
生体分子検出素子、生体分子検出素子の製造方法及び生体分子検出方法
【課題】生体分子検出素子の検出精度及び再現性を向上させる。
【解決手段】単一プローブ分子を基板表面の格子点位置に規則的に配列固定する。そのために、(1)生体分子検出用プローブ分子107を規則的な配列で孤立固定し、(2)生体分子検出用プローブ分子以外の領域に非特異吸着防止用のブロッキングを施し、(3)金属微粒子106を用いて蛍光増強させる。
【解決手段】単一プローブ分子を基板表面の格子点位置に規則的に配列固定する。そのために、(1)生体分子検出用プローブ分子107を規則的な配列で孤立固定し、(2)生体分子検出用プローブ分子以外の領域に非特異吸着防止用のブロッキングを施し、(3)金属微粒子106を用いて蛍光増強させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生体分子をセンシングするための生体分子検出素子及びその製造方法並びに生体分子検出方法に関するものである。
【背景技術】
【0002】
近年、ヒトゲノムDNAの解読がほぼ終了したことにより、遺伝子の機能を解明する研究が盛んに行われている。そこでは、生体内の遺伝子やタンパク質を特異的かつ網羅的に検出することが必要であり、遺伝子・タンパク質検出技術の開発が世界的に進められている。一方で、生体内に進入した病原菌やウイルスを、遺伝子やタンパク質レベルで特定する技術も従来から検討されており、実用化されつつある。このような目的に応じて、特定の遺伝子やタンパク質等の生体分子を検出するための手段として、種々のバイオセンサが用いられている。最も一般的なバイオセンサの構造は、生体分子を捕捉するプローブ分子が固体表面上に固定されたものである。核酸を捕捉する場合には、プローブ分子として主に核酸が用いられ、タンパク質を捕捉する場合には、プローブ分子として主にタンパク質が用いられる。基板にプローブ分子を固定したバイオセンサのメリットは、同一の基板に、多種類のプローブ分子をスポッティングやインクジェットなどの方式を用いて固定できることである。このバイオセンサ基板を用いれば、多種類の生体分子に対する網羅的な解析を一度に迅速に行うことができる。そして基板表面を利用したバイオセンサの代表例が、DNAマイクロアレイやプロテインチップのような生体分子検出素子である。
【0003】
このような生体分子検出素子の製造方法には大別して2つの方法がある。一つは、フォトリソグラフィやインクジェットを用いて、基板上でヌクレオチドやアミノ酸を逐次的に反応させ、固定化することにより、一本鎖のDNAやたんぱく質といったプローブ用の生体分子をin-situ合成する方法である(特許文献1)。もう一つは、プローブ用生体分子をex-situで合成した後に、これらを基板上に固定化する方法である(特許文献2)。
【0004】
DNAマイクロアレイに代表される生体分子検出素子は、将来は癌等の病気の診断を含む医療診断に使われると予想されている。医療診断にDNAマイクロアレイを用いる場合には、マイクロアレイから得られるデータに対して高い信頼性が要求される。フォトリソグラフィやインクジェットを用いてプローブ生体分子を表面にin-situ合成する方法では、逐次的に結合させるヌクレオチドやアミノ酸の反応率が各ステップ毎に100%にはなり得ないため、スポット上の全てのプローブ分子の配列が厳密に設計通りであることを保証することが難しく、また操作ミスでヌクレオチドやアミノ酸の種類の取り違いや欠損部が生じた場合、それらを検査し、製造後に除去することが現状では不可能である。更に、長い塩基やたんぱく質を得るためには莫大な製造コストが必要になる。これに対し、DNAやたんぱく質をex-situで合成してから基板上に固定する方法では、合成した後に精製することで、欠損部等を持つ生体分子を事前に除去することができる。よって、純度の高いプローブ用生体分子を表面に固定することができ、信頼性の高いマイクロアレイを製造することが可能である。この方法では、多くの場合、反応活性な官能基を持つ生体分子と表面を反応させ、共有結合を作ることで固定化している。しかしながら、基板表面にDNAやたんぱく質といった分子量の大きい高分子を固定化させる際に、固定化されたプローブ生体分子の量・構造にバラツキが生じやすく、これがデータの再現性を低下させる要因になっている(Nature Vol.21, pp.5-9, 1999)。また、マイクロアレイを用いて生体分子を検出する際に、生体分子が基板表面に非特異的に吸着することや、固定化されたプローブ生体分子の密度が制御されていないために感度が低いことも定量的な解析を困難にする原因である(Nature Biotech. 19, p.342, 2001)。
【0005】
一方、最近、DNAマイクロアレイで行われているような遺伝子発現解析の精度の大幅向上を狙って、特許文献3ないし非特許文献3に示されているように、ポリヌクレオチドの単一分子アレイを用いた単一分子ベースのシーケンシング(SBS(Sequencing by Synthesis)法等)による遺伝子配列決定方法が開示されている。この方法では、基板表面に適当なプライマーで修飾された検体のポリヌクレオチドを固定し、これを鋳型としてポリメラーゼによる一塩基ずつの伸長反応を実行し、検体ポリヌクレオチドの相補鎖を形成させるものである。この一塩基伸長の個々のステップでは、異なる4種のヌクレオチドのそれぞれのプリン又はピリミジン、あるいは3リン酸のリン酸基の末端部にユニークな蛍光色素を導入しておき、伸長ステップ毎に単一分子蛍光検出を行うことで、導入されたヌクレオチドを識別する。このステップを繰り返し、単一ポリヌクレオチド固定サイトごとのシーケンスを読み取って、検体の配列情報を網羅的に取得する。ここでは、高いS/N比で検出し、シーケンシング時の配列決定正確性を向上させることが重要となる。この技術では、膨大な単一ポリヌクレオチド固定サイトからの蛍光の情報をCCDカメラで検出するため、そのCCDカメラの画素サイズに対応してポリヌクレオチド分子の平均的な固定密度を設定している。即ち、出来るだけ一個の画素で一個のポリヌクレオチド上からの蛍光信号を取り込むようにポリヌクレオチドの平均固定密度あるいは画素の解像度を調整している。一画素の大きさは検出光学系の空間分解能を考慮するとサブミクロン角以上となる。
【0006】
一個の画素で一個のポリヌクレオチド上からの蛍光信号を効率良く取り込むようにするために、下記のような試みが行われている。単一DNA分子やRNA分子を基板に固定し、その密度を1分子/μm2以下し、また固定密度を調整することによって、シーケンシング時の遺伝子配列決定の正確性を高める技術が特許文献4に示されている。また、特許文献5において、1分子をマイクロスフェアーを介して固定する方法が紹介されている。隣接する分子同士の分子間距離を充分に保つことで、例えば、1分子のシーケンシングを蛍光を用いて行う際、高い正確性を持って配列決定できるとしている。また、マイクロナノスフェアーであるコロイド粒子を格子パターン状に並べる手段として、リソグラフィ技術によって形成した微細開口パターンにコロイド粒子を落とし込んで微粒子を規則的なパターンに整列させる方法が非特許文献4に開示されている。更に、同一種類のプローブ分子群をマトリックス状に並べた基板が特許文献6,7に開示されている。一方、シーケンシング時の検出ノイズを低減するために、デバイス形成プロセスに適合し、かつ十分な吸着防止能を持った吸着阻害剤を領域選択的に形成する必要がある。負電荷を持つPolyacrylic acidでデバイス表面を覆い、dNTPの吸着を抑えた例が非特許文献3で紹介されている。
【0007】
次に、DNAを解析する手段であるマイクロアレイあるいはシーケンシングを用いて少量のサンプルを検出するためには、蛍光検出感度を上げることも必要である。蛍光検出感度を向上させるために、蛍光増強を用いる方法が非特許文献5に報告されている。ここでは、プローブ分子であるDNAを修飾した銀ナノ粒子を基板に固定し、蛍光標識された検体中の分子と反応させている。反応量を検出するために蛍光励起光を照射すると、銀ナノ粒子の自由電子が共鳴振動(局在プラズモン共鳴)を起こし蛍光が増強する。この現象を用いることで、感度を向上させることができるとしている。
【0008】
【特許文献1】米国特許第5,424,186号
【特許文献2】米国特許第5,700,637号
【特許文献3】米国特許第6,787,308号
【特許文献4】特表2002-531808号公報
【特許文献5】特表2002-521064号公報
【特許文献6】特開2001-33458号公報
【特許文献7】特開2002-235474号公報
【非特許文献1】Nature Vol.21, pp.5-9, 1999
【非特許文献2】Nature Biotech. 19, p.342, 2001
【非特許文献3】Proc.Natl.Acad.Sci.USA,Vol.100(7),pp3960,2003
【非特許文献4】Nano Letters,Vol.4(6),1093,2004
【非特許文献5】Biochem. Biophys. Res. Comm. 306, p.213(2003)
【非特許文献6】Jpn.J.Appl.Phys.,41,p.1579(2002)
【非特許文献7】Analytical Biochemistry 298, p.1 (2001)
【発明の開示】
【発明が解決しようとする課題】
【0009】
特許文献4,5の方法では、検出対象となる単一DNA分子の配置がランダムであり、なんら規則性を持たない。したがって、一画素と、シーケンシング時に蛍光を発生する場となるDNA分子の空間配置を1:1で対応させることが困難となる。そこで、検出対象分子を規則的に配置させることが重要になる。非特許文献4の方法では微粒子が表面に物理吸着しただけの状態であり、固定力が不十分であって、このコロイド粒子を単一DNA分子の場所の目印として用いようとしても、そのままではデバイスとして安定に使用することが出来ない。特許文献6,7の方法は、単一DNA分子を規則的に配置させるものではない。非特許文献3の場合、Polyacrylic acidは表面に物理吸着しているため、デバイス上で溶液を用いた種々反応を行うにつれて剥離する可能性が高い。またPolyacrylic acidを吸着させるために、正電荷を持つPolyallylamine等も吸着させているため、これが新たな非特異吸着サイトとなる可能性もある。また、単一分子を規則的に配列するという目的達成に微粒子を利用する場合には、微粒子表面のプローブ固定箇所以外をブロックする吸着阻害剤の形成も必要である。非特許文献5の方法は、蛍光検出の感度向上だけを目的としており、銀ナノ粒子上には無数のプローブ分子を固定しているため、検出精度の点で問題がある。検出精度向上のためには、粒子上のプローブ分子の固定方法に工夫が必要になる。
【0010】
以上のように生体分子検出素子の低精度・再現性に対する問題を解決するためのアイデアは存在するが、いずれも単独では有効な結果を得ることは出来ず、別に新たな工夫を加えた総合的な解決策が必要である。
【0011】
本発明の目的は、DNAマイクロアレイやシーケンシングのような生体分子検出素子の精度・再現性を向上させることである。精度・再現性の低下を引き起こす原因として主に下記の諸点が挙げられる。
(1)プローブ分子の固定密度が定量的に制御されていない。
(2)プローブ分子の相互の間隔がランダムであり、個々のプローブ分子による生体分子検出反応が、独立ではなく相互に干渉しあう。
(3)非特異的に吸着した生体分子が表面に残留し、検出信号の精度を低下させる。
【0012】
上記(1)の原因として、プローブ分子固定反応において、固定密度を制御する工夫がなされていないことが挙げられる。上記(2)は(1)の結果として現れることである。また、上記(3)の原因として、生体分子の非特異的吸着に対する防止対策が不十分であることが挙げられる。
【0013】
特に、シーケンシングによって遺伝子配列を決定する際に精度・再現性の低下を引き起こす原因として、検出対象となる単一分子DNAがある平均密度でランダムに固定された表面を用いていること、すなわち、検体ポリヌクレオチドの固定点の配置に何ら規則性がないことが挙げられる。隣り合う検体ポリヌクレオチドが近接しすぎる場合には、両方のポリヌクレオチド分子の上から検出される蛍光信号が同一画素で検出されるためにデータが誤読されてしまう。従って、このような場所は無効サイトとなる。また、膨大な数の蛍光信号発生箇所(ポリヌクレオチド固定サイト)を検出する必要があるので、センサー表面上をステップ&リピート方式でエリア毎に蛍光検出するが、個々の測定箇所がランダム配置であるため、検出・測定作業がたいへん非能率である。このようにシーケンシングにおいては、生体分子である検体ポリヌクレオチドがランダム固定されていることが原因で、検体ポリヌクレオチド固定点に無駄が発生したり、測定時間が長いなどの性能上の制約が発生する。また単一分子ベースのシーケンシングでは、基本的に単一分子蛍光計測が必要であり、安定した蛍光検出のためには、蛍光検出感度の向上が必要である。
【0014】
以上の問題点を解決するための第一の課題は、生体分子検出のための個々のプローブ分子を基板表面上に予め設計された規則正しい幾何学的配列で、単一分子として固定することである。第二の課題は、基板表面のプローブ分子固定サイト以外の領域には、検体分子やその他の妨害分子が非特異的に吸着しない対策を施すことである。第三の課題は、蛍光検出感度を増強することである。
【0015】
これらの課題を同時に解決することにより、高感度、高精度かつ再現性にすぐれた生体分子検出素子を実現する。
【課題を解決するための手段】
【0016】
本発明は、単一のプローブ分子を担体基板表面の格子点位置に規則的に配列固定した生体分子検出素子を実現する。そのために、まず担体基板表面にリンカー分子の格子状ドットパターンを設置する。ここで、格子とは正方格子、三角格子、六角格子、ハニカム型格子、長方形格子等の様々な形状の格子を指す。このリンカー分子は、担体基板表面に固定され、基板と反対側にはアミノ基やチオール基のような金属と強い相互作用をする官能基を持つ。次に、これらの官能基と金属微粒子を結合させることにより、一つのリンカー分子ドット上に一つの金属微粒子を固定する。この金属微粒子一個の上には、一個のプローブ分子を固定する。そして、担体基板表面上の金属微粒子が固定された領域即ちリンカー分子ドットパターン以外の領域には、種々の生体分子の非特異的な吸着を防止するための吸着阻害分子を固定する。さらに、金属微粒子の表面のうち、プローブ分子が結合していない領域には、第二の吸着阻害分子を固定し、金属微粒子表面への種々の生体分子の非特異的な吸着も防止する。
【0017】
金属微粒子の材料としては、貴金属、貴金属の合金、あるいは貴金属の積層体が良い。
金属微粒子径とリンカー分子ドットの径は互いにほぼ匹敵するサイズが好ましい。
【0018】
また貴金属の微粒子をプローブ分子固定の土台に使うことにより、プローブ分子近傍での蛍光検出の際の励起光による金属微粒子のプラズモン共鳴による近接場の効果を活用して蛍光強度を増強し、蛍光検出感度を向上させ、単一分子の蛍光でも十分安定に検出可能とする。
【発明の効果】
【0019】
本発明によれば、生体分子検出素子表面のプローブ分子が孤立して決められた一定の距離を離れて規則的に固定されるため、検体試料との反応において、どのプローブ分子も均等な反応条件に置かれる。また検体分子とプローブ分子との反応において隣接プローブ分子が干渉することが無い。これらのことは、例えばDNAマイクロアレイに用いた場合には、検出精度の再現性向上に寄与する。また、プローブ分子の固定ピッチは目的に応じて任意に変更でき、かつプローブ分子の固定位置が事前に正確に判るので、例えば単一塩基シーケンシングに用いた場合には、検出系CCDの画素ピッチに揃えて単一プローブ分子を固定することで、個々のプローブ分子固定サイトからの蛍光信号を間違いなく分離して検出でき、また素子表面のステップ&リピートによる繰り返しの蛍光測定の駆動作業が大幅に高速化できる。さらに、プローブ分子の固定場として金属微粒子を用いるので、蛍光検出の励起光と金属粒子内自由電子の共鳴に基づく近接場を活用して蛍光の増強が達成でき、単一蛍光分子からの蛍光でも安定に検出することが可能となる。
【0020】
即ち、生体分子検出素子として、検出精度、検出データ再現性、検出感度の三つの課題を同時に解決するとともに、検出工程を大幅に高速化できるデバイスを提供できる。
【発明を実施するための最良の形態】
【0021】
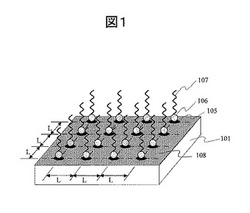
図1に、本発明による生体分子検出素子の表面構造の一例を示す。まず担体基板101の表面にリンカー分子の格子状ドットパターンを設置する。このドットパターンは基板表面のX座標軸、Y座標軸に沿ってピッチLの正方格子に配置してある。これを金属微粒子固定ドット105として用いる。ここでリンカー分子とは、担体基板表面側が共有結合により固定され、基板と反対側にはアミノ基やチオール基のような金属と強い相互作用をする官能基を持つ分子のことである。これらの官能基を用いて金属微粒子106を結合することにより、一つの金属微粒子固定ドットの上に一つの金属微粒子を固定する。この金属微粒子一個の上には、一個のプローブ分子107を固定する。そして、担体基板表面上の金属微粒子が固定された領域即ち金属微粒子固定ドットパターン以外の領域には、種々の生体分子の非特異的な吸着を防止するための吸着阻害分子を固定し、吸着阻害層108を形成する。さらに、金属微粒子の表面のうち、プローブ分子が結合していない領域には、第二の吸着阻害分子を固定し、金属微粒子表面への種々の生体分子の非特異的な吸着も防止する。なお金属微粒子表面の吸着阻害層は図1では省略してある。
【0022】
以上の構成によって、単一のプローブ分子を担体基板表面の格子点位置に規則的に配列固定した生体分子検出素子を実現する。
【0023】
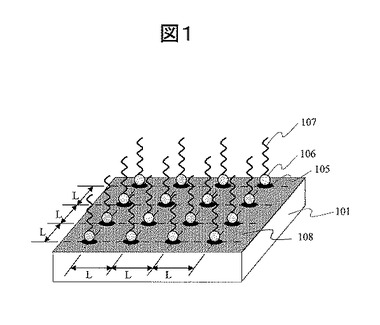
次に、本発明の生体分子検出素子を製造するプロセス概念の一例を、図2を用いて説明する。ここでは、ポジ型電子線レジストを用いた電子線直接描画によるパターン形成を利用したプロセスを取り上げる。製造プロセスは下記の9工程からなる。
(1) 基板表面へのポジ型電子線レジストの塗布
(2) 電子線描画及び現像による開口形成
(3) リンカー分子層の形成I(金属微粒子固定ドット:リンカー分子A)
(4) 金属微粒子の固定
(5) レジスト剥離
(6) リンカー分子層の形成II(微粒子固定ドット外表面:リンカー分子B)
(7) 吸着阻害分子の形成I(吸着阻害分子C)
(8) プローブ分子の固定
(9) 表面吸着阻害分子の形成II(金属微粒子表面:吸着阻害分子D)
【0024】
各工程について以下に説明する。
(1) 基板表面へのポジ型電子線レジストの塗布:図2(a)
