
環式エステルおよび環式カボネートのイモータル開環重合用触媒系
環式エステルおよび環式カボネートのイモータル開環重合用のキレート用フェノキシリガンドで担持された二価金属錯体をベースにした新規な触媒系。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、環式エステルおよび環状カボネートのイモータル(immortal、永続する、不死の)開環重合用のキレート用フェノキシリガンドに担持された二価金属錯体をベースにした新規な触媒系に関するものである。
【背景技術】
【0002】
大部分の汎用ポリマーの生産に必要な化石資源の枯渇および不安定な原油価格に加えて、環境問題に対する関心の高まりによって、産業界および学会の両方で既存の合成材料を代替するバイオ・フレンドリーなポリマーの使用に対する研究が盛んに行われている。その結果、過去10年間はバイオ資源に由来するモノマーの重合分野と生物分解性ポリマーの製法分野に研究が濃縮してきた。
【0003】
環式エステルの開環重合(ROP)は例えば非特許文献1〜4に記載のように生物分解可能な脂肪族ポリエステルを作る最も便利な方法として研究が始まった。
【0004】
先ず最初に、例えば非特許文献5〜8に記載のように、ε-カプロラクトン(CL)とグリコシド(GL)とを(共)重合して生医学分野の用途に適したポリマーを作る方法が注目された。
【0005】
しかし、最近では例えば非特許文献9、10に記載のように、多くの研究者は乳酸に由来する環式ジエステルの重合、特にラクチド(LA)の重合へ関心を移している。このLAは生物-再生可能な資源であり、砂糖やトウモロコシの発酵で製造できる。LA、その他の環式モノマーのROPには工業的には錫ベースの開始剤、一般には錫(ll)2-カプロン酸エチルをベースにしたものが共通して使われている。これらの系は、例えば非特許文献11、12に記載のように、制御性が悪く、重元素の錫に関連する大きな問題がある。
【0006】
最近になって、例えば非特許文献13〜18に記載のようなrac-、S,S-およびR,R-LAのようなLAの各種異性体の制御されたリビングROPのための金属開始剤が開発された。これらは主して下記をベースにしたものである:
(1)非毒性の亜鉛(非特許文献19〜23)
(2)アルミニウム(非特許文献24〜28)
(3)3族金属およびランタニド(非特許文献29〜31)
【0007】
これらのシングルサイト錯体はアイソタクチック立体異性体としてのある種の海草およびバクテリアが製造する極めて結晶性の高い天然に生じる熱可塑性樹脂のポリ(3-ヒドロキシブチレート)を製造するためのβ-ブチロラクトン(BBL)のROPに対しても効率的であり、いくつかの触媒系はシンジオタクチックポリマーをつくる(非特許文献32〜35)。
【0008】
トリメチレンカボネート(TMC)のROPも過去3年間に大きな注意をひき始めた。非特許文献36〜39、特許文献1に記載のように、TMCはグリセリンに直接由来するバイオ資源のモノマーであり、それ自体はトリグリセライドの副生成物である。、この分子はLAとは違って資源採鉱に由来せず、非特許文献40、41に記載のように食物連鎖で使われる。
【0009】
金属-ベースの系の他に、非特許文献42、43に記載の環式モノマーの制御されたROPのための有機触媒の開拓についても言及する必要がある。
【0010】
これらのモノマーのROP、特に一つ以上の立体中心を含むLAやBBLのようなモノマーの場合の立体化学のコントロールおよび得られたポリエステルおよびポリカボネートの分子量に関して最も大きな前進が成し遂げられた。これらの系は一般に「リビング」であるが、工業的用途は含まれない。すなわち、活性中心によって単一のポリマーのみしか作れず、一つの活性サイトで少量のモノマー、一般には100-2000当量したトランスホームできない。工業的触媒系は生産性が極めて良いことが必要で、1活性中心当たり数千当量のモノマーを重合でき、何百ポリマー鎖を生ずることができなければならない。ROPの分野でこの目的を信頼性良く成し遂げる一つの方法は、特許文献1または非特許文献44〜48に記載のように、いわゆる「イモータル」リビング重合時に連鎖移動剤を添加して連鎖移動を操作することである。例えば、特許文献1では、50当量のベンジルアルコールの存在下で二元系(BDI)ZnN(SiMe3)2Bn-OHを使用してTMCのROPで極めて大きい効率でTMCを50000当量まで制御された重合することができる(ここで、BDI=(2,6-iPr2-C6H3)N=C(Me)-CH=C(Me)-N(2,6-iPr2-C6H3)であり、Bn-=C6H5CH2-である)。この方法では最終ポリマー中に使用した20〜100ppmの金属触媒の金属残留物が残る。それに加えて、触媒系が亜鉛ベースの「バイオ-金属」であるため、錫ベースの触媒系とは違った潜在的な毒性問題がある。
【0011】
そのほかのチャレンジとしては、従来の汎用合成ポリマー、すなわちポリ(α-オレフィン)、特にポリ(スチレン)の場合にかなり多量の生物資源を使うものがある。環式エステル(LA、CL、GL)またはカボネート(TMC)とスチレン(S)とのコポリマーを製造する方法は例えば特許文献2や非特許文献49〜63に記載されている。
【0012】
LAおよびTMCはポリスチレンの機械特性および物理特性を改良したスチレンのコポリマーの製造で利用され、コポリマーにはバイオ-モノマーが最高で50%含まれる。
【0013】
特許文献2にはLAの比率が大きイモータル重合方法が開示されている。これは末端官能化されたポリラクチドを作るために、(BDl)ZnN(SiMe3)2のような安全な金属ベースの開始剤と二価アルコール、例えば4-ヒドロキシ-2,2,6,6-テトラメチルピペリジノオキシ(TEMPO-OH)または2-ヒドロキシエチル−メタクリレート(HEMA)とを用いてニートなスチレン中で実行する。このPLAを使用してポリ(ラクチド−ブロック−スチレン)の制御された重合を行う。各ブロックの長さは自由に調整できる。
しかし、さらに改良する余地は十分にある。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】欧州特許出願第08290187.7号公報
【特許文献2】欧州特許出願第08290732.0号公報
【非特許文献】
【0015】
【非特許文献1】Uhrich et al. (K. E. Uhrich, S. M. Cannizzaro, R. S. Langer, K. M. Shakesheff, Chem. Rev., 1999, 99, 3181-3198)
【非特許文献2】Ikada and Tsujt (Y. ikada, H. Tsuji, Macromol. Rapid. Commun., 2000, 21, 117-132)
【非特許文献3】Langer (R. Langer, Ace. Chem. Res., 2000, 33, 94-101)
【非特許文献4】Okada (M. Okada, Prog. Polym. Sci., 2002, 27, 87-133)
【非特許文献5】Vert (M. Vert, Biomacromolecules 2005, 6, 538-546)
【非特許文献6】Albertsson and Varma (A. -C. Albertsson, I.K. Varma, Biomacromolecules 2003, 4, 1466-1486)
【非特許文献7】Sudesh et al. (K. Sudesh, H. Abe, Y. Doi Prog.Polym.Sci.2000,25,1503-1555)
【非特許文献8】Nair and Laurence (L. S. Nair, C. T. Laurence, Prog. polym. Sci. 2007, 32, 762-798)
【非特許文献9】Mecking (S. Mecking, Angew. Chem. Int. Ed, 2004, 43, 1078-1085)
【非特許文献10】Dechy-Cabaret et al. (O. Dechy- Cabaret, B. Martin-Vaca, D. Bourissou, Chem. Rev., 2004, 104, 6147-6176)
【非特許文献11】Drumright et al. (R, E. Drumright, P. R. Gruber, D. E. Henton, Adv. Mater., 2000, 12, 1841-1846)
【非特許文献12】Okada (M. Okada, Prog. Polym, Sci., 2002, 27, 87-133)
【非特許文献13】O'Keefe et al. (B. J. O'Keefe, M. A. Hilimyer, W. B. Tolman, J. Chem. Soc, Dalton Trans., 2001, 2215-2224)
【非特許文献14】Lou et al. (Lou, C. Detrembleur, R. Jerome, Macromol. Rapid. Commun., 2003, 24, 161-172)
【非特許文献15】Nakano et al. (K. Nakano, N. Kosaka, T. Hiyama, K.. Nozaki, J. Chem. Soc, DaltonTrans., 2003, 4039-4050)
【非特許文献16】Dechy-Cabaret et ai. (O. Dechy-Cabaret, B. Martin- Vaca, D. Bourissou, Chem. Rev., 2004, 104, 6147-6176)
【非特許文献17】Wu et al. (Wu, T.-L Yu, C-T. Chen, C-C. Lin, Coord. Chem. Rev., 2006, 250, 602-626)
【非特許文献18】Amgoune et al. (Amgoune, C. M. Thomas, J.-F. Carpentier, Pure Appl. Chem. 2007, 79, 2013- 2030)
【非特許文献19】M. Cheng, A. B. Attygalle, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 1999, 121, 11583-11584
【非特許文献20】B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 2001, 123, 3229-3238
【非特許文献21】C K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr., M. A. Hillmyer, W. B.Tolman, J. Am. Chem. Soc, 2003, 125, 11350-11359
【非特許文献22】G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova, P. Mehrkhodavandi, Organometallics, 2009, 28, 1309-1319
【非特許文献23】Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande Jr., J. -F. Carpentier, New J. Chem., 2008, 32, 2279-2291)
【非特許文献24】N. Spassky, M. Wisniewski, C. Pluta, A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627-2637
【非特許文献25】T. M. Ovitt, G. W. Coates, J. Am. Chem. Soc, 1999, 121, 4072-4073
【非特許文献26】M. Ovitt, G. W. Coates, J, Am. Chem. Soc, 2002, 124, 1316-1326
【非特許文献27】N. Nomura, R. ishii, Y. Yamamoto, T. Kondo, Chem. Eur. J., 2007, 13, 4433-4451
【非特許文献28】H. Zhu, E. Y.-X. Chen, Organometallics, 2007, 26, 5395-5405)
【非特許文献29】C-X. Cai, A. Amgoune, C. W. Lehmann, J.-F. Carpentier, Chem. Commun., 2004, 330-331
【非特許文献30】A, Amgoune, C. M. Thomas, T. Roisnel, J -F. Carpentier, Chem. Eur. J., 2006, 12, 169-179
【非特許文献31】A. Amgoune, C. M. Thomas, S. llinca, T. Roisnel, J.-F. Carpentier, Angew. Chem. Int. Ed., 2006, 45, 2782-2784
【非特許文献32】Amgoume et al. A. (Amgoune, C. M. Thomas, S. ilinca, T. Roisnel, J.-F. Carpentier, ngew. Chem. Int. Ed, 2006, 45, 2782-2784)
【非特許文献33】Rieth et al. (L. R. Rieth, D. R. Moore, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 2002, 124, 15239-15248)
【非特許文献34】Ajellal et al. (N. Ajellal, D. M.Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier, A. A. Trifonov, Chem. Eur. J., 2008, 14, 5440-5448)
【非特許文献35】Ajellal et al. (N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Piliin, Y. Grohens, J.-F. Carpentier, Macromolecules, 2009, 42, 987-993
【非特許文献36】S. Matsumura Adv. Polym. Sci 2005, 194, 95-132
【非特許文献37】Hellaye et al. (M.Le Hellaye, N. Fortin, J. Guilloteau, A. Soum, S. Lecommandoux, S. M. Guillaume Blomacromolecules, 2008, 9, 1924-1933)
【非特許文献38】Darensbourg et al. (D. J. Darensbourg, W. Choi, P. Ganguly, C. P. Richers Macromolecules, 2006, 39, 4374-4379)
【非特許文献39】Helou et al. (M. Helou, O. Miserque, J.- M. Brusson, J.-F. Carpentier, S. M. Guillaume, Chem. Eur. J., 2008, 14, 8772-8775)
【非特許文献40】Zhou et al. (C-H. Zhou, J. N. Beltramini, Y -X. Fan, G. Q. Lu Chem. Soc. Rev. 2008, 37, 527-549)
【非特許文献41】Behr et al. (A. Behr, J. Eilting, K. Irawadi, J. Leschinski, F. Lindner Green Chem. 2008, 10, 13-30)
【非特許文献42】Kamber et al. (N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer, J. L. Hedrick, Chem. Rev., 2007, 107, 5813-5840
【非特許文献43】Bourissou et al. (D. Bourissou, S. Moebs-Sanchez, B. Martin-Vaca, C. R. Chimie, 2007, 10, 775-794)
【非特許文献44】Asano et al. (S. Asano, T. Aida, S. Inoue, J. Chem. Soc, Chem.Commum., 1985, 1148-1149)
【非特許文献45】Aida et al. (T. Aida, Y. Maekawa, S. Asano, S. Snoue, Macromolecules, 1988, 21, 1195-1202)
【非特許文献46】Aida and Inoue (T. Aida, S. Inoue, Acc. Chem. Res., 1996, 29, 39-48)
【非特許文献47】Martin et al. (E. Martin, P. Dubois, R. Jerome, Macromolecuies, 2000, 33, 1530-1535)
【非特許文献48】Amgoume et al. (A. Amgoune, C. M. Thomas, J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693-697)
【非特許文献49】Zalusky et al. (A. S. Zalusky, R. Olayo-Valies, J. H. Wolf, M.A. Hillmyer, J. Am. Chem. Soc, 2002, 124, 12761-12773)
【非特許文献50】Barakat et al. (I. Barakat, P. Dubois, R. Jerome, P. Teyssie, J. Pol. Sci Part A: Polym. Chem., 1993, 31, 505-514
【非特許文献51】L. Barakt, P. Dubois, R. Jerome,P. Teyssie, E. Goethals, J. Pol.Sci. Part A: Polym. Chem., 1994, 32, 2099-2110
【非特許文献52】I. Barakat, P. Dubois, C, Grandfils, R. Jerome, J. Pol. ScL Part A: Polym. Chem., 1999, 37, 2401-2411)
【非特許文献53】Furch et al. (M. Furch, J. L. Eguiburu, M. J. Fernandez-Berridi, J. San Roman, Polymer, 1998, 39, 1977-1982)
【非特許文献54】Eguiburu et al. (J. L Eguiburu, M. J. Fernandez-Berridi, F. P. Cossio, J. San Roman, Macromolecul.theta.s, 1999, 32, 8252-8258
【非特許文献55】J. L. Eguiburu, M. J. Fernandez-Berridi, J. San Roman, Polymer, 2000, 41, 6439-6445)
【非特許文献56】Qiu et al. (H. Qiu, J. Rieger, B. Gilbert, R. Jer.delta.me, C. Jerome, Chem. Mater, 2004, 16, 850-856)
【非特許文献57】Kricheldorf et al. (H. R. Kricheldorf, S.-R. Lee, S. Bush, Macromolec.upsilon.les, 1996, 29, 1375-1381)
【非特許文献58】Trollsas et al. (M. Trollsas, C. J. Hawker, J. L. Hedrick, G. Carrot, J. Hiiborn, Macromolecules, 1998, 31, 5960-5963)
【非特許文献59】Hawker et al. (C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Bakarat, P. Dubois, R. Jerome, W. Volsken, Macromol. Chem. Phys., 1997, 198, 155-166)
【非特許文献60】Yoshida and Osagawa (E. Yoshida, Y. Osagawa, Macromolecules, 1998, 31, 1446-1453)
【非特許文献61】Wang et al. (Y. Wang, G. Lu, J. Huang, J. Pol. Sci Part A: Polym. Chem., 2004, 42, 2093-2099)
