
熱硬化性光反射用樹脂組成物及びその製造方法、並びにその樹脂組成物を用いた光半導体素子搭載用基板及び光半導体装置
【課題】硬化後の、可視光から近紫外光の反射率が高く、耐熱劣化性やタブレット成型性に優れ、なおかつトランスファー成型時にバリが生じ難い熱硬化性光反射用樹脂組成物及びその製造方法、並びに当該樹脂組成物を用いた光半導体素子搭載用基板及び光半導体装置を提供する。
【解決手段】熱硬化性成分と白色顔料とを含む熱硬化性光反射用樹脂組成物110であって、成型温度100℃〜200℃、成型圧力20MPa以下、成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ熱硬化後の、波長350nm〜800nmにおける光反射率が80%以上であることを特徴とする熱硬化性光反射用樹脂組成物110を調製し、そのような樹脂組成物を使用して光半導体素子搭載用基板および光半導体装置を構成する。
【解決手段】熱硬化性成分と白色顔料とを含む熱硬化性光反射用樹脂組成物110であって、成型温度100℃〜200℃、成型圧力20MPa以下、成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ熱硬化後の、波長350nm〜800nmにおける光反射率が80%以上であることを特徴とする熱硬化性光反射用樹脂組成物110を調製し、そのような樹脂組成物を使用して光半導体素子搭載用基板および光半導体装置を構成する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、光半導体素子と蛍光体などの波長変換手段とを組合せた光半導体装置に用いる熱硬化性光反射用樹脂組成物及びその製造方法、並びに当該樹脂組成物を用いた光半導体素子搭載用基板及び光半導体装置に関する。
【背景技術】
【0002】
LED(Light Emitting Diode:発光ダイオード)などの光半導体素子と蛍光体とを組合せた光半導体装置は、高エネルギー効率および長寿命などの利点から、屋外用ディスプレイ、携帯液晶バックライトおよび車載用途など様々な用途に適用され、その需要が拡大しつつある。それに伴って、LEDデバイスの高輝度化が進み、素子の発熱量増大によるジャンクション温度の上昇、あるいは直接的な光エネルギーの増大による素子材料の劣化が問題視され、近年、熱劣化および光劣化に対して耐性を有する素子材料の開発が課題となっている。日本国特開2006−140207号公報では、耐熱試験後の光反射特性に優れる光半導体素子搭載用基板を開示している。
【発明の概要】
【発明が解決しようとする課題】
【0003】
しかし、上記公報で開示された熱硬化性光反射用樹脂組成物を用いて、トランスファー成型によって基板を製造する場合、成型時に成型金型の上型と下型との隙間に樹脂組成物が染み出し、樹脂汚れが発生しやすい傾向がある。加熱成型時に樹脂汚れが発生すると、光半導体素子搭載領域となる基板の開口部(凹部)に樹脂汚れが張り出し、光半導体素子を搭載する際の障害になる。また、開口部に光半導体素子を搭載できたとしても、樹脂汚れは、光半導体素子と金属配線とをボンディングワイヤなどの公知の方法によって電気的に接続する際の障害になる。すなわち、樹脂汚れは、素子搭載およびワイヤボンディングといった半導体素子製造時の作業性を低下させるために望ましくない。半導体素子製造時に上述の障害が起こらないように、基板の開口部に樹脂汚れが存在する場合には、一般に、光半導体素子搭載用基板の製造プロセスに樹脂汚れの除去工程が追加される。しかし、そのような除去工程は、コストや製造時間のロスとなるため、改善が望まれている。
【0004】
本発明は、上記に鑑みてなされたものであり、樹脂硬化後の可視光から近紫外光領域における反射率が高く、その一方で、トランスファー成型時に樹脂汚れが発生し難い熱硬化性光反射用樹脂組成物を提供する。また本発明は、上述の樹脂組成物を用いて、ワイヤボンディング性に優れ、光劣化および熱劣化に対して耐性を有する半導体素子搭載用基板および光半導体装置を提供する。さらに本発明は、上述の光半導体素子搭載用基板および光半導体装置を効率良く製造するための製造方法を提供する。
【課題を解決するための手段】
【0005】
すなわち、本発明は、以下(1)〜(26)に記載の事項をその特徴とするものである。
(1)熱硬化性成分と(E)白色顔料とを含む熱硬化性光反射用樹脂組成物であって、
成型温度100℃〜200℃、成型圧力20MPa以下、及び成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ
熱硬化後の、波長350nm〜800nmにおける光反射率が80%以上である
ことを特徴とする熱硬化性光反射用樹脂組成物。
(2)上記熱硬化性成分が、(A)エポキシ樹脂を含むことを特徴とする上記(1)に記載の熱硬化性光反射用樹脂組成物。
(3)上記(A)エポキシ樹脂が、(A’)エポキシ樹脂と(B’)硬化剤とを混練することによって得られ、かつ100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを含むことを特徴とする上記(2)に記載の熱硬化性光反射用樹脂組成物。
(4)上記熱硬化性成分が、上記(A)エポキシ樹脂とともに用いられる(B)硬化剤をさらに含み、上記(A)エポキシ樹脂と上記(B)硬化剤との配合比が、上記(A)エポキシ樹脂中のエポキシ基1当量に対して、該エポキシ基と反応可能な上記(B)硬化剤中の活性基が0.5〜0.7当量となる比であることを特徴とする、上記(3)に記載の熱硬化性光反射用樹脂組成物。
(5)上記(B)硬化剤が、イソシアヌル酸骨格を有する化合物を含むことを特徴とする上記(4)に記載の熱硬化性光反射用樹脂組成物。
【0006】
(6)上記(B)硬化剤が、35℃以上の融点を有する酸無水物をさらに含むことを特徴とする上記(5)に記載の熱硬化性光反射用樹脂組成物。
(7)上記(B)硬化剤が、シクロへキサントリカルボン酸無水物を含むことを特徴とする上記(4)に記載の熱硬化性光反射用樹脂組成物。
(8)上記シクロへキサントリカルボン酸無水物が、下記構造式(I)で示される化合物であることを特徴とする上記(7)に記載の熱硬化性光反射用樹脂組成物。
【化1】

(9)さらに(H)増粘剤を含み、該(H)増粘剤が、中心粒径が1nm〜1000nmのナノ粒子フィラーを含有することを特徴とする上記(1)〜(8)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(10)さらに(D)無機充填剤を含み、該(D)無機充填剤が、多孔質充填剤または吸油性を有する化合物を含有することを特徴とする上記(1)〜(8)のいずれかに記載の熱硬化性光反射用樹脂組成物。
【0007】
(11)上記多孔質充填剤または吸油性を有する化合物の形状が、真球状、破砕状、円盤状、棒状、繊維状からなる群から選択される少なくとも1種であることを特徴とする上記(10)に記載の熱硬化性光反射用樹脂組成物。
(12)上記多孔質充填剤または吸油性を有する化合物の表面が、疎水化処理または親水化処理されていることを特徴とする上記(10)または(11)に記載の熱硬化性光反射用樹脂組成物。
(13)上記多孔質充填剤または吸油性を有する化合物の見掛け密度が、0.4g/cm3以上であることを特徴とする上記(10)〜(12)のいずれか1つに記載の熱硬化性光反射用樹脂組成物。
(14)上記(D)無機充填剤における上記多孔質充填剤または吸油性を有する化合物の含有量が、0.1体積%〜20体積%の範囲であることを特徴とする上記(10)〜(13)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(15)さらに(D)無機充填剤として、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群の中から選ばれる少なくとも1種を含むことを特徴とする上記(1)〜(14)のいずれかに記載の熱硬化性光反射用樹脂組成物。
【0008】
(16)上記(E)白色顔料が、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子からなる群の中から選ばれる少なくとも1種であることを特徴とする上記(1)〜(15)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(17)上記(E)白色顔料の中心粒径が、0.1〜50μmの範囲にあることを特徴とする上記(1)〜(16)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(18)上記(D)無機充填剤と上記(E)白色顔料とを合計した配合量が、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることを特徴とする上記(1)〜(17)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(19)少なくとも上記各種構成成分を、混練温度20〜100℃、混練時間10〜30分の条件下で混練することによって得られる混練物を含むことを特徴とする上記(1)〜(18)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(20)上記混練物が上記混練後に0〜30℃で1〜72時間にわたってエージングされたものであることを特徴とする上記(19)に記載の熱硬化性光反射用樹脂組成物。
【0009】
(21)上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物を製造する方法であって、少なくとも樹脂組成物の各成分を混練して混練物を形成する混練工程と、上記混練物を0〜30℃で1〜72時間にわたってエージングする工程とを有することを特徴とする熱硬化性光反射用樹脂組成物の製造方法。
(22)上記混練工程を、混練温度20〜100℃、及び混練時間10〜30分の条件下で行うことを特徴とする上記(21)に記載の熱硬化性光反射用樹脂組成物の製造方法。
(23)上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物を用いて構成されることを特徴とする光半導体素子搭載用基板。
(24)光半導体素子搭載領域となる凹部が1つ以上形成されている光半導体素子搭載用基板であって、少なくとも上記凹部の内周側面が上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物から構成されることを特徴とする光半導体素子搭載用基板。
(25)光半導体素子搭載領域となる凹部が1つ以上形成されている光半導体素子搭載用基板の製造方法であって、少なくとも上記凹部を上記(1)〜(20)のいずれかに記載の光反射用熱硬化性樹脂組成物を用いたトランスファー成型によって形成することを特徴とする光半導体搭載用基板の製造方法。
(26)上記(24)に記載の光半導体素子搭載用基板と、上記光半導体素子搭載用基板の凹部底面に搭載された光半導体素子と、上記光半導体素子を覆うように上記凹部内に形成された蛍光体含有透明封止樹脂層とを少なくとも備える光半導体装置。
【0010】
なお、本出願は、同出願人により2006年11月15日に出願された日本国特願2006−309052号、同じく2007年4月4日に出願された日本国特願2007−098354号に基づく優先権主張を伴うものであって、これらの明細書を参照することにより、本明細書の一部に組み込むものとする。
【図面の簡単な説明】
【0011】
【図1】本発明の光半導体素子搭載用基板の一実施形態を示す図であり、(a)は斜視図、(b)は側面断面図である。
【図2】本発明の光半導体素子搭載用基板の製造方法を説明する概略図であり、(a)〜(c)はトランスファー成型によって基板を製造する場合の各工程に対応する。
【図3】本発明の光半導体装置の一実施形態を示す図であり、(a)および(b)はそれぞれ装置の構造を模式的に示す側面断面図である。
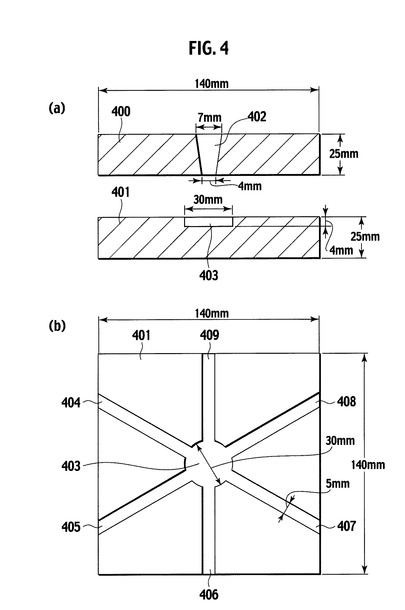
【図4】実施例において使用したバリ測定用金型を模式的に示した図であり、(a)は側面断面図、(b)は平面図である。
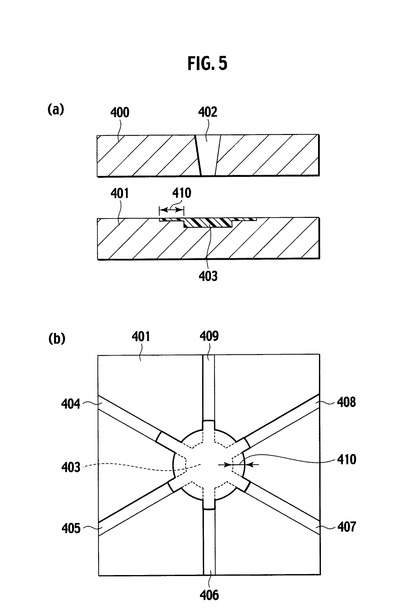
【図5】図4に示したバリ測定用金型を用いた成型時に生じたバリを模式的に示した図であり、(a)は側面断面図、(b)は平面図である。
【発明を実施するための形態】
【0012】
以下、本発明の実施の形態を説明する。本発明の熱硬化性光反射用樹脂組成物は、熱硬化性樹脂成分および白色顔料を含み、実際に成型の際に適用される成型条件下で成型した時の、例えば成型温度100℃〜200℃、成型圧力20MPa以下、及び60〜120秒の条件下でトランスファー成型した時のバリ長さが5mm以下となることを特徴とする。また、それらの熱硬化後の波長350nm〜800nmにおける光反射率が80%以上であることを特徴とする。
【0013】
本明細書において使用する用語、「成型時のバリ長さ」とは、図4に示したバリ測定用金型を用いてトランスファー成型を行った際に、金型中心部のキャビティから、金型の上型と下型との合せ目の隙間に放射方向にはみ出した樹脂硬化物の最大長さを意味する。このようなバリ長さが5mmを超えると、光半導体素子搭載領域の開口部(凹部)に樹脂汚れが張り出し、光半導体素子を搭載する際の障害となる可能性がある。あるいは、光半導体素子と金属配線とをボンディングワイヤなど公知の方法によって電気的に接続する際の障害になる可能性がある。半導体装置製造時の作業性の観点から、本発明による樹脂組成物のバリ長さは、3mm以下がより好ましく、1mm以下がさらに好ましい。
【0014】
本発明の硬化性光反射用樹脂組成物は、トランスファー成型に適用することを考慮して、熱硬化前は室温下(0〜30℃)で加圧成型可能であることが望ましい。より具体的には、例えば、室温下、5〜50MPa、1〜5秒程度の条件下で成型可能であればよい。熱硬化後は、光半導体装置の用途で使用する観点から、波長350nm〜800nmにおける光反射率が好ましくは80%以上、より好ましくは90%以上であることが望ましい。光反射率が80%未満である場合は、光半導体装置の輝度向上に十分に寄与できない可能性がある。
【0015】
以下、本発明の熱硬化性光反射用樹脂組成物の主な構成成分について説明する。
本発明の一実施形態では、熱硬化性樹脂成分として(A)エポキシ樹脂を含むことが好ましい。(A)エポキシ樹脂は、特に限定されることなく、エポキシ樹脂成型材料として一般に使用されている樹脂を用いることができる。例えば、フェノールノボラック型エポキシ樹脂、オルソクレゾールノボラック型エポキシ樹脂をはじめとするフェノール類とアルデヒド類のノボラック樹脂をエポキシ化したもの; ビスフェノールA、ビスフェノールF、ビスフェノールS、アルキル置換ビフェノール等のジグリシジエーテル; ジアミノジフェニルメタン、イソシアヌル酸等のポリアミンとエピクロルヒドリンとの反応により得られるグリシジルアミン型エポキシ樹脂; オレフィン結合を過酢酸等の過酸で酸化して得られる線状脂肪族エポキシ樹脂;及び脂環族エポキシ樹脂等が挙げられる。これらを単独で用いても、または2種以上併用してもよい。使用するエポキシ樹脂は無色または例えば淡黄色の比較的着色していないものが好ましい。そのようなエポキシ樹脂として、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、ジグリシジルイソシアヌレート、トリグリシジルイソシアヌレートを挙げることができる。
