
生体付着性のコンパクトマトリックスの製法
生体付着性のコンパクトマトリックスを製造する方法であって、1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーを含んでなる粉末の均一な混合物の調製、前記粉末混合物を原料とする、直接圧縮による圧縮ユニットの調製、最後に、このようにして得られた圧縮ユニットの、80〜250℃の範囲の温度における1〜60分間の加熱を含んでなる、生体付着性コンパクトマトリックの製法に係る。前記粉末混合物は、少なくとも1つの活性物質も含むことができ、このようにして得られた圧縮ユニットは、延長された放出性によって特徴付けられ、pH4〜8の水溶液中において、実質的に0オーダーの活性物質放出速度を有する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、一般に、化学工業の分野、特に医薬品工業の分野に関する。
【0002】
特に、本発明は、活性物質の放出のために使用される錠剤又はデバイスのような、持続性放出によって特徴付けられるコンパクトマトリックスの製法及びこのようにして得られたコンパクトマトリックスに係る。さらに詳述すれば、本発明は、活性物質の放出のために使用される錠剤又はデバイスを構成できるコンパクトマトリックスの製法であって、特殊な成分の直接圧縮工程及び加熱処理工程を含んでなる製法に係る。
【背景技術】
【0003】
約40年に亘って、医薬工業の研究者は研究を行い、生体への活性物質の放出を修飾及び制御するための新規なシステムを開発してきた。
【0004】
このような修正は、投与回数を減らし、活性物質の放出速度を可及的に制御するために、薬剤の生体への放出を延長するもの(延長された又は持続性の放出)であり、0オーダーの、すなわち、投与剤形内に含まれる薬剤投与量とは無関係の放出速度を得ることを追求するものであった(延長された放出及び標的薬剤送達システム, Remington The Science and Practice of Pharmacy 21版, 47章, p. 939-936)。
【0005】
他の修正は、特殊な刺激(pH、温度、酵素活性、イオン強度)を関数として(Morishita Mら, J. Drug Deliv. Sci. Technol., 16(1): 19-24, 2006)又は特定の期間後又は所定の時間間隔で(遅延化又はパルス化放出;Gazzanigaら, European Journal of Pharmaceutics and Biopharmaceutics, 68(1): 11-18, 2008))、薬剤の放出が生体の特殊なゾーンで生ずるようにすることを目標とするものである。
【0006】
特に、経口投与剤形では、薬剤の持続性放出は、コーティングフィルムとして少量で、又はマトリックスシステムを形成するために、より多い量で使用される好適なポリマーの使用を介して達成される。いずれのケースにおいても、フィルム又はマトリックスの組成は、活性物質の放出に影響を及ぼすことができ、多くのケースにおいて、放出速度は、このようにして事前に設計され、好適なインビトロ溶解テストを介して確認される(Kanjickal DG, Lopina ST, ポリマー送達システムからの薬剤放出のモデル化, Critical Reviews in Therapeutic Drug Carrier Systems, 21(5): 345-386, 2004)。
【0007】
フィルムは、錠剤のコーティングにおいて、又はそのままで又はカプセル化又は錠剤へ変換して投与される顆粒又はペレットのコーティングにおいて直接使用される。
【0008】
マトリックスを形成するために使用されるポリマーは、その異なる溶解又は浸食速度を介して又はマトリックスにおける活性物質の拡散を介して(親水性ポリマーのケースでは、膨潤及びゲル化でき、多少容易に浸食される)、薬剤の放出を制御する(Brazel CA, Peppas NA, リラクシング、膨潤性、親水性、ガラス質のポリマーにおける溶質及び薬剤の輸送のメカニズム, Polymer, 40(12): 3383-3398, 1999)。
【0009】
薬剤送達システム(DDS)と呼ばれる、「エンジニアード」システム又はより高価かつより複雑な工業的方法を介して得られるデバイスがある(Hilt JZ, Peppas NA, International Journal of Pharmaceutics, 306(1-2): 15-23, 2005)。DDSプロトタイプは、半透過性膜、及び薬剤の放出を延長するために、これらの膜内において発生された浸透圧を利用する、いわゆる、「浸透」システムである。溶液又は懸濁液中の薬剤の放出は、錠剤の表面においてレーザー光によって生ずるミクロ孔を介して、0オーダーの放出速度論に従って一定の速度で生ずる(米国特許第4,160,020号;WO 03/075894)。このようなシステムは、含まれる投与量の全部の多量放出(「不当投与」として知られている現象)の欠点をもたらし、輸送される活性物質のタイプに関連して、生体に対して毒作用をもたらす。このような観点から、「ペレット化」剤形は、工業的生産の品質制御工程においても、より多くの保証を提供する。
【0010】
マトリックスシステムは、水性液体の存在下で膨潤性のエチルセルロース又は親水性ポリマー、例えば、ヒドロキシプロピルメチルセルロース(その分子量及び飽和度を関数として、あまり浸食性ではないゲルを形成することもできる)のような非水分散性又は疎水性のシステムを利用する。
【0011】
これらのマトリックスシステムは、一般に、造粒又は「ペレット化」プロセス(いずれも、組成物の改良された均一化を得るため、及び粉末混合物における及びそれらの圧縮の間における偏析現象を回避するため、及び薬剤の放出を効果的に制御するマトリックスの形成を可能にするためのものである)を介して製造される。このようなプロセスは、湿潤条件(湿式造粒、スプレー乾燥)又は乾燥条件(乾燥造粒、又はローラー圧縮及びホットメルト押し出し)で行われる(固状の経口投与剤形, Remington The Science and Practice of Pharmacy, 21版, 45章, p. 889-928)。
【0012】
当然のことながら、造粒工程を含む工業的プロセスは、かなり非経済的であり、従って、工業的には、この目的のために開発され、市販されている賦形剤が存在するため、直接圧縮プロセスを利用する傾向にある(Gohel MC, Jogani PD, Journal of Pharmacy and Pharmaceutical Sciences, 8(1): 76-93, 2005;Goto Kら, Drug Development and Industrial Pharmacy, 25(8) 869-878, 1999;Michoel Aら, Pharmaceutical Development and Technology, 7(1) 79-87, 2002)。
【0013】
市販のCRシステム(Colomboら, 制御された薬剤送達用の膨潤性マトリックス; ゲル層の挙動, メカニズム及び最適パフォーマンス, Pharm. Sci. Techno. Today. 3(6), 2000)の多くは、親水性ポリマーの使用に基づくシステムを介して薬剤の放出を制御するものである(Peppas NAら, 医薬品処方におけるハイドロゲル, European Journal of Pharmaceutics and Biopharmaceutics, 50(1), 27-46, 2000)。
【0014】
上述のように、ポリマーマトリックスを介する活性物質の放出を効果的に制御するシステムは、一般的には、湿式造粒法を介して得られる(ヨーロッパ特許公開1 681 051号;米国特許第5,549,913号)。
【0015】
実際には、直接圧縮によって得られたポリマーマトリックスを介する制御された放出の例は多くはない。例えば、M.E. Pinaらは、主に水性媒体にて膨潤する親水性ポリマー(ヒドロキシプロピルメチルセルロース(HPMC))からなるマトリックス(直接圧縮を介して得られたもの)を介するイブプロフェン(あまり強い水溶性ではない薬剤)についての調節された放出を報告している(Pharmaceutical Development and Technology, 11(2): 213-228, 2006)。Peppas及びSiepmannは、HPMCからなるマトリックスからの薬剤放出のモデル化を充分に検討している(Advanced Drug Delivery Reviews, 48(2-3): 139-157, 2001)。
【0016】
E Crowleyらは、疎水性ポリマー(エチルセルロース)からなるマトリックス(直接圧縮を介して得られたもの)を介するグアイフェネシン(水溶性の薬剤)についての調節された放出を報告している(International Journal of Pharmaceutics, 269(2): 509-522, 2004)。
【0017】
熱処理を含むCR用のマトリックスを得る方法、特に、顆粒を得るため又は「錠剤」として使用される規則的な幾何形状を直接的に得るために利用されるモノリシックマトリックを生成する、ホットメルト押し出し(HME)として知られている熱可塑性ポリマーの押し出し成形の新たな技術は言及に値するものである。
【0018】
この方法は、非晶質ポリマーのガラス転移点(Tg)又は半結晶質ポリマーの融点よりも10〜60℃高い温度に加熱する手段によって、可能なプロセスアジュバントの存在下、ポリマーを溶融することを含む。好適な粘性を獲得した後、このようなメルトを、規則的な断面のスリットを強制的に流動通過させ、このようにして、続く冷却のための前記断面の成形品を生成する(Repka MAら, Drug Development and Industry Pharmacy, Part I, 33(9): 909-926及びPart II, 33(10): 1043-1057, 2007)。
【0019】
従来技術においては、いくつかのケースで、錠剤を直接加熱処理工程に供することによって、錠剤からの活性物質の放出に影響を及ぼすことできることが知見されている。
【0020】
Omelczuckらは、ポリ(dl-乳酸)(PLA)及び微結晶性セルロースを含有する錠剤の加熱処理(40〜80℃において24時間)がテオフィリンの放出を延長させることを報告している(Pharmaceutical Research, 10, 542-548, 1993)。報告された溶解度曲線から、このような放出は、0オーダーの速度論とは異なる複雑な速度論に従うことによって生ずることが推察される。
【0021】
Azarmi S.らは、3:3:4の比で薬剤をEudragit RS PO又はRL PO及び乳糖と共に直接圧縮することを介して調製し、50又は60℃よりも高い温度における2〜24時間の加熱処理に供したインドメタシン錠剤は、引っ張り強さの目に見えるほどの変更を生ずることなく、熱処理に供していない錠剤に対して、延長された放出を示すことを証明している(International Journal of Pharmaceutics, 246 (2002), 171-177)。
【0022】
また、50〜70℃における2〜24時間の加熱処理に供したジクロフェナクナトリウム錠剤(ジクロフェナクナトリウム、Eudragit RS PO又はRL PO及び乳糖(3:4:3)の直接圧縮を介して調製した)について同様の結果が、Azarmi S.らによって得られている(Pharmaceutical Development and Technology, 10: 233-239, 2005)。
【0023】
既に、Billaらは、ジクロフェナクナトリウム/Eudragit NE40D/微結晶性セルロースの錠剤に対する60℃での加熱処理の影響を検討し、錠剤の引っ張り強さの増大と共に、持続性の放出が達成されることを見出している(Drug Development and Industrial Pharmacy, 24(1), 45-50, 1998)。
【0024】
薬剤放出特性に対する錠剤の加熱処理の影響に関する観察は、上述の例、すなわち、Eudragit系の錠剤又はPLAを含有する錠剤に限られているように思われ、長い加熱時間後に得られた結果は、放出時間の延長及び得られた放出速度論の両方について、かなり限られたものである。
【0025】
制御された放出については、非水溶性の架橋ポリマー(しかし、親水性であり、水性媒体中で膨潤性である)の使用も知られている(Brazel CS, Peppas NA 1999, Mechanisms of solute and drug transport in relaxing, swellable, hydrophilic glassy polymers, Polymer 40(12): 3383-3398)。このカテゴリーに属するものとして、ジビニルグリコールにて架橋したポリアクリル酸のポリマーがあり、例えば、錠剤、ディスク又は生体付着特性を備えたフィルムの形のCR剤形の製造において使用されることが知られているポリカルボフィル(CAS RN 9003-01-4)(Handbook of Pharmaceutical Excipients, 5版, Pharmaceutical Press, p. 539-541, 2006)がある。例として、国際公開WO 2005/065685及びWO 01/95888及び米国特許第5,102,666号を参照できる。
【0026】
ポリカルボフィルは、中でも、その生体付着特性のため、剤形内で使用される。Robinsonらは、剤形内で使用されるポリカルボフィル及び他のポリマーの生体付着特性を調査している(Journal of Pharmaceutical Sciences, 89(7): 850-866, 2000)。Repkaらは、ポリカルボフィルを含有し、HMEを介して得られたバッカルフィルムの生体付着特性を研究している(Journal of Controlled Release, 70(3): 341-351, 2001)。
【0027】
ポリカルボフィルが、pHに依存して水を吸収し、その元の容積の1000倍及びその元の直径の10倍まで膨潤する能力は、注目に値するものである。ポリカルボフィルに関連する米国薬局方31のモノグラフの処方によれば、炭酸水素ナトリウム溶液に対する吸収力は、乾燥ポリマー1g当たり62g未満であってはならない。これらの特性のため、ポリカルボフィルは腸の機能不全、慢性の便秘、憩室炎及び過敏性腸症候群の治療のために、医薬品だけでなく、食品サプリメントにおいても使用される。
【0028】
国際公開WO01/95888は、5α-リダクターゼによって代謝される活性成分、例えば、ヒドロキシプロピルメチルセルロースのような水溶性ポリマー、及び非水溶性、水膨潤性の架橋ポリカルボン酸ポリマー、特にポリカルボフィルを含んでなる生体付着性の持続放出性錠剤を開示している。このような錠剤の製法は、加熱工程を含んではいない。国際公開WO 2005/065685は、活性成分及び少なくとも2つのポリマー(1つは酸不溶性ポリマーであり、他は生体付着性ポリマーである)からなるポリマー系(ポリマー系は、例えば、エチルセルロース、ポリカルボフィル及び微結晶性セルロースを含むことができる)を含んでなる生体付着性の放出持続性錠剤を開示している。この明細書によれば、錠剤の製造において、加熱工程は認められない。
【発明の概要】
【発明が解決しようとする課題】
【0029】
本発明の第1の態様では、本発明は、架橋ポリカルボン酸ポリマーを含有し、非浸食性及び生体付着性が付与されており、水の吸収によって膨潤可能であり、活性物質の持続性放出のために使用可能な非浸食性ゲルを形成するコンパクトマトリックスを提供するとの目的を有する。
【課題を解決するための手段】
【0030】
このような目的は、生体付着性のコンパクトマトリックスを製造する方法であって、
−少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋したポリカルボン酸ポリマーを含んでなる粉末の均一な混合物を調製する工程:
−前記粉末混合物を原料として、直接圧縮又は乾燥圧密化によって、圧縮した又は圧密化したユニットを調製する工程;
−このようにして得られた圧縮した又は圧密化したユニットを、80〜250℃の範囲の温度、1〜60分の範囲の時間の加熱処理に供する工程
を含んでなる生体付着性のコンパクトマトリックスの製法によって達成される。
【0031】
他の態様では、本発明は、持続性の又は制御された放出によって特徴付けられる、水中で膨潤できる、上述の生体付着性のコンパクトマトリックスを含んでなる活性物質放出用の圧縮ユニットを提供するとの目的を有する。このような目的は、上述のような、コンパクトマトリックスの製法(当該方法においても、上述の均一な粉末混合物は少なくとも1つの活性物質を含んでなる)によって達成される。
【0032】
用語「圧縮したユニット」について、圧縮したユニットは、医薬品用の一般的な錠剤、特に、活性物質又は生理的状態を回復させる物質を放出できる経口投与のための錠剤だけでなく、粉末圧縮によって得られる他のデバイス、例えば、尿道座剤、錠剤及び膣、口腔、鼻、歯、耳鼻咽喉、目、又は表皮適用のディスクも意味するものである。このような錠剤又はディスクの用途は、ヒト又は動物用の医薬品の分野(ここで、活性物質とは、それぞれ、EU指令2004/27 CE(art. 1)及び2004/28 CE(art. 1)に記載の定義による医薬品を意味する)に限定されるものではなく、EU指令93/42/CEEに記載の定義による医療用具の分野、EU規則(CE)第178/2002の第2項に記載の定義による食品の分野、指令2002/46/CEによって定義された食品サプリメントの分野、EU指令CE第89/398によって定義された食事製品及び幼児用製品の分野、EU指令91/414/CEの定義による植物保護製品の分野、EU規則(CE)第2003/2003の定義及び分類による肥料又は化学肥料の分野、EU指令98/8/CEに記載の定義による消毒剤及び害虫駆除剤及び殺生剤の分野、洗剤の分野のような他の分野にも及びものである。
【0033】
また、診断、治療及び一般的な殺生を目的として、放射性医薬品、放射性核種及び放射性核種によってマーク付けされた分子も、このようなマトリックスによって担持され、放出される。
【0034】
上述の粉末混合物は希釈剤を含むこともできる。希釈剤は、好ましくは、Tabettose(登録商標)及びPharmatose DLC 15(登録商標)と同等の無水の乳糖(CAS RN 63-42-3)又は乳糖1水和物(CAS RN 64044-51-5)(いずれも公知の非晶質又は結晶性の物理的形状であり、スプレー乾燥又は凝集によって得られたもの)及び/又は微結晶性セルロース(例えば、Avicel PH、Ecomocel、Tabulose)からなるものである。例えば、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac(登録商標))又はCellactose(登録商標)のような予め形成された乳糖/微結晶性セルロース混合物、又はLudipress、Starlac、Pharmatose DLC 40、Avicel CE 15、Celocal、Prosolvのような直接圧縮用に同時加工された他の賦形剤も使用できる。
【0035】
上述のアルキルセルロースは、例えば、メチルセルロース(CAS RN 9004-97-5)及びエチルセルロース(CAS RN 90004-57-3)からなる群から選ばれ、上述のヒドロキシアルキルセルロースは、例えば、ヒドロキシプロピルセルロース(CAS RN 9004-65-2及びRN 78214-41-2)、ヒドロキシプロピルメチルセルロース(CAS RN 9004-65-3)、ヒドロキシエチルセルロース(CAS RN 9004-62-0)、ヒドロキシエチルメチルセルロース(CAS RN 9004-42-2)からなる群から選ばれる。
【0036】
アルキル-又はヒドロキシアルキルセルロースの一部の代わりに、次の物質(相互に組み合わせてもよい):クロスポビドン、ポビドン(9003-39-8)、ビニルピロリドン-酢酸ビニルコポリマー(Kollidon(登録商標)VA64)、セルロースアセテートフタレート(CAS 9004-38-0)、ハイプロメロースフタレート(CAS RN 9050-31-1)、ポリビニルアルコール(CAS RN 9002-89-5)、ポリビニルアセテートフタレート(CA RN 34481-48-6)、各種のシクロデキストリン(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、Eudragit E、L、S、RS、RL、PO、NE、RSPMのようなEudragit(Rohm GmbH)の商品名で販売されている各種のタイプのメタクリレートポリマー(Eastman Chemical Company及びBASFによっても製造された各種のもの)、グリセリルトリアセテート、トリエチルシトレート、トリブチルシトレート、アセチルトリエチルシトレート、アセチルトリブチルシトレート、ジブチルセバケート、ジエチルフタレート、ジブチルフタレート、ジオクチルホスフェート、ポリエチレングリコール、ポリエチレンオキシド(CAS RN 25322-68-3)、カルシウムカルボキシメチルセルロース(CAS RN 9050-04-8)、ナトリウムカルボキシメチルセルロース(CAS RN 9004-32-4)、イヌリン(CAS RN 9005-80-5)、キトサン(CAS RN 9012-76-4)及びその誘導体、ガーゴム(CAS RN 9000-30-0)、キサンタンガム(11138-66-2)及びトラガカントゴム(CAS RN 900-65-1)、カルボマー(CAS RN 9003-01-04及び96827-24-6)、カラゲナン(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、アルギン酸(CAS RN 9005-32-7)、ポロキサマー(CAS RN 9003-11-6)、脂肪族ポリエステル(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、セルロースアセテートブチレート、キトサンラクテート、ペクチン、ポリエチレン-コビニルアセテート、ポリエチレン、ポリビニルアセテート-コメタクリル酸、カルナウバワックス、ブチルヒドロキシアニソール、ブチルヒドロキシトルエン、アスコルビルパルミテート、グリセリルパルミトステアレート、水素化大豆油及びヒマシ油(Sterotek(登録商標)K)、グリセリルモノステアレート、d-α-トコフェロール(ビタミンE)、ビタミンEスクシネート、ビタミンE及びTPGS、メチルパラベン、ブチルステアレート、ステアリルアルコール、サッカロースモノパルミテート(Sucroester)、グリセロールエステル及びPEGエステル(Gelucire 44/14)、ポリオキシエチレンアルキルエステル、グリセリルパルミトステアレートPrecirol(登録商標)ATO 5、鉱油、ヒマシ油及び発泡性の混合物又は系を形成することが知られている賦形剤を使用することもできる。水に膨潤可能である上述の非水溶性の架橋ポリカルボン酸ポリマーは、好ましくは、ポリカルボフィル(CAS RN 9003-01-04)からなる。
