
生物学的活性剤を含有する生分解性微粒子の製造
【課題】生物学的または薬学的活性剤を含有する微粒子の適度に定めた狭いサイズ分布を達成しつつ、実験室から工業的規模へのバッチサイズを精密に信頼性よくスケールアップすることができる微粒子製造方法を提供する
【解決手段】生分解性ポリマー封入結合剤、生物学的活性剤および相互に混和性の低水溶性有機溶媒の混合物を含有する溶液から成り、ハロゲン化炭化水素を含有しない第1相を調製し、親水性コロイドまたは界面活性剤の水溶液から成る第2相を調製し、第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相、第2相が連続相となったエマルジョンを形成し、不連続第1相を微粒子の形態で分離し、クエンチ液を用いて、先の工程の微粒子から残留溶媒を抽出することを含んで成る生分解性微粒子の製法。
【解決手段】生分解性ポリマー封入結合剤、生物学的活性剤および相互に混和性の低水溶性有機溶媒の混合物を含有する溶液から成り、ハロゲン化炭化水素を含有しない第1相を調製し、親水性コロイドまたは界面活性剤の水溶液から成る第2相を調製し、第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相、第2相が連続相となったエマルジョンを形成し、不連続第1相を微粒子の形態で分離し、クエンチ液を用いて、先の工程の微粒子から残留溶媒を抽出することを含んで成る生分解性微粒子の製法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、微粒子の製造に関する。本発明はとりわけ、スタティックミキサーの使用により、活性剤を封入して調節的放出性微粒子を形成する方法に関する。本発明は、活性剤を封入して調節的放出性微粒子を形成する方法において有用な溶媒系にも関する。「微粒子」または「小球」とは、該粒子のマトリックスとして機能する生分解性ポリマー中に分散または溶解した活性剤を含有する固体粒子を意味する。
【背景技術】
【0002】
化合物を微粒子の形態に封入することのできる方法は、種々知られている。生物学的活性剤または薬剤を、生体適合性、生分解性、壁形成材料(例えば、ポリマー)中に封入して、薬物または他の活性剤の徐放または遅延放出を提供することは、特に有利である。そのような方法において、封入する材料(薬物または他の活性剤)を、通例、撹拌機または他の動的混合法によって、壁形成材料含有溶媒中に、溶解、分散または乳化する。次いで、溶媒を微粒子から除去した後、微粒子生成物が得られる。
【0003】
従来のマイクロカプセル化方法の一例は、米国特許第3,737,337号に開示されている。それによると、溶媒中の壁または殻形成ポリマー材料の溶液を調製する。溶媒は、部分的にしか水混和性でない。そのポリマー含有溶液中に、固体またはコア材料を溶解または分散した後、そのコア材料含有溶液を、微粒子から溶媒を除去するために、有機溶媒非混和性の水性液体中に分散する。封入または包埋する物質は、従来の混合機(バイブレーターおよび高速撹拌機などを包含する)を用いて(分散液の調製において)、ポリマーの有機溶液(相A)中に溶解または分散する。相(A)(溶液または分散液中に、コア材料を含有する)の、水相
(B)中の分散を、やはり従来の混合機、例えば高速ミキサー、バイブレーションミキサーまたは噴霧ノズル(この場合、微粒子の粒子サイズは、相(A)の濃度だけでなく、得られる粒子サイズによっても決まる)を用いて行う。
【0004】
物質を含有する微粒子から溶媒を除去する方法のもう一つの例は、米国特許第3,523,906号に開示されている。その方法においては、封入する材料を水非混和性の溶媒中のポリマー材料の溶液中に乳化し、次いで、そのエマルジョンを、親水性コロイド含有水溶液中で乳化する。次いで、蒸発によって微粒子から溶媒を除去して、生成物を得る。
【0005】
米国特許第3,691,090号に開示された更に別の方法においては、水性媒体中の微粒子分散液から、好ましくは減圧下に、有機溶媒を蒸発する。
【0006】
同様に、米国特許第3,891,570号が開示する方法においては、誘電率10またはそれ以下で、多価アルコール難混和性の溶媒中に溶解した壁材料の溶液に、コア材料を溶解または分散し、次いで、多価アルコールへの分散または溶解によって小滴に乳化し、最後に加熱または減圧によって溶媒を除去することによって、微粒子を調製する。
【0007】
活性剤を封入し得る方法の別の例が、に開示されている。カプセル用の壁材料を、水難混和性で、沸点100℃未満で、蒸気圧が水よりも高く、誘電率が約10未満の少なくとも1種の有機溶媒に溶解し;得られた溶液に、水不溶または難溶性の薬物を溶解または分散し;得られた溶液または分散液を、親水性コロイドまたは界面活性剤の水溶液から成る液体ビヒクル中で分散して小滴を形成した後、有機溶媒を蒸発により除去することによって、封入薬物を調製する。小滴サイズは、撹拌速度、薬物および壁材料を含有する有機溶媒の粘度、並びにビヒクルの粘度および表面張力によって決まる。
【0008】
タイス(Tice)らの米国特許第4,389,330号には、2段階溶媒除去法による、活性剤含有微粒子の調製が記載されている。この2段階溶媒除去法は、1段階で溶媒を除去する方法と比べて、活性剤封入率がより高く、より高品質の微粒子が得られるので、有利である。タイスらの方法においては、活性剤およびポリマーを溶媒に溶解する。次いで、溶媒中の成分混合物を、該溶媒と非混和性の連続相加工媒体中で乳化する。混合した材料の機械的撹拌により、連続相媒体中に、所定成分含有微粒子の分散液を形成する。その分散液から、溶媒除去法の第1段階において、有機溶媒を部分的に除去する。第1段階後、何らかの従来の分離法により、連続相加工媒体から、分散した微粒子を分離する。この分離後、微粒子中の残りの溶媒を、抽出により除去する。残りの溶媒を微粒子から除去した後、風乾、または他の従来の乾燥法で乾燥する。
【0009】
タイスらの米国特許第4,530,840号には、抗炎症活性剤を含有する微粒子の製法であって、(a)抗炎症剤を溶媒に溶解または分散し、その溶媒に生体適合性および生分解性の壁形成材料を溶解し;(b)抗炎症剤および壁形成材料を含有する溶媒を、連続相加工媒体中に分散し;(c)工程(b)の分散液から、溶媒の一部を蒸発し、それによって、懸濁液中に抗炎症剤を含有する微粒子を形成し;(d)残りの溶媒を微粒子から除去することを含んで成る方法が記載されている。
【0010】
国際特許出願公開90/13361号には、剤をマイクロカプセル化してマイクロカプセル化生成物を形成する方法であって、壁形成材料を溶解した溶媒中に、有効量の剤を分散して、分散液を形成し;分散液を、有効量の連続加工媒体と組み合わせて、加工媒体と、剤、溶媒および壁形成材料から成る小滴とを含有するエマルジョンを形成し;エマルジョンを、有効量の抽出媒体に短時間で加えて、小滴から溶媒を抽出することにより、マイクロカプセル化生成物を形成するという工程を含んで成る方法が開示されている。
【0011】
ボトマイヤー(Bodmeier, R.)らのインターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)43:179−186(1988)には、薬物含量を高めるために、塩化メチレン、クロロホルムおよびベンゼン、並びに塩化メチレンと水非混和性液体、例えばアセトン、酢酸エチル、メタノール、ジメチルスルホキシド、クロロホルムまたはベンゼンとの混合物を包含する種々の溶媒を用いて、活性剤としてのキニジンまたはキニジンスルフェート、および結合剤としてのポリ(D,L−ラクチド)を含有する微粒子の製造が開示されている。
【0012】
ベック(Beck, L.R.)らのバイオロジー・オブ・リプロダクション(Biology of Reproduction)、28:186−195(1983)には、ノルエチステロンを、D,L−ラクチド/グリコリドコポリマー中に封入する方法であって、コポリマーとノルエチステロンの両方を、クロロホルム/アセトン混合物に溶解し、それを、撹拌したポリビニルアルコールの冷水溶液に加えてエマルジョンを形成し、揮発性溶媒を減圧下に除去してマイクロカプセルを得る方法が開示されている。
【0013】
生分解性ポリマーマトリックスおよび生物学的活性剤から成る微粒子の調製には、相分離または非溶媒誘導コアセルベーションも用いられている。ラクチド/グリコリドコポリマーでマイクロカプセル化する文献記載の方法の多くでは、溶媒蒸発/抽出法を採用しているが、そのような方法は、水不溶性薬物に主として適当なものであり、水溶性薬物は、調製工程中に、水相中に一部分配され得る。そのような活性剤の封入方法としては、親水性活性剤も不溶の、ポリマーに対する非溶媒を用いる相分離方法が有効である。
【0014】
従来の相分離法においては、既知量のポリマー、例えばポリ(ラクチド−コ−グリコリド)(PLGA, ラクチド:グリコリドモノマー比100:0ないし50:50)を、適当な有機溶媒に溶解する。固体薬物(好ましくは凍結乾燥または粉砕したもの)を、ポリマー溶液に分散し得る(有機溶媒に不溶または難溶)。あるいは、活性剤を、水、または何らかの添加剤を含有する水に溶解し、ポリマー溶液中で乳化して(好ましくは主として超音波処理による)、油中水型エマルジョンを形成する。次いで、得られた懸濁液またはエマルジョンを反応器に入れ、所定の速度で第1非溶媒の添加を開始する。反応器に備えたタービンミキサーにより、緩やかに混合する。相分離の完了時に、混合物を、第2非溶媒を入れたクエンチタンクに移し、半固形小球を凝固する。硬化した小球を篩過により集め、洗浄し、減圧オーブン内に入れて更に乾燥する。
【0015】
既知のマイクロカプセル化に使用する溶媒は通例、ハロゲン化炭化水素、とりわけクロロホルムまたは塩化メチレンであり、これは活性剤および封入ポリマーのいずれの溶媒としても機能する。しかし、最終生成物中に少量だが検出可能な量で残留するハロゲン化炭化水素は、一般に毒性であり、発癌性を有し得るので、望ましくない。すなわち、既知のマイクロカプセル化方法を、より低毒性の許容し得る溶媒を使用するよう改善する必要がある。
【0016】
前記のような生物学的または薬学的活性剤のマイクロカプセル化のための従来の方法においては、活性剤およびポリマーを含有する溶媒を、撹拌、振動または他の動的混合法によって、しばしば比較的長時間、非混和性溶液中に乳化または分散することによって、微粒子を形成する。上記のような動的混合法は、いくつかの欠点を有する。例えば、得られる微粒子のサイズ、またはサイズ分布を調節しにくい。それ故、動的混合を行うと、生物学的または薬学的活性剤含有微粒子の工業的規模での製造には問題が生じる。とりわけ、製造装置は高価なエマルジョンタンク(液体を撹拌する装置を含む)を包含する。全工程時間の調節因子の一つは、均一なエマルジョンの形成に要する時間である。大きなタンクでバッチサイズが大きいほど、エマルジョン形成に長い時間がかかり、全体的な製造時間が長くなる。活性剤を加工溶媒およびポリマー溶液にさらす時間が長いほど、活性剤の分解または不活性化が起こり易い。生物学的または薬学的活性剤のマイクロカプセル化の場合、実験室エマルジョン法から製造法へのスケールアップは特に困難である。なぜなら、バッチおよびタンクサイズを大きくすると、大きいタンク内での撹拌速度および粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。同様に、相分離法も、微粒子の工業規模の製造工程に容易に変換できない。なぜなら、工程パラメータ、すなわち非溶媒添加速度、撹拌条件、並びに活性剤/ポリマー溶液および非溶媒の粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。すなわち、従来のマイクロカプセル化法のスケールアップは、時間がかかる上に、精密でない。
【0017】
エストラジオールベンゾエート含有微粒子製造において、実験室エマルジョン形成工程を小撹拌ガラス製反応器から製造装置にスケールアップする試みにおいて、試験を行った。ミキサー翼により生じる剪断力が、エマルジョンの粒子サイズを決定した。剪断力が大きいほど、粒子は小さい。エストラジオールベンゾエートの工程においては、油(有機)相が低粘度である故に、所望の大きいエマルジョン粒子を形成するのに、小さい剪断力が必要であった。大きい反応器においては、小さい剪断力を保ち、かつ、均一な混合を行うことが困難である。均一なタンク組成物を提供する撹拌機速度では、サイズ分布の広い小さいサイズの粒子が生成する。混合翼直径を増し、シャフトに沿って複数の混合翼を用いると、小さい剪断力での混合性は向上するが、まだサイズ分布は非常に広い。バッチサイズを大きくすると、粒子サイズ調節の信頼性が低下する。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】米国特許第3,737,337号
【特許文献2】米国特許第3,523,906号
【特許文献3】米国特許第3,691,090号
【特許文献4】米国特許第3,891,570号
【特許文献5】米国特許第3,960,757号
【特許文献6】米国特許第4,389,330号
【特許文献7】米国特許第4,530,840号
【特許文献8】国際特許出願公開90/13361号
【非特許文献】
【0019】
【非特許文献1】International Journal of Pharmaceutics, 43:179−186(1988)
【非特許文献2】Biology of Reproduction, 28:186−195(1983)
【発明の概要】
【発明が解決しようとする課題】
【0020】
本発明の目的は、生物学的または薬学的活性剤を含有する微粒子の適度に定めた狭いサイズ分布を達成しつつ、実験室から工業的規模へのバッチサイズを精密に信頼性よくスケールアップすることができる微粒子製造方法を提供することである。このことは、いずれの適当な封入法(溶媒抽出および相分離を包含するが、それに限定されない)においても達成し得る。
【0021】
本発明の別の目的は、適当に定めたサイズ分布を有する活性剤含有微粒子を、種々のバッチサイズで形成するのに、同一の装置を使用し得る方法を提供することである。
【0022】
本発明の方法のもう一つの目的は、一工程で溶媒を除去するか、または相分離方法によって、活性剤濃度の高い高品質の微粒子が得られる方法を提供することである。
【課題を解決するための手段】
【0023】
本発明は、微粒子の製法に関する
【0024】
とりわけ、本発明は、生分解性ポリマー結合剤および生物学的活性剤から成る生分解性微粒子の製法に関する。本発明の一態様においては、第1相(活性剤およびポリマーを含有する)および第2相を、スタティックミキサーを通してクエンチ液体にポンプ輸送して、活性剤含有微粒子を形成する。他の態様においては、第1相と第2相とは、実質的に非混和性である。他の態様においては、第2相はポリマーおよび活性剤に対する溶媒を含有せず、乳化剤の水溶液から成り得る。スタティックミキサーを使用して微粒子を製造する本発明の方法は、いずれの従来の封入法(溶媒抽出および相分離を包含するが、それに限定されない)にも適用し得る。
【0025】
本発明の更なる態様においては、第1相は、活性剤をポリマー含有溶液に溶解することによって、活性剤含有分散液を調製するとによって、および活性剤含有エマルジョンを調製することによって調製する。
【0026】
本発明の更なる態様においては、次のような活性剤を含有する微粒子を製造するために本発明の方法を用いる:リスペリドン(risperidone)、トレンボロンアセテート、ノルエチンドロン、テストステロン、エストラジオールベンゾエート、ヒト血清アルブミン、ブタアルブミンおよび組換えウシインターフェロン−α。
【0027】
本発明の好ましい一態様においては、活性剤およびポリマーを溶解するのに、少なくとも2種の実質的に無毒性の溶媒の混合物(ハロゲン化炭化水素不含有)を使用する。溶解した活性剤およびポリマーを含有する溶媒混合物を、水溶液に分散して、液滴を形成する。得られるエマルジョンを、次いで、水性抽出媒体(好ましくは溶媒混合物の少なくとも1種の溶媒を含有し、それにより、各溶媒の抽出速度を調節する)に加え、生物学的活性剤を含有する生分解性微粒子を形成する。この方法は、1種の溶媒の水溶性は実質的に他方とは独立しているので、抽出媒体が必要量が少なくてよく、溶媒選択性が広い(とりわけ、特に抽出困難な溶媒を含む)という利点を有する。
【0028】
本発明は好ましい一態様において、有効量の薬物を長時間にわたって調節的に放出するよう設計した微粒子の形態の薬剤組成物を製造する方法において有用な溶媒系に関する。この組成物は、少なくとも1種の薬剤および少なくとも1種の生体適合性、生分解性封入ポリマーを含有する。
【0029】
とりわけ、本発明の更なる態様においては、本発明は、微粒子の製法であって、
A.ハロゲン化炭化水素不含有の、水溶性の低い相互に混和性の有機溶媒少なくとも2種の混合物中に溶解または分散した生分解性ポリマー封入結合剤および活性剤から成る第1相を調製し、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とを混合手段の作用下に組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離する
