
無水(メタ)アクリル酸の改良された製造方法
【課題】(メタ)アクリル酸と無水酢酸とを少なくとも一種の重合禁止剤の存在下でトランス無水化して無水(メタ)アクリル酸A(M)A2Oを製造する方法の改良。
【解決手段】反応物が部分的に変換し、次いで連続的に蒸留する。反応装置の汚れの問題なしに従来方法よりもA(M)A2Oを高純度、高生産性で製造できる。
【解決手段】反応物が部分的に変換し、次いで連続的に蒸留する。反応装置の汚れの問題なしに従来方法よりもA(M)A2Oを高純度、高生産性で製造できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、反応物が部分的に転換されるまで反応は実行し、その後に連続蒸留する、(メタ)アクリル酸と無水酢酸との間のトランス無水化(transanhydrization)によって無水(メタ)アクリル酸を製造する方法の改良に関するものである。
【背景技術】
【0002】
「無水(メタ)アクリル酸」という用語はA(M)A2Oを表し、無水メタアクリル酸と無水アクリル酸とを意味する。
【0003】
無水酢酸と(メタ)アクリル酸とを重合禁止剤の存在下で反応させて無水(メタ)アクリル酸を製造する方法は古くから公知であり、例えば特許文献1(欧州特許第EP 231 689号公報)に記載されている。この方法では反応中に、形成された酢酸を留出し、その後、形成された無水(メタ)アクリル酸を蒸留によって分離する。この方法を実行する場合の問題は、反応装置の上部に載置された蒸留塔から直接蒸留をする段階工程重合が起こる点にある。さらに、得られる無水(メタ)アクリル酸の量が反応装置の大きさと反応装置に導入する反応物とによって制限され、従って、反応容積を最適化することができない。
【0004】
この問題を解決するために特許文献2(欧州特許第EP 1 273 565号公報)では、生成する酢酸の少なくとも一部を除去し、反応中に反応媒体中に無水酢酸および/または(メタ)アクリル酸を連続的に導入して除去した酢酸の少なくとも一部と交換している。この方法で反応媒体の安定性は大幅に改良されるが、懸濁液中にポリマーが形成される問題を完全に解決することはできず、残渣反応物および副産物を除去した後、得られた粗A(M)A2Oを濾過によって精製する必要がある。しかし、A(M)A2Oは催涙性が極めて高いためこの濾過には問題点が多い。
【0005】
特許文献3(米国特許公開第US 2002/0161260号明細書)ではカルボン酸塩の形をしたCr、Zn、Cu、Ca、Zr、Li、La、NaまたはHfをベースにした触媒を使用する。しかし、これらの触媒を使用しても上記方法に比べて生産性が大きく向上させることはできない。
【0006】
(メタ)アクリル酸(A(M)A)と無水酢酸(AC2O)とを反応させて(メタ)アクリル酸無水物(A(M)A2O)を製造するトランス無水化(transanhydrization)のスキームは2つの主反応を用いて以下のように要約できる:
【0007】
反応1:
無水酢酸/(メタ)アクリル酸混合物と酢酸(AcOH)の形成:
H2C=C(R1)COOH+CH3C(O)OC(O)CH3 <-> H2C=C(R1)C(O)OC(O)CH3+CH3COOH
(ここで、R1=HまたはCH3)
この反応1は非常に迅速に進む。
反応2:
上記無水混合物とA(M)Aとの間の反応:
H2C=C(R1)COOH + H2C=C(R1)C(O)OC(O)CH3 <-> H2C=C(R1)C(O)OC(O)C(R1)=CH2+CH3COOH
【0008】
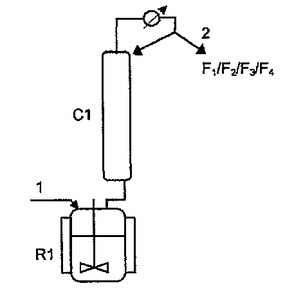
従来は、この合成は[図1]に記載のような装置でバッチ条件下に実行されてきた。後で添加する反応物、例えば特許文献2(欧州特許第EP 1 273 565号公報)に記載の反応物のための配位はこの図には示していない。反応物、A(M)AおよびAC20は、少なくとも一種の重合禁止剤と一緒に蒸留塔C1が上に乗っている反応装置R1に1から導入される。酢酸が形成されるとともに酢酸を蒸留2で除去することによって平衡をシフトさせる。蒸留塔C1は反応で生じた酢酸を除去して、反応平衡をシフトさせる役目と同時に、反応後に未反応物と、軽質無水混合物の型をした副産物と、場合によっては所望生成物A(M)A2Oとを蒸留する役目をする。
【0009】
一般に、回収される蒸留画分F1は酢酸から成り、画分F2は主として酢酸、(メタ)アクリル酸および無水酢酸/(メタ)アクリル酸混合物から成り、画分F3は主として少量のA(M)A2Oを含む無水酢酸/(メタ)アクリル酸混合物から成り、画分F4は主としてA(M)A2Oに富んだ流れから成る。また、A(M)A2Oは画分F1、F2およびF3を蒸留した後に反応装置から直接回収することもできるが、一般に、高品質の生成物を得るためには反応装置からのものを濾過する必要がある。画分F2とF3は一般にバッチまたは連続的に反応装置へ再循環される(図示せず)。
【0010】
