
環状アルファ−コノトキシンペプチド
本発明は、遊離N末端およびC末端を有さないようなアミド環状骨格を有する少なくとも1種の合成環状α-コノトキシンペプチドを、医薬として経口または経腸投与に適したビヒクル中に含む経口または経腸医薬製剤であって、前記ペプチドは、ニコチン性アセチルコリン受容体を抑制する能力を有し、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端はペプチドリンカーを介してC末端に連結されている、医薬製剤に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はα-コノトキシンペプチドに関し、詳細にはヒトの治療的処置に有用な環状α-コノトキシンペプチドに関する。本発明は特に、これらのペプチドを含む経口および経腸製剤、医薬製剤の製造におけるこれらのペプチドの使用、およびヒトの状態または疾患の予防または治療におけるこれらの医薬製剤の使用に関する。
【背景技術】
【0002】
Conus属(イモガイ)のマリンスネイル(marine snail)は、巧みな生化学的戦略を使用して獲物を捕獲する。魚、虫、または他の軟体動物の捕食者として、イモガイは小さな生物活性ペプチドのカクテルを含有する毒液を獲物に注入する。これらの毒素分子はコノトキシンと呼ばれ、種々のイオンチャネルまたは受容体を標的にすることによって神経伝達を阻害する。コノトキシンは、通常、直鎖状に配列された12〜30個のアミノ酸を含有する。Conus属の任意の1種の毒液は、100種を超えるさまざまなペプチドを含有しうる。コノトキシンはその生理学的標的に基づいて分類される。例えば、α-コノトキシンおよびψ-コノトキシンはニコチン性ACh受容体を標的として神経節および神経筋遮断を引き起こし、一方ω-コノトキシンクラスのペプチドは電位感受性Ca2+チャネルを標的として神経伝達物質放出を抑制する。
【0003】
ほとんどのコノトキシンペプチドは、2個または3個のジスルフィド結合をそれぞれ形成するように対になって結合した4個または6個のシステイン残基を含有するが、1個のジスルフィド結合を形成するように結合した2個のシステイン残基を有する例(すなわちコノプレシン)に加えて、3個を超えるジスルフィド結合を有するものもあれば、システイン残基またはジスルフィド結合を含有しないものもある。前述の「活性」クラスのペプチドの一部は、構造モチーフが共通であり、同数のシステイン残基および同じジスルフィド結合連結性を有している。このため、新しい「スーパーファミリー」分類体系が展開された。例えば、ω-コノトキシンならびにδおよびμ-コノトキシンクラスのメンバーは、システイン残基IとIV、およびIIとV、およびIIIとVI(6個のローマ数字はN末端から番号付けした6個のシステイン残基を表す)間に3個のジスルフィド結合を形成するように対になって結合した6個のシステイン残基を有する。この構造モチーフを有するコノトキシンペプチドはコノトキシンのO-およびM-スーパーファミリーに属する。同様に、ρ-コノトキシンおよびほとんどのα-コノトキシンは、システイン残基IとIII、およびIIとIV間に2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を有する。これらのコノトキシンペプチドは、コノトキシンの「A-スーパーファミリー」に属する。本発明は、A-スーパーファミリーのα-コノトキシン、すなわちシステイン残基IとIII、およびIIとIV間に形成された2個のジスルフィド結合を有するα-コノトキシンペプチドに関する。前述したように、コノトキシンペプチドは哺乳動物のさまざまな異なるイオンチャネル受容体に結合するので、したがって、ヒトの疼痛軽減を含めてさまざまな治療に応用できる可能性を有している。しかしながら、一般にペプチドは、その薬剤としての使用に伴い、一般にバイオアベイラビリティが低いこと、プロテアーゼによる切断の受けやすさ、および望ましくない副作用を含むいくつかの問題を有している。
【0004】
1種のω-コノトキシン、MVIIA(SNX-111、ZiconotideおよびPrialtとしても知られる)は最近、癌、AIDSおよび神経障害に伴う難治性疼痛の治療に関して米国食品医薬品局の承認を取得した。投与経路は、いくつかの前述の問題のため、およびこの薬剤が標的とする受容体がCNS内に位置するために、現在のところ脊髄への髄腔内注入に制限されている。
【0005】
臨床試験を開始した他のω-コノトキシンは、Ziconotideと比較してP/Q型チャネルに対するN型カルシウムチャネルへの選択性が向上したと報告されているCVID(AM-336、Zenyth Pharmaceuticals)である。しかしながら、特定の受容体/チャネルへの選択性が向上しても、ωクラスのコノトキシンは一部の患者において望ましくない副作用をやはり伴う。2種の他のコノトキシンペプチド(アンボイナ(Conus geographus)から単離されたCGX-1160およびCGX-1007)も臨床試験中であるが、これらのペプチドの投与も髄腔内経路に制限されている。疼痛に関して研究されている他のペプチドは、χ-クラスのコノトキシン(Xen 2174、Xenome社)である。しかしこの場合も、このペプチドの投与は髄腔内経路に制限されている。
【0006】
薬剤の髄腔内または脊髄投与の主な不利点は、投与が医師または看護師によって行われなければならないこと、および長期にわたる治療のために、機械的送達システムおよび脊髄カテーテルを患者の脊柱に挿入することが好ましいことである。したがって、このような治療は通常、末期および/または入院患者にのみ用いられる。
【0007】
α-コノトキシンペプチドは、典型的には大きさが12個から16個のアミノ酸であり、アミド化されたC末端を通常有するコノトキシンのサブファミリーである。α-コノトキシンは、ニコチン性アセチルコリン受容体を抑制することが知られている。α-コノトキシンは、鎮痛および中毒において役割を果たすだけでなく、アルツハイマー病、統合失調症、うつ病および小細胞肺癌を含むさまざまな障害に関与している筋肉とニューロンのニコチン性アセチルコリン受容体(nAChR)の両方と相互作用する。α-コノトキシンペプチドおよびその使用の可能性は、文献において広く記載されている。(LloydおよびWilliams、2000年、J Pharmacol Exp Ther 292 (2) 461頁参照。)
【0008】
システイン残基間の残基数を使用して、異なるクラスのα-コノトキシンを識別する。第2と第3システイン残基の間(ループ1)および第3と第4残基の間(ループ2)の残基数に基づいて、α-コノトキシンはα3/5、α4/3、α4/4、α4/6およびα4/7構造サブファミリーに分類される。4/7ループ配列を有するα-コノトキシンペプチドの2つの例は、MIIおよびVc1.1(Metabolic Pharmaceuticals社、ACV1としても知られる)である。最も小さいα-コノトキシンペプチドの1つは、4/3ループ配列を有するIM1である。
【0009】
さまざまなサブタイプのニコチン性アセチルコリン受容体への選択性、特に、中枢サブタイプに対する末梢nAChRサブタイプへの選択性を解明するために、いくつかのα-コノトキシンペプチドが研究されてきた。nAChRは、CNSおよびPNS全体にわたって低レベルで発現されているが、各種サブタイプは異なる分布を有する。哺乳動物のnAChRはサブユニットの組合せからなっている。これらのサブユニット型の7種はリガンド結合に関与する主な構成要素(α2、α3、α4、α6、α7、α9およびα10)であり、4種のサブユニット型(α5、β2、β3およびβ4)は、受容体に機能的および薬理学的特性を付与する構造であると考えられている。異なるサブタイプがさまざまな形で(一般にヘテロ五量体として)結合し、特定の薬理学的および電気生理学的特性を有する受容体を形成する。α3サブユニットは(PNSに存在することから)末梢サブユニットであると考えられ、一方α7サブユニットはCNSで優勢なサブユニットであると考えられている。
【0010】
α-コノトキシンVc1.1は、ビクトリアジョウオウイモ(Conus victoriae)の毒管由来cDNAのPCRスクリーニングを使用して最初に発見された(Sandallら、Biochemistry、2003年、42、6904頁)。Vc1.1配列内のシステインの間隔は、Vc1.1が、広く研究されているコノトキシンMII、EpIおよびPnIBを含むα-コノトキシンの4/7サブクラスのメンバーであることを示している。Vc1.1の3次元構造は、小さな膜貫通α-へリックス残基P6からD11を含み、他のα-コノトキシンで見られるI-II、III-IVジスルフィド連結性により支持されている(Clarkら、J. Biol. Chem.2006年、281、23254頁)。ほとんどのα-コノトキシンに共通するアミド化されたC末端に加えて、直鎖Vc1.1のPro6およびGlu14残基をそれぞれヒドロキシプロリンおよびγ-カルボキシグルタミン酸に翻訳後修飾することも可能である。この翻訳後修飾されたVc1.1アナログは、神経再生に関与しているが疼痛には関与していない。(WO02/079236)
【0011】
ウシクロム親和細胞中のニューロンnAChRのアンタゴニストである直鎖Vc1.1は、ヒト神経因性疼痛の3種のラットモデルにおいて神経因性疼痛を軽減し、損傷したニューロンの機能回復を促進することが示されている(Satkunanathanら、2005年、Brain Research 1059 (2) 149〜158頁)。加えて、糖尿病性神経障害の動物モデルにおいてACV-1が神経因性疼痛の軽減に有効であることがLivettらによって報告されている。特に、末梢神経障害のストレプトゾトシン誘導糖尿病ラットモデルにおいて、ACV-1の抗異痛作用が30および300μg/kgの用量で投薬1時間以内に観察された(IDrugs、15th World Congress on Animal、Plant and Microbial Toxins、2006年 9、679〜681頁)。鎮痛剤として、Vc1.1はZiconotideより活性が高いことが報告されている(Sandallら、2003年、Biochemistry、42、6904〜6911頁)。より最近では、Vc1.1は、ヒト末梢神経の遊離切片において、ニコチンに誘導される無髄C線維軸索の軸索興奮性の増大を弱めることが示された(Langら、2005年、Neuroreport 16、479〜483頁)。前述したように、ニューロンnAChRは、中枢および末梢神経系全体で見られるα(α2〜α10)およびβ(β2〜β4)サブユニットの組合せからなる五量体のリガンド依存性イオンチャネルである。電気生理学的および免疫組織化学的データは、無髄C線維の軸索における、α4、β2またはα7ではなくα3、α5およびβ4サブユニットからなるnAChRの機能発現を示す(Langら、2005年、Neuroreport 16、479〜483頁;およびLangら、2003年、Neurophysiol 90、3295〜3303頁)。無髄末梢神経線維でのnAChR遮断は、無髄交感神経および/または感覚軸索に対して鎮痛作用を有する場合がある。興味深いことに、翻訳後修飾された合成Vc1.1(ptm Vc1.1)は、クロム親和細胞においてニューロン型ニコチン反応を抑制しないことが報告され、2つのラット神経因性疼痛試験において不活性であった。直鎖Vc1.1またはACV1は、第2相ヒト臨床試験を開始している。ニコチン作動薬は鎮痛作用を有することが以前に報告されている。このようなニコチン作動薬の例は、エピバチジンおよびABT-594である。これらの作用物質は、ニコチン性受容体の感受性を減じ、受容体を通過するイオン流の減少をもたらすことにより作用すると想定されている。このような状況下で、ニコチン作動薬はnAChR拮抗薬として効率的に作用しており、このためnAChR拮抗薬は鎮痛化合物候補として探求されてきた。コノトキシンペプチドVc1.1はこのような拮抗薬であるといわれている。
【0012】
興味深いことに、nAChRのα3、β4サブタイプのVc1.1のIC50値はマイクロモルの範囲である(4.2μM; Vinclerら、2006年、PNAS、103、17881頁)。Vc1.1の鎮痛作用はin vivoにおいてナノモル濃度で現れるようである。
【0013】
Vc1.1が、α9、α10nAChRの調節によって鎮痛をもたらしうることが最近報告されている(Vinclerら、上記)。Vc1.1のα9、α10nAChRに対する親和性は、α3、β4nAChRに対してよりも100倍高く、ナノモル範囲に入る(22.9nM)。したがって、Vc1.1の生理学的標的の1つは、広範に分布していることが知られ、後根神経節ニューロン、下垂体、リンパ球、皮膚ケラチノサイトおよび精子中に存在するα9、α10nAChRとすることができるようである。しかしながら、Vc1.1の疼痛緩和作用の原因となる、まだ発見されていない他の標的が存在しうると考えられる。
【0014】
疼痛および他の状態の治療のために研究されてきた以前のコノトキシンペプチドとは異なり、Vc1.1は、髄腔内ではなく皮下または筋肉内投与が可能であるという利点を有するといわれている。このことは、Ziconotideを含む以前のコノトキシンペプチドと比べてVc1.1に大きな利益をもたらすといわれている。しかしながら、コノトキシンペプチドVc1.1は経口バイオアベイラビリティを欠くといわれている。2006年1月付のMetabolic Pharmaceuticalのウェブサイトで発表された文書「ACV1、神経因性疼痛の新規治療、前臨床データの技術概要(Technical Summary of Preclinical Data)」によると、ACV1は経口で使用できないが、皮下注射処置用として現在開発されている。Vc1.1はまた、炎症性疼痛の動物モデルにおいて有効であり、損傷した神経および組織の回復を促進するとも報告されている。
【0015】
Metabolic Pharmaceuticals社によるWO2007/014432は、ヒト成長ホルモンおよびそのアナログのC末端配列をペプチド薬のC末端に連結させることによってペプチド薬の経口送達を向上させる方法を記載している。ペプチド薬へのヒト成長ホルモンのC末端配列の付加は、ペプチド薬に経口バイオアベイラビリティ特性を付与するとされている。この特許出願は、神経因性疼痛の動物モデルにおいて活性であるらしい、経口で使用できるペプチドを記載している。ペプチドはVc1.1のC末端に連結したアミノ酸配列Tyr-Leu-Arg-Ile-Valを有するVc1.1を含む。
【特許文献1】WO02/079236
【特許文献2】WO2007/014432
【特許文献3】1999年9月14日に出願された国際特許出願第PCT/AU99/00769号(WO00/15654)
【非特許文献1】LloydおよびWilliams、2000年、J Pharmacol Exp Ther 292 (2) 461頁
【非特許文献2】Sandallら、Biochemistry、2003年、42、6904頁
【非特許文献3】Clarkら、J. Biol. Chem.2006年、281、23254頁
【非特許文献4】Satkunanathanら、2005年、Brain Research 1059 (2) 149〜158頁
【非特許文献5】IDrugs、15th World Congress on Animal、Plant and Microbial Toxins、2006年 9、679〜681頁
【非特許文献6】Sandallら、2003年、Biochemistry、42、6904〜6911頁
【非特許文献7】Langら、2005年、Neuroreport 16、479〜483頁
【非特許文献8】Langら、2003年、Neurophysiol 90、3295〜3303頁
【非特許文献9】Vinclerら、2006年、PNAS、103、17881頁
【非特許文献10】2006年1月付のMetabolic Pharmaceuticalのウェブサイトで発表された文書「ACV1、神経因性疼痛の新規治療法、前臨床データの技術概要(Technical Summary of Preclinical Data)」
【非特許文献11】Hoggら、1999年、J Biol Chem. 274(51):36559〜64頁
【非特許文献12】Hoggら、 2003年、Reviews of Physiology, Biochemistry and Pharmacology 1: 1〜46頁
【非特許文献13】Schnolzerら、(1992) Int. J. Pept. Protein Res. 40、180〜193頁
【非特許文献14】Dawsonら、(1994) Science 266、776〜9頁
【非特許文献15】Tamら、(1999) J. Am. Chem. Soc. 121、4316〜4324頁
【非特許文献16】Yan, L.Z.およびDawson, P.E. (2001) J Am Chem Soc 123、526〜33頁
【非特許文献17】Ingenito, R.、 Bianchi, E.、Fattori, D.およびPessi, A. (1999) J Am Chem Soc 121、11369〜74頁
【非特許文献18】Shin, Y.、Winans, K.A.、Backes, B.J.、Kent, S.B.H.、Ellman, J.A.およびBertozzi, C.R. (1999) J Am Chem Soc 121、11684〜89頁
【非特許文献19】Clippingdale, A.B.、Barrow, C.J.およびWade, J.D. (2000) J Pept Sci 6、225〜34頁
【非特許文献20】Camarero, J.A.、Hackel, B.J.、De Yoreo, J.J.およびMitchell, A.R. (2004) J Org Chem 69、4145〜511頁
【非特許文献21】Rance, M.、Sorensen, Q.W.、Bodenhausen, G.、Wagner, G.、Ernst, R.R.およびWiithrich, K. (1983) Biochem. Biophys. Res. Commun. 117、479〜485頁
【非特許文献22】Bax, A.およびDavis, D.G. (1985) J. Magn. Reson. 65、355〜360頁
【非特許文献23】Griesinger, C.、Sorensen, O.W.およびErnst, R.R. (1987) J. Magn. Reson. 75、474〜492頁
【非特許文献24】Jeener, J,、Meier, B.H.、Bachmann, P.およびErnst, R.R. (1979) J. Chem. Phys. 71、4546〜4553頁
【非特許文献25】Kumar, A.、Ernst, R.R.およびWitthrich, K. (1980) Biochem. Biophys. Res. Commun. 95、1-6頁
【非特許文献26】Piotto, M.、Saudek, VおよびSklenar, V. (1992) J. Biomol. NMR 2、661〜665頁
【非特許文献27】Eccles, C.、Guntert, P.、Billeter, M.およびWuthrich, K. (1991) J. Biomol. NMR 1、111〜30頁
【非特許文献28】Wuthrich, K. (1986) Wiley-Interscience、New York
【非特許文献29】Guntert, P.、 Mumenthaler, C.およびWuthrich, K. (1997) J. Mol. Biol. 273、283〜98頁
【非特許文献30】Brunger, A.T.、Adams, P.D.およびRice, L.M. (1997) Structure 5、325〜336頁
【非特許文献31】Linge, J.P.およびNilges, M. (1999) J. Biomol. NMR 13、51〜9頁
【非特許文献32】Hutchinson, E.G.およびThornton, J.M. (1996) Protein Sci. 5、212〜220頁
【非特許文献33】Laskowski, R.A.、Rullmannn, J.A.、MacArthur, M.W.、Kaptein, R.およびThornton, J.M. (1996) J. Biomol. NMR 8、477〜86頁
【非特許文献34】Lawrence, G.W.、Weller, U.およびDolly, J.O. (1994) Eur J Biochem 222、325〜33頁
【非特許文献35】Meunier, F.A.、Mattei, C.、Chameau, P.、Lawrence, G.、Colasante, C.、Kreger, A.S.、Dolly, J.O.およびMolgo, J. (2000) J Cell Sci 113 (Pt 7)、1119〜25頁
【非特許文献36】Meunier, F.A.、Feng, Z.P.、Molgo, J.、Zamponi, G.W.およびSchiavo, G. (2002) Embo J 21、6733-43頁
【非特許文献37】Sandall DW、Satkunanathan N、Keays DA、Polidano MA、Liping X、Pham V、Down .JG、Khalil Z、Livett BG、Gayler KR、Biochemistry 2003 42(22):6904〜11頁
【非特許文献38】Clarkら、J. Biol. Chem. (2006) 281 23254-23263頁
【非特許文献39】Goransson, U.およびCraik, D.J. (2003) J. Biol. Chem. 278、48188〜96頁
【発明の開示】
【発明が解決しようとする課題】
【0016】
したがって、特に、鎮痛の惹起、神経因性疼痛の治療または予防に関して、および神経損傷からの回復促進において、経口または経腸経路を介してα-コノトキシンペプチドを用いて患者を治療する有効な方法が依然として必要である。
【課題を解決するための手段】
【0017】
本発明は、α-コノトキシンペプチドを環化することによりアミド環状骨格を有する化合物を生成すると、経口有効性を有する環状ペプチドをもたらすという驚くべき発見に基づく。
【0018】
したがって第1の態様では、本発明は、遊離N末端およびC末端を有さないようなアミド環状骨格を有する少なくとも1種の合成環状α-コノトキシンペプチドを、医薬として経口または経腸投与に適したビヒクル中に含む経口または経腸医薬製剤であって、このペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端はペプチドリンカーを介してC末端に連結されている、医薬製剤を提供する。
【0019】
ペプチドリンカーが、6個から8個の天然または非天然アミノ酸がシステイン残基IおよびIVの間にわたる長さであることが好ましい。
【0020】
本発明はさらに、前述の医薬製剤を経口または経腸投与する段階を含む、疼痛の治療または予防方法を提供する。
【0021】
さらなる実施形態では、本発明は、前述の医薬製剤を経口または経腸投与する段階を含む、神経損傷の回復を促進する方法を提供する。
【0022】
