
生物学的材料およびそれらの使用法
【課題】生物学的材料およびそれらの使用法を提供する。
【解決手段】本発明は、標的細胞の細胞表面抗原に選択的に結合する抗体分子を含めた結合分子を提供し、結合分子は、細胞表面抗原との結合により標的細胞のアポトーシスを誘導する。また、アポトーシス誘導の方法およびアポトーシス誘導のための医薬組成物、ならびにその使用も提供される。
【解決手段】本発明は、標的細胞の細胞表面抗原に選択的に結合する抗体分子を含めた結合分子を提供し、結合分子は、細胞表面抗原との結合により標的細胞のアポトーシスを誘導する。また、アポトーシス誘導の方法およびアポトーシス誘導のための医薬組成物、ならびにその使用も提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アポトーシス誘導に関与する分子、アポトーシス誘導のための方法および医薬組成物、ならびにその使用に関する。
【背景技術】
【0002】
抗体は最近、癌を標的とするタンパク質治療法として選択されるようになったが、他の適応症の治療に関しても選択されるようになった(Brekkeら、Nat Rev Drug Discov、2003年:52〜62頁)。抗体工学の到来により、ヒトの性質およびより大きな多様性による免疫原性の減少ならびに特異性および親和性の増強を示す合成ファージライブラリーからヒト抗体を作出するツールが提供されている(Weinerら、Nat Biotechnol、2005年;23:556〜7頁)。未処置ライブラリーは、免疫化および新規ライブラリーの再構築とは独立して、自己抗原などの何らかの特異性に関して抗体の単離に使用できるため、特に魅力的である(Griffithsら、Embo J、1993年;12:725〜34頁)。現在まで、細胞表面受容体は、小型分子阻害剤および抗体などの現代の治療薬によって標的化された最も成功した抗原群を構成している。特に興味深いのは、ユニークな発現をするか、または標的細胞上に増加した発現レベルを示し、さらに、細胞に対する死または生存のシグナルを中継することのできる細胞表面受容体である。このような内因性のシグナル伝達特性を有する差異的発現受容体により、微生物に感染した、形質転換した、または機能不良の細胞の抗体ベースの標的化が可能になる。
【0003】
腫瘍の治療に関して、標的腫瘍細胞にアポトーシスを誘導する一方、正常組織を避ける能力を有する抗体は特に興味深い。このようないくつかの抗体が使用されており、米国食品医薬品局(FDA)に登録されており、例えば、リンパ腫に対する(CD20を標的にするリツキシマブ)または乳癌に対する(Her-2およびEGFRをそれぞれ標的にするトラスツズマブまたはセツキシマブ)従来の癌治療の代替法を提供する。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】WO 2004/023140
【非特許文献】
【0005】
【非特許文献1】Brekkeら、Nat Rev Drug Discov、2003年:52〜62頁
【非特許文献2】Weinerら、Nat Biotechnol、2005年;23:556〜7頁
【非特許文献3】Griffithsら、Embo J、1993年;12:725〜34頁
【非特許文献4】Millerら、N Engl J Med、1982年;306:517〜22頁
【非特許文献5】Riechmannら、Nature、1988年;332:323〜7頁
【非特許文献6】Kwakら、N Engl J Med、1992年;327:1209〜15頁
【非特許文献7】Suarezら、Mol Immunol、2004年;41:519〜26頁
【非特許文献8】Soderlindら、Nat Biotechnol、2000年;18:852〜6頁
【非特許文献9】Clackson Tら、Nature、1991年8月15日;352(6336):624〜8頁
【非特許文献10】Marks JDら、J Mol Biol. 1991年12月5日;222(3):581〜97頁
【非特許文献11】Weng Sら、Proteomics、2002年1月;2(1):48〜57頁
【非特許文献12】Nord Kら、Nat Biotechnol、1997年8月;15(8):772〜7頁
【非特許文献13】Hogbom Mら、Proc Natl Acad Sci U S A、2003年3月18日;100(6):3191〜6頁
【非特許文献14】Borrebaeck CAおよびCarlson R、Curr Opin Pharmacol.2001年8月;1(4):404〜8頁
【非特許文献15】Nagyら、J Mol Med、2003年;81:757〜65頁
【非特許文献16】Smithら、J Clin Pathol、1990年;43:893〜900頁
【非特許文献17】Cosimiら、J Immunol、1990年;144:4604〜12頁
【非特許文献18】Kavanaughら、Arthritis Rheum、1994年;37:992〜9頁
【非特許文献19】Haugら、Transplantation、1993年;55:766〜72頁
【非特許文献20】Huangら(1993) Hybridoma 12、661〜75頁
【非特許文献21】Huangら、(1995) Cancer Res 55、610〜6頁
【非特許文献22】Smallshawら、(2004) J Immunother 27、419〜24頁
【非特許文献23】Maruoら、(2002) Int J Cancer 100、486〜90頁
【非特許文献24】Rosetteら、(2005) Carcinogenesis 26、943〜50頁
【非特許文献25】Sunら、(1999) J Cancer Res Clin Oncol 125、28〜34頁
【非特許文献26】Grotheyら、(1998) Br J Cancer 77、801〜7頁
【非特許文献27】Wangら、(2005) Int J Cancer 27、419〜24頁
【非特許文献28】Rocheら、(2003) Thromb Haemost 89、1089〜97頁
【非特許文献29】Aalinkeelら、(2004) Cancer Res 64、5311〜21頁
【非特許文献30】Nagyら、Nat Med、2002年;8:801〜7頁
【非特許文献31】Uyttenhoveら、J.Exp.Med.、1983年;157:1040〜52頁
【非特許文献32】Kennedyら、Br J Haematol、2002年;119:412〜6頁
【非特許文献33】Weinerら、J Immunol、1989年;142:343〜51頁
【非特許文献34】Baiら、J.Clin.Invest.、2003年;111:1487〜96頁
【非特許文献35】Robertら、Lancet Oncol、2005年;6:491〜500頁
【非特許文献36】Harlowら(編)、Antibodies A Laboratory Manual;Cold Spring Harbor Laboratory:コールドスプリングハーバー、ニューヨーク(1988)、第6章
【非特許文献37】Daibataら、Cancer 1989年;64:1248〜53頁
【非特許文献38】Saltmanら、Blood 1988年;72:2026〜30頁
【非特許文献39】Menezesら、Biomedicine 1975年;22:276〜84頁
【非特許文献40】Ceruttiら、J Immunol 1998年;160:2145〜57頁
【非特許文献41】Soderlindら、Nat Biotechnol、2000年;18:852〜6頁
【非特許文献42】Hallborn Biotechniques、2002年;補遺:30〜7頁
【非特許文献43】Norderhaugら、J Immunol Methods、1997年;204:77〜87頁
【非特許文献44】Edvardssonら、Electrophoresis、1999年;20:935〜42頁
【非特許文献45】Rosenthalら、Anal Biochem、1967年;20:525〜32頁
【非特許文献46】BylundおよびYamamura、Methods in Neurotransmitter Analysed、ニューヨーク、Raven Press社、1990年
【非特許文献47】Marquardt、J.Soc.Indust.Appl.Math、1963年;11:431〜41頁
【非特許文献48】Brixら、J.Clin.Invest.1998年;102:283〜93頁
【非特許文献49】Kimら、Mol Biol Cell、2004年;15:420〜34頁
【非特許文献50】Marlinら、Cell、1987年;51:813〜9頁
【非特許文献51】Rothleinら、J Immunol、1994年;152:2488〜95頁
【非特許文献52】Vyth-Dreeseら、Blood、1995年;85:2802〜12頁
【発明の概要】
【発明が解決しようとする課題】
【0006】
アポトーシス誘導作用を有する他の抗体も、現在、臨床的に開発されている。しかし、たとえこれらの抗体が、患者または動物試験において有益な効果を示すとしても、臨床的に満たされていない必要性が依然として存在している。
【0007】
B細胞腫瘍を標的にする抗イディオタイプの免疫グロブリンは、ヒトにおいて実施された最初のモノクローナル抗体療法であった(Millerら、N Engl J Med、1982年;306:517〜22頁)。受動的抗体投与(Riechmannら、Nature、1988年;332:323〜7頁)、または患者自身の主要免疫グロブリンタンパク質による活性ワクチン療法(Kwakら、N Engl J Med、1992年;327:1209〜15頁)のこのような手段による腫瘍細胞の破壊により、それ以来、種々のB細胞悪性疾患に罹っている患者における腫瘍退縮または腫瘍静止をもたらすことが実証されている。最近の報告には、マウスの抗体産生が欠失していて選択されたヒト抗体鎖の遺伝子座を発現する形質転換マウスを用いてヒト抗イデオタイプ完全抗体の作製が記載されている(Suarezら、Mol Immunol、2004年;41:519〜26頁)。
【0008】
本発明においては、細胞全体の形態における標的細胞抗原、および膜小胞の形態における過剰のサブトラクター細胞抗原を、同時に無処置n-CoDeR(登録商標)抗体ファージライブラリー(WO 2004/023140; Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)に曝露し、回収して、Bリンパ腫標的細胞に対して優れた選択性を有する抗体断片を続いて試験する競合バイオパニング法が用いられている。さらに、選択された結合分子における機能性は、標的細胞にアポトーシスを誘導するが、非標的細胞には誘導しない被試験抗体の能力によって実証されている。
【0009】
同定された抗体特異性としては、HLA-DR/DP(本発明のB1抗体)および表面IgM(本発明のC11抗体)、ならびにICAM-1(本発明のB11抗体)、以前はアポトーシス誘導に関連づけられていない接着分子が挙げられる。単離抗体は、ナノモル以下からナノモルの範囲に親和性を有しているため、それらは直接、標的抗体療法にとっての可能な選択肢となる。
【課題を解決するための手段】
【0010】
本発明の第1の態様において、
a.細胞表面抗原ICAM-1を提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する1種または複数の結合分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法が提供される。
【0011】
結合分子は、抗体分子であることが好ましい。
【0012】
本発明の第2の態様において、
a.細胞表面抗原HLA-DR/DPおよび/または表面IgMを提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合し、HLA-DR/DPおよび/または表面IgMとの結合により標的細胞のアポトーシスを誘導する1種または複数の抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の抗体分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法が提供される。
【0013】
本発明の第3の態様において、細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する結合分子が提供される。あるいは、結合分子は、細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合する抗体分子である。
【0014】
ICAM-1は、指定されたCD54でもあるが、本出願の目的にはICAM-1が用いられる。
【0015】
結合分子は、多くのライブラリーで広範囲に使用されている、抗体由来で、抗体スカフォードに基づくものであり得る[Clackson Tら、Nature、1991年8月15日;352(6336):624〜8頁、Marks JDら、J Mol Biol. 1991年12月5日;222(3):581〜97頁]が、結合分子は、フィブロネクチンスカフォード[Weng Sら、Proteomics、2002年1月;2(1):48〜57頁]およびタンパク質Aスカフォード[Nord Kら、Nat Biotechnol、1997年8月;15(8):772〜7頁、Hogbom Mら、Proc Natl Acad Sci U S A、2003年3月18日;100(6):3191〜6頁]などの他の分子スカフォードに由来するものであってもよい。これらのスカフォードの各々は、適用に依存してそれらの利点を有し得るが、一例として、抗体スカフォードは、天然の可変性から識別できる可変性を作出する目的で、有利に使用できる。
【0016】
抗体の基本的構造、最も一般的に用いられるスカフォードは、十分に認識されている。原則として、2つのシートに配列されたベータ鎖を含むフレーム構造が、可変ループのセット、抗原分子に結合する能力を有する、いわゆる相補性決定領域(CDR)を提示する。抗体はスカフォード構造において変わり得るが、最も大きな変動はCDRにおいて見られる。抗体内抗体間の大きな変動が、原則的にはあらゆるタイプの分子構造と特異的様式で相互作用する能力の基礎である。この能力によって、抗体は、研究、疾患の診断/予後における適用性を有する特異的結合剤の創製のため、また、一定の標的構造に特異的な治療薬として、広範囲に用いられている[Borrebaeck CAおよびCarlson R、Curr Opin Pharmacol.2001年8月;1(4):404〜8頁]。
【0017】
本発明に有用な他の非抗体結合分子は、安定性の程度が高く、しかも一定の位置に変動性を導入することを可能にするスカフォード構造を有するものである。他の結合分子の一例は、変動性を許容するフィブロネクチンドメインおよび58個のアミノ酸の大型タンパク質Aドメインである。ある程度の変動を許容する他の分子フォールドもある。このような例としては、腫瘍組織適合性複合体(MHC)クラスIおよびII分子が挙げられ、最近、新規クラスの分子、いわゆるデフェンシン類が、基本構造は同様であるが、遺伝子ファミリーメンバー内メンバー間の大きな配列変動性を有したままであることが確認されており、分子多様性を有するためのスカフォードとして好適であることが示されている。また、標的分子がICAM-1の場合は、天然リガンド(複数可)、例えば、LFA-1またはそれらの組換え変異体が、標的細胞におけるアポトーシスを誘導することのできる特異的結合分子を構成できる。
【0018】
さらに、結合分子は、標的細胞の細胞表面ICAM-1に選択的に結合し、その結合により標的細胞のアポトーシスを誘導する任意の分子であり得る。
【0019】
結合分子は抗体分子であることが好ましい。
【0020】
一実施形態において、細胞表面抗原はICAM-1である。
【0021】
スクリーニングによって、以前はアポトーシスに関連づけられておらず、細胞における内因性の負のシグナル伝達特性に帰せられていなかった受容体であるICAM-1に特異的な抗体(B11)が回収された。
【0022】
それぞれの性質についての前もっての知見に関わらず、またその知見なしに標的細胞と非標的細胞との間で差異的に発現した全ての表面受容体に対する特異性を単離するために設計されているスクリーニングの直接的結果が、アポトーシス誘導性分子としてのICAM-1の同定であった。ICAM-1誘導細胞死は、ミトコンドリア膜の脱分極を含む活性アポトーシス過程として検証された。ミトコンドリア膜の脱分極は、カスパーゼ依存性アポトーシスとカスパーゼ非依存性アポトーシス双方に関して以前に記載されている(Nagyら、J Mol Med、2003年;81:757〜65頁)。
【0023】
本発見によってさらに、B11抗体によって結合されたエピトープは、種々の出所のBリンパ腫組織に発現し、静止末梢血の白血球に比較して、一定のBリンパ腫細胞でアップレギュレートされることが示されている。Bリンパ腫細胞に加えて、ICAM-1を発現する癌腫細胞もまた、インビトロでICAM-1特異的なB11抗体に供されるとアポトーシスを受けることは重要である(実施例6を参照)。
【0024】
正常なヒト組織上ではICAM-1の発現が制限されていることが、先行の研究で実証されている(Smithら、J Clin Pathol、1990年;43:893〜900頁)。ICAM-1は、細胞対細胞の接着に関与し、その受容体、LFA-1に対する結合を介して、免疫応答および炎症に重要な役割を果たしている。病理的な免疫応答および炎症に干渉するために、ICAM-1に特異的な抗体が用いられている。カニクイザルにおけるマウス抗ICAM-1 mAbのインビボ投与(Cosimiら、J Immunol、1990年;144:4604〜12頁)、またはリウマチ様関節炎を患っているヒト患者または腎臓移植を受けている患者における臨床試験での使用でも、顕在的な毒性は見られなかった(Kavanaughら、Arthritis Rheum、1994年;37:992〜9頁;Haugら、Transplantation、1993年;55:766〜72頁)。
【0025】
ICAM-1標的化がアポトーシスを導くことができるという新たな発見により、抗原を発現するという条件での種々の出所の癌の治療用抗体などのICAM-1特異的結合分子の使用の可能性が示される。
【0026】
ICAM-1の発現に基づき、B11などのアポトーシス誘導性抗ICAM-1抗体によって治療できる可能性を有する癌のタイプとしては: Bリンパ腫、骨髄腫(Huangら(1993) Hybridoma 12、661〜75頁; Huangら、(1995) Cancer Res 55、610〜6頁; Smallshawら、(2004) J Immunother 27、419〜24頁)、胃癌(Maruoら、(2002) Int J Cancer 100、486〜90頁)、乳癌(Rosetteら、(2005) Carcinogenesis 26、943〜50頁)、肝臓癌(Sunら、(1999) J Cancer Res Clin Oncol 125、28〜34頁)、肺癌(Grotheyら、(1998) Br J Cancer 77、801〜7頁)、黒色腫(Wangら、(2005) Int J Cancer 27、419〜24頁)、膀胱癌(Rocheら、(2003) Thromb Haemost 89、1089〜97頁)および前立腺癌(Aalinkeelら、(2004) Cancer Res 64、5311〜21頁)が挙げられる。ICAM-1の発現はまた、ICAM-1 特異的抗体を用いて転移過程に介入する可能性を指摘している(Maruoら、2002年)、(Rosetteら、2005年)、(Sunら、1999年)、(Grotheyら、1998年)、(Aalinkeelら、2004年)によって実証されているように、腫瘍転移においても確認されている。
【0027】
さらなる実施形態において、細胞表面抗原はHLA-DR/DPである。
【0028】
HLA-DR/DPは通常、例えば、B細胞上に存在し、Bリンパ腫細胞上でアップレギュレートされるのを見ることができる。
【0029】
現在までに、3種の異なるHLA-DR特異的モノクローナル抗体が臨床相試験に入っている。その中で一番最近になって加わったのは、n-CoDeR(登録商標)に比較して、同様なサイズだが、精製抗原上にパニングしている固体相を用いる無処置ファージライブラリーから単離された完全ヒトIgG41D09C3である(Nagyら、Nat Med、2002年;8:801〜7頁)。
【0030】
本発明において、多数のBリンパ腫細胞系において、速やかに、そして高い効力でアポトーシスを誘導する、HLA-DR/DPに特異的な新規なヒト抗体(B1)が同定され、それによって、HLA-DR/DPが、標的細胞におけるアポトーシスの誘導に関連していることが実証された。
【0031】
B1抗体は、種々の出所の多数のBリンパ腫細胞系に結合し(実施例1および表1を参照)、HLA-DR/DP抗原発現細胞にアポトーシスを誘導することが示された。さらに、B1抗体は、ラージBリンパ腫細胞系で試験した際、リツキシマブよりも高い効力を示した。B1抗体のIgG4フォーマットが使用された際は、これが特に明瞭であった(図8)。
【0032】
得られたデータによると、この抗体は、HLA-DR/DP発現Bリンパ腫細胞の治療に好適な特性を有する。また、リツキシマブによるリウマチ様関節炎およびSLEの標的化と同様に、B1抗体は、HLA-DR/DP発現B細胞が有害である障害における活性化B細胞の除去に有効であることを立証し得る。
【0033】
さらなる実施形態において、細胞表面抗原は表面IgMである。
【0034】
遊離形態におけるIgMは、大型の五量体構造として存在し、その高分子量によって、血管内に限定される傾向がある。
【0035】
単量体IgMは、Bリンパ球の細胞壁上に見ることができ、抗原認識に関する抗体受容体として働く。
【0036】
本発明のC11抗体は、Bリンパ腫細胞上に発現した表面IgMに結合すると、迅速で効率的な様式でアポトーシスを誘導する(実施例1および表1を参照)。Bリンパ腫の治療のために、以前臨床で用いられたイディオタイプ特異的な抗IgM抗体とは対照的に、C11抗体は、種々のドナーからのB細胞上に発現した非多型エピトープに結合し、したがって、Bリンパ腫のイディオタイプに関わりなく、Bリンパ腫を患っている患者の治療に好適である。
【0037】
特に、エフェクター分子、例えば、抗IgM抗体はまた、種々のイディオタイプのIgM発現細胞におけるアポトーシスを引き起こす、任意の種類の特異的結合分子であり得る。
【0038】
B1、B11およびC11 IgG誘導アポトーシスの動態は速やかであり、いくつかの細胞系においては、3時間後にすでに最高の効力が見られた。速やかなエフェクター機能は、例えば、腫瘍抗原の発現不足(Uyttenhoveら、J.Exp.Med.、1983年;157:1040〜52頁;Kennedyら、Br J Haematol、2002年;119:412〜6頁)またはエピトープ成熟(Weinerら、J Immunol、1989年;142:343〜51頁;Baiら、J.Clin.Invest.、2003年;111:1487〜96頁)から生じる腫瘍脱出の危険性を最小化し、治療期間および副作用を制限する可能性がある(Robertら、Lancet Oncol、2005年;6:491〜500頁)ため、治療の有効性にとって重要である。
【0039】
好ましくは、標的細胞は免疫細胞または上皮細胞であり、有利には、免疫細胞はBリンパ球である。
【0040】
好都合には、標的細胞は疾患に関連している。疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択されることが好ましい。
【0041】
有利には、疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0042】
本出願の定義の節で定義されるとおり、抗体分子という語句は、便宜上用いられ、他にある中でも、抗体、抗体断片、および抗体の誘導体を包含する。
【0043】
好都合には、抗体分子はIgGである。好ましくは、IgGは、IgG1、IgG2、IgG3またはIgG4のいずれかであり得るが、IgG1およびIgG4のいずれかである。好ましくは、抗体分子はヒト化抗体またはヒト抗体である。
【0044】
好都合には、本発明の結合分子または抗体分子は、図9から11の可変領域配列のいずれか一つの配列または機能的に等価なその相同体を有する。
【0045】
本発明の一実施形態において、結合分子または抗体分子は、図9の可変領域配列または機能的に等価なその相同体を有する。
【0046】
本発明のさらなる実施形態において、結合分子または抗体分子は、図10の可変領域配列または機能的に等価なその相同体を有する。
【0047】
本発明のさらなる実施形態において、結合分子または抗体分子は、図11の可変領域配列または機能的に等価なその相同体を有する。
【0048】
本発明の第4の態様において、前記請求項のいずれかに記載の結合分子または抗体分子をコードするヌクレオチド配列を有する核酸が提供される。
【0049】
好都合には、核酸は、図9から11のいずれか一つのヌクレオチド配列を有する。
【0050】
本発明の第5の態様において、標的細胞の破壊を必要とする疾患の診断および/または治療および/または予防における、本発明の第1の態様また第2の態様に記載の結合分子または抗体分子の使用が提供される。また、標的細胞の破壊を必要とする疾患の治療および/または予防のための薬剤の製造における、本発明の第1の態様また第2の態様に記載の結合分子または抗体分子の使用が提供される。
【0051】
好ましい一実施形態において、結合分子は抗体分子である。
【0052】
好都合には、治療される疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される。
【0053】