しかし、反応は常に反応物が高転化するように実行されるため、現在のプロセスには汚染(汚れ、encrassement)に大なり小なり課題がある。反応物の重合率を高くするために反応時間を長くすると、重合禁止剤が存在していても重質副産物(ミカエル付加物タイプ)の含有量とA(M)AおよびA(M)A2Oのポリマーの生成量が増加し選択性が低下し、反応装置の汚れが増加する。
【0011】
蒸留相を反応装置と同じ装置で実行するとポリマーの生成現象が顕著になり、粗反応混合液中に細かく懸濁する。蒸留相に反応装置中の液面が順次低下すると、蒸留塔から還流してくる不安定なモノマーまたは未安定化モノマーまたは低温の壁と接触して凝縮したモノマー(これは例えば反応装置のドームの加熱が不十分な場合である)と長時間接触することになる。この不安定なモノマーまたは未安定化モノマーが流れてジャケットで加熱された壁と接触するとポリマーが形成され、反応装置が汚れる。画分F1/F2/F3を単に分離して粗のA(M)A2Oを回収する場合には、濾過を行って懸濁したポリマーを除去する必要がある。しかし、A(M)A2Oは催涙作用が強いためこの濾過作業、特に濾過装置の清掃作業は困難な作業である。蒸留によってA(M)A2Oを純粋な状態で回収すると、反応装置が完全に閉塞する。
【0012】
さらに、蒸留塔、一般的には反応中に生成した酢酸の蒸留に適した蒸留塔はキャパシティおよび効率が画分F2/F3/F4の蒸留にも理想的にバランスしたものではない。また、加熱された反応生成物が蒸留の間に反応装置中に長時間存在していることは上記理由で非常に有害である。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】欧州特許第EP 231 689号公報
【特許文献2】欧州特許第EP 1 273 565号公報
【特許文献3】米国特許公開第US 2002/0161260号明細書
【発明の概要】
【発明が解決しようとする課題】
【0014】
従って、本発明の目的は上記の各欠点を解決し、反応装置汚染汚の問題を減らし、粗A(M)A2Oの濾過操作を避けることができ、しかも、得られる生成物の純度および選択性に優れた無水(メタ)アクリル酸の改良された製造方法を提供することにある。
【課題を解決するための手段】
【0015】
本発明の対象は、(メタ)アクリル酸と無水酢酸とを少なくとも一種の重合禁止剤の存在下でトランス無水化(transanhydrization)して無水(メタ)アクリル酸を製造する方法において、下記(a)〜(d)の段階から成ることを特徴とする方法にある:
(a)反応物が部分的に変換するまで、蒸留塔Clが載った反応装置Rl中で反応を実行し、
(b)段階(a)で得られた粗反応物を中間貯蔵タンクSl中に送り、そこから第2の蒸留塔C2中に連続的に供給し、
(c)第2の蒸留塔C2の底部から無水(メタ)アクリル酸を回収し、頂部から基本的に未変換反応物と副産物とから成る画分を回収し、
(d)第2の蒸留塔C2の頂部画分を反応装置Rlにバッチまたは連続的に再循環する。
【図面の簡単な説明】
【0016】
【図1】従来装置の単純化した概念図。
【図2】本発明方法を実施するための単純化した操作ダイアグラム。
【発明を実施するための形態】
【0017】
本発明方法の特徴は、反応物の変換が部分的である粗反応混合物を連続的に蒸留し、反応および蒸留の操作を2つの別々の装置で別々に行う点にある。
本発明方法を用いることで従来方法と比較して生産性を約50%増加させることができる。
【0018】
以下、添付図面を参照して本発明の上記以外の特徴および効果を説明する。
本発明方法では、重質副産物およびポリマーの形成を制限して生産性を増加させるために反応物の変換を意図的に完成させなかった粗反応生成物を中間貯蔵装置中に移送する。この粗反応生成物は残留反応物および副産物を除去するのに適した段数を有する蒸留塔で連続的に蒸留される。
この蒸留塔の底部で回収されるA(M)A2Oは97%以上の純度を有して、懸濁ポリマーは全くないので濾過の必要はない。
【0019】
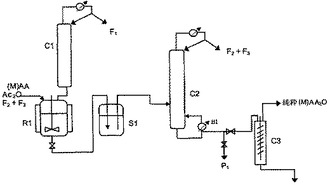
[図2]では反応物A(M)AおよびAc20が反応装置R1に送られる。反応装置R1の上には生成した酢酸を順次除去するための蒸留塔Clが載置されている。メタアクリル酸MAAを用いるのが好ましい。反応は反応装置の許容最大充填量を入れてバッチで(不連続に)行うことができる。A(M)AとAc20のモル比は一般に0.5〜5の間、好ましくは1.8〜2.2の間にするのが好ましい。また、出発材料をバッチ(不連続に)で入れ、次いで、反応中に酢酸回収(除去)によって得られた分を利用して反応物をバッチまたは連続的に添加して反応を行うこともできる。反応の好ましい操作条件は特許文献2(欧州特許第EP 1 273 565号公報)に詳細に記載されている。特に、反応装置に入れる出発材料のモル比A(M)A/Ac20は2.5〜11、好ましくは9〜11であり、添加反応物はAc20であり、全体のモル比A(M)A/Ac20は0.5〜5の間、好ましくは1.8〜2.2の間である。
【0020】
また、粗反応生成物を連続的に抜き出して、反応を連続的で実行することもできる。この場合、懸濁固体はなく、濾過も不必要である。
