
特異的結合タンパク質およびその使用
【課題】特異的抗EGFRvIIIモノクローナル抗体、および該特異的抗EGFRvIIIモノクローナル抗体の製造方法を提供することにある。
【解決手段】本発明のモノクローナル抗体のVH鎖は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とする。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
【解決手段】本発明のモノクローナル抗体のVH鎖は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とする。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医学分野に関する。具体的に、抗上皮成長因子受容体変異体III(EGFRvIII)特異的モノクローナル抗体およびその応用に関する。本発明の抗体は、EGFRvIIIと効率的結合し、或いは細胞が過剰発現した上皮成長因子受容体(EGFR)と部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。本発明の抗体は、EGFRvIIIを発現する腫瘍細胞系の治療に有用である。
【背景技術】
【0002】
上皮成長因子受容体(EGFR)は癌原遺伝子c-erbBの170kDaの膜糖タンパク質の産物である(1)。EGFR遺伝子は、最初、トリ赤芽球症ウイルスで同定されたerbB癌遺伝子の細胞類縁物である(1,2)。各種のヒト腫瘍において、既にこの癌遺伝子の遺伝子増幅による活性化が見られた(3-6)。
【0003】
EGFRが多くの種類のヒト固形腫瘍で過剰発現されることが既に文献で明らかになった(7)。このような腫瘍は、肺癌、大腸癌、乳癌、胃癌、脳癌、膀胱癌、頭頸部腫瘍、卵巣癌、腎癌および前立腺癌を含む(7)。v-erbB癌遺伝子と正常のEGFR遺伝子との違いは、主にウイルスの癌遺伝子が正常の受容体の切断アミノ基の変異型であることにある。細胞質外ドメインの多くが欠損したが、膜貫通およびチロシンキナーゼのドメインが残った(8-11)。よって、上皮成長因子(EGF)と結合することができないが、他のタンパク質をリン酸化させることができる(14-15)。
【0004】
ウイルス性erbB癌遺伝子には色々な遺伝子の変化、例えば、遺伝子のカルボキシ末端におけるアミノ酸の置換や欠失が起こる可能性がある。なかでも、アミノ酸の欠失は、癌誘発作用において重要である。アミノ酸の欠失は大部分のv-erbB癌遺伝子の特徴の一つで、プロモーターの挿入やレトロウイルスの形質導入によるアミノ末端の欠失を含む(13,16)。逆に、カルボキシ末端の欠失はレトロウイルスの形質導入による腫瘍だけに関与し、宿主の範囲および腫瘍の種類の特異性で決まるとされている(11,15)。アミノ末端の欠失のトリc-erbB遺伝子又はヒトEGF受容体のトランスフェクション実験によって、その欠失は細胞を転換させることができることがわかった(16-17)。
【0005】
EGFR遺伝子の増幅は40%のヒト悪性神経膠腫に発生し(3,7)、受容体の遺伝子の組換えが多くの遺伝子増幅の腫瘍で顕著である。組換えは遺伝子のアミノ末端に影響することが多い(6,18)。
【0006】
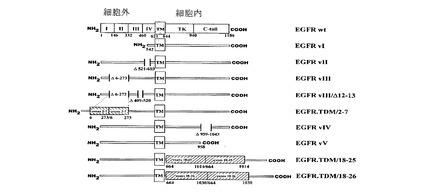
今まで、8種類のEGFR変異体が発見された(図5)。1種類目は、EGFRvIはEGFRの大半の細胞外ドメインが欠失したものである。2種類目は、EGFRvIIはEGFRの細胞外ドメインにおける83aaのインフレーム欠失からなるものである。3種類目は、EGFRvIIIはEGFRの細胞外ドメインにおける267aaのインフレーム欠失からなるものである。4種類目は、EGFRvIVはEGFRの細胞質ドメインにおける欠失を含むものである。5種類目は、EGFRvVはEGFRの細胞質ドメインにおける欠失を含むものである。6種類目はEGFR.TDM/2〜7はEGFRの細胞外ドメインのエクソン2〜7の重複を含むものである。7種類目はEGFR.TDM/18〜26はEGFRの細胞外ドメインのエクソン18〜26の重複を含むものである。8種類目としては、さらに、エクソン11と14の連結部に新規なヒスチジン残基を導入した、より珍しい第2種のEGFRvIII変異体(EGFRvIII/Δ12〜13)が存在する(24)。
【0007】
EGFRvIIIは、ヒトの癌における上皮成長因子(EGF)受容体の最も一般的に存在する変異体である(24)。遺伝子増幅の過程において、細胞外ドメインで267のアミノ酸欠失が起こり、新しい連結部(グリシン)が形成される。EGFRvIIIは、正常の組織に発現されないことが知られている(19,20)。しかしながら、EGFRvIIIは、多くの腫瘍細胞で発現され、例えば27〜76%の乳癌バイオプシーに(21)、50〜70%の神経膠腫に(19,22)、16%の非小細胞肺癌に(23)、75%の卵巣癌に(22)、EGFRvIIIが発現される。
【0008】
一つのEGFRvIIIが過剰発現する癌を治療する方法では、特異的に未分離の正常EGFRの変異体の受容体をターゲットとする腫瘍特異的リボザイムに関する。胸腺欠損ヌードマウスでは、リボザイムは顕著に乳癌の成長を抑制することがわかった。
【0009】
また、267のアミノ酸が欠失してグリシンに置換した特有の連結は抗EGFRvIII特異的モノクローナル抗体の製造に有用である。また、EGFRvIIIは、ある腫瘍で発現され、かつ正常の組織での発現がないことによって、腫瘍の薬物の理想的なターゲットである。具体的に、EGFRvIIIは、腫瘍の免疫抱合体による治療の理想的な候補として有用である。抗EGFRvIII特異的モノクローナル抗体(或いは抗がん剤又は毒素結合の抱合体)は、体内において抗体に依存する細胞溶解又は細胞致死作用を起こすことで、EGFRvIIIを発現する腫瘍細胞を除去する。
【0010】
現在、国内外では複種の抗EGFR抗原の抗体が得られたが、これらの抗体は、例えばEGFRvIIIに対する特異性がない又は低いなどと、満足できるとはいえない。
【0011】
従って、本分野では、EGFRvIIIを認識する特異性が高く、かつ野生型EGFRを認識しない、他の優れた特性を持つ抗EGFRvIIIモノクローナル抗体を開発することによって、治療効果がより顕著な薬物を開発することが切望されている。
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の目的は、特異的抗EGFRvIIIモノクローナル抗体を提供することにある。また、本発明の目的は、前述特異的抗EGFRvIIIモノクローナル抗体の製造方法を提供することにある。
【0013】
さらに、本発明の目的は、前述特異的抗EGFRvIIIモノクローナル抗体を含む薬物組成物を提供することにある。
【課題を解決するための手段】
【0014】
本発明の第一は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有するモノクローナル抗体のVH鎖を提供する。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
他の好ましい例において、前述のVH鎖はSEQ ID NO:2で示されるアミノ酸配列を有する。
【0015】
本発明の第二は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有するモノクローナル抗体のVL鎖を提供する。
SEQ ID NO:8で示されるCDR1、
SEQ ID NO:9で示されるCDR2、および
SEQ ID NO:10で示されるCDR3。
他の好ましい例において、前述のVL鎖はSEQ ID NO:4で示されるアミノ酸配列を有する。
【0016】
本発明の第三は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有するモノクローナル抗体或いはその抱合体を提供する。
【0017】
他の好ましい例において、前述の抗体はヒト上皮成長因子受容体変異体(EGFRvIII)と結合し、かつ細胞が過剰発現するEGFRと部分的に結合するが、細胞が正常発現するEGFRとの結合作用がない。
【0018】
より好ましくは、前述の抗体はA431細胞およびU87-EGFRvIII細胞と結合するが、U87細胞と結合しない。
【0019】
他の好ましい例において、前述の抗体はマウス抗体、ヒト化抗体或いはキメラ抗体である。
【0020】
他の好ましい例において、前述の抱合体は、抗体と抗がん剤或いは毒素(例えば、ジフテリア毒素、リシン、緑膿菌外毒素)の抱合体である。
【0021】
本発明の第四は、以下の群から選ばれるタンパク質をコードする核酸分子(例えばDNA分子)を提供する。
本発明の第一に記載のモノクローナル抗体のVH鎖、
本発明の第二に記載のモノクローナル抗体のVL鎖、
本発明の第三に記載のモノクローナル抗体。
他の好ましい例において、前述の核酸分子はSEQ ID NO:1、3、11或いは13から選ばれるDNA配列を有する。
【0022】
本発明の第五は、VH鎖がSEQ ID NO:5〜7で示される相補性決定領域を有し、かつVL鎖がSEQ ID NO:8〜10で示される相補性決定領域を有するモノクローナル抗体と薬学的に許容される担体とを含む薬物組成物を提供する。
他の好ましい例において、前述モノクローナル抗体は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有する。
【0023】
本発明の第六は、(a)上皮成長因子受容体変異体IIIを発現する細胞の成長の抑制又は致死、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長の抑制に用いられる組成物を製造するための本発明のモノクローナル抗体或いはその抱合体の用途を提供する。
他の好ましい例において、前述の細胞は、腫瘍細胞、例えば、肝癌細胞、肺癌細胞である。
【0024】
本発明の第七は、治療しようとする対象に本発明のモノクローナル抗体或いはその抱合体を給与することを含む、(a)上皮成長因子受容体変異体IIIを発現する細胞の成長抑制又は致死方法、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長抑制方法を提供する。
【0025】
好ましくは、前述対象は、ヒト、マウス、ラットなどを含む哺乳動物である。
【発明の効果】
【0026】
本発明のモノクローナル抗体の特異性および生理活性が顕著に向上する。このモノクローナル抗体は、EGFRvIIIと効率的に結合し、かつ細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。
【0027】
本発明のモノクローナル抗体の親和力、腫瘍抑制率はいずれも既存の抗体(例えばCH806抗体)よりも高く、かつ抗体のアミノ酸配列の構成(特にCDR領域)も違う。
よって、本発明の高親和力、高特異性のモノクローナル抗体は臨床では重要な価値がある。
【図面の簡単な説明】
【0028】
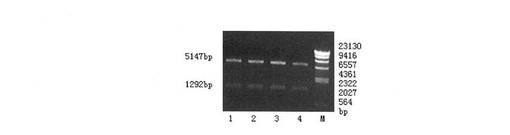
【図1】組換えプラスミドpET28a-EGFRvIIIexがBglII和SalIの酵素切断による同定を示すものである。ただし、レーン1〜4はプラスミドが二つの酵素によって切断されたもので、MはDNA分子量マーカーλHindIIIである。
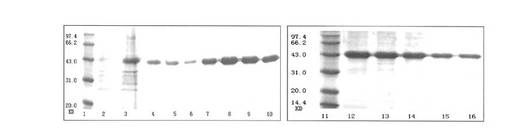
【図2】EGFRvIIIの細胞外ドメインのタンパクを精製した結果を示すものである。レーン1、11はタンパク質の分子量マーカー、レーン2は未誘導の細菌沈殿、レーン3は流出液、レーン4〜6は緩衝液Cの溶離液、レーン7〜10は緩衝液Dの溶離液、レーン12〜16は緩衝液Eの溶離液である。
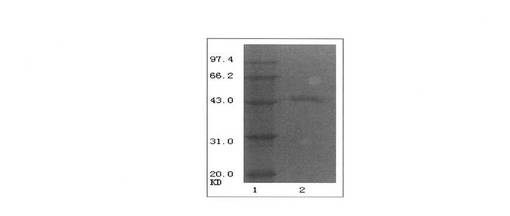
【図3】復元したタンパク質のSDS-PAGEのグラフを示すものである。ただし、レーン1はタンパク質の分子量マーカー、レーン2は復元したタンパク質である。
【図4】復元したタンパク質のウェスタンブロットを示すものである。ただし、レーン1は復元したタンパク質、レーン2はBL21(DE3)-RP菌液の全タンパク質である。
【図5】野生型EGFRおよびその各種の変異体を示すものである。
【図6】12H23抗体サブタイプのELISA分析を示すものである。
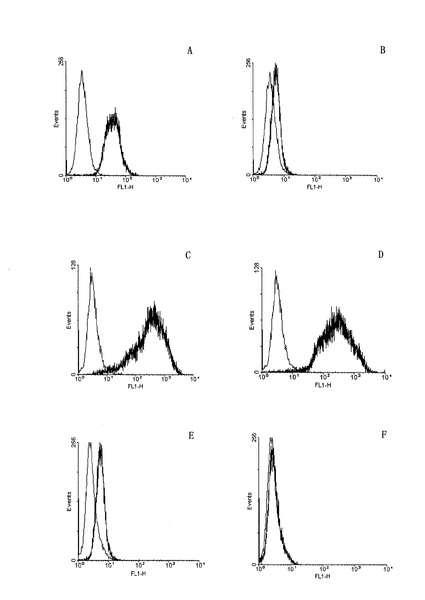
【図7】本発明の抗体12H23および対照抗体C225がそれぞれA431細胞(EGFR過剰発現)、U87-EGFRvIII細胞(EGFRvIIIの安定な高発現)およびU87細胞(EGFR正常発現)とのフローサイトメトリー図を示すものである。ただし、各図は以下の通りである。A:C225とA431細胞の結合図、B:12H23とA431細胞の結合図、C:C225とU87-EGFRvIII細胞系の結合図、D:12H23とU87-EGFRvIII細胞系の結合図、E:C225とU87細胞の結合図、F:12H23とU87細胞の結合図。
【図8】12H23と抗原rEGFRvIIIex細胞の親和力の測定を示すものである。
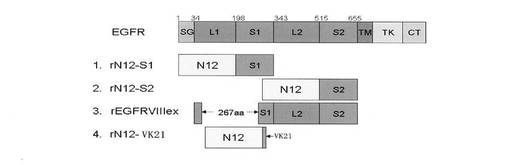
【図9】各組換えタンパク質の配列構造を示す概略図である。
【図10】組換えタンパク質のSDS-PAGE分析を示すものである。ただし、レーンMはタンパク質のマーカー、1はrN12-S1、2はrN12-S2、3はrEGFRvIIIex、4はrN12-VK21である。
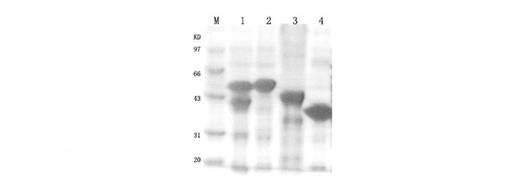
【図11】ELISA法で12H23の結合エピトープを測定した結果を示すものである。ただし、S1はrN12-S1、S2はrN12-S2、EGFRvIIIは組換えEGFRvIII細胞外タンパク質、VK21はrN12-VK21、Negは空白対照である。
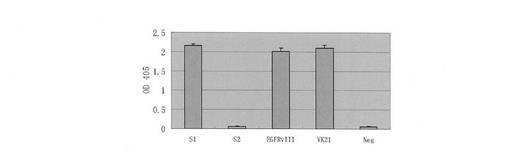
【図12】12H23モノクローナル抗体のヌードマウスの播種腫瘍に対する抑制作用を示すものである。
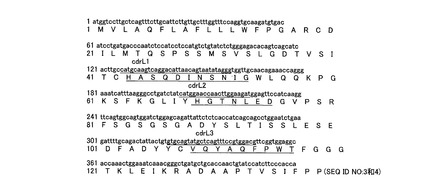
【図13】12H23モノクローナル抗体の重鎖のヌクレオチドコード配列およびアミノ酸配列(下線はCDR)を示すものである。
【図14】12H23モノクローナル抗体の軽鎖のヌクレオチドコード配列およびアミノ酸配列(下線はCDR)を示すものである。
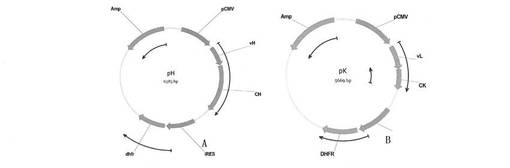
【図15】プラスミドpHおよびpKを示す概略図である。
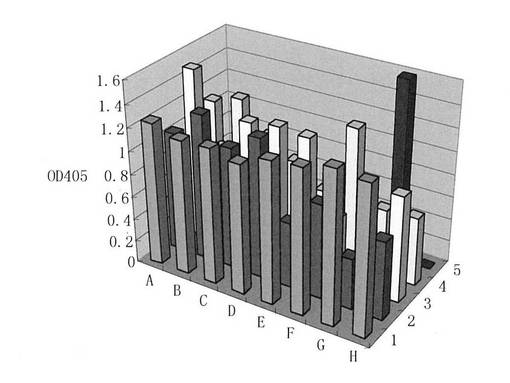
【図16】ヒト-マウスキメラ抗体CH12と組換えEGFRvIII細胞外タンパク質との結合を示すものである。図中において、1〜5はそれぞれCH12を発現する各細胞クローン株を示す。
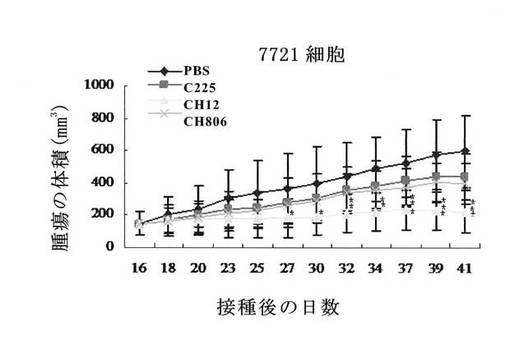
【図17】キメラ体CH12のヌードマウスの播種腫瘍に対する抑制作用を示すものである。
【発明を実施するための形態】
【0029】
発明者らは、幅広く深く研究したところ、EGFRvIIIと効率的に結合し、或いは細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない、EGFRvIIIに対する特異性が高いモノクローナル抗体の製造に成功した。また、本発明の抗体はEGFRvIIIを発現する腫瘍細胞系に対する治療作用が顕著である。これに基づき、本発明を完成させた。
【0030】
本発明は組換え抗EGFRvIIIモノクローナル抗体を提供する。前述の抗体は、マウス抗体、ヒト化抗体或いはキメラ抗体でもよい。例えば、ヒト化抗体はヒト定常領域(例えばヒト定常領域IgG1-Fc)、本発明の重鎖可変領域および軽鎖可変領域を含む。
【0031】