本発明はさらに、前述の医薬製剤を経口または経腸投与する段階を含む、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、気分変調、トゥレット症候群および季節性情動障害など)または炎症の治療または予防方法を提供する。
【0023】
さらなる実施形態では、本発明は、疼痛の治療もしくは予防のための、または神経損傷からの回復促進における経口または経腸投与用の医薬の製造における環状α-コノトキシンペプチドの使用を提供する。
【0024】
本発明はまた、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、トゥレット症候群、気分変調および季節性情動障害など)または炎症の治療または予防のための経口または経腸投与用の医薬の製造における環状α-コノトキシンペプチドの使用も提供する。
【0025】
好ましい実施形態の説明
α-コノトキシンクラスは、文献中に数百の例が記載されている非常に大きなコノトキシンペプチドのクラスである。大部分のα-コノトキシンペプチドは、対になって結合した4個のシステイン残基を有し、システイン残基IとIII、およびシステイン残基IIとIVの間で2個のジスルフィド結合を形成するA-スーパーファミリーのメンバーである。文献中に記載されているα-コノトキシンペプチドの一部の例を以下の表1に示す。
【0026】
【表1】

【0027】
上記表において、γはγ-カルボキシグルタミン酸を、Oは4-ヒドロキシプロリンを表す。その他の記号は、天然アミノ酸を指定するのに一般に使用されるものである。上記ペプチドの大部分はアミド化C末端を有する。
【0028】
安定性を向上させるためのコノトキシンペプチドの環化は、1999年9月14日に出願された国際特許出願第PCT/AU99/00769号(WO00/15654)においてCraikらによって最初に記載された。したがって、本発明により処方される環状α-コノトキシンペプチドは、その内容全体が参照により本明細書に組み込まれるその特許出願に記載された方法を使用して調製することができる。
【0029】
本明細書において、文脈上他の意味に解すべき場合を除き、コノトキシンペプチドに関連して使用される場合の「直鎖」という用語は、ペプチドが非環状状態にある、すなわち、アミド環状骨格を形成するためにN末端とC末端が(直接またはリンカーを用いて)連結されていないことを意味する。α-コノトキシンペプチド中に1個または複数個のジスルフィド結合が存在するとペプチドはある程度環状になるはずであるが、N末端とC末端の連結によりペプチド骨格が環化されていなければ、そのようなジスルフィド結合を有するペプチドであってもやはり「直鎖」とみなされるべきである。
【0030】
本明細書において使用される「経口/経腸バイオアベイラビリティ」、「経口/経腸投与可能な」、「経口/経腸投与に適した」および「経口有効性」という用語は、環状ペプチドが経口または経腸経路によって投与され、疼痛、アルツハイマー病などの治療もしくは予防、または神経損傷の回復促進などの薬学的に適切な効果をもたらす能力を指す。この効果は、ペプチドとその想定された標的の相互作用によって起こると考えられるが、用語はそのような制限を本発明の範囲に課すものではない。他のまたは代替の標的が生理学的にin vivoで関与してもよく、ペプチド代謝産物が薬学的に適切な効果をもたらすのに関与してもよい。例えば、モルヒネ鎮痛剤は、オピオイド受容体に結合し、その後オピオイド受容体がカルシウムおよび/またはカリウムチャネルを調節することにより作用することが知られている。
【0031】
直鎖α-コノトキシンペプチドは、環化可能な任意のα-コノトキシンペプチドとすることができる。直鎖α-コノトキシンペプチドは、天然α-コノトキシンペプチドの配列を有してもよく、またはその誘導体でもよい。α-コノトキシンペプチドは、非環状の形態において、ヒトなどの哺乳動物の治療的処置に関連する活性を有することが好ましい。ペプチドの環化はペプチドの活性を変化させる、または新たな活性を導入する可能性があるので、一部の環状コノトキシンペプチドが、「直鎖」コノトキシンと比較して改変され向上した治療特性を有しうる可能性がある。場合によっては、環状コノトキシンペプチドは、直鎖α-コノトキシンペプチドと異なるジスルフィド連結性、例えば、CysI-IV、II-III(リボン状)連結性およびCysI-II、III-IV(数珠状)連結性を有するであろう。ペプチドはまた、異性体の組合せとして存在してもよい。
【0032】
本発明の一実施形態によると、環化を受ける直鎖α-コノトキシンペプチドは、以下に示す配列を含む4/7または4/6ペプチドである:
Xaa1 CCS Xaa2 P Xaa3 C Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 C 配列番号1
(配列中、Xaa1はグリシンまたはアスパラギン酸であり、Xaa2からXaa7は任意の天然または非天然アミノ酸を表し、Xaa8はプロリン、ヒドロキシプロリンまたはグルタミンを表し、Xaa9はアスパラギン酸、グルタミン酸またはγ-カルボキシグルタミン酸を表し、Xaa10は任意の天然または非天然アミノ酸を表すか、なくてもよい)。
【0033】
より好ましくは、Xaa1からXaa10は以下のように選択される:
Xaa1はグリシンまたはアスパラギン酸であり、
Xaa2はアスパラギン酸、アスパラギン、ヒスチジン、チロシン、アルギニンまたはリシンから、さらにより好ましくはアスパラギン酸、アスパラギン、およびヒスチジン、最も好ましくはアスパラギン酸から選択され、
Xaa3はアルギニン、プロリン、アラニン、バリンまたはセリン、より好ましくはアルギニン、プロリンまたはアラニン、最も好ましくはアルギニンから選択され、
Xaa4はアスパラギン、アラニン、アルギニン、チロシン、ヒスチジン、フェニルアラニン、セリン、イソロイシンまたはリシン、より好ましくはアスパラギン、アラニンまたはチロシン、および最も好ましくはアスパラギンから選択され、
Xaa5はチロシン、ヒスチジン、アラニン、バリン、グルタミン、グリシン、ロイシン、セリン、チオニン、アスパラギン、アスパラギン酸、グルタミン酸、リシンまたはアルギニン、より好ましくは親水性アミノ酸残基、および最も好ましくはチロシンから選択され、
Xaa6はアスパラギン酸、アスパラギン、セリン、チロシン、グルタミン酸、グリシン、アルギニンまたはヒスチジン、より好ましくはアスパラギン酸またはアスパラギン、および最も好ましくはアスパラギン酸から選択され、
Xaa7はヒスチジン、アスパラギンまたはチロシン、より好ましくはヒスチジンから選択され、
Xaa8はプロリン、ヒドロキシプロリン、グルタミンまたはセリン、より好ましくはプロリンから選択され、
Xaa9はグルタミン酸、γ-カルボキシグルタミン酸、アスパラギン酸、グリシンまたはアスパラギン、より好ましくはグルタミン酸、γ-カルボキシグルタミン酸、および最も好ましくはグルタミン酸から選択され、
Xaa10はイソロイシン、チロシン、ロイシンまたはdes-Xaa10、より好ましくはイソロイシンから選択される。
【0034】
特に好ましい実施形態において、
Xaa1はグリシンまたはアスパラギン酸であり、
Xaa2はアスパラギン酸であり、
Xaa3はアルギニンであり、
Xaa4はアスパラギンであり、
Xaa5はチロシンであり、
Xaa6はアスパラギン酸であり、
Xaa7はヒスチジンであり、
Xaa8はプロリンであり、
Xaa9はグルタミン酸であり、
Xaa10はイソロイシンである。
【0035】
好ましい実施形態において、環化を受ける直鎖アルファコノトキシンペプチドは、α4/7コノトキシンペプチドである。このペプチドは、特定のサブタイプのnAChRに対する選択性を有しうる。例えば、このペプチドはα7サブタイプのnAChRよりもα3またはα9/α10サブタイプに対する選択性を有しうる。
【0036】
さらに好ましい実施形態において、環化を受ける直鎖アルファコノトキシンペプチドはVc1.1である。
【0037】
本発明による環状コノトキシンペプチドは、通常、N末端とC末端が連結部分を介して連結されたα-コノトキシンペプチドからなるはずであるが、場合によっては、天然α-コノトキシンペプチドまたはその誘導体のN末端とC末端を、さらなる連結部分を必要とせずに、直接連結することが可能でありうる。連結部分は、存在する場合、環化によりアミド環状ペプチド骨格が生成されるようなペプチドリンカーとすることができる。これらのペプチドは、遊離N末端およびC末端を有さないはずである。
【0038】
連結部分のペプチド配列は大きく変化させることが可能である。この連結部分は、α-コノトキシンの主要活性部位に必ずしも結合するか、またはかみ合うわけではないので、この部分を改変して生理化学的特性を変化させ、可能性としてコノトキシンの副作用を低減させることができる。
【0039】
コノトキシンのN末端とC末端を連結するとき、場合によってはN末端またはC末端残基の1つまたは複数を除去または改変することが必要であるか、または望ましいことがある。直鎖コノトキシン配列のそのような改変は、本発明の範囲内である。例えば、直鎖コノトキシン末端の正または負電荷が活性に重要であった場合、環化によるその欠損は、それぞれリシンまたはグルタミン酸などの正または負の側鎖を有するアミノ酸の置換により救済することができるだろう。
【0040】
連結部分は必ず、コノトキシンペプチドのN末端とC末端の間にわたるのに十分な長さであるはずである。ペプチドリンカーの場合、長さは一般に2から10アミノ酸程度であろう。場合によっては、より長いまたは短いペプチドリンカーが必要とされることがある。一実施形態において、連結部分は、直鎖α-コノトキシンにすでに存在する任意のアミノ酸残基に加えてグリシンおよび/またはアラニン残基からなる。
【0041】
A-スーパーファミリーのα-コノトキシンペプチドについて、システイン残基IおよびIV間の距離が実質的に保存されていることが見出された。そのようなコノトキシンペプチドに関しては、結合角および距離を考慮して、システイン残基IとIVの間のアミノ酸数が6個〜8個になるようにリンカーの長さを選択することが好ましい。したがって、α-コノトキシンVc1.1およびMIIに関して、リンカーに必要な追加のアミノ酸残基は、N末端にすでに存在する1個のアミノ酸を考慮して5個〜7個ということになる。
【0042】
したがって、さらなる態様において本発明は、遊離N末端およびC末端を有さないようなアミド環状骨格を有する合成環状α-コノトキシンペプチドを利用し、前記ペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端は、6個〜8個のアミノ酸残基がシステイン残基IとIVの間にわたるようなペプチドリンカーを介してC末端に連結されている。
【0043】
リンカー中のアミノ酸数は、第1と第4のシステイン残基の間に7個のアミノ酸が存在するように選択することが好ましい。直鎖ペプチドの配列によっては、これらの残基の一部または全てが直鎖配列に由来してもよい。したがって、コノトキシンペプチドが第1のシステイン残基近辺のN末端に1個のアミノ酸を有する場合、リンカーに必要な追加のアミノ酸数は6個となる。
【0044】
当然ながら、N末端残基の1個または複数個を、リンカーを形成することになる他の残基で置換することも可能であろう。
【0045】
本発明によれば、特定の大きさおよび/またはタイプのペプチドリンカーを選択することにより、コノトキシンペプチドの活性を改変または増強することが可能である。連結基の導入により生じるコノトキシンのわずかな立体構造変化によって、特定の結合部位へのペプチドの結合親和性を変化させることできる。反対に、活性が親コノトキシンペプチドの活性に限りなく近い場合は、立体構造変化を最小限にするリンカーが選択されよう。
【0046】
リンカーはまた、主要な生物学的作用を実質的に阻害しない「ハンドル」をペプチドに提供する。リンカーは、分子を機能的にするための空間を提供することにより、例えば経口送達生物活性をもたらすために必要である可能性があるような、生体膜を通過する能力を含む、生物物理学的または生物薬剤学的特性を向上させることができる。
【0047】
直鎖コノトキシンを環化できるいくつかの方法がある。これらは以下のものを含む。
【0048】
1.還元ペプチドの環化に続く酸化による必要なジスルフィド結合の形成
このアプローチでは、伸長された直鎖ペプチドが、固相ペプチド合成法を使用して「on resin」で最初に合成される。この伸長直鎖ペプチドは、新たな連結部分を含むN末端とC末端伸長部の、またはその近くのシステイン残基から始まる天然配列を含む。固相合成は実際には逆の順序で、すなわち、伸長直鎖ペプチドのC末端で開始する。樹脂からの切断後、伸長コノトキシンはチオエステル中間体に環化され、続いてアミド環状ペプチドに再配置される。この還元ペプチドは次に酸化されてジスルフィド結合を形成する。環化に関する反応の概略図を図1に示す。樹脂(R)へのリンカーが付着したまま、直鎖ペプチドを樹脂から切断した。Rはペプチドと樹脂間のリンカーに相当し、環化で使用される連結部分とは異なる。第1の反応は、N末端システインのチオールとカルボキシ末端との間のチオエステルの形成を伴う。これは次に、S、Nアシル基転移を受け、天然ペプチド結合を有する環状ペプチドを形成する。
【0049】
2.還元直鎖ペプチドの酸化とそれに続く環化
このアプローチでは、固相ペプチド合成を使用して伸長ペプチドをアセンブルする。伸長直鎖ペプチドは、Nおよび/またはC末端に付加された追加の残基を有する天然コノトキシン配列を含む。(新しい)N末端とC末端は、好ましくはグリシン残基とするべきである。ペプチドは折り畳まれ、コノトキシン様ペプチドの場合、折り畳まれた分子の両末端は一般に間隙を介してすぐそばにある。このことは、標準的な化学反応を使用した溶液中でのペプチドの環化を容易にする。配列中に多数のリシン、グルタミン酸またはアスパラギン酸残基が存在する場合は問題が起こりうるので、そのときは方法1が好ましい。
【0050】
3.既存のコノトキシンへのリンカーライゲーションとそれに続く環化
この方法では、出発物質は成熟コノトキシンである。ペプチドリンカーは、ペプチドライゲーションに関する公開された手順を使用して、コノトキシンを用いて合成および連結される。伸長ペプチドは次いで環化および酸化される。
【0051】
前述のプロセスにおいて、生成物が所要のジスルフィド結合を有する環状α-コノトキシンペプチドである限り、各段階は任意の順序で実施することができる。例えば、プロセス1において、切断および環化段階は同時に、またはいずれの順序でも実施することができる。同様にプロセス2において、環化および折り畳み段階は同時に、またはいずれの順序でも実施してよい。
【0052】
システイン残基に保護基を選択的に使用してジスルフィド結合を形成することも可能である。このような方法でシステイン残基を選択的に保護することにより、特定のジスルフィド結合パターンの生成が可能である。システイン残基を保護することができる基の例は、アセタミドメチル(Acm)、4-メチルベンジル(MeBzl)および4-メトキシベンジル(Mob)を含む。
【0053】
また最終生成物の環状性を考慮して、合成手順は、α-コノトキシンペプチドの、または伸長ペプチド/リンカー配列の円順列置換変異を含むことができる。例えば、α-コノトキシンのための伸長直鎖ペプチドの設計は、α-コノトキシンのC末端残基にリンカーを付加することにより開始して、N末端残基をC末端に円順列置換してN末端システインおよび前述の環化をもたらすことができよう。
【0054】
天然コノトキシンペプチドに関連して本明細書で使用されるMIIなどの「誘導体」という用語は、1つまたは複数のアミノ酸欠失、付加、置換、または側鎖修飾によって天然ペプチドとは異なるペプチドを指す。
【0055】
置換は、アミノ酸が異なる天然または非従来型アミノ酸残基で置換されているアミノ酸変化を包含する。そのような置換は、「保存的」として分類されてよく、その場合、ポリペプチド中に含有されるアミノ酸残基は、極性、側鎖官能基または大きさのいずれかに関して特徴の類似した他の天然アミノ酸、例えば
【0056】
【化1】
【0057】
で置換される。いくつかの非従来型アミノ酸もまた天然アミノ酸の置換に適しうることが理解されるべきである。例えば、オルニチン、ホモアルギニンおよびジメチルリシンはHis、ArgおよびLysに関係している。
【0058】
本発明に包含される置換はまた、ポリペプチド中に存在するアミノ酸残基が、異なる群の天然アミノ酸などの異なる特性を有するアミノ酸で置換されているか(例えば、荷電または疎水性アミノ酸とアラニンの置換)、あるいは、天然アミノ酸が非従来型アミノ酸で置換されている、「非保存的」置換であってもよい。
【0059】
アミノ酸置換は、典型的に単一残基のものであるが、集合または分散した多数の残基のものであってもよい。
【0060】
アミノ酸置換は保存的であることが好ましい。
【0061】
付加は1つまたは複数の天然または非従来型アミノ酸残基の付加を包含する。欠失は1つまたは複数のアミノ酸残基の欠失を包含する。
【0062】
前述のように、本発明は1個または複数個のアミノ酸が側鎖修飾を受けたペプチドを含む。本発明に意図される側鎖修飾の例は、アルデヒドとの反応による還元的アルキル化に続くNaBH4による還元;メチルアセチミデートによるアミジン化;無水酢酸によるアシル化;シアン酸塩によるアミノ基のカルバモイル化;2,4,6-トリニトロベンゼンスルホン酸(TNBS)によるアミノ基のトリニトロベンジル化;無水コハク酸および無水テトラヒドロフタル酸によるアミノ基のアシル化;およびピリドキサール-5-リン酸によるリシンのピリドキシル化に続くNaBH4による還元などによるアミノ基修飾を含む。
【0063】
アルギニン残基のグアニジン基は、2,3-ブタンジオン、フェニルグリオキサルおよびグリオキサルなどの試薬を用いて複素環縮合生成物の形成により修飾することができる。
【0064】
カルボキシル基は、O-アシルイソ尿素の形成を介するカルボジイミドの活性化に続く、例えば対応するアミドへの誘導体化によって修飾することができる。
【0065】
スルフヒドリル基は、ヨード酢酸またはヨードアセタミドによるカルボキシメチル化;システイン酸への過ギ酸酸化;他のチオール化合物による混合ジスルフィドの形成;マレイミド、無水マレイン酸、または他の置換マレイミドとの反応;4-クロロ安息香酸水銀、4-クロロフェニルスルホン酸水銀、塩化フェニル水銀、2-クロロ水銀-4-ニトロフェノール、および他の水銀剤を使用する水銀誘導体の形成;アルカリ性pHにおけるシアン酸塩によるカルバモイル化などの方法で修飾することができる。システイン残基のいかなる修飾も、ペプチドが必要なジスルフィド結合を形成する能力に影響を及ぼしてはならない。ペプチドが1個または複数個のジスルフィド結合の代わりにジセレニウム結合を形成するように、システインのスルフヒドリル基をセレニウム等価物と置換することも可能である。
【0066】
トリプトファン残基は、例えば、N-ブロモスクシンイミドでの酸化またはインドール環の2-ヒドロキシ-5-ニトロベンジルブロミドまたはスルフェニルハライドでのアルキル化により修飾することができる。一方、チロシン残基は、テトラニトロメタンでニトロ化して3-ニトロチロシン誘導体を形成することにより変化させることができる。
【0067】
ヒスチジン残基のイミダゾール環の修飾は、ヨード酢酸誘導体でのアルキル化またはジエチルピロカーボネートでのN-カルベトキシル化により達成することができる。
【0068】
プロリン残基は、例えば、4位におけるヒドロキシル化により修飾することができる。
【0069】
修飾された側鎖を有するいくつかのアミノ酸および他の非天然アミノ酸の一覧表を表2に示す。
【0070】
【表2】
【0071】
これらのタイプの修飾は、個体に、または診断試薬として使用するために投与される場合、ペプチドを安定化させるのに重要でありうる。
【0072】
本発明に意図される他の誘導体は、全くグリコシル化されていない分子から修飾されたグリコシル化分子まで、さまざまなグリコシル化変異体を含む。変化したグリコシル化パターンは、異なる宿主細胞における組換え分子の発現に由来してもよい。
【0073】
環状α-コノトキシンペプチドは、Cys残基および特徴的なジスルフィド結合パターンを保持することが好ましい。誘導体は、ジスルフィド結合形成の際に保護されるという条件で、さらなるCys残基を含んでもよい。
【0074】
本発明によるコノトキシンペプチドは12〜40個、より好ましくは15〜30個のアミノ酸を有することが好ましい。
【0075】
本発明による環状コノトキシンペプチドは、治療薬として有用である。
【0076】
α-コノトキシンは、疼痛の治療および神経損傷の回復促進に関わるニコチン性アセチルコリン受容体(nAChR)に結合する。このような受容体は、統合失調症、うつ病、中毒、てんかん、アルツハイマー病、パーキンソン病およびトゥレット症候群を含むいくつかの神経精神障害の病態生理学にも関わっており、そのためα-コノトキシンはこれらの疾患の治療指標となる可能性を有する。本発明による環状α-コノトキシンペプチドが他の受容体またはイオンチャネルを標的とすることも可能である。
【0077】
環状コノトキシンペプチドは、神経因性疼痛の治療的処置または予防に特に有用であると考えられている。疼痛は、神経系自体の内部から生じる場合、神経因性であるとみなされる。神経系はさまざまなレベル:神経、神経根ならびに脊髄および脳の中枢痛経路で関与しうる。神経因性疼痛は、損傷した神経の異常な作用に起因する場合があり、最も治療が困難な形態の疼痛である。神経因性疼痛は、持続的に生じる、罹患個体に有益な機能を果たさない、慢性疼痛の形態である。神経因性疼痛を患う患者は典型的に、異痛症(通常痛みを伴わない刺激による疼痛)および痛覚過敏症(痛みを伴う刺激に対する応答の増大)に苦しむ。
【0078】
神経障害、異痛症および痛覚過敏症の特徴的な疼痛症状を例証する、いくつかのよく確立された疼痛動物モデルがある。神経因性疼痛の絞扼性神経損傷(CCI)ラットモデルでは、坐骨神経に緩い結紮を適用し、神経圧迫および神経障害を引き起こす炎症をもたらす。神経部分結紮(PNL)モデルでは、坐骨神経の一部周辺にきつい結紮を作成し、残りの神経は損傷のないままにしておき、異痛症および神経過敏症の疼痛反応をもたらす。ストレプトゾトシン(STZ)モデルでは、膵臓β細胞に糖尿病を誘発し、機械的痛覚過敏および接触性異痛などの神経因性疼痛症状をもたらす。
【0079】
前述の活性を有する化合物を評価するのに有用な試験は、in vitroまたはin vivoでもよく、当業者に知られている。例えば、nAChRでの活性を評価するのに有用な試験は、WO00/15654 (Hoggら、1999年、J Biol Chem. 274(51):36559〜64頁;およびHoggら、2003年、Reviews of Physiology, Biochemistry and Pharmacology 1: 1〜46頁)に記載のものを含む。
【0080】
ペプチドは予防の意味で投与されてもよいが、哺乳動物がこのような治療を必要としていることが好ましい。
【0081】
当業者には容易に理解されるように、投与経路(経口および経腸)および薬学的に許容されるビヒクルの性質は、治療される状態および哺乳動物の性質によって決定するはずである。特定の担体または送達システム、および投与経路の選択は当業者によって容易に決定されうると考えられる。
【0082】
