
生物医学的及び医学的使用のための新規な両親媒性(ampiphilic)フッ素化炭化水素分子ベクター
有効成分のためのベクターとして適用可能な新規の両親媒性(ampiphilic)フッ素化炭化水素分子、その様なベクターを含む活性分子及び薬学的分野における、特に医薬品の製造のためのその使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、有効成分のためのベクターとして使用され得る新規分子、そのようなベクターを含む活性分子、及び薬学的分野における、特に医薬品の調製のためのその使用に関する。
【背景技術】
【0002】
有効成分の送達に関する最近の研究は、外傷性の(traumatic)投与経路を最小にすることによって患者の快適性だけでなく、(通常は欠如しており、しばしば副作用の原因にもなる)細胞親和性を付与することによって医薬品の総体的な有効性を顕著に高める傾向にある。従って、治療的活性の慣習的な考え方に、実際に基質のバイオアベイラビリティーを調節する、作用の特異性の考え方が加えられる。
【0003】
実際のところ、例えば、抗癌化学療法といった細心の注意を払うべき分野(sensitive field)において、ここ数年に亘って観察された進歩(新規な薬物、新規な投与方法、in vitro又はin vivoでのケモプレディクティビティ(chemopredictivity)、新規な治療スキームへの統合等)は、現在から、輸送に関する新規な考え方に基づく新規な道具及び抗癌剤の細胞ターゲッティング(targeting)で装備されることができれば、化学療法の質が進歩し続けるであろうと考えることができる。癌性部位に運ばれるであろう有効成分の有効性及び有効量を増加するため、実際に、これらの医薬品に本当の特異性を付与すること、及びそれらの副作用を軽減することが必要なようである。
【0004】
現在、医薬品の化学構造は、その生理化学的特性及び生物学的特性の両方、ならびに、特に膜受容体に対する親和性を条件付ける。分子構造の修飾によるこの結果の調節は、薬学的特性を変える危険がある。そのため、この生成物を“製剤(galenic)”の観点から処理すること(すなわち、投与に使用される製剤形態に関心を持つこと)、或いは、より良くは、宿主構造中にカプセル化するか、又はこのベクター化(vectorization)を提供することが可能な分子にそれをグラフトすることが導かれる。
【0005】
有効成分のベクター化は、成功するためにいくつかのパラメーターを考慮に入れなくてはならない:
すでに上記に示したように、有効成分は、医薬品又は生物に対して有害なあらゆる相互作用を予防するために、可能な限り、生理学的なメディウムから分離されるべきである。
【0006】
担体は、医薬品のバイオアベイラビリティーを損なうべきではなく、むしろ改善するべきである。言い換えれば、治療剤は、標的分子中に(好ましくは、細胞質内レベルにて、特定の場合は、核内レベルにおいて)放出されるべきであり、その全ての活性をそこに維持すべきである。
【0007】
しかしながら、医薬品(細胞の反応を引き起こすことができ、その結果、欠陥を是正することができる、受容体に特異的な薬剤)のこの構想は、一般的とするにはほど遠い。それは、ホルモン又は鎮痛タイプの有効成分に対して的確である一方、例えば、抗癌治療といった、他の分野において全く適用されない。そのような基質は、この方法によってデザインされていなかった:それらは、いかなる細胞認識特異性も有していない。それらの目的は、細胞増殖を阻害することである。それらは、一般的に抗有糸分裂剤であり、この結果、細胞が癌性か正常かを問わず、全ての細胞のDNAに対して作用し得、そのようにして特定の厄介な状態を生み出す可能性がある(その最も一般的な兆候は、造血組織無形成(hematopoietic tissue aplasia)であり、その次に、免疫阻害及び消化障害を併発する)。化学療法の確立により、開発された有効成分は、ますます強力になってきたが、残念ながら、癌細胞と正常細胞を区別しない。従って、癌治療の観点から、有効性及び選択性を、同一の有効成分上に結合する(conjugated)ことができないことが悔やまれる。
【0008】
既知のこれらの様々な考え及び観察により、本質的にマクロ分子タイプ(合成又は天然ポリマー)及び超分子タイプ(リポソーム)といった、様々なベクターモデルが提案されてきた。全てのこれらのベクターモデルのなかで、特に、有効成分の親水性−親油性バランス(従って、その本質的な物理化学的特性)を調節することができ、その細胞内侵入を促進し及び適切に選択された認識因子(recognition agent)を用いて細胞ターゲッティングを付与することもできる、テロマーと呼ばれる、小さな両親媒性ポリマーの開発について言及する。
【0009】
“Synthesis of new cotelomers derived from tris(hydroxymethyl)aminomethane bearing arabinofuranosylcytosine moieties. Preliminary results on their in vitro and in vivo antitumoral activities” C. Contino, J.C. Maurizis, M. Ollier, M. Rapp, J.M. Lacombe, B. Pucci. Eur. J. Med. Chem., (1998), 33, 809-816.
“Synthesis and preliminary biological assessments of a new class of amphiphilic telomers bearing 5-fluorouracil moieties” C. Contino, J.C. Maurizis and B. Pucci. Macromol. Chem., (1999), 200, 1351-1355.
“A new strategy in biomedical and medical field: the synthesis and applications of telomeric structures”. P. Barthelemy, A. Polidori, B. Pucci. Transworld Research Network, Recent developments in organic chemistry, Trivandrum, (1999), 3, 117-140.
“Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers. Sylvain Jasseron, Christiane Contino-Pepin, Jean Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070.
“Synthesis and preliminary biological assessments of a new class of amphiphilic telomers bearing 5-fluorouracil moieties” C. Contino, J.C. Maurizis and B. Pucci. Macromol. Chem., (1999), 200, 1351-1355.
“Amphiphilic telomers: a new kind of antimitotic drugs macromolecular carriers.” Christiane Contino-Pepin, Jean-Claude Maurizis, Bernard Pucci. Curr. Med. Chem.-Anti-Cancer Agents, (2002), 2, 645-665.
【0010】
これら一連の研究において得られた結果は、様々な主要な点を明示することを可能にした:
基質の親水性−親油性バランスの制御は、その膜貫通通過を促進するが、(“Uptake and subcellular distribution of a new fluorinated telomeric carrier: study on cultivated B16 melanoma and skin rat fibroblastic cells”. F. Chehade, J.C. Maurizis, B. Pucci, A.A. Pavia, M. Ollier, A. Veyre, F. Escaig, C. Jeanguillaume, R. Dennebouy, G. Slodzian, E. Hindie, Cellular and Molecular Biology, (1996), 42, 335-342)界面活性剤、従って毒性の性質の導入を伴わない(“Efficiency of new non ionic telomeric surfactants towards the solubilization of subcellular fractions proteins” B. Pucci, J.C. Maurizis and A.A. Pavia, BioOrg. Med. Chem Lett. (1993), 3, 161-164)。
【0011】
これらの両親媒性ポリマーは、全ての分子、従って有効成分に効果的な細胞ターゲッティングを付与することを可能にする(“Cell targeting by glycosidic telomers - Recognition ability of galactosylated telomers by the yeast Kluyveromyces Bulgaricus” J.Coulon, R. Bonaly, B. Pucci, A. Polidori, P. Barthelemy, C. Contino, Bioconjugate Chem. (1998), 9, 152-159. “Permeability of yeast cell enveloppe to fluorescent galactosylated telomers derived from THAM”. C. Contino, M. Briot, J. Coulon, A. Polidori, R. Bonaly and B. Pucci. Bioconjugate Chem., (2000), 11, 461-468. “Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers”. Sylvain Jasseron, Christiane Contino-Pepin, Jean-Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070)。
【0012】
好適な(細胞質の酵素によって加水分解され得る)ペプチドスペーサーアームを用いてベクター上にグラフトされた有効成分は、ベクターが細胞膜を通過した後に、細胞内レベルにおいて放出される(“Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers”. Sylvain Jasseron, Christiane Contino-Pepin, Jean Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070)。
【0013】
このベクター化の方法は、転移の増殖(proliferation)を阻害し、腫瘍の成長を遅らせ、そして処置されたマウスの生存期間を、コントロールマウスに比べて3倍を超えて延長することから、抗癌剤の効果を極めて顕著に増加することを可能にする。
【0014】
WO 92/02560において記載される、これらのテロマーによってもたらされる明らかな利点にも関わらず、その様なベクターが直面し、商品化及びその使用を困難にし得る主要な問題の1つは、多分散性、すなわち、明確な質量及び構造の欠如である。
【発明の開示】
【発明が解決しようとする課題】
【0015】
従って、本出願人は、有効成分のベクターとなり、明確な構造を有し、調製が容易であって、有効成分をその標的に送達することを容易にすることが可能な分子のデザイン及び調製を目的として設定する。
【課題を解決するための手段】
【0016】
従って、本発明の主題は、下記式(I):
【0017】
【化1】

【0018】
(式中:
APは、生物学的標的に作用することが可能であり、有効成分のその生物学的標的への送達を促進することが望まれる、有効成分を表し;
xは、0及び1から選択される整数を表す
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、ターゲッティング剤(targeting agent)および可溶化剤(solubilizing agent)から選択される基を表す;に相当する分子である。本発明の目的のため、用語“ターゲッティング剤(targeting agent)”は、以下:式(I)の全ての分子のその標的への輸送を促進する分子又は有効成分APの標的によって認識されることが可能なあらゆる分子を意味することを意図する。用語“可溶化剤(solubilizing agent)”は、式(I)の分子のHLBバランスの調節のための薬剤、特に親水性の薬剤を意味することを意図する。本発明において使用され得るターゲッティング剤のうち、単糖類、糖類のアミノ化誘導体、多糖類、天然又は合成ホルモン、ペプチド、抗体及び一般的に、有効成分APの標的によって認識され得るあらゆる分子が挙げられる。本発明において使用され得る可溶化剤のうち、特に、ポリオール、ポリエーテル、ペプチド及び多糖類が挙げられる。
【0019】
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2又はAA3の1つの側鎖のいずれかへの結合(ダッシュ記号−−−によって示される)を可能にする基
【0020】
【化2】
【0021】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表す;
AP−(X)xと鎖[AA3]a3−[AA2]a2−[AA1]の間のダッシュ記号−−−は、AP−(X)xと残りの分子の結合が、アミノ酸AA1、AA2又はAA3の1つの側鎖を介して、或いは必要に応じて、ペプチド鎖の末端にて生じることを示す。
【0022】
より詳しくは、有効成分は、認識される生物学的活性を有し、官能基−O−CO−、−CO−NH−、NH−CO−NH−、−NH−CO−O−、O−CO−O−、−O−、−S−、
【0023】
【化3】
【0024】
から選択され得る結合を用いてアミノ酸に結合され得る全ての有機分子から選択される。
【0025】
これらの有効成分のうち、特に、抗癌、抗炎症、殺菌、鎮痛、神経弛緩、抗真菌作用を
有するもの、及びフリーラジカルスカベンジャー活性を有する分子が挙げられる。
【0026】
一般に、有効成分は、1〜30の炭素原子、1以上の不飽和、特に1以上の芳香環、及び以下:
【0027】
【化4】
【0028】
から選択される1以上の官能基を含む直鎖、分岐又は環状分子からなり得る。
【0029】
有効成分APは、上記の性質の結合によりアミノ酸AA1、AA2又はAA3の1つの側鎖、或いはペプチド鎖の末端のいずれかに、必要に応じてペプチド鎖X(この場合、X=1)により結合される。
【0030】
2つの基、Y及び−(X)x−APの1つとの結合は、アミノ酸AA1、AA2又はAA3のうち1つの側鎖上において行われる。AP−(X)x−又はYにその側鎖を介して結合したアミノ酸は、それらの側鎖上に酸、アミド、アミン、チオール又はアルコール官能基を含むものから選択される。これらのうち、特に、リシン、アルギニン、オルニチン、アスパラギン酸、グルタミン酸、アスパラギン、グルタミン、セリン、チロシン又はシステインが挙げられ得る。好ましくは、AP−(X)x−又はYにその側鎖を介して結合されるアミノ酸は、以下:アスパラギン酸又はリシンから選択される。
【0031】
スペーサーアームX(存在する場合)は、1つの末端にて、アミノ酸AA1、AA2又はAA3の側鎖又は1つの末端との結合、そして他の末端にて有効成分APとの結合に関与するペプチド鎖からなる。
【0032】
このスペーサーアームは、1〜5アミノ酸、好ましくは1〜3アミノ酸を含む。
【0033】
スペーサーアームX及び/又はペプチド鎖[AA3]a3−[AA2]a2−[AA1]は、有効成分APの標的に対するそれらの親和性について選択され得る。また、それらは、125Iで標識した後、in vivoにおいて式(I)の分子を追跡することを可能にするチロシン残基を含む又はチロシン残基からなり得る。
【0034】
Rは、細胞標的に従って選択される;天然の糖類(特定の組織に存在し、肝臓、骨、ある癌性腫瘍の場合ガラクトース、或いはマクロファージ、心臓の場合マンノース、或いは赤血球の場合シアル酸のいずれかを選択的に認識する特定の膜レクチンのターゲッティング等)、キナーゼ、特定の抗体、特にペプチドをターゲッティングするための天然のホルモン(ステロイド等)又は実際の合成物(メシル酸イマチニブ(ST571、Gleevek(登録商標))等)であり得る。Rは、先の研究がそれに対する認識特異性を示した、あらゆる基質から選択され得る。Rが、単糖又は多糖、或いは親水性ペプチドである場合、加えて、そのIV及びIP投与に必要な水溶性(water−solubility)をこの分子に付与し得る。
【0035】
Rがペプチド鎖である場合、Rは、有利には3〜15のアミノ酸を含有し、さらに有利には3〜10のアミノ酸を含有する。また、125Iで標識した後、in vivoにおいて式(I)の分子を追跡することを可能にする1以上のチロシン残基を含み得る。
【0036】
スペーサーアームXを構成するアミノ酸は、鎖[AA3]a3−[AA2]a2−[AA1]又は基Rを構成するものと同様に、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシン又はバリン等の天然アミノ酸、或いは、ヒドロキシプロリン、ノルロイシン、オルニチン、シトルリン又はシクロヘキシルアラニン等の非天然アミノ酸から選択される。
【0037】
3−アミノプロピオン酸及び4−アミノ酪酸等のΩ−アミノ酸の使用も、想定され得る。
【0038】
Rがペプチドの場合、ペプチド鎖Rは、APの生物学的標的に対して顕著な親和性を有するエピトープ又は抗体断片であり得る。
【0039】
本発明において使用され得るペプチドのうち、例えば:そのαVβ3インテグリンに対する親和性で知られるRGD配列が挙げられる。
【0040】
また、Rは、有効成分APの標的に関する限り、式(I)分子に水への溶解性及び細胞内への侵入を促進する親水性/親油性バランスを付与するために、ポリオール又はポリエーテル、特にポリ(エチレンオキサイド)から選択され得る。
【0041】
Rがポリオールで構成される場合、前記ポリオールは、有利には、4〜16の炭素原子及び4〜16の水酸基を含むアルキル鎖からなる。
【0042】
Rが可溶化単位(solubilization unit)としてポリ(エチレンオキサイド)鎖から構成される場合、前記鎖は、有利には、5〜30のエチレンオキサイド単位を含む。
【0043】
Rは、特に、単糖類、糖類のアミノ化誘導体及び多糖類から選択され得る。
【0044】
本発明において使用され得る単糖類のうち、以下:グルコース、フルクトース、マンノース、ガラクトース及びリボースが挙げられる。糖類のアミノ化誘導体のうち、特に、グルコサミンが挙げられる。本発明において使用され得る多糖類のうち、ラクトース、セロビオース又はマルトース、ラクトビオナミド(lactobionamid)及びスクロースが挙げられる。好ましくは、本発明において使用される多糖鎖は、二糖類(bisacchride)である。
【0045】
鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つへのRの結合は、R上にグラフトされ得る官能基(functionality)に従って以下:エーテル、アミド、カルバメート、チオエーテル、エステル、ウレア、ウレタンの好適な結合を用いて行われる
フッ素化炭化水素ベース鎖は、好ましくは、式A−Y’(式中、Aは、以下:
【0046】
【化5】
【0047】
−NH−、−O−CO−NH−、S及びOから選択される基を表し、ならびにY’は式-(CH2)t-(CF2)rF(式中、r及びtは、12≧r+t≧4である2つの整数を表す)に相当する分子を表し、例えば、以下:
-(CF2)4F; -(CF2)5F; -(CF2)6F; -(CF2)7F; -(CF2)8F; -(CF2)9F; -(CF2)10F; -(CF2)11F; -(CF2)12F; -(CF2)13F; -(CF2)14F; -CH2-(CF2)3F; -CH2-(CF2)4F; -CH2-(CF2)5F; -CH2-(CF2)6F; -CH2-(CF2)7F; -CH2-(CF2)8F; -CH2-(CF2)9F; -CH2-(CF2)10F; -CH2-(CF2)11F; -CH2-(CF2)12F; -CH2-(CF2)13F; -(CH2)2-(CF2)2F; -(CH2)2-(CF2)3F; -(CH2)2-(CF2)4F; -(CH2)2-(CF2)5F; -(CH2)2-(CF2)6F; -(CH2)2-(CF2)7F; -(CH2)2-(CF2)8F; -(CH2)2-(CF2)9F; -(CH2)2-(CF2)10F; -(CH2)2-(CF2)11F; -(CH2)2-(CF2)12F; -(CH2)3-(CF2)1F; ...............-(CH2)11-(CF2)F
等に相当するものから選択される。好ましくは、t≧2である。好ましくは12≧r≧4、より好ましくは10≧r≧6である。
【0048】
また、本発明の主題は、式(II)
【0049】
【化6】
【0050】
(式中、R、AA1、AA2、AA3、a2、a3、Y、X及びxは、上記式(I)と同じ定義を有する)の断片を含む、あらゆる生物学的に活性な分子である。
【0051】
実際に、本発明は、この有効成分のヒト又は動物内への侵入を促進し、そしてこの有効成分がその生物学的標的に到達すること可能とするために、好適な結合により上記のあらゆる性質の有効成分を結合することが可能な式(II)の分子断片を提供する。
【0052】
とりわけ、分子の両親媒性の性質は、膜の通過を促進し、有効成分と関連する特異的な標的の認識のための薬剤の任意的な存在が、この標的への送達を促進する。
【0053】
従って、本発明の主題は、上記式(II)の分子断片の活性薬剤のバイオアベイラビリティーを促進するための使用でもある。
【0054】
式(I)の分子の調製は、いくつかの本発明のバリアントに相当する例によって、以下に示される。より一般的には、ペプチド合成の保護、脱保護及びカップリングの方法が使用され、これらの方法は、当業者に周知であり、特に“The peptides” Gross and Meienhofer, 3 vols, Academic Press, New York, 1979-1981の研究において開示されている。
【0055】
式(I)に相当する分子のうち、本発明に特有の主題の1つは、下記式(Ia):
【0056】
【化7】
【0057】
(式中、Suは、上記定義のように単糖、アミノ化単糖誘導体、多糖、ポリオール又は、必要に応じて、ポリエーテルから選択される基Rのバリアントを表す;
AA1は、その側鎖上に酸、アミン、アルコール又はチオール官能基を有するアミノ酸を表し、その側鎖によって(X)x−AP又はYのいずれかに結合される;AA1は、そのN末端及びC末端を介して、Su、及び(X)x−AP又はYのいずれかに結合される)
に相当する分子から構成される。
【0058】
X、x、AP及びYは、上記式(I)と同じ定義を有する。Yは、AA1のアミノ末端又は酸末端、或いは、任意にその側鎖に結合される。
【0059】
好ましくは、下記の1以上の条件を確認する:
−Suは、単糖又は多糖を表す;
−Xは、少なくとも1つのチロシン残基を含む、天然のペプチドであるスペーサーアームを表す;好ましくは、Xはチロシンを表す;
−AA1は、アルギニン及びリシンから選択されるアミノ酸を表す;
−Yは、−NH−官能基を介してアミノ酸AA1に結合した、5〜23のフッ素原子を含むフッ素化C6−C12炭化水素ベース鎖を表す。
【0060】
式(Ia)の2つの化合物の例は、下記及び実施例において示される:
a)例1:血管新生部位のターゲッティング