先ず担体基板201を洗浄する。具体的には例えば、NaOH水溶液等のアルカリ性水溶液で洗浄した後、HCl水溶液等の酸性水溶液で洗浄し、純水ですすいだ後に乾燥する。あるいは、硫酸と過酸化水素を約4:1で混合した溶液で有機物汚染を洗浄する。基板としては、ガラス基板(スライドガラス)、石英基板、プラスチック基板等を用いることができる。また、金属コーティング基板等でもよい。基板の材質は、表面にシラノール基を有するものが好ましい。
【0025】
次いで、基板にポジ型電子線レジスト202をスピンコートし、所定の温度で乾燥ベークする。本発明では、レジスト膜厚は塗膜の均一性が確保できることを前提としてできるだけ薄いことが望ましいが、スループロセス検討結果から少なくとも100nm以下が望ましく、さらに好ましくは60nm以下が好適である。
【0026】
なお、図2のプロセスでは酸化膜付きSiウェハのような導電性基板の上で電子線描画を実施する場合を想定しているが、酸化膜が例えば10nm以上と厚い場合や、石英、ガラスのような絶縁性基板の上で電子線描画を施す場合には、基板のチャージアップを防ぐために電子線レジストの表面にさらに水溶性導電性樹脂の極薄膜を形成する。この導電性樹脂薄膜もスピンコートで容易に形成でき、レジストの電子線感度にはほとんど影響を与えない。また描画後、水洗によって容易に溶解除去され、次のレジスト現像工程には一切影響を与えない。
ここでは、ポジ型電子線レジストを用いているが、描画パターンによってはネガ型レジストを用いても良い。
【0027】
(2) 電子線描画及び現像による開口形成:図2(b)
電子線描画は、目的の解像度を満たす電子線描画装置によって実行する。ここでは、実効解像度(最小加工寸法)10nmの電界放射型電子線描画装置を用い、ドットパターンをXY座標方向ともに一定ピッチの格子状パターンを描画する。基本パターンは円形であり、サイズは最小20nmφから100nmφ程度が主要な値である。電子線走査範囲は、75μm−2400μm角の範囲であり、必要に応じて走査フィールドを繋ぎ合わせ、照射面積を確保する。レーザ干渉計を用いた高精度アライメント機構を用い、走査フィールド繋ぎ精度は3σで50nm以下(走査フィールド600μmの場合)程度を確保する。電子線描画パターンはCADデータとして設計し、コンピュータ制御により描画を実行する。描画パターンは穴加工であり、スループットの観点からポジ型レジストが向いている。ポジ型電子線レジスト塗布基板を電子線描画した後、所定の有機溶剤系現像液に浸漬して、照射部のレジストを除去し、さらにリンス液で洗浄して開口パターン203を得る。20nmφから100nmφの開口パターンは光学顕微鏡では観察できないので、電子顕微鏡及び原子間力顕微鏡(AFM)を用いてパターン解像検査を行う。
【0028】
(3) リンカー分子層の形成I(金属部粒子固定ドット):図2(c)
電子線レジスト開口パターンを形成した基板を、リンカー分子Aの溶液に浸漬し、開口パターン底の酸化膜(SiO2)表面に反応させる。リンカー分子Aとしては、金属微粒子と結合する活性基を持つシランカップリング剤などが使える。シランカップリング剤としては、例えば、基板表面にアミノ基を固定する場合には、3-アミノプロピルトリメトキシシラン(3-aminopropyltrimthoxysilane)、3-アミノプロピルトリエトキシシラン(3-aminopropyltriethoxysilane)、N-(2-aminoethyl)-3-aminopropyltrimethoxysilane)、(aminoethyl-aminomethyl) phenethyltrimethoxysilane等を用いることができる。一方、基板表面にチオール基を固定する場合には、(3-mercaptopropyltrimethoxysilane)を用いることができる。溶媒としては、例えば、エタノール、メタノール、トルエン、ベンゼン、水等を用いることができる。反応温度は、通常、20℃〜85℃の範囲である。ここで、リンカー分子Aは、開口部の酸化膜表面とは表面シラノール基と共有結合で固定されるが、レジスト表面部では表面に結合基が存在しないため物理吸着しているだけである。リンカー分子Aのアミノ基又はチオール基が基板と反対側に露出し、次工程の金属微粒子と結合する。
【0029】
ここでは、工程(1)(2)で電子線レジスト開口パターンを形成させた後に、工程(3)でリンカー分子層を形成させたが、基板を洗浄した後に基板全面に工程(3)でリンカー分子を結合させ、その後(1)(2)の工程を経て電子線レジスト開口パターンを形成させても良い。
【0030】
(4) 金属微粒子の固定:図2(d)
基板表面の活性基と金属微粒子の相互作用により、基板表面に金属微粒子を固定する。図2(d)は、金属微粒子205が固定化された担体表面の様子を示す。金属微粒子材料としては、貴金属類である金、銀、白金、パラジウム、ロジウム、イリジウム、ルテニウム、オスミウムのいずれか、あるいはそれらの合金を用いることができる。あるいは、これらの貴金属類で作られた微粒子上に他の貴金属がコーティングされたもの、例えば金微粒子上に銀がコーティングされた金属微粒子を用いてもよい。用いることができる金属微粒子径は、金属微粒子が安定に存在できる0.6nm以上である。金属微粒子の基板への固定安定性という観点から、金属微粒子径は10nm以上が望ましい。一方、蛍光を増強させるという観点から、金属微粒子径は蛍光増強効果が得られる10nm以上1μm以下を用いるのが良い。一方、後の工程で金属微粒子表面に単一生体分子を効果的に固定するという観点から、金属微粒子径は100nm以下が望ましい。以上の3つの観点から整理すると、固定する金属微粒子径は0.6nm以上1μm以下が良いが、望ましくは10nm以上100nm以下が良い。
【0031】
金属微粒子の固定反応溶媒として、水、あるいはエタノール、トルエンを用いることができる。溶液中での金属微粒子の凝集を防ぐために、保護剤を用いる。保護剤として、クエン酸、メルカプトコハク酸、ポリビニルピロリドン、ポリアクリル酸、テトラメチルアンモニウム、ポリエチレンイミン、1−デカンチオール、1−オクタンチオール、デシルアミン、ホスフィン(bis(p-sulfonatophenyl)phenylphosphine)等を用いることができる。
【0032】
金属微粒子の濃度は、通常、30wt%以下であり、反応温度は、通常、20℃−85℃の範囲である。また反応時間は、0.5時間から50時間の間である。これらの反応条件を変えることによって、金属微粒子の固定密度を制御することができる。例えば、金属微粒子の固定反応は、固定溶液中の金属微粒子濃度に対する1次反応であり、Langmuir型の反応となる。従って、固定溶液中の金属微粒子濃度や反応時間を変えることで、所望の固定密度を得ることができる。金属微粒子の反応時間は、レジストパターンの無い、リンカー分子だけの単純表面における反応時間を参考に決める。ここでは、あくまでレジスト開口部203の底面に金属微粒子205を到達させ、表面活性基と金属微粒子を反応させて固定することが目的である。
【0033】
ここで、ドットパターン上の金属微粒子が、各サイトに複数個入らずに一個づつ固定され、かつドットパターンへの金属微粒子の固定充填率を高く保つために、金属微粒子径と電子線レジスト開口部の径の関係は以下の条件を満たすのが良い。金属微粒子径/電子線レジスト開口部径の比をγとすると0.5≦γ≦1を満たすのが良い。γが0.5より小さい場合、電子線レジスト開口径に比べて金属微粒子径が充分小さいため、各サイトに金属微粒子が複数個入ってしまう。一方、γが1より大きい場合、電子線レジスト開口孔底部に金属微粒子が拡散できず、充填率が著しく低くなる。
【0034】
(5) レジスト剥離:図2(e)
専用レジスト剥離液に工程(4)が終了した基板を浸漬し、レジストを溶解除去する。この際、レジスト上の吸着している金属微粒子及びリンカー分子Aがリフトオフにより除去され、基板表面に共有結合した表面反応分子Aからなる微粒子固定ドットとその上に固定された金属微粒子だけが残る。この段階で重要なことは、ドットパターン上の金属微粒子が、各サイトに一個づつ固定されていることと、ドットパターンへの金属微粒子固定充填率が高いこと、ドット外のエリアには金属微粒子の残留付着が無いことである。
【0035】
ここでは、工程(4)の金属微粒子の固定後に工程(5)のレジスト剥離を行っているが、工程(5)のレジスト剥離を行った後に工程(4)で金属微粒子を固定しても良い。すなわち、レジストを剥離し、リンカー分子層のみがドット状に並んだ表面と、金属微粒子溶液を反応させて、金属微粒子を固定してもよい。また、ここでは、工程(3)でリンカー分子を結合させてから工程(4)で金属微粒子の固定を行ったが、レジスト開口パターンに工程(4)で金属微粒子をキャピラリーフォース等を用いて埋め込んだ後に、工程(3)でリンカー分子を結合させても良い。
【0036】
(6) リンカー分子層の形成II(微粒子固定ドット外表面):図2(f)
基板表面のうち、金属微粒子が固定されていない表面に、後のプロセスで生体分子やdNTPといった試薬が非特異的に吸着する可能性が高い。この非特異吸着は、デバイスとして用いた場合のノイズとなるため、徹底的に防止する必要がある。このため金属微粒子固定ドットパターン以外のエリアに吸着阻害分子を固定し、対策する。図2(f)は、この目的のために、まずリンカー分子Bを形成した様子を示す。ここで、金属微粒子固定ドットに用いた、アミノ基やチオール基を含むシランカップリング剤を用いると、既に表面に固定されている金属微粒子表面に対しても反応するために、最終的な金属微粒子表面へのプローブ固定の障害にもなりかねない。そこで、ここでは金属微粒子とは相互作用せず、基板表面とだけ反応するリンカー分子が望ましい。このような物質として、例えばエポキシ基やカルボキシル基を持つシランカップリング剤が有望である。
【0037】
(7) 吸着阻害分子の形成I(吸着阻害分子C):図2(g)
工程(6)で形成したリンカー分子Bの上に、生体分子の非特異吸着を防止する分子として吸着阻害分子Cを結合させる。リンカー分子Bが、上記エポキシ基を持つシランカップリング剤の場合は、エポキシ基やカルボキシル基と反応するアミノ基や水酸基等を持つ吸着阻害分子が有効であり、具体的には、アミノ基を末端にもつ低分子量のポリエチレングリコール(PEG)や水酸基を持つカルボキシメチルデキストラン(CM-Dextran)などが使える。ただし、リンカー分子Bのみで充分非特異吸着を防止できる場合には、吸着阻害分子Cは必ずしも必要ではない。
【0038】
ここまでの工程で作成した基板には、電子線リソグラフィによって高精度に形成された微粒子固定ドットの格子状パターン(グリッドパターン)の上に金属微粒子が一個ずつ固定されており、かつ微粒子固定ドット以外のエリアには非特異吸着を防止する吸着阻害分子がコーティングされている。ここまで出来上がった基板を、金属微粒子グリッドアレイ基板と呼ぶことにする。
【0039】
(8) 単一プローブ分子の固定工程:図2(h)
金属微粒子グリッドアレイ基板に固定された金属微粒子上に、この金属微粒子と結合することができる官能基を持つプローブ分子を反応させ、金属微粒子上にプローブ分子を一個だけ固定することがこの工程の目的である。図2(h)は、プローブ分子をPとして、基板表面の様子を示す。
【0040】
ここで、金属微粒子として金ナノ粒子を用い、プローブ分子として、チオール基を5’末端に持つプローブDNAを用いた場合について説明する。金ナノ粒子を均一にランダム分散固定し、上記(6)(7)に示した工程で非特異吸着を防止する吸着阻害分子がコーティングされた基板を用い、この基板に対してチオール末端基付きDNAを反応させると、チオールは金ナノ粒子表面だけに反応して吸着する。この吸着挙動を、吸着反応を計測できるSPR法(Surface plasmon resonance:表面プラズモン共鳴)を用いて定量化した。SPR法の測定感度では、金ナノ粒子1個当りプローブ分子1個のみ吸着した時の吸着量を測定するのは困難である。そこで、金ナノ粒子1個当りに1分子より多くのプローブ分子を固定させ、DNA溶液濃度と固定されたプローブ分子数の関係を測定したところ、単純なLangmuir型吸着曲線示すことが判った。この曲線から、金ナノ粒子一個当りプローブ分子一個だけを吸着するプローブ分子溶液濃度を推定することができる。この吸着挙動評価実験から推定されたプローブ分子の最適濃度に調整したプローブDNA溶液に、同じ直径の金微粒子を規則的な格子点に固定した金微粒子グリッドアレイ基板を浸漬し、所定温度で所定時間反応させた後、洗浄液で洗浄し、乾燥させた。
【0041】
この基板上で、各金ナノ粒子毎のプローブDNA分子の固定数を計測した。計測方法は単一分子蛍光測定であり、非特許文献6に開示された手法に則り、高感度イメージインテンシファイヤーとCCDカメラの組合せを用い、光学顕微鏡ステージ上で実行した。この方法により、サンプル基板表面の観測視野内に固定された個々の蛍光色素分子からの蛍光(単一分子蛍光)をリアルタイムで画像として検出することができる。ここで、単なるプローブDNAには蛍光性が無いため、検出ができない。そこで単一プローブDNA分子固定の立証のためには、金ナノ粒子上に固定したチオール末端DNAと完全相補的な配列を持つ蛍光色素付きのDNAをハイブリダイゼーションさせた状態で蛍光計測を実施した。また単一分子蛍光の立証のためには、チオール基を5’末端に持つとともに3’末端にCy3、Cy5、Rhodamine Bのような蛍光分子を持つプローブDNAを用いて実験してもよい。いずれにしても、単一分子蛍光の特徴である、蛍光強度の繰返し点滅と階段状の蛍光退色が確認できた。ここで階段状の蛍光退色とは、蛍光検出時に蛍光強度をモニターし続けると、蛍光色素が一個一個酸化分解するため、そのたび毎に蛍光強度が階段状に低下する現象である。単一蛍光色素分子の蛍光を観測している場合には、分子の分解とともに蛍光が一挙に消えることで確認できる。以上の計測方法を用いることによって、金ナノ粒子グリッドアレイ表面の金ナノ粒子上に単一のプローブDNA分子を固定する最適プロセス条件(プローブDNA溶液濃度・反応温度・反応時間)を決定した。
【0042】
プローブDNAを溶解させる溶液としては、リン酸バッファなどの中性付近の水溶液を用いることができる。この溶液にプローブDNAを溶解させる。この時のプローブDNAの濃度は、例えば従来のDNAチップでは通常、0.5μM−100μMであるが、本発明のような金ナノ粒子グリッドアレイ基板上の金ナノ粒子に単一プローブDNAを固定する場合は、プローブDNAの濃度は数nM以下程度が最適である。なおプローブDNAの最適濃度は、固定表面の状態に依存し、具体的には金微粒子のサイズ・固定密度(グリッドアレイピッチ)によって変動するため、金微粒子グリッドアレイの設計に応じてそれぞれプローブDNA最適濃度を決定する。また、反応時間は、充分反応が平衡に達するまでの時間よりも長い時間とした。
【0043】
担体としてSiウェハやガラス基板を用いた金ナノ粒子グリッドアレイ基板の場合、基板の所望の位置に、プローブDNAを溶解した反応液を所望のサイズにスポッティングできる。この時、基板に多種類のプローブDNAをスポッティングすることが可能であり、こうすると例えばDNAマイクロアレイとして使える。反応温度は、通常、25℃から40℃の範囲である。また、反応時間は、通常、2時間から24時間の範囲である。反応させる時に溶液が乾燥しないよう、充分湿度を保った環境で反応させる。
【0044】
金とチオール基は結合し易いため、チオール基を末端に持つプローブDNAが金ナノ粒子上のみに固定される。金ナノ粒子が固定されていない領域(金微粒子固定ドット外のエリヤ)は、吸着阻害分子Cで覆われているため、プローブDNAはほとんど吸着しない。以後、吸着阻害分子でコーティングすることをブロッキングと呼ぶ。
【0045】
ここでは、金属微粒子を基板に固定した後に、金属微粒子が固定された部分以外の基板表面をブロッキングし、その後にプローブ分子を固定したが、金属微粒子にプローブ分子を固定した後に、この金属微粒子を表面に固定し、その後に金属微粒子が固定された部分以外の基板表面をブロッキングしても良い。
【0046】
(9) 吸着阻害分子の形成工程II(金属微粒子表面):図2(i)
金属微粒子表面で、プローブDNAが固定されなかった領域は、検体の生体分子を吸着させる可能性がある。よって、このプローブDNA固定部以外の金属微粒子表面をブロッキングする。図2(i)は、吸着阻害分子Dを用いて、金属微粒子表面をブロッキング処理した後の基板表面の様子を示す。
【0047】
ここでは、金属微粒子として金ナノ粒子を用いた場合について説明する。金と反応し易く、かつ生体分子を吸着し難いブロッキング剤として、1−メルカプトヘキサノール、2−メルカプトエタノール等を用いることができる。これらのブロッキング剤を溶解した水溶液と担体表面を反応させ、ブロッキング材料を固定する。
【0048】
反応温度は、通常、4℃〜35℃の範囲であり、反応時間は、通常、0.5時間〜10時間である。この反応では、水溶液中のブロッキング剤の濃度が高い場合、ブロッキング剤が金ナノ粒子と反応し金ナノ粒子を覆うことで、金属微粒子と担体の間の結合力を弱める。その結果、金属微粒子が担体表面で拡散し金属微粒子が表面で凝集する。したがって、ブロッキング剤反応溶液の濃度を100μM以下の範囲とした。
【0049】
ここでは、金属微粒子にプローブDNAを固定した後に吸着阻害分子Dを固定したが、プローブDNAと吸着阻害分子Dを同時に固定しても良い。すなわち、プローブDNAを溶解させた溶液に吸着阻害分子Dを混合し、その溶液と金属微粒子を反応させても良い。
【0050】
ここで、金ナノ粒子グリッドアレイ基板において、金ナノ粒子上に固定したプローブ分子がプローブDNAであるアレイ基板を、DNAマイクロアレイとして用いる場合について、以下(10)(11)で説明する。
【0051】
(10) DNAマイクロアレイのハイブリダイゼーション反応評価工程:図3、図4
前述の(1)〜(9)に述べた工程を経て作成した金属微粒子グリッドアレイを基板とする生体分子検出素子表面に、蛍光修飾された検体試料溶液を反応させる。ここではプローブ分子としてプローブDNAを用い、検出用生体分子としても核酸を用いた場合について図3を用いて説明する。簡略化のためリンカー分子、吸着阻害分子などは図示を省略した。基板301上に金属微粒子302が等ピッチLで整列固定され、金属微粒子302上にプローブDNA303が固定されている。これに対して、蛍光分子305によって蛍光修飾されたターゲットDNA304が検体試料として供給された状況を図3(a)に示す。ここで、プローブDNAとターゲットDNAの塩基配列が完全に相補的である場合は互いに速やかに反応し、図3(b)のように相補的水素結合307で結合した2本鎖DNAを形成する。これがハイブリダイゼーション反応である。ここで重要なことは、個々のプローブDNAは均等な環境条件にありかつ一定距離で正確に分離・固定されているので、相互干渉することがない。このため、このような基板で構成した生体分子検出素子では、反応時間や反応効率のバラツキが発生しにくい。
【0052】