本発明の経口および経腸製剤において、活性ペプチドは不活性希釈剤と、または同化可能な食用担体と一緒に処方してもよく、または硬質または軟質ゼラチンカプセルに封入してもよく、または錠剤に圧縮してもよく、または日常食の食物に直接組み入れてもよい。治療的経口投与のために、活性化合物は賦形剤と一緒に組み入れて、摂取可能な錠剤、口腔または舌下錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、オブラートなどの形態で使用してもよい。口腔および舌下錠など、これらの経口製剤タイプのいくつかは、肝臓で代謝されない可能性があることが理解されるであろう。しかしながら、本発明の環状ペプチドは、肝臓代謝に伴う可能性が高い胃にも送達されうる。そのような組成物および調製物は、少なくとも1重量%の活性化合物を含有することが好ましい。当然ながら、組成物および調製物の割合は変化することがあり、好都合には構成単位の約5〜約80重量%とすることができる。このような治療上有用な組成物における活性化合物の量は、適した投薬量が得られるような量である。
【0083】
錠剤、トローチ、丸剤、カプセルなどはまた、以下に記載の成分も含有することができる:ガム、アラビアゴム、コーンスターチまたはゼラチンなどの結合剤;第二リン酸カルシウムなどの賦形剤;コーンスターチ、ジャガイモデンプン、アルギン酸などの崩壊剤;ステアリン酸マグネシウムなどの滑剤;およびショ糖、乳糖またはサッカリンなどの甘味剤を加えてもよく、またはペパーミント、冬緑油またはチェリー風味などの香料。単位剤形がカプセルの場合、上記タイプの材料に加えて液体担体を含んでもよい。種々の他の材料が、コーティングとして、または投薬単位の物理的形態を別の方法で修正するために存在してもよい。例えば、錠剤、丸剤またはカプセルをシェラック、糖または両方で被覆してもよい。シロップまたはエリキシルは活性化合物、甘味剤としてショ糖、保存料としてメチルおよびプロピルパラベン、色素ならびにオレンジまたはチェリー風味などの香料を含有してもよい。当然ながら、任意の単位剤形の調製で使用した任意の材料は、薬学的に純粋で、使用される量で実質的に非毒性であるべきである。加えて、活性化合物は持続放出調製物および製剤に組み込まれてもよい。
【0084】
液体製剤もまた、胃または食道管を通って経腸投与することができる。
【0085】
経腸製剤は、乳化基剤または水溶性基剤などの適切な基剤と混合することにより坐剤の形態で調製することができる。本発明の環状ペプチドが、局所的、鼻腔内、膣内、眼内などに投与されることも可能であるが、必須ではない。
【0086】
薬学的に許容されるビヒクルは、任意のおよび全ての溶剤、分散媒、コーティング、抗菌および抗真菌剤、等張および吸収遅延剤などを含む。薬学的に活性な物質にこのような媒体および作用物質を使用することは、当技術分野においてよく知られている。任意の従来の媒体または作用物質が活性成分と配合禁忌である場合を除いて、治療組成物におけるその使用は予期されている。また補助的な活性成分を組成物中に組み込むこともできる。
【0087】
投与および投薬量の均一化を容易にするために、単位剤形中に経口または経腸組成物を処方することは特に好都合である。本明細書で使用される単位剤形は、治療される哺乳動物対象の単位投薬量に適合させた物理的に分離した単位を指し、各単位は、所要の薬学的に許容されるビヒクルを伴って、所望の治療効果を生み出すために算出された所定量の活性物質を含有する。本発明の新規な単位剤形の仕様は、以下の(a)(b)によって規定され、直接決定される:(a)活性物質特有の特徴および達成されるべき特定の治療効果、および(b)本明細書中で詳細に開示されているように身体的健康が損なわれた病的状態を有する生体対象における疾患の治療用の活性物質を配合する当技術分野に固有の制限。
【0088】
前述のように、主要活性成分は、簡便で有効な投与のために、有効量で、適した薬学的に許容されるビヒクルと一緒に単位剤形中に配合される。例えば、単位剤形は、主要活性成分を0.25μg〜約2000mgの量で含有することができる。割合で表すと、活性化合物は一般に、担体1ml当たり0.25μg〜約2000mgで存在する。補助的な活性成分を含有する組成物の場合、投薬量は、前記成分の通常の用量および投与様式を参照することにより決定される。
【0089】
本発明のいくつかの態様では、経口または経腸以外の投与形態および経路は、例えば、クリーム、ローション、経皮パッチ、スプレーおよびゲル、または、例えば溶液もしくは乾燥粉末などの吸入もしくは鼻腔内送達に適した組成物などの局所適用が予期される。
【発明を実施するための最良の形態】
【0090】
発明の詳細な説明
本発明を、いくつかの環状コノトキシンペプチドの生成およびその生物活性を説明する添付の実施例および図を参照して、以下に説明する。しかしながら、以下の説明の独自性は、本発明の前述の説明の一般性に取ってかわるものではないことが理解されるべきである。
【実施例1】
【0091】
環状MIIアナログ
(a)設計および合成
天然MIIペプチドのN末端とC末端をつなぐために必要なリンカーの大きさは、周知の分子モデリング法を使用して決定した。MIIの3次元モデルは、Protein Data Bank (PDB)からダウンロードした。分子モデリングプログラムAccelrys (Insight II Modeling Environment、2000年リリース、サンディエゴ、Accelrys社 2001年)を使用して、適切なリンカーの長さを決定した。つまり、N末端とC末端間にアミノ酸リンカーを組み込み、次いで親コノトキシン構造の立体構造をロックし、簡易なエネルギー最小化を行った。次いで、親コノトキシンの立体構造をロック解除し、ペプチド構造全体を再度最小化した。リンカーの大きさが小さすぎるモデルは、天然構造を乱さない長さのリンカーを有するものよりもエネルギーが高い構造をもたらす。最小で5残基を含有するリンカーが、N末端とC末端をつなぐために必要とされることが決定された。そのため、N末端グリシンに加えて、5(GGAAG)、6(GGAAGG)、および7(GGAGAAG)残基のリンカーを含有するペプチドを合成した。これは、6〜8残基にわたるシステイン間の全長に相当する。距離の測定によって、リンカー中の各残基が約2.0〜2.4オングストローム(これは、約3.5オングストロームにわたるであろう、つまり緩んだ立体構造を表す伸長立体構造よりも短いことに注意されたい)にわたる必要となることが明らかになった。大きな、または高度に官能化されたアミノ酸残基と比較して、天然ペプチドの他の残基と相互作用しにくい、またはフォールディングを阻害しにくい小さな側鎖を有する比較的不活性なアミノ酸であるため、グリシンおよびアラニンをリンカーアミノ酸に選択した。
【0092】
(b)環状MIIアナログの合成
チオエステルリンカーを利用するin situ中和と一緒にBOC/HBTU化学反応を使用して、ペプチドを合成した(Schnolzerら、(1992) Int. J. Pept. Protein Res. 40、180〜193頁; Dawsonら、(1994) Science 266、776〜9頁; Tamら、(1999) J. Am. Chem. Soc. 121、4316〜4324頁; Yan, L.Z.およびDawson, P.E. (2001) J Am Chem Soc 123、526〜33頁)。
【0093】
Gly PAM樹脂をDMF中で4当量のS-トリチル-β-メルカプトプロピオン酸、4当量のHBTU、および5当量のDIEAで処理する(2×30分間)ことにより、このリンカーを合成した。次いで、樹脂をTFA/トリイソプロピルシラン/H2O(96:2:2)で2×20分間処理することにより、トリチル基を除去する。次いで、標準的なカップリング条件を使用して、ペプチド鎖のC末端残基をリンカーに付加した。C末端チオエステルを有するペプチドはまた、以下の標準的な文献の手順に従ってFMOC化学反応を使用して製造することもできる[Ingenito, R.、 Bianchi, E.、Fattori, D.およびPessi, A. (1999) J Am Chem Soc 121、11369〜74頁; Shin, Y.、Winans, K.A.、Backes, B.J.、Kent, S.B.H.、Ellman, J.A.およびBertozzi, C.R. (1999) J Am Chem Soc 121、11684〜89頁; Clippingdale, A.B.、Barrow, C.J.およびWade, J.D. (2000) J Pept Sci 6、225〜34頁; Camarero, J.A.、Hackel, B.J.、De Yoreo, J.J.およびMitchell, A.R. (2004) J Org Chem 69、4145〜511頁]。ペプチド鎖をいったん組み立て、スカベンジャーとしてクレゾールおよびチオクレゾールと一緒にHFを使用して、樹脂から切断した。次いで粗ペプチド混合物を、C18カラムを使用して、80分間の0〜80%B勾配(A=0.05%TFAを含む水、B=90%アセトニトリル、9.95%水、0.05%TFA)を利用するRP-HPLCにより精製した。画分を集め、MSで分析し、所望の生成物を含有するものをプールおよび凍結乾燥した。
【0094】
精製された直鎖還元ペプチドの酸化および環化は、約0.3mg/mLの濃度で、0.1M NH4HCO3(pH 8.1)または50/50 0.1M NH4HCO3/iPrOHにペプチドを溶解させることにより達成した。反応混合物を室温で一晩撹拌し、得られた混合物を前述の条件を使用してRP-HPLCにより精製した。ペプチドは以下に示す配列を有する。
【0095】
6残基リンカー環状MIIは、約45:55の比率で2つの異なる異性体を形成した。対照的に、5残基リンカー環状MIIの粗酸化HPLCプロファイルは、ほとんど分散していない一連のピークとして現れた。最後に、7残基リンカー環状MIIは、1H NMRデータによると秩序だった構造を有する1種の優勢な(>90%)異性体を形成した。(N末端グリシンに加えて)6残基の大きさのリンカーを有するMIIアナログの、粗酸化混合物におけるわずかなピークは、これがMIIにとって最適な最小サイズであるという予備的な指標を与える。粗混合物について、個々のピークを集め、MSにより分析した。環状および酸化ペプチドに相当する質量のペプチドを含有するものを分析RP-HPLCにかけた。次いで、画分をHPLC分析に基づいて混合し、凍結乾燥した。
【0096】
十分な量で得られた環状ペプチドの1D 1H NMRスペクトルを測定した。1H NMRスペクトルによると、5残基リンカーペプチドの1つの異性体が秩序だった構造を有するようであった。1H NMRスペクトルによると、6残基リンカーペプチドの場合は、遅れて溶出する、より量の多い異性体が秩序だっているようであった。1H NMRスペクトルによると、7残基環状MIIペプチドもまた秩序だっていた。
【0097】
MIIの環状ペプチドアナログ配列を以下に示す。
【0098】
【化2】
【0099】
【化3】
【0100】
【化4】
【0101】
(c)環状MIIアナログの構造特性決定
次いで、3つの秩序だったアナログ(これよりcMII-5、cMII-6およびcMII-7と称する)について、2次元NMRスペクトルデータを得た。NMR完全帰属を行い、HαおよびHNプロトンの化学シフトを天然MIIのものと比較した。化学シフトデータの比較により、これらの構造がいかに類似しているかが示された。結果により、cMII-5の化学シフトがMIIのものとほとんど一致せず、構造の変化を示したことが明らかとなった。対照的に、cMII-6およびcMII-7の化学シフトは、天然コノトキシンのものと高度に相関性があった。このことは、cMII-6およびcMII-7が天然MIIに類似の構造を採用し、cMII-5が天然の構造から明らかに逸脱していることを示す。
【0102】
このことから、酸化プロファイルおよびNMRデータに基づいて、5個の付加残基を有するリンカーは短すぎて、コノトキシンの正しいフォールディングを阻害したが、6個または7個の付加残基を有するリンカーは、環状アナログに天然様の構造を形成させるのに十分な大きさであったことが明らかであった。cMII-6およびcMII-7の完全な3次元(3D)構造を決定し、天然MIIおよびその環状同等物の構造の密接な相関を確認した。
【0103】
cMII-6の3D構造を、NMR分光法を使用して決定した。280および287KにおいてBruker DMX 750分光計においてスペクトルを得た。記録された等核スペクトルは、2量子フィルターDQF-COSY[Rance, M.、Sorensen, Q.W.、Bodenhausen, G.、Wagner, G.、Ernst, R.R.およびWuthrich, K. (1983) Biochem. Biophys. Res. Commun. 117、479〜485頁]、等方性混合時間が80msの、MLEV17スピンロックシーケンスを使用するTOCSY[Bax, A.およびDavis, D.G. (1985) J. Magn. Reson. 65、355〜360頁]、ECOSY[Griesinger, C.、Sorensen, O.W.およびErnst, R.R. (1987) J. Magn. Reson. 75、474〜492頁]、および混合時間が150および350msのNOESY[Jeener, J,、Meier, B.H.、Bachmann, P.およびErnst, R.R. (1979) J. Chem. Phys. 71、4546〜4553頁; Kumar, A.、Ernst, R.R.およびWuthrich, K. (1980) Biochem. Biophys. Res. Commun. 95、1-6頁]を含んでいた。DQF-COSYおよびECOSYの実験において、緩和遅延の間、低出力照射によって水共鳴を抑制した。TOCSYおよびNOESYの実験では、修正WATERGATEシーケンスを使用して水抑制を達成した[Piotto, M.、Saudek, VおよびSklenar, V. (1992) J. Biomol. NMR 2、661〜665頁]。2次元スペクトルは通常、12ppmに相当するスペクトル幅にわたって、f2軸におけるデータ点4096、f1軸における増分512で収集した。ゆっくりと交換するアミドを同定するために、サンプルをD2O中に溶解させた後、一連の1次元およびTOCSYスペクトルを直ちに行った。全てのスペクトルを、XWINNMR (Bruker)を使用してSilicon Graphicsワークステーションで処理した。f1軸は0詰め〜2048の実際のデータ点であり、f1およびf2軸は、フーリエ変換の前に90°シフトしたサイン2乗関数を掛けた。処理したスペクトルはプログラムXEASY[Eccles, C.、Guntert, P.、Billeter, M.およびWuthrich, K. (1991) J. Biomol. NMR 1、111〜30頁]を使用して分析および帰属した。スペクトルは連続帰属法を使用して帰属した[Wuthrich, K. (1986) Wiley-Interscience、New York]。処理は一部、DYANAパッケージ[Guntert, P.、 Mumenthaler, C.およびWuthrich, K. (1997) J. Mol. Biol. 273、283〜98頁]の一部である自動帰属プログラムNOAHを使用して簡便化された。90% H2O、10% D2O中、混合時間350および150msで記録されたNOESYスペクトルの交差ピークは、XEASYで積分およびキャリブレーションを行い、距離拘束はDYANAを使用して導いた。主鎖二面角拘束は、DQF-COSYスペクトルで分裂する逆位相交差ピークの線形分析により測定した3JHNHα結合定数から導いた。角度は、8.5Hzより大きい3JHNHαでは-120°±30°に、5Hzより小さい3JHNHαでは-60°±30°に制約された。β-メチレンプロトンおよびχ1二面角の立体特異的帰属は、NOEピーク強度と組み合わせて、ECOSYスペクトルから測定した3Jαβ結合定数から導いた。初期構造は、DYANAでねじれ角模擬アニーリング法を使用して算出した。最終構造は、CNS version 1.0で模擬アニーリングおよびエネルギー最小化法を使用して算出した[Brunger, A.T.、Adams, P.D.およびRice, L.M. (1997) Structure 5、325〜336頁]。出発構造は、無作為のファイ、プサイ二面角を使用して生成し、エネルギーを最小化することにより正しい局所構造をもたらした。この手順は、4000ステップ、0.015psのねじれ角動力学を含む高温相、4000ステップ、0.015psのねじれ角動力学を含む、温度を0Kまで低下させる冷却相、最後に、5000ステップのパウエル最小化を含むエネルギー最小化相を含む。構造を、LingeおよびNilges[Linge, J.P.およびNilges, M. (1999) J. Biomol. NMR 13、51〜9頁]により記載されたように、ウォーターシェル中でさらなる分子動力学およびエネルギー最小化を使用して精密化した。explicit water中での精密化は、以下の段階を含む。第1に、それぞれ50ステップ、0.005psのデカルト動力学を含む100K毎の段階を介した500Kへの加熱。次に、500Kで2500ステップ、0.005psのデカルト動力学の後に、それぞれ2500ステップ、0.005psのデカルト動力学を含む100K毎の段階で温度を低下させる冷却相。最後に、2000ステップのパウエル最小化で構造を最小化した。構造を、PromotifおよびProcheckを使用して分析した[Hutchinson, E.G.およびThornton, J.M. (1996) Protein Sci. 5、212〜220頁; Laskowski, R.A.、Rullmannn, J.A.、MacArthur, M.W.、Kaptein, R.およびThornton, J.M. (1996) J. Biomol. NMR 8、477〜86頁]。
【0104】
cMII-6とMIIの比較は、MIIの天然立体構造がcMII-6において全体として保持されていることを示す。2つのペプチド間の顕著な類似性を図4で例示する。
【0105】
(d)cMII-6の選択的合成
非選択的酸化が、環状コノトキシンの合成で使用される好ましい方法であるが、cMII-6について例証した特異的ジスルフィド結合形成を使用して製造することもできる。ペプチドは前述のように合成したが、システイン3および16の側鎖をAcm基で保護した。ペプチドを樹脂から切断し、前述の条件を使用して精製および酸化/環化した。第2のジスルフィド結合を、I2を使用した脱保護/酸化によって、3および16位のシステイン間に選択的に形成した。ペプチド(20mg)を、1mg/mLの濃度で50%酢酸水溶液に溶解し、フラスコを窒素でフラッシュした。フラスコへ、2mLの1M HCl、続いて十分量の50%酢酸水溶液中の0.1M I2(約1mL)を加え溶液を淡黄色にした。次いで反応物を窒素下、室温で1.5時間撹拌した。反応混合物が無色になるまで1Mアスコルビン酸を加えることにより、反応をクエンチした。次いで、混合物を緩衝液Aで希釈し、RP-HPLCにより精製し、完全に酸化された環状ペプチドを得た。このペプチドはcMII-6と共溶出され、したがって天然ペプチドのジスルフィド連結性、すなわちCys2-Cys8およびCys3-Cys16(1-3、2-4)に対応するとしてcMII-6のジスルフィド連結性を確認した。
【0106】
(e)cMII-6の生物活性
クロム親和細胞をウシ副腎から調製し、標準的な文献の手順に記載のとおり24ウェルプレート(Nunc)に維持した[Lawrence, G.W.、Weller, U.およびDolly, J.O. (1994) Eur J Biochem 222、325〜33頁; Meunier, F.A.、Mattei, C.、Chameau, P.、Lawrence, G.、Colasante, C.、Kreger, A.S.、Dolly, J.O.およびMolgo, J. (2000) J Cell Sci 113 (Pt 7)、1119〜25頁; Meunier, F.A.、Feng, Z.P.、Molgo, J.、Zamponi, G.W.およびSchiavo, G. (2002) Embo J 21、6733-43頁]。無傷細胞を緩衝液A(mM): NaCl、145; KCl、5、Na2HPO4、1.2;グルコース、10; HEPES-NaOH、20(pH7.4)で簡単に1回洗い、天然および環状コノトキシンと一緒に20分間、2mM CaCl2の存在下でインキュベートし、ニコチン(5μM)で20分間刺激した。各実験の最後に上清のアリコートを採取し、細胞を1%(v/v)Triton X-100(Shigma)で溶解させた。両方のサンプルの組を、カテコールアミンについて蛍光分析により試験し、放出量を文献中に記載のように対照の割合として表した(上記参照)。
【0107】
生物活性試験の結果を図3に示し、環状ペプチドの活性が直鎖MIIの活性と同等であることを示す。
【0108】
(f)cMII-6の安定性
cMII-6およびcMII-7のタンパク質分解酵素による作用に対する安定性を、ペプチドをエンドプロティナーゼGlu-C(Endo Glu-C)と一緒にインキュベートすることにより試験した。MII、cMII-6およびcMII-7は、末端(MIIの場合)およびリンカー(cMII-6およびcMII-7の場合)の対面に同じプロセシング部位を有する可能性がある。ペプチドを、20μg/mLの濃度で0.1M NH4HCO3(pH8.0)緩衝液に溶解させた。次いで、Endo GluCを50:1のペプチド:酵素比(wt/wt)で加え、溶液を37℃でインキュベートした。アリコート(3μL)を抜き取り、0〜10の毎時間5%ギ酸(57μL)でクエンチした。次いで、サンプルをLC/MSにより分析し、各時点において残存する無傷のペプチド量を測定した。各試験は、適切な陽性(Endo GluC切断点を有する直鎖の非ジスルフィドペプチド)および陰性(酵素を含まない緩衝液中のペプチド)対照を用いて3連で行った。
【0109】
安定性試験の結果を図4に示す。環状ペプチドは全10時間にわたって完全に無傷のままであり、一方、天然MIIは約10時間の半減期を有する。cMII-6およびcMII-7により例証された安定性の強化は、MIIおよび環状ペプチドの推定プロセシング部位が同一(すなわち、グルタミン酸のC末端側)であり、末端(MIIの場合)ならびにcMII-6およびcMII-7の場合はペプチドリンカーから離れていることを考えると、驚くべきことである。さらに、任意の強化された安定性は、Glu-Cはエンドプロテアーゼであるが、N末端(MIIはC末端がアミド化されている)に対して活性なエキソプロテアーゼに対してのみであろうことが予想されうる。したがって、環状コノトキシンは、末端単独の保護により予想されうるよりも強化された安定性を有するようである。
【0110】
(g)ヒト血漿中の酵素に対するcMII-6およびcMII-7の安定性
タンパク質分解作用に対するcMII-6およびcMII-7の抵抗性を試験し、これをMIIと比較するために、これらのコノトキシンをヒト血漿中でインキュベートした。MII、cMII-6およびcMII-7(10μM)を、50%ヒト血漿中で24時間インキュベートした。いくつかの時点でアリコートを採取し、15%トリクロロ酢酸水溶液でクエンチし、遠心分離機にかけた。次いで上清をRP-HPLCにより分析した。