血管新生は、前から存在する小静脈から新たな血液微細血管をつくり出すための自然な生物学的プロセスである。それは、通常、成人においては、創傷の治癒、炎症又は排卵周期の間の黄体成長等のある特定の状態でのみ起きる、複合的な現象である。正常な状態では、適当な時間の後、血管新生のプロセスは停止し、これは、促進因子及び抑制因子の正しい調節を示唆する。固形腫瘍増殖、関節リュウマチ、乾癬又は糖尿病性網膜症といった、ある特定の病的状態では、血管新生は、明らかにより制御されていない方法で発達する(“Antiangiogenic agents and their promising potential in combined therapy”, P.A. Burke, S.J. DeNardo, Crit. Rew. In Oncology/Hematology, (2001), 39, 155-171)。30年以上前、J.Folkmanは、固形腫瘍増殖は、血管新生の発達と密接に関連しているという仮説を提唱し、それ以降、極めて多くの研究チームが、この現象に興味を示し、血管新生プロセスを阻害することができる基質の開発を試みてきた(“Tumor angiogenic therapeutic applications” J. Folkman Engl. J. Med.(1971), 285, 1182-1186 and “Tumor angiogenis past, present and the near future”. R.S. Kerbel Carcinogenesis (2000), 21, 505-521)。試験された様々な構造のうち、当初は鎮静剤として妊婦に処方され、四肢形成(tetratogenesis)の問題の原因であるサリドマイドが、血管の成長を妨げるのに極めて有利であることが立証された。行われた研究において広まった考えは、あらかじめヨウ素125−標識チロシン等の放射性単位が付与されたベクター上にサリドマイドをグラフトすることであった。この例において模索された目標は、in vivoにて血管新生部位、従って固形腫瘍を、容易に視覚化し、それらの増殖を妨げることである。
【0061】
【化8】
【0062】
この目標により、中心リシン単位に、第一級酸官能基上のフッ素化炭素鎖、該分子に静脈内又は腹腔内投与に必要な水溶性を付与することが可能なラクトースタイプの単位、ならびに後にヨウ素125で標識され、その上に、あらかじめ3位に反応性の酸官能基が付与されたサリドマイドがグラフトされるチロシンが付与された。
【0063】
本発明の好ましいバリアントによれば、式(Ia)に相当する分子において、有効成分は、血管新生プロセスを妨げることができる分子、特にサリドマイドから選択される。
b)例2:スピン−トラップベクター化
ミトコンドリア細胞症は、様々な疾患を含み、その一般的な共通の特徴は、ミトコンドリアの呼吸鎖における欠陥である。生物においてミトコンドリアが随所に存在することにより、この機能障害は、あらゆる臓器に影響を与え得る。この影響は、分離され得、又は一方で多臓器に作用するもの(plurivisceral)であり、その場合は一般に、神経筋系において優勢を示す。現在のところ、これらの疾患に対する治療は存在せず、“オーファン疾患”の枠内に分類され得る。
【0064】
しかしながら、現在、細胞においてミトコンドリアは、フリーラジカルの産生のための優先的な部位であることから、呼吸鎖における欠陥は、極めて一般的に、フリーラジカルの過剰生産と関連することが明らかであり、その結果は、影響を受けた組織における細胞死を加速する。Dr.P.RustinのチームでNecker病院において行われた最近の研究(“Increased apoptosis in vivo in cells lacking mitochondrial DNA gene expression”, Wang J, Silva JP, Gustafsson C, Rustin P, Larsson NG. Proc Natl Acad Sci USA (2001) (in press))は、一連のヒト培養細胞で、この酸素のフリーラジカルの産生の重要性を示すことを可能にした。これらの細胞培養は、ヒトにおいて知られている様々な呼吸鎖の複合体に影響する、全てのタイプの欠陥を表す。それらは、呼吸鎖に影響する欠陥の観点、ならびにフリーラジカル産生及びその細胞生存に対する結果の観点の両方から特徴付けられた。この細胞の収集は、標的が呼吸鎖欠陥と関連するフリーラジカル反応であるあらゆる分子の効果を研究するための、かけがえのない道具を表す。
【0065】
我々のチームにおける、最近の呼吸鎖によって産生されたフリーラジカルで誘導されたアポトーシスの細胞モデルにおける細胞死を妨げることができる“スピン−トラップ”分子の同定は、さらに増強された効果をあらわす同様の分子の開発の基礎を我々に与えた(“Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA”. Geromel V, Kadhom N, Ceballos-Picot I, Ouari O, Polidori A, Munnich A, Roetig A, Rustin P. Hum Mol Genet (2001) (in press))。ここで追跡された目標は、これらの基質の構造を精緻化し簡略化する一方、同時に、工業段階に容易に適用し得る合成プロセスを開発するために、それらの生物学的活性を保存することであった。以下:ミトコンドリアの呼吸鎖において欠陥をあらわす細胞培養(培養線維芽細胞)、フリーラジカルの作用を受けたニューロン/筋細胞共培養及び、最後に、3度の熱傷を受けた皮膚から抽出された細胞の、様々な細胞モデルに対して試験が行われた。
【0066】
行われた調査の目的は、アポトーシスの現象、さらに一般的に、フリーラジカルの過剰産生に起因する細胞死の現象を処置するために臨床的に使用され得るフリーラジカルトラップを得ることであった。これらの細胞のタイプに関して得られた大いに希望を与える結果は、これらの両親媒性ベクターモデルの開発を充分に保証する。
【0067】
前の分子上に構築された分子Eに、この特定の場合において、周知であって、PBNの誘導体を含む効果的なスピントラップが付与される。
【0068】
【化9】
【0069】
最初の試験は、Necker病院にて、in vitroで、NARP突然変異を示す子供からの皮膚生検由来の線維芽細胞に対して行われた。製品TA1PBN(“Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA”, Geromel V, Kadhom N, Ceballos-Picot I, Ouari O, Polidori A, Munnich A, Roetig A, Rustin P. Hum Mol Genet(2001) (in press) and “Synthesis of a glycolipidic amphiphile nitrone as a new spin trap for biological applications”), O. Ouari, A. Polidori, F. Chalier, P. Tordo, B. Pucci. J. Org. Chem., (1994), 64, 3554-3556)に対して同様の方法であらかじめ試験した場合、分子Eは、細胞保護能力を示し、アポトーシスのプロセスを阻害する。このタイプの製品に関し、培養されたいかなる細胞に対しても、毒性は測定されなかった。
【0070】
これらの結果は、その様なベクター化の概念の利点を、再度立証し、全く異なる適用分野における、その潜在的可能性を明確に示す。
【0071】
本発明の他の好ましいバリアントによれば、式(Ia)に相当する分子において、有効成分は、フリーラジカルスカベンジャー、特にN−ベンジリデン−tert−ブチルアミンオキサイド誘導体から選択される。
【0072】
式(II)に相当する分子のうち、本発明の他の特定の主題は、式(Ib)に相当する分子から構成される:
Pep−[AA1]−Y
(Ib)
式中、Y及びAA1は、上記式(I)、特に式(Ia)と同様に定義され、RのバリアントであるPepは、2〜10、好ましくは4〜6アミノ酸を含むペプチド鎖を表す。有利には、Pep又はAA1は、少なくとも1つのチロシン単位を含む。
【0073】
有利には、Pepは、既知の生物学的標的に対する親和性について選択され;特に、このペプチド鎖は、αVβ3インテグリンによって認識されることが知られているRGD(アルギニン−グリシン−アスパラギン酸)配列を含み得る。
【0074】
本発明の他の主題は、式(Ic)に相当する分子から構成される:
【0075】
【化10】
【0076】
式中、x、X、AP、AA1及びYは、上記式(I)と同様に定義され;特に、分子中、x、X、AP、AA1及びYは、上記式(Ia)と同様に定義される;Pepは、上記式(Ib)と同様に定義される。
【0077】
好ましくは、1以上の以下の条件を確認する:
−Pepは、αVβ3インテグリンによって確認されるペプチドであり、APは抗有糸分裂剤である;
−X、Pep又はAA1は、少なくとも1つのチロシン残基を含む;
−Xは、1〜3アミノ酸の鎖を表す。
【0078】
式(Ib)及び(Ic)の化合物の例は、下記及び実験の部に示される。
c)例3:抗癌治療
現在、腫瘍細胞と正常細胞の有効な区別、従って、適切なターゲッティングのための基準を示すことが可能な技術はない。しかしながら、知られているように、癌性腫瘍の増殖は、脈管化率(rate of vascularization)、従って、それに伴う血管新生の現象と密接に結びついている。これらの観察は、知られているように、血管新生を阻害することができ、従って、この方法によって、腫瘍の増殖を妨げることができる基質を、科学団体に開発させた。実際、現在は、血管新生細胞によって輸送される膜タンパク質(インテグリンと呼ばれる)は、これらの細胞の増殖のプロセスに活発に加わっていることが、一般的に認められている。より詳しくは、αVβ3インテグリンは、特定のペプチド配列(RGD(アルギニン−グリシン−アスパラギン酸)配列)を認識する。従って、この単位の提案されたベクター上へのグラフトは、特異的に血管新生部位、従って、最終的には腫瘍部位をターゲッティングする能力をそれに付与するはずである。この同じベクターに、抗有糸分裂剤を付加することは、癌性細胞の選択的な破壊を可能にするはずである。この予想において、分子Bは、最初、ベクターが無害であることを確認するため、及びその特異性を測定するために調製された。ターゲッティング薬剤により大きな自由度を付与するため、前記ターゲッティング薬剤を、中心リシン上に、疎水性アミノ酸であるセリンを用いてグラフトした。この分子は、水に可溶であり、両親媒性の性質である。
【0079】
【化11】
【0080】
これらの肯定的な結果を考慮すれば、さらに、RGDペプチド配列及びメルファラン(分子D)又はAra−C(分子C及びF)等の抗有糸分裂剤を有するベクターは、その後、合成され、それらの抗癌活性についての解析の過程にある。
【0081】
【化12】
【0082】
選択された抗有糸分裂剤は、ここでは単にモデルであって、与えられた抗有糸分裂剤を導入すること及び輸送することの利便性を簡単に説明するに過ぎない。このタイプのベクターは、アドリアマイシン(この特定の場合において、分子(Ic)は、X又はPepのいずれかにおいて、Gly−Phe−Leu−Glyタイプのペプチド断片を含む)、5−Fu(5−フルオロウラシル)、メルファラン、又はメシル酸イマチニブ(STI573、Glivec(登録商標))等のチロシンキナーゼ阻害剤等(例えば、より一般的にはこれらの担体上にグラフトされ得るあらゆる抗癌剤)基質のベクター化のための薬剤としても使用され得、また使用されるであろう。疎水性フッ素化炭化水素鎖の存在は、膜貫通性の通過を促進する。有効成分の放出は、適切な細胞質酵素によるペプチド結合の加水分解によって提供される。
【0083】
すでに得られた、最初のin vitroの結果は、完全にこの概念を立証する。
【0084】
本発明の主題は、上記式(I)に相当する化合物の医薬品を調製するための使用でもある。
【0085】
実際、本発明による式(I)に相当する化合物は、従来技術の化合物のものよりも大きいか、又は同等の生物学的標的に到達するためのバイオアベイラビリティー及び能力を有することが示されている。
【0086】
この特性は、多様な分野における本発明の分子の使用を想定することを可能にする:
−治療分野において、本発明のプロダクトは、全て種類の病状、特に様々な形態の癌、ならびに酸化ストレス及び含酸素フリーラジカル種の形成に関連する病状の予防及び/又は治療に使用され得る。
【0087】
このため、本発明の主題は、薬学的に活性な担体中に本発明のよる化合物を含有する薬学的組成物である。
【0088】
本発明の主題は、癌の予防及び/又は治療を意図した薬学的組成物の調製のための、式A、C、D又はFの化合物の使用でもある。
【0089】
本発明の主題は、癌性細胞の存在を検出することを意図した薬学的組成物を調製するための式Bの化合物の使用でもある。
【0090】
本発明の主題は、酸化ストレス及び含酸素フリーラジカル種の形成に関連する病状、特に、免疫及び炎症性疾患、虚血再灌流症候群、動脈硬化、アルツハイマー病、パーキンソン病、UV及び電離放射線による損傷、メラノーマ等の特定の癌及び細胞老化を予防及び/又は治療することを意図した薬学的組成物の調製のための、式Eの化合物の使用でもある。
【0091】
本発明のプロダクトは、当業者にとって公知のあらゆる経路、特に、静脈注射又は筋肉注射、或いは経口又は経皮投与によって投与され得る。それらは、単独又は他の活性な薬剤と組み合わせて使用され得る。その用量及び1日投与量は、関係する分子について測定された活性及び患者の体重に従って適応される;
−化粧品分野において、式Eの化合物は、老化の影響を予防及び/又は処置するために使用され得る。
【0092】
従って、本発明の主題は、美容上(cosmetically)許容される担体中に式Eの化合物を含有する化粧品組成物でもある。
【0093】
前記組成物は、皮膚又は外皮(爪、髪)への適用のためのものであってもよい。
【0094】
それは、水性又は油性溶液、油中水型乳剤又は水中油型乳剤、トリプル乳剤又は軟膏の形態であってもよい。
【0095】
本発明の化合物は、フリーラジカルスカベンジャー活性が望まれる、以下:スキンケアクリーム、日焼け止め(antisun)製品、化粧落とし製品、皮膚又は髪用パック、シャンプー、口紅、ほお紅、ファンデーション、マニキュア液等の化粧品等のあらゆる化粧品組成物中に取り入れられ得る
それらの多様なメディウムへの溶解性のため、本発明の化合物は、使用が容易であり、様々な条件下で採用され得る。
【発明を実施するための最良の形態】
【0096】
1/実施例1:
A−分子Aの調製
30mlの無水メタノール中に溶解された、2g(4mmol)の1H,1H,2H,2H−ペルフルオロデカンアジド化合物を、パラジウム−炭の存在下にて水素化に供する。4時間の反応の後、メディウムをセライト(celite)を通して濾過し、溶媒を減圧下にて蒸発させる。相当する(corresponding)アミン2は、精製せずに単離される(定量的収率)。
【0097】
化合物2を、30mlのジクロロメタン中で、2.2g(4mmol)のBoc−Lys−(Z)−OPhF5 3.の存在下で、再度反応させる。溶液のpHを、数滴のDIEAを添加することによって8にする。
【0098】
周囲温度にて16時間攪拌した後、反応メディウムを、減圧下にて濃縮する。
【0099】
このメディウムを、シリカゲルカラムクロマトグラフィー(溶離液:3/7酢酸エチル/シクロヘキサン)によって精製する。酢酸エチル/ヘキサン混合溶液からの結晶化によって、フッ素化化合物4(2.68g;3.24mmol;80%)を、白色粉末の形態で得る。
【0100】
【化13】
【0101】
rf: 5/5 シクロヘキサン/酢酸エチル中に0.36
[α]D = -8.2 (c: 1; CHCl3).
融点: 86.5-88.3°C.
1H NMR (250 MHz, CDCl3): δ 7.36 (5H, s, CH arom), 6.85 (1H, m, NH アミド), 5.27 (1H, d, J = 6.65 Hz, NH ウレタン), 5.11 (2H, s, CH2O), 4.99 (1H, t, J = 5.8 Hz, NH ウレタン), 4.06 (1H, m, CHCO), 3.58 (2H, dd, J = 6.5 Hz, CH2NH), 3.20 (2H, dd, J = 6.2 Hz, CH2NH), 2.35 (2H, m, CH2CF2), 2.0〜1.0 (15H, m, Boc由来CH3 及び CH2 Lys).
13C NMR (62.86 MHz, CDCl3): δ 172.5 (CONH), 156.7; 155.9 (OCONH), 136.6 (CIV arom.), 128.5; 128.1 (CH arom.), 80.4 (CIV), 66.7 (CH2OCONH), 54.4 (CHCO), 40.2 (CH2NH), 31.9 (CH2NH), 31.3 (CH2), 30.7 (CH2Rf), 29.5 (CH2), 28.2 (tert-ブチル由来CH3), 22.4 (CH2).
19F NMR (235 MHz, CDCl3): δ -80.7 (3F, s, CF3), -113.9 (2F, s, CF2CH2), -121.9 (6F, s, (CF2)3), -122.7 (2F, s, CF2), -123.5 (2F, s, CF2), -126.6 (2F, s, CF2CF3).
【0102】
30mlのジオキサン中に溶解された0.5g(0.6mmol)の化合物4を、パラジウム−炭の存在下において水素化に供する。
【0103】
15時間の反応の後、メディウムを、セライトを通して濾過し、溶媒を減圧下にて蒸発させる。得られたアミン5を、0.21g(0.44mmol)の新しく調製されたラクトビオノラクトン(lactobionolacton)の存在下でジクロロメタン中にて反応させ、そしてメディウムに、この溶液のpHを8にするためにDIEAを添加する。
【0104】
アミン5が完全になくなった後(TLC)、反応メディウムを、減圧下にて濃縮する。低温条件下にて、反応粗生成物に40mlの1:1無水酢酸/ピリジン混合溶液を加える。攪拌は、周囲温度にて18時間維持され、その後、反応混合溶液を150mlの1N HCl上に注ぐ。水相を、50mlのジクロロメタンで3回抽出する。有機相を、60mlの1N HCl、その後60mlの塩水で、それぞれ2回洗浄し、最後に、Na2SO4上で乾燥する。この溶媒を、減圧下にて除去し、粗生成物を、シリカゲル上のフラッシュ・クロマトグラフィー(溶離液:6/4その後7/3 酢酸エチル/シクロヘキサン)によって精製して白色粉末の形態で化合物6(0.55g;0.39mmol;65%)を生成する。
【0105】
【化14】
【0106】
rf: 6/4 酢酸エチル/シクロヘキサン中に0.22
[α]D = +2.9 (c, 1; CHCl3).
融点: 65°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.07 (2H, m, NH), 7.34 (5H, s, CH arom.), 7.01 (1H, m, NH), 5.47 (1H, m, 糖由来H), 5.30 〜 5.10 (2H, m, 糖由来H), 5.02 〜 4.79 (5H, m, CH2-O 及び糖由来H), 4.50 〜 3.90 (8H, m, 糖由来H及びリシン由来CHα), 3.38 (2H, m, CH2-NH), 2.97 (2H, m, CH2-NH), 2.30 (2H, m, CH2-CF2), 2.14, 2.09, 2.04, 2.01, 1.96, 1.92 (24H, 6s, アセチル由来CH3), 1.65 〜 1.10 (6H, m, リシン由来CH2)
13C NMR (62.86 MHz, CDCl3): δ 171.3 (CO-NH), 170.5, 170.5, 170.1, 170.0, 170.0, 169.7, 169.2 (7s, CO-O), 167.9 (CO-NH), 156.7 (O-CO-NH), 136.7 (CIVarom.), 128.4, 128.0, 127.9 (CH arom.), 101.6 (CH-1’), 77.9 (CH-4), 72.7 (CH-2), 71.1, 70.9 (CH-5’ 及び CH-3’), 70.0 (CH-5), 69.4 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 66.5 (CH2-O), 61.6, 61.0 (CH2-6及び CH2-6’), 52.6 (CH-CO), 40.4 (CH2-NH), 31.9 (CH2-NH), 31.2 (CH2-), 30.5 (CH2-Rf), 29.1 (CH2-), 22.2 (CH2-), 20.6, 20.5, 20.4, 20.4, 20.3 (アセチル由来CH3)
19F NMR (235 MHz, DMSO-d6): δ -80.2 (3F, s, CF3), -113.0 (2F, s, CF2-CH2), -121.4 (6F, s, 3CF2), -122.2 (2F, s, CF2), -123.0 (2F, s, CF2), -125.4 (2F, s, CF2-CH2).
【0107】
化合物6のベンジルオキシカルボニル基を、化合物4から化合物5に通過(pass)する際にすでに記載された実験方法に従って、脱保護する。0.5g(0.36mmol)の化合物6を用い、アミン7を、定量的収率で得る。
【0108】
得られたアミン7を、0.21(0.44mmol)のZ−Tyr−OPhF5(化合物8)の存在下で30mlのジクロロメタン中にて反応させ、溶液のpHを8にするためにDIEAを添加する。
【0109】
アミン7が完全になくなった後(TLC)、反応メディウムを減圧下で濃縮し、シリカゲルカラムクロマトグラフィー(溶離液:7 酢酸エチル/3 シクロヘキサン)によって精製する。化合物9(0.32g;0.21mmol;58%)を、白色粉末の形態で得る。
【0110】
【化15】
【0111】
rf: 7 酢酸エチル/3 シクロヘキサン中に0.35
[α]D = +1.5 (c: 1; CHCl3).
融点: 37.6°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.01 (2H, m, NH), 7.71 (1H, m, NH), 7.21 (5H, s, CH arom.), 7.05 (3H, m, NH, 2H arom tyr), 6.89 (2H, d, J = 8.29 Hz 2H arom tyr), 5.22 (1H, d, J = 5.35 Hz糖由来H), 5.02 (2H, s, CH2Ph), 4.99 (2H, s, CH2Ph), 4.92 (2H, m, 糖由来H), 4.70 〜 4.55 (5H, m, 糖由来H), 4.01 〜 3.75 (9H, m, CHα tyr, CHα lys, CH2NH, 糖由来5H), 2.76 (2H, m, CH2-NH), 2.16 (2H, m, CH2-CF2), 1.90 〜 1.68 (24H, 6s, アセチル由来CH3), 1.29 〜 0.91 (6H, m, リシン由来CH2)
13C NMR (62.86 MHz, CDCl3): δ 171.85; 171.48 (CO-NH), 170.67, 170.32, 170.18, 170.0, 169.63; 169.42 (6s, CO-O), 156.29 (CO-NH), 137.47; 136.61; 135.54 (CIV arom.), 130.71; 129.03; 128.80; 128.71; 128.12; 127.96; 121.17 (CH arom.), 101.07 (CH-1’), 78.69 (CH-4), 72.22 (CH-2), 70.86; 70.21 (CH-5’ 及び CH-3’), 70.06 (CH-5), 69.61 (CH-3), 69.3 (CH-2’), 67.60 (CH-4’), 65.69 (CH2-O), 61.72, 61.48 (CH2-6 及び CH2-6’), 56.62 (CHαtyr); 52.68 (CHαlys); 37.48 (CH2-NH), 31.86 (CH2-NH), 31.43 (CH2-), 30.08 (CH2-Rf), 29.09 (CH2-), 22.77 (CH2-), 21.08, 21.03, 20.94, 20.86, 20.76 (アセチル由来CH3)
19F NMR (235 MHz, DMSO-d6): δ -80.19 (3F, s, CF3), -113.38 (2F, s, CF2-CH2), -121.67 (6F, s, 3CF2), -122.45 (2F, s, CF2), -123.24 (2F, s, CF2), -125.70 (2F, s, CF2-CH2).
【0112】
同じように、すでに記載された実験方法に従って、30mlのエタノールに溶解された0.3g(0.19mmol)の化合物9を、パラジウム−炭の存在下にて 水素化に供する。
【0113】
得られたアミン10を、0.107g(0.23mmol)のサリドマイドの活性エステル11の存在下にてジクロロメタン中で反応させ、溶液のpHを8にするためにDIEAを添加する。
【0114】
【化16】
【0115】
Rf= 7EtOAc/3シクロヘキサン中に0.65
[αD] = +3.9 (c: 1; DMF).
1H NMR (250 MHz, CDCl3): 8.63 (1H, s, Ph); 8.61 (1H, d, Ph); 8.09 (1H, d, Ph); 5.04 (1H, m, NCH); 2.86 (3H, m, CH2CO, CHCH2CO); 2.19 (2H, m, CHCH2).
