
生物学的エフェクターの細胞内送達
【課題】エフェクターの生体膜を通した移送を容易にすることができるアミノ酸配列を
提供することを課題とする。
【解決手段】上記課題は、多量体トランスポーターペプチドを提供することによって、
解決された。この多量体トランスポーターペプチドは、[P]v−[スペーサー]x−コ
ア−[リンカー]y−[レポーター]zの式を有し、ここでPは、(XmRXoRXn)、
(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)からなる群から選
ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチド、Xは非塩基性アミノ酸、mは0〜14の整数、nはmとは独立に0と14との間の整数、oはmおよびnとは独立に0と5との間の整数、vは2〜8の整数、x、yおよびzは独立に0または1、各Pは同じでも異なっていてもよい。
提供することを課題とする。
【解決手段】上記課題は、多量体トランスポーターペプチドを提供することによって、
解決された。この多量体トランスポーターペプチドは、[P]v−[スペーサー]x−コ
ア−[リンカー]y−[レポーター]zの式を有し、ここでPは、(XmRXoRXn)、
(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)からなる群から選
ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチド、Xは非塩基性アミノ酸、mは0〜14の整数、nはmとは独立に0と14との間の整数、oはmおよびnとは独立に0と5との間の整数、vは2〜8の整数、x、yおよびzは独立に0または1、各Pは同じでも異なっていてもよい。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の技術分野)
本発明は分子生物学の分野に関する。
【背景技術】
【0002】
(発明の背景)
対象物質を外部培地から細胞内、特に細胞核へ効率的に移入することを可能にする技術
は、バイオテクノロジーの分野において極めて重要である。このような技術は、タンパク
質もしくはペプチド産生、遺伝子発現の調節、細胞内シグナル伝達チャネルの解析、およ
び細胞(もしくは細胞核)内への種々多様な物質の移送効果の解析に有用であり得る。現
在利用できる技術は、移入用ベクターに対し、生物活性のある物質を宿主細胞の細胞質(
もしくは核)内に移入させて、宿主ゲノムに影響を与えずに、またはこの活性物質の生物
学的性質を変えずに処理することができないことにより制約を受ける場合が多い。
【0003】
以前は、高分子活性物質の細胞内への導入は、この移送すべき物質のサイズおよび生化
学的性質によって制約されていた。しかしながら、最近、トランスポーターペプチドもし
くはタンパク質を用いることにより、この分野に進歩がもたらされた。例えば、Schw
arzeとその共同研究者は、複合ペプチドもしくはタンパク質を組織の細胞内へ、およ
び血液−脳関門を通過させて移送させることができるHIV、TAT48−57由来10
マーのペプチドについて報告している(非特許文献1参照)。この複合ペプチドもしくは
タンパク質は、エンドサイトーシスを伴わないタンパク質導入プロセスにより細胞に吸収
されるものと考えられている。この発見により、生物医学研究および患者体内への薬物の
直接的な送達に新しい方法論の突破口が開かれた。しかしながら、この方法の1つの重要
な制限は、これらのタイプのトランスポーターに細胞特異性がないことである。従って、
標的としない正常な組織との相互作用により有害な副作用が生じるため、こうした非特異
的トランスポーターペプチドの多くは、有用性に制限がある。このような制限は、糖尿病
などの慢性疾患の処置において特に問題となる。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Schwarzeら、Science 285:1573(1999)
【発明の概要】
【発明が解決しようとする課題】
【0005】
薬物および治療剤をペプチド移送を介して細胞内に送達するための、種々の細胞型を特
異的に標的とするための効率的で安全な組成物および方法は、当該分野でなお求められて
いる。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、生体膜を通過して移行することができるトランスポーターペプチドを提供す
る。また、本発明は、エフェクターに対し生体膜を通過させて移行させるこのようなトラ
ンスポーターペプチドを使用する方法に関する。このトランスポーターペプチドは単体も
しくは多量体であり得る。
本発明は、例えば、以下を提供する:
(項目1)
式I、
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
による多量体トランスポーターペプチドであって:
ここで、Pは
a)(XmRXoRXn)、
b)(XmRRRXn)、
c)(XmRRXRXn)および
d)(XmRXRRXn)
からなる群から選ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチ
ドであり、
Xは非塩基性アミノ酸であり、
mは0〜14の整数であり、
nはmとは独立に0と14との間の整数であり、
oはmおよびnとは独立に0と5との間の整数であり、
vは2〜8の整数であり、
x、yおよびzは独立に0または1であり、
各Pは同じでも異なっていてもよく、該トランスポーターペプチドは生体膜を通過して移
行し得る、トランスポーターペプチド。
(項目2)
項目1に記載のトランスポーターペプチドであって、少なくとも1つのPがアミノ酸
配列R−X−X−Rを有する、トランスポーターペプチド。
(項目3)
項目1に記載のトランスポーターペプチドであって、各Pが配列番号1〜34からな
る群から選ばれる、トランスポーターペプチド。
(項目4)
項目1に記載のトランスポーターペプチドであって、vが2、4、6および8からな
る群から選ばれる、トランスポーターペプチド。
(項目5)
項目4に記載のトランスポーターペプチドであって、vが4である、トランスポータ
ーペプチド。
(項目6)
項目4に記載のトランスポーターペプチドであって、vが8である、トランスポータ
ーペプチド。
(項目7)
項目1に記載のトランスポーターペプチドであって、前記スペーサーがスクシンイミ
ジル−ポリエチレングリコールである、トランスポーターペプチド。
(項目8)
項目1に記載のトランスポーターペプチドであって、前記コアがC4−K2K−K(
succ−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg
−S)−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミド
および(NH2OCH2CO)8−K4K2K−K(GGG)−アミドからなる群から選
ばれる、トランスポーターペプチド。
(項目9)
エフェクターと結合体化されている項目1に記載のトランスポーターペプチドを含む
、トランスポーターユニット。
(項目10)
式II:
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
のトランスポーターユニットであって:
ここで、Eはエフェクターであり、
Pは
e)(XmRXoRXn)、
f)(XmRRRXn)、
g)(XmRRXRXn)および
h)(XmRXRRXn)
からなる群から選ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチド
であり、
Xは非塩基性アミノ酸であり、
mは0〜14の整数であり、
nはmとは独立に0と14との間の整数であり、
oはmおよびnとは独立に0と5との間の整数であり、
vは2〜8の整数であり、
x、yおよびzは独立に0もしくは1であり、
各Pは同じでも異なっていてもよいものとし、ならびに該トランスポーターペプチドは生
体膜を通過して移行し得る、トランスポーターユニット。
(項目11)
項目10に記載のトランスポーターユニットであって、少なくとも1つのPがアミノ
酸配列R−X−X−Rを有する、トランスポーターユニット。
(項目12)
項目10に記載のトランスポーターユニットであって、各Pが配列番号1〜34から
なる群から選ばれる、トランスポーターユニット。
(項目13)
項目10に記載のトランスポーターユニットであって、vが2、4、6および8から
なる群から選ばれる、トランスポーターユニット。
(項目14)
項目13に記載のトランスポーターユニットであって、vが4である、トランスポー
ターユニット。
(項目15)
項目13に記載のトランスポーターユニットであって、vが8である、トランスポー
ターユニット。
(項目16)
項目10のトランスポーターユニットであって、該コアがC4−K2K−K(suc
c−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S)
−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよび
(NH2OCH2CO)8−K4K2K−K(GGG)−アミドからなる群から選ばれる
、トランスポーターユニット。
(項目17)
項目10に記載のトランスポーターユニットであって、前記エフェクターが前記リン
カーに共有結合により融合されている、トランスポーターユニット。
(項目18)
項目10に記載のトランスポーターユニットであって、前記エフェクターが核酸、ペ
プチドおよび薬学的に活性な薬剤からなる群から選ばれる、トランスポーターユニット。
(項目19)
項目18に記載のトランスポーターユニットであって、前記エフェクターが核酸であ
る、トランスポーターユニット。
(項目20)
項目19に記載のトランスポーターユニットであって、前記核酸がDNAである、ト
ランスポーターユニット。
(項目21)
項目19に記載のトランスポーターユニットであって、前記核酸がRNAである、ト
ランスポーターユニット。
(項目22)
項目18に記載のトランスポーターユニットであって、前記エフェクターがペプチド
である、トランスポーターユニット。
(項目23)
項目18に記載のトランスポーターユニットであって、前記エフェクターが薬学的に
活性な薬剤である、トランスポーターユニット。
(項目24)
項目23に記載のトランスポーターユニットであって、前記薬学的に活性な薬剤が、
毒素、抗生物質、抗病原体剤、抗原、抗体断片、免疫調節剤、酵素および治療剤からなる
群から選ばれる、トランスポーターユニット。
(項目25)
項目10に記載のトランスポーターユニットであって、各ペプチドが50アミノ酸長
未満である、トランスポーターユニット。
(項目26)
項目10に記載のトランスポーターユニットであって、各ペプチドが25アミノ酸長
未満である、トランスポーターユニット。
(項目27)
項目10に記載のトランスポーターユニットであって、各ペプチドが15アミノ酸長
未満である、トランスポーターユニット。
(項目28)
項目10に記載のトランスポーターユニットであって、移行が膵B細胞、肝細胞、結
腸細胞、筋肉細胞および肺細胞からなる群から選ばれる組織内へ行われる、トランスポー
ターユニット。
(項目29)
項目12に記載のトランスポーターユニットを膵β細胞の膜を通過させて移行させる
方法であって、少なくとも1種のトランスポーターペプチドが配列番号1〜6からなる群
から選ばれる、方法。
(項目30)
項目29に記載の方法であって、少なくとも1種のトランスポーターペプチドが配列
番号1である、方法。
(項目31)
項目12に記載のトランスポーターユニットを肝細胞の膜を通過させて移行させる方
法であって、少なくとも1種のペプチドが配列番号7〜10からなる群から選ばれる、方
法。
(項目32)
項目12に記載のトランスポーターユニットを結腸細胞の膜を通過させて移行させる
方法であって、少なくとも1種のペプチドが配列番号11である、方法。
(項目33)
項目12に記載のトランスポーターユニットを筋肉細胞の膜を通過させて移行させる
方法であって、少なくとも1種のペプチドが配列番号12〜20からなる群から選ばれる
方法。
(項目34)
項目12に記載のトランスポーターユニットを肺細胞の膜を通過させて移行させる方
法であって、少なくとも1種のペプチドが配列番号21〜34からなる群から選ばれる方
法。
(項目35)
治療的もしくは予防的に有効な量の項目10に記載のトランスポーターユニット、お
よび薬学的受容可能なキャリアを含む、薬学的組成物。
(項目36)
項目1に記載の該トランスポーターペプチドとエフェクターとの間で移行可能な結合
体を生成する方法であって、該エフェクターを該トランスポーターペプチドに結合させて
トランスポーターペプチド−エフェクター結合体を形成することを含む方法。
(項目37)
エフェクターを真核細胞の細胞質および核の中へ移行させる方法であって、
該エフェクターを項目1に記載のトランスポーターペプチドに結合させてトランスポ
ーターペプチド−エフェクター結合体を形成する工程、および
該トランスポーターペプチド−エフェクター結合体を該細胞に導入する工程
を包含する、方法。
(項目38)
項目37に記載の方法であって、該導入工程が、前記トランスポーターペプチド−エ
フェクター結合体の存在下で細胞培養物をインキュベートすることにより、または前記ト
ランスポーターペプチド−エフェクター結合体を前記細胞内に注入することにより達成さ
れる方法。
(項目39)
項目37に記載の方法であって、該真核細胞がヒト細胞である、方法。
(項目40)
項目37に記載の方法であって、該真核細胞がβ細胞である、方法。
(項目41)
真核細胞内のエフェクターの細胞内濃度を増大させる方法であって、
該エフェクターを項目1に記載のトランスポーターペプチドに結合させてトランスポ
ーターペプチド−エフェクター結合体を形成する工程、および
該真核細胞の活性な代謝を促進する条件の下、該トランスポーターペプチド−エフェク
ター結合体の存在下で該細胞をインキュベートする工程
を包含する、方法。
(項目42)
項目41に記載の方法であって、前記真核細胞がヒト細胞である、方法。
(項目43)
1つ以上の容器に項目35に記載の治療的もしくは予防的に有効な量の該薬学的組成
物を含む、キット。
(項目44)
疾患を処置または予防する方法であって、そのような処置または予防が所望されている
被験体に対し、項目35に記載の薬学的組成物を該被験体の該疾患を処置または予防す
るのに十分な量で投与する工程を包含する、方法。
(項目45)
項目44に記載の方法であって、該疾患が、糖尿病、結腸癌、呼吸器疾患、神経変性
障害、心臓麻痺、およびウイルス感染症からなる群から選ばれる、方法。
(項目46)
多量体トランスポーターペプチドであって、各トランスポーターペプチドのアミノ酸配
列が配列番号1〜34からなる群から選ばれる、トランスポーターペプチド。
【0007】
一局面として、本発明は、(XmRXoRXn)、(XmRRRXn)、(XmRRX
RXn)および(XmRXRRXn)から選ばれ、Xが非塩基性アミノ酸であり、mが0
〜14の整数であり、nがmとは独立に0と14との間の整数であり、oがmおよびnと
は独立に0と5との間の整数である少なくとも1種のアミノ酸を有するトランスポーター
ペプチドであって、生体膜を通過して移行することができるトランスポーターペプチドを
含む。
【0008】
一実施形態として、本発明は、アミノ酸配列R−X−X−Rを有するトランスポーター
ペプチドを提供する。他の実施形態として、本発明は、配列番号1−34のうちの任意の
1つのアミノ酸配列を有するトランスポーターペプチドを提供する。種々の別の実施形態
として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来のもので
ある。さらに他の実施形態として、このトランスポーターペプチドは、タンパク質転換酵
素の切断部位から得られるものである。
【0009】
別の局面として、本発明は、各単量体トランスポーターペプチドが(XmRXoRXn
)、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)から選ばれ
、Xが非塩基性アミノ酸であり、mが0〜14の整数であり、nがmとは独立に0と14
との間の整数であり、oがmおよびnとは独立に0と5との間の整数であり、生体膜を通
過して移行することができる2個、3個、4個、5個、6個、7個、8個もしくはそれ以
上の単量体トランスポーターペプチドを含む多量体トランスポーターペプチドを含む。多
量体トランスポーターペプチド内の個々の単量体トランスポーターペプチドは同一もしく
は異なるものであり得る。
【0010】
別の実施形態として、本発明は、アミノ酸配列R−X−X−Rを有する多量体トランス
ポーターペプチドを提供する。他の実施形態として、本発明は、配列番号1−34のうち
の任意の1つのアミノ酸配列を有する多量体トランスポーターペプチドを提供する。種々
の他の実施形態として、この多量体トランスポーターペプチドは、タンパク質転換酵素の
リガンド由来のものである。さらに別の実施形態として、この多量体トランスポーターペ
プチドは、タンパク質転換酵素の切断部位から得られるものである。
【0011】
一部の実施形態として、この多量体トランスポーターペプチドは、スペーサー部分およ
びリンカー部分を含む。好ましくは、このスペーサー部分およびリンカー部分は、可動性
、両親媒性、非免疫原性、およびタンパク質分解酵素に対し非感受性である。このリンカ
ーは、例えば、ポリエチレングリコールであり得る。
【0012】
他の実施形態として、この単量体もしくは重合体トランスポーターペプチドは、レポー
ター基を含むことができる。本明細書に用いている「レポーター基」とは、検出可能な任
意の基である。非限定的な例としては、放射性同位体、蛍光性部分、リン光性部分、化学
発光性部分、および量子ドットが挙げられる。その他のレポーター基としては、ビオチン
、シンテイン、ヒスチジン、赤血球凝集素、mycもしくはフラッグ・タグが挙げられる
。
【0013】
一部の実施形態として、本発明のトランスポーターペプチドは、式I:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
によって表され、式中、Pは前述のコンセンサスモチーフ(consensus mot
if)(XmRXoRXn)、(XmRRRXn)、(XmRRXRXn)および(Xm
RXRRXn)に包含されるペプチドである。例えば、このペプチドは、配列X1−X2
−X3−X4を含むことができ、式中、X1およびX4はRもしくはKであり得、X1お
よびX4は任意のアミノ酸であり得る。一実施形態として、本発明は、アミノ酸配列R−
X−X−Rを有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する
。他の実施形態として、本発明は、配列番号1−34のうちの任意の1つのアミノ酸配列
を有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。種々の実
施形態として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来の
ものである。さらに他の実施形態として、このトランスポーターペプチドは、タンパク質
転換酵素の切断部位から得られるものである。vによって表される所与のトランスポータ
ー内のペプチド数は、整数とする。単量体トランスポーターでは、vは1である。多量体
トランスポーターでは、vは2〜8以上の整数とする。任意の所与の多量体結合体におい
て、それぞれのペプチドは互いに同じであっても異なっていてもよい。
【0014】
単量体のトランスポーター構築物では、コアは存在していてもしていなくてもよいもの
とする。
【0015】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は0または1である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−PEG(succ−
peg)などのポリエチレングリコール(PEG)が挙げられる。
【0016】
本明細書に用いている「コア」とは、リンカー、スペーサーおよびレポーター基の接続
ポイントとなる構造体である。また、コアは多量体トランスポーター内のペプチド間を結
合する役割を果たす。一部の実施形態として、このコアを分枝型とすることによって多く
のペプチドを同一トランスポーターユニット内で結合させることができる。通常、コアに
対し結合に利用可能な多くの官能性を持たせることによって、この多量体構築物の種々の
異なる部分(ペプチド、ならびに存在する場合にはスペーサー、リンカーおよびレポータ
ー)をこのコアと融合させて1つの分子を形成させることができる。企図しているコア部
分としては、以下に詳細を説明するポリリジン(K)が挙げられ、また、リンカーの組み
込まれた構造のものも挙げられる。コアを選択することにより、所与の結合体内に存在す
るペプチドの数が決まる。代表的なコアとしては、C4−K2K−K(succ−peg
−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S)−アミド、
(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよび(NH2O
CH2CO)8−K4K2K−K(GGG)−アミドが挙げられる。
【0017】
多量体のトランスポーター構築物では、リンカーおよびレポーター基は存在していても
していなくてもよく、従って、yおよびzは、式Iにおいて独立に1もしくは0である。
【0018】
本明細書に用いている「トランスポーターペプチド」とは、物質が生体膜を通過して移
行するのを容易にするペプチドである。特に明記しない限り、または用いる文脈において
不正確でない限り、トランスポーターペプチドを意味するものは単量体もしくは多量体の
トランスポーターペプチドを包含する。
【0019】
一部の実施形態として、このトランスポーターペプチドは、1つ以上のエフェクターと
融合させる。「エフェクター」は、DNA、RNA、タンパク質、ペプチド、または薬学
的に活性な薬剤、例えば、毒素、抗生物質、抗病原体剤(antipathogenic
agent)、抗原、抗体、抗体断片、免疫調節剤、酵素もしくは治療剤などを含む任
意の適切な分子であり得る。2つ以上のエフェクターが存在する場合、これらのエフェク
ターは同一もしくは異なるものであり得る。
【0020】
「融合」もしくは「融合した」という用語は、何らかの第3の分子よりも互いを好む2
種以上の分子をもたらすような特定の相互作用全てを含むものである。これには、共有結
合、イオン結合、疎水性結合、水素結合などの作用が含まれるが、溶媒選択性などの非特
異的結合は含まれない。
【0021】
本発明の多量体結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学の法則通りに、エフェクターはコア、もしくはリンカー、またはこれら
の両方に結合させることができる。
【0022】
本明細書に用いている「結合体(conjugate)」もしくは「結合体化(con
jugation)」とは、エフェクターと単量体もしくは多量体トランスポーターペプ
チドとの間の物理的な結合を可能にするあらゆるタイプの相互作用のことを意味する。こ
の結合の性質は、共有もしくは非共有結合であり得るが、その結合は、細胞移行の前もし
くは移行中に結合体が解離しないように十分強力なものである必要がある。結合体化は、
当業者に公知の任意の化学的、生化学的、酵素的もしくは遺伝子的カップリング法を用い
て達成することができる。目的のエフェクターは、トランスポーターペプチドのN末端も
しくはC末端にカップリングまたは融合させることができる。一部の実施形態として、こ
のエフェクターは、リンカー基に、もしくは直接コアに、またはレポーター基にカップリ
ングあるいは融合させる。
【0023】
種々の実施形態として、単量体および/または多量体トランスポーターペプチドは、長
さをアミノ酸長として50個未満、25個未満もしくは15個未満であり得る。
【0024】
さらなる実施形態として、移行は膵B細胞、肝細胞、大腸細胞、筋肉細胞および/また
は肺細胞内で行われる。
【0025】
別の実施形態として、本発明は、トランスポーターペプチドを生体膜を通過させて移行
させる方法を含む。例えば、配列番号1−6の配列のうちの1種以上を含む単量体もしく
は多量体ペプチドは膵B細胞の膜を通過させて移行させることができ、配列番号7−10
の配列のうちの1種以上を含む単量体もしくは多量体ペプチドは肝細胞の膜を通過させて
移行させることができ、配列番号11のペプチドうちの1種以上を含む単量体もしくは多
量体ペプチドは大腸細胞の膜を通過させて移行させることができ、配列番号12−20の
配列のうちの1種以上を含む単量体もしくは多量体ペプチドは筋肉細胞の膜を通過させて
移行させることができ、および配列番号21−34の配列のうちの1種以上を含む単量体
もしくは多量体ペプチドは肺細胞の膜を通過させて移行させることができる。
【0026】
なおさらなる実施形態として、本発明は、1種以上のエフェクターに結合させた単量体
もしくは多量体トランスポーターペプチドであるトランスポーターユニットを含む。種々
の他の実施形態として、このエフェクターは核酸、ペプチドもしくは薬学的に活性な薬剤
であり得る。
【0027】
さらに別の実施形態として、本発明は、トランスポーターペプチドと1種以上のエフェ
クターとの間で移行可能な単量体もしくは多量体結合体を作製し、従ってトランスポータ
ーペプチド−エフェクター結合体を形成する方法を含む。
【0028】
別の実施形態として、本発明は、エフェクターを単量体もしくは多量体トランスポータ
ーペプチドに結合させて真核細胞内に導入することによる、真核細胞の細胞質および/ま
たは核内に1種以上のエフェクターを移行させる方法を含む。例えば、このトランスポー
ターペプチド−エフェクター結合体は、この結合体の存在下で培養細胞をインキュベート
することにより、またはこの結合体を細胞内に注入することにより細胞内に導入すること
ができる。
【0029】
種々の他の実施形態として、本発明は、エフェクターを単量体もしくは多量体のトラン
スポーターペプチドに結合させ、細胞の活性状態の代謝を亢進させる条件の下、この細胞
の存在下にインキュベートすることによる、真核細胞におけるエフェクターの細胞内濃度
を上昇させる方法を含む。本発明の好ましい実施形態は、この真核細胞としてヒト細胞を
使用することを含む。
【0030】
さらなる実施形態として、本発明は、治療的もしくは予防的に有効な量の単量体もしく
は多量体トランスポーターユニット、および薬学的に受容可能な担体を含む薬学的組成物
を含む。
【0031】
好ましい「薬学的組成物」としては、a)希釈剤、例えば、乳糖、ブドウ糖、蔗糖、マ
ンニトール、ソルビトール、セルロースおよび/またはグリシン;b)滑沢剤、例えば、
シリカ、滑石、ステアリン酸、そのマグネシウム塩もしくはカルシウム塩および/または
ポリエチレングリコール;また、錠剤用としてc)結合剤、例えば、ケイ酸マグネシウム
・アルミニウム、でんぷん糊、ゼラチン、トラガント、メチルセルロース、カルボキシメ
チルセルロースナトリウムおよび/またはポリビニルピロリドン;必要に応じてd)崩壊
剤、例えば、でんぷん、寒天、アルギン酸またはそのナトリウム塩もしくは発泡性混合物
;ならびに/あるいはe)吸収剤、着色剤、矯味矯臭剤および甘味剤とともに有効成分を
含む錠剤およびゼラチンカプセル剤が挙げられる。注射用組成物は、好ましくは等張水溶
液もしくは懸濁液であり、坐剤は脂肪性乳濁液もしくは懸濁液から調製するのが有利であ
る。これらの組成物は、滅菌することができ、および/または保存剤、安定化剤、湿潤剤
または乳化剤などのアジュバント、溶解促進剤、浸透圧調節塩および/または緩衝液を含
むことができる。また、さらに、これらは他の治療上有用な物質を含むこともできる。こ
れらの組成物は、それぞれ、従来の混和、顆粒化またはコーティング法によって調製され
、有効成分を約0.1%〜75%、好ましくは約1%〜50%含有する。
【0032】
なおさらなる実施形態として、本発明は、1個以上の容器が本発明の薬学的組成物を治
療的もしくは予防的有効量で含有するキットを含む。
【0033】
本発明の別の実施形態は、疾患の処置もしくは予防を必要とする被験体に対し、疾患を
処置もしくは予防するのに十分な量の薬学的組成物を投与することにより、その疾患を処
置もしくは予防する方法を含む。処置対象の疾患としては、例えば、糖尿病、大腸癌、呼
吸器疾患、神経変性障害、心臓麻痺、および/またはウイルス感染症が挙げられ得る。
【0034】
別の局面として、本発明は、特定の細胞型に対してファージ・ライブラリをスクリーニ
ングした後、どの細胞がファージを取り込んだかを評価する、トランスポーターペプチド
用ファージ・ライブラリのスクリーニング方法を含む。
【0035】
別の実施形態として、この方法は、取り込まれたファージのDNAを同定し、これから
発現されるペプチドを推定することを含む。
【0036】
なおさらなる実施形態として、この方法は、ファージ・ライブラリを少なくとも3サイ
クルにわたって選択するスクリーニング工程を含む。
【0037】
さらに別の実施形態として、本発明は、ペプチドの多価(multivalent)デ
ィスプレイを有するファージを含む。
【図面の簡単な説明】
【0038】
【図1】図1はオキシム1〜3の構造を示す。コアの分枝Lys残基のαおよびεアミノ基は、RRTKペプチドのN末端でアシル化(スペーサがあってもなくてもよい)されている。スペーサーと共に、レポータ・タグFITCが各構築体のコアに導入されている。
【図2】図2はオキシム4〜6の構造を示す。コアの分枝Lys残基のαおよびεアミノ基は、RRTKペプチドのN末端でスペーサーを有してアシル化されている。スペーサーと共に、レポータ・タグのビオチンが各構築体のコアに導入されている。
【発明を実施するための形態】
【0039】
(発明の詳細な説明)
本発明は、薬物および治療剤を細胞内へ送達するための、種々の細胞型を特異的に標的
とするペプチドトランスポーターならびにペプチド移送システムを提供する。当該分野で
公知の既存の移送システムには、これらが非効率的で、宿主ゲノムに影響を与え、活性物
質(例えば、エフェクタ)の生物学的性質を変え、標的細胞を殺傷し、あるいは(例えば
、ウイルス性結合体を使用することにより)極めて高いリスクを生じるためヒト対象には
使用できないので、制約がある場合が多い。本発明のペプチド移送システムでは、治療剤
となり得るものを細胞内へ送達するためにプロタンパク質転換酵素およびその特異的なリ
ガンドを用いることによって当該分野で公知のトランスポーターシステムの限界を克服し
ている。本発明のシステムでは、宿主のゲノムに影響を与えず、その他の点でも、非侵襲
性である、変化を受けていない生物活性物質が効率的に送達される。
【0040】
受容体媒介性のエンドサイトーシスは、細胞内への治療剤の送達を狙った実験系におい
て広く利用されてきた。KatoおよびSugiyama、Crit.Rev.Ther
.Drug Carrier.Syst.14:287(1997)。従って、細胞特異
的なエンドサイトーシス受容体を首尾良く標的にすることによって、ペプチドを細胞型特
異的に送達することができる。プロタンパク質転換酵素は、受容体媒介性エンドサイトー
シスにより取り込まれる細胞表面受容体の一例である。こうしたタンパク質は、ペプチド
ホルモン、神経ペプチドその他の多くのタンパク質の前駆体をその生物活性のある形に転
換する役割を果たしていることが明らかにされている。プロタンパク質転換酵素ファミリ
ーの切断部位にはコンセンサス配列R−X−X−Rが含まれている。発現および局在性に
関する情報は、プロタンパク質転換酵素は細胞外のリガンドを細胞内空間に移送すること
を示す。
【0041】
例えば、哺乳類のプロタンパク質転換酵素は、その組織分布に基づいて3グループに分
類することができる。1つのクラスのフューリン(Furin)、PACE4、PC5/
PC6およびLPCIPC7/PC8/SPC7は広範囲の組織および細胞株において発
現される。第2のクラスの神経内分泌特異的転換酵素PC2およびPC1/PC3の発現
は、膵島、下垂体、副腎髄質および多くの脳領域などの内分泌組織に限定されている。さ
らに、第3のクラスのPC4の発現は、精巣の精子形成細胞に高度に制限されている。神
経内分泌特異的転換酵素は、主として分泌顆粒内に局在している。また、PC5/PC6
Aも、分泌顆粒に局在していると報告されている。さらに、間接的な証拠ではあるが、一
部のプロタンパク質転換酵素分子は細胞表面に存在することが示唆されており、フューリ
ンはTGNと細胞表面との間を循環することが明らかにされている。Mandrup−P
oulsen、BMJ.316:1221(1998)を参照されたい。
【0042】
天然リガンドの働きをするように見えるペプチドリガンドは、ファージ・ディスプレイ
法によって同定される場合が多い。効率的な受容体媒介性エンドサイトーシスを誘導する
ペプチド配列の単離は、ファージ・ディスプレイ法を用いることによって向上する。Iv
anenkovら、Biochem.Biophys.Acta1448:450、p4
63(1999)を参照されたい。ファージ・ディスプレイ・ライブラリは、細胞受容体
に対する天然リガンドの修飾体を含む変異体分子(Cabibboら、Gene 167
:41(1995))および短鎖ペプチド(Zwickら、Curr.Opin.Bio
technol.9:427(1998))の膨大な供給源となる極めて強力なツールで
ある。また、ライブラリを直接注射したマウスにおいて、脳および腎臓に13倍の選択性
を示すペプチド配列が首尾良く単離されている。PasqualiniおよびRuosl
ahti,Nature 380:364(1996)ならびにPasqualini、
Ruoslahti,Mol.Psychiatry 1:423(1996)を参照さ
れたい。
【0043】
時として、この方法により得られたリガンドは、短鎖ペプチド分子内の立体構造の自由
度が高く接触残基数が少ないため、低親和性(マイクロモル程度)となることがある。ペ
プチドリガンドの結合親和性を改善する1つの方法は、低親和性ペプチドのいくつかのコ
ピーを単一の多量体分子の形に結合させることである。この多量体リガンドの親和力(お
よび生物活性)は、単量体リガンドに対して大きく向上させることができる。一部の実施
形態として、多量体構築物は、オキシム化学を用いて、構築ブロックとしての細胞を標的
とするペプチドから作製する。例えば、このペプチドは、特定の細胞(例えば、膵β細胞
)に特異的に結合し、受容体媒介性エンドサイトーシスを介してこの細胞による取り込み
を誘発させることができるペプチドリガンドを選択するファージ・ディスプレイ実験から
得ることができる。
【0044】
例えば、単量体としてのIgMは低親和性タンパク質であるが、5量体の形をとると、
細菌もしくはウイルスの表面に存在する反復性抗原決定基に対する親和力が大きく向上す
る。Roitt、Essential Immunology(Oxford/Blac
kwell,London),p65−84(1991)を参照されたい。
【0045】
同様に、組換えDNA技術により作製されるタンパク質であるこのペンタボディでは、
105倍の向上がみられる。Alexeyら、Proc.Natl.Acad.Sci.