【0037】
圧縮ユニットを加熱する温度は、好ましくは、90〜160℃であり、加熱時間は、1〜30分の範囲であり、特に1〜20分である。圧縮ユニットを処理温度とするための加熱速度は1〜50℃/分の範囲である。
【0038】
加熱処理に供される粉末の圧縮は、100〜500 MPaの圧力で操作することによって行われる。5kPa〜100 MPaの圧力で操作することによって、続く加熱処理に供される、低い引っ張り強さを有する圧縮体を得ることもできる。圧縮ユニットの形状は、各種の規則的な立体幾何形状であり、質量は、要求及び用途(ヒト又は動物)に応じて100g以下であるが、農業用には、100g以上である。
【0039】
これらの圧縮ユニットは、その組成において、必要に応じて、一般的に圧縮プロセスにおいて使用され、当業者には公知であるアジュバント(滑択剤、潤滑剤、抗接着性剤、崩壊剤及び超崩壊剤、芳香剤、甘味料及び吸着剤)のすべてが添加される。
【0040】
このような錠剤は、胃溶解抵抗性、腸溶性又は環境保全性を活性物質に付与するために、ポリマーフィルムコーティング及び/又はドライコーティングの従来の方法にて被覆される(医薬品剤形: Tablets Volume 1,2,3, H.A. Lieberman, L. Lachman, J.B. Schwartz編, Dekker, 第2版, US 1989)。
【0041】
本発明の対象である錠剤は、インレイ錠剤、多層錠剤及びコア錠剤として知られている錠剤を得るために、コア又は層(活性物質を含有する又は含有しない)として使用される(医薬品剤形: Tablets Volume 1, H.A. Lieberman, L. Lachman, J.B. Schwartz編, Dekker, 第2版, US 1989)。各種の追加の層は、ここに述べるものと同一の質的組成及び/又は異なる活性物質又は追加の活性物質を異なる含量で有することができ、あるいは、これらは、既に述べた又は当分野において使用されている異なる物質でもよい。
【0042】
多層錠剤の場合には、本発明によるマトリックスは、多層錠剤の少なくとも1つの層に相当する。
【0043】
コア錠剤の場合には、ここに記載のマトリックスは、コア又は外層と呼ばれるクラウン層に相当し、活性物質を含むか、又は含まず、又は各層ごとに異なった活性物質を含む。
【0044】
インレイ錠剤の場合には、本発明によるマトリックスは、外層及びインレイの両方に相当し、活性物質又はいくつかの活性物質を含有でき、又は含有しなくてもよい。本発明による方法が、好ましくは、直接圧縮法である場合であっても、特定の状況では、特定の活性物質の粒径によって生ずる欠点を解消するために、錠剤又はコア/インレイの調製又はいくつかの層の調製において、当業者にとって公知の技術をなお使用できる。このような公知の技術は、湿式造粒(湿式造粒、流動床造粒、スプレー乾燥、スプレー凝固)又は乾式造粒(乾式造粒又はローラー圧縮)と呼ばれるか;又は、プロセスに供されるペレットに対して、球形化(球形で、制御されたサイズをもつ顆粒を生成する)として知られている圧縮プロセスを施す(Remington, 21版, 45章, p. 903)。当然のことながら、各種のタイプの造粒は、これらのタイプの方法に必要であり、当業者に知られている最少量のアジュバント又は賦形剤を要求する。酸化に対して敏感な活性物質の場合には、不活性雰囲気、例えば、窒素雰囲気において、加熱を行うことができる。
【0045】
揮発性又は昇華性の活性物質については、又は必要な場合には、加熱処理を、自然の雰囲気又は不活性雰囲気において、このような雰囲気を環境圧力よりも0.5MPaまで高い圧力まで加圧して、実施できる。加熱後の冷却工程は、自然に又は強制的に、例えば、室温の乾燥空気又は不活性ガス(N2、Ar、He)又は室温以下の温度に冷却した乾燥空気又は不活性ガスの通気を介して制御して行われる。
【0046】
加熱後、充填前に、雰囲気条件下でのコンディショニング時間が必要であり、この時間は、選択した組成に応じて、24時間であってもよい。一般に、このような待ち時間は、生産の質には影響を及ぼさないが、いずれのケースにおいても、標準的に5分間待つことが好ましい。
【0047】
本発明による方法における使用のための好適な粉末組成物は、活性物質、MicroceLac、エチルセルロース及びポリカルボフィルを含んでなる。ポリカルボフィルは、一般に、圧縮及び加熱処理前の粉末混合物の総質量の5〜35質量%、好ましくは10〜25質量%を構成する。
【0048】
エチルセルロース及びMicroceLacは、粉末混合物中に、一般に、質量比1:2〜2:1、好ましくは0.8:1〜1.2:1で存在し、圧縮及び加熱処理前の粉末混合物の総質量の45〜95質量%、好ましくは60〜80質量%を構成する。
【0049】
エチルセルロース及びポリカルボフィルは、粉末混合物中に、一般に、質量比1:5〜5:1で存在する。活性物質は、粉末混合物中に、0.001 ppm〜圧縮及び加熱処理前の粉末混合物の総質量の50質量%の量で含有される。さらに、この百分率の範囲内において、活性物質を、溶解プロセス、屈水性錯体又は包接錯体を形成するために好適なアジュバント物質、又は薬剤の消化管吸収又はいずれにしても薬剤の経粘膜吸収のプロセスを促進する物質(エンハンサーとして知られている);又は活性物質を物理的に又は化学的に安定化する物質と混合される。
【0050】
活性物質は、ヒト又は動物において、治療、診断又は予防のために使用可能な、薬理活性を有する天然、合成又は半合成のものであるか、あるいは、生物学的、物理学的又は化学的に活性であり、植物のケア(植物保護製品)用に、肥料として、殺菌剤及び/又はヒト又は環境の害虫駆除剤として使用可能な、天然、合成又は半合成の物質、又はEUによって定義されたような殺生剤のカテゴリーに属する物質である。
【0051】
本発明による方法において活性物質の使用に関して要求される唯一の条件は、方法自体によって予測される加熱条件(温度、時間)下において、活性物質が充分な安定性を有していることである。
【0052】
本発明による圧縮ユニットから、健常な対象又は慢性又は急性の病性状態にある対象を含むヒト又は動物用のダイエット用栄養補助食品として、栄養作用を有する物質が放出される。
【0053】
これらの圧縮ユニットは、化粧用に許可される物質と一緒に好適に処方される場合、EUにおいて正当なものとして許される「化粧品」の定義に従って、化粧品としての用途を有する。
【0054】
本発明は、生体付着特性を備え、活性物質、少なくとも1つのアルキルセルロース又はヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋ポリカルボン酸ポリマーを含んでなる持続放出性錠剤にも関する。好ましくは、このような錠剤は、制御された放出性及びpH4〜8の水溶液において実質的に0オーダーの活性物質放出速度を有する。
【0055】
上述の錠剤は、好ましくは、無水の又は1水和物の乳糖(いずれも、公知の結晶性又は非晶質の物理的形である)、及び/又は微結晶性セルロースからなる希釈剤も含有する。また、当業者にとっては一体形賦形剤(例えば、MicroceLac(登録商標))として知られている微結晶セルロース/乳糖の予め形成した混合物を含有することもできる。
【0056】
上述の成分間の比、粉末又は顆粒の圧縮条件、加熱温度及び加熱処理時間の各条件を変更することによって、活性物質の放出が生ずる速度(一般に、pH4〜8において0オーダー速度論に従う)を制御することができる。
【0057】
活性物質が存在しない(0%)場合、このような圧縮し、熱処理されたマトリックスは、その水性媒体中で膨潤する能力及びその生体付着特性のため、腸の機能不全又は慢性の便秘、憩室炎、過敏性腸症候群のようないくつかの病状における治療作用を実行できる。
【0058】
このような錠剤は、上述の方法によって得られる。
【0059】
これらのマトリックス(水中で膨潤した後、乾燥を介して、元の形状及びサイズを再び獲得できる)の特性を利用することによって、いくつかの活性物質(特に、熱不安定性であり、固状で得ることが困難なもの)を、不動化、すなわち、本発明に従って調製したマトリックス(医薬品含まない)を、好適な濃度の活性物質含有する水溶液に浸漬することによって、このマトリックスに負荷させることができる。一定時間後、膨潤した錠剤を取り出し、元の形状及びサイズを再獲得するため、空気中で又は好適な乾燥法を介して、空気又は不活性ガスの強制換気及び温和な加熱を介して、又はIRランプの照射を介して又は凍結乾燥プロセスを介して乾燥できる。
【図面の簡単な説明】
【0060】
【図1a】本発明の方法によって得られた錠剤(T)(実施例1)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を、同一の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図1b】本発明の方法によって得られた錠剤(T)(実施例1)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図1c】本発明の方法によって得られた、熱処理した(150℃、15分間)錠剤(実施例1)のDSCプロフィール(実線)を、同一の組成の組成を有するが、加熱処理に供していない錠剤のDSCプロフィール(一点差線)と対比して示すグラフである。
【図2a】本発明の方法によって得られた錠剤(T)(実施例2)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィールを、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図2b】本発明の方法によって得られた錠剤(T)(実施例2)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールを、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図2c】下側のグラフは、本発明の方法によって得られた錠剤(実施例2)のDSCトレース(実線)を、同一の組成の組成を有するが、熱処理に供していない錠剤のトレース(二点差線)と対比して示すグラフである。上側のグラフは、ジルチアゼム(破線)及びMicroceLac(点線)のDSCトレースを、実施例2による錠剤と同じ組成を有するが、熱処理していない錠剤のDSCトレース(実線)と対比して示すグラフである。
【図3a】本発明の方法によって得られた錠剤(T)(実施例3)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図3b】本発明の方法によって得られた錠剤(T)(実施例3)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図3c】実施例3による錠剤6個から及び実施例2による錠剤6個から放出されるジルチアゼム(DZT)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)の対比を示すグラフである。
【図3d】実施例3による錠剤(実線)及び同一の組成の組成を有するが、熱処理に供していない錠剤(一点差線)のDSCトレースを、相互に対比して示すグラフである。
【図4a】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の方法によって得られた錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)の対比を示すグラフである。
【図4b】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の方法によって得られた錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)の対比を示すグラフである。
【図5a】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の錠剤(T)(実施例2)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)の対比を示すグラフである。
【図5b】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の錠剤(T)(実施例2)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールの対比を示すグラフである。
【図5c】調製直後(t=0)及びブリスター包装して13ヶ月間保存した後の、本発明の錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールの対比を示すグラフである。バーは信頼区間95%を示す。
【図5d】実施例2によるバッチから得られ、熱処理し、ブリスター包装して32ヶ月間保存した錠剤(T)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を示すグラフである。バーは信頼区間95%を示す。
【図5e】実施例2によるバッチから得られ、熱処理し、ブリスター包装して32ヶ月間保存した錠剤(T)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を示すグラフである。バーは信頼区間95%を示す。
【図5f】調製直後(t=0)及びブリスター包装して13ヶ月間(t=13ヶ月)及び32ヶ月間(t=32ヶ月)保存した、実施例2による熱処理した錠剤(T)及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)の異なるバッチの、リン酸塩緩衝液(pH7.2)における、600分の時点での、溶液中に存在する量に関する溶解ジルチアゼム(DTZ)フラクションの平均溶解プロフィール(n=6)の対比を示すグラフである。
【図6】各種の熱処理に供した本発明による錠剤(実施例2)のリン酸塩緩衝液(pH7.2)におけるジルチアゼム(DZT)の溶解プロフィールを示すグラフである。特に、このグラフは、実施例2による錠剤からのリン酸塩緩衝液(pH7.2)におけるジルチアゼム(DZT)の放出に対する各種の熱処理の影響の対比を示す(錠剤の平均溶解プロフィール(n=6):nt=処理していない;150-5=150℃における5分間の処理;90-15=90℃における15分間の処理;150-15=150℃における15分間の処理;130-15=130℃における15分間の処理)。
【図7】本発明による錠剤の膨潤百分率レベル(S%)を示すグラフである。特に、実施例1及び2による熱処理した錠剤の平均膨潤プロフィール(n=3)の対比を示す。バーは標準偏差値を示す。
【図7a】リン酸塩緩衝液における溶解テスト前及び溶解テストの終了時(最大の膨潤度に達した時点)の本発明による熱処理した錠剤を示す3つの写真である。左から右へ順に、溶解テスト前の実施例1又は2の元のサイズの錠剤;リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による錠剤;リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例1による錠剤の写真である。
【図7b】溶解テストの終了時において達した最大膨潤度の本発明による熱処理した錠剤の拡大写真である。特に、図7aの中央に示す写真(リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による錠剤)の拡大である。
【図7c】次の2つの写真を示す。上の写真は、最大膨潤度の本発明による3つの錠剤の写真であり、下の写真は、最大膨潤度の本発明による錠剤を斜めから見た写真である。特に、リン酸塩緩衝液(pH=7.2)、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による熱処理した錠剤の写真である。
【図7d】本発明による圧縮マトリックス(活性物質を含まない)の膨潤を示すグラフである。このグラフは、特に、実施例2による成分間の割合をもち、2種類の異なる加熱処理(150℃において5分間又は15分間)に供したマトリックス及び加熱処理に供していないマトリックス(NT)(いずれも、活性物質を含有していない)の平均膨潤プロフィール(n=3)の対比を示す。
【図8】本発明の方法によって得られた錠剤(T)(150℃において、5又は15分間熱処理したもの)(実施例4)から放出されるグリクラジドのリン酸塩緩衝液(pH=7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図9】ブリスター包装して38ヶ月間保存した本発明による熱処理(150℃において15分間)した錠剤(実施例2)のサイズと、同じバッチの錠剤6個(熱処理(150℃において15分間)及びブリスター包装での13ヶ月間の保存後、0.05 N HClにおける溶解テストに供し、ついで、室温における空気乾燥に供し、最後に、室温において乾燥した後に、さらに、空気中で25ヶ月間保存した)のサイズとの間の対比を示す写真である。
【図10】0.05 N HClにおける溶解テストの終了時に達した最大膨潤度の実施例2による熱処理(150℃において15分間)錠剤と、元のサイズの錠剤との対比を示す写真である。詳しくは、写真の左には、37℃、0.05 N HClにおける溶解テストの終了時に達した最大膨潤度の実施例2による錠剤が示されている。右には、元のサイズの実施例2の錠剤が示されている。
【図11】各種の等温加熱に供したポリカルボフィル粉末の平面収縮率のホットステージ顕微鏡調査の結果を示すグラフである。
【図12】ポリカルボフィル粉末の、DSCプロフィール(endo up)、TGAプロフィール(温度に対する質量%)及び温度に対する平面収縮(PS)%のホットステージ顕微鏡調査(HSM)プロフィールの対比を示すグラフである。
【図13】ポリカルボフィル成形体(熱処理に供していない)の断面のSEM顕微鏡写真である。成形体は、ポリカルボフィル粉末100 mgに圧力750 kPaを15分間かけることによって得られたものである。
【図14】熱処理したポリカルボフィル成形体の断面のSEM顕微鏡写真である。成形体は、ポリカルボフィル粉末100 mgに圧力750 kPaを15分間かけることによって得られたものである。ついで、成形体を、熱空気オーブン内で、150℃、15分間の加熱に供した。
【図15】図14の同じサンプルを、より高い倍率で見たSEM顕微鏡写真である。
【図16】熱処理したエチルセルロース/ポリカルボフィル(3/2)成形体の断面のSEM顕微鏡写真である。成形体は、エチルセルロース/ポリカルボフィル(3/2)の粉末混合物100 mgに圧力750 kPaを15分間かけることによって得られたものである。ついで、成形体を、熱空気オーブン内で、150℃、15分間の加熱に供した。
【図17】各種の熱処理に供したポリカルボフィルのSEM顕微鏡写真である。
【図18】熱空気オーブンにおいて、150℃で15分間加熱したポリカルボフィル粉末の断面のSEM顕微鏡写真である。
【発明を実施するための形態】
【0061】
本発明は、錠剤の構成に有用な各種の賦形剤の混合物について行った実験研究(活性物質の放出が、エネルギー処理によって影響を受ける)に基づくものである。主な目的は、大気条件下での加熱を介して、処方中の成分が分解することなく、放出時間の延長を示す錠剤を得ることにある。
【0062】
また、活性物質の放出が、0オーダー速度論に従って、非〜易浸食性マトリックスから生じ、これによって、時間に対する放出速度が、処方中の活性物質の残量には非依存性である(これは、制御された放出性の医薬剤形の必須の要件である)処方を得ることを追求するものである。
【0063】
第1のステップでは、薬剤を含まない各種の処方を扱い、得られた崩壊時間の遅延に基づいて、開発する錠剤において使用する賦形剤を選択した。その後、エネルギー処理について使用した温度に対するこれら賦形剤の抵抗性を評価するためにテストを行った。
【0064】
