
生体組織の直接質量分析法
【課題】生体組織の解剖学的構造を保持したまま、高い解析感度及び高い解析効率で組織を構成するタンパク質の質量分析を行うことができる、質量分析用試料の前処理方法、及び、生体組織内の解剖学的構造を保持したまま、多段階質量分析を行うことができる、質量分析用試料の前処理方法を提供する。
【解決手段】生体組織標本に対し、変性剤を供給することによって、前記生体組織内のタンパク質を変性させる工程と、染色剤を供給することによって、前記タンパク質を染色する工程と、消化酵素を分注することによって、前記タンパク質を消化する工程と、マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【解決手段】生体組織標本に対し、変性剤を供給することによって、前記生体組織内のタンパク質を変性させる工程と、染色剤を供給することによって、前記タンパク質を染色する工程と、消化酵素を分注することによって、前記タンパク質を消化する工程と、マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ライフサイエンス分野、具体的には、細胞生物学、病理学、生化学などの医学・生物学分野に関する。特に本発明は、生体組織標本上で直接的に生体組織を質量分析する方法に関する。
【背景技術】
【0002】
生体組織を直接質量分析を行った例として、例えば、Stoeckli, M.; Chaurand, P.; Hallahan, E. D.; Caprioli, M. R. Nature Med. 7, 493-496 (2001)、Chaurand, P.; Schwartz, A. S.; Caprioli, M. R. Anal. Chem., 76, 86A-93A (2004)などにおいて、凍結生体組織切片を、直接質量分析ターゲットプレート上で融解し、70(v/v)%エタノール水溶液で洗浄し、酵素消化を行うことなくMSスペクトルを取得した技術が報告されている。
【0003】
また、例えば、Chaurand, P.; Schwartz, A. S.; Billheimer, D.; Xu, J. B; Crecelius, A.; Caprioli, M. R. Anal. Chem., 76, 1145-1155 (2004)においては、伝導性スライドガラスを用いて、上記技術におけるものと同様のMSスペクトルを取得した報告がされている。
【0004】
さらに、例えば、特開2004−347594号公報には、生体組織標本に対し、タンパク質染色、トリプシン消化、及びMALDI質量分析を行い、MSスペクトルを得たことが報告されている。
【0005】
一方、二次元電気泳動においては、等電点電気泳動(IEF)を行った後、SDS(ドデシル硫酸ナトリウム)ポリアクリルアミドゲル電気泳動(SDS−PAGE)により分子量に基づいて分離が行われる。SDS−PAGEを行う際に、一次元目のゲルをSDSを含む溶液で平衡化することにより、ゲル中のタンパク質をSDS化する。
【0006】
【非特許文献1】ストックリ・M(Stoeckli, M.)、ショーラン・P(Chaurand, P.)、ハラハン・E・D(Hallahan, E. D.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「ネイチャー・メディスン(Nature Medicine)」第7巻、p.493−496、2001年
【非特許文献2】ショーラン・P(Chaurand, P.)、シュワルツ・A・S(Schwartz, A. S.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「アナリティカル・ケミストリー(Analytical Chemistry)」、第76巻、p.86A−93A、2004年
【非特許文献3】ショーラン・P(Chaurand, P.)、シュワルツ・A・S(Schwartz, A. S.)、ビルハイマー・D(Billheimer, D.)、スー・J・B(Xu, J. B.)、クレセリウス・A(Crecelius, A.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「アナリティカル・ケミストリー(Analytical Chemistry)」、第76巻、p.1145−1155、2004年
【特許文献1】特開2004−347594号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
上記の従来法では、MSスペクトルを取得することは可能であるが、酵素消化を行っていないため意味のあるMS/MSスペクトルを取得することはできない。また、単純に消化酵素を塗布しても消化を行うことはできないため、やはり意味のあるMS/MSスペクトルを取得することはできない。
【0008】
従って、質量分析を用いて生体組織内のタンパク質の同定を行うためには、予め、これまでの生化学的手法により組織の分離精製を行わなければならない。しかしながら、微量な物質を解析対象の試料とした場合、操作の途中で試料が失われる可能性もある。また、マトリックスの添加がコントロールされていないため、質量分析において解像度や再現性に問題がある。
【0009】
一般的にタンパク質の同定を質量分析によって行うには、多段階の質量分析によって得られたスペクトル情報が必要である。このためには、試料タンパク質を電気泳動などの手法で精製し、その後、変性させ消化酵素でペプチド化する。従って、生体組織中のタンパク質の同定を質量分析によって行うためには、上記に準じた手法で多段階の質量分析を行わなければならない。しかしながら、組織内では、タンパク質は立体構造を保っているため、消化酵素による消化効率が悪い。
【0010】
そこで本発明の目的は、生体組織の解剖学的構造を保持したまま、高い解析感度及び高い解析効率で組織を構成するタンパク質の質量分析を行うことができる、質量分析用試料の前処理方法を提供することにある。また、本発明の目的は、生体組織内の解剖学的構造を保持したまま、多段階質量分析を行うことができる、質量分析用試料の前処理方法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者は、二次元電気泳動において平衡化のために用いられる手法を応用することによって、上記本発明の目的が達成されることを見出し、本発明を完成するに至った。
【0012】
本発明は、以下の発明を含む。
(1)生体組織標本に対し、
変性剤を供給させることによって、前記生体組織内のタンパク質を変性させる工程と、
染色剤を供給することによって、前記タンパク質を染色する工程と、
消化酵素を分注することによって、前記タンパク質を消化する工程と、
マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【0013】
下記(2)及び(3)は、上記生体組織標本について記載する。
