
生殖系幹細胞の分化誘導および増幅方法、並びにそのための培地
【課題】iPS細胞などの多能性幹細胞から、自己再生能および精子形成能を有する生殖系幹細胞を分化誘導し、該生殖系幹細胞を維持増幅する方法、並びにそのための培地を提供すること。
【解決手段】(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)の製造方法。GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、GR細胞を培養することを含む、GR細胞の増幅方法。
【解決手段】(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)の製造方法。GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、GR細胞を培養することを含む、GR細胞の増幅方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、多能性幹細胞から始原生殖細胞(PGC)様細胞を分化誘導する方法および該PGC様細胞を維持増幅する方法、並びにそれらの方法に用いられる培地に関する。
【背景技術】
【0002】
抗がん療法に使用される抗がん剤あるいは放射線は、がん細胞を攻撃するだけでなく、細胞活動の活発な細胞や組織にも重篤な副作用をもたらすことが知られている。抗がん剤の多くは、がん細胞が正常細胞に比べて増殖活性が高いことを利用した選択毒性により治療効果を発揮する。そのため、消化管粘膜、毛根細胞、骨髄細胞、精原細胞等の増殖力が旺盛な正常細胞をも攻撃してしまう。例えば、アドリアマイシンは精原細胞の壊死を特徴とする精巣障害を引き起こすことが知られている。
遺伝子組換えで抗がん剤による副作用を緩和する医薬品が次々と開発された。例えば、顆粒球コロニー刺激因子(G-CSF)は骨髄抑制の副作用を防ぐ画期的な医薬であり、より積極的ながん化学療法を可能とした。しかしながら,抗がん剤あるいは放射線療法の副作用に基づく精巣障害を予防・治療するための有効な薬剤は未だ見出されていない。
【0003】
一方、不妊症の3割は男性側に原因がある(即ち精液中に精子がない無精子症や、あっても十分量がない乏精子症による)といわれている。日本産科婦人科学会は、夫婦間の不妊治療の場合に限り他人から精子をもらう非配偶者間人工授精を認めているが、未婚女性はもとより夫婦間の不妊治療であっても第三者からの精子提供について倫理上の問題性を指摘する声も依然として高い。
【0004】
抗がん療法の副作用に起因する男性不妊症の解決策の1つとして、治療前に精子を採取し凍結保存しておく方法があるが、精子保存ができない小児がん患者には有効でない。Brinsterらは、1994年にin vivoで精子幹細胞を移植することに成功した(非特許文献1)。即ち、幹細胞を含むドナーマウスの精巣細胞を不妊マウスの精細管内に移植すると、ドナー細胞由来の精子形成を起こし、メスとの交配によりドナー由来の仔をつくることができた。さらに、篠原らは、グリア細胞由来神経栄養因子(GDNF)、白血病抑制因子(LIF)、上皮細胞成長因子(EGF)および塩基性線維芽細胞成長因子(bFGF)を含む培地を用いることにより、精子幹細胞のin vitro長期培養法を確立し、当該細胞株をGermline Stem(GS)細胞と名づけた(特許文献1、非特許文献2)。このGS細胞を精巣に移植すると精子形成が起こり、子孫を作ることができた。これにより、治療前に生検により採取した精巣組織からGS細胞を樹立して保存しておけば、治療後に該GS細胞を精巣に自家移植することで、がん治療の副作用による不妊を回避することが可能となった。この方法は精子保存ができない小児がん患者にも有効である。
しかしながら、既に精巣障害を発症し精子形成が不可能な患者については、GS細胞を誘導するための精子幹細胞を十分に採取できない可能性があり、多数箇所生検が必要となる場合があるが、大量の精巣組織標本の採取は精巣萎縮をもたらすリスクがある。
【0005】
近年、マウスおよびヒトの人工多能性幹細胞(iPS細胞)が相次いで樹立された(特許文献2および3、非特許文献3〜6)。ヒトiPS細胞は、治療対象となる患者由来の細胞を用いて作製された後、各組織の細胞へと分化させることができるため、再生医学の領域において、拒絶反応のない移植材料として期待されている。c-Myc遺伝子を除いた3因子によるiPS細胞の作製(非特許文献3)や、プラスミドやエピソーマルベクターを用いて初期化因子がゲノムに組み込まれることなくiPS細胞を誘導できるようになったことで(非特許文献7、8)、ヒトiPS細胞の臨床応用への期待がより高まりつつある。iPS細胞は、例えば皮膚線維芽細胞などから容易に樹立することができるので、ヒトiPS細胞から精子もしくはその前駆細胞を効率よく分化誘導し、増幅することができれば、精巣の多数箇所生検といった危険を伴う細胞ソースの採取を回避することができ、より安全に男性不妊症患者からの精子形成の実現が期待される。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開WO 2004/092357号パンフレット
【特許文献2】国際公開WO 2007/069666号パンフレット
【特許文献3】国際公開WO 2008/118820号パンフレット
【非特許文献】
【0007】
【非特許文献1】Brinster, R.L. et al., Proc. Natl. Acad. Sci. USA, 91: 11298-11302 (1994)
【非特許文献2】Kanatsu-Shinohara, M. et al., Biol. Reprod., 69: 612-616 (2003)
【非特許文献3】Takahashi, K. and Yamanaka, S., Cell, 126: 663-676 (2006)
【非特許文献4】Nakagawa, M. et al., Nat. Biotethnol., 26: 101-106 (2008)
【非特許文献5】Takahashi, K. et al., Cell, 131: 861-872 (2007)
【非特許文献6】Yu, J. et al., Science, 318: 1917-1920 (2007)
【非特許文献7】Okita, K. et al., Science, 322: 949-953 (2008)
【非特許文献8】Yu, J. et al., Science, 324: 797-801 (2009)
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、iPS細胞などの多能性幹細胞から、自己再生能および精子形成能を有する生殖系幹細胞を分化誘導し、該生殖系幹細胞を維持増幅する方法、並びにそのための培地を提供することである。
【課題を解決するための手段】
【0009】
本発明者らは、未分化細胞特異的なOct3/4(Oct4)遺伝子と、生殖細胞系譜特異的なVasa遺伝子のマウスホモログ(Mouse vasa homolog; 以下、Mvhという。)の各発現制御領域の下流にレポーター遺伝子(GFPおよびRFP)を挿入することにより、未分化細胞と生殖細胞がそれぞれ可視化されたトランスジェニック(Tg)マウスを作製し、該マウスより誘導したiPS細胞およびES細胞を実験系として用い、Oct4-GFP陽性かつMvh-RFP陽性(Oct4+/Mvh+)の細胞を効率よく誘導し得る培養条件、並びに当該細胞をOct4+/Mvh+の状態で維持増幅させ得る培養条件を確立すべく鋭意検討を重ねた。その結果、多能性幹細胞をBMP4、GDNF、EGFおよび幹細胞因子(SCF)の存在下で培養することにより、効率よくOct4+/Mvh+細胞の分化を誘導し得ること、並びにGDNF、EGF、bFGFおよびSCFの存在下、好ましくは肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)からなる群より選択される1以上の因子がさらに存在する条件下で、Oct4+/Mvh+細胞を培養することにより、ダブルポジティブの状態を維持したまま、当該細胞を増幅し得ることを見出した。得られたOct4+/Mvh+細胞(GR細胞と名づけた)の特性解析の結果、当該細胞は移動後期の始原生殖細胞(PGC)の特性を反映した細胞株であることが判明した。さらに、本発明者らは、このGR細胞を不妊マウスの精管内に移植した結果、長期にわたって生着し、かつ腫瘍形成が認められないことを見出し、当該細胞が生殖系細胞として運命決定された単能性幹細胞であることを確認して、本発明を完成させるに至った。
【0010】
即ち、本発明は以下に関する。
(1)(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞の製造方法。
(2)BMP4、GDNF、EGFおよびSCFの存在下で多能性幹細胞を培養することを含む、上記(1)記載の方法。
(3)フィーダー細胞の存在下で多能性幹細胞を培養することを特徴とする、上記(1)または(2)記載の方法。
(4)前記成長因子がフィーダー細胞から提供されることを特徴とする、上記(3)記載の方法。
(5)多能性幹細胞がiPS細胞またはES細胞である、上記(1)〜(4)のいずれかに記載に方法。
(6)多能性幹細胞がヒトまたはマウス由来である、上記(1)〜(5)のいずれかに記載の方法。
(7)上記(1)〜(6)のいずれかに記載の方法により得られた、iPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞。
(8)(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
(9)BMP4、GDNF、EGFおよびSCFを組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
(10)前記成長因子を産生する細胞を含有してなる、上記(8)または(9)記載の剤。
(11)上記(8)または(9)記載の剤が添加されてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導用培地。
(11b)基本培地および表1から選ばれる1以上の成分を含有する培地と、上記(8)〜(10)のいずれかに記載の剤とを含んでなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導キット。
(12)Oct4陽性かつVasa陽性の生殖系幹細胞の増幅方法であって、GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、該生殖系幹細胞を培養することを含む、方法。
(13)肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)から選ばされる1以上の因子がさらに存在する条件下で、前記生殖系幹細胞を培養することを含む、上記(12)記載の方法。
(14)前記生殖系幹細胞をフィーダー細胞の存在下で培養することを特徴とする、上記(12)または(13)記載の方法。
(15)前記生殖系幹細胞が多能性幹細胞から分化誘導されたものである、上記(12)〜(14)のいずれかに記載の方法。
(16)GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅支持剤。
(17)HGF、IL-2およびFGF9から選ばされる1以上の因子をさらに組み合わせてなる、上記(16)記載の剤。
(18)上記(16)または(17)記載の剤が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
(18b)基本培地および任意で、表1から選ばれる1以上の成分を含有する培地に、表2から選ばれる1以上の成分が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
(19)上記(1)〜(6)のいずれかに記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、上記(7)記載のOct4陽性かつVasa陽性の生殖系幹細胞、または上記(12)〜(15)のいずれかに記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を、該細胞と同種のレシピエント動物の精巣に移植することを含む、該不妊動物に精子を形成させる方法。
(20)生殖系幹細胞が不妊動物の体細胞から作製したiPS細胞由来である、上記(19)記載の方法。
(21)レシピエント動物がヒトまたはマウスである、上記(19)または(20)記載の方法。
(22)上記(1)〜(6)のいずれかに記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、上記(7)記載のOct4陽性かつVasa陽性の生殖系幹細胞、または上記(12)〜(15)のいずれかに記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を含有してなる、雄性不妊治療剤。
(23)精子幹細胞の採取が困難な個体を投与対象とする、上記(22)記載の剤。
【発明の効果】
【0011】
本発明によれば、皮膚細胞などの体細胞から容易に作製できるiPS細胞から精子形成可能な生殖系幹細胞を誘導し、維持増幅することができる。したがって、精子幹細胞の採取が困難および/または危険を伴う非閉塞性無精子症の患者についても、自己の遺伝情報を有する精子を形成させることが可能となる。
【図面の簡単な説明】
【0012】
【図1】Oct4-GFP/Mvh-RFP Tgマウス胎仔由来のMEFから樹立したiPS細胞の写真である。上図:Oct3/4, Sox2およびKlf4の3種の遺伝子導入により樹立したiPSクローン522A3。下図:Oct3/4, Sox2, Klf4およびNanogの4種の遺伝子導入により樹立したiPSクローン522B2。
【図2】樹立したiPSクローン522A1〜A4および522B1〜522B4についてGenomic PCR解析を行った結果を示す電気泳動の写真である。
【図3】樹立したiPSクローン522A1〜A4および522B1〜522B4における未分化マーカーの発現をRT-PCR解析により調べた結果を示す電気泳動の写真である。
【図4】樹立したiPSクローンを免疫不全マウスの皮下に注射して形成させたテラトーマを組織学的に解析した結果を示す写真である。
【図5】ES細胞およびiPS細胞(クローン522B2)を従来の分化誘導方法により形成させた胚様体(EB)の写真である。
【図6】左図:M15-BMP4細胞にGDNF、mSCFおよびEGF遺伝子を導入して得られたM15-4GF細胞の形態を示す写真である。右図:M15-4GFについてRT-PCR解析を行った結果を示す電気泳動の写真である。
【図7】上図:樹立したiPS細胞とM15-4GF細胞とを図27の培地(Supplement無し)中で浮遊培養して得られた細胞塊の写真である。下図:前記細胞塊をトリプシンおよびコラゲナーゼで解離し、図27の培地(Supplement有り)中、フィーダー細胞上で培養して得られたOct4-GFP陽性/Mvh-RFP陽性コロニーの写真である。
【図8】図27の培養条件下で安定に増殖するOct4-GFP陽性/Mvh-RFP陽性コロニーの写真である。
【図9】Oct4-GFP陽性/Mvh-RFP陽性コロニーが未分化細胞と生殖細胞に共通する細胞表面マーカーであるalkaline phosphatase活性を示した写真である。
【図10】上図:Oct4-GFP陽性/Mvh-RFP陽性コロニーをiPS細胞用の培養条件で培養した際の細胞の写真である。下図:Oct4-GFP陽性/Mvh-RFP陽性コロニーを生殖幹(GS)細胞用の培養条件で培養した際の細胞の写真である。
【図11】上図:未分化iPS細胞をiPS細胞用の培養条件で培養した際の細胞の写真である。下図:未分化iPS細胞を、分化誘導過程を経ずに直接図27の維持培養条件下で培養した際の細胞の写真である。
【図12】上図:Oct4-GFP陽性/Mvh-RFP陽性の生殖細胞様の細胞(GR細胞)の写真である。下図:Oct4-GFP陽性/Mvh-RFP陰性の未分化様の細胞(Gsp細胞)の写真である。
【図13】GR細胞とGsp細胞に対してGenomic PCR解析を行った結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図14】GR細胞とGsp細胞に対してサザンブロット解析を行った結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図15】iPS細胞およびGR細胞を培養し、2日毎に総細胞数を測定した結果を示すグラフである。
【図16】フィーダー細胞(MEF)上、またはフィーダーフリーでゲラチン、ラミニン、またはファイブロネクチンコーティング上でGR細胞を培養した結果を示す写真である。
【図17】GR細胞およびGsp細胞を免疫不全のnu/nuマウス皮下に移植し、移植部位において腫瘍を形成するか否かを調べた結果を示す写真である。コントロールとしてiPS細胞およびEK細胞についても同様の移植を行った。
【図18】GR細胞およびGsp細胞における導入外来遺伝子およびそれに対応する内在性遺伝子の発現をRT-PCR解析した結果を示す電気泳動の写真である。コントロールとしてiPS細胞、EK細胞およびFbx-iPS細胞(Cell, 126, 663-676 (2006))についても解析を行った。
【図19】GR細胞およびGsp細胞における導入外来遺伝子およびそれに対応する内在性遺伝子の発現を、リアルタイムPCRによって定量化した結果を示すグラフである。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図20】GR細胞およびGsp細胞における生殖細胞マーカー遺伝子の発現をRT-PCRで調べた結果を示す電気泳動の写真である。コントロールとしてiPS細胞、EK細胞および精巣についても解析を行った。
【図21】図20と同様に生殖細胞マーカー遺伝子の発現をRT-PCRで調べた結果を示す電気泳動の写真である。
【図22】DNAマイクロアレイを用いてiPS細胞とGR細胞の網羅的な遺伝子発現の比較を行った結果を示す図である。「Up」はiPS細胞に比べてGR細胞で発現上昇していた遺伝子を、「Down」はiPS細胞に比べてGR細胞で発現低下していた遺伝子を、それぞれ示す。
【図23】GR細胞およびGsp細胞における生殖細胞マーカー遺伝子のタンパク質発現をウエスタンブロットによって解析した結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図24】GR細胞およびGsp細胞におけるDNAのメチル化状態をBisulphite genomic sequencingによって解析した結果を示す図である。雌性インプリント遺伝子であるIgf2r、Lit1およびSNRPNと、雄性インプリント遺伝子であるH19について調べた。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図25】W/Wvマウス精巣へGR細胞の移植を行い、得られた移植個体からの精巣を組織学的に解析した結果を示す写真である。「Empty」は移植細胞非生着部位の組織像を、「Engraftment」は移植細胞生着部位の組織像を示す。
【図26】上図:図27の培養条件で培養したOct4-GFP陰性/Mvh-RFP陽性のEK細胞の写真である。下図:図27の培養条件で培養した、ES細胞由来のOct4-GFP陽性/Mvh-RFP陽性GR細胞の写真である。
【図27】iPS細胞やES細胞からOct4-GFP陽性/Mvh-RFP陽性細胞を分化誘導(Supplement無し)および維持培養(Supplement有り)するのに用いた培地の成分表を示す図である。
【発明を実施するための形態】
【0013】
本発明は、多能性幹細胞から生殖系幹細胞を誘導する方法、並びに該生殖系幹細胞を維持増幅する方法を提供する。本発明により多能性幹細胞から分化誘導される生殖系幹細胞は、未分化細胞マーカーであるOct3/4(Oct4)遺伝子と、生殖細胞系譜特異的なVasa遺伝子とを、ともに発現している(Oct4陽性かつVasa陽性、Oct4+/Vasa+と略記する場合がある)ことを特徴とする。Vasa遺伝子はショウジョウバエの生殖細胞形成不能変異の解析から同定された、DEADボックスを有するATP依存性RNAヘリカーゼをコードする遺伝子で、マウス(Proc. Natl. Acad. Sci. USA, 91: 12258-62, 1994)、ラット(Rvh; Biochem. Biophys. Res. Commun., 207: 405-10, 1995)、ブタ(Mol. Reprod. Dev., 72: 320-8, 2005)、ヒト(Proc. Natl. Acad. Sci. USA, 97: 9585-90, 2000)を含む種々の哺乳動物でそのホモログがクローニングされており(チンパンジー(XM_517757)やイヌ(XM_544339)でもアノテーション解析によりホモログの存在が推定されている。)、いずれも生殖細胞で特異的に発現することが知られている。哺乳動物vasaホモログはDEAD (Asp-Glu-Ala-Asp) box polypeptide 4(DDX4)遺伝子とも呼ばれる。本明細書では、ショウジョウバエvasa遺伝子のホモログをVasa遺伝子と総称し、特定のホモログを、例えばマウスの場合、mouse vasa homolog(Mvh)というように表記することとする。
後述の実施例における種々の特性解析の結果から示されるように、本発明の生殖系幹細胞(GR細胞ともいう)は、移動後期(胎仔生殖原基に進入する直前もしくは直後)の始原生殖細胞(PGC)の特性を反映したPGC様細胞である。該生殖系幹細胞は、本発明の維持培養条件下で維持増幅可能な(即ち、自己再生能を有する)幹細胞であり、しかも精巣に移植した場合に腫瘍を形成せず、かつ排除されることなく長期生着することから生殖系細胞として運命決定されている。
【0014】
I. 多能性幹細胞
本発明において出発材料となる多能性幹細胞は、未分化状態を保持したまま増殖できる「自己再生能」と三胚葉系列すべてに分化できる「分化多能性」とを有する未分化細胞であれば特に制限されず、例えば、iPS細胞、ES細胞の他、始原生殖細胞に由来する胚性生殖(EG)細胞、精巣組織からのGS細胞の樹立培養過程で単離されるmutipotent germline stem(mGS)細胞、骨髄から単離されるmultipotent adult progenitor cell(MAPC)等が挙げられる。ES細胞は体細胞から核初期化されて生じたES細胞であってもよい。好ましくはiPS細胞またES細胞であるが、出生後の個体から取得できる点でmGS細胞やMAPCもまた好ましい。本発明の方法は、いずれかの多能性幹細胞が樹立されているか、樹立可能である、任意の哺乳動物において適用することができ、例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等が挙げられるが、好ましくはヒトまたはマウスである。
【0015】
II. 多能性幹細胞の製造方法
本発明における多能性幹細胞として好適なiPS細胞の製造例を以下に示すが、これらに限定されない。
(A) 体細胞ソース
iPS細胞作製のための出発材料として用いることのできる体細胞は、哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット等)由来の生殖細胞以外のいかなる細胞であってもよく、例えば、角質化する上皮細胞(例、角質化表皮細胞)、粘膜上皮細胞(例、舌表層の上皮細胞)、外分泌腺上皮細胞(例、乳腺細胞)、ホルモン分泌細胞(例、副腎髄質細胞)、代謝・貯蔵用の細胞(例、肝細胞)、境界面を構成する内腔上皮細胞(例、I型肺胞細胞)、内鎖管の内腔上皮細胞(例、血管内皮細胞)、運搬能をもつ繊毛のある細胞(例、気道上皮細胞)、細胞外マトリックス分泌用細胞(例、線維芽細胞)、収縮性細胞(例、平滑筋細胞)、血液と免疫系の細胞(例、Tリンパ球)、感覚に関する細胞(例、桿細胞)、自律神経系ニューロン(例、コリン作動性ニューロン)、感覚器と末梢ニューロンの支持細胞(例、随伴細胞)、中枢神経系の神経細胞とグリア細胞(例、星状グリア細胞)、色素細胞(例、網膜色素上皮細胞)、およびそれらの前駆細胞(組織前駆細胞)等が挙げられる。細胞の分化の程度や細胞を採取する動物の齢などに特に制限はなく、未分化な前駆細胞(体性幹細胞も含む)であっても、最終分化した成熟細胞であっても、同様に本発明における体細胞の起源として使用することができる。ここで未分化な前駆細胞としては、たとえば神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)が挙げられる。
【0016】
体細胞を採取するソースとなる哺乳動物個体は特に制限されないが、目的の生殖系幹細胞(GR細胞)が不妊治療用途に使用される場合には、患者本人から体細胞を採取することが望ましい。一方、GR細胞が生殖細胞に対する遺伝子治療などを目的とする場合には、拒絶反応が起こらないという観点から、患者本人またはHLAの型が同一もしくは実質的に同一である他人から体細胞を採取することが好ましい。ここでHLAの型が「実質的に同一」とは、免疫抑制剤などの使用により、該体細胞由来のiPS細胞から分化誘導することにより得られた細胞を患者に移植した場合に移植細胞が生着可能な程度にHLAの型が一致していることをいう。たとえば主たるHLA(例えばHLA-A、HLA-BおよびHLA-DRの3遺伝子座や、さらにHLA-Cwを含む4遺伝子座)が同一である場合などが挙げられる(以下同じ)。また、GR細胞をヒトに投与(移植)しない場合、例えば、患者の精巣における薬剤感受性や副作用の有無を評価するためのスクリーニング用の細胞のソースとしてGR細胞を使用する場合には、同様に患者本人または薬剤感受性や副作用と相関する遺伝子多型が同一である他人から体細胞を採取することが望ましい。
【0017】
哺乳動物から分離した体細胞は、核初期化工程に供するに先立って、細胞の種類に応じてその培養に適した自体公知の培地で前培養することができる。そのような培地としては、例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地などが挙げられるが、それらに限定されない。核初期化物質(さらに必要に応じて、iPS細胞の樹立効率改善物質)との接触に際し、例えば、カチオニックリポソームなど導入試薬を用いる場合には、導入効率の低下を防ぐため、無血清培地に交換しておくことが好ましい場合がある。
【0018】
(B) 核初期化物質
本発明において「核初期化物質」とは、体細胞からiPS細胞を誘導することができるタンパク性因子(群)またはそれをコードする核酸(ベクターに組み込まれた形態を含む)でありうる。本発明に用いられる核初期化物質は、WO 2007/069666に記載の遺伝子であってもよい。より詳細には、Oct3/4, Klf4, Klf1, Klf2, Klf5, Sox2, Sox1, Sox3, Sox15, Sox17, Sox18, c-Myc, L-Myc, N-Myc, TERT, SV40 Large T antigen, HPV16 E6, HPV16 E7, Bmil, Lin28, Lin28b, Nanog, EsrrbまたはEsrrgが例示される。これらの初期化物質は、iPS細胞樹立の際には、組み合わされて使用されてもよく、上記初期化物質を、少なくとも1つ、2つもしくは3つ含む組み合わせであり、好ましくは4つを含む組み合わせである。具体的には、以下の組み合わせが例示される(以下においては、タンパク性因子の名称のみを記載する)。
(1) Oct3/4, Klf4, Sox2, c-Myc(ここで、Sox2はSox1, Sox3, Sox15, Sox17またはSox18で置換可能である。Klf4はKlf1, Klf2またはKlf5で置換可能である。また、c-MycはL-MycまたはN-Mycで置換可能である。)
(2) Oct3/4, Klf4, Sox2, c-Myc, TERT, SV40 Large T antigen(以下、SV40LT)
(3) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E6
(4) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E7
(5) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E6, HPV16 E7
(6) Oct3/4, Klf4, Sox2, c-Myc, TERT, Bmi1
(7) Oct3/4, Klf4, Sox2, c-Myc, Lin28
(8) Oct3/4, Klf4, Sox2, c-Myc, Lin28, SV40LT
(9) Oct3/4, Klf4, Sox2, c-Myc, Lin28, TERT, SV40LT
(10) Oct3/4, Klf4, Sox2, c-Myc, SV40LT
(11) Oct3/4, Esrrb, Sox2, c-Myc (EsrrbはEsrrgで置換可能である。)
(12) Oct3/4, Klf4, Sox2
(13) Oct3/4, Klf4, Sox2, TERT, SV40LT
(14) Oct3/4, Klf4, Sox2, TERT, HPV16 E6
(15) Oct3/4, Klf4, Sox2, TERT, HPV16 E7
(16) Oct3/4, Klf4, Sox2, TERT, HPV16 E6, HPV16 E7
(17) Oct3/4, Klf4, Sox2, TERT, Bmi1
(18) Oct3/4, Klf4, Sox2, Lin28
(19) Oct3/4, Klf4, Sox2, Lin28, SV40LT
(20) Oct3/4, Klf4, Sox2, Lin28, TERT, SV40LT
(21) Oct3/4, Klf4, Sox2, SV40LT
(22) Oct3/4, Esrrb, Sox2 (EsrrbはEsrrgで置換可能である。)
上記において、c-Mycに代えてL-Mycを、Lin28に代えてLin28bを用いることもできる。
【0019】
また、上記(1)-(22)には該当しないが、それらのいずれかにおける構成要素をすべて含み、且つ任意の他の物質をさらに含む組み合わせも、本発明における「核初期化物質」の範疇に含まれ得る。また、核初期化の対象となる体細胞が上記(1)-(22)のいずれかにおける構成要素の一部を、核初期化のために十分なレベルで内在的に発現している条件下にあっては、当該構成要素を除いた残りの構成要素のみの組み合わせもまた、本発明における「核初期化物質」の範疇に含まれ得る。
【0020】
これらの組み合わせの中で、Oct3/4, Sox2, Klf4およびc-Myc(もしくはL-Myc)の4因子並びにOct3/4, Sox2, およびKlf4の3因子が、好ましい核初期化物質の例として挙げられる。これらの組み合わせにLin28(もしくはLin28b)を加えた5因子または4因子、さらにSV40 Large T antigenを加えた6因子または5因子も好ましい。
【0021】
上記の各核初期化物質のマウスおよびヒトcDNA配列情報は、WO 2007/069666に記載のNCBI accession numbersを参照することにより取得することができ(Nanogは当該公報中では「ECAT4」との名称で記載されている。尚、Lin28、Lin28b、Esrrb、Esrrg、L-MycのマウスおよびヒトcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得できる。)、当業者は容易にこれらのcDNAを単離することができる。
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
L-Myc NM_008506 NM_001033081
核初期化物質としてタンパク性因子自体を用いる場合には、得られたcDNAを適当な発現ベクターに挿入して宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク性因子を回収することにより調製することができる。一方、核初期化物質としてタンパク性因子をコードする核酸を用いる場合、得られたcDNAを、ウイルスベクター、プラスミドベクター、エピソーマルベクター等に挿入して発現ベクターを構築し、核初期化工程に供される。
【0022】
(C) 核初期化物質の体細胞への導入方法
核初期化物質の体細胞への導入は、該物質がタンパク性因子である場合、自体公知の細胞へのタンパク質導入方法を用いて実施することができる。そのような方法としては、例えば、タンパク質導入試薬を用いる方法、タンパク質導入ドメイン(PTD)もしくは細胞透過性ペプチド(CPP)融合タンパク質を用いる方法、マイクロインジェクション法などが挙げられる。タンパク質導入試薬としては、カチオン性脂質をベースとしたBioPOTER Protein Delivery Reagent(Gene Therapy Systmes)、Pro-JectTM Protein Transfection Reagent(PIERCE)及びProVectin(IMGENEX)、脂質をベースとしたProfect-1(Targeting Systems)、膜透過性ペプチドをベースとしたPenetrain Peptide(Q biogene)及びChariot Kit(Active Motif)、HVJエンベロープ(不活化センダイウイルス)を利用したGenomONE(石原産業)等が市販されている。導入はこれらの試薬に添付のプロトコルに従って行うことができるが、一般的な手順は以下の通りである。核初期化物質を適当な溶媒(例えば、PBS、HEPES等の緩衝液)に希釈し、導入試薬を加えて室温で5-15分程度インキュベートして複合体を形成させ、これを無血清培地に交換した細胞に添加して37℃で1ないし数時間インキュベートする。その後培地を除去して血清含有培地に交換する。
PTDとしては、ショウジョウバエ由来のAntP、HIV由来のTAT (Frankel, A. et al, Cell 55, 1189-93 (1988); Green, M. & Loewenstein, P.M. Cell 55, 1179-88 (1988))、Penetratin (Derossi, D. et al, J. Biol. Chem. 269, 10444-50 (1994))、Buforin II (Park, C. B. et al. Proc. Natl Acad. Sci. USA 97, 8245-50 (2000))、Transportan (Pooga, M. et al. FASEB J. 12, 67-77 (1998))、MAP (model amphipathic peptide) (Oehlke, J. et al. Biochim. Biophys. Acta. 1414, 127-39 (1998))、K-FGF (Lin, Y. Z. et al. J. Biol. Chem. 270, 14255-14258 (1995))、Ku70 (Sawada, M. et al. Nature Cell Biol. 5, 352-7 (2003))、Prion (Lundberg, P. et al. Biochem. Biophys. Res. Commun. 299, 85-90 (2002))、pVEC (Elmquist, A. et al. Exp. Cell Res. 269, 237-44 (2001))、Pep-1 (Morris, M. C. et al. Nature Biotechnol. 19, 1173-6 (2001))、Pep-7 (Gao, C. et al. Bioorg. Med. Chem. 10, 4057-65 (2002))、SynBl (Rousselle, C. et al. MoI. Pharmacol. 57, 679-86 (2000))、HN-I (Hong, F. D. & Clayman, G L. Cancer Res. 60, 6551-6 (2000))、HSV由来のVP22等のタンパク質の細胞通過ドメインを用いたものが開発されている。PTD由来のCPPとしては、11R (Cell Stem Cell, 4:381-384(2009)) や9R (Cell Stem Cell, 4:472-476(2009))等のポリアルギニンが挙げられる。
【0023】
核初期化物質のcDNAとPTDもしくはCPP配列とを組み込んだ融合タンパク質発現ベクターを作製して組換え発現させ、融合タンパク質を回収して導入に用いる。導入は、タンパク質導入試薬を添加しない以外は上記と同様にして行うことができる。
【0024】
マイクロインジェクションは、先端径1μm程度のガラス針にタンパク質溶液を入れ、細胞に穿刺導入する方法であり、確実に細胞内にタンパク質を導入することができる。
【0025】
タンパク質導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下等)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。導入操作を繰り返し行う場合の間隔としては、例えば6〜48時間、好ましくは12〜24時間が挙げられる。
【0026】
iPS細胞の樹立効率を重視するのであれば、核初期化物質を、タンパク性因子自体としてではなく、それをコードする核酸の形態で用いることが好ましい。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、また、該核酸は二本鎖であっても、一本鎖であってもよい。好ましくは該核酸は二本鎖DNA、特にcDNAである。
核初期化物質のcDNAは、宿主となる体細胞で機能し得るプロモーターを含む適当な発現ベクターに挿入される。発現ベクターとしては、例えば、レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクター、動物細胞発現プラスミド(例、pA1-11,pXT1,pRc/CMV,pRc/RSV,pcDNAI/Neo)などが用いられ得る。
【0027】
用いるベクターの種類は、得られるiPS細胞から分化誘導されるGR細胞の用途に応じて適宜選択することができる。例えば、アデノウイルスベクター、プラスミドベクター、アデノ随伴ウイルスベクター、レトロウイルスベクター、レンチウイルスベクター、センダイウイルスベクター、エピソーマルベクターなどが使用され得る。
【0028】
発現ベクターにおいて使用されるプロモーターとしては、例えばEF1αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、EF1αプロモーター、CAGプロモーター、MoMuLV LTR、CMVプロモーター、SRαプロモーターなどが好ましい。
【0029】
発現ベクターは、プロモーターの他に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子、SV40複製起点などを含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子等が挙げられる。
【0030】
核初期化物質である核酸(初期化遺伝子)は、各々別個の発現ベクター上に組み込んでもよいし、1つの発現ベクターに2種類以上、好ましくは2〜3種類の遺伝子を組み込んでもよい。遺伝子導入効率の高いレトロウイルスやレンチウイルスベクターを用いる場合は前者が、プラスミド、アデノウイルス、エピソーマルベクターなどを用いる場合は後者を選択することが好ましい。さらに、2種類以上の遺伝子を組み込んだ発現ベクターと、1遺伝子のみを組み込んだ発現ベクターとを併用することもできる。
【0031】