有利には、治療される疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0054】
本発明の一実施形態において、結合分子または抗体分子は、ICAM-1に特異的に結合し、および/または図10の配列を有し、上記に挙げられた疾患に関連して使用される。
【0055】
本発明のさらなる実施形態において、抗体分子は、HLA-DR/DPに特異的に結合し、および/または図9の配列を有し、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌の疾患に関連して使用される。
【0056】
本発明のさらなる実施形態において、抗体分子は、表面IgMに特異的に結合し、および/または図11の配列を有し、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌の疾患に関連して使用される。
【0057】
本発明の第6の態様において、本発明の結合分子または抗体分子と、薬学的に許容される担体、賦形剤または希釈剤とを含む医薬組成物が提供される。
【0058】
好ましい一実施形態において、結合分子は抗体分子である。
【0059】
本発明の第7の態様において、
(i)1種または複数の標的細胞を用意するステップと、
(ii)本発明の第1の実施形態に記載の1種または複数の結合分子または抗体分子を用意するステップと、
(iii)(i)の標的細胞を(ii)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと
を含む、標的細胞にアポトーシスを誘導するインビトロ方法が提供される。
【0060】
好ましい一実施形態において、結合分子は抗体分子である。
【0061】
好ましくは、ステップ(i)で用意される標的細胞は、免疫細胞または上皮細胞である。有利には、免疫細胞はBリンパ球である。
【0062】
好都合には、該標的細胞は疾患に関連しており、該疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される。
【0063】
有利には、疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0064】
使用される用語の意味
用語「抗体分子」は、抗体、抗体断片、または抗体の誘導体のいずれか一つを称することとする。これは、野生型抗体、合成抗体、組換え抗体または、それだけに限定されないが、免疫グロブリンの軽鎖および/または重鎖可変領域および/または定常領域のファージディスプレーによって製造される一本鎖修飾抗体分子などの抗体ハイブリッド、または当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる他の免疫相互作用分子を包含することが意図されている。
【0065】
用語「抗体断片」は、抗体、抗体断片、または抗体誘導体のいずれか一つを称することとする。これは、野生型抗体(すなわち、4本のポリペプチド鎖を含む分子)、合成抗体、組換え抗体または、それだけに限定されないが、免疫グロブリンの軽鎖および/または重鎖可変領域および/または定常領域のファージディスプレーによって製造される一本鎖修飾抗体分子などの抗体ハイブリッド、または当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる他の免疫相互作用分子を包含することが意図されている。
【0066】
用語「抗体誘導体」とは、当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる、抗体の断片(例えば、Fab断片またはFv断片)、または、他のペプチドまたはポリペプチド、大型担体タンパク質または固体支持体への該抗体の結合を促進させるために、1種または複数のアミノ酸または他の分子(例えば、中でも、アミノ酸、チロシン、リシン、グルタミン酸、アスパラギン酸、システインおよびそれらの誘導体、NH2-アセチル基またはCOOH-末端アミド基)を付加することによって修飾される修飾抗体分子などの任意の修飾抗体分子を称する。
【0067】
用語「ScFv分子」とは、VHおよびVLパートナードメインが、可撓的オリゴペプチドを介して結合している任意の分子を称する。
【0068】
用語「ヌクレオチド配列」または「核酸」または「ポリヌクレオチド」または「オリゴヌクレオチド」は、交換可能に用いられ、ヌクレオチドのヘテロポリマーまたはこれらのヌクレオチドの配列を称する。これらの語句はまた、一本鎖または二本鎖であり得、センス鎖またはアンチセンス鎖を表し得る、ゲノム起源または合成起源のDNAまたはRNA、ペプチド核酸(PNA)または任意のDNA様またはRNA様物質も称する。本明細書の配列において、Aはアデニンであり、Cはシトシンであり、Tはチミンであり、Gはグアニンであり、NはA、C、GまたはT(U)である。該ポリヌクレオチドがRNAである場合、本明細書に提供される配列内のT(チミン)は、U(ウラシル)で置換されることが考慮されている。一般に、本発明により提供される核酸セグメントは、ゲノムおよび短いオリゴヌクレオチドリンカーの断片から、または一連のオリゴヌクレオチドから、または個々のヌクレオチドから組み立てることができ、微生物もしくはウィルスのオペロン、または真核生物の遺伝子に由来する調節要素を含む組換え転写単位において発現され得る合成核酸を提供することができる。
【0069】
用語「ポリペプチド」または「ペプチド」または「アミノ酸配列」とは、オリゴペプチド、ペプチド、ポリペプチドまたはタンパク質の配列またはそれらの断片、ならびに天然または合成分子を称する。ポリペプチドの「断片」、「部分」、または「セグメント」は、少なくとも約5個のアミノ酸、好ましくは、少なくとも約7個のアミノ酸、より好ましくは、少なくとも約9個のアミノ酸、最も好ましくは、少なくとも約17個以上のアミノ酸のアミノ酸残基のひと続きである。活性であるためには、いずれのポリペプチドも、生物学的および/または免疫学的活性を示すための十分な長さを有する必要がある。
【0070】
本明細書で用いられる用語「精製した」または「実質的に精製した」は、指示された核酸またはポリペプチドが、他の生物学的高分子、例えば、ポリヌクレオチド、タンパク質などの実質的な不在下に存在することを意味する。一実施形態において、ポリヌクレオチドまたはポリペプチドは、存在する指示された生物学的高分子(しかし、水、緩衝液、および他の小型分子、特に、1000ダルトン未満の分子量を有する分子は存在し得る)の少なくとも95重量%、より好ましくは、少なくとも99重量%を構成するように精製される。
【0071】
本明細書で用いられる用語「単離された」とは、天然源における核酸またはポリペプチドと共に存在する少なくとも1種の他の成分(例えば、核酸またはポリペプチド)から分離された核酸またはポリペプチドを称する。一実施形態において、核酸またはポリペプチドは、該核酸またはポリペプチドの溶液中に通常存在する溶媒、緩衝液、イオン、または他の成分がある場合、それらのみの存在下で見られる。用語「単離された」および「精製された」は、その天然源に存在する核酸またはポリペプチドを包含しない。
【0072】
本明細書に用いられる場合の用語「組換え」とは、ポリペプチドまたはタンパク質を称し、ポリペプチドまたはタンパク質が、組換え(例えば、微生物、昆虫、または哺乳動物)発現系に由来することを意味する。「微生物」とは、細菌または真菌(例えば、酵母)発現系において作製された組換えポリペプチドまたは組換えタンパク質を称する。生成物としての「組換え微生物」は、本質的に天然の内因性物質が無く、関連した天然グリコシル化が付随していないポリペプチドまたはタンパク質として定義される。多くの細菌、例えば、大腸菌の培養物中に発現するポリペプチドまたはタンパク質は、グリコシル化修飾が無く、酵母中に発現するポリペプチドまたはタンパク質は、哺乳動物細胞中に発現するものとは一般に異なるグリコシル化パターンを有する。
【0073】
用語「選択的結合」および「結合選択性」は、本発明の抗体の可変領域が、本発明のポリペプチドを排他的に認識して結合する(すなわち、ポリペプチドファミリーに見られる配列同一性、相同性、または類似性に関わらず、他の類似ポリペプチドから本発明のポリペプチドを識別できる)が、該抗体の可変領域の外側、特に、該分子の定常領域における配列との相互作用を介して、他のタンパク質(例えば、黄色ブドウ球菌のタンパク質AまたはELISA法における他の抗体)と相互作用できることを表す。本発明の抗体の結合選択性を判定するスクリーニングアッセイはよく知られており、当業界でルーチンに実施されている。このようなアッセイの包括的考察に関しては、Harlowら(編)、Antibodies A Laboratory Manual;Cold Spring Harbor Laboratory:コールドスプリングハーバー、ニューヨーク(1988)、第6章を参照されたい。本発明のポリペプチドの断片を認識し結合する抗体も、該抗体が上記に定義したとおり、本発明の完全長ポリペプチドに対して、一番に最も選択的であるという条件で、考慮されている。本発明の完全長ポリペプチドに対して選択的な抗体であれば、断片を認識する本発明の抗体は、タンパク質のファミリーに見られる固有な配列同一性、相同性、または類似性に関わらず、ポリペプチドの同じファミリーからポリペプチドを識別できる抗体である。
【0074】
用語「結合親和性」は、抗体分子と抗原との間の結合の強さの意味を含む。
【0075】
用語「免疫細胞」は、それだけに限定されないが、B細胞およびT細胞などの宿主の免疫または炎症応答に関与している任意の細胞を意味する。
【0076】
用語「上皮細胞」は、上皮の細胞を意味する。上皮は、細胞の層からなる組織である。上皮は、身体の内張りの内部(例えば、血管の内側を内張りしている内皮)または外部(例えば、皮膚)の開放表面に見ることができる。
【0077】
口腔および体腔の内側が粘膜に裏打ちされているように、我々の皮膚の最外側層は扁平上皮細胞からなっている。他の上皮細胞は、肺、胃腸管、生殖管および尿管の内側を裏打ちし、エポクリン腺および内分泌腺を構成している。上皮細胞の機能としては、分泌、吸収および保護が挙げられる。上皮細胞は基底層上にある。
【0078】
次に、本発明の一定の好ましい態様を具体化している実施例を、下記の図を参照にして記載する。
【図面の簡単な説明】
【0079】
【図1】図1A及びBは、差異的全細胞/細胞膜小胞バイオパニングによって単離されたscFvが、標的細胞高特異性を示す図である。差異的バイオパニングによって単離されたscFvクローンを、大腸菌TOP10細胞中に発現させ、ラモス細胞またはジャーカット細胞および一次スクリーニングのために発現させたscFvクローンと共にインキュベートした。結合したscFvを、抗His MAb、およびCy5-抗マウスポリクローナルAbによって検出した。細胞結合を、FMAT Macroconfocal High Throughput Screening機器において検出した。細胞結合は、標的ラモス細胞(Y軸)対非標的ジャーカット細胞(X軸)に対する平均蛍光強度として示した。図1Cは、差異的全細胞/細胞膜小胞バイオパニングによって単離されたscFvが、標的細胞高特異性を示す図である。ラモス細胞(黒色バー)およびジャーカット細胞(白色バー)に対する7つのユニークなscFvクローンの結合性。対照scFv(ctrl)は、いずれの細胞にも結合しなかった。
【図2A】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2B】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2C】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2D】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2E】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図3】図3Aは、HLA-DR/DP、IgM、およびICAM-1を含む単離抗体の特異性を示す図である。50〜600×106ラージBリンパ腫細胞を、非イオン性洗浄剤、0.5%v/vでのトリトンX-100により溶解し、B1(レーン1)抗体およびB11(レーン2)抗体の完全ヒトIgG1フォーマット100μgによって免疫沈降させ、続いて、Protein A Sepharoseによって架橋した。C11(レーン3)20μgの免疫沈降に、50×106細胞からのラモスBリンパ腫細胞ライセートを用いた。抗体特異的なバンドを切除し、トリプシン消化に供し、MALDI-TOFにより分析した。図3Bは、HLA-DR/DP、IgM、およびICAM-1を含む単離抗体の特異性を示す図である。Bリンパ腫細胞に対するB1 IgG、B11 IgG、およびC11 IgGの結合を、抗HLA-DR/DP抗体、抗ICAM-1抗体または抗IgM抗体、それぞれと共にプレインキュベートすることによって、特異的にブロックする。抗体クローンB1、B11、およびC11の回収したMALDI-TOF抗原の特性を確認するために、市販の抗体を用いたブロック試験を実施し、フローサイトメトリーによって分析した。種に適合させたブロック用抗体の10倍モル過剰(ヒト抗体に比較して)により細胞を1時間プレブロックし、続いて、単離されたヒト抗体クローンのいずれかを添加した。30分後、細胞を洗浄し、細胞へのヒト抗体の結合を、PE結合ヤギ抗ヒトIgG(Caltag Laboratories、バーリンガム、カリフォルニア州、米国)によって検出した。この試験に用いられたブロック用抗体は:B1に関しては、マウスモノクローナル抗HLA DR(Sigma、クローンHK14)または抗CD40(Beckton Dickinson、クローン5C3);B11に関しては、ウサギポリクローナル抗ICAM-1(Abcam、ab 7815-250)または抗CD22(Abcam、ab25135-100);C11に関しては、ヤギポリクローナル抗IgM(Zymed、サウスサンフランシスコ、カリフォルニア州、米国62-7500)または抗IgG(Zymed、62-8400)であった。
【図4A】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図である。2μg/mlのB11または抗FITC-8(対照)IgGを、4×105のCL-01 Bリンパ腫細胞に加え、氷上で2時間インキュベートし、引き続いて、10μg/mlの架橋用二次Fab'2ヤギ抗ヒトFc抗体を加えた。細胞を、37℃で6時間インキュベートし、抗体インキュベーションの効果を2つの独立したアポトーシスアッセイによって判定した。上記と同様に、AV/PI(上枠)によるか、または5μg/mlのミトコンドリア膜脱局在化試薬JC-1と共に、室温で30分間インキュベーションすることにより(下枠)、細胞を染色した。アポトーシスの誘導は、赤色(y軸)/緑色(x軸)蛍光強度比の低下によって検出可能である。
【図4B】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図であり、Bリンパ腫組織に対するB11抗体の代表的な結合性を示す組織学的切片を示す図である。未分化大型細胞Bリンパ腫を患っている患者から得られた凍結保存組織を、B11またはFITC-8(対照) scFv抗体によって染色した。抗体結合を、DAB(褐色)によって検出した。差込図は、対照scFvによる染色を示す。
【図4C】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図である。CD45-PerCp-Cy5.5 mAb予備標識ラモス細胞を、ドナー由来のPBMCsと混合し、異なる細胞集団を、蛍光色素結合CD特異的抗体およびAlexa Flour647 Zenon予備標識B11 IgG1または対照FITC-8 IgG1によって染色した。異なる細胞集団に対するIgG B11結合を、FL4チャネルに記録した。
【図5】Bリンパ腫細胞に対するIgG B1およびIgG B11の親和性を示す図である。増量させた放射ヨウ素化IgG B1タンパク質または放射ヨウ素化IgG B11タンパク質と共に、0.2mg/mlの対応する非標識IgGタンパク質の存在下、または非存在下、ラージ細胞(左枠、IgG B1)またはラモス細胞(右枠、IgG B11)をインキュベートした。全体の結合から、非標識競合タンパク質の存在下での結合を差し引くことによって、特異的結合を判定した。遊離IgGタンパク質増量させることにより、結合IgG B1タンパク質またはIgG B11タンパク質の量は増加し、それぞれ、約30nM IgG B1および約1nM IgG B11で飽和に達した(上枠)。ローゼンタール-スキャッチャードプロット分析(下枠)により、IgG B1に関しては、ラージ細胞1個当たり、400,000の機能的結合部位で約3nMの解離定数であり、IgG B11(ラージ細胞)に関しては、47,400の機能的結合部位で約0.3nMの解離定数であることが実証された。
【図6】種々の出所の腫瘍細胞系に対するB1、B11、C11 Ig G1の結合を示す図である。種々の癌細胞系について、B1、B11、およびC11抗体により標的にされた抗原の抗原分布を、フローサイトメトリーによって調べた。示されるように、MCF-7乳癌、HPAC膵臓癌、PC-3前立腺癌、LS174 T結腸直腸癌、およびTHP-1単球白血病細胞に対するリツキシマブ抗CD20 Mab(1列目、最も前方のピーク)、B1 IgG1(2列目のピーク)、B11 IgG1(3列目のピーク)、またはC11 Ig G1(4列目、最も背部のピーク)の結合が、ヒストグラムによって示されている。
【図7A】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。輪郭プロットは、2μg/mlのIgG B11またはIgG B1と共に上記のとおりインキュベーションした後の、Annexin Vおよびヨウ化プロピジウム陽性の関数としてのPC-3細胞の相対的分布を示す。
【図7B】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、連続希釈したB11 IgG1または20μg/mlのB1 IgG1と共にインキュベーションした後のアポトーシスPC-3細胞の平均パーセンテージを示す。
【図7C】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、抗体無しの対照、10μg/mlの陰性抗体対照、連続希釈B11 IgG1、または10μg/mlのトラスツズマブIgG1と共にインキュベーションした後のアポトーシスMDA MB 435細胞の平均パーセンテージを示す。
【図7D】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、抗体無しの対照、10μg/mlの陰性抗体対照、連続希釈B11 IgG1、または10μg/mlのトラスツズマブIgG1と共にインキュベーションした後のアポトーシスDU145細胞の平均パーセンテージを示す。
【図8】架橋試薬の非存在下で、B1 IgG 1およびB1 IgG4が、ラージBリンパ腫細胞に直接細胞細胞毒性を誘導することを示す図である。ラージ細胞を、B1 IgG 1、B1 IgG4、リツキシマブIgG 1、または対照CT-17 IgG 1と共に、20、6.7または2.2μg/mlで、24時間インキュベートした。細胞を採集し、生存度を、Annexin Vおよびヨウ化プロピジウム二重陰性細胞のパーセンテージとして判定した。
【図9A】AB1抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図9B】図9Aの続きである。
【図10A】B11抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図10B】図10Aの続きである。
【図11A】C11抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図11B】図11Aの続きである。
【発明を実施するための形態】
【0080】
実施例1
Bリンパ腫関連細胞表面受容体に特異性を有するアポトーシス誘導性抗体に関する選択およびスクリーニング(バイオパニング)
細胞培養
別に記述しない限り、この試験に用いられる細胞系は、ATCC(マナサス、バージニア州、米国)またはDeutsche Sammlung von Mikroorganismen und Zellkulturen(DSMZ)GmbH(Braunschweig、ドイツ国)から入手し、10% FCS、2mMのL-グルタミン、10mMのHEPESおよび1mMのピルビン酸Na(全て、Invitrogen、カールスバッド、カリフォルニア州、米国から)を添加したRPMI 1640培地中で培養した。ジャーカットT白血病細胞系(クローンE6-1、TIB-152、ATCC)、Bリンパ腫細胞系DOHH-2 (ACC47、DSMZ)、SC-1 (ACC558、DSMZ)、WSU-NHL (ACC58、DSMZ)、JVM-2 (ACC12 DSMZ)、Jeko-1 (ACC553、DSMZ 、20%FCS中で増殖)、Rec-1 (ACC 584、DSMZ)、SP-53 (Daibataら、Cancer 1989年;64:1248〜53頁)、RL (CRL-2261、ATCC)、Granta 519 (DSMZ)、NCEB-1 (Saltmanら、Blood 1988年;72:2026〜30頁)、BJAB (Menezesら、Biomedicine 1975年;22:276〜84頁)、ラモス(CRL-1596、ATCC)、ラージ(CCL-86、ATCC)、Daudi (CCL-213、ATCC)、CL-01 (Ceruttiら、J Immunol 1998年;160:2145〜57頁)、プレB細胞リンパ腫KM-3/Reh (CRL-8286、ATCC)および多発性骨髄腫MC/CAR (CRL-8083、ATCC、20% FCSで添加されたIMDM (Invitrogen)中で増殖)は全て、マイコプラズマを含有せず、5% CO2雰囲気を用い、37℃で加湿雰囲気中で培養した。この細胞を2×105〜1×106細胞/mlで維持した。
【0081】
ジャーカット細胞膜小胞の調製
ジャーカット細胞を、500mlのバケット(Corning社、Life Sciences、ニューヨーク、米国)中、300×gで15分間の遠心分離により採集し、ダルベッコーPBS(Invitrogen)中で洗浄し、緩衝液A(1mMのNaHCO3、1.5mMのMgAc、pH7.4)中に再懸濁した。細胞濃度は、およそ、5×107ジャーカット細胞/ml(100mlの緩衝液A中、5×109細胞)であった。
【0082】
細胞破壊は、氷上、10〜30分間の低浸透圧ショック処理(緩衝液A)、および引き続く窒素キャビテーションボンベ(Parr Instrument社、モリン、イリノイ州、米国)中の窒素キャビテーションによって達成した。細胞は、40バール(4,000kPa)の一定圧で、0℃に15分間保持した。
【0083】
破壊細胞を、0.5MのEDTA 500μlを含有する250mlのザルステット管(Sarstedt AG & Co、ニンブレヒト、ドイツ国)内に採集し、2.5mMの最終EDTA濃度を得た。EDTAの添加により、膜小胞の凝集が防止される。ホモジェネート(100ml)を、4×25mlのBeckman厚壁ローター管(Beckman Coulter社、フラートン、カリフォルニア州、米国)に分け、これらを、4℃で10分間、1900×g(Sorvall SS34ローターにおいて4,000rpm)で遠心分離して、破壊されなかった細胞、核、および重いミトコンドリアを除去した。
【0084】
上澄み液を回収し、ペレット化した物質を1mMのEDTAを含有する1mMのNaHCO3緩衝液25ml中に再懸濁し、再遠心分離した(ペレット化した粗製ジャーカット膜のさらなる回収)。ジャーカット膜を最初の遠心分離からの膜と共にプールした。粗製ジャーカット膜小胞を含有する上澄み液を、Beckman 45Ti型ローターを用いて、4℃で2.5時間、40,000rpm(およそ200,000×g)で超遠心分離した。上澄み液を捨て、組織(例えば、Kleenex(商標))に対して該管縁を傾けることによって残りの緩衝液を除去した。
【0085】
粗製膜ペレットを金属棒の補助でDounceホモジェナイザーに移し、該ホモジェナイザー内で数回慎重になでつけることによって、2.5mlのHES緩衝液(10mMのHepes、1mMのEDTA、0.25Mのスクロース、pH7.4)中に再懸濁した。このようにして、80〜100mgのタンパク質を含有する2×109細胞/mlに等価の膜懸濁液濃度が得られた。
【0086】
全細胞/細胞膜小胞競合バイオスパニングによるファージAbsの選択
およそ2×1013のファージ粒子を、断続的に混合しながら、37℃で15分間予備加温し、14,000×gで15分間遠心分離して、沈殿物を除去し、上澄み液を新鮮なエッペンドルフ管に移した。無脂肪乾燥乳を、2%(w/v)の最終濃度まで加えた。2×109細胞から誘導されたジャーカット膜小胞調製物(1回目選択;2×108細胞2回目および3回目選択)を氷上で解凍し、ブロックしたファージ粒子と共に混合した。混合物を氷上で15分間インキュベートした。
【0087】
5×107 (5×106 2回目および3回目)のラモス細胞を4℃で6分間、1,200rpmでの遠心分離により回収した。上澄み液を廃棄し、ラモス細胞を乳-ファージ-ジャーカット膜小胞混合物中に再懸濁した。該懸濁液を、4時間の緩やかな転倒型回転下、10℃でインキュベートした。
【0088】
2%(w/v) BSA/PBS(フィコール-ピラー)中、下部に、0.5mlの100%(トリパンブルー染色)、フィコール-パックPLUS(Amersham Biosciences、ウップサラ、スウェーデン国)、および9.5mlのオーバーレイド40%(v/v)フィコールを含有する15mlのファルコン管(BD Biosciences、ベッドフォード、マサチューセッツ州、米国)に移した。該管を、4℃で10分間、1,500rpmで遠心分離した。該管を遠心分離機から取り出し、管のキャップをねじ締めし、気密シールした。