ことを含んで成る方法に関する。
【0030】
低水溶性とは、20℃における水溶性が約0.1〜25重量%であることを意味する。
【0031】
好ましい態様においては、本発明は、微粒子の製法であって、溶媒混合物中に固体を約5〜50重量%含有する第1「油」相を調製し、その約5〜95重量%は生分解性ポリマー封入結合剤の溶液であり、第1相はポリマー結合剤に対して約5〜95重量%の活性剤を組み合わせたものであり、溶媒混合物は、ハロゲン化炭化水素不含有の、相互に混和性の第1および第2溶媒から成り、各溶媒の水溶性は20℃において約0.1〜25重量%であり;第1相をエマルジョン加工媒体中に1:1ないし1:10の比で含有するエマルジョンを形成して、連続水性第2相加工媒体中に第1相組成物の小滴を形成し;合した第1および第2相を、ポリマーおよび活性剤1g当たり水性クエンチ液約0.1〜20リットルのレベルの水性抽出クエンチ液に加え、クエンチ液は、溶媒混合物の溶媒の水溶性の大きい方を、使用温度でクエンチ液に対して約20〜70%の飽和レベルで含有し;クエンチ液から微粒子を回収することを含んで成る方法に関する。
【0032】
他の態様においては、本発明は、微粒子の製法であって、第1相を調製し、第1相は、生物学的活性剤、生分解性ポリマー、並びにハロゲン化炭化水素不含有の、少なくとも2種の相互に混和性の、活性剤およびポリマーに対する溶媒の混合物から成り;第2相を調製し、第1相は第2相とは実質的に非混和性であり;第1相をスタティックミキサーに第1流速で流し;第2相をスタティックミキサーに第2流速で流すことによって、第1相と第2相とを同時にスタティックミキサーに流して、活性剤含有微粒子を形成し;微粒子を分離することを含んで成る方法に関する。
【0033】
他の態様においては、本発明は、微粒子の製法であって、第1相を調製し、第1相は、生物学的活性剤、生分解性ポリマー、並びにハロゲン化炭化水素不含有の、少なくとも2種の相互に混和性の、活性剤およびポリマーに対する溶媒の混合物から成り;第2相を調製し、第1相は第2相とは実質的に非混和性であり;クエンチ液を調製し;第1相と第2相とをスタティックミキサーを通してクエンチ液にポンプ輸送することによって、活性剤含有微粒子を形成することを含んで成る方法に関する。
【0034】
本発明の更なる態様においては、(1)ハロゲン化炭化水素不含有の少なくとも2種の相互に混和性の溶媒に溶解したポリマーの溶液に、生物学的活性剤を溶解し、または(2)前記溶媒中に活性剤を含有する分散液を調製し、または(3)前記溶媒中に活性剤を含有するエマルジョンを調製することによって、第1相を調製する。
【図面の簡単な説明】
【0035】
【図1】スタティックミキサー内の流れを示す図である。
【図2】本発明の方法において使用し得るスタティックミキサーを示す図である。
【図3】本発明の好ましい微粒子製造方法を行うための実験室用装置を示す図である。
【図4】ノルエチンドロン含有微粒子の2製剤の時間放出動物実験データのグラフを示す。
【図5】バッチプロデックス(Podex)3のリスペリドン微粒子(製造したままのものと、凍結乾燥したものの両方)の、インビトロ溶解データのグラフを示す。
【図6】バッチプロデックス2のリスペリドン微粒子(製造したままのものと、凍結乾燥したものの両方)の、インビトロ溶解データのグラフを示す。
【図7】バッチプロデックス3およびプロデックス2のリスペリドン微粒子の、促進インビトロ溶解データのグラフを示す。
【図8】リスペリドンのデポ製剤を約2.5mg/kgの用量でビーグル犬に1回筋肉内投与した後の、活性物(リスペリドンおよび9−ヒドロキシリスペリドン)の平均(n=2)血漿濃度−時間曲線のグラフを示す。 アポモルヒネ催吐試験における制吐活性(少なくとも3匹中2匹の犬に見られる)時間を、各製剤の解説において示す。星印(*)は、試験開始後に3匹中少なくとも2匹の犬において制吐活性の中断があったことを意味する。点線は、制吐効果に必要なおよその最小血漿濃度を示す。「//」印は、製剤プロデックス2に対しては、14日、18日および21日目に血液サンプリングを行わなかったことを意味する。
【図9】エストラジオールベンゾエート含有微粒子の粒子サイズの累積%のグラフを示す。
【図10】エストラジオールベンゾエート含有微粒子の粒子サイズの%微分のグラフを示す。
【図11】エストラジオールベンゾエート含有微粒子の、時間放出動物実験データのグラフを示す。
【図12】トレンボロンアセテート含有微粒子の粒子サイズの累積%のグラフを示す。
【図13】テストステロン含有微粒子の、時間放出動物実験データのグラフを示す。
【図14A】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図14B】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図14C】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図15A】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【図15B】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【図15C】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【発明を実施するための形態】
【0036】
本発明においては、少なくとも1種の生物学的活性剤を含有する生分解性微粒子を製造するために、少なくとも2種の溶媒から成る、ハロゲン化炭化水素不含有の溶媒混合物を使用する。溶媒混合物の第1溶媒成分は、活性剤に対しては難溶媒であるが、本発明に使用する生分解性ポリマーに対しては良好な溶媒である。溶媒混合物の第2溶媒成分は、活性剤およびポリマーの両方に対して良好な溶媒である。
【0037】
本発明の方法は、既知の方法にまさる利点を有する。本発明の方法はとりわけ、生分解系、処置中の用量損失を防いだ注射系、異種薬物含有微粒子の混合性、ハロゲン化炭化水素残留のない微粒子、並びに必要に応じてより速くまたは遅く薬物を放出する調節的放出性(多相放出パターン)を提供する。
【0038】
本発明の方法で製造した生成物は、選択した微粒子の種類に応じて、作用期間が30ないし200日以上であるという利点を有する。好ましい態様においては、微粒子は、30〜60日間にわたって患者の処置を提供するよう設計する。作用期間は、ポリマー組成、ポリマー:薬物比、および微粒子サイズの変更によって、容易に調節し得る。
【0039】
本発明の方法により製造した微粒子のもう一つの重要な利点は、本発明の方法に使用するポリマーは生分解性であり、封入した剤を全部患者に放出するので、実質的に全部の活性剤が患者にデリバーされるということである。
【0040】
本発明の方法においては、ハロゲン化炭化水素不含有溶媒混合物に活性剤を溶解または分散し、その活性剤含有媒体に、ポリマーマトリックス材料を、活性剤に対し相対的に、所望の活性剤含有量の生成物を与えるような量で加える。要すれば、微粒子生成物の全成分を同時に溶媒混合物媒体中に混合し得る。
【0041】
本発明において使用する溶媒系は、少なくとも2種の溶媒の混合物である。そのような溶媒は、
(1)相互に混和性であり、
(2)混合した状態で、活性剤を溶解または分散でき、
(3)混合物した状態で、ポリマーマトリックス材料を溶解でき、
(4)活性剤に対して化学的に不活性であり、
(5)生体適合性であり、
(6)実質的にクエンチ液と非混和性であり(例えば、溶解度約0.1〜25%以下)、
(7)ハロゲン化炭化水素以外の溶媒でなくてはならない。
【0042】
「ハロゲン化炭化水素」とは、ハロゲン化有機溶媒、すなわちC1−C4ハロゲン化アルカン、例えば塩化メチレン、クロロホルム、塩化メチル、四塩化炭素、二塩化エチレン、塩化エチレン、2,2,2−トリクロロエタンなどを意味する。
【0043】
活性剤封入のための好ましい溶媒混合物は、ポリマー封入剤の溶解度が、通例、20℃で少なくとも約5重量%、好ましくは少なくとも約20重量%と、高くなければならない。溶解度の上限は重要でないが、封入ポリマーが溶液の約50重量%を超えると、溶液が非常に粘性となって、有効かつ好都合に扱うことが困難になり得る。このことは当然、封入ポリマーの性質および分子量によって異なる。
【0044】
溶媒系は、連続相加工媒体およびクエンチ液(通例、水性)と実質的に非混和性であるが、好ましくは水に少し溶解する。溶媒系が加工媒体にいくらでも溶解するならば、エマルジョン相において小滴が生成し得ないであろう。しかし、抽出クエンチ媒体中の溶媒系の溶解度が低すぎると、クエンチ媒体が大量に必要となる。通例、加工媒体およびクエンチ媒体中に約0.1〜25%溶解する溶媒が、本発明において許容し得る。クエンチ媒体が、第1溶媒(すなわち、クエンチ媒体に溶解し易い方の溶媒)を飽和点の約70〜20重量%の量で含有すると、微粒子からクエンチ媒体への第1溶媒の損失率を調節するためにしばしば有利であり得る。
【0045】
本発明の溶媒混合物の成分の選択において更に考慮することは、沸点(すなわち、蒸発による最終生成物の与え易さ)、および比重(乳化およびクエンチング中に、「油相」が浮く傾向)である。また、溶媒系は低毒性であるべきである。
【0046】
通例、溶媒混合物組成物は、第1溶媒を約25〜75重量%、第2溶媒を約75〜25重量%含有し得る。
【0047】
本発明の溶媒混合物は好ましくは、エステル、アルコールおよびケトンの少なくとも2種の混合物である。好ましいエステルは、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基、すなわちメチル、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択する。]で示される。本発明において、溶媒混合物の一成分として使用するのに最も好ましいエステルは、酢酸エチルである。好ましいアルコールは、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示される。R3は、より好ましくはアリールである。本発明において溶媒混合物の一成分として使用するのに最も好ましいアルコールは、ベンジルアルコールである。好ましいケトンは、式R4COR5[式中、R4はC1−C4アルキル基、すなわち、メチル、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択し、R5はC2−C4アルキル基、すなわち、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択する。]で示される。本発明において溶媒混合物の一成分として使用するのに最も好ましいケトンは、メチルエチルケトンである。
【0048】
本発明の方法によって製造する微粒子のポリマーマトリックス材料は、生体適合性および生分解性のポリマー材料である。「生体適合性」とは、ポリマー材料が人体に無毒で、非発癌性で、生体組織中で顕著な炎症を誘導しないことと定義する。マトリックス材料は、生体の代謝によって生体が容易に排泄し得る物質に分解され、生体中に蓄積しないという意味において、生分解性でなければならない。生分解生成物も、ポリマーマトリックスが生体適合性であるのと同様の意味で生体適合性であるべきであり、微粒子中に残留し得る溶媒も同様である。
【0049】
ポリマーマトリックス材料の適当な例は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、並びに天然ポリマー、例えばアルブミン、カゼイン、および蝋、例えばグリセロールモノ−およびジステアレートなどを包含する。種々の市販のポリ(ラクチド−コ−グリコリド)材料(PLGA)を、本発明の方法において使用し得る。例えば、ポリ(d,l−乳酸−コ−グリコール酸)が、メデソーブ・テクノロジーズ・インターナショナルL.P.(Medisorb Technologies International L.P.;オハイオ州シンシナチ)から市販されている。メディソーブから市販の適当な生成物は、メディソーブ(MEDISORB、商標)5050DLとして知られる50:50ポリ(D,L)乳酸−コ−グリコール酸である。この生成物のモル%組成は、ラクチド50%およびグリコリド50%である。他の適当な市販生成物は、メディソーブ65:35DL、75:25DL、85:15DL、およびポリ(d,l−乳酸)(d,l−PLA)である。ポリ(ラクチド−コ−グリコリド)は、ベーリンガー・インゲルハイム(Boehringer Ingelheim、ドイツ)からもレゾマー(Resomer)の商品名で市販されており[例えば、PLGA50:50(レゾマーRG502)、PLGA75:25(レゾマーRG752)およびd,l−PLA(レゾマーRG206)]、バーミンガム・ポリマーズ(Birmingham Polymers、アラバマ州バーミンガム)からも市販されている。このようなポリマーは、種々の分子量、および乳酸:グリコール酸比のものが市販されている。
【0050】
本発明に最も好ましいポリマーは、ポリ(d,l−ラクチド−コ−グリコリド)である。そのようなコポリマー中、ラクチド:グリコリドのモル比は、好ましくは約85:15ないし50:50である。
【0051】
ポリマーマトリックス材料の分子量は、ある程度重要である。分子量は、充分なポリマーコーティングの形成のために充分大きくなくてはならない(すなわち、ポリマーは良好なフィルムポリマーでなくてはならない)。通例、充分な分子量は、5000〜500000ダルトンの範囲で、好ましくは約150000ダルトンである。しかし、フィルムの性質は、使用するポリマー材料の種類によってもいくぶん変化するので、すべてのポリマーについて適当な分子量範囲を特定することは非常に困難である。ポリマー分子量は、ポリマーの生分解速度の点からも重要である。薬物放出の拡散メカニズムのために、ポリマーは薬物全部が微粒子から放出されてしまうまで変化せず、その後、分解されるべきである。薬物は、ポリマー賦形剤が生体により浸食される間に放出されてもよい。ポリマー材料を適当に選択することによって、拡散放出および生分解放出の両方を行う微粒子を形成するように微粒子製剤を製造してもよい。これは、多相放出パターンに有用である。
【0052】
本発明の方法によって製造する製剤は、微粒子のポリマーマトリックス材料中に分散した活性剤を含有する。微粒子中に含まれる活性剤の量は通例、約1〜90重量%、好ましくは30〜50重量%、より好ましくは35〜40重量%である。重量%は、微粒子全重量に対する剤の割合である。例えば、剤10重量%は、剤が10重量部、ポリマーが90重量部存在することを意味する。
【0053】
本発明の方法の実施において、溶媒混合物中に封入ポリマーが実質的に100%溶解した溶液を乳化に用いる。活性剤は、その溶液を連続相加工媒体に加える際に、溶媒混合物に分散または溶解し得る。最初の乳化の際の、溶媒混合物中の通例固体の材料(活性剤と封入ポリマー)の含量は、少なくとも5重量%、好ましくは少なくとも20重量%であるべきである。「油相」中の溶媒を少なくすると、良質の微粒子が得られ、抽出媒体必要量は少なくてよい。
【0054】
本発明の方法によって封入し得る好ましい活性剤の一つは、ノルエチンドロン(NET)である。リスペリドンおよびテストステロンも好ましい。
【0055】
酢酸エチル単独ではNETの難溶媒であり、従来のクロロホルム法よりも多くの溶媒および高い温度を要する。生成物微粒子のコア含有は許容し得るが、収率(とくに63〜90μm範囲のもの)は低い。走査電子顕微鏡写真によると、そのようなより大きい粒子は割れ、崩壊する。その証拠に、そのような粒子は正常な放出速度よりも放出速度が大きい。
【0056】
ベンジルアルコールを単独で溶媒として使用すると、クエンチタンク内容物を光学顕微鏡で観察してわかるように、微粒子サイズを調節し易い。しかし、乾燥すると通例、品質の低いことがわかる。しばしば、付着性の故に、回収困難である。また、溶媒残留量が増加する傾向にある。酢酸エチルとベンジルアルコールとを「油相」の溶媒系として使用すると、微粒子の品質および放出性が改善された。
【0057】
「油相」溶媒系中の成分混合物を、連続相加工媒体中で乳化する;連続相媒体は、所定成分を含有する微粒子の分散液を連続相媒体中に形成するものである。
【0058】
絶対に必要というわけではないが、連続相加工媒体を、「油相」溶媒系を形成する溶媒の少なくとも一種で飽和することが好ましい。これにより安定なエマルジョンが生成し、クエンチング前に微粒子から溶媒が排出されるのを防ぐ。米国特許第4389330号記載のように、減圧を適用してもよい。酢酸エチルおよびベンジルアルコールが溶媒系成分である場合、エマルジョンの水相は好ましくは、酢酸エチル1〜8重量%およびベンジルアルコール1〜4重量%を含有する。
【0059】