【0016】
本発明の一実施形態では、(A)エポキシ樹脂として、100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを使用することが好ましい。混練後の樹脂組成物の溶融粘度が低く流動性が高くなりすぎると、成型金型のエアベントが塞がれてしまい、金型キャビティ内に空気や揮発成分が残ってしまう可能性がある。キャビティ内に残存した空気や揮発成分は、成形ボイドやウェルドマーク等の外観不具合の原因となる。また、成型回数の増加に伴って、硬化物表面に残ったモノマー成分が成型金型に付着して金型を汚染することになる。金型にモノマーの付着物が堆積した結果、金型からの成型物の離型性が悪化する不具合が生じる。これに対し、本発明では、特定の粘度を有する(G)オリゴマーを使用することによって、混練後の樹脂組成物の溶融粘度を増加させ流動性を低下させることが可能である。また、そのような(G)オリゴマーの使用によって、金型の汚染原因となる残存モノマー成分を低減させることが可能である。その結果、溶融粘度が低い場合に生じる不具合を回避し、樹脂組成物のトランスファー成型性を向上させ、外観の優れた成型物を得ることが可能となる。
本発明で使用する(G)オリゴマーは、硬化性光反射用樹脂組成物の調製に先立って、少なくとも(A’)エポキシ樹脂および(B’)硬化剤、さらに必要に応じて(C’)硬化促進剤を配合することによって調製される。上記(A’)エポキシ樹脂、上記(B’)硬化剤、上記(C’)硬化促進剤は、それぞれ先に説明した(A)エポキシ樹脂、後述する(B)硬化剤および(C)硬化促進剤と同様のものを用いることができる。
【0017】
より具体的には、上記(G)オリゴマーは、例えば(A’)エポキシ樹脂および(B’)硬化剤を、当該(A’)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な当該(B’)硬化剤中の活性基(酸無水物基や水酸基)が0.3当量以下となるように配合し、粘土状になるまで混練する工程を経て得ることができる。得られた粘土状混練物を、引き続き、温度25〜60℃の範囲で1〜6時間にわたってエージングする工程を設けることが好ましい。また、(C’)硬化促進剤を使用する場合には、(A’)エポキシ樹脂と(B’)硬化剤との総和100重量部に対し、0.005〜0.05重量部となるように配合することが好ましい。
【0018】
上述のようにして調製された(G)オリゴマーは、成型時のバリ長さを短く調整するために、100〜150℃における粘度が100〜2500mPa・sの範囲にあることが好ましい。100℃における粘度が100〜2500mPa・sの範囲にあることがより好ましい。(G)オリゴマーの粘度が100mPa・s未満であると、トランスファー成型時にバリが発生しやすくなる。一方、粘度が2500mPa・sを超えると、成型時の流動性が低下し、成型性に乏しくなる傾向がある。なお、本明細書で使用する用語「粘度」は、ICIコーンプレート型粘度計を用いた測定によって得られた値である。(G)オリゴマーは、その粒径が1mm以下になるまで粉砕し、温度0℃以下の環境で保存することによって、粘度の上昇を抑制または停止させることができる。オリゴマーの粉砕方法は、陶器製乳鉢による粉砕等、公知のいかなる手法を用いても良い。
【0019】
本発明の一実施形態では、熱硬化性樹脂成分として(A)エポキシ樹脂とともに(B)硬化剤を含む。(B)硬化剤は、特に制限されることなく、エポキシ樹脂と反応可能な化合物であればよいが、その分子量は100〜400程度のものが好ましい。また、無色、または例えば淡黄色の比較的着色していないものが好ましい。具体的な化合物として、酸無水物系硬化剤、イソシアヌル酸誘導体、フェノール系硬化剤などが挙げられる。
【0020】
酸無水物系硬化剤としては、35℃以上の融点を有するものが好ましい。そのような酸無水物としては、例えば、無水フタル酸、無水マレイン酸、無水トリメリット酸、無水ピロメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、無水メチルナジック酸、無水ナジック酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、無水コハク酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸などが挙げられる。
【0021】
イソシアヌル酸誘導体としては、1,3,5−トリス(1−カルボキシメチル)イソシアヌレート、1,3,5−トリス(2−カルボキシエチル)イソシアヌレート、1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート、1,3−ビス(2−カルボキシエチル)イソシアヌレートなどが挙げられる。
フェノール系硬化剤としては、フェノール、クレゾール、レゾルシン、カテコール、ビスフェノールA、ビスフェノールF、フェニルフェノール、アミノフェノール等のフェノール類及び/又はα−ナフトール、β−ナフトール、ジヒドロキシナフタレン等のナフトール類と、ホルムアルデヒド、ベンズアルデヒド、サリチルアルデヒド等のアルデヒド基を有する化合物とを酸性触媒下で縮合又は共縮合させて得られるノボラック型フェノール樹脂; フェノール類及び/又はナフトール類とジメトキシパラキシレン又はビス(メトキシメチル)ビフェニルとから合成されるフェノール・アラルキル樹脂; ビフェニレン型フェノール・アラルキル樹脂、ナフトール・アラルキル樹脂等のアラルキル型フェノール樹脂; フェノール類及び/又はナフトール類とジシクロペンタジエンとの共重合によって合成される、ジシクロベンタジエン型フェノールノボラック樹脂、ジシクロペンタジエン型ナフトールノボラック樹脂等のジシクロペンタジエン型フェノール樹脂; トリフェニルメタン型フェノール樹脂; テルペン変性フェノール樹脂; パラキシリレン及び/又はメタキシリレン変性フェノール樹脂; メラミン変性フェノール樹脂; シクロペンタジエン変性フェノール樹脂; およびこれら2種以上を共重合して得られるフェノール樹脂などが挙げられる。
【0022】
先に例示した硬化剤の中でも、無水フタル酸、無水トリメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、およびシクロへキサントリカルボン酸無水物からなる群から選択される酸無水物、および1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート等のイソシアヌル酸誘導体の少なくとも一方を使用することが好ましい。これらは、単独で用いるだけでなく、二種以上併用しても良い。
【0023】
本発明の一実施態様では、(B)硬化剤として、少なくともイソシアヌル酸誘導体を使用することが好ましく、それらを酸無水物、特に35℃以上の融点を有する酸無水物を組合せて使用することがより好ましい。イソシアヌル酸誘導体のトリアジン骨格は、通常の環状メチレン骨格と比較して、活性酸素によって酸化されにくい特徴を有する。そのため、イソシアヌル酸誘導体を使用し、先の特徴を樹脂組成物に付与することで、成型後の樹脂組成物の耐熱性を向上させることが可能である。また、トリアジン骨格と3官能性の反応基によって成型体の機械特性を向上させることも可能となる。さらに、イソシアヌル酸誘導体を酸無水物と組合せることによって、樹脂組成物の溶融粘度を増加させることができ、成型時に金型から張り出すバリ長さを抑制することが可能である。イソシアヌル酸誘導体と酸無水物との配合比は、1:0〜1:10の範囲で適宜調整することが可能である。コスト削減と、樹脂の黄変による反射率の低下を抑制する観点から、1:1〜1:3の配合比とすることが好ましい。
【0024】
本発明の別の実施態様では、(B)硬化剤として、少なくともシクロへキサントリカルボン酸無水物を使用することが好ましい。シクロへキサントリカルボン酸無水物の使用によって、樹脂組成物の溶融粘度を増加させることができ、成型時のバリ長さを短くすることが可能である。また、樹脂組成物の硬化時間を短縮化できるため、成型効率を向上させることも可能である。シクロへキサントリカルボン酸無水物の具体例としては、下記構造式(I)で示される化合物が挙げられる。
【0025】
【化2】
【0026】
シクロへキサントリカルボン酸無水物と共に(B)硬化剤として先に説明した他の酸無水物、イソシアヌル酸誘導体およびフェノール系硬化剤等を併用してもよい。併用する硬化剤としては、成型時の流動性および成型物の着色の観点から、無水フタル酸、無水トリメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、または1,3,5−トリス(3−カルボキシプロピル)イソシアヌレートが好ましい。(B)硬化剤におけるシクロへキサントリカルボン酸無水物の含有率は、本発明の目的が達成できる範囲であれば特に限定されるものではないが、5質量%以上100質量%以下の範囲で調整することが好ましい。コストと性能とのバランスの観点から、上記含有率は25質量%以上75質量%以下の範囲であることが好ましい。
【0027】
(A)エポキシ樹脂と(B)硬化剤との配合比は、(A)エポキシ樹脂が(G)オリゴマーを含有しない場合、(A)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な(B)硬化剤中の活性基(酸無水物基や水酸基)が0.5〜1.5当量となるような割合であることが好ましい。0.7〜1.2当量となるような割合であることがより好ましい。上記活性基が0.5当量未満の場合には、エポキシ樹脂組成物の硬化速度が遅くなるとともに、得られる硬化物のガラス転移温度が低くなり、充分な弾性率が得られない場合がある。また、上記活性基が1.2当量を超える場合には、硬化後の強度が減少する場合がある。
【0028】
一方、(A)エポキシ樹脂として(G)オリゴマーを単独で使用するか、または(G)オリゴマーと(A)エポキシ樹脂とを併用する場合、(A)エポキシ樹脂(もしくは(G)オリゴマー)と(B)硬化剤との配合比は、(A)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な(B)硬化剤中の活性基(酸無水物基や水酸基)が0.5〜0.7当量となるような割合であることが好ましい。0.6〜0.7当量となるような割合であることがより好ましい。上記活性基が0.5当量未満の場合には、エポキシ樹脂組成物の硬化速度が遅くなるとともに、得られる硬化体のガラス転移温度が低くなり、充分な弾性率が得られない場合がある。また上記活性基が0.7当量を超える場合には、硬化後の強度が減少する場合がある。なお、(A)エポキシ樹脂が(G)オリゴマーを含む場合の(B)硬化剤の当量数は、(G)オリゴマーに含まれる(A’)エポキシ樹脂と(A)エポキシ樹脂のそれぞれに含まれるエポキシ基の総量を1当量とし、それに対して(B’)硬化剤と(B)硬化剤中に含まれる上記エポキシ基と反応可能な活性基の総和を当量数として換算する。
【0029】
本発明の熱硬化性光反射用樹脂組成物は、必要に応じて、(C)硬化促進剤として適切な化合物を含有してもよい。例えば、1,8−ジアザ−ビシクロ(5,4,0)ウンデセン−7、トリエチレンジアミンおよびトリ−2,4,6−ジメチルアミノメチルフェノールなどの3級アミン類; 2−エチル−4メチルイミダゾールおよび2−メチルイミダゾールなどのイミダゾール類; トリフェニルホスフィン、テトラフェニルホスホニウムテトラフェニルボレート、テトラ−n−ブチルホスホニウム−o,o−ジエチルホスホロジチオエート、テトラ−n−ブチルホスホニウム−テトラフルオロボレート、テトラ−n−ブチルホスホニウム−テトラフェニルボレートなどのリン化合物; 4級アンモニウム塩; 有機金属塩類; およびこれらの誘導体などを使用することができる。これらは単独で使用してもよく、あるいは併用してもよい。これらの硬化促進剤の中では、3級アミン類、イミダゾール類、リン化合物を用いることが好ましい。
【0030】
上記(C)硬化促進剤の含有率は、(A)エポキシ樹脂に対して、0.01〜8.0重量%であることが好ましく、0.1〜3.0重量%であることがより好ましい。硬化促進剤の含有率が0.01重量%未満では、十分な硬化促進効果が得られない場合がある。また、含有率が8.0重量%を超えると、得られる成型体に変色が見られる場合がある。
【0031】
本発明の熱硬化性光反射用樹脂組成物は、その成形性を調整するために、(D)無機充填材を含むことが好ましい。(D)無機充填剤は、特に限定されないが、例えば、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群から選ばれる少なくとも1種を使用することができる。熱伝導性、光反射特性および成型性の点から、少なくともシリカを含むことが好ましい。また、難燃性を高めるために、水酸化アルミニウムを組合せて使用することが好ましい。
【0032】
本発明の一実施形態では、(D)無機充填剤として、多孔質充填剤または吸油性を有する化合物を含有することが好ましい。(D)無機充填剤として、多孔質充填剤または吸油性を有するシリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群から選ばれる少なくとも1種を使用することも可能である。また、多孔質構造を有し、さらに吸油性を有する化合物を用いることも可能である。多孔質充填剤または吸油性を有する化合物の形状としては、特に限定されず、例えば、真球状、破砕状、円盤状、棒状、繊維状等のものを用いることができる。トランスファー成型時の金型内の流動性を考慮すると真球状、破砕状のものが好ましく、真球状のものがより好ましい。
【0033】
また、上記多孔質充填剤または吸油性を有する化合物は、その表面が物理的または化学的に親水化処理または疎水化処理されていてもよい。表面が疎水化処理されたものであることが好ましく、吸油量(JIS K5101に準ずる規定量)が50ml/100g以上となるように化学的に疎水化処理されたものであることがより好ましい。表面が疎水化処理された多孔質充填剤または吸油性を有する化合物を用いることで、(A)エポキシ樹脂や(B)硬化剤との接着性が増加し、結果として熱硬化物の機械強度やトランスファー成型時の流動性が向上する。また、吸油量50ml/100g以上となるように表面が疎水化処理された多孔質充填剤または吸油性を有する化合物を用いることで、(A)エポキシ樹脂との接着性が向上するとともに、混錬後の樹脂組成物のポットライフ低下を抑制することができ、また、熱硬化時に着色を抑制することもできる。このような疎水化処理が施された多孔質充填剤としては、例えば、富士シリシア化学株式会社で販売されているサイロホービック702を挙げることができる。
【0034】
