していても良い。さらには、第1の官能基のX2、X3、Y5、及びY6の何れかが、この第1の官能基に隣接する第2の官能基のY2、Y3、Y5、及びY6の何れかと−O−にて結合していても良い。
【0030】
第1の官能基と第2の官能基との存在数比が1:12〜1:60であれば、シリカ粒子材料の表面にX1とRとがバランス良く存在する。このため、第1の官能基と第2の官能基との存在数比が1:12〜1:60であるシリカ粒子材料は、樹脂に対する親和性及び凝集抑制効果に特に優れる。また、X1がシリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個であれば、シリカ粒子材料の表面に充分な数の第1の官能基が結合し、第1の官能基及び第2の官能基に由来するRもまた充分な数存在する。従ってこの場合にも、樹脂に対する親和性及びシリカ粒子材料の凝集抑制効果が充分に発揮される。
【0031】
何れの場合にも、シリカ粒子材料の単位表面積(nm2)あたりのRは、1個〜10個であるのが好ましい。この場合には、シリカ粒子材料の表面に存在するX1の数とRの数とのバランスが良くなり、樹脂に対する親和性及びシリカ粒子材料の凝集抑制効果との両方がバランス良く発揮される。
【0032】
シリカ粒子の表面に存在していた水酸基の全部が第1の官能基又は第2の官能基で置換されていることが好ましい。第1の官能基と第2の官能基との和が、シリカ粒子材料の単位表面積(nm2)あたり2.0個以上であれば、シリカ粒子材料において、シリカ粒子の表面に存在していた水酸基のほぼ全部が第1の官能基又は第2の官能基で置換されているといえる。
【0033】
シリカ粒子材料は、表面にRを持つ。これは、赤外線吸収スペクトルによって確認できる。詳しくは、シリカ粒子材料の赤外線吸収スペクトルを固体拡散反射法で測定すると、2962±2cm−1にC−H伸縮振動の極大吸収がある。このため、本実施形態の無機粉体混合物がもつシリカ粒子材料であるか否かは、赤外線吸収スペクトルによって確認できる。
【0034】
また、上述したように本発明の無機粉体混合物がもつシリカ粒子材料は凝集し難い。従って、シリカ粒子材料としては粒径の小さなものに採用できる。例えば、シリカ粒子材料は、平均粒径3nm〜5000nm程度にできる。平均粒径3〜200nmのシリカ粒子材料に適用するのが好ましい。
【0035】
なお、シリカ粒子材料は、例え僅かに凝集した場合にも、超音波処理することによって再度分散可能である。詳しくは、シリカ粒子材料をメチルエチルケトンに分散させたものに、発振周波数39kHz、出力500Wの超音波を照射することで、シリカ粒子材料を実質的に一次粒子にまで分散できる。このときの超音波照射時間は10分間以下で良い。シリカ粒子材料が一次粒子にまで分散したか否かは、粒度分布を測定することで確認できる。詳しくは、このシリカ粒子材料のメチルエチルケトン分散材料をマイクロトラック装置等の粒度分布測定装置で測定し、シリカ粒子材料の粒度分布があれば、シリカ粒子材料が一次粒子にまで分散したといえる。
【0036】
このシリカ粒子材料は、凝集し難いため、水やアルコール等の液状媒体に分散されていないシリカ粒子材料として提供できる。また、シリカ粒子材料は凝集し難いために、水で容易に洗浄できる。
・シリカ粒子材料(その2)
その1に示すシリカ粒子材料に代えて、以下に示す表面処理を行ったシリカ粒子材料を採用することもできる。なお、以下の方法によりシリカ粒子材料(その1)を得ることもできるため、その1とその2とは排他的なものではない。
【0037】
シリカ粒子材料の表面処理方法は、水を含む液状媒体中で、シランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する工程(表面処理工程)を持つ。シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基(すなわち上記のX1)とを持つ。
【0038】
シランカップリング剤で表面処理することで、シリカ粒子の表面に存在する水酸基がシランカップリング剤に由来する官能基で置換される。シランカップリング剤に由来する官能基は式(3);−OSiX1X4X5で表される。式(3)で表される官能基を第3の官能基と呼ぶ。第3の官能基におけるX1は式(1)で表される官能基におけるX1と同じである。X4、X5は、それぞれ、アルキコキシ基である。オルガノシラザンで表面処理することで、第3の官能基のX4、X5がオルガノシラザンに由来する−OSiY1Y2Y3(式(2)で表される官能基、第2の官能基)で置換される。シリカ粒子の表面に存在する水酸基の全てが第3の官能基で置換されていない場合には、シリカ粒子の表面に残存する水酸基が第2の官能基で置換される。このため、表面処理されたシリカ粒子材料の表面には、式(1):−OSiX1X2X3で表される官能基(すなわち第1の官能基)と、式(2):−OSiY1Y2Y3で表される官能基と(すなわち第2の官能基)が結合する。なお、シランカップリング剤とオルガノシラザンとのモル比は、シランカップリング剤:オルガノシラザン=1:2〜1:10であるため、得られたシリカ粒子材料における第1の官能基と第2の官能基との存在数比は理論上1:12〜1:60となる。
【0039】
表面処理工程においては、シリカ粒子をシランカップリング剤及びオルガノシラザンで同時に表面処理しても良い。又は、先ずシリカ粒子をシランカップリング剤で表面処理し、次いでオルガノシラザンで表面処理しても良い。又は、先ずシリカ粒子をオルガノシラザンで表面処理し、次いでシランカップリング剤で表面処理し、さらにその後にオルガノシラザンで表面処理しても良い。何れの場合にも、シリカ粒子の表面に存在する水酸基全てが第2の官能基で置換されないように、オルガノシラザンの量を調整すれば良い。なお、シリカ粒子の表面に存在する水酸基は、全てが第3の官能基で置換されても良いし、一部のみが第3の官能基で置換され、他の部分が第2の官能基で置換されても良い。第3の官能基に含まれるX4、X5は、全て第2の官能基で置換されるのが良い。
【0040】
