
独自のクローン形質のリンパ球受容体に結合する試薬および方法
【課題】少なくとも1種のリンパ球に影響する分子、および抗原に結合した場合に独自のクローン形質リンパ球受容体に結合する少なくとも1種の分子複合体を含むプラットフォームを用い、治療的に有用な数の特異的リンパ球集団を誘導および拡大する。
【解決手段】T細胞に影響する分子および抗原提示複合体を含む抗原提示プラットフォームは、関連ペプチドの存在下で抗原特異的T細胞を誘導および拡大することができ、治療的数のそのような細胞を作成するための再現可能で経済的な方法を提供する。B細胞に影響する分子およびB細胞表面のMHC-抗原複合体に結合する分子複合体を含む抗体誘導プラットフォームを用い、特定の特異性を伴う抗体を産生するB細胞を誘導および拡大することができる。
【解決手段】T細胞に影響する分子および抗原提示複合体を含む抗原提示プラットフォームは、関連ペプチドの存在下で抗原特異的T細胞を誘導および拡大することができ、治療的数のそのような細胞を作成するための再現可能で経済的な方法を提供する。B細胞に影響する分子およびB細胞表面のMHC-抗原複合体に結合する分子複合体を含む抗体誘導プラットフォームを用い、特定の特異性を伴う抗体を産生するB細胞を誘導および拡大することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、本明細書に参照として組み入れられている2002年7月12日に出願された同時係属中の米国特許仮出願第60/395,781号の利益を請求するものである。
【0002】
本発明は、一部、米国立衛生研究所(NIH)助成金番号AI-29575および同AI-44129により資金提供された研究の結果である。米国政府は、本発明に対し一定の権利を有するものである。
【0003】
発明の技術分野
本発明は、独自のクローン形質のリンパ球受容体に結合する試薬および方法に関する。
【背景技術】
【0004】
発明の背景
養子および能動の両方の免疫療法の開発は、治療できる数の特異的TまたはBリンパ球を作成するための、再現性があり、経済的で実行可能な方法が欠如しているために妨げられている。例えば、養子免疫療法のための抗原特異的細胞傷害性Tリンパ球(CTL)を作成する現在の標準的方法は、CTLを拡大するための単球由来樹状細胞(DC)の作成を必要としている。この工程は、時間および経費の両方がかかる。CTLの臨床的に関連のある量へのCTL拡大のためのDCの使用は、十分な自家DCを得るために、複数回の白血球搬出を必要としている。得られたDCの量および質の両方について認められる変動性は、恐らく基礎をなす疾患および患者の前処置に関連し、これは同じくDCを用いたエクスビボ療法の変動性にも大きい影響を及ぼす。これらの理由により、DCの使用は、T細胞のエクスビボ拡大の工程に限られている。
【0005】
濃縮された集団から抗原特異的CTLを拡大する他の方法は、非特異的抗CD3を用いた技術を使用している。Levineら、J. Hematother.、7:437-48(1998)(非特許文献1)。しかしふたつの問題点がある。第一に、抗CD3/抗CD28ビーズは、CD4 T細胞の長期増殖を支持するが、CD8 T細胞の長期増殖は維持しない。DeethsおよびMescher、Eur. J. Immunol.、27:598-608(1997)(非特許文献2)。加えて、抗CD3を用いた刺激を使用する方法は、例え高度に濃縮された抗原特異的CTL集団を用い開始する場合であっても、抗原特異性の低下を伴う。Mausら、Nature Biotechnol.、20:143-48(2002)(非特許文献3)。これらの問題点は、治療に関連するリンパ球の送達を実質的に制限している。
【0006】
従って当技術分野において、抗原特異的T細胞の、更には特異的抗体産生B細胞の治療的に有用な集団を作成する効果的な手段の必要性が存在する。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Levineら、J. Hematother.、7:437-48(1998)
【非特許文献2】DeethsおよびMescher、Eur. J. Immunol.、27:598-608(1997)
【非特許文献3】Mausら、Nature Biotechnol.、20:143-48(2002)
【発明の概要】
【0008】
発明の簡単な概要
本発明は、少なくとも下記の態様を提供する。本発明のひとつの態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、固形支持体である。
【0009】
別の本発明の態様は、(A)少なくとも1種のB細胞に影響する分子、および(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、固形支持体を提供する。
【0010】
別の本発明の態様は、(A)少なくとも1種のT細胞共刺激分子、および(B)少なくとも2種の融合蛋白質を含む少なくとも1種のMHCクラスI分子を含む粒子を提供する。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。
【0011】
更に別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、複数の粒子を含有する調製物を提供する。
【0012】
更なる本発明の態様は、複数の粒子を含有する調製物を提供する。複数の粒子は、(A)少なくとも1種のB細胞に影響する分子、および(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0013】
更に別の本発明の態様は、抗原特異的T細胞の形成を誘導する方法を提供する。複数の前駆体T細胞を含有する単離された調製物は、少なくとも1種の第一の固形支持体と接触される。この固形支持体は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。抗原は、この抗原結合窩に結合される。これにより、複数の前駆体T細胞のメンバーは、その抗原を認識する抗原特異的T細胞を含む第一の細胞集団の形成を誘導する。第一の集団内の抗原特異的T細胞の数または割合は、前駆体T細胞が、CD3に特異的に結合する抗体は含むが抗原提示複合体は含まない固形支持体と共にインキュベーションされる場合に形成される抗原特異的T細胞の数または割合よりも多い。この抗原特異的T細胞は、患者へ投与することができる。
【0014】
更に別の本発明の態様は、細胞の集団内の抗原特異的T細胞の数または割合を増加する方法を提供する。抗原特異的T細胞を含む第一の細胞集団は、少なくとも1種の第一の固形支持体と共にインキュベーションされる。固形支持体は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。抗原は、抗原結合窩に結合される。インキュベーション工程は、第一の細胞集団内の抗原特異的T細胞の数または割合に比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される。この抗原特異的T細胞は、患者へ投与することができる。
【0015】
更なる本発明の態様は、患者における免疫応答を調節する方法を提供する。(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物が、患者に投与される。複数の粒子のメンバーは、(1)少なくとも1種のT細胞に影響する分子、および(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体は、少なくとも1個の抗原結合窩を含む。抗原は、この少なくとも1個の抗原結合窩に結合される。
【0016】
更に別の本発明の態様は、患者における免疫応答を抑制する方法を提供する。(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物が、患者に投与される。複数の粒子のメンバーは、(1)少なくとも1種のアポトーシス誘導分子、および(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体は、少なくとも1個の抗原結合窩を含む。抗原は、この少なくとも1個の抗原結合窩に結合される。
【0017】
別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、抗原を認識する特異的クローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む細胞を提供する。
【0018】
更に別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、クローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む複数の細胞を含有する調製物を提供する。
【0019】
更に別の本発明の態様は、抗原特異的T細胞の形成を誘導する方法を提供する。複数の前駆体T細胞を含有する単離された調製物は、第一の複数の細胞と接触される。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質である。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖および(ii)MHCクラスIlα鎖の細胞外ドメインを含む。2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、ペプチド結合窩に結合される。これにより複数の前駆体T細胞のメンバーは、抗原性ペプチドを認識する抗原特異的T細胞を含む第一の細胞集団を形成する。
【0020】
更に別の本発明の態様は、細胞の集団内の抗原特異的T細胞の数または割合を増加する方法を提供する。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。この2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、ペプチド結合窩に結合される。インキュベーション工程は、第一の細胞集団内の抗原特異的T細胞の数または割合と比べ、増大した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される。この抗原特異的T細胞を、患者へ投与することができる。
【0021】
別の本発明の態様は、患者の免疫応答を調節する方法を提供する。複数の細胞および薬学的に許容される担体を含有する調製物を、患者に投与することができる。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。この抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、このペプチド結合窩に結合される。
【0022】
更に別の本発明の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。複数の前駆体B細胞を含有する単離された調製物は、少なくとも1種の第一の固形支持体と接触される。固形支持体は、少なくとも1種のB細胞に影響する分子、およびB細胞表面の免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。これにより複数の前駆体B細胞のメンバーは、抗原性ペプチドに特異的に結合する抗体を産生するB細胞を含む第一の細胞集団を形成するように誘導される。
【0023】
本発明の別の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。抗体産生B細胞を含む第一の細胞集団は、少なくとも1種の第一の固形支持体とインキュベーションされる。この固形支持体は、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。インキュベーション工程は、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される。
【0024】
更に別の本発明の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。複数の前駆体B細胞を含む単離された調製物を、調製物と接触させ、これにより第一の細胞集団を形成する。この調製物は、複数の粒子を含む。複数の粒子は、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原に結合する少なくとも1種の分子複合体を含む。第一の細胞集団の細胞は、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む。
【0025】
本発明の別の態様は、患者における免疫応答を調節する方法を提供する。複数の粒子および薬学的に許容される担体を含む調製物が、患者に投与される。複数の粒子のメンバーは、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0026】
従って本発明は、独自のクローン形質のリンパ球受容体に結合する様々な試薬および方法を提供する。本発明は、治療目的で使用することができる、抗原特異的T細胞および抗体特異的B細胞を得るための試薬および方法も提供する。
【図面の簡単な説明】
【0027】
【図1】(図1)自家DCまたはaAPCのいずれかによる、ペプチド特異的CTLの誘導および拡大の概略である。
【図2−1】(図2−1)aAPCにより刺激されたMart-1特異的CD8+ T細胞の誘導および増殖能である。図2Aは、aAPCによる刺激の結果である。図2Bは、DCによる刺激の結果である。
【図2−2】(図2−2)aAPCにより刺激されたMart-1特異的CD8+ T細胞の誘導および増殖能である。図2Cは、T細胞の拡大を示すグラフである。図2Dは、拡大されたT細胞集団内の抗原特異的CTLの割合を示すグラフである。
【図3】(図3)aAPC誘導した抗原特異的CTLは、内因性黒色腫または標的細胞上のpp65抗原を認識することを示す。図3Aは、ペプチド特異的CD8+ T細胞の割合(%)が、Mart-1+/HLA-A2-黒色腫細胞株(左側)、またはMart-1+/HLA-A2+黒色腫細胞株(右側)により刺激されたMart-1特異的T細胞について示されている。図3Bは、Mart-1特異的CTL株による%特異的溶解を、以下の標的について示している:非特異的CMVペプチド

もしくは特異的Mart-1ペプチド
のいずれか、または自家HLA-A2+黒色腫細胞株(□)もしくは自家HLA-A2-黒色腫細胞株(■)
のいずれかにより、パルスされたT2細胞。値は、エフェクター:標的の比25:1、5:1およ
び1:1の3つ組で表している。図3Cは、ペプチド特異的CD8+ T細胞の割合(%)を、pp65-対
照でトランスフェクションされたHLA-A2+ A293細胞(左側)またはpp65+でトランスフェク
ションされたHLA-A2+ A293細胞(右側)のいずれかにより刺激されたCMV特異的T細胞につい
て示している。図3Dは、内因性抗原を発現している標的細胞に対するCMV特異的CTL細胞傷
害活性に関する、51Cr-放出アッセイ法の結果である。CMV特異的CTL株による%特異的溶
解を、以下の標的について示している。pp65でトランスフェクションされたA293細胞
、トランスフェクションされないHLA-A2+ A293細胞(□)、およびIE(CMVの前初期蛋白質)
対照でトランスフェクションされたA293細胞(■)。全てのアッセイ法に関して、抗原特異
的CD8+ T細胞を、ペプチド負荷したaAPCと一緒のインビトロ培養物において3〜7週後に得
た。
【図4】(図4)抗CD3ビーズまたはaAPCによる拡大後の抗原特異的CTLの頻度を示す。T細胞は、実施例1に示したように、単離および精製した。図4Aは、T細胞を、CMVペプチドでパルスした自家単球-由来DCで刺激し、抗原特異的T細胞拡大を誘導する。図4Bでは、誘導の3週間後に、T細胞集団が、抗CD3/抗CD28ビーズ上で拡大された。図4Cでは、DCでの誘導の3週間後に、T細胞集団は、ペプチド負荷されたHLA-Igを用いたaAPCで拡大された。両方の場合において、10日の培養後に、約7倍の拡大が認められた。実施例1に説明したように、細胞は、FITC結合した抗CD8 mAbで、ならびにpp65で負荷したCMV-ペプチドパルスしたA2-Ig(上側パネル)または対照ペプチドMart-1で負荷したA2-Igで染色した。%ペプチド特異的CD8+ CTLは、上部右隅に示している。
【図5】(図5)aAPC誘導したMart-1 CTLは、黒色腫標的細胞上の内因性抗原を認識することを示す。Mart-1 特異的CD8+細胞は、Mart-1負荷したaAPCと一緒のインビトロ培養後に得た。Mart-1特異的T細胞は、Mart-1+/HLA-A2-黒色腫細胞株(第一カラム)またはMart-1+/HLA-A2+黒色腫細胞株(第二カラム)のいずれかにより刺激した。ICS染色に関して、これらの細胞は、サイトカインを含まない通常の培地において黒色腫細胞と共に培養した。ベースラインを評価するために、低用量のPMAおよびイオノマイシンを培地に添加した。1時間後、モネンシン(ゴルジ停止)を培地に添加した。6時間後、T細胞を収集し、および細胞内サイトカイン染色について分析した。ペプチド特異的IL-4+/CD8+ T細胞の割合(%)を示した。
【発明を実施するための形態】
【0028】
発明の詳細な説明
本発明は、独自のクローン形質のリンパ球受容体と結合する(すなわち、結合および生理的反応の引き金を引く)多種多様な道具および方法を提供する。独自のクローン形質の受容体は、例えば、特異的抗原を認識するT細胞受容体を含む。本発明の一部の態様(「抗原提示プラットフォームおよび方法」)を用い、治療または診断目的の抗原特異的T細胞の形成および/または拡大を誘導することができる。抗原特異的T細胞は、細胞傷害性Tリンパ球、ヘルパーT細胞(例えばTh1、Th2)、および調節T細胞を含む。更に他の本発明の態様(「抗体誘導プラットフォームおよび方法」)を用い、特定の抗原に対する抗体を産生するBリンパ球の形成および/または拡大を誘導することができる。
【0029】
抗原提示プラットフォームおよび方法
本発明の抗原提示プラットフォーム(本明細書において「人工的抗原提示細胞」または「aAPC」とも称される)は、以下により詳細に説明されるように、真核細胞または人工的固形支持体を基にすることができる。本発明の抗原提示プラットフォームは、少なくとも1種のT細胞に影響する分子(例えば、T細胞共刺激分子、T細胞増殖因子、接着分子、調節T細胞誘導分子、またはアポトーシス誘導分子)および少なくとも1種の抗原提示複合体を含む。
【0030】
抗体誘導プラットフォーム
本発明の抗体誘導プラットフォームも、以下により詳細に説明されるように、真核細胞または人工的固形支持体を基にすることができる。本発明の抗体誘導プラットフォームは、少なくとも1種のB細胞に影響する分子(例えば、以下に説明されるような、CD40リガンド、サイトカイン、またはサイトカイン分子複合体)およびB細胞表面免疫グロブリンに結合するかもしくはB細胞の表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0031】
各々、抗原特異的T細胞および抗体特異的B細胞のエクスビボ拡大のための本発明の抗原提示プラットフォームおよび抗体誘導プラットフォームの使用は、現在使用される方法に勝る多数の重要な利点を有する。両型のプラットフォームを実行することができ、これらは再現性のある抗原提示または抗体誘導の活性を有し、ならびに大きい患者集団に使用することができる。抗原提示プラットフォームの使用は、抗原特異的T細胞のエクスビボ拡大プロセスを、樹状細胞を使用する現在の方法と比べ、劇的に簡略化および短縮する。加えて、これらのプラットフォームにより拡大された抗原特異的T細胞集団は、現在の方法(例えば抗CD3抗体単独による刺激)により得られる5〜20%と比べ、最大80%の抗原特異的T細胞を含むと思われる。これらのプラットフォームは、治療的用途に適した数までの前駆体TまたはB細胞の拡大を誘導することができる。これらのプラットフォームは、一工程において、前駆体TまたはB細胞の単離を抗原特異性刺激と組合わせることができる。人工の粒子を基にしたプラットフォームの態様は、これらは高密度であることができおよび重力により沈降することができる点、これらは磁石による分離が望ましい場合は磁気特性を有することができる点、これらはコーティングおよび蛋白質複合のための理想的表面化学を有する点、ならびに増大した表面積および標的細胞との増大した接触を提供するために異なる粒子サイズおよび幾何学的形が利用可能である点で、特異的なTまたはB細胞集団を誘導する現在利用可能な手段に勝っている。
【0032】
本発明のプラットフォームの成分は以下に詳細に説明されている。
【0033】
固形支持体
本発明のプラットフォームのための固形支持体は、蛋白質分子が付着することができる固形の人工表面(すなわち、非細胞)のいずれかであることができる。適当な固形支持体は、硬質支持体(例えば、フラスコ、試験管、培養皿、マルチウェルプレート、スライド、粒子)に加え、軟質支持体(例えば、注入バッグ)を含む。
【0034】
軟質支持体
軟質支持体は、注入バッグを含む。これらのバッグは、単回使用のために作成されるか、または再使用可能であることができる。バッグは、滅菌に適した材料で製造されることが好ましい。このような材料は、当技術分野において周知でありおよび広範に使用されている。
【0035】
硬質支持体
硬質支持体の例は、試験管;組織培養器、例えば、フラスコ(例えば、10cm2、25cm2、75cm2、150cm2、285cm2、300cm2、または420cm2)、ペトリ皿(例えば、9.2cm2、22.1cm2、60cm2、147.8cm2)、マルチウェルプレート(例えば、6ウェル、12ウェル、24ウェル、48ウェル、もしくは96ウェル、または384ウェルのプレート);スライド;ならびに、粒子を含む。硬質支持体は、例えば、鉄、ニッケル、アルミニウム、銅、亜鉛、カドミニウム、チタン、ジルコニウム、スズ、鉛、クロム、マンガンおよびコバルトなどの、金属;酸化アルミニウム、酸化クロム、酸化鉄、酸化亜鉛、および酸化コバルトなどの、金属酸化物および水和された酸化物;マグネシウム、アルミニウム、亜鉛、鉛、クロム、銅、鉄、コバルト、およびニッケルなどの、金属のケイ酸塩;青銅、真鍮、ステンレス綱のような合金などで製造することができる。硬質支持体は、非金属または有機物質、例えばセルロース、セラミック、ガラス、ナイロン、ポリスチレン、ゴム、プラスチックまたはラテックスで製造することもできる。または、硬質支持体は、金属および非金属または有機化合物の組合せ、例えばメタクリレート-またはスチレン-コートした金属およびケイ酸塩コートした金属であることができる。基本材料は、物質に浸漬し、その物理的または化学的特性を変更することができる。例えば、希土酸化物は、高密度の常磁性ガラス材料を作成するために、アルミノケイ酸塩ガラスに含むことができる(WhiteおよびDay、Key Engineering Materials、94-95:181-208(1994)参照)。
【0036】
粒子
態様のひとつのセットにおいて、本発明のプラットフォームは、人工の粒子を基にしている。人工の粒子は、前述の多くの材料のいずれかにより製造することができる。望ましい場合には、粒子は全体を、セルロース、デキストランなどの生分解性有機物質で製造することができる。適当な市販の粒子は、例えば、ニッケル粒子(Novamet Specialty Products社(ワイコフ、N.J.)から販売されているタイプ123、VM 63、18/209A、10/585A、347355およびHDNP;Spex社から販売されている、08841R;Aldrich社から販売されている、01509BW)、ステンレス綱粒子(Ametek社から販売されている、P316L)、亜鉛粉(Aldrich社)、パラジウム粒子(D13A17、John Matthey Elec.社)、M-450エポキシビーズ(Dynal社)、およびTiO2、SiO2、またはMnO2粒子(Aldrich社)がある。
【0037】
粒子が、細胞よりもより迅速に試料懸濁液を通じ示差的に沈降するように、粒子密度を選択することができる。従って粒子は、細胞分離および粒子の操作を促進するために、高密度材料で構成されることが好ましい。このような粒子の使用は、抗原特異的T細胞、T細胞前駆体、B細胞前駆体、B細胞、または他の細胞からの粒子の分離を促進するために重力下で粒子が沈降することを可能にする。
【0038】
高密度粒子を使用する更なる利点は、大量の非標的細胞を、治療適用に有用である標的細胞を失うことなく、除くことができることである。複数の細胞分離サイクルを、Kenyonらの論文に記されたように行うことができ(「High Density Particles: A Novel, Highly
Efficient Cell Separation Technology」、CELL SEPARATION METHODS AND APPLICATIONS、Recktenwald およびRadbruch編集、Marcel Dekker社、2000年、103-32頁)、その結果1回の枯渇サイクルにつきわずかに2〜3%の非特異的細胞が喪失される。複数回サイクル法を用い、非標的細胞を、標的細胞(例えばTまたはB細胞前駆体)を著しく喪失することなく、血液産物から除くことができる。標的細胞の回収は、90%を越えることができる。例えば、高密度の粒子は、平均4.7 logにまで動員された成分採血産物(mobilized apheresis product)において正常B細胞を減少するが、3回の枯渇サイクルを使用したシステムにおいてはCD34+細胞の90%よりも多くを維持する。Houdeら、Blood、96:187a(2000)。
【0039】
ひとつの態様において、粒子は、密度約9gm/km3を有しおよび磁気を帯びたニッケル粒子(例えば、Novamet社のタイプ123ニッケル粒子、これはサイズ範囲3〜7μm)である。粒子標的細胞複合体を捕獲するために磁石を使用しなければならないような他の市販の粒子とは異なり、高密度ニッケル粒子は重力により沈降する。沈降後、磁石を用い、懸濁液中の細胞から望ましくない粒子を分離することができる。ニッケル粒子は、リガンドカップリング化学にとって有用である官能基部分を伴う様々な高分子および無機分子の付着を可能にする化学特性も有する。
【0040】
粒子の形状は、不規則な形から球形へ、および/または平らでないかもしくは不規則な表面を有するものから平坦な表面を有するものまで変動することができる。粒子の好ましい特性は、抗原提示プラットフォームが調製および/または使用される特定の条件に応じて選択することができる。例えば、球形粒子は、不規則なサイズの粒子と比べ、より少ない表面積を有する。球形粒子が使用される場合は、減少した表面積のために、より少ない試薬が必要となる。他方で、不規則な形状の粒子は、球形粒子よりも有意に大きい表面積を有し、このことは複合した蛋白質含量/表面積および細胞の表面積接触に関して利点を提供する。
【0041】
粒子サイズも変動することができる。粒子サイズ(公称直径)は、本発明には重要ではないが、典型的には0.05〜50μm、より典型的には3〜35μmの範囲であり、および好ましくは約5μmである。これらの粒子は、サイズが均一であることができるか、またはサイズが変動することができ、平均粒子サイズは、好ましくは0.05〜50μmの範囲である。他の粒子は、微粉砕された粉末または超微細粒子であることができる。公称直径約5μmを有するニッケル粉末の粒子は、優れた蛋白質吸着特性を有する。ひとつの態様において、これらの粒子は、表面積が少なくとも0.4m2/g、好ましくは約0.4m2/g〜約0.5m2/gを有する。粒子サイズ分布は都合の良いことに、例えば、動的光散乱を基にしたMicrotrak装置を用い、決定することができる。
【0042】
固形支持体コーティング
固形支持体は、蛋白質がその表面に結合する前にコートすることができる。一旦コーティング化学が選択されたならば、固形支持体の表面は活性化され、特定の蛋白質分子の特異的付着を可能にすることができる。従ってコーティングは、様々なTもしくはB細胞集団またはTもしくはB前駆体細胞集団との最適な反応性および生体適合性を考慮し選択することができる。いかなるコーティング化学が使用されようと、更なる活性化化学のために適当なマトリックスを提供することが好ましい。多くのこのようなコーティングが、当技術分野において周知である。例えば、固形支持体は、ヒト血清アルブミン、トリス(3-メルカプトプロピル)-N-グリシルアミノ)メタン(米国特許第6,074,884号)、ゼラチン-アミノデキストラン(米国特許第5,466,609号)、またはアミノ酸ホモポリマーもしくはランダムコポリマーでコートすることができる。ひとつの態様において、ポリ(グルタミン酸、リシン、チロシン)[6:3:1]を含有するランダムアミノ酸コポリマーが使用され;このコポリマーは、Sigma Chemical社から、製品番号No.P8854として入手できる。これは、6部のグルタミン酸、3部のリシン、および1部のチロシンの比である、アミノ酸グルタミン酸・リシン・チロシンの線状ランダムポリマーである。別の態様において、4部のリシン対1部のチロシンの比である、リシンおよびチロシンを含むアミノ酸コポリマーが使用される。更に別の態様において、1部のリシン対1部のアラニンの比である、リシンおよびアラニンを含むアミノ酸コポリマーが使用される。
【0043】
別の態様において、固形支持体は、合成ポリマーによりコートされ、次にこの合成ポリマーは活性化され、その後TまたはB細胞に影響する分子、抗原提示複合体、またはB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する分子複合体を含むが、これらに限定されるものではない蛋白質分子に連結される。
【0044】
シリカ(SiO2)によるコーティング
別の態様、特にニッケル表面(特に粒子)に良く適した態様において、固形支持体は、シリカによりコートされている。シリカ表面は、通常使用される有機ポリマー表面に勝るいくつかの利点を有する。これは高度に均質であり、化学的に定義され、ならびに化学的および熱的に安定しており、シラノール残基がその表面全体を被覆し、ならびに蛋白質および他の生体分子の付着のために、トリエトキシシランのアミノ誘導体またはエポキシ誘導体との安定した共有結合に使用することができる。シラン誘導体は、表面全体を被覆することができ、表面との特異的および非特異的相互作用に高度の制御が可能である二次元ポリマーの単層を形成する。
【0045】
様々な固形支持体のシリカによるコーティングの方法は、米国特許第2,885,399に開示されており;同じく、Birkmeyerら、Clin Chem.、33(9):1543-7(1987年9月)を参照のこと。例えば、固形支持体は、メタケイ酸ナトリウム、アルミン酸ナトリウム、およびホウ酸の溶液と共にインキュベーションし、表面に沈着した重合されたシリカを形成することができる。シリカコーティングの別の方法は、ケイ酸ナトリウムを固形支持体と混合し、95℃で硫酸を用いpHを低下し、その後水で洗浄する。米国特許第2,885,366号;Eagerton、KONA、16:46-58(1998)を参照のこと。例えば、ニッケル表面は、最初に0.2N NaSO4溶液中にそれらを分散し、この溶液を95℃に加熱することにより、コーティングすることができる。このpHは、NaOHにより10に調節される。その後硫酸中のケイ酸ナトリウムが添加され、95℃で0.5時間混合される。この支持体は、蒸留水により数回洗浄される。コーティングの程度は、支持体の硝酸消化に対する抵抗を決定することにより試験することができる。
【0046】
X線散乱を基にした、表面化学組成に関するESCA分析を用い、支持体表面の元素組成を得ることができ、活性残基による表面コーティングおよびシラン化の程度に関する情報を提供する。
【0047】
酸化アルミニウムによるコーティング
別の態様において、固形支持体上の表面マトリックスが、ニッケル表面の、例えば酸化アルミニウムなどの、無毒の金属酸化物コーティングによる「不動態化」により提供される。コーティングの他の方法は、酸化アルミニウムのような金属酸化物の、固形支持体の表面への沈着を含む。酸化アルミニウムは、蛋白質複合のために官能基化することができる低い非特異的結合特性を伴う不活性表面を提供するので、これは有用なマトリックスである。
【0048】
酸化アルミニウムコーティングは、ゾル-ゲル法のような、多くの方法により提供され、ここで非晶質酸化アルミニウムの薄い連続層が、固形支持体上へのアルミニウムゾル-ゲルの蒸発により形成され、その後空気中で焼成(baking)され、酸化物が形成される。Ozerら、SPIE、3789:77-83(1999)参照のこと。別の態様において、通常の物理的蒸着法(Smidt、Inter Mat. Rev.、35:21-27(1990))または化学蒸着(Kohら、Thin Solid Films、304:222-24(1997))を使用することができる。ニッケル固形支持体が使用される場合、このようなコーティングの厚さは、適切な安定性を提供すると同時に、ニッケルの滲出を最小化するように制御することができる。ニッケル封印の成功は、ニッケルイオンの定量的化学アッセイ法により試験することができる。固形支持体は、様々な緩衝液および生物学的液体中で、様々な温度で、インキュベーションすることができ、これらの媒体中のニッケルイオンのレベルを測定することができる。
【0049】
表面コーティング効率
表面コーティングの完全性は、表面滲出アッセイ法により決定することができる。例えば、ニッケル固形支持体の表面をガラスまたは他の非反応性金属により完全にコーティングする場合、この固形支持体は、酸性条件下でのニッケル滲出に対して抵抗性である。例えば、被覆されたニッケル固形支持体の既知の塊を、10%硝酸中でインキュベーションし、24時間観察することができる。ニッケルは溶解するので、この溶液は緑色になっていく。未処理のニッケルは、直ぐに溶液を緑色にする。それらの表面上に酸化ニッケルの層を有するニッケル固形支持体は、溶液を約20分間で緑色に変える。先に説明されたようなシリカの層によりコーティングされた固形支持体は、8時間を越える時間硝酸に対し抵抗性があり、これはシリカの厚い層が表面上に沈着されたことを示している。固形支持体は、支持体を、BまたはT細胞活性化に使用した培養条件(下記に説明)に類似した細胞培養培地中でインキュベーションすることにより、水性条件下で試験することもできる。この溶液へ滲出されたニッケルの量は、原子吸光分光測定により測定することができる。
【0050】
コーティング前の予備処理
望ましい場合には、固形支持体は、コーティング前に予備処理することができる。固形支持体の予備処理は、例えば、支持体の滅菌およびパイロジェン除去に加え、支持体の表面上に酸化物の層を形成することができる。この予備処理は、金属の固形支持体が使用される場合に特に有益である。ひとつの態様において、予備処理は、ニッケル固形支持体の約2〜6時間、好ましくは約5時間の、温度範囲約200〜350℃、好ましくは約250℃での加熱が関連する。
【0051】
蛋白質分子の固形支持体への付着
分子は、吸着によるか、または共有結合を含む、直接の化学結合により、固形支持体に直接付着することができる。例えば、Hermanson、BIOCONJUGATE TECHNIQUES、Academic Press社、ニューヨーク、1996年参照。分子それ自身は、求核基、脱離基、または求電子基を含む、様々な化学官能基により、直接活性化することができる。官能基の活性化は、アルキルおよびアシルのハロゲン化物、アミン、スルフヒドリル、アルデヒド、不飽和結合、ヒドラジド、イソシアナート、イソチオシアナート、ケトン、および化学結合を活性化することが分かっている他の基を含む。または分子は、小分子-カップリング試薬の使用を介して、固形支持体に結合することができる。カップリング試薬の非限定的例は、カルボジイミド、マレイミド、N-ヒドロキシスクシンイミドエステル、ビスクロロエチルアミン、グルタルアルデヒドのような二官能性アルデヒド、酸無水物などを含む。別の態様において、分子は、当技術分野において周知である、ビオチンストレプトアビジン連結またはカップリングのような、親和性結合を介して、固形支持体にカップリングされ得る。例えば、ストレプトアビジンは、共有的または非共有的付着により、固形支持体に結合することができ、ビオチン化された分子は、当技術分野において周知の方法を用いて、合成することができる。例えば、Hermansonの論文(1996)を参照のこと。
【0052】
固形支持体への共有結合が企図される場合は、この支持体は、典型的にはリンカーを介し、適当な反応物への共有的付着に利用可能な1種または複数の化学的部分または官能基を含むポリマーによりコーティングされる。例えば、アミノ酸ポリマーは、適当なリンカーを共有的に介した分子のカップリングに使用することができる、リシンのε-アミノ基などの基を有することができる。本発明は、固形支持体の上に第二のコーティングを配置し、これらの官能基を提供することも企図している。