(2)前記生体組織標本は凍結切片である、(1)に記載の質量分析手法。
(3)前記生体組織標本は、ポリビニリデンジフルオリド膜、ニトロセルロース膜、ナイロン膜から選ばれる膜の表面に固着されたものである、(1)又は(2)に記載の質量分析手法。
【0014】
下記(4)及び(5)は、上記変性剤について記載する。
(4)前記変性剤は、界面活性剤、尿素、及びグアジニン塩酸から選ばれる、(1)〜(3)のいずれかに記載の質量分析手法。
(5)前記界面活性剤が、ドデシル硫酸ナトリウム、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸、ポリオキシエチレン−p−t−オクチルフェニルエーテル、モノラウリン酸ソルビタン、及び1−O−n−オクチル−β−D−グルコピラノシドから選ばれる、(4)に記載の質量分析手法。
【0015】
下記(6)及び(7)は、上記変性剤とともに用いられうる還元剤について記載する。
(6)前記変性剤を、還元剤とともに用いる、(1)〜(5)のいずれかに記載の質量分析手法。
(7)前記還元剤は、2−メルカプトエタノール、ジチオスレイトール、トリス[2−カルボキシエチル]ホスフィン、及び2−メルカプトエタノールアミンから選ばれる、(6)に記載の質量分析手法。
【0016】
下記(8)は、上記分注の操作について記載する。
(8)前記タンパク質を消化する工程及び/又は前記質量分析を行う工程において、インクジェット技術を用いて分注操作を行う、(1)〜(7)のいずれかに記載の質量分析手法。
【0017】
下記(9)は、上記質量分析について記載する。
(9)前記質量分析を行う工程において、多段階質量分析を行う、(1)〜(8)のいずれかに記載の質量分析手法。
【発明の効果】
【0018】
本発明によると、生体組織の解剖学的構造を保持したまま、高い解析感度及び高い解析効率で組織を構成するタンパク質の質量分析を行うことができる、質量分析用試料の前処理方法を提供することができる。また、本発明によると、生体組織内の解剖学的構造を保持したまま、多段階質量分析を行うことができる、質量分析用試料の前処理方法を提供することができる。
【発明を実施するための最良の形態】
【0019】
本発明において直接とは、生体組織標本における解剖学的構造を保持した状態で、ということを意味する。本発明においては、生体組織標本に対し各処理液を直接作用させることにより、組織内タンパク質の処理を組織内にて行い、得られた処理済生体組織を質量分析用試料として直接質量分析を行う。具体的には、本発明においては、組織内タンパク質の変性、タンパク質の染色、及びタンパク質の消化の各処理を行い、MALDI質量分析を行う。
【0020】
本発明における生体組織標本は、生体内における解剖学的構造を保持した状態を有するものであれば、特に限定されない。このような状態を得るために、生体組織が適当な包埋剤によって包埋されたものを用いることができる。包埋剤としては、生体試料の適性に応じて特に限定することなく用いることができる。例えば、水、パラフィン、セロイジン、カーボワックス、ゼラチン、アルブミン、アガロース、エポキシ樹脂、ポリエステル樹脂等や、グリコールメタクリレート等の水溶性樹脂等を用いることができ、これら包埋剤は、当業者が適宜選択することができる。このような生体標本としては、例えば凍結切片等の未固定試料、パラフィン包埋切片等の固定化試料等を用いることができる。
【0021】
前記生体標本は、膜及び/又は支持体の表面に固着させて使用することができる。膜としては、ポリビニリデンジフルオリド(PVDF)、ニトロセルロース、ポリアミド、ポリエチレン等の有機合成高分子及びその誘導体を材料とした膜を挙げることができる。ポリアミドとしては、ナイロン等が挙げられる。支持体としては、ガラス製支持体、樹脂製支持体、金属製支持体等が挙げられる。本発明では、質量分析法を用いて解析するため、前記支持体には好ましくは金属製支持体、例えば質量分析用サンプルプレートを用い、生体標本を膜に固着したものを前記サンプルプレートに貼り付けるか、又は前記サンプルプレートに生体標本を固着して用いることができる。凍結切片を生体組織標本とする場合、凍結切片を膜へ載せ融解させれば固着が可能となる。また、サンプルプレートへの固着には導電性両面粘着テープ等を用いると良い。
【0022】
上記の生体標本は、公知の方法を用いて作製することができる。生体標本は、作製後、適宜、洗浄などを行い、乾燥させておくことができる。
【0023】
<変性工程>
このような生体組織標本に対し、適当な変性剤を供給することによって、生体組織内のタンパク質を変性させる。変性剤としては、タンパク質の一次構造を維持したまま高次構造のみを破壊するものであれば、特に限定されない。例えば、変性剤は、各種界面活性剤、尿素、グアジニン塩酸等から選ばれる。界面活性剤としては、陰イオン性界面活性剤、陽イオン性界面活性剤、両性界面活性剤、及び非イオン性界面活性剤のいずれも用いることができる。例えば、陰イオン界面活性剤としては、ドデシル硫酸ナトリウム(SDS)などが挙げられる。両性界面活性剤としては、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸(CHAPS)などが挙げられる。非イオン性界面活性剤としては、Triton X-100 (ポリオキシエチレン−p−t−オクチルフェニルエーテル)、Tween 20 (モノラウリン酸ソルビタン)、1−O−n−オクチル−β−D−グルコピラノシドなどが挙げられる。
【0024】
なお、これら変性剤は、1種又は複数種を選ぶことができる。複数種を選ぶ場合は、その組み合わせは、選ばれた変性剤が互いに相互作用しないものであれば特に限定されない。また、変性剤は、通常水溶液の形態で供給することができる。
【0025】
変性剤濃度としては、1〜10M、好ましくは4〜8Mとすることができる。上記濃度より低い濃度の場合、タンパク質の変性が不十分となる傾向がある。また、上記濃度より高い濃度の場合、組織切片が剥がれ落ちる傾向がある。界面活性剤濃度としては0.05%〜2%、好ましくは0.1〜1%とすることができる(単位%は、界面活性剤の形態によりw/v%或いはv/v%となりうる)。上記濃度より低い濃度の場合、タンパク質の変性が不十分となる傾向がある。また、上記濃度より高い濃度の場合、組織切片上に界面活性剤が残留し質量スペクトルにポリマー由来のピークが検出される傾向がある。
【0026】
変性剤水溶液には、通常、トリス塩酸緩衝液、リン酸緩衝液、リン酸緩衝食塩水、酢酸緩衝液などから選ばれるバッファーが含まれる。トリス塩酸は10mM〜100mM、リン酸緩衝液は0.1M〜0.3M、リン酸緩衝食塩水は0.05M〜0.2M、酢酸緩衝液は0.1M〜0.2Mの濃度のものを使用することができる。上記濃度より低い濃度の場合、組織切片が剥離する傾向がある。また、上記濃度より高い濃度の場合、質量分析において塩の影響によりバックグラウンドが大きくなる傾向がある。なお、これらバッファーは、1種又は複数種を選ぶことができる。またバッファーのpHはトリス塩酸、リン酸緩衝液、リン酸緩衝食塩水においては6〜9、好ましくはpH6.5〜8.0、酢酸緩衝液ではpH3〜4とすることができる。