一方、図4に示すような従来型の生体分子検出素子の表面では、図4(a)のように、プローブDNA(402)の固定密度がランダムであり、基板401の面内にプローブ固定の粗密が存在する。このため、同じハイブリダイゼーション反応を実行すると、ターゲットDNA(403)との反応の速いものと遅いものが共存し、図4(b)、図4(c)のようにハイブリダイゼーション反応の進行度のバラツキが発生しやすい。
【0053】
以上のことから、本発明の生体分子検出素子を用いると、目的の表面反応が均一かつ効率よく進行するので、検出データの精度・再現性が大幅に向上する。
【0054】
なお、ハイブリダイゼーション反応の具体的条件としては、検出用の蛍光修飾された核酸を、界面活性剤を添加したSSC(Standard Saline Citrate)溶液に溶解し、生体分子検出素子表面にこの溶液を接触させる。溶液中の核酸量は0.1amol〜1nmolである、反応温度は、通常、25℃−60℃、反応時間は、通常、1時間〜24時間である。この条件で、検出用の核酸がプローブDNAの配列と完全相補的であった場合、速やかに反応して相補的水素結合により形成された2本鎖DNAを生成する。DNAマイクロアレイの場合には、基板表面の多数のスポットにおいて、多種類の配列のプローブDNAを検体試料とハイブリダイゼーション反応させ、各スポットからの蛍光強度をスキャナーで計測し、データ化する。
【0055】
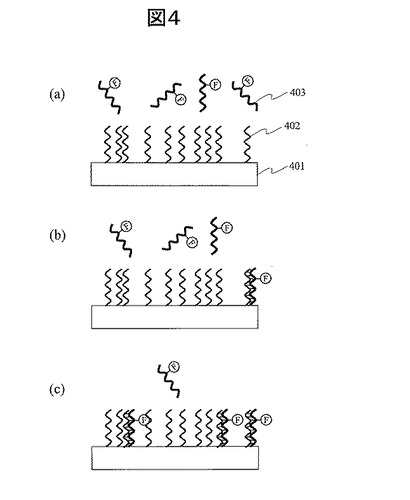
(11) 蛍光増強:図5
本発明では、金属微粒子をプローブ分子の固定場に用いることで、もう一つの特徴である蛍光増強現象が利用できる。本発明において、蛍光増強現象が発現する様子を図5で説明する。図5は、図2で説明した本発明の生体分子検出素子の製造方法によって作成した素子を示しており、図2(i)においてPで表記したプローブ分子としてDNAを想定したものである。図5(a)は、基板501上の金属微粒子に固定化されたプローブDNA(502)と蛍光分子504で蛍光修飾されたターゲットDNA(503)とのハイブリダイゼーション反応前の状態であり、図5(b)は反応後の状態を示す。この図は、完全相補鎖DNA同士の反応として描いてある。
【0056】
金属微粒子固有の局在プラズモン共鳴により、蛍光強度が増強される。まず局在プラズモン共鳴と蛍光増強について説明する。この現象については非特許文献7に詳しい記載がある。金属微粒子は、光が入射されると、金属微粒子内の自由電子が分極し振動する。この金属微粒子内の自由電子の振動と、入射光の振動電場が共鳴することを局在プラズモン共鳴と呼ぶ。局在プラズモン共鳴が起こると、金属微粒子表面での電場強度が入射光の電場強度に比べて数桁大きくなる。次に、上述の蛍光増強について、その二つの要因を説明する。蛍光増強する要因の一つは、蛍光分子の量子効率の向上である。蛍光分子の近傍に金属微粒子が存在する場合、蛍光分子のエネルギー吸収過程で、局在プラズモン共鳴によって金属微粒子近傍の電場増強効果による吸収遷移が生じる。更に、発光過程で、金属微粒子が存在すると発光過程の速度が加速される。したがって、吸収遷移と発光が増すことから、蛍光分子の量子効率が上がる。ただし、量子効率は1を超えることはないため、量子効率1を持つ蛍光分子では、金属微粒子による量子効率の増加は期待できない。しかし、実際にバイオセンサで用いられる蛍光分子は、量子効率が0.04−0.3程度のものが多く、これらの蛍光分子に対して金属微粒子による量子効率の向上が期待できる。
【0057】
第二の要因は、金属微粒子による光散乱強度の増加である。局在プラズモン共鳴により金属微粒子の分極率が増加し近傍の電場が増強されると、金属微粒子からの散乱光強度も増強される。これは、散乱光強度が金属微粒子の分極率の2乗に比例するからである。散乱光強度が増加すれば、蛍光分子を励起させるための入射エネルギー密度が増加し、従って蛍光発光強度も増加する。
【0058】
これらの蛍光増強効果は、金属微粒子と蛍光分子の距離が約5nmから約100nmの間で見られる。蛍光増強効果が現れる領域を近接場506として図5(b)に示した。プローブ分子として、金属微粒子に固定されたプローブ分子端と、プローブ分子に修飾された蛍光分子間のプローブ分子に沿った距離dが、蛍光増強効果が得られる長さである5nmから100nmの範囲に収まる場合、検体分子と反応して相補的水素結合505した後に、蛍光増強効果を利用することができる。
【0059】
以上のメカニズムによって、金属微粒子をプローブ分子の固定場に用いた場合には、蛍光修飾された検体分子を超高感度に蛍光検出することができ、生体分子検出素子としての大幅な感度向上が達成できる。
【0060】
次に、前述の(1)〜(9)に述べた工程を経て作製した金属微粒子グリッドアレイ基板をDNAシーケンシングに用いる場合について説明する。
【0061】
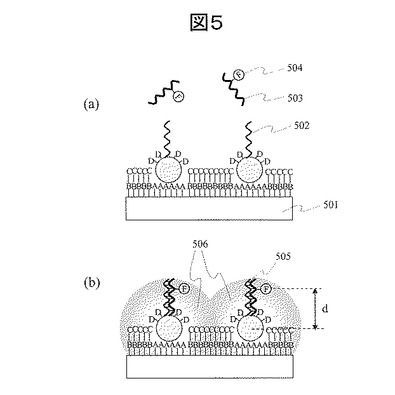
(12) DNAシーケンシング用解析対象分子固定:図6
まず、前述の(1)〜(9)に述べた工程によって、配列解析対象となる単一分子DNA604を固定するためのプローブ分子603を基板601上に金属微粒子602を介して配列させる。次に、配列解析対象の単一分子DNA604をプローブ分子と1対1で反応させて基板に固定する。解析対象の単一分子DNAをプローブ分子と反応させる際の条件として、単一分子DNAを、NaCl等の塩を含む溶液に溶かし、この溶液をプローブ分子を固定したアレイ基板表面と接触させる。反応温度は、通常、20℃〜80℃、反応時間は、通常1時間〜24時間程度である。解析対象の単一分子DNAとプローブ分子を反応させるために、プローブ分子は単一分子DNAを固定するための反応サイトを持つ必要がある。例えば、解析対象の単一分子DNAがAAAAAAAAAといったPolyA配列を持つ場合、これと相補的な配列であるTTTTTTTT等のTが連続したPolyT配列を有するプローブ分子を使用する。
【0062】
次に、図6に示した金ナノ粒子602グリッド配列のピッチLの大きさについて述べる。DNAシーケンシングでは、背景技術で述べたように、蛍光強度を測定することによってDNA配列を読み取る。シーケンシング時には、固定されたある単一分子からの蛍光と、その分子と隣接する単一分子DNAからの蛍光を独立して測定する必要がある。したがって、ピッチLは、蛍光読取光学系の解像度と同等あるいはより大きい値とする必要がある。また、同じ理由によってCCDカメラ読取の1画素のサイズよりも大きい値とする必要がある。
【0063】
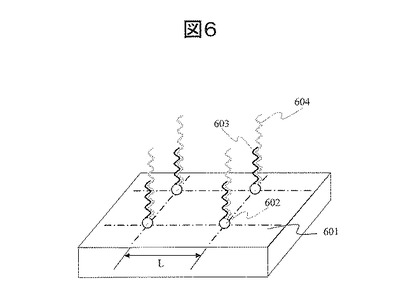
(13) シーケンシング:図7
図7に示すように、プローブ分子702をプライマーとして、ポリメラーゼ704によるポリメラーゼ反応により、プローブ分子702の先端にヌクレオチド705を一塩基伸長させる。この反応時には、ヌクレオチドを、例えば、塩化マグネシウム等の塩を含む溶液に溶解し、この溶液を基板と接触させる。酵素であるポリメラーゼの失活を防止するため、ジチオスレイトール(DTT)やグリセロール、界面活性剤等を上記溶液に混合しても良い。図7に示すように、伸長したヌクレオチドには蛍光分子706を結合している。この蛍光分子からの蛍光を読み取ることで、ヌクレオチドが伸長したか否か、あるいは、伸長したヌクレオチドの種類、例えば、A,T,C,Gを判別する。
【0064】
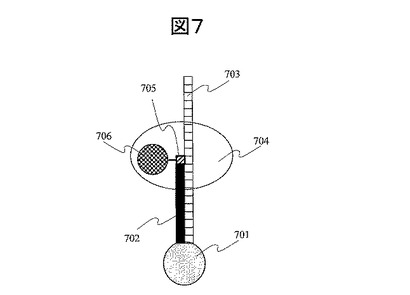
図8にシーケンシング読取システムを示す。このシステムでは、励起レーザ光801を基板803の裏面から石英プリズム802を通して全反射条件で入射させる。一塩基伸長時に、伸長したヌクレオチド808に結合した蛍光分子809を、全反射条件で入射したレーザ光のうち単一分子DNA806が固定された側に染み出したエバネッセント光によって励起する。生じた蛍光810を、基板上面に載置した高感度CCDカメラ811によって測定する。ヌクレオチドが結合された後、例えば、ポリメラーゼによって取り込まれたヌクレオチドのリン酸基が切断される。蛍光分子がヌクレオチドのリン酸基の末端に結合している場合には、リン酸基の切断と共に、蛍光分子が解析対象の単一分子DNA806部から離れ除去される。除去された後に、別種類のヌクレオチドを反応させる。例えば、まずヌクレオチドAを含む溶液と基板を接触させ、ポリメラーゼ反応によってAが一塩基伸長した場合には、上記の読取システムによって蛍光が検出される。ヌクレオチドAが一塩基伸長しなかった場合には蛍光は検出されない。この測定結果から、蛍光が検出されたグリッドサイトでは、解析対象の単一分子DNAがAと相補的であるTを解析対象部分に持つことがわかる。一方、蛍光が検出されなかったグリッドサイトでは、解析対象部分にTを持たないことがわかる。次に例えばヌクレオチドCを含む溶液と基板を接触させた後に、蛍光を検出し、各グリッドサイトにおいて、単一分子DNAの解析対象部分にCと相補的なGを有するか否かを判別する。次に、ヌクレオチドTあるいはGを反応させる。これらの作業を繰り返すことで、各グリッドサイトに固定した単一分子DNAの配列を読み取る。
【0065】
ここで重要なことは、本発明の個々の単一分子DNA806は一定距離で分離・固定されているので、検出した蛍光信号が、隣接する単一分子DNAから発せられる信号と相互干渉することがない。このため、例えば図9に示すように、蛍光を検出するCCDカメラの一画素903の大きさと金ナノ粒子のグリッド格子の大きさ904が等しい場合、グリッドサイト毎に、独立に、蛍光強度を測定できる。すなわち、グリッドサイト毎に、独立に、DNAの配列を読むことができる。したがって、正確性高くDNA配列を読むことができる。画素数をより細かくし、例えば4画素で一つのグリッドサイトからの蛍光信号を読み取る、あるいは、9画素で一つのグリッドサイトからの蛍光信号を読み取っても良い。更には、DNAの固定サイトとなる金属微粒子902効果で、蛍光強度が増強される。その理由については前述の(11)に述べた通りである。通常、単一分子蛍光からの蛍光を測定する際には、高感度に測定するために、励起光のパワー密度を上げ、検出系に光子を増幅するデバイスを用いる等の工夫が必要であり、蛍光励起および検出系が大掛かりで複雑な系になる。しかし、本発明の蛍光増強効果を用いることで、蛍光検出系をコンパクトに設計することが可能になる。
【0066】
一方、図10に示すように、解析対象の単一分子DNA1004が基板1001にランダムに固定されている場合、単一分子DNA間の距離を制御することができないため、一画素1005で2つ以上の単一分子DNAからの蛍光を計測する場合もある。この場合、2つのDNAからの信号を分離することが困難であり、結果として誤読する、あるいは解析できないDNAが存在することになる。更には、単一分子DNA(1004)がランダムに固定される場合、解析の開始時点で、解析対象のDNA(1004)の位置が不明であるため、解析開始前に位置確認を行う必要がある。例えば、蛍光分子を結合させた単一分子DNAを固定した後に蛍光マッピングを行うことで、単一分子DNAが固定された場所を特定し、システムに記憶させるなどのプロセスが必須となる。また、金属微粒子による蛍光増強効果を利用できないため、蛍光検出系が大掛かりで複雑な系になる。
【0067】
本発明の金属微粒子グリッドアレイ基板を用いれば、解析の正確性が各段に向上し、また、解析不可能な無効なDNAを生じさせることなく、全てのDNAの配列を読み取ることができる。更には、固定された解析対象DNAの場所(座標)が予めわかっているため、場所を特定するためのプロセスを省略できる。
【0068】
本発明では、リソグラフィのプロセスを用いて担体基板表面にリンカー分子からなる規則的なドットパターンを形成し、このリンカー分子が保有する活性基と金属微粒子との結合を利用して、まず微粒子金属グリッドアレイを形成した。リソグラフィの方法としては電子線描画に限ったものではなく、電子線リソグラフィの代わりに近接場リソグラフィを用いることも可能である。但しその場合には、メンブレンマスクを電子線リソグラフィによって作製し、このマスクを用いたコンタクト露光によってレジスト加工を実行する。この場合、同じパターンであれば繰り返しハイスループットで作製できるので、量産性の高いプロセスを達成できる。また、リンカー分子の規則的ドットパターンをリソグラフィを用いることなくナノコンタクトプリント技術やナノインプリント技術を用いて行うことも可能である。
【0069】
次に、本発明を実施例により詳細に説明するが、特にDNAマイクロアレイおよびDNAシーケンサに適用した例について説明する。なお本発明は、下記の実施例に限定されるものではなく、核酸、タンパク、糖鎖などのあらゆる生体分子検出素子の性能向上に寄与する基本技術である。
【実施例1】
【0070】
(工程1)基板表面へのポジ型電子線レジストの塗布工程:図11(a)
担体基板1101として、平坦性の高い熱酸化膜100nm付き4インチSi基板を用いた。基板を0.1wt%のNaOH水溶液で洗浄し、更に0.1wt%のHCl水溶液で洗浄し、純水で充分すすいだ後に乾燥した。ここで、レジストとして用いたポジ型レジストは、主鎖切断型のレジストである。レジストをアニソールで希釈し、スピンコーターを用いてコーティングした。コーティング後、溶剤を除去するためにN2フロー中 180℃、20minベークした。本実施例では、レジスト膜厚は充分薄く、かつEB加工による膜減りが見られない60nm膜厚(レジスト:アニソール=1:3希釈)とした。また、EB描画中の基板の帯電を防止するため、塗布したレジスト膜1102の上に導電性ポリマー(ポリイソチアナフテンスルホネート)溶液をコーティングした。この溶液は、導電性ポリマーをコロイド粒子にし、界面活性剤を用いて分散させたものである。同じスピンコーターを用いて導電性ポリマー1103をコーティングした後、溶剤を除去するためにN2フロー中で100℃、10minベークした。
【0071】
(工程2)電子線描画及び現像による開口形成:図11(b)
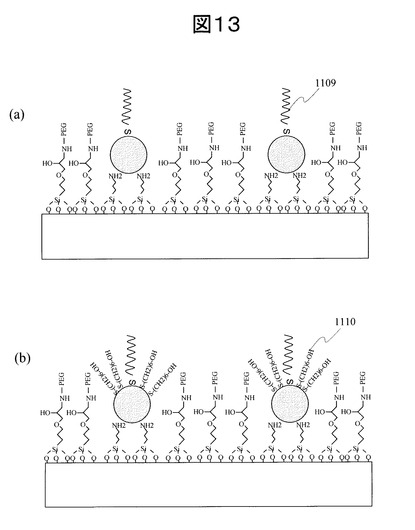
電子線走査フィールド内ではx方向、y方向それぞれ60,000ステップに分割され、各ステップにてスポット径約2〜3nmの電子線をパルス照射する。どのステップ位置で電子線照射をするかをCADソフトを用いて指定することにより、所望のパターンを形成する。電子線加工開口径は、固定させる金属微粒子と同等である必要がある。本実施例では、安定に固定でき、かつ蛍光増強効果を得ることができる30nmの金ナノ粒子を用いた。したがって、EB開口径を40nmφとした。本実施例で用いた描画パターンを図14に示す。40nmφの電子線照射領域が格子状にそれぞれ100nmピッチで並んだパターンとした。この描画パターンを200μm角のエリアに加工した。またこの200μm角の加工エリアを基板上に100点作成した。加工エリアは基板上に2mmピッチで格子状に並べた。
【0072】
電子線ドーズ方式として、電子線を同一箇所に連続照射し、レジスト横方向に徐々に染み出したエネルギーによってレジストポリマー主鎖を切断する照射法を用いた。
フィールドサイズは150μm角とし、電子線ビーム電流5×10-11Aで電子線描画を行った。
【0073】
電子線照射によって主鎖が切断された部分を溶解する現像液として、n-アミルアセテートを用いた。最表面に導電性ポリマーが塗布されているため、まずこれを純水で除去し、次に、25℃、15秒n-アミルアセテート溶液に浸漬して現像後、リンス溶媒Methyl isobutyl ketone 89%/ Isopropyl alcohol 11%溶液でリンス洗浄した。これら一連のプロセスによって、50nmφの金ナノ粒子固定用の開口孔1104を加工した。開口孔は電子顕微鏡で確認した。
【0074】
(工程3)アミノ化層の形成(金属微粒子固定ドット):図11(c)
電子線レジスト開口パターンを形成した基板を、シランカップリング剤である3-アミノプロピルトリメトキシシラン(3-aminopropyltrimthoxysilane, APTMS)溶液に浸漬した。なおAPTMSを希釈する溶媒としてメタノールを用いた。反応温度は室温、反応時間は5分とし、反応後、メタノールで充分洗浄し乾燥した。この結果、開口孔底にAPTMSが吸着する。この基板を80℃2hrアニールした。このアニールプロセスによって、APTMSと開口孔底のSiO2の間にシロキサン結合が形成され、開口孔底が安定にアミノ化される。一方、レジスト上にもAPTMSが吸着するため、レジスト上にもAPTMSが残留する。
【0075】
(工程4)金ナノ粒子の固定:図11(d)
次に、開口孔底部をアミノ化した基板に、直径が30nmの金ナノ粒子クエン酸溶液を作用させた。なお、金ナノ粒子1106の濃度は、約0.4nMである。また、反応温度は室温であり、反応時間は20時間である。この時、金ナノ粒子表面はクエン酸で覆われ負電荷を持つ。一方、アミノ化された表面は正電荷を持つため、金ナノ粒子は表面のアミノ基1105と引き合うため、金ナノ粒子1106が固定される。金ナノ粒子を反応させた後、充分純水洗浄した後に乾燥させた。
【0076】
(工程5)レジスト剥離:図12(a)
基板をジメチルアセトアミドに3分浸漬させ、レジストを除去した。この際、レジスト上に付着した金ナノ粒子が基板に再付着しないよう、充分量のジメチルアセトアミドを使用した。ジメチルアセトアミドに浸漬後、エタノール洗浄と純水洗浄を充分に行った。このプロセスによって、レジスト上に吸着された金ナノ粒子もリフトオフされ、開口部に固定された金ナノ粒子のみが残留する。従って、金ナノ粒子30nmを格子状に配列することができた。
【0077】
(工程6)吸着阻害分子層の固定:図12(b)
金ナノ粒子30nmを格子状に配列させた基板をエタノールに浸漬し乾燥後、エポキシ基1107をコーティングした。Diisopropylethylamine 0.8%が添加された3-Glycidoxypropyltrimethoxysilane(GOPS) 7.7%の脱水トルエン溶液中で、上記基板を85℃下で2hr反応させることによってエポキシ基を金ナノ粒子が固定されていない領域にコーティングした。反応後、脱水トルエン及びエタノールで洗浄し乾燥させた。
【0078】
(工程7)PEGの固定:図12(c)
工程6で固定したエポキシ基1107とアミノ基を末端に持つPEG(分子量2k)1108を弱アルカリ下で反応させた。具体的には、4mMアミノ化PEG溶液に溶解し、基板を室温で1hr浸漬した。その後、純水で充分洗浄し乾燥させた。この工程によって、図7(c)に示すように、金ナノ粒子が固定されていない領域がPEG1108でブロッキングされた。
【0079】
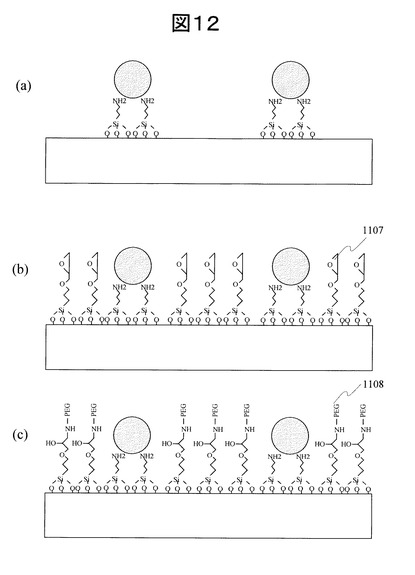
(工程8)単一プローブDNAの固定:図13(a)
工程6及び7でエポキシ基とPEGをコーティングした基板をエタノール洗浄した後、基板表面とチオール基を末端に持つ50mer 1本鎖プローブDNA溶液を金ナノ粒子コーティング領域であるそれぞれ250μm角エリアにスポッティングした。用いたDNAの塩基配列は1種類であり、1種類のプローブDNA(1109)を100点にスポットした。これは、各スポットでの検出バラツキを調べるためである。その5’末端側からの塩基配列を下記に示す。
AGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG
【0080】
用いたプローブDNA反応溶液は、チオール基を5’末端に持つ50mer 1本鎖DNA1 nMを1M Phosphate buffer (pH 6.7)に溶解した溶液である。この濃度は、「発明を実施するための最良の形態」の(8)で述べたSPR法による吸着挙動評価実験から推定し、単一分子蛍光測定によって単一分子固定を確認できた濃度である。このプローブDNA反応溶液と格子状に配列させた金ナノ粒子を室温24hr、100%湿度下で反応させた後、2×SSC、0.1%SDS溶液ですすぎ、純水で2回洗浄した後に乾燥した。
【0081】
(工程9)金ナノ粒子表面メルカプトヘキサノールブロッキング工程:図13(b)
メルカプトヘキサノール水溶液1μMを作成し、この水溶液中にプローブDNAが固定された基板を浸漬した。反応温度は室温、反応時間は1時間とした。反応させた後に純水洗浄を行い、デシケータ中で減圧乾燥し、図13(b)に示すように、金ナノ粒子のプローブDNA固定部以外の表面をメルカプトヘキサノール1110でブロッキングした基板を得た。
【0082】
(工程10)ハイブリダイゼーション
プローブDNAを固定した基板に、プローブDNAと完全相補的な配列をもち、3’末端に蛍光分子Cy3を標識した一本鎖のターゲットDNAをハイブリダイゼーションさせた。ハイブリダイゼーション溶液として5×SSC(Standard Saline Citrate)と0.5%SDS溶液Sodium Dodecyl Sulphate)の混合液を用い、ターゲットDNA量1fmolを42℃で20時間ハイブリダイゼーションさせた。その後、2×SSC、0.1%SDS溶液、2×SSC溶液で洗浄を行い、乾燥した。乾燥させた基板表面に対し、蛍光スキャナーを用いて励起光を入射し、表面からの蛍光強度を測定した。この蛍光強度は、ハイブリダイゼーションによって反応したターゲットDNAの量に比例するものである。
【0083】
一方、Si基板上に一般的な従来からある1本鎖DNAの固定方法で、プローブDNAを固定した基板を作製した。Si基板を上記(工程1)に示した方法と同様に洗浄した後に、(工程3)に示した方法でシランカップリング剤であるAPTMSを全面にコーティングし基板表面をアミノ化した。その後に、末端のアミノ基に、イソチオシアナート基を有するPDC(phenylenediisothiocyanate)を反応させた。更に、このイソチオシアナート基とアミノ基を5’末端に持つプローブDNAを反応させ固定した。用いたプローブDNAの配列は、(工程8)で示したDNA配列と同じである。プローブDNAを反応させる際に用いた反応溶液は、弱アルカリの炭酸バッファにプローブDNAを1μM溶解させた溶液である。この溶液をイソチオシアナート化した基板にスポットした。スポット点数は100点であり、スポット径は200μmφである。室温24hr、100%湿度下で反応させた後、基板を2×SSC、0.1%SDS溶液ですすぎ、純水で2回洗浄した後に乾燥した。この基板を前述の(工程10)に示した方法で蛍光分子付きのターゲットDNAとハイブリダイゼーションさせた後に、蛍光強度を測定した。測定した蛍光強度はハイブリダイゼーション反応したターゲットDNA量を表すものである。
【0084】
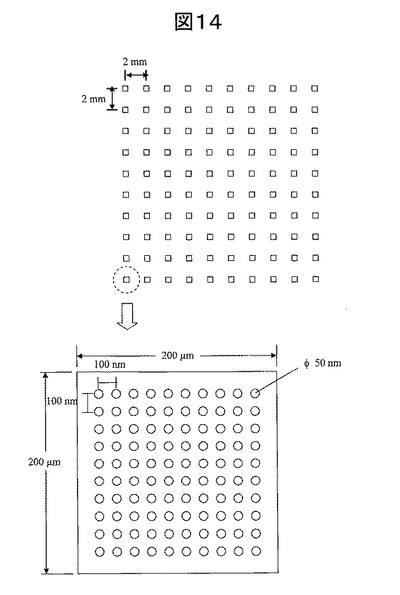
金ナノ粒子を格子状に配列させ単一プローブDNAを金ナノ粒子上に固定して形成したグリッドアレイの100スポットの蛍光強度平均値およびバラツキと、従来の固定方法でプローブDNAを固定したアレイの100スポットの蛍光強度平均値およびバラツキを図15に示す。蛍光強度は、蛍光スキャナーを用いて励起光を入射し、表面からの蛍光強度を測定した。励起レーザ光の波長は532nmであり、このレーザ光をスポットエリアでスキャンさせた。発生した蛍光を光電子倍増管で検出した。
【0085】
本発明の金ナノ粒子グリッドアレイは、従来方法で作製したアレイよりも蛍光強度のバラツキが±50%以上から±5%未満へと大幅に低減する。これは、プローブDNAが100nmピッチで孤立固定されているため、個々のプローブDNAが干渉し合うことなく自由運動しており、ターゲットDNAとのハイブリダイゼーション反応速度や反応効率がプローブDNA間及びスポット間で同等だからである。またプローブDNAの固定密度が均質であり、ターゲットDNAの供給量も場所による差が見られない。一方、従来の基板では、プローブDNAの固定密度が面内でばらついているため、プローブDNA同士の干渉やターゲットDNAの供給量のバラツキがあり、これがハイブリダイゼーション反応速度や反応効率の面内バラツキを発生させる。
【0086】
また、本発明の金ナノ粒子グリッドアレイで検出した蛍光強度は、従来のアレイで検出した蛍光強度よりも大きくなった。金ナノ粒子グリッドアレイの場合、プローブDNA固定密度は100分子/μm2である。これに対し従来のアレイの場合、プローブDNA固定密度はX線反射率等の計測手段により測定すると約5,000分子/μm2と約50倍の固定密度を持つ。したがって、固定密度から考えると、従来アレイの方がより多くのターゲットDNAが反応し、蛍光強度も高いはずである。しかし、前述したように、金ナノ粒子グリッドアレイはプローブDNAが孤立しているため、従来アレイに比べてハイブリダイゼーション反応効率が高い。更には、金ナノ粒子近傍の近接場により、図5に示すように蛍光が増強される。50merDNAが2本鎖を形成した場合、その長さは約17nmである。金ナノ粒子近傍では、近接場光はナノ粒子表面から粒子径程度まで届く。したがって、金ナノ粒子30nmの表面から17nmの距離では、蛍光が増強される領域であると言える。以上の反応効率の上昇及び蛍光増強によって、金ナノ粒子グリッドアレイでは、DNA固定密度は従来型アレイよりも低いにもかかわらず、従来アレイよりもむしろ蛍光強度が高くなった。
【実施例2】
【0087】
DNAマイクロアレイの検出濃度限界を測定するために、実施例1の工程1から工程9と同様の方法で単一プローブDNAを固定した金ナノ粒子グリッドアレイを得た。一方で、実施例1に示した方法と同様に従来のアレイを作製した。実施例1の工程10で示した方法と同様にハイブリダイゼーションを行った。
【0088】
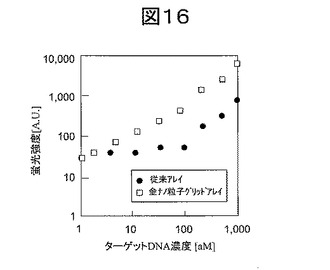
本実施例では、ハイブリダイゼーション時の蛍光分子付き完全相補的ターゲットDNA量を1amolから1fmolまで変化させた。それぞれのアレイにおける蛍光強度とターゲットDNA量の関係を調べた。その結果、図16に示すように、金ナノ粒子グリッドアレイは従来アレイに比べてターゲットDNA検出感度が向上した。従来のアレイでは、100amolが検出限界であったのに対し、金ナノ粒子グリッドアレイでは1amolが検出限界となり感度が大幅に向上した。これは、本発明の金ナノ粒子グリッドアレイでは、孤立プローブDNAの反応効率が向上し、また蛍光増強効果を得ることができるためである。
【実施例3】
【0089】
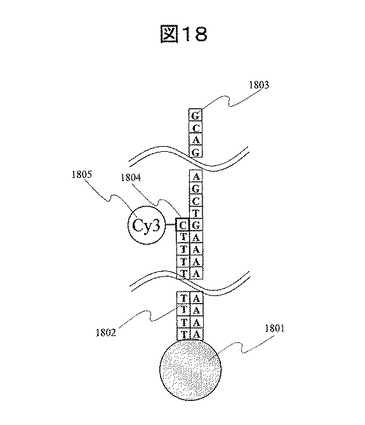
本実施例は、DNAシーケンシングに用いた例を示す。DNAシーケンシングを行うために、実施例1に示した工程1から工程8に従ってDNAシーケンシング基板を作製した。本実施例の場合、基板にSi基板ではなく石英基板を用いた。工程2の電子線描画及び現像による開口形成において、40nmφが格子状にそれぞれ1μmピッチで並んだパターンを作製した。このパターンを用いることで、金ナノ粒子を1μmピッチ格子状に並べた。作製した金ナノ粒子グリッドアレイ基板のSEM観察結果を図17に示す。30nm径の金ナノ粒子が1μmピッチで配列していることを確認した。蛍光強度を検出するCCDカメラの1画素を1μm角とすると、シーケンシング時に、一画素で、DNA一分子からの蛍光信号を読み取ることができる。画素数をより細かくし、例えば4画素でDNA一分子からの蛍光信号を読み取る、あるいは9画素でDNA一分子からの蛍光信号を読み取っても良いが、本実施例では1画素の大きさを1μm角とした。また、工程8で固定するプローブDNAとして、図18に示すように、3’末端にチオール基を持ち、かつT配列を20連続で持つ(TTTTT TTTTT TTTTT TTTTT TTTTT (PolyT 20mer))オリゴヌクレオチド1802を用いた。この場合、このオリゴヌクレオチド末端のチオール基と金ナノ粒子を結合させることによって、オリゴヌクレオチドを固定する。
【0090】
その後、工程10に従って、解析対象となるDNA(1803)をプローブDNAにハイブリダイゼーションさせる。具体的には、解析対象のDNAとして、5’末端からA配列を連続で持ち、下記の配列を持つDNA(1803)をハイブリダイゼーションさせた(図18参照)。
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG
【0091】
次に、プローブDNAである上記PolyT20mer配列をプライマーとして、解析対象DNAのシーケンシングを行った。作製した金ナノ粒子グリッドアレイ基板と、ヌクレオチド3リン酸のリン酸基末端に蛍光標識であるCy3(1805)を持つ4種類のヌクレオチド(1804)をポリメラーゼを用いて反応させた。反応時に用いた反応溶液は、10mM Tris-HCl、5mM MgCl2溶液である。ヌクレオチドの濃度は1μMである。ポリメラーゼ反応によって、DNAの塩基配列と相補的な塩基を持つヌクレオチドがDNAに結合する。本実施例の場合、ヌクレオチドCを溶解した溶液と基板を反応させた場合、ヌクレオチドCが結合する。ヌクレオチドCは解析対象DNAの配列中のAが連続した部分に隣接するGに結合する。図8に示した蛍光検出系を用いて、ポリメラーゼ反応過程における蛍光画像を取得する。蛍光の強度は基板上面から高感度CCDカメラで測定した。検出する際に蛍光分子の励起光を全反射条件で当てると、蛍光分子付きヌクレオチドC(1804)が結合したグリッドサイトから蛍光が検出される。ヌクレオチドがDNAに結合すると、ポリメラーゼによって取り込まれたヌクレオチドのリン酸基が切断される。これによって、そのリン酸基の末端に結合していた蛍光分子が測定対象DNA部から除去される。次に、ヌクレオチドAを溶解した溶液と基板を反応させた場合、ヌクレオチドCの結合点の隣であるTにヌクレオチドAが結合する。結合した際に、蛍光を検出することができる。次に、ヌクレオチドTを溶解した溶液と基板を反応させた場合、ヌクレオチドTはDNAに結合しない。DNA側の次の結合サイトはCであり、ヌクレオチドTとは相補的でないため、ヌクレオチドTが結合せずに蛍光が検出されないことを確認した。
【0092】
これらの手順を繰返し、各グリッドにおいて、同様にDNA配列を読むことが可能であった。すなわち、個々のDNA固定サイトからの蛍光信号を他DNAサイトからの信号と分離して検出でき、無効になるグリッドDNAを生じさせることなく、繰返しDNAの配列を読むことが可能であった。
【0093】
また、図18の状態を蛍光検出した時の結果を図19に示す。検出蛍光強度は、従来方法で測定した蛍光強度に比べて高くなった。これは金ナノ粒子による蛍光増強効果によって、蛍光強度が増加したためである。したがって、本発明の素子を用いることでより高感度にシーケンシングを行うことができる。あるいは、簡易な蛍光検出系を用いてシーケンシングを行うことができる。
【産業上の利用可能性】
【0094】
DNAやmRNAの定量解析を行うDNAマイクロアレイや、DNA又はmRNAの配列を読むDNAシーケンシング、また蛋白質を分析するプロテインチップ、及び解析対象となる蛋白質や糖鎖を解析前に固定させる前処理基板としても適用できる。
【図面の簡単な説明】
【0095】
【図1】本発明による生体分子検出素子の表面の基本構成を示す図。
【図2】本発明による生体分子検出素子の製造工程を示す図。
【図3】本発明による生体分子検出素子を用いたDNAハイブリダイゼーション反応をイメージした図。
【図4】従来型の生体分子検出素子を用いたDNAハイブリダイゼーション反応をイメージした図。
【図5】本発明による生体分子検出素子を用いたDNAハイブリダイゼーション反応における蛍光増強を説明する図。
【図6】本発明によるDNAシーケンシング用解析対象の単一分子DNAをグリッド配列固定した表面の基本構成を示す図。
【図7】DNAシーケンシング反応をイメージした図。
【図8】DNAシーケンシング検出系を示す図。
【図9】グリッド配列された1金属微粒子と1画素の関係を示す図。
【図10】従来型の生体分子検出素子を用いたDNAシーケンシング系をイメージした図。
【図11】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図12】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図13】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図14】本発明で用いた電子線描画パターンを示す図。
【図15】本発明で得られた金ナノ粒子グリッドアレイと従来型アレイの完全相補的なDNAをハイブリダイゼーションさせた時の検出蛍光強度と強度バラツキを示す図。
【図16】金ナノ粒子グリッドアレイと従来型アレイにおいて、完全相補的なターゲットDNAをハイブリダイゼーションさせた時の検出蛍光強度とターゲットDNA濃度の関係を示す図。
【図17】金ナノ粒子を格子状に固定した一例を示す図。
【図18】DNAシーケンシング配列とシーケンシング反応を示す図。
【図19】DNAシーケンシング時の蛍光検出結果を示す図。
【符号の説明】
【0096】
101:担体基板、105:金属微粒子固定ドット、106:金属微粒子、107:プローブ分子、201:担体基板、202:電子線レジスト、203:レジスト開口部、205:金属微粒子、301:基板、302:金属微粒子、303:プローブDNA、304:ターゲットDNA、305:蛍光分子、307:相補的水素結合、401:基板、402:プローブDNA、403:ターゲットDNA、501:基板、502:プローブDNA、503:ターゲットDNA、504:蛍光分子、505:相補的水素結合、506:近接場、601:基板、602:金属微粒子、603:プローブ分子、604:解析対象単一分子DNA、701:金属微粒子、702:プローブ分子、703:解析対象単一分子DNA、704:ポリメラーゼ、705:ヌクレオチド、706:蛍光分子、801:励起レーザ光、802:プリズム、803:基板、804:金属微粒子、805:プローブ分子、806:解析対象単一分子DNA、807:ポリメラーゼ、808:ヌクレオチド、809:蛍光分子、810:蛍光、811:CCDカメラ、901:基板、902:金属微粒子、903:一画素、904:グリッド格子、1001:基板、1003:プローブ分子、1004:解析対象単一分子DNA、1005:一画素、1101:担体基板、1102:電子線レジスト、1103:導電性ポリマー、1104:レジスト開口部、1105:アミノ基、1106:金ナノ粒子、1107:エポキシ基、1108:PEG、1109:プローブDNA、1110:メルカプトヘキサノール、1801:金ナノ粒子、1802:プローブ分子PoyT連続20配列、1803:解析対象単一分子DNA、1804:ヌクレオチドC、1805:蛍光分子Cy3
【技術分野】
【0001】
本発明は、生体分子をセンシングするための生体分子検出素子及びその製造方法並びに生体分子検出方法に関するものである。
【背景技術】
【0002】
近年、ヒトゲノムDNAの解読がほぼ終了したことにより、遺伝子の機能を解明する研究が盛んに行われている。そこでは、生体内の遺伝子やタンパク質を特異的かつ網羅的に検出することが必要であり、遺伝子・タンパク質検出技術の開発が世界的に進められている。一方で、生体内に進入した病原菌やウイルスを、遺伝子やタンパク質レベルで特定する技術も従来から検討されており、実用化されつつある。このような目的に応じて、特定の遺伝子やタンパク質等の生体分子を検出するための手段として、種々のバイオセンサが用いられている。最も一般的なバイオセンサの構造は、生体分子を捕捉するプローブ分子が固体表面上に固定されたものである。核酸を捕捉する場合には、プローブ分子として主に核酸が用いられ、タンパク質を捕捉する場合には、プローブ分子として主にタンパク質が用いられる。基板にプローブ分子を固定したバイオセンサのメリットは、同一の基板に、多種類のプローブ分子をスポッティングやインクジェットなどの方式を用いて固定できることである。このバイオセンサ基板を用いれば、多種類の生体分子に対する網羅的な解析を一度に迅速に行うことができる。そして基板表面を利用したバイオセンサの代表例が、DNAマイクロアレイやプロテインチップのような生体分子検出素子である。
【0003】
このような生体分子検出素子の製造方法には大別して2つの方法がある。一つは、フォトリソグラフィやインクジェットを用いて、基板上でヌクレオチドやアミノ酸を逐次的に反応させ、固定化することにより、一本鎖のDNAやたんぱく質といったプローブ用の生体分子をin-situ合成する方法である(特許文献1)。もう一つは、プローブ用生体分子をex-situで合成した後に、これらを基板上に固定化する方法である(特許文献2)。
【0004】
DNAマイクロアレイに代表される生体分子検出素子は、将来は癌等の病気の診断を含む医療診断に使われると予想されている。医療診断にDNAマイクロアレイを用いる場合には、マイクロアレイから得られるデータに対して高い信頼性が要求される。フォトリソグラフィやインクジェットを用いてプローブ生体分子を表面にin-situ合成する方法では、逐次的に結合させるヌクレオチドやアミノ酸の反応率が各ステップ毎に100%にはなり得ないため、スポット上の全てのプローブ分子の配列が厳密に設計通りであることを保証することが難しく、また操作ミスでヌクレオチドやアミノ酸の種類の取り違いや欠損部が生じた場合、それらを検査し、製造後に除去することが現状では不可能である。更に、長い塩基やたんぱく質を得るためには莫大な製造コストが必要になる。これに対し、DNAやたんぱく質をex-situで合成してから基板上に固定する方法では、合成した後に精製することで、欠損部等を持つ生体分子を事前に除去することができる。よって、純度の高いプローブ用生体分子を表面に固定することができ、信頼性の高いマイクロアレイを製造することが可能である。この方法では、多くの場合、反応活性な官能基を持つ生体分子と表面を反応させ、共有結合を作ることで固定化している。しかしながら、基板表面にDNAやたんぱく質といった分子量の大きい高分子を固定化させる際に、固定化されたプローブ生体分子の量・構造にバラツキが生じやすく、これがデータの再現性を低下させる要因になっている(Nature Vol.21, pp.5-9, 1999)。また、マイクロアレイを用いて生体分子を検出する際に、生体分子が基板表面に非特異的に吸着することや、固定化されたプローブ生体分子の密度が制御されていないために感度が低いことも定量的な解析を困難にする原因である(Nature Biotech. 19, p.342, 2001)。