【0053】
活性化化学
活性化化学を使用し、分子を固形支持体の表面へ特異的で安定して付着させることができる。蛋白質を官能基に付けるために使用することができる多くの方法が存在する;例えば、Hermansonの論文(1996)参照。例えば、二工程法において、共通の架橋剤グルタルアルデヒドを用い、蛋白質アミン基を、アミン化された固形支持体表面に付着することができる。得られた連結は、加水分解的に安定である。他の方法は、蛋白質上のアミンと反応するn-ヒドロ-スクシンイミド(NHS)エステルを含む架橋剤、アミン含有、スルフヒドリル含有、またはヒスチジン含有の蛋白質と反応する活性ハロゲンを含む架橋剤、アミンまたはスルフヒドリル基と反応するエポキシドを含む架橋剤の使用、マレイミド基とスルフヒドリド基の間の結合、ならびにペンダント糖部分の過ヨウ素酸酸化による蛋白質アルデヒド基の形成、それに続く還元性アミノ化を含む。
【0054】
ひとつの態様において、蛋白質分子は、3-アミノプロピルトリエトキシシランを用い、シリカコーティングに付着される(WeetallおよびFilbert、Methods Enzymol.、34:59-72(1974))。この化合物は、シリカ表面と安定した共有結合を形成し、同時にその表面をより疎水性にする。シラン化反応は、水性の低いpHの媒質内で行うことができ、これは結合に利用可能なアミノ基を伴う単層を形成することができることがわかっている。蛋白質の付着は、ホモ二官能性カップリング剤グルタルアルデヒドによるか、またはSMCCのようなヘテロ二官能性物質によることができる。蛋白質付着後、残留する表面に会合したカップリング剤は、様々な蛋白質、親水性ポリマー、およびアミノ酸と共にインキュベーションすることにより活性化されることができる。アルブミンおよびポリエチレングリコールが特に適しており、その理由はこれらは、蛋白質および細胞の固相への非特異的結合をブロックするからである。
【0055】
別の態様において、アミノシラン化を用い、酸化アルミニウムで被覆された固形支持体の表面が活性化される。米国特許第4,554,088号(1985)を参照のこと。酸化アルミニウムで被覆された固形支持体の表面を活性化する別の方法は、glu-lys-tyrトリペプチドのような、強力に接着するポリマーの吸着である。このトリペプチドポリマーは、ジフルオロジニトロベンゼンのようなホモ二官能性架橋剤との反応によるか、またはグルタルアルデヒドとの反応により、リシンアミンを介して活性化することができる。その後蛋白質を、活性化された表面へ直接付着することができる。
【0056】
機能性蛋白質複合体の最適化
特異的蛋白質の固形支持体表面への付着は、蛋白質の直接の連結によるか、または間接的方法の使用により、実現することができる。ある種の蛋白質は、それら自身を、直接的付着または複合に適合させるが、他方で他の蛋白質または抗体は、抗マウスIgGまたはストレプトアビジンのような、リンカーまたはスペーサー蛋白質に連結された場合に、より良い機能活性を保持する。望ましい場合には、リンカーまたは付着蛋白質を使用することができる。
【0057】
固形支持体に連結された機能性蛋白質の比の最適化
同じ固形支持体上の特定の蛋白質の比は、抗原または抗体の提示における固形支持体の有効性を増大するように変動することができる。例えば、A2-Ig(下記実施例1に説明)(シグナル1)の抗CD28(シグナル2)に対する最適比は、下記のように試験することができる。固形支持体は、A2-Igおよび抗CD28に30:1、10:1、3:1、1:1、0.3:1;0.1:1、および0.03:1のような様々な比で連結している。支持体に連結した蛋白質の総量は、一定に保たれる(例えば、粒子150mg/mlで)か、または変動することができる。サイトカイン放出および増殖のようなエフェクター機能は、T細胞活性化および分化と比べ、シグナル1対シグナル2の必要要件が異なるので、これらの機能は個別にアッセイすることができる。
【0058】
分析的アッセイ法
固形支持体は、支持体が製造される時に生じる添加(addition)および反応を評価するいくつかの分析的アッセイ法により特徴付けられる。これらは、アミンおよびアルデヒドのような官能基のアッセイ法、ならびに特定の型の蛋白質分子の結合のアッセイ法を含む。加えて機能アッセイ法を用い、固形支持体の生物学的活性を評価することができる。固形支持体の表面に結合した蛋白質の量は、当技術分野において公知の方法のいずれかにより決定することができる。例えば、結合した蛋白質は、280nmでの吸光度を用い、反応液から取り除かれる蛋白質量を決定することにより、間接的に測定することができる。この態様において、固形支持体の添加の前後の反応液の蛋白質含量は、280nmでの吸光度で測定され、および比較される。いずれかの洗浄液に含まれる蛋白質量も測定され、反応後溶液中に認められた量に追加される。この差は、固形支持体表面に結合した量の指標である。この方法を使用し、異なる反応条件の結合効率を迅速にスクリーニングすることができる。
【0059】
別の態様において、固形支持体に結合した蛋白質量は、標識された抗原および抗体の結合アッセイ法による、より直接的アッセイ法で測定することができる。例えば、様々な濃度の抗体結合した固形支持体を、一定濃度のHRP標識した抗原またはヤギ-抗マウスIgGとインキュベーションすることができる。この支持体は、緩衝液で洗浄し、未結合の標識された蛋白質を除去する。支持体-会合したHRPを、OPD基質を用いて測定し、結合した標識された蛋白質の濃度を得る。スキャッチャードプロット解析は、固定された蛋白質の濃度および親和性を提供することができる。HRP標識された抗体は、商業的に入手できるか、または抗体は、AvrameasおよびTernync(Immunochemisty、8:1175-79(1971))のグルタルアルデヒド法を用い、HRPで標識することができる。
【0060】
前述の方法は、共有結合した蛋白質および非共有的に結合した蛋白質の両方を測定する。これら2種の結合の間を識別するために、固形支持体は、6M塩酸グアニジンまたは8M尿素のような、強力なカオトロピック剤で洗浄することができる。非特異的結合は、これらの条件により破壊され、および固形支持体から洗浄除去された蛋白質の量は、280nmでの吸光度により測定することができる。結合した蛋白質総量およびカオトロピック剤で洗浄除去された量の間の差は、密に結合されおよび恐らく共有結合されている蛋白質の量を表す。
【0061】
細胞
本発明の抗原提示プラットフォームおよび抗体誘導プラットフォームの両方は、細胞を基にすることができる。これらの細胞は、好ましくは真核細胞であり、より好ましくは哺乳類細胞であり、更により好ましくは霊長類の細胞であり、最も好ましくはヒト細胞である。
【0062】
本発明のプラットフォーム表面の多くの分子は、クローニングすることができる。従って細胞は、このような分子をコードしている構築物によりトランスフェクションすることができる。細胞をトランスフェクションする方法は、当技術分野において周知であり、トランスフェリン-ポリカチオン-媒介したDNA導入、裸のまたは被包した核酸によるトランスフェクション、リポソーム-媒介型細胞融合、DNA-被覆したラテックスビーズの細胞内輸送、プロトプラスト融合、ウイルス感染、電気穿孔、およびリン酸カルシウム-媒介型トランスフェクションを含むが、これらに限定されるものではない。
【0063】
または、蛋白質は、細胞表面に化学的に結合することができる。この目的のために、蛋白質の細胞表面へのいずれかの連結法を使用することができ、例えば様々なリンカー(例えば、ペプチドリンカー、ストレプトアビジン-ビオチンリンカー)の使用である。
【0064】
抗原提示プラットフォームに連結した分子
抗原提示プラットフォームに連結した分子は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。任意で、抗原は、この抗原結合窩に結合することができる。これらの構成要素を、以下に説明する。
【0065】
抗原提示複合体
抗原提示複合体は、抗原結合窩を含み、ならびにT細胞またはT細胞前駆体への提示のために抗原を結合することができる。抗原提示複合体は、例えば、MHCクラスIまたはクラスII分子、MHCクラスIもしくはクラスII分子の機能的抗原結合窩を含む融合蛋白質、MHCクラスIまたはクラスII「分子複合体」(後述)、またはCD1ファミリーメンバー(例えば、CD1a、CD1b、CD1c、CD1d、およびCD1e)などの非古典的MHC様分子であることができる。
【0066】
一部の態様において、抗原提示複合体は、MHCクラスIおよび/またはMHCクラスII分子複合体である。MHCクラスIおよびクラスII分子複合体は、多くの有用な特徴を有する。例えばこれらは、免疫グロブリン骨格によりもたらされる安定性および分泌効率を基に、極めて安定しており、ならびに作成が容易である。更に、免疫グロブリンのFc部分を変更することにより、異なる生物学的機能を、Fc部分によりもたらされる生物学的機能を基に、分子に提供することができる。ひとつの型の免疫グロブリン分子のFc部分の別のものへの置換は、当技術分野の技術の範囲内である。
【0067】
MHCクラスI分子複合体
「MHCクラスI分子複合体」は、米国特許第6,268,411号に開示されている。MHCクラスI分子複合体は、免疫グロブリン重鎖の末端に高次構造的に無傷の様式で形成される(概略的説明は、米国特許第6,268,411号の図IA参照)。抗原性ペプチドが結合しているMHCクラスI分子複合体は、独自のクローン形質のリンパ球受容体(例えば、T細胞受容体)に安定して結合することができる。
【0068】
MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成し、これは2個のMHCクラスIペプチド結合窩を含む。免疫グロブリン重鎖は、IgM、IgD、IgG1、IgG3、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG重鎖を使用し、MHCクラスI分子複合体を形成する。多価のMHCクラスI分子複合体が望ましい場合は、IgMまたはIgA重鎖を用い、各々、五価または四価の分子を提供することができる。他の価数を伴うMHCクラスI分子複合体も、複数の免疫グロブリン重鎖を用い構築することができる。MHCクラスI分子複合体の構築は、米国特許第6,268,411号に詳細に開示されている。
【0069】
MHCクラスII分子複合体
「MHCクラスII分子複合体」は、米国特許第6,458,354号、米国特許第6,015,884号、米国特許第6,140,113号、および米国特許第6,448,071号に開示されている。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。これら2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各々の第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各々の第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。
【0070】
免疫グロブリン重鎖は、IgM、IgD、IgG3、IgG1、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG1重鎖を使用し、2個の抗原結合窩を含む二価の分子複合体を形成する。任意で、重鎖の可変領域を含むことができる。IgMまたはIgA重鎖を用い、各々、五価または四価の分子複合体を提供することができる。他の価数を伴う分子複合体も、複数の免疫グロブリン鎖を用い構築することができる。
【0071】
MHCクラスII分子複合体の融合蛋白質は、免疫グロブリン鎖とMHCクラスIIポリペプチドの細胞外ドメインの間に挿入されたペプチドリンカーを含むことができる。リンカー配列の長さは、抗原結合および受容体架橋の程度を調節するために必要な可変性に応じて、変動することができる。MHCクラスIIポリペプチドの細胞外ドメインが、リンカー領域を追加することなく、免疫グロブリン分子に直接的および共有的に付着されるように、構築物を設計することもできる。
【0072】
リンカー領域が含まれる場合は、この領域は、好ましくは少なくとも3種および30種を超えないアミノ酸を含むと思われる。より好ましくは、このリンカーは、約5個および20個を超えないアミノ酸であり;最も好ましくは、リンカーは、10個未満のアミノ酸である。一般にリンカーは、短いグリシン/セリンスペーサーからなるが、あらゆるアミノ酸を使用することができる。免疫グロブリン重鎖をMHCクラスIIβ鎖の細胞外ドメインに接続するのに好ましいリンカーは、
(配列番号:1)である。免疫グロブリン軽鎖をMHCクラスIIα鎖の細胞外ドメインに接続するのに好ましいリンカーは、
(配列番号:2)である。
【0073】
T細胞に影響する分子
T細胞に影響する分子は、前駆体T細胞または抗原特異的T細胞に生物学的作用を有する分子である。このような生物学的作用は、例えば、前駆体T細胞の、CTL、ヘルパーT細胞(例えば、Th1、Th2)、または調節T細胞への分化;T細胞の増殖;ならびに、T細胞アポトーシスの誘導を含むが、これらに限定されるものではない。従って、T細胞に影響する分子は、T細胞共刺激分子、接着分子、T細胞増殖因子、調節T細胞誘導分子、およびアポトーシス誘導分子を含む。本発明の抗原提示プラットフォームは、このような分子の少なくとも1種を含み;任意で、抗原提示プラットフォームは、少なくとも2種、3種または4種のこのような分子を、いずれかの組合せで含む。
【0074】
T細胞共刺激分子は、抗原特異的T細胞の活性化に貢献している。このような分子は、CD28(抗体を含む)、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体を含むが、これらに限定されるものではない。
【0075】
本発明の抗原提示プラットフォームに有用な接着分子は、T細胞またはT細胞前駆体のこのプラットフォームへの接着を媒介する。本発明に有用な接着分子は、例えば、ICAM-1およびLFA-3を含む。
【0076】
T細胞増殖因子は、T細胞の増殖および/または分化に影響を及ぼす。T細胞増殖因子の例は、サイトカイン(例えば、インターロイキン、インターフェロン)およびスーパー抗原である。特に有用なサイトカインは、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンである。望ましい場合には、サイトカインは、融合蛋白質を含む分子複合体中に存在することができる。ひとつの態様において、サイトカイン分子複合体は、少なくとも2種の融合蛋白質を含むことができ:第一の融合蛋白質は、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のサイトカインおよび第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、サイトカイン分子複合体を形成する。別の態様において、サイトカイン分子複合体は、少なくとも4種の融合蛋白質を含み:2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)第一のサイトカインを含み、ならびに2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)第二のサイトカインを含む。2種の第一および2種の第二の融合蛋白質は会合し、サイトカイン分子複合体を形成する。いずれかの型のサイトカイン分子複合体中の第一および第二のサイトカインは、同じまたは異なることができる。
【0077】
スーパー抗原は、強力なT細胞マイトジェンである。スーパー抗原は、最初にクラスII主要組織適合(MHC)分子へ結合し、その後T細胞抗原受容体(TCR)へVβ特異的様式でバイナリー複合体として結合することにより、T細胞の有糸分裂を刺激する。スーパー抗原は、細菌エンテロトキシン、例えばブドウ球菌エンテロトキシン(例えば、SEAおよびそれらの活性部分、米国特許第5,859,207号に開示;SEB、SEC、SEDおよびSEEレトロウイルススーパー抗原(米国特許第5,519,114号に開示);化膿性連鎖球菌(Streptococcus pyogenos)の菌体外毒素(SPE)、黄色ブドウ球菌(Staphylococcus aureus)中毒性ショック症候群毒素(TSST-1)、連鎖球菌性マイトジェン性菌体外毒素(SME)、および連鎖球菌性スーパー抗原(SSA)(米国特許第2003/0039655号に開示);ならびに、米国特許第2003/0036644号および米国特許第2003/0009015号に開示されたスーパー抗原を含むが、これらに限定されるものではない。
【0078】
調節T細胞誘導分子は、調節T細胞の分化および/または維持を誘導する分子である。このような分子は、TGFβ、IL-10、インターフェロン-α、およびIL-15を含むが、これらに限定されるものではない。例えば、米国特許第2003/0049696号、米国特許第2002/0090724号、米国特許第2002/0090357号、米国特許第2002/0034500号、および米国特許第2003/0064067号を参照のこと。
【0079】
アポトーシス誘導する分子は、細胞死を引き起こす。アポトーシス誘導する分子は、毒性物質(例えば、リシンA鎖、突然変異体シュードモナス(Pseudomonas)菌体外毒素、ジフテリア毒素、ストレプトニグリン、ボアマイシン(boamycin)、サポリン、ゲロニン、およびアメリカヤマゴボウ抗ウイルス蛋白質)、TNFα、およびFasリガンドを含む。
【0080】
抗原
様々な抗原は、抗原提示複合体に結合することができる。抗原の性質は、使用される抗原提示複合体の種類によって決まる。例えば、ペプチド抗原は、MHCクラスIおよびクラスIIペプチド結合窩に結合することができる。非古典的MHC様分子は、リン脂質、複合糖質など(例えば、ミコール酸およびリポアラビノマンナンのような、細菌の膜成分)のような、非ペプチド抗原を提示するために使用することができる。本明細書において使用される「抗原」は、「抗原性ペプチド」も含む。
【0081】
抗原性ペプチド
免疫応答を誘導することが可能であるいずれかのペプチドは、抗原提示複合体に結合することができる。抗原性ペプチドは、腫瘍関連抗原、自己抗原、アロ抗原、および感染物質の抗原を含む。
【0082】
腫瘍関連抗原
腫瘍関連抗原は、それらが由来した腫瘍により専ら発現され、多くの腫瘍において発現されたが正常な成人組織においては発現されない腫瘍抗原(腫瘍胎児性抗原)を共有した独自の腫瘍抗原、ならびに同じくそれから腫瘍が生じる正常組織により発現された組織特異抗原を含む。腫瘍関連抗原は、例えば、胚性抗原(embryonic antigen)、異常な翻訳後修飾を伴う抗原、分化抗原、突然変異した癌遺伝子または腫瘍抑制因子の産物、融合蛋白質、またはオンコウイルス蛋白質であることができる。
【0083】
様々な腫瘍関連抗原が当技術分野において公知であり、それらの多くは市販されている。腫瘍胎児性抗原および胚性抗原は、癌胎児性抗原およびα-フェトプロテイン(通常発生期の胚においてのみ高発現されるが、各々、肝臓および結腸の腫瘍において頻繁に高発現される)、MAGE-1およびMAGE-3(黒色腫、乳癌、および神経膠腫において発現される)、胎盤アルカリホスファターゼシアリル-Lewis X(腺癌において発現される)、CA-125およびCA-19(消化器、肝臓、および婦人科の腫瘍において発現される)、TAG-72(結腸直腸癌において発現される)、上皮糖蛋白質2(多くの癌腫において発現される)、膵臓癌胎児性抗原、5T4(胃癌において発現される)、αフェトプロテイン受容体(複数の腫瘍型、特に乳癌において発現される)、およびM2A(生殖細胞腫瘍において発現される)を含む。
【0084】
腫瘍関連分化抗原は、チロシナーゼ(黒色腫において発現される)および特に表面免疫グロブリン(リンパ腫において発現される)を含む。
【0085】
突然変異した癌遺伝子または腫瘍抑制遺伝子の産物は、両方とも多くの腫瘍型において発現されるRasおよびp53、Her-2/neu(乳癌および婦人科の癌において発現される)、EGF-R、エストロゲン受容体、プロゲステロン受容体、網膜芽細胞腫産物、myc(肺癌に関連)、ras、p53、乳癌に関連した非突然変異体、MAGE-1、およびMAGE-3(黒色腫、肺癌、および他の癌に関連)を含む。
【0086】
融合蛋白質は、慢性骨髄性白血病において発現されるBCR-ABLを含む。
【0087】
オンコウイルス蛋白質は、HPV型16、E6、およびE7を含み、これらは頸癌において認められる。
【0088】
組織特異抗原は、メラノトランスフェリンおよびMUCI(膵臓癌および乳癌において発現される);CD10(コモン急性リンパ芽性白血病抗原、またはCALLAとして以前は知られている)または表面免疫グロブリン(B細胞白血病およびリンパ腫において発現される);IL-2受容体α鎖、T細胞受容体、CD45R、CD4+/CD8+(T細胞白血病およびリンパ腫において発現される);前立腺特異的抗原および前立腺酸性ホスファターゼ(前立腺癌において発現される);GP100、MelanA/Mart-1、チロシナーゼ、gp75/ブラウン、BAGE、およびS-100(黒色腫において発現される);サイトケラチン(様々な癌において発現される);ならびに、CD19、CD20、およびCD37(リンパ腫において発現される)を含む。
【0089】
腫瘍関連抗原は、変更された糖脂質抗原および糖蛋白質抗原、例えば、ノイラミン酸含有スフィンゴ糖脂質(例えば、GM2およびGD2、黒色腫および一部の脳腫瘍において発現される);癌腫において異常に発現される血液型抗原、特にT抗原およびシアリル化されたTn抗原;ならびに、ムチン、例えばCA-125およびCA-19-9(卵巣癌において発現される)または低グリコシル化されたMUC-1(乳癌および膵臓癌において発現される)なども含む。
【0090】
組織特異抗原は、上皮膜抗原(多発性上皮癌)、CYFRA 21-1(肺癌において発現される)、Ep-CAM(汎-癌腫(pan-carcinoma)において発現される)、CA125(卵巣癌において発現される)、無傷のモノクローナル免疫グロブリン断片または軽鎖断片(骨髄腫において発現される)、ならびに、ヒト絨毛性ゴナドトロピンβサブユニット(HCG、生殖細胞腫瘍において発現される)を含む。
【0091】
自己抗原
自己抗原は、生体が免疫応答において産生する、生体自身の「自分の抗原」である。自己抗原は、グッドパスチャー症候群、多発性硬化症、グレーヴス症、重症筋無力症、全身性紅斑狼瘡、インスリン依存型真性糖尿病、リウマチ様関節炎、尋常性天疱瘡、アジソン病、疱疹状皮膚炎、セリアック病、および橋本甲状腺炎のような、自己免疫疾患に関連している。
【0092】
糖尿病に関連した自己抗原は、インスリン、グルタミン酸脱炭酸酵素(GAD)および他の島細胞の自己抗原、例えばICA 512/IA-2蛋白質チロシンホスファターゼ、ICA12、ICA69、プレプロインスリンまたはそれらの免疫学的活性断片(例えば、インスリンB-鎖、A-鎖、Cペプチドもしくはそれらの免疫学的活性断片)、HSP60、カルボキシペプチダーゼH、ペリフェリン、ガングリオシド(例えば、GM1-2、GM3)またはそれらの免疫学的活性断片を含む。
【0093】
黄斑変性に関連した自己抗原は、補体系分子、ならびにRPE、脈絡膜および網膜からの様々な自己抗原、ビトロネクチン、βクリスタリン、カルレチキュリン、セロトランスフェリン、ケラチン、ピルビン酸カルボキシラーゼ、C1、およびビリン2を含む。
【0094】
他の自己抗原は、ヌクレオソーム(ヒストンおよびDNAを含む粒子);リボヌクレオ蛋白質(RNP)粒子(RNP粒子内でRNAおよび特定機能を媒介する蛋白質を含む)、および2本鎖DNAを含む。更に他の自己抗原は、ミエリン希突起膠細胞糖蛋白質(MOG)、ミエリン結合糖蛋白質(MAG)、ミエリン/稀突起膠細胞塩基性蛋白質(MOBP)、稀突起膠細胞特異的蛋白質(Osp)、ミエリン塩基性蛋白質(MBP)、蛋白脂質アポ蛋白質(PLP)、ガラクトースセレブロシド(GalC)、糖脂質、スフィンゴ脂質、リン脂質、ガングリオシド、および他の神経抗原を含む。
【0095】
アロ抗原
アロ抗原は、同じ種の別のメンバーにより抗原として検出される対立遺伝子の直接的または間接的産物である。このような対立遺伝子の直接的産物は、コードされたポリペプチドを含み;間接的産物は、対立遺伝子がコードしている酵素により合成された多糖および脂質を含む。アロ抗原は、主要および非主要組織適合性抗原(ヒトにおいてHLAとして知られている)を含み、これは、クラスIおよびクラスII抗原、ABO、Lewis血液型のような血液型抗原、TおよびB細胞上の抗原、ならびに単球/内皮細胞抗原を含む。HLA特異性は、A(例えば、A1-A74、特にA1、A2、A3、A11、A23、A24、A28、A30、A33)、B(例えば、B1-B77、特にB7、B8、B35、B44、B53、B60、B62)、C(例えばC1-C11)、D(例えばD1-D26)、DR(例えばDR1、DR2、DR3、DR4、DR7、DR8、およびDR11)、DQ(例えばDQ1-DQ9)、ならびにDP(例えば、DP1-DP6)を含む。
【0096】
感染物質の抗原
感染物質の抗原は、原生動物、細菌、真菌(単細胞および多細胞の両方)、ウイルス、プリオン、細胞内寄生体、蠕虫、および免疫応答を誘導する他の感染物質の構成要素を含む。
【0097】
細菌抗原は、グラム陽性球菌、グラム陽性桿菌、グラム陰性菌、好気性菌、例えば、放線菌科(Actinomycetaceae)、バシラス科(Bacillaceae)、バルトネラ科(Burtonellaceae)、ボルデテラ科(Bordetellae)、カプトファーガ科(Captophagaceae)、コリネバクテリウム科(Corynebacteriaceae)、エンテロバクテリア科(Enterobacteriaceae)、レジオネラ科(Legionellaceae)、ミクロコッカス科(Micrococcaceae)、マイコバクテリウム科(Mycobacteriaceae)、ノカルジア科(Nocardiaceae)、パスツレラ科(Pasteurellaeceae)、シュードモナス科(Pseudomonadaceae)、スピロヘータ科(Spirochaetaceae)、ビブリオ科(Vibrionaceae)の生物、ならびにアシネトバクター(Acinotobacter)属、ブルセラ(Brucella)属、カンピロバクター(Campylobacter)属、エリジペロトリックス(Erysipelothrix)属、ユーインゲラ(Ewingella)属、フランシセラ(Francisella)属、ガルドネラ(Gardnerella)属、ヘリコバクター(Helicobacter)属、レビネア(Levinea)属、リステリア(Listeria)属、ストレプトバシラス(Streptobacillus)属およびトロフェリーマ(Tropheryma)属の生物の抗原を含む。
【0098】
原生動物の感染物質の抗原は、マラリア原虫、リーシュマニア(Leishmania)種、トリパノソーマ(Trypanosoma)種および住血吸虫(Schistosoma)種の抗原を含む。
【0099】
真菌抗原は、アスペルギルス(Aspergillus)、ブラストミセス(Blastomyces)、カンジダ(Candida)、コクシジオイデス(Coccidioides)、クリプトコッカス(Cryptococcus)、ヒストプラスマ(Histoplasma)、パラコクシジオイデス(Paracoccicioides)、スポロスリックス(Sporothrix)、ムコール(Mucoraiea)目の生物、クロマイコシス(choromycosis)および菌腫(mycetoma)を含む生物、ならびに白癬菌(Trichophyton)属、小胞子菌(Microsporum)属、表皮菌(Epidermophton)属およびマラセジア(Malassezia)属の生物の抗原を含む。
【0100】
プリオンの抗原は、スクレイピー、ウシ海綿状脳症(BSE)、ネコ海綿状脳症、クールー病、クロイツフェルト-ヤコブ病(CJD)、ゲルストマン-シュトロイスラー-シャインカー病(GSS)、および致命的家族性不眠症(FFI)を引き起こす、プリオンのシアロ糖蛋白質PrP 27-30を含む。
【0101】
抗原性ペプチドが得られる細胞内寄生体は、クラミジア科(chlamydiaceae)、マイコプラズマ科(Mycoplasmataceae)、アコレプラスマ科(Acholeplasmataceae)、リケッチア科(Richettsiae)、ならびにコクシエラ科(Coxiella)およびエーリッヒア属(Ehrlishia)の生物を含むが、これらに限定されるものではない。
【0102】
抗原性ペプチドは、線虫、吸虫または条虫のような蠕虫から得られる。
【0103】
ウイルスのペプチド抗原は、アデノウイルス、単純ヘルペスウイルス、パピローマウイルス、呼吸シンシチアウイルス、ポックスウイルス、HIV、インフルエンザウイルス、およびCMVのものを含むが、これらに限定されるものではない。特に有用なウイルスのペプチド抗原は、HIV gag蛋白質(膜アンカー(MA)蛋白質、コアキャプシド(CA)蛋白質およびヌクレオキャプシド(NC)蛋白質を含むが、これらに限定されるものではない)、HIVポリメラーゼのような、HIV蛋白質、インフルエンザウイルスマトリックス(M)蛋白質およびインフルエンザウイルスヌクレオキャプシド(NP)蛋白質、B型肝炎表面抗原(HBsAg)、B型肝炎コア蛋白質(HBcAg)、B型肝炎e蛋白質(HBeAg)、B型肝炎DNAポリメラーゼ、およびC型肝炎抗原などを含む。
【0104】
抗原の抗原提示複合体への結合
抗原性ペプチドを含む抗原は、米国特許第6,268,411号に開示されたように、能動的又は受動的のいずれかで、抗原提示複合体の抗原結合窩に結合することができる。任意で、抗原性ペプチドは、ペプチド結合窩に共有結合することができる。
【0105】
望ましい場合には、ペプチドテザー(tether)を用い、抗原性ペプチドをペプチド結合窩に連結することができる。例えば、複数のクラスI MHC分子の結晶学的解析は、MHCペプチド結合窩内に存在する抗原性ペプチドのカルボキシ末端と、β2Mのアミノ末端が非常に近接し、約20.5Å離れていることを示している。従って、およそ13個のアミノ酸長のような比較的短いリンカー配列を用い、ペプチドを、β2Mのアミノ末端につなぎ止めることができる。この配列が適している場合には、そのペプチドは、MHC結合溝に結合すると思われる(米国特許第6,268,411号参照)。
【0106】
抗体誘導プラットフォームに連結した分子
抗体誘導プラットフォームに連結した分子は、少なくとも1種のB細胞に影響する分子、ならびにB細胞表面免疫グロブリンに結合することができるかまたはB細胞表面の抗原含有MHC複合体に結合することができる、少なくとも1種の分子複合体を含む。
【0107】
B細胞に影響する分子
B細胞に影響する分子は、B細胞またはB細胞前駆体に対し、例えば増殖の誘導または抗体形成のような生物学的作用を有する分子である。このような分子は、CD40リガンドに加え、前述のサイトカインおよびサイトカイン分子複合体を含む。使用されるサイトカイン分子の種類に応じて、B細胞は、特定の種類の抗体を産生するようにしむけられる。例えば、IL-4はIgEの産生を誘導するのに対し、IL-5はIgAの産生を誘導する。
【0108】
分子複合体
抗体誘導プラットフォームにおいて使用するための分子複合体は、B細胞表面免疫グロブリンに結合するか、またはB細胞表面のMHC-抗原複合体に結合する複合体である。B細胞表面免疫グロブリンに結合している分子複合体は、プラットフォーム表面に複合された抗原を含む。B細胞表面のMHC-抗原複合体に結合している分子複合体は、T細胞受容体(TCR)およびTCR分子複合体を含む。抗体誘導プラットフォームは、このような分子複合体の一方または両方の型を含むことができる(すなわち、B細胞表面免疫グロブリン結合またはMHC-抗原結合)。
【0109】
特定の抗原に関するTCR特異性は、当技術分野において周知の方法を用いクローニングすることができる。例えば、米国特許第2002/0064521号を参照のこと。クローニングされた抗原特異的TCRを、そのように用いることができるか、または下記のTCR分子複合体を形成するために使用することができる。
【0110】
TCR分子複合体
「TCR分子複合体」は、米国特許第6,458,354号、米国特許第6,015,884号、米国特許第6,140,113号、および米国特許第6,448,071号において開示されている。TCR分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)TCRα鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)TCRβ鎖の細胞外ドメインを含む。または、2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)TCRγ鎖の細胞外ドメインを含み、ならびに2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)TCRδ鎖の細胞外ドメインを含む。この2種の第一および2種の第二の融合蛋白質は会合し、TCR分子複合体を形成する。各第一の融合蛋白質のTCR鎖の細胞外ドメイン、および各第二の融合蛋白質のTCR鎖の細胞外ドメインは、抗原認識窩を形成する。
【0111】
免疫グロブリン重鎖は、IgM、IgD、IgG3、IgG1、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG1重鎖を用い、2個の抗原認識窩を含む二価のTCR分子複合体を形成する。任意で、重鎖の可変領域を含むことができる。IgMまたはIgA重鎖を使用し、各々、五価または四価のTCR分子複合体を提供する。他の価数を有するTCR分子複合体も、複数の免疫グロブリン鎖を用い構築することができる。
【0112】
TCR分子複合体の融合蛋白質は、免疫グロブリン鎖とTCRポリペプチドの細胞外ドメインの間に挿入されたペプチドリンカーを含むことができる。このリンカー配列の長さは、抗原結合および架橋の程度を調節するために必要である可変性に応じて、変動することができる。構築物は、TCRポリペプチドの細胞外ドメインが、追加のリンカー領域を伴うことなく、免疫グロブリン分子に直接および共有的に付着するように、設計することもできる。リンカー領域が含まれる場合は、この領域は、少なくとも3個および30個を超えないアミノ酸を含むことが好ましい。より好ましくは、このリンカーは、約5個および20個を超えないアミノ酸であり;最も好ましくは、このリンカーは、10個未満のアミノ酸である。一般に、このリンカーは、短いグリシン/セリンスペーサーからなるが、いずれかのアミノ酸を使用することができる。免疫グロブリン重鎖をTCRαまたはγ鎖の細胞外ドメインへ結合するために好ましいリンカーは、
(配列番号:1)である。免疫グロブリン重鎖をTCRβまたはδ鎖の細胞外ドメインへ結合するために好ましいリンカーは、
(配列番号:2)である。
【0113】
特異的細胞集団を誘導および拡大するための本発明のプラットフォームの使用法
抗原特異的T細胞の誘導および拡大
本発明は、CTL、ヘルパーT細胞、および調節T細胞を含む、抗原特異的T細胞の形成を誘導しおよび拡大する方法を提供する。これらの方法は、複数の前駆体T細胞を含有する単離された調製物を、抗原が抗原結合窩に結合した本発明の抗原提示プラットフォームと接触することが関連している。この調製物の抗原提示プラットフォームと一緒のインキュベーションは、集団内の前駆体細胞を誘導し、その抗原を認識する抗原特異的T細胞を形成する。抗原特異的T細胞は、前駆体T細胞の、後述の本発明の抗原提示プラットフォームと一緒のインキュベーションにより得ることができるか、または例えば樹状細胞とのインキュベーション、もしくは当技術分野において公知の人工的抗原提示細胞とのインキュベーションのような、常法により得ることができる。
【0114】
典型的には、第一の細胞集団内の抗原特異的T細胞の数または割合のいずれかは、前駆体T細胞が、CD3に特異的に結合する抗体を含むが、抗原提示複合体を含まないような粒子と共にインキュベーションされた場合に形成された抗原特異的T細胞の数または割合よりも大きい。
【0115】
抗原提示プラットフォームが使用される本明細書において開示されたいずれかの態様において、抗原提示複合体、結合した抗原、およびT細胞に影響する分子のいずれかの組合せを使用することができる。例えば、抗原提示プラットフォームは、1種または複数のT細胞共刺激分子(同じまたは異なるのいずれか)、1種または複数の調節T細胞誘導する分子(同じまたは異なるのいずれか)、1種または複数の接着分子(同じまたは異なるのいずれか)、および/または1種または複数のT細胞増殖因子(同じまたは異なるのいずれか)を含むことができる。同様に、いずれか特定の抗原提示プラットフォームは、同じまたは異なるのいずれかの1種または複数の抗原提示複合体を含むことができ、これに抗原の組合せが結合することができる。ひとつの態様において、いくつかの異なる黒色腫-関連抗原(例えば、チロシナーゼ、MAGE-1、MAGE-3、GP-100、Melan A/Mart-1、gp75/ブラウン、BAGE、およびS-100のいずれかまたは全て)は、1種または複数のプラットフォーム上の抗原提示複合体に結合することができる。