【0027】
変性剤水溶液には、さらに、ジスルフィド結合の切断やSH基の保護などのために、還元剤を含んでいても良い。還元剤としては、例えば、2−メルカプトエタノール、DTT(ジチオスレイトール(dithiothreitol))、TCEP(トリス[2−カルボキシエチル]ホスフィン(Tris[2-carboxyethyl]phosphine))、2−MEA(2−メルカプトエタノールアミン(2-Mercaptoethanolamine))等が挙げられる。これら還元剤は、1種又は複数種を選ぶことができる。
【0028】
変性剤水溶液中の還元剤濃度としては、10〜70mM、好ましくは40mM〜50mMとすることができる。上記濃度より低い濃度の場合、酵素消化において、反応の効率が低下する傾向がある。また、上記濃度より高い濃度の場合、質量分析において塩の影響によりバックグラウンドが大きくなる傾向がある。
【0029】
変性剤水溶液には、さらに、安定剤を含んでいても良い。安定剤としては、例えば、グリセロール(グリセリン)等が挙げられる。
【0030】
変性剤水溶液中の安定剤濃度としては、0.1〜50(v/v)%、好ましくは20〜50(v/v)%とすることができる。上記濃度より高い濃度の場合、上記濃度より高い濃度の場合、変性剤の粘性により取扱が困難になる傾向がある。
【0031】
生体標本中のタンパク質の変性は、上記の変性剤水溶液を、生体組織標本に供給することにより行う。具体的には、生体組織標本を上記の変性剤水溶液に浸漬し振盪するか、エアブラシ等を用いて霧状にした変性剤水溶液を生体組織標本上へ噴霧し、湿潤条件で放置するとよい。振盪時間および湿潤条件での放置時間は3〜24時間とすることができる。変性を行った生体標本は、適宜洗浄などを行うことができる。
【0032】
<染色工程>
上記工程によりタンパク質が変性した生体組織標本に対し、適当な染色剤を供給することによって、生体組織内のタンパク質を染色する。なお、生体組織標本作製の際にパラフィンなど疎水性の包埋剤を用いた場合は、染色工程に先立って包埋剤を除去しておく。
【0033】
染色剤としては、通常タンパク質の染色に用いられるものを、特に限定することなく用いることができる。例えば、メチルレッド等のモノアゾ色素;ポンソーSなどのジアゾ色素;Direct Blue71などのトリアゾ色素;ファストレッドなどのアゾイック色素;オーラミンなどのジフェニルメタン色素;ブリリアントグリーン等のジアミノトリフェニルメタン色素;ファストグリーン等のトリアミノトリフェニルメタン色素;ローダミンBなどのキサンテン色素;ローダミン3Gなどのフルオロン色素;アクリジンオレンジ等のアクリジン色素;クレシルファストバイオレット等のオキサジン色素;トルイジンブルー等のチアジン色素;アリザリンレッドS等のアントラキノン色素;ヘマトキシリン等の天然色素等を用いることができる。これら染色剤を用い、通常の方法に従った操作を行うことによって染色を行うことができる。例えば、エタノール−酢酸水溶液中に適当な濃度で溶解させた染色剤水溶液へ生体組織標本の浸漬及び振盪を行い、その後洗浄する操作を繰り返すと良い。Direct Blue 71を用いる場合、上記エタノール−酢酸水溶液は、10〜70(v/v)%エタノール−1〜20(v/v)%酢酸を含む水溶液とすることが好ましく、染色剤濃度は上記エタノール−酢酸水溶液中0.005〜0.01(w/v)%とすることが好ましく、上記振盪は2.5〜7分行うことが好ましく、振盪及び洗浄の操作は、3〜6回繰り返して行うことが好ましい。
染色を行った生体標本は、十分に乾燥させると良い。
【0034】
<消化工程>
上記工程によりタンパク質が染色された生体組織標本に対し、適当なタンパク質消化酵素を分注することによって、生体組織内のタンパク質を消化する。本発明において分注とは、生体組織標本上の特定の領域に対して、試薬溶液を供給することをいう(後述の質量分析工程においても同じ)。すなわち、消化工程では、上記工程によりタンパク質が染色された生体組織標本の特定の領域に対し、適当なタンパク質消化酵素を供給することによって、特定の領域における生体組織内のタンパク質を消化する。生体組織標本の有効活用及び試薬の消費量の軽減という観点から、特定の領域の面積及び分注量はできるだけ抑え、且つ、後述の質量分析を可能にするために十分な程度であることが好ましい。本発明においては、分注を行うためには、試薬溶液の微量供給が可能である公知の装置を特に限定することなく用いることができる。特に、インクジェット技術による機構を備えた分注装置を用いると良い。このような分注装置としては、ケミカルプリンタCHIP-1000(島津製作所製)等が挙げられる。
【0035】
なお、膜に酵素が吸着しないように、酵素の分注に先立ってポリビニルピロリドンなどを分注する処理を行うことが好ましい。この場合、メタノール水溶液に適当な濃度で溶解させたポリビニルピロリドン水溶液を用いると良い。ポリビニルピロリドンとしては、PVP-40などを用いることが好ましく、上記メタノール水溶液は10〜80(v/v)%とすることが好ましく、ポリビニルピロリドンは上記メタノール水溶液中0.1〜0.5(w/v)%とすることが好ましい。ポリビニルピロリドン水溶液の分注量としては、0.1〜10nlとすることができる。
【0036】
タンパク質消化酵素としては特に限定されないが、通常、トリプシンが用いられる。消化の条件としては、当業者が適宜決定することができる。トリプシン溶液の分注量としては、10〜50nlとすることができる。
【0037】
効率よく組織内消化を行うためには、消化酵素を供給された試料は20〜40℃に設定した恒温槽内に1〜24時間保存すると良い。例えば、保存温度と時間とを、それぞれ37℃、12時間とすることが好ましい。上記温度より低い場合は酵素の活性が低くなる傾向があり、上記温度より高い場合は酵素の活性が失われる傾向がある。また、上記保存時間より短い場合は、消化が十分に行われず、イオン化効率が低くなる傾向があり、上記保存時間より長い場合は、酵素の自己消化産物が多くなり、目的物質の検出強度が低くなる傾向がある。
【0038】
本発明では、組織内タンパク質の変性を行っているため、消化酵素による消化効率が従来に比べて優れている。
【0039】
<質量分析工程>
まず、生体組織標本上の消化された領域に対し、マトリックスを重ねて分注する。マトリックスとしては、タンパク質のMALDI質量分析に用いられるものを、特に限定することなく用いることができる。例えば、2,5−ジヒドロキシ安息香酸(DHBA)、5−メトキシサリチル酸を混合したDHBA(sDHB)、a−シアノ−4−ヒドロキシケイ皮酸(a-CHCA)、3,5−ジメトキシ−4−ヒドロキシケイ皮酸(シナピン酸)等を用いることができる。これらマトリックスは、アセトニトリル−トリフルオロ酢酸(TFA)水溶液などの適当な溶媒に溶解し、マトリックス溶液として用いる。例えばDHBAを用いる場合、上記アセトニトリル−TFA水溶液は、25〜50(v/v)%アセトニトリル−0.05〜1(v/v)% TFAを含む水溶液とすることが好ましく、マトリックス濃度は、上記アセトニトリル−TFA水溶液中5〜30mg/mlとすることが好ましい。マトリックス溶液の分注量としては、50〜200nlとすることができる。
