
生分解性多孔性繊維およびその製造方法

【課題】簡便に機能性薬剤を内包する多孔性繊維が得られる方法;所望の繊維の孔のサイズを有する多孔性繊維が得られる方法;同方法により得られる機能性薬剤を内包する多孔性繊維;及び同方法により得られる機能性薬剤を内包する薬剤徐放用多孔性繊維を提供すること。
【解決手段】生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、前記多孔性繊維に機能性薬剤を内包させる工程、とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
【解決手段】生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、前記多孔性繊維に機能性薬剤を内包させる工程、とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、機能性薬剤を内包する、生分解性ポリエステルを原料とする多孔性繊維およびその製造方法に関する。
【背景技術】
【0002】
保温等の機能付与や軽量化等を目的とした孔(ポア)を有する繊維の作製方法として、樹脂中に、易溶解性ポリマー、発泡性物質等のポアを形成する物質をあらかじめ混合した後に紡糸し、その後の工程で、ポアを形成する物質を取り除く方法が知られており、このような方法が特許文献1〜3等に開示されている。
【0003】
例えば、特許文献1には、難溶解性ポリマーと易溶解性ポリマーとの混合物を溶融紡糸し、糸を作製後に易溶解性ポリマーを除去することで、ポアを有する繊維を作製する方法が記載されている。しかしながら、この方法では、紡糸前に難溶解性ポリマーと易溶解性ポリマーとを混合及び分散させるための技術が必要である。また、ポアサイズの制御は困難である。
【0004】
特許文献2には、樹脂中に発泡性カプセルを混合した後に紡糸することで、多孔性繊維を作製する方法が記載されている。この方法により、多孔性繊維の作製は可能であるが、ポアサイズやポアの含有量の制御は困難であると考えられる。
【0005】
上記技術は、ともに易溶解性ポリマー、発泡性物質等のポアとなりうるものをあらかじめ樹脂に混合しておく技術であり、事前の混合や除去処理が必要になる。また、繊維作製後に、易溶解性ポリマー、発泡性物質等を除去するため、繊維自体の強度は低下する方向になる。
【0006】
ところで、生分解性ポリエステルの一つとして、ポリヒドロキシアルカン酸(PHA)類が知られている。PHA類は、生分解性及び生体適合性を有することから、繊維やフィルム等の各種成形品への利用が検討されている。例えば、PHA類を原料とする繊維は、手術用縫合糸等の医療用用具、釣り糸、漁網等の水産業用用具、繊維等の衣料用材料、不織布、ロープ等の建築用材料、食品その他の包装用材料等として大きな需要を見込むことができる。
【0007】
ポリ(3−ヒドロキシブタン酸)(以下、「P(3HB)」ともいう。)等のPHA類は、自然界に存在する多くの微生物により菌体内貯蔵物質として合成される。このようなP(3HB)産生微生物から得られるP(3HB)は、生分解性製品の原料として期待されている。しかしながら、P(3HB)は硬くて脆いため、物性の向上が求められていた。
【0008】
PHA類の繊維の物性を向上させる方法として、P(3HB)の共重合体(コポリマー)化による方法が知られている。PHA類のコポリマーは、モノマーの種類や組成を変化させることで、多様な物性を示すことが知られている。PHA類のコポリマーとして、弾性に富むゴム状で生体適合性に優れるポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)[P(3HB−co−4HB)]、結晶性の硬いプラスチック状のポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)[P(3HB−co−3HV)]、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシへキサン酸)[P(3HB−co−3HH)]等が知られている。
【0009】
また、PHA類の紡糸技術により、繊維の物性を向上させる方法も開発されており、このような方法が特許文献4〜8等に開示されている。特許文献4には、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷・固化させて非晶質の繊維を作製し、非晶質の繊維をガラス転移点温度+15℃以下に保冷して結晶化繊維を作製し、結晶化繊維を延伸、熱処理することによる繊維の作製方法が記載されている。この方法により、由来によって異なるPHA類の分子量、ポリマー組成等に関わらず、高強度な繊維を得ることができる。しかしながら、この方法により作製された繊維の内部構造の詳細については、研究されていなかった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2005−213688号公報
【特許文献2】特開2004−238760号公報
【特許文献3】特許第3808400号公報
【特許文献4】国際公開WO2006/038373号パンフレット
【特許文献5】特許第4562316号公報
【特許文献6】特開2010−126852号公報
【特許文献7】特開2009−506861号公報
【特許文献8】特開2004−084118号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、簡便に機能性薬剤を内包する多孔性繊維が得られる方法;所望の繊維の孔のサイズを有する多孔性繊維が得られる方法;同方法により得られる機能性薬剤を内包する多孔性繊維;及び同方法により得られる機能性薬剤を内包する薬剤徐放用多孔性繊維を提供することを課題とする。
【課題を解決するための手段】
【0012】
本発明者等は、上記課題を解決すべく鋭意検討を行った結果、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷・固化させて非晶質の繊維を作製し、非晶質の繊維をガラス転移点温度+15℃以下に保冷して結晶化繊維を作製し、結晶化繊維を延伸、熱処理することによる繊維の作製方法において、繊維内部に多数のポアが形成されていることを知見した。また、このポアのサイズや数といったポアの特性を、保冷時間を調整することにより、制御できることを知見した。さらに、このような繊維の作製方法により得られた繊維に機能性薬剤を内包させることにより、上記の課題に合致した機能性薬剤を内包する多孔性繊維及びその製造方法が達成されることを知見し、本発明を完成した。
【0013】
すなわち、本発明の要旨は以下の通りである。
(1)生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
(2)さらに、前記多孔性繊維を延伸する工程を有する、(1)に記載の方法。
(3)前記多孔性繊維を得る工程において、保冷時間が所望の繊維の孔のサイズを与えるように調整されている、(1)又は(2)に記載の方法。
(4)前記生分解性ポリエステルがポリヒドロキシアルカン酸である、(1)〜(3)の
何れかに記載の方法。
(5)(1)〜(4)の何れかに記載の方法により製造される、機能性薬剤を内包する多孔性繊維。
(6)薬剤徐放用である、(5)に記載の繊維。
【発明の効果】
【0014】
本発明により、簡便に機能性薬剤を内包する多孔性繊維が得られる方法及び所望の繊維の孔のサイズを有する多孔性繊維が得られる方法を提供することができる。このような方法により、簡便に機能性薬剤を内包する多孔性繊維及び機能性薬剤を内包する薬剤徐放用多孔性繊維を得ることができる。
【図面の簡単な説明】
【0015】
【図1】微結晶核延伸法を経て作製した繊維の走査型電子顕微鏡像を示す図(写真)である。(a)繊維の横断面像、(b)繊維の横断面像の拡大像、(c)繊維の縦断面像である。
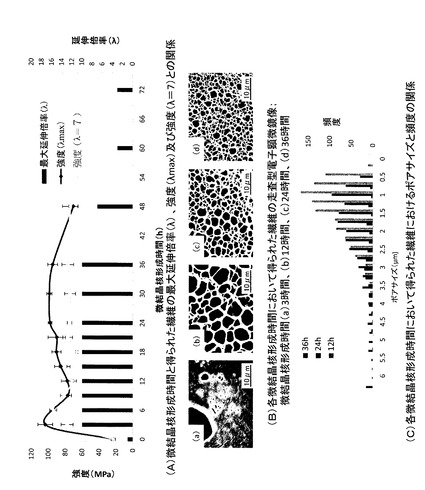
【図2】微結晶核形成時間の繊維の物性への影響を示す図(写真)である。(A)微結晶核形成時間と得られた繊維の最大延伸倍率(λ)、強度(λmax)及び強度(λ=7)との関係を示すグラフである。(B)各微結晶核形成時間において得られた繊維の走査型電子顕微鏡像(横断面像)である。(a)微結晶核形成時間3時間の繊維の横断面像、(b)微結晶核形成時間12時間の繊維の横断面像、(c)微結晶核形成時間24時間の繊維の横断面像、(d)微結晶核形成時間36時間の繊維の横断面像である。(C)各微結晶核形成時間において得られた繊維におけるポアサイズと頻度との関係を示すグラフである。
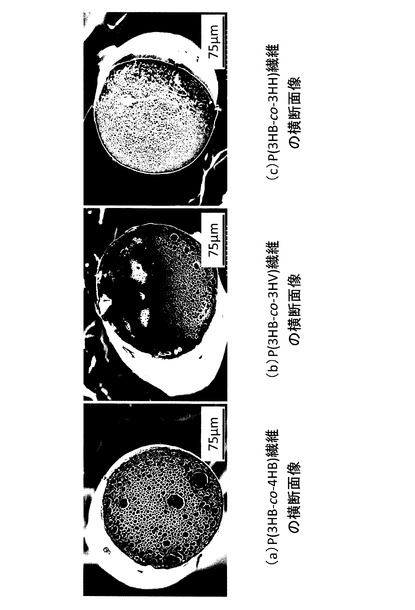
【図3】各種共重合体の繊維の走査型電子顕微鏡像を示す図(写真)である。(a)P(3HB−co−4HB)繊維の横断面像、(b)P(3HB−co−3HV)繊維の横断面像、(c)P(3HB−co−3HH)繊維の横断面像である。
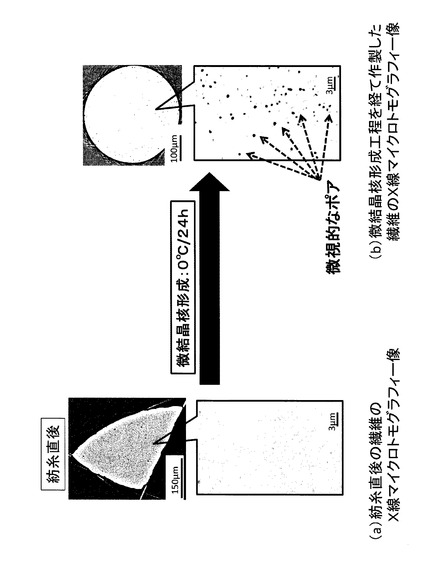
【図4】紡糸直後の繊維及び微結晶核形成工程を経て作製した繊維のX線マイクロトモグラフィー像を示す図(写真)である。(a)紡糸直後の繊維のX線マイクロトモグラフィー像、(b)微結晶核形成工程を経て作製した繊維のX線マイクロトモグラフィー像である。
【図5】40℃,60℃,80℃における、繊維への各濃度の薬剤溶液の浸透性を光学顕微鏡で観察した図(写真)である。DMSOに溶解させたバンコマイシン塩酸塩の濃度は0,1,3,5,10,15,25%である。薬剤溶液が繊維内へ浸透し、空孔を埋めると光が透過して透明になる。
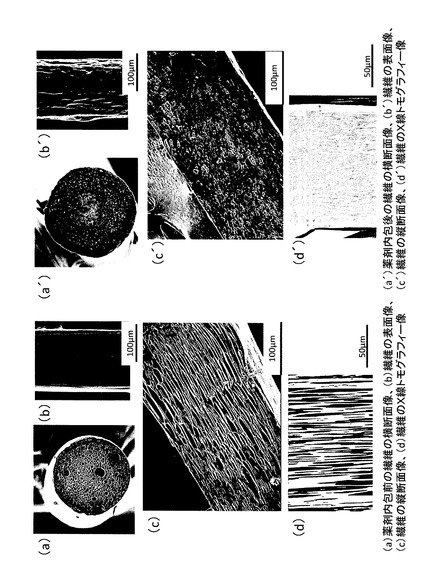
【図6】薬剤内包前及び薬剤内包後の繊維の走査型電子顕微鏡像(a〜c,a’〜c’)とX線マイクロトモグラフィー像(d,d’)を示す図(写真)である。(a)薬剤内包前の繊維の横断面像、(b)繊維の表面像、(c)繊維の縦断面像、(d)繊維の縦断面のX線トモグラフィー像である。(a’)薬剤内包後の繊維の横断面像、(b’)繊維の表面像、(c’)繊維の縦断面像、(d’)繊維の縦断面のX線トモグラフィー像である。
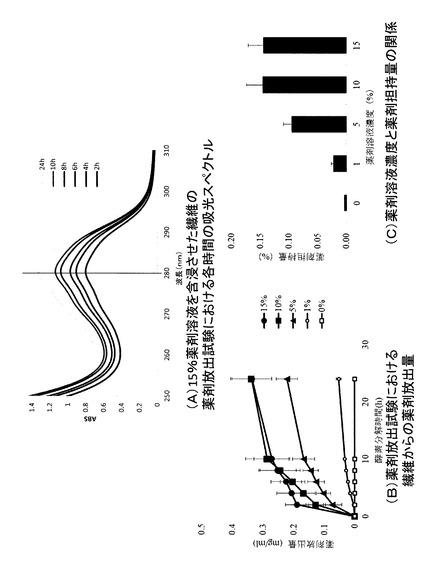
【図7】薬剤内包繊維の酵素分解による薬剤徐放性能及び薬剤担持量を示す図である。(A)15%薬剤溶液を内包させた繊維の薬剤放出試験における各時間の吸光スペクトルを示すグラフである。(B)各濃度の薬剤溶液を内包させた繊維の薬剤放出試験における各時間の薬剤放出量を示すグラフである。(C)内包させた薬剤溶液濃度と薬剤担持量との関係を示すグラフである。
【発明を実施するための形態】
【0016】
以下、本発明の実施の形態について、説明する。
(1)繊維の製造方法
【0017】
本発明の繊維の製造方法は、
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法である。
【0018】
以下、本発明の方法につき、各工程毎に説明する。
(溶融紡糸工程)
本発明の製造方法では、生分解性ポリエステルを含む組成物を繊維の原料として用いる。「生分解性ポリエステルを含む組成物」とは、生分解性ポリエステルを必須成分とし、それ以外の任意成分を含んでもよい組成物を意味する。好ましい生分解性ポリエステルとしては、ポリヒドロキシアルカン酸(PHA)類等が挙げられる。好ましいポリヒドロキシアルカン酸のモノマーとしては、3−ヒドロキシブタン酸、4−ヒドロキシブタン酸、3−ヒドロキシバレリル酸、3−ヒドロキシヘキサン酸、6−ヒドロキシヘキサン酸等が挙げられる。
【0019】
本発明に用いるPHA類としては、上記ヒドロキシアルカン酸のモノマーから選ばれる1種からなるホモポリマーであってよく、また、これらのヒドロキシアルカン酸のモノマーから選ばれる2種以上からなるコポリマーであってもよい。好ましいホモポリマーとしては、P(3HB)が挙げられる。好ましいコポリマーとしては、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシヘキサン酸)、ポリ(3−ヒドロキシブタン酸−co−6−ヒドロキシヘキサン酸)、ポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)等の3−ヒドロキシブタン酸とその他のヒドロキシアルカン酸からなるコポリマーが挙げられる。