USA 94:1663−1668(1997)。ファージ・ライブラリから得られるポ
リペプチドの低親和性はペンタボディの5量体構造によって補われ、その標的に対する高
い親和力が得られる。
【0046】
また、最近、Roseは同様な分子を「ケモボディ(chemobody)」と名付け
た。RoseおよびVizzavona,J.Am.Chem.Soc.121:703
4−7038(1999)を参照されたい。このケモボディは、相補性構造に非共有結合
することにより抗体分子の働きを模倣することができるペプチドサブユニットの複数のコ
ピーを提示する合成分子である。その結合ペプチドのアミノ酸配列は、ファージ・ライブ
ラリ法により同定され、サブユニット自体そのものは、オキシム化学によって構築された
。また、同時に2つ以上のペプチドサブユニットによって標的に結合することができるよ
うに、適切なスペーサーおよびリンカーもこのケモボディに導入された。Roseは、可
動性リンカーを有するインフルエンザ・ウイルスのファージ由来ペプチドのコピーを4個
有するモデルケモボディを作製した。
【0047】
特定の細胞型を選択的に標的とする低分子トランスポーターペプチドは、大きなファー
ジ・ディスプレイ・ライブラリから得て、単量体もしくは多量体トランスポーター分子と
して用いることができる。ファージ・ディスプレイ・ライブラリを用いて得られるような
低分子ペプチド担体の有利な点としては、高品質、高純度、低免疫原性であり、生体の全
ての細胞に極めて効率的に送達することを可能にすることが挙げられる。Schwarz
eら、Science 285:1573(1999)。従って、ペプチド担体では、種
々の高分子を効率的に送達するためのリポソーム、ウイルスなどの従来のトランスポータ
ーを改良することができる可能性がある。例えば、Mahatoら、J.Drug Ta
rget.4:337(1997)、およびMahatoら、Crit.Rev.The
r.Drug Carrier.Syst.14:133 4:337(1997)を参
照されたい。
【0048】
本明細書に記載したトランスポーターペプチドは、種々の疾患の治療に用いられ、この
ような疾患としては、糖尿病が挙げられるが、これに限定されるものではない。正常な生
物体(では、インスリン分泌により正常血糖が維持されるようにβ細胞集団は厳格に制御
されている。β細胞集団は、乳児もしくは成熟生物体の要求に反応して、特に、特定の生
理的および生理病理学的状態への反応として調節されている。β細胞集団の適切性は、基
本的にβ細胞の死と再生との動的バランスによって達成されている。このバランスは、未
熟β細胞の分化および既存のインスリン分泌細胞の増殖によって達成されている。Sch
arfmannおよびCzernichow、Diabetes Metab.22:2
23(1996)ならびにBonner−Weir,Recent Prog.Horm
.Res.49:91(1994)を参照されたい。I型糖尿病におけるβ細胞集団の障
害されたバランスは、膵β細胞を標的にする免疫系の特異的攻撃により惹起される過程で
あるβ細胞破壊の促進によって生じる。従って、β細胞破壊の速度を抑制もしくは低下さ
せると、糖尿病が安定化されるばかりではなく、島が再生されてβ細胞集団の機能不全を
是正することも可能となる。
【0049】
I型糖尿病の実験モデルにおいてβ細胞の喪失速度を低下させるのに用いられる効力の
あるツールとして数種の化合物を確立した。これらのペプチドの多くは本質的にペプチジ
ルであり、従ってペプチド担体に容易に結合する。本明細書に記載したこれらのペプチド
は、治療用「積み荷(cargo)」のデザイン、即ち、治療剤(「エフェクタ」)を担
体(単量体もしくは多量体「トランスポーターペプチド」)とカップリングさせるための
基剤となる。
【0050】
本発明の移送システムの1つの実施形態は、I型糖尿病の治療のためにβ細胞の細胞内
メカニズムを標的にしたものである。I型糖尿病の症状は、免疫系の分泌による膵β細胞
の破壊に続発するものである。Mandrup−Poulsen,BMJ 316:12
21(1998)を参照されたい。ヒトおよび齧歯類における結論的なデータは、サイト
カインのインターロイキン−1β(IL−1β)は、マクロファージおよびT細胞により
分泌されるTNFαおよびINFγと共に、β細胞の機能不全および破壊ならびにI型糖
尿病をもたらす最終的な転帰の原因となる主要な成分であることを示す。Mandrup
−Poulsen,Diabetologia 39:1005(1996)、Neru
pら、Diabetes Care 11 Suppl 1:16(1988)、ならび
にMauricioおよびMandrup−Poulsen,Diabetes 47:
1537(1998)を参照されたい。これらの分泌サイトカインは、膵β細胞において
シグナル伝達およびエフェクター分子の高度に複雑なネットワークに関与している。この
シグナル伝達は、細胞の挙動を改変し、細胞の運命に決定的な影響力を与える。これまで
に蓄積された証拠から、この調節性細胞内ネットワークは新規な治療方法の開発のための
有望な標的となることが分かる。Iwahashiら、Cytokines.Cell
Mol.Ther.4:45(1998)、Sjoholm,Cell Death.D
iffer.5:461(1998)、Stephensら、J.Autoimmun.
10:293(1997)、Rabinovitchら、Diabetes 48:12
23(1999)、Bleichら、J.Clin.Invest.103:1431(
1999)、Welshら、Mol.Med.5:169(1999)、およびChen
ら、Diabetes 49:562(2000)を参照されたい。細胞内サイトカイン
・シグナル伝達の処理および形成に関与するこれらの分子はそれぞれ、トランスポーター
−薬物設計の標的とすることができる。
【0051】
β細胞内でIL−1βによって動員される最も重要なシグナル伝達分子としては、セラ
ミド、プロスタグランジン、熱ショックタンパク質、誘導性NOシンターゼ酵素(iNO
S)、転写因子NF−κB(Torgersonら、J.Immunol.161:60
84(1998)参照)、ならびに3種のMAPキナーゼERK1/2、p38およびJ
NKがある。これらの分子の多くは、β細胞の生残および機能の改善をもたらした既存の
阻害剤によるブロックの標的である。iNOSノックアウト(KO)マウスはIL−1β
の細胞毒性に対して抵抗性であり(Flodstromら、Diabetes 48:7
06(1999)参照)、iNOS作用のブロッカーはNOの細胞毒性の種々の側面を抑
制する(Sjoholm,Cell Death.Differ.5:461(1998
)に概説されている)。島および細胞株による研究は、Ca2+チャネルのブロッカーお
よびカスパーゼ阻害剤は齧歯類のβ細胞死を防止することを示す。Wangら、Endo
crinology 140:1200(1999)、およびYamadaら、Diab
etes 48:478(1999)を参照されたい。p38阻害剤は、グルコース賦活
性インスリン放出のIL−1β媒介性阻害を減弱させる。Larsenら、J.Biol
.Chem.273:15294(1998)を参照されたい。アンチセンスGADトラ
ンスジェニックNODマウスにおいてGADの発現をβ細胞特異的に抑制すると、自己免
疫性糖尿病が阻止された。Yoonら、Science 284:1183(1999)
を参照されたい。膵β細胞株においてbcl−2、IL−1RaおよびJBD(c−Ju
n N末端キナーゼJNKの優性阻害因子)を発現させると、細胞はアポトーシスに抵抗
性となった。Bonnyら、Diabetes 50:77−82(2000)、Iwa
hashiら、Diabetologia 39:530(1996)、Duprazら
、Gene Ther.6:1160(1999)、およびGiannoukakisら
、Diabetes 48:1730(1999)を参照されたい。以上のデータを総合
すると、特定の手段により細胞内事象を操作することはI型糖尿病の治療にかなり有望で
あることを示す。
【0052】
疾病治療の1つの主要な課題は、生物学的に重要な分子をインビボで有効な細胞透過性
の生物活性化合物に変換することである。Gibbs,Science 287:196
9(2000)を参照されたい。例えば、β細胞の喪失を防止するための最も有望なツー
ルは、多くの高分子タンパク質(例えば、Bcl−2(Rabinovitchら、Di
abetes 48:1223(1999)参照)、ドミナントネガティブ型のMyD8
8、TRAF、FADDもしくはIRAK(Burnsら、J.Biol.Chem.2
73:12203(1998)、およびStephensら、Endocrinolog
y 140:3219(1999)参照)などのサイトカイン・シグナル伝達の阻害因子
、または現在インビボで組織および膵β細胞を含む細胞型に送達させることができないJ
NK阻害剤JBD280(Ammendrupら、Diabetes 49:1468−
1476(2000)参照)である。
【0053】
最近の研究は、高分子タンパク質を、細胞および器官に容易に送達させることができる
低分子生物活性化合物に変換する試みの前進を示す。Hawiger,Curr.Opi
n.Chem.Biol.3:89(1999)を参照されたい。これらの方法には基本
的に2つの条件が必要とされる:即ち、1)細胞内に効率的に送達させたい分子に特定の
トランスポーターもしくはその化学的修飾体を結合させること(例えば、Schwarz
eら、Science 285:1573(1999)、Brugidouら、Bioc
hem.Biophys.Res.Commun.214:685(1995)、Oeh
lkeら、Biochim.Biophys.Acta 1414:127(1998)
およびTerskikhら、Proc.Natl.Acad.Sci.U.S.A.94
:1663(1997)に報告されている効率的な短鎖ペプチドトランスポーターを参照
されたい);2)トランスポーターに低分子ペプチド配列を結合させることができるよう
に、タンパク質の活性部分を狭小化する必要があること。手短に言えば、概してこれらの
条件では、元のタンパク質の不可欠な生物学的性質を保ちながら細胞内に入ることができ
る3〜30アミノ酸長の二連(bi−partite)ペプチドが設定される。癌研究の
場合のように(Gibbs、Oliff、Cell 79:193(1994)参照)、
β細胞には操作することによりサイトカイン誘発性アポトーシスからβ細胞を保護するこ
とができる多数の細胞内事象があり、こうした操作は薬剤設計の有望な標的となると考え
られる。
【0054】
受容体媒介性エンドサイトーシスは、細胞内への治療剤の送達を狙った実験系において
広く利用されている。Kato、Sugiyama、Crit.Rev.Ther.Dr
ug Carrier.Syst.14:287(1997)を参照されたい。エンドサ
イトーシス作用は、IgG Fc、ソマトスタチン、インスリン、IGF−IおよびII
、トランスフェリン、EGF、GLP−1、VLDLもしくはインテグリン受容体を含む
多くの受容体について報告されている共通的性質である。Hoflandら、Proc.
Assoc.Am.Physicians.111:63(1999)、Anderso
n、Clin.Immunol.Immunopathol.53:S63(1989)
、Lundら、J.Biol.Chem.265:15713(1990)、Smith
およびJarett、Lab.Invest.58:613(1988)、Solerら
、Endocrinology 127:595(1990)、Widmannら、Bi
ochem.J.310:203(1995)、Yorkら、J.Biol.Chem.
274:1164(1999)、およびMukherjeeら、Physiol.Rev
.77:759(1997)を参照されたい。エンドサイトーシスを媒介する細胞型特異
的受容体についても報告されている。Ivanenkovら、Biochem.Biop
hys.Acta 1448:463(1999)を参照されたい。
【0055】
強い実験的バックグラウンドは、膵β細胞を選択的に標的にするトランスポーターペプ
チドが大きなファージ・ディスプレイ・ライブラリから得られることも考えられることを
示しているが、そのような試みについては報告されていない。ファージ・ディスプレイ・
ライブラリを用いて得られるような低分子ペプチド担体の利点は数多くあり、例えば、化
学合成による作製が容易であること、高品質、高純度、低免疫原性であること、および生
物体の全ての細胞に極めて効率的に送達することが可能であることが挙げられる。Sch
warzeら、Science 285:1573(1999)を参照されたい。従って
、本発明のペプチド担体には、多くの高分子の効率的な送達において、リポソームもしく
はウイルスなどの従来のトランスポーターよりもうまく機能する可能性がある。例えば、
Mahatoら、J.Drug Target 4:337(1997)およびMaha
toら、Crit.Rev.Ther.Drug Carrier.Syst.14:1
33(1997)を参照されたい。
【0056】
ファージ・ペプチド・ライブラリは、従来より、線維状ファージM13の誘導体として
構築されている。ペプチド・ライブラリは、このペプチドモチーフの1〜5個のコピーを
提示するキャプシドのマイナーコートタンパク質pIIIに融合される。Zwickら、
Curr.Opin.Biotechnol.9:427(1998)を参照されたい。
あるいは、メジャーコートタンパク質pVIIIを用いることによって高価(high−
valent)提示(display)が達成される。しかしながら、これらのタイプの
ライブラリは、受容体媒介性エンドサイトーシスペプチド配列の単離のために最適化され
ていない。高い内部移行効率を有する担体を回収することに関連して考慮すべき事柄は以
下の通りである:1)ペプチドの1価もしくは低価(low−valent)提示は、線
維状ファージのような大きな構造体を効率的に取り込ませるには基本的に不十分であるが
、多価提示では効率的取り込みが可能となること(Ivanenkovら、Bioche
m.Biophys.Acta 1448:450(1999)参照);および2)受容
体結合リガンドが内部移行が、原形質膜の特定領域における細胞表面受容体の濃縮、続く
クラスリン被覆小胞の形成に関連すること(Damke、FEBS Lett.389:
48(1996))。
【0057】
M13誘導体のサイズは大きく(1〜1.5μm)(Zacherら、Gene 9:
127(1980)参照)、古典的なクラスリン被覆ピットの通常サイズ(150nM)
を上回る。クラスリン被覆ピットは、膜表面の約2%を占める原形質膜上の陥入構造体で
ある。この特殊な構造は、インスリン、EGFなどの細胞外タンパク質もしくはペプチド
を10〜50%/分という極めて速い速度で除去する高度に効率的な受容体媒介性内部移
行プロセスを指向する。Mukherjeeら、Physiol.Rev.77:759
(1997)を参照されたい。従って、従来のM13ファージでは、こうした特殊で高度
に効率的な構造による受容体媒介性内部移行が行われるとは考えられない。
【0058】
従って、報告されている試みでは、ペプチド担持ファージが高い内部移行率を示すペプ
チドは得られていない。これまで、こうした研究から、特定の細胞型に特異的なコンセン
サス取り込みモチーフは出現していない。
【0059】
一部の局面として、本明細書に開示した本発明は、ペプチド担持エフェクターの取り込
みを促進するトランスポーターペプチドの同定に関する。一旦このペプチド配列が決まる
と、このペプチドをエフェクター分子に結合させることによりエフェクター分子を生体膜
を通過させて移送させる。例えば、ファージ・ディスプレイ・パニングにより膵インスリ
ン分泌β細胞を標的にする一連のペプチドを単離した。こうした細胞を標的にするペプチ
ドを構築ブロックとする単量体もしくは多量体構築物を合成した。これらの構築物は、治
療剤を特異的に送達して糖尿病をコントロールするように設計した新規化学物質である。
【0060】
本発明のトランスポーターペプチドは式Iで表すことができる:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
式中、Pは前述のコンセンサス配列(consensus motif)(XmRXoR
Xn)、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)に包含
されるペプチドである。例えば、このペプチドは、配列X1−X2−X3−X4を含むこ
とができ、式中、X1およびX4はRもしくはKとすることができ、X1およびX4は任
意のアミノ酸とすることができる。一実施形態として、本発明は、アミノ酸配列R−X−
X−Rを有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。別
の実施形態として、本発明は、配列番号1−34のうちの任意の1つのアミノ酸配列を有
する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。種々の別の実
施形態として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来の
ものである。さらに別の実施形態として、このトランスポーターペプチドは、タンパク質
転換酵素の切断部位から得られるものである。vによって表される所与のトランスポータ
ー内のペプチド数は、整数とする。単量体トランスポーターでは、vは1である。多量体
トランスポーターでは、vは2〜8またはそれ以上の整数とする。任意の所与の多量体結
合体において、それぞれのペプチドは互いに同じか、異なるものとすることができる。
【0061】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は1または0である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−ポリエチレングリコ
ール(succ−peg)などのポリエチレングリコール(PEG)が挙げられる。
【0062】
本明細書に用いている「コア」とは、多量体トランスポーターペプチド内のペプチド間
を結合する役割を果たす構造体である。また、コアは、リンカー、スペーサーおよびレポ
ーター基の接続ポイントとなる。通常、コアに対し結合に利用可能な多くの機能性を持た
せることによって、この多量体構築物の種々の異なる部分(ペプチド、および存在する場
合にはスペーサー、リンカーおよびレポーター)をこのコアと融合させて1つの分子を形
成させることができる。企図しているコア部分としては、以下に詳細を説明するポリリジ
ン(K)が挙げられ、また、リンカーの組み込まれた構造のものも挙げられる。コアを選
択することにより、所与の結合体内に存在するペプチドの数が決まる。代表的なコアとし
ては、C4−K2K−K(succ−peg−S)−アミド、C−GGG−[K(C)]
3−K(succ−peg−S)−アミド、(NH2OCH2CO)4−K2K−K(s
ucc−peg)−アミドおよび(NH2OCH2CO)8−K4K2K−K(GGG)
−アミドが挙げられる。単量体トランスポーターペプチドでは、コアは存在していてもし
ていなくてもよい。
【0063】
このトランスポーター構築物では、リンカーおよびレポーター基は存在していてもして
いなくてもよく、従って、yおよびzは、式Iにおいて独立に1もしくは0である。
【0064】
本明細書に用いている「トランスポーターペプチド」とは、物質が生体膜を通過して移
行するのを促進するペプチドである。特に明記しない限り、または用いる文脈において不
正確でない限り、トランスポーターペプチドを意味するものは単量体もしくは多量体のト
ランスポーターペプチドを包含する。
【0065】
トランスポーターペプチドは、物質が細胞の生体膜を横切って、特にその細胞質もしく
は核内へ通過または移行するのを容易にするペプチドである。移行の検出は、例えば、P
CT出願第WO97/02840号に記載されている細胞透過アッセイ(cellula
r penetration assay)を含む種々の方法によって行うことができる
。一般に、細胞透過アッセイは、a)培養細胞を、移行させるペプチドとインキュベート
し、b)この細胞を固定して透過処理し、およびc)細胞内のペプチドの有無を検出する
ことによって実施する。検出工程は、固定、透過処理した細胞をこのペプチドに対する標
識抗体とインキュベートした後、ペプチドと標識抗体との免疫反応を検出することによっ
て行うことができる。あるいは、検出可能な標識ペプチドを用い、細胞コンパートメント
内の標識の有無を直接検出することによっても、検出を行うことができる。この標識は、
例えば、放射性標識もしくは蛍光標識、色素、または検出可能な任意の標識とすることが
できる。例えば、de Jongら、J.Nucl.Med.40:2081(1999
)およびBreemanら、Int.J.Cancer 81:658(1999)を参
照されたい。
【0066】
さらに、本発明は、トランスポーターペプチドをエフェクターにカップリングもしくは
融合させた結合体である移送ユニット、もしくは結合体を含む。本明細書に用いている「
カップリングさせた」とは、エフェクターと前記ペプチドとの物理的な結合を可能にする
任意のタイプの相互作用のことを意味する。この結合の性質は、共有もしくは非共有結合
とすることができるが、その結合は、細胞移行の前もしくは移行中にこのベクターが解離
しないように十分強力なものである必要がある。カップリングは、当業者に既知の任意の
化学的、生化学的、酵素的もしくは遺伝子的カップリング法を用いて達成することができ
る。対象とするエフェクターは、このペプチド・ベクターのN末端もしくはC末端にカッ
プリングさせることができる。
【0067】
「融合した」もしくは「融合」または「結合する」あるいは「相互作用する」という用
語は、2種以上の分子同士が何らかの第3の分子に対するよりも高い選択性を示すような
特定の相互作用全てを含むものであり、これらには、共有結合、イオン結合、疎水性結合
、水素結合などの作用が含まれるが、溶媒選択性などの非特異的結合は含まれない。
【0068】
一部の実施形態として、このトランスポーターペプチドは、1つ以上のエフェクターと
カップリングもしくは融合させる。このカップリングは、直接的なもの、もしくはコアを
介したものとすることができる。「エフェクター」は、DNA、RNA、タンパク質、ペ
プチド、または薬学的に活性な薬剤、例えば、毒素、抗生物質、抗病原体剤(antip
athogenic agent)、抗原、抗体、抗体断片、免疫調節剤、酵素もしくは
治療剤などを含む任意の適切な分子とすることができる。2つ以上のエフェクターが存在
する場合、これらのエフェクターは同一もしくは異なるものとすることができる。エフェ
クターとは、例えば、生物学、医薬、診断、トレーシングもしくは食品加工の対象になる
任意の分子または化合物のことを意味する。エフェクターは、種々の由来の核酸、特にヒ
ト、ウイルス、動物、真核もしくは原核生物、植物または合成由来等の核酸(リボ核酸、
デオキシリボ核酸)で構成することができる。対象とする核酸は、例えば、単純な微量ヌ
クレオチドからゲノム断片もしくはゲノム全体に至る種々のサイズのものとすることがで
きる。同様に、これはウイルスゲノムもしくはプラスミドとすることができる。あるいは
、対象とするエフェクターは、例えば、酵素、ホルモン、サイトカイン、アポリポタンパ
ク質、成長因子、抗原、抗体などのタンパク質とすることもできる。さらに、このエフェ
クターは、例えば、毒素、治療剤、もしくは抗生物質、抗ウイルス剤、抗真菌剤、抗寄生
生物剤などの抗病原体剤のような薬学的に活性な物質とすることができる。この対象とす
るエフェクターは、それ自体そのままで活性があるものとすることができ、またはインサ
イチュでペプチドもしくは別の物質または環境条件により活性化されるものとすることが
できる。
【0069】
本発明のトランスポーターペプチド結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学の法則通りに、エフェクターはコア、もしくはリンカー、またはこれら
の両方に結合させることができる。単量体トランスポーターペプチド結合体(v=1)で
は、コアは存在してもしていなくてもよい。
【0070】
本明細書に用いている「薬学的に活性な物質」とは、ヒトもしくは動物の生物体に投与
したときに、検出可能な薬理学的および/または生理学的効果を誘起する化学物質もしく
は化合物のことを意味する。本明細書に用いている「治療剤」とは、ヒトもしくは動物の
生物体に投与したときに、目的とする薬理学的および/または生理学的効果を誘起する化
学物質もしくは化合物のことを意味する。
【0071】
本発明によるトランスポーターペプチドは、その透過能がこれにカップリングさせる対
象物質(エフェクター)の性質とは事実上無関係であるという事実に特徴がある。
【0072】
また、本発明は、対象物質を細胞もしくは細胞の核内に導入する方法を含む。この方法
は、細胞内への効率的な透過を可能にするのに十分な量のトランスポーターペプチド−エ
フェクター結合体もしくはトランスポーターユニットと細胞とを接触させることを含む。
一般に、本方法は、この結合体をインビボまたはインビトロで取り込ませるために用いる
ことができる。例えば、この結合体は、インビトロ、エキソビボ、またはインビボで与え
ることができる。さらに、本発明によるトランスポーターペプチドは、カップリングさせ
た物質に生物活性を持たせることを可能にすることができる。従って、トランスポーター
ペプチドはカップリングさせたエフェクターの生物活性を増強させることができる。イン
ビトロによる方法では、エフェクターをトランスポーターとカップリングさせ、得られた
結合体を、細胞代謝の活性化を可能にする温度で細胞とインキュベートする。場合によっ
ては、このトランスポーター−エフェクター結合体を特定の細胞に注入する。この結合体
を細胞内に導入する他の任意の方法を用いることもできることは当業者によって認められ
よう。
【0073】
また、以上のペプチド−エフェクター結合体のほかに、本発明は、薬学的に受容可能な
塩基もしくは酸付加塩、水和物、エステル、溶媒和物、プロドラッグ、代謝体、立体異性
体もしくはこれらの混合物を提供する。また、本発明は、薬学的に受容可能な担体、希釈
剤もしくは賦形剤と共に、ペプチド−エフェクター結合体を含む薬学的組成物を含む。
【0074】
「薬学的に受容可能な塩」という用語に包含される塩とは、一般に、この遊離塩基と適
切な有機もしくは無機酸とを反応させて本明細書に開示した化合物の「薬学的に受容可能
な酸付加塩」を作製することにより調製する本発明の化合物の毒性のない塩のことを意味
する。こうした化合物は、この遊離塩基の生物学的効果および性質を保持するものである
。このような塩の代表例は、酢酸塩、アムソン酸塩(4,4−ジアミノスチルベン−2,
2’−ジスルホネート)、ベンゼンスルホン酸塩、ベンゾネート、重炭酸塩、重硫酸塩、
酒石酸水素塩、ホウ酸塩、臭化物、酪酸塩、エデト酸カルシウム塩、カンシル酸塩、炭酸
塩、塩化物、クエン酸塩、クラブラリエート(clavulariate)、二塩化水素
化物、エデト酸塩、エディシレート、エストレート、エシレート、フマル酸塩、グルセプ
ト酸塩、グルコン酸塩、グルタミン酸塩、グリコリルアルサニレート、ヘキサフルオロリ
ン酸塩、ヘキシルレゾルシネート、ヒドラバミン塩、臭化水素酸塩、塩酸塩、ヒドロキシ
ナフトエート、ヨウ化物、イソチオネート、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、
リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシル酸塩、臭化メチル塩、メチル硝酸塩、
メチル硫酸塩、ムチン酸塩、ナプシル酸塩、硝酸塩、N−メチルグルカミン・アンモニウ
ム塩、3−ヒドロキシ−2−ナフトエート、オレイン酸塩、シュウ酸塩、パルミチン酸塩
、パモン酸塩(1,1−メチレン−ビス−2−ヒドロキシ−3−ナフトエート、エンボネ
ート)、パントテン酸塩、リン酸塩/二リン酸塩、ピクリン酸塩、ポリガラクツロン酸塩
、プロピオン酸塩、p−トルエンスルホン酸塩、サリチル酸塩、ステアリン酸塩、塩基性
酢酸塩、コハク酸塩、硫酸塩、スルホサリキュレート、スラメート、タンニン酸塩、酒石
酸塩、テオクル酸塩、トシル酸塩、トリエチオダイド、および吉草酸塩などの水溶性なら
びに非水溶性塩である。
【0075】
本発明の方法によれば、ヒト患者に対し、薬理学的に有効な量のペプチドもしくは結合
体を投与することができる。「薬理学的に有効な量」という用語は、研究者もしくは臨床
家によって求められている組織、器官系、動物もしくはヒトの生物学的または医学的反応
を誘起する薬物もしくは薬学的組成物(エフェクター)の量のことを意味する。
【0076】
また、本発明は、対象エフェクターを細胞もしくは細胞核内に導入するのに適した薬学
的組成物を含む。この組成物は、内服に適したものであることが好ましく、有効量の本発
明の薬理学的に活性な化合物を単独、もしくは1種以上の薬学的に受容可能な担体との組
合せで含む。この化合物は、毒性があるとしても極めて少ないという点で、特に有用であ
る。
【0077】
好ましい薬学的組成物は、a)希釈剤、例えば、乳糖、ブドウ糖、蔗糖、マンニトール
、ソルビトール、セルロースおよび/またはグリシン;b)滑沢剤、例えば、シリカ、滑
石、ステアリン酸、そのマグネシウムもしくはカルシウム塩および/またはポリエチレン
グリコール;錠剤用としてc)カップリング剤、例えば、ケイ酸マグネシウム・アルミニ
ウム、でんぷん糊、ゼラチン、トラガント、メチルセルロース、カルボキシメチルセルロ
ースナトリウムおよび/またはポリビニルピロリドン;必要に応じてd)崩壊剤、例えば
、でんぷん、寒天、アルギン酸またはそのナトリウム塩もしくは発泡性混合物;および/
またはe)吸収剤、着色剤、矯味矯臭剤および甘味剤とともに有効成分を含む錠剤ならび
にゼラチンカプセル剤である。注射用組成物は、好ましくは等張水溶液もしくは懸濁液で
あり、坐剤は脂肪性乳濁液もしくは懸濁液から調製するのが有利である。これらの組成物
は、滅菌することができ、および/または保存剤、安定化剤、湿潤剤または乳化剤などの
佐剤、溶解促進剤、浸透圧調節塩および/または緩衝液を含むことができる。また、さら
に、これらは別の治療上有用な物質を含むことができる。これらの組成物は、それぞれ、
従来の混和、顆粒化またはコーティング法によって調製することができ、有効成分を約0
.1〜75%、好ましくは約1〜50%含有する。
【0078】
本明細書に開示した活性化合物および塩の投与は、一般に認められている治療剤の投与
方法のうちの任意の方法によることができる。これらの方法としては、経口、鼻腔内、非
経口、経皮、皮下もしくは局所性投与様式などの全身性または局所投与が挙げられる。
【0079】
目的とする投与様式に応じて、この組成物は、例えば、好ましくは単位投与量の注射用
剤、錠剤、坐剤、丸剤、徐放性カプセル剤、散剤、液剤、懸濁剤などのような固体もしく
は半固体または液状の投与形態とすることができる。この組成物は、有効量の活性化合物
もしくはその薬学的に受容可能な塩を含むことになり、また、さらに、薬学において通例
用いられている任意の従来の薬学的賦形剤および他の治療薬もしくは薬学的組成物、担体
、アジュバント、希釈剤なども含むことができる。
【0080】
固体組成物の賦形剤としては、医薬用品質のマンニトール、乳糖、でんぷん、ステアリ
ン酸マグネシウム、サッカリン・ナトリウム、滑石、セルロース、グルコース、蔗糖、炭
酸マグネシウムなどを用いることができる。また、上記の活性化合物は、例えば、プロピ
レングリコールなどのポリアルキレングリコールを担体とする坐剤として製剤化すること
ができる。
【0081】
液体、特に注射用組成物は、例えば、溶解、分散などにより調製することができる。活
性化合物は、例えば、水、生理食塩液、ブドウ糖水溶液、グリセロール、エタノールなど
の医薬用純品溶媒に溶解もしくは混合することにより、注射用溶液もしくは懸濁液を作製
する。
【0082】
また、投与することになる薬学的組成物は、必要に応じて、湿潤剤、乳化剤、pH緩衝
剤、および例えば、酢酸ナトリウム、オレイン酸トリエタノールアミンのような他の物質
などの毒性のない補助剤を少量含むことができる。
【0083】
一般に、非経口注射用剤は、皮下、筋肉内もしくは静脈内注射もしくは注入により投与
する。注射用剤は、溶液もしくは懸濁液、または液に溶かした後、注射するのに適した固
体形態のような通常の形態として調製することができる。
【0084】
非経口投与の1つの方法として、本明細書に引用により組み込まれている米国特許第3
,710,795号に記載の方法に従い、投与量の一定のレベルが維持されるのを確実な
ものにする徐放性もしくは持続放出性製剤の埋め込み法を用いる。
【0085】
本発明の化合物は、錠剤、カプセル剤(各々が徐放性および持続放出性製剤を含む)、
丸剤、散剤、顆粒剤、エリキシル剤、チンキ剤、懸濁剤、シロップ剤および乳剤のような
経口剤形として投与することができる。また、同様に、この化合物は、医薬分野の当業者
に周知の形態により静脈内(急速および点滴の両方)、腹腔内、皮下もしくは筋肉内形態
で投与することができる。
【0086】
この化合物の用法・用量は、患者の体型、種類、年齢、体重、性別および病状;治療対
象の症状の重症度;投与経路;患者の腎および肝機能;ならびに使用する特定の化合物も
しくはその塩を含む種々の要素に基づいて選択する。当該分野の医師もしくは獣医師であ
れば、症状の進行を防止し、遅らせ、または止めるのに必要とされる薬剤の有効量を容易
に決定し、処方することができる。
【0087】
本発明の経口用量は、適応となる効果に用いる場合、有効成分を0.5、1.0、2.