また、上記多孔質充填剤または吸油性を有する化合物の見掛け密度は、特に限定されないが、0.4g/cm3以上であることが好ましく、0.4〜2.0g/cm3であることがより好ましい。なお、見掛け密度とは、多孔質充填剤または吸油性を有する化合物の素原料が占める密度と微細孔の占める空間(即ち細孔容積)とを考慮した密度のことである。この見掛け密度が0.4g/cm3に満たない場合は、充填剤粒子の機械的強度が小さく、ミキシングロールミルなどのせん断力を生じるような溶融混錬時において、粒子が破壊されてしまう恐れがある。一方、見掛け密度が2.0g/cm3を超える場合は、トランスファー成型のためのタブレットを成型する際に、臼型と杵型とからなる金型の表面に樹脂組成物が付着し易くなる傾向にある。
【0035】
また、上記多孔質充填剤または吸油性を有する化合物の平均粒径は、0.1〜100μmであることが好ましく、白色顔料とのパッキング効率を考慮すると1〜10μmの範囲であることがより好ましい。平均粒径が100μmよりも大きく、または0.1μmよりも小さくなると、トランスファー成型する際の溶融時に樹脂組成物の流動性が悪くなる傾向にある。
【0036】
また、上記多孔質充填剤または吸油性を有する化合物の比表面積は、100〜1000m2/gであることが好ましく、300〜700m2/gであることがより好ましい。比表面積が100m2/gよりも小さくなると充填剤による樹脂の吸油量が小さくなり、タブレット成型時に杵型に樹脂が付着し易くなる傾向にあり、比表面積が1000m2/gよりも大きくなると、トランスファー成型する際の溶融時に樹脂組成物の流動性が悪くなる傾向にある。
【0037】
また、上記多孔質充填剤または吸油性を有する化合物の含有量は、特に限定されないが、(D)無機充填剤全体に対し、0.1体積%〜20体積%の範囲であることが好ましい。溶融時の樹脂組成物の成型性を考慮すると、1体積%〜5体積%であることがより好ましい。この含有量が0.1体積%よりも小さい場合は、樹脂組成物の一部が臼型と杵型の成型金型の表面に付着し易くなり、20体積%よりも大きい場合は、トランスファー成型する際の溶融時に樹脂組成物の流動性が低下する傾向にある。例えば、上記多孔質充填剤として上記サイロホービック702を用いる場合には、その含有量を、樹脂組成物の溶融時の流動性や樹脂硬化物の強度の観点から5体積%以下とすることが好ましい。
【0038】
本発明で用いられる(E)白色顔料としては、特に限定されるものではなく、公知のものを使用することができる。例えば、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子などを用いることができ、これらは単独でも併用しても構わない。無機中空粒子は、例えば、珪酸ソーダガラス、アルミ珪酸ガラス、硼珪酸ソーダガラス、シラス等が挙げられる。熱伝導性、光反射特性の点からは、少なくともアルミナまたは酸化マグネシウムを使用するか、またはそれらを組合せて使用することが好ましい。(E)白色顔料の粒径は、中心粒径が0.1〜50μmの範囲にあることが好ましい。この中心粒径が0.1μm未満であると、粒子が凝集しやすく分散性が悪くなる傾向にある。一方、中心粒径が50μmを超えると、硬化物の光反射特性が十分に得られない恐れがある。(E)白色顔料の配合量は、特に限定されないが、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることが好ましい。
【0039】
上記(D)無機充填剤と上記(E)白色顔料との合計配合量は、特に限定されないが、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることが好ましい。この合計配合量が10体積%未満であると、硬化物の光反射特性が十分に得られない恐れがある。また、合計配合量が85体積%を超えると、樹脂組成物の成型性が悪くなり、光半導体搭載用基板の作製が困難となる傾向がある。
【0040】
本発明の熱硬化性光反射用樹脂組成物には、必要に応じて(F)カップリング剤を加えることができる。(F)カップリング剤は、特に限定されないが、例えば、シラン系カップリング剤やチタネート系カップリング剤等を用いることができる。シランカップリング剤としては、例えば、エポキシシラン系、アミノシラン系、カチオニックシラン系、ビニルシラン系、アクリルシラン系、メルカプトシラン系及びこれらの複合系等を用いることができる。(F)カップリング剤の種類や処理条件は特に限定されるものではなく、樹脂組成物にそのまま添加する方法や、無機充填剤または白色顔料と予め混合して添加する方法等、従来から用いられている手法を適用してもよい。(F)カップリング剤の配合量は樹脂組成物に対して5重量%以下が好ましい。
【0041】
また、本発明の熱硬化性光反射用樹脂組成物には、溶融粘度調整を目的として(H)増粘剤を添加しても良い。(H)増粘剤としては、特に限定されるものではないが、例えば、トクヤマ(株)で販売されているレオロシールCP−102として入手可能なナノシリカを用いることができる。(H)増粘剤の添加量としては、樹脂組成物の総体積の0.15体積%以下であることが好ましい。(H)増粘剤の添加量が0.15体積%よりも多くなると、樹脂組成物の溶融時の流動性が損なわれるとともに、硬化後に充分な材料強度が得られなくなる恐れがある。また、(H)増粘剤は、中心粒径が1nm〜1000nmであるようなナノ粒子フィラーであることが好ましく、中心粒径が10nm〜1000nmであるようなナノ粒子フィラーであることがより好ましい。中心粒径1nmよりも小さいフィラーは、粒子が凝集しやすく分散性が低下する傾向にあり、特性上好ましくない。このような(H)増粘剤を用いる場合には、(D)無機充填剤の一部としてナノシリカを用いてもよい。一方、1000nmよりも大きなフィラーを添加すると、バリ長さが低減しない傾向にあり、特性上好ましくない。なお、本発明の樹脂組成物には、(H)増粘剤以外に、必要に応じて、酸化防止剤、離型剤、イオン補足剤等の各種添加剤を添加してもよい。
【0042】
本発明の熱硬化性光反射用樹脂組成物は、上記した各成分を均一に分散混合することによって得ることができ、その調製手段や条件等は特に限定されない。一般的な手法として、所定配合量の各種成分をミキサー等によって十分に均一に撹拌および混合した後、ミキシングロール、押出機、ニーダー、ロールおよびエクストルーダー等を用いて混練し、さらに得られた混練物を冷却および粉砕する方法を挙げることができる。なお、混練形式についても特に限定されるものではないが、溶融混練が好ましい。溶融混練の条件は、使用した成分の種類や配合量によって適宜決定すればよく、特に限定されない。例えば、15〜100℃の範囲で5〜40分間にわたって溶融混練することが好ましく、20〜100℃の範囲で10〜30分間にわたって溶融混練することがより好ましい。溶融混練時の温度が15℃未満であると、各成分を溶融混練させることが困難であり、分散性も低下する傾向にある。一方、100℃よりも高温であると、樹脂組成物の高分子量化が進行し、樹脂組成物が硬化してしまう恐れがある。また、溶融混練の時間が5分未満であると、バリ長さを効果的に抑制することができない傾向にあり、40分よりも長いと、樹脂組成物の高分子量化が進行し、成型前に樹脂組成物が硬化してしまう恐れがある。
【0043】
本発明の熱硬化性光反射用樹脂組成物は、上記各成分を配合、混練した後、成型時の溶融粘度を上昇させることを目的として熟成放置(エージング)することが好ましい。より具体的には、エージングは0℃〜30℃で1〜72時間にわたって実施することが好ましい。エージングは、より好ましくは15℃〜30℃で12〜72時間にわたって、さらに好ましくは25℃〜30℃で24〜72時間にわたって、実施することが望ましい。1時間よりも短時間のエージングでは、バリ長さを効果的に抑制できない傾向にあり、72時間より長くエージングすると、トランスファー成型時に充分な流動性を確保できない恐れがある。また、0℃未満の温度でエージングを実施した場合には、(C)硬化促進剤が不活性化されて、樹脂組成物の三次元架橋反応が十分に進行せず、溶融時の粘度が上昇しない恐れがある。また、30℃よりも高温でエージングを実施した場合には、樹脂組成物が水分を吸収してしまい、硬化物の強度や弾性率などの機械的物性が悪くなる傾向にある。
【0044】
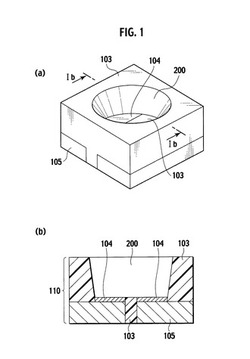
本発明の光半導体素子搭載用基板は、本発明の熱硬化性光反射用樹脂組成物を使用して構成されることを特徴とする。具体的には、光半導体素子搭載領域となる1つ以上の凹部を有し、少なくとも上記凹部の内周側面が本発明の熱硬化性光反射用樹脂組成物から構成される基板が挙げられる。図1は、本発明の光半導体素子搭載用基板の一実施形態を示すものであり、(a)は斜視図、(b)は側面断面図である。図1に示したように、本発明の光半導体素子搭載用基板110は、リフレクター103と、Ni/Agメッキ104および金属配線105を含む配線パターン(リードフレーム)とが一体化され、光半導体素子搭載領域となる凹部200が形成された構造を有し、少なくとも上記凹部200の内周側面は本発明の熱硬化性光反射用樹脂組成物から構成されていることを特徴とする。
【0045】
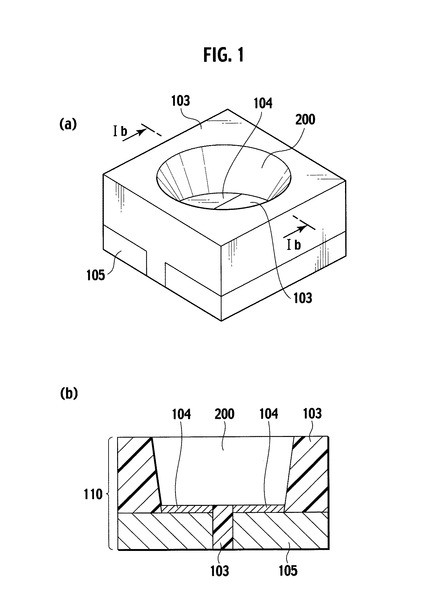
本発明の光半導体素子搭載用基板の製造方法は、特に限定されないが、例えば、本発明の熱硬化性光反射用樹脂組成物またはそのタブレット成型体をトランスファー成型によって製造することができる。図2は、本発明の光半導体素子搭載用基板の製造方法を説明する概略図であり、図2(a)〜(c)はトランスファー成型によって基板を製造する場合の各工程に対応する。より具体的には、光半導体素子搭載用基板は、図2(a)に示すように、金属箔から打ち抜きやエッチング等の公知の方法により金属配線105を形成し、次いで該金属配線105を所定形状の金型301に配置し(図2(b))、金型301の樹脂注入口300から本発明の熱硬化性光反射用樹脂組成物(タブレット成型体の溶融物)を注入する。次いで、注入した樹脂組成物を、好ましくは金型温度170〜190℃、成形圧力2〜8MPaで60〜120秒にわたって硬化させた後に金型301を外し、アフターキュア温度120℃〜180℃で1〜3時間にわたって熱硬化させる。次いで、硬化した熱硬化性光反射用樹脂組成物から構成されるリフレクター103に周囲を囲まれた光半導体素子搭載領域となる凹部200の所定位置に、Ni/銀メッキ104を施すことによって製造することが可能である(図2(c))。
【0046】
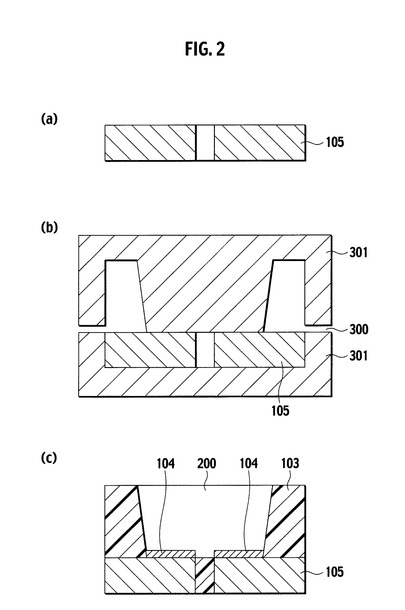
本発明の光半導体素子搭載用基板を用いた光半導体装置は、本発明の光半導体素子搭載用基板と、光半導体素子搭載用基板の凹部底面に搭載される光半導体素子と、光半導体素子を覆うように凹部内に形成される蛍光体含有透明封止樹脂層とを少なくとも備えることを特徴とする。図3(a)および図3(b)は、それぞれ本発明の光半導体装置の一実施形態を示す側面断面図である。より具体的には、図3に示した光半導体装置は、本発明の光半導体素子搭載用基板110の光半導体素子搭載領域となる凹部(図2の参照符号200)の底部所定位置に光半導体素子100が搭載され、該光半導体素子100と金属配線105とがボンディングワイヤ102やはんだバンプ107などの公知の方法によりNi/銀メッキ104を介して電気的に接続され、該光半導体素子100が公知の蛍光体106を含む透明封止樹脂101により覆われている。
【実施例】
【0047】
以下、本発明を実施例により詳述するが、本発明はこれらに限定されるものではない。なお、以下の各実施例および各比較例で使用した成分の詳細は以下のとおりである。
*1:トリグリシジルイソシアヌレート(エポキシ当量100、日産化学社製、商品名TEPIC−S)
*2:ヘキサヒドロ無水フタル酸(和光純薬社製)
*3:1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート(四国化成工業社製、製品名C3CIC酸)
*4:シクロヘキサントリカルボン酸無水物(三菱ガス化学社製、製品名H−TMAn)
*5:テトラヒドロ無水フタル酸(アルドリッチ社製)
*6:メチルヒドロ無水フタル酸(日立化成工業社製 商品名HN5500)
*7:テトラ−n−ブチルホスホニウム−0,0−ジエチルホスホロジチエート(日本化学工業社製、商品名ヒシコーリンPX−4ET)
*8:トリメトキシエポキシシラン(東レダウコーニング社製、商品名SH6040)
*9:脂肪酸エステル(クラリアント社製、商品名ヘキストワックスE)
*10:脂肪族エーテル(東洋ペトロライト社製、商品名ユニトックス420)
*11:溶融シリカ(電気化学工業社製、商品名FB−301)
*12:溶融シリカ(電気化学工業社製、商品名FB−950)
*13:溶融シリカ(アドマテックス社製、商品名SO−25R)
*14:多孔質球状シリカ(富士シリシア化学社製、製品名サイロスフィアC−1504)平均粒径:3μm、見掛け密度:0.58g/ml、比表面積:300m2/g)
*15:多孔質無定形状シリカ(富士シリシア化学社製、製品名サイロホービック702、平均粒径:4μm 見掛け密度:0.48g/ml、比表面積:300m2/g)
*16:多孔質無定形状シリカ(富士シリシア化学社製、製品名サイリシア430、平均粒径:4μm、見掛け密度:0.48g/ml、比表面積:300m2/g)
*17:中空粒子(住友3M社製、商品名S60−HS)
*18:アルミナ(アドマテックス社製、商品名AO−802)
*19:ナノシリカ(トクヤマ社製、CP−102)
【0048】
(実施例A1〜A16および比較例A1〜A7)
1.熱硬化性光反射用樹脂組成物の調製
下記表A1から表A3に示した配合割合に従って各成分を配合し、ミキサーによって十分に混練した後にミキシングロールによって所定条件下で溶融混練して混練物を得た。さらに、得られた混練物を粉砕することによって、実施例A1〜A16および比較例A1〜A7の熱硬化性光反射用樹脂組成物を各々調製した。なお、表A1から表A3に示した各成分の配合量の単位は重量部である。表における空欄は該当する成分の配合が無いことを意味する。
【0049】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例A1〜A16および比較例A1〜A7の各々の樹脂組成物について、以下の手順に従って、光反射率およびバリ長さを測定した。また、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、評価した。それらの結果を下記表A1から表A3に示す。
【0050】
(光反射率)
先に調製した各々の熱硬化性光反射用樹脂組成物を、成型金型温度180℃、成型圧力6.9MPa、硬化時間90秒の条件下でトランスファー成型した後、150℃で2時間にわたって後硬化することによって、厚み1.0mmの試験片をそれぞれ作製した。次いで、積分球型分光光度計V−750型(日本分光株式会社製)を用いて、波長400nmにおける各試験片の光反射率を測定した。
【0051】
(バリ長さ)