また、本発明は、抗EGFRvIIIモノクローナル抗体のアミノ酸配列およびその可変領域鎖、並びにこれらの鎖を有する他のタンパク質或いは融合発現産物を提供する。具体的に、本発明は、超可変領域(相補性決定領域、CDR)が本発明の軽鎖及び重鎖の超可変領域と同様で、或いは少なくとも90%、好ましくは95%の相同性を持つものであれば、この超可変領域を含有する軽鎖及び重鎖を有する任意のタンパク質又はタンパク質抱合体と融合発現産物(すなわち、免疫抱合体と融合発現産物)を含む。
【0032】
抗体の抗原結合特性は、重鎖および軽鎖可変領域に位置する3つの特定の領域によって特徴付けられ、超可変領域(CDR)と呼ばれる。且つ、該領域は4つのフレームワーク領域(FR)に分かれ、4つのFRのアミノ酸配列が比較的に保守的で、直接結合反応に関与しない。これらのCDRは環状構造を形成し、その中のFRで形成されるβシートによって空間構造上で近づき、重鎖におけるCDRおよび相応の軽鎖におけるCDRが抗体の抗原結合部位を構成する。同類の抗体のアミノ酸配列の比較によってどのアミノ酸がFR或いはCDR領域を構成するか確認することが出来る。
【0033】
また、最近、軽鎖可変領域から構成する関連構造は、相応の重鎖可変領域と比べ、結合の動力が小さいため、単離の重鎖可変領域自身が抗原結合活性を持つことがわかった。
【0034】
ここで同定されるV鎖の超可変領域或いは相補性決定領域(complementarity determining region、CDR)は、その少なくとも一部が抗原結合に関与することで、注目されている。そのため、CDR含有モノクローナル抗体の軽鎖および重鎖可変領域鎖を有する分子は、そのCDRはここで同定されるCDRと90%以上(好ましくは95%以上、最も好ましくは98%以上)の相同性を持つものであれば、本発明に含まれる。
【0035】
本発明は、完全のモノクローナル抗体だけではなく、免疫活性を有するFab又は(Fab')2断片のような抗体断片、抗体の重鎖、抗体の軽鎖、遺伝子工学によって改造された一本鎖Fv分子やマウス抗体結合特異性を持つがヒト由来の抗体部分が残った抗体のようなキメラ抗体も含む。
【0036】
さらに、本発明は、上述のモノクローナル抗体又はその断片をコードするDNA分子を提供する。本発明のモノクローナル抗体のヌクレオチド全長配列或いはその断片は、通常、PCR増幅法、組換え法又は人工合成の方法で得られる。適用できる方法として、特に断片の長さが短い場合、人工合成の方法で関連配列を合成する。通常、まず多数の小さい断片を合成し、そして連接させることにより、配列の長い断片を得ることができる。また、軽鎖及び重鎖のコード配列を一体に融合し、一本鎖抗体を形成してもよい。
【0037】
関連の配列を獲得すれば、組換え法で大量に関連配列を獲得することができる。この場合、通常、その配列をベクターにクローンした後、細胞に導入し、さらに通常の方法で増殖させた宿主細胞から関連配列を分離して得る。
【0038】
現在、本発明のタンパク質(又はその断片、或いはその誘導体)をコードするDNA配列を全部化学合成で獲得することがすでに可能である。さらに、このDNA配列を本分野で周知の各種の既知のDNA分子(或いはベクターなど)や細胞に導入してもよい。また、化学合成で本発明のタンパク質配列に変異を導入することもできる。
【0039】
さらに、本発明は、上述の適当なDNA配列及び適当なプロモーター或いは制御配列を含むベクターに関する。これらのベクターは、タンパク質を発現するように、適当な宿主細胞の形質転換に用いることができる。
【0040】
宿主細胞は、原核細胞、例えば細菌細胞、或いは、低等真核細胞、例えば酵母細胞、或いは、高等真核細胞、例えば哺乳動物細胞でもよい。代表例として、大腸菌、ストレプトマイセス属、ネズミチフス菌のような細菌細胞、酵母のような真菌細胞、ミバエS2若しくはSf9のような昆虫細胞、CHO、COS7、293細胞のような動物細胞などがある。
【0041】
DNA組換えによる宿主細胞の転換は当業者に熟知の通常の技術で行っても良い。宿主が原核細胞、例えば大腸菌である場合、DNAを吸収できるコンピテントセルは指数成長期後収集でき、CaCl2法で処理し、用いられる工程は本分野では周知のものである。もう一つの方法は、MgCl2を使用する。必要により、転化はエレクトロポレーションの方法を用いてもよい。宿主が真核生物の場合、リン酸カルシウム沈殿法、マイクロインジェクション、エレクトロポレーションのような通常の機械方法、リポフェクションなどのDNAトランスフェクションの方法が用いられる。
【0042】
得られる形質転換体は通常の方法で培養し、本発明の遺伝子がコードするポリペプチドを発現することが出来る。用いられる宿主細胞によって、培養に用いられる培地は通常の培地を選んでも良い。宿主細胞の成長に適する条件で培養する。宿主細胞が適当の細胞密度に成長したら、適切な方法(例えば温度転換もしくは化学誘導)で選んだプロモーターを誘導し、さらに、細胞を所定の時間培養する。
【0043】
上述の方法における組換えポリペプチドは細胞内または細胞膜で発現し、或いは細胞外に分泌することが出来る。必要であれば、その物理・化学的特性及び他の特性を利用して各種の分離方法で組換えタンパク質を分離・精製することができる。これらの方法は、本分野の当業者に熟知されている。これらの方法の例として、通用の再生処理、タンパク質沈殿剤による処理(塩析法)、遠心分離、浸透圧ショック、超音波処理、超遠心分離、分子篩クロマトグラフィー(ゲルろ過)、吸着クロマトグラフィー、イオン交換クロマトグラフィー、高速液体クロマトグラフィー(HPLC)及び他の各種の液体クロマトグラフィー技術、並びにこれらの方法の組合せを含むが、これらに限定されない。
【0044】
さらに、本発明は組成物を提供する。好ましい例において、前述の組成物が薬物組成物で、上述のモノクローナル抗体或いは免疫抱合体、および薬学的に許容される担体を含む。通常、これらの物質を、無毒で不活性であり薬学的に許容される水系担体で配合し、pH値は配合される物質の性質および治療しようとする疾患にもよるが、通常5〜8程度、好ましくは6〜8程度である。配合された薬物組成物は通常の経路で給与することができ、腫瘍内、腹膜内、静脈内、或いは局部給与が含まれるが、これらに限られない。
【0045】
本発明の薬物組成物は直接腫瘍の予防および治療に用いられる。また、他の治療剤と併用してもよい。
【0046】
本発明の薬物組成物は、安全有効量(例えば0.001〜99wt%、好ましくは0.01〜90wt%、より好ましくは0.1〜80wt%)の本発明の上述のモノクローナル抗体(或いはその抱合体)および薬学的に許容される担体或いは賦形剤を含む。このような担体は、食塩水、緩衝液、ブドウ糖、水、グリセリン、エタノール、及びその組合せを含むが、これらに限定されない。薬物の製剤は給与様態に相応しなければならない。本発明の薬物組成物は、注射剤としてもよく、例えば生理食塩水或いはグルコースおよび他の助剤を含む水溶液を使用し通常の方法で製造することができる。薬物組成物は、注射剤、溶液の場合、無菌条件で製造する。活性成分の給与量は治療有効量、例えば、毎日約1μg/kg体重〜約5mg/kg体重である。また、本発明のポリペプチドは他の治療剤と併用することが出来る。
【0047】
薬物組成物の使用時、安全有効量の免疫抱合体を哺乳動物に使用するが、その安全有効量は、通常、少なくとも約10μg/kg体重で、多くの場合に、約8mg/kg体重未満であり、好ましくはこの投与量が約10μg/kg体重〜約1mg/kg体重である。勿論、具体的な投与量は、さらに投与の様態、患者の健康状況などの要素を考えるべきで、すべて熟練の医者の技能範囲以内である。
【0048】
本発明の利点は以下の通りである。
(a)本発明のモノクローナル抗体の特異性および生理活性が顕著に向上する。このモノクローナル抗体は、EGFRvIIIと効率的に結合し、かつ細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。
(b)本発明のモノクローナル抗体の親和力、腫瘍抑制率はいずれも既存の抗体(例えばCH806抗体)よりも高く、かつ抗体のアミノ酸配列の構成(特にCDR領域)も違う。
よって、本発明の高親和力、高特異性のモノクローナル抗体は臨床では重要な価値がある。
【実施例】
【0049】
以下、具体的な実施例によって、さらに本発明を説明する。これらの実施例は本発明を説明するために用いられるものだけで、本発明の範囲の制限にはならないと理解されるものである。
【0050】
下述の実施例で具体的な条件が示されていない実験方法は、通常、例えばSambrookらによる、「モレキュラー・クローニング:研究室マニュアル」(ニューヨーク、コールド・スプリング・ハーバー研究所出版社、1989)に記載の条件などの通常の条件に、或いは、メーカー推薦の条件に従う。特に断らない限り、%と部は、重量で計算される。
【0051】
実施例1 抗原の製造
1.1 EGFRvIIIタンパク質の細胞外ドメインの原核発現および精製
1.1.1 ベクターの構築および同定
pLNRNL(コード全長EGFRvIII、Ludwig Instituteから購入, San Diego, CA)を鋳型とし、PCR法で両末端にBamHIおよびSalIの切断部位を有するEGFRvIIIex増幅産物を得、BamHIおよびSalIで同時切断し、目的の断片を得た。BglIIとSalIで市販のベクターpET28a(Novagen社から購入)を切断し、アガロースゲル電気泳動後、目的の断片を回収し、T4リガーゼの作用によって連結し、ベクターpET28a-EGFRvIIIexを形成した。さらに市販の大腸菌TOP10(Invitrogenから購入)に導入し、カナマイシン耐性でスクリーンし、BglII和SalIによる切断により、挿入断片を含有する陽性クローンと同定された。
【0052】
1.1.2 発現細菌のスクリーニング
正確と同定された組換えプラスミドでそれぞれ通常の大腸菌Bl21(DE3)、Bl21(DE3)-RP、 HMS174(DE3) (Novagen社から購入)に形質転換を行い、且つカナマイシン耐性プレートに塗布して37℃で一晩倒立培養を行った。モノクローンを選択してODが0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、30℃で4時間誘導した後、菌液を収集し、遠心分離して沈殿物を取って、SDS-PAGE電気泳動でタンパク質の発現量を解析した。
【0053】
1.1.3 融合タンパク質の誘導条件の分析
目的遺伝子の原核細胞における発現を向上させるため、いくつかの発現誘導条件を試験で探った。
(1)誘導時間:細菌をLB培地に接種し、37℃でODが0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、30℃で振とう培養し、それぞれ1、2、3、4、5、6時間の時に菌液を収集した。
(2)誘導濃度:発現菌株を0.6〜0.8になるまで培養し、それぞれ最終濃度が0.2、0.5、0.8、1mMになるようにIPTGを入れ、30℃で4時間振とう培養し、細菌を収集した。
(3)誘導温度:発現菌株を0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、それぞれ37℃、30℃、25℃で4時間誘導した後、細菌を収集した。
【0054】
1.1.4 融合タンパク質の発現様態
上述条件で大量に誘導した後、沈殿物を収集して1/10体積の緩衝液A(50mM NaH2PO4、300mM NaCl、10mM Imidozole(イミダゾール)、pH8.0)で再懸濁させ、PMSF(最終濃度が1mM)を入れて氷の上で超音波処理(超音波3秒、間隔10秒、1サークル99回、計4サークル)を行った後、遠心分離(4℃、12000g)し、上清と沈殿物をそれぞれ収集し、12%SDS-PAGE電気泳動を行い、0.25%クマシーブルーで3時間染色した後、脱色をして観察した。
【0055】
1.1.5 封入体の洗浄と変性
超音波による崩壊、遠心分離を経た沈殿物を十分に洗浄液I(100mM NaH2PO4、10mM Tris.Cl、2M 尿素(urea)、pH8.0)に再懸濁させ、4℃で30分間撹拌した後、15分間遠心分離(4℃、12000g)して沈殿物を収集し、洗浄液II(100mM NaH2PO4,10mM Tris.Cl,2M GuHCl,pH8.0)を入れ、上述操作を繰り返して、精製された封入体を得た。最後、封入体を8M尿素含有溶液(100mM NaH2PO4、10mM Tris.Cl、8M 尿素、pH8.0)に再懸濁させ、氷の上で超音波処理し、15分間遠心分離(4℃、12000g)し、沈殿物を捨て、上清を残した。
【0056】
1.1.6 融合タンパク質の精製
上清をNi-NTA アガロースと4℃で1時間(一晩)均一に混合し、親和クロマトグラフィーにかけて流出液を収集し、4mlの緩衝液C(100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH6.3)で3回洗浄し、さらに0.5mlの緩衝液D(100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH15.9)で4回洗浄し、最後に0.5mlの緩衝液E (100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH4.5)で4回洗浄した。それぞれの脱離液を収集して12%SDS-PAGEで純度を分析し、A280でその含有量を検出した。
【0057】
1.1.7 融合タンパク質のアニーリング
精製されたタンパク質を一滴ずつ10倍体積の冷やしておいたアニーリング緩衝液(25mM Tris-Cl、0.1M NaCl、10%グリセリン、1.0M 尿素、0.01M アルギニン、1mM還元性グルタチオン、0.5mM 酸化性グルタチオン、pH8.0)を入れ、4℃で24時間インキュベートした後、透析袋に入れて、それぞれ0.5M、0.25M、0.125M尿素を含有する緩衝液(PBS、pH7.4)にて4℃で4時間以上透析し、最後に大量のPBSに4℃で24時間透析し、遠心分離して上清を取った。
【0058】
1.3.8 アニーリングタンパク質のウェスタンブロッティングによる同定
Bl21(DE3)-RP菌における全タンパク質を陰性対照とし、アニーリング後の融合タンパク質に12%SDS-PAGEを行い、且つNC膜に転写した。1:1000のモノクローンのウサギ抗DGFRvIII(Zymed社から購入)(4℃で一晩インキュベートし、PBSTで膜を3回、毎回10分間洗浄したもの)および1:5000のHRPマウス抗ウサギIgG(37℃で1時間インキュベートし、膜洗浄は上述と同様なもの)滴下した。最後、ECL化学発光キットの試薬で現像し、且つ暗室でXフィルムを用いて露光した。
【0059】
結果:
1.pET28a-EGFRvIIIex組換え発現プラスミドの同定
プラスミドpET28a-EGFRvIIIexはBglII及びSalIの酵素切断後1292bp、5147bpの電気泳動バンド(図1を参照)が現れ、予想の大きさと一致し、ベクターの構築が正しいことが証明された。
【0060】
2.EGFRvIIIの細胞外領域のタンパク質の精製
図2に示すように、精製されたEGFRvIIIの細胞外領域のタンパク質を得た。
【0061】
3.EGFRvIIIのタンパク質アニーリング後の同定
図3及び4に示すように、EGFRvIIIの細胞外領域のタンパク質の純度が非常に高いことがわかった。
【0062】
実施例2. 抗原免疫及びハイブリドーマスクリーニング
2.1 免疫
(1)組換えタンパク質による免疫:
EGFRvIIIの細胞外領域の組換えタンパク質と等量の完全フロイントアジュバント(Sigma)と充分に乳化混合して6週齢のBALB/cマウスに1匹100μgずつ皮膚下免疫した。4週後組換え抗原を不完全フロイントアジュバントと充分に乳化混合し、免疫マウスに1匹50μgずつ腹腔注射し、その後2週間おきに腹腔強化免疫を続けた。4回目の強化免疫の1週間後、組換え抗原で被覆し、ELISA法でマウスの抗血清力価を検出したところ、>105であった。
【0063】
(2)脾臓内注射による強化免疫:
最後の強化の3週間後、20μgの組換え抗原で脾臓内免疫した。
【0064】
2.2 ハイブリドーマ細胞株の構築