5、5.0、10.0、15.0、25.0、50.0、100.0、250.0、50
0.0もしくは1000.0mg含有する分割錠の形態で提供し得る。
【0088】
本発明の化合物は、1日1回の用量として投与することができ、あるいは1日当たりの
総投与量を1日2回もしくは3回または4回の分割用量として投与することができる。さ
らに、本発明に好ましい化合物は、適切な鼻腔内投与用器具(intranasal v
ehicle)の局所使用による鼻腔内投与、もしくは当業者に周知の形態の経皮投与用
皮膚パッチ剤による経皮投与を行うことができる。経皮送達システムの形態で投与する場
合には、勿論、適量投与(dosage administration)は用法・用量
全体を通して間欠的ではなく連続的なものとなる。その他の好ましい局所用製剤としては
、クリーム剤、軟膏剤、ローション剤、エアロゾル・スプレー剤およびゲル剤が挙げられ
、これらの場合、有効成分の濃度は0.1%〜15%w/wまたはw/vの範囲となろう
。
【0089】
本明細書に詳細に述べた化合物は、有効成分を構成することができ、通常、対象とする
投与形態、即ち、経口用としての錠剤、カプセル剤、エリキシル剤、シロップ剤などに対
して適切に選ばれ、従来の調剤慣行に合った適切な薬学的希釈剤、賦形剤もしくは担体(
本明細書ではこれらを総称して「担体」物質と呼ぶ)と混合して投与する。
【0090】
例えば、錠剤もしくはカプセル剤として経口投与する場合、有効薬物成分は、エタノー
ル、グリセロール、水などの経口用で毒性がなく薬学的に受容可能な不活性担体と混合す
ることができる。さらに、所望もしくは必要に応じて、この混合物に適切な結合剤、滑沢
剤、崩壊剤および着色剤を混和することもできる。好適な結合剤としては、でんぷん、ゼ
ラチン、グルコースもしくはベータ乳糖のような天然糖、コーン甘味剤、アカシア、トラ
ガカントもしくはアルギン酸ナトリウムのような天然および合成ゴム、カルボキシメチル
セルロース、ポリエチレングリコール、ろうなどが挙げられる。こうした剤形に用いる滑
沢剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシ
ウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウムなどが挙げられる。崩壊剤
としては、でんぷん、メチルセルロース、寒天、ベントナイト、キサンタン・ガムなどが
挙げられるが、これらに限定されない。
【0091】
また、本発明の化合物は、小単層小胞、大単層小胞および多重層小胞などのリポソーム
送達システムの形で投与することもできる。リポソームは、コレステロール、ステアリル
アミンもしくはホスファチジルコリンを含有する種々のリン脂質から形成することができ
る。一部の実施形態として、米国特許第5,262,564号に記載されているようにし
て、脂質成分の膜を薬剤の水溶液で水和させて、薬剤を内包する脂質層を形成する。
【0092】
また、本発明の化合物は、標的を定めることができる薬物担体としての可溶性ポリマー
とカップリングさせることもできる。このようなポリマーとしては、ポリビニルピロリド
ン、ピラン・コポリマー、ポリヒドロキシプロピル−メタクリルアミド−フェノール、ポ
リヒドロキシエチルアスパンアミドフェノール(polyhydroxyaspanam
idephenol)、もしくはパルミトイル残基で置換したポリエチレンオキシドポリ
リジンを挙げることができる。さらに、本発明の化合物は、薬剤の放出を制御するのに有
用なクラスの生分解性ポリマー、例えば、ポリ乳酸、ポリεカプロラクトン、ポリヒドロ
キシ酪酸、ポリオルソエステル、ポリアセタール、ポリジヒドロピラン、ポリシアノアク
リレート、およびヒドロゲルの架橋もしくは両親媒性ブロックコポリマーとカップリング
させることができる。
【0093】
上記薬学的組成物はいずれも、活性化合物、特に有効成分としての式Iの化合物を0.
1〜99%、好ましくは1〜70%含有することができる。
【0094】
(その他の実施形態)
本発明の1つ以上の実施形態についてその詳細を前記の添付明細書において説明した。
本発明の特定の実施形態についての前記詳細な説明から、生体膜を通過させて移行させる
ための独自の方法および組成物が開示されたことが明瞭に理解されよう。本明細書では特
定の実施形態についてその詳細を説明したが、これは単に例示を目的として例として説明
したものであり、以下に添付した特許請求の範囲に対して限定しているものではない。特
に、この特許請求の範囲に定義したような本発明の精神および範囲から逸脱することなく
、本発明に対し、種々の置換、変更および変形を行うことができることは、本発明者によ
り企図されている。例えば、特定のタイプの細胞、または移行させるべき特定のエフェク
ターの選択については、本明細書に開示した実施形態を知った当業者には日常的な事柄で
あると考えられる。
【0095】
本明細書および添付の特許請求の範囲では、文脈上明らかに別の意味に解すべき場合を
除き、単数形は複数の対象を含む。特に定義しない限り、本明細書に用いている科学技術
用語は全て、本発明が属する分野の当業者により一般に理解されている意味と同じ意味を
有する。また、本明細書に引用されている全ての特許および刊行物は、引用により本明細
書に組み込まれている。
【0096】
以下に示した実施例は、本発明の好ましい実施形態についてさらに十分に説明するため
のものである。これらの実施例は、添付の特許請求の範囲により定義した本発明の範囲を
限定するものと決して解釈されるべきではない。
【実施例】
【0097】
(実施例1:内部移行ペプチドモチーフの特定)
ファージ・ペプチド・ライブラリは、古典的な方法では、線維状ファージM13の誘導
体として構築される。ペプチド・ライブラリを、ペプチドモチーフの1〜5個のコピーを
提示するキャプシドのマイナーコートタンパク質pIIIに融合する。Zwickら、C
urr.Opin.Biotechnol.9:427−436(1998)を参照され
たい。あるいは、メジャーコートタンパク質pVIIIを用いることによって高価(hi
gh valent)提示が得られる。しかしながら、これらのタイプのライブラリは、
受容体媒介性エンドサイトーシスの対象となるペプチド配列の単離のために最適化されて
いない。何故なら、ペプチドの1価もしくは低価提示は、線維状ファージのような巨大な
構造体を効率的に取り込ませるには不十分であるからである。多価提示のみが効率的取り
込みを可能にする。Ivanenkovら、Biochem.Biophys.Acta
1448:450−462(1999)を参照されたい。さらに、受容体結合リガンド
の内部移行は、原形質膜の特定領域に細胞表面受容体が濃縮され、次いでクラスリン被覆
小胞が形成される必要があり(Damke、FEBS Lett.389:48−51(
1996)参照)、従来のM13ファージでは、こうした特殊で高度に効率的な構造によ
る受容体媒介性内部移行が行われるとは考えられない。
【0098】
T7 415ファージ系およびT7 413bファージ系のファージ・ディスプレイ・
ライブラリを構築した。このファージでは、減少した容量内にこの提示ペプチドが415
個のコピーとして生じる(キャプシドの直径は約50nMである)。従って、本発明に含
まれるのは、以下の基準を満たす新規なファージ系のファージ・ディスプレイ・ライブラ
リである:4〜50merペプチドの多価提示(>400個のコピー/ファージ);サイ
ズが小さい(50nM);内部移行したファージを効率的に回収できる;内部移行しなか
った結合ファージを除去できる;および個別ペプチド配列の数が大である(>109個の
ヘプタペプチド配列を示す3×108個の独立クローン)。
【0099】
このライブラリを用いて、βTC−3細胞モデル内への高分子の効率的、特異的な細胞
内送達を誘導するペプチドモチーフの単離に成功した。さらに、このライブラリを5種の
異なる(非−β)細胞株に対して用い、それぞれの場合で、各細胞型に特異的なペプチド
モチーフの濃縮(enrichment)について観察した。この方法の概括的な全体像
を以下に説明する。
【0100】
(選択/濃縮(enrichment)方法)
多くのインスリン分泌細胞株、齧歯類およびヒト単離島、およびFACSにより精製し
たβ細胞に対するファージ・ディスプレイ・ライブラリをパニングし、最終的に、これを
直接動物(マウス、ラット、ブタ)に注射した後、島を抽出して内部移行したファージを
回収する。パニング方法は、ファージの添加、回収および増幅の少なくとも3サイクルか
らなる。あるいは、最も選択的なリガンドを単離するために、このライブラリを種々のイ
ンスリン非分泌細胞と共にインキュベートした後にβ細胞に対するライブラリを選択する
ことにより、他の細胞型に結合するファージを除去する。記載されるように、クロロキン
を用いてリソソームによる分解をブロックする実験を実施する。Ivanenkovら、
Biochem.Biophys.Acta 1448:450(1999)を参照され
たい。
【0101】
(ファージ特異性の測定)
選択したファージを単離し、多くの種々の細胞および器官と共にインキュベートする。
例えば、一部の実験として、選択したファージをインスリン分泌およびインスリン非分泌
細胞および器官とインキュベートする。取り込みについては、回収されたファージ数をカ
ウントすることにより測定する。抗ファージ抗体を用いて免疫細胞化学的研究を実施する
。
【0102】
(ファージ由来(pharge−beard)ペプチドの特徴付け)
単離ファージからのDNAを配列決定し、発現されるペプチドを推定する。直接の内部
移行を誘導するペプチド、およびこれらのペプチドの変異型を化学的に合成し、N末端を
FITCで標識し、もしくはヨウ素化する。標識ペプチドを種々の細胞型、齧歯類および
ヒト単離島に加え、そして、マウスに直接注射する。取り込みの特異性、細胞内局在性、
クリアランスおよび安定性について評価する。Widmannら、Biochem.J.
310:203(1995)を参照されたい。
【0103】
(生化学的アッセイ)
インスリン分泌細胞およびインスリン非分泌細胞を分析するため、特徴付けを行ったペ
プチドを3種の既知配列:YVAD(カスパーゼ阻害因子、配列番号35;Rouque
tら、Curr.Biol.6:1192(1996))、VQRKRQKLMP(NF
−κB核内局在化阻害因子(Linら、J.Biol.Chem.270:14255(
1995)、配列番号36)もしくはRPKRPTTLNLFPQVPRSQDT(JN
K阻害因子、Bonnyら、Diabetes 50:77−82(2000)、配列番
号37)に結合させる。これらのペプチドは、化学的に合成し、インスリン分泌細胞およ
びインスリン非分泌細胞に加える。カスパーゼ、NF−κBおよびJNKは、一般的活性
化剤エトポシド(Kimら、Anticancer Res.20:439(2000)
参照)もしくはアニソマイシン(Usamiら、Biochem.Pharmacol.
55:185(1998)参照)により活性化する。上記ペプチドによるカスパーゼ、N
F−κBおよびJNKの阻害についてはβ細胞および非β細胞を用いて検討する。以上の
実験によって、これらのペプチド担体が薬剤となり得る物質を活性のある形態で特にβ細
胞内に移送することができるかどうかが分かる。
【0104】
(治療剤となり得る物質のGLP−1受容体による取り込み)
GLP−1受容体(GLP−1R)の発現は、主として脳および膵臓に限定されている
。Yamatoら、Horm.Metab.Res.29:56(1997)を参照され
たい。この受容体は、アゴニストに結合した後、内部移行される。Widmannら、B
iochem.J.310:203(1995)を参照されたい。こうした性質があるた
め、GLP−1Rは膵β細胞への治療剤の選択的送達を媒介する魅力的なツールとなる。
この特性は、上に記載されるように評価される。GLP−1Rを用いて集められた情報が
、例えば、高められた選択性を有する二重特異的ダイマーの設計において、補助する。
【0105】
(GLP−1受容体に対する他の内部移行モチーフの特定)
GLP−1Rを用いてトランスフェクションしたCOS−7細胞は、上記のような選択
実験の基材(substrate)となる。新規に特定したモチーフについてその特異性
およびエンドサイトーシス誘導能を評価する。
【0106】
(全−D−レトロ−インベルソ型(all−D−retro−inverso)ペプチ
ドの作製)
一部の実施形態として、上記ペプチドはレトロ−インベルソ型ペプチドとして合成する
ことができる。安定性が高く、免疫原性の少ない全D−レトロ−インベルソ型ペプチド(
Sela、Zisman,FASEB J.11:449(1997)参照)について、
前述の方法により、分析する。
【0107】
進化によって、天然のタンパク質にはほぼL型アミノ酸のみが存在することになった。
従って、事実上全てのタンパク質分解酵素は、隣接するL型アミノ酸間のペプチド結合を
切断するので、D型アミノ酸からなる人工的なタンパク質もしくはペプチドは、タンパク
質分解に対してかなり抵抗性である。この抵抗性は薬剤の設計者には魅力的であったが、
L−アミノ酸からなるタンパク質の生体系が排他的であるため、このようなタンパク質は
、鏡像異性タンパク質により形成される鏡像面と相互作用することができないということ
になる。従って、通常、全D(all−D)型アミノ酸タンパク質には生物効果もしくは
活性がない。
【0108】
線状の修飾レトロ−ペプチド構造については長い間研究されてきており(Goodma
nら、Accounts of Chemical Research,12:1−7(
1979)参照)、「レトロ−アイソマー」という用語は、親ペプチドに対して配列の方
向が逆であるアイソマーを含むとされた。「レトロ−インベルソ・アイソマー」とは、配
列の方向が逆で、各アミノ酸残基の対掌性が逆であり、従って末端基相補性が生じ得ない
線状ペプチドのアイソマーのことを意味する。
【0109】
さらに最近、Jamesonらは、これら2つの性質、即ち、逆合成および対掌性の変
化を組み合わせることにより、CD4受容体のヘアピンループのアナログを設計した。J
amesonら、Nature 368:744−746(1994)およびBrady
ら、Nature,368:692−693(1994)を参照されたい。D−鏡像異性
体と逆合成を組み合わせたことによる最終的な結果は、各アミド結合のカルボニル基とア
ミノ基とが交換されるが、各α炭素の側鎖基の位置は保存されたことである。James
onらは、従来の全−L鏡像異性体の限定的なインビボ活性(タンパク質分解を受けやす
いことに起因する)とは対照的に、逆Dペプチドの生物活性が増大することを明らかにし
た。
【0110】
ヒト・クラスI組織適合性分子HLA−A2の非天然リガンドとして使用することがで
きる部分修飾したレトロ−インベルソ偽ペプチドが報告されている。Guichardら
、Med.Chem.39:2030−2039(1996)を参照されたい。このよう
な非天然リガンドでは、安定性およびMHC結合能が増大した。
【0111】
レトロ−インベルソ・ペプチドを、以下のようにして配列が既知のペプチドに対して作
製する。レトロ−インベルソ・ペプチドアナログをデザインし、合成するためのモデル・
ペプチドとして、既知の配列を有するペプチド(例えば、腫瘍抗原ペプチド)を選択する
。このアナログは、D−アミノ酸を用い、このレトロ−インベルソ・ペプチド内のアミノ
酸の配列がモデルとして働く選択したペプチド内の配列と正確に逆になるように、D−ア
ミノ酸を結合させてペプチド鎖とすることにより合成する。例えば、このペプチド・モデ
ルがABCの配列を有するL−アミノ酸で形成されているペプチドである場合、D−アミ
ノ酸で形成されるそのレトロ−インベルソ・ペプチドアナログはCBAの配列を有するこ
とになる。D−アミノ酸の鎖を合成してレトロ−インベルソ・ペプチドを作製する方法は
、当該分野で公知であり、前記文献において説明されている。
【0112】
天然ペプチドの場合の固有の問題が天然のタンパク質分解酵素により分解されることに
あるので、本発明のペプチドは、目的ペプチドの「レトロ−インベルソ・アイソマー」を
含むように作製することができる。従って、天然性のタンパク質分解からペプチドを保護
することにより、この特定のヘテロ2価(heterobivalent)もしくはヘテ
ロ多価(heteromultivalent)化合物の有効性は増大するはずである。
【0113】
レトロ−インベルソを含むペプチドでは、天然のタンパク質分解酵素による分解から保
護されるため、レトロ−インベルソを含まないアナログと比べて、生物活性が増大すると
予測される。
【0114】
(修飾ペプチドの作製)
一部の実施形態として、このペプチドは、修飾ペプチドとして合成することができる。
この修飾ペプチドは前述の方法により分析する。
【0115】
アナログは、アミノ酸配列により、またはアミノ酸配列に影響を与えない修飾により、
あるいはこれらの両者により天然のペプチドと異なるものとすることができる。好ましい
アナログとしては、その配列が、単に保存的アミノ酸置換、好ましくは1、2もしくは3
置換のみ、例えば、1つのアミノ酸の同様な特徴を有する別のアミノ酸による置換(例え
ば、グリシンをバリンに、リジンをアルギニンに置換など)によって、またはペプチドの
生物活性を消失させない1つ以上の非保存的アミノ酸置換、欠失もしくは挿入によって、
その野性型配列(すなわち、天然に生じるペプチドの相同な部分の配列)と異なるペプチ
ドが挙げられる。
【0116】
(普通一次配列に影響を与えない)修飾としては、インビボもしくはインビトロにおけ
るペプチドの化学的誘導体化、例えば、アセチル化もしくはカルボキシル化が挙げられる
。また、糖鎖形成の修飾、例えば、ペプチドの合成およびプロセッシング過程において、
または、例えば、糖鎖形成に影響を与える酵素、例えば、哺乳類のグリコシル化もしくは
脱グリコシル化酵素をペプチドに作用させることによる別のプロセッシング工程において
、ペプチドの糖鎖形成パターンを修飾することによるものも挙げることができる。また、
リン酸化されたアミノ酸残基、例えば、ホスホチロシン、ホスホセリンもしくはホスホス
レオニンを有する配列を挙げることもできる。
【0117】
本発明は、1つ以上のペプチド結合が、ペプチダーゼにより切断されにくい別のタイプ
の共有結合(「類似ペプチド結合」)に置換されたアナログを含む。対象に注射後のペプ
チドの蛋白性分解が問題となる場合、特定の切断されやすいペプチド結合を切断不能の類
似ペプチド結合に置換すると、得られるペプチドは安定性が増し、治療剤としてより有用
なものとなる。このような類似ペプチド結合およびこれをペプチドに組み込む方法は、当
該分野では周知である。また、t−ブチルオキシカルボニル、アセチル、テイル(the
yl)、スクシニル、メトキシスクシニル、スベリル、アジピル、アゼライル、ダンシル
、ベンジルオキシカルボニル、フルオレニルメトキシカルボニル、メトキシアゼライル、
メトキシアジピル、メトキシスベリル、および2,4−ジニトロフェニルなどのアミノ末
端ブロック基も有用である。ペプチドの荷電アミノ−末端およびカルボキシ−末端をブロ
ックすると、ペプチドが疎水性細胞膜を通過して細胞内へ入るのが促進されるという別の
メリットがある。
【0118】
(多量体ペプチドの作製)
多量体リガンドでは、内部移行速度の増大につながる最大数桁の結合力の向上がみられ
る。Terskikhら、Proc.Natl.Acad.Sci.U.S.A.94:
1663(1997)およびYorkら、J.Biol.Chem.274:1164(
1999)を参照されたい。単一特異性ダイマーは、強い結合力を示し、二重特異性ダイ
マーでは、特異的に細胞を標的にする物質としての実用面の潜在能力を向上させることが
できる選択性が高まると考えられる。Caruthers and Lerner,Ch
em.Biol.3:537(1996)を参照されたい。単量体および多量体ペプチド
(単一および多重特異性)は、ペプチドとして、あるいは例えば、柔軟性のあるペプチジ
ルまたは糖ベースの主鎖を有するペプチド類似体として合成することができる。Caru
thers and Lerner,Chem.Biol.3:537(1996)、Z
engら、J.Pept.Sci.2:66(1996)およびUlbrichら、J.