19F NMR (235 MHz, CDCl3) : -152.6 (2F, d, CF); -156.67 (1F, t, CF); -161.87 (2F, t, CF).
13C NMR (62.86, DMSO): 177.97; 174.93; 171.64; 171.61; 170.96 (5 CO); 142.01; 140.94; 139.68; 136.89; 129.09; 128.64 (芳香族 C); 54.44 (NCH); 36.13 (CH2CO); 27.12 (NCHCH2) 11
【0116】
アミン10が完全になくなった後(TLC)、反応メディウムを、減圧下にて濃縮し、シリカゲルカラムクロマトグラフィー(溶離液:8/2の後9/1 酢酸エチル/シクロヘキサン)によって精製する。
【0117】
化合物12を、極めて低い収率(11mg;6.4μmol;4.5%)で得る。
【0118】
rf: 酢酸エチル中に0.63
該分子の糖部分の脱アセチル化を、触媒量のナトリウムメトキシドを含有するメタノール中で周囲温度にて行う。
【0119】
H+樹脂(amberlite IRC 50)上での処理、濾過及び溶媒の蒸発の後、脱アセチル化された生成物Aを、定量的収率で単離する。
【0120】
【化17】
【0121】
B−生物学的アッセイ
そのような基質は、線維芽細胞及びB16メラノーマの細胞培養物に対して、完全に許容できない毒性を示さない。In vivoにおいて、この分子は、腫瘍のストローマ(stroma)中に濃縮され、それを極めてはっきりと視覚化することを可能にする(表1参照)。
【0122】
【表1】
【0123】
現在進行中の研究は、サリドマイド単独と比べ、腫瘍成長の阻害におけるその有効性を明示することを可能にするであろう。ニワトリの胚に対する血管成長アッセイ(ニワトリ大動脈輪アッセイ(chick aortic ring assays))において得られた最初の結果は、微小血管の成長の阻害におけるこの構造の有効性を示した。分子Aは、20μMにて有効であり、200μMにて完全に成長を妨げることが見出されたのに対し、その様な濃度において、サリドマイドは、効果がないことが見出される。
【0124】
この最初の結果は、提案された構造の全面的な無害、及び診断のための可能性のある有利な点を示す(ここで、サリドマイドは、有効成分の1つの例にすぎない)。
【0125】
2/実施例2:分子Eの調製
この合成例は、図1に示される。
活性エステルHOOCPBNの合成
【0126】
【化18】
【0127】
N−(tert−ブチル)ヒドロキシルアミンアセテートを、飽和炭酸ナトリウム水溶液中に溶解する。ヒドロキシルアミンを、エーテルで抽出する。有機相を、Na2SO4上で乾燥し、その後、この溶媒を減圧下にて除去して粉体白色結晶の形態で遊離N−tert−ブチルヒドロキシルアミンを生成する。
【0128】
1.00gの4−カルボキシベンズアルデヒド(6.67mmol−1当量)及びスパチュラの先端の4Åモレキュラーシーブを、アルゴン雰囲気下にて5mlの脱気無水エタノール中に懸濁する。5mlのエタノール中の0.570gのヒドロキシルアミン(6.37mmol−0.95当量)溶液を、ベンズアルデヒド溶液に添加し、そしてこのメディウムを、暗所で60℃にする。18時間後、0.200gのヒドロキシルアミン(2.25mmol−0.33当量)を、このメディウムに加え、さらに18時間攪拌を続ける。反応混合溶液を、セライトの層を通して濾過し、減圧下にて溶媒を除去し、そして粗生成物を、シリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4 酢酸エチル/シクロヘキサン)によって精製する。メタノール/エーテル混合溶液からの再結晶後、ニトロンX(0.590g−2.67mmol−40%)を、白色粉末の形態で得る。
【0129】
モル質量 (C12H15NO3): 221.3 g・mol-1
融点: 214.5 - 215.7°C.
1H NMR (250 MHz, DMSO-d6): δ 8.42 (2H, m, J = 8.5 Hz, H arom.), 7.95 (3H, m, H arom. 及び CH=N(O)), 1.51 (9H, s, tert−ブチル由来CH3).
13C NMR (62.86 MHz, DMSO-d6): δ 166.9 (CO), 135.3 (CIVarom), 131.0 (CH=N(O)), 129.2 (CH arom.), 128.3 (CIV arom), 127.9 (CH arom.), 71.1 (CIV), 27.8 (tert−ブチル由来CH3).
UV (MeOH, nm): λmax = 287.
【0130】
0.260gのニトロンX(1.18mmol−1当量)を、アルゴン雰囲気下にて15mlのジオキサン中に溶解する。0.290gのDCC(1.41mmol−1.2当量)及び0.16gのHOSu(1.41mmol−1.2当量)を、反応メディウムに添加する。48時間の攪拌後、この反応メディウムを、焼結ガラス(ポロシティ 4)を通して濾過し、この溶媒を、その後、減圧下にて除去する。シリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4 酢酸エチル/シクロヘキサン)による精製後の、酢酸エチル/ヘキサンの混合溶液からの再結晶の後、化合物Y(0.25g−0.79mol− 67%)を、白色粉末の形態で得る。
【0131】
モル質量 (C16H18N2O5): 318.3 g・mol-1
融点: 177.4 - 178.3°C.
1H NMR (250 MHz, CDCl3): δ 8.37 (2H, m, J = 8.6 Hz, H arom.), 8.12 (2H, d, J = 8.6 Hz, H arom.), 7.65 (1H, s, CH=N(O)), 2.89 (4H, s, CH2-CO), 1.61 (9H, s, tert−ブチル由来CH3).
13C NMR (62.86 MHz, CDCl3): δ 169.3, 161.3 (CO), 136.8 (CIVarom), 130.7 (CH arom.), 128.6 (CIV arom), 128.6 (CH arom.), 125.5 (CH=N(O)), 72.2 (CIV), 28.4 (CH2-CO), 25.7 (tert−ブチル由来CH3).
【0132】
[5−tert−ブトキシカルボニルアミノ−5−(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10−ヘプタデカフルオロデシルカルバモイル)ペンチル]カルバミン酸ベンジルエステル3の合成
【0133】
【化19】
【0134】
2.26gのアジドC8F17CH2CH2N3(4.62mmol−1当量)を、20mlのエーテル中に溶解する。このメディウムを0℃にし、300mgのパラジウム−炭(10% − 65mg/mmol)を、フラクションごとに添加する。水素化ボンベ(圧力8バール)において6時間の攪拌の後、このメディウムをセライトの層を通して濾過し、溶媒を、減圧下にて除去する。アミンC8F17CH2CH2NH2 2を精製せずに得る。
【0135】
1.79gのBoc−Lys(Z)OH(4.70mmol−1当量)、1.07gのDCC(5.18mmol − 1.1当量)及び0.64gのHOBt(4.70mmol − 1当量)を、20mlの無水ジクロロメタン中に溶解する。15分の攪拌後、10mlの無水ジクロロメタン中のアミン2の溶液を、この混合溶液に添加する。攪拌を24時間続ける。反応粗生成物を、焼結ガラス(ポロシティ4)を通して濾過し、その後、溶媒を減圧下にて除去した。シリカゲル上のフラッシュクロマトグラフィー(溶離液:7:3〜6:4 シクロヘキサン/酢酸エチル)による精製の後、フッ素化化合物3(3.11g − 3.70mmol − 79%)を、白色粉末の形態で得る。
【0136】
モル質量 (C29H32F17N3O5): 825.6 g・mol-1
融点: 86.5 - 88.3°C.
1H NMR (250 MHz, CDCl3): δ 7.36 (5H, s, CH arom.), 6.85 (1H, m, NH アミド), 5.27 (1H, d, J = 6.65 Hz, NH ウレタン), 5.11 (2H, s, CH2-O), 4.99 (1H, t, J = 5.8 Hz, NH ウレタン), 4.06 (1H, m, CH-CO), 3.58 (2H, dd, J = 6.5 Hz, CH2-NH), 3.20 (2H, dd, J = 6.2 Hz, CH2-NH), 2.35 (2H, m, CH2-CF2), 2.0 〜 1.0 (15 H, m, Boc由来CH3及びCH2 lys).
13C NMR (62.86 MHz, CDCl3): δ 172.5 (CO-NH), 156.7, 155.9 (O-CO-NH), 136.6 (CIV arom.), 128.5, 128.1 (CH arom.), 80.4 (CIV), 66.7 (CH2-O-CO-NH), 54.4 (CH-CO), 40.2 (CH2-NH), 31.9 (トリプレット, CH2-NH), 31.3 (CH2), 30.7 (CH2-Rf), 29.5 (CH2), 28.2 (tert−ブチル由来CH3), 22.4 (CH2).
19F NMR (235 MHz, DMSO-d6): δ -80.7 (CF3, s), -113.9 (CF2-CF3, s), -121.9 (3 CF2, m), -122.7 (CF2, s), -123.5 (CF2, s), 126.0 (CF2-CH2, s).
[α]D = -8.2 (c, l, CHCl3).
【0137】
ラクトLys(Z)C8F17(OAc)8 5の合成
【0138】
【化20】
【0139】
2.03gの化合物3(2.45mmol − 1当量)を、20mlの無水ジクロロメタン中に溶解する。このメディウムを0℃にし、40mlの8.5:1.5 CH2Cl2/TFA混合溶液を、添加の間温度を0℃に保持しながら滴下する。4時間の攪拌の後、溶媒を減圧下にて除去する。粗生成物をエーテル中に回収し、その後蒸発させて、共蒸発(co−evaporation)によってTFAの残存トレースを除去する。この操作を数回繰り返し、遊離アミン4を生成する。
【0140】
並行して、1.15gのラクトビオン酸(3.19mmol − 1.3当量)を、3滴のTFAで酸性にした40mlの1:1 メトキシエタノール/トルエン混合溶液中に懸濁する。
【0141】
減圧下にて45℃での蒸発の後、メディウムを、30mlの2:1 メトキシエタノール/トルエン混合溶液中に回収し、その後、乾燥するまで蒸発させる。ラクトビオノラクトンを生成するように、後者の操作を2度繰り返す。
【0142】
ラクトビオノラクトン及びアミン4を、アルゴン雰囲気下で、40mlのメタノール中に溶解する。この溶液のpHを、TEAを添加することによって9にし、その後、メディウムを24時間の還流にかける。減圧下でのメタノールの除去後、40mlの1:1 無水酢酸/ピリジン混合溶液を、冷却条件下で、粗生成物に添加する。攪拌を18時間の間維持し、その後、反応混合溶液を150mlの1N HClに注ぐ。水相を、50mlのジクロロメタンで3回抽出する。有機相を、60mlの1N HClで2回、その後60mlの塩水でそれぞれ洗浄し、そして最後に、Na2SO4上で乾燥する。溶媒を減圧下にて除去し、粗生成物をシリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4〜7:3 酢酸エチル/シクロヘキサン)によって精製して化合物5(2.23g − 1.59mmol − 65%)を白色粉末の形態で生成する。
【0143】
モル質量 (C52H60F17N3O22): 1402.0 g・mol-1
融点: 65°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.07 (2H, m, NH), 7.34 (5H, s, CH arom.), 7.01 (1H, m, NH), 5.47 (1H, m, 糖由来H), 5.30 〜 5.10 (2H, m, 糖由来H), 5.02 〜 4.79 (5H, m, CH2-O 及び糖由来 H), 4.50 〜 3.90 (8H, m, 糖由来H及びリシン由来CH), 3.38 (2H, m, CH2-NH), 2.97 (2H, m, CH2-NH), 2.30 (2H, m, CH2-CF2), 2.14, 2.09, 2.04, 2.01, 1.96, 1.92 (24H, 6s, アセチル由来CH3), 1.65 〜 1.10 (6H, m, リシン由来CH2).
13C NMR (62.86 MHz, CDCl3): δ 171.3 (CO-NH), 170.5, 170.5, 170.1, 170.0, 170.0, 169.7, 169.2 (7s, CO-O), 167.9 (CO-NH), 156.7 (O-CO-NH), 136.7 (CIV arom.), 128.4, 128.0, 127.9, (CH arom.), 101.6 (CH-1’), 77.9 (CH-4), 72.7 (CH-2), 71.1, 70.9 (CH-5’ 及び CH-3’), 70.0 (CH-5), 69.4 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 66.5 (CH2-O), 61.6, 61.0 (CH2-6 及び CH2-6’), 52.6 (CH-CO), 40.4 (CH2-NH), 31.9 (トリプレット, CH2-NH), 31.2 (CH2-), 30.5 (トリプレット, CH2-Rf), 29.1 (CH2-), 22.2 (CH2-), 20.6, 20.5, 20.4, 20.4, 20.3 (5s, アセチル由来CH3).
19F NMR (235 MHz, DMSO-d6): δ -80.2 (CF3, s), -113.0 (CF2-CF3, s), -121.4 (3 CF2, s), -122.2 (CF2, s), -123.0 (CF2, s), -125.4 (CF2-CH2, s).
[α]D = +2.9 (c, 1, CHCl3).
【0144】
ラクトLys(PBN)C8F17(OAc)8 7の合成
【0145】
【化21】
【0146】
0.400gの化合物5(0.28mmol−1当量)を、10mlのジオキサン中に溶解する。メディウムを0℃にし、0.190gのパラジウム−炭(10%−65mg/mmol)をフラクションごとに添加する。水素化ボンベ(圧力8バール)において20時間の攪拌の後、メディウムをセライトの層を通して濾過し、溶媒を減圧下にて除去する。アミン6を、精製せずに白色粉末の形態で得る。
【0147】
アミン6を、アルゴン気流の下、5mlの無水ジクロロメタン中に溶解する。0.090gの活性エステルX(0.28mmol−1当量)を、メディウムに添加し、DIEAを添加することによってpHを9にする。
【0148】
攪拌を、アルゴン雰囲気下にて24時間継続する。この溶媒を、減圧下にて蒸発させ、粗生成物をシリカゲル上のフラッシュクロマトグラフィー(溶離液:酢酸エチル)によって精製する。Sephadex LH−20樹脂上でのサイズ排除クロマトグラフィー(溶離液:1:1 ジクロロメタン/エタノール)によるさらなる精製は、ニトロン7(0.230g − 0.156mmol − 54%)を白色粉末の形態で得ることを可能にする。
【0149】
モル質量 (C56H67F17N4O22): 1471.1 g・mol-1
融点: 75°C (分解の開始).
1H NMR (250 MHz, CDCl3): δ 8.36 (2H, d, J = 8.3 Hz, H arom.), 7.92 (2H, d, J = 8.4 Hz, H arom.), 7.68 (1H, s, CH=N(O)), 6.94 (3H, m, NH), 5.51 (1H, dd, J = 2 Hz 及び J = 4.4 Hz, H-4’), 5.32 (2H, m, H-2 及び H-3), 5.17 〜 4.95 (3H, m, H-2’, H-5 及び H-3’), 4.62 (1H, d, J = 7.7 Hz, H-1’), 4.50 (1H, dd, J = 2.7 Hz 及び J = 12.5 Hz, 糖由来H), 4.33 (1H, m, リシン由来CH), 4.18 (1H, dd, J = 1.6 Hz 及び J = 6.3 Hz, 糖由来H), 4.12 〜 3.87 (4H, m, 糖由来H 及び H-5’), 3.50 (2H, m, CH2-NH), 3.39 (2H, m, CH2-NH), 2.35 (2H, m, CH2-CF2), 2.15, 2.09, 2.08, 2.04, 2.02, 2.00, 1.96 (24H, 7s, アセチル由来CH3), 1.60 (11H, m, tert-ブチル由来CH3及び CH2 Lys), 1.85 (2H, m, CH2Lys), 1.35 (2H, m, CH2 Lys).
13C NMR (62.86 MHz, CDCl3): δ 171.5 (CO-NH), 170.6, 170.5, 170.3, 170.3, 170.0, 169.7, 169.32 (7s, CO-O), 168.2 (CO-NH), 167.1 (CO-NH), 135.2 (CIV arom.), 133.7 (CIV arom.), 129.3 (CH=N(O)), 128.6, 127.3 (CH arom.), 101.7 (CH-1’), 78.5 (CH-4), 72.9 (CH-2), 71.4 (CIV), 71.0, 70.9 (CH-5’ 及び CH-3’), 68.9 (CH-5), 69.3 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 61.6, 61.1 (CH2-6及びCH2-6’), 52.7 (CH-CO), 39.4 (CH2-NH), 31.9 (m, CH2-NH), 31.1 (CH2-), 30.5 (トリプレット, CH2-Rf), 28.7 (CH2-), 28.2 (tert-ブチル由来CH3), 22.2 (CH2-), 20.7, 20.7, 20.7, 20.6, 20.5, 20.5, 20.4, (7s, アセチル由来CH3).
19F NMR (235 MHz, CDCl3): δ -80.7 (s, CF3), -114.2 (s, CF2-CF3), -121.9 (s, 3 CF2), -122.7 (s, CF2), -123.5 (s, CF2), -126.1 (s, CF2-CH2).
[α]D = +1.6 (c, 1, CHCl3).
【0150】
3/実施例3:
A−化合物Bの合成
【0151】
【化22】
【0152】
0.99g(3×10−3mol)のCl−+H3N Lys(Z)OMeを、100mlの丸底フラスコ中にて、10mlのジクロロメタン中に溶解する。pHを、TEAを用いて8にする。その後、1.15g(3×10−3mol、1当量)のFmoc Ser(OtBu)OHを、1.72(3.9×10−3mol、1.3当量)のBOPと共に添加する。
【0153】
pHを、反応の間8に保持する。メディウムを、周囲温度にて24時間攪拌し続ける。反応が完了すれば(TLC)、有機相を1N HClで洗浄し、その後、pH7を回復(reestabish)するためにNaHCO3で洗浄する。有機相をNa2SO4上で乾燥し、濾過し、その後減圧下にて蒸発させる。結晶化を、ジクロロメタン/Et2O混合溶液から行うことができる。1.93gの化合物1を、白色粉末の形態で得る。
【0154】
【表2】
【0155】
【化23】
【0156】
0.68g(1.53×10−3mol)のFmoc Asp(OtBu)OHを、0.34g(1.83×10−3mol、1.2当量)のペンタフルオロフェノール及び0.38g(1.83×10−3mol、1.2当量)のDCCと共に、50mlの丸底フラスコ中にて10mlのジクロロメタン中に、溶解する。反応を、周囲温度にて15時間の間攪拌しながら置く。濾過の後、メディウムを減圧下にて濃縮する。残存オイルをシリカゲル上にてクロマトグラフにかける(溶離液:2/8 酢酸エチル/シクロヘキサン)。結晶化を、酢酸エチル/ヘキサンから行う。760mgの2を、白色粉末の形態で得る。
【0157】
【表3】
【0158】
【化24】
【0159】
ジペプチド及び活性化アミノ酸間のカップリングの前に、化合物2の脱保護からなる行程が必要である。このため、1.3g(1.97mmol)のジペプチド1を、15mlの10% v/v ピペリジン/ジクロロメタン混合溶液中に溶解する。メディウムを、周囲温度にて攪拌したまま2時間放置し、その後、分液漏斗を用いて1N HClで洗浄する。その後、有機相を飽和炭酸水素ナトリウム溶液で洗浄する。有機相を、その後、硫酸ナトリウム上で乾燥した後、減圧下にて濃縮する。その後、カップリングを行うことができる。
【0160】
上記の脱保護されたジペプチドを、100mlのシングルネック(single−necked)丸底フラスコ中にて、丸底フラスコに加えられた1.138g(1.97mmol、1当量)の化合物2の存在下で、20mlのジクロロメタン中に溶解する。反応を、窒素気流のもと周囲温度にて、暗所で、DIEAで固定されたpH8にて行う。15時間後、反応を完了する(TLC)。反応メディウムを濃縮し、残渣をシリカゲル上にて、5/5酢酸エチル/シクロヘキサン溶出混合溶液中でクロマトグラフにかける。溶媒の蒸発の後、970mgの化合物3を、半透明ゲルの形態で得る。
【0161】
【表4】
【化25】
【0162】
1g(3.36×10−3mol)のFmoc Gly OH,0.681gのペンタフルオロフェノール(3.7×10−3mol、1.1当量)及び0.764gのDCC(3.7×10−3mol、1.1当量)を、10mlのジクロロメタン中に溶解する。このメディウムを、周囲温度にて24時間の間放置する。反応メディウムを、その後濾過し、その後、この濾過液を減圧下での蒸発によって濃縮する。残渣を、5/5酢酸エチル/シクロヘキサン溶出混合溶液を用いたシリカゲル上のフラッシュクロマトグラフィーにかける。結晶化を、酢酸エチル/シクロヘキサン混合溶液から行う。955mgの4を、白色粉末の形態で得る。収率:61.3%
【0163】
【表5】
【0164】
【化26】
【0165】
この合成は、このトリペプチド(化合物3)の合成と同様のプロトコルに従って、トリペプチドの脱保護によって進められる。500mg(6×10−3mol)の3を、脱保護する。脱保護するとすぐに、このペプチドを100mlの丸底フラスコ中の10mlのジクロロメタン中に溶解する。278mg(6×10−4mol、1当量)の化合物4を、反応メディウムに添加する。反応を、周囲温度にて窒素気流下に行い、DIEAを用いてpH8に維持する。16時間の後、反応を完了する(TLC)。減圧下でのメディウムの蒸発の後、残存オイルを、6/3 酢酸エチル/シクロヘキサン溶出混合溶液を用いてシリカゲル上にてクロマトグラフにかける。酢酸エチル/シクロヘキサン混合溶液から、426mgの5を、白色粉末の形態で得る。収率:80%
【0166】
【表6】
【0167】
【化27】
【0168】
ペンタペプチド5の脱保護は、化合物1の合成と同じ条件下で行われる。400mg(4.5×10−4 mol)のテトラペプチドを脱保護する。脱保護が完了したらすぐ、このテトラペプチドを、50mlのシングルネック丸底フラスコ中の15mlのジクロロメタン中に溶解する。219mg(4.5×10−4mol、1当量)のBocArg(Mtr)OH及び188g(5.85×10−4mol、1.3当量)のTBTUを加える。反応を、周囲温度にて窒素気流下、暗所で行い、DIEAを用いてpH8に維持する。16時間の後、この反応を完了する(TLC)。減圧下での蒸発の後、残存オイルを9/1酢酸エチル/シクロヘキサン溶離液を用いたシリカゲル上のクロマトグラフにかける。酢酸エチル/シクロヘキサンから結晶化を行う。417mgの6を、白色粉末の形態で得る。収率:92.5%
【0169】
【表7】
【0170】
【化28】
【0171】
500mg(1.01×10−3mol)のC8F17CH2CH2COOH、278mgのH2N TYR(OH)OtBu(1.01×10−3mol、1当量)及び251mgのDCC(1.2×10−3mol、1.2当量)を、50mlの丸底フラスコ中の10mlのDMF中に溶解する。反応を、周囲温度にて24時間、窒素気流下、暗所で行い、DIEAを用いてpH8に維持する。減圧下でのDMFの蒸発の後、残渣を2/8 酢酸エチル/シクロヘキサン溶離液を用いたシリカゲル上のクロマトグラフにかける。
【0172】
この生成物を、酢酸エチル/n−ヘプタンから結晶化することができる。550mgの7を、白色粉末の形態で得る。収率:76%
【0173】
【表8】
【0174】
【化29】
【0175】
380mg(5.34×10−4mol)の化合物7を、冷却条件下にて、TFA/CH2Cl2(3/7)溶液中に溶解する。2時間の攪拌の後、脱保護を完了する。その後、反応メディウムを濃縮した後、エーテルから数回にわたって沈殿させる。蒸発の後、白色粉末を得る(化合物8)。
【0176】
150mg(1.32×10−4mol)の化合物6を、メタノール中に溶解する。冷却条件下で8mgの10%パラジウム−炭を添加する。反応メディウムを、水素8気圧の下に置く。2時間30分後、この反応を完了する。この混合溶液を、セライト521を通して濾過し、この濾過液を減圧下にて濃縮する(化合物9)。
【0177】
化合物9(1.32×10−4mol)を、50mlの丸底フラスコ中の10mlのDMF中に溶解する。95mgの化合物8(1.45×10−4mol、1.1当量)及び76mgのTBTU(1.72×10−4mol、1.3当量)を添加する。反応を、周囲温度にて24時間、窒素気流下で暗所にて行い、DIEAを用いてpH8に維持する。反応メディウムを、減圧下にて濃縮する。残渣を酢酸エチル溶離液を用いてシリカゲル上のクロマトグラフにかける。141mgの生成物10を、白色粉末の形態で得る。収率 65.3%。
【0178】
【表9】
【0179】
チオ−アニソール(3;5;2)の存在下にて24時間の間、CH2Cl2中においてトリフルオロ酢酸溶液の反応にかけられる化合物10は、化合物Bを生成する(化合物Bはこの溶液へのエーテルの添加による沈殿、及びSephadex G50カラム(溶離液:H2O)上のクロマトグラフィーの後、純粋な形態で単離される)。凍結乾燥の後、生成物Bは、白色粉末の形態である。
【0180】
B−生物学的アッセイ
この分子の細胞毒性を、B16メラノーマ細胞に対して試験した;100μMを超える濃度まで、毒性を測定することはできなかった。ヨウ素125で標識した後、メラノーマを有している1群のマウスに静脈内に注射されたこの分子は、腫瘍のストローマに蓄積し、その後、徐々に腫瘍中に拡散する(表2参照)。
【0181】
【表10】
【0182】
4/実施例4:
メチルN−(t−ブトキシカルボニル)−N−γ−(2,3,6−トリメチル−4−メトキシ-ベンゼンスルフォニル)−L−アルギニルグリシニル−O−(t−ブチル)−L−アスパルチル−O−(t−ブチル)−L−セリニル−N−ε−((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11−ヘプタデカフルオロウンデカノイル)−N−ε−(4−(4−[ビス(2−クロロエチル)アミノ]フェニル)−ブチルアミド)−L−リシニル−L−チロシンアミド)−L−リシネート
【0183】
化合物Dの合成は、上記化合物に対して上記のものと類似であり、図2A及び2Bに要約される。
【0184】
Dの物理化学的特徴
Rf:酢酸エチル/メタノール: 98/2中に0.49.