先に調製した各々の熱硬化性光反射用樹脂組成物を、ポットを用いて、バリ測定用金型(図4を参照)に流し込み、次いで硬化させることによって樹脂組成物を成型した。なお、成型時の金型温度は180℃、成型圧力は6.9MPa、樹脂の流し込み時間(トランスファー時間)は10秒であり、硬化温度は180℃、硬化時間は90秒とした。成型後、バリ測定用金型の上型を外し、成型時に金型の上型と下型との隙間を流れて生じたバリの長さの最大値を、ノギスを使用して測定した。
【0052】
図4は、先のバリ長さの測定時に使用するバリ測定用金型の構造を模式的に示した図であり、(a)は側面断面図、(b)は平面図である。図4に示したように、バリ測定用金型は、一対の上型400と下型401とから構成され、上型400は樹脂注入口402を有する。また、下型401は、樹脂注入口402に対向するキャビティ403と、キャビティ403から金型外周部に向かって伸びる6本のスリット404、405、406、407、408および409を有する。実際に使用したバリ測定用金型の寸法は、図4に示したように上型400および下型401の外形が140mm×140mm、樹脂注入口402の上径が7mmおよび下径が4mm、キャビティ403の径が30mmおよび深さが4mmである。また、キャビティ403から延びる6本のスリット404から409の幅はそれぞれ5mmであり、深さは順に75、50、30、20、10および2μmであった。図5は、図4に示したバリ測定用金型を用いた成型時に生じたバリを模式的に示す図であり、(a)は側面断面図、(b)は平面図である。バリは、図5に示したように、樹脂組成物がキャビティ403の外延から各スリットに沿って流れ込み硬化した部分410を意味する。本発明で規定する「バリ長さ」は、参照符号410で示されるバリの最大値を、ノギスで測定した値である。
【0053】
(ワイヤボンディング性の評価)
最初に、図2に示した製造工程に従い、先に調製した各々の熱硬化性光反射用樹脂組成物をトランスファー成型することによって、光半導体素子搭載用基板を作製した。なお、成型条件は、成型金型温度180℃、成型圧力6.9MPa、硬化時間90秒であり、150℃で2時間にわたって後硬化を実施した。
次に、作製した上記基板の光半導体素子搭載領域となる凹部に光半導体素子を搭載した後、ワイヤボンダ(HW22U−H、九州松下電器株式会社製、商品名)および直径28μmのボンディングワイヤを使用して、光半導体素子と基板とをワイヤボンディングすることによって、電気的に接続した。ワイヤボンディング時の基板の加熱温度は180℃とした。ワイヤボンディング性は、光半導体素子と基板とを電気的に接続するワイヤボンディングするワイヤの引っ張り強度を、プルテスターPTR−01(株式会社レスカ製、商品名)を使用して測定し、その値について下記の評価基準に従って評価した。
【0054】
ワイヤボンディング性の評価基準
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0055】
【表1】
【0056】
【表2】
【0057】
【表3】
【0058】
表A1から表A3によれば、実施例A1〜A16の樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。したがって、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0059】
(実施例B1〜B11および比較例B1〜B8)
1.熱硬化性光反射用樹脂組成物の調製
下記表B1および表B2に示した配合割合に従って各成分を配合し、ミキサーによって十分に混練した後にミキシングロールによって所定条件下で溶融混練して混練物を得た。さらに、得られた混練物に必要に応じてエージングを施した後に冷却し、それらを粉砕することによって、実施例B1〜B11および比較例B1〜B8の熱硬化性光反射用樹脂組成物を各々調製した。
【0060】
なお、表B1およびB2に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いこと、または該当する工程を実施しないことを意味する。各実施例の詳細は以下のとおりである。
実施例B1は、特定の(G)オリゴマーを使用する手法に関する。
実施例B2は、(A)エポキシ樹脂中のエポキシ基1当量に対する硬化剤中の活性基を0.5〜0.7当量の範囲とする手法に関する。
実施例B3は、(H)増粘剤としてナノフィラーを追加する手法に関する。
実施例B4は、樹脂組成物を所定条件下でエージングする手法に関する。
実施例B5は、溶融混練条件を調整する手法(混練時間を15分から30分に延長)に関する。
実施例B6〜B11は、上述の手法のうちいずれか2つの手法を併用した場合に関する。
【0061】
なお、実施例B1、B6、B7およびB9において使用した(G)オリゴマーは、以下に説明する手順で調製した。また、(G)オリゴマーの粘度は、Reseach Equipment LTD.(London)製のICIコーンプレート型粘度計を使用し、試料量0.155±0.01g、100℃で測定したところ、1000mPa・sであった。
【0062】
(オリゴマーの作製方法)
表B1に示した配合条件に従って各成分を配合し、ミキシングロールによって25℃で10分間にわたって溶融混練を行った。なお、表B1に示した配合は、エポキシ基1当量に対して酸無水物基0.1当量となる割合になっている。次に、溶融混練によって得られた粘土状組成物(混練物)を温度55℃で4時間にわたってエージングした。そのようなエージング後に、混練物を口径300mmの陶器製乳鉢を用いて、粒径が1mm以下になるまで粉砕することにより所望のオリゴマーを得た。得られたオリゴマーは、温度0℃以下の環境下で保存した。
【0063】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例B1〜B6および比較例B1〜B8の各々の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表B1および表B2に示す。
【0064】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0065】
【表4】
【0066】
【表5】
【0067】
表B1および表B2によれば、実施例B1〜B11の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。そのため、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。また、(G)オリゴマーを使用した場合には、モノマー成分の残存量を低減させることが可能であるため、ワイヤボンディング性の向上に加えて、基板成型時の金型汚れおよび成型品の離型性の低下といった不具合を回避することができ、優れた外観を有する成型基板が得られる。
【0068】
(実施例C1〜C3および比較例C1、C2)
1.熱硬化性光反射用樹脂組成物の調製
下記表C1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例C1〜C3、比較例C1およびC2の熱硬化性光反射用樹脂組成物を各々調製した。表C1に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いことを意味する。
【0069】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例C1〜C3および比較例C1、C2の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表C1に示す。
【0070】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0071】
【表6】
【0072】
注記:(*)金型内における樹脂の流動性不良のため、測定不可
【0073】
表C1によれば、実施例C1〜C3の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。C3CIC酸は、その特徴的な化学構造によって、成型体の機械的強度を向上させることが可能であるだけでなく、それらはバリの抑制にも効果的であることが明らかである。但し、硬化剤としてC3CIC酸を単独で使用すると組成物の流動性が悪化する傾向にあるため、HHPAなどの他の硬化剤と組合せて使用することが好ましいことが分かる。本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0074】
(実施例D1〜D5および比較例D1〜D3)
1.熱硬化性光反射用樹脂組成物の調製
表D1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例D1〜D5、比較例D1〜D3の熱硬化性光反射用樹脂組成物を各々調製した。なお、表D1に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いことを意味する。
【0075】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例D1〜D5および比較例D1〜D3の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。なお、耐久性について検討するために、光反射率の測定は、試験片の成型後と、150℃で72時間加熱した後に実施した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表D1に示す。
【0076】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0077】
【表7】
【0078】
表D1によれば、実施例D1〜D5の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。さらに、硬化物(成型基板)の光反射特性が劣化し難いことも分かる。そのため、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。また、本発明の熱硬化性光反射用樹脂組成物を使用することによって、可視光から近紫外光領域において高い反射率を保持することが可能な光半導体素子搭載用基板を効率良く製造することも可能であることが分かる。
【0079】
(実施例E1〜E8)
表E1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例E1〜E8の熱硬化性光反射用樹脂組成物を各々調製した。なお、表E1に示した各成分の詳細は先に説明したとおりであり、各配合量の単位は重量部である。
【0080】
得られた実施例E1〜E8の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表E1に示す。
【0081】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0082】
【表8】
【0083】
表E1によれば、実施例E1〜E8の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、トランスファー成型時のバリの抑制効果が高く、より優れたワイヤボンディング性を示すことが分かる。このように、各構成成分を適切に組合せて配合することによって、ワイヤボンディング性をさらに向上させることも可能である。したがって、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0084】
以上の説明からして、本発明の精神と範囲に反することなしに、広範に異なる実施態様を構成することができることは明白であり、本発明は請求の範囲において限定した以外は、その特定の実施態様によって制約されるものではない。
【技術分野】
【0001】
本発明は、光半導体素子と蛍光体などの波長変換手段とを組合せた光半導体装置に用いる熱硬化性光反射用樹脂組成物及びその製造方法、並びに当該樹脂組成物を用いた光半導体素子搭載用基板及び光半導体装置に関する。
【背景技術】
【0002】
LED(Light Emitting Diode:発光ダイオード)などの光半導体素子と蛍光体とを組合せた光半導体装置は、高エネルギー効率および長寿命などの利点から、屋外用ディスプレイ、携帯液晶バックライトおよび車載用途など様々な用途に適用され、その需要が拡大しつつある。それに伴って、LEDデバイスの高輝度化が進み、素子の発熱量増大によるジャンクション温度の上昇、あるいは直接的な光エネルギーの増大による素子材料の劣化が問題視され、近年、熱劣化および光劣化に対して耐性を有する素子材料の開発が課題となっている。日本国特開2006−140207号公報では、耐熱試験後の光反射特性に優れる光半導体素子搭載用基板を開示している。
【発明の概要】
【発明が解決しようとする課題】
【0003】
しかし、上記公報で開示された熱硬化性光反射用樹脂組成物を用いて、トランスファー成型によって基板を製造する場合、成型時に成型金型の上型と下型との隙間に樹脂組成物が染み出し、樹脂汚れが発生しやすい傾向がある。加熱成型時に樹脂汚れが発生すると、光半導体素子搭載領域となる基板の開口部(凹部)に樹脂汚れが張り出し、光半導体素子を搭載する際の障害になる。また、開口部に光半導体素子を搭載できたとしても、樹脂汚れは、光半導体素子と金属配線とをボンディングワイヤなどの公知の方法によって電気的に接続する際の障害になる。すなわち、樹脂汚れは、素子搭載およびワイヤボンディングといった半導体素子製造時の作業性を低下させるために望ましくない。半導体素子製造時に上述の障害が起こらないように、基板の開口部に樹脂汚れが存在する場合には、一般に、光半導体素子搭載用基板の製造プロセスに樹脂汚れの除去工程が追加される。しかし、そのような除去工程は、コストや製造時間のロスとなるため、改善が望まれている。
【0004】
本発明は、上記に鑑みてなされたものであり、樹脂硬化後の可視光から近紫外光領域における反射率が高く、その一方で、トランスファー成型時に樹脂汚れが発生し難い熱硬化性光反射用樹脂組成物を提供する。また本発明は、上述の樹脂組成物を用いて、ワイヤボンディング性に優れ、光劣化および熱劣化に対して耐性を有する半導体素子搭載用基板および光半導体装置を提供する。さらに本発明は、上述の光半導体素子搭載用基板および光半導体装置を効率良く製造するための製造方法を提供する。
【課題を解決するための手段】
【0005】
すなわち、本発明は、以下(1)〜(26)に記載の事項をその特徴とするものである。
(1)熱硬化性成分と(E)白色顔料とを含む熱硬化性光反射用樹脂組成物であって、
成型温度100℃〜200℃、成型圧力20MPa以下、及び成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ
熱硬化後の、波長350nm〜800nmにおける光反射率が80%以上である
ことを特徴とする熱硬化性光反射用樹脂組成物。
(2)上記熱硬化性成分が、(A)エポキシ樹脂を含むことを特徴とする上記(1)に記載の熱硬化性光反射用樹脂組成物。