【0111】
各試験は3連で行った。図5はこの実験の結果を示す。cMII-6およびcMII-7の安定性は、MIIを劇的に上回っている。天然MIIは約16時間の半減期を有したが、24時間後に残存するcMII-6およびcMII-7の量は90%近くであった。
【実施例2】
【0112】
環状Vc1.1の合成および特性決定
(a)Vc1.1の設計
α-コノトキシンVc1.1は、MIIを含むα-コノトキシンの4/7クラスのメンバーであり、疼痛治療の可能性を有する(Sandall DW、Satkunanathan N、Keays DA、Polidano MA、Liping X、Pham V、Down .JG、Khalil Z、Livett BG、Gayler KR、Biochemistry 2003 42(22):6904〜11頁)。文献中にVc1.1の利用できる構造データがなかったので、環状MIIアナログの設計のための前述の手順に従うことができなかった。しかしながら、この同じ骨格を有する他のコノトキシンが構造的に研究されていたため、Vc1.1のリンカーの最適な長さを、MIIおよびPnIAを含む同じファミリーの他のメンバーの距離の簡易測定により推定した。Protein Data Bankには、重複構造、およびN末端に伸長された柔軟性の「テール」を有するコノトキシンGIDを除いて、4つのα4/7コノトキシン構造がある。これらの4つの化合物のこれらの構造のN末端とC末端間の距離を平均することにより得た値は、よく一致していた(12.2±0.8Å)。後に、Vc1.1の3次元構造が報告され(Clarkら、J. Biol. Chem. (2006) 281 23254-23263頁)、Vc1.1のN末端とC末端間の距離は、11.9±0.5Åであり、α4/7コノトキシンのこの算出された平均の範囲内に入ることが明らかになった。したがって、環状Vc1.1に適したリンカーの長さは、環状MIIに使用されるものと同様、例えば6〜7残基であろうと推定することができる。この場合もまた、これらの6残基を既存のグリシン残基のN末端に付加する。
【0113】
(b)環状Vc1.1およびVc1.1の合成および構造特性決定
環状Vc1.1の合成を、環状MII(実施例1)について記載した合成手順を使用して行った。環状Vc1.1に使用した環化/酸化緩衝液は、0.1M NH4HCO3(pH8.1)であった。環化/酸化により1種の優勢な異性体(これよりcVc1.1-6と称する)が生じ、これを精製し、1H NMR分光法により分析した。Vc1.1(C末端がアミド化されている)およびcVc1.1-6の配列を以下に示す。
【0114】
【化5】
【0115】
【化6】
【0116】
直鎖Vc1.1もまた、MBHA-アミド樹脂上でin situ中和と一緒にBOC/HBTU化学反応を使用して合成した。ペプチドを0.1M NH4HCO3中、室温で一晩フォールディングさせ、単一異性体を得た。合成Vc1.1およびcVc1.1-6のジスルフィド連結性は、標準的な還元/アルキル化法[Goransson, U.およびCraik, D.J. (2003) J. Biol. Chem. 278、48188〜96頁]と一緒にMS/MSシーケンシングを使用して、両方ともCys2-Cys8、Cys3-Cys16であることが確認された。次いで、cVc1.1-6と比較するために3次元NMR構造を決定した。Vc1.1およびcVc1.1-6の両方の3次元構造を、環状MIIについて前述したNMR分光法を使用して決定した。図6に示すように、Vc1.1の天然立体構造はcVc1.1-6において保持されている。
【0117】
(c)Vc1.1のジスルフィドマッピング
Vc1.1を、TCEPと一緒にクエン酸緩衝液中、低pHでインキュベートすることにより部分的に還元した。反応混合物をRP-HPLCにより精製し、1つのジスルフィド種をN-エチルマレイミドでアルキル化した。次いで、アルキル化されたペプチドを完全に還元し、MS/MSにより分析した。MS/MSデータは、C2およびC8がN-エチルマレイミドでアルキル化されたことをはっきりと示した。ペプチド鎖の両末端からのフラグメンテーションパターンを観察すると、そのパターンは推定アルキル化パターンを完全に支持した。したがって、Vc1.1のジスルフィド連結性はC2-C8およびC3-C16であるという結論に達した。これは、他のα-コノトキシンに見られるI-III、II-IVジスルフィド結合パターンに一致する。
【0118】
(d)cVc1.1-6の生物活性
cVc1.1-6の生物活性を、環状MIIの実施例で記載したように、ウシ副腎クロム親和細胞からのカテコールアミン放出を測定することにより分析した。cVc1.1-6活性が直鎖Vc1.1の活性と実験誤差の範囲内で等しいことを例証する試験結果を図7に示す。
【0119】
(e)cVc1.1-6の安定性
cVc1.1-6がタンパク質分解酵素による攻撃に抵抗する能力を、環状MIIについて概説した実験を使用して評価することができる。哺乳動物の胃液および血漿中での分解に対する抵抗性もまた、実施例1で示した手順を使用して評価することができる。
【0120】
(f)組換えnAChRサブタイプのα-コノトキシンVc1.1抑制の選択性
AChに誘導される流れのVc1.1抑制を、種々のnAChRサブユニットの組合せを発現するアフリカツメガエル卵母細胞において調べた。Achに誘発される応答を10分毎に調べ、毒素を4分間、溶液添加した後、アゴニストと毒素を同時適用した。Vc1.1(10μM)は、中枢型nAChRサブタイプであるα4β2およびα4β4、または骨格筋型nAChRサブタイプであるαβγδ(n=7〜12)のいずれかが仲介する、AChに誘発される流れを抑制できなかった。同様に、10μM Vc1.1は、ホモ五量体ニューロンnAChRであるα7(n=11)が仲介するAChに誘発される流れをわずか14±2%しか抑制しなかった。しかしながら、10μM Vc1.1は、末梢nAChRサブタイプα3β2およびα3β4を同程度、それぞれ対照の58±7%(n=8)および56±7%(n=12)抑制した。α3α5β2(n=7)のnAChRの組合せにα5サブユニットを加えると、同様の有効性が観察されたが、Vc1.1は5倍以上低いα3α5β4(n=15)抑制の有効性を示した。AChに誘導される流れの大きさは、nAChRサブタイプα3α6β2(n=13)を含有するα6で、30μM Vc1.1によって約50%しか抑制されなかった。100nM以下の濃度でVc1.1を溶液添加しても、AChに誘発される流れを遮断せず、α3含有nAChRに対して検出可能な応答すら(すなわち50nAより大きい)導くこともなかった。
【0121】
(g)cVc1.1-6の経口および皮下有効性
よく定義された神経因性疼痛モデル(CCIラット)を使用して、皮下(s.c.)および経口ボーラス投与により投与された場合のVc1.1およびcVc1.1-6の抗異痛(疼痛緩和)作用を比較するために、試験を行った。
【0122】
神経因性疼痛の決定的な症状である接触性異痛を、ラットの左脚の坐骨神経周辺に4個の緩い結紮を結ぶことによって誘導したことにより、絞扼性神経損傷(CCI)を誘導し、同じ側の後肢(すなわち同側後肢)における軽い接触に対する過敏性をもたらした。接触性異痛を、目盛り付きvon Freyフィラメントを使用して、CCIラットの同側後肢へ段階的な不快でない圧力を加えることにより、試験した。損傷のないラットおよび損傷のない(反対側の)CCIラット後肢において、von Frey足引っ込め閾値(PWT)は約12gであるが、CCI手術の14日後には同側PWTは6g以下である。治療の目的は、接触性異痛を軽減して同側後肢のPWTを6〜12gに増加させることである。
【0123】
s.c.または経口経路により単回ボーラス投与(100μg/kg)として投与されたVc1.1およびcVc1.1-6は、平均最大同側PWTが6g未満から約10gまで増加したことによって例証されるように、接触性異痛の著しい緩和をもたらした。Vc1.1の環化は、投与経路に関係なく、親直鎖ペプチドと比較して長い作用時間をもたらすようであった(図8および9)。s.c.または経口投与された場合、Vc1.1もcVc1.1-6も、反対側の後肢のPWTを増加させず、予想と一致した(図10)。
【0124】
結果は、このin vivo神経因性疼痛モデルにおいて、cVc1.1-6が直鎖Vc1.1よりも著しく活性であったことを例証する。cVc1.1-6はまた、直鎖Vc1.1よりも長い半減期を有し、より安定で、動物において経口利用可能である。
【0125】
(h)環状Vc1.1の抗異痛有効性および効能
CCIラットにおけるcVc1.1-6の単回経口ボーラス投与の抗異痛有効性を実証するために、試験を行った。
【0126】
有効用量範囲を特定するために、各CCIラット(n=6)はcVc1.1-6の単回経口ボーラス投与を、投与間に最低2日間のウォッシュアウト期間を設けて最大5回受けた。この方法を使用して、環状Vc1.1の抗異痛有効用量は0.3mg/kg〜3mg/kgであった。十分量のペプチドが入手できなかったため、これより多い用量は試験しなかった。対照動物はビヒクルの経口ボーラス投与を受けた(n=3)。環状Vc1.1およびビヒクルは、1ml Hamiltonガラスシリンジを使用して500μLの量で投与し、PWTは上記2(h)に概説した手順を利用して決定した。
【0127】
CCIラットは、cVc1.1-6の単回経口ボーラス投与を1mg/kgで受けた(n=3)。対照動物は、ビヒクルの単回経口ボーラス投与を受けた(n=3)。cVc1.1-6またはビヒクルは、Hamiltonガラスシリンジを使用して500μLの量で投与し、PWTは上記2(h)に概説した手順を利用して決定した。
【0128】
実験手順の終了後、ラットを100% CO2、続いて頸椎脱臼により安楽死させた。ラット死体は、クイーンズランド大学バイオ廃棄物回収サービスにより回収されるまで冷凍した。
【0129】
平均(±SEM)PWT対時間曲線を、CCIラットにおけるcVc1.1-6およびビヒクルの各用量についてプロットした。またPWT値を、それぞれの投与前基準値を引くことにより正規化し、正規化された応答対時間曲線(AUC)の下面積を、台形積分を使用して概算した。AUC値を用量に対してプロットし、ED50値を概算した(図12)。
【0130】
予想通り、接触性異痛は、反対側の(損傷していない)後肢の各平均(±SEM)PWT(10.6±0.2g)に対する、CCI手術から21日後の同側後肢の平均(±SEM)PWT(4.8±0.2g)の顕著な減少(p<0.05)として現れた。治療の目標は、接触性異痛の完全な回復、すなわち、約11gの同側後肢のvon Frey PWTである。
【0131】
「ウォッシュアウト」法に従うCCIラットへのcVc1.1-6の単回経口ボーラス投与(0.3〜3mg/kg)の後に、同側後肢において用量依存的な接触性異痛の緩和が速やかに開始した。試験した用量全てにおいて、同側後肢の抗異痛は投与後約0.5時間で最大になり、対応する作用持続時間は1〜1.25hであった。試験したcVc1.1-6の最大用量(3mg/kg)において、平均(±SEM)PWTは、投与前の5.2(±0.4g)から投与後0.5hで最大効果(9.5±1g)まで増加した。CCIラットの同側後肢における抗異痛の平均(±SEM)範囲および持続時間(ΔPWT AUC値)は、用量依存的に増加した。cVc1.1-6のおおよそのED50値は、1mg/kgと概算し、この用量を薬物未投与CCIラットへの投与に選択した。CCIラットにおける最大3mg/kgまでの用量でのcVc1.1-6経口投与は、反対側の後肢にわずかな抗侵害受容を引き起こした(図11)。
【0132】
薬物未投与CCIラットへcVc1.1-6を1mg/kgで単回経口ボーラス投与した後に、同側後肢において接触性異痛の緩和が速やかに開始した。「ウォッシュアウト」法の結果と一致して、同側後肢の抗異痛は投与後約0.5時間で最大になり、対応する平均作用持続時間は1.5hであった。特に、CCIラットの同側後肢の平均(±SEM)PWT値は、投与前の4(±0g)から最大効果時(0.5h)の8.7(±0.7)gまで増加した。薬物未投与CCIラットにおける1mg/kgのcVc1.1-6経口投与は、反対側の後肢にわずかな抗侵害受容を引き起こした。
【0133】
試験において、試験した用量(0.3〜3mg/kg)でのcVc1.1-6単回経口ボーラス投与により、CCIラットの行動的副作用は認められなかった。
【0134】
CCIラットへのcVc1.1-6の0.3〜3mg/kgでの単回経口ボーラス投与は、対応する反対側の後肢に抗侵害受容を引き起こすことなく、同側(損傷した)後肢に用量依存的抗異痛応答を引き起こした。cVc1.1-6が反対側の(損傷していない)後肢に抗侵害受容を引き起こさなかったという観察結果は、cVc1.1-6が、内因性の疼痛抑制機構を増強することではなく侵害受容促進(疼痛促進)経路を調節することによりその疼痛緩和作用をもたらす、という見解と一致する。
【0135】
本明細書中の、任意の先行出版物(またはそこから得られる情報)、または任意の既知の事実に関する参考文献は、その先行出版物(またはそこから得られる情報)または既知の事実が、本明細書が関わる試みの分野に共通の一般知識の一部を形成するという認識または承認または任意の形態の示唆としてみなされず、またみなされるべきではない。
【0136】
当業者であれば、本明細書中に記載の発明は、具体的に記載された以外の変形および修正が可能であることを理解するであろう。本発明は、本発明の精神および範囲に入る全てのそのような変形および修正を含むことを理解されたい。本発明はまた、本明細書で個別に、またはまとめて言及された、または示された全ての段階、特徴、組成物および化合物、ならびに前記段階および特徴の任意の2つ以上の任意のおよび全ての組合せを含む。
【図面の簡単な説明】
【0137】
【図1】C末端チオエステルを介したペプチド環化のスキームを示す図である。N末端システインの遊離硫黄が、C末端チオエステルと相互作用して中間体を形成し、この中間体がS、Nアシル基転移を受けることにより天然ペプチド結合を有する環状ペプチドを形成する。
【図2】コノトキシンMII、cMII-6およびcMII-7の3次元構造を表す図であり、これらの構造の重ね合わせを含む。ジスルフィド結合をボールアンドスティックで表す。構造はNMR分光法により決定した。
【図3】MII、cMII-5、cMII-6およびcMII-7の濃度の増加による単離ウシクロム親和細胞からのニコチン(5μM)誘発性カテコールアミン放出の阻害に関する濃度-応答曲線を図示するグラフである。結果は、環状ペプチドの活性が直鎖MIIと同等であることを示す。
【図4】ペプチドをエンドプロティナーゼ、Endo Glu-Cと一緒にインキュベートすることにより試験したときの、タンパク質分解酵素による攻撃に対するMII、cMII-6およびcMII-7の相対的安定性を図示するグラフである。残存する無傷のペプチド量はRP-HPLCにより測定した。
【図5】50%ヒト血漿中の酵素に対するMII、cMII-6およびcMII-7の相対的安定性を図示するグラフである。残存する無傷のペプチド量はRP-HPLCにより測定した。
【図6】Vc1.1(黒)および環状Vc1.1-6(灰色)の3次元構造を表す図である。構造はNMR分光法により決定した。各ペプチドについて、20個の最小エネルギー構造の骨格を重ね合わせて示し、Vc1.1の天然立体構造がcVc1.1-6で保持されていることを示す。
【図7】ウシ副腎クロム親和細胞からのカテコールアミン放出を測定することにより試験したときの、cVc1.1-6およびVc1.1の相対的生物活性を図示するグラフである。クロム親和細胞を、示した量のペプチドと一緒に20分間インキュベートし、次いで阻害剤が引き続き存在する中で10pMニコチンで20分間刺激した。アリコートを採取し、以前に記載されたように、蛍光分析によりカテコールアミン分泌について試験した(n=6実験)(Meunierら、2002年)。
【図8】Vc1.1およびcVc1.1-6単回ボーラスs.c.投与(100μg/kg)での6g未満から約10gまでのCCIラット同側後肢における平均足引っ込め閾値(PWT)の増加を図示するグラフである。
【図9】Vc1.1およびcVc1.1-6単回ボーラス経口投与(100μg/kg)での6g未満から約10gまでのCCIラット同側後肢における平均足引っ込め閾値(PWT)の増加を図示するグラフである。
【図10】CCIラットにおけるVc1.1およびcVc1.1-6単回ボーラスs.c.または経口投与(100μg/kg)でのCCIラット反対側後肢における平均足引っ込め閾値(PWT)を図示するグラフである。
【図11】CCIラット同側後肢において用量依存的な接触性異痛の緩和をもたらした、Vc1.1単回経口ボーラス投与(0.03〜3mg/kg)での平均PWTを図示するグラフである。
【図12】用量に対して抗異痛の程度および持続時間(ΔPWT AUC値)をプロットして作成した用量-応答曲線を図示するグラフである;おおよそのED50値は1mg/kgであると概算された。
【技術分野】
【0001】
本発明はα-コノトキシンペプチドに関し、詳細にはヒトの治療的処置に有用な環状α-コノトキシンペプチドに関する。本発明は特に、これらのペプチドを含む経口および経腸製剤、医薬製剤の製造におけるこれらのペプチドの使用、およびヒトの状態または疾患の予防または治療におけるこれらの医薬製剤の使用に関する。
【背景技術】
【0002】
Conus属(イモガイ)のマリンスネイル(marine snail)は、巧みな生化学的戦略を使用して獲物を捕獲する。魚、虫、または他の軟体動物の捕食者として、イモガイは小さな生物活性ペプチドのカクテルを含有する毒液を獲物に注入する。これらの毒素分子はコノトキシンと呼ばれ、種々のイオンチャネルまたは受容体を標的にすることによって神経伝達を阻害する。コノトキシンは、通常、直鎖状に配列された12〜30個のアミノ酸を含有する。Conus属の任意の1種の毒液は、100種を超えるさまざまなペプチドを含有しうる。コノトキシンはその生理学的標的に基づいて分類される。例えば、α-コノトキシンおよびψ-コノトキシンはニコチン性ACh受容体を標的として神経節および神経筋遮断を引き起こし、一方ω-コノトキシンクラスのペプチドは電位感受性Ca2+チャネルを標的として神経伝達物質放出を抑制する。
【0003】
ほとんどのコノトキシンペプチドは、2個または3個のジスルフィド結合をそれぞれ形成するように対になって結合した4個または6個のシステイン残基を含有するが、1個のジスルフィド結合を形成するように結合した2個のシステイン残基を有する例(すなわちコノプレシン)に加えて、3個を超えるジスルフィド結合を有するものもあれば、システイン残基またはジスルフィド結合を含有しないものもある。前述の「活性」クラスのペプチドの一部は、構造モチーフが共通であり、同数のシステイン残基および同じジスルフィド結合連結性を有している。このため、新しい「スーパーファミリー」分類体系が展開された。例えば、ω-コノトキシンならびにδおよびμ-コノトキシンクラスのメンバーは、システイン残基IとIV、およびIIとV、およびIIIとVI(6個のローマ数字はN末端から番号付けした6個のシステイン残基を表す)間に3個のジスルフィド結合を形成するように対になって結合した6個のシステイン残基を有する。この構造モチーフを有するコノトキシンペプチドはコノトキシンのO-およびM-スーパーファミリーに属する。同様に、ρ-コノトキシンおよびほとんどのα-コノトキシンは、システイン残基IとIII、およびIIとIV間に2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を有する。これらのコノトキシンペプチドは、コノトキシンの「A-スーパーファミリー」に属する。本発明は、A-スーパーファミリーのα-コノトキシン、すなわちシステイン残基IとIII、およびIIとIV間に形成された2個のジスルフィド結合を有するα-コノトキシンペプチドに関する。前述したように、コノトキシンペプチドは哺乳動物のさまざまな異なるイオンチャネル受容体に結合するので、したがって、ヒトの疼痛軽減を含めてさまざまな治療に応用できる可能性を有している。しかしながら、一般にペプチドは、その薬剤としての使用に伴い、一般にバイオアベイラビリティが低いこと、プロテアーゼによる切断の受けやすさ、および望ましくない副作用を含むいくつかの問題を有している。
【0004】
1種のω-コノトキシン、MVIIA(SNX-111、ZiconotideおよびPrialtとしても知られる)は最近、癌、AIDSおよび神経障害に伴う難治性疼痛の治療に関して米国食品医薬品局の承認を取得した。投与経路は、いくつかの前述の問題のため、およびこの薬剤が標的とする受容体がCNS内に位置するために、現在のところ脊髄への髄腔内注入に制限されている。
【0005】
臨床試験を開始した他のω-コノトキシンは、Ziconotideと比較してP/Q型チャネルに対するN型カルシウムチャネルへの選択性が向上したと報告されているCVID(AM-336、Zenyth Pharmaceuticals)である。しかしながら、特定の受容体/チャネルへの選択性が向上しても、ωクラスのコノトキシンは一部の患者において望ましくない副作用をやはり伴う。2種の他のコノトキシンペプチド(アンボイナ(Conus geographus)から単離されたCGX-1160およびCGX-1007)も臨床試験中であるが、これらのペプチドの投与も髄腔内経路に制限されている。疼痛に関して研究されている他のペプチドは、χ-クラスのコノトキシン(Xen 2174、Xenome社)である。しかしこの場合も、このペプチドの投与は髄腔内経路に制限されている。
【0006】
薬剤の髄腔内または脊髄投与の主な不利点は、投与が医師または看護師によって行われなければならないこと、および長期にわたる治療のために、機械的送達システムおよび脊髄カテーテルを患者の脊柱に挿入することが好ましいことである。したがって、このような治療は通常、末期および/または入院患者にのみ用いられる。
【0007】
α-コノトキシンペプチドは、典型的には大きさが12個から16個のアミノ酸であり、アミド化されたC末端を通常有するコノトキシンのサブファミリーである。α-コノトキシンは、ニコチン性アセチルコリン受容体を抑制することが知られている。α-コノトキシンは、鎮痛および中毒において役割を果たすだけでなく、アルツハイマー病、統合失調症、うつ病および小細胞肺癌を含むさまざまな障害に関与している筋肉とニューロンのニコチン性アセチルコリン受容体(nAChR)の両方と相互作用する。α-コノトキシンペプチドおよびその使用の可能性は、文献において広く記載されている。(LloydおよびWilliams、2000年、J Pharmacol Exp Ther 292 (2) 461頁参照。)
【0008】
システイン残基間の残基数を使用して、異なるクラスのα-コノトキシンを識別する。第2と第3システイン残基の間(ループ1)および第3と第4残基の間(ループ2)の残基数に基づいて、α-コノトキシンはα3/5、α4/3、α4/4、α4/6およびα4/7構造サブファミリーに分類される。4/7ループ配列を有するα-コノトキシンペプチドの2つの例は、MIIおよびVc1.1(Metabolic Pharmaceuticals社、ACV1としても知られる)である。最も小さいα-コノトキシンペプチドの1つは、4/3ループ配列を有するIM1である。