MM: 2051.87 g・mol-1.
分解温度: 134°C.
[α]D20 = -0.77 (c, 0.1, CH3OH).
MS (FAB): m/z 2052 [M+H]+, 2074 [M+Na]+.
1H NMR (CD3OD):
δ 7.03 (4H, m, 2H arom. Tyr, 2H arom. Chloramb.); 6.67 (5H, m, 2H arom. Tyr, 2H arom. Chloramb., H arom Mtr); 4.81; 4.47; 4.20; 4.01 (6H, 3m, Hα Tyr, 2Hα Lys, Hα Asp, Hα Ser, Hα Arg); 3.82 (3H, s, CH3 エーテル Mtr Arg); 3.68 (3H, s, CH3 メチルエステル Lys); 3.83-3.60 (12H, m, 2Hα Gly, 2Hβ Ser, 8H Chloramb.); 3.14-2.75 (10H, m, 4Hε Lys, 2Hδ Arg, 2Hβ Asp, 2Hβ Tyr); 2.67-2.48 (8H, m, 2Hα 及び 2Hβ フッ素化鎖, 2Hα Chloramb., 2Hγ Chloramb.); 2.67; 2.61 (6H, 2m, 2CH3 Mtr Arg); 2.13 (5H, m, 2Hβ Chloramb., CH3 Mtr Arg); 1.93-1.16 (16H, m, 4Hβ Lys, 4Hγ Lys, 4Hδ Lys, 2Hβ Arg, 2Hγ Arg); 1.44; 1.42; 1.16 (27H, 3s, 9 CH3 tert-ブチルエステル Asp, tert-ブチルエーテル Ser, tert-ブチルウレタン Arg).
13C NMR (CD3OD):
δ 174.67; 174.14; 172.57; 172.31; 171.80; 171.75; 171.26; 170.80; 170.17 (CO Tyr, 2CO Lys, CO Ser, 2 CO Asp, CO Gly, CO Arg, CO フッ素化鎖, CO Chloramb.); 158.46; 156.80; 156.67; 155.93 (CO ウレタン Boc, C-OCH3 arom. Mtr Arg, C-OH arom. Tyr, C グアニジン Arg); 144.55 (C-N arom. Chloramb.); 138.10; 136.48; 133.42; 130.29; 129.97; 129.22; 127.51; 124.29; 114.83; 112.06; 111.38; 110.73 (C arom. Mtr, C arom. Tyr, C arom. Chloramb.); 81.17; 79.43 (C tert-ブチルエステル Asp, C tert-ブチルウレタンArg); 73.40 (C tert-ブチルエーテル Ser); 61.14 (Cβ Ser); 54.96; 54.60; 54.53; 53.89; 53.14; 52.26; 51.31; 49.90 (CH3 メチルエステル Mtr Arg, Cα Ser, Cα Arg, Cα Tyr, CH3 メチルエステル Lys, 2Cα Lys, Cα Asp, 2C-N Chloramb.); 42.30; 40.29; 38.58; 36.67; 36.52; 35.20 (Cα Gly, 2Cε Lys, Cδ Arg, Cβ Asp, Cβ Tyr, 2C-Cl Chloramb.); 33.85; 31.02; 30.69; 28.91; 28.67; 28.15; 27.69; 27.41; 26.96; 26.36; 25.82; 25.46 (2Cδ Lys, 2Cβ Lys, Cβ Arg, Cγ Arg, 9 CH3 tert-ブチルAsp, Ser, Arg, Cα-CF2, 2Cγ Lys, Cα, Cγ Chloramb.); 23.00; 22.66 (Cβ フッ素化鎖, Cγ Chloramb.); 22.52; 17.47; 10.74 (3 CH3 メチル Mtr Arg).
19F NMR (CD3OD):
δ -82.26 (3F); -115.39 (2F); -122.77 (6F); -123.62 (2F); -124.30 (2F); -127.17 (2F).
【0185】
5/実施例5:
メチル L−アルギニル−グリシニル−L−アスパルチル−L−セリニル−N−ε−(―O−(N−4−(N−(4,4,5,5,6,6,7,7,8,8,9,9−トリデカフルオロノナノイル)−L−セリニル)−1−β−D−アラビノフラノシルシトシン)−オキシカルボニル)−L−リシネート
【0186】
【化30】
【0187】
手法
Fを調製する手法は、前述の工程と同一である;この合成は、図3A、3B及び3Cに要約される。
【0188】
Fの物理化学的特徴
MM: 1390.45 g・mol-1. MS (FAB): m/z 1391 [M+H]+.
1H NMR (DMSO, D6):
δ 8.66 (1H, s, H 酸 Asp); 8.27-8.16; 7.94-7.84; 7.65; 7.21-6.92 (15H, 3m, Ha, NH GABA, 2NH Ser, 4H グアニジン Arg, NH2 Arg, NH Gly, NH Asp, 2NH Lys, NH Ara-C); 5.84 (1H, m, Hb); 5.31 (2H, m, 1Hα Ser, H1); 4.92 (1H, m, Hα Asp); 4.37-3.71 (17H, m, Hα Lys, Hα Ser, Hα Arg, 2Hα Gly, 4Hβ Ser, H2, H3, H4, CH3 メチルエステル Lys, 2 H5); 3.18-2.53 (14H, m, 2Hε Lys, CH2-NH GABA, 4OH, 2Hβ Asp, 2 Hα及び 2 Hβフッ素化鎖); 2.23 (2H, m, CH2-CO GABA); 1.46-1.03 (14H, m, 2Hβ, 2Hγ, 2Hδ Lys, 2Hβ, 2Hγ, 2Hδ Arg, CH2 GABA).
13C NMR (DMSO, D6):
δ 173.9; 173.6; 173.4; 172.8; 171.1; 170.4; 170.1; 169.6; 169.1; 168.6 (CO Lys, 2 CO Ser, 2 CO Asp, CO Gly, CO Arg, CO フッ素化鎖, CO GABA); 162.6; 157.4; 156.2; 155.0 (C グアニジン Arg, CO ウレタン Ser, N=C-NH, N-CO-N); 147.2 (Ca); 94.8 (Cb); 87.5; 86.2 (C1, C4); 76.6; 75.0 (C2, C3); 65.4; 62.0; 61.5 (2 Cβ Ser, C5); 55.6; 53.0; 52.5; 52.3; 51.7; 50.1 (2 Cα Ser, Cα Arg, CH3 メチルエステル Lys, Cα Lys, Cα Asp); 42.5 (Cα Gly); 40.8; 38.5; 37.3; 34.8; 34.1; 30.9; 29.4; 29.0; 26.3; 24.8; 24.3; 23.0 (Cε Lys, Cδ Arg, CH2-NH GABA, Cβ Asp, Cδ Lys, Cβ Lys, CH2-CO GABA, Cβ Arg, Cα及び Cβフッ素化鎖, Cγ Arg, CH2 GABA, Cγ Lys).
19F NMR (DMSO, D6):
δ -82.30 (3F); -115.51 (2F); -122.81 (6F); -123.82 (2F); -124.46 (2F); -127.21 (2F).
【図面の簡単な説明】
【0189】
【図1】PBN由来のスピントラップを備えたベクターの化学合成
【図2A】分子Dの合成
【図2B】分子Dの合成(続き)
【図3A】分子Fの合成
【図3B】分子Fの合成
【図3C】分子Fの合成
【技術分野】
【0001】
本発明は、有効成分のためのベクターとして使用され得る新規分子、そのようなベクターを含む活性分子、及び薬学的分野における、特に医薬品の調製のためのその使用に関する。
【背景技術】
【0002】
有効成分の送達に関する最近の研究は、外傷性の(traumatic)投与経路を最小にすることによって患者の快適性だけでなく、(通常は欠如しており、しばしば副作用の原因にもなる)細胞親和性を付与することによって医薬品の総体的な有効性を顕著に高める傾向にある。従って、治療的活性の慣習的な考え方に、実際に基質のバイオアベイラビリティーを調節する、作用の特異性の考え方が加えられる。
【0003】
実際のところ、例えば、抗癌化学療法といった細心の注意を払うべき分野(sensitive field)において、ここ数年に亘って観察された進歩(新規な薬物、新規な投与方法、in vitro又はin vivoでのケモプレディクティビティ(chemopredictivity)、新規な治療スキームへの統合等)は、現在から、輸送に関する新規な考え方に基づく新規な道具及び抗癌剤の細胞ターゲッティング(targeting)で装備されることができれば、化学療法の質が進歩し続けるであろうと考えることができる。癌性部位に運ばれるであろう有効成分の有効性及び有効量を増加するため、実際に、これらの医薬品に本当の特異性を付与すること、及びそれらの副作用を軽減することが必要なようである。
【0004】
現在、医薬品の化学構造は、その生理化学的特性及び生物学的特性の両方、ならびに、特に膜受容体に対する親和性を条件付ける。分子構造の修飾によるこの結果の調節は、薬学的特性を変える危険がある。そのため、この生成物を“製剤(galenic)”の観点から処理すること(すなわち、投与に使用される製剤形態に関心を持つこと)、或いは、より良くは、宿主構造中にカプセル化するか、又はこのベクター化(vectorization)を提供することが可能な分子にそれをグラフトすることが導かれる。
【0005】
有効成分のベクター化は、成功するためにいくつかのパラメーターを考慮に入れなくてはならない:
すでに上記に示したように、有効成分は、医薬品又は生物に対して有害なあらゆる相互作用を予防するために、可能な限り、生理学的なメディウムから分離されるべきである。
【0006】
担体は、医薬品のバイオアベイラビリティーを損なうべきではなく、むしろ改善するべきである。言い換えれば、治療剤は、標的分子中に(好ましくは、細胞質内レベルにて、特定の場合は、核内レベルにおいて)放出されるべきであり、その全ての活性をそこに維持すべきである。
【0007】
しかしながら、医薬品(細胞の反応を引き起こすことができ、その結果、欠陥を是正することができる、受容体に特異的な薬剤)のこの構想は、一般的とするにはほど遠い。それは、ホルモン又は鎮痛タイプの有効成分に対して的確である一方、例えば、抗癌治療といった、他の分野において全く適用されない。そのような基質は、この方法によってデザインされていなかった:それらは、いかなる細胞認識特異性も有していない。それらの目的は、細胞増殖を阻害することである。それらは、一般的に抗有糸分裂剤であり、この結果、細胞が癌性か正常かを問わず、全ての細胞のDNAに対して作用し得、そのようにして特定の厄介な状態を生み出す可能性がある(その最も一般的な兆候は、造血組織無形成(hematopoietic tissue aplasia)であり、その次に、免疫阻害及び消化障害を併発する)。化学療法の確立により、開発された有効成分は、ますます強力になってきたが、残念ながら、癌細胞と正常細胞を区別しない。従って、癌治療の観点から、有効性及び選択性を、同一の有効成分上に結合する(conjugated)ことができないことが悔やまれる。
【0008】
既知のこれらの様々な考え及び観察により、本質的にマクロ分子タイプ(合成又は天然ポリマー)及び超分子タイプ(リポソーム)といった、様々なベクターモデルが提案されてきた。全てのこれらのベクターモデルのなかで、特に、有効成分の親水性−親油性バランス(従って、その本質的な物理化学的特性)を調節することができ、その細胞内侵入を促進し及び適切に選択された認識因子(recognition agent)を用いて細胞ターゲッティングを付与することもできる、テロマーと呼ばれる、小さな両親媒性ポリマーの開発について言及する。
【0009】
“Synthesis of new cotelomers derived from tris(hydroxymethyl)aminomethane bearing arabinofuranosylcytosine moieties. Preliminary results on their in vitro and in vivo antitumoral activities” C. Contino, J.C. Maurizis, M. Ollier, M. Rapp, J.M. Lacombe, B. Pucci. Eur. J. Med. Chem., (1998), 33, 809-816.
“Synthesis and preliminary biological assessments of a new class of amphiphilic telomers bearing 5-fluorouracil moieties” C. Contino, J.C. Maurizis and B. Pucci. Macromol. Chem., (1999), 200, 1351-1355.
“A new strategy in biomedical and medical field: the synthesis and applications of telomeric structures”. P. Barthelemy, A. Polidori, B. Pucci. Transworld Research Network, Recent developments in organic chemistry, Trivandrum, (1999), 3, 117-140.
“Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers. Sylvain Jasseron, Christiane Contino-Pepin, Jean Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070.
“Synthesis and preliminary biological assessments of a new class of amphiphilic telomers bearing 5-fluorouracil moieties” C. Contino, J.C. Maurizis and B. Pucci. Macromol. Chem., (1999), 200, 1351-1355.
“Amphiphilic telomers: a new kind of antimitotic drugs macromolecular carriers.” Christiane Contino-Pepin, Jean-Claude Maurizis, Bernard Pucci. Curr. Med. Chem.-Anti-Cancer Agents, (2002), 2, 645-665.