【非特許文献62】Dubois et al. (P. Dubois, R. Jerome, P. Teyssie, Macromolecules, 1991, 24, 977-981)
【非特許文献63】Jabbar et al. (R. Jabbar, A. Graffe, B. Lessard, M. Maric, J. Pol. Sci Part A: Polym. Chem., 2008, 109, 3185-3195)
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、新規なフェノキシ-ベースのリガンドを提供することにある。
本発明の他の目的は、このフェノキシ-ベースのリガンドを使用して二価金属錯体を製造することにある。
本発明のさらに他の目的は、この金属錯体を使用して環式エステルおよび環状カボネートの制御されたイモータルROP用の触媒系を提供することにある。
本発明のさらに他の目的は、末端が官能化されたPLAを製造することにある。
本発明のさらに他の目的は、ラクチドおよびスチレンのコポリマーのインサイチュー(in situ)合成を促進することにある。
本発明の上記目的の少なくとも一部は本発明によって達成される。
【課題を解決するための手段】
【0017】
本発明は、下記の式で表されるフェノール-ベースのプロ-リガンドのクラスを開示する:
【0018】
【化1】

【0019】
[ここで、
R1は
【化2】
であり(ここで、mは1、2または3であり、nは≧1である)、
【0020】
R2は1〜10の炭素原子を有するヒドロカルビル基であり、好ましくはメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択され、
R3はR1と同じか、1〜20の炭素原子を有するヒドロカルビル基、好ましくはメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されアルキルか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルの中から選択されるアリールである]
【0021】
置換パターンのキーとなる要素はR1で、これは環に含まれる窒素官能基と酸素原子を同時に有しなければならない点にあり、それはシクロアゾエーテルである。
【図面の簡単な説明】
【0022】
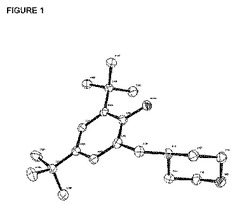
【図1】はリガンド[LO2]HのX線構造を表す図で、図を明瞭にするために水素原子は省略されている。
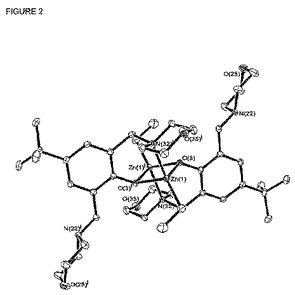
【図2】は錯体[LO1]ZnEtのX線構造を表し、図を明瞭にするために水素原子およびベンゼン分子は省略されている。
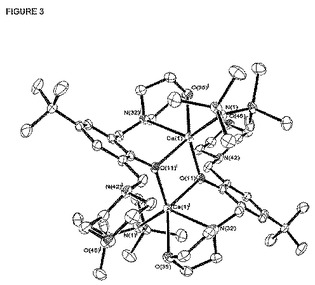
【図3】はダイマー[LO1]CaN(SiMe3)2のX線構造を表し、図を明瞭にするために水素原子は省略されている。
【図4】はL-LA/[LO1]ZnEt/iPrOHを相対量100/1/10、転化率20%で用いて製造した低分子量PLLAの1H NMRスペクトル(500.13MHz、CDCI3、25℃、16走査、D1=0.50秒)を表す。
【図5a】はL-LA/[LO1]ZnEt/iPrOHを相対量1000/1/10で用いて製造した数平均分子量MnGpcが4700g/モルの低分子量PLLAの高解像度MALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群:K+)を表す。
【図5b】もL-LA/[LO1]ZnEt/iPrOHを相対量1000/1/10で用いて製造した数平均分子量MnGpcが4700g/モルの低分子量PLLAの高解像度MALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群:K+)を表す。
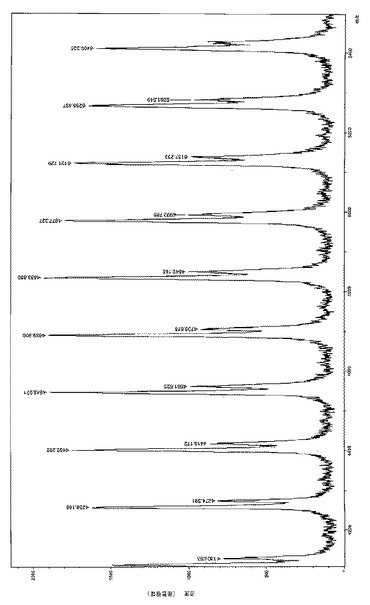
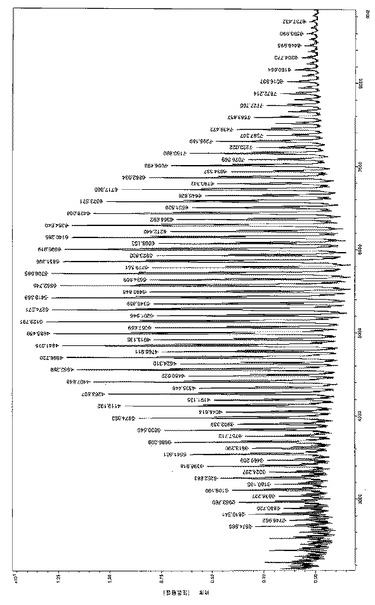
【図6a】はL-LA/[LO1]ZnEt/iPrOHを相対量2500/1/25、転化率98%で用いて製造した数平均分子量MnGpc が13200g/モルの中分子量PLLAのMALDl-TOFマススペクトル(マイナー個体群:Na+、主個体群:K+)を表す。
【図6b】もL-LA/[LO1]ZnEt/iPrOHを相対量2500/1/25、転化率98%で用いて製造した数平均分子量MnGpc が13200g/モルの中分子量PLLAのMALDl-TOFマススペクトル(マイナー個体群:Na+、主個体群:K+)を表す。
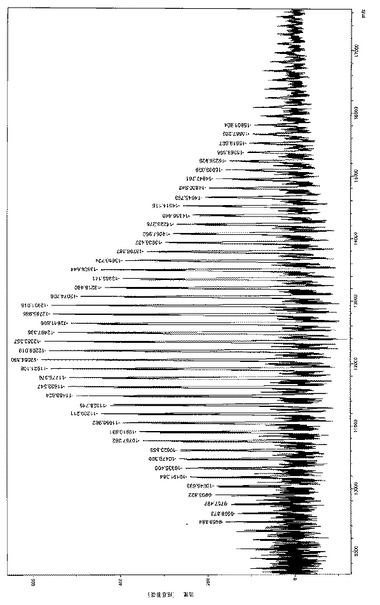
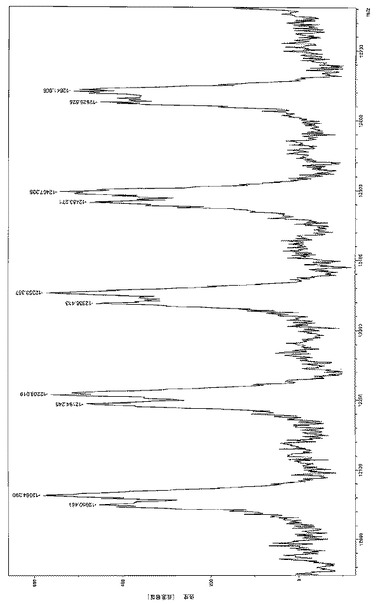
【図7a】はL-UV[LO1]MgBu/iPrOHを相対量5000/1/100、転化率71%で用いて製造した数平均分子量MnGpc が4600g/モルの低分子量PLLAのMALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群、K+)を表す。
【図7b】はL-UV[LO1]MgBu/iPrOHを相対量5000/1/100、転化率71%で用いて製造した数平均分子量MnGpc が4600g/モルの低分子量PLLAのMALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群、K+)を表す。
【図8】はL-LA/[LO1]CaN(SiMe3)2/iPrOH =500/1/25を用いて製造した低分子量PLLA(MnGpc=3000 g/モル、表4、エントリー32)の1H NMR(500.13MHz、CDCl3、25℃、64走査、D1=0.50秒)スペクトルを表す。
【図9a】はL-UV[LO1ICaN(SiMe3)2/iPrOH=500/1/25(転化率86%、Mn the=2500g/モル)を用いて製造した低分子性PLLA(MnGpc=3 000 g/モル、表4、エントリー34)のMALDI-TOFマススペクトル(主な個体群:Na+、マイナー個体群:K+)を表す。
【図9b】もL-UV[LO1ICaN(SiMe3)2/iPrOH=500/1/25(転化率86%、Mn the=2500g/モル)を用いて製造した低分子性PLLA(MnGpc=3 000 g/モル、表4、エントリー34)のMALDI-TOFマススペクトル(主な個体群:Na+、マイナー個体群:K+)を表す。
【発明を実施するための形態】
【0023】
本発明のリガンドはモルホリンまたはアザ-エーテル中に酸素が存在するため特に安定している。従来のリガンド、例えば下記文献に記載のリガンドは酸素がピペラジン環中に存在しないため、本発明のリガンドより性能が悪い。
【非特許文献63】Zheng et al. (Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, CM. Thomas, T. Roisnel, O. Casagrande, J.-F. Carpentie、New Journal of Chemistry, 32, 2279, 2008)
【0024】
従って、たれらは本発明のリガンドより金属中心に対して安定していない。そのため従来のリガンドは本発明のリガンドより早く分解し、従って、生産性および重合反応でのコントロール性が低い。
【0025】
これらのプロリガンドは公知の任意の方法で製造できる。
プロ-リガンドおよび金属複合体を製造するための本発明方法は下記文献に記載の方法の一変形例である。
【非特許文献64】Schanmuga et al. (S. Shanmuga Sundara Raj, M. N. Ponnuswamy, G. Shanmugam, M. Kandaswamy, J. Crystallogr, Sp.beta.ctrosc. Res., 1993, 23, 607-610)
【非特許文献65】Teipel et al. (S. Teipel, K. Griesar, W. Haase, B. Krebs, Inorg. Chem., 1994, 33, 456-464)
【0026】
リガンドの完全な合成方法および金属複合体の製造方法では数グラムのオーダーで分析的に純粋な化合物を長くても48時間でえることができる。比較のために示すと、LA、BBLまたはTMCのROP用の非常に効果的な亜鉛ベースの開始剤である(BDI)ZnN(SiMe3)2の合成には2週間と厳しい条件が必要である。
【0027】
このプロ−リガンドを用いて周期律表の第2族および第12族の二価金属の錯体を製造する。好ましい金属はマグネシウム、カルシウム、亜鉛、ストロンチウムおよびバリウムであり、好ましくはマグネシウム、カルシウムおよび亜鉛である。重量錯体は、プローリガンドを前駆体M(X)2と反応させて得られる。ここで、Xは1〜6個の炭素原子を有するアルキル、例えばメチル、エチル、n-ブチル、フェニルまたはアミド基、例えば、N(SiMe3)2、NMe2、NEt2、NiPr2またはアルコキシド基、例えばOEt、OiPr、OtBu、OCH2Ph、OSiPh3である。
【0028】
好ましい前駆体はZnEt2、Mg(nBu)2、Mg(N(SiMe3)2)2、Ca(N(SiMe3)2)2(THF)2である。
【0029】
本発明はさらに、式[LO]−M−Xの金属錯体を提供する。
ここで、
MはZn、Mg、Ca、SrまたはBaであり、
Xはヒドロカルビルまたはアルコキシド基OR''であり(ここでR''はヒドロカルビル、アリール、シリルまたはアミノ基NR*2であり)(ここで、R*はSiMe3、イソ−プロピル、メチルまたはエチルであり)(好ましいヒドロカルビルはエチルである)
[LO]は2−R1、4−R2、6−R3−C6H2Oである(ここで、R1、R2およびR3は上記で記載のもの)
【0030】
本発明はさらに、アルコールとキレート用フェノキシ・リガンドで支持された二価の金属錯体から成る系の存在下でROPによって環式エステルおよび5員環−、6員環−、7員環−環状カボネートを重合する方法を開示する。
【0031】
これらの金属錯体は1〜10,000当量、好ましくは5〜5000当量、より好ましくは5〜1,000当量のアルコールまたはポリアルコールの存在下で非常に活性で、ラクチド、環式エステルおよび5〜7員の環状カボネートを制御されたイモータルROPするための生産性に優れた触媒系になる。重合は有機溶媒の溶液中または無溶媒の溶融状態で20℃〜200℃、好ましくは25℃〜110℃の温度で行うことができる。
【0032】
一般に、1つの金属中心当たり千当量までのアルコールの存在下で、少なくとも50000〜500000当量、好ましくは50 000〜100000当量のモノマーの変換ができる。
【0033】
アルコールは式R'OHで表すことができる。R'は1〜20の炭素原子を有する直鎖または分岐鎖のヒドロカルビルである。R'は第1または第2アルキル残基か、ベンジル基であるのが好ましく、イソプロピル(iPr)またはベンジルである(Bn)であるのが好ましい。また、ポリオール、例えばジオール、トリオール、さらに高次の官能性ポリハイドリックアルコールでもよく、一般に1,3- プロパンジオールまたはトリメチロールプロパンから選択され、バイオマス、例えばグリセリン、その他任意の糖ベースのアルコール、例えばエリトリトールまたはシクロデキストリンに由来するものでもよい。各アルコールを個別に使用するか、組み合わせて使用できる。アルコールはイソプロパノール、sec-ブタノールまたはベンジルアルコールから選択するのが好ましい。
【0034】
重合反応は下記で表すことができる:
【化3】
【0035】
R1、R2=成長中のポリマー鎖、
[M]:有機金属フラグメント
ktr:変換速度定数
kp:成長速度定数
【0036】
本発明の重合シェーマではアルコールはリバーシブルなトランスファー剤の役目をする。ポリマー鎖の成長に急速なアルコキシド/アルコール交換が起こる。アルコール/金属比が増加し、ポリマー鎖の分子量も同程度に減少することが観察される。
【0037】
変換速度定数ktrが重合速度kpに対して十分に速ければ、形成されるポリマーのモル質量分布の幅は狭くなる。
【0038】
アルコール/金属比が一定の場合、ポリカボネートの分子量はアルコール/ポリオールの種類に依存する。
【0039】
また、官能化されたアルコールを本発明の開始剤と一緒に使用してLLAおよびrac−LAのイモータルROPおよびスチレンのTMCを効率的に促進することができ、それによって末端が官能化されたポリマーを製造することができる。この官能化基はLAまたはTMCとスチレンのコポリマのインサイチュー(in situ)合成で使用できる。
【0040】
このための好ましい官能化されたアルコールはTEMPO−OH、HEMAまたは各種のヒドロキシ−アルコキシアミン、例えばAA−OHの中からは選択するのが好ましい。
【0041】
環式エステルはL-ラクチド(LLA)、rac-ラクチド(rac-LA)またはrac-β-ブチロラクトン(rac-BBL)の中から選択するのが好ましい。
【0042】
好ましい環状カボネートはTMCそその置換誘導体の中から選択される。非制限的な例としては下記が挙げられる:
【化4】
【0043】
重合は20℃〜200℃、好ましくは25〜110℃の温度で実施される。圧力は0.5から20気圧、好ましくは1気圧である。
【0044】
こうして得られたポリマーは一般に1.1〜5.0、より典型的には1.1〜1.7の単峰形の(モノモダルな)分子量分布を有する。
【0045】
数平均分子量Mnはモノマー-to-アルコール比で調整でき、1000〜1000000 g/モル、より典型的には10000〜250000 g/モルである。さらに、サイズ除外クロマトグラフで求めた実験分子量はモノマー-to-アルコール比とモノマー転化率から計算した分子量とよく一致する。
【実施例】
【0046】
実施例での触媒充填のための全ての操作はシュレンク(シュレンク)ラインおよび標準的なシュレンク方法を使用したベンチか、無溶剤のドライグローブ・ボックス中(Jacomex、O2 <1ppm、H2O<5ppm)で不活性雰囲気下で実行した。
【0047】
1-(ベンジルオキシ)-2-フェニル-2-(2',2',6',6'-テトラメチル-1'−ピペリジニルオキシ)−エタン(AA−OH)、BDI−H (ここでBDIは[2,6-iPr2-C6H3)N=C(Me)-CH=C(Me)-N(2,6-iPr2-C6H3)]である)、錯体Zn[N(SiMe3)2]2、{Mg[N(SiMe3)2]2、Ca[N(SiMe3)2]2(THF)2および[BDI]ZnN(SiMe3)2は文献記載の方法で調製した。
【0048】
ZnEt2(ヘキサン中1.0M)およびMgBu2(ヘプタン中1.0M)はAldrich社から入手し、密封アンプル中に入れ、貯蔵した。
【0049】
2,4-ジ-t-ブチル-フェノール(97%、Acros)、4- t-ブチル-フェノール(99%、Alfa Aesar)、ホルムアルデヒド(37重量%水溶液、Acros)、モルホリン(99%、Acro)および1-アザ-15-クラウン-5(97%、Aldrich)は供給者からのものをそのまま使用した。
【0050】
ベンジルアルコール(>99.0%)はAldrich社から購入し、活性3・モレキュラーシーブ上で保存し、精製なしで使用した。
【0051】
iPrOH(HPLCグレード、VWR)はMg粉末上で乾燥、蒸留し、活性3・モレキュラーシーブ上に貯蔵した。
【0052】
4-ヒドロキシ−2,2,6,6-テトラメチルピペリジノオキシ(TEMPO−OH)フリーラジカル(98%、Acros)は4℃で保存した濃縮トルエン溶液から再結晶化し、常に暗闇で取り扱った。
【0053】
スチレン(99+%)はAldrich社から入手し、CaH2上で数日間乾燥し、動的減圧下で約45℃の温度でゆっくり加熱し蒸留し、-24℃で保存した。ポリスチレンによる汚染を避けるために2週間以内に使用した。
【0054】
トルエンはナトリウム上で予備乾燥し、使用前に溶融ナトリウムからアルゴン下に蒸留した。
【0055】
THFは最初に水酸化ナトリウム上で乾燥し、CaH2上でアルゴン下に蒸留し、使用の前に新たにアルゴン下にナトリウムミラー/ベンゾフェノンから第2回目の蒸留をした。
【0056】
ジオキサンはナトリウム・ミラー/ベンゾフェノンから蒸留した。
【0057】
全ての重水素溶剤(Euriso-top、Saclay、フランス)は密封アンプル中で活性3・モレキュラーシーブ上に保存し、使用前に複数回の冷凍−ソウ回路で完全に脱気した。
【0058】
テクニカルグレードのL−ラクチド(LLA)はTotal Petrochemicals社から提供され、rac-ラクチド(99%、rac−LA)はAcrosから入手した。これらラクチド(LA)の異性体の精製は3階段の手順、すなわち熱い濃縮iPrOH溶液(80℃)からの再結晶化と、それに続く2回のトルエン(105℃)からの再結晶化によって行った。LLAをより短時間でより低効率で精製する必要がある場合には、モノマーを単に一回だけiPrOHから再結晶化した。
【0059】
トリメチレンカボネート(TMC)はLabso Chimie Fine、Blanquefort(フランス)から提供された。乾燥した結晶のTMCは3階段、すなわちモノマーを最低24時間、水素化カルシウム上で濃縮THF溶液中で攪拌し、濾過してCaH2を除去し、-24℃の温度で再結晶して得た。
【0060】
精製後、LAおよびTMCは常に-30℃の温度でグローブ・ボックス中で不活性雰囲気下に保存した。異性体のβ-ブチルラクトン(rac-BBL、TCI Europe、97%)は水素化カルシウムから減圧蒸留で精製し、活性3・モレキュラーシーブ上に保存した。
【0061】
NMRスペクトルはBruker AC-200、AC-300およびAM-500分光計で記録した。重水素溶剤の残留信号を用いて全てのケミカルシフトを決定し、SiMe4対に対して較正した。信号のアサインメントは1D(1H 、13C{1H})および2D(COSY、HMBC、HMQC)NMR実験を用いて行った。
【0062】
元素分析はLondon Metropolitan UniversityのCarlo Erba 1108のElemental Analyser機器で実行し、最低でも2つの独立した計測値の平均をとった。
【0063】
ゲル浸透クロマトグラフ(GPC)計測はPLgel 5A MIXED-Cカラムを備えたPolymer Laboratories PL-GPC 50機器と屈折率検出器で実行した。GPCカラムは室温でTHFを1ml/分に流して溶出した。較正は580〜380000 g.モル-1の範囲の5つの単分散ポリスチレン標準品を使用して行った。例えば下記文献66介して68の推薦に従って、ポリスチレン標準品に対するポリ(ラクチド)、低、中および高分子量ポリ(トリメチレンカーボネート)の全ての分子量はMark-Houwinkファクタ0.58、0.58、0.73および0.88で修正した。ポリ(3-ヒドロキシブチレート)の分子量は対ポリ(スチレン)当量で直接得た。
【非特許文献66】M. Jalabert, C. Fraschini, R. E. Prud'homme, J. Pol. Sci. Part A: Polym. Chem., 2007, 45, 1944-1955
【非特許文献67】M. Save, M. Schappacher, A. Soum, Macromol. Chem. Phys., 2002, 203, 889-899
【非特許文献68】I. Palard, M. Schappacher, B. Belloncle, A. Soum, S. M. Guillaume, Chem. Eur. J., 2007, 13, 1511-152
【0064】
ポリ(ラクチド)サンプルのミクロ構造はポリマーのメチン領域のホモデカップリング1H NMRスペクトルを室温でCDCl3中の1.0〜2.0mg/ml濃度でBruker AM-50G分光計で記録して決定した。
【0065】
MALDI-TOF MSスペクトルは窒素レーザ源(337nm、3ns)を使用し、2OkVの正の加速度電圧の直鎖モードで、Bruker Daltonic MicroFlex LTで得た。サンプルの調製法は以下の通り:HPLCグレードのα-シアノ-4-ヒドロキシケイ皮酸(Bruker Care)アセトニトリル飽和溶液とトリフルオロ酢酸の0.1%超純水溶液との2:1混合液の1μmlをサンプルプレート上に付ける。蒸発後、HPLCグレードのTHF中に1〜10mg/mlのポリマー溶液1μmlを付ける。外部較正はBruker Care Peptide Calibration StandardおよびProtein Calibration Standard Iを用いて行った。