【0009】
さまざまなサブタイプのニコチン性アセチルコリン受容体への選択性、特に、中枢サブタイプに対する末梢nAChRサブタイプへの選択性を解明するために、いくつかのα-コノトキシンペプチドが研究されてきた。nAChRは、CNSおよびPNS全体にわたって低レベルで発現されているが、各種サブタイプは異なる分布を有する。哺乳動物のnAChRはサブユニットの組合せからなっている。これらのサブユニット型の7種はリガンド結合に関与する主な構成要素(α2、α3、α4、α6、α7、α9およびα10)であり、4種のサブユニット型(α5、β2、β3およびβ4)は、受容体に機能的および薬理学的特性を付与する構造であると考えられている。異なるサブタイプがさまざまな形で(一般にヘテロ五量体として)結合し、特定の薬理学的および電気生理学的特性を有する受容体を形成する。α3サブユニットは(PNSに存在することから)末梢サブユニットであると考えられ、一方α7サブユニットはCNSで優勢なサブユニットであると考えられている。
【0010】
α-コノトキシンVc1.1は、ビクトリアジョウオウイモ(Conus victoriae)の毒管由来cDNAのPCRスクリーニングを使用して最初に発見された(Sandallら、Biochemistry、2003年、42、6904頁)。Vc1.1配列内のシステインの間隔は、Vc1.1が、広く研究されているコノトキシンMII、EpIおよびPnIBを含むα-コノトキシンの4/7サブクラスのメンバーであることを示している。Vc1.1の3次元構造は、小さな膜貫通α-へリックス残基P6からD11を含み、他のα-コノトキシンで見られるI-II、III-IVジスルフィド連結性により支持されている(Clarkら、J. Biol. Chem.2006年、281、23254頁)。ほとんどのα-コノトキシンに共通するアミド化されたC末端に加えて、直鎖Vc1.1のPro6およびGlu14残基をそれぞれヒドロキシプロリンおよびγ-カルボキシグルタミン酸に翻訳後修飾することも可能である。この翻訳後修飾されたVc1.1アナログは、神経再生に関与しているが疼痛には関与していない。(WO02/079236)
【0011】
ウシクロム親和細胞中のニューロンnAChRのアンタゴニストである直鎖Vc1.1は、ヒト神経因性疼痛の3種のラットモデルにおいて神経因性疼痛を軽減し、損傷したニューロンの機能回復を促進することが示されている(Satkunanathanら、2005年、Brain Research 1059 (2) 149〜158頁)。加えて、糖尿病性神経障害の動物モデルにおいてACV-1が神経因性疼痛の軽減に有効であることがLivettらによって報告されている。特に、末梢神経障害のストレプトゾトシン誘導糖尿病ラットモデルにおいて、ACV-1の抗異痛作用が30および300μg/kgの用量で投薬1時間以内に観察された(IDrugs、15th World Congress on Animal、Plant and Microbial Toxins、2006年 9、679〜681頁)。鎮痛剤として、Vc1.1はZiconotideより活性が高いことが報告されている(Sandallら、2003年、Biochemistry、42、6904〜6911頁)。より最近では、Vc1.1は、ヒト末梢神経の遊離切片において、ニコチンに誘導される無髄C線維軸索の軸索興奮性の増大を弱めることが示された(Langら、2005年、Neuroreport 16、479〜483頁)。前述したように、ニューロンnAChRは、中枢および末梢神経系全体で見られるα(α2〜α10)およびβ(β2〜β4)サブユニットの組合せからなる五量体のリガンド依存性イオンチャネルである。電気生理学的および免疫組織化学的データは、無髄C線維の軸索における、α4、β2またはα7ではなくα3、α5およびβ4サブユニットからなるnAChRの機能発現を示す(Langら、2005年、Neuroreport 16、479〜483頁;およびLangら、2003年、Neurophysiol 90、3295〜3303頁)。無髄末梢神経線維でのnAChR遮断は、無髄交感神経および/または感覚軸索に対して鎮痛作用を有する場合がある。興味深いことに、翻訳後修飾された合成Vc1.1(ptm Vc1.1)は、クロム親和細胞においてニューロン型ニコチン反応を抑制しないことが報告され、2つのラット神経因性疼痛試験において不活性であった。直鎖Vc1.1またはACV1は、第2相ヒト臨床試験を開始している。ニコチン作動薬は鎮痛作用を有することが以前に報告されている。このようなニコチン作動薬の例は、エピバチジンおよびABT-594である。これらの作用物質は、ニコチン性受容体の感受性を減じ、受容体を通過するイオン流の減少をもたらすことにより作用すると想定されている。このような状況下で、ニコチン作動薬はnAChR拮抗薬として効率的に作用しており、このためnAChR拮抗薬は鎮痛化合物候補として探求されてきた。コノトキシンペプチドVc1.1はこのような拮抗薬であるといわれている。
【0012】
興味深いことに、nAChRのα3、β4サブタイプのVc1.1のIC50値はマイクロモルの範囲である(4.2μM; Vinclerら、2006年、PNAS、103、17881頁)。Vc1.1の鎮痛作用はin vivoにおいてナノモル濃度で現れるようである。
【0013】
Vc1.1が、α9、α10nAChRの調節によって鎮痛をもたらしうることが最近報告されている(Vinclerら、上記)。Vc1.1のα9、α10nAChRに対する親和性は、α3、β4nAChRに対してよりも100倍高く、ナノモル範囲に入る(22.9nM)。したがって、Vc1.1の生理学的標的の1つは、広範に分布していることが知られ、後根神経節ニューロン、下垂体、リンパ球、皮膚ケラチノサイトおよび精子中に存在するα9、α10nAChRとすることができるようである。しかしながら、Vc1.1の疼痛緩和作用の原因となる、まだ発見されていない他の標的が存在しうると考えられる。
【0014】
疼痛および他の状態の治療のために研究されてきた以前のコノトキシンペプチドとは異なり、Vc1.1は、髄腔内ではなく皮下または筋肉内投与が可能であるという利点を有するといわれている。このことは、Ziconotideを含む以前のコノトキシンペプチドと比べてVc1.1に大きな利益をもたらすといわれている。しかしながら、コノトキシンペプチドVc1.1は経口バイオアベイラビリティを欠くといわれている。2006年1月付のMetabolic Pharmaceuticalのウェブサイトで発表された文書「ACV1、神経因性疼痛の新規治療、前臨床データの技術概要(Technical Summary of Preclinical Data)」によると、ACV1は経口で使用できないが、皮下注射処置用として現在開発されている。Vc1.1はまた、炎症性疼痛の動物モデルにおいて有効であり、損傷した神経および組織の回復を促進するとも報告されている。
【0015】
Metabolic Pharmaceuticals社によるWO2007/014432は、ヒト成長ホルモンおよびそのアナログのC末端配列をペプチド薬のC末端に連結させることによってペプチド薬の経口送達を向上させる方法を記載している。ペプチド薬へのヒト成長ホルモンのC末端配列の付加は、ペプチド薬に経口バイオアベイラビリティ特性を付与するとされている。この特許出願は、神経因性疼痛の動物モデルにおいて活性であるらしい、経口で使用できるペプチドを記載している。ペプチドはVc1.1のC末端に連結したアミノ酸配列Tyr-Leu-Arg-Ile-Valを有するVc1.1を含む。
【特許文献1】WO02/079236
【特許文献2】WO2007/014432
【特許文献3】1999年9月14日に出願された国際特許出願第PCT/AU99/00769号(WO00/15654)
【非特許文献1】LloydおよびWilliams、2000年、J Pharmacol Exp Ther 292 (2) 461頁
【非特許文献2】Sandallら、Biochemistry、2003年、42、6904頁
【非特許文献3】Clarkら、J. Biol. Chem.2006年、281、23254頁
【非特許文献4】Satkunanathanら、2005年、Brain Research 1059 (2) 149〜158頁
【非特許文献5】IDrugs、15th World Congress on Animal、Plant and Microbial Toxins、2006年 9、679〜681頁
【非特許文献6】Sandallら、2003年、Biochemistry、42、6904〜6911頁
【非特許文献7】Langら、2005年、Neuroreport 16、479〜483頁
【非特許文献8】Langら、2003年、Neurophysiol 90、3295〜3303頁
【非特許文献9】Vinclerら、2006年、PNAS、103、17881頁
【非特許文献10】2006年1月付のMetabolic Pharmaceuticalのウェブサイトで発表された文書「ACV1、神経因性疼痛の新規治療法、前臨床データの技術概要(Technical Summary of Preclinical Data)」
【非特許文献11】Hoggら、1999年、J Biol Chem. 274(51):36559〜64頁
【非特許文献12】Hoggら、 2003年、Reviews of Physiology, Biochemistry and Pharmacology 1: 1〜46頁
【非特許文献13】Schnolzerら、(1992) Int. J. Pept. Protein Res. 40、180〜193頁
【非特許文献14】Dawsonら、(1994) Science 266、776〜9頁
【非特許文献15】Tamら、(1999) J. Am. Chem. Soc. 121、4316〜4324頁
【非特許文献16】Yan, L.Z.およびDawson, P.E. (2001) J Am Chem Soc 123、526〜33頁
【非特許文献17】Ingenito, R.、 Bianchi, E.、Fattori, D.およびPessi, A. (1999) J Am Chem Soc 121、11369〜74頁
【非特許文献18】Shin, Y.、Winans, K.A.、Backes, B.J.、Kent, S.B.H.、Ellman, J.A.およびBertozzi, C.R. (1999) J Am Chem Soc 121、11684〜89頁
【非特許文献19】Clippingdale, A.B.、Barrow, C.J.およびWade, J.D. (2000) J Pept Sci 6、225〜34頁
【非特許文献20】Camarero, J.A.、Hackel, B.J.、De Yoreo, J.J.およびMitchell, A.R. (2004) J Org Chem 69、4145〜511頁
【非特許文献21】Rance, M.、Sorensen, Q.W.、Bodenhausen, G.、Wagner, G.、Ernst, R.R.およびWiithrich, K. (1983) Biochem. Biophys. Res. Commun. 117、479〜485頁
【非特許文献22】Bax, A.およびDavis, D.G. (1985) J. Magn. Reson. 65、355〜360頁
【非特許文献23】Griesinger, C.、Sorensen, O.W.およびErnst, R.R. (1987) J. Magn. Reson. 75、474〜492頁
【非特許文献24】Jeener, J,、Meier, B.H.、Bachmann, P.およびErnst, R.R. (1979) J. Chem. Phys. 71、4546〜4553頁
【非特許文献25】Kumar, A.、Ernst, R.R.およびWitthrich, K. (1980) Biochem. Biophys. Res. Commun. 95、1-6頁
【非特許文献26】Piotto, M.、Saudek, VおよびSklenar, V. (1992) J. Biomol. NMR 2、661〜665頁
【非特許文献27】Eccles, C.、Guntert, P.、Billeter, M.およびWuthrich, K. (1991) J. Biomol. NMR 1、111〜30頁
【非特許文献28】Wuthrich, K. (1986) Wiley-Interscience、New York
【非特許文献29】Guntert, P.、 Mumenthaler, C.およびWuthrich, K. (1997) J. Mol. Biol. 273、283〜98頁
【非特許文献30】Brunger, A.T.、Adams, P.D.およびRice, L.M. (1997) Structure 5、325〜336頁
【非特許文献31】Linge, J.P.およびNilges, M. (1999) J. Biomol. NMR 13、51〜9頁
【非特許文献32】Hutchinson, E.G.およびThornton, J.M. (1996) Protein Sci. 5、212〜220頁
【非特許文献33】Laskowski, R.A.、Rullmannn, J.A.、MacArthur, M.W.、Kaptein, R.およびThornton, J.M. (1996) J. Biomol. NMR 8、477〜86頁
【非特許文献34】Lawrence, G.W.、Weller, U.およびDolly, J.O. (1994) Eur J Biochem 222、325〜33頁
【非特許文献35】Meunier, F.A.、Mattei, C.、Chameau, P.、Lawrence, G.、Colasante, C.、Kreger, A.S.、Dolly, J.O.およびMolgo, J. (2000) J Cell Sci 113 (Pt 7)、1119〜25頁
【非特許文献36】Meunier, F.A.、Feng, Z.P.、Molgo, J.、Zamponi, G.W.およびSchiavo, G. (2002) Embo J 21、6733-43頁
【非特許文献37】Sandall DW、Satkunanathan N、Keays DA、Polidano MA、Liping X、Pham V、Down .JG、Khalil Z、Livett BG、Gayler KR、Biochemistry 2003 42(22):6904〜11頁
【非特許文献38】Clarkら、J. Biol. Chem. (2006) 281 23254-23263頁
【非特許文献39】Goransson, U.およびCraik, D.J. (2003) J. Biol. Chem. 278、48188〜96頁
【発明の開示】
【発明が解決しようとする課題】
【0016】
したがって、特に、鎮痛の惹起、神経因性疼痛の治療または予防に関して、および神経損傷からの回復促進において、経口または経腸経路を介してα-コノトキシンペプチドを用いて患者を治療する有効な方法が依然として必要である。
【課題を解決するための手段】
【0017】
本発明は、α-コノトキシンペプチドを環化することによりアミド環状骨格を有する化合物を生成すると、経口有効性を有する環状ペプチドをもたらすという驚くべき発見に基づく。
【0018】
したがって第1の態様では、本発明は、遊離N末端およびC末端を有さないようなアミド環状骨格を有する少なくとも1種の合成環状α-コノトキシンペプチドを、医薬として経口または経腸投与に適したビヒクル中に含む経口または経腸医薬製剤であって、このペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端はペプチドリンカーを介してC末端に連結されている、医薬製剤を提供する。
【0019】
ペプチドリンカーが、6個から8個の天然または非天然アミノ酸がシステイン残基IおよびIVの間にわたる長さであることが好ましい。
【0020】
本発明はさらに、前述の医薬製剤を経口または経腸投与する段階を含む、疼痛の治療または予防方法を提供する。
【0021】
さらなる実施形態では、本発明は、前述の医薬製剤を経口または経腸投与する段階を含む、神経損傷の回復を促進する方法を提供する。
【0022】
本発明はさらに、前述の医薬製剤を経口または経腸投与する段階を含む、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、気分変調、トゥレット症候群および季節性情動障害など)または炎症の治療または予防方法を提供する。
【0023】
さらなる実施形態では、本発明は、疼痛の治療もしくは予防のための、または神経損傷からの回復促進における経口または経腸投与用の医薬の製造における環状α-コノトキシンペプチドの使用を提供する。
【0024】
本発明はまた、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、トゥレット症候群、気分変調および季節性情動障害など)または炎症の治療または予防のための経口または経腸投与用の医薬の製造における環状α-コノトキシンペプチドの使用も提供する。
【0025】
好ましい実施形態の説明
α-コノトキシンクラスは、文献中に数百の例が記載されている非常に大きなコノトキシンペプチドのクラスである。大部分のα-コノトキシンペプチドは、対になって結合した4個のシステイン残基を有し、システイン残基IとIII、およびシステイン残基IIとIVの間で2個のジスルフィド結合を形成するA-スーパーファミリーのメンバーである。文献中に記載されているα-コノトキシンペプチドの一部の例を以下の表1に示す。
【0026】
【表1】
【0027】
上記表において、γはγ-カルボキシグルタミン酸を、Oは4-ヒドロキシプロリンを表す。その他の記号は、天然アミノ酸を指定するのに一般に使用されるものである。上記ペプチドの大部分はアミド化C末端を有する。
【0028】
安定性を向上させるためのコノトキシンペプチドの環化は、1999年9月14日に出願された国際特許出願第PCT/AU99/00769号(WO00/15654)においてCraikらによって最初に記載された。したがって、本発明により処方される環状α-コノトキシンペプチドは、その内容全体が参照により本明細書に組み込まれるその特許出願に記載された方法を使用して調製することができる。
【0029】
本明細書において、文脈上他の意味に解すべき場合を除き、コノトキシンペプチドに関連して使用される場合の「直鎖」という用語は、ペプチドが非環状状態にある、すなわち、アミド環状骨格を形成するためにN末端とC末端が(直接またはリンカーを用いて)連結されていないことを意味する。α-コノトキシンペプチド中に1個または複数個のジスルフィド結合が存在するとペプチドはある程度環状になるはずであるが、N末端とC末端の連結によりペプチド骨格が環化されていなければ、そのようなジスルフィド結合を有するペプチドであってもやはり「直鎖」とみなされるべきである。
【0030】
本明細書において使用される「経口/経腸バイオアベイラビリティ」、「経口/経腸投与可能な」、「経口/経腸投与に適した」および「経口有効性」という用語は、環状ペプチドが経口または経腸経路によって投与され、疼痛、アルツハイマー病などの治療もしくは予防、または神経損傷の回復促進などの薬学的に適切な効果をもたらす能力を指す。この効果は、ペプチドとその想定された標的の相互作用によって起こると考えられるが、用語はそのような制限を本発明の範囲に課すものではない。他のまたは代替の標的が生理学的にin vivoで関与してもよく、ペプチド代謝産物が薬学的に適切な効果をもたらすのに関与してもよい。例えば、モルヒネ鎮痛剤は、オピオイド受容体に結合し、その後オピオイド受容体がカルシウムおよび/またはカリウムチャネルを調節することにより作用することが知られている。
【0031】
直鎖α-コノトキシンペプチドは、環化可能な任意のα-コノトキシンペプチドとすることができる。直鎖α-コノトキシンペプチドは、天然α-コノトキシンペプチドの配列を有してもよく、またはその誘導体でもよい。α-コノトキシンペプチドは、非環状の形態において、ヒトなどの哺乳動物の治療的処置に関連する活性を有することが好ましい。ペプチドの環化はペプチドの活性を変化させる、または新たな活性を導入する可能性があるので、一部の環状コノトキシンペプチドが、「直鎖」コノトキシンと比較して改変され向上した治療特性を有しうる可能性がある。場合によっては、環状コノトキシンペプチドは、直鎖α-コノトキシンペプチドと異なるジスルフィド連結性、例えば、CysI-IV、II-III(リボン状)連結性およびCysI-II、III-IV(数珠状)連結性を有するであろう。ペプチドはまた、異性体の組合せとして存在してもよい。
【0032】
本発明の一実施形態によると、環化を受ける直鎖α-コノトキシンペプチドは、以下に示す配列を含む4/7または4/6ペプチドである:
Xaa1 CCS Xaa2 P Xaa3 C Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 C 配列番号1
(配列中、Xaa1はグリシンまたはアスパラギン酸であり、Xaa2からXaa7は任意の天然または非天然アミノ酸を表し、Xaa8はプロリン、ヒドロキシプロリンまたはグルタミンを表し、Xaa9はアスパラギン酸、グルタミン酸またはγ-カルボキシグルタミン酸を表し、Xaa10は任意の天然または非天然アミノ酸を表すか、なくてもよい)。
【0033】
より好ましくは、Xaa1からXaa10は以下のように選択される:
Xaa1はグリシンまたはアスパラギン酸であり、
Xaa2はアスパラギン酸、アスパラギン、ヒスチジン、チロシン、アルギニンまたはリシンから、さらにより好ましくはアスパラギン酸、アスパラギン、およびヒスチジン、最も好ましくはアスパラギン酸から選択され、
Xaa3はアルギニン、プロリン、アラニン、バリンまたはセリン、より好ましくはアルギニン、プロリンまたはアラニン、最も好ましくはアルギニンから選択され、
Xaa4はアスパラギン、アラニン、アルギニン、チロシン、ヒスチジン、フェニルアラニン、セリン、イソロイシンまたはリシン、より好ましくはアスパラギン、アラニンまたはチロシン、および最も好ましくはアスパラギンから選択され、
Xaa5はチロシン、ヒスチジン、アラニン、バリン、グルタミン、グリシン、ロイシン、セリン、チオニン、アスパラギン、アスパラギン酸、グルタミン酸、リシンまたはアルギニン、より好ましくは親水性アミノ酸残基、および最も好ましくはチロシンから選択され、
Xaa6はアスパラギン酸、アスパラギン、セリン、チロシン、グルタミン酸、グリシン、アルギニンまたはヒスチジン、より好ましくはアスパラギン酸またはアスパラギン、および最も好ましくはアスパラギン酸から選択され、
Xaa7はヒスチジン、アスパラギンまたはチロシン、より好ましくはヒスチジンから選択され、
Xaa8はプロリン、ヒドロキシプロリン、グルタミンまたはセリン、より好ましくはプロリンから選択され、
Xaa9はグルタミン酸、γ-カルボキシグルタミン酸、アスパラギン酸、グリシンまたはアスパラギン、より好ましくはグルタミン酸、γ-カルボキシグルタミン酸、および最も好ましくはグルタミン酸から選択され、