【0010】
これら一連の研究において得られた結果は、様々な主要な点を明示することを可能にした:
基質の親水性−親油性バランスの制御は、その膜貫通通過を促進するが、(“Uptake and subcellular distribution of a new fluorinated telomeric carrier: study on cultivated B16 melanoma and skin rat fibroblastic cells”. F. Chehade, J.C. Maurizis, B. Pucci, A.A. Pavia, M. Ollier, A. Veyre, F. Escaig, C. Jeanguillaume, R. Dennebouy, G. Slodzian, E. Hindie, Cellular and Molecular Biology, (1996), 42, 335-342)界面活性剤、従って毒性の性質の導入を伴わない(“Efficiency of new non ionic telomeric surfactants towards the solubilization of subcellular fractions proteins” B. Pucci, J.C. Maurizis and A.A. Pavia, BioOrg. Med. Chem Lett. (1993), 3, 161-164)。
【0011】
これらの両親媒性ポリマーは、全ての分子、従って有効成分に効果的な細胞ターゲッティングを付与することを可能にする(“Cell targeting by glycosidic telomers - Recognition ability of galactosylated telomers by the yeast Kluyveromyces Bulgaricus” J.Coulon, R. Bonaly, B. Pucci, A. Polidori, P. Barthelemy, C. Contino, Bioconjugate Chem. (1998), 9, 152-159. “Permeability of yeast cell enveloppe to fluorescent galactosylated telomers derived from THAM”. C. Contino, M. Briot, J. Coulon, A. Polidori, R. Bonaly and B. Pucci. Bioconjugate Chem., (2000), 11, 461-468. “Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers”. Sylvain Jasseron, Christiane Contino-Pepin, Jean-Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070)。
【0012】
好適な(細胞質の酵素によって加水分解され得る)ペプチドスペーサーアームを用いてベクター上にグラフトされた有効成分は、ベクターが細胞膜を通過した後に、細胞内レベルにおいて放出される(“Synthesis and Preliminary biological assessments of RGD bearing biocompatible telomers”. Sylvain Jasseron, Christiane Contino-Pepin, Jean Claude Maurizis, Maryse Rapp, Bernard Pucci. Bio. Med. Chem. Letters, (2002), 12, 1067-1070)。
【0013】
このベクター化の方法は、転移の増殖(proliferation)を阻害し、腫瘍の成長を遅らせ、そして処置されたマウスの生存期間を、コントロールマウスに比べて3倍を超えて延長することから、抗癌剤の効果を極めて顕著に増加することを可能にする。
【0014】
WO 92/02560において記載される、これらのテロマーによってもたらされる明らかな利点にも関わらず、その様なベクターが直面し、商品化及びその使用を困難にし得る主要な問題の1つは、多分散性、すなわち、明確な質量及び構造の欠如である。
【発明の開示】
【発明が解決しようとする課題】
【0015】
従って、本出願人は、有効成分のベクターとなり、明確な構造を有し、調製が容易であって、有効成分をその標的に送達することを容易にすることが可能な分子のデザイン及び調製を目的として設定する。
【課題を解決するための手段】
【0016】
従って、本発明の主題は、下記式(I):
【0017】
【化1】
【0018】
(式中:
APは、生物学的標的に作用することが可能であり、有効成分のその生物学的標的への送達を促進することが望まれる、有効成分を表し;
xは、0及び1から選択される整数を表す
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、ターゲッティング剤(targeting agent)および可溶化剤(solubilizing agent)から選択される基を表す;に相当する分子である。本発明の目的のため、用語“ターゲッティング剤(targeting agent)”は、以下:式(I)の全ての分子のその標的への輸送を促進する分子又は有効成分APの標的によって認識されることが可能なあらゆる分子を意味することを意図する。用語“可溶化剤(solubilizing agent)”は、式(I)の分子のHLBバランスの調節のための薬剤、特に親水性の薬剤を意味することを意図する。本発明において使用され得るターゲッティング剤のうち、単糖類、糖類のアミノ化誘導体、多糖類、天然又は合成ホルモン、ペプチド、抗体及び一般的に、有効成分APの標的によって認識され得るあらゆる分子が挙げられる。本発明において使用され得る可溶化剤のうち、特に、ポリオール、ポリエーテル、ペプチド及び多糖類が挙げられる。
【0019】
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2又はAA3の1つの側鎖のいずれかへの結合(ダッシュ記号−−−によって示される)を可能にする基
【0020】
【化2】
【0021】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表す;
AP−(X)xと鎖[AA3]a3−[AA2]a2−[AA1]の間のダッシュ記号−−−は、AP−(X)xと残りの分子の結合が、アミノ酸AA1、AA2又はAA3の1つの側鎖を介して、或いは必要に応じて、ペプチド鎖の末端にて生じることを示す。
【0022】
より詳しくは、有効成分は、認識される生物学的活性を有し、官能基−O−CO−、−CO−NH−、NH−CO−NH−、−NH−CO−O−、O−CO−O−、−O−、−S−、
【0023】
【化3】
【0024】
から選択され得る結合を用いてアミノ酸に結合され得る全ての有機分子から選択される。
【0025】
これらの有効成分のうち、特に、抗癌、抗炎症、殺菌、鎮痛、神経弛緩、抗真菌作用を
有するもの、及びフリーラジカルスカベンジャー活性を有する分子が挙げられる。
【0026】
一般に、有効成分は、1〜30の炭素原子、1以上の不飽和、特に1以上の芳香環、及び以下:
【0027】
【化4】
【0028】
から選択される1以上の官能基を含む直鎖、分岐又は環状分子からなり得る。
【0029】
有効成分APは、上記の性質の結合によりアミノ酸AA1、AA2又はAA3の1つの側鎖、或いはペプチド鎖の末端のいずれかに、必要に応じてペプチド鎖X(この場合、X=1)により結合される。
【0030】
2つの基、Y及び−(X)x−APの1つとの結合は、アミノ酸AA1、AA2又はAA3のうち1つの側鎖上において行われる。AP−(X)x−又はYにその側鎖を介して結合したアミノ酸は、それらの側鎖上に酸、アミド、アミン、チオール又はアルコール官能基を含むものから選択される。これらのうち、特に、リシン、アルギニン、オルニチン、アスパラギン酸、グルタミン酸、アスパラギン、グルタミン、セリン、チロシン又はシステインが挙げられ得る。好ましくは、AP−(X)x−又はYにその側鎖を介して結合されるアミノ酸は、以下:アスパラギン酸又はリシンから選択される。
【0031】
スペーサーアームX(存在する場合)は、1つの末端にて、アミノ酸AA1、AA2又はAA3の側鎖又は1つの末端との結合、そして他の末端にて有効成分APとの結合に関与するペプチド鎖からなる。
【0032】
このスペーサーアームは、1〜5アミノ酸、好ましくは1〜3アミノ酸を含む。
【0033】
スペーサーアームX及び/又はペプチド鎖[AA3]a3−[AA2]a2−[AA1]は、有効成分APの標的に対するそれらの親和性について選択され得る。また、それらは、125Iで標識した後、in vivoにおいて式(I)の分子を追跡することを可能にするチロシン残基を含む又はチロシン残基からなり得る。
【0034】
Rは、細胞標的に従って選択される;天然の糖類(特定の組織に存在し、肝臓、骨、ある癌性腫瘍の場合ガラクトース、或いはマクロファージ、心臓の場合マンノース、或いは赤血球の場合シアル酸のいずれかを選択的に認識する特定の膜レクチンのターゲッティング等)、キナーゼ、特定の抗体、特にペプチドをターゲッティングするための天然のホルモン(ステロイド等)又は実際の合成物(メシル酸イマチニブ(ST571、Gleevek(登録商標))等)であり得る。Rは、先の研究がそれに対する認識特異性を示した、あらゆる基質から選択され得る。Rが、単糖又は多糖、或いは親水性ペプチドである場合、加えて、そのIV及びIP投与に必要な水溶性(water−solubility)をこの分子に付与し得る。
【0035】
Rがペプチド鎖である場合、Rは、有利には3〜15のアミノ酸を含有し、さらに有利には3〜10のアミノ酸を含有する。また、125Iで標識した後、in vivoにおいて式(I)の分子を追跡することを可能にする1以上のチロシン残基を含み得る。
【0036】
スペーサーアームXを構成するアミノ酸は、鎖[AA3]a3−[AA2]a2−[AA1]又は基Rを構成するものと同様に、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、スレオニン、トリプトファン、チロシン又はバリン等の天然アミノ酸、或いは、ヒドロキシプロリン、ノルロイシン、オルニチン、シトルリン又はシクロヘキシルアラニン等の非天然アミノ酸から選択される。
【0037】
3−アミノプロピオン酸及び4−アミノ酪酸等のΩ−アミノ酸の使用も、想定され得る。
【0038】
Rがペプチドの場合、ペプチド鎖Rは、APの生物学的標的に対して顕著な親和性を有するエピトープ又は抗体断片であり得る。
【0039】
本発明において使用され得るペプチドのうち、例えば:そのαVβ3インテグリンに対する親和性で知られるRGD配列が挙げられる。
【0040】
また、Rは、有効成分APの標的に関する限り、式(I)分子に水への溶解性及び細胞内への侵入を促進する親水性/親油性バランスを付与するために、ポリオール又はポリエーテル、特にポリ(エチレンオキサイド)から選択され得る。
【0041】
Rがポリオールで構成される場合、前記ポリオールは、有利には、4〜16の炭素原子及び4〜16の水酸基を含むアルキル鎖からなる。
【0042】
Rが可溶化単位(solubilization unit)としてポリ(エチレンオキサイド)鎖から構成される場合、前記鎖は、有利には、5〜30のエチレンオキサイド単位を含む。
【0043】
Rは、特に、単糖類、糖類のアミノ化誘導体及び多糖類から選択され得る。
【0044】
本発明において使用され得る単糖類のうち、以下:グルコース、フルクトース、マンノース、ガラクトース及びリボースが挙げられる。糖類のアミノ化誘導体のうち、特に、グルコサミンが挙げられる。本発明において使用され得る多糖類のうち、ラクトース、セロビオース又はマルトース、ラクトビオナミド(lactobionamid)及びスクロースが挙げられる。好ましくは、本発明において使用される多糖鎖は、二糖類(bisacchride)である。
【0045】
鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つへのRの結合は、R上にグラフトされ得る官能基(functionality)に従って以下:エーテル、アミド、カルバメート、チオエーテル、エステル、ウレア、ウレタンの好適な結合を用いて行われる
フッ素化炭化水素ベース鎖は、好ましくは、式A−Y’(式中、Aは、以下:
【0046】
【化5】
【0047】
−NH−、−O−CO−NH−、S及びOから選択される基を表し、ならびにY’は式-(CH2)t-(CF2)rF(式中、r及びtは、12≧r+t≧4である2つの整数を表す)に相当する分子を表し、例えば、以下:
-(CF2)4F; -(CF2)5F; -(CF2)6F; -(CF2)7F; -(CF2)8F; -(CF2)9F; -(CF2)10F; -(CF2)11F; -(CF2)12F; -(CF2)13F; -(CF2)14F; -CH2-(CF2)3F; -CH2-(CF2)4F; -CH2-(CF2)5F; -CH2-(CF2)6F; -CH2-(CF2)7F; -CH2-(CF2)8F; -CH2-(CF2)9F; -CH2-(CF2)10F; -CH2-(CF2)11F; -CH2-(CF2)12F; -CH2-(CF2)13F; -(CH2)2-(CF2)2F; -(CH2)2-(CF2)3F; -(CH2)2-(CF2)4F; -(CH2)2-(CF2)5F; -(CH2)2-(CF2)6F; -(CH2)2-(CF2)7F; -(CH2)2-(CF2)8F; -(CH2)2-(CF2)9F; -(CH2)2-(CF2)10F; -(CH2)2-(CF2)11F; -(CH2)2-(CF2)12F; -(CH2)3-(CF2)1F; ...............-(CH2)11-(CF2)F
等に相当するものから選択される。好ましくは、t≧2である。好ましくは12≧r≧4、より好ましくは10≧r≧6である。
【0048】
また、本発明の主題は、式(II)
【0049】
【化6】
【0050】
(式中、R、AA1、AA2、AA3、a2、a3、Y、X及びxは、上記式(I)と同じ定義を有する)の断片を含む、あらゆる生物学的に活性な分子である。
【0051】
実際に、本発明は、この有効成分のヒト又は動物内への侵入を促進し、そしてこの有効成分がその生物学的標的に到達すること可能とするために、好適な結合により上記のあらゆる性質の有効成分を結合することが可能な式(II)の分子断片を提供する。
【0052】
とりわけ、分子の両親媒性の性質は、膜の通過を促進し、有効成分と関連する特異的な標的の認識のための薬剤の任意的な存在が、この標的への送達を促進する。
【0053】
従って、本発明の主題は、上記式(II)の分子断片の活性薬剤のバイオアベイラビリティーを促進するための使用でもある。
【0054】
式(I)の分子の調製は、いくつかの本発明のバリアントに相当する例によって、以下に示される。より一般的には、ペプチド合成の保護、脱保護及びカップリングの方法が使用され、これらの方法は、当業者に周知であり、特に“The peptides” Gross and Meienhofer, 3 vols, Academic Press, New York, 1979-1981の研究において開示されている。
【0055】
式(I)に相当する分子のうち、本発明に特有の主題の1つは、下記式(Ia):
【0056】
【化7】
【0057】
(式中、Suは、上記定義のように単糖、アミノ化単糖誘導体、多糖、ポリオール又は、必要に応じて、ポリエーテルから選択される基Rのバリアントを表す;
AA1は、その側鎖上に酸、アミン、アルコール又はチオール官能基を有するアミノ酸を表し、その側鎖によって(X)x−AP又はYのいずれかに結合される;AA1は、そのN末端及びC末端を介して、Su、及び(X)x−AP又はYのいずれかに結合される)
に相当する分子から構成される。
【0058】
X、x、AP及びYは、上記式(I)と同じ定義を有する。Yは、AA1のアミノ末端又は酸末端、或いは、任意にその側鎖に結合される。
【0059】
好ましくは、下記の1以上の条件を確認する:
−Suは、単糖又は多糖を表す;
−Xは、少なくとも1つのチロシン残基を含む、天然のペプチドであるスペーサーアームを表す;好ましくは、Xはチロシンを表す;
−AA1は、アルギニン及びリシンから選択されるアミノ酸を表す;
−Yは、−NH−官能基を介してアミノ酸AA1に結合した、5〜23のフッ素原子を含むフッ素化C6−C12炭化水素ベース鎖を表す。
【0060】
式(Ia)の2つの化合物の例は、下記及び実施例において示される:
a)例1:血管新生部位のターゲッティング
血管新生は、前から存在する小静脈から新たな血液微細血管をつくり出すための自然な生物学的プロセスである。それは、通常、成人においては、創傷の治癒、炎症又は排卵周期の間の黄体成長等のある特定の状態でのみ起きる、複合的な現象である。正常な状態では、適当な時間の後、血管新生のプロセスは停止し、これは、促進因子及び抑制因子の正しい調節を示唆する。固形腫瘍増殖、関節リュウマチ、乾癬又は糖尿病性網膜症といった、ある特定の病的状態では、血管新生は、明らかにより制御されていない方法で発達する(“Antiangiogenic agents and their promising potential in combined therapy”, P.A. Burke, S.J. DeNardo, Crit. Rew. In Oncology/Hematology, (2001), 39, 155-171)。30年以上前、J.Folkmanは、固形腫瘍増殖は、血管新生の発達と密接に関連しているという仮説を提唱し、それ以降、極めて多くの研究チームが、この現象に興味を示し、血管新生プロセスを阻害することができる基質の開発を試みてきた(“Tumor angiogenic therapeutic applications” J. Folkman Engl. J. Med.(1971), 285, 1182-1186 and “Tumor angiogenis past, present and the near future”. R.S. Kerbel Carcinogenesis (2000), 21, 505-521)。試験された様々な構造のうち、当初は鎮静剤として妊婦に処方され、四肢形成(tetratogenesis)の問題の原因であるサリドマイドが、血管の成長を妨げるのに極めて有利であることが立証された。行われた研究において広まった考えは、あらかじめヨウ素125−標識チロシン等の放射性単位が付与されたベクター上にサリドマイドをグラフトすることであった。この例において模索された目標は、in vivoにて血管新生部位、従って固形腫瘍を、容易に視覚化し、それらの増殖を妨げることである。
【0061】
【化8】
【0062】
この目標により、中心リシン単位に、第一級酸官能基上のフッ素化炭素鎖、該分子に静脈内又は腹腔内投与に必要な水溶性を付与することが可能なラクトースタイプの単位、ならびに後にヨウ素125で標識され、その上に、あらかじめ3位に反応性の酸官能基が付与されたサリドマイドがグラフトされるチロシンが付与された。
【0063】
本発明の好ましいバリアントによれば、式(Ia)に相当する分子において、有効成分は、血管新生プロセスを妨げることができる分子、特にサリドマイドから選択される。
b)例2:スピン−トラップベクター化
ミトコンドリア細胞症は、様々な疾患を含み、その一般的な共通の特徴は、ミトコンドリアの呼吸鎖における欠陥である。生物においてミトコンドリアが随所に存在することにより、この機能障害は、あらゆる臓器に影響を与え得る。この影響は、分離され得、又は一方で多臓器に作用するもの(plurivisceral)であり、その場合は一般に、神経筋系において優勢を示す。現在のところ、これらの疾患に対する治療は存在せず、“オーファン疾患”の枠内に分類され得る。
【0064】
しかしながら、現在、細胞においてミトコンドリアは、フリーラジカルの産生のための優先的な部位であることから、呼吸鎖における欠陥は、極めて一般的に、フリーラジカルの過剰生産と関連することが明らかであり、その結果は、影響を受けた組織における細胞死を加速する。Dr.P.RustinのチームでNecker病院において行われた最近の研究(“Increased apoptosis in vivo in cells lacking mitochondrial DNA gene expression”, Wang J, Silva JP, Gustafsson C, Rustin P, Larsson NG. Proc Natl Acad Sci USA (2001) (in press))は、一連のヒト培養細胞で、この酸素のフリーラジカルの産生の重要性を示すことを可能にした。これらの細胞培養は、ヒトにおいて知られている様々な呼吸鎖の複合体に影響する、全てのタイプの欠陥を表す。それらは、呼吸鎖に影響する欠陥の観点、ならびにフリーラジカル産生及びその細胞生存に対する結果の観点の両方から特徴付けられた。この細胞の収集は、標的が呼吸鎖欠陥と関連するフリーラジカル反応であるあらゆる分子の効果を研究するための、かけがえのない道具を表す。
【0065】
我々のチームにおける、最近の呼吸鎖によって産生されたフリーラジカルで誘導されたアポトーシスの細胞モデルにおける細胞死を妨げることができる“スピン−トラップ”分子の同定は、さらに増強された効果をあらわす同様の分子の開発の基礎を我々に与えた(“Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA”. Geromel V, Kadhom N, Ceballos-Picot I, Ouari O, Polidori A, Munnich A, Roetig A, Rustin P. Hum Mol Genet (2001) (in press))。ここで追跡された目標は、これらの基質の構造を精緻化し簡略化する一方、同時に、工業段階に容易に適用し得る合成プロセスを開発するために、それらの生物学的活性を保存することであった。以下:ミトコンドリアの呼吸鎖において欠陥をあらわす細胞培養(培養線維芽細胞)、フリーラジカルの作用を受けたニューロン/筋細胞共培養及び、最後に、3度の熱傷を受けた皮膚から抽出された細胞の、様々な細胞モデルに対して試験が行われた。
【0066】
行われた調査の目的は、アポトーシスの現象、さらに一般的に、フリーラジカルの過剰産生に起因する細胞死の現象を処置するために臨床的に使用され得るフリーラジカルトラップを得ることであった。これらの細胞のタイプに関して得られた大いに希望を与える結果は、これらの両親媒性ベクターモデルの開発を充分に保証する。
【0067】
前の分子上に構築された分子Eに、この特定の場合において、周知であって、PBNの誘導体を含む効果的なスピントラップが付与される。
【0068】
【化9】
【0069】
最初の試験は、Necker病院にて、in vitroで、NARP突然変異を示す子供からの皮膚生検由来の線維芽細胞に対して行われた。製品TA1PBN(“Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA”, Geromel V, Kadhom N, Ceballos-Picot I, Ouari O, Polidori A, Munnich A, Roetig A, Rustin P. Hum Mol Genet(2001) (in press) and “Synthesis of a glycolipidic amphiphile nitrone as a new spin trap for biological applications”), O. Ouari, A. Polidori, F. Chalier, P. Tordo, B. Pucci. J. Org. Chem., (1994), 64, 3554-3556)に対して同様の方法であらかじめ試験した場合、分子Eは、細胞保護能力を示し、アポトーシスのプロセスを阻害する。このタイプの製品に関し、培養されたいかなる細胞に対しても、毒性は測定されなかった。
【0070】
これらの結果は、その様なベクター化の概念の利点を、再度立証し、全く異なる適用分野における、その潜在的可能性を明確に示す。
【0071】
本発明の他の好ましいバリアントによれば、式(Ia)に相当する分子において、有効成分は、フリーラジカルスカベンジャー、特にN−ベンジリデン−tert−ブチルアミンオキサイド誘導体から選択される。
【0072】
式(II)に相当する分子のうち、本発明の他の特定の主題は、式(Ib)に相当する分子から構成される:
Pep−[AA1]−Y
(Ib)
式中、Y及びAA1は、上記式(I)、特に式(Ia)と同様に定義され、RのバリアントであるPepは、2〜10、好ましくは4〜6アミノ酸を含むペプチド鎖を表す。有利には、Pep又はAA1は、少なくとも1つのチロシン単位を含む。
【0073】
有利には、Pepは、既知の生物学的標的に対する親和性について選択され;特に、このペプチド鎖は、αVβ3インテグリンによって認識されることが知られているRGD(アルギニン−グリシン−アスパラギン酸)配列を含み得る。
【0074】
本発明の他の主題は、式(Ic)に相当する分子から構成される:
【0075】
【化10】
【0076】
式中、x、X、AP、AA1及びYは、上記式(I)と同様に定義され;特に、分子中、x、X、AP、AA1及びYは、上記式(Ia)と同様に定義される;Pepは、上記式(Ib)と同様に定義される。
【0077】
好ましくは、1以上の以下の条件を確認する:
−Pepは、αVβ3インテグリンによって確認されるペプチドであり、APは抗有糸分裂剤である;
−X、Pep又はAA1は、少なくとも1つのチロシン残基を含む;
−Xは、1〜3アミノ酸の鎖を表す。
【0078】
式(Ib)及び(Ic)の化合物の例は、下記及び実験の部に示される。
c)例3:抗癌治療
現在、腫瘍細胞と正常細胞の有効な区別、従って、適切なターゲッティングのための基準を示すことが可能な技術はない。しかしながら、知られているように、癌性腫瘍の増殖は、脈管化率(rate of vascularization)、従って、それに伴う血管新生の現象と密接に結びついている。これらの観察は、知られているように、血管新生を阻害することができ、従って、この方法によって、腫瘍の増殖を妨げることができる基質を、科学団体に開発させた。実際、現在は、血管新生細胞によって輸送される膜タンパク質(インテグリンと呼ばれる)は、これらの細胞の増殖のプロセスに活発に加わっていることが、一般的に認められている。より詳しくは、αVβ3インテグリンは、特定のペプチド配列(RGD(アルギニン−グリシン−アスパラギン酸)配列)を認識する。従って、この単位の提案されたベクター上へのグラフトは、特異的に血管新生部位、従って、最終的には腫瘍部位をターゲッティングする能力をそれに付与するはずである。この同じベクターに、抗有糸分裂剤を付加することは、癌性細胞の選択的な破壊を可能にするはずである。この予想において、分子Bは、最初、ベクターが無害であることを確認するため、及びその特異性を測定するために調製された。ターゲッティング薬剤により大きな自由度を付与するため、前記ターゲッティング薬剤を、中心リシン上に、疎水性アミノ酸であるセリンを用いてグラフトした。この分子は、水に可溶であり、両親媒性の性質である。
【0079】
【化11】
【0080】
これらの肯定的な結果を考慮すれば、さらに、RGDペプチド配列及びメルファラン(分子D)又はAra−C(分子C及びF)等の抗有糸分裂剤を有するベクターは、その後、合成され、それらの抗癌活性についての解析の過程にある。
【0081】
【化12】
【0082】
選択された抗有糸分裂剤は、ここでは単にモデルであって、与えられた抗有糸分裂剤を導入すること及び輸送することの利便性を簡単に説明するに過ぎない。このタイプのベクターは、アドリアマイシン(この特定の場合において、分子(Ic)は、X又はPepのいずれかにおいて、Gly−Phe−Leu−Glyタイプのペプチド断片を含む)、5−Fu(5−フルオロウラシル)、メルファラン、又はメシル酸イマチニブ(STI573、Glivec(登録商標))等のチロシンキナーゼ阻害剤等(例えば、より一般的にはこれらの担体上にグラフトされ得るあらゆる抗癌剤)基質のベクター化のための薬剤としても使用され得、また使用されるであろう。疎水性フッ素化炭化水素鎖の存在は、膜貫通性の通過を促進する。有効成分の放出は、適切な細胞質酵素によるペプチド結合の加水分解によって提供される。
【0083】
すでに得られた、最初のin vitroの結果は、完全にこの概念を立証する。
【0084】
本発明の主題は、上記式(I)に相当する化合物の医薬品を調製するための使用でもある。
【0085】
実際、本発明による式(I)に相当する化合物は、従来技術の化合物のものよりも大きいか、又は同等の生物学的標的に到達するためのバイオアベイラビリティー及び能力を有することが示されている。
【0086】
この特性は、多様な分野における本発明の分子の使用を想定することを可能にする:
−治療分野において、本発明のプロダクトは、全て種類の病状、特に様々な形態の癌、ならびに酸化ストレス及び含酸素フリーラジカル種の形成に関連する病状の予防及び/又は治療に使用され得る。
【0087】
このため、本発明の主題は、薬学的に活性な担体中に本発明のよる化合物を含有する薬学的組成物である。
【0088】
本発明の主題は、癌の予防及び/又は治療を意図した薬学的組成物の調製のための、式A、C、D又はFの化合物の使用でもある。
【0089】
本発明の主題は、癌性細胞の存在を検出することを意図した薬学的組成物を調製するための式Bの化合物の使用でもある。
【0090】
本発明の主題は、酸化ストレス及び含酸素フリーラジカル種の形成に関連する病状、特に、免疫及び炎症性疾患、虚血再灌流症候群、動脈硬化、アルツハイマー病、パーキンソン病、UV及び電離放射線による損傷、メラノーマ等の特定の癌及び細胞老化を予防及び/又は治療することを意図した薬学的組成物の調製のための、式Eの化合物の使用でもある。
【0091】
本発明のプロダクトは、当業者にとって公知のあらゆる経路、特に、静脈注射又は筋肉注射、或いは経口又は経皮投与によって投与され得る。それらは、単独又は他の活性な薬剤と組み合わせて使用され得る。その用量及び1日投与量は、関係する分子について測定された活性及び患者の体重に従って適応される;
−化粧品分野において、式Eの化合物は、老化の影響を予防及び/又は処置するために使用され得る。
【0092】
従って、本発明の主題は、美容上(cosmetically)許容される担体中に式Eの化合物を含有する化粧品組成物でもある。
【0093】
前記組成物は、皮膚又は外皮(爪、髪)への適用のためのものであってもよい。
【0094】
それは、水性又は油性溶液、油中水型乳剤又は水中油型乳剤、トリプル乳剤又は軟膏の形態であってもよい。
【0095】
本発明の化合物は、フリーラジカルスカベンジャー活性が望まれる、以下:スキンケアクリーム、日焼け止め(antisun)製品、化粧落とし製品、皮膚又は髪用パック、シャンプー、口紅、ほお紅、ファンデーション、マニキュア液等の化粧品等のあらゆる化粧品組成物中に取り入れられ得る
それらの多様なメディウムへの溶解性のため、本発明の化合物は、使用が容易であり、様々な条件下で採用され得る。
【発明を実施するための最良の形態】
【0096】
1/実施例1:
A−分子Aの調製
30mlの無水メタノール中に溶解された、2g(4mmol)の1H,1H,2H,2H−ペルフルオロデカンアジド化合物を、パラジウム−炭の存在下にて水素化に供する。4時間の反応の後、メディウムをセライト(celite)を通して濾過し、溶媒を減圧下にて蒸発させる。相当する(corresponding)アミン2は、精製せずに単離される(定量的収率)。
【0097】
化合物2を、30mlのジクロロメタン中で、2.2g(4mmol)のBoc−Lys−(Z)−OPhF5 3.の存在下で、再度反応させる。溶液のpHを、数滴のDIEAを添加することによって8にする。
【0098】
周囲温度にて16時間攪拌した後、反応メディウムを、減圧下にて濃縮する。
【0099】
このメディウムを、シリカゲルカラムクロマトグラフィー(溶離液:3/7酢酸エチル/シクロヘキサン)によって精製する。酢酸エチル/ヘキサン混合溶液からの結晶化によって、フッ素化化合物4(2.68g;3.24mmol;80%)を、白色粉末の形態で得る。
【0100】
【化13】
【0101】
rf: 5/5 シクロヘキサン/酢酸エチル中に0.36
[α]D = -8.2 (c: 1; CHCl3).
