
無間質条件下におけるヒト胚性幹細胞からの機能的巨核球および血小板の大規模生成
本発明は、巨核球及び血小板を生成する方法を提供する。種々の実施形態では、方法は、巨核球及び血小板へと分化させるために、ヒト胚性幹細胞由来の血管芽細胞を無血清及び無間質条件下で使用することを伴う。この系では、hESCは、胚様体形成及び血管芽細胞分化を介して巨核球に向けられる。血小板輸血を必要とする対象体を治療する方法が更に提供される。本発明の他の実施形態は、血小板を生成する方法を提供し、その方法は:巨核球(MK)を提供すること;およびMKを培養して血小板に分化させることを含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、機能的巨核球および血小板の作製、ならびにそれらの使用に関する。
【背景技術】
【0002】
本明細書におけるすべての刊行物は、個々の刊行物または特許出願が、参照により本明細書に組み込まれることが具体的かつ各々明確に示されている場合と同程度にて、参照により本明細書に組み込まれる。以下の記述は、本発明の理解に有用であり得る情報を含む。それは、本明細書で提供されるいずれの情報についても、それが請求される本発明の先行技術であるかもしくはそれに関連するものであること、または明確にもしくは暗に参照されるいずれの刊行物についても、それが先行技術であること、を承認するものではない。
【0003】
血管損傷が起こると、血小板は、損傷した血管に迅速に接着し、血栓の形成をもたらす複雑な事象のカスケードを引き起こす。ここ数十年の間、血小板輸血に対する需要は増加し続けている51(非特許文献1)。血小板を保存できるのはわずかに1週間未満であり、このことが、ドナーに依存するプログラムを継続的に困難なものとしている。血小板の供給が不足すると、特に複数の輸血を必要とする患者において、命にかかわる可能性のある結果となり得る。繰り返し輸血を行うこともまた、免疫媒介宿主反応に繋がる抵抗性応答を引き起こし得るものであり、費用のかかる患者適合が必要となり得る52;53(非特許文献2;3)。患者に適合した血小板を生体外にて生成することができれば、このような臨床シナリオにおいて大きな利点が提供されることになる。
【0004】
血小板の供給が限られることは、特殊な/稀な血液型を有する輸血依存患者、特に、同種免疫された患者、および癌または白血病の患者で、これらで多く見られる血小板同種免疫を発生させた患者において、命にかかわる可能性のある結果となり得る。輸血されたヒト血小板の半減期は4〜5日間であるため、臨床上、血小板の輸血を頻繁に行う必要がある。さらに、献血プログラムからの血小板は、種々の病原体による汚染のリスクが常に存在する。血小板は凍結保存することができないため、血小板を生体外で生成することができれば、臨床セッティングにおける血小板交換療法にとって大きな進歩となる。ここ10年以上にわたって、骨髄(BM)、臍帯血(CB)、または末梢血(PB)からのヒト造血幹細胞(HSC、CD34+)が、巨核球(MK)および血小板の生成について研究されてきた。サイトカイン、成長因子、および/または間質フィーダー細胞と組み合わせることにより、機能的血小板のHSCからの作製は大きな成功を収めてきた1;2(非特許文献4;5).しかし、それでもHSCはドナーからのものであり、現行の培養条件下での増殖能は限られており、このことは、大規模作製および将来的な臨床適用を妨げる可能性が高い。
【0005】
ヒト胚性幹細胞(hESC)は、生体外にて無限に繁殖および増殖することができ、ヒトの治療法に無尽蔵でドナーを必要としない細胞源を提供する可能性を有する。hESCの造血細胞への分化は、この十年の間に生体外にて広く研究されてきた。指向させたhESCの血球分化は、生体外にて、2つの異なる種類の培養系により成功している。このうちの一方は、血清含有培地中でのhESCと間質フィーダー細胞との共培養を採用している3;4(非特許文献6;7)。2つ目の種類の手順では、血清有り/無しでのサイトカインの存在下における、超低細胞結合プレート(ultra−low cell binding plates)での懸濁培養条件を採用しており5‐7(非特許文献8−10);そのエンドポイントはEBの形成である。造血前駆体、ならびに赤血球、骨髄、マクロファージ、巨核球、およびリンパ球系列を代表する成熟した機能的な後代が、上記の両方の分化hESC培養系において識別された3‐6;8‐14(非特許文献6−9;11−17)。これまでの研究ではまた、血清の存在下にて間質細胞と共に共培養することにより、hESCから巨核球/血小板も生成された15;16(非特許文献18;19)。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Reems JA, Pineault N, and Sun S. In vitro megakaryocyte production and platelet biogenesis: state of the art. Transfus. Med.Rev. 2010; 24: 33-43.
【非特許文献2】Chockalingam P, Sacher RA. Management of patients’ refractory to platelet transfusion. J.Infus.Nurs. 2007; 30 : 220-225.
【非特許文献3】Hod E, Schwartz J. Platelet transfusion refractoriness. Br.J.Haematol. 2008; 142: 348-360.
【非特許文献4】Guerriero R, Mattia G, Testa U et al. Stromal cell-derived factor 1alpha increases polyploidization of megakaryocytes generated by human hematopoietic progenitor cells. Blood 2001;97:2587-2595.
【非特許文献5】Matsunaga T, Tanaka I, Kobune M et al. Ex vivo large-scale generation of human platelets from cord blood CD34+ cells. Stem Cells 2006;24:2877-2887.
【非特許文献6】Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc.Natl.Acad.Sci.U.S.A. 2001;98.:10716-10721.
【非特許文献7】Lu S-J, Li F, Vida L, Honig GR. CD34+CD38- hematopoietic precursors derived from human embryonic stem cells exhibit an embryonic gene expression pattern. Blood 2004;103:4134-4141.
【非特許文献8】Chadwick K, Wang L, Li L et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003 2003;102:906-915.
【非特許文献9】Chang KH, Nelson AM, Cao H et al. Definitive-like erythroid cells derived from human embryonic stem cells coexpress high levels of embryonic and fetal globins with little or no adult globin. Blood 2006;108:1515-1523.
【非特許文献10】Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Exp.Hematol. 2004;32:1000-1009.
【非特許文献11】Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005;105:617-626.
【非特許文献12】Wang L, Menendez P, Shojaei F et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005;201:1603-1614.
【非特許文献13】Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J.Immunol. 2005;175:5095- 5103.
【非特許文献14】Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood 2005;106:860-870.
【非特許文献15】Qiu C, Hanson E, Olivier E et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Exp.Hematol. 2005;33:1450-1458.
【非特許文献16】Zhan X, Dravid G, Ye Z et al. Functional antigen-presenting leucocytes derived from human embryonic stem cells in vitro. Lancet 2004;364:163-171.
【非特許文献17】Ledran MH, Krassowska A, Armstrong L et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008;3:85-98.
【非特許文献18】Gaur M, Kamata T, Wang S et al. Megakaryocytes derived from human embryonic stem cells: a genetically tractable system to study megakaryocytopoiesis and integrin function. J.Thromb.Haemost. 2006;4:436-442.
【非特許文献19】Takayama N, Nishikii H, Usui J et al. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008;111:5298-5306.
【発明の概要】
【発明が解決しようとする課題】
【0007】
hESC由来の血小板は、効率的および大規模に生成することができれば、輸血医療の目的のための可能性を有する。より重要なことには、血小板は、核を持たず、最小限の遺伝物質しか含有せず、輸血前に放射線照射することで、未分化hESCなどの不純物としての細胞のいずれをも効果的に除去することができる。従って、安全性は問題とならないはずである。しかし、上記の研究における巨核球/血小板の収率は低かった15;16。hESCを、均質な巨核球集団、およびそれに続く機能的血小板へ、効率的および制御された形で分化させることが、本技術分野において依然として求められている。
【課題を解決するための手段】
【0008】
以下の実施形態およびその態様は、例示および説明のためであることが意図される組成物および方法と合わせて記載および説明され、範囲を限定するものではない。
【0009】
本発明は、巨核球を生成する方法を提供し、その方法は:血管芽細胞を提供すること;および血管芽細胞を培養して巨核球(MK)に分化させることを含む。特定の実施形態では、血管芽細胞は、多能性幹細胞から生体外で分化させてよい。特定の実施形態では、多能性幹細胞は、ヒト胚性幹細胞(hESC)であってよい。特定の実施形態では、多能性幹細胞は、誘導多能性幹細胞(iPSC)であってよい。特定の実施形態では、iPSCは、ヒト由来の体細胞から誘導されてよい。特定の実施形態では、体細胞は、成人組織からのものであってよい。特定の実施形態では、体細胞は、胎児(仔)組織からのものであってよい。特定の実施形態では、多能性幹細胞の分化は、多能性幹細胞を解離させること、ならびに、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で多能性幹細胞を培養して胚様体(EB)を形成させることを含んでよい。特定の実施形態では、培地は、BMP‐4およびVEGFを含んでよく、多能性幹細胞の培養は、約48時間行ってEBを形成させてよい。特定の実施形態では、BMP‐4の濃度は、50ng/mlであってよく、VEGFは、50ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。
【0010】
特定の実施形態では、形成されたEBは、化学的および/または機械的に解離してよく、培地の少なくとも一部分を、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて、血管芽細胞を生成してよい。特定の実施形態では、bFGFの濃度は、20ng/mlであってよく、TPOは、50〜100ng/mlであってよく、Flt3リガンドは、50ng/mlであってよく、およびSCFは、50ng/mlであってよい。特定の実施形態では、培地は、IL6、エストラジオール、ビタミンB3、1つ以上の細胞外マトリックスタンパク質、またはこれらの組み合わせをさらに含む。特定の実施形態では、解離されたEBは、芽細胞コロニー増殖培地中にて約3から4日間培養して、血管芽細胞を生成してよい。
【0011】
本発明の他の実施形態は、巨核球を生成する方法を提供し、その方法は:血管芽細胞を提供すること;および血管芽細胞を培養して巨核球(MK)に分化させることを含む。特定の実施形態では、血管芽細胞のMKへの分化は、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて、血管芽細胞を約2から8日間培養することを含んでよい。特定の実施形態では、TPOの濃度は、50ng/mlであってよく、SCFは、20ng/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。特定の実施形態では、この方法は、2から3日ごとに、培地の少なくとも一部分を、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて血管芽細胞をMKへ分化させることをさらに含んでよい。
【0012】
本発明はまた、血小板を生成する方法も提供し、その方法は:巨核球(MK)を提供すること;およびMKを培養して血小板に分化させることを含む。特定の実施形態では、MKは、血管芽細胞から分化させてよい。特定の実施形態では、血管芽細胞のMKへの分化は、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて、血管芽細胞を約2から8日間培養することを含んでよい。特定の実施形態では、TPOの濃度は、50ng/mlであってよく、SCFは、20ng/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。特定の実施形態では、この方法は、2から3日ごとに、培地の少なくとも一部分を、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて血管芽細胞をMKへ分化させることをさらに含んでよい。
【0013】
特定の実施形態では、血管芽細胞は、多能性幹細胞から生体外で分化させてよい。特定の実施形態では、多能性幹細胞は、ヒト胚性幹細胞(hESC)であってよい。特定の実施形態では、多能性幹細胞は、誘導多能性幹細胞(iPSC)であってよい。特定の実施形態では、iPSCは、ヒト由来の体細胞から誘導されてよい。特定の実施形態では、体細胞は、成人組織からのものであってよい。特定の実施形態では、体細胞は、胎児(仔)組織からのものであってよい。特定の実施形態では、多能性幹細胞の分化は、多能性幹細胞を解離させること、ならびに、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で多能性幹細胞を培養して胚様体(EB)を形成させることを含んでよい。特定の実施形態では、培地は、BMP‐4およびVEGFを含んでよく、多能性幹細胞の培養は、約48時間行ってEBを形成させてよい。特定の実施形態では、BMP‐4の濃度は、50ng/mlであってよく、VEGFは、50ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。
【0014】
特定の実施形態では、形成されたEBは、化学的および/または機械的に解離してよく、培地の少なくとも一部分を、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて、血管芽細胞を生成してよい。特定の実施形態では、bFGFの濃度は、20ng/mlであってよく、TPOは、50〜100ng/mlであってよく、Flt3リガンドは、50ng/mlであってよく、およびSCFは、50ng/mlであってよい。特定の実施形態では、培地は、IL6、エストラジオール、ビタミンB3、1つ以上の細胞外マトリックスタンパク質、またはこれらの組み合わせをさらに含んでよい。特定の実施形態では、解離されたEBは、芽細胞コロニー増殖培地中にて約3から4日間培養して、血管芽細胞を生成してよい。
【0015】
本発明の他の実施形態は、血小板を生成する方法を提供し、その方法は:巨核球(MK)を提供すること;およびMKを培養して血小板に分化させることを含む。
【0016】
特定の実施形態では、MKを再懸濁させ、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて少なくとも4日間培養して、血小板へ分化させてよい。特定の実施形態では、TPOの濃度は、100ng/mlであってよく、SCFは、50ng/mlであってよく、ヘパリンナトリウムは、25ユニット/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、この方法は、MKを少なくとも4日間培養した後に、GM6001またはIL3を添加することをさらに含んでよい。特定の実施形態では、この方法は、少なくとも2日間ごとに、培地の少なくとも一部分を、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えることをさらに含んでよい。特定の実施形態では、培地は、IMDM培地であってよい。
【0017】
特定の実施形態では、MKは、解離し、ならびにTPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中、有糸分裂停止させたフィーダー細胞層上にて少なくとも4日間培養して血小板へ分化させてよい。特定の実施形態では、フィーダー細胞層は、OP9細胞を含んでいてよい。特定の実施形態では、フィーダー細胞層は、C3H 10T1/2細胞を含んでいてよい。特定の実施形態では、この方法は、MKを少なくとも4日間培養した後に、GM6001またはIL3を添加することをさらに含んでよい。特定の実施形態では、この方法は、少なくとも2日間ごとに、培地の少なくとも一部分を、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えることをさらに含んでよい。特定の実施形態では、培地は、IMDM培地であってよい。
【0018】
本発明はまた、本発明の方法のいずれかによって生成された大量の巨核球も提供する。
【0019】
本発明はまた、本発明の方法のいずれかによって生成された大量の血小板も提供する。特定の実施形態では、血小板は、対象に血小板を投与した際の対象におけるHLA同種免疫原性応答に寄与しない溶液中に保存することができる。特定の実施形態では、血小板は、アピラーゼおよび/またはEDTAの存在下では活性化することができない。
【0020】
本発明はまた、細胞分化の修飾因子のためのスクリーニング方法も提供し、その方法は:大量の巨核球(MK)を提供すること;MKを試験化合物と接触させること;MKと試験化合物との接触からの機能的効果が存在するかまたは存在しないかを判定することを含み;ここで、機能的効果が存在する場合、それは試験化合物が、細胞分化を調節する巨核球形成、血小板新生、および/または造血因子である可能性があることを示しており、機能的効果が存在しない場合、それは試験化合物が、細胞分化を調節する巨核球形成、血小板新生、および/または造血因子ではない可能性があることを示している。本発明はまた、血小板輸血を必要とする対象を治療する方法も提供し、その方法は:大量の血管芽細胞を提供すること;血管芽細胞を培養して巨核球(MK)へ分化させること;MKを生体外で培養して血小板へ分化させること;生体外で分化させた大量の血小板を提供すること;および血小板輸血を必要とする対象へ、生体外で分化させたその量の血小板を投与し、それによって血小板輸血を必要とする対象を治療することを含む。特定の実施形態では、生体外で分化させた血小板は、対象に適合させて、免疫媒介宿主反応を低減または除去することができる。
【0021】
本発明のその他の特徴および利点は、本発明の実施形態の種々の特徴を例として説明する添付の図面と合わせて、以下の詳細な説明から明らかとなるであろう。
【0022】
代表的な実施形態を、参照図面で示す。本明細書で開示される実施形態および図面は、限定のためではなく、説明のためのものとして見なされるべきであることを意図している。
【図面の簡単な説明】
【0023】
【図1−1】図1は、hESC由来の芽細胞が、本発明の種々の実施形態に従って巨核球前駆細胞を生成することを示す。A.は、血清および間質のない条件下にてhESCから巨核球を生成するフローチャートである。前述のように、hESCは、懸濁培養し、EBを発生させた。EBは、第4日目に回収し、トリプシンで単一細胞へ解離し、その後メチルセルロースを主体とする半固体血管芽細胞培養を行った。芽細胞は、第6日目から第8日目の培養物から回収してMK培養に用い、そしてこれは、続く4から6日間の間に懸濁培養にて成熟巨核球を生成した。B.は、第6日目の芽細胞コロニー(位相差、左)および芽細胞(ギムザ染色、右)の代表的なイメージである。C.は、HUES3およびH1 hESC由来BCについてのCFU‐MKコロニーアッセイを示す棒グラフである。D.は、H1 hESC由来BCから生成されたCD41染色CFU‐MKコロニーを示す顕微鏡イメージである。挿入図は、巨核球からの細胞プロセスを示す。E.は、3種類の異なるhESC系統:HUES3、MA01、およびMA09からの芽細胞培養物におけるCD41a+細胞のパーセントの違いを示すFACS分析である。
【図1−2】図1は、hESC由来の芽細胞が、本発明の種々の実施形態に従って巨核球前駆細胞を生成することを示す。A.は、血清および間質のない条件下にてhESCから巨核球を生成するフローチャートである。前述のように、hESCは、懸濁培養し、EBを発生させた。EBは、第4日目に回収し、トリプシンで単一細胞へ解離し、その後メチルセルロースを主体とする半固体血管芽細胞培養を行った。芽細胞は、第6日目から第8日目の培養物から回収してMK培養に用い、そしてこれは、続く4から6日間の間に懸濁培養にて成熟巨核球を生成した。B.は、第6日目の芽細胞コロニー(位相差、左)および芽細胞(ギムザ染色、右)の代表的なイメージである。C.は、HUES3およびH1 hESC由来BCについてのCFU‐MKコロニーアッセイを示す棒グラフである。D.は、H1 hESC由来BCから生成されたCD41染色CFU‐MKコロニーを示す顕微鏡イメージである。挿入図は、巨核球からの細胞プロセスを示す。E.は、3種類の異なるhESC系統:HUES3、MA01、およびMA09からの芽細胞培養物におけるCD41a+細胞のパーセントの違いを示すFACS分析である。
【図2−1】図2は、芽細胞培養物からの二分化能前駆細胞が、本発明の種々の実施形態に従って赤血球および巨核球系列の両方を発生させることを示す。A.は、CD41aおよびCD235aマーカーの発現で定められる別々の集団(左)およびソートされたCD41a+CD235a+細胞(右)を示す、第6日目のHUES3由来芽細胞のFACS分析である。B.は、CD41a+CD235a−またはCD41a+CD235a+細胞のいずれか由来のCFU‐MKコロニーのイメージである。棒グラフは、CFU‐MKコロニーアッセイの定量化を示す。C.は、CD41a−CD235a+またはCD41a+CD235a+細胞のいずれか由来のCFU‐Eコロニーのイメージである。棒グラフは、CFU‐Eコロニーアッセイの定量化を示す。
【図2−2】図2は、芽細胞培養物からの二分化能前駆細胞が、本発明の種々の実施形態に従って赤血球および巨核球系列の両方を発生させることを示す。A.は、CD41aおよびCD235aマーカーの発現で定められる別々の集団(左)およびソートされたCD41a+CD235a+細胞(右)を示す、第6日目のHUES3由来芽細胞のFACS分析である。B.は、CD41a+CD235a−またはCD41a+CD235a+細胞のいずれか由来のCFU‐MKコロニーのイメージである。棒グラフは、CFU‐MKコロニーアッセイの定量化を示す。C.は、CD41a−CD235a+またはCD41a+CD235a+細胞のいずれか由来のCFU‐Eコロニーのイメージである。棒グラフは、CFU‐Eコロニーアッセイの定量化を示す。
【図3】図3は、本発明の種々の実施形態に従う無間質条件下でのhESC由来芽細胞からの指向された巨核球分化を示す。A.は、TPO、SCF、およびその他のサイトカイン/因子の巨核球分化および増殖に対する効果である。B.は、6日間培養の間のCD41a+巨核球の増殖を示す経時実験である。C.は、CD41a、CD42b、およびCD235a系列マーカーの発現レベルの変化を示す、第6日目の芽細胞培養物および第4日目のMK培養物のFACS分析である。MK培養物中にて、CD235a+細胞が消滅した一方、生存細胞の90%超がCD41a+となった。第6日目の芽細胞培養物中では、少数のCD41a+細胞がCD42bに対して陽性であった(左下パネル)。MK培養物中のCD41a+細胞の大部分も、第4日目までに高レベルのCD42bを発現し、このことは巨核球の成熟を示している(右下パネル)。
【図4−1】図4は、本発明の種々の実施形態に従って生体外で生成された巨核球の特性評価を示す。A.は、6日間のMK培養の間における細胞サイズの増加(上パネル)および細胞サイズ分布の変化(下パネル)を示す経時実験である。細胞サイズ(直径)は、ギムザ染色したサイトスピンスライドを用いて測定した。B.は、倍数体巨核球を示すギムザ染色イメージである。C.は、第6日目のMK培養物からのCD41a+巨核球内における、CD42bおよびCD42a巨核球/血小板特異的糖タンパク質の両方の発現を示すFACSドットプロットである。D.は、第4日目のMK培養物からの倍数体巨核球における、CD41染色およびvWF染色を示す免疫蛍光イメージである。E.は、生体外MK生成の種々のステージにおける、赤血球新生/巨核球形成を制御する転写因子の発現レベルを示すRT‐PCRの結果である。全RNAを、ES、第4日目のEB、BC(第6日目から第8日目)、および第4日目のMKから、RNA easy mini‐prep kit(キアジェン(Qiagen))を用いて単離した。2ugの全RNAを、iScript kit(バイオラッド(Biorad))によるcDNA合成に用いた。各サンプルについて、投入cDNAの量に対するコントロールとしてGAPDH RT‐PCRを行った。F.は、第4日目のMK培養物において胞体突起(proplatelet)形成巨核球を示す代表的な位相差イメージである。
【図4−2】図4は、本発明の種々の実施形態に従って生体外で生成された巨核球の特性評価を示す。A.は、6日間のMK培養の間における細胞サイズの増加(上パネル)および細胞サイズ分布の変化(下パネル)を示す経時実験である。細胞サイズ(直径)は、ギムザ染色したサイトスピンスライドを用いて測定した。B.は、倍数体巨核球を示すギムザ染色イメージである。C.は、第6日目のMK培養物からのCD41a+巨核球内における、CD42bおよびCD42a巨核球/血小板特異的糖タンパク質の両方の発現を示すFACSドットプロットである。D.は、第4日目のMK培養物からの倍数体巨核球における、CD41染色およびvWF染色を示す免疫蛍光イメージである。E.は、生体外MK生成の種々のステージにおける、赤血球新生/巨核球形成を制御する転写因子の発現レベルを示すRT‐PCRの結果である。全RNAを、ES、第4日目のEB、BC(第6日目から第8日目)、および第4日目のMKから、RNA easy mini‐prep kit(キアジェン(Qiagen))を用いて単離した。2ugの全RNAを、iScript kit(バイオラッド(Biorad))によるcDNA合成に用いた。各サンプルについて、投入cDNAの量に対するコントロールとしてGAPDH RT‐PCRを行った。F.は、第4日目のMK培養物において胞体突起(proplatelet)形成巨核球を示す代表的な位相差イメージである。
【図5】図5は、本発明の種々の実施形態に従う、hiPSCおよびhESCの血管芽細胞/芽細胞および造血細胞への分化を示す。A.では、hESC(i、×100)から得られた血管芽細胞/芽細胞(ii、×400)を、CD71(×1000)、CXCR4(×1000)、およびTpo受容体(×1000)で染色した。B.では、hiPSC(i、×100)から得られた血管芽細胞/芽細胞(ii、×400)を、CD71(×1000)、CXCR4(×1000)、およびTpo受容体(×1000)で染色した。C.は、再播種の14日後におけるhiPSC‐芽細胞から発生した複数種類の造血コロニー形成単位(CFU)であり、CFU‐E、CFU‐GM、およびCFU‐Mkについては×100、CFU‐Gについては×40である。(D)では、hiPSCから得られた血管芽細胞/芽細胞(i、×200)は、内皮細胞へ分化し、;hiPSC由来内皮細胞は、マトリゲル上で血管様ネットワークを形成し(ii、×40)、vWF(赤色)およびCD31(iii、緑色、×400)、さらにはLDL取り込み能(iv、赤色 ×400)を有するVE‐カドヘリン(iv、緑色)を発現した。
【図6】図6は、本発明の種々の実施形態に従う、機能的ES由来血小板(ES‐PLT)の生成を示す。A.は、OP9共培養から得られたES‐PLTの前方散乱(FSC)および側方散乱(SSC)特性を示すドットプロットである(左上)。ヒト血液の血小板に基づいてゲーティングを設定した。右上パネルは、ES‐PLTでのCD41a(y軸)およびCD42b(x軸)の発現を示す。休止ES‐PLT(中央パネル)と比較すると、トロンビン(1u/ml)処理ES‐PLTは、PAC1結合の増加を示した。平均蛍光強度は、トロンビン処理ES‐PLTにておよそ2倍に増加した。B.は、臨床グレードhESCであるMA01およびMA09から得られたES‐PLTも、トロンビン処理に応答してPAC1結合を示している。
【図7−1】図7は、本発明の種々の実施形態に従って、ES‐PLTが固定化フィブリノゲンと結合すること、および固定化フィブリノゲンと結合したES‐PLTが微小凝集体を形成することを示す。A.は、血液血小板(左パネル)およびES‐PLT(右パネル)が、フィブリノゲンでコーティングしたスライドグラス上にて伸展可能であることを示す伸展アッセイ(Spreading assay)である。RGDSの添加によって伸展が阻止されることから、このプロセスは、インテグリン受容体シグナル伝達に依存している。ADP(+ADP)またはトロンビン(+T)の添加により、フィブリノゲン単独(Con)と比較して、フィブリノゲン上での伸展プロセスが増強される。インテグリンに依存するF‐アクチン線維の再構築および糸状仮足の形成が、CD41a(緑色)およびファロイジン(F‐アクチン、赤色)の蛍光染色によって示される。B.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識血液血小板(上パネル)またはES‐PLT(下パネル)を、トロンビン(0.5u/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差および蛍光イメージより、ヒト血液血小板コントロールと同等である標識ES‐PLTの微小凝集体への関与が示される。C.は、ES‐PLT中でのHLAの発現を示すFACS分析のヒストグラムである。血液血小板をコントロールとして用いた(左)。
【図7−2】図7は、本発明の種々の実施形態に従って、ES‐PLTが固定化フィブリノゲンと結合すること、および固定化フィブリノゲンと結合したES‐PLTが微小凝集体を形成することを示す。A.は、血液血小板(左パネル)およびES‐PLT(右パネル)が、フィブリノゲンでコーティングしたスライドグラス上にて伸展可能であることを示す伸展アッセイ(Spreading assay)である。RGDSの添加によって伸展が阻止されることから、このプロセスは、インテグリン受容体シグナル伝達に依存している。ADP(+ADP)またはトロンビン(+T)の添加により、フィブリノゲン単独(Con)と比較して、フィブリノゲン上での伸展プロセスが増強される。インテグリンに依存するF‐アクチン線維の再構築および糸状仮足の形成が、CD41a(緑色)およびファロイジン(F‐アクチン、赤色)の蛍光染色によって示される。B.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識血液血小板(上パネル)またはES‐PLT(下パネル)を、トロンビン(0.5u/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差および蛍光イメージより、ヒト血液血小板コントロールと同等である標識ES‐PLTの微小凝集体への関与が示される。C.は、ES‐PLT中でのHLAの発現を示すFACS分析のヒストグラムである。血液血小板をコントロールとして用いた(左)。
【図8−1】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図8−2】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図8−3】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図9−1】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図9−2】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図9−3】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図10−1】図10は、本発明の種々の実施形態に従う、生体マウスのレーザー誘発細動脈損傷部位で発生するマウス血小板血栓へのhESC‐PLTの組み込みを示す。カルセインAM標識ヒト血液血小板またはESC由来血小板、50〜100μl(5〜10×105血小板)を、レーザー誘発血管損傷の直後に大腿動脈カニューレを通して注入した。発生するマウス血小板血栓を、Dylight 649標識抗CD42(0.05μg/g体重)の注入によってモニタリングした。2〜3つの血栓の生成後、標識血小板を200μl中2×106のヒト血小板に対して20μgのReoProで前処理し、同一のマウスの血管損傷後に注入した。さらに2〜3つの血栓が生成した。データは、血管損傷後3分間に収集した。A.は、血管損傷後の3つの時間点(0、1/2Tmax、およびTmax)での代表的な蛍光イメージを示す。白色四角内の領域の拡大イメージを下部に示す。スケールバー=10μm。BおよびC.では、微小血管へ循環する標識ヒト血小板の数(B)および血管損傷部位で発生するマウス血小板血栓中に組み込まれた数(C)を、血管損傷後3分間にわたってカウントした。データは、平均±S.E.Mで表す(n=5〜8血栓、3体のマウス)。スチューデントT検定、コントロールに対して*P<0.05および**P<0.01。
【図10−2】図10は、本発明の種々の実施形態に従う、生体マウスのレーザー誘発細動脈損傷部位で発生するマウス血小板血栓へのhESC‐PLTの組み込みを示す。カルセインAM標識ヒト血液血小板またはESC由来血小板、50〜100μl(5〜10×105血小板)を、レーザー誘発血管損傷の直後に大腿動脈カニューレを通して注入した。発生するマウス血小板血栓を、Dylight 649標識抗CD42(0.05μg/g体重)の注入によってモニタリングした。2〜3つの血栓の生成後、標識血小板を200μl中2×106のヒト血小板に対して20μgのReoProで前処理し、同一のマウスの血管損傷後に注入した。さらに2〜3つの血栓が生成した。データは、血管損傷後3分間に収集した。A.は、血管損傷後の3つの時間点(0、1/2Tmax、およびTmax)での代表的な蛍光イメージを示す。白色四角内の領域の拡大イメージを下部に示す。スケールバー=10μm。BおよびC.では、微小血管へ循環する標識ヒト血小板の数(B)および血管損傷部位で発生するマウス血小板血栓中に組み込まれた数(C)を、血管損傷後3分間にわたってカウントした。データは、平均±S.E.Mで表す(n=5〜8血栓、3体のマウス)。スチューデントT検定、コントロールに対して*P<0.05および**P<0.01。
【図11】図11は、本発明の種々の実施形態に従って、フィーダーおよび血清のない条件下で生成されるhESC‐血小板の伸展アッセイを示す。マイクロタイターチャンバースライドを100μg/mLのフィブリノゲン(上パネル)または30μg/mLのvWF(中央および下パネル)でコーティングした。hESC‐血小板を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した。
【図12】図12は、本発明の種々の実施形態に従う、血液血小板に対するPAC‐1結合アッセイを示す。血液血小板は、トロンビン処理(1U/ml)あり(右)またはなし(左)で処理し、次にFACS分析によってPAC‐1結合を実施した。
【図13−1】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【図13−2】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【図13−3】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【発明を実施するための形態】
【0024】
本明細書で引用されるすべての参考文献は、その全内容が完全に記載されているかのように、参照により本明細書に組み込まれる。それ以外の定義がなされない限りにおいて、本明細書で用いられる技術的および科学的用語は、本発明が属する技術分野の当業者によって一般的に理解されるものと同一の意味を有する。Singleton et al., Dictionary of Microbiology and Molecular Biology 3rd ed., J. Wiley & Sons (New York, NY 2001);March, Advanced Organic Chemistry Reactions, Mechanisms and Structure 5th ed., J. Wiley & Sons (New York, NY 2001);および、Sambrook and Russel, Molecular Cloning: A Laboratory Manual 3rd ed., Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY 2001)は、本出願に用いられる用語の多くに対する一般的なガイドを当業者に提供するものである。
【0025】
当業者であれば、本発明の実践に用いることが可能である本明細書で述べるものと類似または同等である多くの方法および物質を識別する。実際、本発明は、記載される方法および物質に限定されるものではまったくない。本発明の目的のために、以下の用語を次のように定義する。
【0026】
本明細書およびそれに続く特許請求の範囲を通して用いる場合、「1つの(a)」、「1つの(an)」、および「その(the)」は、文脈から明らかにそうでないことが示されていない限りにおいて、複数の言及を含む。また、本明細書で用いる場合、「中で(in)」の意味は、文脈から明らかにそうでないことが示されていない限りにおいて、「中で(in)」および「上で(on)」を含む。
【0027】
「胚性幹細胞」(ES細胞)の用語は、本技術分野で用いられるように本明細書において用いられる。この用語は、細胞系統として段階的に継代培養されたものを含む、ヒト胚盤胞または桑実胚の内部細胞塊から得られる細胞を含む。ES細胞は、卵細胞と精子との受精から、さらにはDNA、核移植、単為生殖を用いて、またはHLA領域においてホモ接合性を有するES細胞を生成させる手段によって得られるものであってよい。ES細胞はまた、精子と卵細胞との融合、核移植、単為生殖、雄性生殖、またはクロマチンの再プログラム化およびそれに続く細胞作製のための再プログラム化されたクロマチンの細胞膜への組み込みによって発生された接合体、卵割球、もしくは胚盤胞期哺乳類の胚から得られる細胞でもある。胚性幹細胞は、その作製源またはそれを作製するために用いられる特定の方法に関わらず、(i)3つすべての胚葉の細胞へ分化する能力、(ii)少なくともOct4およびアルカリホスファターゼの発現、ならびに(iii)免疫不全動物へ移植された場合にテラトーマを発生させる能力、に基づいて識別することができる。
【0028】
本明細書で用いる場合、「多能性幹細胞」の用語は、多能性幹細胞が得られる方法に関わらず、胚性幹細胞、胚由来幹細胞、および誘導多能性幹細胞を含む。多能性幹細胞は、機能的に:(a)免疫不全(SCID)マウスに移植された場合にテラトーマを誘発する能力を有する;(b)3つすべての胚葉の細胞型へ分化する能力を有する(例:外胚葉、中胚葉、および内胚葉細胞型に分化することができる);ならびに(c)胚性幹細胞の1つ以上のマーカーを発現する(例:Oct4、アルカリホスファターゼ、SSEA‐3表面抗原、SSEA‐4表面抗原、nanog、TRA‐1‐60、TRA‐1‐81、SOX2、REX1などを発現する)、幹細胞として定義される。代表的な多能性幹細胞は、例えば本技術分野で公知の方法を用いて生成することができる。代表的な多能性幹細胞としては、胚盤胞期胚のICMから得られる胚性幹細胞、さらには卵割期または桑実期胚の1つ以上の卵割球から得られる胚性幹細胞(所望に応じて、胚の残り部分を破壊することなく行ってよい)が挙げられる。そのような胚性幹細胞は、受精によって、または体細胞核移植(SCNT)、単為生殖、および雄性生殖を含む無性生殖的手段によって作製される胚性物質から生成してよい。さらなる代表的な多能性幹細胞としては、因子の組み合わせ(以降本明細書にて再プログラム化因子と称する)を発現することによる体細胞の再プログラム化によって生成される誘導多能性幹細胞(iPSC)が挙げられる。iPSCは、胎児(仔)、出生後、新生児(仔)、若齢、または成体の体細胞を用いて生成してよい。
【0029】
特定の実施形態において、体細胞を再プログラム化して多能性幹細胞とするために用いてよい因子としては、例えば、Oct4(Oct3/4と称される場合もある)、Sox2、c‐Myc、およびKlf4の組み合わせが挙げられる。他の実施形態において、体細胞を再プログラム化して多能性幹細胞とするために用いてよい因子としては、例えば、Oct4、Sox2、Nanog、およびLin28の組み合わせが挙げられる。特定の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも2つの再プログラム化因子が体細胞中で発現される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも3つの再プログラム化因子が体細胞中で発現される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも4つの再プログラム化因子が体細胞中で発現される。他の実施形態では、さらなる再プログラム化因子が識別され、それが単独で、または1つ以上の既知の再プログラム化因子と組み合わせて用いられて、体細胞が多能性幹細胞へと再プログラム化される。誘導多能性幹細胞は、機能的に明らかにされており、様々な方法(組み込みベクター、非組み込みベクター、化学的手段など)のいずれを用いて再プログラム化された細胞も含む。
【0030】
誘導多能性幹細胞は、体細胞における再プログラム化因子のタンパク質形質導入によって作製することができる。特定の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも2つの再プログラム化タンパク質が体細胞内へ形質導入される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも3つの再プログラム化タンパク質が体細胞内へ形質導入される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも4つの再プログラム化タンパク質が体細胞内へ形質導入される。
【0031】
多能性幹細胞は、いかなる種からのものであってもよい。胚性幹細胞は、例えば、マウス、非ヒト霊長類の複数の種、およびヒトで得ることに成功しており、胚性幹様細胞(embryonic stem−like cells)は、数多くのさらなる種から生成されている。従って、当業者であれば、胚性幹細胞および胚由来幹細胞を、これらに限定されないが、ヒト、非ヒト霊長類、げっ歯類(マウス、ラット)、有蹄類(ウシ、ヒツジなど)、イヌ(飼育用および野生のイヌ)、ネコ類(ライオン、トラ、チーターなど、飼育用および野生のネコ類)、ウサギ、ハムスター、アレチネズミ、リス、モルモット、ヤギ、ゾウ、パンダ(ジャイアントパンダを含む)、ブタ、アライグマ、ウマ、シマウマ、海洋哺乳類(イルカ、クジラなど)などを含むいずれの種からも生成することができる。特定の実施形態では、種は、絶滅危惧種である。特定の実施形態では、種は、現在絶滅した種である。