通例、界面活性剤または親水性コロイドを連続相加工媒体に加えて、溶媒小滴の凝集を防止し、エマルジョン中の溶媒小滴のサイズを調節する。界面活性剤または親水性コロイドとして使用し得る化合物の例は、ポリ(ビニルアルコール)、カルボキシメチルセルロース、ゼラチン、ポリ(ビニルピロリドン)、トゥイーン(Tween)80、トゥイーン20などを包含するが、それらに限定されない。加工媒体中の界面活性剤または親水性コロイドの濃度は、エマルジョンの安定化に充分な量であるべきであり、微粒子の最終サイズに影響し得る。通例、加工媒体中の界面活性剤または親水性コロイドの濃度は、界面活性剤または親水性コロイド、「油相」溶媒系、および加工媒体の種類によって異なるが、加工媒体に対して約0.1〜10重量%であり得る。好ましい分散媒体組み合わせは、水中のポリ(ビニルアルコール)の0.1〜10重量%、より好ましくは0.5〜2重量%溶液である。
【0060】
エマルジョンは、混合相の機械的撹拌によって、または活性剤と壁形成材料とを含有する有機相の小滴を連続相加工媒体に加えることによって形成し得る。エマルジョン形成中の温度は特に重要ではないが、微粒子のサイズおよび品質、並びに連続相中の活性剤の溶解度に影響し得る。当然、連続相中に入る活性剤は少ないほどよい。更に、使用する溶媒混合物および連続相加工媒体によっては、温度が低すぎてはならない。なぜなら、溶媒および加工媒体が凝固または不適当に増粘し得るからである。また、加工媒体が蒸発し得るか、または液体加工媒体が維持できなくなり得るほど高温にしてもいけない。更に、エマルジョン温度は、微粒子中に組み合わせる活性剤の安定性を損なうほど高くしてはならない。従って、分散工程は、安定な操作条件を保つ温度で、選択した活性剤および賦形剤によるが、好ましくは約20〜60℃で行い得る。
【0061】
前記のように、活性剤含有微粒子を生成するように、有機相と水相を組み合わせる。有機相と水相とは概ね、または実質的に非混和性であり、水相はエマルジョンの連続相を構成する。有機相は、活性剤と、壁形成ポリマー(すなわちポリマーマトリックス材料)を含有する。有機相は、本発明の有機溶媒系に活性剤を溶解または分散することによって調製する。有機相と水相は、混合手段の作用下に組み合わせる。
【0062】
好ましい混合手段はスタティックミキサーであり、活性剤の封入により調節的放出微粒子を形成する本発明の方法は、スタティックミキサーの使用を伴う。好ましくは、合した有機および水相を、ポンプ輸送によりスタティックミキサーを通してエマルジョン形成し、大量のクエンチ液に入れて、ポリマーマトリックス材料中に封入した活性剤を含有する微粒子を得る。
【0063】
生物学的または薬学的剤をマイクロカプセル化する多くの既知の方法においては、活性剤およびポリマーを含有する溶媒を、非混和性の第2の溶媒中で、撹拌、混合、振動、または何らかの他の動的混合法によって、しばしば比較的長時間、乳化または分散することによって、微粒子を形成する。上記のような動的混合法は、いくつかの欠点を有する。例えば、得られる微粒子のサイズ、またはサイズ分布を調節しにくい。それ故、動的混合を行うと、生物学的または薬学的活性剤含有微粒子の工業的規模での製造には問題が生じる。とりわけ、製造装置は高価なエマルジョンタンク(液体を撹拌する装置を含む)を包含する。全工程時間の調節因子の一つは、均一なエマルジョンの形成に要する時間である。大きなタンクでバッチサイズが大きいほど、エマルジョン形成に長い時間がかかり、全体的な製造時間が長くなる。活性剤を加工溶媒およびポリマー溶液にさらす時間が長いほど、活性剤の分解または不活性化が起こり易い。生物学的または薬学的活性剤のマイクロカプセル化の場合、実験室エマルジョン法から製造法へのスケールアップは特に困難である。なぜなら、バッチおよびタンクサイズを大きくすると、大きいタンク内での撹拌速度および粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。このような方法は、時間がかかる上に、精密でない。
【0064】
すなわち、スタティックミキサーを使用する微粒子製造方法の一つの利点は、生物学的または薬学的活性剤を含有する微粒子の適度に定めた狭いサイズ分布を達成しつつ、実験室から工業的規模へのバッチサイズを精密に信頼性よくスケールアップすることができるということである。本発明の方法の更なる利点は、適当に定めたサイズ分布を有する活性剤含有微粒子を、種々のバッチサイズで形成するのに、同一の装置を使用し得るということである。本発明の方法のもう一つの利点は、一工程で溶媒を除去して、活性剤濃度の高い高品質の微粒子が得られるということである。前記タイスらの米国特許第4389330号に記載のような2段階溶媒除去方法を行う必要がない。方法の改良に加えて、スタティックミキサーは維持費が安く、小さいので動的ミキサーよりもスペースが狭くてよく、エネルギー消費が少なく、比較的投資額が少なくてよい。
【0065】
スタティックまたは静的ミキサーは、導管または管を有し、その内部に複数の静的混合要素を有する。スタティックミキサーは、比較的短い導管内で、比較的短時間のうちに均一な混合を行う。スタティックミキサーの場合、ミキサーの一部(例えば翼)が液体中で動くのではなく、液体がミキサー内を動く。一種のスタティックミキサー内の流れを、図1に示す。ポンプ(図示せず)が、1種またはそれ以上の液流(1)を、スタティックミキサー(10)内に導入する。液流は分れて、向き合った外壁に当たる(2)。スタティックミキサー(10)の中心線を軸とする渦が生じる(3)。渦は途切れて、その過程が繰り返される(ただし逆回転)(4)。右回り/左回りの動きによって、確実に均一な生成物が得られる。
【0066】
スタティックミキサーの例を図2に示す。スタティックミキサー(10)は、導管またはパイプ(12)内に連続的に配置された複数の静止または静的混合要素(14)を有する。要素数は4〜32またはそれ以上であり得る。導管(12)は円形断面を有し、液体を導入および排出するよう、両端(18および20)が開口している。混合要素(14)は、セグメント(142)から成る。各セグメント(142)は、複数の、通例平らな板または羽(144)から成る。2つの実質的に同じセグメント(142)を、好ましくは相互にずらして軸方向に配置する。図2に示すスタティックミキサーは、米国特許第4511258号に、より詳細に記載されており、該特許を引用により本発明の一部とする。
【0067】
スタティックミキサーを用いてエマルジョンを形成する場合、種々の因子がエマルジョン液滴サイズを決定する。そのような因子は、混合する溶液または相の密度および粘度、相の体積比、相間界面張力、スタティックミキサーパラメータ(導管直径、混合要素の長さと数)、並びにスタティックミキサー内の流速を包含する。温度は、密度、粘度および界面張力に影響するので、変えることができる。主な調節因子は、流速である。スタティックミキサー単位長さ当たりの剪断速度および圧力低下も、重要なパラメータである。特に、流速が高まると液滴サイズが小さくなり、流速(および圧力低下)が低下すると液滴サイズが大きくなる。所定の流速で一定数の要素を通って動いた後、液滴サイズは平衡化する。流速が大きいほど、要素の必要数は少ない。このような関係の故に、実験室バッチサイズから工業的バッチサイズへのスケールアップは信頼性があり、精密であり、実験室および工業的のいずれのバッチサイズにも同じ装置を使用し得る。
【0068】
本発明の方法において、有機相および水相を、それらが同時にスタティックミキサー内に流れ、ポリマーマトリックス材料中に封入した活性剤を含有する微粒子を含むエマルジョンを形成するように、ポンプ輸送する。有機相および水相を、スタティックミキサーを経て大量のクエンチ液へポンプ輸送する。クエンチ液は、単なる水、水溶液、または他の適当な液体であり得る。クエンチ液中で洗浄または撹拌する間に、微粒子から有機溶媒を除去し得る。本発明の方法によって有機相および水相をスタティックミキサーを経て溶媒除去用クエンチ液にポンプ輸送すると、前記タイスらの特許(第4389330号)に記載の2段階溶媒除去を行わなくても、活性剤濃度の高い高品質の微粒子が生成する。微粒子をクエンチ液中で洗って有機溶媒を抽出または除去した後、例えば篩過により分離し、乾燥する。
【0069】
本発明の他の方法においては、有機相は、ポリマー溶液と、乾燥粉末として懸濁または水溶液中に溶解し、乳化した活性剤とから成る。ポリマーは、適当な有機溶媒、好ましくは非ハロゲン化溶媒、例えば酢酸エチルに溶解する。有機相をスタティックミキサー内で非溶媒と組み合わせると、活性剤粒子または液滴周囲にポリマー液滴のコアセルベーションまたは沈降(すなわち相分離)が起こる。スタティックミキサー内を流れる溶媒および非溶媒の滞留時間は、本発明の方法において重要な因子である。滞留時間は、混合要素および導管の寸法、並びにスタティックミキサー内を流れる溶液の線速度を変えることによって変化し得る。微粒子の形成に影響し得る他の重要な因子は、混合する2相の密度および粘度、相の体積比、並びに相間の界面張力である。しかし、微粒子サイズ調節は主として、最初の有機相中の活性剤懸濁液またはエマルジョンのサイズおよび均一性によって行う。
【0070】
スタティックミキサー法を行うための実験室用装置を、図3に示す。有機または油相(30)は、撹拌ポット(32)内で活性剤およびポリマーマトリックス材料を溶解することによって調製する。しかし、有機相(30)の調製法は、活性剤の溶解に限定されない。ポリマーマトリックス材料を含有する溶液に活性剤を分散することによって、有機相(30)を調製してもよい。そのような分散液中では、活性剤は有機相(30)に少ししか溶解しない。また、有機相(30)は、活性剤およびポリマーマトリックス材料を含有するエマルジョンの調製によっても調製し得る(ダブルエマルジョン法)。ダブルエマルジョン法においては、活性剤およびポリマーマトリックス材料を含有する第1エマルジョン(有機相30)を調製する。第1エマルジョンは、油中水型エマルジョン、水中油型エマルジョン、または他の適当なエマルジョンであり得る。第1エマルジョン(有機相30)および水相を、次いで、スタティックミキサーにポンプ輸送して、第2エマルジョンを形成する。第2エマルジョンは、ポリマーマトリックス材料中に封入された活性剤を含有する小滴を含有する。本発明のダブルエマルジョン法を用いて活性剤含有微粒子を調製する例は、実施例8に示す。
【0071】
有機相(30)を、磁気駆動ギアポンプ(34)によって撹拌ポット(32)からくみ出す。しかし、いずれの適当なポンプ手段を用いてもよい。ポンプ(34)からの輸送物はY字型連結具(36)に送られる。Y字型連結具(36)の1つの枝管(361)は、ポット(32)に戻る再循環流である。他の枝管(362)は、インラインスタティックミキサー(10)に至る。水相(40)も同様に、撹拌ポット(42)、磁気駆動ギアポンプ(44)およびY字型連結具(46)によって調製する。Y字型連結具(46)の1つの枝管(461)は、ポット(42)に戻る再循環流である。他の枝管(462)は、インラインスタティックミキサー(10)に至る。
【0072】
インラインスタティックミキサー(10)に送られる各溶液の枝流(362および462)は、別のY字形連結具(50)によって合流し、ミキサー導入ライン(51)を通ってスタティックミキサー(10)に送られる。スタティックミキサー(10)からの排出物は、ミキサー排出ライン(52)を通って洗浄タンク(60)に送られる。図3に示す装置において、シリコーンチューブおよびポリプロピレンフィッティングを使用する。ミキサー排出ライン(52)以外のすべてのラインに、内径3/8インチのシリコーンチューブを使用する。ミキサー排出ライン(52)には、ミキサー排出ライン(52)内、および洗浄タンク(60)への導入時の両方で、エマルジョンの破壊を防止するために、より径の小さいチューブ(内径3/16インチ)を使用する。
【0073】
本発明の方法の一態様においては、ポンプ(34および44)は再循環モードで始動し、有機相(30)および水相(40)の所望の流速を設定する。水相(40)の流速は、好ましくは有機相(30)の流速よりも大きい。しかし、2つの流速が実質的に等しくてもよい。水相(40)流速と有機相(30)流速の比は、好ましくは1:1ないし10:1である。次いで、Y字形連結具(46)を、水相(40)が枝管(462)を通ってスタティックミキサー(10)に流れるように切り替える。水相(40)でミキサー導入ライン(51)、スタティックミキサー(10)、およびミキサー排出ライン(52)が満たされたら、Y字形連結具(36)を、有機相(30)が枝管(362)を通ってスタティックミキサー(10)に流れるように切り替える。この時点で、有機相(30)と水相(40)とが、同時にスタティックミキサー(10)に流れる。所望量の有機相をスタティックミキサー(10)にポンプ輸送した後、Y字形連結具(36)を枝管(361)からの再循環に切り替える。水相(40)は、ミキサー導入ライン(51)、スタティックミキサー(10)およびミキサー排出ライン(52)内に残った有機相を洗い流すように、短時間流し続ける。Y字形連結具(46)を、次いで、枝管(461)からの再循環に切り替える。
【0074】
有機相(30)および水相(40)をスタティックミキサー(10)内で混合して、エマルジョンを形成する。生成したエマルジョンは、ポリマーマトリックス材料中に封入した活性剤を含有する小滴を含有する。小滴から有機溶媒を除去するために、クエンチ溶液の入った洗浄タンク(60)内で小滴を撹拌して、硬化した微粒子を形成する。次いで、いずれの従来の分離方法で、水性クエンチ溶液から微粒子を分離してもよい。液体を微粒子からデカントするか、微粒子懸濁液を濾過するか、または篩カラムを使用し得る。要すれば、種々の他の分離法を組み合わせてもよい。次いで、微粒子を、従来の乾燥法で乾燥し、更にサイズ分別を行い得る。
【0075】
小滴がスタティックミキサーから洗浄タンクに移ると、連続相加工媒体は希釈され、小滴中の残留溶媒が抽出によって除去される。この抽出クエンチ工程において、微粒子を、親水性コロイドまたは界面活性剤の存在または不存在下に、乳化に用いたのと同じ連続相加工媒体中に懸濁、または他の液体中に懸濁し得る。抽出媒体は、微粒子から溶媒を除去するが、微粒子を溶解しない。抽出中、溶解した溶媒を含有する抽出媒体を、要すれば除去し、新たな抽出媒体を補充し得る。これは、断続的または連続的に行うことが好ましく、その場合、抽出媒体補充速
度が重要である。速度が小さすぎると、活性剤結晶が微粒子から突出するか、抽出媒体中で成長し得る。適当な抽出媒体補充速度は方法によって変化し得、方法を行う際に決定し得、厳密に速度範囲を予め決める必要はない。残留溶媒を除去後、微粒子を前記のように分離し、風乾するか、または他の従来の乾燥法(例えば減圧乾燥、乾燥剤による乾燥など)によって乾燥する。本発明の方法は、約80重量%まで、好ましくは約50重量%までのコア含量を達成し得るので、活性剤の封入に非常に有効である。
【0076】
エマルジョン中の「油相」液滴の形成に使用した溶媒混合物の一方の溶媒(例えば、好ましい酢酸エチル/ベンジルアルコール混合物の場合は、第1溶媒の酢酸エチル)が、他方の溶媒よりも速く抽出され得る。すなわち、第2溶媒(ベンジルアルコール)が高残留量で残る。ベンジルアルコールは沸点が高いので、微粒子を空気または他の従来の蒸発手段に付しても容易に除去できない。この問題を克服するために、エマルジョンを入れる前の抽出媒体に、抽出の速い方の溶媒をいくらか加える。抽出の速い方の溶媒の、抽出媒体中の濃度は通例、抽出温度における媒体中の該溶媒の飽和点の約20〜70%である。すなわち、エマルジョンをクエンチ液に入れる際、抽出の速い方の溶媒の抽出は遅延され、抽出の遅い方の第2溶媒がより多く除去される。
【0077】
この抽出の速い方の溶媒の「スパイク(添加)」量は、最終的な微粒子の品質に重要である。溶媒が多すぎる(すなわち飽和点に近い)と、活性剤含有微粒子表面に目に見える孔が生じ、望ましくない高放出速度をもたらし得る。抽出媒体中の溶媒が少なすぎると、抽出の遅い方の溶媒の残留量が多くなり、微粒子の品質が低下し得る。抽出媒体の温度も、溶媒溶解度および抽出速度に影響するので重要である。
【0078】
温度および溶媒スパイク量のいずれも、望ましい性質を有する最終生成物(すなわち非常に多孔性で、放出の速い微粒子、または多孔性が低く、放出の遅い微粒子)が得られるように調節し得る。
【0079】
クエンチ液は、純粋な水、水溶液、または他の適当な液体であり得、その体積、量および種類は、エマルジョン相に使用した溶媒に応じて変化し得る。クエンチ液は、好ましくは水である。通例、クエンチ液体積は、飽和体積の10倍のオーダーである(すなわち、エマルジョン中の溶媒体積の完全な吸収に、10倍体積のクエンチ液を要する)。しかし、溶媒系によって、クエンチ体積は飽和体積の約2〜20倍の範囲で変化し得る。更に、バッチサイズ(微粒子生成物)に対するクエンチ体積の条件を説明することが好都合である。この比は、抽出工程の効率の指標となり、場合によっては一定の装置に対するバッチサイズを決定する。比が大きいほど、生成物重量当たり大きい体積が必要である。比が小さいほど、一定のクエンチ体積から、より多くの生成物を得ることができる。この比は、生成する微粒子1g当たり、クエンチ体積約0.1〜10lである。1l/gよりも小さい比が好ましい。
【0080】
