
特異的キナーゼ阻害剤
C5-C6シス二重結合及びC7にケトンを有するレゾルシル酸ラクトン並びにマイケル付加物を形成することができる他の化合物は、ATP結合部位に特異的なシステイン残基を有するタンパク質キナーゼサブセットの強力で安定な阻害剤である。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定のタンパク質キナーゼを阻害し、さらにヒトの疾患の治療に有用である化合物を提供する。本発明は、化学、生化学、分子生物学、医学、及び薬理学分野に関する。
【背景技術】
【0002】
分子を標的とする癌治療薬は、現時点及び将来の癌治療に極めて有望である。複数のタンパク質が、細胞シグナル伝達の特定の工程で決定的な役割を果たすと認定された。これらシグナル伝達経路のタンパク質は、正常な健常細胞に対してある程度の選択性を許容するので癌治療薬の魅力的な標的である(Sausville et al. Annu Rev Pharmacol Toxicol (2003) 43:199-231)。しかしながら、細胞シグナル伝達は典型的には複数の経路を必要とするので、ある特定のシグナル伝達経路のタンパク質の特異的な阻害剤は、所望の治療結果を得るためには不充分であるかもしれない。逆に、複数のシグナル伝達経路の非特異的な阻害は正常細胞に対して有害な結果を有し、したがってシグナル伝達経路のタンパク質を標的にするという目的の出鼻をくじくであろう。
この領域における有効な医薬の開発はしたがって困難であり予想不可能である。特定の細胞シグナル伝達経路を阻害するその能力を規準に開発された化合物は、それがまた別の細胞シグナル伝達経路のタンパク質を同様に阻害する場合にのみ特定の適応症について機能することができる。しかし前記機能は従来技術では予測することができない特性である。例えば、グリーベック(Gleevec)(イマチニブメシレート、STI-571, Novartis)は、Bcr-Ablチロシンキナーゼの特異的阻害剤として設計されたが、その有効性はc-Kit及び他のチロシンキナーゼを同様に阻害するその能力に左右される。したがって、グリーベックは実際、慢性骨髄性白血病(CML)細胞の機能の維持に重要なBcr-Ablチロシンキナーゼを阻害し(Hernandez-Boluda et al. Drugs Today (Barc) (2002) 38:601-13)、CMLに対して有効であるが、その有効性はまた部分的にはc-Kitチロシンキナーゼを阻害するその能力に左右される(前記能力はまた、変異によってc-Kitチロシンキナーゼが上昇する胃腸の間質腫瘍に対してグリーベックを有効にする)(Blanke et al. Curr. Treat Options Oncol (2001) 2:485-91)。
【0003】
グリーベックはまた、癌治療薬の開発でタンパク質キナーゼを標的にすることの重要性を例証する。500を超えるタンパク質キナーゼの大きなファミリーの中のメンバーは、全てではないとしてもほとんどが重要な細胞シグナル伝達経路で必要とされる。4つの主要なシグナル伝達経路又はカスケード(1つは細胞外マイトジェンに応答性であり、他のものはストレスシグナルに応答性であり、それぞれはタンパク質キナーゼによって制御され、さらに各々は複数の他のタンパク質キナーゼを含む)は、癌細胞分裂及び細胞のストレス応答で不可欠の役割を果たし、したがって抗癌剤及び抗炎症剤の開発に極めて重要である。しかしながら、どのようにしてある化合物が、複数の異なるシグナル伝達経路において多くの異なるタンパク質キナーゼに影響を及ぼすかについての予想不可能な性質は、医薬の開発を遅らせ続けている。
異常なマイトジェン活性化タンパク質キナーゼ、いわゆるMAP(マイトジェン活性化タンパク質)キナーゼ又はMAPK酵素が中心的に関与する細胞シグナル伝達経路の遮断(Chen et al. Chem Rev (2001) 101:2449-76;Pearson et al. Endocr Rev (2001) 22:153-83)は、新しいタイプの癌治療薬の発見及び開発に重要な方向として出現してきた(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Sebolt-Leopold, Oncogene (2000) 19:6594-99)。MAPK依存経路の1つは、細胞外シグナルに由来するシグナルの伝達を可能にする。前記細胞外シグナルは、例えば表皮増殖因子(EGF)及び血管内皮由来増殖因子(VEGF)である。前記因子は、細胞膜の対応するレセプター(それぞれEGFR(HER)及びVEGFR)と結合し、前記レセプターは、中継キナーゼ及びキナーゼ標的(例えばERK経路:Ras、Raf-1、A-Raf(BRAF)、MEK1及びMEK2(前記は包括的に本明細書ではMEK1/2と称する)、並びにERK1及びERK2(前記は本明細書では包括的にERK1/2と称する))を介して細胞の核にシグナルを送る。後者のタンパク質は、最終的には生存のための細胞機能、例えば分裂、増殖、運動性及び生存を制御する遺伝子の発現を支配する。2〜3つの他のタンパク質キナーゼ経路は“ストレスシグナル”に応答する。
【0004】
特定のタンパク質キナーゼを標的とする小分子の非タンパク質性医薬が開発中であり(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Sebolt-Leopold, Oncogene (2000) 19:6594-99;Noble et al. Science (2004) 303:1800-05)、3つが使用を承認された。すなわちグリーベック、ゲフチニブ(イレッサ;Barker et al. Bioorg Med Chem Lett (2001) 11:1911-14)及びエルロチニブ(タルセバ)である。医薬として承認された小分子キナーゼ阻害剤の不足は、従来の方法の非予測性を例証した。特定のタンパク質キナーゼを阻害する化合物を設計し、それらの標的の3D構造の助けを借りて前記を評価することができるが(Noble et al. Science (2004) 303:1800-05)、一方、臨床的知見は、多くの化合物が前臨床活性を基準にした楽観的期待に応えることができないことを示した(Sausville et al. Annu Rev Pharmacol Toxicol (2003) 43:199-231;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。この失敗は、部分的には、単純に特定のキナーゼを阻害するその能力を基準にして、重要な細胞シグナル伝達経路における無数の他のタンパク質キナーゼに対する阻害剤の作用を予測することの難しさから生じる。したがって、特異的なタンパク質キナーゼ及び特異的なタンパク質キナーゼサブセットを標的とする新規で改善された医薬及び同定方法、並びに癌及び他の疾患の治療で既知の阻害剤を使用する方法が強く希求されている。
そのような医薬はヒトの疾患の治療に重大な影響を与えることができるだろう。例えば、癌の治療では、MAPK経路の薬理学的阻害剤は、シグナル伝達過程におけるいくつかの異なるタンパク質のいずれも標的とすることができるだろう(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24)。癌治療のために特に興味深いタンパク質にはMAPK/細胞外シグナル関連(ERK)キナーゼ(MEK又はMKKと称される)が含まれ、前記は、特にMAPKシグナル伝達のERK支流(Ras/Raf-1、A-Raf及び/又はB-Raf、MEK1/2、及びERK1/2(図1参照)を含む)で作用するキナーゼである。G-タンパク質のRasは、マイトジェン活性化増殖因子レセプターのシグナルをRaf-1、A-Raf及び/又はB-Rafへ伝達する。後者は二重特異的セリン/スレオニン及びチロシンキナーゼMEK1/2をリン酸化し、したがってこれを活性化し、前記は続いてERK1/2を活性化する。Ras/Raf/MEK/ERK経路は、報告によればもっとも詳しく特徴付けられたシグナル伝達経路であるヒトの癌の発生及び増殖に必要とされ、抗癌剤開発の標的として提唱されてきた(Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。
【0005】
しかしながら、細胞分裂及び癌の移動並びに正常細胞の生命機能を制御する複雑な経路群は、ただ1つの経路又は経路複合体の支流のみを阻害する化合物は有効ではない可能性を示唆した。正常細胞にとって有害な非特異的な活性を示すことなく、複数の経路を正確に阻害する化合物を設計し、これを試験することは困難である。MEK1/2キナーゼを標的とする化合物は前記問題の例証である。
MEK1/2は抗腫瘍薬(抗癌剤)の開発のための標的として以下の2つの優れた特徴を有する:(1)それらは、Rasを経過する多様なマイトジェン活性化タンパク質キナーゼの入力を統合する経路集合点の決定的地点に存在し;さらに(2)それらは、制限的な基質特異性を有し、MAPK ERK1/2は重要な唯一の公知の基質である。MEK1/2の構成的活性化又は活性強化は、複数のヒト原発性腫瘍で検出され(Hoshino et al. Oncogene (1999) 18:813-22)、実際、B-Rafのただ1つの変異がERK経路を構成的に活性化し、変異遺伝子は腫瘍原性である。主要なB-Raf変異はV599E(この変異の正確な名称はV600Eであるが、ほとんどの文献、特に古い文献は前記をV599Eと称している)である(Davies et al. Nature (2002) 417:949-54)。しかしながら、ほんのわずかの小分子又はMEK1/2のアンチセンス阻害剤(PD184352/CI-1040(Pfizer)、U-0126(Promega)及びWyeth-Ayerstの化合物(Zhang et al. Bioorg Med Chem Lett (2000)10:2825-28)又はRaf-1/B-Rafのアンチセンス阻害剤(BAY-439006)(Lyons et al. Endocr Relat Cancer (2001) 8:219-25)が前臨床開発段階又は臨床試験中であると報告されている(Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。これまでのところ、特異的で強力なERK1/2阻害剤は報告されていない。
【0006】
公知のMEK1阻害化合物のいくつかの特性の調査によって、それらの有効性は、部分的には複数の経路を阻害するその能力に左右され得ることが明らかになった。PD184352及びU-0126はMEK1を阻害しATPと非競合的であり、おそらくはATP結合部位の外側で結合するアロステリック阻害剤として機能するのであろう。これらの化合物はまた、同様な濃度でMEK5-ERK-5経路の活性化を阻害する。両化合物は動物で抗腫瘍活性を有し、特にERK経路が構成的に活性化される腫瘍に対して活性を有し、報告によれば臨床試験中である(Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。
しかしながら、たとえ開発中のこれらのMEK1阻害化合物が複数のシグナル経路を標的とすることができるとしても、医薬としてのそれらの成功は決して確実とはいえない。複数のシグナル伝達経路の阻害が要求されるならば、前記医薬は、各経路で少なくとも1つのタンパク質キナーゼを充分な能力で阻害し、所望の治療結果を生じなければならない。さらにまた、そのような医薬はしばしば原則的に細胞分裂抑制剤であり、腫瘍細胞を効率的に殺さず、耐性及び再発をもたらす可能性は高い。迅速な可逆性を有する阻害剤である医薬の場合、それらの除去又は細胞レベルでのそれらの減少は、腫瘍細胞分裂の再開を許容する。共有結合により結合する阻害剤は、可逆的なタンパク質キナーゼ阻害剤よりも有効であり得る(Noble et al. Science (2004) 303:1800-05)。前記はEGFR及びHer-2を阻害する医薬について示されたとおりで、この場合、化合物は、ATPポケットでシステイン残基とマイケル付加によって共有結合を形成する(Wissner et al. Bioorg Med Chem Lett (2002) 12:2893-97;Baslega et al. Oncology (2002) 63 Suppl 1:6-16;Wissner et al. J Med Chem (2003) 46:49-63)。医薬として開発することができるタンパク質キナーゼ阻害剤がなお希求されており、それらの標的を共有結合により改変して前記標的を阻害する阻害剤はヒトの疾患の治療で特に有用である。
【0007】
タンパク質キナーゼ阻害剤の探索では天然の生成物が研究された。なぜならば、そのような化合物は、シグナル伝達経路に影響を与える医薬の先導物質として貴重であることが証明されたからである(Newman et al. Curr Cancer Drug Targets (2002) 2:279-308)。“レゾルシル酸ラクトン” (本明細書ではまた“RAL”と称される)として知られる菌類のこのクラスの天然の生成物には、ゼアラレノン(前記はエストロゲン様作用を有し、動物で同化作用促進剤(例えばゼアララノール)として用いられている)の他に(5Z)-7-オキソゼアネオール、ヒポテマイシン、Ro-09-2210、及びL-783,277(前記は細胞分裂を阻害し(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Camacho et al. Immunopharmacology (1999) 44:255-65)、さらに抗腫瘍特性を有すると報告されている(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Tanaka et al. Jpn J Cancer Res (1999) 90:1139-45))が含まれる。さらにまた興味深いことは、ナノモル濃度の低いIC50値でin vitroで、細胞内でのJNK/p38シグナル伝達(Tanaka et al. Biochem Biophys Res Commun (1999) 257:19-23)、血小板由来増殖因子(PDGF)レセプターの自己リン酸化(Giese et al. US5,728,726(1998))MEK1/2(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Dombrowski et al. J Antibiot (Tokyou) (1999) 52:1077-85;Williams et al. Biochemistry (1998) 37:9579-85)又はTAK1(MEKK)(Ninomiya-Tsuji et al. J Biol Chem (2003) 278:18485-90)を阻害するそれらの能力である。しかしながら、それらの興味深い活性にもかかわらず、レゾルシル酸ラクトンはこれまでヒトで調べられたことはなく、医薬としても承認されていない。
【0008】
レゾルシル酸ラクトンL-783,277は、精製MEK1(IC50は4nM)のリン酸化を阻害するが、PKA、PKC又はRafは阻害しない。前記阻害はATPと競合し、60分のプレインキュベーションはMEK1に対するIC50値を10倍低下させる(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94)。MEK1をL-783,277と30分プレインキュベートし、続いてゲルろ過することによって、不活性なMEK1タンパク質が回収され、L-783,277は緊密にMEK1と結合することを示している。しかしながら、L-783,277よりも、5E C=C異性体は能力が100倍低く、さらに7-ジヒドロヒドロキシル異性体は400〜5000倍能力が低いが、明瞭なSARは出現しない(Zhao et al.上掲書)。ヒポテマイシン(図2参照)(前記は構造的にL-783,277に類似するが、11,12-エポキシド成分を有する)はMEK1阻害剤としての能力は4倍低い(Zhao et al.上掲書)。Ro-09-2210はMEK1の強力な阻害剤であり(IC50は59nM)、未発表研究において(Williams et al. Biochemistry (1998) 37:9579-85を参照されたい)、MEK4、6及び7を4〜10倍高いIC50値で阻害すると主張されている。(5Z)-7-オキソゼアネオールはTAK1 MEKK酵素に対して同様な能力を有し(IC50は8nM)、ラットのMEK1に対しはより弱い阻害を示した(IC50は411nM)(Ninomiya-Tsuji et al. J. Biol Chem (2003) 278:18485-90)。
前記のような類似体によるこれら標的キナーゼの強力な阻害の理由は、本発明より前には明らかではなく、ヒトゲノムでコードされるおよそ500を超えるタンパク質キナーゼ(“キノム(kinome)”)に対する総合的な評価は、これらの化合物又は他のいずれの化合物についてもこれまで実施されてなかった。そのような評価は、タンパク質キナーゼアッセイが、これらキナーゼのおよそ150についてのみ開発されてきたためにこれまでは可能ではなかった。化合物がキナーゼを阻害することができるか否かをアッセイする方法、及び化合物がどのキナーゼを阻害するかを決定する方法がなお希求されている。そのような方法が存在しなければ、さらに複数のキナーゼのin vitro判定が存在しなければ(前記はこれまでいずれのRAL化合物についても報告されていない)、タンパク質キナーゼファミリーメンバー間における化合物の相対的な選択性を決定することができないし、さらにヒトの疾患における化合物の有用性を容易に評価することもできない。
したがって、タンパク質キナーゼ阻害剤を同定する方法、及びそれらのキノムにおける相対的選択性及び特にヒトの疾患に中心的に関与する種々のタンパク質キナーゼに対する相対的選択性を判定する方法がなお希求されている。そのような方法を用いて、ある疾患の生物学に直接関係する複数の細胞シグナル伝達経路のタンパク質キナーゼを生産的に阻害する化合物を同定及び選別することが可能であろう。特異的な標的およびシグナルトランスダクション経路のみを阻害する阻害剤を選別し、それらを医薬品として処方し、さらにそれらを投与して、そのような標的の阻害が治療効果(例えば癌、炎症及び他の症状に対抗することを含む)を提供する疾患を治療することが可能であろう。本発明はこれらの要求を満たし、下記に開示する方法、化合物及び医薬品を提供する。
【発明の開示】
【0009】
(発明の要旨)
第一の観点では、本発明は、タンパク質キナーゼの識別可能なサブクラスを、タンパク質キナーゼとマイケル付加物を形成することができる化合物とともに用いて、タンパク質キナーゼを阻害する方法を提供する。タンパク質キナーゼの前記サブクラスは、リン酸化標的及びATPのリン酸と複合体を形成するMg2+と相互作用するタンパク質キナーゼ内の高度に保存された2つのアスパラギン酸残基(Asp)の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン残基(Cys)を有するタンパク質キナーゼを含む。前記タンパク質キナーゼ内のこれらのアミノ酸はATP結合部位として知られる領域に存在する。本発明の方法では、そのようなシステイン残基を有するタンパク質キナーゼは、前記システイン残基でマイケル付加物を形成することができる化合物と接触させることによって阻害される。マイケル付加物形成の結果、前記阻害剤とキナーゼの間で共有結合が生じ、したがって前記阻害は本質的に不可逆になる。
ある実施態様では、該Cysを含むサブクラスに由来する1つ以上のタンパク質キナーゼと前記の重要なCys残基を欠くキナーゼに由来する1つ以上のタンパク質キナーゼとを含むタンパク質キナーゼの混合物は、該Cys残基を含む酵素と可逆的な複合体を形成し、続いて該Cys残基とマイケル付加物を形成することができる部分を含む化合物と接触させられる。ある実施態様では、前記化合物の前記部分はZ-エノン(Z-α、β-不飽和カルボニル部分)である。ある実施態様では、前記化合物の前記部分はレゾルシル酸ラクトンに含まれているか、又は7位のカルボニルと共役している5−6位のシス炭素-炭素二重結合を含む誘導体であるか(α、β-不飽和ケトン;図2参照)、又はそのような部分のバイオイソスター(bioisostere)(例えばエステル、アミド、ビス-ラクトン、スルホンアミド又はスルホン)である。本方法では、重要なCys残基を含むキナーゼのサブクラス由来の1以上のタンパク質キナーゼだけがマイケル付加物形成によって阻害され、該Cys残基を欠くタンパク質キナーゼは阻害されないか(又は同じ程度には阻害されないか)、又はマイケル付加物形成を必要としない異なるメカニズムによって阻害される。
【0010】
本発明の方法は多様な混合物を用いて実施することができる。ある実施態様では、前記混合物はin vitroアッセイで用いられる反応混合物である。別の実施態様では、前記混合物は細胞又は細胞の部分である。また別の実施態様では、前記混合物は、細胞培養アッセイから得られる細胞及び培養液を含む。また別の実施態様では、前記混合物は体液又は体組織である。ある重要な実施態様では、前記混合物は、医学的治療を受けているヒト又は他の哺乳動物の病変組織を含む。
本発明の方法で有用なタンパク質キナーゼ阻害剤は、それらの治療効果を発揮するときに、典型的には少なくとも2以上の異なるタンパク質キナーゼを阻害する。本発明の方法で有用な化合物は、それらの所望される効果を達成するときに、例えば、2以上の異なるタンパク質キナーゼ、少なくとも2つの異なるシグナル伝達経路の各々から1つを阻害することができるか、又は同じ経路又は両方の経路の2以上の異なるタンパク質キナーゼを阻害することができる。いくつかの実施態様では、本発明の方法で用いられる化合物は、それらの所望される効果を達成するときに、少なくとも3つの異なるタンパク質キナーゼを阻害する。
ある実施態様では、本発明の化合物はERK経路の複数の酵素を阻害するために投与され、所望の治療効果を達成する。ある実施態様では、これらの酵素はMEK1/2及びERK1/2である。ある実施態様では、本発明の化合物は、ERK経路の複数の酵素をマイトジェンレセプターキナーゼと同様に阻害する。ある実施態様では、前記化合物はVEGFレセプターを阻害し、さらにERK経路の阻害を介してVEGFの生成を阻害する。本発明のそのような化合物は、血管形成を伴う疾患(癌及び黄斑変性を含むが、ただしこれらに限定されない)の治療に特に有用である。なぜならばそれら化合物はVEGFの産生につながる経路の阻害を介してその産生を阻害するだけでなく、そのレセプターVEGFRもまた直接阻害するからである。
【0011】
ある実施態様では、本発明の化合物によって阻害されるタンパク質はMAPキナーゼである。ある実施態様では、阻害される別個のシグナル伝達経路は、少なくとも1つのマイトジェン誘導経路及び1つのストレス誘導経路を含む。ある実施態様では、少なくとも1つのタンパク質キナーゼはMEKである。ある実施態様では、少なくとも1つのタンパク質キナーゼは、MAPKKファミリーのメンバーである。ある実施態様では、少なくとも1つのタンパク質キナーゼはチロシンレセプターキナーゼであり、前記には、野生型及び変異体PDGFRA、PDGFRB、FLT-3、c-KIT、並びにVEGFレセプターが含まれるが、ただしこれらに限定されない。ある実施態様では、少なくとも1つのタンパク質キナーゼはVEGFレセプターであり、前記にはVEGFR1、VEGFR2(KDRとしてもまた知られている)、及びVEGFR3が含まれる。ある実施態様では、少なくとも1つのタンパク質キナーゼはFLT3である。ある実施態様では、少なくとも1つのタンパク質キナーゼはc-KITである。ある実施態様では、少なくとも1つのタンパク質キナーゼはPDGFRA又はPDGFRBである。
ある実施態様では、本発明の方法で有用な化合物によって阻害されるタンパク質キナーゼは以下から成る群より選択される:AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAK。
ある実施態様では、本発明の方法で用いられた化合物は少なくとも2つの前述のタンパク質を阻害する。別の実施態様では、少なくとも3つのタンパク質キナーゼが阻害される。
【0012】
第二の観点では、本発明は、標的Cys残基を含むタンパク質キナーゼとマイケル付加物を形成することができる化合物を治療の必要がある対象者に投与することを含む、疾患の治療方法を提供する。ある実施態様では、前記対象者はヒトである。ある実施態様では、前記化合物はレゾルシル酸ラクトン又は誘導化合物である。本発明以前には、キノム中の各々別個のキナーゼについていずれのレゾルシル酸ラクトン又はいずれのキナーゼ阻害剤の特異性も先験的に予測することは不可能であった。キナーゼの標的の知識は実験的な試験を必要とし、in vitroアッセイは、今日までにキノム中の500を越えるキナーゼのわずか約150について開発されただけである。タンパク質キナーゼ及び多様な正常及び疾患過程におけるそれらの基本的役割の数の大きさ故に、そのような化合物又は他の化合物(たとえ特定のキナーゼを阻害することが示されたとしても)が、キナーゼの阻害及び疾患の治療に要求される特異性を有しているのか、そうではなくて非常に非特異的で生命に関わる正常な過程を害するのかを誰も決定することができなかった。対照的に、本発明のキナーゼ標的は、マイケル付加物を形成することができるか否かのようなそれらの分子構造によって同定されるので、標的の全レパートリーをキノムの利用可能な配列データから同定することができる。
本発明はまた、医薬組成物及び疾患の治療のためにそれらを投与する方法を提供する。ある実施態様では、本方法は、タンパク質キナーゼ阻害剤とともに別の薬剤を同時投与することを含む。ある実施態様では、前記他の薬剤は抗癌剤である。別の実施態様では、前記薬剤は抗炎症剤である。また別の実施態様では、前記薬剤は別のタンパク質キナーゼである。ある実施態様では、前記医薬組成物は、重要なCys残基を含むタンパク質キナーゼサブクラスの1つ以上に対して特異性を有し、さらに前記とマイケル付加物を形成することができ、さらに疾患又は症状を標的とする化合物(レゾルシル酸ラクトン又は誘導体を含むが、ただしこれらに限定されない)を含む。ある実施態様では、前記医薬組成物を投与して、標的Cys残基(タンパク質キナーゼのATP結合部位内の保存された2つのAsp残基の間に位置し、Asp残基の1つと隣接する)を含まないタンパク質キナーゼの阻害の場合には生じ得る望ましくない副作用をもたらすことなく治療効果が達成される。
【0013】
発明の詳細な説明
ヒトゲノムは、これまでのところ、標準的な真核細胞タンパク質キナーゼ型の510の識別可能な遺伝子(ヒト“キノム”と称される)を有すると報告されている(Kositch et al. Genome Biology 2002, 3(9):RESEARCH 0043)。前記タンパク質キナーゼファミリーは治療的処置のための標的の豊富な供給源を提供する。なぜならば、前記のメンバーは、多くの疾患過程(炎症及び癌を含む)において重要な役割を果たすからである。しかしながら、このファミリーに含まれるタンパク質が数多く存在し、且つそれらを包含する種々の細胞シグナル伝達経路も数多く存在するため、医学的利用のため充分に活性な且つ特異的である医薬の発見を困難にし、かつ予想のつかないものにしている。本発明は、多様な生物の多様な異なる細胞シグナル伝達経路に由来する識別可能で特異的なタンパク質キナーゼサブセットを阻害する化合物、組成物及び方法を提供し、したがって疾患の治療においてタンパク質キナーゼを標的とする試みにおいて重要な進歩を提示する。
前記タンパク質キナーゼファミリーでは、2つの高度に保存されたAsp残基(D167及びD185、PKA-Calpha(NP_002721)の残基番号を使用)は以下の役割を割り振られている:第一のAspは、ATPのリン酸基のOHからH+を受け入れ、第二のAspは、ATPのリン酸基と複合体を形成するMg2+と相互作用する(したがって移転のためにγ-リン酸基の位置決定に寄与する)。この領域では、第二のAspの直前(PKA-Calphaの184位に一致する)は可変性の位置であり、ヒトのキナーゼの約10%(およそ50/510)では前記位置がCysである。この位置は、第二のAspに接近しているため、必然的にATP結合部位領域内に存在する。
【0014】
本発明は、一つには、これらのCys含有キナーゼを阻害するある種のレゾルシル酸ラクトンが共通の構造的特徴を共有するという発見から生じている。これらの化合物は、一般的に、5−7位のカルボニルと共役しているシス二重結合を有する(例えば図2の最初の4つの構造を参照されたい)。そのような化合物は以下の分子骨組みを有する(本明細書で用いられる番号付与もまた示されている)
【0015】
【化1】
【0016】
最初の可逆的な酵素-阻害剤複合体の形成後、その複合体内の上述する構造とキナーゼドメイン/ATP結合部位内のCys側鎖とが接近していることによって、続いて非常に緩慢な可逆的、又は事実上不可逆的なマイケル付加物の形成がもたらされ、極めて強力な阻害のメカニズムが提供される。
マイケル付加物は、下記の反応式によって示されるように、形式的には、求核種の共役求電子二重結合への1,4-付加の生成物である:
【0017】
【化2】
【0018】
式中、Xは典型的にはO又はNRであり、Nuは、典型的には炭素、窒素、酸素又は硫黄をベースとする求核基である。前記共役求電子二重結合は、典型的にはα,β-不飽和ケトン、アルデヒド、又はエステル部分に存在するが、または不飽和ニトリル、スルホン、又はニトロ部分にも存在し得る。本出願の目的のために、“マイケル付加物”という用語は、前記生成物生成の正確なメカニズムには関係なく、そのような1,4-付加をもつ形式的生成物を指し、さらにそのような形式的生成物の互変異性形(例えばエノール化形を含む)も包含する。
入手可能な発表されたデータを本発明の観点から調査することによって、前記Cysは、そのような構造を有するレゾルシル酸ラクトンに対して感受性を有すると報告されているいくつかのMAPキナーゼに存在し、非感受性と報告されたものには存在しないことが明らかになった。例えば、シス-エノンレゾルシル酸ラクトンは、MEK1、4、6及び7をMAPKKK TAK1及びマイトジェンレセプターチロシンキナーゼPDGFRと同様に阻害することが報告された。前記標的Cys残基を有しない約10のキナーゼは、ある種のシス-エノンレゾルシル酸ラクトンによって阻害されなかった。
したがって、本発明は、前記構造を含むレゾルシル酸ラクトン及び類似体、並びにキナーゼドメインATP結合部位内の又は前記部位に近接するシステイン残基を含む50までのキナーゼを選択的に阻害するときに前記構造を含むレゾルシル酸ラクトン及び類似体を使用する方法を提供する。本発明はまた、そのような化合物を含む医薬組成物及びそれらを用いて疾患を治療する方法を提供する。特に、本発明の化合物の特異性は、特定の症状と関係を有する複数のキナーゼ標的について予測することができる。本発明の方法は、阻害される標的が原因的又は促進的役割を果たす疾患の治療を提供する。
【0019】
このモデルが適切なことは、キナーゼEKR2とヒポテマイシンとの共有結合複合体のX線構造によって実証された。図3は、2.5オングストロームの解析度の構造において、上部にERK2のN末端突出部、下部にC末端突出部、及びヒンジ領域で共有結合しているヒポテマイシンを有する複合体を示す。図4は、ヒンジ領域のクローズアップ図であり、マイケル反応でヒポテマイシンのエノン二重結合にわたって付加されたシステインの硫黄を示している。
本明細書に開示するレゾルシル酸ラクトン阻害剤によるタンパク質キナーゼの強力な阻害のためには、前記阻害剤は、標的酵素が課す2つの“選択性フィルター”を通過することを要求される。第一に、前記阻害剤は、相応に堅固な結合定数で酵素と可逆的に結合しなければならない。この可逆的な結合フィルターは、可逆的なエネルギー形成結合(例えば水素結合、イオン相互作用、疎水性相互作用)と同様に、阻害剤と酵素の相補的なトポロジーに依存する。第二のフィルターは、酵素の標的チオールと阻害剤のエノン部分のβ-炭素との間で共有結合が形成されることを必要とする。このフィルターは、酵素-阻害剤複合体内に適切なCys残基が存在することを必要とし、前記フィルターの有効性は、マイケル受容体である炭素原子を有する反応性チオールが適切に並列するか否かに左右される。いくつかのレゾルシル酸ラクトンは第一のキナーゼフィルター(可逆的結合)を通過することができず、したがって第二のフィルター(共有結合)に遭遇することはあり得ない。いくつかのレゾルシル酸ラクトンは第一のキナーゼフィルターを通過するが、そのキナーゼは、共有結合を形成するCys残基を持たないであろう。実際に両者の例は本明細書で言及される。本発明のレゾルシル酸ラクトンに対する興味対象の標的は、この両方のフィルターを通過するものである。
【0020】
ほとんどのキナーゼ阻害剤は、慣例的なスクリーニングで発見され、続いて1つ又はいくつかのキナーゼに対して最適化された。結果として、それらは第一のフィルター(上記に記載)を通過するように開発され、それらの特異性は、どれほど多くの異なるキナーゼがそれらの結合部位で類似するトポロジーと可逆的相互作用とを共有するかに左右される。タンパク質キナーゼのATP部位は高度に保存されているので、この部位と結合する可逆的な阻害剤はおそらく多くのキナーゼを阻害するが、しかし予想不能で明らかに無差別的な態様で阻害するわけではない。例えば、およそ120のキナーゼのパネルでは、Sugen11248と同定された化合物は、およそ79のキナーゼを0.002〜6.6μMのKi値の範囲で阻害し(Fabian et al. Nat. Biotechnol. (2005) 23(3):329-36)、これらのうちで、およそ56のキナーゼは0.1μM未満のKi値を示し、したがってin vivoの標的に対応し得る。第二のフィルターとしてのレゾルシル酸ラクトンとの共有結合に関しては、標的Cys残基を含むサブセットのみが不可逆的に阻害されるため、キナーゼ間の識別はユニークなものであり極めて増強される。
キナーゼ-レゾルシル酸ラクトン相互作用の共有結合性と事実上の不可逆性によって、薬剤作用及び本発明によって提供される投与方法に関して更なる利点が提供される。例えば、レゾルシル酸ラクトンは種々のキナーゼと種々の可逆的親和性(Ki)及び種々の共有結合不活化速度(Kinact)を有するので、キナーゼ混合物のレゾルシル酸ラクトンへの曝露(濃度x時間)を制御することによって、ある種のキナーゼの選択的阻害を達成することができる。これは、特定のレゾルシル酸ラクトンの特定のキナーゼに対する“特異性定数”によって反映される。前記は、非常に希薄な阻害剤及びキナーゼ濃度の共有結合については実質的には2桁の速度定数である。例えば、ヒポテマイシンはERK2に対して2μMのKiを有し(Kinact/Ki=1.9E+03)、不活化のt1/2は3分であり、KDRに対してはKiは0.01μMで、不活化のt1/2は約1分である(Kinact/Ki=5E+05)。2つのキナーゼを0.1μMで約10分処理することによって((Ki ERK>0.1um>Ki KDR)、5%未満のERK活性が阻害される条件下で、98%を超えるKDRを阻害することができると計算できる。さらにまた、曝露が、共有結合阻害を完了させるのに充分である場合、薬剤の投与を停止して非Cysキナーゼの一切の可逆的阻害を軽減することができるが、共有結合によって改変されたキナーゼの特異的なセットの阻害は維持される。
【0021】
本発明は、一つには、ヒポテマイシン(マイケル付加物形成構造を含む)とゼアラレノン及び5,6-ジヒドロヒポテマイシン(マイケル付加物形成構造を含まない、図2を参照)を比較することによって、さらにそれら各々のERK1/2の活性化を阻害する能力を比較することによって、理解することができる。化合物を0.3〜3μMで調べたとき、ヒポテマイシンは、ヒトのT細胞でERK1/2の活性化を阻害するが、ゼアラレノンは阻害しないと報告されている(Camacho et al. 1999 Immunopharmacology 44(3):255-265)。対応するヒトタンパク質キナーゼのアミノ酸配列の調査によって、ERK1/2内に適切に配置されているCys残基が示されている。
任意の多様なタンパク質キナーゼ、例えばMEK1/2又はERK1/2についての相同性モデルの調査によって、タンパク質キナーゼのATP結合部位領域内でのレゾルシル酸ラクトンの適切な配置がマイケル付加物の形成を可能にするであろうということが例証される。例えば、MEK1 ATP結合部位の相同性モデルは、α、β-不飽和カルボニル含有レゾルシル酸ラクトン又は誘導体が、前記重要なCys残基を含むタンパク質キナーゼをマイケル付加物の形成によって阻害することができるメカニズムを支持する。
そのようなモデルは、本発明の観点から、マイケル付加物形成による阻害に感受性を有するタンパク質キナーゼを阻害することができる新規なキナーゼ阻害剤の構造を予測することを可能にするだけでなく、本発明の方法で有用な構造を有する既知の化合物を同定することもまた可能にする。ある実施態様では、本発明の方法で有用な化合物は、公知で以前に試験された化合物であり、それらは、用いられる混合物が、それら既知の化合物のキナーゼ阻害の特異性が未だ試験されたり決定されたりしていないキナーゼを含む、本発明の方法で用いられる。別の実施態様では、本発明の化合物は、以前には生成又は試験されたことがない新規な化合物である。
【0022】
本発明によって提供される進歩を理解するためには、本質的に全てのタンパク質キナーゼ阻害剤は複数のキナーゼを阻害することが充分に証明されていることと、特定の阻害剤に対する細胞の応答は、2つの又は通常はそれ以上のキナーゼの同時阻害を伴うということとを理解しなければならない。続いて、いずれの阻害剤の特異性及び有効性も、そのキナーゼ阻害プロフィールに依存し、また、異なるプロフィールは異なる効果を細胞にもたらす。ほとんどの既知のキナーゼ阻害剤のキナーゼプロフィールは実験的にのみ決定することができ、したがってアッセイで利用可能な酵素の数によって制限される。例えば、120キナーゼの大きなパネルに対する複数のキナーゼ阻害剤の阻害活性プロフィールが報告されている(Fabian et al. Nat Biotechnol 2005, 23(3):329-36)。イマチニブ(グリーベック)は120キナーゼのうち10個を阻害し、BAY43-9006は120キナーゼのうちKi<0.1μMの19キナーゼを阻害したが、スクリーニングにこれまで利用可能でなかった残りの300キナーゼのどれくらいが、又はどれがこれら化合物によって阻害されるかは不明である。対照的に、本発明は、本発明のレゾルシル酸ラクトン(RAL)によって阻害される全ヒトキノムにおける標的の信頼できるリストを提供する(前記RALは、その標的が含む重要なCys残基でこれら標的とマイケル付加物を形成することができる)。
阻害剤の完全なキナーゼプロフィールの知識は、ある種の細胞タイプに対するその有効性及び特異性に関する有用な情報を提供する。例えば、前記プロフィールを他の阻害剤のプロフィールを比較することができる。新規な阻害剤の標的キナーゼサブセットが、公知の有効な阻害剤に対して関連があると考えられているサブセットとオーバーラップするならば、前記新規な阻害剤は類似の活性及び効果を示すはずである。本発明の方法で有用なレゾルシル酸ラクトンは固有のキナーゼ阻害プロフィールを有するが、その標的キナーゼのある種のサブセットは他の有効なキナーゼ阻害剤によって阻害されるサブセットとオーバーラップする。例えば、キナーゼ阻害剤SU11248は、FLT3内在縦列重複変異(ITD)を含むAMLの阻害で有効である。なぜならば、FLT3(野生型及びITD)、PDGFR、VEGFR及びcKITを含むキナーゼサブセットを標的とするからである。ヒポテマイシンは、キナーゼの同じサブセットを阻害し、したがって、本発明によって提供されるように、AML細胞の阻害で有効である。下記実施例に記載するある試験では、AML(FLT3 ITD)細胞株MV-4-11に対するSU11248のGI50は12nMであり、ヒポテマイシンは6nMのGI50を有していた。
【0023】
経路の初期に酵素が過剰産生されるか又はアミノ酸変異のために、ある種のキナーゼ及びキナーゼ経路は過剰に活性であるか又は構成的に活性であり、したがって、(直接的又は経路内のより初期の別の酵素を介する間接的な)阻害は、前記活性な経路から生じる表現型の選択的な阻害又は調節をもたらすことができると期待することができる。例えば、B-Raf V599E(V600E)変異体は、およそ70%のメラノーマ及びおよそ20%の結腸癌で見出され、細胞分裂に必要なERK経路の構成的活性化をもたらす。BAY 43-9006は、最初はメラノーマ細胞のこの経路を阻害するためにRaf阻害剤として開発された。ヒポテマイシン及び本発明の方法で有用な他のRALは、前記経路の2つの点(MEK1/2及びERK1/2)を不可逆的に阻害し、したがって前記経路を完全に阻害してERK/RSKリン酸化の下流のシグナル伝達を閉鎖するはずである。
下記実施例に記載したin vitro試験は、B-Raf V599E(V600E)変異体はRAL阻害剤に非常に感受性が高いことを示している。メラノーマ細胞株COLO829に関しては、ヒポテマイシンは50nMのGI50を有し、BAY 43-9006は6,000nMのGI50を有し、さらにSU11248は7,100nMのGI50を有している。活性化されたERK経路はまた、細胞生物学的研究及びMEK1/2阻害剤の効果によって実証されるように、腫瘍の広範囲(乳房、大腸、卵巣、前立腺及び膵臓の腫瘍を含む)で関与している。MEK及びRaf阻害剤は、ERK/RSK経路に依存する細胞に対して有効であり、本発明のRALはこれらの細胞に対して同様に有効である。
【0024】
ただ1つの酵素に対する可逆的阻害剤による100%阻害の達成は非常に困難であり、一方、ある経路の複数の工程を阻害する阻害剤は経路のほぼ完全な遮断を引き起こすことができる。キナーゼプロフィールが、直線的経路の2以上の連続的工程の阻害を示すならば、経路全体に対する薬剤の影響は協同的でないとしても少なくとも累積的であろう。本発明の方法で有用なRAL阻害剤は、それらがERK経路の少なくとも2つの点を不可逆的に阻害するという点で固有である。それらはまた、ERK経路を刺激するチロシンキナーゼマイトジェンレセプターの多くを不可逆的に阻害し、直線的経路の3点阻害及びそれに続くマイトジェン刺激分裂経路の強力な阻害を提供する。例えば、下記の実施例に示すように、変異体のマイトジェンレセプターFlt3及び構成的に活性なERK経路を含むAML細胞株MV-4-11に関して、ヒポテマイシンは6nMのGI50を有し、ヒポテマイシンは、VEGFRの非常に強力な不可逆的阻害剤であり、VEGFRを必要とする細胞の治療はVEGFR、MEK及びERKを閉鎖する。さらにまた、ERKリン酸化はVEGF分泌に要求されるので、VEGF産生細胞での産生及びVEGF応答細胞でのVEGFに対する応答の両方が阻害される。これらの理由のために、ヒポテマイシン及び前記必須のCys残基をもつタンパク質キナーゼとマイケル付加物を形成することができる本明細書に開示の他のRALは、血管形成の極めて有効な阻害剤である。別の例は基底細胞癌(BCC)の治療である。この適応症では、BCC腫瘍の90%が、ERK経路及び細胞分裂を駆動するPDGFRを過剰発現する。本発明の方法で有用なRALはPDGFR経路とERK経路の2つの点とを阻害し、したがって直線的経路の3点阻害を提供する。
【0025】
ほとんどのキナーゼ阻害剤は可逆的阻害剤であり、したがって標的の阻害は濃度の関数であり、完全阻害には阻害定数Kiをはるかに超える阻害剤濃度が要求される。さらにまた、いったん阻害剤が除去されたならば酵素活性は急速に復帰するので、細胞は持続的な曝露を要求する。本発明の方法で用いられる化合物は、タンパク質キナーゼの不可逆的阻害剤である(ただし標的キナーゼサブセットのみを不可逆的に阻害する)。ヒポテマイシン及び本発明の方法で有用な他のRALによる標的阻害は、濃度及び/又は時間の関数であるので、曝露時間が増加されるならば、完全阻害は低濃度の阻害剤で達成することができる。本発明は、これらの特性の利点を有する本発明のRALの単位用量形態及び前記RALを投与する方法を提供する。したがって、ある実施態様では、疾患を治療する本発明の方法は、充分な化合物を投与して、阻害定数にあるか又は阻害定数以下の該化合物の血中又は腫瘍中レベルを提供するか、及び/又は充分な時間にわたって前記血中又は腫瘍中レベルの維持を提供し、その結果、標的タンパク質キナーゼの少なくとも50%、より好ましくは90%を超える(例えば99%又は100%)不可逆的阻害を達成することを含む。ある実施態様では、前記薬剤の第二の投与(多くの実施態様では、薬剤は同じ患者に複数回投与されるであろう)は、不可逆的に阻害されたキナーゼのde novo合成による交換を基準にすると、前記薬剤の最初の投与後1又は2日以内である。
例えば、下記実施例で示すように、B-Raf V599E(V600E)細胞株COLO829(及び調査されたBRAF変異体を有する他の細胞)中のERK経路は、前記酵素に対して数倍低いKd濃度でヒポテマイシンに10分曝露した後で閉鎖された。さらにまた、前記阻害剤の除去は活性の即時復活を伴わず、それどころか、リン酸化された活性なERKは何時間(約24時間)も存在せず、その復帰は新規な酵素合成を明らかに必要とする。したがって、本発明は、これら化合物を投与して、正常細胞に対する毒性を低下させる方法を提供する。ある実施態様では、前記化合物は、腫瘍又は他の癌細胞若しくは組織から得られる測定値によって決定して、標的キナーゼ活性が完全に阻害されるまで投与される。この時点で、治療効果を低下させることなく投与を停止することができ、有意なレベルの標的キナーゼ活性が回復した後でのみ投与を再開することができる。
【0026】
ある実施態様では、本発明の方法で有用で、さらに本発明の医薬組成物に含まれる化合物は、以下の一般構造式(I)を有するもの、並びに医薬的に許容できるその塩及び誘導体である。
【0027】
【化3】
【0028】
式中、
R1は、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、各々独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、場合によって置換されたアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子又は酸素原子を含む飽和若しくは不飽和環式環を形成し;R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になってN3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、又はアルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は、
R8及びR9は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子又は酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であるか、又はR20及びR2が組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であるか、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり、式中R19は水素又はアルキルである。
【0029】
好ましくは式(I)の化合物では、以下の条件の少なくとも1つが当てはまる:(i)R6は水素又はヒドロキシルである、(ii)nは1である、(iii)R8は水素以外のものである、(iv)R10は水素である、及び(v)R11は保護されたヒドロキシル以外のものである。
好ましい実施態様では、前記化合物は下記式(Ia)の構造を有する:
【0030】
【化4】
【0031】
式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
ここで、R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されたアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;ここでR17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ia)の化合物の好ましい実施態様では、R9のOR12はOMe以外のものである。
【0032】
別の好ましい実施態様では、前記化合物は下記式(Ib)の構造を有する:
【0033】
【化5】
【0034】
式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
ここでR12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;さらに
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
【0035】
別の好ましい実施態様では、前記化合物は下記式(Ic)の構造を有する:
【0036】
【化6】
【0037】
式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ic)の化合物の好ましい実施態様では、R8は水素又はハロゲン以外のものである。
別の好ましい実施態様では、前記化合物は下記式(Id)の構造を有する:
【0038】
【化7】
【0039】
式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
別の好ましい実施態様では、前記化合物は下記式(Ie)の構造を有する:
【0040】
【化8】
【0041】
式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ie)の化合物の好ましい実施態様では、R5は水素以外のものである。
【0042】
別の好ましい実施態様では、前記化合物は下記式(If)の構造を有する:
【0043】
【化9】
【0044】
式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
別の好ましい実施態様では、前記化合物は下記式(Ig)の構造を有する:
【0045】
【化10】
【0046】
式中、
R4はH又はFであり;
R8はHであり;さらに
R9は以下の基から成る群より選択されるか、
【0047】
【化11】
【0048】
又は、R8及びR9が一緒になって以下の基を形成する。
【0049】
【化12】
【0050】
“脂肪族”とは直鎖又は分枝鎖の飽和若しくは不飽和の非芳香族炭化水素部分を指し、特定の数の炭素原子(例えば“C3脂肪族”、C1〜C5脂肪族”又は“C1からC5脂肪族”のように、後者の2つの語句は1〜5の炭素原子を有する脂肪族部分についての同義語である)を有するか、又は炭素原子の数が特定されない場合は1〜4の炭素原子を有する。不飽和脂肪族部分は必然的に少なくとも2つの炭素原子を含むことは当業者には理解されよう。
“アルキル”は飽和脂肪族部分を意味し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C1〜C4アルキル部分は、メチル、エチル、プロピル、イソプロピル、イソブチル、t-ブチル、1-ブチル、2-ブチルなどが含まれるが、ただしこれらに限定されない。
“アルケニル”は、少なくとも1つの炭素-炭素二重結合を有する脂肪族部分を指し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C2〜C4アルケニル部分は、エテニル(ビニル)、2-プロペニル(アリル又はプロパ-2-エニル)、シス-1-プロペニル、トランス-1-プロペニル、E-(又はZ-)2-ブテニル、3-ブテニル、1,3-ブタジエニル(ブタ-1,3-ジエニル)などを含むが、ただしこれらに限定されない。