【0089】
100%フィコールを含有する該ファルコン管の底部「先端」をシガーチョッパーを用いて切り開いた。このようにして、膜小胞シートおよび細胞核を含むきわめて高濃度の物質を該管から排出させた。次いで、管のキャップを慎重に開けると、管内部の減圧状態が破られ、(切り開かれた)管底部の開口部を通って液体が滴状に放出される。
【0090】
回収した細胞懸濁液をPBS中で1回洗浄して過剰のフィコールを除去した。該ペレットを、1mlのPBS中に再懸濁し(最終洗浄後に実施しなかった)、該懸濁液を、新鮮なフィコール-ピラー上部に再充填し、洗浄操作を繰り返した(2回目および3回目に2回)。
【0091】
PBS中76mMのクエン酸(pH2.5) 150μlの添加により、細胞からファージを溶出させ、続いて、室温で5分間インキュベートした。1Mのトリス-HCl、pH7.4 200μlの添加により、該混合物を中和した。溶出したファージを含有する上澄み液を保存し、300×gで5分間、細胞のペレット化を行った。再懸濁によりファージをさらに溶出させ、1mlのトリプシン中、RTで10分間、細胞ペレットをインキュベートした。
【0092】
1mg/mlのアプロチニン40μlによる活性化の後、細胞を遠心分離し、溶出したファージを含有する上澄み液を保存した。溶出したファージを用いて、大腸菌HB101F'に感染させ、該細菌を適切な抗生物質およびグルコースを含有するTB培地に塗布した。細菌コロニーをカウントし、プレートからかき取り、次回のパニング用種菌として用いた。
【0093】
scFvフォーマットへの転換、scFv発現、精製、および細胞結合の解析
3回選択後に得られたファージミドプールを、EagIにより消化して、遺伝子IIIを除去した。得られたベクターを再結合した。再結合させた非切断遺伝子III断片を含有するベクターを、EcoRI酵素を用いた消化によって線状化した。このようにして作出したscFvベクタープールを用いて、基本的には先に記載したとおり、大腸菌TOP10を形質転換した。(Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)
【0094】
細菌を、大型の500cm2の寒天プレートに塗布し、個々のクローンを取り出して96ウェルプレートに移し、自動システム(Hallborn Biotechniques、2002年;補遺:30〜7頁)を用いて、TB培地中、37℃、220rpmで一晩培養することにより発現させた。適切な抗生物質を含有するTB培地中、組換えscFv断片を作製した。
【0095】
標的ラモス細胞およびジャーカット非標的細胞に結合しているscFvクローンの一次スクリーニングのために、5,000のラモス細胞またはジャーカット細胞を、960のscFvクローンのいずれかと共にインキュベートし、3回目の選択から誘導し、上記のとおり作製した。0.5μg/mlの抗6xHis mAb(R&D Systems、ミネアポリス、ミネソタ州、米国)および0.7μg/mlのCy5結合ヤギ抗マウス試薬(Amersham Biosciences)と共に、細胞をインキュベートした。8200 Cellular Detection System Fluorescence Macroconfocal High Throughput Screening(FMAT)装置(Applied Biosystems、フォスターシティー、カリフォルニア州、米国)において、細胞結合を解析した。
【0096】
一次スクリーニング後、以前記載されたDNA配列決定(Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)(Soderlindら、2000年)のために、72の細菌クローンをランダムに取り出した(すなわち、一次スクリーニングにおける標的細胞対非標的細胞の反応性に関わりなく)。フローサイトメトリーによる細胞表面結合の評価のため、ラモス細胞およびジャーカット細胞(双方とも、1試験当たり、2×105細胞で加える)を0.5%w/vのBSA(DPBS-B)を含有するPBS(Invitrogen)中、2〜10μg/mlの濃度で、個々のscFvクローンと共に、1時間インキュベートした。
【0097】
300×gで6分間の遠心分離により細胞を洗浄した。次いで、FITC結合CD19mAbおよびPE結合CD3mAb(BD)と共に細胞をインキュベートし、引き続く標的細胞および非標的細胞それぞれの同定を可能にした。scFv結合の検出は、RPE-Cy5-ストレプトアビジン(Dako Cytomation、グロストラップ、デンマーク国)とのインキュベーション、続いてビオチン化抗6xHis mAbとのインキュベーションにより達成した。第2および第3の試薬により、それぞれ40分間と15分間、細胞をインキュベートした。全てのインキュベーションは、氷冷溶液を用いて氷上で実施した。
【0098】
細胞全体/細胞膜小胞の差異的パニング
本試験は、天然の細胞表面構造において差異的に発現した抗原を標的にする抗体を単離するために、新規のパニングプロトコルを利用した。3回のバイオパニングの後、ラモスBリンパ腫細胞全体およびジャーカットT白血病細胞から誘導した膜小胞を用いて、組換えファージscFvを単離した。これらを可溶性scFvに変換し、大腸菌TOP10細胞内で発現させた。
【0099】
組換えscFvを標的(ラモス) 全細胞または全非標的(ジャーカット) 細胞と共にインキュベートし、細胞結合に関して調べた。発現した482のscFvクローンが、弱からきわめて強の範囲の強度でラモス標的細胞に選択的に結合することが示された(図1A)ため、該抗体クローンの標的細胞抗原に対する特異性は著しかった。2種のクローンだけが、弱く染色された非標的ジャーカット細胞として同定された(図1A)。
【0100】
次に我々は、単離されたファージディスプレーされたscFvの遺伝子型の多様性を判定した。DNA配列決定のために、72のscFvクローンをランダムに取り出した(すなわち、一次スクリーニングで判定された結合親和性に関わりなく)。
【0101】
記載された(図1B)とおり、FMAT法により、標的細胞特異性(ラモス対ジャーカット)に関して、該クローンを同時に再発現および再評価した。7つの異なる抗体遺伝子型を、それらの異なるCDRH3配列およびCDRL3配列によって判定して同定した(データは示していない)。
【0102】
等しい数のラモス細胞およびジャーカット細胞とのインキュベーションおよび抗タグ抗体によるscFv結合の検出の後、3色フローサイトメトリー分析によって、抗ラモスscFvの高特異性が確認された(図1C)。
【0103】
蛍光色素結合CD特異的モノクローナル抗体を用い、CD19およびCD3それぞれの発現により、標的および非標的細胞を確定した。陰性対照scFvに比較して、7種のユニークな遺伝子型のscFvクローンは、標的ラモス細胞に対して高く可変的な結合強度を示したが、非標的ジャーカット細胞に対する結合は示さなかった。
【0104】
アポトーシスアッセイ
製造元(Pierce Biotechnology、ロックフォード、イリノイ州、米国)の指示に従い、Detoxigelカラムを用いて、組換え作製されたscFv断片のリポ多糖濃度を減少させた。残りのエンドトキシン濃度を、LALアメーバ様細胞ライセートアッセイにより定量化した(Cambrex Bioscience、ウオーカースビレ、メリーランド州、米国)。
【0105】
全てのscFvサンプルが、0.1IU/ml未満のリポ多糖を含有することが判明した。キメラ抗CD20抗体のMabthera(商標)(リツキシマブ)を、Lund大学病院(Lund、スウェーデン国)から購入した。2×105のBリンパ腫細胞(ラージまたはラモス)またはジャーカットT細胞を連続希釈して解毒したscFvと共に、氷上で培養培地中、1時間インキュベートした。
【0106】
細胞を、二次抗6xHis mAb(%μg/ml)、および三次ヤギFab'2抗マウスFab'2抗体(Jackson Immuno Research Laboratories社、ウェストグローブ、ペンシルベニア州、米国)と共に経時的にインキュベートした。断続的洗浄により、過剰な非結合抗体試薬の除去を確実にした。細胞を、5% CO2の加湿雰囲気中、37℃で24時間インキュベートした。
【0107】
全IgGsをアポトーシス誘導に用いる場合、架橋剤を、非IgG抗体アイソタイプと最小の交差反応性を有するヤギFab'2抗ヒトFcγ抗体(Jackson ImmunoResearch)と置き換え(内因性Bリンパ腫関連表面免疫グロブリンの非特異的架橋を避けるため)、上記のとおり、6時間インキュベートした。
【0108】
別に記述しない限り、アポトーシス細胞は、Annexin V Alexa Fluor 488(AV)およびPropidium Iodide(PI)(双方とも、Molecular Probes、Invitrogenから)および引き続くフローサイトメトリー分析によって検出し、細胞を、生存(AV-/PI-)、早期アポトーシス(AV+/PI-)または後期アポトーシス/壊死(AV+/PI+)として確定した。AVおよびPIシグナルを、FACSCalibur機器(BD Bioscience)を用いて、FL1およびFL2またはFL3チャネルにそれぞれ記録した(本文に指示されているとおり)。
【0109】
単離されたscFvの機能性を調べるために、我々は、細胞の経時的インキュベーションおよびscFvおよび架橋剤による洗浄に基づき、ハイスループットアポトーシススクリーニングアッセイを設定した。scFvクローンへの依存性および該アポトーシスアッセイにおける濃度は、図2A〜2Cに示されており、選択されたscFvクローンB1のアポトーシス作用が、アポトーシス誘導を示さないscFvクローンF1の作用と比較されている。標的抗原発現を欠失しているジャーカット細胞は、試験したscFvのいずれの処理の後でもアポトーシスにより死滅することはなく、アポトーシス誘導が標的抗原への結合に依存することを実証した(データは示していない)。
【0110】
確立されたscFv-アポトーシスアッセイを用いて、ラモスおよびラージBリンパ腫細胞におけるアポトーシスに関して、クローンをスクリーンした。scFv誘導アポトーシス作用を、リツキシマブ抗CD20 mAbにより誘導されたものと比較した(図2D)。ラモスおよびラージ細胞双方に有意なアポトーシスを誘導した3種のscFvクローン、B1、B11およびC11が同定された(図2DおよびE)。ラージ細胞に結合しなかったscFvクローンはアポトーシスを誘導しなかったため、ラージ細胞におけるscFvによるアポトーシスの誘導は、これらの細胞への結合性に相関的であった(図2D)。
【0111】
B1、B11およびC11クローンを、完全ヒトIgG1抗体に移入した。広範囲群のBリンパ腫細胞系に対する強力な結合性および強力な細胞毒性によって実証される(表1)ように、再フォーマット後もそれらの特異性および機能性は損なわれないままであった。特に、いくつかの細胞系において、3時間から6時間後にはすでに、アネキシンV陽性アポトーシス細胞の最高パーセンテージに達しており、アポトーシス誘導は迅速であった(表1、データは示していない)。
【0112】
IgG生産およびエンドトキシンスクリーニングアッセイ
scFv抗体断片は、修飾pCDNA3ベクター(Norderhaugら、J Immunol Methods、1997年;204:77〜87頁)内へのクローニングにより、完全長ヒトIgG1λフォーマットに転換させ、製造元(Invitrogen)の指示に従い、Lipofectamine2000試薬を用いて、HEK293細胞内へ一時的にトランスフェクトした。
【0113】
使用ずみ培養培地からのヒトIgGを、MabSelectタンパク質Aカラム(Amersham Bioscience)上で精製した。調製物の純度は、SDS-PAGE分析により判定すると、>98%であった。抗体調製物をスクリーンし、本試験に使用した濃度で、LALアメーバ様細胞ライセート試験(Cambrex Bioscience)によって判定すると、<0.1IU/mlのエンドトキシンを含有することが分かった。
【0114】
実施例2
抗体特異性の分析
抗原同定
標的抗原の確認を、Bリンパ腫細胞ライセートの免疫沈降によって行った。細胞(抗体および細胞系によって、1mlの溶解用緩衝液当たり、50〜600×106)を遠心分離により採集し、PBS中で2回洗浄し、0.5%v/vの洗浄剤TritonX-100(Sigma-Aldrich、セントルイス、ミズーリ州、米国)を含有するLysis Buffer(25mMのトリス-HCl、pH7.5、150mMのNaCl、5mMのEDTA、および完全無EDTAプロテアーゼ阻害剤カクテル(Roche Diagnostics GmbH、マンハイム、ドイツ国))中で、15分間インキュベートした。
【0115】
細胞デブリを慣例的なテーブルトップ遠心分離機において、16,000×gで15分間、遠心沈殿させ、可溶性タンパク質を、Protein A Sepharose4 Fast Flow (Amersham Bioscience)(1/10容量の反応液)によって、1時間回転させて予備洗浄した。各サンプルに関し、1mlの予備洗浄した細胞ライセートを、いずれかのヒト抗体20〜100μgによって2時間免疫沈降させた。再度、Protein A Sepharose4 Fast Flowを加え、30分間インキュベートし、免疫複合体を溶解用緩衝液中で広範囲に洗浄し、5分間煮沸し、最後にSample Buffer(1X NuPAGE LDS Sample Buffer、1X NuPAGE Sample Reducing Agent)中に再懸濁し、NuPAGE Novex 4〜12% Bis-Tris Gel(全てInvitrogenから)中で分離させた。
【0116】
染色(Simply Blue Safestain、Invitrogen)後、対象となっているタンパク質バンドをSDS-PAGEから切除し、記載 (Edvardssonら、Electrophoresis、1999年;20:935〜42頁) されたとおり、トリプシン消化に供した。
【0117】
簡単に述べると、ゲルプラグを脱染色し、攪拌下、200μlの50%アセトニトリル(ACN)による3回洗浄によって平衡化した。SpeedVac濃縮機(Savant、ファーミングデール、ニューヨーク、米国)中で15分間乾燥後、10mMのDTT/100mMのNH4HCO3 25μlの添加により還元し、56℃で1時間インキュベートし、55mMのヨードアセトアミド/100mMのNH4HCO3 を25μl添加することによりアルキル化し、続いて、室温で45分間インキュベートした。
【0118】
100mMのNH4HCO3 中でさらに2回、10分間の洗浄ステップ、続いて50%v/v ACN中、1回の洗浄後、該ゲル片を、SpeedVac濃縮機中で乾燥し、再膨張させ、25mMのNH4HCO3 における15ng/μlのトリプシン(Promega社、マジソン、ウィスコンシン州、米国) 15μl中、37℃で一晩消化させた。50%v/vACN/1%v/vTFAの添加により、ペプチドを抽出し、室温で10分間インキュベートした。1μlの抽出物をMALDIサンプルプレート上にスポットし、乾燥させた。1μlのマトリックス溶液(70%v/vACN/1%v/vTFA中、5mg/mlのアルファ-シアノ-4-ヒドロキシ桂皮酸(CHCA))を、該ペプチドの上部にスポットした。
【0119】
ペプチド質量は、Applied Biosystems 4700 Maldi Workstationを用いて決定した。該タンパク質は、Mascot探索ツール(Matrixscience、英国)を用い、ペプチド質量フィンガープリントデータベース探索によって同定した。クローンB10、C10、およびG12の抗原特異性は、免疫沈降にscFvおよび抗Hisコーティングした磁気マイクロビーズ(Miltenyi Biotec GmbH、ベルギッシュグラッドバッハ、ドイツ国)を使用した以外は、同様の方法論を用いて確認した。
【0120】
完全抗体フォーマットB1、B11およびC11に変換した後、IgGを用いて、ラージおよびラモスリンパ腫細胞から抗原を沈降させた。IgG B1は、それぞれ、およそ28kDaおよび34kDaの2つのバンドを沈降させた(図3A、レーン1)。これらのバンドを含有するゲルスライスを調製し、トリプシンで消化し、質量分光により分析し、標的抗原としてHLADR/DPを同定した。
【0121】
HLA-DR/DPに対するIgG B1の特異性は、市販のモノクローナル抗体を用いたウェスタンブロッティングおよびHLA-DR/DPタンパク質の検出双方、ならびに市販のHLA-DR特異性モノクローナル抗体を用いた細胞のプレインキュベーション後にB1 IgG結合のブロックにより検証した(図3B)。
【0122】
IgG B11およびC11規定抗原の同定を、同様な方法論を用いて確立した。IgG C11は、B細胞受容体μ鎖の膜結合形態として同定される68kDaのタンパク質バンドを沈降させることが分かった(図3A、レーン3)。IgG B11は、細胞間細胞接着分子-1(ICAM-1)として同定される90kDaのタンパク質バンドを沈降させた(図3A、レーン2)。ICAM-1に対するIgG B11、およびIgMに対するC11 IgGの特異性は、市販の抗体を用い、MS-MS分析、抗体ブロック試験(図3B)、およびウェスタンブロット分析(データは示していない)によって確認した。
【0123】
クローンB10、C10、およびG12の特異性は、scFvおよび免疫沈降用の抗Hisコーティングした磁気マイクロビーズを用いて判定した。3種のscFvクローンは、68kDaのタンパク質バンドを沈降させ、これらのバンドを含有するトリプシン消化ずみゲルスライスのMS分析により、表面IgMに対するそれらの特異性が判明した。これらの抗体はいずれも、末梢血Bリンパ球または他のIgM陽性B細胞系と交差反応しないため、ラモスIgMイディオタイプを認識すると考えられる。
【0124】
実施例3
抗体親和性の分析
B1およびB11免疫グロブリンのインビトロヨウ素化
[125I] NaIによる1mg/mlのIgG1B1またはIgG1B11タンパク質のヨウ素化は、Iodogen予備コーティングしたヨウ素化管(Pierce)を用いて、PBS中、10分間実施した。PD-10カラム(Amersham Biosciences)上での脱塩により、遊離[125I] NaIを除去し、タンパク質1ng当たり、1000〜1600cpmの範囲における特異的放射活性を得た。抗体親和性の判定には、[125I] IgG1 B1および[125I] IgG1 B11を用いた。
【0125】
IgG B1およびIgG B11親和性定数の決定
放射ヨウ素化IgG B1またはIgG B11をDPBS-B-hIgG(0.2mg/mlのヒトIgGを含有するDPBS-B)中、Bリンパ腫細胞と共に、氷上で2時間、断続的に混合しながらインキュベートした。適切な場合、0.2mg/mlの非標識IgG B1またはIgG B11タンパク質の存在下、非特異的結合を判定した。分析は三重で実施した。
【0126】
個々の管内の上部の40%v/v Ficoll/DPBS-Bクッション上に細胞を入れ、4℃で6分間、400×gで遠心分離した。サンプルを-80℃で凍結させた。細胞ペレットおよび細胞上澄み液を単離し、ガンマカウンターで125I-IgGタンパク質定数に関して別々に分析してから、該管を半分に切断した。
【0127】
以前記載された(Brixら、J.Clin.Invest.1998年;102:283〜93頁)とおり、Rosenthalら(Anal Biochem、1967年;20:525〜32頁)、BylundおよびYamamura(Methods in Neurotransmitter Analysed、ニューヨーク: Raven Press社、1990年)、およびMarquardt(J.Soc.Indust.Appl.Math、1963年;11:431〜41頁)に従って、抗体親和性定数(Kd値)および細胞1個当たりのエピトープ数を決定した。
【0128】
0.2mg/mlの対応する非標識IgGタンパク質の存在または非存在下、放射ヨウ素化タンパク質を、ラージまたはラモス細胞と共に、4℃でインキュベートすることにより、HLA-DRおよびICAM-1に対するIgG B1およびIgG B11の結合を特性化した。細胞表面に対する[125I]IgGの特異的結合を、総結合から非特異的結合 (過剰の非標識IgG存在下での結合) を差し引くことにより算出した。
【0129】
約30nMのIgG B1で、ラージ細胞に対するIgG B1の特異的結合の飽和に達した(図5)。Rosenthal-Scatchardプロット分析により、ラージ細胞1個当たり、400,000の機能的結合部位で、約3nMの解離定数が判明し、二価のエピトープ-IgG相互作用が考えられた(図5)。同様に、IgG B11の解離定数は、ラモス細胞1個当たり、47,400の受容体で約0.2nMと判定された。
【0130】
【表1】

【0131】
実施例4
ICAM-1は、アポトーシス誘導特性を有するBリンパ腫関連抗原である
ラモス細胞に対するIgG結合のフローサイトメトリー分析
氷上で45分間のインキュベーションにより、ラモス細胞(13×106)をCD45-PerCp-Cy5.5mAbによって染色し、DPBS-B中で洗浄し、非修飾精製PBLsと混合するまで氷上に保存した。
【0132】
Lund大学病院から、2人の健常志願者からのバッフィーコートを入手した。バッフィーコートを、PBS中、1:2に希釈し、500×g(1500rpm Beckman Spinchron 遠心分離機)で10分間の遠心分離によって洗浄し、上澄み液を完全吸引し、1%熱不活化FCSを含有するDPBS(DPBS-HI)中へ再懸濁した。洗浄は2回繰り返した。赤血球溶解溶液(BD Bioscience)と共に室温で15分間のインキュベーションによって、赤血球を溶解した。細胞を、60×g(667rpm Beckman Spinchron 遠心分離機)で10分間の遠心分離により洗浄し、上澄み液を慎重に吸引した。細胞をトリパンブルー試薬(Invitrogen)により染色して死細胞を排除した後、ビュルケルチャンバー内でカウントし、DPBS-HI中で洗浄し、ペレット化し、200μg/mlのヒト精製IgGを含有するDPBS-B(Fc受容体のブロック)中に再懸濁した。
【0133】
各ドナーおよび試験条件に関して、およそ2.5×106の白血球を、1.6×105のPerCpCy5.5予備標識ラモス細胞と混合した。マウスモノクローナルCD3-FITC、CD56-PE、およびCD19- PerCpCy5.5抗体(BD Bioscience)を加え、標識ヒトIgGを添加するまで、その混合物を氷上でインキュベートした。n-CoDeRヒトIgG抗体および陽性対照の抗CD20マウス-ヒトキメラ抗体リツキシマブの標識化を、製造元の指示に従い、AF647 Fab断片(Molecular Probes、Invitrogen)によって実施した。
【0134】
簡単に述べると、IgG B1、B11、C11、およびリツキシマブ抗体各々の4μgを、20μlのAF647-Fab標識試薬と共に、室温で5分間インキュベートした。20μlのヒトIgGブロック試薬を添加してさらに5分間のインキュベーション後、AF647標識化IgGをDBPS-B中に3倍連続希釈し、希釈したIgGタンパク質を、混合ラモス/PBL細胞溶液に加えた。
【0135】
サンプルを1時間インキュベートし、洗浄し、DPBS-B中に再懸濁し、4色分析用機器の適切な検量および補正の後、フローサイトメトリーにより種々の細胞亜集団に対する結合に関して分析した。ラモス細胞は、Bリンパ球PerCpCy5.5low集団から識別できるPerCpCy5.5high集団として同定された。
【0136】
免疫組織化学
未分化大型細胞Bリンパ腫(1人)、中心芽細胞性/中心細胞性B非ホジキンリンパ腫(3人)、およびB細胞慢性リンパ性白血病(1人)の凍結保存リンパ節生検を、Lund大学の病理学部(Lund、スウェーデン国)から入手した。凍結保存組織の8マイクロメートル切片をアセトン中、4℃で10分間固定した。アビジンおよびビオチン(Avidin/Biotin blockingキット、Invitrogen)による各20分間の連続処理により、内因性ビオチン結合活性をブロックした。
【0137】
5μg/mlの対照scFvまたはB11scFvと共に、組織を1時間インキュベートした。洗浄後、切片をビオチン結合マウス抗His mAb(R&D Systems)と共に30分間インキュベートした。ABC Complex/HRP試薬(Dako Cytomation)による30分間の処理、引き続き、DABとの5分間のインキュベーション後、scFv結合を検出した。
【0138】
Leica DMR光/蛍光顕微鏡の上部に据え付けたLeica DC 300Fデジタルカメラを用いて、切片の写真を撮った。
【0139】
ヒト組織の取り扱いは、Lund大学病院の地域倫理委員会の推奨に従った。
【0140】
ミトコンドリア膜脱分極アッセイ
以前記載された(Kimら、Mol Biol Cell、2004年;15:420〜34頁)とおり、ミトコンドリア膜脱分極を分析した。簡単に述べると、抗体処置細胞を5μg/mlのJC-1試薬(Molecular Probes)と混合し、室温で30分間インキュベートした。細胞を氷冷PBS中で2回洗浄し、300μlのPBS中に再懸濁し、FACS Aria(BD Biosciences)で分析した。494/518nm(FL-1)および595/615nm(FL-2)バンド通過フィルターを通して、緑色蛍光および赤色蛍光をそれぞれを採集した。
【0141】