なお、オルガノシラザンの一部を、第2のシランカップリング剤で置き換えても良い。第2のシランカップリング剤としては、3つのアルコキシ基と、1つのアルキル基とを持つものを用いることができる。この場合には、第3の官能基に含まれるX4、X5が、第2のシランカップリング剤に由来する第4の官能基で置換される。第4の官能基は式(4);−OSiY1X6X7で表される。Y1は第2の官能基におけるY1と同じRであり、X6、X7はそれぞれアルコキシ基又は水酸基である。第4の官能基に含まれるX6、X7は、オルガノシラザンに由来する第2の官能基で置換されるか、又は、別の第4の官能基で置換される。この場合には、シリカ粒子材料の表面に存在するRの量をさらに多くする事ができる。なお、オルガノシラザンの一部を、第2のシランカップリング剤に置き換える場合、第2のシランカップリング剤で表面処理した後に、再度オルガノシラザンで表面処理する必要がある。第4の官能基に含まれるX6、X7を、最終的にはオルガノシラザンに由来する第2の官能基で置換するためである。
【0041】
オルガノシラザンの一部を第2のシランカップリング剤で置き換える場合、上述した第1の官能基に含まれるX4、X5は、オルガノシラザンに由来する第2の官能基で置換されるか、第2のシランカップリング剤に由来する第4の官能基で置換される。X4、X5が第4の官能基で置換された場合、第4の官能基に含まれるX6、X7は、第2の官能基で置換されるか、別の第4の官能基によって置換される。第4の官能基に含まれるX6、X7が別の第4の官能基によって置換された場合、第4の官能基に含まれるX6、X7は、第2の官能基で置換される。このため第2のシランカップリング剤は、第1のカップリング剤及びオルガノシラザンのみで表面処理する場合(オルガノシラザンを第2のシランカップリング剤で置き換えなかった場合)に設定されるオルガノシラザンの量(a)molに対して、最大限5a/3mol置き換えることができる。この場合に必要になるオルガノシラザンの量は、8a/3molである。
【0042】
シランカップリング剤及び第2のシランカップリング剤のアルコキシ基は特に限定しないが、比較的炭素数の小さなものが好ましく、炭素数1〜12であることが好ましい。アルコキシ基の加水分解性を考慮すると、アルコキシ基はメトキシ基、エトキシ基、プロポキシ基、ブトキシ基の何れかであることがより好ましい。
【0043】
シランカップリング剤として、具体的には、フェニルトリメトキシシラン、ビニルトリメトキシシラン、ビニルトリエトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、3−グリシドキシプロピルトリメトキシシラン、3−グリシドキシプロピルトリエトキシシラン、p−スチリルトリメトキシシラン、3−メタクリロキシプロピルトリメトキシシラン、3−メタクリロキシプロピルトリエトキシシラン、3−アクリロキシプロピルトリメトキシシラン、N−フェニル−3−アミノプロピルトリメトキシシランが挙げられる。
【0044】
オルガノシラザンとしては、シリカ粒子の表面に存在する水酸基及びシランカップリング剤に由来するアルコキシ基を、上述した第2の官能基で置換できるものであれば良いが、分子量の小さなものを用いるのが好ましい。具体的には、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザン等が挙げられる。
【0045】
第2のシランカップリング剤としては、メチルトリメトキシシラン、メチルトリエトキシシラン、n−プロピルトリメトキシシラン、n−プロピルトリエトキシシラン、ヘキシルトリメトキシシラン、ヘキシルトリエトキシシラン等が挙げられる。
【0046】
なお、表面処理工程において、シランカップリング剤の重合や第2のシランカップリング剤の重合を抑制するため、重合禁止剤を加えても良い。重合禁止剤としては、3,5−ジブチル−4−ヒドロキシトルエン(BHT)、p−メトキシフェノール(メトキノン)等の一般的なものを用いることができる。
【0047】
シリカ粒子材料を得るための表面処理について説明する。本表面処理方法は、表面処理工程後に固形化工程を備えても良い。固形化工程は、表面処理後のシリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る工程である。上述したように、一般的なシリカ粒子材料は非常に凝集し易いため、一旦固形化したシリカ粒子材料を再度分散するのは非常に困難である。しかし、シリカ粒子材料は凝集し難いため、固形化しても凝集し難く、また、例え凝集しても再分散し易い。なお、上述したように、シリカ粒子材料を水で洗浄することで、電子部品等の用途に用いられるシリカ粒子材料を容易に製造できる。なお、洗浄工程においては、シリカ粒子材料の抽出水(詳しくは、シリカ粒子材料を121℃で24時間浸漬した水)の電気伝導度が50μS/cm以下となるまで、洗浄を繰り返すのが好ましい。
【0048】
固形化工程で用いる鉱酸としては塩酸、硝酸、硫酸、リン酸などが例示でき、特に塩酸が望ましい。鉱酸はそのまま用いても良いが、鉱酸水溶液として用いるのが好ましい。鉱酸水溶液における鉱酸の濃度は0.1質量%以上が望ましく、0.5質量%以上が更に望ましい。鉱酸水溶液の量は、洗浄対象であるシリカ粒子材料の質量を基準として6〜12倍程度にすることができる。
【0049】
鉱酸水溶液による洗浄は複数回数行うことも可能である。鉱酸水溶液による洗浄はシリカ粒子材料を鉱酸水溶液に浸漬後、撹拌することが望ましい。また、浸漬した状態で1時間から24時間、更には72時間程度放置することができる。放置する際には撹拌を継続することもできるし、撹拌しないこともできる。鉱酸含有液中にて洗浄する際には常温以上に加熱することもできる。
【0050】