【0066】
典型的な重合手順
全ての取扱いは、不活性雰囲気の下で実行した。グローブ・ボックス中で金属ベースの開始剤と精製モノマーとを直ちに大きいシュレンクチューブに入れた。管を封止してグローブ・ボックスから取り出す。以下の操作は全て標準的なシュレンクテクニックを使用しシュレンクラインで実行した。必要に応じて、トルエン、THFまたはスチレンから選択されるドライな脱気した溶剤の必要量をシュリンジで加えた。次に、iPrOH、ベンジルアルコール、HEMA、AA−OHまたはTEMPO−OHの中から選択されるアルコールを、開始剤とモノマーを収容したシュレンクチューブに添加して金属錯塩活性化させた。迅速に添加後、シュレンクチューブを所望温度に予熱されたオイルバス中に漬けた。この時から重合時間を計測する。MeOH(1%、HCl)を添加して反応を停止し、ポリマーはメタノール中に沈澱させる。ジクロロメタンまたはTHFを溶剤として使用し、メタノールを非溶媒として使用て再沈殿で精製した。次いで、10-2 mbar以下の動的減圧下でポリマーを恒量乾燥した。
【0067】
リガンドの合成
プロ−リガンド:2,6-ビス(モルホリノメチル)-4-t−ブチル−フェノール([L01]H)2,4-ジ−t−ブチル-6-(モルホリノメチル)−フェノール([LO2]H)および2,4−ジt−ブチル-6−[(1-アザ-15-クラウン-5)メチル]−フェノール([LO3]H)の製造方法は下記のシェーマ1に示す。
【0068】
【化5】
【0069】
プロ-リガンド:2,6-ビス(モルホリノメチル)-4-t−ブチル−フェノール([L01]H)
11.7mlのホルムアルデヒド(138.3mmol、37重量%水溶液)を9.0gの4-t−ブチルフェノール(60.6mmol)と10.3mlのモルホリン(10.2g、118.2mmol)のジオキサン溶液60mlに加えた。得られた混合液を120℃の温度で一晩還流した。揮発性画分は減圧除去し、得られた固体をトルエン/水で抽出した。トルエン層を合せ、硫酸マグネシウム硫酸マグネシウム上で乾燥した。濾過後、黄色の溶液を減圧濃縮し、-24の温度で一晩保存した。大きな無色の結晶[LO1]Hが収率77%で得られた。この化合物の(1Hおよび13C{1H}NMR)の分光分析データは文献報告値と一致した。純度は元素分析で確認した。[LO1]Hはエーテル、塩素化溶剤および芳香族炭化水素に完全溶解し、脂肪族炭化水素にはわずかに溶ける。
【0070】
プロ−リガンド: 2,4-ジ-t−ブチル-6-(モルホリノメチル)−フェノール([LO2]H)
12.2gの2,4-ジ- t−ブチル−フェノール(59.1mmol)と、5.9mlのホルムアルデヒド(67.5mmol、水中37重量%)と、6.2mlのモルホリン(6.2g、70.9mmol)と黄色溶液を120℃の温度で一晩、90mlのジオキサン中で還流した。揮発油はポンプで排気し、得られた粘着性の固形物をトルエンおよびNaCl飽和水溶液で抽出した。有機層を合せ、MgSO4上で乾燥し、トルエンを排気するとオフホワイトの固形物が生ずる。それを減圧下に恒量乾燥して17.0gを得た(収率94%)。X線回折に適した単結晶[LO2]Hは +4℃の温度に一晩維持した濃縮ペンタン溶液から成長させて、その構造を決定した。それを[図1]に示す。
【0071】
元素分析:C19H31NO2(305.46グラム/モル):
理論値:C 74.71、H 10.23、N 4.59%
分析値:C 75.18、H 10.23、N 5.12%
【0072】
[LO2]Hは脂肪族炭化水素を含む全ての普通の有機溶媒に完全に溶ける。
【0073】
2,4- ジ-tブチル-6[1-アザ-15-クラウン-5]メチル]- フェノール([LO3]H)
1.03gの2,4-ジ-t−ブチル−フェノール(5.0mmol)、0.5mlのホルムアルデヒド(6.2mmol、37重量%水溶液)および1.25gの1-アザ-15-クラウン-5(5.7mmol)の混合液を20mlのジオキサン中で120℃の温度で24時間還流した。溶剤を減圧除去して得られるオレンジ油を恒量乾燥して2.23gの粗生成物を得る。移動層として純粋なクロロホルムを使用した薄層クロマトグラフィで精製して所望生成物を完全に精製した。クロロホルムを蒸発後、1.78gの化合物が81%の収率で得られた。[LO3]Hの分光分析データは公知の文献値と一致した。[LO3]Hは黄色の粘性のあるオイルで、全ての有機溶媒に充分に溶ける。
【0074】
二価金属ヘテロレプティック錯体の合成
[2,6-ビス(モルホリノメチル)-4-tBu-フェノキシ]亜鉛-エチル[[LQ1]ZnEt] の合成
3.5gの[LO1]-H(10.0mmol)のトルエン溶液75mlを-45℃の温度で20分かけて10.2mlのZnEt2(ヘキサン中1.0のM溶液、10.2mモル)のトルエン溶液125mlに加えた。得られた混合液を-45℃の温度で60分間攪拌し、さらに室温で2時間攪拌すると白い懸濁が生じる。沈降物を濾過分離し、減圧乾燥して分析的に純粋な4gの白い粉末を91%の収率で得た。{[LO1]ZnEt}2 C6H5の無色単結晶を濃縮ベンゼン溶液から室温で成長させ、その固体物理構造をX線結晶学で決定した。
元素分析:C22H36N2O3Zn(440.20グラム/モル):
理論値:C 59.79、H 8.21、N 6.34%
分析値:C 59.78)H 8.21、N 6.05%
【0075】
CD2Cl2またはC6D6中の[LO1]ZnEtの1H NMRスペクトルは複雑に見えたが、1−D(1H、13C{1H}および2−D(COSY、HMBCおよびHMQC)後、その1Hおよび13C(1H) NMR信号が得られた。NMR実験はトルエン-d8中で−60℃で実した。その固体構造を室温で濃縮ベンゼン溶液から成長させたX線品質結晶を使用して評価した。その結果、[LO1]ZnEtは[図2]に示すように2つの亜鉛原子がフェノキシ部分の酸素原子を介してブリッジしたダイマー種として存在することが示された。[LO1]ZnEtはエーテルおよびジクロロメタンに溶け、ベンゼンおよびトルエンに少し溶け、脂肪族炭化水素には溶解しない。
【0076】
[2,4-ジ-t−ブチル-6-(モルホリノメチル)-フェノキシ]亜鉛-エチル[[LQ2]ZnEt] の合成
1.06gの[LO2]-H、3.47mmol)のトルエン溶液20mlトルエン溶液を-25℃の温度で3.50mlのZnEt2(3.50mmol、1.0Mヘキサン溶液)のトルエン40mlに20分かけてゆっくり加える。さらに-25℃の温度で40分間攪拌し、アルカン除去後に無色溶液を得る。溶剤を蒸発させて得られた白い固形物を20mlのペンタンで3回洗浄し、減圧乾燥して1.18gの錯体を85%の収率で得た。
元素分析:C21H35NO2Zn(397.20グラム/モル):
理論値:C 63.23、H 8.84、N 3.51%
分析値:C 63.09、H 8.73、N 3.51%
【0077】
この錯体はTHFおよびジエチルエーテルに溶けるが、トルエンへの溶解性をは限られ、軽質石油エーテルには溶けない。
【0078】
[2.4-ジ-t−ブチル-6-(モルホリノメチル)-フェノキシ]マグネシウム-ブチル[[LQ1]MgBu] の合成
[LQ2]ZnEtの合成で記載したのと同じ手順で、0.94gの[LO1]H(2.70mmol)と3.0mlの MgBu2(3.50mmol、1.0Mヘプタン溶液)のトルエン22.0mlとの反応によって、[LQ1]MgBuを82%の収率で得た。
元素分析:C24H40N2O3Mg(428.90グラム/モル):
理論値:C 67.21、H 9.40、N 5.67%
分析値:C 67.32、H 9.89、N 6.19%
【0079】
この錯体はTHFおよびジエチルエーテルに溶け、トルエンには一定溶解性を有し、軽質石油エーテルに溶けない。
【0080】
[2,6-ビス(モルホリノメチル)-4-tBu-フェノキシ]カルシウム−[ビス(トリメチルシリル)アミド][[LO1]N(SiMe3)2の合成
1.32gの[LO1]H(3.79mmol)のTHF溶液20mlを室温で45分かけて1.71gのCa[N(SiMe3)2]2(THF)2(3.39mmol)のTHF溶液20mlに加えた。黄色の溶液を室温で一晩攪拌し、溶剤を減圧蒸発させると白い粉末が得られる。熱いヘキサン(ヘプタンまたは高級炭化水素も使用できる)での抽出を繰り返した後、溶剤を真空乾燥して77%の収率で分析的に純粋なヘテロレプティック(heteroleptic)な化合物が得られる。[[LO1]N(SiMe3)2の単結晶は室温でTHF溶液中にヘキサンをゆっくり拡散させて成長させて得た。その固体物理構造はX線回折で確認した。
元素分析:026H49N3O3Si2Ca(547.29グラム/モル:
理論値:C 56.99、H 9.01、N 7.67%
分析値:C 56.88、H 8.95、N 7.51%
【0081】
[[LO1]N(SiMe3)2の固体物理の構造は[図3]に示す。この図はこの化合物はフェノキシ部分の酸素原子を解してブリッジしたダイマー種の形で存在することを示している。
普通の有機溶媒への[[LO1]N(SiMe3)2の溶解性は脂肪族炭化水素でも良から優である。溶液中での安定正も非常に良く、NMRチューブでCeDe溶液中に5日間貯蔵した後でも劣化の兆候は見られない。しかも、そのヘテロレプティック性は溶液中でも保存される。すなわち、[[LO1]N(SiMe3)2のみでシュレンク-タイプの平衡無しに80℃でC6D6中で長時間反応させた後でも保存される。Ca[N(SiMe3)2および[[LO1]2Caが生じる兆候はない。
【0082】
本発明の本質的な特徴は、商用的な供給材料から出発して、リガンド[LO1]Hおよび[LO2]Hを完全合成でき、しかも、錯体の‖[LO1]ZnEt、[LO2]ZnEtおよび[LO1]JMgBuの合成を48時間で数グラムのスケールで分析的に純粋な化合物として得ることができる点にある。これに対して、効率的なLA、BBLまたはTMCのROPで使用される亜鉛-ベースの開始剤(BDI)ZnN(SiMe3)2の合成には2週間を要し、厳しい条件が必要である。
【0083】
連鎖移動剤の合成
1-(ベンジルオキシ)-2-フェニル-2-(2',2',6',6−テトラメチル-1'−ピペリジニルオキシ)エタン(AA−QHV)の合成
AA-OHの合成法を下記シェーマ3に示す。
【化6】
【0084】
下記文献に記載の公知の手順を変更して実行した。
【非特許文献69】Hawker et al. (C. J. Hawker, G. G. Barclay, A. Oreliana, J. Dao, W. Devonport, Macromotecules, 1996, 29, 5245-5254
【非特許文献70】Asri et al. (M. Asri Abd Ghani, D. Abdaliah, P. M. Kazmaier, B. Keoshkerian, E. Buncel, Can. J. Chem., 2004, 82, 1403-1412
【0085】
蒸留済みスチレン中のTEMPO溶液に、1.14当量の過酸化ベンゾイル(水中に75%)をゆっくり加えた。80℃の温度で30分間加熱すると、反応液は順次、赤、黄になり、最後に緑になる。揮発分を減圧除去し、緑のオイル材料からペンタンを加てて沈澱させて白い粉末が得られる。粉末を濾別後、溶剤を蒸発させると緑のオイルが得られる。それをメタノールに溶かす。-4℃の温度で再結晶して純粋なベンジル化物Aを55%の収率で得た。
【0086】
3.1gのこの化合物Aと15mlの2N−NaOH水溶液との混合をエチルアルコール中で3時間還流した。揮発性画分を蒸発させると油性の物質が得られる。ジクロロメタン/水で抽出後、合せた有機層はMgSO4を通して乾燥し、溶剤を減圧除去して恒量乾燥すると80%の収率でオレンジ油が得られる。この物質の特性のNMR分光(1H、13C(1H)およびCOSY実験)および元素分析の結果はAA−OHの予想組成および純度と一致した。
【0087】
環式エステルおよび環状カボネートの重合
トルエンまたはTHF中でのラクチド重合
一般的な重合手順をシェーマ4に示す。
【化7】
【0088】
1. [LO1]ZnEt
アルコールのトランスファー剤の存在下での上記の新規な開始剤[LO1]ZnEtを用いたLAのイモータルROPは極めて迅速であり、良く制御され、これに匹敵する公知のROP触媒系は文献にはない。重合条件および結果は[表1]にまとめた。
【0089】
【表1】
【0090】
[表1]での収率はメタノール中での沈殿後に測定し、理論的数平均分子量Mnは式[LA]o/[POH]o×モノマー変換率×MLA+MiproHを使用して計算した(ここで、MLA=144g/モルおよびMiproH=60g/モル)。MnGPCはゲル透過クロマトグラフィで対ポリスチレン標準品で決定し、mark-Houwinkファクタ0.58で修正した。Pmはラクチド単位のメソ結合確率で、CDCl3中、室温で記録したポリマーのホモデカップル1H−NMRスペクトルのメチン領域で決定した。
【0091】
[LO1]ZnEtを用いたアルコール不存在下のトルエン中でのL−LAの1000当量の重合([表1]の実施例1)は80℃では急速である(60分で転化率91%)であるが、制御性が悪く、PDIが1.50と多分散性の幅が広く、数平均分子量Mnの理論表し達と観測値の相関指数が悪い。しかし、60℃ではさらに遅く、制御性も非常に悪い(例えば1bis参照)。10当量のi PrOHを添加すると(例4)、ROPはアルコール無しより速くなり、制御性が良くなり、PDIは1.10になり、数平均分子量Mnの理論値と実験値とが良く一致する。さらに、これらの条件下では、PLLA4のホモデカップル1H−NMRスペクトルのメチン領域の調査値Pm = 1.00で示されるように、上記触媒系で予備実験しないで光学活性なモノマーを完全に重合できる。PLLAの後の指数4は実施例の番号を表す。
【0092】
実施例3と実施例4との比較から、[LO1]ZnEt /iPrOH系を用いた重合の最初の段階は、実施例3と他の条件は全く同じに時間を60分にした実施例4の結果から、Zn/アルコール比はが1:10では遅く、20分後に転化率は20%に過ぎないことが分かる。これはこの触媒の誘導期の存在を示す。
【0093】
[表1]全体から、アルコールの存在下ではLO1]ZnEtで極めて多当量のLLAを制御された重合できることが分かる。モノマー供給量を増加させた時の多分散性指数および分子量の予想値と観測値との間の相関も良〜優であり、L-LA/[LO1]ZnEt比は最高で50000にもなる。転化は定量的で、使用した条件下ではL-LA/[LO1]ZnEt比は500〜50000の間にある。さらに、トランスファー剤(一般にiPrOH)を非常に大過剰に使用できることは注目に値する。重合パラメータの制御性に目に見える有害な効果なしに、一つの金属センター当たりiPrOHを1〜1000当量の範囲で使用できる。本発明の触媒系は制御性と生産性との組合せを著しく良くする。
【0094】
トルエン中のモノマー濃度を最高6.0Mにして使用でき、モノマーの迅速な転化で芳香族溶剤中への完全な溶解が容易になる。転化率が高いので全てのモノマーが反応媒体中に溶解し、得られたポリマーは不溶性で高転化率で沈殿する。従って、反応の進化は速く、視覚で容易にモニターできる。
【0095】
実施例4(トルエン、97%転化)と実施例7(THF、57%転化)とを比較することによって分かるように、THFのようなコーディネート溶剤中でのLAのROPは、トルエンのような非コーディネート溶剤中より遅い。重合速度のこの減少は、溶剤をコーディネートする際に、モノマーによる金属中心上への配位が溶剤によって妨げられるためである。それにもかかわらず、THF中での重合は非常によく制御された状態に維持される。
【0096】
実施例6(トルエン、Pm = 0.50)と実施例8(THF、Pm = 0.45)に示すように、[LO1]ZnEt/iPrOH のZn/アルコール比1:10を用いた異性体(rac−LA)のラセミ混合物の重合は立体−選択性(stereo-selectiv)でない。
【0097】
単一の開始基の種類、すなわち[LO1]ZnEt/iPrOH触媒系中のイソプロポキシ基OCH(CH3)2-がNMR分光およびMALDI−TOFマススペクトル分析で示された。[図4]はL-LA/[L01]ZnEt/iPr0H(相対量100/1/10)で製造した低分子量PLLAの1H−NMRスペクトルを示す。-OCH(CH3)2 末端基の存在はδ5.06および1.24ppmでの信号で証明される。Et-または[LO1]−基によって開始されるPLLA鎖の存在は示されない。この解析結果は[LO1]ZnEt/iPrOHを用いて製造した[図5]に示す分子量4700g/モルおよび[図6]に示す分子量13200g/モルを有する2つのPLLAのMALDS−TOF質量スペクトル検査でも確認される。両方のスペクトルでイソプロポキシ-末端のPLLA鎖の分子量はGPC実験結果と理論値との間で非常に良く一致している。
【0098】
各スペクトルで2つのガウス分布が観測され、その第1および第2の2つの個体群はイオン化Na+およびK+に対応する[[図5(b)]および[図6(b)]に示すように2つの個体群間のΔ(m/z)]=16Da]。各ピークの分子量はオンマトリックス(on-matrix)化合物(H)(C6H8O4)n(OiPr)(Na)および(H)(C6H8O4)n(OiPr)(K)の計算された分子量と一致している(nは重合度である)。同じ個体群で各スペクトルの連続ピーク間に144Daの増加分が繰り返してみられ、これは一般に亜鉛錯体によって促進される望ましくないトランス-エステル化反応は、重合中でもモノマーが完全に転化した後でも、ないことを示す決定的な証拠である。従って、これらのポリマーは環状ポリマーを含まない。
【0099】
[表1]に示すように、iPrOHは[LO1]ZnEtを用いてLAの大過剰のイモータル制御されたROPでの極めて効率的な連鎖移動剤であることが証明されたが、本発明方法はiPrOHに限定されのものではなく、その代わりに他のアルコール、例えばベンジルアルコール、TEMPO−OH、HEMAまたは各種のヒドロキシ-アルコキシアミンを効率的に使用できる。このことは実施例4および実施例5に示されている。両方の場合で、重合パラメータの反応速度および制御性は同一である。従って、iPrOHを例えばAA−OHに完全に置換できる。
【0100】
2. [LO2IZnEt
重合条件および結果は[表2]にまとめた。
【表2】
【0101】
連鎖移動剤としてZnEtを用いたアルコール存在下での開始剤[LO2]を用いたLAのイモータル開環重合は極めて迅速かつ良く制御されて実行できる。。[表2]の実施例17で分かるように、10当量のiPrOHを用いたトルエン中での1000当量のLLAの重合は60℃の温度で60分以内に完了する。25当量のiPrOHを用いたトルエン中での5000当量のLLAは60℃の温度で90分でほぼ定量的に転化する。モノマーまたはアルコールの供給量が高くなると、重合パラメターの制御性にわずかな影響がでるが、多分散性指数はほぼ1.10に維持される。また、数平均分子量Mnの理論値と観測値はほぼ完全に一致する。[LO1]ZnEt錯体のROPでも述べたように、実施例16(反応時間=1分、転化なし)と実施例17(反応時間=60分、定量転化)との比較から明らかなように、[LO2]ZnEt/iPrOH触媒系でも少なくとも15分の賦活時間が存在することが明らかである。1000当量のrac−LAの重合はトルエン中(実施例18、完全転化)でもTHF中(実施例19、92%転化)でも非常に速い。両方のケースで制御性が良いが、後者の場合には分子量の予想値と観測値にわずかな差があた、分布がわずかに広い。この触媒系はシンジオタクチックPLAを形成する傾向をわずかに示し、それはトルエン中でのPm = 0.45よりTHF中でのPm = 0.40でより強くなる。
【0102】
3.[LO1]MgBu
重合条件および結果は[表3]にまとめた。
【表3】
【0103】
アルコールと組み合わせたMg錯体[LO1]MgBuはLAのROPの非常に効率的な開始剤を構成し、環式ジエステルの迅速かつ制御されたイモータル重合を促進する。例えば[表3]の実施例21に示すように、連鎖移動剤としての10当量のiPrOHは1000当量のLLAをほぼ定量的に重合し、Pm=1.00で示されるようにステレオセンターの目立つたepimerisation無しに非常によく制御されて重合は15分で完了する。
【0104】