ICAM-1は、二方向性シグナル伝達を誘導できる(Rothleinら、J Immunol、1994年;152:2488〜95頁; Vyth-Dreeseら、Blood、1995年;85:2802〜12頁)免疫グロブリンスーパーファミリーの糖タンパク質である(Marlinら、Cell、1987年;51:813〜9頁)。ICAM-1がBリンパ腫細胞におけるプログラム死に関与しているという実証は以前されていない。
【0142】
したがって、我々は、IgG B11誘導細胞死が、細胞膜ホスファチジルセリンの移動以外の方法による活性過程であることを確認したかった。
【0143】
ミトコンドリア膜の脱分極をアポトーシスの確認として選択した。これが、ヨウ化5,5',6,6'-テトラクロロ-1,1',3,3'-テトラエチル-ベンジミダゾイル-カルボシアニン(JC-1試薬)による細胞染色によりモニターできるカスパーゼ依存性およびカスパーゼ非依存性アポトーシスの共通の特徴であるためである。
【0144】
我々のAnnexin V/ヨウ化プロピジウムアッセイ(図4A、上枠)に従い、JC-1試薬による染色後、フローサイトメトリー分析によって判定すると、IgG B11は、CL-01 Bリンパ腫細胞において、ミトコンドリア膜の脱分極を誘導することが分かった(図4A、下枠)。
【0145】
ICAM-1の発現が、細胞培養中の一般的なアップレギュレーションから生じるインビトロアーチファクトであった可能性を排除するために、種々のBリンパ腫腫瘍に罹っている5人の異なる患者から得た組織に対するIgG B11の結合を調べた。
【0146】
免疫組織化学により、IgG B11は、抗HLA-DR/DP抗体IgG B1と同等か、またはわずかに低い強度で(表2)、5つのリンパ腫組織に対する強い結合性を示した(図4B)。
【0147】
次に、Bリンパ腫対静止末梢血白血球に対するIgG B11の結合性を調べた。低端エピトープ発現であって、しかもB11誘導アポトーシスに対する有意な感受性があることに基づき、ラモスが代表的なBリンパ腫細胞系として選択された。予備標識したラモス細胞と全血末梢血白血球ならびにIgG B1、B11またはC11抗体のいずれかとの混合インキュベーション後のフローサイトメトリー分析によって、IgG B11は、ラモス細胞に対する強い結合性を示した。
【0148】
さらに重要なことに、B11は、ラモスBリンパ腫対正常末梢血白血球に対する3腫の抗体の結合性の大きな差異(最強の抗原アップレギュレーション)を示した(図4C、データは示していない)。IgG B11の結合は、すでに0.1μg/mlで最高となり、ラモス対単球細胞で3.7倍アップレギュレートされ(MFI654対176)、ラモス対末梢血Bリンパ球で8.3倍アップレギュレートされ(MFI654対78)、NK細胞に比較すると、23倍アップレギュレートされた。モニターされた他の末梢血白血球サブセットに対する結合性は負であった。
【0149】
【表2】
【0150】
実施例5
フローサイトメトリーにより判定された、種々の出所の腫瘍細胞系におけるB1、B11、C11 IgG1の抗原分布
主にB11に関するヒト抗体標的抗原の抗原分布を、種々の癌細胞系について調べた。細胞(MCF-7およびMDA MB 435S乳癌、JARおよびJEG-3絨毛癌、A549肺癌、TCC-SUP膀胱癌、MDA MB 435黒色腫、HPAC、PANC-1およびBxPC-3膵臓癌、PC-3およびDU145前立腺癌、LS174T、CaCo2、およびLovo結腸直腸癌、およびTHP-1単球白血病細胞)をPBS中で洗浄し、完全培地(200,000細胞/50μlサンプル)中、4×106細胞/mlで再懸濁した。B1 Ig G1、B11 Ig G1、C11 Ig G1、陰性対照FITC-8 Ig G1、およびリツキシマブ抗CD20 mAbを、完全培地(50μl/サンプル)中、3〜10倍に連続希釈した(10〜0.1pg/ml)。細胞を、いずれかの抗体と共に、1時間、氷上でインキュベートし、PBS/BSA 0.5%中に再懸濁することにより洗浄し、1200rpmで5分間遠心分離し、上澄み液の完全吸引を行った。細胞を、PBS/BSA 0.5%中、1/50に希釈したPE結合ヤギF(ab')2抗ヒトIgG(Caltag Laboratories、カタログ番号:H10104)と共に、氷上で30分間インキュベートした。300μlのPBS/BSA 0.5%中に再懸濁した後、FACScan機器を用いて、細胞を、IgG結合性に関して分析した。
【0151】
PC-3前立腺癌細胞は、これらの細胞に対するB11 IgGの強い結合性によって実証されるように、ICAM-1の強い発現を示した(図6)。MCF-7乳癌細胞、HPAC膵臓癌細胞、およびLS174T結腸直腸癌細胞もまた、前立腺癌に比較して強度はより低いが、ICAM-1を発現することが分かった。対照的に、THP-1単球白血病細胞は、ICAM-1を発現しなかった。最初に試験した全ての癌細胞系は、リツキシマブIgG、B1 IgG、およびC11 IgGそれぞれの結合が欠如していることから実証されるように、CD20、HLA-DR/DP、およびIgM発現に関して陰性であることが分かった。追加の癌細胞系におけるさらなる試験により、調べられた全ての癌細胞が、ICAM-1発現に関して陽性であることが示された(表3)。
【0152】
【表3】
【0153】
実施例6
癌細胞におけるB11 IgG1アポトーシス誘導
実施例5において、B11 IgG1は、癌細胞に強く結合することが示された。本実施例では、癌細胞に対するこの抗体のアポトーシス誘導特性を調べた。
【0154】
実験開始の3日前に、完全増殖培地を有する6ウェルプレートにおいて、細胞を接種した。実験時、細胞は50〜75%の間の集密度であった。細胞を氷冷PBSにより洗浄し、指示されたとおり、連続希釈した(完全増殖培地1ml中、図に示されるように、20〜0.02μg/ml)B11 IgG1、20μg/mlの対照B1 IgG1、10μg/mlの陰性対照IgG1または10μg/mlのトラスツズマブIgG1と共に、4℃で1〜2時間インキュベートした。細胞を氷冷PBSにより洗浄し、二次F(ab'2)ヤギ抗ヒトF(ab'2)抗体(完全増殖培地中、10μg/mlに希釈)を加えた。5% CO2の加湿雰囲気下、細胞を37℃で16〜24時間インキュベートした。先ず、上澄み液を単離し、続いてPBS洗浄し、残りの接着細胞をトリプシン処理することによって総細胞を採集した。熱不活化した10%ウシ胎仔血清を含有するPBS中に再懸濁することによって酵素反応を終結させた。氷冷PBS中で細胞を洗浄し、Annexin V/ヨウ化プロピジウム染色に供し、上記実施例5に記載されたとおり、生存度/アポトーシスに関して分析した。
【0155】
B11 IgG1は、特定の滴定可能な様式で、癌細胞系にアポトーシスを誘導することが示された(図7)。PC-3細胞に結合しなかった対照IgG B1(実施例5を参照)は、PC-3細胞にアポトーシスを誘導することもなかった。陰性対照IgG1またはトラスツマブIgG1は、DU145細胞またはMDA MB435細胞にアポトーシスを誘導することができなかった。
【0156】
実施例7
医薬製剤および投与
本発明のさらなる態様では、薬学的にまたは獣医学的に許容されるアジュバント、希釈または担体と混合させて、本発明の第1の態様による化合物を含む医薬製剤が提供される。
【0157】
該製剤は、活性成分の1日用量または単位、1日のサブ用量またはその適切な画分を含有する単位用量であることが好ましい。
【0158】
本発明の化合物は通常、薬学的に許容される剤形において、活性成分を、任意に、非毒性の有機または無機の酸または塩基付加塩の形態で含む医薬製剤の形態で、経口で、または任意の非経口経路により投与される。治療される障害および患者、ならびに投与経路により、該組成物は用量を変化させて投与できる。
【0159】
ヒトの治療において、本発明の化合物は、単独でも投与できるが、一般に、意図された投与経路および標準的な医薬上の実践を考慮して選択される好適な医薬用の賦形剤、希釈剤または担体と混合して投与される。
【0160】
例えば、本発明の化合物は、即時、遅延または制御放出適用のために、風味剤または着色剤を含有し得る、錠剤、カプセル剤、卵形剤、エレキシル剤、液剤または懸濁剤の形態で、経口で、頬側に、または舌下に投与できる。本発明の化合物は、鼻腔内注入によっても投与できる。
【0161】
このような錠剤は、微結晶性セルロース、乳糖、クエン酸ナトリウム、炭酸カルシウム、第二リン酸カルシウムおよびグリシンなどの賦形剤、澱粉(好ましくは、トウモロコシ、ジャガイモまたはタピオカ澱粉)、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウムおよび一定の錯体ケイ酸塩などの崩壊剤、ならびにポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ-プロピルセルロース(HPC)、ショ糖、ゼラチンおよびアラビアゴムなどの顆粒化結合剤を含有してもよい。また、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリルおよびタルクなどの滑剤を含めることができる。
【0162】
同様なタイプの固体組成物は、ゼラチンカプセル剤中の充填剤としても使用できる。これに関して好ましい賦形剤としては、乳糖、澱粉、セルロース、ミルクシュガーまたは高分子量のポリエチレングリコールが挙げられる。水性懸濁剤および/またはエリキシル剤では、本発明の化合物を、種々の甘味剤または風味剤、着色物質または色素と、乳化剤および/または懸濁化剤と、水、エタノール、プロピレングリコールおよびグリセリンなどの希釈剤と、およびそれらの組み合わせと組み合わせることができる。
【0163】
本発明の化合物は、非経口で、例えば、静脈内、動脈内、腹腔内、鞘内、心室内、ステム内、頭蓋内、筋肉内または皮下に投与できるか、または、それらを注入法により投与できる。それらは、他の物質、例えば、該溶液を血液と等張にする上で十分な塩またはグルコースを含有し得る、滅菌水性溶液の形態で最良に用いられる。該水性溶液は、必要ならば、好適に緩衝化(好ましくは、pH3から9に)される必要がある。滅菌条件下での好適な非経口製剤の調製は、当業者によく知られた標準的な製薬技法によって容易に達成される。
【0164】
非経口投与に好適な製剤としては、抗酸化剤、緩衝剤、静菌剤および該製剤を意図されたレシピエントの血液と等張にする溶質を含有し得る水性および非水性の滅菌注射液剤;ならびに懸濁化剤および増粘剤を含み得る水性および非水性の懸濁剤が挙げられる。該製剤は、単位用量または多用量容器、例えば、密封アンプルおよびバイアルにおいて提供でき、使用直前に滅菌液体担体、例えば注射用の水の添加のみを必要とするフリーズドライ(凍結乾燥)条件下で保存できる。即時注射液および懸濁液は、以前記載された種類の滅菌した粉末、顆粒および錠剤から調製できる。
【0165】
ヒト患者への経口および非経口投与に関して、本発明の化合物の1日用量レベルは通常、1mg/kgから30mg/kgである。したがって、例えば、本発明の化合物の錠剤またはカプセル剤は、適宜、一度に一回、または2回以上の投与のための活性化合物の用量を含有できる。何らかの事象において、医師は、個々の患者にとって最も好適な実際の用量を決定し、この用量は、具体的な患者の年齢、体重および応答によって変化する。上記の投与量は、平均的な場合の代表的なものである。勿論、より高いまたは低い投与量の範囲が有利である個々の例があり得、このようなことは本発明の範囲内にある。
【0166】
本発明の化合物はまた、鼻腔内に、または吸入によっても投与でき、ドライパウダー吸入器の形態、または好適な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、1,1,1,2-テトラフルオロエタン(HFA 134A3または1,1,1,2,3,3,3-ヘプタフルオロプロパン(HFA227EA3)、二酸化炭素または他の好適な気体の使用と共に、加圧容器、ポンプ、スプレーまたはネブライザーからエアゾールスプレー提供の形態で簡便に送達される。加圧エアゾールの場合、計測量を送達するためにバルブを提供することによって、用量単位を決定できる。加圧容器、ポンプ、スプレーまたはネブライザーは、例えば、溶媒としてエタノールと噴射剤との混合物を用いて、活性化合物の溶液または懸濁液を含有でき、さらに、滑剤、例えば、ソルビタントリオレエートを含有できる。吸入器または注入器に使用するためのカプセルおよびカートリッジ(例えば、ゼラチンから作製)は、本発明の化合物と乳糖または澱粉などの好適な粉末基材との粉末混合物を含有するように製剤化できる。
【0167】
エアゾール製剤またはドライパウダー製剤は、各計測用量または「パフ」が、患者への送達のために、本発明の化合物の適切な用量を送達するように設定されていることが好ましい。エアゾールによる1日用量の合計は、患者によって異なり、単一用量で、より通常的には、1日の分割用量で投与できる。
【0168】
あるいは、本発明の化合物を、座剤または膣座剤の形態で投与でき、それらは、ローション、溶液、クリーム、軟膏または粉剤の形態で局所に適用できる。本発明の化合物は、例えば、皮膚パッチの使用により、経皮投与することもできる。それらはまた、特に眼の疾患を治療するために、眼の経路により投与できる。
【0169】
眼科使用に関して、本発明の化合物は、等張でpH調整した滅菌生理食塩水におけるミクロ化懸濁剤として、または好ましくは、等張でpH調整した滅菌生理食塩水における液剤として、任意に、塩化ベンジルアルコニウムなどの保存剤と組み合わせて製剤化できる。あるいは、それらを、ペトロラタムなどの軟膏に製剤化できる。
【0170】
皮膚への局所適用に関して、本発明の化合物は、例えば、以下:鉱油、流動パラフィン、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックスおよび水、の1種または複数の混合物に懸濁または溶解させた活性化合物を含有する好適な軟膏として製剤化できる。あるいは、それらを、例えば、以下:鉱油、モノステアリン酸ソルビタン、ポリエチレングリコール、流動パラフィン、ポリソルベート60、セチルエステル類、ワックス、セテアリールアルコール、2-オクチルドデカノール、ベンジルアルコールおよび水、の1種または複数の混合物に、懸濁または溶解させた好適なローションまたはクリームとして製剤化できる。
【0171】
口腔における局所投与のために好適な製剤としては、風味づけされた基材、通常は、スクロースおよびアラビアゴム、またはトラガントゴム中に該活性成分を含む舐剤;ゼラチンおよびグリセリン、またはスクロースおよびアラビアゴムなどの不活性基材中に該活性成分を含むパステル剤;および好適な液体担体中に該活性成分を含む口腔洗浄剤が挙げられる。
【0172】
一般に、ヒトにおいて、本発明の化合物の経口投与または局所投与が、最も簡便であるので、好ましい経路である。レシピエントが嚥下障害または経口投与後の薬剤吸収の障害を患っている状況では、該薬剤は腸管外的に、例えば、舌下または頬側に投与できる。
【0173】
獣医学使用に関しては、本発明の化合物は、通常の獣医学実践に従って、好適に許容される製剤として投与され、獣医学医師は、具体的な動物にとって最も適切である投与療法および投与経路を決定する。
【技術分野】
【0001】
本発明は、アポトーシス誘導に関与する分子、アポトーシス誘導のための方法および医薬組成物、ならびにその使用に関する。
【背景技術】
【0002】
抗体は最近、癌を標的とするタンパク質治療法として選択されるようになったが、他の適応症の治療に関しても選択されるようになった(Brekkeら、Nat Rev Drug Discov、2003年:52〜62頁)。抗体工学の到来により、ヒトの性質およびより大きな多様性による免疫原性の減少ならびに特異性および親和性の増強を示す合成ファージライブラリーからヒト抗体を作出するツールが提供されている(Weinerら、Nat Biotechnol、2005年;23:556〜7頁)。未処置ライブラリーは、免疫化および新規ライブラリーの再構築とは独立して、自己抗原などの何らかの特異性に関して抗体の単離に使用できるため、特に魅力的である(Griffithsら、Embo J、1993年;12:725〜34頁)。現在まで、細胞表面受容体は、小型分子阻害剤および抗体などの現代の治療薬によって標的化された最も成功した抗原群を構成している。特に興味深いのは、ユニークな発現をするか、または標的細胞上に増加した発現レベルを示し、さらに、細胞に対する死または生存のシグナルを中継することのできる細胞表面受容体である。このような内因性のシグナル伝達特性を有する差異的発現受容体により、微生物に感染した、形質転換した、または機能不良の細胞の抗体ベースの標的化が可能になる。
【0003】
腫瘍の治療に関して、標的腫瘍細胞にアポトーシスを誘導する一方、正常組織を避ける能力を有する抗体は特に興味深い。このようないくつかの抗体が使用されており、米国食品医薬品局(FDA)に登録されており、例えば、リンパ腫に対する(CD20を標的にするリツキシマブ)または乳癌に対する(Her-2およびEGFRをそれぞれ標的にするトラスツズマブまたはセツキシマブ)従来の癌治療の代替法を提供する。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】WO 2004/023140
【非特許文献】
【0005】
【非特許文献1】Brekkeら、Nat Rev Drug Discov、2003年:52〜62頁
【非特許文献2】Weinerら、Nat Biotechnol、2005年;23:556〜7頁
【非特許文献3】Griffithsら、Embo J、1993年;12:725〜34頁
【非特許文献4】Millerら、N Engl J Med、1982年;306:517〜22頁
【非特許文献5】Riechmannら、Nature、1988年;332:323〜7頁
【非特許文献6】Kwakら、N Engl J Med、1992年;327:1209〜15頁
【非特許文献7】Suarezら、Mol Immunol、2004年;41:519〜26頁
【非特許文献8】Soderlindら、Nat Biotechnol、2000年;18:852〜6頁
【非特許文献9】Clackson Tら、Nature、1991年8月15日;352(6336):624〜8頁
【非特許文献10】Marks JDら、J Mol Biol. 1991年12月5日;222(3):581〜97頁
【非特許文献11】Weng Sら、Proteomics、2002年1月;2(1):48〜57頁
【非特許文献12】Nord Kら、Nat Biotechnol、1997年8月;15(8):772〜7頁
【非特許文献13】Hogbom Mら、Proc Natl Acad Sci U S A、2003年3月18日;100(6):3191〜6頁
【非特許文献14】Borrebaeck CAおよびCarlson R、Curr Opin Pharmacol.2001年8月;1(4):404〜8頁
【非特許文献15】Nagyら、J Mol Med、2003年;81:757〜65頁
【非特許文献16】Smithら、J Clin Pathol、1990年;43:893〜900頁
【非特許文献17】Cosimiら、J Immunol、1990年;144:4604〜12頁
【非特許文献18】Kavanaughら、Arthritis Rheum、1994年;37:992〜9頁
【非特許文献19】Haugら、Transplantation、1993年;55:766〜72頁
【非特許文献20】Huangら(1993) Hybridoma 12、661〜75頁
【非特許文献21】Huangら、(1995) Cancer Res 55、610〜6頁
【非特許文献22】Smallshawら、(2004) J Immunother 27、419〜24頁
【非特許文献23】Maruoら、(2002) Int J Cancer 100、486〜90頁
【非特許文献24】Rosetteら、(2005) Carcinogenesis 26、943〜50頁
【非特許文献25】Sunら、(1999) J Cancer Res Clin Oncol 125、28〜34頁
【非特許文献26】Grotheyら、(1998) Br J Cancer 77、801〜7頁
【非特許文献27】Wangら、(2005) Int J Cancer 27、419〜24頁
【非特許文献28】Rocheら、(2003) Thromb Haemost 89、1089〜97頁
【非特許文献29】Aalinkeelら、(2004) Cancer Res 64、5311〜21頁
【非特許文献30】Nagyら、Nat Med、2002年;8:801〜7頁
【非特許文献31】Uyttenhoveら、J.Exp.Med.、1983年;157:1040〜52頁
【非特許文献32】Kennedyら、Br J Haematol、2002年;119:412〜6頁
【非特許文献33】Weinerら、J Immunol、1989年;142:343〜51頁
【非特許文献34】Baiら、J.Clin.Invest.、2003年;111:1487〜96頁
【非特許文献35】Robertら、Lancet Oncol、2005年;6:491〜500頁
【非特許文献36】Harlowら(編)、Antibodies A Laboratory Manual;Cold Spring Harbor Laboratory:コールドスプリングハーバー、ニューヨーク(1988)、第6章
【非特許文献37】Daibataら、Cancer 1989年;64:1248〜53頁
【非特許文献38】Saltmanら、Blood 1988年;72:2026〜30頁
【非特許文献39】Menezesら、Biomedicine 1975年;22:276〜84頁
【非特許文献40】Ceruttiら、J Immunol 1998年;160:2145〜57頁
【非特許文献41】Soderlindら、Nat Biotechnol、2000年;18:852〜6頁
【非特許文献42】Hallborn Biotechniques、2002年;補遺:30〜7頁
【非特許文献43】Norderhaugら、J Immunol Methods、1997年;204:77〜87頁
【非特許文献44】Edvardssonら、Electrophoresis、1999年;20:935〜42頁
【非特許文献45】Rosenthalら、Anal Biochem、1967年;20:525〜32頁
【非特許文献46】BylundおよびYamamura、Methods in Neurotransmitter Analysed、ニューヨーク、Raven Press社、1990年
【非特許文献47】Marquardt、J.Soc.Indust.Appl.Math、1963年;11:431〜41頁
【非特許文献48】Brixら、J.Clin.Invest.1998年;102:283〜93頁
【非特許文献49】Kimら、Mol Biol Cell、2004年;15:420〜34頁
【非特許文献50】Marlinら、Cell、1987年;51:813〜9頁
【非特許文献51】Rothleinら、J Immunol、1994年;152:2488〜95頁
【非特許文献52】Vyth-Dreeseら、Blood、1995年;85:2802〜12頁
【発明の概要】
【発明が解決しようとする課題】
【0006】
アポトーシス誘導作用を有する他の抗体も、現在、臨床的に開発されている。しかし、たとえこれらの抗体が、患者または動物試験において有益な効果を示すとしても、臨床的に満たされていない必要性が依然として存在している。
【0007】
B細胞腫瘍を標的にする抗イディオタイプの免疫グロブリンは、ヒトにおいて実施された最初のモノクローナル抗体療法であった(Millerら、N Engl J Med、1982年;306:517〜22頁)。受動的抗体投与(Riechmannら、Nature、1988年;332:323〜7頁)、または患者自身の主要免疫グロブリンタンパク質による活性ワクチン療法(Kwakら、N Engl J Med、1992年;327:1209〜15頁)のこのような手段による腫瘍細胞の破壊により、それ以来、種々のB細胞悪性疾患に罹っている患者における腫瘍退縮または腫瘍静止をもたらすことが実証されている。最近の報告には、マウスの抗体産生が欠失していて選択されたヒト抗体鎖の遺伝子座を発現する形質転換マウスを用いてヒト抗イデオタイプ完全抗体の作製が記載されている(Suarezら、Mol Immunol、2004年;41:519〜26頁)。
【0008】
本発明においては、細胞全体の形態における標的細胞抗原、および膜小胞の形態における過剰のサブトラクター細胞抗原を、同時に無処置n-CoDeR(登録商標)抗体ファージライブラリー(WO 2004/023140; Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)に曝露し、回収して、Bリンパ腫標的細胞に対して優れた選択性を有する抗体断片を続いて試験する競合バイオパニング法が用いられている。さらに、選択された結合分子における機能性は、標的細胞にアポトーシスを誘導するが、非標的細胞には誘導しない被試験抗体の能力によって実証されている。
【0009】
同定された抗体特異性としては、HLA-DR/DP(本発明のB1抗体)および表面IgM(本発明のC11抗体)、ならびにICAM-1(本発明のB11抗体)、以前はアポトーシス誘導に関連づけられていない接着分子が挙げられる。単離抗体は、ナノモル以下からナノモルの範囲に親和性を有しているため、それらは直接、標的抗体療法にとっての可能な選択肢となる。
【課題を解決するための手段】
【0010】
本発明の第1の態様において、
a.細胞表面抗原ICAM-1を提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する1種または複数の結合分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法が提供される。
【0011】