【0040】
次に、マトリックス溶液を分注した箇所にレーザー照射し、MALDI質量分析を行う。MALDI質量分析装置としては、例えばAXIMA-QIT(島津製作所製)等を用いることができる。前述のように、本発明においては、組織内タンパク質の変性を行っているため、消化酵素による消化効率が従来に比べて優れている。このため、MS解析において、タンパク質のピークを比較的強い強度で検出することが可能である。このことは、MS/MS及びそれ以上の多段階質量分析をも可能にする。本発明では、多段階質量分析を行うことができるため、タンパク質のアミノ酸配列の決定や、翻訳後修飾の解析などに必要なスペクトル情報を得ることができる。
【実施例】
【0041】
<実施例1>
マウス脳組織の凍結切片をPVDF膜上で融解し、固着を行った。凍結切片が固着された膜を70(v/v)%エタノール水溶液中で30秒間振盪した。2回洗浄を行った後、乾燥させた。乾燥後、変性剤溶液中で12時間振盪した。ここで、変性剤の溶液の組成は、0.5M Tris/HCl(pH6.8)2ml、10(v/v)%SDS水溶液4ml、50(v/v)%グリセロール水溶液7ml、尿素8.4g、及びDTT0.1gである。この変性剤溶液に、凍結切片を融解接着させたPVDF膜を12時間浸した。この組織切片部分が透明になった後、洗浄溶液(40(v/v)%エタノール及び10(v/v)%酢酸を水中に含む溶液)で30秒間洗浄した。その後、染色溶液に7分間浸し、組織切片を染色した。ここで、染色溶液は、Direct blue(アルドリッチ、12、240−7)を終濃度が0.008(v/v)%になるように、前記の洗浄溶液に溶解したものである。染色を施した組織切片を、5分間洗浄溶液中で振盪した。この、染色及び洗浄の一連の作業を合計3回行った。その後、組織切片を超純水中で5分間振盪し、十分に乾燥させた。
【0042】
上記の処理を行った組織切片に対し、ケミカルプリンタ(CHIP-1000、島津製作所)を用いて以下の処理を行った。
ポリビニルピロリドン溶液(ポリビニルピロリドンを、60%(v/v)メタノール水溶液中に、0.25%(v/v)の濃度で含む溶液)をプリントした。プリント全量は、7nlであった。
消化酵素溶液(トリプシンを、25mM炭酸水素ナトリウム及び10(v/v)%2−プロパノールを含む水溶液中に、200μg/mlの濃度で含む溶液)をプリントした。プリント全量は、50nlであった。その後、組織切片を、温度を37℃に設定した恒温槽内で12時間湿潤条件で保存した。
マトリックス溶液(2,5−ジヒドロキシ安息香酸を、25%(v/v)アセトニトリル及び0.1%(v/v)TFAを含む水溶液中に、8mg/mlの濃度で含む溶液)をプリントした。プリント全量は、100nlであった。
試薬をプリントした箇所を、質量分析計(AXIMA-QIT、島津製作所)で測定を行った。
【0043】
<比較例1>
変性剤溶液の代わりに70(v/v)%エタノール水溶液を用いた以外は、実施例1と同じ操作を行った。
【0044】
<結果>
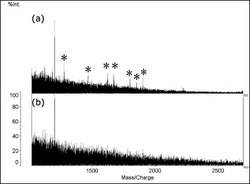
図1(a)に、実施例1で得られたMSスペクトルを、図1(b)に、比較例1で得られたMSスペクトルを示す。図1において、横軸は質量/電荷(Mass/Charge)、縦軸は相対強度を示し、*印は、それが付されているピークが、組織中のタンパク質に由来するピークであることを示す。図1が示すように、変性処理が行われた(a)では、比較的強度の強いピークが多く検出されていることがわかる。
【0045】
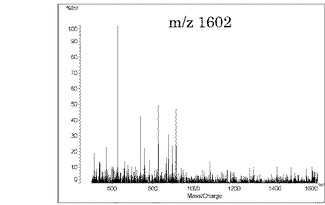
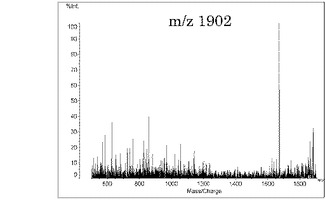
本発明の方法によって、上記のようにMSスペクトルでピークが多く検出されたため、これらのピークについてのMS/MS測定を行った。図1(a)における、m/z 1602をプレカーサイオンとしてMS/MS測定を行った結果を図2に、m/z 1902をプレカーサイオンとしてMS/MS測定を行った結果を図3に示す。図2及び図3において、横軸は質量/電荷(Mass/Charge)、縦軸は相対強度を示す。これらのスペクトルが示すように、本発明により非常に良好なMS/MSスペクトルの取得が可能になったことが分かる。
【図面の簡単な説明】
【0046】
【図1】変性剤溶液に12時間浸漬する前処理を行った実施例1で得られたMSスペクトル(a)、及び、エタノール溶液に12時間浸漬する前処理を行った比較例1で得られたMSスペクトル(b)である。
【図2】図1(a)におけるm/z 1602をプリカーサーイオンとしたMS/MSスペクトルである。
【図3】図1(a)におけるm/z 1902をプリカーサーイオンとしたMS/MSスペクトルである。
【技術分野】
【0001】
本発明は、ライフサイエンス分野、具体的には、細胞生物学、病理学、生化学などの医学・生物学分野に関する。特に本発明は、生体組織標本上で直接的に生体組織を質量分析する方法に関する。
【背景技術】
【0002】
生体組織を直接質量分析を行った例として、例えば、Stoeckli, M.; Chaurand, P.; Hallahan, E. D.; Caprioli, M. R. Nature Med. 7, 493-496 (2001)、Chaurand, P.; Schwartz, A. S.; Caprioli, M. R. Anal. Chem., 76, 86A-93A (2004)などにおいて、凍結生体組織切片を、直接質量分析ターゲットプレート上で融解し、70(v/v)%エタノール水溶液で洗浄し、酵素消化を行うことなくMSスペクトルを取得した技術が報告されている。
【0003】
また、例えば、Chaurand, P.; Schwartz, A. S.; Billheimer, D.; Xu, J. B; Crecelius, A.; Caprioli, M. R. Anal. Chem., 76, 1145-1155 (2004)においては、伝導性スライドガラスを用いて、上記技術におけるものと同様のMSスペクトルを取得した報告がされている。
【0004】
さらに、例えば、特開2004−347594号公報には、生体組織標本に対し、タンパク質染色、トリプシン消化、及びMALDI質量分析を行い、MSスペクトルを得たことが報告されている。
【0005】