1金属中心当たり1000〜5000当量の高い供給量にし、連鎖移動剤(ここではiPrOH)をMgに対して10〜100当量の大過剰にし、60〜80℃の温度で一般に2時間でLLAを重合した。全てのケースで多分散性指数は非常に狭く、1.11〜1.21の間にあり、分子量は予想値と実験値との相関が近似した。Zn-ベースの類似物[LO1]ZnEtおよび[LO2]ZnEtとは違って、[LO1]MgBuはモノマーの純度と連鎖移動剤の供給量にかなり敏感であった。60℃の温度でiPrOHの当量数を25(実施例24)から100(実施例例26)へ増やすと、同じ反応時間で、L-LA/[LO1]MgBu比を5000に固定した場合、活性がわずかに低下する。これはMgベース錯体は対応する亜鉛錯体と比較して高いオキソ毒性(oxophiiicit)を反映したものと考えられる。この反応速度の減少は反応温度を増やすことで部分的に補償できる。実施例25と実施例27を比較すると、5000当量のLLAを2時間で完全転化できないことがわかる。同様に、実施例24、28と実施例27、29とを比較することによって分かるように、連鎖移動剤を変えずにモノマー供給量を増加すると活性がかなり大きく低下する。これはモノマーをいかに完全に精製しても反応液中に存在する不純物に対するMg錯体の感度に起因する。
【0105】
アルコールおよび/またはLLAの量を増やすと反応速度が遅くなるのが観測されるが、分子量およびその分布はこれらのプロセスによってほとんど変化しない。従って、これらの現象はモノマーに対して不活性な種が同時に生成して触媒を部分的に失活させる結果であることを示唆している。観測事実とは逆に2つの活性種の存在が多分散性指数の広がりに有意な結果をもたらすと思われる。
【0106】
実施例22のPm=0.54が示すように、錯体[LO1]MgBuはトルエン中でのrac−LAの重合中に立体制御を殆どしない。THF中ではシンジオタクチックの方へのわずかな傾向が観測され、実施例23のPmは0.41である。[LO1]MgBuのステレオ選択性は非常に制限されるがトルエン中で逆方向へ生じ、メゾ二回対称軸性(diads)か観測され、THF中ではrac二回対称性が強くなることを強調しなければならない。実施例21と実施例22とを比較することで、LLAおよびrac−LAのROP中に活性種に起因する重合速度および品質は実質的に同一である。連鎖移動剤としてはiPrOH、BnOH、TEMPO−OH、AA−OH、HEMのような広範囲のアルコールを使用して[LO1]MgBuでLAの制御されたイモータルROPで種々の末端官能化したPLAを合成できる。L-LA/[LO1]MgBu/iPrOH(相対比5000/1/100)を用いて、5000当量のL-LAのイモータルのROPによってイソプロポキシ末端を付けたPLLAサンプルを製造できる。
【0107】
得られた低分子量PLLAのMALDI−TOFマススペクトルを[図7a]に示す。全てのポリマー鎖が-Oi/Pr単位で終っていることが明らかに示し、これはこの系でのROP機構の本当のイモータル性を決定的に証明している。[図7b]に示すように、不均一な強度の2つの個体群を全ガウス分布を通じた一連のピーク間の72Daの増加分はエステル転移反応プロセスがMg錯体による触媒作用でLAの重合中にかなりの範囲で起こることを示している。
【0108】
[LO1]CaN(SiMe3)2
重合条件および結果は[表4]にまとめた。
【表4】
【0109】
LAのイモータル重合はトルエン中またはTHF中で触媒系[LO1] CaN(SiMe3)2 /ROHを用い、ROHをiPrOH、BnOH、TEMPO−OH、AA−OH、HEMAの中から選択することで効率的に促進された。これは溶液および固相の両方で制御された状態で多量のモノマーを重合することを可能にするヘテロレプティックなフェノキシ-ベースのカルシウム錯体の最初の例であることを示している。
【0110】
アルコールが存在しない場合(実施例30)では、開始剤[LO1] CaN(SiMe3)2はアルコールが存在する場合に実行される他の実施例と比較して遅くなる。10〜100当量のiPrOHの存在下では、L-LAの最大2500当量を60℃の温度で重合できる。例えば、実施例33に示すように、iPrOH/[LO1]CaN(SiMe3)2比>10の場合、500当量のモノマーは1分以内に定量的に転化される。実施例35〜37に示すように、同じ条件下で1000当量のモノマーを15分以下で変換できる。多分散性指数が1.20〜1.40の範囲にあることから重合は良く制御されており、iPrOH/[LO1]CaN(SiMe3)2比が50の場合でも、分子量の実験値と理論値は近似している。多分散性指数が1.6〜1.7へ広がるのが観測されるのは、モノマーの完全転化後に反応を長く続けて、望ましくないエステル転移反応プロセスを起こすときであることを触れておく。
【0111】
触媒[L01]CaN(SiMe3)2/iPr0Hは低温で反応速度および制御性の両方で非常に性能が良い。従って、実施例40に示すように、500当量のモノマーは室温で1分後には70%が転化され、実施例33に示すように、60℃の温度では完全に添加される。トルエン中でのrac-LAの重合(実施例41)はL−LA(実施例33)のそれよりかなり遅く、立体制御性がなく、Pm = 0.50である。THF中で実行する(実施例42)と、rac-LAの重合はトルエン中よりも遅くなり、立体選択性もなく、Pm=0.50である。単一開始剤のグループ、すなわち触媒系[LO1]CaN(SiMe3)2/iPrOHdの−イソプロポキシ基OCH(CH3)2の種類はNMR分光およびMALDi−TOFマススペクトル分析で示された。低分子量のPLLAの1H NMRスペクトルは、実施例34に示すように、L-LA/[LO1]CaN(SiMe3)2/iPrOH(相対量は500/1/25)で得られ、(OCH(CH3)2の末端基は[図8]に示すようにδ55.06(OCH(CH3)2)および1.24 OCH(CH3)2)PPmの特徴的信号によって確認され、-N(SiMe3)2または[LO1]−基にり始まるPLLA鎖の存在は示されなかった。
【0112】
この解析は、[図9]に示すように、上記PLLAサンプルの高解像度MALDI−TOF質量スペクトル検査でも更に確認した。イソプロポキシ-末端を有するPLLA鎖の理論分子量とGPC実験結果とは非常に良く一致した:2つのガウス分布が観測された(それぞれNa+およびK+ のイオン化に対応した第1および第2の個体群[2つの個体群環のΔ(m/z)=16Da]が観察され、各ピークはオンマトリックス化合物(H)(C6H8O4)n(OiPr)(Na)および(H)(C6H8O4)n(OiPr)(K)の分子量(ここで、nは重合度)と良く一致する。この事実は個体群(Na+に対応する主個体群)で連続ピーク間の増加分が144Daでなく72Daのみであるということは重合中にカルシウム錯体によって望ましくないトランスエステル反応化合物が極めて迅速に促進されたことを強く証明している。これはモノマー完全転化後に反応を続けることでPLLAの多分散性指数が大きく広がることを示した上記観察と一致している。
【0113】
iPrOH存在下での開始剤の比較
トルエン中でiPrOHを使用した大量のL-LAのイモータルROPでの開始剤(BDI)ZnN(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuおよび[LO1]CaN(SiMe3)2の有効度を決定するための一連の実験を実施した。重合条件および結果は[表5]にまとめてある。
【0114】
【表5】
【0115】
[表5]に示すように、テストした全ての金属開始剤で、重合パラメータの制御性は常に良〜優であり、多分散性指数はの狭く、分子量は理論値と観測値でよく一致した。従って、ここでの触媒系の比較は単に重合速度をベースにしたものである。
【0116】
L−LA/[M]/iPrOH比が1000/1/10を用いて60℃の温度で15分間の実施した実施例43〜47は、錯体 [LO1]ZnEtおよび[LO2]ZnEtは他の3つの前駆体よりはるかに活性が低いように見え、これらは活性増加の基準では以下のようにランク付けられる:[LO2]ZnEt<<[LO2]ZnEt<<(BDI)ZnN(SiMe3)2=[LO1]CaN(SiMe3)2 <[LO1]MgBu。上記条件下での[LO2]ZnEtと[LO2]ZnEtの効率不足および短い反応時間は、これら2つの前駆体をベースにした触媒は[表1]および[票2]で観測されたように、10〜15分の賦活時間を必要とするという事実を反映していると言える。反応時間をより長くし、モノマー供給量を高くすると効率は異なったものになる。例えば、5000/1/25のL-LA/[M]/iPrOHの場合、60℃の温度、90分での効率順番は[LO1]MgBu < [LO1]ZnEt 〜 [LO2]ZnEt 〜(BDi)ZnN(SiMe3)2になる。亜鉛錯体では完全転化し、[LO1]MgBu錯体では80%である。これはZn-ベースの類似物に比べたMgベース錯体の特徴である高い感度を反映している。
【0117】
3つのZn-ベースの触媒系を区別するために、3つの全ての触媒でモノマーの完全転化前の60分後に反応を故意に止めた。以下の結果が観測された:[LO2]ZnEt(45%)< [LO1]ZnEt(71%)<(BDI)ZnN(SiMe3)2(95%)。これは実施例48〜50に示されている。
【0118】
結論として以下のことが言える:
(1) 連鎖移動剤の最大100〜500当量用いてLAを数万当量の量でイモータルROPする場合の最高の候補はZn-ベースの開始剤である。
(2) 条件が全く同じ場合、[LO1]ZnEtが[LO2]ZnEtより効率的である。
(3) 重合速度、[LO1]ZnEt(および[LO2]ZnEt)を活性化するために必要な誘導時間に対して有害効果は反応時間が90分以上では無視できる。
【0119】
第2の実験シリーズは、実施例例55〜60に示すように、最も有望な開始剤である[LO1]ZnEtと(BDI)ZnN(SiMe3)2に焦点をあてて、LLAのイモータルのROPを工業的な実験条件下で実行したものである。両方の実験セットで20000当量のモノマーをそれぞれ100当量のアルコール(実施例57、57bis、58および59)および250当量ののアルコール(実施例55および56)を用いて重合した。結果は、この条件下では活性の順番が逆になることを示した:(BDI) ZnN(SiMe3)2 <[LO1]ZnEt。しかし、両方とも実施例57bisの単純なZnEt2前駆体よりはるかに優れたものであった。
【0120】
全く驚くことであるが、[LO1]ZnEt/iPrOH=1/250を用いて60℃で50000当量のLLAをイモータルROPしても、非常に制御性がよく、8時間(エントリー59)とほぼ同程度に定量的であった。一方、500当量のアルコールの存在下(エントリー60)では16時間後に完全な転化が達成された。LAのROPでの[LO1]ZnEt錯体によって提供される範囲は大きなもので、この系でこれまでに達成された最大の性能である。
【0121】
スチレン中のラクチドの重合
反応階段はシェーマ5に、重合条件および結果は[表6]に示してある。
【0122】
【化8】
【表6】
【0123】
この表でMnの理論値は [LA]0/[ROH]0×モノマー転化率×MLA+MTEMPO−OHで計算した。MLA=144グラム/モル、MTEMPO−OH=162グラム/モル。
【0124】
iPrOH、BnOH、TEMPO−OH、AA−OH、HEMAから選択される広範囲の連鎖移動剤で、本発明の金属開始剤は[表6]に示すように、スチレン中のLAのイモータルROPに適している。例えば、1000当量のL-LAのスチレン中での重合はよく制御された状態でスチレンの重合干渉なしに進み、15分以内に完了する。そのため、この系は官能化PLLAの大規模合成およびその後のポリ(LA−ブロック-スチレン)コポリマーの製造に適している。
【0125】
ベンジルアルコールの存在下でのトリメチレンカボネート(TMC)のバルク重合
反応階段はシェーマ6に、重合条件および結果は[表7]に示した。
【0126】
【化9】
【表7】
【0127】
この表でMnの理論値は[LA]0/[ROH]0×モノマー転化率×MLA+MTEMPO−OHで計算した。MLA=144グラム/モル、MTEMPO−OH=162グラム/モル。
【0128】
iPrOH、BnOH、TEMPO−OH、AA−OH、HEMAから選択される広範囲の連鎖移動剤で、本発明の金属開始剤はバルクモノマー中でのTMCのイモータルROPに適している。
【0129】
例えば、5〜20当量のベンジルアルコールの添加で、[LO1]ZnEtは実施例例66、68および70に示すように60℃の温度で2、3時間以内に最大で25000当量のTMCを完全に転化できる。このタイプのTMCの亜鉛促進ROPの一般的な多分散性指数は1.50〜1.65で、分子量の観測値と計算値との相関は非常によい。
【0130】
制御性、活性および生産性に関しては、[LO1]ZnEtは(BDI)ZnN(SiMe3)2 より優れ、実施例68および実施例70に示すように、前者は10000〜25000当量のTMCを完全に転化できるが、後者は実施例67および実施例69に示すようにモノマーのそれぞれ89%および75%を変えるだけである。これによって二元系[LO1]ZnEt/BnOHは、欧州特許出願第EP-08 290 187.7号公報に記載の系(BDI)ZnN(SiMe3)2よりはるかに優れた、TMCのイモータルROPに対して最大の活性および生産性のある触媒系となる。さらに、この新規な[LO1]ZnEt/BnOH触媒は、既に述べたように、重合の制御性か良く、PDIの幅が狭く、Mnの実験値と理論値とが良くマッチする。
【0131】
驚くことに、100の当量のBnOHの添加で、実施例71に示すように、60℃の温度で48時間で最高100000当量のTMCをほぼ完全に転化できる。得られたポリマーPDIの幅は狭く、実験値と理論値は完全に一致した。実施例72に示すように、110℃まで加熱すると反応時間は有害効果無しに8時間に短縮できる。
【0132】
スチレン中でのトリメチレンカボネート(TMC)の重合
反応階段はシェーマ7に、重合条件および結果は[表8]に示した。
【0133】
【化10】
【表8】
【0134】
TMCは(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、溶剤からの目立った有害効果無しにスチレン中で制御されたイモータル重合される。5000当量のTMCを完全転化でき、分子量は理論値と実験値で一致した。但し、多分散性指数は類似条件下でLAのROPで観測されたもの(1.20の1.40)より大きくなり、一般に1.8〜1.9になる。
【0135】
ベンジルアルコール存在下での各種6-員カボネートの隗重合
反応階段はシェーマ8に、重合条件および結果は[表9]に示した。
【化11】
【0136】
官能化した6員環カボネートを、[表9]に示すように、(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、制御されたイモータル重合した。
【0137】
【表9】
【0138】
5当量の連鎖移動剤の存在下で500当量のモノマーを15〜60分以内に完全に転化できた。多分散性指数は一般に1.20〜1.70と狭く、分子量の観測値と計測値は近似した。ここでは分子量は対ポリスチレンで直接与えられるので、Mark-Houwink係数は修正のために加えなかった。
【0139】
β-ブチルラクトンラセミ体の重合
反応階段はシェーマ9に、重合条件および結果は[表10]に示した。
【0140】
【化12】
【表10】
【0141】
rac-BBLの隗重合を、[表10]に示すように、(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、制御されたイモータル重合した。
10当量のiPrOHの添加で、[LO1]ZnEtは200〜500当量のrac-BBLを定量的に数時間以内に容易に変換した。得られたポリマーのPDIは非常に狭く、一般に約1.10であり、分子量(GPCまたはMALDl-TOF質量分析で決定)は実験値と理論値とで極めて良く一致した。実施例84および実施例85に示すように、公知の(BDi)Zn-N(SiMe3)2と比較して、[LO1]ZnEtは活性および制御性の両方に優れている。
【技術分野】
【0001】
本発明は、環式エステルおよび環状カボネートのイモータル(immortal、永続する、不死の)開環重合用のキレート用フェノキシリガンドに担持された二価金属錯体をベースにした新規な触媒系に関するものである。
【背景技術】
【0002】
大部分の汎用ポリマーの生産に必要な化石資源の枯渇および不安定な原油価格に加えて、環境問題に対する関心の高まりによって、産業界および学会の両方で既存の合成材料を代替するバイオ・フレンドリーなポリマーの使用に対する研究が盛んに行われている。その結果、過去10年間はバイオ資源に由来するモノマーの重合分野と生物分解性ポリマーの製法分野に研究が濃縮してきた。
【0003】
環式エステルの開環重合(ROP)は例えば非特許文献1〜4に記載のように生物分解可能な脂肪族ポリエステルを作る最も便利な方法として研究が始まった。
【0004】
先ず最初に、例えば非特許文献5〜8に記載のように、ε-カプロラクトン(CL)とグリコシド(GL)とを(共)重合して生医学分野の用途に適したポリマーを作る方法が注目された。
【0005】
しかし、最近では例えば非特許文献9、10に記載のように、多くの研究者は乳酸に由来する環式ジエステルの重合、特にラクチド(LA)の重合へ関心を移している。このLAは生物-再生可能な資源であり、砂糖やトウモロコシの発酵で製造できる。LA、その他の環式モノマーのROPには工業的には錫ベースの開始剤、一般には錫(ll)2-カプロン酸エチルをベースにしたものが共通して使われている。これらの系は、例えば非特許文献11、12に記載のように、制御性が悪く、重元素の錫に関連する大きな問題がある。
【0006】
最近になって、例えば非特許文献13〜18に記載のようなrac-、S,S-およびR,R-LAのようなLAの各種異性体の制御されたリビングROPのための金属開始剤が開発された。これらは主して下記をベースにしたものである:
(1)非毒性の亜鉛(非特許文献19〜23)
(2)アルミニウム(非特許文献24〜28)
(3)3族金属およびランタニド(非特許文献29〜31)
【0007】
これらのシングルサイト錯体はアイソタクチック立体異性体としてのある種の海草およびバクテリアが製造する極めて結晶性の高い天然に生じる熱可塑性樹脂のポリ(3-ヒドロキシブチレート)を製造するためのβ-ブチロラクトン(BBL)のROPに対しても効率的であり、いくつかの触媒系はシンジオタクチックポリマーをつくる(非特許文献32〜35)。
【0008】
トリメチレンカボネート(TMC)のROPも過去3年間に大きな注意をひき始めた。非特許文献36〜39、特許文献1に記載のように、TMCはグリセリンに直接由来するバイオ資源のモノマーであり、それ自体はトリグリセライドの副生成物である。、この分子はLAとは違って資源採鉱に由来せず、非特許文献40、41に記載のように食物連鎖で使われる。
【0009】
金属-ベースの系の他に、非特許文献42、43に記載の環式モノマーの制御されたROPのための有機触媒の開拓についても言及する必要がある。
【0010】
これらのモノマーのROP、特に一つ以上の立体中心を含むLAやBBLのようなモノマーの場合の立体化学のコントロールおよび得られたポリエステルおよびポリカボネートの分子量に関して最も大きな前進が成し遂げられた。これらの系は一般に「リビング」であるが、工業的用途は含まれない。すなわち、活性中心によって単一のポリマーのみしか作れず、一つの活性サイトで少量のモノマー、一般には100-2000当量したトランスホームできない。工業的触媒系は生産性が極めて良いことが必要で、1活性中心当たり数千当量のモノマーを重合でき、何百ポリマー鎖を生ずることができなければならない。ROPの分野でこの目的を信頼性良く成し遂げる一つの方法は、特許文献1または非特許文献44〜48に記載のように、いわゆる「イモータル」リビング重合時に連鎖移動剤を添加して連鎖移動を操作することである。例えば、特許文献1では、50当量のベンジルアルコールの存在下で二元系(BDI)ZnN(SiMe3)2Bn-OHを使用してTMCのROPで極めて大きい効率でTMCを50000当量まで制御された重合することができる(ここで、BDI=(2,6-iPr2-C6H3)N=C(Me)-CH=C(Me)-N(2,6-iPr2-C6H3)であり、Bn-=C6H5CH2-である)。この方法では最終ポリマー中に使用した20〜100ppmの金属触媒の金属残留物が残る。それに加えて、触媒系が亜鉛ベースの「バイオ-金属」であるため、錫ベースの触媒系とは違った潜在的な毒性問題がある。
【0011】