【0032】
同様に、iPSCもいかなる種からのものであってもよい。これらのiPSCは、マウスおよびヒト細胞(hiPSC)を用いての生成に成功している。さらに、iPSCは、胚、胎児(胎仔)、新生児(仔)、および成体組織を用いての生成に成功している。従って、iPSCは、いかなる種からのドナー細胞を用いても容易に生成することができる。従って、iPSCは、これらに限定されないが、ヒト、非ヒト霊長類、げっ歯類(マウス、ラット)、有蹄類(ウシ、ヒツジなど)、イヌ(飼育用および野生のイヌ)、ネコ類(ライオン、トラ、チーターなど、飼育用および野生のネコ類)、ウサギ、ハムスター、ヤギ、ゾウ、パンダ(ジャイアントパンダを含む)、ブタ、アライグマ、ウマ、シマウマ、海洋哺乳類(イルカ、クジラなど)などを含むいずれの種からも生成することができる。特定の実施形態では、種は、絶滅危惧種である。特定の実施形態では、種は、現在絶滅した種である。
【0033】
誘導多能性幹細胞は、出発点として、実質的にいかなる細胞発生段階のいかなる体細胞を用いて生成してもよい。例えば、細胞は、胚、胎児(仔)、新生児(仔)、若齢、または成体ドナーからのものであってよい。用いてよい代表的な体細胞としては、皮膚サンプルもしくは生検によって得られる皮膚線維芽細胞などの線維芽細胞、滑膜組織からの滑膜細胞、包皮細胞、頬細胞、または肺線維芽細胞が挙げられる。皮膚および頬は、容易に利用可能であり、簡単に入手可能である適切な細胞源を提供するが、実質的にいかなる細胞を用いてもよい。特定の実施形態では、体細胞は線維芽細胞ではない。
【0034】
「血管芽細胞」および「血管コロニー形成細胞(hemangio−colony forming cell)」の用語は、本出願全体を通して交換可能に用いられる。これらの細胞は、数多くの構造的および機能的特徴を有する。これらの細胞の特徴の中で、宿主へ投与された場合に骨髄へ生着する能力がある。これらの細胞は、これらに限定されないが、1つ以上のマーカーの発現(RNAまたはタンパク質)、または発現(RNAまたはタンパク質)がないこと、を含む数多くの構造的および機能的特性に基づいて説明することができる。血管コロニー形成細胞は、分化して、少なくとも造血細胞型または内皮細胞型を発生させる能力を有する。好ましくは、血管コロニー形成細胞は、二分化能であり、分化して、少なくとも造血細胞型および内皮細胞型を発生させる能力を有する。従って、本発明の血管コロニー形成細胞は、少なくとも単分化能であり、好ましくは二分化能である。しかし、加えて、血管コロニー形成細胞は、より高い度合いの発生能を有していてよく、特定の実施形態では、分化して、その他の系列の細胞型を発生させてよい。特定の実施形態では、血管コロニー形成細胞は、分化して、心臓細胞(例えば、心筋細胞)および/または平滑筋細胞などのその他の中胚葉誘導体を発生させる能力を有する。
【0035】
「非生着血管芽細胞(non−engrafting hemangioblasts)」および「非生着血管細胞(non−engrafting hemangio cells)」の用語は、本出願全体を通して、血管コロニー形成細胞の特徴の一部を共有する細胞の集団を意味するために用いられる。しかし、非生着血管細胞は、免疫不全宿主に投与された場合に骨髄へ生着しないという点で、区別することができる。この違いにも関わらず、非生着血管細胞は、血管コロニー形成細胞の機能的または構造的特徴および特性の1もしくは2つ以上(2、3、4、5、6、7、8、9、10)を共有し得る。例えば、特定の実施形態では、非生着血管細胞は、互いに緩やかに接着している。他の実施形態では、非生着血管細胞は、以下のタンパク質:CD34、KDR、CD133、CD31、の1もしくは2つ以上(2、3、4)を発現しない。理論に束縛されるものではないが、非生着血管細胞は、血管コロニー形成細胞よりもある程度特殊化された独特の幹細胞集団を提供し得るが、それでも、一定範囲の造血細胞型を生成する能力を有する。
【0036】
血小板の供給が限られることは、輸血依存患者にとって命にかかわる可能性のある結果となり得る。hESCは、生体外にて無限に繁殖させることができ、ヒトの治療法にとっての無尽蔵でドナーを必要としない血小板源とすることができる。適合した、または不適合性が低減されたhESC系統のバンクを作り出すことができることは、免疫抑制薬および/または免疫調節プロトコルの必要性を低減または除去することができる可能性を有する。誘導多能性幹細胞(iPSC)の出現により41;42;43;44、このような技術を、血小板同種免疫が発生している輸血依存患者のための患者特異的血小板の作製に適応させることが可能であり得る。
【0037】
ドナーから収集される血小板は、保存期間が非常に限られており、患者の予防的輸血のための需要は高まってきている。ドナーに依存する臍帯血または骨髄CD34+ヒト造血幹細胞とは対照的に、ヒト胚性幹細胞(hESC)は、制御された条件下にて血小板を生体外で連続的に作製するための有望な別の選択肢としての作製源であり得る。発明者らは、血清および間質のない条件下にてhESCから巨核球(MK)を生成する新規な系を開発した。この系では、hESCは、胚様体形成および血管芽細胞分化を通してhESCを巨核球へと指向される。CD41aおよびCD235aマーカーの両方を発現する一過性の二分化能細胞集団が、血管芽細胞培養後に識別された。これらの細胞は、FACSソーティングおよびCFUアッセイで示されるように、MKおよび赤血球細胞の両方を生成する能力を有する。無血清懸濁培養におけるTPO、SCF、およびその他のサイトカインの存在下にて、hESCからMKへ最大で100倍までの増殖を14〜15日間で達成することができる。この系は、hESCからMKを生成する強力な生体外法である。OP9細胞上に播種されると、hESC由来MKは、血小板様粒子(ES‐PLT)を生成することができる。このようなES‐PLTは、トロンビン刺激に対して応答性であり、微小凝集体形成に関与することができる。
【0038】
血清および間質のない条件下にてhESCから機能的巨核球を生成する効率的な方法について述べる。本系では、hESCを、血管芽細胞(本明細書にて以降芽細胞(BC)とも称する)を中間体として用いて巨核球分化へと指向させたものであり17、1×106細胞のhESCから最大1×108の巨核球が生成され、これは、Takayama et al.16による最も最近報告された方法よりもおよそ10〜20倍高い効率である。さらなる精製を行うことなく、懸濁培養物からの生存細胞の>90%がCD41a+であり、これらの細胞の大多数はCD42aおよびCD42bも発現している。このような生体外で得られたMK細胞は、細胞質顆粒中のvWFのギムザ染色および免疫蛍光染色で示されるように、核内分裂を起こして成熟した倍数体MKと成ることができる。重要なことには、MK培養の後期において胞体突起形成細胞が終始観察されており、このことは、本系で生成されたMKが、無フィーダー条件下にて最終分化を起こすことができることを示している。これらの結果はまた、OP9間質細胞上に播種された場合、これらの巨核球が、血液血小板の機能的特性を共有する血小板様粒子を生成したことも示す。
【0039】
本明細書では、制御された条件下、hESCを作製源細胞として用いる生体外での巨核球の大量作製に適応可能である効率的な系について述べる。この細胞は、CD41a,CD42a,およびCD42bを発現し、核内分裂を起こして、成熟した倍数体MKを形成した。さらに成熟すると、それらは、トロンビン刺激によって活性化可能であり、フィブリノゲンおよびフォンウィルブランド因子(vWF)の表面上で伸展することができる血小板を生成した。このhESC由来血小板はまた、微小凝集体を形成し、正常なヒト血液血小板と同等な形でクロットの形成および退縮を促進した。重要なことには、これらの結果はまた、このような生体外で得られた巨核球が、その血液中対応物と機能的に同等である血小板を生成する能力を有していることも示した。このようなES‐PLTは、その接着および伸展する能力、ならびに正常血液血小板と共に微小凝集体を形成する能力を示すことによって、血小板アゴニストに対して応答性を有する。間質細胞や手選プロセス(hand−picking process)が巨核球生成に関与しないことから、本プラットフォームは、無間質血小板生成の条件の確立も可能とする。これは、最近発表された3Dバイオリアクター系30を含む生体外血小板生成のための無間質条件が他所にて記述されていることから、技術的に可能である。エストラジオール、ビタミンB3、および細胞外マトリックスタンパク質を含む因子が血小板生成を向上させることは、既に報告されている31‐33。これらの因子による、間質細胞なしでの巨核球成熟および血小板生成の刺激について、本系で試験される。
【0040】
無血清および無間質条件下で巨核球が作製されることにより、十分に定義された条件下での巨核球形成および血小板新生の制御に不可欠である因子のスクリーニングが可能となる。このような今後の研究で識別される因子は、将来的な臨床適用に寄与する可能性がある。この分野での進歩はまた、系列拘束(lineage commitment)、増殖、および成熟を含む巨核球形成の種々の側面を制御する細胞および分子メカニズムに関する見識も恐らくは提供することになるであろう。芽細胞培養物中に二分化能CD41a+CD235a+集団を発明者らが識別したことは、赤血球対巨核球細胞の系列特定化(lineage specifications)に関するメカニズムの研究を確実に促進させるであろう。最近、Klimchenko et al.は、OP9間質細胞上でのヒトES細胞の造血分化からの、CD41aおよびCD235aの両方を共発現する類似の巨核球/赤血球二分化能細胞を報告した34。これらの細胞は、赤血球新生対巨核球形成の分岐点である可能性が高い。この集団についての詳細な研究は、造血の過程でのこの重要なプロセスの制御に関与する事象に光を当てる可能性が高い。
【0041】
この系は、hESCからの巨核球分化の段階的な誘導を一体化するものである。工程内制御のさらなる最適化および確立を行うことで、臨床適用のための本系の一貫性および効率性を改善することができる。現段階では、巨核球成熟を制御する基本的な細胞または細胞外メカニズムは、完全には明らかにされていない。MK培養物中においてこれらの細胞の大部分が依然として二倍体であることから、生体外で得られた巨核球の最終分化を促進するためには、倍数体化および細胞質成熟を促進するその他の因子を識別して本系に含める必要があり得る。例えば、系において、ROCKキナーゼ阻害剤は、初期段階で巨核球の核内分裂を誘発することができる。しかし、この効果は、分化する巨核球の細胞および核成熟を調整した結果というよりは、染色体分配および細胞質分裂の人工的なブロッキングに起因する可能性が高い。生体外巨核球収率、最終分化状態、および下流での明らかな条件下における機能的血小板の作製について最良の結果を達成するために、増殖、核内分裂、および細胞質成熟の間のバランスを得ることが重要である。
【0042】
これらの実績は、生体外で得られた血小板が、アゴニスト刺激によって誘発されるインテグリン受容体リモデリングおよび血小板伸展に対して応答性であることを示した。さらに、生体外で得られた血小板は、血漿血小板と一緒に作用して、凝集体を形成することができる。このような生体外で得られたヒト血小板は、生体内でも機能することができる。ヒトES細胞由来血小板に対して発明者らが実施したFACS特性評価および3つの機能性試験(PAC1結合アッセイ、伸展アッセイ、および微小凝集体アッセイ)は、生体外で作製された血小板が、正常血液血小板の形態的および機能的特性を共有していることを示している。Nichii et al.による最近の報告によると、生体外で得られたマウスES細胞由来血小板の生体内機能が試験され、有望な結果が得られた35。
【0043】
生体外研究では、生物内での血小板血栓の形成および広がりの過程で発生する無数の血行動態的事象を模倣することはできない。新しい生体内イメージング技術が利用可能となったことにより、複雑な生体内系において血管損傷後に発生する血小板依存性の血栓プロセスを直接調べ、定量化する手段が提供される。高速広視野生体内顕微鏡法を用いることで、発明者らは、hESC由来血小板が、正常ヒト血液血小板に類似して、生体マウスにおけるレーザー誘発細動脈壁損傷の部位で発生するマウス血小板血栓中に組み込まれることを示した。hESC由来血小板およびコントロール血小板をReoProで前処理すると、血栓中に組み込まれる血小板の数が大きく減少しており、このことから、この結合がαIIbβ3インテグリンによって媒介されることが確認された。これらの結果は、hESC由来血小板が、生体動物の血管損傷部位において機能性であるという貴重な証拠を提供するものである。
【0044】
血小板は、血管損傷に応答して、組織に、および互いに接着する無核細胞である。このプロセスは、主として血小板インテグリンαIIbβ3によって媒介され、これは、vWFおよびフィブリノゲンなどのいくつかの接着性基質と結合して、成長する血栓中の血小板を架橋し、これをさらに活性化する36。本明細書の結果は、hESCから生成された血小板が、生体外および生体動物内の両方において、正常血液血小板と機能的に類似していることを示している。hESC‐PLTは、フィブリノゲンおよびvWFコーティングした表面上で接着および伸展する能力、ならびに生理学的アゴニストによって刺激されると凝集する能力を含む、止血に関与する重要な機能を有することが示された。重要なことには、hESCから得られた血小板が、フィブリンクロットを退縮させることができるということが初めて示されている。この結果は、一次止血での中心的な役割に加えて、hESC‐PLTが、フィブリンとの相互作用によって凝固を促進することができ、インテグリンの媒介による退縮性の挙動によって創傷治癒を促進することを示している。
【0045】
重要な科学的及び臨床的問題は、hESC由来の血小板が、複雑な生体内の設定において機能的であるか否かということである。マウスの血栓形成を研究するための多数の実験モデルが、過去十年間に確立されており、それらには、近年幾つかのグループにより使用されたレーザー損傷血栓モデルが含まれる37;38;39。レーザー誘導性血栓モデルでは、損傷後5〜30秒の速さで血小板血栓形成が開始される。従って、このモデルは、迅速に除去されたヒト血小板及びhESC−PLTが、発生中のマウス血小板血栓に取り込まれるのをリアルタイムでモニタリングすることを可能にし、取り込みには、多数のシグナル伝達経路、酵素カスケード、並びに無数の細胞成分及びタンパク質成分の相互作用が伴う。また、このモデルは、トロンビン誘導性血栓症に関連する炎症反応を忠実に表す。
【0046】
レーザー誘導性血管損傷モデルを使用した生体顕微鏡分析は、hESC−PLTが、血液血小板のように、血管損傷後にαIIbβ3インテグリンにより、発生中のマウス血小板血栓に取り込まれることを示す。マウス血小板血栓と相互作用するhESC−PLTの数は、ヒト血液血小板と相互作用するhESC−PLTの数より少なく(図9C)、これは、活性化hESC−PLTに対するPAC−1の結合が、活性化ヒト血液血小板と比較して低いことよる可能性がある(図7G及び図11)。これらの結果は、hESC−PLTが、生体内の血管損傷部位で機能的であるという明らかな根拠を提供する。
【0047】
2つの先行研究は、MKがhESCから生成されることを報告している。これらの系での収量は非常に低く(本明細書で報告された収量より100倍を超えて低い)、現行の系と異なり、血清で補完された動物性間質細胞との共培養に依存する15;16。更に、生体外で機能的だったhESC−血小板の生成を示したのは、そのうちの1つだけであるが、それらの生体内での機能性は報告されなかった16。hESC分化におけるこれらの2つの変数を排除することにより、動物性産物と接触させずに血小板を生成することが可能になる。本研究で使用された無フィーダー系は、MKを高効率で生成することが観察されたが、無フィーダー条件下で機能的血小板を生成する最終ステップは、OP9細胞共培養系ほど効率的ではない。
【0048】
従って、これらの結果は、OP9間質細胞が、hESC−MKからの機能的血小板の生成を増強することができる追加的な支援因子を提供することを示唆する。根底にある機序は、まだ研究されていない。血小板新生は、膜及び微小管が精巧に再編成され、顆粒及び細胞小器官が正確に分布される高度に複雑なプロセスである40。血小板生合成の理解が近年進歩したにもかかわらず、膜再編成、胞体突起の開始、血小板細胞小器官及び分泌性顆粒の輸送、及び血小板サイズの制御の根底にある機械論的詳細は、まだ解明されていない。無血清及び無フィーダー条件下でMKを生成する能力は、明確に規定された条件下で、系列拘束、増殖、及び成熟を含む巨核球形成の様々な側面を制御するのに重要な因子のスクリーニングを支援するはずである。
【0049】
巨核球
本発明の種々の実施形態は、無間質条件下で、ヒト胚性幹細胞及び多能性幹細胞(iPSC及びhiPSCを含む)から機能的な巨核球を生成する方法を提供する。hiPSC及びhESCを含む多能性幹細胞は、血管芽細胞/芽球細胞を中間体として使用して、巨核球分化へと向けられた。
【0050】
1つの実施形態では、本方法は、血管芽細胞(芽球細胞とも呼ばれる)を準備すること、及び血管芽細胞を巨核球へと分化させることを含む。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。別の実施形態では、血管芽細胞は、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0051】
1つの実施形態では、血管芽細胞を巨核球へと分化させることは、6日目〜8日目の血管芽細胞を精製し、TPO、SCFで補完された培地に播種して、MKへのBC分化を誘導することを含む。種々の実施形態では、濃度は、TPO(50ng/ml)及びSCF(20ng/ml)である。別の実施形態では、濃度は、TPO(100ng/ml)及びSCF(50ng/ml)である。別の実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。更なる実施形態では、MKは、CD41a、CD42a、及びCD42bを発現する。代替的な実施形態では、培地は、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。更なる実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地を、2〜3日毎に新しい培地と交換することを更に含む。別の実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを含む。代替的な実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。種々の実施形態では、約1〜5×105細胞/mlが、播種される。種々の実施形態では、Stemline II培地が使用される。
【0052】
別の実施形態では、本発明は、細胞分化の修飾因子をスクリーニングする方法であって、ある量の巨核球(MK)を準備すること、MKを試験化合物と接触させること、及びMKと試験化合物との接触に由来する機能的効果の存在又は非存在を決定することを含み、機能的効果の存在が、試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子(hematopoeitic factor)であることを示し、機能的効果の非存在が、試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子でないことを示す方法を提供する。他の実施形態では、巨核球形成、血小板新生、及び/又は造血因子は、機能的血小板の増殖、核内分裂、細胞質成熟、及び最終分化に関連する。
【0053】
血小板
本発明の他の実施形態は、ヒト胚性幹細胞及び多能性幹細胞(iPSC及びhiPSCを含む)から血小板を生成する方法を提供する。1つの実施形態では、本方法は、ヒト胚性幹細胞(hESC)を準備すること、胚様体(EB)を形成すること、血管芽細胞(芽球細胞(BC)とも呼ばれる)を生成すること、BCを巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0054】
本発明の他の実施形態は、多能性幹細胞(pluipotent stem cell)から血小板を生成する方法を提供する。1つの実施形態では、本方法は、多能性幹細胞を準備すること、胚様体(EB)を形成すること、血管芽細胞(芽球細胞(BC)とも呼ばれる)を生成すること、BCを巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。1つの実施形態では、iPSCは、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0055】
別の実施形態では、血小板を生成する方法は、血管芽細胞を準備すること、血管芽細胞を巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。血管芽細胞を巨核球へと分化させるプロセスは、上述のように実施することができる。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。1つの実施形態では、iPSCは、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0056】
種々の実施形態では、巨核球を血小板へと分化させるプロセスは、巨核球の培養を継続して、巨核球が血小板へと分化することを可能にすることを含む。種々の実施形態では、巨核球を血小板へと分化させるプロセスは、無フィーダー条件下であり、4日目〜6日目の巨核球培養物から巨核球を収集すること、及びTPO、SCF、及びヘパリンナトリウムで補完された培地に巨核球を再懸濁することを含む。ある実施形態では、TPO(100ng/ml)、SCF(50ng/ml)、及びヘパリンナトリウム(25単位/ml)の濃度である。ある実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。代替的な実施形態では、培地は、IL3、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。ある実施形態では、IL3が、4日目〜7日目の巨核球培養物に添加される。別の実施形態では、メタロプロテイナーゼ阻害剤GM6001が、4日目以降の巨核球(megakaryoyte)培養物に添加される。ある実施形態では、培地は、IMDM培地である。更なる実施形態では、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地は、2日毎に交換される。別の実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを更に含む。代替的な実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。更なる実施形態では、血小板は、4日目〜12日目の巨核球培養物から収集される。種々の実施形態では、巨核球を血小板へと分化させるプロセスは、巨核球の培養を継続して、巨核球が血小板へと分化することを可能にすることを含む。種々の実施形態では、様々な因子が、巨核球からの血小板形成の効率を向上させる。種々の実施形態では、IL6、IL3、Notch、又は他のタイプのサイトカイン、インターロイキン、成長因子、低分子、及びそれらの組み合わせは、巨核球からの血小板分化の効率を向上させる。
【0057】
他の実施形態では、巨核球を血小板へと分化させる方法は、OP9間質細胞又はC3H 10T1/2細胞と共に巨核球を共培養することを含む。種々の実施形態では、OP9間質細胞又はC3H 10T1/2細胞と共に巨核球を共培養することは、4日目〜6日目の巨核球培養物から巨核球を収集すること、及びTPO、SCF、及びヘパリンナトリウムで補完された培地に巨核球を再懸濁することを含む。ある実施形態では、TPO(100ng/ml)、SCF(50ng/ml)、及びヘパリンナトリウム(25単位/ml)の濃度である。ある実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。代替的な実施形態では、培地は、IL3、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。ある実施形態では、IL3が、4日目〜7日目の巨核球培養物に添加される。別の実施形態では、メタロプロテイナーゼ阻害剤GM6001が、4日目以降の巨核球培養物に添加される。更なる実施形態では、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地は、2日毎に交換される。別の実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを更に含む。代替的な実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。種々の実施形態では、様々な因子が、巨核球からの血小板形成の効率を向上させる。種々の実施形態では、IL6、IL3、Notch、又は他のタイプのサイトカイン、インターロイキン、成長因子、低分子、及びそれらの組み合わせは、巨核球からの血小板分化の効率を向上させる。
【0058】
更なる実施形態では、血小板は、4日目〜12日目の巨核球培養物から収集される。ある実施形態では、血小板は、密度勾配遠心分離を使用して精製される。更なる実施形態では、密度勾配遠心分離は、パーコール培地を使用する。別の実施形態では、血小板精製法は、CD41a陰性の粒子を分離する。別の実施形態では、血小板精製法は、細胞生存能及び形態学的完全性を保持する。他の実施形態では、血小板は、CD41a及びCD42bを発現する。他の実施形態では、血小板は、トロンビン刺激に応答する。別の実施形態では、血小板は、フィブリノゲン及びフォンビルブラント因子(vWF)表面に伸展する(spread)ことができる。代替的な実施形態では、血小板は、PAC−1結合及びインテグリン活性化の能力を有する。別の実施形態では、血小板は、微小凝集体を形成し、クロット形成及び退縮を促進する。別の実施形態では、血小板は、アピラーゼ及び/又はEDTAの存在下で活性化されない。
【0059】
本発明の種々の実施形態は、hESC由来の血小板を使用する方法を提供する。ある実施形態では、hESC由来の血小板は、血小板輸血で使用される。本方法は、ある量のhESC由来血小板を準備すること、及びある量のhESC由来血小板をその必要がある対象体に投与することを含んでいてもよい。種々の実施形態では、hESC由来の血小板は、患者と一致した血小板であってもよい。別の実施形態では、血小板は、iPSCに由来する。ある実施形態では、血小板は、hiPSCに由来する。他の実施形態では、血小板は、血小板を対象体に投与した際に、対象体においてHLA同種免疫原性反応に寄与しない溶液中で保管される。
【0060】
血管芽細胞
種々の実施形態では、血管芽細胞は、ヒト胚性幹細胞(hESC)を準備すること、胚様体(EB)を形成すること、及び芽球細胞(BC)を生成することを含む方法より生成することができる。代替的な実施形態では、本方法は、多能性幹細胞(iPSC及びhiPSCを含む)を準備すること、胚様体(EB)を形成すること、及び芽球細胞(BC)を生成することを含む。ある実施形態では、iPSCは、体細胞の再プログラミングに由来する。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。
【0061】
1つの実施形態では、EBを形成することは、hESCを解離すること、解離されたhESCを、BMP−4及びVEGFで補完された培地に約48時間播種すること、及びこの培地の半分を、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoを含む新しい培地と交換することを含む。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地は、2日毎に交換される。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の少なくとも一部は、2日毎に交換される。代替的な実施形態では、多能性幹細胞(iPSC及びhiPSCを含む)が、hESCの代りに使用される。
【0062】
種々の実施形態では、hESCを解離することは、トリプシンによりhESCを解離することを含む。1つの実施形態では、トリプシンの濃度は、0.05%である。種々の実施形態では、培地は、Stemline II培地である。種々の実施形態では、BMP−4及びVEGFの濃度は両方とも、50ng/mlである。種々の実施形態では、bFGF、SCF、Flt3リガンド、及びTpoの濃度は、bFGF(20ng/ml)、SCF(50ng/ml)、Flt3リガンド(50ng/ml)、及びTpo(50ng/ml)である。代替的な実施形態では、培地は、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。別の実施形態では、EBを形成することは、細胞の継続的培養を更に含む。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地は、2日毎に交換される。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の少なくとも一部は、2日毎に交換される。1つの実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の半分は、2日毎に交換される。
【0063】
1つの実施形態では、血管芽細胞を生成することは、4日目にEB培養物から胚様体(EB)を収集すること、EBを解離すること、単一細胞懸濁物を得ること、生細胞を芽球コロニー増殖培地(BGM)と混合すること、及び生細胞を播種して血管芽細胞を生成することを含む。
【0064】
種々の実施形態では、EBを解離することは、トリプシンにより解離することを含む。種々の実施形態では、単一細胞懸濁物を得ることは、22ゲージ針及び40μm細胞ストレーナーに細胞を通すこと、遠心分離により細胞を収集すること、及び細胞を培地に再懸濁することを含む。1つの実施形態では、培地は、Stemline II培地である。種々の実施形態では、生細胞を芽球コロニー増殖培地(BGM)と混合することは、1〜2×105細胞/mlをBGMと混合することを含む。更なる実施形態では、芽球コロニーは、生細胞を播種した3〜4日後に形成され、血管芽細胞を生成する。更なる実施形態では、血管芽細胞は、急速に増殖する。別の実施形態では、血管芽細胞は、CD71陽性であり、CXCR4陽性であり、TPO受容体を発現する。別の実施形態では、血管芽細胞は、CD31陰性、CD34陰性、及びKDR陰性である。
【0065】
国際出願PCT/US09/43050号及び同PCT/US09/43043号は、双方ともに、2009年5月6日に出願され、その全内容が完全に記載されているかのように参照により本明細書に組み込まれ、血管芽細胞および非生着血管芽細胞を生成する際の追加的な手引きを提供する。
【実施例】
【0066】
以下の例は、特許請求されている発明をよりよく説明するために提供されており、本発明の範囲を限定すると解釈されるべきではない。特定の物質が言及される場合、それは例示目的に過ぎず、本発明を限定することは意図されていない。当業者であれば、発明的な能力を行使せず、本発明の範囲から逸脱することなく、同等の手段又は反応物を開発することができる。
【0067】
実施例1
hESCの培養
この研究で使用したヒトESC系統(HUES3、H1、MA01、及びMA09)を、以前に記述されているように維持した18。手短に言えば、8ng/mlのbFGFで補完された完全hESC増殖培地中のマイトマイシンC処理マウス胚線維芽細胞(MEF)上で、hESCを増殖させた。0.05%トリプシン−EDTAを使用して、60〜80%コンフルエンスで、3〜4日毎にhESCを継代した。37℃、5%CO2下で細胞を維持し、培地を毎日補充した。
【0068】
実施例2
胚様体(EB)形成及び芽球細胞(BC)の生成
EB形成及びBC生成の手順は、以前に報告されているように実施した17;19。手短に述べると、70%コンフルエンスのhESCを0.05%トリプシンで解離し、BMP−4、VEGF(50ng/ml)で補完されたStemline II(Sigma社)培地を有するUltra−Lowディッシュ(Corning社、ニューヨーク州)に播種した。48時間後に、培地の半分を、同じサイトカインに加えて、bFGF 20ng/ml、SCF、Flt3リガンド(FL)、及びTpo(各50ng/ml、R&D System社)を含有する新しい培地に交換した。4日目にEBを収集し、0.05%トリプシンで解離した。単一細胞懸濁物を、22ゲージ針及び40μm細胞ストレーナーに細胞を通すことにより得、遠心分離により収集し、Stemline II培地に再懸濁した。生細胞をトリパンブルー排除法により計数し、1〜2×105個/mlを、以前に記述されているように芽球コロニー増殖培地(BGM)と混合し、Ultra−Low皿に播種した。播種した3〜4日後に芽球コロニーが観察され、その後急速に増殖した。
【0069】
実施例3
巨核球分化培養
6〜8日目の芽球培養物に由来するBCを精製し、50ng/mlのTPO、20ng/mlのSCF、及び他のサイトカインで補完されたStemline II培地に播種して(1〜5×105個/ml)、MKへのBC分化を誘導した。MK培地の半分を、2日又は3日毎に新しい培地と交換した。
【0070】
OP9間質細胞を、15%ウシ胎仔血清(Hyclone社)を有するα−MEM中で維持した。コンフルエントなOP9細胞を、共培養の前日に100ng/mlマイトマイシンCで処理した。細胞をPBSで穏やかに2回洗浄し、共培養の前にOP9培地中で一晩回復させた。OP9共培養の場合、4〜6日目のMK培養物に由来する巨核球を収集し、Takayamaらによる記載のように16、100ng/mlのTPO、50ng/mlのSCF、及び25単位/mlのヘパリンナトリウムで補完されたIMDM培地に再懸濁し、2日毎に新しくした。血小板様粒子を、4日目から12日目まで分析用に収集した。
【0071】
実施例4
FACS分析
芽球培養又は巨核球培養に由来する細胞を、FACS Caliberでフローサイトメトリー分析により日常的にモニターする。系列マーカー、CD41a、CD42a、CD42b、及びCD235aに対する蛍光色素(Flurochrome)結合抗体(BD Biosciences社)を使用して、巨核球及び赤血球系列を特徴付けた。抗体は、5%新生仔ウシ血清を有するPBSで新しく調製した(CD42a及びCD42b抗体の場合は1:100希釈、CD41a抗体の場合は1:250希釈、CD235a抗体の場合は1:2000)。典型的には、1〜2×105細胞を抗体標識に使用した。細胞を、氷上で1時間100μl抗体混合液中で染色し、その後緩衝液で2回洗浄し、1μg/mlヨウ化プロピジウムで補完された250μl緩衝液に再懸濁した。HLA−ABCの発現レベルを検出するために、血小板を、フルオレセインイソチオシアネート(FITC)結合抗ヒトHLA−ABC抗体又は対照としてのFITC結合マウス免疫グロブリンG(IgG)と共にインキュベートした。その後、フローサイトメーター(FACSCalibur、Becton Dickinson社製)を使用して試料を分析し、データは、Cellquest又はFlowjoソフトウェアを使用して分析した。細胞選別は、UMASSメディカルスクールコア施設のBD FACS Ariaシステムで実施した。選別した細胞を、1000rpmで10分間遠心分離することにより収集し、コロニー形成アッセイ用の適切な培地に再懸濁した。
【0072】
実施例5
Megacult−C及びメチルセルロースを用いたコロニー形成アッセイ
Megacult−C及びH4436メチルセルロース培地(Stem Cell Technology社製)を、CFU−MK及びCFU−Eコロニー形成アッセイに使用した。播種の10日後に、製造業者の指示に従って、CFU−MK培養物を脱水し、固定し、抗CD41抗体で染色した。Megacult−Cプロトコルに提供されている基準に従って、CFU−MKコロニーをスコア化した。CFU−Eコロニーを、12日目にスコア化した。
【0073】
実施例6
サイトスピン調製、ギムザ染色、及び免疫蛍光顕微鏡法
芽球培養物又は巨核球培養物のいずれかに由来する細胞(1〜2×104個)を、サイト遠心分離(Cytopro社製)により、ポリリシンをコーティングしたスライドにサイトスピンした。スライドは、Wright−Giemsa(Sigma社製)又は免疫蛍光染色のいずれかに使用した。抗CD41(DAKO社製、1:100)及び抗vWF(DAKO社製、1:200)抗体を、サイトスピン調製で巨核球を特定するために使用した。インキュベーションは全て室温で実施した。細胞を、無動物性ブロッカー(Vector Laboratories社製)で30分間ブロッキングした。細胞を、一次抗体と共に1時間インキュベートし、その後PBSで3回洗浄した。以下のインキュベーション及び洗浄は、暗所で実施した。その後、細胞を二次抗体(各々、1:200)と共に30分間インキュベートした。細胞を再びPBSで3回洗浄した。PBS中のヘキスト染料(1μg/ml)を使用して核DNAを5分間染色し、その後追加的な3×PBS洗浄を行った。その後、スライドを蛍光顕微鏡(オリンパスBXV)下にマウントし、検査した。蛍光画像を、QICAM Fastカメラ(Qimaging社製、カナダ)を使用することにより取り込み、QCapture Proバージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で分析した。Nikon社製顕微鏡、PAXCAMデジタルカメラ、及びPAX−itソフトウェアを使用して、位相差生細胞画像を取り込んだ。
【0074】
実施例7
血小板の調製
ヒト末梢血小板を、1U/mLアピラーゼ及び5mM EDTA(Sigma−Aldrich社製、セントルイス、ミズーリ州)と共に、血小板豊富な血漿(AllCells社製、エメリービル、カリフォルニア州)を分画遠心分離することにより単離した。血小板を洗浄し、改変タイロード緩衝液(12mM NaHCO3、138mM NaCl、5.5mMグルコース、2.9mM KCl、0.42mM NaHPO4、10mM HEPES、1mM CaCl2、1mM MgCl2)に再懸濁した。hESC由来の血小板(ES−PLT)を含有する培地を穏やかに収集し、アピラーゼ(1U/ml)及びEDTA(5mM)(Sigma−Aldrich社製、セントルイス、ミズーリ州)を添加して血小板活性化を防止し、その後細胞を300gで10分間遠心して細胞をペレットにした。上清を新しいチューブに移し、アピラーゼ(1U/ml)及びEDTA(5mM)の存在下で10分間1000gで遠心分離して、血小板を収集した。その後、血小板を1回洗浄し、改変タイロード緩衝液に再懸濁した。洗浄した血小板を、機能性アッセイの前に1〜2時間37℃でインキュベートした。
【0075】
実施例8
固定化されたフィブリノゲン上での血小板の伸展
マイクロタイターウエルを有するチャンバースライド(Nalgen Nunc社製、ロチェスター、ニューヨーク州)を、0.1M NaHCO3(pH8.3)中の100pg/mLフィブリノゲン(Sigma社製)で一晩4℃でコーティングした。洗浄したヒト末梢血小板又はhESC−血小板(1×107個/mL)を、フィブリノゲンをコーティングしたウエルに90分間37℃で付着及び伸展させた。幾つかの試料では、血小板を、負荷前に5分間、インテグリンアンタゴニスト、RGDSペプチドと共にプレインキュベートした。他の試料では、血小板を、ADP(10μM)又はトロンビン(1U/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)と混合し、フィブリノゲンをコーティングしたウエルに直ちに負荷した。PBSで洗浄した後、細胞を固定し、透過処理し、Alexa Fluor568ファロイジン(Molecular Probes社製、ユージーン、オレゴン州)、FITC結合抗ヒトCD41a抗体(Dako cytomation社製、カーピンテリア、カリフォルニア州)、及びDAPIで染色した。付着血小板を、100×/1.40オイル対物レンズのPlanApoレンズを使用して、オリンパスBX51蛍光顕微鏡(MVI社、エイボン、マサチューセッツ州)で観察した。画像を、QICAM Fastカメラ(QImaging社製、サリー、ブリティッシュコロンビア州、カナダ)を使用することにより取得し、Q Capture バージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で処理した。
【0076】
実施例9
血小板凝集
洗浄したヒト末梢血小板及びhESC−血小板を、改変タイロード緩衝液に再懸濁し、PKH67緑色蛍光細胞リンカー(10μM、Sigma社製、セントルイス、ミズーリ州)で標識した。ヒト末梢血小板(6×107個)を、450μlキュベット(Chronolog社製、ヘイバータウン、ペンシルベニア州)中で、蛍光標識したヒト末梢血小板(3×105個)又はhESC−血小板(3×105個)と混合し、その後トロンビン(0.5U/mL)を添加し、1200rpm、37℃で撹拌して血小板凝集を開始させた。対照実験では、トロンビンを添加する前に、RGDSペプチドを37℃で5分間血小板と共にプレインキュベートし、上記のように凝集アッセイを実施した。50μL緩衝液中の血小板微小凝集体を、スライドガラスに塗布し、蛍光顕微鏡で視覚化した。
【0077】
実施例10
PAC−1結合アッセイ
トロンビン刺激(1U/ml、室温で20分間のインキュベーション)をした又はしなかったES−PLTを、改変タイロード緩衝液中のAPC結合CD41a、PE結合CD42b、及びFITC結合PAC−1抗体で染色した。その後、フローサイトメーター(FACSCalibur、Becton Dickinson社製)を使用して試料を分析し、ヒト血液血小板を対照として使用して、前方散乱及び側方散乱ゲーティングを決定した。FACSデータは、Cellquest又はFlowjoソフトウェアを使用して分析した。
【0078】
実施例11
hESCに由来する芽球細胞(BC)は巨核球を生成した
本発明者らの以前の研究は、適切な条件下で、hESCに由来するBCを赤血球細胞へと生体外で効率的に分化させることができることを示した20。巨核球(MK)および赤血球は、哺乳動物造血中に共通の前駆体を共有するため、本発明者らは、hESCに由来するBCも、TPO及び他のサイトカインを有する巨核球促進条件下で巨核球に分化することができると考えた。図1Aで示されているのと類似した戦略を考案して、hESCからMKを分化させた。以前に報告されるように17;19、初期EBを、モルフォゲン及び初期造血サイトカインの組み合わせで補完された無血清培地中で培養されたhESCから生成した。その後、EBを解離し、個々の細胞を、BCの成長及び増殖用の無血清半固形芽球コロニー増殖培地(BGM)に播種した。ブドウ様芽球コロニーが、3日間の培養の始めに最初に出現し、4日目から6日目まで急速に増殖した。この段階で、BCは、サイトスピン調製では形態が比較的均質であり、直径が約15μmのサイズを有する(図1B)。これらのBCを、コロニー形成単位(CFU)−MKアッセイにより、それらが巨核球になる能力について最初に試験した。図1C及び1Dに示されているように、HUES3及びH1細胞の両方に由来する6日目のBCは、再播種後10〜12日目にCFU−MKコロニーを発生させ、これらのコロニーは、CD41に陽性染色された。幾つかのCFU−MKコロニーでは、細胞プロセスが類似しており、CD41陽性細胞で胞体突起が観察された(図1D挿入部分)。これらの結果は、hESC由来のBCが、巨核球に分化することができることを示した。同時に、それらのCFU−MK能力における有意差が、HUES3及びH1細胞系統間で観察された。HUES3に由来するBCは、同数のH1芽球細胞より、およそ3.5倍より多くのCFU−MKコロニーを生成した。
【0079】
初期BGM培地は、効率的なBC生成のためのEPOを含有しており、赤血球細胞(CD235a+)は、芽球培養の後期で優勢になる20。本発明者らの実験の結果は、EB形成の第2期(hESC播種後2日間)並びに芽球コロニー成長及び増殖物中のより高濃度のTPO(100ng/ml)及びSCF(50ng/ml)が、芽球培養の終了時のCD41a+細胞の割合を増加させたことを示した。BGM培地にIL−11(20ng/ml)を添加することにより、6〜8日目の芽球培養中のCD41a+細胞の比率が更に増加された。従って、本発明者らは、本研究において、これらの改変を芽球培養に応用して、巨核球系列拘束を向上させた。HUES3、MA01、及びMA09 hESC系統を、この改変条件を用いて試験した。3つの系統は全て、巨核球になる能力を示した。CFU−MKアッセイでなされた観察と同様に(図1C)、様々なhESC系統から生成されたBC(6〜8日目)中のCD41a+細胞の割合における有意差が、FACS分析により観察された。例えば、HUES3細胞に由来するBCの32.9+8.4%(n=13)は、MA01及びMA09細胞系統よりも有意に多かった(P<0.001、スチューデント検定)。MA01又はMA09由来の芽球細胞では、これらの細胞の15.3%+5.8%(n=5)又は17.2%+3.7%(n=5)が、それぞれCD41a+だった(図1E)。
【0080】
実施例12
二分化能芽球細胞は、赤血球及び巨核球を両方とも生じさせた
主な増殖は、芽球培養では5日目〜8日目まで生じ、幾つかの芽球コロニーは、この期間中に赤みを帯び、赤血球拘束及びヘモグロビン合成を示した。FACS分析は、巨核球マーカーCD41a及び赤血球マーカーCD235aの発現により規定されるBCに、4つの異なる細胞集団:CD41a+CD235a−、CD41a−CD235a+、CD41a+CD235a+、及びCD41a−CD235a−集団が存在したことを示した(図2A)。CD41a及びCD235a二重陽性集団は、赤血球系列及び巨核球系列の両方のマーカーを共有するため、これらの細胞が両系列の二分化能前駆体であるか否かを試験した。