【0021】
本発明方法では、反応物が部分的に転換されるまで反応を実行する。換言すれば粗反応生成物中のA(M)A2Oの含有量が75%以下、好ましくは粗反応生成物中のA(M)A2Oの含有量が50%〜70%の間、好ましくは50%〜60%の間で、残りがA(M)A、Ac20および実変換混合無水物となるように反応を実行する。
【0022】
反応は一般に6〜8時間行う。この時間は反応物をより強力に変換するのに必要な時間より大幅に短い。従って、上記時間後に反応装置を開放し、次に合成を迅速に実行する。結果的にプラントの生産性が増加する。
【0023】
反応温度は一般に50℃〜120℃の間、有利には85℃〜105℃の間である。圧力は選択した反応温度に従って調節するが、一般には20〜200mmHg(0.0267〜0.2666バール)の間である。蒸留塔のプレート(敏なプレート)の温度は酢酸の蒸留温度にあわせて反応中の圧力に応じて調節するのが有利である。こうした運転を行うことでカラムC1の頂部で酢酸純度が90%以上、さらには99%以上である画分F1を得ることができる。
【0024】
本発明反応は反応装置中および蒸留塔中に導入された少なくとも一種の重合禁止剤の存在下で実行される。
【0025】
重合禁止剤の例としては当業者に周知の化合物、特にハイドロキノン、ハイドロキノン・モノメチルエーテル、フェノチアジン、ジ (tert−ブチル)−パラ−クレゾール(BHT)、2,4-ジメチル-6-(tert-ブチル)フェノール(Topanol A)、パラ−フェニレンジアミン、ジ(tert-ブチル)カテコール(BHT)、4-OH-TEMPO(4-ヒドロキシ−2,2,6,6-テトラメチル−1-ピペリジンオキシル)またはTEMPO誘導体を使用でき、これらは単独または混合物として反応混合物に対して100〜5000ppmの比率で使用される。
【0026】
本発明の好ましい実施例では反応は触媒なしで実行される。しかし、触媒の存在下で反応を実行することができ、例えば欧州特許第EP 196 520号公報およびドイツ特許第DE 102006029320号公報に記載のポリマー支持体に担持された遊離型スルホン酸のような触媒または米国特許公開第US 2002/0161260号明細書に記載の触媒を使用できる。変換を完全には行わない本発明では触媒の使用は必須ではない。
【0027】
反応全体にわたって空気または酸素を8%に減らした空気をバブリングすることができる。
【0028】
反応の終わりに得られる粗生成物は一般にポリマーを全く含まない透明で、反応で生じる酢酸は除去されている。
【0029】
本発明方法の段階(b)では反応物が所望の変換度に達した時に上記の粗反応生成物が中間貯蔵タンクS1へ移される。本発明の特定実施例では、反応は連続的に実行され、粗反応生成物は運転条件、特に反応温度および酢酸を回収するカラムC1の還流比に合わせて連続的に抜き出される。
【0030】
中間貯蔵タンクS1は残った反応物と副産物、特に1モルのA(M)Aと1モルのAc2Oとの反応によって形成される混合無水物の回収に適した蒸留塔C2へ連続的に供給するために用いられる。蒸留塔C2は理論段数が10以上、好ましくは理論段数が15以上の分離効率を有するのが好ましい。カラムのパッキングは従来のパッキング、ランダムまたは組み立てた充填材または2つのタイプのパッキングの混合物にすることができる。カラムの加熱は強制循環加熱で行うことができる。
【0031】
蒸留塔C2への供給流速は設備およびカラムの寸法に応じて広範囲で変えることができる。
【0032】
蒸留塔C2の頂部では未変換反応物と副産物とから成る画分F2+F3が連続的に回収される。これらは反応装置R1へ連続的に直接再循環されるか、貯蔵および一種以上の重合禁止剤で安定化した後に再循環される(段階d)。驚くことに、従来法の対応方法では重合前の貯蔵時間が相対的に短いにもかかわらず、貯蔵時の画分F2+F3の安定性は優れていることが確認された。これは反応装置中で再循環できない画分F2が破壊されることを意味する。これは経済的なロスと成る。これに対して本発明方法ではこれはなく、出発材料が節減できることが観測された。
【0033】
蒸留塔C2の底部では濾過を必要としない無水(メタ)アクリル酸Plが97%以上の純度、すなわち従来方法で一般的に得られる純度(約90%〜94%)より高い純度で回収される。
【0034】
本発明方法で得られたA(M)A2Oは合成の反応物として直接使用でき、例えばジメチルアミノプロピルアミンとの反応によってジメチルアミノプロパノール(メタ)アクリレートを合成できる。
【0035】
本発明の特定実施例は蒸留塔C2の底部から回収されたA(M)A2Oを滞流時間が短い装置、例えば、存在する可能性のある重質副産物および重合防止剤を除去するために、[図]2に装置C3で表したような薄膜蒸発器を使用して精製する追加の段階(e)をさらに有する。こうして精製されたA(M)A2Oは従来方法で達成可能な純度より著しく高い少なくとも99%の純度を有する。
以下、本発明の実施例を図面を参照して説明するが、方法はが下記実施例に限定されるものではない。