結合分子は、抗体分子であることが好ましい。
【0012】
本発明の第2の態様において、
a.細胞表面抗原HLA-DR/DPおよび/または表面IgMを提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合し、HLA-DR/DPおよび/または表面IgMとの結合により標的細胞のアポトーシスを誘導する1種または複数の抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の抗体分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法が提供される。
【0013】
本発明の第3の態様において、細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する結合分子が提供される。あるいは、結合分子は、細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合する抗体分子である。
【0014】
ICAM-1は、指定されたCD54でもあるが、本出願の目的にはICAM-1が用いられる。
【0015】
結合分子は、多くのライブラリーで広範囲に使用されている、抗体由来で、抗体スカフォードに基づくものであり得る[Clackson Tら、Nature、1991年8月15日;352(6336):624〜8頁、Marks JDら、J Mol Biol. 1991年12月5日;222(3):581〜97頁]が、結合分子は、フィブロネクチンスカフォード[Weng Sら、Proteomics、2002年1月;2(1):48〜57頁]およびタンパク質Aスカフォード[Nord Kら、Nat Biotechnol、1997年8月;15(8):772〜7頁、Hogbom Mら、Proc Natl Acad Sci U S A、2003年3月18日;100(6):3191〜6頁]などの他の分子スカフォードに由来するものであってもよい。これらのスカフォードの各々は、適用に依存してそれらの利点を有し得るが、一例として、抗体スカフォードは、天然の可変性から識別できる可変性を作出する目的で、有利に使用できる。
【0016】
抗体の基本的構造、最も一般的に用いられるスカフォードは、十分に認識されている。原則として、2つのシートに配列されたベータ鎖を含むフレーム構造が、可変ループのセット、抗原分子に結合する能力を有する、いわゆる相補性決定領域(CDR)を提示する。抗体はスカフォード構造において変わり得るが、最も大きな変動はCDRにおいて見られる。抗体内抗体間の大きな変動が、原則的にはあらゆるタイプの分子構造と特異的様式で相互作用する能力の基礎である。この能力によって、抗体は、研究、疾患の診断/予後における適用性を有する特異的結合剤の創製のため、また、一定の標的構造に特異的な治療薬として、広範囲に用いられている[Borrebaeck CAおよびCarlson R、Curr Opin Pharmacol.2001年8月;1(4):404〜8頁]。
【0017】
本発明に有用な他の非抗体結合分子は、安定性の程度が高く、しかも一定の位置に変動性を導入することを可能にするスカフォード構造を有するものである。他の結合分子の一例は、変動性を許容するフィブロネクチンドメインおよび58個のアミノ酸の大型タンパク質Aドメインである。ある程度の変動を許容する他の分子フォールドもある。このような例としては、腫瘍組織適合性複合体(MHC)クラスIおよびII分子が挙げられ、最近、新規クラスの分子、いわゆるデフェンシン類が、基本構造は同様であるが、遺伝子ファミリーメンバー内メンバー間の大きな配列変動性を有したままであることが確認されており、分子多様性を有するためのスカフォードとして好適であることが示されている。また、標的分子がICAM-1の場合は、天然リガンド(複数可)、例えば、LFA-1またはそれらの組換え変異体が、標的細胞におけるアポトーシスを誘導することのできる特異的結合分子を構成できる。
【0018】
さらに、結合分子は、標的細胞の細胞表面ICAM-1に選択的に結合し、その結合により標的細胞のアポトーシスを誘導する任意の分子であり得る。
【0019】
結合分子は抗体分子であることが好ましい。
【0020】
一実施形態において、細胞表面抗原はICAM-1である。
【0021】
スクリーニングによって、以前はアポトーシスに関連づけられておらず、細胞における内因性の負のシグナル伝達特性に帰せられていなかった受容体であるICAM-1に特異的な抗体(B11)が回収された。
【0022】
それぞれの性質についての前もっての知見に関わらず、またその知見なしに標的細胞と非標的細胞との間で差異的に発現した全ての表面受容体に対する特異性を単離するために設計されているスクリーニングの直接的結果が、アポトーシス誘導性分子としてのICAM-1の同定であった。ICAM-1誘導細胞死は、ミトコンドリア膜の脱分極を含む活性アポトーシス過程として検証された。ミトコンドリア膜の脱分極は、カスパーゼ依存性アポトーシスとカスパーゼ非依存性アポトーシス双方に関して以前に記載されている(Nagyら、J Mol Med、2003年;81:757〜65頁)。
【0023】
本発見によってさらに、B11抗体によって結合されたエピトープは、種々の出所のBリンパ腫組織に発現し、静止末梢血の白血球に比較して、一定のBリンパ腫細胞でアップレギュレートされることが示されている。Bリンパ腫細胞に加えて、ICAM-1を発現する癌腫細胞もまた、インビトロでICAM-1特異的なB11抗体に供されるとアポトーシスを受けることは重要である(実施例6を参照)。
【0024】
正常なヒト組織上ではICAM-1の発現が制限されていることが、先行の研究で実証されている(Smithら、J Clin Pathol、1990年;43:893〜900頁)。ICAM-1は、細胞対細胞の接着に関与し、その受容体、LFA-1に対する結合を介して、免疫応答および炎症に重要な役割を果たしている。病理的な免疫応答および炎症に干渉するために、ICAM-1に特異的な抗体が用いられている。カニクイザルにおけるマウス抗ICAM-1 mAbのインビボ投与(Cosimiら、J Immunol、1990年;144:4604〜12頁)、またはリウマチ様関節炎を患っているヒト患者または腎臓移植を受けている患者における臨床試験での使用でも、顕在的な毒性は見られなかった(Kavanaughら、Arthritis Rheum、1994年;37:992〜9頁;Haugら、Transplantation、1993年;55:766〜72頁)。
【0025】
ICAM-1標的化がアポトーシスを導くことができるという新たな発見により、抗原を発現するという条件での種々の出所の癌の治療用抗体などのICAM-1特異的結合分子の使用の可能性が示される。
【0026】
ICAM-1の発現に基づき、B11などのアポトーシス誘導性抗ICAM-1抗体によって治療できる可能性を有する癌のタイプとしては: Bリンパ腫、骨髄腫(Huangら(1993) Hybridoma 12、661〜75頁; Huangら、(1995) Cancer Res 55、610〜6頁; Smallshawら、(2004) J Immunother 27、419〜24頁)、胃癌(Maruoら、(2002) Int J Cancer 100、486〜90頁)、乳癌(Rosetteら、(2005) Carcinogenesis 26、943〜50頁)、肝臓癌(Sunら、(1999) J Cancer Res Clin Oncol 125、28〜34頁)、肺癌(Grotheyら、(1998) Br J Cancer 77、801〜7頁)、黒色腫(Wangら、(2005) Int J Cancer 27、419〜24頁)、膀胱癌(Rocheら、(2003) Thromb Haemost 89、1089〜97頁)および前立腺癌(Aalinkeelら、(2004) Cancer Res 64、5311〜21頁)が挙げられる。ICAM-1の発現はまた、ICAM-1 特異的抗体を用いて転移過程に介入する可能性を指摘している(Maruoら、2002年)、(Rosetteら、2005年)、(Sunら、1999年)、(Grotheyら、1998年)、(Aalinkeelら、2004年)によって実証されているように、腫瘍転移においても確認されている。
【0027】
さらなる実施形態において、細胞表面抗原はHLA-DR/DPである。
【0028】
HLA-DR/DPは通常、例えば、B細胞上に存在し、Bリンパ腫細胞上でアップレギュレートされるのを見ることができる。
【0029】
現在までに、3種の異なるHLA-DR特異的モノクローナル抗体が臨床相試験に入っている。その中で一番最近になって加わったのは、n-CoDeR(登録商標)に比較して、同様なサイズだが、精製抗原上にパニングしている固体相を用いる無処置ファージライブラリーから単離された完全ヒトIgG41D09C3である(Nagyら、Nat Med、2002年;8:801〜7頁)。
【0030】
本発明において、多数のBリンパ腫細胞系において、速やかに、そして高い効力でアポトーシスを誘導する、HLA-DR/DPに特異的な新規なヒト抗体(B1)が同定され、それによって、HLA-DR/DPが、標的細胞におけるアポトーシスの誘導に関連していることが実証された。
【0031】
B1抗体は、種々の出所の多数のBリンパ腫細胞系に結合し(実施例1および表1を参照)、HLA-DR/DP抗原発現細胞にアポトーシスを誘導することが示された。さらに、B1抗体は、ラージBリンパ腫細胞系で試験した際、リツキシマブよりも高い効力を示した。B1抗体のIgG4フォーマットが使用された際は、これが特に明瞭であった(図8)。
【0032】
得られたデータによると、この抗体は、HLA-DR/DP発現Bリンパ腫細胞の治療に好適な特性を有する。また、リツキシマブによるリウマチ様関節炎およびSLEの標的化と同様に、B1抗体は、HLA-DR/DP発現B細胞が有害である障害における活性化B細胞の除去に有効であることを立証し得る。
【0033】
さらなる実施形態において、細胞表面抗原は表面IgMである。
【0034】
遊離形態におけるIgMは、大型の五量体構造として存在し、その高分子量によって、血管内に限定される傾向がある。
【0035】
単量体IgMは、Bリンパ球の細胞壁上に見ることができ、抗原認識に関する抗体受容体として働く。
【0036】
本発明のC11抗体は、Bリンパ腫細胞上に発現した表面IgMに結合すると、迅速で効率的な様式でアポトーシスを誘導する(実施例1および表1を参照)。Bリンパ腫の治療のために、以前臨床で用いられたイディオタイプ特異的な抗IgM抗体とは対照的に、C11抗体は、種々のドナーからのB細胞上に発現した非多型エピトープに結合し、したがって、Bリンパ腫のイディオタイプに関わりなく、Bリンパ腫を患っている患者の治療に好適である。
【0037】
特に、エフェクター分子、例えば、抗IgM抗体はまた、種々のイディオタイプのIgM発現細胞におけるアポトーシスを引き起こす、任意の種類の特異的結合分子であり得る。
【0038】
B1、B11およびC11 IgG誘導アポトーシスの動態は速やかであり、いくつかの細胞系においては、3時間後にすでに最高の効力が見られた。速やかなエフェクター機能は、例えば、腫瘍抗原の発現不足(Uyttenhoveら、J.Exp.Med.、1983年;157:1040〜52頁;Kennedyら、Br J Haematol、2002年;119:412〜6頁)またはエピトープ成熟(Weinerら、J Immunol、1989年;142:343〜51頁;Baiら、J.Clin.Invest.、2003年;111:1487〜96頁)から生じる腫瘍脱出の危険性を最小化し、治療期間および副作用を制限する可能性がある(Robertら、Lancet Oncol、2005年;6:491〜500頁)ため、治療の有効性にとって重要である。
【0039】
好ましくは、標的細胞は免疫細胞または上皮細胞であり、有利には、免疫細胞はBリンパ球である。
【0040】
好都合には、標的細胞は疾患に関連している。疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択されることが好ましい。
【0041】
有利には、疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0042】
本出願の定義の節で定義されるとおり、抗体分子という語句は、便宜上用いられ、他にある中でも、抗体、抗体断片、および抗体の誘導体を包含する。
【0043】
好都合には、抗体分子はIgGである。好ましくは、IgGは、IgG1、IgG2、IgG3またはIgG4のいずれかであり得るが、IgG1およびIgG4のいずれかである。好ましくは、抗体分子はヒト化抗体またはヒト抗体である。
【0044】
好都合には、本発明の結合分子または抗体分子は、図9から11の可変領域配列のいずれか一つの配列または機能的に等価なその相同体を有する。
【0045】
本発明の一実施形態において、結合分子または抗体分子は、図9の可変領域配列または機能的に等価なその相同体を有する。
【0046】
本発明のさらなる実施形態において、結合分子または抗体分子は、図10の可変領域配列または機能的に等価なその相同体を有する。
【0047】
本発明のさらなる実施形態において、結合分子または抗体分子は、図11の可変領域配列または機能的に等価なその相同体を有する。
【0048】
本発明の第4の態様において、前記請求項のいずれかに記載の結合分子または抗体分子をコードするヌクレオチド配列を有する核酸が提供される。
【0049】
好都合には、核酸は、図9から11のいずれか一つのヌクレオチド配列を有する。
【0050】
本発明の第5の態様において、標的細胞の破壊を必要とする疾患の診断および/または治療および/または予防における、本発明の第1の態様また第2の態様に記載の結合分子または抗体分子の使用が提供される。また、標的細胞の破壊を必要とする疾患の治療および/または予防のための薬剤の製造における、本発明の第1の態様また第2の態様に記載の結合分子または抗体分子の使用が提供される。
【0051】
好ましい一実施形態において、結合分子は抗体分子である。
【0052】
好都合には、治療される疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される。
【0053】
有利には、治療される疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0054】
本発明の一実施形態において、結合分子または抗体分子は、ICAM-1に特異的に結合し、および/または図10の配列を有し、上記に挙げられた疾患に関連して使用される。
【0055】
本発明のさらなる実施形態において、抗体分子は、HLA-DR/DPに特異的に結合し、および/または図9の配列を有し、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌の疾患に関連して使用される。
【0056】
本発明のさらなる実施形態において、抗体分子は、表面IgMに特異的に結合し、および/または図11の配列を有し、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌の疾患に関連して使用される。
【0057】
本発明の第6の態様において、本発明の結合分子または抗体分子と、薬学的に許容される担体、賦形剤または希釈剤とを含む医薬組成物が提供される。
【0058】
好ましい一実施形態において、結合分子は抗体分子である。
【0059】
本発明の第7の態様において、
(i)1種または複数の標的細胞を用意するステップと、
(ii)本発明の第1の実施形態に記載の1種または複数の結合分子または抗体分子を用意するステップと、
(iii)(i)の標的細胞を(ii)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと
を含む、標的細胞にアポトーシスを誘導するインビトロ方法が提供される。
【0060】
好ましい一実施形態において、結合分子は抗体分子である。
【0061】
好ましくは、ステップ(i)で用意される標的細胞は、免疫細胞または上皮細胞である。有利には、免疫細胞はBリンパ球である。
【0062】
好都合には、該標的細胞は疾患に関連しており、該疾患は、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性ならびに慢性の炎症性障害、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される。
【0063】
有利には、疾患は、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である。
【0064】
使用される用語の意味
用語「抗体分子」は、抗体、抗体断片、または抗体の誘導体のいずれか一つを称することとする。これは、野生型抗体、合成抗体、組換え抗体または、それだけに限定されないが、免疫グロブリンの軽鎖および/または重鎖可変領域および/または定常領域のファージディスプレーによって製造される一本鎖修飾抗体分子などの抗体ハイブリッド、または当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる他の免疫相互作用分子を包含することが意図されている。
【0065】
用語「抗体断片」は、抗体、抗体断片、または抗体誘導体のいずれか一つを称することとする。これは、野生型抗体(すなわち、4本のポリペプチド鎖を含む分子)、合成抗体、組換え抗体または、それだけに限定されないが、免疫グロブリンの軽鎖および/または重鎖可変領域および/または定常領域のファージディスプレーによって製造される一本鎖修飾抗体分子などの抗体ハイブリッド、または当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる他の免疫相互作用分子を包含することが意図されている。
【0066】
用語「抗体誘導体」とは、当業者に知られている免疫アッセイフォーマットにおいて抗原に結合することのできる、抗体の断片(例えば、Fab断片またはFv断片)、または、他のペプチドまたはポリペプチド、大型担体タンパク質または固体支持体への該抗体の結合を促進させるために、1種または複数のアミノ酸または他の分子(例えば、中でも、アミノ酸、チロシン、リシン、グルタミン酸、アスパラギン酸、システインおよびそれらの誘導体、NH2-アセチル基またはCOOH-末端アミド基)を付加することによって修飾される修飾抗体分子などの任意の修飾抗体分子を称する。
【0067】
用語「ScFv分子」とは、VHおよびVLパートナードメインが、可撓的オリゴペプチドを介して結合している任意の分子を称する。
【0068】
用語「ヌクレオチド配列」または「核酸」または「ポリヌクレオチド」または「オリゴヌクレオチド」は、交換可能に用いられ、ヌクレオチドのヘテロポリマーまたはこれらのヌクレオチドの配列を称する。これらの語句はまた、一本鎖または二本鎖であり得、センス鎖またはアンチセンス鎖を表し得る、ゲノム起源または合成起源のDNAまたはRNA、ペプチド核酸(PNA)または任意のDNA様またはRNA様物質も称する。本明細書の配列において、Aはアデニンであり、Cはシトシンであり、Tはチミンであり、Gはグアニンであり、NはA、C、GまたはT(U)である。該ポリヌクレオチドがRNAである場合、本明細書に提供される配列内のT(チミン)は、U(ウラシル)で置換されることが考慮されている。一般に、本発明により提供される核酸セグメントは、ゲノムおよび短いオリゴヌクレオチドリンカーの断片から、または一連のオリゴヌクレオチドから、または個々のヌクレオチドから組み立てることができ、微生物もしくはウィルスのオペロン、または真核生物の遺伝子に由来する調節要素を含む組換え転写単位において発現され得る合成核酸を提供することができる。
【0069】
用語「ポリペプチド」または「ペプチド」または「アミノ酸配列」とは、オリゴペプチド、ペプチド、ポリペプチドまたはタンパク質の配列またはそれらの断片、ならびに天然または合成分子を称する。ポリペプチドの「断片」、「部分」、または「セグメント」は、少なくとも約5個のアミノ酸、好ましくは、少なくとも約7個のアミノ酸、より好ましくは、少なくとも約9個のアミノ酸、最も好ましくは、少なくとも約17個以上のアミノ酸のアミノ酸残基のひと続きである。活性であるためには、いずれのポリペプチドも、生物学的および/または免疫学的活性を示すための十分な長さを有する必要がある。
【0070】
本明細書で用いられる用語「精製した」または「実質的に精製した」は、指示された核酸またはポリペプチドが、他の生物学的高分子、例えば、ポリヌクレオチド、タンパク質などの実質的な不在下に存在することを意味する。一実施形態において、ポリヌクレオチドまたはポリペプチドは、存在する指示された生物学的高分子(しかし、水、緩衝液、および他の小型分子、特に、1000ダルトン未満の分子量を有する分子は存在し得る)の少なくとも95重量%、より好ましくは、少なくとも99重量%を構成するように精製される。
【0071】
本明細書で用いられる用語「単離された」とは、天然源における核酸またはポリペプチドと共に存在する少なくとも1種の他の成分(例えば、核酸またはポリペプチド)から分離された核酸またはポリペプチドを称する。一実施形態において、核酸またはポリペプチドは、該核酸またはポリペプチドの溶液中に通常存在する溶媒、緩衝液、イオン、または他の成分がある場合、それらのみの存在下で見られる。用語「単離された」および「精製された」は、その天然源に存在する核酸またはポリペプチドを包含しない。
【0072】
本明細書に用いられる場合の用語「組換え」とは、ポリペプチドまたはタンパク質を称し、ポリペプチドまたはタンパク質が、組換え(例えば、微生物、昆虫、または哺乳動物)発現系に由来することを意味する。「微生物」とは、細菌または真菌(例えば、酵母)発現系において作製された組換えポリペプチドまたは組換えタンパク質を称する。生成物としての「組換え微生物」は、本質的に天然の内因性物質が無く、関連した天然グリコシル化が付随していないポリペプチドまたはタンパク質として定義される。多くの細菌、例えば、大腸菌の培養物中に発現するポリペプチドまたはタンパク質は、グリコシル化修飾が無く、酵母中に発現するポリペプチドまたはタンパク質は、哺乳動物細胞中に発現するものとは一般に異なるグリコシル化パターンを有する。
【0073】
用語「選択的結合」および「結合選択性」は、本発明の抗体の可変領域が、本発明のポリペプチドを排他的に認識して結合する(すなわち、ポリペプチドファミリーに見られる配列同一性、相同性、または類似性に関わらず、他の類似ポリペプチドから本発明のポリペプチドを識別できる)が、該抗体の可変領域の外側、特に、該分子の定常領域における配列との相互作用を介して、他のタンパク質(例えば、黄色ブドウ球菌のタンパク質AまたはELISA法における他の抗体)と相互作用できることを表す。本発明の抗体の結合選択性を判定するスクリーニングアッセイはよく知られており、当業界でルーチンに実施されている。このようなアッセイの包括的考察に関しては、Harlowら(編)、Antibodies A Laboratory Manual;Cold Spring Harbor Laboratory:コールドスプリングハーバー、ニューヨーク(1988)、第6章を参照されたい。本発明のポリペプチドの断片を認識し結合する抗体も、該抗体が上記に定義したとおり、本発明の完全長ポリペプチドに対して、一番に最も選択的であるという条件で、考慮されている。本発明の完全長ポリペプチドに対して選択的な抗体であれば、断片を認識する本発明の抗体は、タンパク質のファミリーに見られる固有な配列同一性、相同性、または類似性に関わらず、ポリペプチドの同じファミリーからポリペプチドを識別できる抗体である。
【0074】
用語「結合親和性」は、抗体分子と抗原との間の結合の強さの意味を含む。
【0075】
用語「免疫細胞」は、それだけに限定されないが、B細胞およびT細胞などの宿主の免疫または炎症応答に関与している任意の細胞を意味する。
【0076】
用語「上皮細胞」は、上皮の細胞を意味する。上皮は、細胞の層からなる組織である。上皮は、身体の内張りの内部(例えば、血管の内側を内張りしている内皮)または外部(例えば、皮膚)の開放表面に見ることができる。
【0077】
口腔および体腔の内側が粘膜に裏打ちされているように、我々の皮膚の最外側層は扁平上皮細胞からなっている。他の上皮細胞は、肺、胃腸管、生殖管および尿管の内側を裏打ちし、エポクリン腺および内分泌腺を構成している。上皮細胞の機能としては、分泌、吸収および保護が挙げられる。上皮細胞は基底層上にある。
【0078】
次に、本発明の一定の好ましい態様を具体化している実施例を、下記の図を参照にして記載する。
【図面の簡単な説明】
【0079】
【図1】図1A及びBは、差異的全細胞/細胞膜小胞バイオパニングによって単離されたscFvが、標的細胞高特異性を示す図である。差異的バイオパニングによって単離されたscFvクローンを、大腸菌TOP10細胞中に発現させ、ラモス細胞またはジャーカット細胞および一次スクリーニングのために発現させたscFvクローンと共にインキュベートした。結合したscFvを、抗His MAb、およびCy5-抗マウスポリクローナルAbによって検出した。細胞結合を、FMAT Macroconfocal High Throughput Screening機器において検出した。細胞結合は、標的ラモス細胞(Y軸)対非標的ジャーカット細胞(X軸)に対する平均蛍光強度として示した。図1Cは、差異的全細胞/細胞膜小胞バイオパニングによって単離されたscFvが、標的細胞高特異性を示す図である。ラモス細胞(黒色バー)およびジャーカット細胞(白色バー)に対する7つのユニークなscFvクローンの結合性。対照scFv(ctrl)は、いずれの細胞にも結合しなかった。