上記において複数の初期化遺伝子(例えば、Oct3/4、Sox2、Klf4、c-Mycから選択される2つ以上、好ましくは2〜3遺伝子)を1つの発現ベクターに組み込む場合、これら複数の遺伝子は、好ましくはポリシストロニック発現を可能にする配列を介して発現ベクターに組み込むことができる。ポリシストロニック発現を可能にする配列を用いることにより、1種類の発現ベクターに組み込まれている複数の遺伝子をより効率的に発現させることが可能になる。ポリシストロニック発現を可能にする配列としては、例えば、口蹄疫ウイルスの2A配列(PLoS ONE3, e2532, 2008、Stem Cells 25, 1707, 2007)、IRES配列(U.S. Patent No. 4,937,190)など、好ましくは2A配列を用いることができる。
【0032】
初期化遺伝子を含む発現ベクターは、ベクターの種類に応じて、自体公知の手法により細胞に導入することができる。例えば、ウイルスベクターの場合、該核酸を含むプラスミドを適当なパッケージング細胞(例、Plat-E細胞)や相補細胞株(例、293細胞)に導入して、培養上清中に産生されるウイルスベクターを回収し、各ウイルスベクターに応じた適切な方法により、該ベクターを細胞に感染させる。例えば、ベクターとしてレトロウイルスベクターを用いる具体的手段が WO2007/69666、Cell, 126, 663-676 (2006) 及び Cell, 131, 861-872 (2007) に開示されており、ベクターとしてレンチウイルスベクターを用いる場合については、Science, 318, 1917-1920 (2007) に開示がある。iPS細胞から分化誘導されるGR細胞を不妊治療や生殖細胞の遺伝子治療等の医療用途に利用する場合、初期化遺伝子の発現(再活性化)は、iPS細胞由来のGR細胞から再生された精子や精巣組織における発癌リスクを高める可能性があるので、初期化遺伝子は細胞の染色体に組み込まれず、一過的に発現することが好ましい。かかる観点からは、染色体への組込みが稀なアデノウイルスベクターの使用が好ましい。アデノウイルスベクターを用いる具体的手段は、Science, 322, 945-949 (2008)に開示されている。また、アデノ随伴ウイルスも染色体への組込み頻度が低く、アデノウイルスベクターと比べて細胞毒性や炎症惹起作用が低いので、別の好ましいベクターとして挙げられる。センダイウイルスベクターは染色体外で安定に存在することができ、必要に応じてsiRNAにより分解除去することができるので、同様に好ましく利用され得る。センダイウイルスベクターについては、J. Biol. Chem., 282, 27383-27391 (2007) や特許第3602058号に記載のものを用いることができる。
【0033】
レトロウイルスベクターやレンチウイルスベクターを用いる場合は、いったん導入遺伝子のサイレンシングが起こったとしても、後に再活性化される可能性があるので、例えば、Cre/loxPシステムを用いて、不要となった時点で核初期化物質をコードする核酸を切り出す方法が好ましく用いられ得る。即ち、該核酸の両端にloxP配列を配置しておき、iPS細胞が誘導された後で、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞にCreリコンビナーゼを作用させ、loxP配列に挟まれた領域を切り出すことができる。また、LTR U3領域のエンハンサー−プロモーター配列は、挿入突然変異によって近傍の宿主遺伝子を上方制御する可能性があるので、当該配列を欠失、もしくはSV40などのポリアデニル化配列で置換した3’-自己不活性化(SIN)LTRを使用して、切り出されずゲノム中に残存するloxP配列より外側のLTRによる内因性遺伝子の発現制御を回避することがより好ましい。Cre-loxPシステムおよびSIN LTRを用いる具体的手段は、Chang et al., Stem Cells, 27: 1042-1049 (2009) に開示されている。
【0034】
一方、非ウイルスベクターであるプラスミドベクターの場合には、リポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。ベクターとしてプラスミドを用いる具体的手段は、例えばScience, 322, 949-953 (2008) 等に記載されている。
【0035】
プラスミドベクターやアデノウイルスベクター等を用いる場合、遺伝子導入は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができる。2種以上の発現ベクターを体細胞に導入する場合には、これらの全ての種類の発現ベクターを同時に体細胞に導入することが好ましいが、この場合においても、導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。
【0036】
尚、アデノウイルスやプラスミドを用いる場合でも、導入遺伝子が染色体に組み込まれることがあるので、結局はサザンブロットやPCRにより染色体への遺伝子挿入がないことを確認する必要がある。そのため、上記Cre-loxPシステムのように、いったん染色体に導入遺伝子を組み込んだ後に、該遺伝子を除去する手段を用いることは好都合であり得る。別の好ましい一実施態様においては、トランスポゾンを用いて染色体に導入遺伝子を組み込んだ後に、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞に転移酵素を作用させ、導入遺伝子を完全に染色体から除去する方法が用いられ得る。好ましいトランスポゾンとしては、例えば、鱗翅目昆虫由来のトランスポゾンであるpiggyBac等が挙げられる。piggyBacトランスポゾンを用いる具体的手段は、Kaji, K. et al., Nature, 458: 771-775 (2009)、Woltjen et al., Nature, 458: 766-770 (2009) に開示されている。
別の好ましい非組込み型ベクターとして、染色体外で自律複製可能なエピソーマルベクターが挙げられる。エピソーマルベクターを用いる具体的手段は、Yu et al., Science, 324, 797-801 (2009)に開示されている。
【0037】
本発明に用いられるエピソーマルベクターとしては、例えば、EBV、SV40等に由来する自律複製に必要な配列をベクター要素として含むベクターが挙げられる。自律複製に必要なベクター要素としては、具体的には、複製開始点と、複製開始点に結合して複製を制御するタンパク質をコードする遺伝子であり、例えば、EBVにあっては複製開始点oriPとEBNA-1遺伝子、SV40にあっては複製開始点oriとSV40 large T antigen遺伝子が挙げられる。
【0038】
また、エピソーマル発現ベクターは、初期化遺伝子の転写を制御するプロモーターを含む。該プロモーターとしては、前記と同様のプロモーターが用いられ得る。また、エピソーマル発現ベクターは、前記と同様に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子などをさらに含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0039】
エピソーマルベクターは、例えばリポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。具体的には、例えばScience, 324: 797-801 (2009)等に記載される方法を用いることができる。
【0040】
iPS細胞からエピソーマルベクターが除去されたか否かの確認は、該ベクターの一部をプローブまたはプライマーとして用い、iPS細胞から単離したエピソーム画分を鋳型としてサザンブロット分析またはPCR分析を行い、バンドの有無または検出バンドの長さを調べることにより実施することができる。エピソーム画分の調製は当該分野で周知の方法を用いて行えばよく、例えば、Science, 324: 797-801 (2009)等に記載される方法を用いることができる。
【0041】
(D) p53の機能阻害物質
本発明は、上記の核初期化物質に加えて、p53の機能阻害物質を接触させることがより好ましい。本明細書において「p53の機能阻害物質」とは、(a)p53タンパク質の機能もしくは(b)p53遺伝子の発現を阻害し得る限り、いかなる物質であってもよい。すなわち、p53タンパク質に直接作用してその機能を阻害する物質や、p53遺伝子に直接作用してその発現を阻害する物質のみならず、p53のシグナル伝達に関与する因子に作用することにより、結果的にp53タンパク質の機能やp53遺伝子の発現を阻害する物質も、本明細書における「p53の機能阻害物質」に含まれる。好ましくは、p53の機能阻害物質は、p53遺伝子の発現を阻害する物質であり、より好ましくはp53に対するsiRNAやshRNAをコードする発現ベクターである。
【0042】
p53タンパク質の機能を阻害する物質としては、例えば、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、抗p53アンタゴニスト抗体もしくはそれをコードする核酸、p53応答エレメントのコンセンサス配列を含むデコイ核酸、p53経路を阻害する物質などが挙げられるが、これらに限定されない。好ましくは、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、p53経路阻害物質が挙げられる。
【0043】
(D1) p53の化学的阻害物質
p53の化学的阻害物質としては、例えば、WO 00/44364に開示されるpifithrin(PFT)-α及び-βに代表されるp53阻害剤、Stormら(Nat. Chem. Biol. 2, 474 (2006))に開示されるPFT-μ、それらの類縁体及びそれらの塩(例えば、塩酸酸、臭素酸塩等の酸付加塩など)等が挙げられるが、これらに限定されない。これらのうち、PFT-α及びその類縁体[2-(2-Imino-4,5,6,7-tetrahydrobenzothiazol-3-yl)-1-p-tolylethanone, HBr (製品名:Pifithrin-α)及び1-(4-Nitrophenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzothiazolyl)ethanone, HBr(製品名:Pifithrin-α, p-Nitro)]、PFT-β及びその類縁体[2-(4-Methylphenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole, HBr(製品名:Pifithrin-α, Cyclic)及び2-(4-Nitrophenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole(製品名:Pifithrin-α, p-Nitro, Cyclic)]、PFT-μ[Phenylacetylenylsulfonamide(製品名:Pifithrin-μ)]は、Merck社より市販されている。
【0044】
体細胞へのp53の化学的阻害物質の接触は、該阻害物質を適当な濃度で水性もしくは非水性溶媒に溶解し、ヒトまたはマウスより単離した体細胞の培養に適した培地(例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地など)中に、阻害物質濃度がp53の機能阻害に十分で且つ細胞毒性がみられない範囲となるように該阻害物質溶液を添加して、細胞を一定期間培養することにより実施することができる。阻害物質濃度は用いる阻害物質の種類によって異なるが、約0.1nM〜約100nMの範囲で適宜選択される。接触期間は細胞の核初期化が達成されるのに十分な時間であれば特に制限はないが、通常は陽性コロニーが出現するまで培地に共存させておけばよい。
【0045】
p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性がある。p53の化学的阻害物質は、培地に添加するだけで細胞への導入が可能であるという利点に加えて、iPS細胞の誘導後に該阻害物質を含む培地を除去することにより、容易かつ迅速にp53の機能阻害を解除できる点でも有用である。
【0046】
(D2) p53のドミナントネガティブ変異体
p53のドミナントネガティブ変異体としては、体細胞に内在する野生型p53タンパク質と競合的に作用して、その機能を阻害し得る限り特に制限はないが、例えば、マウスp53のDNA結合領域に位置する275位(ヒトの場合は278位)のプロリンをセリンに点変異させたp53P275S(de Vries, A., Proc. Natl. Acad. Sci. USA, 99, 2948-2953 (2002))、マウスp53の14-301位(ヒトp53では11-304位に対応)のアミノ酸を欠失させたp53DD(Bowman, T., Genes Develop., 10, 826-835 (1996))などが挙げられる。その他にも、例えば、マウスp53の58位(ヒトの場合は61位)のセリンをアラニンに点変異させたp53S58A、ヒトp53の135位(マウスの場合は132位)のシステインをチロシンに点変異させたp53C135Y、マウスp53の135位(ヒトの場合は138位)のアラニンをバリンに点変異させたp53A135V、172位(ヒトの場合は175位)のアルギニンをヒスチジンに点変異させたp53R172H、270位(ヒトの場合は273位)のアルギニンをヒスチジンに点変異させたp53R270H、マウスp53の278位(ヒトの場合は281位)のアスパラギン酸をアスパラギンに点変異させたp53D278Nなどが知られており、同様に使用することができる。
【0047】
p53のドミナントネガティブ変異体は、例えば、以下の手法により得ることができる。まず、マウスまたはヒトのp53 cDNA配列情報に基づいて適当なオリゴヌクレオチドをプローブもしくはプライマーとして合成し、マウスまたはヒトの細胞・組織由来のmRNA、cDNAもしくはcDNAライブラリーから、ハイブリダイゼーション法や(RT-)PCR法を用いてマウスまたはヒトp53 cDNAをクローニングし、適当なプラスミドにサブクローニングする。変異を導入しようとする部位のコドン(例えば、p53P275Sの場合、275位のProをコードするコドンcct)を所望の他のアミノ酸をコードするコドン(例えば、p53P275Sの場合、Serをコードするコドンtct)に置換した形で、当該部位を含むプライマーを合成し、これを用いてp53 cDNAを挿入したプラスミドを鋳型とするインバースPCRを行うことにより、目的のドミナントネガティブ変異体をコードする核酸を取得する。p53DDのような欠失変異体の場合には、欠失させる部位の外側にプライマーを設計して、同様にインバースPCRを行えばよい。このようにして得られたドミナントネガティブ変異体をコードする核酸を宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク質を回収することにより、所望のドミナントネガティブ変異体を取得することができる。
【0048】
体細胞へのドミナントネガティブ変異体の接触は、上記タンパク性の核初期化物質の場合と同様に実施することができる。上述のように、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるが、p53のドミナントネガティブ変異体は、導入された細胞内でプロテアーゼによる分解を受けて徐々に消失し、それに応じて細胞に内在するp53の機能が回復することから、該変異体タンパク質の使用は、得られるiPS細胞を治療用途で利用する場合のように、高度な安全性を要求される場合に好適であり得る。
【0049】
(D3) p53のドミナントネガティブ変異体をコードする核酸
本発明の別の好ましい実施態様において、p53機能阻害物質は、p53のドミナントネガティブ変異体をコードする核酸である。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、好ましくはDNAである。また、該核酸は二本鎖であっても、一本鎖であってもよい。p53のドミナントネガティブ変異体をコードするcDNAは、該変異体タンパク質の作製について上記した手法によりクローニングすることができる。
単離されたcDNAは、前記核初期化物質である核酸(初期化遺伝子)の場合と同様に、適当な発現ベクターに挿入され、体細胞に導入され得る。
【0050】
(D4) p53経路阻害物質
ここでp53経路とは、p53を活性化し得るあらゆる上流のシグナルカスケードおよび活性化p53によって媒介されるあらゆる下流のシグナルカスケードを包含する意味で用いられる。したがって、p53経路阻害物質には、上記シグナル伝達経路のいずれかを阻害するいかなる物質も含まれるが、好ましい一実施態様においては、p53経路阻害物質はp53によりその転写が活性化されるp21の発現もしくは機能(Myc阻害活性)を阻害する物質であり、例えば、p21に対するsiRNA、shRNA、アンチセンス核酸、リボザイム等が挙げられる。p21の発現を阻害するこれらの核酸は、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムと同様の方法により設計・合成し、体細胞に導入することができる。当該核酸は、それらを発現するベクターの形態で提供されてもよく、該ベクターは、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムを発現するベクターと同様の方法により構築し、体細胞に導入することができる。
【0051】
別の好ましい一実施態様においては、p53経路阻害物質はARF-MDM2-p53経路を阻害する物質であり、例えば、ARF-MDM2-p53経路阻害物質として、p53に直接結合してその核外輸送やユビキチン化を促進するMDM2もしくはそれをコードする核酸、p53へのMDM2の作用を阻害するp19ARFやATM(ataxia-telangiectasia mutated)の発現もしくは機能を阻害する物質(例えば、これらの因子に対するsiRNAやshRNA)等が挙げられる。
【0052】
(D5) その他の物質
p53タンパク質の機能を阻害するその他の物質として、例えば、抗p53アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p53アンタゴニスト抗体はポリクローナル抗体、モノクローナル抗体の何れであってもよい。抗体のアイソタイプは特に限定されないが、好ましくはIgG、IgMまたはIgA、特に好ましくはIgGが挙げられる。また、該抗体は、完全抗体分子の他、例えばFab、Fab'、F(ab’)2等のフラグメント、scFv、scFv-Fc、ミニボディー、ダイアボディー等の遺伝子工学的に作製されたコンジュゲート分子、あるいはポリエチレングリコール(PEG)等の蛋白質安定化作用を有する分子等で修飾されたそれらの誘導体などであってもよい。抗p53アンタゴニスト抗体は、p53またはその部分ペプチドを抗原として用い、自体公知の抗体または抗血清の製造法に従って製造することができる。また、公知の抗p53アンタゴニスト抗体として、例えば、PAb1801(Oncogene Science Ab-2)及びDO-1(Oncogene Science Ab-6)(Gire and Wynford-Thomas, Mol. Cell. Biol., 18, 1611-1621 (1998))等が挙げられる。抗p53アンタゴニスト抗体をコードする核酸は、抗p53モノクローナル抗体産生ハイブリドーマから常法により単離することができる。得られるH鎖及びL鎖遺伝子を連結して単鎖抗体をコードする核酸を作製することもできる。
【0053】
p53タンパク質の機能を阻害する別の物質として、抗p21アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p21アンタゴニスト抗体及びそれをコードする核酸も、上記抗p53アンタゴニスト抗体及びそれをコードする核酸と同様にして作製することができる。
p53タンパク質の機能を阻害するさらに別の物質は、p53応答エレメントのコンセンサス配列(例、Pu-Pu-Pu-G-A/T-T/A-C-Py-Py-Py (Pu: プリン塩基, Py: ピリミジン塩基))を含むデコイ核酸である。このような核酸は上記塩基配列情報に基づいてDNA/RNA自動合成機で合成することができる。あるいはそのようなデコイ核酸は市販されている(例、p53 transcription factor decoy (GeneDetect.com))。
抗p53アンタゴニスト抗体及び抗p21アンタゴニスト抗体はp53のドミナントネガティブ変異体と同様に、また、該抗体をコードする核酸は該変異体をコードする核酸と同様にして、それぞれ細胞に導入することができる。また、上記デコイ核酸は、リポフェクション法などにより細胞に導入することができる。
【0054】
一方、p53遺伝子の発現を阻害する物質としては、例えば、p53に対するsiRNAもしくはshRNA、p53に対するsiRNAもしくはshRNAを発現するベクター、p53に対するアンチセンス核酸及びp53に対するリボザイム等が挙げられるが、好ましくはp53に対するsiRNA、shRNA及びsiRNA、shRNAを発現するベクターである。
【0055】
(D6) p53に対するsiRNA及びshRNA
p53に対するsiRNAは、マウスまたはヒトのp53 cDNA配列情報に基づいて、例えば、Elbashirら(Genes Dev., 15, 188-200 (2001))の提唱する規則に従って設計することができる。siRNAの標的配列としては、原則的にはAA+(N)19であるが、AA+(N)21もしくはNA+(N)21であってもよい。また、センス鎖の5’末端がAAである必要はない。標的配列の位置は特に制限されるわけではないが、5’-UTR及び開始コドンから約50塩基まで、並びに3’-UTR以外の領域から標的配列を選択することが望ましい。標的配列のGC含量も特に制限はないが、約30-約50%が好ましく、GC分布に偏りがなく繰り返しが少ない配列が望ましい。尚、下記(b2)のsiRNAもしくはshRNAを発現するベクターの設計において、プロモーターとしてpolIII系プロモーターを使用する場合、ポリメラーゼの転写が停止しないように、4塩基以上TまたはAが連続する配列は選択しないようにすべきである。
上述の規則に基づいて選択された標的配列の候補群について、標的以外のmRNAにおいて16-17塩基の連続した配列に相同性がないかどうかを、BLAST(http://www.ncbi.nlm.nih.gov/BLAST/)等のホモロジー検索ソフトを用いて調べ、選択した標的配列の特異性を確認する。特異性の確認された標的配列について、AA(もしくはNA)以降の19-21塩基にTTもしくはUUの3’末端オーバーハングを有するセンス鎖と、該19-21塩基に相補的な配列及びTTもしくはUUの3’末端オーバーハングを有するアンチセンス鎖とからなる2本鎖RNAをsiRNAとして設計する。また、shRNAは、ループ構造を形成しうる任意のリンカー配列(例えば、8-25塩基程度)を適宜選択し、上記センス鎖とアンチセンス鎖とを該リンカー配列を介して連結することにより設計することができる。
【0056】
siRNA及び/又はshRNAの配列は、種々のwebサイト上に無料で提供される検索ソフトを用いて検索が可能である。このようなサイトとしては、例えば、Ambionが提供するsiRNA Target Finder(http://www.ambion.com/jp/techlib/misc/siRNA_finder.html)及びpSilencerTM Expression Vector用 インサート デザインツール(http://www.ambion.com/jp/techlib/misc/psilencer_converter.html)、RNAi Codexが提供するGeneSeer(http://codex.cshl.edu/scripts/newsearchhairpin.cgi)がこれらに限定されず、QIAGEN、タカラバイオ、SiSearch、Dharmacon、Whitehead Institute、Invitrogen、Promega等のwebサイト上でも同様に検索が可能である。
【0057】
p53に対するsiRNAは、上記のようにして設計されたセンス鎖及びアンチセンス鎖オリゴヌクレオチドをDNA/RNA自動合成機でそれぞれ合成し、例えば、適当なアニーリング緩衝液中、約90〜約95℃で約1分程度変性させた後、約30〜約70℃で約1〜約8時間アニーリングさせることにより調製することができる。また、p53に対するshRNAは、上記のようにして設計されたshRNA配列を有するオリゴヌクレオチドをDNA/RNA自動合成機で合成し、上記と同様にしてセルフアニーリングさせることによって調製することができる。
【0058】
siRNA及びshRNAを構成するヌクレオチド分子は、天然型のRNAでもよいが、安定性(化学的および/または対酵素)や比活性(mRNAとの親和性)を向上させるために、種々の化学修飾を含むことができる。例えば、ヌクレアーゼなどの加水分解酵素による分解を防ぐために、アンチセンス核酸を構成する各ヌクレオチドのリン酸残基(ホスフェート)を、例えば、ホスホロチオエート(PS)、メチルホスホネート、ホスホロジチオネートなどの化学修飾リン酸残基に置換することができる。また、各ヌクレオチドの糖(リボース)の2'位の水酸基を、-OR(Rは、例えばCH3(2'-O-Me)、CH2CH2OCH3(2'-O-MOE)、CH2CH2NHC(NH)NH2、CH2CONHCH3、CH2CH2CN等を示す)に置換してもよい。さらに、塩基部分(ピリミジン、プリン)に化学修飾を施してもよく、例えば、ピリミジン塩基の5位へのメチル基やカチオン性官能基の導入、あるいは2位のカルボニル基のチオカルボニルへの置換などが挙げられる。
【0059】
RNAの糖部のコンフォーメーションはC2'-endo(S型)とC3'-endo(N型)の2つが支配的であり、一本鎖RNAではこの両者の平衡として存在するが、二本鎖を形成するとN型に固定される。したがって、標的RNAに対して強い結合能を付与するために、2'酸素と4’炭素を架橋することにより、糖部のコンフォーメーションをN型に固定したRNA誘導体であるBNA(LNA)(Imanishi, T. et al., Chem. Commun., 1653-9, 2002; Jepsen, J.S. et al., Oligonucleotides, 14, 130-46, 2004)やENA(Morita, K. et al., Nucleosides Nucleotides Nucleic Acids, 22, 1619-21, 2003)もまた、好ましく用いられ得る。
但し、天然型RNA中のすべてのリボヌクレオシド分子を修飾型で置換すると、RNAi活性が失われる場合があるので、RISC複合体が機能できる最小限の修飾ヌクレオシドの導入が必要である。
【0060】
p53に対するsiRNAは、例えば、Ambion(例、Ambion Cat# AM16708, siRNA ID# 69659, 69753, 69843, 187424, 187425, 187426)やSanta Cruz(例、Santa Cruz Cat# sc-29436, 44219)等から購入することもできる。
また、ヒトp53に対するsiRNAおよびshRNAも、上記のいずれかの検索ソフトを用いて、ヒトp53 cDNAの配列情報(例、Refseq. No. NM_000546)等をクエリーとして入力することにより設計し、合成することができ、あるいはAmbion等から購入することもできる。具体的には、Science, 296, 550-553 (2002) に記載されるp53に対するshRNAなどが例示される。
【0061】
p53に対するsiRNAもしくはshRNAの体細胞への接触は、プラスミドDNAの場合と同様に、リポソーム法、ポリアミン法、エレクトロポレーション法、ビーズ法等を用いて、該核酸を細胞内へ導入することにより実施することができる。カチオニックリポソームを用いた方法が最も一般的で、導入効率も高い。Lipofectamine2000やOligofectamine(Invitrogen)などの一般的な遺伝子導入試薬の他、例えば、GeneEraserTMsiRNA transfection reagent(Stratagene)等のsiRNA導入に適した導入試薬も市販されている。
【0062】
(D7) p53に対するsiRNAもしくはshRNAを発現するベクター
siRNAを発現するベクターには、タンデムタイプとステムループ(ヘアピン)タイプとがある。前者はsiRNAのセンス鎖の発現カセットとアンチセンス鎖の発現カセットをタンデムに連結したもので、細胞内で各鎖が発現してアニーリングすることにより2本鎖のsiRNA(dsRNA)を形成するというものである。一方、後者はshRNAの発現カセットをベクターに挿入したもので、細胞内でshRNAが発現しdicerによるプロセシングを受けてdsRNAを形成するというものである。プロモーターとしては、polII系プロモーター(例えば、CMV前初期プロモーター)を使用することもできるが、短いRNAの転写を正確に行わせるために、polIII系プロモーターを使用するのが一般的である。polIII系プロモーターとしては、マウスおよびヒトのU6-snRNAプロモーター、ヒトH1-RNase P RNAプロモーター、ヒトバリン-tRNAプロモーターなどが挙げられる。また、転写終結シグナルとして4個以上Tが連続した配列が用いられる。
【0063】
このようにして構築したsiRNAもしくはshRNA発現カセットを、次いでプラスミドベクターやエピソーマルベクター、ウイルスベクター等に挿入する。このようなベクターとしては、核初期化物質である核酸(初期化遺伝子)について上記したと同様のものが、好ましく利用され得る(レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクターや、動物細胞発現プラスミド、エピソーマルベクターなど)。使用するベクターは、初期化遺伝子の場合と同様、得られるiPS細胞から分化誘導されるGR細胞の用途に応じて適宜選択され得る。上述の通り、p53の恒常的な機能阻害は発癌リスクを高める可能性があるので、GR細胞をヒトの医療用途に用いる場合、p53を一過的に発現し、iPS細胞樹立後は速やかに細胞から脱落し得るベクター(例えば、プラスミドベクターなど)を用いることが好ましい。あるいは、p53に対するshRNAをコードする発現ベクターとして、市販のプラスミド(例えば、Addgene社から市販されるpMKO.1-puro p53 shRNA2: #10672等)をもとに作製したレトロウイルス等のウイルスベクター、プラスミドベクター、エピソーマルベクターなどを使用することもできる。必要に応じて、上記Cre-loxPシステムやpiggyBacトランスポゾンシステムを利用することもできる。
【0064】
p53に対するsiRNAもしくはshRNAを発現するベクターの体細胞への接触は、上記のようにして調製されるプラスミドベクター、エピソーマルベクターもしくはウイルスベクターを細胞に導入することにより行われる。これらの遺伝子導入は、初期化遺伝子について上記したと同様の手法で行うことができる。
【0065】
(D8) その他の物質
p53遺伝子の発現を阻害する他の物質として、p53に対するアンチセンス核酸やリボザイムが挙げられる。
アンチセンス核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよい。アンチセンス核酸がDNAの場合、標的RNAとアンチセンスDNAとによって形成されるRNA:DNAハイブリッドは、内在性RNase Hに認識されて標的RNAの選択的な分解を引き起こすことができる。したがって、RNase Hによる分解を指向するアンチセンスDNAの場合、標的配列は、p53 mRNA中の配列だけでなく、p53遺伝子の初期転写産物におけるイントロン領域の配列であってもよい。アンチセンス核酸の標的領域は、該アンチセンス核酸がハイブリダイズすることにより、結果としてp53蛋白質への翻訳が阻害されるものであればその長さに特に制限はなく、p53 mRNAの全配列であっても部分配列であってもよく、短いもので約15塩基程度、長いものでmRNAもしくは初期転写産物の全配列が挙げられる。合成の容易さや抗原性、細胞内移行性の問題等を考慮すれば、約15〜約40塩基、特に約18〜約30塩基からなるオリゴヌクレオチドが好ましい。標的配列の位置としては、5’-及び3’-UTR、開始コドン近傍などが挙げられるが、それらに限定されない。
リボザイムとは、狭義には、核酸を切断する酵素活性を有するRNAをいうが、本明細書では配列特異的な核酸切断活性を有する限りDNAをも包含する概念として用いるものとする。リボザイムとして最も汎用性の高いものとしては、ウイロイドやウイルソイド等の感染性RNAに見られるセルフスプライシングRNAがあり、ハンマーヘッド型やヘアピン型等が知られている。ハンマーヘッド型は約40塩基程度で酵素活性を発揮し、ハンマーヘッド構造をとる部分に隣接する両端の数塩基ずつ(合わせて約10塩基程度)をmRNAの所望の切断部位と相補的な配列にすることにより、標的mRNAのみを特異的に切断することが可能である。
【0066】
アンチセンス核酸やリボザイムはDNA/RNA自動合成機を用いて合成することができる。これらを構成するヌクレオチド分子もまた、安定性、比活性などを向上させるために、上記のsiRNAの場合と同様の修飾を受けていてもよい。
あるいは、アンチセンス核酸やリボザイムは、siRNAの場合と同様に、それらをコードする核酸の形態で使用することもできる。
【0067】
p53の機能阻害物質は、体細胞の核初期化工程においてp53の機能を阻害するのに十分な様式で体細胞に接触させる必要がある。この条件が満たされる限り、核初期化物質とp53の機能阻害物質とは、同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよい。一実施態様において、例えば、核初期化物質がタンパク性因子をコードする核酸であり、p53の機能阻害物質が化学的阻害物質である場合には、前者は遺伝子導入処理からタンパク性因子を大量発現するまでに一定期間のラグがあるのに対し、後者は速やかにp53の機能を阻害しうることから、遺伝子導入処理から一定期間細胞を培養した後に、p53の化学的阻害物質を培地に添加することができる。別の実施態様において、例えば、核初期化物質とp53の機能阻害物質とがいずれもウイルスベクター、プラスミドベクター、エピソーマルベクター等の形態で用いられる場合には、両者を同時に細胞に導入してもよい。
【0068】
(E) 他のiPS細胞の樹立効率改善物質
上記の初期化因子やp53機能阻害物質に加え、公知の他のiPS細胞樹立効率改善物質を体細胞に接触させることにより、iPS細胞の樹立効率をさらに高めることが期待できる。そのようなiPS細胞の樹立効率改善物質としては、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA Smartpool(登録商標)(Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-calcium channel agonist (例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、Wnt Signaling(例えばsoluble Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、2i/LIF (2iはmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤、PloS Biology, 6(10), 2237-2247 (2008))、ES細胞特異的miRNA(例えば、miR-302-367クラスター (Mol. Cell. Biol. doi:10.1128/MCB.00398-08、WO2009/075119)、miR-302 (RNA (2008) 14: 1-10)、miR-291-3p, miR-294およびmiR-295 (以上、Nat. Biotechnol. 27: 459-461 (2009)))等が挙げられるが、それらに限定されない。前記において核酸性の発現阻害剤はsiRNAもしくはshRNAをコードするDNAを含む発現ベクターの形態であってもよい。
【0069】
尚、前記核初期化物質の構成要素のうち、例えば、SV40 Large T antigen等は、体細胞の核初期化のために必須ではなく補助的な因子であるという点において、iPS細胞の樹立効率改善物質の範疇にも含まれ得る。核初期化の機序が明らかでない現状においては、核初期化に必須の因子以外の補助的な因子について、それらを核初期化物質として位置づけるか、あるいはiPS細胞の樹立効率改善物質として位置づけるかは便宜的であってもよい。即ち、体細胞の核初期化プロセスは、体細胞への核初期化物質およびiPS細胞の樹立効率改善物質の接触によって生じる全体的事象として捉えられるので、当業者にとって両者を必ずしも明確に区別する必要性はないであろう。
【0070】
これら他のiPS細胞樹立効率改善物質の体細胞への接触は、該物質が(a) タンパク性因子である場合、(b) 該タンパク性因子をコードする核酸である場合、あるいは(c) 低分子化合物である場合に応じて、p53の機能阻害物質についてそれぞれ上記したと同様の方法により、実施することができる。
他のiPS細胞の樹立効率改善物質は、該物質の非存在下と比較して体細胞からのiPS細胞樹立効率が有意に改善される限り、核初期化物質と同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよく、該物質の物性に応じて、p53の機能阻害物質について上記したと同様のタイミングで体細胞と接触させることができる。
【0071】
(F) 培養条件による樹立効率の改善
体細胞の核初期化工程において低酸素条件下で細胞を培養することにより、iPS細胞の樹立効率をさらに改善することができる。本明細書において「低酸素条件」とは、細胞を培養する際の雰囲気中の酸素濃度が、大気中のそれよりも有意に低いことを意味する。具体的には、通常の細胞培養で一般的に使用される5-10% CO2/95-90%大気の雰囲気中の酸素濃度よりも低い酸素濃度の条件が挙げられ、例えば雰囲気中の酸素濃度が18%以下の条件が該当する。好ましくは、雰囲気中の酸素濃度は15%以下(例、14%以下、13%以下、12%以下、11%以下など)、10%以下(例、9%以下、8%以下、7%以下、6%以下など)、または5%以下(例、4%以下、3%以下、2%以下など)である。また、雰囲気中の酸素濃度は、好ましくは0.1%以上(例、0.2%以上、0.3%以上、0.4%以上など)、0.5%以上(例、0.6%以上、0.7%以上、0.8%以上、0.95以上など)、または1%以上(例、1.1%以上、1.2%以上、1.3%以上、1.4%以上など)である。
【0072】
細胞の環境において低酸素状態を創出する手法は特に制限されないが、酸素濃度の調節可能なCO2インキュベーター内で細胞を培養する方法が最も容易であり、好適な例として挙げられる。酸素濃度の調節可能なCO2インキュベーターは、種々の機器メーカーから販売されている(例えば、Thermo scientific社、池本理化学工業、十慈フィールド、和研薬株式会社などのメーカー製の低酸素培養用CO2インキュベーターを用いることができる)。
【0073】
低酸素条件下で細胞培養を開始する時期は、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、体細胞への核初期化物質の接触より前であっても、該接触と同時であっても、該接触より後であってもよいが、例えば、体細胞に核初期化物質を接触させた直後から、あるいは接触後一定期間(例えば、1ないし10(例、2,3,4,5,6,7,8または9)日)おいた後に低酸素条件下で培養することが好ましい。
【0074】