マウスの脾臓内強化免疫の4日後、無菌の条件で脾臓を採取し、100メッシュのフィルターでリンパ細胞を分離し、骨髄腫瘍細胞系SP2/0と融合させた。ピポキサンチン-アミノプテリン-チミジン(hypoxathine, aminopterin and thymidine、HAT)で選択培養を3日行った後、HT培地を追加し、1週間培養を続けた。組換え抗原で被覆し、ELISAで陽性クローンを選択し、限界希釈法でサブクローニングし、2ヵ月培養を続けて、最後に安定したハイブリドーマ細胞系を得た。
その結果、陽性クローンを複数得たが、中でも12H23の活性が一番高かった。
【0065】
2.3 抗体の精製
2.3.1 オクタン酸/硫酸アンモニウム沈殿法による粗精製
腹水100mlを2倍体積の0.06MのpH 4.0の酢酸ナトリウム緩衝液で希釈し、4%オクタン酸をゆっくり滴下しながら、撹拌した。30分間撹拌した後、混濁液を10000gで30分間遠心分離した。沈殿物を捨て、上清液を0.01MのpH7.4のリン酸緩衝液で一晩透析した。透析液を取り、等体積の飽和硫酸アンモニウム液をゆっくり入れ、2時間置いた。混濁液を10000gで10分間遠心分離した。上清液を捨て、0.01M、pH7.4のPBSで溶解させた。溶解した溶液を0.01M、pH7.4のPBSで透析し、その期間で液を2回入れ替え、2回の入れ替えの間隔が5時間以上であった。透析溶液を10000gで10分間遠心分離し、沈殿物を捨て、上清液を収集した。
【0066】
2.3.2 プロテインG親和性精製
プロテインG親和性カラムを室温に戻し、5倍のカラム体積のPBSでバランスを取った。上述モノクローナル抗体溶液をカラムにかけ、5倍のカラム体積のPBSで洗った。pH2.3の0.1Mグリシン塩酸溶液で脱離させ、脱離液を1/10体積の1Mリン酸水素二ナトリウム溶液(pH9.0)で中和した。溶液を0.01M、pH7.4のPBSで透析し、その期間で液を2回入れ替え、2回の入れ替えの間隔が5時間以上であった。透析溶液を10000gで10分間遠心分離し、上清を0.22μmろ過膜でろ過してモノクローナル抗体溶液として保存した。上述の精製で、純度>95%の抗体を得た。
【0067】
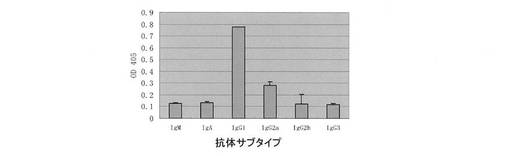
2.4 12H23モノクローナル抗体の亜型の同定
rEGFRvIIIexを被覆緩衝液(NaHCO3pH9.6)で1.0mg/Lに希釈し、ELISAマイクロプレートに各孔50μLずつ入れた。4℃で24時間被覆した。5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。初期濃度が1mg/Lの12H23モノクローナル抗体50μLを入れ、37℃で1時間結合させた。PBSで3回洗浄し、それぞれヒツジ抗マウス亜型ポリクローナル抗体(1:1000希釈)100μLを入れ、37℃で1時間結合させた。PBSで3回洗浄した。HRP標識のロバ抗ヒツジのポリクローナル抗体、37℃で1/2時間結合させ、PBSで5回洗浄した。ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。
結果は図6に示すように、この抗体12H23の亜型はIgG1型であった。
【0068】
実施例3 モノクローナル抗体の結合能力の測定
3.1 12H23の受容体結合特異性のFACS解析
ベクターの構築:
細胞:U87細胞(脳膠腫細胞系、EGFRが正常発現、ATCC細胞バンクから購入)、U87-EGFRvIII細胞(pLERNLベクターをトランスフェクションしたU87細胞系)およびA431細胞(人扁平上皮癌、EGFRが過剰発現、ATCC細胞バンクから購入)
抗体:実施例2で調製した抗体12H23、及び市販のC225モノクローナル抗体(対照として)。抗体濃度はいずれも2mg/mLで、1:100で希釈した。
【0069】
1)対数増殖期の細胞を約90%の接種細胞密度で6孔プレートに接種し、インキュベーターを用い37℃で一晩培養した。
2)次の日、10mMのEDTAで消化した細胞を使用し、5000rpm×3分間で遠心分離し、細胞を2mLのエッペンドルフチューブに収集した。
3)0.5〜1mlのPBSで細胞を再懸濁させ、4%パラホルムアルデヒドで細胞を固定し、37℃で10分間インキュベートした。
4)細胞を氷の上に置いて1分間冷却した。
【0070】
5)5000rpm×3分間で遠心分離し、上清を捨てた。
6)細胞を氷冷した90%メタノールに再懸濁させ、氷の上に30分間置いた。
7)5000rpm×3分間で遠心分離し、上清を捨てた。
8)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
【0071】
9)細胞をそれぞれ各EP管に0.5〜1×106細胞/管(各細胞は8管に分け、その中で、1管は空白細胞、2管は第二抗体のみ)入れた。
10)各管に0.5%BSA(PBSで調製)を入れ、室温で10分間ブロックした。
11)5000rpm×3分間で遠心分離し、ブロック液を捨てた。
【0072】
12)各細胞に相応の第一抗体100μL入れ、室温で30〜60分間インキュベートした。
13)遠心分離し、第一抗体インキュベート液を捨てた。
14)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
【0073】
15)各細胞にFITC標識のヒツジ抗マウス第二抗体(12H23)或いはロバ抗ヒト第二抗体(C225)100μL入れ、室温で30分間インキュベートした。
16)遠心分離し、第二抗体インキュベート液を捨てた。
【0074】
17)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
18)最後に、0.5〜1mLのPBSで細胞を再懸濁させ、フローサイトメトリー専用試験管に移した。
19)フローサイトメーターで解析した。
【0075】
結果:
図7A〜7Fに示すように、抗体12H23はEGFRvIIIを高発現する細胞系と効率的に結合することができ、EGFRを過剰発現するA431細胞とも一部結合したが、EGFRを正常発現する細胞系U87だけでほとんど結合しなかった。一方、製品化の抗体C225(Erbitux)はEGFRvIIIを高発現する細胞系だけでなく、EGFRを正常発現する細胞系U87とも結合できた。本発明の抗体12H23がより優れた特異性を有することがわかった。
【0076】
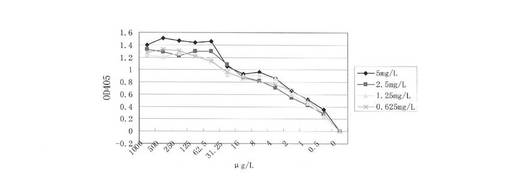
3.2 非競争法による12H23の親和力の測定
rEGFRvIIIexを被覆緩衝液(NaHCO3pH9.6)で5.0mg/L、2.5mg/L、1.25mg/Lおよび0.625mg/Lに希釈し、ELISAマイクロプレートに各孔100μLずつ入れた。4℃で24時間被覆した。被覆液を捨て、PBSで1回洗浄し、5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。異なる濃度の抗原を被覆したマイクロプレートに、初期濃度が1mg/Lの12H23モノクローナル抗体を入れ、勾配希釈(希釈液が5%脱脂粉乳を含有するPBS)で12級に希釈した。37℃で1時間結合させ、PBSで3回洗浄し、ヒツジ抗マウスの第二抗体100μLを入れ、37℃で1時間結合させた、PBSで5回洗浄し、ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。吸収係数の結果によって結合反応曲線を作り、作図法でその最高のOD値の半分(すなわち、OD:50%)の時の抗体濃度を得た。
【0077】
結果:
図8に示すように、12H23は、5.0mg/L被覆曲線では、OD50%抗体濃度が3μg/L(2×10-11mol/L)で、2.5mg/L被覆曲線では、OD50%抗体濃度が2.5μg/L(1.7×10-11mol/L)で、1.25mg/L被覆曲線では、OD50%抗体濃度が2.3μg/L(1.5×10-11mol/L)で、0.625mg/L被覆曲線では、OD50%抗体濃度が2μg/L(1.5×10-11mol/L)であった。(式)K=(n−1)/2(nAb′-Ab)によって親和定数を算出した。ここで、Ab′とAbはそれぞれ抗原濃度がAg′とAgの時OD50%抗体濃度になることを表し、n=Ag/Ag′である。そして、それぞれ比較すると、6つのK値を算出し、6つのK値の平均を最終の結果とした。算出した12H23の親和定数が3.8×10-10mol/Lで、解離定数Kd値が2.6×10-11mol/Lであった。
【0078】
実施例4:モノクローナル抗体の結合エピトープの解析
4.1 組換えタンパク質の調製:
通常の方法で、それぞれEGFRの細胞外領域のS1ドメイン、S2ドメインおよびS1ドメインにおけるVK21ポリペプチドとM13ファージのpIIIタンパクのN12タンパクドメインが融合したもの(1.1の方法と同じ方法で)を構築し、EGFRvIIIの細胞外領域の組換えタンパク質を陽性対照とした(図9)。
【0079】
電気泳動の結果から、各組換えタンパク質rN12-S1、rN12-S2、EGFRvIIIex、rN12-VK21が得られたことがわかった(図10)。
【0080】
4.2 ELISA法による12H23の結合エピトープの測定
組換えタンパク質rN12-S1、rN12-S2、EGFRvIIIex、rN12-VK21をそれぞれ被覆緩衝液(NaHCO3 pH9.6)で1.0mg/Lに希釈し、順にELISAマイクロプレートに各孔100μLずつ入れた。4℃で24時間被覆した。被覆液を捨て、PBSで1回洗浄し、5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。それぞれ初期濃度が1mg/Lの12H23モノクローナル抗体を入れ、37℃で1時間結合させ、PBSで3回洗浄した。更に、ヒツジ抗マウスの第二抗体100μLを入れ、37℃で1時間結合させた後、PBSで5回洗浄した。ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。
【0081】
結果:
ELISAの結果は図11に示すように、抗体12H23はEGFRvIII、rN12-S1およびrN12-VK21と結合することができた。構造および配列によれば、12H23が結合する領域はこれらのタンパク質の共有領域、すなわち、VK21であることが推測された。VK21のポリペプチド配列はVRACGADSYEMEEDGVRKCKK(SEQ ID NO:11)である。
【0082】
実施例5:モノクローナル抗体の生体内における腫瘍生長を抑制する能力
1)3×106個の通常のHuh7-EGFRvIII腫瘍細胞(pLRNLをトランスフェクションしたHuH-7肝臓癌細胞系、Huh-7は米国ATCC細胞バンクから購入)をそれぞれ18匹のBalb/cヌードマウスの右側肩甲の皮下に注射した。
【0083】
2)数日後、皮下の腫瘍体積が約80〜100mm3になったとき、それぞれC225抗体と12H23抗体を腹腔内注射した。1匹あたりの抗体注射量が0.5mgであり、同時にPBSを陰性対照として注射した。各群は6匹であった。
【0084】
3)その後、1日おきに抗体を腹腔内注射し、2週間続けた。
4)抗体注射と同時に、腫瘍の体積を1日おきに測定し、抗体注射が終わった後、腫瘍の体積の測定を2週間続けた。腫瘍の体積は(式)腫瘍の体積=腫瘍の長さ×腫瘍の幅2/2で算出した。
5)腫瘍の生長状況を観察した。
【0085】
結果:
12H23はHuh7-EGFRvIIIのヌードマウスの播種腫瘍に顕著な抑制作用があり(約70%)、且つその抑制率もC225抗体より優れていた(図12)。
【0086】
実施例6 モノクローナル抗体の配列決定
5'RACE法で5'配列が未知の遺伝子を大体以下のようにクローンした(具体的な操作はTakara 5'-full RACE Kitの説明書に従った)。
【0087】
1)アルカリホスファターゼ(CIAP)で全RNAにおける露出した5'リン酸基を脱リン酸反応を行った。全RNAの使用量は2μgで、反応後フェノール/クロロホルムで抽出して回収した。
2)タバコ酸性ピロホスファターゼ(Tobacco Acid Pyrophosphatase、TAP)でmRNAの5'キャップ構造を除去し、一つのリン酸基を残した。
【0088】
3)T4 RNAリガーゼで5'RACEアダプター(Adaptor)をmRNAに連結し、反応後フェノール/クロロホルムで抽出して回収した。
4)逆転写酵素で逆転写反応を行った。使用されたプライマーはキットで提供されたランダムの9量体のプライマーであった。
【0089】
5)逆転写産物を鋳型として、高忠実度の酵素で目的遺伝子を増幅した。使用されたプライマーは以下の通りである。
5':5'RACE Outer Primer(CATGGCTACATGCTGACAGCCTA) (SEQ ID NO: 12)
3':重鎖:CCAGAGTTCCAGGTCACTGTCACT (SEQ ID NO: 13)
軽鎖:ACACGACTGAGGCACCTCCA (SEQ ID NO: 14)
【0090】
6)上述PCR産物を鋳型として、ネステッドPCRを行った。使用されたプライマーは以下の通りである。
5':5'RACE Inner Primer(CGCGGATCCACAGCCTACTGATGATCAGTCGATG) (SEQ ID NO: 15)
3':重鎖:CCAGGGTCACCATGGAGTTAGTTT (SEQ ID NO: 16)
軽鎖:TGGATGGTGGGAAGATGGATACA (SEQ ID NO: 17)
7)TAクローンを行い、配列を測定した。
【0091】
配列決定の結果
12H23モノクローナル抗体の重鎖、軽鎖および各CDRの配列は図13〜14および下記表1に示す。
【0092】
【表1】

【0093】
実施例7
1.抗体可変領域コード配列を含有するヒト-マウスキメラ抗体の発現ベクターのクローニング
それぞれhCMVプロモーター、重鎖可変領域のクローン部位NheIとApaIおよびヒトIgG1の重鎖定常領域、IRESリボソーム挿入部位、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)及びアンピシリン耐性遺伝子を含む抗体の重鎖の発現ベクターpHを構築した(図15Aを参照)。
【0094】
それぞれhCMVプロモーター、軽鎖可変領域のクローン部位EcoRVとBsiWIおよびヒトIgG1の軽鎖定常領域、IRESリボソーム挿入部位、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)及びアンピシリン耐性遺伝子を含む抗体の軽鎖の発現ベクターpKを構築した(図15Bを参照)。
【0095】
実施例6で測定した軽鎖および重鎖の配列に基づき、重鎖および軽鎖の可変領域コード配列を人工合成し、重鎖コード配列の両末端にNheIとApaIの酵素切断部位を、軽鎖コード配列の両末端にEcoRVとBsiWIの酵素切断部位をつけた。NheIとApaIの酵素で重鎖可変領域コード配列を、EcoRVとBsiWIの酵素で軽鎖可変領域コード配列を切断した。
【0096】
その後、上述重鎖および軽鎖の可変領域コード配列を発現ベクターpHおよびpKに挿入し、キメラの抗EGFRvIIIの抗体遺伝子の発現ベクターを構築した。
【0097】
2.CHO細胞のトランスフェクション及び組換えクローンのスクリーニング
上述構築された抗体遺伝子をもつ発現ベクターを大腸菌DH5α菌株に導入し、そして100mLのLB培地に接種して増幅させ、Qiagen社の高純度プラスミドDNA精製キット(Ultrapure Plasmid DNA Purification Kit)でプラスミドDNAを抽出、精製した。上述精製されたプラスミドDNAをInvitrogen社のリプソーム法キットでCHO細胞にトランスフェクションし、操作はメーカーの説明書を参照して行った。
【0098】
形質変換されたCHO細胞を濃度が高くなっていくMTX選択培地で培養を9週間続け、最後は96孔プレートで勾配希釈培養を行い、連続して3回を行い、モノクローナル化した。
【0099】
選択されたモノクローナル細胞系はRPM1640培地で培養し、上清にELISA実験をし、発色反応で発現結合強度を判断した。測定によってこれらのクローンもrEGFRvIII抗原と特異的に結合する活性を有し(図16を参照)、幾つかの発現の高いクローンを細胞株の候補として選択し、キメラ抗体を調製した。得られた抗体をCH12と呼ぶ。