【図2A】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2B】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2C】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2D】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図2E】抗ラモスscFvのアポトーシス誘導を示す図である。ラモスBリンパ腫細胞を、抗ラモスscFv、抗His mAb、および抗マウスポリクローナルAbと共に、氷上で、経時的にインキュベートし(過剰の非結合抗体を除去するために断続的に洗浄しながら)、5% CO2の加湿雰囲気下、37℃で24時間インキュベートした。次いで、細胞を採集し、Annexin V-AF488(AV)およびヨウ化プロピジウム(PI)による組み合わせ染色に供した。細胞を、AVに関する陽性の差異およびPI染色に基づいて(図2Bの四角ゲートにより規定された)、生存(AV-PI-、図2Cの黒丸)、早期アポトーシス(AV+PI-、図2Cの白色三角)、または後期アポトーシス/壊死(AV+PI+、図2Cの白色菱形)としてスコア化した。結果は、(A)側方散乱に対する前方散乱(FSC-高さ) および(B) PI (FL-3)に対するAV (FL-1)をプロットすることによって示される。scFv B1およびF1の滴定効果もまた示される(C)。7種のユニークなscFvクローンを、種々の濃度で、(D)ラモスまたは(E)ラージBリンパ腫細胞と共に、37℃で24時間インキュベートし、アポトーシス誘導に及ぼす効果を調べた。3種のscFv:B1、B11、およびC11は、双方の細胞系に対して、滴定可能な活性を示すが、scFvB10、C10、およびG12のアポトーシス誘導能力は、ラモスBリンパ腫細胞に限定されている。
【図3】図3Aは、HLA-DR/DP、IgM、およびICAM-1を含む単離抗体の特異性を示す図である。50〜600×106ラージBリンパ腫細胞を、非イオン性洗浄剤、0.5%v/vでのトリトンX-100により溶解し、B1(レーン1)抗体およびB11(レーン2)抗体の完全ヒトIgG1フォーマット100μgによって免疫沈降させ、続いて、Protein A Sepharoseによって架橋した。C11(レーン3)20μgの免疫沈降に、50×106細胞からのラモスBリンパ腫細胞ライセートを用いた。抗体特異的なバンドを切除し、トリプシン消化に供し、MALDI-TOFにより分析した。図3Bは、HLA-DR/DP、IgM、およびICAM-1を含む単離抗体の特異性を示す図である。Bリンパ腫細胞に対するB1 IgG、B11 IgG、およびC11 IgGの結合を、抗HLA-DR/DP抗体、抗ICAM-1抗体または抗IgM抗体、それぞれと共にプレインキュベートすることによって、特異的にブロックする。抗体クローンB1、B11、およびC11の回収したMALDI-TOF抗原の特性を確認するために、市販の抗体を用いたブロック試験を実施し、フローサイトメトリーによって分析した。種に適合させたブロック用抗体の10倍モル過剰(ヒト抗体に比較して)により細胞を1時間プレブロックし、続いて、単離されたヒト抗体クローンのいずれかを添加した。30分後、細胞を洗浄し、細胞へのヒト抗体の結合を、PE結合ヤギ抗ヒトIgG(Caltag Laboratories、バーリンガム、カリフォルニア州、米国)によって検出した。この試験に用いられたブロック用抗体は:B1に関しては、マウスモノクローナル抗HLA DR(Sigma、クローンHK14)または抗CD40(Beckton Dickinson、クローン5C3);B11に関しては、ウサギポリクローナル抗ICAM-1(Abcam、ab 7815-250)または抗CD22(Abcam、ab25135-100);C11に関しては、ヤギポリクローナル抗IgM(Zymed、サウスサンフランシスコ、カリフォルニア州、米国62-7500)または抗IgG(Zymed、62-8400)であった。
【図4A】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図である。2μg/mlのB11または抗FITC-8(対照)IgGを、4×105のCL-01 Bリンパ腫細胞に加え、氷上で2時間インキュベートし、引き続いて、10μg/mlの架橋用二次Fab'2ヤギ抗ヒトFc抗体を加えた。細胞を、37℃で6時間インキュベートし、抗体インキュベーションの効果を2つの独立したアポトーシスアッセイによって判定した。上記と同様に、AV/PI(上枠)によるか、または5μg/mlのミトコンドリア膜脱局在化試薬JC-1と共に、室温で30分間インキュベーションすることにより(下枠)、細胞を染色した。アポトーシスの誘導は、赤色(y軸)/緑色(x軸)蛍光強度比の低下によって検出可能である。
【図4B】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図であり、Bリンパ腫組織に対するB11抗体の代表的な結合性を示す組織学的切片を示す図である。未分化大型細胞Bリンパ腫を患っている患者から得られた凍結保存組織を、B11またはFITC-8(対照) scFv抗体によって染色した。抗体結合を、DAB(褐色)によって検出した。差込図は、対照scFvによる染色を示す。
【図4C】ICAM-1が、プログラム細胞死を媒介できるB細胞種関連細胞表面受容体であることを示す図である。CD45-PerCp-Cy5.5 mAb予備標識ラモス細胞を、ドナー由来のPBMCsと混合し、異なる細胞集団を、蛍光色素結合CD特異的抗体およびAlexa Flour647 Zenon予備標識B11 IgG1または対照FITC-8 IgG1によって染色した。異なる細胞集団に対するIgG B11結合を、FL4チャネルに記録した。
【図5】Bリンパ腫細胞に対するIgG B1およびIgG B11の親和性を示す図である。増量させた放射ヨウ素化IgG B1タンパク質または放射ヨウ素化IgG B11タンパク質と共に、0.2mg/mlの対応する非標識IgGタンパク質の存在下、または非存在下、ラージ細胞(左枠、IgG B1)またはラモス細胞(右枠、IgG B11)をインキュベートした。全体の結合から、非標識競合タンパク質の存在下での結合を差し引くことによって、特異的結合を判定した。遊離IgGタンパク質増量させることにより、結合IgG B1タンパク質またはIgG B11タンパク質の量は増加し、それぞれ、約30nM IgG B1および約1nM IgG B11で飽和に達した(上枠)。ローゼンタール-スキャッチャードプロット分析(下枠)により、IgG B1に関しては、ラージ細胞1個当たり、400,000の機能的結合部位で約3nMの解離定数であり、IgG B11(ラージ細胞)に関しては、47,400の機能的結合部位で約0.3nMの解離定数であることが実証された。
【図6】種々の出所の腫瘍細胞系に対するB1、B11、C11 Ig G1の結合を示す図である。種々の癌細胞系について、B1、B11、およびC11抗体により標的にされた抗原の抗原分布を、フローサイトメトリーによって調べた。示されるように、MCF-7乳癌、HPAC膵臓癌、PC-3前立腺癌、LS174 T結腸直腸癌、およびTHP-1単球白血病細胞に対するリツキシマブ抗CD20 Mab(1列目、最も前方のピーク)、B1 IgG1(2列目のピーク)、B11 IgG1(3列目のピーク)、またはC11 Ig G1(4列目、最も背部のピーク)の結合が、ヒストグラムによって示されている。
【図7A】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。輪郭プロットは、2μg/mlのIgG B11またはIgG B1と共に上記のとおりインキュベーションした後の、Annexin Vおよびヨウ化プロピジウム陽性の関数としてのPC-3細胞の相対的分布を示す。
【図7B】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、連続希釈したB11 IgG1または20μg/mlのB1 IgG1と共にインキュベーションした後のアポトーシスPC-3細胞の平均パーセンテージを示す。
【図7C】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、抗体無しの対照、10μg/mlの陰性抗体対照、連続希釈B11 IgG1、または10μg/mlのトラスツズマブIgG1と共にインキュベーションした後のアポトーシスMDA MB 435細胞の平均パーセンテージを示す。
【図7D】癌細胞におけるB11 IgG1アポトーシス誘導を示す図である。前立腺癌細胞系PC-3を、完全増殖培地(10% FCS、10mMのHEPES、および2mMのL-グルタミンを添加したRPMI 1640)中、6ウェルプレートで80%の集密度まで増殖させた。前立腺癌細胞系DU145を、10% FCS、1mMのピルビン酸ナトリウム、および1mMの非必須アミノ酸を添加したアール塩と共に、MEM中で増殖させ、黒色腫細胞系MDA MB435の派生株を、10% FCSを添加したDMEM中、増殖させた。アポトーシスアッセイのため、細胞をPBS中で洗浄し、個々のウェルに、連続希釈したB11 IgG1(またはB1 IgG1、対照として、トラスツズマブまたは陰性抗体対照)を加え、4℃で1〜2時間のインキュベーションの間、結合させた。該細胞を洗浄し、架橋用抗体、Fab'2ヤギ抗ヒトFab'2を10μg/mlで含有した完全増殖培地を加えた。細胞を、5% CO2の加湿雰囲気下、37℃で16〜24時間インキュベートした。細胞をトリプシン処理により回収し、Alexa Fluor 488- Annexin V (AF488-AV)およびヨウ化プロピジウム(PI)により、製造元の指示に従って染色した。式: %アポトーシス細胞= 100-%AF488-AV/PI-/-によって、アポトーシス細胞のパーセンテージを判定した。棒グラフは、抗体無しの対照、10μg/mlの陰性抗体対照、連続希釈B11 IgG1、または10μg/mlのトラスツズマブIgG1と共にインキュベーションした後のアポトーシスDU145細胞の平均パーセンテージを示す。
【図8】架橋試薬の非存在下で、B1 IgG 1およびB1 IgG4が、ラージBリンパ腫細胞に直接細胞細胞毒性を誘導することを示す図である。ラージ細胞を、B1 IgG 1、B1 IgG4、リツキシマブIgG 1、または対照CT-17 IgG 1と共に、20、6.7または2.2μg/mlで、24時間インキュベートした。細胞を採集し、生存度を、Annexin Vおよびヨウ化プロピジウム二重陰性細胞のパーセンテージとして判定した。
【図9A】AB1抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図9B】図9Aの続きである。
【図10A】B11抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図10B】図10Aの続きである。
【図11A】C11抗体に関するVH配列およびVL配列(ヌクレオチドおよびアミノ酸)の配列を示す図である。
【図11B】図11Aの続きである。
【発明を実施するための形態】
【0080】
実施例1
Bリンパ腫関連細胞表面受容体に特異性を有するアポトーシス誘導性抗体に関する選択およびスクリーニング(バイオパニング)
細胞培養
別に記述しない限り、この試験に用いられる細胞系は、ATCC(マナサス、バージニア州、米国)またはDeutsche Sammlung von Mikroorganismen und Zellkulturen(DSMZ)GmbH(Braunschweig、ドイツ国)から入手し、10% FCS、2mMのL-グルタミン、10mMのHEPESおよび1mMのピルビン酸Na(全て、Invitrogen、カールスバッド、カリフォルニア州、米国から)を添加したRPMI 1640培地中で培養した。ジャーカットT白血病細胞系(クローンE6-1、TIB-152、ATCC)、Bリンパ腫細胞系DOHH-2 (ACC47、DSMZ)、SC-1 (ACC558、DSMZ)、WSU-NHL (ACC58、DSMZ)、JVM-2 (ACC12 DSMZ)、Jeko-1 (ACC553、DSMZ 、20%FCS中で増殖)、Rec-1 (ACC 584、DSMZ)、SP-53 (Daibataら、Cancer 1989年;64:1248〜53頁)、RL (CRL-2261、ATCC)、Granta 519 (DSMZ)、NCEB-1 (Saltmanら、Blood 1988年;72:2026〜30頁)、BJAB (Menezesら、Biomedicine 1975年;22:276〜84頁)、ラモス(CRL-1596、ATCC)、ラージ(CCL-86、ATCC)、Daudi (CCL-213、ATCC)、CL-01 (Ceruttiら、J Immunol 1998年;160:2145〜57頁)、プレB細胞リンパ腫KM-3/Reh (CRL-8286、ATCC)および多発性骨髄腫MC/CAR (CRL-8083、ATCC、20% FCSで添加されたIMDM (Invitrogen)中で増殖)は全て、マイコプラズマを含有せず、5% CO2雰囲気を用い、37℃で加湿雰囲気中で培養した。この細胞を2×105〜1×106細胞/mlで維持した。
【0081】
ジャーカット細胞膜小胞の調製
ジャーカット細胞を、500mlのバケット(Corning社、Life Sciences、ニューヨーク、米国)中、300×gで15分間の遠心分離により採集し、ダルベッコーPBS(Invitrogen)中で洗浄し、緩衝液A(1mMのNaHCO3、1.5mMのMgAc、pH7.4)中に再懸濁した。細胞濃度は、およそ、5×107ジャーカット細胞/ml(100mlの緩衝液A中、5×109細胞)であった。
【0082】
細胞破壊は、氷上、10〜30分間の低浸透圧ショック処理(緩衝液A)、および引き続く窒素キャビテーションボンベ(Parr Instrument社、モリン、イリノイ州、米国)中の窒素キャビテーションによって達成した。細胞は、40バール(4,000kPa)の一定圧で、0℃に15分間保持した。
【0083】
破壊細胞を、0.5MのEDTA 500μlを含有する250mlのザルステット管(Sarstedt AG & Co、ニンブレヒト、ドイツ国)内に採集し、2.5mMの最終EDTA濃度を得た。EDTAの添加により、膜小胞の凝集が防止される。ホモジェネート(100ml)を、4×25mlのBeckman厚壁ローター管(Beckman Coulter社、フラートン、カリフォルニア州、米国)に分け、これらを、4℃で10分間、1900×g(Sorvall SS34ローターにおいて4,000rpm)で遠心分離して、破壊されなかった細胞、核、および重いミトコンドリアを除去した。
【0084】
上澄み液を回収し、ペレット化した物質を1mMのEDTAを含有する1mMのNaHCO3緩衝液25ml中に再懸濁し、再遠心分離した(ペレット化した粗製ジャーカット膜のさらなる回収)。ジャーカット膜を最初の遠心分離からの膜と共にプールした。粗製ジャーカット膜小胞を含有する上澄み液を、Beckman 45Ti型ローターを用いて、4℃で2.5時間、40,000rpm(およそ200,000×g)で超遠心分離した。上澄み液を捨て、組織(例えば、Kleenex(商標))に対して該管縁を傾けることによって残りの緩衝液を除去した。
【0085】
粗製膜ペレットを金属棒の補助でDounceホモジェナイザーに移し、該ホモジェナイザー内で数回慎重になでつけることによって、2.5mlのHES緩衝液(10mMのHepes、1mMのEDTA、0.25Mのスクロース、pH7.4)中に再懸濁した。このようにして、80〜100mgのタンパク質を含有する2×109細胞/mlに等価の膜懸濁液濃度が得られた。
【0086】
全細胞/細胞膜小胞競合バイオスパニングによるファージAbsの選択
およそ2×1013のファージ粒子を、断続的に混合しながら、37℃で15分間予備加温し、14,000×gで15分間遠心分離して、沈殿物を除去し、上澄み液を新鮮なエッペンドルフ管に移した。無脂肪乾燥乳を、2%(w/v)の最終濃度まで加えた。2×109細胞から誘導されたジャーカット膜小胞調製物(1回目選択;2×108細胞2回目および3回目選択)を氷上で解凍し、ブロックしたファージ粒子と共に混合した。混合物を氷上で15分間インキュベートした。
【0087】
5×107 (5×106 2回目および3回目)のラモス細胞を4℃で6分間、1,200rpmでの遠心分離により回収した。上澄み液を廃棄し、ラモス細胞を乳-ファージ-ジャーカット膜小胞混合物中に再懸濁した。該懸濁液を、4時間の緩やかな転倒型回転下、10℃でインキュベートした。
【0088】
2%(w/v) BSA/PBS(フィコール-ピラー)中、下部に、0.5mlの100%(トリパンブルー染色)、フィコール-パックPLUS(Amersham Biosciences、ウップサラ、スウェーデン国)、および9.5mlのオーバーレイド40%(v/v)フィコールを含有する15mlのファルコン管(BD Biosciences、ベッドフォード、マサチューセッツ州、米国)に移した。該管を、4℃で10分間、1,500rpmで遠心分離した。該管を遠心分離機から取り出し、管のキャップをねじ締めし、気密シールした。
【0089】
100%フィコールを含有する該ファルコン管の底部「先端」をシガーチョッパーを用いて切り開いた。このようにして、膜小胞シートおよび細胞核を含むきわめて高濃度の物質を該管から排出させた。次いで、管のキャップを慎重に開けると、管内部の減圧状態が破られ、(切り開かれた)管底部の開口部を通って液体が滴状に放出される。
【0090】
回収した細胞懸濁液をPBS中で1回洗浄して過剰のフィコールを除去した。該ペレットを、1mlのPBS中に再懸濁し(最終洗浄後に実施しなかった)、該懸濁液を、新鮮なフィコール-ピラー上部に再充填し、洗浄操作を繰り返した(2回目および3回目に2回)。
【0091】
PBS中76mMのクエン酸(pH2.5) 150μlの添加により、細胞からファージを溶出させ、続いて、室温で5分間インキュベートした。1Mのトリス-HCl、pH7.4 200μlの添加により、該混合物を中和した。溶出したファージを含有する上澄み液を保存し、300×gで5分間、細胞のペレット化を行った。再懸濁によりファージをさらに溶出させ、1mlのトリプシン中、RTで10分間、細胞ペレットをインキュベートした。
【0092】
1mg/mlのアプロチニン40μlによる活性化の後、細胞を遠心分離し、溶出したファージを含有する上澄み液を保存した。溶出したファージを用いて、大腸菌HB101F'に感染させ、該細菌を適切な抗生物質およびグルコースを含有するTB培地に塗布した。細菌コロニーをカウントし、プレートからかき取り、次回のパニング用種菌として用いた。
【0093】
scFvフォーマットへの転換、scFv発現、精製、および細胞結合の解析
3回選択後に得られたファージミドプールを、EagIにより消化して、遺伝子IIIを除去した。得られたベクターを再結合した。再結合させた非切断遺伝子III断片を含有するベクターを、EcoRI酵素を用いた消化によって線状化した。このようにして作出したscFvベクタープールを用いて、基本的には先に記載したとおり、大腸菌TOP10を形質転換した。(Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)
【0094】
細菌を、大型の500cm2の寒天プレートに塗布し、個々のクローンを取り出して96ウェルプレートに移し、自動システム(Hallborn Biotechniques、2002年;補遺:30〜7頁)を用いて、TB培地中、37℃、220rpmで一晩培養することにより発現させた。適切な抗生物質を含有するTB培地中、組換えscFv断片を作製した。
【0095】
標的ラモス細胞およびジャーカット非標的細胞に結合しているscFvクローンの一次スクリーニングのために、5,000のラモス細胞またはジャーカット細胞を、960のscFvクローンのいずれかと共にインキュベートし、3回目の選択から誘導し、上記のとおり作製した。0.5μg/mlの抗6xHis mAb(R&D Systems、ミネアポリス、ミネソタ州、米国)および0.7μg/mlのCy5結合ヤギ抗マウス試薬(Amersham Biosciences)と共に、細胞をインキュベートした。8200 Cellular Detection System Fluorescence Macroconfocal High Throughput Screening(FMAT)装置(Applied Biosystems、フォスターシティー、カリフォルニア州、米国)において、細胞結合を解析した。
【0096】
一次スクリーニング後、以前記載されたDNA配列決定(Soderlindら、Nat Biotechnol、2000年;18:852〜6頁)(Soderlindら、2000年)のために、72の細菌クローンをランダムに取り出した(すなわち、一次スクリーニングにおける標的細胞対非標的細胞の反応性に関わりなく)。フローサイトメトリーによる細胞表面結合の評価のため、ラモス細胞およびジャーカット細胞(双方とも、1試験当たり、2×105細胞で加える)を0.5%w/vのBSA(DPBS-B)を含有するPBS(Invitrogen)中、2〜10μg/mlの濃度で、個々のscFvクローンと共に、1時間インキュベートした。
【0097】
300×gで6分間の遠心分離により細胞を洗浄した。次いで、FITC結合CD19mAbおよびPE結合CD3mAb(BD)と共に細胞をインキュベートし、引き続く標的細胞および非標的細胞それぞれの同定を可能にした。scFv結合の検出は、RPE-Cy5-ストレプトアビジン(Dako Cytomation、グロストラップ、デンマーク国)とのインキュベーション、続いてビオチン化抗6xHis mAbとのインキュベーションにより達成した。第2および第3の試薬により、それぞれ40分間と15分間、細胞をインキュベートした。全てのインキュベーションは、氷冷溶液を用いて氷上で実施した。
【0098】
細胞全体/細胞膜小胞の差異的パニング
本試験は、天然の細胞表面構造において差異的に発現した抗原を標的にする抗体を単離するために、新規のパニングプロトコルを利用した。3回のバイオパニングの後、ラモスBリンパ腫細胞全体およびジャーカットT白血病細胞から誘導した膜小胞を用いて、組換えファージscFvを単離した。これらを可溶性scFvに変換し、大腸菌TOP10細胞内で発現させた。
【0099】
組換えscFvを標的(ラモス) 全細胞または全非標的(ジャーカット) 細胞と共にインキュベートし、細胞結合に関して調べた。発現した482のscFvクローンが、弱からきわめて強の範囲の強度でラモス標的細胞に選択的に結合することが示された(図1A)ため、該抗体クローンの標的細胞抗原に対する特異性は著しかった。2種のクローンだけが、弱く染色された非標的ジャーカット細胞として同定された(図1A)。
【0100】
次に我々は、単離されたファージディスプレーされたscFvの遺伝子型の多様性を判定した。DNA配列決定のために、72のscFvクローンをランダムに取り出した(すなわち、一次スクリーニングで判定された結合親和性に関わりなく)。
【0101】
記載された(図1B)とおり、FMAT法により、標的細胞特異性(ラモス対ジャーカット)に関して、該クローンを同時に再発現および再評価した。7つの異なる抗体遺伝子型を、それらの異なるCDRH3配列およびCDRL3配列によって判定して同定した(データは示していない)。
【0102】
等しい数のラモス細胞およびジャーカット細胞とのインキュベーションおよび抗タグ抗体によるscFv結合の検出の後、3色フローサイトメトリー分析によって、抗ラモスscFvの高特異性が確認された(図1C)。
【0103】
蛍光色素結合CD特異的モノクローナル抗体を用い、CD19およびCD3それぞれの発現により、標的および非標的細胞を確定した。陰性対照scFvに比較して、7種のユニークな遺伝子型のscFvクローンは、標的ラモス細胞に対して高く可変的な結合強度を示したが、非標的ジャーカット細胞に対する結合は示さなかった。
【0104】
アポトーシスアッセイ
製造元(Pierce Biotechnology、ロックフォード、イリノイ州、米国)の指示に従い、Detoxigelカラムを用いて、組換え作製されたscFv断片のリポ多糖濃度を減少させた。残りのエンドトキシン濃度を、LALアメーバ様細胞ライセートアッセイにより定量化した(Cambrex Bioscience、ウオーカースビレ、メリーランド州、米国)。
【0105】
全てのscFvサンプルが、0.1IU/ml未満のリポ多糖を含有することが判明した。キメラ抗CD20抗体のMabthera(商標)(リツキシマブ)を、Lund大学病院(Lund、スウェーデン国)から購入した。2×105のBリンパ腫細胞(ラージまたはラモス)またはジャーカットT細胞を連続希釈して解毒したscFvと共に、氷上で培養培地中、1時間インキュベートした。
【0106】
細胞を、二次抗6xHis mAb(%μg/ml)、および三次ヤギFab'2抗マウスFab'2抗体(Jackson Immuno Research Laboratories社、ウェストグローブ、ペンシルベニア州、米国)と共に経時的にインキュベートした。断続的洗浄により、過剰な非結合抗体試薬の除去を確実にした。細胞を、5% CO2の加湿雰囲気中、37℃で24時間インキュベートした。
【0107】
全IgGsをアポトーシス誘導に用いる場合、架橋剤を、非IgG抗体アイソタイプと最小の交差反応性を有するヤギFab'2抗ヒトFcγ抗体(Jackson ImmunoResearch)と置き換え(内因性Bリンパ腫関連表面免疫グロブリンの非特異的架橋を避けるため)、上記のとおり、6時間インキュベートした。
【0108】