一方、二次元電気泳動においては、等電点電気泳動(IEF)を行った後、SDS(ドデシル硫酸ナトリウム)ポリアクリルアミドゲル電気泳動(SDS−PAGE)により分子量に基づいて分離が行われる。SDS−PAGEを行う際に、一次元目のゲルをSDSを含む溶液で平衡化することにより、ゲル中のタンパク質をSDS化する。
【0006】
【非特許文献1】ストックリ・M(Stoeckli, M.)、ショーラン・P(Chaurand, P.)、ハラハン・E・D(Hallahan, E. D.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「ネイチャー・メディスン(Nature Medicine)」第7巻、p.493−496、2001年
【非特許文献2】ショーラン・P(Chaurand, P.)、シュワルツ・A・S(Schwartz, A. S.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「アナリティカル・ケミストリー(Analytical Chemistry)」、第76巻、p.86A−93A、2004年
【非特許文献3】ショーラン・P(Chaurand, P.)、シュワルツ・A・S(Schwartz, A. S.)、ビルハイマー・D(Billheimer, D.)、スー・J・B(Xu, J. B.)、クレセリウス・A(Crecelius, A.)、及びカプリオーリ・M・R(Caprioli, M. R.)、「アナリティカル・ケミストリー(Analytical Chemistry)」、第76巻、p.1145−1155、2004年
【特許文献1】特開2004−347594号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
上記の従来法では、MSスペクトルを取得することは可能であるが、酵素消化を行っていないため意味のあるMS/MSスペクトルを取得することはできない。また、単純に消化酵素を塗布しても消化を行うことはできないため、やはり意味のあるMS/MSスペクトルを取得することはできない。
【0008】
従って、質量分析を用いて生体組織内のタンパク質の同定を行うためには、予め、これまでの生化学的手法により組織の分離精製を行わなければならない。しかしながら、微量な物質を解析対象の試料とした場合、操作の途中で試料が失われる可能性もある。また、マトリックスの添加がコントロールされていないため、質量分析において解像度や再現性に問題がある。
【0009】
一般的にタンパク質の同定を質量分析によって行うには、多段階の質量分析によって得られたスペクトル情報が必要である。このためには、試料タンパク質を電気泳動などの手法で精製し、その後、変性させ消化酵素でペプチド化する。従って、生体組織中のタンパク質の同定を質量分析によって行うためには、上記に準じた手法で多段階の質量分析を行わなければならない。しかしながら、組織内では、タンパク質は立体構造を保っているため、消化酵素による消化効率が悪い。
【0010】
そこで本発明の目的は、生体組織の解剖学的構造を保持したまま、高い解析感度及び高い解析効率で組織を構成するタンパク質の質量分析を行うことができる、質量分析用試料の前処理方法を提供することにある。また、本発明の目的は、生体組織内の解剖学的構造を保持したまま、多段階質量分析を行うことができる、質量分析用試料の前処理方法を提供することにある。
【課題を解決するための手段】
【0011】
本発明者は、二次元電気泳動において平衡化のために用いられる手法を応用することによって、上記本発明の目的が達成されることを見出し、本発明を完成するに至った。
【0012】
本発明は、以下の発明を含む。
(1)生体組織標本に対し、
変性剤を供給させることによって、前記生体組織内のタンパク質を変性させる工程と、
染色剤を供給することによって、前記タンパク質を染色する工程と、
消化酵素を分注することによって、前記タンパク質を消化する工程と、
マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【0013】
下記(2)及び(3)は、上記生体組織標本について記載する。
(2)前記生体組織標本は凍結切片である、(1)に記載の質量分析手法。
(3)前記生体組織標本は、ポリビニリデンジフルオリド膜、ニトロセルロース膜、ナイロン膜から選ばれる膜の表面に固着されたものである、(1)又は(2)に記載の質量分析手法。
【0014】
下記(4)及び(5)は、上記変性剤について記載する。
(4)前記変性剤は、界面活性剤、尿素、及びグアジニン塩酸から選ばれる、(1)〜(3)のいずれかに記載の質量分析手法。
(5)前記界面活性剤が、ドデシル硫酸ナトリウム、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸、ポリオキシエチレン−p−t−オクチルフェニルエーテル、モノラウリン酸ソルビタン、及び1−O−n−オクチル−β−D−グルコピラノシドから選ばれる、(4)に記載の質量分析手法。
【0015】
下記(6)及び(7)は、上記変性剤とともに用いられうる還元剤について記載する。
(6)前記変性剤を、還元剤とともに用いる、(1)〜(5)のいずれかに記載の質量分析手法。
(7)前記還元剤は、2−メルカプトエタノール、ジチオスレイトール、トリス[2−カルボキシエチル]ホスフィン、及び2−メルカプトエタノールアミンから選ばれる、(6)に記載の質量分析手法。
【0016】
下記(8)は、上記分注の操作について記載する。
(8)前記タンパク質を消化する工程及び/又は前記質量分析を行う工程において、インクジェット技術を用いて分注操作を行う、(1)〜(7)のいずれかに記載の質量分析手法。
【0017】
下記(9)は、上記質量分析について記載する。
(9)前記質量分析を行う工程において、多段階質量分析を行う、(1)〜(8)のいずれかに記載の質量分析手法。
【発明の効果】
【0018】
本発明によると、生体組織の解剖学的構造を保持したまま、高い解析感度及び高い解析効率で組織を構成するタンパク質の質量分析を行うことができる、質量分析用試料の前処理方法を提供することができる。また、本発明によると、生体組織内の解剖学的構造を保持したまま、多段階質量分析を行うことができる、質量分析用試料の前処理方法を提供することができる。
【発明を実施するための最良の形態】
【0019】
本発明において直接とは、生体組織標本における解剖学的構造を保持した状態で、ということを意味する。本発明においては、生体組織標本に対し各処理液を直接作用させることにより、組織内タンパク質の処理を組織内にて行い、得られた処理済生体組織を質量分析用試料として直接質量分析を行う。具体的には、本発明においては、組織内タンパク質の変性、タンパク質の染色、及びタンパク質の消化の各処理を行い、MALDI質量分析を行う。
【0020】