【実施例】
【0036】
以下に記載の百分比は重量百分比を表す。また、以下では下記の略号を使用する:
AMA2O: 無水メタアクリル酸
AMA: メタアクリル酸
AC20: 無水酢酸
ACOH: 酢酸
混合無水物: H2C=C(CH3)C(O)OC(O)CH3
Topanol A: 2、4-ジメチル-6-(tert-ブチル)フェノール
BHT: 2、6-ジ(tert-ブチル)−パラ−クレゾール
【0037】
実施例1(比較例)
頂部に凝縮器と還流ヘッドとを有する理論段数15の蒸留塔Clが載置された1リットル容の機械撹拌(4−枚プロペラ混合機)式のジャケット付きガラス製反応装置Rl中に、302gのAC20と、576gのAMAと、0.64gのTopanol Aと、0.19gのBHTとを入れた。合成時間全体にわたって反応媒体を減酸素空気のバブリングを維持した。反応中、反応装置の温度を90℃〜95℃に維持し、運転圧力を150mmHg〜50mmHgに徐々に下げた。
反応終了時すなわち10時間後に、AMA2Oを70%含む478gの粗反応生成物が得られる。この段階中375gのACOHが生じ、それは形成と同時に蒸留によって除去した。次いで、カラム頂部の圧力を15mmHgまで下げ、温度を102℃まで上げて、軽質物の蒸留(画分F2:AC20の除去、画分F3:AMAおよび混合無水物の除去)を行う。得られたF2およびF3の量は100gである。これらの画分を合せ、720ppmのTopanol Aで安定して次回の反応へ一度で再循環した。画分F4の蒸留によってAMA2O(300g)が得られる。画分F4の平均組成はAMA2Oが95.5%、重質化合物が2.5%、AMAが1%、混合無水物が0.7%である。蒸留終了時に反応装置はきわめて汚れていた。
【0038】
実施例2(比較例)
実施例1と同じ反応は実行したが、画分F1、F2およびF3の回収後にボトムとして反応装置RlからAMA2O(300g)が得られ、反応装置を排出させ、ポリマーを除去するための加圧濾過を行った。平均組成はAMA2Oが94%、重産物が4%、AMAが1%、混合無水物が0.7%である。
【0039】
実施例3(本発明)
反応で使用した反応装置と、反応物の供給および操作条件は実施例1に記載のものである。6時間反応させた後に676gの粗反応生成物(組成:60%のAMA2O、10%のMAA、15%のAC20、11%の混合無水物、1%の重質物+安定化剤)を得た。それを冷却し、中間貯蔵タンクS1へ送った。輸送は容易である。中間貯蔵タンクから80グラム/時の流量でカラムC2へ供給した。反応装置の汚れは観測されなかった。
【0040】
上記カラムC2は直径が30mmで、Multiknit型充填材を収容しており、理論段数は20である。加熱は熱サイホンで行ない、パレットポンプによって減圧した。上記80グラム/時の流量の粗生成物を分けて、塔底から48.8 g/時で取り出し、軽質物である残りは塔頂から取り出す。塔頂生成物はタンクに貯蔵し、次の反応べリサイクルした。上記凝縮器および還流はAMA2O中に5%のTopanol A溶液を入れて安定した。減酸素空気をリボイラに注入した。運転圧力はカラム頂部で15mmHg、塔底温度は91℃である。得られた塔低生成物中のAMA2Oの純度は97.5%である。
比較例1および2と、実施例3で得られたAMA2Oとジメチルアミノプロピルアミンとからジメチルアミノプロピルアミドを合成した。本発明の実施例3で得られた品質のAMA2Oを用いた場合に純度がより良いという結果が得られた。
【技術分野】
【0001】
本発明は、反応物が部分的に転換されるまで反応は実行し、その後に連続蒸留する、(メタ)アクリル酸と無水酢酸との間のトランス無水化(transanhydrization)によって無水(メタ)アクリル酸を製造する方法の改良に関するものである。
【背景技術】
【0002】
「無水(メタ)アクリル酸」という用語はA(M)A2Oを表し、無水メタアクリル酸と無水アクリル酸とを意味する。
【0003】
無水酢酸と(メタ)アクリル酸とを重合禁止剤の存在下で反応させて無水(メタ)アクリル酸を製造する方法は古くから公知であり、例えば特許文献1(欧州特許第EP 231 689号公報)に記載されている。この方法では反応中に、形成された酢酸を留出し、その後、形成された無水(メタ)アクリル酸を蒸留によって分離する。この方法を実行する場合の問題は、反応装置の上部に載置された蒸留塔から直接蒸留をする段階工程重合が起こる点にある。さらに、得られる無水(メタ)アクリル酸の量が反応装置の大きさと反応装置に導入する反応物とによって制限され、従って、反応容積を最適化することができない。
【0004】
この問題を解決するために特許文献2(欧州特許第EP 1 273 565号公報)では、生成する酢酸の少なくとも一部を除去し、反応中に反応媒体中に無水酢酸および/または(メタ)アクリル酸を連続的に導入して除去した酢酸の少なくとも一部と交換している。この方法で反応媒体の安定性は大幅に改良されるが、懸濁液中にポリマーが形成される問題を完全に解決することはできず、残渣反応物および副産物を除去した後、得られた粗A(M)A2Oを濾過によって精製する必要がある。しかし、A(M)A2Oは催涙性が極めて高いためこの濾過には問題点が多い。