融点: 86.5-88.3°C.
1H NMR (250 MHz, CDCl3): δ 7.36 (5H, s, CH arom), 6.85 (1H, m, NH アミド), 5.27 (1H, d, J = 6.65 Hz, NH ウレタン), 5.11 (2H, s, CH2O), 4.99 (1H, t, J = 5.8 Hz, NH ウレタン), 4.06 (1H, m, CHCO), 3.58 (2H, dd, J = 6.5 Hz, CH2NH), 3.20 (2H, dd, J = 6.2 Hz, CH2NH), 2.35 (2H, m, CH2CF2), 2.0〜1.0 (15H, m, Boc由来CH3 及び CH2 Lys).
13C NMR (62.86 MHz, CDCl3): δ 172.5 (CONH), 156.7; 155.9 (OCONH), 136.6 (CIV arom.), 128.5; 128.1 (CH arom.), 80.4 (CIV), 66.7 (CH2OCONH), 54.4 (CHCO), 40.2 (CH2NH), 31.9 (CH2NH), 31.3 (CH2), 30.7 (CH2Rf), 29.5 (CH2), 28.2 (tert-ブチル由来CH3), 22.4 (CH2).
19F NMR (235 MHz, CDCl3): δ -80.7 (3F, s, CF3), -113.9 (2F, s, CF2CH2), -121.9 (6F, s, (CF2)3), -122.7 (2F, s, CF2), -123.5 (2F, s, CF2), -126.6 (2F, s, CF2CF3).
【0102】
30mlのジオキサン中に溶解された0.5g(0.6mmol)の化合物4を、パラジウム−炭の存在下において水素化に供する。
【0103】
15時間の反応の後、メディウムを、セライトを通して濾過し、溶媒を減圧下にて蒸発させる。得られたアミン5を、0.21g(0.44mmol)の新しく調製されたラクトビオノラクトン(lactobionolacton)の存在下でジクロロメタン中にて反応させ、そしてメディウムに、この溶液のpHを8にするためにDIEAを添加する。
【0104】
アミン5が完全になくなった後(TLC)、反応メディウムを、減圧下にて濃縮する。低温条件下にて、反応粗生成物に40mlの1:1無水酢酸/ピリジン混合溶液を加える。攪拌は、周囲温度にて18時間維持され、その後、反応混合溶液を150mlの1N HCl上に注ぐ。水相を、50mlのジクロロメタンで3回抽出する。有機相を、60mlの1N HCl、その後60mlの塩水で、それぞれ2回洗浄し、最後に、Na2SO4上で乾燥する。この溶媒を、減圧下にて除去し、粗生成物を、シリカゲル上のフラッシュ・クロマトグラフィー(溶離液:6/4その後7/3 酢酸エチル/シクロヘキサン)によって精製して白色粉末の形態で化合物6(0.55g;0.39mmol;65%)を生成する。
【0105】
【化14】
【0106】
rf: 6/4 酢酸エチル/シクロヘキサン中に0.22
[α]D = +2.9 (c, 1; CHCl3).
融点: 65°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.07 (2H, m, NH), 7.34 (5H, s, CH arom.), 7.01 (1H, m, NH), 5.47 (1H, m, 糖由来H), 5.30 〜 5.10 (2H, m, 糖由来H), 5.02 〜 4.79 (5H, m, CH2-O 及び糖由来H), 4.50 〜 3.90 (8H, m, 糖由来H及びリシン由来CHα), 3.38 (2H, m, CH2-NH), 2.97 (2H, m, CH2-NH), 2.30 (2H, m, CH2-CF2), 2.14, 2.09, 2.04, 2.01, 1.96, 1.92 (24H, 6s, アセチル由来CH3), 1.65 〜 1.10 (6H, m, リシン由来CH2)
13C NMR (62.86 MHz, CDCl3): δ 171.3 (CO-NH), 170.5, 170.5, 170.1, 170.0, 170.0, 169.7, 169.2 (7s, CO-O), 167.9 (CO-NH), 156.7 (O-CO-NH), 136.7 (CIVarom.), 128.4, 128.0, 127.9 (CH arom.), 101.6 (CH-1’), 77.9 (CH-4), 72.7 (CH-2), 71.1, 70.9 (CH-5’ 及び CH-3’), 70.0 (CH-5), 69.4 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 66.5 (CH2-O), 61.6, 61.0 (CH2-6及び CH2-6’), 52.6 (CH-CO), 40.4 (CH2-NH), 31.9 (CH2-NH), 31.2 (CH2-), 30.5 (CH2-Rf), 29.1 (CH2-), 22.2 (CH2-), 20.6, 20.5, 20.4, 20.4, 20.3 (アセチル由来CH3)
19F NMR (235 MHz, DMSO-d6): δ -80.2 (3F, s, CF3), -113.0 (2F, s, CF2-CH2), -121.4 (6F, s, 3CF2), -122.2 (2F, s, CF2), -123.0 (2F, s, CF2), -125.4 (2F, s, CF2-CH2).
【0107】
化合物6のベンジルオキシカルボニル基を、化合物4から化合物5に通過(pass)する際にすでに記載された実験方法に従って、脱保護する。0.5g(0.36mmol)の化合物6を用い、アミン7を、定量的収率で得る。
【0108】
得られたアミン7を、0.21(0.44mmol)のZ−Tyr−OPhF5(化合物8)の存在下で30mlのジクロロメタン中にて反応させ、溶液のpHを8にするためにDIEAを添加する。
【0109】
アミン7が完全になくなった後(TLC)、反応メディウムを減圧下で濃縮し、シリカゲルカラムクロマトグラフィー(溶離液:7 酢酸エチル/3 シクロヘキサン)によって精製する。化合物9(0.32g;0.21mmol;58%)を、白色粉末の形態で得る。
【0110】
【化15】
【0111】
rf: 7 酢酸エチル/3 シクロヘキサン中に0.35
[α]D = +1.5 (c: 1; CHCl3).
融点: 37.6°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.01 (2H, m, NH), 7.71 (1H, m, NH), 7.21 (5H, s, CH arom.), 7.05 (3H, m, NH, 2H arom tyr), 6.89 (2H, d, J = 8.29 Hz 2H arom tyr), 5.22 (1H, d, J = 5.35 Hz糖由来H), 5.02 (2H, s, CH2Ph), 4.99 (2H, s, CH2Ph), 4.92 (2H, m, 糖由来H), 4.70 〜 4.55 (5H, m, 糖由来H), 4.01 〜 3.75 (9H, m, CHα tyr, CHα lys, CH2NH, 糖由来5H), 2.76 (2H, m, CH2-NH), 2.16 (2H, m, CH2-CF2), 1.90 〜 1.68 (24H, 6s, アセチル由来CH3), 1.29 〜 0.91 (6H, m, リシン由来CH2)
13C NMR (62.86 MHz, CDCl3): δ 171.85; 171.48 (CO-NH), 170.67, 170.32, 170.18, 170.0, 169.63; 169.42 (6s, CO-O), 156.29 (CO-NH), 137.47; 136.61; 135.54 (CIV arom.), 130.71; 129.03; 128.80; 128.71; 128.12; 127.96; 121.17 (CH arom.), 101.07 (CH-1’), 78.69 (CH-4), 72.22 (CH-2), 70.86; 70.21 (CH-5’ 及び CH-3’), 70.06 (CH-5), 69.61 (CH-3), 69.3 (CH-2’), 67.60 (CH-4’), 65.69 (CH2-O), 61.72, 61.48 (CH2-6 及び CH2-6’), 56.62 (CHαtyr); 52.68 (CHαlys); 37.48 (CH2-NH), 31.86 (CH2-NH), 31.43 (CH2-), 30.08 (CH2-Rf), 29.09 (CH2-), 22.77 (CH2-), 21.08, 21.03, 20.94, 20.86, 20.76 (アセチル由来CH3)
19F NMR (235 MHz, DMSO-d6): δ -80.19 (3F, s, CF3), -113.38 (2F, s, CF2-CH2), -121.67 (6F, s, 3CF2), -122.45 (2F, s, CF2), -123.24 (2F, s, CF2), -125.70 (2F, s, CF2-CH2).
【0112】
同じように、すでに記載された実験方法に従って、30mlのエタノールに溶解された0.3g(0.19mmol)の化合物9を、パラジウム−炭の存在下にて 水素化に供する。
【0113】
得られたアミン10を、0.107g(0.23mmol)のサリドマイドの活性エステル11の存在下にてジクロロメタン中で反応させ、溶液のpHを8にするためにDIEAを添加する。
【0114】
【化16】
【0115】
Rf= 7EtOAc/3シクロヘキサン中に0.65
[αD] = +3.9 (c: 1; DMF).
1H NMR (250 MHz, CDCl3): 8.63 (1H, s, Ph); 8.61 (1H, d, Ph); 8.09 (1H, d, Ph); 5.04 (1H, m, NCH); 2.86 (3H, m, CH2CO, CHCH2CO); 2.19 (2H, m, CHCH2).
19F NMR (235 MHz, CDCl3) : -152.6 (2F, d, CF); -156.67 (1F, t, CF); -161.87 (2F, t, CF).
13C NMR (62.86, DMSO): 177.97; 174.93; 171.64; 171.61; 170.96 (5 CO); 142.01; 140.94; 139.68; 136.89; 129.09; 128.64 (芳香族 C); 54.44 (NCH); 36.13 (CH2CO); 27.12 (NCHCH2) 11
【0116】
アミン10が完全になくなった後(TLC)、反応メディウムを、減圧下にて濃縮し、シリカゲルカラムクロマトグラフィー(溶離液:8/2の後9/1 酢酸エチル/シクロヘキサン)によって精製する。
【0117】
化合物12を、極めて低い収率(11mg;6.4μmol;4.5%)で得る。
【0118】
rf: 酢酸エチル中に0.63
該分子の糖部分の脱アセチル化を、触媒量のナトリウムメトキシドを含有するメタノール中で周囲温度にて行う。
【0119】
H+樹脂(amberlite IRC 50)上での処理、濾過及び溶媒の蒸発の後、脱アセチル化された生成物Aを、定量的収率で単離する。
【0120】
【化17】
【0121】
B−生物学的アッセイ
そのような基質は、線維芽細胞及びB16メラノーマの細胞培養物に対して、完全に許容できない毒性を示さない。In vivoにおいて、この分子は、腫瘍のストローマ(stroma)中に濃縮され、それを極めてはっきりと視覚化することを可能にする(表1参照)。
【0122】
【表1】
【0123】
現在進行中の研究は、サリドマイド単独と比べ、腫瘍成長の阻害におけるその有効性を明示することを可能にするであろう。ニワトリの胚に対する血管成長アッセイ(ニワトリ大動脈輪アッセイ(chick aortic ring assays))において得られた最初の結果は、微小血管の成長の阻害におけるこの構造の有効性を示した。分子Aは、20μMにて有効であり、200μMにて完全に成長を妨げることが見出されたのに対し、その様な濃度において、サリドマイドは、効果がないことが見出される。
【0124】
この最初の結果は、提案された構造の全面的な無害、及び診断のための可能性のある有利な点を示す(ここで、サリドマイドは、有効成分の1つの例にすぎない)。
【0125】
2/実施例2:分子Eの調製
この合成例は、図1に示される。
活性エステルHOOCPBNの合成
【0126】
【化18】
【0127】
N−(tert−ブチル)ヒドロキシルアミンアセテートを、飽和炭酸ナトリウム水溶液中に溶解する。ヒドロキシルアミンを、エーテルで抽出する。有機相を、Na2SO4上で乾燥し、その後、この溶媒を減圧下にて除去して粉体白色結晶の形態で遊離N−tert−ブチルヒドロキシルアミンを生成する。
【0128】
1.00gの4−カルボキシベンズアルデヒド(6.67mmol−1当量)及びスパチュラの先端の4Åモレキュラーシーブを、アルゴン雰囲気下にて5mlの脱気無水エタノール中に懸濁する。5mlのエタノール中の0.570gのヒドロキシルアミン(6.37mmol−0.95当量)溶液を、ベンズアルデヒド溶液に添加し、そしてこのメディウムを、暗所で60℃にする。18時間後、0.200gのヒドロキシルアミン(2.25mmol−0.33当量)を、このメディウムに加え、さらに18時間攪拌を続ける。反応混合溶液を、セライトの層を通して濾過し、減圧下にて溶媒を除去し、そして粗生成物を、シリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4 酢酸エチル/シクロヘキサン)によって精製する。メタノール/エーテル混合溶液からの再結晶後、ニトロンX(0.590g−2.67mmol−40%)を、白色粉末の形態で得る。
【0129】
モル質量 (C12H15NO3): 221.3 g・mol-1
融点: 214.5 - 215.7°C.
1H NMR (250 MHz, DMSO-d6): δ 8.42 (2H, m, J = 8.5 Hz, H arom.), 7.95 (3H, m, H arom. 及び CH=N(O)), 1.51 (9H, s, tert−ブチル由来CH3).
13C NMR (62.86 MHz, DMSO-d6): δ 166.9 (CO), 135.3 (CIVarom), 131.0 (CH=N(O)), 129.2 (CH arom.), 128.3 (CIV arom), 127.9 (CH arom.), 71.1 (CIV), 27.8 (tert−ブチル由来CH3).
UV (MeOH, nm): λmax = 287.
【0130】
0.260gのニトロンX(1.18mmol−1当量)を、アルゴン雰囲気下にて15mlのジオキサン中に溶解する。0.290gのDCC(1.41mmol−1.2当量)及び0.16gのHOSu(1.41mmol−1.2当量)を、反応メディウムに添加する。48時間の攪拌後、この反応メディウムを、焼結ガラス(ポロシティ 4)を通して濾過し、この溶媒を、その後、減圧下にて除去する。シリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4 酢酸エチル/シクロヘキサン)による精製後の、酢酸エチル/ヘキサンの混合溶液からの再結晶の後、化合物Y(0.25g−0.79mol− 67%)を、白色粉末の形態で得る。
【0131】
モル質量 (C16H18N2O5): 318.3 g・mol-1
融点: 177.4 - 178.3°C.
1H NMR (250 MHz, CDCl3): δ 8.37 (2H, m, J = 8.6 Hz, H arom.), 8.12 (2H, d, J = 8.6 Hz, H arom.), 7.65 (1H, s, CH=N(O)), 2.89 (4H, s, CH2-CO), 1.61 (9H, s, tert−ブチル由来CH3).
13C NMR (62.86 MHz, CDCl3): δ 169.3, 161.3 (CO), 136.8 (CIVarom), 130.7 (CH arom.), 128.6 (CIV arom), 128.6 (CH arom.), 125.5 (CH=N(O)), 72.2 (CIV), 28.4 (CH2-CO), 25.7 (tert−ブチル由来CH3).
【0132】
[5−tert−ブトキシカルボニルアミノ−5−(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10−ヘプタデカフルオロデシルカルバモイル)ペンチル]カルバミン酸ベンジルエステル3の合成
【0133】
【化19】
【0134】
2.26gのアジドC8F17CH2CH2N3(4.62mmol−1当量)を、20mlのエーテル中に溶解する。このメディウムを0℃にし、300mgのパラジウム−炭(10% − 65mg/mmol)を、フラクションごとに添加する。水素化ボンベ(圧力8バール)において6時間の攪拌の後、このメディウムをセライトの層を通して濾過し、溶媒を、減圧下にて除去する。アミンC8F17CH2CH2NH2 2を精製せずに得る。
【0135】
1.79gのBoc−Lys(Z)OH(4.70mmol−1当量)、1.07gのDCC(5.18mmol − 1.1当量)及び0.64gのHOBt(4.70mmol − 1当量)を、20mlの無水ジクロロメタン中に溶解する。15分の攪拌後、10mlの無水ジクロロメタン中のアミン2の溶液を、この混合溶液に添加する。攪拌を24時間続ける。反応粗生成物を、焼結ガラス(ポロシティ4)を通して濾過し、その後、溶媒を減圧下にて除去した。シリカゲル上のフラッシュクロマトグラフィー(溶離液:7:3〜6:4 シクロヘキサン/酢酸エチル)による精製の後、フッ素化化合物3(3.11g − 3.70mmol − 79%)を、白色粉末の形態で得る。
【0136】
モル質量 (C29H32F17N3O5): 825.6 g・mol-1
融点: 86.5 - 88.3°C.
1H NMR (250 MHz, CDCl3): δ 7.36 (5H, s, CH arom.), 6.85 (1H, m, NH アミド), 5.27 (1H, d, J = 6.65 Hz, NH ウレタン), 5.11 (2H, s, CH2-O), 4.99 (1H, t, J = 5.8 Hz, NH ウレタン), 4.06 (1H, m, CH-CO), 3.58 (2H, dd, J = 6.5 Hz, CH2-NH), 3.20 (2H, dd, J = 6.2 Hz, CH2-NH), 2.35 (2H, m, CH2-CF2), 2.0 〜 1.0 (15 H, m, Boc由来CH3及びCH2 lys).
13C NMR (62.86 MHz, CDCl3): δ 172.5 (CO-NH), 156.7, 155.9 (O-CO-NH), 136.6 (CIV arom.), 128.5, 128.1 (CH arom.), 80.4 (CIV), 66.7 (CH2-O-CO-NH), 54.4 (CH-CO), 40.2 (CH2-NH), 31.9 (トリプレット, CH2-NH), 31.3 (CH2), 30.7 (CH2-Rf), 29.5 (CH2), 28.2 (tert−ブチル由来CH3), 22.4 (CH2).
19F NMR (235 MHz, DMSO-d6): δ -80.7 (CF3, s), -113.9 (CF2-CF3, s), -121.9 (3 CF2, m), -122.7 (CF2, s), -123.5 (CF2, s), 126.0 (CF2-CH2, s).
[α]D = -8.2 (c, l, CHCl3).
【0137】
ラクトLys(Z)C8F17(OAc)8 5の合成
【0138】
【化20】
【0139】
2.03gの化合物3(2.45mmol − 1当量)を、20mlの無水ジクロロメタン中に溶解する。このメディウムを0℃にし、40mlの8.5:1.5 CH2Cl2/TFA混合溶液を、添加の間温度を0℃に保持しながら滴下する。4時間の攪拌の後、溶媒を減圧下にて除去する。粗生成物をエーテル中に回収し、その後蒸発させて、共蒸発(co−evaporation)によってTFAの残存トレースを除去する。この操作を数回繰り返し、遊離アミン4を生成する。
【0140】
並行して、1.15gのラクトビオン酸(3.19mmol − 1.3当量)を、3滴のTFAで酸性にした40mlの1:1 メトキシエタノール/トルエン混合溶液中に懸濁する。
【0141】
減圧下にて45℃での蒸発の後、メディウムを、30mlの2:1 メトキシエタノール/トルエン混合溶液中に回収し、その後、乾燥するまで蒸発させる。ラクトビオノラクトンを生成するように、後者の操作を2度繰り返す。
【0142】
ラクトビオノラクトン及びアミン4を、アルゴン雰囲気下で、40mlのメタノール中に溶解する。この溶液のpHを、TEAを添加することによって9にし、その後、メディウムを24時間の還流にかける。減圧下でのメタノールの除去後、40mlの1:1 無水酢酸/ピリジン混合溶液を、冷却条件下で、粗生成物に添加する。攪拌を18時間の間維持し、その後、反応混合溶液を150mlの1N HClに注ぐ。水相を、50mlのジクロロメタンで3回抽出する。有機相を、60mlの1N HClで2回、その後60mlの塩水でそれぞれ洗浄し、そして最後に、Na2SO4上で乾燥する。溶媒を減圧下にて除去し、粗生成物をシリカゲル上のフラッシュクロマトグラフィー(溶離液:6:4〜7:3 酢酸エチル/シクロヘキサン)によって精製して化合物5(2.23g − 1.59mmol − 65%)を白色粉末の形態で生成する。
【0143】
モル質量 (C52H60F17N3O22): 1402.0 g・mol-1
融点: 65°C (分解の開始).