【0020】
一般に、PHA類を合成する方法としては、発酵合成法と化学合成法とがある。化学合成法は、通常の有機合成の手法に従って、PHA類を化学合成する方法である。化学合成法として、具体的には、例えば、(R)-β-ブチロラクトン、ε-カプロラクトン等の脂
肪酸ラクトンを、触媒下で開環重合すること等により合成することができる(Abe et al., Macromolecules, 28, 7630 (1995))。
【0021】
発酵合成法は、PHA類生産能を有する微生物を培養し、その菌体内に蓄積されるPHA類を取り出す方法である。発酵合成法で利用できる微生物としては、PHA類生産能を有する微生物であれば特に限定されない。ポリヒドロキシブタン酸(以下、「PHB」ともいう)生産菌としては、ラルストニア・ユートロファ(Ralstonia eutropha)等のラルストニア属、アルカリゲネス・ラタス(Alcaligenes latus)、アルカリゲネス・ファエ
カリス(Alcaligenes faecalis)等のアルカリゲネス属をはじめ60種以上の天然微生物が知られており、これらの微生物ではPHBが菌体内に蓄積される。また、ヒドロキシブタン酸とその他のヒドロキシアルカン酸とのコポリマー生産菌としては、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)及びポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシヘキサン酸)生産菌であるアエロモナス・キャビエ(Aeromonas caviae)、ポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)生産菌であるラルストニア・ユートロファ(Ralstonia eutropha)等が知られている。
【0022】
発酵合成法においては、通常これらの微生物を、炭素源、窒素源、無機イオン及び必要に応じその他の有機成分を含有する通常の培地で培養することにより、菌体内にPHAを蓄積させることができる。菌体からのPHAの採取は、クロロホルム等の有機溶媒による
抽出や、菌体成分をリゾチーム等の酵素で分解した後、PHAグラニュールを濾別する方法等により実施できる。
【0023】
また、発酵合成法の一態様として、PHA合成遺伝子を含む組換えDNAを導入して形質転換させた微生物を培養し、その菌体内に生成したPHAを採取する方法が挙げられる。この方法においては、ラルストニア・ユートロファ等のPHA生産菌を直接培養する場合と異なり、形質転換体は菌体内にPHA分解酵素を持たないため、格段に高分子量のPHAを蓄積することができる。
【0024】
このような形質転換株として、例えば、特開平10−176070号において、Escherichia coli XL1-Blueに、ラルストニア・ユートロファのPHB合成遺伝子であるphbCAB
を含むプラスミドpSYL105を導入して得られる形質転換株Escherichia coli XL1-Blue(pSYL105)が開示されている。また、該形質転換株Escherichia coli XL1-Blue(pSYL105)は、Stratagene Cloning System(11011 North Torrey Pines Road La Jolla CA92037, USA)
から入手することができる。
【0025】
形質転換体を好適な培地で培養することにより、PHAを菌体内に蓄積させることができる。使用する培地としては、炭素源、窒素源、無機イオン及び必要に応じその他の有機成分を含有する通常の培地が挙げられる。大腸菌を用いる場合、炭素源としてはグルコース等が挙げられ、窒素源としてはイーストエキス、トリプトン等の天然物由来のものが挙げられる。その他、アンモニウム塩等の無機の窒素化合物等が含まれていてもよい。培養は通常、好気的条件下で12〜20時間、培養温度は30〜37℃、培養中のpHは6.0〜8.0に制御する。菌体からのPHAの採取は、クロロホルム等の有機溶媒による抽出や、菌体成分をリゾチーム等の酵素で分解した後、PHAグラニュールを濾別する方法等により実施できる。具体的には、例えば培養液から分離回収した乾燥菌体からPHAを適当な貧溶媒で抽出した後、沈殿剤で沈殿させることにより実施できる。
【0026】
また、本発明に用いられるPHA類としては、カネカ社、テレス(Telles)社により製造されているP(3HB)、P(3HB−co−3HV)、P(3HB−co−3HH)、P(3HB−co−4HB)等のPHA類を用いてもよい。本発明に用いられるPHA類としては、PHA類を含むグラニュールを精製せずに用いてもよく、下記実施例に記載する精製方法等により精製してポリマー化したものを用いてもよい。
【0027】
本発明に用いられるPHA類の分子量としては、本発明の効果を損なわない限り特に制限されないが、通常Mw=10万以上、好ましくはMw=20万以上である。なお、高い物性が得られるという点で50万以上であることも好ましい。分子量の上限は、特に制限されない。分子量は、ゲルパーミエーションクロマトグラフィー(GPC)等により、測定することができる。
【0028】
本発明における繊維の成形材料においては、上記生分解性ポリエステル以外に、任意成分として、通常繊維に用いられる各種添加剤、例えば滑剤、紫外線吸収剤、耐候剤、帯電防止剤、酸化防止剤、熱安定剤、核剤、流動改良剤、着色剤等を必要に応じて含有させることができる。
【0029】
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する。生分解性ポリエステルを含む組成物からの繊維の溶融紡糸方法としては、通常のプラスチック繊維の溶融技術を用いて行うことができ、例えば、生分解性ポリエステルを押出器に充填し、加熱溶融させ、加重をかけて、押出口より押し出すことにより行うことができる。
【0030】
溶融の温度としては、通常、溶融させる生分解性ポリエステルの融点以上であり、好ま
しくは融点+10℃以上、より好ましくは融点+15〜20℃以上である。PHBの場合、融点は170〜180℃付近である。コポリマーの場合は、その組成、及び組成比により異なるが、例えばP(3HB−co−3HV)の場合、140〜170℃付近、例えばP(3HB−co−3HH)の場合、100〜170℃付近、P(3HB−co−4HB)の場合、100〜170℃付近である。
【0031】
(急冷工程)
溶融紡糸した繊維を、生分解性ポリエステルのガラス転移点温度+15℃以下に急冷し、非晶質の繊維を作製する。急冷の温度としては、通常ガラス転移点温度+15℃以下、好ましくはガラス転移点温度+10℃以下、更に好ましくはガラス転移点温以下である。また、特に下限はないが、経済性の点から通常−180℃以上で行うことができる。同急冷工程により、溶融紡糸した生分解性ポリエステルは、非晶質の繊維となる。
【0032】
ガラス転移点温度は、例えば、示差走査熱量測定(DSC)により測定することができる。示差走査熱量測定は、例えば、示差走査熱量測定機(DSC8000,パーキンエルマー社製)を用い窒素雰囲気下の条件で測定することができる。
【0033】
例えば、PHBでは、ガラス転移点温度は4℃付近である。コポリマーの場合は、その組成、及び組成比により異なるが、−10℃〜5℃である。なお、ガラス転移点温度は高い方が、加工しやすいという点で有用である。
【0034】
冷却媒体としては、例えば、空気、水(氷水)、不活性気体等が挙げられる。本発明において、急冷は、例えば、溶融した生分解性ポリエステルをガラス転移点温度+15℃以下の空気又は氷水等の媒体中に押出し、巻き取りながら同媒体中を通過させて行うことができる。巻き取りの速度としては、通常3〜150m/min、好ましくは5〜20m/minである。
【0035】
非晶質の繊維であることは、例えば、X線回折等の方法により確認することができる。X線回折において、結晶に由来するピークが確認できなければ、非晶質であるといえる。
【0036】
(保冷工程)
急冷した繊維を、生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を作製する。保冷は、通常ガラス転移点温度+15℃以下、好ましくはガラス転移点温度+10℃以下、さらに好ましくはガラス転移点温度以下、最も望ましくはガラス転移点温度未満で行うことができる。保冷の温度としては、特に下限はないが、経済性の点から通常−180℃以上で行うことができる。同保冷工程により、急冷した繊維内部に、ポアが形成され、多孔性繊維が得られる。また、結晶化の観点からは、同保冷工程により、結晶化繊維が形成(微結晶核が形成)される。
【0037】
保冷の時間が短すぎる場合には、繊維内部にポアが十分に形成されにくく、保冷の時間が長すぎる場合には、繊維の加工性が低下する傾向にある。したがって、保冷の時間は、通常3〜72時間、好ましくは12〜48時間程度である。本発明の製造方法においては、このポアのサイズ、数、均一性といったポアの特性を、保冷時間を調整することにより、制御することが可能である。すなわち、最終繊維製品、内包させる機能性薬剤、必要な薬剤序放性能等に応じて、所望の繊維の孔のサイズを与える保冷時間を設定し、適用することができる。
【0038】
ポアのサイズは、ポリマーの種類、組成等により異なるが、コポリマーの場合は、例えばP(3HB−co−4HB)の場合、12時間未満では平均3.4μm程度以上、12〜24時間未満では平均3.4±1.6μm程度、24〜36時間未満では平均1.7±
0.9μm程度、36時間〜48時間未満では平均1.3±0.7μm程度、48時間以上では平均1.3μm程度以下である。例えばP(3HB−co−3HV)の場合、12時間未満では平均2.3μm程度以上、12〜24時間未満では平均2.3±1.3μm程度、24〜36時間未満では平均2.2±1.0μm程度、36時間〜48時間未満では平均1.1±0.6μm程度、48時間以上では平均1.1μm程度以下である。例えばP(3HB−co−3HH)の場合、12時間未満では1.2μm程度以上、12〜24時間未満では平均1.2±0.7μm程度、24〜36時間未満では平均0.9±0.6μm程度、36時間〜48時間未満では平均0.7±0.5μm程度、48時間以上では平均0.7μm程度以下である。ホモポリマーの場合は、P(3HB−co−4HV)の場合とほぼ同様である。
【0039】
なお、結晶化の観点からは、保冷工程は繊維の等温結晶化(微結晶核形成)工程ということもでき、このガラス転移点温度+15℃以下での保冷工程により、繊維における結晶化が非常にゆっくり進む。また、生成される結晶は非常に小さいものである。その小さな結晶が延伸の基点(延伸核)となり、1段階の延伸(比較的低倍率の延伸)で分子鎖が高度に配向するものと考えられる。
【0040】
上記工程に加えて、所望の繊維物性等に応じて、さらに延伸工程、熱処理工程を行うことができる。
(延伸工程)
多孔性繊維の延伸は、生分解性ポリエステルのガラス転移点温度以上で行うことができ、例えば室温で行うことができる。延伸の温度としては、特に上限はないが、通常融点以下で行うことができる。
【0041】
延伸は、例えば、延伸機等に固定して行うことができる。また、巻き取りローラーにより巻き取りながら張力をかけて行うことができる。延伸機等に固定して延伸する場合、延伸倍率は通常2倍以上、好ましくは5倍以上、より好ましくは10倍以上である。延伸倍率としては、特に上限はなく、破断しない程度であればよい。
【0042】
(熱処理工程)
多孔性繊維の熱処理は、例えば、緊張下で、温風熱処理、乾燥機熱処理等により行うことができる。熱処理は、通常25〜150℃、好ましくは40℃〜100℃程度で、通常5秒〜120分、好ましくは10秒〜30分程度行うことができる。
【0043】