【0100】
実施例8
抱合体の調製
キメラモノクローナル抗体CH12を直接ジフテリア毒素(武漢生物製品研究所から購入)と共役結合で結合させた。Huh7-EGFRvIIIの細胞に結合物を入れると、特異的な細胞毒性が観察される。EGFRvIIIを発現しないHuh7細胞は、非常に高濃度の抗体に曝されるときだけ殺傷される。
【0101】
実施例9
注射液の調製
実施例5で調製された抗体12H23或いは実施例7で調製されたキメラモノクローナル抗体CH12を注射用生理食塩水に入れ、均一に混合した後、0.22uMの無菌フィルターで除菌し、注射液を小瓶に分け(50mL/瓶)、包装して使用に供する。ここで、50mLあたりの注射液にモノクローナル抗体50mgが含有される。
【0102】
実施例10
キメラ抗体CH12とマウス由来のモノクローナル抗体12H23の競争結合実験
ELISA法によって、HRP標識のCH12抗体(1.0μg/mL)を固定し、さらに異なる濃度の未標識CH12抗体或いは12H23抗体(0、3、9、27、81、243、729μg/mL)を入れた。
【0103】
結果から、競争するCH12或いは12H23抗体濃度の増加に従い、OD405の値が低くなっていくことがわかった。しかも、二種の抗体の異なる濃度における競争抑制率がほぼ同じであることがわかった。キメラ抗体CH12とマウス由来のモノクローナル抗体12H23は、抗原に対する親和力が同等程度で、且つ結合部位も一致することが示唆される。
【0104】
実施例11
キメラ抗体CH12の細胞免疫組織化学的検出
通常の免疫蛍光の方法によってCH12キメラ抗体とHuh-7細胞、Huh7-EGFRおよびHuh7-EGFRvIIIの結合状況を検出した。
【0105】
結果から、CH12抗体は顕著にHuh7-EGFRvIIIと結合することができ、またEGFRを増幅的に発現するHuh7-EGFRとも結合することができるが、Huh-7細胞とほとんど結合しないことがわかった。キメラ改造後、CH12抗体の細胞に対する結合能力及び特異性は変わらないことが示唆される。
【0106】
実施例12
CH12キメラ抗体の生体内における腫瘍生長を抑制する能力
1)Huh7-EGFRvIII腫瘍細胞或いはSMMC-7721肝臓癌細胞系(中科院細胞バンクから購入)をそれぞれBalb/cヌードマウスの右側肩甲の皮下に注射した。1匹あたりのマウスの播種細胞量は3×106 個であった。(注:SMMC-7721細胞が肝臓癌細胞系で、本発明者らは実験によって内因的にEGFRvIIIが存在することを証明した。)
【0107】
2)数日後、皮下の腫瘍体積が約150mm3になったとき、それぞれC225抗体、CH806抗体或いはCH12抗体を腹腔内注射した。1匹あたりの抗体注射量が0.5mgであり、同時にPBSを陰性対照として注射した。各群は6匹であった。
【0108】
3)毎週3回給与し、2週間続けた。
4)抗体注射と同時に、腫瘍の体積を測定し、抗体注射が終わった後腫瘍の体積の測定を2週間続けた。腫瘍の体積は、(式)腫瘍の体積=腫瘍の長さ×腫瘍の幅2/2で算出した。
5)腫瘍の生長状況を観察した。
【0109】
結果(図17)から、CH12モノクローナル抗体は、効率的にSMMC-7721の播種腫瘍のヌードマウスの体内における生長を抑制することができ、給与後25日(腫瘍細胞を接種後41日)のときの腫瘍抑制率が62.94%で、一方、対照抗体C225とCH806の腫瘍抑制率がそれぞれ25.97%と33.11%であることがわかった。
【0110】
CH12処理群では、腫瘍接種後27日はPBS対照群と比べて顕著な差(p<0.05)があり、腫瘍接種後32日はC225処理群と比べて顕著な差(p<0.05)があり、腫瘍接種後37日はCH806処理群と比べて顕著な差(p<0.05)があった。CH12はSMMC-7721肝臓癌を治療する場合C225とCH806よりも優れることが示唆される。
【0111】
また、CH12はHuh7-EGFRvIII播種腫瘍のヌードマウスの体内における生長を抑制することもでき、PBS群(p=0.0001)とC225治療群(抑制率32.9%だけ)よりも優れた。
【0112】
各文献がそれぞれ単独に引用されるように、本発明に係るすべての文献は本出願で参考として引用する。また、本発明の上記の内容を読み終わった後、この分野の技術者が本発明を各種の変動や修正をすることができるが、それらの等価の様態のものは本発明の請求の範囲に含まれることが理解されるべきである。
【0113】
参考文献
1) Ullrich A.ら、 Human Epidermal Growth Factor Receptor cDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature, 1984, 309: 418-425
2) Downwardら、 Close similarity of Epidermal Growth Factor Receptor and v-erb B oncogene Protein Sequence. Nature 1984, 307:521-527
3) Libermannら、 Amplification,Enhanced Expression and Possible Rearrangement of EGF Receptor Gene in Primary Human Brain Tumors of Glial Origin. Nature 1985,313:144-147
【0114】
4) Wongら、 Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas is Invariably Associated with Gene Amplification. Proc. Natl. Acad. Sci. USA. 1987, 84:6899-6903
5) Yamazakiら、 Amplification of the Structurally and Functionally Altered Epidermal Growth Factor Receptor Gene (c-erbB) in Human Brain Tumors. Molecular and Cellular Biology 1988,8:1816-1820
6) Maidenら、 Selective Amplification of the Cytoplasmic Domain of the Epidermal Growth Factor Receptor Gene in Glioblastoma Multifome. Cancer Research 1988(4):2711-2714
【0115】
7) Modjtahedi H, and Dean C. The receptor for EGF and its ligands Expression, prognostic value and target for therapy in cancer. International Journal of Oncology 1994 (4): 277-296
8) Fung YKT,ら、 Activation of the Cellular Oncogene c-erb B by LTR Insertion: Molecular Basis for Induction of Erythroblastosis by Avian Leukosis Virus. Cell 1983, 33:357-368
9) Yamamotoら、 A New Avian Erythroblastosis Virus, AEV-H Carries erbB Gene Responsible for the Induction of Both Erythroblastosis and Sarcoma. Cell 1983,34:225-232
【0116】
10) Nilsenら、 c-erbB Activation in ALV-Induced Erythroblastosis: Novel RNA Processing and Promoter Insertion Results in Expression of an Amino-Truncated EGF Receptor. Cell 1985, 41: 719-726
11) Gammettら、 Differences in Sequences Encoding the Carboxy-Terminal Domain of the Epidermal Growth Factor Receptor Correlate with Differences in the Disease Potential of Viral erbB Genes.”Proc. Natl. Acad. Sci. USA 83:6053-6057(1986)
12) Gilmoreら、 Protein Phosphorylation at Tyrosine is Induced by the v-erb B Gene Product in Vivo and in Vitro. Cell, 1985,40:609-618, (1985)
【0117】
13) Krisら、 Antibodies Against a Synthetic Peptide as a Probe for the Kinase Activity of the Avian EGF Receptor and v-erbB Protein. Cell, 40:619-625(1985)
14) Nilsenら、 c-erbB Activation in ALV-Induced Erythroblastosis: Novel RNA Processing and Promoter Insertion Results in Expression of an Amino-Truncated EGR Receptor. Cell, 1985,41:719-726
15) Rainesら、 c-erbB Activation in Avian Leukosis Virus-Induced Erythroblastosis: Clustered Integration Sites and the Arrangement of Provirus in the c-erbB Alleles. Proc. Natl. Acad. Sci. USA,1985, 82:2287-2291
【0118】
16) Pelleyら、 Proviral-Activated c-erbB is Leukemogenic but not Sarcomagenic: Characterization of a Replication-Competent Retrovirus Containing the Activated c-erbB. Journal of Virology 1988, 62: 1840-1844
17) Wellsら、 Genetic Determinant of Neoplastic Transformation by the Retroviral Oncogene v-erbB. Proc. Natl. Acad. Sci. USA 1988, 85:7597-7601
18) Yamazakiら、 Amplification, Enhanced Expression and Possible Rearrangement of EGF Receptor Gene in Primary Human Brain Tumours of Glial Origin. Nature 1985, 313:144-147
【0119】
19) Wikstrand CJ,ら、 Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas malignant gliomas. Cancer Research 1995, 55(14):3140-3148
20) Olapade-Olaopa EO,ら、 Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer. 2000, 82(1):186-94
21) Ge H,ら Evidence of High incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J cancer. 2002, 98(3):357-61
【0120】
22) Moscatello G.,ら、 Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 55(23):5536-9(1,995)
23) Garcia de Palazzo, IE.,ら、 Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993, 53(14):3217-20
24) Moscatello,G. et al,Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer. 2000, 82(1):186-94
【0121】
25) Luo XY,et al. Suppression of EGFRvIII-Mediated Proliferation and Tumorigenesis of Breast Cancer Cells by Ribozyme. Int. J. Cancer. 2003, 104(6):716-21
26) Kuan CT,et al. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001, 8(2):83-96
27) Scott A,ら、 A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. PNAS 2007, 104(10): 4071-4076
【技術分野】
【0001】
本発明は、医学分野に関する。具体的に、抗上皮成長因子受容体変異体III(EGFRvIII)特異的モノクローナル抗体およびその応用に関する。本発明の抗体は、EGFRvIIIと効率的結合し、或いは細胞が過剰発現した上皮成長因子受容体(EGFR)と部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。本発明の抗体は、EGFRvIIIを発現する腫瘍細胞系の治療に有用である。
【背景技術】
【0002】
上皮成長因子受容体(EGFR)は癌原遺伝子c-erbBの170kDaの膜糖タンパク質の産物である(1)。EGFR遺伝子は、最初、トリ赤芽球症ウイルスで同定されたerbB癌遺伝子の細胞類縁物である(1,2)。各種のヒト腫瘍において、既にこの癌遺伝子の遺伝子増幅による活性化が見られた(3-6)。
【0003】
EGFRが多くの種類のヒト固形腫瘍で過剰発現されることが既に文献で明らかになった(7)。このような腫瘍は、肺癌、大腸癌、乳癌、胃癌、脳癌、膀胱癌、頭頸部腫瘍、卵巣癌、腎癌および前立腺癌を含む(7)。v-erbB癌遺伝子と正常のEGFR遺伝子との違いは、主にウイルスの癌遺伝子が正常の受容体の切断アミノ基の変異型であることにある。細胞質外ドメインの多くが欠損したが、膜貫通およびチロシンキナーゼのドメインが残った(8-11)。よって、上皮成長因子(EGF)と結合することができないが、他のタンパク質をリン酸化させることができる(14-15)。
【0004】
ウイルス性erbB癌遺伝子には色々な遺伝子の変化、例えば、遺伝子のカルボキシ末端におけるアミノ酸の置換や欠失が起こる可能性がある。なかでも、アミノ酸の欠失は、癌誘発作用において重要である。アミノ酸の欠失は大部分のv-erbB癌遺伝子の特徴の一つで、プロモーターの挿入やレトロウイルスの形質導入によるアミノ末端の欠失を含む(13,16)。逆に、カルボキシ末端の欠失はレトロウイルスの形質導入による腫瘍だけに関与し、宿主の範囲および腫瘍の種類の特異性で決まるとされている(11,15)。アミノ末端の欠失のトリc-erbB遺伝子又はヒトEGF受容体のトランスフェクション実験によって、その欠失は細胞を転換させることができることがわかった(16-17)。