そのほかのチャレンジとしては、従来の汎用合成ポリマー、すなわちポリ(α-オレフィン)、特にポリ(スチレン)の場合にかなり多量の生物資源を使うものがある。環式エステル(LA、CL、GL)またはカボネート(TMC)とスチレン(S)とのコポリマーを製造する方法は例えば特許文献2や非特許文献49〜63に記載されている。
【0012】
LAおよびTMCはポリスチレンの機械特性および物理特性を改良したスチレンのコポリマーの製造で利用され、コポリマーにはバイオ-モノマーが最高で50%含まれる。
【0013】
特許文献2にはLAの比率が大きイモータル重合方法が開示されている。これは末端官能化されたポリラクチドを作るために、(BDl)ZnN(SiMe3)2のような安全な金属ベースの開始剤と二価アルコール、例えば4-ヒドロキシ-2,2,6,6-テトラメチルピペリジノオキシ(TEMPO-OH)または2-ヒドロキシエチル−メタクリレート(HEMA)とを用いてニートなスチレン中で実行する。このPLAを使用してポリ(ラクチド−ブロック−スチレン)の制御された重合を行う。各ブロックの長さは自由に調整できる。
しかし、さらに改良する余地は十分にある。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】欧州特許出願第08290187.7号公報
【特許文献2】欧州特許出願第08290732.0号公報
【非特許文献】
【0015】
【非特許文献1】Uhrich et al. (K. E. Uhrich, S. M. Cannizzaro, R. S. Langer, K. M. Shakesheff, Chem. Rev., 1999, 99, 3181-3198)
【非特許文献2】Ikada and Tsujt (Y. ikada, H. Tsuji, Macromol. Rapid. Commun., 2000, 21, 117-132)
【非特許文献3】Langer (R. Langer, Ace. Chem. Res., 2000, 33, 94-101)
【非特許文献4】Okada (M. Okada, Prog. Polym. Sci., 2002, 27, 87-133)
【非特許文献5】Vert (M. Vert, Biomacromolecules 2005, 6, 538-546)
【非特許文献6】Albertsson and Varma (A. -C. Albertsson, I.K. Varma, Biomacromolecules 2003, 4, 1466-1486)
【非特許文献7】Sudesh et al. (K. Sudesh, H. Abe, Y. Doi Prog.Polym.Sci.2000,25,1503-1555)
【非特許文献8】Nair and Laurence (L. S. Nair, C. T. Laurence, Prog. polym. Sci. 2007, 32, 762-798)
【非特許文献9】Mecking (S. Mecking, Angew. Chem. Int. Ed, 2004, 43, 1078-1085)
【非特許文献10】Dechy-Cabaret et al. (O. Dechy- Cabaret, B. Martin-Vaca, D. Bourissou, Chem. Rev., 2004, 104, 6147-6176)
【非特許文献11】Drumright et al. (R, E. Drumright, P. R. Gruber, D. E. Henton, Adv. Mater., 2000, 12, 1841-1846)
【非特許文献12】Okada (M. Okada, Prog. Polym, Sci., 2002, 27, 87-133)
【非特許文献13】O'Keefe et al. (B. J. O'Keefe, M. A. Hilimyer, W. B. Tolman, J. Chem. Soc, Dalton Trans., 2001, 2215-2224)
【非特許文献14】Lou et al. (Lou, C. Detrembleur, R. Jerome, Macromol. Rapid. Commun., 2003, 24, 161-172)
【非特許文献15】Nakano et al. (K. Nakano, N. Kosaka, T. Hiyama, K.. Nozaki, J. Chem. Soc, DaltonTrans., 2003, 4039-4050)
【非特許文献16】Dechy-Cabaret et ai. (O. Dechy-Cabaret, B. Martin- Vaca, D. Bourissou, Chem. Rev., 2004, 104, 6147-6176)
【非特許文献17】Wu et al. (Wu, T.-L Yu, C-T. Chen, C-C. Lin, Coord. Chem. Rev., 2006, 250, 602-626)
【非特許文献18】Amgoune et al. (Amgoune, C. M. Thomas, J.-F. Carpentier, Pure Appl. Chem. 2007, 79, 2013- 2030)
【非特許文献19】M. Cheng, A. B. Attygalle, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 1999, 121, 11583-11584
【非特許文献20】B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 2001, 123, 3229-3238
【非特許文献21】C K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr., M. A. Hillmyer, W. B.Tolman, J. Am. Chem. Soc, 2003, 125, 11350-11359
【非特許文献22】G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova, P. Mehrkhodavandi, Organometallics, 2009, 28, 1309-1319
【非特許文献23】Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande Jr., J. -F. Carpentier, New J. Chem., 2008, 32, 2279-2291)
【非特許文献24】N. Spassky, M. Wisniewski, C. Pluta, A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627-2637
【非特許文献25】T. M. Ovitt, G. W. Coates, J. Am. Chem. Soc, 1999, 121, 4072-4073
【非特許文献26】M. Ovitt, G. W. Coates, J, Am. Chem. Soc, 2002, 124, 1316-1326
【非特許文献27】N. Nomura, R. ishii, Y. Yamamoto, T. Kondo, Chem. Eur. J., 2007, 13, 4433-4451
【非特許文献28】H. Zhu, E. Y.-X. Chen, Organometallics, 2007, 26, 5395-5405)
【非特許文献29】C-X. Cai, A. Amgoune, C. W. Lehmann, J.-F. Carpentier, Chem. Commun., 2004, 330-331
【非特許文献30】A, Amgoune, C. M. Thomas, T. Roisnel, J -F. Carpentier, Chem. Eur. J., 2006, 12, 169-179
【非特許文献31】A. Amgoune, C. M. Thomas, S. llinca, T. Roisnel, J.-F. Carpentier, Angew. Chem. Int. Ed., 2006, 45, 2782-2784
【非特許文献32】Amgoume et al. A. (Amgoune, C. M. Thomas, S. ilinca, T. Roisnel, J.-F. Carpentier, ngew. Chem. Int. Ed, 2006, 45, 2782-2784)
【非特許文献33】Rieth et al. (L. R. Rieth, D. R. Moore, E. B. Lobkovsky, G. W. Coates, J. Am. Chem. Soc, 2002, 124, 15239-15248)
【非特許文献34】Ajellal et al. (N. Ajellal, D. M.Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier, A. A. Trifonov, Chem. Eur. J., 2008, 14, 5440-5448)
【非特許文献35】Ajellal et al. (N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Piliin, Y. Grohens, J.-F. Carpentier, Macromolecules, 2009, 42, 987-993
【非特許文献36】S. Matsumura Adv. Polym. Sci 2005, 194, 95-132
【非特許文献37】Hellaye et al. (M.Le Hellaye, N. Fortin, J. Guilloteau, A. Soum, S. Lecommandoux, S. M. Guillaume Blomacromolecules, 2008, 9, 1924-1933)
【非特許文献38】Darensbourg et al. (D. J. Darensbourg, W. Choi, P. Ganguly, C. P. Richers Macromolecules, 2006, 39, 4374-4379)
【非特許文献39】Helou et al. (M. Helou, O. Miserque, J.- M. Brusson, J.-F. Carpentier, S. M. Guillaume, Chem. Eur. J., 2008, 14, 8772-8775)
【非特許文献40】Zhou et al. (C-H. Zhou, J. N. Beltramini, Y -X. Fan, G. Q. Lu Chem. Soc. Rev. 2008, 37, 527-549)
【非特許文献41】Behr et al. (A. Behr, J. Eilting, K. Irawadi, J. Leschinski, F. Lindner Green Chem. 2008, 10, 13-30)
【非特許文献42】Kamber et al. (N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer, J. L. Hedrick, Chem. Rev., 2007, 107, 5813-5840
【非特許文献43】Bourissou et al. (D. Bourissou, S. Moebs-Sanchez, B. Martin-Vaca, C. R. Chimie, 2007, 10, 775-794)
【非特許文献44】Asano et al. (S. Asano, T. Aida, S. Inoue, J. Chem. Soc, Chem.Commum., 1985, 1148-1149)
【非特許文献45】Aida et al. (T. Aida, Y. Maekawa, S. Asano, S. Snoue, Macromolecules, 1988, 21, 1195-1202)
【非特許文献46】Aida and Inoue (T. Aida, S. Inoue, Acc. Chem. Res., 1996, 29, 39-48)
【非特許文献47】Martin et al. (E. Martin, P. Dubois, R. Jerome, Macromolecuies, 2000, 33, 1530-1535)
【非特許文献48】Amgoume et al. (A. Amgoune, C. M. Thomas, J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693-697)
【非特許文献49】Zalusky et al. (A. S. Zalusky, R. Olayo-Valies, J. H. Wolf, M.A. Hillmyer, J. Am. Chem. Soc, 2002, 124, 12761-12773)
【非特許文献50】Barakat et al. (I. Barakat, P. Dubois, R. Jerome, P. Teyssie, J. Pol. Sci Part A: Polym. Chem., 1993, 31, 505-514
【非特許文献51】L. Barakt, P. Dubois, R. Jerome,P. Teyssie, E. Goethals, J. Pol.Sci. Part A: Polym. Chem., 1994, 32, 2099-2110
【非特許文献52】I. Barakat, P. Dubois, C, Grandfils, R. Jerome, J. Pol. ScL Part A: Polym. Chem., 1999, 37, 2401-2411)
【非特許文献53】Furch et al. (M. Furch, J. L. Eguiburu, M. J. Fernandez-Berridi, J. San Roman, Polymer, 1998, 39, 1977-1982)
【非特許文献54】Eguiburu et al. (J. L Eguiburu, M. J. Fernandez-Berridi, F. P. Cossio, J. San Roman, Macromolecul.theta.s, 1999, 32, 8252-8258
【非特許文献55】J. L. Eguiburu, M. J. Fernandez-Berridi, J. San Roman, Polymer, 2000, 41, 6439-6445)
【非特許文献56】Qiu et al. (H. Qiu, J. Rieger, B. Gilbert, R. Jer.delta.me, C. Jerome, Chem. Mater, 2004, 16, 850-856)
【非特許文献57】Kricheldorf et al. (H. R. Kricheldorf, S.-R. Lee, S. Bush, Macromolec.upsilon.les, 1996, 29, 1375-1381)
【非特許文献58】Trollsas et al. (M. Trollsas, C. J. Hawker, J. L. Hedrick, G. Carrot, J. Hiiborn, Macromolecules, 1998, 31, 5960-5963)
【非特許文献59】Hawker et al. (C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Bakarat, P. Dubois, R. Jerome, W. Volsken, Macromol. Chem. Phys., 1997, 198, 155-166)
【非特許文献60】Yoshida and Osagawa (E. Yoshida, Y. Osagawa, Macromolecules, 1998, 31, 1446-1453)
【非特許文献61】Wang et al. (Y. Wang, G. Lu, J. Huang, J. Pol. Sci Part A: Polym. Chem., 2004, 42, 2093-2099)
【非特許文献62】Dubois et al. (P. Dubois, R. Jerome, P. Teyssie, Macromolecules, 1991, 24, 977-981)
【非特許文献63】Jabbar et al. (R. Jabbar, A. Graffe, B. Lessard, M. Maric, J. Pol. Sci Part A: Polym. Chem., 2008, 109, 3185-3195)
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、新規なフェノキシ-ベースのリガンドを提供することにある。
本発明の他の目的は、このフェノキシ-ベースのリガンドを使用して二価金属錯体を製造することにある。
本発明のさらに他の目的は、この金属錯体を使用して環式エステルおよび環状カボネートの制御されたイモータルROP用の触媒系を提供することにある。
本発明のさらに他の目的は、末端が官能化されたPLAを製造することにある。
本発明のさらに他の目的は、ラクチドおよびスチレンのコポリマーのインサイチュー(in situ)合成を促進することにある。
本発明の上記目的の少なくとも一部は本発明によって達成される。
【課題を解決するための手段】
【0017】
本発明は、下記の式で表されるフェノール-ベースのプロ-リガンドのクラスを開示する:
【0018】
【化1】
【0019】
[ここで、
R1は
【化2】
であり(ここで、mは1、2または3であり、nは≧1である)、
【0020】
R2は1〜10の炭素原子を有するヒドロカルビル基であり、好ましくはメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択され、
R3はR1と同じか、1〜20の炭素原子を有するヒドロカルビル基、好ましくはメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されアルキルか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルの中から選択されるアリールである]
【0021】
置換パターンのキーとなる要素はR1で、これは環に含まれる窒素官能基と酸素原子を同時に有しなければならない点にあり、それはシクロアゾエーテルである。
【図面の簡単な説明】
【0022】
【図1】はリガンド[LO2]HのX線構造を表す図で、図を明瞭にするために水素原子は省略されている。
【図2】は錯体[LO1]ZnEtのX線構造を表し、図を明瞭にするために水素原子およびベンゼン分子は省略されている。
【図3】はダイマー[LO1]CaN(SiMe3)2のX線構造を表し、図を明瞭にするために水素原子は省略されている。
【図4】はL-LA/[LO1]ZnEt/iPrOHを相対量100/1/10、転化率20%で用いて製造した低分子量PLLAの1H NMRスペクトル(500.13MHz、CDCI3、25℃、16走査、D1=0.50秒)を表す。
【図5a】はL-LA/[LO1]ZnEt/iPrOHを相対量1000/1/10で用いて製造した数平均分子量MnGpcが4700g/モルの低分子量PLLAの高解像度MALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群:K+)を表す。
【図5b】もL-LA/[LO1]ZnEt/iPrOHを相対量1000/1/10で用いて製造した数平均分子量MnGpcが4700g/モルの低分子量PLLAの高解像度MALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群:K+)を表す。
【図6a】はL-LA/[LO1]ZnEt/iPrOHを相対量2500/1/25、転化率98%で用いて製造した数平均分子量MnGpc が13200g/モルの中分子量PLLAのMALDl-TOFマススペクトル(マイナー個体群:Na+、主個体群:K+)を表す。
【図6b】もL-LA/[LO1]ZnEt/iPrOHを相対量2500/1/25、転化率98%で用いて製造した数平均分子量MnGpc が13200g/モルの中分子量PLLAのMALDl-TOFマススペクトル(マイナー個体群:Na+、主個体群:K+)を表す。
【図7a】はL-UV[LO1]MgBu/iPrOHを相対量5000/1/100、転化率71%で用いて製造した数平均分子量MnGpc が4600g/モルの低分子量PLLAのMALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群、K+)を表す。
【図7b】はL-UV[LO1]MgBu/iPrOHを相対量5000/1/100、転化率71%で用いて製造した数平均分子量MnGpc が4600g/モルの低分子量PLLAのMALDI-TOFマススペクトル(主個体群:Na+、マイナー個体群、K+)を表す。