ベンゾイルアルコールおよび酢酸エチルの好ましい溶媒組み合わせを使用した場合、クエンチ液の酢酸エチルが、微粒子生成物中の残留溶媒レベルに影響すると考えられる。クエンチ液の酢酸エチル含量が低いと、微粒子中のベンジルアルコール残留量が多く、酢酸エチルは殆んど検出されない。クエンチ液の酢酸エチル含量が高いと(5〜7重量%またはそれ以上)、微粒子は酢酸エチルをベンジルアルコールよりも多く含有し得る。クエンチングする活性剤およびポリマー封入材料1g当たりクエンチ体積約1lでは、クエンチ液体中の酢酸エチル量は、0〜4℃で約3〜4重量%が最適である。微粒子のコア含量は、クエンチ液中の酢酸エチル濃度によって少し変化し、酢酸エチル濃度が高いか、低い場合、少なくなる。微粒子からのインビトロ放出速度は、クエンチ液の酢酸エチル含量の変化によって実質的に変化する。NETの場合、酢酸エチル含量が多すぎると、NET放出が速まる。走査電子顕微鏡で観察すると、クエンチ液中の酢酸エチル量が多すぎた場合は、微粒子表面上にNETおよび孔の存在が見られる。
【0081】
クエンチ液体の体積を変えても、微粒子中の相対溶媒残留量が大きく影響される。体積が小さいと、ベンジルアルコール:酢酸エチル比が大きく、クエンチ体積を、クエンチングする活性剤およびポリマー封入材料1g当たり約1.5lに増加すると、前記比は1未満に低下する。微粒子生成物からの活性剤放出速度は、顕著に大きい。(NETおよびポリマー封入材料の溶液1g当たり、クエンチ液0.125lでは、走査電子顕微鏡写真によると、微粒子生成物は非常に多孔性である。NETおよびポリマー封入材料の溶液1g当たり、クエンチ液0.25〜1.5lでは、微粒子生成物からのNET放出速度はほぼ同程度で、クエンチ液1l/g(クエンチングするNET+ポリマー材料)の場合に最も小さい速度となり得る。)
【0082】
スタティックミキサーを使用して微粒子を製造する本発明の方法は、種々の活性剤封入法に適用し得る。本発明の方法は、前記溶媒抽出法に限定されず、他の封入法にも適用し得る。例えば、本発明の方法は、相分離封入法にも適用し得る。そのためには、ポリマー溶液に懸濁または分散した活性剤を含有する有機相を調製する。非溶媒第2相は、ポリマーおよび活性剤に対する溶媒を含有しない。好ましい非溶媒第2相は、シリコーン油である。有機相および非溶媒相を、スタティックミキサーを通して非溶媒クエンチ液(例えばヘプタン)にポンプ輸送する。半固体粒子をクエンチングして、充分硬化および洗浄する。このような方法の例を、実施例11〜14に挙げる。
【0083】
微粒子生成物は通例、球形粒子から成るが、不規則な形を有することもある。微粒子サイズは、直径サブミクロンないしミリメートルの範囲で変化し得る。好ましくは1〜500μ、より好ましくは25〜180μの微粒子を製造し、そうすると、標準的ゲージの針を用いて患者に投与することができる。
【0084】
好ましくは、薬物含有微粒子は、患者に1回投与する。薬物が連続的または不連続的に患者に放出されるので、反復注射の必要がない。
【0085】
活性剤含有微粒子は、乾燥物として得られ、貯蔵する。患者に投与する前に、乾燥微粒子を、許容し得る薬剤液体ビヒクル、例えば2.5重量%カルボキシメチルセルロース溶液に懸濁し、微粒子懸濁液を患者に注射し得る。
【0086】
活性剤を多相様式で患者にデリバーするように、および/または異なる活性剤を異なる時点でデリバーするように、または活性剤混合物を同時にデリバーするように、大きさまたは種類の異なる微粒子を混合し得る。例えば、主活性剤を、他の抗体、ワクチンまたは所望の活性剤(微粒子形態、または従来の非封入形態のもの)と混合して、患者に投与し得る。
【0087】
適当な活性剤は、エストロゲン、例えばジエチルスチルベストロール、17−β−エストラジオール、エストロン、エチニルエストラジオール、メストラノールなど;プロゲスチン、例えばノルエチンドロン、ノルゲストリル、エチノジオールジアセテート、リネステノール、メドロキシプロゲステロンアセテート、ジメスチステロン、メゲストロールアセテート、クロルマジノンアセテート、ノルゲスチマート、ノルエチステロン、エチステロン、メレンゲストロール、ノルエチノドレルなど; および殺精子剤化合物、例えばノニルフェノキシポリオキシエチレングリコール、ベンゼトニウムクロリド、クロルインダノールなどを包含する。
【0088】
本発明の方法によって組み合わせ得る他の生物学的活性剤は、胃腸処置剤、例えば水酸化アルミニウム、炭酸カルシウム、炭酸マグネシウム、炭酸ナトリウムなど;非ステロイド抗受精剤;副交感神経興奮剤;精神療法剤;リスペリドン;メジャートランキライザー、例えばクロルプロマジンHCl、クロザピン、メソリダジン、メチアピン、レセルピン、チオリダジンなど;マイナートランキライザー、例えばクロルジアゼポキシド、ジアゼパム、メプロバメート、テマゼパムなど;鼻科学的充血除去剤;鎮静−催眠剤、例えばコデイン、フェノバルビタール、ナトリウムペントバルビタール、ナトリウムセコバルビタールなど;ステロイド、例えばテストステロンおよびテストステロンプロピオネート;スルホンアミド;交感神経興奮剤;ワクチン;ビタミンおよび栄養剤、例えば必須アミノ酸;必須脂肪など;抗マラリア剤、例えば4−アミノキノリン、8−アミノキノリン、ピリメタミンなど;抗片頭痛剤、例えばマジンドール、フェンテリニンなど; 抗パーキンソン病剤、例えばL−ドーパ;抗けいれん剤、例えばアトロピン、メトスコポラミンブロミドなど;抗けいれんおよび抗コリン剤、例えば担汁療法剤、消化剤、酵素など;鎮咳剤、例えばデキストロメトルファン、ノスカピンなど;気管支拡張剤;心血管剤、例えば抗高血圧剤、ラウオルフィアアルカロイド、冠血管拡張剤、ニトログリセリン、有機ナイトレート、ペンタエリスリトテトラナイトレートなど;電解質、例えば塩化カリウム;麦角アルカロイド、例えばエルゴタミン(カフェイン有および無)、水素化麦角アルカロイド、ジヒドロエルゴクリスチンメタンスルフェート、ジヒドロエルゴコルニンメタンスルフェート、ジヒドロエルゴクロイプチンメタンスルフェートおよびそれらの組み合わせ;アルカロイド、例えばアトロピンスルフェート、ベラドンナ、ヒヨスチンヒドロブロミドなど;鎮痛剤;麻酔剤、例えばコデイン、ジヒドロコデイン、メペリジン、モルヒネなど;非麻酔剤、例えばサリチレート、アスピリン、アセトアミノフェン、d−プロポキシフェンなど;抗生物質、例えばセファロスポリン、クロラムフェニコール、ゲンタマイシン、カナマイシンA、カナマイシンB、ペニシリン、アンピシリン、ストレプトマイシンA、アンチマイシンA、クロロパンテニオール、メトロミダゾール、オキシテトラサイクリンペニシリンG、テトラサイクリンなど;抗癌剤;抗けいれん剤、例えばメフェニトイン、フェノバルビタール、トリメタジオン;鎮吐剤、例えばチエチルペラジン;抗ヒスタミン剤、例えばクロロフィナジン、ジメンヒドリネート、ジフェンヒドラミン、パーフェナジン、トリペレナミンなど;抗炎症剤、例えばホルモン剤、ヒドロコルチゾン、プレドニゾロン、プレドニゾン、非ホルモン剤、アロプリノール、アスピリン、インドメタシン、フェニルブタゾンなど;プロスタグランジン;細胞毒性剤、例えばチオテパ、クロラムブシル、シクロホスファミド、メルファラン、ナイトロジエンマスタード、メトトレキセートなど;下記のような微生物の抗原:ナイセリア・ゴノレア(Neisseria gonorrhea)、ミコバクテリウム・ツベルクローシス(Mycobacterium tuberculosis)、ヘルペスウイルス(ユモニス(humonis)、タイプ1および2)、カンジダ・アルビカンス(Candida albicans)、カンジダ・トロピカリス(Candida tropicalis)、トリコモナス・バギナリス(Trichomonas vaginalis)、ヘモフィルス・バギナリス(Haemophilus vaginalis)、グループBストレプトコッカス・エコリ(Streptococcus ecoli)、ミクロプラズマ・ホミニス(Microplasma hominis)、ヘモフィルス・ドゥクレイ(Haemophilus ducreyi)、グラヌロマ・インギナレ(Granuloma inguinale)、リンホパチア・ベネレウム(Lymphopathia venereum)、トレポネマ・パリドゥム(Treponema pallidum)、ブルセラ・アボルツス(Brucella abortus)、ブルセラ・メリテンシス(Brucellamelitensis)、ブルセラ・スイス(Brucella suis)、ブルセラ・カニス(Brucellacanis)、カンピロバクター・フェツス(Campylobacter fetus)、カンピロバクター・フェツス・インテスティナリス(Campylobacter fetus intestinalis)、レプトスピラ・ポノマ(Leptospira pomona)、リステリア・モノシトゲネス(Listeria monocytogenes)、ブルセラ・オビス(Brucella ovis)、ウマヘルペスウイルス1、ウマ動脈炎ウイルス、IBR−IBPウイルス、BVD−MBウイルス、クラミジア・プシタキ(Chlamydia psittaci)、トリコモナス・フェツス(Trichomonas foetus)、トキソプラズマ・ゴンジ(Toxoplasma gondii)、エシェリシア・コリ(Escherichia coli)、アクチノバチルス・エクリ(Actinobacillus equuli)、サルモネラ・アボルツス・オビス(Salmonella abortus ovis)、サルモネラ・アボルツス・エキ(Salmonella abortus aqui)、シュードモナス・エルジノサ(Pseudomonas aeruginosa)、コリネバクテリウム・エキ(Corynebacterium equi)、コリネバクテリウム・ピオゲネス(Corynebacterium pyogenes)、アクチノバチルス・セミニス(Actinobacillus seminis)、マイコプラズマ・ボビゲニタリウム(Mycoplasma bovigenitalium)、アスペルギルス・フミガーツス(Aspergillus fumigatus)、アブシジア・ラモサ(Absidia ramosa)、トリパノソマ・エキペルドゥム(Trypanosoma equiperdum)、バベシア・カバリ(Babesia caballi)、クロストリジウム・テタニ(Clostridiumtetani)など;上記微生物に対する抗体;酵素、例えばリボヌクレアーゼ、ノイラミニダーゼ、トリプシン、グリコーゲンホスホリラーゼ、精子乳酸デヒドロゲナーゼ、精子ヒアルロニダーゼ、アドノシントリホスファターゼ、アルカリ性ホスファターゼ、アルカリ性ホスファターゼエステラーゼ、アミノペプチダーゼ、トリプシン、キモトリプシン、アミラーゼ、ムラミダーゼ、先体プロテイナーゼ、ジエステラーゼ、グルタミン酸デヒドロゲナーゼ、コハク酸デヒドロゲナーゼ、β−グリコホスファターゼ、リパーゼ、ATPアーゼα−ペプテートγ−グルタミロトランスペプチダーゼ、ステロール−3−β−オール−デヒドロゲナーゼおよびDPN−ジ−アプロラーゼを包含する。
【0089】
組み合わせ得る更に別の巨大分子生物学的活性剤は、血液凝固因子、造血因子、サイトカイン、インターロイキン、コロニー刺激因子、成長因子、並びにそれらの類似体およびフラグメントを包含するが、それらに限定されない。
【0090】
以下の実施例は、本発明の実施に用いる材料および方法を更に説明するものである。実施例は、本発明を制限するものではない。
【実施例】
【0091】
(実施例1)
ノルエチンドロンを30%、33%および50%理論的に含有する微粒子の製造
ノルエチンドロン30%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ(Koflo)、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(インヘレント粘度(IV)=0.65dl/g)(770g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(329g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(注射用水)(27.27kg)にポリビニルアルコール[PVA−デュポン・エルバノール(DuPont Elvanol、商標)51−05](150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。冷WFI(750l)に、酢酸エチルNF(0.2μm濾過)(26.25kg)を溶解し、2〜4℃に保つ。
【0092】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子650gが得られる。
【0093】
ノルエチンドロン33%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(IV=0.62dl/g)(737g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(363g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(27.27kg)にPVA(デュポン・エルバノール51−05)(150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。WFIに溶解した3.5%酢酸エチルNF(0.2μm濾過)(750l)を2〜4℃に保つ。
【0094】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子630gが得られる。
【0095】
ノルエチンドロン50%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(85モル%乳酸/15モル%グリコール酸コポリマー;ポリ(ラクチド−コ−グリコリド);IV=0.62dl/g)(550g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(546g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(27.27kg)にPVA(デュポン・エルバノール51−05)(150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。冷WFI(750l)に酢酸エチルNF(0.2μm濾過)(26.25kg)を溶解し、2〜4℃に保つ。
【0096】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子685gが得られる。
【0097】
30%および50%含有粒子を、次いで、ヒヒに注射する2種の65mg(NET)製剤の調製に使用した。ヒヒ製剤1は、50%含有粒子35%、および30%含有粒子65%から成っていた。ヒヒ製剤2は、30%および50%含有粒子50%ずつから成っていた。ヒヒ製剤1および2の放出データを図4に示す。
【0098】
(実施例2)
リスペリドンを35%理論的に含有する微粒子の製造(バッチプロデックス2)
まず、1%ポリ(ビニルアルコール)[ヴィノール(Vinol)205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社(Air Products and Chemical Inc.)](906.1g)、ベンジルアルコール[ジェイ・ティ・ベイカー(J.T.Baker)、ニュージャージー州フィリプスバーグ](29.7g)、および酢酸エチル[フィッシャー・サイエンティフィク(Fisher Scientific)、ニュージャージー州フェア・ローン](65.3g)を計量し、混合することにより、水相(溶液A)を調製する。次いで、有機相(溶液B)を調製するために、酢酸エチル(108.7g)およびベンジルアルコール(108.4g)に、高粘度75:25 dl(ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナル、L.P.、オハイオ州シンシナチ)(29.3g)を溶解する。ポリマーが完全に溶解したら、リスペリドン塩基[ヤンセン・ファルマセウティカ(Janssen Pharmaceutica)、ベルギー国ベルセ](15.7g)を加え、ポリマー溶液に溶解する。溶解したリスペリドンのポリマー曝露時間は、できるだけ短く(<10分間)する。次いで、溶液AおよびBを、それぞれ198および24ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマー(Cole−Parmer)L07149−04、L07002−16]から、直径1/4インチのスタティックミキサー(コール−パーマーL−04667−14)を経て、クエンチ媒体(洗浄液)にポンプ輸送する。クエンチ液は、酢酸エチル(1276.0g)、無水炭酸水素ナトリウム(92.3g、0.02モル)、および無水炭酸ナトリウム[マリンクロット・スペシャルティ・ケミカルズ(Mallinckrodt Specialty Chemicals)、ケンタッキー州パリス](116.2g、0.02モル)を含有する注射用水(55l)から成る(11℃)。微粒子を、この第1洗浄液中で1時間45分撹拌した後、25μ篩を通して分離する。篩に残った生成物を第2のWFIの洗浄液(20l、13℃)に移す。第2洗浄液中で2時間15分撹拌後、微粒子を分離し、25および180μメッシュサイズから成るステンレススチール篩カラムに通してサイズ分別する。微粒子を一晩乾燥し、回収し、計量する。