緊張は、例えば、固定、加重、張力等によって行うことができる。固定熱処理とは、繊維の両端を固定した状態で熱処理を行うことである。また、繊維の先に重りを吊して加重して熱処理を行う場合、加重は繊維が切断しなければ、重ければ重い程良い。加重は、繊維に加重をかけて切断しない程度までの範囲で決定することができる。また、巻き取りローラー等により、送りと巻き取りのローラー速度を変えて、張力をかけながら熱処理を行うことができる。この場合、繊維を張力により延伸しながら熱処理することが可能である。巻き取りローラーにより張力をかけて熱処理を行う場合、通常延伸倍率1倍以上、好ましくは3倍以上、より好ましくは10倍以上で行うことができる。なお、倍率1倍での延伸とは、繊維が伸びないように巻き取ることである。延伸倍率としては、特に上限はなく、破断しない程度であればよい。
【0044】
(薬剤内包工程)
多孔性繊維に、機能性薬剤を内包し、薬剤内包多孔性繊維を作製する。本発明の繊維は、繊維内部に多数のポアを有する多孔性繊維であるため、このポア内に機能性薬剤を担持させることができる。
多孔性繊維へ機能性薬剤を内包させる方法は、特に限定されないが、例えば、多孔性繊
維へ機能性薬剤を含浸させる方法が挙げられる。含浸方法は、特に限定されず、公知の含浸方法を適宜選択することができる。
【0045】
含浸方法としては、例えば、機能性薬剤を適当な溶媒に溶解させて機能性薬剤溶液を調製し、多孔性繊維を同機能性薬剤溶液中に浸漬する方法、多孔性繊維の表面に同機能性薬剤溶液を、リバースコート法、ダイコート法、コンマコート法、グラビアコート法、マイクログラビアコート法、ナイフコート法、スピンコート法、刷毛塗り法など各種の塗工方法を用いて塗工する方法、多孔性繊維の表面に同機能性薬剤溶液をスプレーする方法等が挙げられる。これらは1種または2種以上を併用してもよい。
【0046】
なお、通常上記のような含浸方法を経れば、多孔性繊維のポア内だけに選択的に機能性薬剤が内包されるものではなく、多孔性繊維の表面にも機能性薬剤が付着することが予想される。本発明の薬剤内包多孔性繊維は、多孔性繊維のポア内に機能性薬剤が内包される限り、多孔性繊維の表面にも機能性薬剤が付着していてもよい。
【0047】
以下、多孔性繊維へ機能性薬剤を内包させる方法として、多孔性繊維を機能性薬剤溶液中に浸漬する方法を例として、より詳細に説明する。
機能性薬剤溶液の調製に用いる溶媒としては、生分解性ポリエステルや薬剤の薬効に悪影響を及ぼさない溶媒を使用できる。溶媒としては、極性溶媒又は非極性溶媒単独、あるいは極性溶媒と非極性溶媒との混合溶媒等を使用できる。極性溶媒として、具体的には、水、メタノール、エタノール等のアルコール類、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン類、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル等のエステル類、ジエチルエーテル、テトラヒドロフラン等のエーテル類、ジメチルホルムアミド、ジメチルイミダゾリジノン、ジメチルスルフォキシド等の非プロトン性極性溶媒類等が挙げられる。非極性溶媒として、具体的には、ベンゼン、トルエン、キシレン等の芳香族炭化水素類及びそのハロゲン置換体類、ヘキサン、シクロヘキサン等の脂肪族炭化水素類、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類等が挙げられる。溶媒として、好ましくは極性溶媒であり、より好ましくは水、メタノール、ジメチルスルフォキシド(DMSO)であり、さらに好ましくはジメチルスルフォキシドである。
【0048】
機能性薬剤溶液の濃度が低すぎると、多孔性繊維へ十分な薬剤が担持されない場合があり、機能性薬剤溶液の濃度が高すぎると、多孔性繊維への薬剤担持量が頭打ちになる場合がある。薬剤や溶媒の種類等によっても異なるが、機能性薬剤溶液の濃度としては、通常1〜50%(質量%)程度、好ましくは3〜30%程度、さらに好ましくは5〜15%程度である。
【0049】
浸漬させる機能性薬剤溶液の温度が高いほうが、多孔性繊維への機能性薬剤溶液の浸透性が高い傾向にある。薬剤や溶媒の種類等によっても異なるが、浸漬させる際の機能性薬剤溶液の温度としては、通常15〜100℃程度、好ましくは20〜90℃程度、さらに好ましくは25〜80℃程度である。
機能性薬剤溶液中に浸漬する時間としては、所望の薬剤担持量が得られるように調整できるが、通常24時間以下程度、好ましくは12時間以下程度、さらに好ましくは30分以下程度である。
【0050】
(2)繊維
本発明の繊維は、上記本発明の製造方法によって製造された繊維である。
本発明の繊維は、繊維内部に多数のポアを有する多孔性繊維であり、このポア内に機能性薬剤を内包させたものである。なお、本発明の薬剤内包多孔性繊維は、多孔性繊維のポア内に機能性薬剤が内包される限り、さらに、多孔性繊維を形成する基材に、機能性薬剤
をあらかじめ混合し、製造されたものであってもよい。
機能性薬剤としては、特に限定されず、例えば抗菌、殺菌、防虫、殺虫、防かび、消臭、芳香、光触媒、抗炎症等の機能を有する物質が挙げられ、繊維の用途に応じて用いることができる。これらの物質は、1種または2種以上を併用してもよい。
【0051】
本発明の繊維は、生分解性ポリエステルを繊維の形成基材として使用するものであり、分解酵素、水等の条件が揃った場合、生分解性及び生体吸収性を発揮する。このような条件下において、本発明の繊維は繊維表面から徐々に分解され、それに伴い、多孔性繊維のポア内に内包されていた薬剤が露出し、繊維の外に徐々に放出される。このようにして、本発明の繊維は、薬剤徐放性を有するものである。したがって、本発明の繊維は、薬剤徐放性が求められる用途に応用することができる。
【0052】
薬剤徐放性能は、例えば、以下のようにして評価することができる。
薬剤内包多孔性繊維を、繊維を分解する能力を有する酵素を溶解した酵素溶液中に浸漬する。経時的に酵素溶液を採取し、酵素溶液中に放出された薬剤量を測定することにより、薬剤徐放性能を評価することができる。
例えば、薬剤が紫外・可視領域に吸光性を有する物質であれば、UV−Vis検出器(U-2910形分光光度計,日立ハイテクノロジーズ製)等を用いたUV−Vis測定により、経時的に酵素溶液の吸光スペクトルを測定することにより、評価することができる。
【0053】
薬剤徐放性能は、薬剤担時量や繊維の生分解条件等によって異なる。薬剤担時量が多ければ、単位時間当りに放出される薬剤量が多くなり、繊維の生分解速度が速ければ、単位時間当りに放出される薬剤量が多くなる。
薬剤担時量は、薬剤や溶媒の種類、ポリマーの種類、繊維の製造条件によっても異なるが、薬剤含浸前の繊維に対する薬剤量として、通常0.01〜10%(質量%)程度、好ましくは0.05〜1%程度、より好ましくは0.1〜0.5%程度である。例えば、バンコマイシン塩酸塩の場合、0.2%(質量%)程度である。
なお、所望の単位時間当りの薬剤放出量を得るために、適用用途における繊維の生分解条件等を考慮して、薬剤担時量等を調整することができる。
【0054】
本発明の繊維は、上述したように十分な強度を有し、かつ生分解性及び生体適合性に優れた生分解性ポリエステルからなるものであり、ドア基材、パッケージトレー、ピラーガーニッシュ、スイッチベース、クオーターパネル、アームレストの芯材、ドアトリム、シート・マット構造材、コンソールボックス、ダッシュボード、各種インストルメントパネル、デッキトリム、スポイラー、カウリング等の車両用材料、パッケージトレー、アームレストの芯材、シート・マット構造材、コンソールボックス、ダッシュボード、各種インストルメントパネル等の船舶及び航空機用材料、手術用縫合糸等の医療用用具、釣り糸、漁網等の水産業用用具、フィルム、シート、不織布等の農業用材料、繊維等の衣料用材料、不織布、ロープ、壁材等の建築用材料、食品その他の包装用材料、メンブレンフィルター等の各種分離材料等に有用である。
【実施例】
【0055】
以下、実施例により、本発明をさらに具体的に説明するが、本発明はその要旨を超えない限り、これらの実施例に限定されるものではない。
【0056】
<試験例1>
本試験では、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷、保冷及び延伸することを特徴とする方法(微結晶核延伸法)による繊維の作製及びその物性評価を行った。
(1)ポリマーの調製
ポリ[(R)−3−ヒドロキシブチレート−co−4−ヒドロキシブチレート][P(3HB−co−4HB)]グラニュールをクロロホルム中に溶解させ、濾過後、ヘキサンに再沈殿させて、精製したP(3HB−co−9.4mol%−4HB)を得た。P(3HB−co−4HB)の分子量はMw=2.3×105、Mn=1.2×105、多分散度はMw/Mn=2.0(GPC測定による)であった。融点とガラス転移点はTm=170℃、Tg=−2.6℃(DSC測定による)であった。
【0057】
(2)繊維の作製
押出装置の内径5mm、長さ120mmの芯柱にP(3HB−co−4HB)試料を詰め込み、溶融温度(180℃)にて一定時間保ち、試料が完全溶融した後に押出を開始した。押出口のノズルは1mmのものを使用した。
【0058】
溶融押出により紡糸した繊維を、氷水浴中で巻き取り(30rpm:ロッド径114mm)、非晶質の繊維を得た。この非晶質の繊維を、氷水中(0℃)に24時間保冷し、微結晶核形成を行なった。その後、手回し延伸機を用いて室温で表1に示す倍率に延伸した後、50℃で5分間の定張(倍率100%)熱処理を行い、繊維を作製した。
【0059】
(3)物性・構造解析及び結果
得られた繊維について、引張強度、破壊伸び及びヤング率を測定した。引張強度、破壊伸び及びヤング率は、JIS−K−6301に沿って、島津製作所製小型卓上試験機EZTestを用いて測定した。引張速度は20mm/分とした。
【0060】
結果を表1に示す。表1に示される結果から、延伸により、繊維の強度が増加することが分かった。
【0061】
繊維の構造解析を、延伸前の繊維及び延伸後の繊維について、広角X線回折(WAXD)測定及び小角X線散乱(SAXS)測定により行った。
広角X線回折測定及び小角X線散乱測定は、BL45XUビームライン(SPring-8大型放射光施設)を用いて、0.09nmの波長で行った。単繊維をX線ビームに垂直に、検出器に平行に設置し、回折パターンをCCDカメラ(C7300-10-12NR,浜松ホトニクス株式会社製)にて、露出時間76−1058ミリ秒で記録した。CCDカメラのピクセルサイズは125×125μm、12ビット/ピクセルである。SAXS測定の場合、カメラ長は2200mm、WAXD測定の場合、カメラ長は120mmとした。
【0062】
広角X線回折測定の結果、延伸前の繊維では、2回らせん構造(α構造)に起因した回折が見られたが、平面ジグザグ構造(β構造)に起因した回折は見られなかった。対して、延伸後の繊維の解析結果では、α構造に起因した回折及びβ構造に起因した回折が見られた。延伸により、β構造が形成されていることが分かった。β構造は、繊維の強度向上に重要な因子である。さらに、X線回折測定の結果より、結晶化度及び配向度を算出した。
【0063】
結果を表2に示す。表2に示される結果から、延伸により、結晶化度は減少し、配向度は増加することが分かった。
【0064】
【表1】

【0065】
【表2】
【0066】
小角X線散乱測定の結果、延伸前の繊維では、ラメラ結晶の長周期に由来する回折が見られた。対して、延伸後の繊維では、ラメラ結晶の長周期に由来する回折が消失し、ストリーク回折が見られた。このことから、繊維の内部構造が変化したことが示唆された。
【0067】
小角X線散乱測定の結果、繊維の内部構造が変化したことが示唆されたため、走査型電子顕微鏡により、繊維内部の構造を観察した。試料としては、10倍延伸の繊維を用いた。
走査型電子顕微鏡観察は、電界放射型走査型電子顕微鏡(FE-SEM S-4000,株式会社日立製作所製)を用いて行った。なお、試料は観察前に、イオンスパッター装置(E-1030,株式会社日立製作所製)を使用して白金を蒸着した。
【0068】