【図8】はL-LA/[LO1]CaN(SiMe3)2/iPrOH =500/1/25を用いて製造した低分子量PLLA(MnGpc=3000 g/モル、表4、エントリー32)の1H NMR(500.13MHz、CDCl3、25℃、64走査、D1=0.50秒)スペクトルを表す。
【図9a】はL-UV[LO1ICaN(SiMe3)2/iPrOH=500/1/25(転化率86%、Mn the=2500g/モル)を用いて製造した低分子性PLLA(MnGpc=3 000 g/モル、表4、エントリー34)のMALDI-TOFマススペクトル(主な個体群:Na+、マイナー個体群:K+)を表す。
【図9b】もL-UV[LO1ICaN(SiMe3)2/iPrOH=500/1/25(転化率86%、Mn the=2500g/モル)を用いて製造した低分子性PLLA(MnGpc=3 000 g/モル、表4、エントリー34)のMALDI-TOFマススペクトル(主な個体群:Na+、マイナー個体群:K+)を表す。
【発明を実施するための形態】
【0023】
本発明のリガンドはモルホリンまたはアザ-エーテル中に酸素が存在するため特に安定している。従来のリガンド、例えば下記文献に記載のリガンドは酸素がピペラジン環中に存在しないため、本発明のリガンドより性能が悪い。
【非特許文献63】Zheng et al. (Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, CM. Thomas, T. Roisnel, O. Casagrande, J.-F. Carpentie、New Journal of Chemistry, 32, 2279, 2008)
【0024】
従って、たれらは本発明のリガンドより金属中心に対して安定していない。そのため従来のリガンドは本発明のリガンドより早く分解し、従って、生産性および重合反応でのコントロール性が低い。
【0025】
これらのプロリガンドは公知の任意の方法で製造できる。
プロ-リガンドおよび金属複合体を製造するための本発明方法は下記文献に記載の方法の一変形例である。
【非特許文献64】Schanmuga et al. (S. Shanmuga Sundara Raj, M. N. Ponnuswamy, G. Shanmugam, M. Kandaswamy, J. Crystallogr, Sp.beta.ctrosc. Res., 1993, 23, 607-610)
【非特許文献65】Teipel et al. (S. Teipel, K. Griesar, W. Haase, B. Krebs, Inorg. Chem., 1994, 33, 456-464)
【0026】
リガンドの完全な合成方法および金属複合体の製造方法では数グラムのオーダーで分析的に純粋な化合物を長くても48時間でえることができる。比較のために示すと、LA、BBLまたはTMCのROP用の非常に効果的な亜鉛ベースの開始剤である(BDI)ZnN(SiMe3)2の合成には2週間と厳しい条件が必要である。
【0027】
このプロ−リガンドを用いて周期律表の第2族および第12族の二価金属の錯体を製造する。好ましい金属はマグネシウム、カルシウム、亜鉛、ストロンチウムおよびバリウムであり、好ましくはマグネシウム、カルシウムおよび亜鉛である。重量錯体は、プローリガンドを前駆体M(X)2と反応させて得られる。ここで、Xは1〜6個の炭素原子を有するアルキル、例えばメチル、エチル、n-ブチル、フェニルまたはアミド基、例えば、N(SiMe3)2、NMe2、NEt2、NiPr2またはアルコキシド基、例えばOEt、OiPr、OtBu、OCH2Ph、OSiPh3である。
【0028】
好ましい前駆体はZnEt2、Mg(nBu)2、Mg(N(SiMe3)2)2、Ca(N(SiMe3)2)2(THF)2である。
【0029】
本発明はさらに、式[LO]−M−Xの金属錯体を提供する。
ここで、
MはZn、Mg、Ca、SrまたはBaであり、
Xはヒドロカルビルまたはアルコキシド基OR''であり(ここでR''はヒドロカルビル、アリール、シリルまたはアミノ基NR*2であり)(ここで、R*はSiMe3、イソ−プロピル、メチルまたはエチルであり)(好ましいヒドロカルビルはエチルである)
[LO]は2−R1、4−R2、6−R3−C6H2Oである(ここで、R1、R2およびR3は上記で記載のもの)
【0030】
本発明はさらに、アルコールとキレート用フェノキシ・リガンドで支持された二価の金属錯体から成る系の存在下でROPによって環式エステルおよび5員環−、6員環−、7員環−環状カボネートを重合する方法を開示する。
【0031】
これらの金属錯体は1〜10,000当量、好ましくは5〜5000当量、より好ましくは5〜1,000当量のアルコールまたはポリアルコールの存在下で非常に活性で、ラクチド、環式エステルおよび5〜7員の環状カボネートを制御されたイモータルROPするための生産性に優れた触媒系になる。重合は有機溶媒の溶液中または無溶媒の溶融状態で20℃〜200℃、好ましくは25℃〜110℃の温度で行うことができる。
【0032】
一般に、1つの金属中心当たり千当量までのアルコールの存在下で、少なくとも50000〜500000当量、好ましくは50 000〜100000当量のモノマーの変換ができる。
【0033】
アルコールは式R'OHで表すことができる。R'は1〜20の炭素原子を有する直鎖または分岐鎖のヒドロカルビルである。R'は第1または第2アルキル残基か、ベンジル基であるのが好ましく、イソプロピル(iPr)またはベンジルである(Bn)であるのが好ましい。また、ポリオール、例えばジオール、トリオール、さらに高次の官能性ポリハイドリックアルコールでもよく、一般に1,3- プロパンジオールまたはトリメチロールプロパンから選択され、バイオマス、例えばグリセリン、その他任意の糖ベースのアルコール、例えばエリトリトールまたはシクロデキストリンに由来するものでもよい。各アルコールを個別に使用するか、組み合わせて使用できる。アルコールはイソプロパノール、sec-ブタノールまたはベンジルアルコールから選択するのが好ましい。
【0034】
重合反応は下記で表すことができる:
【化3】
【0035】
R1、R2=成長中のポリマー鎖、
[M]:有機金属フラグメント
ktr:変換速度定数
kp:成長速度定数
【0036】
本発明の重合シェーマではアルコールはリバーシブルなトランスファー剤の役目をする。ポリマー鎖の成長に急速なアルコキシド/アルコール交換が起こる。アルコール/金属比が増加し、ポリマー鎖の分子量も同程度に減少することが観察される。
【0037】
変換速度定数ktrが重合速度kpに対して十分に速ければ、形成されるポリマーのモル質量分布の幅は狭くなる。
【0038】
アルコール/金属比が一定の場合、ポリカボネートの分子量はアルコール/ポリオールの種類に依存する。
【0039】
また、官能化されたアルコールを本発明の開始剤と一緒に使用してLLAおよびrac−LAのイモータルROPおよびスチレンのTMCを効率的に促進することができ、それによって末端が官能化されたポリマーを製造することができる。この官能化基はLAまたはTMCとスチレンのコポリマのインサイチュー(in situ)合成で使用できる。
【0040】
このための好ましい官能化されたアルコールはTEMPO−OH、HEMAまたは各種のヒドロキシ−アルコキシアミン、例えばAA−OHの中からは選択するのが好ましい。
【0041】
環式エステルはL-ラクチド(LLA)、rac-ラクチド(rac-LA)またはrac-β-ブチロラクトン(rac-BBL)の中から選択するのが好ましい。
【0042】
好ましい環状カボネートはTMCそその置換誘導体の中から選択される。非制限的な例としては下記が挙げられる:
【化4】
【0043】
重合は20℃〜200℃、好ましくは25〜110℃の温度で実施される。圧力は0.5から20気圧、好ましくは1気圧である。
【0044】
こうして得られたポリマーは一般に1.1〜5.0、より典型的には1.1〜1.7の単峰形の(モノモダルな)分子量分布を有する。
【0045】
数平均分子量Mnはモノマー-to-アルコール比で調整でき、1000〜1000000 g/モル、より典型的には10000〜250000 g/モルである。さらに、サイズ除外クロマトグラフで求めた実験分子量はモノマー-to-アルコール比とモノマー転化率から計算した分子量とよく一致する。
【実施例】
【0046】
実施例での触媒充填のための全ての操作はシュレンク(シュレンク)ラインおよび標準的なシュレンク方法を使用したベンチか、無溶剤のドライグローブ・ボックス中(Jacomex、O2 <1ppm、H2O<5ppm)で不活性雰囲気下で実行した。
【0047】
1-(ベンジルオキシ)-2-フェニル-2-(2',2',6',6'-テトラメチル-1'−ピペリジニルオキシ)−エタン(AA−OH)、BDI−H (ここでBDIは[2,6-iPr2-C6H3)N=C(Me)-CH=C(Me)-N(2,6-iPr2-C6H3)]である)、錯体Zn[N(SiMe3)2]2、{Mg[N(SiMe3)2]2、Ca[N(SiMe3)2]2(THF)2および[BDI]ZnN(SiMe3)2は文献記載の方法で調製した。
【0048】
ZnEt2(ヘキサン中1.0M)およびMgBu2(ヘプタン中1.0M)はAldrich社から入手し、密封アンプル中に入れ、貯蔵した。
【0049】
2,4-ジ-t-ブチル-フェノール(97%、Acros)、4- t-ブチル-フェノール(99%、Alfa Aesar)、ホルムアルデヒド(37重量%水溶液、Acros)、モルホリン(99%、Acro)および1-アザ-15-クラウン-5(97%、Aldrich)は供給者からのものをそのまま使用した。
【0050】
ベンジルアルコール(>99.0%)はAldrich社から購入し、活性3・モレキュラーシーブ上で保存し、精製なしで使用した。
【0051】
iPrOH(HPLCグレード、VWR)はMg粉末上で乾燥、蒸留し、活性3・モレキュラーシーブ上に貯蔵した。
【0052】
4-ヒドロキシ−2,2,6,6-テトラメチルピペリジノオキシ(TEMPO−OH)フリーラジカル(98%、Acros)は4℃で保存した濃縮トルエン溶液から再結晶化し、常に暗闇で取り扱った。
【0053】
スチレン(99+%)はAldrich社から入手し、CaH2上で数日間乾燥し、動的減圧下で約45℃の温度でゆっくり加熱し蒸留し、-24℃で保存した。ポリスチレンによる汚染を避けるために2週間以内に使用した。
【0054】
トルエンはナトリウム上で予備乾燥し、使用前に溶融ナトリウムからアルゴン下に蒸留した。
【0055】
THFは最初に水酸化ナトリウム上で乾燥し、CaH2上でアルゴン下に蒸留し、使用の前に新たにアルゴン下にナトリウムミラー/ベンゾフェノンから第2回目の蒸留をした。
【0056】
ジオキサンはナトリウム・ミラー/ベンゾフェノンから蒸留した。
【0057】
全ての重水素溶剤(Euriso-top、Saclay、フランス)は密封アンプル中で活性3・モレキュラーシーブ上に保存し、使用前に複数回の冷凍−ソウ回路で完全に脱気した。
【0058】
テクニカルグレードのL−ラクチド(LLA)はTotal Petrochemicals社から提供され、rac-ラクチド(99%、rac−LA)はAcrosから入手した。これらラクチド(LA)の異性体の精製は3階段の手順、すなわち熱い濃縮iPrOH溶液(80℃)からの再結晶化と、それに続く2回のトルエン(105℃)からの再結晶化によって行った。LLAをより短時間でより低効率で精製する必要がある場合には、モノマーを単に一回だけiPrOHから再結晶化した。
【0059】
トリメチレンカボネート(TMC)はLabso Chimie Fine、Blanquefort(フランス)から提供された。乾燥した結晶のTMCは3階段、すなわちモノマーを最低24時間、水素化カルシウム上で濃縮THF溶液中で攪拌し、濾過してCaH2を除去し、-24℃の温度で再結晶して得た。
【0060】
精製後、LAおよびTMCは常に-30℃の温度でグローブ・ボックス中で不活性雰囲気下に保存した。異性体のβ-ブチルラクトン(rac-BBL、TCI Europe、97%)は水素化カルシウムから減圧蒸留で精製し、活性3・モレキュラーシーブ上に保存した。
【0061】
NMRスペクトルはBruker AC-200、AC-300およびAM-500分光計で記録した。重水素溶剤の残留信号を用いて全てのケミカルシフトを決定し、SiMe4対に対して較正した。信号のアサインメントは1D(1H 、13C{1H})および2D(COSY、HMBC、HMQC)NMR実験を用いて行った。
【0062】
元素分析はLondon Metropolitan UniversityのCarlo Erba 1108のElemental Analyser機器で実行し、最低でも2つの独立した計測値の平均をとった。
【0063】
ゲル浸透クロマトグラフ(GPC)計測はPLgel 5A MIXED-Cカラムを備えたPolymer Laboratories PL-GPC 50機器と屈折率検出器で実行した。GPCカラムは室温でTHFを1ml/分に流して溶出した。較正は580〜380000 g.モル-1の範囲の5つの単分散ポリスチレン標準品を使用して行った。例えば下記文献66介して68の推薦に従って、ポリスチレン標準品に対するポリ(ラクチド)、低、中および高分子量ポリ(トリメチレンカーボネート)の全ての分子量はMark-Houwinkファクタ0.58、0.58、0.73および0.88で修正した。ポリ(3-ヒドロキシブチレート)の分子量は対ポリ(スチレン)当量で直接得た。
【非特許文献66】M. Jalabert, C. Fraschini, R. E. Prud'homme, J. Pol. Sci. Part A: Polym. Chem., 2007, 45, 1944-1955
【非特許文献67】M. Save, M. Schappacher, A. Soum, Macromol. Chem. Phys., 2002, 203, 889-899
【非特許文献68】I. Palard, M. Schappacher, B. Belloncle, A. Soum, S. M. Guillaume, Chem. Eur. J., 2007, 13, 1511-152
【0064】
ポリ(ラクチド)サンプルのミクロ構造はポリマーのメチン領域のホモデカップリング1H NMRスペクトルを室温でCDCl3中の1.0〜2.0mg/ml濃度でBruker AM-50G分光計で記録して決定した。
【0065】
MALDI-TOF MSスペクトルは窒素レーザ源(337nm、3ns)を使用し、2OkVの正の加速度電圧の直鎖モードで、Bruker Daltonic MicroFlex LTで得た。サンプルの調製法は以下の通り:HPLCグレードのα-シアノ-4-ヒドロキシケイ皮酸(Bruker Care)アセトニトリル飽和溶液とトリフルオロ酢酸の0.1%超純水溶液との2:1混合液の1μmlをサンプルプレート上に付ける。蒸発後、HPLCグレードのTHF中に1〜10mg/mlのポリマー溶液1μmlを付ける。外部較正はBruker Care Peptide Calibration StandardおよびProtein Calibration Standard Iを用いて行った。
【0066】
典型的な重合手順
全ての取扱いは、不活性雰囲気の下で実行した。グローブ・ボックス中で金属ベースの開始剤と精製モノマーとを直ちに大きいシュレンクチューブに入れた。管を封止してグローブ・ボックスから取り出す。以下の操作は全て標準的なシュレンクテクニックを使用しシュレンクラインで実行した。必要に応じて、トルエン、THFまたはスチレンから選択されるドライな脱気した溶剤の必要量をシュリンジで加えた。次に、iPrOH、ベンジルアルコール、HEMA、AA−OHまたはTEMPO−OHの中から選択されるアルコールを、開始剤とモノマーを収容したシュレンクチューブに添加して金属錯塩活性化させた。迅速に添加後、シュレンクチューブを所望温度に予熱されたオイルバス中に漬けた。この時から重合時間を計測する。MeOH(1%、HCl)を添加して反応を停止し、ポリマーはメタノール中に沈澱させる。ジクロロメタンまたはTHFを溶剤として使用し、メタノールを非溶媒として使用て再沈殿で精製した。次いで、10-2 mbar以下の動的減圧下でポリマーを恒量乾燥した。
【0067】
リガンドの合成
プロ−リガンド:2,6-ビス(モルホリノメチル)-4-t−ブチル−フェノール([L01]H)2,4-ジ−t−ブチル-6-(モルホリノメチル)−フェノール([LO2]H)および2,4−ジt−ブチル-6−[(1-アザ-15-クラウン-5)メチル]−フェノール([LO3]H)の製造方法は下記のシェーマ1に示す。
【0068】
【化5】
【0069】
プロ-リガンド:2,6-ビス(モルホリノメチル)-4-t−ブチル−フェノール([L01]H)
11.7mlのホルムアルデヒド(138.3mmol、37重量%水溶液)を9.0gの4-t−ブチルフェノール(60.6mmol)と10.3mlのモルホリン(10.2g、118.2mmol)のジオキサン溶液60mlに加えた。得られた混合液を120℃の温度で一晩還流した。揮発性画分は減圧除去し、得られた固体をトルエン/水で抽出した。トルエン層を合せ、硫酸マグネシウム硫酸マグネシウム上で乾燥した。濾過後、黄色の溶液を減圧濃縮し、-24の温度で一晩保存した。大きな無色の結晶[LO1]Hが収率77%で得られた。この化合物の(1Hおよび13C{1H}NMR)の分光分析データは文献報告値と一致した。純度は元素分析で確認した。[LO1]Hはエーテル、塩素化溶剤および芳香族炭化水素に完全溶解し、脂肪族炭化水素にはわずかに溶ける。
【0070】
プロ−リガンド: 2,4-ジ-t−ブチル-6-(モルホリノメチル)−フェノール([LO2]H)
12.2gの2,4-ジ- t−ブチル−フェノール(59.1mmol)と、5.9mlのホルムアルデヒド(67.5mmol、水中37重量%)と、6.2mlのモルホリン(6.2g、70.9mmol)と黄色溶液を120℃の温度で一晩、90mlのジオキサン中で還流した。揮発油はポンプで排気し、得られた粘着性の固形物をトルエンおよびNaCl飽和水溶液で抽出した。有機層を合せ、MgSO4上で乾燥し、トルエンを排気するとオフホワイトの固形物が生ずる。それを減圧下に恒量乾燥して17.0gを得た(収率94%)。X線回折に適した単結晶[LO2]Hは +4℃の温度に一晩維持した濃縮ペンタン溶液から成長させて、その構造を決定した。それを[図1]に示す。
【0071】
元素分析:C19H31NO2(305.46グラム/モル):
理論値:C 74.71、H 10.23、N 4.59%
分析値:C 75.18、H 10.23、N 5.12%
【0072】
[LO2]Hは脂肪族炭化水素を含む全ての普通の有機溶媒に完全に溶ける。
【0073】
2,4- ジ-tブチル-6[1-アザ-15-クラウン-5]メチル]- フェノール([LO3]H)
1.03gの2,4-ジ-t−ブチル−フェノール(5.0mmol)、0.5mlのホルムアルデヒド(6.2mmol、37重量%水溶液)および1.25gの1-アザ-15-クラウン-5(5.7mmol)の混合液を20mlのジオキサン中で120℃の温度で24時間還流した。溶剤を減圧除去して得られるオレンジ油を恒量乾燥して2.23gの粗生成物を得る。移動層として純粋なクロロホルムを使用した薄層クロマトグラフィで精製して所望生成物を完全に精製した。クロロホルムを蒸発後、1.78gの化合物が81%の収率で得られた。[LO3]Hの分光分析データは公知の文献値と一致した。[LO3]Hは黄色の粘性のあるオイルで、全ての有機溶媒に充分に溶ける。
【0074】
二価金属ヘテロレプティック錯体の合成
[2,6-ビス(モルホリノメチル)-4-tBu-フェノキシ]亜鉛-エチル[[LQ1]ZnEt] の合成
3.5gの[LO1]-H(10.0mmol)のトルエン溶液75mlを-45℃の温度で20分かけて10.2mlのZnEt2(ヘキサン中1.0のM溶液、10.2mモル)のトルエン溶液125mlに加えた。得られた混合液を-45℃の温度で60分間攪拌し、さらに室温で2時間攪拌すると白い懸濁が生じる。沈降物を濾過分離し、減圧乾燥して分析的に純粋な4gの白い粉末を91%の収率で得た。{[LO1]ZnEt}2 C6H5の無色単結晶を濃縮ベンゼン溶液から室温で成長させ、その固体物理構造をX線結晶学で決定した。
元素分析:C22H36N2O3Zn(440.20グラム/モル):
理論値:C 59.79、H 8.21、N 6.34%
分析値:C 59.78)H 8.21、N 6.05%
【0075】
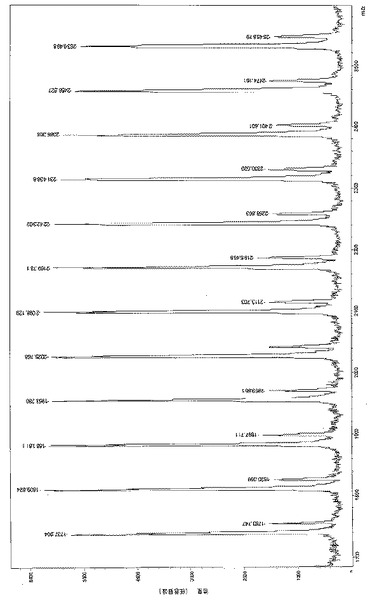