Xaa10はイソロイシン、チロシン、ロイシンまたはdes-Xaa10、より好ましくはイソロイシンから選択される。
【0034】
特に好ましい実施形態において、
Xaa1はグリシンまたはアスパラギン酸であり、
Xaa2はアスパラギン酸であり、
Xaa3はアルギニンであり、
Xaa4はアスパラギンであり、
Xaa5はチロシンであり、
Xaa6はアスパラギン酸であり、
Xaa7はヒスチジンであり、
Xaa8はプロリンであり、
Xaa9はグルタミン酸であり、
Xaa10はイソロイシンである。
【0035】
好ましい実施形態において、環化を受ける直鎖アルファコノトキシンペプチドは、α4/7コノトキシンペプチドである。このペプチドは、特定のサブタイプのnAChRに対する選択性を有しうる。例えば、このペプチドはα7サブタイプのnAChRよりもα3またはα9/α10サブタイプに対する選択性を有しうる。
【0036】
さらに好ましい実施形態において、環化を受ける直鎖アルファコノトキシンペプチドはVc1.1である。
【0037】
本発明による環状コノトキシンペプチドは、通常、N末端とC末端が連結部分を介して連結されたα-コノトキシンペプチドからなるはずであるが、場合によっては、天然α-コノトキシンペプチドまたはその誘導体のN末端とC末端を、さらなる連結部分を必要とせずに、直接連結することが可能でありうる。連結部分は、存在する場合、環化によりアミド環状ペプチド骨格が生成されるようなペプチドリンカーとすることができる。これらのペプチドは、遊離N末端およびC末端を有さないはずである。
【0038】
連結部分のペプチド配列は大きく変化させることが可能である。この連結部分は、α-コノトキシンの主要活性部位に必ずしも結合するか、またはかみ合うわけではないので、この部分を改変して生理化学的特性を変化させ、可能性としてコノトキシンの副作用を低減させることができる。
【0039】
コノトキシンのN末端とC末端を連結するとき、場合によってはN末端またはC末端残基の1つまたは複数を除去または改変することが必要であるか、または望ましいことがある。直鎖コノトキシン配列のそのような改変は、本発明の範囲内である。例えば、直鎖コノトキシン末端の正または負電荷が活性に重要であった場合、環化によるその欠損は、それぞれリシンまたはグルタミン酸などの正または負の側鎖を有するアミノ酸の置換により救済することができるだろう。
【0040】
連結部分は必ず、コノトキシンペプチドのN末端とC末端の間にわたるのに十分な長さであるはずである。ペプチドリンカーの場合、長さは一般に2から10アミノ酸程度であろう。場合によっては、より長いまたは短いペプチドリンカーが必要とされることがある。一実施形態において、連結部分は、直鎖α-コノトキシンにすでに存在する任意のアミノ酸残基に加えてグリシンおよび/またはアラニン残基からなる。
【0041】
A-スーパーファミリーのα-コノトキシンペプチドについて、システイン残基IおよびIV間の距離が実質的に保存されていることが見出された。そのようなコノトキシンペプチドに関しては、結合角および距離を考慮して、システイン残基IとIVの間のアミノ酸数が6個〜8個になるようにリンカーの長さを選択することが好ましい。したがって、α-コノトキシンVc1.1およびMIIに関して、リンカーに必要な追加のアミノ酸残基は、N末端にすでに存在する1個のアミノ酸を考慮して5個〜7個ということになる。
【0042】
したがって、さらなる態様において本発明は、遊離N末端およびC末端を有さないようなアミド環状骨格を有する合成環状α-コノトキシンペプチドを利用し、前記ペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端は、6個〜8個のアミノ酸残基がシステイン残基IとIVの間にわたるようなペプチドリンカーを介してC末端に連結されている。
【0043】
リンカー中のアミノ酸数は、第1と第4のシステイン残基の間に7個のアミノ酸が存在するように選択することが好ましい。直鎖ペプチドの配列によっては、これらの残基の一部または全てが直鎖配列に由来してもよい。したがって、コノトキシンペプチドが第1のシステイン残基近辺のN末端に1個のアミノ酸を有する場合、リンカーに必要な追加のアミノ酸数は6個となる。
【0044】
当然ながら、N末端残基の1個または複数個を、リンカーを形成することになる他の残基で置換することも可能であろう。
【0045】
本発明によれば、特定の大きさおよび/またはタイプのペプチドリンカーを選択することにより、コノトキシンペプチドの活性を改変または増強することが可能である。連結基の導入により生じるコノトキシンのわずかな立体構造変化によって、特定の結合部位へのペプチドの結合親和性を変化させることできる。反対に、活性が親コノトキシンペプチドの活性に限りなく近い場合は、立体構造変化を最小限にするリンカーが選択されよう。
【0046】
リンカーはまた、主要な生物学的作用を実質的に阻害しない「ハンドル」をペプチドに提供する。リンカーは、分子を機能的にするための空間を提供することにより、例えば経口送達生物活性をもたらすために必要である可能性があるような、生体膜を通過する能力を含む、生物物理学的または生物薬剤学的特性を向上させることができる。
【0047】
直鎖コノトキシンを環化できるいくつかの方法がある。これらは以下のものを含む。
【0048】
1.還元ペプチドの環化に続く酸化による必要なジスルフィド結合の形成
このアプローチでは、伸長された直鎖ペプチドが、固相ペプチド合成法を使用して「on resin」で最初に合成される。この伸長直鎖ペプチドは、新たな連結部分を含むN末端とC末端伸長部の、またはその近くのシステイン残基から始まる天然配列を含む。固相合成は実際には逆の順序で、すなわち、伸長直鎖ペプチドのC末端で開始する。樹脂からの切断後、伸長コノトキシンはチオエステル中間体に環化され、続いてアミド環状ペプチドに再配置される。この還元ペプチドは次に酸化されてジスルフィド結合を形成する。環化に関する反応の概略図を図1に示す。樹脂(R)へのリンカーが付着したまま、直鎖ペプチドを樹脂から切断した。Rはペプチドと樹脂間のリンカーに相当し、環化で使用される連結部分とは異なる。第1の反応は、N末端システインのチオールとカルボキシ末端との間のチオエステルの形成を伴う。これは次に、S、Nアシル基転移を受け、天然ペプチド結合を有する環状ペプチドを形成する。
【0049】
2.還元直鎖ペプチドの酸化とそれに続く環化
このアプローチでは、固相ペプチド合成を使用して伸長ペプチドをアセンブルする。伸長直鎖ペプチドは、Nおよび/またはC末端に付加された追加の残基を有する天然コノトキシン配列を含む。(新しい)N末端とC末端は、好ましくはグリシン残基とするべきである。ペプチドは折り畳まれ、コノトキシン様ペプチドの場合、折り畳まれた分子の両末端は一般に間隙を介してすぐそばにある。このことは、標準的な化学反応を使用した溶液中でのペプチドの環化を容易にする。配列中に多数のリシン、グルタミン酸またはアスパラギン酸残基が存在する場合は問題が起こりうるので、そのときは方法1が好ましい。
【0050】
3.既存のコノトキシンへのリンカーライゲーションとそれに続く環化
この方法では、出発物質は成熟コノトキシンである。ペプチドリンカーは、ペプチドライゲーションに関する公開された手順を使用して、コノトキシンを用いて合成および連結される。伸長ペプチドは次いで環化および酸化される。
【0051】
前述のプロセスにおいて、生成物が所要のジスルフィド結合を有する環状α-コノトキシンペプチドである限り、各段階は任意の順序で実施することができる。例えば、プロセス1において、切断および環化段階は同時に、またはいずれの順序でも実施することができる。同様にプロセス2において、環化および折り畳み段階は同時に、またはいずれの順序でも実施してよい。
【0052】
システイン残基に保護基を選択的に使用してジスルフィド結合を形成することも可能である。このような方法でシステイン残基を選択的に保護することにより、特定のジスルフィド結合パターンの生成が可能である。システイン残基を保護することができる基の例は、アセタミドメチル(Acm)、4-メチルベンジル(MeBzl)および4-メトキシベンジル(Mob)を含む。
【0053】
また最終生成物の環状性を考慮して、合成手順は、α-コノトキシンペプチドの、または伸長ペプチド/リンカー配列の円順列置換変異を含むことができる。例えば、α-コノトキシンのための伸長直鎖ペプチドの設計は、α-コノトキシンのC末端残基にリンカーを付加することにより開始して、N末端残基をC末端に円順列置換してN末端システインおよび前述の環化をもたらすことができよう。
【0054】
天然コノトキシンペプチドに関連して本明細書で使用されるMIIなどの「誘導体」という用語は、1つまたは複数のアミノ酸欠失、付加、置換、または側鎖修飾によって天然ペプチドとは異なるペプチドを指す。
【0055】
置換は、アミノ酸が異なる天然または非従来型アミノ酸残基で置換されているアミノ酸変化を包含する。そのような置換は、「保存的」として分類されてよく、その場合、ポリペプチド中に含有されるアミノ酸残基は、極性、側鎖官能基または大きさのいずれかに関して特徴の類似した他の天然アミノ酸、例えば
【0056】
【化1】
【0057】
で置換される。いくつかの非従来型アミノ酸もまた天然アミノ酸の置換に適しうることが理解されるべきである。例えば、オルニチン、ホモアルギニンおよびジメチルリシンはHis、ArgおよびLysに関係している。
【0058】
本発明に包含される置換はまた、ポリペプチド中に存在するアミノ酸残基が、異なる群の天然アミノ酸などの異なる特性を有するアミノ酸で置換されているか(例えば、荷電または疎水性アミノ酸とアラニンの置換)、あるいは、天然アミノ酸が非従来型アミノ酸で置換されている、「非保存的」置換であってもよい。
【0059】
アミノ酸置換は、典型的に単一残基のものであるが、集合または分散した多数の残基のものであってもよい。
【0060】
アミノ酸置換は保存的であることが好ましい。
【0061】
付加は1つまたは複数の天然または非従来型アミノ酸残基の付加を包含する。欠失は1つまたは複数のアミノ酸残基の欠失を包含する。
【0062】
前述のように、本発明は1個または複数個のアミノ酸が側鎖修飾を受けたペプチドを含む。本発明に意図される側鎖修飾の例は、アルデヒドとの反応による還元的アルキル化に続くNaBH4による還元;メチルアセチミデートによるアミジン化;無水酢酸によるアシル化;シアン酸塩によるアミノ基のカルバモイル化;2,4,6-トリニトロベンゼンスルホン酸(TNBS)によるアミノ基のトリニトロベンジル化;無水コハク酸および無水テトラヒドロフタル酸によるアミノ基のアシル化;およびピリドキサール-5-リン酸によるリシンのピリドキシル化に続くNaBH4による還元などによるアミノ基修飾を含む。
【0063】
アルギニン残基のグアニジン基は、2,3-ブタンジオン、フェニルグリオキサルおよびグリオキサルなどの試薬を用いて複素環縮合生成物の形成により修飾することができる。
【0064】
カルボキシル基は、O-アシルイソ尿素の形成を介するカルボジイミドの活性化に続く、例えば対応するアミドへの誘導体化によって修飾することができる。
【0065】
スルフヒドリル基は、ヨード酢酸またはヨードアセタミドによるカルボキシメチル化;システイン酸への過ギ酸酸化;他のチオール化合物による混合ジスルフィドの形成;マレイミド、無水マレイン酸、または他の置換マレイミドとの反応;4-クロロ安息香酸水銀、4-クロロフェニルスルホン酸水銀、塩化フェニル水銀、2-クロロ水銀-4-ニトロフェノール、および他の水銀剤を使用する水銀誘導体の形成;アルカリ性pHにおけるシアン酸塩によるカルバモイル化などの方法で修飾することができる。システイン残基のいかなる修飾も、ペプチドが必要なジスルフィド結合を形成する能力に影響を及ぼしてはならない。ペプチドが1個または複数個のジスルフィド結合の代わりにジセレニウム結合を形成するように、システインのスルフヒドリル基をセレニウム等価物と置換することも可能である。
【0066】
トリプトファン残基は、例えば、N-ブロモスクシンイミドでの酸化またはインドール環の2-ヒドロキシ-5-ニトロベンジルブロミドまたはスルフェニルハライドでのアルキル化により修飾することができる。一方、チロシン残基は、テトラニトロメタンでニトロ化して3-ニトロチロシン誘導体を形成することにより変化させることができる。
【0067】
ヒスチジン残基のイミダゾール環の修飾は、ヨード酢酸誘導体でのアルキル化またはジエチルピロカーボネートでのN-カルベトキシル化により達成することができる。
【0068】
プロリン残基は、例えば、4位におけるヒドロキシル化により修飾することができる。
【0069】
修飾された側鎖を有するいくつかのアミノ酸および他の非天然アミノ酸の一覧表を表2に示す。
【0070】
【表2】
【0071】
これらのタイプの修飾は、個体に、または診断試薬として使用するために投与される場合、ペプチドを安定化させるのに重要でありうる。
【0072】
本発明に意図される他の誘導体は、全くグリコシル化されていない分子から修飾されたグリコシル化分子まで、さまざまなグリコシル化変異体を含む。変化したグリコシル化パターンは、異なる宿主細胞における組換え分子の発現に由来してもよい。
【0073】
環状α-コノトキシンペプチドは、Cys残基および特徴的なジスルフィド結合パターンを保持することが好ましい。誘導体は、ジスルフィド結合形成の際に保護されるという条件で、さらなるCys残基を含んでもよい。
【0074】
本発明によるコノトキシンペプチドは12〜40個、より好ましくは15〜30個のアミノ酸を有することが好ましい。
【0075】
本発明による環状コノトキシンペプチドは、治療薬として有用である。
【0076】
α-コノトキシンは、疼痛の治療および神経損傷の回復促進に関わるニコチン性アセチルコリン受容体(nAChR)に結合する。このような受容体は、統合失調症、うつ病、中毒、てんかん、アルツハイマー病、パーキンソン病およびトゥレット症候群を含むいくつかの神経精神障害の病態生理学にも関わっており、そのためα-コノトキシンはこれらの疾患の治療指標となる可能性を有する。本発明による環状α-コノトキシンペプチドが他の受容体またはイオンチャネルを標的とすることも可能である。
【0077】
環状コノトキシンペプチドは、神経因性疼痛の治療的処置または予防に特に有用であると考えられている。疼痛は、神経系自体の内部から生じる場合、神経因性であるとみなされる。神経系はさまざまなレベル:神経、神経根ならびに脊髄および脳の中枢痛経路で関与しうる。神経因性疼痛は、損傷した神経の異常な作用に起因する場合があり、最も治療が困難な形態の疼痛である。神経因性疼痛は、持続的に生じる、罹患個体に有益な機能を果たさない、慢性疼痛の形態である。神経因性疼痛を患う患者は典型的に、異痛症(通常痛みを伴わない刺激による疼痛)および痛覚過敏症(痛みを伴う刺激に対する応答の増大)に苦しむ。
【0078】
神経障害、異痛症および痛覚過敏症の特徴的な疼痛症状を例証する、いくつかのよく確立された疼痛動物モデルがある。神経因性疼痛の絞扼性神経損傷(CCI)ラットモデルでは、坐骨神経に緩い結紮を適用し、神経圧迫および神経障害を引き起こす炎症をもたらす。神経部分結紮(PNL)モデルでは、坐骨神経の一部周辺にきつい結紮を作成し、残りの神経は損傷のないままにしておき、異痛症および神経過敏症の疼痛反応をもたらす。ストレプトゾトシン(STZ)モデルでは、膵臓β細胞に糖尿病を誘発し、機械的痛覚過敏および接触性異痛などの神経因性疼痛症状をもたらす。
【0079】
前述の活性を有する化合物を評価するのに有用な試験は、in vitroまたはin vivoでもよく、当業者に知られている。例えば、nAChRでの活性を評価するのに有用な試験は、WO00/15654 (Hoggら、1999年、J Biol Chem. 274(51):36559〜64頁;およびHoggら、2003年、Reviews of Physiology, Biochemistry and Pharmacology 1: 1〜46頁)に記載のものを含む。
【0080】
ペプチドは予防の意味で投与されてもよいが、哺乳動物がこのような治療を必要としていることが好ましい。
【0081】
当業者には容易に理解されるように、投与経路(経口および経腸)および薬学的に許容されるビヒクルの性質は、治療される状態および哺乳動物の性質によって決定するはずである。特定の担体または送達システム、および投与経路の選択は当業者によって容易に決定されうると考えられる。
【0082】
本発明の経口および経腸製剤において、活性ペプチドは不活性希釈剤と、または同化可能な食用担体と一緒に処方してもよく、または硬質または軟質ゼラチンカプセルに封入してもよく、または錠剤に圧縮してもよく、または日常食の食物に直接組み入れてもよい。治療的経口投与のために、活性化合物は賦形剤と一緒に組み入れて、摂取可能な錠剤、口腔または舌下錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、オブラートなどの形態で使用してもよい。口腔および舌下錠など、これらの経口製剤タイプのいくつかは、肝臓で代謝されない可能性があることが理解されるであろう。しかしながら、本発明の環状ペプチドは、肝臓代謝に伴う可能性が高い胃にも送達されうる。そのような組成物および調製物は、少なくとも1重量%の活性化合物を含有することが好ましい。当然ながら、組成物および調製物の割合は変化することがあり、好都合には構成単位の約5〜約80重量%とすることができる。このような治療上有用な組成物における活性化合物の量は、適した投薬量が得られるような量である。
【0083】
錠剤、トローチ、丸剤、カプセルなどはまた、以下に記載の成分も含有することができる:ガム、アラビアゴム、コーンスターチまたはゼラチンなどの結合剤;第二リン酸カルシウムなどの賦形剤;コーンスターチ、ジャガイモデンプン、アルギン酸などの崩壊剤;ステアリン酸マグネシウムなどの滑剤;およびショ糖、乳糖またはサッカリンなどの甘味剤を加えてもよく、またはペパーミント、冬緑油またはチェリー風味などの香料。単位剤形がカプセルの場合、上記タイプの材料に加えて液体担体を含んでもよい。種々の他の材料が、コーティングとして、または投薬単位の物理的形態を別の方法で修正するために存在してもよい。例えば、錠剤、丸剤またはカプセルをシェラック、糖または両方で被覆してもよい。シロップまたはエリキシルは活性化合物、甘味剤としてショ糖、保存料としてメチルおよびプロピルパラベン、色素ならびにオレンジまたはチェリー風味などの香料を含有してもよい。当然ながら、任意の単位剤形の調製で使用した任意の材料は、薬学的に純粋で、使用される量で実質的に非毒性であるべきである。加えて、活性化合物は持続放出調製物および製剤に組み込まれてもよい。
【0084】
液体製剤もまた、胃または食道管を通って経腸投与することができる。
【0085】
経腸製剤は、乳化基剤または水溶性基剤などの適切な基剤と混合することにより坐剤の形態で調製することができる。本発明の環状ペプチドが、局所的、鼻腔内、膣内、眼内などに投与されることも可能であるが、必須ではない。
【0086】
薬学的に許容されるビヒクルは、任意のおよび全ての溶剤、分散媒、コーティング、抗菌および抗真菌剤、等張および吸収遅延剤などを含む。薬学的に活性な物質にこのような媒体および作用物質を使用することは、当技術分野においてよく知られている。任意の従来の媒体または作用物質が活性成分と配合禁忌である場合を除いて、治療組成物におけるその使用は予期されている。また補助的な活性成分を組成物中に組み込むこともできる。
【0087】
投与および投薬量の均一化を容易にするために、単位剤形中に経口または経腸組成物を処方することは特に好都合である。本明細書で使用される単位剤形は、治療される哺乳動物対象の単位投薬量に適合させた物理的に分離した単位を指し、各単位は、所要の薬学的に許容されるビヒクルを伴って、所望の治療効果を生み出すために算出された所定量の活性物質を含有する。本発明の新規な単位剤形の仕様は、以下の(a)(b)によって規定され、直接決定される:(a)活性物質特有の特徴および達成されるべき特定の治療効果、および(b)本明細書中で詳細に開示されているように身体的健康が損なわれた病的状態を有する生体対象における疾患の治療用の活性物質を配合する当技術分野に固有の制限。
【0088】
前述のように、主要活性成分は、簡便で有効な投与のために、有効量で、適した薬学的に許容されるビヒクルと一緒に単位剤形中に配合される。例えば、単位剤形は、主要活性成分を0.25μg〜約2000mgの量で含有することができる。割合で表すと、活性化合物は一般に、担体1ml当たり0.25μg〜約2000mgで存在する。補助的な活性成分を含有する組成物の場合、投薬量は、前記成分の通常の用量および投与様式を参照することにより決定される。
【0089】
本発明のいくつかの態様では、経口または経腸以外の投与形態および経路は、例えば、クリーム、ローション、経皮パッチ、スプレーおよびゲル、または、例えば溶液もしくは乾燥粉末などの吸入もしくは鼻腔内送達に適した組成物などの局所適用が予期される。
【発明を実施するための最良の形態】
【0090】
発明の詳細な説明
本発明を、いくつかの環状コノトキシンペプチドの生成およびその生物活性を説明する添付の実施例および図を参照して、以下に説明する。しかしながら、以下の説明の独自性は、本発明の前述の説明の一般性に取ってかわるものではないことが理解されるべきである。
【実施例1】
【0091】
環状MIIアナログ
(a)設計および合成
天然MIIペプチドのN末端とC末端をつなぐために必要なリンカーの大きさは、周知の分子モデリング法を使用して決定した。MIIの3次元モデルは、Protein Data Bank (PDB)からダウンロードした。分子モデリングプログラムAccelrys (Insight II Modeling Environment、2000年リリース、サンディエゴ、Accelrys社 2001年)を使用して、適切なリンカーの長さを決定した。つまり、N末端とC末端間にアミノ酸リンカーを組み込み、次いで親コノトキシン構造の立体構造をロックし、簡易なエネルギー最小化を行った。次いで、親コノトキシンの立体構造をロック解除し、ペプチド構造全体を再度最小化した。リンカーの大きさが小さすぎるモデルは、天然構造を乱さない長さのリンカーを有するものよりもエネルギーが高い構造をもたらす。最小で5残基を含有するリンカーが、N末端とC末端をつなぐために必要とされることが決定された。そのため、N末端グリシンに加えて、5(GGAAG)、6(GGAAGG)、および7(GGAGAAG)残基のリンカーを含有するペプチドを合成した。これは、6〜8残基にわたるシステイン間の全長に相当する。距離の測定によって、リンカー中の各残基が約2.0〜2.4オングストローム(これは、約3.5オングストロームにわたるであろう、つまり緩んだ立体構造を表す伸長立体構造よりも短いことに注意されたい)にわたる必要となることが明らかになった。大きな、または高度に官能化されたアミノ酸残基と比較して、天然ペプチドの他の残基と相互作用しにくい、またはフォールディングを阻害しにくい小さな側鎖を有する比較的不活性なアミノ酸であるため、グリシンおよびアラニンをリンカーアミノ酸に選択した。
【0092】
(b)環状MIIアナログの合成