ついで、錠剤からの放出の特徴評価に有用と思われるモデル薬剤を選択した。迅速な又は標準的な放出形の処方及び変性した放出形の処方の両方において、市販のジルチアゼム塩酸塩を選択した。
【0065】
予備テストに基づき、及びこのモデル薬剤を使用して、各種組成の錠剤を調製した。
【0066】
これらの錠剤からの活性物質の放出に対する加熱処理の影響を、酸性媒体中(胃環境をシミュレートする)及びリン酸塩緩衝液中(一部、腸管環境をシミュレートする)の両方で実施した溶解テストによって評価した。錠剤が受けた変性を、熱分析技術、顕微鏡分析を介して及びこのタイプの医薬剤形について一般的な各種の物理的テストを介して調査した。
【0067】
活性物質の各種賦形剤との混合物の調製
それぞれ任意に篩通した成分の混合を、テフロン(登録商標)ストッパーを具備するねじ付きキャップをもつ琥珀色の円筒状ガラス容器において又は好適なステンレス鋼容器において行い、Turbula(登録商標)ミキサーにおいて、成分の混合物が完全に均質になるまで、一般に下記のようにして実施した:
1.少数の成分及び同一質量の活性物質からなるコアを形成する;
2.残りの活性物質の全量を添加する;
3.多数の賦形剤を、容器内に収容された粉末の量と同じ量で添加する;
4.多数の賦形剤の全量を添加する。
全ての混合物について、粉末の各一定量を、量にに応じた時間で混合し、一般に、最大量に関して最長30〜40分間以下である。
【0068】
錠剤の調製
150〜170 mgの範囲の質量をもつ錠剤を、凹形のモノパンチを具備する交互打錠機によって調製した。
【0069】
錠剤及び粉末の処理
処理に供する錠剤を金属支持体上に置き、各々を、小さい金網バスケットによって保護した。処理をガスクロマトグラフ(HP 5890シリーズII)のオーブン内で行い、処理は、所定の処理温度への加熱及び係る温度における所定時間の維持からなる。使用した温度プログラムは次のとおりである:25℃において0.1分、温度勾配30℃/分で最終温度に到達;及び係る温度における所定時間の維持、ついて、室温への錠剤の強制又は自然冷却。
各錠剤の処理後、質量損失率を、下記の式に従って評価した:
Δm%=(m0−m)/m0*100
ここで、m0は錠剤の初期質量であり、及びmは加熱処理後の錠剤の質量である。
比較用の粉末の処理を、ガスクロマトグラフにおいて、パイレックス(登録商標)ガラス管において行った。
【0070】
錠剤の保存
非処理及び処理済の錠剤を、PVCブリスター包装して、室温において、各種の期間:47日間、13ヵ月間及び32ヶ月間保存した。保存期間の終了時に、質量増加率(Δw%)を、式:
Δw%=(mc−m0)/m0*100
ここで、mcは保存期間後の錠剤の質量であり、及びm0は錠剤の初期質量である。処理済の錠剤について、m0は熱処理後の質量を表す。
【0071】
錠剤の硬さの測定
処理済及び非処理の錠剤について、好適な器具によってテストを行い、錠剤の実際の半径方向の崩壊によるもののみを有効な結果とみなし、変形現象又はキャッピングによるものについては、有効な結果とみなさなかった。得られた結果は、半径方向の引っ張り強さを示し、kp(キロポンド=キログラムf=9.80665ニュートン)で示す。
【0072】
錠剤中に存在する水分量の測定
好適な自動装置(Metter-Roledo DL38)によるKarl Fischer(KF)滴定によって測定を行った。滴定剤として、酒石酸ナトリウム・2水和物(Riedl-deHaen)にて標準化したHydranal Composite5(Riedel-deHaen)を使用した。得られた結果を、ガラス製乳鉢において粉砕した錠剤に由来する粉末サンプル55.0 mg(正確に秤量した)に含有される水分率(m/m)として表す。この場合にも、処理済及び非処理の錠剤についてテストを行った。
【0073】
示差走査熱量測定法(DSC)
錠剤の処方において使用した活性物質及び賦形剤についての物理的安定性の測定
正確に秤量した各賦形剤/物質5.0mgをアルミニウム製の皿に入れ、好適なプレスにて閉じ、窒素流下、DSC(Perkin Elmer 7)によって分析した。分析を、熱処理した粉末について実施した。使用した操作条件は次のとおりである:初期温度(T開始時)=50℃;最終温度(T終了時)=250℃;温度勾配=10℃/分。
活性物質を含有する錠剤についてのDSCコントロール
ガラス製乳鉢において錠剤を粉砕し、各錠剤から得られた粉末5.0mg(正確に秤量した)を、上述のようにして分析した。このテストにおいても、非処理の錠剤及び熱処理した錠剤の両方についてテストを行った。全てのスキャンを窒素流下で実施した。
加熱の間の質量変動の測定法−熱質量分析(TGA)
活性物質及び賦形剤、錠剤を粉砕することによって得られた粉末混合物又は粉末の質量変動の測定を、加熱処理に関して使用したものと同じ温度及び加熱勾配を使用し、Perkin ElmerのTGA 7を介して、窒素雰囲気下で行った。
【0074】
崩壊テスト
ヨーロッパ薬局方第6版のモノグラフ「錠剤及びカプセルの崩壊」による器具を使用することによって、テストを行った。使用した媒体、脱イオン水1Lを温度37±0.1℃に維持した。一度に錠剤6個についてテストを行った。
【0075】
溶解テスト
ヨーロッパ薬局方第6版のモノグラフ「固体剤形に関する溶解テスト:パドル装置」による装置(Distek)においてテストを行った。ガラス容器内に収容した溶解媒体1Lを温度37±0.1℃に維持し、パドルの回転速度を50rpmに一定化した。溶解した活性物質の測定を、蠕動ポンプ及び試験管キャリヤーシステム"Multicell Trasport for Agilent 8453"にて自動化し、関連するソフトによって制御したDAD UV-visible Agilent Technologies 8453を介して行った。各読み取り後、溶解媒体を元の容器に戻した。サンプリング時間を、最初の20分については5分、及びついで200分までは10分に固定し、その後は、総テスト時間に応じた経過で設定した。450〜600 nmにセットしたバックグラウンド除去法にて、分析波長236 nmにおいて分析を行った。錠剤における活性物質の理論的含量の1%〜100%の溶解を考慮する濃度範囲において較正曲線を作成することによって、測定を行った。
酸性の溶解媒体は、脱イオン水を、0.05 N溶液が得られるような容量で、好適な量の37%HClに添加することによって調製された緩衝液からなる。
pH7.2の溶解媒体は、脱イオン水に好適な量のリン酸水素ナトリウム・2水和物を溶解し、好適な量のリン酸又は水酸化ナトリウムにてpHを調整することによって得られた0.05 Mリン酸塩緩衝液からなる。
各テストについて、溶解プロフィールを、錠剤6個について評価した。
【0076】
IR分光分析
各種の物質及び混合物のIRスペクトルを、KBrディスクにおいてサンプルを調製して、分光計Perkin Elmer 1310を介して得た。
【0077】
付着テスト
粘膜付着測定用に変更を加えた(Russo E, Parodi B, Caviglioli G, Cafaggi S, Bignardiら, J Drug Deliv. Sci. Technol. 14(6): 489-494, 2004)引っ張りテスター(LLOYD LRX)を介して、係る測定を行った。
係るテストを平らな表面において行うことができるように、手動式の油圧プレスにて1分当たり2トンの荷重をかけることによって、質量約200 mg及び直径約13mmを有する円筒状の錠剤を調製した。このようなプレスは、IR分光分析用のKBrのディスクを調製するために販売されている。付着用の基材は、1分当たり5トンの荷重をかけることによってIR用のプレスにて調製された質量約250 mg及び直径13mmを有するムチンタブレット(Sigma)からなる。
付着用の基材をロードセルに固定した;37℃で恒温サポート上に固定したサンプルを、37℃に維持した0.05 Mリン酸塩緩衝液(pH7.2)200μlにて1分間湿らせた。
1Nの先行荷重を速度10mm/分で2分間かけた;付着性の評価のため、伸張3mmを速度0.1mm/秒でセットした。
得られたトレースの解釈
得られたグラフから、下記のパラメーターが得られた:
最大荷重[N];
伸張×加重面積の積算として仕事[N・mm];
最大荷重と錠剤面積(132.73 mm2)との間の比から単位荷重[MPa]
付着テストを、賦形剤のみの錠剤(非処理又は熱処理済)について、及び活性物質を含有する錠剤(非処理又は処理済)について行った。
【0078】
膨潤度の評価
使用した錠剤を、37℃で恒温とし、ブレードを50rpmにて回転させることによって撹拌下に維持した0.05 Mリン酸塩緩衝液(pH7.2)中に浸漬した。定期的に(30分又は60分)、錠剤を媒体から取り出し、金属格子の上で30秒間乾燥し、化学天秤にて秤量した。
膨潤度(S%)を、下記の式に従って算定した:
S%=(mt−m0)/m0*100
ここで、mtは時間tで取り出した錠剤の質量であり、m0は錠剤の初期質量である。
活性物質を含有する又は含有しない錠剤(熱処理したもので又は熱処理していないもの)について、このテストを行った。
【0079】
錠剤の容積の評価
既知容積のワセリンオイルを収容する目盛り付きシリンダーに浸漬した。このような錠剤の容積を、初期液体容積との差を介して評価した。
このテストを、処理した錠剤及び溶解テスト後の同じ錠剤について行った。
【0080】
上述の技術的課題を解決するために、活性物質モデルとしてジルチアゼム塩酸塩(DTZ)を使用し、異なる賦形剤の多くの混合物をテストした。
【0081】
テストした賦形剤のいくつかを下記の表1に示す。
【表1】
【0082】
表に示した賦形剤及びジルチアゼム塩酸塩を使用して、3成分、4成分及び5成分の混合物を調製し、これら混合物から、上述の方法にて錠剤を調製した。ついで、錠剤を上述のテスト及び測定に供した。
【0083】
粉末混合物が特許請求の範囲の請求項1に示す成分の少なくとも1つを含んでなる場合に、制御された放出についての所望の結果が得られることが証明された。
【0084】
さらに、得られる生体付着性のコンパクトマトリックスの特定成分に対する本発明の方法に含まれる熱処理の影響を調査した。
【0085】
図11において、ポリカルボフィルの熱収縮(収縮対加熱時間)についての調査の結果を示す。この図から理解されるように、ポリカルボフィルサンプルを160℃において5分間加熱する場合に最大の平面収縮が生ずる。
【0086】
粉末の平面収縮の結果は、図17及び18のSEM顕微鏡写真から理解される。図17では、ブドウ様形態の群が示されており、一方、図18では、個々のブドウ様形態が消失し、むしろ、連続したマトリックス(バラのように見える)及びより小さい全体容積を有する構成が認められる。これは、粉末が受けた熱処理の結果である。さらに、図18では、各顆粒を結合するブリッジが観察され、一方、図17の顆粒は、明らかに、相互に離れている。
【0087】
図12では、一緒に示された3つのプロフィール(DSC、TGA及びHSM)は、収縮現象(顕微鏡の焦点面における平面収縮として測定される)は、ポリカルボフィルの分解現象には何ら関連せず、おそらく、ポリカルボフィルにおいて50℃以上で生じ、128〜147℃の小さい吸熱曲線でそのピークに達する吸熱に関連するものであろう。
【0088】
図13及び14のSEM顕微鏡写真の対比から、ポリカルボフィル圧縮ユニットを熱処理(150℃、15分間)に供する際、その構造が劇的に変化することが明確に理解される:図14では、小柱状マトリックスが、見た目に極めて明らかであるが、図13では、同様の小柱状マトリックスは全く見られない。
【0089】
小柱状マトリックス及びこのようなマトリックスに含まれる孔は、さらに、図15の拡大した倍率の写真から認められる。圧縮及び加熱の併用による効果を、エチルセルロース及びポリカルボフィルの混合物(3:2)についても調査した。図16の顕微鏡写真から理解されるように、2つのポリマーの相互作用は、新たに形成されたポリカルボフィルマトリックスの孔への2つのエチルセルロース微粒の部分的吸蔵を生ずる。
【0090】
本発明による方法での使用のために採用される組成(ジルチアゼム塩酸塩(DTZ)に加えて、グリクラジド(GLZ)も含有する)のいくつか例を以下に例示するが、これらは、単なる例示であり、非限定的なものである。
【実施例1】
【0091】
上述の操作法に従って、均一な粉末が得られるまで、下記の成分を混合した:
ジルチアゼム塩酸塩 20%
エチルセルロース 35%
MicroceLac 35%
ポリカルボフィル 10%
本明細書において表示する百分率は、他の特定しない限り、圧縮前及び加熱処理前の粉末混合物の総質量の質量%として理解されなければならない。
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述の方法に従い、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)2.57%(標準偏差(sd)=0.09)質量/質量(錠剤)及び平均硬さ307.8 N(31.4 kp;sd=1.2)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量0.88%(sd=0.08)及び硬さ405.9 N(41.4 kp;sd=1.1)を有していた。
熱処理した錠剤(Tと表示する)は、図1a(対応する非熱処理錠剤(NTと表示する)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、曲線は非処理錠剤6個の平均値及び加熱処理錠剤6個の平均値である。
錠剤の熱処理によって生じた変性は、酸性環境におけるジルチアゼム塩酸塩の放出に関して、図1aから明白である。加熱処理は錠剤の成分に対して作用して、薬剤の放出をかなり減速させる(0オーダー速度論に従うことによって生ずる)マトリックスを生成する。
初期フェーズにおいて、薬剤放出20%に達する前では、両錠剤からの放出は、おそらく錠剤Tのマトリックスの水化作用及びゲル化に先行する崩壊作用のために、相互に重複するものと思われる。第1フェーズにおいて、処理済錠剤の放出は、いずれのケースにおいても、若干早いものと思われ、実際に、30分後では、マトリックスは11%以上放出する。
図1bには、錠剤Tのリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応するNTの溶解プロフィールと対比して示されている。ここでも、曲線は、非処理錠剤6個の平均値及び熱処理錠剤6個の平均値である。
リン酸塩緩衝液におけるDTZの放出に対する処理の影響はさらに明白である。係る処理後に生成されたマトリックスは、薬剤をさらにゆっくりと放出する。実際、100〜850分では、担持された薬剤の約47%を放出する。t50(薬剤の50%が放出される時間)は、NTについては4時間であり、一方、Tについては、t50は約14時間である。実際、Tは、4時間後では、担持した薬剤のわずかに33%を放出し、850分後でも、担持したDTZの最大放出には達しない。
緩衝液においても、錠剤Tは、崩壊効果につながる調整の短い初期期間の後、0オーダー速度論に従う放出を有する。
リン酸塩緩衝液のpHにおいて、熱処理を行った錠剤Tにおいて形成されたマトリックスは、水性溶解媒体を吸収し、ゲル化し及びこのようにして、その容積を増大させ、ゼリー状の外層を形成し(図7a、右端の写真参照)、このため、薬剤の放出メカニズムが、0オーダー速度論に従って作動される。さらに、加熱処理を受けた錠剤の膨潤したマトリクスは一体性を保持しており、すなわち、マトリックスは、溶解テストの全期間浸食現象を受けず(膨潤も参照)、及び一旦回収され及び乾燥されると、マトリックスは、実施例2について生ずるものと同様に(図9)、マトリックスが由来する錠剤の形状及びサイズを再度獲得する。
実施例2について生ずるものと同様に(図10)、酸性環境においても、ゼリー状クラウン(多少程度は劣る)が観察される。
NT錠剤との対比により、成分混合物についての加熱処理が、放出速度(%(放出された薬剤)/放出時間)を一定に維持することによって、薬剤を延長された期間にわたって放出する非浸食性のマトリックスを形成することが認められる。加えて、ゲル化した半透明のクラウン(おそらく薬剤放出の制御に関与する及びこのマトリックスから、漸次の膨潤現象を介して形成される)が目視的に観察される。
放出速度の制御は、マトリックスの膨潤現象及びマトリックスのゲル化した層を介する薬剤(マトリックスを膨潤させる水性媒体中に溶解している)の分子拡散の両方によるものである。このような現象は、NT錠剤(溶解テストの終了時には、完全に崩壊されている)においては観察されない。
マトリックスの形成に関連する変性の発現も、錠剤硬さ98.04 N(10kp)の増大から明らかである。
【実施例2】
【0092】
上述の操作法に従って、均一な粉末が得られるまで、下記の成分を混合した:
ジルチアゼム塩酸塩 20%
エチルセルロース 30%
MicroceLac 30%
ポリカルボフィル 20%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)2.57%(標準偏差(sd)=0.08)質量/質量(錠剤)及び平均硬さ247.1 N(25.2 kp)(sd=1.3)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量1.26%(sd=0.05)及び硬さ401.0 N(40.9 kp)(sd=1.1)を有していた。
熱処理した錠剤(Tと表示する)は、図2a(対応する非熱処理錠剤(NT)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、曲線は非処理錠剤6個の平均値及び加熱処理錠剤6個の平均値である。
図2aにおいて、DTZ放出のかなりの延長が観察された(錠剤の加熱処理を行ったものについて達成された)。この実施例の量的に異なる組成(この実施例では、ポリカルボフィル含量は2倍である)に対する加熱処理の影響は明らかに認められる。これは、酸性環境におけるDTZの放出について観察される。より長い放出の延長が観察され、10時間後であっても、その最大には達しないが、NT錠剤では5時間後に最大に達する。同時に、初期放出は、NT錠剤よりも早くなり、40〜210分では、0オーダー速度論に従う放出が認められる。
図2bには、錠剤6個のリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応する非処理錠剤NT6個の溶解プロフィールと対比して示されている。
各曲線は、非処理錠剤6個の平均値であり、垂直のバーは信頼区間95%を示す。
先の実施例のように、処理した錠剤は、短い初期調整期間の後において、0オーダー速度論に従う放出を有する。ポリカルボフィル含量の増大は、先の実施例に対して、さらに放出速度の低下を生じさせ、実際、溶解840分後では、NTは担持した薬剤の80%を放出するが、Tは約41%を放出する。Tは、1400分後、担持した薬剤の約66%を放出する(図3c)。
このケースでも、テストの間に、熱処理した錠剤では、未だ溶解していない薬剤を含有する極めて明確な固体コア(図7aの中央の写真;図7b、図7c参照)を包囲する半透明のゲル化した物質のクラウンの形成と共に、容積の増大が観察された。
ゲル化したクラウン(pH7.2での溶解において形成されるものよりも小さい)は、酸性pHでの溶解においても認められる(図10)。図9(この実施例によって得られた錠剤に関する)は、上述のように、本発明による錠剤が、水中での膨潤後、乾燥時には、元の形状及びサイズを再度獲得することを示している。これは、本発明による方法の用途を、熱不安定性又は固体状態で得ることが困難な活性物質にも拡大することを可能にするものであり、このような活性物質は、本発明の方法によって得られる圧縮したマトリックス(活性物質を含まない)の当該活性物質の水溶液による膨潤、続く、含浸及び膨潤したマトリックスの乾燥によって担持される。
加熱処理した錠剤の硬さの増大は、実施例1のように、錠剤内におけるマトリックスの形成の証拠である。この実施例において、硬さの増大の平均値は、先の実施例のものよりの大きく(135.3 N(13.8 kp))、おそらく、固状コアを包囲し及び溶解テストによって示される放出速度の減少をもたらす水化マトリックスのゲルの異なるコンシステンシーと関連するものであろう。
図2cは、NT錠剤のDSCのトレースと対比する、上述の様式で得られた錠剤TのDSCのトレースである。図の上側には、当該実施例の粉末の物理的混合物のDSCトレース及びMicrceLac(ML)及びジルチアゼム塩酸塩(DTZ)のDSCトレースが示されている。
図2cの下側部分に示されるように、マトリックスを製造する加熱処理によって誘発される変化がDSCトレースによって示されている。
【実施例3】
【0093】
上述の操作法に従って、均一な粉末を得るため、下記の成分を混合した:
ジルチアゼム塩酸塩 40%
エチルセルロース 22.5%
MicroceLac 22.5%
ポリカルボフィル 15%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)3.14%(標準偏差(sd)=0.1)質量/質量(錠剤)及び平均硬さ236.3 N(24.1 kp)(sd=0.8)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量1.26%(sd=0.09)及び硬さ259.8 N(26.5 kp)(sd=1.4)を有していた。
熱処理した錠剤(T)は、図3a(対応する非熱処理錠剤(NT)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、各曲線は錠剤6個の溶解プロフィールの平均値であり、一方、垂直のバーは信頼区間95%の値を示す。