【0116】
前駆体T細胞は、患者または適当なドナーから得ることができる。ドナーは、同一の双子である必要はなく、更に患者に関連する必要もない。しかし、ドナーおよび患者は、少なくとも1種のHLA分子を共有していることが好ましい。前駆体T細胞は、末梢血単核細胞、骨髄、リンパ節組織、脾臓組織、および腫瘍を含む、多くの給源から得ることができる。または、当技術分野においてT細胞系が使用される。
【0117】
ひとつの態様において、前駆体T細胞は、フィコール(Ficoll)分離のような、当業者に公知の多くの技術を用い、対象から収集された血液単位から得られる。例えば、個人の循環血からの前駆体T細胞は、成分採血または白血球搬出により得ることができる。成分採血産物は、典型的には、T細胞および前駆体T細胞を含むリンパ球、単球、顆粒球、B細胞、他の有核白血球、赤血球、および血小板を含む。成分採血により収集された細胞は、血漿画分を除去するため、および引き続きの処理工程のために細胞を適当な緩衝液または培地中に配置するために、洗浄することができる。洗浄工程は、例えば半-自動化された「フロー-スルー」遠心分離(例えば、Cobe 2991細胞プロセッサー)を製造業者の指示に従い使用することにより、当業者に公知の方法で行うことができる。洗浄後、細胞を、例えば、Ca非含有、Mg非含有PBSのような、様々な生体適合性緩衝液中に再懸濁してもよい。または、成分採血試料中の望ましくない成分を除去することができ、および細胞を直接培養培地中に懸濁することができる。望ましい場合には、赤血球の溶解、および例えばパーコール(PERCOLL)(商標)勾配による遠心分離による単球の枯渇により、前駆体T細胞を、末梢血リンパ球から単離することができる。
【0118】
任意で、抗原特異的T細胞を含む細胞集団は、第一の細胞集団内の抗原特異的T細胞の数に対して増加した数の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な期間、同じ抗原提示プラットフォームまたは第二の抗原提示プラットフォームのいずれかと共に、インキュベーションを継続することができる。典型的には、このようなインキュベーションは、3〜21日間、好ましくは7〜10日間実行される。
【0119】
適当なインキュベーション条件(培養培地、温度など)は、T細胞またはT細胞前駆体の培養に使用されるものに加え、DCもしくは人工の抗原提示細胞を使用する抗原特異的T細胞の形成を誘導するために当業者に公知のものを含む。例えば、LatoucheおよびSadelain、Nature Biotechnol.、18:405-09、4月(2000);Levineら、J. Immunol.、159:5921-30(1997);Mausら、Nature Biotechnol.、20:143-48、2月(2002)を参照のこと。同じく下記具体例を参照のこと。
【0120】
抗原提示プラットフォームとT細胞の間の相互作用期間の最適化
本発明の抗原提示プラットフォームによるT細胞刺激と、通常の正常な樹状細胞によるものの間の差のひとつは、必要とされる刺激期間である。例えば、正常なDCのCTLによる認識は、最終的には溶解および活性化されたT細胞による抗原性刺激の排除につながる。対照的に、T細胞は、抗原提示プラットフォーム上の抗原、特に人工的な生分解性でない表面を基にしたものを排除する効果的方法を有さないことがある。従って、このプラットフォームによる刺激は、数日ではない場合は、数時間行われると思われる。
【0121】
増殖シグナルの大きさを評価するために、抗原特異的T細胞集団は、CFSEで標識され、および細胞分裂の速度および数が分析される。T細胞は、抗原が結合している本発明の抗原提示プラットフォームのいずれかにより1〜2回の刺激ラウンドが行われた後、CFSEで標識された。その時点で、抗原特異的T細胞は、総細胞集団の2〜10%を占める。この抗原特異的T細胞は、抗原特異的染色を用いて検出することができ、その結果抗原特異的T細胞の分裂の速度および数は、CFSEの喪失に従う。刺激後異なる時間で(例えば、12時間、24時間、36時間、48時間および72時間など)、細胞を、抗原提示複合体の染色およびCFSEの両方について分析することができる。抗原が結合していない抗原提示プラットフォームによる刺激を用い、増殖のベースラインレベルを決定することができる。任意に当技術分野において公知のように、増殖は、3H-チミジンの取込みのモニタリングにより検出することができる。
【0122】
培養物は、本発明の抗原提示プラットフォームにより、異なる時間(例えば、0.5時間、2時間、6時間、12時間、36時間に加え、連続する刺激)刺激することができる。粒子または細胞を用いたプラットフォームは、あらゆる複合体を破壊するような激しいピペッティングにより、T細胞から分離することができる。人工の粒子を用いたプラットフォームは、重力により単離することができ;細胞を用いたプラットフォームは、例えばFACSを用い、単離することができる。高度に濃縮された抗原特異的T細胞培養物における刺激時間の作用を、評価することができ、ならびに条件は、プラットフォームの大きい割合(例えば、50%、70%、75%、80%、85%、90%、95%、または98%)を、ほとんど細胞が喪失されることなく回収することができるものと同じである。その後抗原特異的T細胞は、培養物中に戻され、および細胞増殖、増殖速度、アポトーシスに対する作用、様々なエフェクター機能などについて、当技術分野において公知のように分析される。このような条件は、望ましい抗原特異的T細胞反応に応じて変動しても良い。
【0123】
抗原特異的T細胞の検出
本発明の抗原提示プラットフォームのT細胞前駆体の拡大、活性化および分化に対する作用は、かなり多くの当業者に公知の方法においてアッセイすることができる。機能の迅速な決定は、増殖アッセイ法を用い、各T細胞型に特異的マーカーを検出し、培養物中のCTL、ヘルパーT細胞、または調節T細胞の増加を、決定することにより実現することができる。このようなマーカーは、当技術分野において公知である。CTLは、クロム放出アッセイ法を用い、サイトカイン産生または細胞溶解活性をアッセイすることにより検出することができる。
【0124】
プラットフォーム誘導/拡大した抗原特異的T細胞のホーミング受容体の分析
適当なエフェクター機能を伴う抗原特異的T細胞の作成に加え、抗原特異的T細胞の有効性の別のパラメータは、T細胞の病巣への移動を可能にするホーミング受容体の発現である(Sallustoら、Nature、401:708-12(1999);LanzavecchiaおよびSallusto、Science、290:92-97(2000))。適当なホーミング受容体の非存在は、慢性CMVおよびEBV感染症の状況に関与している(Chenら、Blood、98:156-64(2001))。加えて、抗原特異的T細胞を拡大するためのプロフェッショナルAPCの使用と非プロフェッショナルAPCの間の注目されたひとつの差異は、適当なホーミング受容体の発現であり、これはインビボにおける機能不全CTLの存在を説明し得る(Salioら、J. Immunol.、167:1188-97(2001))。
【0125】
例えば、エフェクターCTLの有効性は、ホーミング受容体の下記の表現型、CD62L+、CD45RO+、およびCCR7-に関連されている。従ってプラットフォーム誘導および/または拡大したCTL集団は、これらのホーミング受容体の発現について特徴付けることができる。ホーミング受容体発現は、最初の刺激条件に関連づけられた複合体の特質である。恐らく、これは、共刺激複合体に加え、サイトカイン環境の両方により制御される。関係のあるひとつの重要なサイトカインは、IL-12である(Salioら、2001)。後述のように、本発明のプラットフォームは、生物学的結果のパラメータを最適化するために、個々に個別の構成要素(例えば、T細胞エフェクター分子および抗原提示複合体)を変動する可能性をもたらす。任意で、IL-12などのサイトカインは、抗原特異的T細胞集団におけるホーミング受容体プロファイルに影響を及ぼすために、最初の誘導培地中に含むことができる。
【0126】
誘導および/または拡大された抗原特異的T細胞集団内のオフレート分析
二次抗体反応の進行は、TCR「オフレート(off-rate)」分析により決定される、親和性の焦点化に関係している(Savageら、Immunity、10:485-92:1999;Buschら、J. Exp. Med.、188:61-70(1998);BuschおよびPamer、J. Exp. Med.、189:701-09(1999))。TCRオフレートの減少(すなわち、増加したTCR親和性の結果)は、少ない量の抗原を認識する増加した能力、および関心のあるT細胞集団の生物学的有効性と良く相関するパラメーターである。オフレートは、抗原提示プラットフォーム-媒介した刺激の大きさおよび/または期間を変動することにより、最適化することができる。
【0127】
他の細胞からの抗原特異的T細胞の分離
抗原に結合した抗原特異的T細胞は、結合していない細胞から分離することができる。血漿搬出、フローサイトメトリー、ディファレンシャル遠心分離を含む、当技術分野において公知の方法のいずれかを用い、この分離を実現することができる。ひとつの態様において、T細胞は、例えば、DYNABEADS(登録商標)M-450 CD3/CD28 Tなどの、抗CD3/抗CD28結合したビーズのような、ビーズとの、所望のT細胞の陽性選択に十分な期間のインキュベーションにより、単離される。
【0128】
望ましい場合には、抗原特異的T細胞の亜集団は、存在し得る他の細胞から分離することができる。例えば、CD28+、CD4+、CD8+、CD45RA+、およびCD45RO+T細胞のような、T細胞の特異的亜集団は、陽性または陰性選択技術により、更に単離され得る。ひとつの方法は、負に選択された細胞上に存在する細胞表面マーカーに対するモノクローナル抗体のカクテルを使用する負の磁気免疫接着またはフローサイトメトリーによる、細胞選別および/または選択である。例えば、負の選択によりCD4+細胞を濃縮するために、モノクローナル抗体カクテルは典型的には、CD14、CD20、CD11b、CD16、HLA-DR、およびCD8に対する抗体を含む。抗原特異的調節T細胞は、マーカーFoxp3を用い、検出および/または他の細胞から分離することができる。その時間は、30分〜36時間、もしくは10〜24時間の範囲であることができ、または少なくとも1時間、2時間、3時間、4時間、5時間、または6時間、もしくは少なくとも24時間であることができる。より長いインキュベーション時間を用い、他の細胞と比較しほとんどT細胞が存在しないいずれかの状況、例えば腫瘍組織または免疫無防備状態の個人から、腫瘍浸潤するリンパ球(TIL)を単離するような状況で、T細胞を単離することができる。
【0129】
抗体産生B細胞の誘導および拡大
本発明は、抗体産生B細胞の形成を誘導する方法も提供する。これらの方法は、複数の前駆体B細胞を含む単離された調製物を、本発明の抗体誘導プラットフォームと接触することに関する。調製物の抗体誘導プラットフォームとのインキュベーションは、集団内の前駆体細胞を誘導し、抗原を特異的に認識する抗体を産生する抗体産生B細胞を形成する。典型的には、第一の細胞集団における抗体産生B細胞の数または割合のいずれかは、前駆体B細胞が、非特異性刺激、例えばフィトヘマグルチニン(PHA)、リポ多糖(LPS)、またはヤマゴボウなどと共にインキュベーションされる場合に形成される抗体産生細胞の数または割合よりも大きい。抗体誘導プラットフォームが使用される本明細書において説明したいずれかの態様において、B細胞に影響する分子とB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する複合体との組合せを使用することができる。
【0130】
前駆体B細胞は、患者または適当なドナーから得ることができる。ドナーおよび患者は、関連する必要はないが、少なくとも1種のHLA分子を共有することが好ましい。または、当技術分野において入手可能なB細胞を使用することができる。ひとつの態様において、前駆体B細胞は、フィコール分離のような、当業者に公知のかなり多くの技術を用い、対象から収集した血液単位から得られる。例えば、個人の循環血液からの前駆体B細胞は、前述のように、成分採血または白血球搬出により得ることができる。
【0131】
B細胞またはそれらの前駆体は、当技術分野において公知の方法を用い、培養することができる。例えば、Schultzeら、J. Clin. Invest.、100:2757-65(1997);von Bergwelt-Baildonら、Blood、99:3319-25(2002)参照。このような条件は、B細胞前駆体の、本発明の抗体誘導プラットフォームとのインキュベーションにも適している。
【0132】
任意で、抗体産生B細胞を含む細胞集団は、第一の細胞集団内の抗体産生B細胞の数と比べ増大した数の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な期間、同じ抗体誘導プラットフォームまたは第二の抗体誘導プラットフォームのいずれかと、インキュベーションするように継続することができる。典型的には、このようなインキュベーションは、3〜21日間、好ましくは7〜10日間実行される。
【0133】
抗体誘導プラットフォームとB細胞の間の相互作用期間の最適化
前述のT細胞刺激のように、抗体産生B細胞の集団を誘導または拡大するために必要な刺激の期間は、特に人工的で生分解性でない表面がプラットフォームに使用される場合、通常生じるものとは異なる。従ってプラットフォームによる刺激は、おそらく数日ではない場合は、数時間行われると思われる。本発明の様々な抗体誘導プラットフォームと前駆体もしくは抗体産生B細胞の間の相互作用の期間は、抗原特異的T細胞について先に説明したものに類似した方法を用いて決定することができる。
【0134】
抗体産生B細胞の検出
本発明の抗体産生プラットフォームのB細胞前駆体の拡大、活性化および分化に対する作用は、当業者に公知のかなり多くの方法によりアッセイすることができる。機能の迅速決定は、増殖アッセイ法を用いるか、B細胞特異的マーカーを検出することによるか、または特異的抗体産生をアッセイすることにより実現することができる。
【0135】
薬学的調製物
本発明の粒子または細胞を用いた抗原提示プラットフォームもしくは抗体誘導プラットフォームに加え、このようなプラットフォームを用い得られた抗原特異的T細胞または抗体特異的B細胞を含有する薬学的調製物を、患者への直接注射用に、製剤化することができる。このような薬学的調製物は、本発明の組成物の患者への送達に適した薬学的に許容される担体、例えば生理食塩水、緩衝した生理食塩水(例えば、リン酸緩衝生理食塩水)、またはリン酸緩衝生理食塩グルコース溶液などを含有する。
【0136】
免疫療法
投与経路
本発明の粒子または細胞を用いた抗原提示プラットフォームまたは抗体誘導プラットフォームに加え、このようなプラットフォームを用いて得られた抗原特異的T細胞または抗体特異的B細胞を、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与を含む、いずれか適当な経路により、患者へ投与することができる。患者は、ヒトおよび獣医学上の患者の両方を含む。
【0137】
治療法
本発明のプラットフォームを用い、当技術分野において公知の診断法および治療法において使用することができる、治療に有用な数の抗原特異的T細胞または抗体産生B細胞を作成することができる。例えば、国際公開公報第01/94944号;米国特許第2002/0004041号;米国特許第5,583,031号;米国特許第2002/0119121号;米国特許第2002/0122818号;米国特許第5,635,363号;米国特許第2002/0090357号;米国特許第6,458,354号;米国特許第2002/0034500号を参照。
【0138】
特に、抗原特異的T細胞または抗体産生B細胞を使用し、感染症、癌、もしくは自己免疫疾患を伴う患者を治療するか、または免疫抑制状態の患者に予防的保護を提供することができる。
【0139】
治療することができる感染症は、細菌、ウイルス、プリオン、真菌、寄生体、蠕虫などにより引き起こされるものを含む。このような疾患は、AIDS、肝炎、CMV感染症、および移植後リンパ増殖性障害(PTLD)を含む。例えばCMVは、臓器移植した患者において認められる最も一般的なウイルス病原体であり、ならびに骨髄移植もしくは末梢血幹細胞移植を受ける患者における有病および死亡の主因である(Zaia、Hematol. Oncol. Clin. North Am.、4:603-23(1990))。これは、これらの患者の免疫無防備状態が原因であり、このことは血清陽性患者における潜在ウイルスの再活性化または血清陰性の個人での日和見感染をもたらす。現在の治療は、薬物耐性CMVの発達が最も重大である欠点を有する、ガンシクロビルのような抗ウイルス化合物の使用に焦点が当てられている。これらの治療の有用な代替は、移植手技の開始前の、患者または適当なドナー由来のウイルス特異的CTLの作成に関連する予防的免疫療法的投薬法である。
【0140】
PTLDは、移植患者の大きい割合において生じ、およびエプスタイン-バーウイルス(EBV)感染症から生じる。EBV感染は、米国の成人集団の約90%に存在する(AnagnostopoulosおよびHummel、Histopathology、29:297-315(1996))。活性ウイルス複製および感染は、免疫系によりチェックされ続けるが、CMVの場合のように、移植療法により免疫無防備状態となった個人は、制御するT細胞集団を喪失し、このことはウイルス再活性化をもたらす。これは、移植プロトコールの重大な障害を表している。EBVは、様々な血液学的癌および非血液学的癌における腫瘍のプロモーションにも関連することがある。EBVと鼻咽頭癌の間の強力な関係も存在する。従って、EBV特異的T細胞による予防的治療は、現在の療法に対する優れた代替を提供する。
【0141】
本発明に従い治療することができる癌は、黒色腫、癌腫、例えば結腸癌、十二指腸癌、前立腺癌、乳癌、卵巣癌、腺管癌、肝臓癌、膵臓癌、腎臓癌、子宮内膜癌、胃癌、口内粘膜の形成異常、ポリープ症、侵襲性口内癌、非小細胞肺癌、転移性および扁平上皮細胞尿路癌など;神経学的悪性疾患、例えば、神経芽細胞腫、神経膠腫など;血液学的悪性疾患、例えば、慢性骨髄性白血病、小児急性白血病、非ホジキンリンパ腫、慢性リンパ球性白血病、悪性皮膚T細胞リンパ腫(malignant cutaneous)、菌状息肉腫、非MF皮膚T細胞リンパ腫、リンパ腫様丘疹症、T細胞豊富な皮膚リンパ球様過形成、水疱性類天疱瘡、円板状紅斑狼瘡、扁平苔癬;などを含む。例えば、Mackensenら、Int. J. Cancer、86:385-92(2000);Jonuleitら、Int. J. Cancer、93:243-51(2001);Lanら、J. Immunotherapy、24:66-78(2001);Meidenbauerら、J. Immunol.、170(4):2161-69(2003)参照。
【0142】
治療することができる自己免疫疾患は、喘息、全身性紅斑狼瘡、リウマチ様関節炎、I型糖尿病、多発性硬化症、クローン病、潰瘍性大腸炎、乾癬、重症筋無力症、グッドバスチャー症候群、グレーヴス病、尋常性天疱瘡、アジソン病、疱疹状皮膚炎、セリアック病、および橋本甲状腺炎を含む。
【0143】
抗原特異的ヘルパーT細胞を使用し、マクロファージを活性化、またはB細胞を活性化し、例えば、感染症および癌の治療に使用することができる特異的抗体を産生することができる。抗体産生B細胞それ自身も、この目的に使用することができる。
【0144】
抗原特異的調節T細胞を使用し、例えば、移植患者における移植片対宿主疾患を治療もしくは予防するために、または前述のような自己免疫疾患、もしくはアレルギーを治療もしくは予防するために、免疫抑制作用を達成することができる。調節T細胞の使用は、例えば、米国特許第2003/0049696号、米国特許第2002/0090724号、米国特許第2002/0090357号、米国特許第2002/0034500号、および米国特許第2003/0064067号に開示されている。T細胞影響する分子がアポトーシス誘導する分子である抗原提示プラットフォームは、免疫応答の抑制に使用することができる。
【0145】
用量
抗原特異的T細胞は、用量範囲約5〜10x106CTL/kgの体重(〜7x108CTL/処置)から、最大約3.3x109CTL/m2(〜6x109CTL/処置)で、患者へ投与することができる(Walterら、New England Journal of Medicine、333:1038-44(1995);Yeeら、J. Exp. Med.、192:1637-44(2000))。他の態様において、患者は、静脈内投与される1回投与量あたり、103個、5x103個、104個、5x104個、105個、5x105個、106個、5x106個、107個、5x107個、108個、5x108個、109個、5x109個または1010個の細胞を受け取ることができる。更に別の態様において、患者は、200μlのボーラス中に例えば8x106個または12x106個の細胞の結節内注射を受け取ることができる。抗体産生B細胞それ自身に加え、細胞を用いた抗原提示プラットフォームまたは抗体誘導プラットフォームを、同様の用量で患者に投与することができる。
【0146】
粒子を用いたプラットフォームが投与される場合、典型量は、1回投与量あたり103個、5x103個、104個、5x104個、105個、5x105個、106個、5x106個、107個、5x107個、108個、5x108個、109個、5x109個または1010個の粒子を含む。
【0147】
動物モデル
多くのマウスモデルが、腫瘍治療に関する養子免疫療法プロトコールを評価するために利用可能である。黒色腫治療を評価するためには、ふたつのモデルが特に適している。ひとつのモデルは、皮下移植されたヒト黒色腫株、例えばBMLを有する、ヒト/SCIDマウスを使用する。このようなモデルにおいて、エクスビボで拡大したMart-1特異的CTLの移植は、この腫瘍の発症および/または増殖を遅延する。第二のモデルは、マウスA2-トランスジェニックマウス、およびAADと称される、HLA-A2様分子がトランスフェクションされたマウスB16黒色腫を使用する。A2-トランスジェニックの基礎でもあるこの分子は、マウスα3ドメインに融合したα1-2ドメイン中のヒトHLA-A2である。これらのトランスジェニックマウスを使用し、マウスのB16-AAD黒色腫は、チロシナーゼおよびgp100に由来した詳細に明らかにされたA2拘束された黒色腫エピトープを超えた拒絶反応に対し感受性がある。
【0148】
キット
本発明の抗原提示プラットフォームまたは抗体誘導プラットフォームのいずれかは、キットで提供することができる。粒子または細胞を用いた抗原提示または抗体誘導プラットフォームに適した容器は、例えば、ボトル、バイアル、注射器、および試験管を含む。容器は、ガラスまたはプラスチックを含む、様々な材料で製造することができる。容器は、無菌のアクセスポートを有しても良い(例えば、容器は、皮下注射針により、剥離可能なストッパーを備えている、静注液用バッグまたはバイアルであることができる。)。またはキットは、前述のような、硬質または軟質の抗原提示または抗体誘導プラットフォームを備えることができる。任意で、1種または複数の異なる抗原を、プラットフォームに結合するか、または個別に供給することができる。
【0149】
キットは更に、リン酸緩衝生理食塩水、リンゲル液、またはデキストロース液などの、薬学的に許容される緩衝液を含む第二の容器を備えることができる。これは、その他の緩衝液、希釈液、フィルター、針、および注射器などの、最終使用者に有用な他の材料も備えることができる。キットは、例えば、最終使用者により抗原提示プラットフォームの抗原提示複合体に結合することができる、化学療法剤または感染症治療薬など、または特定の抗原を含むもののような別の活性物質を伴う第二または第三の容器を備えることもできる。
【0150】
キットは、抗原特異的T細胞または抗体産生B細胞の誘導または拡大、例えば、特異的マーカー蛋白質に対する抗体、MHCクラスIもしくはクラスII分子複合体、TCR分子複合体、抗クローン形質の抗体などの程度および有効性を評価するための試薬も備えることができる。
【0151】
キットは、様々な治療プロトコールにおいて、キット内の抗原特異的T細胞を誘導、抗原特異的T細胞を拡大、抗原提示プラットフォームもしくは抗体誘導プラットフォームを使用する方法について書面による指示を記載した添付文書も備えることができる。添付文書は、未承認の草稿段階の添付文書であるか、または米国食品医薬品局(FDA)または他の規制機関により承認された添付文書であることができる。
【0152】
本開示に引用された全ての特許、特許出願、および参考文献は、明白に本明細書に参照として組み入れられている。前記開示は一般に、本発明を説明している。より完全な理解を、単に本発明の例証を目的として提供され、限定することは意図していない、下記の具体例を参照し得られる。
【実施例】
【0153】
実施例1
材料および方法
細胞株
TAP-欠損174CEM.T2(T2)細胞株および黒色腫細胞株は、10%FCSを補充したM'培地(Oelkeら、Scand. J. Immunol.、52:544-49(2000))において維持した。
【0154】
ペプチド
本試験において使用したペプチド(Mart-1、ELAGIGILTV、配列番号:3;CMVpp65、NLVPMVATV、配列番号:4)は、JHU中核施設が調製した。各ペプチドの純度(>98%)は、質量分析およびHPLCにより確認した。
【0155】
HLA A2.1+リンパ球
Johns Hopkins Universityの施設内倫理委員会(IEC)は、下記実施例において考察した試験を承認した。全てのドナーは、本試験に登録する前に、書面によるインフォームドコンセントを提出した。健常志願者および黒色腫患者のドナー#7は、サイトメトリーにより表現型がHLA-A2.1であった。この黒色腫患者は、肺、肝臓およびリンパ節転移を伴う広範囲の転移性疾患を有した。PBMCは、フィコール-ハイパーク(Hypaque)密度勾配遠心分離により単離した。
【0156】
aAPCの作成
aAPCは、「HLA-Ig」(米国特許第6,268,411号に開示)および抗CD28のミクロビーズ(Dynal社、レークサクセス、NY)上への連結により作成した。簡単に述べると、ビーズを、滅菌した0.1Mホウ酸緩衝液(「ビーズ洗浄緩衝液」)中で2回洗浄した。これらのビーズを、ホウ酸緩衝液中のHLA-A2-Igおよび抗CD28 mAb 9.3の1:1混合液と共に、ローター上で24時間4℃でインキュベーションし、ならびにビーズ洗浄緩衝液で2回洗浄した。ビーズ洗浄緩衝液中で更に24時間4℃でインキュベーションし、この緩衝液を交換した。得られるaAPCは、A2-Ig/ビーズの0.9x105個分子、および抗CD28分子/ビーズの1.9x105個を有することがわかった。aAPCビーズは、4℃で3ヶ月を越えて貯蔵し、活性は失われなかった。ペプチド負荷に関して、HLA-Ig被覆したaAPCを、PBSで2回洗浄し、およびペプチド30mg/ml中に107個ビーズ/mlとなるよう調節した(最終濃度)。aAPCビーズは、4℃でペプチド溶液中に貯蔵した。
【0157】
樹状細胞のインビトロ作成
単球は、CD14+磁気分離(Miltenyi、オーバーン、CA)により、PBMCから単離した。CD14+細胞は、2%自家血清、100ng/mlヒトGM-CSF、50ng/ml IL-4、および5ng/ml TGF-β1を含む、M'培地において培養した。5〜7日培養した後、10ng/ml TNF-βおよびIL-1β、1000U/ml IL-6(BD-Pharmingen社、サンディエゴ、CA)および1mg/ml PGE2(Sigma社、セントルイス、MO)を含有する成熟カクテルに24時間添加した。細胞は、DCの典型的細胞表面マーカー(CD1a+、CD14-、CD86+)を展示した。ペプチド負荷のために、DCを収集し、および密度1〜2x106個細胞/mlでM'培地中の30mg/mlでインキュベーションした。
【0158】
インビトロCTL誘導
CD8+ Tリンパ球は、CD8単離キット(Miltenyi社、オーバーン、CA)を使用するCD8-細胞の枯渇により、PBMCにより濃縮した。90%を超えるCD8+ T細胞を含む得られる集団は、レスポンダー細胞として使用し、ペプチドパルスしたDCまたはペプチドパルスしたaAPCのいずれかで刺激した。10000個のレスポンダー細胞/ウェルを、5%自家血漿および3%TCGFを補充した200μlのM'培地/ウェルの96ウェル丸底プレートにおいて、5x103個DC/ウェルまたは104ペプチドパルスしたaAPC/ウェルのいずれかと共培養した。追加した同種異系のフィーダー細胞は、CTLの誘導または拡大のいずれにも使用しなかった。TCGFは、Oelkeらの論文に記されたように調製した(Clin. Canc. Res.、6:1997-2005(2000))。培地およびTCGFは、1週間に2回補充した。7日目およびその後毎週、T細胞を収集し、計数し、ならびに3%TCGFを補充した完全培地内で、5x103個ペプチドパルスした自家DC/ウェルまたは104個ペプチドパルスしたaAPC/ウェルのいずれかと共に、104個T細胞/ウェルとなるよう再び播種した。
【0159】
二量体染色および細胞内サイトカイン染色(ICS)分析
Gretenらの論文(Proc. Natl. Acad. Sci. USA、95:7568-73(1998))に記されたように、細胞は、FITC結合したCD8 mAbおよびMart-1もしくはCMVパルスしたA2-Igで、ならびに第二工程において、抗マウスIg-PEで染色し、Ig-A2二量体を検出した。対照染色について、記したように、A2-Igを、不適切なペプチドで負荷するかまたは負荷しないかのいずれかであった。負荷しないA2-Ig(図2)または不適切なA2-結合ペプチドを負荷したA2-Ig(図4)のいずれかを用い、同様のバックグランド染色が観察された。分析のために、本発明者らはCD8+細胞をゲーティングした(gate on)。
【0160】
ICSは、下記を変更し、説明されたように行った(BD-Pharmingen、サンディエゴ、CA、ICSプロトコール)。1000000個のエフェクター細胞は、2x105個ペプチドパルスしたT2細胞(30μg/ml)または106個黒色腫細胞で、37℃で5時間刺激した。黒色腫細胞が標的として使用される場合、0.5ng/mlホルボール12-ミリスチン酸13-酢酸(PMA)および4ng/mlイオノマイシンを添加した。対照実験は、低用量のPMAおよびイオノマイシンは、エフェクター細胞中のサイトカイン産生を誘導しないことを明らかにした。細胞内染色を、FITC標識されたIFN-gまたはIL4 mAb(BD、サンディエゴ、CA)で行った。
【0161】
51Cr-放出アッセイ法
51Cr-放出-アッセイ法は、Oelkeらの論文(2000)に説明されたように行った。CTL活性は、下記式を用い、特異的51Cr-放出の割合として計算した:%特異的殺傷=(試料放出−自然発生的放出)÷(最大放出−自然発生的放出)×100%。
【0162】
実施例2
aAPCによるMart-1およびCMV特異的CTLの誘導および拡大
これは、ふたつの臨床的関連した標的であるCMV-ペプチドpp65およびMart-1による抗原特異的CTLの誘導および拡大を説明している。これらのペプチドは、それらの同族TCRに関して広範に変動する親和性を有する。CMV-ペプチドpp65は、高親和性ペプチドであるのに対し、メラニン形成細胞自己抗原に由来した修飾されたMart-1ペプチドは、低親和性ペプチドであることが知られている(Valmoriら、Int. Immnunol.、11:1971-80(1999))。
【0163】
現在の方法は、自家ペプチドパルスしたDCを使用し、正常なPBMCから抗原特異的CTLを誘導している(図1)。これらの方法は、DCまたはCD40Lで刺激された自家B細胞を使用し、抗原特異的CTLが培養物の主要部分になるまで、2-4刺激サイクルにわたり、抗原特異的CTLを誘導することが多い(図1、工程2)。従って本発明者らは、aAPC誘導を、DCによる誘導と比較した。T細胞を、単離し、精製し、およびMart-1負荷したaAPCまたはMart-1ペプチドでパルスした単球-由来した自家DCのいずれかで誘導した。CD8+ T細胞は、DCまたはaAPCのいずれかにより、週1回、合計3ラウンド刺激した。
【0164】
代表的実験において、T細胞の総数は、DC誘導した培養物中において1x106個から20x106個に増大し、aAPC誘導した培養物中において1x106個から14x106個に増大した。この培養物の抗原特異性は、A2-Ig二量体染色およびICSの両方により3週間後に分析した。本発明者らの状況においては、おそらく誘導されたCTL集団における異質性により、ICS染色は、二量体染色の最大2倍の感度であることができる。ICSは、二量体染色よりも高いおよび低い親和性CTLの境界集団を検出すると思われる。細胞は、前述のようにFITC結合したCD8 mAbおよびMart-1パルスしたA2-Igにより染色した。ICSに関して、細胞は、サイトカインを含まない通常培地において、ペプチドパルスしたT2細胞と共にインキュベーションした。1時間後、モネンシン(ゴルジ停止)をこの培養物に添加した。6時間後、T細胞を収集し、ICSにより分析した。ペプチド特異的CD8+ CTLの割合は、右上隅に示した。
【0165】
MART-1ペプチド負荷したaAPCによる3ラウンド刺激後、Mart-1A2-Ig二量体染色により決定すると、全CD8 CTLの62.3%はMart-1特異性であり(図2A、左側)、および細胞内サイトカイン染色(ICS)により決定すると、84.3%がそうであった(図2A、右側)。抗原特異的CTLのHLA-Ig二量体染色およびICS分析の間の差異は、恐らくDCまたはaAPC誘導したCTL集団により使用されたTCRレパートリーの多様性に関連すると思われる。ペプチド誘導した抗原特異的CTL集団中の異質性は、先に報告されている。Valmoriら、J. Immunol.、168:4231-40(2002)。レパートリーの多様性は、一方のアッセイ法により認識されるが他方のアッセイ法により認識されない、より高いおよびより低い親和性のCTLに関連していると思われる。
【0166】
aAPCにより得られた結果と比べ、自家DCは、わずかに29.7%のMART-1特異的細胞を誘導し、およびICS Mart-1特異的CTLは61.1%誘導した(図2B)。
【0167】
aAPC刺激したPBMCの増殖可能性を検討するために、T細胞を、aAPCで7週間刺激した。0.05%未満がMart-1特異性である1x106個総CD8+ T細胞から開始し、細胞を、85%よりも多くがMart-1特異性である約109個のCTL細胞に拡大した(図2Cおよび2D)。これは、2ヶ月足らずのうちの少なくとも106倍の抗原特異的T細胞の拡大を示している。
【0168】
aAPC媒介した刺激は、Mart-1およびCMV誘導したCTLの両方について、より良いものではないとしても、DCによる刺激と同等であった(表1)。
【0169】
【表1】
【0170】
これは、3名の異なる健常ドナーおよび転移性黒色腫の患者由来の細胞(Mart-1負荷したaAPC)ならびに3名の異なるドナー由来の細胞(CMV負荷したaAPC)を用いる、5つの実験中4つにおいて認められた。Mart-1誘導に関して、HLA-Ig二量体染色およびICSの両方により認められるように、aAPCは、DCよりも、約2〜3倍多く抗原特異的細胞を誘導した。このことは、転移性黒色腫の患者においても認められた。CMV特異的CTLの誘導は、Mart-1特異的CTLよりもより豊富であった。例えaAPCによる1回刺激後であっても、最大90%のCTL集団がCMV特異性であった。わずかに少ないCMV特異的CTLが、DCを用いて得られた。従ってaAPCは一般に、複数の健常ドナーに加え黒色腫患者由来のふたつの異なるCTLシステムにおいて抗原特異的CTLの誘導について、より良いものではないとしても、DCと同等であった。
【0171】