チオエステルリンカーを利用するin situ中和と一緒にBOC/HBTU化学反応を使用して、ペプチドを合成した(Schnolzerら、(1992) Int. J. Pept. Protein Res. 40、180〜193頁; Dawsonら、(1994) Science 266、776〜9頁; Tamら、(1999) J. Am. Chem. Soc. 121、4316〜4324頁; Yan, L.Z.およびDawson, P.E. (2001) J Am Chem Soc 123、526〜33頁)。
【0093】
Gly PAM樹脂をDMF中で4当量のS-トリチル-β-メルカプトプロピオン酸、4当量のHBTU、および5当量のDIEAで処理する(2×30分間)ことにより、このリンカーを合成した。次いで、樹脂をTFA/トリイソプロピルシラン/H2O(96:2:2)で2×20分間処理することにより、トリチル基を除去する。次いで、標準的なカップリング条件を使用して、ペプチド鎖のC末端残基をリンカーに付加した。C末端チオエステルを有するペプチドはまた、以下の標準的な文献の手順に従ってFMOC化学反応を使用して製造することもできる[Ingenito, R.、 Bianchi, E.、Fattori, D.およびPessi, A. (1999) J Am Chem Soc 121、11369〜74頁; Shin, Y.、Winans, K.A.、Backes, B.J.、Kent, S.B.H.、Ellman, J.A.およびBertozzi, C.R. (1999) J Am Chem Soc 121、11684〜89頁; Clippingdale, A.B.、Barrow, C.J.およびWade, J.D. (2000) J Pept Sci 6、225〜34頁; Camarero, J.A.、Hackel, B.J.、De Yoreo, J.J.およびMitchell, A.R. (2004) J Org Chem 69、4145〜511頁]。ペプチド鎖をいったん組み立て、スカベンジャーとしてクレゾールおよびチオクレゾールと一緒にHFを使用して、樹脂から切断した。次いで粗ペプチド混合物を、C18カラムを使用して、80分間の0〜80%B勾配(A=0.05%TFAを含む水、B=90%アセトニトリル、9.95%水、0.05%TFA)を利用するRP-HPLCにより精製した。画分を集め、MSで分析し、所望の生成物を含有するものをプールおよび凍結乾燥した。
【0094】
精製された直鎖還元ペプチドの酸化および環化は、約0.3mg/mLの濃度で、0.1M NH4HCO3(pH 8.1)または50/50 0.1M NH4HCO3/iPrOHにペプチドを溶解させることにより達成した。反応混合物を室温で一晩撹拌し、得られた混合物を前述の条件を使用してRP-HPLCにより精製した。ペプチドは以下に示す配列を有する。
【0095】
6残基リンカー環状MIIは、約45:55の比率で2つの異なる異性体を形成した。対照的に、5残基リンカー環状MIIの粗酸化HPLCプロファイルは、ほとんど分散していない一連のピークとして現れた。最後に、7残基リンカー環状MIIは、1H NMRデータによると秩序だった構造を有する1種の優勢な(>90%)異性体を形成した。(N末端グリシンに加えて)6残基の大きさのリンカーを有するMIIアナログの、粗酸化混合物におけるわずかなピークは、これがMIIにとって最適な最小サイズであるという予備的な指標を与える。粗混合物について、個々のピークを集め、MSにより分析した。環状および酸化ペプチドに相当する質量のペプチドを含有するものを分析RP-HPLCにかけた。次いで、画分をHPLC分析に基づいて混合し、凍結乾燥した。
【0096】
十分な量で得られた環状ペプチドの1D 1H NMRスペクトルを測定した。1H NMRスペクトルによると、5残基リンカーペプチドの1つの異性体が秩序だった構造を有するようであった。1H NMRスペクトルによると、6残基リンカーペプチドの場合は、遅れて溶出する、より量の多い異性体が秩序だっているようであった。1H NMRスペクトルによると、7残基環状MIIペプチドもまた秩序だっていた。
【0097】
MIIの環状ペプチドアナログ配列を以下に示す。
【0098】
【化2】
【0099】
【化3】
【0100】
【化4】
【0101】
(c)環状MIIアナログの構造特性決定
次いで、3つの秩序だったアナログ(これよりcMII-5、cMII-6およびcMII-7と称する)について、2次元NMRスペクトルデータを得た。NMR完全帰属を行い、HαおよびHNプロトンの化学シフトを天然MIIのものと比較した。化学シフトデータの比較により、これらの構造がいかに類似しているかが示された。結果により、cMII-5の化学シフトがMIIのものとほとんど一致せず、構造の変化を示したことが明らかとなった。対照的に、cMII-6およびcMII-7の化学シフトは、天然コノトキシンのものと高度に相関性があった。このことは、cMII-6およびcMII-7が天然MIIに類似の構造を採用し、cMII-5が天然の構造から明らかに逸脱していることを示す。
【0102】
このことから、酸化プロファイルおよびNMRデータに基づいて、5個の付加残基を有するリンカーは短すぎて、コノトキシンの正しいフォールディングを阻害したが、6個または7個の付加残基を有するリンカーは、環状アナログに天然様の構造を形成させるのに十分な大きさであったことが明らかであった。cMII-6およびcMII-7の完全な3次元(3D)構造を決定し、天然MIIおよびその環状同等物の構造の密接な相関を確認した。
【0103】
cMII-6の3D構造を、NMR分光法を使用して決定した。280および287KにおいてBruker DMX 750分光計においてスペクトルを得た。記録された等核スペクトルは、2量子フィルターDQF-COSY[Rance, M.、Sorensen, Q.W.、Bodenhausen, G.、Wagner, G.、Ernst, R.R.およびWuthrich, K. (1983) Biochem. Biophys. Res. Commun. 117、479〜485頁]、等方性混合時間が80msの、MLEV17スピンロックシーケンスを使用するTOCSY[Bax, A.およびDavis, D.G. (1985) J. Magn. Reson. 65、355〜360頁]、ECOSY[Griesinger, C.、Sorensen, O.W.およびErnst, R.R. (1987) J. Magn. Reson. 75、474〜492頁]、および混合時間が150および350msのNOESY[Jeener, J,、Meier, B.H.、Bachmann, P.およびErnst, R.R. (1979) J. Chem. Phys. 71、4546〜4553頁; Kumar, A.、Ernst, R.R.およびWuthrich, K. (1980) Biochem. Biophys. Res. Commun. 95、1-6頁]を含んでいた。DQF-COSYおよびECOSYの実験において、緩和遅延の間、低出力照射によって水共鳴を抑制した。TOCSYおよびNOESYの実験では、修正WATERGATEシーケンスを使用して水抑制を達成した[Piotto, M.、Saudek, VおよびSklenar, V. (1992) J. Biomol. NMR 2、661〜665頁]。2次元スペクトルは通常、12ppmに相当するスペクトル幅にわたって、f2軸におけるデータ点4096、f1軸における増分512で収集した。ゆっくりと交換するアミドを同定するために、サンプルをD2O中に溶解させた後、一連の1次元およびTOCSYスペクトルを直ちに行った。全てのスペクトルを、XWINNMR (Bruker)を使用してSilicon Graphicsワークステーションで処理した。f1軸は0詰め〜2048の実際のデータ点であり、f1およびf2軸は、フーリエ変換の前に90°シフトしたサイン2乗関数を掛けた。処理したスペクトルはプログラムXEASY[Eccles, C.、Guntert, P.、Billeter, M.およびWuthrich, K. (1991) J. Biomol. NMR 1、111〜30頁]を使用して分析および帰属した。スペクトルは連続帰属法を使用して帰属した[Wuthrich, K. (1986) Wiley-Interscience、New York]。処理は一部、DYANAパッケージ[Guntert, P.、 Mumenthaler, C.およびWuthrich, K. (1997) J. Mol. Biol. 273、283〜98頁]の一部である自動帰属プログラムNOAHを使用して簡便化された。90% H2O、10% D2O中、混合時間350および150msで記録されたNOESYスペクトルの交差ピークは、XEASYで積分およびキャリブレーションを行い、距離拘束はDYANAを使用して導いた。主鎖二面角拘束は、DQF-COSYスペクトルで分裂する逆位相交差ピークの線形分析により測定した3JHNHα結合定数から導いた。角度は、8.5Hzより大きい3JHNHαでは-120°±30°に、5Hzより小さい3JHNHαでは-60°±30°に制約された。β-メチレンプロトンおよびχ1二面角の立体特異的帰属は、NOEピーク強度と組み合わせて、ECOSYスペクトルから測定した3Jαβ結合定数から導いた。初期構造は、DYANAでねじれ角模擬アニーリング法を使用して算出した。最終構造は、CNS version 1.0で模擬アニーリングおよびエネルギー最小化法を使用して算出した[Brunger, A.T.、Adams, P.D.およびRice, L.M. (1997) Structure 5、325〜336頁]。出発構造は、無作為のファイ、プサイ二面角を使用して生成し、エネルギーを最小化することにより正しい局所構造をもたらした。この手順は、4000ステップ、0.015psのねじれ角動力学を含む高温相、4000ステップ、0.015psのねじれ角動力学を含む、温度を0Kまで低下させる冷却相、最後に、5000ステップのパウエル最小化を含むエネルギー最小化相を含む。構造を、LingeおよびNilges[Linge, J.P.およびNilges, M. (1999) J. Biomol. NMR 13、51〜9頁]により記載されたように、ウォーターシェル中でさらなる分子動力学およびエネルギー最小化を使用して精密化した。explicit water中での精密化は、以下の段階を含む。第1に、それぞれ50ステップ、0.005psのデカルト動力学を含む100K毎の段階を介した500Kへの加熱。次に、500Kで2500ステップ、0.005psのデカルト動力学の後に、それぞれ2500ステップ、0.005psのデカルト動力学を含む100K毎の段階で温度を低下させる冷却相。最後に、2000ステップのパウエル最小化で構造を最小化した。構造を、PromotifおよびProcheckを使用して分析した[Hutchinson, E.G.およびThornton, J.M. (1996) Protein Sci. 5、212〜220頁; Laskowski, R.A.、Rullmannn, J.A.、MacArthur, M.W.、Kaptein, R.およびThornton, J.M. (1996) J. Biomol. NMR 8、477〜86頁]。
【0104】
cMII-6とMIIの比較は、MIIの天然立体構造がcMII-6において全体として保持されていることを示す。2つのペプチド間の顕著な類似性を図4で例示する。
【0105】
(d)cMII-6の選択的合成
非選択的酸化が、環状コノトキシンの合成で使用される好ましい方法であるが、cMII-6について例証した特異的ジスルフィド結合形成を使用して製造することもできる。ペプチドは前述のように合成したが、システイン3および16の側鎖をAcm基で保護した。ペプチドを樹脂から切断し、前述の条件を使用して精製および酸化/環化した。第2のジスルフィド結合を、I2を使用した脱保護/酸化によって、3および16位のシステイン間に選択的に形成した。ペプチド(20mg)を、1mg/mLの濃度で50%酢酸水溶液に溶解し、フラスコを窒素でフラッシュした。フラスコへ、2mLの1M HCl、続いて十分量の50%酢酸水溶液中の0.1M I2(約1mL)を加え溶液を淡黄色にした。次いで反応物を窒素下、室温で1.5時間撹拌した。反応混合物が無色になるまで1Mアスコルビン酸を加えることにより、反応をクエンチした。次いで、混合物を緩衝液Aで希釈し、RP-HPLCにより精製し、完全に酸化された環状ペプチドを得た。このペプチドはcMII-6と共溶出され、したがって天然ペプチドのジスルフィド連結性、すなわちCys2-Cys8およびCys3-Cys16(1-3、2-4)に対応するとしてcMII-6のジスルフィド連結性を確認した。
【0106】
(e)cMII-6の生物活性
クロム親和細胞をウシ副腎から調製し、標準的な文献の手順に記載のとおり24ウェルプレート(Nunc)に維持した[Lawrence, G.W.、Weller, U.およびDolly, J.O. (1994) Eur J Biochem 222、325〜33頁; Meunier, F.A.、Mattei, C.、Chameau, P.、Lawrence, G.、Colasante, C.、Kreger, A.S.、Dolly, J.O.およびMolgo, J. (2000) J Cell Sci 113 (Pt 7)、1119〜25頁; Meunier, F.A.、Feng, Z.P.、Molgo, J.、Zamponi, G.W.およびSchiavo, G. (2002) Embo J 21、6733-43頁]。無傷細胞を緩衝液A(mM): NaCl、145; KCl、5、Na2HPO4、1.2;グルコース、10; HEPES-NaOH、20(pH7.4)で簡単に1回洗い、天然および環状コノトキシンと一緒に20分間、2mM CaCl2の存在下でインキュベートし、ニコチン(5μM)で20分間刺激した。各実験の最後に上清のアリコートを採取し、細胞を1%(v/v)Triton X-100(Shigma)で溶解させた。両方のサンプルの組を、カテコールアミンについて蛍光分析により試験し、放出量を文献中に記載のように対照の割合として表した(上記参照)。
【0107】
生物活性試験の結果を図3に示し、環状ペプチドの活性が直鎖MIIの活性と同等であることを示す。
【0108】
(f)cMII-6の安定性
cMII-6およびcMII-7のタンパク質分解酵素による作用に対する安定性を、ペプチドをエンドプロティナーゼGlu-C(Endo Glu-C)と一緒にインキュベートすることにより試験した。MII、cMII-6およびcMII-7は、末端(MIIの場合)およびリンカー(cMII-6およびcMII-7の場合)の対面に同じプロセシング部位を有する可能性がある。ペプチドを、20μg/mLの濃度で0.1M NH4HCO3(pH8.0)緩衝液に溶解させた。次いで、Endo GluCを50:1のペプチド:酵素比(wt/wt)で加え、溶液を37℃でインキュベートした。アリコート(3μL)を抜き取り、0〜10の毎時間5%ギ酸(57μL)でクエンチした。次いで、サンプルをLC/MSにより分析し、各時点において残存する無傷のペプチド量を測定した。各試験は、適切な陽性(Endo GluC切断点を有する直鎖の非ジスルフィドペプチド)および陰性(酵素を含まない緩衝液中のペプチド)対照を用いて3連で行った。
【0109】
安定性試験の結果を図4に示す。環状ペプチドは全10時間にわたって完全に無傷のままであり、一方、天然MIIは約10時間の半減期を有する。cMII-6およびcMII-7により例証された安定性の強化は、MIIおよび環状ペプチドの推定プロセシング部位が同一(すなわち、グルタミン酸のC末端側)であり、末端(MIIの場合)ならびにcMII-6およびcMII-7の場合はペプチドリンカーから離れていることを考えると、驚くべきことである。さらに、任意の強化された安定性は、Glu-Cはエンドプロテアーゼであるが、N末端(MIIはC末端がアミド化されている)に対して活性なエキソプロテアーゼに対してのみであろうことが予想されうる。したがって、環状コノトキシンは、末端単独の保護により予想されうるよりも強化された安定性を有するようである。
【0110】
(g)ヒト血漿中の酵素に対するcMII-6およびcMII-7の安定性
タンパク質分解作用に対するcMII-6およびcMII-7の抵抗性を試験し、これをMIIと比較するために、これらのコノトキシンをヒト血漿中でインキュベートした。MII、cMII-6およびcMII-7(10μM)を、50%ヒト血漿中で24時間インキュベートした。いくつかの時点でアリコートを採取し、15%トリクロロ酢酸水溶液でクエンチし、遠心分離機にかけた。次いで上清をRP-HPLCにより分析した。
【0111】
各試験は3連で行った。図5はこの実験の結果を示す。cMII-6およびcMII-7の安定性は、MIIを劇的に上回っている。天然MIIは約16時間の半減期を有したが、24時間後に残存するcMII-6およびcMII-7の量は90%近くであった。
【実施例2】
【0112】
環状Vc1.1の合成および特性決定
(a)Vc1.1の設計
α-コノトキシンVc1.1は、MIIを含むα-コノトキシンの4/7クラスのメンバーであり、疼痛治療の可能性を有する(Sandall DW、Satkunanathan N、Keays DA、Polidano MA、Liping X、Pham V、Down .JG、Khalil Z、Livett BG、Gayler KR、Biochemistry 2003 42(22):6904〜11頁)。文献中にVc1.1の利用できる構造データがなかったので、環状MIIアナログの設計のための前述の手順に従うことができなかった。しかしながら、この同じ骨格を有する他のコノトキシンが構造的に研究されていたため、Vc1.1のリンカーの最適な長さを、MIIおよびPnIAを含む同じファミリーの他のメンバーの距離の簡易測定により推定した。Protein Data Bankには、重複構造、およびN末端に伸長された柔軟性の「テール」を有するコノトキシンGIDを除いて、4つのα4/7コノトキシン構造がある。これらの4つの化合物のこれらの構造のN末端とC末端間の距離を平均することにより得た値は、よく一致していた(12.2±0.8Å)。後に、Vc1.1の3次元構造が報告され(Clarkら、J. Biol. Chem. (2006) 281 23254-23263頁)、Vc1.1のN末端とC末端間の距離は、11.9±0.5Åであり、α4/7コノトキシンのこの算出された平均の範囲内に入ることが明らかになった。したがって、環状Vc1.1に適したリンカーの長さは、環状MIIに使用されるものと同様、例えば6〜7残基であろうと推定することができる。この場合もまた、これらの6残基を既存のグリシン残基のN末端に付加する。
【0113】
(b)環状Vc1.1およびVc1.1の合成および構造特性決定
環状Vc1.1の合成を、環状MII(実施例1)について記載した合成手順を使用して行った。環状Vc1.1に使用した環化/酸化緩衝液は、0.1M NH4HCO3(pH8.1)であった。環化/酸化により1種の優勢な異性体(これよりcVc1.1-6と称する)が生じ、これを精製し、1H NMR分光法により分析した。Vc1.1(C末端がアミド化されている)およびcVc1.1-6の配列を以下に示す。
【0114】
【化5】
【0115】
【化6】
【0116】
直鎖Vc1.1もまた、MBHA-アミド樹脂上でin situ中和と一緒にBOC/HBTU化学反応を使用して合成した。ペプチドを0.1M NH4HCO3中、室温で一晩フォールディングさせ、単一異性体を得た。合成Vc1.1およびcVc1.1-6のジスルフィド連結性は、標準的な還元/アルキル化法[Goransson, U.およびCraik, D.J. (2003) J. Biol. Chem. 278、48188〜96頁]と一緒にMS/MSシーケンシングを使用して、両方ともCys2-Cys8、Cys3-Cys16であることが確認された。次いで、cVc1.1-6と比較するために3次元NMR構造を決定した。Vc1.1およびcVc1.1-6の両方の3次元構造を、環状MIIについて前述したNMR分光法を使用して決定した。図6に示すように、Vc1.1の天然立体構造はcVc1.1-6において保持されている。
【0117】
(c)Vc1.1のジスルフィドマッピング
Vc1.1を、TCEPと一緒にクエン酸緩衝液中、低pHでインキュベートすることにより部分的に還元した。反応混合物をRP-HPLCにより精製し、1つのジスルフィド種をN-エチルマレイミドでアルキル化した。次いで、アルキル化されたペプチドを完全に還元し、MS/MSにより分析した。MS/MSデータは、C2およびC8がN-エチルマレイミドでアルキル化されたことをはっきりと示した。ペプチド鎖の両末端からのフラグメンテーションパターンを観察すると、そのパターンは推定アルキル化パターンを完全に支持した。したがって、Vc1.1のジスルフィド連結性はC2-C8およびC3-C16であるという結論に達した。これは、他のα-コノトキシンに見られるI-III、II-IVジスルフィド結合パターンに一致する。
【0118】
(d)cVc1.1-6の生物活性
cVc1.1-6の生物活性を、環状MIIの実施例で記載したように、ウシ副腎クロム親和細胞からのカテコールアミン放出を測定することにより分析した。cVc1.1-6活性が直鎖Vc1.1の活性と実験誤差の範囲内で等しいことを例証する試験結果を図7に示す。
【0119】
(e)cVc1.1-6の安定性
cVc1.1-6がタンパク質分解酵素による攻撃に抵抗する能力を、環状MIIについて概説した実験を使用して評価することができる。哺乳動物の胃液および血漿中での分解に対する抵抗性もまた、実施例1で示した手順を使用して評価することができる。
【0120】
(f)組換えnAChRサブタイプのα-コノトキシンVc1.1抑制の選択性
AChに誘導される流れのVc1.1抑制を、種々のnAChRサブユニットの組合せを発現するアフリカツメガエル卵母細胞において調べた。Achに誘発される応答を10分毎に調べ、毒素を4分間、溶液添加した後、アゴニストと毒素を同時適用した。Vc1.1(10μM)は、中枢型nAChRサブタイプであるα4β2およびα4β4、または骨格筋型nAChRサブタイプであるαβγδ(n=7〜12)のいずれかが仲介する、AChに誘発される流れを抑制できなかった。同様に、10μM Vc1.1は、ホモ五量体ニューロンnAChRであるα7(n=11)が仲介するAChに誘発される流れをわずか14±2%しか抑制しなかった。しかしながら、10μM Vc1.1は、末梢nAChRサブタイプα3β2およびα3β4を同程度、それぞれ対照の58±7%(n=8)および56±7%(n=12)抑制した。α3α5β2(n=7)のnAChRの組合せにα5サブユニットを加えると、同様の有効性が観察されたが、Vc1.1は5倍以上低いα3α5β4(n=15)抑制の有効性を示した。AChに誘導される流れの大きさは、nAChRサブタイプα3α6β2(n=13)を含有するα6で、30μM Vc1.1によって約50%しか抑制されなかった。100nM以下の濃度でVc1.1を溶液添加しても、AChに誘発される流れを遮断せず、α3含有nAChRに対して検出可能な応答すら(すなわち50nAより大きい)導くこともなかった。
【0121】
(g)cVc1.1-6の経口および皮下有効性