【0099】
(実施例3)
リスペリドンを40%理論的に含有する微粒子の製造(バッチプロデックス3)
まず、1%ポリ(ビニルアルコール)[ヴィノール205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社](904.4g)、ベンジルアルコール[ジェイ・ティ・ベイカー、ニュージャージー州フィリプスバーグ](30.1g)、および酢酸エチル[フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン)(65.8g)を計量し、混合することにより、水相(溶液A)を調製する。次いで、有機相(溶液B)を調製するために、酢酸エチル(99.3g)およびベンジルアルコール(99.1g)に、高粘度75:25 dl (ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナル、L.P.、オハイオ州シンシナチ)(27.1g)を溶解する。ポリマーが完全に溶解したら、リスペリドン塩基[ヤンセン・ファルマセウティカ、ベルギー国ベルセ](18.1g)を加え、ポリマー溶液に溶解する。溶解したリスペリドンのポリマー曝露時間は、できるだけ短く(<10分間)する。次いで、溶液AおよびBを、それぞれ198および24ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマーL07149−04、L07002−16]から、直径1/4インチのスタティックミキサー(コール−パーマーL−04667−14)を経て、クエンチ媒体(洗浄液)にポンプ輸送する。クエンチ液は、酢酸エチル(1375.6g)、無水炭酸水素ナトリウム(92.4g、0.02モル)、および無水炭酸ナトリウム[マリンクロット・スペシャルティ・ケミカルズ、ケンタッキー州パリス](116.6g、0.02モル)を含有する注射用水(55l)から成る(12℃)。微粒子を、この第1洗浄液中で2時間撹拌した後、25μ篩を通して分離する。篩に残った生成物を第2のWFIの洗浄液(20l、12℃)に移す。第2洗浄液中で3時間撹拌後、微粒子を分離し、25および180μメッシュサイズから成るステンレススチール篩カラムに通してサイズ分別する。微粒子を一晩乾燥し、回収し、計量する。
【0100】
(実施例4)
バッチプロデックス2およびプロデックス3の微粒子の凍結乾燥およびγ線照射(サンプルプロデックス4A、プロデックス4Bおよびプロデックス4C)
バッチプロデックス2および3の微粒子を、次のようにして凍結乾燥した。微粒子を5cc血清バイアルに量り入れた。次いで、0.75%CMC、5%マンニトールおよび0.1%トゥイーン80から成る水性ビヒクルを、バイアルに入れた。微粒子を撹拌によりビヒクルに懸濁し、ドライアイス/アセトン浴内で急速に凍結した。バイアルを、次いで、実験室用凍結乾燥器[デュラ・ストップ・マイクロプロセッサー・コントロール(Dura Stop Microprocessor Control)、ニューヨーク州ストーン・リッジのFTSシステムズ社]内で、勾配30℃最大温度サイクルを用いて50時間凍結乾燥した。サンプルプロデックス4Aおよび4Cはそれぞれ、プロデックス2および3の凍結乾燥サンプルであった。サンプルプロデックス4Bは、プロデックス2を60Co源から2.2Mラドγ線照射によって滅菌し、凍結乾燥したものであった。
【0101】
インビトロ溶解試験
プロデックス2、3、4A、4Bおよび4Cに対し、インビトロ溶解試験を行った。リアルタイムおよび促進法を用いた。装置は、分光光度計およびデータステーションに接したハンソン(Hanson)リサーチ9−セルUSPパドル(方法II)溶解装置から成っていた。受容媒体は、各セルから分光光度計内のフローセルに連続的に循環した。受容媒体の吸光度を236nmでモニターして、リスペリドンを定量した。
【0102】
リアルタイムモデルでは、37℃の50mMトリス緩衝液(pH7.4)から成る受容媒体への、微粒子からの放出速度を測定した。リスペリドンは、前記受容媒体を用いるインビトロ実験を行うための充分な溶解度(≧0.5mg/ml)を有することがわかった。溶解条件を無限にするために、リスペリドンの量は、飽和の20%未満に保った。データを図5および図6に示す。
【0103】
促進モデルも行った。受容媒体として、WFI中の27.5重量%エタノールを使用した。結果を図7に示す。
動物への投与および血液サンプリング
乾燥微粒子(プロデックス2、3)および凍結乾燥形態(プロデックス4A、4B、4C)の生成物に対し、イヌにおいてインビボ試験を行った。乾燥微粒子を注射器に入れ、注射器内で、2.5重量%カルボキシメチルセルロース(CMC)から成る注射ビヒクルに再懸濁した。凍結乾燥サンプル(プロデックス4A、4B、4C)は、注射前にWFI中で再構成した。
【0104】
雄および雌のイヌ(体重11.6±2.3kg)を、3匹ずつの群に分けた。イヌを3匹の群として、標準的な実験室条件で飼育および餌を与えた。
【0105】
イヌの左後肢の腿の位置の二頭股筋に、各デポ製剤の適量を、リスペリドン約2.5mg/kgの用量で筋肉内投与した。
【0106】
アポモルモネ催吐試験において、製剤投与の0(投与前)、1、5および24時間後、並びに4、7、11、14、18、23、25、28、32、35、39、42、46、49、53および56日目に、一つの頸静脈から血液サンプル(EDTA上5ml)を採った。アポモルモネ試験は、ヤンセン(P.A.J.Janssen)およびニーメゲルス(C.J.E.Niemegeers)、アルツナイミッテルフォルシュング(Arzneim.−Forsch.)(Drug Res.)、9:765−767(1959)に記載されている。試験中に一つの群の3匹のイヌのいずれにも、アポモルモネ催吐に対する保護が見られなくなったら、血液サンプリングは中断した。血液サンプルは、3000rpmで10分間遠心し、血漿を分離した。血漿サンプルを、分析するまで≦20℃で貯蔵した。
【0107】
血漿サンプルのリスペリドン(RISP)および9−ヒドロキシリスペリドン(9−OH RISP)を、ラジオイムノアッセイ(RIA)によって分析した。RIAで分析する血漿サンプルに、2つの異なるRIA法を用いた。一つは未変化リスペリドンに、もう一つは活性物(リスペリドンと9−ヒドロキシ−リスペリドンの合計;「活性剤」と混同しないよう注意されたい)に対するものである。後者の血漿サンプルでは、9−ヒドロキシ−リスペリドン濃度を、活性物濃度とリスペリドン濃度との差として計算した。RIA法の定量限界は、リスペリドン0.20ng/ml、活性物0.50ng/mlであった。
【0108】
各製剤に対して、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の平均(±S.D、n=3)血漿濃度を計算した。9−ヒドロキシ−リスペリドン血漿濃度とリスペリドン血漿濃度との比を、可能なら計算した。データの目視により、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の血漿濃度ピークおよびピーク時間を求めた。リスペリドンおよび9−ヒドロキシ−リスペリドンのAUC(「曲線下面積」)値を、0時間および時間tの間で、台形則により計算した。時間tは、3匹中少なくとも1匹のイヌにおいて、リスペリドンまたは9−ヒドロキシ−リスペリドンの濃度が定量限界を上回った最終時点である。同じ製剤群に属するイヌには、AUCは同じ最終時間tまで計算した(濃度が定量限界よりも低かったものには、定量限界の値を用いた)。2濃度連続して定量限界を下回ったら、先のサンプリング点の濃度を定量限界とし、後のサンプリング点の濃度を0とた。AUCは、無限に外挿しなかった。活性物のAUCは、リスペリドンと9−ヒドロキシ−リスペリドンとのAUCの合計として計算した。
【0109】
製剤プロデックス2/3/4A/4B/4Cについて、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の平均もしくは中央血漿濃度および/または薬物動態パラメータを第1表に示す。製剤プロデックス2/3/4A/4B/4Cについて、平均血漿濃度−時間曲線を、図8に示す。
【0110】
各製剤群について、最初にリスペリドン、次いで9−ヒドロキシ−リスペリドン、最後に活性物に関して結果を考察する。活性物については、血漿濃度とアポモルモネ催吐試験における制吐作用とを関連付ける。
【0111】
製剤プロデックス2〜4Cの投与後、リスペリドンの平均血漿レベルのピークは低かった。非常に異なった時点でピークに達した。異なる製剤からの更なるリスペリドン放出が徐々に起こり、長時間続いた。その結果、リスペリドンおよびその代謝産物のいずれの血漿濃度も低かった。9−ヒドロキシ−リスペリドンの平均ピーク時間はいずれも、26〜30日目の間であった。活性物の血漿濃度−時間曲線は、製剤プロデックス2〜4Cのいずれでも同様であった。試験開始後1または2日以内に、活性物の血漿濃度は、リスペリドンの急速な初期放出の故にピークに達した。そのピークの後、5〜8日目に、濃度は下降線をたどった。8日目から、濃度は再び20日目まで上昇し、20日目から平均15日間は、概ね一定のレベルにあった。この間、各製剤について、活性物濃度は第2のピークを示し、濃度は第1のピークよりも高かった。製剤プロデックス2、4Aおよび4Bの制吐作用は35〜42日間持続した。製剤プロデックス4Cでは制吐作用は49日間持続し、いずれのイヌにおいても中断が無かった。製剤プロデックス4Cの最長作用は、活性物の最高Cmax、TmaxおよびAUC0−tと対応していた(同じ群の他の4製剤との比較)。
【0112】
イヌにおけるアポモルモネ催吐試験において、微粒子リスペリドン製剤の作用時間をも調べた。神経遮断剤が、第四脳室の最後野においてドーパミンD2レセプターを遮断することにより、アポモルモネ催吐に拮抗した。この試験は通例、ヒトにおける神経遮断剤の抗精神病薬作用の開始および作用時間を予測するために用いられる[ヤンセンら、アルツナイミッテルフォルシュング、10:1196−1206(1965);ニーメゲルスら、ライフ・サイエンシーズ(Lefe Sci.)、24:2201−2216(1979)]。9−OH−リスペリドンは、実質的に親化合物と同じ薬理学的性質を有する。親化合物と活性代謝産物とが、リスペリドンの生物学的性質を決定する「活性物」を構成する。
【0113】
試験期間中、アポモルモネを週2回、イヌに0.31mg/kgの量で皮下投与した。アポモルモネ投与後1時間の間、イヌに嘔吐が見られた。アポモルモネ投与後1時間の間に嘔吐の全く無いことが、明らかな制吐活性の反映と考えられる。制吐作用時間は、3匹中2匹のイヌに制吐が見られる時間の長さと定義した。
【0114】
一方の後肢の腿の位置の二頭股筋に、製剤0.5mlを注射した。この筋肉注射から種々の時間後に血液サンプルを採り、その後すぐにイヌにアポモルモネを投与した。アポモルモネ投与1時間以内に嘔吐(対照動物には決して見られない;n>1000)が全く無いことが、明らかな制吐作用の反映と考えられる。
【0115】
第2表には、イヌにデポ製剤を筋肉注射してから種々の時間後に、アポモルモネ催吐に対する保護が見られた(+)か、見られなかった(−)かを示す。いずれの製剤も、短時間で制吐作用を開始した。
【0116】
【表1−1】

【0117】
【表1−2】
【0118】
【表1−3】
【0119】
【表1−4】
【0120】
【表2−1】
【0121】
【表2−2】
【0122】
(実施例5)
エストラジオールベンゾエートを20%および30%理論的に含有する微粒子の製造
第3表には、実験的に製造した20%および30%含有微粒子の粒子サイズ分布を示す。粒子サイズ分布は、図9にも示す。図9は、20%含有微粒子の一つのバッチの微粒子サイズの累積%「より大きい体積%」)を示す。図10は、20%含有微粒子の一つのバッチの粒子サイズによる%微分(「体積微分」)を示す。このデータから、流速が増すと、粒子サイズが小さくなることがわかる。12および24要素ミキサーの間には、顕著な差は無かった。水相:有機相比が高いほど、粒子サイズ分布は狭くなる。遷移流速範囲、レイノルズ数(Re)2000〜4000で、良好な微粒子が得られた。
【0123】
次いで、20%および30%含有微粒子の更なるバッチを製造した。薬物20%含有微粒子7kgバッチの製造に、直径1/2"×24要素スタティックミキサー(コール−パーマー・ポリプロピレン・ディスポーザブル04667;イリノイ州シカゴのコール−パーマー・インストゥルメント社)を使用した。有機相は、4.0%エストラジオールベンゾエート(活性剤)、16.0%85:15dl PLGA(85モル%乳酸/15%グリコール酸コポリマー;ポリラクチド−コ−グリコリド)、および80%酢酸エチルから成っていた(60〜70℃)。水相は、1.0%ポリビニルアルコール、5.0%酢酸エチル、および94.0%水から成っていた(60〜70℃)。有機相流速も水相流速も、1100ml/分であった。有機相および水相のいずれにも、7001−80ヘッドを有するコール−パーマー6231−26ポンプを使用した。得られた粒子サイズ分布は、15%<25μm、14%25〜45μm、56%45〜90μm、12%90〜150μm、および3%>150μmであった。
【0124】
薬物30%含有微粒子5kgバッチの製造に、直径1/2"×24要素スタティックミキサー並びに直径3/8"×11、12および24要素スタティックミキサー(コール−パーマー・ディスポーザブル)を使用した。有機相は、4.3%エストラジオールベンゾエート(活性剤)、10.0%85:15dl PLGA、および85.7%酢酸エチルから成っていた(60〜70℃)。水相は、20%含有微粒子の場合と同じものを使用した。有機相流速は880ml/分、水相流速は1650ml/分であった。得られた粒子サイズ分布は、44%<25μm、31%25〜45μm、および25%>45μmであった。
【0125】
エストラジオールベンゾエート含有微粒子を、従来の注射器に量り入れ、若いホルスタイン雄子牛に、活性薬物40mg/動物の用量で投与した。微粒子を水性カルボキシメチルセルロースビヒクルに懸濁し、耳の基部に注射した。血清を採り、エストラジオールレベルをラジオイムノアッセイによって測定した;結果を図11に示す。
【0126】
【表3】
【0127】
(実施例6)
トレンボロンアセテートを40%理論的に含有する微粒子の製造
トレンボロンアセテート(TBA)微粒子9バッチを、スタティックミキサーを用いて製造した。TBA40%およびブチル化ヒドロキシトルエン(BHT、抗酸化剤として使用)5%を含有する微粒子を、次のような方法で製造した。PLGA(D,L−ラクチド:グリコリドをモル比85:15;GPCによる分子量Mw=86810;Mn=36417)、TBA、およびBHTを、55℃で酢酸エチル(EtOAc)に溶解した。(EtOAc:PLGA比10〜20:1。)EtOAcを55℃に加熱し、高速攪拌下にEtOAcにPLGAを加えた。ポリマーの溶解後、ポリマー溶液に、BHT、および次いでTBAを溶解した。同時に、EtOAcを加えたポリ(ビニルアルコール)(PVA)水溶液(3重量%)を、ジャケット付フラスコに入れ、55℃に加熱した(PVA溶液:EtOAc比10:1)。ポリマー/薬物溶液(有機相)およびPVA溶液(水相)の温度が一定になったら、ポリマー/薬物溶液およびPVA溶液を別々にスタティックミキサー(直径1cm、長さ12cm)にポンプ輸送した。スタティックミキサーは、コール−パーマー製のモデル番号6−04667−06で、混合要素を12個有していた。ポリマー/薬物溶液とPVA溶液の流速比は、1:1.2ないし1:2の間で変化した。クエンチ水とEtOAcとの比は、100〜150:1であった。クエンチ水温度は約1℃であった。6時間洗浄後、微粒子を篩カラム(25μm、90μm、150μm、および212μm)で篩過した後、乾燥した。
【0128】
一つのバッチの微粒子の粒子サイズ分布を図12に示す。
【0129】
(実施例7)
テストステロンを46%理論的に含有する微粒子の製造
テストステロン46%含有微粒子1kgのバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(3.683kg)中のメディソーブ75:25dl PLGA(乳酸75モル%/グリコール酸25モル%コポリマー;ポリラクチド−コ−グリコリド; IV=0.68dl/g)(594g)の加熱(65〜70℃)した溶液に、テストステロンUSP(506g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(9.75kg)にPVA(デュポン・エルバノール51−05)(52g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)した酢酸エチル(626g)を加える。溶液を65〜70℃に保つ。クエンチ液はWFI(750l)から成り、0〜4℃に保つ。
【0130】