【0005】
EGFR遺伝子の増幅は40%のヒト悪性神経膠腫に発生し(3,7)、受容体の遺伝子の組換えが多くの遺伝子増幅の腫瘍で顕著である。組換えは遺伝子のアミノ末端に影響することが多い(6,18)。
【0006】
今まで、8種類のEGFR変異体が発見された(図5)。1種類目は、EGFRvIはEGFRの大半の細胞外ドメインが欠失したものである。2種類目は、EGFRvIIはEGFRの細胞外ドメインにおける83aaのインフレーム欠失からなるものである。3種類目は、EGFRvIIIはEGFRの細胞外ドメインにおける267aaのインフレーム欠失からなるものである。4種類目は、EGFRvIVはEGFRの細胞質ドメインにおける欠失を含むものである。5種類目は、EGFRvVはEGFRの細胞質ドメインにおける欠失を含むものである。6種類目はEGFR.TDM/2〜7はEGFRの細胞外ドメインのエクソン2〜7の重複を含むものである。7種類目はEGFR.TDM/18〜26はEGFRの細胞外ドメインのエクソン18〜26の重複を含むものである。8種類目としては、さらに、エクソン11と14の連結部に新規なヒスチジン残基を導入した、より珍しい第2種のEGFRvIII変異体(EGFRvIII/Δ12〜13)が存在する(24)。
【0007】
EGFRvIIIは、ヒトの癌における上皮成長因子(EGF)受容体の最も一般的に存在する変異体である(24)。遺伝子増幅の過程において、細胞外ドメインで267のアミノ酸欠失が起こり、新しい連結部(グリシン)が形成される。EGFRvIIIは、正常の組織に発現されないことが知られている(19,20)。しかしながら、EGFRvIIIは、多くの腫瘍細胞で発現され、例えば27〜76%の乳癌バイオプシーに(21)、50〜70%の神経膠腫に(19,22)、16%の非小細胞肺癌に(23)、75%の卵巣癌に(22)、EGFRvIIIが発現される。
【0008】
一つのEGFRvIIIが過剰発現する癌を治療する方法では、特異的に未分離の正常EGFRの変異体の受容体をターゲットとする腫瘍特異的リボザイムに関する。胸腺欠損ヌードマウスでは、リボザイムは顕著に乳癌の成長を抑制することがわかった。
【0009】
また、267のアミノ酸が欠失してグリシンに置換した特有の連結は抗EGFRvIII特異的モノクローナル抗体の製造に有用である。また、EGFRvIIIは、ある腫瘍で発現され、かつ正常の組織での発現がないことによって、腫瘍の薬物の理想的なターゲットである。具体的に、EGFRvIIIは、腫瘍の免疫抱合体による治療の理想的な候補として有用である。抗EGFRvIII特異的モノクローナル抗体(或いは抗がん剤又は毒素結合の抱合体)は、体内において抗体に依存する細胞溶解又は細胞致死作用を起こすことで、EGFRvIIIを発現する腫瘍細胞を除去する。
【0010】
現在、国内外では複種の抗EGFR抗原の抗体が得られたが、これらの抗体は、例えばEGFRvIIIに対する特異性がない又は低いなどと、満足できるとはいえない。
【0011】
従って、本分野では、EGFRvIIIを認識する特異性が高く、かつ野生型EGFRを認識しない、他の優れた特性を持つ抗EGFRvIIIモノクローナル抗体を開発することによって、治療効果がより顕著な薬物を開発することが切望されている。
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の目的は、特異的抗EGFRvIIIモノクローナル抗体を提供することにある。また、本発明の目的は、前述特異的抗EGFRvIIIモノクローナル抗体の製造方法を提供することにある。
【0013】
さらに、本発明の目的は、前述特異的抗EGFRvIIIモノクローナル抗体を含む薬物組成物を提供することにある。
【課題を解決するための手段】
【0014】
本発明の第一は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有するモノクローナル抗体のVH鎖を提供する。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
他の好ましい例において、前述のVH鎖はSEQ ID NO:2で示されるアミノ酸配列を有する。
【0015】
本発明の第二は、相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有するモノクローナル抗体のVL鎖を提供する。
SEQ ID NO:8で示されるCDR1、
SEQ ID NO:9で示されるCDR2、および
SEQ ID NO:10で示されるCDR3。
他の好ましい例において、前述のVL鎖はSEQ ID NO:4で示されるアミノ酸配列を有する。
【0016】
本発明の第三は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有するモノクローナル抗体或いはその抱合体を提供する。
【0017】
他の好ましい例において、前述の抗体はヒト上皮成長因子受容体変異体(EGFRvIII)と結合し、かつ細胞が過剰発現するEGFRと部分的に結合するが、細胞が正常発現するEGFRとの結合作用がない。
【0018】
より好ましくは、前述の抗体はA431細胞およびU87-EGFRvIII細胞と結合するが、U87細胞と結合しない。
【0019】
他の好ましい例において、前述の抗体はマウス抗体、ヒト化抗体或いはキメラ抗体である。
【0020】
他の好ましい例において、前述の抱合体は、抗体と抗がん剤或いは毒素(例えば、ジフテリア毒素、リシン、緑膿菌外毒素)の抱合体である。
【0021】
本発明の第四は、以下の群から選ばれるタンパク質をコードする核酸分子(例えばDNA分子)を提供する。
本発明の第一に記載のモノクローナル抗体のVH鎖、
本発明の第二に記載のモノクローナル抗体のVL鎖、
本発明の第三に記載のモノクローナル抗体。
他の好ましい例において、前述の核酸分子はSEQ ID NO:1、3、11或いは13から選ばれるDNA配列を有する。
【0022】
本発明の第五は、VH鎖がSEQ ID NO:5〜7で示される相補性決定領域を有し、かつVL鎖がSEQ ID NO:8〜10で示される相補性決定領域を有するモノクローナル抗体と薬学的に許容される担体とを含む薬物組成物を提供する。
他の好ましい例において、前述モノクローナル抗体は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有する。
【0023】
本発明の第六は、(a)上皮成長因子受容体変異体IIIを発現する細胞の成長の抑制又は致死、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長の抑制に用いられる組成物を製造するための本発明のモノクローナル抗体或いはその抱合体の用途を提供する。
他の好ましい例において、前述の細胞は、腫瘍細胞、例えば、肝癌細胞、肺癌細胞である。
【0024】
本発明の第七は、治療しようとする対象に本発明のモノクローナル抗体或いはその抱合体を給与することを含む、(a)上皮成長因子受容体変異体IIIを発現する細胞の成長抑制又は致死方法、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長抑制方法を提供する。
【0025】
好ましくは、前述対象は、ヒト、マウス、ラットなどを含む哺乳動物である。
【発明の効果】
【0026】
本発明のモノクローナル抗体の特異性および生理活性が顕著に向上する。このモノクローナル抗体は、EGFRvIIIと効率的に結合し、かつ細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。
【0027】
本発明のモノクローナル抗体の親和力、腫瘍抑制率はいずれも既存の抗体(例えばCH806抗体)よりも高く、かつ抗体のアミノ酸配列の構成(特にCDR領域)も違う。
よって、本発明の高親和力、高特異性のモノクローナル抗体は臨床では重要な価値がある。
【図面の簡単な説明】
【0028】
【図1】組換えプラスミドpET28a-EGFRvIIIexがBglII和SalIの酵素切断による同定を示すものである。ただし、レーン1〜4はプラスミドが二つの酵素によって切断されたもので、MはDNA分子量マーカーλHindIIIである。
【図2】EGFRvIIIの細胞外ドメインのタンパクを精製した結果を示すものである。レーン1、11はタンパク質の分子量マーカー、レーン2は未誘導の細菌沈殿、レーン3は流出液、レーン4〜6は緩衝液Cの溶離液、レーン7〜10は緩衝液Dの溶離液、レーン12〜16は緩衝液Eの溶離液である。
【図3】復元したタンパク質のSDS-PAGEのグラフを示すものである。ただし、レーン1はタンパク質の分子量マーカー、レーン2は復元したタンパク質である。
【図4】復元したタンパク質のウェスタンブロットを示すものである。ただし、レーン1は復元したタンパク質、レーン2はBL21(DE3)-RP菌液の全タンパク質である。
【図5】野生型EGFRおよびその各種の変異体を示すものである。
【図6】12H23抗体サブタイプのELISA分析を示すものである。
【図7】本発明の抗体12H23および対照抗体C225がそれぞれA431細胞(EGFR過剰発現)、U87-EGFRvIII細胞(EGFRvIIIの安定な高発現)およびU87細胞(EGFR正常発現)とのフローサイトメトリー図を示すものである。ただし、各図は以下の通りである。A:C225とA431細胞の結合図、B:12H23とA431細胞の結合図、C:C225とU87-EGFRvIII細胞系の結合図、D:12H23とU87-EGFRvIII細胞系の結合図、E:C225とU87細胞の結合図、F:12H23とU87細胞の結合図。
【図8】12H23と抗原rEGFRvIIIex細胞の親和力の測定を示すものである。
【図9】各組換えタンパク質の配列構造を示す概略図である。
【図10】組換えタンパク質のSDS-PAGE分析を示すものである。ただし、レーンMはタンパク質のマーカー、1はrN12-S1、2はrN12-S2、3はrEGFRvIIIex、4はrN12-VK21である。
【図11】ELISA法で12H23の結合エピトープを測定した結果を示すものである。ただし、S1はrN12-S1、S2はrN12-S2、EGFRvIIIは組換えEGFRvIII細胞外タンパク質、VK21はrN12-VK21、Negは空白対照である。
【図12】12H23モノクローナル抗体のヌードマウスの播種腫瘍に対する抑制作用を示すものである。
【図13】12H23モノクローナル抗体の重鎖のヌクレオチドコード配列およびアミノ酸配列(下線はCDR)を示すものである。
【図14】12H23モノクローナル抗体の軽鎖のヌクレオチドコード配列およびアミノ酸配列(下線はCDR)を示すものである。
【図15】プラスミドpHおよびpKを示す概略図である。
【図16】ヒト-マウスキメラ抗体CH12と組換えEGFRvIII細胞外タンパク質との結合を示すものである。図中において、1〜5はそれぞれCH12を発現する各細胞クローン株を示す。
【図17】キメラ体CH12のヌードマウスの播種腫瘍に対する抑制作用を示すものである。
【発明を実施するための形態】
【0029】
発明者らは、幅広く深く研究したところ、EGFRvIIIと効率的に結合し、或いは細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない、EGFRvIIIに対する特異性が高いモノクローナル抗体の製造に成功した。また、本発明の抗体はEGFRvIIIを発現する腫瘍細胞系に対する治療作用が顕著である。これに基づき、本発明を完成させた。
【0030】
本発明は組換え抗EGFRvIIIモノクローナル抗体を提供する。前述の抗体は、マウス抗体、ヒト化抗体或いはキメラ抗体でもよい。例えば、ヒト化抗体はヒト定常領域(例えばヒト定常領域IgG1-Fc)、本発明の重鎖可変領域および軽鎖可変領域を含む。
【0031】
また、本発明は、抗EGFRvIIIモノクローナル抗体のアミノ酸配列およびその可変領域鎖、並びにこれらの鎖を有する他のタンパク質或いは融合発現産物を提供する。具体的に、本発明は、超可変領域(相補性決定領域、CDR)が本発明の軽鎖及び重鎖の超可変領域と同様で、或いは少なくとも90%、好ましくは95%の相同性を持つものであれば、この超可変領域を含有する軽鎖及び重鎖を有する任意のタンパク質又はタンパク質抱合体と融合発現産物(すなわち、免疫抱合体と融合発現産物)を含む。
【0032】
抗体の抗原結合特性は、重鎖および軽鎖可変領域に位置する3つの特定の領域によって特徴付けられ、超可変領域(CDR)と呼ばれる。且つ、該領域は4つのフレームワーク領域(FR)に分かれ、4つのFRのアミノ酸配列が比較的に保守的で、直接結合反応に関与しない。これらのCDRは環状構造を形成し、その中のFRで形成されるβシートによって空間構造上で近づき、重鎖におけるCDRおよび相応の軽鎖におけるCDRが抗体の抗原結合部位を構成する。同類の抗体のアミノ酸配列の比較によってどのアミノ酸がFR或いはCDR領域を構成するか確認することが出来る。
【0033】
また、最近、軽鎖可変領域から構成する関連構造は、相応の重鎖可変領域と比べ、結合の動力が小さいため、単離の重鎖可変領域自身が抗原結合活性を持つことがわかった。
【0034】
ここで同定されるV鎖の超可変領域或いは相補性決定領域(complementarity determining region、CDR)は、その少なくとも一部が抗原結合に関与することで、注目されている。そのため、CDR含有モノクローナル抗体の軽鎖および重鎖可変領域鎖を有する分子は、そのCDRはここで同定されるCDRと90%以上(好ましくは95%以上、最も好ましくは98%以上)の相同性を持つものであれば、本発明に含まれる。
【0035】
本発明は、完全のモノクローナル抗体だけではなく、免疫活性を有するFab又は(Fab')2断片のような抗体断片、抗体の重鎖、抗体の軽鎖、遺伝子工学によって改造された一本鎖Fv分子やマウス抗体結合特異性を持つがヒト由来の抗体部分が残った抗体のようなキメラ抗体も含む。
【0036】
さらに、本発明は、上述のモノクローナル抗体又はその断片をコードするDNA分子を提供する。本発明のモノクローナル抗体のヌクレオチド全長配列或いはその断片は、通常、PCR増幅法、組換え法又は人工合成の方法で得られる。適用できる方法として、特に断片の長さが短い場合、人工合成の方法で関連配列を合成する。通常、まず多数の小さい断片を合成し、そして連接させることにより、配列の長い断片を得ることができる。また、軽鎖及び重鎖のコード配列を一体に融合し、一本鎖抗体を形成してもよい。
【0037】
関連の配列を獲得すれば、組換え法で大量に関連配列を獲得することができる。この場合、通常、その配列をベクターにクローンした後、細胞に導入し、さらに通常の方法で増殖させた宿主細胞から関連配列を分離して得る。
【0038】
現在、本発明のタンパク質(又はその断片、或いはその誘導体)をコードするDNA配列を全部化学合成で獲得することがすでに可能である。さらに、このDNA配列を本分野で周知の各種の既知のDNA分子(或いはベクターなど)や細胞に導入してもよい。また、化学合成で本発明のタンパク質配列に変異を導入することもできる。
【0039】
さらに、本発明は、上述の適当なDNA配列及び適当なプロモーター或いは制御配列を含むベクターに関する。これらのベクターは、タンパク質を発現するように、適当な宿主細胞の形質転換に用いることができる。
【0040】
宿主細胞は、原核細胞、例えば細菌細胞、或いは、低等真核細胞、例えば酵母細胞、或いは、高等真核細胞、例えば哺乳動物細胞でもよい。代表例として、大腸菌、ストレプトマイセス属、ネズミチフス菌のような細菌細胞、酵母のような真菌細胞、ミバエS2若しくはSf9のような昆虫細胞、CHO、COS7、293細胞のような動物細胞などがある。
【0041】