“アルキニル”は、少なくとも1つの炭素-炭素三重結合を有する脂肪族部分を指し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C2〜C4アルキニル基は、エチニル(アセチレニル)、プロパルギル(プロパ-2-イニル)、1-プロピニル、ブタ-2-イニルなどを含む。
“環式脂肪族”は、飽和又は不飽和の非芳香族炭化水素部分を指し、1〜3の環を有し、各環は3〜8(好ましくは3〜6)の炭素原子を有する。“シクロアルキル”は、各環が飽和されている環式脂肪族部分を指す。“シクロアルケニル”は、少なくとも1つの環が少なくとも1つの炭素-炭素二重結合を有する環式脂肪族部分を指す。“シクロアルキニル”は、少なくとも1つの環が少なくとも1つの炭素-炭素三重結合を有する環式脂肪族部分を指す。例示すれば、環式脂肪族部分には、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプチル、シクロオクチル及びアダマンチルが含まれるが、ただしこれらに限定されない。好ましい環式脂肪族部分は、シクロアルキル部分、特にシクロプロピル、シクロブチル、シクロペンチル、及びシクロヘキシルである。
【0051】
“複素環式脂肪族”は環式脂肪族部分であって、その少なくとも1つの環において、3つまで(好ましくは1〜2)の炭素が、N、O、又はSから独立に選択されるヘテロ原子で置き換えられているものを指し、ここでN及びSは場合によって酸化されてもよく、さらにNは場合によって四級化されてもよい。同様に、“ヘテロシクロアルキル”、“ヘテロシクロアルケニル”、及び“ヘテロシクロアルキニル”は、それぞれシクロアルキル、シクロアルケニル、又はシクロアルキニル部分であって、前記の少なくとも1つの環が上記のように改変されているものを指す。例示的な複素環式脂肪族部分には、アジリジニル、アゼチジニル、1,3-ジオキサニル、オキセタニル、テトラヒドロフリル、ピロリジニル、ピペリジニル、ピペラジニル、テトラヒドロピラニル、テトラヒドロチオピラニル、テトラヒドロチオピラニルスルホン、モルホリニル、チオモルホリニル、チオモルホリニルスルホキシド、チオモルホリニルスルホン、1,3-ジオキソラニル、テトラヒドロ-1,1-ジオキソチエニル、1,4-ジオキサニル、チエタニルなどが含まれる。
“アルコキシ”、“アリールオキシ”、“アルキルチオ”及び“アリールチオ”は、それぞれ−O(アルキル)、−O(アリール)、−S(アルキル)及び−S(アリール)を意味する。例は、それぞれメトキシ、フェノキシ、メチルチオ及びフェニルチオである。
“ハロゲン”又は“ハロ”は、フッ素、塩素、臭素又はヨウ素を指す。
“アリール”は、一環式、二環式、又は三環式環系を有する炭化水素部分を指し、各環は3〜7の炭素原子を有し、さらに少なくとも1つの環は芳香族である。前記環系の環は互いに縮合(ナフチルにおけるように)していてもよく、又は互いに結合(ビフェニルにおけるように)していてもよく、さらに(インダニル又はシクロヘキシルフェニルにおけるように)非芳香環と縮合又は非芳香環と結合してもよい。さらに例示すれば、アリール部分には、フェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニル、フェナントリル、アントラセニル及びアセナフチルが含まれるが、ただしこれらに限定されない。
【0052】
“ヘテロアリール”は、一環式、二環式、又は三環式環系を有する部分を指し、各環は3〜7個の炭素原子を有し、さらに少なくとも1つの環は、N、O又はSから独立に選択される1〜4のヘテロ原子を含む芳香環である。ここで、前記N及びSは場合によって酸化されてもよく、Nは場合によって四級化されてもよい。そのようなヘテロ原子含有芳香環は、(ベンゾフラニル又はテトラヒドロイソキノリルにおけるように)他のタイプの環と縮合してもよく、又は(フェニルピリジル又は2-シクロペンチルピリジルにおけるように)他のタイプの環と直接結合してもよい。さらに例示すれば、ヘテロアリール部分には、ピロリル、フラニル、チオフェニル(チエニル)、イミダゾリル、ピラゾリル、オキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、ピリジル、N-オキソピリジル、ピリダジニル、ピリミジニル、ピラジニル、キノリニル、イソキノリニル、キナゾリニル、シンノリニル、キノザリニル、ナフチリジニル、ベンゾフラニル、インドリル、ベンゾチオフェニル、ベンゾイミダゾリル、ベンゾトリアゾリル、ジベンゾフラニル、カルバゾリル、ジベンゾチオフェニル、アクリジニルなどが含まれる。
部分が置換され得ると表示されている場合(例えば“置換又は非置換C1〜C5アルキル”又は“ヘテロアリールで置換されていてもよい”におけるように表現される、“置換又は非置換”又は“置換されていてもよい”を使用することにより)、そのような部分は、1以上の独立して選択される置換基を、好ましくは個数として1〜5、より好ましくは個数として1又は2有することができる。置換基及び置換パターンは、置換基が結合される部分について考慮しながら、化学的に安定であり、且つ本技術分野で公知の技術及び本明細書で述べる方法によって合成することができる化合物を提供するように、当業者が選択することができる。
【0053】
“アリールアルキル、(複素環式脂肪族)アルキル”、“アリールアルケニル”、“アリールアルキニル”、“ビアリールアルキル”などは、事情に応じてアリール、複素環式脂肪族、ビアリールなどの部分で置換されたアルキル部分、アルケニル部分、又はアルキニル部分を指し、事情に応じて前記アルキル部分、アルケニル部分、又はアルキニル部分で(例えばベンジル、フェネチル、N-イミダゾリルエチル、N-モルホリノエチルなどのように)開いた(未飽和の)原子価を有する。逆に、“アルキルアリール”、“アルケニルシクロアルキル”、ハロヘテロアリールなどは、例えば、事情に応じてメチルフェニル(トリル)、又はアリルシクロヘキシルにおけるように、事情に応じてアルキル、アルケニル、ハロなどの部分で置換されたアリール、シクロアルキル、ヘテロアリールなどの部分を指す。“ヒドロキシアルキル”、“ハロアルキル”、“アミノアルキル”、“アルキルアリール”、“シアノアリール”などは、事情に応じて特定された置換基(事情に応じてヒドロキシル、ハロ、アミノなど)で置換されたアルキル、アリールなどの部分を指す。例示すれば、許容できる置換基には、アルキル(特にメチル又はエチル)、アルケニル(特にアリル)、アルキニル、アリール、ヘテロアリール、環式脂肪族基、複素環式脂肪族基、ハロ(特にフルオロ)、ハロアルキル(特にアリルトリフルオロメチル)、ヒドロキシル、ヒドロキシアルキル(特にヒドロキシエチル)、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)(特に−OCF3)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)、−SO2N(アルキル)2などが含まれるが、ただしこれらに限定されない。
【0054】
前記置換される部分が脂肪族部分である場合、好ましい置換基は、アリール、ヘテロアリール、環式脂肪族基、複素環式脂肪族基、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)、及び−SO2N(アルキル)2である。より好ましい置換基は、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、−O(アリール)、=O、=NOH、=NO(アルキル)、−OC(=O)(アルキル)、−OC(=O)O(アルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、及び−NHC(=NH)NH2である。
前記置換される部分が環式脂肪族部分、複素環式脂肪族部分、アリール部分又はヘテロアリール部分である場合は、好ましい置換基は、アルキル、アルケニル、アルキニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)及び−SO2N(アルキル)2である。より好ましい置換基は、アルキル、アルケニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2及び−NHC(=NH)NH2である。
【0055】
範囲が“C1〜C5アルキル”又は“5〜10%”のように記載されている場合、そのような範囲は前記範囲の両端の値を含む。
具体的な立体異性体が特に(例えば構造式中の対応する立体中心で太字又は点線による結合によって、構造式中のE又はZ構造を有する二重結合の描写によって、又は立体化学の呼称の使用によって)指定されないかぎり、全ての立体異性体が、その混合物と同様に純粋な化合物として本発明の範囲内に含まれる。特段に規定されないかぎり、個々の鏡像体、ジアステレオマー、幾何学異性体並びにそれらの組合せ及び混合物は全て本発明に包含される。多形性結晶形及び溶媒和物もまた本発明の範囲内に包含される。
“医薬的に許容される塩”は、塩として医薬処方物に適した化合物の塩を指す。化合物が1以上の塩基性官能性を有する場合、前記塩は酸付加塩、例えば硫酸塩、臭化水素酸塩、酒石酸塩、メシレート、マレイン酸塩、クエン酸塩、リン酸塩、酢酸塩、パモポエート(エンボネート)、ヨウ化水素酸塩、硝酸塩、塩酸塩、乳酸塩、メチルスルフェート、フマル酸塩、安息香酸塩、コハク酸塩、メシレート、ラクトビオネート、スブレート、トシレートなどであり得る。化合物が1つ又は2つ以上の酸性部分を有する場合は、前記塩は、例えばカルシウム塩、カリウム塩、マグネシウム塩、メグルミン塩、アンモニウム塩、亜鉛塩、ピペラジン塩、トロメタミン塩、リチウム塩、コリン塩、ジエチルアミン塩、4-フェニルシクロヘキシルアミン塩、ベンザチン塩、ナトリウム塩、テトラメチルアンモニウム塩などのような塩であり得る。
【0056】
本発明は、その範囲内に本発明の化合物のプロドラッグを含む。そのようなプロドラッグは前記化合物の一般的な機能的誘導体であり、要求される化合物にin vivoで容易に変換され得る。したがって、本発明の治療方法では、“投与する”という用語は、具体的に開示された化合物を用いて、又は具体的に開示されてないがその必要がある対象者に投与後n vivoで特定された化合物に変換される化合物を用いて、開示の種々の疾患を治療することを包含するであろう。適切なプロドラッグ誘導体の選択及び調製のための通常的な方法は、例えば以下に記載されている:Wermuth, “Designing Prodrugs and Bioprecursors”, in Wermuth, ed., The Practice of Medical Chemistry, 2nd Ed., pp.561-586, Academic Press 2003(前記文献の内容は参照により本明細書に含まれる)。プロドラッグは、in vivoで(例えばヒトの体内で)加水分解されて本発明の化合物又はその塩を生成するエステルを含む。適切なエステル群には、医薬的に許容できる脂肪族カルボン酸、特にアルカン酸、アルケン酸、シクロアルカン酸及びアルカンジオン酸(前記で各アルキル又はアルケニル部分は好ましくは6未満の炭素原子を有する)から誘導されるものが含まれるが、ただし前記に限定されない。例示的なエステルには、ギ酸エステル、酢酸エステル、プロピオン酸エステル、酪酸エステル、アクリル酸エステル、クエン酸エステル、コハク酸エステル及びエチルコハク酸エステルが含まれるが、ただしこれらに限定されない。
ある重要な実施態様では、本発明の化合物には、本発明の方法で用いられる経口投与に適したレゾルシル酸ラクトンのプロドラッグエステルが含まれる。ある実施態様では、これらのプロドラッグは、本発明の方法で有用なレゾルシル酸ラクトンのアミノ酸エステル(ジメチルグリシンエステル及びバリンエステルが含まれるが、ただしこれらに限定されない)である。
【0057】
“保護基”は、特定の官能部分(例えばO、S又はN)を一時的に遮断し、それによって反応が多官能性化合物の別の反応部位で選択的に実施され得る部分を指す。好ましい実施態様では、保護基は、(a)良好な収量で選択的に反応して、計画の反応にとって安定な保護基質を生じ、(b)他の官能基を攻撃しない、容易に入手可能で、好ましくは毒性のない試薬によって良好な収量で選択的に除去でき、(c)容易に分離することができる誘導体を生成し(より好ましくは新規な立体中心を生じることなく)、さらに(d)更なる反応部位を回避するために付加される官能性が最小限である。“酸素保護基”は酸素に結合される保護基を指し、メチルエーテル、置換メチルエーテル(例えばMOM(メトキシメチルエーテル)、MTM(メチルチオメチルエーテル)、BOM(ベンジルオキシメチルエーテル)、PMBM又はMPM(p-メトキシベンジルオキシメチルエーテル))、置換エチルエーテル、置換ベンジルエーテル、シリルエーテル(例えばTMS(トリメチルシリルエーテル)、TES(トリエチルシリルエーテル)、TIPS(トリイソプロピルシリルエーテル)、TBDMS(t-ブチルジメチルシリルエーテル)、トリベンジルシリルエーテル、TBDPS(t-ブチルジフェニルシリルエーテル))、エステル(例えばギ酸エステル、酢酸エステル、安息香酸エステル(Bz)、トリフルオロ酢酸エステル、ジクロロ酢酸エステル)、カルボネート、環式アセタールおよびケタールが含まれるが、ただし前記に限定されない。“窒素保護基”はアミンの窒素に結合される保護基を指し、カルバメート(例えばメチル、エチル及び置換エチルカルバメート(例えばTroc))アミド、環式イミド誘導体、N-アルキル及びN-アリールアミン、イミン誘導体、及びエナミン誘導体が含まれるが、ただしこれらに限定されない。保護基の多くの例は、それらの使用態様に関する教示とともに以下の文献で見出すことができる:Greene and Wuts, Protective Groups in Organic Synthesis, 3rd edition, pp. 17-245(John Wiley & Sons, New York, 1999)(前記文献の内容は参照により本明細書に含まれる)。したがって、“保護されたヒドロキシル”は、水素が酸素保護基によって置き換えられたヒドロキシル基を指し、“保護アミン”は、水素が窒素保護基によって置き換えられた1級又は2級アミン基を指す。
【0058】
5−7位のカルボニル(又はビオイソステア)と共役した前記重要なシス二重結合を保持する上記構造に包含される化合物の類似体及び誘導体もまた本発明の方法で有用である。一般的には、レゾルシル酸ラクトンであろうと、誘導体であろうと、他の化合物であろうと、前記重要なCys残基とマイケル付加物を形成することができるいずれの化合物も本発明の1以上の方法で用いることができる。例えば、本発明の化合物が、本質的にこれら酵素の1つに対する既知の阻害剤の適切な位置に付加されたマイケル受容体から成るように、結晶構造を用いて本発明の化合物を設計することができる。得られた化合物は可逆的な複合体を前記酵素と形成することができ、共有結合形成はその後で生じるであろう。
したがって、本発明の方法で有用な化合物は、保存された2つのAsp残基の間に位置し、その1つと隣接するATP結合部位内のCys残基を有するタンパク質キナーゼを特異的に阻害し、さらに重要なことには、ATP結合部位内のこの位置にこのCysを欠くタンパク質キナーゼに対してはほとんど阻害活性をもたない。したがって、そのようなものは、特定のタンパク質キナーゼを特異的に阻害するために用いることができ、前記は、ヒトの疾患を治療するために新規で重要な方法を提供する。さらにまた、そのようなタンパク質キナーゼは複数のシグナル伝達経路に存在するので、本発明の方法で有用な化合物は、治療活性のために要求される多経路遮断作用を提供することができる。
【0059】
この重要なCysを含むタンパク質キナーゼには、AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAKが含まれるが、ただしこれらに限定されない。
本発明の方法は、種々の細胞シグナル伝達経路で特異的なタンパク質キナーゼを阻害することによって多シグナル伝達経路阻害を達成することができるRAL又は誘導体を投与することを含む。このタイプの阻害は、グリーベックに関して上記で例示したように所望の効果を達成するために所望され、必要ですらある。また別の例示は、ゲルダマイシン、17-AAG及び17-DMAGのような阻害剤によるHsp90の阻害である。この阻害は、Hsp90の阻害が複数の細胞シグナル伝達経路の複数の配下のタンパク質キナーゼの分解/阻害を生じるので複数の経路に影響を与える。
【0060】
しかしながら、普遍的に多くのキナーゼを阻害することなく、複数のタンパク質キナーゼを特異的に阻害する阻害剤を設計することは困難である。同様に、たとえタンパク質キナーゼを同定したとしても、500を超える他のタンパク質キナーゼのどれを前記阻害剤が阻害するかを予測することは困難である。対照的に、本発明の方法で有用な化合物のコア構造(タンパク質キナーゼ内の重要なCysとマイケル付加物を形成することができるエノン又はα、β-不飽和ケトン部分)は、見事な特異性及び治療結果の改善を提供する。ある実施態様では、本発明のこれらの化合物は、レゾルシル酸ラクトン構造の5−7位にエノン部分を含む。そのような化合物を用いて、全てのキナーゼのうち特定のサブセットを予測的に阻害することができる。本発明の化合物はまた、治療の適応症のために望ましい特定のキナーゼサブセット内でのキナーゼ阻害の均衡を示す特定の阻害剤を選択することができる、コア構造が構造的に改変された複数の化合物を含む。
多タンパク質キナーゼ阻害は、(a)ネットワークの種々の支流を阻害しネットワーク全体を阻害する潜在能力を生じるか、又は(b)ネットワークのただ1つの直線的支流上の種々のキナーゼを阻害するか、又は(c)前記の両方を阻害することができる。これらのタイプの多タンパク質キナーゼ阻害は、ただ1つのキナーゼを阻害する化合物よりも累積的な阻害効果を提供し、協同的な阻害をもたらす潜在能力を有する。ある種のレゾルシル酸ラクトン阻害剤は、いかにして本発明の方法が、どちらかのアプローチ又は両方のアプローチを包含することができるかを説明するために有用である。例えば、これらの阻害剤はERKシグナル伝達経路及びJNKシグナル伝達経路を阻害し、したがって、細胞分裂及び炎症の両方で重要な種々の均衡のとれたシグナル伝達経路に影響を与え、ネットワーク阻害アプローチを例証する。
【0061】
本発明の方法で有用なある種のレゾルシル酸ラクトンはまた、ただ1つの経路で複数の酵素を阻害する(協同的経路阻害アプローチ)。例えば、ある種のレゾルシル酸ラクトン化合物はMEK1/2及びERK1/2を阻害する。本発明のそのような阻害剤及び他の化合物を投与して、たとえ個々のプロテインキナーゼに対するそれらの潜在能力が特に高くなくても、疾患過程で臨床的に相応な阻害を達成することができる。
例えば、阻害剤が、両酵素の活性化(すなわちリン酸化)型に対して等しく強力であると仮定した場合、MEK1/2の50%を阻害するために必要な前記阻害剤濃度は、ERK1/2のリン酸化型(阻害剤が存在しない場合と比較して)の単に50%の生成しかもたらさない。同じ濃度では、前記阻害剤が活性化ERK1/2の50%を同時に阻害するとしたら、前記経路は75%阻害される(経路の協同的阻害)。さらにまた、本発明の方法で有用なある種の化合物は、ERK経路の複数のキナーゼを阻害するだけでなく、VEGFR(前記は活性化されたときERK経路の活性化を引き起こす)もまた阻害する。阻害剤が、3つの酵素の全てに対して同じ潜在能力を有するならば、VEGFRからERK1/2のシグナル伝達経路(抗分裂作用のための阻害剤の標的)は、いずれかの単一酵素を単に50%阻害する濃度で87.5%阻害される。
この多タンパク質キナーゼ阻害は、所望の治療効果を達成するためにPDGFRB、PDGFRA及びKITの阻害を必要とする治療方法に関連して本発明のある実施態様で例示される。これらの標的はグリーベックによって阻害され、グリーベックは、慢性骨髄性単球性白血病及び多形型神経芽細胞腫とともにGIST及び転移性GISTの治療で治療的価値を有する(グリーベックはまたBcr-Ablを阻害し、Bcr-Ablは本発明の方法で有用な化合物とのマイケル付加物形成に感受性ではない)。したがって、本発明の方法で有用な化合物及び医薬組成物はこれら疾患に対して治療適用性を有する。しかしながら、重要なことには、本発明で有用な化合物のタンパク質キナーゼとの結合は、グリーベック耐性を付与するタンパク質キナーゼの変異は本発明の化合物に対する耐性を付与できないというものである。したがって、本発明の方法は、グリーベック耐性症状(グリーベックが投与される癌のグリーベック耐性型を含む)を治療する方法を含む。本発明の方法はまた、以下の節(それぞれ個々の癌又は他の適応症状に焦点を当てる)で考察されるように、他の癌適応症及び疾患を治療する方法を含む。
【0062】
消化管間質腫瘍:
消化管間質腫瘍(GIST)はもっぱら胃(60%)及び小腸(25%)で見出されるが、また低頻度で直腸、食道、及び他の場所にも出現する。GISTは過去にはしばしば誤診され、それらの出現の正確な組織像を得ることは困難である。ほぼ5000の新規症例が毎年合衆国で発生すると概算される(www.orpha.net/data/patho/GB/uk-GIST.pdf)。ほぼ95%のGISTがc-KITについて免疫組織化学的に陽性に染色され、GISTの85%までがc-KITチロシンキナーゼの活性化変異を保有している(Hirota et al. Science 1998; 279(5350):577-80)。さらにまた、c-KITの遺伝性活性化変異を有するいくつかの血縁群が同定された(Nishida et al. Nat Genet 1998; 19(4):323-4)。これらの家族は、多発性良性及び悪性GISTの発症を示す。c-KITについて野生型であることが判明したGISTのうち、ほぼ5%はPDGFRAに変異を保有している(Heinrich et al. Science 2003; 299(5607):708-10)。c-KIT及びPDGFRAチロシンキナーゼの活性化変異は、下流のシグナル伝達経路(MEK1/2及びERK1/2酵素経路を含む)の活性化を伴う。本明細書に記載の、ヒポテマイシン並びにその誘導体及び類似体は、前記レセプターキナーゼc-KIT及びPDGFRと同様に前記連続するERK経路のMEK1/2及びERK1/2の強力な阻害剤であり、本発明の方法にしたがってGISTの治療のために患者に投与することができる。
【0063】
急性骨髄性白血病:
本発明の方法で有用な化合物にはまた、FLT3(急性骨髄性白血病(ALM)でもっとも一般的な分子異常(変異))を阻害する化合物が含まれる。AMLは、成人のもっとも一般的な白血病であるとともに小児のもっとも一般的な癌形態である。2003年に合衆国ではほぼ10,000件の新規症例及び8,000件の死亡がAMLによってもたらされた。ほぼ同じ数の症例がヨーロッパ及びオーストラリアで発生した。いくつかのキナーゼがAMLで役割を演じていると示唆されてきた。AML治療の臨床試験における従来の薬剤の治療標的にはFLT3、c-KIT及びVEGFRが含まれる。FLT3は正常な造血作用で重要な役割を果たす。前記は、AMLの患者の70%〜100%で異常に活性化されるか、又はアップレギュレートされる(Spiekermann et al. Clin Cancer Res 2003; 9(6):2140-50;及びBlood 2003; 101(4):1494-504)。c-KITタンパク質キナーゼはAML患者の60%〜80%で高レベルで見出され、分裂及び抗アポトーシス作用を仲介すると考えられる(Heinrich et al. J Clin Oncol 2002; 20(6):1692-703)。VEGF及びVEGFRは骨髄の血管形成で役割を果たすと示唆されてきた(Aguayo et al. Blood 2000; 96(6):2240-5)。AML患者の骨髄生検によって、VEGF及びVEGFRレベルの変化は毛細血管密度の変化と平行することが示された(Kuzu et al. Leuk Lymphoma 2004; 45(6):1185-90)。VEGFレベルは、AML患者の生存と反比例するようである(Brunner et al. J Hematother Stem Cell Res 2002; 11(1):119-25)。ヒポテマイシンはFLT3、c-KIT、VGFR及びVEGF産生(ERK経路のMEK1/2及びERK1/2の阻害を介する)の強力な阻害剤であり、本発明の方法にしたがって、本明細書に記載のようにヒポテマイシン並びにその誘導体及び類似体をAMLの治療のために患者に投与することができる。
したがって、本発明の方法はAMLを治療する方法を含む。ある実施態様では、これらの方法は、病変組織がAML又は他の癌タイプの指標となるFLT3変異を有する細胞を含むか否かを同定する初期工程を含む。FLT3変異はAMLで発生する(患者のほぼ41%)。これらの変異には、活性化ループ内のAsp835、及びD835->Y又はV又はH又はE又はNが含まれるが、ただしこれらに限定されない(前記変異は公知の方法にしたがって検出することができる)。
【0064】
B-Raf変異を伴う癌:
特異的なB-Raf変異V599E(V600E)は、悪性メラノーマの70%及び結腸癌の約20%に見出される。本発明のある実施態様では、癌患者の腫瘍の生検を実施して、腫瘍細胞が、これらERK経路依存癌に特徴的なB-Raf変異を示すか否かが決定され、B-Raf変異が存在する場合は、続いて本発明の方法で有用な化合物が癌治療のために投与される。
この診断/治療一体化方法(又は“テラノスチック(theranostic)”)の有効性は図5のデータによって部分的に例証される。ヒポテマイシン(本発明のある種の方法で有用なレゾルシル酸ラクトン)を60細胞株のNCIパネルで試験し、その結果のlog GI50値(50%の増殖低下の達成に要求される薬剤量)が図5で棒グラフの形で示される。前記化合物に対してもっとも感受性が強い細胞株は、垂直線の平均活性から右を指す棒線を用いて示される。この結果は、感受生細胞株は、異常なMAPKシグナル伝達経路に含まれるタンパク質キナーゼ(例えばMEK1/2、ERK1/2)とともにB-Raf変異V599E(V600E)を有するB-Raf依存癌に由来し、前記は、これらの変異体キナーゼに重要なCys残基が存在し、さらに可逆的結合及び必須のマイケル付加物形成に必要な構造が存在することから、ヒポテマイシンに対して感受性であることは本明細書での開示から予想され得るとおりである。
表1は、上記で考察した本発明の有用性を支持するデータを表形態で提示する。
【0065】
表1:B-Raf変異癌細胞のキナーゼ阻害剤に対する感受性
表1のデータは、ヒポテマイシンによって例証されるように、変異B-Rafを有する癌細胞株は、マイケル付加物形成に馴染みやすいエノン構造を有するレゾルシル酸ラクトンに特に感受性が高いことを示している。対照的に、野生型B-Rafを有するA549細胞株は、感受性は低いが、ただしその増殖はなお顕著に阻害される。ベンゾピラン-4-オンの骨組みを土台とするMEK阻害剤であるPD98059、及び水素添加されたエノン炭素-炭素二重結合を有し、したがってマイケル反応に参加することができない5,6-ジヒドロヒポテマイシンはともに阻害剤として効果は貧弱である。さらにまた、エノンレゾルシル酸ラクトンは、B-Rafをもつ細胞に対してBayer43-9006(ソフラニブ)よりも活性は顕著に高い。Bayer43-9006は、最初Raf-1阻害剤として開発され、現在はメラノーマに対するヒトの臨床試験中である。同様にエノンレゾルシル酸ラクトンはSU11248(臨床試験で精査されたまた別のキナーゼ阻害剤である)よりもはるかに強力である。
【0066】
B-Raf変異癌細胞株のRALに対する感受性は、B-Raf変異体メラノーマ(A375)異種移植モデルで確認された。図6に示されているように、15mg/kg又は20mg/kgのヒポテマイシンを毎日投与することによって、A375異種移植の増殖は賦形剤単独と比較して顕著に阻害される。さらにまた、両投与量のヒポテマイシンは、25mg/kg又は50mg/kgで1日おきに投与されたBayer43-9006(非RAL、非シスエノンキナーゼ阻害剤)よりも顕著に有効である(前記のBayer43-9006の投与計画は、以前に有効であると報告されたBayer43-9006用の計画である(Sharma et al. Cancer Res 2005; 65(6):2412-2421))。したがって、in vitro及びin vivoの両分析によって、活性化B-Raf変異をもつ癌細胞株は特にRALによる増殖阻害に感受性を有することが示された。
メラノーマの治療で本発明の化合物を使用することは特に有益である。悪性メラノーマのほぼ70%が変異B-Rafを有し、さらにメラノーマは、外科的処置によって治療可能な時期を過ぎて進行した場合、治療は極めて困難である。同様に、本発明の化合物は結腸癌の治療に有用である。結腸癌のほぼ20%が変異B-Rafを有し、ある実施態様では、本発明の方法にしたがい、本発明の化合物による治療に適した患者を特定するために、BRAF変異について生検標本の予備スクリーニングが実施される。したがって、本発明の化合物は、変異B-Raf、特にV599E(従来の命名法ではV600E)及びV599D(従来の命名法ではV600D)を特徴とする細胞の分裂の阻害に有効である。
【0067】
腎細胞癌:
本発明の方法は、腎細胞癌(RCC)を治療する方法を含み、RCCは成人の全ての悪性疾患の約3%を占め、合衆国では毎年約31,000件の新規症例が診断される。サイトカインによる免疫学的療法が従来の標準的治療であるが、ほんの限定的患者群が応答するだけである。RCCの生物学的研究によって、治療標的としてVEGF及びそのレセプター(VEGFR)(血管内皮増殖因子レセプター;vascular endothelial growth factor receptor)が同定された(Rathmell et al. Curr Opin Oncol 2005; 17(3):261-7)。複数の企業(Onyx及びSugenを含む)が、VEGFRをRCCの治療に用いることができるか否かを研究している。そのような化合物は、本発明で有用な化合物よりも一般的には劣っている。なぜならば、それらはレセプターのみを阻害するだけであるが、一方、本発明の化合物はレセプターだけでなくVEGFの産生もまた阻害するからである。
フォン・ヒッペル・リンダウ症候群は家族性異常であり、フォン・ヒッペル・リンダウ(VHL)腫瘍サプレッサーの変異を特徴とする。前記は明細胞RCCに対する感受性増加を伴い、RCC発症の生涯リスクはほぼ50%である。VHLタンパク質は、正常な酸素条件下ではユビキチン依存タンパク質分解のために転写因子HIFαを標的とする。機能的なVHLの非存在下では、HIFαは蓄積して、HIFαの下流の転写標的(VEGF及びPDGFを含む)の構成的発現をもたらす。VHLの不活化はまた、明細胞RCCの散発性症例の60〜80%で発生することが示され、VEGF過剰発現は解析されたRCCサンプルの大半で明示された(Rini et al. J Clin Oncol 2005; 23(5):1028-43)。VEGFを標的とするモノクローナル抗体並びに小分子VEGFR及びPDGFR阻害剤(例えばBayer43-9006)は、RCC臨床試験において進行時間を遅らせることについて有望な結果を示すか、又は顕著な百分率の患者で部分的応答若しくは疾患の安定の証拠を示した(Rini et al. 上掲書)。両増殖因子レセプターVEGFR及びPDGFRの阻害に加えて、本発明の方法で有用なレゾルシル酸ラクトンキナーゼ阻害剤はまた、MEK1/2及びERK1/2の阻害を介して下流のERKシグナル伝達経路の4つの酵素を同時に標的とする(ERKシグナル伝達経路はRCCでは構成的に活性であることが示された(Ahmad et al. Clin Cancer Res 2004; 10(18Pt2):6388S-92S;及びOka et al. Cancer Res 1995; 55(18)4182-7)。なぜならばVEGFはERK経路によって刺激されるので、本発明の方法で有用な阻害剤はまたVEGF産生を低下させる。本明細書で提供されるヒポテマイシン並びにその類似体及び誘導体は、RCCの治療のために本発明の方法にしたがって患者に投与することができる。
【0068】
Ras依存性癌腫:
本発明の方法はRas依存性癌腫を治療する方法を含む。マイトジェン活性化タンパク質キナーゼ(MAPK)シグナル伝達経路又はERK経路は、多くのヒトの腫瘍で細胞の増殖及び生存を調節する(Sebolt-Leopold et al. Nat Rev Cancer 2004; 4(12):937-47)。癌細胞の多くのタイプが、Rasの活性化変異によって引き起こされるMAPKシグナル伝達経路の構成的活性化を示す。これらの変異は、MAPK経路を介するシグナル伝達の増加及び細胞分裂の増加をもたらし、さらにK-Rasにおける変異(結腸癌で45%、膵臓癌で90%及び非小細胞肺癌で35%の陽性率);N-Rasにおける変異(メラノーマで15%、ALL及びAMLで30%の陽性率);及びH-Rasにおける変異(K-Ras及びN-Rasと併せて、甲状腺乳頭癌で60%の陽性率)を含む。Raf阻害剤(例えばBAY 43-9006)又はMEK阻害剤(例えばPD184352)は、活性化Ras変異を保有するヒト腫瘍細胞株で増殖及びMAPK経路の両方を阻害することが示され、さらにマウス腫瘍モデルで、腫瘍増殖を阻害することが示された(Sebolt-Leopold et al. Nat Med 1999; 5(7):810-6;及びSebolt-Leopold, Oncogene 2000; 19(56):6594-9)。ヒポテマイシン並びにその誘導体及び類似体は、カスケードの2つのレベル(MEK1/2及びERK1/2)での阻害を介するMAPKシグナル伝達経路の強力な阻害剤であり、Ras活性化変異を保持する腫瘍の治療のために本発明の方法にしたがって用いることができる。
【0069】
前立腺癌:
本発明の化合物及び方法はまた前立腺癌の治療で有用である。前立腺癌は男性でもっとも普遍的な癌であり、合衆国単独で130万人を超える患者が存在する。2003年には221,000件の前立腺癌の新規症例が発生し、アンドロゲン除去療法に使用にもかかわらず、転移性前立腺癌により29,000人が死亡するであろうと概算された。アンドロゲン除去は進行癌の患者の唯一の有効な療法であり、ほぼ80%の患者がアンドロゲン除去後に症状的及び/又は客観的応答を達成する。しかしながら、最終的にはアンドロゲン非依存性への進行がほぼ全ての患者で発生する。複数の非ホルモン性薬剤がホルモン不応性前立腺癌をもつ患者で判定されたが、これらの薬剤は限定的な抗腫瘍活性を有し、客観的応答率は20%であり、いずれも生存効果を示さなかった。前立腺癌の進行を仲介する分子標的の同定及び選択的阻害は、この疾患の将来の治療に多大な影響を与えるであろう。
マイトジェン活性化タンパク質キナーゼ(MAPK)活性の増加は、前立腺癌のヒトでの症状の進行と相関性を有する(Gioeli et al. Cancer Res 1999; 59:279-84)。これらの結果は、Ras活性が異種移植における前立腺腫瘍増殖のアンドロゲン要求性を調節するという観察と併せて、MAPK経路が前立腺癌の分裂に重要な役割を果たすということを示している(Bakin et al. Cancer Res 2003; 63:1981-9;Bakin et al. Cancer Res 2003; 63:1975-80)。セリン/スレオニンタンパク質キナーゼファミリー、p90リボソームS6キナーゼ(RSK)はMAPKの下流エフェクターとして機能する。RSKファミリーは4つのアイソフォームから成り、それらは別個の遺伝子の生成物である。RSKは、重要な基質(いくつかの転写因子及びキナーゼ、サイクリン依存キナーゼ阻害剤、p27Kip1、腫瘍サプレッサー、チューブリン及び前アポトーシスタンパク質、Badを含む)をリン酸化し、その活性を調節するというそれらの能力を介して体細胞における細胞生存及び分裂で重要な役割を果たす。これらの観察は前立腺癌におけるMAPKの公知の重要性とあいまって、RSKはまた前立腺癌の進行にも寄与することを示している。
ヒト前立腺癌株LNCaPにおけるRSLアイソフォーム2(RSK2)レベルの増加は前立腺特異的抗原(PSA)の発現を強化し、一方、RSK阻害剤、3Ac-SL0101を用いたRSK活性の阻害はPSA発現を低下させることが最近示された(Clark et al. Cancer Res 2005; 65(8):3108-16)。RSKレベルは、正常な前立腺組織と比較してヒトの前立腺癌の約50%で高く、RSKレベルの増加は、前立腺癌で生じるPSA発現の上昇に関与することを示している。さらにまた、3Ac-SL0101は、LNCaP株及びアンドロゲン非依存性ヒト前立腺癌株PC-3の分裂を阻害する。これらの結果は、いくらかの前立腺癌細胞の分裂はRSK活性に依存し、RSKは前立腺癌の重要な化学療法標的であることを示している。
ヒポテマイシン並びにその誘導体及び類似体は、ERK経路の2つの重要な点及びRSKアイソフォームのC-末端キナーゼドメインを強力に阻害する。したがって、本発明のマイケル付加物形成RALは、前立腺癌及び転移性前立腺癌の単一療法及びアンドロゲン除去療法との併用による治療で、本発明の方法にしたがって有用である。
【0070】
乳癌:
本発明の方法及び化合物はまた乳癌の治療で有用である。2003年の女性の乳癌症例は210,000件と概算され、40,000人が死亡し、有病率がもっとも高い癌形態となっている。乳癌は、エストロゲンレセプター-α(ERα)陽性又はERα陰性として存在する。ERαの存在は、全快生存及び全体的生存の増加の両観点で良好な予後と相関する。ERα陰性乳癌は、増殖因子レセプター(例えばEGFR及びerbB-2(HER2))を過剰発現する傾向がある。Raf-1は、これらレセプターのシグナルトランスダクション経路における主要な中間体である。高レベルの構成的Rafキナーゼ又は下流のMAPキナーゼ活性は、ERα陽性乳癌細胞に、エストロゲンの非存在下で増殖する能力を付与し、ERα陰性表現型と類似する。MEK阻害剤を用いる治療によるRafシグナル伝達の除去は、ERα陽性態様を回復させることができる(Oh et al. Mol Endocrinol 2001; 15(8):1344-59)。抗エストロゲン(例えばタモキシフェン)による治療が、細胞周期停止及びアポトーシス誘導によってERα癌細胞の増殖を阻害するために一般的に用いられる。前記は、細胞周期阻害剤、p27Kip1の作用を必要とする。ERα陽性細胞でのMAPKシグナル伝達経路の構成的活性化は、p27リン酸化及び残りのp27のcdk2阻害活性を低下させ、前記は一緒になって抗エストロゲン耐性を促進する(Donovan et al. J Biol Chem 2001; 276(44):40888-95)。パクリタキセル、ドキソルビシン及び5-フルオロウラシルのような細胞傷害性薬剤に対する耐性は、部分的にはRas-シグナル伝達(Rafの上流エフェクター)によって仲介される。MEKキナーゼ阻害剤を用いる治療によるRas/Rafシグナル伝達の阻害は、顕著な程度で前記耐性と対抗する(Jin et al. Br J Cancer 2003; 89(1):185-91)。これらの事実は、乳癌の治療でシグナルトランスダクション阻害剤を使用することを正当化し(Nahta et al. Curr Med Chem Anti-Canc Agents 2003; 3(3):201-16)、前記は、MEK及びEGFR阻害剤の二重使用は、どちらかの薬剤の単独使用よりも乳癌細胞の顕著に強い増殖阻害及びアポトーシスをもたらすという報告によって強調される(Lev et al. Br J Cancer 2004; 91(4):795-802)。さらにまた、EGFR及びHER2(乳癌のための標的として立証済み)は、ERK経路を介してそれらの分裂活性を伝達する。最後に、モノクローナル抗体アバスチンによるVEGF作用の阻害は、化学療法に対する乳癌の応答率を劇的に改善した。本明細書に開示したようにマイケル付加物を形成することができるヒポテマイシン並びにその誘導体及び類似体は、ERK経路の4つの酵素(MEK1/2及びERK1/2)とその後に続くVEGF産生(VEGFRも同様に)の強力な阻害剤であり、乳癌の治療のために本発明の方法にしたがって用いることができる。
【0071】
膵臓癌:
本発明の方法はまた膵臓癌を治療する方法を含む。膵臓癌は100,000人に約10症例の発症率しかもたないが、前記は西欧では4番目〜5番目の癌関連死因である。新規に診断される患者の大半は既に切除不能の腫瘍病期にいる。これら患者の5年生存率は1%未満であり、平均生存期間は腫瘍検出後ほぼ5〜6ヶ月である。近年では、ヒト腫瘍の病理発生における増殖因子の役割にますます関心が向けられている。ヒトの膵臓癌は、複数の重要なチロシンキナーゼ増殖因子レセプター及びそれらのリガンド(例えば表皮増殖因子(EGF)、線維芽細胞増殖因子(FGF)、インスリン様増殖因子(IGF-1)、血管内皮増殖因子(VEGF)及び血小板由来増殖因子(PDGF)ファミリーに属するもの)を過剰発現する(Korc, Surg Oncol Clin N Am 1998; 7(1):25-41;Ozawa et al. Teratog Mutagen 2001; 21(1):27-44;及びEbert et al. Int J Cancer 1995; 62(5):529-35)。これらの増殖因子は、オートクライン及び/又はパラクライン的態様で作用し、ERK経路の活性化を介して膵臓癌の増殖を刺激すると考えられている。K-Rasオンコジーンの変異は膵臓癌では75〜90%の頻度で生じ(Li, Cancer J 2001; 7(4):259-65)、前記はこの癌の活発な増殖を強調している。レセプターチロシンキナーゼ及び下流のシグナル伝達キナーゼ(MEK及びp38)に対する小分子阻害剤は、膵臓癌培養細胞の分裂を阻止することが報告された(Matsuda et al. Cancer Res 2002; 62(19):5611-7;及びDing et al. Biochem Biophys Res Commun 2001; 282(2):447-53)。ヒポテマイシン並びにその誘導体及び類似体は、PDGFR、VEGFR、MEK及びERKキナーゼの強力な阻害剤であるとともに、変異K-rasによる過剰なマイトジェンシグナル伝達の阻害剤であり、膵臓癌の治療で本発明の方法にしたがって用いることができる。
【0072】
上皮性卵巣癌:
本発明の化合物及び方法はまた卵巣がんの治療で有用である。上皮性卵巣癌(EOC)は婦人科の悪性腫瘍の中で主要な死亡原因であり、女性の癌関連死の5番目に主要な原因である。2003年には、24,000件の新規な症例が発生し、14,000人が死亡すると予想される。ほとんどの患者は卵巣腫瘍の進行期にあり、治療は徹底的な外科手術とそれに続く化学療法を土台にしている。化学療法計画の骨格は、相変わらず、近年タキサンが加えられた白金誘導体である。MAPKシグナル伝達経路(特にERK1/2セリン-スレオニンキナーゼ)が卵巣癌において主要な役割を果たす(Choi et al. Reprod Biol Endocrinol 2003; 1(1):71)。この経路は、卵巣癌の治療に一般的に用いられる白金含有-又はタキサン系-化学療法薬剤(例えばシスプラチン、カルバプラチン、ドセタキセル及びパクリタキセル)、並びにゴナドトロピン及び卵胞細胞刺激ホルモンによって活性化される。薬剤耐性細胞は、MEK1/2阻害剤による治療によって薬剤感受性細胞に回復させることができる。したがって、ある実施態様では、本発明は卵巣癌を治療する方法を提供し、前記方法は、MEK1/2及びERK1/2タンパク質キナーゼとマイケル付加物を形成することができるタンパク質キナーゼ阻害剤を、白金含有抗癌剤又はタキサンと一緒に又はその投与後に投与することを含む。
卵巣癌細胞の転移はERK経路阻害剤による治療によって抑制することができる。約39%の卵巣腫瘍がPDGFRを発現し、したがって活性なERK経路及びその発現レベルは、卵巣腫瘍のより進んだ組織学的等級及び進行した外科的病期と相関する。さらにまた、PDGFR-A陽性腫瘍をもつ患者は、陰性腫瘍を有する患者よりその生存時間は短い。イマチニブ(グリーベック)は、PDGFR-Aの阻害に依存するメカニズムを介して、卵巣癌細胞の増殖を臨床的に相応な濃度で阻害する(Matei et al. Clin Cancer Res 2004; 10(2):681-90)。腹腔内播種は卵巣癌の進行にとって決定的である。肝細胞増殖因子は、Ras/Raf/MEK/ERKシグナル伝達経路の活性化によって、卵巣癌細胞の遊走および侵襲を誘導し(Ueoka et al. Br J Cancer 2000; 82(4):891-9)、前記は、この疾患の治療のために本発明によって提供されるMEK及びERK阻害剤を使用することを支持する。ヒポテマイシン並びにその誘導体及び類似体は、PDGFRと同様に、PDGFRが活性化する下流の酵素の強力な共有結合阻害剤であり、卵巣癌の治療で本発明の方法にしたがって用いることができる。
【0073】
肺癌:
本発明の方法はまた肺癌を治療する方法を含む。肺癌は合衆国では癌死亡率の主要原因である。2003年の調査では、171,000件の新規症例の発生が予想され、その年には157,000人が死亡した。最近の治療の進歩にもかかわらず、局所的に進行した転移症例についての成果はなお貧弱である。合衆国では非小細胞肺癌(NSCLC)は全ての肺癌の75%を超える。化学療法は、この疾患の進行期の管理に重要な役割を果たす。従来の薬剤には白金を土台にする併用療法及び第二の治療のためのドセタキセルが含まれる。EGFRは、肺を含むほとんどの上皮性腫瘍で発現されるか又は過剰発現される。NSCLC扁平上皮癌は80%の過剰発現を示す。
ゲフチニブ(イレッサ)が他の化学療法が有効でなかった患者でのNSCLCの治療用に合衆国で承認されたが、EGFR由来シグナル伝達におけるMAPK経路の関与は、他の標的がこの困難な癌の治療に利用することができることを示している。VEGFR-2(KDR)及びVEGFR-3(Flt-4)はNSCLCで発現され(Tanno et al. Lung Cancer 2004; 46(1):11-9)、それらのリガンド量の増加及び低酸素状態は、培養NSCLC癌細胞タイプの分裂及び遊走を刺激した。KDR及びFlt-4の刺激はまた、MAPK経路の活性の強化をもたらした。同様に、NSCLCをもつ患者由来の組織サンプルの34%はERK経路の高活性化を示した(Vicent et al. Br J Cancer 2004; 90(5):1047-52)。ERK2及びAkt(EGFRによって制御される2つのシグナル伝達キナーゼ)のリン酸化状態とゲフチニブ療法との間の強い相関関係もまた報告されている(Cappuzzo et al. J Natl Cancer Inst 2004; 96(15):1133-41)。
これらの観察及び他の最近の臨床的観察(Cesario et al. Curr Med Chem Anti-Canc Agents 2004; 4(3):231-45)は、NSCLCの治療でシグナル伝達タンパク質キナーゼの阻害剤を拡大的に使用することを正当化する(前記治療では、トポイソメラーゼ阻害剤(Maulik et al. J Environ Pathol Toxicol Oncol 2004; 23(4)237-51)及び確立された他のタイプの癌治療薬を用いる併用療法が含まれる)。最後に、モノクローナル抗体アバスチンによるVEGF作用の阻害は、パクリタキセル及びカルボプラチンによる化学療法に対するNSCLC癌の応答率における劇的な改善をもたらした。本明細書で提供するヒポテマイシン並びにその誘導体及び類似体は、ERK経路の4つの酵素(MEK1/2及びERK1/2、前記はその後に続くVEGF産生を調節する)と同様、肺癌で重要であることが示されたレセプターキナーゼKDR(VEGFR)、Flt-4及びcKITの強力な阻害剤であり、肺癌の治療のために単一療法及び併用療法で本発明の方法にしたがって用いることができる。
【0074】
結直腸癌:
結直腸癌は合衆国では癌の第二の主要な死因であり、ヒトの悪性疾患の約15%を占める。アメリカ癌学会は、2003年にはほぼ150,000件の結直腸癌の新規症例が診断されるであろうと概算した(Jemal et al. CA Cancer J Clin 2003, 53:5-26)。進行した結直腸癌をもつ患者の大半は、最終的には癌の再発を経験し、結直腸癌は治癒不能と考えられる。標準的な治療は外科的切除及び時に放射線治療を必要とし、一方、化学療法(例えば標準的なカンプトサール(Camptosar(商標))(イリノテカンHCIの注射)/5フルオロウラシル/ロイコボリン投与計画)は満足というには程遠い。
疫学的及び遺伝子マッピング研究によって、多くのタイプの結腸癌は細胞シグナル伝達経路の異常を伴うことが示された。例えば、MAPK経路でB-Raf V599E(V600E)変異が〜15%の結腸癌で見出され、細胞分裂に必要なERK経路の構成的活性化に至る(Sebolt-Leopold et al. Nat Rev Cancer 2004, 4:937-47)。MAPKシグナル伝達の特異的阻害剤はしたがって、Raf V599E(V600E)変異をもつ細胞の分裂の阻害に有効である(Sebolt-Leopold et al. 上掲書;同書、Nat Med 1999, 5:810-6)下記の実施例5に示すように、B-Raf V599E(V600E)細胞株COLO829のERK経路は、マイクロモル濃度以下でMEK1/2及びERK1/2阻害剤のヒポテマイシンに曝露後10分で完全に遮断される。同様な結果が、B-Raf V599E変異結腸癌細胞株HT29で観察される。CI-1040、PD0325901及びARRY-142886のような有効性がより低いMEK1/2阻害剤は結腸癌の動物モデルで有効である(Sebolt-Leopold et al.上掲書)。
結腸癌の転移はマトリックスメタロプロテアーゼ(MMP)の分泌を必要とし、MEK1/2阻害剤は結腸癌細胞でMMP-7遺伝子の発現を遮断することができ(Lynch et al. Int J Oncol 2004, 24:1565-72)、ERK1/2阻害剤はまたこの特性を有する(なぜならばERK2は、結腸癌細胞によるインテグリンα(V)β6仲介MMP-9発現に中心的に関与するからである)(Gu et al. Br J Cancer 2002, 87:348-51)。ERK及び/又はp38依存MAPKシグナル伝達経路の特異的阻害剤はまた、他の状況における結腸癌の治療に、すなわち非ステロイド系抗炎症薬の結腸癌細胞のアポトーシス刺激能力の強化に(Nishihara et al. J Biol Chem 2004, 279:26176-83;Sun and Sinicrope, Mol Cancer Ther 2005, 4:51-9)、CCK-2レセプター仲介プロスタグランジンE2産生刺激による結腸癌細胞の増殖を促進するガストリン-17の能力の阻害に((Colucci et al. Br. J. Pharmacol 2005, 144:338-48)、及びTNFレセプター関連因子(TRAF1)誘導(前記はNFkB経路を介する結腸癌の腫瘍促進の一局面である)の阻害に(Wang et al. Oncogene 2004, 23:1885-95)本発明の方法にしたがって有用である。
VEGFレセプターの刺激は血管形成を強化することができる。細胞から遊離されるVEGFと結合し、VEGFの作用を阻害するアバスタチンのようなモノクローナル抗体は、非常に有効で、転移性結腸癌の治療のために2004年に承認された。最近の臨床試験の結果は、初期療法として一般的化学療法計画である5-フルオロウラシル/ロイコボリンへのアバスタチンの添加は進行結腸癌の進行停止生存を改善することを示している(http://patient.cancerconsultants.mom/colon_cancer_news.aspx?id=17462)。以前の臨床試験では、この疾患の治療で、カンプトサール(Camptosar(商標))/5フルオロウラシル/ロイコボリンへのアバスタチンの添加の化学療法計画による利点が示された。ニューロピリン-1はヒト結腸癌細胞のVEGFコレセプターであり、前記の生成(したがって血管形成及び細胞増殖を刺激する能力)はまたERK1/2及びp38阻害剤によって阻害され得ることが示された(Parikh et al. Am J Pathol 2004, 164:2139-51)。