その後、洗浄して懸濁させたシリカ粒子材料をろ取した後、水にて洗浄する。使用する水はアルカリ金属などのイオンを含まない(例えば質量基準で1ppm以下)ことが望ましい。例えば、イオン交換水、蒸留水、純水などである。水による洗浄は鉱酸水溶液による洗浄と同じく、シリカ粒子材料を分散、懸濁させた後、ろ過することもできるし、ろ取したシリカ粒子材料に対して水を継続的に通過させることによっても可能である。水による洗浄の終了時期は、上述した抽出水の電気伝導度で判断しても良いし、シリカ粒子材料を洗浄した後の排水中のアルカリ金属濃度が1ppm以下になった時点としても良いし、抽出水のアルカリ金属濃度が5ppm以下になった時点としても良い。なお、水で洗浄する際には常温以上に加熱することもできる。
【0051】
シリカ粒子材料の乾燥は、常法により行うことができる。例えば、加熱や、減圧(真空)下に放置する等である。
・フィラー含有組成物
本実施形態のフィラー含有組成物は、上述した無機粉体混合物と、樹脂材料及び/又は樹脂材料前駆体とを有する。無機粉体混合物は乾燥状態のまま樹脂材料及び/又は樹脂材料前駆体に混合される。フィラーの含有量は特に限定されず必要な量が添加される。上述した無機粉体混合物は流動性が高いため、大量に樹脂材料(又は樹脂材料前駆体)中に混合することができる。例えば、フィラー含有組成物全体の質量を基準として80%以上含有させることができる。また、90%以上含有させることもできる。このように大量にフィラーを含有させても流動性を示す組成物が得られる。
【0052】
樹脂材料は高分子材料であって、樹脂材料前駆体は高分子又は低分子の材料であり、更に反応が進行することにより分子量が増大したり、架橋が進行したりして樹脂材料を形成できる材料である。樹脂材料、並びに、樹脂材料前駆体により形成される樹脂材料は熱可塑性樹脂、熱硬化性樹脂、光硬化性樹脂などの一般的な樹脂材料が例示できる。樹脂材料前駆体は、エポキシ基、オキセタン基、水酸基、ブロックされたイソシアネート基、アミノ基、ハーフエステル基、アミック基、カルボキシ基、及び炭素-炭素二重結合基を化学構造中に有することが望ましい。これらの官能基は好適な反応条件を設定することで互いに結合可能な官能基(重合性官能基)であり、混合材料の分子量を向上できる。好適な反応条件としては単純に加熱や光照射を行ったり、熱や光照射によりラジカルやイオン(アニオン、カチオン)などの反応性種を生成したり、それらの官能基間を結合する反応開始剤(重合開始剤)を添加して加熱や光照射を行うことなどである。重合反応に際して必要な化合物を硬化剤として添加したり、その反応に対する触媒を添加することもできる。
【0053】
樹脂材料前駆体としては重合により高分子材料を形成する単量体や、上述したような重合性官能基により修飾した高分子材料が好ましいものとして挙げられる。例えば、硬化前の、エポキシ樹脂、アクリル樹脂、ウレタン樹脂などのプレポリマーが好適である。
【0054】
樹脂材料前駆体に対して無機粉体混合物を混合する場合にはそのまま混合することもできる。樹脂材料として熱可塑性樹脂を採用する場合に、その樹脂材料に混合するときには熱可塑性樹脂を加熱により溶融させた後に混合することができる。
【実施例】
【0055】
以下、本発明の無機粉体混合物について実施例に基づき詳細に説明する。
(本発明の無機粉体混合物が含有するシリカ粒子材料について)
(実施例1)
(材料)
シリカ粒子として、コロイダルシリカの一種であるスノーテックスOS(日産化学工業株式会社製、平均粒径10nm、水中に分散されており固形分濃度20%)を準備した。
【0056】
アルコールとして、イソプロパノールを準備した。
【0057】
シランカップリング剤として、フェニルトリメトキシシラン(信越化学工業株式会社製、KBM−103)を準備した。
【0058】
オルガノシラザンとして、ヘキサメチルジシラザン(HMDS、信越化学工業株式会社製、HDMS−1)を準備した。
【0059】
(表面処理工程)
(1)準備工程
シリカ粒子が20質量%の濃度で水に分散したスラリー100質量部にイソプロパノール60質量部を加え、室温(約25℃)で混合することで、シリカ粒子が液状媒体に分散されてなる分散液を得た。
【0060】
(2)第1工程
この分散液にフェニルトリメトキシシラン1.8質量部を加え、40℃で72時間混合した。この工程により、シリカ粒子の表面に存在する水酸基をシランカップリング剤で表面処理した。なお、このときフェニルトリメトキシシランは、必要な量の水酸基(一部)が表面処理されず残存するように計算して加えた。
【0061】
(3)第2工程
次いで、この混合物に、ヘキサメチルジシラザン3.7質量部を加え、40℃で72時間放置した。この工程によって、シリカ粒子が表面処理され、シリカ粒子材料が得られた。表面処理の進行に伴い、疎水性になったシリカ粒子が水及びイソプロパノールの中で安定に存在できなくなり、凝集・沈殿した。なお、フェニルトリメトキシシランとヘキサメチルジシラザンとのモル比は2:5であった。
【0062】
(固形化工程)
表面処理工程で得られた混合物全量に35%塩酸水溶液を5質量部を加え、シリカ粒子材料を沈殿させた。沈殿物をろ紙(アドバンテック社製 5A)で濾過した。濾過残渣(固形分)を純水で洗浄した後に100℃で真空乾燥して、シリカ粒子材料の固形物を得た。
【0063】
(実施例2)
実施例2のシリカ粒子の表面処理方法は、フェニルトリメトキシシランにかえてビニルトリメトキシシランを用い、ビニルトリメトキシシランとヘキサメチルジシラザンとのモル比が2:5であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、ビニルトリメトキシシラン1.36質量部を加え、第2工程においてはヘキサメチルジシラザン3.7質量部を加えた。
【0064】
なおビニルトリメトキシシランとしては、信越化学工業株式会社製 KBM−1003を用いた。
【0065】
(実施例3)
実施例3のシリカ粒子の表面処理方法は、フェニルトリメトキシシランにかえてビニルトリメトキシシランを用い、ビニルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:5であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、ビニルトリメトキシシラン1.36質量部を加え、第2工程においてはヘキサメチルジシラザン7.41質量部を加えた。
【0066】