低酸素条件下で細胞を培養する期間も、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、例えば3日以上、5日以上、7日以上または10日以上で、50日以下、40日以下、35日以下または30日以下の期間等が挙げられるが、それらに限定されない。低酸素条件下での好ましい培養期間は、雰囲気中の酸素濃度によっても変動し、当業者は用いる酸素濃度に応じて適宜当該培養期間を調整することができる。また、一実施態様において、iPS細胞の候補コロニーの選択を、薬剤耐性を指標にして行う場合には、薬剤選択を開始する迄に低酸素条件から正常酸素濃度に戻すことが好ましい。
さらに、低酸素条件下で細胞培養を開始する好ましい時期および好ましい培養期間は、用いられる核初期化物質の種類、正常酸素濃度条件下でのiPS細胞樹立効率などによっても変動する。
【0075】
核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞を、例えばES細胞の培養に適した条件下で培養することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、LIFの代わりに塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加することが望ましい。
また通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞の共存下で培養される。マウス胎仔由来の線維芽細胞としては、通常STO細胞株(ATCC CRL-1503)等がフィーダーとしてよく使われるが、iPS細胞の誘導には、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。しかしながら、マウス胎仔由来の初代線維芽細胞(MEF)を用いた方がヒトiPS細胞の樹立効率がより改善される場合もあるので、MEFの使用もまた好ましい。マイトマイシンC処理済のMEFは、ミリポア社やリプロセル社から市販されている。これらのフィーダー細胞との共培養は、核初期化物質の接触より前から開始してもよいし、該接触時から、あるいは該接触より後(例えば1-10日後)から開始してもよい。
【0076】
(G) iPS細胞の選択・確認
iPS細胞の候補コロニーの選択は、薬剤耐性とレポーター活性を指標とする方法と目視による形態観察による方法とが挙げられる。前者としては、例えば、分化多能性細胞において特異的に高発現する遺伝子(例えば、Fbx15、Nanog、Oct3/4など、好ましくはNanog又はOct3/4)の遺伝子座に、薬剤耐性遺伝子及び/又はレポーター遺伝子をターゲッティングした組換え体細胞を用い、薬剤耐性及び/又はレポーター活性陽性のコロニーを選択するというものである。そのような組換え体細胞としては、例えばFbx15遺伝子座にβgeo(β-ガラクトシダーゼとネオマイシンホスホトランスフェラーゼとの融合タンパク質をコードする)遺伝子をノックインしたマウス由来のMEF(Takahashi & Yamanaka, Cell, 126, 663-676 (2006))、あるいはNanog遺伝子座に緑色蛍光タンパク質(GFP)遺伝子とピューロマイシン耐性遺伝子を組み込んだトランスジェニックマウス由来のMEF(Okita et al., Nature, 448, 313-317 (2007))等が挙げられる。本発明のGR細胞はOct3/4遺伝子を高発現する(Oct4陽性)ことを特徴とするので、レポーター活性を指標とする方法においては、Oct3/4遺伝子座にGFPやRFP等の可視化タンパク質をコードするレポーター遺伝子をノックインした細胞を用いることがより好ましい。一方、目視による形態観察で候補コロニーを選択する方法としては、例えばTakahashi et al., Cell, 131, 861-872 (2007)に記載の方法が挙げられる。レポーター細胞を用いる方法は簡便で効率的ではあるが、iPS細胞から分化誘導されるGR細胞がヒトの治療用途を目的として作製される場合、安全性の観点から目視によるコロニー選択が望ましい。
【0077】
選択されたコロニーの細胞がiPS細胞であることの確認は、上記したOct4(もしくはNanog)レポーター陽性(ピューロマイシン耐性、GFP陽性など)および目視によるES細胞様コロニーの形成によっても行い得るが、より正確を期すために、アルカリフォスファターゼ染色や、各種ES細胞特異的遺伝子の発現を解析したり、選択された細胞をマウスに移植してテラトーマ形成を確認する等の試験を実施することもできる。
【0078】
(H) 他の多能性幹細胞の製造方法
ES細胞の作製方法としては、例えば、哺乳動物の胚盤胞ステージにおける内部細胞塊を培養する方法(例えば、Manipulating the Mouse Embryo A Laboratory Manual,Second Edition,Cold Spring Harbor Laboratory Press (1994) を参照)、体細胞核移植によって作製された初期胚を培養する方法(Wilmut et al., Nature, 385, 810 (1997); Cibelli et al., Science, 280, 1256 (1998); 入谷明ら, 蛋白質核酸酵素, 44, 892 (1999); Baguisi et al., Nature Biotechnology,17, 456 (1999); Wakayama et al., Nature, 394, 369 (1998); Wakayama et al., Nature Genetics, 22, 127 (1999); Wakayama et al., Proc. Natl. Acad. Sci. USA, 96, 14984 (1999); RideoutIII et al., Nature Genetics, 24, 109 (2000))などが挙げられるが、これらに限定されない。また、ES細胞は、所定の機関より入手でき、さらには市販品を購入することもできる。例えば、ヒトES細胞であるKhES-1、KhES-2およびKhES-3は、京都大学再生医科学研究所より入手可能である。
体細胞核移植による場合、体細胞の種類や体細胞を採取するソースは上記iPS細胞の場合に準ずる。
EG細胞は、常法に従って始原生殖細胞を単離し、これをLIF、bFGFおよびSCFの存在下で培養することにより誘導することができる。また、mGS細胞はWO 2005/100548に記載される方法に従って、精巣細胞から作製することができる。多能性成体前駆細胞(MAPC)はJ. Clin. Invest. 109:337-346 (2002) に記載される方法に従って、骨髄から単離することができる。
【0079】
III. 多能性幹細胞からGR細胞への分化誘導
GR細胞の分化誘導用の基本培地としては、例えば、Neurobasal培地、Neural Progenitor Basal培地、NS-A培地、BME培地、BGJb培地、CMRL 1066培地、Glasgow MEM培地、Improved MEM Zinc Option培地、IMDM培地、Medium 199培地、Eagle MEM培地、αMEM培地、DMEM培地、DMEM/F12培地、ハム培地、RPMI 1640培地、Fischer’s培地、およびこれらの混合培地など、動物細胞の培養に用いることのできる培地であれば特に限定されない。より好ましくは、Neurobasal培地である。これらの培地は、血清(例えば、胎仔ウシ血清(FCS)、ヒト血清等)を含有してもしなくてもよい。あるいは、血清の代替添加物(例えばKnockout Serum Replacement (KSR)(Invitrogen社製) 等)を用いてもよい。血清の添加濃度としては0〜20%の範囲で適宜選択することができるが、好ましくは無血清もしくは低血清(例えば、0〜5%、好ましくは0〜2%)の培地が使用され得る。
【0080】
前記いずれかの培地に、例えば、血清タンパク質(例えば、ウシ血清アルブミン(BSA)、ヒト血清アルブミン(HSA)等のアルブミンなど)、還元剤(例えば、2-メルカプトエタノール等)、成長因子(例えば、インスリン、bFGF、HGF、FGF9等)、幹細胞の分化抑制剤(例えば、LIF、Wnt、TGF-β等)、鉄源(例えば、トランスフェリン等)、ミネラル(例えば、亜セレン酸ナトリウム)、アミノ酸(例えば、グルタミン、アラニン、アスパラギン、セリン、アスパラギン酸、システイン、グルタミン酸、グリシン、プロリン、チロシン等の非必須アミノ酸)、ビタミン類(例えば、塩化コリン、パントテン酸、葉酸、ニコチンアミド、塩酸ピリドキサル、リボフラビン、塩酸チアミン、アスコルビン酸、ビオチン、イノシトール等)、糖類(例えば、グルコース等)、有機アミン類(例えば、プトレシン等)、ステロイド(例えば、プロゲステロン、β-エストラジオール等)、抗生物質(例えば、ペニシリン、ストレプトマイシン等)、インターロイキン類(例えば、IL-1、IL-2、IL-3、IL-6等)、接着因子(例えば、ヘパリン、ヘパラン硫酸、コラーゲン、フィブロネクチン等)、有機酸(例えば、ピルビン酸、コハク酸、乳酸等)もしくはその塩、緩衝剤(例えば、HEPES等)、栄養添加物(例えば、B27 supplement、N2 supplement、StemPro supplement等)などの培地添加物を適宜補充して、使用することができる。
好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子が、上記いずれかの基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27の「Medium component」に記載された濃度(最終濃度)を選択することができる。
【0081】
【表1】

【0082】
本発明の多能性幹細胞からGR細胞への分化誘導方法は、(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを特徴とする。好ましくは、多能性幹細胞は、BMP4と、GDNF、EGFおよびSCFから選ばれる2以上の成長因子との3因子の存在下で培養され、より好ましくは、BMP4、GDNF、EGFおよびSCFの4因子の存在下で培養される。これらの成長因子は、上記のいずれかの培地に添加して分化誘導用培地としてもよいし、あるいは、当該因子を産生する細胞をフィーダー細胞として用い、多能性幹細胞と共培養することによって提供されてもよい。
【0083】
BMP4、GDNF、EGFおよびSCFは任意の哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等、好ましくはヒトまたはマウス)由来のものを用いることができるが、多能性幹細胞と同種由来のものを用いることが好ましい。特に、ヒトGR細胞をヒトの治療目的で使用する場合には、異種成分不含(xeno-free)条件下でヒトGR細胞を誘導することが望ましいので、これらの成長因子としてヒト由来タンパク質を用いることが好ましい。
【0084】
BMP4、GDNF、EGFまたはSCFを培地に添加して用いる場合、これらの成長因子は、それらを産生する哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等)の細胞[例えば、神経細胞、グリア細胞、肝細胞、脾細胞、膵β細胞、骨髄細胞、メサンギウム細胞、ランゲルハンス細胞、表皮細胞、上皮細胞、内皮細胞、平滑筋細胞、線維芽細胞、線維細胞、筋細胞、脂肪細胞、免疫細胞(例、マクロファージ、T細胞、B細胞、ナチュラルキラー細胞、肥満細胞、好中球、好塩基球、好酸球、単球)、巨核球、滑膜細胞、軟骨細胞、骨細胞、骨芽細胞、破骨細胞、乳腺細胞もしくは間質細胞、またはこれら細胞の前駆細胞、幹細胞もしくは癌細胞など]もしくはそれらの細胞が存在するあらゆる組織[例えば、脳、脳の各部位(例、嗅球、扁桃核、大脳基底球、海馬、視床、視床下部、大脳皮質、延髄、小脳)、脊髄、下垂体、胃、膵臓、腎臓、肝臓、生殖腺、甲状腺、胆のう、骨髄、副腎、皮膚、筋肉(例、平滑筋、骨格筋)、肺、消化管(例、大腸、小腸)、血管、心臓、胸腺、脾臓、顎下腺、末梢血、前立腺、睾丸、卵巣、胎盤、子宮、骨、関節、脂肪組織(例、白色脂肪組織、褐色脂肪組織)など]等から、自体公知のタンパク質分離精製技術(例えば、塩析や溶媒沈澱法などの溶解度を利用する方法;透析法、限外ろ過法、ゲルろ過法、およびSDS-ポリアクリルアミドゲル電気泳動法などの主として分子量の差を利用する方法;イオン交換クロマトグラフィーなどの荷電の差を利用する方法;アフィニティークロマトグラフィーなどの特異的親和性を利用する方法;逆相高速液体クロマトグラフィーなどの疎水性の差を利用する方法;等電点電気泳動法などの等電点の差を利用する方法など。これらの方法は適宜組み合わせることもできる。)により単離・精製されたものであってよい。また、化学合成もしくは無細胞翻訳系で生化学的に合成されたタンパク質であってもよいが、好ましくは、これらのタンパク質をコードする核酸を導入した形質転換体から産生される組換えタンパク質である。
【0085】
例えば、BMP4、GDNF、EGFおよびSCFのヒトおよびマウスcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得でき、当業者はそれをもとに常法により容易にこれらのcDNAを単離することができる。
遺伝子名 ヒト マウス
BMP4 NM_001202 NM_007554
GDNF NM_000514 NM_010275
EGF NM_001963 NM_010113
SCF NM_000899 NM_013598
得られたcDNAを適当な発現ベクターに挿入して宿主細胞(例えば、動物細胞、昆虫細胞、枯草菌、酵母、大腸菌など)に導入し、得られた形質転換体を培養して、その培養上清から、上記のタンパク質分離精製技術を適宜組み合わせて目的の組換えタンパク質を単離・精製することができる。また、BMP4、GDNF、EGFおよびSCFの組換えタンパク質は市販されている。
【0086】
本発明のGR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持する限り、上記cDNA配列によりコードされるアミノ酸配列と異なるアミノ酸配列を有するタンパク質であってもよい。具体的には、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、
(a) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約90%以上の相同性を有するアミノ酸配列;
(b) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列において、1〜20個のアミノ酸が置換および/または欠失および/または挿入および/または付加されたアミノ酸配列;
(c) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質の他の哺乳動物におけるオルソログのアミノ酸配列;
(d) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質または上記(c)のオルソログのスプライスバリアント、アレル変異体もしくは多型(例、SNPなど)におけるアミノ酸配列;あるいは
(e) 上記(a)〜(d)のアミノ酸配列の一部(フラグメント)
を含み、かつ他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持するタンパク質を意味する。
ここで「相同性」とは、当該技術分野において公知の数学的アルゴリズムを用いて2つのアミノ酸配列をアラインさせた場合の、最適なアラインメント(好ましくは、該アルゴリズムは最適なアラインメントのために配列の一方もしくは両方へのギャップの導入を考慮し得るものである)における、オーバーラップする全アミノ酸残基に対する同一アミノ酸および類似アミノ酸残基の割合(%)を意味する。「類似アミノ酸」とは物理化学的性質において類似したアミノ酸を意味し、例えば、芳香族アミノ酸(Phe、Trp、Tyr)、脂肪族アミノ酸(Ala、Leu、Ile、Val)、極性アミノ酸(Gln、Asn)、塩基性アミノ酸(Lys、Arg、His)、酸性アミノ酸(Glu、Asp)、水酸基を有するアミノ酸(Ser、Thr)、側鎖の小さいアミノ酸(Gly、Ala、Ser、Thr、Met)などの同じグループに分類されるアミノ酸が挙げられる。このような類似アミノ酸による置換は蛋白質の表現型に変化をもたらさない(即ち、保存的アミノ酸置換である)ことが予測される。保存的アミノ酸置換の具体例は当該技術分野で周知であり、種々の文献に記載されている(例えば、Bowieら,Science, 247:1306-1310 (1990)を参照)。本明細書におけるアミノ酸配列の相同性は、相同性計算アルゴリズムNCBI BLAST(National Center for Biotechnology Information Basic Local Alignment Search Tool)を用い、以下の条件(期待値=10;ギャップを許す;マトリクス=BLOSUM62;フィルタリング=OFF)にて計算することができる。
【0087】
上記(a)において、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、より好ましくは、上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約95%以上、より好ましくは約97%以上、特に好ましくは約98%以上の同一性を有するアミノ酸配列を含むタンパク質である。
【0088】
また、上記(b)において、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、好ましくは、(i)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が欠失したアミノ酸配列、(ii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が付加したアミノ酸配列、(iii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が挿入されたアミノ酸配列、(iv)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または(v)それらを組み合わせたアミノ酸配列を含み、かつ他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持するタンパク質などであり得る。
上記のようにアミノ酸配列が挿入、欠失または置換されている場合、その挿入、欠失または置換の位置は、GR細胞の分化誘導活性が保持される限り特に限定されない。
【0089】
本発明の一実施態様においては、多能性幹細胞からGR細胞を分化誘導する際に、BMP4に代えてあるいはそれに加えて、他のBMPファミリータンパク質、例えば、BMP8b、BMP2、BMP7等を用いることができる。好ましくはBMP4とBMP8bとの併用が挙げられる。また、本発明の別の一実施態様においては、多能性幹細胞からGR細胞を分化誘導する際に、GDNFに代えてあるいはそれに加えて、WO 2004/092357に記載されるGDNFの均等物を用いることも可能である。
【0090】
BMP4を培地に添加して用いる場合、BMP4の濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、BMP4の濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。他のBMPファミリータンパク質を併用する場合、BMP全体として上記の濃度範囲となるように添加することが好ましい。
【0091】
GDNFを培地に添加して用いる場合、GDNFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、GDNFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。GDNFの均等物を併用する場合、GDNFおよび均等物全体として上記の濃度範囲となるように添加することが好ましい。
【0092】
EGFを培地に添加して用いる場合、EGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、EGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0093】
SCFを培地に添加して用いる場合、SCFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、SCFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0094】
(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子を培地に添加して用いる場合、(a)および(b)成分は、上記の基本培地もしくは該基本培地に上記の培地添加物が添加された培地とは別個に提供され、用時培地中に添加して用いてもよいし、各成長因子や他の培地成分の安定性などに悪影響を与えない限り、予め培地中に含有された形態で分化誘導用培地として提供されてもよい。
【0095】
本発明の好ましい一実施態様においては、本発明のGR細胞の分化誘導において用いられる(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子のうちの少なくとも1つの因子、好ましくは用いられるすべての因子が、培地に添加されるのではなく、フィーダー細胞から供給される。フィーダー細胞はこれらの成長因子を生来産生する哺乳動物細胞であってもよいが、該成長因子をコードする遺伝子が導入され、該成長因子を過剰発現する組換え細胞を用いることがより好ましい。異なる成長因子を発現する複数種の細胞を組み合わせて用いることも可能であるが、フィーダーからの供給が意図されるすべての成長因子を発現する1種類の細胞を用いることが好ましい。宿主となる細胞としては、従来よりフィーダー細胞として好適に使用されている細胞を用いることができるが、それらに限定されない。例えば、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞 (MEF)(例、STO細胞株(ATCC CRL-1503)等) などがフィーダーとしてよく使われるが、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)なども好ましい。また、多能性幹細胞と同種の細胞をフィーダーとして用いることも好ましい。例えば、ヒト多能性幹細胞からGR細胞を誘導する場合には、ヒト皮膚由来線維芽細胞(HDF)やヒト歯髄幹細胞などをフィーダーとして用いることができる。
【0096】
例えば、BMP4をフィーダー細胞から供給しようとする場合、例えば、M15-BMP4細胞(Proc. Natl. Acad. Sci. USA, 100: 11457-11462 (2003))をフィーダー細胞として用いることができる。さらに、GDNF、EGFおよびSCFから選ばれる1以上の成長因子もフィーダー細胞から供給しようとする場合、例えば、M15-BMP4細胞に、GDNF、EGFおよびSCFから選ばれる1以上の成長因子をコードする核酸を含む発現ベクターを導入して得られる形質転換細胞を用いることもできる。例えば、後述の実施例において、M15-BMP4細胞にGDNF、EGFおよびSCFをコードする各核酸を含むウイルスベクターを導入して、BMP4、GDNF、EGFおよびSCFの4成長因子を過剰発現する細胞(M15-4GF)を作製している。
【0097】
多能性幹細胞からのGR細胞の分化誘導培養は、例えば以下のようにして行うことができる。
本分化誘導工程に用いられる培養器は、細胞培養用であれば特に限定されないが、例えば、フラスコ、組織培養用フラスコ、デッシュ、ペトリデッシュ、組織培養用デッシュ、マルチデッシュ、マイクロプレート、マイクロウエルプレート、マルチプレート、マルチウエルプレート、チャンバースライド、シャーレ、チューブ、トレイ、培養バック、ローラーボトルが挙げられる。培養器は、培養法(浮遊培養法もしくは接着培養法)に応じて、細胞非接着性(低接着性)または細胞接着性とすることができる。細胞接着性の培養器は、培養器の表面が、細胞(多能性幹細胞またはフィーダー細胞)との接着性を向上させる目的で、細胞支持用基質でコーティングされたものであり、そのような細胞支持用基質としては、例えば、コラーゲン、ゼラチン、マトリゲル、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチンなどが挙げられる。
【0098】
培養は、上記培養器中に、多能性幹細胞を、例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように播種し、例えば、CO2インキュベーター中、約1〜約10%、好ましくは約2〜約5%のCO2濃度の雰囲気下、約30〜約40℃、好ましくは約37℃で、約1〜約8週間、好ましくは約2〜約6週間行われる。培養は、浮遊培養で行っても、接着培養で行ってもよいが、好ましくは浮遊培養である。(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子(本発明の分化誘導成長因子)のうちの少なくとも1つの因子がフィーダー細胞から供給される場合、多能性幹細胞とフィーダー細胞とを1:10〜10:1、好ましくは1:5〜5:1、より好ましくは1:2〜2:1の割合で混合し、培養器中に播種して浮遊培養する。あるいは、予めフィーダー細胞を播種した培養器に多能性幹細胞を播種して共培養を行うこともできる。また、本発明の分化誘導成長因子がすべて培地に添加して用いられる場合であっても、フィーダー細胞の存在下で多能性幹細胞を培養してもよい。この場合のフィーダー細胞としては、放射線や抗生物質で処理して細胞分裂を停止させた、通常のMEF (例、STO細胞株(ATCC CRL-1503)等)、SNL細胞(SNL76/7 STO細胞; ECACC 07032801)、HDF、歯髄幹細胞などが挙げられる。
【0099】
多能性幹細胞からGR細胞への分化は、Oct4陽性かつVasa陽性を指標として確認することができる。例えば、両遺伝子の発現制御領域の下流に可視化されるレポーター遺伝子が挿入された多能性幹細胞(例えば、後述の実施例に示されるOct4-GFP/Vasa-RFPノックインマウス由来の多能性幹細胞)を用いた場合、該レポーター遺伝子の発現による発色や蛍光などを検出することにより、Oct4陽性かつVasa陽性の生殖系幹細胞、即ちGR細胞が誘導されていることを確認することができる。Oct4陽性かつVasa陽性(Oct4+/Vasa+)細胞は、フローサイトメトリー(FACS)により、他の未分化細胞(Oct4+/Vasa-)および体細胞分化の進んだ細胞(Oct4-)と選別し、単離することができる。フィーダー細胞を用いて浮遊培養を行った場合、フィーダー細胞を含む浮遊細胞塊が形成されるので、例えば、トリプシン/EDTA、コラゲナーゼ等を加えて細胞塊を解離させ、得られる細胞懸濁液を細胞支持用基質でコーティングされた培養器に播種してインキュベートした後、浮遊細胞を回収することによって、フィーダー細胞を分離除去することができる。
一方、ヒトの治療用途を念頭においた場合、Oct4およびVasa遺伝子座にレポーター遺伝子がノックインした多能性幹細胞の使用は望ましくないので、Oct4遺伝子およびVasa遺伝子自体の発現を検出する必要がある。この場合、Oct4発現に対応する未分化細胞表面マーカーおよびVasaタンパク質もしくはVasa発現に対応する生殖系細胞表面マーカーに対する各抗体とセルソータを用いて、細胞の表面抗原の表現型を解析することにより行うことができる。例えば、未分化細胞表面マーカーとしてはSSEA-1、フォルスマン抗原、β1-およびα6-インテグリン等が、生殖系細胞表面マーカーとしてはEpCAM、CD9、EE2、c-kit等が挙げられる。必要に応じて、Oct4や他の転写因子の発現についても調べることができる。
【0100】
本発明はまた、上記の方法により得られたiPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞、即ちGR細胞を提供する。これまで、ES細胞からVasa陽性の始原生殖細胞(PGC)様細胞を誘導したり(Proc. Natl. Acad. Sci. USA, 100: 11457-11462 (2003))、精子幹細胞から長期培養可能な細胞株(GS細胞)を誘導したりして(例えば、WO 2004/092357など)、不妊マウスの精巣に移植することにより該マウスにおける精子形成を可能にした例はあるが、後述の実施例に示されるとおり、これら従来の培養条件では、iPS細胞から精子形成を引き起こし得る生殖系幹細胞を樹立することはできず、本発明の分化誘導方法を用いることにより、初めてiPS細胞由来の生殖系幹細胞を得ることができた。
【0101】
上述のように、本発明のGR細胞の分化誘導方法においては、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子が、培地に添加されるか、あるいはフィーダー細胞から供給される。従って、本発明はまた、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)への分化誘導剤を提供する。好ましくは、本発明のGR細胞の分化誘導剤には、GDNF、EGFおよびSCFのうちの2因子以上、より好ましくは3因子すべてが含まれる。これらの成長因子は、水もしくは適当な緩衝液中に溶解した形態で提供されてもよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いることもできる。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
本発明のGR細胞の分化誘導剤は、さらに生理学的に許容される担体、賦形剤、防腐剤、安定剤、結合剤、溶解補助剤、非イオン性界面活性剤、緩衝剤、保存剤、酸化防止剤などを含むこともできる。
【0102】
別の好ましい実施態様においては、本発明のGR細胞の分化誘導剤は、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を産生する細胞を含有する。当該細胞としては、例えば、これらの成長因子をコードする核酸を含む発現ベクターを宿主細胞に導入して得られる、当該成長因子を過剰発現する組換え細胞が挙げられる。当該細胞はフィーダー細胞として多能性幹細胞と共培養されるので、当該技術分野で、従来よりフィーダー細胞として使用されている細胞を宿主細胞として用いることが好ましい。具体的には上記した通りの細胞が例示される。(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を産生する細胞は、適当な培地(例えば、上記基本培地など)に適当な細胞密度で懸濁した状態、あるいは常法により凍結保存された状態で提供され得る。
【0103】
本発明はまた、上記のいずれかの基本培地もしくは該基本培地に上記のいずれかの培地添加物が補充された培地に、上記本発明のGR細胞の分化誘導剤が添加されてなる、GR細胞の分化誘導用培地を提供する。該分化誘導用培地は、分化誘導剤が添加された状態で提供されてもよいし、分化誘導剤が別個の試薬として提供され、用時培地に添加されるようにキット化されたものであってもよい。
【0104】
IV. GR細胞の維持増幅方法
上記のようにして得られるOct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)を、GDNF、EGF、SCFおよび塩基性線維芽細胞成長因子(bFGF)の存在下で培養することにより、Oct4陽性かつVasa陽性の性質を維持したまま、長期培養可能である。したがって、本発明はまた、GDNF、EGF、SCFおよびbFGFの存在下でGR細胞を培養することを含む、GR細胞の増幅方法を提供する。
【0105】
本発明の維持増幅方法に用いられるGR細胞は、Oct4陽性かつVasa陽性であり、精巣に移植した場合に腫瘍を形成せず、かつ排除されることなく長期生着可能な、生殖系細胞として運命決定された細胞である限り、その由来に特に制限はないが、好ましくは、多能性幹細胞由来のGR細胞であり、より好ましくは、上記の本発明の分化誘導方法により誘導されたGR細胞である。GR細胞は単離精製された均一なOct4陽性かつVasa陽性細胞集団として提供されてもよいし、Oct4陰性もしくはVasa陰性の細胞が混在した不均一な細胞集団として提供されてもよい(以下、特にことわらない限り、GR細胞を含む不均一な細胞集団も含めてGR細胞と記載する)。
【0106】
本発明のGR細胞の維持増幅方法に用いられる基本培地および任意の培地添加物としては、上記の多能性幹細胞からGR細胞への分化誘導方法において使用されるものと同じものが挙げられる。但し、後述するように、本発明のGR細胞の維持増幅方法はSCFおよびbFGFの存在下で行われるため、LIFの併用はPGC様細胞であるGR細胞のEG細胞様細胞への脱分化を誘導する可能性があり、使用しないことが好ましい場合がある。
好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子が、基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27の「Medium component」に記載された濃度(最終濃度)を選択することができる。
【0107】
本発明のGR細胞の維持増幅方法において、GR細胞はGDNF、EGF、SCFおよびbFGFの存在下で培養される。これらの成長因子は、上記のいずれかの培地に添加して分化誘導用培地としてもよいし、あるいは、当該因子を産生する細胞をフィーダー細胞として用い、多能性幹細胞と共培養することによって提供されてもよいが、好ましくは培地に添加して用いられる。
【0108】
GDNF、EGF、SCFおよびbFGFは任意の哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等、好ましくはヒトまたはマウス)由来のものを用いることができるが、GR細胞と同種由来のものを用いることが好ましい。特に、ヒトGR細胞をヒトの治療目的で使用する場合には、異種成分不含(xeno-free)条件下でヒトGR細胞を誘導することが望ましいので、これらの成長因子としてヒト由来タンパク質を用いることが好ましい。
【0109】
GDNF、EGF、SCFまたはbFGFを培地に添加して用いる場合、これらの成長因子は、それらを産生する哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等)の細胞もしくはそれらの細胞が存在するあらゆる組織等から、自体公知のタンパク質分離精製技術により単離・精製されたものであってよい。また、化学合成もしくは無細胞翻訳系で生化学的に合成されたタンパク質であってもよいが、好ましくは、これらのタンパク質をコードする核酸を導入した形質転換体から産生される組換えタンパク質である。GDNF、EGFおよびSCFの組換えタンパク質は、上述の方法により調製することができる。また、bFGFのヒトおよびマウスcDNA配列情報はNCBI accession numbers NM_002006およびNM_008006を参照することにより取得でき、当業者はそれをもとに常法によりbFGFのcDNAを単離し、上記と同様の方法により、容易に組換えタンパク質を調製することができる。また、GDNF、EGF、SCFおよびbFGFの組換えタンパク質は市販されている。
【0110】
本発明のGR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持する限り、上記cDNA配列によりコードされるアミノ酸配列と異なるアミノ酸配列を有するタンパク質であってもよい。具体的には、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、
(a) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約90%以上の相同性を有するアミノ酸配列;
(b) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列において、1〜20個のアミノ酸が置換および/または欠失および/または挿入および/または付加されたアミノ酸配列;
(c) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質の他の哺乳動物におけるオルソログのアミノ酸配列;
(d) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質または上記(c)のオルソログのスプライスバリアント、アレル変異体もしくは多型(例、SNPなど)におけるアミノ酸配列;あるいは
(e) 上記(a)〜(d)のアミノ酸配列の一部(フラグメント)
を含み、かつ他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持するタンパク質を意味する。
ここで「相同性」とは、GR細胞の分化誘導に用いられるBMP4などについて上記したのと同義である。上記(a)において、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、より好ましくは、上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約95%以上、より好ましくは約97%以上、特に好ましくは約98%以上の同一性を有するアミノ酸配列を含むタンパク質である。
【0111】
また、上記(b)において、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、好ましくは、(i)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が欠失したアミノ酸配列、(ii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が付加したアミノ酸配列、(iii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が挿入されたアミノ酸配列、(iv)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または(v)それらを組み合わせたアミノ酸配列を含み、かつ他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持するタンパク質などであり得る。
上記のようにアミノ酸配列が挿入、欠失または置換されている場合、その挿入、欠失または置換の位置は、GR細胞の維持増幅活性が保持される限り特に限定されない。
【0112】
本発明の一実施態様においては、GR細胞を維持増幅する際に、GDNFに代えてあるいはそれに加えて、WO 2004/092357に記載されるGDNFの均等物を用いることも可能である。
【0113】
GDNFを培地に添加して用いる場合、GDNFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、GDNFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。GDNFの均等物を併用する場合、GDNFおよび均等物全体として上記の濃度範囲となるように添加することが好ましい。
【0114】
EGFを培地に添加して用いる場合、EGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、EGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0115】
SCFを培地に添加して用いる場合、SCFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、SCFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0116】