CD2Cl2またはC6D6中の[LO1]ZnEtの1H NMRスペクトルは複雑に見えたが、1−D(1H、13C{1H}および2−D(COSY、HMBCおよびHMQC)後、その1Hおよび13C(1H) NMR信号が得られた。NMR実験はトルエン-d8中で−60℃で実した。その固体構造を室温で濃縮ベンゼン溶液から成長させたX線品質結晶を使用して評価した。その結果、[LO1]ZnEtは[図2]に示すように2つの亜鉛原子がフェノキシ部分の酸素原子を介してブリッジしたダイマー種として存在することが示された。[LO1]ZnEtはエーテルおよびジクロロメタンに溶け、ベンゼンおよびトルエンに少し溶け、脂肪族炭化水素には溶解しない。
【0076】
[2,4-ジ-t−ブチル-6-(モルホリノメチル)-フェノキシ]亜鉛-エチル[[LQ2]ZnEt] の合成
1.06gの[LO2]-H、3.47mmol)のトルエン溶液20mlトルエン溶液を-25℃の温度で3.50mlのZnEt2(3.50mmol、1.0Mヘキサン溶液)のトルエン40mlに20分かけてゆっくり加える。さらに-25℃の温度で40分間攪拌し、アルカン除去後に無色溶液を得る。溶剤を蒸発させて得られた白い固形物を20mlのペンタンで3回洗浄し、減圧乾燥して1.18gの錯体を85%の収率で得た。
元素分析:C21H35NO2Zn(397.20グラム/モル):
理論値:C 63.23、H 8.84、N 3.51%
分析値:C 63.09、H 8.73、N 3.51%
【0077】
この錯体はTHFおよびジエチルエーテルに溶けるが、トルエンへの溶解性をは限られ、軽質石油エーテルには溶けない。
【0078】
[2.4-ジ-t−ブチル-6-(モルホリノメチル)-フェノキシ]マグネシウム-ブチル[[LQ1]MgBu] の合成
[LQ2]ZnEtの合成で記載したのと同じ手順で、0.94gの[LO1]H(2.70mmol)と3.0mlの MgBu2(3.50mmol、1.0Mヘプタン溶液)のトルエン22.0mlとの反応によって、[LQ1]MgBuを82%の収率で得た。
元素分析:C24H40N2O3Mg(428.90グラム/モル):
理論値:C 67.21、H 9.40、N 5.67%
分析値:C 67.32、H 9.89、N 6.19%
【0079】
この錯体はTHFおよびジエチルエーテルに溶け、トルエンには一定溶解性を有し、軽質石油エーテルに溶けない。
【0080】
[2,6-ビス(モルホリノメチル)-4-tBu-フェノキシ]カルシウム−[ビス(トリメチルシリル)アミド][[LO1]N(SiMe3)2の合成
1.32gの[LO1]H(3.79mmol)のTHF溶液20mlを室温で45分かけて1.71gのCa[N(SiMe3)2]2(THF)2(3.39mmol)のTHF溶液20mlに加えた。黄色の溶液を室温で一晩攪拌し、溶剤を減圧蒸発させると白い粉末が得られる。熱いヘキサン(ヘプタンまたは高級炭化水素も使用できる)での抽出を繰り返した後、溶剤を真空乾燥して77%の収率で分析的に純粋なヘテロレプティック(heteroleptic)な化合物が得られる。[[LO1]N(SiMe3)2の単結晶は室温でTHF溶液中にヘキサンをゆっくり拡散させて成長させて得た。その固体物理構造はX線回折で確認した。
元素分析:026H49N3O3Si2Ca(547.29グラム/モル:
理論値:C 56.99、H 9.01、N 7.67%
分析値:C 56.88、H 8.95、N 7.51%
【0081】
[[LO1]N(SiMe3)2の固体物理の構造は[図3]に示す。この図はこの化合物はフェノキシ部分の酸素原子を解してブリッジしたダイマー種の形で存在することを示している。
普通の有機溶媒への[[LO1]N(SiMe3)2の溶解性は脂肪族炭化水素でも良から優である。溶液中での安定正も非常に良く、NMRチューブでCeDe溶液中に5日間貯蔵した後でも劣化の兆候は見られない。しかも、そのヘテロレプティック性は溶液中でも保存される。すなわち、[[LO1]N(SiMe3)2のみでシュレンク-タイプの平衡無しに80℃でC6D6中で長時間反応させた後でも保存される。Ca[N(SiMe3)2および[[LO1]2Caが生じる兆候はない。
【0082】
本発明の本質的な特徴は、商用的な供給材料から出発して、リガンド[LO1]Hおよび[LO2]Hを完全合成でき、しかも、錯体の‖[LO1]ZnEt、[LO2]ZnEtおよび[LO1]JMgBuの合成を48時間で数グラムのスケールで分析的に純粋な化合物として得ることができる点にある。これに対して、効率的なLA、BBLまたはTMCのROPで使用される亜鉛-ベースの開始剤(BDI)ZnN(SiMe3)2の合成には2週間を要し、厳しい条件が必要である。
【0083】
連鎖移動剤の合成
1-(ベンジルオキシ)-2-フェニル-2-(2',2',6',6−テトラメチル-1'−ピペリジニルオキシ)エタン(AA−QHV)の合成
AA-OHの合成法を下記シェーマ3に示す。
【化6】
【0084】
下記文献に記載の公知の手順を変更して実行した。
【非特許文献69】Hawker et al. (C. J. Hawker, G. G. Barclay, A. Oreliana, J. Dao, W. Devonport, Macromotecules, 1996, 29, 5245-5254
【非特許文献70】Asri et al. (M. Asri Abd Ghani, D. Abdaliah, P. M. Kazmaier, B. Keoshkerian, E. Buncel, Can. J. Chem., 2004, 82, 1403-1412
【0085】
蒸留済みスチレン中のTEMPO溶液に、1.14当量の過酸化ベンゾイル(水中に75%)をゆっくり加えた。80℃の温度で30分間加熱すると、反応液は順次、赤、黄になり、最後に緑になる。揮発分を減圧除去し、緑のオイル材料からペンタンを加てて沈澱させて白い粉末が得られる。粉末を濾別後、溶剤を蒸発させると緑のオイルが得られる。それをメタノールに溶かす。-4℃の温度で再結晶して純粋なベンジル化物Aを55%の収率で得た。
【0086】
3.1gのこの化合物Aと15mlの2N−NaOH水溶液との混合をエチルアルコール中で3時間還流した。揮発性画分を蒸発させると油性の物質が得られる。ジクロロメタン/水で抽出後、合せた有機層はMgSO4を通して乾燥し、溶剤を減圧除去して恒量乾燥すると80%の収率でオレンジ油が得られる。この物質の特性のNMR分光(1H、13C(1H)およびCOSY実験)および元素分析の結果はAA−OHの予想組成および純度と一致した。
【0087】
環式エステルおよび環状カボネートの重合
トルエンまたはTHF中でのラクチド重合
一般的な重合手順をシェーマ4に示す。
【化7】
【0088】
1. [LO1]ZnEt
アルコールのトランスファー剤の存在下での上記の新規な開始剤[LO1]ZnEtを用いたLAのイモータルROPは極めて迅速であり、良く制御され、これに匹敵する公知のROP触媒系は文献にはない。重合条件および結果は[表1]にまとめた。
【0089】
【表1】
【0090】
[表1]での収率はメタノール中での沈殿後に測定し、理論的数平均分子量Mnは式[LA]o/[POH]o×モノマー変換率×MLA+MiproHを使用して計算した(ここで、MLA=144g/モルおよびMiproH=60g/モル)。MnGPCはゲル透過クロマトグラフィで対ポリスチレン標準品で決定し、mark-Houwinkファクタ0.58で修正した。Pmはラクチド単位のメソ結合確率で、CDCl3中、室温で記録したポリマーのホモデカップル1H−NMRスペクトルのメチン領域で決定した。
【0091】
[LO1]ZnEtを用いたアルコール不存在下のトルエン中でのL−LAの1000当量の重合([表1]の実施例1)は80℃では急速である(60分で転化率91%)であるが、制御性が悪く、PDIが1.50と多分散性の幅が広く、数平均分子量Mnの理論表し達と観測値の相関指数が悪い。しかし、60℃ではさらに遅く、制御性も非常に悪い(例えば1bis参照)。10当量のi PrOHを添加すると(例4)、ROPはアルコール無しより速くなり、制御性が良くなり、PDIは1.10になり、数平均分子量Mnの理論値と実験値とが良く一致する。さらに、これらの条件下では、PLLA4のホモデカップル1H−NMRスペクトルのメチン領域の調査値Pm = 1.00で示されるように、上記触媒系で予備実験しないで光学活性なモノマーを完全に重合できる。PLLAの後の指数4は実施例の番号を表す。
【0092】
実施例3と実施例4との比較から、[LO1]ZnEt /iPrOH系を用いた重合の最初の段階は、実施例3と他の条件は全く同じに時間を60分にした実施例4の結果から、Zn/アルコール比はが1:10では遅く、20分後に転化率は20%に過ぎないことが分かる。これはこの触媒の誘導期の存在を示す。
【0093】
[表1]全体から、アルコールの存在下ではLO1]ZnEtで極めて多当量のLLAを制御された重合できることが分かる。モノマー供給量を増加させた時の多分散性指数および分子量の予想値と観測値との間の相関も良〜優であり、L-LA/[LO1]ZnEt比は最高で50000にもなる。転化は定量的で、使用した条件下ではL-LA/[LO1]ZnEt比は500〜50000の間にある。さらに、トランスファー剤(一般にiPrOH)を非常に大過剰に使用できることは注目に値する。重合パラメータの制御性に目に見える有害な効果なしに、一つの金属センター当たりiPrOHを1〜1000当量の範囲で使用できる。本発明の触媒系は制御性と生産性との組合せを著しく良くする。
【0094】
トルエン中のモノマー濃度を最高6.0Mにして使用でき、モノマーの迅速な転化で芳香族溶剤中への完全な溶解が容易になる。転化率が高いので全てのモノマーが反応媒体中に溶解し、得られたポリマーは不溶性で高転化率で沈殿する。従って、反応の進化は速く、視覚で容易にモニターできる。
【0095】
実施例4(トルエン、97%転化)と実施例7(THF、57%転化)とを比較することによって分かるように、THFのようなコーディネート溶剤中でのLAのROPは、トルエンのような非コーディネート溶剤中より遅い。重合速度のこの減少は、溶剤をコーディネートする際に、モノマーによる金属中心上への配位が溶剤によって妨げられるためである。それにもかかわらず、THF中での重合は非常によく制御された状態に維持される。
【0096】
実施例6(トルエン、Pm = 0.50)と実施例8(THF、Pm = 0.45)に示すように、[LO1]ZnEt/iPrOH のZn/アルコール比1:10を用いた異性体(rac−LA)のラセミ混合物の重合は立体−選択性(stereo-selectiv)でない。
【0097】
単一の開始基の種類、すなわち[LO1]ZnEt/iPrOH触媒系中のイソプロポキシ基OCH(CH3)2-がNMR分光およびMALDI−TOFマススペクトル分析で示された。[図4]はL-LA/[L01]ZnEt/iPr0H(相対量100/1/10)で製造した低分子量PLLAの1H−NMRスペクトルを示す。-OCH(CH3)2 末端基の存在はδ5.06および1.24ppmでの信号で証明される。Et-または[LO1]−基によって開始されるPLLA鎖の存在は示されない。この解析結果は[LO1]ZnEt/iPrOHを用いて製造した[図5]に示す分子量4700g/モルおよび[図6]に示す分子量13200g/モルを有する2つのPLLAのMALDS−TOF質量スペクトル検査でも確認される。両方のスペクトルでイソプロポキシ-末端のPLLA鎖の分子量はGPC実験結果と理論値との間で非常に良く一致している。
【0098】
各スペクトルで2つのガウス分布が観測され、その第1および第2の2つの個体群はイオン化Na+およびK+に対応する[[図5(b)]および[図6(b)]に示すように2つの個体群間のΔ(m/z)]=16Da]。各ピークの分子量はオンマトリックス(on-matrix)化合物(H)(C6H8O4)n(OiPr)(Na)および(H)(C6H8O4)n(OiPr)(K)の計算された分子量と一致している(nは重合度である)。同じ個体群で各スペクトルの連続ピーク間に144Daの増加分が繰り返してみられ、これは一般に亜鉛錯体によって促進される望ましくないトランス-エステル化反応は、重合中でもモノマーが完全に転化した後でも、ないことを示す決定的な証拠である。従って、これらのポリマーは環状ポリマーを含まない。
【0099】
[表1]に示すように、iPrOHは[LO1]ZnEtを用いてLAの大過剰のイモータル制御されたROPでの極めて効率的な連鎖移動剤であることが証明されたが、本発明方法はiPrOHに限定されのものではなく、その代わりに他のアルコール、例えばベンジルアルコール、TEMPO−OH、HEMAまたは各種のヒドロキシ-アルコキシアミンを効率的に使用できる。このことは実施例4および実施例5に示されている。両方の場合で、重合パラメータの反応速度および制御性は同一である。従って、iPrOHを例えばAA−OHに完全に置換できる。
【0100】
2. [LO2IZnEt
重合条件および結果は[表2]にまとめた。
【表2】
【0101】
連鎖移動剤としてZnEtを用いたアルコール存在下での開始剤[LO2]を用いたLAのイモータル開環重合は極めて迅速かつ良く制御されて実行できる。。[表2]の実施例17で分かるように、10当量のiPrOHを用いたトルエン中での1000当量のLLAの重合は60℃の温度で60分以内に完了する。25当量のiPrOHを用いたトルエン中での5000当量のLLAは60℃の温度で90分でほぼ定量的に転化する。モノマーまたはアルコールの供給量が高くなると、重合パラメターの制御性にわずかな影響がでるが、多分散性指数はほぼ1.10に維持される。また、数平均分子量Mnの理論値と観測値はほぼ完全に一致する。[LO1]ZnEt錯体のROPでも述べたように、実施例16(反応時間=1分、転化なし)と実施例17(反応時間=60分、定量転化)との比較から明らかなように、[LO2]ZnEt/iPrOH触媒系でも少なくとも15分の賦活時間が存在することが明らかである。1000当量のrac−LAの重合はトルエン中(実施例18、完全転化)でもTHF中(実施例19、92%転化)でも非常に速い。両方のケースで制御性が良いが、後者の場合には分子量の予想値と観測値にわずかな差があた、分布がわずかに広い。この触媒系はシンジオタクチックPLAを形成する傾向をわずかに示し、それはトルエン中でのPm = 0.45よりTHF中でのPm = 0.40でより強くなる。
【0102】
3.[LO1]MgBu
重合条件および結果は[表3]にまとめた。
【表3】
【0103】
アルコールと組み合わせたMg錯体[LO1]MgBuはLAのROPの非常に効率的な開始剤を構成し、環式ジエステルの迅速かつ制御されたイモータル重合を促進する。例えば[表3]の実施例21に示すように、連鎖移動剤としての10当量のiPrOHは1000当量のLLAをほぼ定量的に重合し、Pm=1.00で示されるようにステレオセンターの目立つたepimerisation無しに非常によく制御されて重合は15分で完了する。
【0104】
1金属中心当たり1000〜5000当量の高い供給量にし、連鎖移動剤(ここではiPrOH)をMgに対して10〜100当量の大過剰にし、60〜80℃の温度で一般に2時間でLLAを重合した。全てのケースで多分散性指数は非常に狭く、1.11〜1.21の間にあり、分子量は予想値と実験値との相関が近似した。Zn-ベースの類似物[LO1]ZnEtおよび[LO2]ZnEtとは違って、[LO1]MgBuはモノマーの純度と連鎖移動剤の供給量にかなり敏感であった。60℃の温度でiPrOHの当量数を25(実施例24)から100(実施例例26)へ増やすと、同じ反応時間で、L-LA/[LO1]MgBu比を5000に固定した場合、活性がわずかに低下する。これはMgベース錯体は対応する亜鉛錯体と比較して高いオキソ毒性(oxophiiicit)を反映したものと考えられる。この反応速度の減少は反応温度を増やすことで部分的に補償できる。実施例25と実施例27を比較すると、5000当量のLLAを2時間で完全転化できないことがわかる。同様に、実施例24、28と実施例27、29とを比較することによって分かるように、連鎖移動剤を変えずにモノマー供給量を増加すると活性がかなり大きく低下する。これはモノマーをいかに完全に精製しても反応液中に存在する不純物に対するMg錯体の感度に起因する。
【0105】
アルコールおよび/またはLLAの量を増やすと反応速度が遅くなるのが観測されるが、分子量およびその分布はこれらのプロセスによってほとんど変化しない。従って、これらの現象はモノマーに対して不活性な種が同時に生成して触媒を部分的に失活させる結果であることを示唆している。観測事実とは逆に2つの活性種の存在が多分散性指数の広がりに有意な結果をもたらすと思われる。
【0106】
実施例22のPm=0.54が示すように、錯体[LO1]MgBuはトルエン中でのrac−LAの重合中に立体制御を殆どしない。THF中ではシンジオタクチックの方へのわずかな傾向が観測され、実施例23のPmは0.41である。[LO1]MgBuのステレオ選択性は非常に制限されるがトルエン中で逆方向へ生じ、メゾ二回対称軸性(diads)か観測され、THF中ではrac二回対称性が強くなることを強調しなければならない。実施例21と実施例22とを比較することで、LLAおよびrac−LAのROP中に活性種に起因する重合速度および品質は実質的に同一である。連鎖移動剤としてはiPrOH、BnOH、TEMPO−OH、AA−OH、HEMのような広範囲のアルコールを使用して[LO1]MgBuでLAの制御されたイモータルROPで種々の末端官能化したPLAを合成できる。L-LA/[LO1]MgBu/iPrOH(相対比5000/1/100)を用いて、5000当量のL-LAのイモータルのROPによってイソプロポキシ末端を付けたPLLAサンプルを製造できる。
【0107】
得られた低分子量PLLAのMALDI−TOFマススペクトルを[図7a]に示す。全てのポリマー鎖が-Oi/Pr単位で終っていることが明らかに示し、これはこの系でのROP機構の本当のイモータル性を決定的に証明している。[図7b]に示すように、不均一な強度の2つの個体群を全ガウス分布を通じた一連のピーク間の72Daの増加分はエステル転移反応プロセスがMg錯体による触媒作用でLAの重合中にかなりの範囲で起こることを示している。
【0108】
[LO1]CaN(SiMe3)2
重合条件および結果は[表4]にまとめた。
【表4】
【0109】
LAのイモータル重合はトルエン中またはTHF中で触媒系[LO1] CaN(SiMe3)2 /ROHを用い、ROHをiPrOH、BnOH、TEMPO−OH、AA−OH、HEMAの中から選択することで効率的に促進された。これは溶液および固相の両方で制御された状態で多量のモノマーを重合することを可能にするヘテロレプティックなフェノキシ-ベースのカルシウム錯体の最初の例であることを示している。
【0110】
アルコールが存在しない場合(実施例30)では、開始剤[LO1] CaN(SiMe3)2はアルコールが存在する場合に実行される他の実施例と比較して遅くなる。10〜100当量のiPrOHの存在下では、L-LAの最大2500当量を60℃の温度で重合できる。例えば、実施例33に示すように、iPrOH/[LO1]CaN(SiMe3)2比>10の場合、500当量のモノマーは1分以内に定量的に転化される。実施例35〜37に示すように、同じ条件下で1000当量のモノマーを15分以下で変換できる。多分散性指数が1.20〜1.40の範囲にあることから重合は良く制御されており、iPrOH/[LO1]CaN(SiMe3)2比が50の場合でも、分子量の実験値と理論値は近似している。多分散性指数が1.6〜1.7へ広がるのが観測されるのは、モノマーの完全転化後に反応を長く続けて、望ましくないエステル転移反応プロセスを起こすときであることを触れておく。
【0111】
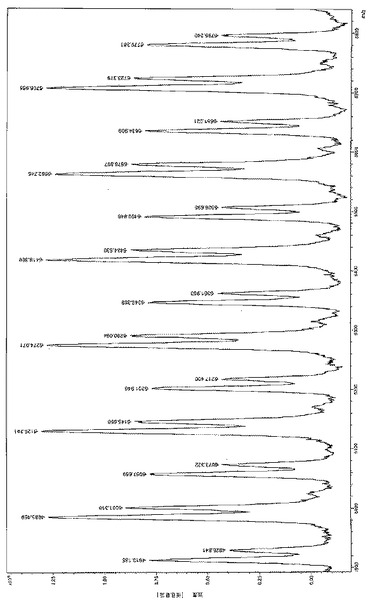
触媒[L01]CaN(SiMe3)2/iPr0Hは低温で反応速度および制御性の両方で非常に性能が良い。従って、実施例40に示すように、500当量のモノマーは室温で1分後には70%が転化され、実施例33に示すように、60℃の温度では完全に添加される。トルエン中でのrac-LAの重合(実施例41)はL−LA(実施例33)のそれよりかなり遅く、立体制御性がなく、Pm = 0.50である。THF中で実行する(実施例42)と、rac-LAの重合はトルエン中よりも遅くなり、立体選択性もなく、Pm=0.50である。単一開始剤のグループ、すなわち触媒系[LO1]CaN(SiMe3)2/iPrOHdの−イソプロポキシ基OCH(CH3)2の種類はNMR分光およびMALDi−TOFマススペクトル分析で示された。低分子量のPLLAの1H NMRスペクトルは、実施例34に示すように、L-LA/[LO1]CaN(SiMe3)2/iPrOH(相対量は500/1/25)で得られ、(OCH(CH3)2の末端基は[図8]に示すようにδ55.06(OCH(CH3)2)および1.24 OCH(CH3)2)PPmの特徴的信号によって確認され、-N(SiMe3)2または[LO1]−基にり始まるPLLA鎖の存在は示されなかった。