有機相を2150cc/分、水相を4300cc/分の流速でスタティックミキサーを通してポンプ輸送する。1時間クエンチ後、材料を45および150μm篩に通す。45〜150μmクラクションを周囲温度で攪拌下に36時間減圧乾燥する。テストステロン含有微粒子875gが得られる。
【0131】
46%含有微粒子を用いて125mgテストステロン製剤(懸濁液充填および凍結乾燥)を調製し、ヒヒに投与した。時間放出データを図13に示す。
【0132】
(実施例8)
rgp120を1.7%理論的に含有する微粒子の製造
rgp120(糖タンパク質)1.7%含有微粒子10gバッチを製造するために、直径1/4"×5要素のスタティックミキサー[コッホ(Koch)、1/4"SMV−DY;直径1/4"、5要素、316SS、カンザス州ウィチタのコッホ・エンジニアリング社(Koch Engineering Company, Inc.)]を使用する。ポリマー/薬物溶液(有機相)は、次のようにして調製する。酢酸エチル(47g)中のメディソーブ65:35dl PLGA(乳酸65モル%/グリコール酸35%コポリマー;ポリラクチド−コ−グリコリド)(IV=0.61)(10g)に、トリス緩衝液(20mMトリス、120mM NaCl、pH7.4)中の56mg/mlのrgp120(3cc)を加えることにより、第1のエマルジョンを調製する。このエマルジョンは、超音波処理[ビブラ(Vibra)セル・ソニケーター、600W、20秒、50%パワー、1/2"プローブ]により形成する。エマルジョンを0〜4℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(2025g)に、PVA(エア・プロダクツ、ヴィノール205)(225g)を加え、溶解するまで加熱(65〜70℃)する。この溶液に、酢酸エチル(250g)を加える。溶液を0〜4℃に保つ。クエンチ液は水(12l)から成り、0〜4℃に保つ。
【0133】
有機相を40cc/分、水相を1500cc/分の流速でスタティックミキサーを通してポンプ輸送する。1時間クエンチ後、材料を20および150μm篩に通す。20〜150μmフラクションを0〜4℃で0.1%トゥイーン20溶液(13l)で洗い、凍結乾燥する。rgp120含有微粒子3.09gが得られる。
【0134】
(実施例9)
イベルメクチンを60%理論的に含有する微粒子の製造
イベルメクチンを60%理論的に含有する微粒子35gバッチを製造するために、65:35dl(ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナルL.P.、オハイオ州シンシナチ)9.5%および酢酸エチル(フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン)90.5%から成る52℃のポリマー溶液(116.72g)に、イベルメクチン(21.03g)を溶解することにより、分散相(溶液A)を調製する。次いで、ポリ(ビニルアルコール)(ヴィノール205、ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社)(1%)およびWFI(99%)から成る連続相(溶液B)(1300g)を計量し、52℃に加熱する。次いで、溶液AおよびBを、それぞれ88および165ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマーL07149−04、L07002−16]から、スタティックミキサー(コール−パーマーL04667−14)を経て、クエンチ液にポンプ輸送する。クエンチ液は、CWFI(冷注射用水)(35l)から成る(8℃)。微粒子を、約20分間攪拌した後、25μ篩を通して分離する。篩に残った生成物をCWFIの洗浄液(20l、8℃)に移す。2時間攪拌後、洗浄液を25および212μ篩に通して、微粒子を分離し、サイズ分別する。微粒子を一晩風乾し、回収する。
【0135】
(実施例10)
ブピバカイン塩基を60%理論的に含有する微粒子の製造
5%ポリ(ビニルアルコール)水溶液[ヴィノール205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社](3780.3g)に、酢酸エチル[フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン](216.1g)を加え、周囲温度で1N−NaOHでpH8.5に調節することにより、過剰の連続水相を調製する。次いで、有機相を調製するために、酢酸エチル(92.1g)に低粘度50:50dlポリラクチド−コ−グリコリド(乳酸50モル%/グリコール酸50モル%コポリマー)(メディソーブ・テクノロジーズ・インターナショナル、L.P.)(8.05g)およびブピバカイン塩基[アセト社(Aceto Corporation)、ニューヨーク州レーク・サクセス](12.05g)を室温で溶解する。次いで水溶液および有機溶液を、それぞれ233および116ml/分の流速で、ギアドライブポンプおよびヘッド(ポンプ:コール−パーマーL07144−05; 水溶液用ヘッド:L7002−16;有機溶液用ヘッド:L07002−26)から、24要素、直径1/4"のスタティックミキサー(コール−パーマーL04667)を経て、pH8.5に調節した注射用水(12l)(室温)から成るクエンチ液に、同時にポンプ輸送する。微粒子をクエンチ液中で1時間攪拌後、25、45および90μmメッシュステンレンスチール篩で篩過する。微粒子を一晩乾燥し、計量まで遮光する。
【0136】
(実施例11)
ブタアルブミンを17.5%理論的に含有する微粒子の製造
ブタアルブミン含有微粒子2gバッチを製造するために、直径1/2"×長さ12"のスタティックミキサーを使用した。ポリマー/薬物溶液(有機相)は、次のようにして調製した。酢酸エチル(70g)に、メディソーブ75:25dl PLGA(インヘレント粘度0.65dl/g)(1.65g)を溶解した。有機相中のポリマー濃度は、2.36%であった。粉砕したブタアルブミン[シグマ(Sigma)、ロット番号95F−9538](0.35g)を、2×10秒間の超音波処理[テクマー・ソニケーション・ディスラプター(Tekmar Sonication Disruptor)、モデル350、マイクロチップ付]によってポリマー溶液に懸濁した。均一で細かい分散液が得られた。懸濁液を、底部に排出バルブを有する50ml反応器に入れた。タービン6翼プロペラにより、約700rpmで混合を開始した。蠕動ポンプを反応器の排出口に連結し、有機相を7.0g/分の速度でスタティックミキサーにポンプ輸送した。350csシリコーン油[ダウ・コーニング(Dow Corning)、ロット番号HH121209]を9.0g/分の速度でスタティックミキサーの導入口に供給するために、もう一つの蠕動ポンプを設置した。
【0137】
まずシリコーン油流を流し、約2〜5秒後に有機相を流した。クエンチタンク内では、ヘプタン[ケム・ピュア(Chem Pure)、ロット番号M138KLAP](1.2l)を、空気作動インペラーで穏やかに攪拌した。スタティックミキサーの排出ラインは、ヘプタン表面から1/2"下に配置した。有機相をスタティックミキサーから移し終った後、残留有機相をミキサーから流し出すために、酢酸エチル(8〜10g)を用いた。この流し出しの間にも、シリコーン油を流し続けた。半固形微粒子をヘプタン中で室温で1.5時間クエンチして、硬化および洗浄を完了した。次いで、バーサポル(Versapor)−3000メンブランフィルター(孔サイズ3μ)[ゲルマン(Gelman)]を用いて、微粒子を減圧濾取した。回収した微粒子を、フィルター上で新たなヘプタン(300ml)で洗い、更に乾燥するために減圧デシケーターに入れた。ブタアルブミン含有微粒子1.91gが得られた。微粒子は概ね球形で、サイズ範囲25〜200μであった。
【0138】
第2の製剤を調製して、より短いスタティックミキサー(直径1/2"×長さ6")を、より低い総流量で使用し得ることを示した。スタティックミキサー内への有機相流量は、第1の製剤の場合7.0g/分であったが、6.5g/分とした。シリコーン油流量は、第1の製剤の場合9.0g/分であったが、8.8g/分とした。しかし、有機相:シリコーン油の流速比は、第2の製剤の場合も、第1の製剤の場合とほぼ同じであった。この製剤の固体収率は90%であった。
【0139】
(実施例12)
rBoインターフェロン−αを10.0%理論的に含有する微粒子の製造
rBoインターフェロン−α(組換ウシインターフェロンα)含有微粒子1.89g、バッチを製造するために、1/2"×12"スタティックミキサーを使用した。有機相中のポリマー(酢酸エチルに溶解した75:25dl PLGA)濃度は、2.70%であった。注射用水(0.5ml)にタンパク質を溶解し、ポリマー溶液中で超音波処理により乳化して、有機相(ポリマー/活性剤相)を形成した。エマルジョンを反応器に入れ、実施例11と同様に小球形成を行った。有機相流速は6.6g/分、シリコーン油流速は10.9g/分であった。半固形微粒子を、ヘプタン(1.0l)中で1.5時間クエンチングした。
【0140】
(実施例13)
HSAを17.5%理論的に含有する微粒子の製造
HSA(ヒト血清アルブミン)含有微粒子のバッチを製造するために、1/2"×6"スタティックミキサーを使用した。有機相中のポリマー(酢酸エチルに溶解した75:25dl PLGA)濃度は、2.36%であった。水の内相(1.0ml)を用いて、タンパク質を完全に溶解した。溶解したタンパク質を、ポリマー溶液中で超音波処理により乳化して、有機相を形成した。6"のより短い流路内で非溶媒(シリコーン油)によるコアセルベーションを誘導するのに必要な滞留時間を保つために、より低い総流速を採用した。有機相流速は4.9g/分、シリコーン油流速は5.6g/分であった。より低い総流速の結果、このサンプルで65%の固体収率が得られた。このサンプルでは、シリコーン油流速と有機相流速との比は、実施例12よりも少し小さかった(1.4に対し、1.1)。半固形微粒子を、ヘプタン(0.8l)中で1.5時間クエンチングした。非溶媒(シリコーン油)をより少なく使用したので、ヘプタン(第2の非溶媒)の必要量もより少なかった。
【0141】
(実施例14)
ブタアルブミンを10.0%理論的に含有する微粒子の製造
ブタアルブミンをモデルタンパク質(封入する活性剤)として用いて、実施例12と同様に製剤を製造した。スタティックミキサー内の2相の総流速は、このサンプルではより低かった。有機相流速は6.0g/分、シリコーン油流速は8.8g/分であった。微粒子をヘプタン(1.2l)中で1.5時間クエンチングした。この製剤でも、実施例13と同様の総固体収率が達成された。
実施例11〜14の製剤の性質を第4表にまとめる。
【0142】
【表4】
【0143】
(実施例15)
酢酸エチルおよびベンジルアルコールの50:50(重量)混合物(80g)に、85:15D,L−ラクチド/グリコール酸コポリマー(10.6g)およびノルエチンドロンUSP(9.4g)を順次溶解した(「油相」)。溶解したら、溶液を、ポリ(ビニルアルコール)(ヴィノール205、エア・プロダクツ;数平均分子量15000〜27000、加水分解度87〜89%)0.5重量%、酢酸エチル5.9重量%、ベンジルアルコール2.7重量%、および水90.9重量%から成る60〜65℃のエマルジョン浴混合物(500g)の入った、タービン攪拌機およびサーモスタットヒーターを取り付けたジャケット付1000mlビーカーに入れた。このエマルジョン浴混合物は、60℃で酢酸エチルおよびベンジルアルコールの両方の飽和溶液に近いものである。すなわち、エマルジョン形成中、「油相」からの溶媒抽出が防止でき、この工程における時間効果を最少にし得る。油滴サイズが約90μmとなるよう、攪拌速度を調節した。得られたエマルジョンを、図14および図15に示すように水および酢酸エチルを種々の量で含有する冷却(2〜4℃)水タンクに移した。1時間後、微粒子を篩スタック(25、45、63および90μm)上に回収し、フード内で一晩乾燥した。翌日、微粒子を混合し(25〜45μm15%、45〜63μm50%、63〜90μm35%)、サンプリングした。結果を図14および図15に示す。
【0144】
(実施例16)
実施例15を繰り返した。ただし、NETおよびポリマーの「油相」溶液の量をいずれの場合も5gとし、エマルジョン浴は、実施例15において使用したポリ(ビニルアルコール)を0.5重量%含有する水300mlであった。結果を第5表に示す。
【0145】
【表5】
【0146】
(実施例17)
テストステロン含有微粒子の20gバッチを、次のようにして製造した。酢酸エチルおよびベンジルアルコールの75:25混合物(67g)に、実施例15のポリマー(10.8g)およびテストステロン(9.2g)を溶解し、約65℃に加熱した。次いで、溶液を、ポリ(ビニルアルコール)0.5%、および酢酸エチル6.5%の水性混合物(500g)の入った、タービン攪拌機を取り付けたジャケット付1000mlガラス製反応器に入れた。攪拌速度を約245rpmに調節した。5分後、エマルジョンを、酢酸エチルを5%の濃度で含有する水(20l)の入った冷却(0〜4℃)タンクに移した。1時間後、微粒子を篩スタック(25、および150μm)上に回収し、実験室フード内で一晩乾燥した。翌日、25μ篩上の微粒子を回収し、サンプリングした。生成物は、テストステロン39.7%、酢酸エチル3.67%およびベンジルアルコール0.89%を含有していた。促進インビトロ放出モデルによると、18時間後に15%の薬物が受容液体に放出された。
【0147】
本発明の種々の態様を説明したが、それらは説明のためのもので、制限するものではないと理解すべきである。すなわち、本発明の範囲は、上記例示態様ではなく、以下の特許請求の範囲によってのみ制限すべきである。
【符号の説明】
【0148】
1 液流
10 スタティックミキサー
12 パイプ
14 静的混合要素
30 油相または有機相
32 攪拌ポット
40 水相
50 Y字形連結具
60 洗浄タンク
【技術分野】
【0001】
本発明は、微粒子の製造に関する。本発明はとりわけ、スタティックミキサーの使用により、活性剤を封入して調節的放出性微粒子を形成する方法に関する。本発明は、活性剤を封入して調節的放出性微粒子を形成する方法において有用な溶媒系にも関する。「微粒子」または「小球」とは、該粒子のマトリックスとして機能する生分解性ポリマー中に分散または溶解した活性剤を含有する固体粒子を意味する。
【背景技術】
【0002】
化合物を微粒子の形態に封入することのできる方法は、種々知られている。生物学的活性剤または薬剤を、生体適合性、生分解性、壁形成材料(例えば、ポリマー)中に封入して、薬物または他の活性剤の徐放または遅延放出を提供することは、特に有利である。そのような方法において、封入する材料(薬物または他の活性剤)を、通例、撹拌機または他の動的混合法によって、壁形成材料含有溶媒中に、溶解、分散または乳化する。次いで、溶媒を微粒子から除去した後、微粒子生成物が得られる。
【0003】
従来のマイクロカプセル化方法の一例は、米国特許第3,737,337号に開示されている。それによると、溶媒中の壁または殻形成ポリマー材料の溶液を調製する。溶媒は、部分的にしか水混和性でない。そのポリマー含有溶液中に、固体またはコア材料を溶解または分散した後、そのコア材料含有溶液を、微粒子から溶媒を除去するために、有機溶媒非混和性の水性液体中に分散する。封入または包埋する物質は、従来の混合機(バイブレーターおよび高速撹拌機などを包含する)を用いて(分散液の調製において)、ポリマーの有機溶液(相A)中に溶解または分散する。相(A)(溶液または分散液中に、コア材料を含有する)の、水相
(B)中の分散を、やはり従来の混合機、例えば高速ミキサー、バイブレーションミキサーまたは噴霧ノズル(この場合、微粒子の粒子サイズは、相(A)の濃度だけでなく、得られる粒子サイズによっても決まる)を用いて行う。
【0004】
物質を含有する微粒子から溶媒を除去する方法のもう一つの例は、米国特許第3,523,906号に開示されている。その方法においては、封入する材料を水非混和性の溶媒中のポリマー材料の溶液中に乳化し、次いで、そのエマルジョンを、親水性コロイド含有水溶液中で乳化する。次いで、蒸発によって微粒子から溶媒を除去して、生成物を得る。
【0005】
米国特許第3,691,090号に開示された更に別の方法においては、水性媒体中の微粒子分散液から、好ましくは減圧下に、有機溶媒を蒸発する。
【0006】
同様に、米国特許第3,891,570号が開示する方法においては、誘電率10またはそれ以下で、多価アルコール難混和性の溶媒中に溶解した壁材料の溶液に、コア材料を溶解または分散し、次いで、多価アルコールへの分散または溶解によって小滴に乳化し、最後に加熱または減圧によって溶媒を除去することによって、微粒子を調製する。
【0007】
活性剤を封入し得る方法の別の例が、に開示されている。カプセル用の壁材料を、水難混和性で、沸点100℃未満で、蒸気圧が水よりも高く、誘電率が約10未満の少なくとも1種の有機溶媒に溶解し;得られた溶液に、水不溶または難溶性の薬物を溶解または分散し;得られた溶液または分散液を、親水性コロイドまたは界面活性剤の水溶液から成る液体ビヒクル中で分散して小滴を形成した後、有機溶媒を蒸発により除去することによって、封入薬物を調製する。小滴サイズは、撹拌速度、薬物および壁材料を含有する有機溶媒の粘度、並びにビヒクルの粘度および表面張力によって決まる。