(実施例4)
実施例4のシリカ粒子の表面処理方法においては、シリカ粒子として、コロイダルシリカの一種であるスノーテックスOL(日産化学工業株式会社製、平均粒径50nm、水中に分散されており固形分濃度20%)を用いた。また、第1工程においてシランカップリング剤として3−メタクリロキシプロピルトリメトキシシラン(信越化学工業株式会社製、KBM−503)0.48質量部を加えた。さらに、このシランカップリング剤に加えて重合禁止剤(3,5−ジブチル−4−ヒドロキシトルエン(BHT)、関東化学株式会社製)を0.01質量部加えた。また、第2工程において、ヘキサメチルジシラザン0.78質量部を加えた。さらに、固形化工程においては、表面処理工程で得られた混合物全量に35%塩酸水溶液2.6質量部を加えてシリカ粒子材料を沈殿させた。これ以外は、実施例4のシリカ粒子の表面処理方法は、実施例1のシリカ粒子の表面処理方法と同じであった。なお、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比は2:5であった。
【0067】
(比較例1)
比較例1のシリカ粒子の表面処理方法は、フェニルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:1であったこと以外は、実施例1のシリカ粒子の表面処理方法と同じである。第1工程においては、フェニルトリメトキシシラン4.5質量部を加え、第2工程においてはヘキサメチルジシラザン3.7質量部を加えた。
【0068】
(比較例2)
比較例2のシリカ粒子の表面処理方法は、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比が2:1であったこと以外は、実施例4のシリカ粒子の表面処理方法と同じである。第1工程においては、3−メタクリロキシプロピルトリメトキシシラン0.48質量部を加え、第2工程においてはヘキサメチルジシラザン0.16質量部を加えた。
【0069】
(比較例3)
比較例3のシリカ粒子の表面処理方法は、3−メタクリロキシプロピルトリメトキシシランとヘキサメチルジシラザンとのモル比が1:1であったこと以外は、実施例4のシリカ粒子の表面処理方法と同じである。第1工程においては、3−メタクリロキシプロピルトリメトキシシラン0.48質量部を加え、第2工程においてはヘキサメチルジシラザン0.31質量部を加えた。
【0070】
(凝集性評価試験)
実施例1〜4及び比較例1〜3のシリカ粒子材料について、液状媒体中における凝集性を測定した。
【0071】
詳しくは、実施例1〜3及び比較例1については、シリカ粒子材料10gとメチルエチルケトン40gとの混合物を攪拌し、シリカ粒子材料の分散試料を得た。
【0072】
実施例4及び比較例2、3については、シリカ粒子材料10gとメチルエチルケトン10gとの混合物を攪拌し、シリカ粒子材料の分散試料を得た。得られた各分散試料に含まれるシリカ粒子材料の粒度分布を、粒祖分布測定装置(日機装株式会社製、マイクロトラック)により測定した。凝集性評価試験の結果を図1〜4に示す。なお、図1は実施例1のシリカ粒子材料の粒度分布を表す。図2は実施例2のシリカ粒子材料の粒度分布を表す。図3は実施例3のシリカ粒子材料の粒度分布を表す。図4は実施例4のシリカ粒子材料の粒度分布を表す。
【0073】
図1〜4に示すように、実施例1〜4のシリカ粒子材料は、凝集のない一次粒子の状態で分散している。これは、図1〜4のグラフにおいて、各シリカ粒子材料の粒度分布(ピーク)が、粒子径10nm程度の位置に一つのみ現れていることから裏付けられる。シリカ粒子材料が二次粒子であれば(すなわち、少しでも凝集があれば)、粒子径100nm以上の位置に少なくとも一つのピークが現れる。このため、実施例1〜4のシリカ粒子材料は、一旦固形化したにもかかわらず、その殆どが一次粒子であり、殆ど凝集していないことがわかる。これに対して、比較例1〜3のシリカ粒子材料は、攪拌するだけでは分散せず、攪拌後に発振周波数39kHz、出力500Wで1時間以上超音波照射しても、肉眼で凝集が確認でき、一次粒子にまで分散しなかった。この結果から、シランカップリング剤とオルガノシラザンとのモル比を1:2〜1:10の範囲にすることで、固形化しても凝集し難いシリカ粒子材料を製造できることがわかる。なお、実施例1のシリカ粒子材料の平均粒径は10nm、実施例2のシリカ粒子材料の平均粒径は10nm、実施例3のシリカ粒子材料の平均粒径は10nm、実施例4のシリカ粒子材料の平均粒径は50nmであった。この結果から、凝集抑制のためには、シランカップリング剤とオルガノシラザンとのモル比を1:5〜2:5の範囲にするのが好ましいことがわかる。
【0074】
(極大吸収測定試験)
実施例1〜4及び比較例1〜3のシリカ粒子材料を準備し、この試料の赤外線吸収スペクトルを、サーモニコレット社製 FT−IR Avatorを用いた粉体拡散反射法で測定した。このときの測定条件は、分解能4、スキャン回数64であった。極大吸収測定試験の結果を表すグラフを図5〜図6に示す。図5〜図6に示すように、実施例1〜4及び比較例1〜3のシリカ粒子材料の赤外吸収スペクトルは、何れも、2962cm−1にC-H伸縮振動の極大吸収(ピーク)を持つ。このため、実施例1〜4及び比較例1〜3のシリカ粒子材料は、アルキル基を持つこと(すなわち、アルキル基を持つオルガノシラザンで表面処理されていること)がわかる。なお、比較例1〜3のシリカ粒子材料のピーク高さは実施例1〜4のシリカ粒子材料のピーク高さに比べて低かった。この結果は、比較例1〜3のシリカ粒子材料においては、充分な量のアルキル基を持たないことを示唆している。詳しくは、実施例1〜4のシリカ粒子材料の赤外線吸収スペクトルにおいては、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが3倍以上であった。比較例1〜3のシリカ粒子材料の赤外線吸収スペクトルにおいては、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが2倍以下であった。上述したように、実施例1〜4のシリカ粒子材料は凝集し難く、比較例1〜3のシリカ粒子材料は凝集し易かった。これらの結果から、シランカップリング剤に由来する各官能基固有のC−Hのピーク高さに対してオルガノシラザンに由来するメチル基(2962cm−1)のピーク高さが3倍以上であるシリカ粒子材料は凝集し難いといえる。