DNA組換えによる宿主細胞の転換は当業者に熟知の通常の技術で行っても良い。宿主が原核細胞、例えば大腸菌である場合、DNAを吸収できるコンピテントセルは指数成長期後収集でき、CaCl2法で処理し、用いられる工程は本分野では周知のものである。もう一つの方法は、MgCl2を使用する。必要により、転化はエレクトロポレーションの方法を用いてもよい。宿主が真核生物の場合、リン酸カルシウム沈殿法、マイクロインジェクション、エレクトロポレーションのような通常の機械方法、リポフェクションなどのDNAトランスフェクションの方法が用いられる。
【0042】
得られる形質転換体は通常の方法で培養し、本発明の遺伝子がコードするポリペプチドを発現することが出来る。用いられる宿主細胞によって、培養に用いられる培地は通常の培地を選んでも良い。宿主細胞の成長に適する条件で培養する。宿主細胞が適当の細胞密度に成長したら、適切な方法(例えば温度転換もしくは化学誘導)で選んだプロモーターを誘導し、さらに、細胞を所定の時間培養する。
【0043】
上述の方法における組換えポリペプチドは細胞内または細胞膜で発現し、或いは細胞外に分泌することが出来る。必要であれば、その物理・化学的特性及び他の特性を利用して各種の分離方法で組換えタンパク質を分離・精製することができる。これらの方法は、本分野の当業者に熟知されている。これらの方法の例として、通用の再生処理、タンパク質沈殿剤による処理(塩析法)、遠心分離、浸透圧ショック、超音波処理、超遠心分離、分子篩クロマトグラフィー(ゲルろ過)、吸着クロマトグラフィー、イオン交換クロマトグラフィー、高速液体クロマトグラフィー(HPLC)及び他の各種の液体クロマトグラフィー技術、並びにこれらの方法の組合せを含むが、これらに限定されない。
【0044】
さらに、本発明は組成物を提供する。好ましい例において、前述の組成物が薬物組成物で、上述のモノクローナル抗体或いは免疫抱合体、および薬学的に許容される担体を含む。通常、これらの物質を、無毒で不活性であり薬学的に許容される水系担体で配合し、pH値は配合される物質の性質および治療しようとする疾患にもよるが、通常5〜8程度、好ましくは6〜8程度である。配合された薬物組成物は通常の経路で給与することができ、腫瘍内、腹膜内、静脈内、或いは局部給与が含まれるが、これらに限られない。
【0045】
本発明の薬物組成物は直接腫瘍の予防および治療に用いられる。また、他の治療剤と併用してもよい。
【0046】
本発明の薬物組成物は、安全有効量(例えば0.001〜99wt%、好ましくは0.01〜90wt%、より好ましくは0.1〜80wt%)の本発明の上述のモノクローナル抗体(或いはその抱合体)および薬学的に許容される担体或いは賦形剤を含む。このような担体は、食塩水、緩衝液、ブドウ糖、水、グリセリン、エタノール、及びその組合せを含むが、これらに限定されない。薬物の製剤は給与様態に相応しなければならない。本発明の薬物組成物は、注射剤としてもよく、例えば生理食塩水或いはグルコースおよび他の助剤を含む水溶液を使用し通常の方法で製造することができる。薬物組成物は、注射剤、溶液の場合、無菌条件で製造する。活性成分の給与量は治療有効量、例えば、毎日約1μg/kg体重〜約5mg/kg体重である。また、本発明のポリペプチドは他の治療剤と併用することが出来る。
【0047】
薬物組成物の使用時、安全有効量の免疫抱合体を哺乳動物に使用するが、その安全有効量は、通常、少なくとも約10μg/kg体重で、多くの場合に、約8mg/kg体重未満であり、好ましくはこの投与量が約10μg/kg体重〜約1mg/kg体重である。勿論、具体的な投与量は、さらに投与の様態、患者の健康状況などの要素を考えるべきで、すべて熟練の医者の技能範囲以内である。
【0048】
本発明の利点は以下の通りである。
(a)本発明のモノクローナル抗体の特異性および生理活性が顕著に向上する。このモノクローナル抗体は、EGFRvIIIと効率的に結合し、かつ細胞が過剰発現したEGFRと部分的に結合するが、細胞が正常に発現したEGFRとの結合作用がない。
(b)本発明のモノクローナル抗体の親和力、腫瘍抑制率はいずれも既存の抗体(例えばCH806抗体)よりも高く、かつ抗体のアミノ酸配列の構成(特にCDR領域)も違う。
よって、本発明の高親和力、高特異性のモノクローナル抗体は臨床では重要な価値がある。
【実施例】
【0049】
以下、具体的な実施例によって、さらに本発明を説明する。これらの実施例は本発明を説明するために用いられるものだけで、本発明の範囲の制限にはならないと理解されるものである。
【0050】
下述の実施例で具体的な条件が示されていない実験方法は、通常、例えばSambrookらによる、「モレキュラー・クローニング:研究室マニュアル」(ニューヨーク、コールド・スプリング・ハーバー研究所出版社、1989)に記載の条件などの通常の条件に、或いは、メーカー推薦の条件に従う。特に断らない限り、%と部は、重量で計算される。
【0051】
実施例1 抗原の製造
1.1 EGFRvIIIタンパク質の細胞外ドメインの原核発現および精製
1.1.1 ベクターの構築および同定
pLNRNL(コード全長EGFRvIII、Ludwig Instituteから購入, San Diego, CA)を鋳型とし、PCR法で両末端にBamHIおよびSalIの切断部位を有するEGFRvIIIex増幅産物を得、BamHIおよびSalIで同時切断し、目的の断片を得た。BglIIとSalIで市販のベクターpET28a(Novagen社から購入)を切断し、アガロースゲル電気泳動後、目的の断片を回収し、T4リガーゼの作用によって連結し、ベクターpET28a-EGFRvIIIexを形成した。さらに市販の大腸菌TOP10(Invitrogenから購入)に導入し、カナマイシン耐性でスクリーンし、BglII和SalIによる切断により、挿入断片を含有する陽性クローンと同定された。
【0052】
1.1.2 発現細菌のスクリーニング
正確と同定された組換えプラスミドでそれぞれ通常の大腸菌Bl21(DE3)、Bl21(DE3)-RP、 HMS174(DE3) (Novagen社から購入)に形質転換を行い、且つカナマイシン耐性プレートに塗布して37℃で一晩倒立培養を行った。モノクローンを選択してODが0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、30℃で4時間誘導した後、菌液を収集し、遠心分離して沈殿物を取って、SDS-PAGE電気泳動でタンパク質の発現量を解析した。
【0053】
1.1.3 融合タンパク質の誘導条件の分析
目的遺伝子の原核細胞における発現を向上させるため、いくつかの発現誘導条件を試験で探った。
(1)誘導時間:細菌をLB培地に接種し、37℃でODが0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、30℃で振とう培養し、それぞれ1、2、3、4、5、6時間の時に菌液を収集した。
(2)誘導濃度:発現菌株を0.6〜0.8になるまで培養し、それぞれ最終濃度が0.2、0.5、0.8、1mMになるようにIPTGを入れ、30℃で4時間振とう培養し、細菌を収集した。
(3)誘導温度:発現菌株を0.6〜0.8になるまで振とう培養し、最終濃度が1mMになるようにIPTGを入れ、それぞれ37℃、30℃、25℃で4時間誘導した後、細菌を収集した。
【0054】
1.1.4 融合タンパク質の発現様態
上述条件で大量に誘導した後、沈殿物を収集して1/10体積の緩衝液A(50mM NaH2PO4、300mM NaCl、10mM Imidozole(イミダゾール)、pH8.0)で再懸濁させ、PMSF(最終濃度が1mM)を入れて氷の上で超音波処理(超音波3秒、間隔10秒、1サークル99回、計4サークル)を行った後、遠心分離(4℃、12000g)し、上清と沈殿物をそれぞれ収集し、12%SDS-PAGE電気泳動を行い、0.25%クマシーブルーで3時間染色した後、脱色をして観察した。
【0055】
1.1.5 封入体の洗浄と変性
超音波による崩壊、遠心分離を経た沈殿物を十分に洗浄液I(100mM NaH2PO4、10mM Tris.Cl、2M 尿素(urea)、pH8.0)に再懸濁させ、4℃で30分間撹拌した後、15分間遠心分離(4℃、12000g)して沈殿物を収集し、洗浄液II(100mM NaH2PO4,10mM Tris.Cl,2M GuHCl,pH8.0)を入れ、上述操作を繰り返して、精製された封入体を得た。最後、封入体を8M尿素含有溶液(100mM NaH2PO4、10mM Tris.Cl、8M 尿素、pH8.0)に再懸濁させ、氷の上で超音波処理し、15分間遠心分離(4℃、12000g)し、沈殿物を捨て、上清を残した。
【0056】
1.1.6 融合タンパク質の精製
上清をNi-NTA アガロースと4℃で1時間(一晩)均一に混合し、親和クロマトグラフィーにかけて流出液を収集し、4mlの緩衝液C(100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH6.3)で3回洗浄し、さらに0.5mlの緩衝液D(100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH15.9)で4回洗浄し、最後に0.5mlの緩衝液E (100mM NaH2PO4、10mM Tris.Cl、2M 尿素、pH4.5)で4回洗浄した。それぞれの脱離液を収集して12%SDS-PAGEで純度を分析し、A280でその含有量を検出した。
【0057】
1.1.7 融合タンパク質のアニーリング
精製されたタンパク質を一滴ずつ10倍体積の冷やしておいたアニーリング緩衝液(25mM Tris-Cl、0.1M NaCl、10%グリセリン、1.0M 尿素、0.01M アルギニン、1mM還元性グルタチオン、0.5mM 酸化性グルタチオン、pH8.0)を入れ、4℃で24時間インキュベートした後、透析袋に入れて、それぞれ0.5M、0.25M、0.125M尿素を含有する緩衝液(PBS、pH7.4)にて4℃で4時間以上透析し、最後に大量のPBSに4℃で24時間透析し、遠心分離して上清を取った。
【0058】
1.3.8 アニーリングタンパク質のウェスタンブロッティングによる同定
Bl21(DE3)-RP菌における全タンパク質を陰性対照とし、アニーリング後の融合タンパク質に12%SDS-PAGEを行い、且つNC膜に転写した。1:1000のモノクローンのウサギ抗DGFRvIII(Zymed社から購入)(4℃で一晩インキュベートし、PBSTで膜を3回、毎回10分間洗浄したもの)および1:5000のHRPマウス抗ウサギIgG(37℃で1時間インキュベートし、膜洗浄は上述と同様なもの)滴下した。最後、ECL化学発光キットの試薬で現像し、且つ暗室でXフィルムを用いて露光した。
【0059】
結果:
1.pET28a-EGFRvIIIex組換え発現プラスミドの同定
プラスミドpET28a-EGFRvIIIexはBglII及びSalIの酵素切断後1292bp、5147bpの電気泳動バンド(図1を参照)が現れ、予想の大きさと一致し、ベクターの構築が正しいことが証明された。
【0060】
2.EGFRvIIIの細胞外領域のタンパク質の精製
図2に示すように、精製されたEGFRvIIIの細胞外領域のタンパク質を得た。
【0061】
3.EGFRvIIIのタンパク質アニーリング後の同定
図3及び4に示すように、EGFRvIIIの細胞外領域のタンパク質の純度が非常に高いことがわかった。
【0062】
実施例2. 抗原免疫及びハイブリドーマスクリーニング
2.1 免疫
(1)組換えタンパク質による免疫:
EGFRvIIIの細胞外領域の組換えタンパク質と等量の完全フロイントアジュバント(Sigma)と充分に乳化混合して6週齢のBALB/cマウスに1匹100μgずつ皮膚下免疫した。4週後組換え抗原を不完全フロイントアジュバントと充分に乳化混合し、免疫マウスに1匹50μgずつ腹腔注射し、その後2週間おきに腹腔強化免疫を続けた。4回目の強化免疫の1週間後、組換え抗原で被覆し、ELISA法でマウスの抗血清力価を検出したところ、>105であった。
【0063】
(2)脾臓内注射による強化免疫:
最後の強化の3週間後、20μgの組換え抗原で脾臓内免疫した。
【0064】
2.2 ハイブリドーマ細胞株の構築
マウスの脾臓内強化免疫の4日後、無菌の条件で脾臓を採取し、100メッシュのフィルターでリンパ細胞を分離し、骨髄腫瘍細胞系SP2/0と融合させた。ピポキサンチン-アミノプテリン-チミジン(hypoxathine, aminopterin and thymidine、HAT)で選択培養を3日行った後、HT培地を追加し、1週間培養を続けた。組換え抗原で被覆し、ELISAで陽性クローンを選択し、限界希釈法でサブクローニングし、2ヵ月培養を続けて、最後に安定したハイブリドーマ細胞系を得た。
その結果、陽性クローンを複数得たが、中でも12H23の活性が一番高かった。
【0065】
2.3 抗体の精製
2.3.1 オクタン酸/硫酸アンモニウム沈殿法による粗精製
腹水100mlを2倍体積の0.06MのpH 4.0の酢酸ナトリウム緩衝液で希釈し、4%オクタン酸をゆっくり滴下しながら、撹拌した。30分間撹拌した後、混濁液を10000gで30分間遠心分離した。沈殿物を捨て、上清液を0.01MのpH7.4のリン酸緩衝液で一晩透析した。透析液を取り、等体積の飽和硫酸アンモニウム液をゆっくり入れ、2時間置いた。混濁液を10000gで10分間遠心分離した。上清液を捨て、0.01M、pH7.4のPBSで溶解させた。溶解した溶液を0.01M、pH7.4のPBSで透析し、その期間で液を2回入れ替え、2回の入れ替えの間隔が5時間以上であった。透析溶液を10000gで10分間遠心分離し、沈殿物を捨て、上清液を収集した。
【0066】
2.3.2 プロテインG親和性精製
プロテインG親和性カラムを室温に戻し、5倍のカラム体積のPBSでバランスを取った。上述モノクローナル抗体溶液をカラムにかけ、5倍のカラム体積のPBSで洗った。pH2.3の0.1Mグリシン塩酸溶液で脱離させ、脱離液を1/10体積の1Mリン酸水素二ナトリウム溶液(pH9.0)で中和した。溶液を0.01M、pH7.4のPBSで透析し、その期間で液を2回入れ替え、2回の入れ替えの間隔が5時間以上であった。透析溶液を10000gで10分間遠心分離し、上清を0.22μmろ過膜でろ過してモノクローナル抗体溶液として保存した。上述の精製で、純度>95%の抗体を得た。
【0067】
2.4 12H23モノクローナル抗体の亜型の同定
rEGFRvIIIexを被覆緩衝液(NaHCO3pH9.6)で1.0mg/Lに希釈し、ELISAマイクロプレートに各孔50μLずつ入れた。4℃で24時間被覆した。5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。初期濃度が1mg/Lの12H23モノクローナル抗体50μLを入れ、37℃で1時間結合させた。PBSで3回洗浄し、それぞれヒツジ抗マウス亜型ポリクローナル抗体(1:1000希釈)100μLを入れ、37℃で1時間結合させた。PBSで3回洗浄した。HRP標識のロバ抗ヒツジのポリクローナル抗体、37℃で1/2時間結合させ、PBSで5回洗浄した。ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。
結果は図6に示すように、この抗体12H23の亜型はIgG1型であった。
【0068】
実施例3 モノクローナル抗体の結合能力の測定
3.1 12H23の受容体結合特異性のFACS解析
ベクターの構築:
細胞:U87細胞(脳膠腫細胞系、EGFRが正常発現、ATCC細胞バンクから購入)、U87-EGFRvIII細胞(pLERNLベクターをトランスフェクションしたU87細胞系)およびA431細胞(人扁平上皮癌、EGFRが過剰発現、ATCC細胞バンクから購入)
抗体:実施例2で調製した抗体12H23、及び市販のC225モノクローナル抗体(対照として)。抗体濃度はいずれも2mg/mLで、1:100で希釈した。
【0069】
1)対数増殖期の細胞を約90%の接種細胞密度で6孔プレートに接種し、インキュベーターを用い37℃で一晩培養した。
2)次の日、10mMのEDTAで消化した細胞を使用し、5000rpm×3分間で遠心分離し、細胞を2mLのエッペンドルフチューブに収集した。
3)0.5〜1mlのPBSで細胞を再懸濁させ、4%パラホルムアルデヒドで細胞を固定し、37℃で10分間インキュベートした。