aAPCは、A2拘束されたサブドミナントな黒色腫エピトープNY-ESO-1(Jagerら、J. Exp. Med.、187:265-70(1998))、およびLMP-2由来のサブドミナントなEBVエピトープ (Leeら、J. Virol.、67:7428-35(1993))について、CTL特異的な大きい拡大も媒介した(表2参照)。全てのCD8+細胞の約1.2%は、3回のaAPC刺激の後NY-ESO-1特異性であった。これは、免疫ドミナントなエピトープに特異的であるCTL拡大において認められるものよりも明らかに低かったが、サブドミナントなエピトープのCTL特異性の拡大を分析する場合には、より少ない数の抗原特異的CTLが予想された。
【0172】
NY-ESO-1特異的CTLは、同族特異的標的細胞の溶解を媒介したが、無関係の標的細胞は溶解しなかった(表2)。これらの実験において、CTLは、分析前に3〜4週間分析した。抗原特異的CTLの頻度は、LMP-2特異的CTLの二量体染色によるか、またはNY-ESO-1特異的CTLに関する四量体染色および51Cr放出アッセイ法により分析した。表2は、E:T比25:1で観察された特異的溶解率(%)、およびA2-Ig二量体もしくは四量体のいずれかを用いるフローサイトメーターにより決定されたペプチド特異的CD8+ T細胞の割合(%)を示している。対照的に、DCを用いた刺激は、標準の51Cr放出アッセイ法において、検出可能な細胞傷害活性を伴わない有意に低い頻度のNY-ESO-1特異的CTLを生じた。
【0173】
【表2】
【0174】
実施例3
aAPC誘導したPBMCにより内因性にプロセッシングされた抗原の認識
CTL機能の評価における有用な判定基準は、内因性抗原-HLA複合体を発現する標的の認識である。黒色腫特異的CTLの拡大のためのペプチドパルスしたDCを使用する最初の作業は、同族抗原によりパルスした標的の溶解を媒介した低親和性CTLを生じたが、しばしば抗原-HLA複合体を内因性に発現した黒色腫標的を認めることができなかった。Yeeら、J. Immunol.、162:2227-34(1999)。従って本発明者らは、aAPC誘導したCTLの内因性Mart-1またはpp65 CMV抗原を認識する能力を試験した(図3)。
【0175】
ICS染色に関して、これらの細胞は、サイトカインを伴わない通常培地中の標的細胞と共にインキュベーションした。ICSアッセイ法の感度を増加するために、低用量PMAおよびイオノマイシンを、培地に添加した。Perez-Diezらの説明したように(Cancer Res.、58:5305-09(1998))、この方法は、本発明者らが集団中のより抗原特異的のT細胞を検出することを可能にした。この追加の刺激を伴うまたは伴わないこれらの結果における差異は、これらの刺激に応じて決まった。低用量のPMAおよびイオノマイシンにより認められる増強は、自家腫瘍細胞が刺激細胞として使用される場合により顕著であった(最大3〜4倍)が、抗原特異的T細胞の刺激にペプチドパルスしたT2細胞またはA293細胞が使用される場合は、大きくはなかった。低用量PMAおよびイオノマイシンの添加は、図3Aおよび3Cに認められるように、バックグラウンド活性を変化しなかった。古典的クロム放出溶解アッセイ法を、PMAおよびイオノマシンを添加せずに行った(図3Bおよび3D)。
【0176】
Mart-1特異的aAPC誘導した細胞は、黒色腫標的細胞により刺激した場合、約37%がIFN-γを産生した(図3A)。同等数がIL-4を産生した(図5)。対照Mart-1+/HLA-A2-黒色腫標的は、有意なエフェクターサイトカイン産物を刺激しなかった。更に、aAPC誘導したエフェクターCTL集団は、標的Mart-1+/HLA-A2+黒色腫標的細胞を用量依存的に溶解したが、対照Mart-1+/HLA-A2-標的は溶解しなかった(図3B)。進行した黒色腫患者から得たPBMC由来のaAPC誘導したMart-1特異的CTLは、HLA-A2+ Mart-1発現する黒色腫細胞の溶解を媒介することもでき、14.7%の溶解が、E:T比25:1で認められた。
【0177】
aAPCも、内因性にプロセッシングされおよび提示されたpp65抗原を認識したCMV特異的CTLを誘導することができた(図3Cおよび3D)。A293-N pp65+標的で刺激した場合(pp65でトランスフェクションしたA293細胞)、約45%の細胞がIFNγを産生した。aAPC誘導したエフェクターCTL集団も、トランスフェクションされた標的細胞を用量依存的に媒介したが、対照標的は媒介しなかった(図3D)。従って、正常な健常ドナーに加え黒色腫患者の両方から得たaAPC誘導したCTL培養物は、内因性にプロセッシングされた抗原-HLA複合体を認識しなかった。
【0178】
抗原特異的CTLの一部は、aAPCまたはDCのいずれにより誘導されようと、IFN-γおよびIL4のいずれかまたは両方を産生した(図5)。IFN-γおよびIL4の両方を産生するヒトCD8+細胞が、DCを用いたエクスビボ拡大において報告されている。Oelkeら、2000。本発明者らのaAPCによる結果は、これらの興味深いDCを用いた知見を確認し、ならびにaAPC媒介した刺激は明らかに同様の抗原特異的CTLを生じることを示している。
【0179】
実施例4
CMV特異的CTLのaAPCによる拡大
DCの使用に関連したひとつの制限は、臨床に関連した数へのCTLの拡大は、十分なDCを得るための白血球搬出または抗CD3ビーズなどの非特異性拡大の使用のいずれかを必要とすることである(図1、工程3参照)。従って本発明者らは、aAPCまたは抗CD3/抗CD28-ビーズを使用する「拡大」相を比較した。CMV特異的CTLの拡大時に、抗CD3/抗CD28ビーズを使用するCTLの総数が7倍増加した。しかし、抗原特異的細胞の割合は、87.9%から7.3%に減少した(図4Aおよび4Bの比較)。この問題点は、多様なCTL集団の抗CD3を用いた拡大の使用の有用性を制限した。Mausら、Nature Biotechnol.、20:143-48(2002)。対照的に、CMV特異的aAPCが抗原特異的CTLの拡大に使用される場合、抗原特異性の同時喪失はなかった。CMV特異的CTLの割合は、それらの培養物中で86%であり続け、そこには依然T細胞数の7倍の増加があった(図4Aおよび4Cの比較)。従って、HLA-Igを用いたaAPC支持体は、抗原特異的様式でのCTLの拡大を継続し、抗CD3を用いた拡大に勝る有意な利点を表している。
【技術分野】
【0001】
本出願は、本明細書に参照として組み入れられている2002年7月12日に出願された同時係属中の米国特許仮出願第60/395,781号の利益を請求するものである。
【0002】
本発明は、一部、米国立衛生研究所(NIH)助成金番号AI-29575および同AI-44129により資金提供された研究の結果である。米国政府は、本発明に対し一定の権利を有するものである。
【0003】
発明の技術分野
本発明は、独自のクローン形質のリンパ球受容体に結合する試薬および方法に関する。
【背景技術】
【0004】
発明の背景
養子および能動の両方の免疫療法の開発は、治療できる数の特異的TまたはBリンパ球を作成するための、再現性があり、経済的で実行可能な方法が欠如しているために妨げられている。例えば、養子免疫療法のための抗原特異的細胞傷害性Tリンパ球(CTL)を作成する現在の標準的方法は、CTLを拡大するための単球由来樹状細胞(DC)の作成を必要としている。この工程は、時間および経費の両方がかかる。CTLの臨床的に関連のある量へのCTL拡大のためのDCの使用は、十分な自家DCを得るために、複数回の白血球搬出を必要としている。得られたDCの量および質の両方について認められる変動性は、恐らく基礎をなす疾患および患者の前処置に関連し、これは同じくDCを用いたエクスビボ療法の変動性にも大きい影響を及ぼす。これらの理由により、DCの使用は、T細胞のエクスビボ拡大の工程に限られている。
【0005】
濃縮された集団から抗原特異的CTLを拡大する他の方法は、非特異的抗CD3を用いた技術を使用している。Levineら、J. Hematother.、7:437-48(1998)(非特許文献1)。しかしふたつの問題点がある。第一に、抗CD3/抗CD28ビーズは、CD4 T細胞の長期増殖を支持するが、CD8 T細胞の長期増殖は維持しない。DeethsおよびMescher、Eur. J. Immunol.、27:598-608(1997)(非特許文献2)。加えて、抗CD3を用いた刺激を使用する方法は、例え高度に濃縮された抗原特異的CTL集団を用い開始する場合であっても、抗原特異性の低下を伴う。Mausら、Nature Biotechnol.、20:143-48(2002)(非特許文献3)。これらの問題点は、治療に関連するリンパ球の送達を実質的に制限している。
【0006】
従って当技術分野において、抗原特異的T細胞の、更には特異的抗体産生B細胞の治療的に有用な集団を作成する効果的な手段の必要性が存在する。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Levineら、J. Hematother.、7:437-48(1998)
【非特許文献2】DeethsおよびMescher、Eur. J. Immunol.、27:598-608(1997)
【非特許文献3】Mausら、Nature Biotechnol.、20:143-48(2002)
【発明の概要】
【0008】
発明の簡単な概要
本発明は、少なくとも下記の態様を提供する。本発明のひとつの態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、固形支持体である。
【0009】
別の本発明の態様は、(A)少なくとも1種のB細胞に影響する分子、および(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、固形支持体を提供する。
【0010】
別の本発明の態様は、(A)少なくとも1種のT細胞共刺激分子、および(B)少なくとも2種の融合蛋白質を含む少なくとも1種のMHCクラスI分子を含む粒子を提供する。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。
【0011】
更に別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、複数の粒子を含有する調製物を提供する。
【0012】
更なる本発明の態様は、複数の粒子を含有する調製物を提供する。複数の粒子は、(A)少なくとも1種のB細胞に影響する分子、および(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0013】
更に別の本発明の態様は、抗原特異的T細胞の形成を誘導する方法を提供する。複数の前駆体T細胞を含有する単離された調製物は、少なくとも1種の第一の固形支持体と接触される。この固形支持体は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。抗原は、この抗原結合窩に結合される。これにより、複数の前駆体T細胞のメンバーは、その抗原を認識する抗原特異的T細胞を含む第一の細胞集団の形成を誘導する。第一の集団内の抗原特異的T細胞の数または割合は、前駆体T細胞が、CD3に特異的に結合する抗体は含むが抗原提示複合体は含まない固形支持体と共にインキュベーションされる場合に形成される抗原特異的T細胞の数または割合よりも多い。この抗原特異的T細胞は、患者へ投与することができる。
【0014】
更に別の本発明の態様は、細胞の集団内の抗原特異的T細胞の数または割合を増加する方法を提供する。抗原特異的T細胞を含む第一の細胞集団は、少なくとも1種の第一の固形支持体と共にインキュベーションされる。固形支持体は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。抗原は、抗原結合窩に結合される。インキュベーション工程は、第一の細胞集団内の抗原特異的T細胞の数または割合に比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される。この抗原特異的T細胞は、患者へ投与することができる。
【0015】
更なる本発明の態様は、患者における免疫応答を調節する方法を提供する。(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物が、患者に投与される。複数の粒子のメンバーは、(1)少なくとも1種のT細胞に影響する分子、および(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体は、少なくとも1個の抗原結合窩を含む。抗原は、この少なくとも1個の抗原結合窩に結合される。
【0016】
更に別の本発明の態様は、患者における免疫応答を抑制する方法を提供する。(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物が、患者に投与される。複数の粒子のメンバーは、(1)少なくとも1種のアポトーシス誘導分子、および(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体は、少なくとも1個の抗原結合窩を含む。抗原は、この少なくとも1個の抗原結合窩に結合される。
【0017】
別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、抗原を認識する特異的クローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む細胞を提供する。
【0018】
更に別の本発明の態様は、(A)少なくとも1種のリンパ球に影響する分子、および(B)抗原に結合した場合に、クローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む複数の細胞を含有する調製物を提供する。
【0019】
更に別の本発明の態様は、抗原特異的T細胞の形成を誘導する方法を提供する。複数の前駆体T細胞を含有する単離された調製物は、第一の複数の細胞と接触される。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質である。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖および(ii)MHCクラスIlα鎖の細胞外ドメインを含む。2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、ペプチド結合窩に結合される。これにより複数の前駆体T細胞のメンバーは、抗原性ペプチドを認識する抗原特異的T細胞を含む第一の細胞集団を形成する。
【0020】
更に別の本発明の態様は、細胞の集団内の抗原特異的T細胞の数または割合を増加する方法を提供する。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。この2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、ペプチド結合窩に結合される。インキュベーション工程は、第一の細胞集団内の抗原特異的T細胞の数または割合と比べ、増大した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される。この抗原特異的T細胞を、患者へ投与することができる。
【0021】
別の本発明の態様は、患者の免疫応答を調節する方法を提供する。複数の細胞および薬学的に許容される担体を含有する調製物を、患者に投与することができる。これらの細胞は、少なくとも1種のT細胞に影響する分子および少なくとも1種の抗原提示複合体を含む。この抗原提示複合体は、MHCクラスI分子複合体またはMHCクラスII分子複合体のいずれかである。MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成する。MHCクラスI分子複合体は、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。抗原性ペプチドは、このペプチド結合窩に結合される。
【0022】
更に別の本発明の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。複数の前駆体B細胞を含有する単離された調製物は、少なくとも1種の第一の固形支持体と接触される。固形支持体は、少なくとも1種のB細胞に影響する分子、およびB細胞表面の免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。これにより複数の前駆体B細胞のメンバーは、抗原性ペプチドに特異的に結合する抗体を産生するB細胞を含む第一の細胞集団を形成するように誘導される。
【0023】
本発明の別の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。抗体産生B細胞を含む第一の細胞集団は、少なくとも1種の第一の固形支持体とインキュベーションされる。この固形支持体は、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。インキュベーション工程は、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される。
【0024】
更に別の本発明の態様は、集団内の抗体産生B細胞の数または割合を増加する方法を提供する。複数の前駆体B細胞を含む単離された調製物を、調製物と接触させ、これにより第一の細胞集団を形成する。この調製物は、複数の粒子を含む。複数の粒子は、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原に結合する少なくとも1種の分子複合体を含む。第一の細胞集団の細胞は、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む。
【0025】
本発明の別の態様は、患者における免疫応答を調節する方法を提供する。複数の粒子および薬学的に許容される担体を含む調製物が、患者に投与される。複数の粒子のメンバーは、少なくとも1種のB細胞に影響する分子、およびB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0026】
従って本発明は、独自のクローン形質のリンパ球受容体に結合する様々な試薬および方法を提供する。本発明は、治療目的で使用することができる、抗原特異的T細胞および抗体特異的B細胞を得るための試薬および方法も提供する。
【図面の簡単な説明】
【0027】
【図1】(図1)自家DCまたはaAPCのいずれかによる、ペプチド特異的CTLの誘導および拡大の概略である。
【図2−1】(図2−1)aAPCにより刺激されたMart-1特異的CD8+ T細胞の誘導および増殖能である。図2Aは、aAPCによる刺激の結果である。図2Bは、DCによる刺激の結果である。
【図2−2】(図2−2)aAPCにより刺激されたMart-1特異的CD8+ T細胞の誘導および増殖能である。図2Cは、T細胞の拡大を示すグラフである。図2Dは、拡大されたT細胞集団内の抗原特異的CTLの割合を示すグラフである。
【図3】(図3)aAPC誘導した抗原特異的CTLは、内因性黒色腫または標的細胞上のpp65抗原を認識することを示す。図3Aは、ペプチド特異的CD8+ T細胞の割合(%)が、Mart-1+/HLA-A2-黒色腫細胞株(左側)、またはMart-1+/HLA-A2+黒色腫細胞株(右側)により刺激されたMart-1特異的T細胞について示されている。図3Bは、Mart-1特異的CTL株による%特異的溶解を、以下の標的について示している:非特異的CMVペプチド
もしくは特異的Mart-1ペプチド
のいずれか、または自家HLA-A2+黒色腫細胞株(□)もしくは自家HLA-A2-黒色腫細胞株(■)
のいずれかにより、パルスされたT2細胞。値は、エフェクター:標的の比25:1、5:1およ
び1:1の3つ組で表している。図3Cは、ペプチド特異的CD8+ T細胞の割合(%)を、pp65-対
照でトランスフェクションされたHLA-A2+ A293細胞(左側)またはpp65+でトランスフェク
ションされたHLA-A2+ A293細胞(右側)のいずれかにより刺激されたCMV特異的T細胞につい
て示している。図3Dは、内因性抗原を発現している標的細胞に対するCMV特異的CTL細胞傷
害活性に関する、51Cr-放出アッセイ法の結果である。CMV特異的CTL株による%特異的溶
解を、以下の標的について示している。pp65でトランスフェクションされたA293細胞
、トランスフェクションされないHLA-A2+ A293細胞(□)、およびIE(CMVの前初期蛋白質)
対照でトランスフェクションされたA293細胞(■)。全てのアッセイ法に関して、抗原特異
的CD8+ T細胞を、ペプチド負荷したaAPCと一緒のインビトロ培養物において3〜7週後に得
た。
【図4】(図4)抗CD3ビーズまたはaAPCによる拡大後の抗原特異的CTLの頻度を示す。T細胞は、実施例1に示したように、単離および精製した。図4Aは、T細胞を、CMVペプチドでパルスした自家単球-由来DCで刺激し、抗原特異的T細胞拡大を誘導する。図4Bでは、誘導の3週間後に、T細胞集団が、抗CD3/抗CD28ビーズ上で拡大された。図4Cでは、DCでの誘導の3週間後に、T細胞集団は、ペプチド負荷されたHLA-Igを用いたaAPCで拡大された。両方の場合において、10日の培養後に、約7倍の拡大が認められた。実施例1に説明したように、細胞は、FITC結合した抗CD8 mAbで、ならびにpp65で負荷したCMV-ペプチドパルスしたA2-Ig(上側パネル)または対照ペプチドMart-1で負荷したA2-Igで染色した。%ペプチド特異的CD8+ CTLは、上部右隅に示している。
【図5】(図5)aAPC誘導したMart-1 CTLは、黒色腫標的細胞上の内因性抗原を認識することを示す。Mart-1 特異的CD8+細胞は、Mart-1負荷したaAPCと一緒のインビトロ培養後に得た。Mart-1特異的T細胞は、Mart-1+/HLA-A2-黒色腫細胞株(第一カラム)またはMart-1+/HLA-A2+黒色腫細胞株(第二カラム)のいずれかにより刺激した。ICS染色に関して、これらの細胞は、サイトカインを含まない通常の培地において黒色腫細胞と共に培養した。ベースラインを評価するために、低用量のPMAおよびイオノマイシンを培地に添加した。1時間後、モネンシン(ゴルジ停止)を培地に添加した。6時間後、T細胞を収集し、および細胞内サイトカイン染色について分析した。ペプチド特異的IL-4+/CD8+ T細胞の割合(%)を示した。
【発明を実施するための形態】
【0028】
発明の詳細な説明
本発明は、独自のクローン形質のリンパ球受容体と結合する(すなわち、結合および生理的反応の引き金を引く)多種多様な道具および方法を提供する。独自のクローン形質の受容体は、例えば、特異的抗原を認識するT細胞受容体を含む。本発明の一部の態様(「抗原提示プラットフォームおよび方法」)を用い、治療または診断目的の抗原特異的T細胞の形成および/または拡大を誘導することができる。抗原特異的T細胞は、細胞傷害性Tリンパ球、ヘルパーT細胞(例えばTh1、Th2)、および調節T細胞を含む。更に他の本発明の態様(「抗体誘導プラットフォームおよび方法」)を用い、特定の抗原に対する抗体を産生するBリンパ球の形成および/または拡大を誘導することができる。
【0029】
抗原提示プラットフォームおよび方法
本発明の抗原提示プラットフォーム(本明細書において「人工的抗原提示細胞」または「aAPC」とも称される)は、以下により詳細に説明されるように、真核細胞または人工的固形支持体を基にすることができる。本発明の抗原提示プラットフォームは、少なくとも1種のT細胞に影響する分子(例えば、T細胞共刺激分子、T細胞増殖因子、接着分子、調節T細胞誘導分子、またはアポトーシス誘導分子)および少なくとも1種の抗原提示複合体を含む。
【0030】
抗体誘導プラットフォーム
本発明の抗体誘導プラットフォームも、以下により詳細に説明されるように、真核細胞または人工的固形支持体を基にすることができる。本発明の抗体誘導プラットフォームは、少なくとも1種のB細胞に影響する分子(例えば、以下に説明されるような、CD40リガンド、サイトカイン、またはサイトカイン分子複合体)およびB細胞表面免疫グロブリンに結合するかもしくはB細胞の表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む。
【0031】
各々、抗原特異的T細胞および抗体特異的B細胞のエクスビボ拡大のための本発明の抗原提示プラットフォームおよび抗体誘導プラットフォームの使用は、現在使用される方法に勝る多数の重要な利点を有する。両型のプラットフォームを実行することができ、これらは再現性のある抗原提示または抗体誘導の活性を有し、ならびに大きい患者集団に使用することができる。抗原提示プラットフォームの使用は、抗原特異的T細胞のエクスビボ拡大プロセスを、樹状細胞を使用する現在の方法と比べ、劇的に簡略化および短縮する。加えて、これらのプラットフォームにより拡大された抗原特異的T細胞集団は、現在の方法(例えば抗CD3抗体単独による刺激)により得られる5〜20%と比べ、最大80%の抗原特異的T細胞を含むと思われる。これらのプラットフォームは、治療的用途に適した数までの前駆体TまたはB細胞の拡大を誘導することができる。これらのプラットフォームは、一工程において、前駆体TまたはB細胞の単離を抗原特異性刺激と組合わせることができる。人工の粒子を基にしたプラットフォームの態様は、これらは高密度であることができおよび重力により沈降することができる点、これらは磁石による分離が望ましい場合は磁気特性を有することができる点、これらはコーティングおよび蛋白質複合のための理想的表面化学を有する点、ならびに増大した表面積および標的細胞との増大した接触を提供するために異なる粒子サイズおよび幾何学的形が利用可能である点で、特異的なTまたはB細胞集団を誘導する現在利用可能な手段に勝っている。
【0032】
本発明のプラットフォームの成分は以下に詳細に説明されている。
【0033】
固形支持体
本発明のプラットフォームのための固形支持体は、蛋白質分子が付着することができる固形の人工表面(すなわち、非細胞)のいずれかであることができる。適当な固形支持体は、硬質支持体(例えば、フラスコ、試験管、培養皿、マルチウェルプレート、スライド、粒子)に加え、軟質支持体(例えば、注入バッグ)を含む。
【0034】
軟質支持体
軟質支持体は、注入バッグを含む。これらのバッグは、単回使用のために作成されるか、または再使用可能であることができる。バッグは、滅菌に適した材料で製造されることが好ましい。このような材料は、当技術分野において周知でありおよび広範に使用されている。
【0035】
硬質支持体
硬質支持体の例は、試験管;組織培養器、例えば、フラスコ(例えば、10cm2、25cm2、75cm2、150cm2、285cm2、300cm2、または420cm2)、ペトリ皿(例えば、9.2cm2、22.1cm2、60cm2、147.8cm2)、マルチウェルプレート(例えば、6ウェル、12ウェル、24ウェル、48ウェル、もしくは96ウェル、または384ウェルのプレート);スライド;ならびに、粒子を含む。硬質支持体は、例えば、鉄、ニッケル、アルミニウム、銅、亜鉛、カドミニウム、チタン、ジルコニウム、スズ、鉛、クロム、マンガンおよびコバルトなどの、金属;酸化アルミニウム、酸化クロム、酸化鉄、酸化亜鉛、および酸化コバルトなどの、金属酸化物および水和された酸化物;マグネシウム、アルミニウム、亜鉛、鉛、クロム、銅、鉄、コバルト、およびニッケルなどの、金属のケイ酸塩;青銅、真鍮、ステンレス綱のような合金などで製造することができる。硬質支持体は、非金属または有機物質、例えばセルロース、セラミック、ガラス、ナイロン、ポリスチレン、ゴム、プラスチックまたはラテックスで製造することもできる。または、硬質支持体は、金属および非金属または有機化合物の組合せ、例えばメタクリレート-またはスチレン-コートした金属およびケイ酸塩コートした金属であることができる。基本材料は、物質に浸漬し、その物理的または化学的特性を変更することができる。例えば、希土酸化物は、高密度の常磁性ガラス材料を作成するために、アルミノケイ酸塩ガラスに含むことができる(WhiteおよびDay、Key Engineering Materials、94-95:181-208(1994)参照)。
【0036】
粒子
態様のひとつのセットにおいて、本発明のプラットフォームは、人工の粒子を基にしている。人工の粒子は、前述の多くの材料のいずれかにより製造することができる。望ましい場合には、粒子は全体を、セルロース、デキストランなどの生分解性有機物質で製造することができる。適当な市販の粒子は、例えば、ニッケル粒子(Novamet Specialty Products社(ワイコフ、N.J.)から販売されているタイプ123、VM 63、18/209A、10/585A、347355およびHDNP;Spex社から販売されている、08841R;Aldrich社から販売されている、01509BW)、ステンレス綱粒子(Ametek社から販売されている、P316L)、亜鉛粉(Aldrich社)、パラジウム粒子(D13A17、John Matthey Elec.社)、M-450エポキシビーズ(Dynal社)、およびTiO2、SiO2、またはMnO2粒子(Aldrich社)がある。
【0037】
粒子が、細胞よりもより迅速に試料懸濁液を通じ示差的に沈降するように、粒子密度を選択することができる。従って粒子は、細胞分離および粒子の操作を促進するために、高密度材料で構成されることが好ましい。このような粒子の使用は、抗原特異的T細胞、T細胞前駆体、B細胞前駆体、B細胞、または他の細胞からの粒子の分離を促進するために重力下で粒子が沈降することを可能にする。
【0038】
高密度粒子を使用する更なる利点は、大量の非標的細胞を、治療適用に有用である標的細胞を失うことなく、除くことができることである。複数の細胞分離サイクルを、Kenyonらの論文に記されたように行うことができ(「High Density Particles: A Novel, Highly
Efficient Cell Separation Technology」、CELL SEPARATION METHODS AND APPLICATIONS、Recktenwald およびRadbruch編集、Marcel Dekker社、2000年、103-32頁)、その結果1回の枯渇サイクルにつきわずかに2〜3%の非特異的細胞が喪失される。複数回サイクル法を用い、非標的細胞を、標的細胞(例えばTまたはB細胞前駆体)を著しく喪失することなく、血液産物から除くことができる。標的細胞の回収は、90%を越えることができる。例えば、高密度の粒子は、平均4.7 logにまで動員された成分採血産物(mobilized apheresis product)において正常B細胞を減少するが、3回の枯渇サイクルを使用したシステムにおいてはCD34+細胞の90%よりも多くを維持する。Houdeら、Blood、96:187a(2000)。
【0039】
ひとつの態様において、粒子は、密度約9gm/km3を有しおよび磁気を帯びたニッケル粒子(例えば、Novamet社のタイプ123ニッケル粒子、これはサイズ範囲3〜7μm)である。粒子標的細胞複合体を捕獲するために磁石を使用しなければならないような他の市販の粒子とは異なり、高密度ニッケル粒子は重力により沈降する。沈降後、磁石を用い、懸濁液中の細胞から望ましくない粒子を分離することができる。ニッケル粒子は、リガンドカップリング化学にとって有用である官能基部分を伴う様々な高分子および無機分子の付着を可能にする化学特性も有する。
【0040】
粒子の形状は、不規則な形から球形へ、および/または平らでないかもしくは不規則な表面を有するものから平坦な表面を有するものまで変動することができる。粒子の好ましい特性は、抗原提示プラットフォームが調製および/または使用される特定の条件に応じて選択することができる。例えば、球形粒子は、不規則なサイズの粒子と比べ、より少ない表面積を有する。球形粒子が使用される場合は、減少した表面積のために、より少ない試薬が必要となる。他方で、不規則な形状の粒子は、球形粒子よりも有意に大きい表面積を有し、このことは複合した蛋白質含量/表面積および細胞の表面積接触に関して利点を提供する。
【0041】
粒子サイズも変動することができる。粒子サイズ(公称直径)は、本発明には重要ではないが、典型的には0.05〜50μm、より典型的には3〜35μmの範囲であり、および好ましくは約5μmである。これらの粒子は、サイズが均一であることができるか、またはサイズが変動することができ、平均粒子サイズは、好ましくは0.05〜50μmの範囲である。他の粒子は、微粉砕された粉末または超微細粒子であることができる。公称直径約5μmを有するニッケル粉末の粒子は、優れた蛋白質吸着特性を有する。ひとつの態様において、これらの粒子は、表面積が少なくとも0.4m2/g、好ましくは約0.4m2/g〜約0.5m2/gを有する。粒子サイズ分布は都合の良いことに、例えば、動的光散乱を基にしたMicrotrak装置を用い、決定することができる。
【0042】
固形支持体コーティング
固形支持体は、蛋白質がその表面に結合する前にコートすることができる。一旦コーティング化学が選択されたならば、固形支持体の表面は活性化され、特定の蛋白質分子の特異的付着を可能にすることができる。従ってコーティングは、様々なTもしくはB細胞集団またはTもしくはB前駆体細胞集団との最適な反応性および生体適合性を考慮し選択することができる。いかなるコーティング化学が使用されようと、更なる活性化化学のために適当なマトリックスを提供することが好ましい。多くのこのようなコーティングが、当技術分野において周知である。例えば、固形支持体は、ヒト血清アルブミン、トリス(3-メルカプトプロピル)-N-グリシルアミノ)メタン(米国特許第6,074,884号)、ゼラチン-アミノデキストラン(米国特許第5,466,609号)、またはアミノ酸ホモポリマーもしくはランダムコポリマーでコートすることができる。ひとつの態様において、ポリ(グルタミン酸、リシン、チロシン)[6:3:1]を含有するランダムアミノ酸コポリマーが使用され;このコポリマーは、Sigma Chemical社から、製品番号No.P8854として入手できる。これは、6部のグルタミン酸、3部のリシン、および1部のチロシンの比である、アミノ酸グルタミン酸・リシン・チロシンの線状ランダムポリマーである。別の態様において、4部のリシン対1部のチロシンの比である、リシンおよびチロシンを含むアミノ酸コポリマーが使用される。更に別の態様において、1部のリシン対1部のアラニンの比である、リシンおよびアラニンを含むアミノ酸コポリマーが使用される。
【0043】
別の態様において、固形支持体は、合成ポリマーによりコートされ、次にこの合成ポリマーは活性化され、その後TまたはB細胞に影響する分子、抗原提示複合体、またはB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する分子複合体を含むが、これらに限定されるものではない蛋白質分子に連結される。
【0044】
シリカ(SiO2)によるコーティング
別の態様、特にニッケル表面(特に粒子)に良く適した態様において、固形支持体は、シリカによりコートされている。シリカ表面は、通常使用される有機ポリマー表面に勝るいくつかの利点を有する。これは高度に均質であり、化学的に定義され、ならびに化学的および熱的に安定しており、シラノール残基がその表面全体を被覆し、ならびに蛋白質および他の生体分子の付着のために、トリエトキシシランのアミノ誘導体またはエポキシ誘導体との安定した共有結合に使用することができる。シラン誘導体は、表面全体を被覆することができ、表面との特異的および非特異的相互作用に高度の制御が可能である二次元ポリマーの単層を形成する。
【0045】
様々な固形支持体のシリカによるコーティングの方法は、米国特許第2,885,399に開示されており;同じく、Birkmeyerら、Clin Chem.、33(9):1543-7(1987年9月)を参照のこと。例えば、固形支持体は、メタケイ酸ナトリウム、アルミン酸ナトリウム、およびホウ酸の溶液と共にインキュベーションし、表面に沈着した重合されたシリカを形成することができる。シリカコーティングの別の方法は、ケイ酸ナトリウムを固形支持体と混合し、95℃で硫酸を用いpHを低下し、その後水で洗浄する。米国特許第2,885,366号;Eagerton、KONA、16:46-58(1998)を参照のこと。例えば、ニッケル表面は、最初に0.2N NaSO4溶液中にそれらを分散し、この溶液を95℃に加熱することにより、コーティングすることができる。このpHは、NaOHにより10に調節される。その後硫酸中のケイ酸ナトリウムが添加され、95℃で0.5時間混合される。この支持体は、蒸留水により数回洗浄される。コーティングの程度は、支持体の硝酸消化に対する抵抗を決定することにより試験することができる。
【0046】
X線散乱を基にした、表面化学組成に関するESCA分析を用い、支持体表面の元素組成を得ることができ、活性残基による表面コーティングおよびシラン化の程度に関する情報を提供する。
【0047】
酸化アルミニウムによるコーティング