bFGFを培地に添加して用いる場合、bFGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、bFGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0117】
GDNF、EGF、SCFおよびbFGFを培地に添加して用いる場合、これらの成分は、上記の基本培地もしくは該基本培地に上記の任意成分である培地添加物が添加された培地とは別個に提供され、用時培地中に添加して用いてもよいし、各成長因子や他の培地成分の安定性などに悪影響を与えない限り、予め培地中に含有された形態でGR細胞の維持増幅用培地として提供されてもよい。
【0118】
本発明の別の一実施態様においては、本発明のGR細胞の維持増幅において用いられるGDNF、EGF、SCFおよびbFGFのうちの少なくとも1つの因子が、培地に添加されるのではなく、フィーダー細胞から供給され得る。フィーダー細胞はこれらの成長因子を生来産生する哺乳動物細胞であってもよいが、該成長因子をコードする遺伝子が導入され、該成長因子を過剰発現する組換え細胞を用いることがより好ましい。異なる成長因子を発現する複数種の細胞を組み合わせて用いることも可能であるが、フィーダーからの供給が意図されるすべての成長因子を発現する1種類の細胞を用いることが好ましい。宿主となる細胞としては、GR細胞の分化誘導において上記したのと同様の細胞を用いることができる。組換え細胞の製造についても、GR細胞の分化誘導において上記したのと同様の手法で行うことができる。
【0119】
本発明のGR細胞の維持増幅方法においては、上記の4因子(GDNF、EGF、SCFおよびbFGF)に加えて、さらにHGF、FGF9等の他の成長因子、IL-2等のインターロイキン類から選ばれる1以上の培地添加物の存在下でGR細胞を培養することが好ましい。
HGFやFGF9などの成長因子はそれぞれ、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上で、例えば約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下の濃度で、培地に添加することができる。また、IL-2等のインターロイキン類は、例えば約0.01 ng/ml以上、好ましくは約0.1 ng/ml以上、より好ましくは約0.5 ng/ml以上、特に好ましくは約1 ng/ml以上で、例えば約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下の濃度で、培地に添加することができる。
【0120】
本発明のGR細胞の維持増幅方法においては、さらに男性ホルモン(例、テストステロン等の)および/またはその誘導物質(例、フォルスコリン)の存在下で、GR細胞を培養することもまた好ましい。これらの成分は、GR細胞の維持増幅を支持し、かつ細胞の生存に悪影響を及ぼさない範囲で培地に添加することができるが、例えば約0.001 μM以上、好ましくは0.01 μM以上、より好ましくは0.05 μM以上で、約1000 μM以下、好ましくは約500 μM以下、より好ましくは約100 μM以下の濃度で培地に添加することができる。
【0121】
別の好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子と、表2に記載される培地添加物から選ばれる1以上の因子とが、上記いずれかの基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27に記載された濃度(最終濃度)を選択することができる。
【0122】
【表2】
【0123】
GR細胞の維持増幅培養は、例えば以下のようにして行うことができる。
本培養工程に用いられる培養器は、細胞培養用であれば特に限定されず、GR細胞の分化誘導工程で用いられるものと同様の培養器が例示される。培養器は、培養法(浮遊培養法もしくは接着培養法)に応じて、細胞非接着性(低接着性)または細胞接着性とすることができる。細胞接着性の培養器は、培養器の表面が、細胞(多能性幹細胞またはフィーダー細胞)との接着性を向上させる目的で、細胞支持用基質でコーティングされたものであり、そのような細胞支持用基質としては、例えば、コラーゲン、ゼラチン、マトリゲル、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチンなどが挙げられる。
【0124】
培養は、上記培養器中に、GR細胞を、例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように播種し、例えば、CO2インキュベーター中、約1〜約10%、好ましくは約2〜約5%のCO2濃度の雰囲気下、約30〜約40℃、好ましくは約37℃で行われる。GDNF、EGF、SCFおよびbFGFのうちの少なくとも1つの因子がフィーダー細胞から供給される場合、フィーダー細胞を例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように培養器中に播種し、その上にGR細胞を播種する。また、GDNF、EGF、SCFおよびbFGFがすべて培地に添加して用いられる場合であっても、フィーダー細胞の存在下で多能性幹細胞を培養することが好ましい。この場合のフィーダー細胞としては、放射線や抗生物質で処理して細胞分裂を停止させた、通常のMEF (例、STO細胞株(ATCC CRL-1503)等)、SNL細胞(SNL76/7 STO細胞; ECACC 07032801)、HDF、歯髄幹細胞などが挙げられる。細胞がコンフルエントもしくはサブコンフルエントに達した段階で常法に従って細胞を継代培養することにより、GR細胞をその分化状態を維持したまま、長期維持、増幅することができる。
【0125】
GR細胞がOct4陰性もしくはVasa陰性の細胞を含む不均一な細胞集団として提供された場合、増幅された細胞集団から、Oct4陽性およびVasa陽性を指標としてGR細胞を分離回収することにより、均一なGR細胞を得ることができる。GR細胞の分離方法としては、多能性幹細胞から誘導されたGR細胞を分離する際に用いるのと同様の方法が挙げられる。
【0126】
本発明のGR細胞の分化誘導方法により、多能性幹細胞から誘導された細胞集団の中には、GR細胞に加えて、Oct4陽性かつVasa陰性の未分化細胞が含まれており、分離された当該未分化細胞を、上記GR細胞の維持増幅条件下で培養すると、Oct4陽性かつVasa陰性の分化状態を維持したまま安定に増幅させることができたので、この未分化細胞株をGsp細胞と名づけた。Gsp細胞を免疫不全マウスに移植するとテラトーマを形成し、また種々の特性解析の結果からも、Gsp細胞がもとの多能性幹細胞とは異なる、新規に樹立された多能性幹細胞であることが示された。したがって、本発明はまた、GR細胞の分化誘導過程で誘導され、かつGR細胞の維持増幅条件下で安定に維持増幅される新規多能性幹細胞であるGsp細胞並びにその製造および維持方法を提供する。Gsp細胞は、GS細胞樹立過程で見出されたmGS細胞と同様、ES細胞やiPS細胞などの既知多能性幹細胞の代替物として、種々の用途に利用可能である。
【0127】
上述のように、本発明のGR細胞の維持増幅方法においては、GDNF、EGF、SCFおよびbFGFが培地に添加される。従って、本発明はまた、GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)の増幅支持剤を提供する。なお、本発明のGR細胞の維持増幅条件下では、Oct4陽性かつVasa陰性の多能性幹細胞(Gsp細胞)も安定に維持されるので、当該増幅支持剤はまたGsp細胞の増幅支持剤でもある。GDNF、EGF、SCF、bFGFの各因子は、水もしくは適当な緩衝液中に溶解した形態で提供されてもよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いることもできる。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
本発明のGR細胞の増幅支持剤は、さらに生理学的に許容される担体、賦形剤、防腐剤、安定剤、結合剤、溶解補助剤、非イオン性界面活性剤、緩衝剤、保存剤、酸化防止剤などを含むこともできる。
【0128】
別の好ましい実施態様においては、本発明のGR細胞の増幅支持剤には、GDNF、EGF、SCFおよびbFGFの4因子に加えて、HGF、IL-2およびFGF9から選ばされる1以上、好ましくは2以上の因子、より好ましくは3因子すべてがさらに組み合わされる。これらの各因子も、水もしくは適当な緩衝液中に溶解した形態で提供されてよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いてもよい。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
【0129】
本発明はまた、上記のいずれかの基本培地もしくは該基本培地に上記の任意成分である培地添加物が補充された培地に、上記本発明のGR細胞の増幅支持剤が添加されてなる、GR細胞の維持増幅用培地を提供する。該維持増幅用培地は、増幅支持剤が添加された状態で提供されてもよいし、増幅支持剤が別個の試薬として提供され、用時培地に添加されるようにキット化されたものであってもよい。
【0130】
本発明また、上記の分化誘導方法により得られたGR細胞、または上記の維持増幅方法により維持されたGR細胞を、該細胞と同種の不妊動物の精巣に移植することによる、該不妊動物に精子を形成させる方法を提供する。当該方法は、本発明の分化誘導方法および/または維持増幅方法によりGR細胞を樹立することができる任意の哺乳動物に対して適用可能であるが、不妊治療や遺伝子治療への応用を念頭におく場合、対象動物として好ましくはヒトが挙げられる。また、研究目的においては、従来より実験動物として繁用されているマウス、ラット、モルモット、ハムスター、スナネズミ、ウサギ、イヌ、サルなどが挙げられる。さらに、育種目的においては、種々の家畜動物(例、ウシ、ウマ、ブタ、ヒツジ、ヤギ等)や愛玩動物(例、イヌ、ネコ等)などが挙げられる。
【0131】
GR細胞の精巣への移植は、例えばWO 2004/092357やBiol. Reprod., 69: 612-616 (2003) に記載の方法において、GS細胞の代わりに本発明のGR細胞を用いることにより実施することができる。
【0132】
本発明の精子形成方法を不妊治療に用いる場合、移植されるGR細胞はレシピエント動物個体から誘導されたものであることが特に望ましい。患者由来の精子を作るためには、精子形成のために移植される生殖系幹細胞は、出生後に採取可能な細胞から誘導可能なものであることが必要であるが、iPS細胞は安全面を考慮してタンパク質導入やウイルスフリーの遺伝子導入により作製しようとする場合、樹立効率が低いことが欠点である。そのため、患者本人の精子幹細胞を採取できる場合は、精子幹細胞からGS細胞を誘導して用いる方が有利な場合もある。しかしながら、精子幹細胞の採取が不可能か、もしくは多数回精巣生検を必要とするなどの理由で精子幹細胞を採取することに相当程度のリスクを伴う場合などでは、皮膚細胞などから容易に誘導可能なiPS細胞を材料として本発明の方法により作製されたGR細胞を用いることはきわめて有意義である。したがって、本発明の精子形成方法を不妊治療に用いる場合、GR細胞の好ましい投与対象として、精子幹細胞の採取が困難な不妊動物が挙げられる。
【0133】
また、本発明の精子形成方法を遺伝子治療に用いる場合、遺伝病患者から誘導されるiPS細胞やmGS細胞から分化誘導されたGR細胞をそのまま移植することはできない。そのため、患者から誘導した多能性幹細胞もしくは該細胞から本発明の方法により分化誘導したGR細胞に遺伝子操作を施して、変異遺伝子を正常な遺伝子と置換するなどの処理をしたGR細胞を作製して、これを患者に移植する必要がある。このようにして正常な遺伝子機能を有する患者由来の精子を形成させることにより、患者の変異遺伝子が後代に伝わり子孫が遺伝病を発症するのを予防することができる。
【0134】
また、育種分野において、優良な血統が途絶えた場合でも、当該優良な遺伝情報を有する個体から多能性幹細胞を樹立しておけば、本発明の方法により該多能性幹細胞からGR細胞を誘導、増幅して別の個体の精巣に移植することにより、該個体に多能性幹細胞由来の精子を形成させることができ、該個体を用いて繁殖を行うことにより優良な血統を維持させることができる。
【0135】
本発明はまた、上記の分化誘導方法により得られたGR細胞、または上記の維持増幅方法により維持されたGR細胞を含有してなる、雄性不妊治療剤を提供する。
本発明のGR細胞は、常套手段にしたがって医薬上許容される担体と混合するなどして、注射剤、懸濁剤、点滴剤等の非経口製剤として製造される。当該非経口製剤に含まれ得る医薬上許容される担体としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液(例えば、D-ソルビトール、D-マンニトール、塩化ナトリウムなど)などの注射用の水性液を挙げることができる。本発明の剤は、例えば、緩衝剤(例えば、リン酸塩緩衝液、酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩化ベンザルコニウム、塩酸プロカインなど)、安定剤(例えば、ヒト血清アルブミン、ポリエチレングリコールなど)、保存剤、酸化防止剤などと配合しても良い。もちろん、GR細胞は、上記のGR細胞の維持増幅用培地に懸濁したものをそのまま製剤として用いてもよい。本発明の不妊治療剤を水性懸濁液剤として製剤化する場合、上記水性液に約1×106〜約1×108細胞/mLとなるように、GR細胞を懸濁させればよい。
本発明の雄性不妊治療剤は、幹細胞の凍結保存に通常使用される条件で凍結保存された状態で提供され、用時融解して用いることもできる。その場合、血清もしくはその代替物、有機溶剤(例、DMSO)等をさらに含んでいてもよい。この場合、血清もしくはその代替物の濃度は、特に限定されるものではないが約1〜約30% (v/v)、好ましくは約5〜約20% (v/v) であり得る。有機溶剤の濃度は、特に限定されるものではないが0〜約50% (v/v)、好ましくは約5〜約20% (v/v) であり得る。
【0136】
このようにして得られる製剤は、安定で低毒性であるので、ヒトなどの哺乳動物に対して安全に投与することができる。投与方法は特に限定されないが、好ましくは注射もしくは点滴投与であり、精細管内に投与され得る。本発明の不妊治療剤は、ヒト不妊患者においては、例えば、1回につきGR細胞量として約1.0×105〜約1×107細胞を、単回もしくは約1〜約2週間隔で、複数回(例、2〜10回)投与され得る。
【0137】
以下、実施例を示して本発明をより具体的に説明するが、本発明は以下に示す実施例によって何ら限定されるものではない。
【実施例】
【0138】
実施例1 iPS細胞の樹立
実験系として、レポーター遺伝子であるOct4-GFPとMvh (mouse vasa homolog)-RFPの導入により未分化細胞と生殖細胞がそれぞれ可視化されたトランスジェニックマウス(Oct4-GFP/Mvh-RFP Tgマウス)を使用した。Oct3/4(Oct4)は未分化細胞特異的発現遺伝子であり、マウスvasaは生殖細胞系譜特異的発現遺伝子であることが知られている。このマウス系統は、マウスOct4遺伝子の発現制御領域にGFP遺伝子を連結したレポータープラスミドDNAを受精卵に顕微注入して作製されたTgマウス (Oct4-GFP)、およびマウスMvhゲノム遺伝子にRFP遺伝子を挿入したBACクローンDNAを顕微注入したTgマウス (Mvh-RFP) の両者を自然交配することによって作製した。
初期化に使用するレトロウイルスは、前日に6 well培養プレート (Falcon) に1 well当り0.6 x 106で播種したPlat-E細胞(Morita, S. et al., Gene Ther. 7, 1063-1066)にレトロウイルス発現ベクター (pMXs-Oct3/4, pMXs-Sox2, pMXs-Klf4, pMXs-Nanog) を個々に導入して作製した。培養液はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎仔血清を10%加えたもの) を使用し、37℃、5% CO2で培養した。ベクターの導入のためにFuGene6 transfection reagent (Roche) 4.5 μLをOpti-MEM I Reduced-Serum Medium (Invitrogen) 100 μLに入れ、室温で5分間静置した。その後、各発現ベクターを1.5 μg加え、さらに室温で15分静置してからPlat-E細胞の培養液に加えた。2日目にPlat-E細胞の培養上清を新しい培地に換え、3日目に培養上清を回収して0.45 μm sterile filter (Whatman) で濾過し、polybrene (Nacalai) を4 μg/mLとなるように加えてウイルス液とした。
前述のOct4-GFP/Mvh-RFP Tgマウスの胎仔(受精後13.5日)から、線維芽細胞(MEF)を単離した。このMEFを0.1% ゼラチン (Sigma) でコートした6 well培養プレート (Falcon) に1 well当り1 x 105で播種した。培養液はDMEM/10% FCSを使用し、37℃、5% CO2で培養した。翌日、レトロウイルス液を加え、一晩感染させて遺伝子を導入した。遺伝子導入は、Oct3/4, Sox2, Klf4およびNanogの4種の遺伝子導入と、Oct3/4, Sox2およびKlf4の3種の遺伝子導入を行った。
ウイルス感染後3日目からLIFを加えたES細胞用培地(DMEM (Nacarai tesque) に15% ウシ胎仔血清、2 mM L-グルタミン (Invitrogen)、100 μM 非必須アミノ酸(Invitrogen)、100 μM 2-メルカプトエタノール (Invitrogen)、50 U/mL ペニシリン (Invitrogen) と50 μg/mL ストレプトマイシン (Invitrogen) を加えたもの)を用いて培養した。感染後5日目にMEFの培地を除き、PBS 1 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらES細胞用培地を加えて懸濁し、5 x 103個の細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞にはマイトマイシンCで処理して、細胞分裂を止めたSNL細胞(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))を用いた。以後コロニーが観察できるようになるまで2日ごとにES細胞用培地の交換を行った。感染から17日目のiPS細胞コロニーの写真を図1に示す。4遺伝子導入、3遺伝子導入いずれの場合も、Oct3/4-GFP陽性で、かつMvh-RFP陰性のiPS細胞コロニーが樹立できた。
これらのiPSクローンについて常法によりGenomic PCR解析を行った結果、いずれのクローンにおいても導入した外来遺伝子の組み込みが確認された(図2)。
また、各iPSクローンにおける未分化マーカーの発現を、Rever Tra Ace kit(Takara)を使用してRT-PCR解析を行うことにより調べた。結果を図3に示す。各iPSクローンは、ES細胞やNanog-iPS(Nature, 448, 313-317 (2007))と同等の未分化マーカーの発現を示した。
次に、Cell, 126, 663-676 (2006) に記載の方法に従ってテラトーマを形成させた。具体的には、1 x 106個のiPS細胞を免疫不全マウスの皮下に注射し、4週間後にテラトーマを単離した。テラトーマを切り刻んで4% フォルムアルデヒドを含有するPBS(-)で固定した。パラフィン包埋組織をスライスし、ヘマトキシリン・エオシンで染色した。結果を図4に示す。組織学的に見ると、腫瘍は複数の種類の細胞から構成されており、神経組織、表皮組織、筋肉組織、軟骨組織、脂肪組織、および腸管様上皮組織が認められたことから、樹立したiPS細胞の多能性が証明された。
【0139】
実施例2 生殖系幹細胞の分化維持培地の確立
樹立したiPS細胞の分化誘導を行うために、DMEM/10% FCSを使用して胚様体 (EB) 形成を行った。具体的には、培養液1 mL当り5 x 105個のiPS細胞を6 wellの低細胞接着性培養プレート (Nunc) に播種し、2日毎に培地交換を行った。培養14日目の写真を図5に示す。同じOct4-GFP/Mvh-RFP TgのES細胞由来のEB中には部分的にMvh-RFP陽性の生殖細胞の分化が認められたが、iPS細胞由来のEB中にはMvh-RFP陽性の細胞は確認されなかった。
そこで、iPS細胞の生殖細胞分化を誘導する培養条件を確立するために、分化支持細胞としてPNAS, 100, 11457-11462 (2003) で使用されたM15-BMP4細胞の改良を行った。前述の方法でPlat-E細胞にグリア細胞由来神経栄養因子 (GDNF)、細胞膜結合型幹細胞因子 (mSCF)、上皮細胞成長因子 (EGF)の レトロウイルス発現ベクター (pMXs-GDNF-IP, pMXs-mSCF-IP, pMXs-EGF-IP) を導入し、得られたウイルス液をM15-BMP4細胞に感染させた。ウイルス感染細胞を選択するために0.2 mg/mL ネオマイシンおよび2.5 μg/mL ピューロマイシン存在下で培養を行い、増殖し続ける細胞を新たにM15-4GFと命名した。M15-4GFの形態とRT-PCRの結果を図6に示す。RT-PCRによって、M15-4GFではBMP4に加えて遺伝子導入したGDNF、mSCF、EGFが高発現していることを確認した。
さらに、生殖細胞の分化誘導に適した培地作製を行った。組成を図27に示す。
Neurobasal medium (Invitrogen) を基礎培地として、最終濃度で1x B-27 Supplement (Invitrogen)、1 x Penicillin-Streptomycin-Glutamine (Invitrogen)、5 mg/mL Bovine Albumin (MP Biomedicals)、0.1 mM Non-Essential Amino Acids (Invitrogen)、1 mM Sodium Pyruvate (Invitrogen)、1x Vitamin Solution (Invitrogen)、1x Insulin-Transferrin-Selenium Supplement (Invitrogen)、6 mg/mL D-(+)-Glucose (Sigma)、60 ng/mL Progesteron (Sigma)、30 ng/mL β-Estradiol (Sigma)、55 μM 2-Mercaptoethanol (Invitorgen)、0.34 μL/mL Sodium DL-lactate (Sigma)、60 μg/mL Putrescine dihydrochloride (Sigma)、0.1% FCS (Invitorgen) を添加した(図中「Medium component」)。また、分化誘導後の細胞の維持培養には、上記培養液に最終濃度で15 ng/mL GDNF (R&D systems)、20 ng/mL EGF (AUSTRAL Biologicals)、12.5 ng/mL basic fibroblast growth factor (bFGF) (Wako)、10 ng/mL SCF (R&D systems)、10 ng/mL hepatocyte growth factor (HGF) (PEPROTECH)、5 ng/mL interleukin-2 (IL2) (Roche)、12.5 ng/mL fibroblast growth factor 9 (R&D systems)、10 μM Forskolin (Sigma)、0.1 μM Testosterone (Sigma)、0.4x StemPro Supplement (Invitrogen)(図中「Supplement」)を添加した。
生殖細胞分化誘導を行うために、iPS細胞とM15-4GF細胞を培養液1 mL当り各々5×105個となるように増殖因子(Supplement)無添加の上記培養液中で混合し、6 wellの低細胞接着性培養プレートに播種した。2日毎に培地交換を行いながら浮遊培養を行ったところ、細胞塊中にMvh-RFP陽性細胞の分化が確認された。培養28日目に浮遊細胞塊を15 mLチューブに回収し、PBSで洗浄後に0.25% Trypsin/1 mM EDTAと0.2 mg/mL collagenase IV (Invitrogen) を加えて、37℃で15分間程度反応させた。DMEM/10% FCSを加えて懸濁後に遠心 (1000 rpm) し、維持培養用の増殖因子(Supplement)添加培養液に置換した。M15-4GF細胞を除去するために、細胞懸濁液をゲラチンコートした培養プレート上に播種して30分から1時間程度静置した後に浮遊細胞を回収し、マイトマイシンC処理したMEF feeder細胞 (Millipore) 上に播種して培養を行った。その結果、Oct4-GFP陽性/Mvh-RFP陽性のコロニーの形成が観察された(図7)。
Oct4-GFP陽性/Mvh-RFP陽性のコロニーを形成する細胞は上記培養条件下で安定に増殖し、未分化iPS細胞とは異なる形態を示した(図8)。また、未分化細胞と生殖細胞に共通する細胞表面マーカーであるalkaline phosphataseの活性が認められた(図9)。
このOct4-GFP陽性/Mvh-RFP陽性コロニーを、iPS細胞用の培養条件(DMEM/10% FCS)やBiol. Reprod, 69, 612-616 (2003) に記載されているGS細胞用の培養条件(GDNF、LIF、EGFおよびbFGF添加)で培養した場合、上記培養条件下に比べて増殖速度は低下し、部分的な細胞死やMvh-RFP蛍光強度の低下が認められた (図10)。また、未分化iPS細胞を分化誘導過程を経ずに直接上記維持培養条件下で培養した場合には、体細胞分化に伴うOct4-GFPの縮退や細胞死は観察されるものの、Mvh-RFPの誘導は認められなかった (図11)。
【0140】
実施例3 Oct4-GFP陽性/Mvh-RFP陽性細胞の特徴
上記培養条件で浮遊細胞塊を解離、培養した際に、Oct4-GFP陽性/Mvh-RFP陽性の生殖細胞様細胞に加えて、Oct4-GFP陽性/Mvh-RFP陰性の未分化様の細胞も分離、安定増殖することが出来た(図12)。以降、Oct4-GFP陽性/Mvh-RFP陽性細胞を「GR細胞」と称し、Oct4-GFP陽性/Mvh-RFP陰性細胞を「Gsp細胞」と称することとした。
GR細胞とGsp細胞に対してGenomic PCR解析を行ったところ、iPS細胞作製時に導入した外来遺伝子の組み込みが確認された。過去にES細胞から分化誘導、樹立されたOct4-GFP陰性/Mvh-RFP陽性の生殖細胞様細胞株であるEK細胞では外来遺伝子は認められなかったのに対して、GR細胞とGsp細胞はiPS細胞由来であることが示された (図13)。また、サザンブロット解析が示す外来遺伝子のバンドパターンから、GR細胞とGsp細胞がiPS細胞と同一クローン由来であることも確認された (図14)。しかし、GR細胞ではSox2の外来遺伝子が一つ欠損していることが明らかとなった。
24 well培養プレートにあらかじめ播種したマイトマイシンC処理MEF feeder細胞上に1 well当たり2.5×103個となるように未分化iPS細胞とGR細胞をそれぞれ播種し、2日毎に総細胞数を測定した結果、GR細胞はiPS細胞に比べて増殖速度が1/2程度であることが分った (図15)。また、GR細胞の増殖におけるfeeder細胞依存性を調べる為に、6 well培養プレートをゲラチン、ラミニン、またはファイブロネクチンでコーティングし、1 well当たり1×104個となるようにGR細胞を播種した。その結果、MEF feeder細胞上に播種した際にはGR細胞はコロニーを形成しながら増殖するのに対し、feeder freeの条件ではどの細胞外マトリックスを用いた場合においても細胞がほとんど接着することがなく、コロニー形成も認められなかった (図16)。
腫瘍細胞や未分化なiPS細胞を免疫不全のnu/nuマウス皮下に移植すると、移植部位において腫瘍を形成する。一方、精子幹細胞株であるGS細胞の移植においては腫瘍の形成は認められないことが知られている。そこでGR細胞およびGsp細胞を免疫不全マウスに移植し、腫瘍形成の有無を調べた。その結果、Gsp細胞を移植した場合、iPS細胞に比べてサイズは小さいが腫瘍化することが分かった。一方、GR細胞を移植した場合には、ES細胞由来のEK細胞と同様に腫瘍形成は認められず、GR細胞は胚性腫瘍 (EC) 細胞のように腫瘍化した細胞ではないことが示された (図17)。
分化誘導に使用したiPS細胞はレトロウイルスによって外来遺伝子を導入していることから、レトロウイルスの再活性化によって不死化した細胞が得られた可能性も考えられる。そこで、RT-PCRによって導入した外来遺伝子とそれに対応する内在性遺伝子の発現を解析した。その結果、導入した外来遺伝子の異常な遺伝子発現の向上は認められなかった (図18)。また、リアルタイムPCRによって遺伝子発現を定量化したところ、GR細胞における外来遺伝子の発現は未分化iPS細胞と比較してむしろ低下していることが分かった (図19)。
続いて、GR細胞における生殖細胞マーカー遺伝子の発現をRT-PCRで調べた。BMC Dev. Biol., 6, 34 (2006) にあるように、GS細胞ではECAT1やFgf4の発現が抑制されていることが判明しているが、GR細胞ではそれらの遺伝子発現はまだ抑制されていなかった。また、生体内の細胞では発現が認められないERasが発現しており、GR細胞が株化された培養細胞であることが示された。GR細胞ではPrdm14、Mvh、Plzf、c-Retなどの発現が上昇している一方で、Stra8、Ngn3などの発現は低下していた。結果を図20、21に示す。
また、DNAマイクロアレイを用いてiPS細胞とGR細胞の網羅的な遺伝子発現の比較を行ったところ、マトリックスメタロプロテアーゼやケモカイン関連の遺伝子の発現が上昇し、細胞外マトリックスであるケラチンや細胞接着因子のクローディンなどの発現が低下していることが分かった。この結果から、GR細胞では移動期の始原生殖細胞のように細胞の移動活性が高まっていることが示唆された (図22)。
また、生殖細胞マーカー遺伝子のタンパク質発現をウエスタンブロットによって解析した。結果を図23に示す。GR細胞はOct4-GFP陽性であり、RT-PCRによって内在性Oct4遺伝子の転写も確認されていたにも関わらず、タンパク質レベルでは抑制を受けていることが分かった。また、GR細胞ではNanog、ECAT1、Mvh、Dnmt3Lのタンパク質発現が上昇していた。
続いて、Bisulphite genomic sequencingによってDNAのメチル化状態を解析した。結果を図24に示す。EK細胞では雌性インプリント遺伝子であるIgf2r、SNRPN、Lit1のメチル化制御領域が脱メチル化状態であり、雄性インプリント遺伝子であるH19は高メチル化状態であることから、雄型のインプリント状態を獲得していることが分かった。一方、GR細胞では雌性インプリント遺伝子のメチル化制御領域は体細胞パターンを示しており、胎仔生殖原基において誘導されるゲノムインプリントの消去が未だ行われていないことが分かった。以上の遺伝子発現とDNAのメチル化状態を総合的に解釈すると、GR細胞は移動後期(胎仔生殖原基に進入直前、または直後)の始原生殖細胞の特性を反映した細胞株であることが推測された。
GR細胞の精子形成能の有無を調べるために、Biol. Reprod, 69, 612-616 (2003) に記載されている方法に従って、c-kit遺伝子の変異によって生殖細胞を欠損した不妊モデルマウスであるW/Wvマウスの精巣へのGR細胞の移植を行った(京都大学医学部・篠原隆司教授の協力による)。具体的には、GR細胞を3×107個/mLとなるようにDMEM/10% FCS中に懸濁し、W/Wvマウス新生仔の精細管内に注射した。4カ月後に移植個体から精巣を摘出し、4% フォルムアルデヒドを含有するPBS(-)で固定した。パラフィン包埋組織をスライスし、ヘマトキシリン・エオシンで染色した。結果を図25に示す。細胞移植を行っていないW/Wvマウス精巣は、セルトリオンリーと呼ばれる生殖細胞を欠損した組織像を示すが、GR細胞を移植した場合には移植細胞の生着が認められた。生着した移植細胞はテラトーマ形成を示さず、また明瞭な腫瘍化も観察されなかった。
非生殖細胞を移植した場合は、精巣内環境に適応できずに死滅、あるいは排除されてしまう。一方、未分化なiPS細胞や腫瘍細胞を移植した場合には腫瘍が形成されてしまう。前記のようにGR細胞の移植においては、4ヶ月という長期に渡って細胞が精巣内に維持されており、かつ腫瘍化が認められなかったことから、GR細胞が生殖系細胞として運命決定された細胞であることが示された。しかしながら正常な精子形成像とも異なっており、TUNEL染色の結果、本来減数分裂後の成熟した精子細胞が存在する管腔において、アポトーシスによる細胞死が誘導されていることが明らかとなった。
【0141】
実施例4 ES細胞からOct4-GFP陽性/Mvh-RFP陽性細胞への分化誘導
本培養条件を用いて、これまでにiPS細胞から計5株のGR細胞を独立した実験から樹立することに成功している。また、本条件はiPS細胞に対してだけではなくES細胞にも適用可能であり、ES細胞を用いて同様に分化誘導を行った場合においてもOct4-GFP陽性/Mvh-RFP陽性細胞を樹立することが出来た(図26)。
【産業上の利用可能性】
【0142】
本発明のGR細胞の分化誘導および維持増幅方法によれば、皮膚細胞などの体細胞から容易に作製できるiPS細胞から精子形成可能な生殖系幹細胞を誘導し、維持増幅することができるので、精子幹細胞の採取が困難および/または危険を伴う非閉塞性無精子症の患者の不妊治療や遺伝子治療に特に有用である。また、育種分野における優良動物品種の保存の目的においても、本発明により作製されたGR細胞は極めて有用である。
【技術分野】
【0001】
本発明は、多能性幹細胞から始原生殖細胞(PGC)様細胞を分化誘導する方法および該PGC様細胞を維持増幅する方法、並びにそれらの方法に用いられる培地に関する。
【背景技術】
【0002】
抗がん療法に使用される抗がん剤あるいは放射線は、がん細胞を攻撃するだけでなく、細胞活動の活発な細胞や組織にも重篤な副作用をもたらすことが知られている。抗がん剤の多くは、がん細胞が正常細胞に比べて増殖活性が高いことを利用した選択毒性により治療効果を発揮する。そのため、消化管粘膜、毛根細胞、骨髄細胞、精原細胞等の増殖力が旺盛な正常細胞をも攻撃してしまう。例えば、アドリアマイシンは精原細胞の壊死を特徴とする精巣障害を引き起こすことが知られている。
遺伝子組換えで抗がん剤による副作用を緩和する医薬品が次々と開発された。例えば、顆粒球コロニー刺激因子(G-CSF)は骨髄抑制の副作用を防ぐ画期的な医薬であり、より積極的ながん化学療法を可能とした。しかしながら,抗がん剤あるいは放射線療法の副作用に基づく精巣障害を予防・治療するための有効な薬剤は未だ見出されていない。
【0003】
一方、不妊症の3割は男性側に原因がある(即ち精液中に精子がない無精子症や、あっても十分量がない乏精子症による)といわれている。日本産科婦人科学会は、夫婦間の不妊治療の場合に限り他人から精子をもらう非配偶者間人工授精を認めているが、未婚女性はもとより夫婦間の不妊治療であっても第三者からの精子提供について倫理上の問題性を指摘する声も依然として高い。
【0004】
抗がん療法の副作用に起因する男性不妊症の解決策の1つとして、治療前に精子を採取し凍結保存しておく方法があるが、精子保存ができない小児がん患者には有効でない。Brinsterらは、1994年にin vivoで精子幹細胞を移植することに成功した(非特許文献1)。即ち、幹細胞を含むドナーマウスの精巣細胞を不妊マウスの精細管内に移植すると、ドナー細胞由来の精子形成を起こし、メスとの交配によりドナー由来の仔をつくることができた。さらに、篠原らは、グリア細胞由来神経栄養因子(GDNF)、白血病抑制因子(LIF)、上皮細胞成長因子(EGF)および塩基性線維芽細胞成長因子(bFGF)を含む培地を用いることにより、精子幹細胞のin vitro長期培養法を確立し、当該細胞株をGermline Stem(GS)細胞と名づけた(特許文献1、非特許文献2)。このGS細胞を精巣に移植すると精子形成が起こり、子孫を作ることができた。これにより、治療前に生検により採取した精巣組織からGS細胞を樹立して保存しておけば、治療後に該GS細胞を精巣に自家移植することで、がん治療の副作用による不妊を回避することが可能となった。この方法は精子保存ができない小児がん患者にも有効である。
しかしながら、既に精巣障害を発症し精子形成が不可能な患者については、GS細胞を誘導するための精子幹細胞を十分に採取できない可能性があり、多数箇所生検が必要となる場合があるが、大量の精巣組織標本の採取は精巣萎縮をもたらすリスクがある。
【0005】
近年、マウスおよびヒトの人工多能性幹細胞(iPS細胞)が相次いで樹立された(特許文献2および3、非特許文献3〜6)。ヒトiPS細胞は、治療対象となる患者由来の細胞を用いて作製された後、各組織の細胞へと分化させることができるため、再生医学の領域において、拒絶反応のない移植材料として期待されている。c-Myc遺伝子を除いた3因子によるiPS細胞の作製(非特許文献3)や、プラスミドやエピソーマルベクターを用いて初期化因子がゲノムに組み込まれることなくiPS細胞を誘導できるようになったことで(非特許文献7、8)、ヒトiPS細胞の臨床応用への期待がより高まりつつある。iPS細胞は、例えば皮膚線維芽細胞などから容易に樹立することができるので、ヒトiPS細胞から精子もしくはその前駆細胞を効率よく分化誘導し、増幅することができれば、精巣の多数箇所生検といった危険を伴う細胞ソースの採取を回避することができ、より安全に男性不妊症患者からの精子形成の実現が期待される。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開WO 2004/092357号パンフレット
【特許文献2】国際公開WO 2007/069666号パンフレット
【特許文献3】国際公開WO 2008/118820号パンフレット
【非特許文献】
【0007】
【非特許文献1】Brinster, R.L. et al., Proc. Natl. Acad. Sci. USA, 91: 11298-11302 (1994)
【非特許文献2】Kanatsu-Shinohara, M. et al., Biol. Reprod., 69: 612-616 (2003)
【非特許文献3】Takahashi, K. and Yamanaka, S., Cell, 126: 663-676 (2006)
【非特許文献4】Nakagawa, M. et al., Nat. Biotethnol., 26: 101-106 (2008)
【非特許文献5】Takahashi, K. et al., Cell, 131: 861-872 (2007)
【非特許文献6】Yu, J. et al., Science, 318: 1917-1920 (2007)
【非特許文献7】Okita, K. et al., Science, 322: 949-953 (2008)
【非特許文献8】Yu, J. et al., Science, 324: 797-801 (2009)
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の目的は、iPS細胞などの多能性幹細胞から、自己再生能および精子形成能を有する生殖系幹細胞を分化誘導し、該生殖系幹細胞を維持増幅する方法、並びにそのための培地を提供することである。
【課題を解決するための手段】
【0009】