よく定義された神経因性疼痛モデル(CCIラット)を使用して、皮下(s.c.)および経口ボーラス投与により投与された場合のVc1.1およびcVc1.1-6の抗異痛(疼痛緩和)作用を比較するために、試験を行った。
【0122】
神経因性疼痛の決定的な症状である接触性異痛を、ラットの左脚の坐骨神経周辺に4個の緩い結紮を結ぶことによって誘導したことにより、絞扼性神経損傷(CCI)を誘導し、同じ側の後肢(すなわち同側後肢)における軽い接触に対する過敏性をもたらした。接触性異痛を、目盛り付きvon Freyフィラメントを使用して、CCIラットの同側後肢へ段階的な不快でない圧力を加えることにより、試験した。損傷のないラットおよび損傷のない(反対側の)CCIラット後肢において、von Frey足引っ込め閾値(PWT)は約12gであるが、CCI手術の14日後には同側PWTは6g以下である。治療の目的は、接触性異痛を軽減して同側後肢のPWTを6〜12gに増加させることである。
【0123】
s.c.または経口経路により単回ボーラス投与(100μg/kg)として投与されたVc1.1およびcVc1.1-6は、平均最大同側PWTが6g未満から約10gまで増加したことによって例証されるように、接触性異痛の著しい緩和をもたらした。Vc1.1の環化は、投与経路に関係なく、親直鎖ペプチドと比較して長い作用時間をもたらすようであった(図8および9)。s.c.または経口投与された場合、Vc1.1もcVc1.1-6も、反対側の後肢のPWTを増加させず、予想と一致した(図10)。
【0124】
結果は、このin vivo神経因性疼痛モデルにおいて、cVc1.1-6が直鎖Vc1.1よりも著しく活性であったことを例証する。cVc1.1-6はまた、直鎖Vc1.1よりも長い半減期を有し、より安定で、動物において経口利用可能である。
【0125】
(h)環状Vc1.1の抗異痛有効性および効能
CCIラットにおけるcVc1.1-6の単回経口ボーラス投与の抗異痛有効性を実証するために、試験を行った。
【0126】
有効用量範囲を特定するために、各CCIラット(n=6)はcVc1.1-6の単回経口ボーラス投与を、投与間に最低2日間のウォッシュアウト期間を設けて最大5回受けた。この方法を使用して、環状Vc1.1の抗異痛有効用量は0.3mg/kg〜3mg/kgであった。十分量のペプチドが入手できなかったため、これより多い用量は試験しなかった。対照動物はビヒクルの経口ボーラス投与を受けた(n=3)。環状Vc1.1およびビヒクルは、1ml Hamiltonガラスシリンジを使用して500μLの量で投与し、PWTは上記2(h)に概説した手順を利用して決定した。
【0127】
CCIラットは、cVc1.1-6の単回経口ボーラス投与を1mg/kgで受けた(n=3)。対照動物は、ビヒクルの単回経口ボーラス投与を受けた(n=3)。cVc1.1-6またはビヒクルは、Hamiltonガラスシリンジを使用して500μLの量で投与し、PWTは上記2(h)に概説した手順を利用して決定した。
【0128】
実験手順の終了後、ラットを100% CO2、続いて頸椎脱臼により安楽死させた。ラット死体は、クイーンズランド大学バイオ廃棄物回収サービスにより回収されるまで冷凍した。
【0129】
平均(±SEM)PWT対時間曲線を、CCIラットにおけるcVc1.1-6およびビヒクルの各用量についてプロットした。またPWT値を、それぞれの投与前基準値を引くことにより正規化し、正規化された応答対時間曲線(AUC)の下面積を、台形積分を使用して概算した。AUC値を用量に対してプロットし、ED50値を概算した(図12)。
【0130】
予想通り、接触性異痛は、反対側の(損傷していない)後肢の各平均(±SEM)PWT(10.6±0.2g)に対する、CCI手術から21日後の同側後肢の平均(±SEM)PWT(4.8±0.2g)の顕著な減少(p<0.05)として現れた。治療の目標は、接触性異痛の完全な回復、すなわち、約11gの同側後肢のvon Frey PWTである。
【0131】
「ウォッシュアウト」法に従うCCIラットへのcVc1.1-6の単回経口ボーラス投与(0.3〜3mg/kg)の後に、同側後肢において用量依存的な接触性異痛の緩和が速やかに開始した。試験した用量全てにおいて、同側後肢の抗異痛は投与後約0.5時間で最大になり、対応する作用持続時間は1〜1.25hであった。試験したcVc1.1-6の最大用量(3mg/kg)において、平均(±SEM)PWTは、投与前の5.2(±0.4g)から投与後0.5hで最大効果(9.5±1g)まで増加した。CCIラットの同側後肢における抗異痛の平均(±SEM)範囲および持続時間(ΔPWT AUC値)は、用量依存的に増加した。cVc1.1-6のおおよそのED50値は、1mg/kgと概算し、この用量を薬物未投与CCIラットへの投与に選択した。CCIラットにおける最大3mg/kgまでの用量でのcVc1.1-6経口投与は、反対側の後肢にわずかな抗侵害受容を引き起こした(図11)。
【0132】
薬物未投与CCIラットへcVc1.1-6を1mg/kgで単回経口ボーラス投与した後に、同側後肢において接触性異痛の緩和が速やかに開始した。「ウォッシュアウト」法の結果と一致して、同側後肢の抗異痛は投与後約0.5時間で最大になり、対応する平均作用持続時間は1.5hであった。特に、CCIラットの同側後肢の平均(±SEM)PWT値は、投与前の4(±0g)から最大効果時(0.5h)の8.7(±0.7)gまで増加した。薬物未投与CCIラットにおける1mg/kgのcVc1.1-6経口投与は、反対側の後肢にわずかな抗侵害受容を引き起こした。
【0133】
試験において、試験した用量(0.3〜3mg/kg)でのcVc1.1-6単回経口ボーラス投与により、CCIラットの行動的副作用は認められなかった。
【0134】
CCIラットへのcVc1.1-6の0.3〜3mg/kgでの単回経口ボーラス投与は、対応する反対側の後肢に抗侵害受容を引き起こすことなく、同側(損傷した)後肢に用量依存的抗異痛応答を引き起こした。cVc1.1-6が反対側の(損傷していない)後肢に抗侵害受容を引き起こさなかったという観察結果は、cVc1.1-6が、内因性の疼痛抑制機構を増強することではなく侵害受容促進(疼痛促進)経路を調節することによりその疼痛緩和作用をもたらす、という見解と一致する。
【0135】
本明細書中の、任意の先行出版物(またはそこから得られる情報)、または任意の既知の事実に関する参考文献は、その先行出版物(またはそこから得られる情報)または既知の事実が、本明細書が関わる試みの分野に共通の一般知識の一部を形成するという認識または承認または任意の形態の示唆としてみなされず、またみなされるべきではない。
【0136】
当業者であれば、本明細書中に記載の発明は、具体的に記載された以外の変形および修正が可能であることを理解するであろう。本発明は、本発明の精神および範囲に入る全てのそのような変形および修正を含むことを理解されたい。本発明はまた、本明細書で個別に、またはまとめて言及された、または示された全ての段階、特徴、組成物および化合物、ならびに前記段階および特徴の任意の2つ以上の任意のおよび全ての組合せを含む。
【図面の簡単な説明】
【0137】
【図1】C末端チオエステルを介したペプチド環化のスキームを示す図である。N末端システインの遊離硫黄が、C末端チオエステルと相互作用して中間体を形成し、この中間体がS、Nアシル基転移を受けることにより天然ペプチド結合を有する環状ペプチドを形成する。
【図2】コノトキシンMII、cMII-6およびcMII-7の3次元構造を表す図であり、これらの構造の重ね合わせを含む。ジスルフィド結合をボールアンドスティックで表す。構造はNMR分光法により決定した。
【図3】MII、cMII-5、cMII-6およびcMII-7の濃度の増加による単離ウシクロム親和細胞からのニコチン(5μM)誘発性カテコールアミン放出の阻害に関する濃度-応答曲線を図示するグラフである。結果は、環状ペプチドの活性が直鎖MIIと同等であることを示す。
【図4】ペプチドをエンドプロティナーゼ、Endo Glu-Cと一緒にインキュベートすることにより試験したときの、タンパク質分解酵素による攻撃に対するMII、cMII-6およびcMII-7の相対的安定性を図示するグラフである。残存する無傷のペプチド量はRP-HPLCにより測定した。
【図5】50%ヒト血漿中の酵素に対するMII、cMII-6およびcMII-7の相対的安定性を図示するグラフである。残存する無傷のペプチド量はRP-HPLCにより測定した。
【図6】Vc1.1(黒)および環状Vc1.1-6(灰色)の3次元構造を表す図である。構造はNMR分光法により決定した。各ペプチドについて、20個の最小エネルギー構造の骨格を重ね合わせて示し、Vc1.1の天然立体構造がcVc1.1-6で保持されていることを示す。
【図7】ウシ副腎クロム親和細胞からのカテコールアミン放出を測定することにより試験したときの、cVc1.1-6およびVc1.1の相対的生物活性を図示するグラフである。クロム親和細胞を、示した量のペプチドと一緒に20分間インキュベートし、次いで阻害剤が引き続き存在する中で10pMニコチンで20分間刺激した。アリコートを採取し、以前に記載されたように、蛍光分析によりカテコールアミン分泌について試験した(n=6実験)(Meunierら、2002年)。
【図8】Vc1.1およびcVc1.1-6単回ボーラスs.c.投与(100μg/kg)での6g未満から約10gまでのCCIラット同側後肢における平均足引っ込め閾値(PWT)の増加を図示するグラフである。
【図9】Vc1.1およびcVc1.1-6単回ボーラス経口投与(100μg/kg)での6g未満から約10gまでのCCIラット同側後肢における平均足引っ込め閾値(PWT)の増加を図示するグラフである。
【図10】CCIラットにおけるVc1.1およびcVc1.1-6単回ボーラスs.c.または経口投与(100μg/kg)でのCCIラット反対側後肢における平均足引っ込め閾値(PWT)を図示するグラフである。
【図11】CCIラット同側後肢において用量依存的な接触性異痛の緩和をもたらした、Vc1.1単回経口ボーラス投与(0.03〜3mg/kg)での平均PWTを図示するグラフである。
【図12】用量に対して抗異痛の程度および持続時間(ΔPWT AUC値)をプロットして作成した用量-応答曲線を図示するグラフである;おおよそのED50値は1mg/kgであると概算された。
【特許請求の範囲】
【請求項1】
遊離N末端およびC末端を有さないようなアミド環状骨格を有する少なくとも1種の合成環状α-コノトキシンペプチドを、医薬として経口または経腸投与に適したビヒクル中に含む経口または経腸医薬製剤であって、前記ペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端はペプチドリンカーを介してC末端に連結されている、医薬製剤。
【請求項2】
対応する直鎖/非環状α-コノトキシンペプチドが、
Xaa1 CCS Xaa2 P Xaa3 C Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 C 配列番号1
(配列中、Xaa1はグリシンまたはアスパラギン酸であり、Xaa2からXaa7は任意の天然または非天然アミノ酸を表し、Xaa8はプロリン、ヒドロキシプロリンまたはグルタミンを表し、Xaa9はアスパラギン酸、グルタミン酸またはγ-カルボキシグルタミン酸を表し、Xaa10は任意の天然または非天然アミノ酸を表すか、なくてもよい)から選択される、請求項1に記載の医薬製剤。
【請求項3】
Xaa1からXaa10が以下:
Xaa1がグリシンまたはアスパラギン酸であり、
Xaa2がアスパラギン酸、アスパラギン、ヒスチジン、チロシン、アルギニンまたはリシンから選択され、
Xaa3がアルギニン、プロリン、アラニン、バリンまたはセリンから選択され、
Xaa4がアスパラギン、アラニン、アルギニン、チロシン、ヒスチジン、フェニルアラニン、セリン、イソロイシンまたはリシンから選択され、
Xaa5がチロシン、ヒスチジン、アラニン、バリン、グルタミン、グリシン、ロイシン、セリン、チオニン、アスパラギン、アスパラギン酸、グルタミン酸、リシンまたはアルギニンから選択され、
Xaa6がアスパラギン酸、アスパラギン、セリン、チロシン、グルタミン酸、グリシン、アルギニンまたはヒスチジンから選択され、
Xaa7がヒスチジン、アスパラギンまたはチロシンから選択され、
Xaa8がプロリン、ヒドロキシプロリン、グルタミンまたはセリンから選択され、
Xaa9がグルタミン酸、γ-カルボキシグルタミン酸、アスパラギン酸、グリシンまたはアスパラギンから選択され、
Xaa10がイソロイシン、チロシン、ロイシンまたはdes-Xaa10から選択される、
のように選択される、請求項2に記載の医薬製剤。
【請求項4】
Xaa1からXaa10が以下:
Xaa1がグリシンまたはアスパラギン酸であり、
Xaa2がアスパラギン酸であり、
Xaa3がアルギニンであり、
Xaa4がアスパラギンであり、
Xaa5がチロシンであり、
Xaa6がアスパラギン酸であり、
Xaa7がヒスチジンであり、
Xaa8がプロリンであり、
Xaa9がグルタミン酸であり、
Xaa10がイソロイシンである、
のように選択される、請求項2に記載の医薬製剤。
【請求項5】
対応する直鎖/非環状α-コノトキシンペプチドがVc1.1である、請求項1に記載の医薬製剤。
【請求項6】
リンカーが、第1と第4のシステイン残基の間に7個のアミノ酸残基が存在するように選択される、請求項1から5のいずれか一項に記載の医薬製剤。
【請求項7】
リンカーが、直鎖α-コノトキシン中にすでに存在する任意のアミノ酸残基に加えてグリシンおよび/またはアラニン残基からなる、請求項6に記載の医薬製剤。
【請求項8】
ペプチドリンカーが、6個から8個の天然または非天然アミノ酸がシステイン残基IとIVの間にわたるようなリンカーである、請求項1に記載の医薬製剤。
【請求項9】
合成環状ペプチドが、
【化1】
【化2】
【化3】
から選択される、請求項1に記載の医薬製剤。
【請求項10】
合成環状ペプチドが、
【化4】
である、請求項8に記載の医薬製剤。
【請求項11】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、疼痛の治療または予防方法。
【請求項12】
疼痛が神経因性疼痛である、請求項11に記載の方法。
【請求項13】
疼痛が炎症性疼痛である、請求項11に記載の方法。
【請求項14】
神経因性疼痛が糖尿病性神経因性疼痛である、請求項12に記載の方法。
【請求項15】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、神経損傷の回復を促進する方法。
【請求項16】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、気分変調、および季節性情動障害など)、または炎症の治療または予防方法。
【請求項17】
疼痛の治療または予防のための経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【請求項18】
疼痛が神経因性疼痛である、請求項17に記載の使用。
【請求項19】
疼痛が糖尿病性神経因性疼痛である、請求項18に記載の使用。
【請求項20】
疼痛が炎症性疼痛である、請求項17に記載の使用。
【請求項21】
神経損傷からの回復促進における経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【請求項22】
アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、トゥレット症候群、気分変調、および季節性情動障害など)、または炎症の治療または予防のための経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【請求項1】
遊離N末端およびC末端を有さないようなアミド環状骨格を有する少なくとも1種の合成環状α-コノトキシンペプチドを、医薬として経口または経腸投与に適したビヒクル中に含む経口または経腸医薬製剤であって、前記ペプチドは、2個のジスルフィド結合を形成するように対になって結合した4個のシステイン残基を含み、対応する直鎖/非環状コノトキシンペプチドのN末端はペプチドリンカーを介してC末端に連結されている、医薬製剤。
【請求項2】
対応する直鎖/非環状α-コノトキシンペプチドが、
Xaa1 CCS Xaa2 P Xaa3 C Xaa4 Xaa5 Xaa6 Xaa7 Xaa8 Xaa9 Xaa10 C 配列番号1
(配列中、Xaa1はグリシンまたはアスパラギン酸であり、Xaa2からXaa7は任意の天然または非天然アミノ酸を表し、Xaa8はプロリン、ヒドロキシプロリンまたはグルタミンを表し、Xaa9はアスパラギン酸、グルタミン酸またはγ-カルボキシグルタミン酸を表し、Xaa10は任意の天然または非天然アミノ酸を表すか、なくてもよい)から選択される、請求項1に記載の医薬製剤。
【請求項3】
Xaa1からXaa10が以下:
Xaa1がグリシンまたはアスパラギン酸であり、
Xaa2がアスパラギン酸、アスパラギン、ヒスチジン、チロシン、アルギニンまたはリシンから選択され、
Xaa3がアルギニン、プロリン、アラニン、バリンまたはセリンから選択され、
Xaa4がアスパラギン、アラニン、アルギニン、チロシン、ヒスチジン、フェニルアラニン、セリン、イソロイシンまたはリシンから選択され、
Xaa5がチロシン、ヒスチジン、アラニン、バリン、グルタミン、グリシン、ロイシン、セリン、チオニン、アスパラギン、アスパラギン酸、グルタミン酸、リシンまたはアルギニンから選択され、
Xaa6がアスパラギン酸、アスパラギン、セリン、チロシン、グルタミン酸、グリシン、アルギニンまたはヒスチジンから選択され、
Xaa7がヒスチジン、アスパラギンまたはチロシンから選択され、
Xaa8がプロリン、ヒドロキシプロリン、グルタミンまたはセリンから選択され、
Xaa9がグルタミン酸、γ-カルボキシグルタミン酸、アスパラギン酸、グリシンまたはアスパラギンから選択され、
Xaa10がイソロイシン、チロシン、ロイシンまたはdes-Xaa10から選択される、
のように選択される、請求項2に記載の医薬製剤。
【請求項4】
Xaa1からXaa10が以下:
Xaa1がグリシンまたはアスパラギン酸であり、
Xaa2がアスパラギン酸であり、
Xaa3がアルギニンであり、
Xaa4がアスパラギンであり、
Xaa5がチロシンであり、
Xaa6がアスパラギン酸であり、
Xaa7がヒスチジンであり、
Xaa8がプロリンであり、
Xaa9がグルタミン酸であり、
Xaa10がイソロイシンである、
のように選択される、請求項2に記載の医薬製剤。
【請求項5】
対応する直鎖/非環状α-コノトキシンペプチドがVc1.1である、請求項1に記載の医薬製剤。
【請求項6】
リンカーが、第1と第4のシステイン残基の間に7個のアミノ酸残基が存在するように選択される、請求項1から5のいずれか一項に記載の医薬製剤。
【請求項7】
リンカーが、直鎖α-コノトキシン中にすでに存在する任意のアミノ酸残基に加えてグリシンおよび/またはアラニン残基からなる、請求項6に記載の医薬製剤。
【請求項8】
ペプチドリンカーが、6個から8個の天然または非天然アミノ酸がシステイン残基IとIVの間にわたるようなリンカーである、請求項1に記載の医薬製剤。
【請求項9】
合成環状ペプチドが、
【化1】
【化2】
【化3】
から選択される、請求項1に記載の医薬製剤。
【請求項10】
合成環状ペプチドが、
【化4】
である、請求項8に記載の医薬製剤。
【請求項11】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、疼痛の治療または予防方法。
【請求項12】
疼痛が神経因性疼痛である、請求項11に記載の方法。
【請求項13】
疼痛が炎症性疼痛である、請求項11に記載の方法。
【請求項14】
神経因性疼痛が糖尿病性神経因性疼痛である、請求項12に記載の方法。
【請求項15】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、神経損傷の回復を促進する方法。
【請求項16】
請求項1から10のいずれか一項に記載の医薬製剤を経口または経腸投与する段階を含む、アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、気分変調、および季節性情動障害など)、または炎症の治療または予防方法。
【請求項17】
疼痛の治療または予防のための経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【請求項18】
疼痛が神経因性疼痛である、請求項17に記載の使用。
【請求項19】
疼痛が糖尿病性神経因性疼痛である、請求項18に記載の使用。
【請求項20】
疼痛が炎症性疼痛である、請求項17に記載の使用。
【請求項21】
神経損傷からの回復促進における経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【請求項22】
アルツハイマー病、統合失調症、うつ病、てんかん、小細胞肺癌、心血管障害、胃運動障害、尿失禁、ニコチン中毒、気分障害(双極性障害、単極性うつ病、トゥレット症候群、気分変調、および季節性情動障害など)、または炎症の治療または予防のための経口または経腸投与用の医薬の製造における合成環状α-コノトキシンペプチドの使用。
【図1】

【図2】
【図3】
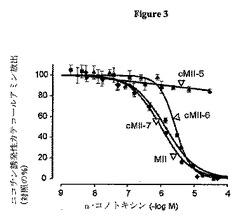
【図4】
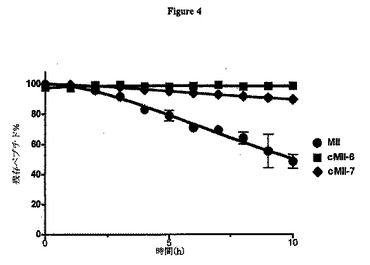
【図5】
【図6】
【図7】
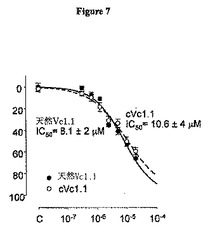
【図8】
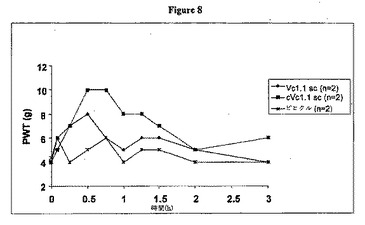
【図9】
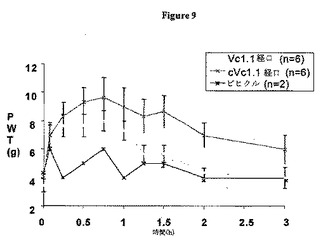
【図10】
【図11】
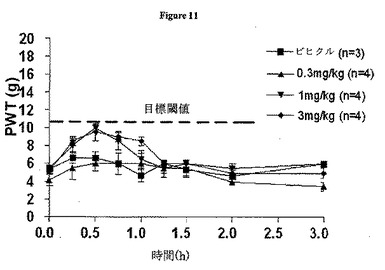
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2009−533355(P2009−533355A)
【公表日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願番号】特願2009−504526(P2009−504526)
【出願日】平成19年4月3日(2007.4.3)
【国際出願番号】PCT/AU2007/000439
【国際公開番号】WO2007/118270
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(503319102)ザ ユニバーシティ オブ クィーンズランド (4)
【Fターム(参考)】
【公表日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願日】平成19年4月3日(2007.4.3)
【国際出願番号】PCT/AU2007/000439
【国際公開番号】WO2007/118270
【国際公開日】平成19年10月25日(2007.10.25)
【出願人】(503319102)ザ ユニバーシティ オブ クィーンズランド (4)
【Fターム(参考)】
[ Back to top ]