図3bには、熱処理した錠剤(T)のリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応する非処理錠剤(NT)の溶解プロフィールと対比して示されている。ここでも、各曲線は、非処理錠剤6個及び処理した錠剤6個の平均値である。
溶解プロフィールから、担持される薬剤が2倍であっても(先の実施例と比べて)、いかに、本発明のマトリックスが薬剤の放出を制御し続けられるかが理解される。加熱処理の効果は、先のケースのように、HClおけるよりも、リン酸塩緩衝液において、より明らかである。放出の速度論は、リン酸塩緩衝液におけるプロフィールが、初期フェーズの後、おそらく錠剤の完全崩壊に先立つ浸食現象により、直線性から逸脱するものである非処理錠剤(NT)を除いて、溶解プロフィール直線性の進行過程によって証明されるように、0オーダーとなる。
酸性環境では、錠剤Tは、初めの30分では、より容易に薬剤を放出し、ついで、0オーダー速度論が確立され、担持されたDTZの約50%が放出される。錠剤Tについての放出は約400分後に完了し、一方、NT錠剤では、230分内で完了する
リン酸塩緩衝液では、NT錠剤は、約560分後に完全な放出を達成し、一方、錠剤Tは1400分後に完全な放出を達成する。
このケースでも、処理された錠剤は、HCl中での溶解テスト後も、一体性を保持していることが観察され、リン酸塩緩衝液中でのテスト後、錠剤はより多くの程度まで膨潤され、ゼリー状の層によって包囲されており、及び回収、乾燥後では、その元の形状を回復することができる。
この実施例のリン酸塩緩衝液における溶解プロフィールと、実施例2の同様のプロフィールとの間の比較は、極めて興味深い(図3c)。2つのプロフィール線状区域の平行度から、実施例2と同じEC/ポリカルボフィル比率を有する組成物にて製造された実施例3のマトリックスが、薬剤の担持量が2倍であっても、実施例2と同じ速度でDTZの放出を制御できることが推察される。もちろん、崩壊効果は、実施例3において、より大きい。
図3dは、NT錠剤のDSCのトレースと対比する、上述の様式で得られた、錠剤TのDSCのトレースである。マトリックスを製造する加熱処理によって誘発される変化が、図3dから見られるように、DSCトレースによって、再現可能に強調されている。
【0094】
保存安定性の評価
実施例1によって得られた錠剤(図4a及び4b)及び実施例2によって得られた錠剤(図5a及び5b)を、処理効果が経時的に可逆的であるか否かを証明するため、ブリスター包装して47日間保存した後に検討した。錠剤T及びNT6個ずつの平均として、0.05 N HCl(4a)及びリン酸塩緩衝液(4b)における関連する溶解プロフィールを、t=0及びt=47日の時点て比較した。
下記の表2に、ブリスター包装にて保存した実施例1及び2による錠剤から得られたいくつかのパラメーターを示す。
【表2】
図4及び5から見られるように、表2において示されるように、錠剤が特定の量の水を再度獲得するものであったとしても、保存後、2つのタイプの錠剤の放出においては有意の差異はない。ブリスター包装し、室温において保存した実施例2の錠剤について32ヶ月間で行った安定性テストでは、Karl Fisherによって測定した水分含量は2.1〜2.5%であり、このように、実際には、元の値の水分含量の復元が経時的に確認された。これは、加熱処理による放出の延長が、マトリックスによる水の損失によるものではなく、このようなマトリックスを製造する成分の物理的状態の変化(保存期間中の不可逆性によって証明される)によるものであるとの概念につながる。2〜3年の保存時間後に得られた他のテストデータも上記の事実を証明している。
図5cには、調製直後、及びブリスター包装して室温において13ヶ月間保存した後の実施例1の錠剤6個の0.05 N HClにおける平均溶解プロフィールが示されている。2つのプロフィールは、実質的に重なり合うことが観察された。
図5dには、ブリスター包装して32ヶ月間保存した後の実施例2の錠剤6個の0.05 N HClにおける平均溶解プロフィールが示されており、図5eには、同じ保存期間後、実施例2による錠剤6個のリン酸塩緩衝液における平均溶解プロフィールが示されている。
図5fには、600分後の溶解量に対する溶解したDTZフラクションとして、溶解プロフィールが報告されている。このような表示様式は、放出メカニズムを明確に示すためには有用であるが、放出速度及び活性物質の放出の延長に関する情報を与えるものではない。係る図では、実施例2による異なる錠剤バッチ及び同じ組成物から調製されたものではあるが、熱処理していない錠剤(NT)のリン酸塩緩衝液における平均溶解プロフィール(n=6)を比較している。
この比較から、各種のバッチのマトリックスからの薬剤の放出メカニズムは全体に重ね合わせることができ、ブリスター包装して32ヶ月間保存した後であっても変わらないことが明らかである。また、この表示モードは、非処理錠剤と対比して、本発明による圧縮マトリックスからの異なる放出メカニズムを強調するものである。加えて、非処理錠剤に関する放出メカニズムは再現性に乏しい。
表2に硬さテストの結果を、保存前に得られた結果と比較すると、実施例1による錠剤の引っ張り強さの減少及び実施例2による錠剤NTのケースのその増大が認められる。表3に、32ヶ月間保存した実施例2による錠剤の硬さのデータを報告する。
【表3】
【0095】
熱処理についての調査
ついで、錠剤の加熱処理によって得られた活性物質の放出に関する延長が、予め賦形剤組成物又は賦形剤単独を同じ加熱処理に供し、続いて活性物質を添加し、ついで圧縮を行うことによっても得られるか否かを判断するために調査を行った。
圧縮前に、実施例2による全ての成分の物理的混合物を、150℃における15分間の加熱に供することによって、又は1つの賦形剤(例えば、ポリカルボフィル、エチルセルロース又はMicroceLac)を同じ加熱処理に供することによっては、続く他の成分の添加の後、ジルチアゼムの所望の放出プロフィールを有していない錠剤が得られた。ジルチアゼム塩酸塩の150℃における15分間の加熱は、HPLC、熱分析(DSC、TGA及びHSM)、及び分光分析によれば、このような活性物質の物理的又は化学的変性を全く伴うものではない。このように前処理され及び実施例2の錠剤を調製するために使用されたジルチアゼム塩酸塩は、このようなマトリックスからの溶解プロフィールを変性させず、同じ組成の錠剤NTの放出も変性させなかった。
これらの観察から、加熱処理は、相互に均一な混合物における成分が、圧密化又は圧縮プロセスの間に発生される密集又は付着力によって相互に密着された錠剤について行われる場合にのみ、本発明によるマトリックスの構成を生ずるものと推察される。
ついで、実施例2による錠剤の加熱処理の様式を変更した:いくつかについては、90℃又は130℃において15分間で処理し、他では、150℃において5分間で処理した。得られた溶解プロフィールを、実施例2において報告した様式で処理した錠剤及びNT錠剤に係る曲線と対比して、図6に報告している。
図6から、150℃において5又は15分間処理した錠剤は、130℃において処理したものよりも活性物質の放出の顕著な延長を有するものであっても、全ての錠剤が0オーダー放出速度を示すことが認められる。130℃で処理した錠剤は、いずれのケースでも、NT錠剤に対して活性物質の放出の延長及び溶解テスト後の感知できる膨潤度を示した(放出の延長及び膨潤度は、いずれも、150℃で処理した錠剤について認められるものよりも、程度は劣る)。
このデータは、リン酸塩緩衝液における溶解テストの後、130℃において15分間処理した錠剤において、膨潤が最小であり及びゼリー状の層は評価が困難であるとの観察と一致するものであり、及び90℃において、ただし、わずか15分間処理したものでは、NTについては、目に見える変性観察されない。
このデータは、活性物質の放出は、適切な加熱処理温度及び時間のパラメーターの適切な選択を介しても調整されることを表している。
【0096】
付着テスト
実施例2の3つの賦形剤を同じ相互割合で含有するが、DTZを含有しない錠剤を調製した:これらの錠剤及び上述操作法に従って調製した実施例2の錠剤について、張力計を使用して、付着テストを行った。表3bis及び表4は、分析した錠剤の水分含量及び係るテストから得られた結果を示す。
【表3bis】
【表4】
表4に示すデータから理解されるように、DTZを含有しないNT錠剤は、胃腸粘膜をシミュレートする基材(ムチン)への特定の付着(次の処理、すなわち、おそらく加熱処理によって生成されるマトリックスの構造の変化に関連する現象を低減させる)を示す。
NTに関しては、薬剤を含有する錠剤は、賦形剤のみにて処方されたものよりも低い付着性を示す。これは、組成物の相違によると思われる:実際、成分間の割合が一定であったとしても、DTZを含有しない錠剤は、実施例2に対して25%多い絶対量のポリカルボフィルを含有している。処理した錠剤から得られる結果は、130℃での処理の場合、薬剤を含有するNT錠剤に対して、付着性の低減を示し、一方、150℃において15分間の処理後では、付着特性は低減しない。
【0097】
膨潤テスト
実施例1及び2に従って得られた錠剤について、37℃、pH7.2(このpH条件では、現象が特に明瞭である)のリン酸塩緩衝液において膨潤テストを行った。図7は、実施例1及び2による2つのタイプの錠剤の膨潤度(S%)を示す。得られた曲線は、各タイプ当たり錠剤3個の平均膨潤度を示しており、バーは標準偏差を表す。
表5に、次の事項:リン酸塩緩衝液に24時間浸漬した後のS%60、S%max、質量及び容積及び関連する密度を報告している。
【表5】
図7において理解されるように、実施例2に従って処理した錠剤は、表5においても示されているように、実施例1に従って処理した錠剤に対して、より高い膨潤度及び速度を有する:この特性は、混合物中におけるポリカルボフィルの濃度に関係するものと思われる。いずれの錠剤についても、初めの120分間を除いて、経時的に一定である質量増加(質量増加に基づき、Sが算定される)が存在することが注目されなければならない。
実施例1による錠剤は、最大膨潤時間(これを越えると、さらには膨潤しない時間)1440分(24時間)に達するが、1830分(30.5時間)までは、浸漬されたままでも崩壊することなく、一方、実施例2による錠剤については、最大24時間(最後の測定を行った時間)に達して後では、錠剤は24〜30時間の間で完全に崩壊する傾向にあるため、このような現象は目視観察されない。これらの現象は、インビトロ測定条件(高容積の水性媒体及び連続撹拌を含む)下において観察される。
図7には、実施例1及び2による錠剤の、37℃、リン酸塩緩衝液における膨潤プロフィールを報告している。図7aには、2つの実施例による錠剤を、それぞれの最大膨潤度において撮影した写真が、溶解テスト前の本来のサイズの錠剤の写真と対比して示されている。
図7bには、実施例2による錠剤のその最大膨潤度における拡大写真が報告されており、固状の中心コアが明確に認められる。
図7cには、実施例2による膨潤した錠剤を、異なる視野から見た写真が報告されており、しっかりしたマトリックスの形成が明確に認められる。
膨潤を、活性物質を含有しないマトリックスについても測定した。図7dには、DTZを除く実施例2の全ての成分を含有する(ただし、実施例2と同じ割合である)錠剤の膨潤プロフィールを報告している。最長の加熱処理、すなわち、150℃において15分間の加熱処理にて生成したマトリックスによって最大の膨潤が達成される:このようなマトリックスは、5時間後、1744%の最大質量増加値を達成し、さらに2時間、浸食(連続撹拌によって生ずる)に耐えることができる。
5分間の加熱によって形成されたマトリックスは、2時間後に、最大質量増加値(1282%)に達し、さらに60分間、浸食に耐えることができる。
加熱を介して得られるマトリックスにおける薬剤の存在は、膨潤するマトリックスにおける水性媒体の拡散のため、膨潤現象を減速させる。減速は、逆のマスプロセス、すなわち、活性物質の外側方向への拡散に関連する。2つの現象の同時性は、0オーダーモデルによる活性物質放出速度論を説明するものである。
マトリックスの形成に対する加熱処理の影響は、膨潤テストの間に示されるNT錠剤の挙動からも明らかである。実際、これらの錠剤は、初めの1時間では非常にわずかに膨潤し(わずかに80%)、ついで、120分後には、完全に崩壊する。
【実施例4】
【0098】
グリクラジド 20%
エチルセルロース 30%
MicroceLac 30%
ポリカルボフィル 20%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間5分間又は15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
図8において、各処理のタイプ(150℃において15分間及び150℃において5分間)について、pH7.2のリン酸塩緩衝液における錠剤6個の平均溶解プロフィールを、NT錠剤の平均溶解プロフィールと対比して報告している。
マトリックスの形成に対する加熱処理の影響は、グリクラジドの放出速度についてのコントロールから明白であり、コンパクトゲルクラウンの形成に関するユニットの膨潤から目視的に明白である。
加熱処理の効果は、マトリックス成分との相互作用が異なるため、活性物質のタイプによっても変化することは明白である。
【技術分野】
【0001】
本発明は、一般に、化学工業の分野、特に医薬品工業の分野に関する。
【0002】
特に、本発明は、活性物質の放出のために使用される錠剤又はデバイスのような、持続性放出によって特徴付けられるコンパクトマトリックスの製法及びこのようにして得られたコンパクトマトリックスに係る。さらに詳述すれば、本発明は、活性物質の放出のために使用される錠剤又はデバイスを構成できるコンパクトマトリックスの製法であって、特殊な成分の直接圧縮工程及び加熱処理工程を含んでなる製法に係る。
【背景技術】
【0003】
約40年に亘って、医薬工業の研究者は研究を行い、生体への活性物質の放出を修飾及び制御するための新規なシステムを開発してきた。
【0004】
このような修正は、投与回数を減らし、活性物質の放出速度を可及的に制御するために、薬剤の生体への放出を延長するもの(延長された又は持続性の放出)であり、0オーダーの、すなわち、投与剤形内に含まれる薬剤投与量とは無関係の放出速度を得ることを追求するものであった(延長された放出及び標的薬剤送達システム, Remington The Science and Practice of Pharmacy 21版, 47章, p. 939-936)。
【0005】
他の修正は、特殊な刺激(pH、温度、酵素活性、イオン強度)を関数として(Morishita Mら, J. Drug Deliv. Sci. Technol., 16(1): 19-24, 2006)又は特定の期間後又は所定の時間間隔で(遅延化又はパルス化放出;Gazzanigaら, European Journal of Pharmaceutics and Biopharmaceutics, 68(1): 11-18, 2008))、薬剤の放出が生体の特殊なゾーンで生ずるようにすることを目標とするものである。
【0006】
特に、経口投与剤形では、薬剤の持続性放出は、コーティングフィルムとして少量で、又はマトリックスシステムを形成するために、より多い量で使用される好適なポリマーの使用を介して達成される。いずれのケースにおいても、フィルム又はマトリックスの組成は、活性物質の放出に影響を及ぼすことができ、多くのケースにおいて、放出速度は、このようにして事前に設計され、好適なインビトロ溶解テストを介して確認される(Kanjickal DG, Lopina ST, ポリマー送達システムからの薬剤放出のモデル化, Critical Reviews in Therapeutic Drug Carrier Systems, 21(5): 345-386, 2004)。
【0007】
フィルムは、錠剤のコーティングにおいて、又はそのままで又はカプセル化又は錠剤へ変換して投与される顆粒又はペレットのコーティングにおいて直接使用される。
【0008】
マトリックスを形成するために使用されるポリマーは、その異なる溶解又は浸食速度を介して又はマトリックスにおける活性物質の拡散を介して(親水性ポリマーのケースでは、膨潤及びゲル化でき、多少容易に浸食される)、薬剤の放出を制御する(Brazel CA, Peppas NA, リラクシング、膨潤性、親水性、ガラス質のポリマーにおける溶質及び薬剤の輸送のメカニズム, Polymer, 40(12): 3383-3398, 1999)。
【0009】
薬剤送達システム(DDS)と呼ばれる、「エンジニアード」システム又はより高価かつより複雑な工業的方法を介して得られるデバイスがある(Hilt JZ, Peppas NA, International Journal of Pharmaceutics, 306(1-2): 15-23, 2005)。DDSプロトタイプは、半透過性膜、及び薬剤の放出を延長するために、これらの膜内において発生された浸透圧を利用する、いわゆる、「浸透」システムである。溶液又は懸濁液中の薬剤の放出は、錠剤の表面においてレーザー光によって生ずるミクロ孔を介して、0オーダーの放出速度論に従って一定の速度で生ずる(米国特許第4,160,020号;WO 03/075894)。このようなシステムは、含まれる投与量の全部の多量放出(「不当投与」として知られている現象)の欠点をもたらし、輸送される活性物質のタイプに関連して、生体に対して毒作用をもたらす。このような観点から、「ペレット化」剤形は、工業的生産の品質制御工程においても、より多くの保証を提供する。
【0010】
マトリックスシステムは、水性液体の存在下で膨潤性のエチルセルロース又は親水性ポリマー、例えば、ヒドロキシプロピルメチルセルロース(その分子量及び飽和度を関数として、あまり浸食性ではないゲルを形成することもできる)のような非水分散性又は疎水性のシステムを利用する。
【0011】
これらのマトリックスシステムは、一般に、造粒又は「ペレット化」プロセス(いずれも、組成物の改良された均一化を得るため、及び粉末混合物における及びそれらの圧縮の間における偏析現象を回避するため、及び薬剤の放出を効果的に制御するマトリックスの形成を可能にするためのものである)を介して製造される。このようなプロセスは、湿潤条件(湿式造粒、スプレー乾燥)又は乾燥条件(乾燥造粒、又はローラー圧縮及びホットメルト押し出し)で行われる(固状の経口投与剤形, Remington The Science and Practice of Pharmacy, 21版, 45章, p. 889-928)。
【0012】
当然のことながら、造粒工程を含む工業的プロセスは、かなり非経済的であり、従って、工業的には、この目的のために開発され、市販されている賦形剤が存在するため、直接圧縮プロセスを利用する傾向にある(Gohel MC, Jogani PD, Journal of Pharmacy and Pharmaceutical Sciences, 8(1): 76-93, 2005;Goto Kら, Drug Development and Industrial Pharmacy, 25(8) 869-878, 1999;Michoel Aら, Pharmaceutical Development and Technology, 7(1) 79-87, 2002)。
【0013】
市販のCRシステム(Colomboら, 制御された薬剤送達用の膨潤性マトリックス; ゲル層の挙動, メカニズム及び最適パフォーマンス, Pharm. Sci. Techno. Today. 3(6), 2000)の多くは、親水性ポリマーの使用に基づくシステムを介して薬剤の放出を制御するものである(Peppas NAら, 医薬品処方におけるハイドロゲル, European Journal of Pharmaceutics and Biopharmaceutics, 50(1), 27-46, 2000)。
【0014】
上述のように、ポリマーマトリックスを介する活性物質の放出を効果的に制御するシステムは、一般的には、湿式造粒法を介して得られる(ヨーロッパ特許公開1 681 051号;米国特許第5,549,913号)。
【0015】
実際には、直接圧縮によって得られたポリマーマトリックスを介する制御された放出の例は多くはない。例えば、M.E. Pinaらは、主に水性媒体にて膨潤する親水性ポリマー(ヒドロキシプロピルメチルセルロース(HPMC))からなるマトリックス(直接圧縮を介して得られたもの)を介するイブプロフェン(あまり強い水溶性ではない薬剤)についての調節された放出を報告している(Pharmaceutical Development and Technology, 11(2): 213-228, 2006)。Peppas及びSiepmannは、HPMCからなるマトリックスからの薬剤放出のモデル化を充分に検討している(Advanced Drug Delivery Reviews, 48(2-3): 139-157, 2001)。
【0016】
E Crowleyらは、疎水性ポリマー(エチルセルロース)からなるマトリックス(直接圧縮を介して得られたもの)を介するグアイフェネシン(水溶性の薬剤)についての調節された放出を報告している(International Journal of Pharmaceutics, 269(2): 509-522, 2004)。
【0017】