本発明の方法で有用なレゾルシル酸ラクトンは、BRAF V599E変異を有する結腸癌の治療に、前記変異をもたない結腸癌の治療と同様、特に有用である。全ての細胞に存在するMEK1/2及びERK1/2でERK経路の2点阻害に加えて、前記は、VEGF産生(ERK経路の阻害を介する)を、VEGFR同様に阻害し、さらにTAK1を阻害してNFkB経路を阻害する。
【0075】
基底細胞癌及び他のソニック・ヘッジホッグ経路関連癌:
本発明の方法はまた基底細胞癌及び他の活性化ヘッジホッグ(Hh)経路関連癌を治療する方法を含む。Hh-シグナル伝達経路は3つの主要な成分を含む:1)Hhリガンド;2)負の調節因子パッチド(Ptch)及びアクチベーターであるスムーザンド(Smoothend)(Smo)で構成される膜貫通型レセプターサーキット;及び3)最後にキュビツス・インタラプツス(Cubitus interruptus)(Ci)を調節する細胞質複合体又は転写エフェクターのGliファミリー(Frank-Kamenetsky et al. J Biol 2002, 1:10)。Gli1のような転写レベルでの正及び負のフィードバックが存在し、Ptch1遺伝子は前記経路の活性化の直接的転写標的である。Hhリガンドはおよそ45kDaの前駆体として合成され、前記前駆体は自己プロセッシングを経て、前記前駆体のアミノ末端半分へのコレステロール部分の共有結合付加がもたらされる。Smoは、Gタンパク質共役レセプター(GPCR)と相同性をもつ7回膜貫通型タンパク質であり、一方、Ptch1は12回膜貫通型タンパク質で、チャネル又はトランスポーターに類似する。極めて重要な経路阻害剤としてのその役割と合致して、Ptch1の除去は、Hhリガンドとは別個に機能する構成的に活性なHh経路を生じる。同様に、Smoの膜貫通型ヘリックスの特異的な点変異は、Ptch1阻害を効果的に迂回しながら、前記経路を構成的に刺激することができる。
動物の適切な発育にとって必須であるが、一方、変異によるか、又はPtch1を不活化するか若しくはSmoを活性化する他の事象による不適切なヘッジホッグ経路シグナル伝達は、いくつかのタイプの腫瘍(基底細胞癌、髄芽細胞腫、横紋筋肉腫、グリオーマ、表層膀胱癌、胃腸管腫瘍、小細胞肺癌(SCLC)、膵臓癌、及び前立腺癌を含む)を生じる(di Magliano and Hebrok, Nat Rev Cancer 2003, 3:901-11;Ruiz et al. Nat Rev Cancer 2002, 2:361-72;Fan et al. Nat Rev Cancer 2002, 2:361-72;Fan et al. Endocrinology 2004, 145:3961-70;Sanchez et al. Proc Natl Acad Sci USA 2004, 101:12561-6)。したがって、Hhシグナル伝達の阻害剤は、抗癌剤の開発のための重要な先導物質を提供する(Romer et al. Cancer Res 2005, 65:4975-8;Taipale et al. Nature 2000, 406:1005-9;Williams, Drug News Perspect 2003, 16:657-62)。
Hh応答細胞株C3H10T1/2を用いて、Gli1は血清応答成分を誘導し、さらにPDGFRを活性化することが示された(前記PDGFRは順を追ってRas-ERK経路を活性化し、細胞分裂を刺激する(Xie et al. Proc Natl Acad Sci USA, 2001, 98:9255-9289)。したがって、PDGFR又はERK経路の阻害はHh経路活性化作用の遮断を提供し、Hh活性化のメカニズムにもかかわらずHh経路の終点をもたらす(すなわち刺激又は阻害の解除)。
基底細胞癌(BCC)はもっとも一般的なヒトの癌であり、合衆国では毎年750,000件の新規症例がある。パッチド遺伝子(Ptch1又は2)の変異は、遺伝性異常である基底細胞母斑症候群に、散発性BCCと同様付随することは既に確立されている。下流分子Gli1は前記経路の生物学的作用を仲介し、前記は約90%のBCCでアップレギュレートする。Gil1は、続いてPDGFRαをアップレギュレートし、前記は細胞分裂を誘導するERK経路の活性化を引き起こす。その後のERK経路の活性化によるPDGFRαの過剰産生は重要なメカニズムであり、前記メカニズムによってヘッジホッグ経路の変異がBCCを惹起する(Xie et al. Proc Natl Acad Sci USA 2001, 98:9255-9)。
腫瘍内IFNαはBCCの治療には不便であるが有効であり、回復率は約50〜80%である。イミキモド(サイトカイン、例えばIFNαの分泌を刺激する)もまた有効である。最近、IFNαによって仲介されるヘッジホッグ経路活性化BCC細胞の殺細胞は、そのERK経路との干渉からもたらされることが示された(前記干渉はFas発現を上昇させ続いてアポトーシスをもたらす)(Li et al. Oncogene 2004, 23:1608-17)。
上記の考察は、PDGFR又はERK経路の阻害はHh経路活性化作用の遮断を提供し、Hh活性化のメカニズムにもかかわらずHh経路の終点をもたらすことを示している。本明細書で開示するヒポテマイシン並びにその誘導体及び類似体は、PDGFRα及びERK経路の2つの酵素の強力な阻害剤である。下記表4に示すように、それらは、培養BCC細胞の強力な阻害剤であり、BCC及び活性化ヘッジホッグ経路によって惹起される他の腫瘍の治療のため本発明の方法にしたがって用いることができる。したがって、ヒポテマイシンは、培養BCC細胞株ASZ001に対して約100nMのIC50を有する(表4)。比較すれば、タザロテン(Ptc±マウス(So et al. Cancer Res 2004, 64, 4385-9)で85%を超えるBCC発症阻害をもたらす局所用アセチレンレチノイドでありBCCの治療に臨床で用いられる)は、ASZ001 BCC細胞をおよそ10,000nMのIC50で阻害する。
【0076】
再狭窄:
本発明の化合物及び方法はまた、それらは再狭窄を予防することができるという点で血管形成術及びステントの使用で有用である。平滑筋細胞分裂は、血管形成術後の新生内膜形成における重要な事象である。PDGFは、損傷に対する血管平滑筋細胞の応答に必要な有糸分裂促進因子であり、平滑筋細胞でERK経路を活性化し、前記は遊走に必須である。MEK阻害剤は、血管平滑筋細胞の分裂及び遊走を妨げるために有効な薬理学的物質である。なぜならば、それらはERK活性化を遮断し、それによってPDGFに対する細胞性応答を遮断するからである。ストレス活性化MAPK p38はまた、血管損傷に対する応答に必要とされ、p38及びその活性を調節する上流のキナーゼを標的とする阻害剤は再狭窄の治療で有効である。PDGFレセプターは平滑筋細胞の遊走及び分裂を刺激し、VEGFレセプターは新生血管形成を刺激する。本発明の方法で有用な化合物は、PDGFR及びVEGFRとともにERK及びJNK経路の複数のキナーゼを阻害するので、それらは強力な再狭窄の阻害剤であり、したがってステント及び有害な平滑筋細胞遊走を刺激する他の装置の調製で一般的に有用である。
したがって、ある実施態様では、本発明は、ステント又はin vivoでの使用を意図する他の装置を提供し、前記は、望ましくない平滑筋細胞の分裂及び前記ステントへの遊走を妨げるか又は遅らせる、本発明の方法で有用な化合物で被覆されているか、前記化合物とともに包埋されるか、そうでなければ前記化合物を含む。平滑筋細胞のこれらステントへの無制御な遊走は、本発明の方法にしたがって治療可能な症状を生じる。したがって、本発明によって提供されるステントは、再狭窄にとって極めて重要な複数のレセプター及び細胞シグナル伝達経路の強力で不可逆的な阻害剤を含むので、従来の技術を超える顕著な進歩を提供する。ある実施態様では、ステントを調製するために用いられるRALは、ヒポテマイシン又はUS2004/0243224 A1(Tremble, 2004)に開示されたRAL以外の本発明の方法で有用なRALである。
【0077】
慢性関節リウマチ:
慢性関節リウマチ(RA)は、合衆国では1,000,000人を超える人々が罹患している結合組織疾患である。この自己免疫異常は、罹患関節への活性化免疫細胞(T及びB細胞)及びマクロファージの補充によってもっぱら引き起こされる。前記関節で、これら細胞によって産生されるサイトカインIL-1及びTNF-αがRAで観察される不可逆的な関節の破壊を仲介する。これらサイトカインによって活性化される下流の遺伝子(NFkB及びMAPKシグナル伝達経路によって誘導されるNFkB及びAP-1転写因子を介する)は、炎症性分子及びマトリックスメタロプロテイナーゼ(MMP)ファミリーの分泌プロテイナーゼの両者をコードする(前記はRAで高レベルで見出される)。サイトカイン誘導性MMP遺伝子発現を阻害し、さらにまたNFkB及びMAPKシグナル伝達経路を遮断することができる化合物は新規な関節炎薬を提供することができる(Vincenti and Brinckerhoff, J Clin. Invest. 2001, 108:181)。IL-1はMEKKK TAK1の活性化を誘導する。TAK1は、NFkBの活性化、さらにJNKを介してAP-1の活性化を制御し(Ninomiya-Tsuji et al. Nature 1999, 398:252)、したがって特異的なTAK1阻害剤は、NFkB、p38及びJNK経路のIL-1誘導性活性化を遮断することによって炎症を予防することができる。JNK及び炎症細胞(RA細胞を含む)に多いp38アイソフォームの特異的阻害剤は、培養細胞でJNK及びp38経路によって制御される遺伝子の発現を効果的に阻止し、動物でコラゲナーゼ遺伝子発現及び関節破壊の低下を示す。MEK1/2阻害剤はまた、培養細胞でIL-1刺激応答を効果的に遮断する(Barchowsky et al. Cyrokine 2000, 12:1469)。
ある実施態様では、本発明は、p38経路を阻害するためにTAK1及びMEK3/6と、JNK経路を阻害するためにTAK1及びMEK4/7と、さらにERK経路を阻害するためにMEK1/2及びERK1/2とマイケル付加物を形成することができる阻害剤を用いてRAを治療する方法を提供する。この広範囲の連続的及びネットワーク的阻害を介して、NFkB及びAP-1依存シグナル伝達経路は効果的に阻害され、前記疾患は治療される。
【0078】
乾癬:
本発明の方法で有用な化合物による乾癬の治療は、連続多シグナル伝達経路阻害アプローチの威力を例証する。世界中で1千万人を超える人々が乾癬に苦しみ、多くの治療が存在するが長期にわたって有効なものは少なく、完治方法は開発されていない(Geilen and Orfanos, Clin Exp Rheumatol 2002, 20(6 Suppl 28):S81-7;Gniadecki et al. Acta Derm Venereol 2002, 82(6):401-10)。
乾癬は、表皮の過剰分裂、分化障害、炎症及び過剰な表皮血管形成を特徴とする遺伝性皮膚疾患群である。乾癬の発生病理は、免疫学的メカニズム、不完全な増殖制御メカニズム又は前記メカニズムの組合せを土台にしている。表皮の過剰分裂、異常なケラチン化、血管形成及び炎症は乾癬斑の確立された特徴であり、前記乾癬斑は一般的には関節、四肢及び頭皮に生じるが、ただし身体のいずれの場所にも出現し得る。
免疫抑制剤及び抗炎症剤はしばしば乾癬の治療に用いられる。前記薬剤の使用は、乾癬の病因論で重要であると考えられている、自己免疫応答でのT細胞の関与(直接的作用、又は種々のケモカイン及びサイトカイン(Erk経路の活性化を介してケラチノサイトに過剰増殖シグナルを伝えるTNFαを含む)の放出を介する間接的作用による関与)を根拠にしている(Bowcock et al. Hum Mol Genet 2001, 10(17):1793-805)。インテグリン及び他の粘着分子もまた中心的に関与する。トランスジェニックマウスを用いた研究によって、インテグリンの過剰発現はMAPKシグナル伝達経路(ERK経路)を活性化してケラチノサイトの増殖速度を高め、さらに乾癬の組織学的特徴を再生させることが示された。さらにまた、特にIL-1αレベルの上昇下でのMEK1の構成的活性化は、多くの乾癬の特徴を有する過剰分裂性及び炎症性皮膚病巣を生じるために充分である。最近、タンパク質キナーゼSTAT3が乾癬に必須であり、この酵素の阻害が症状の緩和に有効であることが示された(Sano et al. Nat Med 2005, 11(1):43-49)。
乾癬の治療のために本発明の方法で有用な化合物は、MEK1、ERK1/2、VEGFR、PDGFR、JNK(インテグリン)経路のMEK4/7並びにp38ストレス経路のTAK1及びMEK3/6を含むキナーゼサブセットを阻害する。上記に記載したように、乾癬の細胞分裂は活性なERK経路と密接に関連し、VEGFが乾癬の皮膚病層で高レベルで見出される。本発明の方法で有用な化合物は乾癬の多くの特徴に影響を与える。すなわち、それらは、ERK経路の阻害を介して細胞分裂を阻害する。それらは、VEGFRを阻害することによって、さらにERKの阻害を介してVEGF及びSTAT3の産生を阻害することによって血管形成を阻害する。それらはEGFRを直接阻害しないが、それらは、EGFRと細胞分裂とを結びつけるものとして機能するERK経路を阻害し、さらにそれらは、p38ストレス経路の二重阻害(TAK1及びMEK3/6)を提供する。最後に、3つのシグナル経路の統合は、T細胞によるサイトカインの分泌及びそれに続く以下のエフェクター機能の獲得をもたらす:(i)カルシニューリンの活性化、(ii)ERK経路の活性化、及び(iii)JNK経路の活性化。本発明の方法で有用な化合物は、MEK及びERKをJNK経路と同様に阻害し、したがってT細胞活性化に必要な3つの経路のうちの2つを阻害する。したがって、本発明のRALは乾癬の生物学的特徴の原因となる経路の各々で標的を阻害し、乾癬を治療する本発明の方法は、この疾患の治療で実質的な有望性を提供する。
【0079】
炎症性腸疾患:
本発明の方法はまた、本明細書に記載のマイケル付加物形成タンパク質キナーゼ阻害剤の治療的に有効な用量を投与することによって、炎症性腸疾患(IBD)(クローン病及び潰瘍性大腸炎を含む)を治療する方法を提供する。前記疾患は、腹痛及び慢性の下痢をもたらす胃腸管の慢性再発性炎症を特徴とする、病因が不明な疾患である。それらは、遺伝的、環境的、及び免疫学的因子が相互に影響し合って惹起される他因子性疾患である。IBD(特にクローン病)のためのいくつかの治療選択肢が、特定のシグナルトランスダクションエレメントの阻害を基にして開発された。
例えば、モノクローナル抗TNF-α抗体(インフリキシマブ)による中心的前炎症サイトカイン、腫瘍壊死因子-α(TNF-α)の特異的阻害が、ステロイド耐性クローン病治療の本流となった。炎症性シグナルトランスダクションにおけるそれらの重要性のために、MAPK経路は、急性及び慢性炎症抑制のための標的である。複数のMAPK経路が、IBDの病因と密接に結びつく炎症応答を統制する。例えば、ERK1/2、p38、JNK/SAPKタンパク質キナーゼ及びそれらと密接に関係するシグナル伝達経路は、クローン病で顕著に活性化されることが知られている。これらの経路のタンパク質キナーゼ及びそれらの活性を調節する上流のキナーゼの阻害剤による治療は、IBDの臨床治療で有効である。ある実施態様では、本発明は、炎症及び炎症性疾患(IBDを含む)を、これらの経路で複数の酵素とマイケル付加物を形成することができるレゾルシル酸ラクトンを用いて治療する方法を提供する。本発明は、ERK経路の2ヶ所(MEK1/2及びERK1/2)、JNK/SAPK経路の1ヶ所(MEK4/7)及びp38経路の2ヶ所(TAK1及びMEK3/6)の強力な阻害剤を治療を要する患者に投与する、これら疾患の治療方法を提供する。
【0080】
肥満細胞症:
本発明の方法はまた、肥満細胞症(過剰な肥満細胞を伴う分裂増殖性異常)を治療する方法を含む。2つの主要な形態は皮膚型(肥満細胞が皮膚に蓄積する)及び全身型(肥満細胞は多くの異なる組織に蓄積し得る)である(www.niai.nih.gov/factsheets/masto.htm)。前記の両形態は、この疾患のより攻撃的な形態、悪性肥満細胞症に進行し、悪性肥満細胞症は続いて白血病の一形態に進行し得る(Longley, Cutis 1999, 64(4):281-2及びLongkey et al. Nat Genet 1996, 12(3):312-4)。従来の肥満細胞症治療は症状の緩和に焦点をあて、この症状に対する利用可能な治療薬は現在のところ存在しない。
cKITタンパク質は、肥満細胞増殖因子の存在下で活性化される肥満細胞膜貫通型レセプターチロシンキナーゼであり、ERK経路の活性化を介して肥満細胞の分裂を刺激する。構成的に活性なcKITの発現をもたらすcKITの変異(通常D816V)が、全身性及び皮膚性肥満細胞症で観察された(Longley et al. Proc Natl Acad Sci USA 1999, 96(4):1609-14)。前記疾患のこの形態はイマチニブに対して耐性を示す(グリーベック;Ma et al. J Invest. Dekmarol 1999, 112(2):165-70)(イマチニブはヒトの医薬としての使用が承認された最初のキナーゼ阻害剤である)。本明細書に記載のヒポテマイシン並びにその誘導体及び類似体は、野生型KIT及び構成的に活性なKIT(D816V)とともにERK経路の2ヶ所(MEK1/2及びERK1/2)の強力な阻害剤であり、肥満細胞症療法として本発明の方法にしたがって患者に投与することができる。in vitro試験によって、肥満細胞腫細胞株はヒポテマイシンに感受性であることが示された。構成的に活性なcKIT(D814Y、前記はヒトのD816V変異に対応する)を発現する、マウスの肥満細胞腫細胞株p815を用いたとき、ヒポテマイシンは310nMのGI50を示し、一方、他の既知のcKIT 阻害剤、BAY 43-9006及びSU11248は、それぞれ310nM及び320nMのGI50を示す。
【0081】
肥満細胞成分を有する炎症性疾患:
本発明の方法で有用な化合物はまた肥満細胞関連炎症性疾患の治療のために投与することができる。肥満細胞はまた、本発明の方法及び化合物で治療することができる他の疾患及び症状の発達に必要とされる。肥満細胞は、IgEのためのそれらの表面レセプター(FcqRI)の架橋を介してアレルギー反応の発達に必要である(前記架橋は脱顆粒並びに血管作用性、前炎症性及び侵害受容性仲介物質の放出をもたらす)。肥満細胞生理学の主要な特徴(最近までほとんど省みられなかった)は、明白な脱顆粒を示すことなく、弁別的又は選択的放出を介して仲介物質を分泌することができるということである。この過程は、別個のタンパク質キナーゼの作用によって調節されると考えられている(Theoharides et al. J Neuroimmunol 2004, 146(1-2):1-12)。
アレルギー反応とは異なり、自己免疫又は炎症過程時には肥満細胞の脱顆粒はほとんど認められない。その代わりに、肥満細胞は、明白な脱顆粒を示すことなく、分泌の表示である、それらの高電子密度顆粒コアの超微細構造変化(“活性化”、“顆粒内活性化”又は“断片的”脱顆粒と称される過程)を経るように見える。肥満細胞は、喘息、アトピー性皮膚炎、心脈管系疾患、慢性乾癬、線維筋痛、炎症性腸症候群、間質性膀胱炎、片頭痛、多発性硬化症(MS)、神経線維腫症、変形性関節症、慢性関節リウマチ、強皮症を含む炎症性疾患に必要である(Theoharides et al.上掲書)。実際、これら疾患の多くが、間質性膀胱炎の場合のように同時に発生するようである。肥満細胞は、自己免疫関節炎に要求され、皮膚の過敏反応で決定的な役割を果たし、心脈管系症状の発生(特に不安定狭心症および無症状心筋虚血)でその関与が強く疑われる。さらにまた、神経終末とのそれらの緊密な物理的結合は、多くのストレス誘導性炎症性疾患の病因における肥満細胞の関与を示唆する。
レセプターチロシンキナーゼ、c-Kit(CD117)は肥満細胞の生存に必須である(Tsujimura, Pathol Int 1996, 46(12):933-8)。c-Kitリガンド(幹細胞因子(SCF))は、ヒト肥満細胞の分裂及び成熟に重要であり、前記の除去は肥満細胞のアポトーシスをもたらす。c-Kitの構成的発現は肥満細胞の疾患で生じる(Mol et al. J Biol Chem 2003, 278(34):3146-4)。本明細書に記載のヒポテマイシン並びにその誘導体及び類似体は、c-Kitとともに、c-Kit活性化ERK経路の下流の2ヶ所(MEK1/2、ERK1/2)の強力な不可逆性阻害剤であり、本発明は、感受性を有するプロテアーゼとマイケル付加物を形成することができるRALの治療的に有効な用量を投与することによって、肥満細胞によって影響を受けるか、又は前記によって惹起される炎症性疾患(特に上記に列挙した疾患を含む)を治療する方法を提供する。
肺線維症:
本発明はまた肺線維症を治療する方法を提供する。特発性肺線維症(IPF)は、病因が不明の間質性肺疾患の仮借なき進行型である。IPFと診断されたヒトの平均生存は3年未満である。従来の治療は抗炎症性ステロイド及び免疫抑制剤による治療を含むが、応答率は非常に低い。前線維細胞性サイトカイン(例えばIPFのTGF-β及びPDGF)の役割における興味は、そのようなサイトカインは線維芽細胞の形質転換、分裂及び蓄積を引き起こし、細胞外マトリックスの産生及び堆積、組織破壊及び肺機能の低下をもたらすという事実に集中した(Lasky et al. Environ Health Perspect 2000, 108:Suppl 4:751-62;及びSime et al. Clin Immunol 2001, 99(3):308-19)。最近の研究は、イマチニブは、PDGFRのリン酸化を阻害することによって(Aono et al. Am J Respir Crit. CareMed 2005)、及びおそらくc-Ab1タンパク質キナーゼのリン酸化を阻害することによって(Daniels et al. J Clin Invest 2004, 114(9):1308-16)、マウスのブレオマイシン誘導性肺線維症の進行を阻止することができることを示している。本明細書に記載したヒポテマイシン並びにその誘導体及び類似体は、PDGFRとともに、PDGFシグナルを伝達するERK経路の強力な阻害剤であり、本発明は、マイケル付加物形成を介してそのようなタンパク質キナーゼを阻害することができるRALの治療的に有効な用量を投与することによって肺線維症を治療する方法を提供する。
【0082】
黄斑変性:
本発明はまた、病因としてVEGF(VEGFRは本発明の方法で有用な化合物の標的である)及びERK経路が中心的に関与する糖尿病関連黄斑変性及び緑内障のみならず、加齢関連黄斑変性もまた治療する方法を提供する。本発明のこれらの方法で有用な化合物は、ERK経路の多キナーゼ阻害によるVEGF産生阻害だけでなく、またERK経路阻害を介するVEGF産生とともに内皮細胞内VEGF産生の阻害によっても、VEGF仲介血管形成を阻害する。ある実施態様では、本発明の方法で有用な化合物は、この消耗性症状の治療のために、黄斑変性治療用のまた別の薬剤と一緒に同時投与される。
アレルギー性皮膚炎:
本発明の方法はまた、アレルギー性皮膚炎及び免疫抑制が所望される他の疾患を治療する方法を含む。上記に記載したように、3つのシグナル経路の統合は、T細胞によるサイトカインの分泌及びそれに続く以下のエフェクター機能の獲得をもたらす:(i)カルシニューリンの活性化、(ii)ERK経路の活性化、及び(iii)JNK経路の活性化。ヒポテマイシンは、ERK経路を2ヶ所(MEK1/2及びERK1/2)で阻害するとともにJNK経路をMEK4/7で阻害し、したがってT細胞活性化に必要な3つの経路のうちの2つを阻害する。FK506は、カルシニューリンの作用を阻害する周知の免疫抑制剤で、アトピー性皮膚炎の治療で用いられる。本発明の方法にしたがえば、本明細書で提供される本発明の化合物の投与はアトピー性皮膚炎の治療に用いられる。ある実施態様では、本発明の化合物は、カルシニューリンを阻害するか又はその作用を阻害する化合物又は薬剤とともに同時投与される。そのような化合物には、FK506並びに学術文献及び特許文献に報告された前記の複数の誘導体が含まれるが、ただしそれらに限定されない。この治療によって、サイトカイン分泌をもたらす3つのシグナル伝達経路の全てが阻害され、アレルギー性皮膚炎及び免疫抑制が所望される他の疾患の効果的な治療が提供される。
【0083】
痛み:
本発明はまた痛みを治療する方法を提供する。合衆国の人口の9%が中等度から重篤な癌に関連しない全てのタイプの痛みに苦しんでいる。前記には慢性の痛みを抱える1,500万人を超える人々が含まれる。世界中でほぼ2,600万人の患者(合衆国では1,000万人)が何らかの形態の神経障害性の痛み(痛みが刺激に対して不適切である慢性の疼痛)を有する。末梢神経障害性疼痛は、典型的には末梢神経が、外科手術、骨の圧迫(種々の疾患で)、糖尿病、及び感染により損傷を受けたときに発生する。神経障害性疼痛症状の2つの一般的で重度の消耗性症状は痛覚過敏及び異痛である。痛覚過敏は、疼痛刺激によって生じる疼痛応答過多であり、異痛は、通常痛みを感じない刺激から生じる疼痛である。両者はしばしば通常の鎮痛剤に耐性を示す。これらの症状の治療に鎮痛剤が一般的に有効でないのは、脊髄内の神経プロセッシングにおける長期的変化の結果であり得る。実際、脊髄及び後根神経節における多様な神経伝達物質、それらのレセプター、及び他の遺伝子の発現の変化は痛覚過敏と密接に関連していることが示された(以下を参照されたい:Woolf and Costigan, Proc Natl Acad Sci USA, 1999, Jul 6, 96(14):7723-30)。
高い発生率及び神経障害性疼痛の従来治療の貧弱な有効性のために、この症状に対する新規な標的が希求されている。タンパク質キナーゼは種々のタイプの痛みにおいて重要な役割を果たしている。薬剤誘導性神経障害性疼痛における遺伝子発現変化の研究によって、ストレプトズーシン誘導性糖尿病性神経障害及び慢性絞窄損傷の疼痛動物モデルの両方で変化する細胞外シグナル調節キナーゼ(ERK)カスケードの重要な成分が同定された(以下を参照されたい:Ciruela et al. 2003, Br J Pharmacol 138(5):751-6)。脊髄内のERK1/2活性レベルの上昇は痛覚過敏と相関性を示した。MEK1/2阻害剤、PD198306の脊髄内投与によって、定常的異痛(疼痛応答の一般的な実験的測定)は、両神経障害疼痛モデルにおいて用量依存態様で遮断された。PD198306の足底内投与は痛覚過敏のどちらのモデルでも効果はなかった。したがって、ERK1/2の活性における対応する変化(MEK1/2阻害作用の主要な結果である)は中枢神経系に局在しなければならない。他の研究は、炎症性疼痛過敏(Ji et al. 2002, J Neurosci 22(2):478-85)の結果として、又は脊髄内の代謝向性グルタミン酸レセプターアゴニストの作用(Adwanikar et al. 2004, Pain 11(1-2):125-35)の結果として、脊髄後角ニューロン内の活性化ERK1/2キナーゼの中心的関与を示した。各事例で、MEK阻害剤は疼痛応答を緩和することができた。リン酸化ERKが核内に入るとき、前記はキナーゼのRSK2タイプを活性化する。前記は続いて、疼痛応答の開始に必要な種々の遺伝子のcAMP仲介転写をもたらすCREBを活性化する(Ji et al. 2002, Neurosci 2(2):478-85)。他のMAPKシグナル伝達経路もまた神経障害性疼痛に関与していた。例えば、p38ストレス活性化MAPKは、成獣ラットのL5脊髄神経の結紮後1日以内で活性化され、その効果は3週間より長く持続する(Jin et al. 2003, J Neurosci 23(10):4017-20)。p38阻害剤、SB203580の脊髄内注射は疼痛応答を、特に神経障害誘導の初期時点で投与したとき顕著に低下させた。
本明細書に開示したレゾルシル酸ラクトン阻害剤の各々は痛みに関連する複数のタンパク質キナーゼを阻害することができ、したがって有益な鎮痛剤である。それぞれは、MEK/ERKシグナル伝達経路の中心的部分の2カ所において強力な阻害剤であり、ほぼ4つの酵素(MEK1/2及びERK1/2)を阻害する(それぞれは、TAK1及びMEK3/6を阻害することによってp38経路を阻害する)。さらにまた、ERK経路の2ヶ所の阻害に加えて、それぞれは下流のRSK2型のキナーゼを阻害し、したがってCREB活性化につながる経路の複数の工程を遮断する。したがって、本発明は、感受性タンパク質とマイケル付加物を形成することができるRAL阻害剤の治療的に有効な用量を投与することを含む、痛みを治療する方法を提供する。
【0084】
併用療法:
ある種の抗癌化合物はある種の細胞タイプでERK経路を活性化することが知られており、したがって、本発明の方法のある観点では、本発明の方法で有用なRALと同時投与される。タキソール及び他のチューブリン相互作用剤は癌細胞でERK経路の活性化を誘導することができる(Stone and Chambers, Exp Cell Res 2000, 254:110-119;MacKeigan et al. J Biol Chem 2000, 275:38953-38956;McDaid and Horwitz Mol Pharmacol 2001, 60:290-301)。前記はいくつかの細胞、例えばHeLa及びCHO細胞で生じるが、他の細胞、例えばMCF-7細胞(McDaid and Horwitz(2001)上掲書)では生じない。さらにまた、パクリタキセル誘導性ERK活性化を示す細胞をMEK阻害剤U0126で処理すると、アポトーシス及び細胞傷害性の累積性が観察される。同様に、ERK経路はカルボプラチン及びシスプラチンによって活性化される(Choi et al. Reprod Biol Endocrinol 2003, 1(1):71)。ある種の癌細胞は、基本的には耐性メカニズムをもたらすある種の薬剤ストレスに対する調節的応答でERK経路を活性化すると考えられる。そのような事例では、薬剤耐性細胞は、ERK経路阻害剤による処置によって薬剤感受生細胞に変換することができる(Choi et al Reprod Biol Endocrinol 2003, 1(1):71)。したがって、ある実施態様では、癌又は特定の癌適応症を治療する本発明の方法は、ERK経路を活性化する抗癌化合物(タキサン(例えばドセタキセル又はパクリタキセル)又は他の微小管安定化剤若しくは微小管脱安定化剤、エポチロン(例えばエポチロンB若しくは D又はエポチロン誘導体)、又は白金剤(例えばシスプラチン又はカルボプラチン)を含むが、ただしこれらに限定されない)を、本明細書に開示のRALと併用して患者に投与して、ERK経路依存癌を治療することを含む。
本発明の別の併用療法では、それ自体Hsp90のクライアントタンパク質であるか、又はHsp90のクライアントタンパク質によって活性化されるキナーゼとマイケル付加物を形成することができるRALタンパク質キナーゼ阻害剤が、Hsp90阻害剤とともに同時投与される。ここで、RALエノンはその特異的キナーゼを阻害し、Hsp90阻害剤は同じ又は別個のキナーゼサブセット(前記はHsp90クライアントタンパク質として機能する)の破壊をもたらす。ある実施態様では、Hsp90阻害剤はゲルダマイシン又はゲルダマイシン類似体(例えば17-AAG又は17-DMAG)である。本発明のまた別の併用療法では、その標的タンパク質キナーゼとマイケル付加物を形成することができるRALタンパク質キナーゼ阻害剤は、トポイソメラーゼ阻害剤と一緒に同時投与される。
【0085】
したがって、ヒトの疾患の治療に用いられるときは、本発明の方法で有用な化合物は他の薬剤と併用して投与することができる。例えば、MAPK経路阻害剤として期待されるものは典型的には細胞に対して細胞増殖抑制作用を示す。前記細胞では、ERK、JNK又は他のMAPK経路は、マイトジェン、異常に機能が亢進したマイトジェンレセプター(例えばVEGFR又はPDGFR)、変異Ras又はRafタンパク質、異常に活性化されたMEKK酵素、又は構成的に発現されたERK遺伝子によって活性化されている。対照的に、一般的に用いられる癌の化学療法薬は典型的には細胞傷害性作用を示す。したがって、本発明のMAPK経路阻害剤は、既に確立された細胞傷害性薬剤又はより新しい薬剤、例えばHsp90阻害性ゲルダマイシン類似体、17-AAG及び17-DMAG(それらの抗腫瘍作用はMAPK経路阻害剤の抗腫瘍作用を補完する)と併用して投与することができる。
本発明の方法で有用な化合物と同時投与することができる抗癌剤又は細胞傷害性薬剤には以下が含まれる:アルキル化剤、血管形成阻害剤、抗代謝薬、DNA分割剤、DNA架橋剤、DNA挿入剤、DNA小溝結合剤、エネディーン(enediynes)、熱ショックタンパク質90阻害剤、ヒストンデアセチラーゼ阻害剤、微小管安定化剤、ヌクレオシド(プリン又はピリミジン)類似体、核外搬出(nuclear export)阻害剤、プロテアソーム阻害剤、トポイソメラーゼ(I又はII)阻害剤、チロシンキナーゼ阻害剤。具体的な抗癌剤又は細胞傷害性薬剤には以下が含まれる:β-ラパコン、アンサミトシンP3、アウリスタチン、ビカルタミド、ブレオマイシン、ボルテゾミブ、ブスルファン、カリスタチンA、カンプトテシン、カペシタビン、CC-1065、シスプラチン、クリプトフィシン、ダウノルビシン、ディソラゾール、ドセタキセル、ドキソルビシン、デュオカルマイシン、ダイネマイシンA、エポシロン、エトポシド、フロキシウリジン、フルダラビン、フルオロウラシル、ゲフィチニブ、ゲルダナマイシン、17-アリルアミノ-17-デメトキシゲルダマイシン(17-AAG)、17-(2-ジメチルアミノエチル)アミノ17-デメトキシゲルダマイシン(17-DMAG)、ゲムシタビン、ヒドロキシウレア、イマチニブ、インターフェロン、インターロイキン、イリノテカン、マイタンシン、メトトレキセート、マイトマイシンC、オキサリプラチン、パクリタキセル、スベロイルアニリドヒドロキサム酸(SAHA)、チオテパ、トポテカン、トリコスタチンA、ビンブラスチン、ビンクリスチン、及びビンデシン。
【0086】
一般的癌治療:
本発明の化合物は、例えば以下のような疾患の治療に用いることができる(ただしこれらに限定されない):以下を含む過剰増殖性疾患:頭部、頸部、副鼻腔、鼻咽頭、口腔、口腔咽頭、喉頭、下咽頭、唾液腺の腫瘍、パラガングリオーマを含む頭部及び頸部の癌;肝臓及び胆管樹の癌、特に肝細胞癌;胃腸の癌、特に結直腸癌;卵巣癌;小細胞及び非小細胞肺癌;乳癌肉腫、例えば線維肉腫、悪性線維性組織球腫、胎児性横紋筋肉腫、平滑筋肉腫、神経線維肉腫、骨肉腫、滑膜肉腫、脂肪肉腫、及び肺胞軟部肉腫;中枢神経系の新形成、特に脳の癌;リンパ腫、例えばホジキンリンパ腫、リンパプラズマ細胞性リンパ腫、濾胞性リンパ腫、粘膜結合リンパ組織性リンパ腫、被膜細胞リンパ腫、B系列大細胞型リンパ腫、バーキットリンパ腫、T-細胞脱分化大細胞型リンパ腫。臨床的には、本明細書に開示した方法及び組成物の実施並びに使用は、癌性増殖のサイズ又は数の減少、及び/又は(該当する場合は)付随症状の減少をもたらす。病理学的には、本明細書に開示した方法及び組成物の実施並びに使用は、病理学的に対応する応答を生じるであろう。例えば癌細胞分裂の阻害、癌又は腫瘍のサイズの減少、更なる転移の予防、及び腫瘍の血管形成の阻害である。そのような疾患を治療する方法は、本明細書に開示したRALの治療的に有効な量を、単独で、又は別の抗癌剤と一緒に対象者に投与することを含む。前記方法は、治療的利益のために必要に応じて反復することができる。
【0087】
細胞の過剰増殖を示す非癌性疾患:
本発明はまた、細胞の過剰増殖を特徴とする非癌性疾患を治療する方法を提供する。前記方法は、本明細書に開示したRAL化合物を、そのような治療の必要がある患者に投与することを含む。そのような疾患の例には以下が含まれる(ただしこれらに限定されない):萎縮性胃炎、炎症性溶血性貧血、移植片拒絶、炎症性好中球減少症、水疱性類天疱瘡、腹腔の疾患、脱髄神経障害、皮膚筋炎、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、多発性硬化症、心筋炎、筋炎、鼻腔ポリープ、慢性副鼻腔炎、尋常性天疱瘡、一次性糸球体腎炎、乾癬、手術後癒着、狭窄又は再狭窄、強膜炎、強皮症、湿疹(アトピー性皮膚炎、過敏性皮膚炎、アレルギー性皮膚炎を含む)、歯周の疾患(すなわち歯周炎)、多嚢胞性腎疾患及びI型糖尿病。他の例には以下が含まれる:脈管炎、例えば巨細胞動脈炎(側頭動脈炎、タカヤス動脈炎)、結節性多発性動脈炎、アレルギー性血管炎及び肉芽腫症(チュルグ-シュトラウス病)、多発性血管炎オーバーラップ症候群、高血圧性血管炎(ヘノッホ-シェーンライン紫斑病)、血清病、薬剤誘導性血管炎、感染性血管炎、新形成血管炎、結合組織疾患に付随する血管炎、補体系の先天的欠損に付随する血管炎、ヴェーゲナー肉芽腫症、川崎病、中枢神経系の血管炎、ベルガー病及び全身性硬化症;胃腸管の病気(例えば、膵炎、クローン病、潰瘍性結腸炎、潰瘍性直腸炎、一次性硬化性胆管炎、特発性を含む任意の原因による良性狭窄症(例えば胆管、食堂、十二指腸、小腸又は結腸の狭窄);気道の病気(例えば喘息、過敏性肺実質炎、石綿肺症、珪肺症及び塵肺症の他の形態、慢性気管支炎、並びに慢性閉塞性気道疾患);鼻涙管の病気(例えば特発性を含む全ての原因による狭窄);及び耳管の病気(例えば特発性を含む全ての原因による狭窄)。
【0088】
医薬組成物及び投与量:
本発明は、本発明の方法で有用な化合物を含む医薬組成物及び調製物を提供する。これらの組成物及び調製物には種々の形態、例えば固体、半固体及び液体形が含まれる。一般的には、前記医薬調製物は、活性成分として本発明の方法で有用な1つ又は2つ以上の化合物、及び医薬的に許容できる担体又は賦形剤を含む。典型的には、前記活性成分は、外部適用、腸内適用又は非経口適用に適した有機又は無機担体若しくは賦形剤と混合されてある。前記活性成分は、例えば、錠剤、ペレット、カプセル、座薬、ペッサリー、溶液、乳濁液、懸濁液、及び使用に適した他の任意の形態のための通常の無毒な医薬的に許容できる担体との化合物でもよい。特に、静脈内投与及び経口投与が意図され、本発明はそのような態様に適した医薬組成物を提供する。
使用することができる賦形剤には、担体、表面活性剤、濃縮剤又は乳化剤、固体結合剤、分散剤又は分散補助剤、可溶化剤、着色剤、香料、被覆剤、崩壊剤、滑沢剤、甘味剤、保存料、等張剤及び前記の組合せが含まれる。適切な賦形剤の選択及び使用は以下の成書に教示されている:Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003)、前記文献の内容は参照により本明細書に含まれる。
前記担体材料と混合して単一投薬形を生成することができる活性成分の量は、治療される宿主及び具体的な投与態様にしたがって変動するであろう。例えば、ヒトへの経口投与が意図される処方物は担体物質を含むことができ、前記は、全組成物の約5%〜約95%で変動し得る。ユニット投薬形は一般的に約5mg〜約500mgの活性成分を含むであろう。
本発明の治療的に有効量の化合物は、単回投与量又は分割投与量で対象者に投与することができる。投与頻度は毎日であっても、又は他のなんらかの規則的スケジュールに従っても(例えば3日目毎)、又は不規則なスケジュールでもよい。投薬量は、例えば体重1kgあたり約0.01〜10mg(約0.01〜10mg/kg)、又はより通常的には約0.1〜2mg/kgであり得る。
【0089】
しかしながら任意の個々の患者に対する具体的な用量レベルは多様な因子に左右され得ることは理解されよう。これらの因子には、用いられる具体的な化合物、体重、一般的な健康状態、性別、対象者の食事、投与時間及び投与経路、薬剤の排出速度、薬剤併用が治療に用いられるか否か、並びに治療が試みられている個々の疾患又は症状の重篤度が含まれる。
不可逆的阻害剤(例えば本明細書で考察された化合物)は、それらが投与される治療計画に強い影響を与えるある種の格別な特徴を有する。標的キナーゼは急速に阻害され、阻害効果は長期に及び、シグナル伝達活性の回復のためにそれらキナーゼの再合成が要求される。したがって、可逆的阻害剤と比較して、不可逆的阻害剤は有効性のために必ずしも高い血中濃度又は長時間の血中半減期の達成を必要としない(例えば、Calvoらの文献中のCC-1033(EGFR機能の不可逆的阻害剤)の考察を参照されたい)。さらにまた、不可逆的阻害剤はより低い頻度で投与することができる。なぜならば、それらの阻害作用はより長く続くからである。腫瘍の増殖を阻害するために要求される曝露の短縮はまた毒性を低下させることができる。不可逆的阻害剤の前記固有の特徴は、標準的な曝露に関する薬理学的動態実験ではなく又は前記に加えて、標的キナーゼの阻害及び回復を基準にした投与計画の最適化を推進する。
必要な場合には、本発明の化合物はマイクロカプセル及びナノ粒子として処方することができる。一般的なプロトコルは例えば以下に記載されている: US5,510,118(Bosch et al., 1996);US5,534,270(De Castro, 1996);及びUS5,662,883(Bagchi et al., 1997)(前記文献は全て参照により本明細書に含まれる)。表面積対容積比を増すことによって、これらの処方物は、経口送達には適さない化合物の経口送達を可能にする。
上記に記載したように、本発明の化合物は、他の医薬(特に他の抗癌剤)と併用して同時投与することができる。前記同時投与は、同時でも又は連続的でもよい。
上記に記載したように、本発明は、その範囲内に本発明の化合物のプロドラッグを含み、本発明はそのようなプロドラッグを含む医薬組成物を提供する。そのようなプロドラッグは、一般的に、要求される化合物にin vivoで容易に変換され得る化合物の機能的誘導体である。したがって、本発明の治療方法では、“投与する”という用語は、具体的に開示した化合物で、又は具体的には開示されなかったが、その必要がある対象者に投与後にin vivoで明示の化合物に変換され得る化合物で、記載の種々の疾患を治療することを含む。適切なプロドラッグ誘導体の選択及び調製のための一般的な方法は例えば以下に記載されている:Wermuth, “Designing Prodrugs and Bioprecursors,” in Wermuth, ed., The Practice of Medicinal Chemistry, 2nd Ed., pp.561-586(Academic Press 2003)。プロドラッグには、in vivoで(例えばヒトの体内で)加水分解して本発明の化合物を生じるエステル、又はその塩が含まれる。適切なエステル基には、医薬的に許容できる脂肪族カルボン酸、特にアルカン酸、アルケン酸、シクロアルカン酸及びアルカン二酸から誘導され、前記の各アルキル又はアルケニル部分が好ましくは6未満の炭素原子を有するものが含まれるが、ただしこれらに限定されない。例示的エステルには、ギ酸エステル、酢酸エステル、プロピオン酸エステル、酪酸エステル、アクリル酸エステル、クエン酸エステル及びエチルコハク酸エステルが含まれる。
【0090】
本発明の方法で有用な化合物:
本明細書に記載したように感受性を有するタンパク質キナーゼとマイケル付加物を形成することができる複数のレゾルシル酸ラクトンが存在するということを、本発明の開示から当業者は理解しえよう。種々のRAL化合物が生成され試験され、さらに多くのものが関連する広範囲の特許文献に記載されてきた。本発明の方法は、部分的には、現時点で存在し、さらに考えられ得るレゾルシル酸ラクトン及び誘導化合物クラスのうち極めてわずかなセットが、ほんのわずかなキナーゼタンパク質ファミリーセット中の重要なCys残基と緩徐な可逆的マイケル付加を介して阻害を達成するために用いることができるという発見から得られた。これらの発見は、以前にはそのような化合物による治療には適しないと考えられた疾患を治療するための薬剤として前臨床試験で既知の化合物を再調査しようとする、さらに、有用であると今日まで単に特許文献で予想されていただけであった化合物を作成し試験しようとする強力な原動力を提供する。
したがって、以前に公知であり試験された化合物が本発明のある種の方法では有用であり得るが、本発明の他の方法はそのような化合物の使用を含まない。
ある実施態様では、本発明の治療方法で投与される化合物及び医薬組成物は、エーザイ株式会社の特許公開公報US2004/0224936 A1(2004)、WO03/076424 A1(2003)及びWO2005/023792 A1(2005)(前記文献は参照により本明細書に含まれる)に記載された化合物であるか、又はそのような刊行物においてある種のジェネリック化合物の範囲に含まれる化合物である。これらの刊行物は、その中に記載されている化合物は、NF-kB及びAP-1活性並びにタンパク質キナーゼ(例えばMEKK、MEK1、VEGFR、PDGFR)の阻害剤としての活性を示すことができるが、特定の疾患及び症状で重要な役割を果たす、キノム中の他のタンパク質キナーゼに関してはサイレントであると述べている。これらの刊行物は、前記化合物は癌並びに炎症性及び免疫疾患の治療に適用できるかもしれないと述べ、RA、乾癬、血管形成及びステント技術の記載を含んでいる。しかしながら、利用可能な限定的データから見れば、開示された化合物の治療潜在能力をこれらの刊行物から見つけることはできない。さらにまた、本明細書に開示したように、そのような化合物はマイケル付加物形成によってMEKK1を阻害することはできない(MEKK1は本発明の化合物とマイケル付加物を形成することができない)。さらにまた、上記で考察し、下記実施例で述べるように、これら特許公開公報のジェネリック化合物の記載に含まれるある種の化合物はマイケル付加物を形成せず、したがって本発明の方法で有用な化合物は、これら刊行物中の化合物の記載に包括的に含まれる化合物の新規なサブセットを含む。
【0091】
エーザイの特許公開公報に開示された化合物を用いて以下を含む多様な癌の治療に用いることができることを本発明は教示するが、それら疾患の全ては、エーザイの特許公開公報で開示された化合物による治療に対して感受性であると記載されなかった以下の症状である:AML、基底細胞癌、B-Raf変異依存癌(大腸癌及びメラノーマを含むがただしこれらに限定されない)、乳癌、GI間質腫瘍、Ras依存癌、腎細胞癌、及び前立腺癌並びに他の症状(肺線維症、肥満細胞症、炎症性腸疾患及びアレルギー性皮膚炎)。本発明はまた、2つ以上のキナーゼを阻害する化合物を投与することによって、種々の症状(特にMEKK、MEK1、VEGFR及びPDGFRに加えてまた別のキナーゼを、MEKK以外のキナーゼと同様に阻害することが治療の有効性を高めるであろうと期待される疾患及び症状)を治療する方法を提供する。他の実施態様では、本発明は、MEKK、MEK1、VEGFR及びPDGFR以外のキナーゼにおける変異のためにある種の薬剤に対して耐性を有する癌を、エーザイの特許公開公報に記載された化合物を投与して変異キナーゼを阻害することによって治療する方法を提供する。本発明の方法の他の実施態様では、エーザイの特許公開公報に具体的に記載された化合物以外の化合物が、本明細書で特定した疾患又は症状を治療するために投与される。
別の実施態様では、本発明の治療方法で投与される化合物及び医薬組成物は、Cor Therapeutics, Incの米国特許5,674,892号(1997)、5,795,910号(1998)及び5,728,726号(1998)(前記文献は参照により本明細書に含まれる)に記載された化合物のサブセットである。これらの刊行物は、多様なRLA(本明細書に記載したマイケル付加物を形成することができるもの、及びできないものを含む)がキナーゼ阻害剤として一般的に有用であることを記載している。くり返せば、他の重要なキナーゼ(Cor社の特許公開では3つのキナーゼしか記載されていない)に対する化合物の作用についての情報が存在しないこと、及びこれらの刊行物に記載されたいくつかのキナーゼに関して利用可能なデータが限られているために、Cor Therapeutics社の特許だけからでは、それら化合物の治療の潜在能力を判定することは不可能である。本発明は、本明細書に開示したマイケル付加物を形成することができる、これらCor Therapeutics社の特許に開示された化合物が、多様な癌適応症並びに他の疾患及び症状の治療に用いることができることを教示し、Cor Therapeutics社の特許に記載されたもの以外の化合物もタンパク質キナーゼを標的とすることを示すデータを提供する。本発明の方法の他の実施態様では、Cor Therapeutics社の特許に具体的に記載された化合物以外の化合物が投与され、本明細書で特定した疾患又は症状が治療される。
本発明の方法のまた別の実施態様では、本発明の方法で有用な化合物は、天然に存在するレゾルシル酸ラクトン、ヒポテマイシン、(5Z)-7-オキソゼアネオール、Ro-09-2210及びL-783,277から成る群より選択される以外の化合物であり、AML、基底細胞癌、B-Raf変異依存癌(大腸癌及びメラノーマを含むがただしこれらに限定されない)、乳癌、GI間質腫瘍、Ras依存癌、腎細胞癌、及び前立腺癌、肺線維症、肥満細胞症、炎症性腸疾患及びアレルギー性皮膚から成る群より選択される疾患又は症状のために治療の必要がある患者に投与される。
以下の実施例は、本発明の方法で有用な化合物を生成し、試験し、さらに使用する種々の方法を例示する。
【0092】
(実施例)
以下の実施例は、ヒポミセス・スビキュロスス(Hypomyces subiculosus)、ATCC44392又はアイギアルス・パルブス(Aigialus parvus)の発酵から得られるヒポテマイシン及び(5Z)-7-オキソゼアネオールの精製を述べる。これら実施例は、放射能、蛍光若しくはビオチン標識したラクトン又は質量分析を用いて、化合物(本実施例では例示的化合物、ヒポテマイシン及び(5Z)-7-オキソゼアネオールが用いられる)が、MEK1又は他のCys標的キナーゼと共有結合付加物を形成するか否かを示すために、酵素の動力学的解析をどのように用いることができるか示す。さらにまた、これら実施例は、MAPKキナーゼシグナル伝達経路を阻害するラクトンの能力をどのように細胞利用アッセイによって決定することができるか、さらにどのようにしてERK依存腫瘍培養由来の癌細胞でラクトンの抗分裂態様を提示することができるかを示す。
【実施例1】
【0093】
実施例1:レゾルシル酸ラクトンの産生:
ヒポテマイシン又は(5Z)-7-オキソゼアネオールは、文献の方法にしたがってヒポミセス・スビキュロスス(Hypomyces subiculosus)、ATCC44392の発酵物から精製することができる。これら及び近縁レゾルシル酸ラクトン(アイギアロマイシンとして知られている)のまた別の供給源は、アイギアルス・パルブス(Aigialus parvus)株の発酵物である。本発明の他のレゾルシル酸ラクトン化合物は、本明細書の開示および文献に記載された方法にしたがって合成することができる。単離した化合物の構造は、精製物質のNMR及びMS分析によって確認することができる。ラクトン又はその類似体の1つの3H若しくは14C形は、化学的又は酵素的半合成方法によって業者(例えばMoravek Biochemicals, Brea, CA)により調製され、その構造はクロマトグラフィー及び分光分析により立証することができる。本発明はまた、以下のように、ヒポテマイシンの代わりに、(5Z)-7-オキソゼアネオール又は15-デスメチルヒポテマイシンを産生する変異株を得る方法を提供する。ヒポテマイシンのための生合成遺伝子クラスターは、末端配列決定法を用いて、モノモデュラーI型ポリケチドシンターゼ(PKS)及び必須のテーラー酵素をコードする遺伝子を同定した後で、H.スビキュロススのゲノムDNAから作成したコスミドライブラリーからサブクローニングされる。候補コスミドの配列を決定し予想される特徴を有するコスミド、すなわちPKS遺伝子プラス少なくとも1つのオキシダーゼ遺伝子(O-メチルトランスフェラーゼ遺伝子)及び付随する調節遺伝子を含むオーバーラップコスミドを見つける。遺伝子破壊を実施し、正確な合成遺伝子セットが同定されていることを確認する。最後に、オキシダーゼ遺伝子の破壊は、(5Z)-7オキソゼアネオール(ヒポテマイシンの前駆体)の破壊をもたらすか、又はO-メチルトランスフェラーゼ遺伝子の破壊は15-デスメチルヒポテマイシンの生成をもたらす。本発明の方法で有用な化合物はまた全化学合成によっても調製することができる(以下を参照されたい:Selles et al. Tetrahedron Lett. 2002, 43(26):4621-5;Selles et al. Tetrahedron Lett 2002, 43(26):4627-31;Geng et al. Org Lett 2004, 6(3):413-6)。
【実施例2】
【0094】
実施例2:ラクトンによる標的Cysキナーゼ阻害の動力学的解析
本実施例は、例示的タンパク質キナーゼとしてMEK1、MEK2及びいくつかのマイトジェンレセプターキナーゼを用い、標的タンパク質キナーゼと化合物がマイケル付加物を形成することができることを示す方法を例証する。阻害剤と酵素間の共有結合付加物形成の特徴は、酵素活性の“時間依存阻害”である。
典型的には、阻害剤の存在下で時間の経過にしたがいタンパク質キナーゼ活性の阻害の増加が測定される。ある方法では、酵素及び阻害剤を含む“プレインキュベーション”反応混合物のアリコートを時間の経過とともに活性についてアッセイする。阻害の増加又は初期速度の低下が、マイケル付加物が形成されるために時間の経過につれて観察されるであろう(C. Walsh, Enzyme Reaction Mechanisms, W.H. Freeman & Co., 1979, pp 86-94)。第二の方法では、活性の時間依存低下は、形成される生成物(例えばADP)対時間を測定及び分析する“進行曲線”として測定される(Morrison & Walsh, Adv. Enzymol Relat Areas Mol Biol 1988, 61:201-301)。いずれの事例でも、時間依存不活化は、競合する基質(この場合はATP)の存在によって鈍化させることができる。
決定される可逆的解離定数(Kd)及び不活化のための速度定数(Kinact)値は、阻害メカニズムの解析に用いられる基礎的データである。ヒポテマイシンとコントロールとしての前記の非反応性5,6-ジヒドロ形を用いるこれらのアッセイの実施は、酵素阻害のためのα,β-不飽和ケトンの重要性を示す。
他のMEK1阻害剤(例えばPD184352及びUO0126)の確立されたメカニズム(前記はともにATPと非競合的に機能する)から、本発明の方法で有用なラクトン化合物は、MEKによってERK1のリン酸化を阻害するはずである。時間依存性酵素阻害は、堅固で緩徐な結合の阻害剤又は共有結合形成阻害剤で観察され、上記に記載した標準的なアプローチで検出することができる。
本発明の方法で有用な化合物の標的である得るMEK1及び多くの他のタンパク質キナーゼは業者(Invitrogen, Carlsbad, CA)から入手するか、又は標準的な分子生物学的技術を用いて調製することができる。リン酸化による活性化の後で、それらは、標的キナーゼ又は代用基質をリン酸化するその能力についてアッセイされる。例えば、MEK1は、Mops緩衝液(pH7.6)中のMEK1(30nM)及びERK1(2μM)、[γ-32P]ATP(10uM)及びMgCl2を含む混合物でアッセイすることができる。リン酸化は、[γ-32P]リン酸化ERK1をホスホセルロース紙上で単離し、放射性生成物を計測することによって測定することができる。また別には、共役酵素系を用いてもよい。前記系では、キナーゼ反応生成物(例えばADP)は、容易に測定できる物質(例えばNADH)に前記生成物(例えばADP)を変換する二次系を用いて解析される。そのような共役酵素系は便利な分光分析アッセイによって測定することができる。
“プレインキュベーション法”を用いて時間依存阻害を測定するために、MEK1(又は他のキナーゼ)は、ラクトン阻害剤の量を変化させながら一緒にインキュベートされる。コントロールは阻害剤を含まないか、又は競合阻害剤を含む(例えばUO126(IC50は72nM)、EMD Biosciences(San Diego, CA)から入手できる)。アリコートを種々の時間で取り出し、基質[γ-32P]ATP、ERK1(又は他の基質)及び他の反応成分を含む溶液に添加し、初期速度を残存酵素活性の測定として決定する。
共有結合阻害剤の場合、酵素活性の時間依存低下が存在し、一方、可逆的阻害剤の場合、活性は時間につれて変化しない(Morrison & Walsh, Adv. Enzymol Relat Areas Mol Biol 1988, 61:201-301;Sculey et al. Biochem Biophys Acta 1996, 1298(1):78-86)。共有結合阻害剤の場合、log(活性)対時間のプロットは、酵素活性低下の見かけの一次速度定数(Kobsd)を提供する。これらのアッセイが種々の濃度の阻害剤において実施されるならば、一連の一次プロットが得られ、各阻害剤濃度でKobsdが得られる。“進行曲線法”(上記に引用した参考文献及び以下の文献(Kuzmic et al. Methods Enzymol 2004, 383:366-81)を参照されたい)を用いて時間依存阻害を測定するために、ERK(又は他のキナーゼ)を種々の濃度のラクトン阻害剤で処理し、ADPの生成を共役アッセイによって持続的に測定する。得られた生成物対時間の曲線は以下の等式に適合する:[P]=(vi/kobs)* (1-exp(-kobs*t)、式中、Pは時間tで形成される生成物であり、viは初期速度であり、kobsは見かけの阻害一次速度定数であり、kobs値は種々の阻害剤濃度の各々について決定される。1/kobs対1/[I]の再プロットはKd(初期可逆性結合定数)及びkinact(可逆的に結合したE-Iの共有結合したE-Iへの変換のための一次速度定数)の決定を可能にする(0.693を前記数値で割ることによって、前記数値を用いて不活化の半減期を産出することができる)。コントロール実験は、α,β-不飽和カルボニルをもたず(例えば5,6-ジヒドロヒポテマイシン)、したがってマイケル付加物を形成することができないヒポテマイシン類似体を用いて実施することができる。そのような分子は競合的阻害剤ではあり得るが、時間依存性不活化を示すことはない。
表2は、いくつかのキナーゼに対するヒポテマイシンの対応する阻害定数を示す(前記には、対応する場合は、時間依存不活化のための動力学的定数が含まれる)。パラメータは、別個のキナーゼについて顕著に異なり、“選択性定数”(kinact/Ki)における100倍を超える相違は、例えばKDR(VEGFR)及びMEK1のようなキナーゼは、低い濃度×時間(細胞又は生物では低い投与量×曝露)で他のものよりも選択的に阻害され得ることを示唆している。本発明の方法で有用な化合物のセット内では、特異性における顕著な多様性が達成され、異なる適用について最適な化合物の同定を可能にすることは当業者には理解されよう。阻害進行曲線解析は、連続的比色分析又は蛍光測定分析を用いて実施した。全ての実験は、KDR(5mMのATP)を除いて1mMのATPで実施した。
【0095】
表2:種々のタンパク質キナーゼに対するレゾルシル酸ラクトンの阻害データ
a 阻害剤の存在なしに時間の経過とともに酵素活性の顕著な低下
b 最高の阻害剤濃度で想定される阻害の5%
c 最高の阻害剤濃度での阻害率(初期速度)から算出されるKi
下記の表3に示される結果は、ヒポテマイシンは必須のCys残基を欠くキナーゼを顕著には阻害せず、前記Cysを有するキナーゼは様々な程度でこれを阻害することを示している。124キナーゼのこのパネルでは、活性部位のシステインを有すると識別された19キナーゼのうちで18が、0.2μM及び/又は2μMの濃度のヒポテマイシンによってある程度まで阻害された。非活性部位システインに関しては、わずかに2つがヒポテマイシンによる顕著な阻害を示した。(これらの値は、種々のアッセイ(すなわち種々のサンプル操作技術を用いる)で達成し得る値と異なる可能性がある。なぜならば、それらの値は、共有結合性阻害の時間依存特性を考慮しない単一点アッセイだからである)。
【0096】
表3:ヒポテマイシン(0.2μM、カッコ内の場合は2.0μM)で処理したタンパク質キナーゼのパーセント残留活性
a 活性部位システインを有する
放射性[32P]ATP及び生成物のフィルター結合によるプレインキュベーション実験を用いてMEK1アッセイを実施した。連続的分光分析アッセイ由来の進行曲線分析を用いて他の全てのキナーゼを解析した。
TRKA及びBは、上記に記載した単一点スクリーニングアッセイ(表3)でヒポテマイシンによる阻害を示したが、マイケル付加物形成のための標的Cysを含まない。これらの酵素をこのより正確な方法によってアッセイした時、ヒポテマイシンは、ATPと競合する可逆的な阻害を示し、KiはTRKAについては2.2μMで、TRKBについては0.37μMであったが、前記酵素の時間依存不活化(すなわち共有結合の形成)は示さなかった(表2)。このことは、共有結合性阻害は標的Cys残基を要求することを立証し、標的キナーゼの共有結合酵素阻害のための規準として時間依存阻害の正当性を実証する。
【実施例3】
【0097】
実施例3:共有結合形成の決定
マイケル反応(前記は基本的には可逆反応である)では、遊離リガンドと共有結合リガンドとの間の見かけの親和性は、反応に中心的に関与する2つの解離定数(Kreversible x Kcovalent)の産物であり、複合体の形成/破壊は、該当タンパク質は両方向で反応を触媒するので理論的には可逆的である。タンパク質の変性は両方向の触媒を停止し、変性マイケル付加物は物理的に単離及び定量ができるほど通常は充分に安定である。例えば、天然のFdUMP-チミジレートシンターゼマイケル付加物は緩徐ではあるが完全に不可逆性であるが、SDS変性は安定で単離可能な複合体を提供する(D.V. Santi et al. Biochemistry 1974, 13:471;Methods in Enzymol 1977, 46:307-312)。
もちろん、複合体が共有結合付加物を含まない場合は、タンパク質の変性は阻害物の迅速な解離をもたらす。したがって、[3H]-リガンド-タンパク質複合体を変性させ、さらにSDS-PAGEによりタンパク質-結合放射能を検出することによって、複数のマイケル付加物が簡単に単離された。そのような複合体の検出は以下を提供する:(a)共有結合付加物形成の証拠、及び(b)平衡(Kd)及び動力学定数(Koff及びKon)を決定するための相互反応定量用ツール。
例えば、MEK1及び他の標的Cys含有キナーゼの種々の利用可能な形態は、蛍光若しくは [3H]-ヒポテマイシン又は同様な類似体で処理し、SDS-PAGE又は変性ゲル浸透カラムに付し、前記ゲル又はカラムをタンパク質結合蛍光又は放射能について分析することができる。安定な複合体が形成される場合、複数の重要な試験を実施することができる。例えば、複合体をSDS-PAGEから単離し、トリプシンで消化し、クロマトグラフィー又は質量スペクトル(MS)分析によってタンパク質の共有結合ペプチドを同定することができる。 [3H]標識又は蛍光標識エノンの濃度を変化させ、複合体をSDS-PAGEによって単離/定量することによって、複合体形成の平衡及び動力学的特性を決定することができる。培養哺乳動物細胞又はそのような細胞から得た可溶性細胞抽出物を[3H]標識又は蛍光標識したヒポテマイシンで処理し、2Dゲルで解析し、放射能スポット中のタンパク質をMALDI MSによって同定することができる。例えば、MEK1がヒポテマイシンとの共有結合付加物形成のための唯一の標的であるならば、MEK1は標識されるただ1つのタンパク質であろう。複数のタンパク質が標識されるならば、さらに別の標的が存在すると結論することができ、それらを同定することができる。
例えば、キナーゼの必須システインとの共有結合形成は、共有結合複合体のタンパク分解消化によって得られるペプチドの質量スペクトル分析によって示すことができる。図7は、ヒポテマイシン処理又は非処理ERK2のトリプシン消化物の質量スペクトルを示す。951の質量ピークは、標的Cys172残基を含む最小のトリプシン消化ペプチドの質量に対応する。以前にヒポテマイシンで処理されたERK2の非活性化及び活性化形のトリプシン消化物は、標的Cysペプチドの質量の増加は1273(ヒポテマイシンの質量と正確に一致する量)であることを示している。
【実施例4】
【0098】
実施例4:Cysキナーゼ依存癌から培養された細胞のラクトンによる分裂阻害
マイケル付加物形成に感受性を有する活性部位Cys残基を含むタンパク質キナーゼを有するか、又は前記タンパク質キナーゼによって活性化される、活性なシグナル伝達経路を必要とする腫瘍に由来する細胞株の細胞分裂を阻害する、本発明の方法で有用な化合物の能力は、細胞分裂アッセイ及び細胞株(例えばHT-29(ヒト結腸癌)、COLO829(メラノーマ)、MV-4-11(急性骨髄性白血病)及びP815(マウスの肥満細胞腫))を用いて示すことができる。ある例示的な方法では、細胞を96ウェルプレートで種々の濃度の阻害剤で処理し、37℃/5%CO2で3日間インキュベートし、さらにセルタイターグロ(Cell Titer Glo)キット(Promega)を用いて分析する。
表4は、マイケル付加物形成に感受性を有する活性部位Cys残基を含むタンパク質キナーゼを有するか、又は前記タンパク質キナーゼによって活性化される、活性なシグナル伝達経路を必要とする腫瘍に由来する細胞株に対する、本発明の方法で有用な化合物の増殖阻害特性を示す。疾患の感受性が主としてそれによってもたらされる変異キナーゼだけでなく、感受性に寄与すると合理的に推測される他のタンパク質キナーゼもまた示される。
【0099】
表4:キナーゼ変異細胞株におけるキナーゼ阻害剤の細胞傷害性
* タザロテンの場合10
【実施例5】
【0100】
実施例5:全細胞のシグナル伝達経路に対するラクトン阻害剤の影響
個々のキナーゼ(例えばMEK)の阻害による下流の影響は前記キナーゼをリン酸化のために必要とするいくつかのタンパク質(例えばERK1)のリン酸化状態を測定することによって確立することができる。培養細胞をヒポテマイシン又は本明細書に記載した他のラクトン類似体で処理し、細胞抽出物のウェスタンブロットを、下流の標的の非改変形及びリン酸化形に特異的な抗体をプローブとして用いて調べた。例として、MEK1/2に対するヒポテマイシンの影響はERK1/2のリン酸化レベルを測定することによって決定することができる。図8は、COLO829細胞(BRAF V599E変異を含む)のヒポテマイシンによる処理は、ERKのリン酸化形の急速な(10分以内)枯渇をもたらすことを示している。同様に、高レベルのマイトジェンレセプターキナーゼヒポテマイシン標的(例えばMV-4-11、前記はFLT3(ITD)変異を有する)を含む細胞の処理は、FLT3のリン酸化形だけでなくレセプターチロシンキナーゼMEK及びERKの下流の両標的のリン酸化形の低下ももたらす。
図8を参照すれば、B-Raf V599E変異メラノーマ細胞株COLO829を1μMのヒポテマイシンとともに2、5、10、15、30及び60分間インキュベートした。続いて前記細胞を溶解させ、タンパク質を抽出した。各サンプルから取った等量の全タンパク質をSDS-PAGEで分離し、続いてPVDF膜に電気的にブロッティングした。前記の膜を抗リン酸化ERK抗体(Cell Signaling Technologies)とともにインキュベートし、続いてHRP結合二次抗体とともにインキュベートすることによって、各抽出物に存在するリン酸化ERKレベルを可視化した。ECLウェスタン検出キット(Amersham)を用いてリン酸化ERK含有バンドをオートラジオグラフィーによって検出した。ERK抗体をプローブとして用いてこのブロットを再度調べて、等レベルの全ERKが各レーンにロードされていることを示した(データは示さず)。
可逆性阻害剤に関しては、作用を受けた下流のキナーゼのリン酸化によって測定されるように、標的Cysキナーゼを阻害する作用は迅速に達成される。しかしながら、可逆性競合性阻害剤とは異なり、図9に示すように、ラクトンは1時間未満の短時間曝露後に細胞から除去することができ、阻害を受けたキナーゼは長時間(24時間まで)にわたって回復しない。したがって、in vitroの場合のように、細胞では、共有結合阻害剤-キナーゼ付加物が迅速に形成され、長時間にわたって結合が維持される。したがって、これら阻害剤の薬剤として尋常ではない特性は、薬剤の標的への短時間曝露によって長時間持続する作用を示すことができるということであり、これは、オフターゲット作用により毒性を回避しつつ最大効果を達成するためにスケジュールを立てるということに関連して所望の選択肢を提供する。このことはまた、標的キナーゼの顕著な阻害を担保するために用量及び半減期が充分であることを条件として、比較的短いin vivo半減期を有するRALを本発明の方法で有効に用いることができることを意味する。
図9を参照すれば、B-Raf V599E変異細胞株HT29をDMSO、1μMのU0126又はヒポテマイシンのいずれかとともに1時間インキュベートした。前記1時間のインキュベーションに続いて、細胞を培養液で2回洗浄し、さらにインキュベートした。薬剤処理並びに洗浄の3、6及び24時間後に直ちにタンパク質抽出物を調製した。各サンプルから取った等量の全タンパク質をSDS-PAGEで分離し、続いてPVDF膜に電気的にブロッティングした。前記の膜を抗リン酸化ERK抗体(Cell Signaling Technologies)とともにインキュベートし、続いてHRP結合二次抗体とともにインキュベートすることによって、各抽出物に存在するリン酸化ERKレベルを可視化した。ECLウェスタン検出キット(Amersham)を用いてリン酸化ERK含有バンドをオートラジオグラフィーによって検出した。
薬剤の開発の前に、化合物の薬理学的動力学、生体利用性、動物における抗腫瘍活性及び急性毒性試験を実施した。レゾルシル酸ラクトンについての現時点で存在する知識を基準にすれば、本発明の方法で有用な化合物は顕著な毒性をもたず、良好な生体利用性を有するはずである。そのような薬剤に対する感受性が予測される変異対立遺伝子座をもつ患者のタイピングもまた、イレッサによる肺癌患者の最近の研究によって実施されたように、いくつかの実施態様で実施される(例えば悪性メラノーマにおけるB-Raf変異)。
【実施例6】
【0101】
実施例6:本発明の化合物の調製及び特性
本実施例は、下記式(II)の構造を有する本発明の化合物、すなわち4-O-デスメチルヒポテマイシンの調製を述べる。特にこの化合物は、その精製形及び単離形で提供される:
【0102】
【化13】
【0103】
接種調製物:以下の機関(Deutsch Sammlung von Mikroorganismen und Zellkulturen (DSMZ ))から入手した、20%(v/v)グリセロール中で維持されたヒポミセス・スビキュロスス(Hypomyces subiculosus)DSM11931の凍結細胞の1mLを、バッフル無しの250mLエーレンマイヤーフラスコ中の50mLの種培養液に接種した。前記種培養液は、水に30g/Lのクォーカーひき割りエンバクから成り、オートクレーブの前に70〜80℃で10分間加熱した。前記種培養物を22℃の暗所で、ロータリーシェーカー(5.08cm(2インチ)のストローク、190rpm)で3日間インキュベートした。50mLのエンバクフレーク培養液を含むバッフル無しの50mLエーレンマイヤーフラスコに2mLの一次種培養物を移し、二次種培養物を作製した。
ファーメンター生産:12LのCYS80培養液(Dombrowski et al. J Antibiot 1999, 52(12):1077-1085)を含む20Lのバイオリアクターをそのまま121℃で30分滅菌した。前記培養液は、80g/Lのスクロース、50g/Lのコーンミール(Sigma)及び1g/Lのバクト酵母抽出物(BD)から成っていた。続いて前記培養液を480mLのH.スビキュロススDSM1193の二次種培養物とともにインキュベートした。発酵は、通気速度0.4v/v/m及び初期攪拌速度200rpmで22℃で実施した。培養の溶解酸素は、200〜400rpmの攪拌カスケードによって30%の空気飽和で管理した。泡立ちは、100%のUCON LB-625の自動添加で管理した。培養のpHはモニターしたが、制御はしなかった。D,L-エチオニンを接種のときに50mg/Lの濃度で添加した。発酵は、最大のKOSN-2176産生が得られるまで35日間継続させた。サンプルは必要に応じて抜き取り、後の分析のために-20℃で保存した。
当業者はCYS80培養液組成の変型は有用であることを理解しえよう。例えば、前記は約30〜120g/Lのスクロース、約20〜80g/Lのコーンミール、及び約0(好ましくは約0.1)〜10g/Lの酵母抽出物を含むことができる。同様に、D,L-エチオニン濃度は、例えば約10〜10mg/L培養液で変動し得る。
化合物IIの蓄積を促進するために、C-4ヒドロキシル基のメチル化を触媒してヒポテマイシンを生成するために必要なメチルトランスフェラーゼの阻害剤として種々の化合物を判定した。D,L-エチオニン(前記はメチルトランスフェラーゼ阻害剤であると文献で報告された)は化合物IIの産生の増加に有効であることが見出されたが、一方メチルトランスフェラーゼ阻害剤は有効ではないとも報告された。さらにまた、複数の培養液を判定し、CYS80が他のものよりも化合物IIの産生を促進した。化合物IIの力価は、40mg/mL〜540mg/mL(20リットルバイオリアクター)又は900mg/mL(振盪フラスコ)に改善された。
化合物IIの定量: 1mLのメタノールを用いて500μLの発酵ブロス抽出することによって、化合物II及びヒポテマイシンの産生をモニターした。続いて、混合物を13,000gで3分遠心した。ヒューレットパッカード1090HPLCを用い、220、267及び307nmでのUV検出により上清中の2つの生成物の定量を実施した。5μLの上清を4.6x10mmのガードカラム(Inertsil, ODS-3, 5μm)及びより長い4.6x150mmのカラム(Inertsil, ODS-3, 5μm)に注入した。最終的なヒポテマイシン濃度が1g/Lになるまでサンプルをメタノールで希釈した。前記アッセイ方法は周囲温度で1mL/分の流速で実施した。前記は、8分にわたる40:60〜80:20のアセトニトリル:水の勾配から成り、続いて4分間の100%アセトニトリル洗浄を実施した。両移動相は0.1%(v/v)の酢酸を含んでいた。精製した化合物II及びヒポテマイシンを用いて標準物を調製した。
化合物IIの精製: H.スビキュロススDSM11931の2つの12リットル発酵物の発酵ブロス24リットルを24リットルの100%メタノールで1時間抽出した。前記混合物を薄層セリットを含む真空フィルター(Hyflo)に通し、前記フィルターケーキを1Lの50:50メタノール:水で洗浄した。ろ液を水で希釈し最終メタノール濃度を30%(v/v)にした。精製方法に用いた全ての溶媒は0.1%(v/v)の酢酸を含んでいた。
ミリポアモジュリン(50cmx9cm)プロセスカラムに1.3LのHP-20SS樹脂(Mitubishi)を充填し、30:70メタノール:水の3カラム容積で700mL/分で平衡化した。生成物プールを同じ流速でカラムにロードした。カラムを1CVの30:70メタノール:水で洗浄し、段階勾配(3CVの45:55メタノール:水、9CVの50:50メタノール:水、及び3CVの60:40メタノール:水)で300mL/分で溶出させた。画分(1.5CV)を採集し、HPLCで上述のように分析した。画分3〜15を生成物プールとして一緒にした。
ミリポアモジュリン(50cmx9cm)プロセスカラムに2.3LのC18ソルベント(Bakerbond, 40μm)を充填し、30:70メタノール:水の3CVで180mL/分で平衡化した。HP-20SSクロマトグラフィー工程から得た生成物プールを水で希釈し、最終メタノール濃度を30:70メタノール:水にし、C18カラムに180mL/分でロードした。カラムを30:70メタノール:水の1CVで洗浄し、さらに9CVの42:58メタノール:水で180mL/分180mL/分で溶出させた。画分(0.4CV)を採集し、上述のようにHPLCで分析した。画分10〜16を生成物プールとして一緒にした。
化合物IIの結晶化を促進するために、前記生成物プールを40℃での回転蒸発で濃縮し、その容積を36%に減少させた。続いて、−20℃に冷却した。形成された化合物IIの白色結晶をワットマン#5のろ紙を設置したブッフナーロートに通し、100mLの冷却水で洗浄した。最終生成物を40℃の真空オーブンで一晩乾燥させ、4℃で保存した。精製方法の全体的収量はほぼ60%であった。精製方法終了時の化合物IIの純度はほぼ95%であった。
化合物IIの特徴付け:精製された化合物IIは白色の結晶として得られた;UV(MeOH)λmax 219, 266, 306nm;HRESIMS m/z 363.1074 [M-H]-(C18H19O8で計算、363.1065);1H及び13C NMRデータ(表5及び6参照)。
【0104】
表5:化合物IIの1H NMR
【0105】
表6:化合物IIの13C NMR
【実施例7】
【0106】
実施例7:化合物の合成
本実施例は、本発明の方法で有用な別の化合物の合成について述べる。
【0107】
【化14】
【0108】
THF(4.0mL)中の化合物II(12mg、0.033mmol)の攪拌溶液に、3-モルホリノプロパン-1-オール(10.0μL、0.072mmol)、トリフェニルホスフィン(22.6mg、0.086mmol)及びジエチルアゾジカルボキシレート(13.4μL、0.086mmol)を添加した。室温で3時間攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、1.2mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液の10%〜90%勾配を用いた40分の溶出によって、化合物III(11mg、70%収量)が提供された:LRMS m/z (M+H) C25H34NO9による計算値492.2;実測値492.2。
【0109】
【化15】
【0110】
THF(2.0mL)中の化合物II(6mg、0.017mmol)の攪拌溶液に、3-(4-メチルピエラジン-1-イル)プロパン-1-オール(5.0μL、0.036mmol)、トリフェニルホスフィン(12mg、0.043mmol)及びジエチルアゾジカルボキシレート(7.0μL、0.043mmol)を添加した。室温で45分攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、0.8mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液に10%〜90%勾配を用いた40分の溶出によって、化合物IV(3.8mg、45%収量)が提供された:LRMS m/z (M+H) C26H37N2O8による計算値505.2;実測値505.2。
【0111】
【化16】
【0112】
THF(1.0mL)中の化合物II(3mg、0.009mmol)の攪拌溶液に、(1-メチルピエラジン-3-イル)メタノール(5.0μL、0.036mmol)、トリフェニルホスフィン(16mg、0.048mmol)及びジエチルアゾジカルボキシレート(6.3μL、0.048mmol)を添加した。室温で3時間攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、0.8mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液に10%〜90%勾配を用いた40分の溶出によって、化合物IV(1.7mg、40%収量)が提供された:LRMS m/z (M+H) C25H34NO8による計算値476.2;実測値476.2。
化合物II〜Vの生物学的特性をアッセイし、ヒポテマイシンの特性と比較した。COLO829はヒトのメラノーマ細胞株である。HT29はヒト大腸癌細胞株である。両細胞株はV600E B-Raf変異を有する。SKOV3は野生型B-Rafをもつ卵巣癌細胞株である。EKR2(細胞外シグナルによって調製されるキナーゼ)はRas/B-Raf MAPキナーゼカスケード経路のキナーゼである。結果は表7に示されている。
【0113】
表7:化合物II〜Vの特性
本発明をこれまで記述による詳細な説明及び実施例で述べてきたが、前記は多様な態様で実施され得ること、及び前述の記載及び実施例は例示を目的とし、特許請求の範囲を制限するものではないことは、当業者には理解されよう。
【図面の簡単な説明】
【0114】
【図1】ERK/MAPKシグナル伝達経路の模式図を示す。
【図2】ある種のレゾルシル酸ラクトンの化学構造を示す。
【図3】ヒポテマイシンが共有結合したキナーゼERK2のX線構造を示す。
【図4】ヒポテマイシンが共有結合したキナーゼERK2のX線構造を示す。
【図5】60細胞株のNCIパネルに対するヒポテマイシンのlog GI50値(50%増殖低下を達成するために必要な薬剤の量)を棒グラフの形で示す。前記化合物に対してもっとも感受性を有する細胞株は、垂直線の平均活性から右を指す棒線で表されている。
【図6】ヒポテマイシン及び非RAL薬剤についての異種移植比較データを示す。
【図7】ヒポテマイシンの存在下及び非存在下におけるキナーゼERK2のトリプシン消化の質量スペクトルの比較である。
【図8】BRAFV599E変異を有するColo829細胞におけるキナーゼERKのリン酸化に対するヒポテマイシンの作用を示す。
【図9】B-Raf V599E変異を有するHT29細胞におけるヒポテマイシンによるキナーゼERKリン酸化阻害の時間を示す。
【技術分野】
【0001】
本発明は、特定のタンパク質キナーゼを阻害し、さらにヒトの疾患の治療に有用である化合物を提供する。本発明は、化学、生化学、分子生物学、医学、及び薬理学分野に関する。
【背景技術】
【0002】
分子を標的とする癌治療薬は、現時点及び将来の癌治療に極めて有望である。複数のタンパク質が、細胞シグナル伝達の特定の工程で決定的な役割を果たすと認定された。これらシグナル伝達経路のタンパク質は、正常な健常細胞に対してある程度の選択性を許容するので癌治療薬の魅力的な標的である(Sausville et al. Annu Rev Pharmacol Toxicol (2003) 43:199-231)。しかしながら、細胞シグナル伝達は典型的には複数の経路を必要とするので、ある特定のシグナル伝達経路のタンパク質の特異的な阻害剤は、所望の治療結果を得るためには不充分であるかもしれない。逆に、複数のシグナル伝達経路の非特異的な阻害は正常細胞に対して有害な結果を有し、したがってシグナル伝達経路のタンパク質を標的にするという目的の出鼻をくじくであろう。
この領域における有効な医薬の開発はしたがって困難であり予想不可能である。特定の細胞シグナル伝達経路を阻害するその能力を規準に開発された化合物は、それがまた別の細胞シグナル伝達経路のタンパク質を同様に阻害する場合にのみ特定の適応症について機能することができる。しかし前記機能は従来技術では予測することができない特性である。例えば、グリーベック(Gleevec)(イマチニブメシレート、STI-571, Novartis)は、Bcr-Ablチロシンキナーゼの特異的阻害剤として設計されたが、その有効性はc-Kit及び他のチロシンキナーゼを同様に阻害するその能力に左右される。したがって、グリーベックは実際、慢性骨髄性白血病(CML)細胞の機能の維持に重要なBcr-Ablチロシンキナーゼを阻害し(Hernandez-Boluda et al. Drugs Today (Barc) (2002) 38:601-13)、CMLに対して有効であるが、その有効性はまた部分的にはc-Kitチロシンキナーゼを阻害するその能力に左右される(前記能力はまた、変異によってc-Kitチロシンキナーゼが上昇する胃腸の間質腫瘍に対してグリーベックを有効にする)(Blanke et al. Curr. Treat Options Oncol (2001) 2:485-91)。
【0003】
グリーベックはまた、癌治療薬の開発でタンパク質キナーゼを標的にすることの重要性を例証する。500を超えるタンパク質キナーゼの大きなファミリーの中のメンバーは、全てではないとしてもほとんどが重要な細胞シグナル伝達経路で必要とされる。4つの主要なシグナル伝達経路又はカスケード(1つは細胞外マイトジェンに応答性であり、他のものはストレスシグナルに応答性であり、それぞれはタンパク質キナーゼによって制御され、さらに各々は複数の他のタンパク質キナーゼを含む)は、癌細胞分裂及び細胞のストレス応答で不可欠の役割を果たし、したがって抗癌剤及び抗炎症剤の開発に極めて重要である。しかしながら、どのようにしてある化合物が、複数の異なるシグナル伝達経路において多くの異なるタンパク質キナーゼに影響を及ぼすかについての予想不可能な性質は、医薬の開発を遅らせ続けている。
異常なマイトジェン活性化タンパク質キナーゼ、いわゆるMAP(マイトジェン活性化タンパク質)キナーゼ又はMAPK酵素が中心的に関与する細胞シグナル伝達経路の遮断(Chen et al. Chem Rev (2001) 101:2449-76;Pearson et al. Endocr Rev (2001) 22:153-83)は、新しいタイプの癌治療薬の発見及び開発に重要な方向として出現してきた(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Sebolt-Leopold, Oncogene (2000) 19:6594-99)。MAPK依存経路の1つは、細胞外シグナルに由来するシグナルの伝達を可能にする。前記細胞外シグナルは、例えば表皮増殖因子(EGF)及び血管内皮由来増殖因子(VEGF)である。前記因子は、細胞膜の対応するレセプター(それぞれEGFR(HER)及びVEGFR)と結合し、前記レセプターは、中継キナーゼ及びキナーゼ標的(例えばERK経路:Ras、Raf-1、A-Raf(BRAF)、MEK1及びMEK2(前記は包括的に本明細書ではMEK1/2と称する)、並びにERK1及びERK2(前記は本明細書では包括的にERK1/2と称する))を介して細胞の核にシグナルを送る。後者のタンパク質は、最終的には生存のための細胞機能、例えば分裂、増殖、運動性及び生存を制御する遺伝子の発現を支配する。2〜3つの他のタンパク質キナーゼ経路は“ストレスシグナル”に応答する。
【0004】
特定のタンパク質キナーゼを標的とする小分子の非タンパク質性医薬が開発中であり(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Sebolt-Leopold, Oncogene (2000) 19:6594-99;Noble et al. Science (2004) 303:1800-05)、3つが使用を承認された。すなわちグリーベック、ゲフチニブ(イレッサ;Barker et al. Bioorg Med Chem Lett (2001) 11:1911-14)及びエルロチニブ(タルセバ)である。医薬として承認された小分子キナーゼ阻害剤の不足は、従来の方法の非予測性を例証した。特定のタンパク質キナーゼを阻害する化合物を設計し、それらの標的の3D構造の助けを借りて前記を評価することができるが(Noble et al. Science (2004) 303:1800-05)、一方、臨床的知見は、多くの化合物が前臨床活性を基準にした楽観的期待に応えることができないことを示した(Sausville et al. Annu Rev Pharmacol Toxicol (2003) 43:199-231;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。この失敗は、部分的には、単純に特定のキナーゼを阻害するその能力を基準にして、重要な細胞シグナル伝達経路における無数の他のタンパク質キナーゼに対する阻害剤の作用を予測することの難しさから生じる。したがって、特異的なタンパク質キナーゼ及び特異的なタンパク質キナーゼサブセットを標的とする新規で改善された医薬及び同定方法、並びに癌及び他の疾患の治療で既知の阻害剤を使用する方法が強く希求されている。
そのような医薬はヒトの疾患の治療に重大な影響を与えることができるだろう。例えば、癌の治療では、MAPK経路の薬理学的阻害剤は、シグナル伝達過程におけるいくつかの異なるタンパク質のいずれも標的とすることができるだろう(English et al. Trends Pharmacol Sci (2002) 23:40-45;Kohno et al Prog Cell Cycle Res (2003) 5:219-24)。癌治療のために特に興味深いタンパク質にはMAPK/細胞外シグナル関連(ERK)キナーゼ(MEK又はMKKと称される)が含まれ、前記は、特にMAPKシグナル伝達のERK支流(Ras/Raf-1、A-Raf及び/又はB-Raf、MEK1/2、及びERK1/2(図1参照)を含む)で作用するキナーゼである。G-タンパク質のRasは、マイトジェン活性化増殖因子レセプターのシグナルをRaf-1、A-Raf及び/又はB-Rafへ伝達する。後者は二重特異的セリン/スレオニン及びチロシンキナーゼMEK1/2をリン酸化し、したがってこれを活性化し、前記は続いてERK1/2を活性化する。Ras/Raf/MEK/ERK経路は、報告によればもっとも詳しく特徴付けられたシグナル伝達経路であるヒトの癌の発生及び増殖に必要とされ、抗癌剤開発の標的として提唱されてきた(Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。
【0005】
しかしながら、細胞分裂及び癌の移動並びに正常細胞の生命機能を制御する複雑な経路群は、ただ1つの経路又は経路複合体の支流のみを阻害する化合物は有効ではない可能性を示唆した。正常細胞にとって有害な非特異的な活性を示すことなく、複数の経路を正確に阻害する化合物を設計し、これを試験することは困難である。MEK1/2キナーゼを標的とする化合物は前記問題の例証である。
MEK1/2は抗腫瘍薬(抗癌剤)の開発のための標的として以下の2つの優れた特徴を有する:(1)それらは、Rasを経過する多様なマイトジェン活性化タンパク質キナーゼの入力を統合する経路集合点の決定的地点に存在し;さらに(2)それらは、制限的な基質特異性を有し、MAPK ERK1/2は重要な唯一の公知の基質である。MEK1/2の構成的活性化又は活性強化は、複数のヒト原発性腫瘍で検出され(Hoshino et al. Oncogene (1999) 18:813-22)、実際、B-Rafのただ1つの変異がERK経路を構成的に活性化し、変異遺伝子は腫瘍原性である。主要なB-Raf変異はV599E(この変異の正確な名称はV600Eであるが、ほとんどの文献、特に古い文献は前記をV599Eと称している)である(Davies et al. Nature (2002) 417:949-54)。しかしながら、ほんのわずかの小分子又はMEK1/2のアンチセンス阻害剤(PD184352/CI-1040(Pfizer)、U-0126(Promega)及びWyeth-Ayerstの化合物(Zhang et al. Bioorg Med Chem Lett (2000)10:2825-28)又はRaf-1/B-Rafのアンチセンス阻害剤(BAY-439006)(Lyons et al. Endocr Relat Cancer (2001) 8:219-25)が前臨床開発段階又は臨床試験中であると報告されている(Kohno et al Prog Cell Cycle Res (2003) 5:219-24;Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。これまでのところ、特異的で強力なERK1/2阻害剤は報告されていない。
【0006】
公知のMEK1阻害化合物のいくつかの特性の調査によって、それらの有効性は、部分的には複数の経路を阻害するその能力に左右され得ることが明らかになった。PD184352及びU-0126はMEK1を阻害しATPと非競合的であり、おそらくはATP結合部位の外側で結合するアロステリック阻害剤として機能するのであろう。これらの化合物はまた、同様な濃度でMEK5-ERK-5経路の活性化を阻害する。両化合物は動物で抗腫瘍活性を有し、特にERK経路が構成的に活性化される腫瘍に対して活性を有し、報告によれば臨床試験中である(Dancey et al. Nat Rev Drug Discov (2003) 2:296-313)。
しかしながら、たとえ開発中のこれらのMEK1阻害化合物が複数のシグナル経路を標的とすることができるとしても、医薬としてのそれらの成功は決して確実とはいえない。複数のシグナル伝達経路の阻害が要求されるならば、前記医薬は、各経路で少なくとも1つのタンパク質キナーゼを充分な能力で阻害し、所望の治療結果を生じなければならない。さらにまた、そのような医薬はしばしば原則的に細胞分裂抑制剤であり、腫瘍細胞を効率的に殺さず、耐性及び再発をもたらす可能性は高い。迅速な可逆性を有する阻害剤である医薬の場合、それらの除去又は細胞レベルでのそれらの減少は、腫瘍細胞分裂の再開を許容する。共有結合により結合する阻害剤は、可逆的なタンパク質キナーゼ阻害剤よりも有効であり得る(Noble et al. Science (2004) 303:1800-05)。前記はEGFR及びHer-2を阻害する医薬について示されたとおりで、この場合、化合物は、ATPポケットでシステイン残基とマイケル付加によって共有結合を形成する(Wissner et al. Bioorg Med Chem Lett (2002) 12:2893-97;Baslega et al. Oncology (2002) 63 Suppl 1:6-16;Wissner et al. J Med Chem (2003) 46:49-63)。医薬として開発することができるタンパク質キナーゼ阻害剤がなお希求されており、それらの標的を共有結合により改変して前記標的を阻害する阻害剤はヒトの疾患の治療で特に有用である。
【0007】
タンパク質キナーゼ阻害剤の探索では天然の生成物が研究された。なぜならば、そのような化合物は、シグナル伝達経路に影響を与える医薬の先導物質として貴重であることが証明されたからである(Newman et al. Curr Cancer Drug Targets (2002) 2:279-308)。“レゾルシル酸ラクトン” (本明細書ではまた“RAL”と称される)として知られる菌類のこのクラスの天然の生成物には、ゼアラレノン(前記はエストロゲン様作用を有し、動物で同化作用促進剤(例えばゼアララノール)として用いられている)の他に(5Z)-7-オキソゼアネオール、ヒポテマイシン、Ro-09-2210、及びL-783,277(前記は細胞分裂を阻害し(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Camacho et al. Immunopharmacology (1999) 44:255-65)、さらに抗腫瘍特性を有すると報告されている(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Tanaka et al. Jpn J Cancer Res (1999) 90:1139-45))が含まれる。さらにまた興味深いことは、ナノモル濃度の低いIC50値でin vitroで、細胞内でのJNK/p38シグナル伝達(Tanaka et al. Biochem Biophys Res Commun (1999) 257:19-23)、血小板由来増殖因子(PDGF)レセプターの自己リン酸化(Giese et al. US5,728,726(1998))MEK1/2(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94;Dombrowski et al. J Antibiot (Tokyou) (1999) 52:1077-85;Williams et al. Biochemistry (1998) 37:9579-85)又はTAK1(MEKK)(Ninomiya-Tsuji et al. J Biol Chem (2003) 278:18485-90)を阻害するそれらの能力である。しかしながら、それらの興味深い活性にもかかわらず、レゾルシル酸ラクトンはこれまでヒトで調べられたことはなく、医薬としても承認されていない。
【0008】
レゾルシル酸ラクトンL-783,277は、精製MEK1(IC50は4nM)のリン酸化を阻害するが、PKA、PKC又はRafは阻害しない。前記阻害はATPと競合し、60分のプレインキュベーションはMEK1に対するIC50値を10倍低下させる(Zhao et al. J Antibiot (Tokyo) (1995) 52:1086-94)。MEK1をL-783,277と30分プレインキュベートし、続いてゲルろ過することによって、不活性なMEK1タンパク質が回収され、L-783,277は緊密にMEK1と結合することを示している。しかしながら、L-783,277よりも、5E C=C異性体は能力が100倍低く、さらに7-ジヒドロヒドロキシル異性体は400〜5000倍能力が低いが、明瞭なSARは出現しない(Zhao et al.上掲書)。ヒポテマイシン(図2参照)(前記は構造的にL-783,277に類似するが、11,12-エポキシド成分を有する)はMEK1阻害剤としての能力は4倍低い(Zhao et al.上掲書)。Ro-09-2210はMEK1の強力な阻害剤であり(IC50は59nM)、未発表研究において(Williams et al. Biochemistry (1998) 37:9579-85を参照されたい)、MEK4、6及び7を4〜10倍高いIC50値で阻害すると主張されている。(5Z)-7-オキソゼアネオールはTAK1 MEKK酵素に対して同様な能力を有し(IC50は8nM)、ラットのMEK1に対しはより弱い阻害を示した(IC50は411nM)(Ninomiya-Tsuji et al. J. Biol Chem (2003) 278:18485-90)。
前記のような類似体によるこれら標的キナーゼの強力な阻害の理由は、本発明より前には明らかではなく、ヒトゲノムでコードされるおよそ500を超えるタンパク質キナーゼ(“キノム(kinome)”)に対する総合的な評価は、これらの化合物又は他のいずれの化合物についてもこれまで実施されてなかった。そのような評価は、タンパク質キナーゼアッセイが、これらキナーゼのおよそ150についてのみ開発されてきたためにこれまでは可能ではなかった。化合物がキナーゼを阻害することができるか否かをアッセイする方法、及び化合物がどのキナーゼを阻害するかを決定する方法がなお希求されている。そのような方法が存在しなければ、さらに複数のキナーゼのin vitro判定が存在しなければ(前記はこれまでいずれのRAL化合物についても報告されていない)、タンパク質キナーゼファミリーメンバー間における化合物の相対的な選択性を決定することができないし、さらにヒトの疾患における化合物の有用性を容易に評価することもできない。
したがって、タンパク質キナーゼ阻害剤を同定する方法、及びそれらのキノムにおける相対的選択性及び特にヒトの疾患に中心的に関与する種々のタンパク質キナーゼに対する相対的選択性を判定する方法がなお希求されている。そのような方法を用いて、ある疾患の生物学に直接関係する複数の細胞シグナル伝達経路のタンパク質キナーゼを生産的に阻害する化合物を同定及び選別することが可能であろう。特異的な標的およびシグナルトランスダクション経路のみを阻害する阻害剤を選別し、それらを医薬品として処方し、さらにそれらを投与して、そのような標的の阻害が治療効果(例えば癌、炎症及び他の症状に対抗することを含む)を提供する疾患を治療することが可能であろう。本発明はこれらの要求を満たし、下記に開示する方法、化合物及び医薬品を提供する。
【発明の開示】
【0009】
(発明の要旨)
第一の観点では、本発明は、タンパク質キナーゼの識別可能なサブクラスを、タンパク質キナーゼとマイケル付加物を形成することができる化合物とともに用いて、タンパク質キナーゼを阻害する方法を提供する。タンパク質キナーゼの前記サブクラスは、リン酸化標的及びATPのリン酸と複合体を形成するMg2+と相互作用するタンパク質キナーゼ内の高度に保存された2つのアスパラギン酸残基(Asp)の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン残基(Cys)を有するタンパク質キナーゼを含む。前記タンパク質キナーゼ内のこれらのアミノ酸はATP結合部位として知られる領域に存在する。本発明の方法では、そのようなシステイン残基を有するタンパク質キナーゼは、前記システイン残基でマイケル付加物を形成することができる化合物と接触させることによって阻害される。マイケル付加物形成の結果、前記阻害剤とキナーゼの間で共有結合が生じ、したがって前記阻害は本質的に不可逆になる。
ある実施態様では、該Cysを含むサブクラスに由来する1つ以上のタンパク質キナーゼと前記の重要なCys残基を欠くキナーゼに由来する1つ以上のタンパク質キナーゼとを含むタンパク質キナーゼの混合物は、該Cys残基を含む酵素と可逆的な複合体を形成し、続いて該Cys残基とマイケル付加物を形成することができる部分を含む化合物と接触させられる。ある実施態様では、前記化合物の前記部分はZ-エノン(Z-α、β-不飽和カルボニル部分)である。ある実施態様では、前記化合物の前記部分はレゾルシル酸ラクトンに含まれているか、又は7位のカルボニルと共役している5−6位のシス炭素-炭素二重結合を含む誘導体であるか(α、β-不飽和ケトン;図2参照)、又はそのような部分のバイオイソスター(bioisostere)(例えばエステル、アミド、ビス-ラクトン、スルホンアミド又はスルホン)である。本方法では、重要なCys残基を含むキナーゼのサブクラス由来の1以上のタンパク質キナーゼだけがマイケル付加物形成によって阻害され、該Cys残基を欠くタンパク質キナーゼは阻害されないか(又は同じ程度には阻害されないか)、又はマイケル付加物形成を必要としない異なるメカニズムによって阻害される。
【0010】
本発明の方法は多様な混合物を用いて実施することができる。ある実施態様では、前記混合物はin vitroアッセイで用いられる反応混合物である。別の実施態様では、前記混合物は細胞又は細胞の部分である。また別の実施態様では、前記混合物は、細胞培養アッセイから得られる細胞及び培養液を含む。また別の実施態様では、前記混合物は体液又は体組織である。ある重要な実施態様では、前記混合物は、医学的治療を受けているヒト又は他の哺乳動物の病変組織を含む。
本発明の方法で有用なタンパク質キナーゼ阻害剤は、それらの治療効果を発揮するときに、典型的には少なくとも2以上の異なるタンパク質キナーゼを阻害する。本発明の方法で有用な化合物は、それらの所望される効果を達成するときに、例えば、2以上の異なるタンパク質キナーゼ、少なくとも2つの異なるシグナル伝達経路の各々から1つを阻害することができるか、又は同じ経路又は両方の経路の2以上の異なるタンパク質キナーゼを阻害することができる。いくつかの実施態様では、本発明の方法で用いられる化合物は、それらの所望される効果を達成するときに、少なくとも3つの異なるタンパク質キナーゼを阻害する。