【0005】
一方、最近、DNAマイクロアレイで行われているような遺伝子発現解析の精度の大幅向上を狙って、特許文献3ないし非特許文献3に示されているように、ポリヌクレオチドの単一分子アレイを用いた単一分子ベースのシーケンシング(SBS(Sequencing by Synthesis)法等)による遺伝子配列決定方法が開示されている。この方法では、基板表面に適当なプライマーで修飾された検体のポリヌクレオチドを固定し、これを鋳型としてポリメラーゼによる一塩基ずつの伸長反応を実行し、検体ポリヌクレオチドの相補鎖を形成させるものである。この一塩基伸長の個々のステップでは、異なる4種のヌクレオチドのそれぞれのプリン又はピリミジン、あるいは3リン酸のリン酸基の末端部にユニークな蛍光色素を導入しておき、伸長ステップ毎に単一分子蛍光検出を行うことで、導入されたヌクレオチドを識別する。このステップを繰り返し、単一ポリヌクレオチド固定サイトごとのシーケンスを読み取って、検体の配列情報を網羅的に取得する。ここでは、高いS/N比で検出し、シーケンシング時の配列決定正確性を向上させることが重要となる。この技術では、膨大な単一ポリヌクレオチド固定サイトからの蛍光の情報をCCDカメラで検出するため、そのCCDカメラの画素サイズに対応してポリヌクレオチド分子の平均的な固定密度を設定している。即ち、出来るだけ一個の画素で一個のポリヌクレオチド上からの蛍光信号を取り込むようにポリヌクレオチドの平均固定密度あるいは画素の解像度を調整している。一画素の大きさは検出光学系の空間分解能を考慮するとサブミクロン角以上となる。
【0006】
一個の画素で一個のポリヌクレオチド上からの蛍光信号を効率良く取り込むようにするために、下記のような試みが行われている。単一DNA分子やRNA分子を基板に固定し、その密度を1分子/μm2以下し、また固定密度を調整することによって、シーケンシング時の遺伝子配列決定の正確性を高める技術が特許文献4に示されている。また、特許文献5において、1分子をマイクロスフェアーを介して固定する方法が紹介されている。隣接する分子同士の分子間距離を充分に保つことで、例えば、1分子のシーケンシングを蛍光を用いて行う際、高い正確性を持って配列決定できるとしている。また、マイクロナノスフェアーであるコロイド粒子を格子パターン状に並べる手段として、リソグラフィ技術によって形成した微細開口パターンにコロイド粒子を落とし込んで微粒子を規則的なパターンに整列させる方法が非特許文献4に開示されている。更に、同一種類のプローブ分子群をマトリックス状に並べた基板が特許文献6,7に開示されている。一方、シーケンシング時の検出ノイズを低減するために、デバイス形成プロセスに適合し、かつ十分な吸着防止能を持った吸着阻害剤を領域選択的に形成する必要がある。負電荷を持つPolyacrylic acidでデバイス表面を覆い、dNTPの吸着を抑えた例が非特許文献3で紹介されている。
【0007】
次に、DNAを解析する手段であるマイクロアレイあるいはシーケンシングを用いて少量のサンプルを検出するためには、蛍光検出感度を上げることも必要である。蛍光検出感度を向上させるために、蛍光増強を用いる方法が非特許文献5に報告されている。ここでは、プローブ分子であるDNAを修飾した銀ナノ粒子を基板に固定し、蛍光標識された検体中の分子と反応させている。反応量を検出するために蛍光励起光を照射すると、銀ナノ粒子の自由電子が共鳴振動(局在プラズモン共鳴)を起こし蛍光が増強する。この現象を用いることで、感度を向上させることができるとしている。
【0008】
【特許文献1】米国特許第5,424,186号
【特許文献2】米国特許第5,700,637号
【特許文献3】米国特許第6,787,308号
【特許文献4】特表2002-531808号公報
【特許文献5】特表2002-521064号公報
【特許文献6】特開2001-33458号公報
【特許文献7】特開2002-235474号公報
【非特許文献1】Nature Vol.21, pp.5-9, 1999
【非特許文献2】Nature Biotech. 19, p.342, 2001
【非特許文献3】Proc.Natl.Acad.Sci.USA,Vol.100(7),pp3960,2003
【非特許文献4】Nano Letters,Vol.4(6),1093,2004
【非特許文献5】Biochem. Biophys. Res. Comm. 306, p.213(2003)
【非特許文献6】Jpn.J.Appl.Phys.,41,p.1579(2002)
【非特許文献7】Analytical Biochemistry 298, p.1 (2001)
【発明の開示】
【発明が解決しようとする課題】
【0009】
特許文献4,5の方法では、検出対象となる単一DNA分子の配置がランダムであり、なんら規則性を持たない。したがって、一画素と、シーケンシング時に蛍光を発生する場となるDNA分子の空間配置を1:1で対応させることが困難となる。そこで、検出対象分子を規則的に配置させることが重要になる。非特許文献4の方法では微粒子が表面に物理吸着しただけの状態であり、固定力が不十分であって、このコロイド粒子を単一DNA分子の場所の目印として用いようとしても、そのままではデバイスとして安定に使用することが出来ない。特許文献6,7の方法は、単一DNA分子を規則的に配置させるものではない。非特許文献3の場合、Polyacrylic acidは表面に物理吸着しているため、デバイス上で溶液を用いた種々反応を行うにつれて剥離する可能性が高い。またPolyacrylic acidを吸着させるために、正電荷を持つPolyallylamine等も吸着させているため、これが新たな非特異吸着サイトとなる可能性もある。また、単一分子を規則的に配列するという目的達成に微粒子を利用する場合には、微粒子表面のプローブ固定箇所以外をブロックする吸着阻害剤の形成も必要である。非特許文献5の方法は、蛍光検出の感度向上だけを目的としており、銀ナノ粒子上には無数のプローブ分子を固定しているため、検出精度の点で問題がある。検出精度向上のためには、粒子上のプローブ分子の固定方法に工夫が必要になる。
【0010】
以上のように生体分子検出素子の低精度・再現性に対する問題を解決するためのアイデアは存在するが、いずれも単独では有効な結果を得ることは出来ず、別に新たな工夫を加えた総合的な解決策が必要である。
【0011】
本発明の目的は、DNAマイクロアレイやシーケンシングのような生体分子検出素子の精度・再現性を向上させることである。精度・再現性の低下を引き起こす原因として主に下記の諸点が挙げられる。
(1)プローブ分子の固定密度が定量的に制御されていない。
(2)プローブ分子の相互の間隔がランダムであり、個々のプローブ分子による生体分子検出反応が、独立ではなく相互に干渉しあう。
(3)非特異的に吸着した生体分子が表面に残留し、検出信号の精度を低下させる。
【0012】
上記(1)の原因として、プローブ分子固定反応において、固定密度を制御する工夫がなされていないことが挙げられる。上記(2)は(1)の結果として現れることである。また、上記(3)の原因として、生体分子の非特異的吸着に対する防止対策が不十分であることが挙げられる。
【0013】
特に、シーケンシングによって遺伝子配列を決定する際に精度・再現性の低下を引き起こす原因として、検出対象となる単一分子DNAがある平均密度でランダムに固定された表面を用いていること、すなわち、検体ポリヌクレオチドの固定点の配置に何ら規則性がないことが挙げられる。隣り合う検体ポリヌクレオチドが近接しすぎる場合には、両方のポリヌクレオチド分子の上から検出される蛍光信号が同一画素で検出されるためにデータが誤読されてしまう。従って、このような場所は無効サイトとなる。また、膨大な数の蛍光信号発生箇所(ポリヌクレオチド固定サイト)を検出する必要があるので、センサー表面上をステップ&リピート方式でエリア毎に蛍光検出するが、個々の測定箇所がランダム配置であるため、検出・測定作業がたいへん非能率である。このようにシーケンシングにおいては、生体分子である検体ポリヌクレオチドがランダム固定されていることが原因で、検体ポリヌクレオチド固定点に無駄が発生したり、測定時間が長いなどの性能上の制約が発生する。また単一分子ベースのシーケンシングでは、基本的に単一分子蛍光計測が必要であり、安定した蛍光検出のためには、蛍光検出感度の向上が必要である。
【0014】
以上の問題点を解決するための第一の課題は、生体分子検出のための個々のプローブ分子を基板表面上に予め設計された規則正しい幾何学的配列で、単一分子として固定することである。第二の課題は、基板表面のプローブ分子固定サイト以外の領域には、検体分子やその他の妨害分子が非特異的に吸着しない対策を施すことである。第三の課題は、蛍光検出感度を増強することである。
【0015】
これらの課題を同時に解決することにより、高感度、高精度かつ再現性にすぐれた生体分子検出素子を実現する。
【課題を解決するための手段】
【0016】
本発明は、単一のプローブ分子を担体基板表面の格子点位置に規則的に配列固定した生体分子検出素子を実現する。そのために、まず担体基板表面にリンカー分子の格子状ドットパターンを設置する。ここで、格子とは正方格子、三角格子、六角格子、ハニカム型格子、長方形格子等の様々な形状の格子を指す。このリンカー分子は、担体基板表面に固定され、基板と反対側にはアミノ基やチオール基のような金属と強い相互作用をする官能基を持つ。次に、これらの官能基と金属微粒子を結合させることにより、一つのリンカー分子ドット上に一つの金属微粒子を固定する。この金属微粒子一個の上には、一個のプローブ分子を固定する。そして、担体基板表面上の金属微粒子が固定された領域即ちリンカー分子ドットパターン以外の領域には、種々の生体分子の非特異的な吸着を防止するための吸着阻害分子を固定する。さらに、金属微粒子の表面のうち、プローブ分子が結合していない領域には、第二の吸着阻害分子を固定し、金属微粒子表面への種々の生体分子の非特異的な吸着も防止する。
【0017】
金属微粒子の材料としては、貴金属、貴金属の合金、あるいは貴金属の積層体が良い。
金属微粒子径とリンカー分子ドットの径は互いにほぼ匹敵するサイズが好ましい。
【0018】
また貴金属の微粒子をプローブ分子固定の土台に使うことにより、プローブ分子近傍での蛍光検出の際の励起光による金属微粒子のプラズモン共鳴による近接場の効果を活用して蛍光強度を増強し、蛍光検出感度を向上させ、単一分子の蛍光でも十分安定に検出可能とする。
【発明の効果】
【0019】
本発明によれば、生体分子検出素子表面のプローブ分子が孤立して決められた一定の距離を離れて規則的に固定されるため、検体試料との反応において、どのプローブ分子も均等な反応条件に置かれる。また検体分子とプローブ分子との反応において隣接プローブ分子が干渉することが無い。これらのことは、例えばDNAマイクロアレイに用いた場合には、検出精度の再現性向上に寄与する。また、プローブ分子の固定ピッチは目的に応じて任意に変更でき、かつプローブ分子の固定位置が事前に正確に判るので、例えば単一塩基シーケンシングに用いた場合には、検出系CCDの画素ピッチに揃えて単一プローブ分子を固定することで、個々のプローブ分子固定サイトからの蛍光信号を間違いなく分離して検出でき、また素子表面のステップ&リピートによる繰り返しの蛍光測定の駆動作業が大幅に高速化できる。さらに、プローブ分子の固定場として金属微粒子を用いるので、蛍光検出の励起光と金属粒子内自由電子の共鳴に基づく近接場を活用して蛍光の増強が達成でき、単一蛍光分子からの蛍光でも安定に検出することが可能となる。
【0020】
即ち、生体分子検出素子として、検出精度、検出データ再現性、検出感度の三つの課題を同時に解決するとともに、検出工程を大幅に高速化できるデバイスを提供できる。
【発明を実施するための最良の形態】
【0021】
図1に、本発明による生体分子検出素子の表面構造の一例を示す。まず担体基板101の表面にリンカー分子の格子状ドットパターンを設置する。このドットパターンは基板表面のX座標軸、Y座標軸に沿ってピッチLの正方格子に配置してある。これを金属微粒子固定ドット105として用いる。ここでリンカー分子とは、担体基板表面側が共有結合により固定され、基板と反対側にはアミノ基やチオール基のような金属と強い相互作用をする官能基を持つ分子のことである。これらの官能基を用いて金属微粒子106を結合することにより、一つの金属微粒子固定ドットの上に一つの金属微粒子を固定する。この金属微粒子一個の上には、一個のプローブ分子107を固定する。そして、担体基板表面上の金属微粒子が固定された領域即ち金属微粒子固定ドットパターン以外の領域には、種々の生体分子の非特異的な吸着を防止するための吸着阻害分子を固定し、吸着阻害層108を形成する。さらに、金属微粒子の表面のうち、プローブ分子が結合していない領域には、第二の吸着阻害分子を固定し、金属微粒子表面への種々の生体分子の非特異的な吸着も防止する。なお金属微粒子表面の吸着阻害層は図1では省略してある。
【0022】
以上の構成によって、単一のプローブ分子を担体基板表面の格子点位置に規則的に配列固定した生体分子検出素子を実現する。
【0023】
次に、本発明の生体分子検出素子を製造するプロセス概念の一例を、図2を用いて説明する。ここでは、ポジ型電子線レジストを用いた電子線直接描画によるパターン形成を利用したプロセスを取り上げる。製造プロセスは下記の9工程からなる。
(1) 基板表面へのポジ型電子線レジストの塗布
(2) 電子線描画及び現像による開口形成
(3) リンカー分子層の形成I(金属微粒子固定ドット:リンカー分子A)
(4) 金属微粒子の固定
(5) レジスト剥離
(6) リンカー分子層の形成II(微粒子固定ドット外表面:リンカー分子B)
(7) 吸着阻害分子の形成I(吸着阻害分子C)
(8) プローブ分子の固定
(9) 表面吸着阻害分子の形成II(金属微粒子表面:吸着阻害分子D)
【0024】
各工程について以下に説明する。
(1) 基板表面へのポジ型電子線レジストの塗布:図2(a)
先ず担体基板201を洗浄する。具体的には例えば、NaOH水溶液等のアルカリ性水溶液で洗浄した後、HCl水溶液等の酸性水溶液で洗浄し、純水ですすいだ後に乾燥する。あるいは、硫酸と過酸化水素を約4:1で混合した溶液で有機物汚染を洗浄する。基板としては、ガラス基板(スライドガラス)、石英基板、プラスチック基板等を用いることができる。また、金属コーティング基板等でもよい。基板の材質は、表面にシラノール基を有するものが好ましい。
【0025】
次いで、基板にポジ型電子線レジスト202をスピンコートし、所定の温度で乾燥ベークする。本発明では、レジスト膜厚は塗膜の均一性が確保できることを前提としてできるだけ薄いことが望ましいが、スループロセス検討結果から少なくとも100nm以下が望ましく、さらに好ましくは60nm以下が好適である。
【0026】
なお、図2のプロセスでは酸化膜付きSiウェハのような導電性基板の上で電子線描画を実施する場合を想定しているが、酸化膜が例えば10nm以上と厚い場合や、石英、ガラスのような絶縁性基板の上で電子線描画を施す場合には、基板のチャージアップを防ぐために電子線レジストの表面にさらに水溶性導電性樹脂の極薄膜を形成する。この導電性樹脂薄膜もスピンコートで容易に形成でき、レジストの電子線感度にはほとんど影響を与えない。また描画後、水洗によって容易に溶解除去され、次のレジスト現像工程には一切影響を与えない。
ここでは、ポジ型電子線レジストを用いているが、描画パターンによってはネガ型レジストを用いても良い。
【0027】
(2) 電子線描画及び現像による開口形成:図2(b)
電子線描画は、目的の解像度を満たす電子線描画装置によって実行する。ここでは、実効解像度(最小加工寸法)10nmの電界放射型電子線描画装置を用い、ドットパターンをXY座標方向ともに一定ピッチの格子状パターンを描画する。基本パターンは円形であり、サイズは最小20nmφから100nmφ程度が主要な値である。電子線走査範囲は、75μm−2400μm角の範囲であり、必要に応じて走査フィールドを繋ぎ合わせ、照射面積を確保する。レーザ干渉計を用いた高精度アライメント機構を用い、走査フィールド繋ぎ精度は3σで50nm以下(走査フィールド600μmの場合)程度を確保する。電子線描画パターンはCADデータとして設計し、コンピュータ制御により描画を実行する。描画パターンは穴加工であり、スループットの観点からポジ型レジストが向いている。ポジ型電子線レジスト塗布基板を電子線描画した後、所定の有機溶剤系現像液に浸漬して、照射部のレジストを除去し、さらにリンス液で洗浄して開口パターン203を得る。20nmφから100nmφの開口パターンは光学顕微鏡では観察できないので、電子顕微鏡及び原子間力顕微鏡(AFM)を用いてパターン解像検査を行う。
【0028】
(3) リンカー分子層の形成I(金属部粒子固定ドット):図2(c)
電子線レジスト開口パターンを形成した基板を、リンカー分子Aの溶液に浸漬し、開口パターン底の酸化膜(SiO2)表面に反応させる。リンカー分子Aとしては、金属微粒子と結合する活性基を持つシランカップリング剤などが使える。シランカップリング剤としては、例えば、基板表面にアミノ基を固定する場合には、3-アミノプロピルトリメトキシシラン(3-aminopropyltrimthoxysilane)、3-アミノプロピルトリエトキシシラン(3-aminopropyltriethoxysilane)、N-(2-aminoethyl)-3-aminopropyltrimethoxysilane)、(aminoethyl-aminomethyl) phenethyltrimethoxysilane等を用いることができる。一方、基板表面にチオール基を固定する場合には、(3-mercaptopropyltrimethoxysilane)を用いることができる。溶媒としては、例えば、エタノール、メタノール、トルエン、ベンゼン、水等を用いることができる。反応温度は、通常、20℃〜85℃の範囲である。ここで、リンカー分子Aは、開口部の酸化膜表面とは表面シラノール基と共有結合で固定されるが、レジスト表面部では表面に結合基が存在しないため物理吸着しているだけである。リンカー分子Aのアミノ基又はチオール基が基板と反対側に露出し、次工程の金属微粒子と結合する。
【0029】
ここでは、工程(1)(2)で電子線レジスト開口パターンを形成させた後に、工程(3)でリンカー分子層を形成させたが、基板を洗浄した後に基板全面に工程(3)でリンカー分子を結合させ、その後(1)(2)の工程を経て電子線レジスト開口パターンを形成させても良い。
【0030】
(4) 金属微粒子の固定:図2(d)