4)細胞を氷の上に置いて1分間冷却した。
【0070】
5)5000rpm×3分間で遠心分離し、上清を捨てた。
6)細胞を氷冷した90%メタノールに再懸濁させ、氷の上に30分間置いた。
7)5000rpm×3分間で遠心分離し、上清を捨てた。
8)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
【0071】
9)細胞をそれぞれ各EP管に0.5〜1×106細胞/管(各細胞は8管に分け、その中で、1管は空白細胞、2管は第二抗体のみ)入れた。
10)各管に0.5%BSA(PBSで調製)を入れ、室温で10分間ブロックした。
11)5000rpm×3分間で遠心分離し、ブロック液を捨てた。
【0072】
12)各細胞に相応の第一抗体100μL入れ、室温で30〜60分間インキュベートした。
13)遠心分離し、第一抗体インキュベート液を捨てた。
14)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
【0073】
15)各細胞にFITC標識のヒツジ抗マウス第二抗体(12H23)或いはロバ抗ヒト第二抗体(C225)100μL入れ、室温で30分間インキュベートした。
16)遠心分離し、第二抗体インキュベート液を捨てた。
【0074】
17)0.5%BSA(PBSで調製)を入れて細胞を再懸濁させ、5000rpm×3分間で遠心分離し、3回洗浄した。
18)最後に、0.5〜1mLのPBSで細胞を再懸濁させ、フローサイトメトリー専用試験管に移した。
19)フローサイトメーターで解析した。
【0075】
結果:
図7A〜7Fに示すように、抗体12H23はEGFRvIIIを高発現する細胞系と効率的に結合することができ、EGFRを過剰発現するA431細胞とも一部結合したが、EGFRを正常発現する細胞系U87だけでほとんど結合しなかった。一方、製品化の抗体C225(Erbitux)はEGFRvIIIを高発現する細胞系だけでなく、EGFRを正常発現する細胞系U87とも結合できた。本発明の抗体12H23がより優れた特異性を有することがわかった。
【0076】
3.2 非競争法による12H23の親和力の測定
rEGFRvIIIexを被覆緩衝液(NaHCO3pH9.6)で5.0mg/L、2.5mg/L、1.25mg/Lおよび0.625mg/Lに希釈し、ELISAマイクロプレートに各孔100μLずつ入れた。4℃で24時間被覆した。被覆液を捨て、PBSで1回洗浄し、5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。異なる濃度の抗原を被覆したマイクロプレートに、初期濃度が1mg/Lの12H23モノクローナル抗体を入れ、勾配希釈(希釈液が5%脱脂粉乳を含有するPBS)で12級に希釈した。37℃で1時間結合させ、PBSで3回洗浄し、ヒツジ抗マウスの第二抗体100μLを入れ、37℃で1時間結合させた、PBSで5回洗浄し、ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。吸収係数の結果によって結合反応曲線を作り、作図法でその最高のOD値の半分(すなわち、OD:50%)の時の抗体濃度を得た。
【0077】
結果:
図8に示すように、12H23は、5.0mg/L被覆曲線では、OD50%抗体濃度が3μg/L(2×10-11mol/L)で、2.5mg/L被覆曲線では、OD50%抗体濃度が2.5μg/L(1.7×10-11mol/L)で、1.25mg/L被覆曲線では、OD50%抗体濃度が2.3μg/L(1.5×10-11mol/L)で、0.625mg/L被覆曲線では、OD50%抗体濃度が2μg/L(1.5×10-11mol/L)であった。(式)K=(n−1)/2(nAb′-Ab)によって親和定数を算出した。ここで、Ab′とAbはそれぞれ抗原濃度がAg′とAgの時OD50%抗体濃度になることを表し、n=Ag/Ag′である。そして、それぞれ比較すると、6つのK値を算出し、6つのK値の平均を最終の結果とした。算出した12H23の親和定数が3.8×10-10mol/Lで、解離定数Kd値が2.6×10-11mol/Lであった。
【0078】
実施例4:モノクローナル抗体の結合エピトープの解析
4.1 組換えタンパク質の調製:
通常の方法で、それぞれEGFRの細胞外領域のS1ドメイン、S2ドメインおよびS1ドメインにおけるVK21ポリペプチドとM13ファージのpIIIタンパクのN12タンパクドメインが融合したもの(1.1の方法と同じ方法で)を構築し、EGFRvIIIの細胞外領域の組換えタンパク質を陽性対照とした(図9)。
【0079】
電気泳動の結果から、各組換えタンパク質rN12-S1、rN12-S2、EGFRvIIIex、rN12-VK21が得られたことがわかった(図10)。
【0080】
4.2 ELISA法による12H23の結合エピトープの測定
組換えタンパク質rN12-S1、rN12-S2、EGFRvIIIex、rN12-VK21をそれぞれ被覆緩衝液(NaHCO3 pH9.6)で1.0mg/Lに希釈し、順にELISAマイクロプレートに各孔100μLずつ入れた。4℃で24時間被覆した。被覆液を捨て、PBSで1回洗浄し、5%脱脂粉乳を含有するPBS 350μLを入れて一晩ブロックした。PBSで2回洗浄した。それぞれ初期濃度が1mg/Lの12H23モノクローナル抗体を入れ、37℃で1時間結合させ、PBSで3回洗浄した。更に、ヒツジ抗マウスの第二抗体100μLを入れ、37℃で1時間結合させた後、PBSで5回洗浄した。ABTS基質を入れて15分間発色させ、マイクロプレートリーダーで405nmの吸収係数の値を測定した。
【0081】
結果:
ELISAの結果は図11に示すように、抗体12H23はEGFRvIII、rN12-S1およびrN12-VK21と結合することができた。構造および配列によれば、12H23が結合する領域はこれらのタンパク質の共有領域、すなわち、VK21であることが推測された。VK21のポリペプチド配列はVRACGADSYEMEEDGVRKCKK(SEQ ID NO:11)である。
【0082】
実施例5:モノクローナル抗体の生体内における腫瘍生長を抑制する能力
1)3×106個の通常のHuh7-EGFRvIII腫瘍細胞(pLRNLをトランスフェクションしたHuH-7肝臓癌細胞系、Huh-7は米国ATCC細胞バンクから購入)をそれぞれ18匹のBalb/cヌードマウスの右側肩甲の皮下に注射した。
【0083】
2)数日後、皮下の腫瘍体積が約80〜100mm3になったとき、それぞれC225抗体と12H23抗体を腹腔内注射した。1匹あたりの抗体注射量が0.5mgであり、同時にPBSを陰性対照として注射した。各群は6匹であった。
【0084】
3)その後、1日おきに抗体を腹腔内注射し、2週間続けた。
4)抗体注射と同時に、腫瘍の体積を1日おきに測定し、抗体注射が終わった後、腫瘍の体積の測定を2週間続けた。腫瘍の体積は(式)腫瘍の体積=腫瘍の長さ×腫瘍の幅2/2で算出した。
5)腫瘍の生長状況を観察した。
【0085】
結果:
12H23はHuh7-EGFRvIIIのヌードマウスの播種腫瘍に顕著な抑制作用があり(約70%)、且つその抑制率もC225抗体より優れていた(図12)。
【0086】
実施例6 モノクローナル抗体の配列決定
5'RACE法で5'配列が未知の遺伝子を大体以下のようにクローンした(具体的な操作はTakara 5'-full RACE Kitの説明書に従った)。
【0087】
1)アルカリホスファターゼ(CIAP)で全RNAにおける露出した5'リン酸基を脱リン酸反応を行った。全RNAの使用量は2μgで、反応後フェノール/クロロホルムで抽出して回収した。
2)タバコ酸性ピロホスファターゼ(Tobacco Acid Pyrophosphatase、TAP)でmRNAの5'キャップ構造を除去し、一つのリン酸基を残した。
【0088】
3)T4 RNAリガーゼで5'RACEアダプター(Adaptor)をmRNAに連結し、反応後フェノール/クロロホルムで抽出して回収した。
4)逆転写酵素で逆転写反応を行った。使用されたプライマーはキットで提供されたランダムの9量体のプライマーであった。
【0089】
5)逆転写産物を鋳型として、高忠実度の酵素で目的遺伝子を増幅した。使用されたプライマーは以下の通りである。
5':5'RACE Outer Primer(CATGGCTACATGCTGACAGCCTA) (SEQ ID NO: 12)
3':重鎖:CCAGAGTTCCAGGTCACTGTCACT (SEQ ID NO: 13)
軽鎖:ACACGACTGAGGCACCTCCA (SEQ ID NO: 14)
【0090】
6)上述PCR産物を鋳型として、ネステッドPCRを行った。使用されたプライマーは以下の通りである。
5':5'RACE Inner Primer(CGCGGATCCACAGCCTACTGATGATCAGTCGATG) (SEQ ID NO: 15)
3':重鎖:CCAGGGTCACCATGGAGTTAGTTT (SEQ ID NO: 16)
軽鎖:TGGATGGTGGGAAGATGGATACA (SEQ ID NO: 17)
7)TAクローンを行い、配列を測定した。
【0091】
配列決定の結果
12H23モノクローナル抗体の重鎖、軽鎖および各CDRの配列は図13〜14および下記表1に示す。
【0092】
【表1】
【0093】
実施例7
1.抗体可変領域コード配列を含有するヒト-マウスキメラ抗体の発現ベクターのクローニング
それぞれhCMVプロモーター、重鎖可変領域のクローン部位NheIとApaIおよびヒトIgG1の重鎖定常領域、IRESリボソーム挿入部位、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)及びアンピシリン耐性遺伝子を含む抗体の重鎖の発現ベクターpHを構築した(図15Aを参照)。
【0094】
それぞれhCMVプロモーター、軽鎖可変領域のクローン部位EcoRVとBsiWIおよびヒトIgG1の軽鎖定常領域、IRESリボソーム挿入部位、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)及びアンピシリン耐性遺伝子を含む抗体の軽鎖の発現ベクターpKを構築した(図15Bを参照)。
【0095】
実施例6で測定した軽鎖および重鎖の配列に基づき、重鎖および軽鎖の可変領域コード配列を人工合成し、重鎖コード配列の両末端にNheIとApaIの酵素切断部位を、軽鎖コード配列の両末端にEcoRVとBsiWIの酵素切断部位をつけた。NheIとApaIの酵素で重鎖可変領域コード配列を、EcoRVとBsiWIの酵素で軽鎖可変領域コード配列を切断した。
【0096】
その後、上述重鎖および軽鎖の可変領域コード配列を発現ベクターpHおよびpKに挿入し、キメラの抗EGFRvIIIの抗体遺伝子の発現ベクターを構築した。
【0097】
2.CHO細胞のトランスフェクション及び組換えクローンのスクリーニング
上述構築された抗体遺伝子をもつ発現ベクターを大腸菌DH5α菌株に導入し、そして100mLのLB培地に接種して増幅させ、Qiagen社の高純度プラスミドDNA精製キット(Ultrapure Plasmid DNA Purification Kit)でプラスミドDNAを抽出、精製した。上述精製されたプラスミドDNAをInvitrogen社のリプソーム法キットでCHO細胞にトランスフェクションし、操作はメーカーの説明書を参照して行った。
【0098】
形質変換されたCHO細胞を濃度が高くなっていくMTX選択培地で培養を9週間続け、最後は96孔プレートで勾配希釈培養を行い、連続して3回を行い、モノクローナル化した。
【0099】
選択されたモノクローナル細胞系はRPM1640培地で培養し、上清にELISA実験をし、発色反応で発現結合強度を判断した。測定によってこれらのクローンもrEGFRvIII抗原と特異的に結合する活性を有し(図16を参照)、幾つかの発現の高いクローンを細胞株の候補として選択し、キメラ抗体を調製した。得られた抗体をCH12と呼ぶ。
【0100】
実施例8
抱合体の調製
キメラモノクローナル抗体CH12を直接ジフテリア毒素(武漢生物製品研究所から購入)と共役結合で結合させた。Huh7-EGFRvIIIの細胞に結合物を入れると、特異的な細胞毒性が観察される。EGFRvIIIを発現しないHuh7細胞は、非常に高濃度の抗体に曝されるときだけ殺傷される。
【0101】
実施例9
注射液の調製
実施例5で調製された抗体12H23或いは実施例7で調製されたキメラモノクローナル抗体CH12を注射用生理食塩水に入れ、均一に混合した後、0.22uMの無菌フィルターで除菌し、注射液を小瓶に分け(50mL/瓶)、包装して使用に供する。ここで、50mLあたりの注射液にモノクローナル抗体50mgが含有される。
【0102】
実施例10
キメラ抗体CH12とマウス由来のモノクローナル抗体12H23の競争結合実験
ELISA法によって、HRP標識のCH12抗体(1.0μg/mL)を固定し、さらに異なる濃度の未標識CH12抗体或いは12H23抗体(0、3、9、27、81、243、729μg/mL)を入れた。
【0103】
結果から、競争するCH12或いは12H23抗体濃度の増加に従い、OD405の値が低くなっていくことがわかった。しかも、二種の抗体の異なる濃度における競争抑制率がほぼ同じであることがわかった。キメラ抗体CH12とマウス由来のモノクローナル抗体12H23は、抗原に対する親和力が同等程度で、且つ結合部位も一致することが示唆される。
【0104】
実施例11
キメラ抗体CH12の細胞免疫組織化学的検出
通常の免疫蛍光の方法によってCH12キメラ抗体とHuh-7細胞、Huh7-EGFRおよびHuh7-EGFRvIIIの結合状況を検出した。
【0105】
結果から、CH12抗体は顕著にHuh7-EGFRvIIIと結合することができ、またEGFRを増幅的に発現するHuh7-EGFRとも結合することができるが、Huh-7細胞とほとんど結合しないことがわかった。キメラ改造後、CH12抗体の細胞に対する結合能力及び特異性は変わらないことが示唆される。
【0106】
実施例12
CH12キメラ抗体の生体内における腫瘍生長を抑制する能力
1)Huh7-EGFRvIII腫瘍細胞或いはSMMC-7721肝臓癌細胞系(中科院細胞バンクから購入)をそれぞれBalb/cヌードマウスの右側肩甲の皮下に注射した。1匹あたりのマウスの播種細胞量は3×106 個であった。(注:SMMC-7721細胞が肝臓癌細胞系で、本発明者らは実験によって内因的にEGFRvIIIが存在することを証明した。)
【0107】
2)数日後、皮下の腫瘍体積が約150mm3になったとき、それぞれC225抗体、CH806抗体或いはCH12抗体を腹腔内注射した。1匹あたりの抗体注射量が0.5mgであり、同時にPBSを陰性対照として注射した。各群は6匹であった。
【0108】
3)毎週3回給与し、2週間続けた。
4)抗体注射と同時に、腫瘍の体積を測定し、抗体注射が終わった後腫瘍の体積の測定を2週間続けた。腫瘍の体積は、(式)腫瘍の体積=腫瘍の長さ×腫瘍の幅2/2で算出した。
5)腫瘍の生長状況を観察した。
【0109】
結果(図17)から、CH12モノクローナル抗体は、効率的にSMMC-7721の播種腫瘍のヌードマウスの体内における生長を抑制することができ、給与後25日(腫瘍細胞を接種後41日)のときの腫瘍抑制率が62.94%で、一方、対照抗体C225とCH806の腫瘍抑制率がそれぞれ25.97%と33.11%であることがわかった。
【0110】
CH12処理群では、腫瘍接種後27日はPBS対照群と比べて顕著な差(p<0.05)があり、腫瘍接種後32日はC225処理群と比べて顕著な差(p<0.05)があり、腫瘍接種後37日はCH806処理群と比べて顕著な差(p<0.05)があった。CH12はSMMC-7721肝臓癌を治療する場合C225とCH806よりも優れることが示唆される。
【0111】
また、CH12はHuh7-EGFRvIII播種腫瘍のヌードマウスの体内における生長を抑制することもでき、PBS群(p=0.0001)とC225治療群(抑制率32.9%だけ)よりも優れた。
【0112】