【0081】
その後、6日目の芽球培養に由来する、選別されたCD41a+CD235a−、CD41a−CD235a+、及びCD41a+CD235a+細胞を、2つのコロニー形成アッセイにより、CFU−MK又はCFU−E前駆体のいずれかについて試験した。これらのコロニーアッセイのために、等数の選別された細胞を播種した。予想通り、CD41a+CD235a−及びCD41a−CD235a+は、それぞれCFU−MK及びCFU−Eコロニーを生成した。同時に、類似した数のCFU−MKコロニーが、CD41a+CD235a−及びCD41a+CD235a+細胞により生成され、同程度の数のCFU−Eが、CD41a−CD235a+及びCD41a+CD235a+細胞により発生された(図2B及びC)。従って、これらの結果は、CD41a+CD235a+細胞が、巨核球系列及び赤血球系列の両方の二分化能前駆体であることを示す。これらのCD41a+CD235a+細胞は、一時的な集団であると考えられ、巨核球分化培養で培養された直後に消失した。
【0082】
実施例13
hESC由来BCからの巨核球増殖及び成熟を指図する明確な培養条件の確立
巨核球の分化を更に指図するために、メチルセルロース半固形培養に由来する6日目〜8日目のBCを回収し、その後、無血清培地、つまりMK培養用のサイトカインで補完されたStemline IIに再懸濁した。CD41a+細胞の細胞数を測定値として使用して、懸濁培養中に増殖巨核球を決定した。それは、CD41a+(FACS)の割合と総生細胞数(トリパンブルー排除法)を乗算することにより計算した。初期試験は、高濃度TPO(50〜100μg)単独では、BCからのCD41a+巨核球の増殖を支援するのに十分でなかったことを示した。培地にSCF(20ng/ml)を添加することにより、CD41a+巨核球の収量が有意に増加した(図3A)。BCからの巨核球の高い純度及び総収量を達成するために、一群のサイトカイン及び因子も試験した。IL9及びBMP4を含むこれらの因子は、他の系において巨核球促進因子として以前に報告されていた21;22。
【0083】
図3Aに示されているように、IL9及びBMP4の添加は、TPO及びSCFのみと比較して、CD41+細胞の収量を向上させる明らかな効果を示さなかった。しかしながら、ある因子は、CD41+細胞の割合をわずかに増加させた(非表示)。IL6、IL11、VEGF、及びbFGFは全て、追加的栄養補給をしない4日間の液体培養で巨核球の収量をわずかに増加させた。しかしながら、これらの因子を一緒に混合することは、一貫した相乗効果を示さなかった(非表示)。加えて、血清又はメチルセルロースマトリックスを培養物に添加することも試みられた。これらの改変は、CD41巨核球の収量を有意には向上させなかった。注目すべきには、巨核球核内分裂及び胞体突起形成を促進することが示された23;24特定のROCKシグナル伝達阻害剤(Y27632)も試験した。4日間のMK培養中に10μMのROCK阻害剤を包含させることは、倍数体細胞の割合を劇的に増加させた(データ非表示)。しかしながら、ROCK阻害剤は、同時に4日間の培養の終了時のCD41+細胞数を低減させた。従って、ROCK阻害剤は、最終MK培養条件から除外された。
【0084】
初期試験の結果に基づき、BCからの至適化巨核球分化及び増殖を達成するために、TPO 50ng/ml、SCF 20ng/ml、及びIL11 20ng/mlをMK培地に含めた。IL11は、巨核球の増殖及び成熟の両方を増強することが以前に報告された25;26。図3Bは、液体培養の経時的実験を示す。6日間の培養期間にわたって、CD41+巨核球の細胞数が5倍増加したことが、本条件下で観察された。6日目のBCS及び4日目のMK培養物に対するFACS分析を、2つの巨核球マーカーCD41a、CD42b、及び赤血球マーカーCD235aについて実施した。MK培養では、赤血球細胞(CD235a+)は、2〜3日で急速に死滅する。それとは際立って対照的に、CD41a+巨核球は、劇的に増加した。MK培養の4日目までに、90%を超える生細胞がCD41aを発現し、これらの細胞のおよそ80%は、成熟中の巨核球に重要な機能的役割を果たす27CD42bにも陽性であった(図3C)。
【0085】
実施例14
BCに由来する巨核球の特徴付け
巨核球の分化及び成熟を示す細胞表面マーカーの変化と一致して、平均細胞サイズは、MK培養中に徐々に増加した。成熟中の巨核球は、細胞サイズがより大きく、より不均一になった(図4A)。1日目のMK培養では、約65%の細胞は、10〜20μmの直径を有していた。残り細胞は、20〜30μmだった。6日目までに、20%を超える細胞は、サイトスピン調製での直径が40ミクロンよりも大きかった。数パーセントの細胞は、MK培養で倍数体細胞の表現型を示した(図4B)。CD42b(GP1b−アルファ)に加えて、糖タンパク質CD42a(GPIX)も、MK培養に由来するCD41a+巨核球内で高度に発現された(図4C)。CD41及びvWFの免疫蛍光染色も実施して、巨核球細胞質顆粒での特徴的なvWF発現を検査した。図4Dに示されているように、成熟巨核球表現型を有する大型の倍数体細胞は、CD41に陽性染色され、細胞質におけるvWFの顆粒状蓄積が検出された。
【0086】
更に、RT−PCR分析を実施して、GATA1、GATA2、Fli1、FOG、及びNFE2(図4E)を含む、赤血球/巨核球形成の重要な転写修飾因子の発現レベルを検査した。これらの転写因子は全て、胚様体段階で強力に誘導され、6日目の芽球培養で発現持続を示した。興味深いことには、BCが巨核球分化及び成熟に向けられた場合、GATA2及びFOGのRNAレベルは、4日目のMK培養で下方制御された。しかしながら、この観察の機械論的意味は、まだ判明していない。RT−PCRの結果は、ES、EB、BC、及びMK培養中に5つの赤血球−巨核球転写因子GATA1、GATA2、Fli1、FOG1、及びNF−E2の発現を示した。
【0087】
実施例15
hESCから巨核球への増殖
細胞系統間の効率は様々であるが、試験したhESC系統は全て、無血清及び無間質培養条件下で巨核球(CD41a+と規定された)を生成することができた。hESCsからEB(増殖、1.5〜6倍)、芽球(5〜12倍)、及びMK培養(1.5〜4倍)を経由して増殖した後、この系は、ヒトES細胞から開始して、100万個から最大1億個のCD41a+巨核球を産生することが可能である。HUES3、及び2つの臨床グレードの単一卵割球由来hESC系統、MA01及びMA09の巨核球産生の能力は、表1に要約されている。
【0088】
表1.hESCからの巨核球の生成
MK培養に由来するCD41a+細胞の割合を、FACSにより決定した。hESCから巨核球への総増殖倍数を、EB、芽球、及びMK培養までの各ステップの増殖倍数を乗算することにより計算した。HuES3実験Bは、100万個のES細胞からのMK細胞の実測計数値である。
【表1】

【0089】
実施例16
hiPSCは、血管芽細胞/BC、造血及び内皮細胞に生体外で分化することが可能である
本発明者らは、Thomson再プログラム法(Oct−4、Sox−2、Nanog、及びLin28のレンチウイルス発現)により生成されたhiPSCを使用して、同様の結果を達成した。これは、細胞系統IMR90−1、Foreskin−1−1、Foreskin−4−1、及びForeskin−4−3を含む。それとは別に、Yamanaka再プログラム法(Oct−4、Sox−2、Klf4、及びc−Mycのレトロウイルス発現)により生成されたhiPSCを同様に実施した。これは、細胞系統rv−hiPS01、rv−hiPS02、rvhiPS03、及びrv−hiPS04を含む。これらのhiPSC系統は、多分化能の標準的マーカーを発現し、報告されているようにSCIDマウスへの接種後に奇形腫を形成し、この系統は全て、hESCと形態学的に区別不能だった(データ非表示)。本明細書に記載の方法を使用して、hiPSC系統は、BCを生成することが可能だった。まず、hiPSCを、BC発生用に至適化された条件下でEBに分化させ、個々のEB細胞を、芽球コロニー発生用の芽球成長/増殖培地(BGM)に播種した。全てのhiPSC系統に由来するEB細胞は、播種の6日後に芽球コロニーを発生させた。様々な効率が、様々なhiPSC系統で観察された(表2)。にもかかわらず、hESCに由来するBCで観察されたように(図5A)、hiPSCに由来するこれらのBCは、血管芽細胞マーカーCD71、CXCR−4、及びTpo受容体を発現したが(図5B)、これらのBCの大多数は、免疫細胞化学的分析により示されたように、CD31、CD34、及びKDRを発現しなかった。広範なサイトカインで補完された造血コロニー形成培地に10〜14日間再播種した後、赤血球(CFU−E)、骨髄(CFU−G及びCFU−GM)、及びマクロファージ(CFU−M、非表示)造血細胞コロニーが発生した(図5C)。また、BCは、巨核球へと分化する能力を有するCFU−MKを形成した(図5C−iv)。更に、再プログラムタンパク質Oct4、Sox2、Klf4、及びc−Mycの形質移入等の、無ウイルスベクターを使用して生成されたhiPSCは、BC発育、MK及び血小板生成について同様の能力を示す。これらの結果は、様々な送達系(レンチウイルス、レトロウイルス、形質移入)を使用して様々な組み合わせの再プログラム因子を用いて生成されたhiPSCが、無血清条件下で血管芽細胞、造血及び内皮系列に分化することが可能であることを明らかに示す。
【表2】
*播種した100,000個のEB細胞当たり、(±)は、ほとんど0個のコロニー又は時には10個未満のコロニーが得られる結果を表し、(+)は、少数のコロニー(10〜30個)が一貫して得られる結果を表し、(++)は、最大50個の芽球コロニーの中程度の効率の収量を表す。
【0090】
実施例17
hESC由来の巨核球は、OP9間質細胞上で機能的な血小板を生成する
MK培養での4日目から開始して、少数(およそ1〜2%)のhESC由来巨核球は、胞体突起様プロセスを形成することが顕微鏡で一貫して観察された(図4F)。この観察は、無間質系で生成された巨核球が、最終分化プロセスまで進行することが可能であることを示唆する。
【0091】
近年、Takayamaらは、機能的なES由来血小板(ES−PLT)を産生するための、間質細胞OP9に基づく共培養系を記述した16。本発明者らは、細胞が血小板を機能的に製作可能であることを示すために、このOP9系上の巨核球を原理の証明として試験することにした。MK培養から生成された(HUES3、MA01、及びMA09に由来した)巨核球を、原報に記載のように、TPO 100ng/ml、SCF 50ng/ml、及びヘパリン 25U/mlで補完された同じ培地中の有系分裂的に停止されたOP9細胞上に播種した。胞体突起形成細胞(PFC)及び血小板様粒子(ES−PLT)が、OP9間質層上に播種した後4日以内に出現した。ESP−LTを、6日目から12日目まで2日毎に培地から収集した。100万個のhESC由来MKから、およそ20万〜50万個のES−PLTを、この期間中に培養物から回収することができる。以前の報告より低いES−PLT産生効率は、培養条件の抜本的な変更の結果であり得る。また、OP9との共培養は、播種した巨核球の倍数性を著しくは増加させなかった。これも低い血小板収量/巨核球を説明することができる。にもかかわらず、OP9共培養物から回収されたES−PLTを、正常な血液血小板に重要な機能性について更に試験した。
【0092】
実施例18
トロンビン活性化ES−PLTは、機能的なインテグリン受容体結合活性を有する
インテグリン媒介性血小板凝集は、生体内での血栓形成に不可欠である。循環中では、血小板は、その接着性受容体を介して損傷血管壁に付着する。初期の付着は、血小板インテグリンαIIbβ3の活性化に結び付くシグナル伝達カスケードを開始させ、血小板の安定的な接着、凝集、及び血栓形成に結び付く。トロンビン等の血小板アゴニストは、インテグリンαIIbβ3のリガンド結合機能を活性化する。その主要リガンド、フィブリノゲンの結合は、インテグリンに依存する血小板の安定的な接着及び凝集を媒介する。また、αIIbβ3の活性化は、細胞外から内部へのインテグリンシグナル伝達を媒介し、血小板細胞骨格再編成および伸展に結び付く。
【0093】
OP9共培養から生成されたES−PLTを、血小板の主要アゴニストであるトロンビンによる活性化について試験した。インテグリンαIIbβ3活性化を、FACS分析により、活性化形態のインテグリンαIIbβ3受容体にのみ結合するFITC結合PAC−1モノクローナル抗体を用いた結合アッセイにより検査した。タイロード緩衝液に再懸濁されたES−PLTを、抗ヒトCD41a(インテグリンαIIb)抗体、CD42b抗体、及びPAC−1抗体の混合液と共にインキュベートした。その後、それらの表面にCD41a及びCD42b糖タンパク質を両方とも発現するES−PLTを、PAC−1結合活性についてゲート制御した。トロンビン刺激に際して、PAC−1結合は、休止対照と比較して、トロンビン処理ES−PLTで増加した。この結果は、ES−PLTが、血小板アゴニストからの刺激に応答することを示した(図6A)。ES−PLTは、2つの臨床グレードのhESC系統、MA01及びMA09を含む、3つのhESC系統に由来した。3つの系統は全て、トロンビン刺激に応答してPAC−1結合が可能なES−PLTを生じさせることができた(図6B)。
【0094】
ES−PLTにおける細胞外から内部へのインテグリンシグナル伝達の機能的特性を、スライドガラスに固定化されたフィブリノゲン上での伸展アッセイにより更に評価した。スライドを抗ヒトCD41a抗体及びDAPIで染色して、血小板を特定した。ヒト血漿血小板対照と同様に、ES−PLTは、フィブリノゲンをコーティングした表面に接着及び伸展し、機能的な血小板と同様のF−アクチンフィラメントの再編成を示した。RGDSペプチドは、血小板インテグリン受容体に対するフィブリノゲン結合を阻止し、フィブリノゲンをコーティングした表面へのES−PLTの接着及び伸展を消失させ、伸展がインテグリン依存性であることを示した。アゴニストの非存在下で、幾つかの血小板は、主として糸状仮足形成を有するフィブリノゲン上で不完全な伸展を示した。血小板アゴニストADP又はトロンビンで刺激した場合、対照血漿血小板及びES−PLTは両方とも、葉状仮足及びアクチンストレス線維形成の増強を示した(図7A)。
【0095】
実施例19
ES−PLTは、微小血栓形成に寄与することができる
アゴニスト誘導性凝集を形成するそれらの能力、血小板の重要な機能を検査するために、ES−PLTを、緑色蛍光染料で標識し、血漿由来血小板と混合した。撹拌条件下でのトロンビン刺激に応答して、血小板微小凝集体が、位相差及び蛍光顕微鏡で観察された。蛍光標識hESC−血小板は、ヒト血漿由来血小板と一緒にそれら微小凝集体に均等に組み込まれた。トロンビン誘導性血小板凝集は、インテグリンアンタゴニスト、RGDSにより阻害され、凝集がインテグリン依存性であることを示した(図7B)これらの結果は、ES−PLTが、アゴニスト誘導性微小凝集体形成において血液血小板と一緒に作用することができることを示した。要約すると、これらの結果は、培養系に由来するES−PLTが、ヒト末梢血液血小板と機能的に同等であり、これらのES−PLTは、生理学的な血小板アゴニストにより刺激することができることを示した。
【0096】
実施例20
ES−PLT上でのHLAクラスIの発現
ヒト末梢血小板は、免疫関連血小板輸血不応状態(reflactive platelet transfusion)に寄付する場合があるクラスIヒト白血球抗原(HLA−ABC)を合成及び発現する28。従って、ES−PLT上でのHLAクラスIの発現も、フローサイトメトリーにより検査した。正常な血漿由来血小板は、高レベルのHLA−ABC発現を示した。ES−PLTは、HLAクラスIの発現レベル(平均蛍光強度)が同等であることを示した。血小板におけるHLAの新規合成に加えて、可溶性HLA抗原が、血漿から吸収され得ることも報告されている29。培養条件下でhESCに由来する血小板は、他の血液細胞に由来するHLA抗原を含有する血漿を回避し、従って血小板同種異系免疫化の頻度を低減することができる。
【0097】
実施例21
hESC由来の血管芽細胞/BCからのMKの大規模生成
実験を実施して、hESC由来BCも、MKに分化するように誘導することができるか否かを決定した。TPO、SCF、及びIL−11を含有する無血清Stemline II培地中で芽球培養物を6日間分化させた後、CD41+MKの5倍の増加が得られた。また、FACS分析は、分化の初期段階中に、CD235a+細胞(赤血球細胞)が急速に減少したが、CD41a+及びCD42b+細胞(MK)は劇的に増加したことを示した。4日目までに、90%を超える細胞がCD41aを発現し、およそ80%の細胞が、CD42b陽性だった(CD42bは、MKの成熟に重要な機能的役割を果たす)27(図8A)。CD42bに加えて、糖タンパク質CD42aも、CD41a+MKの表面に高度に発現された(データ非表示)。6日目までに、20%を超える細胞が40μmのサイズよりも大きく、30%を超える細胞が、4Nを超えるDNA含有量を有する倍数体だった(図8B及び8D)。大型の倍数体細胞上のCD41及びvWFについての免疫蛍光染色は、CD41陽性膜染色を有する細胞質におけるvWFの特徴的な顆粒状蓄積を示し(図8C)、細胞が成熟MKだったことを確認した。
【0098】
試験した全てのhESC系統(WA07[H7]、HuES−3、MA01、及びMA09)は、これらの無血清及び無フィーダー培養条件下でMKを生成したが、様々な効率が観察された(表3)。8つの別々な実験では、hESCからCD41a+MK細胞への総細胞増殖は、HuES−3及びMA09 hESC系統の場合、それぞれ30〜111倍及び18〜113倍の範囲であり、MA09 hESCの6つのウエルプレート当たり(1.0×107個のhESCとほぼ等しい、表3)最大6×108個のCD41a+MKが生成された。全体的な効率は、以前に発表された方法よりもおよそ2マグニチュード(two magnitudes)高い16。
【表3】
【0099】
培養に由来するCD41+MK細胞の数は、生細胞(トリパンブルー排除法)の総数とCD41a+細胞の割合(FACS分析)を乗算することにより計算した。hESCからMKへの総増殖倍数は、全てのEB細胞又はBCがその後の培養に使用された訳ではないため、EB、BC、及びMK培養までの各ステップの増殖倍数を乗算することにより計算した。
【0100】
実施例22
hESC−MKからの機能的血小板の生成
無血清及び無フィーダーMK培養で4日目から開始して、およそ1〜2%のMKが、胞体突起様細胞プロセスを形成することが観察された(図8E)。機能的な血小板が無フィーダー条件下で生成されたか否かを検査するために、本発明者らは、FACS分析により、hESC由来血小板(hESC−PLT)上でのCD41a/CD42b発現を検査した。本発明者らの結果は、無フィーダー条件下で生成されたhESC−PLT(血液血小板対照の前方散乱及び側方散乱パターンに従ってゲート制御された)の大多数が、CD41aを発現したことを示した。しかしながら、これらのhESC−PLTの5%未満が、CD42b陽性だった。最近の研究は、メタロプロテイナーゼ阻害剤GM6001が、マウスESC由来血小板上でのCD42bの発現を著しく増加させたことを示した35。ヒトESC−PLT分化の後期中にGM6001(100μM)を補完することにより、CD41a及びCD42b二重陽性hESC−PLTが、約15%に増加した。無フィーダーで生成されたhESC−PLTの機能を、固定化されたフィブリノゲン及びvWF上のインテグリン依存性伸展アッセイにより評価した。抗ヒトCD41a抗体及びDAPI染色を使用して、血小板を特定した。無フィーダーで生成されたhESC−PLTは、フィブリノゲン及びvWFをコーティングした表面上に伸展することができ(図11)、これらのhESC−PLTが生体外で機能的であることを示した。
【0101】
OP9又はC3H 10T1/2マウス間質細胞は、生体外血小板生合成を支援することが示された。OP9間質細胞との共培養が、hESC−MKからの機能的血小板の産生を促進したか否かを試験した。HuES−3、MA01、及びMA09細胞から生成されたMKを、以前に記述されているように16、TPO、SCF、及びヘパリンで補完された同じ培地中の有系分裂的に停止されたOP9細胞に播種した。胞体突起形成細胞及びhESC−PLTが、OP9間質層への播種後4日以内に出現した。hESC−PLTを、6日目から12日目まで2日毎に培地から収集し、CD41a及びCD42b発現について分析した。無間質条件下で生成されたhESC−PLTとは対照的に、大多数のCD41a+hESC−PLTも、CD42bを発現した(図8F、右パネル)。
【0102】
無フィーダー条件下で生成されたhESC−PLTと同様に、OP9細胞共培養で産生されたhESC−PLTは、フィブリノゲンをコーティングした表面(図9B)及びvWFをコーティングした表面に接着及び伸展した。RGDSペプチドは、血小板インテグリン受容体に対するフィブリノゲン結合を阻止し、フィブリノゲンをコーティングした表面へのヒト血液血小板及びhESC−PLTの両方の接着及び伸展を消失させた(図9B)。ファロイジン染色は、ヒト血液血小板での形成と同様に(図9A)、ADP及びトロンビンで刺激されたhESC−PLTでのF−アクチンストレス線維の形成を示した(図9B)。無フィーダー条件下で又はOP9間質細胞上で生成されたhESC−PLTのサイズは、ヒト血液血小板よりもわずかに大きく、マウスES細胞を用いて生体外で生成された血小板と類似する35。
【0103】
OP9共培養でHuES−3から生成されたhESC−PLTも、血小板の主要アゴニスト、トロンビンによる活性化について試験した。フィブリノゲン模倣体としてのPAC−1モノクローナル抗体は、活性化形態のインテグリンαIIbβ3受容体にのみ結合する。トロンビン刺激時に、PAC−1結合アッセイを、OP9共培養から生成されたhESC−PLTに対して実施した。トロンビン処理hESC−PLTでのPAC−1結合は、休止対照と比較しておよそ5倍数増加した(図8G)。トロンビン刺激後のPAC−1結合の増加は、末梢血小板対照の増加よりも弱いが(hESC−PLTの24.8%対血液血小板の58.6%、図12)、Takayamaらにより以前に報告されたhESCから生成された血小板と類似している16。2つの臨床グレードhESC系統、MA01及びMA09に由来するhESC−PLTも、CD41a及びCD42b抗原を発現し(図13A)、トロンビン刺激に応答してPAC−1に結合することができた(図13B)。また、hESC−PLTは、それらの表面にHLAクラスI抗原を発現した(図13C)。
【0104】
実施例23
hESC−PLTは、微小凝集体形成及びクロット形成/退縮に寄与する
損傷血管壁への血小板接着は、血小板インテグリンαIIbβ3の活性化に結び付くシグナル伝達カスケードを開始させる。活性化インテグリンは、血小板凝集及び血栓形成を媒介する45。アゴニスト誘導性凝集を形成するそれらの能力、血小板の重要な機能を検査するために、hESC−PLTを緑色蛍光染料で標識し、ヒト血液血小板と混合した。撹拌条件下でのトロンビン刺激に応答して、hESC−PLT及び血液血小板による微小凝集体の形成が、位相差及び蛍光顕微鏡法を使用して示された(図9D)。標識hESC−PLTは、ヒト血液血小板と一緒に微小凝集体に組み込まれた。これらの微小凝集体におけるhESC−PLTの分布は、蛍光染料で標識された同数の血液血小板の分布と類似していた(図9C)。これらの結果は、hESC−PLTが、アゴニスト誘導性微小凝集体形成においてヒト血液血小板と一緒に作用することを示す。
【0105】
血管損傷部位では、活性化された血小板が、血漿フィブリノゲンから生成されたフィブリンと相互作用し、血栓形成及び血餅退縮に結び付く46。血餅形成及び退縮を促進するそれらの能力を検査するために、hESC−PLTを血小板枯渇血漿に懸濁し、トロンビンを添加してクロット形成及び退縮を誘導した。追加的な血小板の補完をしない血小板枯渇血漿中では、1時間を超えるトロンビン刺激でさえクロットは形成されなかった。対照的に、血小板枯渇血漿へのhESC−PLTの添加は、トロンビン刺激後の30分未満でクロット形成及び退縮に結び付いた(図9E、左パネル)。hESC−PLT血栓の凍結切片の免疫蛍光染色は、ヒト血液血小板で形成されたクロット切片と同様に(図9F)、フィブリン及びCD41aの両方に陽性染色されたメッシュ様ネットワークを示した(図9E、右パネル)。これらの結果は、hESC−PLTが、フィブリンクロット形成を促進し、小型の血小板栓を形成するインテグリンαIIbβ3依存性収縮能力を有することを示す。
【0106】
実施例24
hESC−PLTは、生存マウスのレーザー誘導性細動脈損傷部位で、発生中の血小板血栓に組み込まれる
hESC−PLTが生体内で機能的か否かを決定するために、本発明者らは、生存マウスでの蛍光生体顕微鏡法を実施した。精巣挙筋細動脈を、micropointレーザアブレーションにより損傷させた。以前に記述されているように47、マウス血小板血栓が急速に形成され、Tmaxは血管損傷の70〜95秒後だった。ヒト末梢血小板、5〜10×105個を損傷後に注入したところ、41.0±2.8の循環血小板が、微小循環中で検出された(図10B)。それらの中で、6.4±1.1の血小板が、血管損傷後のTmax(73秒)で血管損傷部位にて、発生中のマウス血小板血栓に組み込まれた(図10A及び10C、ビデオ1(非表示))。発生中のマウス血小板血栓へのヒト血液血小板の組み込みが、αIIbβ3インテグリンにより媒介されることを確認するために、ReoPro(ヒト血小板上のαIIbβ3インテグリンに結合し、その機能を阻害するヒト−マウスキメラモノクローナル抗体のFab断片)48を用いて、注入前にヒト血小板を前処理した。循環血小板の数は、ReoProにより有意には影響を受けなかったが(32.4±4.4、図10B)、発生中の血小板血栓に結合するヒト血小板の数は、ReoProによる処置により、血管損傷後のTmax(67秒)で1.6±0.5に有意に低減された(図10A及び10C、ビデオ2(非表示)、P<0.01)。これらの結果は、ヒト血小板が、血管損傷部位で、発生中のマウス血小板血栓に結合し、そのような結合がαIIbβ3インテグリンにより媒介されることを示す。ReoProは、血管損傷部位でのマウス血小板血栓形成に影響を及ぼさなかった。
5〜10×105個のhESC−PLTを血管損傷後に注入したところ、34.8±1.7の循環血小板が、微小循環中で検出された(図10B)。それらの中で、4.3±0.7の血小板が、損傷後のTmax(72秒)で血管損傷部位にて、成長中のマウス血小板血栓に組み込まれた(図10A及び10C、ビデオ3(非表示))。損傷部位で発生中のマウス血小板血栓に組み込まれるヒト血液血小板及びhESC−PLTの数は、統計的に有意ではなかった(P=0.28)。ReoProによるhESC−PLTの前処理は、マウス血小板血栓に組み込まれた血小板の数を、血管損傷後のTmax(99秒)で1.2±0.4に有意に低減された(図10A及びC、付録ビデオ4、P<0.05)。これらの結果は、hESC−PLTが、正常対照血液血小板のように、生体内の血管損傷部位で機能的であることを示す。
【0107】
実施例25
動物
雄野生型マウス(C57BL/6、6〜8週齢)を、Jackson Laboratory社(バーハーバー、メーン州)から購入した。イリノイ大学の施設内動物実験委員会は、全ての動物管理および実験手順を承認した。
【0108】
実施例26
hESCから血管芽細胞/BCを介したMKの生成
hESC系統(HuES−3、H7[WA07]、MA01、及びMA09)からの血管芽細胞/BC生成を、以前に報告されているように実施した17;20。6〜8日目の培養物に由来するBCを精製し、50ng/mlのTPO、20ng/mlのSCF、及び20ng/mlのIL−11(Peprotech社製)で補完されたStemline II培地に播種して(1〜5×105個/ml)、MKへのBC分化を誘導した。MK培地の半分を、2日又は3日毎に新しい培地と交換した。MKからの血小板生成は、無フィーダー条件下で、又はOP9若しくはC3H 10T1/2間質細胞との共培養で実施した。無フィーダー条件の場合、4〜6日目のMK培養物に由来するMKを収集し、記述されているように16、100ng/mlのTPO、50ng/mlのSCF、及び25単位/mlのヘパリンナトリウムで補完されたIMDM培地に再懸濁し、2日毎に新しくした。血小板を、4日目から12日目まで分析用に収集した。幾つかの実験では、GM6001(100μM)を後期分化培養物に添加した。共培養実験の場合、OP9又はC3H 10T1/2間質細胞を、15%ウシ胎仔血清(Hyclone社製)を有するα−MEM中で維持した。コンフルエントなOP9又はC3H 10T1/2細胞を、共培養の前日に100ng/mlマイトマイシンCで処理した。細胞をPBSで穏やかに2回洗浄し、共培養の前にOP9培地中で一晩回復させた。4〜6日目のMK培養物に由来するMKを収集し、上述のよう培地を有するOP9又はCH1/2間質細胞上に播種した。血小板を、4日目から12日目まで分析用に収集した。
【0109】
実施例27
FACS分析
芽球培養またはMK培養に由来する細胞を、FACSCalibur(Becton Dickinson社製)またはAccuri C6血球計算器(Accuri Cytometers社製)でフローサイトメトリー分析により日常的にモニターする。系列マーカー、CD41a、CD42a、CD42b、及びCD235aに対する蛍光色素結合抗体(BD Biosciences社)を使用して、MK及び赤血球系列を特徴付けた。抗体は、5%胎仔ウシ血清(FBS)を有するPBS緩衝液で新しく調製した(CD42a及びCD42b抗体の場合は1:100希釈、CD41a抗体の場合は1:250希釈、CD235a抗体の場合は1:2000)。典型的には、1〜2×105細胞を抗体標識に使用した。細胞を氷上で1時間100μl抗体混合液中で染色し、その後緩衝液で2回洗浄し、1μg/mlヨウ化プロピジウムで補完された250μl緩衝液に再懸濁した。HLA−ABCの発現レベルを検出するために、血小板を、フルオレセインイソチオシアネート(FITC)結合抗ヒトHLA−ABC抗体又は対照としてのFITC結合マウス免疫グロブリンG(IgG)と共にインキュベートした。その後、試料を、FACSCaliburで分析し、データを、Cellquest又はFlowjoソフトウェアを使用して分析した。細胞選別は、UMASSメディカルスクールコア施設のBD FACSAriaシステムで実施した。選別した細胞を、1000rpmで10分間遠心分離することにより収集し、コロニー形成アッセイに適切な培地に再懸濁した。倍数性分析の場合、4日目のMK培養物に由来する細胞を、70%エタノール中で2時間固定した。その後、細胞をPBS緩衝液で1回洗浄した後で、PBS緩衝液中20μg/mlのヨウ化プロピジウム(propidum iodide)(Sigma社製)、20μg/mlのRNase A(Sigma社製)で一晩4℃にて染色した。細胞内DNA含有量を、Accuri C6血球計算器で分析した。
【0110】
実施例28
サイトスピン調製、ギムザ染色、及び免疫蛍光染色
芽球培養又はMK培養のいずれかに由来する細胞(1〜2×104個)を、ポリリシンでコーティングしたスライド(Wessco)にサイトスピンした。スライドを、Wright−Giemsa(Sigma社製)又は免疫蛍光(immunofluroscent)染色のいずれかに使用した。抗CD41(DAKO社製、1:100)及び抗vWF(DAKO社製、1:200)抗体を、サイトスピン調製でMKを特定するために使用した。インキュベーションは全て室温で実施した。細胞を、無動物性ブロッカー(Vector Laboratories社製)で30分間ブロッキングし、一次抗体と共に1時間インキュベートし、その後PBSで3回洗浄した。その後のインキュベーション及び洗浄は、暗所で実施した。細胞を、二次抗体(各々、1:200)と共に30分間インキュベートし、再度PBSで3回洗浄した。PBS中のDAPI(1μg/ml)を使用して核DNAを5分間染色し、その後追加的な3×PBS洗浄を行った。その後、スライドをオリンパスBX51蛍光顕微鏡(MVI社、エーボン、マサチューセッツ州)にマウントし、検査した。蛍光画像を、QICAM Fastカメラ(Qimaging社製、サリー、ブリティッシュコロンビア州、カナダ)を使用して取り込み、Q Capture Proバージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で分析した。Nikon社製顕微鏡、PAXCAMデジタルカメラ、及びPAX−itソフトウェアを使用して、位相差生細胞画像を取り込んだ。
【0111】
実施例29
ヒト血液血小板及びhESC由来血小板の調製
ヒト血小板を、以前に記述されているように単離した49。手短に言えば、クエン酸ナトリウムで処理したヒト血液を200gで20分間遠心分離することにより、ヒト血小板豊富な血漿を調製した。上清を収集し、0.5μM PGE1及び10%クエン酸ナトリウム緩衝液の存在下で10分間700gで遠心分離した。ペレットを、0.15μM PGE1を含有するHEPES−タイロード緩衝液(12mM NaHCO3、138mM NaCl、5.5mMグルコース、2.9mM KCl、0.42mM NaHPO4、10mM HEPES、1mM CaCl2、1mM MgCl2)に再懸濁し、800gで5分間遠心分離した。ペレットを、0.1%無脂肪酸ウシ血清アルブミン、2mM CaCl2、及び1mM MgCl2を含有するRPMI1640に再懸濁した。洗浄した血小板の最終懸濁物を、1×107血小板/mlに調節した。血液試料を取得する承認は、イリノイ大学−シカゴ審査委員会から得た。インフォームドコンセントが与えられた。幾つかの実験では、ヒト血液試料を、商業的供給源(AllCells社、エメリービル、カリフォルニア州)からも取得した。hESC−PLTの場合、hESC−PLTを含有する培地を穏やかに収集し、アピラーゼ(1U/ml)及びEDTA(5mM)(Sigma−Aldrich社製、セントルイス、ミズーリ州)を添加して、血小板活性化を防止した。hESC−PLTを、上述のように濃縮及び洗浄した。洗浄した血液血小板及びhESC−PLTを、機能性アッセイの前に0.5〜2時間37℃でインキュベートした。
【0112】
実施例30
固定化されたフィブリノゲン及びvWF上での血小板の伸展
マイクロタイターウエルを有するチャンバースライド(Nalgen Nunc社製、ロチェスター、ニューヨーク州)を、0.1M NaHCO3(pH8.3)中の100μg/mLフィブリノゲン又はvWF(30g/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)で、一晩4℃でコーティングした。洗浄したヒト血液血小板又はhESC−PLT(1×107個/mL)を、フィブリノゲンでコーティングしたウエルに90分間37℃で付着及び伸展させた。幾つかの実験では、血小板を、インテグリンアンタゴニスト、RGDSペプチドと共に、負荷前に5分間プレインキュベートした。他の実験では、血小板を、ADP(20μM)又はトロンビン(1U/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)と混合し、フィブリノゲン又はvWFでコーティングしたウエルに直ちに負荷した。PBS緩衝液で洗浄した後、細胞を固定し、透過処理し、Alexa Fluor568ファロイジン(Molecular Probes社製、ユージーン、オレゴン州)、FITC結合抗ヒトCD41a抗体(Dako cytomation社製、カーピンテリア、カリフォルニア州)、及びDAPIで染色した。付着血小板を、100×/1.40オイル対物レンズのPlanApoレンズを使用して、オリンパスBX51蛍光顕微鏡で観察した。画像を、QICAM Fastカメラを使用して取得し、Q Capture バージョン5.1ソフトウェアで処理した。
【0113】
実施例31
血小板微小凝集体の形成
洗浄したヒト血液血小板及びhESC−PLTを、改変タイロード緩衝液に再懸濁し、PKH67緑色蛍光細胞リンカー(10μM、Sigma社製、セントルイス、ミズーリ州)で標識した。ヒト血液血小板(6×107個)を、450μlキュベット(Chronolog社製、ヘイバータウン、ペンシルベニア州)中で、蛍光標識したヒト血液血小板(3×105個)又はhESC−PLT(3×105個)と混合し、トロンビン(0.5U/mL)で処理し、1200rpm、37℃で撹拌して血小板凝集を開始させた。対照実験では、トロンビンを添加する前に、血小板をRGDSペプチドと共に5分間37℃でプレインキュベートし、凝集アッセイを上記のように実施した。50μL緩衝液中の血小板微小凝集体を、スライドガラスに塗布し、オリンパスBX51蛍光顕微鏡で視覚化した。
【0114】
実施例32
PAC−1結合アッセイ
トロンビン刺激(1U/ml、室温で20分間のインキュベーション)をした又はしなかったヒト血液血小板又はhESC−PLTを、改変タイロード緩衝液中のAPC結合CD41a、PE結合CD42b、及びFITC結合PAC−1抗体で染色した。その後、試料をFACSCaliburを使用して分析した。前方散乱及び側方散乱ゲーティングを、ヒト血液血小板を対照として使用して決定した。フローサイトメトリーデータは、Cellquest又はFlowjoソフトウェアを使用して分析した。
【0115】
実施例33
クロット形成及び退縮
ヒト血液血小板又はhESC−PLT(約1.5×107個/ml)を、シリコン処理したガラス管(Kimble Chase社製、バインランド、ニュージャージー州)中の50μL血小板枯渇血漿に再懸濁した。トロンビン(2U/ml)及び10mM CaCl2を細胞に添加して、クロット形成及び退縮を誘導した。クロットを37℃で1時間退縮させ、画像を撮影した。クロットを、Tissue−Tek OCT化合物及びTissue−Tek Cryomolds(Sakura Finetek社製、トランス、カリフォルニア州)に埋込み、その後ドライアイスで凍結した。10μmのクロット切片を、凍結ミクロトームMicrom HM560(Thermo Scientific社製、カラマズー、ミシガン州)を使用して製作した。
【0116】
スライドをメタノール/アセトン(1:3)で30分間固定し、PBSで洗浄し、0.1% Triton X−100、0.1M Tris、10mM EGTA、0.15 NaCl、5mM MgCl2、及び1%ウシ血清アルブミン(BSA)、pH7.5で透過処理した。5%BSAでブロッキングし洗浄した後、切片を、ウサギ抗ヒトインテグリンαIIb(クローンH−160)及びマウス抗ヒトフィブリン(クローンUC45)抗体(Santa Cruz Biotech社製、サンタクルーズ、カリフォルニア州)で免疫染色した。その後、切片を、ローダミンに結合された1:200の二次抗ウサギIgG抗体及びFITCに結合された抗マウスIgM抗体(Jackson Immunoresearch Lab社製)中で1時間インキュベートした。上述のように画像を撮影した。
【0117】
実施例34
生体顕微鏡法
精巣挙筋微小循環の広視野多チャンネル生体顕微鏡法を、以前に記述されているように実施した47;50。ケタミン(125mg/kg、Bedford Laboratories社製、ベッドフォード、オハイオ州)及びキシラジン(25mg/kg、Akorn Inc.社製、ディケーター、イリノイ州)の腹腔内注射により、雄マウスに麻酔をかけた。気管チューブを挿入し、マウスを温度制御毛布(37℃)に配置した。麻酔を維持するために、頚静脈に設置したカニューレ(cannulus)で、ケタミン及びキシラジン溶液50μlを30分毎に投与した。更に、10U/mlヘパリンを含有する生理食塩水で満たされたカニューレを、大腿動脈に設置した。陰嚢を切開した後、精巣挙筋を生体顕微鏡トレーに肱置した。筋肉調製物を、温度調節し(37℃)通気した(95%N2、5%CO2)重炭酸緩衝生理食塩水を用いて、実験の全体にわたって表面還流した。
【0118】
精巣挙筋細動脈壁を、以前に記述されているように47、Micropointレーザーシステム(Photonics Instruments社製、サウスウィンザー、コネティカット州)を使用してレーザアブレーションにより損傷した。発生中のマウス血小板血栓を、Dylight649標識抗CD42(Emfret Analytics社製、体重1g当たり0.05μg)の注入によりモニターした。1回又は2回のパルスを血管壁内側でアブレーションさせて、血小板血栓形成を開始させた。カルセインAM(Invitrogen社製)で標識されたヒト血液血小板又はhESC−PLT、50〜100μl(5〜10×105血小板)を、血管損傷の直後に大腿動脈カニューレで注入した。1匹のマウス中で複数の血栓を研究し、より初期の血栓の上流に沿って新しい血栓を形成させ、以前に生成された血栓からのあらゆる寄与を回避した。以前に注入された標識ヒト血小板が完全に除去された後で、新しい血栓を生成した。2〜3個の血栓を生成した後、ReoPro(Centocor社製)、200μl中2×106個のヒト血小板毎に20μgで前処理した標識ヒト血小板50〜100μlを、血管損傷後に同じマウスに注入した。別の2〜3個の血栓を生成した。ヒト血液血小板及びhESC−PLTを、異なるマウスで研究した。60×対物レンズを備えたオリンパスBX61WI顕微鏡を使用して、微小血管データを得た。デジタル画像は、倍増管(Video Scope International社製、ダレス、バージニア州)により高速デジタルカメラ(camara)(Hamamatsu社製C9300)で取り込んだ。蛍光画像は、Slidebook v5.0(Intelligent Imaging Innovations社製、デンバー(Denvor)、コロラド州)を使用して分析した。蛍光画像は、10〜100ミリ秒の露光時間で取り込み、明視野画像は、20ミリ秒の露光時間で取り込んだ。データは、血管壁損傷後3分間収集した。画像分析を単純化するために、各擬似カラーの強度のダイナミックレンジを2値化した。微小血管を循環し、血管損傷部位で、発生中のマウス血小板血栓に組み込まれたヒト血小板の数を、血管損傷後3分間計数した。
【0119】
実施例35
統計分析
データは、GraphPad Prism(GraphPad Software社製)を使用して、2群を比較するためにスチューデントt検定により統計的に分析した。違いは、P<0.05を有意とみなした。
【0120】
実施例36
血管芽細胞から生成された血小板の密度遠心分離を使用した血小板精製法
MK分化培養に由来する血小板は、CD41a陰性である粒子を含んでいる。これらの粒子は、MK分化培養に由来する粒子の最大50%を構成する場合があり、血小板調製物の純度を増加させることが望ましい。低粘性パーコール培地等の、生存能及び形態学的完全性(indensity)を保存する培地を使用した密度遠心分離法を使用して、血小板からCD41a陰性粒子を分離した。PAC−1結合及びインテグリン活性化の能力を無効にせずに、薄層に分離されたヒト末梢血小板を、CD41a陰性粒子から取り出した。
【0121】
本発明の種々の実施形態は、発明を実施するための形態に記述されている。これらの説明は、上記の実施形態を直接記述するが、当業者であれば、本明細書に表示及び記述されている特定の実施形態の改変及び/又は変異を企図することができることが理解される。本説明の範囲以内にあるあらゆるそのような改変又は変異も同様に本説明の範囲に含まれることが意図される。特別な注記がない限り、本明細書及び特許請求の範囲にある単語及び語句には、該当する技術分野(複数可)の当業者にとって通常の習慣的な意味が与えられることが、本発明者らの意図である。
【0122】
本出願の出願時に出願者に知られている本発明の種々の実施形態の先術の説明が示されており、それは、例示及び説明のためであることが意図されている。本説明は、網羅的であることも、開示された詳細な形態に本発明を限定することも意図されておらず、上記の教示に照らして多数の改変及び変異が可能である。記載された実施形態は、本発明の原理及びその実用的応用を説明し、他の当業者が、企図した特定の使用に好適なように種々の改変を施して、本発明を種々の実施形態で使用することを可能にする役目を果たす。従って、本発明は、本発明を実施するための開示された特定の実施形態に限定されないことが意図されている。
【0123】
本発明の特定の実施形態が表示及び記述されているが、当業者であれば、本明細書の教示に基づき、本発明及びそのより広範な態様から逸脱せずに変更及び改変を為すことができることは明らかであり、従って添付の特許請求の範囲は、本発明の真の趣旨及び範囲内にあるそのような変更及び改変を全て、それらの範囲に包含することになる。本明細書で使用されている用語は、一般的に「非限定的」な用語であると意図されていることが、当業者により一般的に理解されるだろう(例えば、用語「含む(including)」は、「〜を含むが、限定されない」と解釈されるべきであり、用語「有する(having)」は、「〜を少なくとも有する」と解釈されるべきであり、用語「含む(includes)」は、「〜を含むが、限定されない」と解釈されるべきである等)。
参考文献
1. Guerriero R, Mattia G, Testa U et al. Stromal cell-derived factor 1alpha increases polyploidization of megakaryocytes generated by human hematopoietic progenitor cells. Blood 2001;97:2587-2595.