基板表面の活性基と金属微粒子の相互作用により、基板表面に金属微粒子を固定する。図2(d)は、金属微粒子205が固定化された担体表面の様子を示す。金属微粒子材料としては、貴金属類である金、銀、白金、パラジウム、ロジウム、イリジウム、ルテニウム、オスミウムのいずれか、あるいはそれらの合金を用いることができる。あるいは、これらの貴金属類で作られた微粒子上に他の貴金属がコーティングされたもの、例えば金微粒子上に銀がコーティングされた金属微粒子を用いてもよい。用いることができる金属微粒子径は、金属微粒子が安定に存在できる0.6nm以上である。金属微粒子の基板への固定安定性という観点から、金属微粒子径は10nm以上が望ましい。一方、蛍光を増強させるという観点から、金属微粒子径は蛍光増強効果が得られる10nm以上1μm以下を用いるのが良い。一方、後の工程で金属微粒子表面に単一生体分子を効果的に固定するという観点から、金属微粒子径は100nm以下が望ましい。以上の3つの観点から整理すると、固定する金属微粒子径は0.6nm以上1μm以下が良いが、望ましくは10nm以上100nm以下が良い。
【0031】
金属微粒子の固定反応溶媒として、水、あるいはエタノール、トルエンを用いることができる。溶液中での金属微粒子の凝集を防ぐために、保護剤を用いる。保護剤として、クエン酸、メルカプトコハク酸、ポリビニルピロリドン、ポリアクリル酸、テトラメチルアンモニウム、ポリエチレンイミン、1−デカンチオール、1−オクタンチオール、デシルアミン、ホスフィン(bis(p-sulfonatophenyl)phenylphosphine)等を用いることができる。
【0032】
金属微粒子の濃度は、通常、30wt%以下であり、反応温度は、通常、20℃−85℃の範囲である。また反応時間は、0.5時間から50時間の間である。これらの反応条件を変えることによって、金属微粒子の固定密度を制御することができる。例えば、金属微粒子の固定反応は、固定溶液中の金属微粒子濃度に対する1次反応であり、Langmuir型の反応となる。従って、固定溶液中の金属微粒子濃度や反応時間を変えることで、所望の固定密度を得ることができる。金属微粒子の反応時間は、レジストパターンの無い、リンカー分子だけの単純表面における反応時間を参考に決める。ここでは、あくまでレジスト開口部203の底面に金属微粒子205を到達させ、表面活性基と金属微粒子を反応させて固定することが目的である。
【0033】
ここで、ドットパターン上の金属微粒子が、各サイトに複数個入らずに一個づつ固定され、かつドットパターンへの金属微粒子の固定充填率を高く保つために、金属微粒子径と電子線レジスト開口部の径の関係は以下の条件を満たすのが良い。金属微粒子径/電子線レジスト開口部径の比をγとすると0.5≦γ≦1を満たすのが良い。γが0.5より小さい場合、電子線レジスト開口径に比べて金属微粒子径が充分小さいため、各サイトに金属微粒子が複数個入ってしまう。一方、γが1より大きい場合、電子線レジスト開口孔底部に金属微粒子が拡散できず、充填率が著しく低くなる。
【0034】
(5) レジスト剥離:図2(e)
専用レジスト剥離液に工程(4)が終了した基板を浸漬し、レジストを溶解除去する。この際、レジスト上の吸着している金属微粒子及びリンカー分子Aがリフトオフにより除去され、基板表面に共有結合した表面反応分子Aからなる微粒子固定ドットとその上に固定された金属微粒子だけが残る。この段階で重要なことは、ドットパターン上の金属微粒子が、各サイトに一個づつ固定されていることと、ドットパターンへの金属微粒子固定充填率が高いこと、ドット外のエリアには金属微粒子の残留付着が無いことである。
【0035】
ここでは、工程(4)の金属微粒子の固定後に工程(5)のレジスト剥離を行っているが、工程(5)のレジスト剥離を行った後に工程(4)で金属微粒子を固定しても良い。すなわち、レジストを剥離し、リンカー分子層のみがドット状に並んだ表面と、金属微粒子溶液を反応させて、金属微粒子を固定してもよい。また、ここでは、工程(3)でリンカー分子を結合させてから工程(4)で金属微粒子の固定を行ったが、レジスト開口パターンに工程(4)で金属微粒子をキャピラリーフォース等を用いて埋め込んだ後に、工程(3)でリンカー分子を結合させても良い。
【0036】
(6) リンカー分子層の形成II(微粒子固定ドット外表面):図2(f)
基板表面のうち、金属微粒子が固定されていない表面に、後のプロセスで生体分子やdNTPといった試薬が非特異的に吸着する可能性が高い。この非特異吸着は、デバイスとして用いた場合のノイズとなるため、徹底的に防止する必要がある。このため金属微粒子固定ドットパターン以外のエリアに吸着阻害分子を固定し、対策する。図2(f)は、この目的のために、まずリンカー分子Bを形成した様子を示す。ここで、金属微粒子固定ドットに用いた、アミノ基やチオール基を含むシランカップリング剤を用いると、既に表面に固定されている金属微粒子表面に対しても反応するために、最終的な金属微粒子表面へのプローブ固定の障害にもなりかねない。そこで、ここでは金属微粒子とは相互作用せず、基板表面とだけ反応するリンカー分子が望ましい。このような物質として、例えばエポキシ基やカルボキシル基を持つシランカップリング剤が有望である。
【0037】
(7) 吸着阻害分子の形成I(吸着阻害分子C):図2(g)
工程(6)で形成したリンカー分子Bの上に、生体分子の非特異吸着を防止する分子として吸着阻害分子Cを結合させる。リンカー分子Bが、上記エポキシ基を持つシランカップリング剤の場合は、エポキシ基やカルボキシル基と反応するアミノ基や水酸基等を持つ吸着阻害分子が有効であり、具体的には、アミノ基を末端にもつ低分子量のポリエチレングリコール(PEG)や水酸基を持つカルボキシメチルデキストラン(CM-Dextran)などが使える。ただし、リンカー分子Bのみで充分非特異吸着を防止できる場合には、吸着阻害分子Cは必ずしも必要ではない。
【0038】
ここまでの工程で作成した基板には、電子線リソグラフィによって高精度に形成された微粒子固定ドットの格子状パターン(グリッドパターン)の上に金属微粒子が一個ずつ固定されており、かつ微粒子固定ドット以外のエリアには非特異吸着を防止する吸着阻害分子がコーティングされている。ここまで出来上がった基板を、金属微粒子グリッドアレイ基板と呼ぶことにする。
【0039】
(8) 単一プローブ分子の固定工程:図2(h)
金属微粒子グリッドアレイ基板に固定された金属微粒子上に、この金属微粒子と結合することができる官能基を持つプローブ分子を反応させ、金属微粒子上にプローブ分子を一個だけ固定することがこの工程の目的である。図2(h)は、プローブ分子をPとして、基板表面の様子を示す。
【0040】
ここで、金属微粒子として金ナノ粒子を用い、プローブ分子として、チオール基を5’末端に持つプローブDNAを用いた場合について説明する。金ナノ粒子を均一にランダム分散固定し、上記(6)(7)に示した工程で非特異吸着を防止する吸着阻害分子がコーティングされた基板を用い、この基板に対してチオール末端基付きDNAを反応させると、チオールは金ナノ粒子表面だけに反応して吸着する。この吸着挙動を、吸着反応を計測できるSPR法(Surface plasmon resonance:表面プラズモン共鳴)を用いて定量化した。SPR法の測定感度では、金ナノ粒子1個当りプローブ分子1個のみ吸着した時の吸着量を測定するのは困難である。そこで、金ナノ粒子1個当りに1分子より多くのプローブ分子を固定させ、DNA溶液濃度と固定されたプローブ分子数の関係を測定したところ、単純なLangmuir型吸着曲線示すことが判った。この曲線から、金ナノ粒子一個当りプローブ分子一個だけを吸着するプローブ分子溶液濃度を推定することができる。この吸着挙動評価実験から推定されたプローブ分子の最適濃度に調整したプローブDNA溶液に、同じ直径の金微粒子を規則的な格子点に固定した金微粒子グリッドアレイ基板を浸漬し、所定温度で所定時間反応させた後、洗浄液で洗浄し、乾燥させた。
【0041】
この基板上で、各金ナノ粒子毎のプローブDNA分子の固定数を計測した。計測方法は単一分子蛍光測定であり、非特許文献6に開示された手法に則り、高感度イメージインテンシファイヤーとCCDカメラの組合せを用い、光学顕微鏡ステージ上で実行した。この方法により、サンプル基板表面の観測視野内に固定された個々の蛍光色素分子からの蛍光(単一分子蛍光)をリアルタイムで画像として検出することができる。ここで、単なるプローブDNAには蛍光性が無いため、検出ができない。そこで単一プローブDNA分子固定の立証のためには、金ナノ粒子上に固定したチオール末端DNAと完全相補的な配列を持つ蛍光色素付きのDNAをハイブリダイゼーションさせた状態で蛍光計測を実施した。また単一分子蛍光の立証のためには、チオール基を5’末端に持つとともに3’末端にCy3、Cy5、Rhodamine Bのような蛍光分子を持つプローブDNAを用いて実験してもよい。いずれにしても、単一分子蛍光の特徴である、蛍光強度の繰返し点滅と階段状の蛍光退色が確認できた。ここで階段状の蛍光退色とは、蛍光検出時に蛍光強度をモニターし続けると、蛍光色素が一個一個酸化分解するため、そのたび毎に蛍光強度が階段状に低下する現象である。単一蛍光色素分子の蛍光を観測している場合には、分子の分解とともに蛍光が一挙に消えることで確認できる。以上の計測方法を用いることによって、金ナノ粒子グリッドアレイ表面の金ナノ粒子上に単一のプローブDNA分子を固定する最適プロセス条件(プローブDNA溶液濃度・反応温度・反応時間)を決定した。
【0042】
プローブDNAを溶解させる溶液としては、リン酸バッファなどの中性付近の水溶液を用いることができる。この溶液にプローブDNAを溶解させる。この時のプローブDNAの濃度は、例えば従来のDNAチップでは通常、0.5μM−100μMであるが、本発明のような金ナノ粒子グリッドアレイ基板上の金ナノ粒子に単一プローブDNAを固定する場合は、プローブDNAの濃度は数nM以下程度が最適である。なおプローブDNAの最適濃度は、固定表面の状態に依存し、具体的には金微粒子のサイズ・固定密度(グリッドアレイピッチ)によって変動するため、金微粒子グリッドアレイの設計に応じてそれぞれプローブDNA最適濃度を決定する。また、反応時間は、充分反応が平衡に達するまでの時間よりも長い時間とした。
【0043】
担体としてSiウェハやガラス基板を用いた金ナノ粒子グリッドアレイ基板の場合、基板の所望の位置に、プローブDNAを溶解した反応液を所望のサイズにスポッティングできる。この時、基板に多種類のプローブDNAをスポッティングすることが可能であり、こうすると例えばDNAマイクロアレイとして使える。反応温度は、通常、25℃から40℃の範囲である。また、反応時間は、通常、2時間から24時間の範囲である。反応させる時に溶液が乾燥しないよう、充分湿度を保った環境で反応させる。
【0044】
金とチオール基は結合し易いため、チオール基を末端に持つプローブDNAが金ナノ粒子上のみに固定される。金ナノ粒子が固定されていない領域(金微粒子固定ドット外のエリヤ)は、吸着阻害分子Cで覆われているため、プローブDNAはほとんど吸着しない。以後、吸着阻害分子でコーティングすることをブロッキングと呼ぶ。
【0045】
ここでは、金属微粒子を基板に固定した後に、金属微粒子が固定された部分以外の基板表面をブロッキングし、その後にプローブ分子を固定したが、金属微粒子にプローブ分子を固定した後に、この金属微粒子を表面に固定し、その後に金属微粒子が固定された部分以外の基板表面をブロッキングしても良い。
【0046】
(9) 吸着阻害分子の形成工程II(金属微粒子表面):図2(i)
金属微粒子表面で、プローブDNAが固定されなかった領域は、検体の生体分子を吸着させる可能性がある。よって、このプローブDNA固定部以外の金属微粒子表面をブロッキングする。図2(i)は、吸着阻害分子Dを用いて、金属微粒子表面をブロッキング処理した後の基板表面の様子を示す。
【0047】
ここでは、金属微粒子として金ナノ粒子を用いた場合について説明する。金と反応し易く、かつ生体分子を吸着し難いブロッキング剤として、1−メルカプトヘキサノール、2−メルカプトエタノール等を用いることができる。これらのブロッキング剤を溶解した水溶液と担体表面を反応させ、ブロッキング材料を固定する。
【0048】
反応温度は、通常、4℃〜35℃の範囲であり、反応時間は、通常、0.5時間〜10時間である。この反応では、水溶液中のブロッキング剤の濃度が高い場合、ブロッキング剤が金ナノ粒子と反応し金ナノ粒子を覆うことで、金属微粒子と担体の間の結合力を弱める。その結果、金属微粒子が担体表面で拡散し金属微粒子が表面で凝集する。したがって、ブロッキング剤反応溶液の濃度を100μM以下の範囲とした。
【0049】
ここでは、金属微粒子にプローブDNAを固定した後に吸着阻害分子Dを固定したが、プローブDNAと吸着阻害分子Dを同時に固定しても良い。すなわち、プローブDNAを溶解させた溶液に吸着阻害分子Dを混合し、その溶液と金属微粒子を反応させても良い。
【0050】
ここで、金ナノ粒子グリッドアレイ基板において、金ナノ粒子上に固定したプローブ分子がプローブDNAであるアレイ基板を、DNAマイクロアレイとして用いる場合について、以下(10)(11)で説明する。
【0051】
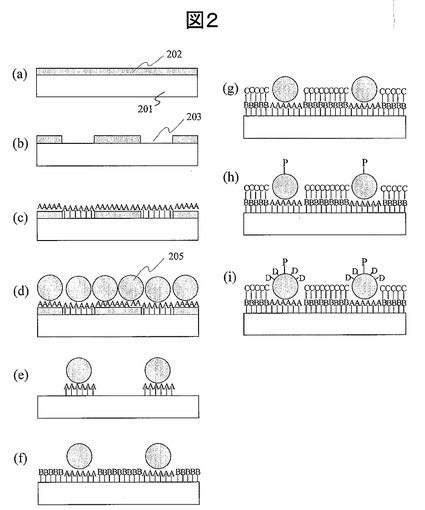
(10) DNAマイクロアレイのハイブリダイゼーション反応評価工程:図3、図4
前述の(1)〜(9)に述べた工程を経て作成した金属微粒子グリッドアレイを基板とする生体分子検出素子表面に、蛍光修飾された検体試料溶液を反応させる。ここではプローブ分子としてプローブDNAを用い、検出用生体分子としても核酸を用いた場合について図3を用いて説明する。簡略化のためリンカー分子、吸着阻害分子などは図示を省略した。基板301上に金属微粒子302が等ピッチLで整列固定され、金属微粒子302上にプローブDNA303が固定されている。これに対して、蛍光分子305によって蛍光修飾されたターゲットDNA304が検体試料として供給された状況を図3(a)に示す。ここで、プローブDNAとターゲットDNAの塩基配列が完全に相補的である場合は互いに速やかに反応し、図3(b)のように相補的水素結合307で結合した2本鎖DNAを形成する。これがハイブリダイゼーション反応である。ここで重要なことは、個々のプローブDNAは均等な環境条件にありかつ一定距離で正確に分離・固定されているので、相互干渉することがない。このため、このような基板で構成した生体分子検出素子では、反応時間や反応効率のバラツキが発生しにくい。
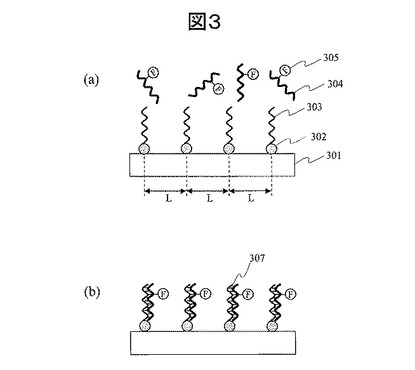
【0052】
一方、図4に示すような従来型の生体分子検出素子の表面では、図4(a)のように、プローブDNA(402)の固定密度がランダムであり、基板401の面内にプローブ固定の粗密が存在する。このため、同じハイブリダイゼーション反応を実行すると、ターゲットDNA(403)との反応の速いものと遅いものが共存し、図4(b)、図4(c)のようにハイブリダイゼーション反応の進行度のバラツキが発生しやすい。
【0053】
以上のことから、本発明の生体分子検出素子を用いると、目的の表面反応が均一かつ効率よく進行するので、検出データの精度・再現性が大幅に向上する。
【0054】
なお、ハイブリダイゼーション反応の具体的条件としては、検出用の蛍光修飾された核酸を、界面活性剤を添加したSSC(Standard Saline Citrate)溶液に溶解し、生体分子検出素子表面にこの溶液を接触させる。溶液中の核酸量は0.1amol〜1nmolである、反応温度は、通常、25℃−60℃、反応時間は、通常、1時間〜24時間である。この条件で、検出用の核酸がプローブDNAの配列と完全相補的であった場合、速やかに反応して相補的水素結合により形成された2本鎖DNAを生成する。DNAマイクロアレイの場合には、基板表面の多数のスポットにおいて、多種類の配列のプローブDNAを検体試料とハイブリダイゼーション反応させ、各スポットからの蛍光強度をスキャナーで計測し、データ化する。
【0055】
(11) 蛍光増強:図5
本発明では、金属微粒子をプローブ分子の固定場に用いることで、もう一つの特徴である蛍光増強現象が利用できる。本発明において、蛍光増強現象が発現する様子を図5で説明する。図5は、図2で説明した本発明の生体分子検出素子の製造方法によって作成した素子を示しており、図2(i)においてPで表記したプローブ分子としてDNAを想定したものである。図5(a)は、基板501上の金属微粒子に固定化されたプローブDNA(502)と蛍光分子504で蛍光修飾されたターゲットDNA(503)とのハイブリダイゼーション反応前の状態であり、図5(b)は反応後の状態を示す。この図は、完全相補鎖DNA同士の反応として描いてある。
【0056】
金属微粒子固有の局在プラズモン共鳴により、蛍光強度が増強される。まず局在プラズモン共鳴と蛍光増強について説明する。この現象については非特許文献7に詳しい記載がある。金属微粒子は、光が入射されると、金属微粒子内の自由電子が分極し振動する。この金属微粒子内の自由電子の振動と、入射光の振動電場が共鳴することを局在プラズモン共鳴と呼ぶ。局在プラズモン共鳴が起こると、金属微粒子表面での電場強度が入射光の電場強度に比べて数桁大きくなる。次に、上述の蛍光増強について、その二つの要因を説明する。蛍光増強する要因の一つは、蛍光分子の量子効率の向上である。蛍光分子の近傍に金属微粒子が存在する場合、蛍光分子のエネルギー吸収過程で、局在プラズモン共鳴によって金属微粒子近傍の電場増強効果による吸収遷移が生じる。更に、発光過程で、金属微粒子が存在すると発光過程の速度が加速される。したがって、吸収遷移と発光が増すことから、蛍光分子の量子効率が上がる。ただし、量子効率は1を超えることはないため、量子効率1を持つ蛍光分子では、金属微粒子による量子効率の増加は期待できない。しかし、実際にバイオセンサで用いられる蛍光分子は、量子効率が0.04−0.3程度のものが多く、これらの蛍光分子に対して金属微粒子による量子効率の向上が期待できる。
【0057】
第二の要因は、金属微粒子による光散乱強度の増加である。局在プラズモン共鳴により金属微粒子の分極率が増加し近傍の電場が増強されると、金属微粒子からの散乱光強度も増強される。これは、散乱光強度が金属微粒子の分極率の2乗に比例するからである。散乱光強度が増加すれば、蛍光分子を励起させるための入射エネルギー密度が増加し、従って蛍光発光強度も増加する。
【0058】
これらの蛍光増強効果は、金属微粒子と蛍光分子の距離が約5nmから約100nmの間で見られる。蛍光増強効果が現れる領域を近接場506として図5(b)に示した。プローブ分子として、金属微粒子に固定されたプローブ分子端と、プローブ分子に修飾された蛍光分子間のプローブ分子に沿った距離dが、蛍光増強効果が得られる長さである5nmから100nmの範囲に収まる場合、検体分子と反応して相補的水素結合505した後に、蛍光増強効果を利用することができる。
【0059】
以上のメカニズムによって、金属微粒子をプローブ分子の固定場に用いた場合には、蛍光修飾された検体分子を超高感度に蛍光検出することができ、生体分子検出素子としての大幅な感度向上が達成できる。