本発明における生体組織標本は、生体内における解剖学的構造を保持した状態を有するものであれば、特に限定されない。このような状態を得るために、生体組織が適当な包埋剤によって包埋されたものを用いることができる。包埋剤としては、生体試料の適性に応じて特に限定することなく用いることができる。例えば、水、パラフィン、セロイジン、カーボワックス、ゼラチン、アルブミン、アガロース、エポキシ樹脂、ポリエステル樹脂等や、グリコールメタクリレート等の水溶性樹脂等を用いることができ、これら包埋剤は、当業者が適宜選択することができる。このような生体標本としては、例えば凍結切片等の未固定試料、パラフィン包埋切片等の固定化試料等を用いることができる。
【0021】
前記生体標本は、膜及び/又は支持体の表面に固着させて使用することができる。膜としては、ポリビニリデンジフルオリド(PVDF)、ニトロセルロース、ポリアミド、ポリエチレン等の有機合成高分子及びその誘導体を材料とした膜を挙げることができる。ポリアミドとしては、ナイロン等が挙げられる。支持体としては、ガラス製支持体、樹脂製支持体、金属製支持体等が挙げられる。本発明では、質量分析法を用いて解析するため、前記支持体には好ましくは金属製支持体、例えば質量分析用サンプルプレートを用い、生体標本を膜に固着したものを前記サンプルプレートに貼り付けるか、又は前記サンプルプレートに生体標本を固着して用いることができる。凍結切片を生体組織標本とする場合、凍結切片を膜へ載せ融解させれば固着が可能となる。また、サンプルプレートへの固着には導電性両面粘着テープ等を用いると良い。
【0022】
上記の生体標本は、公知の方法を用いて作製することができる。生体標本は、作製後、適宜、洗浄などを行い、乾燥させておくことができる。
【0023】
<変性工程>
このような生体組織標本に対し、適当な変性剤を供給することによって、生体組織内のタンパク質を変性させる。変性剤としては、タンパク質の一次構造を維持したまま高次構造のみを破壊するものであれば、特に限定されない。例えば、変性剤は、各種界面活性剤、尿素、グアジニン塩酸等から選ばれる。界面活性剤としては、陰イオン性界面活性剤、陽イオン性界面活性剤、両性界面活性剤、及び非イオン性界面活性剤のいずれも用いることができる。例えば、陰イオン界面活性剤としては、ドデシル硫酸ナトリウム(SDS)などが挙げられる。両性界面活性剤としては、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸(CHAPS)などが挙げられる。非イオン性界面活性剤としては、Triton X-100 (ポリオキシエチレン−p−t−オクチルフェニルエーテル)、Tween 20 (モノラウリン酸ソルビタン)、1−O−n−オクチル−β−D−グルコピラノシドなどが挙げられる。
【0024】
なお、これら変性剤は、1種又は複数種を選ぶことができる。複数種を選ぶ場合は、その組み合わせは、選ばれた変性剤が互いに相互作用しないものであれば特に限定されない。また、変性剤は、通常水溶液の形態で供給することができる。
【0025】
変性剤濃度としては、1〜10M、好ましくは4〜8Mとすることができる。上記濃度より低い濃度の場合、タンパク質の変性が不十分となる傾向がある。また、上記濃度より高い濃度の場合、組織切片が剥がれ落ちる傾向がある。界面活性剤濃度としては0.05%〜2%、好ましくは0.1〜1%とすることができる(単位%は、界面活性剤の形態によりw/v%或いはv/v%となりうる)。上記濃度より低い濃度の場合、タンパク質の変性が不十分となる傾向がある。また、上記濃度より高い濃度の場合、組織切片上に界面活性剤が残留し質量スペクトルにポリマー由来のピークが検出される傾向がある。
【0026】
変性剤水溶液には、通常、トリス塩酸緩衝液、リン酸緩衝液、リン酸緩衝食塩水、酢酸緩衝液などから選ばれるバッファーが含まれる。トリス塩酸は10mM〜100mM、リン酸緩衝液は0.1M〜0.3M、リン酸緩衝食塩水は0.05M〜0.2M、酢酸緩衝液は0.1M〜0.2Mの濃度のものを使用することができる。上記濃度より低い濃度の場合、組織切片が剥離する傾向がある。また、上記濃度より高い濃度の場合、質量分析において塩の影響によりバックグラウンドが大きくなる傾向がある。なお、これらバッファーは、1種又は複数種を選ぶことができる。またバッファーのpHはトリス塩酸、リン酸緩衝液、リン酸緩衝食塩水においては6〜9、好ましくはpH6.5〜8.0、酢酸緩衝液ではpH3〜4とすることができる。
【0027】
変性剤水溶液には、さらに、ジスルフィド結合の切断やSH基の保護などのために、還元剤を含んでいても良い。還元剤としては、例えば、2−メルカプトエタノール、DTT(ジチオスレイトール(dithiothreitol))、TCEP(トリス[2−カルボキシエチル]ホスフィン(Tris[2-carboxyethyl]phosphine))、2−MEA(2−メルカプトエタノールアミン(2-Mercaptoethanolamine))等が挙げられる。これら還元剤は、1種又は複数種を選ぶことができる。
【0028】
変性剤水溶液中の還元剤濃度としては、10〜70mM、好ましくは40mM〜50mMとすることができる。上記濃度より低い濃度の場合、酵素消化において、反応の効率が低下する傾向がある。また、上記濃度より高い濃度の場合、質量分析において塩の影響によりバックグラウンドが大きくなる傾向がある。
【0029】
変性剤水溶液には、さらに、安定剤を含んでいても良い。安定剤としては、例えば、グリセロール(グリセリン)等が挙げられる。
【0030】
変性剤水溶液中の安定剤濃度としては、0.1〜50(v/v)%、好ましくは20〜50(v/v)%とすることができる。上記濃度より高い濃度の場合、上記濃度より高い濃度の場合、変性剤の粘性により取扱が困難になる傾向がある。
【0031】
生体標本中のタンパク質の変性は、上記の変性剤水溶液を、生体組織標本に供給することにより行う。具体的には、生体組織標本を上記の変性剤水溶液に浸漬し振盪するか、エアブラシ等を用いて霧状にした変性剤水溶液を生体組織標本上へ噴霧し、湿潤条件で放置するとよい。振盪時間および湿潤条件での放置時間は3〜24時間とすることができる。変性を行った生体標本は、適宜洗浄などを行うことができる。
【0032】
<染色工程>
上記工程によりタンパク質が変性した生体組織標本に対し、適当な染色剤を供給することによって、生体組織内のタンパク質を染色する。なお、生体組織標本作製の際にパラフィンなど疎水性の包埋剤を用いた場合は、染色工程に先立って包埋剤を除去しておく。
【0033】