Controlled Release 64.(1.−3.):63−79.、64:
63(2000)を参照されたい。
【0119】
(細胞内局在性)
単離した種々のペプチド配列は、種々の細胞コンパートメント(例えば、核、ミトコン
ドリア、細胞質ゾルなど)に局在し得る。これについては、標識(例えば、ヨウ素化もし
くはFITC標識)ペプチドを用いて調べる。この局在性情報を利用して機能性研究をデ
ザインする。例えば、細胞質ゾル内に蓄積するペプチドは、NF−κBの核移行を阻害す
るのに好ましく、核内に移行するペプチドは、JNKを阻害するのに最も適している。一
部の実施形態として、核局在化モチーフのような配列を結合体に加えて、担体を適切な細
胞コンパートメントへ再誘導することができる。
【0120】
(機能性研究)
カスパーゼ、NF−κBもしくはJNK阻害剤に結合させた、β細胞を標的とするトラ
ンスポーターペプチド(例えば、単量体もしくは多量体のL型もしくはD型エナンチオマ
ー)をβ細胞株、FACS精製β細胞ならびに単離ヒトおよび齧歯類の島(islet)
に加える。IL−β(TNFαおよびIFNγを併用)によりアポトーシスを誘発させ、
アポトーシスに対する抵抗性を調べる。
【0121】
(インビボ糖尿病実験)
糖尿病発症前および発症後の状態のNODマウスにエフェクタ・ペプチド(カスパーゼ
、NF−κBもしくはJNK阻害剤に結合させたβ細胞を標的とするペプチド)を注射す
る。用量および注射頻度は、前述のようにして決定する。次いで、糖尿病の発生を測定す
る。
【0122】
(免疫原性に関するアッセイ)
齧歯類およびウサギを用い、ペプチドの免疫原性の潜在性について評価する。
【0123】
(クローニング)
特定の細胞による効率的な内部移行を誘導するペプチドモチーフについては実施例II
IおよびIVにおいて説明する。このペプチドを用いて、確立された手順により、例えば
、INS−1、βTC−3およびヒト島cDNAライブラリからの同族(cognate
)受容体のクローニングならびに特徴付けを行う。Thorens、Proc.Natl
.Acad.Sci.U.S.A.89:8641(1992)およびVolzら、FE
ES Lett.373:23(1995)を参照されたい。
【0124】
(特徴付け)
上記のクローニングした受容体の組織分布について、インスリン分泌およびインスリン
非分泌細胞および器官のノーザンブロッティングおよびウェスタンブロッティングにより
調べる。結合速度、クリアランスおよび内部移行の特異性についてはCOS−7細胞にこ
の受容体を一時的にトランスフェクションさせることにより調べる。対照ペプチドは、変
異配列、および例えば、GLP−1、GIP、グルカゴン、セクレチンなどの既知ペプチ
ドとする。これらの受容体に対する別の内部移行モチーフについては上記のトランスフェ
クションしたCOS−7細胞において前記ライブラリをパニングすることにより特徴付け
る。
【0125】
(実施例II:トランスポーターペプチドのスクリーニング方法)
特に指定した場合を除き、溶媒および試薬は全てフルカ社(Fluka)、ブックス(
Buchs)、スイスから入手し、分析品質以上のものとし、さらに精製することなく使
用した。アミノ酸は全てペプチド・インスティテュート社(Peptide Insti
tute Inc.)(日本)から購入した。樹脂はアプライド・バイオシステムズ社(
Applied Biosystems)、米国;ノバビオケム社(Novabioch
em)、スイスもしくはバッヘム社(Bachem)、スイスから入手した。水は、Mi
lli−Qシステム(ミリポア社(Millipore,Inc.)を用いて再精製した
。バイオアッセイにはインスリン分泌細胞株βTC−3を用いた。Efratら、Pro
c.Natl.Acad.Sci.USA 85:9037−9041を参照されたい。
【0126】
(ファージの調製および濃縮(enrichment)方法)
標準的な手順により(例えば、ノバジェン社(Novagen)のT7 414ファー
ジもしくはT7 413bファージを用い)、キャブシドの表面にランダムな15マーエ
ピトープを提示する3×108個の単独(independent)ファージのライブラ
リを作製した。SmithおよびScott,Methods Enzymol.217
:228(1993)を参照されたい。(Smith、Scottの上記文献に)記載さ
れるように、ファージを増幅させた後、ポリエチレングリコール(PEG)沈殿法により
精製し、最終的に、Tris−EDTA緩衝液(10:1mM、TE)中に1010個感
染性粒子/μlの濃度で再懸濁した。培養培地中の細胞にファージ(1012個)を1〜
24時間加えておいた。エンドサイトーシス小胞内でタンパク質分解を免れたファージの
単離を容易にするには、インキュベーション時間をさらに長くすることが好ましかった。
結合および内部移行を行わせた後、細胞を洗浄し、内部移行されなかったファージを、サ
ブチリシン(3mg/ml)(44)で消化させて破壊した。充分に洗った後、2%デオ
キシコール酸塩、10mM Tris−HClおよび2mM EDTAを含有するpH8
.0の緩衝液で細胞を溶解させることにより、内部移行されたファージを回収した。回収
したファージは、最終的にE.coli細胞(XL−1−Blue)を用いて増幅させ、
前述のようにして精製した。次いで、この選択したファージの調製物を用いて第2ラウン
ドのパニングを行った。3〜5ラウンドを順次実施することにより特定のファージ保有ペ
プチド配列を濃縮した。
【0127】
(免疫細胞化学および蛍光による検討)
上記の濃縮スキームによって単離した単一ファージを増幅させた後、培養培地中の細胞
に24時間加えておいた。次いで、培地を洗い流し、細胞を冷メタノール−アセトン(1
:1)で5分間固定した。ファージ・キャプシドに対する抗体は、蛍光結合二次抗体と共
に用いた。古典的な蛍光顕微鏡による検討および共焦点顕微鏡によるアッセイを実施した
。組織は、処理の前にパラフィン包埋した。
【0128】
(ペプチド)
C−末端アミド基を有し、必要に応じてFITC標識もしくはヨウ素化したペプチドを
、古典的なF−moc化学(オースペップ社(Auspep)、オーストラリア)を用い
て合成した。ペプチドは全てHPLCにより精製し、質量分析法によって分析した。
【0129】
(生化学的検討)
種々の細胞株、例えばβTC−3、INS−1、HeLa、WiDr、HepG2、N
IH3T3、COS−7などにおいて、ペプチド添加の1時間後からJNK、NF−κB
およびカスパーゼをエトポシド(VP−16,Alexis)により1時間賦活化する。
細胞抽出物を処理して(c−Junを基質として用いる固相JNKアッセイ(Bonny
ら、J.Biol.Chem.275:16466(2000)参照)により)JNK活
性、(電気泳動移動度シフトアッセイ(Linら、J.Biol.Chem.270:1
4255(1995)参照)により)NF−κBの核移行および(アップステート・バイ
オケミカルス社(Upstate Biochemicals)から市販のキットおよび
抗体により)カスパーゼ活性を測定する。
【0130】
(アポトーシスの測定)
アポトーシスは、以前に報告された方法に従い、Hoechst33342およびヨウ
化プロピジウムの組合せを用いて測定する。Bonnyら、J.Biol.Chem.2
75:16466(2000)、Hoorensら、J.Clin.Invest.98
:1568(1996)を参照されたい。
【0131】
(島)
島は、Gotohらの方法(Gotohら、Transplantation 43:
725(1987)参照)により単離する。ヒト島は、例えば、「Insel Spit
al」、ベルン(Bern)、スイスから入手することができる。
【0132】
(マウス)
正確な投与量および注射の時間枠は、各ペプチドごとに最適化する。しかしながら、J
NKIを用いたこれまでの経験から、ペプチドの1mM PBS溶液100μlを2日お
きに注射するのが合理的な出発点であることが分かっている。
【0133】
(λZAP発現ライブラリ)
λZAP発現原核/真核発現ベクター中のINS−1 cDNAライブラリを用いてI
B1 cDNAおよびIB2 cDNAをクローン化した。Bonnyら、J.Biol
.Chem.273:1843(1998)およびNegriら、Genomics 6
4:324(2000)参照。このライブラリは、真核CMVプロモータの制御下で、単
純なヘルパー・ファージ切り出し(excision)(ストラタジーン社(Strat
agene)によって容易にプラスミド・ライブラリに変換される。
【0134】
(実施例III:ファージ・ディスプレイ・ライブラリのパニングおよび内部移行され
たペプチドモチーフの特徴付け)
β細胞を特異的に標的にして薬剤を送達させることができれば、I型糖尿病の治療に極
めて大きな影響を与えることになろう。基本的にβ細胞の機能(すなわち、インスリン分
泌)を変えないβ細胞破壊のブロッカーはすでに存在する(例えば、JBD、bel−2
)。こうした分子の1つ(JBD)を低分子ペプチドに変換しても全生物活性を保持させ
ることができることが示されている。
【0135】
膵β細胞株βTC−3は、本明細書に記載したファージ・ディスプレイ・ライブラリを
用いてパニングした。下記の表1に示すように、選択の各サイクルにおいて、回収ファー
ジ数の選択的な濃縮が認められた。βTC−3細胞を用いるパニング実験を、濃縮手順の
各工程で109個のファージを用いて実施した。0℃(エンドサイトーシスはみられない
)で回収されたファージ数は100個未満であり、このことから、本明細書に記載した条
件下では、内部移行されなかった細胞外の結合ファージのバックグラウンドは極めて低い
ことが分かる。
【0136】
【表1】

【0137】
βTC−3細胞株における3工程のパニング後のファージ回収率を表2に示す。
【0138】
【表2】
【0139】
表3に示した時間βTC−3細胞とインキュベートしたファージP1(配列番号1)を
用い、滴定実験を実施した。また、投入/回収ファージの比率も示す。滴定実験から、最
初の投入P1ファージの10%も回収することができることが分かった。
【0140】
【表3】
【0141】
取り込みの特異性についての測定は、5種の細胞株における回収ファージ数をタイトレ
ートすることにより行った。ファージ(108個)を、示した細胞株と16時間インキュ
ベートした後、表4に示したように、内部移行されたファージ数および回収されたファー
ジ数を算定した。インテグリン内部移行モチーフを提示する対照ファージは、全ての細胞
株に対して同様な回収ファージ数(1〜3×106個)を示した。このことから、P1(
配列番号1、表5参照)は、βTC−3細胞によって、テストした他のどの細胞株より1
0,000〜1,000,000倍効率的に内部移行されることが分かる。
【0142】
【表4】
【0143】
次いで、ファージP1の提示ペプチドの配列に基づいてペプチドを合成した。FITC
で標識した10アミノ酸ランダム配列にP1 5マーペプチドの配列を結合させた。対照
配列は、P1 5マー配列を(Ala)5で置き換えた以外は同一であった。細胞にペプ
チド(10μM)を1時間加え、細胞を洗い、冷メタノール−アセトン(1:1)で固定
した。βTC−3細胞内のFITC標識P1ペプチドは可視化することができたが、他の
細胞型ではできなかった。
【0144】
最後のサイクルの濃縮(enrichment)時の20個の回収ファージの配列解析
結果を表5に示す。重要なことであるが、全ての配列は、5個のアミノ酸の同じ保存コン
センサス配列に厳密に従った。このことから、ファージの効率的な取り込みを誘導する保
存モチーフが特異的に選択/濃縮されることが示唆される。こうして得られたペプチドモ
チーフの大部分は、プロタンパク質転換酵素のコンセンサスなR−X−X−Rに従う。こ
の知見は、潜在的薬物および高分子を特定の細胞型の細胞内に送達するためのビヒクルと
してプロタンパク質転換酵素を使用する提案の根拠を成すものである。
【0145】
【表5−1】
【0146】
【表5−2】
【0147】
(実施例IV:多量体結合体)
ファージ・ライブラリから得られるリガンド・ペプチドは低親和性(マイクロモル程度
)とすることができる。その結合親和性もしくは結合力は、ペプチド分子のコピーを多量
体化することにより、例えば、ペプタボディ(peptabody)を形成させることに
より向上させることができる。ScottおよびSmith,Science 249:
386−90(1990)、Renschlerら、Proc.Natl.Acad.S
ci.USA 91:3623−3627(1994)、およびAlexeyら、Pro
c.Natl.Acad.Sci.USA 94:1663−1668(1997)を参
照されたい。ファージを用いた実験から、ファージ・ペプチドRRTK(配列番号1、上
記参照)は受容体媒介性エンドサイトーシスを介してβTC−3細胞により特異的に取り
込まれるが、その内部移行効率は低い(これはその低親和性結合(ほぼマイクロモルの程
度)に起因し得る)ことが分かった。レポーター・タグ(FITCまたはビオチン)と共
にこのペプチドの複数(4もしくは8個)のコピーを有する一連の多量体(図1および2
)を構築した。
【0148】
本発明の多量体トランスポーターペプチドは、式I:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
によって表すことができ、式中、Pは前述のコンセンサスモチーフ(XmRXoRXn)
、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)のトランスポ
ーターペプチドである。例えば、このペプチドは、配列X1−X2−X3−X4を含むこ
とができ、式中、X1およびX4はRもしくはKとすることができ、X1およびX4は任
意のアミノ酸とすることができる。vによって表される所与のトランスポーターペプチド
内のペプチド数は、2〜8以上の整数とする。任意の所与の多量体トランスポーターペプ
チドにおいて、それぞれのペプチドは互いに同じか、異なるものとすることができる。好
ましいペプチドとしては、例えば、表5に挙げたものがある。
【0149】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は1または0である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−PEG(succ−
peg)などのポリエチレングリコール(PEG)が挙げられる。
【0150】
企図されるコア部分としては、表7に記載したポリリジン(K)コアが挙げられ、また
、組み込まれたリンカーを有するものも挙げられる。選択されるコアにより、所与の結合
体内に存在するペプチドの数が決まる。代表的なコアとしては、C4−K2K−K(su
cc−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S
)−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよ
び(NH2OCH2CO)8−K4K2K−K(GGG)−アミドが挙げられる。リンカ
ーおよびレポーター基は存在していてもしていなくてもよく、従って、yおよびzは、式
Iにおいて独立に1もしくは0である。
【0151】
リンカーは存在していてもしていなくてもよい(即ち、yは1もしくは0である)。リ
ンカーとしては、コアをレポーターに接続する部分が挙げられ、例えば、succ−PE
GもしくはNH2OCH2CO−Lysとすることができる。
【0152】
レポーター基は存在していてもしていなくてもよい(zは1もしくは0である)。レポ
ーター基は検出可能な任意の基である。例としては、放射性同位体、蛍光性部分、リン光
性部分、化学発光性部分、および量子ドットが挙げられるが、これらに限定されるもので
はない。その他のレポーター基としては、ビオチン、シンテイン、ヒスチジン、赤血球凝
集素、mycもしくはフラッグ・タグが挙げられる。本明細書において用いる代表的な蛍
光レポーター基はFITC(フルオレセインイソチオシアネート)である。
【0153】
代表的な多量体トランスポーターペプチドは図1および図2に示した。
【0154】
多量体トランスポーターペプチド・ユニットもしくは結合体においては、多量体トラン
スポーターペプチドを、1つ以上のエフェクターと融合させる。このエフェクターは、D
NA、RNA、タンパク質、ペプチド、または薬学的に活性な薬剤、例えば、毒素、抗生
物質、抗病原体剤(antipathogenic agent)、抗原、抗体、抗体断
片、免疫調節剤、酵素もしくは治療剤などを含む任意の適切な分子とすることができる。
2つ以上のエフェクターが存在する場合、これらのエフェクターは同一もしくは異なるも
のとすることができる。
【0155】
従って、本発明の多量体結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学結合の法則通りに、エフェクターはコア、もしくはリンカー、またはこ
れらの両方に結合させることができる。
【0156】
(ペプチド合成)
ペプチド配列RRTK(P1、配列番号1)を、膵β細胞に結合させるためのペプチド
コンセンサスモチーフX1−X2−X3−X4(ここで、X1およびX4はRもしくはK
とすることができ、X1およびX4は任意のアミノ酸とすることができる)を明らかにす
るファージ・ディスプレイ実験から得た。表6に示されるように修飾したペプチドを、B
oc化学により0.5mmolのBoc−Lys(CIZ)−PAM樹脂を用いて手動で
合成した。イタリック体は、対応するタンパク質配列には存在しないリンカー残基および
スペースを示す。この実験に用いたいくつかのコア構造を表7に示す。合成を、Boc化
学により0.5mmolのメチルベンズヒドリルアミン樹脂(MBHA樹脂)を用いて行
った。
【0157】
ペプチドは全て、(5%のp−クレゾールの存在下に0℃で1時間)HFを作用させて
樹脂から切断し、冷エーテルで沈殿させ、50%アセトニトリルで抽出し、ろ過して凍結
乾燥した。粗製ペプチドを分取HPLCにより精製し、ESIMSにより特性を決定した
。
【0158】
【表6】
【0159】
【表7】
【0160】
(フルオレセイン・リンカーの調製)
フルオレセイン・リンカーNH2OCH2CO−Lys(FITC)−OHを、オフォ
ード(Offord)の方法により調製した。Offordら、Methods Enz
ymol.287:348−69(1997)を参照されたい。簡単に言えば、2mmo
lのBoc−アミノオキシアセチル(Boc−AOA−OSu)および1.2mmolの
Nε−(TFA)−Lysを3mlのDMSO(ジメチルスルホキシド)溶液に加え、N
−エチルモルホリンをpH値が8〜9となるまで加えた。室温で15時間、次いで37℃
で1時間インキュベートした後、絶えず撹拌しながら氷酢酸を注意深く加えることによっ
てpH値を3.0に下げた。生成物[Boc−AOA−Lys(TFA)−OH]を、分
取スケールのHPLCにより単離し、次いで乾燥した。この乾燥した化合物に4mlの水
、次いで0.44mlのピペリジン(最終濃度1M)を加え、これを室温で4時間インキ
ュベートした後、氷上でpHを3.0に調整した。次に、この混合物を0.1%のTFA
10mlで希釈した。分取HPLCにより脱保護物質(Boc−AOA−Lys−OH)
を単離し、再度乾燥した。この乾燥物質を300μlのN,N−ジメチルホルムアミド(
DMF)に溶かし、40mgのフルオレセインイソチオシアネート(FITC)を加え、
得られた混合物をN−エチルモルホリンでpH8.0に調整した。暗所で15時間インキ
ュベートした後、生成物を、メタノール/CH3Cl(1:1、v/v)で平衡させたシ
リカ・カラム[Kieselgel 60(フルカ・ケミー社(Fluka Chemi
e)、ブックス(Buchs)、スイス)、1.5×20cm]を用いたクロマトグラフ
ィーにより精製した。過剰のFITCを貫流(flow−through)画分に溶出さ
せ、予測される生成物を含む第二の黄色画分をメタノール/CH3Cl(4:1、v/v
)で溶出させた。回転式蒸発により溶媒を除去した後、この乾燥化合物Boc−AOA−
Lys(FITC)−OHを(20mg/ml以下の濃度の)TFA中で脱保護し、室温
で45分間置いておいた。次に、TFAの大部分を蒸発させ、凍結乾燥により乾燥を完了
させた。最終生成物[AOA−Lys(FITC)−OH]は、分取HPLCにより単離
し、ESI−MSにより特性を決定した。
【0161】
(コアへのペプチドリガンドのアルキル化)
0.9μmolのCys−コア・ペプチド(1.19mg)を50μlのアセトニトリ
ルおよび100μlの水に溶かした(ペプチドは完全には溶けない)。ブロモアセチル−
ペプチドの新鮮溶液(pH7.0の0.1Mリン酸ナトリウム緩衝液の3.6μmol溶
液)を調製した。次いで、2つの溶液を室温の暗所で混合することによりアルキル化を開
始させた。40分後に生成物を半分取RP−HPLCにより精製し、凍結乾燥し、そして
特性を決定した。
【0162】
(過ヨウ素酸酸化)
過ヨウ素酸酸化を行うため、0.375μmolのペプチド(表6のペプチド3もしく
は4、または表8に挙げたアルキル化ペプチド)を433μlアセトニトリルおよび1.