結果を図1に示す。繊維内部に多数の孔(ポア)が認められた。画像解析ソフトウェア(Image J, アメリカ国立衛生研究所(NIH)開発)により、一定範囲(30μm×30
μm)における平均ポアサイズを算出したところ、1.7±0.9μmであった。
【0069】
<試験例2>
本試験では、繊維の製造における微結晶核形成時間のポアサイズへの影響を検討した。(1)構造解析
繊維の製造方法において、氷水中(0℃)への保冷時間を0〜72時間とした以外は、試験例1と同様の方法で繊維を製造した。なお、延伸倍率は10倍としたが、繊維の破断により延伸不可能であった場合は、10倍未満で最大の延伸倍率を採用した。
試験例1と同様の方法で、引張強度を測定した。保冷時間が3,12,24,36時間の繊維については、試験例1と同様の方法で、走査型電子顕微鏡(SEM)により、繊維内部の構造を観察し、平均ポアサイズを算出した。
【0070】
(2)結果
結果を図2に示す。なお、保冷時間が12,24,36時間の繊維の平均ポアサイズは、それぞれ3.4±1.6μm、1.7±0.9μm、1.3±0.7μmであった。保冷時間を長くするに従って、ポアの数が増加することが分かった。また、保冷時間を長くするに従って、ポアサイズが減少し、より均一になることが分かった。
【0071】
<試験例3>
本試験では、繊維のポアサイズにおける共重合体の種類の影響を検討した。
(1)構造解析
繊維の製造方法において、氷水中(0℃)への保冷時間を24時間の他、12,36時間とし、P(3HB−co−4HB)試料の他、P(3HB−co−3HV)試料又はP(3HB−co−3HH)試料を使用した以外は、試験例1と同様の方法で繊維を製造した。なお、延伸倍率は10倍とした。
試験例1と同様の方法で、走査型電子顕微鏡により、繊維内部の構造を観察し、平均ポアサイズを算出した。
【0072】
(2)結果
結果を表3及び図3に示す。P(3HB−co−4HB)以外の繊維においても、P(3HB−co−4HB)と同様、内部にポアが形成され、保冷時間を長くするに従って、ポアサイズが減少する傾向が見られた。保冷時間が同じ場合、共重合体の種類によって、ポアサイズに違いが見られた。
【0073】
【表3】
【0074】
<試験例4>
本試験では、繊維のポア形成機構を検討した。紡糸直後の繊維及び微結晶核形成(紡糸後、0℃で24時間保冷)後の繊維(未延伸)を試料として、X線マイクロトモグラフィー測定により、構造解析を行った。
(1)構造解析
X線マイクロトモグラフィー測定は、BL47XUビームライン(SPring-8大型放射光施設)を用いて、0.15nmの波長にて行った。試料を、X線ビーム中に配置された回
転ロッドに固定した。繊維の3D解析のため、測定は繊維軸に垂直に、試料を0.2度ずつ回転して行った。CCDカメラの有効ピクセルサイズは0.2μm、形式は2×2ビニングモードにおいて2000×1312である。ピクセル当たりのビット数は、12ビットである。
【0075】
(2)結果
結果を図4に示す。紡糸直後の繊維には、ポアは認められなかった。対して、保冷を行った繊維には、微視的なポアが認められた。したがって、ガラス転移温度+15℃以下に保冷することにより、繊維内部にポアが形成されることが確認された。
【0076】
<実施例1>
本発明の製造方法に従い、薬剤内包繊維を作製し、その薬剤徐放性能を評価した。
(薬剤内包繊維の作製)
試験例1で作製した10倍延伸の繊維を試料とした。薬剤として、バンコマイシン塩酸塩を使用した。溶媒として、ジメチルスルフォキシド(DMSO)を使用した。
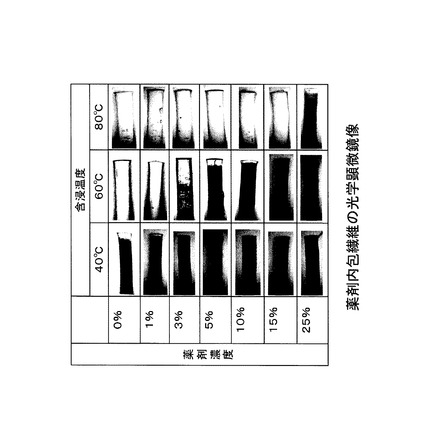
DMSOを溶媒として、バンコマイシン塩酸塩を薬剤濃度が0〜25%(質量%)となるように溶解させ、薬剤溶液を調製した。80℃に保温した薬剤溶液に、繊維を浸し、20分間保持し、繊維に薬剤を含浸させた。光学顕微鏡(ニコン社製)(5〜10倍)で、
繊維を観察した。15%の薬剤溶液を含浸させた繊維を真空乾燥(室温、24時間)し、試験例1と同様の方法で、走査型電子顕微鏡及びX線トモグラフィー測定により繊維内部の構造を観察した。
【0077】
(結果)
光学顕微鏡による観察結果を図5に示す。繊維内への薬剤の含浸により、繊維の光透過性が向上し、透明な像として観察される。繊維内への薬剤の含浸が認められた。温度を高くするにつれて、より高濃度の薬剤溶液が浸透可能であることが確認された。
走査型電子顕微鏡及びX線マイクロトモグラフィーによる観察結果を図6に示す。薬剤含浸後の繊維中央部の断面を走査型電子顕微鏡で観察すると、繊維のポア内に薬剤が内包されていることが確認された。
また、X線トモグラフィー測定によっても、繊維のポア内に内包された薬剤が白い筋状に観察され(図6(d’))、薬剤が内包されていることが確認された。
【0078】
(薬剤徐放性評価)
上記方法で作製した、0〜15%の薬剤溶液を含浸させた繊維を試料とした。酵素溶液として、Ralstonia pickettii T1に由来するPHB分解酵素を、0.1M リン酸緩衝液(pH7.4)に溶解させ、反応溶液として使用した。
薬剤含浸した繊維を酵素溶液の入ったセルに浸し、37℃で0〜24時間保持し、繊維を石英セル(3.5mL)中で分解した。繊維分解開始24時間後に、繊維は完全に分解した。繊維分解開始0〜24時間後に、UV−Vis検出器(U-2910形分光光度計,日立ハイテクノロジーズ社製)を用いて、280nmにおける吸光度を測定することにより、放出薬剤量を測定した。バンコマイシン塩酸塩は、λ=280nmに吸収ピークを有するので、280nmにおける吸光度(ABS)を測定することにより、放出薬剤量を測定することができる。
【0079】
(結果)
結果を図7に示す。15%の薬剤溶液を含浸させた繊維のUV−Vis測定結果(280nmにおける各時間の吸光度)を図7(A)に示す。280nmにおけるABSが高いほうから、24時間、10時間、8時間、6時間、4時間、2時間のスペクトルである。試験開始後、繊維が完全に分解した24時間まで、ABSが徐々に増加し続けた。酵素分解により、繊維が表面から徐々に分解され、それに伴い繊維内からの経時的な薬剤の徐放が確認された。
繊維分解開始後の各時間における、薬剤放出量のプロファイルを図7(B)に示す。いずれの薬剤溶液濃度においても、繊維の酵素分解に伴って、徐々に薬剤が放出される挙動が確認された。
繊維が完全に分解した24時間後の薬剤放出量と薬剤含浸前の繊維の重量より、下記式に基づいて、薬剤担持量(%)を算出した。各薬剤溶液濃度と繊維における薬剤担持量を図7(C)に示す。繊維に含浸する薬剤溶液の濃度を増加(〜10%)させることにより、繊維の薬剤担持量を増加させることができることが分かった。薬剤溶液の濃度が10%を越えると、繊維の薬剤担持量は一定になった。
【0080】
【数1】
【0081】
<実施例2>
本発明の薬剤内包繊維について、繊維の分解により放出された薬剤の抗菌活性を評価した。
試料及び被検菌は、以下のとおりとした。
(試料)
サンプル1:薬剤溶液濃度0%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル2:薬剤溶液濃度1%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル3:薬剤溶液濃度5%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル4:薬剤溶液濃度10%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル5:薬剤溶液濃度15%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル6(陽性対照):バンコマイシン塩酸塩溶液1.0mg(薬剤)/ml(溶媒)
サンプル7(陽性対照):バンコマイシン塩酸塩溶液0.5mg(薬剤)/ml(溶媒)
サンプル8(陽性対照):バンコマイシン塩酸塩溶液0.1mg(薬剤)/ml(溶媒)
(被検菌)
エシェリヒア・コリ(Eschrichia coli JM109)(グラム陰性菌)(抗菌活性測定条件
:LB培地、37℃、18〜24時間)
バチルス・サブチリス(Bacillus subtilis JCM1465T)(グラム陽性細菌)(抗菌活性測定条件:NB培地、30℃、18〜24時間)
【0082】
(バンコマイシン塩酸塩の抗菌活性評価試験)
上記被検菌2種類を用いたspot−on−lawn法によるバイオアッセイを行った。サンプル6〜8(陽性対照)について、フィルター除菌した0.1Mリン酸緩衝液を用いて2倍希釈系列(〜28倍)を作製し、培地上にスポットした。
【0083】
(結果)
1)E. coliを被検菌とした場合
サンプル6・・・21倍希釈液まで薄い生育阻止円を確認(静菌的)。
サンプル7・・・原液(20倍)でごく薄い阻止円が観察された。
サンプル8・・・阻止円は観察されなかった。
以上より、E. coli JM109に対するバンコマイシン塩酸塩のMIC(最小発育阻止濃度
:Minimum Inhibitory Concentration)は、0.5mg/mlであった。
2)B. subtilisを被検菌とした場合
サンプル6・・・27倍希釈まで明瞭な生育阻止円を確認(殺菌的)。28倍希釈液まで活性有りと判断(28倍希釈液は薄い)。MICは3.9μg/ml。
サンプル7・・・27倍希釈液まで活性有りと判断。MICは3.9μg/ml。
サンプル8・・・27倍希釈液まで活性有り。
以上より、B. subtilis JCM1465Tに対するMICは、約4μg/mlと考えられた。
【0084】
(サンプル1〜5の抗菌活性評価試験)
B. subtilisを被検菌としたバイオアッセイにより、サンプル1〜5(薬剤内包繊維の
酵素分解物)について抗菌活性の測定及びバンコマイシン塩酸塩の濃度の算出を行った。
【0085】
(結果)
サンプル1・・・原液の活性なし。
サンプル2・・・25倍希釈液まで明瞭な活性あり。(26倍希釈液には活性なし。)
サンプル3・・・27倍希釈液まで活性あり。(28倍希釈液には活性なし。)
サンプル4・・・27倍希釈液まで活性あり。(28倍以上の希釈液には活性なし。)
サンプル5・・・27倍希釈液まで活性あり。(28倍希釈液はごく薄く、活性なしと判断。)
【0086】
これに関して、上記バンコマイシン塩酸塩の抗菌活性評価試験の結果よりB. subtilis JCM1465Tに対するMICが約4μg/mlであることを適用すると、サンプル2のバンコマイシン塩酸塩濃度は、128μg/ml(4μg/ml×25)と算出された。同様に、サンプル3では、512μg/ml、サンプル4では、512μg/ml、サンプル5では、512μg/mlと算出された。
【0087】
上記実施例1の結果から算出すると、サンプル5において、繊維の分解により放出されたバンコマイシン塩酸塩濃度は、約0.4mg/mlと推定される。バイオアッセイの結果により推定されたバンコマイシン塩酸塩濃度も、ほぼ同様な結果を示していた。
【0088】
本試験結果から、繊維から徐放されたバンコマイシン塩酸塩は抗菌活性を有すること、さらには抗菌活性の力価から判断して、バンコマイシン塩酸塩の失活や分解は認められず、抗菌活性を維持していることが確認された。
【産業上の利用可能性】
【0089】
本発明は、自動車関連分野において、芳香、消臭、光触媒等の機能を有する繊維製品として、応用が可能である。また、医療、農業分野において、医薬や農薬等の薬剤徐放用繊維製品として、応用が可能である。さらに、高強度な多孔性繊維を、繊維強化プラスチック(FRP)に応用することで、部材等の軽量化の効果が期待できる。
【技術分野】
【0001】
本発明は、機能性薬剤を内包する、生分解性ポリエステルを原料とする多孔性繊維およびその製造方法に関する。
【背景技術】
【0002】
保温等の機能付与や軽量化等を目的とした孔(ポア)を有する繊維の作製方法として、樹脂中に、易溶解性ポリマー、発泡性物質等のポアを形成する物質をあらかじめ混合した後に紡糸し、その後の工程で、ポアを形成する物質を取り除く方法が知られており、このような方法が特許文献1〜3等に開示されている。
【0003】