各文献がそれぞれ単独に引用されるように、本発明に係るすべての文献は本出願で参考として引用する。また、本発明の上記の内容を読み終わった後、この分野の技術者が本発明を各種の変動や修正をすることができるが、それらの等価の様態のものは本発明の請求の範囲に含まれることが理解されるべきである。
【0113】
参考文献
1) Ullrich A.ら、 Human Epidermal Growth Factor Receptor cDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature, 1984, 309: 418-425
2) Downwardら、 Close similarity of Epidermal Growth Factor Receptor and v-erb B oncogene Protein Sequence. Nature 1984, 307:521-527
3) Libermannら、 Amplification,Enhanced Expression and Possible Rearrangement of EGF Receptor Gene in Primary Human Brain Tumors of Glial Origin. Nature 1985,313:144-147
【0114】
4) Wongら、 Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas is Invariably Associated with Gene Amplification. Proc. Natl. Acad. Sci. USA. 1987, 84:6899-6903
5) Yamazakiら、 Amplification of the Structurally and Functionally Altered Epidermal Growth Factor Receptor Gene (c-erbB) in Human Brain Tumors. Molecular and Cellular Biology 1988,8:1816-1820
6) Maidenら、 Selective Amplification of the Cytoplasmic Domain of the Epidermal Growth Factor Receptor Gene in Glioblastoma Multifome. Cancer Research 1988(4):2711-2714
【0115】
7) Modjtahedi H, and Dean C. The receptor for EGF and its ligands Expression, prognostic value and target for therapy in cancer. International Journal of Oncology 1994 (4): 277-296
8) Fung YKT,ら、 Activation of the Cellular Oncogene c-erb B by LTR Insertion: Molecular Basis for Induction of Erythroblastosis by Avian Leukosis Virus. Cell 1983, 33:357-368
9) Yamamotoら、 A New Avian Erythroblastosis Virus, AEV-H Carries erbB Gene Responsible for the Induction of Both Erythroblastosis and Sarcoma. Cell 1983,34:225-232
【0116】
10) Nilsenら、 c-erbB Activation in ALV-Induced Erythroblastosis: Novel RNA Processing and Promoter Insertion Results in Expression of an Amino-Truncated EGF Receptor. Cell 1985, 41: 719-726
11) Gammettら、 Differences in Sequences Encoding the Carboxy-Terminal Domain of the Epidermal Growth Factor Receptor Correlate with Differences in the Disease Potential of Viral erbB Genes.”Proc. Natl. Acad. Sci. USA 83:6053-6057(1986)
12) Gilmoreら、 Protein Phosphorylation at Tyrosine is Induced by the v-erb B Gene Product in Vivo and in Vitro. Cell, 1985,40:609-618, (1985)
【0117】
13) Krisら、 Antibodies Against a Synthetic Peptide as a Probe for the Kinase Activity of the Avian EGF Receptor and v-erbB Protein. Cell, 40:619-625(1985)
14) Nilsenら、 c-erbB Activation in ALV-Induced Erythroblastosis: Novel RNA Processing and Promoter Insertion Results in Expression of an Amino-Truncated EGR Receptor. Cell, 1985,41:719-726
15) Rainesら、 c-erbB Activation in Avian Leukosis Virus-Induced Erythroblastosis: Clustered Integration Sites and the Arrangement of Provirus in the c-erbB Alleles. Proc. Natl. Acad. Sci. USA,1985, 82:2287-2291
【0118】
16) Pelleyら、 Proviral-Activated c-erbB is Leukemogenic but not Sarcomagenic: Characterization of a Replication-Competent Retrovirus Containing the Activated c-erbB. Journal of Virology 1988, 62: 1840-1844
17) Wellsら、 Genetic Determinant of Neoplastic Transformation by the Retroviral Oncogene v-erbB. Proc. Natl. Acad. Sci. USA 1988, 85:7597-7601
18) Yamazakiら、 Amplification, Enhanced Expression and Possible Rearrangement of EGF Receptor Gene in Primary Human Brain Tumours of Glial Origin. Nature 1985, 313:144-147
【0119】
19) Wikstrand CJ,ら、 Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas malignant gliomas. Cancer Research 1995, 55(14):3140-3148
20) Olapade-Olaopa EO,ら、 Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer. 2000, 82(1):186-94
21) Ge H,ら Evidence of High incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J cancer. 2002, 98(3):357-61
【0120】
22) Moscatello G.,ら、 Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 55(23):5536-9(1,995)
23) Garcia de Palazzo, IE.,ら、 Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993, 53(14):3217-20
24) Moscatello,G. et al,Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer. 2000, 82(1):186-94
【0121】
25) Luo XY,et al. Suppression of EGFRvIII-Mediated Proliferation and Tumorigenesis of Breast Cancer Cells by Ribozyme. Int. J. Cancer. 2003, 104(6):716-21
26) Kuan CT,et al. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001, 8(2):83-96
27) Scott A,ら、 A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. PNAS 2007, 104(10): 4071-4076
【特許請求の範囲】
【請求項1】
相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とするモノクローナル抗体のVH鎖。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
【請求項2】
SEQ ID NO:2で示されるアミノ酸配列を有することを特徴とする請求項1に記載のモノクローナル抗体のVH鎖。
【請求項3】
相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とするモノクローナル抗体のVL鎖。
SEQ ID NO:8で示されるCDR1、
SEQ ID NO:9で示されるCDR2、および
SEQ ID NO:10で示されるCDR3。
【請求項4】
SEQ ID NO:4で示されるアミノ酸配列を有することを特徴とする請求項3に記載のモノクローナル抗体のVL鎖。
【請求項5】
VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有することを特徴とするモノクローナル抗体或いはその抱合体。
【請求項6】
前記抗体が、マウス抗体、ヒト化抗体或いはキメラ抗体であることを特徴とする請求項5に記載のモノクローナル抗体。
【請求項7】
以下の群から選ばれるタンパク質をコードすることを特徴とするDNA分子。
請求項1に記載のモノクローナル抗体のVH鎖、
請求項3に記載のモノクローナル抗体のVL鎖、
請求項5に記載のモノクローナル抗体。
【請求項8】
SEQ ID NO:1、3、11或いは13から選ばれることを特徴とする請求項7に記載のDNA分子。
【請求項9】
VH鎖がSEQ ID NO:5〜7で示される相補性決定領域を有し、かつVL鎖がSEQ ID NO:8〜10で示される相補性決定領域を有するモノクローナル抗体と薬学的に許容される担体とを含むこと、好ましくは、前述モノクローナル抗体は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有することを特徴とする医薬組成物。
【請求項10】
(a)上皮成長因子受容体変異体IIIを発現する細胞の成長の抑制又は致死、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長の抑制のための組成物の製造に用いられることを特徴とする請求項5に記載のモノクローナル抗体或いはその抱合体の用途。
【請求項1】
相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とするモノクローナル抗体のVH鎖。
SEQ ID NO:5で示されるCDR1、
SEQ ID NO:6で示されるCDR2、および
SEQ ID NO:7で示されるCDR3。
【請求項2】
SEQ ID NO:2で示されるアミノ酸配列を有することを特徴とする請求項1に記載のモノクローナル抗体のVH鎖。
【請求項3】
相補性決定領域CDRが以下の群から選ばれるCDRのアミノ酸配列を有することを特徴とするモノクローナル抗体のVL鎖。
SEQ ID NO:8で示されるCDR1、
SEQ ID NO:9で示されるCDR2、および
SEQ ID NO:10で示されるCDR3。
【請求項4】
SEQ ID NO:4で示されるアミノ酸配列を有することを特徴とする請求項3に記載のモノクローナル抗体のVL鎖。
【請求項5】
VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有することを特徴とするモノクローナル抗体或いはその抱合体。
【請求項6】
前記抗体が、マウス抗体、ヒト化抗体或いはキメラ抗体であることを特徴とする請求項5に記載のモノクローナル抗体。
【請求項7】
以下の群から選ばれるタンパク質をコードすることを特徴とするDNA分子。
請求項1に記載のモノクローナル抗体のVH鎖、
請求項3に記載のモノクローナル抗体のVL鎖、
請求項5に記載のモノクローナル抗体。
【請求項8】
SEQ ID NO:1、3、11或いは13から選ばれることを特徴とする請求項7に記載のDNA分子。
【請求項9】
VH鎖がSEQ ID NO:5〜7で示される相補性決定領域を有し、かつVL鎖がSEQ ID NO:8〜10で示される相補性決定領域を有するモノクローナル抗体と薬学的に許容される担体とを含むこと、好ましくは、前述モノクローナル抗体は、VH鎖がSEQ ID NO:2で示されるアミノ酸配列を有し、かつVL鎖がSEQ ID NO:4で示されるアミノ酸配列を有することを特徴とする医薬組成物。
【請求項10】
(a)上皮成長因子受容体変異体IIIを発現する細胞の成長の抑制又は致死、或いは(b)上皮成長因子受容体を過剰発現する細胞の成長の抑制のための組成物の製造に用いられることを特徴とする請求項5に記載のモノクローナル抗体或いはその抱合体の用途。
【図5】

【図7】
【図13】

【図14】
【図1】
【図2】
【図3】
【図4】

【図6】
【図8】
【図9】
【図10】
【図11】
【図12】
【図15】
【図16】
【図17】
【図7】
【図13】
【図14】
【図1】
【図2】
【図3】
【図4】
【図6】
【図8】
【図9】
【図10】
【図11】
【図12】
【図15】
【図16】
【図17】
【公表番号】特表2013−505026(P2013−505026A)
【公表日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−530073(P2012−530073)
【出願日】平成21年9月22日(2009.9.22)
【国際出願番号】PCT/CN2009/074090
【国際公開番号】WO2011/035465
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(504200870)上海市腫瘤研究所 (1)
【Fターム(参考)】
【公表日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願日】平成21年9月22日(2009.9.22)
【国際出願番号】PCT/CN2009/074090
【国際公開番号】WO2011/035465
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(504200870)上海市腫瘤研究所 (1)
【Fターム(参考)】
[ Back to top ]