2. Matsunaga T, Tanaka I, Kobune M et al. Ex vivo large-scale generation of human platelets from cord blood CD34+ cells. Stem Cells 2006;24:2877-2887.
3. Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc.Natl.Acad.Sci.U.S.A. 2001;98.:10716-10721.
4. Lu S-J, Li F, Vida L, Honig GR. CD34+CD38- hematopoietic precursors derived from human embryonic stem cells exhibit an embryonic gene expression pattern. Blood 2004;103:4134-4141.
5, Chadwick K, Wang L, Li L et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003 2003;102:906-915.
6. Chang KH, Nelson AM, Cao H et al. Definitive-like erythroid cells derived from human embryonic stem cells coexpress high levels of embryonic and fetal globins with little or no adult globin. Blood 2006;108:1515-1523.
7. Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Exp.Hematol. 2004;32:1000-1009.
8. Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005;105:617-626.
9. Wang L, Menendez P, Shojaei F et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005;201:1603-1614.
10. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J.Immunol. 2005;175:5095- 5103.
11. Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood 2005;106:860-870.
12. Qiu C, Hanson E, Olivier E et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Exp.Hematol. 2005;33:1450-1458.
13. Zhan X, Dravid G, Ye Z et al. Functional antigen-presenting leucocytes derived from human embryonic stem cells in vitro. Lancet 2004;364:163-171.
14. Ledran MH, Krassowska A, Armstrong L et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008;3:85-98.
15. Gaur M, Kamata T, Wang S et al. Megakaryocytes derived from human embryonic stem cells: a genetically tractable system to study megakaryocytopoiesis and integrin function. J.Thromb.Haemost. 2006;4:436-442.
16. Takayama N, Nishikii H, Usui J et al. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008;111:5298-5306.
17. Lu SJ, Feng Q, Caballero S et al. Generation of functional hemangioblasts from human embryonic stem cells. Nat.Methods 2007;4:501-509.
18. Klimanskaya I, McMahon J. Approaches of derivation and maintenance of human ES cells: Detailed procedures and alternatives. In: Lanza Rea, ed. Handbook of Stem Cells. Volume 1: Embryonic Stem Cells. New York, USA: Elsevier/Academic Press; 2004:437- 449.
19. Lu SJ, Luo C, Holton K et al. Robust generation of hemangioblastic progenitors from human embryonic stem cells. Regen.Med. 2008;3:693-704.
20. Lu SJ, Feng Q, Park JS et al. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood 2008;112:4475-4484.
21. Fujiki H, Kimura T, Minamiguchi H et al. Role of human interleukin-9 as a megakaryocyte potentiator in culture. Exp.Hematol. 2002;30:1373-1380.
22. Jeanpierre S, Nicolini FE, Kaniewski B et al. BMP4 regulation of human megakaryocytic differentiation is involved in thrombopoietin signaling. Blood 2008;112:3154-3163.
23. Lordier L, Jalil A, Aurade F et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood 2008;112:3164-3174.
24. Chang Y, Aurade F, Larbret F et al. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood 2007;109:4229-4236.
25. Taguchi K, Saitoh M, Arai Y et al. Disparate effects of interleukin 11 and thrombopoietin on megakaryocytopoiesis in vitro. Cytokine 2001;15:241-249.
26. Philipp CS, Remmler J, Zucker-Franklin D. The effects of Mpl-ligand, interleukin-6 and interleukin-11 on megakaryocyte and platelet alpha-granule proteins. Thromb.Haemost. 1998;80:968-975.
27. Kanaji T, Russell S, Cunningham J et al. Megakaryocyte proliferation and ploidy regulated by the cytoplasmic tail of glycoprotein Ibalpha. Blood 2004;104:3161-3168.
28. Santoso S, Kalb R, Kiefel V, Mueller-Eckhardt C. The presence of messenger RNA for HLA class I in human platelets and its capability for protein biosynthesis. Br.J.Haematol. 1993;84:451-456.
29. Lalezari P, Driscoll AM. Ability of thrombocytes to acquire HLA specificity from plasma. Blood 1982;59:167-170.
30. Sullenbarger B, Bahng JH, Gruner R, Kotov N, Lasky LC. Prolonged continuous in vitro human platelet production using three-dimensional scaffolds. Exp.Hematol. 2009;37:101-110.
31. Giammona LM, Fuhrken PG, Papoutsakis ET, Miller WM. Nicotinamide (vitamin B3) increases the polyploidisation and proplatelet formation of cultured primary human megakaryocytes. Br.J.Haematol. 2006;135:554-566.
32. Nagata Y, Yoshikawa J, Hashimoto A et al. Proplatelet formation of megakaryocytes is triggered by autocrine-synthesized estradiol. Genes Dev. 2003;17:2864-2869.
33. Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood 2006;108:1509-1514.
34. Klimchenko O, Mori M, Distefano A et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood 2009;114:1506-1517.
35. Nishikii H, Eto K, Tamura N et al. Metalloproteinase regulation improves in vitro generation of efficacious platelets from mouse embryonic stem cells. J.Exp.Med. 2008;205:1917-1927.
36. Furie B, Furie BC. Mechanisms of thrombus formation. N. Engl.J.Med. 2008; 359: 938-949.
37. Day SM, Reeve JL, et al. Murine thrombosis models. Thromb.Haemost. 2004; 92: 486-494.
38. Sachs UJ and Nieswandt B. In vivo thrombus formation in murine models. Circ.Res. 2007; 100: 979-991.
39. Furie B and Furie BC. In vivo thrombus formation. J.Thromb.Haemost. 2007; 5 Suppl 1: 12-17.
40. Junt T, Schulze H, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007; 317: 1767-1770.
41. Yu J, Hu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009; 324: 797-801.
42. Takahashi K, Tanabe K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861-872.
43. Yu J, Vodyanik MA, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318: 1917-1920.
44. Kim D, Kim CH, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009; 4: 472-476.
45. Ginsberg MH, Du X, Plow EF. Inside-out integrin signalling. Curr.Opin.Cell Biol. 1992; 4: 766-771.
46. Chen YP, O'Toole TE, et al. A point mutation in the integrin beta 3 cytoplasmic domain (S752-->P) impairs bidirectional signaling through alpha IIb beta 3 (platelet glycoprotein IIb-IIIa). Blood 1994; 84: 1857-1865.
47. Cho J, Furie BC, et al. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J.Clin.Invest 2008; 118: 1123-1131.
48. Coller BS. Anti-GPIIb/IIIa drugs: current strategies and future directions. Thromb. Haemost. 2001; 86: 427-443.
49. Cho J, Mosher DF. Enhancement of thrombogenesis by plasma fibronectin cross-linked to fibrin and assembled in platelet thrombi. Blood 2006: 107; 3555-3563.
50. Falati S, Gross P, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat.Med. 2002; 8: 1175-1181.
51. Reems JA, Pineault N, and Sun S. In vitro megakaryocyte production and platelet biogenesis: state of the art. Transfus. Med.Rev. 2010; 24: 33-43.
52. Chockalingam P, Sacher RA. Management of patients’ refractory to platelet transfusion. J.Infus.Nurs. 2007; 30 : 220-225.
53. Hod E, Schwartz J. Platelet transfusion refractoriness. Br.J.Haematol. 2008; 142: 348-360.
54. Tian X, Kaufman DS. Differentiation of embryonic stem cells towards hematopoietic cells: progress and pitfalls. Curr.Opin.Hematol. 2008; 15: 312-318.
55. Wang L, Menendez P, et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005; 201: 1603-1614.
【技術分野】
【0001】
本発明は、機能的巨核球および血小板の作製、ならびにそれらの使用に関する。
【背景技術】
【0002】
本明細書におけるすべての刊行物は、個々の刊行物または特許出願が、参照により本明細書に組み込まれることが具体的かつ各々明確に示されている場合と同程度にて、参照により本明細書に組み込まれる。以下の記述は、本発明の理解に有用であり得る情報を含む。それは、本明細書で提供されるいずれの情報についても、それが請求される本発明の先行技術であるかもしくはそれに関連するものであること、または明確にもしくは暗に参照されるいずれの刊行物についても、それが先行技術であること、を承認するものではない。
【0003】
血管損傷が起こると、血小板は、損傷した血管に迅速に接着し、血栓の形成をもたらす複雑な事象のカスケードを引き起こす。ここ数十年の間、血小板輸血に対する需要は増加し続けている51(非特許文献1)。血小板を保存できるのはわずかに1週間未満であり、このことが、ドナーに依存するプログラムを継続的に困難なものとしている。血小板の供給が不足すると、特に複数の輸血を必要とする患者において、命にかかわる可能性のある結果となり得る。繰り返し輸血を行うこともまた、免疫媒介宿主反応に繋がる抵抗性応答を引き起こし得るものであり、費用のかかる患者適合が必要となり得る52;53(非特許文献2;3)。患者に適合した血小板を生体外にて生成することができれば、このような臨床シナリオにおいて大きな利点が提供されることになる。
【0004】
血小板の供給が限られることは、特殊な/稀な血液型を有する輸血依存患者、特に、同種免疫された患者、および癌または白血病の患者で、これらで多く見られる血小板同種免疫を発生させた患者において、命にかかわる可能性のある結果となり得る。輸血されたヒト血小板の半減期は4〜5日間であるため、臨床上、血小板の輸血を頻繁に行う必要がある。さらに、献血プログラムからの血小板は、種々の病原体による汚染のリスクが常に存在する。血小板は凍結保存することができないため、血小板を生体外で生成することができれば、臨床セッティングにおける血小板交換療法にとって大きな進歩となる。ここ10年以上にわたって、骨髄(BM)、臍帯血(CB)、または末梢血(PB)からのヒト造血幹細胞(HSC、CD34+)が、巨核球(MK)および血小板の生成について研究されてきた。サイトカイン、成長因子、および/または間質フィーダー細胞と組み合わせることにより、機能的血小板のHSCからの作製は大きな成功を収めてきた1;2(非特許文献4;5).しかし、それでもHSCはドナーからのものであり、現行の培養条件下での増殖能は限られており、このことは、大規模作製および将来的な臨床適用を妨げる可能性が高い。
【0005】
ヒト胚性幹細胞(hESC)は、生体外にて無限に繁殖および増殖することができ、ヒトの治療法に無尽蔵でドナーを必要としない細胞源を提供する可能性を有する。hESCの造血細胞への分化は、この十年の間に生体外にて広く研究されてきた。指向させたhESCの血球分化は、生体外にて、2つの異なる種類の培養系により成功している。このうちの一方は、血清含有培地中でのhESCと間質フィーダー細胞との共培養を採用している3;4(非特許文献6;7)。2つ目の種類の手順では、血清有り/無しでのサイトカインの存在下における、超低細胞結合プレート(ultra−low cell binding plates)での懸濁培養条件を採用しており5‐7(非特許文献8−10);そのエンドポイントはEBの形成である。造血前駆体、ならびに赤血球、骨髄、マクロファージ、巨核球、およびリンパ球系列を代表する成熟した機能的な後代が、上記の両方の分化hESC培養系において識別された3‐6;8‐14(非特許文献6−9;11−17)。これまでの研究ではまた、血清の存在下にて間質細胞と共に共培養することにより、hESCから巨核球/血小板も生成された15;16(非特許文献18;19)。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Reems JA, Pineault N, and Sun S. In vitro megakaryocyte production and platelet biogenesis: state of the art. Transfus. Med.Rev. 2010; 24: 33-43.
【非特許文献2】Chockalingam P, Sacher RA. Management of patients’ refractory to platelet transfusion. J.Infus.Nurs. 2007; 30 : 220-225.
【非特許文献3】Hod E, Schwartz J. Platelet transfusion refractoriness. Br.J.Haematol. 2008; 142: 348-360.
【非特許文献4】Guerriero R, Mattia G, Testa U et al. Stromal cell-derived factor 1alpha increases polyploidization of megakaryocytes generated by human hematopoietic progenitor cells. Blood 2001;97:2587-2595.
【非特許文献5】Matsunaga T, Tanaka I, Kobune M et al. Ex vivo large-scale generation of human platelets from cord blood CD34+ cells. Stem Cells 2006;24:2877-2887.
【非特許文献6】Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc.Natl.Acad.Sci.U.S.A. 2001;98.:10716-10721.
【非特許文献7】Lu S-J, Li F, Vida L, Honig GR. CD34+CD38- hematopoietic precursors derived from human embryonic stem cells exhibit an embryonic gene expression pattern. Blood 2004;103:4134-4141.
【非特許文献8】Chadwick K, Wang L, Li L et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003 2003;102:906-915.
【非特許文献9】Chang KH, Nelson AM, Cao H et al. Definitive-like erythroid cells derived from human embryonic stem cells coexpress high levels of embryonic and fetal globins with little or no adult globin. Blood 2006;108:1515-1523.
【非特許文献10】Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Exp.Hematol. 2004;32:1000-1009.
【非特許文献11】Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005;105:617-626.
【非特許文献12】Wang L, Menendez P, Shojaei F et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005;201:1603-1614.
【非特許文献13】Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J.Immunol. 2005;175:5095- 5103.
【非特許文献14】Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood 2005;106:860-870.
【非特許文献15】Qiu C, Hanson E, Olivier E et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Exp.Hematol. 2005;33:1450-1458.
【非特許文献16】Zhan X, Dravid G, Ye Z et al. Functional antigen-presenting leucocytes derived from human embryonic stem cells in vitro. Lancet 2004;364:163-171.
【非特許文献17】Ledran MH, Krassowska A, Armstrong L et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008;3:85-98.
【非特許文献18】Gaur M, Kamata T, Wang S et al. Megakaryocytes derived from human embryonic stem cells: a genetically tractable system to study megakaryocytopoiesis and integrin function. J.Thromb.Haemost. 2006;4:436-442.
【非特許文献19】Takayama N, Nishikii H, Usui J et al. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008;111:5298-5306.
【発明の概要】
【発明が解決しようとする課題】
【0007】
hESC由来の血小板は、効率的および大規模に生成することができれば、輸血医療の目的のための可能性を有する。より重要なことには、血小板は、核を持たず、最小限の遺伝物質しか含有せず、輸血前に放射線照射することで、未分化hESCなどの不純物としての細胞のいずれをも効果的に除去することができる。従って、安全性は問題とならないはずである。しかし、上記の研究における巨核球/血小板の収率は低かった15;16。hESCを、均質な巨核球集団、およびそれに続く機能的血小板へ、効率的および制御された形で分化させることが、本技術分野において依然として求められている。
【課題を解決するための手段】
【0008】
以下の実施形態およびその態様は、例示および説明のためであることが意図される組成物および方法と合わせて記載および説明され、範囲を限定するものではない。
【0009】
本発明は、巨核球を生成する方法を提供し、その方法は:血管芽細胞を提供すること;および血管芽細胞を培養して巨核球(MK)に分化させることを含む。特定の実施形態では、血管芽細胞は、多能性幹細胞から生体外で分化させてよい。特定の実施形態では、多能性幹細胞は、ヒト胚性幹細胞(hESC)であってよい。特定の実施形態では、多能性幹細胞は、誘導多能性幹細胞(iPSC)であってよい。特定の実施形態では、iPSCは、ヒト由来の体細胞から誘導されてよい。特定の実施形態では、体細胞は、成人組織からのものであってよい。特定の実施形態では、体細胞は、胎児(仔)組織からのものであってよい。特定の実施形態では、多能性幹細胞の分化は、多能性幹細胞を解離させること、ならびに、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で多能性幹細胞を培養して胚様体(EB)を形成させることを含んでよい。特定の実施形態では、培地は、BMP‐4およびVEGFを含んでよく、多能性幹細胞の培養は、約48時間行ってEBを形成させてよい。特定の実施形態では、BMP‐4の濃度は、50ng/mlであってよく、VEGFは、50ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。
【0010】
特定の実施形態では、形成されたEBは、化学的および/または機械的に解離してよく、培地の少なくとも一部分を、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて、血管芽細胞を生成してよい。特定の実施形態では、bFGFの濃度は、20ng/mlであってよく、TPOは、50〜100ng/mlであってよく、Flt3リガンドは、50ng/mlであってよく、およびSCFは、50ng/mlであってよい。特定の実施形態では、培地は、IL6、エストラジオール、ビタミンB3、1つ以上の細胞外マトリックスタンパク質、またはこれらの組み合わせをさらに含む。特定の実施形態では、解離されたEBは、芽細胞コロニー増殖培地中にて約3から4日間培養して、血管芽細胞を生成してよい。
【0011】
本発明の他の実施形態は、巨核球を生成する方法を提供し、その方法は:血管芽細胞を提供すること;および血管芽細胞を培養して巨核球(MK)に分化させることを含む。特定の実施形態では、血管芽細胞のMKへの分化は、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて、血管芽細胞を約2から8日間培養することを含んでよい。特定の実施形態では、TPOの濃度は、50ng/mlであってよく、SCFは、20ng/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。特定の実施形態では、この方法は、2から3日ごとに、培地の少なくとも一部分を、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて血管芽細胞をMKへ分化させることをさらに含んでよい。
【0012】
本発明はまた、血小板を生成する方法も提供し、その方法は:巨核球(MK)を提供すること;およびMKを培養して血小板に分化させることを含む。特定の実施形態では、MKは、血管芽細胞から分化させてよい。特定の実施形態では、血管芽細胞のMKへの分化は、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて、血管芽細胞を約2から8日間培養することを含んでよい。特定の実施形態では、TPOの濃度は、50ng/mlであってよく、SCFは、20ng/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。特定の実施形態では、この方法は、2から3日ごとに、培地の少なくとも一部分を、TPO、SCF、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて血管芽細胞をMKへ分化させることをさらに含んでよい。
【0013】
特定の実施形態では、血管芽細胞は、多能性幹細胞から生体外で分化させてよい。特定の実施形態では、多能性幹細胞は、ヒト胚性幹細胞(hESC)であってよい。特定の実施形態では、多能性幹細胞は、誘導多能性幹細胞(iPSC)であってよい。特定の実施形態では、iPSCは、ヒト由来の体細胞から誘導されてよい。特定の実施形態では、体細胞は、成人組織からのものであってよい。特定の実施形態では、体細胞は、胎児(仔)組織からのものであってよい。特定の実施形態では、多能性幹細胞の分化は、多能性幹細胞を解離させること、ならびに、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で多能性幹細胞を培養して胚様体(EB)を形成させることを含んでよい。特定の実施形態では、培地は、BMP‐4およびVEGFを含んでよく、多能性幹細胞の培養は、約48時間行ってEBを形成させてよい。特定の実施形態では、BMP‐4の濃度は、50ng/mlであってよく、VEGFは、50ng/mlであってよい。特定の実施形態では、培地は、Stemline II培地であってよい。
【0014】
特定の実施形態では、形成されたEBは、化学的および/または機械的に解離してよく、培地の少なくとも一部分を、BMP‐4、VEGF、bFGF、TPO、Flt3リガンド、SCF、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えて、血管芽細胞を生成してよい。特定の実施形態では、bFGFの濃度は、20ng/mlであってよく、TPOは、50〜100ng/mlであってよく、Flt3リガンドは、50ng/mlであってよく、およびSCFは、50ng/mlであってよい。特定の実施形態では、培地は、IL6、エストラジオール、ビタミンB3、1つ以上の細胞外マトリックスタンパク質、またはこれらの組み合わせをさらに含んでよい。特定の実施形態では、解離されたEBは、芽細胞コロニー増殖培地中にて約3から4日間培養して、血管芽細胞を生成してよい。
【0015】
本発明の他の実施形態は、血小板を生成する方法を提供し、その方法は:巨核球(MK)を提供すること;およびMKを培養して血小板に分化させることを含む。
【0016】
特定の実施形態では、MKを再懸濁させ、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中にて少なくとも4日間培養して、血小板へ分化させてよい。特定の実施形態では、TPOの濃度は、100ng/mlであってよく、SCFは、50ng/mlであってよく、ヘパリンナトリウムは、25ユニット/mlであってよく、IL11は、20ng/mlであってよい。特定の実施形態では、この方法は、MKを少なくとも4日間培養した後に、GM6001またはIL3を添加することをさらに含んでよい。特定の実施形態では、この方法は、少なくとも2日間ごとに、培地の少なくとも一部分を、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えることをさらに含んでよい。特定の実施形態では、培地は、IMDM培地であってよい。
【0017】
特定の実施形態では、MKは、解離し、ならびにTPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地中、有糸分裂停止させたフィーダー細胞層上にて少なくとも4日間培養して血小板へ分化させてよい。特定の実施形態では、フィーダー細胞層は、OP9細胞を含んでいてよい。特定の実施形態では、フィーダー細胞層は、C3H 10T1/2細胞を含んでいてよい。特定の実施形態では、この方法は、MKを少なくとも4日間培養した後に、GM6001またはIL3を添加することをさらに含んでよい。特定の実施形態では、この方法は、少なくとも2日間ごとに、培地の少なくとも一部分を、TPO、SCF、ヘパリンナトリウム、IL11、およびこれらの組み合わせから成る群より選択される成長因子またはサイトカインを含む培地で置き換えることをさらに含んでよい。特定の実施形態では、培地は、IMDM培地であってよい。
【0018】
本発明はまた、本発明の方法のいずれかによって生成された大量の巨核球も提供する。
【0019】
本発明はまた、本発明の方法のいずれかによって生成された大量の血小板も提供する。特定の実施形態では、血小板は、対象に血小板を投与した際の対象におけるHLA同種免疫原性応答に寄与しない溶液中に保存することができる。特定の実施形態では、血小板は、アピラーゼおよび/またはEDTAの存在下では活性化することができない。
【0020】
本発明はまた、細胞分化の修飾因子のためのスクリーニング方法も提供し、その方法は:大量の巨核球(MK)を提供すること;MKを試験化合物と接触させること;MKと試験化合物との接触からの機能的効果が存在するかまたは存在しないかを判定することを含み;ここで、機能的効果が存在する場合、それは試験化合物が、細胞分化を調節する巨核球形成、血小板新生、および/または造血因子である可能性があることを示しており、機能的効果が存在しない場合、それは試験化合物が、細胞分化を調節する巨核球形成、血小板新生、および/または造血因子ではない可能性があることを示している。本発明はまた、血小板輸血を必要とする対象を治療する方法も提供し、その方法は:大量の血管芽細胞を提供すること;血管芽細胞を培養して巨核球(MK)へ分化させること;MKを生体外で培養して血小板へ分化させること;生体外で分化させた大量の血小板を提供すること;および血小板輸血を必要とする対象へ、生体外で分化させたその量の血小板を投与し、それによって血小板輸血を必要とする対象を治療することを含む。特定の実施形態では、生体外で分化させた血小板は、対象に適合させて、免疫媒介宿主反応を低減または除去することができる。
【0021】
本発明のその他の特徴および利点は、本発明の実施形態の種々の特徴を例として説明する添付の図面と合わせて、以下の詳細な説明から明らかとなるであろう。
【0022】
代表的な実施形態を、参照図面で示す。本明細書で開示される実施形態および図面は、限定のためではなく、説明のためのものとして見なされるべきであることを意図している。
【図面の簡単な説明】
【0023】
【図1−1】図1は、hESC由来の芽細胞が、本発明の種々の実施形態に従って巨核球前駆細胞を生成することを示す。A.は、血清および間質のない条件下にてhESCから巨核球を生成するフローチャートである。前述のように、hESCは、懸濁培養し、EBを発生させた。EBは、第4日目に回収し、トリプシンで単一細胞へ解離し、その後メチルセルロースを主体とする半固体血管芽細胞培養を行った。芽細胞は、第6日目から第8日目の培養物から回収してMK培養に用い、そしてこれは、続く4から6日間の間に懸濁培養にて成熟巨核球を生成した。B.は、第6日目の芽細胞コロニー(位相差、左)および芽細胞(ギムザ染色、右)の代表的なイメージである。C.は、HUES3およびH1 hESC由来BCについてのCFU‐MKコロニーアッセイを示す棒グラフである。D.は、H1 hESC由来BCから生成されたCD41染色CFU‐MKコロニーを示す顕微鏡イメージである。挿入図は、巨核球からの細胞プロセスを示す。E.は、3種類の異なるhESC系統:HUES3、MA01、およびMA09からの芽細胞培養物におけるCD41a+細胞のパーセントの違いを示すFACS分析である。
【図1−2】図1は、hESC由来の芽細胞が、本発明の種々の実施形態に従って巨核球前駆細胞を生成することを示す。A.は、血清および間質のない条件下にてhESCから巨核球を生成するフローチャートである。前述のように、hESCは、懸濁培養し、EBを発生させた。EBは、第4日目に回収し、トリプシンで単一細胞へ解離し、その後メチルセルロースを主体とする半固体血管芽細胞培養を行った。芽細胞は、第6日目から第8日目の培養物から回収してMK培養に用い、そしてこれは、続く4から6日間の間に懸濁培養にて成熟巨核球を生成した。B.は、第6日目の芽細胞コロニー(位相差、左)および芽細胞(ギムザ染色、右)の代表的なイメージである。C.は、HUES3およびH1 hESC由来BCについてのCFU‐MKコロニーアッセイを示す棒グラフである。D.は、H1 hESC由来BCから生成されたCD41染色CFU‐MKコロニーを示す顕微鏡イメージである。挿入図は、巨核球からの細胞プロセスを示す。E.は、3種類の異なるhESC系統:HUES3、MA01、およびMA09からの芽細胞培養物におけるCD41a+細胞のパーセントの違いを示すFACS分析である。
【図2−1】図2は、芽細胞培養物からの二分化能前駆細胞が、本発明の種々の実施形態に従って赤血球および巨核球系列の両方を発生させることを示す。A.は、CD41aおよびCD235aマーカーの発現で定められる別々の集団(左)およびソートされたCD41a+CD235a+細胞(右)を示す、第6日目のHUES3由来芽細胞のFACS分析である。B.は、CD41a+CD235a−またはCD41a+CD235a+細胞のいずれか由来のCFU‐MKコロニーのイメージである。棒グラフは、CFU‐MKコロニーアッセイの定量化を示す。C.は、CD41a−CD235a+またはCD41a+CD235a+細胞のいずれか由来のCFU‐Eコロニーのイメージである。棒グラフは、CFU‐Eコロニーアッセイの定量化を示す。
【図2−2】図2は、芽細胞培養物からの二分化能前駆細胞が、本発明の種々の実施形態に従って赤血球および巨核球系列の両方を発生させることを示す。A.は、CD41aおよびCD235aマーカーの発現で定められる別々の集団(左)およびソートされたCD41a+CD235a+細胞(右)を示す、第6日目のHUES3由来芽細胞のFACS分析である。B.は、CD41a+CD235a−またはCD41a+CD235a+細胞のいずれか由来のCFU‐MKコロニーのイメージである。棒グラフは、CFU‐MKコロニーアッセイの定量化を示す。C.は、CD41a−CD235a+またはCD41a+CD235a+細胞のいずれか由来のCFU‐Eコロニーのイメージである。棒グラフは、CFU‐Eコロニーアッセイの定量化を示す。
【図3】図3は、本発明の種々の実施形態に従う無間質条件下でのhESC由来芽細胞からの指向された巨核球分化を示す。A.は、TPO、SCF、およびその他のサイトカイン/因子の巨核球分化および増殖に対する効果である。B.は、6日間培養の間のCD41a+巨核球の増殖を示す経時実験である。C.は、CD41a、CD42b、およびCD235a系列マーカーの発現レベルの変化を示す、第6日目の芽細胞培養物および第4日目のMK培養物のFACS分析である。MK培養物中にて、CD235a+細胞が消滅した一方、生存細胞の90%超がCD41a+となった。第6日目の芽細胞培養物中では、少数のCD41a+細胞がCD42bに対して陽性であった(左下パネル)。MK培養物中のCD41a+細胞の大部分も、第4日目までに高レベルのCD42bを発現し、このことは巨核球の成熟を示している(右下パネル)。
【図4−1】図4は、本発明の種々の実施形態に従って生体外で生成された巨核球の特性評価を示す。A.は、6日間のMK培養の間における細胞サイズの増加(上パネル)および細胞サイズ分布の変化(下パネル)を示す経時実験である。細胞サイズ(直径)は、ギムザ染色したサイトスピンスライドを用いて測定した。B.は、倍数体巨核球を示すギムザ染色イメージである。C.は、第6日目のMK培養物からのCD41a+巨核球内における、CD42bおよびCD42a巨核球/血小板特異的糖タンパク質の両方の発現を示すFACSドットプロットである。D.は、第4日目のMK培養物からの倍数体巨核球における、CD41染色およびvWF染色を示す免疫蛍光イメージである。E.は、生体外MK生成の種々のステージにおける、赤血球新生/巨核球形成を制御する転写因子の発現レベルを示すRT‐PCRの結果である。全RNAを、ES、第4日目のEB、BC(第6日目から第8日目)、および第4日目のMKから、RNA easy mini‐prep kit(キアジェン(Qiagen))を用いて単離した。2ugの全RNAを、iScript kit(バイオラッド(Biorad))によるcDNA合成に用いた。各サンプルについて、投入cDNAの量に対するコントロールとしてGAPDH RT‐PCRを行った。F.は、第4日目のMK培養物において胞体突起(proplatelet)形成巨核球を示す代表的な位相差イメージである。