熱処理を含むCR用のマトリックスを得る方法、特に、顆粒を得るため又は「錠剤」として使用される規則的な幾何形状を直接的に得るために利用されるモノリシックマトリックを生成する、ホットメルト押し出し(HME)として知られている熱可塑性ポリマーの押し出し成形の新たな技術は言及に値するものである。
【0018】
この方法は、非晶質ポリマーのガラス転移点(Tg)又は半結晶質ポリマーの融点よりも10〜60℃高い温度に加熱する手段によって、可能なプロセスアジュバントの存在下、ポリマーを溶融することを含む。好適な粘性を獲得した後、このようなメルトを、規則的な断面のスリットを強制的に流動通過させ、このようにして、続く冷却のための前記断面の成形品を生成する(Repka MAら, Drug Development and Industry Pharmacy, Part I, 33(9): 909-926及びPart II, 33(10): 1043-1057, 2007)。
【0019】
従来技術においては、いくつかのケースで、錠剤を直接加熱処理工程に供することによって、錠剤からの活性物質の放出に影響を及ぼすことできることが知見されている。
【0020】
Omelczuckらは、ポリ(dl-乳酸)(PLA)及び微結晶性セルロースを含有する錠剤の加熱処理(40〜80℃において24時間)がテオフィリンの放出を延長させることを報告している(Pharmaceutical Research, 10, 542-548, 1993)。報告された溶解度曲線から、このような放出は、0オーダーの速度論とは異なる複雑な速度論に従うことによって生ずることが推察される。
【0021】
Azarmi S.らは、3:3:4の比で薬剤をEudragit RS PO又はRL PO及び乳糖と共に直接圧縮することを介して調製し、50又は60℃よりも高い温度における2〜24時間の加熱処理に供したインドメタシン錠剤は、引っ張り強さの目に見えるほどの変更を生ずることなく、熱処理に供していない錠剤に対して、延長された放出を示すことを証明している(International Journal of Pharmaceutics, 246 (2002), 171-177)。
【0022】
また、50〜70℃における2〜24時間の加熱処理に供したジクロフェナクナトリウム錠剤(ジクロフェナクナトリウム、Eudragit RS PO又はRL PO及び乳糖(3:4:3)の直接圧縮を介して調製した)について同様の結果が、Azarmi S.らによって得られている(Pharmaceutical Development and Technology, 10: 233-239, 2005)。
【0023】
既に、Billaらは、ジクロフェナクナトリウム/Eudragit NE40D/微結晶性セルロースの錠剤に対する60℃での加熱処理の影響を検討し、錠剤の引っ張り強さの増大と共に、持続性の放出が達成されることを見出している(Drug Development and Industrial Pharmacy, 24(1), 45-50, 1998)。
【0024】
薬剤放出特性に対する錠剤の加熱処理の影響に関する観察は、上述の例、すなわち、Eudragit系の錠剤又はPLAを含有する錠剤に限られているように思われ、長い加熱時間後に得られた結果は、放出時間の延長及び得られた放出速度論の両方について、かなり限られたものである。
【0025】
制御された放出については、非水溶性の架橋ポリマー(しかし、親水性であり、水性媒体中で膨潤性である)の使用も知られている(Brazel CS, Peppas NA 1999, Mechanisms of solute and drug transport in relaxing, swellable, hydrophilic glassy polymers, Polymer 40(12): 3383-3398)。このカテゴリーに属するものとして、ジビニルグリコールにて架橋したポリアクリル酸のポリマーがあり、例えば、錠剤、ディスク又は生体付着特性を備えたフィルムの形のCR剤形の製造において使用されることが知られているポリカルボフィル(CAS RN 9003-01-4)(Handbook of Pharmaceutical Excipients, 5版, Pharmaceutical Press, p. 539-541, 2006)がある。例として、国際公開WO 2005/065685及びWO 01/95888及び米国特許第5,102,666号を参照できる。
【0026】
ポリカルボフィルは、中でも、その生体付着特性のため、剤形内で使用される。Robinsonらは、剤形内で使用されるポリカルボフィル及び他のポリマーの生体付着特性を調査している(Journal of Pharmaceutical Sciences, 89(7): 850-866, 2000)。Repkaらは、ポリカルボフィルを含有し、HMEを介して得られたバッカルフィルムの生体付着特性を研究している(Journal of Controlled Release, 70(3): 341-351, 2001)。
【0027】
ポリカルボフィルが、pHに依存して水を吸収し、その元の容積の1000倍及びその元の直径の10倍まで膨潤する能力は、注目に値するものである。ポリカルボフィルに関連する米国薬局方31のモノグラフの処方によれば、炭酸水素ナトリウム溶液に対する吸収力は、乾燥ポリマー1g当たり62g未満であってはならない。これらの特性のため、ポリカルボフィルは腸の機能不全、慢性の便秘、憩室炎及び過敏性腸症候群の治療のために、医薬品だけでなく、食品サプリメントにおいても使用される。
【0028】
国際公開WO01/95888は、5α-リダクターゼによって代謝される活性成分、例えば、ヒドロキシプロピルメチルセルロースのような水溶性ポリマー、及び非水溶性、水膨潤性の架橋ポリカルボン酸ポリマー、特にポリカルボフィルを含んでなる生体付着性の持続放出性錠剤を開示している。このような錠剤の製法は、加熱工程を含んではいない。国際公開WO 2005/065685は、活性成分及び少なくとも2つのポリマー(1つは酸不溶性ポリマーであり、他は生体付着性ポリマーである)からなるポリマー系(ポリマー系は、例えば、エチルセルロース、ポリカルボフィル及び微結晶性セルロースを含むことができる)を含んでなる生体付着性の放出持続性錠剤を開示している。この明細書によれば、錠剤の製造において、加熱工程は認められない。
【発明の概要】
【発明が解決しようとする課題】
【0029】
本発明の第1の態様では、本発明は、架橋ポリカルボン酸ポリマーを含有し、非浸食性及び生体付着性が付与されており、水の吸収によって膨潤可能であり、活性物質の持続性放出のために使用可能な非浸食性ゲルを形成するコンパクトマトリックスを提供するとの目的を有する。
【課題を解決するための手段】
【0030】
このような目的は、生体付着性のコンパクトマトリックスを製造する方法であって、
−少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋したポリカルボン酸ポリマーを含んでなる粉末の均一な混合物を調製する工程:
−前記粉末混合物を原料として、直接圧縮又は乾燥圧密化によって、圧縮した又は圧密化したユニットを調製する工程;
−このようにして得られた圧縮した又は圧密化したユニットを、80〜250℃の範囲の温度、1〜60分の範囲の時間の加熱処理に供する工程
を含んでなる生体付着性のコンパクトマトリックスの製法によって達成される。
【0031】
他の態様では、本発明は、持続性の又は制御された放出によって特徴付けられる、水中で膨潤できる、上述の生体付着性のコンパクトマトリックスを含んでなる活性物質放出用の圧縮ユニットを提供するとの目的を有する。このような目的は、上述のような、コンパクトマトリックスの製法(当該方法においても、上述の均一な粉末混合物は少なくとも1つの活性物質を含んでなる)によって達成される。
【0032】
用語「圧縮したユニット」について、圧縮したユニットは、医薬品用の一般的な錠剤、特に、活性物質又は生理的状態を回復させる物質を放出できる経口投与のための錠剤だけでなく、粉末圧縮によって得られる他のデバイス、例えば、尿道座剤、錠剤及び膣、口腔、鼻、歯、耳鼻咽喉、目、又は表皮適用のディスクも意味するものである。このような錠剤又はディスクの用途は、ヒト又は動物用の医薬品の分野(ここで、活性物質とは、それぞれ、EU指令2004/27 CE(art. 1)及び2004/28 CE(art. 1)に記載の定義による医薬品を意味する)に限定されるものではなく、EU指令93/42/CEEに記載の定義による医療用具の分野、EU規則(CE)第178/2002の第2項に記載の定義による食品の分野、指令2002/46/CEによって定義された食品サプリメントの分野、EU指令CE第89/398によって定義された食事製品及び幼児用製品の分野、EU指令91/414/CEの定義による植物保護製品の分野、EU規則(CE)第2003/2003の定義及び分類による肥料又は化学肥料の分野、EU指令98/8/CEに記載の定義による消毒剤及び害虫駆除剤及び殺生剤の分野、洗剤の分野のような他の分野にも及びものである。
【0033】
また、診断、治療及び一般的な殺生を目的として、放射性医薬品、放射性核種及び放射性核種によってマーク付けされた分子も、このようなマトリックスによって担持され、放出される。
【0034】
上述の粉末混合物は希釈剤を含むこともできる。希釈剤は、好ましくは、Tabettose(登録商標)及びPharmatose DLC 15(登録商標)と同等の無水の乳糖(CAS RN 63-42-3)又は乳糖1水和物(CAS RN 64044-51-5)(いずれも公知の非晶質又は結晶性の物理的形状であり、スプレー乾燥又は凝集によって得られたもの)及び/又は微結晶性セルロース(例えば、Avicel PH、Ecomocel、Tabulose)からなるものである。例えば、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac(登録商標))又はCellactose(登録商標)のような予め形成された乳糖/微結晶性セルロース混合物、又はLudipress、Starlac、Pharmatose DLC 40、Avicel CE 15、Celocal、Prosolvのような直接圧縮用に同時加工された他の賦形剤も使用できる。
【0035】
上述のアルキルセルロースは、例えば、メチルセルロース(CAS RN 9004-97-5)及びエチルセルロース(CAS RN 90004-57-3)からなる群から選ばれ、上述のヒドロキシアルキルセルロースは、例えば、ヒドロキシプロピルセルロース(CAS RN 9004-65-2及びRN 78214-41-2)、ヒドロキシプロピルメチルセルロース(CAS RN 9004-65-3)、ヒドロキシエチルセルロース(CAS RN 9004-62-0)、ヒドロキシエチルメチルセルロース(CAS RN 9004-42-2)からなる群から選ばれる。
【0036】
アルキル-又はヒドロキシアルキルセルロースの一部の代わりに、次の物質(相互に組み合わせてもよい):クロスポビドン、ポビドン(9003-39-8)、ビニルピロリドン-酢酸ビニルコポリマー(Kollidon(登録商標)VA64)、セルロースアセテートフタレート(CAS 9004-38-0)、ハイプロメロースフタレート(CAS RN 9050-31-1)、ポリビニルアルコール(CAS RN 9002-89-5)、ポリビニルアセテートフタレート(CA RN 34481-48-6)、各種のシクロデキストリン(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、Eudragit E、L、S、RS、RL、PO、NE、RSPMのようなEudragit(Rohm GmbH)の商品名で販売されている各種のタイプのメタクリレートポリマー(Eastman Chemical Company及びBASFによっても製造された各種のもの)、グリセリルトリアセテート、トリエチルシトレート、トリブチルシトレート、アセチルトリエチルシトレート、アセチルトリブチルシトレート、ジブチルセバケート、ジエチルフタレート、ジブチルフタレート、ジオクチルホスフェート、ポリエチレングリコール、ポリエチレンオキシド(CAS RN 25322-68-3)、カルシウムカルボキシメチルセルロース(CAS RN 9050-04-8)、ナトリウムカルボキシメチルセルロース(CAS RN 9004-32-4)、イヌリン(CAS RN 9005-80-5)、キトサン(CAS RN 9012-76-4)及びその誘導体、ガーゴム(CAS RN 9000-30-0)、キサンタンガム(11138-66-2)及びトラガカントゴム(CAS RN 900-65-1)、カルボマー(CAS RN 9003-01-04及び96827-24-6)、カラゲナン(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、アルギン酸(CAS RN 9005-32-7)、ポロキサマー(CAS RN 9003-11-6)、脂肪族ポリエステル(Handbook of Pharmaceutical Excipients, 第5版, Pharmaceutical Pressの関連するモノグラフに記載されている)、セルロースアセテートブチレート、キトサンラクテート、ペクチン、ポリエチレン-コビニルアセテート、ポリエチレン、ポリビニルアセテート-コメタクリル酸、カルナウバワックス、ブチルヒドロキシアニソール、ブチルヒドロキシトルエン、アスコルビルパルミテート、グリセリルパルミトステアレート、水素化大豆油及びヒマシ油(Sterotek(登録商標)K)、グリセリルモノステアレート、d-α-トコフェロール(ビタミンE)、ビタミンEスクシネート、ビタミンE及びTPGS、メチルパラベン、ブチルステアレート、ステアリルアルコール、サッカロースモノパルミテート(Sucroester)、グリセロールエステル及びPEGエステル(Gelucire 44/14)、ポリオキシエチレンアルキルエステル、グリセリルパルミトステアレートPrecirol(登録商標)ATO 5、鉱油、ヒマシ油及び発泡性の混合物又は系を形成することが知られている賦形剤を使用することもできる。水に膨潤可能である上述の非水溶性の架橋ポリカルボン酸ポリマーは、好ましくは、ポリカルボフィル(CAS RN 9003-01-04)からなる。
【0037】
圧縮ユニットを加熱する温度は、好ましくは、90〜160℃であり、加熱時間は、1〜30分の範囲であり、特に1〜20分である。圧縮ユニットを処理温度とするための加熱速度は1〜50℃/分の範囲である。
【0038】
加熱処理に供される粉末の圧縮は、100〜500 MPaの圧力で操作することによって行われる。5kPa〜100 MPaの圧力で操作することによって、続く加熱処理に供される、低い引っ張り強さを有する圧縮体を得ることもできる。圧縮ユニットの形状は、各種の規則的な立体幾何形状であり、質量は、要求及び用途(ヒト又は動物)に応じて100g以下であるが、農業用には、100g以上である。
【0039】
これらの圧縮ユニットは、その組成において、必要に応じて、一般的に圧縮プロセスにおいて使用され、当業者には公知であるアジュバント(滑択剤、潤滑剤、抗接着性剤、崩壊剤及び超崩壊剤、芳香剤、甘味料及び吸着剤)のすべてが添加される。
【0040】
このような錠剤は、胃溶解抵抗性、腸溶性又は環境保全性を活性物質に付与するために、ポリマーフィルムコーティング及び/又はドライコーティングの従来の方法にて被覆される(医薬品剤形: Tablets Volume 1,2,3, H.A. Lieberman, L. Lachman, J.B. Schwartz編, Dekker, 第2版, US 1989)。
【0041】
本発明の対象である錠剤は、インレイ錠剤、多層錠剤及びコア錠剤として知られている錠剤を得るために、コア又は層(活性物質を含有する又は含有しない)として使用される(医薬品剤形: Tablets Volume 1, H.A. Lieberman, L. Lachman, J.B. Schwartz編, Dekker, 第2版, US 1989)。各種の追加の層は、ここに述べるものと同一の質的組成及び/又は異なる活性物質又は追加の活性物質を異なる含量で有することができ、あるいは、これらは、既に述べた又は当分野において使用されている異なる物質でもよい。
【0042】
多層錠剤の場合には、本発明によるマトリックスは、多層錠剤の少なくとも1つの層に相当する。
【0043】
コア錠剤の場合には、ここに記載のマトリックスは、コア又は外層と呼ばれるクラウン層に相当し、活性物質を含むか、又は含まず、又は各層ごとに異なった活性物質を含む。
【0044】
インレイ錠剤の場合には、本発明によるマトリックスは、外層及びインレイの両方に相当し、活性物質又はいくつかの活性物質を含有でき、又は含有しなくてもよい。本発明による方法が、好ましくは、直接圧縮法である場合であっても、特定の状況では、特定の活性物質の粒径によって生ずる欠点を解消するために、錠剤又はコア/インレイの調製又はいくつかの層の調製において、当業者にとって公知の技術をなお使用できる。このような公知の技術は、湿式造粒(湿式造粒、流動床造粒、スプレー乾燥、スプレー凝固)又は乾式造粒(乾式造粒又はローラー圧縮)と呼ばれるか;又は、プロセスに供されるペレットに対して、球形化(球形で、制御されたサイズをもつ顆粒を生成する)として知られている圧縮プロセスを施す(Remington, 21版, 45章, p. 903)。当然のことながら、各種のタイプの造粒は、これらのタイプの方法に必要であり、当業者に知られている最少量のアジュバント又は賦形剤を要求する。酸化に対して敏感な活性物質の場合には、不活性雰囲気、例えば、窒素雰囲気において、加熱を行うことができる。
【0045】
揮発性又は昇華性の活性物質については、又は必要な場合には、加熱処理を、自然の雰囲気又は不活性雰囲気において、このような雰囲気を環境圧力よりも0.5MPaまで高い圧力まで加圧して、実施できる。加熱後の冷却工程は、自然に又は強制的に、例えば、室温の乾燥空気又は不活性ガス(N2、Ar、He)又は室温以下の温度に冷却した乾燥空気又は不活性ガスの通気を介して制御して行われる。
【0046】
加熱後、充填前に、雰囲気条件下でのコンディショニング時間が必要であり、この時間は、選択した組成に応じて、24時間であってもよい。一般に、このような待ち時間は、生産の質には影響を及ぼさないが、いずれのケースにおいても、標準的に5分間待つことが好ましい。
【0047】
本発明による方法における使用のための好適な粉末組成物は、活性物質、MicroceLac、エチルセルロース及びポリカルボフィルを含んでなる。ポリカルボフィルは、一般に、圧縮及び加熱処理前の粉末混合物の総質量の5〜35質量%、好ましくは10〜25質量%を構成する。
【0048】
エチルセルロース及びMicroceLacは、粉末混合物中に、一般に、質量比1:2〜2:1、好ましくは0.8:1〜1.2:1で存在し、圧縮及び加熱処理前の粉末混合物の総質量の45〜95質量%、好ましくは60〜80質量%を構成する。
【0049】
エチルセルロース及びポリカルボフィルは、粉末混合物中に、一般に、質量比1:5〜5:1で存在する。活性物質は、粉末混合物中に、0.001 ppm〜圧縮及び加熱処理前の粉末混合物の総質量の50質量%の量で含有される。さらに、この百分率の範囲内において、活性物質を、溶解プロセス、屈水性錯体又は包接錯体を形成するために好適なアジュバント物質、又は薬剤の消化管吸収又はいずれにしても薬剤の経粘膜吸収のプロセスを促進する物質(エンハンサーとして知られている);又は活性物質を物理的に又は化学的に安定化する物質と混合される。
【0050】
活性物質は、ヒト又は動物において、治療、診断又は予防のために使用可能な、薬理活性を有する天然、合成又は半合成のものであるか、あるいは、生物学的、物理学的又は化学的に活性であり、植物のケア(植物保護製品)用に、肥料として、殺菌剤及び/又はヒト又は環境の害虫駆除剤として使用可能な、天然、合成又は半合成の物質、又はEUによって定義されたような殺生剤のカテゴリーに属する物質である。
【0051】
本発明による方法において活性物質の使用に関して要求される唯一の条件は、方法自体によって予測される加熱条件(温度、時間)下において、活性物質が充分な安定性を有していることである。
【0052】
本発明による圧縮ユニットから、健常な対象又は慢性又は急性の病性状態にある対象を含むヒト又は動物用のダイエット用栄養補助食品として、栄養作用を有する物質が放出される。
【0053】
これらの圧縮ユニットは、化粧用に許可される物質と一緒に好適に処方される場合、EUにおいて正当なものとして許される「化粧品」の定義に従って、化粧品としての用途を有する。
【0054】
本発明は、生体付着特性を備え、活性物質、少なくとも1つのアルキルセルロース又はヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋ポリカルボン酸ポリマーを含んでなる持続放出性錠剤にも関する。好ましくは、このような錠剤は、制御された放出性及びpH4〜8の水溶液において実質的に0オーダーの活性物質放出速度を有する。
【0055】
上述の錠剤は、好ましくは、無水の又は1水和物の乳糖(いずれも、公知の結晶性又は非晶質の物理的形である)、及び/又は微結晶性セルロースからなる希釈剤も含有する。また、当業者にとっては一体形賦形剤(例えば、MicroceLac(登録商標))として知られている微結晶セルロース/乳糖の予め形成した混合物を含有することもできる。
【0056】