別に記述しない限り、アポトーシス細胞は、Annexin V Alexa Fluor 488(AV)およびPropidium Iodide(PI)(双方とも、Molecular Probes、Invitrogenから)および引き続くフローサイトメトリー分析によって検出し、細胞を、生存(AV-/PI-)、早期アポトーシス(AV+/PI-)または後期アポトーシス/壊死(AV+/PI+)として確定した。AVおよびPIシグナルを、FACSCalibur機器(BD Bioscience)を用いて、FL1およびFL2またはFL3チャネルにそれぞれ記録した(本文に指示されているとおり)。
【0109】
単離されたscFvの機能性を調べるために、我々は、細胞の経時的インキュベーションおよびscFvおよび架橋剤による洗浄に基づき、ハイスループットアポトーシススクリーニングアッセイを設定した。scFvクローンへの依存性および該アポトーシスアッセイにおける濃度は、図2A〜2Cに示されており、選択されたscFvクローンB1のアポトーシス作用が、アポトーシス誘導を示さないscFvクローンF1の作用と比較されている。標的抗原発現を欠失しているジャーカット細胞は、試験したscFvのいずれの処理の後でもアポトーシスにより死滅することはなく、アポトーシス誘導が標的抗原への結合に依存することを実証した(データは示していない)。
【0110】
確立されたscFv-アポトーシスアッセイを用いて、ラモスおよびラージBリンパ腫細胞におけるアポトーシスに関して、クローンをスクリーンした。scFv誘導アポトーシス作用を、リツキシマブ抗CD20 mAbにより誘導されたものと比較した(図2D)。ラモスおよびラージ細胞双方に有意なアポトーシスを誘導した3種のscFvクローン、B1、B11およびC11が同定された(図2DおよびE)。ラージ細胞に結合しなかったscFvクローンはアポトーシスを誘導しなかったため、ラージ細胞におけるscFvによるアポトーシスの誘導は、これらの細胞への結合性に相関的であった(図2D)。
【0111】
B1、B11およびC11クローンを、完全ヒトIgG1抗体に移入した。広範囲群のBリンパ腫細胞系に対する強力な結合性および強力な細胞毒性によって実証される(表1)ように、再フォーマット後もそれらの特異性および機能性は損なわれないままであった。特に、いくつかの細胞系において、3時間から6時間後にはすでに、アネキシンV陽性アポトーシス細胞の最高パーセンテージに達しており、アポトーシス誘導は迅速であった(表1、データは示していない)。
【0112】
IgG生産およびエンドトキシンスクリーニングアッセイ
scFv抗体断片は、修飾pCDNA3ベクター(Norderhaugら、J Immunol Methods、1997年;204:77〜87頁)内へのクローニングにより、完全長ヒトIgG1λフォーマットに転換させ、製造元(Invitrogen)の指示に従い、Lipofectamine2000試薬を用いて、HEK293細胞内へ一時的にトランスフェクトした。
【0113】
使用ずみ培養培地からのヒトIgGを、MabSelectタンパク質Aカラム(Amersham Bioscience)上で精製した。調製物の純度は、SDS-PAGE分析により判定すると、>98%であった。抗体調製物をスクリーンし、本試験に使用した濃度で、LALアメーバ様細胞ライセート試験(Cambrex Bioscience)によって判定すると、<0.1IU/mlのエンドトキシンを含有することが分かった。
【0114】
実施例2
抗体特異性の分析
抗原同定
標的抗原の確認を、Bリンパ腫細胞ライセートの免疫沈降によって行った。細胞(抗体および細胞系によって、1mlの溶解用緩衝液当たり、50〜600×106)を遠心分離により採集し、PBS中で2回洗浄し、0.5%v/vの洗浄剤TritonX-100(Sigma-Aldrich、セントルイス、ミズーリ州、米国)を含有するLysis Buffer(25mMのトリス-HCl、pH7.5、150mMのNaCl、5mMのEDTA、および完全無EDTAプロテアーゼ阻害剤カクテル(Roche Diagnostics GmbH、マンハイム、ドイツ国))中で、15分間インキュベートした。
【0115】
細胞デブリを慣例的なテーブルトップ遠心分離機において、16,000×gで15分間、遠心沈殿させ、可溶性タンパク質を、Protein A Sepharose4 Fast Flow (Amersham Bioscience)(1/10容量の反応液)によって、1時間回転させて予備洗浄した。各サンプルに関し、1mlの予備洗浄した細胞ライセートを、いずれかのヒト抗体20〜100μgによって2時間免疫沈降させた。再度、Protein A Sepharose4 Fast Flowを加え、30分間インキュベートし、免疫複合体を溶解用緩衝液中で広範囲に洗浄し、5分間煮沸し、最後にSample Buffer(1X NuPAGE LDS Sample Buffer、1X NuPAGE Sample Reducing Agent)中に再懸濁し、NuPAGE Novex 4〜12% Bis-Tris Gel(全てInvitrogenから)中で分離させた。
【0116】
染色(Simply Blue Safestain、Invitrogen)後、対象となっているタンパク質バンドをSDS-PAGEから切除し、記載 (Edvardssonら、Electrophoresis、1999年;20:935〜42頁) されたとおり、トリプシン消化に供した。
【0117】
簡単に述べると、ゲルプラグを脱染色し、攪拌下、200μlの50%アセトニトリル(ACN)による3回洗浄によって平衡化した。SpeedVac濃縮機(Savant、ファーミングデール、ニューヨーク、米国)中で15分間乾燥後、10mMのDTT/100mMのNH4HCO3 25μlの添加により還元し、56℃で1時間インキュベートし、55mMのヨードアセトアミド/100mMのNH4HCO3 を25μl添加することによりアルキル化し、続いて、室温で45分間インキュベートした。
【0118】
100mMのNH4HCO3 中でさらに2回、10分間の洗浄ステップ、続いて50%v/v ACN中、1回の洗浄後、該ゲル片を、SpeedVac濃縮機中で乾燥し、再膨張させ、25mMのNH4HCO3 における15ng/μlのトリプシン(Promega社、マジソン、ウィスコンシン州、米国) 15μl中、37℃で一晩消化させた。50%v/vACN/1%v/vTFAの添加により、ペプチドを抽出し、室温で10分間インキュベートした。1μlの抽出物をMALDIサンプルプレート上にスポットし、乾燥させた。1μlのマトリックス溶液(70%v/vACN/1%v/vTFA中、5mg/mlのアルファ-シアノ-4-ヒドロキシ桂皮酸(CHCA))を、該ペプチドの上部にスポットした。
【0119】
ペプチド質量は、Applied Biosystems 4700 Maldi Workstationを用いて決定した。該タンパク質は、Mascot探索ツール(Matrixscience、英国)を用い、ペプチド質量フィンガープリントデータベース探索によって同定した。クローンB10、C10、およびG12の抗原特異性は、免疫沈降にscFvおよび抗Hisコーティングした磁気マイクロビーズ(Miltenyi Biotec GmbH、ベルギッシュグラッドバッハ、ドイツ国)を使用した以外は、同様の方法論を用いて確認した。
【0120】
完全抗体フォーマットB1、B11およびC11に変換した後、IgGを用いて、ラージおよびラモスリンパ腫細胞から抗原を沈降させた。IgG B1は、それぞれ、およそ28kDaおよび34kDaの2つのバンドを沈降させた(図3A、レーン1)。これらのバンドを含有するゲルスライスを調製し、トリプシンで消化し、質量分光により分析し、標的抗原としてHLADR/DPを同定した。
【0121】
HLA-DR/DPに対するIgG B1の特異性は、市販のモノクローナル抗体を用いたウェスタンブロッティングおよびHLA-DR/DPタンパク質の検出双方、ならびに市販のHLA-DR特異性モノクローナル抗体を用いた細胞のプレインキュベーション後にB1 IgG結合のブロックにより検証した(図3B)。
【0122】
IgG B11およびC11規定抗原の同定を、同様な方法論を用いて確立した。IgG C11は、B細胞受容体μ鎖の膜結合形態として同定される68kDaのタンパク質バンドを沈降させることが分かった(図3A、レーン3)。IgG B11は、細胞間細胞接着分子-1(ICAM-1)として同定される90kDaのタンパク質バンドを沈降させた(図3A、レーン2)。ICAM-1に対するIgG B11、およびIgMに対するC11 IgGの特異性は、市販の抗体を用い、MS-MS分析、抗体ブロック試験(図3B)、およびウェスタンブロット分析(データは示していない)によって確認した。
【0123】
クローンB10、C10、およびG12の特異性は、scFvおよび免疫沈降用の抗Hisコーティングした磁気マイクロビーズを用いて判定した。3種のscFvクローンは、68kDaのタンパク質バンドを沈降させ、これらのバンドを含有するトリプシン消化ずみゲルスライスのMS分析により、表面IgMに対するそれらの特異性が判明した。これらの抗体はいずれも、末梢血Bリンパ球または他のIgM陽性B細胞系と交差反応しないため、ラモスIgMイディオタイプを認識すると考えられる。
【0124】
実施例3
抗体親和性の分析
B1およびB11免疫グロブリンのインビトロヨウ素化
[125I] NaIによる1mg/mlのIgG1B1またはIgG1B11タンパク質のヨウ素化は、Iodogen予備コーティングしたヨウ素化管(Pierce)を用いて、PBS中、10分間実施した。PD-10カラム(Amersham Biosciences)上での脱塩により、遊離[125I] NaIを除去し、タンパク質1ng当たり、1000〜1600cpmの範囲における特異的放射活性を得た。抗体親和性の判定には、[125I] IgG1 B1および[125I] IgG1 B11を用いた。
【0125】
IgG B1およびIgG B11親和性定数の決定
放射ヨウ素化IgG B1またはIgG B11をDPBS-B-hIgG(0.2mg/mlのヒトIgGを含有するDPBS-B)中、Bリンパ腫細胞と共に、氷上で2時間、断続的に混合しながらインキュベートした。適切な場合、0.2mg/mlの非標識IgG B1またはIgG B11タンパク質の存在下、非特異的結合を判定した。分析は三重で実施した。
【0126】
個々の管内の上部の40%v/v Ficoll/DPBS-Bクッション上に細胞を入れ、4℃で6分間、400×gで遠心分離した。サンプルを-80℃で凍結させた。細胞ペレットおよび細胞上澄み液を単離し、ガンマカウンターで125I-IgGタンパク質定数に関して別々に分析してから、該管を半分に切断した。
【0127】
以前記載された(Brixら、J.Clin.Invest.1998年;102:283〜93頁)とおり、Rosenthalら(Anal Biochem、1967年;20:525〜32頁)、BylundおよびYamamura(Methods in Neurotransmitter Analysed、ニューヨーク: Raven Press社、1990年)、およびMarquardt(J.Soc.Indust.Appl.Math、1963年;11:431〜41頁)に従って、抗体親和性定数(Kd値)および細胞1個当たりのエピトープ数を決定した。
【0128】
0.2mg/mlの対応する非標識IgGタンパク質の存在または非存在下、放射ヨウ素化タンパク質を、ラージまたはラモス細胞と共に、4℃でインキュベートすることにより、HLA-DRおよびICAM-1に対するIgG B1およびIgG B11の結合を特性化した。細胞表面に対する[125I]IgGの特異的結合を、総結合から非特異的結合 (過剰の非標識IgG存在下での結合) を差し引くことにより算出した。
【0129】
約30nMのIgG B1で、ラージ細胞に対するIgG B1の特異的結合の飽和に達した(図5)。Rosenthal-Scatchardプロット分析により、ラージ細胞1個当たり、400,000の機能的結合部位で、約3nMの解離定数が判明し、二価のエピトープ-IgG相互作用が考えられた(図5)。同様に、IgG B11の解離定数は、ラモス細胞1個当たり、47,400の受容体で約0.2nMと判定された。
【0130】
【表1】
【0131】
実施例4
ICAM-1は、アポトーシス誘導特性を有するBリンパ腫関連抗原である
ラモス細胞に対するIgG結合のフローサイトメトリー分析
氷上で45分間のインキュベーションにより、ラモス細胞(13×106)をCD45-PerCp-Cy5.5mAbによって染色し、DPBS-B中で洗浄し、非修飾精製PBLsと混合するまで氷上に保存した。
【0132】
Lund大学病院から、2人の健常志願者からのバッフィーコートを入手した。バッフィーコートを、PBS中、1:2に希釈し、500×g(1500rpm Beckman Spinchron 遠心分離機)で10分間の遠心分離によって洗浄し、上澄み液を完全吸引し、1%熱不活化FCSを含有するDPBS(DPBS-HI)中へ再懸濁した。洗浄は2回繰り返した。赤血球溶解溶液(BD Bioscience)と共に室温で15分間のインキュベーションによって、赤血球を溶解した。細胞を、60×g(667rpm Beckman Spinchron 遠心分離機)で10分間の遠心分離により洗浄し、上澄み液を慎重に吸引した。細胞をトリパンブルー試薬(Invitrogen)により染色して死細胞を排除した後、ビュルケルチャンバー内でカウントし、DPBS-HI中で洗浄し、ペレット化し、200μg/mlのヒト精製IgGを含有するDPBS-B(Fc受容体のブロック)中に再懸濁した。
【0133】
各ドナーおよび試験条件に関して、およそ2.5×106の白血球を、1.6×105のPerCpCy5.5予備標識ラモス細胞と混合した。マウスモノクローナルCD3-FITC、CD56-PE、およびCD19- PerCpCy5.5抗体(BD Bioscience)を加え、標識ヒトIgGを添加するまで、その混合物を氷上でインキュベートした。n-CoDeRヒトIgG抗体および陽性対照の抗CD20マウス-ヒトキメラ抗体リツキシマブの標識化を、製造元の指示に従い、AF647 Fab断片(Molecular Probes、Invitrogen)によって実施した。
【0134】
簡単に述べると、IgG B1、B11、C11、およびリツキシマブ抗体各々の4μgを、20μlのAF647-Fab標識試薬と共に、室温で5分間インキュベートした。20μlのヒトIgGブロック試薬を添加してさらに5分間のインキュベーション後、AF647標識化IgGをDBPS-B中に3倍連続希釈し、希釈したIgGタンパク質を、混合ラモス/PBL細胞溶液に加えた。
【0135】
サンプルを1時間インキュベートし、洗浄し、DPBS-B中に再懸濁し、4色分析用機器の適切な検量および補正の後、フローサイトメトリーにより種々の細胞亜集団に対する結合に関して分析した。ラモス細胞は、Bリンパ球PerCpCy5.5low集団から識別できるPerCpCy5.5high集団として同定された。
【0136】
免疫組織化学
未分化大型細胞Bリンパ腫(1人)、中心芽細胞性/中心細胞性B非ホジキンリンパ腫(3人)、およびB細胞慢性リンパ性白血病(1人)の凍結保存リンパ節生検を、Lund大学の病理学部(Lund、スウェーデン国)から入手した。凍結保存組織の8マイクロメートル切片をアセトン中、4℃で10分間固定した。アビジンおよびビオチン(Avidin/Biotin blockingキット、Invitrogen)による各20分間の連続処理により、内因性ビオチン結合活性をブロックした。
【0137】
5μg/mlの対照scFvまたはB11scFvと共に、組織を1時間インキュベートした。洗浄後、切片をビオチン結合マウス抗His mAb(R&D Systems)と共に30分間インキュベートした。ABC Complex/HRP試薬(Dako Cytomation)による30分間の処理、引き続き、DABとの5分間のインキュベーション後、scFv結合を検出した。
【0138】
Leica DMR光/蛍光顕微鏡の上部に据え付けたLeica DC 300Fデジタルカメラを用いて、切片の写真を撮った。
【0139】
ヒト組織の取り扱いは、Lund大学病院の地域倫理委員会の推奨に従った。
【0140】
ミトコンドリア膜脱分極アッセイ
以前記載された(Kimら、Mol Biol Cell、2004年;15:420〜34頁)とおり、ミトコンドリア膜脱分極を分析した。簡単に述べると、抗体処置細胞を5μg/mlのJC-1試薬(Molecular Probes)と混合し、室温で30分間インキュベートした。細胞を氷冷PBS中で2回洗浄し、300μlのPBS中に再懸濁し、FACS Aria(BD Biosciences)で分析した。494/518nm(FL-1)および595/615nm(FL-2)バンド通過フィルターを通して、緑色蛍光および赤色蛍光をそれぞれを採集した。
【0141】
ICAM-1は、二方向性シグナル伝達を誘導できる(Rothleinら、J Immunol、1994年;152:2488〜95頁; Vyth-Dreeseら、Blood、1995年;85:2802〜12頁)免疫グロブリンスーパーファミリーの糖タンパク質である(Marlinら、Cell、1987年;51:813〜9頁)。ICAM-1がBリンパ腫細胞におけるプログラム死に関与しているという実証は以前されていない。
【0142】
したがって、我々は、IgG B11誘導細胞死が、細胞膜ホスファチジルセリンの移動以外の方法による活性過程であることを確認したかった。
【0143】
ミトコンドリア膜の脱分極をアポトーシスの確認として選択した。これが、ヨウ化5,5',6,6'-テトラクロロ-1,1',3,3'-テトラエチル-ベンジミダゾイル-カルボシアニン(JC-1試薬)による細胞染色によりモニターできるカスパーゼ依存性およびカスパーゼ非依存性アポトーシスの共通の特徴であるためである。
【0144】
我々のAnnexin V/ヨウ化プロピジウムアッセイ(図4A、上枠)に従い、JC-1試薬による染色後、フローサイトメトリー分析によって判定すると、IgG B11は、CL-01 Bリンパ腫細胞において、ミトコンドリア膜の脱分極を誘導することが分かった(図4A、下枠)。
【0145】
ICAM-1の発現が、細胞培養中の一般的なアップレギュレーションから生じるインビトロアーチファクトであった可能性を排除するために、種々のBリンパ腫腫瘍に罹っている5人の異なる患者から得た組織に対するIgG B11の結合を調べた。
【0146】
免疫組織化学により、IgG B11は、抗HLA-DR/DP抗体IgG B1と同等か、またはわずかに低い強度で(表2)、5つのリンパ腫組織に対する強い結合性を示した(図4B)。
【0147】
次に、Bリンパ腫対静止末梢血白血球に対するIgG B11の結合性を調べた。低端エピトープ発現であって、しかもB11誘導アポトーシスに対する有意な感受性があることに基づき、ラモスが代表的なBリンパ腫細胞系として選択された。予備標識したラモス細胞と全血末梢血白血球ならびにIgG B1、B11またはC11抗体のいずれかとの混合インキュベーション後のフローサイトメトリー分析によって、IgG B11は、ラモス細胞に対する強い結合性を示した。
【0148】
さらに重要なことに、B11は、ラモスBリンパ腫対正常末梢血白血球に対する3腫の抗体の結合性の大きな差異(最強の抗原アップレギュレーション)を示した(図4C、データは示していない)。IgG B11の結合は、すでに0.1μg/mlで最高となり、ラモス対単球細胞で3.7倍アップレギュレートされ(MFI654対176)、ラモス対末梢血Bリンパ球で8.3倍アップレギュレートされ(MFI654対78)、NK細胞に比較すると、23倍アップレギュレートされた。モニターされた他の末梢血白血球サブセットに対する結合性は負であった。
【0149】
【表2】
【0150】
実施例5
フローサイトメトリーにより判定された、種々の出所の腫瘍細胞系におけるB1、B11、C11 IgG1の抗原分布
主にB11に関するヒト抗体標的抗原の抗原分布を、種々の癌細胞系について調べた。細胞(MCF-7およびMDA MB 435S乳癌、JARおよびJEG-3絨毛癌、A549肺癌、TCC-SUP膀胱癌、MDA MB 435黒色腫、HPAC、PANC-1およびBxPC-3膵臓癌、PC-3およびDU145前立腺癌、LS174T、CaCo2、およびLovo結腸直腸癌、およびTHP-1単球白血病細胞)をPBS中で洗浄し、完全培地(200,000細胞/50μlサンプル)中、4×106細胞/mlで再懸濁した。B1 Ig G1、B11 Ig G1、C11 Ig G1、陰性対照FITC-8 Ig G1、およびリツキシマブ抗CD20 mAbを、完全培地(50μl/サンプル)中、3〜10倍に連続希釈した(10〜0.1pg/ml)。細胞を、いずれかの抗体と共に、1時間、氷上でインキュベートし、PBS/BSA 0.5%中に再懸濁することにより洗浄し、1200rpmで5分間遠心分離し、上澄み液の完全吸引を行った。細胞を、PBS/BSA 0.5%中、1/50に希釈したPE結合ヤギF(ab')2抗ヒトIgG(Caltag Laboratories、カタログ番号:H10104)と共に、氷上で30分間インキュベートした。300μlのPBS/BSA 0.5%中に再懸濁した後、FACScan機器を用いて、細胞を、IgG結合性に関して分析した。
【0151】
PC-3前立腺癌細胞は、これらの細胞に対するB11 IgGの強い結合性によって実証されるように、ICAM-1の強い発現を示した(図6)。MCF-7乳癌細胞、HPAC膵臓癌細胞、およびLS174T結腸直腸癌細胞もまた、前立腺癌に比較して強度はより低いが、ICAM-1を発現することが分かった。対照的に、THP-1単球白血病細胞は、ICAM-1を発現しなかった。最初に試験した全ての癌細胞系は、リツキシマブIgG、B1 IgG、およびC11 IgGそれぞれの結合が欠如していることから実証されるように、CD20、HLA-DR/DP、およびIgM発現に関して陰性であることが分かった。追加の癌細胞系におけるさらなる試験により、調べられた全ての癌細胞が、ICAM-1発現に関して陽性であることが示された(表3)。
【0152】
【表3】
【0153】
実施例6
癌細胞におけるB11 IgG1アポトーシス誘導
実施例5において、B11 IgG1は、癌細胞に強く結合することが示された。本実施例では、癌細胞に対するこの抗体のアポトーシス誘導特性を調べた。
【0154】
実験開始の3日前に、完全増殖培地を有する6ウェルプレートにおいて、細胞を接種した。実験時、細胞は50〜75%の間の集密度であった。細胞を氷冷PBSにより洗浄し、指示されたとおり、連続希釈した(完全増殖培地1ml中、図に示されるように、20〜0.02μg/ml)B11 IgG1、20μg/mlの対照B1 IgG1、10μg/mlの陰性対照IgG1または10μg/mlのトラスツズマブIgG1と共に、4℃で1〜2時間インキュベートした。細胞を氷冷PBSにより洗浄し、二次F(ab'2)ヤギ抗ヒトF(ab'2)抗体(完全増殖培地中、10μg/mlに希釈)を加えた。5% CO2の加湿雰囲気下、細胞を37℃で16〜24時間インキュベートした。先ず、上澄み液を単離し、続いてPBS洗浄し、残りの接着細胞をトリプシン処理することによって総細胞を採集した。熱不活化した10%ウシ胎仔血清を含有するPBS中に再懸濁することによって酵素反応を終結させた。氷冷PBS中で細胞を洗浄し、Annexin V/ヨウ化プロピジウム染色に供し、上記実施例5に記載されたとおり、生存度/アポトーシスに関して分析した。
【0155】
B11 IgG1は、特定の滴定可能な様式で、癌細胞系にアポトーシスを誘導することが示された(図7)。PC-3細胞に結合しなかった対照IgG B1(実施例5を参照)は、PC-3細胞にアポトーシスを誘導することもなかった。陰性対照IgG1またはトラスツマブIgG1は、DU145細胞またはMDA MB435細胞にアポトーシスを誘導することができなかった。
【0156】
実施例7
医薬製剤および投与
本発明のさらなる態様では、薬学的にまたは獣医学的に許容されるアジュバント、希釈または担体と混合させて、本発明の第1の態様による化合物を含む医薬製剤が提供される。
【0157】
該製剤は、活性成分の1日用量または単位、1日のサブ用量またはその適切な画分を含有する単位用量であることが好ましい。
【0158】
本発明の化合物は通常、薬学的に許容される剤形において、活性成分を、任意に、非毒性の有機または無機の酸または塩基付加塩の形態で含む医薬製剤の形態で、経口で、または任意の非経口経路により投与される。治療される障害および患者、ならびに投与経路により、該組成物は用量を変化させて投与できる。
【0159】
ヒトの治療において、本発明の化合物は、単独でも投与できるが、一般に、意図された投与経路および標準的な医薬上の実践を考慮して選択される好適な医薬用の賦形剤、希釈剤または担体と混合して投与される。
【0160】
例えば、本発明の化合物は、即時、遅延または制御放出適用のために、風味剤または着色剤を含有し得る、錠剤、カプセル剤、卵形剤、エレキシル剤、液剤または懸濁剤の形態で、経口で、頬側に、または舌下に投与できる。本発明の化合物は、鼻腔内注入によっても投与できる。
【0161】
このような錠剤は、微結晶性セルロース、乳糖、クエン酸ナトリウム、炭酸カルシウム、第二リン酸カルシウムおよびグリシンなどの賦形剤、澱粉(好ましくは、トウモロコシ、ジャガイモまたはタピオカ澱粉)、澱粉グリコール酸ナトリウム、クロスカルメロースナトリウムおよび一定の錯体ケイ酸塩などの崩壊剤、ならびにポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ-プロピルセルロース(HPC)、ショ糖、ゼラチンおよびアラビアゴムなどの顆粒化結合剤を含有してもよい。また、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリルおよびタルクなどの滑剤を含めることができる。
【0162】