【0060】
次に、前述の(1)〜(9)に述べた工程を経て作製した金属微粒子グリッドアレイ基板をDNAシーケンシングに用いる場合について説明する。
【0061】
(12) DNAシーケンシング用解析対象分子固定:図6
まず、前述の(1)〜(9)に述べた工程によって、配列解析対象となる単一分子DNA604を固定するためのプローブ分子603を基板601上に金属微粒子602を介して配列させる。次に、配列解析対象の単一分子DNA604をプローブ分子と1対1で反応させて基板に固定する。解析対象の単一分子DNAをプローブ分子と反応させる際の条件として、単一分子DNAを、NaCl等の塩を含む溶液に溶かし、この溶液をプローブ分子を固定したアレイ基板表面と接触させる。反応温度は、通常、20℃〜80℃、反応時間は、通常1時間〜24時間程度である。解析対象の単一分子DNAとプローブ分子を反応させるために、プローブ分子は単一分子DNAを固定するための反応サイトを持つ必要がある。例えば、解析対象の単一分子DNAがAAAAAAAAAといったPolyA配列を持つ場合、これと相補的な配列であるTTTTTTTT等のTが連続したPolyT配列を有するプローブ分子を使用する。
【0062】
次に、図6に示した金ナノ粒子602グリッド配列のピッチLの大きさについて述べる。DNAシーケンシングでは、背景技術で述べたように、蛍光強度を測定することによってDNA配列を読み取る。シーケンシング時には、固定されたある単一分子からの蛍光と、その分子と隣接する単一分子DNAからの蛍光を独立して測定する必要がある。したがって、ピッチLは、蛍光読取光学系の解像度と同等あるいはより大きい値とする必要がある。また、同じ理由によってCCDカメラ読取の1画素のサイズよりも大きい値とする必要がある。
【0063】
(13) シーケンシング:図7
図7に示すように、プローブ分子702をプライマーとして、ポリメラーゼ704によるポリメラーゼ反応により、プローブ分子702の先端にヌクレオチド705を一塩基伸長させる。この反応時には、ヌクレオチドを、例えば、塩化マグネシウム等の塩を含む溶液に溶解し、この溶液を基板と接触させる。酵素であるポリメラーゼの失活を防止するため、ジチオスレイトール(DTT)やグリセロール、界面活性剤等を上記溶液に混合しても良い。図7に示すように、伸長したヌクレオチドには蛍光分子706を結合している。この蛍光分子からの蛍光を読み取ることで、ヌクレオチドが伸長したか否か、あるいは、伸長したヌクレオチドの種類、例えば、A,T,C,Gを判別する。
【0064】
図8にシーケンシング読取システムを示す。このシステムでは、励起レーザ光801を基板803の裏面から石英プリズム802を通して全反射条件で入射させる。一塩基伸長時に、伸長したヌクレオチド808に結合した蛍光分子809を、全反射条件で入射したレーザ光のうち単一分子DNA806が固定された側に染み出したエバネッセント光によって励起する。生じた蛍光810を、基板上面に載置した高感度CCDカメラ811によって測定する。ヌクレオチドが結合された後、例えば、ポリメラーゼによって取り込まれたヌクレオチドのリン酸基が切断される。蛍光分子がヌクレオチドのリン酸基の末端に結合している場合には、リン酸基の切断と共に、蛍光分子が解析対象の単一分子DNA806部から離れ除去される。除去された後に、別種類のヌクレオチドを反応させる。例えば、まずヌクレオチドAを含む溶液と基板を接触させ、ポリメラーゼ反応によってAが一塩基伸長した場合には、上記の読取システムによって蛍光が検出される。ヌクレオチドAが一塩基伸長しなかった場合には蛍光は検出されない。この測定結果から、蛍光が検出されたグリッドサイトでは、解析対象の単一分子DNAがAと相補的であるTを解析対象部分に持つことがわかる。一方、蛍光が検出されなかったグリッドサイトでは、解析対象部分にTを持たないことがわかる。次に例えばヌクレオチドCを含む溶液と基板を接触させた後に、蛍光を検出し、各グリッドサイトにおいて、単一分子DNAの解析対象部分にCと相補的なGを有するか否かを判別する。次に、ヌクレオチドTあるいはGを反応させる。これらの作業を繰り返すことで、各グリッドサイトに固定した単一分子DNAの配列を読み取る。
【0065】
ここで重要なことは、本発明の個々の単一分子DNA806は一定距離で分離・固定されているので、検出した蛍光信号が、隣接する単一分子DNAから発せられる信号と相互干渉することがない。このため、例えば図9に示すように、蛍光を検出するCCDカメラの一画素903の大きさと金ナノ粒子のグリッド格子の大きさ904が等しい場合、グリッドサイト毎に、独立に、蛍光強度を測定できる。すなわち、グリッドサイト毎に、独立に、DNAの配列を読むことができる。したがって、正確性高くDNA配列を読むことができる。画素数をより細かくし、例えば4画素で一つのグリッドサイトからの蛍光信号を読み取る、あるいは、9画素で一つのグリッドサイトからの蛍光信号を読み取っても良い。更には、DNAの固定サイトとなる金属微粒子902効果で、蛍光強度が増強される。その理由については前述の(11)に述べた通りである。通常、単一分子蛍光からの蛍光を測定する際には、高感度に測定するために、励起光のパワー密度を上げ、検出系に光子を増幅するデバイスを用いる等の工夫が必要であり、蛍光励起および検出系が大掛かりで複雑な系になる。しかし、本発明の蛍光増強効果を用いることで、蛍光検出系をコンパクトに設計することが可能になる。
【0066】
一方、図10に示すように、解析対象の単一分子DNA1004が基板1001にランダムに固定されている場合、単一分子DNA間の距離を制御することができないため、一画素1005で2つ以上の単一分子DNAからの蛍光を計測する場合もある。この場合、2つのDNAからの信号を分離することが困難であり、結果として誤読する、あるいは解析できないDNAが存在することになる。更には、単一分子DNA(1004)がランダムに固定される場合、解析の開始時点で、解析対象のDNA(1004)の位置が不明であるため、解析開始前に位置確認を行う必要がある。例えば、蛍光分子を結合させた単一分子DNAを固定した後に蛍光マッピングを行うことで、単一分子DNAが固定された場所を特定し、システムに記憶させるなどのプロセスが必須となる。また、金属微粒子による蛍光増強効果を利用できないため、蛍光検出系が大掛かりで複雑な系になる。
【0067】
本発明の金属微粒子グリッドアレイ基板を用いれば、解析の正確性が各段に向上し、また、解析不可能な無効なDNAを生じさせることなく、全てのDNAの配列を読み取ることができる。更には、固定された解析対象DNAの場所(座標)が予めわかっているため、場所を特定するためのプロセスを省略できる。
【0068】
本発明では、リソグラフィのプロセスを用いて担体基板表面にリンカー分子からなる規則的なドットパターンを形成し、このリンカー分子が保有する活性基と金属微粒子との結合を利用して、まず微粒子金属グリッドアレイを形成した。リソグラフィの方法としては電子線描画に限ったものではなく、電子線リソグラフィの代わりに近接場リソグラフィを用いることも可能である。但しその場合には、メンブレンマスクを電子線リソグラフィによって作製し、このマスクを用いたコンタクト露光によってレジスト加工を実行する。この場合、同じパターンであれば繰り返しハイスループットで作製できるので、量産性の高いプロセスを達成できる。また、リンカー分子の規則的ドットパターンをリソグラフィを用いることなくナノコンタクトプリント技術やナノインプリント技術を用いて行うことも可能である。
【0069】
次に、本発明を実施例により詳細に説明するが、特にDNAマイクロアレイおよびDNAシーケンサに適用した例について説明する。なお本発明は、下記の実施例に限定されるものではなく、核酸、タンパク、糖鎖などのあらゆる生体分子検出素子の性能向上に寄与する基本技術である。
【実施例1】
【0070】
(工程1)基板表面へのポジ型電子線レジストの塗布工程:図11(a)
担体基板1101として、平坦性の高い熱酸化膜100nm付き4インチSi基板を用いた。基板を0.1wt%のNaOH水溶液で洗浄し、更に0.1wt%のHCl水溶液で洗浄し、純水で充分すすいだ後に乾燥した。ここで、レジストとして用いたポジ型レジストは、主鎖切断型のレジストである。レジストをアニソールで希釈し、スピンコーターを用いてコーティングした。コーティング後、溶剤を除去するためにN2フロー中 180℃、20minベークした。本実施例では、レジスト膜厚は充分薄く、かつEB加工による膜減りが見られない60nm膜厚(レジスト:アニソール=1:3希釈)とした。また、EB描画中の基板の帯電を防止するため、塗布したレジスト膜1102の上に導電性ポリマー(ポリイソチアナフテンスルホネート)溶液をコーティングした。この溶液は、導電性ポリマーをコロイド粒子にし、界面活性剤を用いて分散させたものである。同じスピンコーターを用いて導電性ポリマー1103をコーティングした後、溶剤を除去するためにN2フロー中で100℃、10minベークした。
【0071】
(工程2)電子線描画及び現像による開口形成:図11(b)
電子線走査フィールド内ではx方向、y方向それぞれ60,000ステップに分割され、各ステップにてスポット径約2〜3nmの電子線をパルス照射する。どのステップ位置で電子線照射をするかをCADソフトを用いて指定することにより、所望のパターンを形成する。電子線加工開口径は、固定させる金属微粒子と同等である必要がある。本実施例では、安定に固定でき、かつ蛍光増強効果を得ることができる30nmの金ナノ粒子を用いた。したがって、EB開口径を40nmφとした。本実施例で用いた描画パターンを図14に示す。40nmφの電子線照射領域が格子状にそれぞれ100nmピッチで並んだパターンとした。この描画パターンを200μm角のエリアに加工した。またこの200μm角の加工エリアを基板上に100点作成した。加工エリアは基板上に2mmピッチで格子状に並べた。
【0072】
電子線ドーズ方式として、電子線を同一箇所に連続照射し、レジスト横方向に徐々に染み出したエネルギーによってレジストポリマー主鎖を切断する照射法を用いた。
フィールドサイズは150μm角とし、電子線ビーム電流5×10-11Aで電子線描画を行った。
【0073】
電子線照射によって主鎖が切断された部分を溶解する現像液として、n-アミルアセテートを用いた。最表面に導電性ポリマーが塗布されているため、まずこれを純水で除去し、次に、25℃、15秒n-アミルアセテート溶液に浸漬して現像後、リンス溶媒Methyl isobutyl ketone 89%/ Isopropyl alcohol 11%溶液でリンス洗浄した。これら一連のプロセスによって、50nmφの金ナノ粒子固定用の開口孔1104を加工した。開口孔は電子顕微鏡で確認した。
【0074】
(工程3)アミノ化層の形成(金属微粒子固定ドット):図11(c)
電子線レジスト開口パターンを形成した基板を、シランカップリング剤である3-アミノプロピルトリメトキシシラン(3-aminopropyltrimthoxysilane, APTMS)溶液に浸漬した。なおAPTMSを希釈する溶媒としてメタノールを用いた。反応温度は室温、反応時間は5分とし、反応後、メタノールで充分洗浄し乾燥した。この結果、開口孔底にAPTMSが吸着する。この基板を80℃2hrアニールした。このアニールプロセスによって、APTMSと開口孔底のSiO2の間にシロキサン結合が形成され、開口孔底が安定にアミノ化される。一方、レジスト上にもAPTMSが吸着するため、レジスト上にもAPTMSが残留する。
【0075】
(工程4)金ナノ粒子の固定:図11(d)
次に、開口孔底部をアミノ化した基板に、直径が30nmの金ナノ粒子クエン酸溶液を作用させた。なお、金ナノ粒子1106の濃度は、約0.4nMである。また、反応温度は室温であり、反応時間は20時間である。この時、金ナノ粒子表面はクエン酸で覆われ負電荷を持つ。一方、アミノ化された表面は正電荷を持つため、金ナノ粒子は表面のアミノ基1105と引き合うため、金ナノ粒子1106が固定される。金ナノ粒子を反応させた後、充分純水洗浄した後に乾燥させた。
【0076】
(工程5)レジスト剥離:図12(a)
基板をジメチルアセトアミドに3分浸漬させ、レジストを除去した。この際、レジスト上に付着した金ナノ粒子が基板に再付着しないよう、充分量のジメチルアセトアミドを使用した。ジメチルアセトアミドに浸漬後、エタノール洗浄と純水洗浄を充分に行った。このプロセスによって、レジスト上に吸着された金ナノ粒子もリフトオフされ、開口部に固定された金ナノ粒子のみが残留する。従って、金ナノ粒子30nmを格子状に配列することができた。
【0077】
(工程6)吸着阻害分子層の固定:図12(b)
金ナノ粒子30nmを格子状に配列させた基板をエタノールに浸漬し乾燥後、エポキシ基1107をコーティングした。Diisopropylethylamine 0.8%が添加された3-Glycidoxypropyltrimethoxysilane(GOPS) 7.7%の脱水トルエン溶液中で、上記基板を85℃下で2hr反応させることによってエポキシ基を金ナノ粒子が固定されていない領域にコーティングした。反応後、脱水トルエン及びエタノールで洗浄し乾燥させた。
【0078】
(工程7)PEGの固定:図12(c)
工程6で固定したエポキシ基1107とアミノ基を末端に持つPEG(分子量2k)1108を弱アルカリ下で反応させた。具体的には、4mMアミノ化PEG溶液に溶解し、基板を室温で1hr浸漬した。その後、純水で充分洗浄し乾燥させた。この工程によって、図7(c)に示すように、金ナノ粒子が固定されていない領域がPEG1108でブロッキングされた。
【0079】
(工程8)単一プローブDNAの固定:図13(a)
工程6及び7でエポキシ基とPEGをコーティングした基板をエタノール洗浄した後、基板表面とチオール基を末端に持つ50mer 1本鎖プローブDNA溶液を金ナノ粒子コーティング領域であるそれぞれ250μm角エリアにスポッティングした。用いたDNAの塩基配列は1種類であり、1種類のプローブDNA(1109)を100点にスポットした。これは、各スポットでの検出バラツキを調べるためである。その5’末端側からの塩基配列を下記に示す。
AGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG
【0080】
用いたプローブDNA反応溶液は、チオール基を5’末端に持つ50mer 1本鎖DNA1 nMを1M Phosphate buffer (pH 6.7)に溶解した溶液である。この濃度は、「発明を実施するための最良の形態」の(8)で述べたSPR法による吸着挙動評価実験から推定し、単一分子蛍光測定によって単一分子固定を確認できた濃度である。このプローブDNA反応溶液と格子状に配列させた金ナノ粒子を室温24hr、100%湿度下で反応させた後、2×SSC、0.1%SDS溶液ですすぎ、純水で2回洗浄した後に乾燥した。
【0081】
(工程9)金ナノ粒子表面メルカプトヘキサノールブロッキング工程:図13(b)
メルカプトヘキサノール水溶液1μMを作成し、この水溶液中にプローブDNAが固定された基板を浸漬した。反応温度は室温、反応時間は1時間とした。反応させた後に純水洗浄を行い、デシケータ中で減圧乾燥し、図13(b)に示すように、金ナノ粒子のプローブDNA固定部以外の表面をメルカプトヘキサノール1110でブロッキングした基板を得た。
【0082】
(工程10)ハイブリダイゼーション
プローブDNAを固定した基板に、プローブDNAと完全相補的な配列をもち、3’末端に蛍光分子Cy3を標識した一本鎖のターゲットDNAをハイブリダイゼーションさせた。ハイブリダイゼーション溶液として5×SSC(Standard Saline Citrate)と0.5%SDS溶液Sodium Dodecyl Sulphate)の混合液を用い、ターゲットDNA量1fmolを42℃で20時間ハイブリダイゼーションさせた。その後、2×SSC、0.1%SDS溶液、2×SSC溶液で洗浄を行い、乾燥した。乾燥させた基板表面に対し、蛍光スキャナーを用いて励起光を入射し、表面からの蛍光強度を測定した。この蛍光強度は、ハイブリダイゼーションによって反応したターゲットDNAの量に比例するものである。
【0083】
一方、Si基板上に一般的な従来からある1本鎖DNAの固定方法で、プローブDNAを固定した基板を作製した。Si基板を上記(工程1)に示した方法と同様に洗浄した後に、(工程3)に示した方法でシランカップリング剤であるAPTMSを全面にコーティングし基板表面をアミノ化した。その後に、末端のアミノ基に、イソチオシアナート基を有するPDC(phenylenediisothiocyanate)を反応させた。更に、このイソチオシアナート基とアミノ基を5’末端に持つプローブDNAを反応させ固定した。用いたプローブDNAの配列は、(工程8)で示したDNA配列と同じである。プローブDNAを反応させる際に用いた反応溶液は、弱アルカリの炭酸バッファにプローブDNAを1μM溶解させた溶液である。この溶液をイソチオシアナート化した基板にスポットした。スポット点数は100点であり、スポット径は200μmφである。室温24hr、100%湿度下で反応させた後、基板を2×SSC、0.1%SDS溶液ですすぎ、純水で2回洗浄した後に乾燥した。この基板を前述の(工程10)に示した方法で蛍光分子付きのターゲットDNAとハイブリダイゼーションさせた後に、蛍光強度を測定した。測定した蛍光強度はハイブリダイゼーション反応したターゲットDNA量を表すものである。
【0084】
金ナノ粒子を格子状に配列させ単一プローブDNAを金ナノ粒子上に固定して形成したグリッドアレイの100スポットの蛍光強度平均値およびバラツキと、従来の固定方法でプローブDNAを固定したアレイの100スポットの蛍光強度平均値およびバラツキを図15に示す。蛍光強度は、蛍光スキャナーを用いて励起光を入射し、表面からの蛍光強度を測定した。励起レーザ光の波長は532nmであり、このレーザ光をスポットエリアでスキャンさせた。発生した蛍光を光電子倍増管で検出した。
【0085】
本発明の金ナノ粒子グリッドアレイは、従来方法で作製したアレイよりも蛍光強度のバラツキが±50%以上から±5%未満へと大幅に低減する。これは、プローブDNAが100nmピッチで孤立固定されているため、個々のプローブDNAが干渉し合うことなく自由運動しており、ターゲットDNAとのハイブリダイゼーション反応速度や反応効率がプローブDNA間及びスポット間で同等だからである。またプローブDNAの固定密度が均質であり、ターゲットDNAの供給量も場所による差が見られない。一方、従来の基板では、プローブDNAの固定密度が面内でばらついているため、プローブDNA同士の干渉やターゲットDNAの供給量のバラツキがあり、これがハイブリダイゼーション反応速度や反応効率の面内バラツキを発生させる。
【0086】