上述の成分間の比、粉末又は顆粒の圧縮条件、加熱温度及び加熱処理時間の各条件を変更することによって、活性物質の放出が生ずる速度(一般に、pH4〜8において0オーダー速度論に従う)を制御することができる。
【0057】
活性物質が存在しない(0%)場合、このような圧縮し、熱処理されたマトリックスは、その水性媒体中で膨潤する能力及びその生体付着特性のため、腸の機能不全又は慢性の便秘、憩室炎、過敏性腸症候群のようないくつかの病状における治療作用を実行できる。
【0058】
このような錠剤は、上述の方法によって得られる。
【0059】
これらのマトリックス(水中で膨潤した後、乾燥を介して、元の形状及びサイズを再び獲得できる)の特性を利用することによって、いくつかの活性物質(特に、熱不安定性であり、固状で得ることが困難なもの)を、不動化、すなわち、本発明に従って調製したマトリックス(医薬品含まない)を、好適な濃度の活性物質含有する水溶液に浸漬することによって、このマトリックスに負荷させることができる。一定時間後、膨潤した錠剤を取り出し、元の形状及びサイズを再獲得するため、空気中で又は好適な乾燥法を介して、空気又は不活性ガスの強制換気及び温和な加熱を介して、又はIRランプの照射を介して又は凍結乾燥プロセスを介して乾燥できる。
【図面の簡単な説明】
【0060】
【図1a】本発明の方法によって得られた錠剤(T)(実施例1)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を、同一の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図1b】本発明の方法によって得られた錠剤(T)(実施例1)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図1c】本発明の方法によって得られた、熱処理した(150℃、15分間)錠剤(実施例1)のDSCプロフィール(実線)を、同一の組成の組成を有するが、加熱処理に供していない錠剤のDSCプロフィール(一点差線)と対比して示すグラフである。
【図2a】本発明の方法によって得られた錠剤(T)(実施例2)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィールを、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図2b】本発明の方法によって得られた錠剤(T)(実施例2)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールを、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図2c】下側のグラフは、本発明の方法によって得られた錠剤(実施例2)のDSCトレース(実線)を、同一の組成の組成を有するが、熱処理に供していない錠剤のトレース(二点差線)と対比して示すグラフである。上側のグラフは、ジルチアゼム(破線)及びMicroceLac(点線)のDSCトレースを、実施例2による錠剤と同じ組成を有するが、熱処理していない錠剤のDSCトレース(実線)と対比して示すグラフである。
【図3a】本発明の方法によって得られた錠剤(T)(実施例3)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図3b】本発明の方法によって得られた錠剤(T)(実施例3)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)から放出されるDTZと対比して示すグラフである。バーは信頼区間95%を示す。
【図3c】実施例3による錠剤6個から及び実施例2による錠剤6個から放出されるジルチアゼム(DZT)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)の対比を示すグラフである。
【図3d】実施例3による錠剤(実線)及び同一の組成の組成を有するが、熱処理に供していない錠剤(一点差線)のDSCトレースを、相互に対比して示すグラフである。
【図4a】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の方法によって得られた錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)の対比を示すグラフである。
【図4b】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の方法によって得られた錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)の対比を示すグラフである。
【図5a】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の錠剤(T)(実施例2)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)の対比を示すグラフである。
【図5b】調製直後(t=0)及びブリスター包装して47日間保存した後の、本発明の錠剤(T)(実施例2)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールの対比を示すグラフである。
【図5c】調製直後(t=0)及びブリスター包装して13ヶ月間保存した後の、本発明の錠剤(T)(実施例1)から及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィールの対比を示すグラフである。バーは信頼区間95%を示す。
【図5d】実施例2によるバッチから得られ、熱処理し、ブリスター包装して32ヶ月間保存した錠剤(T)から放出されるジルチアゼム(DTZ)の0.05 N HClにおける平均溶解プロフィール(n=6)を示すグラフである。バーは信頼区間95%を示す。
【図5e】実施例2によるバッチから得られ、熱処理し、ブリスター包装して32ヶ月間保存した錠剤(T)から放出されるジルチアゼム(DTZ)のリン酸塩緩衝液(pH7.2)における平均溶解プロフィール(n=6)を示すグラフである。バーは信頼区間95%を示す。
【図5f】調製直後(t=0)及びブリスター包装して13ヶ月間(t=13ヶ月)及び32ヶ月間(t=32ヶ月)保存した、実施例2による熱処理した錠剤(T)及び同一の組成の組成を有するが、熱処理に供していない錠剤(NT)の異なるバッチの、リン酸塩緩衝液(pH7.2)における、600分の時点での、溶液中に存在する量に関する溶解ジルチアゼム(DTZ)フラクションの平均溶解プロフィール(n=6)の対比を示すグラフである。
【図6】各種の熱処理に供した本発明による錠剤(実施例2)のリン酸塩緩衝液(pH7.2)におけるジルチアゼム(DZT)の溶解プロフィールを示すグラフである。特に、このグラフは、実施例2による錠剤からのリン酸塩緩衝液(pH7.2)におけるジルチアゼム(DZT)の放出に対する各種の熱処理の影響の対比を示す(錠剤の平均溶解プロフィール(n=6):nt=処理していない;150-5=150℃における5分間の処理;90-15=90℃における15分間の処理;150-15=150℃における15分間の処理;130-15=130℃における15分間の処理)。
【図7】本発明による錠剤の膨潤百分率レベル(S%)を示すグラフである。特に、実施例1及び2による熱処理した錠剤の平均膨潤プロフィール(n=3)の対比を示す。バーは標準偏差値を示す。
【図7a】リン酸塩緩衝液における溶解テスト前及び溶解テストの終了時(最大の膨潤度に達した時点)の本発明による熱処理した錠剤を示す3つの写真である。左から右へ順に、溶解テスト前の実施例1又は2の元のサイズの錠剤;リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による錠剤;リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例1による錠剤の写真である。
【図7b】溶解テストの終了時において達した最大膨潤度の本発明による熱処理した錠剤の拡大写真である。特に、図7aの中央に示す写真(リン酸塩緩衝液、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による錠剤)の拡大である。
【図7c】次の2つの写真を示す。上の写真は、最大膨潤度の本発明による3つの錠剤の写真であり、下の写真は、最大膨潤度の本発明による錠剤を斜めから見た写真である。特に、リン酸塩緩衝液(pH=7.2)、37℃における溶解テストの終了時において達した最大膨潤度の実施例2による熱処理した錠剤の写真である。
【図7d】本発明による圧縮マトリックス(活性物質を含まない)の膨潤を示すグラフである。このグラフは、特に、実施例2による成分間の割合をもち、2種類の異なる加熱処理(150℃において5分間又は15分間)に供したマトリックス及び加熱処理に供していないマトリックス(NT)(いずれも、活性物質を含有していない)の平均膨潤プロフィール(n=3)の対比を示す。
【図8】本発明の方法によって得られた錠剤(T)(150℃において、5又は15分間熱処理したもの)(実施例4)から放出されるグリクラジドのリン酸塩緩衝液(pH=7.2)における平均溶解プロフィール(n=6)を、同一の組成の組成を有するが、加熱処理に供していない錠剤(NT)と対比して示すグラフである。バーは信頼区間95%を示す。
【図9】ブリスター包装して38ヶ月間保存した本発明による熱処理(150℃において15分間)した錠剤(実施例2)のサイズと、同じバッチの錠剤6個(熱処理(150℃において15分間)及びブリスター包装での13ヶ月間の保存後、0.05 N HClにおける溶解テストに供し、ついで、室温における空気乾燥に供し、最後に、室温において乾燥した後に、さらに、空気中で25ヶ月間保存した)のサイズとの間の対比を示す写真である。
【図10】0.05 N HClにおける溶解テストの終了時に達した最大膨潤度の実施例2による熱処理(150℃において15分間)錠剤と、元のサイズの錠剤との対比を示す写真である。詳しくは、写真の左には、37℃、0.05 N HClにおける溶解テストの終了時に達した最大膨潤度の実施例2による錠剤が示されている。右には、元のサイズの実施例2の錠剤が示されている。
【図11】各種の等温加熱に供したポリカルボフィル粉末の平面収縮率のホットステージ顕微鏡調査の結果を示すグラフである。
【図12】ポリカルボフィル粉末の、DSCプロフィール(endo up)、TGAプロフィール(温度に対する質量%)及び温度に対する平面収縮(PS)%のホットステージ顕微鏡調査(HSM)プロフィールの対比を示すグラフである。
【図13】ポリカルボフィル成形体(熱処理に供していない)の断面のSEM顕微鏡写真である。成形体は、ポリカルボフィル粉末100 mgに圧力750 kPaを15分間かけることによって得られたものである。
【図14】熱処理したポリカルボフィル成形体の断面のSEM顕微鏡写真である。成形体は、ポリカルボフィル粉末100 mgに圧力750 kPaを15分間かけることによって得られたものである。ついで、成形体を、熱空気オーブン内で、150℃、15分間の加熱に供した。
【図15】図14の同じサンプルを、より高い倍率で見たSEM顕微鏡写真である。
【図16】熱処理したエチルセルロース/ポリカルボフィル(3/2)成形体の断面のSEM顕微鏡写真である。成形体は、エチルセルロース/ポリカルボフィル(3/2)の粉末混合物100 mgに圧力750 kPaを15分間かけることによって得られたものである。ついで、成形体を、熱空気オーブン内で、150℃、15分間の加熱に供した。
【図17】各種の熱処理に供したポリカルボフィルのSEM顕微鏡写真である。
【図18】熱空気オーブンにおいて、150℃で15分間加熱したポリカルボフィル粉末の断面のSEM顕微鏡写真である。
【発明を実施するための形態】
【0061】
本発明は、錠剤の構成に有用な各種の賦形剤の混合物について行った実験研究(活性物質の放出が、エネルギー処理によって影響を受ける)に基づくものである。主な目的は、大気条件下での加熱を介して、処方中の成分が分解することなく、放出時間の延長を示す錠剤を得ることにある。
【0062】
また、活性物質の放出が、0オーダー速度論に従って、非〜易浸食性マトリックスから生じ、これによって、時間に対する放出速度が、処方中の活性物質の残量には非依存性である(これは、制御された放出性の医薬剤形の必須の要件である)処方を得ることを追求するものである。
【0063】
第1のステップでは、薬剤を含まない各種の処方を扱い、得られた崩壊時間の遅延に基づいて、開発する錠剤において使用する賦形剤を選択した。その後、エネルギー処理について使用した温度に対するこれら賦形剤の抵抗性を評価するためにテストを行った。
【0064】
ついで、錠剤からの放出の特徴評価に有用と思われるモデル薬剤を選択した。迅速な又は標準的な放出形の処方及び変性した放出形の処方の両方において、市販のジルチアゼム塩酸塩を選択した。
【0065】
予備テストに基づき、及びこのモデル薬剤を使用して、各種組成の錠剤を調製した。
【0066】
これらの錠剤からの活性物質の放出に対する加熱処理の影響を、酸性媒体中(胃環境をシミュレートする)及びリン酸塩緩衝液中(一部、腸管環境をシミュレートする)の両方で実施した溶解テストによって評価した。錠剤が受けた変性を、熱分析技術、顕微鏡分析を介して及びこのタイプの医薬剤形について一般的な各種の物理的テストを介して調査した。
【0067】
活性物質の各種賦形剤との混合物の調製
それぞれ任意に篩通した成分の混合を、テフロン(登録商標)ストッパーを具備するねじ付きキャップをもつ琥珀色の円筒状ガラス容器において又は好適なステンレス鋼容器において行い、Turbula(登録商標)ミキサーにおいて、成分の混合物が完全に均質になるまで、一般に下記のようにして実施した:
1.少数の成分及び同一質量の活性物質からなるコアを形成する;
2.残りの活性物質の全量を添加する;
3.多数の賦形剤を、容器内に収容された粉末の量と同じ量で添加する;
4.多数の賦形剤の全量を添加する。
全ての混合物について、粉末の各一定量を、量にに応じた時間で混合し、一般に、最大量に関して最長30〜40分間以下である。
【0068】
錠剤の調製
150〜170 mgの範囲の質量をもつ錠剤を、凹形のモノパンチを具備する交互打錠機によって調製した。
【0069】
錠剤及び粉末の処理
処理に供する錠剤を金属支持体上に置き、各々を、小さい金網バスケットによって保護した。処理をガスクロマトグラフ(HP 5890シリーズII)のオーブン内で行い、処理は、所定の処理温度への加熱及び係る温度における所定時間の維持からなる。使用した温度プログラムは次のとおりである:25℃において0.1分、温度勾配30℃/分で最終温度に到達;及び係る温度における所定時間の維持、ついて、室温への錠剤の強制又は自然冷却。
各錠剤の処理後、質量損失率を、下記の式に従って評価した:
Δm%=(m0−m)/m0*100
ここで、m0は錠剤の初期質量であり、及びmは加熱処理後の錠剤の質量である。
比較用の粉末の処理を、ガスクロマトグラフにおいて、パイレックス(登録商標)ガラス管において行った。
【0070】
錠剤の保存
非処理及び処理済の錠剤を、PVCブリスター包装して、室温において、各種の期間:47日間、13ヵ月間及び32ヶ月間保存した。保存期間の終了時に、質量増加率(Δw%)を、式:
Δw%=(mc−m0)/m0*100
ここで、mcは保存期間後の錠剤の質量であり、及びm0は錠剤の初期質量である。処理済の錠剤について、m0は熱処理後の質量を表す。
【0071】
錠剤の硬さの測定
処理済及び非処理の錠剤について、好適な器具によってテストを行い、錠剤の実際の半径方向の崩壊によるもののみを有効な結果とみなし、変形現象又はキャッピングによるものについては、有効な結果とみなさなかった。得られた結果は、半径方向の引っ張り強さを示し、kp(キロポンド=キログラムf=9.80665ニュートン)で示す。
【0072】
錠剤中に存在する水分量の測定
好適な自動装置(Metter-Roledo DL38)によるKarl Fischer(KF)滴定によって測定を行った。滴定剤として、酒石酸ナトリウム・2水和物(Riedl-deHaen)にて標準化したHydranal Composite5(Riedel-deHaen)を使用した。得られた結果を、ガラス製乳鉢において粉砕した錠剤に由来する粉末サンプル55.0 mg(正確に秤量した)に含有される水分率(m/m)として表す。この場合にも、処理済及び非処理の錠剤についてテストを行った。
【0073】
示差走査熱量測定法(DSC)
錠剤の処方において使用した活性物質及び賦形剤についての物理的安定性の測定
正確に秤量した各賦形剤/物質5.0mgをアルミニウム製の皿に入れ、好適なプレスにて閉じ、窒素流下、DSC(Perkin Elmer 7)によって分析した。分析を、熱処理した粉末について実施した。使用した操作条件は次のとおりである:初期温度(T開始時)=50℃;最終温度(T終了時)=250℃;温度勾配=10℃/分。
活性物質を含有する錠剤についてのDSCコントロール
ガラス製乳鉢において錠剤を粉砕し、各錠剤から得られた粉末5.0mg(正確に秤量した)を、上述のようにして分析した。このテストにおいても、非処理の錠剤及び熱処理した錠剤の両方についてテストを行った。全てのスキャンを窒素流下で実施した。
加熱の間の質量変動の測定法−熱質量分析(TGA)
活性物質及び賦形剤、錠剤を粉砕することによって得られた粉末混合物又は粉末の質量変動の測定を、加熱処理に関して使用したものと同じ温度及び加熱勾配を使用し、Perkin ElmerのTGA 7を介して、窒素雰囲気下で行った。
【0074】
崩壊テスト
ヨーロッパ薬局方第6版のモノグラフ「錠剤及びカプセルの崩壊」による器具を使用することによって、テストを行った。使用した媒体、脱イオン水1Lを温度37±0.1℃に維持した。一度に錠剤6個についてテストを行った。
【0075】
溶解テスト
ヨーロッパ薬局方第6版のモノグラフ「固体剤形に関する溶解テスト:パドル装置」による装置(Distek)においてテストを行った。ガラス容器内に収容した溶解媒体1Lを温度37±0.1℃に維持し、パドルの回転速度を50rpmに一定化した。溶解した活性物質の測定を、蠕動ポンプ及び試験管キャリヤーシステム"Multicell Trasport for Agilent 8453"にて自動化し、関連するソフトによって制御したDAD UV-visible Agilent Technologies 8453を介して行った。各読み取り後、溶解媒体を元の容器に戻した。サンプリング時間を、最初の20分については5分、及びついで200分までは10分に固定し、その後は、総テスト時間に応じた経過で設定した。450〜600 nmにセットしたバックグラウンド除去法にて、分析波長236 nmにおいて分析を行った。錠剤における活性物質の理論的含量の1%〜100%の溶解を考慮する濃度範囲において較正曲線を作成することによって、測定を行った。
酸性の溶解媒体は、脱イオン水を、0.05 N溶液が得られるような容量で、好適な量の37%HClに添加することによって調製された緩衝液からなる。
pH7.2の溶解媒体は、脱イオン水に好適な量のリン酸水素ナトリウム・2水和物を溶解し、好適な量のリン酸又は水酸化ナトリウムにてpHを調整することによって得られた0.05 Mリン酸塩緩衝液からなる。
各テストについて、溶解プロフィールを、錠剤6個について評価した。
【0076】
IR分光分析
各種の物質及び混合物のIRスペクトルを、KBrディスクにおいてサンプルを調製して、分光計Perkin Elmer 1310を介して得た。
【0077】
付着テスト
粘膜付着測定用に変更を加えた(Russo E, Parodi B, Caviglioli G, Cafaggi S, Bignardiら, J Drug Deliv. Sci. Technol. 14(6): 489-494, 2004)引っ張りテスター(LLOYD LRX)を介して、係る測定を行った。
係るテストを平らな表面において行うことができるように、手動式の油圧プレスにて1分当たり2トンの荷重をかけることによって、質量約200 mg及び直径約13mmを有する円筒状の錠剤を調製した。このようなプレスは、IR分光分析用のKBrのディスクを調製するために販売されている。付着用の基材は、1分当たり5トンの荷重をかけることによってIR用のプレスにて調製された質量約250 mg及び直径13mmを有するムチンタブレット(Sigma)からなる。
付着用の基材をロードセルに固定した;37℃で恒温サポート上に固定したサンプルを、37℃に維持した0.05 Mリン酸塩緩衝液(pH7.2)200μlにて1分間湿らせた。
1Nの先行荷重を速度10mm/分で2分間かけた;付着性の評価のため、伸張3mmを速度0.1mm/秒でセットした。
得られたトレースの解釈
得られたグラフから、下記のパラメーターが得られた:
最大荷重[N];
伸張×加重面積の積算として仕事[N・mm];
最大荷重と錠剤面積(132.73 mm2)との間の比から単位荷重[MPa]
付着テストを、賦形剤のみの錠剤(非処理又は熱処理済)について、及び活性物質を含有する錠剤(非処理又は処理済)について行った。
【0078】
膨潤度の評価
使用した錠剤を、37℃で恒温とし、ブレードを50rpmにて回転させることによって撹拌下に維持した0.05 Mリン酸塩緩衝液(pH7.2)中に浸漬した。定期的に(30分又は60分)、錠剤を媒体から取り出し、金属格子の上で30秒間乾燥し、化学天秤にて秤量した。
膨潤度(S%)を、下記の式に従って算定した:
S%=(mt−m0)/m0*100
ここで、mtは時間tで取り出した錠剤の質量であり、m0は錠剤の初期質量である。