【0005】
特許文献3(米国特許公開第US 2002/0161260号明細書)ではカルボン酸塩の形をしたCr、Zn、Cu、Ca、Zr、Li、La、NaまたはHfをベースにした触媒を使用する。しかし、これらの触媒を使用しても上記方法に比べて生産性が大きく向上させることはできない。
【0006】
(メタ)アクリル酸(A(M)A)と無水酢酸(AC2O)とを反応させて(メタ)アクリル酸無水物(A(M)A2O)を製造するトランス無水化(transanhydrization)のスキームは2つの主反応を用いて以下のように要約できる:
【0007】
反応1:
無水酢酸/(メタ)アクリル酸混合物と酢酸(AcOH)の形成:
H2C=C(R1)COOH+CH3C(O)OC(O)CH3 <-> H2C=C(R1)C(O)OC(O)CH3+CH3COOH
(ここで、R1=HまたはCH3)
この反応1は非常に迅速に進む。
反応2:
上記無水混合物とA(M)Aとの間の反応:
H2C=C(R1)COOH + H2C=C(R1)C(O)OC(O)CH3 <-> H2C=C(R1)C(O)OC(O)C(R1)=CH2+CH3COOH
【0008】
従来は、この合成は[図1]に記載のような装置でバッチ条件下に実行されてきた。後で添加する反応物、例えば特許文献2(欧州特許第EP 1 273 565号公報)に記載の反応物のための配位はこの図には示していない。反応物、A(M)AおよびAC20は、少なくとも一種の重合禁止剤と一緒に蒸留塔C1が上に乗っている反応装置R1に1から導入される。酢酸が形成されるとともに酢酸を蒸留2で除去することによって平衡をシフトさせる。蒸留塔C1は反応で生じた酢酸を除去して、反応平衡をシフトさせる役目と同時に、反応後に未反応物と、軽質無水混合物の型をした副産物と、場合によっては所望生成物A(M)A2Oとを蒸留する役目をする。
【0009】
一般に、回収される蒸留画分F1は酢酸から成り、画分F2は主として酢酸、(メタ)アクリル酸および無水酢酸/(メタ)アクリル酸混合物から成り、画分F3は主として少量のA(M)A2Oを含む無水酢酸/(メタ)アクリル酸混合物から成り、画分F4は主としてA(M)A2Oに富んだ流れから成る。また、A(M)A2Oは画分F1、F2およびF3を蒸留した後に反応装置から直接回収することもできるが、一般に、高品質の生成物を得るためには反応装置からのものを濾過する必要がある。画分F2とF3は一般にバッチまたは連続的に反応装置へ再循環される(図示せず)。
【0010】
しかし、反応は常に反応物が高転化するように実行されるため、現在のプロセスには汚染(汚れ、encrassement)に大なり小なり課題がある。反応物の重合率を高くするために反応時間を長くすると、重合禁止剤が存在していても重質副産物(ミカエル付加物タイプ)の含有量とA(M)AおよびA(M)A2Oのポリマーの生成量が増加し選択性が低下し、反応装置の汚れが増加する。
【0011】
蒸留相を反応装置と同じ装置で実行するとポリマーの生成現象が顕著になり、粗反応混合液中に細かく懸濁する。蒸留相に反応装置中の液面が順次低下すると、蒸留塔から還流してくる不安定なモノマーまたは未安定化モノマーまたは低温の壁と接触して凝縮したモノマー(これは例えば反応装置のドームの加熱が不十分な場合である)と長時間接触することになる。この不安定なモノマーまたは未安定化モノマーが流れてジャケットで加熱された壁と接触するとポリマーが形成され、反応装置が汚れる。画分F1/F2/F3を単に分離して粗のA(M)A2Oを回収する場合には、濾過を行って懸濁したポリマーを除去する必要がある。しかし、A(M)A2Oは催涙作用が強いためこの濾過作業、特に濾過装置の清掃作業は困難な作業である。蒸留によってA(M)A2Oを純粋な状態で回収すると、反応装置が完全に閉塞する。
【0012】
さらに、蒸留塔、一般的には反応中に生成した酢酸の蒸留に適した蒸留塔はキャパシティおよび効率が画分F2/F3/F4の蒸留にも理想的にバランスしたものではない。また、加熱された反応生成物が蒸留の間に反応装置中に長時間存在していることは上記理由で非常に有害である。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】欧州特許第EP 231 689号公報
【特許文献2】欧州特許第EP 1 273 565号公報
【特許文献3】米国特許公開第US 2002/0161260号明細書
【発明の概要】
【発明が解決しようとする課題】
【0014】
従って、本発明の目的は上記の各欠点を解決し、反応装置汚染汚の問題を減らし、粗A(M)A2Oの濾過操作を避けることができ、しかも、得られる生成物の純度および選択性に優れた無水(メタ)アクリル酸の改良された製造方法を提供することにある。
【課題を解決するための手段】
【0015】
本発明の対象は、(メタ)アクリル酸と無水酢酸とを少なくとも一種の重合禁止剤の存在下でトランス無水化(transanhydrization)して無水(メタ)アクリル酸を製造する方法において、下記(a)〜(d)の段階から成ることを特徴とする方法にある:
(a)反応物が部分的に変換するまで、蒸留塔Clが載った反応装置Rl中で反応を実行し、
(b)段階(a)で得られた粗反応物を中間貯蔵タンクSl中に送り、そこから第2の蒸留塔C2中に連続的に供給し、
(c)第2の蒸留塔C2の底部から無水(メタ)アクリル酸を回収し、頂部から基本的に未変換反応物と副産物とから成る画分を回収し、
(d)第2の蒸留塔C2の頂部画分を反応装置Rlにバッチまたは連続的に再循環する。
【図面の簡単な説明】
【0016】
【図1】従来装置の単純化した概念図。
【図2】本発明方法を実施するための単純化した操作ダイアグラム。
【発明を実施するための形態】
【0017】
本発明方法の特徴は、反応物の変換が部分的である粗反応混合物を連続的に蒸留し、反応および蒸留の操作を2つの別々の装置で別々に行う点にある。
本発明方法を用いることで従来方法と比較して生産性を約50%増加させることができる。
【0018】
以下、添付図面を参照して本発明の上記以外の特徴および効果を説明する。
本発明方法では、重質副産物およびポリマーの形成を制限して生産性を増加させるために反応物の変換を意図的に完成させなかった粗反応生成物を中間貯蔵装置中に移送する。この粗反応生成物は残留反応物および副産物を除去するのに適した段数を有する蒸留塔で連続的に蒸留される。
この蒸留塔の底部で回収されるA(M)A2Oは97%以上の純度を有して、懸濁ポリマーは全くないので濾過の必要はない。
【0019】
[図2]では反応物A(M)AおよびAc20が反応装置R1に送られる。反応装置R1の上には生成した酢酸を順次除去するための蒸留塔Clが載置されている。メタアクリル酸MAAを用いるのが好ましい。反応は反応装置の許容最大充填量を入れてバッチで(不連続に)行うことができる。A(M)AとAc20のモル比は一般に0.5〜5の間、好ましくは1.8〜2.2の間にするのが好ましい。また、出発材料をバッチ(不連続に)で入れ、次いで、反応中に酢酸回収(除去)によって得られた分を利用して反応物をバッチまたは連続的に添加して反応を行うこともできる。反応の好ましい操作条件は特許文献2(欧州特許第EP 1 273 565号公報)に詳細に記載されている。特に、反応装置に入れる出発材料のモル比A(M)A/Ac20は2.5〜11、好ましくは9〜11であり、添加反応物はAc20であり、全体のモル比A(M)A/Ac20は0.5〜5の間、好ましくは1.8〜2.2の間である。
【0020】
また、粗反応生成物を連続的に抜き出して、反応を連続的で実行することもできる。この場合、懸濁固体はなく、濾過も不必要である。
【0021】
本発明方法では、反応物が部分的に転換されるまで反応を実行する。換言すれば粗反応生成物中のA(M)A2Oの含有量が75%以下、好ましくは粗反応生成物中のA(M)A2Oの含有量が50%〜70%の間、好ましくは50%〜60%の間で、残りがA(M)A、Ac20および実変換混合無水物となるように反応を実行する。
【0022】
反応は一般に6〜8時間行う。この時間は反応物をより強力に変換するのに必要な時間より大幅に短い。従って、上記時間後に反応装置を開放し、次に合成を迅速に実行する。結果的にプラントの生産性が増加する。
【0023】
反応温度は一般に50℃〜120℃の間、有利には85℃〜105℃の間である。圧力は選択した反応温度に従って調節するが、一般には20〜200mmHg(0.0267〜0.2666バール)の間である。蒸留塔のプレート(敏なプレート)の温度は酢酸の蒸留温度にあわせて反応中の圧力に応じて調節するのが有利である。こうした運転を行うことでカラムC1の頂部で酢酸純度が90%以上、さらには99%以上である画分F1を得ることができる。
【0024】
本発明反応は反応装置中および蒸留塔中に導入された少なくとも一種の重合禁止剤の存在下で実行される。
【0025】
重合禁止剤の例としては当業者に周知の化合物、特にハイドロキノン、ハイドロキノン・モノメチルエーテル、フェノチアジン、ジ (tert−ブチル)−パラ−クレゾール(BHT)、2,4-ジメチル-6-(tert-ブチル)フェノール(Topanol A)、パラ−フェニレンジアミン、ジ(tert-ブチル)カテコール(BHT)、4-OH-TEMPO(4-ヒドロキシ−2,2,6,6-テトラメチル−1-ピペリジンオキシル)またはTEMPO誘導体を使用でき、これらは単独または混合物として反応混合物に対して100〜5000ppmの比率で使用される。
【0026】
本発明の好ましい実施例では反応は触媒なしで実行される。しかし、触媒の存在下で反応を実行することができ、例えば欧州特許第EP 196 520号公報およびドイツ特許第DE 102006029320号公報に記載のポリマー支持体に担持された遊離型スルホン酸のような触媒または米国特許公開第US 2002/0161260号明細書に記載の触媒を使用できる。変換を完全には行わない本発明では触媒の使用は必須ではない。
【0027】
反応全体にわたって空気または酸素を8%に減らした空気をバブリングすることができる。