【図4−2】図4は、本発明の種々の実施形態に従って生体外で生成された巨核球の特性評価を示す。A.は、6日間のMK培養の間における細胞サイズの増加(上パネル)および細胞サイズ分布の変化(下パネル)を示す経時実験である。細胞サイズ(直径)は、ギムザ染色したサイトスピンスライドを用いて測定した。B.は、倍数体巨核球を示すギムザ染色イメージである。C.は、第6日目のMK培養物からのCD41a+巨核球内における、CD42bおよびCD42a巨核球/血小板特異的糖タンパク質の両方の発現を示すFACSドットプロットである。D.は、第4日目のMK培養物からの倍数体巨核球における、CD41染色およびvWF染色を示す免疫蛍光イメージである。E.は、生体外MK生成の種々のステージにおける、赤血球新生/巨核球形成を制御する転写因子の発現レベルを示すRT‐PCRの結果である。全RNAを、ES、第4日目のEB、BC(第6日目から第8日目)、および第4日目のMKから、RNA easy mini‐prep kit(キアジェン(Qiagen))を用いて単離した。2ugの全RNAを、iScript kit(バイオラッド(Biorad))によるcDNA合成に用いた。各サンプルについて、投入cDNAの量に対するコントロールとしてGAPDH RT‐PCRを行った。F.は、第4日目のMK培養物において胞体突起(proplatelet)形成巨核球を示す代表的な位相差イメージである。
【図5】図5は、本発明の種々の実施形態に従う、hiPSCおよびhESCの血管芽細胞/芽細胞および造血細胞への分化を示す。A.では、hESC(i、×100)から得られた血管芽細胞/芽細胞(ii、×400)を、CD71(×1000)、CXCR4(×1000)、およびTpo受容体(×1000)で染色した。B.では、hiPSC(i、×100)から得られた血管芽細胞/芽細胞(ii、×400)を、CD71(×1000)、CXCR4(×1000)、およびTpo受容体(×1000)で染色した。C.は、再播種の14日後におけるhiPSC‐芽細胞から発生した複数種類の造血コロニー形成単位(CFU)であり、CFU‐E、CFU‐GM、およびCFU‐Mkについては×100、CFU‐Gについては×40である。(D)では、hiPSCから得られた血管芽細胞/芽細胞(i、×200)は、内皮細胞へ分化し、;hiPSC由来内皮細胞は、マトリゲル上で血管様ネットワークを形成し(ii、×40)、vWF(赤色)およびCD31(iii、緑色、×400)、さらにはLDL取り込み能(iv、赤色 ×400)を有するVE‐カドヘリン(iv、緑色)を発現した。
【図6】図6は、本発明の種々の実施形態に従う、機能的ES由来血小板(ES‐PLT)の生成を示す。A.は、OP9共培養から得られたES‐PLTの前方散乱(FSC)および側方散乱(SSC)特性を示すドットプロットである(左上)。ヒト血液の血小板に基づいてゲーティングを設定した。右上パネルは、ES‐PLTでのCD41a(y軸)およびCD42b(x軸)の発現を示す。休止ES‐PLT(中央パネル)と比較すると、トロンビン(1u/ml)処理ES‐PLTは、PAC1結合の増加を示した。平均蛍光強度は、トロンビン処理ES‐PLTにておよそ2倍に増加した。B.は、臨床グレードhESCであるMA01およびMA09から得られたES‐PLTも、トロンビン処理に応答してPAC1結合を示している。
【図7−1】図7は、本発明の種々の実施形態に従って、ES‐PLTが固定化フィブリノゲンと結合すること、および固定化フィブリノゲンと結合したES‐PLTが微小凝集体を形成することを示す。A.は、血液血小板(左パネル)およびES‐PLT(右パネル)が、フィブリノゲンでコーティングしたスライドグラス上にて伸展可能であることを示す伸展アッセイ(Spreading assay)である。RGDSの添加によって伸展が阻止されることから、このプロセスは、インテグリン受容体シグナル伝達に依存している。ADP(+ADP)またはトロンビン(+T)の添加により、フィブリノゲン単独(Con)と比較して、フィブリノゲン上での伸展プロセスが増強される。インテグリンに依存するF‐アクチン線維の再構築および糸状仮足の形成が、CD41a(緑色)およびファロイジン(F‐アクチン、赤色)の蛍光染色によって示される。B.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識血液血小板(上パネル)またはES‐PLT(下パネル)を、トロンビン(0.5u/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差および蛍光イメージより、ヒト血液血小板コントロールと同等である標識ES‐PLTの微小凝集体への関与が示される。C.は、ES‐PLT中でのHLAの発現を示すFACS分析のヒストグラムである。血液血小板をコントロールとして用いた(左)。
【図7−2】図7は、本発明の種々の実施形態に従って、ES‐PLTが固定化フィブリノゲンと結合すること、および固定化フィブリノゲンと結合したES‐PLTが微小凝集体を形成することを示す。A.は、血液血小板(左パネル)およびES‐PLT(右パネル)が、フィブリノゲンでコーティングしたスライドグラス上にて伸展可能であることを示す伸展アッセイ(Spreading assay)である。RGDSの添加によって伸展が阻止されることから、このプロセスは、インテグリン受容体シグナル伝達に依存している。ADP(+ADP)またはトロンビン(+T)の添加により、フィブリノゲン単独(Con)と比較して、フィブリノゲン上での伸展プロセスが増強される。インテグリンに依存するF‐アクチン線維の再構築および糸状仮足の形成が、CD41a(緑色)およびファロイジン(F‐アクチン、赤色)の蛍光染色によって示される。B.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識血液血小板(上パネル)またはES‐PLT(下パネル)を、トロンビン(0.5u/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差および蛍光イメージより、ヒト血液血小板コントロールと同等である標識ES‐PLTの微小凝集体への関与が示される。C.は、ES‐PLT中でのHLAの発現を示すFACS分析のヒストグラムである。血液血小板をコントロールとして用いた(左)。
【図8−1】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図8−2】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図8−3】図8は、本発明の種々の実施形態に従ってhESCから生成された巨核球および血小板の特性評価を示す。A.は、第4日目のMK培養物からの、CD41aおよび42b抗原の発現についての細胞のFACS分析である。B.は、倍数体巨核球を示すギムザ染色イメージである(100×)。C.は、第4日目の培養物からの倍数体MKにおけるCD41、vWF、およびDAPI染色を示す免疫蛍光イメージである(上パネル 40×、下パネル 100×)。D.は、DNA含有量についてのMKのFACS分析である。E.は、第4日目のMK培養物において胞体突起形成巨核球を示す代表的な位相差イメージである(40×)。F.は、hESCから得られた血小板についての前方散乱(FSC)、側方散乱(SSC)特性(左パネル)、ならびにCD41aおよびCD42bの発現(右パネル)を示すFACSドットプロットである。ヒト血液の血小板に基づいてゲーティングを設定した。G.は、休止(左パネル)およびトロンビン(1U/ml)処理hESC‐血小板(右パネル)に対するPAC1結合アッセイである。
【図9−1】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図9−2】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図9−3】図9は、本発明の種々の実施形態に従ってhESCから生成された血小板の機能的特性評価を示す。AおよびB.では、マイクロタイターチャンバースライドを100μg/mLのフィブリノゲンでコーティングした。ヒト血液血小板(A)およびhESC‐血小板(B)を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した(100×)。血小板はまた、示したように、RGDS(1mM)、ADP(20μM)、またはトロンビン(T、1U/ml)での処理も行った。CおよびD.は、微小凝集体形成アッセイである。同数のPKH67(緑色)標識ヒト血液血小板(C)またはhESC‐血小板(D)を、トロンビン(0.5U/ml)の刺激下、非標識血液血小板と混合して凝集体を形成させた。位相差(左パネル)および蛍光イメージ(中央パネル)は、これを合わせることにより(右パネル)、標識血液血小板(C、右パネル)およびhESC血小板(D、右パネル)の微小凝集体への関与を示している。EおよびF.は、hESC‐血小板(1.5×107/ml、E)および血液血小板(1.5×107/ml、F)を、血小板欠乏血漿中に再懸濁させた。トロンビン(2U/ml)およびCaCl2(10mM)を血小板懸濁液に添加し、クロット形成/退縮を誘発した(EおよびF、左パネル)。血漿のみを用いてネガティブコントロールとした。クロットの凍結切片を、抗ヒトCD41および抗ヒトフィブリン抗体で免疫染色し、続いて、それぞれ、二次ローダミン結合抗ウサギIgGおよびFITC結合抗マウスIgM抗体で免疫染色した。蛍光顕微鏡下にてイメージを撮影した(20×)。
【図10−1】図10は、本発明の種々の実施形態に従う、生体マウスのレーザー誘発細動脈損傷部位で発生するマウス血小板血栓へのhESC‐PLTの組み込みを示す。カルセインAM標識ヒト血液血小板またはESC由来血小板、50〜100μl(5〜10×105血小板)を、レーザー誘発血管損傷の直後に大腿動脈カニューレを通して注入した。発生するマウス血小板血栓を、Dylight 649標識抗CD42(0.05μg/g体重)の注入によってモニタリングした。2〜3つの血栓の生成後、標識血小板を200μl中2×106のヒト血小板に対して20μgのReoProで前処理し、同一のマウスの血管損傷後に注入した。さらに2〜3つの血栓が生成した。データは、血管損傷後3分間に収集した。A.は、血管損傷後の3つの時間点(0、1/2Tmax、およびTmax)での代表的な蛍光イメージを示す。白色四角内の領域の拡大イメージを下部に示す。スケールバー=10μm。BおよびC.では、微小血管へ循環する標識ヒト血小板の数(B)および血管損傷部位で発生するマウス血小板血栓中に組み込まれた数(C)を、血管損傷後3分間にわたってカウントした。データは、平均±S.E.Mで表す(n=5〜8血栓、3体のマウス)。スチューデントT検定、コントロールに対して*P<0.05および**P<0.01。
【図10−2】図10は、本発明の種々の実施形態に従う、生体マウスのレーザー誘発細動脈損傷部位で発生するマウス血小板血栓へのhESC‐PLTの組み込みを示す。カルセインAM標識ヒト血液血小板またはESC由来血小板、50〜100μl(5〜10×105血小板)を、レーザー誘発血管損傷の直後に大腿動脈カニューレを通して注入した。発生するマウス血小板血栓を、Dylight 649標識抗CD42(0.05μg/g体重)の注入によってモニタリングした。2〜3つの血栓の生成後、標識血小板を200μl中2×106のヒト血小板に対して20μgのReoProで前処理し、同一のマウスの血管損傷後に注入した。さらに2〜3つの血栓が生成した。データは、血管損傷後3分間に収集した。A.は、血管損傷後の3つの時間点(0、1/2Tmax、およびTmax)での代表的な蛍光イメージを示す。白色四角内の領域の拡大イメージを下部に示す。スケールバー=10μm。BおよびC.では、微小血管へ循環する標識ヒト血小板の数(B)および血管損傷部位で発生するマウス血小板血栓中に組み込まれた数(C)を、血管損傷後3分間にわたってカウントした。データは、平均±S.E.Mで表す(n=5〜8血栓、3体のマウス)。スチューデントT検定、コントロールに対して*P<0.05および**P<0.01。
【図11】図11は、本発明の種々の実施形態に従って、フィーダーおよび血清のない条件下で生成されるhESC‐血小板の伸展アッセイを示す。マイクロタイターチャンバースライドを100μg/mLのフィブリノゲン(上パネル)または30μg/mLのvWF(中央および下パネル)でコーティングした。hESC‐血小板を90分間伸展させた。接着血小板を、Alexa Fluor 568ファロイジン、FITC結合抗ヒトCD41a抗体、およびDAPIで染色し、蛍光顕微鏡下で撮影した。
【図12】図12は、本発明の種々の実施形態に従う、血液血小板に対するPAC‐1結合アッセイを示す。血液血小板は、トロンビン処理(1U/ml)あり(右)またはなし(左)で処理し、次にFACS分析によってPAC‐1結合を実施した。
【図13−1】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【図13−2】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【図13−3】図13は、本発明の種々の実施形態に従って、異なるhESC系統から生成されたhESC‐血小板の特性評価を示す。A.は、臨床グレードのMA01およびMA09 hESC細胞から得られたhESC‐血小板でのCD41aおよびCD42bの発現である。B.において、トロンビン処理(1U/ml)に応答して、MA01およびMA09 hESC由来血小板は、休止コントロール(上パネル)と比較してPAC‐1結合の増加を示している(下パネル)。C.では、hESC‐血小板は、IgGアイソタイプコントロール染色と比較して、HLA‐ABCの発現を示す。FACS分析のヒストグラムは、hESC‐血小板におけるHLA発現を示している。血液血小板をコントロールとして用いた(左)。
【発明を実施するための形態】
【0024】
本明細書で引用されるすべての参考文献は、その全内容が完全に記載されているかのように、参照により本明細書に組み込まれる。それ以外の定義がなされない限りにおいて、本明細書で用いられる技術的および科学的用語は、本発明が属する技術分野の当業者によって一般的に理解されるものと同一の意味を有する。Singleton et al., Dictionary of Microbiology and Molecular Biology 3rd ed., J. Wiley & Sons (New York, NY 2001);March, Advanced Organic Chemistry Reactions, Mechanisms and Structure 5th ed., J. Wiley & Sons (New York, NY 2001);および、Sambrook and Russel, Molecular Cloning: A Laboratory Manual 3rd ed., Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY 2001)は、本出願に用いられる用語の多くに対する一般的なガイドを当業者に提供するものである。
【0025】
当業者であれば、本発明の実践に用いることが可能である本明細書で述べるものと類似または同等である多くの方法および物質を識別する。実際、本発明は、記載される方法および物質に限定されるものではまったくない。本発明の目的のために、以下の用語を次のように定義する。
【0026】
本明細書およびそれに続く特許請求の範囲を通して用いる場合、「1つの(a)」、「1つの(an)」、および「その(the)」は、文脈から明らかにそうでないことが示されていない限りにおいて、複数の言及を含む。また、本明細書で用いる場合、「中で(in)」の意味は、文脈から明らかにそうでないことが示されていない限りにおいて、「中で(in)」および「上で(on)」を含む。
【0027】
「胚性幹細胞」(ES細胞)の用語は、本技術分野で用いられるように本明細書において用いられる。この用語は、細胞系統として段階的に継代培養されたものを含む、ヒト胚盤胞または桑実胚の内部細胞塊から得られる細胞を含む。ES細胞は、卵細胞と精子との受精から、さらにはDNA、核移植、単為生殖を用いて、またはHLA領域においてホモ接合性を有するES細胞を生成させる手段によって得られるものであってよい。ES細胞はまた、精子と卵細胞との融合、核移植、単為生殖、雄性生殖、またはクロマチンの再プログラム化およびそれに続く細胞作製のための再プログラム化されたクロマチンの細胞膜への組み込みによって発生された接合体、卵割球、もしくは胚盤胞期哺乳類の胚から得られる細胞でもある。胚性幹細胞は、その作製源またはそれを作製するために用いられる特定の方法に関わらず、(i)3つすべての胚葉の細胞へ分化する能力、(ii)少なくともOct4およびアルカリホスファターゼの発現、ならびに(iii)免疫不全動物へ移植された場合にテラトーマを発生させる能力、に基づいて識別することができる。
【0028】
本明細書で用いる場合、「多能性幹細胞」の用語は、多能性幹細胞が得られる方法に関わらず、胚性幹細胞、胚由来幹細胞、および誘導多能性幹細胞を含む。多能性幹細胞は、機能的に:(a)免疫不全(SCID)マウスに移植された場合にテラトーマを誘発する能力を有する;(b)3つすべての胚葉の細胞型へ分化する能力を有する(例:外胚葉、中胚葉、および内胚葉細胞型に分化することができる);ならびに(c)胚性幹細胞の1つ以上のマーカーを発現する(例:Oct4、アルカリホスファターゼ、SSEA‐3表面抗原、SSEA‐4表面抗原、nanog、TRA‐1‐60、TRA‐1‐81、SOX2、REX1などを発現する)、幹細胞として定義される。代表的な多能性幹細胞は、例えば本技術分野で公知の方法を用いて生成することができる。代表的な多能性幹細胞としては、胚盤胞期胚のICMから得られる胚性幹細胞、さらには卵割期または桑実期胚の1つ以上の卵割球から得られる胚性幹細胞(所望に応じて、胚の残り部分を破壊することなく行ってよい)が挙げられる。そのような胚性幹細胞は、受精によって、または体細胞核移植(SCNT)、単為生殖、および雄性生殖を含む無性生殖的手段によって作製される胚性物質から生成してよい。さらなる代表的な多能性幹細胞としては、因子の組み合わせ(以降本明細書にて再プログラム化因子と称する)を発現することによる体細胞の再プログラム化によって生成される誘導多能性幹細胞(iPSC)が挙げられる。iPSCは、胎児(仔)、出生後、新生児(仔)、若齢、または成体の体細胞を用いて生成してよい。
【0029】
特定の実施形態において、体細胞を再プログラム化して多能性幹細胞とするために用いてよい因子としては、例えば、Oct4(Oct3/4と称される場合もある)、Sox2、c‐Myc、およびKlf4の組み合わせが挙げられる。他の実施形態において、体細胞を再プログラム化して多能性幹細胞とするために用いてよい因子としては、例えば、Oct4、Sox2、Nanog、およびLin28の組み合わせが挙げられる。特定の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも2つの再プログラム化因子が体細胞中で発現される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも3つの再プログラム化因子が体細胞中で発現される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも4つの再プログラム化因子が体細胞中で発現される。他の実施形態では、さらなる再プログラム化因子が識別され、それが単独で、または1つ以上の既知の再プログラム化因子と組み合わせて用いられて、体細胞が多能性幹細胞へと再プログラム化される。誘導多能性幹細胞は、機能的に明らかにされており、様々な方法(組み込みベクター、非組み込みベクター、化学的手段など)のいずれを用いて再プログラム化された細胞も含む。
【0030】
誘導多能性幹細胞は、体細胞における再プログラム化因子のタンパク質形質導入によって作製することができる。特定の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも2つの再プログラム化タンパク質が体細胞内へ形質導入される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも3つの再プログラム化タンパク質が体細胞内へ形質導入される。他の実施形態では、体細胞の再プログラム化を成功させるために、少なくとも4つの再プログラム化タンパク質が体細胞内へ形質導入される。
【0031】
多能性幹細胞は、いかなる種からのものであってもよい。胚性幹細胞は、例えば、マウス、非ヒト霊長類の複数の種、およびヒトで得ることに成功しており、胚性幹様細胞(embryonic stem−like cells)は、数多くのさらなる種から生成されている。従って、当業者であれば、胚性幹細胞および胚由来幹細胞を、これらに限定されないが、ヒト、非ヒト霊長類、げっ歯類(マウス、ラット)、有蹄類(ウシ、ヒツジなど)、イヌ(飼育用および野生のイヌ)、ネコ類(ライオン、トラ、チーターなど、飼育用および野生のネコ類)、ウサギ、ハムスター、アレチネズミ、リス、モルモット、ヤギ、ゾウ、パンダ(ジャイアントパンダを含む)、ブタ、アライグマ、ウマ、シマウマ、海洋哺乳類(イルカ、クジラなど)などを含むいずれの種からも生成することができる。特定の実施形態では、種は、絶滅危惧種である。特定の実施形態では、種は、現在絶滅した種である。
【0032】
同様に、iPSCもいかなる種からのものであってもよい。これらのiPSCは、マウスおよびヒト細胞(hiPSC)を用いての生成に成功している。さらに、iPSCは、胚、胎児(胎仔)、新生児(仔)、および成体組織を用いての生成に成功している。従って、iPSCは、いかなる種からのドナー細胞を用いても容易に生成することができる。従って、iPSCは、これらに限定されないが、ヒト、非ヒト霊長類、げっ歯類(マウス、ラット)、有蹄類(ウシ、ヒツジなど)、イヌ(飼育用および野生のイヌ)、ネコ類(ライオン、トラ、チーターなど、飼育用および野生のネコ類)、ウサギ、ハムスター、ヤギ、ゾウ、パンダ(ジャイアントパンダを含む)、ブタ、アライグマ、ウマ、シマウマ、海洋哺乳類(イルカ、クジラなど)などを含むいずれの種からも生成することができる。特定の実施形態では、種は、絶滅危惧種である。特定の実施形態では、種は、現在絶滅した種である。
【0033】
誘導多能性幹細胞は、出発点として、実質的にいかなる細胞発生段階のいかなる体細胞を用いて生成してもよい。例えば、細胞は、胚、胎児(仔)、新生児(仔)、若齢、または成体ドナーからのものであってよい。用いてよい代表的な体細胞としては、皮膚サンプルもしくは生検によって得られる皮膚線維芽細胞などの線維芽細胞、滑膜組織からの滑膜細胞、包皮細胞、頬細胞、または肺線維芽細胞が挙げられる。皮膚および頬は、容易に利用可能であり、簡単に入手可能である適切な細胞源を提供するが、実質的にいかなる細胞を用いてもよい。特定の実施形態では、体細胞は線維芽細胞ではない。
【0034】
「血管芽細胞」および「血管コロニー形成細胞(hemangio−colony forming cell)」の用語は、本出願全体を通して交換可能に用いられる。これらの細胞は、数多くの構造的および機能的特徴を有する。これらの細胞の特徴の中で、宿主へ投与された場合に骨髄へ生着する能力がある。これらの細胞は、これらに限定されないが、1つ以上のマーカーの発現(RNAまたはタンパク質)、または発現(RNAまたはタンパク質)がないこと、を含む数多くの構造的および機能的特性に基づいて説明することができる。血管コロニー形成細胞は、分化して、少なくとも造血細胞型または内皮細胞型を発生させる能力を有する。好ましくは、血管コロニー形成細胞は、二分化能であり、分化して、少なくとも造血細胞型および内皮細胞型を発生させる能力を有する。従って、本発明の血管コロニー形成細胞は、少なくとも単分化能であり、好ましくは二分化能である。しかし、加えて、血管コロニー形成細胞は、より高い度合いの発生能を有していてよく、特定の実施形態では、分化して、その他の系列の細胞型を発生させてよい。特定の実施形態では、血管コロニー形成細胞は、分化して、心臓細胞(例えば、心筋細胞)および/または平滑筋細胞などのその他の中胚葉誘導体を発生させる能力を有する。
【0035】
「非生着血管芽細胞(non−engrafting hemangioblasts)」および「非生着血管細胞(non−engrafting hemangio cells)」の用語は、本出願全体を通して、血管コロニー形成細胞の特徴の一部を共有する細胞の集団を意味するために用いられる。しかし、非生着血管細胞は、免疫不全宿主に投与された場合に骨髄へ生着しないという点で、区別することができる。この違いにも関わらず、非生着血管細胞は、血管コロニー形成細胞の機能的または構造的特徴および特性の1もしくは2つ以上(2、3、4、5、6、7、8、9、10)を共有し得る。例えば、特定の実施形態では、非生着血管細胞は、互いに緩やかに接着している。他の実施形態では、非生着血管細胞は、以下のタンパク質:CD34、KDR、CD133、CD31、の1もしくは2つ以上(2、3、4)を発現しない。理論に束縛されるものではないが、非生着血管細胞は、血管コロニー形成細胞よりもある程度特殊化された独特の幹細胞集団を提供し得るが、それでも、一定範囲の造血細胞型を生成する能力を有する。
【0036】
血小板の供給が限られることは、輸血依存患者にとって命にかかわる可能性のある結果となり得る。hESCは、生体外にて無限に繁殖させることができ、ヒトの治療法にとっての無尽蔵でドナーを必要としない血小板源とすることができる。適合した、または不適合性が低減されたhESC系統のバンクを作り出すことができることは、免疫抑制薬および/または免疫調節プロトコルの必要性を低減または除去することができる可能性を有する。誘導多能性幹細胞(iPSC)の出現により41;42;43;44、このような技術を、血小板同種免疫が発生している輸血依存患者のための患者特異的血小板の作製に適応させることが可能であり得る。
【0037】
ドナーから収集される血小板は、保存期間が非常に限られており、患者の予防的輸血のための需要は高まってきている。ドナーに依存する臍帯血または骨髄CD34+ヒト造血幹細胞とは対照的に、ヒト胚性幹細胞(hESC)は、制御された条件下にて血小板を生体外で連続的に作製するための有望な別の選択肢としての作製源であり得る。発明者らは、血清および間質のない条件下にてhESCから巨核球(MK)を生成する新規な系を開発した。この系では、hESCは、胚様体形成および血管芽細胞分化を通してhESCを巨核球へと指向される。CD41aおよびCD235aマーカーの両方を発現する一過性の二分化能細胞集団が、血管芽細胞培養後に識別された。これらの細胞は、FACSソーティングおよびCFUアッセイで示されるように、MKおよび赤血球細胞の両方を生成する能力を有する。無血清懸濁培養におけるTPO、SCF、およびその他のサイトカインの存在下にて、hESCからMKへ最大で100倍までの増殖を14〜15日間で達成することができる。この系は、hESCからMKを生成する強力な生体外法である。OP9細胞上に播種されると、hESC由来MKは、血小板様粒子(ES‐PLT)を生成することができる。このようなES‐PLTは、トロンビン刺激に対して応答性であり、微小凝集体形成に関与することができる。
【0038】
血清および間質のない条件下にてhESCから機能的巨核球を生成する効率的な方法について述べる。本系では、hESCを、血管芽細胞(本明細書にて以降芽細胞(BC)とも称する)を中間体として用いて巨核球分化へと指向させたものであり17、1×106細胞のhESCから最大1×108の巨核球が生成され、これは、Takayama et al.16による最も最近報告された方法よりもおよそ10〜20倍高い効率である。さらなる精製を行うことなく、懸濁培養物からの生存細胞の>90%がCD41a+であり、これらの細胞の大多数はCD42aおよびCD42bも発現している。このような生体外で得られたMK細胞は、細胞質顆粒中のvWFのギムザ染色および免疫蛍光染色で示されるように、核内分裂を起こして成熟した倍数体MKと成ることができる。重要なことには、MK培養の後期において胞体突起形成細胞が終始観察されており、このことは、本系で生成されたMKが、無フィーダー条件下にて最終分化を起こすことができることを示している。これらの結果はまた、OP9間質細胞上に播種された場合、これらの巨核球が、血液血小板の機能的特性を共有する血小板様粒子を生成したことも示す。
【0039】
本明細書では、制御された条件下、hESCを作製源細胞として用いる生体外での巨核球の大量作製に適応可能である効率的な系について述べる。この細胞は、CD41a,CD42a,およびCD42bを発現し、核内分裂を起こして、成熟した倍数体MKを形成した。さらに成熟すると、それらは、トロンビン刺激によって活性化可能であり、フィブリノゲンおよびフォンウィルブランド因子(vWF)の表面上で伸展することができる血小板を生成した。このhESC由来血小板はまた、微小凝集体を形成し、正常なヒト血液血小板と同等な形でクロットの形成および退縮を促進した。重要なことには、これらの結果はまた、このような生体外で得られた巨核球が、その血液中対応物と機能的に同等である血小板を生成する能力を有していることも示した。このようなES‐PLTは、その接着および伸展する能力、ならびに正常血液血小板と共に微小凝集体を形成する能力を示すことによって、血小板アゴニストに対して応答性を有する。間質細胞や手選プロセス(hand−picking process)が巨核球生成に関与しないことから、本プラットフォームは、無間質血小板生成の条件の確立も可能とする。これは、最近発表された3Dバイオリアクター系30を含む生体外血小板生成のための無間質条件が他所にて記述されていることから、技術的に可能である。エストラジオール、ビタミンB3、および細胞外マトリックスタンパク質を含む因子が血小板生成を向上させることは、既に報告されている31‐33。これらの因子による、間質細胞なしでの巨核球成熟および血小板生成の刺激について、本系で試験される。
【0040】
無血清および無間質条件下で巨核球が作製されることにより、十分に定義された条件下での巨核球形成および血小板新生の制御に不可欠である因子のスクリーニングが可能となる。このような今後の研究で識別される因子は、将来的な臨床適用に寄与する可能性がある。この分野での進歩はまた、系列拘束(lineage commitment)、増殖、および成熟を含む巨核球形成の種々の側面を制御する細胞および分子メカニズムに関する見識も恐らくは提供することになるであろう。芽細胞培養物中に二分化能CD41a+CD235a+集団を発明者らが識別したことは、赤血球対巨核球細胞の系列特定化(lineage specifications)に関するメカニズムの研究を確実に促進させるであろう。最近、Klimchenko et al.は、OP9間質細胞上でのヒトES細胞の造血分化からの、CD41aおよびCD235aの両方を共発現する類似の巨核球/赤血球二分化能細胞を報告した34。これらの細胞は、赤血球新生対巨核球形成の分岐点である可能性が高い。この集団についての詳細な研究は、造血の過程でのこの重要なプロセスの制御に関与する事象に光を当てる可能性が高い。
【0041】
この系は、hESCからの巨核球分化の段階的な誘導を一体化するものである。工程内制御のさらなる最適化および確立を行うことで、臨床適用のための本系の一貫性および効率性を改善することができる。現段階では、巨核球成熟を制御する基本的な細胞または細胞外メカニズムは、完全には明らかにされていない。MK培養物中においてこれらの細胞の大部分が依然として二倍体であることから、生体外で得られた巨核球の最終分化を促進するためには、倍数体化および細胞質成熟を促進するその他の因子を識別して本系に含める必要があり得る。例えば、系において、ROCKキナーゼ阻害剤は、初期段階で巨核球の核内分裂を誘発することができる。しかし、この効果は、分化する巨核球の細胞および核成熟を調整した結果というよりは、染色体分配および細胞質分裂の人工的なブロッキングに起因する可能性が高い。生体外巨核球収率、最終分化状態、および下流での明らかな条件下における機能的血小板の作製について最良の結果を達成するために、増殖、核内分裂、および細胞質成熟の間のバランスを得ることが重要である。
【0042】
これらの実績は、生体外で得られた血小板が、アゴニスト刺激によって誘発されるインテグリン受容体リモデリングおよび血小板伸展に対して応答性であることを示した。さらに、生体外で得られた血小板は、血漿血小板と一緒に作用して、凝集体を形成することができる。このような生体外で得られたヒト血小板は、生体内でも機能することができる。ヒトES細胞由来血小板に対して発明者らが実施したFACS特性評価および3つの機能性試験(PAC1結合アッセイ、伸展アッセイ、および微小凝集体アッセイ)は、生体外で作製された血小板が、正常血液血小板の形態的および機能的特性を共有していることを示している。Nichii et al.による最近の報告によると、生体外で得られたマウスES細胞由来血小板の生体内機能が試験され、有望な結果が得られた35。
【0043】
生体外研究では、生物内での血小板血栓の形成および広がりの過程で発生する無数の血行動態的事象を模倣することはできない。新しい生体内イメージング技術が利用可能となったことにより、複雑な生体内系において血管損傷後に発生する血小板依存性の血栓プロセスを直接調べ、定量化する手段が提供される。高速広視野生体内顕微鏡法を用いることで、発明者らは、hESC由来血小板が、正常ヒト血液血小板に類似して、生体マウスにおけるレーザー誘発細動脈壁損傷の部位で発生するマウス血小板血栓中に組み込まれることを示した。hESC由来血小板およびコントロール血小板をReoProで前処理すると、血栓中に組み込まれる血小板の数が大きく減少しており、このことから、この結合がαIIbβ3インテグリンによって媒介されることが確認された。これらの結果は、hESC由来血小板が、生体動物の血管損傷部位において機能性であるという貴重な証拠を提供するものである。
【0044】
血小板は、血管損傷に応答して、組織に、および互いに接着する無核細胞である。このプロセスは、主として血小板インテグリンαIIbβ3によって媒介され、これは、vWFおよびフィブリノゲンなどのいくつかの接着性基質と結合して、成長する血栓中の血小板を架橋し、これをさらに活性化する36。本明細書の結果は、hESCから生成された血小板が、生体外および生体動物内の両方において、正常血液血小板と機能的に類似していることを示している。hESC‐PLTは、フィブリノゲンおよびvWFコーティングした表面上で接着および伸展する能力、ならびに生理学的アゴニストによって刺激されると凝集する能力を含む、止血に関与する重要な機能を有することが示された。重要なことには、hESCから得られた血小板が、フィブリンクロットを退縮させることができるということが初めて示されている。この結果は、一次止血での中心的な役割に加えて、hESC‐PLTが、フィブリンとの相互作用によって凝固を促進することができ、インテグリンの媒介による退縮性の挙動によって創傷治癒を促進することを示している。
【0045】
重要な科学的及び臨床的問題は、hESC由来の血小板が、複雑な生体内の設定において機能的であるか否かということである。マウスの血栓形成を研究するための多数の実験モデルが、過去十年間に確立されており、それらには、近年幾つかのグループにより使用されたレーザー損傷血栓モデルが含まれる37;38;39。レーザー誘導性血栓モデルでは、損傷後5〜30秒の速さで血小板血栓形成が開始される。従って、このモデルは、迅速に除去されたヒト血小板及びhESC−PLTが、発生中のマウス血小板血栓に取り込まれるのをリアルタイムでモニタリングすることを可能にし、取り込みには、多数のシグナル伝達経路、酵素カスケード、並びに無数の細胞成分及びタンパク質成分の相互作用が伴う。また、このモデルは、トロンビン誘導性血栓症に関連する炎症反応を忠実に表す。
【0046】
レーザー誘導性血管損傷モデルを使用した生体顕微鏡分析は、hESC−PLTが、血液血小板のように、血管損傷後にαIIbβ3インテグリンにより、発生中のマウス血小板血栓に取り込まれることを示す。マウス血小板血栓と相互作用するhESC−PLTの数は、ヒト血液血小板と相互作用するhESC−PLTの数より少なく(図9C)、これは、活性化hESC−PLTに対するPAC−1の結合が、活性化ヒト血液血小板と比較して低いことよる可能性がある(図7G及び図11)。これらの結果は、hESC−PLTが、生体内の血管損傷部位で機能的であるという明らかな根拠を提供する。
【0047】
2つの先行研究は、MKがhESCから生成されることを報告している。これらの系での収量は非常に低く(本明細書で報告された収量より100倍を超えて低い)、現行の系と異なり、血清で補完された動物性間質細胞との共培養に依存する15;16。更に、生体外で機能的だったhESC−血小板の生成を示したのは、そのうちの1つだけであるが、それらの生体内での機能性は報告されなかった16。hESC分化におけるこれらの2つの変数を排除することにより、動物性産物と接触させずに血小板を生成することが可能になる。本研究で使用された無フィーダー系は、MKを高効率で生成することが観察されたが、無フィーダー条件下で機能的血小板を生成する最終ステップは、OP9細胞共培養系ほど効率的ではない。
【0048】
従って、これらの結果は、OP9間質細胞が、hESC−MKからの機能的血小板の生成を増強することができる追加的な支援因子を提供することを示唆する。根底にある機序は、まだ研究されていない。血小板新生は、膜及び微小管が精巧に再編成され、顆粒及び細胞小器官が正確に分布される高度に複雑なプロセスである40。血小板生合成の理解が近年進歩したにもかかわらず、膜再編成、胞体突起の開始、血小板細胞小器官及び分泌性顆粒の輸送、及び血小板サイズの制御の根底にある機械論的詳細は、まだ解明されていない。無血清及び無フィーダー条件下でMKを生成する能力は、明確に規定された条件下で、系列拘束、増殖、及び成熟を含む巨核球形成の様々な側面を制御するのに重要な因子のスクリーニングを支援するはずである。
【0049】
巨核球
本発明の種々の実施形態は、無間質条件下で、ヒト胚性幹細胞及び多能性幹細胞(iPSC及びhiPSCを含む)から機能的な巨核球を生成する方法を提供する。hiPSC及びhESCを含む多能性幹細胞は、血管芽細胞/芽球細胞を中間体として使用して、巨核球分化へと向けられた。
【0050】
1つの実施形態では、本方法は、血管芽細胞(芽球細胞とも呼ばれる)を準備すること、及び血管芽細胞を巨核球へと分化させることを含む。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。別の実施形態では、血管芽細胞は、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0051】
1つの実施形態では、血管芽細胞を巨核球へと分化させることは、6日目〜8日目の血管芽細胞を精製し、TPO、SCFで補完された培地に播種して、MKへのBC分化を誘導することを含む。種々の実施形態では、濃度は、TPO(50ng/ml)及びSCF(20ng/ml)である。別の実施形態では、濃度は、TPO(100ng/ml)及びSCF(50ng/ml)である。別の実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。更なる実施形態では、MKは、CD41a、CD42a、及びCD42bを発現する。代替的な実施形態では、培地は、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。更なる実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地を、2〜3日毎に新しい培地と交換することを更に含む。別の実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを含む。代替的な実施形態では、本方法は、TPO、SCF、及び/又はIL11で補完された培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。種々の実施形態では、約1〜5×105細胞/mlが、播種される。種々の実施形態では、Stemline II培地が使用される。
【0052】
別の実施形態では、本発明は、細胞分化の修飾因子をスクリーニングする方法であって、ある量の巨核球(MK)を準備すること、MKを試験化合物と接触させること、及びMKと試験化合物との接触に由来する機能的効果の存在又は非存在を決定することを含み、機能的効果の存在が、試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子(hematopoeitic factor)であることを示し、機能的効果の非存在が、試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子でないことを示す方法を提供する。他の実施形態では、巨核球形成、血小板新生、及び/又は造血因子は、機能的血小板の増殖、核内分裂、細胞質成熟、及び最終分化に関連する。
【0053】
血小板
本発明の他の実施形態は、ヒト胚性幹細胞及び多能性幹細胞(iPSC及びhiPSCを含む)から血小板を生成する方法を提供する。1つの実施形態では、本方法は、ヒト胚性幹細胞(hESC)を準備すること、胚様体(EB)を形成すること、血管芽細胞(芽球細胞(BC)とも呼ばれる)を生成すること、BCを巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0054】
本発明の他の実施形態は、多能性幹細胞(pluipotent stem cell)から血小板を生成する方法を提供する。1つの実施形態では、本方法は、多能性幹細胞を準備すること、胚様体(EB)を形成すること、血管芽細胞(芽球細胞(BC)とも呼ばれる)を生成すること、BCを巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。1つの実施形態では、iPSCは、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0055】
別の実施形態では、血小板を生成する方法は、血管芽細胞を準備すること、血管芽細胞を巨核球へと分化させること、及び巨核球を血小板へと分化させることを含む。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。血管芽細胞を巨核球へと分化させるプロセスは、上述のように実施することができる。1つの実施形態では、血管芽細胞は、ヒト血管芽細胞である。別の実施形態では、血管芽細胞は、hESCに由来する。別の実施形態では、血管芽細胞は、誘導多能性幹細胞(iPSC)に由来する。1つの実施形態では、iPSCは、体細胞の再プログラミングに由来するヒトiPSC(hiPSC)である。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。代替的な実施形態では、血管芽細胞は、非生着血管芽細胞であってもよい。
【0056】