活性物質を含有する又は含有しない錠剤(熱処理したもので又は熱処理していないもの)について、このテストを行った。
【0079】
錠剤の容積の評価
既知容積のワセリンオイルを収容する目盛り付きシリンダーに浸漬した。このような錠剤の容積を、初期液体容積との差を介して評価した。
このテストを、処理した錠剤及び溶解テスト後の同じ錠剤について行った。
【0080】
上述の技術的課題を解決するために、活性物質モデルとしてジルチアゼム塩酸塩(DTZ)を使用し、異なる賦形剤の多くの混合物をテストした。
【0081】
テストした賦形剤のいくつかを下記の表1に示す。
【表1】
【0082】
表に示した賦形剤及びジルチアゼム塩酸塩を使用して、3成分、4成分及び5成分の混合物を調製し、これら混合物から、上述の方法にて錠剤を調製した。ついで、錠剤を上述のテスト及び測定に供した。
【0083】
粉末混合物が特許請求の範囲の請求項1に示す成分の少なくとも1つを含んでなる場合に、制御された放出についての所望の結果が得られることが証明された。
【0084】
さらに、得られる生体付着性のコンパクトマトリックスの特定成分に対する本発明の方法に含まれる熱処理の影響を調査した。
【0085】
図11において、ポリカルボフィルの熱収縮(収縮対加熱時間)についての調査の結果を示す。この図から理解されるように、ポリカルボフィルサンプルを160℃において5分間加熱する場合に最大の平面収縮が生ずる。
【0086】
粉末の平面収縮の結果は、図17及び18のSEM顕微鏡写真から理解される。図17では、ブドウ様形態の群が示されており、一方、図18では、個々のブドウ様形態が消失し、むしろ、連続したマトリックス(バラのように見える)及びより小さい全体容積を有する構成が認められる。これは、粉末が受けた熱処理の結果である。さらに、図18では、各顆粒を結合するブリッジが観察され、一方、図17の顆粒は、明らかに、相互に離れている。
【0087】
図12では、一緒に示された3つのプロフィール(DSC、TGA及びHSM)は、収縮現象(顕微鏡の焦点面における平面収縮として測定される)は、ポリカルボフィルの分解現象には何ら関連せず、おそらく、ポリカルボフィルにおいて50℃以上で生じ、128〜147℃の小さい吸熱曲線でそのピークに達する吸熱に関連するものであろう。
【0088】
図13及び14のSEM顕微鏡写真の対比から、ポリカルボフィル圧縮ユニットを熱処理(150℃、15分間)に供する際、その構造が劇的に変化することが明確に理解される:図14では、小柱状マトリックスが、見た目に極めて明らかであるが、図13では、同様の小柱状マトリックスは全く見られない。
【0089】
小柱状マトリックス及びこのようなマトリックスに含まれる孔は、さらに、図15の拡大した倍率の写真から認められる。圧縮及び加熱の併用による効果を、エチルセルロース及びポリカルボフィルの混合物(3:2)についても調査した。図16の顕微鏡写真から理解されるように、2つのポリマーの相互作用は、新たに形成されたポリカルボフィルマトリックスの孔への2つのエチルセルロース微粒の部分的吸蔵を生ずる。
【0090】
本発明による方法での使用のために採用される組成(ジルチアゼム塩酸塩(DTZ)に加えて、グリクラジド(GLZ)も含有する)のいくつか例を以下に例示するが、これらは、単なる例示であり、非限定的なものである。
【実施例1】
【0091】
上述の操作法に従って、均一な粉末が得られるまで、下記の成分を混合した:
ジルチアゼム塩酸塩 20%
エチルセルロース 35%
MicroceLac 35%
ポリカルボフィル 10%
本明細書において表示する百分率は、他の特定しない限り、圧縮前及び加熱処理前の粉末混合物の総質量の質量%として理解されなければならない。
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述の方法に従い、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)2.57%(標準偏差(sd)=0.09)質量/質量(錠剤)及び平均硬さ307.8 N(31.4 kp;sd=1.2)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量0.88%(sd=0.08)及び硬さ405.9 N(41.4 kp;sd=1.1)を有していた。
熱処理した錠剤(Tと表示する)は、図1a(対応する非熱処理錠剤(NTと表示する)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、曲線は非処理錠剤6個の平均値及び加熱処理錠剤6個の平均値である。
錠剤の熱処理によって生じた変性は、酸性環境におけるジルチアゼム塩酸塩の放出に関して、図1aから明白である。加熱処理は錠剤の成分に対して作用して、薬剤の放出をかなり減速させる(0オーダー速度論に従うことによって生ずる)マトリックスを生成する。
初期フェーズにおいて、薬剤放出20%に達する前では、両錠剤からの放出は、おそらく錠剤Tのマトリックスの水化作用及びゲル化に先行する崩壊作用のために、相互に重複するものと思われる。第1フェーズにおいて、処理済錠剤の放出は、いずれのケースにおいても、若干早いものと思われ、実際に、30分後では、マトリックスは11%以上放出する。
図1bには、錠剤Tのリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応するNTの溶解プロフィールと対比して示されている。ここでも、曲線は、非処理錠剤6個の平均値及び熱処理錠剤6個の平均値である。
リン酸塩緩衝液におけるDTZの放出に対する処理の影響はさらに明白である。係る処理後に生成されたマトリックスは、薬剤をさらにゆっくりと放出する。実際、100〜850分では、担持された薬剤の約47%を放出する。t50(薬剤の50%が放出される時間)は、NTについては4時間であり、一方、Tについては、t50は約14時間である。実際、Tは、4時間後では、担持した薬剤のわずかに33%を放出し、850分後でも、担持したDTZの最大放出には達しない。
緩衝液においても、錠剤Tは、崩壊効果につながる調整の短い初期期間の後、0オーダー速度論に従う放出を有する。
リン酸塩緩衝液のpHにおいて、熱処理を行った錠剤Tにおいて形成されたマトリックスは、水性溶解媒体を吸収し、ゲル化し及びこのようにして、その容積を増大させ、ゼリー状の外層を形成し(図7a、右端の写真参照)、このため、薬剤の放出メカニズムが、0オーダー速度論に従って作動される。さらに、加熱処理を受けた錠剤の膨潤したマトリクスは一体性を保持しており、すなわち、マトリックスは、溶解テストの全期間浸食現象を受けず(膨潤も参照)、及び一旦回収され及び乾燥されると、マトリックスは、実施例2について生ずるものと同様に(図9)、マトリックスが由来する錠剤の形状及びサイズを再度獲得する。
実施例2について生ずるものと同様に(図10)、酸性環境においても、ゼリー状クラウン(多少程度は劣る)が観察される。
NT錠剤との対比により、成分混合物についての加熱処理が、放出速度(%(放出された薬剤)/放出時間)を一定に維持することによって、薬剤を延長された期間にわたって放出する非浸食性のマトリックスを形成することが認められる。加えて、ゲル化した半透明のクラウン(おそらく薬剤放出の制御に関与する及びこのマトリックスから、漸次の膨潤現象を介して形成される)が目視的に観察される。
放出速度の制御は、マトリックスの膨潤現象及びマトリックスのゲル化した層を介する薬剤(マトリックスを膨潤させる水性媒体中に溶解している)の分子拡散の両方によるものである。このような現象は、NT錠剤(溶解テストの終了時には、完全に崩壊されている)においては観察されない。
マトリックスの形成に関連する変性の発現も、錠剤硬さ98.04 N(10kp)の増大から明らかである。
【実施例2】
【0092】
上述の操作法に従って、均一な粉末が得られるまで、下記の成分を混合した:
ジルチアゼム塩酸塩 20%
エチルセルロース 30%
MicroceLac 30%
ポリカルボフィル 20%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)2.57%(標準偏差(sd)=0.08)質量/質量(錠剤)及び平均硬さ247.1 N(25.2 kp)(sd=1.3)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量1.26%(sd=0.05)及び硬さ401.0 N(40.9 kp)(sd=1.1)を有していた。
熱処理した錠剤(Tと表示する)は、図2a(対応する非熱処理錠剤(NT)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、曲線は非処理錠剤6個の平均値及び加熱処理錠剤6個の平均値である。
図2aにおいて、DTZ放出のかなりの延長が観察された(錠剤の加熱処理を行ったものについて達成された)。この実施例の量的に異なる組成(この実施例では、ポリカルボフィル含量は2倍である)に対する加熱処理の影響は明らかに認められる。これは、酸性環境におけるDTZの放出について観察される。より長い放出の延長が観察され、10時間後であっても、その最大には達しないが、NT錠剤では5時間後に最大に達する。同時に、初期放出は、NT錠剤よりも早くなり、40〜210分では、0オーダー速度論に従う放出が認められる。
図2bには、錠剤6個のリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応する非処理錠剤NT6個の溶解プロフィールと対比して示されている。
各曲線は、非処理錠剤6個の平均値であり、垂直のバーは信頼区間95%を示す。
先の実施例のように、処理した錠剤は、短い初期調整期間の後において、0オーダー速度論に従う放出を有する。ポリカルボフィル含量の増大は、先の実施例に対して、さらに放出速度の低下を生じさせ、実際、溶解840分後では、NTは担持した薬剤の80%を放出するが、Tは約41%を放出する。Tは、1400分後、担持した薬剤の約66%を放出する(図3c)。
このケースでも、テストの間に、熱処理した錠剤では、未だ溶解していない薬剤を含有する極めて明確な固体コア(図7aの中央の写真;図7b、図7c参照)を包囲する半透明のゲル化した物質のクラウンの形成と共に、容積の増大が観察された。
ゲル化したクラウン(pH7.2での溶解において形成されるものよりも小さい)は、酸性pHでの溶解においても認められる(図10)。図9(この実施例によって得られた錠剤に関する)は、上述のように、本発明による錠剤が、水中での膨潤後、乾燥時には、元の形状及びサイズを再度獲得することを示している。これは、本発明による方法の用途を、熱不安定性又は固体状態で得ることが困難な活性物質にも拡大することを可能にするものであり、このような活性物質は、本発明の方法によって得られる圧縮したマトリックス(活性物質を含まない)の当該活性物質の水溶液による膨潤、続く、含浸及び膨潤したマトリックスの乾燥によって担持される。
加熱処理した錠剤の硬さの増大は、実施例1のように、錠剤内におけるマトリックスの形成の証拠である。この実施例において、硬さの増大の平均値は、先の実施例のものよりの大きく(135.3 N(13.8 kp))、おそらく、固状コアを包囲し及び溶解テストによって示される放出速度の減少をもたらす水化マトリックスのゲルの異なるコンシステンシーと関連するものであろう。
図2cは、NT錠剤のDSCのトレースと対比する、上述の様式で得られた錠剤TのDSCのトレースである。図の上側には、当該実施例の粉末の物理的混合物のDSCトレース及びMicrceLac(ML)及びジルチアゼム塩酸塩(DTZ)のDSCトレースが示されている。
図2cの下側部分に示されるように、マトリックスを製造する加熱処理によって誘発される変化がDSCトレースによって示されている。
【実施例3】
【0093】
上述の操作法に従って、均一な粉末を得るため、下記の成分を混合した:
ジルチアゼム塩酸塩 40%
エチルセルロース 22.5%
MicroceLac 22.5%
ポリカルボフィル 15%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
非熱処理錠剤(サンプル数(n)=20)は、平均水分含量(KFによる)3.14%(標準偏差(sd)=0.1)質量/質量(錠剤)及び平均硬さ236.3 N(24.1 kp)(sd=0.8)を有しており、一方、熱処理した錠剤(サンプル数(n)=20)は、水分含量1.26%(sd=0.09)及び硬さ259.8 N(26.5 kp)(sd=1.4)を有していた。
熱処理した錠剤(T)は、図3a(対応する非熱処理錠剤(NT)の溶解プロフィールの示している)の0.05 N HClにおける溶解プロフィールを示した。なお、各曲線は錠剤6個の溶解プロフィールの平均値であり、一方、垂直のバーは信頼区間95%の値を示す。
図3bには、熱処理した錠剤(T)のリン酸塩緩衝液(pH=7.2)における溶解プロフィールが、対応する非処理錠剤(NT)の溶解プロフィールと対比して示されている。ここでも、各曲線は、非処理錠剤6個及び処理した錠剤6個の平均値である。
溶解プロフィールから、担持される薬剤が2倍であっても(先の実施例と比べて)、いかに、本発明のマトリックスが薬剤の放出を制御し続けられるかが理解される。加熱処理の効果は、先のケースのように、HClおけるよりも、リン酸塩緩衝液において、より明らかである。放出の速度論は、リン酸塩緩衝液におけるプロフィールが、初期フェーズの後、おそらく錠剤の完全崩壊に先立つ浸食現象により、直線性から逸脱するものである非処理錠剤(NT)を除いて、溶解プロフィール直線性の進行過程によって証明されるように、0オーダーとなる。
酸性環境では、錠剤Tは、初めの30分では、より容易に薬剤を放出し、ついで、0オーダー速度論が確立され、担持されたDTZの約50%が放出される。錠剤Tについての放出は約400分後に完了し、一方、NT錠剤では、230分内で完了する
リン酸塩緩衝液では、NT錠剤は、約560分後に完全な放出を達成し、一方、錠剤Tは1400分後に完全な放出を達成する。
このケースでも、処理された錠剤は、HCl中での溶解テスト後も、一体性を保持していることが観察され、リン酸塩緩衝液中でのテスト後、錠剤はより多くの程度まで膨潤され、ゼリー状の層によって包囲されており、及び回収、乾燥後では、その元の形状を回復することができる。
この実施例のリン酸塩緩衝液における溶解プロフィールと、実施例2の同様のプロフィールとの間の比較は、極めて興味深い(図3c)。2つのプロフィール線状区域の平行度から、実施例2と同じEC/ポリカルボフィル比率を有する組成物にて製造された実施例3のマトリックスが、薬剤の担持量が2倍であっても、実施例2と同じ速度でDTZの放出を制御できることが推察される。もちろん、崩壊効果は、実施例3において、より大きい。
図3dは、NT錠剤のDSCのトレースと対比する、上述の様式で得られた、錠剤TのDSCのトレースである。マトリックスを製造する加熱処理によって誘発される変化が、図3dから見られるように、DSCトレースによって、再現可能に強調されている。
【0094】
保存安定性の評価
実施例1によって得られた錠剤(図4a及び4b)及び実施例2によって得られた錠剤(図5a及び5b)を、処理効果が経時的に可逆的であるか否かを証明するため、ブリスター包装して47日間保存した後に検討した。錠剤T及びNT6個ずつの平均として、0.05 N HCl(4a)及びリン酸塩緩衝液(4b)における関連する溶解プロフィールを、t=0及びt=47日の時点て比較した。
下記の表2に、ブリスター包装にて保存した実施例1及び2による錠剤から得られたいくつかのパラメーターを示す。
【表2】
図4及び5から見られるように、表2において示されるように、錠剤が特定の量の水を再度獲得するものであったとしても、保存後、2つのタイプの錠剤の放出においては有意の差異はない。ブリスター包装し、室温において保存した実施例2の錠剤について32ヶ月間で行った安定性テストでは、Karl Fisherによって測定した水分含量は2.1〜2.5%であり、このように、実際には、元の値の水分含量の復元が経時的に確認された。これは、加熱処理による放出の延長が、マトリックスによる水の損失によるものではなく、このようなマトリックスを製造する成分の物理的状態の変化(保存期間中の不可逆性によって証明される)によるものであるとの概念につながる。2〜3年の保存時間後に得られた他のテストデータも上記の事実を証明している。
図5cには、調製直後、及びブリスター包装して室温において13ヶ月間保存した後の実施例1の錠剤6個の0.05 N HClにおける平均溶解プロフィールが示されている。2つのプロフィールは、実質的に重なり合うことが観察された。
図5dには、ブリスター包装して32ヶ月間保存した後の実施例2の錠剤6個の0.05 N HClにおける平均溶解プロフィールが示されており、図5eには、同じ保存期間後、実施例2による錠剤6個のリン酸塩緩衝液における平均溶解プロフィールが示されている。
図5fには、600分後の溶解量に対する溶解したDTZフラクションとして、溶解プロフィールが報告されている。このような表示様式は、放出メカニズムを明確に示すためには有用であるが、放出速度及び活性物質の放出の延長に関する情報を与えるものではない。係る図では、実施例2による異なる錠剤バッチ及び同じ組成物から調製されたものではあるが、熱処理していない錠剤(NT)のリン酸塩緩衝液における平均溶解プロフィール(n=6)を比較している。
この比較から、各種のバッチのマトリックスからの薬剤の放出メカニズムは全体に重ね合わせることができ、ブリスター包装して32ヶ月間保存した後であっても変わらないことが明らかである。また、この表示モードは、非処理錠剤と対比して、本発明による圧縮マトリックスからの異なる放出メカニズムを強調するものである。加えて、非処理錠剤に関する放出メカニズムは再現性に乏しい。
表2に硬さテストの結果を、保存前に得られた結果と比較すると、実施例1による錠剤の引っ張り強さの減少及び実施例2による錠剤NTのケースのその増大が認められる。表3に、32ヶ月間保存した実施例2による錠剤の硬さのデータを報告する。
【表3】
【0095】
熱処理についての調査
ついで、錠剤の加熱処理によって得られた活性物質の放出に関する延長が、予め賦形剤組成物又は賦形剤単独を同じ加熱処理に供し、続いて活性物質を添加し、ついで圧縮を行うことによっても得られるか否かを判断するために調査を行った。
圧縮前に、実施例2による全ての成分の物理的混合物を、150℃における15分間の加熱に供することによって、又は1つの賦形剤(例えば、ポリカルボフィル、エチルセルロース又はMicroceLac)を同じ加熱処理に供することによっては、続く他の成分の添加の後、ジルチアゼムの所望の放出プロフィールを有していない錠剤が得られた。ジルチアゼム塩酸塩の150℃における15分間の加熱は、HPLC、熱分析(DSC、TGA及びHSM)、及び分光分析によれば、このような活性物質の物理的又は化学的変性を全く伴うものではない。このように前処理され及び実施例2の錠剤を調製するために使用されたジルチアゼム塩酸塩は、このようなマトリックスからの溶解プロフィールを変性させず、同じ組成の錠剤NTの放出も変性させなかった。
これらの観察から、加熱処理は、相互に均一な混合物における成分が、圧密化又は圧縮プロセスの間に発生される密集又は付着力によって相互に密着された錠剤について行われる場合にのみ、本発明によるマトリックスの構成を生ずるものと推察される。
ついで、実施例2による錠剤の加熱処理の様式を変更した:いくつかについては、90℃又は130℃において15分間で処理し、他では、150℃において5分間で処理した。得られた溶解プロフィールを、実施例2において報告した様式で処理した錠剤及びNT錠剤に係る曲線と対比して、図6に報告している。
図6から、150℃において5又は15分間処理した錠剤は、130℃において処理したものよりも活性物質の放出の顕著な延長を有するものであっても、全ての錠剤が0オーダー放出速度を示すことが認められる。130℃で処理した錠剤は、いずれのケースでも、NT錠剤に対して活性物質の放出の延長及び溶解テスト後の感知できる膨潤度を示した(放出の延長及び膨潤度は、いずれも、150℃で処理した錠剤について認められるものよりも、程度は劣る)。
このデータは、リン酸塩緩衝液における溶解テストの後、130℃において15分間処理した錠剤において、膨潤が最小であり及びゼリー状の層は評価が困難であるとの観察と一致するものであり、及び90℃において、ただし、わずか15分間処理したものでは、NTについては、目に見える変性観察されない。
このデータは、活性物質の放出は、適切な加熱処理温度及び時間のパラメーターの適切な選択を介しても調整されることを表している。
【0096】
付着テスト
実施例2の3つの賦形剤を同じ相互割合で含有するが、DTZを含有しない錠剤を調製した:これらの錠剤及び上述操作法に従って調製した実施例2の錠剤について、張力計を使用して、付着テストを行った。表3bis及び表4は、分析した錠剤の水分含量及び係るテストから得られた結果を示す。
【表3bis】
【表4】
表4に示すデータから理解されるように、DTZを含有しないNT錠剤は、胃腸粘膜をシミュレートする基材(ムチン)への特定の付着(次の処理、すなわち、おそらく加熱処理によって生成されるマトリックスの構造の変化に関連する現象を低減させる)を示す。
NTに関しては、薬剤を含有する錠剤は、賦形剤のみにて処方されたものよりも低い付着性を示す。これは、組成物の相違によると思われる:実際、成分間の割合が一定であったとしても、DTZを含有しない錠剤は、実施例2に対して25%多い絶対量のポリカルボフィルを含有している。処理した錠剤から得られる結果は、130℃での処理の場合、薬剤を含有するNT錠剤に対して、付着性の低減を示し、一方、150℃において15分間の処理後では、付着特性は低減しない。