26mlイミダゾール緩衝液(50mM、pH6.95、塩化物対イオン)に溶かした。
メチオニン(113μl、200mM)を加えた。次に、17μlのNaO4(0.1M
水溶液)を加えて酸化を開始させた。5分後、40μlのエチレングリコール(500m
M水溶液)を加えることにより反応を停止させた。得られた生成物は分取HPLCにより
精製し、質量分析法により特性を決定した。
【0163】
【表8】
【0164】
(オキシム化)
次に、過ヨウ素酸酸化により得られたアルデヒドをコア3もしくは4(各コアにAOA
基を含む)、またはAOA−Lys(FITC)−OHと反応させてオキシムを形成した
。図1および2に示したオキシム1〜6をこうして作製した。また、表9に予測および実
測質量分析データを示した。図1に示されるように、オキシム1〜3については、コアの
分枝Lys残基を、αアミノ基とεアミノ基との両方において、スペーサーありまたはな
しで、RRTK(配列番号1)ペプチドのN末端でアシル化した。スペーサーと共に、レ
ポータ・タグFITCを各構築体のコアに導入した。図2のオキシム4〜6では、コアの
分枝Lys残基を、αアミノ基とεアミノ基との両方において、スペーサーとともにRR
TK(配列番号1)ペプチドのN末端でアシル化した。スペーサーと共に、レポータ・タ
グのビオチンを各構築体のコアに導入した。
【0165】
【表9】
【0166】
AOALys(FITC)−OHでオキシム化するために、0.129μmolの酸化
構築物を5μlのアセトニトリルおよび200μlのNaOAc緩衝液(酢酸0.57m
lおよび水100mlの混合物と、酢酸ナトリウム0.82gを水100mlに溶かした
溶液とをpH4.0となるように混合した溶液)に溶解させた。この混合物に0.516
μmolのAOA−Lys(FITC)−OH(100μlのアセトニトリルおよび10
0μlのNaOAc緩衝液に8.8mg/mlの濃度で溶かしたもの)を加えた後、酢酸
を最終濃度が4%となるよう加えた。コア3もしくは4をオキシム化するために、酸化し
たペプチド3もしくは4の3.8μmolを100μlのアセトニトリルおよび200μ
lのNaOAc緩衝液に溶解させた。この溶液に0.475μmolのコア3もしくは0
.237μmolのコア4を加えた後、酢酸を最終濃度が4%となるよう加えた。室温で
24時間反応させた後、生成物を半分取RP−HPLCで精製し、質量分析により特性を
決定した。
【0167】
4本の分枝のそれぞれの末端にRRTK(配列番号1)ペプチド(ペプチド1)のコピ
ーを有する円形のトリリジン・コア(コア1)にオキシム1(図1)を形成させた。各ペ
プチドのN末端を、ブロモアセチル基とチオール基との特異的な反応により生じるチオエ
ーテル結合を介して結合させた。また、このトリリジン・コアにはスクシンイミジル−P
EGスペーサー、および酸化を受けてアルデヒドを形成することができるセリンを含ませ
たので、レポーター基アミノオキシアセチル−Lys(FITC)を、オキシム形成を介
してこのテトラマーに導入した。HPLCおよびESI−MS分析により、目的生成物で
あることが示された:質量分析データ:Mr実測値4289.31、Mr理論値4289
.04。オキシム1は酸性および中性pHで24時間安定であった。
【0168】
オキシム2および3(図1)を、同様にして形成したが、オキシム3ではRRTKペプ
チド(配列番号1)とコアとの間に2ユニットのPEGスペーサーを配置し、オキシム2
ではRRTKペプチド(配列番号1)のN末端はコア1ではなくコア2と結合させた。質
量分析データ:オキシム2:Mr実測値4459.48、Mr理論値4460.19;オ
キシム3:Mr実測値6708.29、Mr理論値6708.00。
【0169】
オキシム4(図2)を、オキシム3と同様にして合成したが、FITC基をレポーター
とするのではなく、ビオチン基をトリリジン・コアのアミド結合を介して直接結合させた
。質量分析データ:Mr実測値6160.0、Mr理論値6160.3。オキシム5はオ
キシム4と同様であったが、RRTK(配列番号1)残基は全てD型であった。質量分析
データ:Mr実測値6158.6、Mr理論値6160.3。
【0170】
オキシム6を、オキシム4と同様に合成したが、RRTK(配列番号1)ペプチドのコ
ピーを4個ではなく、8個含む。質量分析データ:Mr実測値11646.1、Mr理論
値11643.6。
【0171】
(免疫細胞化学および蛍光による検討)
前述の濃縮スキームによって単離した単一ファージを増幅させた。このファージ・ペプ
チドおよびオキシム1〜6のうちの1種を培養培地中の細胞に加え、24時間インキュベ
ートした。次に、培地を洗い流し、細胞を冷メタノール−アセトン(1:1)で5分間固
定した。ファージ・キャプシドに対する抗体もしくはビオチン(ペプチド)に対する抗体
を、フルオレセイン結合(ファージ)もしくはテキサス・レッド標識(ペプチド)二次抗
体と共に用いた。古典的な蛍光顕微鏡による検討および共焦点顕微鏡によるアッセイを実
施した。
【0172】
まず、オキシム1〜3のβTC−3細胞内への侵入について調べた。蛍光による検討で
は、構築ブロックとして2ユニットのPEGスペースを有するRRTKペプチドの4つの
コピーを用いるテトラオキシム(樹状形)のオキシム3は、βTC−3細胞表面の受容体
に明確に結合し、細胞内へ侵入可能であることが明らかになった。オキシム3が優れてい
るのは、細胞表面の受容体への結合を増強するPEGスペーサーの可動性に起因し得る。
【0173】
次いで、オキシム3に類似のオキシム4〜6を合成し、結合親和力および侵入効率につ
いてアッセイした。RRTK(配列番号1)ペプチドのコピー数を、オキシム6では8個
まで増やし、D−ペプチドが高い安定性を有し、免疫原性がより低いことが実証されてい
るので(Ivanenkovら、Biochem.Biophys.Acta 1448
:450(1999)を参照)、オキシム5では上記ペプチドをD−エナンチオマー型に
変更した。顕微鏡による検査では、8個のペプチドコピーを有するオキシム6(オクタオ
キシム)は細胞内へ効率的に侵入することができることが明らかになった。ビオチンに対
するテキサス・レッド標識二次抗体によって、このオクタオキシムは、「空胞状」構造体
としての細胞の内側に、または細胞膜に局在することが明らかになった。従って、ペプチ
ドコピー数を最大8個まで増加させた多量体は、結合ペプチドの向上した結合活性および
移送効率を示す。
【技術分野】
【0001】
(発明の技術分野)
本発明は分子生物学の分野に関する。
【背景技術】
【0002】
(発明の背景)
対象物質を外部培地から細胞内、特に細胞核へ効率的に移入することを可能にする技術
は、バイオテクノロジーの分野において極めて重要である。このような技術は、タンパク
質もしくはペプチド産生、遺伝子発現の調節、細胞内シグナル伝達チャネルの解析、およ
び細胞(もしくは細胞核)内への種々多様な物質の移送効果の解析に有用であり得る。現
在利用できる技術は、移入用ベクターに対し、生物活性のある物質を宿主細胞の細胞質(
もしくは核)内に移入させて、宿主ゲノムに影響を与えずに、またはこの活性物質の生物
学的性質を変えずに処理することができないことにより制約を受ける場合が多い。
【0003】
以前は、高分子活性物質の細胞内への導入は、この移送すべき物質のサイズおよび生化
学的性質によって制約されていた。しかしながら、最近、トランスポーターペプチドもし
くはタンパク質を用いることにより、この分野に進歩がもたらされた。例えば、Schw
arzeとその共同研究者は、複合ペプチドもしくはタンパク質を組織の細胞内へ、およ
び血液−脳関門を通過させて移送させることができるHIV、TAT48−57由来10
マーのペプチドについて報告している(非特許文献1参照)。この複合ペプチドもしくは
タンパク質は、エンドサイトーシスを伴わないタンパク質導入プロセスにより細胞に吸収
されるものと考えられている。この発見により、生物医学研究および患者体内への薬物の
直接的な送達に新しい方法論の突破口が開かれた。しかしながら、この方法の1つの重要
な制限は、これらのタイプのトランスポーターに細胞特異性がないことである。従って、
標的としない正常な組織との相互作用により有害な副作用が生じるため、こうした非特異
的トランスポーターペプチドの多くは、有用性に制限がある。このような制限は、糖尿病
などの慢性疾患の処置において特に問題となる。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Schwarzeら、Science 285:1573(1999)
【発明の概要】
【発明が解決しようとする課題】
【0005】
薬物および治療剤をペプチド移送を介して細胞内に送達するための、種々の細胞型を特
異的に標的とするための効率的で安全な組成物および方法は、当該分野でなお求められて
いる。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、生体膜を通過して移行することができるトランスポーターペプチドを提供す
る。また、本発明は、エフェクターに対し生体膜を通過させて移行させるこのようなトラ
ンスポーターペプチドを使用する方法に関する。このトランスポーターペプチドは単体も
しくは多量体であり得る。
本発明は、例えば、以下を提供する:
(項目1)
式I、
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
による多量体トランスポーターペプチドであって:
ここで、Pは
a)(XmRXoRXn)、
b)(XmRRRXn)、
c)(XmRRXRXn)および
d)(XmRXRRXn)
からなる群から選ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチ
ドであり、
Xは非塩基性アミノ酸であり、
mは0〜14の整数であり、
nはmとは独立に0と14との間の整数であり、
oはmおよびnとは独立に0と5との間の整数であり、
vは2〜8の整数であり、
x、yおよびzは独立に0または1であり、
各Pは同じでも異なっていてもよく、該トランスポーターペプチドは生体膜を通過して移
行し得る、トランスポーターペプチド。
(項目2)
項目1に記載のトランスポーターペプチドであって、少なくとも1つのPがアミノ酸
配列R−X−X−Rを有する、トランスポーターペプチド。
(項目3)
項目1に記載のトランスポーターペプチドであって、各Pが配列番号1〜34からな
る群から選ばれる、トランスポーターペプチド。
(項目4)
項目1に記載のトランスポーターペプチドであって、vが2、4、6および8からな
る群から選ばれる、トランスポーターペプチド。
(項目5)
項目4に記載のトランスポーターペプチドであって、vが4である、トランスポータ
ーペプチド。
(項目6)
項目4に記載のトランスポーターペプチドであって、vが8である、トランスポータ
ーペプチド。
(項目7)
項目1に記載のトランスポーターペプチドであって、前記スペーサーがスクシンイミ
ジル−ポリエチレングリコールである、トランスポーターペプチド。
(項目8)
項目1に記載のトランスポーターペプチドであって、前記コアがC4−K2K−K(
succ−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg
−S)−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミド
および(NH2OCH2CO)8−K4K2K−K(GGG)−アミドからなる群から選
ばれる、トランスポーターペプチド。
(項目9)
エフェクターと結合体化されている項目1に記載のトランスポーターペプチドを含む
、トランスポーターユニット。
(項目10)
式II:
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
のトランスポーターユニットであって:
ここで、Eはエフェクターであり、
Pは
e)(XmRXoRXn)、
f)(XmRRRXn)、
g)(XmRRXRXn)および
h)(XmRXRRXn)
からなる群から選ばれる少なくとも1種のアミノ酸配列を含むトランスポーターペプチド
であり、
Xは非塩基性アミノ酸であり、
mは0〜14の整数であり、
nはmとは独立に0と14との間の整数であり、
oはmおよびnとは独立に0と5との間の整数であり、
vは2〜8の整数であり、
x、yおよびzは独立に0もしくは1であり、
各Pは同じでも異なっていてもよいものとし、ならびに該トランスポーターペプチドは生
体膜を通過して移行し得る、トランスポーターユニット。
(項目11)
項目10に記載のトランスポーターユニットであって、少なくとも1つのPがアミノ
酸配列R−X−X−Rを有する、トランスポーターユニット。
(項目12)
項目10に記載のトランスポーターユニットであって、各Pが配列番号1〜34から
なる群から選ばれる、トランスポーターユニット。
(項目13)
項目10に記載のトランスポーターユニットであって、vが2、4、6および8から
なる群から選ばれる、トランスポーターユニット。
(項目14)
項目13に記載のトランスポーターユニットであって、vが4である、トランスポー
ターユニット。
(項目15)
項目13に記載のトランスポーターユニットであって、vが8である、トランスポー
ターユニット。
(項目16)
項目10のトランスポーターユニットであって、該コアがC4−K2K−K(suc
c−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S)
−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよび
(NH2OCH2CO)8−K4K2K−K(GGG)−アミドからなる群から選ばれる
、トランスポーターユニット。
(項目17)
項目10に記載のトランスポーターユニットであって、前記エフェクターが前記リン
カーに共有結合により融合されている、トランスポーターユニット。
(項目18)
項目10に記載のトランスポーターユニットであって、前記エフェクターが核酸、ペ
プチドおよび薬学的に活性な薬剤からなる群から選ばれる、トランスポーターユニット。
(項目19)
項目18に記載のトランスポーターユニットであって、前記エフェクターが核酸であ
る、トランスポーターユニット。
(項目20)
項目19に記載のトランスポーターユニットであって、前記核酸がDNAである、ト
ランスポーターユニット。
(項目21)
項目19に記載のトランスポーターユニットであって、前記核酸がRNAである、ト
ランスポーターユニット。
(項目22)
項目18に記載のトランスポーターユニットであって、前記エフェクターがペプチド
である、トランスポーターユニット。
(項目23)
項目18に記載のトランスポーターユニットであって、前記エフェクターが薬学的に
活性な薬剤である、トランスポーターユニット。
(項目24)
項目23に記載のトランスポーターユニットであって、前記薬学的に活性な薬剤が、
毒素、抗生物質、抗病原体剤、抗原、抗体断片、免疫調節剤、酵素および治療剤からなる
群から選ばれる、トランスポーターユニット。
(項目25)
項目10に記載のトランスポーターユニットであって、各ペプチドが50アミノ酸長
未満である、トランスポーターユニット。
(項目26)
項目10に記載のトランスポーターユニットであって、各ペプチドが25アミノ酸長
未満である、トランスポーターユニット。
(項目27)
項目10に記載のトランスポーターユニットであって、各ペプチドが15アミノ酸長
未満である、トランスポーターユニット。
(項目28)
項目10に記載のトランスポーターユニットであって、移行が膵B細胞、肝細胞、結
腸細胞、筋肉細胞および肺細胞からなる群から選ばれる組織内へ行われる、トランスポー
ターユニット。
(項目29)
項目12に記載のトランスポーターユニットを膵β細胞の膜を通過させて移行させる
方法であって、少なくとも1種のトランスポーターペプチドが配列番号1〜6からなる群
から選ばれる、方法。
(項目30)
項目29に記載の方法であって、少なくとも1種のトランスポーターペプチドが配列
番号1である、方法。
(項目31)
項目12に記載のトランスポーターユニットを肝細胞の膜を通過させて移行させる方
法であって、少なくとも1種のペプチドが配列番号7〜10からなる群から選ばれる、方
法。
(項目32)
項目12に記載のトランスポーターユニットを結腸細胞の膜を通過させて移行させる
方法であって、少なくとも1種のペプチドが配列番号11である、方法。
(項目33)
項目12に記載のトランスポーターユニットを筋肉細胞の膜を通過させて移行させる
方法であって、少なくとも1種のペプチドが配列番号12〜20からなる群から選ばれる
方法。
(項目34)
項目12に記載のトランスポーターユニットを肺細胞の膜を通過させて移行させる方
法であって、少なくとも1種のペプチドが配列番号21〜34からなる群から選ばれる方
法。
(項目35)
治療的もしくは予防的に有効な量の項目10に記載のトランスポーターユニット、お
よび薬学的受容可能なキャリアを含む、薬学的組成物。
(項目36)
項目1に記載の該トランスポーターペプチドとエフェクターとの間で移行可能な結合
体を生成する方法であって、該エフェクターを該トランスポーターペプチドに結合させて
トランスポーターペプチド−エフェクター結合体を形成することを含む方法。
(項目37)
エフェクターを真核細胞の細胞質および核の中へ移行させる方法であって、
該エフェクターを項目1に記載のトランスポーターペプチドに結合させてトランスポ
ーターペプチド−エフェクター結合体を形成する工程、および
該トランスポーターペプチド−エフェクター結合体を該細胞に導入する工程
を包含する、方法。
(項目38)
項目37に記載の方法であって、該導入工程が、前記トランスポーターペプチド−エ
フェクター結合体の存在下で細胞培養物をインキュベートすることにより、または前記ト
ランスポーターペプチド−エフェクター結合体を前記細胞内に注入することにより達成さ
れる方法。
(項目39)
項目37に記載の方法であって、該真核細胞がヒト細胞である、方法。
(項目40)
項目37に記載の方法であって、該真核細胞がβ細胞である、方法。
(項目41)
真核細胞内のエフェクターの細胞内濃度を増大させる方法であって、
該エフェクターを項目1に記載のトランスポーターペプチドに結合させてトランスポ
ーターペプチド−エフェクター結合体を形成する工程、および
該真核細胞の活性な代謝を促進する条件の下、該トランスポーターペプチド−エフェク
ター結合体の存在下で該細胞をインキュベートする工程
を包含する、方法。
(項目42)
項目41に記載の方法であって、前記真核細胞がヒト細胞である、方法。
(項目43)
1つ以上の容器に項目35に記載の治療的もしくは予防的に有効な量の該薬学的組成
物を含む、キット。
(項目44)
疾患を処置または予防する方法であって、そのような処置または予防が所望されている
被験体に対し、項目35に記載の薬学的組成物を該被験体の該疾患を処置または予防す
るのに十分な量で投与する工程を包含する、方法。
(項目45)
項目44に記載の方法であって、該疾患が、糖尿病、結腸癌、呼吸器疾患、神経変性
障害、心臓麻痺、およびウイルス感染症からなる群から選ばれる、方法。
(項目46)
多量体トランスポーターペプチドであって、各トランスポーターペプチドのアミノ酸配
列が配列番号1〜34からなる群から選ばれる、トランスポーターペプチド。
【0007】
一局面として、本発明は、(XmRXoRXn)、(XmRRRXn)、(XmRRX
RXn)および(XmRXRRXn)から選ばれ、Xが非塩基性アミノ酸であり、mが0
〜14の整数であり、nがmとは独立に0と14との間の整数であり、oがmおよびnと
は独立に0と5との間の整数である少なくとも1種のアミノ酸を有するトランスポーター
ペプチドであって、生体膜を通過して移行することができるトランスポーターペプチドを
含む。
【0008】
一実施形態として、本発明は、アミノ酸配列R−X−X−Rを有するトランスポーター
ペプチドを提供する。他の実施形態として、本発明は、配列番号1−34のうちの任意の
1つのアミノ酸配列を有するトランスポーターペプチドを提供する。種々の別の実施形態
として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来のもので
ある。さらに他の実施形態として、このトランスポーターペプチドは、タンパク質転換酵
素の切断部位から得られるものである。
【0009】
別の局面として、本発明は、各単量体トランスポーターペプチドが(XmRXoRXn
)、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)から選ばれ
、Xが非塩基性アミノ酸であり、mが0〜14の整数であり、nがmとは独立に0と14
との間の整数であり、oがmおよびnとは独立に0と5との間の整数であり、生体膜を通
過して移行することができる2個、3個、4個、5個、6個、7個、8個もしくはそれ以
上の単量体トランスポーターペプチドを含む多量体トランスポーターペプチドを含む。多
量体トランスポーターペプチド内の個々の単量体トランスポーターペプチドは同一もしく
は異なるものであり得る。
【0010】
別の実施形態として、本発明は、アミノ酸配列R−X−X−Rを有する多量体トランス
ポーターペプチドを提供する。他の実施形態として、本発明は、配列番号1−34のうち
の任意の1つのアミノ酸配列を有する多量体トランスポーターペプチドを提供する。種々
の他の実施形態として、この多量体トランスポーターペプチドは、タンパク質転換酵素の
リガンド由来のものである。さらに別の実施形態として、この多量体トランスポーターペ
プチドは、タンパク質転換酵素の切断部位から得られるものである。
【0011】
一部の実施形態として、この多量体トランスポーターペプチドは、スペーサー部分およ
びリンカー部分を含む。好ましくは、このスペーサー部分およびリンカー部分は、可動性
、両親媒性、非免疫原性、およびタンパク質分解酵素に対し非感受性である。このリンカ
ーは、例えば、ポリエチレングリコールであり得る。
【0012】
他の実施形態として、この単量体もしくは重合体トランスポーターペプチドは、レポー
ター基を含むことができる。本明細書に用いている「レポーター基」とは、検出可能な任
意の基である。非限定的な例としては、放射性同位体、蛍光性部分、リン光性部分、化学
発光性部分、および量子ドットが挙げられる。その他のレポーター基としては、ビオチン
、シンテイン、ヒスチジン、赤血球凝集素、mycもしくはフラッグ・タグが挙げられる
。
【0013】
一部の実施形態として、本発明のトランスポーターペプチドは、式I:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
によって表され、式中、Pは前述のコンセンサスモチーフ(consensus mot
if)(XmRXoRXn)、(XmRRRXn)、(XmRRXRXn)および(Xm
RXRRXn)に包含されるペプチドである。例えば、このペプチドは、配列X1−X2
−X3−X4を含むことができ、式中、X1およびX4はRもしくはKであり得、X1お
よびX4は任意のアミノ酸であり得る。一実施形態として、本発明は、アミノ酸配列R−
X−X−Rを有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する
。他の実施形態として、本発明は、配列番号1−34のうちの任意の1つのアミノ酸配列
を有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。種々の実
施形態として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来の
ものである。さらに他の実施形態として、このトランスポーターペプチドは、タンパク質
転換酵素の切断部位から得られるものである。vによって表される所与のトランスポータ
ー内のペプチド数は、整数とする。単量体トランスポーターでは、vは1である。多量体
トランスポーターでは、vは2〜8以上の整数とする。任意の所与の多量体結合体におい
て、それぞれのペプチドは互いに同じであっても異なっていてもよい。
【0014】
単量体のトランスポーター構築物では、コアは存在していてもしていなくてもよいもの
とする。
【0015】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は0または1である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−PEG(succ−
peg)などのポリエチレングリコール(PEG)が挙げられる。
【0016】
本明細書に用いている「コア」とは、リンカー、スペーサーおよびレポーター基の接続
ポイントとなる構造体である。また、コアは多量体トランスポーター内のペプチド間を結
合する役割を果たす。一部の実施形態として、このコアを分枝型とすることによって多く
のペプチドを同一トランスポーターユニット内で結合させることができる。通常、コアに
対し結合に利用可能な多くの官能性を持たせることによって、この多量体構築物の種々の
異なる部分(ペプチド、ならびに存在する場合にはスペーサー、リンカーおよびレポータ
ー)をこのコアと融合させて1つの分子を形成させることができる。企図しているコア部
分としては、以下に詳細を説明するポリリジン(K)が挙げられ、また、リンカーの組み
込まれた構造のものも挙げられる。コアを選択することにより、所与の結合体内に存在す
るペプチドの数が決まる。代表的なコアとしては、C4−K2K−K(succ−peg
−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S)−アミド、
(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよび(NH2O
CH2CO)8−K4K2K−K(GGG)−アミドが挙げられる。
【0017】
多量体のトランスポーター構築物では、リンカーおよびレポーター基は存在していても
していなくてもよく、従って、yおよびzは、式Iにおいて独立に1もしくは0である。
【0018】
本明細書に用いている「トランスポーターペプチド」とは、物質が生体膜を通過して移
行するのを容易にするペプチドである。特に明記しない限り、または用いる文脈において
不正確でない限り、トランスポーターペプチドを意味するものは単量体もしくは多量体の
トランスポーターペプチドを包含する。
【0019】
一部の実施形態として、このトランスポーターペプチドは、1つ以上のエフェクターと
融合させる。「エフェクター」は、DNA、RNA、タンパク質、ペプチド、または薬学
的に活性な薬剤、例えば、毒素、抗生物質、抗病原体剤(antipathogenic
agent)、抗原、抗体、抗体断片、免疫調節剤、酵素もしくは治療剤などを含む任
意の適切な分子であり得る。2つ以上のエフェクターが存在する場合、これらのエフェク
ターは同一もしくは異なるものであり得る。
【0020】
「融合」もしくは「融合した」という用語は、何らかの第3の分子よりも互いを好む2
種以上の分子をもたらすような特定の相互作用全てを含むものである。これには、共有結
合、イオン結合、疎水性結合、水素結合などの作用が含まれるが、溶媒選択性などの非特
異的結合は含まれない。
【0021】
本発明の多量体結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学の法則通りに、エフェクターはコア、もしくはリンカー、またはこれら
の両方に結合させることができる。
【0022】
本明細書に用いている「結合体(conjugate)」もしくは「結合体化(con
jugation)」とは、エフェクターと単量体もしくは多量体トランスポーターペプ
チドとの間の物理的な結合を可能にするあらゆるタイプの相互作用のことを意味する。こ
の結合の性質は、共有もしくは非共有結合であり得るが、その結合は、細胞移行の前もし
くは移行中に結合体が解離しないように十分強力なものである必要がある。結合体化は、
当業者に公知の任意の化学的、生化学的、酵素的もしくは遺伝子的カップリング法を用い
て達成することができる。目的のエフェクターは、トランスポーターペプチドのN末端も
しくはC末端にカップリングまたは融合させることができる。一部の実施形態として、こ
のエフェクターは、リンカー基に、もしくは直接コアに、またはレポーター基にカップリ
ングあるいは融合させる。
【0023】
種々の実施形態として、単量体および/または多量体トランスポーターペプチドは、長
さをアミノ酸長として50個未満、25個未満もしくは15個未満であり得る。
【0024】
さらなる実施形態として、移行は膵B細胞、肝細胞、大腸細胞、筋肉細胞および/また
は肺細胞内で行われる。
【0025】
別の実施形態として、本発明は、トランスポーターペプチドを生体膜を通過させて移行
させる方法を含む。例えば、配列番号1−6の配列のうちの1種以上を含む単量体もしく
は多量体ペプチドは膵B細胞の膜を通過させて移行させることができ、配列番号7−10
の配列のうちの1種以上を含む単量体もしくは多量体ペプチドは肝細胞の膜を通過させて
移行させることができ、配列番号11のペプチドうちの1種以上を含む単量体もしくは多
量体ペプチドは大腸細胞の膜を通過させて移行させることができ、配列番号12−20の
配列のうちの1種以上を含む単量体もしくは多量体ペプチドは筋肉細胞の膜を通過させて
移行させることができ、および配列番号21−34の配列のうちの1種以上を含む単量体
もしくは多量体ペプチドは肺細胞の膜を通過させて移行させることができる。
【0026】
なおさらなる実施形態として、本発明は、1種以上のエフェクターに結合させた単量体
もしくは多量体トランスポーターペプチドであるトランスポーターユニットを含む。種々
の他の実施形態として、このエフェクターは核酸、ペプチドもしくは薬学的に活性な薬剤
であり得る。
【0027】
さらに別の実施形態として、本発明は、トランスポーターペプチドと1種以上のエフェ
クターとの間で移行可能な単量体もしくは多量体結合体を作製し、従ってトランスポータ
ーペプチド−エフェクター結合体を形成する方法を含む。
【0028】
別の実施形態として、本発明は、エフェクターを単量体もしくは多量体トランスポータ
ーペプチドに結合させて真核細胞内に導入することによる、真核細胞の細胞質および/ま
たは核内に1種以上のエフェクターを移行させる方法を含む。例えば、このトランスポー
ターペプチド−エフェクター結合体は、この結合体の存在下で培養細胞をインキュベート
することにより、またはこの結合体を細胞内に注入することにより細胞内に導入すること
ができる。
【0029】
種々の他の実施形態として、本発明は、エフェクターを単量体もしくは多量体のトラン
スポーターペプチドに結合させ、細胞の活性状態の代謝を亢進させる条件の下、この細胞
の存在下にインキュベートすることによる、真核細胞におけるエフェクターの細胞内濃度
を上昇させる方法を含む。本発明の好ましい実施形態は、この真核細胞としてヒト細胞を
使用することを含む。
【0030】
さらなる実施形態として、本発明は、治療的もしくは予防的に有効な量の単量体もしく
は多量体トランスポーターユニット、および薬学的に受容可能な担体を含む薬学的組成物
を含む。
【0031】
好ましい「薬学的組成物」としては、a)希釈剤、例えば、乳糖、ブドウ糖、蔗糖、マ
ンニトール、ソルビトール、セルロースおよび/またはグリシン;b)滑沢剤、例えば、
シリカ、滑石、ステアリン酸、そのマグネシウム塩もしくはカルシウム塩および/または
ポリエチレングリコール;また、錠剤用としてc)結合剤、例えば、ケイ酸マグネシウム
・アルミニウム、でんぷん糊、ゼラチン、トラガント、メチルセルロース、カルボキシメ
チルセルロースナトリウムおよび/またはポリビニルピロリドン;必要に応じてd)崩壊
剤、例えば、でんぷん、寒天、アルギン酸またはそのナトリウム塩もしくは発泡性混合物
;ならびに/あるいはe)吸収剤、着色剤、矯味矯臭剤および甘味剤とともに有効成分を
含む錠剤およびゼラチンカプセル剤が挙げられる。注射用組成物は、好ましくは等張水溶
液もしくは懸濁液であり、坐剤は脂肪性乳濁液もしくは懸濁液から調製するのが有利であ
る。これらの組成物は、滅菌することができ、および/または保存剤、安定化剤、湿潤剤
または乳化剤などのアジュバント、溶解促進剤、浸透圧調節塩および/または緩衝液を含
むことができる。また、さらに、これらは他の治療上有用な物質を含むこともできる。こ
れらの組成物は、それぞれ、従来の混和、顆粒化またはコーティング法によって調製され
、有効成分を約0.1%〜75%、好ましくは約1%〜50%含有する。
【0032】
なおさらなる実施形態として、本発明は、1個以上の容器が本発明の薬学的組成物を治
療的もしくは予防的有効量で含有するキットを含む。
【0033】
本発明の別の実施形態は、疾患の処置もしくは予防を必要とする被験体に対し、疾患を
処置もしくは予防するのに十分な量の薬学的組成物を投与することにより、その疾患を処
置もしくは予防する方法を含む。処置対象の疾患としては、例えば、糖尿病、大腸癌、呼
吸器疾患、神経変性障害、心臓麻痺、および/またはウイルス感染症が挙げられ得る。
【0034】
別の局面として、本発明は、特定の細胞型に対してファージ・ライブラリをスクリーニ
ングした後、どの細胞がファージを取り込んだかを評価する、トランスポーターペプチド
用ファージ・ライブラリのスクリーニング方法を含む。
【0035】
別の実施形態として、この方法は、取り込まれたファージのDNAを同定し、これから
発現されるペプチドを推定することを含む。
【0036】
なおさらなる実施形態として、この方法は、ファージ・ライブラリを少なくとも3サイ
クルにわたって選択するスクリーニング工程を含む。
【0037】
さらに別の実施形態として、本発明は、ペプチドの多価(multivalent)デ
ィスプレイを有するファージを含む。
【図面の簡単な説明】
【0038】
【図1】図1はオキシム1〜3の構造を示す。コアの分枝Lys残基のαおよびεアミノ基は、RRTKペプチドのN末端でアシル化(スペーサがあってもなくてもよい)されている。スペーサーと共に、レポータ・タグFITCが各構築体のコアに導入されている。
【図2】図2はオキシム4〜6の構造を示す。コアの分枝Lys残基のαおよびεアミノ基は、RRTKペプチドのN末端でスペーサーを有してアシル化されている。スペーサーと共に、レポータ・タグのビオチンが各構築体のコアに導入されている。
【発明を実施するための形態】
【0039】
(発明の詳細な説明)
本発明は、薬物および治療剤を細胞内へ送達するための、種々の細胞型を特異的に標的
とするペプチドトランスポーターならびにペプチド移送システムを提供する。当該分野で
公知の既存の移送システムには、これらが非効率的で、宿主ゲノムに影響を与え、活性物
質(例えば、エフェクタ)の生物学的性質を変え、標的細胞を殺傷し、あるいは(例えば
、ウイルス性結合体を使用することにより)極めて高いリスクを生じるためヒト対象には
使用できないので、制約がある場合が多い。本発明のペプチド移送システムでは、治療剤
となり得るものを細胞内へ送達するためにプロタンパク質転換酵素およびその特異的なリ
ガンドを用いることによって当該分野で公知のトランスポーターシステムの限界を克服し
ている。本発明のシステムでは、宿主のゲノムに影響を与えず、その他の点でも、非侵襲
性である、変化を受けていない生物活性物質が効率的に送達される。
【0040】
受容体媒介性のエンドサイトーシスは、細胞内への治療剤の送達を狙った実験系におい
て広く利用されてきた。KatoおよびSugiyama、Crit.Rev.Ther
.Drug Carrier.Syst.14:287(1997)。従って、細胞特異
的なエンドサイトーシス受容体を首尾良く標的にすることによって、ペプチドを細胞型特
異的に送達することができる。プロタンパク質転換酵素は、受容体媒介性エンドサイトー
シスにより取り込まれる細胞表面受容体の一例である。こうしたタンパク質は、ペプチド
ホルモン、神経ペプチドその他の多くのタンパク質の前駆体をその生物活性のある形に転
換する役割を果たしていることが明らかにされている。プロタンパク質転換酵素ファミリ
ーの切断部位にはコンセンサス配列R−X−X−Rが含まれている。発現および局在性に
関する情報は、プロタンパク質転換酵素は細胞外のリガンドを細胞内空間に移送すること
を示す。
【0041】
例えば、哺乳類のプロタンパク質転換酵素は、その組織分布に基づいて3グループに分
類することができる。1つのクラスのフューリン(Furin)、PACE4、PC5/
PC6およびLPCIPC7/PC8/SPC7は広範囲の組織および細胞株において発
現される。第2のクラスの神経内分泌特異的転換酵素PC2およびPC1/PC3の発現
は、膵島、下垂体、副腎髄質および多くの脳領域などの内分泌組織に限定されている。さ
らに、第3のクラスのPC4の発現は、精巣の精子形成細胞に高度に制限されている。神
経内分泌特異的転換酵素は、主として分泌顆粒内に局在している。また、PC5/PC6
Aも、分泌顆粒に局在していると報告されている。さらに、間接的な証拠ではあるが、一
部のプロタンパク質転換酵素分子は細胞表面に存在することが示唆されており、フューリ
ンはTGNと細胞表面との間を循環することが明らかにされている。Mandrup−P
oulsen、BMJ.316:1221(1998)を参照されたい。
【0042】
天然リガンドの働きをするように見えるペプチドリガンドは、ファージ・ディスプレイ
法によって同定される場合が多い。効率的な受容体媒介性エンドサイトーシスを誘導する
ペプチド配列の単離は、ファージ・ディスプレイ法を用いることによって向上する。Iv
anenkovら、Biochem.Biophys.Acta1448:450、p4
63(1999)を参照されたい。ファージ・ディスプレイ・ライブラリは、細胞受容体
に対する天然リガンドの修飾体を含む変異体分子(Cabibboら、Gene 167
:41(1995))および短鎖ペプチド(Zwickら、Curr.Opin.Bio
technol.9:427(1998))の膨大な供給源となる極めて強力なツールで
ある。また、ライブラリを直接注射したマウスにおいて、脳および腎臓に13倍の選択性
を示すペプチド配列が首尾良く単離されている。PasqualiniおよびRuosl
ahti,Nature 380:364(1996)ならびにPasqualini、
Ruoslahti,Mol.Psychiatry 1:423(1996)を参照さ
れたい。
【0043】
時として、この方法により得られたリガンドは、短鎖ペプチド分子内の立体構造の自由
度が高く接触残基数が少ないため、低親和性(マイクロモル程度)となることがある。ペ
プチドリガンドの結合親和性を改善する1つの方法は、低親和性ペプチドのいくつかのコ
ピーを単一の多量体分子の形に結合させることである。この多量体リガンドの親和力(お
よび生物活性)は、単量体リガンドに対して大きく向上させることができる。一部の実施
形態として、多量体構築物は、オキシム化学を用いて、構築ブロックとしての細胞を標的
とするペプチドから作製する。例えば、このペプチドは、特定の細胞(例えば、膵β細胞
)に特異的に結合し、受容体媒介性エンドサイトーシスを介してこの細胞による取り込み
を誘発させることができるペプチドリガンドを選択するファージ・ディスプレイ実験から
得ることができる。
【0044】
例えば、単量体としてのIgMは低親和性タンパク質であるが、5量体の形をとると、
細菌もしくはウイルスの表面に存在する反復性抗原決定基に対する親和力が大きく向上す
る。Roitt、Essential Immunology(Oxford/Blac
kwell,London),p65−84(1991)を参照されたい。
【0045】
同様に、組換えDNA技術により作製されるタンパク質であるこのペンタボディでは、
105倍の向上がみられる。Alexeyら、Proc.Natl.Acad.Sci.