種々の実施形態では、巨核球を血小板へと分化させるプロセスは、巨核球の培養を継続して、巨核球が血小板へと分化することを可能にすることを含む。種々の実施形態では、巨核球を血小板へと分化させるプロセスは、無フィーダー条件下であり、4日目〜6日目の巨核球培養物から巨核球を収集すること、及びTPO、SCF、及びヘパリンナトリウムで補完された培地に巨核球を再懸濁することを含む。ある実施形態では、TPO(100ng/ml)、SCF(50ng/ml)、及びヘパリンナトリウム(25単位/ml)の濃度である。ある実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。代替的な実施形態では、培地は、IL3、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。ある実施形態では、IL3が、4日目〜7日目の巨核球培養物に添加される。別の実施形態では、メタロプロテイナーゼ阻害剤GM6001が、4日目以降の巨核球(megakaryoyte)培養物に添加される。ある実施形態では、培地は、IMDM培地である。更なる実施形態では、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地は、2日毎に交換される。別の実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを更に含む。代替的な実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。更なる実施形態では、血小板は、4日目〜12日目の巨核球培養物から収集される。種々の実施形態では、巨核球を血小板へと分化させるプロセスは、巨核球の培養を継続して、巨核球が血小板へと分化することを可能にすることを含む。種々の実施形態では、様々な因子が、巨核球からの血小板形成の効率を向上させる。種々の実施形態では、IL6、IL3、Notch、又は他のタイプのサイトカイン、インターロイキン、成長因子、低分子、及びそれらの組み合わせは、巨核球からの血小板分化の効率を向上させる。
【0057】
他の実施形態では、巨核球を血小板へと分化させる方法は、OP9間質細胞又はC3H 10T1/2細胞と共に巨核球を共培養することを含む。種々の実施形態では、OP9間質細胞又はC3H 10T1/2細胞と共に巨核球を共培養することは、4日目〜6日目の巨核球培養物から巨核球を収集すること、及びTPO、SCF、及びヘパリンナトリウムで補完された培地に巨核球を再懸濁することを含む。ある実施形態では、TPO(100ng/ml)、SCF(50ng/ml)、及びヘパリンナトリウム(25単位/ml)の濃度である。ある実施形態では、培地は、IL11(例えば、20ng/ml)ででも補完される。代替的な実施形態では、培地は、IL3、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。ある実施形態では、IL3が、4日目〜7日目の巨核球培養物に添加される。別の実施形態では、メタロプロテイナーゼ阻害剤GM6001が、4日目以降の巨核球培養物に添加される。更なる実施形態では、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地は、2日毎に交換される。別の実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の少なくとも一部を、2〜3日毎に新しい培地と交換することを更に含む。代替的な実施形態では、本方法は、TPO、SCF、及びヘパリンナトリウム、並びに随意にIL11を含む培地の半分を、2〜3日毎に新しい培地と交換することを更に含む。種々の実施形態では、様々な因子が、巨核球からの血小板形成の効率を向上させる。種々の実施形態では、IL6、IL3、Notch、又は他のタイプのサイトカイン、インターロイキン、成長因子、低分子、及びそれらの組み合わせは、巨核球からの血小板分化の効率を向上させる。
【0058】
更なる実施形態では、血小板は、4日目〜12日目の巨核球培養物から収集される。ある実施形態では、血小板は、密度勾配遠心分離を使用して精製される。更なる実施形態では、密度勾配遠心分離は、パーコール培地を使用する。別の実施形態では、血小板精製法は、CD41a陰性の粒子を分離する。別の実施形態では、血小板精製法は、細胞生存能及び形態学的完全性を保持する。他の実施形態では、血小板は、CD41a及びCD42bを発現する。他の実施形態では、血小板は、トロンビン刺激に応答する。別の実施形態では、血小板は、フィブリノゲン及びフォンビルブラント因子(vWF)表面に伸展する(spread)ことができる。代替的な実施形態では、血小板は、PAC−1結合及びインテグリン活性化の能力を有する。別の実施形態では、血小板は、微小凝集体を形成し、クロット形成及び退縮を促進する。別の実施形態では、血小板は、アピラーゼ及び/又はEDTAの存在下で活性化されない。
【0059】
本発明の種々の実施形態は、hESC由来の血小板を使用する方法を提供する。ある実施形態では、hESC由来の血小板は、血小板輸血で使用される。本方法は、ある量のhESC由来血小板を準備すること、及びある量のhESC由来血小板をその必要がある対象体に投与することを含んでいてもよい。種々の実施形態では、hESC由来の血小板は、患者と一致した血小板であってもよい。別の実施形態では、血小板は、iPSCに由来する。ある実施形態では、血小板は、hiPSCに由来する。他の実施形態では、血小板は、血小板を対象体に投与した際に、対象体においてHLA同種免疫原性反応に寄与しない溶液中で保管される。
【0060】
血管芽細胞
種々の実施形態では、血管芽細胞は、ヒト胚性幹細胞(hESC)を準備すること、胚様体(EB)を形成すること、及び芽球細胞(BC)を生成することを含む方法より生成することができる。代替的な実施形態では、本方法は、多能性幹細胞(iPSC及びhiPSCを含む)を準備すること、胚様体(EB)を形成すること、及び芽球細胞(BC)を生成することを含む。ある実施形態では、iPSCは、体細胞の再プログラミングに由来する。1つの実施形態では、体細胞は、胎児(仔)組織由来である。別の実施形態では、体細胞は、成体組織由来である。
【0061】
1つの実施形態では、EBを形成することは、hESCを解離すること、解離されたhESCを、BMP−4及びVEGFで補完された培地に約48時間播種すること、及びこの培地の半分を、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoを含む新しい培地と交換することを含む。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地は、2日毎に交換される。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の少なくとも一部は、2日毎に交換される。代替的な実施形態では、多能性幹細胞(iPSC及びhiPSCを含む)が、hESCの代りに使用される。
【0062】
種々の実施形態では、hESCを解離することは、トリプシンによりhESCを解離することを含む。1つの実施形態では、トリプシンの濃度は、0.05%である。種々の実施形態では、培地は、Stemline II培地である。種々の実施形態では、BMP−4及びVEGFの濃度は両方とも、50ng/mlである。種々の実施形態では、bFGF、SCF、Flt3リガンド、及びTpoの濃度は、bFGF(20ng/ml)、SCF(50ng/ml)、Flt3リガンド(50ng/ml)、及びTpo(50ng/ml)である。代替的な実施形態では、培地は、IL6、IL11、VEGF、bFGF、エストラジオール、ビタミンB3、及び/又は細胞外マトリックスタンパク質ででも補完される。別の実施形態では、EBを形成することは、細胞の継続的培養を更に含む。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地は、2日毎に交換される。別の実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の少なくとも一部は、2日毎に交換される。1つの実施形態では、BMP−4、VEGF、bFGF、SCF、Flt3リガンド、及び/又はTpoで補完された培地の半分は、2日毎に交換される。
【0063】
1つの実施形態では、血管芽細胞を生成することは、4日目にEB培養物から胚様体(EB)を収集すること、EBを解離すること、単一細胞懸濁物を得ること、生細胞を芽球コロニー増殖培地(BGM)と混合すること、及び生細胞を播種して血管芽細胞を生成することを含む。
【0064】
種々の実施形態では、EBを解離することは、トリプシンにより解離することを含む。種々の実施形態では、単一細胞懸濁物を得ることは、22ゲージ針及び40μm細胞ストレーナーに細胞を通すこと、遠心分離により細胞を収集すること、及び細胞を培地に再懸濁することを含む。1つの実施形態では、培地は、Stemline II培地である。種々の実施形態では、生細胞を芽球コロニー増殖培地(BGM)と混合することは、1〜2×105細胞/mlをBGMと混合することを含む。更なる実施形態では、芽球コロニーは、生細胞を播種した3〜4日後に形成され、血管芽細胞を生成する。更なる実施形態では、血管芽細胞は、急速に増殖する。別の実施形態では、血管芽細胞は、CD71陽性であり、CXCR4陽性であり、TPO受容体を発現する。別の実施形態では、血管芽細胞は、CD31陰性、CD34陰性、及びKDR陰性である。
【0065】
国際出願PCT/US09/43050号及び同PCT/US09/43043号は、双方ともに、2009年5月6日に出願され、その全内容が完全に記載されているかのように参照により本明細書に組み込まれ、血管芽細胞および非生着血管芽細胞を生成する際の追加的な手引きを提供する。
【実施例】
【0066】
以下の例は、特許請求されている発明をよりよく説明するために提供されており、本発明の範囲を限定すると解釈されるべきではない。特定の物質が言及される場合、それは例示目的に過ぎず、本発明を限定することは意図されていない。当業者であれば、発明的な能力を行使せず、本発明の範囲から逸脱することなく、同等の手段又は反応物を開発することができる。
【0067】
実施例1
hESCの培養
この研究で使用したヒトESC系統(HUES3、H1、MA01、及びMA09)を、以前に記述されているように維持した18。手短に言えば、8ng/mlのbFGFで補完された完全hESC増殖培地中のマイトマイシンC処理マウス胚線維芽細胞(MEF)上で、hESCを増殖させた。0.05%トリプシン−EDTAを使用して、60〜80%コンフルエンスで、3〜4日毎にhESCを継代した。37℃、5%CO2下で細胞を維持し、培地を毎日補充した。
【0068】
実施例2
胚様体(EB)形成及び芽球細胞(BC)の生成
EB形成及びBC生成の手順は、以前に報告されているように実施した17;19。手短に述べると、70%コンフルエンスのhESCを0.05%トリプシンで解離し、BMP−4、VEGF(50ng/ml)で補完されたStemline II(Sigma社)培地を有するUltra−Lowディッシュ(Corning社、ニューヨーク州)に播種した。48時間後に、培地の半分を、同じサイトカインに加えて、bFGF 20ng/ml、SCF、Flt3リガンド(FL)、及びTpo(各50ng/ml、R&D System社)を含有する新しい培地に交換した。4日目にEBを収集し、0.05%トリプシンで解離した。単一細胞懸濁物を、22ゲージ針及び40μm細胞ストレーナーに細胞を通すことにより得、遠心分離により収集し、Stemline II培地に再懸濁した。生細胞をトリパンブルー排除法により計数し、1〜2×105個/mlを、以前に記述されているように芽球コロニー増殖培地(BGM)と混合し、Ultra−Low皿に播種した。播種した3〜4日後に芽球コロニーが観察され、その後急速に増殖した。
【0069】
実施例3
巨核球分化培養
6〜8日目の芽球培養物に由来するBCを精製し、50ng/mlのTPO、20ng/mlのSCF、及び他のサイトカインで補完されたStemline II培地に播種して(1〜5×105個/ml)、MKへのBC分化を誘導した。MK培地の半分を、2日又は3日毎に新しい培地と交換した。
【0070】
OP9間質細胞を、15%ウシ胎仔血清(Hyclone社)を有するα−MEM中で維持した。コンフルエントなOP9細胞を、共培養の前日に100ng/mlマイトマイシンCで処理した。細胞をPBSで穏やかに2回洗浄し、共培養の前にOP9培地中で一晩回復させた。OP9共培養の場合、4〜6日目のMK培養物に由来する巨核球を収集し、Takayamaらによる記載のように16、100ng/mlのTPO、50ng/mlのSCF、及び25単位/mlのヘパリンナトリウムで補完されたIMDM培地に再懸濁し、2日毎に新しくした。血小板様粒子を、4日目から12日目まで分析用に収集した。
【0071】
実施例4
FACS分析
芽球培養又は巨核球培養に由来する細胞を、FACS Caliberでフローサイトメトリー分析により日常的にモニターする。系列マーカー、CD41a、CD42a、CD42b、及びCD235aに対する蛍光色素(Flurochrome)結合抗体(BD Biosciences社)を使用して、巨核球及び赤血球系列を特徴付けた。抗体は、5%新生仔ウシ血清を有するPBSで新しく調製した(CD42a及びCD42b抗体の場合は1:100希釈、CD41a抗体の場合は1:250希釈、CD235a抗体の場合は1:2000)。典型的には、1〜2×105細胞を抗体標識に使用した。細胞を、氷上で1時間100μl抗体混合液中で染色し、その後緩衝液で2回洗浄し、1μg/mlヨウ化プロピジウムで補完された250μl緩衝液に再懸濁した。HLA−ABCの発現レベルを検出するために、血小板を、フルオレセインイソチオシアネート(FITC)結合抗ヒトHLA−ABC抗体又は対照としてのFITC結合マウス免疫グロブリンG(IgG)と共にインキュベートした。その後、フローサイトメーター(FACSCalibur、Becton Dickinson社製)を使用して試料を分析し、データは、Cellquest又はFlowjoソフトウェアを使用して分析した。細胞選別は、UMASSメディカルスクールコア施設のBD FACS Ariaシステムで実施した。選別した細胞を、1000rpmで10分間遠心分離することにより収集し、コロニー形成アッセイ用の適切な培地に再懸濁した。
【0072】
実施例5
Megacult−C及びメチルセルロースを用いたコロニー形成アッセイ
Megacult−C及びH4436メチルセルロース培地(Stem Cell Technology社製)を、CFU−MK及びCFU−Eコロニー形成アッセイに使用した。播種の10日後に、製造業者の指示に従って、CFU−MK培養物を脱水し、固定し、抗CD41抗体で染色した。Megacult−Cプロトコルに提供されている基準に従って、CFU−MKコロニーをスコア化した。CFU−Eコロニーを、12日目にスコア化した。
【0073】
実施例6
サイトスピン調製、ギムザ染色、及び免疫蛍光顕微鏡法
芽球培養物又は巨核球培養物のいずれかに由来する細胞(1〜2×104個)を、サイト遠心分離(Cytopro社製)により、ポリリシンをコーティングしたスライドにサイトスピンした。スライドは、Wright−Giemsa(Sigma社製)又は免疫蛍光染色のいずれかに使用した。抗CD41(DAKO社製、1:100)及び抗vWF(DAKO社製、1:200)抗体を、サイトスピン調製で巨核球を特定するために使用した。インキュベーションは全て室温で実施した。細胞を、無動物性ブロッカー(Vector Laboratories社製)で30分間ブロッキングした。細胞を、一次抗体と共に1時間インキュベートし、その後PBSで3回洗浄した。以下のインキュベーション及び洗浄は、暗所で実施した。その後、細胞を二次抗体(各々、1:200)と共に30分間インキュベートした。細胞を再びPBSで3回洗浄した。PBS中のヘキスト染料(1μg/ml)を使用して核DNAを5分間染色し、その後追加的な3×PBS洗浄を行った。その後、スライドを蛍光顕微鏡(オリンパスBXV)下にマウントし、検査した。蛍光画像を、QICAM Fastカメラ(Qimaging社製、カナダ)を使用することにより取り込み、QCapture Proバージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で分析した。Nikon社製顕微鏡、PAXCAMデジタルカメラ、及びPAX−itソフトウェアを使用して、位相差生細胞画像を取り込んだ。
【0074】
実施例7
血小板の調製
ヒト末梢血小板を、1U/mLアピラーゼ及び5mM EDTA(Sigma−Aldrich社製、セントルイス、ミズーリ州)と共に、血小板豊富な血漿(AllCells社製、エメリービル、カリフォルニア州)を分画遠心分離することにより単離した。血小板を洗浄し、改変タイロード緩衝液(12mM NaHCO3、138mM NaCl、5.5mMグルコース、2.9mM KCl、0.42mM NaHPO4、10mM HEPES、1mM CaCl2、1mM MgCl2)に再懸濁した。hESC由来の血小板(ES−PLT)を含有する培地を穏やかに収集し、アピラーゼ(1U/ml)及びEDTA(5mM)(Sigma−Aldrich社製、セントルイス、ミズーリ州)を添加して血小板活性化を防止し、その後細胞を300gで10分間遠心して細胞をペレットにした。上清を新しいチューブに移し、アピラーゼ(1U/ml)及びEDTA(5mM)の存在下で10分間1000gで遠心分離して、血小板を収集した。その後、血小板を1回洗浄し、改変タイロード緩衝液に再懸濁した。洗浄した血小板を、機能性アッセイの前に1〜2時間37℃でインキュベートした。
【0075】
実施例8
固定化されたフィブリノゲン上での血小板の伸展
マイクロタイターウエルを有するチャンバースライド(Nalgen Nunc社製、ロチェスター、ニューヨーク州)を、0.1M NaHCO3(pH8.3)中の100pg/mLフィブリノゲン(Sigma社製)で一晩4℃でコーティングした。洗浄したヒト末梢血小板又はhESC−血小板(1×107個/mL)を、フィブリノゲンをコーティングしたウエルに90分間37℃で付着及び伸展させた。幾つかの試料では、血小板を、負荷前に5分間、インテグリンアンタゴニスト、RGDSペプチドと共にプレインキュベートした。他の試料では、血小板を、ADP(10μM)又はトロンビン(1U/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)と混合し、フィブリノゲンをコーティングしたウエルに直ちに負荷した。PBSで洗浄した後、細胞を固定し、透過処理し、Alexa Fluor568ファロイジン(Molecular Probes社製、ユージーン、オレゴン州)、FITC結合抗ヒトCD41a抗体(Dako cytomation社製、カーピンテリア、カリフォルニア州)、及びDAPIで染色した。付着血小板を、100×/1.40オイル対物レンズのPlanApoレンズを使用して、オリンパスBX51蛍光顕微鏡(MVI社、エイボン、マサチューセッツ州)で観察した。画像を、QICAM Fastカメラ(QImaging社製、サリー、ブリティッシュコロンビア州、カナダ)を使用することにより取得し、Q Capture バージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で処理した。
【0076】
実施例9
血小板凝集
洗浄したヒト末梢血小板及びhESC−血小板を、改変タイロード緩衝液に再懸濁し、PKH67緑色蛍光細胞リンカー(10μM、Sigma社製、セントルイス、ミズーリ州)で標識した。ヒト末梢血小板(6×107個)を、450μlキュベット(Chronolog社製、ヘイバータウン、ペンシルベニア州)中で、蛍光標識したヒト末梢血小板(3×105個)又はhESC−血小板(3×105個)と混合し、その後トロンビン(0.5U/mL)を添加し、1200rpm、37℃で撹拌して血小板凝集を開始させた。対照実験では、トロンビンを添加する前に、RGDSペプチドを37℃で5分間血小板と共にプレインキュベートし、上記のように凝集アッセイを実施した。50μL緩衝液中の血小板微小凝集体を、スライドガラスに塗布し、蛍光顕微鏡で視覚化した。
【0077】
実施例10
PAC−1結合アッセイ
トロンビン刺激(1U/ml、室温で20分間のインキュベーション)をした又はしなかったES−PLTを、改変タイロード緩衝液中のAPC結合CD41a、PE結合CD42b、及びFITC結合PAC−1抗体で染色した。その後、フローサイトメーター(FACSCalibur、Becton Dickinson社製)を使用して試料を分析し、ヒト血液血小板を対照として使用して、前方散乱及び側方散乱ゲーティングを決定した。FACSデータは、Cellquest又はFlowjoソフトウェアを使用して分析した。
【0078】
実施例11
hESCに由来する芽球細胞(BC)は巨核球を生成した
本発明者らの以前の研究は、適切な条件下で、hESCに由来するBCを赤血球細胞へと生体外で効率的に分化させることができることを示した20。巨核球(MK)および赤血球は、哺乳動物造血中に共通の前駆体を共有するため、本発明者らは、hESCに由来するBCも、TPO及び他のサイトカインを有する巨核球促進条件下で巨核球に分化することができると考えた。図1Aで示されているのと類似した戦略を考案して、hESCからMKを分化させた。以前に報告されるように17;19、初期EBを、モルフォゲン及び初期造血サイトカインの組み合わせで補完された無血清培地中で培養されたhESCから生成した。その後、EBを解離し、個々の細胞を、BCの成長及び増殖用の無血清半固形芽球コロニー増殖培地(BGM)に播種した。ブドウ様芽球コロニーが、3日間の培養の始めに最初に出現し、4日目から6日目まで急速に増殖した。この段階で、BCは、サイトスピン調製では形態が比較的均質であり、直径が約15μmのサイズを有する(図1B)。これらのBCを、コロニー形成単位(CFU)−MKアッセイにより、それらが巨核球になる能力について最初に試験した。図1C及び1Dに示されているように、HUES3及びH1細胞の両方に由来する6日目のBCは、再播種後10〜12日目にCFU−MKコロニーを発生させ、これらのコロニーは、CD41に陽性染色された。幾つかのCFU−MKコロニーでは、細胞プロセスが類似しており、CD41陽性細胞で胞体突起が観察された(図1D挿入部分)。これらの結果は、hESC由来のBCが、巨核球に分化することができることを示した。同時に、それらのCFU−MK能力における有意差が、HUES3及びH1細胞系統間で観察された。HUES3に由来するBCは、同数のH1芽球細胞より、およそ3.5倍より多くのCFU−MKコロニーを生成した。
【0079】
初期BGM培地は、効率的なBC生成のためのEPOを含有しており、赤血球細胞(CD235a+)は、芽球培養の後期で優勢になる20。本発明者らの実験の結果は、EB形成の第2期(hESC播種後2日間)並びに芽球コロニー成長及び増殖物中のより高濃度のTPO(100ng/ml)及びSCF(50ng/ml)が、芽球培養の終了時のCD41a+細胞の割合を増加させたことを示した。BGM培地にIL−11(20ng/ml)を添加することにより、6〜8日目の芽球培養中のCD41a+細胞の比率が更に増加された。従って、本発明者らは、本研究において、これらの改変を芽球培養に応用して、巨核球系列拘束を向上させた。HUES3、MA01、及びMA09 hESC系統を、この改変条件を用いて試験した。3つの系統は全て、巨核球になる能力を示した。CFU−MKアッセイでなされた観察と同様に(図1C)、様々なhESC系統から生成されたBC(6〜8日目)中のCD41a+細胞の割合における有意差が、FACS分析により観察された。例えば、HUES3細胞に由来するBCの32.9+8.4%(n=13)は、MA01及びMA09細胞系統よりも有意に多かった(P<0.001、スチューデント検定)。MA01又はMA09由来の芽球細胞では、これらの細胞の15.3%+5.8%(n=5)又は17.2%+3.7%(n=5)が、それぞれCD41a+だった(図1E)。
【0080】
実施例12
二分化能芽球細胞は、赤血球及び巨核球を両方とも生じさせた
主な増殖は、芽球培養では5日目〜8日目まで生じ、幾つかの芽球コロニーは、この期間中に赤みを帯び、赤血球拘束及びヘモグロビン合成を示した。FACS分析は、巨核球マーカーCD41a及び赤血球マーカーCD235aの発現により規定されるBCに、4つの異なる細胞集団:CD41a+CD235a−、CD41a−CD235a+、CD41a+CD235a+、及びCD41a−CD235a−集団が存在したことを示した(図2A)。CD41a及びCD235a二重陽性集団は、赤血球系列及び巨核球系列の両方のマーカーを共有するため、これらの細胞が両系列の二分化能前駆体であるか否かを試験した。
【0081】
その後、6日目の芽球培養に由来する、選別されたCD41a+CD235a−、CD41a−CD235a+、及びCD41a+CD235a+細胞を、2つのコロニー形成アッセイにより、CFU−MK又はCFU−E前駆体のいずれかについて試験した。これらのコロニーアッセイのために、等数の選別された細胞を播種した。予想通り、CD41a+CD235a−及びCD41a−CD235a+は、それぞれCFU−MK及びCFU−Eコロニーを生成した。同時に、類似した数のCFU−MKコロニーが、CD41a+CD235a−及びCD41a+CD235a+細胞により生成され、同程度の数のCFU−Eが、CD41a−CD235a+及びCD41a+CD235a+細胞により発生された(図2B及びC)。従って、これらの結果は、CD41a+CD235a+細胞が、巨核球系列及び赤血球系列の両方の二分化能前駆体であることを示す。これらのCD41a+CD235a+細胞は、一時的な集団であると考えられ、巨核球分化培養で培養された直後に消失した。
【0082】
実施例13
hESC由来BCからの巨核球増殖及び成熟を指図する明確な培養条件の確立
巨核球の分化を更に指図するために、メチルセルロース半固形培養に由来する6日目〜8日目のBCを回収し、その後、無血清培地、つまりMK培養用のサイトカインで補完されたStemline IIに再懸濁した。CD41a+細胞の細胞数を測定値として使用して、懸濁培養中に増殖巨核球を決定した。それは、CD41a+(FACS)の割合と総生細胞数(トリパンブルー排除法)を乗算することにより計算した。初期試験は、高濃度TPO(50〜100μg)単独では、BCからのCD41a+巨核球の増殖を支援するのに十分でなかったことを示した。培地にSCF(20ng/ml)を添加することにより、CD41a+巨核球の収量が有意に増加した(図3A)。BCからの巨核球の高い純度及び総収量を達成するために、一群のサイトカイン及び因子も試験した。IL9及びBMP4を含むこれらの因子は、他の系において巨核球促進因子として以前に報告されていた21;22。
【0083】
図3Aに示されているように、IL9及びBMP4の添加は、TPO及びSCFのみと比較して、CD41+細胞の収量を向上させる明らかな効果を示さなかった。しかしながら、ある因子は、CD41+細胞の割合をわずかに増加させた(非表示)。IL6、IL11、VEGF、及びbFGFは全て、追加的栄養補給をしない4日間の液体培養で巨核球の収量をわずかに増加させた。しかしながら、これらの因子を一緒に混合することは、一貫した相乗効果を示さなかった(非表示)。加えて、血清又はメチルセルロースマトリックスを培養物に添加することも試みられた。これらの改変は、CD41巨核球の収量を有意には向上させなかった。注目すべきには、巨核球核内分裂及び胞体突起形成を促進することが示された23;24特定のROCKシグナル伝達阻害剤(Y27632)も試験した。4日間のMK培養中に10μMのROCK阻害剤を包含させることは、倍数体細胞の割合を劇的に増加させた(データ非表示)。しかしながら、ROCK阻害剤は、同時に4日間の培養の終了時のCD41+細胞数を低減させた。従って、ROCK阻害剤は、最終MK培養条件から除外された。
【0084】
初期試験の結果に基づき、BCからの至適化巨核球分化及び増殖を達成するために、TPO 50ng/ml、SCF 20ng/ml、及びIL11 20ng/mlをMK培地に含めた。IL11は、巨核球の増殖及び成熟の両方を増強することが以前に報告された25;26。図3Bは、液体培養の経時的実験を示す。6日間の培養期間にわたって、CD41+巨核球の細胞数が5倍増加したことが、本条件下で観察された。6日目のBCS及び4日目のMK培養物に対するFACS分析を、2つの巨核球マーカーCD41a、CD42b、及び赤血球マーカーCD235aについて実施した。MK培養では、赤血球細胞(CD235a+)は、2〜3日で急速に死滅する。それとは際立って対照的に、CD41a+巨核球は、劇的に増加した。MK培養の4日目までに、90%を超える生細胞がCD41aを発現し、これらの細胞のおよそ80%は、成熟中の巨核球に重要な機能的役割を果たす27CD42bにも陽性であった(図3C)。
【0085】
実施例14
BCに由来する巨核球の特徴付け
巨核球の分化及び成熟を示す細胞表面マーカーの変化と一致して、平均細胞サイズは、MK培養中に徐々に増加した。成熟中の巨核球は、細胞サイズがより大きく、より不均一になった(図4A)。1日目のMK培養では、約65%の細胞は、10〜20μmの直径を有していた。残り細胞は、20〜30μmだった。6日目までに、20%を超える細胞は、サイトスピン調製での直径が40ミクロンよりも大きかった。数パーセントの細胞は、MK培養で倍数体細胞の表現型を示した(図4B)。CD42b(GP1b−アルファ)に加えて、糖タンパク質CD42a(GPIX)も、MK培養に由来するCD41a+巨核球内で高度に発現された(図4C)。CD41及びvWFの免疫蛍光染色も実施して、巨核球細胞質顆粒での特徴的なvWF発現を検査した。図4Dに示されているように、成熟巨核球表現型を有する大型の倍数体細胞は、CD41に陽性染色され、細胞質におけるvWFの顆粒状蓄積が検出された。
【0086】
更に、RT−PCR分析を実施して、GATA1、GATA2、Fli1、FOG、及びNFE2(図4E)を含む、赤血球/巨核球形成の重要な転写修飾因子の発現レベルを検査した。これらの転写因子は全て、胚様体段階で強力に誘導され、6日目の芽球培養で発現持続を示した。興味深いことには、BCが巨核球分化及び成熟に向けられた場合、GATA2及びFOGのRNAレベルは、4日目のMK培養で下方制御された。しかしながら、この観察の機械論的意味は、まだ判明していない。RT−PCRの結果は、ES、EB、BC、及びMK培養中に5つの赤血球−巨核球転写因子GATA1、GATA2、Fli1、FOG1、及びNF−E2の発現を示した。
【0087】
実施例15
hESCから巨核球への増殖
細胞系統間の効率は様々であるが、試験したhESC系統は全て、無血清及び無間質培養条件下で巨核球(CD41a+と規定された)を生成することができた。hESCsからEB(増殖、1.5〜6倍)、芽球(5〜12倍)、及びMK培養(1.5〜4倍)を経由して増殖した後、この系は、ヒトES細胞から開始して、100万個から最大1億個のCD41a+巨核球を産生することが可能である。HUES3、及び2つの臨床グレードの単一卵割球由来hESC系統、MA01及びMA09の巨核球産生の能力は、表1に要約されている。
【0088】
表1.hESCからの巨核球の生成
MK培養に由来するCD41a+細胞の割合を、FACSにより決定した。hESCから巨核球への総増殖倍数を、EB、芽球、及びMK培養までの各ステップの増殖倍数を乗算することにより計算した。HuES3実験Bは、100万個のES細胞からのMK細胞の実測計数値である。
【表1】
【0089】
実施例16
hiPSCは、血管芽細胞/BC、造血及び内皮細胞に生体外で分化することが可能である
本発明者らは、Thomson再プログラム法(Oct−4、Sox−2、Nanog、及びLin28のレンチウイルス発現)により生成されたhiPSCを使用して、同様の結果を達成した。これは、細胞系統IMR90−1、Foreskin−1−1、Foreskin−4−1、及びForeskin−4−3を含む。それとは別に、Yamanaka再プログラム法(Oct−4、Sox−2、Klf4、及びc−Mycのレトロウイルス発現)により生成されたhiPSCを同様に実施した。これは、細胞系統rv−hiPS01、rv−hiPS02、rvhiPS03、及びrv−hiPS04を含む。これらのhiPSC系統は、多分化能の標準的マーカーを発現し、報告されているようにSCIDマウスへの接種後に奇形腫を形成し、この系統は全て、hESCと形態学的に区別不能だった(データ非表示)。本明細書に記載の方法を使用して、hiPSC系統は、BCを生成することが可能だった。まず、hiPSCを、BC発生用に至適化された条件下でEBに分化させ、個々のEB細胞を、芽球コロニー発生用の芽球成長/増殖培地(BGM)に播種した。全てのhiPSC系統に由来するEB細胞は、播種の6日後に芽球コロニーを発生させた。様々な効率が、様々なhiPSC系統で観察された(表2)。にもかかわらず、hESCに由来するBCで観察されたように(図5A)、hiPSCに由来するこれらのBCは、血管芽細胞マーカーCD71、CXCR−4、及びTpo受容体を発現したが(図5B)、これらのBCの大多数は、免疫細胞化学的分析により示されたように、CD31、CD34、及びKDRを発現しなかった。広範なサイトカインで補完された造血コロニー形成培地に10〜14日間再播種した後、赤血球(CFU−E)、骨髄(CFU−G及びCFU−GM)、及びマクロファージ(CFU−M、非表示)造血細胞コロニーが発生した(図5C)。また、BCは、巨核球へと分化する能力を有するCFU−MKを形成した(図5C−iv)。更に、再プログラムタンパク質Oct4、Sox2、Klf4、及びc−Mycの形質移入等の、無ウイルスベクターを使用して生成されたhiPSCは、BC発育、MK及び血小板生成について同様の能力を示す。これらの結果は、様々な送達系(レンチウイルス、レトロウイルス、形質移入)を使用して様々な組み合わせの再プログラム因子を用いて生成されたhiPSCが、無血清条件下で血管芽細胞、造血及び内皮系列に分化することが可能であることを明らかに示す。
【表2】
*播種した100,000個のEB細胞当たり、(±)は、ほとんど0個のコロニー又は時には10個未満のコロニーが得られる結果を表し、(+)は、少数のコロニー(10〜30個)が一貫して得られる結果を表し、(++)は、最大50個の芽球コロニーの中程度の効率の収量を表す。
【0090】
実施例17
hESC由来の巨核球は、OP9間質細胞上で機能的な血小板を生成する
MK培養での4日目から開始して、少数(およそ1〜2%)のhESC由来巨核球は、胞体突起様プロセスを形成することが顕微鏡で一貫して観察された(図4F)。この観察は、無間質系で生成された巨核球が、最終分化プロセスまで進行することが可能であることを示唆する。
【0091】
近年、Takayamaらは、機能的なES由来血小板(ES−PLT)を産生するための、間質細胞OP9に基づく共培養系を記述した16。本発明者らは、細胞が血小板を機能的に製作可能であることを示すために、このOP9系上の巨核球を原理の証明として試験することにした。MK培養から生成された(HUES3、MA01、及びMA09に由来した)巨核球を、原報に記載のように、TPO 100ng/ml、SCF 50ng/ml、及びヘパリン 25U/mlで補完された同じ培地中の有系分裂的に停止されたOP9細胞上に播種した。胞体突起形成細胞(PFC)及び血小板様粒子(ES−PLT)が、OP9間質層上に播種した後4日以内に出現した。ESP−LTを、6日目から12日目まで2日毎に培地から収集した。100万個のhESC由来MKから、およそ20万〜50万個のES−PLTを、この期間中に培養物から回収することができる。以前の報告より低いES−PLT産生効率は、培養条件の抜本的な変更の結果であり得る。また、OP9との共培養は、播種した巨核球の倍数性を著しくは増加させなかった。これも低い血小板収量/巨核球を説明することができる。にもかかわらず、OP9共培養物から回収されたES−PLTを、正常な血液血小板に重要な機能性について更に試験した。
【0092】
実施例18
トロンビン活性化ES−PLTは、機能的なインテグリン受容体結合活性を有する
インテグリン媒介性血小板凝集は、生体内での血栓形成に不可欠である。循環中では、血小板は、その接着性受容体を介して損傷血管壁に付着する。初期の付着は、血小板インテグリンαIIbβ3の活性化に結び付くシグナル伝達カスケードを開始させ、血小板の安定的な接着、凝集、及び血栓形成に結び付く。トロンビン等の血小板アゴニストは、インテグリンαIIbβ3のリガンド結合機能を活性化する。その主要リガンド、フィブリノゲンの結合は、インテグリンに依存する血小板の安定的な接着及び凝集を媒介する。また、αIIbβ3の活性化は、細胞外から内部へのインテグリンシグナル伝達を媒介し、血小板細胞骨格再編成および伸展に結び付く。
【0093】
OP9共培養から生成されたES−PLTを、血小板の主要アゴニストであるトロンビンによる活性化について試験した。インテグリンαIIbβ3活性化を、FACS分析により、活性化形態のインテグリンαIIbβ3受容体にのみ結合するFITC結合PAC−1モノクローナル抗体を用いた結合アッセイにより検査した。タイロード緩衝液に再懸濁されたES−PLTを、抗ヒトCD41a(インテグリンαIIb)抗体、CD42b抗体、及びPAC−1抗体の混合液と共にインキュベートした。その後、それらの表面にCD41a及びCD42b糖タンパク質を両方とも発現するES−PLTを、PAC−1結合活性についてゲート制御した。トロンビン刺激に際して、PAC−1結合は、休止対照と比較して、トロンビン処理ES−PLTで増加した。この結果は、ES−PLTが、血小板アゴニストからの刺激に応答することを示した(図6A)。ES−PLTは、2つの臨床グレードのhESC系統、MA01及びMA09を含む、3つのhESC系統に由来した。3つの系統は全て、トロンビン刺激に応答してPAC−1結合が可能なES−PLTを生じさせることができた(図6B)。
【0094】
ES−PLTにおける細胞外から内部へのインテグリンシグナル伝達の機能的特性を、スライドガラスに固定化されたフィブリノゲン上での伸展アッセイにより更に評価した。スライドを抗ヒトCD41a抗体及びDAPIで染色して、血小板を特定した。ヒト血漿血小板対照と同様に、ES−PLTは、フィブリノゲンをコーティングした表面に接着及び伸展し、機能的な血小板と同様のF−アクチンフィラメントの再編成を示した。RGDSペプチドは、血小板インテグリン受容体に対するフィブリノゲン結合を阻止し、フィブリノゲンをコーティングした表面へのES−PLTの接着及び伸展を消失させ、伸展がインテグリン依存性であることを示した。アゴニストの非存在下で、幾つかの血小板は、主として糸状仮足形成を有するフィブリノゲン上で不完全な伸展を示した。血小板アゴニストADP又はトロンビンで刺激した場合、対照血漿血小板及びES−PLTは両方とも、葉状仮足及びアクチンストレス線維形成の増強を示した(図7A)。
【0095】
実施例19
ES−PLTは、微小血栓形成に寄与することができる
アゴニスト誘導性凝集を形成するそれらの能力、血小板の重要な機能を検査するために、ES−PLTを、緑色蛍光染料で標識し、血漿由来血小板と混合した。撹拌条件下でのトロンビン刺激に応答して、血小板微小凝集体が、位相差及び蛍光顕微鏡で観察された。蛍光標識hESC−血小板は、ヒト血漿由来血小板と一緒にそれら微小凝集体に均等に組み込まれた。トロンビン誘導性血小板凝集は、インテグリンアンタゴニスト、RGDSにより阻害され、凝集がインテグリン依存性であることを示した(図7B)これらの結果は、ES−PLTが、アゴニスト誘導性微小凝集体形成において血液血小板と一緒に作用することができることを示した。要約すると、これらの結果は、培養系に由来するES−PLTが、ヒト末梢血液血小板と機能的に同等であり、これらのES−PLTは、生理学的な血小板アゴニストにより刺激することができることを示した。
【0096】
実施例20
ES−PLT上でのHLAクラスIの発現
ヒト末梢血小板は、免疫関連血小板輸血不応状態(reflactive platelet transfusion)に寄付する場合があるクラスIヒト白血球抗原(HLA−ABC)を合成及び発現する28。従って、ES−PLT上でのHLAクラスIの発現も、フローサイトメトリーにより検査した。正常な血漿由来血小板は、高レベルのHLA−ABC発現を示した。ES−PLTは、HLAクラスIの発現レベル(平均蛍光強度)が同等であることを示した。血小板におけるHLAの新規合成に加えて、可溶性HLA抗原が、血漿から吸収され得ることも報告されている29。培養条件下でhESCに由来する血小板は、他の血液細胞に由来するHLA抗原を含有する血漿を回避し、従って血小板同種異系免疫化の頻度を低減することができる。
【0097】
実施例21
hESC由来の血管芽細胞/BCからのMKの大規模生成
実験を実施して、hESC由来BCも、MKに分化するように誘導することができるか否かを決定した。TPO、SCF、及びIL−11を含有する無血清Stemline II培地中で芽球培養物を6日間分化させた後、CD41+MKの5倍の増加が得られた。また、FACS分析は、分化の初期段階中に、CD235a+細胞(赤血球細胞)が急速に減少したが、CD41a+及びCD42b+細胞(MK)は劇的に増加したことを示した。4日目までに、90%を超える細胞がCD41aを発現し、およそ80%の細胞が、CD42b陽性だった(CD42bは、MKの成熟に重要な機能的役割を果たす)27(図8A)。CD42bに加えて、糖タンパク質CD42aも、CD41a+MKの表面に高度に発現された(データ非表示)。6日目までに、20%を超える細胞が40μmのサイズよりも大きく、30%を超える細胞が、4Nを超えるDNA含有量を有する倍数体だった(図8B及び8D)。大型の倍数体細胞上のCD41及びvWFについての免疫蛍光染色は、CD41陽性膜染色を有する細胞質におけるvWFの特徴的な顆粒状蓄積を示し(図8C)、細胞が成熟MKだったことを確認した。
【0098】
試験した全てのhESC系統(WA07[H7]、HuES−3、MA01、及びMA09)は、これらの無血清及び無フィーダー培養条件下でMKを生成したが、様々な効率が観察された(表3)。8つの別々な実験では、hESCからCD41a+MK細胞への総細胞増殖は、HuES−3及びMA09 hESC系統の場合、それぞれ30〜111倍及び18〜113倍の範囲であり、MA09 hESCの6つのウエルプレート当たり(1.0×107個のhESCとほぼ等しい、表3)最大6×108個のCD41a+MKが生成された。全体的な効率は、以前に発表された方法よりもおよそ2マグニチュード(two magnitudes)高い16。
【表3】
【0099】
培養に由来するCD41+MK細胞の数は、生細胞(トリパンブルー排除法)の総数とCD41a+細胞の割合(FACS分析)を乗算することにより計算した。hESCからMKへの総増殖倍数は、全てのEB細胞又はBCがその後の培養に使用された訳ではないため、EB、BC、及びMK培養までの各ステップの増殖倍数を乗算することにより計算した。
【0100】
実施例22
hESC−MKからの機能的血小板の生成