本発明者らは、未分化細胞特異的なOct3/4(Oct4)遺伝子と、生殖細胞系譜特異的なVasa遺伝子のマウスホモログ(Mouse vasa homolog; 以下、Mvhという。)の各発現制御領域の下流にレポーター遺伝子(GFPおよびRFP)を挿入することにより、未分化細胞と生殖細胞がそれぞれ可視化されたトランスジェニック(Tg)マウスを作製し、該マウスより誘導したiPS細胞およびES細胞を実験系として用い、Oct4-GFP陽性かつMvh-RFP陽性(Oct4+/Mvh+)の細胞を効率よく誘導し得る培養条件、並びに当該細胞をOct4+/Mvh+の状態で維持増幅させ得る培養条件を確立すべく鋭意検討を重ねた。その結果、多能性幹細胞をBMP4、GDNF、EGFおよび幹細胞因子(SCF)の存在下で培養することにより、効率よくOct4+/Mvh+細胞の分化を誘導し得ること、並びにGDNF、EGF、bFGFおよびSCFの存在下、好ましくは肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)からなる群より選択される1以上の因子がさらに存在する条件下で、Oct4+/Mvh+細胞を培養することにより、ダブルポジティブの状態を維持したまま、当該細胞を増幅し得ることを見出した。得られたOct4+/Mvh+細胞(GR細胞と名づけた)の特性解析の結果、当該細胞は移動後期の始原生殖細胞(PGC)の特性を反映した細胞株であることが判明した。さらに、本発明者らは、このGR細胞を不妊マウスの精管内に移植した結果、長期にわたって生着し、かつ腫瘍形成が認められないことを見出し、当該細胞が生殖系細胞として運命決定された単能性幹細胞であることを確認して、本発明を完成させるに至った。
【0010】
即ち、本発明は以下に関する。
(1)(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞の製造方法。
(2)BMP4、GDNF、EGFおよびSCFの存在下で多能性幹細胞を培養することを含む、上記(1)記載の方法。
(3)フィーダー細胞の存在下で多能性幹細胞を培養することを特徴とする、上記(1)または(2)記載の方法。
(4)前記成長因子がフィーダー細胞から提供されることを特徴とする、上記(3)記載の方法。
(5)多能性幹細胞がiPS細胞またはES細胞である、上記(1)〜(4)のいずれかに記載に方法。
(6)多能性幹細胞がヒトまたはマウス由来である、上記(1)〜(5)のいずれかに記載の方法。
(7)上記(1)〜(6)のいずれかに記載の方法により得られた、iPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞。
(8)(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
(9)BMP4、GDNF、EGFおよびSCFを組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
(10)前記成長因子を産生する細胞を含有してなる、上記(8)または(9)記載の剤。
(11)上記(8)または(9)記載の剤が添加されてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導用培地。
(11b)基本培地および表1から選ばれる1以上の成分を含有する培地と、上記(8)〜(10)のいずれかに記載の剤とを含んでなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導キット。
(12)Oct4陽性かつVasa陽性の生殖系幹細胞の増幅方法であって、GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、該生殖系幹細胞を培養することを含む、方法。
(13)肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)から選ばされる1以上の因子がさらに存在する条件下で、前記生殖系幹細胞を培養することを含む、上記(12)記載の方法。
(14)前記生殖系幹細胞をフィーダー細胞の存在下で培養することを特徴とする、上記(12)または(13)記載の方法。
(15)前記生殖系幹細胞が多能性幹細胞から分化誘導されたものである、上記(12)〜(14)のいずれかに記載の方法。
(16)GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅支持剤。
(17)HGF、IL-2およびFGF9から選ばされる1以上の因子をさらに組み合わせてなる、上記(16)記載の剤。
(18)上記(16)または(17)記載の剤が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
(18b)基本培地および任意で、表1から選ばれる1以上の成分を含有する培地に、表2から選ばれる1以上の成分が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
(19)上記(1)〜(6)のいずれかに記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、上記(7)記載のOct4陽性かつVasa陽性の生殖系幹細胞、または上記(12)〜(15)のいずれかに記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を、該細胞と同種のレシピエント動物の精巣に移植することを含む、該不妊動物に精子を形成させる方法。
(20)生殖系幹細胞が不妊動物の体細胞から作製したiPS細胞由来である、上記(19)記載の方法。
(21)レシピエント動物がヒトまたはマウスである、上記(19)または(20)記載の方法。
(22)上記(1)〜(6)のいずれかに記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、上記(7)記載のOct4陽性かつVasa陽性の生殖系幹細胞、または上記(12)〜(15)のいずれかに記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を含有してなる、雄性不妊治療剤。
(23)精子幹細胞の採取が困難な個体を投与対象とする、上記(22)記載の剤。
【発明の効果】
【0011】
本発明によれば、皮膚細胞などの体細胞から容易に作製できるiPS細胞から精子形成可能な生殖系幹細胞を誘導し、維持増幅することができる。したがって、精子幹細胞の採取が困難および/または危険を伴う非閉塞性無精子症の患者についても、自己の遺伝情報を有する精子を形成させることが可能となる。
【図面の簡単な説明】
【0012】
【図1】Oct4-GFP/Mvh-RFP Tgマウス胎仔由来のMEFから樹立したiPS細胞の写真である。上図:Oct3/4, Sox2およびKlf4の3種の遺伝子導入により樹立したiPSクローン522A3。下図:Oct3/4, Sox2, Klf4およびNanogの4種の遺伝子導入により樹立したiPSクローン522B2。
【図2】樹立したiPSクローン522A1〜A4および522B1〜522B4についてGenomic PCR解析を行った結果を示す電気泳動の写真である。
【図3】樹立したiPSクローン522A1〜A4および522B1〜522B4における未分化マーカーの発現をRT-PCR解析により調べた結果を示す電気泳動の写真である。
【図4】樹立したiPSクローンを免疫不全マウスの皮下に注射して形成させたテラトーマを組織学的に解析した結果を示す写真である。
【図5】ES細胞およびiPS細胞(クローン522B2)を従来の分化誘導方法により形成させた胚様体(EB)の写真である。
【図6】左図:M15-BMP4細胞にGDNF、mSCFおよびEGF遺伝子を導入して得られたM15-4GF細胞の形態を示す写真である。右図:M15-4GFについてRT-PCR解析を行った結果を示す電気泳動の写真である。
【図7】上図:樹立したiPS細胞とM15-4GF細胞とを図27の培地(Supplement無し)中で浮遊培養して得られた細胞塊の写真である。下図:前記細胞塊をトリプシンおよびコラゲナーゼで解離し、図27の培地(Supplement有り)中、フィーダー細胞上で培養して得られたOct4-GFP陽性/Mvh-RFP陽性コロニーの写真である。
【図8】図27の培養条件下で安定に増殖するOct4-GFP陽性/Mvh-RFP陽性コロニーの写真である。
【図9】Oct4-GFP陽性/Mvh-RFP陽性コロニーが未分化細胞と生殖細胞に共通する細胞表面マーカーであるalkaline phosphatase活性を示した写真である。
【図10】上図:Oct4-GFP陽性/Mvh-RFP陽性コロニーをiPS細胞用の培養条件で培養した際の細胞の写真である。下図:Oct4-GFP陽性/Mvh-RFP陽性コロニーを生殖幹(GS)細胞用の培養条件で培養した際の細胞の写真である。
【図11】上図:未分化iPS細胞をiPS細胞用の培養条件で培養した際の細胞の写真である。下図:未分化iPS細胞を、分化誘導過程を経ずに直接図27の維持培養条件下で培養した際の細胞の写真である。
【図12】上図:Oct4-GFP陽性/Mvh-RFP陽性の生殖細胞様の細胞(GR細胞)の写真である。下図:Oct4-GFP陽性/Mvh-RFP陰性の未分化様の細胞(Gsp細胞)の写真である。
【図13】GR細胞とGsp細胞に対してGenomic PCR解析を行った結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図14】GR細胞とGsp細胞に対してサザンブロット解析を行った結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図15】iPS細胞およびGR細胞を培養し、2日毎に総細胞数を測定した結果を示すグラフである。
【図16】フィーダー細胞(MEF)上、またはフィーダーフリーでゲラチン、ラミニン、またはファイブロネクチンコーティング上でGR細胞を培養した結果を示す写真である。
【図17】GR細胞およびGsp細胞を免疫不全のnu/nuマウス皮下に移植し、移植部位において腫瘍を形成するか否かを調べた結果を示す写真である。コントロールとしてiPS細胞およびEK細胞についても同様の移植を行った。
【図18】GR細胞およびGsp細胞における導入外来遺伝子およびそれに対応する内在性遺伝子の発現をRT-PCR解析した結果を示す電気泳動の写真である。コントロールとしてiPS細胞、EK細胞およびFbx-iPS細胞(Cell, 126, 663-676 (2006))についても解析を行った。
【図19】GR細胞およびGsp細胞における導入外来遺伝子およびそれに対応する内在性遺伝子の発現を、リアルタイムPCRによって定量化した結果を示すグラフである。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図20】GR細胞およびGsp細胞における生殖細胞マーカー遺伝子の発現をRT-PCRで調べた結果を示す電気泳動の写真である。コントロールとしてiPS細胞、EK細胞および精巣についても解析を行った。
【図21】図20と同様に生殖細胞マーカー遺伝子の発現をRT-PCRで調べた結果を示す電気泳動の写真である。
【図22】DNAマイクロアレイを用いてiPS細胞とGR細胞の網羅的な遺伝子発現の比較を行った結果を示す図である。「Up」はiPS細胞に比べてGR細胞で発現上昇していた遺伝子を、「Down」はiPS細胞に比べてGR細胞で発現低下していた遺伝子を、それぞれ示す。
【図23】GR細胞およびGsp細胞における生殖細胞マーカー遺伝子のタンパク質発現をウエスタンブロットによって解析した結果を示す電気泳動の写真である。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図24】GR細胞およびGsp細胞におけるDNAのメチル化状態をBisulphite genomic sequencingによって解析した結果を示す図である。雌性インプリント遺伝子であるIgf2r、Lit1およびSNRPNと、雄性インプリント遺伝子であるH19について調べた。コントロールとしてiPS細胞およびEK細胞についても解析を行った。
【図25】W/Wvマウス精巣へGR細胞の移植を行い、得られた移植個体からの精巣を組織学的に解析した結果を示す写真である。「Empty」は移植細胞非生着部位の組織像を、「Engraftment」は移植細胞生着部位の組織像を示す。
【図26】上図:図27の培養条件で培養したOct4-GFP陰性/Mvh-RFP陽性のEK細胞の写真である。下図:図27の培養条件で培養した、ES細胞由来のOct4-GFP陽性/Mvh-RFP陽性GR細胞の写真である。
【図27】iPS細胞やES細胞からOct4-GFP陽性/Mvh-RFP陽性細胞を分化誘導(Supplement無し)および維持培養(Supplement有り)するのに用いた培地の成分表を示す図である。
【発明を実施するための形態】
【0013】
本発明は、多能性幹細胞から生殖系幹細胞を誘導する方法、並びに該生殖系幹細胞を維持増幅する方法を提供する。本発明により多能性幹細胞から分化誘導される生殖系幹細胞は、未分化細胞マーカーであるOct3/4(Oct4)遺伝子と、生殖細胞系譜特異的なVasa遺伝子とを、ともに発現している(Oct4陽性かつVasa陽性、Oct4+/Vasa+と略記する場合がある)ことを特徴とする。Vasa遺伝子はショウジョウバエの生殖細胞形成不能変異の解析から同定された、DEADボックスを有するATP依存性RNAヘリカーゼをコードする遺伝子で、マウス(Proc. Natl. Acad. Sci. USA, 91: 12258-62, 1994)、ラット(Rvh; Biochem. Biophys. Res. Commun., 207: 405-10, 1995)、ブタ(Mol. Reprod. Dev., 72: 320-8, 2005)、ヒト(Proc. Natl. Acad. Sci. USA, 97: 9585-90, 2000)を含む種々の哺乳動物でそのホモログがクローニングされており(チンパンジー(XM_517757)やイヌ(XM_544339)でもアノテーション解析によりホモログの存在が推定されている。)、いずれも生殖細胞で特異的に発現することが知られている。哺乳動物vasaホモログはDEAD (Asp-Glu-Ala-Asp) box polypeptide 4(DDX4)遺伝子とも呼ばれる。本明細書では、ショウジョウバエvasa遺伝子のホモログをVasa遺伝子と総称し、特定のホモログを、例えばマウスの場合、mouse vasa homolog(Mvh)というように表記することとする。
後述の実施例における種々の特性解析の結果から示されるように、本発明の生殖系幹細胞(GR細胞ともいう)は、移動後期(胎仔生殖原基に進入する直前もしくは直後)の始原生殖細胞(PGC)の特性を反映したPGC様細胞である。該生殖系幹細胞は、本発明の維持培養条件下で維持増幅可能な(即ち、自己再生能を有する)幹細胞であり、しかも精巣に移植した場合に腫瘍を形成せず、かつ排除されることなく長期生着することから生殖系細胞として運命決定されている。
【0014】
I. 多能性幹細胞
本発明において出発材料となる多能性幹細胞は、未分化状態を保持したまま増殖できる「自己再生能」と三胚葉系列すべてに分化できる「分化多能性」とを有する未分化細胞であれば特に制限されず、例えば、iPS細胞、ES細胞の他、始原生殖細胞に由来する胚性生殖(EG)細胞、精巣組織からのGS細胞の樹立培養過程で単離されるmutipotent germline stem(mGS)細胞、骨髄から単離されるmultipotent adult progenitor cell(MAPC)等が挙げられる。ES細胞は体細胞から核初期化されて生じたES細胞であってもよい。好ましくはiPS細胞またES細胞であるが、出生後の個体から取得できる点でmGS細胞やMAPCもまた好ましい。本発明の方法は、いずれかの多能性幹細胞が樹立されているか、樹立可能である、任意の哺乳動物において適用することができ、例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等が挙げられるが、好ましくはヒトまたはマウスである。
【0015】
II. 多能性幹細胞の製造方法
本発明における多能性幹細胞として好適なiPS細胞の製造例を以下に示すが、これらに限定されない。
(A) 体細胞ソース
iPS細胞作製のための出発材料として用いることのできる体細胞は、哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット等)由来の生殖細胞以外のいかなる細胞であってもよく、例えば、角質化する上皮細胞(例、角質化表皮細胞)、粘膜上皮細胞(例、舌表層の上皮細胞)、外分泌腺上皮細胞(例、乳腺細胞)、ホルモン分泌細胞(例、副腎髄質細胞)、代謝・貯蔵用の細胞(例、肝細胞)、境界面を構成する内腔上皮細胞(例、I型肺胞細胞)、内鎖管の内腔上皮細胞(例、血管内皮細胞)、運搬能をもつ繊毛のある細胞(例、気道上皮細胞)、細胞外マトリックス分泌用細胞(例、線維芽細胞)、収縮性細胞(例、平滑筋細胞)、血液と免疫系の細胞(例、Tリンパ球)、感覚に関する細胞(例、桿細胞)、自律神経系ニューロン(例、コリン作動性ニューロン)、感覚器と末梢ニューロンの支持細胞(例、随伴細胞)、中枢神経系の神経細胞とグリア細胞(例、星状グリア細胞)、色素細胞(例、網膜色素上皮細胞)、およびそれらの前駆細胞(組織前駆細胞)等が挙げられる。細胞の分化の程度や細胞を採取する動物の齢などに特に制限はなく、未分化な前駆細胞(体性幹細胞も含む)であっても、最終分化した成熟細胞であっても、同様に本発明における体細胞の起源として使用することができる。ここで未分化な前駆細胞としては、たとえば神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)が挙げられる。
【0016】
体細胞を採取するソースとなる哺乳動物個体は特に制限されないが、目的の生殖系幹細胞(GR細胞)が不妊治療用途に使用される場合には、患者本人から体細胞を採取することが望ましい。一方、GR細胞が生殖細胞に対する遺伝子治療などを目的とする場合には、拒絶反応が起こらないという観点から、患者本人またはHLAの型が同一もしくは実質的に同一である他人から体細胞を採取することが好ましい。ここでHLAの型が「実質的に同一」とは、免疫抑制剤などの使用により、該体細胞由来のiPS細胞から分化誘導することにより得られた細胞を患者に移植した場合に移植細胞が生着可能な程度にHLAの型が一致していることをいう。たとえば主たるHLA(例えばHLA-A、HLA-BおよびHLA-DRの3遺伝子座や、さらにHLA-Cwを含む4遺伝子座)が同一である場合などが挙げられる(以下同じ)。また、GR細胞をヒトに投与(移植)しない場合、例えば、患者の精巣における薬剤感受性や副作用の有無を評価するためのスクリーニング用の細胞のソースとしてGR細胞を使用する場合には、同様に患者本人または薬剤感受性や副作用と相関する遺伝子多型が同一である他人から体細胞を採取することが望ましい。
【0017】
哺乳動物から分離した体細胞は、核初期化工程に供するに先立って、細胞の種類に応じてその培養に適した自体公知の培地で前培養することができる。そのような培地としては、例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地などが挙げられるが、それらに限定されない。核初期化物質(さらに必要に応じて、iPS細胞の樹立効率改善物質)との接触に際し、例えば、カチオニックリポソームなど導入試薬を用いる場合には、導入効率の低下を防ぐため、無血清培地に交換しておくことが好ましい場合がある。
【0018】
(B) 核初期化物質
本発明において「核初期化物質」とは、体細胞からiPS細胞を誘導することができるタンパク性因子(群)またはそれをコードする核酸(ベクターに組み込まれた形態を含む)でありうる。本発明に用いられる核初期化物質は、WO 2007/069666に記載の遺伝子であってもよい。より詳細には、Oct3/4, Klf4, Klf1, Klf2, Klf5, Sox2, Sox1, Sox3, Sox15, Sox17, Sox18, c-Myc, L-Myc, N-Myc, TERT, SV40 Large T antigen, HPV16 E6, HPV16 E7, Bmil, Lin28, Lin28b, Nanog, EsrrbまたはEsrrgが例示される。これらの初期化物質は、iPS細胞樹立の際には、組み合わされて使用されてもよく、上記初期化物質を、少なくとも1つ、2つもしくは3つ含む組み合わせであり、好ましくは4つを含む組み合わせである。具体的には、以下の組み合わせが例示される(以下においては、タンパク性因子の名称のみを記載する)。
(1) Oct3/4, Klf4, Sox2, c-Myc(ここで、Sox2はSox1, Sox3, Sox15, Sox17またはSox18で置換可能である。Klf4はKlf1, Klf2またはKlf5で置換可能である。また、c-MycはL-MycまたはN-Mycで置換可能である。)
(2) Oct3/4, Klf4, Sox2, c-Myc, TERT, SV40 Large T antigen(以下、SV40LT)
(3) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E6
(4) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E7
(5) Oct3/4, Klf4, Sox2, c-Myc, TERT, HPV16 E6, HPV16 E7
(6) Oct3/4, Klf4, Sox2, c-Myc, TERT, Bmi1
(7) Oct3/4, Klf4, Sox2, c-Myc, Lin28
(8) Oct3/4, Klf4, Sox2, c-Myc, Lin28, SV40LT
(9) Oct3/4, Klf4, Sox2, c-Myc, Lin28, TERT, SV40LT
(10) Oct3/4, Klf4, Sox2, c-Myc, SV40LT
(11) Oct3/4, Esrrb, Sox2, c-Myc (EsrrbはEsrrgで置換可能である。)
(12) Oct3/4, Klf4, Sox2
(13) Oct3/4, Klf4, Sox2, TERT, SV40LT
(14) Oct3/4, Klf4, Sox2, TERT, HPV16 E6
(15) Oct3/4, Klf4, Sox2, TERT, HPV16 E7
(16) Oct3/4, Klf4, Sox2, TERT, HPV16 E6, HPV16 E7
(17) Oct3/4, Klf4, Sox2, TERT, Bmi1
(18) Oct3/4, Klf4, Sox2, Lin28
(19) Oct3/4, Klf4, Sox2, Lin28, SV40LT
(20) Oct3/4, Klf4, Sox2, Lin28, TERT, SV40LT
(21) Oct3/4, Klf4, Sox2, SV40LT
(22) Oct3/4, Esrrb, Sox2 (EsrrbはEsrrgで置換可能である。)
上記において、c-Mycに代えてL-Mycを、Lin28に代えてLin28bを用いることもできる。
【0019】
また、上記(1)-(22)には該当しないが、それらのいずれかにおける構成要素をすべて含み、且つ任意の他の物質をさらに含む組み合わせも、本発明における「核初期化物質」の範疇に含まれ得る。また、核初期化の対象となる体細胞が上記(1)-(22)のいずれかにおける構成要素の一部を、核初期化のために十分なレベルで内在的に発現している条件下にあっては、当該構成要素を除いた残りの構成要素のみの組み合わせもまた、本発明における「核初期化物質」の範疇に含まれ得る。
【0020】
これらの組み合わせの中で、Oct3/4, Sox2, Klf4およびc-Myc(もしくはL-Myc)の4因子並びにOct3/4, Sox2, およびKlf4の3因子が、好ましい核初期化物質の例として挙げられる。これらの組み合わせにLin28(もしくはLin28b)を加えた5因子または4因子、さらにSV40 Large T antigenを加えた6因子または5因子も好ましい。
【0021】
上記の各核初期化物質のマウスおよびヒトcDNA配列情報は、WO 2007/069666に記載のNCBI accession numbersを参照することにより取得することができ(Nanogは当該公報中では「ECAT4」との名称で記載されている。尚、Lin28、Lin28b、Esrrb、Esrrg、L-MycのマウスおよびヒトcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得できる。)、当業者は容易にこれらのcDNAを単離することができる。
遺伝子名 マウス ヒト
Lin28 NM_145833 NM_024674
Lin28b NM_001031772 NM_001004317
Esrrb NM_011934 NM_004452
Esrrg NM_011935 NM_001438
L-Myc NM_008506 NM_001033081
核初期化物質としてタンパク性因子自体を用いる場合には、得られたcDNAを適当な発現ベクターに挿入して宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク性因子を回収することにより調製することができる。一方、核初期化物質としてタンパク性因子をコードする核酸を用いる場合、得られたcDNAを、ウイルスベクター、プラスミドベクター、エピソーマルベクター等に挿入して発現ベクターを構築し、核初期化工程に供される。
【0022】
(C) 核初期化物質の体細胞への導入方法
核初期化物質の体細胞への導入は、該物質がタンパク性因子である場合、自体公知の細胞へのタンパク質導入方法を用いて実施することができる。そのような方法としては、例えば、タンパク質導入試薬を用いる方法、タンパク質導入ドメイン(PTD)もしくは細胞透過性ペプチド(CPP)融合タンパク質を用いる方法、マイクロインジェクション法などが挙げられる。タンパク質導入試薬としては、カチオン性脂質をベースとしたBioPOTER Protein Delivery Reagent(Gene Therapy Systmes)、Pro-JectTM Protein Transfection Reagent(PIERCE)及びProVectin(IMGENEX)、脂質をベースとしたProfect-1(Targeting Systems)、膜透過性ペプチドをベースとしたPenetrain Peptide(Q biogene)及びChariot Kit(Active Motif)、HVJエンベロープ(不活化センダイウイルス)を利用したGenomONE(石原産業)等が市販されている。導入はこれらの試薬に添付のプロトコルに従って行うことができるが、一般的な手順は以下の通りである。核初期化物質を適当な溶媒(例えば、PBS、HEPES等の緩衝液)に希釈し、導入試薬を加えて室温で5-15分程度インキュベートして複合体を形成させ、これを無血清培地に交換した細胞に添加して37℃で1ないし数時間インキュベートする。その後培地を除去して血清含有培地に交換する。
PTDとしては、ショウジョウバエ由来のAntP、HIV由来のTAT (Frankel, A. et al, Cell 55, 1189-93 (1988); Green, M. & Loewenstein, P.M. Cell 55, 1179-88 (1988))、Penetratin (Derossi, D. et al, J. Biol. Chem. 269, 10444-50 (1994))、Buforin II (Park, C. B. et al. Proc. Natl Acad. Sci. USA 97, 8245-50 (2000))、Transportan (Pooga, M. et al. FASEB J. 12, 67-77 (1998))、MAP (model amphipathic peptide) (Oehlke, J. et al. Biochim. Biophys. Acta. 1414, 127-39 (1998))、K-FGF (Lin, Y. Z. et al. J. Biol. Chem. 270, 14255-14258 (1995))、Ku70 (Sawada, M. et al. Nature Cell Biol. 5, 352-7 (2003))、Prion (Lundberg, P. et al. Biochem. Biophys. Res. Commun. 299, 85-90 (2002))、pVEC (Elmquist, A. et al. Exp. Cell Res. 269, 237-44 (2001))、Pep-1 (Morris, M. C. et al. Nature Biotechnol. 19, 1173-6 (2001))、Pep-7 (Gao, C. et al. Bioorg. Med. Chem. 10, 4057-65 (2002))、SynBl (Rousselle, C. et al. MoI. Pharmacol. 57, 679-86 (2000))、HN-I (Hong, F. D. & Clayman, G L. Cancer Res. 60, 6551-6 (2000))、HSV由来のVP22等のタンパク質の細胞通過ドメインを用いたものが開発されている。PTD由来のCPPとしては、11R (Cell Stem Cell, 4:381-384(2009)) や9R (Cell Stem Cell, 4:472-476(2009))等のポリアルギニンが挙げられる。
【0023】
核初期化物質のcDNAとPTDもしくはCPP配列とを組み込んだ融合タンパク質発現ベクターを作製して組換え発現させ、融合タンパク質を回収して導入に用いる。導入は、タンパク質導入試薬を添加しない以外は上記と同様にして行うことができる。
【0024】
マイクロインジェクションは、先端径1μm程度のガラス針にタンパク質溶液を入れ、細胞に穿刺導入する方法であり、確実に細胞内にタンパク質を導入することができる。
【0025】
タンパク質導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下等)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。導入操作を繰り返し行う場合の間隔としては、例えば6〜48時間、好ましくは12〜24時間が挙げられる。
【0026】
iPS細胞の樹立効率を重視するのであれば、核初期化物質を、タンパク性因子自体としてではなく、それをコードする核酸の形態で用いることが好ましい。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、また、該核酸は二本鎖であっても、一本鎖であってもよい。好ましくは該核酸は二本鎖DNA、特にcDNAである。
核初期化物質のcDNAは、宿主となる体細胞で機能し得るプロモーターを含む適当な発現ベクターに挿入される。発現ベクターとしては、例えば、レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクター、動物細胞発現プラスミド(例、pA1-11,pXT1,pRc/CMV,pRc/RSV,pcDNAI/Neo)などが用いられ得る。
【0027】
用いるベクターの種類は、得られるiPS細胞から分化誘導されるGR細胞の用途に応じて適宜選択することができる。例えば、アデノウイルスベクター、プラスミドベクター、アデノ随伴ウイルスベクター、レトロウイルスベクター、レンチウイルスベクター、センダイウイルスベクター、エピソーマルベクターなどが使用され得る。
【0028】
発現ベクターにおいて使用されるプロモーターとしては、例えばEF1αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、EF1αプロモーター、CAGプロモーター、MoMuLV LTR、CMVプロモーター、SRαプロモーターなどが好ましい。
【0029】
発現ベクターは、プロモーターの他に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子、SV40複製起点などを含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子等が挙げられる。
【0030】
核初期化物質である核酸(初期化遺伝子)は、各々別個の発現ベクター上に組み込んでもよいし、1つの発現ベクターに2種類以上、好ましくは2〜3種類の遺伝子を組み込んでもよい。遺伝子導入効率の高いレトロウイルスやレンチウイルスベクターを用いる場合は前者が、プラスミド、アデノウイルス、エピソーマルベクターなどを用いる場合は後者を選択することが好ましい。さらに、2種類以上の遺伝子を組み込んだ発現ベクターと、1遺伝子のみを組み込んだ発現ベクターとを併用することもできる。
【0031】
上記において複数の初期化遺伝子(例えば、Oct3/4、Sox2、Klf4、c-Mycから選択される2つ以上、好ましくは2〜3遺伝子)を1つの発現ベクターに組み込む場合、これら複数の遺伝子は、好ましくはポリシストロニック発現を可能にする配列を介して発現ベクターに組み込むことができる。ポリシストロニック発現を可能にする配列を用いることにより、1種類の発現ベクターに組み込まれている複数の遺伝子をより効率的に発現させることが可能になる。ポリシストロニック発現を可能にする配列としては、例えば、口蹄疫ウイルスの2A配列(PLoS ONE3, e2532, 2008、Stem Cells 25, 1707, 2007)、IRES配列(U.S. Patent No. 4,937,190)など、好ましくは2A配列を用いることができる。
【0032】
初期化遺伝子を含む発現ベクターは、ベクターの種類に応じて、自体公知の手法により細胞に導入することができる。例えば、ウイルスベクターの場合、該核酸を含むプラスミドを適当なパッケージング細胞(例、Plat-E細胞)や相補細胞株(例、293細胞)に導入して、培養上清中に産生されるウイルスベクターを回収し、各ウイルスベクターに応じた適切な方法により、該ベクターを細胞に感染させる。例えば、ベクターとしてレトロウイルスベクターを用いる具体的手段が WO2007/69666、Cell, 126, 663-676 (2006) 及び Cell, 131, 861-872 (2007) に開示されており、ベクターとしてレンチウイルスベクターを用いる場合については、Science, 318, 1917-1920 (2007) に開示がある。iPS細胞から分化誘導されるGR細胞を不妊治療や生殖細胞の遺伝子治療等の医療用途に利用する場合、初期化遺伝子の発現(再活性化)は、iPS細胞由来のGR細胞から再生された精子や精巣組織における発癌リスクを高める可能性があるので、初期化遺伝子は細胞の染色体に組み込まれず、一過的に発現することが好ましい。かかる観点からは、染色体への組込みが稀なアデノウイルスベクターの使用が好ましい。アデノウイルスベクターを用いる具体的手段は、Science, 322, 945-949 (2008)に開示されている。また、アデノ随伴ウイルスも染色体への組込み頻度が低く、アデノウイルスベクターと比べて細胞毒性や炎症惹起作用が低いので、別の好ましいベクターとして挙げられる。センダイウイルスベクターは染色体外で安定に存在することができ、必要に応じてsiRNAにより分解除去することができるので、同様に好ましく利用され得る。センダイウイルスベクターについては、J. Biol. Chem., 282, 27383-27391 (2007) や特許第3602058号に記載のものを用いることができる。
【0033】
レトロウイルスベクターやレンチウイルスベクターを用いる場合は、いったん導入遺伝子のサイレンシングが起こったとしても、後に再活性化される可能性があるので、例えば、Cre/loxPシステムを用いて、不要となった時点で核初期化物質をコードする核酸を切り出す方法が好ましく用いられ得る。即ち、該核酸の両端にloxP配列を配置しておき、iPS細胞が誘導された後で、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞にCreリコンビナーゼを作用させ、loxP配列に挟まれた領域を切り出すことができる。また、LTR U3領域のエンハンサー−プロモーター配列は、挿入突然変異によって近傍の宿主遺伝子を上方制御する可能性があるので、当該配列を欠失、もしくはSV40などのポリアデニル化配列で置換した3’-自己不活性化(SIN)LTRを使用して、切り出されずゲノム中に残存するloxP配列より外側のLTRによる内因性遺伝子の発現制御を回避することがより好ましい。Cre-loxPシステムおよびSIN LTRを用いる具体的手段は、Chang et al., Stem Cells, 27: 1042-1049 (2009) に開示されている。
【0034】
一方、非ウイルスベクターであるプラスミドベクターの場合には、リポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。ベクターとしてプラスミドを用いる具体的手段は、例えばScience, 322, 949-953 (2008) 等に記載されている。
【0035】
プラスミドベクターやアデノウイルスベクター等を用いる場合、遺伝子導入は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができる。2種以上の発現ベクターを体細胞に導入する場合には、これらの全ての種類の発現ベクターを同時に体細胞に導入することが好ましいが、この場合においても、導入操作は1回以上の任意の回数(例えば、1回以上10回以下、又は1回以上5回以下など)行うことができ、好ましくは導入操作を2回以上(たとえば3回又は4回)繰り返して行うことができる。
【0036】
尚、アデノウイルスやプラスミドを用いる場合でも、導入遺伝子が染色体に組み込まれることがあるので、結局はサザンブロットやPCRにより染色体への遺伝子挿入がないことを確認する必要がある。そのため、上記Cre-loxPシステムのように、いったん染色体に導入遺伝子を組み込んだ後に、該遺伝子を除去する手段を用いることは好都合であり得る。別の好ましい一実施態様においては、トランスポゾンを用いて染色体に導入遺伝子を組み込んだ後に、プラスミドベクターもしくはアデノウイルスベクターを用いて細胞に転移酵素を作用させ、導入遺伝子を完全に染色体から除去する方法が用いられ得る。好ましいトランスポゾンとしては、例えば、鱗翅目昆虫由来のトランスポゾンであるpiggyBac等が挙げられる。piggyBacトランスポゾンを用いる具体的手段は、Kaji, K. et al., Nature, 458: 771-775 (2009)、Woltjen et al., Nature, 458: 766-770 (2009) に開示されている。
別の好ましい非組込み型ベクターとして、染色体外で自律複製可能なエピソーマルベクターが挙げられる。エピソーマルベクターを用いる具体的手段は、Yu et al., Science, 324, 797-801 (2009)に開示されている。
【0037】
本発明に用いられるエピソーマルベクターとしては、例えば、EBV、SV40等に由来する自律複製に必要な配列をベクター要素として含むベクターが挙げられる。自律複製に必要なベクター要素としては、具体的には、複製開始点と、複製開始点に結合して複製を制御するタンパク質をコードする遺伝子であり、例えば、EBVにあっては複製開始点oriPとEBNA-1遺伝子、SV40にあっては複製開始点oriとSV40 large T antigen遺伝子が挙げられる。
【0038】