(3)上記(A)エポキシ樹脂が、(A’)エポキシ樹脂と(B’)硬化剤とを混練することによって得られ、かつ100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを含むことを特徴とする上記(2)に記載の熱硬化性光反射用樹脂組成物。
(4)上記熱硬化性成分が、上記(A)エポキシ樹脂とともに用いられる(B)硬化剤をさらに含み、上記(A)エポキシ樹脂と上記(B)硬化剤との配合比が、上記(A)エポキシ樹脂中のエポキシ基1当量に対して、該エポキシ基と反応可能な上記(B)硬化剤中の活性基が0.5〜0.7当量となる比であることを特徴とする、上記(3)に記載の熱硬化性光反射用樹脂組成物。
(5)上記(B)硬化剤が、イソシアヌル酸骨格を有する化合物を含むことを特徴とする上記(4)に記載の熱硬化性光反射用樹脂組成物。
【0006】
(6)上記(B)硬化剤が、35℃以上の融点を有する酸無水物をさらに含むことを特徴とする上記(5)に記載の熱硬化性光反射用樹脂組成物。
(7)上記(B)硬化剤が、シクロへキサントリカルボン酸無水物を含むことを特徴とする上記(4)に記載の熱硬化性光反射用樹脂組成物。
(8)上記シクロへキサントリカルボン酸無水物が、下記構造式(I)で示される化合物であることを特徴とする上記(7)に記載の熱硬化性光反射用樹脂組成物。
【化1】
(9)さらに(H)増粘剤を含み、該(H)増粘剤が、中心粒径が1nm〜1000nmのナノ粒子フィラーを含有することを特徴とする上記(1)〜(8)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(10)さらに(D)無機充填剤を含み、該(D)無機充填剤が、多孔質充填剤または吸油性を有する化合物を含有することを特徴とする上記(1)〜(8)のいずれかに記載の熱硬化性光反射用樹脂組成物。
【0007】
(11)上記多孔質充填剤または吸油性を有する化合物の形状が、真球状、破砕状、円盤状、棒状、繊維状からなる群から選択される少なくとも1種であることを特徴とする上記(10)に記載の熱硬化性光反射用樹脂組成物。
(12)上記多孔質充填剤または吸油性を有する化合物の表面が、疎水化処理または親水化処理されていることを特徴とする上記(10)または(11)に記載の熱硬化性光反射用樹脂組成物。
(13)上記多孔質充填剤または吸油性を有する化合物の見掛け密度が、0.4g/cm3以上であることを特徴とする上記(10)〜(12)のいずれか1つに記載の熱硬化性光反射用樹脂組成物。
(14)上記(D)無機充填剤における上記多孔質充填剤または吸油性を有する化合物の含有量が、0.1体積%〜20体積%の範囲であることを特徴とする上記(10)〜(13)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(15)さらに(D)無機充填剤として、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群の中から選ばれる少なくとも1種を含むことを特徴とする上記(1)〜(14)のいずれかに記載の熱硬化性光反射用樹脂組成物。
【0008】
(16)上記(E)白色顔料が、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子からなる群の中から選ばれる少なくとも1種であることを特徴とする上記(1)〜(15)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(17)上記(E)白色顔料の中心粒径が、0.1〜50μmの範囲にあることを特徴とする上記(1)〜(16)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(18)上記(D)無機充填剤と上記(E)白色顔料とを合計した配合量が、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることを特徴とする上記(1)〜(17)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(19)少なくとも上記各種構成成分を、混練温度20〜100℃、混練時間10〜30分の条件下で混練することによって得られる混練物を含むことを特徴とする上記(1)〜(18)のいずれかに記載の熱硬化性光反射用樹脂組成物。
(20)上記混練物が上記混練後に0〜30℃で1〜72時間にわたってエージングされたものであることを特徴とする上記(19)に記載の熱硬化性光反射用樹脂組成物。
【0009】
(21)上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物を製造する方法であって、少なくとも樹脂組成物の各成分を混練して混練物を形成する混練工程と、上記混練物を0〜30℃で1〜72時間にわたってエージングする工程とを有することを特徴とする熱硬化性光反射用樹脂組成物の製造方法。
(22)上記混練工程を、混練温度20〜100℃、及び混練時間10〜30分の条件下で行うことを特徴とする上記(21)に記載の熱硬化性光反射用樹脂組成物の製造方法。
(23)上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物を用いて構成されることを特徴とする光半導体素子搭載用基板。
(24)光半導体素子搭載領域となる凹部が1つ以上形成されている光半導体素子搭載用基板であって、少なくとも上記凹部の内周側面が上記(1)〜(20)のいずれかに記載の熱硬化性光反射用樹脂組成物から構成されることを特徴とする光半導体素子搭載用基板。
(25)光半導体素子搭載領域となる凹部が1つ以上形成されている光半導体素子搭載用基板の製造方法であって、少なくとも上記凹部を上記(1)〜(20)のいずれかに記載の光反射用熱硬化性樹脂組成物を用いたトランスファー成型によって形成することを特徴とする光半導体搭載用基板の製造方法。
(26)上記(24)に記載の光半導体素子搭載用基板と、上記光半導体素子搭載用基板の凹部底面に搭載された光半導体素子と、上記光半導体素子を覆うように上記凹部内に形成された蛍光体含有透明封止樹脂層とを少なくとも備える光半導体装置。
【0010】
なお、本出願は、同出願人により2006年11月15日に出願された日本国特願2006−309052号、同じく2007年4月4日に出願された日本国特願2007−098354号に基づく優先権主張を伴うものであって、これらの明細書を参照することにより、本明細書の一部に組み込むものとする。
【図面の簡単な説明】
【0011】
【図1】本発明の光半導体素子搭載用基板の一実施形態を示す図であり、(a)は斜視図、(b)は側面断面図である。
【図2】本発明の光半導体素子搭載用基板の製造方法を説明する概略図であり、(a)〜(c)はトランスファー成型によって基板を製造する場合の各工程に対応する。
【図3】本発明の光半導体装置の一実施形態を示す図であり、(a)および(b)はそれぞれ装置の構造を模式的に示す側面断面図である。
【図4】実施例において使用したバリ測定用金型を模式的に示した図であり、(a)は側面断面図、(b)は平面図である。
【図5】図4に示したバリ測定用金型を用いた成型時に生じたバリを模式的に示した図であり、(a)は側面断面図、(b)は平面図である。
【発明を実施するための形態】
【0012】
以下、本発明の実施の形態を説明する。本発明の熱硬化性光反射用樹脂組成物は、熱硬化性樹脂成分および白色顔料を含み、実際に成型の際に適用される成型条件下で成型した時の、例えば成型温度100℃〜200℃、成型圧力20MPa以下、及び60〜120秒の条件下でトランスファー成型した時のバリ長さが5mm以下となることを特徴とする。また、それらの熱硬化後の波長350nm〜800nmにおける光反射率が80%以上であることを特徴とする。
【0013】
本明細書において使用する用語、「成型時のバリ長さ」とは、図4に示したバリ測定用金型を用いてトランスファー成型を行った際に、金型中心部のキャビティから、金型の上型と下型との合せ目の隙間に放射方向にはみ出した樹脂硬化物の最大長さを意味する。このようなバリ長さが5mmを超えると、光半導体素子搭載領域の開口部(凹部)に樹脂汚れが張り出し、光半導体素子を搭載する際の障害となる可能性がある。あるいは、光半導体素子と金属配線とをボンディングワイヤなど公知の方法によって電気的に接続する際の障害になる可能性がある。半導体装置製造時の作業性の観点から、本発明による樹脂組成物のバリ長さは、3mm以下がより好ましく、1mm以下がさらに好ましい。
【0014】
本発明の硬化性光反射用樹脂組成物は、トランスファー成型に適用することを考慮して、熱硬化前は室温下(0〜30℃)で加圧成型可能であることが望ましい。より具体的には、例えば、室温下、5〜50MPa、1〜5秒程度の条件下で成型可能であればよい。熱硬化後は、光半導体装置の用途で使用する観点から、波長350nm〜800nmにおける光反射率が好ましくは80%以上、より好ましくは90%以上であることが望ましい。光反射率が80%未満である場合は、光半導体装置の輝度向上に十分に寄与できない可能性がある。
【0015】
以下、本発明の熱硬化性光反射用樹脂組成物の主な構成成分について説明する。
本発明の一実施形態では、熱硬化性樹脂成分として(A)エポキシ樹脂を含むことが好ましい。(A)エポキシ樹脂は、特に限定されることなく、エポキシ樹脂成型材料として一般に使用されている樹脂を用いることができる。例えば、フェノールノボラック型エポキシ樹脂、オルソクレゾールノボラック型エポキシ樹脂をはじめとするフェノール類とアルデヒド類のノボラック樹脂をエポキシ化したもの; ビスフェノールA、ビスフェノールF、ビスフェノールS、アルキル置換ビフェノール等のジグリシジエーテル; ジアミノジフェニルメタン、イソシアヌル酸等のポリアミンとエピクロルヒドリンとの反応により得られるグリシジルアミン型エポキシ樹脂; オレフィン結合を過酢酸等の過酸で酸化して得られる線状脂肪族エポキシ樹脂;及び脂環族エポキシ樹脂等が挙げられる。これらを単独で用いても、または2種以上併用してもよい。使用するエポキシ樹脂は無色または例えば淡黄色の比較的着色していないものが好ましい。そのようなエポキシ樹脂として、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、ジグリシジルイソシアヌレート、トリグリシジルイソシアヌレートを挙げることができる。
【0016】
本発明の一実施形態では、(A)エポキシ樹脂として、100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを使用することが好ましい。混練後の樹脂組成物の溶融粘度が低く流動性が高くなりすぎると、成型金型のエアベントが塞がれてしまい、金型キャビティ内に空気や揮発成分が残ってしまう可能性がある。キャビティ内に残存した空気や揮発成分は、成形ボイドやウェルドマーク等の外観不具合の原因となる。また、成型回数の増加に伴って、硬化物表面に残ったモノマー成分が成型金型に付着して金型を汚染することになる。金型にモノマーの付着物が堆積した結果、金型からの成型物の離型性が悪化する不具合が生じる。これに対し、本発明では、特定の粘度を有する(G)オリゴマーを使用することによって、混練後の樹脂組成物の溶融粘度を増加させ流動性を低下させることが可能である。また、そのような(G)オリゴマーの使用によって、金型の汚染原因となる残存モノマー成分を低減させることが可能である。その結果、溶融粘度が低い場合に生じる不具合を回避し、樹脂組成物のトランスファー成型性を向上させ、外観の優れた成型物を得ることが可能となる。
本発明で使用する(G)オリゴマーは、硬化性光反射用樹脂組成物の調製に先立って、少なくとも(A’)エポキシ樹脂および(B’)硬化剤、さらに必要に応じて(C’)硬化促進剤を配合することによって調製される。上記(A’)エポキシ樹脂、上記(B’)硬化剤、上記(C’)硬化促進剤は、それぞれ先に説明した(A)エポキシ樹脂、後述する(B)硬化剤および(C)硬化促進剤と同様のものを用いることができる。
【0017】
より具体的には、上記(G)オリゴマーは、例えば(A’)エポキシ樹脂および(B’)硬化剤を、当該(A’)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な当該(B’)硬化剤中の活性基(酸無水物基や水酸基)が0.3当量以下となるように配合し、粘土状になるまで混練する工程を経て得ることができる。得られた粘土状混練物を、引き続き、温度25〜60℃の範囲で1〜6時間にわたってエージングする工程を設けることが好ましい。また、(C’)硬化促進剤を使用する場合には、(A’)エポキシ樹脂と(B’)硬化剤との総和100重量部に対し、0.005〜0.05重量部となるように配合することが好ましい。
【0018】
上述のようにして調製された(G)オリゴマーは、成型時のバリ長さを短く調整するために、100〜150℃における粘度が100〜2500mPa・sの範囲にあることが好ましい。100℃における粘度が100〜2500mPa・sの範囲にあることがより好ましい。(G)オリゴマーの粘度が100mPa・s未満であると、トランスファー成型時にバリが発生しやすくなる。一方、粘度が2500mPa・sを超えると、成型時の流動性が低下し、成型性に乏しくなる傾向がある。なお、本明細書で使用する用語「粘度」は、ICIコーンプレート型粘度計を用いた測定によって得られた値である。(G)オリゴマーは、その粒径が1mm以下になるまで粉砕し、温度0℃以下の環境で保存することによって、粘度の上昇を抑制または停止させることができる。オリゴマーの粉砕方法は、陶器製乳鉢による粉砕等、公知のいかなる手法を用いても良い。
【0019】
本発明の一実施形態では、熱硬化性樹脂成分として(A)エポキシ樹脂とともに(B)硬化剤を含む。(B)硬化剤は、特に制限されることなく、エポキシ樹脂と反応可能な化合物であればよいが、その分子量は100〜400程度のものが好ましい。また、無色、または例えば淡黄色の比較的着色していないものが好ましい。具体的な化合物として、酸無水物系硬化剤、イソシアヌル酸誘導体、フェノール系硬化剤などが挙げられる。
【0020】
酸無水物系硬化剤としては、35℃以上の融点を有するものが好ましい。そのような酸無水物としては、例えば、無水フタル酸、無水マレイン酸、無水トリメリット酸、無水ピロメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、無水メチルナジック酸、無水ナジック酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、無水コハク酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸などが挙げられる。
【0021】
イソシアヌル酸誘導体としては、1,3,5−トリス(1−カルボキシメチル)イソシアヌレート、1,3,5−トリス(2−カルボキシエチル)イソシアヌレート、1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート、1,3−ビス(2−カルボキシエチル)イソシアヌレートなどが挙げられる。