ある実施態様では、本発明の化合物はERK経路の複数の酵素を阻害するために投与され、所望の治療効果を達成する。ある実施態様では、これらの酵素はMEK1/2及びERK1/2である。ある実施態様では、本発明の化合物は、ERK経路の複数の酵素をマイトジェンレセプターキナーゼと同様に阻害する。ある実施態様では、前記化合物はVEGFレセプターを阻害し、さらにERK経路の阻害を介してVEGFの生成を阻害する。本発明のそのような化合物は、血管形成を伴う疾患(癌及び黄斑変性を含むが、ただしこれらに限定されない)の治療に特に有用である。なぜならばそれら化合物はVEGFの産生につながる経路の阻害を介してその産生を阻害するだけでなく、そのレセプターVEGFRもまた直接阻害するからである。
【0011】
ある実施態様では、本発明の化合物によって阻害されるタンパク質はMAPキナーゼである。ある実施態様では、阻害される別個のシグナル伝達経路は、少なくとも1つのマイトジェン誘導経路及び1つのストレス誘導経路を含む。ある実施態様では、少なくとも1つのタンパク質キナーゼはMEKである。ある実施態様では、少なくとも1つのタンパク質キナーゼは、MAPKKファミリーのメンバーである。ある実施態様では、少なくとも1つのタンパク質キナーゼはチロシンレセプターキナーゼであり、前記には、野生型及び変異体PDGFRA、PDGFRB、FLT-3、c-KIT、並びにVEGFレセプターが含まれるが、ただしこれらに限定されない。ある実施態様では、少なくとも1つのタンパク質キナーゼはVEGFレセプターであり、前記にはVEGFR1、VEGFR2(KDRとしてもまた知られている)、及びVEGFR3が含まれる。ある実施態様では、少なくとも1つのタンパク質キナーゼはFLT3である。ある実施態様では、少なくとも1つのタンパク質キナーゼはc-KITである。ある実施態様では、少なくとも1つのタンパク質キナーゼはPDGFRA又はPDGFRBである。
ある実施態様では、本発明の方法で有用な化合物によって阻害されるタンパク質キナーゼは以下から成る群より選択される:AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAK。
ある実施態様では、本発明の方法で用いられた化合物は少なくとも2つの前述のタンパク質を阻害する。別の実施態様では、少なくとも3つのタンパク質キナーゼが阻害される。
【0012】
第二の観点では、本発明は、標的Cys残基を含むタンパク質キナーゼとマイケル付加物を形成することができる化合物を治療の必要がある対象者に投与することを含む、疾患の治療方法を提供する。ある実施態様では、前記対象者はヒトである。ある実施態様では、前記化合物はレゾルシル酸ラクトン又は誘導化合物である。本発明以前には、キノム中の各々別個のキナーゼについていずれのレゾルシル酸ラクトン又はいずれのキナーゼ阻害剤の特異性も先験的に予測することは不可能であった。キナーゼの標的の知識は実験的な試験を必要とし、in vitroアッセイは、今日までにキノム中の500を越えるキナーゼのわずか約150について開発されただけである。タンパク質キナーゼ及び多様な正常及び疾患過程におけるそれらの基本的役割の数の大きさ故に、そのような化合物又は他の化合物(たとえ特定のキナーゼを阻害することが示されたとしても)が、キナーゼの阻害及び疾患の治療に要求される特異性を有しているのか、そうではなくて非常に非特異的で生命に関わる正常な過程を害するのかを誰も決定することができなかった。対照的に、本発明のキナーゼ標的は、マイケル付加物を形成することができるか否かのようなそれらの分子構造によって同定されるので、標的の全レパートリーをキノムの利用可能な配列データから同定することができる。
本発明はまた、医薬組成物及び疾患の治療のためにそれらを投与する方法を提供する。ある実施態様では、本方法は、タンパク質キナーゼ阻害剤とともに別の薬剤を同時投与することを含む。ある実施態様では、前記他の薬剤は抗癌剤である。別の実施態様では、前記薬剤は抗炎症剤である。また別の実施態様では、前記薬剤は別のタンパク質キナーゼである。ある実施態様では、前記医薬組成物は、重要なCys残基を含むタンパク質キナーゼサブクラスの1つ以上に対して特異性を有し、さらに前記とマイケル付加物を形成することができ、さらに疾患又は症状を標的とする化合物(レゾルシル酸ラクトン又は誘導体を含むが、ただしこれらに限定されない)を含む。ある実施態様では、前記医薬組成物を投与して、標的Cys残基(タンパク質キナーゼのATP結合部位内の保存された2つのAsp残基の間に位置し、Asp残基の1つと隣接する)を含まないタンパク質キナーゼの阻害の場合には生じ得る望ましくない副作用をもたらすことなく治療効果が達成される。
【0013】
発明の詳細な説明
ヒトゲノムは、これまでのところ、標準的な真核細胞タンパク質キナーゼ型の510の識別可能な遺伝子(ヒト“キノム”と称される)を有すると報告されている(Kositch et al. Genome Biology 2002, 3(9):RESEARCH 0043)。前記タンパク質キナーゼファミリーは治療的処置のための標的の豊富な供給源を提供する。なぜならば、前記のメンバーは、多くの疾患過程(炎症及び癌を含む)において重要な役割を果たすからである。しかしながら、このファミリーに含まれるタンパク質が数多く存在し、且つそれらを包含する種々の細胞シグナル伝達経路も数多く存在するため、医学的利用のため充分に活性な且つ特異的である医薬の発見を困難にし、かつ予想のつかないものにしている。本発明は、多様な生物の多様な異なる細胞シグナル伝達経路に由来する識別可能で特異的なタンパク質キナーゼサブセットを阻害する化合物、組成物及び方法を提供し、したがって疾患の治療においてタンパク質キナーゼを標的とする試みにおいて重要な進歩を提示する。
前記タンパク質キナーゼファミリーでは、2つの高度に保存されたAsp残基(D167及びD185、PKA-Calpha(NP_002721)の残基番号を使用)は以下の役割を割り振られている:第一のAspは、ATPのリン酸基のOHからH+を受け入れ、第二のAspは、ATPのリン酸基と複合体を形成するMg2+と相互作用する(したがって移転のためにγ-リン酸基の位置決定に寄与する)。この領域では、第二のAspの直前(PKA-Calphaの184位に一致する)は可変性の位置であり、ヒトのキナーゼの約10%(およそ50/510)では前記位置がCysである。この位置は、第二のAspに接近しているため、必然的にATP結合部位領域内に存在する。
【0014】
本発明は、一つには、これらのCys含有キナーゼを阻害するある種のレゾルシル酸ラクトンが共通の構造的特徴を共有するという発見から生じている。これらの化合物は、一般的に、5−7位のカルボニルと共役しているシス二重結合を有する(例えば図2の最初の4つの構造を参照されたい)。そのような化合物は以下の分子骨組みを有する(本明細書で用いられる番号付与もまた示されている)
【0015】
【化1】
【0016】
最初の可逆的な酵素-阻害剤複合体の形成後、その複合体内の上述する構造とキナーゼドメイン/ATP結合部位内のCys側鎖とが接近していることによって、続いて非常に緩慢な可逆的、又は事実上不可逆的なマイケル付加物の形成がもたらされ、極めて強力な阻害のメカニズムが提供される。
マイケル付加物は、下記の反応式によって示されるように、形式的には、求核種の共役求電子二重結合への1,4-付加の生成物である:
【0017】
【化2】
【0018】
式中、Xは典型的にはO又はNRであり、Nuは、典型的には炭素、窒素、酸素又は硫黄をベースとする求核基である。前記共役求電子二重結合は、典型的にはα,β-不飽和ケトン、アルデヒド、又はエステル部分に存在するが、または不飽和ニトリル、スルホン、又はニトロ部分にも存在し得る。本出願の目的のために、“マイケル付加物”という用語は、前記生成物生成の正確なメカニズムには関係なく、そのような1,4-付加をもつ形式的生成物を指し、さらにそのような形式的生成物の互変異性形(例えばエノール化形を含む)も包含する。
入手可能な発表されたデータを本発明の観点から調査することによって、前記Cysは、そのような構造を有するレゾルシル酸ラクトンに対して感受性を有すると報告されているいくつかのMAPキナーゼに存在し、非感受性と報告されたものには存在しないことが明らかになった。例えば、シス-エノンレゾルシル酸ラクトンは、MEK1、4、6及び7をMAPKKK TAK1及びマイトジェンレセプターチロシンキナーゼPDGFRと同様に阻害することが報告された。前記標的Cys残基を有しない約10のキナーゼは、ある種のシス-エノンレゾルシル酸ラクトンによって阻害されなかった。
したがって、本発明は、前記構造を含むレゾルシル酸ラクトン及び類似体、並びにキナーゼドメインATP結合部位内の又は前記部位に近接するシステイン残基を含む50までのキナーゼを選択的に阻害するときに前記構造を含むレゾルシル酸ラクトン及び類似体を使用する方法を提供する。本発明はまた、そのような化合物を含む医薬組成物及びそれらを用いて疾患を治療する方法を提供する。特に、本発明の化合物の特異性は、特定の症状と関係を有する複数のキナーゼ標的について予測することができる。本発明の方法は、阻害される標的が原因的又は促進的役割を果たす疾患の治療を提供する。
【0019】
このモデルが適切なことは、キナーゼEKR2とヒポテマイシンとの共有結合複合体のX線構造によって実証された。図3は、2.5オングストロームの解析度の構造において、上部にERK2のN末端突出部、下部にC末端突出部、及びヒンジ領域で共有結合しているヒポテマイシンを有する複合体を示す。図4は、ヒンジ領域のクローズアップ図であり、マイケル反応でヒポテマイシンのエノン二重結合にわたって付加されたシステインの硫黄を示している。
本明細書に開示するレゾルシル酸ラクトン阻害剤によるタンパク質キナーゼの強力な阻害のためには、前記阻害剤は、標的酵素が課す2つの“選択性フィルター”を通過することを要求される。第一に、前記阻害剤は、相応に堅固な結合定数で酵素と可逆的に結合しなければならない。この可逆的な結合フィルターは、可逆的なエネルギー形成結合(例えば水素結合、イオン相互作用、疎水性相互作用)と同様に、阻害剤と酵素の相補的なトポロジーに依存する。第二のフィルターは、酵素の標的チオールと阻害剤のエノン部分のβ-炭素との間で共有結合が形成されることを必要とする。このフィルターは、酵素-阻害剤複合体内に適切なCys残基が存在することを必要とし、前記フィルターの有効性は、マイケル受容体である炭素原子を有する反応性チオールが適切に並列するか否かに左右される。いくつかのレゾルシル酸ラクトンは第一のキナーゼフィルター(可逆的結合)を通過することができず、したがって第二のフィルター(共有結合)に遭遇することはあり得ない。いくつかのレゾルシル酸ラクトンは第一のキナーゼフィルターを通過するが、そのキナーゼは、共有結合を形成するCys残基を持たないであろう。実際に両者の例は本明細書で言及される。本発明のレゾルシル酸ラクトンに対する興味対象の標的は、この両方のフィルターを通過するものである。
【0020】
ほとんどのキナーゼ阻害剤は、慣例的なスクリーニングで発見され、続いて1つ又はいくつかのキナーゼに対して最適化された。結果として、それらは第一のフィルター(上記に記載)を通過するように開発され、それらの特異性は、どれほど多くの異なるキナーゼがそれらの結合部位で類似するトポロジーと可逆的相互作用とを共有するかに左右される。タンパク質キナーゼのATP部位は高度に保存されているので、この部位と結合する可逆的な阻害剤はおそらく多くのキナーゼを阻害するが、しかし予想不能で明らかに無差別的な態様で阻害するわけではない。例えば、およそ120のキナーゼのパネルでは、Sugen11248と同定された化合物は、およそ79のキナーゼを0.002〜6.6μMのKi値の範囲で阻害し(Fabian et al. Nat. Biotechnol. (2005) 23(3):329-36)、これらのうちで、およそ56のキナーゼは0.1μM未満のKi値を示し、したがってin vivoの標的に対応し得る。第二のフィルターとしてのレゾルシル酸ラクトンとの共有結合に関しては、標的Cys残基を含むサブセットのみが不可逆的に阻害されるため、キナーゼ間の識別はユニークなものであり極めて増強される。
キナーゼ-レゾルシル酸ラクトン相互作用の共有結合性と事実上の不可逆性によって、薬剤作用及び本発明によって提供される投与方法に関して更なる利点が提供される。例えば、レゾルシル酸ラクトンは種々のキナーゼと種々の可逆的親和性(Ki)及び種々の共有結合不活化速度(Kinact)を有するので、キナーゼ混合物のレゾルシル酸ラクトンへの曝露(濃度x時間)を制御することによって、ある種のキナーゼの選択的阻害を達成することができる。これは、特定のレゾルシル酸ラクトンの特定のキナーゼに対する“特異性定数”によって反映される。前記は、非常に希薄な阻害剤及びキナーゼ濃度の共有結合については実質的には2桁の速度定数である。例えば、ヒポテマイシンはERK2に対して2μMのKiを有し(Kinact/Ki=1.9E+03)、不活化のt1/2は3分であり、KDRに対してはKiは0.01μMで、不活化のt1/2は約1分である(Kinact/Ki=5E+05)。2つのキナーゼを0.1μMで約10分処理することによって((Ki ERK>0.1um>Ki KDR)、5%未満のERK活性が阻害される条件下で、98%を超えるKDRを阻害することができると計算できる。さらにまた、曝露が、共有結合阻害を完了させるのに充分である場合、薬剤の投与を停止して非Cysキナーゼの一切の可逆的阻害を軽減することができるが、共有結合によって改変されたキナーゼの特異的なセットの阻害は維持される。
【0021】
本発明は、一つには、ヒポテマイシン(マイケル付加物形成構造を含む)とゼアラレノン及び5,6-ジヒドロヒポテマイシン(マイケル付加物形成構造を含まない、図2を参照)を比較することによって、さらにそれら各々のERK1/2の活性化を阻害する能力を比較することによって、理解することができる。化合物を0.3〜3μMで調べたとき、ヒポテマイシンは、ヒトのT細胞でERK1/2の活性化を阻害するが、ゼアラレノンは阻害しないと報告されている(Camacho et al. 1999 Immunopharmacology 44(3):255-265)。対応するヒトタンパク質キナーゼのアミノ酸配列の調査によって、ERK1/2内に適切に配置されているCys残基が示されている。
任意の多様なタンパク質キナーゼ、例えばMEK1/2又はERK1/2についての相同性モデルの調査によって、タンパク質キナーゼのATP結合部位領域内でのレゾルシル酸ラクトンの適切な配置がマイケル付加物の形成を可能にするであろうということが例証される。例えば、MEK1 ATP結合部位の相同性モデルは、α、β-不飽和カルボニル含有レゾルシル酸ラクトン又は誘導体が、前記重要なCys残基を含むタンパク質キナーゼをマイケル付加物の形成によって阻害することができるメカニズムを支持する。
そのようなモデルは、本発明の観点から、マイケル付加物形成による阻害に感受性を有するタンパク質キナーゼを阻害することができる新規なキナーゼ阻害剤の構造を予測することを可能にするだけでなく、本発明の方法で有用な構造を有する既知の化合物を同定することもまた可能にする。ある実施態様では、本発明の方法で有用な化合物は、公知で以前に試験された化合物であり、それらは、用いられる混合物が、それら既知の化合物のキナーゼ阻害の特異性が未だ試験されたり決定されたりしていないキナーゼを含む、本発明の方法で用いられる。別の実施態様では、本発明の化合物は、以前には生成又は試験されたことがない新規な化合物である。
【0022】
本発明によって提供される進歩を理解するためには、本質的に全てのタンパク質キナーゼ阻害剤は複数のキナーゼを阻害することが充分に証明されていることと、特定の阻害剤に対する細胞の応答は、2つの又は通常はそれ以上のキナーゼの同時阻害を伴うということとを理解しなければならない。続いて、いずれの阻害剤の特異性及び有効性も、そのキナーゼ阻害プロフィールに依存し、また、異なるプロフィールは異なる効果を細胞にもたらす。ほとんどの既知のキナーゼ阻害剤のキナーゼプロフィールは実験的にのみ決定することができ、したがってアッセイで利用可能な酵素の数によって制限される。例えば、120キナーゼの大きなパネルに対する複数のキナーゼ阻害剤の阻害活性プロフィールが報告されている(Fabian et al. Nat Biotechnol 2005, 23(3):329-36)。イマチニブ(グリーベック)は120キナーゼのうち10個を阻害し、BAY43-9006は120キナーゼのうちKi<0.1μMの19キナーゼを阻害したが、スクリーニングにこれまで利用可能でなかった残りの300キナーゼのどれくらいが、又はどれがこれら化合物によって阻害されるかは不明である。対照的に、本発明は、本発明のレゾルシル酸ラクトン(RAL)によって阻害される全ヒトキノムにおける標的の信頼できるリストを提供する(前記RALは、その標的が含む重要なCys残基でこれら標的とマイケル付加物を形成することができる)。
阻害剤の完全なキナーゼプロフィールの知識は、ある種の細胞タイプに対するその有効性及び特異性に関する有用な情報を提供する。例えば、前記プロフィールを他の阻害剤のプロフィールを比較することができる。新規な阻害剤の標的キナーゼサブセットが、公知の有効な阻害剤に対して関連があると考えられているサブセットとオーバーラップするならば、前記新規な阻害剤は類似の活性及び効果を示すはずである。本発明の方法で有用なレゾルシル酸ラクトンは固有のキナーゼ阻害プロフィールを有するが、その標的キナーゼのある種のサブセットは他の有効なキナーゼ阻害剤によって阻害されるサブセットとオーバーラップする。例えば、キナーゼ阻害剤SU11248は、FLT3内在縦列重複変異(ITD)を含むAMLの阻害で有効である。なぜならば、FLT3(野生型及びITD)、PDGFR、VEGFR及びcKITを含むキナーゼサブセットを標的とするからである。ヒポテマイシンは、キナーゼの同じサブセットを阻害し、したがって、本発明によって提供されるように、AML細胞の阻害で有効である。下記実施例に記載するある試験では、AML(FLT3 ITD)細胞株MV-4-11に対するSU11248のGI50は12nMであり、ヒポテマイシンは6nMのGI50を有していた。
【0023】
経路の初期に酵素が過剰産生されるか又はアミノ酸変異のために、ある種のキナーゼ及びキナーゼ経路は過剰に活性であるか又は構成的に活性であり、したがって、(直接的又は経路内のより初期の別の酵素を介する間接的な)阻害は、前記活性な経路から生じる表現型の選択的な阻害又は調節をもたらすことができると期待することができる。例えば、B-Raf V599E(V600E)変異体は、およそ70%のメラノーマ及びおよそ20%の結腸癌で見出され、細胞分裂に必要なERK経路の構成的活性化をもたらす。BAY 43-9006は、最初はメラノーマ細胞のこの経路を阻害するためにRaf阻害剤として開発された。ヒポテマイシン及び本発明の方法で有用な他のRALは、前記経路の2つの点(MEK1/2及びERK1/2)を不可逆的に阻害し、したがって前記経路を完全に阻害してERK/RSKリン酸化の下流のシグナル伝達を閉鎖するはずである。
下記実施例に記載したin vitro試験は、B-Raf V599E(V600E)変異体はRAL阻害剤に非常に感受性が高いことを示している。メラノーマ細胞株COLO829に関しては、ヒポテマイシンは50nMのGI50を有し、BAY 43-9006は6,000nMのGI50を有し、さらにSU11248は7,100nMのGI50を有している。活性化されたERK経路はまた、細胞生物学的研究及びMEK1/2阻害剤の効果によって実証されるように、腫瘍の広範囲(乳房、大腸、卵巣、前立腺及び膵臓の腫瘍を含む)で関与している。MEK及びRaf阻害剤は、ERK/RSK経路に依存する細胞に対して有効であり、本発明のRALはこれらの細胞に対して同様に有効である。
【0024】
ただ1つの酵素に対する可逆的阻害剤による100%阻害の達成は非常に困難であり、一方、ある経路の複数の工程を阻害する阻害剤は経路のほぼ完全な遮断を引き起こすことができる。キナーゼプロフィールが、直線的経路の2以上の連続的工程の阻害を示すならば、経路全体に対する薬剤の影響は協同的でないとしても少なくとも累積的であろう。本発明の方法で有用なRAL阻害剤は、それらがERK経路の少なくとも2つの点を不可逆的に阻害するという点で固有である。それらはまた、ERK経路を刺激するチロシンキナーゼマイトジェンレセプターの多くを不可逆的に阻害し、直線的経路の3点阻害及びそれに続くマイトジェン刺激分裂経路の強力な阻害を提供する。例えば、下記の実施例に示すように、変異体のマイトジェンレセプターFlt3及び構成的に活性なERK経路を含むAML細胞株MV-4-11に関して、ヒポテマイシンは6nMのGI50を有し、ヒポテマイシンは、VEGFRの非常に強力な不可逆的阻害剤であり、VEGFRを必要とする細胞の治療はVEGFR、MEK及びERKを閉鎖する。さらにまた、ERKリン酸化はVEGF分泌に要求されるので、VEGF産生細胞での産生及びVEGF応答細胞でのVEGFに対する応答の両方が阻害される。これらの理由のために、ヒポテマイシン及び前記必須のCys残基をもつタンパク質キナーゼとマイケル付加物を形成することができる本明細書に開示の他のRALは、血管形成の極めて有効な阻害剤である。別の例は基底細胞癌(BCC)の治療である。この適応症では、BCC腫瘍の90%が、ERK経路及び細胞分裂を駆動するPDGFRを過剰発現する。本発明の方法で有用なRALはPDGFR経路とERK経路の2つの点とを阻害し、したがって直線的経路の3点阻害を提供する。
【0025】
ほとんどのキナーゼ阻害剤は可逆的阻害剤であり、したがって標的の阻害は濃度の関数であり、完全阻害には阻害定数Kiをはるかに超える阻害剤濃度が要求される。さらにまた、いったん阻害剤が除去されたならば酵素活性は急速に復帰するので、細胞は持続的な曝露を要求する。本発明の方法で用いられる化合物は、タンパク質キナーゼの不可逆的阻害剤である(ただし標的キナーゼサブセットのみを不可逆的に阻害する)。ヒポテマイシン及び本発明の方法で有用な他のRALによる標的阻害は、濃度及び/又は時間の関数であるので、曝露時間が増加されるならば、完全阻害は低濃度の阻害剤で達成することができる。本発明は、これらの特性の利点を有する本発明のRALの単位用量形態及び前記RALを投与する方法を提供する。したがって、ある実施態様では、疾患を治療する本発明の方法は、充分な化合物を投与して、阻害定数にあるか又は阻害定数以下の該化合物の血中又は腫瘍中レベルを提供するか、及び/又は充分な時間にわたって前記血中又は腫瘍中レベルの維持を提供し、その結果、標的タンパク質キナーゼの少なくとも50%、より好ましくは90%を超える(例えば99%又は100%)不可逆的阻害を達成することを含む。ある実施態様では、前記薬剤の第二の投与(多くの実施態様では、薬剤は同じ患者に複数回投与されるであろう)は、不可逆的に阻害されたキナーゼのde novo合成による交換を基準にすると、前記薬剤の最初の投与後1又は2日以内である。
例えば、下記実施例で示すように、B-Raf V599E(V600E)細胞株COLO829(及び調査されたBRAF変異体を有する他の細胞)中のERK経路は、前記酵素に対して数倍低いKd濃度でヒポテマイシンに10分曝露した後で閉鎖された。さらにまた、前記阻害剤の除去は活性の即時復活を伴わず、それどころか、リン酸化された活性なERKは何時間(約24時間)も存在せず、その復帰は新規な酵素合成を明らかに必要とする。したがって、本発明は、これら化合物を投与して、正常細胞に対する毒性を低下させる方法を提供する。ある実施態様では、前記化合物は、腫瘍又は他の癌細胞若しくは組織から得られる測定値によって決定して、標的キナーゼ活性が完全に阻害されるまで投与される。この時点で、治療効果を低下させることなく投与を停止することができ、有意なレベルの標的キナーゼ活性が回復した後でのみ投与を再開することができる。
【0026】
ある実施態様では、本発明の方法で有用で、さらに本発明の医薬組成物に含まれる化合物は、以下の一般構造式(I)を有するもの、並びに医薬的に許容できるその塩及び誘導体である。
【0027】
【化3】
【0028】
式中、
R1は、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、各々独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、場合によって置換されたアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子又は酸素原子を含む飽和若しくは不飽和環式環を形成し;R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になってN3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、又はアルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は、
R8及びR9は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子又は酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であるか、又はR20及びR2が組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であるか、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり、式中R19は水素又はアルキルである。
【0029】
好ましくは式(I)の化合物では、以下の条件の少なくとも1つが当てはまる:(i)R6は水素又はヒドロキシルである、(ii)nは1である、(iii)R8は水素以外のものである、(iv)R10は水素である、及び(v)R11は保護されたヒドロキシル以外のものである。
好ましい実施態様では、前記化合物は下記式(Ia)の構造を有する:
【0030】
【化4】
【0031】
式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
ここで、R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されたアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;ここでR17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ia)の化合物の好ましい実施態様では、R9のOR12はOMe以外のものである。
【0032】
別の好ましい実施態様では、前記化合物は下記式(Ib)の構造を有する:
【0033】
【化5】
【0034】
式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
ここでR12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;さらに
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
【0035】
別の好ましい実施態様では、前記化合物は下記式(Ic)の構造を有する:
【0036】
【化6】
【0037】
式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ic)の化合物の好ましい実施態様では、R8は水素又はハロゲン以外のものである。
別の好ましい実施態様では、前記化合物は下記式(Id)の構造を有する:
【0038】
【化7】
【0039】
式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
別の好ましい実施態様では、前記化合物は下記式(Ie)の構造を有する:
【0040】
【化8】
【0041】
式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
式(Ie)の化合物の好ましい実施態様では、R5は水素以外のものである。
【0042】
別の好ましい実施態様では、前記化合物は下記式(If)の構造を有する:
【0043】
【化9】
【0044】
式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である。
別の好ましい実施態様では、前記化合物は下記式(Ig)の構造を有する:
【0045】
【化10】
【0046】
式中、
R4はH又はFであり;
R8はHであり;さらに
R9は以下の基から成る群より選択されるか、
【0047】
【化11】
【0048】
又は、R8及びR9が一緒になって以下の基を形成する。
【0049】
【化12】
【0050】
“脂肪族”とは直鎖又は分枝鎖の飽和若しくは不飽和の非芳香族炭化水素部分を指し、特定の数の炭素原子(例えば“C3脂肪族”、C1〜C5脂肪族”又は“C1からC5脂肪族”のように、後者の2つの語句は1〜5の炭素原子を有する脂肪族部分についての同義語である)を有するか、又は炭素原子の数が特定されない場合は1〜4の炭素原子を有する。不飽和脂肪族部分は必然的に少なくとも2つの炭素原子を含むことは当業者には理解されよう。
“アルキル”は飽和脂肪族部分を意味し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C1〜C4アルキル部分は、メチル、エチル、プロピル、イソプロピル、イソブチル、t-ブチル、1-ブチル、2-ブチルなどが含まれるが、ただしこれらに限定されない。
“アルケニル”は、少なくとも1つの炭素-炭素二重結合を有する脂肪族部分を指し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C2〜C4アルケニル部分は、エテニル(ビニル)、2-プロペニル(アリル又はプロパ-2-エニル)、シス-1-プロペニル、トランス-1-プロペニル、E-(又はZ-)2-ブテニル、3-ブテニル、1,3-ブタジエニル(ブタ-1,3-ジエニル)などを含むが、ただしこれらに限定されない。
“アルキニル”は、少なくとも1つの炭素-炭素三重結合を有する脂肪族部分を指し、炭素原子の数を示すために同じ取り決めを適用することができる。例示すれば、C2〜C4アルキニル基は、エチニル(アセチレニル)、プロパルギル(プロパ-2-イニル)、1-プロピニル、ブタ-2-イニルなどを含む。
“環式脂肪族”は、飽和又は不飽和の非芳香族炭化水素部分を指し、1〜3の環を有し、各環は3〜8(好ましくは3〜6)の炭素原子を有する。“シクロアルキル”は、各環が飽和されている環式脂肪族部分を指す。“シクロアルケニル”は、少なくとも1つの環が少なくとも1つの炭素-炭素二重結合を有する環式脂肪族部分を指す。“シクロアルキニル”は、少なくとも1つの環が少なくとも1つの炭素-炭素三重結合を有する環式脂肪族部分を指す。例示すれば、環式脂肪族部分には、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプチル、シクロオクチル及びアダマンチルが含まれるが、ただしこれらに限定されない。好ましい環式脂肪族部分は、シクロアルキル部分、特にシクロプロピル、シクロブチル、シクロペンチル、及びシクロヘキシルである。
【0051】
“複素環式脂肪族”は環式脂肪族部分であって、その少なくとも1つの環において、3つまで(好ましくは1〜2)の炭素が、N、O、又はSから独立に選択されるヘテロ原子で置き換えられているものを指し、ここでN及びSは場合によって酸化されてもよく、さらにNは場合によって四級化されてもよい。同様に、“ヘテロシクロアルキル”、“ヘテロシクロアルケニル”、及び“ヘテロシクロアルキニル”は、それぞれシクロアルキル、シクロアルケニル、又はシクロアルキニル部分であって、前記の少なくとも1つの環が上記のように改変されているものを指す。例示的な複素環式脂肪族部分には、アジリジニル、アゼチジニル、1,3-ジオキサニル、オキセタニル、テトラヒドロフリル、ピロリジニル、ピペリジニル、ピペラジニル、テトラヒドロピラニル、テトラヒドロチオピラニル、テトラヒドロチオピラニルスルホン、モルホリニル、チオモルホリニル、チオモルホリニルスルホキシド、チオモルホリニルスルホン、1,3-ジオキソラニル、テトラヒドロ-1,1-ジオキソチエニル、1,4-ジオキサニル、チエタニルなどが含まれる。
“アルコキシ”、“アリールオキシ”、“アルキルチオ”及び“アリールチオ”は、それぞれ−O(アルキル)、−O(アリール)、−S(アルキル)及び−S(アリール)を意味する。例は、それぞれメトキシ、フェノキシ、メチルチオ及びフェニルチオである。
“ハロゲン”又は“ハロ”は、フッ素、塩素、臭素又はヨウ素を指す。
“アリール”は、一環式、二環式、又は三環式環系を有する炭化水素部分を指し、各環は3〜7の炭素原子を有し、さらに少なくとも1つの環は芳香族である。前記環系の環は互いに縮合(ナフチルにおけるように)していてもよく、又は互いに結合(ビフェニルにおけるように)していてもよく、さらに(インダニル又はシクロヘキシルフェニルにおけるように)非芳香環と縮合又は非芳香環と結合してもよい。さらに例示すれば、アリール部分には、フェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニル、フェナントリル、アントラセニル及びアセナフチルが含まれるが、ただしこれらに限定されない。
【0052】
“ヘテロアリール”は、一環式、二環式、又は三環式環系を有する部分を指し、各環は3〜7個の炭素原子を有し、さらに少なくとも1つの環は、N、O又はSから独立に選択される1〜4のヘテロ原子を含む芳香環である。ここで、前記N及びSは場合によって酸化されてもよく、Nは場合によって四級化されてもよい。そのようなヘテロ原子含有芳香環は、(ベンゾフラニル又はテトラヒドロイソキノリルにおけるように)他のタイプの環と縮合してもよく、又は(フェニルピリジル又は2-シクロペンチルピリジルにおけるように)他のタイプの環と直接結合してもよい。さらに例示すれば、ヘテロアリール部分には、ピロリル、フラニル、チオフェニル(チエニル)、イミダゾリル、ピラゾリル、オキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、ピリジル、N-オキソピリジル、ピリダジニル、ピリミジニル、ピラジニル、キノリニル、イソキノリニル、キナゾリニル、シンノリニル、キノザリニル、ナフチリジニル、ベンゾフラニル、インドリル、ベンゾチオフェニル、ベンゾイミダゾリル、ベンゾトリアゾリル、ジベンゾフラニル、カルバゾリル、ジベンゾチオフェニル、アクリジニルなどが含まれる。
部分が置換され得ると表示されている場合(例えば“置換又は非置換C1〜C5アルキル”又は“ヘテロアリールで置換されていてもよい”におけるように表現される、“置換又は非置換”又は“置換されていてもよい”を使用することにより)、そのような部分は、1以上の独立して選択される置換基を、好ましくは個数として1〜5、より好ましくは個数として1又は2有することができる。置換基及び置換パターンは、置換基が結合される部分について考慮しながら、化学的に安定であり、且つ本技術分野で公知の技術及び本明細書で述べる方法によって合成することができる化合物を提供するように、当業者が選択することができる。
【0053】
“アリールアルキル、(複素環式脂肪族)アルキル”、“アリールアルケニル”、“アリールアルキニル”、“ビアリールアルキル”などは、事情に応じてアリール、複素環式脂肪族、ビアリールなどの部分で置換されたアルキル部分、アルケニル部分、又はアルキニル部分を指し、事情に応じて前記アルキル部分、アルケニル部分、又はアルキニル部分で(例えばベンジル、フェネチル、N-イミダゾリルエチル、N-モルホリノエチルなどのように)開いた(未飽和の)原子価を有する。逆に、“アルキルアリール”、“アルケニルシクロアルキル”、ハロヘテロアリールなどは、例えば、事情に応じてメチルフェニル(トリル)、又はアリルシクロヘキシルにおけるように、事情に応じてアルキル、アルケニル、ハロなどの部分で置換されたアリール、シクロアルキル、ヘテロアリールなどの部分を指す。“ヒドロキシアルキル”、“ハロアルキル”、“アミノアルキル”、“アルキルアリール”、“シアノアリール”などは、事情に応じて特定された置換基(事情に応じてヒドロキシル、ハロ、アミノなど)で置換されたアルキル、アリールなどの部分を指す。例示すれば、許容できる置換基には、アルキル(特にメチル又はエチル)、アルケニル(特にアリル)、アルキニル、アリール、ヘテロアリール、環式脂肪族基、複素環式脂肪族基、ハロ(特にフルオロ)、ハロアルキル(特にアリルトリフルオロメチル)、ヒドロキシル、ヒドロキシアルキル(特にヒドロキシエチル)、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)(特に−OCF3)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)、−SO2N(アルキル)2などが含まれるが、ただしこれらに限定されない。
【0054】
前記置換される部分が脂肪族部分である場合、好ましい置換基は、アリール、ヘテロアリール、環式脂肪族基、複素環式脂肪族基、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)、及び−SO2N(アルキル)2である。より好ましい置換基は、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、−O(アリール)、=O、=NOH、=NO(アルキル)、−OC(=O)(アルキル)、−OC(=O)O(アルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、及び−NHC(=NH)NH2である。
前記置換される部分が環式脂肪族部分、複素環式脂肪族部分、アリール部分又はヘテロアリール部分である場合は、好ましい置換基は、アルキル、アルケニル、アルキニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−O(ハロアルキル)、−O(シクロアルキル)、−O(ヘテロシクロアルキル)、−O(アリール)、アルキルチオ、アリールチオ、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、アジド、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NH(ヒドロキシアルキル)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2、−NHC(=NH)NH2、−OSO2(アルキル)、−SH、−S(アルキル)、−S(アリール)、−S(シクロアルキル)、−S(=O)アルキル、−SO2(アルキル)、−SO2NH2、−SO2NH(アルキル)及び−SO2N(アルキル)2である。より好ましい置換基は、アルキル、アルケニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、−O(ヒドロキシアルキル)、−C(=O)(アルキル)、−C(=O)H、−CO2H、−C(=O)NHOH、−C(=O)O(アルキル)、−C(=O)O(ヒドロキシアルキル)、−C(=O)NH2、−C(=O)NH(アルキル)、−C(=O)N(アルキル)2、−OC(=O)(アルキル)、−OC(=O)(ヒドロキシアルキル)、−OC(=O)O(アルキル)、−OC(=O)O(ヒドロキシアルキル)、−OC(=O)NH2、−OC(=O)NH(アルキル)、−OC(=O)N(アルキル)2、−NH2、−NH(アルキル)、−N(アルキル)2、−NH(アリール)、−NHC(=O)(アルキル)、−NHC(=O)H、−NHC(=O)NH2、−NHC(=O)NH(アルキル)、−NHC(=O)N(アルキル)2及び−NHC(=NH)NH2である。
【0055】
範囲が“C1〜C5アルキル”又は“5〜10%”のように記載されている場合、そのような範囲は前記範囲の両端の値を含む。
具体的な立体異性体が特に(例えば構造式中の対応する立体中心で太字又は点線による結合によって、構造式中のE又はZ構造を有する二重結合の描写によって、又は立体化学の呼称の使用によって)指定されないかぎり、全ての立体異性体が、その混合物と同様に純粋な化合物として本発明の範囲内に含まれる。特段に規定されないかぎり、個々の鏡像体、ジアステレオマー、幾何学異性体並びにそれらの組合せ及び混合物は全て本発明に包含される。多形性結晶形及び溶媒和物もまた本発明の範囲内に包含される。
“医薬的に許容される塩”は、塩として医薬処方物に適した化合物の塩を指す。化合物が1以上の塩基性官能性を有する場合、前記塩は酸付加塩、例えば硫酸塩、臭化水素酸塩、酒石酸塩、メシレート、マレイン酸塩、クエン酸塩、リン酸塩、酢酸塩、パモポエート(エンボネート)、ヨウ化水素酸塩、硝酸塩、塩酸塩、乳酸塩、メチルスルフェート、フマル酸塩、安息香酸塩、コハク酸塩、メシレート、ラクトビオネート、スブレート、トシレートなどであり得る。化合物が1つ又は2つ以上の酸性部分を有する場合は、前記塩は、例えばカルシウム塩、カリウム塩、マグネシウム塩、メグルミン塩、アンモニウム塩、亜鉛塩、ピペラジン塩、トロメタミン塩、リチウム塩、コリン塩、ジエチルアミン塩、4-フェニルシクロヘキシルアミン塩、ベンザチン塩、ナトリウム塩、テトラメチルアンモニウム塩などのような塩であり得る。
【0056】
本発明は、その範囲内に本発明の化合物のプロドラッグを含む。そのようなプロドラッグは前記化合物の一般的な機能的誘導体であり、要求される化合物にin vivoで容易に変換され得る。したがって、本発明の治療方法では、“投与する”という用語は、具体的に開示された化合物を用いて、又は具体的に開示されてないがその必要がある対象者に投与後n vivoで特定された化合物に変換される化合物を用いて、開示の種々の疾患を治療することを包含するであろう。適切なプロドラッグ誘導体の選択及び調製のための通常的な方法は、例えば以下に記載されている:Wermuth, “Designing Prodrugs and Bioprecursors”, in Wermuth, ed., The Practice of Medical Chemistry, 2nd Ed., pp.561-586, Academic Press 2003(前記文献の内容は参照により本明細書に含まれる)。プロドラッグは、in vivoで(例えばヒトの体内で)加水分解されて本発明の化合物又はその塩を生成するエステルを含む。適切なエステル群には、医薬的に許容できる脂肪族カルボン酸、特にアルカン酸、アルケン酸、シクロアルカン酸及びアルカンジオン酸(前記で各アルキル又はアルケニル部分は好ましくは6未満の炭素原子を有する)から誘導されるものが含まれるが、ただし前記に限定されない。例示的なエステルには、ギ酸エステル、酢酸エステル、プロピオン酸エステル、酪酸エステル、アクリル酸エステル、クエン酸エステル、コハク酸エステル及びエチルコハク酸エステルが含まれるが、ただしこれらに限定されない。
ある重要な実施態様では、本発明の化合物には、本発明の方法で用いられる経口投与に適したレゾルシル酸ラクトンのプロドラッグエステルが含まれる。ある実施態様では、これらのプロドラッグは、本発明の方法で有用なレゾルシル酸ラクトンのアミノ酸エステル(ジメチルグリシンエステル及びバリンエステルが含まれるが、ただしこれらに限定されない)である。
【0057】
“保護基”は、特定の官能部分(例えばO、S又はN)を一時的に遮断し、それによって反応が多官能性化合物の別の反応部位で選択的に実施され得る部分を指す。好ましい実施態様では、保護基は、(a)良好な収量で選択的に反応して、計画の反応にとって安定な保護基質を生じ、(b)他の官能基を攻撃しない、容易に入手可能で、好ましくは毒性のない試薬によって良好な収量で選択的に除去でき、(c)容易に分離することができる誘導体を生成し(より好ましくは新規な立体中心を生じることなく)、さらに(d)更なる反応部位を回避するために付加される官能性が最小限である。“酸素保護基”は酸素に結合される保護基を指し、メチルエーテル、置換メチルエーテル(例えばMOM(メトキシメチルエーテル)、MTM(メチルチオメチルエーテル)、BOM(ベンジルオキシメチルエーテル)、PMBM又はMPM(p-メトキシベンジルオキシメチルエーテル))、置換エチルエーテル、置換ベンジルエーテル、シリルエーテル(例えばTMS(トリメチルシリルエーテル)、TES(トリエチルシリルエーテル)、TIPS(トリイソプロピルシリルエーテル)、TBDMS(t-ブチルジメチルシリルエーテル)、トリベンジルシリルエーテル、TBDPS(t-ブチルジフェニルシリルエーテル))、エステル(例えばギ酸エステル、酢酸エステル、安息香酸エステル(Bz)、トリフルオロ酢酸エステル、ジクロロ酢酸エステル)、カルボネート、環式アセタールおよびケタールが含まれるが、ただし前記に限定されない。“窒素保護基”はアミンの窒素に結合される保護基を指し、カルバメート(例えばメチル、エチル及び置換エチルカルバメート(例えばTroc))アミド、環式イミド誘導体、N-アルキル及びN-アリールアミン、イミン誘導体、及びエナミン誘導体が含まれるが、ただしこれらに限定されない。保護基の多くの例は、それらの使用態様に関する教示とともに以下の文献で見出すことができる:Greene and Wuts, Protective Groups in Organic Synthesis, 3rd edition, pp. 17-245(John Wiley & Sons, New York, 1999)(前記文献の内容は参照により本明細書に含まれる)。したがって、“保護されたヒドロキシル”は、水素が酸素保護基によって置き換えられたヒドロキシル基を指し、“保護アミン”は、水素が窒素保護基によって置き換えられた1級又は2級アミン基を指す。
【0058】
5−7位のカルボニル(又はビオイソステア)と共役した前記重要なシス二重結合を保持する上記構造に包含される化合物の類似体及び誘導体もまた本発明の方法で有用である。一般的には、レゾルシル酸ラクトンであろうと、誘導体であろうと、他の化合物であろうと、前記重要なCys残基とマイケル付加物を形成することができるいずれの化合物も本発明の1以上の方法で用いることができる。例えば、本発明の化合物が、本質的にこれら酵素の1つに対する既知の阻害剤の適切な位置に付加されたマイケル受容体から成るように、結晶構造を用いて本発明の化合物を設計することができる。得られた化合物は可逆的な複合体を前記酵素と形成することができ、共有結合形成はその後で生じるであろう。
したがって、本発明の方法で有用な化合物は、保存された2つのAsp残基の間に位置し、その1つと隣接するATP結合部位内のCys残基を有するタンパク質キナーゼを特異的に阻害し、さらに重要なことには、ATP結合部位内のこの位置にこのCysを欠くタンパク質キナーゼに対してはほとんど阻害活性をもたない。したがって、そのようなものは、特定のタンパク質キナーゼを特異的に阻害するために用いることができ、前記は、ヒトの疾患を治療するために新規で重要な方法を提供する。さらにまた、そのようなタンパク質キナーゼは複数のシグナル伝達経路に存在するので、本発明の方法で有用な化合物は、治療活性のために要求される多経路遮断作用を提供することができる。
【0059】
この重要なCysを含むタンパク質キナーゼには、AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAKが含まれるが、ただしこれらに限定されない。
本発明の方法は、種々の細胞シグナル伝達経路で特異的なタンパク質キナーゼを阻害することによって多シグナル伝達経路阻害を達成することができるRAL又は誘導体を投与することを含む。このタイプの阻害は、グリーベックに関して上記で例示したように所望の効果を達成するために所望され、必要ですらある。また別の例示は、ゲルダマイシン、17-AAG及び17-DMAGのような阻害剤によるHsp90の阻害である。この阻害は、Hsp90の阻害が複数の細胞シグナル伝達経路の複数の配下のタンパク質キナーゼの分解/阻害を生じるので複数の経路に影響を与える。
【0060】
しかしながら、普遍的に多くのキナーゼを阻害することなく、複数のタンパク質キナーゼを特異的に阻害する阻害剤を設計することは困難である。同様に、たとえタンパク質キナーゼを同定したとしても、500を超える他のタンパク質キナーゼのどれを前記阻害剤が阻害するかを予測することは困難である。対照的に、本発明の方法で有用な化合物のコア構造(タンパク質キナーゼ内の重要なCysとマイケル付加物を形成することができるエノン又はα、β-不飽和ケトン部分)は、見事な特異性及び治療結果の改善を提供する。ある実施態様では、本発明のこれらの化合物は、レゾルシル酸ラクトン構造の5−7位にエノン部分を含む。そのような化合物を用いて、全てのキナーゼのうち特定のサブセットを予測的に阻害することができる。本発明の化合物はまた、治療の適応症のために望ましい特定のキナーゼサブセット内でのキナーゼ阻害の均衡を示す特定の阻害剤を選択することができる、コア構造が構造的に改変された複数の化合物を含む。
多タンパク質キナーゼ阻害は、(a)ネットワークの種々の支流を阻害しネットワーク全体を阻害する潜在能力を生じるか、又は(b)ネットワークのただ1つの直線的支流上の種々のキナーゼを阻害するか、又は(c)前記の両方を阻害することができる。これらのタイプの多タンパク質キナーゼ阻害は、ただ1つのキナーゼを阻害する化合物よりも累積的な阻害効果を提供し、協同的な阻害をもたらす潜在能力を有する。ある種のレゾルシル酸ラクトン阻害剤は、いかにして本発明の方法が、どちらかのアプローチ又は両方のアプローチを包含することができるかを説明するために有用である。例えば、これらの阻害剤はERKシグナル伝達経路及びJNKシグナル伝達経路を阻害し、したがって、細胞分裂及び炎症の両方で重要な種々の均衡のとれたシグナル伝達経路に影響を与え、ネットワーク阻害アプローチを例証する。
【0061】
本発明の方法で有用なある種のレゾルシル酸ラクトンはまた、ただ1つの経路で複数の酵素を阻害する(協同的経路阻害アプローチ)。例えば、ある種のレゾルシル酸ラクトン化合物はMEK1/2及びERK1/2を阻害する。本発明のそのような阻害剤及び他の化合物を投与して、たとえ個々のプロテインキナーゼに対するそれらの潜在能力が特に高くなくても、疾患過程で臨床的に相応な阻害を達成することができる。
例えば、阻害剤が、両酵素の活性化(すなわちリン酸化)型に対して等しく強力であると仮定した場合、MEK1/2の50%を阻害するために必要な前記阻害剤濃度は、ERK1/2のリン酸化型(阻害剤が存在しない場合と比較して)の単に50%の生成しかもたらさない。同じ濃度では、前記阻害剤が活性化ERK1/2の50%を同時に阻害するとしたら、前記経路は75%阻害される(経路の協同的阻害)。さらにまた、本発明の方法で有用なある種の化合物は、ERK経路の複数のキナーゼを阻害するだけでなく、VEGFR(前記は活性化されたときERK経路の活性化を引き起こす)もまた阻害する。阻害剤が、3つの酵素の全てに対して同じ潜在能力を有するならば、VEGFRからERK1/2のシグナル伝達経路(抗分裂作用のための阻害剤の標的)は、いずれかの単一酵素を単に50%阻害する濃度で87.5%阻害される。
この多タンパク質キナーゼ阻害は、所望の治療効果を達成するためにPDGFRB、PDGFRA及びKITの阻害を必要とする治療方法に関連して本発明のある実施態様で例示される。これらの標的はグリーベックによって阻害され、グリーベックは、慢性骨髄性単球性白血病及び多形型神経芽細胞腫とともにGIST及び転移性GISTの治療で治療的価値を有する(グリーベックはまたBcr-Ablを阻害し、Bcr-Ablは本発明の方法で有用な化合物とのマイケル付加物形成に感受性ではない)。したがって、本発明の方法で有用な化合物及び医薬組成物はこれら疾患に対して治療適用性を有する。しかしながら、重要なことには、本発明で有用な化合物のタンパク質キナーゼとの結合は、グリーベック耐性を付与するタンパク質キナーゼの変異は本発明の化合物に対する耐性を付与できないというものである。したがって、本発明の方法は、グリーベック耐性症状(グリーベックが投与される癌のグリーベック耐性型を含む)を治療する方法を含む。本発明の方法はまた、以下の節(それぞれ個々の癌又は他の適応症状に焦点を当てる)で考察されるように、他の癌適応症及び疾患を治療する方法を含む。