また、エピソーマル発現ベクターは、初期化遺伝子の転写を制御するプロモーターを含む。該プロモーターとしては、前記と同様のプロモーターが用いられ得る。また、エピソーマル発現ベクターは、前記と同様に、所望によりエンハンサー、ポリA付加シグナル、選択マーカー遺伝子などをさらに含有していてもよい。選択マーカー遺伝子としては、例えば、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0039】
エピソーマルベクターは、例えばリポフェクション法、リポソーム法、エレクトロポレーション法、リン酸カルシウム共沈殿法、DEAEデキストラン法、マイクロインジェクション法、遺伝子銃法などを用いて該ベクターを細胞に導入することができる。具体的には、例えばScience, 324: 797-801 (2009)等に記載される方法を用いることができる。
【0040】
iPS細胞からエピソーマルベクターが除去されたか否かの確認は、該ベクターの一部をプローブまたはプライマーとして用い、iPS細胞から単離したエピソーム画分を鋳型としてサザンブロット分析またはPCR分析を行い、バンドの有無または検出バンドの長さを調べることにより実施することができる。エピソーム画分の調製は当該分野で周知の方法を用いて行えばよく、例えば、Science, 324: 797-801 (2009)等に記載される方法を用いることができる。
【0041】
(D) p53の機能阻害物質
本発明は、上記の核初期化物質に加えて、p53の機能阻害物質を接触させることがより好ましい。本明細書において「p53の機能阻害物質」とは、(a)p53タンパク質の機能もしくは(b)p53遺伝子の発現を阻害し得る限り、いかなる物質であってもよい。すなわち、p53タンパク質に直接作用してその機能を阻害する物質や、p53遺伝子に直接作用してその発現を阻害する物質のみならず、p53のシグナル伝達に関与する因子に作用することにより、結果的にp53タンパク質の機能やp53遺伝子の発現を阻害する物質も、本明細書における「p53の機能阻害物質」に含まれる。好ましくは、p53の機能阻害物質は、p53遺伝子の発現を阻害する物質であり、より好ましくはp53に対するsiRNAやshRNAをコードする発現ベクターである。
【0042】
p53タンパク質の機能を阻害する物質としては、例えば、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、抗p53アンタゴニスト抗体もしくはそれをコードする核酸、p53応答エレメントのコンセンサス配列を含むデコイ核酸、p53経路を阻害する物質などが挙げられるが、これらに限定されない。好ましくは、p53の化学的阻害物質、p53のドミナントネガティブ変異体もしくはそれをコードする核酸、p53経路阻害物質が挙げられる。
【0043】
(D1) p53の化学的阻害物質
p53の化学的阻害物質としては、例えば、WO 00/44364に開示されるpifithrin(PFT)-α及び-βに代表されるp53阻害剤、Stormら(Nat. Chem. Biol. 2, 474 (2006))に開示されるPFT-μ、それらの類縁体及びそれらの塩(例えば、塩酸酸、臭素酸塩等の酸付加塩など)等が挙げられるが、これらに限定されない。これらのうち、PFT-α及びその類縁体[2-(2-Imino-4,5,6,7-tetrahydrobenzothiazol-3-yl)-1-p-tolylethanone, HBr (製品名:Pifithrin-α)及び1-(4-Nitrophenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H)-benzothiazolyl)ethanone, HBr(製品名:Pifithrin-α, p-Nitro)]、PFT-β及びその類縁体[2-(4-Methylphenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole, HBr(製品名:Pifithrin-α, Cyclic)及び2-(4-Nitrophenyl)imidazo[2,1-b]-5,6,7,8-tetrahydrobenzothiazole(製品名:Pifithrin-α, p-Nitro, Cyclic)]、PFT-μ[Phenylacetylenylsulfonamide(製品名:Pifithrin-μ)]は、Merck社より市販されている。
【0044】
体細胞へのp53の化学的阻害物質の接触は、該阻害物質を適当な濃度で水性もしくは非水性溶媒に溶解し、ヒトまたはマウスより単離した体細胞の培養に適した培地(例えば、約5〜20%の胎仔ウシ血清を含む最小必須培地(MEM)、ダルベッコ改変イーグル培地(DMEM)、RPMI1640培地、199培地、F12培地など)中に、阻害物質濃度がp53の機能阻害に十分で且つ細胞毒性がみられない範囲となるように該阻害物質溶液を添加して、細胞を一定期間培養することにより実施することができる。阻害物質濃度は用いる阻害物質の種類によって異なるが、約0.1nM〜約100nMの範囲で適宜選択される。接触期間は細胞の核初期化が達成されるのに十分な時間であれば特に制限はないが、通常は陽性コロニーが出現するまで培地に共存させておけばよい。
【0045】
p53遺伝子は癌抑制遺伝子として知られており、p53の恒常的な機能阻害は発癌のリスクを高める可能性がある。p53の化学的阻害物質は、培地に添加するだけで細胞への導入が可能であるという利点に加えて、iPS細胞の誘導後に該阻害物質を含む培地を除去することにより、容易かつ迅速にp53の機能阻害を解除できる点でも有用である。
【0046】
(D2) p53のドミナントネガティブ変異体
p53のドミナントネガティブ変異体としては、体細胞に内在する野生型p53タンパク質と競合的に作用して、その機能を阻害し得る限り特に制限はないが、例えば、マウスp53のDNA結合領域に位置する275位(ヒトの場合は278位)のプロリンをセリンに点変異させたp53P275S(de Vries, A., Proc. Natl. Acad. Sci. USA, 99, 2948-2953 (2002))、マウスp53の14-301位(ヒトp53では11-304位に対応)のアミノ酸を欠失させたp53DD(Bowman, T., Genes Develop., 10, 826-835 (1996))などが挙げられる。その他にも、例えば、マウスp53の58位(ヒトの場合は61位)のセリンをアラニンに点変異させたp53S58A、ヒトp53の135位(マウスの場合は132位)のシステインをチロシンに点変異させたp53C135Y、マウスp53の135位(ヒトの場合は138位)のアラニンをバリンに点変異させたp53A135V、172位(ヒトの場合は175位)のアルギニンをヒスチジンに点変異させたp53R172H、270位(ヒトの場合は273位)のアルギニンをヒスチジンに点変異させたp53R270H、マウスp53の278位(ヒトの場合は281位)のアスパラギン酸をアスパラギンに点変異させたp53D278Nなどが知られており、同様に使用することができる。
【0047】
p53のドミナントネガティブ変異体は、例えば、以下の手法により得ることができる。まず、マウスまたはヒトのp53 cDNA配列情報に基づいて適当なオリゴヌクレオチドをプローブもしくはプライマーとして合成し、マウスまたはヒトの細胞・組織由来のmRNA、cDNAもしくはcDNAライブラリーから、ハイブリダイゼーション法や(RT-)PCR法を用いてマウスまたはヒトp53 cDNAをクローニングし、適当なプラスミドにサブクローニングする。変異を導入しようとする部位のコドン(例えば、p53P275Sの場合、275位のProをコードするコドンcct)を所望の他のアミノ酸をコードするコドン(例えば、p53P275Sの場合、Serをコードするコドンtct)に置換した形で、当該部位を含むプライマーを合成し、これを用いてp53 cDNAを挿入したプラスミドを鋳型とするインバースPCRを行うことにより、目的のドミナントネガティブ変異体をコードする核酸を取得する。p53DDのような欠失変異体の場合には、欠失させる部位の外側にプライマーを設計して、同様にインバースPCRを行えばよい。このようにして得られたドミナントネガティブ変異体をコードする核酸を宿主細胞に導入し、該細胞を培養して得られる培養物から組換えタンパク質を回収することにより、所望のドミナントネガティブ変異体を取得することができる。
【0048】
体細胞へのドミナントネガティブ変異体の接触は、上記タンパク性の核初期化物質の場合と同様に実施することができる。上述のように、p53の恒常的な機能阻害は発癌のリスクを高める可能性があるが、p53のドミナントネガティブ変異体は、導入された細胞内でプロテアーゼによる分解を受けて徐々に消失し、それに応じて細胞に内在するp53の機能が回復することから、該変異体タンパク質の使用は、得られるiPS細胞を治療用途で利用する場合のように、高度な安全性を要求される場合に好適であり得る。
【0049】
(D3) p53のドミナントネガティブ変異体をコードする核酸
本発明の別の好ましい実施態様において、p53機能阻害物質は、p53のドミナントネガティブ変異体をコードする核酸である。該核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよいが、好ましくはDNAである。また、該核酸は二本鎖であっても、一本鎖であってもよい。p53のドミナントネガティブ変異体をコードするcDNAは、該変異体タンパク質の作製について上記した手法によりクローニングすることができる。
単離されたcDNAは、前記核初期化物質である核酸(初期化遺伝子)の場合と同様に、適当な発現ベクターに挿入され、体細胞に導入され得る。
【0050】
(D4) p53経路阻害物質
ここでp53経路とは、p53を活性化し得るあらゆる上流のシグナルカスケードおよび活性化p53によって媒介されるあらゆる下流のシグナルカスケードを包含する意味で用いられる。したがって、p53経路阻害物質には、上記シグナル伝達経路のいずれかを阻害するいかなる物質も含まれるが、好ましい一実施態様においては、p53経路阻害物質はp53によりその転写が活性化されるp21の発現もしくは機能(Myc阻害活性)を阻害する物質であり、例えば、p21に対するsiRNA、shRNA、アンチセンス核酸、リボザイム等が挙げられる。p21の発現を阻害するこれらの核酸は、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムと同様の方法により設計・合成し、体細胞に導入することができる。当該核酸は、それらを発現するベクターの形態で提供されてもよく、該ベクターは、後記p53に対するsiRNA、shRNA、アンチセンス核酸、リボザイムを発現するベクターと同様の方法により構築し、体細胞に導入することができる。
【0051】
別の好ましい一実施態様においては、p53経路阻害物質はARF-MDM2-p53経路を阻害する物質であり、例えば、ARF-MDM2-p53経路阻害物質として、p53に直接結合してその核外輸送やユビキチン化を促進するMDM2もしくはそれをコードする核酸、p53へのMDM2の作用を阻害するp19ARFやATM(ataxia-telangiectasia mutated)の発現もしくは機能を阻害する物質(例えば、これらの因子に対するsiRNAやshRNA)等が挙げられる。
【0052】
(D5) その他の物質
p53タンパク質の機能を阻害するその他の物質として、例えば、抗p53アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p53アンタゴニスト抗体はポリクローナル抗体、モノクローナル抗体の何れであってもよい。抗体のアイソタイプは特に限定されないが、好ましくはIgG、IgMまたはIgA、特に好ましくはIgGが挙げられる。また、該抗体は、完全抗体分子の他、例えばFab、Fab'、F(ab’)2等のフラグメント、scFv、scFv-Fc、ミニボディー、ダイアボディー等の遺伝子工学的に作製されたコンジュゲート分子、あるいはポリエチレングリコール(PEG)等の蛋白質安定化作用を有する分子等で修飾されたそれらの誘導体などであってもよい。抗p53アンタゴニスト抗体は、p53またはその部分ペプチドを抗原として用い、自体公知の抗体または抗血清の製造法に従って製造することができる。また、公知の抗p53アンタゴニスト抗体として、例えば、PAb1801(Oncogene Science Ab-2)及びDO-1(Oncogene Science Ab-6)(Gire and Wynford-Thomas, Mol. Cell. Biol., 18, 1611-1621 (1998))等が挙げられる。抗p53アンタゴニスト抗体をコードする核酸は、抗p53モノクローナル抗体産生ハイブリドーマから常法により単離することができる。得られるH鎖及びL鎖遺伝子を連結して単鎖抗体をコードする核酸を作製することもできる。
【0053】
p53タンパク質の機能を阻害する別の物質として、抗p21アンタゴニスト抗体もしくはそれをコードする核酸が挙げられる。抗p21アンタゴニスト抗体及びそれをコードする核酸も、上記抗p53アンタゴニスト抗体及びそれをコードする核酸と同様にして作製することができる。
p53タンパク質の機能を阻害するさらに別の物質は、p53応答エレメントのコンセンサス配列(例、Pu-Pu-Pu-G-A/T-T/A-C-Py-Py-Py (Pu: プリン塩基, Py: ピリミジン塩基))を含むデコイ核酸である。このような核酸は上記塩基配列情報に基づいてDNA/RNA自動合成機で合成することができる。あるいはそのようなデコイ核酸は市販されている(例、p53 transcription factor decoy (GeneDetect.com))。
抗p53アンタゴニスト抗体及び抗p21アンタゴニスト抗体はp53のドミナントネガティブ変異体と同様に、また、該抗体をコードする核酸は該変異体をコードする核酸と同様にして、それぞれ細胞に導入することができる。また、上記デコイ核酸は、リポフェクション法などにより細胞に導入することができる。
【0054】
一方、p53遺伝子の発現を阻害する物質としては、例えば、p53に対するsiRNAもしくはshRNA、p53に対するsiRNAもしくはshRNAを発現するベクター、p53に対するアンチセンス核酸及びp53に対するリボザイム等が挙げられるが、好ましくはp53に対するsiRNA、shRNA及びsiRNA、shRNAを発現するベクターである。
【0055】
(D6) p53に対するsiRNA及びshRNA
p53に対するsiRNAは、マウスまたはヒトのp53 cDNA配列情報に基づいて、例えば、Elbashirら(Genes Dev., 15, 188-200 (2001))の提唱する規則に従って設計することができる。siRNAの標的配列としては、原則的にはAA+(N)19であるが、AA+(N)21もしくはNA+(N)21であってもよい。また、センス鎖の5’末端がAAである必要はない。標的配列の位置は特に制限されるわけではないが、5’-UTR及び開始コドンから約50塩基まで、並びに3’-UTR以外の領域から標的配列を選択することが望ましい。標的配列のGC含量も特に制限はないが、約30-約50%が好ましく、GC分布に偏りがなく繰り返しが少ない配列が望ましい。尚、下記(b2)のsiRNAもしくはshRNAを発現するベクターの設計において、プロモーターとしてpolIII系プロモーターを使用する場合、ポリメラーゼの転写が停止しないように、4塩基以上TまたはAが連続する配列は選択しないようにすべきである。
上述の規則に基づいて選択された標的配列の候補群について、標的以外のmRNAにおいて16-17塩基の連続した配列に相同性がないかどうかを、BLAST(http://www.ncbi.nlm.nih.gov/BLAST/)等のホモロジー検索ソフトを用いて調べ、選択した標的配列の特異性を確認する。特異性の確認された標的配列について、AA(もしくはNA)以降の19-21塩基にTTもしくはUUの3’末端オーバーハングを有するセンス鎖と、該19-21塩基に相補的な配列及びTTもしくはUUの3’末端オーバーハングを有するアンチセンス鎖とからなる2本鎖RNAをsiRNAとして設計する。また、shRNAは、ループ構造を形成しうる任意のリンカー配列(例えば、8-25塩基程度)を適宜選択し、上記センス鎖とアンチセンス鎖とを該リンカー配列を介して連結することにより設計することができる。
【0056】
siRNA及び/又はshRNAの配列は、種々のwebサイト上に無料で提供される検索ソフトを用いて検索が可能である。このようなサイトとしては、例えば、Ambionが提供するsiRNA Target Finder(http://www.ambion.com/jp/techlib/misc/siRNA_finder.html)及びpSilencerTM Expression Vector用 インサート デザインツール(http://www.ambion.com/jp/techlib/misc/psilencer_converter.html)、RNAi Codexが提供するGeneSeer(http://codex.cshl.edu/scripts/newsearchhairpin.cgi)がこれらに限定されず、QIAGEN、タカラバイオ、SiSearch、Dharmacon、Whitehead Institute、Invitrogen、Promega等のwebサイト上でも同様に検索が可能である。
【0057】
p53に対するsiRNAは、上記のようにして設計されたセンス鎖及びアンチセンス鎖オリゴヌクレオチドをDNA/RNA自動合成機でそれぞれ合成し、例えば、適当なアニーリング緩衝液中、約90〜約95℃で約1分程度変性させた後、約30〜約70℃で約1〜約8時間アニーリングさせることにより調製することができる。また、p53に対するshRNAは、上記のようにして設計されたshRNA配列を有するオリゴヌクレオチドをDNA/RNA自動合成機で合成し、上記と同様にしてセルフアニーリングさせることによって調製することができる。
【0058】
siRNA及びshRNAを構成するヌクレオチド分子は、天然型のRNAでもよいが、安定性(化学的および/または対酵素)や比活性(mRNAとの親和性)を向上させるために、種々の化学修飾を含むことができる。例えば、ヌクレアーゼなどの加水分解酵素による分解を防ぐために、アンチセンス核酸を構成する各ヌクレオチドのリン酸残基(ホスフェート)を、例えば、ホスホロチオエート(PS)、メチルホスホネート、ホスホロジチオネートなどの化学修飾リン酸残基に置換することができる。また、各ヌクレオチドの糖(リボース)の2'位の水酸基を、-OR(Rは、例えばCH3(2'-O-Me)、CH2CH2OCH3(2'-O-MOE)、CH2CH2NHC(NH)NH2、CH2CONHCH3、CH2CH2CN等を示す)に置換してもよい。さらに、塩基部分(ピリミジン、プリン)に化学修飾を施してもよく、例えば、ピリミジン塩基の5位へのメチル基やカチオン性官能基の導入、あるいは2位のカルボニル基のチオカルボニルへの置換などが挙げられる。
【0059】
RNAの糖部のコンフォーメーションはC2'-endo(S型)とC3'-endo(N型)の2つが支配的であり、一本鎖RNAではこの両者の平衡として存在するが、二本鎖を形成するとN型に固定される。したがって、標的RNAに対して強い結合能を付与するために、2'酸素と4’炭素を架橋することにより、糖部のコンフォーメーションをN型に固定したRNA誘導体であるBNA(LNA)(Imanishi, T. et al., Chem. Commun., 1653-9, 2002; Jepsen, J.S. et al., Oligonucleotides, 14, 130-46, 2004)やENA(Morita, K. et al., Nucleosides Nucleotides Nucleic Acids, 22, 1619-21, 2003)もまた、好ましく用いられ得る。
但し、天然型RNA中のすべてのリボヌクレオシド分子を修飾型で置換すると、RNAi活性が失われる場合があるので、RISC複合体が機能できる最小限の修飾ヌクレオシドの導入が必要である。
【0060】
p53に対するsiRNAは、例えば、Ambion(例、Ambion Cat# AM16708, siRNA ID# 69659, 69753, 69843, 187424, 187425, 187426)やSanta Cruz(例、Santa Cruz Cat# sc-29436, 44219)等から購入することもできる。
また、ヒトp53に対するsiRNAおよびshRNAも、上記のいずれかの検索ソフトを用いて、ヒトp53 cDNAの配列情報(例、Refseq. No. NM_000546)等をクエリーとして入力することにより設計し、合成することができ、あるいはAmbion等から購入することもできる。具体的には、Science, 296, 550-553 (2002) に記載されるp53に対するshRNAなどが例示される。
【0061】
p53に対するsiRNAもしくはshRNAの体細胞への接触は、プラスミドDNAの場合と同様に、リポソーム法、ポリアミン法、エレクトロポレーション法、ビーズ法等を用いて、該核酸を細胞内へ導入することにより実施することができる。カチオニックリポソームを用いた方法が最も一般的で、導入効率も高い。Lipofectamine2000やOligofectamine(Invitrogen)などの一般的な遺伝子導入試薬の他、例えば、GeneEraserTMsiRNA transfection reagent(Stratagene)等のsiRNA導入に適した導入試薬も市販されている。
【0062】
(D7) p53に対するsiRNAもしくはshRNAを発現するベクター
siRNAを発現するベクターには、タンデムタイプとステムループ(ヘアピン)タイプとがある。前者はsiRNAのセンス鎖の発現カセットとアンチセンス鎖の発現カセットをタンデムに連結したもので、細胞内で各鎖が発現してアニーリングすることにより2本鎖のsiRNA(dsRNA)を形成するというものである。一方、後者はshRNAの発現カセットをベクターに挿入したもので、細胞内でshRNAが発現しdicerによるプロセシングを受けてdsRNAを形成するというものである。プロモーターとしては、polII系プロモーター(例えば、CMV前初期プロモーター)を使用することもできるが、短いRNAの転写を正確に行わせるために、polIII系プロモーターを使用するのが一般的である。polIII系プロモーターとしては、マウスおよびヒトのU6-snRNAプロモーター、ヒトH1-RNase P RNAプロモーター、ヒトバリン-tRNAプロモーターなどが挙げられる。また、転写終結シグナルとして4個以上Tが連続した配列が用いられる。
【0063】
このようにして構築したsiRNAもしくはshRNA発現カセットを、次いでプラスミドベクターやエピソーマルベクター、ウイルスベクター等に挿入する。このようなベクターとしては、核初期化物質である核酸(初期化遺伝子)について上記したと同様のものが、好ましく利用され得る(レトロウイルス、レンチウイルス、アデノウイルス、アデノ随伴ウイルス、ヘルペスウイルス、センダイウイルスなどのウイルスベクターや、動物細胞発現プラスミド、エピソーマルベクターなど)。使用するベクターは、初期化遺伝子の場合と同様、得られるiPS細胞から分化誘導されるGR細胞の用途に応じて適宜選択され得る。上述の通り、p53の恒常的な機能阻害は発癌リスクを高める可能性があるので、GR細胞をヒトの医療用途に用いる場合、p53を一過的に発現し、iPS細胞樹立後は速やかに細胞から脱落し得るベクター(例えば、プラスミドベクターなど)を用いることが好ましい。あるいは、p53に対するshRNAをコードする発現ベクターとして、市販のプラスミド(例えば、Addgene社から市販されるpMKO.1-puro p53 shRNA2: #10672等)をもとに作製したレトロウイルス等のウイルスベクター、プラスミドベクター、エピソーマルベクターなどを使用することもできる。必要に応じて、上記Cre-loxPシステムやpiggyBacトランスポゾンシステムを利用することもできる。
【0064】
p53に対するsiRNAもしくはshRNAを発現するベクターの体細胞への接触は、上記のようにして調製されるプラスミドベクター、エピソーマルベクターもしくはウイルスベクターを細胞に導入することにより行われる。これらの遺伝子導入は、初期化遺伝子について上記したと同様の手法で行うことができる。
【0065】
(D8) その他の物質
p53遺伝子の発現を阻害する他の物質として、p53に対するアンチセンス核酸やリボザイムが挙げられる。
アンチセンス核酸はDNAであってもRNAであってもよく、あるいはDNA/RNAキメラであってもよい。アンチセンス核酸がDNAの場合、標的RNAとアンチセンスDNAとによって形成されるRNA:DNAハイブリッドは、内在性RNase Hに認識されて標的RNAの選択的な分解を引き起こすことができる。したがって、RNase Hによる分解を指向するアンチセンスDNAの場合、標的配列は、p53 mRNA中の配列だけでなく、p53遺伝子の初期転写産物におけるイントロン領域の配列であってもよい。アンチセンス核酸の標的領域は、該アンチセンス核酸がハイブリダイズすることにより、結果としてp53蛋白質への翻訳が阻害されるものであればその長さに特に制限はなく、p53 mRNAの全配列であっても部分配列であってもよく、短いもので約15塩基程度、長いものでmRNAもしくは初期転写産物の全配列が挙げられる。合成の容易さや抗原性、細胞内移行性の問題等を考慮すれば、約15〜約40塩基、特に約18〜約30塩基からなるオリゴヌクレオチドが好ましい。標的配列の位置としては、5’-及び3’-UTR、開始コドン近傍などが挙げられるが、それらに限定されない。
リボザイムとは、狭義には、核酸を切断する酵素活性を有するRNAをいうが、本明細書では配列特異的な核酸切断活性を有する限りDNAをも包含する概念として用いるものとする。リボザイムとして最も汎用性の高いものとしては、ウイロイドやウイルソイド等の感染性RNAに見られるセルフスプライシングRNAがあり、ハンマーヘッド型やヘアピン型等が知られている。ハンマーヘッド型は約40塩基程度で酵素活性を発揮し、ハンマーヘッド構造をとる部分に隣接する両端の数塩基ずつ(合わせて約10塩基程度)をmRNAの所望の切断部位と相補的な配列にすることにより、標的mRNAのみを特異的に切断することが可能である。
【0066】
アンチセンス核酸やリボザイムはDNA/RNA自動合成機を用いて合成することができる。これらを構成するヌクレオチド分子もまた、安定性、比活性などを向上させるために、上記のsiRNAの場合と同様の修飾を受けていてもよい。
あるいは、アンチセンス核酸やリボザイムは、siRNAの場合と同様に、それらをコードする核酸の形態で使用することもできる。
【0067】
p53の機能阻害物質は、体細胞の核初期化工程においてp53の機能を阻害するのに十分な様式で体細胞に接触させる必要がある。この条件が満たされる限り、核初期化物質とp53の機能阻害物質とは、同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよい。一実施態様において、例えば、核初期化物質がタンパク性因子をコードする核酸であり、p53の機能阻害物質が化学的阻害物質である場合には、前者は遺伝子導入処理からタンパク性因子を大量発現するまでに一定期間のラグがあるのに対し、後者は速やかにp53の機能を阻害しうることから、遺伝子導入処理から一定期間細胞を培養した後に、p53の化学的阻害物質を培地に添加することができる。別の実施態様において、例えば、核初期化物質とp53の機能阻害物質とがいずれもウイルスベクター、プラスミドベクター、エピソーマルベクター等の形態で用いられる場合には、両者を同時に細胞に導入してもよい。
【0068】
(E) 他のiPS細胞の樹立効率改善物質
上記の初期化因子やp53機能阻害物質に加え、公知の他のiPS細胞樹立効率改善物質を体細胞に接触させることにより、iPS細胞の樹立効率をさらに高めることが期待できる。そのようなiPS細胞の樹立効率改善物質としては、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA Smartpool(登録商標)(Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-calcium channel agonist (例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、Wnt Signaling(例えばsoluble Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、2i/LIF (2iはmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤、PloS Biology, 6(10), 2237-2247 (2008))、ES細胞特異的miRNA(例えば、miR-302-367クラスター (Mol. Cell. Biol. doi:10.1128/MCB.00398-08、WO2009/075119)、miR-302 (RNA (2008) 14: 1-10)、miR-291-3p, miR-294およびmiR-295 (以上、Nat. Biotechnol. 27: 459-461 (2009)))等が挙げられるが、それらに限定されない。前記において核酸性の発現阻害剤はsiRNAもしくはshRNAをコードするDNAを含む発現ベクターの形態であってもよい。
【0069】
尚、前記核初期化物質の構成要素のうち、例えば、SV40 Large T antigen等は、体細胞の核初期化のために必須ではなく補助的な因子であるという点において、iPS細胞の樹立効率改善物質の範疇にも含まれ得る。核初期化の機序が明らかでない現状においては、核初期化に必須の因子以外の補助的な因子について、それらを核初期化物質として位置づけるか、あるいはiPS細胞の樹立効率改善物質として位置づけるかは便宜的であってもよい。即ち、体細胞の核初期化プロセスは、体細胞への核初期化物質およびiPS細胞の樹立効率改善物質の接触によって生じる全体的事象として捉えられるので、当業者にとって両者を必ずしも明確に区別する必要性はないであろう。
【0070】
これら他のiPS細胞樹立効率改善物質の体細胞への接触は、該物質が(a) タンパク性因子である場合、(b) 該タンパク性因子をコードする核酸である場合、あるいは(c) 低分子化合物である場合に応じて、p53の機能阻害物質についてそれぞれ上記したと同様の方法により、実施することができる。
他のiPS細胞の樹立効率改善物質は、該物質の非存在下と比較して体細胞からのiPS細胞樹立効率が有意に改善される限り、核初期化物質と同時に体細胞に接触させてもよいし、また、どちらかを先に接触させてもよく、該物質の物性に応じて、p53の機能阻害物質について上記したと同様のタイミングで体細胞と接触させることができる。
【0071】
(F) 培養条件による樹立効率の改善
体細胞の核初期化工程において低酸素条件下で細胞を培養することにより、iPS細胞の樹立効率をさらに改善することができる。本明細書において「低酸素条件」とは、細胞を培養する際の雰囲気中の酸素濃度が、大気中のそれよりも有意に低いことを意味する。具体的には、通常の細胞培養で一般的に使用される5-10% CO2/95-90%大気の雰囲気中の酸素濃度よりも低い酸素濃度の条件が挙げられ、例えば雰囲気中の酸素濃度が18%以下の条件が該当する。好ましくは、雰囲気中の酸素濃度は15%以下(例、14%以下、13%以下、12%以下、11%以下など)、10%以下(例、9%以下、8%以下、7%以下、6%以下など)、または5%以下(例、4%以下、3%以下、2%以下など)である。また、雰囲気中の酸素濃度は、好ましくは0.1%以上(例、0.2%以上、0.3%以上、0.4%以上など)、0.5%以上(例、0.6%以上、0.7%以上、0.8%以上、0.95以上など)、または1%以上(例、1.1%以上、1.2%以上、1.3%以上、1.4%以上など)である。
【0072】
細胞の環境において低酸素状態を創出する手法は特に制限されないが、酸素濃度の調節可能なCO2インキュベーター内で細胞を培養する方法が最も容易であり、好適な例として挙げられる。酸素濃度の調節可能なCO2インキュベーターは、種々の機器メーカーから販売されている(例えば、Thermo scientific社、池本理化学工業、十慈フィールド、和研薬株式会社などのメーカー製の低酸素培養用CO2インキュベーターを用いることができる)。
【0073】
低酸素条件下で細胞培養を開始する時期は、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、体細胞への核初期化物質の接触より前であっても、該接触と同時であっても、該接触より後であってもよいが、例えば、体細胞に核初期化物質を接触させた直後から、あるいは接触後一定期間(例えば、1ないし10(例、2,3,4,5,6,7,8または9)日)おいた後に低酸素条件下で培養することが好ましい。
【0074】
低酸素条件下で細胞を培養する期間も、iPS細胞の樹立効率が正常酸素濃度(20%)の場合に比して改善されることを妨げない限り特に限定されず、例えば3日以上、5日以上、7日以上または10日以上で、50日以下、40日以下、35日以下または30日以下の期間等が挙げられるが、それらに限定されない。低酸素条件下での好ましい培養期間は、雰囲気中の酸素濃度によっても変動し、当業者は用いる酸素濃度に応じて適宜当該培養期間を調整することができる。また、一実施態様において、iPS細胞の候補コロニーの選択を、薬剤耐性を指標にして行う場合には、薬剤選択を開始する迄に低酸素条件から正常酸素濃度に戻すことが好ましい。
さらに、低酸素条件下で細胞培養を開始する好ましい時期および好ましい培養期間は、用いられる核初期化物質の種類、正常酸素濃度条件下でのiPS細胞樹立効率などによっても変動する。
【0075】
核初期化物質およびp53の機能阻害物質(さらに必要に応じて他のiPS細胞の樹立効率改善物質)を接触させた後、細胞を、例えばES細胞の培養に適した条件下で培養することができる。マウス細胞の場合、通常の培地に分化抑制因子としてLeukemia Inhibitory Factor(LIF)を添加して培養を行う。一方、ヒト細胞の場合には、LIFの代わりに塩基性線維芽細胞増殖因子(bFGF)および/または幹細胞因子(SCF)を添加することが望ましい。
また通常、細胞は、フィーダー細胞として、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞の共存下で培養される。マウス胎仔由来の線維芽細胞としては、通常STO細胞株(ATCC CRL-1503)等がフィーダーとしてよく使われるが、iPS細胞の誘導には、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))等がよく使われている。しかしながら、マウス胎仔由来の初代線維芽細胞(MEF)を用いた方がヒトiPS細胞の樹立効率がより改善される場合もあるので、MEFの使用もまた好ましい。マイトマイシンC処理済のMEFは、ミリポア社やリプロセル社から市販されている。これらのフィーダー細胞との共培養は、核初期化物質の接触より前から開始してもよいし、該接触時から、あるいは該接触より後(例えば1-10日後)から開始してもよい。
【0076】
(G) iPS細胞の選択・確認
iPS細胞の候補コロニーの選択は、薬剤耐性とレポーター活性を指標とする方法と目視による形態観察による方法とが挙げられる。前者としては、例えば、分化多能性細胞において特異的に高発現する遺伝子(例えば、Fbx15、Nanog、Oct3/4など、好ましくはNanog又はOct3/4)の遺伝子座に、薬剤耐性遺伝子及び/又はレポーター遺伝子をターゲッティングした組換え体細胞を用い、薬剤耐性及び/又はレポーター活性陽性のコロニーを選択するというものである。そのような組換え体細胞としては、例えばFbx15遺伝子座にβgeo(β-ガラクトシダーゼとネオマイシンホスホトランスフェラーゼとの融合タンパク質をコードする)遺伝子をノックインしたマウス由来のMEF(Takahashi & Yamanaka, Cell, 126, 663-676 (2006))、あるいはNanog遺伝子座に緑色蛍光タンパク質(GFP)遺伝子とピューロマイシン耐性遺伝子を組み込んだトランスジェニックマウス由来のMEF(Okita et al., Nature, 448, 313-317 (2007))等が挙げられる。本発明のGR細胞はOct3/4遺伝子を高発現する(Oct4陽性)ことを特徴とするので、レポーター活性を指標とする方法においては、Oct3/4遺伝子座にGFPやRFP等の可視化タンパク質をコードするレポーター遺伝子をノックインした細胞を用いることがより好ましい。一方、目視による形態観察で候補コロニーを選択する方法としては、例えばTakahashi et al., Cell, 131, 861-872 (2007)に記載の方法が挙げられる。レポーター細胞を用いる方法は簡便で効率的ではあるが、iPS細胞から分化誘導されるGR細胞がヒトの治療用途を目的として作製される場合、安全性の観点から目視によるコロニー選択が望ましい。
【0077】
選択されたコロニーの細胞がiPS細胞であることの確認は、上記したOct4(もしくはNanog)レポーター陽性(ピューロマイシン耐性、GFP陽性など)および目視によるES細胞様コロニーの形成によっても行い得るが、より正確を期すために、アルカリフォスファターゼ染色や、各種ES細胞特異的遺伝子の発現を解析したり、選択された細胞をマウスに移植してテラトーマ形成を確認する等の試験を実施することもできる。
【0078】
(H) 他の多能性幹細胞の製造方法
ES細胞の作製方法としては、例えば、哺乳動物の胚盤胞ステージにおける内部細胞塊を培養する方法(例えば、Manipulating the Mouse Embryo A Laboratory Manual,Second Edition,Cold Spring Harbor Laboratory Press (1994) を参照)、体細胞核移植によって作製された初期胚を培養する方法(Wilmut et al., Nature, 385, 810 (1997); Cibelli et al., Science, 280, 1256 (1998); 入谷明ら, 蛋白質核酸酵素, 44, 892 (1999); Baguisi et al., Nature Biotechnology,17, 456 (1999); Wakayama et al., Nature, 394, 369 (1998); Wakayama et al., Nature Genetics, 22, 127 (1999); Wakayama et al., Proc. Natl. Acad. Sci. USA, 96, 14984 (1999); RideoutIII et al., Nature Genetics, 24, 109 (2000))などが挙げられるが、これらに限定されない。また、ES細胞は、所定の機関より入手でき、さらには市販品を購入することもできる。例えば、ヒトES細胞であるKhES-1、KhES-2およびKhES-3は、京都大学再生医科学研究所より入手可能である。
体細胞核移植による場合、体細胞の種類や体細胞を採取するソースは上記iPS細胞の場合に準ずる。
EG細胞は、常法に従って始原生殖細胞を単離し、これをLIF、bFGFおよびSCFの存在下で培養することにより誘導することができる。また、mGS細胞はWO 2005/100548に記載される方法に従って、精巣細胞から作製することができる。多能性成体前駆細胞(MAPC)はJ. Clin. Invest. 109:337-346 (2002) に記載される方法に従って、骨髄から単離することができる。
【0079】
III. 多能性幹細胞からGR細胞への分化誘導