無血清及び無フィーダーMK培養で4日目から開始して、およそ1〜2%のMKが、胞体突起様細胞プロセスを形成することが観察された(図8E)。機能的な血小板が無フィーダー条件下で生成されたか否かを検査するために、本発明者らは、FACS分析により、hESC由来血小板(hESC−PLT)上でのCD41a/CD42b発現を検査した。本発明者らの結果は、無フィーダー条件下で生成されたhESC−PLT(血液血小板対照の前方散乱及び側方散乱パターンに従ってゲート制御された)の大多数が、CD41aを発現したことを示した。しかしながら、これらのhESC−PLTの5%未満が、CD42b陽性だった。最近の研究は、メタロプロテイナーゼ阻害剤GM6001が、マウスESC由来血小板上でのCD42bの発現を著しく増加させたことを示した35。ヒトESC−PLT分化の後期中にGM6001(100μM)を補完することにより、CD41a及びCD42b二重陽性hESC−PLTが、約15%に増加した。無フィーダーで生成されたhESC−PLTの機能を、固定化されたフィブリノゲン及びvWF上のインテグリン依存性伸展アッセイにより評価した。抗ヒトCD41a抗体及びDAPI染色を使用して、血小板を特定した。無フィーダーで生成されたhESC−PLTは、フィブリノゲン及びvWFをコーティングした表面上に伸展することができ(図11)、これらのhESC−PLTが生体外で機能的であることを示した。
【0101】
OP9又はC3H 10T1/2マウス間質細胞は、生体外血小板生合成を支援することが示された。OP9間質細胞との共培養が、hESC−MKからの機能的血小板の産生を促進したか否かを試験した。HuES−3、MA01、及びMA09細胞から生成されたMKを、以前に記述されているように16、TPO、SCF、及びヘパリンで補完された同じ培地中の有系分裂的に停止されたOP9細胞に播種した。胞体突起形成細胞及びhESC−PLTが、OP9間質層への播種後4日以内に出現した。hESC−PLTを、6日目から12日目まで2日毎に培地から収集し、CD41a及びCD42b発現について分析した。無間質条件下で生成されたhESC−PLTとは対照的に、大多数のCD41a+hESC−PLTも、CD42bを発現した(図8F、右パネル)。
【0102】
無フィーダー条件下で生成されたhESC−PLTと同様に、OP9細胞共培養で産生されたhESC−PLTは、フィブリノゲンをコーティングした表面(図9B)及びvWFをコーティングした表面に接着及び伸展した。RGDSペプチドは、血小板インテグリン受容体に対するフィブリノゲン結合を阻止し、フィブリノゲンをコーティングした表面へのヒト血液血小板及びhESC−PLTの両方の接着及び伸展を消失させた(図9B)。ファロイジン染色は、ヒト血液血小板での形成と同様に(図9A)、ADP及びトロンビンで刺激されたhESC−PLTでのF−アクチンストレス線維の形成を示した(図9B)。無フィーダー条件下で又はOP9間質細胞上で生成されたhESC−PLTのサイズは、ヒト血液血小板よりもわずかに大きく、マウスES細胞を用いて生体外で生成された血小板と類似する35。
【0103】
OP9共培養でHuES−3から生成されたhESC−PLTも、血小板の主要アゴニスト、トロンビンによる活性化について試験した。フィブリノゲン模倣体としてのPAC−1モノクローナル抗体は、活性化形態のインテグリンαIIbβ3受容体にのみ結合する。トロンビン刺激時に、PAC−1結合アッセイを、OP9共培養から生成されたhESC−PLTに対して実施した。トロンビン処理hESC−PLTでのPAC−1結合は、休止対照と比較しておよそ5倍数増加した(図8G)。トロンビン刺激後のPAC−1結合の増加は、末梢血小板対照の増加よりも弱いが(hESC−PLTの24.8%対血液血小板の58.6%、図12)、Takayamaらにより以前に報告されたhESCから生成された血小板と類似している16。2つの臨床グレードhESC系統、MA01及びMA09に由来するhESC−PLTも、CD41a及びCD42b抗原を発現し(図13A)、トロンビン刺激に応答してPAC−1に結合することができた(図13B)。また、hESC−PLTは、それらの表面にHLAクラスI抗原を発現した(図13C)。
【0104】
実施例23
hESC−PLTは、微小凝集体形成及びクロット形成/退縮に寄与する
損傷血管壁への血小板接着は、血小板インテグリンαIIbβ3の活性化に結び付くシグナル伝達カスケードを開始させる。活性化インテグリンは、血小板凝集及び血栓形成を媒介する45。アゴニスト誘導性凝集を形成するそれらの能力、血小板の重要な機能を検査するために、hESC−PLTを緑色蛍光染料で標識し、ヒト血液血小板と混合した。撹拌条件下でのトロンビン刺激に応答して、hESC−PLT及び血液血小板による微小凝集体の形成が、位相差及び蛍光顕微鏡法を使用して示された(図9D)。標識hESC−PLTは、ヒト血液血小板と一緒に微小凝集体に組み込まれた。これらの微小凝集体におけるhESC−PLTの分布は、蛍光染料で標識された同数の血液血小板の分布と類似していた(図9C)。これらの結果は、hESC−PLTが、アゴニスト誘導性微小凝集体形成においてヒト血液血小板と一緒に作用することを示す。
【0105】
血管損傷部位では、活性化された血小板が、血漿フィブリノゲンから生成されたフィブリンと相互作用し、血栓形成及び血餅退縮に結び付く46。血餅形成及び退縮を促進するそれらの能力を検査するために、hESC−PLTを血小板枯渇血漿に懸濁し、トロンビンを添加してクロット形成及び退縮を誘導した。追加的な血小板の補完をしない血小板枯渇血漿中では、1時間を超えるトロンビン刺激でさえクロットは形成されなかった。対照的に、血小板枯渇血漿へのhESC−PLTの添加は、トロンビン刺激後の30分未満でクロット形成及び退縮に結び付いた(図9E、左パネル)。hESC−PLT血栓の凍結切片の免疫蛍光染色は、ヒト血液血小板で形成されたクロット切片と同様に(図9F)、フィブリン及びCD41aの両方に陽性染色されたメッシュ様ネットワークを示した(図9E、右パネル)。これらの結果は、hESC−PLTが、フィブリンクロット形成を促進し、小型の血小板栓を形成するインテグリンαIIbβ3依存性収縮能力を有することを示す。
【0106】
実施例24
hESC−PLTは、生存マウスのレーザー誘導性細動脈損傷部位で、発生中の血小板血栓に組み込まれる
hESC−PLTが生体内で機能的か否かを決定するために、本発明者らは、生存マウスでの蛍光生体顕微鏡法を実施した。精巣挙筋細動脈を、micropointレーザアブレーションにより損傷させた。以前に記述されているように47、マウス血小板血栓が急速に形成され、Tmaxは血管損傷の70〜95秒後だった。ヒト末梢血小板、5〜10×105個を損傷後に注入したところ、41.0±2.8の循環血小板が、微小循環中で検出された(図10B)。それらの中で、6.4±1.1の血小板が、血管損傷後のTmax(73秒)で血管損傷部位にて、発生中のマウス血小板血栓に組み込まれた(図10A及び10C、ビデオ1(非表示))。発生中のマウス血小板血栓へのヒト血液血小板の組み込みが、αIIbβ3インテグリンにより媒介されることを確認するために、ReoPro(ヒト血小板上のαIIbβ3インテグリンに結合し、その機能を阻害するヒト−マウスキメラモノクローナル抗体のFab断片)48を用いて、注入前にヒト血小板を前処理した。循環血小板の数は、ReoProにより有意には影響を受けなかったが(32.4±4.4、図10B)、発生中の血小板血栓に結合するヒト血小板の数は、ReoProによる処置により、血管損傷後のTmax(67秒)で1.6±0.5に有意に低減された(図10A及び10C、ビデオ2(非表示)、P<0.01)。これらの結果は、ヒト血小板が、血管損傷部位で、発生中のマウス血小板血栓に結合し、そのような結合がαIIbβ3インテグリンにより媒介されることを示す。ReoProは、血管損傷部位でのマウス血小板血栓形成に影響を及ぼさなかった。
5〜10×105個のhESC−PLTを血管損傷後に注入したところ、34.8±1.7の循環血小板が、微小循環中で検出された(図10B)。それらの中で、4.3±0.7の血小板が、損傷後のTmax(72秒)で血管損傷部位にて、成長中のマウス血小板血栓に組み込まれた(図10A及び10C、ビデオ3(非表示))。損傷部位で発生中のマウス血小板血栓に組み込まれるヒト血液血小板及びhESC−PLTの数は、統計的に有意ではなかった(P=0.28)。ReoProによるhESC−PLTの前処理は、マウス血小板血栓に組み込まれた血小板の数を、血管損傷後のTmax(99秒)で1.2±0.4に有意に低減された(図10A及びC、付録ビデオ4、P<0.05)。これらの結果は、hESC−PLTが、正常対照血液血小板のように、生体内の血管損傷部位で機能的であることを示す。
【0107】
実施例25
動物
雄野生型マウス(C57BL/6、6〜8週齢)を、Jackson Laboratory社(バーハーバー、メーン州)から購入した。イリノイ大学の施設内動物実験委員会は、全ての動物管理および実験手順を承認した。
【0108】
実施例26
hESCから血管芽細胞/BCを介したMKの生成
hESC系統(HuES−3、H7[WA07]、MA01、及びMA09)からの血管芽細胞/BC生成を、以前に報告されているように実施した17;20。6〜8日目の培養物に由来するBCを精製し、50ng/mlのTPO、20ng/mlのSCF、及び20ng/mlのIL−11(Peprotech社製)で補完されたStemline II培地に播種して(1〜5×105個/ml)、MKへのBC分化を誘導した。MK培地の半分を、2日又は3日毎に新しい培地と交換した。MKからの血小板生成は、無フィーダー条件下で、又はOP9若しくはC3H 10T1/2間質細胞との共培養で実施した。無フィーダー条件の場合、4〜6日目のMK培養物に由来するMKを収集し、記述されているように16、100ng/mlのTPO、50ng/mlのSCF、及び25単位/mlのヘパリンナトリウムで補完されたIMDM培地に再懸濁し、2日毎に新しくした。血小板を、4日目から12日目まで分析用に収集した。幾つかの実験では、GM6001(100μM)を後期分化培養物に添加した。共培養実験の場合、OP9又はC3H 10T1/2間質細胞を、15%ウシ胎仔血清(Hyclone社製)を有するα−MEM中で維持した。コンフルエントなOP9又はC3H 10T1/2細胞を、共培養の前日に100ng/mlマイトマイシンCで処理した。細胞をPBSで穏やかに2回洗浄し、共培養の前にOP9培地中で一晩回復させた。4〜6日目のMK培養物に由来するMKを収集し、上述のよう培地を有するOP9又はCH1/2間質細胞上に播種した。血小板を、4日目から12日目まで分析用に収集した。
【0109】
実施例27
FACS分析
芽球培養またはMK培養に由来する細胞を、FACSCalibur(Becton Dickinson社製)またはAccuri C6血球計算器(Accuri Cytometers社製)でフローサイトメトリー分析により日常的にモニターする。系列マーカー、CD41a、CD42a、CD42b、及びCD235aに対する蛍光色素結合抗体(BD Biosciences社)を使用して、MK及び赤血球系列を特徴付けた。抗体は、5%胎仔ウシ血清(FBS)を有するPBS緩衝液で新しく調製した(CD42a及びCD42b抗体の場合は1:100希釈、CD41a抗体の場合は1:250希釈、CD235a抗体の場合は1:2000)。典型的には、1〜2×105細胞を抗体標識に使用した。細胞を氷上で1時間100μl抗体混合液中で染色し、その後緩衝液で2回洗浄し、1μg/mlヨウ化プロピジウムで補完された250μl緩衝液に再懸濁した。HLA−ABCの発現レベルを検出するために、血小板を、フルオレセインイソチオシアネート(FITC)結合抗ヒトHLA−ABC抗体又は対照としてのFITC結合マウス免疫グロブリンG(IgG)と共にインキュベートした。その後、試料を、FACSCaliburで分析し、データを、Cellquest又はFlowjoソフトウェアを使用して分析した。細胞選別は、UMASSメディカルスクールコア施設のBD FACSAriaシステムで実施した。選別した細胞を、1000rpmで10分間遠心分離することにより収集し、コロニー形成アッセイに適切な培地に再懸濁した。倍数性分析の場合、4日目のMK培養物に由来する細胞を、70%エタノール中で2時間固定した。その後、細胞をPBS緩衝液で1回洗浄した後で、PBS緩衝液中20μg/mlのヨウ化プロピジウム(propidum iodide)(Sigma社製)、20μg/mlのRNase A(Sigma社製)で一晩4℃にて染色した。細胞内DNA含有量を、Accuri C6血球計算器で分析した。
【0110】
実施例28
サイトスピン調製、ギムザ染色、及び免疫蛍光染色
芽球培養又はMK培養のいずれかに由来する細胞(1〜2×104個)を、ポリリシンでコーティングしたスライド(Wessco)にサイトスピンした。スライドを、Wright−Giemsa(Sigma社製)又は免疫蛍光(immunofluroscent)染色のいずれかに使用した。抗CD41(DAKO社製、1:100)及び抗vWF(DAKO社製、1:200)抗体を、サイトスピン調製でMKを特定するために使用した。インキュベーションは全て室温で実施した。細胞を、無動物性ブロッカー(Vector Laboratories社製)で30分間ブロッキングし、一次抗体と共に1時間インキュベートし、その後PBSで3回洗浄した。その後のインキュベーション及び洗浄は、暗所で実施した。細胞を、二次抗体(各々、1:200)と共に30分間インキュベートし、再度PBSで3回洗浄した。PBS中のDAPI(1μg/ml)を使用して核DNAを5分間染色し、その後追加的な3×PBS洗浄を行った。その後、スライドをオリンパスBX51蛍光顕微鏡(MVI社、エーボン、マサチューセッツ州)にマウントし、検査した。蛍光画像を、QICAM Fastカメラ(Qimaging社製、サリー、ブリティッシュコロンビア州、カナダ)を使用して取り込み、Q Capture Proバージョン5.1ソフトウェア(Media Cybernetics Inc.社製、ベセスダ、メリーランド州)で分析した。Nikon社製顕微鏡、PAXCAMデジタルカメラ、及びPAX−itソフトウェアを使用して、位相差生細胞画像を取り込んだ。
【0111】
実施例29
ヒト血液血小板及びhESC由来血小板の調製
ヒト血小板を、以前に記述されているように単離した49。手短に言えば、クエン酸ナトリウムで処理したヒト血液を200gで20分間遠心分離することにより、ヒト血小板豊富な血漿を調製した。上清を収集し、0.5μM PGE1及び10%クエン酸ナトリウム緩衝液の存在下で10分間700gで遠心分離した。ペレットを、0.15μM PGE1を含有するHEPES−タイロード緩衝液(12mM NaHCO3、138mM NaCl、5.5mMグルコース、2.9mM KCl、0.42mM NaHPO4、10mM HEPES、1mM CaCl2、1mM MgCl2)に再懸濁し、800gで5分間遠心分離した。ペレットを、0.1%無脂肪酸ウシ血清アルブミン、2mM CaCl2、及び1mM MgCl2を含有するRPMI1640に再懸濁した。洗浄した血小板の最終懸濁物を、1×107血小板/mlに調節した。血液試料を取得する承認は、イリノイ大学−シカゴ審査委員会から得た。インフォームドコンセントが与えられた。幾つかの実験では、ヒト血液試料を、商業的供給源(AllCells社、エメリービル、カリフォルニア州)からも取得した。hESC−PLTの場合、hESC−PLTを含有する培地を穏やかに収集し、アピラーゼ(1U/ml)及びEDTA(5mM)(Sigma−Aldrich社製、セントルイス、ミズーリ州)を添加して、血小板活性化を防止した。hESC−PLTを、上述のように濃縮及び洗浄した。洗浄した血液血小板及びhESC−PLTを、機能性アッセイの前に0.5〜2時間37℃でインキュベートした。
【0112】
実施例30
固定化されたフィブリノゲン及びvWF上での血小板の伸展
マイクロタイターウエルを有するチャンバースライド(Nalgen Nunc社製、ロチェスター、ニューヨーク州)を、0.1M NaHCO3(pH8.3)中の100μg/mLフィブリノゲン又はvWF(30g/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)で、一晩4℃でコーティングした。洗浄したヒト血液血小板又はhESC−PLT(1×107個/mL)を、フィブリノゲンでコーティングしたウエルに90分間37℃で付着及び伸展させた。幾つかの実験では、血小板を、インテグリンアンタゴニスト、RGDSペプチドと共に、負荷前に5分間プレインキュベートした。他の実験では、血小板を、ADP(20μM)又はトロンビン(1U/ml)(Sigma−Aldrich社製、セントルイス、ミズーリ州)と混合し、フィブリノゲン又はvWFでコーティングしたウエルに直ちに負荷した。PBS緩衝液で洗浄した後、細胞を固定し、透過処理し、Alexa Fluor568ファロイジン(Molecular Probes社製、ユージーン、オレゴン州)、FITC結合抗ヒトCD41a抗体(Dako cytomation社製、カーピンテリア、カリフォルニア州)、及びDAPIで染色した。付着血小板を、100×/1.40オイル対物レンズのPlanApoレンズを使用して、オリンパスBX51蛍光顕微鏡で観察した。画像を、QICAM Fastカメラを使用して取得し、Q Capture バージョン5.1ソフトウェアで処理した。
【0113】
実施例31
血小板微小凝集体の形成
洗浄したヒト血液血小板及びhESC−PLTを、改変タイロード緩衝液に再懸濁し、PKH67緑色蛍光細胞リンカー(10μM、Sigma社製、セントルイス、ミズーリ州)で標識した。ヒト血液血小板(6×107個)を、450μlキュベット(Chronolog社製、ヘイバータウン、ペンシルベニア州)中で、蛍光標識したヒト血液血小板(3×105個)又はhESC−PLT(3×105個)と混合し、トロンビン(0.5U/mL)で処理し、1200rpm、37℃で撹拌して血小板凝集を開始させた。対照実験では、トロンビンを添加する前に、血小板をRGDSペプチドと共に5分間37℃でプレインキュベートし、凝集アッセイを上記のように実施した。50μL緩衝液中の血小板微小凝集体を、スライドガラスに塗布し、オリンパスBX51蛍光顕微鏡で視覚化した。
【0114】
実施例32
PAC−1結合アッセイ
トロンビン刺激(1U/ml、室温で20分間のインキュベーション)をした又はしなかったヒト血液血小板又はhESC−PLTを、改変タイロード緩衝液中のAPC結合CD41a、PE結合CD42b、及びFITC結合PAC−1抗体で染色した。その後、試料をFACSCaliburを使用して分析した。前方散乱及び側方散乱ゲーティングを、ヒト血液血小板を対照として使用して決定した。フローサイトメトリーデータは、Cellquest又はFlowjoソフトウェアを使用して分析した。
【0115】
実施例33
クロット形成及び退縮
ヒト血液血小板又はhESC−PLT(約1.5×107個/ml)を、シリコン処理したガラス管(Kimble Chase社製、バインランド、ニュージャージー州)中の50μL血小板枯渇血漿に再懸濁した。トロンビン(2U/ml)及び10mM CaCl2を細胞に添加して、クロット形成及び退縮を誘導した。クロットを37℃で1時間退縮させ、画像を撮影した。クロットを、Tissue−Tek OCT化合物及びTissue−Tek Cryomolds(Sakura Finetek社製、トランス、カリフォルニア州)に埋込み、その後ドライアイスで凍結した。10μmのクロット切片を、凍結ミクロトームMicrom HM560(Thermo Scientific社製、カラマズー、ミシガン州)を使用して製作した。
【0116】
スライドをメタノール/アセトン(1:3)で30分間固定し、PBSで洗浄し、0.1% Triton X−100、0.1M Tris、10mM EGTA、0.15 NaCl、5mM MgCl2、及び1%ウシ血清アルブミン(BSA)、pH7.5で透過処理した。5%BSAでブロッキングし洗浄した後、切片を、ウサギ抗ヒトインテグリンαIIb(クローンH−160)及びマウス抗ヒトフィブリン(クローンUC45)抗体(Santa Cruz Biotech社製、サンタクルーズ、カリフォルニア州)で免疫染色した。その後、切片を、ローダミンに結合された1:200の二次抗ウサギIgG抗体及びFITCに結合された抗マウスIgM抗体(Jackson Immunoresearch Lab社製)中で1時間インキュベートした。上述のように画像を撮影した。
【0117】
実施例34
生体顕微鏡法
精巣挙筋微小循環の広視野多チャンネル生体顕微鏡法を、以前に記述されているように実施した47;50。ケタミン(125mg/kg、Bedford Laboratories社製、ベッドフォード、オハイオ州)及びキシラジン(25mg/kg、Akorn Inc.社製、ディケーター、イリノイ州)の腹腔内注射により、雄マウスに麻酔をかけた。気管チューブを挿入し、マウスを温度制御毛布(37℃)に配置した。麻酔を維持するために、頚静脈に設置したカニューレ(cannulus)で、ケタミン及びキシラジン溶液50μlを30分毎に投与した。更に、10U/mlヘパリンを含有する生理食塩水で満たされたカニューレを、大腿動脈に設置した。陰嚢を切開した後、精巣挙筋を生体顕微鏡トレーに肱置した。筋肉調製物を、温度調節し(37℃)通気した(95%N2、5%CO2)重炭酸緩衝生理食塩水を用いて、実験の全体にわたって表面還流した。
【0118】
精巣挙筋細動脈壁を、以前に記述されているように47、Micropointレーザーシステム(Photonics Instruments社製、サウスウィンザー、コネティカット州)を使用してレーザアブレーションにより損傷した。発生中のマウス血小板血栓を、Dylight649標識抗CD42(Emfret Analytics社製、体重1g当たり0.05μg)の注入によりモニターした。1回又は2回のパルスを血管壁内側でアブレーションさせて、血小板血栓形成を開始させた。カルセインAM(Invitrogen社製)で標識されたヒト血液血小板又はhESC−PLT、50〜100μl(5〜10×105血小板)を、血管損傷の直後に大腿動脈カニューレで注入した。1匹のマウス中で複数の血栓を研究し、より初期の血栓の上流に沿って新しい血栓を形成させ、以前に生成された血栓からのあらゆる寄与を回避した。以前に注入された標識ヒト血小板が完全に除去された後で、新しい血栓を生成した。2〜3個の血栓を生成した後、ReoPro(Centocor社製)、200μl中2×106個のヒト血小板毎に20μgで前処理した標識ヒト血小板50〜100μlを、血管損傷後に同じマウスに注入した。別の2〜3個の血栓を生成した。ヒト血液血小板及びhESC−PLTを、異なるマウスで研究した。60×対物レンズを備えたオリンパスBX61WI顕微鏡を使用して、微小血管データを得た。デジタル画像は、倍増管(Video Scope International社製、ダレス、バージニア州)により高速デジタルカメラ(camara)(Hamamatsu社製C9300)で取り込んだ。蛍光画像は、Slidebook v5.0(Intelligent Imaging Innovations社製、デンバー(Denvor)、コロラド州)を使用して分析した。蛍光画像は、10〜100ミリ秒の露光時間で取り込み、明視野画像は、20ミリ秒の露光時間で取り込んだ。データは、血管壁損傷後3分間収集した。画像分析を単純化するために、各擬似カラーの強度のダイナミックレンジを2値化した。微小血管を循環し、血管損傷部位で、発生中のマウス血小板血栓に組み込まれたヒト血小板の数を、血管損傷後3分間計数した。
【0119】
実施例35
統計分析
データは、GraphPad Prism(GraphPad Software社製)を使用して、2群を比較するためにスチューデントt検定により統計的に分析した。違いは、P<0.05を有意とみなした。
【0120】
実施例36
血管芽細胞から生成された血小板の密度遠心分離を使用した血小板精製法
MK分化培養に由来する血小板は、CD41a陰性である粒子を含んでいる。これらの粒子は、MK分化培養に由来する粒子の最大50%を構成する場合があり、血小板調製物の純度を増加させることが望ましい。低粘性パーコール培地等の、生存能及び形態学的完全性(indensity)を保存する培地を使用した密度遠心分離法を使用して、血小板からCD41a陰性粒子を分離した。PAC−1結合及びインテグリン活性化の能力を無効にせずに、薄層に分離されたヒト末梢血小板を、CD41a陰性粒子から取り出した。
【0121】
本発明の種々の実施形態は、発明を実施するための形態に記述されている。これらの説明は、上記の実施形態を直接記述するが、当業者であれば、本明細書に表示及び記述されている特定の実施形態の改変及び/又は変異を企図することができることが理解される。本説明の範囲以内にあるあらゆるそのような改変又は変異も同様に本説明の範囲に含まれることが意図される。特別な注記がない限り、本明細書及び特許請求の範囲にある単語及び語句には、該当する技術分野(複数可)の当業者にとって通常の習慣的な意味が与えられることが、本発明者らの意図である。
【0122】
本出願の出願時に出願者に知られている本発明の種々の実施形態の先術の説明が示されており、それは、例示及び説明のためであることが意図されている。本説明は、網羅的であることも、開示された詳細な形態に本発明を限定することも意図されておらず、上記の教示に照らして多数の改変及び変異が可能である。記載された実施形態は、本発明の原理及びその実用的応用を説明し、他の当業者が、企図した特定の使用に好適なように種々の改変を施して、本発明を種々の実施形態で使用することを可能にする役目を果たす。従って、本発明は、本発明を実施するための開示された特定の実施形態に限定されないことが意図されている。
【0123】
本発明の特定の実施形態が表示及び記述されているが、当業者であれば、本明細書の教示に基づき、本発明及びそのより広範な態様から逸脱せずに変更及び改変を為すことができることは明らかであり、従って添付の特許請求の範囲は、本発明の真の趣旨及び範囲内にあるそのような変更及び改変を全て、それらの範囲に包含することになる。本明細書で使用されている用語は、一般的に「非限定的」な用語であると意図されていることが、当業者により一般的に理解されるだろう(例えば、用語「含む(including)」は、「〜を含むが、限定されない」と解釈されるべきであり、用語「有する(having)」は、「〜を少なくとも有する」と解釈されるべきであり、用語「含む(includes)」は、「〜を含むが、限定されない」と解釈されるべきである等)。
参考文献
1. Guerriero R, Mattia G, Testa U et al. Stromal cell-derived factor 1alpha increases polyploidization of megakaryocytes generated by human hematopoietic progenitor cells. Blood 2001;97:2587-2595.
2. Matsunaga T, Tanaka I, Kobune M et al. Ex vivo large-scale generation of human platelets from cord blood CD34+ cells. Stem Cells 2006;24:2877-2887.
3. Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc.Natl.Acad.Sci.U.S.A. 2001;98.:10716-10721.
4. Lu S-J, Li F, Vida L, Honig GR. CD34+CD38- hematopoietic precursors derived from human embryonic stem cells exhibit an embryonic gene expression pattern. Blood 2004;103:4134-4141.
5, Chadwick K, Wang L, Li L et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003 2003;102:906-915.
6. Chang KH, Nelson AM, Cao H et al. Definitive-like erythroid cells derived from human embryonic stem cells coexpress high levels of embryonic and fetal globins with little or no adult globin. Blood 2006;108:1515-1523.
7. Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Exp.Hematol. 2004;32:1000-1009.
8. Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005;105:617-626.
9. Wang L, Menendez P, Shojaei F et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005;201:1603-1614.
10. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J.Immunol. 2005;175:5095- 5103.
11. Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood 2005;106:860-870.
12. Qiu C, Hanson E, Olivier E et al. Differentiation of human embryonic stem cells into hematopoietic cells by coculture with human fetal liver cells recapitulates the globin switch that occurs early in development. Exp.Hematol. 2005;33:1450-1458.
13. Zhan X, Dravid G, Ye Z et al. Functional antigen-presenting leucocytes derived from human embryonic stem cells in vitro. Lancet 2004;364:163-171.
14. Ledran MH, Krassowska A, Armstrong L et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008;3:85-98.
15. Gaur M, Kamata T, Wang S et al. Megakaryocytes derived from human embryonic stem cells: a genetically tractable system to study megakaryocytopoiesis and integrin function. J.Thromb.Haemost. 2006;4:436-442.
16. Takayama N, Nishikii H, Usui J et al. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008;111:5298-5306.
17. Lu SJ, Feng Q, Caballero S et al. Generation of functional hemangioblasts from human embryonic stem cells. Nat.Methods 2007;4:501-509.
18. Klimanskaya I, McMahon J. Approaches of derivation and maintenance of human ES cells: Detailed procedures and alternatives. In: Lanza Rea, ed. Handbook of Stem Cells. Volume 1: Embryonic Stem Cells. New York, USA: Elsevier/Academic Press; 2004:437- 449.
19. Lu SJ, Luo C, Holton K et al. Robust generation of hemangioblastic progenitors from human embryonic stem cells. Regen.Med. 2008;3:693-704.
20. Lu SJ, Feng Q, Park JS et al. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood 2008;112:4475-4484.
21. Fujiki H, Kimura T, Minamiguchi H et al. Role of human interleukin-9 as a megakaryocyte potentiator in culture. Exp.Hematol. 2002;30:1373-1380.
22. Jeanpierre S, Nicolini FE, Kaniewski B et al. BMP4 regulation of human megakaryocytic differentiation is involved in thrombopoietin signaling. Blood 2008;112:3154-3163.
23. Lordier L, Jalil A, Aurade F et al. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood 2008;112:3164-3174.
24. Chang Y, Aurade F, Larbret F et al. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood 2007;109:4229-4236.
25. Taguchi K, Saitoh M, Arai Y et al. Disparate effects of interleukin 11 and thrombopoietin on megakaryocytopoiesis in vitro. Cytokine 2001;15:241-249.
26. Philipp CS, Remmler J, Zucker-Franklin D. The effects of Mpl-ligand, interleukin-6 and interleukin-11 on megakaryocyte and platelet alpha-granule proteins. Thromb.Haemost. 1998;80:968-975.
27. Kanaji T, Russell S, Cunningham J et al. Megakaryocyte proliferation and ploidy regulated by the cytoplasmic tail of glycoprotein Ibalpha. Blood 2004;104:3161-3168.
28. Santoso S, Kalb R, Kiefel V, Mueller-Eckhardt C. The presence of messenger RNA for HLA class I in human platelets and its capability for protein biosynthesis. Br.J.Haematol. 1993;84:451-456.
29. Lalezari P, Driscoll AM. Ability of thrombocytes to acquire HLA specificity from plasma. Blood 1982;59:167-170.
30. Sullenbarger B, Bahng JH, Gruner R, Kotov N, Lasky LC. Prolonged continuous in vitro human platelet production using three-dimensional scaffolds. Exp.Hematol. 2009;37:101-110.
31. Giammona LM, Fuhrken PG, Papoutsakis ET, Miller WM. Nicotinamide (vitamin B3) increases the polyploidisation and proplatelet formation of cultured primary human megakaryocytes. Br.J.Haematol. 2006;135:554-566.
32. Nagata Y, Yoshikawa J, Hashimoto A et al. Proplatelet formation of megakaryocytes is triggered by autocrine-synthesized estradiol. Genes Dev. 2003;17:2864-2869.
33. Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood 2006;108:1509-1514.
34. Klimchenko O, Mori M, Distefano A et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood 2009;114:1506-1517.
35. Nishikii H, Eto K, Tamura N et al. Metalloproteinase regulation improves in vitro generation of efficacious platelets from mouse embryonic stem cells. J.Exp.Med. 2008;205:1917-1927.
36. Furie B, Furie BC. Mechanisms of thrombus formation. N. Engl.J.Med. 2008; 359: 938-949.
37. Day SM, Reeve JL, et al. Murine thrombosis models. Thromb.Haemost. 2004; 92: 486-494.
38. Sachs UJ and Nieswandt B. In vivo thrombus formation in murine models. Circ.Res. 2007; 100: 979-991.
39. Furie B and Furie BC. In vivo thrombus formation. J.Thromb.Haemost. 2007; 5 Suppl 1: 12-17.
40. Junt T, Schulze H, et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007; 317: 1767-1770.
41. Yu J, Hu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009; 324: 797-801.
42. Takahashi K, Tanabe K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861-872.
43. Yu J, Vodyanik MA, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318: 1917-1920.
44. Kim D, Kim CH, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009; 4: 472-476.
45. Ginsberg MH, Du X, Plow EF. Inside-out integrin signalling. Curr.Opin.Cell Biol. 1992; 4: 766-771.
46. Chen YP, O'Toole TE, et al. A point mutation in the integrin beta 3 cytoplasmic domain (S752-->P) impairs bidirectional signaling through alpha IIb beta 3 (platelet glycoprotein IIb-IIIa). Blood 1994; 84: 1857-1865.
47. Cho J, Furie BC, et al. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J.Clin.Invest 2008; 118: 1123-1131.
48. Coller BS. Anti-GPIIb/IIIa drugs: current strategies and future directions. Thromb. Haemost. 2001; 86: 427-443.
49. Cho J, Mosher DF. Enhancement of thrombogenesis by plasma fibronectin cross-linked to fibrin and assembled in platelet thrombi. Blood 2006: 107; 3555-3563.
50. Falati S, Gross P, et al. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat.Med. 2002; 8: 1175-1181.
51. Reems JA, Pineault N, and Sun S. In vitro megakaryocyte production and platelet biogenesis: state of the art. Transfus. Med.Rev. 2010; 24: 33-43.
52. Chockalingam P, Sacher RA. Management of patients’ refractory to platelet transfusion. J.Infus.Nurs. 2007; 30 : 220-225.
53. Hod E, Schwartz J. Platelet transfusion refractoriness. Br.J.Haematol. 2008; 142: 348-360.
54. Tian X, Kaufman DS. Differentiation of embryonic stem cells towards hematopoietic cells: progress and pitfalls. Curr.Opin.Hematol. 2008; 15: 312-318.