フェノール系硬化剤としては、フェノール、クレゾール、レゾルシン、カテコール、ビスフェノールA、ビスフェノールF、フェニルフェノール、アミノフェノール等のフェノール類及び/又はα−ナフトール、β−ナフトール、ジヒドロキシナフタレン等のナフトール類と、ホルムアルデヒド、ベンズアルデヒド、サリチルアルデヒド等のアルデヒド基を有する化合物とを酸性触媒下で縮合又は共縮合させて得られるノボラック型フェノール樹脂; フェノール類及び/又はナフトール類とジメトキシパラキシレン又はビス(メトキシメチル)ビフェニルとから合成されるフェノール・アラルキル樹脂; ビフェニレン型フェノール・アラルキル樹脂、ナフトール・アラルキル樹脂等のアラルキル型フェノール樹脂; フェノール類及び/又はナフトール類とジシクロペンタジエンとの共重合によって合成される、ジシクロベンタジエン型フェノールノボラック樹脂、ジシクロペンタジエン型ナフトールノボラック樹脂等のジシクロペンタジエン型フェノール樹脂; トリフェニルメタン型フェノール樹脂; テルペン変性フェノール樹脂; パラキシリレン及び/又はメタキシリレン変性フェノール樹脂; メラミン変性フェノール樹脂; シクロペンタジエン変性フェノール樹脂; およびこれら2種以上を共重合して得られるフェノール樹脂などが挙げられる。
【0022】
先に例示した硬化剤の中でも、無水フタル酸、無水トリメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、およびシクロへキサントリカルボン酸無水物からなる群から選択される酸無水物、および1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート等のイソシアヌル酸誘導体の少なくとも一方を使用することが好ましい。これらは、単独で用いるだけでなく、二種以上併用しても良い。
【0023】
本発明の一実施態様では、(B)硬化剤として、少なくともイソシアヌル酸誘導体を使用することが好ましく、それらを酸無水物、特に35℃以上の融点を有する酸無水物を組合せて使用することがより好ましい。イソシアヌル酸誘導体のトリアジン骨格は、通常の環状メチレン骨格と比較して、活性酸素によって酸化されにくい特徴を有する。そのため、イソシアヌル酸誘導体を使用し、先の特徴を樹脂組成物に付与することで、成型後の樹脂組成物の耐熱性を向上させることが可能である。また、トリアジン骨格と3官能性の反応基によって成型体の機械特性を向上させることも可能となる。さらに、イソシアヌル酸誘導体を酸無水物と組合せることによって、樹脂組成物の溶融粘度を増加させることができ、成型時に金型から張り出すバリ長さを抑制することが可能である。イソシアヌル酸誘導体と酸無水物との配合比は、1:0〜1:10の範囲で適宜調整することが可能である。コスト削減と、樹脂の黄変による反射率の低下を抑制する観点から、1:1〜1:3の配合比とすることが好ましい。
【0024】
本発明の別の実施態様では、(B)硬化剤として、少なくともシクロへキサントリカルボン酸無水物を使用することが好ましい。シクロへキサントリカルボン酸無水物の使用によって、樹脂組成物の溶融粘度を増加させることができ、成型時のバリ長さを短くすることが可能である。また、樹脂組成物の硬化時間を短縮化できるため、成型効率を向上させることも可能である。シクロへキサントリカルボン酸無水物の具体例としては、下記構造式(I)で示される化合物が挙げられる。
【0025】
【化2】
【0026】
シクロへキサントリカルボン酸無水物と共に(B)硬化剤として先に説明した他の酸無水物、イソシアヌル酸誘導体およびフェノール系硬化剤等を併用してもよい。併用する硬化剤としては、成型時の流動性および成型物の着色の観点から、無水フタル酸、無水トリメリット酸、ヘキサヒドロ無水フタル酸、テトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、メチルテトラヒドロ無水フタル酸、無水グルタル酸、無水ジメチルグルタル酸、無水ジエチルグルタル酸、または1,3,5−トリス(3−カルボキシプロピル)イソシアヌレートが好ましい。(B)硬化剤におけるシクロへキサントリカルボン酸無水物の含有率は、本発明の目的が達成できる範囲であれば特に限定されるものではないが、5質量%以上100質量%以下の範囲で調整することが好ましい。コストと性能とのバランスの観点から、上記含有率は25質量%以上75質量%以下の範囲であることが好ましい。
【0027】
(A)エポキシ樹脂と(B)硬化剤との配合比は、(A)エポキシ樹脂が(G)オリゴマーを含有しない場合、(A)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な(B)硬化剤中の活性基(酸無水物基や水酸基)が0.5〜1.5当量となるような割合であることが好ましい。0.7〜1.2当量となるような割合であることがより好ましい。上記活性基が0.5当量未満の場合には、エポキシ樹脂組成物の硬化速度が遅くなるとともに、得られる硬化物のガラス転移温度が低くなり、充分な弾性率が得られない場合がある。また、上記活性基が1.2当量を超える場合には、硬化後の強度が減少する場合がある。
【0028】
一方、(A)エポキシ樹脂として(G)オリゴマーを単独で使用するか、または(G)オリゴマーと(A)エポキシ樹脂とを併用する場合、(A)エポキシ樹脂(もしくは(G)オリゴマー)と(B)硬化剤との配合比は、(A)エポキシ樹脂中のエポキシ基1当量に対して、当該エポキシ基と反応可能な(B)硬化剤中の活性基(酸無水物基や水酸基)が0.5〜0.7当量となるような割合であることが好ましい。0.6〜0.7当量となるような割合であることがより好ましい。上記活性基が0.5当量未満の場合には、エポキシ樹脂組成物の硬化速度が遅くなるとともに、得られる硬化体のガラス転移温度が低くなり、充分な弾性率が得られない場合がある。また上記活性基が0.7当量を超える場合には、硬化後の強度が減少する場合がある。なお、(A)エポキシ樹脂が(G)オリゴマーを含む場合の(B)硬化剤の当量数は、(G)オリゴマーに含まれる(A’)エポキシ樹脂と(A)エポキシ樹脂のそれぞれに含まれるエポキシ基の総量を1当量とし、それに対して(B’)硬化剤と(B)硬化剤中に含まれる上記エポキシ基と反応可能な活性基の総和を当量数として換算する。
【0029】
本発明の熱硬化性光反射用樹脂組成物は、必要に応じて、(C)硬化促進剤として適切な化合物を含有してもよい。例えば、1,8−ジアザ−ビシクロ(5,4,0)ウンデセン−7、トリエチレンジアミンおよびトリ−2,4,6−ジメチルアミノメチルフェノールなどの3級アミン類; 2−エチル−4メチルイミダゾールおよび2−メチルイミダゾールなどのイミダゾール類; トリフェニルホスフィン、テトラフェニルホスホニウムテトラフェニルボレート、テトラ−n−ブチルホスホニウム−o,o−ジエチルホスホロジチオエート、テトラ−n−ブチルホスホニウム−テトラフルオロボレート、テトラ−n−ブチルホスホニウム−テトラフェニルボレートなどのリン化合物; 4級アンモニウム塩; 有機金属塩類; およびこれらの誘導体などを使用することができる。これらは単独で使用してもよく、あるいは併用してもよい。これらの硬化促進剤の中では、3級アミン類、イミダゾール類、リン化合物を用いることが好ましい。
【0030】
上記(C)硬化促進剤の含有率は、(A)エポキシ樹脂に対して、0.01〜8.0重量%であることが好ましく、0.1〜3.0重量%であることがより好ましい。硬化促進剤の含有率が0.01重量%未満では、十分な硬化促進効果が得られない場合がある。また、含有率が8.0重量%を超えると、得られる成型体に変色が見られる場合がある。
【0031】
本発明の熱硬化性光反射用樹脂組成物は、その成形性を調整するために、(D)無機充填材を含むことが好ましい。(D)無機充填剤は、特に限定されないが、例えば、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群から選ばれる少なくとも1種を使用することができる。熱伝導性、光反射特性および成型性の点から、少なくともシリカを含むことが好ましい。また、難燃性を高めるために、水酸化アルミニウムを組合せて使用することが好ましい。
【0032】
本発明の一実施形態では、(D)無機充填剤として、多孔質充填剤または吸油性を有する化合物を含有することが好ましい。(D)無機充填剤として、多孔質充填剤または吸油性を有するシリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群から選ばれる少なくとも1種を使用することも可能である。また、多孔質構造を有し、さらに吸油性を有する化合物を用いることも可能である。多孔質充填剤または吸油性を有する化合物の形状としては、特に限定されず、例えば、真球状、破砕状、円盤状、棒状、繊維状等のものを用いることができる。トランスファー成型時の金型内の流動性を考慮すると真球状、破砕状のものが好ましく、真球状のものがより好ましい。
【0033】
また、上記多孔質充填剤または吸油性を有する化合物は、その表面が物理的または化学的に親水化処理または疎水化処理されていてもよい。表面が疎水化処理されたものであることが好ましく、吸油量(JIS K5101に準ずる規定量)が50ml/100g以上となるように化学的に疎水化処理されたものであることがより好ましい。表面が疎水化処理された多孔質充填剤または吸油性を有する化合物を用いることで、(A)エポキシ樹脂や(B)硬化剤との接着性が増加し、結果として熱硬化物の機械強度やトランスファー成型時の流動性が向上する。また、吸油量50ml/100g以上となるように表面が疎水化処理された多孔質充填剤または吸油性を有する化合物を用いることで、(A)エポキシ樹脂との接着性が向上するとともに、混錬後の樹脂組成物のポットライフ低下を抑制することができ、また、熱硬化時に着色を抑制することもできる。このような疎水化処理が施された多孔質充填剤としては、例えば、富士シリシア化学株式会社で販売されているサイロホービック702を挙げることができる。
【0034】
また、上記多孔質充填剤または吸油性を有する化合物の見掛け密度は、特に限定されないが、0.4g/cm3以上であることが好ましく、0.4〜2.0g/cm3であることがより好ましい。なお、見掛け密度とは、多孔質充填剤または吸油性を有する化合物の素原料が占める密度と微細孔の占める空間(即ち細孔容積)とを考慮した密度のことである。この見掛け密度が0.4g/cm3に満たない場合は、充填剤粒子の機械的強度が小さく、ミキシングロールミルなどのせん断力を生じるような溶融混錬時において、粒子が破壊されてしまう恐れがある。一方、見掛け密度が2.0g/cm3を超える場合は、トランスファー成型のためのタブレットを成型する際に、臼型と杵型とからなる金型の表面に樹脂組成物が付着し易くなる傾向にある。
【0035】
また、上記多孔質充填剤または吸油性を有する化合物の平均粒径は、0.1〜100μmであることが好ましく、白色顔料とのパッキング効率を考慮すると1〜10μmの範囲であることがより好ましい。平均粒径が100μmよりも大きく、または0.1μmよりも小さくなると、トランスファー成型する際の溶融時に樹脂組成物の流動性が悪くなる傾向にある。
【0036】
また、上記多孔質充填剤または吸油性を有する化合物の比表面積は、100〜1000m2/gであることが好ましく、300〜700m2/gであることがより好ましい。比表面積が100m2/gよりも小さくなると充填剤による樹脂の吸油量が小さくなり、タブレット成型時に杵型に樹脂が付着し易くなる傾向にあり、比表面積が1000m2/gよりも大きくなると、トランスファー成型する際の溶融時に樹脂組成物の流動性が悪くなる傾向にある。
【0037】
また、上記多孔質充填剤または吸油性を有する化合物の含有量は、特に限定されないが、(D)無機充填剤全体に対し、0.1体積%〜20体積%の範囲であることが好ましい。溶融時の樹脂組成物の成型性を考慮すると、1体積%〜5体積%であることがより好ましい。この含有量が0.1体積%よりも小さい場合は、樹脂組成物の一部が臼型と杵型の成型金型の表面に付着し易くなり、20体積%よりも大きい場合は、トランスファー成型する際の溶融時に樹脂組成物の流動性が低下する傾向にある。例えば、上記多孔質充填剤として上記サイロホービック702を用いる場合には、その含有量を、樹脂組成物の溶融時の流動性や樹脂硬化物の強度の観点から5体積%以下とすることが好ましい。
【0038】
本発明で用いられる(E)白色顔料としては、特に限定されるものではなく、公知のものを使用することができる。例えば、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子などを用いることができ、これらは単独でも併用しても構わない。無機中空粒子は、例えば、珪酸ソーダガラス、アルミ珪酸ガラス、硼珪酸ソーダガラス、シラス等が挙げられる。熱伝導性、光反射特性の点からは、少なくともアルミナまたは酸化マグネシウムを使用するか、またはそれらを組合せて使用することが好ましい。(E)白色顔料の粒径は、中心粒径が0.1〜50μmの範囲にあることが好ましい。この中心粒径が0.1μm未満であると、粒子が凝集しやすく分散性が悪くなる傾向にある。一方、中心粒径が50μmを超えると、硬化物の光反射特性が十分に得られない恐れがある。(E)白色顔料の配合量は、特に限定されないが、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることが好ましい。
【0039】
上記(D)無機充填剤と上記(E)白色顔料との合計配合量は、特に限定されないが、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることが好ましい。この合計配合量が10体積%未満であると、硬化物の光反射特性が十分に得られない恐れがある。また、合計配合量が85体積%を超えると、樹脂組成物の成型性が悪くなり、光半導体搭載用基板の作製が困難となる傾向がある。
【0040】
本発明の熱硬化性光反射用樹脂組成物には、必要に応じて(F)カップリング剤を加えることができる。(F)カップリング剤は、特に限定されないが、例えば、シラン系カップリング剤やチタネート系カップリング剤等を用いることができる。シランカップリング剤としては、例えば、エポキシシラン系、アミノシラン系、カチオニックシラン系、ビニルシラン系、アクリルシラン系、メルカプトシラン系及びこれらの複合系等を用いることができる。(F)カップリング剤の種類や処理条件は特に限定されるものではなく、樹脂組成物にそのまま添加する方法や、無機充填剤または白色顔料と予め混合して添加する方法等、従来から用いられている手法を適用してもよい。(F)カップリング剤の配合量は樹脂組成物に対して5重量%以下が好ましい。
【0041】