例えば、特許文献1には、難溶解性ポリマーと易溶解性ポリマーとの混合物を溶融紡糸し、糸を作製後に易溶解性ポリマーを除去することで、ポアを有する繊維を作製する方法が記載されている。しかしながら、この方法では、紡糸前に難溶解性ポリマーと易溶解性ポリマーとを混合及び分散させるための技術が必要である。また、ポアサイズの制御は困難である。
【0004】
特許文献2には、樹脂中に発泡性カプセルを混合した後に紡糸することで、多孔性繊維を作製する方法が記載されている。この方法により、多孔性繊維の作製は可能であるが、ポアサイズやポアの含有量の制御は困難であると考えられる。
【0005】
上記技術は、ともに易溶解性ポリマー、発泡性物質等のポアとなりうるものをあらかじめ樹脂に混合しておく技術であり、事前の混合や除去処理が必要になる。また、繊維作製後に、易溶解性ポリマー、発泡性物質等を除去するため、繊維自体の強度は低下する方向になる。
【0006】
ところで、生分解性ポリエステルの一つとして、ポリヒドロキシアルカン酸(PHA)類が知られている。PHA類は、生分解性及び生体適合性を有することから、繊維やフィルム等の各種成形品への利用が検討されている。例えば、PHA類を原料とする繊維は、手術用縫合糸等の医療用用具、釣り糸、漁網等の水産業用用具、繊維等の衣料用材料、不織布、ロープ等の建築用材料、食品その他の包装用材料等として大きな需要を見込むことができる。
【0007】
ポリ(3−ヒドロキシブタン酸)(以下、「P(3HB)」ともいう。)等のPHA類は、自然界に存在する多くの微生物により菌体内貯蔵物質として合成される。このようなP(3HB)産生微生物から得られるP(3HB)は、生分解性製品の原料として期待されている。しかしながら、P(3HB)は硬くて脆いため、物性の向上が求められていた。
【0008】
PHA類の繊維の物性を向上させる方法として、P(3HB)の共重合体(コポリマー)化による方法が知られている。PHA類のコポリマーは、モノマーの種類や組成を変化させることで、多様な物性を示すことが知られている。PHA類のコポリマーとして、弾性に富むゴム状で生体適合性に優れるポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)[P(3HB−co−4HB)]、結晶性の硬いプラスチック状のポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)[P(3HB−co−3HV)]、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシへキサン酸)[P(3HB−co−3HH)]等が知られている。
【0009】
また、PHA類の紡糸技術により、繊維の物性を向上させる方法も開発されており、このような方法が特許文献4〜8等に開示されている。特許文献4には、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷・固化させて非晶質の繊維を作製し、非晶質の繊維をガラス転移点温度+15℃以下に保冷して結晶化繊維を作製し、結晶化繊維を延伸、熱処理することによる繊維の作製方法が記載されている。この方法により、由来によって異なるPHA類の分子量、ポリマー組成等に関わらず、高強度な繊維を得ることができる。しかしながら、この方法により作製された繊維の内部構造の詳細については、研究されていなかった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2005−213688号公報
【特許文献2】特開2004−238760号公報
【特許文献3】特許第3808400号公報
【特許文献4】国際公開WO2006/038373号パンフレット
【特許文献5】特許第4562316号公報
【特許文献6】特開2010−126852号公報
【特許文献7】特開2009−506861号公報
【特許文献8】特開2004−084118号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、簡便に機能性薬剤を内包する多孔性繊維が得られる方法;所望の繊維の孔のサイズを有する多孔性繊維が得られる方法;同方法により得られる機能性薬剤を内包する多孔性繊維;及び同方法により得られる機能性薬剤を内包する薬剤徐放用多孔性繊維を提供することを課題とする。
【課題を解決するための手段】
【0012】
本発明者等は、上記課題を解決すべく鋭意検討を行った結果、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷・固化させて非晶質の繊維を作製し、非晶質の繊維をガラス転移点温度+15℃以下に保冷して結晶化繊維を作製し、結晶化繊維を延伸、熱処理することによる繊維の作製方法において、繊維内部に多数のポアが形成されていることを知見した。また、このポアのサイズや数といったポアの特性を、保冷時間を調整することにより、制御できることを知見した。さらに、このような繊維の作製方法により得られた繊維に機能性薬剤を内包させることにより、上記の課題に合致した機能性薬剤を内包する多孔性繊維及びその製造方法が達成されることを知見し、本発明を完成した。
【0013】
すなわち、本発明の要旨は以下の通りである。
(1)生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
(2)さらに、前記多孔性繊維を延伸する工程を有する、(1)に記載の方法。
(3)前記多孔性繊維を得る工程において、保冷時間が所望の繊維の孔のサイズを与えるように調整されている、(1)又は(2)に記載の方法。
(4)前記生分解性ポリエステルがポリヒドロキシアルカン酸である、(1)〜(3)の
何れかに記載の方法。
(5)(1)〜(4)の何れかに記載の方法により製造される、機能性薬剤を内包する多孔性繊維。
(6)薬剤徐放用である、(5)に記載の繊維。
【発明の効果】
【0014】
本発明により、簡便に機能性薬剤を内包する多孔性繊維が得られる方法及び所望の繊維の孔のサイズを有する多孔性繊維が得られる方法を提供することができる。このような方法により、簡便に機能性薬剤を内包する多孔性繊維及び機能性薬剤を内包する薬剤徐放用多孔性繊維を得ることができる。
【図面の簡単な説明】
【0015】
【図1】微結晶核延伸法を経て作製した繊維の走査型電子顕微鏡像を示す図(写真)である。(a)繊維の横断面像、(b)繊維の横断面像の拡大像、(c)繊維の縦断面像である。
【図2】微結晶核形成時間の繊維の物性への影響を示す図(写真)である。(A)微結晶核形成時間と得られた繊維の最大延伸倍率(λ)、強度(λmax)及び強度(λ=7)との関係を示すグラフである。(B)各微結晶核形成時間において得られた繊維の走査型電子顕微鏡像(横断面像)である。(a)微結晶核形成時間3時間の繊維の横断面像、(b)微結晶核形成時間12時間の繊維の横断面像、(c)微結晶核形成時間24時間の繊維の横断面像、(d)微結晶核形成時間36時間の繊維の横断面像である。(C)各微結晶核形成時間において得られた繊維におけるポアサイズと頻度との関係を示すグラフである。
【図3】各種共重合体の繊維の走査型電子顕微鏡像を示す図(写真)である。(a)P(3HB−co−4HB)繊維の横断面像、(b)P(3HB−co−3HV)繊維の横断面像、(c)P(3HB−co−3HH)繊維の横断面像である。
【図4】紡糸直後の繊維及び微結晶核形成工程を経て作製した繊維のX線マイクロトモグラフィー像を示す図(写真)である。(a)紡糸直後の繊維のX線マイクロトモグラフィー像、(b)微結晶核形成工程を経て作製した繊維のX線マイクロトモグラフィー像である。
【図5】40℃,60℃,80℃における、繊維への各濃度の薬剤溶液の浸透性を光学顕微鏡で観察した図(写真)である。DMSOに溶解させたバンコマイシン塩酸塩の濃度は0,1,3,5,10,15,25%である。薬剤溶液が繊維内へ浸透し、空孔を埋めると光が透過して透明になる。
【図6】薬剤内包前及び薬剤内包後の繊維の走査型電子顕微鏡像(a〜c,a’〜c’)とX線マイクロトモグラフィー像(d,d’)を示す図(写真)である。(a)薬剤内包前の繊維の横断面像、(b)繊維の表面像、(c)繊維の縦断面像、(d)繊維の縦断面のX線トモグラフィー像である。(a’)薬剤内包後の繊維の横断面像、(b’)繊維の表面像、(c’)繊維の縦断面像、(d’)繊維の縦断面のX線トモグラフィー像である。
【図7】薬剤内包繊維の酵素分解による薬剤徐放性能及び薬剤担持量を示す図である。(A)15%薬剤溶液を内包させた繊維の薬剤放出試験における各時間の吸光スペクトルを示すグラフである。(B)各濃度の薬剤溶液を内包させた繊維の薬剤放出試験における各時間の薬剤放出量を示すグラフである。(C)内包させた薬剤溶液濃度と薬剤担持量との関係を示すグラフである。
【発明を実施するための形態】
【0016】
以下、本発明の実施の形態について、説明する。
(1)繊維の製造方法
【0017】
本発明の繊維の製造方法は、
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法である。
【0018】
以下、本発明の方法につき、各工程毎に説明する。
(溶融紡糸工程)
本発明の製造方法では、生分解性ポリエステルを含む組成物を繊維の原料として用いる。「生分解性ポリエステルを含む組成物」とは、生分解性ポリエステルを必須成分とし、それ以外の任意成分を含んでもよい組成物を意味する。好ましい生分解性ポリエステルとしては、ポリヒドロキシアルカン酸(PHA)類等が挙げられる。好ましいポリヒドロキシアルカン酸のモノマーとしては、3−ヒドロキシブタン酸、4−ヒドロキシブタン酸、3−ヒドロキシバレリル酸、3−ヒドロキシヘキサン酸、6−ヒドロキシヘキサン酸等が挙げられる。
【0019】
本発明に用いるPHA類としては、上記ヒドロキシアルカン酸のモノマーから選ばれる1種からなるホモポリマーであってよく、また、これらのヒドロキシアルカン酸のモノマーから選ばれる2種以上からなるコポリマーであってもよい。好ましいホモポリマーとしては、P(3HB)が挙げられる。好ましいコポリマーとしては、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシヘキサン酸)、ポリ(3−ヒドロキシブタン酸−co−6−ヒドロキシヘキサン酸)、ポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)等の3−ヒドロキシブタン酸とその他のヒドロキシアルカン酸からなるコポリマーが挙げられる。
【0020】
一般に、PHA類を合成する方法としては、発酵合成法と化学合成法とがある。化学合成法は、通常の有機合成の手法に従って、PHA類を化学合成する方法である。化学合成法として、具体的には、例えば、(R)-β-ブチロラクトン、ε-カプロラクトン等の脂
肪酸ラクトンを、触媒下で開環重合すること等により合成することができる(Abe et al., Macromolecules, 28, 7630 (1995))。
【0021】
発酵合成法は、PHA類生産能を有する微生物を培養し、その菌体内に蓄積されるPHA類を取り出す方法である。発酵合成法で利用できる微生物としては、PHA類生産能を有する微生物であれば特に限定されない。ポリヒドロキシブタン酸(以下、「PHB」ともいう)生産菌としては、ラルストニア・ユートロファ(Ralstonia eutropha)等のラルストニア属、アルカリゲネス・ラタス(Alcaligenes latus)、アルカリゲネス・ファエ
カリス(Alcaligenes faecalis)等のアルカリゲネス属をはじめ60種以上の天然微生物が知られており、これらの微生物ではPHBが菌体内に蓄積される。また、ヒドロキシブタン酸とその他のヒドロキシアルカン酸とのコポリマー生産菌としては、ポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシバレリル酸)及びポリ(3−ヒドロキシブタン酸−co−3−ヒドロキシヘキサン酸)生産菌であるアエロモナス・キャビエ(Aeromonas caviae)、ポリ(3−ヒドロキシブタン酸−co−4−ヒドロキシブタン酸)生産菌であるラルストニア・ユートロファ(Ralstonia eutropha)等が知られている。