【0062】
消化管間質腫瘍:
消化管間質腫瘍(GIST)はもっぱら胃(60%)及び小腸(25%)で見出されるが、また低頻度で直腸、食道、及び他の場所にも出現する。GISTは過去にはしばしば誤診され、それらの出現の正確な組織像を得ることは困難である。ほぼ5000の新規症例が毎年合衆国で発生すると概算される(www.orpha.net/data/patho/GB/uk-GIST.pdf)。ほぼ95%のGISTがc-KITについて免疫組織化学的に陽性に染色され、GISTの85%までがc-KITチロシンキナーゼの活性化変異を保有している(Hirota et al. Science 1998; 279(5350):577-80)。さらにまた、c-KITの遺伝性活性化変異を有するいくつかの血縁群が同定された(Nishida et al. Nat Genet 1998; 19(4):323-4)。これらの家族は、多発性良性及び悪性GISTの発症を示す。c-KITについて野生型であることが判明したGISTのうち、ほぼ5%はPDGFRAに変異を保有している(Heinrich et al. Science 2003; 299(5607):708-10)。c-KIT及びPDGFRAチロシンキナーゼの活性化変異は、下流のシグナル伝達経路(MEK1/2及びERK1/2酵素経路を含む)の活性化を伴う。本明細書に記載の、ヒポテマイシン並びにその誘導体及び類似体は、前記レセプターキナーゼc-KIT及びPDGFRと同様に前記連続するERK経路のMEK1/2及びERK1/2の強力な阻害剤であり、本発明の方法にしたがってGISTの治療のために患者に投与することができる。
【0063】
急性骨髄性白血病:
本発明の方法で有用な化合物にはまた、FLT3(急性骨髄性白血病(ALM)でもっとも一般的な分子異常(変異))を阻害する化合物が含まれる。AMLは、成人のもっとも一般的な白血病であるとともに小児のもっとも一般的な癌形態である。2003年に合衆国ではほぼ10,000件の新規症例及び8,000件の死亡がAMLによってもたらされた。ほぼ同じ数の症例がヨーロッパ及びオーストラリアで発生した。いくつかのキナーゼがAMLで役割を演じていると示唆されてきた。AML治療の臨床試験における従来の薬剤の治療標的にはFLT3、c-KIT及びVEGFRが含まれる。FLT3は正常な造血作用で重要な役割を果たす。前記は、AMLの患者の70%〜100%で異常に活性化されるか、又はアップレギュレートされる(Spiekermann et al. Clin Cancer Res 2003; 9(6):2140-50;及びBlood 2003; 101(4):1494-504)。c-KITタンパク質キナーゼはAML患者の60%〜80%で高レベルで見出され、分裂及び抗アポトーシス作用を仲介すると考えられる(Heinrich et al. J Clin Oncol 2002; 20(6):1692-703)。VEGF及びVEGFRは骨髄の血管形成で役割を果たすと示唆されてきた(Aguayo et al. Blood 2000; 96(6):2240-5)。AML患者の骨髄生検によって、VEGF及びVEGFRレベルの変化は毛細血管密度の変化と平行することが示された(Kuzu et al. Leuk Lymphoma 2004; 45(6):1185-90)。VEGFレベルは、AML患者の生存と反比例するようである(Brunner et al. J Hematother Stem Cell Res 2002; 11(1):119-25)。ヒポテマイシンはFLT3、c-KIT、VGFR及びVEGF産生(ERK経路のMEK1/2及びERK1/2の阻害を介する)の強力な阻害剤であり、本発明の方法にしたがって、本明細書に記載のようにヒポテマイシン並びにその誘導体及び類似体をAMLの治療のために患者に投与することができる。
したがって、本発明の方法はAMLを治療する方法を含む。ある実施態様では、これらの方法は、病変組織がAML又は他の癌タイプの指標となるFLT3変異を有する細胞を含むか否かを同定する初期工程を含む。FLT3変異はAMLで発生する(患者のほぼ41%)。これらの変異には、活性化ループ内のAsp835、及びD835->Y又はV又はH又はE又はNが含まれるが、ただしこれらに限定されない(前記変異は公知の方法にしたがって検出することができる)。
【0064】
B-Raf変異を伴う癌:
特異的なB-Raf変異V599E(V600E)は、悪性メラノーマの70%及び結腸癌の約20%に見出される。本発明のある実施態様では、癌患者の腫瘍の生検を実施して、腫瘍細胞が、これらERK経路依存癌に特徴的なB-Raf変異を示すか否かが決定され、B-Raf変異が存在する場合は、続いて本発明の方法で有用な化合物が癌治療のために投与される。
この診断/治療一体化方法(又は“テラノスチック(theranostic)”)の有効性は図5のデータによって部分的に例証される。ヒポテマイシン(本発明のある種の方法で有用なレゾルシル酸ラクトン)を60細胞株のNCIパネルで試験し、その結果のlog GI50値(50%の増殖低下の達成に要求される薬剤量)が図5で棒グラフの形で示される。前記化合物に対してもっとも感受性が強い細胞株は、垂直線の平均活性から右を指す棒線を用いて示される。この結果は、感受生細胞株は、異常なMAPKシグナル伝達経路に含まれるタンパク質キナーゼ(例えばMEK1/2、ERK1/2)とともにB-Raf変異V599E(V600E)を有するB-Raf依存癌に由来し、前記は、これらの変異体キナーゼに重要なCys残基が存在し、さらに可逆的結合及び必須のマイケル付加物形成に必要な構造が存在することから、ヒポテマイシンに対して感受性であることは本明細書での開示から予想され得るとおりである。
表1は、上記で考察した本発明の有用性を支持するデータを表形態で提示する。
【0065】
表1:B-Raf変異癌細胞のキナーゼ阻害剤に対する感受性
表1のデータは、ヒポテマイシンによって例証されるように、変異B-Rafを有する癌細胞株は、マイケル付加物形成に馴染みやすいエノン構造を有するレゾルシル酸ラクトンに特に感受性が高いことを示している。対照的に、野生型B-Rafを有するA549細胞株は、感受性は低いが、ただしその増殖はなお顕著に阻害される。ベンゾピラン-4-オンの骨組みを土台とするMEK阻害剤であるPD98059、及び水素添加されたエノン炭素-炭素二重結合を有し、したがってマイケル反応に参加することができない5,6-ジヒドロヒポテマイシンはともに阻害剤として効果は貧弱である。さらにまた、エノンレゾルシル酸ラクトンは、B-Rafをもつ細胞に対してBayer43-9006(ソフラニブ)よりも活性は顕著に高い。Bayer43-9006は、最初Raf-1阻害剤として開発され、現在はメラノーマに対するヒトの臨床試験中である。同様にエノンレゾルシル酸ラクトンはSU11248(臨床試験で精査されたまた別のキナーゼ阻害剤である)よりもはるかに強力である。
【0066】
B-Raf変異癌細胞株のRALに対する感受性は、B-Raf変異体メラノーマ(A375)異種移植モデルで確認された。図6に示されているように、15mg/kg又は20mg/kgのヒポテマイシンを毎日投与することによって、A375異種移植の増殖は賦形剤単独と比較して顕著に阻害される。さらにまた、両投与量のヒポテマイシンは、25mg/kg又は50mg/kgで1日おきに投与されたBayer43-9006(非RAL、非シスエノンキナーゼ阻害剤)よりも顕著に有効である(前記のBayer43-9006の投与計画は、以前に有効であると報告されたBayer43-9006用の計画である(Sharma et al. Cancer Res 2005; 65(6):2412-2421))。したがって、in vitro及びin vivoの両分析によって、活性化B-Raf変異をもつ癌細胞株は特にRALによる増殖阻害に感受性を有することが示された。
メラノーマの治療で本発明の化合物を使用することは特に有益である。悪性メラノーマのほぼ70%が変異B-Rafを有し、さらにメラノーマは、外科的処置によって治療可能な時期を過ぎて進行した場合、治療は極めて困難である。同様に、本発明の化合物は結腸癌の治療に有用である。結腸癌のほぼ20%が変異B-Rafを有し、ある実施態様では、本発明の方法にしたがい、本発明の化合物による治療に適した患者を特定するために、BRAF変異について生検標本の予備スクリーニングが実施される。したがって、本発明の化合物は、変異B-Raf、特にV599E(従来の命名法ではV600E)及びV599D(従来の命名法ではV600D)を特徴とする細胞の分裂の阻害に有効である。
【0067】
腎細胞癌:
本発明の方法は、腎細胞癌(RCC)を治療する方法を含み、RCCは成人の全ての悪性疾患の約3%を占め、合衆国では毎年約31,000件の新規症例が診断される。サイトカインによる免疫学的療法が従来の標準的治療であるが、ほんの限定的患者群が応答するだけである。RCCの生物学的研究によって、治療標的としてVEGF及びそのレセプター(VEGFR)(血管内皮増殖因子レセプター;vascular endothelial growth factor receptor)が同定された(Rathmell et al. Curr Opin Oncol 2005; 17(3):261-7)。複数の企業(Onyx及びSugenを含む)が、VEGFRをRCCの治療に用いることができるか否かを研究している。そのような化合物は、本発明で有用な化合物よりも一般的には劣っている。なぜならば、それらはレセプターのみを阻害するだけであるが、一方、本発明の化合物はレセプターだけでなくVEGFの産生もまた阻害するからである。
フォン・ヒッペル・リンダウ症候群は家族性異常であり、フォン・ヒッペル・リンダウ(VHL)腫瘍サプレッサーの変異を特徴とする。前記は明細胞RCCに対する感受性増加を伴い、RCC発症の生涯リスクはほぼ50%である。VHLタンパク質は、正常な酸素条件下ではユビキチン依存タンパク質分解のために転写因子HIFαを標的とする。機能的なVHLの非存在下では、HIFαは蓄積して、HIFαの下流の転写標的(VEGF及びPDGFを含む)の構成的発現をもたらす。VHLの不活化はまた、明細胞RCCの散発性症例の60〜80%で発生することが示され、VEGF過剰発現は解析されたRCCサンプルの大半で明示された(Rini et al. J Clin Oncol 2005; 23(5):1028-43)。VEGFを標的とするモノクローナル抗体並びに小分子VEGFR及びPDGFR阻害剤(例えばBayer43-9006)は、RCC臨床試験において進行時間を遅らせることについて有望な結果を示すか、又は顕著な百分率の患者で部分的応答若しくは疾患の安定の証拠を示した(Rini et al. 上掲書)。両増殖因子レセプターVEGFR及びPDGFRの阻害に加えて、本発明の方法で有用なレゾルシル酸ラクトンキナーゼ阻害剤はまた、MEK1/2及びERK1/2の阻害を介して下流のERKシグナル伝達経路の4つの酵素を同時に標的とする(ERKシグナル伝達経路はRCCでは構成的に活性であることが示された(Ahmad et al. Clin Cancer Res 2004; 10(18Pt2):6388S-92S;及びOka et al. Cancer Res 1995; 55(18)4182-7)。なぜならばVEGFはERK経路によって刺激されるので、本発明の方法で有用な阻害剤はまたVEGF産生を低下させる。本明細書で提供されるヒポテマイシン並びにその類似体及び誘導体は、RCCの治療のために本発明の方法にしたがって患者に投与することができる。
【0068】
Ras依存性癌腫:
本発明の方法はRas依存性癌腫を治療する方法を含む。マイトジェン活性化タンパク質キナーゼ(MAPK)シグナル伝達経路又はERK経路は、多くのヒトの腫瘍で細胞の増殖及び生存を調節する(Sebolt-Leopold et al. Nat Rev Cancer 2004; 4(12):937-47)。癌細胞の多くのタイプが、Rasの活性化変異によって引き起こされるMAPKシグナル伝達経路の構成的活性化を示す。これらの変異は、MAPK経路を介するシグナル伝達の増加及び細胞分裂の増加をもたらし、さらにK-Rasにおける変異(結腸癌で45%、膵臓癌で90%及び非小細胞肺癌で35%の陽性率);N-Rasにおける変異(メラノーマで15%、ALL及びAMLで30%の陽性率);及びH-Rasにおける変異(K-Ras及びN-Rasと併せて、甲状腺乳頭癌で60%の陽性率)を含む。Raf阻害剤(例えばBAY 43-9006)又はMEK阻害剤(例えばPD184352)は、活性化Ras変異を保有するヒト腫瘍細胞株で増殖及びMAPK経路の両方を阻害することが示され、さらにマウス腫瘍モデルで、腫瘍増殖を阻害することが示された(Sebolt-Leopold et al. Nat Med 1999; 5(7):810-6;及びSebolt-Leopold, Oncogene 2000; 19(56):6594-9)。ヒポテマイシン並びにその誘導体及び類似体は、カスケードの2つのレベル(MEK1/2及びERK1/2)での阻害を介するMAPKシグナル伝達経路の強力な阻害剤であり、Ras活性化変異を保持する腫瘍の治療のために本発明の方法にしたがって用いることができる。
【0069】
前立腺癌:
本発明の化合物及び方法はまた前立腺癌の治療で有用である。前立腺癌は男性でもっとも普遍的な癌であり、合衆国単独で130万人を超える患者が存在する。2003年には221,000件の前立腺癌の新規症例が発生し、アンドロゲン除去療法に使用にもかかわらず、転移性前立腺癌により29,000人が死亡するであろうと概算された。アンドロゲン除去は進行癌の患者の唯一の有効な療法であり、ほぼ80%の患者がアンドロゲン除去後に症状的及び/又は客観的応答を達成する。しかしながら、最終的にはアンドロゲン非依存性への進行がほぼ全ての患者で発生する。複数の非ホルモン性薬剤がホルモン不応性前立腺癌をもつ患者で判定されたが、これらの薬剤は限定的な抗腫瘍活性を有し、客観的応答率は20%であり、いずれも生存効果を示さなかった。前立腺癌の進行を仲介する分子標的の同定及び選択的阻害は、この疾患の将来の治療に多大な影響を与えるであろう。
マイトジェン活性化タンパク質キナーゼ(MAPK)活性の増加は、前立腺癌のヒトでの症状の進行と相関性を有する(Gioeli et al. Cancer Res 1999; 59:279-84)。これらの結果は、Ras活性が異種移植における前立腺腫瘍増殖のアンドロゲン要求性を調節するという観察と併せて、MAPK経路が前立腺癌の分裂に重要な役割を果たすということを示している(Bakin et al. Cancer Res 2003; 63:1981-9;Bakin et al. Cancer Res 2003; 63:1975-80)。セリン/スレオニンタンパク質キナーゼファミリー、p90リボソームS6キナーゼ(RSK)はMAPKの下流エフェクターとして機能する。RSKファミリーは4つのアイソフォームから成り、それらは別個の遺伝子の生成物である。RSKは、重要な基質(いくつかの転写因子及びキナーゼ、サイクリン依存キナーゼ阻害剤、p27Kip1、腫瘍サプレッサー、チューブリン及び前アポトーシスタンパク質、Badを含む)をリン酸化し、その活性を調節するというそれらの能力を介して体細胞における細胞生存及び分裂で重要な役割を果たす。これらの観察は前立腺癌におけるMAPKの公知の重要性とあいまって、RSKはまた前立腺癌の進行にも寄与することを示している。
ヒト前立腺癌株LNCaPにおけるRSLアイソフォーム2(RSK2)レベルの増加は前立腺特異的抗原(PSA)の発現を強化し、一方、RSK阻害剤、3Ac-SL0101を用いたRSK活性の阻害はPSA発現を低下させることが最近示された(Clark et al. Cancer Res 2005; 65(8):3108-16)。RSKレベルは、正常な前立腺組織と比較してヒトの前立腺癌の約50%で高く、RSKレベルの増加は、前立腺癌で生じるPSA発現の上昇に関与することを示している。さらにまた、3Ac-SL0101は、LNCaP株及びアンドロゲン非依存性ヒト前立腺癌株PC-3の分裂を阻害する。これらの結果は、いくらかの前立腺癌細胞の分裂はRSK活性に依存し、RSKは前立腺癌の重要な化学療法標的であることを示している。
ヒポテマイシン並びにその誘導体及び類似体は、ERK経路の2つの重要な点及びRSKアイソフォームのC-末端キナーゼドメインを強力に阻害する。したがって、本発明のマイケル付加物形成RALは、前立腺癌及び転移性前立腺癌の単一療法及びアンドロゲン除去療法との併用による治療で、本発明の方法にしたがって有用である。
【0070】
乳癌:
本発明の方法及び化合物はまた乳癌の治療で有用である。2003年の女性の乳癌症例は210,000件と概算され、40,000人が死亡し、有病率がもっとも高い癌形態となっている。乳癌は、エストロゲンレセプター-α(ERα)陽性又はERα陰性として存在する。ERαの存在は、全快生存及び全体的生存の増加の両観点で良好な予後と相関する。ERα陰性乳癌は、増殖因子レセプター(例えばEGFR及びerbB-2(HER2))を過剰発現する傾向がある。Raf-1は、これらレセプターのシグナルトランスダクション経路における主要な中間体である。高レベルの構成的Rafキナーゼ又は下流のMAPキナーゼ活性は、ERα陽性乳癌細胞に、エストロゲンの非存在下で増殖する能力を付与し、ERα陰性表現型と類似する。MEK阻害剤を用いる治療によるRafシグナル伝達の除去は、ERα陽性態様を回復させることができる(Oh et al. Mol Endocrinol 2001; 15(8):1344-59)。抗エストロゲン(例えばタモキシフェン)による治療が、細胞周期停止及びアポトーシス誘導によってERα癌細胞の増殖を阻害するために一般的に用いられる。前記は、細胞周期阻害剤、p27Kip1の作用を必要とする。ERα陽性細胞でのMAPKシグナル伝達経路の構成的活性化は、p27リン酸化及び残りのp27のcdk2阻害活性を低下させ、前記は一緒になって抗エストロゲン耐性を促進する(Donovan et al. J Biol Chem 2001; 276(44):40888-95)。パクリタキセル、ドキソルビシン及び5-フルオロウラシルのような細胞傷害性薬剤に対する耐性は、部分的にはRas-シグナル伝達(Rafの上流エフェクター)によって仲介される。MEKキナーゼ阻害剤を用いる治療によるRas/Rafシグナル伝達の阻害は、顕著な程度で前記耐性と対抗する(Jin et al. Br J Cancer 2003; 89(1):185-91)。これらの事実は、乳癌の治療でシグナルトランスダクション阻害剤を使用することを正当化し(Nahta et al. Curr Med Chem Anti-Canc Agents 2003; 3(3):201-16)、前記は、MEK及びEGFR阻害剤の二重使用は、どちらかの薬剤の単独使用よりも乳癌細胞の顕著に強い増殖阻害及びアポトーシスをもたらすという報告によって強調される(Lev et al. Br J Cancer 2004; 91(4):795-802)。さらにまた、EGFR及びHER2(乳癌のための標的として立証済み)は、ERK経路を介してそれらの分裂活性を伝達する。最後に、モノクローナル抗体アバスチンによるVEGF作用の阻害は、化学療法に対する乳癌の応答率を劇的に改善した。本明細書に開示したようにマイケル付加物を形成することができるヒポテマイシン並びにその誘導体及び類似体は、ERK経路の4つの酵素(MEK1/2及びERK1/2)とその後に続くVEGF産生(VEGFRも同様に)の強力な阻害剤であり、乳癌の治療のために本発明の方法にしたがって用いることができる。
【0071】
膵臓癌:
本発明の方法はまた膵臓癌を治療する方法を含む。膵臓癌は100,000人に約10症例の発症率しかもたないが、前記は西欧では4番目〜5番目の癌関連死因である。新規に診断される患者の大半は既に切除不能の腫瘍病期にいる。これら患者の5年生存率は1%未満であり、平均生存期間は腫瘍検出後ほぼ5〜6ヶ月である。近年では、ヒト腫瘍の病理発生における増殖因子の役割にますます関心が向けられている。ヒトの膵臓癌は、複数の重要なチロシンキナーゼ増殖因子レセプター及びそれらのリガンド(例えば表皮増殖因子(EGF)、線維芽細胞増殖因子(FGF)、インスリン様増殖因子(IGF-1)、血管内皮増殖因子(VEGF)及び血小板由来増殖因子(PDGF)ファミリーに属するもの)を過剰発現する(Korc, Surg Oncol Clin N Am 1998; 7(1):25-41;Ozawa et al. Teratog Mutagen 2001; 21(1):27-44;及びEbert et al. Int J Cancer 1995; 62(5):529-35)。これらの増殖因子は、オートクライン及び/又はパラクライン的態様で作用し、ERK経路の活性化を介して膵臓癌の増殖を刺激すると考えられている。K-Rasオンコジーンの変異は膵臓癌では75〜90%の頻度で生じ(Li, Cancer J 2001; 7(4):259-65)、前記はこの癌の活発な増殖を強調している。レセプターチロシンキナーゼ及び下流のシグナル伝達キナーゼ(MEK及びp38)に対する小分子阻害剤は、膵臓癌培養細胞の分裂を阻止することが報告された(Matsuda et al. Cancer Res 2002; 62(19):5611-7;及びDing et al. Biochem Biophys Res Commun 2001; 282(2):447-53)。ヒポテマイシン並びにその誘導体及び類似体は、PDGFR、VEGFR、MEK及びERKキナーゼの強力な阻害剤であるとともに、変異K-rasによる過剰なマイトジェンシグナル伝達の阻害剤であり、膵臓癌の治療で本発明の方法にしたがって用いることができる。
【0072】
上皮性卵巣癌:
本発明の化合物及び方法はまた卵巣がんの治療で有用である。上皮性卵巣癌(EOC)は婦人科の悪性腫瘍の中で主要な死亡原因であり、女性の癌関連死の5番目に主要な原因である。2003年には、24,000件の新規な症例が発生し、14,000人が死亡すると予想される。ほとんどの患者は卵巣腫瘍の進行期にあり、治療は徹底的な外科手術とそれに続く化学療法を土台にしている。化学療法計画の骨格は、相変わらず、近年タキサンが加えられた白金誘導体である。MAPKシグナル伝達経路(特にERK1/2セリン-スレオニンキナーゼ)が卵巣癌において主要な役割を果たす(Choi et al. Reprod Biol Endocrinol 2003; 1(1):71)。この経路は、卵巣癌の治療に一般的に用いられる白金含有-又はタキサン系-化学療法薬剤(例えばシスプラチン、カルバプラチン、ドセタキセル及びパクリタキセル)、並びにゴナドトロピン及び卵胞細胞刺激ホルモンによって活性化される。薬剤耐性細胞は、MEK1/2阻害剤による治療によって薬剤感受性細胞に回復させることができる。したがって、ある実施態様では、本発明は卵巣癌を治療する方法を提供し、前記方法は、MEK1/2及びERK1/2タンパク質キナーゼとマイケル付加物を形成することができるタンパク質キナーゼ阻害剤を、白金含有抗癌剤又はタキサンと一緒に又はその投与後に投与することを含む。
卵巣癌細胞の転移はERK経路阻害剤による治療によって抑制することができる。約39%の卵巣腫瘍がPDGFRを発現し、したがって活性なERK経路及びその発現レベルは、卵巣腫瘍のより進んだ組織学的等級及び進行した外科的病期と相関する。さらにまた、PDGFR-A陽性腫瘍をもつ患者は、陰性腫瘍を有する患者よりその生存時間は短い。イマチニブ(グリーベック)は、PDGFR-Aの阻害に依存するメカニズムを介して、卵巣癌細胞の増殖を臨床的に相応な濃度で阻害する(Matei et al. Clin Cancer Res 2004; 10(2):681-90)。腹腔内播種は卵巣癌の進行にとって決定的である。肝細胞増殖因子は、Ras/Raf/MEK/ERKシグナル伝達経路の活性化によって、卵巣癌細胞の遊走および侵襲を誘導し(Ueoka et al. Br J Cancer 2000; 82(4):891-9)、前記は、この疾患の治療のために本発明によって提供されるMEK及びERK阻害剤を使用することを支持する。ヒポテマイシン並びにその誘導体及び類似体は、PDGFRと同様に、PDGFRが活性化する下流の酵素の強力な共有結合阻害剤であり、卵巣癌の治療で本発明の方法にしたがって用いることができる。
【0073】
肺癌:
本発明の方法はまた肺癌を治療する方法を含む。肺癌は合衆国では癌死亡率の主要原因である。2003年の調査では、171,000件の新規症例の発生が予想され、その年には157,000人が死亡した。最近の治療の進歩にもかかわらず、局所的に進行した転移症例についての成果はなお貧弱である。合衆国では非小細胞肺癌(NSCLC)は全ての肺癌の75%を超える。化学療法は、この疾患の進行期の管理に重要な役割を果たす。従来の薬剤には白金を土台にする併用療法及び第二の治療のためのドセタキセルが含まれる。EGFRは、肺を含むほとんどの上皮性腫瘍で発現されるか又は過剰発現される。NSCLC扁平上皮癌は80%の過剰発現を示す。
ゲフチニブ(イレッサ)が他の化学療法が有効でなかった患者でのNSCLCの治療用に合衆国で承認されたが、EGFR由来シグナル伝達におけるMAPK経路の関与は、他の標的がこの困難な癌の治療に利用することができることを示している。VEGFR-2(KDR)及びVEGFR-3(Flt-4)はNSCLCで発現され(Tanno et al. Lung Cancer 2004; 46(1):11-9)、それらのリガンド量の増加及び低酸素状態は、培養NSCLC癌細胞タイプの分裂及び遊走を刺激した。KDR及びFlt-4の刺激はまた、MAPK経路の活性の強化をもたらした。同様に、NSCLCをもつ患者由来の組織サンプルの34%はERK経路の高活性化を示した(Vicent et al. Br J Cancer 2004; 90(5):1047-52)。ERK2及びAkt(EGFRによって制御される2つのシグナル伝達キナーゼ)のリン酸化状態とゲフチニブ療法との間の強い相関関係もまた報告されている(Cappuzzo et al. J Natl Cancer Inst 2004; 96(15):1133-41)。
これらの観察及び他の最近の臨床的観察(Cesario et al. Curr Med Chem Anti-Canc Agents 2004; 4(3):231-45)は、NSCLCの治療でシグナル伝達タンパク質キナーゼの阻害剤を拡大的に使用することを正当化する(前記治療では、トポイソメラーゼ阻害剤(Maulik et al. J Environ Pathol Toxicol Oncol 2004; 23(4)237-51)及び確立された他のタイプの癌治療薬を用いる併用療法が含まれる)。最後に、モノクローナル抗体アバスチンによるVEGF作用の阻害は、パクリタキセル及びカルボプラチンによる化学療法に対するNSCLC癌の応答率における劇的な改善をもたらした。本明細書で提供するヒポテマイシン並びにその誘導体及び類似体は、ERK経路の4つの酵素(MEK1/2及びERK1/2、前記はその後に続くVEGF産生を調節する)と同様、肺癌で重要であることが示されたレセプターキナーゼKDR(VEGFR)、Flt-4及びcKITの強力な阻害剤であり、肺癌の治療のために単一療法及び併用療法で本発明の方法にしたがって用いることができる。
【0074】
結直腸癌:
結直腸癌は合衆国では癌の第二の主要な死因であり、ヒトの悪性疾患の約15%を占める。アメリカ癌学会は、2003年にはほぼ150,000件の結直腸癌の新規症例が診断されるであろうと概算した(Jemal et al. CA Cancer J Clin 2003, 53:5-26)。進行した結直腸癌をもつ患者の大半は、最終的には癌の再発を経験し、結直腸癌は治癒不能と考えられる。標準的な治療は外科的切除及び時に放射線治療を必要とし、一方、化学療法(例えば標準的なカンプトサール(Camptosar(商標))(イリノテカンHCIの注射)/5フルオロウラシル/ロイコボリン投与計画)は満足というには程遠い。
疫学的及び遺伝子マッピング研究によって、多くのタイプの結腸癌は細胞シグナル伝達経路の異常を伴うことが示された。例えば、MAPK経路でB-Raf V599E(V600E)変異が〜15%の結腸癌で見出され、細胞分裂に必要なERK経路の構成的活性化に至る(Sebolt-Leopold et al. Nat Rev Cancer 2004, 4:937-47)。MAPKシグナル伝達の特異的阻害剤はしたがって、Raf V599E(V600E)変異をもつ細胞の分裂の阻害に有効である(Sebolt-Leopold et al. 上掲書;同書、Nat Med 1999, 5:810-6)下記の実施例5に示すように、B-Raf V599E(V600E)細胞株COLO829のERK経路は、マイクロモル濃度以下でMEK1/2及びERK1/2阻害剤のヒポテマイシンに曝露後10分で完全に遮断される。同様な結果が、B-Raf V599E変異結腸癌細胞株HT29で観察される。CI-1040、PD0325901及びARRY-142886のような有効性がより低いMEK1/2阻害剤は結腸癌の動物モデルで有効である(Sebolt-Leopold et al.上掲書)。
結腸癌の転移はマトリックスメタロプロテアーゼ(MMP)の分泌を必要とし、MEK1/2阻害剤は結腸癌細胞でMMP-7遺伝子の発現を遮断することができ(Lynch et al. Int J Oncol 2004, 24:1565-72)、ERK1/2阻害剤はまたこの特性を有する(なぜならばERK2は、結腸癌細胞によるインテグリンα(V)β6仲介MMP-9発現に中心的に関与するからである)(Gu et al. Br J Cancer 2002, 87:348-51)。ERK及び/又はp38依存MAPKシグナル伝達経路の特異的阻害剤はまた、他の状況における結腸癌の治療に、すなわち非ステロイド系抗炎症薬の結腸癌細胞のアポトーシス刺激能力の強化に(Nishihara et al. J Biol Chem 2004, 279:26176-83;Sun and Sinicrope, Mol Cancer Ther 2005, 4:51-9)、CCK-2レセプター仲介プロスタグランジンE2産生刺激による結腸癌細胞の増殖を促進するガストリン-17の能力の阻害に((Colucci et al. Br. J. Pharmacol 2005, 144:338-48)、及びTNFレセプター関連因子(TRAF1)誘導(前記はNFkB経路を介する結腸癌の腫瘍促進の一局面である)の阻害に(Wang et al. Oncogene 2004, 23:1885-95)本発明の方法にしたがって有用である。
VEGFレセプターの刺激は血管形成を強化することができる。細胞から遊離されるVEGFと結合し、VEGFの作用を阻害するアバスタチンのようなモノクローナル抗体は、非常に有効で、転移性結腸癌の治療のために2004年に承認された。最近の臨床試験の結果は、初期療法として一般的化学療法計画である5-フルオロウラシル/ロイコボリンへのアバスタチンの添加は進行結腸癌の進行停止生存を改善することを示している(http://patient.cancerconsultants.mom/colon_cancer_news.aspx?id=17462)。以前の臨床試験では、この疾患の治療で、カンプトサール(Camptosar(商標))/5フルオロウラシル/ロイコボリンへのアバスタチンの添加の化学療法計画による利点が示された。ニューロピリン-1はヒト結腸癌細胞のVEGFコレセプターであり、前記の生成(したがって血管形成及び細胞増殖を刺激する能力)はまたERK1/2及びp38阻害剤によって阻害され得ることが示された(Parikh et al. Am J Pathol 2004, 164:2139-51)。
本発明の方法で有用なレゾルシル酸ラクトンは、BRAF V599E変異を有する結腸癌の治療に、前記変異をもたない結腸癌の治療と同様、特に有用である。全ての細胞に存在するMEK1/2及びERK1/2でERK経路の2点阻害に加えて、前記は、VEGF産生(ERK経路の阻害を介する)を、VEGFR同様に阻害し、さらにTAK1を阻害してNFkB経路を阻害する。
【0075】
基底細胞癌及び他のソニック・ヘッジホッグ経路関連癌:
本発明の方法はまた基底細胞癌及び他の活性化ヘッジホッグ(Hh)経路関連癌を治療する方法を含む。Hh-シグナル伝達経路は3つの主要な成分を含む:1)Hhリガンド;2)負の調節因子パッチド(Ptch)及びアクチベーターであるスムーザンド(Smoothend)(Smo)で構成される膜貫通型レセプターサーキット;及び3)最後にキュビツス・インタラプツス(Cubitus interruptus)(Ci)を調節する細胞質複合体又は転写エフェクターのGliファミリー(Frank-Kamenetsky et al. J Biol 2002, 1:10)。Gli1のような転写レベルでの正及び負のフィードバックが存在し、Ptch1遺伝子は前記経路の活性化の直接的転写標的である。Hhリガンドはおよそ45kDaの前駆体として合成され、前記前駆体は自己プロセッシングを経て、前記前駆体のアミノ末端半分へのコレステロール部分の共有結合付加がもたらされる。Smoは、Gタンパク質共役レセプター(GPCR)と相同性をもつ7回膜貫通型タンパク質であり、一方、Ptch1は12回膜貫通型タンパク質で、チャネル又はトランスポーターに類似する。極めて重要な経路阻害剤としてのその役割と合致して、Ptch1の除去は、Hhリガンドとは別個に機能する構成的に活性なHh経路を生じる。同様に、Smoの膜貫通型ヘリックスの特異的な点変異は、Ptch1阻害を効果的に迂回しながら、前記経路を構成的に刺激することができる。
動物の適切な発育にとって必須であるが、一方、変異によるか、又はPtch1を不活化するか若しくはSmoを活性化する他の事象による不適切なヘッジホッグ経路シグナル伝達は、いくつかのタイプの腫瘍(基底細胞癌、髄芽細胞腫、横紋筋肉腫、グリオーマ、表層膀胱癌、胃腸管腫瘍、小細胞肺癌(SCLC)、膵臓癌、及び前立腺癌を含む)を生じる(di Magliano and Hebrok, Nat Rev Cancer 2003, 3:901-11;Ruiz et al. Nat Rev Cancer 2002, 2:361-72;Fan et al. Nat Rev Cancer 2002, 2:361-72;Fan et al. Endocrinology 2004, 145:3961-70;Sanchez et al. Proc Natl Acad Sci USA 2004, 101:12561-6)。したがって、Hhシグナル伝達の阻害剤は、抗癌剤の開発のための重要な先導物質を提供する(Romer et al. Cancer Res 2005, 65:4975-8;Taipale et al. Nature 2000, 406:1005-9;Williams, Drug News Perspect 2003, 16:657-62)。
Hh応答細胞株C3H10T1/2を用いて、Gli1は血清応答成分を誘導し、さらにPDGFRを活性化することが示された(前記PDGFRは順を追ってRas-ERK経路を活性化し、細胞分裂を刺激する(Xie et al. Proc Natl Acad Sci USA, 2001, 98:9255-9289)。したがって、PDGFR又はERK経路の阻害はHh経路活性化作用の遮断を提供し、Hh活性化のメカニズムにもかかわらずHh経路の終点をもたらす(すなわち刺激又は阻害の解除)。
基底細胞癌(BCC)はもっとも一般的なヒトの癌であり、合衆国では毎年750,000件の新規症例がある。パッチド遺伝子(Ptch1又は2)の変異は、遺伝性異常である基底細胞母斑症候群に、散発性BCCと同様付随することは既に確立されている。下流分子Gli1は前記経路の生物学的作用を仲介し、前記は約90%のBCCでアップレギュレートする。Gil1は、続いてPDGFRαをアップレギュレートし、前記は細胞分裂を誘導するERK経路の活性化を引き起こす。その後のERK経路の活性化によるPDGFRαの過剰産生は重要なメカニズムであり、前記メカニズムによってヘッジホッグ経路の変異がBCCを惹起する(Xie et al. Proc Natl Acad Sci USA 2001, 98:9255-9)。
腫瘍内IFNαはBCCの治療には不便であるが有効であり、回復率は約50〜80%である。イミキモド(サイトカイン、例えばIFNαの分泌を刺激する)もまた有効である。最近、IFNαによって仲介されるヘッジホッグ経路活性化BCC細胞の殺細胞は、そのERK経路との干渉からもたらされることが示された(前記干渉はFas発現を上昇させ続いてアポトーシスをもたらす)(Li et al. Oncogene 2004, 23:1608-17)。
上記の考察は、PDGFR又はERK経路の阻害はHh経路活性化作用の遮断を提供し、Hh活性化のメカニズムにもかかわらずHh経路の終点をもたらすことを示している。本明細書で開示するヒポテマイシン並びにその誘導体及び類似体は、PDGFRα及びERK経路の2つの酵素の強力な阻害剤である。下記表4に示すように、それらは、培養BCC細胞の強力な阻害剤であり、BCC及び活性化ヘッジホッグ経路によって惹起される他の腫瘍の治療のため本発明の方法にしたがって用いることができる。したがって、ヒポテマイシンは、培養BCC細胞株ASZ001に対して約100nMのIC50を有する(表4)。比較すれば、タザロテン(Ptc±マウス(So et al. Cancer Res 2004, 64, 4385-9)で85%を超えるBCC発症阻害をもたらす局所用アセチレンレチノイドでありBCCの治療に臨床で用いられる)は、ASZ001 BCC細胞をおよそ10,000nMのIC50で阻害する。
【0076】
再狭窄:
本発明の化合物及び方法はまた、それらは再狭窄を予防することができるという点で血管形成術及びステントの使用で有用である。平滑筋細胞分裂は、血管形成術後の新生内膜形成における重要な事象である。PDGFは、損傷に対する血管平滑筋細胞の応答に必要な有糸分裂促進因子であり、平滑筋細胞でERK経路を活性化し、前記は遊走に必須である。MEK阻害剤は、血管平滑筋細胞の分裂及び遊走を妨げるために有効な薬理学的物質である。なぜならば、それらはERK活性化を遮断し、それによってPDGFに対する細胞性応答を遮断するからである。ストレス活性化MAPK p38はまた、血管損傷に対する応答に必要とされ、p38及びその活性を調節する上流のキナーゼを標的とする阻害剤は再狭窄の治療で有効である。PDGFレセプターは平滑筋細胞の遊走及び分裂を刺激し、VEGFレセプターは新生血管形成を刺激する。本発明の方法で有用な化合物は、PDGFR及びVEGFRとともにERK及びJNK経路の複数のキナーゼを阻害するので、それらは強力な再狭窄の阻害剤であり、したがってステント及び有害な平滑筋細胞遊走を刺激する他の装置の調製で一般的に有用である。
したがって、ある実施態様では、本発明は、ステント又はin vivoでの使用を意図する他の装置を提供し、前記は、望ましくない平滑筋細胞の分裂及び前記ステントへの遊走を妨げるか又は遅らせる、本発明の方法で有用な化合物で被覆されているか、前記化合物とともに包埋されるか、そうでなければ前記化合物を含む。平滑筋細胞のこれらステントへの無制御な遊走は、本発明の方法にしたがって治療可能な症状を生じる。したがって、本発明によって提供されるステントは、再狭窄にとって極めて重要な複数のレセプター及び細胞シグナル伝達経路の強力で不可逆的な阻害剤を含むので、従来の技術を超える顕著な進歩を提供する。ある実施態様では、ステントを調製するために用いられるRALは、ヒポテマイシン又はUS2004/0243224 A1(Tremble, 2004)に開示されたRAL以外の本発明の方法で有用なRALである。
【0077】
慢性関節リウマチ:
慢性関節リウマチ(RA)は、合衆国では1,000,000人を超える人々が罹患している結合組織疾患である。この自己免疫異常は、罹患関節への活性化免疫細胞(T及びB細胞)及びマクロファージの補充によってもっぱら引き起こされる。前記関節で、これら細胞によって産生されるサイトカインIL-1及びTNF-αがRAで観察される不可逆的な関節の破壊を仲介する。これらサイトカインによって活性化される下流の遺伝子(NFkB及びMAPKシグナル伝達経路によって誘導されるNFkB及びAP-1転写因子を介する)は、炎症性分子及びマトリックスメタロプロテイナーゼ(MMP)ファミリーの分泌プロテイナーゼの両者をコードする(前記はRAで高レベルで見出される)。サイトカイン誘導性MMP遺伝子発現を阻害し、さらにまたNFkB及びMAPKシグナル伝達経路を遮断することができる化合物は新規な関節炎薬を提供することができる(Vincenti and Brinckerhoff, J Clin. Invest. 2001, 108:181)。IL-1はMEKKK TAK1の活性化を誘導する。TAK1は、NFkBの活性化、さらにJNKを介してAP-1の活性化を制御し(Ninomiya-Tsuji et al. Nature 1999, 398:252)、したがって特異的なTAK1阻害剤は、NFkB、p38及びJNK経路のIL-1誘導性活性化を遮断することによって炎症を予防することができる。JNK及び炎症細胞(RA細胞を含む)に多いp38アイソフォームの特異的阻害剤は、培養細胞でJNK及びp38経路によって制御される遺伝子の発現を効果的に阻止し、動物でコラゲナーゼ遺伝子発現及び関節破壊の低下を示す。MEK1/2阻害剤はまた、培養細胞でIL-1刺激応答を効果的に遮断する(Barchowsky et al. Cyrokine 2000, 12:1469)。
ある実施態様では、本発明は、p38経路を阻害するためにTAK1及びMEK3/6と、JNK経路を阻害するためにTAK1及びMEK4/7と、さらにERK経路を阻害するためにMEK1/2及びERK1/2とマイケル付加物を形成することができる阻害剤を用いてRAを治療する方法を提供する。この広範囲の連続的及びネットワーク的阻害を介して、NFkB及びAP-1依存シグナル伝達経路は効果的に阻害され、前記疾患は治療される。
【0078】
乾癬:
本発明の方法で有用な化合物による乾癬の治療は、連続多シグナル伝達経路阻害アプローチの威力を例証する。世界中で1千万人を超える人々が乾癬に苦しみ、多くの治療が存在するが長期にわたって有効なものは少なく、完治方法は開発されていない(Geilen and Orfanos, Clin Exp Rheumatol 2002, 20(6 Suppl 28):S81-7;Gniadecki et al. Acta Derm Venereol 2002, 82(6):401-10)。
乾癬は、表皮の過剰分裂、分化障害、炎症及び過剰な表皮血管形成を特徴とする遺伝性皮膚疾患群である。乾癬の発生病理は、免疫学的メカニズム、不完全な増殖制御メカニズム又は前記メカニズムの組合せを土台にしている。表皮の過剰分裂、異常なケラチン化、血管形成及び炎症は乾癬斑の確立された特徴であり、前記乾癬斑は一般的には関節、四肢及び頭皮に生じるが、ただし身体のいずれの場所にも出現し得る。
免疫抑制剤及び抗炎症剤はしばしば乾癬の治療に用いられる。前記薬剤の使用は、乾癬の病因論で重要であると考えられている、自己免疫応答でのT細胞の関与(直接的作用、又は種々のケモカイン及びサイトカイン(Erk経路の活性化を介してケラチノサイトに過剰増殖シグナルを伝えるTNFαを含む)の放出を介する間接的作用による関与)を根拠にしている(Bowcock et al. Hum Mol Genet 2001, 10(17):1793-805)。インテグリン及び他の粘着分子もまた中心的に関与する。トランスジェニックマウスを用いた研究によって、インテグリンの過剰発現はMAPKシグナル伝達経路(ERK経路)を活性化してケラチノサイトの増殖速度を高め、さらに乾癬の組織学的特徴を再生させることが示された。さらにまた、特にIL-1αレベルの上昇下でのMEK1の構成的活性化は、多くの乾癬の特徴を有する過剰分裂性及び炎症性皮膚病巣を生じるために充分である。最近、タンパク質キナーゼSTAT3が乾癬に必須であり、この酵素の阻害が症状の緩和に有効であることが示された(Sano et al. Nat Med 2005, 11(1):43-49)。
乾癬の治療のために本発明の方法で有用な化合物は、MEK1、ERK1/2、VEGFR、PDGFR、JNK(インテグリン)経路のMEK4/7並びにp38ストレス経路のTAK1及びMEK3/6を含むキナーゼサブセットを阻害する。上記に記載したように、乾癬の細胞分裂は活性なERK経路と密接に関連し、VEGFが乾癬の皮膚病層で高レベルで見出される。本発明の方法で有用な化合物は乾癬の多くの特徴に影響を与える。すなわち、それらは、ERK経路の阻害を介して細胞分裂を阻害する。それらは、VEGFRを阻害することによって、さらにERKの阻害を介してVEGF及びSTAT3の産生を阻害することによって血管形成を阻害する。それらはEGFRを直接阻害しないが、それらは、EGFRと細胞分裂とを結びつけるものとして機能するERK経路を阻害し、さらにそれらは、p38ストレス経路の二重阻害(TAK1及びMEK3/6)を提供する。最後に、3つのシグナル経路の統合は、T細胞によるサイトカインの分泌及びそれに続く以下のエフェクター機能の獲得をもたらす:(i)カルシニューリンの活性化、(ii)ERK経路の活性化、及び(iii)JNK経路の活性化。本発明の方法で有用な化合物は、MEK及びERKをJNK経路と同様に阻害し、したがってT細胞活性化に必要な3つの経路のうちの2つを阻害する。したがって、本発明のRALは乾癬の生物学的特徴の原因となる経路の各々で標的を阻害し、乾癬を治療する本発明の方法は、この疾患の治療で実質的な有望性を提供する。
【0079】
炎症性腸疾患:
本発明の方法はまた、本明細書に記載のマイケル付加物形成タンパク質キナーゼ阻害剤の治療的に有効な用量を投与することによって、炎症性腸疾患(IBD)(クローン病及び潰瘍性大腸炎を含む)を治療する方法を提供する。前記疾患は、腹痛及び慢性の下痢をもたらす胃腸管の慢性再発性炎症を特徴とする、病因が不明な疾患である。それらは、遺伝的、環境的、及び免疫学的因子が相互に影響し合って惹起される他因子性疾患である。IBD(特にクローン病)のためのいくつかの治療選択肢が、特定のシグナルトランスダクションエレメントの阻害を基にして開発された。
例えば、モノクローナル抗TNF-α抗体(インフリキシマブ)による中心的前炎症サイトカイン、腫瘍壊死因子-α(TNF-α)の特異的阻害が、ステロイド耐性クローン病治療の本流となった。炎症性シグナルトランスダクションにおけるそれらの重要性のために、MAPK経路は、急性及び慢性炎症抑制のための標的である。複数のMAPK経路が、IBDの病因と密接に結びつく炎症応答を統制する。例えば、ERK1/2、p38、JNK/SAPKタンパク質キナーゼ及びそれらと密接に関係するシグナル伝達経路は、クローン病で顕著に活性化されることが知られている。これらの経路のタンパク質キナーゼ及びそれらの活性を調節する上流のキナーゼの阻害剤による治療は、IBDの臨床治療で有効である。ある実施態様では、本発明は、炎症及び炎症性疾患(IBDを含む)を、これらの経路で複数の酵素とマイケル付加物を形成することができるレゾルシル酸ラクトンを用いて治療する方法を提供する。本発明は、ERK経路の2ヶ所(MEK1/2及びERK1/2)、JNK/SAPK経路の1ヶ所(MEK4/7)及びp38経路の2ヶ所(TAK1及びMEK3/6)の強力な阻害剤を治療を要する患者に投与する、これら疾患の治療方法を提供する。
【0080】
肥満細胞症:
本発明の方法はまた、肥満細胞症(過剰な肥満細胞を伴う分裂増殖性異常)を治療する方法を含む。2つの主要な形態は皮膚型(肥満細胞が皮膚に蓄積する)及び全身型(肥満細胞は多くの異なる組織に蓄積し得る)である(www.niai.nih.gov/factsheets/masto.htm)。前記の両形態は、この疾患のより攻撃的な形態、悪性肥満細胞症に進行し、悪性肥満細胞症は続いて白血病の一形態に進行し得る(Longley, Cutis 1999, 64(4):281-2及びLongkey et al. Nat Genet 1996, 12(3):312-4)。従来の肥満細胞症治療は症状の緩和に焦点をあて、この症状に対する利用可能な治療薬は現在のところ存在しない。
cKITタンパク質は、肥満細胞増殖因子の存在下で活性化される肥満細胞膜貫通型レセプターチロシンキナーゼであり、ERK経路の活性化を介して肥満細胞の分裂を刺激する。構成的に活性なcKITの発現をもたらすcKITの変異(通常D816V)が、全身性及び皮膚性肥満細胞症で観察された(Longley et al. Proc Natl Acad Sci USA 1999, 96(4):1609-14)。前記疾患のこの形態はイマチニブに対して耐性を示す(グリーベック;Ma et al. J Invest. Dekmarol 1999, 112(2):165-70)(イマチニブはヒトの医薬としての使用が承認された最初のキナーゼ阻害剤である)。本明細書に記載のヒポテマイシン並びにその誘導体及び類似体は、野生型KIT及び構成的に活性なKIT(D816V)とともにERK経路の2ヶ所(MEK1/2及びERK1/2)の強力な阻害剤であり、肥満細胞症療法として本発明の方法にしたがって患者に投与することができる。in vitro試験によって、肥満細胞腫細胞株はヒポテマイシンに感受性であることが示された。構成的に活性なcKIT(D814Y、前記はヒトのD816V変異に対応する)を発現する、マウスの肥満細胞腫細胞株p815を用いたとき、ヒポテマイシンは310nMのGI50を示し、一方、他の既知のcKIT 阻害剤、BAY 43-9006及びSU11248は、それぞれ310nM及び320nMのGI50を示す。
【0081】
肥満細胞成分を有する炎症性疾患:
本発明の方法で有用な化合物はまた肥満細胞関連炎症性疾患の治療のために投与することができる。肥満細胞はまた、本発明の方法及び化合物で治療することができる他の疾患及び症状の発達に必要とされる。肥満細胞は、IgEのためのそれらの表面レセプター(FcqRI)の架橋を介してアレルギー反応の発達に必要である(前記架橋は脱顆粒並びに血管作用性、前炎症性及び侵害受容性仲介物質の放出をもたらす)。肥満細胞生理学の主要な特徴(最近までほとんど省みられなかった)は、明白な脱顆粒を示すことなく、弁別的又は選択的放出を介して仲介物質を分泌することができるということである。この過程は、別個のタンパク質キナーゼの作用によって調節されると考えられている(Theoharides et al. J Neuroimmunol 2004, 146(1-2):1-12)。
アレルギー反応とは異なり、自己免疫又は炎症過程時には肥満細胞の脱顆粒はほとんど認められない。その代わりに、肥満細胞は、明白な脱顆粒を示すことなく、分泌の表示である、それらの高電子密度顆粒コアの超微細構造変化(“活性化”、“顆粒内活性化”又は“断片的”脱顆粒と称される過程)を経るように見える。肥満細胞は、喘息、アトピー性皮膚炎、心脈管系疾患、慢性乾癬、線維筋痛、炎症性腸症候群、間質性膀胱炎、片頭痛、多発性硬化症(MS)、神経線維腫症、変形性関節症、慢性関節リウマチ、強皮症を含む炎症性疾患に必要である(Theoharides et al.上掲書)。実際、これら疾患の多くが、間質性膀胱炎の場合のように同時に発生するようである。肥満細胞は、自己免疫関節炎に要求され、皮膚の過敏反応で決定的な役割を果たし、心脈管系症状の発生(特に不安定狭心症および無症状心筋虚血)でその関与が強く疑われる。さらにまた、神経終末とのそれらの緊密な物理的結合は、多くのストレス誘導性炎症性疾患の病因における肥満細胞の関与を示唆する。