また、本発明の熱硬化性光反射用樹脂組成物には、溶融粘度調整を目的として(H)増粘剤を添加しても良い。(H)増粘剤としては、特に限定されるものではないが、例えば、トクヤマ(株)で販売されているレオロシールCP−102として入手可能なナノシリカを用いることができる。(H)増粘剤の添加量としては、樹脂組成物の総体積の0.15体積%以下であることが好ましい。(H)増粘剤の添加量が0.15体積%よりも多くなると、樹脂組成物の溶融時の流動性が損なわれるとともに、硬化後に充分な材料強度が得られなくなる恐れがある。また、(H)増粘剤は、中心粒径が1nm〜1000nmであるようなナノ粒子フィラーであることが好ましく、中心粒径が10nm〜1000nmであるようなナノ粒子フィラーであることがより好ましい。中心粒径1nmよりも小さいフィラーは、粒子が凝集しやすく分散性が低下する傾向にあり、特性上好ましくない。このような(H)増粘剤を用いる場合には、(D)無機充填剤の一部としてナノシリカを用いてもよい。一方、1000nmよりも大きなフィラーを添加すると、バリ長さが低減しない傾向にあり、特性上好ましくない。なお、本発明の樹脂組成物には、(H)増粘剤以外に、必要に応じて、酸化防止剤、離型剤、イオン補足剤等の各種添加剤を添加してもよい。
【0042】
本発明の熱硬化性光反射用樹脂組成物は、上記した各成分を均一に分散混合することによって得ることができ、その調製手段や条件等は特に限定されない。一般的な手法として、所定配合量の各種成分をミキサー等によって十分に均一に撹拌および混合した後、ミキシングロール、押出機、ニーダー、ロールおよびエクストルーダー等を用いて混練し、さらに得られた混練物を冷却および粉砕する方法を挙げることができる。なお、混練形式についても特に限定されるものではないが、溶融混練が好ましい。溶融混練の条件は、使用した成分の種類や配合量によって適宜決定すればよく、特に限定されない。例えば、15〜100℃の範囲で5〜40分間にわたって溶融混練することが好ましく、20〜100℃の範囲で10〜30分間にわたって溶融混練することがより好ましい。溶融混練時の温度が15℃未満であると、各成分を溶融混練させることが困難であり、分散性も低下する傾向にある。一方、100℃よりも高温であると、樹脂組成物の高分子量化が進行し、樹脂組成物が硬化してしまう恐れがある。また、溶融混練の時間が5分未満であると、バリ長さを効果的に抑制することができない傾向にあり、40分よりも長いと、樹脂組成物の高分子量化が進行し、成型前に樹脂組成物が硬化してしまう恐れがある。
【0043】
本発明の熱硬化性光反射用樹脂組成物は、上記各成分を配合、混練した後、成型時の溶融粘度を上昇させることを目的として熟成放置(エージング)することが好ましい。より具体的には、エージングは0℃〜30℃で1〜72時間にわたって実施することが好ましい。エージングは、より好ましくは15℃〜30℃で12〜72時間にわたって、さらに好ましくは25℃〜30℃で24〜72時間にわたって、実施することが望ましい。1時間よりも短時間のエージングでは、バリ長さを効果的に抑制できない傾向にあり、72時間より長くエージングすると、トランスファー成型時に充分な流動性を確保できない恐れがある。また、0℃未満の温度でエージングを実施した場合には、(C)硬化促進剤が不活性化されて、樹脂組成物の三次元架橋反応が十分に進行せず、溶融時の粘度が上昇しない恐れがある。また、30℃よりも高温でエージングを実施した場合には、樹脂組成物が水分を吸収してしまい、硬化物の強度や弾性率などの機械的物性が悪くなる傾向にある。
【0044】
本発明の光半導体素子搭載用基板は、本発明の熱硬化性光反射用樹脂組成物を使用して構成されることを特徴とする。具体的には、光半導体素子搭載領域となる1つ以上の凹部を有し、少なくとも上記凹部の内周側面が本発明の熱硬化性光反射用樹脂組成物から構成される基板が挙げられる。図1は、本発明の光半導体素子搭載用基板の一実施形態を示すものであり、(a)は斜視図、(b)は側面断面図である。図1に示したように、本発明の光半導体素子搭載用基板110は、リフレクター103と、Ni/Agメッキ104および金属配線105を含む配線パターン(リードフレーム)とが一体化され、光半導体素子搭載領域となる凹部200が形成された構造を有し、少なくとも上記凹部200の内周側面は本発明の熱硬化性光反射用樹脂組成物から構成されていることを特徴とする。
【0045】
本発明の光半導体素子搭載用基板の製造方法は、特に限定されないが、例えば、本発明の熱硬化性光反射用樹脂組成物またはそのタブレット成型体をトランスファー成型によって製造することができる。図2は、本発明の光半導体素子搭載用基板の製造方法を説明する概略図であり、図2(a)〜(c)はトランスファー成型によって基板を製造する場合の各工程に対応する。より具体的には、光半導体素子搭載用基板は、図2(a)に示すように、金属箔から打ち抜きやエッチング等の公知の方法により金属配線105を形成し、次いで該金属配線105を所定形状の金型301に配置し(図2(b))、金型301の樹脂注入口300から本発明の熱硬化性光反射用樹脂組成物(タブレット成型体の溶融物)を注入する。次いで、注入した樹脂組成物を、好ましくは金型温度170〜190℃、成形圧力2〜8MPaで60〜120秒にわたって硬化させた後に金型301を外し、アフターキュア温度120℃〜180℃で1〜3時間にわたって熱硬化させる。次いで、硬化した熱硬化性光反射用樹脂組成物から構成されるリフレクター103に周囲を囲まれた光半導体素子搭載領域となる凹部200の所定位置に、Ni/銀メッキ104を施すことによって製造することが可能である(図2(c))。
【0046】
本発明の光半導体素子搭載用基板を用いた光半導体装置は、本発明の光半導体素子搭載用基板と、光半導体素子搭載用基板の凹部底面に搭載される光半導体素子と、光半導体素子を覆うように凹部内に形成される蛍光体含有透明封止樹脂層とを少なくとも備えることを特徴とする。図3(a)および図3(b)は、それぞれ本発明の光半導体装置の一実施形態を示す側面断面図である。より具体的には、図3に示した光半導体装置は、本発明の光半導体素子搭載用基板110の光半導体素子搭載領域となる凹部(図2の参照符号200)の底部所定位置に光半導体素子100が搭載され、該光半導体素子100と金属配線105とがボンディングワイヤ102やはんだバンプ107などの公知の方法によりNi/銀メッキ104を介して電気的に接続され、該光半導体素子100が公知の蛍光体106を含む透明封止樹脂101により覆われている。
【実施例】
【0047】
以下、本発明を実施例により詳述するが、本発明はこれらに限定されるものではない。なお、以下の各実施例および各比較例で使用した成分の詳細は以下のとおりである。
*1:トリグリシジルイソシアヌレート(エポキシ当量100、日産化学社製、商品名TEPIC−S)
*2:ヘキサヒドロ無水フタル酸(和光純薬社製)
*3:1,3,5−トリス(3−カルボキシプロピル)イソシアヌレート(四国化成工業社製、製品名C3CIC酸)
*4:シクロヘキサントリカルボン酸無水物(三菱ガス化学社製、製品名H−TMAn)
*5:テトラヒドロ無水フタル酸(アルドリッチ社製)
*6:メチルヒドロ無水フタル酸(日立化成工業社製 商品名HN5500)
*7:テトラ−n−ブチルホスホニウム−0,0−ジエチルホスホロジチエート(日本化学工業社製、商品名ヒシコーリンPX−4ET)
*8:トリメトキシエポキシシラン(東レダウコーニング社製、商品名SH6040)
*9:脂肪酸エステル(クラリアント社製、商品名ヘキストワックスE)
*10:脂肪族エーテル(東洋ペトロライト社製、商品名ユニトックス420)
*11:溶融シリカ(電気化学工業社製、商品名FB−301)
*12:溶融シリカ(電気化学工業社製、商品名FB−950)
*13:溶融シリカ(アドマテックス社製、商品名SO−25R)
*14:多孔質球状シリカ(富士シリシア化学社製、製品名サイロスフィアC−1504)平均粒径:3μm、見掛け密度:0.58g/ml、比表面積:300m2/g)
*15:多孔質無定形状シリカ(富士シリシア化学社製、製品名サイロホービック702、平均粒径:4μm 見掛け密度:0.48g/ml、比表面積:300m2/g)
*16:多孔質無定形状シリカ(富士シリシア化学社製、製品名サイリシア430、平均粒径:4μm、見掛け密度:0.48g/ml、比表面積:300m2/g)
*17:中空粒子(住友3M社製、商品名S60−HS)
*18:アルミナ(アドマテックス社製、商品名AO−802)
*19:ナノシリカ(トクヤマ社製、CP−102)
【0048】
(実施例A1〜A16および比較例A1〜A7)
1.熱硬化性光反射用樹脂組成物の調製
下記表A1から表A3に示した配合割合に従って各成分を配合し、ミキサーによって十分に混練した後にミキシングロールによって所定条件下で溶融混練して混練物を得た。さらに、得られた混練物を粉砕することによって、実施例A1〜A16および比較例A1〜A7の熱硬化性光反射用樹脂組成物を各々調製した。なお、表A1から表A3に示した各成分の配合量の単位は重量部である。表における空欄は該当する成分の配合が無いことを意味する。
【0049】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例A1〜A16および比較例A1〜A7の各々の樹脂組成物について、以下の手順に従って、光反射率およびバリ長さを測定した。また、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、評価した。それらの結果を下記表A1から表A3に示す。
【0050】
(光反射率)
先に調製した各々の熱硬化性光反射用樹脂組成物を、成型金型温度180℃、成型圧力6.9MPa、硬化時間90秒の条件下でトランスファー成型した後、150℃で2時間にわたって後硬化することによって、厚み1.0mmの試験片をそれぞれ作製した。次いで、積分球型分光光度計V−750型(日本分光株式会社製)を用いて、波長400nmにおける各試験片の光反射率を測定した。
【0051】
(バリ長さ)
先に調製した各々の熱硬化性光反射用樹脂組成物を、ポットを用いて、バリ測定用金型(図4を参照)に流し込み、次いで硬化させることによって樹脂組成物を成型した。なお、成型時の金型温度は180℃、成型圧力は6.9MPa、樹脂の流し込み時間(トランスファー時間)は10秒であり、硬化温度は180℃、硬化時間は90秒とした。成型後、バリ測定用金型の上型を外し、成型時に金型の上型と下型との隙間を流れて生じたバリの長さの最大値を、ノギスを使用して測定した。
【0052】
図4は、先のバリ長さの測定時に使用するバリ測定用金型の構造を模式的に示した図であり、(a)は側面断面図、(b)は平面図である。図4に示したように、バリ測定用金型は、一対の上型400と下型401とから構成され、上型400は樹脂注入口402を有する。また、下型401は、樹脂注入口402に対向するキャビティ403と、キャビティ403から金型外周部に向かって伸びる6本のスリット404、405、406、407、408および409を有する。実際に使用したバリ測定用金型の寸法は、図4に示したように上型400および下型401の外形が140mm×140mm、樹脂注入口402の上径が7mmおよび下径が4mm、キャビティ403の径が30mmおよび深さが4mmである。また、キャビティ403から延びる6本のスリット404から409の幅はそれぞれ5mmであり、深さは順に75、50、30、20、10および2μmであった。図5は、図4に示したバリ測定用金型を用いた成型時に生じたバリを模式的に示す図であり、(a)は側面断面図、(b)は平面図である。バリは、図5に示したように、樹脂組成物がキャビティ403の外延から各スリットに沿って流れ込み硬化した部分410を意味する。本発明で規定する「バリ長さ」は、参照符号410で示されるバリの最大値を、ノギスで測定した値である。
【0053】
(ワイヤボンディング性の評価)
最初に、図2に示した製造工程に従い、先に調製した各々の熱硬化性光反射用樹脂組成物をトランスファー成型することによって、光半導体素子搭載用基板を作製した。なお、成型条件は、成型金型温度180℃、成型圧力6.9MPa、硬化時間90秒であり、150℃で2時間にわたって後硬化を実施した。
次に、作製した上記基板の光半導体素子搭載領域となる凹部に光半導体素子を搭載した後、ワイヤボンダ(HW22U−H、九州松下電器株式会社製、商品名)および直径28μmのボンディングワイヤを使用して、光半導体素子と基板とをワイヤボンディングすることによって、電気的に接続した。ワイヤボンディング時の基板の加熱温度は180℃とした。ワイヤボンディング性は、光半導体素子と基板とを電気的に接続するワイヤボンディングするワイヤの引っ張り強度を、プルテスターPTR−01(株式会社レスカ製、商品名)を使用して測定し、その値について下記の評価基準に従って評価した。
【0054】
ワイヤボンディング性の評価基準
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0055】
【表1】
【0056】
【表2】
【0057】
【表3】
【0058】
表A1から表A3によれば、実施例A1〜A16の樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。したがって、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0059】
(実施例B1〜B11および比較例B1〜B8)
1.熱硬化性光反射用樹脂組成物の調製
下記表B1および表B2に示した配合割合に従って各成分を配合し、ミキサーによって十分に混練した後にミキシングロールによって所定条件下で溶融混練して混練物を得た。さらに、得られた混練物に必要に応じてエージングを施した後に冷却し、それらを粉砕することによって、実施例B1〜B11および比較例B1〜B8の熱硬化性光反射用樹脂組成物を各々調製した。
【0060】
なお、表B1およびB2に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いこと、または該当する工程を実施しないことを意味する。各実施例の詳細は以下のとおりである。
実施例B1は、特定の(G)オリゴマーを使用する手法に関する。
実施例B2は、(A)エポキシ樹脂中のエポキシ基1当量に対する硬化剤中の活性基を0.5〜0.7当量の範囲とする手法に関する。
実施例B3は、(H)増粘剤としてナノフィラーを追加する手法に関する。
実施例B4は、樹脂組成物を所定条件下でエージングする手法に関する。
実施例B5は、溶融混練条件を調整する手法(混練時間を15分から30分に延長)に関する。
実施例B6〜B11は、上述の手法のうちいずれか2つの手法を併用した場合に関する。
【0061】
なお、実施例B1、B6、B7およびB9において使用した(G)オリゴマーは、以下に説明する手順で調製した。また、(G)オリゴマーの粘度は、Reseach Equipment LTD.(London)製のICIコーンプレート型粘度計を使用し、試料量0.155±0.01g、100℃で測定したところ、1000mPa・sであった。
【0062】
(オリゴマーの作製方法)
表B1に示した配合条件に従って各成分を配合し、ミキシングロールによって25℃で10分間にわたって溶融混練を行った。なお、表B1に示した配合は、エポキシ基1当量に対して酸無水物基0.1当量となる割合になっている。次に、溶融混練によって得られた粘土状組成物(混練物)を温度55℃で4時間にわたってエージングした。そのようなエージング後に、混練物を口径300mmの陶器製乳鉢を用いて、粒径が1mm以下になるまで粉砕することにより所望のオリゴマーを得た。得られたオリゴマーは、温度0℃以下の環境下で保存した。