【0075】
(実施例5)
実1のシリカ材料100質量部とメチルイソブチルケトン100質量部とを混合し、シリカ粒子材料とメチルイソブチルケトンとの混合物を得た。この混合物4質量部にアクリル樹脂(根上工業株式会社製 アートレジン UK904)6質量部を加え混合した。この混合物100質量部に対して5質量部の重合開始剤(チバ・スペシャルティ・ケミカルズ株式会社製、ダロキュアTPO)を加え、さらに、固形分濃度が30質量%となるようにメチルイソブチルケトンを加えた。この混合物をさらに攪拌して、フィラー含有樹脂組成物(前駆体)を得た。この前駆体は、均一溶液状であった。実施例5のフィラー含有樹脂組成物を、基板に塗布し、実施例5のフィラー含有樹脂組成物(フィルム)を成膜した。
【0076】
(樹脂に対する親和性評価I)
実施例5のフィラー含有樹脂組成物(フィルム)を目視にて観察したところ、このフィルムは硬く透明であった。実施例5のフィルムが硬いのは、シリカ粒子材料が樹脂材料中に略均一に分散しているためだと考えられる。また、実施例5のフィルムが透明なのは、樹脂材料に対するシリカ粒子材料の親和性に優れるためだと考えられる。すなわち、実施例1のシリカ粒子材料は、凝集し難く、かつ、樹脂に対する親和性に優れていた。また、実施例1のシリカ粒子材料を用いた実施例5のフィラー含有樹脂組成物(前駆体)は硬く透明なフィルムを形成できた。さらに、実施例1のシリカ粒子材料を用いた実施例5のフィラー含有樹脂組成物(フィルム)は、硬く透明であった。
【0077】
(実施例6)
実施例4のシリカ粒子材料100質量部とイソプロパノール100質量部とを混合し、シリカ粒子材料とイソプロパノールとの混合物を得た。この混合物に、発振周波数39kHz、出力500Wで15分間超音波照射し、シリカ粒子材料含有スラリーを得た。
【0078】
アドマファイン SE2200(株式会社アドマテックス製、平均粒径約500nmのシリカ粒子材料)をイソプロパノールに等量配合し、シリカ粒子材料を50質量%含むスラリーを準備した。このスラリーをアドマファインスラリーと呼ぶ。
【0079】
上述したシリカ粒子材料含有スラリーとアドマファインスラリーとをシリカ粒子材料含有スラリー:アドマファインスラリー=1:10(シリカ質量比)となるように混合した。その後、減圧加熱することでこの混合物からイソプロパノールを揮発させ、乾燥させた。この乾燥混合物をエポキシ樹脂(東都化成株式会社製、ZX1059)に加えた。このとき、乾燥混合物とエポキシ樹脂との総量を100質量%として、シリカ濃度が70質量%になるようにした。この混合物を三本ロール機(EXAKT社製、80s)にかけて、実施例4のシリカ粒子材料及びアドマファインをエポキシ樹脂中に略均一に分散させた。以上の工程で、実施例6のフィラー含有樹脂組成物を得た。
【0080】
(比較例4)
アドマファインスラリーを減圧加熱することで、アドマファインスラリーからイソプロパノールを揮発させ、乾燥させた。この乾燥物をエポキシ樹脂(東都化成株式会社製、ZX1059)に加えた。このとき、乾燥物とエポキシ樹脂との総量を100質量%として、シリカ濃度が70質量%になるようにした。この混合物を三本ロール機(EXAKT社製、80s)にかけて、アドマファインをエポキシ樹脂中に略均一に分散させた。以上の工程で、比較例4のフィラー含有樹脂組成物を得た。
【0081】
(樹脂に対する親和性評価II)
実施例6のフィラー含有樹脂組成物及び比較例4のフィラー含有樹脂組成物の粘度を、レオメータ(TA Instruments社製、ARES G−2)により測定した。その結果を表すグラフを図7に示す。なお、図7中縦軸は粘度を表し、横軸は剪断速度を表す。
【0082】
図7に示すように、実施例6のフィラー含有樹脂組成物の粘度は、比較例4のフィラー含有樹脂組成物の粘度に比べて低かった。この結果から、実施例4のシリカ粒子材料を樹脂材料用のフィラーとして用いる場合には、従来のシリカ粒子材料を樹脂材料用のフィラーとして用いる場合に比べて、フィラー含有樹脂組成物の粘度を大幅に低下させ得る事がわかる。これは、実施例4のシリカ粒子材料が凝集なく、かつ、樹脂に対する親和性に優れるためだと考えられる。なお、粘度の低いフィラー含有樹脂組成物は、成形性に優れる。
【0083】
(炭素量測定試験)
実施例1〜4のシリカ粒子材料及び比較例1〜3のシリカ粒子材料について、シリカ粒子材料の質量あたりに存在する炭素の量(質量%)を測定した。測定には、有機炭素測定装置(HORIBA社製、EMIA−320V)を用いた。
【0084】
その結果、実施例1のシリカ粒子材料の炭素量は3.5質量%であり、実施例2のシリカ粒子材料の炭素量は2.6質量%であり、実施例3のシリカ粒子材料の炭素量は2.8質量%であり、実施例4のシリカ粒子材料の炭素量は0.96質量%であった。比較例1のシリカ粒子材料の炭素量は4.0質量%であり、比較例2のシリカ粒子材料の炭素量は1.8質量%であり、比較例3のシリカ粒子材料の炭素量は1.0質量%であった。
【0085】
(X1数測定試験)
実施例1〜4のシリカ粒子材料及び比較例1〜3のシリカ粒子材料について、シリカ粒子材料の単位表面積(nm2)あたりのX1の存在数を測定した。実施例1及び比較例1のシリカ粒子材料におけるX1はフェニル基であり、実施例2、3のシリカ粒子材料におけるX1はビニル基であり、実施例4及び比較例2、3のシリカ粒子材料におけるX1はメタクリロキシ基であった。シリカ粒子材料の表面積(比表面積)は窒素を用いたBET法で測定した。X1の存在数はシリカ粒子材料の炭素量を基に算出した。詳しくは、第1工程後のシリカ粒子を、水で洗浄し遠心分離した後に乾燥して、シランカップリング剤処理後のシリカ粒子試料を得た。この試料の炭素量を、有機炭素測定装置を用いて測定し、測定値を基にX1数を算出した。