【0028】
反応の終わりに得られる粗生成物は一般にポリマーを全く含まない透明で、反応で生じる酢酸は除去されている。
【0029】
本発明方法の段階(b)では反応物が所望の変換度に達した時に上記の粗反応生成物が中間貯蔵タンクS1へ移される。本発明の特定実施例では、反応は連続的に実行され、粗反応生成物は運転条件、特に反応温度および酢酸を回収するカラムC1の還流比に合わせて連続的に抜き出される。
【0030】
中間貯蔵タンクS1は残った反応物と副産物、特に1モルのA(M)Aと1モルのAc2Oとの反応によって形成される混合無水物の回収に適した蒸留塔C2へ連続的に供給するために用いられる。蒸留塔C2は理論段数が10以上、好ましくは理論段数が15以上の分離効率を有するのが好ましい。カラムのパッキングは従来のパッキング、ランダムまたは組み立てた充填材または2つのタイプのパッキングの混合物にすることができる。カラムの加熱は強制循環加熱で行うことができる。
【0031】
蒸留塔C2への供給流速は設備およびカラムの寸法に応じて広範囲で変えることができる。
【0032】
蒸留塔C2の頂部では未変換反応物と副産物とから成る画分F2+F3が連続的に回収される。これらは反応装置R1へ連続的に直接再循環されるか、貯蔵および一種以上の重合禁止剤で安定化した後に再循環される(段階d)。驚くことに、従来法の対応方法では重合前の貯蔵時間が相対的に短いにもかかわらず、貯蔵時の画分F2+F3の安定性は優れていることが確認された。これは反応装置中で再循環できない画分F2が破壊されることを意味する。これは経済的なロスと成る。これに対して本発明方法ではこれはなく、出発材料が節減できることが観測された。
【0033】
蒸留塔C2の底部では濾過を必要としない無水(メタ)アクリル酸Plが97%以上の純度、すなわち従来方法で一般的に得られる純度(約90%〜94%)より高い純度で回収される。
【0034】
本発明方法で得られたA(M)A2Oは合成の反応物として直接使用でき、例えばジメチルアミノプロピルアミンとの反応によってジメチルアミノプロパノール(メタ)アクリレートを合成できる。
【0035】
本発明の特定実施例は蒸留塔C2の底部から回収されたA(M)A2Oを滞流時間が短い装置、例えば、存在する可能性のある重質副産物および重合防止剤を除去するために、[図]2に装置C3で表したような薄膜蒸発器を使用して精製する追加の段階(e)をさらに有する。こうして精製されたA(M)A2Oは従来方法で達成可能な純度より著しく高い少なくとも99%の純度を有する。
以下、本発明の実施例を図面を参照して説明するが、方法はが下記実施例に限定されるものではない。
【実施例】
【0036】
以下に記載の百分比は重量百分比を表す。また、以下では下記の略号を使用する:
AMA2O: 無水メタアクリル酸
AMA: メタアクリル酸
AC20: 無水酢酸
ACOH: 酢酸
混合無水物: H2C=C(CH3)C(O)OC(O)CH3
Topanol A: 2、4-ジメチル-6-(tert-ブチル)フェノール
BHT: 2、6-ジ(tert-ブチル)−パラ−クレゾール
【0037】
実施例1(比較例)
頂部に凝縮器と還流ヘッドとを有する理論段数15の蒸留塔Clが載置された1リットル容の機械撹拌(4−枚プロペラ混合機)式のジャケット付きガラス製反応装置Rl中に、302gのAC20と、576gのAMAと、0.64gのTopanol Aと、0.19gのBHTとを入れた。合成時間全体にわたって反応媒体を減酸素空気のバブリングを維持した。反応中、反応装置の温度を90℃〜95℃に維持し、運転圧力を150mmHg〜50mmHgに徐々に下げた。
反応終了時すなわち10時間後に、AMA2Oを70%含む478gの粗反応生成物が得られる。この段階中375gのACOHが生じ、それは形成と同時に蒸留によって除去した。次いで、カラム頂部の圧力を15mmHgまで下げ、温度を102℃まで上げて、軽質物の蒸留(画分F2:AC20の除去、画分F3:AMAおよび混合無水物の除去)を行う。得られたF2およびF3の量は100gである。これらの画分を合せ、720ppmのTopanol Aで安定して次回の反応へ一度で再循環した。画分F4の蒸留によってAMA2O(300g)が得られる。画分F4の平均組成はAMA2Oが95.5%、重質化合物が2.5%、AMAが1%、混合無水物が0.7%である。蒸留終了時に反応装置はきわめて汚れていた。
【0038】
実施例2(比較例)
実施例1と同じ反応は実行したが、画分F1、F2およびF3の回収後にボトムとして反応装置RlからAMA2O(300g)が得られ、反応装置を排出させ、ポリマーを除去するための加圧濾過を行った。平均組成はAMA2Oが94%、重産物が4%、AMAが1%、混合無水物が0.7%である。
【0039】
実施例3(本発明)
反応で使用した反応装置と、反応物の供給および操作条件は実施例1に記載のものである。6時間反応させた後に676gの粗反応生成物(組成:60%のAMA2O、10%のMAA、15%のAC20、11%の混合無水物、1%の重質物+安定化剤)を得た。それを冷却し、中間貯蔵タンクS1へ送った。輸送は容易である。中間貯蔵タンクから80グラム/時の流量でカラムC2へ供給した。反応装置の汚れは観測されなかった。