USA 94:1663−1668(1997)。ファージ・ライブラリから得られるポ
リペプチドの低親和性はペンタボディの5量体構造によって補われ、その標的に対する高
い親和力が得られる。
【0046】
また、最近、Roseは同様な分子を「ケモボディ(chemobody)」と名付け
た。RoseおよびVizzavona,J.Am.Chem.Soc.121:703
4−7038(1999)を参照されたい。このケモボディは、相補性構造に非共有結合
することにより抗体分子の働きを模倣することができるペプチドサブユニットの複数のコ
ピーを提示する合成分子である。その結合ペプチドのアミノ酸配列は、ファージ・ライブ
ラリ法により同定され、サブユニット自体そのものは、オキシム化学によって構築された
。また、同時に2つ以上のペプチドサブユニットによって標的に結合することができるよ
うに、適切なスペーサーおよびリンカーもこのケモボディに導入された。Roseは、可
動性リンカーを有するインフルエンザ・ウイルスのファージ由来ペプチドのコピーを4個
有するモデルケモボディを作製した。
【0047】
特定の細胞型を選択的に標的とする低分子トランスポーターペプチドは、大きなファー
ジ・ディスプレイ・ライブラリから得て、単量体もしくは多量体トランスポーター分子と
して用いることができる。ファージ・ディスプレイ・ライブラリを用いて得られるような
低分子ペプチド担体の有利な点としては、高品質、高純度、低免疫原性であり、生体の全
ての細胞に極めて効率的に送達することを可能にすることが挙げられる。Schwarz
eら、Science 285:1573(1999)。従って、ペプチド担体では、種
々の高分子を効率的に送達するためのリポソーム、ウイルスなどの従来のトランスポータ
ーを改良することができる可能性がある。例えば、Mahatoら、J.Drug Ta
rget.4:337(1997)、およびMahatoら、Crit.Rev.The
r.Drug Carrier.Syst.14:133 4:337(1997)を参
照されたい。
【0048】
本明細書に記載したトランスポーターペプチドは、種々の疾患の治療に用いられ、この
ような疾患としては、糖尿病が挙げられるが、これに限定されるものではない。正常な生
物体(では、インスリン分泌により正常血糖が維持されるようにβ細胞集団は厳格に制御
されている。β細胞集団は、乳児もしくは成熟生物体の要求に反応して、特に、特定の生
理的および生理病理学的状態への反応として調節されている。β細胞集団の適切性は、基
本的にβ細胞の死と再生との動的バランスによって達成されている。このバランスは、未
熟β細胞の分化および既存のインスリン分泌細胞の増殖によって達成されている。Sch
arfmannおよびCzernichow、Diabetes Metab.22:2
23(1996)ならびにBonner−Weir,Recent Prog.Horm
.Res.49:91(1994)を参照されたい。I型糖尿病におけるβ細胞集団の障
害されたバランスは、膵β細胞を標的にする免疫系の特異的攻撃により惹起される過程で
あるβ細胞破壊の促進によって生じる。従って、β細胞破壊の速度を抑制もしくは低下さ
せると、糖尿病が安定化されるばかりではなく、島が再生されてβ細胞集団の機能不全を
是正することも可能となる。
【0049】
I型糖尿病の実験モデルにおいてβ細胞の喪失速度を低下させるのに用いられる効力の
あるツールとして数種の化合物を確立した。これらのペプチドの多くは本質的にペプチジ
ルであり、従ってペプチド担体に容易に結合する。本明細書に記載したこれらのペプチド
は、治療用「積み荷(cargo)」のデザイン、即ち、治療剤(「エフェクタ」)を担
体(単量体もしくは多量体「トランスポーターペプチド」)とカップリングさせるための
基剤となる。
【0050】
本発明の移送システムの1つの実施形態は、I型糖尿病の治療のためにβ細胞の細胞内
メカニズムを標的にしたものである。I型糖尿病の症状は、免疫系の分泌による膵β細胞
の破壊に続発するものである。Mandrup−Poulsen,BMJ 316:12
21(1998)を参照されたい。ヒトおよび齧歯類における結論的なデータは、サイト
カインのインターロイキン−1β(IL−1β)は、マクロファージおよびT細胞により
分泌されるTNFαおよびINFγと共に、β細胞の機能不全および破壊ならびにI型糖
尿病をもたらす最終的な転帰の原因となる主要な成分であることを示す。Mandrup
−Poulsen,Diabetologia 39:1005(1996)、Neru
pら、Diabetes Care 11 Suppl 1:16(1988)、ならび
にMauricioおよびMandrup−Poulsen,Diabetes 47:
1537(1998)を参照されたい。これらの分泌サイトカインは、膵β細胞において
シグナル伝達およびエフェクター分子の高度に複雑なネットワークに関与している。この
シグナル伝達は、細胞の挙動を改変し、細胞の運命に決定的な影響力を与える。これまで
に蓄積された証拠から、この調節性細胞内ネットワークは新規な治療方法の開発のための
有望な標的となることが分かる。Iwahashiら、Cytokines.Cell
Mol.Ther.4:45(1998)、Sjoholm,Cell Death.D
iffer.5:461(1998)、Stephensら、J.Autoimmun.
10:293(1997)、Rabinovitchら、Diabetes 48:12
23(1999)、Bleichら、J.Clin.Invest.103:1431(
1999)、Welshら、Mol.Med.5:169(1999)、およびChen
ら、Diabetes 49:562(2000)を参照されたい。細胞内サイトカイン
・シグナル伝達の処理および形成に関与するこれらの分子はそれぞれ、トランスポーター
−薬物設計の標的とすることができる。
【0051】
β細胞内でIL−1βによって動員される最も重要なシグナル伝達分子としては、セラ
ミド、プロスタグランジン、熱ショックタンパク質、誘導性NOシンターゼ酵素(iNO
S)、転写因子NF−κB(Torgersonら、J.Immunol.161:60
84(1998)参照)、ならびに3種のMAPキナーゼERK1/2、p38およびJ
NKがある。これらの分子の多くは、β細胞の生残および機能の改善をもたらした既存の
阻害剤によるブロックの標的である。iNOSノックアウト(KO)マウスはIL−1β
の細胞毒性に対して抵抗性であり(Flodstromら、Diabetes 48:7
06(1999)参照)、iNOS作用のブロッカーはNOの細胞毒性の種々の側面を抑
制する(Sjoholm,Cell Death.Differ.5:461(1998
)に概説されている)。島および細胞株による研究は、Ca2+チャネルのブロッカーお
よびカスパーゼ阻害剤は齧歯類のβ細胞死を防止することを示す。Wangら、Endo
crinology 140:1200(1999)、およびYamadaら、Diab
etes 48:478(1999)を参照されたい。p38阻害剤は、グルコース賦活
性インスリン放出のIL−1β媒介性阻害を減弱させる。Larsenら、J.Biol
.Chem.273:15294(1998)を参照されたい。アンチセンスGADトラ
ンスジェニックNODマウスにおいてGADの発現をβ細胞特異的に抑制すると、自己免
疫性糖尿病が阻止された。Yoonら、Science 284:1183(1999)
を参照されたい。膵β細胞株においてbcl−2、IL−1RaおよびJBD(c−Ju
n N末端キナーゼJNKの優性阻害因子)を発現させると、細胞はアポトーシスに抵抗
性となった。Bonnyら、Diabetes 50:77−82(2000)、Iwa
hashiら、Diabetologia 39:530(1996)、Duprazら
、Gene Ther.6:1160(1999)、およびGiannoukakisら
、Diabetes 48:1730(1999)を参照されたい。以上のデータを総合
すると、特定の手段により細胞内事象を操作することはI型糖尿病の治療にかなり有望で
あることを示す。
【0052】
疾病治療の1つの主要な課題は、生物学的に重要な分子をインビボで有効な細胞透過性
の生物活性化合物に変換することである。Gibbs,Science 287:196
9(2000)を参照されたい。例えば、β細胞の喪失を防止するための最も有望なツー
ルは、多くの高分子タンパク質(例えば、Bcl−2(Rabinovitchら、Di
abetes 48:1223(1999)参照)、ドミナントネガティブ型のMyD8
8、TRAF、FADDもしくはIRAK(Burnsら、J.Biol.Chem.2
73:12203(1998)、およびStephensら、Endocrinolog
y 140:3219(1999)参照)などのサイトカイン・シグナル伝達の阻害因子
、または現在インビボで組織および膵β細胞を含む細胞型に送達させることができないJ
NK阻害剤JBD280(Ammendrupら、Diabetes 49:1468−
1476(2000)参照)である。
【0053】
最近の研究は、高分子タンパク質を、細胞および器官に容易に送達させることができる
低分子生物活性化合物に変換する試みの前進を示す。Hawiger,Curr.Opi
n.Chem.Biol.3:89(1999)を参照されたい。これらの方法には基本
的に2つの条件が必要とされる:即ち、1)細胞内に効率的に送達させたい分子に特定の
トランスポーターもしくはその化学的修飾体を結合させること(例えば、Schwarz
eら、Science 285:1573(1999)、Brugidouら、Bioc
hem.Biophys.Res.Commun.214:685(1995)、Oeh
lkeら、Biochim.Biophys.Acta 1414:127(1998)
およびTerskikhら、Proc.Natl.Acad.Sci.U.S.A.94
:1663(1997)に報告されている効率的な短鎖ペプチドトランスポーターを参照
されたい);2)トランスポーターに低分子ペプチド配列を結合させることができるよう
に、タンパク質の活性部分を狭小化する必要があること。手短に言えば、概してこれらの
条件では、元のタンパク質の不可欠な生物学的性質を保ちながら細胞内に入ることができ
る3〜30アミノ酸長の二連(bi−partite)ペプチドが設定される。癌研究の
場合のように(Gibbs、Oliff、Cell 79:193(1994)参照)、
β細胞には操作することによりサイトカイン誘発性アポトーシスからβ細胞を保護するこ
とができる多数の細胞内事象があり、こうした操作は薬剤設計の有望な標的となると考え
られる。
【0054】
受容体媒介性エンドサイトーシスは、細胞内への治療剤の送達を狙った実験系において
広く利用されている。Kato、Sugiyama、Crit.Rev.Ther.Dr
ug Carrier.Syst.14:287(1997)を参照されたい。エンドサ
イトーシス作用は、IgG Fc、ソマトスタチン、インスリン、IGF−IおよびII
、トランスフェリン、EGF、GLP−1、VLDLもしくはインテグリン受容体を含む
多くの受容体について報告されている共通的性質である。Hoflandら、Proc.
Assoc.Am.Physicians.111:63(1999)、Anderso
n、Clin.Immunol.Immunopathol.53:S63(1989)
、Lundら、J.Biol.Chem.265:15713(1990)、Smith
およびJarett、Lab.Invest.58:613(1988)、Solerら
、Endocrinology 127:595(1990)、Widmannら、Bi
ochem.J.310:203(1995)、Yorkら、J.Biol.Chem.
274:1164(1999)、およびMukherjeeら、Physiol.Rev
.77:759(1997)を参照されたい。エンドサイトーシスを媒介する細胞型特異
的受容体についても報告されている。Ivanenkovら、Biochem.Biop
hys.Acta 1448:463(1999)を参照されたい。
【0055】
強い実験的バックグラウンドは、膵β細胞を選択的に標的にするトランスポーターペプ
チドが大きなファージ・ディスプレイ・ライブラリから得られることも考えられることを
示しているが、そのような試みについては報告されていない。ファージ・ディスプレイ・
ライブラリを用いて得られるような低分子ペプチド担体の利点は数多くあり、例えば、化
学合成による作製が容易であること、高品質、高純度、低免疫原性であること、および生
物体の全ての細胞に極めて効率的に送達することが可能であることが挙げられる。Sch
warzeら、Science 285:1573(1999)を参照されたい。従って
、本発明のペプチド担体には、多くの高分子の効率的な送達において、リポソームもしく
はウイルスなどの従来のトランスポーターよりもうまく機能する可能性がある。例えば、
Mahatoら、J.Drug Target 4:337(1997)およびMaha
toら、Crit.Rev.Ther.Drug Carrier.Syst.14:1
33(1997)を参照されたい。
【0056】
ファージ・ペプチド・ライブラリは、従来より、線維状ファージM13の誘導体として
構築されている。ペプチド・ライブラリは、このペプチドモチーフの1〜5個のコピーを
提示するキャプシドのマイナーコートタンパク質pIIIに融合される。Zwickら、
Curr.Opin.Biotechnol.9:427(1998)を参照されたい。
あるいは、メジャーコートタンパク質pVIIIを用いることによって高価(high−
valent)提示(display)が達成される。しかしながら、これらのタイプの
ライブラリは、受容体媒介性エンドサイトーシスペプチド配列の単離のために最適化され
ていない。高い内部移行効率を有する担体を回収することに関連して考慮すべき事柄は以
下の通りである:1)ペプチドの1価もしくは低価(low−valent)提示は、線
維状ファージのような大きな構造体を効率的に取り込ませるには基本的に不十分であるが
、多価提示では効率的取り込みが可能となること(Ivanenkovら、Bioche
m.Biophys.Acta 1448:450(1999)参照);および2)受容
体結合リガンドが内部移行が、原形質膜の特定領域における細胞表面受容体の濃縮、続く
クラスリン被覆小胞の形成に関連すること(Damke、FEBS Lett.389:
48(1996))。
【0057】
M13誘導体のサイズは大きく(1〜1.5μm)(Zacherら、Gene 9:
127(1980)参照)、古典的なクラスリン被覆ピットの通常サイズ(150nM)
を上回る。クラスリン被覆ピットは、膜表面の約2%を占める原形質膜上の陥入構造体で
ある。この特殊な構造は、インスリン、EGFなどの細胞外タンパク質もしくはペプチド
を10〜50%/分という極めて速い速度で除去する高度に効率的な受容体媒介性内部移
行プロセスを指向する。Mukherjeeら、Physiol.Rev.77:759
(1997)を参照されたい。従って、従来のM13ファージでは、こうした特殊で高度
に効率的な構造による受容体媒介性内部移行が行われるとは考えられない。
【0058】
従って、報告されている試みでは、ペプチド担持ファージが高い内部移行率を示すペプ
チドは得られていない。これまで、こうした研究から、特定の細胞型に特異的なコンセン
サス取り込みモチーフは出現していない。
【0059】
一部の局面として、本明細書に開示した本発明は、ペプチド担持エフェクターの取り込
みを促進するトランスポーターペプチドの同定に関する。一旦このペプチド配列が決まる
と、このペプチドをエフェクター分子に結合させることによりエフェクター分子を生体膜
を通過させて移送させる。例えば、ファージ・ディスプレイ・パニングにより膵インスリ
ン分泌β細胞を標的にする一連のペプチドを単離した。こうした細胞を標的にするペプチ
ドを構築ブロックとする単量体もしくは多量体構築物を合成した。これらの構築物は、治
療剤を特異的に送達して糖尿病をコントロールするように設計した新規化学物質である。
【0060】
本発明のトランスポーターペプチドは式Iで表すことができる:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
式中、Pは前述のコンセンサス配列(consensus motif)(XmRXoR
Xn)、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)に包含
されるペプチドである。例えば、このペプチドは、配列X1−X2−X3−X4を含むこ
とができ、式中、X1およびX4はRもしくはKとすることができ、X1およびX4は任
意のアミノ酸とすることができる。一実施形態として、本発明は、アミノ酸配列R−X−
X−Rを有する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。別
の実施形態として、本発明は、配列番号1−34のうちの任意の1つのアミノ酸配列を有
する少なくとも1種のペプチドを含む多量体トランスポーターを提供する。種々の別の実
施形態として、このトランスポーターペプチドは、タンパク質転換酵素のリガンド由来の
ものである。さらに別の実施形態として、このトランスポーターペプチドは、タンパク質
転換酵素の切断部位から得られるものである。vによって表される所与のトランスポータ
ー内のペプチド数は、整数とする。単量体トランスポーターでは、vは1である。多量体
トランスポーターでは、vは2〜8またはそれ以上の整数とする。任意の所与の多量体結
合体において、それぞれのペプチドは互いに同じか、異なるものとすることができる。
【0061】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は1または0である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−ポリエチレングリコ
ール(succ−peg)などのポリエチレングリコール(PEG)が挙げられる。
【0062】
本明細書に用いている「コア」とは、多量体トランスポーターペプチド内のペプチド間
を結合する役割を果たす構造体である。また、コアは、リンカー、スペーサーおよびレポ
ーター基の接続ポイントとなる。通常、コアに対し結合に利用可能な多くの機能性を持た
せることによって、この多量体構築物の種々の異なる部分(ペプチド、および存在する場
合にはスペーサー、リンカーおよびレポーター)をこのコアと融合させて1つの分子を形
成させることができる。企図しているコア部分としては、以下に詳細を説明するポリリジ
ン(K)が挙げられ、また、リンカーの組み込まれた構造のものも挙げられる。コアを選
択することにより、所与の結合体内に存在するペプチドの数が決まる。代表的なコアとし
ては、C4−K2K−K(succ−peg−S)−アミド、C−GGG−[K(C)]
3−K(succ−peg−S)−アミド、(NH2OCH2CO)4−K2K−K(s
ucc−peg)−アミドおよび(NH2OCH2CO)8−K4K2K−K(GGG)
−アミドが挙げられる。単量体トランスポーターペプチドでは、コアは存在していてもし
ていなくてもよい。
【0063】
このトランスポーター構築物では、リンカーおよびレポーター基は存在していてもして
いなくてもよく、従って、yおよびzは、式Iにおいて独立に1もしくは0である。
【0064】
本明細書に用いている「トランスポーターペプチド」とは、物質が生体膜を通過して移
行するのを促進するペプチドである。特に明記しない限り、または用いる文脈において不
正確でない限り、トランスポーターペプチドを意味するものは単量体もしくは多量体のト
ランスポーターペプチドを包含する。
【0065】
トランスポーターペプチドは、物質が細胞の生体膜を横切って、特にその細胞質もしく
は核内へ通過または移行するのを容易にするペプチドである。移行の検出は、例えば、P
CT出願第WO97/02840号に記載されている細胞透過アッセイ(cellula
r penetration assay)を含む種々の方法によって行うことができる
。一般に、細胞透過アッセイは、a)培養細胞を、移行させるペプチドとインキュベート
し、b)この細胞を固定して透過処理し、およびc)細胞内のペプチドの有無を検出する
ことによって実施する。検出工程は、固定、透過処理した細胞をこのペプチドに対する標
識抗体とインキュベートした後、ペプチドと標識抗体との免疫反応を検出することによっ
て行うことができる。あるいは、検出可能な標識ペプチドを用い、細胞コンパートメント
内の標識の有無を直接検出することによっても、検出を行うことができる。この標識は、
例えば、放射性標識もしくは蛍光標識、色素、または検出可能な任意の標識とすることが
できる。例えば、de Jongら、J.Nucl.Med.40:2081(1999
)およびBreemanら、Int.J.Cancer 81:658(1999)を参
照されたい。
【0066】
さらに、本発明は、トランスポーターペプチドをエフェクターにカップリングもしくは
融合させた結合体である移送ユニット、もしくは結合体を含む。本明細書に用いている「
カップリングさせた」とは、エフェクターと前記ペプチドとの物理的な結合を可能にする
任意のタイプの相互作用のことを意味する。この結合の性質は、共有もしくは非共有結合
とすることができるが、その結合は、細胞移行の前もしくは移行中にこのベクターが解離
しないように十分強力なものである必要がある。カップリングは、当業者に既知の任意の
化学的、生化学的、酵素的もしくは遺伝子的カップリング法を用いて達成することができ
る。対象とするエフェクターは、このペプチド・ベクターのN末端もしくはC末端にカッ
プリングさせることができる。
【0067】
「融合した」もしくは「融合」または「結合する」あるいは「相互作用する」という用
語は、2種以上の分子同士が何らかの第3の分子に対するよりも高い選択性を示すような
特定の相互作用全てを含むものであり、これらには、共有結合、イオン結合、疎水性結合
、水素結合などの作用が含まれるが、溶媒選択性などの非特異的結合は含まれない。
【0068】
一部の実施形態として、このトランスポーターペプチドは、1つ以上のエフェクターと
カップリングもしくは融合させる。このカップリングは、直接的なもの、もしくはコアを
介したものとすることができる。「エフェクター」は、DNA、RNA、タンパク質、ペ
プチド、または薬学的に活性な薬剤、例えば、毒素、抗生物質、抗病原体剤(antip
athogenic agent)、抗原、抗体、抗体断片、免疫調節剤、酵素もしくは
治療剤などを含む任意の適切な分子とすることができる。2つ以上のエフェクターが存在
する場合、これらのエフェクターは同一もしくは異なるものとすることができる。エフェ
クターとは、例えば、生物学、医薬、診断、トレーシングもしくは食品加工の対象になる
任意の分子または化合物のことを意味する。エフェクターは、種々の由来の核酸、特にヒ
ト、ウイルス、動物、真核もしくは原核生物、植物または合成由来等の核酸(リボ核酸、
デオキシリボ核酸)で構成することができる。対象とする核酸は、例えば、単純な微量ヌ
クレオチドからゲノム断片もしくはゲノム全体に至る種々のサイズのものとすることがで
きる。同様に、これはウイルスゲノムもしくはプラスミドとすることができる。あるいは
、対象とするエフェクターは、例えば、酵素、ホルモン、サイトカイン、アポリポタンパ
ク質、成長因子、抗原、抗体などのタンパク質とすることもできる。さらに、このエフェ
クターは、例えば、毒素、治療剤、もしくは抗生物質、抗ウイルス剤、抗真菌剤、抗寄生
生物剤などの抗病原体剤のような薬学的に活性な物質とすることができる。この対象とす
るエフェクターは、それ自体そのままで活性があるものとすることができ、またはインサ
イチュでペプチドもしくは別の物質または環境条件により活性化されるものとすることが
できる。
【0069】
本発明のトランスポーターペプチド結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学の法則通りに、エフェクターはコア、もしくはリンカー、またはこれら
の両方に結合させることができる。単量体トランスポーターペプチド結合体(v=1)で
は、コアは存在してもしていなくてもよい。
【0070】
本明細書に用いている「薬学的に活性な物質」とは、ヒトもしくは動物の生物体に投与
したときに、検出可能な薬理学的および/または生理学的効果を誘起する化学物質もしく
は化合物のことを意味する。本明細書に用いている「治療剤」とは、ヒトもしくは動物の
生物体に投与したときに、目的とする薬理学的および/または生理学的効果を誘起する化
学物質もしくは化合物のことを意味する。
【0071】
本発明によるトランスポーターペプチドは、その透過能がこれにカップリングさせる対
象物質(エフェクター)の性質とは事実上無関係であるという事実に特徴がある。
【0072】
また、本発明は、対象物質を細胞もしくは細胞の核内に導入する方法を含む。この方法
は、細胞内への効率的な透過を可能にするのに十分な量のトランスポーターペプチド−エ
フェクター結合体もしくはトランスポーターユニットと細胞とを接触させることを含む。
一般に、本方法は、この結合体をインビボまたはインビトロで取り込ませるために用いる
ことができる。例えば、この結合体は、インビトロ、エキソビボ、またはインビボで与え
ることができる。さらに、本発明によるトランスポーターペプチドは、カップリングさせ
た物質に生物活性を持たせることを可能にすることができる。従って、トランスポーター
ペプチドはカップリングさせたエフェクターの生物活性を増強させることができる。イン
ビトロによる方法では、エフェクターをトランスポーターとカップリングさせ、得られた
結合体を、細胞代謝の活性化を可能にする温度で細胞とインキュベートする。場合によっ
ては、このトランスポーター−エフェクター結合体を特定の細胞に注入する。この結合体
を細胞内に導入する他の任意の方法を用いることもできることは当業者によって認められ
よう。
【0073】
また、以上のペプチド−エフェクター結合体のほかに、本発明は、薬学的に受容可能な
塩基もしくは酸付加塩、水和物、エステル、溶媒和物、プロドラッグ、代謝体、立体異性
体もしくはこれらの混合物を提供する。また、本発明は、薬学的に受容可能な担体、希釈
剤もしくは賦形剤と共に、ペプチド−エフェクター結合体を含む薬学的組成物を含む。
【0074】
「薬学的に受容可能な塩」という用語に包含される塩とは、一般に、この遊離塩基と適
切な有機もしくは無機酸とを反応させて本明細書に開示した化合物の「薬学的に受容可能
な酸付加塩」を作製することにより調製する本発明の化合物の毒性のない塩のことを意味
する。こうした化合物は、この遊離塩基の生物学的効果および性質を保持するものである
。このような塩の代表例は、酢酸塩、アムソン酸塩(4,4−ジアミノスチルベン−2,
2’−ジスルホネート)、ベンゼンスルホン酸塩、ベンゾネート、重炭酸塩、重硫酸塩、
酒石酸水素塩、ホウ酸塩、臭化物、酪酸塩、エデト酸カルシウム塩、カンシル酸塩、炭酸
塩、塩化物、クエン酸塩、クラブラリエート(clavulariate)、二塩化水素
化物、エデト酸塩、エディシレート、エストレート、エシレート、フマル酸塩、グルセプ
ト酸塩、グルコン酸塩、グルタミン酸塩、グリコリルアルサニレート、ヘキサフルオロリ
ン酸塩、ヘキシルレゾルシネート、ヒドラバミン塩、臭化水素酸塩、塩酸塩、ヒドロキシ
ナフトエート、ヨウ化物、イソチオネート、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、
リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシル酸塩、臭化メチル塩、メチル硝酸塩、
メチル硫酸塩、ムチン酸塩、ナプシル酸塩、硝酸塩、N−メチルグルカミン・アンモニウ
ム塩、3−ヒドロキシ−2−ナフトエート、オレイン酸塩、シュウ酸塩、パルミチン酸塩
、パモン酸塩(1,1−メチレン−ビス−2−ヒドロキシ−3−ナフトエート、エンボネ
ート)、パントテン酸塩、リン酸塩/二リン酸塩、ピクリン酸塩、ポリガラクツロン酸塩
、プロピオン酸塩、p−トルエンスルホン酸塩、サリチル酸塩、ステアリン酸塩、塩基性
酢酸塩、コハク酸塩、硫酸塩、スルホサリキュレート、スラメート、タンニン酸塩、酒石
酸塩、テオクル酸塩、トシル酸塩、トリエチオダイド、および吉草酸塩などの水溶性なら
びに非水溶性塩である。
【0075】
本発明の方法によれば、ヒト患者に対し、薬理学的に有効な量のペプチドもしくは結合
体を投与することができる。「薬理学的に有効な量」という用語は、研究者もしくは臨床
家によって求められている組織、器官系、動物もしくはヒトの生物学的または医学的反応
を誘起する薬物もしくは薬学的組成物(エフェクター)の量のことを意味する。
【0076】
また、本発明は、対象エフェクターを細胞もしくは細胞核内に導入するのに適した薬学
的組成物を含む。この組成物は、内服に適したものであることが好ましく、有効量の本発
明の薬理学的に活性な化合物を単独、もしくは1種以上の薬学的に受容可能な担体との組
合せで含む。この化合物は、毒性があるとしても極めて少ないという点で、特に有用であ
る。
【0077】
好ましい薬学的組成物は、a)希釈剤、例えば、乳糖、ブドウ糖、蔗糖、マンニトール
、ソルビトール、セルロースおよび/またはグリシン;b)滑沢剤、例えば、シリカ、滑
石、ステアリン酸、そのマグネシウムもしくはカルシウム塩および/またはポリエチレン
グリコール;錠剤用としてc)カップリング剤、例えば、ケイ酸マグネシウム・アルミニ
ウム、でんぷん糊、ゼラチン、トラガント、メチルセルロース、カルボキシメチルセルロ
ースナトリウムおよび/またはポリビニルピロリドン;必要に応じてd)崩壊剤、例えば
、でんぷん、寒天、アルギン酸またはそのナトリウム塩もしくは発泡性混合物;および/
またはe)吸収剤、着色剤、矯味矯臭剤および甘味剤とともに有効成分を含む錠剤ならび
にゼラチンカプセル剤である。注射用組成物は、好ましくは等張水溶液もしくは懸濁液で
あり、坐剤は脂肪性乳濁液もしくは懸濁液から調製するのが有利である。これらの組成物
は、滅菌することができ、および/または保存剤、安定化剤、湿潤剤または乳化剤などの
佐剤、溶解促進剤、浸透圧調節塩および/または緩衝液を含むことができる。また、さら
に、これらは別の治療上有用な物質を含むことができる。これらの組成物は、それぞれ、
従来の混和、顆粒化またはコーティング法によって調製することができ、有効成分を約0
.1〜75%、好ましくは約1〜50%含有する。
【0078】
本明細書に開示した活性化合物および塩の投与は、一般に認められている治療剤の投与
方法のうちの任意の方法によることができる。これらの方法としては、経口、鼻腔内、非
経口、経皮、皮下もしくは局所性投与様式などの全身性または局所投与が挙げられる。
【0079】
目的とする投与様式に応じて、この組成物は、例えば、好ましくは単位投与量の注射用
剤、錠剤、坐剤、丸剤、徐放性カプセル剤、散剤、液剤、懸濁剤などのような固体もしく
は半固体または液状の投与形態とすることができる。この組成物は、有効量の活性化合物
もしくはその薬学的に受容可能な塩を含むことになり、また、さらに、薬学において通例
用いられている任意の従来の薬学的賦形剤および他の治療薬もしくは薬学的組成物、担体
、アジュバント、希釈剤なども含むことができる。
【0080】
固体組成物の賦形剤としては、医薬用品質のマンニトール、乳糖、でんぷん、ステアリ
ン酸マグネシウム、サッカリン・ナトリウム、滑石、セルロース、グルコース、蔗糖、炭
酸マグネシウムなどを用いることができる。また、上記の活性化合物は、例えば、プロピ
レングリコールなどのポリアルキレングリコールを担体とする坐剤として製剤化すること
ができる。
【0081】
液体、特に注射用組成物は、例えば、溶解、分散などにより調製することができる。活
性化合物は、例えば、水、生理食塩液、ブドウ糖水溶液、グリセロール、エタノールなど
の医薬用純品溶媒に溶解もしくは混合することにより、注射用溶液もしくは懸濁液を作製
する。
【0082】
また、投与することになる薬学的組成物は、必要に応じて、湿潤剤、乳化剤、pH緩衝
剤、および例えば、酢酸ナトリウム、オレイン酸トリエタノールアミンのような他の物質
などの毒性のない補助剤を少量含むことができる。
【0083】
一般に、非経口注射用剤は、皮下、筋肉内もしくは静脈内注射もしくは注入により投与
する。注射用剤は、溶液もしくは懸濁液、または液に溶かした後、注射するのに適した固
体形態のような通常の形態として調製することができる。
【0084】
非経口投与の1つの方法として、本明細書に引用により組み込まれている米国特許第3
,710,795号に記載の方法に従い、投与量の一定のレベルが維持されるのを確実な
ものにする徐放性もしくは持続放出性製剤の埋め込み法を用いる。
【0085】
本発明の化合物は、錠剤、カプセル剤(各々が徐放性および持続放出性製剤を含む)、
丸剤、散剤、顆粒剤、エリキシル剤、チンキ剤、懸濁剤、シロップ剤および乳剤のような
経口剤形として投与することができる。また、同様に、この化合物は、医薬分野の当業者
に周知の形態により静脈内(急速および点滴の両方)、腹腔内、皮下もしくは筋肉内形態
で投与することができる。
【0086】
この化合物の用法・用量は、患者の体型、種類、年齢、体重、性別および病状;治療対
象の症状の重症度;投与経路;患者の腎および肝機能;ならびに使用する特定の化合物も
しくはその塩を含む種々の要素に基づいて選択する。当該分野の医師もしくは獣医師であ
れば、症状の進行を防止し、遅らせ、または止めるのに必要とされる薬剤の有効量を容易
に決定し、処方することができる。
【0087】
本発明の経口用量は、適応となる効果に用いる場合、有効成分を0.5、1.0、2.