【0086】
その結果、実施例1のシリカ粒子材料におけるX1数は、約1.2個/nm2であった。実施例2のシリカ粒子材料におけるX1数は、約1.1個/nm2であった。実施例3のシリカ粒子材料におけるX1数は、約1.1個/nm2であった。実施例4のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。比較例1のシリカ粒子材料におけるX1数は、約1.7個/nm2であった。比較例2のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。比較例3のシリカ粒子材料におけるX1数は、約2.0個/nm2であった。参考までに、シランカップリング剤処理後のシリカ粒子試料の炭素量は、実施例1のシリカ粒子材料では3.6質量%、実施例2のシリカ粒子材料では1.1質量%、実施例3のシリカ粒子材料では1.1質量%、実施例4のシリカ粒子材料では1.5質量%であった。また、比較例1のシリカ粒子材料では5.0質量%、比較例2のシリカ粒子材料では1.5質量%、比較例3のシリカ粒子材料では1.5質量%であった。
【0087】
上述したように、シリカ粒子材料の樹脂材料に対する親和性はX1の数及び種類によって異なり、実施例1のシリカ粒子材料及び実施例4のシリカ粒子材料は、樹脂材料に対する親和性に優れていた。この結果から、樹脂材料に対して優れた親和性を発揮するためには、シリカ粒子材料の単位表面積(nm2)あたりのX1は0.5個〜2.5個であるのが好ましく、1.0個〜2.0個であるのがより好ましいといえる。
(高流動性粉体への応用)
以下に本発明の無機粉体混合物について説明する。
(実施例A)
無機粉末としてのシリカ粉末(アドマヒューズFEB24C:体積平均粒径12μm、24μm以上の粒子をカット:株式会社アドマテックス製)を63質量部、無機粉末としてのシリカ粉末(アドマヒューズFEA24A:体積平均粒径6μm、24μm以上の粒子をカット:株式会社アドマテックス製)を10質量部、シリカ粒子材料(アドマナノスラリー(シリカ濃度30質量%):体積平均粒径10nm:株式会社アドマテックス製:ビニルシランで表面処理)を粉末固形分換算で3質量部加え、80℃で乾燥させた後、ミキサーで解砕した。
【0088】
ここに、無機粉末としてのシリカ粉末(アドマファインSE5200−SQV:体積平均粒径1.5μm:24μm以上の粒子をカット:株式会社アドマテックス製)を8質量部、無機粉末としてのシリカ粉末(アドマファインSE2200−SQV体積平均粒径0.5μm:24μm以上の粒子をカット:株式会社アドマテックス製)を16質量部を加え、ミキサーで混合し、実施例Aの無機粉体混合物を得た。この粉体流動特性をパウダーテスター(篩通過性)、Sysmex製パウダーレオメータFT−4(粉体流動性)で測定した。
(実施例B)
実施例Aの無機粉体混合物を、エポキシ樹脂ZX1059に全体の質量を基準として75質量%分散させ、25℃での粘度を測定した。
(実施例C)
無機粉末としてのシリカ粉末(アドマファインSE2050−SEJ:体積平均粒径0.5μm:5μm以上の粒子をカット:エポキシシランで表面処理:株式会社アドマテックス製)を100質量部、シリカ粒子材料(アドマナノYA010C−SM1:体積平均粒径10nm:メタクリルシランにて表面処理)を3質量部混合後、エポキシ樹脂ZX1059に分散させた。シリカ濃度は全体の質量を基準として70%にした。更に、硬化剤を添加し、実施例Cのフィラー含有組成物とした。得られた本実施例のフィラー含有組成物について、110℃での浸透性を測定した。
(比較例A〜C)
それぞれ実施例A〜Cにおけるシリカ粒子材料を添加しない以外は同様の組成及び方法で組成物を調製し、同様の試験を行った。
(比較例D)
シリカ粒子材料に代えて表面処理を行っていないシリカ粒子(IPA−ST:日産化学製)を用いた以外は実施例Aと同様の組成及び方法で組成物を調製し、同様の試験を行った。
(結果)
・篩透過性
測定装置としてホソカワミクロンのTYPE−Eを用い、バイブレーション設定をRheostat7.5として、実施例A、比較例A,Dのそれぞれ100gについて粉体流動特性を測定した。結果を表1に示す。
【0089】
【表1】
【0090】
表1より明らかなように、実施例Aの無機粉体混合物は篩目が250μmであっても残渣もなく全て篩を通過させることができた。それに対して、シリカ粒子材料を含有させていない比較例Aの無機粉体混合物は目詰まりを起こして篩目250μmを通過することができず、篩目355μmでは実施例Aの4倍以上(更には残渣もあった)、篩目710μmでは実施例Aの2倍以上の時間がかかっており、実施例Aの無機粉体混合物の篩通過性は優れたものであることが分かった。
【0091】
また、表面処理が為されていないシリカ粒子を混ぜた比較例Dの無機粉体混合物では乾燥によって凝集が生じてどの篩目においても目詰まりが生じて通過できなかった。
・粉体流動特性
Sysmex製パウダーレオメータFT−4を用い、実施例A及び比較例Aの無機粉体混合物について粉体流動特性を測定した。具体的には円筒状の測定容器内に測定対象の無機粉体混合物を充填し、下部より空気を導入しながら所定の大きさの回転式ブレード(回転翼)をらせん状に回転させたときの回転トルクから粉体流動特性情報を得る方法である。
【0092】
結果を図8に示す。図8より明らかなように、実施例Aの無機粉体混合物の方が比較例Aの無機粉体混合物よりも小さなトルクで回転できることが分かり、流動性に優れていることが明らかになった。なお、空気流速が2〜2.5mm/s程度でトルクが逆転しているが、目視による観察によると、実施例Aの無機粉体混合物では流動性が良すぎるために空気によって吹きこぼれが生じたために、かえってトルクが上昇したものと考えられる。
・粘度測定
実施例B及び比較例Bのフィラー含有組成物について25℃における粘度を測定した。結果を図9に示す。図9より明らかなように、シェアレート2(s−1)〜8(s−1)程度の間で顕著な差が認められた。具体的には実施例Bのフィラー含有組成物の方が比較例Bのフィラー含有組成物よりも常に小さな粘度であった。