GR細胞の分化誘導用の基本培地としては、例えば、Neurobasal培地、Neural Progenitor Basal培地、NS-A培地、BME培地、BGJb培地、CMRL 1066培地、Glasgow MEM培地、Improved MEM Zinc Option培地、IMDM培地、Medium 199培地、Eagle MEM培地、αMEM培地、DMEM培地、DMEM/F12培地、ハム培地、RPMI 1640培地、Fischer’s培地、およびこれらの混合培地など、動物細胞の培養に用いることのできる培地であれば特に限定されない。より好ましくは、Neurobasal培地である。これらの培地は、血清(例えば、胎仔ウシ血清(FCS)、ヒト血清等)を含有してもしなくてもよい。あるいは、血清の代替添加物(例えばKnockout Serum Replacement (KSR)(Invitrogen社製) 等)を用いてもよい。血清の添加濃度としては0〜20%の範囲で適宜選択することができるが、好ましくは無血清もしくは低血清(例えば、0〜5%、好ましくは0〜2%)の培地が使用され得る。
【0080】
前記いずれかの培地に、例えば、血清タンパク質(例えば、ウシ血清アルブミン(BSA)、ヒト血清アルブミン(HSA)等のアルブミンなど)、還元剤(例えば、2-メルカプトエタノール等)、成長因子(例えば、インスリン、bFGF、HGF、FGF9等)、幹細胞の分化抑制剤(例えば、LIF、Wnt、TGF-β等)、鉄源(例えば、トランスフェリン等)、ミネラル(例えば、亜セレン酸ナトリウム)、アミノ酸(例えば、グルタミン、アラニン、アスパラギン、セリン、アスパラギン酸、システイン、グルタミン酸、グリシン、プロリン、チロシン等の非必須アミノ酸)、ビタミン類(例えば、塩化コリン、パントテン酸、葉酸、ニコチンアミド、塩酸ピリドキサル、リボフラビン、塩酸チアミン、アスコルビン酸、ビオチン、イノシトール等)、糖類(例えば、グルコース等)、有機アミン類(例えば、プトレシン等)、ステロイド(例えば、プロゲステロン、β-エストラジオール等)、抗生物質(例えば、ペニシリン、ストレプトマイシン等)、インターロイキン類(例えば、IL-1、IL-2、IL-3、IL-6等)、接着因子(例えば、ヘパリン、ヘパラン硫酸、コラーゲン、フィブロネクチン等)、有機酸(例えば、ピルビン酸、コハク酸、乳酸等)もしくはその塩、緩衝剤(例えば、HEPES等)、栄養添加物(例えば、B27 supplement、N2 supplement、StemPro supplement等)などの培地添加物を適宜補充して、使用することができる。
好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子が、上記いずれかの基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27の「Medium component」に記載された濃度(最終濃度)を選択することができる。
【0081】
【表1】
【0082】
本発明の多能性幹細胞からGR細胞への分化誘導方法は、(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを特徴とする。好ましくは、多能性幹細胞は、BMP4と、GDNF、EGFおよびSCFから選ばれる2以上の成長因子との3因子の存在下で培養され、より好ましくは、BMP4、GDNF、EGFおよびSCFの4因子の存在下で培養される。これらの成長因子は、上記のいずれかの培地に添加して分化誘導用培地としてもよいし、あるいは、当該因子を産生する細胞をフィーダー細胞として用い、多能性幹細胞と共培養することによって提供されてもよい。
【0083】
BMP4、GDNF、EGFおよびSCFは任意の哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等、好ましくはヒトまたはマウス)由来のものを用いることができるが、多能性幹細胞と同種由来のものを用いることが好ましい。特に、ヒトGR細胞をヒトの治療目的で使用する場合には、異種成分不含(xeno-free)条件下でヒトGR細胞を誘導することが望ましいので、これらの成長因子としてヒト由来タンパク質を用いることが好ましい。
【0084】
BMP4、GDNF、EGFまたはSCFを培地に添加して用いる場合、これらの成長因子は、それらを産生する哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等)の細胞[例えば、神経細胞、グリア細胞、肝細胞、脾細胞、膵β細胞、骨髄細胞、メサンギウム細胞、ランゲルハンス細胞、表皮細胞、上皮細胞、内皮細胞、平滑筋細胞、線維芽細胞、線維細胞、筋細胞、脂肪細胞、免疫細胞(例、マクロファージ、T細胞、B細胞、ナチュラルキラー細胞、肥満細胞、好中球、好塩基球、好酸球、単球)、巨核球、滑膜細胞、軟骨細胞、骨細胞、骨芽細胞、破骨細胞、乳腺細胞もしくは間質細胞、またはこれら細胞の前駆細胞、幹細胞もしくは癌細胞など]もしくはそれらの細胞が存在するあらゆる組織[例えば、脳、脳の各部位(例、嗅球、扁桃核、大脳基底球、海馬、視床、視床下部、大脳皮質、延髄、小脳)、脊髄、下垂体、胃、膵臓、腎臓、肝臓、生殖腺、甲状腺、胆のう、骨髄、副腎、皮膚、筋肉(例、平滑筋、骨格筋)、肺、消化管(例、大腸、小腸)、血管、心臓、胸腺、脾臓、顎下腺、末梢血、前立腺、睾丸、卵巣、胎盤、子宮、骨、関節、脂肪組織(例、白色脂肪組織、褐色脂肪組織)など]等から、自体公知のタンパク質分離精製技術(例えば、塩析や溶媒沈澱法などの溶解度を利用する方法;透析法、限外ろ過法、ゲルろ過法、およびSDS-ポリアクリルアミドゲル電気泳動法などの主として分子量の差を利用する方法;イオン交換クロマトグラフィーなどの荷電の差を利用する方法;アフィニティークロマトグラフィーなどの特異的親和性を利用する方法;逆相高速液体クロマトグラフィーなどの疎水性の差を利用する方法;等電点電気泳動法などの等電点の差を利用する方法など。これらの方法は適宜組み合わせることもできる。)により単離・精製されたものであってよい。また、化学合成もしくは無細胞翻訳系で生化学的に合成されたタンパク質であってもよいが、好ましくは、これらのタンパク質をコードする核酸を導入した形質転換体から産生される組換えタンパク質である。
【0085】
例えば、BMP4、GDNF、EGFおよびSCFのヒトおよびマウスcDNA配列情報は、それぞれ下記NCBI accession numbersを参照することにより取得でき、当業者はそれをもとに常法により容易にこれらのcDNAを単離することができる。
遺伝子名 ヒト マウス
BMP4 NM_001202 NM_007554
GDNF NM_000514 NM_010275
EGF NM_001963 NM_010113
SCF NM_000899 NM_013598
得られたcDNAを適当な発現ベクターに挿入して宿主細胞(例えば、動物細胞、昆虫細胞、枯草菌、酵母、大腸菌など)に導入し、得られた形質転換体を培養して、その培養上清から、上記のタンパク質分離精製技術を適宜組み合わせて目的の組換えタンパク質を単離・精製することができる。また、BMP4、GDNF、EGFおよびSCFの組換えタンパク質は市販されている。
【0086】
本発明のGR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持する限り、上記cDNA配列によりコードされるアミノ酸配列と異なるアミノ酸配列を有するタンパク質であってもよい。具体的には、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、
(a) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約90%以上の相同性を有するアミノ酸配列;
(b) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列において、1〜20個のアミノ酸が置換および/または欠失および/または挿入および/または付加されたアミノ酸配列;
(c) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質の他の哺乳動物におけるオルソログのアミノ酸配列;
(d) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質または上記(c)のオルソログのスプライスバリアント、アレル変異体もしくは多型(例、SNPなど)におけるアミノ酸配列;あるいは
(e) 上記(a)〜(d)のアミノ酸配列の一部(フラグメント)
を含み、かつ他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持するタンパク質を意味する。
ここで「相同性」とは、当該技術分野において公知の数学的アルゴリズムを用いて2つのアミノ酸配列をアラインさせた場合の、最適なアラインメント(好ましくは、該アルゴリズムは最適なアラインメントのために配列の一方もしくは両方へのギャップの導入を考慮し得るものである)における、オーバーラップする全アミノ酸残基に対する同一アミノ酸および類似アミノ酸残基の割合(%)を意味する。「類似アミノ酸」とは物理化学的性質において類似したアミノ酸を意味し、例えば、芳香族アミノ酸(Phe、Trp、Tyr)、脂肪族アミノ酸(Ala、Leu、Ile、Val)、極性アミノ酸(Gln、Asn)、塩基性アミノ酸(Lys、Arg、His)、酸性アミノ酸(Glu、Asp)、水酸基を有するアミノ酸(Ser、Thr)、側鎖の小さいアミノ酸(Gly、Ala、Ser、Thr、Met)などの同じグループに分類されるアミノ酸が挙げられる。このような類似アミノ酸による置換は蛋白質の表現型に変化をもたらさない(即ち、保存的アミノ酸置換である)ことが予測される。保存的アミノ酸置換の具体例は当該技術分野で周知であり、種々の文献に記載されている(例えば、Bowieら,Science, 247:1306-1310 (1990)を参照)。本明細書におけるアミノ酸配列の相同性は、相同性計算アルゴリズムNCBI BLAST(National Center for Biotechnology Information Basic Local Alignment Search Tool)を用い、以下の条件(期待値=10;ギャップを許す;マトリクス=BLOSUM62;フィルタリング=OFF)にて計算することができる。
【0087】
上記(a)において、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、より好ましくは、上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約95%以上、より好ましくは約97%以上、特に好ましくは約98%以上の同一性を有するアミノ酸配列を含むタンパク質である。
【0088】
また、上記(b)において、GR細胞の分化誘導に用いられるBMP4、GDNF、EGFまたはSCFは、好ましくは、(i)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が欠失したアミノ酸配列、(ii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が付加したアミノ酸配列、(iii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が挿入されたアミノ酸配列、(iv)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または(v)それらを組み合わせたアミノ酸配列を含み、かつ他の因子と組み合わせることにより、多能性幹細胞からGR細胞を分化誘導する能力を保持するタンパク質などであり得る。
上記のようにアミノ酸配列が挿入、欠失または置換されている場合、その挿入、欠失または置換の位置は、GR細胞の分化誘導活性が保持される限り特に限定されない。
【0089】
本発明の一実施態様においては、多能性幹細胞からGR細胞を分化誘導する際に、BMP4に代えてあるいはそれに加えて、他のBMPファミリータンパク質、例えば、BMP8b、BMP2、BMP7等を用いることができる。好ましくはBMP4とBMP8bとの併用が挙げられる。また、本発明の別の一実施態様においては、多能性幹細胞からGR細胞を分化誘導する際に、GDNFに代えてあるいはそれに加えて、WO 2004/092357に記載されるGDNFの均等物を用いることも可能である。
【0090】
BMP4を培地に添加して用いる場合、BMP4の濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、BMP4の濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。他のBMPファミリータンパク質を併用する場合、BMP全体として上記の濃度範囲となるように添加することが好ましい。
【0091】
GDNFを培地に添加して用いる場合、GDNFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、GDNFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。GDNFの均等物を併用する場合、GDNFおよび均等物全体として上記の濃度範囲となるように添加することが好ましい。
【0092】
EGFを培地に添加して用いる場合、EGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、EGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0093】
SCFを培地に添加して用いる場合、SCFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、SCFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0094】
(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子を培地に添加して用いる場合、(a)および(b)成分は、上記の基本培地もしくは該基本培地に上記の培地添加物が添加された培地とは別個に提供され、用時培地中に添加して用いてもよいし、各成長因子や他の培地成分の安定性などに悪影響を与えない限り、予め培地中に含有された形態で分化誘導用培地として提供されてもよい。
【0095】
本発明の好ましい一実施態様においては、本発明のGR細胞の分化誘導において用いられる(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子のうちの少なくとも1つの因子、好ましくは用いられるすべての因子が、培地に添加されるのではなく、フィーダー細胞から供給される。フィーダー細胞はこれらの成長因子を生来産生する哺乳動物細胞であってもよいが、該成長因子をコードする遺伝子が導入され、該成長因子を過剰発現する組換え細胞を用いることがより好ましい。異なる成長因子を発現する複数種の細胞を組み合わせて用いることも可能であるが、フィーダーからの供給が意図されるすべての成長因子を発現する1種類の細胞を用いることが好ましい。宿主となる細胞としては、従来よりフィーダー細胞として好適に使用されている細胞を用いることができるが、それらに限定されない。例えば、放射線や抗生物質で処理して細胞分裂を停止させたマウス胎仔由来の線維芽細胞 (MEF)(例、STO細胞株(ATCC CRL-1503)等) などがフィーダーとしてよく使われるが、STO細胞にネオマイシン耐性遺伝子とLIF遺伝子を安定に組み込んだSNL細胞(SNL76/7 STO細胞; ECACC 07032801)なども好ましい。また、多能性幹細胞と同種の細胞をフィーダーとして用いることも好ましい。例えば、ヒト多能性幹細胞からGR細胞を誘導する場合には、ヒト皮膚由来線維芽細胞(HDF)やヒト歯髄幹細胞などをフィーダーとして用いることができる。
【0096】
例えば、BMP4をフィーダー細胞から供給しようとする場合、例えば、M15-BMP4細胞(Proc. Natl. Acad. Sci. USA, 100: 11457-11462 (2003))をフィーダー細胞として用いることができる。さらに、GDNF、EGFおよびSCFから選ばれる1以上の成長因子もフィーダー細胞から供給しようとする場合、例えば、M15-BMP4細胞に、GDNF、EGFおよびSCFから選ばれる1以上の成長因子をコードする核酸を含む発現ベクターを導入して得られる形質転換細胞を用いることもできる。例えば、後述の実施例において、M15-BMP4細胞にGDNF、EGFおよびSCFをコードする各核酸を含むウイルスベクターを導入して、BMP4、GDNF、EGFおよびSCFの4成長因子を過剰発現する細胞(M15-4GF)を作製している。
【0097】
多能性幹細胞からのGR細胞の分化誘導培養は、例えば以下のようにして行うことができる。
本分化誘導工程に用いられる培養器は、細胞培養用であれば特に限定されないが、例えば、フラスコ、組織培養用フラスコ、デッシュ、ペトリデッシュ、組織培養用デッシュ、マルチデッシュ、マイクロプレート、マイクロウエルプレート、マルチプレート、マルチウエルプレート、チャンバースライド、シャーレ、チューブ、トレイ、培養バック、ローラーボトルが挙げられる。培養器は、培養法(浮遊培養法もしくは接着培養法)に応じて、細胞非接着性(低接着性)または細胞接着性とすることができる。細胞接着性の培養器は、培養器の表面が、細胞(多能性幹細胞またはフィーダー細胞)との接着性を向上させる目的で、細胞支持用基質でコーティングされたものであり、そのような細胞支持用基質としては、例えば、コラーゲン、ゼラチン、マトリゲル、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチンなどが挙げられる。
【0098】
培養は、上記培養器中に、多能性幹細胞を、例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように播種し、例えば、CO2インキュベーター中、約1〜約10%、好ましくは約2〜約5%のCO2濃度の雰囲気下、約30〜約40℃、好ましくは約37℃で、約1〜約8週間、好ましくは約2〜約6週間行われる。培養は、浮遊培養で行っても、接着培養で行ってもよいが、好ましくは浮遊培養である。(a) BMP4、並びに(b) GDNF、EGFおよびSCFから選ばれる1以上の成長因子(本発明の分化誘導成長因子)のうちの少なくとも1つの因子がフィーダー細胞から供給される場合、多能性幹細胞とフィーダー細胞とを1:10〜10:1、好ましくは1:5〜5:1、より好ましくは1:2〜2:1の割合で混合し、培養器中に播種して浮遊培養する。あるいは、予めフィーダー細胞を播種した培養器に多能性幹細胞を播種して共培養を行うこともできる。また、本発明の分化誘導成長因子がすべて培地に添加して用いられる場合であっても、フィーダー細胞の存在下で多能性幹細胞を培養してもよい。この場合のフィーダー細胞としては、放射線や抗生物質で処理して細胞分裂を停止させた、通常のMEF (例、STO細胞株(ATCC CRL-1503)等)、SNL細胞(SNL76/7 STO細胞; ECACC 07032801)、HDF、歯髄幹細胞などが挙げられる。
【0099】
多能性幹細胞からGR細胞への分化は、Oct4陽性かつVasa陽性を指標として確認することができる。例えば、両遺伝子の発現制御領域の下流に可視化されるレポーター遺伝子が挿入された多能性幹細胞(例えば、後述の実施例に示されるOct4-GFP/Vasa-RFPノックインマウス由来の多能性幹細胞)を用いた場合、該レポーター遺伝子の発現による発色や蛍光などを検出することにより、Oct4陽性かつVasa陽性の生殖系幹細胞、即ちGR細胞が誘導されていることを確認することができる。Oct4陽性かつVasa陽性(Oct4+/Vasa+)細胞は、フローサイトメトリー(FACS)により、他の未分化細胞(Oct4+/Vasa-)および体細胞分化の進んだ細胞(Oct4-)と選別し、単離することができる。フィーダー細胞を用いて浮遊培養を行った場合、フィーダー細胞を含む浮遊細胞塊が形成されるので、例えば、トリプシン/EDTA、コラゲナーゼ等を加えて細胞塊を解離させ、得られる細胞懸濁液を細胞支持用基質でコーティングされた培養器に播種してインキュベートした後、浮遊細胞を回収することによって、フィーダー細胞を分離除去することができる。
一方、ヒトの治療用途を念頭においた場合、Oct4およびVasa遺伝子座にレポーター遺伝子がノックインした多能性幹細胞の使用は望ましくないので、Oct4遺伝子およびVasa遺伝子自体の発現を検出する必要がある。この場合、Oct4発現に対応する未分化細胞表面マーカーおよびVasaタンパク質もしくはVasa発現に対応する生殖系細胞表面マーカーに対する各抗体とセルソータを用いて、細胞の表面抗原の表現型を解析することにより行うことができる。例えば、未分化細胞表面マーカーとしてはSSEA-1、フォルスマン抗原、β1-およびα6-インテグリン等が、生殖系細胞表面マーカーとしてはEpCAM、CD9、EE2、c-kit等が挙げられる。必要に応じて、Oct4や他の転写因子の発現についても調べることができる。
【0100】
本発明はまた、上記の方法により得られたiPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞、即ちGR細胞を提供する。これまで、ES細胞からVasa陽性の始原生殖細胞(PGC)様細胞を誘導したり(Proc. Natl. Acad. Sci. USA, 100: 11457-11462 (2003))、精子幹細胞から長期培養可能な細胞株(GS細胞)を誘導したりして(例えば、WO 2004/092357など)、不妊マウスの精巣に移植することにより該マウスにおける精子形成を可能にした例はあるが、後述の実施例に示されるとおり、これら従来の培養条件では、iPS細胞から精子形成を引き起こし得る生殖系幹細胞を樹立することはできず、本発明の分化誘導方法を用いることにより、初めてiPS細胞由来の生殖系幹細胞を得ることができた。
【0101】
上述のように、本発明のGR細胞の分化誘導方法においては、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子が、培地に添加されるか、あるいはフィーダー細胞から供給される。従って、本発明はまた、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)への分化誘導剤を提供する。好ましくは、本発明のGR細胞の分化誘導剤には、GDNF、EGFおよびSCFのうちの2因子以上、より好ましくは3因子すべてが含まれる。これらの成長因子は、水もしくは適当な緩衝液中に溶解した形態で提供されてもよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いることもできる。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
本発明のGR細胞の分化誘導剤は、さらに生理学的に許容される担体、賦形剤、防腐剤、安定剤、結合剤、溶解補助剤、非イオン性界面活性剤、緩衝剤、保存剤、酸化防止剤などを含むこともできる。
【0102】
別の好ましい実施態様においては、本発明のGR細胞の分化誘導剤は、(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を産生する細胞を含有する。当該細胞としては、例えば、これらの成長因子をコードする核酸を含む発現ベクターを宿主細胞に導入して得られる、当該成長因子を過剰発現する組換え細胞が挙げられる。当該細胞はフィーダー細胞として多能性幹細胞と共培養されるので、当該技術分野で、従来よりフィーダー細胞として使用されている細胞を宿主細胞として用いることが好ましい。具体的には上記した通りの細胞が例示される。(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を産生する細胞は、適当な培地(例えば、上記基本培地など)に適当な細胞密度で懸濁した状態、あるいは常法により凍結保存された状態で提供され得る。
【0103】
本発明はまた、上記のいずれかの基本培地もしくは該基本培地に上記のいずれかの培地添加物が補充された培地に、上記本発明のGR細胞の分化誘導剤が添加されてなる、GR細胞の分化誘導用培地を提供する。該分化誘導用培地は、分化誘導剤が添加された状態で提供されてもよいし、分化誘導剤が別個の試薬として提供され、用時培地に添加されるようにキット化されたものであってもよい。
【0104】
IV. GR細胞の維持増幅方法
上記のようにして得られるOct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)を、GDNF、EGF、SCFおよび塩基性線維芽細胞成長因子(bFGF)の存在下で培養することにより、Oct4陽性かつVasa陽性の性質を維持したまま、長期培養可能である。したがって、本発明はまた、GDNF、EGF、SCFおよびbFGFの存在下でGR細胞を培養することを含む、GR細胞の増幅方法を提供する。
【0105】
本発明の維持増幅方法に用いられるGR細胞は、Oct4陽性かつVasa陽性であり、精巣に移植した場合に腫瘍を形成せず、かつ排除されることなく長期生着可能な、生殖系細胞として運命決定された細胞である限り、その由来に特に制限はないが、好ましくは、多能性幹細胞由来のGR細胞であり、より好ましくは、上記の本発明の分化誘導方法により誘導されたGR細胞である。GR細胞は単離精製された均一なOct4陽性かつVasa陽性細胞集団として提供されてもよいし、Oct4陰性もしくはVasa陰性の細胞が混在した不均一な細胞集団として提供されてもよい(以下、特にことわらない限り、GR細胞を含む不均一な細胞集団も含めてGR細胞と記載する)。
【0106】
本発明のGR細胞の維持増幅方法に用いられる基本培地および任意の培地添加物としては、上記の多能性幹細胞からGR細胞への分化誘導方法において使用されるものと同じものが挙げられる。但し、後述するように、本発明のGR細胞の維持増幅方法はSCFおよびbFGFの存在下で行われるため、LIFの併用はPGC様細胞であるGR細胞のEG細胞様細胞への脱分化を誘導する可能性があり、使用しないことが好ましい場合がある。
好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子が、基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27の「Medium component」に記載された濃度(最終濃度)を選択することができる。
【0107】
本発明のGR細胞の維持増幅方法において、GR細胞はGDNF、EGF、SCFおよびbFGFの存在下で培養される。これらの成長因子は、上記のいずれかの培地に添加して分化誘導用培地としてもよいし、あるいは、当該因子を産生する細胞をフィーダー細胞として用い、多能性幹細胞と共培養することによって提供されてもよいが、好ましくは培地に添加して用いられる。
【0108】
GDNF、EGF、SCFおよびbFGFは任意の哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等、好ましくはヒトまたはマウス)由来のものを用いることができるが、GR細胞と同種由来のものを用いることが好ましい。特に、ヒトGR細胞をヒトの治療目的で使用する場合には、異種成分不含(xeno-free)条件下でヒトGR細胞を誘導することが望ましいので、これらの成長因子としてヒト由来タンパク質を用いることが好ましい。
【0109】
GDNF、EGF、SCFまたはbFGFを培地に添加して用いる場合、これらの成長因子は、それらを産生する哺乳動物(例えば、ヒト、マウス、サル、ブタ、ラット、イヌ等)の細胞もしくはそれらの細胞が存在するあらゆる組織等から、自体公知のタンパク質分離精製技術により単離・精製されたものであってよい。また、化学合成もしくは無細胞翻訳系で生化学的に合成されたタンパク質であってもよいが、好ましくは、これらのタンパク質をコードする核酸を導入した形質転換体から産生される組換えタンパク質である。GDNF、EGFおよびSCFの組換えタンパク質は、上述の方法により調製することができる。また、bFGFのヒトおよびマウスcDNA配列情報はNCBI accession numbers NM_002006およびNM_008006を参照することにより取得でき、当業者はそれをもとに常法によりbFGFのcDNAを単離し、上記と同様の方法により、容易に組換えタンパク質を調製することができる。また、GDNF、EGF、SCFおよびbFGFの組換えタンパク質は市販されている。
【0110】
本発明のGR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持する限り、上記cDNA配列によりコードされるアミノ酸配列と異なるアミノ酸配列を有するタンパク質であってもよい。具体的には、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、
(a) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約90%以上の相同性を有するアミノ酸配列;
(b) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列において、1〜20個のアミノ酸が置換および/または欠失および/または挿入および/または付加されたアミノ酸配列;
(c) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質の他の哺乳動物におけるオルソログのアミノ酸配列;
(d) 上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列からなるヒトもしくはマウスタンパク質または上記(c)のオルソログのスプライスバリアント、アレル変異体もしくは多型(例、SNPなど)におけるアミノ酸配列;あるいは
(e) 上記(a)〜(d)のアミノ酸配列の一部(フラグメント)
を含み、かつ他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持するタンパク質を意味する。
ここで「相同性」とは、GR細胞の分化誘導に用いられるBMP4などについて上記したのと同義である。上記(a)において、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、より好ましくは、上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列と約95%以上、より好ましくは約97%以上、特に好ましくは約98%以上の同一性を有するアミノ酸配列を含むタンパク質である。
【0111】
また、上記(b)において、GR細胞の維持増幅に用いられるGDNF、EGF、SCFまたはbFGFは、好ましくは、(i)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が欠失したアミノ酸配列、(ii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が付加したアミノ酸配列、(iii)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列に1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が挿入されたアミノ酸配列、(iv)上記NCBI accession numbersに示されるcDNA配列によりコードされるアミノ酸配列中の1〜10個、より好ましくは1〜数(5、4、3もしくは2)個のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または(v)それらを組み合わせたアミノ酸配列を含み、かつ他の因子と組み合わせることにより、GR細胞がその分化状態を維持したまま自己複製する能力を支持するタンパク質などであり得る。
上記のようにアミノ酸配列が挿入、欠失または置換されている場合、その挿入、欠失または置換の位置は、GR細胞の維持増幅活性が保持される限り特に限定されない。
【0112】
本発明の一実施態様においては、GR細胞を維持増幅する際に、GDNFに代えてあるいはそれに加えて、WO 2004/092357に記載されるGDNFの均等物を用いることも可能である。
【0113】
GDNFを培地に添加して用いる場合、GDNFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、GDNFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。GDNFの均等物を併用する場合、GDNFおよび均等物全体として上記の濃度範囲となるように添加することが好ましい。
【0114】
EGFを培地に添加して用いる場合、EGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、EGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0115】
SCFを培地に添加して用いる場合、SCFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、SCFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0116】
bFGFを培地に添加して用いる場合、bFGFの濃度は、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上である。また、bFGFの濃度は、例えば、約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下である。
【0117】
GDNF、EGF、SCFおよびbFGFを培地に添加して用いる場合、これらの成分は、上記の基本培地もしくは該基本培地に上記の任意成分である培地添加物が添加された培地とは別個に提供され、用時培地中に添加して用いてもよいし、各成長因子や他の培地成分の安定性などに悪影響を与えない限り、予め培地中に含有された形態でGR細胞の維持増幅用培地として提供されてもよい。
【0118】
本発明の別の一実施態様においては、本発明のGR細胞の維持増幅において用いられるGDNF、EGF、SCFおよびbFGFのうちの少なくとも1つの因子が、培地に添加されるのではなく、フィーダー細胞から供給され得る。フィーダー細胞はこれらの成長因子を生来産生する哺乳動物細胞であってもよいが、該成長因子をコードする遺伝子が導入され、該成長因子を過剰発現する組換え細胞を用いることがより好ましい。異なる成長因子を発現する複数種の細胞を組み合わせて用いることも可能であるが、フィーダーからの供給が意図されるすべての成長因子を発現する1種類の細胞を用いることが好ましい。宿主となる細胞としては、GR細胞の分化誘導において上記したのと同様の細胞を用いることができる。組換え細胞の製造についても、GR細胞の分化誘導において上記したのと同様の手法で行うことができる。
【0119】
本発明のGR細胞の維持増幅方法においては、上記の4因子(GDNF、EGF、SCFおよびbFGF)に加えて、さらにHGF、FGF9等の他の成長因子、IL-2等のインターロイキン類から選ばれる1以上の培地添加物の存在下でGR細胞を培養することが好ましい。
HGFやFGF9などの成長因子はそれぞれ、例えば約0.1 ng/ml以上、好ましくは約0.5 ng/ml以上、より好ましくは約1 ng/ml以上、特に好ましくは約5 ng/ml以上で、例えば約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下の濃度で、培地に添加することができる。また、IL-2等のインターロイキン類は、例えば約0.01 ng/ml以上、好ましくは約0.1 ng/ml以上、より好ましくは約0.5 ng/ml以上、特に好ましくは約1 ng/ml以上で、例えば約100 ng/ml以下、好ましくは約50 ng/ml以下、より好ましくは約30 ng/ml以下、特に好ましくは約20 ng/ml以下の濃度で、培地に添加することができる。
【0120】
本発明のGR細胞の維持増幅方法においては、さらに男性ホルモン(例、テストステロン等の)および/またはその誘導物質(例、フォルスコリン)の存在下で、GR細胞を培養することもまた好ましい。これらの成分は、GR細胞の維持増幅を支持し、かつ細胞の生存に悪影響を及ぼさない範囲で培地に添加することができるが、例えば約0.001 μM以上、好ましくは0.01 μM以上、より好ましくは0.05 μM以上で、約1000 μM以下、好ましくは約500 μM以下、より好ましくは約100 μM以下の濃度で培地に添加することができる。
【0121】
別の好ましい一実施態様においては、表1に記載される培地添加物から選ばれる1以上の因子と、表2に記載される培地添加物から選ばれる1以上の因子とが、上記いずれかの基本培地に添加されて用いられる。当業者はこれらの因子を適当な濃度で基本培地に添加することができるが、例えば、図27に記載された濃度(最終濃度)を選択することができる。
【0122】
【表2】
【0123】
GR細胞の維持増幅培養は、例えば以下のようにして行うことができる。
本培養工程に用いられる培養器は、細胞培養用であれば特に限定されず、GR細胞の分化誘導工程で用いられるものと同様の培養器が例示される。培養器は、培養法(浮遊培養法もしくは接着培養法)に応じて、細胞非接着性(低接着性)または細胞接着性とすることができる。細胞接着性の培養器は、培養器の表面が、細胞(多能性幹細胞またはフィーダー細胞)との接着性を向上させる目的で、細胞支持用基質でコーティングされたものであり、そのような細胞支持用基質としては、例えば、コラーゲン、ゼラチン、マトリゲル、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチンなどが挙げられる。
【0124】
培養は、上記培養器中に、GR細胞を、例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように播種し、例えば、CO2インキュベーター中、約1〜約10%、好ましくは約2〜約5%のCO2濃度の雰囲気下、約30〜約40℃、好ましくは約37℃で行われる。GDNF、EGF、SCFおよびbFGFのうちの少なくとも1つの因子がフィーダー細胞から供給される場合、フィーダー細胞を例えば約0.5〜約50×104細胞/cm2、好ましくは約1〜約10×104細胞/cm2の細胞密度となるように培養器中に播種し、その上にGR細胞を播種する。また、GDNF、EGF、SCFおよびbFGFがすべて培地に添加して用いられる場合であっても、フィーダー細胞の存在下で多能性幹細胞を培養することが好ましい。この場合のフィーダー細胞としては、放射線や抗生物質で処理して細胞分裂を停止させた、通常のMEF (例、STO細胞株(ATCC CRL-1503)等)、SNL細胞(SNL76/7 STO細胞; ECACC 07032801)、HDF、歯髄幹細胞などが挙げられる。細胞がコンフルエントもしくはサブコンフルエントに達した段階で常法に従って細胞を継代培養することにより、GR細胞をその分化状態を維持したまま、長期維持、増幅することができる。
【0125】
GR細胞がOct4陰性もしくはVasa陰性の細胞を含む不均一な細胞集団として提供された場合、増幅された細胞集団から、Oct4陽性およびVasa陽性を指標としてGR細胞を分離回収することにより、均一なGR細胞を得ることができる。GR細胞の分離方法としては、多能性幹細胞から誘導されたGR細胞を分離する際に用いるのと同様の方法が挙げられる。
【0126】