染色剤としては、通常タンパク質の染色に用いられるものを、特に限定することなく用いることができる。例えば、メチルレッド等のモノアゾ色素;ポンソーSなどのジアゾ色素;Direct Blue71などのトリアゾ色素;ファストレッドなどのアゾイック色素;オーラミンなどのジフェニルメタン色素;ブリリアントグリーン等のジアミノトリフェニルメタン色素;ファストグリーン等のトリアミノトリフェニルメタン色素;ローダミンBなどのキサンテン色素;ローダミン3Gなどのフルオロン色素;アクリジンオレンジ等のアクリジン色素;クレシルファストバイオレット等のオキサジン色素;トルイジンブルー等のチアジン色素;アリザリンレッドS等のアントラキノン色素;ヘマトキシリン等の天然色素等を用いることができる。これら染色剤を用い、通常の方法に従った操作を行うことによって染色を行うことができる。例えば、エタノール−酢酸水溶液中に適当な濃度で溶解させた染色剤水溶液へ生体組織標本の浸漬及び振盪を行い、その後洗浄する操作を繰り返すと良い。Direct Blue 71を用いる場合、上記エタノール−酢酸水溶液は、10〜70(v/v)%エタノール−1〜20(v/v)%酢酸を含む水溶液とすることが好ましく、染色剤濃度は上記エタノール−酢酸水溶液中0.005〜0.01(w/v)%とすることが好ましく、上記振盪は2.5〜7分行うことが好ましく、振盪及び洗浄の操作は、3〜6回繰り返して行うことが好ましい。
染色を行った生体標本は、十分に乾燥させると良い。
【0034】
<消化工程>
上記工程によりタンパク質が染色された生体組織標本に対し、適当なタンパク質消化酵素を分注することによって、生体組織内のタンパク質を消化する。本発明において分注とは、生体組織標本上の特定の領域に対して、試薬溶液を供給することをいう(後述の質量分析工程においても同じ)。すなわち、消化工程では、上記工程によりタンパク質が染色された生体組織標本の特定の領域に対し、適当なタンパク質消化酵素を供給することによって、特定の領域における生体組織内のタンパク質を消化する。生体組織標本の有効活用及び試薬の消費量の軽減という観点から、特定の領域の面積及び分注量はできるだけ抑え、且つ、後述の質量分析を可能にするために十分な程度であることが好ましい。本発明においては、分注を行うためには、試薬溶液の微量供給が可能である公知の装置を特に限定することなく用いることができる。特に、インクジェット技術による機構を備えた分注装置を用いると良い。このような分注装置としては、ケミカルプリンタCHIP-1000(島津製作所製)等が挙げられる。
【0035】
なお、膜に酵素が吸着しないように、酵素の分注に先立ってポリビニルピロリドンなどを分注する処理を行うことが好ましい。この場合、メタノール水溶液に適当な濃度で溶解させたポリビニルピロリドン水溶液を用いると良い。ポリビニルピロリドンとしては、PVP-40などを用いることが好ましく、上記メタノール水溶液は10〜80(v/v)%とすることが好ましく、ポリビニルピロリドンは上記メタノール水溶液中0.1〜0.5(w/v)%とすることが好ましい。ポリビニルピロリドン水溶液の分注量としては、0.1〜10nlとすることができる。
【0036】
タンパク質消化酵素としては特に限定されないが、通常、トリプシンが用いられる。消化の条件としては、当業者が適宜決定することができる。トリプシン溶液の分注量としては、10〜50nlとすることができる。
【0037】
効率よく組織内消化を行うためには、消化酵素を供給された試料は20〜40℃に設定した恒温槽内に1〜24時間保存すると良い。例えば、保存温度と時間とを、それぞれ37℃、12時間とすることが好ましい。上記温度より低い場合は酵素の活性が低くなる傾向があり、上記温度より高い場合は酵素の活性が失われる傾向がある。また、上記保存時間より短い場合は、消化が十分に行われず、イオン化効率が低くなる傾向があり、上記保存時間より長い場合は、酵素の自己消化産物が多くなり、目的物質の検出強度が低くなる傾向がある。
【0038】
本発明では、組織内タンパク質の変性を行っているため、消化酵素による消化効率が従来に比べて優れている。
【0039】
<質量分析工程>
まず、生体組織標本上の消化された領域に対し、マトリックスを重ねて分注する。マトリックスとしては、タンパク質のMALDI質量分析に用いられるものを、特に限定することなく用いることができる。例えば、2,5−ジヒドロキシ安息香酸(DHBA)、5−メトキシサリチル酸を混合したDHBA(sDHB)、a−シアノ−4−ヒドロキシケイ皮酸(a-CHCA)、3,5−ジメトキシ−4−ヒドロキシケイ皮酸(シナピン酸)等を用いることができる。これらマトリックスは、アセトニトリル−トリフルオロ酢酸(TFA)水溶液などの適当な溶媒に溶解し、マトリックス溶液として用いる。例えばDHBAを用いる場合、上記アセトニトリル−TFA水溶液は、25〜50(v/v)%アセトニトリル−0.05〜1(v/v)% TFAを含む水溶液とすることが好ましく、マトリックス濃度は、上記アセトニトリル−TFA水溶液中5〜30mg/mlとすることが好ましい。マトリックス溶液の分注量としては、50〜200nlとすることができる。
【0040】
次に、マトリックス溶液を分注した箇所にレーザー照射し、MALDI質量分析を行う。MALDI質量分析装置としては、例えばAXIMA-QIT(島津製作所製)等を用いることができる。前述のように、本発明においては、組織内タンパク質の変性を行っているため、消化酵素による消化効率が従来に比べて優れている。このため、MS解析において、タンパク質のピークを比較的強い強度で検出することが可能である。このことは、MS/MS及びそれ以上の多段階質量分析をも可能にする。本発明では、多段階質量分析を行うことができるため、タンパク質のアミノ酸配列の決定や、翻訳後修飾の解析などに必要なスペクトル情報を得ることができる。
【実施例】
【0041】
<実施例1>
マウス脳組織の凍結切片をPVDF膜上で融解し、固着を行った。凍結切片が固着された膜を70(v/v)%エタノール水溶液中で30秒間振盪した。2回洗浄を行った後、乾燥させた。乾燥後、変性剤溶液中で12時間振盪した。ここで、変性剤の溶液の組成は、0.5M Tris/HCl(pH6.8)2ml、10(v/v)%SDS水溶液4ml、50(v/v)%グリセロール水溶液7ml、尿素8.4g、及びDTT0.1gである。この変性剤溶液に、凍結切片を融解接着させたPVDF膜を12時間浸した。この組織切片部分が透明になった後、洗浄溶液(40(v/v)%エタノール及び10(v/v)%酢酸を水中に含む溶液)で30秒間洗浄した。その後、染色溶液に7分間浸し、組織切片を染色した。ここで、染色溶液は、Direct blue(アルドリッチ、12、240−7)を終濃度が0.008(v/v)%になるように、前記の洗浄溶液に溶解したものである。染色を施した組織切片を、5分間洗浄溶液中で振盪した。この、染色及び洗浄の一連の作業を合計3回行った。その後、組織切片を超純水中で5分間振盪し、十分に乾燥させた。
【0042】
上記の処理を行った組織切片に対し、ケミカルプリンタ(CHIP-1000、島津製作所)を用いて以下の処理を行った。
ポリビニルピロリドン溶液(ポリビニルピロリドンを、60%(v/v)メタノール水溶液中に、0.25%(v/v)の濃度で含む溶液)をプリントした。プリント全量は、7nlであった。
消化酵素溶液(トリプシンを、25mM炭酸水素ナトリウム及び10(v/v)%2−プロパノールを含む水溶液中に、200μg/mlの濃度で含む溶液)をプリントした。プリント全量は、50nlであった。その後、組織切片を、温度を37℃に設定した恒温槽内で12時間湿潤条件で保存した。