55. Wang L, Menendez P, et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J.Exp.Med. 2005; 201: 1603-1614.
【特許請求の範囲】
【請求項1】
巨核球を生成する方法であって、
血管芽細胞を準備すること、及び
前記血管芽細胞を培養して、巨核球(MK)へと分化させることを含む方法。
【請求項2】
前記血管芽細胞が、生体外で多能性幹細胞から分化する、請求項1に記載の方法。
【請求項3】
前記多能性幹細胞が、ヒト胚性幹細胞(hESC)である、請求項2に記載の方法。
【請求項4】
前記多能性幹細胞が、誘導多能性幹細胞(iPSC)である、請求項2に記載の方法。
【請求項5】
多能性幹細胞の前記分化が、前記多能性幹細胞を解離すること、並びにBMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地で前記多能性幹細胞を培養して、胚様体(EB)を形成することを含む、請求項2に記載の方法。
【請求項6】
前記培地が、BMP−4及びVEGFを含み、前記多能性幹細胞の培養が、EBを形成するために約48時間である、請求項5に記載の方法。
【請求項7】
前記BMP−4の濃度が、50ng/mlであり、VEGFが、50ng/mlである、請求項6に記載の方法。
【請求項8】
前記形成されたEBを、化学的に及び/又は機械的に解離し、前記培養培地の少なくとも一部を、BMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と交換して、前記血管芽細胞を生成する、請求項5に記載の方法。
【請求項9】
前記bFGFの濃度が、20ng/mlであり、TPOが、50〜100ng/mlであり、Flt3リガンドが、50ng/mlであり、SCFが、50ng/mlである、請求項8に記載の方法。
【請求項10】
前記解離されたEBを、芽球コロニー増殖培地中で約3〜4日間培養して、前記血管芽細胞を生成する、請求項8に記載の方法。
【請求項11】
前記培地が、IL6、エストラジオール、ビタミンB3、1つ又は複数の細胞外マトリックスタンパク質、又はそれらの組み合わせを更に含む、請求項8に記載の方法。
【請求項12】
前記iPSCが、ヒト由来の体細胞から誘導される、請求項4に記載の方法。
【請求項13】
前記体細胞が、成体組織に由来する、請求項12に記載の方法。
【請求項14】
前記体細胞が、胎児(仔)組織に由来する、請求項12に記載の方法。
【請求項15】
前記培地が、Stemline II培地である、請求項5に記載の方法。
【請求項16】
前記血管芽細胞の前記MKへの分化が、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中で、約2〜8日間培養することを含む、請求項1に記載の方法。
【請求項17】
前記TPOの濃度が、50ng/mlであり、SCFが、20ng/mlであり、IL11が、20ng/mlである、請求項16に記載の方法。
【請求項18】
前記培養培地の少なくとも一部を、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と2〜3日毎に交換して、前記血管芽細胞を前記MKへと分化させることを更に含む、請求項16に記載の方法。
【請求項19】
前記培地が、Stemline II培地である、請求項16に記載の方法。
【請求項20】
請求項1〜19に記載の方法のいずれかから生成される、ある量の巨核球。
【請求項21】
血小板を生成する方法であって、
巨核球(MK)を準備すること、及び
前記MKを培養して、血小板へと分化させることを含む方法。
【請求項22】
前記MKが、血管芽細胞から分化する、請求項21に記載の方法。
【請求項23】
前記血管芽細胞の前記MKへの分化が、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中で、約2〜8日間培養することを含む、請求項22に記載の方法。
【請求項24】
前記TPOの濃度が、50ng/mlであり、SCFが、20ng/mlであり、IL11が、20ng/mlである、請求項23に記載の方法。
【請求項25】
前記培養培地の少なくとも一部を、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と2〜3日毎に交換して、前記血管芽細胞を前記MKへと分化させることを更に含む、請求項23に記載の方法。
【請求項26】
前記培地が、Stemline II培地である、請求項23に記載の方法。
【請求項27】
前記血管芽細胞が、生体外で多能性幹細胞から分化する、請求項22に記載の方法。
【請求項28】
前記多能性幹細胞が、ヒト胚性幹細胞(hESC)である、請求項27に記載の方法。
【請求項29】
前記多能性幹細胞が、誘導多能性幹細胞(iPSC)である、請求項27に記載の方法。
【請求項30】
前記多能性幹細胞の分化が、前記多能性幹細胞を解離すること、並びにBMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地で前記多能性幹細胞を培養して、胚様体(EB)を形成することを含む、請求項27に記載の方法。
【請求項31】
前記培地が、BMP−4及びVEGFを含み、前記多能性幹細胞の培養が、EBを形成するために約48時間である、請求項30に記載の方法。
【請求項32】
前記BMP−4の濃度が、50ng/mlであり、VEGFが、50ng/mlである、請求項31に記載の方法。
【請求項33】
形成された前記EBを、化学的に及び/又は機械的に解離し、前記培養培地の少なくとも一部を、BMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と交換して、前記血管芽細胞を生成する、請求項30に記載の方法。
【請求項34】
前記bFGFの濃度が、20ng/mlであり、TPOが、50〜100ng/mlであり、Flt3リガンドが、50ng/mlであり、SCFが、50ng/mlである、請求項33に記載の方法。
【請求項35】
解離された前記EBを、芽球コロニー増殖培地中で約3〜4日間養殖して、前記血管芽細胞を生成する、請求項33に記載の方法。
【請求項36】
前記培地が、IL6、エストラジオール、ビタミンB3、1つ又は複数の細胞外マトリックスタンパク質、又はそれらの組み合わせを更に含む、請求項33に記載の方法。
【請求項37】
前記iPSCが、ヒト由来の体細胞から誘導される、請求項29に記載の方法。
【請求項38】
前記体細胞が、成体組織に由来する、請求項37に記載の方法。
【請求項39】
前記体細胞が、胎児(仔)組織に由来する、請求項37に記載の方法。
【請求項40】
前記培地が、Stemline II培地である、請求項30に記載の方法。
【請求項41】
前記MKを、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地に再懸濁し、少なくと4日間培養して、前記血小板へと分化させる、請求項21に記載の方法。
【請求項42】
前記TPOの濃度が、100ng/mlであり、SCFが、50ng/mlであり、ヘパリンナトリウムが、25単位/mlであり、IL11が、20ng/mlである、請求項41に記載の方法。
【請求項43】
前記少なくとも4日間の前記MK培養後に、GM6001又はIL3を添加することを更に含む、請求項41に記載の方法。
【請求項44】
前記培地の少なくとも一部を、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と、少なくとも2日毎に交換することを更に含む、請求項41に記載の方法。
【請求項45】
前記培地が、IMDM培地である、請求項41に記載の方法。
【請求項46】
前記MKを解離し、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中の、有糸分裂的に停止されたフィーダー層上で少なくとも4日間培養して、前記血小板へと分化させる、請求項21に記載の方法。
【請求項47】
前記フィーダー層が、OP9細胞を含む、請求項46に記載の方法。
【請求項48】
前記フィーダー層が、C3H 10T1/2細胞を含む、請求項46に記載の方法。
【請求項49】
前記少なくとも4日間の前記MK培養後に、GM6001又はIL3を添加することを更に含む、請求項46に記載の方法
【請求項50】
前記培地の少なくとも一部を、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と、少なくとも2日毎に交換することを更に含む、請求項46に記載の方法。
【請求項51】
前記培地が、IMDM培地である、請求項46に記載の方法。
【請求項52】
請求項21〜51に記載の方法のいずれかから生成される、ある量の血小板。
【請求項53】
前記血小板が、前記血小板を対象体に投与した際に、前記対象体においてHLA同種免疫原性反応に寄与しない溶液中で保管される、請求項52に記載の血小板。
【請求項54】
前記血小板が、アピラーゼ及び/又はEDTAの存在下で活性化されない、請求項52に記載の血小板。
【請求項55】
細胞分化の修飾因子をスクリーニングする方法であって、
ある量の巨核球(MK)を準備すること、
前記MKを試験化合物と接触させること、及び
前記MKと前記試験化合物との接触に由来する機能的効果の存在又は非存在を決定することを含み、
前記機能的効果の存在が、前記試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子であることを示し、前記機能的効果の非存在が、前記試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子でないことを示す方法。
【請求項56】
血小板輸血を必要とする対象体を治療する方法であって、
ある量の血管芽細胞を準備すること、
前記血管芽細胞を培養して、巨核球(MK)へと分化させること、
前記MKを生体外で培養して、血小板へと分化させること、
ある量の前記生体外で分化した血小板を準備すること、及び
前記ある量の前記生体外で分化した血小板を、前記血小板輸血を必要とする前記対象体に投与して、それにより前記血小板輸血を必要とする対象体を治療することを含む方法。
【請求項57】
前記生体外で分化した血小板を前記対象体と一致させて、免疫媒介性宿主反応を低減又は排除する、請求項56に記載の方法。
【請求項1】
巨核球を生成する方法であって、
血管芽細胞を準備すること、及び
前記血管芽細胞を培養して、巨核球(MK)へと分化させることを含む方法。
【請求項2】
前記血管芽細胞が、生体外で多能性幹細胞から分化する、請求項1に記載の方法。
【請求項3】
前記多能性幹細胞が、ヒト胚性幹細胞(hESC)である、請求項2に記載の方法。
【請求項4】
前記多能性幹細胞が、誘導多能性幹細胞(iPSC)である、請求項2に記載の方法。
【請求項5】
多能性幹細胞の前記分化が、前記多能性幹細胞を解離すること、並びにBMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地で前記多能性幹細胞を培養して、胚様体(EB)を形成することを含む、請求項2に記載の方法。
【請求項6】
前記培地が、BMP−4及びVEGFを含み、前記多能性幹細胞の培養が、EBを形成するために約48時間である、請求項5に記載の方法。
【請求項7】
前記BMP−4の濃度が、50ng/mlであり、VEGFが、50ng/mlである、請求項6に記載の方法。
【請求項8】
前記形成されたEBを、化学的に及び/又は機械的に解離し、前記培養培地の少なくとも一部を、BMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と交換して、前記血管芽細胞を生成する、請求項5に記載の方法。
【請求項9】
前記bFGFの濃度が、20ng/mlであり、TPOが、50〜100ng/mlであり、Flt3リガンドが、50ng/mlであり、SCFが、50ng/mlである、請求項8に記載の方法。
【請求項10】
前記解離されたEBを、芽球コロニー増殖培地中で約3〜4日間培養して、前記血管芽細胞を生成する、請求項8に記載の方法。
【請求項11】
前記培地が、IL6、エストラジオール、ビタミンB3、1つ又は複数の細胞外マトリックスタンパク質、又はそれらの組み合わせを更に含む、請求項8に記載の方法。
【請求項12】
前記iPSCが、ヒト由来の体細胞から誘導される、請求項4に記載の方法。
【請求項13】
前記体細胞が、成体組織に由来する、請求項12に記載の方法。
【請求項14】
前記体細胞が、胎児(仔)組織に由来する、請求項12に記載の方法。
【請求項15】
前記培地が、Stemline II培地である、請求項5に記載の方法。
【請求項16】
前記血管芽細胞の前記MKへの分化が、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中で、約2〜8日間培養することを含む、請求項1に記載の方法。
【請求項17】
前記TPOの濃度が、50ng/mlであり、SCFが、20ng/mlであり、IL11が、20ng/mlである、請求項16に記載の方法。
【請求項18】
前記培養培地の少なくとも一部を、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と2〜3日毎に交換して、前記血管芽細胞を前記MKへと分化させることを更に含む、請求項16に記載の方法。
【請求項19】
前記培地が、Stemline II培地である、請求項16に記載の方法。
【請求項20】
請求項1〜19に記載の方法のいずれかから生成される、ある量の巨核球。
【請求項21】
血小板を生成する方法であって、
巨核球(MK)を準備すること、及び
前記MKを培養して、血小板へと分化させることを含む方法。
【請求項22】
前記MKが、血管芽細胞から分化する、請求項21に記載の方法。
【請求項23】
前記血管芽細胞の前記MKへの分化が、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中で、約2〜8日間培養することを含む、請求項22に記載の方法。
【請求項24】
前記TPOの濃度が、50ng/mlであり、SCFが、20ng/mlであり、IL11が、20ng/mlである、請求項23に記載の方法。
【請求項25】
前記培養培地の少なくとも一部を、TPO、SCF、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と2〜3日毎に交換して、前記血管芽細胞を前記MKへと分化させることを更に含む、請求項23に記載の方法。
【請求項26】
前記培地が、Stemline II培地である、請求項23に記載の方法。
【請求項27】
前記血管芽細胞が、生体外で多能性幹細胞から分化する、請求項22に記載の方法。
【請求項28】
前記多能性幹細胞が、ヒト胚性幹細胞(hESC)である、請求項27に記載の方法。
【請求項29】
前記多能性幹細胞が、誘導多能性幹細胞(iPSC)である、請求項27に記載の方法。
【請求項30】
前記多能性幹細胞の分化が、前記多能性幹細胞を解離すること、並びにBMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地で前記多能性幹細胞を培養して、胚様体(EB)を形成することを含む、請求項27に記載の方法。
【請求項31】
前記培地が、BMP−4及びVEGFを含み、前記多能性幹細胞の培養が、EBを形成するために約48時間である、請求項30に記載の方法。
【請求項32】
前記BMP−4の濃度が、50ng/mlであり、VEGFが、50ng/mlである、請求項31に記載の方法。
【請求項33】
形成された前記EBを、化学的に及び/又は機械的に解離し、前記培養培地の少なくとも一部を、BMP−4、VEGF、bFGF、TPO、Flt3リガンド、SCF、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と交換して、前記血管芽細胞を生成する、請求項30に記載の方法。
【請求項34】
前記bFGFの濃度が、20ng/mlであり、TPOが、50〜100ng/mlであり、Flt3リガンドが、50ng/mlであり、SCFが、50ng/mlである、請求項33に記載の方法。
【請求項35】
解離された前記EBを、芽球コロニー増殖培地中で約3〜4日間養殖して、前記血管芽細胞を生成する、請求項33に記載の方法。
【請求項36】
前記培地が、IL6、エストラジオール、ビタミンB3、1つ又は複数の細胞外マトリックスタンパク質、又はそれらの組み合わせを更に含む、請求項33に記載の方法。
【請求項37】
前記iPSCが、ヒト由来の体細胞から誘導される、請求項29に記載の方法。
【請求項38】
前記体細胞が、成体組織に由来する、請求項37に記載の方法。
【請求項39】
前記体細胞が、胎児(仔)組織に由来する、請求項37に記載の方法。
【請求項40】
前記培地が、Stemline II培地である、請求項30に記載の方法。
【請求項41】
前記MKを、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地に再懸濁し、少なくと4日間培養して、前記血小板へと分化させる、請求項21に記載の方法。
【請求項42】
前記TPOの濃度が、100ng/mlであり、SCFが、50ng/mlであり、ヘパリンナトリウムが、25単位/mlであり、IL11が、20ng/mlである、請求項41に記載の方法。
【請求項43】
前記少なくとも4日間の前記MK培養後に、GM6001又はIL3を添加することを更に含む、請求項41に記載の方法。
【請求項44】
前記培地の少なくとも一部を、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と、少なくとも2日毎に交換することを更に含む、請求項41に記載の方法。
【請求項45】
前記培地が、IMDM培地である、請求項41に記載の方法。
【請求項46】
前記MKを解離し、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地中の、有糸分裂的に停止されたフィーダー層上で少なくとも4日間培養して、前記血小板へと分化させる、請求項21に記載の方法。
【請求項47】
前記フィーダー層が、OP9細胞を含む、請求項46に記載の方法。
【請求項48】
前記フィーダー層が、C3H 10T1/2細胞を含む、請求項46に記載の方法。
【請求項49】
前記少なくとも4日間の前記MK培養後に、GM6001又はIL3を添加することを更に含む、請求項46に記載の方法
【請求項50】
前記培地の少なくとも一部を、TPO、SCF、ヘパリンナトリウム、IL11、及びそれらの組み合わせからなる群から選択される成長因子又はサイトカインを含む培地と、少なくとも2日毎に交換することを更に含む、請求項46に記載の方法。
【請求項51】
前記培地が、IMDM培地である、請求項46に記載の方法。
【請求項52】
請求項21〜51に記載の方法のいずれかから生成される、ある量の血小板。
【請求項53】
前記血小板が、前記血小板を対象体に投与した際に、前記対象体においてHLA同種免疫原性反応に寄与しない溶液中で保管される、請求項52に記載の血小板。
【請求項54】
前記血小板が、アピラーゼ及び/又はEDTAの存在下で活性化されない、請求項52に記載の血小板。
【請求項55】
細胞分化の修飾因子をスクリーニングする方法であって、
ある量の巨核球(MK)を準備すること、
前記MKを試験化合物と接触させること、及び
前記MKと前記試験化合物との接触に由来する機能的効果の存在又は非存在を決定することを含み、
前記機能的効果の存在が、前記試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子であることを示し、前記機能的効果の非存在が、前記試験化合物が、細胞分化を調節する巨核球形成、血小板新生、及び/又は造血因子でないことを示す方法。
【請求項56】
血小板輸血を必要とする対象体を治療する方法であって、
ある量の血管芽細胞を準備すること、
前記血管芽細胞を培養して、巨核球(MK)へと分化させること、
前記MKを生体外で培養して、血小板へと分化させること、
ある量の前記生体外で分化した血小板を準備すること、及び
前記ある量の前記生体外で分化した血小板を、前記血小板輸血を必要とする前記対象体に投与して、それにより前記血小板輸血を必要とする対象体を治療することを含む方法。
【請求項57】
前記生体外で分化した血小板を前記対象体と一致させて、免疫媒介性宿主反応を低減又は排除する、請求項56に記載の方法。
【図1−2】

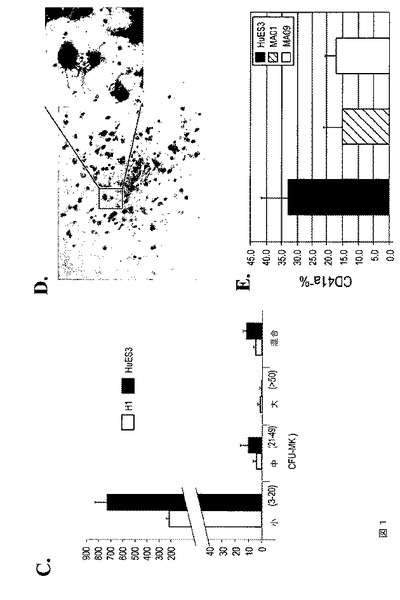
【図2−1】
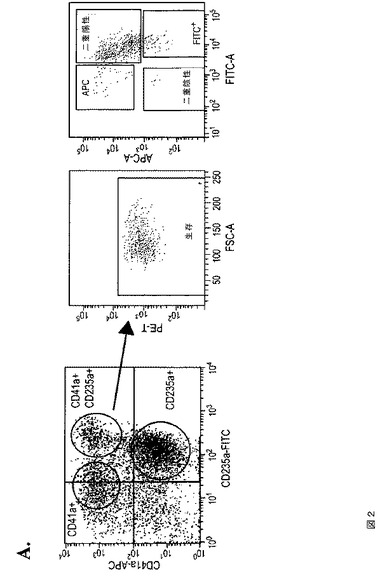
【図3】
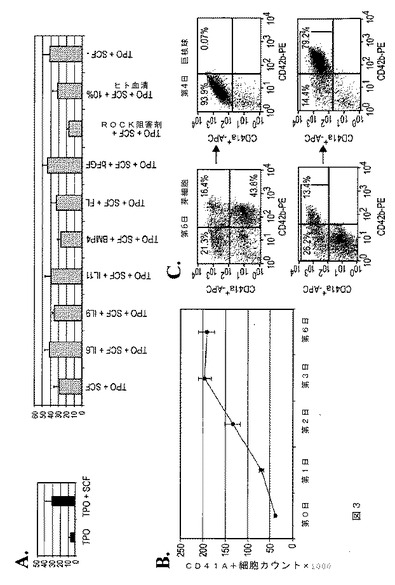
【図4−1】
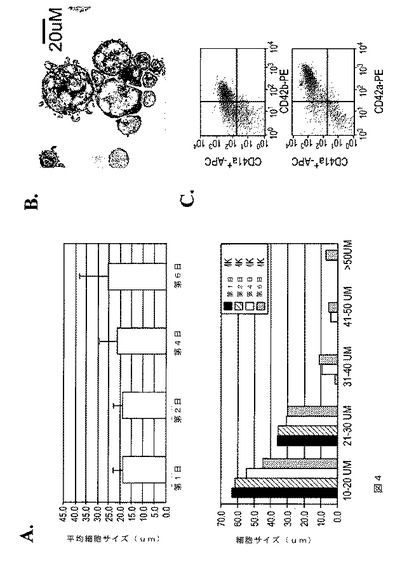
【図5−1】

【図5−2】

【図6】
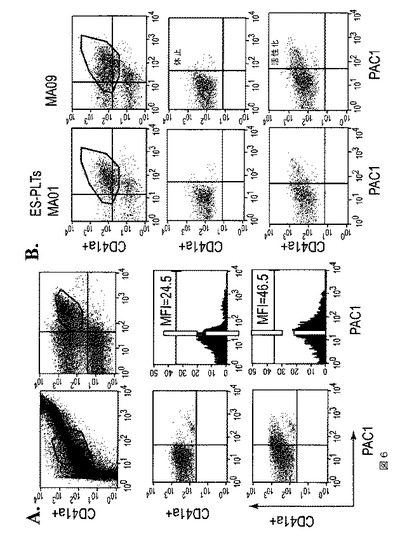
【図7−2】
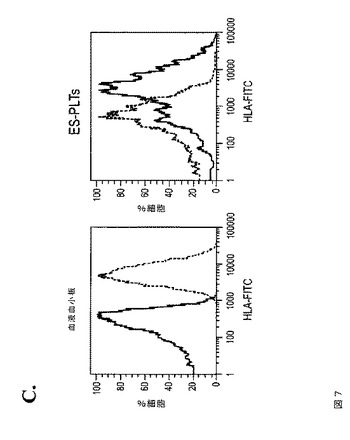
【図8−3】
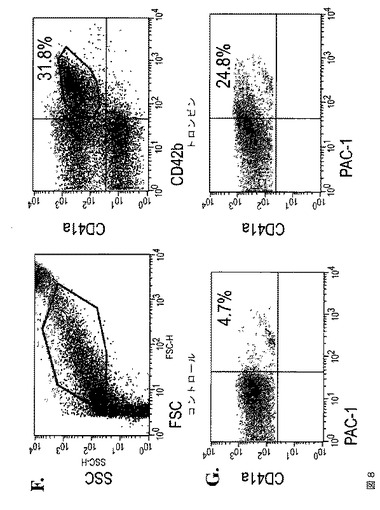
【図10−2】
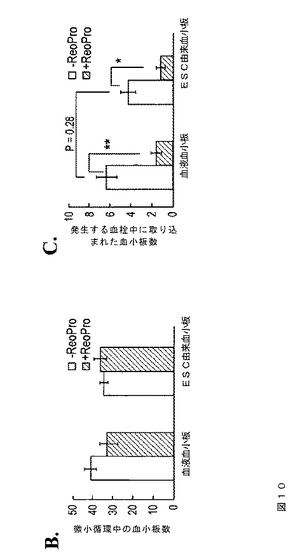
【図12】
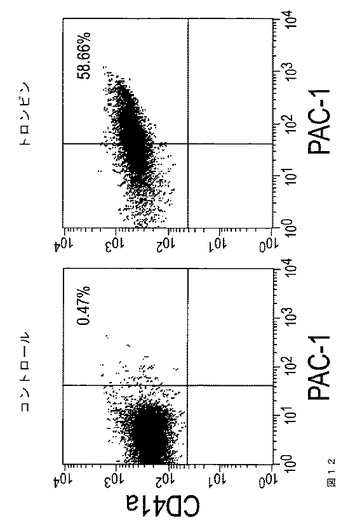
【図13−1】
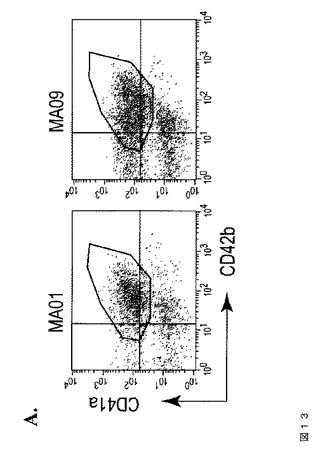
【図13−2】
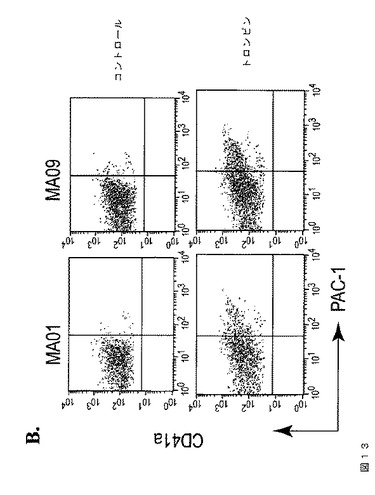
【図13−3】
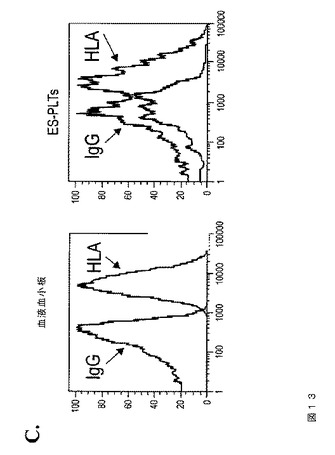
【図1−1】
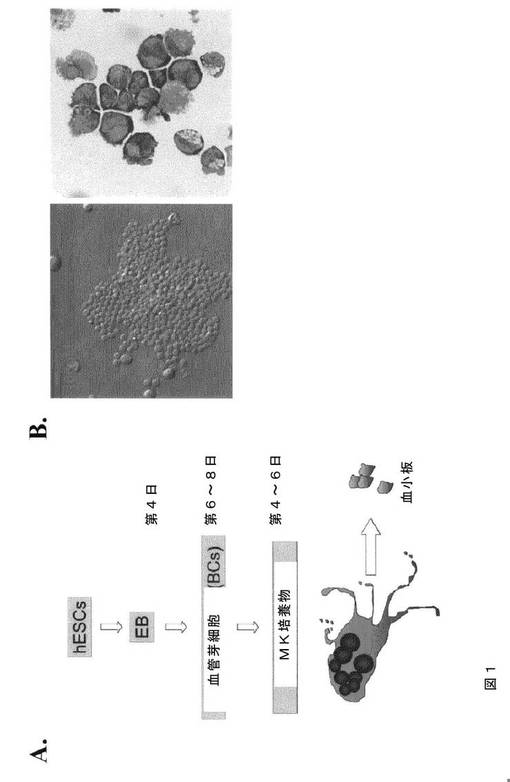
【図2−2】
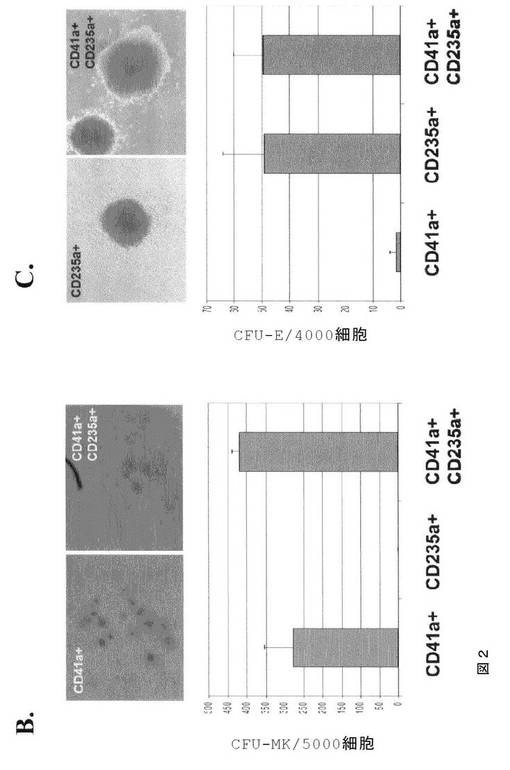
【図4−2】
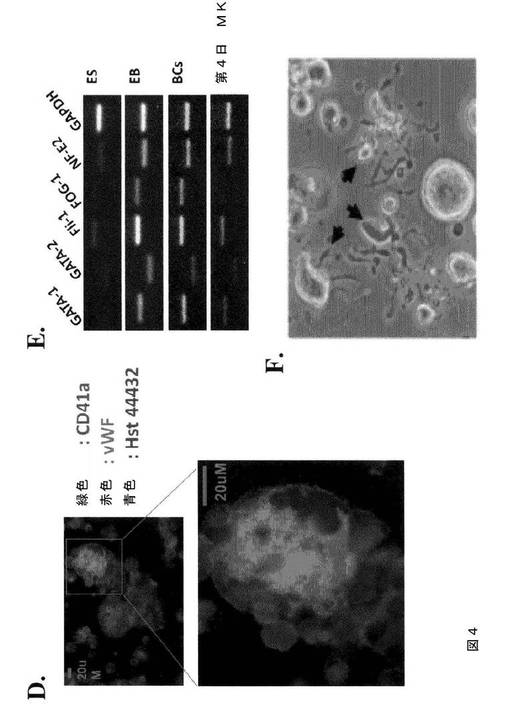
【図7−1】
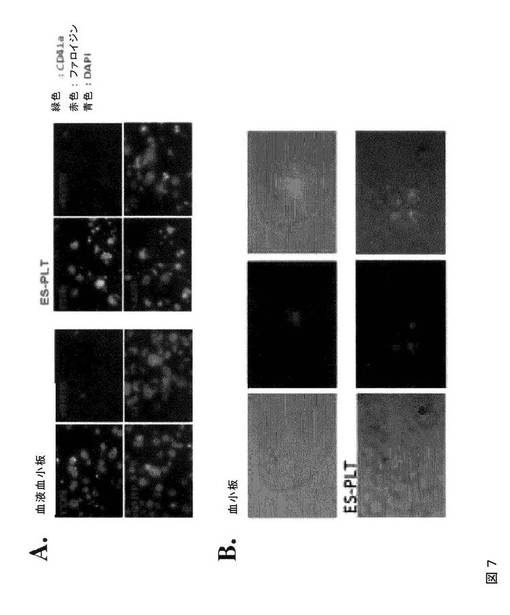
【図8−1】
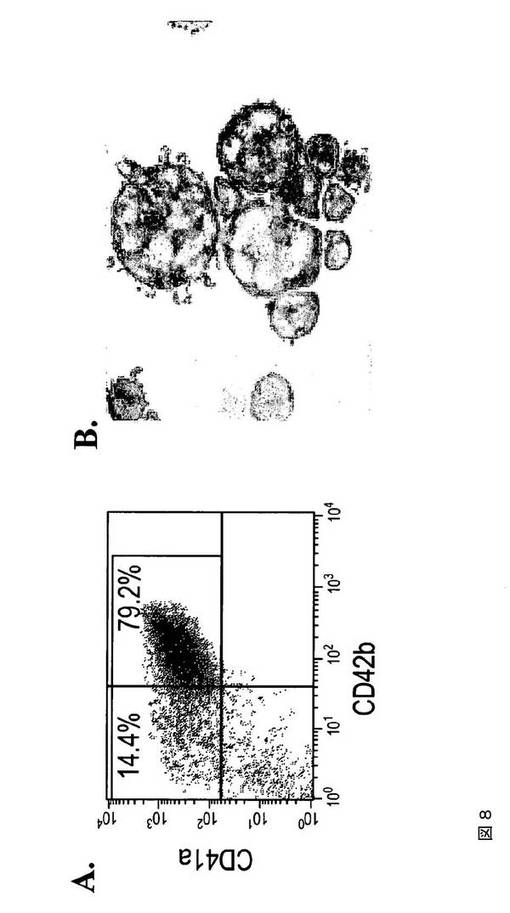
【図8−2】
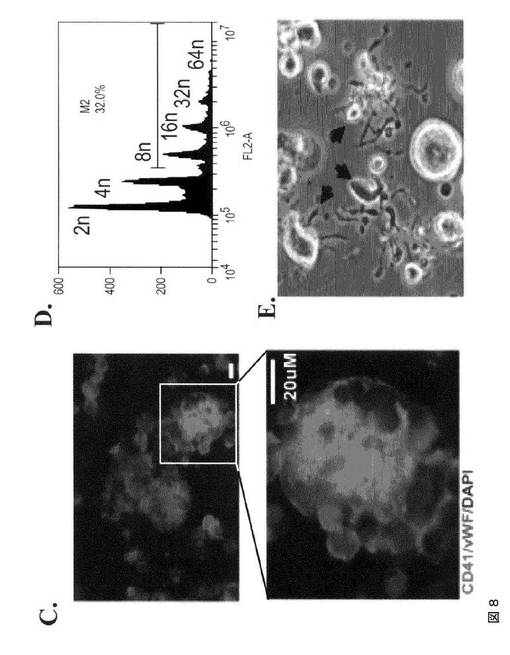
【図9−1】
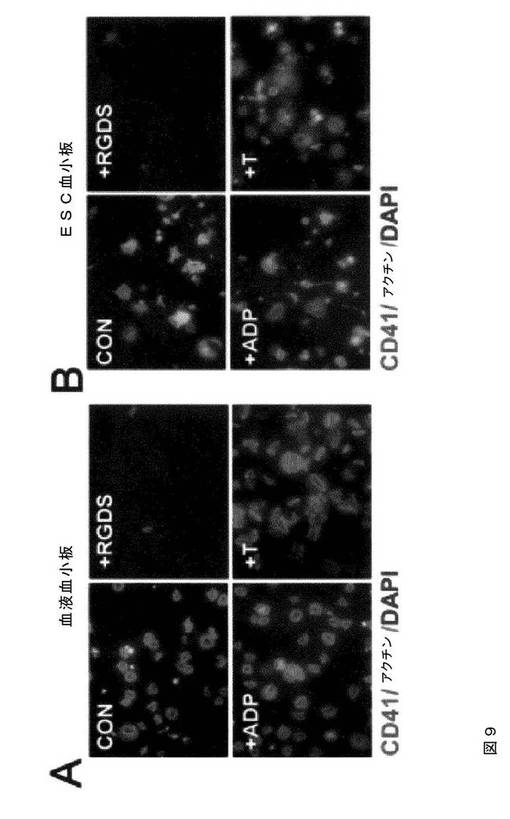
【図9−2】
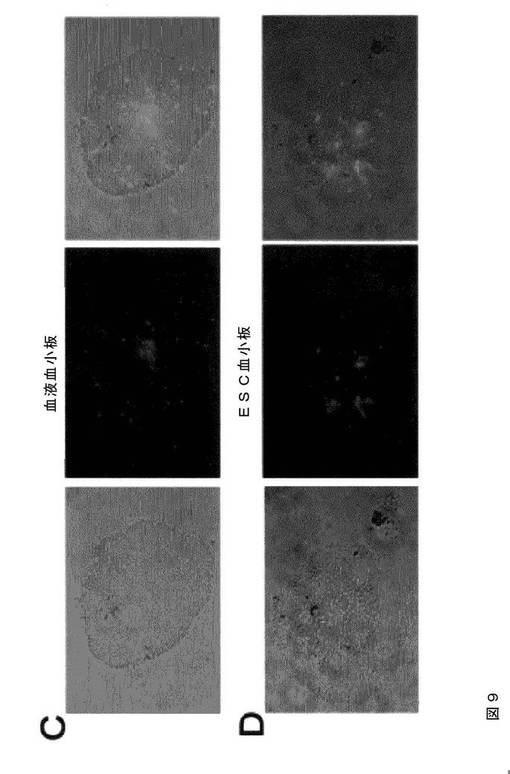
【図9−3】
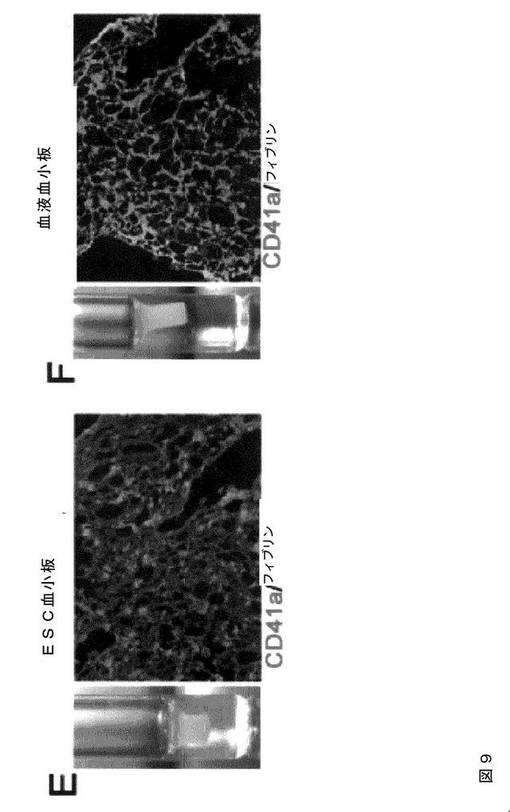
【図10−1】
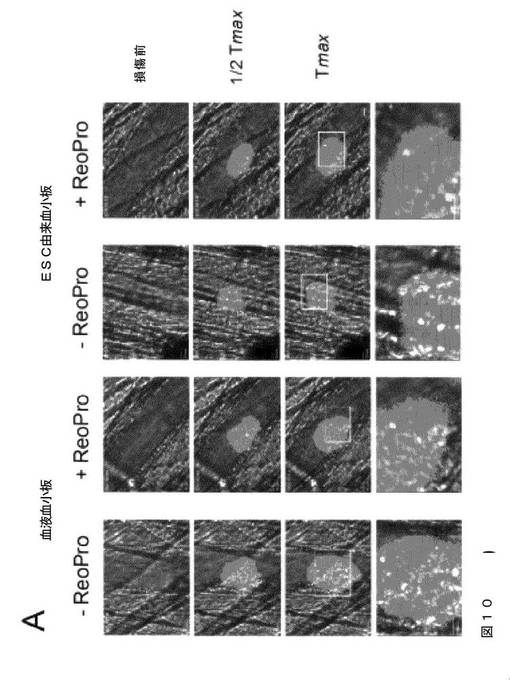
【図11】
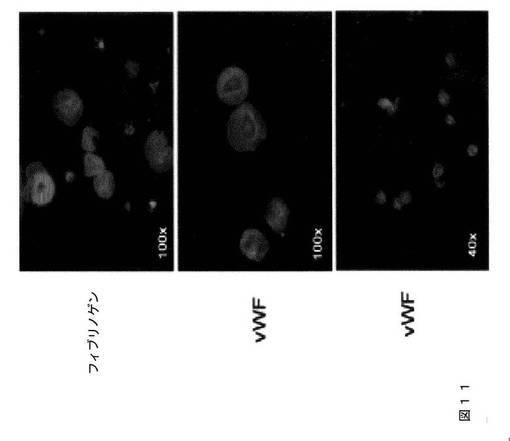
【図2−1】
【図3】
【図4−1】
【図5−1】
【図5−2】
【図6】
【図7−2】
【図8−3】
【図10−2】
【図12】
【図13−1】
【図13−2】
【図13−3】
【図1−1】
【図2−2】
【図4−2】
【図7−1】
【図8−1】
【図8−2】
【図9−1】
【図9−2】
【図9−3】
【図10−1】
【図11】
【公表番号】特表2013−512676(P2013−512676A)
【公表日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2012−542230(P2012−542230)
【出願日】平成22年12月3日(2010.12.3)
【国際出願番号】PCT/US2010/058990
【国際公開番号】WO2011/069127
【国際公開日】平成23年6月9日(2011.6.9)
【出願人】(512144874)ステム セル アンド リジェネレイティブ メディスン インターナショナル, インコーポレイテッド (2)
【Fターム(参考)】
【公表日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願日】平成22年12月3日(2010.12.3)
【国際出願番号】PCT/US2010/058990
【国際公開番号】WO2011/069127
【国際公開日】平成23年6月9日(2011.6.9)
【出願人】(512144874)ステム セル アンド リジェネレイティブ メディスン インターナショナル, インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]