1H NMR (250 MHz, DMSO-d6): δ 8.07 (2H, m, NH), 7.34 (5H, s, CH arom.), 7.01 (1H, m, NH), 5.47 (1H, m, 糖由来H), 5.30 〜 5.10 (2H, m, 糖由来H), 5.02 〜 4.79 (5H, m, CH2-O 及び糖由来 H), 4.50 〜 3.90 (8H, m, 糖由来H及びリシン由来CH), 3.38 (2H, m, CH2-NH), 2.97 (2H, m, CH2-NH), 2.30 (2H, m, CH2-CF2), 2.14, 2.09, 2.04, 2.01, 1.96, 1.92 (24H, 6s, アセチル由来CH3), 1.65 〜 1.10 (6H, m, リシン由来CH2).
13C NMR (62.86 MHz, CDCl3): δ 171.3 (CO-NH), 170.5, 170.5, 170.1, 170.0, 170.0, 169.7, 169.2 (7s, CO-O), 167.9 (CO-NH), 156.7 (O-CO-NH), 136.7 (CIV arom.), 128.4, 128.0, 127.9, (CH arom.), 101.6 (CH-1’), 77.9 (CH-4), 72.7 (CH-2), 71.1, 70.9 (CH-5’ 及び CH-3’), 70.0 (CH-5), 69.4 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 66.5 (CH2-O), 61.6, 61.0 (CH2-6 及び CH2-6’), 52.6 (CH-CO), 40.4 (CH2-NH), 31.9 (トリプレット, CH2-NH), 31.2 (CH2-), 30.5 (トリプレット, CH2-Rf), 29.1 (CH2-), 22.2 (CH2-), 20.6, 20.5, 20.4, 20.4, 20.3 (5s, アセチル由来CH3).
19F NMR (235 MHz, DMSO-d6): δ -80.2 (CF3, s), -113.0 (CF2-CF3, s), -121.4 (3 CF2, s), -122.2 (CF2, s), -123.0 (CF2, s), -125.4 (CF2-CH2, s).
[α]D = +2.9 (c, 1, CHCl3).
【0144】
ラクトLys(PBN)C8F17(OAc)8 7の合成
【0145】
【化21】
【0146】
0.400gの化合物5(0.28mmol−1当量)を、10mlのジオキサン中に溶解する。メディウムを0℃にし、0.190gのパラジウム−炭(10%−65mg/mmol)をフラクションごとに添加する。水素化ボンベ(圧力8バール)において20時間の攪拌の後、メディウムをセライトの層を通して濾過し、溶媒を減圧下にて除去する。アミン6を、精製せずに白色粉末の形態で得る。
【0147】
アミン6を、アルゴン気流の下、5mlの無水ジクロロメタン中に溶解する。0.090gの活性エステルX(0.28mmol−1当量)を、メディウムに添加し、DIEAを添加することによってpHを9にする。
【0148】
攪拌を、アルゴン雰囲気下にて24時間継続する。この溶媒を、減圧下にて蒸発させ、粗生成物をシリカゲル上のフラッシュクロマトグラフィー(溶離液:酢酸エチル)によって精製する。Sephadex LH−20樹脂上でのサイズ排除クロマトグラフィー(溶離液:1:1 ジクロロメタン/エタノール)によるさらなる精製は、ニトロン7(0.230g − 0.156mmol − 54%)を白色粉末の形態で得ることを可能にする。
【0149】
モル質量 (C56H67F17N4O22): 1471.1 g・mol-1
融点: 75°C (分解の開始).
1H NMR (250 MHz, CDCl3): δ 8.36 (2H, d, J = 8.3 Hz, H arom.), 7.92 (2H, d, J = 8.4 Hz, H arom.), 7.68 (1H, s, CH=N(O)), 6.94 (3H, m, NH), 5.51 (1H, dd, J = 2 Hz 及び J = 4.4 Hz, H-4’), 5.32 (2H, m, H-2 及び H-3), 5.17 〜 4.95 (3H, m, H-2’, H-5 及び H-3’), 4.62 (1H, d, J = 7.7 Hz, H-1’), 4.50 (1H, dd, J = 2.7 Hz 及び J = 12.5 Hz, 糖由来H), 4.33 (1H, m, リシン由来CH), 4.18 (1H, dd, J = 1.6 Hz 及び J = 6.3 Hz, 糖由来H), 4.12 〜 3.87 (4H, m, 糖由来H 及び H-5’), 3.50 (2H, m, CH2-NH), 3.39 (2H, m, CH2-NH), 2.35 (2H, m, CH2-CF2), 2.15, 2.09, 2.08, 2.04, 2.02, 2.00, 1.96 (24H, 7s, アセチル由来CH3), 1.60 (11H, m, tert-ブチル由来CH3及び CH2 Lys), 1.85 (2H, m, CH2Lys), 1.35 (2H, m, CH2 Lys).
13C NMR (62.86 MHz, CDCl3): δ 171.5 (CO-NH), 170.6, 170.5, 170.3, 170.3, 170.0, 169.7, 169.32 (7s, CO-O), 168.2 (CO-NH), 167.1 (CO-NH), 135.2 (CIV arom.), 133.7 (CIV arom.), 129.3 (CH=N(O)), 128.6, 127.3 (CH arom.), 101.7 (CH-1’), 78.5 (CH-4), 72.9 (CH-2), 71.4 (CIV), 71.0, 70.9 (CH-5’ 及び CH-3’), 68.9 (CH-5), 69.3 (CH-3), 69.0 (CH-2’), 66.9 (CH-4’), 61.6, 61.1 (CH2-6及びCH2-6’), 52.7 (CH-CO), 39.4 (CH2-NH), 31.9 (m, CH2-NH), 31.1 (CH2-), 30.5 (トリプレット, CH2-Rf), 28.7 (CH2-), 28.2 (tert-ブチル由来CH3), 22.2 (CH2-), 20.7, 20.7, 20.7, 20.6, 20.5, 20.5, 20.4, (7s, アセチル由来CH3).
19F NMR (235 MHz, CDCl3): δ -80.7 (s, CF3), -114.2 (s, CF2-CF3), -121.9 (s, 3 CF2), -122.7 (s, CF2), -123.5 (s, CF2), -126.1 (s, CF2-CH2).
[α]D = +1.6 (c, 1, CHCl3).
【0150】
3/実施例3:
A−化合物Bの合成
【0151】
【化22】
【0152】
0.99g(3×10−3mol)のCl−+H3N Lys(Z)OMeを、100mlの丸底フラスコ中にて、10mlのジクロロメタン中に溶解する。pHを、TEAを用いて8にする。その後、1.15g(3×10−3mol、1当量)のFmoc Ser(OtBu)OHを、1.72(3.9×10−3mol、1.3当量)のBOPと共に添加する。
【0153】
pHを、反応の間8に保持する。メディウムを、周囲温度にて24時間攪拌し続ける。反応が完了すれば(TLC)、有機相を1N HClで洗浄し、その後、pH7を回復(reestabish)するためにNaHCO3で洗浄する。有機相をNa2SO4上で乾燥し、濾過し、その後減圧下にて蒸発させる。結晶化を、ジクロロメタン/Et2O混合溶液から行うことができる。1.93gの化合物1を、白色粉末の形態で得る。
【0154】
【表2】
【0155】
【化23】
【0156】
0.68g(1.53×10−3mol)のFmoc Asp(OtBu)OHを、0.34g(1.83×10−3mol、1.2当量)のペンタフルオロフェノール及び0.38g(1.83×10−3mol、1.2当量)のDCCと共に、50mlの丸底フラスコ中にて10mlのジクロロメタン中に、溶解する。反応を、周囲温度にて15時間の間攪拌しながら置く。濾過の後、メディウムを減圧下にて濃縮する。残存オイルをシリカゲル上にてクロマトグラフにかける(溶離液:2/8 酢酸エチル/シクロヘキサン)。結晶化を、酢酸エチル/ヘキサンから行う。760mgの2を、白色粉末の形態で得る。
【0157】
【表3】
【0158】
【化24】
【0159】
ジペプチド及び活性化アミノ酸間のカップリングの前に、化合物2の脱保護からなる行程が必要である。このため、1.3g(1.97mmol)のジペプチド1を、15mlの10% v/v ピペリジン/ジクロロメタン混合溶液中に溶解する。メディウムを、周囲温度にて攪拌したまま2時間放置し、その後、分液漏斗を用いて1N HClで洗浄する。その後、有機相を飽和炭酸水素ナトリウム溶液で洗浄する。有機相を、その後、硫酸ナトリウム上で乾燥した後、減圧下にて濃縮する。その後、カップリングを行うことができる。
【0160】
上記の脱保護されたジペプチドを、100mlのシングルネック(single−necked)丸底フラスコ中にて、丸底フラスコに加えられた1.138g(1.97mmol、1当量)の化合物2の存在下で、20mlのジクロロメタン中に溶解する。反応を、窒素気流のもと周囲温度にて、暗所で、DIEAで固定されたpH8にて行う。15時間後、反応を完了する(TLC)。反応メディウムを濃縮し、残渣をシリカゲル上にて、5/5酢酸エチル/シクロヘキサン溶出混合溶液中でクロマトグラフにかける。溶媒の蒸発の後、970mgの化合物3を、半透明ゲルの形態で得る。
【0161】
【表4】
【化25】
【0162】
1g(3.36×10−3mol)のFmoc Gly OH,0.681gのペンタフルオロフェノール(3.7×10−3mol、1.1当量)及び0.764gのDCC(3.7×10−3mol、1.1当量)を、10mlのジクロロメタン中に溶解する。このメディウムを、周囲温度にて24時間の間放置する。反応メディウムを、その後濾過し、その後、この濾過液を減圧下での蒸発によって濃縮する。残渣を、5/5酢酸エチル/シクロヘキサン溶出混合溶液を用いたシリカゲル上のフラッシュクロマトグラフィーにかける。結晶化を、酢酸エチル/シクロヘキサン混合溶液から行う。955mgの4を、白色粉末の形態で得る。収率:61.3%
【0163】
【表5】
【0164】
【化26】
【0165】
この合成は、このトリペプチド(化合物3)の合成と同様のプロトコルに従って、トリペプチドの脱保護によって進められる。500mg(6×10−3mol)の3を、脱保護する。脱保護するとすぐに、このペプチドを100mlの丸底フラスコ中の10mlのジクロロメタン中に溶解する。278mg(6×10−4mol、1当量)の化合物4を、反応メディウムに添加する。反応を、周囲温度にて窒素気流下に行い、DIEAを用いてpH8に維持する。16時間の後、反応を完了する(TLC)。減圧下でのメディウムの蒸発の後、残存オイルを、6/3 酢酸エチル/シクロヘキサン溶出混合溶液を用いてシリカゲル上にてクロマトグラフにかける。酢酸エチル/シクロヘキサン混合溶液から、426mgの5を、白色粉末の形態で得る。収率:80%
【0166】
【表6】
【0167】
【化27】
【0168】
ペンタペプチド5の脱保護は、化合物1の合成と同じ条件下で行われる。400mg(4.5×10−4 mol)のテトラペプチドを脱保護する。脱保護が完了したらすぐ、このテトラペプチドを、50mlのシングルネック丸底フラスコ中の15mlのジクロロメタン中に溶解する。219mg(4.5×10−4mol、1当量)のBocArg(Mtr)OH及び188g(5.85×10−4mol、1.3当量)のTBTUを加える。反応を、周囲温度にて窒素気流下、暗所で行い、DIEAを用いてpH8に維持する。16時間の後、この反応を完了する(TLC)。減圧下での蒸発の後、残存オイルを9/1酢酸エチル/シクロヘキサン溶離液を用いたシリカゲル上のクロマトグラフにかける。酢酸エチル/シクロヘキサンから結晶化を行う。417mgの6を、白色粉末の形態で得る。収率:92.5%
【0169】
【表7】
【0170】
【化28】
【0171】
500mg(1.01×10−3mol)のC8F17CH2CH2COOH、278mgのH2N TYR(OH)OtBu(1.01×10−3mol、1当量)及び251mgのDCC(1.2×10−3mol、1.2当量)を、50mlの丸底フラスコ中の10mlのDMF中に溶解する。反応を、周囲温度にて24時間、窒素気流下、暗所で行い、DIEAを用いてpH8に維持する。減圧下でのDMFの蒸発の後、残渣を2/8 酢酸エチル/シクロヘキサン溶離液を用いたシリカゲル上のクロマトグラフにかける。
【0172】
この生成物を、酢酸エチル/n−ヘプタンから結晶化することができる。550mgの7を、白色粉末の形態で得る。収率:76%
【0173】
【表8】
【0174】
【化29】
【0175】
380mg(5.34×10−4mol)の化合物7を、冷却条件下にて、TFA/CH2Cl2(3/7)溶液中に溶解する。2時間の攪拌の後、脱保護を完了する。その後、反応メディウムを濃縮した後、エーテルから数回にわたって沈殿させる。蒸発の後、白色粉末を得る(化合物8)。
【0176】
150mg(1.32×10−4mol)の化合物6を、メタノール中に溶解する。冷却条件下で8mgの10%パラジウム−炭を添加する。反応メディウムを、水素8気圧の下に置く。2時間30分後、この反応を完了する。この混合溶液を、セライト521を通して濾過し、この濾過液を減圧下にて濃縮する(化合物9)。
【0177】
化合物9(1.32×10−4mol)を、50mlの丸底フラスコ中の10mlのDMF中に溶解する。95mgの化合物8(1.45×10−4mol、1.1当量)及び76mgのTBTU(1.72×10−4mol、1.3当量)を添加する。反応を、周囲温度にて24時間、窒素気流下で暗所にて行い、DIEAを用いてpH8に維持する。反応メディウムを、減圧下にて濃縮する。残渣を酢酸エチル溶離液を用いてシリカゲル上のクロマトグラフにかける。141mgの生成物10を、白色粉末の形態で得る。収率 65.3%。
【0178】
【表9】
【0179】
チオ−アニソール(3;5;2)の存在下にて24時間の間、CH2Cl2中においてトリフルオロ酢酸溶液の反応にかけられる化合物10は、化合物Bを生成する(化合物Bはこの溶液へのエーテルの添加による沈殿、及びSephadex G50カラム(溶離液:H2O)上のクロマトグラフィーの後、純粋な形態で単離される)。凍結乾燥の後、生成物Bは、白色粉末の形態である。
【0180】
B−生物学的アッセイ
この分子の細胞毒性を、B16メラノーマ細胞に対して試験した;100μMを超える濃度まで、毒性を測定することはできなかった。ヨウ素125で標識した後、メラノーマを有している1群のマウスに静脈内に注射されたこの分子は、腫瘍のストローマに蓄積し、その後、徐々に腫瘍中に拡散する(表2参照)。
【0181】
【表10】
【0182】
4/実施例4:
メチルN−(t−ブトキシカルボニル)−N−γ−(2,3,6−トリメチル−4−メトキシ-ベンゼンスルフォニル)−L−アルギニルグリシニル−O−(t−ブチル)−L−アスパルチル−O−(t−ブチル)−L−セリニル−N−ε−((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11−ヘプタデカフルオロウンデカノイル)−N−ε−(4−(4−[ビス(2−クロロエチル)アミノ]フェニル)−ブチルアミド)−L−リシニル−L−チロシンアミド)−L−リシネート
【0183】
化合物Dの合成は、上記化合物に対して上記のものと類似であり、図2A及び2Bに要約される。
【0184】
Dの物理化学的特徴
Rf:酢酸エチル/メタノール: 98/2中に0.49.
MM: 2051.87 g・mol-1.
分解温度: 134°C.
[α]D20 = -0.77 (c, 0.1, CH3OH).
MS (FAB): m/z 2052 [M+H]+, 2074 [M+Na]+.
1H NMR (CD3OD):
δ 7.03 (4H, m, 2H arom. Tyr, 2H arom. Chloramb.); 6.67 (5H, m, 2H arom. Tyr, 2H arom. Chloramb., H arom Mtr); 4.81; 4.47; 4.20; 4.01 (6H, 3m, Hα Tyr, 2Hα Lys, Hα Asp, Hα Ser, Hα Arg); 3.82 (3H, s, CH3 エーテル Mtr Arg); 3.68 (3H, s, CH3 メチルエステル Lys); 3.83-3.60 (12H, m, 2Hα Gly, 2Hβ Ser, 8H Chloramb.); 3.14-2.75 (10H, m, 4Hε Lys, 2Hδ Arg, 2Hβ Asp, 2Hβ Tyr); 2.67-2.48 (8H, m, 2Hα 及び 2Hβ フッ素化鎖, 2Hα Chloramb., 2Hγ Chloramb.); 2.67; 2.61 (6H, 2m, 2CH3 Mtr Arg); 2.13 (5H, m, 2Hβ Chloramb., CH3 Mtr Arg); 1.93-1.16 (16H, m, 4Hβ Lys, 4Hγ Lys, 4Hδ Lys, 2Hβ Arg, 2Hγ Arg); 1.44; 1.42; 1.16 (27H, 3s, 9 CH3 tert-ブチルエステル Asp, tert-ブチルエーテル Ser, tert-ブチルウレタン Arg).
13C NMR (CD3OD):
δ 174.67; 174.14; 172.57; 172.31; 171.80; 171.75; 171.26; 170.80; 170.17 (CO Tyr, 2CO Lys, CO Ser, 2 CO Asp, CO Gly, CO Arg, CO フッ素化鎖, CO Chloramb.); 158.46; 156.80; 156.67; 155.93 (CO ウレタン Boc, C-OCH3 arom. Mtr Arg, C-OH arom. Tyr, C グアニジン Arg); 144.55 (C-N arom. Chloramb.); 138.10; 136.48; 133.42; 130.29; 129.97; 129.22; 127.51; 124.29; 114.83; 112.06; 111.38; 110.73 (C arom. Mtr, C arom. Tyr, C arom. Chloramb.); 81.17; 79.43 (C tert-ブチルエステル Asp, C tert-ブチルウレタンArg); 73.40 (C tert-ブチルエーテル Ser); 61.14 (Cβ Ser); 54.96; 54.60; 54.53; 53.89; 53.14; 52.26; 51.31; 49.90 (CH3 メチルエステル Mtr Arg, Cα Ser, Cα Arg, Cα Tyr, CH3 メチルエステル Lys, 2Cα Lys, Cα Asp, 2C-N Chloramb.); 42.30; 40.29; 38.58; 36.67; 36.52; 35.20 (Cα Gly, 2Cε Lys, Cδ Arg, Cβ Asp, Cβ Tyr, 2C-Cl Chloramb.); 33.85; 31.02; 30.69; 28.91; 28.67; 28.15; 27.69; 27.41; 26.96; 26.36; 25.82; 25.46 (2Cδ Lys, 2Cβ Lys, Cβ Arg, Cγ Arg, 9 CH3 tert-ブチルAsp, Ser, Arg, Cα-CF2, 2Cγ Lys, Cα, Cγ Chloramb.); 23.00; 22.66 (Cβ フッ素化鎖, Cγ Chloramb.); 22.52; 17.47; 10.74 (3 CH3 メチル Mtr Arg).
19F NMR (CD3OD):
δ -82.26 (3F); -115.39 (2F); -122.77 (6F); -123.62 (2F); -124.30 (2F); -127.17 (2F).
【0185】
5/実施例5:
メチル L−アルギニル−グリシニル−L−アスパルチル−L−セリニル−N−ε−(―O−(N−4−(N−(4,4,5,5,6,6,7,7,8,8,9,9−トリデカフルオロノナノイル)−L−セリニル)−1−β−D−アラビノフラノシルシトシン)−オキシカルボニル)−L−リシネート
【0186】
【化30】
【0187】
手法
Fを調製する手法は、前述の工程と同一である;この合成は、図3A、3B及び3Cに要約される。
【0188】
Fの物理化学的特徴
MM: 1390.45 g・mol-1. MS (FAB): m/z 1391 [M+H]+.
1H NMR (DMSO, D6):
δ 8.66 (1H, s, H 酸 Asp); 8.27-8.16; 7.94-7.84; 7.65; 7.21-6.92 (15H, 3m, Ha, NH GABA, 2NH Ser, 4H グアニジン Arg, NH2 Arg, NH Gly, NH Asp, 2NH Lys, NH Ara-C); 5.84 (1H, m, Hb); 5.31 (2H, m, 1Hα Ser, H1); 4.92 (1H, m, Hα Asp); 4.37-3.71 (17H, m, Hα Lys, Hα Ser, Hα Arg, 2Hα Gly, 4Hβ Ser, H2, H3, H4, CH3 メチルエステル Lys, 2 H5); 3.18-2.53 (14H, m, 2Hε Lys, CH2-NH GABA, 4OH, 2Hβ Asp, 2 Hα及び 2 Hβフッ素化鎖); 2.23 (2H, m, CH2-CO GABA); 1.46-1.03 (14H, m, 2Hβ, 2Hγ, 2Hδ Lys, 2Hβ, 2Hγ, 2Hδ Arg, CH2 GABA).