同様なタイプの固体組成物は、ゼラチンカプセル剤中の充填剤としても使用できる。これに関して好ましい賦形剤としては、乳糖、澱粉、セルロース、ミルクシュガーまたは高分子量のポリエチレングリコールが挙げられる。水性懸濁剤および/またはエリキシル剤では、本発明の化合物を、種々の甘味剤または風味剤、着色物質または色素と、乳化剤および/または懸濁化剤と、水、エタノール、プロピレングリコールおよびグリセリンなどの希釈剤と、およびそれらの組み合わせと組み合わせることができる。
【0163】
本発明の化合物は、非経口で、例えば、静脈内、動脈内、腹腔内、鞘内、心室内、ステム内、頭蓋内、筋肉内または皮下に投与できるか、または、それらを注入法により投与できる。それらは、他の物質、例えば、該溶液を血液と等張にする上で十分な塩またはグルコースを含有し得る、滅菌水性溶液の形態で最良に用いられる。該水性溶液は、必要ならば、好適に緩衝化(好ましくは、pH3から9に)される必要がある。滅菌条件下での好適な非経口製剤の調製は、当業者によく知られた標準的な製薬技法によって容易に達成される。
【0164】
非経口投与に好適な製剤としては、抗酸化剤、緩衝剤、静菌剤および該製剤を意図されたレシピエントの血液と等張にする溶質を含有し得る水性および非水性の滅菌注射液剤;ならびに懸濁化剤および増粘剤を含み得る水性および非水性の懸濁剤が挙げられる。該製剤は、単位用量または多用量容器、例えば、密封アンプルおよびバイアルにおいて提供でき、使用直前に滅菌液体担体、例えば注射用の水の添加のみを必要とするフリーズドライ(凍結乾燥)条件下で保存できる。即時注射液および懸濁液は、以前記載された種類の滅菌した粉末、顆粒および錠剤から調製できる。
【0165】
ヒト患者への経口および非経口投与に関して、本発明の化合物の1日用量レベルは通常、1mg/kgから30mg/kgである。したがって、例えば、本発明の化合物の錠剤またはカプセル剤は、適宜、一度に一回、または2回以上の投与のための活性化合物の用量を含有できる。何らかの事象において、医師は、個々の患者にとって最も好適な実際の用量を決定し、この用量は、具体的な患者の年齢、体重および応答によって変化する。上記の投与量は、平均的な場合の代表的なものである。勿論、より高いまたは低い投与量の範囲が有利である個々の例があり得、このようなことは本発明の範囲内にある。
【0166】
本発明の化合物はまた、鼻腔内に、または吸入によっても投与でき、ドライパウダー吸入器の形態、または好適な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、1,1,1,2-テトラフルオロエタン(HFA 134A3または1,1,1,2,3,3,3-ヘプタフルオロプロパン(HFA227EA3)、二酸化炭素または他の好適な気体の使用と共に、加圧容器、ポンプ、スプレーまたはネブライザーからエアゾールスプレー提供の形態で簡便に送達される。加圧エアゾールの場合、計測量を送達するためにバルブを提供することによって、用量単位を決定できる。加圧容器、ポンプ、スプレーまたはネブライザーは、例えば、溶媒としてエタノールと噴射剤との混合物を用いて、活性化合物の溶液または懸濁液を含有でき、さらに、滑剤、例えば、ソルビタントリオレエートを含有できる。吸入器または注入器に使用するためのカプセルおよびカートリッジ(例えば、ゼラチンから作製)は、本発明の化合物と乳糖または澱粉などの好適な粉末基材との粉末混合物を含有するように製剤化できる。
【0167】
エアゾール製剤またはドライパウダー製剤は、各計測用量または「パフ」が、患者への送達のために、本発明の化合物の適切な用量を送達するように設定されていることが好ましい。エアゾールによる1日用量の合計は、患者によって異なり、単一用量で、より通常的には、1日の分割用量で投与できる。
【0168】
あるいは、本発明の化合物を、座剤または膣座剤の形態で投与でき、それらは、ローション、溶液、クリーム、軟膏または粉剤の形態で局所に適用できる。本発明の化合物は、例えば、皮膚パッチの使用により、経皮投与することもできる。それらはまた、特に眼の疾患を治療するために、眼の経路により投与できる。
【0169】
眼科使用に関して、本発明の化合物は、等張でpH調整した滅菌生理食塩水におけるミクロ化懸濁剤として、または好ましくは、等張でpH調整した滅菌生理食塩水における液剤として、任意に、塩化ベンジルアルコニウムなどの保存剤と組み合わせて製剤化できる。あるいは、それらを、ペトロラタムなどの軟膏に製剤化できる。
【0170】
皮膚への局所適用に関して、本発明の化合物は、例えば、以下:鉱油、流動パラフィン、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ワックスおよび水、の1種または複数の混合物に懸濁または溶解させた活性化合物を含有する好適な軟膏として製剤化できる。あるいは、それらを、例えば、以下:鉱油、モノステアリン酸ソルビタン、ポリエチレングリコール、流動パラフィン、ポリソルベート60、セチルエステル類、ワックス、セテアリールアルコール、2-オクチルドデカノール、ベンジルアルコールおよび水、の1種または複数の混合物に、懸濁または溶解させた好適なローションまたはクリームとして製剤化できる。
【0171】
口腔における局所投与のために好適な製剤としては、風味づけされた基材、通常は、スクロースおよびアラビアゴム、またはトラガントゴム中に該活性成分を含む舐剤;ゼラチンおよびグリセリン、またはスクロースおよびアラビアゴムなどの不活性基材中に該活性成分を含むパステル剤;および好適な液体担体中に該活性成分を含む口腔洗浄剤が挙げられる。
【0172】
一般に、ヒトにおいて、本発明の化合物の経口投与または局所投与が、最も簡便であるので、好ましい経路である。レシピエントが嚥下障害または経口投与後の薬剤吸収の障害を患っている状況では、該薬剤は腸管外的に、例えば、舌下または頬側に投与できる。
【0173】
獣医学使用に関しては、本発明の化合物は、通常の獣医学実践に従って、好適に許容される製剤として投与され、獣医学医師は、具体的な動物にとって最も適切である投与療法および投与経路を決定する。
【特許請求の範囲】
【請求項1】
a.細胞表面抗原ICAM-1を提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する1種または複数の結合分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法。
【請求項2】
前記結合分子が抗体分子である、請求項1に記載の方法。
【請求項3】
a.細胞表面抗原HLA-DR/DPおよび/または表面IgMを提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合し、細胞表面HLA-DR/DPおよび/または表面IgMとの結合により標的細胞のアポトーシスを誘導する1種または複数の抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の抗体分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法。
【請求項4】
細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する、請求項1または2の方法に使用するための結合分子。
【請求項5】
標的細胞の細胞表面抗原に選択的に結合し、細胞表面抗原との結合により標的細胞のアポトーシスを誘導し、細胞表面抗原がHLA-DR/DPおよび/または表面IgMである、請求項3に記載の方法に使用するための抗体分子。
【請求項6】
前記結合分子が抗体分子である、請求項4に記載の結合分子。
【請求項7】
前記細胞表面抗原がICAM-1である、請求項4または6のいずれかに記載の結合分子。
【請求項8】
前記細胞表面抗原がHLA-DR/DPである、請求項5に記載の抗体分子。
【請求項9】
前記細胞表面抗原が表面IgMである、請求項5に記載の抗体分子。
【請求項10】
前記標的細胞が免疫細胞または上皮細胞である、請求項4から9のいずれか一項に記載の結合分子または抗体分子。
【請求項11】
前記免疫細胞がBリンパ球である、請求項10に記載の結合分子または抗体分子。
【請求項12】
前記標的細胞が疾患に関連している、請求項4から11のいずれか一項に記載の結合分子または抗体分子。
【請求項13】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される、請求項12に記載の結合分子または抗体分子。
【請求項14】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項13に記載の結合分子または抗体分子。
【請求項15】
前記抗体分子がIgGである、請求項6から14のいずれか一項に記載の結合分子または抗体分子。
【請求項16】
一本鎖抗体が、IgG1、IgG2、IgG3またはIgG4の群から選択される、請求項15に記載の結合分子または抗体分子。
【請求項17】
前記抗体分子が、ヒト抗体分子またはヒト化抗体分子である、請求項6から16のいずれか一項に記載の結合分子または抗体分子。
【請求項18】
前記抗体が、図9から11のいずれか一つの配列または機能的に等価なその相同体を有する可変領域を有する、請求項6から17のいずれか一項に記載の結合分子または抗体分子。
【請求項19】
前記抗体が、図9の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項20】
前記抗体が、図10の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項21】
前記抗体が、図11の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項22】
請求項4から21のいずれか一項に記載の抗体分子をコードするヌクレオチド配列を有する核酸。
【請求項23】
図9から11のいずれか一つのヌクレオチド配列を有する、請求項22に記載の核酸。
【請求項24】
標的細胞の破壊を必要とする、疾患の診断および/または治療および/または予防における、請求項4から21のいずれか一項に記載の結合分子または抗体分子の使用。
【請求項25】
標的細胞の破壊を必要とする、疾患の診断および/または治療および/または予防のための薬剤の製造における、請求項4から21のいずれか一項に記載の結合分子または抗体分子の使用。
【請求項26】
前記治療される疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される請求項12に記載の結合分子または抗体分子からなる群から選択される、請求項24または25に記載の使用。
【請求項27】
前記治療される疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項26に記載の使用。
【請求項28】
前記結合分子または抗体分子が、請求項8または19のいずれかに記載のものであり、前記治療される疾患が、請求項24または25のいずれかに記載のものである、請求項24から27のいずれか一項に記載の使用。
【請求項29】
前記結合分子または抗体分子が、請求項7または20のいずれかに記載のものであり、前記治療される疾患が、請求項27に記載のリンパ腫である、請求項24から27のいずれか一項に記載の使用。
【請求項30】
前記結合分子または抗体分子が、請求項9または21のいずれかに記載のものであり、前記治療される疾患が、請求項27に記載のリンパ腫である、請求項24から27のいずれか一項に記載の使用。
【請求項31】
請求項4から21のいずれか一項に記載の結合分子または抗体分子と、薬学的に許容される担体、賦形剤または希釈剤とを含む医薬組成物。
【請求項32】
a.1種または複数の標的細胞を用意するステップと、
b.請求項4から21のいずれか一項に記載の1種または複数の結合分子または抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと
を含む、標的細胞にアポトーシスを誘導するインビトロ方法。
【請求項33】
前記ステップ(a)で用意される標的細胞が、免疫細胞または上皮細胞である、請求項32に記載のインビトロ方法。
【請求項34】
前記免疫細胞がBリンパ球である、請求項33に記載のインビトロ方法。
【請求項35】
前記標的細胞が疾患に関連している、請求項32から34のいずれか一項に記載のインビトロ方法。
【請求項36】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症ならびにそれだけに限定されないが、HIVを含めた感染性疾患からなる群から選択される、請求項35に記載のインビトロ方法。
【請求項37】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項36に記載のインビトロ方法。
【請求項38】
a.1種または複数の標的細胞を用意するステップと、
b.請求項4から21のいずれかに記載の1種または複数の結合分子または抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと、
を含む、標的細胞にアポトーシスを誘導するインビボ方法。
【請求項39】
前記ステップ(a)で用意される標的細胞が、免疫細胞または上皮細胞である、請求項38に記載のインビボ方法。
【請求項40】
前記免疫細胞がBリンパ球である、請求項39に記載のインビボ方法。
【請求項41】
前記標的細胞が疾患に関連している、請求項38から40のいずれか一項に記載のインビボ方法。
【請求項42】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される、請求項41に記載のインビボ方法。
【請求項43】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項42に記載のインビボ方法。
【請求項44】
実質的に実施例および図を参照して本明細書に記載された、アポトーシスを誘導する方法。
【請求項45】
実質的に実施例および図を参照して本明細書に記載された、アポトーシス誘導性の結合分子または抗体分子。
【請求項46】
実質的に実施例および図を参照として本明細書に記載された、アポトーシス誘導性の結合分子または抗体分子の使用。
【請求項47】
実質的に実施例ならびに図を参照として本明細書に記載された、アポトーシス誘導性の医薬組成物。
【請求項1】
a.細胞表面抗原ICAM-1を提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する1種または複数の結合分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法。
【請求項2】
前記結合分子が抗体分子である、請求項1に記載の方法。
【請求項3】
a.細胞表面抗原HLA-DR/DPおよび/または表面IgMを提示する1種または複数の標的細胞を用意するステップと、
b.細胞表面HLA-DR/DPおよび/または表面IgMに選択的に結合し、細胞表面HLA-DR/DPおよび/または表面IgMとの結合により標的細胞のアポトーシスを誘導する1種または複数の抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の抗体分子に曝露して、標的細胞におけるアポトーシスを誘導するステップと
を含む、標的細胞におけるアポトーシスを誘導する方法。
【請求項4】
細胞表面ICAM-1に選択的に結合し、ICAM-1との結合により標的細胞のアポトーシスを誘導する、請求項1または2の方法に使用するための結合分子。
【請求項5】
標的細胞の細胞表面抗原に選択的に結合し、細胞表面抗原との結合により標的細胞のアポトーシスを誘導し、細胞表面抗原がHLA-DR/DPおよび/または表面IgMである、請求項3に記載の方法に使用するための抗体分子。
【請求項6】
前記結合分子が抗体分子である、請求項4に記載の結合分子。
【請求項7】
前記細胞表面抗原がICAM-1である、請求項4または6のいずれかに記載の結合分子。
【請求項8】
前記細胞表面抗原がHLA-DR/DPである、請求項5に記載の抗体分子。
【請求項9】
前記細胞表面抗原が表面IgMである、請求項5に記載の抗体分子。
【請求項10】
前記標的細胞が免疫細胞または上皮細胞である、請求項4から9のいずれか一項に記載の結合分子または抗体分子。
【請求項11】
前記免疫細胞がBリンパ球である、請求項10に記載の結合分子または抗体分子。
【請求項12】
前記標的細胞が疾患に関連している、請求項4から11のいずれか一項に記載の結合分子または抗体分子。
【請求項13】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される、請求項12に記載の結合分子または抗体分子。
【請求項14】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項13に記載の結合分子または抗体分子。
【請求項15】
前記抗体分子がIgGである、請求項6から14のいずれか一項に記載の結合分子または抗体分子。
【請求項16】
一本鎖抗体が、IgG1、IgG2、IgG3またはIgG4の群から選択される、請求項15に記載の結合分子または抗体分子。
【請求項17】
前記抗体分子が、ヒト抗体分子またはヒト化抗体分子である、請求項6から16のいずれか一項に記載の結合分子または抗体分子。
【請求項18】
前記抗体が、図9から11のいずれか一つの配列または機能的に等価なその相同体を有する可変領域を有する、請求項6から17のいずれか一項に記載の結合分子または抗体分子。
【請求項19】
前記抗体が、図9の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項20】
前記抗体が、図10の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項21】
前記抗体が、図11の配列または機能的に等価なその相同体を有する可変領域を有する、請求項18に記載の結合分子または抗体分子。
【請求項22】
請求項4から21のいずれか一項に記載の抗体分子をコードするヌクレオチド配列を有する核酸。
【請求項23】
図9から11のいずれか一つのヌクレオチド配列を有する、請求項22に記載の核酸。
【請求項24】
標的細胞の破壊を必要とする、疾患の診断および/または治療および/または予防における、請求項4から21のいずれか一項に記載の結合分子または抗体分子の使用。
【請求項25】
標的細胞の破壊を必要とする、疾患の診断および/または治療および/または予防のための薬剤の製造における、請求項4から21のいずれか一項に記載の結合分子または抗体分子の使用。
【請求項26】
前記治療される疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される請求項12に記載の結合分子または抗体分子からなる群から選択される、請求項24または25に記載の使用。
【請求項27】
前記治療される疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項26に記載の使用。
【請求項28】
前記結合分子または抗体分子が、請求項8または19のいずれかに記載のものであり、前記治療される疾患が、請求項24または25のいずれかに記載のものである、請求項24から27のいずれか一項に記載の使用。
【請求項29】
前記結合分子または抗体分子が、請求項7または20のいずれかに記載のものであり、前記治療される疾患が、請求項27に記載のリンパ腫である、請求項24から27のいずれか一項に記載の使用。
【請求項30】
前記結合分子または抗体分子が、請求項9または21のいずれかに記載のものであり、前記治療される疾患が、請求項27に記載のリンパ腫である、請求項24から27のいずれか一項に記載の使用。
【請求項31】
請求項4から21のいずれか一項に記載の結合分子または抗体分子と、薬学的に許容される担体、賦形剤または希釈剤とを含む医薬組成物。
【請求項32】
a.1種または複数の標的細胞を用意するステップと、
b.請求項4から21のいずれか一項に記載の1種または複数の結合分子または抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと
を含む、標的細胞にアポトーシスを誘導するインビトロ方法。
【請求項33】
前記ステップ(a)で用意される標的細胞が、免疫細胞または上皮細胞である、請求項32に記載のインビトロ方法。
【請求項34】
前記免疫細胞がBリンパ球である、請求項33に記載のインビトロ方法。
【請求項35】
前記標的細胞が疾患に関連している、請求項32から34のいずれか一項に記載のインビトロ方法。
【請求項36】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症ならびにそれだけに限定されないが、HIVを含めた感染性疾患からなる群から選択される、請求項35に記載のインビトロ方法。
【請求項37】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項36に記載のインビトロ方法。
【請求項38】
a.1種または複数の標的細胞を用意するステップと、
b.請求項4から21のいずれかに記載の1種または複数の結合分子または抗体分子を用意するステップと、
c.(a)の標的細胞を(b)の結合分子または抗体分子に曝露して、標的細胞にアポトーシスを誘導するステップと、
を含む、標的細胞にアポトーシスを誘導するインビボ方法。
【請求項39】
前記ステップ(a)で用意される標的細胞が、免疫細胞または上皮細胞である、請求項38に記載のインビボ方法。
【請求項40】
前記免疫細胞がBリンパ球である、請求項39に記載のインビボ方法。
【請求項41】
前記標的細胞が疾患に関連している、請求項38から40のいずれか一項に記載のインビボ方法。
【請求項42】
前記疾患が、癌、それだけに限定されないがリウマチ様関節炎およびSLEを含めた自己免疫疾患、急性および慢性の炎症性疾患、敗血症、ならびにそれだけに限定されないがHIVを含めた感染性疾患からなる群から選択される、請求項41に記載のインビボ方法。
【請求項43】
前記疾患が、リンパ腫(白血病、骨髄腫)、胃癌、乳癌、肝臓癌、肺癌、黒色腫、膀胱癌、脈絡膜癌、膵臓癌、結腸癌および前立腺癌から選択される癌である、請求項42に記載のインビボ方法。
【請求項44】
実質的に実施例および図を参照して本明細書に記載された、アポトーシスを誘導する方法。
【請求項45】
実質的に実施例および図を参照して本明細書に記載された、アポトーシス誘導性の結合分子または抗体分子。
【請求項46】
実質的に実施例および図を参照として本明細書に記載された、アポトーシス誘導性の結合分子または抗体分子の使用。
【請求項47】
実質的に実施例ならびに図を参照として本明細書に記載された、アポトーシス誘導性の医薬組成物。
【図1】

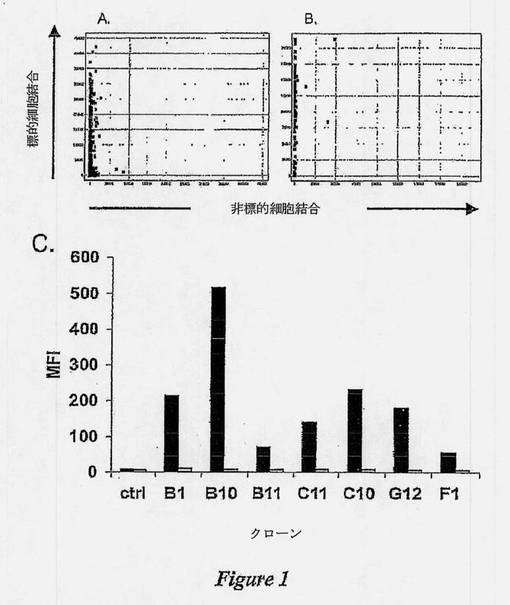
【図2A】
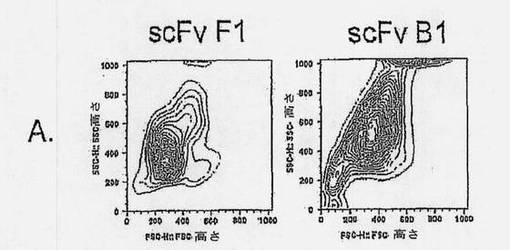
【図2B】
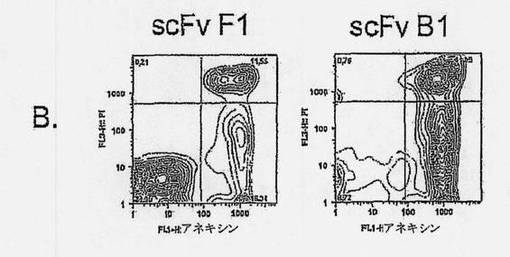
【図2C】
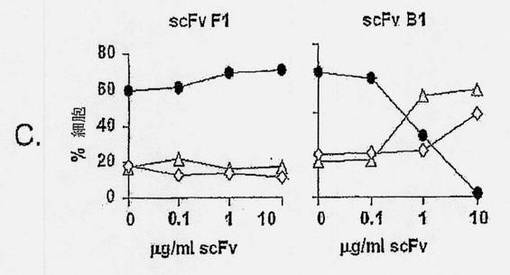
【図2D】
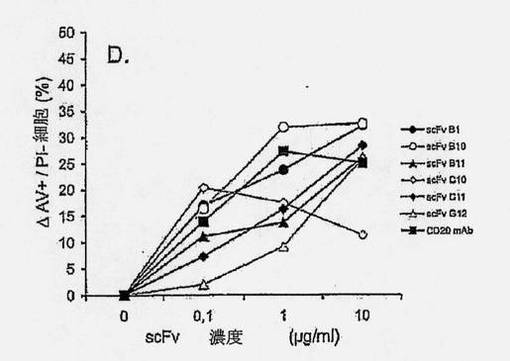
【図2E】
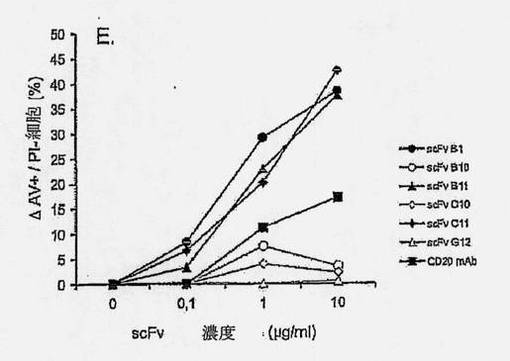
【図3】
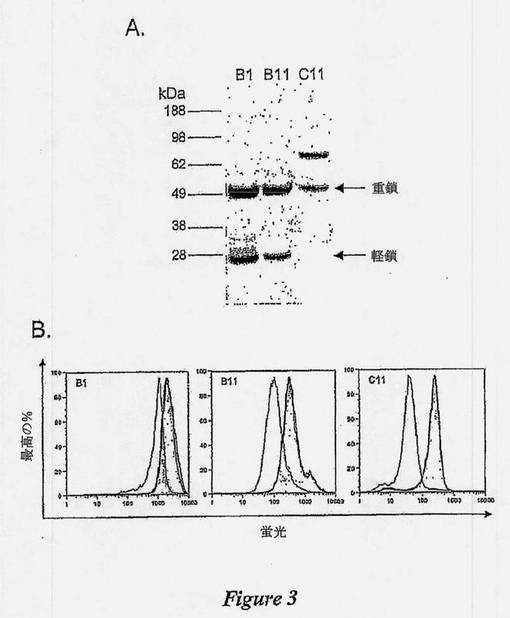
【図4A】
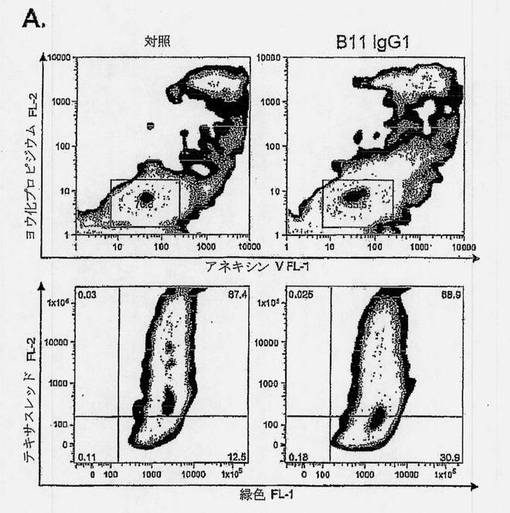
【図4B】
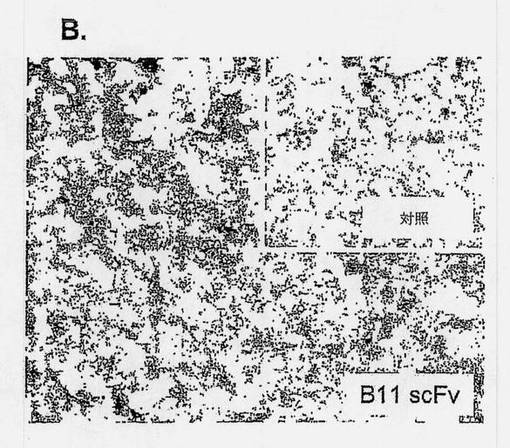
【図4C】
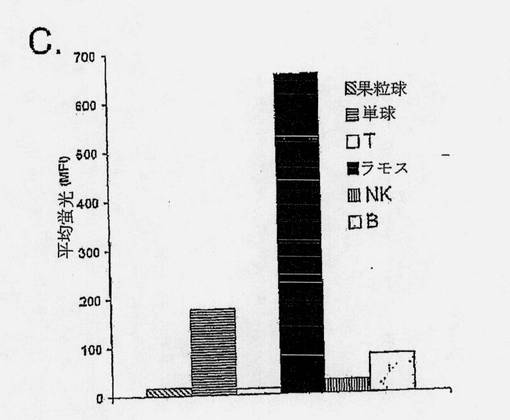
【図5】
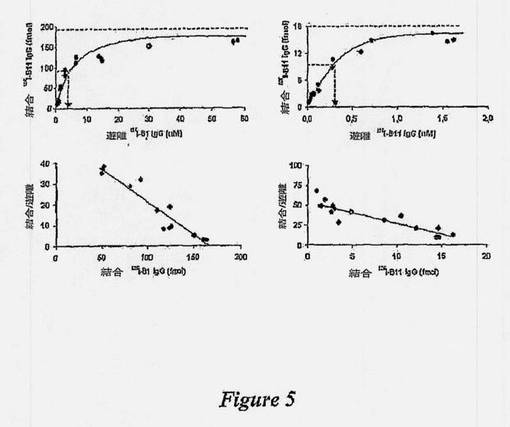
【図6】
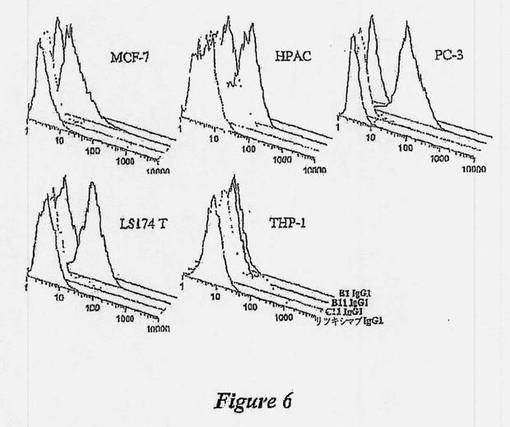
【図7A】

【図7B】
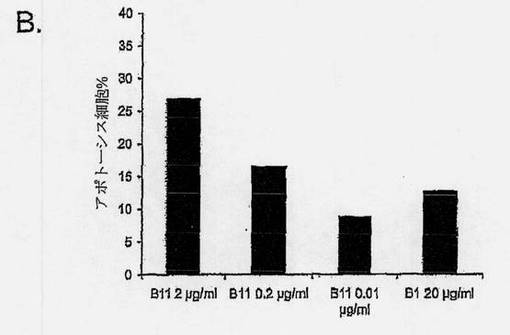
【図7C】
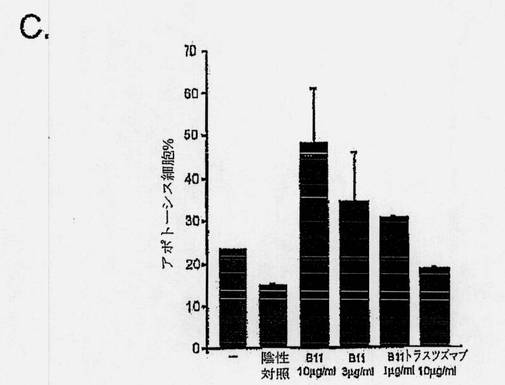
【図7D】
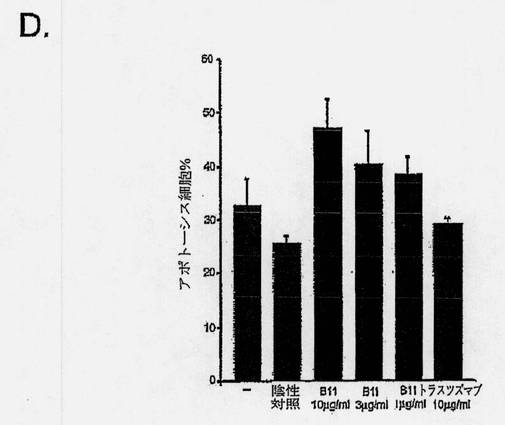
【図8】

【図9A】

【図9B】

【図10A】

【図10B】

【図11A】

【図11B】

【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7A】
【図7B】
【図7C】
【図7D】
【図8】
【図9A】
【図9B】
【図10A】
【図10B】
【図11A】
【図11B】
【公開番号】特開2013−5815(P2013−5815A)
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−207965(P2012−207965)
【出願日】平成24年9月21日(2012.9.21)
【分割の表示】特願2008−544879(P2008−544879)の分割
【原出願日】平成18年12月8日(2006.12.8)
【出願人】(506209743)バイオインヴェント インターナショナル アーベー (9)
【Fターム(参考)】
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願番号】特願2012−207965(P2012−207965)
【出願日】平成24年9月21日(2012.9.21)
【分割の表示】特願2008−544879(P2008−544879)の分割
【原出願日】平成18年12月8日(2006.12.8)
【出願人】(506209743)バイオインヴェント インターナショナル アーベー (9)
【Fターム(参考)】
[ Back to top ]