【0063】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例B1〜B6および比較例B1〜B8の各々の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表B1および表B2に示す。
【0064】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0065】
【表4】
【0066】
【表5】
【0067】
表B1および表B2によれば、実施例B1〜B11の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。そのため、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。また、(G)オリゴマーを使用した場合には、モノマー成分の残存量を低減させることが可能であるため、ワイヤボンディング性の向上に加えて、基板成型時の金型汚れおよび成型品の離型性の低下といった不具合を回避することができ、優れた外観を有する成型基板が得られる。
【0068】
(実施例C1〜C3および比較例C1、C2)
1.熱硬化性光反射用樹脂組成物の調製
下記表C1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例C1〜C3、比較例C1およびC2の熱硬化性光反射用樹脂組成物を各々調製した。表C1に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いことを意味する。
【0069】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例C1〜C3および比較例C1、C2の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表C1に示す。
【0070】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0071】
【表6】
【0072】
注記:(*)金型内における樹脂の流動性不良のため、測定不可
【0073】
表C1によれば、実施例C1〜C3の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。C3CIC酸は、その特徴的な化学構造によって、成型体の機械的強度を向上させることが可能であるだけでなく、それらはバリの抑制にも効果的であることが明らかである。但し、硬化剤としてC3CIC酸を単独で使用すると組成物の流動性が悪化する傾向にあるため、HHPAなどの他の硬化剤と組合せて使用することが好ましいことが分かる。本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0074】
(実施例D1〜D5および比較例D1〜D3)
1.熱硬化性光反射用樹脂組成物の調製
表D1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例D1〜D5、比較例D1〜D3の熱硬化性光反射用樹脂組成物を各々調製した。なお、表D1に示した各成分の配合量の単位は重量部である。また、表における空欄は該当する成分の配合が無いことを意味する。
【0075】
2.熱硬化性光反射用樹脂組成物の評価
先に調製した実施例D1〜D5および比較例D1〜D3の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして、光反射率およびバリ長さを測定した。なお、耐久性について検討するために、光反射率の測定は、試験片の成型後と、150℃で72時間加熱した後に実施した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表D1に示す。
【0076】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0077】
【表7】
【0078】
表D1によれば、実施例D1〜D5の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、またトランスファー成型時のバリ長さが抑制され、優れたワイヤボンディング性を示すことが分かる。さらに、硬化物(成型基板)の光反射特性が劣化し難いことも分かる。そのため、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。また、本発明の熱硬化性光反射用樹脂組成物を使用することによって、可視光から近紫外光領域において高い反射率を保持することが可能な光半導体素子搭載用基板を効率良く製造することも可能であることが分かる。
【0079】
(実施例E1〜E8)
表E1に示した配合割合に従って各成分を配合し、ミキサーによって十分に混合した後、ミキシングロールによって溶融混練することによって混練物を得た。得られた混練物を室温まで冷却し、それらを粉砕することによって、実施例E1〜E8の熱硬化性光反射用樹脂組成物を各々調製した。なお、表E1に示した各成分の詳細は先に説明したとおりであり、各配合量の単位は重量部である。
【0080】
得られた実施例E1〜E8の熱硬化性光反射用樹脂組成物について、先の実施例と同様にして光反射率およびバリ長さを測定した。また、先の実施例と同様にして、各々の樹脂組成物を成型して得られる基板のワイヤボンディング性について検討し、以下の基準に従って評価した。それらの結果を下記表E1に示す。
【0081】
(ワイヤボンディング性の評価基準)
◎:引っ張り強度10g以上
○:引っ張り強度4g以上10g未満
△:引っ張り強度4g未満
×:ボンディング不可
【0082】
【表8】
【0083】
表E1によれば、実施例E1〜E8の熱硬化性光反射用樹脂組成物は、優れた光反射特性を示し、トランスファー成型時のバリの抑制効果が高く、より優れたワイヤボンディング性を示すことが分かる。このように、各構成成分を適切に組合せて配合することによって、ワイヤボンディング性をさらに向上させることも可能である。したがって、本発明の熱硬化性光反射用樹脂組成物を使用して、光半導体素子搭載用基板や光半導体装置を製造した場合、樹脂汚れを除去する工程が不要となるため、コストや製造時間など生産性の点で非常に有利となる。
【0084】
以上の説明からして、本発明の精神と範囲に反することなしに、広範に異なる実施態様を構成することができることは明白であり、本発明は請求の範囲において限定した以外は、その特定の実施態様によって制約されるものではない。
【特許請求の範囲】
【請求項1】
光半導体素子搭載用基板と、前記光半導体素子搭載用基板の凹部底面に搭載された光半導体素子と、前記光半導体素子を覆うように前記凹部内に形成された蛍光体含有透明封止樹脂層とを少なくとも備える光半導体装置であって、
前記光半導体素子搭載用基板の少なくとも前記凹部の内周側面が、熱硬化性光反射用樹脂組成物を用いて構成されたものであり、
前記熱硬化性光反射用樹脂組成物が、熱硬化性成分と、(E)白色顔料と、(H)増粘剤とを含み、成型温度100℃〜200℃、成型圧力20MPa以下、及び成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ熱硬化後の、波長350nm〜800nmにおける光反射率80%以上であり、
前記(H)増粘剤が、中心粒径が1nm〜1000nmのナノ粒子フイラ一を含有することを特徴とする、光半導体装置。
【請求項2】
前記熱硬化性成分が、(A)エポキシ樹脂を含むことを特徴とする請求項1に記載の光半導体装置。
【請求項3】
前記(A)エポキシ樹脂が、(A’)エポキシ樹脂と(B’)硬化剤とを混練することによって得られ、かつ100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを含むことを特徴とする請求項2に記載の光半導体装置。
【請求項4】
前記熱硬化性成分が、前記(A)エポキシ樹脂とともに用いられる(B)硬化剤をさらに含み、前記(A)エポキシ樹脂と前記(B)硬化剤との配合比が、前記(A)エポキシ樹脂中のエポキシ基1当量に対して、該エポキシ基と反応可能な前記(B)硬化剤中の活性基が0.5〜0.7当量となる比であることを特徴とする請求項3に記載の光半導体装置。
【請求項5】
前記(B)硬化剤が、イソシアヌル酸骨格を有する化合物を含むことを特徴とする請求項4に記載の光半導体装置。
【請求項6】
前記(B)硬化剤が、35℃以上の融点を有する酸無水物をさらに含むことを特徴とする請求項5に記載の光半導体装置。
【請求項7】
前記(B)硬化剤が、シクロへキサントリカルボン酸無水物を含むことを特徴とする請求項4に記載の光半導体装置。
【請求項8】
前記シクロへキサントリカルボン酸無水物が、下記構造式(I)で示される化合物であることを特徴とする請求項7に記載の光半導体装置。
【化1】
【請求項9】
前記熱硬化性光反射用樹脂組成物が、さらに(D)無機充填剤を含み、前記(D)無機充填剤が、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群の中から選ばれる少なくとも1種を含むことを特徴とする請求項1〜8のいずれか1項に記載の光半導体装置。
【請求項10】
前記(E)白色顔料が、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子からなる群の中から選ばれる少なくとも1種であることを特徴とする請求項1〜9のいずれか1項に記載の光半導体装置。
【請求項11】
前記(E)白色顔料の中心粒径が0.1〜50μmの範囲にあることを特徴とする請求項1〜10のいずれか1項に記載の光半導体装置。
【請求項12】
前記(D)無機充填剤と前記(E)白色顔料とを合計した配合量が、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることを特徴とする請求項9〜11のいずれか1項に記載の光半導体装置。
【請求項13】
前記熱硬化性光反射用樹脂組成物が、少なくとも各種構成成分を、混練温度20〜100℃、混練時間10〜30分の条件下で混練することによって得られる混練物を含むことを特徴とする請求項1〜12のいずれか1項に記載の光半導体装置。
【請求項1】
光半導体素子搭載用基板と、前記光半導体素子搭載用基板の凹部底面に搭載された光半導体素子と、前記光半導体素子を覆うように前記凹部内に形成された蛍光体含有透明封止樹脂層とを少なくとも備える光半導体装置であって、
前記光半導体素子搭載用基板の少なくとも前記凹部の内周側面が、熱硬化性光反射用樹脂組成物を用いて構成されたものであり、
前記熱硬化性光反射用樹脂組成物が、熱硬化性成分と、(E)白色顔料と、(H)増粘剤とを含み、成型温度100℃〜200℃、成型圧力20MPa以下、及び成型時間60〜120秒の条件下でトランスファー成型した時に生じるバリ長さが5mm以下であり、かつ熱硬化後の、波長350nm〜800nmにおける光反射率80%以上であり、
前記(H)増粘剤が、中心粒径が1nm〜1000nmのナノ粒子フイラ一を含有することを特徴とする、光半導体装置。
【請求項2】
前記熱硬化性成分が、(A)エポキシ樹脂を含むことを特徴とする請求項1に記載の光半導体装置。
【請求項3】
前記(A)エポキシ樹脂が、(A’)エポキシ樹脂と(B’)硬化剤とを混練することによって得られ、かつ100〜150℃における粘度が100〜2500mPa・sの範囲である(G)オリゴマーを含むことを特徴とする請求項2に記載の光半導体装置。
【請求項4】
前記熱硬化性成分が、前記(A)エポキシ樹脂とともに用いられる(B)硬化剤をさらに含み、前記(A)エポキシ樹脂と前記(B)硬化剤との配合比が、前記(A)エポキシ樹脂中のエポキシ基1当量に対して、該エポキシ基と反応可能な前記(B)硬化剤中の活性基が0.5〜0.7当量となる比であることを特徴とする請求項3に記載の光半導体装置。
【請求項5】
前記(B)硬化剤が、イソシアヌル酸骨格を有する化合物を含むことを特徴とする請求項4に記載の光半導体装置。
【請求項6】
前記(B)硬化剤が、35℃以上の融点を有する酸無水物をさらに含むことを特徴とする請求項5に記載の光半導体装置。
【請求項7】
前記(B)硬化剤が、シクロへキサントリカルボン酸無水物を含むことを特徴とする請求項4に記載の光半導体装置。
【請求項8】
前記シクロへキサントリカルボン酸無水物が、下記構造式(I)で示される化合物であることを特徴とする請求項7に記載の光半導体装置。
【化1】
【請求項9】
前記熱硬化性光反射用樹脂組成物が、さらに(D)無機充填剤を含み、前記(D)無機充填剤が、シリカ、水酸化アルミニウム、水酸化マグネシウム、硫酸バリウム、炭酸マグネシウム、炭酸バリウムからなる群の中から選ばれる少なくとも1種を含むことを特徴とする請求項1〜8のいずれか1項に記載の光半導体装置。
【請求項10】
前記(E)白色顔料が、アルミナ、酸化マグネシウム、酸化アンチモン、酸化チタン、酸化ジルコニウム、無機中空粒子からなる群の中から選ばれる少なくとも1種であることを特徴とする請求項1〜9のいずれか1項に記載の光半導体装置。
【請求項11】
前記(E)白色顔料の中心粒径が0.1〜50μmの範囲にあることを特徴とする請求項1〜10のいずれか1項に記載の光半導体装置。
【請求項12】
前記(D)無機充填剤と前記(E)白色顔料とを合計した配合量が、樹脂組成物全体に対して、10体積%〜85体積%の範囲であることを特徴とする請求項9〜11のいずれか1項に記載の光半導体装置。
【請求項13】
前記熱硬化性光反射用樹脂組成物が、少なくとも各種構成成分を、混練温度20〜100℃、混練時間10〜30分の条件下で混練することによって得られる混練物を含むことを特徴とする請求項1〜12のいずれか1項に記載の光半導体装置。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−238876(P2012−238876A)
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2012−162977(P2012−162977)
【出願日】平成24年7月23日(2012.7.23)
【分割の表示】特願2008−544162(P2008−544162)の分割
【原出願日】平成19年11月14日(2007.11.14)
【出願人】(000004455)日立化成工業株式会社 (4,649)
【Fターム(参考)】
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成24年7月23日(2012.7.23)
【分割の表示】特願2008−544162(P2008−544162)の分割
【原出願日】平成19年11月14日(2007.11.14)
【出願人】(000004455)日立化成工業株式会社 (4,649)
【Fターム(参考)】
[ Back to top ]