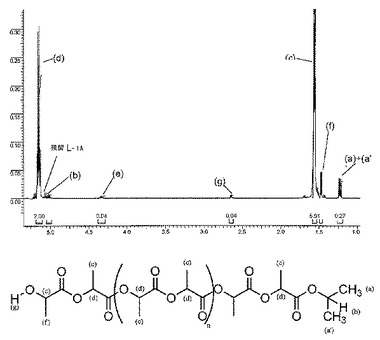
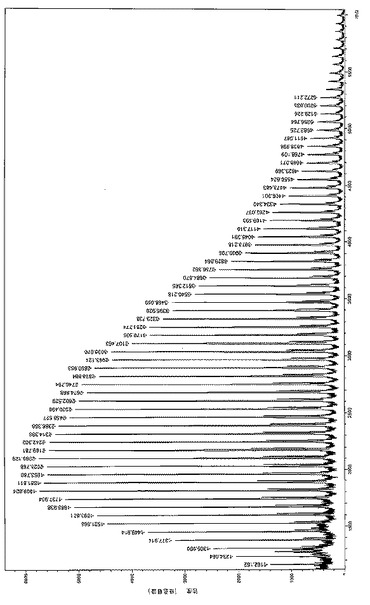
【0112】
この解析は、[図9]に示すように、上記PLLAサンプルの高解像度MALDI−TOF質量スペクトル検査でも更に確認した。イソプロポキシ-末端を有するPLLA鎖の理論分子量とGPC実験結果とは非常に良く一致した:2つのガウス分布が観測された(それぞれNa+およびK+ のイオン化に対応した第1および第2の個体群[2つの個体群環のΔ(m/z)=16Da]が観察され、各ピークはオンマトリックス化合物(H)(C6H8O4)n(OiPr)(Na)および(H)(C6H8O4)n(OiPr)(K)の分子量(ここで、nは重合度)と良く一致する。この事実は個体群(Na+に対応する主個体群)で連続ピーク間の増加分が144Daでなく72Daのみであるということは重合中にカルシウム錯体によって望ましくないトランスエステル反応化合物が極めて迅速に促進されたことを強く証明している。これはモノマー完全転化後に反応を続けることでPLLAの多分散性指数が大きく広がることを示した上記観察と一致している。
【0113】
iPrOH存在下での開始剤の比較
トルエン中でiPrOHを使用した大量のL-LAのイモータルROPでの開始剤(BDI)ZnN(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuおよび[LO1]CaN(SiMe3)2の有効度を決定するための一連の実験を実施した。重合条件および結果は[表5]にまとめてある。
【0114】
【表5】
【0115】
[表5]に示すように、テストした全ての金属開始剤で、重合パラメータの制御性は常に良〜優であり、多分散性指数はの狭く、分子量は理論値と観測値でよく一致した。従って、ここでの触媒系の比較は単に重合速度をベースにしたものである。
【0116】
L−LA/[M]/iPrOH比が1000/1/10を用いて60℃の温度で15分間の実施した実施例43〜47は、錯体 [LO1]ZnEtおよび[LO2]ZnEtは他の3つの前駆体よりはるかに活性が低いように見え、これらは活性増加の基準では以下のようにランク付けられる:[LO2]ZnEt<<[LO2]ZnEt<<(BDI)ZnN(SiMe3)2=[LO1]CaN(SiMe3)2 <[LO1]MgBu。上記条件下での[LO2]ZnEtと[LO2]ZnEtの効率不足および短い反応時間は、これら2つの前駆体をベースにした触媒は[表1]および[票2]で観測されたように、10〜15分の賦活時間を必要とするという事実を反映していると言える。反応時間をより長くし、モノマー供給量を高くすると効率は異なったものになる。例えば、5000/1/25のL-LA/[M]/iPrOHの場合、60℃の温度、90分での効率順番は[LO1]MgBu < [LO1]ZnEt 〜 [LO2]ZnEt 〜(BDi)ZnN(SiMe3)2になる。亜鉛錯体では完全転化し、[LO1]MgBu錯体では80%である。これはZn-ベースの類似物に比べたMgベース錯体の特徴である高い感度を反映している。
【0117】
3つのZn-ベースの触媒系を区別するために、3つの全ての触媒でモノマーの完全転化前の60分後に反応を故意に止めた。以下の結果が観測された:[LO2]ZnEt(45%)< [LO1]ZnEt(71%)<(BDI)ZnN(SiMe3)2(95%)。これは実施例48〜50に示されている。
【0118】
結論として以下のことが言える:
(1) 連鎖移動剤の最大100〜500当量用いてLAを数万当量の量でイモータルROPする場合の最高の候補はZn-ベースの開始剤である。
(2) 条件が全く同じ場合、[LO1]ZnEtが[LO2]ZnEtより効率的である。
(3) 重合速度、[LO1]ZnEt(および[LO2]ZnEt)を活性化するために必要な誘導時間に対して有害効果は反応時間が90分以上では無視できる。
【0119】
第2の実験シリーズは、実施例例55〜60に示すように、最も有望な開始剤である[LO1]ZnEtと(BDI)ZnN(SiMe3)2に焦点をあてて、LLAのイモータルのROPを工業的な実験条件下で実行したものである。両方の実験セットで20000当量のモノマーをそれぞれ100当量のアルコール(実施例57、57bis、58および59)および250当量ののアルコール(実施例55および56)を用いて重合した。結果は、この条件下では活性の順番が逆になることを示した:(BDI) ZnN(SiMe3)2 <[LO1]ZnEt。しかし、両方とも実施例57bisの単純なZnEt2前駆体よりはるかに優れたものであった。
【0120】
全く驚くことであるが、[LO1]ZnEt/iPrOH=1/250を用いて60℃で50000当量のLLAをイモータルROPしても、非常に制御性がよく、8時間(エントリー59)とほぼ同程度に定量的であった。一方、500当量のアルコールの存在下(エントリー60)では16時間後に完全な転化が達成された。LAのROPでの[LO1]ZnEt錯体によって提供される範囲は大きなもので、この系でこれまでに達成された最大の性能である。
【0121】
スチレン中のラクチドの重合
反応階段はシェーマ5に、重合条件および結果は[表6]に示してある。
【0122】
【化8】
【表6】
【0123】
この表でMnの理論値は [LA]0/[ROH]0×モノマー転化率×MLA+MTEMPO−OHで計算した。MLA=144グラム/モル、MTEMPO−OH=162グラム/モル。
【0124】
iPrOH、BnOH、TEMPO−OH、AA−OH、HEMAから選択される広範囲の連鎖移動剤で、本発明の金属開始剤は[表6]に示すように、スチレン中のLAのイモータルROPに適している。例えば、1000当量のL-LAのスチレン中での重合はよく制御された状態でスチレンの重合干渉なしに進み、15分以内に完了する。そのため、この系は官能化PLLAの大規模合成およびその後のポリ(LA−ブロック-スチレン)コポリマーの製造に適している。
【0125】
ベンジルアルコールの存在下でのトリメチレンカボネート(TMC)のバルク重合
反応階段はシェーマ6に、重合条件および結果は[表7]に示した。
【0126】
【化9】
【表7】
【0127】
この表でMnの理論値は[LA]0/[ROH]0×モノマー転化率×MLA+MTEMPO−OHで計算した。MLA=144グラム/モル、MTEMPO−OH=162グラム/モル。
【0128】
iPrOH、BnOH、TEMPO−OH、AA−OH、HEMAから選択される広範囲の連鎖移動剤で、本発明の金属開始剤はバルクモノマー中でのTMCのイモータルROPに適している。
【0129】
例えば、5〜20当量のベンジルアルコールの添加で、[LO1]ZnEtは実施例例66、68および70に示すように60℃の温度で2、3時間以内に最大で25000当量のTMCを完全に転化できる。このタイプのTMCの亜鉛促進ROPの一般的な多分散性指数は1.50〜1.65で、分子量の観測値と計算値との相関は非常によい。
【0130】
制御性、活性および生産性に関しては、[LO1]ZnEtは(BDI)ZnN(SiMe3)2 より優れ、実施例68および実施例70に示すように、前者は10000〜25000当量のTMCを完全に転化できるが、後者は実施例67および実施例69に示すようにモノマーのそれぞれ89%および75%を変えるだけである。これによって二元系[LO1]ZnEt/BnOHは、欧州特許出願第EP-08 290 187.7号公報に記載の系(BDI)ZnN(SiMe3)2よりはるかに優れた、TMCのイモータルROPに対して最大の活性および生産性のある触媒系となる。さらに、この新規な[LO1]ZnEt/BnOH触媒は、既に述べたように、重合の制御性か良く、PDIの幅が狭く、Mnの実験値と理論値とが良くマッチする。
【0131】
驚くことに、100の当量のBnOHの添加で、実施例71に示すように、60℃の温度で48時間で最高100000当量のTMCをほぼ完全に転化できる。得られたポリマーPDIの幅は狭く、実験値と理論値は完全に一致した。実施例72に示すように、110℃まで加熱すると反応時間は有害効果無しに8時間に短縮できる。
【0132】
スチレン中でのトリメチレンカボネート(TMC)の重合
反応階段はシェーマ7に、重合条件および結果は[表8]に示した。
【0133】
【化10】
【表8】
【0134】
TMCは(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、溶剤からの目立った有害効果無しにスチレン中で制御されたイモータル重合される。5000当量のTMCを完全転化でき、分子量は理論値と実験値で一致した。但し、多分散性指数は類似条件下でLAのROPで観測されたもの(1.20の1.40)より大きくなり、一般に1.8〜1.9になる。
【0135】
ベンジルアルコール存在下での各種6-員カボネートの隗重合
反応階段はシェーマ8に、重合条件および結果は[表9]に示した。
【化11】
【0136】
官能化した6員環カボネートを、[表9]に示すように、(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、制御されたイモータル重合した。
【0137】
【表9】
【0138】
5当量の連鎖移動剤の存在下で500当量のモノマーを15〜60分以内に完全に転化できた。多分散性指数は一般に1.20〜1.70と狭く、分子量の観測値と計測値は近似した。ここでは分子量は対ポリスチレンで直接与えられるので、Mark-Houwink係数は修正のために加えなかった。
【0139】
β-ブチルラクトンラセミ体の重合
反応階段はシェーマ9に、重合条件および結果は[表10]に示した。
【0140】
【化12】
【表10】
【0141】
rac-BBLの隗重合を、[表10]に示すように、(BDI)Zn-N(SiMe3)2、[LO1]ZnEt、[LO2]ZnEt、[LO1]MgBuまたは[LO1]CaN(SiMe3)2から選択される開始剤と、iPrOH、BnOH、AA−OH、HEMAおよびTEMPO−OHの中から選択されるアルコールとを用いて、制御されたイモータル重合した。
10当量のiPrOHの添加で、[LO1]ZnEtは200〜500当量のrac-BBLを定量的に数時間以内に容易に変換した。得られたポリマーのPDIは非常に狭く、一般に約1.10であり、分子量(GPCまたはMALDl-TOF質量分析で決定)は実験値と理論値とで極めて良く一致した。実施例84および実施例85に示すように、公知の(BDi)Zn-N(SiMe3)2と比較して、[LO1]ZnEtは活性および制御性の両方に優れている。
【特許請求の範囲】
【請求項1】
式[LO]−M−Xの二価金属錯体:
{ここで、
Mは周期律表の第2族または第12族から選択される、
XはヒドロカルビルまたはアルコキシドOR''であり[ここで、R''はヒドロカルビル、アリール、シリルまたはアミノ基NR*2であり(ここで、R*はSiMe3、イソ−プロピル、メチルまたはエチルである)]、
[LO]は2−R1、4〜R2、6−R3−C6H2Oであるか、下記の式で表され:
【化1】
[ここで、
R1
【化2】
であり(ここで、mは1、2または3であり、nは≧1である)、
R2は1〜10の炭素原子を有するヒドロカルビル基であり、好ましくはメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択され、
R3はR1と同じか、1〜20の炭素原子を有するヒドロカルビル基、好ましくはメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されアルキルか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルの中から選択されるアリールである]}
【請求項2】
MがZn、Mg、Ca、SrまたはBa、好ましくはZnまたはMgまたはCaから選択される請求項1に記載の金属錯体。
【請求項3】
Xがメチル、エチル、n−ブチル、フェニルまたはアミド基、例えばN(SiMe3)2、NMe2、NEt2、NZPr2またはアルコキシド基、例えばOEt、OiPr、OtBu、OCH2Ph、OSiPh3の中から選択される請求項1または2に記載の金属錯体。
【請求項4】
Xがエチル、n−ブチルまたはN(SiMe3)2の中から選択される請求項3に記載の金属錯体。
【請求項5】
R2がメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択される請求項1〜4のいずれか一項に記載の金属錯体。
【請求項6】
R3がメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されるアルキルであるか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルから選択されるアリールである請求項1〜5のいずれか一項に記載の金属錯体。
【請求項7】
前駆体MX2を下記式のプロ−リガンドと反応させる、請求項1〜6のいずれか一項に記載の金属錯体の製造方法:
【化3】
(ここで、M、X、R1、R2、R3は上記定義のもの)
【請求項8】
キレート用フェノキシリガンドで担持された請求項1〜7のいずれか一項に記載の二価金属錯体を含む系とアルコールまたはポリオールとの存在下で、ラクチド、環式エステルおよび5−、6−または7−員環の環状カボネートを制御されたイモータル開環重合する方法。
【請求項9】
アルコールまたはポリオールの量が金属に対して1〜10,000当量である請求項8に記載の方法。
【請求項10】
アルコールが、R'OHである(ここでR'は一級または二級アルキル残基であるか、ベンゾイック基であり、好ましくはiPrまたはベンジルである請求項8または9に記載の方法。
【請求項11】
上記環式エステルがL−ラクチド(LLA)、rac−ラクチド(rac−LA)またはrac−β−ブチロラクトン(rac−BBL)の中から選択され、上記環式カボネートがTMCおよびその下記置換誘導体の中から選択される請求項8〜10のいずれか一項に記載の方法:
【化4】
【請求項12】
アルコールが官能化されており、開環重合がスチレンで実行される、末端官能化されたポリマーを製造するための請求項8〜11のいずれか一項に記載の方法。
【請求項13】
上記の官能化されたアルコールがTEMPO−OH、HEMAまたはヒドロキシ−アルコキシアミン、例えばAA−OHのから選択される製造12に記載の方法。
【請求項14】
請求項12または請求項13に記載の末端が官能化されたポリマーの、ラクチドまたはTMCとスチレンのコポリマーのインサイチュー(in situ)製造での使用。
【請求項15】
請求項8〜13のいずれか一項に記載の方法で得られるポリマーまたはコポリマー。
【請求項1】
式[LO]−M−Xの二価金属錯体:
{ここで、
Mは周期律表の第2族または第12族から選択される、
XはヒドロカルビルまたはアルコキシドOR''であり[ここで、R''はヒドロカルビル、アリール、シリルまたはアミノ基NR*2であり(ここで、R*はSiMe3、イソ−プロピル、メチルまたはエチルである)]、
[LO]は2−R1、4〜R2、6−R3−C6H2Oであるか、下記の式で表され:
【化1】
[ここで、
R1
【化2】
であり(ここで、mは1、2または3であり、nは≧1である)、
R2は1〜10の炭素原子を有するヒドロカルビル基であり、好ましくはメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択され、
R3はR1と同じか、1〜20の炭素原子を有するヒドロカルビル基、好ましくはメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されアルキルか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルの中から選択されるアリールである]}
【請求項2】
MがZn、Mg、Ca、SrまたはBa、好ましくはZnまたはMgまたはCaから選択される請求項1に記載の金属錯体。
【請求項3】
Xがメチル、エチル、n−ブチル、フェニルまたはアミド基、例えばN(SiMe3)2、NMe2、NEt2、NZPr2またはアルコキシド基、例えばOEt、OiPr、OtBu、OCH2Ph、OSiPh3の中から選択される請求項1または2に記載の金属錯体。
【請求項4】
Xがエチル、n−ブチルまたはN(SiMe3)2の中から選択される請求項3に記載の金属錯体。
【請求項5】
R2がメチル、エチル、イソプロピル、tert-ブチルまたはネオペンチルの中から選択される請求項1〜4のいずれか一項に記載の金属錯体。
【請求項6】
R3がメチル、エチル、イソプロピル、tert-ブチル、ネオペンチル、クミル、トリチルの中から選択されるアルキルであるか、フェニル、2,4,6−トリメチルフェニル、2,6−ジイソプロピルフェニルから選択されるアリールである請求項1〜5のいずれか一項に記載の金属錯体。
【請求項7】
前駆体MX2を下記式のプロ−リガンドと反応させる、請求項1〜6のいずれか一項に記載の金属錯体の製造方法:
【化3】
(ここで、M、X、R1、R2、R3は上記定義のもの)
【請求項8】
キレート用フェノキシリガンドで担持された請求項1〜7のいずれか一項に記載の二価金属錯体を含む系とアルコールまたはポリオールとの存在下で、ラクチド、環式エステルおよび5−、6−または7−員環の環状カボネートを制御されたイモータル開環重合する方法。
【請求項9】
アルコールまたはポリオールの量が金属に対して1〜10,000当量である請求項8に記載の方法。
【請求項10】
アルコールが、R'OHである(ここでR'は一級または二級アルキル残基であるか、ベンゾイック基であり、好ましくはiPrまたはベンジルである請求項8または9に記載の方法。
【請求項11】
上記環式エステルがL−ラクチド(LLA)、rac−ラクチド(rac−LA)またはrac−β−ブチロラクトン(rac−BBL)の中から選択され、上記環式カボネートがTMCおよびその下記置換誘導体の中から選択される請求項8〜10のいずれか一項に記載の方法:
【化4】
【請求項12】
アルコールが官能化されており、開環重合がスチレンで実行される、末端官能化されたポリマーを製造するための請求項8〜11のいずれか一項に記載の方法。
【請求項13】
上記の官能化されたアルコールがTEMPO−OH、HEMAまたはヒドロキシ−アルコキシアミン、例えばAA−OHのから選択される製造12に記載の方法。
【請求項14】
請求項12または請求項13に記載の末端が官能化されたポリマーの、ラクチドまたはTMCとスチレンのコポリマーのインサイチュー(in situ)製造での使用。
【請求項15】
請求項8〜13のいずれか一項に記載の方法で得られるポリマーまたはコポリマー。
【図1】

【図2】
【図3】
【図4】
【図5a】

【図5b】
【図6a】
【図6b】
【図7a】
【図7b】
【図8】
【図9a】
【図9b】
【図2】
【図3】
【図4】
【図5a】
【図5b】
【図6a】
【図6b】
【図7a】
【図7b】
【図8】
【図9a】
【図9b】
【公表番号】特表2012−525357(P2012−525357A)
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2012−507764(P2012−507764)
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/EP2010/055794
【国際公開番号】WO2010/125138
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(504469606)トータル・ペトロケミカルズ・リサーチ・フエリユイ (180)
【出願人】(505252333)サントル・ナシヨナル・ド・ラ・ルシエルシユ・シヤンテイフイク (24)
【Fターム(参考)】
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/EP2010/055794
【国際公開番号】WO2010/125138
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(504469606)トータル・ペトロケミカルズ・リサーチ・フエリユイ (180)
【出願人】(505252333)サントル・ナシヨナル・ド・ラ・ルシエルシユ・シヤンテイフイク (24)
【Fターム(参考)】
[ Back to top ]