また、本発明の金ナノ粒子グリッドアレイで検出した蛍光強度は、従来のアレイで検出した蛍光強度よりも大きくなった。金ナノ粒子グリッドアレイの場合、プローブDNA固定密度は100分子/μm2である。これに対し従来のアレイの場合、プローブDNA固定密度はX線反射率等の計測手段により測定すると約5,000分子/μm2と約50倍の固定密度を持つ。したがって、固定密度から考えると、従来アレイの方がより多くのターゲットDNAが反応し、蛍光強度も高いはずである。しかし、前述したように、金ナノ粒子グリッドアレイはプローブDNAが孤立しているため、従来アレイに比べてハイブリダイゼーション反応効率が高い。更には、金ナノ粒子近傍の近接場により、図5に示すように蛍光が増強される。50merDNAが2本鎖を形成した場合、その長さは約17nmである。金ナノ粒子近傍では、近接場光はナノ粒子表面から粒子径程度まで届く。したがって、金ナノ粒子30nmの表面から17nmの距離では、蛍光が増強される領域であると言える。以上の反応効率の上昇及び蛍光増強によって、金ナノ粒子グリッドアレイでは、DNA固定密度は従来型アレイよりも低いにもかかわらず、従来アレイよりもむしろ蛍光強度が高くなった。
【実施例2】
【0087】
DNAマイクロアレイの検出濃度限界を測定するために、実施例1の工程1から工程9と同様の方法で単一プローブDNAを固定した金ナノ粒子グリッドアレイを得た。一方で、実施例1に示した方法と同様に従来のアレイを作製した。実施例1の工程10で示した方法と同様にハイブリダイゼーションを行った。
【0088】
本実施例では、ハイブリダイゼーション時の蛍光分子付き完全相補的ターゲットDNA量を1amolから1fmolまで変化させた。それぞれのアレイにおける蛍光強度とターゲットDNA量の関係を調べた。その結果、図16に示すように、金ナノ粒子グリッドアレイは従来アレイに比べてターゲットDNA検出感度が向上した。従来のアレイでは、100amolが検出限界であったのに対し、金ナノ粒子グリッドアレイでは1amolが検出限界となり感度が大幅に向上した。これは、本発明の金ナノ粒子グリッドアレイでは、孤立プローブDNAの反応効率が向上し、また蛍光増強効果を得ることができるためである。
【実施例3】
【0089】
本実施例は、DNAシーケンシングに用いた例を示す。DNAシーケンシングを行うために、実施例1に示した工程1から工程8に従ってDNAシーケンシング基板を作製した。本実施例の場合、基板にSi基板ではなく石英基板を用いた。工程2の電子線描画及び現像による開口形成において、40nmφが格子状にそれぞれ1μmピッチで並んだパターンを作製した。このパターンを用いることで、金ナノ粒子を1μmピッチ格子状に並べた。作製した金ナノ粒子グリッドアレイ基板のSEM観察結果を図17に示す。30nm径の金ナノ粒子が1μmピッチで配列していることを確認した。蛍光強度を検出するCCDカメラの1画素を1μm角とすると、シーケンシング時に、一画素で、DNA一分子からの蛍光信号を読み取ることができる。画素数をより細かくし、例えば4画素でDNA一分子からの蛍光信号を読み取る、あるいは9画素でDNA一分子からの蛍光信号を読み取っても良いが、本実施例では1画素の大きさを1μm角とした。また、工程8で固定するプローブDNAとして、図18に示すように、3’末端にチオール基を持ち、かつT配列を20連続で持つ(TTTTT TTTTT TTTTT TTTTT TTTTT (PolyT 20mer))オリゴヌクレオチド1802を用いた。この場合、このオリゴヌクレオチド末端のチオール基と金ナノ粒子を結合させることによって、オリゴヌクレオチドを固定する。
【0090】
その後、工程10に従って、解析対象となるDNA(1803)をプローブDNAにハイブリダイゼーションさせる。具体的には、解析対象のDNAとして、5’末端からA配列を連続で持ち、下記の配列を持つDNA(1803)をハイブリダイゼーションさせた(図18参照)。
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG
【0091】
次に、プローブDNAである上記PolyT20mer配列をプライマーとして、解析対象DNAのシーケンシングを行った。作製した金ナノ粒子グリッドアレイ基板と、ヌクレオチド3リン酸のリン酸基末端に蛍光標識であるCy3(1805)を持つ4種類のヌクレオチド(1804)をポリメラーゼを用いて反応させた。反応時に用いた反応溶液は、10mM Tris-HCl、5mM MgCl2溶液である。ヌクレオチドの濃度は1μMである。ポリメラーゼ反応によって、DNAの塩基配列と相補的な塩基を持つヌクレオチドがDNAに結合する。本実施例の場合、ヌクレオチドCを溶解した溶液と基板を反応させた場合、ヌクレオチドCが結合する。ヌクレオチドCは解析対象DNAの配列中のAが連続した部分に隣接するGに結合する。図8に示した蛍光検出系を用いて、ポリメラーゼ反応過程における蛍光画像を取得する。蛍光の強度は基板上面から高感度CCDカメラで測定した。検出する際に蛍光分子の励起光を全反射条件で当てると、蛍光分子付きヌクレオチドC(1804)が結合したグリッドサイトから蛍光が検出される。ヌクレオチドがDNAに結合すると、ポリメラーゼによって取り込まれたヌクレオチドのリン酸基が切断される。これによって、そのリン酸基の末端に結合していた蛍光分子が測定対象DNA部から除去される。次に、ヌクレオチドAを溶解した溶液と基板を反応させた場合、ヌクレオチドCの結合点の隣であるTにヌクレオチドAが結合する。結合した際に、蛍光を検出することができる。次に、ヌクレオチドTを溶解した溶液と基板を反応させた場合、ヌクレオチドTはDNAに結合しない。DNA側の次の結合サイトはCであり、ヌクレオチドTとは相補的でないため、ヌクレオチドTが結合せずに蛍光が検出されないことを確認した。
【0092】
これらの手順を繰返し、各グリッドにおいて、同様にDNA配列を読むことが可能であった。すなわち、個々のDNA固定サイトからの蛍光信号を他DNAサイトからの信号と分離して検出でき、無効になるグリッドDNAを生じさせることなく、繰返しDNAの配列を読むことが可能であった。
【0093】
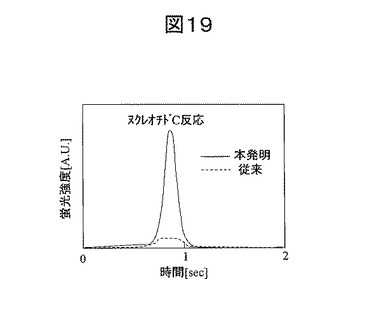
また、図18の状態を蛍光検出した時の結果を図19に示す。検出蛍光強度は、従来方法で測定した蛍光強度に比べて高くなった。これは金ナノ粒子による蛍光増強効果によって、蛍光強度が増加したためである。したがって、本発明の素子を用いることでより高感度にシーケンシングを行うことができる。あるいは、簡易な蛍光検出系を用いてシーケンシングを行うことができる。
【産業上の利用可能性】
【0094】
DNAやmRNAの定量解析を行うDNAマイクロアレイや、DNA又はmRNAの配列を読むDNAシーケンシング、また蛋白質を分析するプロテインチップ、及び解析対象となる蛋白質や糖鎖を解析前に固定させる前処理基板としても適用できる。
【図面の簡単な説明】
【0095】
【図1】本発明による生体分子検出素子の表面の基本構成を示す図。
【図2】本発明による生体分子検出素子の製造工程を示す図。
【図3】本発明による生体分子検出素子を用いたDNAハイブリダイゼーション反応をイメージした図。
【図4】従来型の生体分子検出素子を用いたDNAハイブリダイゼーション反応をイメージした図。
【図5】本発明による生体分子検出素子を用いたDNAハイブリダイゼーション反応における蛍光増強を説明する図。
【図6】本発明によるDNAシーケンシング用解析対象の単一分子DNAをグリッド配列固定した表面の基本構成を示す図。
【図7】DNAシーケンシング反応をイメージした図。
【図8】DNAシーケンシング検出系を示す図。
【図9】グリッド配列された1金属微粒子と1画素の関係を示す図。
【図10】従来型の生体分子検出素子を用いたDNAシーケンシング系をイメージした図。
【図11】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図12】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図13】本発明による生体分子検出素子の製造工程の具体例を示す概念図。
【図14】本発明で用いた電子線描画パターンを示す図。
【図15】本発明で得られた金ナノ粒子グリッドアレイと従来型アレイの完全相補的なDNAをハイブリダイゼーションさせた時の検出蛍光強度と強度バラツキを示す図。
【図16】金ナノ粒子グリッドアレイと従来型アレイにおいて、完全相補的なターゲットDNAをハイブリダイゼーションさせた時の検出蛍光強度とターゲットDNA濃度の関係を示す図。
【図17】金ナノ粒子を格子状に固定した一例を示す図。
【図18】DNAシーケンシング配列とシーケンシング反応を示す図。
【図19】DNAシーケンシング時の蛍光検出結果を示す図。
【符号の説明】
【0096】
101:担体基板、105:金属微粒子固定ドット、106:金属微粒子、107:プローブ分子、201:担体基板、202:電子線レジスト、203:レジスト開口部、205:金属微粒子、301:基板、302:金属微粒子、303:プローブDNA、304:ターゲットDNA、305:蛍光分子、307:相補的水素結合、401:基板、402:プローブDNA、403:ターゲットDNA、501:基板、502:プローブDNA、503:ターゲットDNA、504:蛍光分子、505:相補的水素結合、506:近接場、601:基板、602:金属微粒子、603:プローブ分子、604:解析対象単一分子DNA、701:金属微粒子、702:プローブ分子、703:解析対象単一分子DNA、704:ポリメラーゼ、705:ヌクレオチド、706:蛍光分子、801:励起レーザ光、802:プリズム、803:基板、804:金属微粒子、805:プローブ分子、806:解析対象単一分子DNA、807:ポリメラーゼ、808:ヌクレオチド、809:蛍光分子、810:蛍光、811:CCDカメラ、901:基板、902:金属微粒子、903:一画素、904:グリッド格子、1001:基板、1003:プローブ分子、1004:解析対象単一分子DNA、1005:一画素、1101:担体基板、1102:電子線レジスト、1103:導電性ポリマー、1104:レジスト開口部、1105:アミノ基、1106:金ナノ粒子、1107:エポキシ基、1108:PEG、1109:プローブDNA、1110:メルカプトヘキサノール、1801:金ナノ粒子、1802:プローブ分子PoyT連続20配列、1803:解析対象単一分子DNA、1804:ヌクレオチドC、1805:蛍光分子Cy3
【特許請求の範囲】
【請求項1】
単一プローブ分子を担体基板上の格子点位置に配列固定したことを特徴とする生体分子検出素子。
【請求項2】
請求項1記載の生体分子検出素子において、前記担体基板表面の前記格子点位置にリンカー分子が固定され、前記リンカー分子上に金属微粒子が結合され、さらに前記金属微粒子表面に前記単一プローブ分子が固定されていることを特徴とする生体分子検出素子。
【請求項3】
請求項2記載の生体分子検出素子において、前記金属微粒子が貴金属類に属する金属、あるいは貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものであることを特徴とする生体分子検出素子。
【請求項4】
請求項2記載の生体分子検出素子において、前記金属微粒子の粒子径が0.6nm以上1μm以下であることを特徴とする生体分子検出素子。
【請求項5】
請求項2記載の生体分子検出素子において、前記担体基板表面の前記金属微粒子が固定された部分以外の領域が、前記担体基板表面に共有結合で固定された吸着阻害分子によって被覆されていることを特徴とする生体分子検出素子。
【請求項6】
請求項2記載の生体分子検出素子において、前記金属微粒子表面の前記単一プローブ分子が固定された部分以外の領域が、吸着阻害分子によって被覆されていることを特徴とする生体分子検出素子。
【請求項7】
請求項2記載の生体分子検出素子において、前記金属微粒子の径r1とリンカー分子が固定された部分の径r2との比γが
0.5≦γ(r1/r2)≦1
を満たすことを特徴とする生体分子検出素子。
【請求項8】
請求項2記載の生体分子検出素子において、前記単一プローブ分子が核酸からなることを特徴とする生体分子検出素子。
【請求項9】
単一プローブ分子が担体基板上の格子点位置に配列固定された生体分子検出素子の製造方法において、
担体基板表面の格子点位置に金属微粒子を固定する工程と、
前記基板表面に吸着阻害分子を固定する工程と、
単一プローブ分子を前記金属微粒子表面に固定する工程と、
を有することを特徴とする生体分子検出素子の製造方法。
【請求項10】
請求項9記載の生体分子検出素子の製造方法において、前記基板表面の格子点位置にリンカー分子を固定する工程と、前記リンカー分子に前記金属微粒子を結合させる工程とを有することを特徴とする生体分子検出素子の製造方法。
【請求項11】
請求項9記載の生体分子検出素子の製造方法において、更に、前記金属微粒子表面に吸着阻害分子を固定する工程を有することを特徴とする生体分子検出素子の製造方法。
【請求項12】
担体基板上の格子点位置に金属微粒子が結合され、さらに前記金属微粒子表面に単一プローブ分子が固定された生体分子検出素子を用いて、
前記生体分子検出素子の前記プローブ分子と蛍光標識された試料生体分子とを反応させる工程と、
反応後の生体分子検出素子に励起光を照射する工程と、
前記プローブ分子が固定された領域から発生する蛍光を検出する工程と、
を有することを特徴とする生体分子検出方法。
【請求項13】
担体基板上の格子点位置に金属微粒子が結合され、さらに前記金属微粒子表面に単一プローブ核酸分子が固定された生体分子検出素子を用いて、核酸の配列を検出する方法において、
前記生体分子検出素子の前記プローブ核酸分子と被配列検出核酸分子とを反応させる工程と、
前記被配列検出核酸分子に、蛍光標識されたヌクレオチドを反応させる工程と、
前記生体分子素子に励起光を照射する工程と、
前記標識されたヌクレオチドから発生する蛍光を検出する工程と、
を有することを特徴とする生体分子検出方法。
【請求項1】
単一プローブ分子を担体基板上の格子点位置に配列固定したことを特徴とする生体分子検出素子。
【請求項2】
請求項1記載の生体分子検出素子において、前記担体基板表面の前記格子点位置にリンカー分子が固定され、前記リンカー分子上に金属微粒子が結合され、さらに前記金属微粒子表面に前記単一プローブ分子が固定されていることを特徴とする生体分子検出素子。
【請求項3】
請求項2記載の生体分子検出素子において、前記金属微粒子が貴金属類に属する金属、あるいは貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものであることを特徴とする生体分子検出素子。
【請求項4】
請求項2記載の生体分子検出素子において、前記金属微粒子の粒子径が0.6nm以上1μm以下であることを特徴とする生体分子検出素子。
【請求項5】
請求項2記載の生体分子検出素子において、前記担体基板表面の前記金属微粒子が固定された部分以外の領域が、前記担体基板表面に共有結合で固定された吸着阻害分子によって被覆されていることを特徴とする生体分子検出素子。
【請求項6】
請求項2記載の生体分子検出素子において、前記金属微粒子表面の前記単一プローブ分子が固定された部分以外の領域が、吸着阻害分子によって被覆されていることを特徴とする生体分子検出素子。
【請求項7】
請求項2記載の生体分子検出素子において、前記金属微粒子の径r1とリンカー分子が固定された部分の径r2との比γが
0.5≦γ(r1/r2)≦1
を満たすことを特徴とする生体分子検出素子。
【請求項8】
請求項2記載の生体分子検出素子において、前記単一プローブ分子が核酸からなることを特徴とする生体分子検出素子。
【請求項9】
単一プローブ分子が担体基板上の格子点位置に配列固定された生体分子検出素子の製造方法において、
担体基板表面の格子点位置に金属微粒子を固定する工程と、
前記基板表面に吸着阻害分子を固定する工程と、
単一プローブ分子を前記金属微粒子表面に固定する工程と、
を有することを特徴とする生体分子検出素子の製造方法。
【請求項10】
請求項9記載の生体分子検出素子の製造方法において、前記基板表面の格子点位置にリンカー分子を固定する工程と、前記リンカー分子に前記金属微粒子を結合させる工程とを有することを特徴とする生体分子検出素子の製造方法。
【請求項11】
請求項9記載の生体分子検出素子の製造方法において、更に、前記金属微粒子表面に吸着阻害分子を固定する工程を有することを特徴とする生体分子検出素子の製造方法。
【請求項12】
担体基板上の格子点位置に金属微粒子が結合され、さらに前記金属微粒子表面に単一プローブ分子が固定された生体分子検出素子を用いて、
前記生体分子検出素子の前記プローブ分子と蛍光標識された試料生体分子とを反応させる工程と、
反応後の生体分子検出素子に励起光を照射する工程と、
前記プローブ分子が固定された領域から発生する蛍光を検出する工程と、
を有することを特徴とする生体分子検出方法。
【請求項13】
担体基板上の格子点位置に金属微粒子が結合され、さらに前記金属微粒子表面に単一プローブ核酸分子が固定された生体分子検出素子を用いて、核酸の配列を検出する方法において、
前記生体分子検出素子の前記プローブ核酸分子と被配列検出核酸分子とを反応させる工程と、
前記被配列検出核酸分子に、蛍光標識されたヌクレオチドを反応させる工程と、
前記生体分子素子に励起光を照射する工程と、
前記標識されたヌクレオチドから発生する蛍光を検出する工程と、
を有することを特徴とする生体分子検出方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
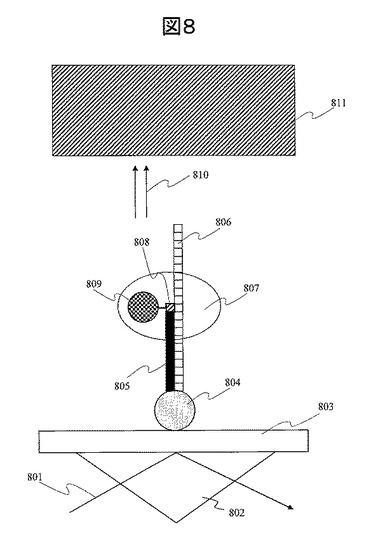
【図9】
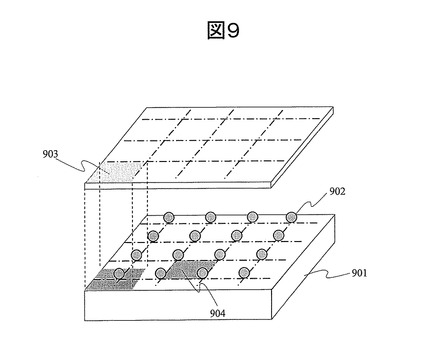
【図10】
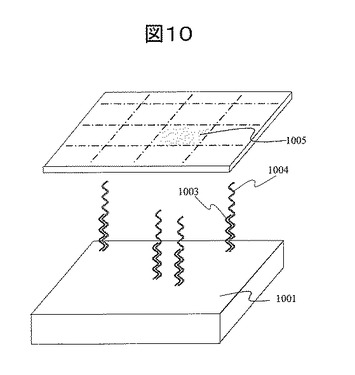
【図11】
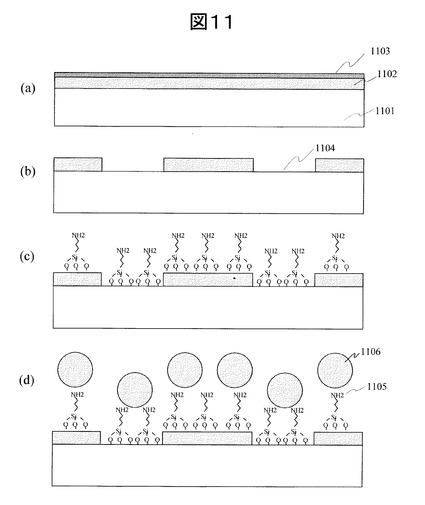
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】

【図18】
【図19】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2008−190937(P2008−190937A)
【公開日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願番号】特願2007−24082(P2007−24082)
【出願日】平成19年2月2日(2007.2.2)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
【公開日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願日】平成19年2月2日(2007.2.2)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
[ Back to top ]