13C NMR (DMSO, D6):
δ 173.9; 173.6; 173.4; 172.8; 171.1; 170.4; 170.1; 169.6; 169.1; 168.6 (CO Lys, 2 CO Ser, 2 CO Asp, CO Gly, CO Arg, CO フッ素化鎖, CO GABA); 162.6; 157.4; 156.2; 155.0 (C グアニジン Arg, CO ウレタン Ser, N=C-NH, N-CO-N); 147.2 (Ca); 94.8 (Cb); 87.5; 86.2 (C1, C4); 76.6; 75.0 (C2, C3); 65.4; 62.0; 61.5 (2 Cβ Ser, C5); 55.6; 53.0; 52.5; 52.3; 51.7; 50.1 (2 Cα Ser, Cα Arg, CH3 メチルエステル Lys, Cα Lys, Cα Asp); 42.5 (Cα Gly); 40.8; 38.5; 37.3; 34.8; 34.1; 30.9; 29.4; 29.0; 26.3; 24.8; 24.3; 23.0 (Cε Lys, Cδ Arg, CH2-NH GABA, Cβ Asp, Cδ Lys, Cβ Lys, CH2-CO GABA, Cβ Arg, Cα及び Cβフッ素化鎖, Cγ Arg, CH2 GABA, Cγ Lys).
19F NMR (DMSO, D6):
δ -82.30 (3F); -115.51 (2F); -122.81 (6F); -123.82 (2F); -124.46 (2F); -127.21 (2F).
【図面の簡単な説明】
【0189】
【図1】PBN由来のスピントラップを備えたベクターの化学合成
【図2A】分子Dの合成
【図2B】分子Dの合成(続き)
【図3A】分子Fの合成
【図3B】分子Fの合成
【図3C】分子Fの合成
【特許請求の範囲】
【請求項1】
下記式(I):
【化1】
(式中:
APは、生物学的標的に作用することが可能な有効成分を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、以下:
−有効成分APの標的によって認識されることが可能なあらゆる分子、
ならびに
−式(I)の分子のHLBバランス(HLB balance)を調節するための親水性薬剤であり、Rが、単糖類、糖類のアミノ化誘導体、多糖類、天然又は合成ホルモン、ペプチド、抗体、ポリエーテル及びポリオール
から選択される基を表し、
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2もしくはAA3の1つの側鎖のいずれかへの結合を可能にする基
【化2】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表す;
AP−(X)xと鎖[AA3]a3−[AA2]a2−[AA1]の間の結合は、アミノ酸AA1、AA2又はもしくはAA3の1つの側鎖を介して、或いはペプチド鎖の末端にて生じる)
に相当する化合物。
【請求項2】
有効成分が、抗癌作用、或いはフリーラジカルスカベンジャー、抗炎症、殺菌、鎮痛、神経弛緩又は抗真菌作用を有するものから選択されることを特徴とする、請求項1に記載の化合物。
【請求項3】
有効成分が、1〜30の炭素原子、1以上の不飽和、特に1以上の芳香環、及び1以上の以下:
−O−、−S−、−OH、−SH、−Cl、−F、−Br、
【化3】
から選択される官能基を含む直鎖、分岐又は環状分子であることを特徴とする、請求項1及び2のいずれかに記載の化合物。
【請求項4】
側鎖を介してAP−(X)x−又はYに結合したアミノ酸が、それらの側鎖上に酸、アミド、アミン、チオール又はアルコール官能基(function)を含むものから選択されることを特徴とする、請求項1〜3のいずれかに記載の化合物。
【請求項5】
スペーサーアームXが1〜3アミノ酸を含むことを特徴とする、請求項1〜4のいずれかに記載の化合物。
【請求項6】
Rが、APの生物学的標的に対して顕著な親和性を有するエピトープ又は抗体断片から選択されるペプチドであることを特徴とする、請求項1〜5のいずれかに記載の化合物。
【請求項7】
Arg−Gly−Asp配列から選択される少なくとも1つのペプチド配列を含むことを特徴とする、請求項6に記載の化合物。
【請求項8】
Rが、5〜30のエチレンオキサイド単位を含むポリ(エチレンオキサイド)鎖或いは4〜16の炭素原子を含むアルキル鎖及び4〜16の水酸基からなるポリオールからなることを特徴とする、請求項1〜7のいずれかに記載の化合物。
【請求項9】
Rが、以下:グルコース、フルクトース、マンノース、ガラクトース、リボース、グルコサミン、ラクトース、セロビオース、マルトース、ラクトビオナミド及びスクロースから選択されることを特徴とする、請求項1〜8のいずれかに記載の化合物。
【請求項10】
スペーサーアームX、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]及びRの少なくとも1つが、少なくとも1つのチロシン残基を含むことを特徴とする、請求項1〜9のいずれかに記載の化合物。
【請求項11】
フッ素化炭化水素ベース鎖Yが、式A−Y’(式中、Aは以下:
【化4】
−NH−、−O−CO−NH−、S及びOから選択される基を表し、Y’は、式−(CH2)t−(CF2)rF(式中、r及びtは、12≧r+t≧4である2つの整数を表す)に相当する分子を表す)に相当するものから選択されることを特徴とする、請求項1〜10のいずれかに記載の化合物。
【請求項12】
式(II):
【化5】
(式中、xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、単糖類、糖類のアミノ化誘導体、多糖類、ポリエーテル、ポリオール、ペプチド、天然又は合成ホルモン、及び抗体から選択される;
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2又はAA3の1つの側鎖のいずれかへの結合を可能にする
【化6】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表し、ならびにスペーサーアームX、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]及びRの少なくとも1つが、少なくとも1つのチロシン残基を含む)
の断片を含む生物学的に活性な分子。
【請求項13】
式(Ia):
【化7】
(式中:
Suは、単糖、アミノ化単糖誘導体、多糖、ポリオール又はポリエーテルから選択される基を表す;
AA1は、その側鎖上に酸、アミン、アルコール又はチオール官能基を保有し、それにより、(X)x−AP又はYのいずれかに結合されるアミノ酸を表す;AA1は、そのN−及びC−末端を介してSu及び(X)x−AP又はYのいずれかに結合される;
APは、生物学的標的に作用することができる有効成分を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化8】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す)
に相当することを特徴とする、請求項1〜9のいずれかに記載の化合物。
【請求項14】
以下:
−Suは、単糖又は多糖を表す;
−Xは、少なくとも1つのチロシン残基を含む、天然のペプチドであるスペーサーアームを表す;
−AA1は、アルギニン及びリシンから選択されるアミノ酸を表す;
−Yは、−NH−官能基を介してアミノ酸AA1に結合した、5〜23のフッ素原子を含むフッ素化C6−C12炭化水素ベース鎖を表す
の1以上の条件が確認されることを特徴とする、請求項13に記載の化合物。
【請求項15】
有効成分が、血管新生プロセスを阻害することができる分子、特にサリドマイドから選択されることを特徴とする、請求項14に記載の化合物。
【請求項16】
式A:
【化9】
に相当することを特徴とする、請求項15に記載の化合物。
【請求項17】
有効成分APが、フリーラジカルスカベンジャー、特にN−ベンジリデン−tert−ブチルアミンオキサイド誘導体から選択されることを特徴とする、請求項15に記載の化合物。
【請求項18】
式E:
【化10】
に相当することを特徴とする、請求項17に記載の化合物。
【請求項19】
式(Ib):
Pep−[AA1]−Y
(Ib)
(式中:
AA1は、酸、アミン、アルコール又はチオール官能基をその側鎖上に有するアミノ酸を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化11】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す、
Pepは、2〜10、好ましくは4〜6のアミノ酸を含むペプチド鎖を表し、Pep及びAA1の少なくとも1つが少なくとも1つのチロシン単位を含む)
に相当することを特徴とする、請求項12に記載の化合物。
【請求項20】
Pepが、アルギニン−グリシン−アスパラギン酸配列を含むことを特徴とする、請求項19に記載の化合物。
【請求項21】
式B:
【化12】
に相当することを特徴とする、請求項19及び20のいずれかに記載の化合物。
【請求項22】
式(Ic):
【化13】
(式中:
APは、生物学的標的に作用することができる有効成分を表す;
Pepは、2〜10アミノ酸を含むペプチド鎖を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1は、酸、アミン、アルコール又はチオール官能基をその側鎖上に有するアミノ酸を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化14】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す)
に相当することを特徴とする、請求項1〜11のいずれかに記載の化合物。
【請求項23】
以下:
PepがαVβ3インテグリンによって認識されるペプチドであり、APが抗有糸分裂剤である;
X、Pep又はAA1が、少なくとも1つのチロシン残基を含む;
Xが、1〜3アミノ酸の鎖を表す
の1以上の条件が確認されることを特徴とする、請求項22に記載の化合物。
【請求項24】
式C、D及びF:
【化15】
の1つに相当することを特徴とする、請求項22又は23に記載の化合物。
【請求項25】
APがアドリアマイシンであり、X又はPepがGly−Phe−Leu−Gly断片を含むことを特徴とする、請求項22に記載の化合物。
【請求項26】
APが、メルファラン、5−フルオロウラシル及びメシル酸イマチニブから選択されることを特徴とする、請求項22に記載の化合物。
【請求項27】
薬学的に許容される担体中に、請求項1〜11及び13〜18のいずれかに記載される化合物を含む薬学的組成物。
【請求項28】
癌の予防及び/又は治療用薬学的組成物を調製するための、請求項16と24のいずれかに記載の式A、C、D又はFの化合物の使用。
【請求項29】
癌性細胞の存在を検出するための薬学的組成物を調製するための、請求項21に記載の式Bの化合物の使用。
【請求項30】
酸化ストレス及び酸化フリーラジカル種の形成に関連する病状を予防及び/又は治療するための薬学的組成物の調製のための、請求項18に記載される式Eの化合物の使用。
【請求項1】
下記式(I):
【化1】
(式中:
APは、生物学的標的に作用することが可能な有効成分を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、以下:
−有効成分APの標的によって認識されることが可能なあらゆる分子、
ならびに
−式(I)の分子のHLBバランス(HLB balance)を調節するための親水性薬剤であり、Rが、単糖類、糖類のアミノ化誘導体、多糖類、天然又は合成ホルモン、ペプチド、抗体、ポリエーテル及びポリオール
から選択される基を表し、
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2もしくはAA3の1つの側鎖のいずれかへの結合を可能にする基
【化2】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表す;
AP−(X)xと鎖[AA3]a3−[AA2]a2−[AA1]の間の結合は、アミノ酸AA1、AA2又はもしくはAA3の1つの側鎖を介して、或いはペプチド鎖の末端にて生じる)
に相当する化合物。
【請求項2】
有効成分が、抗癌作用、或いはフリーラジカルスカベンジャー、抗炎症、殺菌、鎮痛、神経弛緩又は抗真菌作用を有するものから選択されることを特徴とする、請求項1に記載の化合物。
【請求項3】
有効成分が、1〜30の炭素原子、1以上の不飽和、特に1以上の芳香環、及び1以上の以下:
−O−、−S−、−OH、−SH、−Cl、−F、−Br、
【化3】
から選択される官能基を含む直鎖、分岐又は環状分子であることを特徴とする、請求項1及び2のいずれかに記載の化合物。
【請求項4】
側鎖を介してAP−(X)x−又はYに結合したアミノ酸が、それらの側鎖上に酸、アミド、アミン、チオール又はアルコール官能基(function)を含むものから選択されることを特徴とする、請求項1〜3のいずれかに記載の化合物。
【請求項5】
スペーサーアームXが1〜3アミノ酸を含むことを特徴とする、請求項1〜4のいずれかに記載の化合物。
【請求項6】
Rが、APの生物学的標的に対して顕著な親和性を有するエピトープ又は抗体断片から選択されるペプチドであることを特徴とする、請求項1〜5のいずれかに記載の化合物。
【請求項7】
Arg−Gly−Asp配列から選択される少なくとも1つのペプチド配列を含むことを特徴とする、請求項6に記載の化合物。
【請求項8】
Rが、5〜30のエチレンオキサイド単位を含むポリ(エチレンオキサイド)鎖或いは4〜16の炭素原子を含むアルキル鎖及び4〜16の水酸基からなるポリオールからなることを特徴とする、請求項1〜7のいずれかに記載の化合物。
【請求項9】
Rが、以下:グルコース、フルクトース、マンノース、ガラクトース、リボース、グルコサミン、ラクトース、セロビオース、マルトース、ラクトビオナミド及びスクロースから選択されることを特徴とする、請求項1〜8のいずれかに記載の化合物。
【請求項10】
スペーサーアームX、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]及びRの少なくとも1つが、少なくとも1つのチロシン残基を含むことを特徴とする、請求項1〜9のいずれかに記載の化合物。
【請求項11】
フッ素化炭化水素ベース鎖Yが、式A−Y’(式中、Aは以下:
【化4】
−NH−、−O−CO−NH−、S及びOから選択される基を表し、Y’は、式−(CH2)t−(CF2)rF(式中、r及びtは、12≧r+t≧4である2つの整数を表す)に相当する分子を表す)に相当するものから選択されることを特徴とする、請求項1〜10のいずれかに記載の化合物。
【請求項12】
式(II):
【化5】
(式中、xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1、AA2及びAA3は、同一又は異なって、それぞれ1つのアミノ酸を表す;
a2及びa3は、同一又は異なって、それぞれ0及び1から選択される整数を表す;
Rは、単糖類、糖類のアミノ化誘導体、多糖類、ポリエーテル、ポリオール、ペプチド、天然又は合成ホルモン、及び抗体から選択される;
Yは、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]の末端の1つ、或いはアミノ酸AA1、AA2又はAA3の1つの側鎖のいずれかへの結合を可能にする
【化6】
−NH−、−O−CO−NH−、S又はOを含むフッ素化C4−C12炭化水素ベース鎖を表し、ならびにスペーサーアームX、ペプチド鎖[AA3]a3−[AA2]a2−[AA1]及びRの少なくとも1つが、少なくとも1つのチロシン残基を含む)
の断片を含む生物学的に活性な分子。
【請求項13】
式(Ia):
【化7】
(式中:
Suは、単糖、アミノ化単糖誘導体、多糖、ポリオール又はポリエーテルから選択される基を表す;
AA1は、その側鎖上に酸、アミン、アルコール又はチオール官能基を保有し、それにより、(X)x−AP又はYのいずれかに結合されるアミノ酸を表す;AA1は、そのN−及びC−末端を介してSu及び(X)x−AP又はYのいずれかに結合される;
APは、生物学的標的に作用することができる有効成分を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化8】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す)
に相当することを特徴とする、請求項1〜9のいずれかに記載の化合物。
【請求項14】
以下:
−Suは、単糖又は多糖を表す;
−Xは、少なくとも1つのチロシン残基を含む、天然のペプチドであるスペーサーアームを表す;
−AA1は、アルギニン及びリシンから選択されるアミノ酸を表す;
−Yは、−NH−官能基を介してアミノ酸AA1に結合した、5〜23のフッ素原子を含むフッ素化C6−C12炭化水素ベース鎖を表す
の1以上の条件が確認されることを特徴とする、請求項13に記載の化合物。
【請求項15】
有効成分が、血管新生プロセスを阻害することができる分子、特にサリドマイドから選択されることを特徴とする、請求項14に記載の化合物。
【請求項16】
式A:
【化9】
に相当することを特徴とする、請求項15に記載の化合物。
【請求項17】
有効成分APが、フリーラジカルスカベンジャー、特にN−ベンジリデン−tert−ブチルアミンオキサイド誘導体から選択されることを特徴とする、請求項15に記載の化合物。
【請求項18】
式E:
【化10】
に相当することを特徴とする、請求項17に記載の化合物。
【請求項19】
式(Ib):
Pep−[AA1]−Y
(Ib)
(式中:
AA1は、酸、アミン、アルコール又はチオール官能基をその側鎖上に有するアミノ酸を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化11】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す、
Pepは、2〜10、好ましくは4〜6のアミノ酸を含むペプチド鎖を表し、Pep及びAA1の少なくとも1つが少なくとも1つのチロシン単位を含む)
に相当することを特徴とする、請求項12に記載の化合物。
【請求項20】
Pepが、アルギニン−グリシン−アスパラギン酸配列を含むことを特徴とする、請求項19に記載の化合物。
【請求項21】
式B:
【化12】
に相当することを特徴とする、請求項19及び20のいずれかに記載の化合物。
【請求項22】
式(Ic):
【化13】
(式中:
APは、生物学的標的に作用することができる有効成分を表す;
Pepは、2〜10アミノ酸を含むペプチド鎖を表す;
xは、0及び1から選択される整数を表す;
Xは、1〜5アミノ酸を含むペプチド鎖を表す;
AA1は、酸、アミン、アルコール又はチオール官能基をその側鎖上に有するアミノ酸を表す;
Yは、アミノ酸AA1の末端の1つ又はAA1の側鎖のいずれかへの結合を可能にする
【化14】
−NH、−O−CO−NH−、S及びOから選択される官能基を含むフッ素化C4−C12炭化水素ベース鎖を表す)
に相当することを特徴とする、請求項1〜11のいずれかに記載の化合物。
【請求項23】
以下:
PepがαVβ3インテグリンによって認識されるペプチドであり、APが抗有糸分裂剤である;
X、Pep又はAA1が、少なくとも1つのチロシン残基を含む;
Xが、1〜3アミノ酸の鎖を表す
の1以上の条件が確認されることを特徴とする、請求項22に記載の化合物。
【請求項24】
式C、D及びF:
【化15】
の1つに相当することを特徴とする、請求項22又は23に記載の化合物。
【請求項25】
APがアドリアマイシンであり、X又はPepがGly−Phe−Leu−Gly断片を含むことを特徴とする、請求項22に記載の化合物。
【請求項26】
APが、メルファラン、5−フルオロウラシル及びメシル酸イマチニブから選択されることを特徴とする、請求項22に記載の化合物。
【請求項27】
薬学的に許容される担体中に、請求項1〜11及び13〜18のいずれかに記載される化合物を含む薬学的組成物。
【請求項28】
癌の予防及び/又は治療用薬学的組成物を調製するための、請求項16と24のいずれかに記載の式A、C、D又はFの化合物の使用。
【請求項29】
癌性細胞の存在を検出するための薬学的組成物を調製するための、請求項21に記載の式Bの化合物の使用。
【請求項30】
酸化ストレス及び酸化フリーラジカル種の形成に関連する病状を予防及び/又は治療するための薬学的組成物の調製のための、請求項18に記載される式Eの化合物の使用。
【図1】

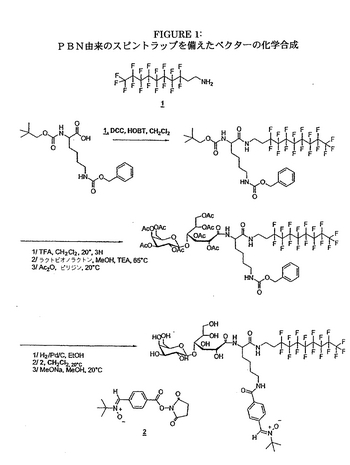
【図2A】
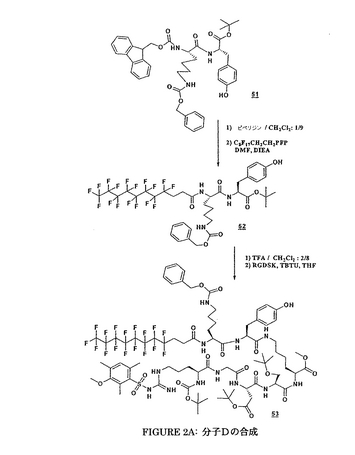
【図2B】
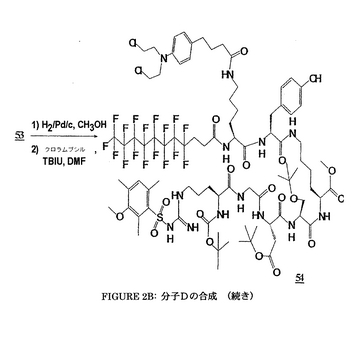
【図3A】
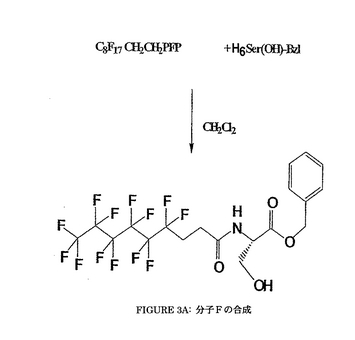
【図3B】
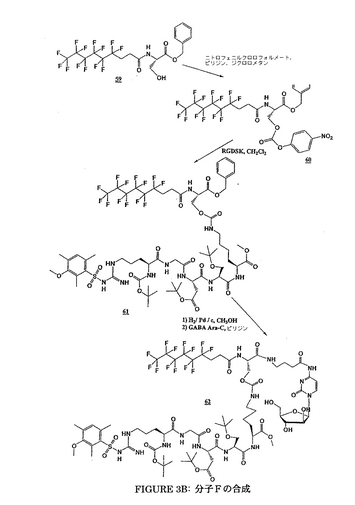
【図3C】
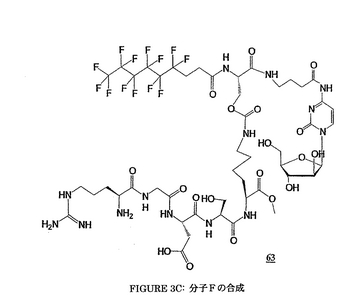
【図2A】
【図2B】
【図3A】
【図3B】
【図3C】
【公表番号】特表2006−520741(P2006−520741A)
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願番号】特願2004−550759(P2004−550759)
【出願日】平成15年11月7日(2003.11.7)
【国際出願番号】PCT/FR2003/003336
【国際公開番号】WO2004/043993
【国際公開日】平成16年5月27日(2004.5.27)
【出願人】(505167048)
【出願人】(505167059)ユニヴェルジット ダヴィニョン エ デ ペイズ デュ ボークリューズ (4)
【Fターム(参考)】
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願日】平成15年11月7日(2003.11.7)
【国際出願番号】PCT/FR2003/003336
【国際公開番号】WO2004/043993
【国際公開日】平成16年5月27日(2004.5.27)
【出願人】(505167048)
【出願人】(505167059)ユニヴェルジット ダヴィニョン エ デ ペイズ デュ ボークリューズ (4)
【Fターム(参考)】
[ Back to top ]