別の態様において、固形支持体上の表面マトリックスが、ニッケル表面の、例えば酸化アルミニウムなどの、無毒の金属酸化物コーティングによる「不動態化」により提供される。コーティングの他の方法は、酸化アルミニウムのような金属酸化物の、固形支持体の表面への沈着を含む。酸化アルミニウムは、蛋白質複合のために官能基化することができる低い非特異的結合特性を伴う不活性表面を提供するので、これは有用なマトリックスである。
【0048】
酸化アルミニウムコーティングは、ゾル-ゲル法のような、多くの方法により提供され、ここで非晶質酸化アルミニウムの薄い連続層が、固形支持体上へのアルミニウムゾル-ゲルの蒸発により形成され、その後空気中で焼成(baking)され、酸化物が形成される。Ozerら、SPIE、3789:77-83(1999)参照のこと。別の態様において、通常の物理的蒸着法(Smidt、Inter Mat. Rev.、35:21-27(1990))または化学蒸着(Kohら、Thin Solid Films、304:222-24(1997))を使用することができる。ニッケル固形支持体が使用される場合、このようなコーティングの厚さは、適切な安定性を提供すると同時に、ニッケルの滲出を最小化するように制御することができる。ニッケル封印の成功は、ニッケルイオンの定量的化学アッセイ法により試験することができる。固形支持体は、様々な緩衝液および生物学的液体中で、様々な温度で、インキュベーションすることができ、これらの媒体中のニッケルイオンのレベルを測定することができる。
【0049】
表面コーティング効率
表面コーティングの完全性は、表面滲出アッセイ法により決定することができる。例えば、ニッケル固形支持体の表面をガラスまたは他の非反応性金属により完全にコーティングする場合、この固形支持体は、酸性条件下でのニッケル滲出に対して抵抗性である。例えば、被覆されたニッケル固形支持体の既知の塊を、10%硝酸中でインキュベーションし、24時間観察することができる。ニッケルは溶解するので、この溶液は緑色になっていく。未処理のニッケルは、直ぐに溶液を緑色にする。それらの表面上に酸化ニッケルの層を有するニッケル固形支持体は、溶液を約20分間で緑色に変える。先に説明されたようなシリカの層によりコーティングされた固形支持体は、8時間を越える時間硝酸に対し抵抗性があり、これはシリカの厚い層が表面上に沈着されたことを示している。固形支持体は、支持体を、BまたはT細胞活性化に使用した培養条件(下記に説明)に類似した細胞培養培地中でインキュベーションすることにより、水性条件下で試験することもできる。この溶液へ滲出されたニッケルの量は、原子吸光分光測定により測定することができる。
【0050】
コーティング前の予備処理
望ましい場合には、固形支持体は、コーティング前に予備処理することができる。固形支持体の予備処理は、例えば、支持体の滅菌およびパイロジェン除去に加え、支持体の表面上に酸化物の層を形成することができる。この予備処理は、金属の固形支持体が使用される場合に特に有益である。ひとつの態様において、予備処理は、ニッケル固形支持体の約2〜6時間、好ましくは約5時間の、温度範囲約200〜350℃、好ましくは約250℃での加熱が関連する。
【0051】
蛋白質分子の固形支持体への付着
分子は、吸着によるか、または共有結合を含む、直接の化学結合により、固形支持体に直接付着することができる。例えば、Hermanson、BIOCONJUGATE TECHNIQUES、Academic Press社、ニューヨーク、1996年参照。分子それ自身は、求核基、脱離基、または求電子基を含む、様々な化学官能基により、直接活性化することができる。官能基の活性化は、アルキルおよびアシルのハロゲン化物、アミン、スルフヒドリル、アルデヒド、不飽和結合、ヒドラジド、イソシアナート、イソチオシアナート、ケトン、および化学結合を活性化することが分かっている他の基を含む。または分子は、小分子-カップリング試薬の使用を介して、固形支持体に結合することができる。カップリング試薬の非限定的例は、カルボジイミド、マレイミド、N-ヒドロキシスクシンイミドエステル、ビスクロロエチルアミン、グルタルアルデヒドのような二官能性アルデヒド、酸無水物などを含む。別の態様において、分子は、当技術分野において周知である、ビオチンストレプトアビジン連結またはカップリングのような、親和性結合を介して、固形支持体にカップリングされ得る。例えば、ストレプトアビジンは、共有的または非共有的付着により、固形支持体に結合することができ、ビオチン化された分子は、当技術分野において周知の方法を用いて、合成することができる。例えば、Hermansonの論文(1996)を参照のこと。
【0052】
固形支持体への共有結合が企図される場合は、この支持体は、典型的にはリンカーを介し、適当な反応物への共有的付着に利用可能な1種または複数の化学的部分または官能基を含むポリマーによりコーティングされる。例えば、アミノ酸ポリマーは、適当なリンカーを共有的に介した分子のカップリングに使用することができる、リシンのε-アミノ基などの基を有することができる。本発明は、固形支持体の上に第二のコーティングを配置し、これらの官能基を提供することも企図している。
【0053】
活性化化学
活性化化学を使用し、分子を固形支持体の表面へ特異的で安定して付着させることができる。蛋白質を官能基に付けるために使用することができる多くの方法が存在する;例えば、Hermansonの論文(1996)参照。例えば、二工程法において、共通の架橋剤グルタルアルデヒドを用い、蛋白質アミン基を、アミン化された固形支持体表面に付着することができる。得られた連結は、加水分解的に安定である。他の方法は、蛋白質上のアミンと反応するn-ヒドロ-スクシンイミド(NHS)エステルを含む架橋剤、アミン含有、スルフヒドリル含有、またはヒスチジン含有の蛋白質と反応する活性ハロゲンを含む架橋剤、アミンまたはスルフヒドリル基と反応するエポキシドを含む架橋剤の使用、マレイミド基とスルフヒドリド基の間の結合、ならびにペンダント糖部分の過ヨウ素酸酸化による蛋白質アルデヒド基の形成、それに続く還元性アミノ化を含む。
【0054】
ひとつの態様において、蛋白質分子は、3-アミノプロピルトリエトキシシランを用い、シリカコーティングに付着される(WeetallおよびFilbert、Methods Enzymol.、34:59-72(1974))。この化合物は、シリカ表面と安定した共有結合を形成し、同時にその表面をより疎水性にする。シラン化反応は、水性の低いpHの媒質内で行うことができ、これは結合に利用可能なアミノ基を伴う単層を形成することができることがわかっている。蛋白質の付着は、ホモ二官能性カップリング剤グルタルアルデヒドによるか、またはSMCCのようなヘテロ二官能性物質によることができる。蛋白質付着後、残留する表面に会合したカップリング剤は、様々な蛋白質、親水性ポリマー、およびアミノ酸と共にインキュベーションすることにより活性化されることができる。アルブミンおよびポリエチレングリコールが特に適しており、その理由はこれらは、蛋白質および細胞の固相への非特異的結合をブロックするからである。
【0055】
別の態様において、アミノシラン化を用い、酸化アルミニウムで被覆された固形支持体の表面が活性化される。米国特許第4,554,088号(1985)を参照のこと。酸化アルミニウムで被覆された固形支持体の表面を活性化する別の方法は、glu-lys-tyrトリペプチドのような、強力に接着するポリマーの吸着である。このトリペプチドポリマーは、ジフルオロジニトロベンゼンのようなホモ二官能性架橋剤との反応によるか、またはグルタルアルデヒドとの反応により、リシンアミンを介して活性化することができる。その後蛋白質を、活性化された表面へ直接付着することができる。
【0056】
機能性蛋白質複合体の最適化
特異的蛋白質の固形支持体表面への付着は、蛋白質の直接の連結によるか、または間接的方法の使用により、実現することができる。ある種の蛋白質は、それら自身を、直接的付着または複合に適合させるが、他方で他の蛋白質または抗体は、抗マウスIgGまたはストレプトアビジンのような、リンカーまたはスペーサー蛋白質に連結された場合に、より良い機能活性を保持する。望ましい場合には、リンカーまたは付着蛋白質を使用することができる。
【0057】
固形支持体に連結された機能性蛋白質の比の最適化
同じ固形支持体上の特定の蛋白質の比は、抗原または抗体の提示における固形支持体の有効性を増大するように変動することができる。例えば、A2-Ig(下記実施例1に説明)(シグナル1)の抗CD28(シグナル2)に対する最適比は、下記のように試験することができる。固形支持体は、A2-Igおよび抗CD28に30:1、10:1、3:1、1:1、0.3:1;0.1:1、および0.03:1のような様々な比で連結している。支持体に連結した蛋白質の総量は、一定に保たれる(例えば、粒子150mg/mlで)か、または変動することができる。サイトカイン放出および増殖のようなエフェクター機能は、T細胞活性化および分化と比べ、シグナル1対シグナル2の必要要件が異なるので、これらの機能は個別にアッセイすることができる。
【0058】
分析的アッセイ法
固形支持体は、支持体が製造される時に生じる添加(addition)および反応を評価するいくつかの分析的アッセイ法により特徴付けられる。これらは、アミンおよびアルデヒドのような官能基のアッセイ法、ならびに特定の型の蛋白質分子の結合のアッセイ法を含む。加えて機能アッセイ法を用い、固形支持体の生物学的活性を評価することができる。固形支持体の表面に結合した蛋白質の量は、当技術分野において公知の方法のいずれかにより決定することができる。例えば、結合した蛋白質は、280nmでの吸光度を用い、反応液から取り除かれる蛋白質量を決定することにより、間接的に測定することができる。この態様において、固形支持体の添加の前後の反応液の蛋白質含量は、280nmでの吸光度で測定され、および比較される。いずれかの洗浄液に含まれる蛋白質量も測定され、反応後溶液中に認められた量に追加される。この差は、固形支持体表面に結合した量の指標である。この方法を使用し、異なる反応条件の結合効率を迅速にスクリーニングすることができる。
【0059】
別の態様において、固形支持体に結合した蛋白質量は、標識された抗原および抗体の結合アッセイ法による、より直接的アッセイ法で測定することができる。例えば、様々な濃度の抗体結合した固形支持体を、一定濃度のHRP標識した抗原またはヤギ-抗マウスIgGとインキュベーションすることができる。この支持体は、緩衝液で洗浄し、未結合の標識された蛋白質を除去する。支持体-会合したHRPを、OPD基質を用いて測定し、結合した標識された蛋白質の濃度を得る。スキャッチャードプロット解析は、固定された蛋白質の濃度および親和性を提供することができる。HRP標識された抗体は、商業的に入手できるか、または抗体は、AvrameasおよびTernync(Immunochemisty、8:1175-79(1971))のグルタルアルデヒド法を用い、HRPで標識することができる。
【0060】
前述の方法は、共有結合した蛋白質および非共有的に結合した蛋白質の両方を測定する。これら2種の結合の間を識別するために、固形支持体は、6M塩酸グアニジンまたは8M尿素のような、強力なカオトロピック剤で洗浄することができる。非特異的結合は、これらの条件により破壊され、および固形支持体から洗浄除去された蛋白質の量は、280nmでの吸光度により測定することができる。結合した蛋白質総量およびカオトロピック剤で洗浄除去された量の間の差は、密に結合されおよび恐らく共有結合されている蛋白質の量を表す。
【0061】
細胞
本発明の抗原提示プラットフォームおよび抗体誘導プラットフォームの両方は、細胞を基にすることができる。これらの細胞は、好ましくは真核細胞であり、より好ましくは哺乳類細胞であり、更により好ましくは霊長類の細胞であり、最も好ましくはヒト細胞である。
【0062】
本発明のプラットフォーム表面の多くの分子は、クローニングすることができる。従って細胞は、このような分子をコードしている構築物によりトランスフェクションすることができる。細胞をトランスフェクションする方法は、当技術分野において周知であり、トランスフェリン-ポリカチオン-媒介したDNA導入、裸のまたは被包した核酸によるトランスフェクション、リポソーム-媒介型細胞融合、DNA-被覆したラテックスビーズの細胞内輸送、プロトプラスト融合、ウイルス感染、電気穿孔、およびリン酸カルシウム-媒介型トランスフェクションを含むが、これらに限定されるものではない。
【0063】
または、蛋白質は、細胞表面に化学的に結合することができる。この目的のために、蛋白質の細胞表面へのいずれかの連結法を使用することができ、例えば様々なリンカー(例えば、ペプチドリンカー、ストレプトアビジン-ビオチンリンカー)の使用である。
【0064】
抗原提示プラットフォームに連結した分子
抗原提示プラットフォームに連結した分子は、少なくとも1種のT細胞に影響する分子および少なくとも1個の抗原結合窩を含む少なくとも1種の抗原提示複合体を含む。任意で、抗原は、この抗原結合窩に結合することができる。これらの構成要素を、以下に説明する。
【0065】
抗原提示複合体
抗原提示複合体は、抗原結合窩を含み、ならびにT細胞またはT細胞前駆体への提示のために抗原を結合することができる。抗原提示複合体は、例えば、MHCクラスIまたはクラスII分子、MHCクラスIもしくはクラスII分子の機能的抗原結合窩を含む融合蛋白質、MHCクラスIまたはクラスII「分子複合体」(後述)、またはCD1ファミリーメンバー(例えば、CD1a、CD1b、CD1c、CD1d、およびCD1e)などの非古典的MHC様分子であることができる。
【0066】
一部の態様において、抗原提示複合体は、MHCクラスIおよび/またはMHCクラスII分子複合体である。MHCクラスIおよびクラスII分子複合体は、多くの有用な特徴を有する。例えばこれらは、免疫グロブリン骨格によりもたらされる安定性および分泌効率を基に、極めて安定しており、ならびに作成が容易である。更に、免疫グロブリンのFc部分を変更することにより、異なる生物学的機能を、Fc部分によりもたらされる生物学的機能を基に、分子に提供することができる。ひとつの型の免疫グロブリン分子のFc部分の別のものへの置換は、当技術分野の技術の範囲内である。
【0067】
MHCクラスI分子複合体
「MHCクラスI分子複合体」は、米国特許第6,268,411号に開示されている。MHCクラスI分子複合体は、免疫グロブリン重鎖の末端に高次構造的に無傷の様式で形成される(概略的説明は、米国特許第6,268,411号の図IA参照)。抗原性ペプチドが結合しているMHCクラスI分子複合体は、独自のクローン形質のリンパ球受容体(例えば、T細胞受容体)に安定して結合することができる。
【0068】
MHCクラスI分子複合体は、少なくとも2種の融合蛋白質を含む。第一の融合蛋白質は、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、MHCクラスI分子複合体を形成し、これは2個のMHCクラスIペプチド結合窩を含む。免疫グロブリン重鎖は、IgM、IgD、IgG1、IgG3、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG重鎖を使用し、MHCクラスI分子複合体を形成する。多価のMHCクラスI分子複合体が望ましい場合は、IgMまたはIgA重鎖を用い、各々、五価または四価の分子を提供することができる。他の価数を伴うMHCクラスI分子複合体も、複数の免疫グロブリン重鎖を用い構築することができる。MHCクラスI分子複合体の構築は、米国特許第6,268,411号に詳細に開示されている。
【0069】
MHCクラスII分子複合体
「MHCクラスII分子複合体」は、米国特許第6,458,354号、米国特許第6,015,884号、米国特許第6,140,113号、および米国特許第6,448,071号に開示されている。MHCクラスII分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含む。これら2種の第一および2種の第二の融合蛋白質は会合し、MHCクラスII分子複合体を形成する。各々の第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各々の第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインは、MHCクラスIIペプチド結合窩を形成する。
【0070】
免疫グロブリン重鎖は、IgM、IgD、IgG3、IgG1、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG1重鎖を使用し、2個の抗原結合窩を含む二価の分子複合体を形成する。任意で、重鎖の可変領域を含むことができる。IgMまたはIgA重鎖を用い、各々、五価または四価の分子複合体を提供することができる。他の価数を伴う分子複合体も、複数の免疫グロブリン鎖を用い構築することができる。
【0071】
MHCクラスII分子複合体の融合蛋白質は、免疫グロブリン鎖とMHCクラスIIポリペプチドの細胞外ドメインの間に挿入されたペプチドリンカーを含むことができる。リンカー配列の長さは、抗原結合および受容体架橋の程度を調節するために必要な可変性に応じて、変動することができる。MHCクラスIIポリペプチドの細胞外ドメインが、リンカー領域を追加することなく、免疫グロブリン分子に直接的および共有的に付着されるように、構築物を設計することもできる。
【0072】
リンカー領域が含まれる場合は、この領域は、好ましくは少なくとも3種および30種を超えないアミノ酸を含むと思われる。より好ましくは、このリンカーは、約5個および20個を超えないアミノ酸であり;最も好ましくは、リンカーは、10個未満のアミノ酸である。一般にリンカーは、短いグリシン/セリンスペーサーからなるが、あらゆるアミノ酸を使用することができる。免疫グロブリン重鎖をMHCクラスIIβ鎖の細胞外ドメインに接続するのに好ましいリンカーは、
(配列番号:1)である。免疫グロブリン軽鎖をMHCクラスIIα鎖の細胞外ドメインに接続するのに好ましいリンカーは、
(配列番号:2)である。
【0073】
T細胞に影響する分子
T細胞に影響する分子は、前駆体T細胞または抗原特異的T細胞に生物学的作用を有する分子である。このような生物学的作用は、例えば、前駆体T細胞の、CTL、ヘルパーT細胞(例えば、Th1、Th2)、または調節T細胞への分化;T細胞の増殖;ならびに、T細胞アポトーシスの誘導を含むが、これらに限定されるものではない。従って、T細胞に影響する分子は、T細胞共刺激分子、接着分子、T細胞増殖因子、調節T細胞誘導分子、およびアポトーシス誘導分子を含む。本発明の抗原提示プラットフォームは、このような分子の少なくとも1種を含み;任意で、抗原提示プラットフォームは、少なくとも2種、3種または4種のこのような分子を、いずれかの組合せで含む。
【0074】
T細胞共刺激分子は、抗原特異的T細胞の活性化に貢献している。このような分子は、CD28(抗体を含む)、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体を含むが、これらに限定されるものではない。
【0075】
本発明の抗原提示プラットフォームに有用な接着分子は、T細胞またはT細胞前駆体のこのプラットフォームへの接着を媒介する。本発明に有用な接着分子は、例えば、ICAM-1およびLFA-3を含む。
【0076】
T細胞増殖因子は、T細胞の増殖および/または分化に影響を及ぼす。T細胞増殖因子の例は、サイトカイン(例えば、インターロイキン、インターフェロン)およびスーパー抗原である。特に有用なサイトカインは、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンである。望ましい場合には、サイトカインは、融合蛋白質を含む分子複合体中に存在することができる。ひとつの態様において、サイトカイン分子複合体は、少なくとも2種の融合蛋白質を含むことができ:第一の融合蛋白質は、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質は、第二のサイトカインおよび第二の免疫グロブリン重鎖を含む。第一および第二の免疫グロブリン重鎖は会合し、サイトカイン分子複合体を形成する。別の態様において、サイトカイン分子複合体は、少なくとも4種の融合蛋白質を含み:2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)第一のサイトカインを含み、ならびに2種の第二の融合蛋白質は、(i)免疫グロブリン軽鎖、および(ii)第二のサイトカインを含む。2種の第一および2種の第二の融合蛋白質は会合し、サイトカイン分子複合体を形成する。いずれかの型のサイトカイン分子複合体中の第一および第二のサイトカインは、同じまたは異なることができる。
【0077】
スーパー抗原は、強力なT細胞マイトジェンである。スーパー抗原は、最初にクラスII主要組織適合(MHC)分子へ結合し、その後T細胞抗原受容体(TCR)へVβ特異的様式でバイナリー複合体として結合することにより、T細胞の有糸分裂を刺激する。スーパー抗原は、細菌エンテロトキシン、例えばブドウ球菌エンテロトキシン(例えば、SEAおよびそれらの活性部分、米国特許第5,859,207号に開示;SEB、SEC、SEDおよびSEEレトロウイルススーパー抗原(米国特許第5,519,114号に開示);化膿性連鎖球菌(Streptococcus pyogenos)の菌体外毒素(SPE)、黄色ブドウ球菌(Staphylococcus aureus)中毒性ショック症候群毒素(TSST-1)、連鎖球菌性マイトジェン性菌体外毒素(SME)、および連鎖球菌性スーパー抗原(SSA)(米国特許第2003/0039655号に開示);ならびに、米国特許第2003/0036644号および米国特許第2003/0009015号に開示されたスーパー抗原を含むが、これらに限定されるものではない。
【0078】
調節T細胞誘導分子は、調節T細胞の分化および/または維持を誘導する分子である。このような分子は、TGFβ、IL-10、インターフェロン-α、およびIL-15を含むが、これらに限定されるものではない。例えば、米国特許第2003/0049696号、米国特許第2002/0090724号、米国特許第2002/0090357号、米国特許第2002/0034500号、および米国特許第2003/0064067号を参照のこと。
【0079】
アポトーシス誘導する分子は、細胞死を引き起こす。アポトーシス誘導する分子は、毒性物質(例えば、リシンA鎖、突然変異体シュードモナス(Pseudomonas)菌体外毒素、ジフテリア毒素、ストレプトニグリン、ボアマイシン(boamycin)、サポリン、ゲロニン、およびアメリカヤマゴボウ抗ウイルス蛋白質)、TNFα、およびFasリガンドを含む。
【0080】
抗原
様々な抗原は、抗原提示複合体に結合することができる。抗原の性質は、使用される抗原提示複合体の種類によって決まる。例えば、ペプチド抗原は、MHCクラスIおよびクラスIIペプチド結合窩に結合することができる。非古典的MHC様分子は、リン脂質、複合糖質など(例えば、ミコール酸およびリポアラビノマンナンのような、細菌の膜成分)のような、非ペプチド抗原を提示するために使用することができる。本明細書において使用される「抗原」は、「抗原性ペプチド」も含む。
【0081】
抗原性ペプチド
免疫応答を誘導することが可能であるいずれかのペプチドは、抗原提示複合体に結合することができる。抗原性ペプチドは、腫瘍関連抗原、自己抗原、アロ抗原、および感染物質の抗原を含む。
【0082】
腫瘍関連抗原
腫瘍関連抗原は、それらが由来した腫瘍により専ら発現され、多くの腫瘍において発現されたが正常な成人組織においては発現されない腫瘍抗原(腫瘍胎児性抗原)を共有した独自の腫瘍抗原、ならびに同じくそれから腫瘍が生じる正常組織により発現された組織特異抗原を含む。腫瘍関連抗原は、例えば、胚性抗原(embryonic antigen)、異常な翻訳後修飾を伴う抗原、分化抗原、突然変異した癌遺伝子または腫瘍抑制因子の産物、融合蛋白質、またはオンコウイルス蛋白質であることができる。
【0083】
様々な腫瘍関連抗原が当技術分野において公知であり、それらの多くは市販されている。腫瘍胎児性抗原および胚性抗原は、癌胎児性抗原およびα-フェトプロテイン(通常発生期の胚においてのみ高発現されるが、各々、肝臓および結腸の腫瘍において頻繁に高発現される)、MAGE-1およびMAGE-3(黒色腫、乳癌、および神経膠腫において発現される)、胎盤アルカリホスファターゼシアリル-Lewis X(腺癌において発現される)、CA-125およびCA-19(消化器、肝臓、および婦人科の腫瘍において発現される)、TAG-72(結腸直腸癌において発現される)、上皮糖蛋白質2(多くの癌腫において発現される)、膵臓癌胎児性抗原、5T4(胃癌において発現される)、αフェトプロテイン受容体(複数の腫瘍型、特に乳癌において発現される)、およびM2A(生殖細胞腫瘍において発現される)を含む。
【0084】
腫瘍関連分化抗原は、チロシナーゼ(黒色腫において発現される)および特に表面免疫グロブリン(リンパ腫において発現される)を含む。
【0085】
突然変異した癌遺伝子または腫瘍抑制遺伝子の産物は、両方とも多くの腫瘍型において発現されるRasおよびp53、Her-2/neu(乳癌および婦人科の癌において発現される)、EGF-R、エストロゲン受容体、プロゲステロン受容体、網膜芽細胞腫産物、myc(肺癌に関連)、ras、p53、乳癌に関連した非突然変異体、MAGE-1、およびMAGE-3(黒色腫、肺癌、および他の癌に関連)を含む。
【0086】
融合蛋白質は、慢性骨髄性白血病において発現されるBCR-ABLを含む。
【0087】
オンコウイルス蛋白質は、HPV型16、E6、およびE7を含み、これらは頸癌において認められる。
【0088】
組織特異抗原は、メラノトランスフェリンおよびMUCI(膵臓癌および乳癌において発現される);CD10(コモン急性リンパ芽性白血病抗原、またはCALLAとして以前は知られている)または表面免疫グロブリン(B細胞白血病およびリンパ腫において発現される);IL-2受容体α鎖、T細胞受容体、CD45R、CD4+/CD8+(T細胞白血病およびリンパ腫において発現される);前立腺特異的抗原および前立腺酸性ホスファターゼ(前立腺癌において発現される);GP100、MelanA/Mart-1、チロシナーゼ、gp75/ブラウン、BAGE、およびS-100(黒色腫において発現される);サイトケラチン(様々な癌において発現される);ならびに、CD19、CD20、およびCD37(リンパ腫において発現される)を含む。
【0089】
腫瘍関連抗原は、変更された糖脂質抗原および糖蛋白質抗原、例えば、ノイラミン酸含有スフィンゴ糖脂質(例えば、GM2およびGD2、黒色腫および一部の脳腫瘍において発現される);癌腫において異常に発現される血液型抗原、特にT抗原およびシアリル化されたTn抗原;ならびに、ムチン、例えばCA-125およびCA-19-9(卵巣癌において発現される)または低グリコシル化されたMUC-1(乳癌および膵臓癌において発現される)なども含む。
【0090】
組織特異抗原は、上皮膜抗原(多発性上皮癌)、CYFRA 21-1(肺癌において発現される)、Ep-CAM(汎-癌腫(pan-carcinoma)において発現される)、CA125(卵巣癌において発現される)、無傷のモノクローナル免疫グロブリン断片または軽鎖断片(骨髄腫において発現される)、ならびに、ヒト絨毛性ゴナドトロピンβサブユニット(HCG、生殖細胞腫瘍において発現される)を含む。
【0091】
自己抗原
自己抗原は、生体が免疫応答において産生する、生体自身の「自分の抗原」である。自己抗原は、グッドパスチャー症候群、多発性硬化症、グレーヴス症、重症筋無力症、全身性紅斑狼瘡、インスリン依存型真性糖尿病、リウマチ様関節炎、尋常性天疱瘡、アジソン病、疱疹状皮膚炎、セリアック病、および橋本甲状腺炎のような、自己免疫疾患に関連している。
【0092】
糖尿病に関連した自己抗原は、インスリン、グルタミン酸脱炭酸酵素(GAD)および他の島細胞の自己抗原、例えばICA 512/IA-2蛋白質チロシンホスファターゼ、ICA12、ICA69、プレプロインスリンまたはそれらの免疫学的活性断片(例えば、インスリンB-鎖、A-鎖、Cペプチドもしくはそれらの免疫学的活性断片)、HSP60、カルボキシペプチダーゼH、ペリフェリン、ガングリオシド(例えば、GM1-2、GM3)またはそれらの免疫学的活性断片を含む。
【0093】
黄斑変性に関連した自己抗原は、補体系分子、ならびにRPE、脈絡膜および網膜からの様々な自己抗原、ビトロネクチン、βクリスタリン、カルレチキュリン、セロトランスフェリン、ケラチン、ピルビン酸カルボキシラーゼ、C1、およびビリン2を含む。
【0094】
他の自己抗原は、ヌクレオソーム(ヒストンおよびDNAを含む粒子);リボヌクレオ蛋白質(RNP)粒子(RNP粒子内でRNAおよび特定機能を媒介する蛋白質を含む)、および2本鎖DNAを含む。更に他の自己抗原は、ミエリン希突起膠細胞糖蛋白質(MOG)、ミエリン結合糖蛋白質(MAG)、ミエリン/稀突起膠細胞塩基性蛋白質(MOBP)、稀突起膠細胞特異的蛋白質(Osp)、ミエリン塩基性蛋白質(MBP)、蛋白脂質アポ蛋白質(PLP)、ガラクトースセレブロシド(GalC)、糖脂質、スフィンゴ脂質、リン脂質、ガングリオシド、および他の神経抗原を含む。
【0095】
アロ抗原
アロ抗原は、同じ種の別のメンバーにより抗原として検出される対立遺伝子の直接的または間接的産物である。このような対立遺伝子の直接的産物は、コードされたポリペプチドを含み;間接的産物は、対立遺伝子がコードしている酵素により合成された多糖および脂質を含む。アロ抗原は、主要および非主要組織適合性抗原(ヒトにおいてHLAとして知られている)を含み、これは、クラスIおよびクラスII抗原、ABO、Lewis血液型のような血液型抗原、TおよびB細胞上の抗原、ならびに単球/内皮細胞抗原を含む。HLA特異性は、A(例えば、A1-A74、特にA1、A2、A3、A11、A23、A24、A28、A30、A33)、B(例えば、B1-B77、特にB7、B8、B35、B44、B53、B60、B62)、C(例えばC1-C11)、D(例えばD1-D26)、DR(例えばDR1、DR2、DR3、DR4、DR7、DR8、およびDR11)、DQ(例えばDQ1-DQ9)、ならびにDP(例えば、DP1-DP6)を含む。
【0096】
感染物質の抗原
感染物質の抗原は、原生動物、細菌、真菌(単細胞および多細胞の両方)、ウイルス、プリオン、細胞内寄生体、蠕虫、および免疫応答を誘導する他の感染物質の構成要素を含む。
【0097】
細菌抗原は、グラム陽性球菌、グラム陽性桿菌、グラム陰性菌、好気性菌、例えば、放線菌科(Actinomycetaceae)、バシラス科(Bacillaceae)、バルトネラ科(Burtonellaceae)、ボルデテラ科(Bordetellae)、カプトファーガ科(Captophagaceae)、コリネバクテリウム科(Corynebacteriaceae)、エンテロバクテリア科(Enterobacteriaceae)、レジオネラ科(Legionellaceae)、ミクロコッカス科(Micrococcaceae)、マイコバクテリウム科(Mycobacteriaceae)、ノカルジア科(Nocardiaceae)、パスツレラ科(Pasteurellaeceae)、シュードモナス科(Pseudomonadaceae)、スピロヘータ科(Spirochaetaceae)、ビブリオ科(Vibrionaceae)の生物、ならびにアシネトバクター(Acinotobacter)属、ブルセラ(Brucella)属、カンピロバクター(Campylobacter)属、エリジペロトリックス(Erysipelothrix)属、ユーインゲラ(Ewingella)属、フランシセラ(Francisella)属、ガルドネラ(Gardnerella)属、ヘリコバクター(Helicobacter)属、レビネア(Levinea)属、リステリア(Listeria)属、ストレプトバシラス(Streptobacillus)属およびトロフェリーマ(Tropheryma)属の生物の抗原を含む。
【0098】
原生動物の感染物質の抗原は、マラリア原虫、リーシュマニア(Leishmania)種、トリパノソーマ(Trypanosoma)種および住血吸虫(Schistosoma)種の抗原を含む。
【0099】
真菌抗原は、アスペルギルス(Aspergillus)、ブラストミセス(Blastomyces)、カンジダ(Candida)、コクシジオイデス(Coccidioides)、クリプトコッカス(Cryptococcus)、ヒストプラスマ(Histoplasma)、パラコクシジオイデス(Paracoccicioides)、スポロスリックス(Sporothrix)、ムコール(Mucoraiea)目の生物、クロマイコシス(choromycosis)および菌腫(mycetoma)を含む生物、ならびに白癬菌(Trichophyton)属、小胞子菌(Microsporum)属、表皮菌(Epidermophton)属およびマラセジア(Malassezia)属の生物の抗原を含む。
【0100】
プリオンの抗原は、スクレイピー、ウシ海綿状脳症(BSE)、ネコ海綿状脳症、クールー病、クロイツフェルト-ヤコブ病(CJD)、ゲルストマン-シュトロイスラー-シャインカー病(GSS)、および致命的家族性不眠症(FFI)を引き起こす、プリオンのシアロ糖蛋白質PrP 27-30を含む。
【0101】
抗原性ペプチドが得られる細胞内寄生体は、クラミジア科(chlamydiaceae)、マイコプラズマ科(Mycoplasmataceae)、アコレプラスマ科(Acholeplasmataceae)、リケッチア科(Richettsiae)、ならびにコクシエラ科(Coxiella)およびエーリッヒア属(Ehrlishia)の生物を含むが、これらに限定されるものではない。
【0102】
抗原性ペプチドは、線虫、吸虫または条虫のような蠕虫から得られる。
【0103】
ウイルスのペプチド抗原は、アデノウイルス、単純ヘルペスウイルス、パピローマウイルス、呼吸シンシチアウイルス、ポックスウイルス、HIV、インフルエンザウイルス、およびCMVのものを含むが、これらに限定されるものではない。特に有用なウイルスのペプチド抗原は、HIV gag蛋白質(膜アンカー(MA)蛋白質、コアキャプシド(CA)蛋白質およびヌクレオキャプシド(NC)蛋白質を含むが、これらに限定されるものではない)、HIVポリメラーゼのような、HIV蛋白質、インフルエンザウイルスマトリックス(M)蛋白質およびインフルエンザウイルスヌクレオキャプシド(NP)蛋白質、B型肝炎表面抗原(HBsAg)、B型肝炎コア蛋白質(HBcAg)、B型肝炎e蛋白質(HBeAg)、B型肝炎DNAポリメラーゼ、およびC型肝炎抗原などを含む。
【0104】
抗原の抗原提示複合体への結合
抗原性ペプチドを含む抗原は、米国特許第6,268,411号に開示されたように、能動的又は受動的のいずれかで、抗原提示複合体の抗原結合窩に結合することができる。任意で、抗原性ペプチドは、ペプチド結合窩に共有結合することができる。
【0105】
望ましい場合には、ペプチドテザー(tether)を用い、抗原性ペプチドをペプチド結合窩に連結することができる。例えば、複数のクラスI MHC分子の結晶学的解析は、MHCペプチド結合窩内に存在する抗原性ペプチドのカルボキシ末端と、β2Mのアミノ末端が非常に近接し、約20.5Å離れていることを示している。従って、およそ13個のアミノ酸長のような比較的短いリンカー配列を用い、ペプチドを、β2Mのアミノ末端につなぎ止めることができる。この配列が適している場合には、そのペプチドは、MHC結合溝に結合すると思われる(米国特許第6,268,411号参照)。
【0106】
抗体誘導プラットフォームに連結した分子
抗体誘導プラットフォームに連結した分子は、少なくとも1種のB細胞に影響する分子、ならびにB細胞表面免疫グロブリンに結合することができるかまたはB細胞表面の抗原含有MHC複合体に結合することができる、少なくとも1種の分子複合体を含む。
【0107】
B細胞に影響する分子
B細胞に影響する分子は、B細胞またはB細胞前駆体に対し、例えば増殖の誘導または抗体形成のような生物学的作用を有する分子である。このような分子は、CD40リガンドに加え、前述のサイトカインおよびサイトカイン分子複合体を含む。使用されるサイトカイン分子の種類に応じて、B細胞は、特定の種類の抗体を産生するようにしむけられる。例えば、IL-4はIgEの産生を誘導するのに対し、IL-5はIgAの産生を誘導する。
【0108】
分子複合体
抗体誘導プラットフォームにおいて使用するための分子複合体は、B細胞表面免疫グロブリンに結合するか、またはB細胞表面のMHC-抗原複合体に結合する複合体である。B細胞表面免疫グロブリンに結合している分子複合体は、プラットフォーム表面に複合された抗原を含む。B細胞表面のMHC-抗原複合体に結合している分子複合体は、T細胞受容体(TCR)およびTCR分子複合体を含む。抗体誘導プラットフォームは、このような分子複合体の一方または両方の型を含むことができる(すなわち、B細胞表面免疫グロブリン結合またはMHC-抗原結合)。