【0097】
膨潤テスト
実施例1及び2に従って得られた錠剤について、37℃、pH7.2(このpH条件では、現象が特に明瞭である)のリン酸塩緩衝液において膨潤テストを行った。図7は、実施例1及び2による2つのタイプの錠剤の膨潤度(S%)を示す。得られた曲線は、各タイプ当たり錠剤3個の平均膨潤度を示しており、バーは標準偏差を表す。
表5に、次の事項:リン酸塩緩衝液に24時間浸漬した後のS%60、S%max、質量及び容積及び関連する密度を報告している。
【表5】
図7において理解されるように、実施例2に従って処理した錠剤は、表5においても示されているように、実施例1に従って処理した錠剤に対して、より高い膨潤度及び速度を有する:この特性は、混合物中におけるポリカルボフィルの濃度に関係するものと思われる。いずれの錠剤についても、初めの120分間を除いて、経時的に一定である質量増加(質量増加に基づき、Sが算定される)が存在することが注目されなければならない。
実施例1による錠剤は、最大膨潤時間(これを越えると、さらには膨潤しない時間)1440分(24時間)に達するが、1830分(30.5時間)までは、浸漬されたままでも崩壊することなく、一方、実施例2による錠剤については、最大24時間(最後の測定を行った時間)に達して後では、錠剤は24〜30時間の間で完全に崩壊する傾向にあるため、このような現象は目視観察されない。これらの現象は、インビトロ測定条件(高容積の水性媒体及び連続撹拌を含む)下において観察される。
図7には、実施例1及び2による錠剤の、37℃、リン酸塩緩衝液における膨潤プロフィールを報告している。図7aには、2つの実施例による錠剤を、それぞれの最大膨潤度において撮影した写真が、溶解テスト前の本来のサイズの錠剤の写真と対比して示されている。
図7bには、実施例2による錠剤のその最大膨潤度における拡大写真が報告されており、固状の中心コアが明確に認められる。
図7cには、実施例2による膨潤した錠剤を、異なる視野から見た写真が報告されており、しっかりしたマトリックスの形成が明確に認められる。
膨潤を、活性物質を含有しないマトリックスについても測定した。図7dには、DTZを除く実施例2の全ての成分を含有する(ただし、実施例2と同じ割合である)錠剤の膨潤プロフィールを報告している。最長の加熱処理、すなわち、150℃において15分間の加熱処理にて生成したマトリックスによって最大の膨潤が達成される:このようなマトリックスは、5時間後、1744%の最大質量増加値を達成し、さらに2時間、浸食(連続撹拌によって生ずる)に耐えることができる。
5分間の加熱によって形成されたマトリックスは、2時間後に、最大質量増加値(1282%)に達し、さらに60分間、浸食に耐えることができる。
加熱を介して得られるマトリックスにおける薬剤の存在は、膨潤するマトリックスにおける水性媒体の拡散のため、膨潤現象を減速させる。減速は、逆のマスプロセス、すなわち、活性物質の外側方向への拡散に関連する。2つの現象の同時性は、0オーダーモデルによる活性物質放出速度論を説明するものである。
マトリックスの形成に対する加熱処理の影響は、膨潤テストの間に示されるNT錠剤の挙動からも明らかである。実際、これらの錠剤は、初めの1時間では非常にわずかに膨潤し(わずかに80%)、ついで、120分後には、完全に崩壊する。
【実施例4】
【0098】
グリクラジド 20%
エチルセルロース 30%
MicroceLac 30%
ポリカルボフィル 20%
この粉末混合物から、上述の操作法に従って、直接圧縮によって錠剤を調製した。
これら錠剤の一部を、上述のように、処理温度150℃に処理時間5分間又は15分間維持することによって加熱処理に供した。前記時間の経過後、強制換気によって、オーブンを直ちに室温に冷却した。包装前における室温での最短コンディショニング時間:5分。
図8において、各処理のタイプ(150℃において15分間及び150℃において5分間)について、pH7.2のリン酸塩緩衝液における錠剤6個の平均溶解プロフィールを、NT錠剤の平均溶解プロフィールと対比して報告している。
マトリックスの形成に対する加熱処理の影響は、グリクラジドの放出速度についてのコントロールから明白であり、コンパクトゲルクラウンの形成に関するユニットの膨潤から目視的に明白である。
加熱処理の効果は、マトリックス成分との相互作用が異なるため、活性物質のタイプによっても変化することは明白である。
【特許請求の範囲】
【請求項1】
生体付着性のコンパクトマトリックスを製造する方法であって、
少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーを含んでなる均一な粉末混合物を調製する工程;
前記粉末混合物を原料として、直接圧縮によって、圧縮した又は圧密化したユニットを調製する工程;
このようにして得られた圧縮した又は圧密化したユニットを、80〜250℃、好ましくは、90〜160℃の範囲の温度における1〜60分間、好ましくは、1〜30分間の加熱に供する工程
を含んでなる生体付着性コンパクトマトリックの製法。
【請求項2】
少なくとも1つの活性物質を含有し、前記少なくとも1つの活性物質の延長された又は制御された放出に適合する生体付着性の圧縮ユニットを調製するものであり、均一な混合物が少なくとも1つの活性物質を含んでなるものである請求項1記載の方法。
【請求項3】
均一な混合物が、さらに、非晶質又は結晶性の物理的形状の無水又は1水和物の乳糖、及び微結晶性セルロース又は任意に予め形成した混合物、特に、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac(登録商標))の中から選ばれる希釈剤を含んでなるものである請求項1又は2記載の方法。
【請求項4】
アルキルセルロースが、メチルセルロース及びエチルセルロースからなる群から選ばれものであり、ヒドロキシアルキルセルロースが、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロースからなる群から選ばれるものである請求項1〜3のいずれかに記載の方法。
【請求項5】
非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーがポリカルボフィルである請求項1〜4のいずれかに記載の方法。
【請求項6】
均一な粉末混合物が、さらに、クロスポビドン、ポビドン、ビニルピロリドン-酢酸ビニルコポリマー(Kollidon(登録商標)VA64)、セルロースアセテートフタレート、ハイプロメロースフタレート、ポリビニルアルコール、ポリビニルアセテートフタレート、シクロデキストリン、メタクリレートポリマー、グリセリルトリアセテート、トリエチルシトレート、トリブチルシトレート、アセチルトリエチルシトレート、アセチルトリブチルシトレート、ジブチルセバケート、ジエチルフタレート、ジブチルフタレート、ジオクチルホスフェート、ポリエチレングリコール、ポリエチレンオキシド、カルシウムカルボキシメチルセルロース、ナトリウムカルボキシメチルセルロース、イヌリン、キトサン、ガーゴム、キサンタンガム、トラガカントゴム、カルボマー、カラゲナン、アルギン酸、ポロキサマー、脂肪族ポリエステル、セルロースアセテートブチレート、キトサンラクテート、ペクチン、ポリエチレン-コビニルアセテート、ポリエチレン、ポリビニルアセテート-コメタクリル酸、カルナウバワックス、ブチルヒドロキシアニソール、ブチルヒドロキシトルエン、アスコルビルパルミテート、グリセリルパルミトステアレート、水素化大豆油及びヒマシ油、グリセリルモノステアレート、d-α-トコフェロール(ビタミンE)、ビタミンEスクシネート、ビタミンE及びTPGS、メチルパラベン、ブチルステアレート、ステアリルアルコール、サッカロースモノパルミテート(Sucroester)、グリセロールエステル及びPEGエステル、ポリオキシエチレンアルキルエステル、グリセリルパルミトステアレート、鉱油、ヒマシ油の中から選ばれる1以上の成分を含むものである請求項1〜5のいずれかに記載の方法。
【請求項7】
均一な粉末混合物が、少なくとも1つの活性物質、スプレー乾燥したα-乳糖・1水和物75%及び微結晶性セルロース25%を含有する化合物(MicroceLac)、エチルセスロース及びポリカルボフィルを含んでなるものである請求項1〜6のいずれかに記載の方法。
【請求項8】
ポリカルボフィルが、粉末混合物の総質量の5〜35%、好ましくは10〜25%を構成する請求項7記載の方法。
【請求項9】
エチルセルロース及びスプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物が、粉末混合物中に、1:2〜2:1、好ましくは、0.8:1〜1.2:1の質量比で存在して、一緒に、粉末混合物の総質量の45〜95%、好ましくは、60〜80%を構成し、及びエチルセルロースは、ポリカルボフィルとの質量比1:5〜5:1で存在する請求項8記載の方法。
【請求項10】
少なくとも1つの活性物質が、粉末混合物中に、0.001 ppm〜混合物の総質量の50%の量で含有される請求項8又は9記載の方法。
【請求項11】
圧縮したユニットが医薬錠剤であり、活性物質が薬理学的に活性な物質である請求項1〜10のいずれかに記載の方法。
【請求項12】
少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び好ましくは、ポリカルボフィルによって構成される、非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーを含んでなる、請求項1記載の方法によって得られた生体付着性の錠剤。
【請求項13】
さらに、少なくとも1つの活性物質を含んでなり、前記少なくとも1つの活性物質の延長された又は制御された放出に適合する、請求項2記載の方法によって得られる請求項12記載の生体付着性の錠剤。
【請求項14】
制御された放出及びpH4〜8の水溶液において実質的に0オーダーの活性物質の放出速度を有することを特徴とする請求項13記載の生体付着性の錠剤。
【請求項15】
少なくとも1つのアルキルセルロースがエチルセルロースであり、及び非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーがポリカルボフィルであり、さらに、非晶質又は結晶性の物理的形状の無水又は1水和物の乳糖、及び微結晶性セルロース又は任意に予め形成した混合物、特に、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac)の中から選ばれる希釈剤を含んでなる請求項13又は14記載の錠剤。
【請求項1】
生体付着性のコンパクトマトリックスを製造する方法であって、
少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーを含んでなる均一な粉末混合物を調製する工程;
前記粉末混合物を原料として、直接圧縮によって、圧縮した又は圧密化したユニットを調製する工程;
このようにして得られた圧縮した又は圧密化したユニットを、80〜250℃、好ましくは、90〜160℃の範囲の温度における1〜60分間、好ましくは、1〜30分間の加熱に供する工程
を含んでなる生体付着性コンパクトマトリックの製法。
【請求項2】
少なくとも1つの活性物質を含有し、前記少なくとも1つの活性物質の延長された又は制御された放出に適合する生体付着性の圧縮ユニットを調製するものであり、均一な混合物が少なくとも1つの活性物質を含んでなるものである請求項1記載の方法。
【請求項3】
均一な混合物が、さらに、非晶質又は結晶性の物理的形状の無水又は1水和物の乳糖、及び微結晶性セルロース又は任意に予め形成した混合物、特に、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac(登録商標))の中から選ばれる希釈剤を含んでなるものである請求項1又は2記載の方法。
【請求項4】
アルキルセルロースが、メチルセルロース及びエチルセルロースからなる群から選ばれものであり、ヒドロキシアルキルセルロースが、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシエチルメチルセルロースからなる群から選ばれるものである請求項1〜3のいずれかに記載の方法。
【請求項5】
非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーがポリカルボフィルである請求項1〜4のいずれかに記載の方法。
【請求項6】
均一な粉末混合物が、さらに、クロスポビドン、ポビドン、ビニルピロリドン-酢酸ビニルコポリマー(Kollidon(登録商標)VA64)、セルロースアセテートフタレート、ハイプロメロースフタレート、ポリビニルアルコール、ポリビニルアセテートフタレート、シクロデキストリン、メタクリレートポリマー、グリセリルトリアセテート、トリエチルシトレート、トリブチルシトレート、アセチルトリエチルシトレート、アセチルトリブチルシトレート、ジブチルセバケート、ジエチルフタレート、ジブチルフタレート、ジオクチルホスフェート、ポリエチレングリコール、ポリエチレンオキシド、カルシウムカルボキシメチルセルロース、ナトリウムカルボキシメチルセルロース、イヌリン、キトサン、ガーゴム、キサンタンガム、トラガカントゴム、カルボマー、カラゲナン、アルギン酸、ポロキサマー、脂肪族ポリエステル、セルロースアセテートブチレート、キトサンラクテート、ペクチン、ポリエチレン-コビニルアセテート、ポリエチレン、ポリビニルアセテート-コメタクリル酸、カルナウバワックス、ブチルヒドロキシアニソール、ブチルヒドロキシトルエン、アスコルビルパルミテート、グリセリルパルミトステアレート、水素化大豆油及びヒマシ油、グリセリルモノステアレート、d-α-トコフェロール(ビタミンE)、ビタミンEスクシネート、ビタミンE及びTPGS、メチルパラベン、ブチルステアレート、ステアリルアルコール、サッカロースモノパルミテート(Sucroester)、グリセロールエステル及びPEGエステル、ポリオキシエチレンアルキルエステル、グリセリルパルミトステアレート、鉱油、ヒマシ油の中から選ばれる1以上の成分を含むものである請求項1〜5のいずれかに記載の方法。
【請求項7】
均一な粉末混合物が、少なくとも1つの活性物質、スプレー乾燥したα-乳糖・1水和物75%及び微結晶性セルロース25%を含有する化合物(MicroceLac)、エチルセスロース及びポリカルボフィルを含んでなるものである請求項1〜6のいずれかに記載の方法。
【請求項8】
ポリカルボフィルが、粉末混合物の総質量の5〜35%、好ましくは10〜25%を構成する請求項7記載の方法。
【請求項9】
エチルセルロース及びスプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物が、粉末混合物中に、1:2〜2:1、好ましくは、0.8:1〜1.2:1の質量比で存在して、一緒に、粉末混合物の総質量の45〜95%、好ましくは、60〜80%を構成し、及びエチルセルロースは、ポリカルボフィルとの質量比1:5〜5:1で存在する請求項8記載の方法。
【請求項10】
少なくとも1つの活性物質が、粉末混合物中に、0.001 ppm〜混合物の総質量の50%の量で含有される請求項8又は9記載の方法。
【請求項11】
圧縮したユニットが医薬錠剤であり、活性物質が薬理学的に活性な物質である請求項1〜10のいずれかに記載の方法。
【請求項12】
少なくとも1つのアルキルセルロース又は1つのヒドロキシアルキルセルロース及び好ましくは、ポリカルボフィルによって構成される、非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーを含んでなる、請求項1記載の方法によって得られた生体付着性の錠剤。
【請求項13】
さらに、少なくとも1つの活性物質を含んでなり、前記少なくとも1つの活性物質の延長された又は制御された放出に適合する、請求項2記載の方法によって得られる請求項12記載の生体付着性の錠剤。
【請求項14】
制御された放出及びpH4〜8の水溶液において実質的に0オーダーの活性物質の放出速度を有することを特徴とする請求項13記載の生体付着性の錠剤。
【請求項15】
少なくとも1つのアルキルセルロースがエチルセルロースであり、及び非水溶性、水膨潤性の架橋ポリカルボン酸系ポリマーがポリカルボフィルであり、さらに、非晶質又は結晶性の物理的形状の無水又は1水和物の乳糖、及び微結晶性セルロース又は任意に予め形成した混合物、特に、スプレー乾燥したα-乳糖75%及び微結晶性セルロース25%を含有する化合物(MicroceLac)の中から選ばれる希釈剤を含んでなる請求項13又は14記載の錠剤。
【図1a】

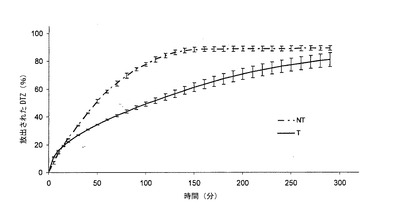
【図1b】
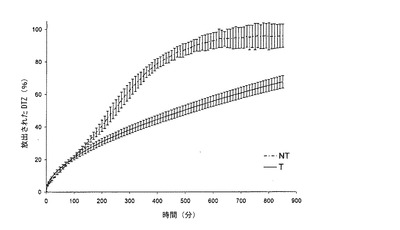
【図1c】
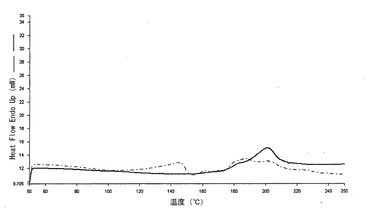
【図2a】
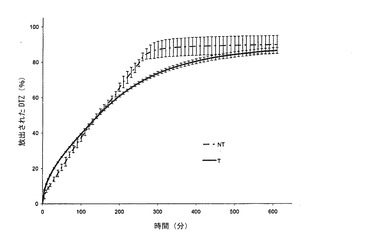
【図2b】
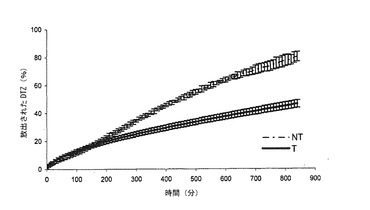
【図2c】
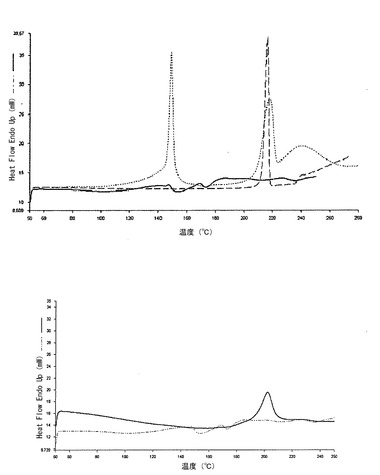
【図3a】
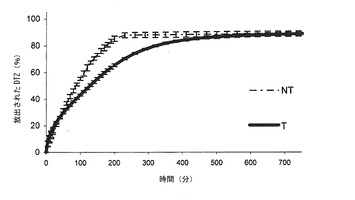
【図3b】
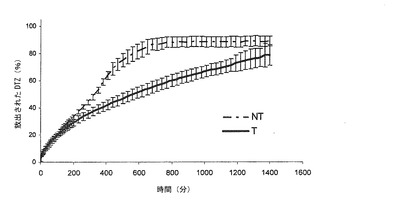
【図3c】
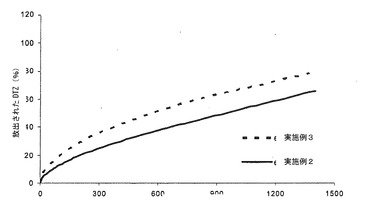
【図3d】
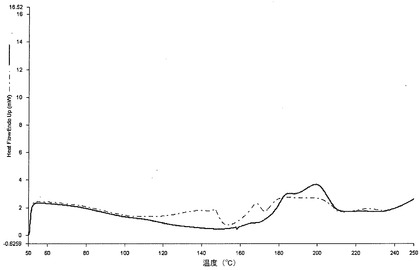
【図4a】
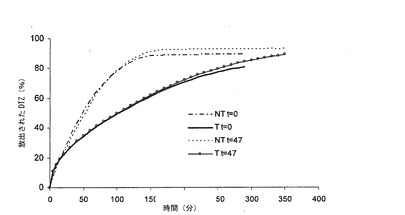
【図4b】
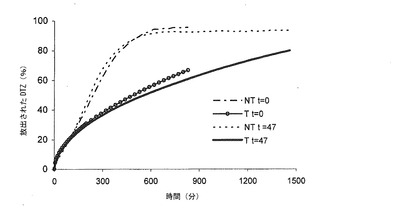
【図5a】
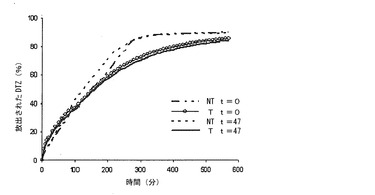
【図5b】
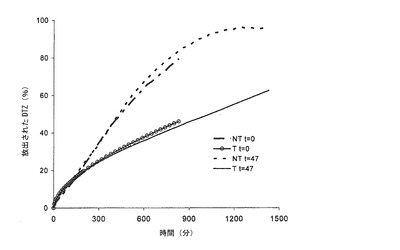
【図5c】
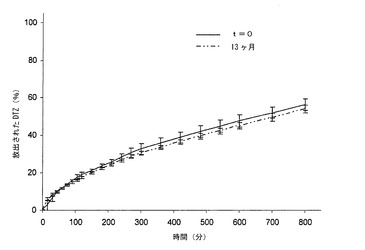
【図5d】
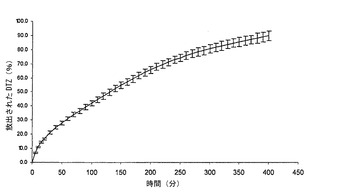
【図5e】
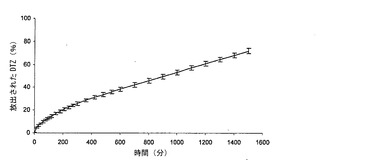
【図5f】
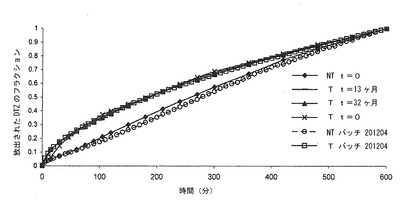
【図6】
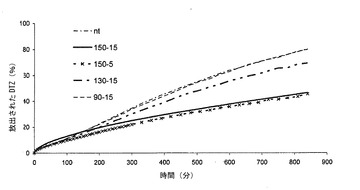
【図7】
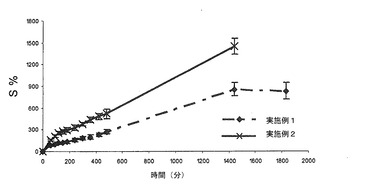
【図7a】

【図7b】

【図7c】
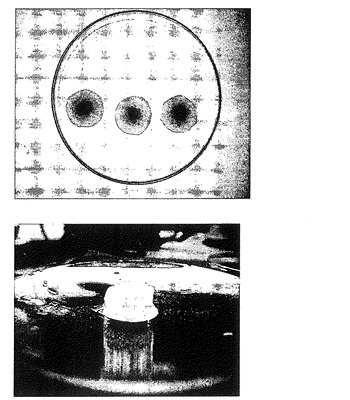
【図7d】
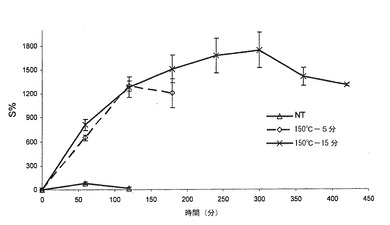
【図8】
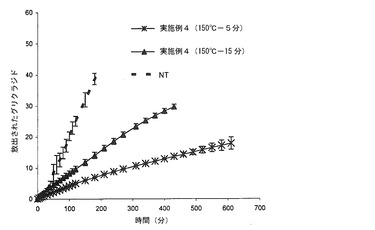
【図9】

【図10】
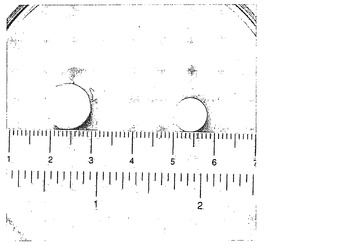
【図11】
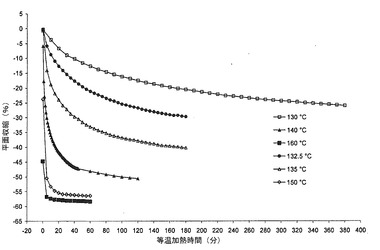
【図12】
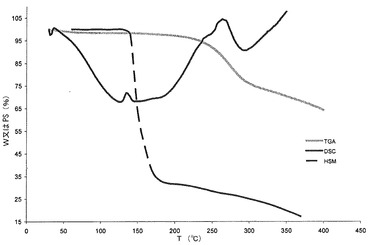
【図13】

【図14】

【図15】
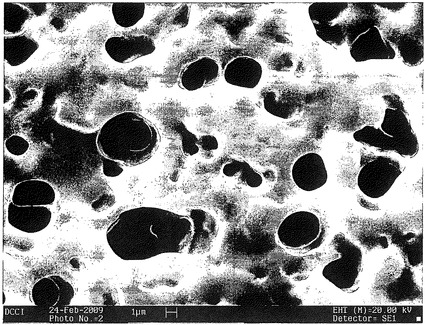
【図16】
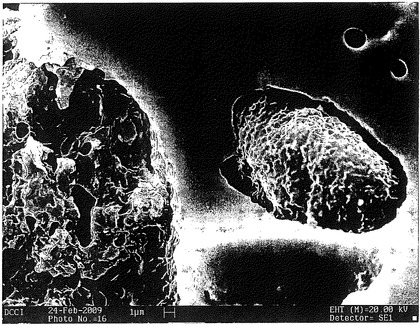
【図17】
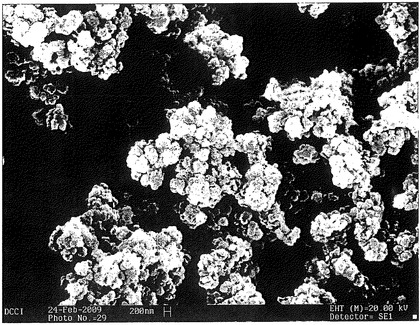
【図18】
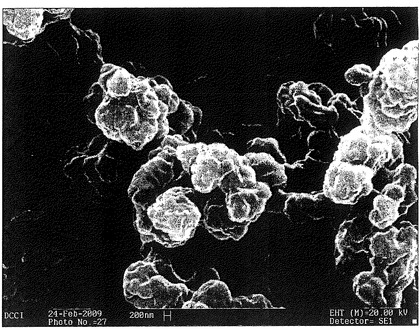
【図1b】
【図1c】
【図2a】
【図2b】
【図2c】
【図3a】
【図3b】
【図3c】
【図3d】
【図4a】
【図4b】
【図5a】
【図5b】
【図5c】
【図5d】
【図5e】
【図5f】
【図6】
【図7】
【図7a】
【図7b】
【図7c】
【図7d】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公表番号】特表2012−511503(P2012−511503A)
【公表日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願番号】特願2011−526408(P2011−526408)
【出願日】平成21年9月10日(2009.9.10)
【国際出願番号】PCT/EP2009/006567
【国際公開番号】WO2010/028826
【国際公開日】平成22年3月18日(2010.3.18)
【出願人】(511065118)ウニヴェルシタ デグリ ステューディ ディ ジェノヴァ (1)
【Fターム(参考)】
【公表日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願日】平成21年9月10日(2009.9.10)
【国際出願番号】PCT/EP2009/006567
【国際公開番号】WO2010/028826
【国際公開日】平成22年3月18日(2010.3.18)
【出願人】(511065118)ウニヴェルシタ デグリ ステューディ ディ ジェノヴァ (1)
【Fターム(参考)】
[ Back to top ]