5、5.0、10.0、15.0、25.0、50.0、100.0、250.0、50
0.0もしくは1000.0mg含有する分割錠の形態で提供し得る。
【0088】
本発明の化合物は、1日1回の用量として投与することができ、あるいは1日当たりの
総投与量を1日2回もしくは3回または4回の分割用量として投与することができる。さ
らに、本発明に好ましい化合物は、適切な鼻腔内投与用器具(intranasal v
ehicle)の局所使用による鼻腔内投与、もしくは当業者に周知の形態の経皮投与用
皮膚パッチ剤による経皮投与を行うことができる。経皮送達システムの形態で投与する場
合には、勿論、適量投与(dosage administration)は用法・用量
全体を通して間欠的ではなく連続的なものとなる。その他の好ましい局所用製剤としては
、クリーム剤、軟膏剤、ローション剤、エアロゾル・スプレー剤およびゲル剤が挙げられ
、これらの場合、有効成分の濃度は0.1%〜15%w/wまたはw/vの範囲となろう
。
【0089】
本明細書に詳細に述べた化合物は、有効成分を構成することができ、通常、対象とする
投与形態、即ち、経口用としての錠剤、カプセル剤、エリキシル剤、シロップ剤などに対
して適切に選ばれ、従来の調剤慣行に合った適切な薬学的希釈剤、賦形剤もしくは担体(
本明細書ではこれらを総称して「担体」物質と呼ぶ)と混合して投与する。
【0090】
例えば、錠剤もしくはカプセル剤として経口投与する場合、有効薬物成分は、エタノー
ル、グリセロール、水などの経口用で毒性がなく薬学的に受容可能な不活性担体と混合す
ることができる。さらに、所望もしくは必要に応じて、この混合物に適切な結合剤、滑沢
剤、崩壊剤および着色剤を混和することもできる。好適な結合剤としては、でんぷん、ゼ
ラチン、グルコースもしくはベータ乳糖のような天然糖、コーン甘味剤、アカシア、トラ
ガカントもしくはアルギン酸ナトリウムのような天然および合成ゴム、カルボキシメチル
セルロース、ポリエチレングリコール、ろうなどが挙げられる。こうした剤形に用いる滑
沢剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシ
ウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウムなどが挙げられる。崩壊剤
としては、でんぷん、メチルセルロース、寒天、ベントナイト、キサンタン・ガムなどが
挙げられるが、これらに限定されない。
【0091】
また、本発明の化合物は、小単層小胞、大単層小胞および多重層小胞などのリポソーム
送達システムの形で投与することもできる。リポソームは、コレステロール、ステアリル
アミンもしくはホスファチジルコリンを含有する種々のリン脂質から形成することができ
る。一部の実施形態として、米国特許第5,262,564号に記載されているようにし
て、脂質成分の膜を薬剤の水溶液で水和させて、薬剤を内包する脂質層を形成する。
【0092】
また、本発明の化合物は、標的を定めることができる薬物担体としての可溶性ポリマー
とカップリングさせることもできる。このようなポリマーとしては、ポリビニルピロリド
ン、ピラン・コポリマー、ポリヒドロキシプロピル−メタクリルアミド−フェノール、ポ
リヒドロキシエチルアスパンアミドフェノール(polyhydroxyaspanam
idephenol)、もしくはパルミトイル残基で置換したポリエチレンオキシドポリ
リジンを挙げることができる。さらに、本発明の化合物は、薬剤の放出を制御するのに有
用なクラスの生分解性ポリマー、例えば、ポリ乳酸、ポリεカプロラクトン、ポリヒドロ
キシ酪酸、ポリオルソエステル、ポリアセタール、ポリジヒドロピラン、ポリシアノアク
リレート、およびヒドロゲルの架橋もしくは両親媒性ブロックコポリマーとカップリング
させることができる。
【0093】
上記薬学的組成物はいずれも、活性化合物、特に有効成分としての式Iの化合物を0.
1〜99%、好ましくは1〜70%含有することができる。
【0094】
(その他の実施形態)
本発明の1つ以上の実施形態についてその詳細を前記の添付明細書において説明した。
本発明の特定の実施形態についての前記詳細な説明から、生体膜を通過させて移行させる
ための独自の方法および組成物が開示されたことが明瞭に理解されよう。本明細書では特
定の実施形態についてその詳細を説明したが、これは単に例示を目的として例として説明
したものであり、以下に添付した特許請求の範囲に対して限定しているものではない。特
に、この特許請求の範囲に定義したような本発明の精神および範囲から逸脱することなく
、本発明に対し、種々の置換、変更および変形を行うことができることは、本発明者によ
り企図されている。例えば、特定のタイプの細胞、または移行させるべき特定のエフェク
ターの選択については、本明細書に開示した実施形態を知った当業者には日常的な事柄で
あると考えられる。
【0095】
本明細書および添付の特許請求の範囲では、文脈上明らかに別の意味に解すべき場合を
除き、単数形は複数の対象を含む。特に定義しない限り、本明細書に用いている科学技術
用語は全て、本発明が属する分野の当業者により一般に理解されている意味と同じ意味を
有する。また、本明細書に引用されている全ての特許および刊行物は、引用により本明細
書に組み込まれている。
【0096】
以下に示した実施例は、本発明の好ましい実施形態についてさらに十分に説明するため
のものである。これらの実施例は、添付の特許請求の範囲により定義した本発明の範囲を
限定するものと決して解釈されるべきではない。
【実施例】
【0097】
(実施例1:内部移行ペプチドモチーフの特定)
ファージ・ペプチド・ライブラリは、古典的な方法では、線維状ファージM13の誘導
体として構築される。ペプチド・ライブラリを、ペプチドモチーフの1〜5個のコピーを
提示するキャプシドのマイナーコートタンパク質pIIIに融合する。Zwickら、C
urr.Opin.Biotechnol.9:427−436(1998)を参照され
たい。あるいは、メジャーコートタンパク質pVIIIを用いることによって高価(hi
gh valent)提示が得られる。しかしながら、これらのタイプのライブラリは、
受容体媒介性エンドサイトーシスの対象となるペプチド配列の単離のために最適化されて
いない。何故なら、ペプチドの1価もしくは低価提示は、線維状ファージのような巨大な
構造体を効率的に取り込ませるには不十分であるからである。多価提示のみが効率的取り
込みを可能にする。Ivanenkovら、Biochem.Biophys.Acta
1448:450−462(1999)を参照されたい。さらに、受容体結合リガンド
の内部移行は、原形質膜の特定領域に細胞表面受容体が濃縮され、次いでクラスリン被覆
小胞が形成される必要があり(Damke、FEBS Lett.389:48−51(
1996)参照)、従来のM13ファージでは、こうした特殊で高度に効率的な構造によ
る受容体媒介性内部移行が行われるとは考えられない。
【0098】
T7 415ファージ系およびT7 413bファージ系のファージ・ディスプレイ・
ライブラリを構築した。このファージでは、減少した容量内にこの提示ペプチドが415
個のコピーとして生じる(キャプシドの直径は約50nMである)。従って、本発明に含
まれるのは、以下の基準を満たす新規なファージ系のファージ・ディスプレイ・ライブラ
リである:4〜50merペプチドの多価提示(>400個のコピー/ファージ);サイ
ズが小さい(50nM);内部移行したファージを効率的に回収できる;内部移行しなか
った結合ファージを除去できる;および個別ペプチド配列の数が大である(>109個の
ヘプタペプチド配列を示す3×108個の独立クローン)。
【0099】
このライブラリを用いて、βTC−3細胞モデル内への高分子の効率的、特異的な細胞
内送達を誘導するペプチドモチーフの単離に成功した。さらに、このライブラリを5種の
異なる(非−β)細胞株に対して用い、それぞれの場合で、各細胞型に特異的なペプチド
モチーフの濃縮(enrichment)について観察した。この方法の概括的な全体像
を以下に説明する。
【0100】
(選択/濃縮(enrichment)方法)
多くのインスリン分泌細胞株、齧歯類およびヒト単離島、およびFACSにより精製し
たβ細胞に対するファージ・ディスプレイ・ライブラリをパニングし、最終的に、これを
直接動物(マウス、ラット、ブタ)に注射した後、島を抽出して内部移行したファージを
回収する。パニング方法は、ファージの添加、回収および増幅の少なくとも3サイクルか
らなる。あるいは、最も選択的なリガンドを単離するために、このライブラリを種々のイ
ンスリン非分泌細胞と共にインキュベートした後にβ細胞に対するライブラリを選択する
ことにより、他の細胞型に結合するファージを除去する。記載されるように、クロロキン
を用いてリソソームによる分解をブロックする実験を実施する。Ivanenkovら、
Biochem.Biophys.Acta 1448:450(1999)を参照され
たい。
【0101】
(ファージ特異性の測定)
選択したファージを単離し、多くの種々の細胞および器官と共にインキュベートする。
例えば、一部の実験として、選択したファージをインスリン分泌およびインスリン非分泌
細胞および器官とインキュベートする。取り込みについては、回収されたファージ数をカ
ウントすることにより測定する。抗ファージ抗体を用いて免疫細胞化学的研究を実施する
。
【0102】
(ファージ由来(pharge−beard)ペプチドの特徴付け)
単離ファージからのDNAを配列決定し、発現されるペプチドを推定する。直接の内部
移行を誘導するペプチド、およびこれらのペプチドの変異型を化学的に合成し、N末端を
FITCで標識し、もしくはヨウ素化する。標識ペプチドを種々の細胞型、齧歯類および
ヒト単離島に加え、そして、マウスに直接注射する。取り込みの特異性、細胞内局在性、
クリアランスおよび安定性について評価する。Widmannら、Biochem.J.
310:203(1995)を参照されたい。
【0103】
(生化学的アッセイ)
インスリン分泌細胞およびインスリン非分泌細胞を分析するため、特徴付けを行ったペ
プチドを3種の既知配列:YVAD(カスパーゼ阻害因子、配列番号35;Rouque
tら、Curr.Biol.6:1192(1996))、VQRKRQKLMP(NF
−κB核内局在化阻害因子(Linら、J.Biol.Chem.270:14255(
1995)、配列番号36)もしくはRPKRPTTLNLFPQVPRSQDT(JN
K阻害因子、Bonnyら、Diabetes 50:77−82(2000)、配列番
号37)に結合させる。これらのペプチドは、化学的に合成し、インスリン分泌細胞およ
びインスリン非分泌細胞に加える。カスパーゼ、NF−κBおよびJNKは、一般的活性
化剤エトポシド(Kimら、Anticancer Res.20:439(2000)
参照)もしくはアニソマイシン(Usamiら、Biochem.Pharmacol.
55:185(1998)参照)により活性化する。上記ペプチドによるカスパーゼ、N
F−κBおよびJNKの阻害についてはβ細胞および非β細胞を用いて検討する。以上の
実験によって、これらのペプチド担体が薬剤となり得る物質を活性のある形態で特にβ細
胞内に移送することができるかどうかが分かる。
【0104】
(治療剤となり得る物質のGLP−1受容体による取り込み)
GLP−1受容体(GLP−1R)の発現は、主として脳および膵臓に限定されている
。Yamatoら、Horm.Metab.Res.29:56(1997)を参照され
たい。この受容体は、アゴニストに結合した後、内部移行される。Widmannら、B
iochem.J.310:203(1995)を参照されたい。こうした性質があるた
め、GLP−1Rは膵β細胞への治療剤の選択的送達を媒介する魅力的なツールとなる。
この特性は、上に記載されるように評価される。GLP−1Rを用いて集められた情報が
、例えば、高められた選択性を有する二重特異的ダイマーの設計において、補助する。
【0105】
(GLP−1受容体に対する他の内部移行モチーフの特定)
GLP−1Rを用いてトランスフェクションしたCOS−7細胞は、上記のような選択
実験の基材(substrate)となる。新規に特定したモチーフについてその特異性
およびエンドサイトーシス誘導能を評価する。
【0106】
(全−D−レトロ−インベルソ型(all−D−retro−inverso)ペプチ
ドの作製)
一部の実施形態として、上記ペプチドはレトロ−インベルソ型ペプチドとして合成する
ことができる。安定性が高く、免疫原性の少ない全D−レトロ−インベルソ型ペプチド(
Sela、Zisman,FASEB J.11:449(1997)参照)について、
前述の方法により、分析する。
【0107】
進化によって、天然のタンパク質にはほぼL型アミノ酸のみが存在することになった。
従って、事実上全てのタンパク質分解酵素は、隣接するL型アミノ酸間のペプチド結合を
切断するので、D型アミノ酸からなる人工的なタンパク質もしくはペプチドは、タンパク
質分解に対してかなり抵抗性である。この抵抗性は薬剤の設計者には魅力的であったが、
L−アミノ酸からなるタンパク質の生体系が排他的であるため、このようなタンパク質は
、鏡像異性タンパク質により形成される鏡像面と相互作用することができないということ
になる。従って、通常、全D(all−D)型アミノ酸タンパク質には生物効果もしくは
活性がない。
【0108】
線状の修飾レトロ−ペプチド構造については長い間研究されてきており(Goodma
nら、Accounts of Chemical Research,12:1−7(
1979)参照)、「レトロ−アイソマー」という用語は、親ペプチドに対して配列の方
向が逆であるアイソマーを含むとされた。「レトロ−インベルソ・アイソマー」とは、配
列の方向が逆で、各アミノ酸残基の対掌性が逆であり、従って末端基相補性が生じ得ない
線状ペプチドのアイソマーのことを意味する。
【0109】
さらに最近、Jamesonらは、これら2つの性質、即ち、逆合成および対掌性の変
化を組み合わせることにより、CD4受容体のヘアピンループのアナログを設計した。J
amesonら、Nature 368:744−746(1994)およびBrady
ら、Nature,368:692−693(1994)を参照されたい。D−鏡像異性
体と逆合成を組み合わせたことによる最終的な結果は、各アミド結合のカルボニル基とア
ミノ基とが交換されるが、各α炭素の側鎖基の位置は保存されたことである。James
onらは、従来の全−L鏡像異性体の限定的なインビボ活性(タンパク質分解を受けやす
いことに起因する)とは対照的に、逆Dペプチドの生物活性が増大することを明らかにし
た。
【0110】
ヒト・クラスI組織適合性分子HLA−A2の非天然リガンドとして使用することがで
きる部分修飾したレトロ−インベルソ偽ペプチドが報告されている。Guichardら
、Med.Chem.39:2030−2039(1996)を参照されたい。このよう
な非天然リガンドでは、安定性およびMHC結合能が増大した。
【0111】
レトロ−インベルソ・ペプチドを、以下のようにして配列が既知のペプチドに対して作
製する。レトロ−インベルソ・ペプチドアナログをデザインし、合成するためのモデル・
ペプチドとして、既知の配列を有するペプチド(例えば、腫瘍抗原ペプチド)を選択する
。このアナログは、D−アミノ酸を用い、このレトロ−インベルソ・ペプチド内のアミノ
酸の配列がモデルとして働く選択したペプチド内の配列と正確に逆になるように、D−ア
ミノ酸を結合させてペプチド鎖とすることにより合成する。例えば、このペプチド・モデ
ルがABCの配列を有するL−アミノ酸で形成されているペプチドである場合、D−アミ
ノ酸で形成されるそのレトロ−インベルソ・ペプチドアナログはCBAの配列を有するこ
とになる。D−アミノ酸の鎖を合成してレトロ−インベルソ・ペプチドを作製する方法は
、当該分野で公知であり、前記文献において説明されている。
【0112】
天然ペプチドの場合の固有の問題が天然のタンパク質分解酵素により分解されることに
あるので、本発明のペプチドは、目的ペプチドの「レトロ−インベルソ・アイソマー」を
含むように作製することができる。従って、天然性のタンパク質分解からペプチドを保護
することにより、この特定のヘテロ2価(heterobivalent)もしくはヘテ
ロ多価(heteromultivalent)化合物の有効性は増大するはずである。
【0113】
レトロ−インベルソを含むペプチドでは、天然のタンパク質分解酵素による分解から保
護されるため、レトロ−インベルソを含まないアナログと比べて、生物活性が増大すると
予測される。
【0114】
(修飾ペプチドの作製)
一部の実施形態として、このペプチドは、修飾ペプチドとして合成することができる。
この修飾ペプチドは前述の方法により分析する。
【0115】
アナログは、アミノ酸配列により、またはアミノ酸配列に影響を与えない修飾により、
あるいはこれらの両者により天然のペプチドと異なるものとすることができる。好ましい
アナログとしては、その配列が、単に保存的アミノ酸置換、好ましくは1、2もしくは3
置換のみ、例えば、1つのアミノ酸の同様な特徴を有する別のアミノ酸による置換(例え
ば、グリシンをバリンに、リジンをアルギニンに置換など)によって、またはペプチドの
生物活性を消失させない1つ以上の非保存的アミノ酸置換、欠失もしくは挿入によって、
その野性型配列(すなわち、天然に生じるペプチドの相同な部分の配列)と異なるペプチ
ドが挙げられる。
【0116】
(普通一次配列に影響を与えない)修飾としては、インビボもしくはインビトロにおけ
るペプチドの化学的誘導体化、例えば、アセチル化もしくはカルボキシル化が挙げられる
。また、糖鎖形成の修飾、例えば、ペプチドの合成およびプロセッシング過程において、
または、例えば、糖鎖形成に影響を与える酵素、例えば、哺乳類のグリコシル化もしくは
脱グリコシル化酵素をペプチドに作用させることによる別のプロセッシング工程において
、ペプチドの糖鎖形成パターンを修飾することによるものも挙げることができる。また、
リン酸化されたアミノ酸残基、例えば、ホスホチロシン、ホスホセリンもしくはホスホス
レオニンを有する配列を挙げることもできる。
【0117】
本発明は、1つ以上のペプチド結合が、ペプチダーゼにより切断されにくい別のタイプ
の共有結合(「類似ペプチド結合」)に置換されたアナログを含む。対象に注射後のペプ
チドの蛋白性分解が問題となる場合、特定の切断されやすいペプチド結合を切断不能の類
似ペプチド結合に置換すると、得られるペプチドは安定性が増し、治療剤としてより有用
なものとなる。このような類似ペプチド結合およびこれをペプチドに組み込む方法は、当
該分野では周知である。また、t−ブチルオキシカルボニル、アセチル、テイル(the
yl)、スクシニル、メトキシスクシニル、スベリル、アジピル、アゼライル、ダンシル
、ベンジルオキシカルボニル、フルオレニルメトキシカルボニル、メトキシアゼライル、
メトキシアジピル、メトキシスベリル、および2,4−ジニトロフェニルなどのアミノ末
端ブロック基も有用である。ペプチドの荷電アミノ−末端およびカルボキシ−末端をブロ
ックすると、ペプチドが疎水性細胞膜を通過して細胞内へ入るのが促進されるという別の
メリットがある。
【0118】
(多量体ペプチドの作製)
多量体リガンドでは、内部移行速度の増大につながる最大数桁の結合力の向上がみられ
る。Terskikhら、Proc.Natl.Acad.Sci.U.S.A.94:
1663(1997)およびYorkら、J.Biol.Chem.274:1164(
1999)を参照されたい。単一特異性ダイマーは、強い結合力を示し、二重特異性ダイ
マーでは、特異的に細胞を標的にする物質としての実用面の潜在能力を向上させることが
できる選択性が高まると考えられる。Caruthers and Lerner,Ch
em.Biol.3:537(1996)を参照されたい。単量体および多量体ペプチド
(単一および多重特異性)は、ペプチドとして、あるいは例えば、柔軟性のあるペプチジ
ルまたは糖ベースの主鎖を有するペプチド類似体として合成することができる。Caru
thers and Lerner,Chem.Biol.3:537(1996)、Z
engら、J.Pept.Sci.2:66(1996)およびUlbrichら、J.