【0109】
特定の抗原に関するTCR特異性は、当技術分野において周知の方法を用いクローニングすることができる。例えば、米国特許第2002/0064521号を参照のこと。クローニングされた抗原特異的TCRを、そのように用いることができるか、または下記のTCR分子複合体を形成するために使用することができる。
【0110】
TCR分子複合体
「TCR分子複合体」は、米国特許第6,458,354号、米国特許第6,015,884号、米国特許第6,140,113号、および米国特許第6,448,071号において開示されている。TCR分子複合体は、少なくとも4種の融合蛋白質を含む。2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)TCRα鎖の細胞外ドメインを含む。2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)TCRβ鎖の細胞外ドメインを含む。または、2種の第一の融合蛋白質は、(i)免疫グロブリン重鎖、および(ii)TCRγ鎖の細胞外ドメインを含み、ならびに2種の第二の融合蛋白質は、(i)免疫グロブリンκまたはλ軽鎖、および(ii)TCRδ鎖の細胞外ドメインを含む。この2種の第一および2種の第二の融合蛋白質は会合し、TCR分子複合体を形成する。各第一の融合蛋白質のTCR鎖の細胞外ドメイン、および各第二の融合蛋白質のTCR鎖の細胞外ドメインは、抗原認識窩を形成する。
【0111】
免疫グロブリン重鎖は、IgM、IgD、IgG3、IgG1、IgG2β、IgG2α、IgE、またはIgAの重鎖であることができる。好ましくは、IgG1重鎖を用い、2個の抗原認識窩を含む二価のTCR分子複合体を形成する。任意で、重鎖の可変領域を含むことができる。IgMまたはIgA重鎖を使用し、各々、五価または四価のTCR分子複合体を提供する。他の価数を有するTCR分子複合体も、複数の免疫グロブリン鎖を用い構築することができる。
【0112】
TCR分子複合体の融合蛋白質は、免疫グロブリン鎖とTCRポリペプチドの細胞外ドメインの間に挿入されたペプチドリンカーを含むことができる。このリンカー配列の長さは、抗原結合および架橋の程度を調節するために必要である可変性に応じて、変動することができる。構築物は、TCRポリペプチドの細胞外ドメインが、追加のリンカー領域を伴うことなく、免疫グロブリン分子に直接および共有的に付着するように、設計することもできる。リンカー領域が含まれる場合は、この領域は、少なくとも3個および30個を超えないアミノ酸を含むことが好ましい。より好ましくは、このリンカーは、約5個および20個を超えないアミノ酸であり;最も好ましくは、このリンカーは、10個未満のアミノ酸である。一般に、このリンカーは、短いグリシン/セリンスペーサーからなるが、いずれかのアミノ酸を使用することができる。免疫グロブリン重鎖をTCRαまたはγ鎖の細胞外ドメインへ結合するために好ましいリンカーは、
(配列番号:1)である。免疫グロブリン重鎖をTCRβまたはδ鎖の細胞外ドメインへ結合するために好ましいリンカーは、
(配列番号:2)である。
【0113】
特異的細胞集団を誘導および拡大するための本発明のプラットフォームの使用法
抗原特異的T細胞の誘導および拡大
本発明は、CTL、ヘルパーT細胞、および調節T細胞を含む、抗原特異的T細胞の形成を誘導しおよび拡大する方法を提供する。これらの方法は、複数の前駆体T細胞を含有する単離された調製物を、抗原が抗原結合窩に結合した本発明の抗原提示プラットフォームと接触することが関連している。この調製物の抗原提示プラットフォームと一緒のインキュベーションは、集団内の前駆体細胞を誘導し、その抗原を認識する抗原特異的T細胞を形成する。抗原特異的T細胞は、前駆体T細胞の、後述の本発明の抗原提示プラットフォームと一緒のインキュベーションにより得ることができるか、または例えば樹状細胞とのインキュベーション、もしくは当技術分野において公知の人工的抗原提示細胞とのインキュベーションのような、常法により得ることができる。
【0114】
典型的には、第一の細胞集団内の抗原特異的T細胞の数または割合のいずれかは、前駆体T細胞が、CD3に特異的に結合する抗体を含むが、抗原提示複合体を含まないような粒子と共にインキュベーションされた場合に形成された抗原特異的T細胞の数または割合よりも大きい。
【0115】
抗原提示プラットフォームが使用される本明細書において開示されたいずれかの態様において、抗原提示複合体、結合した抗原、およびT細胞に影響する分子のいずれかの組合せを使用することができる。例えば、抗原提示プラットフォームは、1種または複数のT細胞共刺激分子(同じまたは異なるのいずれか)、1種または複数の調節T細胞誘導する分子(同じまたは異なるのいずれか)、1種または複数の接着分子(同じまたは異なるのいずれか)、および/または1種または複数のT細胞増殖因子(同じまたは異なるのいずれか)を含むことができる。同様に、いずれか特定の抗原提示プラットフォームは、同じまたは異なるのいずれかの1種または複数の抗原提示複合体を含むことができ、これに抗原の組合せが結合することができる。ひとつの態様において、いくつかの異なる黒色腫-関連抗原(例えば、チロシナーゼ、MAGE-1、MAGE-3、GP-100、Melan A/Mart-1、gp75/ブラウン、BAGE、およびS-100のいずれかまたは全て)は、1種または複数のプラットフォーム上の抗原提示複合体に結合することができる。
【0116】
前駆体T細胞は、患者または適当なドナーから得ることができる。ドナーは、同一の双子である必要はなく、更に患者に関連する必要もない。しかし、ドナーおよび患者は、少なくとも1種のHLA分子を共有していることが好ましい。前駆体T細胞は、末梢血単核細胞、骨髄、リンパ節組織、脾臓組織、および腫瘍を含む、多くの給源から得ることができる。または、当技術分野においてT細胞系が使用される。
【0117】
ひとつの態様において、前駆体T細胞は、フィコール(Ficoll)分離のような、当業者に公知の多くの技術を用い、対象から収集された血液単位から得られる。例えば、個人の循環血からの前駆体T細胞は、成分採血または白血球搬出により得ることができる。成分採血産物は、典型的には、T細胞および前駆体T細胞を含むリンパ球、単球、顆粒球、B細胞、他の有核白血球、赤血球、および血小板を含む。成分採血により収集された細胞は、血漿画分を除去するため、および引き続きの処理工程のために細胞を適当な緩衝液または培地中に配置するために、洗浄することができる。洗浄工程は、例えば半-自動化された「フロー-スルー」遠心分離(例えば、Cobe 2991細胞プロセッサー)を製造業者の指示に従い使用することにより、当業者に公知の方法で行うことができる。洗浄後、細胞を、例えば、Ca非含有、Mg非含有PBSのような、様々な生体適合性緩衝液中に再懸濁してもよい。または、成分採血試料中の望ましくない成分を除去することができ、および細胞を直接培養培地中に懸濁することができる。望ましい場合には、赤血球の溶解、および例えばパーコール(PERCOLL)(商標)勾配による遠心分離による単球の枯渇により、前駆体T細胞を、末梢血リンパ球から単離することができる。
【0118】
任意で、抗原特異的T細胞を含む細胞集団は、第一の細胞集団内の抗原特異的T細胞の数に対して増加した数の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な期間、同じ抗原提示プラットフォームまたは第二の抗原提示プラットフォームのいずれかと共に、インキュベーションを継続することができる。典型的には、このようなインキュベーションは、3〜21日間、好ましくは7〜10日間実行される。
【0119】
適当なインキュベーション条件(培養培地、温度など)は、T細胞またはT細胞前駆体の培養に使用されるものに加え、DCもしくは人工の抗原提示細胞を使用する抗原特異的T細胞の形成を誘導するために当業者に公知のものを含む。例えば、LatoucheおよびSadelain、Nature Biotechnol.、18:405-09、4月(2000);Levineら、J. Immunol.、159:5921-30(1997);Mausら、Nature Biotechnol.、20:143-48、2月(2002)を参照のこと。同じく下記具体例を参照のこと。
【0120】
抗原提示プラットフォームとT細胞の間の相互作用期間の最適化
本発明の抗原提示プラットフォームによるT細胞刺激と、通常の正常な樹状細胞によるものの間の差のひとつは、必要とされる刺激期間である。例えば、正常なDCのCTLによる認識は、最終的には溶解および活性化されたT細胞による抗原性刺激の排除につながる。対照的に、T細胞は、抗原提示プラットフォーム上の抗原、特に人工的な生分解性でない表面を基にしたものを排除する効果的方法を有さないことがある。従って、このプラットフォームによる刺激は、数日ではない場合は、数時間行われると思われる。
【0121】
増殖シグナルの大きさを評価するために、抗原特異的T細胞集団は、CFSEで標識され、および細胞分裂の速度および数が分析される。T細胞は、抗原が結合している本発明の抗原提示プラットフォームのいずれかにより1〜2回の刺激ラウンドが行われた後、CFSEで標識された。その時点で、抗原特異的T細胞は、総細胞集団の2〜10%を占める。この抗原特異的T細胞は、抗原特異的染色を用いて検出することができ、その結果抗原特異的T細胞の分裂の速度および数は、CFSEの喪失に従う。刺激後異なる時間で(例えば、12時間、24時間、36時間、48時間および72時間など)、細胞を、抗原提示複合体の染色およびCFSEの両方について分析することができる。抗原が結合していない抗原提示プラットフォームによる刺激を用い、増殖のベースラインレベルを決定することができる。任意に当技術分野において公知のように、増殖は、3H-チミジンの取込みのモニタリングにより検出することができる。
【0122】
培養物は、本発明の抗原提示プラットフォームにより、異なる時間(例えば、0.5時間、2時間、6時間、12時間、36時間に加え、連続する刺激)刺激することができる。粒子または細胞を用いたプラットフォームは、あらゆる複合体を破壊するような激しいピペッティングにより、T細胞から分離することができる。人工の粒子を用いたプラットフォームは、重力により単離することができ;細胞を用いたプラットフォームは、例えばFACSを用い、単離することができる。高度に濃縮された抗原特異的T細胞培養物における刺激時間の作用を、評価することができ、ならびに条件は、プラットフォームの大きい割合(例えば、50%、70%、75%、80%、85%、90%、95%、または98%)を、ほとんど細胞が喪失されることなく回収することができるものと同じである。その後抗原特異的T細胞は、培養物中に戻され、および細胞増殖、増殖速度、アポトーシスに対する作用、様々なエフェクター機能などについて、当技術分野において公知のように分析される。このような条件は、望ましい抗原特異的T細胞反応に応じて変動しても良い。
【0123】
抗原特異的T細胞の検出
本発明の抗原提示プラットフォームのT細胞前駆体の拡大、活性化および分化に対する作用は、かなり多くの当業者に公知の方法においてアッセイすることができる。機能の迅速な決定は、増殖アッセイ法を用い、各T細胞型に特異的マーカーを検出し、培養物中のCTL、ヘルパーT細胞、または調節T細胞の増加を、決定することにより実現することができる。このようなマーカーは、当技術分野において公知である。CTLは、クロム放出アッセイ法を用い、サイトカイン産生または細胞溶解活性をアッセイすることにより検出することができる。
【0124】
プラットフォーム誘導/拡大した抗原特異的T細胞のホーミング受容体の分析
適当なエフェクター機能を伴う抗原特異的T細胞の作成に加え、抗原特異的T細胞の有効性の別のパラメータは、T細胞の病巣への移動を可能にするホーミング受容体の発現である(Sallustoら、Nature、401:708-12(1999);LanzavecchiaおよびSallusto、Science、290:92-97(2000))。適当なホーミング受容体の非存在は、慢性CMVおよびEBV感染症の状況に関与している(Chenら、Blood、98:156-64(2001))。加えて、抗原特異的T細胞を拡大するためのプロフェッショナルAPCの使用と非プロフェッショナルAPCの間の注目されたひとつの差異は、適当なホーミング受容体の発現であり、これはインビボにおける機能不全CTLの存在を説明し得る(Salioら、J. Immunol.、167:1188-97(2001))。
【0125】
例えば、エフェクターCTLの有効性は、ホーミング受容体の下記の表現型、CD62L+、CD45RO+、およびCCR7-に関連されている。従ってプラットフォーム誘導および/または拡大したCTL集団は、これらのホーミング受容体の発現について特徴付けることができる。ホーミング受容体発現は、最初の刺激条件に関連づけられた複合体の特質である。恐らく、これは、共刺激複合体に加え、サイトカイン環境の両方により制御される。関係のあるひとつの重要なサイトカインは、IL-12である(Salioら、2001)。後述のように、本発明のプラットフォームは、生物学的結果のパラメータを最適化するために、個々に個別の構成要素(例えば、T細胞エフェクター分子および抗原提示複合体)を変動する可能性をもたらす。任意で、IL-12などのサイトカインは、抗原特異的T細胞集団におけるホーミング受容体プロファイルに影響を及ぼすために、最初の誘導培地中に含むことができる。
【0126】
誘導および/または拡大された抗原特異的T細胞集団内のオフレート分析
二次抗体反応の進行は、TCR「オフレート(off-rate)」分析により決定される、親和性の焦点化に関係している(Savageら、Immunity、10:485-92:1999;Buschら、J. Exp. Med.、188:61-70(1998);BuschおよびPamer、J. Exp. Med.、189:701-09(1999))。TCRオフレートの減少(すなわち、増加したTCR親和性の結果)は、少ない量の抗原を認識する増加した能力、および関心のあるT細胞集団の生物学的有効性と良く相関するパラメーターである。オフレートは、抗原提示プラットフォーム-媒介した刺激の大きさおよび/または期間を変動することにより、最適化することができる。
【0127】
他の細胞からの抗原特異的T細胞の分離
抗原に結合した抗原特異的T細胞は、結合していない細胞から分離することができる。血漿搬出、フローサイトメトリー、ディファレンシャル遠心分離を含む、当技術分野において公知の方法のいずれかを用い、この分離を実現することができる。ひとつの態様において、T細胞は、例えば、DYNABEADS(登録商標)M-450 CD3/CD28 Tなどの、抗CD3/抗CD28結合したビーズのような、ビーズとの、所望のT細胞の陽性選択に十分な期間のインキュベーションにより、単離される。
【0128】
望ましい場合には、抗原特異的T細胞の亜集団は、存在し得る他の細胞から分離することができる。例えば、CD28+、CD4+、CD8+、CD45RA+、およびCD45RO+T細胞のような、T細胞の特異的亜集団は、陽性または陰性選択技術により、更に単離され得る。ひとつの方法は、負に選択された細胞上に存在する細胞表面マーカーに対するモノクローナル抗体のカクテルを使用する負の磁気免疫接着またはフローサイトメトリーによる、細胞選別および/または選択である。例えば、負の選択によりCD4+細胞を濃縮するために、モノクローナル抗体カクテルは典型的には、CD14、CD20、CD11b、CD16、HLA-DR、およびCD8に対する抗体を含む。抗原特異的調節T細胞は、マーカーFoxp3を用い、検出および/または他の細胞から分離することができる。その時間は、30分〜36時間、もしくは10〜24時間の範囲であることができ、または少なくとも1時間、2時間、3時間、4時間、5時間、または6時間、もしくは少なくとも24時間であることができる。より長いインキュベーション時間を用い、他の細胞と比較しほとんどT細胞が存在しないいずれかの状況、例えば腫瘍組織または免疫無防備状態の個人から、腫瘍浸潤するリンパ球(TIL)を単離するような状況で、T細胞を単離することができる。
【0129】
抗体産生B細胞の誘導および拡大
本発明は、抗体産生B細胞の形成を誘導する方法も提供する。これらの方法は、複数の前駆体B細胞を含む単離された調製物を、本発明の抗体誘導プラットフォームと接触することに関する。調製物の抗体誘導プラットフォームとのインキュベーションは、集団内の前駆体細胞を誘導し、抗原を特異的に認識する抗体を産生する抗体産生B細胞を形成する。典型的には、第一の細胞集団における抗体産生B細胞の数または割合のいずれかは、前駆体B細胞が、非特異性刺激、例えばフィトヘマグルチニン(PHA)、リポ多糖(LPS)、またはヤマゴボウなどと共にインキュベーションされる場合に形成される抗体産生細胞の数または割合よりも大きい。抗体誘導プラットフォームが使用される本明細書において説明したいずれかの態様において、B細胞に影響する分子とB細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する複合体との組合せを使用することができる。
【0130】
前駆体B細胞は、患者または適当なドナーから得ることができる。ドナーおよび患者は、関連する必要はないが、少なくとも1種のHLA分子を共有することが好ましい。または、当技術分野において入手可能なB細胞を使用することができる。ひとつの態様において、前駆体B細胞は、フィコール分離のような、当業者に公知のかなり多くの技術を用い、対象から収集した血液単位から得られる。例えば、個人の循環血液からの前駆体B細胞は、前述のように、成分採血または白血球搬出により得ることができる。
【0131】
B細胞またはそれらの前駆体は、当技術分野において公知の方法を用い、培養することができる。例えば、Schultzeら、J. Clin. Invest.、100:2757-65(1997);von Bergwelt-Baildonら、Blood、99:3319-25(2002)参照。このような条件は、B細胞前駆体の、本発明の抗体誘導プラットフォームとのインキュベーションにも適している。
【0132】
任意で、抗体産生B細胞を含む細胞集団は、第一の細胞集団内の抗体産生B細胞の数と比べ増大した数の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な期間、同じ抗体誘導プラットフォームまたは第二の抗体誘導プラットフォームのいずれかと、インキュベーションするように継続することができる。典型的には、このようなインキュベーションは、3〜21日間、好ましくは7〜10日間実行される。
【0133】
抗体誘導プラットフォームとB細胞の間の相互作用期間の最適化
前述のT細胞刺激のように、抗体産生B細胞の集団を誘導または拡大するために必要な刺激の期間は、特に人工的で生分解性でない表面がプラットフォームに使用される場合、通常生じるものとは異なる。従ってプラットフォームによる刺激は、おそらく数日ではない場合は、数時間行われると思われる。本発明の様々な抗体誘導プラットフォームと前駆体もしくは抗体産生B細胞の間の相互作用の期間は、抗原特異的T細胞について先に説明したものに類似した方法を用いて決定することができる。
【0134】
抗体産生B細胞の検出
本発明の抗体産生プラットフォームのB細胞前駆体の拡大、活性化および分化に対する作用は、当業者に公知のかなり多くの方法によりアッセイすることができる。機能の迅速決定は、増殖アッセイ法を用いるか、B細胞特異的マーカーを検出することによるか、または特異的抗体産生をアッセイすることにより実現することができる。
【0135】
薬学的調製物
本発明の粒子または細胞を用いた抗原提示プラットフォームもしくは抗体誘導プラットフォームに加え、このようなプラットフォームを用い得られた抗原特異的T細胞または抗体特異的B細胞を含有する薬学的調製物を、患者への直接注射用に、製剤化することができる。このような薬学的調製物は、本発明の組成物の患者への送達に適した薬学的に許容される担体、例えば生理食塩水、緩衝した生理食塩水(例えば、リン酸緩衝生理食塩水)、またはリン酸緩衝生理食塩グルコース溶液などを含有する。
【0136】
免疫療法
投与経路
本発明の粒子または細胞を用いた抗原提示プラットフォームまたは抗体誘導プラットフォームに加え、このようなプラットフォームを用いて得られた抗原特異的T細胞または抗体特異的B細胞を、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与を含む、いずれか適当な経路により、患者へ投与することができる。患者は、ヒトおよび獣医学上の患者の両方を含む。
【0137】
治療法
本発明のプラットフォームを用い、当技術分野において公知の診断法および治療法において使用することができる、治療に有用な数の抗原特異的T細胞または抗体産生B細胞を作成することができる。例えば、国際公開公報第01/94944号;米国特許第2002/0004041号;米国特許第5,583,031号;米国特許第2002/0119121号;米国特許第2002/0122818号;米国特許第5,635,363号;米国特許第2002/0090357号;米国特許第6,458,354号;米国特許第2002/0034500号を参照。
【0138】
特に、抗原特異的T細胞または抗体産生B細胞を使用し、感染症、癌、もしくは自己免疫疾患を伴う患者を治療するか、または免疫抑制状態の患者に予防的保護を提供することができる。
【0139】
治療することができる感染症は、細菌、ウイルス、プリオン、真菌、寄生体、蠕虫などにより引き起こされるものを含む。このような疾患は、AIDS、肝炎、CMV感染症、および移植後リンパ増殖性障害(PTLD)を含む。例えばCMVは、臓器移植した患者において認められる最も一般的なウイルス病原体であり、ならびに骨髄移植もしくは末梢血幹細胞移植を受ける患者における有病および死亡の主因である(Zaia、Hematol. Oncol. Clin. North Am.、4:603-23(1990))。これは、これらの患者の免疫無防備状態が原因であり、このことは血清陽性患者における潜在ウイルスの再活性化または血清陰性の個人での日和見感染をもたらす。現在の治療は、薬物耐性CMVの発達が最も重大である欠点を有する、ガンシクロビルのような抗ウイルス化合物の使用に焦点が当てられている。これらの治療の有用な代替は、移植手技の開始前の、患者または適当なドナー由来のウイルス特異的CTLの作成に関連する予防的免疫療法的投薬法である。
【0140】
PTLDは、移植患者の大きい割合において生じ、およびエプスタイン-バーウイルス(EBV)感染症から生じる。EBV感染は、米国の成人集団の約90%に存在する(AnagnostopoulosおよびHummel、Histopathology、29:297-315(1996))。活性ウイルス複製および感染は、免疫系によりチェックされ続けるが、CMVの場合のように、移植療法により免疫無防備状態となった個人は、制御するT細胞集団を喪失し、このことはウイルス再活性化をもたらす。これは、移植プロトコールの重大な障害を表している。EBVは、様々な血液学的癌および非血液学的癌における腫瘍のプロモーションにも関連することがある。EBVと鼻咽頭癌の間の強力な関係も存在する。従って、EBV特異的T細胞による予防的治療は、現在の療法に対する優れた代替を提供する。
【0141】
本発明に従い治療することができる癌は、黒色腫、癌腫、例えば結腸癌、十二指腸癌、前立腺癌、乳癌、卵巣癌、腺管癌、肝臓癌、膵臓癌、腎臓癌、子宮内膜癌、胃癌、口内粘膜の形成異常、ポリープ症、侵襲性口内癌、非小細胞肺癌、転移性および扁平上皮細胞尿路癌など;神経学的悪性疾患、例えば、神経芽細胞腫、神経膠腫など;血液学的悪性疾患、例えば、慢性骨髄性白血病、小児急性白血病、非ホジキンリンパ腫、慢性リンパ球性白血病、悪性皮膚T細胞リンパ腫(malignant cutaneous)、菌状息肉腫、非MF皮膚T細胞リンパ腫、リンパ腫様丘疹症、T細胞豊富な皮膚リンパ球様過形成、水疱性類天疱瘡、円板状紅斑狼瘡、扁平苔癬;などを含む。例えば、Mackensenら、Int. J. Cancer、86:385-92(2000);Jonuleitら、Int. J. Cancer、93:243-51(2001);Lanら、J. Immunotherapy、24:66-78(2001);Meidenbauerら、J. Immunol.、170(4):2161-69(2003)参照。
【0142】
治療することができる自己免疫疾患は、喘息、全身性紅斑狼瘡、リウマチ様関節炎、I型糖尿病、多発性硬化症、クローン病、潰瘍性大腸炎、乾癬、重症筋無力症、グッドバスチャー症候群、グレーヴス病、尋常性天疱瘡、アジソン病、疱疹状皮膚炎、セリアック病、および橋本甲状腺炎を含む。
【0143】
抗原特異的ヘルパーT細胞を使用し、マクロファージを活性化、またはB細胞を活性化し、例えば、感染症および癌の治療に使用することができる特異的抗体を産生することができる。抗体産生B細胞それ自身も、この目的に使用することができる。
【0144】
抗原特異的調節T細胞を使用し、例えば、移植患者における移植片対宿主疾患を治療もしくは予防するために、または前述のような自己免疫疾患、もしくはアレルギーを治療もしくは予防するために、免疫抑制作用を達成することができる。調節T細胞の使用は、例えば、米国特許第2003/0049696号、米国特許第2002/0090724号、米国特許第2002/0090357号、米国特許第2002/0034500号、および米国特許第2003/0064067号に開示されている。T細胞影響する分子がアポトーシス誘導する分子である抗原提示プラットフォームは、免疫応答の抑制に使用することができる。
【0145】
用量
抗原特異的T細胞は、用量範囲約5〜10x106CTL/kgの体重(〜7x108CTL/処置)から、最大約3.3x109CTL/m2(〜6x109CTL/処置)で、患者へ投与することができる(Walterら、New England Journal of Medicine、333:1038-44(1995);Yeeら、J. Exp. Med.、192:1637-44(2000))。他の態様において、患者は、静脈内投与される1回投与量あたり、103個、5x103個、104個、5x104個、105個、5x105個、106個、5x106個、107個、5x107個、108個、5x108個、109個、5x109個または1010個の細胞を受け取ることができる。更に別の態様において、患者は、200μlのボーラス中に例えば8x106個または12x106個の細胞の結節内注射を受け取ることができる。抗体産生B細胞それ自身に加え、細胞を用いた抗原提示プラットフォームまたは抗体誘導プラットフォームを、同様の用量で患者に投与することができる。
【0146】
粒子を用いたプラットフォームが投与される場合、典型量は、1回投与量あたり103個、5x103個、104個、5x104個、105個、5x105個、106個、5x106個、107個、5x107個、108個、5x108個、109個、5x109個または1010個の粒子を含む。
【0147】
動物モデル
多くのマウスモデルが、腫瘍治療に関する養子免疫療法プロトコールを評価するために利用可能である。黒色腫治療を評価するためには、ふたつのモデルが特に適している。ひとつのモデルは、皮下移植されたヒト黒色腫株、例えばBMLを有する、ヒト/SCIDマウスを使用する。このようなモデルにおいて、エクスビボで拡大したMart-1特異的CTLの移植は、この腫瘍の発症および/または増殖を遅延する。第二のモデルは、マウスA2-トランスジェニックマウス、およびAADと称される、HLA-A2様分子がトランスフェクションされたマウスB16黒色腫を使用する。A2-トランスジェニックの基礎でもあるこの分子は、マウスα3ドメインに融合したα1-2ドメイン中のヒトHLA-A2である。これらのトランスジェニックマウスを使用し、マウスのB16-AAD黒色腫は、チロシナーゼおよびgp100に由来した詳細に明らかにされたA2拘束された黒色腫エピトープを超えた拒絶反応に対し感受性がある。
【0148】
キット
本発明の抗原提示プラットフォームまたは抗体誘導プラットフォームのいずれかは、キットで提供することができる。粒子または細胞を用いた抗原提示または抗体誘導プラットフォームに適した容器は、例えば、ボトル、バイアル、注射器、および試験管を含む。容器は、ガラスまたはプラスチックを含む、様々な材料で製造することができる。容器は、無菌のアクセスポートを有しても良い(例えば、容器は、皮下注射針により、剥離可能なストッパーを備えている、静注液用バッグまたはバイアルであることができる。)。またはキットは、前述のような、硬質または軟質の抗原提示または抗体誘導プラットフォームを備えることができる。任意で、1種または複数の異なる抗原を、プラットフォームに結合するか、または個別に供給することができる。
【0149】
キットは更に、リン酸緩衝生理食塩水、リンゲル液、またはデキストロース液などの、薬学的に許容される緩衝液を含む第二の容器を備えることができる。これは、その他の緩衝液、希釈液、フィルター、針、および注射器などの、最終使用者に有用な他の材料も備えることができる。キットは、例えば、最終使用者により抗原提示プラットフォームの抗原提示複合体に結合することができる、化学療法剤または感染症治療薬など、または特定の抗原を含むもののような別の活性物質を伴う第二または第三の容器を備えることもできる。
【0150】
キットは、抗原特異的T細胞または抗体産生B細胞の誘導または拡大、例えば、特異的マーカー蛋白質に対する抗体、MHCクラスIもしくはクラスII分子複合体、TCR分子複合体、抗クローン形質の抗体などの程度および有効性を評価するための試薬も備えることができる。
【0151】
キットは、様々な治療プロトコールにおいて、キット内の抗原特異的T細胞を誘導、抗原特異的T細胞を拡大、抗原提示プラットフォームもしくは抗体誘導プラットフォームを使用する方法について書面による指示を記載した添付文書も備えることができる。添付文書は、未承認の草稿段階の添付文書であるか、または米国食品医薬品局(FDA)または他の規制機関により承認された添付文書であることができる。
【0152】
本開示に引用された全ての特許、特許出願、および参考文献は、明白に本明細書に参照として組み入れられている。前記開示は一般に、本発明を説明している。より完全な理解を、単に本発明の例証を目的として提供され、限定することは意図していない、下記の具体例を参照し得られる。
【実施例】
【0153】
実施例1
材料および方法
細胞株
TAP-欠損174CEM.T2(T2)細胞株および黒色腫細胞株は、10%FCSを補充したM'培地(Oelkeら、Scand. J. Immunol.、52:544-49(2000))において維持した。
【0154】
ペプチド
本試験において使用したペプチド(Mart-1、ELAGIGILTV、配列番号:3;CMVpp65、NLVPMVATV、配列番号:4)は、JHU中核施設が調製した。各ペプチドの純度(>98%)は、質量分析およびHPLCにより確認した。
【0155】
HLA A2.1+リンパ球
Johns Hopkins Universityの施設内倫理委員会(IEC)は、下記実施例において考察した試験を承認した。全てのドナーは、本試験に登録する前に、書面によるインフォームドコンセントを提出した。健常志願者および黒色腫患者のドナー#7は、サイトメトリーにより表現型がHLA-A2.1であった。この黒色腫患者は、肺、肝臓およびリンパ節転移を伴う広範囲の転移性疾患を有した。PBMCは、フィコール-ハイパーク(Hypaque)密度勾配遠心分離により単離した。
【0156】
aAPCの作成
aAPCは、「HLA-Ig」(米国特許第6,268,411号に開示)および抗CD28のミクロビーズ(Dynal社、レークサクセス、NY)上への連結により作成した。簡単に述べると、ビーズを、滅菌した0.1Mホウ酸緩衝液(「ビーズ洗浄緩衝液」)中で2回洗浄した。これらのビーズを、ホウ酸緩衝液中のHLA-A2-Igおよび抗CD28 mAb 9.3の1:1混合液と共に、ローター上で24時間4℃でインキュベーションし、ならびにビーズ洗浄緩衝液で2回洗浄した。ビーズ洗浄緩衝液中で更に24時間4℃でインキュベーションし、この緩衝液を交換した。得られるaAPCは、A2-Ig/ビーズの0.9x105個分子、および抗CD28分子/ビーズの1.9x105個を有することがわかった。aAPCビーズは、4℃で3ヶ月を越えて貯蔵し、活性は失われなかった。ペプチド負荷に関して、HLA-Ig被覆したaAPCを、PBSで2回洗浄し、およびペプチド30mg/ml中に107個ビーズ/mlとなるよう調節した(最終濃度)。aAPCビーズは、4℃でペプチド溶液中に貯蔵した。
【0157】
樹状細胞のインビトロ作成
単球は、CD14+磁気分離(Miltenyi、オーバーン、CA)により、PBMCから単離した。CD14+細胞は、2%自家血清、100ng/mlヒトGM-CSF、50ng/ml IL-4、および5ng/ml TGF-β1を含む、M'培地において培養した。5〜7日培養した後、10ng/ml TNF-βおよびIL-1β、1000U/ml IL-6(BD-Pharmingen社、サンディエゴ、CA)および1mg/ml PGE2(Sigma社、セントルイス、MO)を含有する成熟カクテルに24時間添加した。細胞は、DCの典型的細胞表面マーカー(CD1a+、CD14-、CD86+)を展示した。ペプチド負荷のために、DCを収集し、および密度1〜2x106個細胞/mlでM'培地中の30mg/mlでインキュベーションした。
【0158】
インビトロCTL誘導
CD8+ Tリンパ球は、CD8単離キット(Miltenyi社、オーバーン、CA)を使用するCD8-細胞の枯渇により、PBMCにより濃縮した。90%を超えるCD8+ T細胞を含む得られる集団は、レスポンダー細胞として使用し、ペプチドパルスしたDCまたはペプチドパルスしたaAPCのいずれかで刺激した。10000個のレスポンダー細胞/ウェルを、5%自家血漿および3%TCGFを補充した200μlのM'培地/ウェルの96ウェル丸底プレートにおいて、5x103個DC/ウェルまたは104ペプチドパルスしたaAPC/ウェルのいずれかと共培養した。追加した同種異系のフィーダー細胞は、CTLの誘導または拡大のいずれにも使用しなかった。TCGFは、Oelkeらの論文に記されたように調製した(Clin. Canc. Res.、6:1997-2005(2000))。培地およびTCGFは、1週間に2回補充した。7日目およびその後毎週、T細胞を収集し、計数し、ならびに3%TCGFを補充した完全培地内で、5x103個ペプチドパルスした自家DC/ウェルまたは104個ペプチドパルスしたaAPC/ウェルのいずれかと共に、104個T細胞/ウェルとなるよう再び播種した。
【0159】
二量体染色および細胞内サイトカイン染色(ICS)分析
Gretenらの論文(Proc. Natl. Acad. Sci. USA、95:7568-73(1998))に記されたように、細胞は、FITC結合したCD8 mAbおよびMart-1もしくはCMVパルスしたA2-Igで、ならびに第二工程において、抗マウスIg-PEで染色し、Ig-A2二量体を検出した。対照染色について、記したように、A2-Igを、不適切なペプチドで負荷するかまたは負荷しないかのいずれかであった。負荷しないA2-Ig(図2)または不適切なA2-結合ペプチドを負荷したA2-Ig(図4)のいずれかを用い、同様のバックグランド染色が観察された。分析のために、本発明者らはCD8+細胞をゲーティングした(gate on)。
【0160】
ICSは、下記を変更し、説明されたように行った(BD-Pharmingen、サンディエゴ、CA、ICSプロトコール)。1000000個のエフェクター細胞は、2x105個ペプチドパルスしたT2細胞(30μg/ml)または106個黒色腫細胞で、37℃で5時間刺激した。黒色腫細胞が標的として使用される場合、0.5ng/mlホルボール12-ミリスチン酸13-酢酸(PMA)および4ng/mlイオノマイシンを添加した。対照実験は、低用量のPMAおよびイオノマイシンは、エフェクター細胞中のサイトカイン産生を誘導しないことを明らかにした。細胞内染色を、FITC標識されたIFN-gまたはIL4 mAb(BD、サンディエゴ、CA)で行った。
【0161】
51Cr-放出アッセイ法
51Cr-放出-アッセイ法は、Oelkeらの論文(2000)に説明されたように行った。CTL活性は、下記式を用い、特異的51Cr-放出の割合として計算した:%特異的殺傷=(試料放出−自然発生的放出)÷(最大放出−自然発生的放出)×100%。
【0162】
実施例2
aAPCによるMart-1およびCMV特異的CTLの誘導および拡大