本発明のGR細胞の分化誘導方法により、多能性幹細胞から誘導された細胞集団の中には、GR細胞に加えて、Oct4陽性かつVasa陰性の未分化細胞が含まれており、分離された当該未分化細胞を、上記GR細胞の維持増幅条件下で培養すると、Oct4陽性かつVasa陰性の分化状態を維持したまま安定に増幅させることができたので、この未分化細胞株をGsp細胞と名づけた。Gsp細胞を免疫不全マウスに移植するとテラトーマを形成し、また種々の特性解析の結果からも、Gsp細胞がもとの多能性幹細胞とは異なる、新規に樹立された多能性幹細胞であることが示された。したがって、本発明はまた、GR細胞の分化誘導過程で誘導され、かつGR細胞の維持増幅条件下で安定に維持増幅される新規多能性幹細胞であるGsp細胞並びにその製造および維持方法を提供する。Gsp細胞は、GS細胞樹立過程で見出されたmGS細胞と同様、ES細胞やiPS細胞などの既知多能性幹細胞の代替物として、種々の用途に利用可能である。
【0127】
上述のように、本発明のGR細胞の維持増幅方法においては、GDNF、EGF、SCFおよびbFGFが培地に添加される。従って、本発明はまた、GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞(GR細胞)の増幅支持剤を提供する。なお、本発明のGR細胞の維持増幅条件下では、Oct4陽性かつVasa陰性の多能性幹細胞(Gsp細胞)も安定に維持されるので、当該増幅支持剤はまたGsp細胞の増幅支持剤でもある。GDNF、EGF、SCF、bFGFの各因子は、水もしくは適当な緩衝液中に溶解した形態で提供されてもよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いることもできる。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
本発明のGR細胞の増幅支持剤は、さらに生理学的に許容される担体、賦形剤、防腐剤、安定剤、結合剤、溶解補助剤、非イオン性界面活性剤、緩衝剤、保存剤、酸化防止剤などを含むこともできる。
【0128】
別の好ましい実施態様においては、本発明のGR細胞の増幅支持剤には、GDNF、EGF、SCFおよびbFGFの4因子に加えて、HGF、IL-2およびFGF9から選ばされる1以上、好ましくは2以上の因子、より好ましくは3因子すべてがさらに組み合わされる。これらの各因子も、水もしくは適当な緩衝液中に溶解した形態で提供されてよく、凍結乾燥粉末として提供され、用時適当な溶媒に溶解して用いてもよい。また、これらの成分はそれぞれ単独の試薬としてキット化されていてもよいし、互いに悪影響を与えない限り、2種以上を混合して1つの試薬として提供することもできる。
【0129】
本発明はまた、上記のいずれかの基本培地もしくは該基本培地に上記の任意成分である培地添加物が補充された培地に、上記本発明のGR細胞の増幅支持剤が添加されてなる、GR細胞の維持増幅用培地を提供する。該維持増幅用培地は、増幅支持剤が添加された状態で提供されてもよいし、増幅支持剤が別個の試薬として提供され、用時培地に添加されるようにキット化されたものであってもよい。
【0130】
本発明また、上記の分化誘導方法により得られたGR細胞、または上記の維持増幅方法により維持されたGR細胞を、該細胞と同種の不妊動物の精巣に移植することによる、該不妊動物に精子を形成させる方法を提供する。当該方法は、本発明の分化誘導方法および/または維持増幅方法によりGR細胞を樹立することができる任意の哺乳動物に対して適用可能であるが、不妊治療や遺伝子治療への応用を念頭におく場合、対象動物として好ましくはヒトが挙げられる。また、研究目的においては、従来より実験動物として繁用されているマウス、ラット、モルモット、ハムスター、スナネズミ、ウサギ、イヌ、サルなどが挙げられる。さらに、育種目的においては、種々の家畜動物(例、ウシ、ウマ、ブタ、ヒツジ、ヤギ等)や愛玩動物(例、イヌ、ネコ等)などが挙げられる。
【0131】
GR細胞の精巣への移植は、例えばWO 2004/092357やBiol. Reprod., 69: 612-616 (2003) に記載の方法において、GS細胞の代わりに本発明のGR細胞を用いることにより実施することができる。
【0132】
本発明の精子形成方法を不妊治療に用いる場合、移植されるGR細胞はレシピエント動物個体から誘導されたものであることが特に望ましい。患者由来の精子を作るためには、精子形成のために移植される生殖系幹細胞は、出生後に採取可能な細胞から誘導可能なものであることが必要であるが、iPS細胞は安全面を考慮してタンパク質導入やウイルスフリーの遺伝子導入により作製しようとする場合、樹立効率が低いことが欠点である。そのため、患者本人の精子幹細胞を採取できる場合は、精子幹細胞からGS細胞を誘導して用いる方が有利な場合もある。しかしながら、精子幹細胞の採取が不可能か、もしくは多数回精巣生検を必要とするなどの理由で精子幹細胞を採取することに相当程度のリスクを伴う場合などでは、皮膚細胞などから容易に誘導可能なiPS細胞を材料として本発明の方法により作製されたGR細胞を用いることはきわめて有意義である。したがって、本発明の精子形成方法を不妊治療に用いる場合、GR細胞の好ましい投与対象として、精子幹細胞の採取が困難な不妊動物が挙げられる。
【0133】
また、本発明の精子形成方法を遺伝子治療に用いる場合、遺伝病患者から誘導されるiPS細胞やmGS細胞から分化誘導されたGR細胞をそのまま移植することはできない。そのため、患者から誘導した多能性幹細胞もしくは該細胞から本発明の方法により分化誘導したGR細胞に遺伝子操作を施して、変異遺伝子を正常な遺伝子と置換するなどの処理をしたGR細胞を作製して、これを患者に移植する必要がある。このようにして正常な遺伝子機能を有する患者由来の精子を形成させることにより、患者の変異遺伝子が後代に伝わり子孫が遺伝病を発症するのを予防することができる。
【0134】
また、育種分野において、優良な血統が途絶えた場合でも、当該優良な遺伝情報を有する個体から多能性幹細胞を樹立しておけば、本発明の方法により該多能性幹細胞からGR細胞を誘導、増幅して別の個体の精巣に移植することにより、該個体に多能性幹細胞由来の精子を形成させることができ、該個体を用いて繁殖を行うことにより優良な血統を維持させることができる。
【0135】
本発明はまた、上記の分化誘導方法により得られたGR細胞、または上記の維持増幅方法により維持されたGR細胞を含有してなる、雄性不妊治療剤を提供する。
本発明のGR細胞は、常套手段にしたがって医薬上許容される担体と混合するなどして、注射剤、懸濁剤、点滴剤等の非経口製剤として製造される。当該非経口製剤に含まれ得る医薬上許容される担体としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液(例えば、D-ソルビトール、D-マンニトール、塩化ナトリウムなど)などの注射用の水性液を挙げることができる。本発明の剤は、例えば、緩衝剤(例えば、リン酸塩緩衝液、酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩化ベンザルコニウム、塩酸プロカインなど)、安定剤(例えば、ヒト血清アルブミン、ポリエチレングリコールなど)、保存剤、酸化防止剤などと配合しても良い。もちろん、GR細胞は、上記のGR細胞の維持増幅用培地に懸濁したものをそのまま製剤として用いてもよい。本発明の不妊治療剤を水性懸濁液剤として製剤化する場合、上記水性液に約1×106〜約1×108細胞/mLとなるように、GR細胞を懸濁させればよい。
本発明の雄性不妊治療剤は、幹細胞の凍結保存に通常使用される条件で凍結保存された状態で提供され、用時融解して用いることもできる。その場合、血清もしくはその代替物、有機溶剤(例、DMSO)等をさらに含んでいてもよい。この場合、血清もしくはその代替物の濃度は、特に限定されるものではないが約1〜約30% (v/v)、好ましくは約5〜約20% (v/v) であり得る。有機溶剤の濃度は、特に限定されるものではないが0〜約50% (v/v)、好ましくは約5〜約20% (v/v) であり得る。
【0136】
このようにして得られる製剤は、安定で低毒性であるので、ヒトなどの哺乳動物に対して安全に投与することができる。投与方法は特に限定されないが、好ましくは注射もしくは点滴投与であり、精細管内に投与され得る。本発明の不妊治療剤は、ヒト不妊患者においては、例えば、1回につきGR細胞量として約1.0×105〜約1×107細胞を、単回もしくは約1〜約2週間隔で、複数回(例、2〜10回)投与され得る。
【0137】
以下、実施例を示して本発明をより具体的に説明するが、本発明は以下に示す実施例によって何ら限定されるものではない。
【実施例】
【0138】
実施例1 iPS細胞の樹立
実験系として、レポーター遺伝子であるOct4-GFPとMvh (mouse vasa homolog)-RFPの導入により未分化細胞と生殖細胞がそれぞれ可視化されたトランスジェニックマウス(Oct4-GFP/Mvh-RFP Tgマウス)を使用した。Oct3/4(Oct4)は未分化細胞特異的発現遺伝子であり、マウスvasaは生殖細胞系譜特異的発現遺伝子であることが知られている。このマウス系統は、マウスOct4遺伝子の発現制御領域にGFP遺伝子を連結したレポータープラスミドDNAを受精卵に顕微注入して作製されたTgマウス (Oct4-GFP)、およびマウスMvhゲノム遺伝子にRFP遺伝子を挿入したBACクローンDNAを顕微注入したTgマウス (Mvh-RFP) の両者を自然交配することによって作製した。
初期化に使用するレトロウイルスは、前日に6 well培養プレート (Falcon) に1 well当り0.6 x 106で播種したPlat-E細胞(Morita, S. et al., Gene Ther. 7, 1063-1066)にレトロウイルス発現ベクター (pMXs-Oct3/4, pMXs-Sox2, pMXs-Klf4, pMXs-Nanog) を個々に導入して作製した。培養液はDMEM/10% FCS (DMEM (Nacalai tesque) にウシ胎仔血清を10%加えたもの) を使用し、37℃、5% CO2で培養した。ベクターの導入のためにFuGene6 transfection reagent (Roche) 4.5 μLをOpti-MEM I Reduced-Serum Medium (Invitrogen) 100 μLに入れ、室温で5分間静置した。その後、各発現ベクターを1.5 μg加え、さらに室温で15分静置してからPlat-E細胞の培養液に加えた。2日目にPlat-E細胞の培養上清を新しい培地に換え、3日目に培養上清を回収して0.45 μm sterile filter (Whatman) で濾過し、polybrene (Nacalai) を4 μg/mLとなるように加えてウイルス液とした。
前述のOct4-GFP/Mvh-RFP Tgマウスの胎仔(受精後13.5日)から、線維芽細胞(MEF)を単離した。このMEFを0.1% ゼラチン (Sigma) でコートした6 well培養プレート (Falcon) に1 well当り1 x 105で播種した。培養液はDMEM/10% FCSを使用し、37℃、5% CO2で培養した。翌日、レトロウイルス液を加え、一晩感染させて遺伝子を導入した。遺伝子導入は、Oct3/4, Sox2, Klf4およびNanogの4種の遺伝子導入と、Oct3/4, Sox2およびKlf4の3種の遺伝子導入を行った。
ウイルス感染後3日目からLIFを加えたES細胞用培地(DMEM (Nacarai tesque) に15% ウシ胎仔血清、2 mM L-グルタミン (Invitrogen)、100 μM 非必須アミノ酸(Invitrogen)、100 μM 2-メルカプトエタノール (Invitrogen)、50 U/mL ペニシリン (Invitrogen) と50 μg/mL ストレプトマイシン (Invitrogen) を加えたもの)を用いて培養した。感染後5日目にMEFの培地を除き、PBS 1 mLを加えて細胞を洗浄した。PBSを除いた後、0.25% Trypsin/1 mM EDTA (Invitrogen) を加えて、37℃で5分間程度反応させた。細胞が浮き上がったらES細胞用培地を加えて懸濁し、5 x 103個の細胞を、あらかじめフィーダー細胞を蒔いておいた100 mm dishに蒔いた。フィーダー細胞にはマイトマイシンCで処理して、細胞分裂を止めたSNL細胞(McMahon, A. P. & Bradley, A. Cell 62, 1073-1085 (1990))を用いた。以後コロニーが観察できるようになるまで2日ごとにES細胞用培地の交換を行った。感染から17日目のiPS細胞コロニーの写真を図1に示す。4遺伝子導入、3遺伝子導入いずれの場合も、Oct3/4-GFP陽性で、かつMvh-RFP陰性のiPS細胞コロニーが樹立できた。
これらのiPSクローンについて常法によりGenomic PCR解析を行った結果、いずれのクローンにおいても導入した外来遺伝子の組み込みが確認された(図2)。
また、各iPSクローンにおける未分化マーカーの発現を、Rever Tra Ace kit(Takara)を使用してRT-PCR解析を行うことにより調べた。結果を図3に示す。各iPSクローンは、ES細胞やNanog-iPS(Nature, 448, 313-317 (2007))と同等の未分化マーカーの発現を示した。
次に、Cell, 126, 663-676 (2006) に記載の方法に従ってテラトーマを形成させた。具体的には、1 x 106個のiPS細胞を免疫不全マウスの皮下に注射し、4週間後にテラトーマを単離した。テラトーマを切り刻んで4% フォルムアルデヒドを含有するPBS(-)で固定した。パラフィン包埋組織をスライスし、ヘマトキシリン・エオシンで染色した。結果を図4に示す。組織学的に見ると、腫瘍は複数の種類の細胞から構成されており、神経組織、表皮組織、筋肉組織、軟骨組織、脂肪組織、および腸管様上皮組織が認められたことから、樹立したiPS細胞の多能性が証明された。
【0139】
実施例2 生殖系幹細胞の分化維持培地の確立
樹立したiPS細胞の分化誘導を行うために、DMEM/10% FCSを使用して胚様体 (EB) 形成を行った。具体的には、培養液1 mL当り5 x 105個のiPS細胞を6 wellの低細胞接着性培養プレート (Nunc) に播種し、2日毎に培地交換を行った。培養14日目の写真を図5に示す。同じOct4-GFP/Mvh-RFP TgのES細胞由来のEB中には部分的にMvh-RFP陽性の生殖細胞の分化が認められたが、iPS細胞由来のEB中にはMvh-RFP陽性の細胞は確認されなかった。
そこで、iPS細胞の生殖細胞分化を誘導する培養条件を確立するために、分化支持細胞としてPNAS, 100, 11457-11462 (2003) で使用されたM15-BMP4細胞の改良を行った。前述の方法でPlat-E細胞にグリア細胞由来神経栄養因子 (GDNF)、細胞膜結合型幹細胞因子 (mSCF)、上皮細胞成長因子 (EGF)の レトロウイルス発現ベクター (pMXs-GDNF-IP, pMXs-mSCF-IP, pMXs-EGF-IP) を導入し、得られたウイルス液をM15-BMP4細胞に感染させた。ウイルス感染細胞を選択するために0.2 mg/mL ネオマイシンおよび2.5 μg/mL ピューロマイシン存在下で培養を行い、増殖し続ける細胞を新たにM15-4GFと命名した。M15-4GFの形態とRT-PCRの結果を図6に示す。RT-PCRによって、M15-4GFではBMP4に加えて遺伝子導入したGDNF、mSCF、EGFが高発現していることを確認した。
さらに、生殖細胞の分化誘導に適した培地作製を行った。組成を図27に示す。
Neurobasal medium (Invitrogen) を基礎培地として、最終濃度で1x B-27 Supplement (Invitrogen)、1 x Penicillin-Streptomycin-Glutamine (Invitrogen)、5 mg/mL Bovine Albumin (MP Biomedicals)、0.1 mM Non-Essential Amino Acids (Invitrogen)、1 mM Sodium Pyruvate (Invitrogen)、1x Vitamin Solution (Invitrogen)、1x Insulin-Transferrin-Selenium Supplement (Invitrogen)、6 mg/mL D-(+)-Glucose (Sigma)、60 ng/mL Progesteron (Sigma)、30 ng/mL β-Estradiol (Sigma)、55 μM 2-Mercaptoethanol (Invitorgen)、0.34 μL/mL Sodium DL-lactate (Sigma)、60 μg/mL Putrescine dihydrochloride (Sigma)、0.1% FCS (Invitorgen) を添加した(図中「Medium component」)。また、分化誘導後の細胞の維持培養には、上記培養液に最終濃度で15 ng/mL GDNF (R&D systems)、20 ng/mL EGF (AUSTRAL Biologicals)、12.5 ng/mL basic fibroblast growth factor (bFGF) (Wako)、10 ng/mL SCF (R&D systems)、10 ng/mL hepatocyte growth factor (HGF) (PEPROTECH)、5 ng/mL interleukin-2 (IL2) (Roche)、12.5 ng/mL fibroblast growth factor 9 (R&D systems)、10 μM Forskolin (Sigma)、0.1 μM Testosterone (Sigma)、0.4x StemPro Supplement (Invitrogen)(図中「Supplement」)を添加した。
生殖細胞分化誘導を行うために、iPS細胞とM15-4GF細胞を培養液1 mL当り各々5×105個となるように増殖因子(Supplement)無添加の上記培養液中で混合し、6 wellの低細胞接着性培養プレートに播種した。2日毎に培地交換を行いながら浮遊培養を行ったところ、細胞塊中にMvh-RFP陽性細胞の分化が確認された。培養28日目に浮遊細胞塊を15 mLチューブに回収し、PBSで洗浄後に0.25% Trypsin/1 mM EDTAと0.2 mg/mL collagenase IV (Invitrogen) を加えて、37℃で15分間程度反応させた。DMEM/10% FCSを加えて懸濁後に遠心 (1000 rpm) し、維持培養用の増殖因子(Supplement)添加培養液に置換した。M15-4GF細胞を除去するために、細胞懸濁液をゲラチンコートした培養プレート上に播種して30分から1時間程度静置した後に浮遊細胞を回収し、マイトマイシンC処理したMEF feeder細胞 (Millipore) 上に播種して培養を行った。その結果、Oct4-GFP陽性/Mvh-RFP陽性のコロニーの形成が観察された(図7)。
Oct4-GFP陽性/Mvh-RFP陽性のコロニーを形成する細胞は上記培養条件下で安定に増殖し、未分化iPS細胞とは異なる形態を示した(図8)。また、未分化細胞と生殖細胞に共通する細胞表面マーカーであるalkaline phosphataseの活性が認められた(図9)。
このOct4-GFP陽性/Mvh-RFP陽性コロニーを、iPS細胞用の培養条件(DMEM/10% FCS)やBiol. Reprod, 69, 612-616 (2003) に記載されているGS細胞用の培養条件(GDNF、LIF、EGFおよびbFGF添加)で培養した場合、上記培養条件下に比べて増殖速度は低下し、部分的な細胞死やMvh-RFP蛍光強度の低下が認められた (図10)。また、未分化iPS細胞を分化誘導過程を経ずに直接上記維持培養条件下で培養した場合には、体細胞分化に伴うOct4-GFPの縮退や細胞死は観察されるものの、Mvh-RFPの誘導は認められなかった (図11)。
【0140】
実施例3 Oct4-GFP陽性/Mvh-RFP陽性細胞の特徴
上記培養条件で浮遊細胞塊を解離、培養した際に、Oct4-GFP陽性/Mvh-RFP陽性の生殖細胞様細胞に加えて、Oct4-GFP陽性/Mvh-RFP陰性の未分化様の細胞も分離、安定増殖することが出来た(図12)。以降、Oct4-GFP陽性/Mvh-RFP陽性細胞を「GR細胞」と称し、Oct4-GFP陽性/Mvh-RFP陰性細胞を「Gsp細胞」と称することとした。
GR細胞とGsp細胞に対してGenomic PCR解析を行ったところ、iPS細胞作製時に導入した外来遺伝子の組み込みが確認された。過去にES細胞から分化誘導、樹立されたOct4-GFP陰性/Mvh-RFP陽性の生殖細胞様細胞株であるEK細胞では外来遺伝子は認められなかったのに対して、GR細胞とGsp細胞はiPS細胞由来であることが示された (図13)。また、サザンブロット解析が示す外来遺伝子のバンドパターンから、GR細胞とGsp細胞がiPS細胞と同一クローン由来であることも確認された (図14)。しかし、GR細胞ではSox2の外来遺伝子が一つ欠損していることが明らかとなった。
24 well培養プレートにあらかじめ播種したマイトマイシンC処理MEF feeder細胞上に1 well当たり2.5×103個となるように未分化iPS細胞とGR細胞をそれぞれ播種し、2日毎に総細胞数を測定した結果、GR細胞はiPS細胞に比べて増殖速度が1/2程度であることが分った (図15)。また、GR細胞の増殖におけるfeeder細胞依存性を調べる為に、6 well培養プレートをゲラチン、ラミニン、またはファイブロネクチンでコーティングし、1 well当たり1×104個となるようにGR細胞を播種した。その結果、MEF feeder細胞上に播種した際にはGR細胞はコロニーを形成しながら増殖するのに対し、feeder freeの条件ではどの細胞外マトリックスを用いた場合においても細胞がほとんど接着することがなく、コロニー形成も認められなかった (図16)。
腫瘍細胞や未分化なiPS細胞を免疫不全のnu/nuマウス皮下に移植すると、移植部位において腫瘍を形成する。一方、精子幹細胞株であるGS細胞の移植においては腫瘍の形成は認められないことが知られている。そこでGR細胞およびGsp細胞を免疫不全マウスに移植し、腫瘍形成の有無を調べた。その結果、Gsp細胞を移植した場合、iPS細胞に比べてサイズは小さいが腫瘍化することが分かった。一方、GR細胞を移植した場合には、ES細胞由来のEK細胞と同様に腫瘍形成は認められず、GR細胞は胚性腫瘍 (EC) 細胞のように腫瘍化した細胞ではないことが示された (図17)。
分化誘導に使用したiPS細胞はレトロウイルスによって外来遺伝子を導入していることから、レトロウイルスの再活性化によって不死化した細胞が得られた可能性も考えられる。そこで、RT-PCRによって導入した外来遺伝子とそれに対応する内在性遺伝子の発現を解析した。その結果、導入した外来遺伝子の異常な遺伝子発現の向上は認められなかった (図18)。また、リアルタイムPCRによって遺伝子発現を定量化したところ、GR細胞における外来遺伝子の発現は未分化iPS細胞と比較してむしろ低下していることが分かった (図19)。
続いて、GR細胞における生殖細胞マーカー遺伝子の発現をRT-PCRで調べた。BMC Dev. Biol., 6, 34 (2006) にあるように、GS細胞ではECAT1やFgf4の発現が抑制されていることが判明しているが、GR細胞ではそれらの遺伝子発現はまだ抑制されていなかった。また、生体内の細胞では発現が認められないERasが発現しており、GR細胞が株化された培養細胞であることが示された。GR細胞ではPrdm14、Mvh、Plzf、c-Retなどの発現が上昇している一方で、Stra8、Ngn3などの発現は低下していた。結果を図20、21に示す。
また、DNAマイクロアレイを用いてiPS細胞とGR細胞の網羅的な遺伝子発現の比較を行ったところ、マトリックスメタロプロテアーゼやケモカイン関連の遺伝子の発現が上昇し、細胞外マトリックスであるケラチンや細胞接着因子のクローディンなどの発現が低下していることが分かった。この結果から、GR細胞では移動期の始原生殖細胞のように細胞の移動活性が高まっていることが示唆された (図22)。
また、生殖細胞マーカー遺伝子のタンパク質発現をウエスタンブロットによって解析した。結果を図23に示す。GR細胞はOct4-GFP陽性であり、RT-PCRによって内在性Oct4遺伝子の転写も確認されていたにも関わらず、タンパク質レベルでは抑制を受けていることが分かった。また、GR細胞ではNanog、ECAT1、Mvh、Dnmt3Lのタンパク質発現が上昇していた。
続いて、Bisulphite genomic sequencingによってDNAのメチル化状態を解析した。結果を図24に示す。EK細胞では雌性インプリント遺伝子であるIgf2r、SNRPN、Lit1のメチル化制御領域が脱メチル化状態であり、雄性インプリント遺伝子であるH19は高メチル化状態であることから、雄型のインプリント状態を獲得していることが分かった。一方、GR細胞では雌性インプリント遺伝子のメチル化制御領域は体細胞パターンを示しており、胎仔生殖原基において誘導されるゲノムインプリントの消去が未だ行われていないことが分かった。以上の遺伝子発現とDNAのメチル化状態を総合的に解釈すると、GR細胞は移動後期(胎仔生殖原基に進入直前、または直後)の始原生殖細胞の特性を反映した細胞株であることが推測された。
GR細胞の精子形成能の有無を調べるために、Biol. Reprod, 69, 612-616 (2003) に記載されている方法に従って、c-kit遺伝子の変異によって生殖細胞を欠損した不妊モデルマウスであるW/Wvマウスの精巣へのGR細胞の移植を行った(京都大学医学部・篠原隆司教授の協力による)。具体的には、GR細胞を3×107個/mLとなるようにDMEM/10% FCS中に懸濁し、W/Wvマウス新生仔の精細管内に注射した。4カ月後に移植個体から精巣を摘出し、4% フォルムアルデヒドを含有するPBS(-)で固定した。パラフィン包埋組織をスライスし、ヘマトキシリン・エオシンで染色した。結果を図25に示す。細胞移植を行っていないW/Wvマウス精巣は、セルトリオンリーと呼ばれる生殖細胞を欠損した組織像を示すが、GR細胞を移植した場合には移植細胞の生着が認められた。生着した移植細胞はテラトーマ形成を示さず、また明瞭な腫瘍化も観察されなかった。
非生殖細胞を移植した場合は、精巣内環境に適応できずに死滅、あるいは排除されてしまう。一方、未分化なiPS細胞や腫瘍細胞を移植した場合には腫瘍が形成されてしまう。前記のようにGR細胞の移植においては、4ヶ月という長期に渡って細胞が精巣内に維持されており、かつ腫瘍化が認められなかったことから、GR細胞が生殖系細胞として運命決定された細胞であることが示された。しかしながら正常な精子形成像とも異なっており、TUNEL染色の結果、本来減数分裂後の成熟した精子細胞が存在する管腔において、アポトーシスによる細胞死が誘導されていることが明らかとなった。
【0141】
実施例4 ES細胞からOct4-GFP陽性/Mvh-RFP陽性細胞への分化誘導
本培養条件を用いて、これまでにiPS細胞から計5株のGR細胞を独立した実験から樹立することに成功している。また、本条件はiPS細胞に対してだけではなくES細胞にも適用可能であり、ES細胞を用いて同様に分化誘導を行った場合においてもOct4-GFP陽性/Mvh-RFP陽性細胞を樹立することが出来た(図26)。
【産業上の利用可能性】
【0142】
本発明のGR細胞の分化誘導および維持増幅方法によれば、皮膚細胞などの体細胞から容易に作製できるiPS細胞から精子形成可能な生殖系幹細胞を誘導し、維持増幅することができるので、精子幹細胞の採取が困難および/または危険を伴う非閉塞性無精子症の患者の不妊治療や遺伝子治療に特に有用である。また、育種分野における優良動物品種の保存の目的においても、本発明により作製されたGR細胞は極めて有用である。
【特許請求の範囲】
【請求項1】
(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞の製造方法。
【請求項2】
BMP4、GDNF、EGFおよびSCFの存在下で多能性幹細胞を培養することを含む、請求項1記載の方法。
【請求項3】
フィーダー細胞の存在下で多能性幹細胞を培養することを特徴とする、請求項1または2記載の方法。
【請求項4】
前記成長因子がフィーダー細胞から提供されることを特徴とする、請求項3記載の方法。
【請求項5】
多能性幹細胞がiPS細胞またはES細胞である、請求項1〜4のいずれか1項に記載に方法。
【請求項6】
多能性幹細胞がヒトまたはマウス由来である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
請求項1〜6のいずれか1項に記載の方法により得られた、iPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞。
【請求項8】
(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
【請求項9】
BMP4、GDNF、EGFおよびSCFを組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
【請求項10】
前記成長因子を産生する細胞を含有してなる、請求項8または9記載の剤。
【請求項11】
請求項8または9記載の剤が添加されてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導用培地。
【請求項12】
Oct4陽性かつVasa陽性の生殖系幹細胞の増幅方法であって、GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、該生殖系幹細胞を培養することを含む、方法。
【請求項13】
肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)から選ばされる1以上の因子がさらに存在する条件下で、前記生殖系幹細胞を培養することを含む、請求項12記載の方法。
【請求項14】
前記生殖系幹細胞をフィーダー細胞の存在下で培養することを特徴とする、請求項12または13記載の方法。
【請求項15】
前記生殖系幹細胞が多能性幹細胞から分化誘導されたものである、請求項12〜14のいずれか1項に記載の方法。
【請求項16】
GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅支持剤。
【請求項17】
HGF、IL-2およびFGF9から選ばされる1以上の因子をさらに組み合わせてなる、請求項16記載の剤。
【請求項18】
請求項16または17記載の剤が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
【請求項19】
請求項1〜6のいずれか1項に記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、請求項7記載のOct4陽性かつVasa陽性の生殖系幹細胞、または請求項12〜15のいずれか1項に記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を、該細胞と同種の不妊動物の精巣に移植することを含む、該不妊動物に精子を形成させる方法。
【請求項20】
生殖系幹細胞が不妊動物の体細胞から作製したiPS細胞由来である、請求項19記載の方法。
【請求項21】
不妊動物がヒトまたはマウスである、請求項19または20記載の方法。
【請求項22】
請求項1〜6のいずれか1項に記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、請求項7記載のOct4陽性かつVasa陽性の生殖系幹細胞、または請求項12〜15のいずれか1項に記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を含有してなる、雄性不妊治療剤。
【請求項23】
精子幹細胞の採取が困難な個体を投与対象とする、請求項22記載の剤。
【請求項1】
(a)骨形成タンパク質4(BMP4)、並びに(b)グリア細胞由来神経栄養因子(GDNF)、上皮細胞成長因子(EGF)および幹細胞因子(SCF)から選ばれる1以上の成長因子の存在下で多能性幹細胞を培養することを含む、Oct4陽性かつVasa陽性の生殖系幹細胞の製造方法。
【請求項2】
BMP4、GDNF、EGFおよびSCFの存在下で多能性幹細胞を培養することを含む、請求項1記載の方法。
【請求項3】
フィーダー細胞の存在下で多能性幹細胞を培養することを特徴とする、請求項1または2記載の方法。
【請求項4】
前記成長因子がフィーダー細胞から提供されることを特徴とする、請求項3記載の方法。
【請求項5】
多能性幹細胞がiPS細胞またはES細胞である、請求項1〜4のいずれか1項に記載に方法。
【請求項6】
多能性幹細胞がヒトまたはマウス由来である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
請求項1〜6のいずれか1項に記載の方法により得られた、iPS細胞由来のOct4陽性かつVasa陽性の生殖系幹細胞。
【請求項8】
(a)BMP4、並びに(b)GDNF、EGFおよびSCFから選ばれる1以上の成長因子を組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
【請求項9】
BMP4、GDNF、EGFおよびSCFを組み合わせてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導剤。
【請求項10】
前記成長因子を産生する細胞を含有してなる、請求項8または9記載の剤。
【請求項11】
請求項8または9記載の剤が添加されてなる、多能性幹細胞からOct4陽性かつVasa陽性の生殖系幹細胞への分化誘導用培地。
【請求項12】
Oct4陽性かつVasa陽性の生殖系幹細胞の増幅方法であって、GDNF、EGF、SCFおよび塩基性繊維芽細胞成長因子(bFGF)の存在下で、該生殖系幹細胞を培養することを含む、方法。
【請求項13】
肝細胞成長因子(HGF)、インターロイキン-2(IL-2)および線維芽細胞成長因子9(FGF9)から選ばされる1以上の因子がさらに存在する条件下で、前記生殖系幹細胞を培養することを含む、請求項12記載の方法。
【請求項14】
前記生殖系幹細胞をフィーダー細胞の存在下で培養することを特徴とする、請求項12または13記載の方法。
【請求項15】
前記生殖系幹細胞が多能性幹細胞から分化誘導されたものである、請求項12〜14のいずれか1項に記載の方法。
【請求項16】
GDNF、EGF、SCFおよびbFGFを組み合わせてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅支持剤。
【請求項17】
HGF、IL-2およびFGF9から選ばされる1以上の因子をさらに組み合わせてなる、請求項16記載の剤。
【請求項18】
請求項16または17記載の剤が添加されてなる、Oct4陽性かつVasa陽性の生殖系幹細胞の増幅用培地。
【請求項19】
請求項1〜6のいずれか1項に記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、請求項7記載のOct4陽性かつVasa陽性の生殖系幹細胞、または請求項12〜15のいずれか1項に記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を、該細胞と同種の不妊動物の精巣に移植することを含む、該不妊動物に精子を形成させる方法。
【請求項20】
生殖系幹細胞が不妊動物の体細胞から作製したiPS細胞由来である、請求項19記載の方法。
【請求項21】
不妊動物がヒトまたはマウスである、請求項19または20記載の方法。
【請求項22】
請求項1〜6のいずれか1項に記載の方法により得られたOct4陽性かつVasa陽性の生殖系幹細胞、請求項7記載のOct4陽性かつVasa陽性の生殖系幹細胞、または請求項12〜15のいずれか1項に記載の方法により増幅されたOct4陽性かつVasa陽性の生殖系幹細胞を含有してなる、雄性不妊治療剤。
【請求項23】
精子幹細胞の採取が困難な個体を投与対象とする、請求項22記載の剤。
【図1】

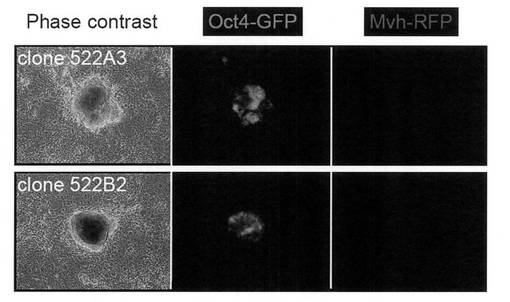
【図2】
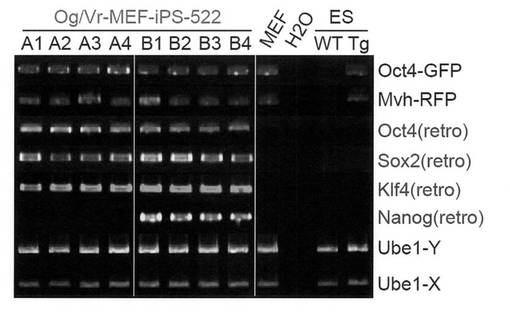
【図3】
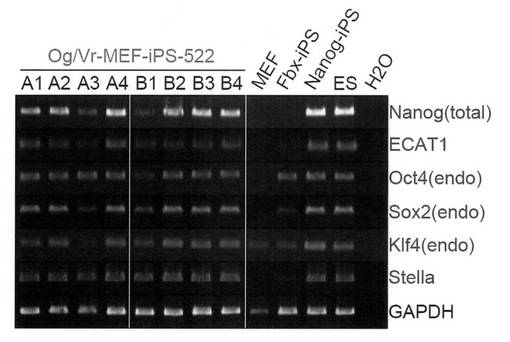
【図4】
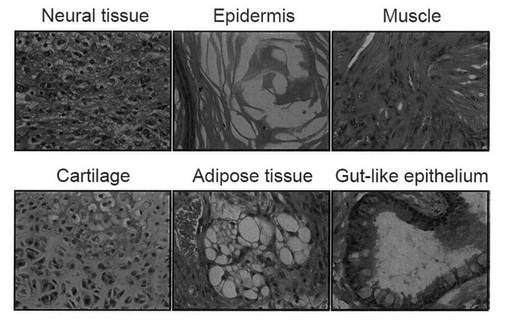
【図5】
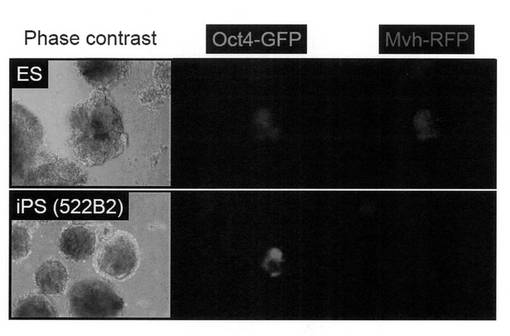
【図6】
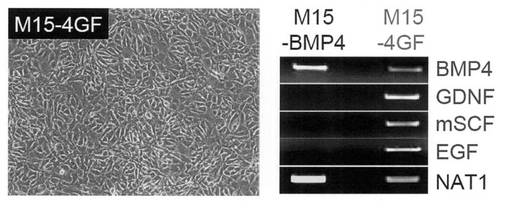
【図7】
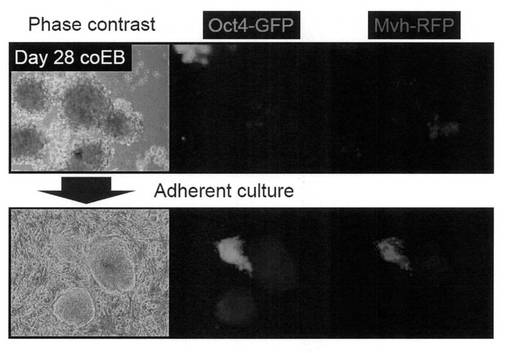
【図8】
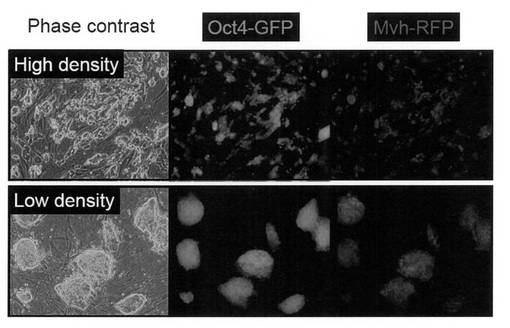
【図9】
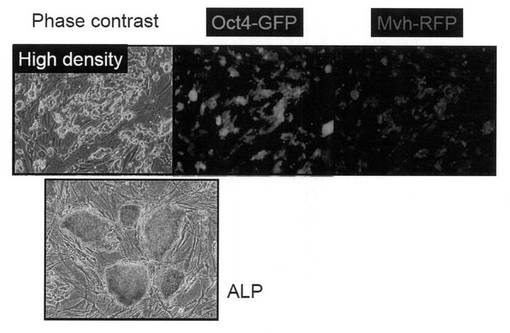
【図10】
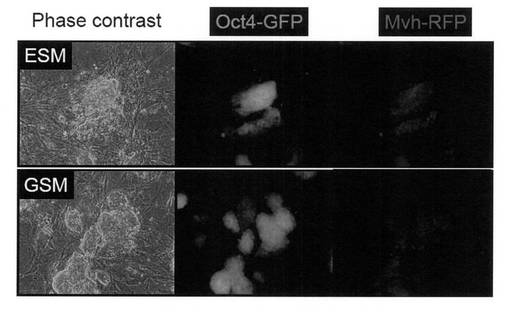
【図11】
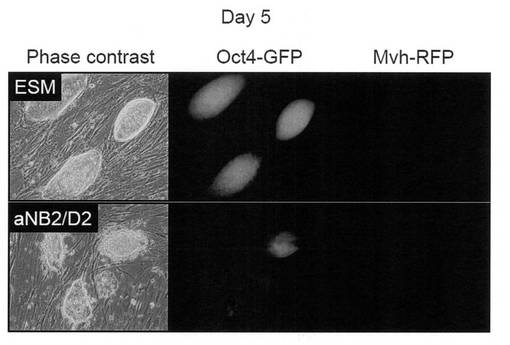
【図12】
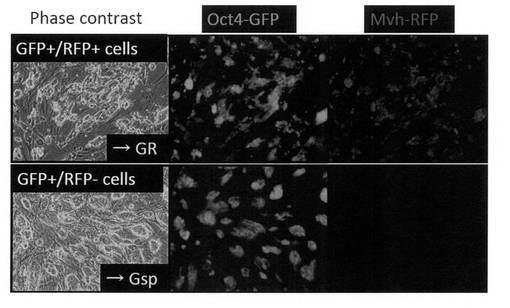
【図13】
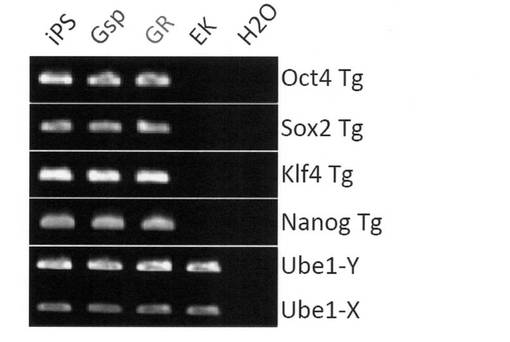
【図14】
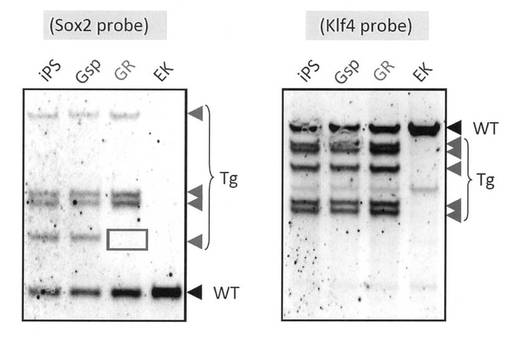
【図15】
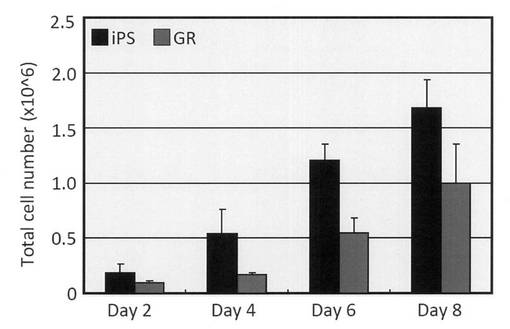
【図16】
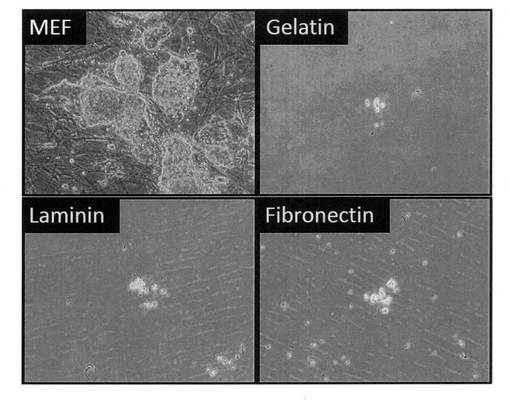
【図17】
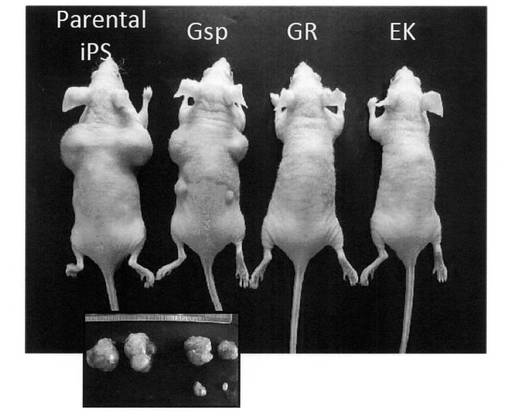
【図18】
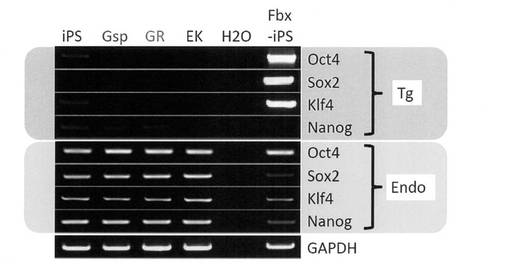
【図19】
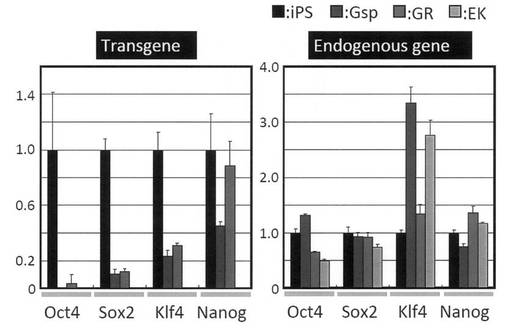
【図20】
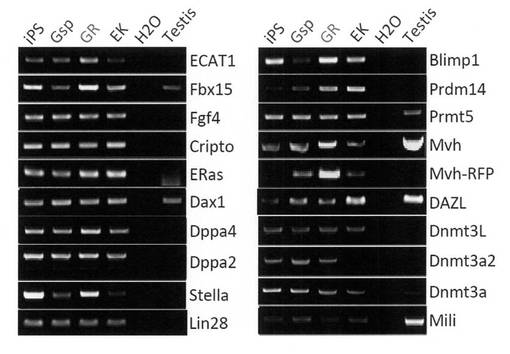
【図21】
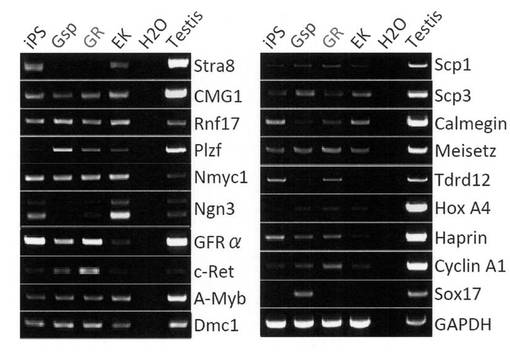
【図22】

【図23】
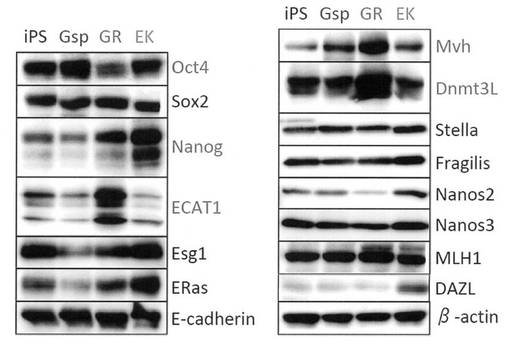
【図24】
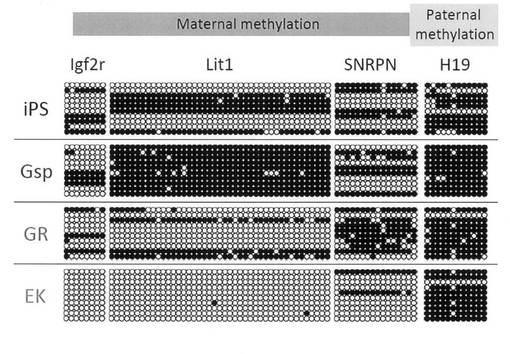
【図25】
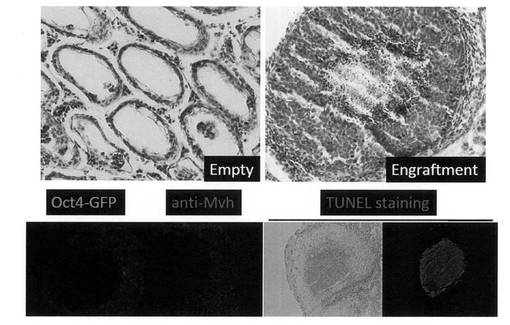
【図26】
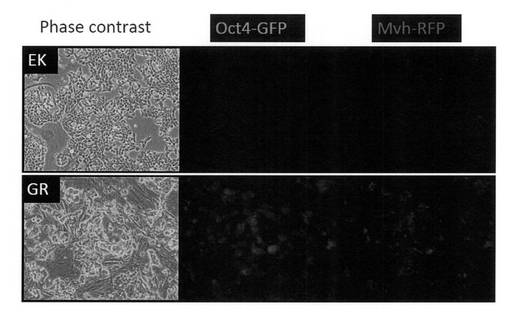
【図27】
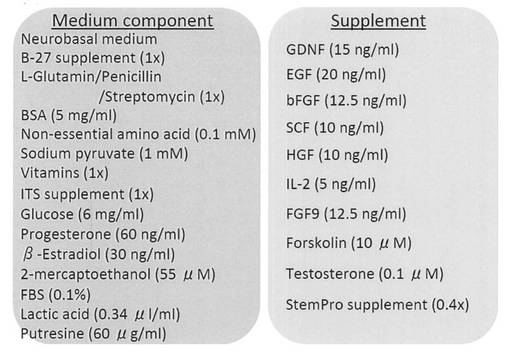
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2011−182720(P2011−182720A)
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願番号】特願2010−52384(P2010−52384)
【出願日】平成22年3月9日(2010.3.9)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【出願人】(504177284)国立大学法人滋賀医科大学 (41)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成23年9月22日(2011.9.22)
【国際特許分類】
【出願日】平成22年3月9日(2010.3.9)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【出願人】(504177284)国立大学法人滋賀医科大学 (41)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]