・浸透性評価
実施例C及び比較例Cのフィラー含有組成物について110℃で浸透性の測定を行った。浸透性の評価は、スライドガラスの上にギャップ70μmでスライドガラスを載せた松波硝子社製MUR−300(セル幅18mm)を用い、試料滴下用の一辺(短辺)にシリンジを用いて満遍なく各実施例及び比較例のフィラー含有組成物を載置した。載置してから対辺まで進んで(17.8mm)到達した時間を計測した。その結果、実施例Cのフィラー含有組成物では2分12秒60であり、比較例Cでは10分15秒39となった。つまり、70μmという狭い隙間のセル内を非常に早く充填することができるため、実施例Cの方が浸透性に優れていることが分かった。
(熱可塑性樹脂への無機粉体混合物の添加)
実施例Aの無機粉体混合物と比較例Aの無機粉体混合物との双方について熱可塑性樹脂(ポリエチレン)に混練した。混練の条件は200℃で行い、全体の質量を基準として40%の量を加えて混練した。
【0093】
その結果、実施例Aの無機粉体混合物では樹脂中に均一に分散されており、得られたフィラー含有組成物についても元の熱可塑性樹脂よりも機械的強度に優れたものになった。それに対して比較例Aの無機粉体混合物では樹脂中において無機粉体混合物が凝集していることが肉眼でも確認可能であり、得られたフィラー含有組成物の強度も元になった熱可塑性樹脂よりも低下することが分かった。
【産業上の利用可能性】
【0094】
本発明の無機粉体混合物は、EMC、ダイアタッチ、フィルム、プリプレーグ、TIM、UF、穴埋め材、ダム材、レジスト、リッドシーツ接着剤、放熱材接着剤、LCD接着剤、LCD封止材、光学接着剤、導波路材、電子ペーパ・有機ELにおける隔壁材、MEMS、インプリンティング転写材、ハードコート、プラスチック塗料、自動車用塗料などの組成物に添加して利用可能である。また、透明性が要求されるディスプレー、発光素子等のハードコート、カラーフィルタ等のオーバーコート、レジストインキ、MEMS等の光ナノインプリンティング材料に用いられる組成物の構成要素としてのシリカ粒子材料としても好適である。これらの組成物はエポキシ樹脂、アクリル樹脂などの熱硬化性樹脂、熱可塑性樹脂などを含む本発明のフィラー含有組成物として用いることもできる。
【特許請求の範囲】
【請求項1】
式(1):−OSiX1X2X3で表される官能基及び式(2):−OSiY1Y2Y3で表される官能基と、両官能基が表面に結合するシリカ粒子とからなり、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、
前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、
前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%である無機粉体混合物。
(上記式(1)、(2)中;X1はフェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基であり;X2、X3は−OSiR3及び−OSiY4Y5Y6よりそれぞれ独立して選択され;Y1はRであり;Y2、Y3はR及び−OSiY4Y5Y6よりそれぞれ独立して選択される。Y4はRであり;Y5及びY6は、R及び−OSiR3からそれぞれ独立して選択され;Rは炭素数1〜3のアルキル基から独立して選択される。なお、X2、X3、Y2、Y3、Y5、及びY6の何れかは、近接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。)
【請求項2】
前記式(1)で表される官能基と前記式(2)で表される官能基との存在数比が1:12〜1:60である請求項1に記載の無機粉体混合物。
【請求項3】
前記X1は前記シリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個である請求項1又は2に記載の無機粉体混合物。
【請求項4】
前記Rは前記シリカ粒子材料の単位表面積(nm2)あたり1〜10個である請求項1〜3の何れか1項に記載の無機粉体混合物。
【請求項5】
水を含む液状媒体中でシランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する表面処理工程をもつ表面処理方法により処理され、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、
前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、
前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%であり、
前記シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基と、を持ち、
前記シランカップリング剤と前記オルガノシラザンとのモル比は、前記シランカップリング剤:前記オルガノシラザン=1:2〜1:10である無機粉体混合物。
【請求項6】
前記表面処理工程は、
前記シリカ粒子を前記シランカップリング剤で処理する第1の処理工程と、
前記シリカ粒子を前記オルガノシラザンで処理する第2の処理工程と、を持ち、
前記第2の処理工程は、前記第1の処理工程後に行う請求項5に記載の無機粉体混合物。
【請求項7】
前記第2の処理工程において、3つのアルコキシ基と炭素数1〜3のアルキル基とを持つ第2のシランカップリング剤で前記オルガノシラザンの一部を置き換え、
前記第2の処理工程後に、さらに前記シリカ粒子を前記オルガノシラザンで処理する第3の処理工程を持つ請求項5又は6に記載の無機粉体混合物。
【請求項8】
前記表面処理工程後に、前記シリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る固形化工程を備える請求項5〜7の何れか1項に記載の無機粉体混合物。
【請求項9】