これは、ふたつの臨床的関連した標的であるCMV-ペプチドpp65およびMart-1による抗原特異的CTLの誘導および拡大を説明している。これらのペプチドは、それらの同族TCRに関して広範に変動する親和性を有する。CMV-ペプチドpp65は、高親和性ペプチドであるのに対し、メラニン形成細胞自己抗原に由来した修飾されたMart-1ペプチドは、低親和性ペプチドであることが知られている(Valmoriら、Int. Immnunol.、11:1971-80(1999))。
【0163】
現在の方法は、自家ペプチドパルスしたDCを使用し、正常なPBMCから抗原特異的CTLを誘導している(図1)。これらの方法は、DCまたはCD40Lで刺激された自家B細胞を使用し、抗原特異的CTLが培養物の主要部分になるまで、2-4刺激サイクルにわたり、抗原特異的CTLを誘導することが多い(図1、工程2)。従って本発明者らは、aAPC誘導を、DCによる誘導と比較した。T細胞を、単離し、精製し、およびMart-1負荷したaAPCまたはMart-1ペプチドでパルスした単球-由来した自家DCのいずれかで誘導した。CD8+ T細胞は、DCまたはaAPCのいずれかにより、週1回、合計3ラウンド刺激した。
【0164】
代表的実験において、T細胞の総数は、DC誘導した培養物中において1x106個から20x106個に増大し、aAPC誘導した培養物中において1x106個から14x106個に増大した。この培養物の抗原特異性は、A2-Ig二量体染色およびICSの両方により3週間後に分析した。本発明者らの状況においては、おそらく誘導されたCTL集団における異質性により、ICS染色は、二量体染色の最大2倍の感度であることができる。ICSは、二量体染色よりも高いおよび低い親和性CTLの境界集団を検出すると思われる。細胞は、前述のようにFITC結合したCD8 mAbおよびMart-1パルスしたA2-Igにより染色した。ICSに関して、細胞は、サイトカインを含まない通常培地において、ペプチドパルスしたT2細胞と共にインキュベーションした。1時間後、モネンシン(ゴルジ停止)をこの培養物に添加した。6時間後、T細胞を収集し、ICSにより分析した。ペプチド特異的CD8+ CTLの割合は、右上隅に示した。
【0165】
MART-1ペプチド負荷したaAPCによる3ラウンド刺激後、Mart-1A2-Ig二量体染色により決定すると、全CD8 CTLの62.3%はMart-1特異性であり(図2A、左側)、および細胞内サイトカイン染色(ICS)により決定すると、84.3%がそうであった(図2A、右側)。抗原特異的CTLのHLA-Ig二量体染色およびICS分析の間の差異は、恐らくDCまたはaAPC誘導したCTL集団により使用されたTCRレパートリーの多様性に関連すると思われる。ペプチド誘導した抗原特異的CTL集団中の異質性は、先に報告されている。Valmoriら、J. Immunol.、168:4231-40(2002)。レパートリーの多様性は、一方のアッセイ法により認識されるが他方のアッセイ法により認識されない、より高いおよびより低い親和性のCTLに関連していると思われる。
【0166】
aAPCにより得られた結果と比べ、自家DCは、わずかに29.7%のMART-1特異的細胞を誘導し、およびICS Mart-1特異的CTLは61.1%誘導した(図2B)。
【0167】
aAPC刺激したPBMCの増殖可能性を検討するために、T細胞を、aAPCで7週間刺激した。0.05%未満がMart-1特異性である1x106個総CD8+ T細胞から開始し、細胞を、85%よりも多くがMart-1特異性である約109個のCTL細胞に拡大した(図2Cおよび2D)。これは、2ヶ月足らずのうちの少なくとも106倍の抗原特異的T細胞の拡大を示している。
【0168】
aAPC媒介した刺激は、Mart-1およびCMV誘導したCTLの両方について、より良いものではないとしても、DCによる刺激と同等であった(表1)。
【0169】
【表1】
【0170】
これは、3名の異なる健常ドナーおよび転移性黒色腫の患者由来の細胞(Mart-1負荷したaAPC)ならびに3名の異なるドナー由来の細胞(CMV負荷したaAPC)を用いる、5つの実験中4つにおいて認められた。Mart-1誘導に関して、HLA-Ig二量体染色およびICSの両方により認められるように、aAPCは、DCよりも、約2〜3倍多く抗原特異的細胞を誘導した。このことは、転移性黒色腫の患者においても認められた。CMV特異的CTLの誘導は、Mart-1特異的CTLよりもより豊富であった。例えaAPCによる1回刺激後であっても、最大90%のCTL集団がCMV特異性であった。わずかに少ないCMV特異的CTLが、DCを用いて得られた。従ってaAPCは一般に、複数の健常ドナーに加え黒色腫患者由来のふたつの異なるCTLシステムにおいて抗原特異的CTLの誘導について、より良いものではないとしても、DCと同等であった。
【0171】
aAPCは、A2拘束されたサブドミナントな黒色腫エピトープNY-ESO-1(Jagerら、J. Exp. Med.、187:265-70(1998))、およびLMP-2由来のサブドミナントなEBVエピトープ (Leeら、J. Virol.、67:7428-35(1993))について、CTL特異的な大きい拡大も媒介した(表2参照)。全てのCD8+細胞の約1.2%は、3回のaAPC刺激の後NY-ESO-1特異性であった。これは、免疫ドミナントなエピトープに特異的であるCTL拡大において認められるものよりも明らかに低かったが、サブドミナントなエピトープのCTL特異性の拡大を分析する場合には、より少ない数の抗原特異的CTLが予想された。
【0172】
NY-ESO-1特異的CTLは、同族特異的標的細胞の溶解を媒介したが、無関係の標的細胞は溶解しなかった(表2)。これらの実験において、CTLは、分析前に3〜4週間分析した。抗原特異的CTLの頻度は、LMP-2特異的CTLの二量体染色によるか、またはNY-ESO-1特異的CTLに関する四量体染色および51Cr放出アッセイ法により分析した。表2は、E:T比25:1で観察された特異的溶解率(%)、およびA2-Ig二量体もしくは四量体のいずれかを用いるフローサイトメーターにより決定されたペプチド特異的CD8+ T細胞の割合(%)を示している。対照的に、DCを用いた刺激は、標準の51Cr放出アッセイ法において、検出可能な細胞傷害活性を伴わない有意に低い頻度のNY-ESO-1特異的CTLを生じた。
【0173】
【表2】
【0174】
実施例3
aAPC誘導したPBMCにより内因性にプロセッシングされた抗原の認識
CTL機能の評価における有用な判定基準は、内因性抗原-HLA複合体を発現する標的の認識である。黒色腫特異的CTLの拡大のためのペプチドパルスしたDCを使用する最初の作業は、同族抗原によりパルスした標的の溶解を媒介した低親和性CTLを生じたが、しばしば抗原-HLA複合体を内因性に発現した黒色腫標的を認めることができなかった。Yeeら、J. Immunol.、162:2227-34(1999)。従って本発明者らは、aAPC誘導したCTLの内因性Mart-1またはpp65 CMV抗原を認識する能力を試験した(図3)。
【0175】
ICS染色に関して、これらの細胞は、サイトカインを伴わない通常培地中の標的細胞と共にインキュベーションした。ICSアッセイ法の感度を増加するために、低用量PMAおよびイオノマイシンを、培地に添加した。Perez-Diezらの説明したように(Cancer Res.、58:5305-09(1998))、この方法は、本発明者らが集団中のより抗原特異的のT細胞を検出することを可能にした。この追加の刺激を伴うまたは伴わないこれらの結果における差異は、これらの刺激に応じて決まった。低用量のPMAおよびイオノマイシンにより認められる増強は、自家腫瘍細胞が刺激細胞として使用される場合により顕著であった(最大3〜4倍)が、抗原特異的T細胞の刺激にペプチドパルスしたT2細胞またはA293細胞が使用される場合は、大きくはなかった。低用量PMAおよびイオノマイシンの添加は、図3Aおよび3Cに認められるように、バックグラウンド活性を変化しなかった。古典的クロム放出溶解アッセイ法を、PMAおよびイオノマシンを添加せずに行った(図3Bおよび3D)。
【0176】
Mart-1特異的aAPC誘導した細胞は、黒色腫標的細胞により刺激した場合、約37%がIFN-γを産生した(図3A)。同等数がIL-4を産生した(図5)。対照Mart-1+/HLA-A2-黒色腫標的は、有意なエフェクターサイトカイン産物を刺激しなかった。更に、aAPC誘導したエフェクターCTL集団は、標的Mart-1+/HLA-A2+黒色腫標的細胞を用量依存的に溶解したが、対照Mart-1+/HLA-A2-標的は溶解しなかった(図3B)。進行した黒色腫患者から得たPBMC由来のaAPC誘導したMart-1特異的CTLは、HLA-A2+ Mart-1発現する黒色腫細胞の溶解を媒介することもでき、14.7%の溶解が、E:T比25:1で認められた。
【0177】
aAPCも、内因性にプロセッシングされおよび提示されたpp65抗原を認識したCMV特異的CTLを誘導することができた(図3Cおよび3D)。A293-N pp65+標的で刺激した場合(pp65でトランスフェクションしたA293細胞)、約45%の細胞がIFNγを産生した。aAPC誘導したエフェクターCTL集団も、トランスフェクションされた標的細胞を用量依存的に媒介したが、対照標的は媒介しなかった(図3D)。従って、正常な健常ドナーに加え黒色腫患者の両方から得たaAPC誘導したCTL培養物は、内因性にプロセッシングされた抗原-HLA複合体を認識しなかった。
【0178】
抗原特異的CTLの一部は、aAPCまたはDCのいずれにより誘導されようと、IFN-γおよびIL4のいずれかまたは両方を産生した(図5)。IFN-γおよびIL4の両方を産生するヒトCD8+細胞が、DCを用いたエクスビボ拡大において報告されている。Oelkeら、2000。本発明者らのaAPCによる結果は、これらの興味深いDCを用いた知見を確認し、ならびにaAPC媒介した刺激は明らかに同様の抗原特異的CTLを生じることを示している。
【0179】
実施例4
CMV特異的CTLのaAPCによる拡大
DCの使用に関連したひとつの制限は、臨床に関連した数へのCTLの拡大は、十分なDCを得るための白血球搬出または抗CD3ビーズなどの非特異性拡大の使用のいずれかを必要とすることである(図1、工程3参照)。従って本発明者らは、aAPCまたは抗CD3/抗CD28-ビーズを使用する「拡大」相を比較した。CMV特異的CTLの拡大時に、抗CD3/抗CD28ビーズを使用するCTLの総数が7倍増加した。しかし、抗原特異的細胞の割合は、87.9%から7.3%に減少した(図4Aおよび4Bの比較)。この問題点は、多様なCTL集団の抗CD3を用いた拡大の使用の有用性を制限した。Mausら、Nature Biotechnol.、20:143-48(2002)。対照的に、CMV特異的aAPCが抗原特異的CTLの拡大に使用される場合、抗原特異性の同時喪失はなかった。CMV特異的CTLの割合は、それらの培養物中で86%であり続け、そこには依然T細胞数の7倍の増加があった(図4Aおよび4Cの比較)。従って、HLA-Igを用いたaAPC支持体は、抗原特異的様式でのCTLの拡大を継続し、抗CD3を用いた拡大に勝る有意な利点を表している。
【特許請求の範囲】
【請求項1】
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、固形支持体。
【請求項2】
硬質固形支持体または軟質固形支持体である、請求項1記載の固形支持体。
【請求項3】
粒子である硬質固形支持体である、請求項2記載の固形支持体。
【請求項4】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、かつ分子複合体が、少なくとも1個の抗原結合窩を含む抗原提示複合体である、請求項1記載の固形支持体。
【請求項5】
少なくとも1種の抗原提示複合体が、MHCクラスIペプチド結合窩を含む、請求項4記載の固形支持体。
【請求項6】
少なくとも1種の抗原提示複合体が、MHCクラスI分子である、請求項5記載の固形支持体。
【請求項7】
少なくとも1種の抗原提示複合体が、少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む、請求項5記載の固形支持体。
【請求項8】
少なくとも1種の抗原提示複合体が、MHCクラスIIペプチド結合窩を含む、請求項4記載の固形支持体。
【請求項9】
抗原提示複合体が、MHCクラスII分子である、請求項8記載の固形支持体。
【請求項10】
抗原提示複合体が、少なくとも4種の融合蛋白質を含む、MHCクラスII分子複合体である、固形支持体であって、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)MHCクラスIIα鎖の細胞外ドメインを含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項8記載の固形支持体。
【請求項11】
免疫グロブリン重鎖が、可変領域を含む、請求項10記載の固形支持体。
【請求項12】
抗原性ペプチドが、少なくとも1個の抗原結合窩に結合される、請求項4記載の固形支持体。
【請求項13】
抗原性ペプチドが、腫瘍関連抗原のペプチド、自己抗原のペプチド、アロ抗原のペプチド、および感染物質抗原のペプチドからなる群より選択される、請求項12記載の固形支持体。
【請求項14】
少なくとも2種の抗原提示複合体を含む、請求項4記載の固形支持体。
【請求項15】
同一抗原が、少なくとも2種の抗原提示複合体の各抗原結合窩に結合されている、請求項14記載の固形支持体。
【請求項16】
異なる抗原が、少なくとも2種の抗原提示複合体の各抗原結合窩に結合されている、請求項14記載の固形支持体。
【請求項17】
第一の抗原提示複合体が、少なくとも1個のMHCクラスIペプチド結合窩を含み、および第二の抗原提示複合体が、少なくとも1個のMHCクラスIIペプチド結合窩を含む、請求項14記載の固形支持体。
【請求項18】
同一の抗原性ペプチドが、少なくとも1個のMHCクラスIペプチド結合窩および少なくとも1個のMHCクラスIIペプチド結合窩に結合される、請求項17記載の固形支持体。
【請求項19】
異なる抗原性ペプチドが、少なくとも1個のMHCクラスIペプチド結合窩および少なくとも1個のMHCクラスIIペプチド結合窩に結合される、請求項17記載の固形支持体。
【請求項20】
少なくとも1種の抗原提示複合体が、非古典的MHC様分子である、請求項4記載の固形支持体。
【請求項21】
非古典的MHC様分子が、CD1ファミリーメンバーである、請求項20記載の固形支持体。
【請求項22】
非古典的MHC様分子が、CD1a、CD1b、CD1c、CD1d、およびCD1eからなる群より選択される、請求項21記載の固形支持体。
【請求項23】
少なくとも1種のT細胞に影響する分子が、T細胞共刺激分子である、請求項4記載の固形支持体。
【請求項24】
T細胞共刺激分子が、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、CD28に特異的に結合する抗体、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体からなる群より選択される、請求項23記載の固形支持体。
【請求項25】
少なくとも1種のT細胞に影響する分子が、接着分子である、請求項4記載の固形支持体。
【請求項26】
接着分子が、ICAM-1およびLFA-3からなる群より選択される、請求項25記載の固形支持体。
【請求項27】
少なくとも1種のT細胞に影響する分子が、T細胞増殖因子である、請求項4記載の固形支持体。
【請求項28】
T細胞増殖因子が、サイトカインおよびスーパー抗原からなる群より選択される、請求項27記載の固形支持体。
【請求項29】
T細胞増殖因子が、サイトカインであり、ならびにサイトカインが、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンからなる群より選択される、請求項28記載の固形支持体。
【請求項30】
T細胞増殖因子が
(A)少なくとも2種の融合蛋白質を含む第一の分子複合体であり、ここで第一の融合蛋白質が、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質が、第二のサイトカインおよび第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、第一の分子複合体を形成するもの;ならびに
(B)少なくとも4種の融合蛋白質を含む第二の分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)第一のサイトカインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)第二のサイトカインを含み、
ここでこれら2種の第一および2種の第二の融合蛋白質が会合し、第二の分子複合体を形成するものからなる群より選択される、請求項27記載の固形支持体。
【請求項31】
T細胞増殖因子が、第一の分子複合体である、請求項30記載の固形支持体。
【請求項32】
第一および第二のサイトカインが同一である、請求項31記載の固形支持体。
【請求項33】
第一および第二のサイトカインが異なる、請求項31記載の固形支持体。
【請求項34】
T細胞増殖因子が、第二の分子複合体である、請求項30記載の固形支持体。
【請求項35】
第一および第二のサイトカインが同一である、請求項34記載の固形支持体。
【請求項36】
第一および第二のサイトカインが異なる、請求項34記載の固形支持体。
【請求項37】
少なくとも1種のT細胞に影響する分子が、調節T細胞誘導分子である、請求項4記載の固形支持体。
【請求項38】
少なくとも1種の調節T細胞誘導分子が、TGFβ、IL-10、インターフェロン-α、およびIL-15からなる群より選択される、請求項37記載の固形支持体。
【請求項39】
少なくとも1種のT細胞に影響する分子が、アポトーシス誘導分子である、請求項4記載の固形支持体。
【請求項40】
アポトーシス誘導分子が、毒性物質、TNFα、およびFasリガンドからなる群より選択される、請求項39記載の固形支持体。
【請求項41】
少なくとも2種の異なるT細胞に影響する分子を含む、請求項4記載の固形支持体。
【請求項42】
(A)少なくとも1種のB細胞に影響する分子;および
(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、固形支持体。
【請求項43】
少なくとも1種のB細胞に影響する分子が、CD40リガンドである、請求項42載の固形支持体。
【請求項44】
分子複合体が、T細胞受容体(TCR)である、請求項42載の固形支持体。
【請求項45】
分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRδ鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合、その結果2種の第二の融合蛋白質がTCRβ鎖を含み、ならびにここで2種の第一の融合蛋白質がTCRγ鎖を含む場合、その結果2種の第二の融合蛋白質がTCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項42記載の固形支持体。
【請求項46】
(A)少なくとも1種のT細胞共刺激分子;および
(B)少なくとも2種の融合蛋白質を含む少なくとも1種のMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含むものを含む、粒子。
【請求項47】
少なくとも1種のT細胞共刺激分子が、CD28に特異的に結合する抗体である、請求項46記載の粒子。
【請求項48】
複数の粒子を含む調製物であって、ここで複数の粒子が
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、調製物。
【請求項49】
薬学的に許容される担体を更に含む、請求項48記載の調製物。
【請求項50】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、および少なくとも1種の分子複合体が、少なくとも1個の抗原結合窩を含む抗原提示複合体である、請求項48記載の調製物。
【請求項51】
複数の粒子が
(A)第一の粒子の少なくとも1個の抗原結合窩が、MHCクラスIペプチド結合窩である、少なくとも1種の第一の粒子;および
(B)少なくとも1個の抗原結合窩が、MHCクラスIIペプチド結合窩である、少なくとも1種の第二の粒子を含む、請求項48記載の調製物。
【請求項52】
抗原性ペプチドが、第一の粒子の少なくとも1個のペプチド結合窩に結合されている、請求項51記載の調製物。
【請求項53】
第一の抗原性ペプチドが、第一の粒子の少なくとも1個のペプチド結合窩に結合され、および第二の抗原性ペプチドが、第二の粒子の少なくとも1個のペプチド結合窩に結合されている、請求項51記載の調製物。
【請求項54】
第一および第二の抗原性ペプチドが同一である、請求項53記載の調製物。
【請求項55】
第一および第二の抗原性ペプチドが異なる、請求項53記載の調製物。
【請求項56】
抗原提示複合体の各抗原結合窩が、MHCクラスIペプチド結合窩である、請求項50記載の調製物。
【請求項57】
抗原性ペプチドが、MHCクラスIペプチド結合窩に結合されている、請求項56記載の調製物。
【請求項58】
抗原性ペプチドが同一である、請求項57記載の調製物。
【請求項59】
抗原性ペプチドが異なる、請求項57記載の調製物。
【請求項60】
抗原提示複合体の各抗原結合窩が、MHCクラスIIペプチド結合窩である、請求項50記載の調製物。
【請求項61】
抗原性ペプチドが、MHCクラスIIペプチド結合窩に結合されている、請求項60記載の調製物。
【請求項62】
抗原性ペプチドが同一である、請求項61記載の調製物。
【請求項63】
抗原性ペプチドが異なる、請求項61記載の調製物。
【請求項64】
抗原提示複合体が、少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体である、調製物であって、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む、請求項50記載の調製物。
【請求項65】
抗原提示複合体が、少なくとも4種の融合蛋白質を含むMHCクラスII分子複合体である調製物であって、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含み、
ここでこれらの2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項50記載の調製物。
【請求項66】
複数の粒子を含む調製物であって、ここで複数の粒子が
(A)少なくとも1種のB細胞に影響する分子;および
(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、調製物。
【請求項67】
分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体である調製物であって、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;および
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRα鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合は、その結果この2種の第二の融合蛋白質がTCRδ鎖を含み、かつこの2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果この2種の第二の融合蛋白質がTCRδ鎖を含み、
ここでこれら2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項66記載の調製物。
【請求項68】
磁気を帯びた、請求項3記載の粒子。
【請求項69】
生分解性である、請求項3記載の粒子。
【請求項70】
プラスチックである、請求項3記載の粒子。
【請求項71】
複数の前駆体T細胞を含む単離された調製物を、請求項4記載の少なくとも1種の第一の固形支持体と接触する工程を含む、抗原特異的T細胞の形成を誘導する方法であって、ここで抗原が、抗原結合窩に結合され、これにより複数の前駆体T細胞のメンバーを、抗原を認識する抗原特異的T細胞を含む第一の細胞集団を形成するように誘導し、ここで第一の細胞集団内の抗原特異的T細胞の数または割合が、前駆体T細胞がCD3に特異的に結合する抗体は含むが抗原提示複合体は含まない固形支持体と共にインキュベーションされた場合に形成される抗原特異的T細胞の数または割合よりも多い、方法。
【請求項72】
抗原特異的T細胞が、細胞傷害性T細胞である、請求項71記載の方法。
【請求項73】
抗原特異的T細胞が、ヘルパーT細胞である、請求項71記載の方法。
【請求項74】
抗原特異的T細胞が、調節T細胞である、請求項71記載の方法。
【請求項75】
抗原特異的T細胞を第一の細胞集団から分離する工程を更に含む、請求項71記載の方法。
【請求項76】
第一の細胞集団を請求項4記載の少なくとも1種の第二の固形支持体と共にインキュベーションする工程を更に含む方法であって、ここで抗原が粒子の抗原結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団内の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項71記載の方法。
【請求項77】
抗原が同一である、請求項71記載の方法。
【請求項78】
抗原が異なる、請求項71記載の方法。
【請求項79】
単離された調製物が、少なくとも2種の第一の固形支持体と接触され、ここで異なる抗原が、第一の固形支持体の各々に結合される、請求項71記載の方法。
【請求項80】
抗原特異的T細胞を含む第一の細胞集団を、請求項4記載の少なくとも1種の第一の固形支持体と共にインキュベーションする工程を含む、細胞集団内の抗原特異的T細胞の数または割合を増加する方法であって、ここで抗原が抗原結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団内の抗原特異的T細胞の数または割合に比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な期間実行される、方法。
【請求項81】
第一の細胞集団が、均質な細胞集団である、請求項80記載の方法。
【請求項82】
抗原特異的T細胞を患者に投与する工程を更に含む、請求項71記載の方法。
【請求項83】
患者が、癌、自己免疫疾患、感染症を有するか、または免疫抑制されている、請求項82記載の方法。
【請求項84】
前駆体T細胞が、患者から得られる、請求項82記載の方法。
【請求項85】
前駆体T細胞が、患者ではないドナーから得られる、請求項82記載の方法。
【請求項86】
抗原特異的T細胞が、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与からなる群より選択される投与経路により投与される、請求項82記載の方法。
【請求項87】
第二の集団の抗原特異的T細胞を、患者へ投与する工程を更に含む、請求項80記載の方法。
【請求項88】
(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物を患者に投与する工程を含む、患者の免疫応答を調節する方法であって、
ここでこの複数の粒子のメンバーが
(1)少なくとも1種のT細胞に影響する分子;および
(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体が、少なくとも1個の抗原結合窩を含み、ここで抗原が少なくとも1個の抗原結合窩に結合している、方法。
【請求項89】
少なくとも1種のT細胞に影響する分子が、(1)アポトーシス誘導分子、(2)調節T細胞誘導分子、(3)T細胞共刺激性分子、(4)接着分子、および(5)T細胞増殖因子からなる群より選択される、請求項88記載の方法。
【請求項90】
(A)複数の粒子、および(B)薬学的に許容される担体を含む調製物を、患者に投与する工程を含む、患者における免疫応答を抑制する方法であって、
ここでこの複数の粒子のメンバーが
(1)少なくとも1種のアポトーシス誘導分子;および
(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体が、少なくとも1個の抗原結合窩を含み、ここで抗原が少なくとも1個の抗原結合窩に結合されている、方法。
【請求項91】
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、細胞。
【請求項92】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、かつこの少なくとも1種の分子複合体が、少なくとも1個のペプチド結合窩を含む抗原提示複合体であり、ここで抗原提示複合体が
(1)少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含むもの;ならびに
(2)少なくとも4種の融合蛋白質を含む、MHCクラスII分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIlα鎖の細胞外ドメインを含むものからなる群より選択され、
ここで2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項91記載の細胞。
【請求項93】
抗原提示複合体が、MHCクラスII分子複合体であり、ここで免疫グロブリン重鎖が可変領域を含む、請求項92記載の細胞。
【請求項94】
抗原性ペプチドが、少なくとも1種のペプチド結合窩に結合されている、請求項92記載の細胞。
【請求項95】
抗原性ペプチドが、腫瘍関連抗原のペプチド、自己抗原のペプチド、アロ抗原のペプチド、および感染物質抗原のペプチドからなる群より選択される、請求項94記載の細胞。
【請求項96】
少なくとも2種の抗原提示複合体を含む、請求項92記載の細胞。
【請求項97】
同一抗原性ペプチドが、少なくとも2種の抗原提示複合体の各ペプチド結合窩に結合される、請求項96記載の細胞。
【請求項98】
異なる抗原性ペプチドが、少なくとも2種の抗原提示複合体の各ペプチド結合窩に結合される、請求項96記載の細胞。
【請求項99】
第一の抗原提示複合体が、MHCクラスI分子複合体であり、ここで第二の抗原提示複合体が、MHCクラスII分子複合体である、請求項96記載の細胞。
【請求項100】
同一の抗原性ペプチドが、少なくとも1個のMHCクラスI分子複合体のペプチド結合窩、および少なくとも1個のMHCクラスII分子複合体のペプチド結合窩に結合される、請求項99記載の細胞。
【請求項101】
異なる抗原性ペプチドが、少なくとも1個のMHCクラスI分子複合体のペプチド結合窩および少なくとも1個のMHCクラスII分子複合体のペプチド結合窩に結合している、請求項99記載の細胞。
【請求項102】
少なくとも1種のT細胞に影響する分子が、T細胞共刺激分子である、請求項92記載の細胞。
【請求項103】
T細胞共刺激分子が、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、CD28に特異的に結合する抗体、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体からなる群より選択される、請求項102記載の細胞。
【請求項104】
少なくとも1種のT細に影響する分子が、接着分子である、請求項92記載の細胞。
【請求項105】
接着分子が、ICAM-1、LFA-3およびLFA-1からなる群より選択される、請求項104記載の細胞。
【請求項106】
少なくとも1種のT細胞に影響する分子が、T細胞増殖因子である、請求項92記載の細胞。
【請求項107】
T細胞増殖因子が、サイトカインおよびスーパー抗原からなる群より選択される、請求項106記載の細胞。
【請求項108】
T細胞増殖因子が、サイトカインであり、およびサイトカインが、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンからなる群より選択される、請求項106記載の細胞。
【請求項109】
少なくとも1種のT細胞に影響する分子が
(A)少なくとも2種の融合蛋白質を含む第一の分子複合体であり、ここで第一の融合蛋白質が、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のサイトカインおよび第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、第一の分子複合体を形成するもの、;ならびに
(B)少なくとも4種の融合蛋白質を含む第二の分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)第一のサイトカインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)第二のサイトカインを含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、第二の分子複合体を形成するものからなる群より選択される、請求項106記載の細胞。
【請求項110】
第一および第二のサイトカインが同一である、請求項109記載の細胞。
【請求項111】
第一および第二のサイトカインが異なる、請求項109記載の細胞。
【請求項112】
少なくとも1種の外因性T細胞に影響する分子が、調節T細胞誘導分子である、請求項92記載の細胞。
【請求項113】
少なくとも1種の調節T細胞誘導分子が、TGFβ、IL-10、インターフェロン-α、およびIL-15からなる群より選択される、請求項112記載の細胞。
【請求項114】
少なくとも2種の異なるT細胞に影響する分子を含む、請求項92記載の細胞。
【請求項115】
少なくとも1種のリンパ球に影響する分子が、B細胞に影響する分子であり、および少なくとも1種の分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)TCRβ鎖またはTCRγ鎖の細胞外ドメインを含み、ここでこれら2種の第一の融合蛋白質がTCRα鎖を含む場合、その結果2種の第二の融合蛋白質が、TCRβ鎖を含み、ここで2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項91記載の細胞。
【請求項116】
請求項91記載の細胞を複数含む、調製物。
【請求項117】
薬学的に許容される担体を更に含む、請求項116記載の調製物。
【請求項118】
複数の前駆体T細胞を含む単離された調製物を、請求項92記載の第一の複数の細胞と接触する工程を含む、抗原特異的T細胞の形成を誘導する方法であって、
ここで抗原性ペプチドが、ペプチド結合窩に結合され、これにより複数の前駆体T細胞のメンバーを、抗原性ペプチドを認識する抗原特異的T細胞を含む第一の細胞集団を形成するように誘導し、ここで第一の細胞集団内の抗原特異的T細胞の数または割合が、前駆体T細胞が第二の複数の細胞とインキュベーションされる場合に形成される抗原特異的T細胞の数または割合よりも大きく、ここで第二の複数の細胞が、CD3に特異的に結合する抗体を含むが、抗原提示複合体は含まない、方法。
【請求項119】
抗原特異的T細胞が、細胞傷害性T細胞である、請求項118記載の方法。
【請求項120】
抗原特異的T細胞が、ヘルパーT細胞である、請求項118記載の方法。
【請求項121】
抗原特異的T細胞が、調節T細胞である、請求項118記載の方法。
【請求項122】
抗原特異的T細胞を第一の細胞集団から分離する工程を更に含む、請求項118記載の方法。
【請求項123】
第一の細胞集団を、請求項1記載の第二の複数の細胞と共にインキュベーションする工程を更に含む方法であって、ここで抗原性ペプチドが、これらの細胞のペプチド結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項118記載の方法。
【請求項124】
抗原特異的T細胞を含む第一の細胞集団を、請求項92記載の複数の細胞と共にインキュベーションする工程を含む、細胞集団内の抗原特異的T細胞の数または割合を増加する方法であって、
ここで抗原性ペプチドが、これらの細胞のペプチド結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、方法。
【請求項125】
第一の細胞集団が、均質な細胞集団である、請求項124記載の方法。
【請求項126】
抗原特異的T細胞を患者に投与する工程を更に含む、請求項118記載の方法。
【請求項127】
患者が、癌、自己免疫疾患、感染症を有するか、または免疫抑制される、請求項126記載の方法。
【請求項128】
前駆体T細胞が、患者から得られる、請求項126記載の方法。
【請求項129】