マトリックス溶液(2,5−ジヒドロキシ安息香酸を、25%(v/v)アセトニトリル及び0.1%(v/v)TFAを含む水溶液中に、8mg/mlの濃度で含む溶液)をプリントした。プリント全量は、100nlであった。
試薬をプリントした箇所を、質量分析計(AXIMA-QIT、島津製作所)で測定を行った。
【0043】
<比較例1>
変性剤溶液の代わりに70(v/v)%エタノール水溶液を用いた以外は、実施例1と同じ操作を行った。
【0044】
<結果>
図1(a)に、実施例1で得られたMSスペクトルを、図1(b)に、比較例1で得られたMSスペクトルを示す。図1において、横軸は質量/電荷(Mass/Charge)、縦軸は相対強度を示し、*印は、それが付されているピークが、組織中のタンパク質に由来するピークであることを示す。図1が示すように、変性処理が行われた(a)では、比較的強度の強いピークが多く検出されていることがわかる。
【0045】
本発明の方法によって、上記のようにMSスペクトルでピークが多く検出されたため、これらのピークについてのMS/MS測定を行った。図1(a)における、m/z 1602をプレカーサイオンとしてMS/MS測定を行った結果を図2に、m/z 1902をプレカーサイオンとしてMS/MS測定を行った結果を図3に示す。図2及び図3において、横軸は質量/電荷(Mass/Charge)、縦軸は相対強度を示す。これらのスペクトルが示すように、本発明により非常に良好なMS/MSスペクトルの取得が可能になったことが分かる。
【図面の簡単な説明】
【0046】
【図1】変性剤溶液に12時間浸漬する前処理を行った実施例1で得られたMSスペクトル(a)、及び、エタノール溶液に12時間浸漬する前処理を行った比較例1で得られたMSスペクトル(b)である。
【図2】図1(a)におけるm/z 1602をプリカーサーイオンとしたMS/MSスペクトルである。
【図3】図1(a)におけるm/z 1902をプリカーサーイオンとしたMS/MSスペクトルである。
【特許請求の範囲】
【請求項1】
生体組織標本に対し、
変性剤を供給することによって、前記生体組織内のタンパク質を変性させる工程と、
染色剤を供給することによって、前記タンパク質を染色する工程と、
消化酵素を分注することによって、前記タンパク質を消化する工程と、
マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【請求項2】
前記生体組織標本は凍結切片である、請求項1に記載の質量分析手法。
【請求項3】
前記生体組織標本は、ポリビニリデンジフルオリド膜、ニトロセルロース膜、ナイロン膜から選ばれる膜の表面に固着されたものである、請求項1又は2に記載の質量分析手法。
【請求項4】
前記変性剤は、界面活性剤、尿素、及びグアジニン塩酸から選ばれる、請求項1〜3のいずれか1項に記載の質量分析手法。
【請求項5】
前記界面活性剤が、ドデシル硫酸ナトリウム、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸、ポリオキシエチレン−p−t−オクチルフェニルエーテル、モノラウリン酸ソルビタン、及び1−O−n−オクチル−β−D−グルコピラノシドから選ばれる、請求項4に記載の質量分析手法。
【請求項6】
前記変性剤を、還元剤とともに用いる、請求項1〜5のいずれか1項に記載の質量分析手法。
【請求項7】
前記還元剤は、2−メルカプトエタノール、ジチオスレイトール、トリス[2−カルボキシエチル]ホスフィン、及び2−メルカプトエタノールアミンから選ばれる、請求項6に記載の質量分析手法。
【請求項8】
前記タンパク質を消化する工程及び/又は前記質量分析を行う工程において、インクジェット技術を用いて分注操作を行う、請求項1〜7のいずれか1項に記載の質量分析手法。
【請求項9】
前記質量分析を行う工程において、多段階質量分析を行う、請求項1〜8のいずれか1項に記載の質量分析手法。
【請求項1】
生体組織標本に対し、
変性剤を供給することによって、前記生体組織内のタンパク質を変性させる工程と、
染色剤を供給することによって、前記タンパク質を染色する工程と、
消化酵素を分注することによって、前記タンパク質を消化する工程と、
マトリックスを分注し、MALDI質量分析装置を用いることによって、質量分析を行う工程とを含む、生体組織の直接質量分析手法。
【請求項2】
前記生体組織標本は凍結切片である、請求項1に記載の質量分析手法。
【請求項3】
前記生体組織標本は、ポリビニリデンジフルオリド膜、ニトロセルロース膜、ナイロン膜から選ばれる膜の表面に固着されたものである、請求項1又は2に記載の質量分析手法。
【請求項4】
前記変性剤は、界面活性剤、尿素、及びグアジニン塩酸から選ばれる、請求項1〜3のいずれか1項に記載の質量分析手法。
【請求項5】
前記界面活性剤が、ドデシル硫酸ナトリウム、3−[(3−コールアミドプロピル) ジメチルアンモニオ]−1−プロパンスルホン酸、ポリオキシエチレン−p−t−オクチルフェニルエーテル、モノラウリン酸ソルビタン、及び1−O−n−オクチル−β−D−グルコピラノシドから選ばれる、請求項4に記載の質量分析手法。
【請求項6】
前記変性剤を、還元剤とともに用いる、請求項1〜5のいずれか1項に記載の質量分析手法。
【請求項7】
前記還元剤は、2−メルカプトエタノール、ジチオスレイトール、トリス[2−カルボキシエチル]ホスフィン、及び2−メルカプトエタノールアミンから選ばれる、請求項6に記載の質量分析手法。
【請求項8】
前記タンパク質を消化する工程及び/又は前記質量分析を行う工程において、インクジェット技術を用いて分注操作を行う、請求項1〜7のいずれか1項に記載の質量分析手法。
【請求項9】
前記質量分析を行う工程において、多段階質量分析を行う、請求項1〜8のいずれか1項に記載の質量分析手法。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2007−51957(P2007−51957A)
【公開日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願番号】特願2005−238037(P2005−238037)
【出願日】平成17年8月18日(2005.8.18)
【出願人】(000001993)株式会社島津製作所 (3,708)
【出願人】(504261077)大学共同利用機関法人自然科学研究機構 (156)
【Fターム(参考)】
【公開日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願日】平成17年8月18日(2005.8.18)
【出願人】(000001993)株式会社島津製作所 (3,708)
【出願人】(504261077)大学共同利用機関法人自然科学研究機構 (156)
【Fターム(参考)】
[ Back to top ]