【0022】
発酵合成法においては、通常これらの微生物を、炭素源、窒素源、無機イオン及び必要に応じその他の有機成分を含有する通常の培地で培養することにより、菌体内にPHAを蓄積させることができる。菌体からのPHAの採取は、クロロホルム等の有機溶媒による
抽出や、菌体成分をリゾチーム等の酵素で分解した後、PHAグラニュールを濾別する方法等により実施できる。
【0023】
また、発酵合成法の一態様として、PHA合成遺伝子を含む組換えDNAを導入して形質転換させた微生物を培養し、その菌体内に生成したPHAを採取する方法が挙げられる。この方法においては、ラルストニア・ユートロファ等のPHA生産菌を直接培養する場合と異なり、形質転換体は菌体内にPHA分解酵素を持たないため、格段に高分子量のPHAを蓄積することができる。
【0024】
このような形質転換株として、例えば、特開平10−176070号において、Escherichia coli XL1-Blueに、ラルストニア・ユートロファのPHB合成遺伝子であるphbCAB
を含むプラスミドpSYL105を導入して得られる形質転換株Escherichia coli XL1-Blue(pSYL105)が開示されている。また、該形質転換株Escherichia coli XL1-Blue(pSYL105)は、Stratagene Cloning System(11011 North Torrey Pines Road La Jolla CA92037, USA)
から入手することができる。
【0025】
形質転換体を好適な培地で培養することにより、PHAを菌体内に蓄積させることができる。使用する培地としては、炭素源、窒素源、無機イオン及び必要に応じその他の有機成分を含有する通常の培地が挙げられる。大腸菌を用いる場合、炭素源としてはグルコース等が挙げられ、窒素源としてはイーストエキス、トリプトン等の天然物由来のものが挙げられる。その他、アンモニウム塩等の無機の窒素化合物等が含まれていてもよい。培養は通常、好気的条件下で12〜20時間、培養温度は30〜37℃、培養中のpHは6.0〜8.0に制御する。菌体からのPHAの採取は、クロロホルム等の有機溶媒による抽出や、菌体成分をリゾチーム等の酵素で分解した後、PHAグラニュールを濾別する方法等により実施できる。具体的には、例えば培養液から分離回収した乾燥菌体からPHAを適当な貧溶媒で抽出した後、沈殿剤で沈殿させることにより実施できる。
【0026】
また、本発明に用いられるPHA類としては、カネカ社、テレス(Telles)社により製造されているP(3HB)、P(3HB−co−3HV)、P(3HB−co−3HH)、P(3HB−co−4HB)等のPHA類を用いてもよい。本発明に用いられるPHA類としては、PHA類を含むグラニュールを精製せずに用いてもよく、下記実施例に記載する精製方法等により精製してポリマー化したものを用いてもよい。
【0027】
本発明に用いられるPHA類の分子量としては、本発明の効果を損なわない限り特に制限されないが、通常Mw=10万以上、好ましくはMw=20万以上である。なお、高い物性が得られるという点で50万以上であることも好ましい。分子量の上限は、特に制限されない。分子量は、ゲルパーミエーションクロマトグラフィー(GPC)等により、測定することができる。
【0028】
本発明における繊維の成形材料においては、上記生分解性ポリエステル以外に、任意成分として、通常繊維に用いられる各種添加剤、例えば滑剤、紫外線吸収剤、耐候剤、帯電防止剤、酸化防止剤、熱安定剤、核剤、流動改良剤、着色剤等を必要に応じて含有させることができる。
【0029】
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する。生分解性ポリエステルを含む組成物からの繊維の溶融紡糸方法としては、通常のプラスチック繊維の溶融技術を用いて行うことができ、例えば、生分解性ポリエステルを押出器に充填し、加熱溶融させ、加重をかけて、押出口より押し出すことにより行うことができる。
【0030】
溶融の温度としては、通常、溶融させる生分解性ポリエステルの融点以上であり、好ま
しくは融点+10℃以上、より好ましくは融点+15〜20℃以上である。PHBの場合、融点は170〜180℃付近である。コポリマーの場合は、その組成、及び組成比により異なるが、例えばP(3HB−co−3HV)の場合、140〜170℃付近、例えばP(3HB−co−3HH)の場合、100〜170℃付近、P(3HB−co−4HB)の場合、100〜170℃付近である。
【0031】
(急冷工程)
溶融紡糸した繊維を、生分解性ポリエステルのガラス転移点温度+15℃以下に急冷し、非晶質の繊維を作製する。急冷の温度としては、通常ガラス転移点温度+15℃以下、好ましくはガラス転移点温度+10℃以下、更に好ましくはガラス転移点温以下である。また、特に下限はないが、経済性の点から通常−180℃以上で行うことができる。同急冷工程により、溶融紡糸した生分解性ポリエステルは、非晶質の繊維となる。
【0032】
ガラス転移点温度は、例えば、示差走査熱量測定(DSC)により測定することができる。示差走査熱量測定は、例えば、示差走査熱量測定機(DSC8000,パーキンエルマー社製)を用い窒素雰囲気下の条件で測定することができる。
【0033】
例えば、PHBでは、ガラス転移点温度は4℃付近である。コポリマーの場合は、その組成、及び組成比により異なるが、−10℃〜5℃である。なお、ガラス転移点温度は高い方が、加工しやすいという点で有用である。
【0034】
冷却媒体としては、例えば、空気、水(氷水)、不活性気体等が挙げられる。本発明において、急冷は、例えば、溶融した生分解性ポリエステルをガラス転移点温度+15℃以下の空気又は氷水等の媒体中に押出し、巻き取りながら同媒体中を通過させて行うことができる。巻き取りの速度としては、通常3〜150m/min、好ましくは5〜20m/minである。
【0035】
非晶質の繊維であることは、例えば、X線回折等の方法により確認することができる。X線回折において、結晶に由来するピークが確認できなければ、非晶質であるといえる。
【0036】
(保冷工程)
急冷した繊維を、生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を作製する。保冷は、通常ガラス転移点温度+15℃以下、好ましくはガラス転移点温度+10℃以下、さらに好ましくはガラス転移点温度以下、最も望ましくはガラス転移点温度未満で行うことができる。保冷の温度としては、特に下限はないが、経済性の点から通常−180℃以上で行うことができる。同保冷工程により、急冷した繊維内部に、ポアが形成され、多孔性繊維が得られる。また、結晶化の観点からは、同保冷工程により、結晶化繊維が形成(微結晶核が形成)される。
【0037】
保冷の時間が短すぎる場合には、繊維内部にポアが十分に形成されにくく、保冷の時間が長すぎる場合には、繊維の加工性が低下する傾向にある。したがって、保冷の時間は、通常3〜72時間、好ましくは12〜48時間程度である。本発明の製造方法においては、このポアのサイズ、数、均一性といったポアの特性を、保冷時間を調整することにより、制御することが可能である。すなわち、最終繊維製品、内包させる機能性薬剤、必要な薬剤序放性能等に応じて、所望の繊維の孔のサイズを与える保冷時間を設定し、適用することができる。
【0038】
ポアのサイズは、ポリマーの種類、組成等により異なるが、コポリマーの場合は、例えばP(3HB−co−4HB)の場合、12時間未満では平均3.4μm程度以上、12〜24時間未満では平均3.4±1.6μm程度、24〜36時間未満では平均1.7±
0.9μm程度、36時間〜48時間未満では平均1.3±0.7μm程度、48時間以上では平均1.3μm程度以下である。例えばP(3HB−co−3HV)の場合、12時間未満では平均2.3μm程度以上、12〜24時間未満では平均2.3±1.3μm程度、24〜36時間未満では平均2.2±1.0μm程度、36時間〜48時間未満では平均1.1±0.6μm程度、48時間以上では平均1.1μm程度以下である。例えばP(3HB−co−3HH)の場合、12時間未満では1.2μm程度以上、12〜24時間未満では平均1.2±0.7μm程度、24〜36時間未満では平均0.9±0.6μm程度、36時間〜48時間未満では平均0.7±0.5μm程度、48時間以上では平均0.7μm程度以下である。ホモポリマーの場合は、P(3HB−co−4HV)の場合とほぼ同様である。
【0039】
なお、結晶化の観点からは、保冷工程は繊維の等温結晶化(微結晶核形成)工程ということもでき、このガラス転移点温度+15℃以下での保冷工程により、繊維における結晶化が非常にゆっくり進む。また、生成される結晶は非常に小さいものである。その小さな結晶が延伸の基点(延伸核)となり、1段階の延伸(比較的低倍率の延伸)で分子鎖が高度に配向するものと考えられる。
【0040】
上記工程に加えて、所望の繊維物性等に応じて、さらに延伸工程、熱処理工程を行うことができる。
(延伸工程)
多孔性繊維の延伸は、生分解性ポリエステルのガラス転移点温度以上で行うことができ、例えば室温で行うことができる。延伸の温度としては、特に上限はないが、通常融点以下で行うことができる。
【0041】
延伸は、例えば、延伸機等に固定して行うことができる。また、巻き取りローラーにより巻き取りながら張力をかけて行うことができる。延伸機等に固定して延伸する場合、延伸倍率は通常2倍以上、好ましくは5倍以上、より好ましくは10倍以上である。延伸倍率としては、特に上限はなく、破断しない程度であればよい。
【0042】
(熱処理工程)
多孔性繊維の熱処理は、例えば、緊張下で、温風熱処理、乾燥機熱処理等により行うことができる。熱処理は、通常25〜150℃、好ましくは40℃〜100℃程度で、通常5秒〜120分、好ましくは10秒〜30分程度行うことができる。
【0043】
緊張は、例えば、固定、加重、張力等によって行うことができる。固定熱処理とは、繊維の両端を固定した状態で熱処理を行うことである。また、繊維の先に重りを吊して加重して熱処理を行う場合、加重は繊維が切断しなければ、重ければ重い程良い。加重は、繊維に加重をかけて切断しない程度までの範囲で決定することができる。また、巻き取りローラー等により、送りと巻き取りのローラー速度を変えて、張力をかけながら熱処理を行うことができる。この場合、繊維を張力により延伸しながら熱処理することが可能である。巻き取りローラーにより張力をかけて熱処理を行う場合、通常延伸倍率1倍以上、好ましくは3倍以上、より好ましくは10倍以上で行うことができる。なお、倍率1倍での延伸とは、繊維が伸びないように巻き取ることである。延伸倍率としては、特に上限はなく、破断しない程度であればよい。
【0044】
(薬剤内包工程)
多孔性繊維に、機能性薬剤を内包し、薬剤内包多孔性繊維を作製する。本発明の繊維は、繊維内部に多数のポアを有する多孔性繊維であるため、このポア内に機能性薬剤を担持させることができる。
多孔性繊維へ機能性薬剤を内包させる方法は、特に限定されないが、例えば、多孔性繊
維へ機能性薬剤を含浸させる方法が挙げられる。含浸方法は、特に限定されず、公知の含浸方法を適宜選択することができる。
【0045】
含浸方法としては、例えば、機能性薬剤を適当な溶媒に溶解させて機能性薬剤溶液を調製し、多孔性繊維を同機能性薬剤溶液中に浸漬する方法、多孔性繊維の表面に同機能性薬剤溶液を、リバースコート法、ダイコート法、コンマコート法、グラビアコート法、マイクログラビアコート法、ナイフコート法、スピンコート法、刷毛塗り法など各種の塗工方法を用いて塗工する方法、多孔性繊維の表面に同機能性薬剤溶液をスプレーする方法等が挙げられる。これらは1種または2種以上を併用してもよい。
【0046】
なお、通常上記のような含浸方法を経れば、多孔性繊維のポア内だけに選択的に機能性薬剤が内包されるものではなく、多孔性繊維の表面にも機能性薬剤が付着することが予想される。本発明の薬剤内包多孔性繊維は、多孔性繊維のポア内に機能性薬剤が内包される限り、多孔性繊維の表面にも機能性薬剤が付着していてもよい。