【0040】
上記カラムC2は直径が30mmで、Multiknit型充填材を収容しており、理論段数は20である。加熱は熱サイホンで行ない、パレットポンプによって減圧した。上記80グラム/時の流量の粗生成物を分けて、塔底から48.8 g/時で取り出し、軽質物である残りは塔頂から取り出す。塔頂生成物はタンクに貯蔵し、次の反応べリサイクルした。上記凝縮器および還流はAMA2O中に5%のTopanol A溶液を入れて安定した。減酸素空気をリボイラに注入した。運転圧力はカラム頂部で15mmHg、塔底温度は91℃である。得られた塔低生成物中のAMA2Oの純度は97.5%である。
比較例1および2と、実施例3で得られたAMA2Oとジメチルアミノプロピルアミンとからジメチルアミノプロピルアミドを合成した。本発明の実施例3で得られた品質のAMA2Oを用いた場合に純度がより良いという結果が得られた。
【特許請求の範囲】
【請求項1】
(メタ)アクリル酸と無水酢酸とを少なくとも一種の重合禁止剤の存在下でトランス無水化して無水(メタ)アクリル酸を製造する方法において、
下記(a)〜(d)の段階から成ることを特徴とする方法:
(a)反応物が部分的に変換するまで、蒸留塔C1が載った反応装置R1中で反応を実行し、
(b)段階(a)で得られた粗反応物を中間貯蔵タンクS1中に送り、そこから第2の蒸留塔C2中に連続的に供給し、
(c)第2の蒸留塔C2の底部から無水(メタ)アクリル酸を回収し、頂部から基本的に未変換反応物と副産物とから成る画分を回収し、
(d)第2の蒸留塔C2の頂部画分を反応装置R1にバッチまたは連続的に再循環する。
【請求項2】
段階(a)で得られる粗反応生成物中の無水(メタ)アクリル酸の含有量が75%以下、好ましくは50%〜70%である請求項1に記載の方法。
【請求項3】
段階(a)をバッチで行う請求項1または2に記載の方法。
【請求項4】
出発材料を初期に入れ、次いで反応中に反応物を連続的またはバッチで添加して段階(a)を実行する請求項1〜3のいずれか一項に記載の方法。
【請求項5】
段階(a)で粗反応生成物の排出を連続的に実行する請求項1または2に記載の方法。
【請求項6】
段階(a)の反応を触媒なしに実行する請求項1〜5のいずれか一項に記載の方法。
【請求項7】
段階(e))から回収したA(M)A2Oを滞在時間が短い装置を使用しての精製する請求項1〜6のいずれか一項に記載の方法。
【請求項1】
(メタ)アクリル酸と無水酢酸とを少なくとも一種の重合禁止剤の存在下でトランス無水化して無水(メタ)アクリル酸を製造する方法において、
下記(a)〜(d)の段階から成ることを特徴とする方法:
(a)反応物が部分的に変換するまで、蒸留塔C1が載った反応装置R1中で反応を実行し、
(b)段階(a)で得られた粗反応物を中間貯蔵タンクS1中に送り、そこから第2の蒸留塔C2中に連続的に供給し、
(c)第2の蒸留塔C2の底部から無水(メタ)アクリル酸を回収し、頂部から基本的に未変換反応物と副産物とから成る画分を回収し、
(d)第2の蒸留塔C2の頂部画分を反応装置R1にバッチまたは連続的に再循環する。
【請求項2】
段階(a)で得られる粗反応生成物中の無水(メタ)アクリル酸の含有量が75%以下、好ましくは50%〜70%である請求項1に記載の方法。
【請求項3】
段階(a)をバッチで行う請求項1または2に記載の方法。
【請求項4】
出発材料を初期に入れ、次いで反応中に反応物を連続的またはバッチで添加して段階(a)を実行する請求項1〜3のいずれか一項に記載の方法。
【請求項5】
段階(a)で粗反応生成物の排出を連続的に実行する請求項1または2に記載の方法。
【請求項6】
段階(a)の反応を触媒なしに実行する請求項1〜5のいずれか一項に記載の方法。
【請求項7】
段階(e))から回収したA(M)A2Oを滞在時間が短い装置を使用しての精製する請求項1〜6のいずれか一項に記載の方法。
【図1】


【図2】
【図2】
【公表番号】特表2011−511048(P2011−511048A)
【公表日】平成23年4月7日(2011.4.7)
【国際特許分類】
【出願番号】特願2010−545529(P2010−545529)
【出願日】平成21年1月27日(2009.1.27)
【国際出願番号】PCT/FR2009/050113
【国際公開番号】WO2009/098422
【国際公開日】平成21年8月13日(2009.8.13)
【出願人】(505005522)アルケマ フランス (335)
【Fターム(参考)】
【公表日】平成23年4月7日(2011.4.7)
【国際特許分類】
【出願日】平成21年1月27日(2009.1.27)
【国際出願番号】PCT/FR2009/050113
【国際公開番号】WO2009/098422
【国際公開日】平成21年8月13日(2009.8.13)
【出願人】(505005522)アルケマ フランス (335)
【Fターム(参考)】
[ Back to top ]