Controlled Release 64.(1.−3.):63−79.、64:
63(2000)を参照されたい。
【0119】
(細胞内局在性)
単離した種々のペプチド配列は、種々の細胞コンパートメント(例えば、核、ミトコン
ドリア、細胞質ゾルなど)に局在し得る。これについては、標識(例えば、ヨウ素化もし
くはFITC標識)ペプチドを用いて調べる。この局在性情報を利用して機能性研究をデ
ザインする。例えば、細胞質ゾル内に蓄積するペプチドは、NF−κBの核移行を阻害す
るのに好ましく、核内に移行するペプチドは、JNKを阻害するのに最も適している。一
部の実施形態として、核局在化モチーフのような配列を結合体に加えて、担体を適切な細
胞コンパートメントへ再誘導することができる。
【0120】
(機能性研究)
カスパーゼ、NF−κBもしくはJNK阻害剤に結合させた、β細胞を標的とするトラ
ンスポーターペプチド(例えば、単量体もしくは多量体のL型もしくはD型エナンチオマ
ー)をβ細胞株、FACS精製β細胞ならびに単離ヒトおよび齧歯類の島(islet)
に加える。IL−β(TNFαおよびIFNγを併用)によりアポトーシスを誘発させ、
アポトーシスに対する抵抗性を調べる。
【0121】
(インビボ糖尿病実験)
糖尿病発症前および発症後の状態のNODマウスにエフェクタ・ペプチド(カスパーゼ
、NF−κBもしくはJNK阻害剤に結合させたβ細胞を標的とするペプチド)を注射す
る。用量および注射頻度は、前述のようにして決定する。次いで、糖尿病の発生を測定す
る。
【0122】
(免疫原性に関するアッセイ)
齧歯類およびウサギを用い、ペプチドの免疫原性の潜在性について評価する。
【0123】
(クローニング)
特定の細胞による効率的な内部移行を誘導するペプチドモチーフについては実施例II
IおよびIVにおいて説明する。このペプチドを用いて、確立された手順により、例えば
、INS−1、βTC−3およびヒト島cDNAライブラリからの同族(cognate
)受容体のクローニングならびに特徴付けを行う。Thorens、Proc.Natl
.Acad.Sci.U.S.A.89:8641(1992)およびVolzら、FE
ES Lett.373:23(1995)を参照されたい。
【0124】
(特徴付け)
上記のクローニングした受容体の組織分布について、インスリン分泌およびインスリン
非分泌細胞および器官のノーザンブロッティングおよびウェスタンブロッティングにより
調べる。結合速度、クリアランスおよび内部移行の特異性についてはCOS−7細胞にこ
の受容体を一時的にトランスフェクションさせることにより調べる。対照ペプチドは、変
異配列、および例えば、GLP−1、GIP、グルカゴン、セクレチンなどの既知ペプチ
ドとする。これらの受容体に対する別の内部移行モチーフについては上記のトランスフェ
クションしたCOS−7細胞において前記ライブラリをパニングすることにより特徴付け
る。
【0125】
(実施例II:トランスポーターペプチドのスクリーニング方法)
特に指定した場合を除き、溶媒および試薬は全てフルカ社(Fluka)、ブックス(
Buchs)、スイスから入手し、分析品質以上のものとし、さらに精製することなく使
用した。アミノ酸は全てペプチド・インスティテュート社(Peptide Insti
tute Inc.)(日本)から購入した。樹脂はアプライド・バイオシステムズ社(
Applied Biosystems)、米国;ノバビオケム社(Novabioch
em)、スイスもしくはバッヘム社(Bachem)、スイスから入手した。水は、Mi
lli−Qシステム(ミリポア社(Millipore,Inc.)を用いて再精製した
。バイオアッセイにはインスリン分泌細胞株βTC−3を用いた。Efratら、Pro
c.Natl.Acad.Sci.USA 85:9037−9041を参照されたい。
【0126】
(ファージの調製および濃縮(enrichment)方法)
標準的な手順により(例えば、ノバジェン社(Novagen)のT7 414ファー
ジもしくはT7 413bファージを用い)、キャブシドの表面にランダムな15マーエ
ピトープを提示する3×108個の単独(independent)ファージのライブラ
リを作製した。SmithおよびScott,Methods Enzymol.217
:228(1993)を参照されたい。(Smith、Scottの上記文献に)記載さ
れるように、ファージを増幅させた後、ポリエチレングリコール(PEG)沈殿法により
精製し、最終的に、Tris−EDTA緩衝液(10:1mM、TE)中に1010個感
染性粒子/μlの濃度で再懸濁した。培養培地中の細胞にファージ(1012個)を1〜
24時間加えておいた。エンドサイトーシス小胞内でタンパク質分解を免れたファージの
単離を容易にするには、インキュベーション時間をさらに長くすることが好ましかった。
結合および内部移行を行わせた後、細胞を洗浄し、内部移行されなかったファージを、サ
ブチリシン(3mg/ml)(44)で消化させて破壊した。充分に洗った後、2%デオ
キシコール酸塩、10mM Tris−HClおよび2mM EDTAを含有するpH8
.0の緩衝液で細胞を溶解させることにより、内部移行されたファージを回収した。回収
したファージは、最終的にE.coli細胞(XL−1−Blue)を用いて増幅させ、
前述のようにして精製した。次いで、この選択したファージの調製物を用いて第2ラウン
ドのパニングを行った。3〜5ラウンドを順次実施することにより特定のファージ保有ペ
プチド配列を濃縮した。
【0127】
(免疫細胞化学および蛍光による検討)
上記の濃縮スキームによって単離した単一ファージを増幅させた後、培養培地中の細胞
に24時間加えておいた。次いで、培地を洗い流し、細胞を冷メタノール−アセトン(1
:1)で5分間固定した。ファージ・キャプシドに対する抗体は、蛍光結合二次抗体と共
に用いた。古典的な蛍光顕微鏡による検討および共焦点顕微鏡によるアッセイを実施した
。組織は、処理の前にパラフィン包埋した。
【0128】
(ペプチド)
C−末端アミド基を有し、必要に応じてFITC標識もしくはヨウ素化したペプチドを
、古典的なF−moc化学(オースペップ社(Auspep)、オーストラリア)を用い
て合成した。ペプチドは全てHPLCにより精製し、質量分析法によって分析した。
【0129】
(生化学的検討)
種々の細胞株、例えばβTC−3、INS−1、HeLa、WiDr、HepG2、N
IH3T3、COS−7などにおいて、ペプチド添加の1時間後からJNK、NF−κB
およびカスパーゼをエトポシド(VP−16,Alexis)により1時間賦活化する。
細胞抽出物を処理して(c−Junを基質として用いる固相JNKアッセイ(Bonny
ら、J.Biol.Chem.275:16466(2000)参照)により)JNK活
性、(電気泳動移動度シフトアッセイ(Linら、J.Biol.Chem.270:1
4255(1995)参照)により)NF−κBの核移行および(アップステート・バイ
オケミカルス社(Upstate Biochemicals)から市販のキットおよび
抗体により)カスパーゼ活性を測定する。
【0130】
(アポトーシスの測定)
アポトーシスは、以前に報告された方法に従い、Hoechst33342およびヨウ
化プロピジウムの組合せを用いて測定する。Bonnyら、J.Biol.Chem.2
75:16466(2000)、Hoorensら、J.Clin.Invest.98
:1568(1996)を参照されたい。
【0131】
(島)
島は、Gotohらの方法(Gotohら、Transplantation 43:
725(1987)参照)により単離する。ヒト島は、例えば、「Insel Spit
al」、ベルン(Bern)、スイスから入手することができる。
【0132】
(マウス)
正確な投与量および注射の時間枠は、各ペプチドごとに最適化する。しかしながら、J
NKIを用いたこれまでの経験から、ペプチドの1mM PBS溶液100μlを2日お
きに注射するのが合理的な出発点であることが分かっている。
【0133】
(λZAP発現ライブラリ)
λZAP発現原核/真核発現ベクター中のINS−1 cDNAライブラリを用いてI
B1 cDNAおよびIB2 cDNAをクローン化した。Bonnyら、J.Biol
.Chem.273:1843(1998)およびNegriら、Genomics 6
4:324(2000)参照。このライブラリは、真核CMVプロモータの制御下で、単
純なヘルパー・ファージ切り出し(excision)(ストラタジーン社(Strat
agene)によって容易にプラスミド・ライブラリに変換される。
【0134】
(実施例III:ファージ・ディスプレイ・ライブラリのパニングおよび内部移行され
たペプチドモチーフの特徴付け)
β細胞を特異的に標的にして薬剤を送達させることができれば、I型糖尿病の治療に極
めて大きな影響を与えることになろう。基本的にβ細胞の機能(すなわち、インスリン分
泌)を変えないβ細胞破壊のブロッカーはすでに存在する(例えば、JBD、bel−2
)。こうした分子の1つ(JBD)を低分子ペプチドに変換しても全生物活性を保持させ
ることができることが示されている。
【0135】
膵β細胞株βTC−3は、本明細書に記載したファージ・ディスプレイ・ライブラリを
用いてパニングした。下記の表1に示すように、選択の各サイクルにおいて、回収ファー
ジ数の選択的な濃縮が認められた。βTC−3細胞を用いるパニング実験を、濃縮手順の
各工程で109個のファージを用いて実施した。0℃(エンドサイトーシスはみられない
)で回収されたファージ数は100個未満であり、このことから、本明細書に記載した条
件下では、内部移行されなかった細胞外の結合ファージのバックグラウンドは極めて低い
ことが分かる。
【0136】
【表1】
【0137】
βTC−3細胞株における3工程のパニング後のファージ回収率を表2に示す。
【0138】
【表2】
【0139】
表3に示した時間βTC−3細胞とインキュベートしたファージP1(配列番号1)を
用い、滴定実験を実施した。また、投入/回収ファージの比率も示す。滴定実験から、最
初の投入P1ファージの10%も回収することができることが分かった。
【0140】
【表3】
【0141】
取り込みの特異性についての測定は、5種の細胞株における回収ファージ数をタイトレ
ートすることにより行った。ファージ(108個)を、示した細胞株と16時間インキュ
ベートした後、表4に示したように、内部移行されたファージ数および回収されたファー
ジ数を算定した。インテグリン内部移行モチーフを提示する対照ファージは、全ての細胞
株に対して同様な回収ファージ数(1〜3×106個)を示した。このことから、P1(
配列番号1、表5参照)は、βTC−3細胞によって、テストした他のどの細胞株より1
0,000〜1,000,000倍効率的に内部移行されることが分かる。
【0142】
【表4】
【0143】
次いで、ファージP1の提示ペプチドの配列に基づいてペプチドを合成した。FITC
で標識した10アミノ酸ランダム配列にP1 5マーペプチドの配列を結合させた。対照
配列は、P1 5マー配列を(Ala)5で置き換えた以外は同一であった。細胞にペプ
チド(10μM)を1時間加え、細胞を洗い、冷メタノール−アセトン(1:1)で固定
した。βTC−3細胞内のFITC標識P1ペプチドは可視化することができたが、他の
細胞型ではできなかった。
【0144】
最後のサイクルの濃縮(enrichment)時の20個の回収ファージの配列解析
結果を表5に示す。重要なことであるが、全ての配列は、5個のアミノ酸の同じ保存コン
センサス配列に厳密に従った。このことから、ファージの効率的な取り込みを誘導する保
存モチーフが特異的に選択/濃縮されることが示唆される。こうして得られたペプチドモ
チーフの大部分は、プロタンパク質転換酵素のコンセンサスなR−X−X−Rに従う。こ
の知見は、潜在的薬物および高分子を特定の細胞型の細胞内に送達するためのビヒクルと
してプロタンパク質転換酵素を使用する提案の根拠を成すものである。
【0145】
【表5−1】
【0146】
【表5−2】
【0147】
(実施例IV:多量体結合体)
ファージ・ライブラリから得られるリガンド・ペプチドは低親和性(マイクロモル程度
)とすることができる。その結合親和性もしくは結合力は、ペプチド分子のコピーを多量
体化することにより、例えば、ペプタボディ(peptabody)を形成させることに
より向上させることができる。ScottおよびSmith,Science 249:
386−90(1990)、Renschlerら、Proc.Natl.Acad.S
ci.USA 91:3623−3627(1994)、およびAlexeyら、Pro
c.Natl.Acad.Sci.USA 94:1663−1668(1997)を参
照されたい。ファージを用いた実験から、ファージ・ペプチドRRTK(配列番号1、上
記参照)は受容体媒介性エンドサイトーシスを介してβTC−3細胞により特異的に取り
込まれるが、その内部移行効率は低い(これはその低親和性結合(ほぼマイクロモルの程
度)に起因し得る)ことが分かった。レポーター・タグ(FITCまたはビオチン)と共
にこのペプチドの複数(4もしくは8個)のコピーを有する一連の多量体(図1および2
)を構築した。
【0148】
本発明の多量体トランスポーターペプチドは、式I:
[P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z (I)
によって表すことができ、式中、Pは前述のコンセンサスモチーフ(XmRXoRXn)
、(XmRRRXn)、(XmRRXRXn)および(XmRXRRXn)のトランスポ
ーターペプチドである。例えば、このペプチドは、配列X1−X2−X3−X4を含むこ
とができ、式中、X1およびX4はRもしくはKとすることができ、X1およびX4は任
意のアミノ酸とすることができる。vによって表される所与のトランスポーターペプチド
内のペプチド数は、2〜8以上の整数とする。任意の所与の多量体トランスポーターペプ
チドにおいて、それぞれのペプチドは互いに同じか、異なるものとすることができる。好
ましいペプチドとしては、例えば、表5に挙げたものがある。
【0149】
ペプチドとコアとの間のスペーサーは存在していてもしていなくてもよく、従って、x
は1または0である。好ましいスペーサーは可動性、両親媒性、非免疫原性、およびタン
パク質分解酵素に対し非感受性であり、例えば、スクシンイミジル−PEG(succ−
peg)などのポリエチレングリコール(PEG)が挙げられる。
【0150】
企図されるコア部分としては、表7に記載したポリリジン(K)コアが挙げられ、また
、組み込まれたリンカーを有するものも挙げられる。選択されるコアにより、所与の結合
体内に存在するペプチドの数が決まる。代表的なコアとしては、C4−K2K−K(su
cc−peg−S)−アミド、C−GGG−[K(C)]3−K(succ−peg−S
)−アミド、(NH2OCH2CO)4−K2K−K(succ−peg)−アミドおよ
び(NH2OCH2CO)8−K4K2K−K(GGG)−アミドが挙げられる。リンカ
ーおよびレポーター基は存在していてもしていなくてもよく、従って、yおよびzは、式
Iにおいて独立に1もしくは0である。
【0151】
リンカーは存在していてもしていなくてもよい(即ち、yは1もしくは0である)。リ
ンカーとしては、コアをレポーターに接続する部分が挙げられ、例えば、succ−PE
GもしくはNH2OCH2CO−Lysとすることができる。
【0152】
レポーター基は存在していてもしていなくてもよい(zは1もしくは0である)。レポ
ーター基は検出可能な任意の基である。例としては、放射性同位体、蛍光性部分、リン光
性部分、化学発光性部分、および量子ドットが挙げられるが、これらに限定されるもので
はない。その他のレポーター基としては、ビオチン、シンテイン、ヒスチジン、赤血球凝
集素、mycもしくはフラッグ・タグが挙げられる。本明細書において用いる代表的な蛍
光レポーター基はFITC(フルオレセインイソチオシアネート)である。
【0153】
代表的な多量体トランスポーターペプチドは図1および図2に示した。
【0154】
多量体トランスポーターペプチド・ユニットもしくは結合体においては、多量体トラン
スポーターペプチドを、1つ以上のエフェクターと融合させる。このエフェクターは、D
NA、RNA、タンパク質、ペプチド、または薬学的に活性な薬剤、例えば、毒素、抗生
物質、抗病原体剤(antipathogenic agent)、抗原、抗体、抗体断
片、免疫調節剤、酵素もしくは治療剤などを含む任意の適切な分子とすることができる。
2つ以上のエフェクターが存在する場合、これらのエフェクターは同一もしくは異なるも
のとすることができる。
【0155】
従って、本発明の多量体結合体は、式(II):
([P]v−[スペーサー]x−コア−[リンカー]y−[レポーター]z)・E
(II)
によって表すことができ、式中、P、コア、スペーサー、リンカー、レポーター、v、x
、yおよびzは前記の通りであり、Eはエフェクターである。共有結合により融合させた
結合体では、化学結合の法則通りに、エフェクターはコア、もしくはリンカー、またはこ
れらの両方に結合させることができる。
【0156】
(ペプチド合成)
ペプチド配列RRTK(P1、配列番号1)を、膵β細胞に結合させるためのペプチド
コンセンサスモチーフX1−X2−X3−X4(ここで、X1およびX4はRもしくはK
とすることができ、X1およびX4は任意のアミノ酸とすることができる)を明らかにす
るファージ・ディスプレイ実験から得た。表6に示されるように修飾したペプチドを、B
oc化学により0.5mmolのBoc−Lys(CIZ)−PAM樹脂を用いて手動で
合成した。イタリック体は、対応するタンパク質配列には存在しないリンカー残基および
スペースを示す。この実験に用いたいくつかのコア構造を表7に示す。合成を、Boc化
学により0.5mmolのメチルベンズヒドリルアミン樹脂(MBHA樹脂)を用いて行
った。
【0157】
ペプチドは全て、(5%のp−クレゾールの存在下に0℃で1時間)HFを作用させて
樹脂から切断し、冷エーテルで沈殿させ、50%アセトニトリルで抽出し、ろ過して凍結
乾燥した。粗製ペプチドを分取HPLCにより精製し、ESIMSにより特性を決定した
。
【0158】
【表6】
【0159】
【表7】
【0160】
(フルオレセイン・リンカーの調製)
フルオレセイン・リンカーNH2OCH2CO−Lys(FITC)−OHを、オフォ
ード(Offord)の方法により調製した。Offordら、Methods Enz
ymol.287:348−69(1997)を参照されたい。簡単に言えば、2mmo
lのBoc−アミノオキシアセチル(Boc−AOA−OSu)および1.2mmolの
Nε−(TFA)−Lysを3mlのDMSO(ジメチルスルホキシド)溶液に加え、N
−エチルモルホリンをpH値が8〜9となるまで加えた。室温で15時間、次いで37℃
で1時間インキュベートした後、絶えず撹拌しながら氷酢酸を注意深く加えることによっ
てpH値を3.0に下げた。生成物[Boc−AOA−Lys(TFA)−OH]を、分
取スケールのHPLCにより単離し、次いで乾燥した。この乾燥した化合物に4mlの水
、次いで0.44mlのピペリジン(最終濃度1M)を加え、これを室温で4時間インキ
ュベートした後、氷上でpHを3.0に調整した。次に、この混合物を0.1%のTFA
10mlで希釈した。分取HPLCにより脱保護物質(Boc−AOA−Lys−OH)
を単離し、再度乾燥した。この乾燥物質を300μlのN,N−ジメチルホルムアミド(
DMF)に溶かし、40mgのフルオレセインイソチオシアネート(FITC)を加え、
得られた混合物をN−エチルモルホリンでpH8.0に調整した。暗所で15時間インキ
ュベートした後、生成物を、メタノール/CH3Cl(1:1、v/v)で平衡させたシ
リカ・カラム[Kieselgel 60(フルカ・ケミー社(Fluka Chemi
e)、ブックス(Buchs)、スイス)、1.5×20cm]を用いたクロマトグラフ
ィーにより精製した。過剰のFITCを貫流(flow−through)画分に溶出さ
せ、予測される生成物を含む第二の黄色画分をメタノール/CH3Cl(4:1、v/v
)で溶出させた。回転式蒸発により溶媒を除去した後、この乾燥化合物Boc−AOA−
Lys(FITC)−OHを(20mg/ml以下の濃度の)TFA中で脱保護し、室温
で45分間置いておいた。次に、TFAの大部分を蒸発させ、凍結乾燥により乾燥を完了
させた。最終生成物[AOA−Lys(FITC)−OH]は、分取HPLCにより単離
し、ESI−MSにより特性を決定した。
【0161】
(コアへのペプチドリガンドのアルキル化)
0.9μmolのCys−コア・ペプチド(1.19mg)を50μlのアセトニトリ
ルおよび100μlの水に溶かした(ペプチドは完全には溶けない)。ブロモアセチル−
ペプチドの新鮮溶液(pH7.0の0.1Mリン酸ナトリウム緩衝液の3.6μmol溶
液)を調製した。次いで、2つの溶液を室温の暗所で混合することによりアルキル化を開
始させた。40分後に生成物を半分取RP−HPLCにより精製し、凍結乾燥し、そして
特性を決定した。
【0162】
(過ヨウ素酸酸化)
過ヨウ素酸酸化を行うため、0.375μmolのペプチド(表6のペプチド3もしく
は4、または表8に挙げたアルキル化ペプチド)を433μlアセトニトリルおよび1.
26mlイミダゾール緩衝液(50mM、pH6.95、塩化物対イオン)に溶かした。
メチオニン(113μl、200mM)を加えた。次に、17μlのNaO4(0.1M
水溶液)を加えて酸化を開始させた。5分後、40μlのエチレングリコール(500m
M水溶液)を加えることにより反応を停止させた。得られた生成物は分取HPLCにより
精製し、質量分析法により特性を決定した。
【0163】
【表8】
【0164】
(オキシム化)
次に、過ヨウ素酸酸化により得られたアルデヒドをコア3もしくは4(各コアにAOA
基を含む)、またはAOA−Lys(FITC)−OHと反応させてオキシムを形成した
。図1および2に示したオキシム1〜6をこうして作製した。また、表9に予測および実
測質量分析データを示した。図1に示されるように、オキシム1〜3については、コアの
分枝Lys残基を、αアミノ基とεアミノ基との両方において、スペーサーありまたはな
しで、RRTK(配列番号1)ペプチドのN末端でアシル化した。スペーサーと共に、レ
ポータ・タグFITCを各構築体のコアに導入した。図2のオキシム4〜6では、コアの
分枝Lys残基を、αアミノ基とεアミノ基との両方において、スペーサーとともにRR
TK(配列番号1)ペプチドのN末端でアシル化した。スペーサーと共に、レポータ・タ
グのビオチンを各構築体のコアに導入した。
【0165】
【表9】
【0166】
AOALys(FITC)−OHでオキシム化するために、0.129μmolの酸化
構築物を5μlのアセトニトリルおよび200μlのNaOAc緩衝液(酢酸0.57m
lおよび水100mlの混合物と、酢酸ナトリウム0.82gを水100mlに溶かした
溶液とをpH4.0となるように混合した溶液)に溶解させた。この混合物に0.516
μmolのAOA−Lys(FITC)−OH(100μlのアセトニトリルおよび10
0μlのNaOAc緩衝液に8.8mg/mlの濃度で溶かしたもの)を加えた後、酢酸
を最終濃度が4%となるよう加えた。コア3もしくは4をオキシム化するために、酸化し
たペプチド3もしくは4の3.8μmolを100μlのアセトニトリルおよび200μ
lのNaOAc緩衝液に溶解させた。この溶液に0.475μmolのコア3もしくは0
.237μmolのコア4を加えた後、酢酸を最終濃度が4%となるよう加えた。室温で
24時間反応させた後、生成物を半分取RP−HPLCで精製し、質量分析により特性を
決定した。
【0167】
4本の分枝のそれぞれの末端にRRTK(配列番号1)ペプチド(ペプチド1)のコピ
ーを有する円形のトリリジン・コア(コア1)にオキシム1(図1)を形成させた。各ペ
プチドのN末端を、ブロモアセチル基とチオール基との特異的な反応により生じるチオエ
ーテル結合を介して結合させた。また、このトリリジン・コアにはスクシンイミジル−P
EGスペーサー、および酸化を受けてアルデヒドを形成することができるセリンを含ませ
たので、レポーター基アミノオキシアセチル−Lys(FITC)を、オキシム形成を介
してこのテトラマーに導入した。HPLCおよびESI−MS分析により、目的生成物で
あることが示された:質量分析データ:Mr実測値4289.31、Mr理論値4289
.04。オキシム1は酸性および中性pHで24時間安定であった。
【0168】
オキシム2および3(図1)を、同様にして形成したが、オキシム3ではRRTKペプ
チド(配列番号1)とコアとの間に2ユニットのPEGスペーサーを配置し、オキシム2
ではRRTKペプチド(配列番号1)のN末端はコア1ではなくコア2と結合させた。質
量分析データ:オキシム2:Mr実測値4459.48、Mr理論値4460.19;オ
キシム3:Mr実測値6708.29、Mr理論値6708.00。
【0169】
オキシム4(図2)を、オキシム3と同様にして合成したが、FITC基をレポーター
とするのではなく、ビオチン基をトリリジン・コアのアミド結合を介して直接結合させた
。質量分析データ:Mr実測値6160.0、Mr理論値6160.3。オキシム5はオ
キシム4と同様であったが、RRTK(配列番号1)残基は全てD型であった。質量分析
データ:Mr実測値6158.6、Mr理論値6160.3。
【0170】
オキシム6を、オキシム4と同様に合成したが、RRTK(配列番号1)ペプチドのコ
ピーを4個ではなく、8個含む。質量分析データ:Mr実測値11646.1、Mr理論
値11643.6。
【0171】
(免疫細胞化学および蛍光による検討)
前述の濃縮スキームによって単離した単一ファージを増幅させた。このファージ・ペプ
チドおよびオキシム1〜6のうちの1種を培養培地中の細胞に加え、24時間インキュベ
ートした。次に、培地を洗い流し、細胞を冷メタノール−アセトン(1:1)で5分間固
定した。ファージ・キャプシドに対する抗体もしくはビオチン(ペプチド)に対する抗体
を、フルオレセイン結合(ファージ)もしくはテキサス・レッド標識(ペプチド)二次抗
体と共に用いた。古典的な蛍光顕微鏡による検討および共焦点顕微鏡によるアッセイを実
施した。
【0172】
まず、オキシム1〜3のβTC−3細胞内への侵入について調べた。蛍光による検討で
は、構築ブロックとして2ユニットのPEGスペースを有するRRTKペプチドの4つの
コピーを用いるテトラオキシム(樹状形)のオキシム3は、βTC−3細胞表面の受容体
に明確に結合し、細胞内へ侵入可能であることが明らかになった。オキシム3が優れてい
るのは、細胞表面の受容体への結合を増強するPEGスペーサーの可動性に起因し得る。
【0173】
次いで、オキシム3に類似のオキシム4〜6を合成し、結合親和力および侵入効率につ
いてアッセイした。RRTK(配列番号1)ペプチドのコピー数を、オキシム6では8個
まで増やし、D−ペプチドが高い安定性を有し、免疫原性がより低いことが実証されてい
るので(Ivanenkovら、Biochem.Biophys.Acta 1448
:450(1999)を参照)、オキシム5では上記ペプチドをD−エナンチオマー型に
変更した。顕微鏡による検査では、8個のペプチドコピーを有するオキシム6(オクタオ
キシム)は細胞内へ効率的に侵入することができることが明らかになった。ビオチンに対
するテキサス・レッド標識二次抗体によって、このオクタオキシムは、「空胞状」構造体
としての細胞の内側に、または細胞膜に局在することが明らかになった。従って、ペプチ
ドコピー数を最大8個まで増加させた多量体は、結合ペプチドの向上した結合活性および
移送効率を示す。
【特許請求の範囲】
【請求項1】
明細書に記載のトランスポーターペプチド。
【請求項1】
明細書に記載のトランスポーターペプチド。
【図1】

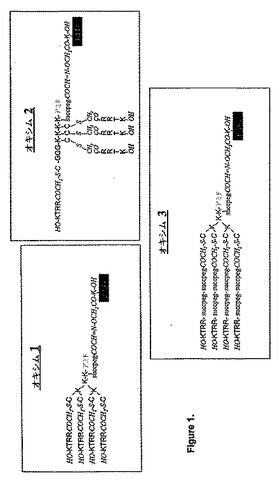
【図2】
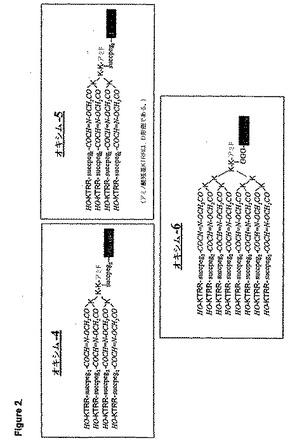
【図2】
【公開番号】特開2010−24250(P2010−24250A)
【公開日】平成22年2月4日(2010.2.4)
【国際特許分類】
【出願番号】特願2009−252478(P2009−252478)
【出願日】平成21年11月2日(2009.11.2)
【分割の表示】特願2004−510837(P2004−510837)の分割
【原出願日】平成15年6月6日(2003.6.6)
【出願人】(507045085)ザイジェン エス.アー. (17)
【Fターム(参考)】
【公開日】平成22年2月4日(2010.2.4)
【国際特許分類】
【出願日】平成21年11月2日(2009.11.2)
【分割の表示】特願2004−510837(P2004−510837)の分割
【原出願日】平成15年6月6日(2003.6.6)
【出願人】(507045085)ザイジェン エス.アー. (17)
【Fターム(参考)】
[ Back to top ]