【0047】
以下、多孔性繊維へ機能性薬剤を内包させる方法として、多孔性繊維を機能性薬剤溶液中に浸漬する方法を例として、より詳細に説明する。
機能性薬剤溶液の調製に用いる溶媒としては、生分解性ポリエステルや薬剤の薬効に悪影響を及ぼさない溶媒を使用できる。溶媒としては、極性溶媒又は非極性溶媒単独、あるいは極性溶媒と非極性溶媒との混合溶媒等を使用できる。極性溶媒として、具体的には、水、メタノール、エタノール等のアルコール類、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン類、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸ブチル等のエステル類、ジエチルエーテル、テトラヒドロフラン等のエーテル類、ジメチルホルムアミド、ジメチルイミダゾリジノン、ジメチルスルフォキシド等の非プロトン性極性溶媒類等が挙げられる。非極性溶媒として、具体的には、ベンゼン、トルエン、キシレン等の芳香族炭化水素類及びそのハロゲン置換体類、ヘキサン、シクロヘキサン等の脂肪族炭化水素類、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素類等が挙げられる。溶媒として、好ましくは極性溶媒であり、より好ましくは水、メタノール、ジメチルスルフォキシド(DMSO)であり、さらに好ましくはジメチルスルフォキシドである。
【0048】
機能性薬剤溶液の濃度が低すぎると、多孔性繊維へ十分な薬剤が担持されない場合があり、機能性薬剤溶液の濃度が高すぎると、多孔性繊維への薬剤担持量が頭打ちになる場合がある。薬剤や溶媒の種類等によっても異なるが、機能性薬剤溶液の濃度としては、通常1〜50%(質量%)程度、好ましくは3〜30%程度、さらに好ましくは5〜15%程度である。
【0049】
浸漬させる機能性薬剤溶液の温度が高いほうが、多孔性繊維への機能性薬剤溶液の浸透性が高い傾向にある。薬剤や溶媒の種類等によっても異なるが、浸漬させる際の機能性薬剤溶液の温度としては、通常15〜100℃程度、好ましくは20〜90℃程度、さらに好ましくは25〜80℃程度である。
機能性薬剤溶液中に浸漬する時間としては、所望の薬剤担持量が得られるように調整できるが、通常24時間以下程度、好ましくは12時間以下程度、さらに好ましくは30分以下程度である。
【0050】
(2)繊維
本発明の繊維は、上記本発明の製造方法によって製造された繊維である。
本発明の繊維は、繊維内部に多数のポアを有する多孔性繊維であり、このポア内に機能性薬剤を内包させたものである。なお、本発明の薬剤内包多孔性繊維は、多孔性繊維のポア内に機能性薬剤が内包される限り、さらに、多孔性繊維を形成する基材に、機能性薬剤
をあらかじめ混合し、製造されたものであってもよい。
機能性薬剤としては、特に限定されず、例えば抗菌、殺菌、防虫、殺虫、防かび、消臭、芳香、光触媒、抗炎症等の機能を有する物質が挙げられ、繊維の用途に応じて用いることができる。これらの物質は、1種または2種以上を併用してもよい。
【0051】
本発明の繊維は、生分解性ポリエステルを繊維の形成基材として使用するものであり、分解酵素、水等の条件が揃った場合、生分解性及び生体吸収性を発揮する。このような条件下において、本発明の繊維は繊維表面から徐々に分解され、それに伴い、多孔性繊維のポア内に内包されていた薬剤が露出し、繊維の外に徐々に放出される。このようにして、本発明の繊維は、薬剤徐放性を有するものである。したがって、本発明の繊維は、薬剤徐放性が求められる用途に応用することができる。
【0052】
薬剤徐放性能は、例えば、以下のようにして評価することができる。
薬剤内包多孔性繊維を、繊維を分解する能力を有する酵素を溶解した酵素溶液中に浸漬する。経時的に酵素溶液を採取し、酵素溶液中に放出された薬剤量を測定することにより、薬剤徐放性能を評価することができる。
例えば、薬剤が紫外・可視領域に吸光性を有する物質であれば、UV−Vis検出器(U-2910形分光光度計,日立ハイテクノロジーズ製)等を用いたUV−Vis測定により、経時的に酵素溶液の吸光スペクトルを測定することにより、評価することができる。
【0053】
薬剤徐放性能は、薬剤担時量や繊維の生分解条件等によって異なる。薬剤担時量が多ければ、単位時間当りに放出される薬剤量が多くなり、繊維の生分解速度が速ければ、単位時間当りに放出される薬剤量が多くなる。
薬剤担時量は、薬剤や溶媒の種類、ポリマーの種類、繊維の製造条件によっても異なるが、薬剤含浸前の繊維に対する薬剤量として、通常0.01〜10%(質量%)程度、好ましくは0.05〜1%程度、より好ましくは0.1〜0.5%程度である。例えば、バンコマイシン塩酸塩の場合、0.2%(質量%)程度である。
なお、所望の単位時間当りの薬剤放出量を得るために、適用用途における繊維の生分解条件等を考慮して、薬剤担時量等を調整することができる。
【0054】
本発明の繊維は、上述したように十分な強度を有し、かつ生分解性及び生体適合性に優れた生分解性ポリエステルからなるものであり、ドア基材、パッケージトレー、ピラーガーニッシュ、スイッチベース、クオーターパネル、アームレストの芯材、ドアトリム、シート・マット構造材、コンソールボックス、ダッシュボード、各種インストルメントパネル、デッキトリム、スポイラー、カウリング等の車両用材料、パッケージトレー、アームレストの芯材、シート・マット構造材、コンソールボックス、ダッシュボード、各種インストルメントパネル等の船舶及び航空機用材料、手術用縫合糸等の医療用用具、釣り糸、漁網等の水産業用用具、フィルム、シート、不織布等の農業用材料、繊維等の衣料用材料、不織布、ロープ、壁材等の建築用材料、食品その他の包装用材料、メンブレンフィルター等の各種分離材料等に有用である。
【実施例】
【0055】
以下、実施例により、本発明をさらに具体的に説明するが、本発明はその要旨を超えない限り、これらの実施例に限定されるものではない。
【0056】
<試験例1>
本試験では、溶融紡糸した繊維を、ポリマーのガラス転移点温度+15℃以下に急冷、保冷及び延伸することを特徴とする方法(微結晶核延伸法)による繊維の作製及びその物性評価を行った。
(1)ポリマーの調製
ポリ[(R)−3−ヒドロキシブチレート−co−4−ヒドロキシブチレート][P(3HB−co−4HB)]グラニュールをクロロホルム中に溶解させ、濾過後、ヘキサンに再沈殿させて、精製したP(3HB−co−9.4mol%−4HB)を得た。P(3HB−co−4HB)の分子量はMw=2.3×105、Mn=1.2×105、多分散度はMw/Mn=2.0(GPC測定による)であった。融点とガラス転移点はTm=170℃、Tg=−2.6℃(DSC測定による)であった。
【0057】
(2)繊維の作製
押出装置の内径5mm、長さ120mmの芯柱にP(3HB−co−4HB)試料を詰め込み、溶融温度(180℃)にて一定時間保ち、試料が完全溶融した後に押出を開始した。押出口のノズルは1mmのものを使用した。
【0058】
溶融押出により紡糸した繊維を、氷水浴中で巻き取り(30rpm:ロッド径114mm)、非晶質の繊維を得た。この非晶質の繊維を、氷水中(0℃)に24時間保冷し、微結晶核形成を行なった。その後、手回し延伸機を用いて室温で表1に示す倍率に延伸した後、50℃で5分間の定張(倍率100%)熱処理を行い、繊維を作製した。
【0059】
(3)物性・構造解析及び結果
得られた繊維について、引張強度、破壊伸び及びヤング率を測定した。引張強度、破壊伸び及びヤング率は、JIS−K−6301に沿って、島津製作所製小型卓上試験機EZTestを用いて測定した。引張速度は20mm/分とした。
【0060】
結果を表1に示す。表1に示される結果から、延伸により、繊維の強度が増加することが分かった。
【0061】
繊維の構造解析を、延伸前の繊維及び延伸後の繊維について、広角X線回折(WAXD)測定及び小角X線散乱(SAXS)測定により行った。
広角X線回折測定及び小角X線散乱測定は、BL45XUビームライン(SPring-8大型放射光施設)を用いて、0.09nmの波長で行った。単繊維をX線ビームに垂直に、検出器に平行に設置し、回折パターンをCCDカメラ(C7300-10-12NR,浜松ホトニクス株式会社製)にて、露出時間76−1058ミリ秒で記録した。CCDカメラのピクセルサイズは125×125μm、12ビット/ピクセルである。SAXS測定の場合、カメラ長は2200mm、WAXD測定の場合、カメラ長は120mmとした。
【0062】
広角X線回折測定の結果、延伸前の繊維では、2回らせん構造(α構造)に起因した回折が見られたが、平面ジグザグ構造(β構造)に起因した回折は見られなかった。対して、延伸後の繊維の解析結果では、α構造に起因した回折及びβ構造に起因した回折が見られた。延伸により、β構造が形成されていることが分かった。β構造は、繊維の強度向上に重要な因子である。さらに、X線回折測定の結果より、結晶化度及び配向度を算出した。
【0063】
結果を表2に示す。表2に示される結果から、延伸により、結晶化度は減少し、配向度は増加することが分かった。
【0064】
【表1】
【0065】
【表2】
【0066】
小角X線散乱測定の結果、延伸前の繊維では、ラメラ結晶の長周期に由来する回折が見られた。対して、延伸後の繊維では、ラメラ結晶の長周期に由来する回折が消失し、ストリーク回折が見られた。このことから、繊維の内部構造が変化したことが示唆された。
【0067】
小角X線散乱測定の結果、繊維の内部構造が変化したことが示唆されたため、走査型電子顕微鏡により、繊維内部の構造を観察した。試料としては、10倍延伸の繊維を用いた。
走査型電子顕微鏡観察は、電界放射型走査型電子顕微鏡(FE-SEM S-4000,株式会社日立製作所製)を用いて行った。なお、試料は観察前に、イオンスパッター装置(E-1030,株式会社日立製作所製)を使用して白金を蒸着した。
【0068】
結果を図1に示す。繊維内部に多数の孔(ポア)が認められた。画像解析ソフトウェア(Image J, アメリカ国立衛生研究所(NIH)開発)により、一定範囲(30μm×30
μm)における平均ポアサイズを算出したところ、1.7±0.9μmであった。
【0069】
<試験例2>
本試験では、繊維の製造における微結晶核形成時間のポアサイズへの影響を検討した。(1)構造解析
繊維の製造方法において、氷水中(0℃)への保冷時間を0〜72時間とした以外は、試験例1と同様の方法で繊維を製造した。なお、延伸倍率は10倍としたが、繊維の破断により延伸不可能であった場合は、10倍未満で最大の延伸倍率を採用した。
試験例1と同様の方法で、引張強度を測定した。保冷時間が3,12,24,36時間の繊維については、試験例1と同様の方法で、走査型電子顕微鏡(SEM)により、繊維内部の構造を観察し、平均ポアサイズを算出した。
【0070】
(2)結果
結果を図2に示す。なお、保冷時間が12,24,36時間の繊維の平均ポアサイズは、それぞれ3.4±1.6μm、1.7±0.9μm、1.3±0.7μmであった。保冷時間を長くするに従って、ポアの数が増加することが分かった。また、保冷時間を長くするに従って、ポアサイズが減少し、より均一になることが分かった。
【0071】
<試験例3>
本試験では、繊維のポアサイズにおける共重合体の種類の影響を検討した。
(1)構造解析
繊維の製造方法において、氷水中(0℃)への保冷時間を24時間の他、12,36時間とし、P(3HB−co−4HB)試料の他、P(3HB−co−3HV)試料又はP(3HB−co−3HH)試料を使用した以外は、試験例1と同様の方法で繊維を製造した。なお、延伸倍率は10倍とした。
試験例1と同様の方法で、走査型電子顕微鏡により、繊維内部の構造を観察し、平均ポアサイズを算出した。
【0072】
(2)結果
結果を表3及び図3に示す。P(3HB−co−4HB)以外の繊維においても、P(3HB−co−4HB)と同様、内部にポアが形成され、保冷時間を長くするに従って、ポアサイズが減少する傾向が見られた。保冷時間が同じ場合、共重合体の種類によって、ポアサイズに違いが見られた。
【0073】
【表3】