レセプターチロシンキナーゼ、c-Kit(CD117)は肥満細胞の生存に必須である(Tsujimura, Pathol Int 1996, 46(12):933-8)。c-Kitリガンド(幹細胞因子(SCF))は、ヒト肥満細胞の分裂及び成熟に重要であり、前記の除去は肥満細胞のアポトーシスをもたらす。c-Kitの構成的発現は肥満細胞の疾患で生じる(Mol et al. J Biol Chem 2003, 278(34):3146-4)。本明細書に記載のヒポテマイシン並びにその誘導体及び類似体は、c-Kitとともに、c-Kit活性化ERK経路の下流の2ヶ所(MEK1/2、ERK1/2)の強力な不可逆性阻害剤であり、本発明は、感受性を有するプロテアーゼとマイケル付加物を形成することができるRALの治療的に有効な用量を投与することによって、肥満細胞によって影響を受けるか、又は前記によって惹起される炎症性疾患(特に上記に列挙した疾患を含む)を治療する方法を提供する。
肺線維症:
本発明はまた肺線維症を治療する方法を提供する。特発性肺線維症(IPF)は、病因が不明の間質性肺疾患の仮借なき進行型である。IPFと診断されたヒトの平均生存は3年未満である。従来の治療は抗炎症性ステロイド及び免疫抑制剤による治療を含むが、応答率は非常に低い。前線維細胞性サイトカイン(例えばIPFのTGF-β及びPDGF)の役割における興味は、そのようなサイトカインは線維芽細胞の形質転換、分裂及び蓄積を引き起こし、細胞外マトリックスの産生及び堆積、組織破壊及び肺機能の低下をもたらすという事実に集中した(Lasky et al. Environ Health Perspect 2000, 108:Suppl 4:751-62;及びSime et al. Clin Immunol 2001, 99(3):308-19)。最近の研究は、イマチニブは、PDGFRのリン酸化を阻害することによって(Aono et al. Am J Respir Crit. CareMed 2005)、及びおそらくc-Ab1タンパク質キナーゼのリン酸化を阻害することによって(Daniels et al. J Clin Invest 2004, 114(9):1308-16)、マウスのブレオマイシン誘導性肺線維症の進行を阻止することができることを示している。本明細書に記載したヒポテマイシン並びにその誘導体及び類似体は、PDGFRとともに、PDGFシグナルを伝達するERK経路の強力な阻害剤であり、本発明は、マイケル付加物形成を介してそのようなタンパク質キナーゼを阻害することができるRALの治療的に有効な用量を投与することによって肺線維症を治療する方法を提供する。
【0082】
黄斑変性:
本発明はまた、病因としてVEGF(VEGFRは本発明の方法で有用な化合物の標的である)及びERK経路が中心的に関与する糖尿病関連黄斑変性及び緑内障のみならず、加齢関連黄斑変性もまた治療する方法を提供する。本発明のこれらの方法で有用な化合物は、ERK経路の多キナーゼ阻害によるVEGF産生阻害だけでなく、またERK経路阻害を介するVEGF産生とともに内皮細胞内VEGF産生の阻害によっても、VEGF仲介血管形成を阻害する。ある実施態様では、本発明の方法で有用な化合物は、この消耗性症状の治療のために、黄斑変性治療用のまた別の薬剤と一緒に同時投与される。
アレルギー性皮膚炎:
本発明の方法はまた、アレルギー性皮膚炎及び免疫抑制が所望される他の疾患を治療する方法を含む。上記に記載したように、3つのシグナル経路の統合は、T細胞によるサイトカインの分泌及びそれに続く以下のエフェクター機能の獲得をもたらす:(i)カルシニューリンの活性化、(ii)ERK経路の活性化、及び(iii)JNK経路の活性化。ヒポテマイシンは、ERK経路を2ヶ所(MEK1/2及びERK1/2)で阻害するとともにJNK経路をMEK4/7で阻害し、したがってT細胞活性化に必要な3つの経路のうちの2つを阻害する。FK506は、カルシニューリンの作用を阻害する周知の免疫抑制剤で、アトピー性皮膚炎の治療で用いられる。本発明の方法にしたがえば、本明細書で提供される本発明の化合物の投与はアトピー性皮膚炎の治療に用いられる。ある実施態様では、本発明の化合物は、カルシニューリンを阻害するか又はその作用を阻害する化合物又は薬剤とともに同時投与される。そのような化合物には、FK506並びに学術文献及び特許文献に報告された前記の複数の誘導体が含まれるが、ただしそれらに限定されない。この治療によって、サイトカイン分泌をもたらす3つのシグナル伝達経路の全てが阻害され、アレルギー性皮膚炎及び免疫抑制が所望される他の疾患の効果的な治療が提供される。
【0083】
痛み:
本発明はまた痛みを治療する方法を提供する。合衆国の人口の9%が中等度から重篤な癌に関連しない全てのタイプの痛みに苦しんでいる。前記には慢性の痛みを抱える1,500万人を超える人々が含まれる。世界中でほぼ2,600万人の患者(合衆国では1,000万人)が何らかの形態の神経障害性の痛み(痛みが刺激に対して不適切である慢性の疼痛)を有する。末梢神経障害性疼痛は、典型的には末梢神経が、外科手術、骨の圧迫(種々の疾患で)、糖尿病、及び感染により損傷を受けたときに発生する。神経障害性疼痛症状の2つの一般的で重度の消耗性症状は痛覚過敏及び異痛である。痛覚過敏は、疼痛刺激によって生じる疼痛応答過多であり、異痛は、通常痛みを感じない刺激から生じる疼痛である。両者はしばしば通常の鎮痛剤に耐性を示す。これらの症状の治療に鎮痛剤が一般的に有効でないのは、脊髄内の神経プロセッシングにおける長期的変化の結果であり得る。実際、脊髄及び後根神経節における多様な神経伝達物質、それらのレセプター、及び他の遺伝子の発現の変化は痛覚過敏と密接に関連していることが示された(以下を参照されたい:Woolf and Costigan, Proc Natl Acad Sci USA, 1999, Jul 6, 96(14):7723-30)。
高い発生率及び神経障害性疼痛の従来治療の貧弱な有効性のために、この症状に対する新規な標的が希求されている。タンパク質キナーゼは種々のタイプの痛みにおいて重要な役割を果たしている。薬剤誘導性神経障害性疼痛における遺伝子発現変化の研究によって、ストレプトズーシン誘導性糖尿病性神経障害及び慢性絞窄損傷の疼痛動物モデルの両方で変化する細胞外シグナル調節キナーゼ(ERK)カスケードの重要な成分が同定された(以下を参照されたい:Ciruela et al. 2003, Br J Pharmacol 138(5):751-6)。脊髄内のERK1/2活性レベルの上昇は痛覚過敏と相関性を示した。MEK1/2阻害剤、PD198306の脊髄内投与によって、定常的異痛(疼痛応答の一般的な実験的測定)は、両神経障害疼痛モデルにおいて用量依存態様で遮断された。PD198306の足底内投与は痛覚過敏のどちらのモデルでも効果はなかった。したがって、ERK1/2の活性における対応する変化(MEK1/2阻害作用の主要な結果である)は中枢神経系に局在しなければならない。他の研究は、炎症性疼痛過敏(Ji et al. 2002, J Neurosci 22(2):478-85)の結果として、又は脊髄内の代謝向性グルタミン酸レセプターアゴニストの作用(Adwanikar et al. 2004, Pain 11(1-2):125-35)の結果として、脊髄後角ニューロン内の活性化ERK1/2キナーゼの中心的関与を示した。各事例で、MEK阻害剤は疼痛応答を緩和することができた。リン酸化ERKが核内に入るとき、前記はキナーゼのRSK2タイプを活性化する。前記は続いて、疼痛応答の開始に必要な種々の遺伝子のcAMP仲介転写をもたらすCREBを活性化する(Ji et al. 2002, Neurosci 2(2):478-85)。他のMAPKシグナル伝達経路もまた神経障害性疼痛に関与していた。例えば、p38ストレス活性化MAPKは、成獣ラットのL5脊髄神経の結紮後1日以内で活性化され、その効果は3週間より長く持続する(Jin et al. 2003, J Neurosci 23(10):4017-20)。p38阻害剤、SB203580の脊髄内注射は疼痛応答を、特に神経障害誘導の初期時点で投与したとき顕著に低下させた。
本明細書に開示したレゾルシル酸ラクトン阻害剤の各々は痛みに関連する複数のタンパク質キナーゼを阻害することができ、したがって有益な鎮痛剤である。それぞれは、MEK/ERKシグナル伝達経路の中心的部分の2カ所において強力な阻害剤であり、ほぼ4つの酵素(MEK1/2及びERK1/2)を阻害する(それぞれは、TAK1及びMEK3/6を阻害することによってp38経路を阻害する)。さらにまた、ERK経路の2ヶ所の阻害に加えて、それぞれは下流のRSK2型のキナーゼを阻害し、したがってCREB活性化につながる経路の複数の工程を遮断する。したがって、本発明は、感受性タンパク質とマイケル付加物を形成することができるRAL阻害剤の治療的に有効な用量を投与することを含む、痛みを治療する方法を提供する。
【0084】
併用療法:
ある種の抗癌化合物はある種の細胞タイプでERK経路を活性化することが知られており、したがって、本発明の方法のある観点では、本発明の方法で有用なRALと同時投与される。タキソール及び他のチューブリン相互作用剤は癌細胞でERK経路の活性化を誘導することができる(Stone and Chambers, Exp Cell Res 2000, 254:110-119;MacKeigan et al. J Biol Chem 2000, 275:38953-38956;McDaid and Horwitz Mol Pharmacol 2001, 60:290-301)。前記はいくつかの細胞、例えばHeLa及びCHO細胞で生じるが、他の細胞、例えばMCF-7細胞(McDaid and Horwitz(2001)上掲書)では生じない。さらにまた、パクリタキセル誘導性ERK活性化を示す細胞をMEK阻害剤U0126で処理すると、アポトーシス及び細胞傷害性の累積性が観察される。同様に、ERK経路はカルボプラチン及びシスプラチンによって活性化される(Choi et al. Reprod Biol Endocrinol 2003, 1(1):71)。ある種の癌細胞は、基本的には耐性メカニズムをもたらすある種の薬剤ストレスに対する調節的応答でERK経路を活性化すると考えられる。そのような事例では、薬剤耐性細胞は、ERK経路阻害剤による処置によって薬剤感受生細胞に変換することができる(Choi et al Reprod Biol Endocrinol 2003, 1(1):71)。したがって、ある実施態様では、癌又は特定の癌適応症を治療する本発明の方法は、ERK経路を活性化する抗癌化合物(タキサン(例えばドセタキセル又はパクリタキセル)又は他の微小管安定化剤若しくは微小管脱安定化剤、エポチロン(例えばエポチロンB若しくは D又はエポチロン誘導体)、又は白金剤(例えばシスプラチン又はカルボプラチン)を含むが、ただしこれらに限定されない)を、本明細書に開示のRALと併用して患者に投与して、ERK経路依存癌を治療することを含む。
本発明の別の併用療法では、それ自体Hsp90のクライアントタンパク質であるか、又はHsp90のクライアントタンパク質によって活性化されるキナーゼとマイケル付加物を形成することができるRALタンパク質キナーゼ阻害剤が、Hsp90阻害剤とともに同時投与される。ここで、RALエノンはその特異的キナーゼを阻害し、Hsp90阻害剤は同じ又は別個のキナーゼサブセット(前記はHsp90クライアントタンパク質として機能する)の破壊をもたらす。ある実施態様では、Hsp90阻害剤はゲルダマイシン又はゲルダマイシン類似体(例えば17-AAG又は17-DMAG)である。本発明のまた別の併用療法では、その標的タンパク質キナーゼとマイケル付加物を形成することができるRALタンパク質キナーゼ阻害剤は、トポイソメラーゼ阻害剤と一緒に同時投与される。
【0085】
したがって、ヒトの疾患の治療に用いられるときは、本発明の方法で有用な化合物は他の薬剤と併用して投与することができる。例えば、MAPK経路阻害剤として期待されるものは典型的には細胞に対して細胞増殖抑制作用を示す。前記細胞では、ERK、JNK又は他のMAPK経路は、マイトジェン、異常に機能が亢進したマイトジェンレセプター(例えばVEGFR又はPDGFR)、変異Ras又はRafタンパク質、異常に活性化されたMEKK酵素、又は構成的に発現されたERK遺伝子によって活性化されている。対照的に、一般的に用いられる癌の化学療法薬は典型的には細胞傷害性作用を示す。したがって、本発明のMAPK経路阻害剤は、既に確立された細胞傷害性薬剤又はより新しい薬剤、例えばHsp90阻害性ゲルダマイシン類似体、17-AAG及び17-DMAG(それらの抗腫瘍作用はMAPK経路阻害剤の抗腫瘍作用を補完する)と併用して投与することができる。
本発明の方法で有用な化合物と同時投与することができる抗癌剤又は細胞傷害性薬剤には以下が含まれる:アルキル化剤、血管形成阻害剤、抗代謝薬、DNA分割剤、DNA架橋剤、DNA挿入剤、DNA小溝結合剤、エネディーン(enediynes)、熱ショックタンパク質90阻害剤、ヒストンデアセチラーゼ阻害剤、微小管安定化剤、ヌクレオシド(プリン又はピリミジン)類似体、核外搬出(nuclear export)阻害剤、プロテアソーム阻害剤、トポイソメラーゼ(I又はII)阻害剤、チロシンキナーゼ阻害剤。具体的な抗癌剤又は細胞傷害性薬剤には以下が含まれる:β-ラパコン、アンサミトシンP3、アウリスタチン、ビカルタミド、ブレオマイシン、ボルテゾミブ、ブスルファン、カリスタチンA、カンプトテシン、カペシタビン、CC-1065、シスプラチン、クリプトフィシン、ダウノルビシン、ディソラゾール、ドセタキセル、ドキソルビシン、デュオカルマイシン、ダイネマイシンA、エポシロン、エトポシド、フロキシウリジン、フルダラビン、フルオロウラシル、ゲフィチニブ、ゲルダナマイシン、17-アリルアミノ-17-デメトキシゲルダマイシン(17-AAG)、17-(2-ジメチルアミノエチル)アミノ17-デメトキシゲルダマイシン(17-DMAG)、ゲムシタビン、ヒドロキシウレア、イマチニブ、インターフェロン、インターロイキン、イリノテカン、マイタンシン、メトトレキセート、マイトマイシンC、オキサリプラチン、パクリタキセル、スベロイルアニリドヒドロキサム酸(SAHA)、チオテパ、トポテカン、トリコスタチンA、ビンブラスチン、ビンクリスチン、及びビンデシン。
【0086】
一般的癌治療:
本発明の化合物は、例えば以下のような疾患の治療に用いることができる(ただしこれらに限定されない):以下を含む過剰増殖性疾患:頭部、頸部、副鼻腔、鼻咽頭、口腔、口腔咽頭、喉頭、下咽頭、唾液腺の腫瘍、パラガングリオーマを含む頭部及び頸部の癌;肝臓及び胆管樹の癌、特に肝細胞癌;胃腸の癌、特に結直腸癌;卵巣癌;小細胞及び非小細胞肺癌;乳癌肉腫、例えば線維肉腫、悪性線維性組織球腫、胎児性横紋筋肉腫、平滑筋肉腫、神経線維肉腫、骨肉腫、滑膜肉腫、脂肪肉腫、及び肺胞軟部肉腫;中枢神経系の新形成、特に脳の癌;リンパ腫、例えばホジキンリンパ腫、リンパプラズマ細胞性リンパ腫、濾胞性リンパ腫、粘膜結合リンパ組織性リンパ腫、被膜細胞リンパ腫、B系列大細胞型リンパ腫、バーキットリンパ腫、T-細胞脱分化大細胞型リンパ腫。臨床的には、本明細書に開示した方法及び組成物の実施並びに使用は、癌性増殖のサイズ又は数の減少、及び/又は(該当する場合は)付随症状の減少をもたらす。病理学的には、本明細書に開示した方法及び組成物の実施並びに使用は、病理学的に対応する応答を生じるであろう。例えば癌細胞分裂の阻害、癌又は腫瘍のサイズの減少、更なる転移の予防、及び腫瘍の血管形成の阻害である。そのような疾患を治療する方法は、本明細書に開示したRALの治療的に有効な量を、単独で、又は別の抗癌剤と一緒に対象者に投与することを含む。前記方法は、治療的利益のために必要に応じて反復することができる。
【0087】
細胞の過剰増殖を示す非癌性疾患:
本発明はまた、細胞の過剰増殖を特徴とする非癌性疾患を治療する方法を提供する。前記方法は、本明細書に開示したRAL化合物を、そのような治療の必要がある患者に投与することを含む。そのような疾患の例には以下が含まれる(ただしこれらに限定されない):萎縮性胃炎、炎症性溶血性貧血、移植片拒絶、炎症性好中球減少症、水疱性類天疱瘡、腹腔の疾患、脱髄神経障害、皮膚筋炎、炎症性腸疾患(潰瘍性大腸炎及びクローン病)、多発性硬化症、心筋炎、筋炎、鼻腔ポリープ、慢性副鼻腔炎、尋常性天疱瘡、一次性糸球体腎炎、乾癬、手術後癒着、狭窄又は再狭窄、強膜炎、強皮症、湿疹(アトピー性皮膚炎、過敏性皮膚炎、アレルギー性皮膚炎を含む)、歯周の疾患(すなわち歯周炎)、多嚢胞性腎疾患及びI型糖尿病。他の例には以下が含まれる:脈管炎、例えば巨細胞動脈炎(側頭動脈炎、タカヤス動脈炎)、結節性多発性動脈炎、アレルギー性血管炎及び肉芽腫症(チュルグ-シュトラウス病)、多発性血管炎オーバーラップ症候群、高血圧性血管炎(ヘノッホ-シェーンライン紫斑病)、血清病、薬剤誘導性血管炎、感染性血管炎、新形成血管炎、結合組織疾患に付随する血管炎、補体系の先天的欠損に付随する血管炎、ヴェーゲナー肉芽腫症、川崎病、中枢神経系の血管炎、ベルガー病及び全身性硬化症;胃腸管の病気(例えば、膵炎、クローン病、潰瘍性結腸炎、潰瘍性直腸炎、一次性硬化性胆管炎、特発性を含む任意の原因による良性狭窄症(例えば胆管、食堂、十二指腸、小腸又は結腸の狭窄);気道の病気(例えば喘息、過敏性肺実質炎、石綿肺症、珪肺症及び塵肺症の他の形態、慢性気管支炎、並びに慢性閉塞性気道疾患);鼻涙管の病気(例えば特発性を含む全ての原因による狭窄);及び耳管の病気(例えば特発性を含む全ての原因による狭窄)。
【0088】
医薬組成物及び投与量:
本発明は、本発明の方法で有用な化合物を含む医薬組成物及び調製物を提供する。これらの組成物及び調製物には種々の形態、例えば固体、半固体及び液体形が含まれる。一般的には、前記医薬調製物は、活性成分として本発明の方法で有用な1つ又は2つ以上の化合物、及び医薬的に許容できる担体又は賦形剤を含む。典型的には、前記活性成分は、外部適用、腸内適用又は非経口適用に適した有機又は無機担体若しくは賦形剤と混合されてある。前記活性成分は、例えば、錠剤、ペレット、カプセル、座薬、ペッサリー、溶液、乳濁液、懸濁液、及び使用に適した他の任意の形態のための通常の無毒な医薬的に許容できる担体との化合物でもよい。特に、静脈内投与及び経口投与が意図され、本発明はそのような態様に適した医薬組成物を提供する。
使用することができる賦形剤には、担体、表面活性剤、濃縮剤又は乳化剤、固体結合剤、分散剤又は分散補助剤、可溶化剤、着色剤、香料、被覆剤、崩壊剤、滑沢剤、甘味剤、保存料、等張剤及び前記の組合せが含まれる。適切な賦形剤の選択及び使用は以下の成書に教示されている:Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003)、前記文献の内容は参照により本明細書に含まれる。
前記担体材料と混合して単一投薬形を生成することができる活性成分の量は、治療される宿主及び具体的な投与態様にしたがって変動するであろう。例えば、ヒトへの経口投与が意図される処方物は担体物質を含むことができ、前記は、全組成物の約5%〜約95%で変動し得る。ユニット投薬形は一般的に約5mg〜約500mgの活性成分を含むであろう。
本発明の治療的に有効量の化合物は、単回投与量又は分割投与量で対象者に投与することができる。投与頻度は毎日であっても、又は他のなんらかの規則的スケジュールに従っても(例えば3日目毎)、又は不規則なスケジュールでもよい。投薬量は、例えば体重1kgあたり約0.01〜10mg(約0.01〜10mg/kg)、又はより通常的には約0.1〜2mg/kgであり得る。
【0089】
しかしながら任意の個々の患者に対する具体的な用量レベルは多様な因子に左右され得ることは理解されよう。これらの因子には、用いられる具体的な化合物、体重、一般的な健康状態、性別、対象者の食事、投与時間及び投与経路、薬剤の排出速度、薬剤併用が治療に用いられるか否か、並びに治療が試みられている個々の疾患又は症状の重篤度が含まれる。
不可逆的阻害剤(例えば本明細書で考察された化合物)は、それらが投与される治療計画に強い影響を与えるある種の格別な特徴を有する。標的キナーゼは急速に阻害され、阻害効果は長期に及び、シグナル伝達活性の回復のためにそれらキナーゼの再合成が要求される。したがって、可逆的阻害剤と比較して、不可逆的阻害剤は有効性のために必ずしも高い血中濃度又は長時間の血中半減期の達成を必要としない(例えば、Calvoらの文献中のCC-1033(EGFR機能の不可逆的阻害剤)の考察を参照されたい)。さらにまた、不可逆的阻害剤はより低い頻度で投与することができる。なぜならば、それらの阻害作用はより長く続くからである。腫瘍の増殖を阻害するために要求される曝露の短縮はまた毒性を低下させることができる。不可逆的阻害剤の前記固有の特徴は、標準的な曝露に関する薬理学的動態実験ではなく又は前記に加えて、標的キナーゼの阻害及び回復を基準にした投与計画の最適化を推進する。
必要な場合には、本発明の化合物はマイクロカプセル及びナノ粒子として処方することができる。一般的なプロトコルは例えば以下に記載されている: US5,510,118(Bosch et al., 1996);US5,534,270(De Castro, 1996);及びUS5,662,883(Bagchi et al., 1997)(前記文献は全て参照により本明細書に含まれる)。表面積対容積比を増すことによって、これらの処方物は、経口送達には適さない化合物の経口送達を可能にする。
上記に記載したように、本発明の化合物は、他の医薬(特に他の抗癌剤)と併用して同時投与することができる。前記同時投与は、同時でも又は連続的でもよい。
上記に記載したように、本発明は、その範囲内に本発明の化合物のプロドラッグを含み、本発明はそのようなプロドラッグを含む医薬組成物を提供する。そのようなプロドラッグは、一般的に、要求される化合物にin vivoで容易に変換され得る化合物の機能的誘導体である。したがって、本発明の治療方法では、“投与する”という用語は、具体的に開示した化合物で、又は具体的には開示されなかったが、その必要がある対象者に投与後にin vivoで明示の化合物に変換され得る化合物で、記載の種々の疾患を治療することを含む。適切なプロドラッグ誘導体の選択及び調製のための一般的な方法は例えば以下に記載されている:Wermuth, “Designing Prodrugs and Bioprecursors,” in Wermuth, ed., The Practice of Medicinal Chemistry, 2nd Ed., pp.561-586(Academic Press 2003)。プロドラッグには、in vivoで(例えばヒトの体内で)加水分解して本発明の化合物を生じるエステル、又はその塩が含まれる。適切なエステル基には、医薬的に許容できる脂肪族カルボン酸、特にアルカン酸、アルケン酸、シクロアルカン酸及びアルカン二酸から誘導され、前記の各アルキル又はアルケニル部分が好ましくは6未満の炭素原子を有するものが含まれるが、ただしこれらに限定されない。例示的エステルには、ギ酸エステル、酢酸エステル、プロピオン酸エステル、酪酸エステル、アクリル酸エステル、クエン酸エステル及びエチルコハク酸エステルが含まれる。
【0090】
本発明の方法で有用な化合物:
本明細書に記載したように感受性を有するタンパク質キナーゼとマイケル付加物を形成することができる複数のレゾルシル酸ラクトンが存在するということを、本発明の開示から当業者は理解しえよう。種々のRAL化合物が生成され試験され、さらに多くのものが関連する広範囲の特許文献に記載されてきた。本発明の方法は、部分的には、現時点で存在し、さらに考えられ得るレゾルシル酸ラクトン及び誘導化合物クラスのうち極めてわずかなセットが、ほんのわずかなキナーゼタンパク質ファミリーセット中の重要なCys残基と緩徐な可逆的マイケル付加を介して阻害を達成するために用いることができるという発見から得られた。これらの発見は、以前にはそのような化合物による治療には適しないと考えられた疾患を治療するための薬剤として前臨床試験で既知の化合物を再調査しようとする、さらに、有用であると今日まで単に特許文献で予想されていただけであった化合物を作成し試験しようとする強力な原動力を提供する。
したがって、以前に公知であり試験された化合物が本発明のある種の方法では有用であり得るが、本発明の他の方法はそのような化合物の使用を含まない。
ある実施態様では、本発明の治療方法で投与される化合物及び医薬組成物は、エーザイ株式会社の特許公開公報US2004/0224936 A1(2004)、WO03/076424 A1(2003)及びWO2005/023792 A1(2005)(前記文献は参照により本明細書に含まれる)に記載された化合物であるか、又はそのような刊行物においてある種のジェネリック化合物の範囲に含まれる化合物である。これらの刊行物は、その中に記載されている化合物は、NF-kB及びAP-1活性並びにタンパク質キナーゼ(例えばMEKK、MEK1、VEGFR、PDGFR)の阻害剤としての活性を示すことができるが、特定の疾患及び症状で重要な役割を果たす、キノム中の他のタンパク質キナーゼに関してはサイレントであると述べている。これらの刊行物は、前記化合物は癌並びに炎症性及び免疫疾患の治療に適用できるかもしれないと述べ、RA、乾癬、血管形成及びステント技術の記載を含んでいる。しかしながら、利用可能な限定的データから見れば、開示された化合物の治療潜在能力をこれらの刊行物から見つけることはできない。さらにまた、本明細書に開示したように、そのような化合物はマイケル付加物形成によってMEKK1を阻害することはできない(MEKK1は本発明の化合物とマイケル付加物を形成することができない)。さらにまた、上記で考察し、下記実施例で述べるように、これら特許公開公報のジェネリック化合物の記載に含まれるある種の化合物はマイケル付加物を形成せず、したがって本発明の方法で有用な化合物は、これら刊行物中の化合物の記載に包括的に含まれる化合物の新規なサブセットを含む。
【0091】
エーザイの特許公開公報に開示された化合物を用いて以下を含む多様な癌の治療に用いることができることを本発明は教示するが、それら疾患の全ては、エーザイの特許公開公報で開示された化合物による治療に対して感受性であると記載されなかった以下の症状である:AML、基底細胞癌、B-Raf変異依存癌(大腸癌及びメラノーマを含むがただしこれらに限定されない)、乳癌、GI間質腫瘍、Ras依存癌、腎細胞癌、及び前立腺癌並びに他の症状(肺線維症、肥満細胞症、炎症性腸疾患及びアレルギー性皮膚炎)。本発明はまた、2つ以上のキナーゼを阻害する化合物を投与することによって、種々の症状(特にMEKK、MEK1、VEGFR及びPDGFRに加えてまた別のキナーゼを、MEKK以外のキナーゼと同様に阻害することが治療の有効性を高めるであろうと期待される疾患及び症状)を治療する方法を提供する。他の実施態様では、本発明は、MEKK、MEK1、VEGFR及びPDGFR以外のキナーゼにおける変異のためにある種の薬剤に対して耐性を有する癌を、エーザイの特許公開公報に記載された化合物を投与して変異キナーゼを阻害することによって治療する方法を提供する。本発明の方法の他の実施態様では、エーザイの特許公開公報に具体的に記載された化合物以外の化合物が、本明細書で特定した疾患又は症状を治療するために投与される。
別の実施態様では、本発明の治療方法で投与される化合物及び医薬組成物は、Cor Therapeutics, Incの米国特許5,674,892号(1997)、5,795,910号(1998)及び5,728,726号(1998)(前記文献は参照により本明細書に含まれる)に記載された化合物のサブセットである。これらの刊行物は、多様なRLA(本明細書に記載したマイケル付加物を形成することができるもの、及びできないものを含む)がキナーゼ阻害剤として一般的に有用であることを記載している。くり返せば、他の重要なキナーゼ(Cor社の特許公開では3つのキナーゼしか記載されていない)に対する化合物の作用についての情報が存在しないこと、及びこれらの刊行物に記載されたいくつかのキナーゼに関して利用可能なデータが限られているために、Cor Therapeutics社の特許だけからでは、それら化合物の治療の潜在能力を判定することは不可能である。本発明は、本明細書に開示したマイケル付加物を形成することができる、これらCor Therapeutics社の特許に開示された化合物が、多様な癌適応症並びに他の疾患及び症状の治療に用いることができることを教示し、Cor Therapeutics社の特許に記載されたもの以外の化合物もタンパク質キナーゼを標的とすることを示すデータを提供する。本発明の方法の他の実施態様では、Cor Therapeutics社の特許に具体的に記載された化合物以外の化合物が投与され、本明細書で特定した疾患又は症状が治療される。
本発明の方法のまた別の実施態様では、本発明の方法で有用な化合物は、天然に存在するレゾルシル酸ラクトン、ヒポテマイシン、(5Z)-7-オキソゼアネオール、Ro-09-2210及びL-783,277から成る群より選択される以外の化合物であり、AML、基底細胞癌、B-Raf変異依存癌(大腸癌及びメラノーマを含むがただしこれらに限定されない)、乳癌、GI間質腫瘍、Ras依存癌、腎細胞癌、及び前立腺癌、肺線維症、肥満細胞症、炎症性腸疾患及びアレルギー性皮膚から成る群より選択される疾患又は症状のために治療の必要がある患者に投与される。
以下の実施例は、本発明の方法で有用な化合物を生成し、試験し、さらに使用する種々の方法を例示する。
【0092】
(実施例)
以下の実施例は、ヒポミセス・スビキュロスス(Hypomyces subiculosus)、ATCC44392又はアイギアルス・パルブス(Aigialus parvus)の発酵から得られるヒポテマイシン及び(5Z)-7-オキソゼアネオールの精製を述べる。これら実施例は、放射能、蛍光若しくはビオチン標識したラクトン又は質量分析を用いて、化合物(本実施例では例示的化合物、ヒポテマイシン及び(5Z)-7-オキソゼアネオールが用いられる)が、MEK1又は他のCys標的キナーゼと共有結合付加物を形成するか否かを示すために、酵素の動力学的解析をどのように用いることができるか示す。さらにまた、これら実施例は、MAPKキナーゼシグナル伝達経路を阻害するラクトンの能力をどのように細胞利用アッセイによって決定することができるか、さらにどのようにしてERK依存腫瘍培養由来の癌細胞でラクトンの抗分裂態様を提示することができるかを示す。
【実施例1】
【0093】
実施例1:レゾルシル酸ラクトンの産生:
ヒポテマイシン又は(5Z)-7-オキソゼアネオールは、文献の方法にしたがってヒポミセス・スビキュロスス(Hypomyces subiculosus)、ATCC44392の発酵物から精製することができる。これら及び近縁レゾルシル酸ラクトン(アイギアロマイシンとして知られている)のまた別の供給源は、アイギアルス・パルブス(Aigialus parvus)株の発酵物である。本発明の他のレゾルシル酸ラクトン化合物は、本明細書の開示および文献に記載された方法にしたがって合成することができる。単離した化合物の構造は、精製物質のNMR及びMS分析によって確認することができる。ラクトン又はその類似体の1つの3H若しくは14C形は、化学的又は酵素的半合成方法によって業者(例えばMoravek Biochemicals, Brea, CA)により調製され、その構造はクロマトグラフィー及び分光分析により立証することができる。本発明はまた、以下のように、ヒポテマイシンの代わりに、(5Z)-7-オキソゼアネオール又は15-デスメチルヒポテマイシンを産生する変異株を得る方法を提供する。ヒポテマイシンのための生合成遺伝子クラスターは、末端配列決定法を用いて、モノモデュラーI型ポリケチドシンターゼ(PKS)及び必須のテーラー酵素をコードする遺伝子を同定した後で、H.スビキュロススのゲノムDNAから作成したコスミドライブラリーからサブクローニングされる。候補コスミドの配列を決定し予想される特徴を有するコスミド、すなわちPKS遺伝子プラス少なくとも1つのオキシダーゼ遺伝子(O-メチルトランスフェラーゼ遺伝子)及び付随する調節遺伝子を含むオーバーラップコスミドを見つける。遺伝子破壊を実施し、正確な合成遺伝子セットが同定されていることを確認する。最後に、オキシダーゼ遺伝子の破壊は、(5Z)-7オキソゼアネオール(ヒポテマイシンの前駆体)の破壊をもたらすか、又はO-メチルトランスフェラーゼ遺伝子の破壊は15-デスメチルヒポテマイシンの生成をもたらす。本発明の方法で有用な化合物はまた全化学合成によっても調製することができる(以下を参照されたい:Selles et al. Tetrahedron Lett. 2002, 43(26):4621-5;Selles et al. Tetrahedron Lett 2002, 43(26):4627-31;Geng et al. Org Lett 2004, 6(3):413-6)。
【実施例2】
【0094】
実施例2:ラクトンによる標的Cysキナーゼ阻害の動力学的解析
本実施例は、例示的タンパク質キナーゼとしてMEK1、MEK2及びいくつかのマイトジェンレセプターキナーゼを用い、標的タンパク質キナーゼと化合物がマイケル付加物を形成することができることを示す方法を例証する。阻害剤と酵素間の共有結合付加物形成の特徴は、酵素活性の“時間依存阻害”である。
典型的には、阻害剤の存在下で時間の経過にしたがいタンパク質キナーゼ活性の阻害の増加が測定される。ある方法では、酵素及び阻害剤を含む“プレインキュベーション”反応混合物のアリコートを時間の経過とともに活性についてアッセイする。阻害の増加又は初期速度の低下が、マイケル付加物が形成されるために時間の経過につれて観察されるであろう(C. Walsh, Enzyme Reaction Mechanisms, W.H. Freeman & Co., 1979, pp 86-94)。第二の方法では、活性の時間依存低下は、形成される生成物(例えばADP)対時間を測定及び分析する“進行曲線”として測定される(Morrison & Walsh, Adv. Enzymol Relat Areas Mol Biol 1988, 61:201-301)。いずれの事例でも、時間依存不活化は、競合する基質(この場合はATP)の存在によって鈍化させることができる。
決定される可逆的解離定数(Kd)及び不活化のための速度定数(Kinact)値は、阻害メカニズムの解析に用いられる基礎的データである。ヒポテマイシンとコントロールとしての前記の非反応性5,6-ジヒドロ形を用いるこれらのアッセイの実施は、酵素阻害のためのα,β-不飽和ケトンの重要性を示す。
他のMEK1阻害剤(例えばPD184352及びUO0126)の確立されたメカニズム(前記はともにATPと非競合的に機能する)から、本発明の方法で有用なラクトン化合物は、MEKによってERK1のリン酸化を阻害するはずである。時間依存性酵素阻害は、堅固で緩徐な結合の阻害剤又は共有結合形成阻害剤で観察され、上記に記載した標準的なアプローチで検出することができる。
本発明の方法で有用な化合物の標的である得るMEK1及び多くの他のタンパク質キナーゼは業者(Invitrogen, Carlsbad, CA)から入手するか、又は標準的な分子生物学的技術を用いて調製することができる。リン酸化による活性化の後で、それらは、標的キナーゼ又は代用基質をリン酸化するその能力についてアッセイされる。例えば、MEK1は、Mops緩衝液(pH7.6)中のMEK1(30nM)及びERK1(2μM)、[γ-32P]ATP(10uM)及びMgCl2を含む混合物でアッセイすることができる。リン酸化は、[γ-32P]リン酸化ERK1をホスホセルロース紙上で単離し、放射性生成物を計測することによって測定することができる。また別には、共役酵素系を用いてもよい。前記系では、キナーゼ反応生成物(例えばADP)は、容易に測定できる物質(例えばNADH)に前記生成物(例えばADP)を変換する二次系を用いて解析される。そのような共役酵素系は便利な分光分析アッセイによって測定することができる。
“プレインキュベーション法”を用いて時間依存阻害を測定するために、MEK1(又は他のキナーゼ)は、ラクトン阻害剤の量を変化させながら一緒にインキュベートされる。コントロールは阻害剤を含まないか、又は競合阻害剤を含む(例えばUO126(IC50は72nM)、EMD Biosciences(San Diego, CA)から入手できる)。アリコートを種々の時間で取り出し、基質[γ-32P]ATP、ERK1(又は他の基質)及び他の反応成分を含む溶液に添加し、初期速度を残存酵素活性の測定として決定する。
共有結合阻害剤の場合、酵素活性の時間依存低下が存在し、一方、可逆的阻害剤の場合、活性は時間につれて変化しない(Morrison & Walsh, Adv. Enzymol Relat Areas Mol Biol 1988, 61:201-301;Sculey et al. Biochem Biophys Acta 1996, 1298(1):78-86)。共有結合阻害剤の場合、log(活性)対時間のプロットは、酵素活性低下の見かけの一次速度定数(Kobsd)を提供する。これらのアッセイが種々の濃度の阻害剤において実施されるならば、一連の一次プロットが得られ、各阻害剤濃度でKobsdが得られる。“進行曲線法”(上記に引用した参考文献及び以下の文献(Kuzmic et al. Methods Enzymol 2004, 383:366-81)を参照されたい)を用いて時間依存阻害を測定するために、ERK(又は他のキナーゼ)を種々の濃度のラクトン阻害剤で処理し、ADPの生成を共役アッセイによって持続的に測定する。得られた生成物対時間の曲線は以下の等式に適合する:[P]=(vi/kobs)* (1-exp(-kobs*t)、式中、Pは時間tで形成される生成物であり、viは初期速度であり、kobsは見かけの阻害一次速度定数であり、kobs値は種々の阻害剤濃度の各々について決定される。1/kobs対1/[I]の再プロットはKd(初期可逆性結合定数)及びkinact(可逆的に結合したE-Iの共有結合したE-Iへの変換のための一次速度定数)の決定を可能にする(0.693を前記数値で割ることによって、前記数値を用いて不活化の半減期を産出することができる)。コントロール実験は、α,β-不飽和カルボニルをもたず(例えば5,6-ジヒドロヒポテマイシン)、したがってマイケル付加物を形成することができないヒポテマイシン類似体を用いて実施することができる。そのような分子は競合的阻害剤ではあり得るが、時間依存性不活化を示すことはない。
表2は、いくつかのキナーゼに対するヒポテマイシンの対応する阻害定数を示す(前記には、対応する場合は、時間依存不活化のための動力学的定数が含まれる)。パラメータは、別個のキナーゼについて顕著に異なり、“選択性定数”(kinact/Ki)における100倍を超える相違は、例えばKDR(VEGFR)及びMEK1のようなキナーゼは、低い濃度×時間(細胞又は生物では低い投与量×曝露)で他のものよりも選択的に阻害され得ることを示唆している。本発明の方法で有用な化合物のセット内では、特異性における顕著な多様性が達成され、異なる適用について最適な化合物の同定を可能にすることは当業者には理解されよう。阻害進行曲線解析は、連続的比色分析又は蛍光測定分析を用いて実施した。全ての実験は、KDR(5mMのATP)を除いて1mMのATPで実施した。
【0095】
表2:種々のタンパク質キナーゼに対するレゾルシル酸ラクトンの阻害データ
a 阻害剤の存在なしに時間の経過とともに酵素活性の顕著な低下
b 最高の阻害剤濃度で想定される阻害の5%
c 最高の阻害剤濃度での阻害率(初期速度)から算出されるKi
下記の表3に示される結果は、ヒポテマイシンは必須のCys残基を欠くキナーゼを顕著には阻害せず、前記Cysを有するキナーゼは様々な程度でこれを阻害することを示している。124キナーゼのこのパネルでは、活性部位のシステインを有すると識別された19キナーゼのうちで18が、0.2μM及び/又は2μMの濃度のヒポテマイシンによってある程度まで阻害された。非活性部位システインに関しては、わずかに2つがヒポテマイシンによる顕著な阻害を示した。(これらの値は、種々のアッセイ(すなわち種々のサンプル操作技術を用いる)で達成し得る値と異なる可能性がある。なぜならば、それらの値は、共有結合性阻害の時間依存特性を考慮しない単一点アッセイだからである)。
【0096】
表3:ヒポテマイシン(0.2μM、カッコ内の場合は2.0μM)で処理したタンパク質キナーゼのパーセント残留活性
a 活性部位システインを有する
放射性[32P]ATP及び生成物のフィルター結合によるプレインキュベーション実験を用いてMEK1アッセイを実施した。連続的分光分析アッセイ由来の進行曲線分析を用いて他の全てのキナーゼを解析した。
TRKA及びBは、上記に記載した単一点スクリーニングアッセイ(表3)でヒポテマイシンによる阻害を示したが、マイケル付加物形成のための標的Cysを含まない。これらの酵素をこのより正確な方法によってアッセイした時、ヒポテマイシンは、ATPと競合する可逆的な阻害を示し、KiはTRKAについては2.2μMで、TRKBについては0.37μMであったが、前記酵素の時間依存不活化(すなわち共有結合の形成)は示さなかった(表2)。このことは、共有結合性阻害は標的Cys残基を要求することを立証し、標的キナーゼの共有結合酵素阻害のための規準として時間依存阻害の正当性を実証する。
【実施例3】
【0097】
実施例3:共有結合形成の決定
マイケル反応(前記は基本的には可逆反応である)では、遊離リガンドと共有結合リガンドとの間の見かけの親和性は、反応に中心的に関与する2つの解離定数(Kreversible x Kcovalent)の産物であり、複合体の形成/破壊は、該当タンパク質は両方向で反応を触媒するので理論的には可逆的である。タンパク質の変性は両方向の触媒を停止し、変性マイケル付加物は物理的に単離及び定量ができるほど通常は充分に安定である。例えば、天然のFdUMP-チミジレートシンターゼマイケル付加物は緩徐ではあるが完全に不可逆性であるが、SDS変性は安定で単離可能な複合体を提供する(D.V. Santi et al. Biochemistry 1974, 13:471;Methods in Enzymol 1977, 46:307-312)。
もちろん、複合体が共有結合付加物を含まない場合は、タンパク質の変性は阻害物の迅速な解離をもたらす。したがって、[3H]-リガンド-タンパク質複合体を変性させ、さらにSDS-PAGEによりタンパク質-結合放射能を検出することによって、複数のマイケル付加物が簡単に単離された。そのような複合体の検出は以下を提供する:(a)共有結合付加物形成の証拠、及び(b)平衡(Kd)及び動力学定数(Koff及びKon)を決定するための相互反応定量用ツール。
例えば、MEK1及び他の標的Cys含有キナーゼの種々の利用可能な形態は、蛍光若しくは [3H]-ヒポテマイシン又は同様な類似体で処理し、SDS-PAGE又は変性ゲル浸透カラムに付し、前記ゲル又はカラムをタンパク質結合蛍光又は放射能について分析することができる。安定な複合体が形成される場合、複数の重要な試験を実施することができる。例えば、複合体をSDS-PAGEから単離し、トリプシンで消化し、クロマトグラフィー又は質量スペクトル(MS)分析によってタンパク質の共有結合ペプチドを同定することができる。 [3H]標識又は蛍光標識エノンの濃度を変化させ、複合体をSDS-PAGEによって単離/定量することによって、複合体形成の平衡及び動力学的特性を決定することができる。培養哺乳動物細胞又はそのような細胞から得た可溶性細胞抽出物を[3H]標識又は蛍光標識したヒポテマイシンで処理し、2Dゲルで解析し、放射能スポット中のタンパク質をMALDI MSによって同定することができる。例えば、MEK1がヒポテマイシンとの共有結合付加物形成のための唯一の標的であるならば、MEK1は標識されるただ1つのタンパク質であろう。複数のタンパク質が標識されるならば、さらに別の標的が存在すると結論することができ、それらを同定することができる。
例えば、キナーゼの必須システインとの共有結合形成は、共有結合複合体のタンパク分解消化によって得られるペプチドの質量スペクトル分析によって示すことができる。図7は、ヒポテマイシン処理又は非処理ERK2のトリプシン消化物の質量スペクトルを示す。951の質量ピークは、標的Cys172残基を含む最小のトリプシン消化ペプチドの質量に対応する。以前にヒポテマイシンで処理されたERK2の非活性化及び活性化形のトリプシン消化物は、標的Cysペプチドの質量の増加は1273(ヒポテマイシンの質量と正確に一致する量)であることを示している。
【実施例4】
【0098】
実施例4:Cysキナーゼ依存癌から培養された細胞のラクトンによる分裂阻害
マイケル付加物形成に感受性を有する活性部位Cys残基を含むタンパク質キナーゼを有するか、又は前記タンパク質キナーゼによって活性化される、活性なシグナル伝達経路を必要とする腫瘍に由来する細胞株の細胞分裂を阻害する、本発明の方法で有用な化合物の能力は、細胞分裂アッセイ及び細胞株(例えばHT-29(ヒト結腸癌)、COLO829(メラノーマ)、MV-4-11(急性骨髄性白血病)及びP815(マウスの肥満細胞腫))を用いて示すことができる。ある例示的な方法では、細胞を96ウェルプレートで種々の濃度の阻害剤で処理し、37℃/5%CO2で3日間インキュベートし、さらにセルタイターグロ(Cell Titer Glo)キット(Promega)を用いて分析する。
表4は、マイケル付加物形成に感受性を有する活性部位Cys残基を含むタンパク質キナーゼを有するか、又は前記タンパク質キナーゼによって活性化される、活性なシグナル伝達経路を必要とする腫瘍に由来する細胞株に対する、本発明の方法で有用な化合物の増殖阻害特性を示す。疾患の感受性が主としてそれによってもたらされる変異キナーゼだけでなく、感受性に寄与すると合理的に推測される他のタンパク質キナーゼもまた示される。
【0099】
表4:キナーゼ変異細胞株におけるキナーゼ阻害剤の細胞傷害性
* タザロテンの場合10
【実施例5】
【0100】
実施例5:全細胞のシグナル伝達経路に対するラクトン阻害剤の影響
個々のキナーゼ(例えばMEK)の阻害による下流の影響は前記キナーゼをリン酸化のために必要とするいくつかのタンパク質(例えばERK1)のリン酸化状態を測定することによって確立することができる。培養細胞をヒポテマイシン又は本明細書に記載した他のラクトン類似体で処理し、細胞抽出物のウェスタンブロットを、下流の標的の非改変形及びリン酸化形に特異的な抗体をプローブとして用いて調べた。例として、MEK1/2に対するヒポテマイシンの影響はERK1/2のリン酸化レベルを測定することによって決定することができる。図8は、COLO829細胞(BRAF V599E変異を含む)のヒポテマイシンによる処理は、ERKのリン酸化形の急速な(10分以内)枯渇をもたらすことを示している。同様に、高レベルのマイトジェンレセプターキナーゼヒポテマイシン標的(例えばMV-4-11、前記はFLT3(ITD)変異を有する)を含む細胞の処理は、FLT3のリン酸化形だけでなくレセプターチロシンキナーゼMEK及びERKの下流の両標的のリン酸化形の低下ももたらす。
図8を参照すれば、B-Raf V599E変異メラノーマ細胞株COLO829を1μMのヒポテマイシンとともに2、5、10、15、30及び60分間インキュベートした。続いて前記細胞を溶解させ、タンパク質を抽出した。各サンプルから取った等量の全タンパク質をSDS-PAGEで分離し、続いてPVDF膜に電気的にブロッティングした。前記の膜を抗リン酸化ERK抗体(Cell Signaling Technologies)とともにインキュベートし、続いてHRP結合二次抗体とともにインキュベートすることによって、各抽出物に存在するリン酸化ERKレベルを可視化した。ECLウェスタン検出キット(Amersham)を用いてリン酸化ERK含有バンドをオートラジオグラフィーによって検出した。ERK抗体をプローブとして用いてこのブロットを再度調べて、等レベルの全ERKが各レーンにロードされていることを示した(データは示さず)。
可逆性阻害剤に関しては、作用を受けた下流のキナーゼのリン酸化によって測定されるように、標的Cysキナーゼを阻害する作用は迅速に達成される。しかしながら、可逆性競合性阻害剤とは異なり、図9に示すように、ラクトンは1時間未満の短時間曝露後に細胞から除去することができ、阻害を受けたキナーゼは長時間(24時間まで)にわたって回復しない。したがって、in vitroの場合のように、細胞では、共有結合阻害剤-キナーゼ付加物が迅速に形成され、長時間にわたって結合が維持される。したがって、これら阻害剤の薬剤として尋常ではない特性は、薬剤の標的への短時間曝露によって長時間持続する作用を示すことができるということであり、これは、オフターゲット作用により毒性を回避しつつ最大効果を達成するためにスケジュールを立てるということに関連して所望の選択肢を提供する。このことはまた、標的キナーゼの顕著な阻害を担保するために用量及び半減期が充分であることを条件として、比較的短いin vivo半減期を有するRALを本発明の方法で有効に用いることができることを意味する。
図9を参照すれば、B-Raf V599E変異細胞株HT29をDMSO、1μMのU0126又はヒポテマイシンのいずれかとともに1時間インキュベートした。前記1時間のインキュベーションに続いて、細胞を培養液で2回洗浄し、さらにインキュベートした。薬剤処理並びに洗浄の3、6及び24時間後に直ちにタンパク質抽出物を調製した。各サンプルから取った等量の全タンパク質をSDS-PAGEで分離し、続いてPVDF膜に電気的にブロッティングした。前記の膜を抗リン酸化ERK抗体(Cell Signaling Technologies)とともにインキュベートし、続いてHRP結合二次抗体とともにインキュベートすることによって、各抽出物に存在するリン酸化ERKレベルを可視化した。ECLウェスタン検出キット(Amersham)を用いてリン酸化ERK含有バンドをオートラジオグラフィーによって検出した。
薬剤の開発の前に、化合物の薬理学的動力学、生体利用性、動物における抗腫瘍活性及び急性毒性試験を実施した。レゾルシル酸ラクトンについての現時点で存在する知識を基準にすれば、本発明の方法で有用な化合物は顕著な毒性をもたず、良好な生体利用性を有するはずである。そのような薬剤に対する感受性が予測される変異対立遺伝子座をもつ患者のタイピングもまた、イレッサによる肺癌患者の最近の研究によって実施されたように、いくつかの実施態様で実施される(例えば悪性メラノーマにおけるB-Raf変異)。
【実施例6】
【0101】
実施例6:本発明の化合物の調製及び特性
本実施例は、下記式(II)の構造を有する本発明の化合物、すなわち4-O-デスメチルヒポテマイシンの調製を述べる。特にこの化合物は、その精製形及び単離形で提供される:
【0102】
【化13】
【0103】
接種調製物:以下の機関(Deutsch Sammlung von Mikroorganismen und Zellkulturen (DSMZ ))から入手した、20%(v/v)グリセロール中で維持されたヒポミセス・スビキュロスス(Hypomyces subiculosus)DSM11931の凍結細胞の1mLを、バッフル無しの250mLエーレンマイヤーフラスコ中の50mLの種培養液に接種した。前記種培養液は、水に30g/Lのクォーカーひき割りエンバクから成り、オートクレーブの前に70〜80℃で10分間加熱した。前記種培養物を22℃の暗所で、ロータリーシェーカー(5.08cm(2インチ)のストローク、190rpm)で3日間インキュベートした。50mLのエンバクフレーク培養液を含むバッフル無しの50mLエーレンマイヤーフラスコに2mLの一次種培養物を移し、二次種培養物を作製した。