【0008】
タイス(Tice)らの米国特許第4,389,330号には、2段階溶媒除去法による、活性剤含有微粒子の調製が記載されている。この2段階溶媒除去法は、1段階で溶媒を除去する方法と比べて、活性剤封入率がより高く、より高品質の微粒子が得られるので、有利である。タイスらの方法においては、活性剤およびポリマーを溶媒に溶解する。次いで、溶媒中の成分混合物を、該溶媒と非混和性の連続相加工媒体中で乳化する。混合した材料の機械的撹拌により、連続相媒体中に、所定成分含有微粒子の分散液を形成する。その分散液から、溶媒除去法の第1段階において、有機溶媒を部分的に除去する。第1段階後、何らかの従来の分離法により、連続相加工媒体から、分散した微粒子を分離する。この分離後、微粒子中の残りの溶媒を、抽出により除去する。残りの溶媒を微粒子から除去した後、風乾、または他の従来の乾燥法で乾燥する。
【0009】
タイスらの米国特許第4,530,840号には、抗炎症活性剤を含有する微粒子の製法であって、(a)抗炎症剤を溶媒に溶解または分散し、その溶媒に生体適合性および生分解性の壁形成材料を溶解し;(b)抗炎症剤および壁形成材料を含有する溶媒を、連続相加工媒体中に分散し;(c)工程(b)の分散液から、溶媒の一部を蒸発し、それによって、懸濁液中に抗炎症剤を含有する微粒子を形成し;(d)残りの溶媒を微粒子から除去することを含んで成る方法が記載されている。
【0010】
国際特許出願公開90/13361号には、剤をマイクロカプセル化してマイクロカプセル化生成物を形成する方法であって、壁形成材料を溶解した溶媒中に、有効量の剤を分散して、分散液を形成し;分散液を、有効量の連続加工媒体と組み合わせて、加工媒体と、剤、溶媒および壁形成材料から成る小滴とを含有するエマルジョンを形成し;エマルジョンを、有効量の抽出媒体に短時間で加えて、小滴から溶媒を抽出することにより、マイクロカプセル化生成物を形成するという工程を含んで成る方法が開示されている。
【0011】
ボトマイヤー(Bodmeier, R.)らのインターナショナル・ジャーナル・オブ・ファーマシューティクス(International Journal of Pharmaceutics)43:179−186(1988)には、薬物含量を高めるために、塩化メチレン、クロロホルムおよびベンゼン、並びに塩化メチレンと水非混和性液体、例えばアセトン、酢酸エチル、メタノール、ジメチルスルホキシド、クロロホルムまたはベンゼンとの混合物を包含する種々の溶媒を用いて、活性剤としてのキニジンまたはキニジンスルフェート、および結合剤としてのポリ(D,L−ラクチド)を含有する微粒子の製造が開示されている。
【0012】
ベック(Beck, L.R.)らのバイオロジー・オブ・リプロダクション(Biology of Reproduction)、28:186−195(1983)には、ノルエチステロンを、D,L−ラクチド/グリコリドコポリマー中に封入する方法であって、コポリマーとノルエチステロンの両方を、クロロホルム/アセトン混合物に溶解し、それを、撹拌したポリビニルアルコールの冷水溶液に加えてエマルジョンを形成し、揮発性溶媒を減圧下に除去してマイクロカプセルを得る方法が開示されている。
【0013】
生分解性ポリマーマトリックスおよび生物学的活性剤から成る微粒子の調製には、相分離または非溶媒誘導コアセルベーションも用いられている。ラクチド/グリコリドコポリマーでマイクロカプセル化する文献記載の方法の多くでは、溶媒蒸発/抽出法を採用しているが、そのような方法は、水不溶性薬物に主として適当なものであり、水溶性薬物は、調製工程中に、水相中に一部分配され得る。そのような活性剤の封入方法としては、親水性活性剤も不溶の、ポリマーに対する非溶媒を用いる相分離方法が有効である。
【0014】
従来の相分離法においては、既知量のポリマー、例えばポリ(ラクチド−コ−グリコリド)(PLGA, ラクチド:グリコリドモノマー比100:0ないし50:50)を、適当な有機溶媒に溶解する。固体薬物(好ましくは凍結乾燥または粉砕したもの)を、ポリマー溶液に分散し得る(有機溶媒に不溶または難溶)。あるいは、活性剤を、水、または何らかの添加剤を含有する水に溶解し、ポリマー溶液中で乳化して(好ましくは主として超音波処理による)、油中水型エマルジョンを形成する。次いで、得られた懸濁液またはエマルジョンを反応器に入れ、所定の速度で第1非溶媒の添加を開始する。反応器に備えたタービンミキサーにより、緩やかに混合する。相分離の完了時に、混合物を、第2非溶媒を入れたクエンチタンクに移し、半固形小球を凝固する。硬化した小球を篩過により集め、洗浄し、減圧オーブン内に入れて更に乾燥する。
【0015】
既知のマイクロカプセル化に使用する溶媒は通例、ハロゲン化炭化水素、とりわけクロロホルムまたは塩化メチレンであり、これは活性剤および封入ポリマーのいずれの溶媒としても機能する。しかし、最終生成物中に少量だが検出可能な量で残留するハロゲン化炭化水素は、一般に毒性であり、発癌性を有し得るので、望ましくない。すなわち、既知のマイクロカプセル化方法を、より低毒性の許容し得る溶媒を使用するよう改善する必要がある。
【0016】
前記のような生物学的または薬学的活性剤のマイクロカプセル化のための従来の方法においては、活性剤およびポリマーを含有する溶媒を、撹拌、振動または他の動的混合法によって、しばしば比較的長時間、非混和性溶液中に乳化または分散することによって、微粒子を形成する。上記のような動的混合法は、いくつかの欠点を有する。例えば、得られる微粒子のサイズ、またはサイズ分布を調節しにくい。それ故、動的混合を行うと、生物学的または薬学的活性剤含有微粒子の工業的規模での製造には問題が生じる。とりわけ、製造装置は高価なエマルジョンタンク(液体を撹拌する装置を含む)を包含する。全工程時間の調節因子の一つは、均一なエマルジョンの形成に要する時間である。大きなタンクでバッチサイズが大きいほど、エマルジョン形成に長い時間がかかり、全体的な製造時間が長くなる。活性剤を加工溶媒およびポリマー溶液にさらす時間が長いほど、活性剤の分解または不活性化が起こり易い。生物学的または薬学的活性剤のマイクロカプセル化の場合、実験室エマルジョン法から製造法へのスケールアップは特に困難である。なぜなら、バッチおよびタンクサイズを大きくすると、大きいタンク内での撹拌速度および粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。同様に、相分離法も、微粒子の工業規模の製造工程に容易に変換できない。なぜなら、工程パラメータ、すなわち非溶媒添加速度、撹拌条件、並びに活性剤/ポリマー溶液および非溶媒の粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。すなわち、従来のマイクロカプセル化法のスケールアップは、時間がかかる上に、精密でない。
【0017】
エストラジオールベンゾエート含有微粒子製造において、実験室エマルジョン形成工程を小撹拌ガラス製反応器から製造装置にスケールアップする試みにおいて、試験を行った。ミキサー翼により生じる剪断力が、エマルジョンの粒子サイズを決定した。剪断力が大きいほど、粒子は小さい。エストラジオールベンゾエートの工程においては、油(有機)相が低粘度である故に、所望の大きいエマルジョン粒子を形成するのに、小さい剪断力が必要であった。大きい反応器においては、小さい剪断力を保ち、かつ、均一な混合を行うことが困難である。均一なタンク組成物を提供する撹拌機速度では、サイズ分布の広い小さいサイズの粒子が生成する。混合翼直径を増し、シャフトに沿って複数の混合翼を用いると、小さい剪断力での混合性は向上するが、まだサイズ分布は非常に広い。バッチサイズを大きくすると、粒子サイズ調節の信頼性が低下する。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】米国特許第3,737,337号
【特許文献2】米国特許第3,523,906号
【特許文献3】米国特許第3,691,090号
【特許文献4】米国特許第3,891,570号
【特許文献5】米国特許第3,960,757号
【特許文献6】米国特許第4,389,330号
【特許文献7】米国特許第4,530,840号
【特許文献8】国際特許出願公開90/13361号
【非特許文献】
【0019】
【非特許文献1】International Journal of Pharmaceutics, 43:179−186(1988)
【非特許文献2】Biology of Reproduction, 28:186−195(1983)
【発明の概要】
【発明が解決しようとする課題】
【0020】
本発明の目的は、生物学的または薬学的活性剤を含有する微粒子の適度に定めた狭いサイズ分布を達成しつつ、実験室から工業的規模へのバッチサイズを精密に信頼性よくスケールアップすることができる微粒子製造方法を提供することである。このことは、いずれの適当な封入法(溶媒抽出および相分離を包含するが、それに限定されない)においても達成し得る。
【0021】
本発明の別の目的は、適当に定めたサイズ分布を有する活性剤含有微粒子を、種々のバッチサイズで形成するのに、同一の装置を使用し得る方法を提供することである。
【0022】
本発明の方法のもう一つの目的は、一工程で溶媒を除去するか、または相分離方法によって、活性剤濃度の高い高品質の微粒子が得られる方法を提供することである。
【課題を解決するための手段】
【0023】
本発明は、微粒子の製法に関する
【0024】
とりわけ、本発明は、生分解性ポリマー結合剤および生物学的活性剤から成る生分解性微粒子の製法に関する。本発明の一態様においては、第1相(活性剤およびポリマーを含有する)および第2相を、スタティックミキサーを通してクエンチ液体にポンプ輸送して、活性剤含有微粒子を形成する。他の態様においては、第1相と第2相とは、実質的に非混和性である。他の態様においては、第2相はポリマーおよび活性剤に対する溶媒を含有せず、乳化剤の水溶液から成り得る。スタティックミキサーを使用して微粒子を製造する本発明の方法は、いずれの従来の封入法(溶媒抽出および相分離を包含するが、それに限定されない)にも適用し得る。
【0025】
本発明の更なる態様においては、第1相は、活性剤をポリマー含有溶液に溶解することによって、活性剤含有分散液を調製するとによって、および活性剤含有エマルジョンを調製することによって調製する。
【0026】
本発明の更なる態様においては、次のような活性剤を含有する微粒子を製造するために本発明の方法を用いる:リスペリドン(risperidone)、トレンボロンアセテート、ノルエチンドロン、テストステロン、エストラジオールベンゾエート、ヒト血清アルブミン、ブタアルブミンおよび組換えウシインターフェロン−α。
【0027】
本発明の好ましい一態様においては、活性剤およびポリマーを溶解するのに、少なくとも2種の実質的に無毒性の溶媒の混合物(ハロゲン化炭化水素不含有)を使用する。溶解した活性剤およびポリマーを含有する溶媒混合物を、水溶液に分散して、液滴を形成する。得られるエマルジョンを、次いで、水性抽出媒体(好ましくは溶媒混合物の少なくとも1種の溶媒を含有し、それにより、各溶媒の抽出速度を調節する)に加え、生物学的活性剤を含有する生分解性微粒子を形成する。この方法は、1種の溶媒の水溶性は実質的に他方とは独立しているので、抽出媒体が必要量が少なくてよく、溶媒選択性が広い(とりわけ、特に抽出困難な溶媒を含む)という利点を有する。
【0028】
本発明は好ましい一態様において、有効量の薬物を長時間にわたって調節的に放出するよう設計した微粒子の形態の薬剤組成物を製造する方法において有用な溶媒系に関する。この組成物は、少なくとも1種の薬剤および少なくとも1種の生体適合性、生分解性封入ポリマーを含有する。
【0029】
とりわけ、本発明の更なる態様においては、本発明は、微粒子の製法であって、
A.ハロゲン化炭化水素不含有の、水溶性の低い相互に混和性の有機溶媒少なくとも2種の混合物中に溶解または分散した生分解性ポリマー封入結合剤および活性剤から成る第1相を調製し、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とを混合手段の作用下に組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離する
ことを含んで成る方法に関する。
【0030】
低水溶性とは、20℃における水溶性が約0.1〜25重量%であることを意味する。
【0031】
好ましい態様においては、本発明は、微粒子の製法であって、溶媒混合物中に固体を約5〜50重量%含有する第1「油」相を調製し、その約5〜95重量%は生分解性ポリマー封入結合剤の溶液であり、第1相はポリマー結合剤に対して約5〜95重量%の活性剤を組み合わせたものであり、溶媒混合物は、ハロゲン化炭化水素不含有の、相互に混和性の第1および第2溶媒から成り、各溶媒の水溶性は20℃において約0.1〜25重量%であり;第1相をエマルジョン加工媒体中に1:1ないし1:10の比で含有するエマルジョンを形成して、連続水性第2相加工媒体中に第1相組成物の小滴を形成し;合した第1および第2相を、ポリマーおよび活性剤1g当たり水性クエンチ液約0.1〜20リットルのレベルの水性抽出クエンチ液に加え、クエンチ液は、溶媒混合物の溶媒の水溶性の大きい方を、使用温度でクエンチ液に対して約20〜70%の飽和レベルで含有し;クエンチ液から微粒子を回収することを含んで成る方法に関する。
【0032】
他の態様においては、本発明は、微粒子の製法であって、第1相を調製し、第1相は、生物学的活性剤、生分解性ポリマー、並びにハロゲン化炭化水素不含有の、少なくとも2種の相互に混和性の、活性剤およびポリマーに対する溶媒の混合物から成り;第2相を調製し、第1相は第2相とは実質的に非混和性であり;第1相をスタティックミキサーに第1流速で流し;第2相をスタティックミキサーに第2流速で流すことによって、第1相と第2相とを同時にスタティックミキサーに流して、活性剤含有微粒子を形成し;微粒子を分離することを含んで成る方法に関する。
【0033】
他の態様においては、本発明は、微粒子の製法であって、第1相を調製し、第1相は、生物学的活性剤、生分解性ポリマー、並びにハロゲン化炭化水素不含有の、少なくとも2種の相互に混和性の、活性剤およびポリマーに対する溶媒の混合物から成り;第2相を調製し、第1相は第2相とは実質的に非混和性であり;クエンチ液を調製し;第1相と第2相とをスタティックミキサーを通してクエンチ液にポンプ輸送することによって、活性剤含有微粒子を形成することを含んで成る方法に関する。
【0034】
本発明の更なる態様においては、(1)ハロゲン化炭化水素不含有の少なくとも2種の相互に混和性の溶媒に溶解したポリマーの溶液に、生物学的活性剤を溶解し、または(2)前記溶媒中に活性剤を含有する分散液を調製し、または(3)前記溶媒中に活性剤を含有するエマルジョンを調製することによって、第1相を調製する。
【図面の簡単な説明】
【0035】
【図1】スタティックミキサー内の流れを示す図である。
【図2】本発明の方法において使用し得るスタティックミキサーを示す図である。
【図3】本発明の好ましい微粒子製造方法を行うための実験室用装置を示す図である。
【図4】ノルエチンドロン含有微粒子の2製剤の時間放出動物実験データのグラフを示す。
【図5】バッチプロデックス(Podex)3のリスペリドン微粒子(製造したままのものと、凍結乾燥したものの両方)の、インビトロ溶解データのグラフを示す。
【図6】バッチプロデックス2のリスペリドン微粒子(製造したままのものと、凍結乾燥したものの両方)の、インビトロ溶解データのグラフを示す。
【図7】バッチプロデックス3およびプロデックス2のリスペリドン微粒子の、促進インビトロ溶解データのグラフを示す。
【図8】リスペリドンのデポ製剤を約2.5mg/kgの用量でビーグル犬に1回筋肉内投与した後の、活性物(リスペリドンおよび9−ヒドロキシリスペリドン)の平均(n=2)血漿濃度−時間曲線のグラフを示す。 アポモルヒネ催吐試験における制吐活性(少なくとも3匹中2匹の犬に見られる)時間を、各製剤の解説において示す。星印(*)は、試験開始後に3匹中少なくとも2匹の犬において制吐活性の中断があったことを意味する。点線は、制吐効果に必要なおよその最小血漿濃度を示す。「//」印は、製剤プロデックス2に対しては、14日、18日および21日目に血液サンプリングを行わなかったことを意味する。
【図9】エストラジオールベンゾエート含有微粒子の粒子サイズの累積%のグラフを示す。
【図10】エストラジオールベンゾエート含有微粒子の粒子サイズの%微分のグラフを示す。
【図11】エストラジオールベンゾエート含有微粒子の、時間放出動物実験データのグラフを示す。
【図12】トレンボロンアセテート含有微粒子の粒子サイズの累積%のグラフを示す。
【図13】テストステロン含有微粒子の、時間放出動物実験データのグラフを示す。