【0074】
<試験例4>
本試験では、繊維のポア形成機構を検討した。紡糸直後の繊維及び微結晶核形成(紡糸後、0℃で24時間保冷)後の繊維(未延伸)を試料として、X線マイクロトモグラフィー測定により、構造解析を行った。
(1)構造解析
X線マイクロトモグラフィー測定は、BL47XUビームライン(SPring-8大型放射光施設)を用いて、0.15nmの波長にて行った。試料を、X線ビーム中に配置された回
転ロッドに固定した。繊維の3D解析のため、測定は繊維軸に垂直に、試料を0.2度ずつ回転して行った。CCDカメラの有効ピクセルサイズは0.2μm、形式は2×2ビニングモードにおいて2000×1312である。ピクセル当たりのビット数は、12ビットである。
【0075】
(2)結果
結果を図4に示す。紡糸直後の繊維には、ポアは認められなかった。対して、保冷を行った繊維には、微視的なポアが認められた。したがって、ガラス転移温度+15℃以下に保冷することにより、繊維内部にポアが形成されることが確認された。
【0076】
<実施例1>
本発明の製造方法に従い、薬剤内包繊維を作製し、その薬剤徐放性能を評価した。
(薬剤内包繊維の作製)
試験例1で作製した10倍延伸の繊維を試料とした。薬剤として、バンコマイシン塩酸塩を使用した。溶媒として、ジメチルスルフォキシド(DMSO)を使用した。
DMSOを溶媒として、バンコマイシン塩酸塩を薬剤濃度が0〜25%(質量%)となるように溶解させ、薬剤溶液を調製した。80℃に保温した薬剤溶液に、繊維を浸し、20分間保持し、繊維に薬剤を含浸させた。光学顕微鏡(ニコン社製)(5〜10倍)で、
繊維を観察した。15%の薬剤溶液を含浸させた繊維を真空乾燥(室温、24時間)し、試験例1と同様の方法で、走査型電子顕微鏡及びX線トモグラフィー測定により繊維内部の構造を観察した。
【0077】
(結果)
光学顕微鏡による観察結果を図5に示す。繊維内への薬剤の含浸により、繊維の光透過性が向上し、透明な像として観察される。繊維内への薬剤の含浸が認められた。温度を高くするにつれて、より高濃度の薬剤溶液が浸透可能であることが確認された。
走査型電子顕微鏡及びX線マイクロトモグラフィーによる観察結果を図6に示す。薬剤含浸後の繊維中央部の断面を走査型電子顕微鏡で観察すると、繊維のポア内に薬剤が内包されていることが確認された。
また、X線トモグラフィー測定によっても、繊維のポア内に内包された薬剤が白い筋状に観察され(図6(d’))、薬剤が内包されていることが確認された。
【0078】
(薬剤徐放性評価)
上記方法で作製した、0〜15%の薬剤溶液を含浸させた繊維を試料とした。酵素溶液として、Ralstonia pickettii T1に由来するPHB分解酵素を、0.1M リン酸緩衝液(pH7.4)に溶解させ、反応溶液として使用した。
薬剤含浸した繊維を酵素溶液の入ったセルに浸し、37℃で0〜24時間保持し、繊維を石英セル(3.5mL)中で分解した。繊維分解開始24時間後に、繊維は完全に分解した。繊維分解開始0〜24時間後に、UV−Vis検出器(U-2910形分光光度計,日立ハイテクノロジーズ社製)を用いて、280nmにおける吸光度を測定することにより、放出薬剤量を測定した。バンコマイシン塩酸塩は、λ=280nmに吸収ピークを有するので、280nmにおける吸光度(ABS)を測定することにより、放出薬剤量を測定することができる。
【0079】
(結果)
結果を図7に示す。15%の薬剤溶液を含浸させた繊維のUV−Vis測定結果(280nmにおける各時間の吸光度)を図7(A)に示す。280nmにおけるABSが高いほうから、24時間、10時間、8時間、6時間、4時間、2時間のスペクトルである。試験開始後、繊維が完全に分解した24時間まで、ABSが徐々に増加し続けた。酵素分解により、繊維が表面から徐々に分解され、それに伴い繊維内からの経時的な薬剤の徐放が確認された。
繊維分解開始後の各時間における、薬剤放出量のプロファイルを図7(B)に示す。いずれの薬剤溶液濃度においても、繊維の酵素分解に伴って、徐々に薬剤が放出される挙動が確認された。
繊維が完全に分解した24時間後の薬剤放出量と薬剤含浸前の繊維の重量より、下記式に基づいて、薬剤担持量(%)を算出した。各薬剤溶液濃度と繊維における薬剤担持量を図7(C)に示す。繊維に含浸する薬剤溶液の濃度を増加(〜10%)させることにより、繊維の薬剤担持量を増加させることができることが分かった。薬剤溶液の濃度が10%を越えると、繊維の薬剤担持量は一定になった。
【0080】
【数1】
【0081】
<実施例2>
本発明の薬剤内包繊維について、繊維の分解により放出された薬剤の抗菌活性を評価した。
試料及び被検菌は、以下のとおりとした。
(試料)
サンプル1:薬剤溶液濃度0%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル2:薬剤溶液濃度1%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル3:薬剤溶液濃度5%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル4:薬剤溶液濃度10%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル5:薬剤溶液濃度15%の薬剤(バンコマイシン塩酸塩)溶液を内包した繊維の酵素分解溶液
サンプル6(陽性対照):バンコマイシン塩酸塩溶液1.0mg(薬剤)/ml(溶媒)
サンプル7(陽性対照):バンコマイシン塩酸塩溶液0.5mg(薬剤)/ml(溶媒)
サンプル8(陽性対照):バンコマイシン塩酸塩溶液0.1mg(薬剤)/ml(溶媒)
(被検菌)
エシェリヒア・コリ(Eschrichia coli JM109)(グラム陰性菌)(抗菌活性測定条件
:LB培地、37℃、18〜24時間)
バチルス・サブチリス(Bacillus subtilis JCM1465T)(グラム陽性細菌)(抗菌活性測定条件:NB培地、30℃、18〜24時間)
【0082】
(バンコマイシン塩酸塩の抗菌活性評価試験)
上記被検菌2種類を用いたspot−on−lawn法によるバイオアッセイを行った。サンプル6〜8(陽性対照)について、フィルター除菌した0.1Mリン酸緩衝液を用いて2倍希釈系列(〜28倍)を作製し、培地上にスポットした。
【0083】
(結果)
1)E. coliを被検菌とした場合
サンプル6・・・21倍希釈液まで薄い生育阻止円を確認(静菌的)。
サンプル7・・・原液(20倍)でごく薄い阻止円が観察された。
サンプル8・・・阻止円は観察されなかった。
以上より、E. coli JM109に対するバンコマイシン塩酸塩のMIC(最小発育阻止濃度
:Minimum Inhibitory Concentration)は、0.5mg/mlであった。
2)B. subtilisを被検菌とした場合
サンプル6・・・27倍希釈まで明瞭な生育阻止円を確認(殺菌的)。28倍希釈液まで活性有りと判断(28倍希釈液は薄い)。MICは3.9μg/ml。
サンプル7・・・27倍希釈液まで活性有りと判断。MICは3.9μg/ml。
サンプル8・・・27倍希釈液まで活性有り。
以上より、B. subtilis JCM1465Tに対するMICは、約4μg/mlと考えられた。
【0084】
(サンプル1〜5の抗菌活性評価試験)
B. subtilisを被検菌としたバイオアッセイにより、サンプル1〜5(薬剤内包繊維の
酵素分解物)について抗菌活性の測定及びバンコマイシン塩酸塩の濃度の算出を行った。
【0085】
(結果)
サンプル1・・・原液の活性なし。
サンプル2・・・25倍希釈液まで明瞭な活性あり。(26倍希釈液には活性なし。)
サンプル3・・・27倍希釈液まで活性あり。(28倍希釈液には活性なし。)
サンプル4・・・27倍希釈液まで活性あり。(28倍以上の希釈液には活性なし。)
サンプル5・・・27倍希釈液まで活性あり。(28倍希釈液はごく薄く、活性なしと判断。)
【0086】
これに関して、上記バンコマイシン塩酸塩の抗菌活性評価試験の結果よりB. subtilis JCM1465Tに対するMICが約4μg/mlであることを適用すると、サンプル2のバンコマイシン塩酸塩濃度は、128μg/ml(4μg/ml×25)と算出された。同様に、サンプル3では、512μg/ml、サンプル4では、512μg/ml、サンプル5では、512μg/mlと算出された。
【0087】
上記実施例1の結果から算出すると、サンプル5において、繊維の分解により放出されたバンコマイシン塩酸塩濃度は、約0.4mg/mlと推定される。バイオアッセイの結果により推定されたバンコマイシン塩酸塩濃度も、ほぼ同様な結果を示していた。
【0088】
本試験結果から、繊維から徐放されたバンコマイシン塩酸塩は抗菌活性を有すること、さらには抗菌活性の力価から判断して、バンコマイシン塩酸塩の失活や分解は認められず、抗菌活性を維持していることが確認された。
【産業上の利用可能性】
【0089】
本発明は、自動車関連分野において、芳香、消臭、光触媒等の機能を有する繊維製品として、応用が可能である。また、医療、農業分野において、医薬や農薬等の薬剤徐放用繊維製品として、応用が可能である。さらに、高強度な多孔性繊維を、繊維強化プラスチック(FRP)に応用することで、部材等の軽量化の効果が期待できる。
【特許請求の範囲】
【請求項1】
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
【請求項2】
さらに、前記多孔性繊維を延伸する工程を有する、請求項1に記載の方法。
【請求項3】
前記多孔性繊維を得る工程において、保冷時間が所望の繊維の孔のサイズを与えるように調整されている、請求項1又は2に記載の方法。
【請求項4】
前記生分解性ポリエステルがポリヒドロキシアルカン酸である、請求項1〜3の何れか1項に記載の方法。
【請求項5】
請求項1〜4の何れか1項に記載の方法により製造される、機能性薬剤を内包する多孔性繊維。
【請求項6】
薬剤徐放用である、請求項5に記載の繊維。
【請求項1】
生分解性ポリエステルを含む組成物から繊維を溶融紡糸する工程と、
前記溶融紡糸した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に急冷する工程と、
前記急冷した繊維を生分解性ポリエステルのガラス転移点温度+15℃以下に保冷し、多孔性繊維を得る工程と、
前記多孔性繊維に機能性薬剤を内包させる工程、
とを有する、機能性薬剤を内包する多孔性繊維の製造方法。
【請求項2】
さらに、前記多孔性繊維を延伸する工程を有する、請求項1に記載の方法。
【請求項3】
前記多孔性繊維を得る工程において、保冷時間が所望の繊維の孔のサイズを与えるように調整されている、請求項1又は2に記載の方法。
【請求項4】
前記生分解性ポリエステルがポリヒドロキシアルカン酸である、請求項1〜3の何れか1項に記載の方法。
【請求項5】
請求項1〜4の何れか1項に記載の方法により製造される、機能性薬剤を内包する多孔性繊維。
【請求項6】
薬剤徐放用である、請求項5に記載の繊維。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−246588(P2012−246588A)
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2011−120631(P2011−120631)
【出願日】平成23年5月30日(2011.5.30)
【出願人】(504137912)国立大学法人 東京大学 (1,942)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【Fターム(参考)】
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願日】平成23年5月30日(2011.5.30)
【出願人】(504137912)国立大学法人 東京大学 (1,942)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【Fターム(参考)】
[ Back to top ]