前記シランカップリング剤は、フェニルトリメトキシシラン、ビニルトリメトキシシラン、エポキシトリメトキシシラン、メタクリルトリメトキシシラン、アミノトリメトキシシラン、ウレイドトリメトキシシラン、メルカプトトリメトキシシラン、イソシアネート、又はアクリルトリメトキシシランから選ばれる少なくとも一種である請求項5〜8の何れか1項に記載の無機粉体混合物。
【請求項10】
前記オルガノシラザンは、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザンから選ばれる少なくとも一種である請求項5〜9の何れか1項に記載の無機粉体混合物。
【請求項11】
前記無機粉体の体積平均粒径が0.5μm〜50μmである請求項1〜10の何れか1項に記載の無機粉体混合物。
【請求項12】
請求項1〜11の何れか1項に記載の無機粉体混合物からなるフィラーと、樹脂材料及び/又は樹脂材料前駆体と、を含むことを特徴とするフィラー含有組成物。
【請求項1】
式(1):−OSiX1X2X3で表される官能基及び式(2):−OSiY1Y2Y3で表される官能基と、両官能基が表面に結合するシリカ粒子とからなり、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、
前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、
前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%である無機粉体混合物。
(上記式(1)、(2)中;X1はフェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基であり;X2、X3は−OSiR3及び−OSiY4Y5Y6よりそれぞれ独立して選択され;Y1はRであり;Y2、Y3はR及び−OSiY4Y5Y6よりそれぞれ独立して選択される。Y4はRであり;Y5及びY6は、R及び−OSiR3からそれぞれ独立して選択され;Rは炭素数1〜3のアルキル基から独立して選択される。なお、X2、X3、Y2、Y3、Y5、及びY6の何れかは、近接する官能基のX2、X3、Y2、Y3、Y5、及びY6の何れかと−O−にて結合しても良い。)
【請求項2】
前記式(1)で表される官能基と前記式(2)で表される官能基との存在数比が1:12〜1:60である請求項1に記載の無機粉体混合物。
【請求項3】
前記X1は前記シリカ粒子材料の単位表面積(nm2)あたり0.5〜2.5個である請求項1又は2に記載の無機粉体混合物。
【請求項4】
前記Rは前記シリカ粒子材料の単位表面積(nm2)あたり1〜10個である請求項1〜3の何れか1項に記載の無機粉体混合物。
【請求項5】
水を含む液状媒体中でシランカップリング剤及びオルガノシラザンによってシリカ粒子を表面処理する表面処理工程をもつ表面処理方法により処理され、体積平均粒径が5nm〜100nmであるシリカ粒子材料と、
前記シリカ粒子材料よりも体積平均粒径が大きい無機粉体と、を乾燥状態で有する混合物であって、
前記シリカ粒子の混合量は、前記無機粒子及び前記シリカ粒子の質量の和を基準として、0.05〜10%であり、
前記シランカップリング剤は、3つのアルコキシ基と、フェニル基、ビニル基、エポキシ基、メタクリル基、アミノ基、ウレイド基、メルカプト基、イソシアネート基、又はアクリル基と、を持ち、
前記シランカップリング剤と前記オルガノシラザンとのモル比は、前記シランカップリング剤:前記オルガノシラザン=1:2〜1:10である無機粉体混合物。
【請求項6】
前記表面処理工程は、
前記シリカ粒子を前記シランカップリング剤で処理する第1の処理工程と、
前記シリカ粒子を前記オルガノシラザンで処理する第2の処理工程と、を持ち、
前記第2の処理工程は、前記第1の処理工程後に行う請求項5に記載の無機粉体混合物。
【請求項7】
前記第2の処理工程において、3つのアルコキシ基と炭素数1〜3のアルキル基とを持つ第2のシランカップリング剤で前記オルガノシラザンの一部を置き換え、
前記第2の処理工程後に、さらに前記シリカ粒子を前記オルガノシラザンで処理する第3の処理工程を持つ請求項5又は6に記載の無機粉体混合物。
【請求項8】
前記表面処理工程後に、前記シリカ粒子材料を鉱酸で沈殿させ、沈殿物を水で洗浄・乾燥して、シリカ粒子材料の固形物を得る固形化工程を備える請求項5〜7の何れか1項に記載の無機粉体混合物。
【請求項9】
前記シランカップリング剤は、フェニルトリメトキシシラン、ビニルトリメトキシシラン、エポキシトリメトキシシラン、メタクリルトリメトキシシラン、アミノトリメトキシシラン、ウレイドトリメトキシシラン、メルカプトトリメトキシシラン、イソシアネート、又はアクリルトリメトキシシランから選ばれる少なくとも一種である請求項5〜8の何れか1項に記載の無機粉体混合物。
【請求項10】
前記オルガノシラザンは、テトラメチルジシラザン、ヘキサメチルジシラザン、ペンタメチルジシラザンから選ばれる少なくとも一種である請求項5〜9の何れか1項に記載の無機粉体混合物。
【請求項11】
前記無機粉体の体積平均粒径が0.5μm〜50μmである請求項1〜10の何れか1項に記載の無機粉体混合物。
【請求項12】
請求項1〜11の何れか1項に記載の無機粉体混合物からなるフィラーと、樹脂材料及び/又は樹脂材料前駆体と、を含むことを特徴とするフィラー含有組成物。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−206886(P2012−206886A)
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願番号】特願2011−73296(P2011−73296)
【出願日】平成23年3月29日(2011.3.29)
【出願人】(501402730)株式会社アドマテックス (82)
【Fターム(参考)】
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願日】平成23年3月29日(2011.3.29)
【出願人】(501402730)株式会社アドマテックス (82)
【Fターム(参考)】
[ Back to top ]