ファーメンター生産:12LのCYS80培養液(Dombrowski et al. J Antibiot 1999, 52(12):1077-1085)を含む20Lのバイオリアクターをそのまま121℃で30分滅菌した。前記培養液は、80g/Lのスクロース、50g/Lのコーンミール(Sigma)及び1g/Lのバクト酵母抽出物(BD)から成っていた。続いて前記培養液を480mLのH.スビキュロススDSM1193の二次種培養物とともにインキュベートした。発酵は、通気速度0.4v/v/m及び初期攪拌速度200rpmで22℃で実施した。培養の溶解酸素は、200〜400rpmの攪拌カスケードによって30%の空気飽和で管理した。泡立ちは、100%のUCON LB-625の自動添加で管理した。培養のpHはモニターしたが、制御はしなかった。D,L-エチオニンを接種のときに50mg/Lの濃度で添加した。発酵は、最大のKOSN-2176産生が得られるまで35日間継続させた。サンプルは必要に応じて抜き取り、後の分析のために-20℃で保存した。
当業者はCYS80培養液組成の変型は有用であることを理解しえよう。例えば、前記は約30〜120g/Lのスクロース、約20〜80g/Lのコーンミール、及び約0(好ましくは約0.1)〜10g/Lの酵母抽出物を含むことができる。同様に、D,L-エチオニン濃度は、例えば約10〜10mg/L培養液で変動し得る。
化合物IIの蓄積を促進するために、C-4ヒドロキシル基のメチル化を触媒してヒポテマイシンを生成するために必要なメチルトランスフェラーゼの阻害剤として種々の化合物を判定した。D,L-エチオニン(前記はメチルトランスフェラーゼ阻害剤であると文献で報告された)は化合物IIの産生の増加に有効であることが見出されたが、一方メチルトランスフェラーゼ阻害剤は有効ではないとも報告された。さらにまた、複数の培養液を判定し、CYS80が他のものよりも化合物IIの産生を促進した。化合物IIの力価は、40mg/mL〜540mg/mL(20リットルバイオリアクター)又は900mg/mL(振盪フラスコ)に改善された。
化合物IIの定量: 1mLのメタノールを用いて500μLの発酵ブロス抽出することによって、化合物II及びヒポテマイシンの産生をモニターした。続いて、混合物を13,000gで3分遠心した。ヒューレットパッカード1090HPLCを用い、220、267及び307nmでのUV検出により上清中の2つの生成物の定量を実施した。5μLの上清を4.6x10mmのガードカラム(Inertsil, ODS-3, 5μm)及びより長い4.6x150mmのカラム(Inertsil, ODS-3, 5μm)に注入した。最終的なヒポテマイシン濃度が1g/Lになるまでサンプルをメタノールで希釈した。前記アッセイ方法は周囲温度で1mL/分の流速で実施した。前記は、8分にわたる40:60〜80:20のアセトニトリル:水の勾配から成り、続いて4分間の100%アセトニトリル洗浄を実施した。両移動相は0.1%(v/v)の酢酸を含んでいた。精製した化合物II及びヒポテマイシンを用いて標準物を調製した。
化合物IIの精製: H.スビキュロススDSM11931の2つの12リットル発酵物の発酵ブロス24リットルを24リットルの100%メタノールで1時間抽出した。前記混合物を薄層セリットを含む真空フィルター(Hyflo)に通し、前記フィルターケーキを1Lの50:50メタノール:水で洗浄した。ろ液を水で希釈し最終メタノール濃度を30%(v/v)にした。精製方法に用いた全ての溶媒は0.1%(v/v)の酢酸を含んでいた。
ミリポアモジュリン(50cmx9cm)プロセスカラムに1.3LのHP-20SS樹脂(Mitubishi)を充填し、30:70メタノール:水の3カラム容積で700mL/分で平衡化した。生成物プールを同じ流速でカラムにロードした。カラムを1CVの30:70メタノール:水で洗浄し、段階勾配(3CVの45:55メタノール:水、9CVの50:50メタノール:水、及び3CVの60:40メタノール:水)で300mL/分で溶出させた。画分(1.5CV)を採集し、HPLCで上述のように分析した。画分3〜15を生成物プールとして一緒にした。
ミリポアモジュリン(50cmx9cm)プロセスカラムに2.3LのC18ソルベント(Bakerbond, 40μm)を充填し、30:70メタノール:水の3CVで180mL/分で平衡化した。HP-20SSクロマトグラフィー工程から得た生成物プールを水で希釈し、最終メタノール濃度を30:70メタノール:水にし、C18カラムに180mL/分でロードした。カラムを30:70メタノール:水の1CVで洗浄し、さらに9CVの42:58メタノール:水で180mL/分180mL/分で溶出させた。画分(0.4CV)を採集し、上述のようにHPLCで分析した。画分10〜16を生成物プールとして一緒にした。
化合物IIの結晶化を促進するために、前記生成物プールを40℃での回転蒸発で濃縮し、その容積を36%に減少させた。続いて、−20℃に冷却した。形成された化合物IIの白色結晶をワットマン#5のろ紙を設置したブッフナーロートに通し、100mLの冷却水で洗浄した。最終生成物を40℃の真空オーブンで一晩乾燥させ、4℃で保存した。精製方法の全体的収量はほぼ60%であった。精製方法終了時の化合物IIの純度はほぼ95%であった。
化合物IIの特徴付け:精製された化合物IIは白色の結晶として得られた;UV(MeOH)λmax 219, 266, 306nm;HRESIMS m/z 363.1074 [M-H]-(C18H19O8で計算、363.1065);1H及び13C NMRデータ(表5及び6参照)。
【0104】
表5:化合物IIの1H NMR
【0105】
表6:化合物IIの13C NMR
【実施例7】
【0106】
実施例7:化合物の合成
本実施例は、本発明の方法で有用な別の化合物の合成について述べる。
【0107】
【化14】
【0108】
THF(4.0mL)中の化合物II(12mg、0.033mmol)の攪拌溶液に、3-モルホリノプロパン-1-オール(10.0μL、0.072mmol)、トリフェニルホスフィン(22.6mg、0.086mmol)及びジエチルアゾジカルボキシレート(13.4μL、0.086mmol)を添加した。室温で3時間攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、1.2mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液の10%〜90%勾配を用いた40分の溶出によって、化合物III(11mg、70%収量)が提供された:LRMS m/z (M+H) C25H34NO9による計算値492.2;実測値492.2。
【0109】
【化15】
【0110】
THF(2.0mL)中の化合物II(6mg、0.017mmol)の攪拌溶液に、3-(4-メチルピエラジン-1-イル)プロパン-1-オール(5.0μL、0.036mmol)、トリフェニルホスフィン(12mg、0.043mmol)及びジエチルアゾジカルボキシレート(7.0μL、0.043mmol)を添加した。室温で45分攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、0.8mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液に10%〜90%勾配を用いた40分の溶出によって、化合物IV(3.8mg、45%収量)が提供された:LRMS m/z (M+H) C26H37N2O8による計算値505.2;実測値505.2。
【0111】
【化16】
【0112】
THF(1.0mL)中の化合物II(3mg、0.009mmol)の攪拌溶液に、(1-メチルピエラジン-3-イル)メタノール(5.0μL、0.036mmol)、トリフェニルホスフィン(16mg、0.048mmol)及びジエチルアゾジカルボキシレート(6.3μL、0.048mmol)を添加した。室温で3時間攪拌後、前記反応混合物を濃縮した。残留物をTHF/水(3:2、0.8mL)に溶解し、0.45μmのフィルターに通し、ヴァリアンイナートシル(Varian Inertsil(商標))5μODS-3(250x100)逆相HPLCカラムでHPLCによって精製した。0.1%AcOH水溶液/0.1%AcOHのCH3CN溶液に10%〜90%勾配を用いた40分の溶出によって、化合物IV(1.7mg、40%収量)が提供された:LRMS m/z (M+H) C25H34NO8による計算値476.2;実測値476.2。
化合物II〜Vの生物学的特性をアッセイし、ヒポテマイシンの特性と比較した。COLO829はヒトのメラノーマ細胞株である。HT29はヒト大腸癌細胞株である。両細胞株はV600E B-Raf変異を有する。SKOV3は野生型B-Rafをもつ卵巣癌細胞株である。EKR2(細胞外シグナルによって調製されるキナーゼ)はRas/B-Raf MAPキナーゼカスケード経路のキナーゼである。結果は表7に示されている。
【0113】
表7:化合物II〜Vの特性
本発明をこれまで記述による詳細な説明及び実施例で述べてきたが、前記は多様な態様で実施され得ること、及び前述の記載及び実施例は例示を目的とし、特許請求の範囲を制限するものではないことは、当業者には理解されよう。
【図面の簡単な説明】
【0114】
【図1】ERK/MAPKシグナル伝達経路の模式図を示す。
【図2】ある種のレゾルシル酸ラクトンの化学構造を示す。
【図3】ヒポテマイシンが共有結合したキナーゼERK2のX線構造を示す。
【図4】ヒポテマイシンが共有結合したキナーゼERK2のX線構造を示す。
【図5】60細胞株のNCIパネルに対するヒポテマイシンのlog GI50値(50%増殖低下を達成するために必要な薬剤の量)を棒グラフの形で示す。前記化合物に対してもっとも感受性を有する細胞株は、垂直線の平均活性から右を指す棒線で表されている。
【図6】ヒポテマイシン及び非RAL薬剤についての異種移植比較データを示す。
【図7】ヒポテマイシンの存在下及び非存在下におけるキナーゼERK2のトリプシン消化の質量スペクトルの比較である。
【図8】BRAFV599E変異を有するColo829細胞におけるキナーゼERKのリン酸化に対するヒポテマイシンの作用を示す。
【図9】B-Raf V599E変異を有するHT29細胞におけるヒポテマイシンによるキナーゼERKリン酸化阻害の時間を示す。
【特許請求の範囲】
【請求項1】
混合物又は細胞において、タンパク質キナーゼ上のATP結合部位に保存された2つのアスパラギン酸残基の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン(Cys)残基を有する1以上のタンパク質キナーゼを阻害する方法であって、
前記混合物は、タンパク質キナーゼ上のATP結合部位に保存された2つのアスパラギン酸残基の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するCys残基を欠く更なるタンパク質キナーゼを含み、
前記Cys残基を有する前記1以上のタンパク質キナーゼを、前記Cys残基とマイケル付加物を形成することができる化合物に、その化合物と前記Cys残基との間でマイケル付加物が形成されて前記1以上のタンパク質キナーゼの阻害がもたらされる条件下において、接触させることを含む、前記方法。
【請求項2】
該Cys残基とマイケル付加物を形成することができる化合物が、該Cys残基とマイケル付加物を形成するエノン部分を含む、請求項1記載の方法。
【請求項3】
該Cys残基とマイケル付加物を形成することができる化合物が、7位のカルボニルと共役している5−6位のシス炭素-炭素二重結合を有するレゾルシル酸ラクトンである、請求項2記載の方法。
【請求項4】
該Cys残基とマイケル付加物を形成することができる化合物が、下記の構造(I)を有する化合物、又はその医薬的に許容できる塩若しくは誘導体である、請求項2記載の方法
【化1】
(式中、
R1は、水素、任意に置換された脂肪族部分、任意に置換された環式脂肪族部分、任意に置換された複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成するか;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C4アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは任意にヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3若しくは複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は
R8及びR9が一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であるか、又はR20及びR2は組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であり、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり;ここでR19は水素又はアルキルである)。
【請求項5】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ia)の構造を有する、請求項4記載の方法
【化2】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;さらに
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項6】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ib)の構造を有する、請求項4記載の方法
【化3】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項7】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ic)の構造を有する、請求項4記載の方法
【化4】
(式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分はヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項8】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Id)の構造を有する、請求項4記載の方法
【化5】
(式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項9】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ie)の構造を有する、請求項4記載の方法
【化6】
(式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項10】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(If)の構造を有する、請求項4記載の方法
【化7】
(式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項11】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ig)の構造を有する、請求項4記載の方法
【化8】
(式中、
R4はH又はFであり;
R8はHであり;
R9は以下の基から成る群より選択されるか、
【化9】
又はR8及びR9が組み合わさって以下の基を形成する)。
【化10】
【請求項12】
混合物がin vitroの反応混合物である、請求項1記載の方法。
【請求項13】
阻害の工程が細胞内で行われる、請求項1記載の方法。
【請求項14】
細胞が病変細胞であるか、又は病変組織に存在する、請求項1記載の方法。
【請求項15】
疾患又は疾患症状の治療を必要とする患者に、タンパク質キナーゼ上のATP結合部位領域に保存された2つのアスパラギン酸残基に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン(Cys)残基を有するタンパク質キナーゼを特異的に阻害する化合物を含む医薬組成物を投与することにより、疾患又は疾患症状を治療する方法であって、
前記タンパク質キナーゼを、前記Cys残基とマイケル付加物を形成する化合物と接触させることを含む、前記方法。
【請求項16】
医薬組成物が、下記の構造(I)の化合物、並びにその医薬的に許容できる塩及び誘導体を含む、請求項15記載の方法
【化11】
(式中、
R1は、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、各々独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C4アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分はヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH、又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は
R8及びR9は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であり、又は、R20及びR2が組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であり、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり、式中R19は水素又はアルキルである)。
【請求項17】
化合物が、下記式(Ia)の構造を有する、請求項16記載の方法
【化12】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項18】
化合物が下記式(Ib)の構造を有する、請求項16記載の方法
【化13】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;ここで
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項19】
化合物が下記式(Ic)の構造を有する、請求項16記載の方法
【化14】
(式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項20】
化合物が下記式(Id)の構造を有する、請求項16記載の方法
【化15】
(式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項21】
化合物が下記式(Ie)の構造を有する、請求項16記載の方法
【化16】
(式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項22】
化合物が下記式(If)の構造を有する、請求項16記載の方法
【化17】
(式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項23】
化合物が下記式(Ig)の構造を有する、請求項16記載の方法
【化18】
(式中、
R4はH又はFであり;
R8はHであり;
R9は以下の基から成る群より選択されるか、
【化19】
又はR8及びR9が一緒になって以下の基を形成する)。
【化20】
【請求項24】
該Cys残基を有するタンパク質キナーゼが以下から成る群より選択される、請求項1記載の方法:AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAK。
【請求項25】
該Cys残基を有するタンパク質キナーゼの少なくとも2つが阻害される、請求項24記載の方法。
【請求項26】
該Cys残基を有するタンパク質キナーゼの少なくとも3つが阻害される、請求項24記載の方法。
【請求項27】
1以上の該Cys残基を有するタンパク質キナーゼがERK経路キナーゼであり、前記ERK経路キナーゼの少なくとも2つが阻害される、請求項1記載の方法。
【請求項28】
ERK経路キナーゼの少なくとも4つが阻害される、請求項27記載の方法。
【請求項29】
該Cys残基を有するタンパク質キナーゼが、MEK1、MEK2、ERK1及びERK2である、請求項28記載の方法。
【請求項30】
阻害される1以上の該Cys残基を有するタンパク質キナーゼが、少なくとも2つのERK MAPKカスケード経路キナーゼとマイトジェンレセプターキナーゼとを含む、請求項1記載の方法。
【請求項31】
マイトジェンレセプターキナーゼが、VEGFレセプター;PDGFレセプター;c-KIT(肥満細胞増殖因子レセプター);FLT3(FL(Flt-3リガンド)のレセプター);及び、VEGFレセプター、PDGFレセプター、cKIT又はFLT3の構成的に活性化された変異体;から成る群より選択される、請求項30記載の方法。
【請求項32】
該Cys残基を有するタンパク質キナーゼの阻害剤が微小管安定化剤又は微小管脱安定化剤と一緒に投与される、請求項15記載の方法。
【請求項33】
該Cys残基を有するタンパク質キナーゼの阻害剤がHsp90阻害剤と一緒に投与される、請求項15記載の方法。
【請求項34】
Hsp90阻害剤が17-AAG又は17-DMAGである、請求項33記載の方法。
【請求項35】
該Cys残基を有するタンパク質キナーゼが、PDGFRα、PDGFRβ、VEGFレセプター(Flt-1、Flt-4及びKdr)、MEK1/2及びERK1/2から成る群より選択され、疾患が、加齢が関係する筋変性又は緑内障である、請求項15記載の方法。
【請求項36】
該Cys残基を有するタンパク質キナーゼが、Flt-3、c-Kit MEK、ERK又はVEGFRのいずれかであり、疾患が急性骨髄性白血病である、請求項15記載の方法。
【請求項37】
該Cys残基を有するタンパク質キナーゼが、c-Kit、PDGFR、MEK1/2又はERK1/2のいずれかであり、疾患が消化管間質腫瘍である、請求項15記載の方法。
【請求項38】
該Cys残基を有するタンパク質キナーゼが、野生型c-Kit、構成的に活性なc-Kit V816D変異体、MEK1/2又はERK1/2のいずれかであり、疾患が肥満細胞症である、請求項15記載の方法。
【請求項39】
該Cys残基を有するタンパク質キナーゼが、MEK1/2、ERK1/2又はTak1のいずれかであり、疾患が炎症性腸疾患である、請求項15記載の方法。
【請求項40】
炎症性腸疾患がクローン病又は潰瘍性大腸炎である、請求項39記載の方法。
【請求項41】
該Cys残基を有するタンパク質キナーゼがc-Kit、MEK又はERKであり、疾患が肥満細胞によって影響される又は引き起こされる炎症性症候群である、請求項15記載の方法。
【請求項42】
該Cys残基を有するタンパク質キナーゼがMEK1/2又はERK1/2のいずれかであり、疾患が乳癌である、請求項15記載の方法。
【請求項43】
該Cys残基を有するタンパク質キナーゼがKdr、c-Kit、MEK1/2、又はERK1/2のいずれかであり、疾患が非小細胞肺癌である、請求項15記載の方法。
【請求項44】
該Cys残基を有するタンパク質キナーゼがPDGFRA、MEK1/2、又はERK1/2のいずれかであり、疾患が卵巣癌である、請求項15記載の方法。
【請求項45】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が膵臓癌である、請求項15記載の方法。
【請求項46】
該Cys残基を有するタンパク質キナーゼが、変異体Raf-1タンパク質キナーゼによって活性化されるキナーゼであり、疾患が前立腺癌である、請求項15記載の方法。
【請求項47】
該Cys残基を有するタンパク質キナーゼがRSK又はMEK/ERKである、請求項46記載の方法。
【請求項48】
該Cys残基を有するタンパク質キナーゼが、VEGFR、PDGFR、MEK1/2、ERK1/2、Tak1、又はJNKシグナル伝達経路及びp38シグナル伝達経路を活性化するキナーゼのいずれかであり、疾患が乾癬である、請求項15記載の方法。
【請求項49】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が基底細胞癌である、請求項15記載の方法。
【請求項50】
該Cys残基を有するタンパク質キナーゼが、MEK1/2、ERK1/2、Tak1、又はJNKシグナル伝達経路を活性化するキナーゼのいずれかであり、疾患が炎症性症候群である、請求項15記載の方法。
【請求項51】
炎症性症候群がアレルギー性皮膚炎である、請求項50記載の方法。
【請求項52】
該Cys残基を有するタンパク質キナーゼがPDGFRであり、疾患が肺線維症である、請求項15記載の方法。
【請求項53】
該Cys残基を有するタンパク質キナーゼがMEK1/2、又はERK1/2のいずれかであり、疾患がRas変異体依存性癌腫である、請求項15記載の方法。
【請求項54】
該Cys残基を有するタンパク質キナーゼがVEGFR、PDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が腎細胞癌である、請求項13記載の方法。
【請求項55】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、ERK1/2、又はTak1のいずれかであり、疾患が再狭窄である、請求項15記載の方法。
【請求項56】
該Cys残基を有するタンパク質キナーゼがMEK1/2、ERK1/2、又はTak1のいずれかであり、疾患が慢性関節リウマチである、請求項15記載の方法。
【請求項57】
該Cys残基を有するタンパク質キナーゼが、変異したB-Rafによって活性化される細胞シグナル伝達経路のキナーゼである、請求項1記載の方法。
【請求項58】
該Cys残基とマイケル付加物を形成することができる化合物がヒポテマイシンである、請求項57記載の方法。
【請求項59】
該Cys残基を有するタンパク質キナーゼがPDGFRB、PDGFRA、MEK/ERK、又はKITのいずれかであり、疾患が慢性骨髄性白血病、多形型グリア芽細胞腫、GIST又は転移性GISTである、請求項15記載の方法。
【請求項60】
該Cys残基を有するタンパク質キナーゼがFLT3である、請求項15記載の方法。
【請求項61】
疾患が急性骨髄性白血病である、請求項15記載の方法。
【請求項62】
該Cys残基を有するタンパク質キナーゼがKDR、FLT4又はFLT1のいずれかである、請求項15記載の方法。
【請求項63】
疾患が血管形成を伴う、請求項15記載の方法。
【請求項64】
疾患がリンパ管形成を伴う、請求項15記載の方法。
【請求項65】
疾患が血管透過性の誘導を伴う、請求項15記載の方法。
【請求項66】
疾患が炎症を伴う、請求項15記載の方法。
【請求項67】
疾患が変異B-Rafを有する細胞の増殖によって特徴付けられる、請求項15記載の方法。
【請求項68】
化合物がヒポテマイシンである、請求項67記載の方法。
【請求項69】
疾患がメラノーマである、請求項15記載の方法。
【請求項70】
化合物がヒポテマイシンである、請求項69記載の方法。
【請求項71】
該Cys残基とマイケル付加物を形成することができる化合物が、天然に存在するレゾルシル酸ラクトン、ヒポテマイシン、(5Z)-7-オキソゼアネオール、Ro-09-2210及びL-783,277以外のものである、請求項1記載の方法。
【請求項72】
下記式(II)の構造を有する精製及び単離された化合物。
【化21】
【請求項73】
生物ヒポミセス・スビキュロサス(Hypomyces subiculosus)DSM 11931を、培養液1Lあたり約10〜100mgの量のD,L-エチオニンを含む培養液中で培養する工程を含む、下記式(II)の構造を有する化合物の調製方法。
【化22】
【請求項74】
培養液が、約30〜120g/Lのスクロース、約20〜80g/Lのコーンミール、及び約0〜10g/Lの酵母抽出物を含む、請求項73記載の方法。
【請求項1】
混合物又は細胞において、タンパク質キナーゼ上のATP結合部位に保存された2つのアスパラギン酸残基の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン(Cys)残基を有する1以上のタンパク質キナーゼを阻害する方法であって、
前記混合物は、タンパク質キナーゼ上のATP結合部位に保存された2つのアスパラギン酸残基の間に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するCys残基を欠く更なるタンパク質キナーゼを含み、
前記Cys残基を有する前記1以上のタンパク質キナーゼを、前記Cys残基とマイケル付加物を形成することができる化合物に、その化合物と前記Cys残基との間でマイケル付加物が形成されて前記1以上のタンパク質キナーゼの阻害がもたらされる条件下において、接触させることを含む、前記方法。
【請求項2】
該Cys残基とマイケル付加物を形成することができる化合物が、該Cys残基とマイケル付加物を形成するエノン部分を含む、請求項1記載の方法。
【請求項3】
該Cys残基とマイケル付加物を形成することができる化合物が、7位のカルボニルと共役している5−6位のシス炭素-炭素二重結合を有するレゾルシル酸ラクトンである、請求項2記載の方法。
【請求項4】
該Cys残基とマイケル付加物を形成することができる化合物が、下記の構造(I)を有する化合物、又はその医薬的に許容できる塩若しくは誘導体である、請求項2記載の方法
【化1】
(式中、
R1は、水素、任意に置換された脂肪族部分、任意に置換された環式脂肪族部分、任意に置換された複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成するか;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C4アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは任意にヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3若しくは複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は
R8及びR9が一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であるか、又はR20及びR2は組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であり、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり;ここでR19は水素又はアルキルである)。
【請求項5】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ia)の構造を有する、請求項4記載の方法
【化2】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;さらに
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項6】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ib)の構造を有する、請求項4記載の方法
【化3】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項7】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ic)の構造を有する、請求項4記載の方法
【化4】
(式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分はヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項8】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Id)の構造を有する、請求項4記載の方法
【化5】
(式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項9】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ie)の構造を有する、請求項4記載の方法
【化6】
(式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項10】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(If)の構造を有する、請求項4記載の方法
【化7】
(式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項11】
該Cys残基とマイケル付加物を形成することができる化合物が下記式(Ig)の構造を有する、請求項4記載の方法
【化8】
(式中、
R4はH又はFであり;
R8はHであり;
R9は以下の基から成る群より選択されるか、
【化9】
又はR8及びR9が組み合わさって以下の基を形成する)。
【化10】
【請求項12】
混合物がin vitroの反応混合物である、請求項1記載の方法。
【請求項13】
阻害の工程が細胞内で行われる、請求項1記載の方法。
【請求項14】
細胞が病変細胞であるか、又は病変組織に存在する、請求項1記載の方法。
【請求項15】
疾患又は疾患症状の治療を必要とする患者に、タンパク質キナーゼ上のATP結合部位領域に保存された2つのアスパラギン酸残基に位置し且つ前記アスパラギン酸残基のうちの1つのすぐ隣に位置するシステイン(Cys)残基を有するタンパク質キナーゼを特異的に阻害する化合物を含む医薬組成物を投与することにより、疾患又は疾患症状を治療する方法であって、
前記タンパク質キナーゼを、前記Cys残基とマイケル付加物を形成する化合物と接触させることを含む、前記方法。
【請求項16】
医薬組成物が、下記の構造(I)の化合物、並びにその医薬的に許容できる塩及び誘導体を含む、請求項15記載の方法
【化11】
(式中、
R1は、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;
R2及びR3は、各々独立して、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、又は任意に置換されていてもよいヘテロアリール部分であり;又は、R1及びR2は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;又は、R1及びR3は、一緒になって、任意に置換されていてもよい炭素数3〜8の飽和若しくは不飽和環式環を形成し;
R4は、水素又はハロゲンであり;
R5は、水素、C2〜C4アルキル、酸素保護基、又はプロドラッグ部分であり;
R6は、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
nは、0、1又は2であり;
R7は、存在する各々が独立して、水素、ヒドロキシル、又は保護されたヒドロキシルであり;
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分はヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH、又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここで、R14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;又は
R8及びR9は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し、前記環は、ヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
R11は、水素、ヒドロキシル、保護されたヒドロキシル、アミノ、又は保護されたアミノであり;
R20は水素であり、又は、R20及びR2が組み合わさって結合を形成し;
Xは、存在しないか、又はO、NH、N−アルキル、CH2若しくはSであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17、O、C=O、CR17又はNR17であり、ZはCHR18、O、C=O、CR18、又はNR18であり;R17及びR18は、存在する各々が独立して、水素又は任意に置換されていてもよい脂肪族部分であり、又はR17及びR18が一緒になって−O−、−CH2−又は−NR19−であり、式中R19は水素又はアルキルである)。
【請求項17】
化合物が、下記式(Ia)の構造を有する、請求項16記載の方法
【化12】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項18】
化合物が下記式(Ib)の構造を有する、請求項16記載の方法
【化13】
(式中、
R9は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、OR12、SR12、NR12R13、−X1(CH2)pX2−R14、又はアルキルであり、前記アルキルは、ヒドロキシル、保護されたヒドロキシル、ハロゲン、アミノ、保護されたアミノ、又は−X1(CH2)pX2−R14で置換されていてもよく;ここで
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここで、R12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
X1及びX2は、各々独立して、存在しないか、酸素、NH又は−N(アルキル)であり;又は、X2−R14が一緒になって、N3又は複素環式脂肪族部分であり;
pは、2〜10の整数であり;
R14は、水素、アリール部分、ヘテロアリール部分、アルキルアリール部分、アルキルヘテロアリール部分、−(C=O)NHR15、−(C=O)OR15、又は−(C=O)R15であり、式中、存在するR15の各々は、独立して、水素、脂肪族部分、環式脂肪族部分、複素環式脂肪族部分、アリール部分、又はヘテロアリール部分であり;又は、R14は−SO2(R16)であり、式中、R16は脂肪族部分であり;ここでR14、R15及びR16の1以上は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;さらに
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項19】
化合物が下記式(Ic)の構造を有する、請求項16記載の方法
【化14】
(式中、
R8は、水素、ハロゲン、ヒドロキシル、保護されたヒドロキシル、アルコキシ、又は脂肪族部分であり、前記脂肪族部分は、ヒドロキシル、保護されたヒドロキシル、SR12又はNR12R13で置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項20】
化合物が下記式(Id)の構造を有する、請求項16記載の方法
【化15】
(式中、
R10は、水素、ヒドロキシル、アルコキシ、ヒドロキシアルキル、ハロゲン、又は保護されたヒドロキシルであり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項21】
化合物が下記式(Ie)の構造を有する、請求項16記載の方法
【化16】
(式中、
R5は、水素、C2〜C5アルキル、酸素保護基、又はプロドラッグ部分であり;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項22】
化合物が下記式(If)の構造を有する、請求項16記載の方法
【化17】
(式中、
R12及びR13は、存在する各々が独立して、水素、任意に置換されていてもよい脂肪族部分、任意に置換されていてもよい環式脂肪族部分、任意に置換されていてもよい複素環式脂肪族部分、任意に置換されていてもよいアリール部分、任意に置換されていてもよいヘテロアリール部分、又はN若しくはS保護基であり;又は、R12及びR13は、一緒になって、1〜4の炭素原子及び1〜3の窒素原子若しくは酸素原子を含む飽和若しくは不飽和環式環を形成し;ここでR12及びR13の各々は、1以上のヒドロキシル、保護されたヒドロキシル、アルコキシ、アミノ、保護されたアミノ、−NH(アルキル)、アミノアルキル、又はハロゲンで置換されていてもよく;
Y及びZは、単結合又は二重結合によって連結されており、YはCHR17であり、ZはCHR18であり;R17及びR18は水素であるか、又はR17及びR18が一緒になって−O−である)。
【請求項23】
化合物が下記式(Ig)の構造を有する、請求項16記載の方法
【化18】
(式中、
R4はH又はFであり;
R8はHであり;
R9は以下の基から成る群より選択されるか、
【化19】
又はR8及びR9が一緒になって以下の基を形成する)。
【化20】
【請求項24】
該Cys残基を有するタンパク質キナーゼが以下から成る群より選択される、請求項1記載の方法:AAK1、キナーゼドメインを有するAPEG1スプライス変種(SPEG)、BMP2K(BIKE)、CDKL1、CDKL2、CDKL3、CDKL4、CDKL5(STK9)、ERK1(MAPK3)、ERK2(MAPK1)、FLT3、GAK、GSK3A、GSK3B、KIT(cKIT)、MAP3K14(NIK)、MAP3K7(TAK1)、MAPK15(ERK8)、MAPKAPK5(PRAK)、MEK1(MKK1, MAP2K1)、MEK2(MKK2, MAP2K2)、MEK3(MKK3, MAP2K3)、MEK4(MKK4, MAP2K4)、MEK5(MKK5, MAP2K5)、MEK6(MKK6, MAP2K6)、MEK7(MKK7, MAP2K7)、MKNK1(MNK1)、MKNK2(MNK2, GPRK7)、NLK、PDGFRα、PDGFRβ、PRKD1(PRKCM)、PRKD2、PRKD3(PRKCN)、PRPF4B(PRP4K)、RPS6KA1(RSK1, MAPKAPK1A)、RPS6KA2(RSK3, MAPKAP1B)、RPS6KA3(RSK2, MAPKAP1C)、RPS6KA6(RSK4)、STK36(FUSED_STK)、STYK1、TGFBR2、TOPK、VEGFR1(FLT1)、VEGFR2(KDR)、VEGFR3(FLT4)及びZAK。
【請求項25】
該Cys残基を有するタンパク質キナーゼの少なくとも2つが阻害される、請求項24記載の方法。
【請求項26】
該Cys残基を有するタンパク質キナーゼの少なくとも3つが阻害される、請求項24記載の方法。
【請求項27】
1以上の該Cys残基を有するタンパク質キナーゼがERK経路キナーゼであり、前記ERK経路キナーゼの少なくとも2つが阻害される、請求項1記載の方法。
【請求項28】
ERK経路キナーゼの少なくとも4つが阻害される、請求項27記載の方法。
【請求項29】
該Cys残基を有するタンパク質キナーゼが、MEK1、MEK2、ERK1及びERK2である、請求項28記載の方法。
【請求項30】
阻害される1以上の該Cys残基を有するタンパク質キナーゼが、少なくとも2つのERK MAPKカスケード経路キナーゼとマイトジェンレセプターキナーゼとを含む、請求項1記載の方法。
【請求項31】
マイトジェンレセプターキナーゼが、VEGFレセプター;PDGFレセプター;c-KIT(肥満細胞増殖因子レセプター);FLT3(FL(Flt-3リガンド)のレセプター);及び、VEGFレセプター、PDGFレセプター、cKIT又はFLT3の構成的に活性化された変異体;から成る群より選択される、請求項30記載の方法。
【請求項32】
該Cys残基を有するタンパク質キナーゼの阻害剤が微小管安定化剤又は微小管脱安定化剤と一緒に投与される、請求項15記載の方法。
【請求項33】
該Cys残基を有するタンパク質キナーゼの阻害剤がHsp90阻害剤と一緒に投与される、請求項15記載の方法。
【請求項34】
Hsp90阻害剤が17-AAG又は17-DMAGである、請求項33記載の方法。
【請求項35】
該Cys残基を有するタンパク質キナーゼが、PDGFRα、PDGFRβ、VEGFレセプター(Flt-1、Flt-4及びKdr)、MEK1/2及びERK1/2から成る群より選択され、疾患が、加齢が関係する筋変性又は緑内障である、請求項15記載の方法。
【請求項36】
該Cys残基を有するタンパク質キナーゼが、Flt-3、c-Kit MEK、ERK又はVEGFRのいずれかであり、疾患が急性骨髄性白血病である、請求項15記載の方法。
【請求項37】
該Cys残基を有するタンパク質キナーゼが、c-Kit、PDGFR、MEK1/2又はERK1/2のいずれかであり、疾患が消化管間質腫瘍である、請求項15記載の方法。
【請求項38】
該Cys残基を有するタンパク質キナーゼが、野生型c-Kit、構成的に活性なc-Kit V816D変異体、MEK1/2又はERK1/2のいずれかであり、疾患が肥満細胞症である、請求項15記載の方法。
【請求項39】
該Cys残基を有するタンパク質キナーゼが、MEK1/2、ERK1/2又はTak1のいずれかであり、疾患が炎症性腸疾患である、請求項15記載の方法。
【請求項40】
炎症性腸疾患がクローン病又は潰瘍性大腸炎である、請求項39記載の方法。
【請求項41】
該Cys残基を有するタンパク質キナーゼがc-Kit、MEK又はERKであり、疾患が肥満細胞によって影響される又は引き起こされる炎症性症候群である、請求項15記載の方法。
【請求項42】
該Cys残基を有するタンパク質キナーゼがMEK1/2又はERK1/2のいずれかであり、疾患が乳癌である、請求項15記載の方法。
【請求項43】
該Cys残基を有するタンパク質キナーゼがKdr、c-Kit、MEK1/2、又はERK1/2のいずれかであり、疾患が非小細胞肺癌である、請求項15記載の方法。
【請求項44】
該Cys残基を有するタンパク質キナーゼがPDGFRA、MEK1/2、又はERK1/2のいずれかであり、疾患が卵巣癌である、請求項15記載の方法。
【請求項45】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が膵臓癌である、請求項15記載の方法。
【請求項46】
該Cys残基を有するタンパク質キナーゼが、変異体Raf-1タンパク質キナーゼによって活性化されるキナーゼであり、疾患が前立腺癌である、請求項15記載の方法。
【請求項47】
該Cys残基を有するタンパク質キナーゼがRSK又はMEK/ERKである、請求項46記載の方法。
【請求項48】
該Cys残基を有するタンパク質キナーゼが、VEGFR、PDGFR、MEK1/2、ERK1/2、Tak1、又はJNKシグナル伝達経路及びp38シグナル伝達経路を活性化するキナーゼのいずれかであり、疾患が乾癬である、請求項15記載の方法。
【請求項49】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が基底細胞癌である、請求項15記載の方法。
【請求項50】
該Cys残基を有するタンパク質キナーゼが、MEK1/2、ERK1/2、Tak1、又はJNKシグナル伝達経路を活性化するキナーゼのいずれかであり、疾患が炎症性症候群である、請求項15記載の方法。
【請求項51】
炎症性症候群がアレルギー性皮膚炎である、請求項50記載の方法。
【請求項52】
該Cys残基を有するタンパク質キナーゼがPDGFRであり、疾患が肺線維症である、請求項15記載の方法。
【請求項53】
該Cys残基を有するタンパク質キナーゼがMEK1/2、又はERK1/2のいずれかであり、疾患がRas変異体依存性癌腫である、請求項15記載の方法。
【請求項54】
該Cys残基を有するタンパク質キナーゼがVEGFR、PDGFR、MEK1/2、又はERK1/2のいずれかであり、疾患が腎細胞癌である、請求項13記載の方法。
【請求項55】
該Cys残基を有するタンパク質キナーゼがPDGFR、MEK1/2、ERK1/2、又はTak1のいずれかであり、疾患が再狭窄である、請求項15記載の方法。
【請求項56】
該Cys残基を有するタンパク質キナーゼがMEK1/2、ERK1/2、又はTak1のいずれかであり、疾患が慢性関節リウマチである、請求項15記載の方法。
【請求項57】
該Cys残基を有するタンパク質キナーゼが、変異したB-Rafによって活性化される細胞シグナル伝達経路のキナーゼである、請求項1記載の方法。
【請求項58】
該Cys残基とマイケル付加物を形成することができる化合物がヒポテマイシンである、請求項57記載の方法。
【請求項59】
該Cys残基を有するタンパク質キナーゼがPDGFRB、PDGFRA、MEK/ERK、又はKITのいずれかであり、疾患が慢性骨髄性白血病、多形型グリア芽細胞腫、GIST又は転移性GISTである、請求項15記載の方法。
【請求項60】
該Cys残基を有するタンパク質キナーゼがFLT3である、請求項15記載の方法。
【請求項61】
疾患が急性骨髄性白血病である、請求項15記載の方法。
【請求項62】
該Cys残基を有するタンパク質キナーゼがKDR、FLT4又はFLT1のいずれかである、請求項15記載の方法。
【請求項63】
疾患が血管形成を伴う、請求項15記載の方法。
【請求項64】
疾患がリンパ管形成を伴う、請求項15記載の方法。
【請求項65】
疾患が血管透過性の誘導を伴う、請求項15記載の方法。
【請求項66】
疾患が炎症を伴う、請求項15記載の方法。
【請求項67】
疾患が変異B-Rafを有する細胞の増殖によって特徴付けられる、請求項15記載の方法。
【請求項68】
化合物がヒポテマイシンである、請求項67記載の方法。
【請求項69】
疾患がメラノーマである、請求項15記載の方法。
【請求項70】
化合物がヒポテマイシンである、請求項69記載の方法。
【請求項71】
該Cys残基とマイケル付加物を形成することができる化合物が、天然に存在するレゾルシル酸ラクトン、ヒポテマイシン、(5Z)-7-オキソゼアネオール、Ro-09-2210及びL-783,277以外のものである、請求項1記載の方法。
【請求項72】
下記式(II)の構造を有する精製及び単離された化合物。
【化21】
【請求項73】
生物ヒポミセス・スビキュロサス(Hypomyces subiculosus)DSM 11931を、培養液1Lあたり約10〜100mgの量のD,L-エチオニンを含む培養液中で培養する工程を含む、下記式(II)の構造を有する化合物の調製方法。
【化22】
【請求項74】
培養液が、約30〜120g/Lのスクロース、約20〜80g/Lのコーンミール、及び約0〜10g/Lの酵母抽出物を含む、請求項73記載の方法。
【図1】

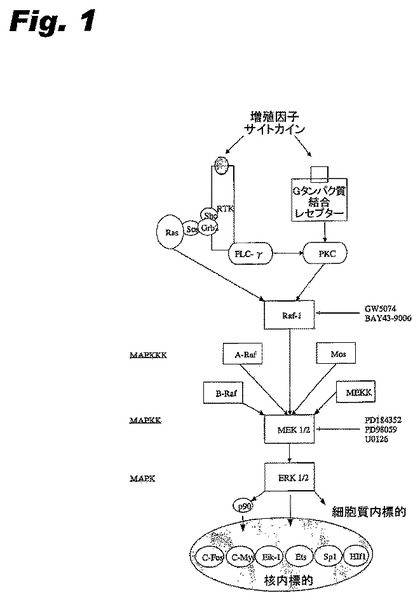
【図2】
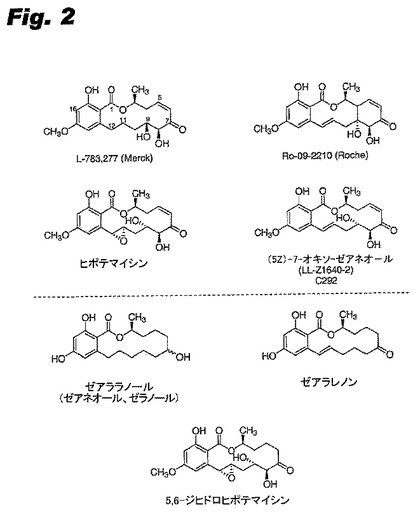
【図3】
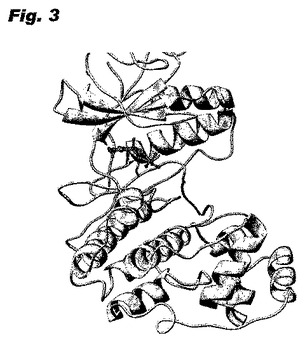
【図4】
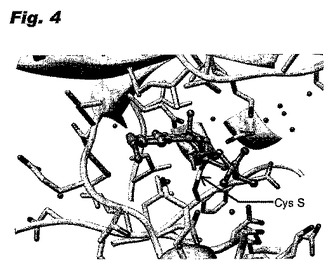
【図5】

【図6】
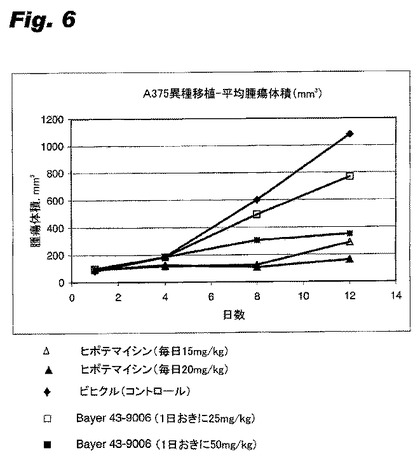
【図7】
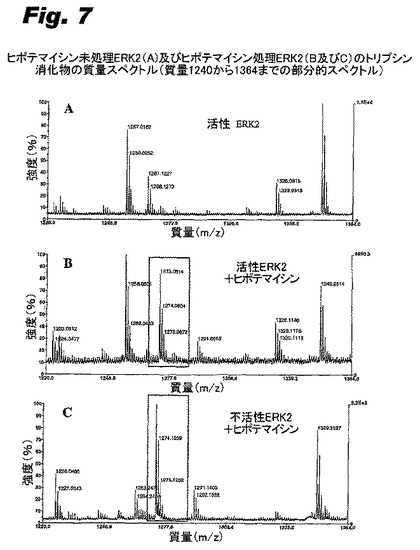
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2008−514635(P2008−514635A)
【公表日】平成20年5月8日(2008.5.8)
【国際特許分類】
【出願番号】特願2007−533734(P2007−533734)
【出願日】平成17年9月26日(2005.9.26)
【国際出願番号】PCT/US2005/034537
【国際公開番号】WO2006/036941
【国際公開日】平成18年4月6日(2006.4.6)
【出願人】(504269110)コーザン バイオサイエンシス インコーポレイテッド (17)
【Fターム(参考)】
【公表日】平成20年5月8日(2008.5.8)
【国際特許分類】
【出願日】平成17年9月26日(2005.9.26)
【国際出願番号】PCT/US2005/034537
【国際公開番号】WO2006/036941
【国際公開日】平成18年4月6日(2006.4.6)
【出願人】(504269110)コーザン バイオサイエンシス インコーポレイテッド (17)
【Fターム(参考)】
[ Back to top ]