前駆体T細胞が、患者ではないドナーから得られる、請求項126記載の方法。
【請求項130】
抗原特異的T細胞が、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与からなる群より選択される投与経路により投与される、請求項126記載の方法。
【請求項131】
第二の集団の抗原特異的T細胞を、患者に投与する工程を更に含む、請求項124記載の方法。
【請求項132】
請求項92記載の複数の細胞および薬学的に許容される担体を含有する調製物を患者に投与する工程を含む、患者における免疫応答を調節する方法であって、
ここで抗原性ペプチドが、少なくとも1種のペプチド結合窩に結合される、方法。
【請求項133】
少なくとも1種のT細胞に影響する分子が、(1)アポトーシス誘導分子、(2)調節T細胞誘導性分子、(3)T細胞共刺激分子、(4)接着分子、および(5)T細胞増殖因子からなる群より選択される、請求項132記載の方法。
【請求項134】
集団内の抗体産生B細胞の数または割合を増加する方法であって、複数の前駆体B細胞を含む単離された調製物を、少なくとも1種の請求項42記載の第一の固形支持体と接触し、これにより複数の前駆体B細胞のメンバーを、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む第一の細胞集団を形成するように誘導する工程を含む、方法。
【請求項135】
抗体を産生するB細胞を、第一の細胞集団から分離する工程を更に含む、請求項134記載の方法。
【請求項136】
第一の細胞集団を、少なくとも1種の請求項42記載の第二の固形支持体と共にインキュベーションする工程を更に含み、ここでインキュベーション工程が、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項134記載の方法。
【請求項137】
抗体産生B細胞を含む第一の細胞集団を、少なくとも1種の請求項42記載の第一の固形支持体と共にインキュベーションする工程を含む、集団内の抗体産生B細胞の数または割合を増加する方法であって、
ここでインキュベーションの工程が、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される、方法。
【請求項138】
第一の細胞集団が、均質な細胞集団である、請求項137記載の方法。
【請求項139】
集団内の抗体産生B細胞の数または割合を増加する方法であって、複数の前駆体B細胞を含む単離された調製物を、請求項66記載の調製物と接触し、これにより複数の前駆体B細胞のメンバーに、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む第一の細胞集団の形成を誘導する工程を含む、方法。
【請求項140】
(A)複数の粒子、および(B)薬学的に許容される担体を含む調製物を、患者に投与する工程を含む、患者の免疫応答を調節する方法であって、
ここで複数の粒子のメンバーが
(1)少なくとも1種のB細胞に影響する分子;および
(2)B細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、方法。
【請求項141】
少なくとも1種のB細胞に影響する分子が、(1)CD40リガンド、(2)サイトカイン、および(3)サイトカイン分子複合体からなる群より選択される、請求項140記載の方法。
【請求項142】
分子複合体が、T細胞受容体および少なくとも4種の融合蛋白質を含むTCR分子複合体からなる群より選択され、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;および
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRδ鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRβ鎖を含み、かつ2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項140記載の方法。
【請求項1】
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、固形支持体。
【請求項2】
硬質固形支持体または軟質固形支持体である、請求項1記載の固形支持体。
【請求項3】
粒子である硬質固形支持体である、請求項2記載の固形支持体。
【請求項4】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、かつ分子複合体が、少なくとも1個の抗原結合窩を含む抗原提示複合体である、請求項1記載の固形支持体。
【請求項5】
少なくとも1種の抗原提示複合体が、MHCクラスIペプチド結合窩を含む、請求項4記載の固形支持体。
【請求項6】
少なくとも1種の抗原提示複合体が、MHCクラスI分子である、請求項5記載の固形支持体。
【請求項7】
少なくとも1種の抗原提示複合体が、少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む、請求項5記載の固形支持体。
【請求項8】
少なくとも1種の抗原提示複合体が、MHCクラスIIペプチド結合窩を含む、請求項4記載の固形支持体。
【請求項9】
抗原提示複合体が、MHCクラスII分子である、請求項8記載の固形支持体。
【請求項10】
抗原提示複合体が、少なくとも4種の融合蛋白質を含む、MHCクラスII分子複合体である、固形支持体であって、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)MHCクラスIIα鎖の細胞外ドメインを含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項8記載の固形支持体。
【請求項11】
免疫グロブリン重鎖が、可変領域を含む、請求項10記載の固形支持体。
【請求項12】
抗原性ペプチドが、少なくとも1個の抗原結合窩に結合される、請求項4記載の固形支持体。
【請求項13】
抗原性ペプチドが、腫瘍関連抗原のペプチド、自己抗原のペプチド、アロ抗原のペプチド、および感染物質抗原のペプチドからなる群より選択される、請求項12記載の固形支持体。
【請求項14】
少なくとも2種の抗原提示複合体を含む、請求項4記載の固形支持体。
【請求項15】
同一抗原が、少なくとも2種の抗原提示複合体の各抗原結合窩に結合されている、請求項14記載の固形支持体。
【請求項16】
異なる抗原が、少なくとも2種の抗原提示複合体の各抗原結合窩に結合されている、請求項14記載の固形支持体。
【請求項17】
第一の抗原提示複合体が、少なくとも1個のMHCクラスIペプチド結合窩を含み、および第二の抗原提示複合体が、少なくとも1個のMHCクラスIIペプチド結合窩を含む、請求項14記載の固形支持体。
【請求項18】
同一の抗原性ペプチドが、少なくとも1個のMHCクラスIペプチド結合窩および少なくとも1個のMHCクラスIIペプチド結合窩に結合される、請求項17記載の固形支持体。
【請求項19】
異なる抗原性ペプチドが、少なくとも1個のMHCクラスIペプチド結合窩および少なくとも1個のMHCクラスIIペプチド結合窩に結合される、請求項17記載の固形支持体。
【請求項20】
少なくとも1種の抗原提示複合体が、非古典的MHC様分子である、請求項4記載の固形支持体。
【請求項21】
非古典的MHC様分子が、CD1ファミリーメンバーである、請求項20記載の固形支持体。
【請求項22】
非古典的MHC様分子が、CD1a、CD1b、CD1c、CD1d、およびCD1eからなる群より選択される、請求項21記載の固形支持体。
【請求項23】
少なくとも1種のT細胞に影響する分子が、T細胞共刺激分子である、請求項4記載の固形支持体。
【請求項24】
T細胞共刺激分子が、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、CD28に特異的に結合する抗体、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体からなる群より選択される、請求項23記載の固形支持体。
【請求項25】
少なくとも1種のT細胞に影響する分子が、接着分子である、請求項4記載の固形支持体。
【請求項26】
接着分子が、ICAM-1およびLFA-3からなる群より選択される、請求項25記載の固形支持体。
【請求項27】
少なくとも1種のT細胞に影響する分子が、T細胞増殖因子である、請求項4記載の固形支持体。
【請求項28】
T細胞増殖因子が、サイトカインおよびスーパー抗原からなる群より選択される、請求項27記載の固形支持体。
【請求項29】
T細胞増殖因子が、サイトカインであり、ならびにサイトカインが、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンからなる群より選択される、請求項28記載の固形支持体。
【請求項30】
T細胞増殖因子が
(A)少なくとも2種の融合蛋白質を含む第一の分子複合体であり、ここで第一の融合蛋白質が、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびに第二の融合蛋白質が、第二のサイトカインおよび第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、第一の分子複合体を形成するもの;ならびに
(B)少なくとも4種の融合蛋白質を含む第二の分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)第一のサイトカインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)第二のサイトカインを含み、
ここでこれら2種の第一および2種の第二の融合蛋白質が会合し、第二の分子複合体を形成するものからなる群より選択される、請求項27記載の固形支持体。
【請求項31】
T細胞増殖因子が、第一の分子複合体である、請求項30記載の固形支持体。
【請求項32】
第一および第二のサイトカインが同一である、請求項31記載の固形支持体。
【請求項33】
第一および第二のサイトカインが異なる、請求項31記載の固形支持体。
【請求項34】
T細胞増殖因子が、第二の分子複合体である、請求項30記載の固形支持体。
【請求項35】
第一および第二のサイトカインが同一である、請求項34記載の固形支持体。
【請求項36】
第一および第二のサイトカインが異なる、請求項34記載の固形支持体。
【請求項37】
少なくとも1種のT細胞に影響する分子が、調節T細胞誘導分子である、請求項4記載の固形支持体。
【請求項38】
少なくとも1種の調節T細胞誘導分子が、TGFβ、IL-10、インターフェロン-α、およびIL-15からなる群より選択される、請求項37記載の固形支持体。
【請求項39】
少なくとも1種のT細胞に影響する分子が、アポトーシス誘導分子である、請求項4記載の固形支持体。
【請求項40】
アポトーシス誘導分子が、毒性物質、TNFα、およびFasリガンドからなる群より選択される、請求項39記載の固形支持体。
【請求項41】
少なくとも2種の異なるT細胞に影響する分子を含む、請求項4記載の固形支持体。
【請求項42】
(A)少なくとも1種のB細胞に影響する分子;および
(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、固形支持体。
【請求項43】
少なくとも1種のB細胞に影響する分子が、CD40リガンドである、請求項42載の固形支持体。
【請求項44】
分子複合体が、T細胞受容体(TCR)である、請求項42載の固形支持体。
【請求項45】
分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRδ鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合、その結果2種の第二の融合蛋白質がTCRβ鎖を含み、ならびにここで2種の第一の融合蛋白質がTCRγ鎖を含む場合、その結果2種の第二の融合蛋白質がTCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項42記載の固形支持体。
【請求項46】
(A)少なくとも1種のT細胞共刺激分子;および
(B)少なくとも2種の融合蛋白質を含む少なくとも1種のMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含むものを含む、粒子。
【請求項47】
少なくとも1種のT細胞共刺激分子が、CD28に特異的に結合する抗体である、請求項46記載の粒子。
【請求項48】
複数の粒子を含む調製物であって、ここで複数の粒子が
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、調製物。
【請求項49】
薬学的に許容される担体を更に含む、請求項48記載の調製物。
【請求項50】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、および少なくとも1種の分子複合体が、少なくとも1個の抗原結合窩を含む抗原提示複合体である、請求項48記載の調製物。
【請求項51】
複数の粒子が
(A)第一の粒子の少なくとも1個の抗原結合窩が、MHCクラスIペプチド結合窩である、少なくとも1種の第一の粒子;および
(B)少なくとも1個の抗原結合窩が、MHCクラスIIペプチド結合窩である、少なくとも1種の第二の粒子を含む、請求項48記載の調製物。
【請求項52】
抗原性ペプチドが、第一の粒子の少なくとも1個のペプチド結合窩に結合されている、請求項51記載の調製物。
【請求項53】
第一の抗原性ペプチドが、第一の粒子の少なくとも1個のペプチド結合窩に結合され、および第二の抗原性ペプチドが、第二の粒子の少なくとも1個のペプチド結合窩に結合されている、請求項51記載の調製物。
【請求項54】
第一および第二の抗原性ペプチドが同一である、請求項53記載の調製物。
【請求項55】
第一および第二の抗原性ペプチドが異なる、請求項53記載の調製物。
【請求項56】
抗原提示複合体の各抗原結合窩が、MHCクラスIペプチド結合窩である、請求項50記載の調製物。
【請求項57】
抗原性ペプチドが、MHCクラスIペプチド結合窩に結合されている、請求項56記載の調製物。
【請求項58】
抗原性ペプチドが同一である、請求項57記載の調製物。
【請求項59】
抗原性ペプチドが異なる、請求項57記載の調製物。
【請求項60】
抗原提示複合体の各抗原結合窩が、MHCクラスIIペプチド結合窩である、請求項50記載の調製物。
【請求項61】
抗原性ペプチドが、MHCクラスIIペプチド結合窩に結合されている、請求項60記載の調製物。
【請求項62】
抗原性ペプチドが同一である、請求項61記載の調製物。
【請求項63】
抗原性ペプチドが異なる、請求項61記載の調製物。
【請求項64】
抗原提示複合体が、少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体である、調製物であって、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含む、請求項50記載の調製物。
【請求項65】
抗原提示複合体が、少なくとも4種の融合蛋白質を含むMHCクラスII分子複合体である調製物であって、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIIα鎖の細胞外ドメインを含み、
ここでこれらの2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項50記載の調製物。
【請求項66】
複数の粒子を含む調製物であって、ここで複数の粒子が
(A)少なくとも1種のB細胞に影響する分子;および
(B)B細胞表面免疫グロブリンまたはB細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、調製物。
【請求項67】
分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体である調製物であって、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;および
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRα鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合は、その結果この2種の第二の融合蛋白質がTCRδ鎖を含み、かつこの2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果この2種の第二の融合蛋白質がTCRδ鎖を含み、
ここでこれら2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項66記載の調製物。
【請求項68】
磁気を帯びた、請求項3記載の粒子。
【請求項69】
生分解性である、請求項3記載の粒子。
【請求項70】
プラスチックである、請求項3記載の粒子。
【請求項71】
複数の前駆体T細胞を含む単離された調製物を、請求項4記載の少なくとも1種の第一の固形支持体と接触する工程を含む、抗原特異的T細胞の形成を誘導する方法であって、ここで抗原が、抗原結合窩に結合され、これにより複数の前駆体T細胞のメンバーを、抗原を認識する抗原特異的T細胞を含む第一の細胞集団を形成するように誘導し、ここで第一の細胞集団内の抗原特異的T細胞の数または割合が、前駆体T細胞がCD3に特異的に結合する抗体は含むが抗原提示複合体は含まない固形支持体と共にインキュベーションされた場合に形成される抗原特異的T細胞の数または割合よりも多い、方法。
【請求項72】
抗原特異的T細胞が、細胞傷害性T細胞である、請求項71記載の方法。
【請求項73】
抗原特異的T細胞が、ヘルパーT細胞である、請求項71記載の方法。
【請求項74】
抗原特異的T細胞が、調節T細胞である、請求項71記載の方法。
【請求項75】
抗原特異的T細胞を第一の細胞集団から分離する工程を更に含む、請求項71記載の方法。
【請求項76】
第一の細胞集団を請求項4記載の少なくとも1種の第二の固形支持体と共にインキュベーションする工程を更に含む方法であって、ここで抗原が粒子の抗原結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団内の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項71記載の方法。
【請求項77】
抗原が同一である、請求項71記載の方法。
【請求項78】
抗原が異なる、請求項71記載の方法。
【請求項79】
単離された調製物が、少なくとも2種の第一の固形支持体と接触され、ここで異なる抗原が、第一の固形支持体の各々に結合される、請求項71記載の方法。
【請求項80】
抗原特異的T細胞を含む第一の細胞集団を、請求項4記載の少なくとも1種の第一の固形支持体と共にインキュベーションする工程を含む、細胞集団内の抗原特異的T細胞の数または割合を増加する方法であって、ここで抗原が抗原結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団内の抗原特異的T細胞の数または割合に比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な期間実行される、方法。
【請求項81】
第一の細胞集団が、均質な細胞集団である、請求項80記載の方法。
【請求項82】
抗原特異的T細胞を患者に投与する工程を更に含む、請求項71記載の方法。
【請求項83】
患者が、癌、自己免疫疾患、感染症を有するか、または免疫抑制されている、請求項82記載の方法。
【請求項84】
前駆体T細胞が、患者から得られる、請求項82記載の方法。
【請求項85】
前駆体T細胞が、患者ではないドナーから得られる、請求項82記載の方法。
【請求項86】
抗原特異的T細胞が、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与からなる群より選択される投与経路により投与される、請求項82記載の方法。
【請求項87】
第二の集団の抗原特異的T細胞を、患者へ投与する工程を更に含む、請求項80記載の方法。
【請求項88】
(A)複数の粒子、および(B)薬学的に許容される担体を含有する調製物を患者に投与する工程を含む、患者の免疫応答を調節する方法であって、
ここでこの複数の粒子のメンバーが
(1)少なくとも1種のT細胞に影響する分子;および
(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体が、少なくとも1個の抗原結合窩を含み、ここで抗原が少なくとも1個の抗原結合窩に結合している、方法。
【請求項89】
少なくとも1種のT細胞に影響する分子が、(1)アポトーシス誘導分子、(2)調節T細胞誘導分子、(3)T細胞共刺激性分子、(4)接着分子、および(5)T細胞増殖因子からなる群より選択される、請求項88記載の方法。
【請求項90】
(A)複数の粒子、および(B)薬学的に許容される担体を含む調製物を、患者に投与する工程を含む、患者における免疫応答を抑制する方法であって、
ここでこの複数の粒子のメンバーが
(1)少なくとも1種のアポトーシス誘導分子;および
(2)少なくとも1種の抗原提示複合体を含み、ここで少なくとも1種の抗原提示複合体が、少なくとも1個の抗原結合窩を含み、ここで抗原が少なくとも1個の抗原結合窩に結合されている、方法。
【請求項91】
(A)少なくとも1種のリンパ球に影響する分子;および
(B)抗原に結合した場合に、独自のクローン形質のリンパ球受容体に結合する、少なくとも1種の分子複合体を含む、細胞。
【請求項92】
少なくとも1種のリンパ球に影響する分子が、T細胞に影響する分子であり、かつこの少なくとも1種の分子複合体が、少なくとも1個のペプチド結合窩を含む抗原提示複合体であり、ここで抗原提示複合体が
(1)少なくとも2種の融合蛋白質を含むMHCクラスI分子複合体であり、ここで第一の融合蛋白質が、第一のMHCクラスIα鎖および第一の免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のMHCクラスIα鎖および第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、MHCクラスI分子複合体を形成し、ここでMHCクラスI分子複合体が、第一のMHCクラスIペプチド結合窩および第二のMHCクラスIペプチド結合窩を含むもの;ならびに
(2)少なくとも4種の融合蛋白質を含む、MHCクラスII分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖、および(ii)MHCクラスIIβ鎖の細胞外ドメインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)MHCクラスIlα鎖の細胞外ドメインを含むものからなる群より選択され、
ここで2種の第一および2種の第二の融合蛋白質が会合し、MHCクラスII分子複合体を形成し、ここで各第一の融合蛋白質のMHCクラスIIβ鎖の細胞外ドメインおよび各第二の融合蛋白質のMHCクラスIIα鎖の細胞外ドメインが、MHCクラスIIペプチド結合窩を形成する、請求項91記載の細胞。
【請求項93】
抗原提示複合体が、MHCクラスII分子複合体であり、ここで免疫グロブリン重鎖が可変領域を含む、請求項92記載の細胞。
【請求項94】
抗原性ペプチドが、少なくとも1種のペプチド結合窩に結合されている、請求項92記載の細胞。
【請求項95】
抗原性ペプチドが、腫瘍関連抗原のペプチド、自己抗原のペプチド、アロ抗原のペプチド、および感染物質抗原のペプチドからなる群より選択される、請求項94記載の細胞。
【請求項96】
少なくとも2種の抗原提示複合体を含む、請求項92記載の細胞。
【請求項97】
同一抗原性ペプチドが、少なくとも2種の抗原提示複合体の各ペプチド結合窩に結合される、請求項96記載の細胞。
【請求項98】
異なる抗原性ペプチドが、少なくとも2種の抗原提示複合体の各ペプチド結合窩に結合される、請求項96記載の細胞。
【請求項99】
第一の抗原提示複合体が、MHCクラスI分子複合体であり、ここで第二の抗原提示複合体が、MHCクラスII分子複合体である、請求項96記載の細胞。
【請求項100】
同一の抗原性ペプチドが、少なくとも1個のMHCクラスI分子複合体のペプチド結合窩、および少なくとも1個のMHCクラスII分子複合体のペプチド結合窩に結合される、請求項99記載の細胞。
【請求項101】
異なる抗原性ペプチドが、少なくとも1個のMHCクラスI分子複合体のペプチド結合窩および少なくとも1個のMHCクラスII分子複合体のペプチド結合窩に結合している、請求項99記載の細胞。
【請求項102】
少なくとも1種のT細胞に影響する分子が、T細胞共刺激分子である、請求項92記載の細胞。
【請求項103】
T細胞共刺激分子が、CD80(B7-1)、CD86(B7-2)、B7-H3、4-1BBL、CD27、CD30、CD134(OX-40L)、B7h(B7RP-1)、CD40、LIGHT、CD28に特異的に結合する抗体、HVEMに特異的に結合する抗体、CD40Lに特異的に結合する抗体、OX40に特異的に結合する抗体、および4-1BBに特異的に結合する抗体からなる群より選択される、請求項102記載の細胞。
【請求項104】
少なくとも1種のT細に影響する分子が、接着分子である、請求項92記載の細胞。
【請求項105】
接着分子が、ICAM-1、LFA-3およびLFA-1からなる群より選択される、請求項104記載の細胞。
【請求項106】
少なくとも1種のT細胞に影響する分子が、T細胞増殖因子である、請求項92記載の細胞。
【請求項107】
T細胞増殖因子が、サイトカインおよびスーパー抗原からなる群より選択される、請求項106記載の細胞。
【請求項108】
T細胞増殖因子が、サイトカインであり、およびサイトカインが、IL-2、IL-4、IL-7、IL-10、IL-12、IL-15、およびγインターフェロンからなる群より選択される、請求項106記載の細胞。
【請求項109】
少なくとも1種のT細胞に影響する分子が
(A)少なくとも2種の融合蛋白質を含む第一の分子複合体であり、ここで第一の融合蛋白質が、第一のサイトカインおよび免疫グロブリン重鎖を含み、ならびにここで第二の融合蛋白質が、第二のサイトカインおよび第二の免疫グロブリン重鎖を含み、ここで第一および第二の免疫グロブリン重鎖が会合し、第一の分子複合体を形成するもの、;ならびに
(B)少なくとも4種の融合蛋白質を含む第二の分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)免疫グロブリン重鎖および(ii)第一のサイトカインを含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)第二のサイトカインを含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、第二の分子複合体を形成するものからなる群より選択される、請求項106記載の細胞。
【請求項110】
第一および第二のサイトカインが同一である、請求項109記載の細胞。
【請求項111】
第一および第二のサイトカインが異なる、請求項109記載の細胞。
【請求項112】
少なくとも1種の外因性T細胞に影響する分子が、調節T細胞誘導分子である、請求項92記載の細胞。
【請求項113】
少なくとも1種の調節T細胞誘導分子が、TGFβ、IL-10、インターフェロン-α、およびIL-15からなる群より選択される、請求項112記載の細胞。
【請求項114】
少なくとも2種の異なるT細胞に影響する分子を含む、請求項92記載の細胞。
【請求項115】
少なくとも1種のリンパ球に影響する分子が、B細胞に影響する分子であり、および少なくとも1種の分子複合体が、少なくとも4種の融合蛋白質を含むTCR分子複合体であり、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;ならびに
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖、および(ii)TCRβ鎖またはTCRγ鎖の細胞外ドメインを含み、ここでこれら2種の第一の融合蛋白質がTCRα鎖を含む場合、その結果2種の第二の融合蛋白質が、TCRβ鎖を含み、ここで2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項91記載の細胞。
【請求項116】
請求項91記載の細胞を複数含む、調製物。
【請求項117】
薬学的に許容される担体を更に含む、請求項116記載の調製物。
【請求項118】
複数の前駆体T細胞を含む単離された調製物を、請求項92記載の第一の複数の細胞と接触する工程を含む、抗原特異的T細胞の形成を誘導する方法であって、
ここで抗原性ペプチドが、ペプチド結合窩に結合され、これにより複数の前駆体T細胞のメンバーを、抗原性ペプチドを認識する抗原特異的T細胞を含む第一の細胞集団を形成するように誘導し、ここで第一の細胞集団内の抗原特異的T細胞の数または割合が、前駆体T細胞が第二の複数の細胞とインキュベーションされる場合に形成される抗原特異的T細胞の数または割合よりも大きく、ここで第二の複数の細胞が、CD3に特異的に結合する抗体を含むが、抗原提示複合体は含まない、方法。
【請求項119】
抗原特異的T細胞が、細胞傷害性T細胞である、請求項118記載の方法。
【請求項120】
抗原特異的T細胞が、ヘルパーT細胞である、請求項118記載の方法。
【請求項121】
抗原特異的T細胞が、調節T細胞である、請求項118記載の方法。
【請求項122】
抗原特異的T細胞を第一の細胞集団から分離する工程を更に含む、請求項118記載の方法。
【請求項123】
第一の細胞集団を、請求項1記載の第二の複数の細胞と共にインキュベーションする工程を更に含む方法であって、ここで抗原性ペプチドが、これらの細胞のペプチド結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項118記載の方法。
【請求項124】
抗原特異的T細胞を含む第一の細胞集団を、請求項92記載の複数の細胞と共にインキュベーションする工程を含む、細胞集団内の抗原特異的T細胞の数または割合を増加する方法であって、
ここで抗原性ペプチドが、これらの細胞のペプチド結合窩に結合され、ここでインキュベーション工程が、第一の細胞集団の抗原特異的T細胞の数または割合と比べ、増加した数または割合の抗原特異的T細胞を含む第二の細胞集団を形成するのに十分な時間実行される、方法。
【請求項125】
第一の細胞集団が、均質な細胞集団である、請求項124記載の方法。
【請求項126】
抗原特異的T細胞を患者に投与する工程を更に含む、請求項118記載の方法。
【請求項127】
患者が、癌、自己免疫疾患、感染症を有するか、または免疫抑制される、請求項126記載の方法。
【請求項128】
前駆体T細胞が、患者から得られる、請求項126記載の方法。
【請求項129】
前駆体T細胞が、患者ではないドナーから得られる、請求項126記載の方法。
【請求項130】
抗原特異的T細胞が、静脈内投与、動脈内投与、皮下投与、皮内投与、リンパ内投与、および腫瘍内投与からなる群より選択される投与経路により投与される、請求項126記載の方法。
【請求項131】
第二の集団の抗原特異的T細胞を、患者に投与する工程を更に含む、請求項124記載の方法。
【請求項132】
請求項92記載の複数の細胞および薬学的に許容される担体を含有する調製物を患者に投与する工程を含む、患者における免疫応答を調節する方法であって、
ここで抗原性ペプチドが、少なくとも1種のペプチド結合窩に結合される、方法。
【請求項133】
少なくとも1種のT細胞に影響する分子が、(1)アポトーシス誘導分子、(2)調節T細胞誘導性分子、(3)T細胞共刺激分子、(4)接着分子、および(5)T細胞増殖因子からなる群より選択される、請求項132記載の方法。
【請求項134】
集団内の抗体産生B細胞の数または割合を増加する方法であって、複数の前駆体B細胞を含む単離された調製物を、少なくとも1種の請求項42記載の第一の固形支持体と接触し、これにより複数の前駆体B細胞のメンバーを、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む第一の細胞集団を形成するように誘導する工程を含む、方法。
【請求項135】
抗体を産生するB細胞を、第一の細胞集団から分離する工程を更に含む、請求項134記載の方法。
【請求項136】
第一の細胞集団を、少なくとも1種の請求項42記載の第二の固形支持体と共にインキュベーションする工程を更に含み、ここでインキュベーション工程が、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される、請求項134記載の方法。
【請求項137】
抗体産生B細胞を含む第一の細胞集団を、少なくとも1種の請求項42記載の第一の固形支持体と共にインキュベーションする工程を含む、集団内の抗体産生B細胞の数または割合を増加する方法であって、
ここでインキュベーションの工程が、第一の細胞集団内の抗体産生B細胞の数または割合と比べ、増加した数または割合の抗体産生B細胞を含む第二の細胞集団を形成するのに十分な時間実行される、方法。
【請求項138】
第一の細胞集団が、均質な細胞集団である、請求項137記載の方法。
【請求項139】
集団内の抗体産生B細胞の数または割合を増加する方法であって、複数の前駆体B細胞を含む単離された調製物を、請求項66記載の調製物と接触し、これにより複数の前駆体B細胞のメンバーに、抗原性ペプチドに特異的に結合する抗体を産生する抗体産生B細胞を含む第一の細胞集団の形成を誘導する工程を含む、方法。
【請求項140】
(A)複数の粒子、および(B)薬学的に許容される担体を含む調製物を、患者に投与する工程を含む、患者の免疫応答を調節する方法であって、
ここで複数の粒子のメンバーが
(1)少なくとも1種のB細胞に影響する分子;および
(2)B細胞表面のMHC-抗原複合体に結合する少なくとも1種の分子複合体を含む、方法。
【請求項141】
少なくとも1種のB細胞に影響する分子が、(1)CD40リガンド、(2)サイトカイン、および(3)サイトカイン分子複合体からなる群より選択される、請求項140記載の方法。
【請求項142】
分子複合体が、T細胞受容体および少なくとも4種の融合蛋白質を含むTCR分子複合体からなる群より選択され、ここで
(a)2種の第一の融合蛋白質が、(i)TCRα鎖またはTCRγ鎖を含み;および
(b)2種の第二の融合蛋白質が、(i)免疫グロブリン軽鎖および(ii)TCRβ鎖またはTCRδ鎖の細胞外ドメインを含み、ここで2種の第一の融合蛋白質がTCRα鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRβ鎖を含み、かつ2種の第一の融合蛋白質がTCRγ鎖を含む場合は、その結果2種の第二の融合蛋白質が、TCRδ鎖を含み、
ここで2種の第一および2種の第二の融合蛋白質が会合し、TCR分子複合体を形成し、ここで各第一の融合蛋白質のTCRαまたはγ鎖の細胞外ドメインおよび各第二の融合蛋白質のTCRβまたはδ鎖の細胞外ドメインが、TCR抗原結合窩を形成する、請求項140記載の方法。
【図1】

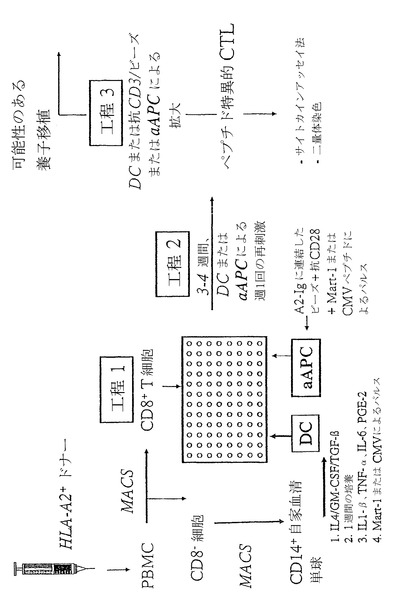
【図2−1】
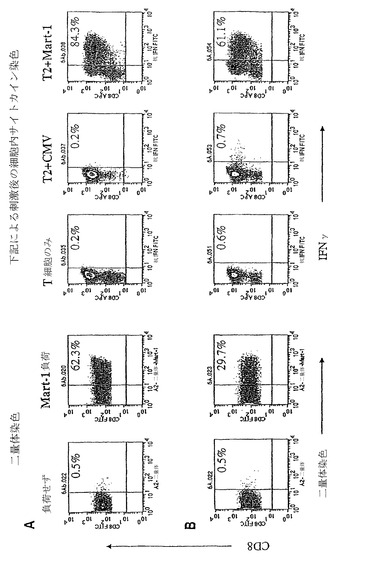
【図2−2】
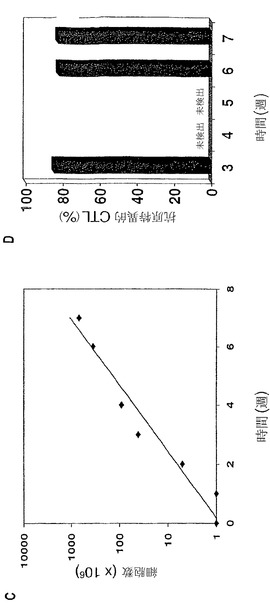
【図3】
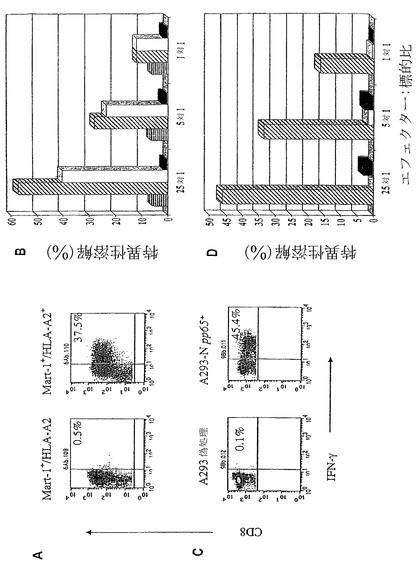
【図4】
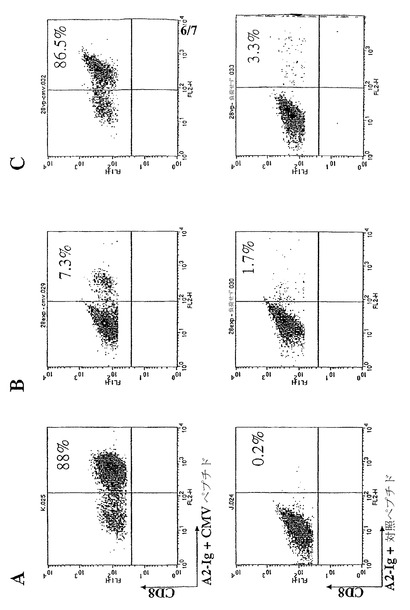
【図5】
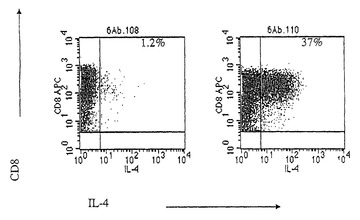
【図2−1】
【図2−2】
【図3】
【図4】
【図5】
【公開番号】特開2012−184233(P2012−184233A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2012−92552(P2012−92552)
【出願日】平成24年4月16日(2012.4.16)
【分割の表示】特願2004−521720(P2004−521720)の分割
【原出願日】平成15年7月14日(2003.7.14)
【出願人】(505011280)ザ ジョンズ ホプキンス ユニバーシティー (12)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成24年4月16日(2012.4.16)
【分割の表示】特願2004−521720(P2004−521720)の分割
【原出願日】平成15年7月14日(2003.7.14)
【出願人】(505011280)ザ ジョンズ ホプキンス ユニバーシティー (12)
【Fターム(参考)】
[ Back to top ]