【図14A】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図14B】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図14C】ノルエチンドロン(NET)微粒子の性質に対する、クエンチ液への酢酸エチル添加効果を示すグラフの1つである。
【図15A】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【図15B】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【図15C】NET微粒子の性質に対する、クエンチ体積の効果を示すグラフの1つである。
【発明を実施するための形態】
【0036】
本発明においては、少なくとも1種の生物学的活性剤を含有する生分解性微粒子を製造するために、少なくとも2種の溶媒から成る、ハロゲン化炭化水素不含有の溶媒混合物を使用する。溶媒混合物の第1溶媒成分は、活性剤に対しては難溶媒であるが、本発明に使用する生分解性ポリマーに対しては良好な溶媒である。溶媒混合物の第2溶媒成分は、活性剤およびポリマーの両方に対して良好な溶媒である。
【0037】
本発明の方法は、既知の方法にまさる利点を有する。本発明の方法はとりわけ、生分解系、処置中の用量損失を防いだ注射系、異種薬物含有微粒子の混合性、ハロゲン化炭化水素残留のない微粒子、並びに必要に応じてより速くまたは遅く薬物を放出する調節的放出性(多相放出パターン)を提供する。
【0038】
本発明の方法で製造した生成物は、選択した微粒子の種類に応じて、作用期間が30ないし200日以上であるという利点を有する。好ましい態様においては、微粒子は、30〜60日間にわたって患者の処置を提供するよう設計する。作用期間は、ポリマー組成、ポリマー:薬物比、および微粒子サイズの変更によって、容易に調節し得る。
【0039】
本発明の方法により製造した微粒子のもう一つの重要な利点は、本発明の方法に使用するポリマーは生分解性であり、封入した剤を全部患者に放出するので、実質的に全部の活性剤が患者にデリバーされるということである。
【0040】
本発明の方法においては、ハロゲン化炭化水素不含有溶媒混合物に活性剤を溶解または分散し、その活性剤含有媒体に、ポリマーマトリックス材料を、活性剤に対し相対的に、所望の活性剤含有量の生成物を与えるような量で加える。要すれば、微粒子生成物の全成分を同時に溶媒混合物媒体中に混合し得る。
【0041】
本発明において使用する溶媒系は、少なくとも2種の溶媒の混合物である。そのような溶媒は、
(1)相互に混和性であり、
(2)混合した状態で、活性剤を溶解または分散でき、
(3)混合物した状態で、ポリマーマトリックス材料を溶解でき、
(4)活性剤に対して化学的に不活性であり、
(5)生体適合性であり、
(6)実質的にクエンチ液と非混和性であり(例えば、溶解度約0.1〜25%以下)、
(7)ハロゲン化炭化水素以外の溶媒でなくてはならない。
【0042】
「ハロゲン化炭化水素」とは、ハロゲン化有機溶媒、すなわちC1−C4ハロゲン化アルカン、例えば塩化メチレン、クロロホルム、塩化メチル、四塩化炭素、二塩化エチレン、塩化エチレン、2,2,2−トリクロロエタンなどを意味する。
【0043】
活性剤封入のための好ましい溶媒混合物は、ポリマー封入剤の溶解度が、通例、20℃で少なくとも約5重量%、好ましくは少なくとも約20重量%と、高くなければならない。溶解度の上限は重要でないが、封入ポリマーが溶液の約50重量%を超えると、溶液が非常に粘性となって、有効かつ好都合に扱うことが困難になり得る。このことは当然、封入ポリマーの性質および分子量によって異なる。
【0044】
溶媒系は、連続相加工媒体およびクエンチ液(通例、水性)と実質的に非混和性であるが、好ましくは水に少し溶解する。溶媒系が加工媒体にいくらでも溶解するならば、エマルジョン相において小滴が生成し得ないであろう。しかし、抽出クエンチ媒体中の溶媒系の溶解度が低すぎると、クエンチ媒体が大量に必要となる。通例、加工媒体およびクエンチ媒体中に約0.1〜25%溶解する溶媒が、本発明において許容し得る。クエンチ媒体が、第1溶媒(すなわち、クエンチ媒体に溶解し易い方の溶媒)を飽和点の約70〜20重量%の量で含有すると、微粒子からクエンチ媒体への第1溶媒の損失率を調節するためにしばしば有利であり得る。
【0045】
本発明の溶媒混合物の成分の選択において更に考慮することは、沸点(すなわち、蒸発による最終生成物の与え易さ)、および比重(乳化およびクエンチング中に、「油相」が浮く傾向)である。また、溶媒系は低毒性であるべきである。
【0046】
通例、溶媒混合物組成物は、第1溶媒を約25〜75重量%、第2溶媒を約75〜25重量%含有し得る。
【0047】
本発明の溶媒混合物は好ましくは、エステル、アルコールおよびケトンの少なくとも2種の混合物である。好ましいエステルは、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基、すなわちメチル、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択する。]で示される。本発明において、溶媒混合物の一成分として使用するのに最も好ましいエステルは、酢酸エチルである。好ましいアルコールは、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示される。R3は、より好ましくはアリールである。本発明において溶媒混合物の一成分として使用するのに最も好ましいアルコールは、ベンジルアルコールである。好ましいケトンは、式R4COR5[式中、R4はC1−C4アルキル基、すなわち、メチル、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択し、R5はC2−C4アルキル基、すなわち、エチル、プロピル、ブチルおよびそれらの異性体から成る群から選択する。]で示される。本発明において溶媒混合物の一成分として使用するのに最も好ましいケトンは、メチルエチルケトンである。
【0048】
本発明の方法によって製造する微粒子のポリマーマトリックス材料は、生体適合性および生分解性のポリマー材料である。「生体適合性」とは、ポリマー材料が人体に無毒で、非発癌性で、生体組織中で顕著な炎症を誘導しないことと定義する。マトリックス材料は、生体の代謝によって生体が容易に排泄し得る物質に分解され、生体中に蓄積しないという意味において、生分解性でなければならない。生分解生成物も、ポリマーマトリックスが生体適合性であるのと同様の意味で生体適合性であるべきであり、微粒子中に残留し得る溶媒も同様である。
【0049】
ポリマーマトリックス材料の適当な例は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、並びに天然ポリマー、例えばアルブミン、カゼイン、および蝋、例えばグリセロールモノ−およびジステアレートなどを包含する。種々の市販のポリ(ラクチド−コ−グリコリド)材料(PLGA)を、本発明の方法において使用し得る。例えば、ポリ(d,l−乳酸−コ−グリコール酸)が、メデソーブ・テクノロジーズ・インターナショナルL.P.(Medisorb Technologies International L.P.;オハイオ州シンシナチ)から市販されている。メディソーブから市販の適当な生成物は、メディソーブ(MEDISORB、商標)5050DLとして知られる50:50ポリ(D,L)乳酸−コ−グリコール酸である。この生成物のモル%組成は、ラクチド50%およびグリコリド50%である。他の適当な市販生成物は、メディソーブ65:35DL、75:25DL、85:15DL、およびポリ(d,l−乳酸)(d,l−PLA)である。ポリ(ラクチド−コ−グリコリド)は、ベーリンガー・インゲルハイム(Boehringer Ingelheim、ドイツ)からもレゾマー(Resomer)の商品名で市販されており[例えば、PLGA50:50(レゾマーRG502)、PLGA75:25(レゾマーRG752)およびd,l−PLA(レゾマーRG206)]、バーミンガム・ポリマーズ(Birmingham Polymers、アラバマ州バーミンガム)からも市販されている。このようなポリマーは、種々の分子量、および乳酸:グリコール酸比のものが市販されている。
【0050】
本発明に最も好ましいポリマーは、ポリ(d,l−ラクチド−コ−グリコリド)である。そのようなコポリマー中、ラクチド:グリコリドのモル比は、好ましくは約85:15ないし50:50である。
【0051】
ポリマーマトリックス材料の分子量は、ある程度重要である。分子量は、充分なポリマーコーティングの形成のために充分大きくなくてはならない(すなわち、ポリマーは良好なフィルムポリマーでなくてはならない)。通例、充分な分子量は、5000〜500000ダルトンの範囲で、好ましくは約150000ダルトンである。しかし、フィルムの性質は、使用するポリマー材料の種類によってもいくぶん変化するので、すべてのポリマーについて適当な分子量範囲を特定することは非常に困難である。ポリマー分子量は、ポリマーの生分解速度の点からも重要である。薬物放出の拡散メカニズムのために、ポリマーは薬物全部が微粒子から放出されてしまうまで変化せず、その後、分解されるべきである。薬物は、ポリマー賦形剤が生体により浸食される間に放出されてもよい。ポリマー材料を適当に選択することによって、拡散放出および生分解放出の両方を行う微粒子を形成するように微粒子製剤を製造してもよい。これは、多相放出パターンに有用である。
【0052】
本発明の方法によって製造する製剤は、微粒子のポリマーマトリックス材料中に分散した活性剤を含有する。微粒子中に含まれる活性剤の量は通例、約1〜90重量%、好ましくは30〜50重量%、より好ましくは35〜40重量%である。重量%は、微粒子全重量に対する剤の割合である。例えば、剤10重量%は、剤が10重量部、ポリマーが90重量部存在することを意味する。
【0053】
本発明の方法の実施において、溶媒混合物中に封入ポリマーが実質的に100%溶解した溶液を乳化に用いる。活性剤は、その溶液を連続相加工媒体に加える際に、溶媒混合物に分散または溶解し得る。最初の乳化の際の、溶媒混合物中の通例固体の材料(活性剤と封入ポリマー)の含量は、少なくとも5重量%、好ましくは少なくとも20重量%であるべきである。「油相」中の溶媒を少なくすると、良質の微粒子が得られ、抽出媒体必要量は少なくてよい。
【0054】
本発明の方法によって封入し得る好ましい活性剤の一つは、ノルエチンドロン(NET)である。リスペリドンおよびテストステロンも好ましい。
【0055】
酢酸エチル単独ではNETの難溶媒であり、従来のクロロホルム法よりも多くの溶媒および高い温度を要する。生成物微粒子のコア含有は許容し得るが、収率(とくに63〜90μm範囲のもの)は低い。走査電子顕微鏡写真によると、そのようなより大きい粒子は割れ、崩壊する。その証拠に、そのような粒子は正常な放出速度よりも放出速度が大きい。
【0056】
ベンジルアルコールを単独で溶媒として使用すると、クエンチタンク内容物を光学顕微鏡で観察してわかるように、微粒子サイズを調節し易い。しかし、乾燥すると通例、品質の低いことがわかる。しばしば、付着性の故に、回収困難である。また、溶媒残留量が増加する傾向にある。酢酸エチルとベンジルアルコールとを「油相」の溶媒系として使用すると、微粒子の品質および放出性が改善された。
【0057】
「油相」溶媒系中の成分混合物を、連続相加工媒体中で乳化する;連続相媒体は、所定成分を含有する微粒子の分散液を連続相媒体中に形成するものである。
【0058】
絶対に必要というわけではないが、連続相加工媒体を、「油相」溶媒系を形成する溶媒の少なくとも一種で飽和することが好ましい。これにより安定なエマルジョンが生成し、クエンチング前に微粒子から溶媒が排出されるのを防ぐ。米国特許第4389330号記載のように、減圧を適用してもよい。酢酸エチルおよびベンジルアルコールが溶媒系成分である場合、エマルジョンの水相は好ましくは、酢酸エチル1〜8重量%およびベンジルアルコール1〜4重量%を含有する。
【0059】
通例、界面活性剤または親水性コロイドを連続相加工媒体に加えて、溶媒小滴の凝集を防止し、エマルジョン中の溶媒小滴のサイズを調節する。界面活性剤または親水性コロイドとして使用し得る化合物の例は、ポリ(ビニルアルコール)、カルボキシメチルセルロース、ゼラチン、ポリ(ビニルピロリドン)、トゥイーン(Tween)80、トゥイーン20などを包含するが、それらに限定されない。加工媒体中の界面活性剤または親水性コロイドの濃度は、エマルジョンの安定化に充分な量であるべきであり、微粒子の最終サイズに影響し得る。通例、加工媒体中の界面活性剤または親水性コロイドの濃度は、界面活性剤または親水性コロイド、「油相」溶媒系、および加工媒体の種類によって異なるが、加工媒体に対して約0.1〜10重量%であり得る。好ましい分散媒体組み合わせは、水中のポリ(ビニルアルコール)の0.1〜10重量%、より好ましくは0.5〜2重量%溶液である。
【0060】
エマルジョンは、混合相の機械的撹拌によって、または活性剤と壁形成材料とを含有する有機相の小滴を連続相加工媒体に加えることによって形成し得る。エマルジョン形成中の温度は特に重要ではないが、微粒子のサイズおよび品質、並びに連続相中の活性剤の溶解度に影響し得る。当然、連続相中に入る活性剤は少ないほどよい。更に、使用する溶媒混合物および連続相加工媒体によっては、温度が低すぎてはならない。なぜなら、溶媒および加工媒体が凝固または不適当に増粘し得るからである。また、加工媒体が蒸発し得るか、または液体加工媒体が維持できなくなり得るほど高温にしてもいけない。更に、エマルジョン温度は、微粒子中に組み合わせる活性剤の安定性を損なうほど高くしてはならない。従って、分散工程は、安定な操作条件を保つ温度で、選択した活性剤および賦形剤によるが、好ましくは約20〜60℃で行い得る。
【0061】
前記のように、活性剤含有微粒子を生成するように、有機相と水相を組み合わせる。有機相と水相とは概ね、または実質的に非混和性であり、水相はエマルジョンの連続相を構成する。有機相は、活性剤と、壁形成ポリマー(すなわちポリマーマトリックス材料)を含有する。有機相は、本発明の有機溶媒系に活性剤を溶解または分散することによって調製する。有機相と水相は、混合手段の作用下に組み合わせる。
【0062】
好ましい混合手段はスタティックミキサーであり、活性剤の封入により調節的放出微粒子を形成する本発明の方法は、スタティックミキサーの使用を伴う。好ましくは、合した有機および水相を、ポンプ輸送によりスタティックミキサーを通してエマルジョン形成し、大量のクエンチ液に入れて、ポリマーマトリックス材料中に封入した活性剤を含有する微粒子を得る。
【0063】
生物学的または薬学的剤をマイクロカプセル化する多くの既知の方法においては、活性剤およびポリマーを含有する溶媒を、非混和性の第2の溶媒中で、撹拌、混合、振動、または何らかの他の動的混合法によって、しばしば比較的長時間、乳化または分散することによって、微粒子を形成する。上記のような動的混合法は、いくつかの欠点を有する。例えば、得られる微粒子のサイズ、またはサイズ分布を調節しにくい。それ故、動的混合を行うと、生物学的または薬学的活性剤含有微粒子の工業的規模での製造には問題が生じる。とりわけ、製造装置は高価なエマルジョンタンク(液体を撹拌する装置を含む)を包含する。全工程時間の調節因子の一つは、均一なエマルジョンの形成に要する時間である。大きなタンクでバッチサイズが大きいほど、エマルジョン形成に長い時間がかかり、全体的な製造時間が長くなる。活性剤を加工溶媒およびポリマー溶液にさらす時間が長いほど、活性剤の分解または不活性化が起こり易い。生物学的または薬学的活性剤のマイクロカプセル化の場合、実験室エマルジョン法から製造法へのスケールアップは特に困難である。なぜなら、バッチおよびタンクサイズを大きくすると、大きいタンク内での撹拌速度および粘度を、スケールアップの段階毎に試行錯誤により実験的に最適化しなければならないからである。このような方法は、時間がかかる上に、精密でない。
【0064】
すなわち、スタティックミキサーを使用する微粒子製造方法の一つの利点は、生物学的または薬学的活性剤を含有する微粒子の適度に定めた狭いサイズ分布を達成しつつ、実験室から工業的規模へのバッチサイズを精密に信頼性よくスケールアップすることができるということである。本発明の方法の更なる利点は、適当に定めたサイズ分布を有する活性剤含有微粒子を、種々のバッチサイズで形成するのに、同一の装置を使用し得るということである。本発明の方法のもう一つの利点は、一工程で溶媒を除去して、活性剤濃度の高い高品質の微粒子が得られるということである。前記タイスらの米国特許第4389330号に記載のような2段階溶媒除去方法を行う必要がない。方法の改良に加えて、スタティックミキサーは維持費が安く、小さいので動的ミキサーよりもスペースが狭くてよく、エネルギー消費が少なく、比較的投資額が少なくてよい。
【0065】
スタティックまたは静的ミキサーは、導管または管を有し、その内部に複数の静的混合要素を有する。スタティックミキサーは、比較的短い導管内で、比較的短時間のうちに均一な混合を行う。スタティックミキサーの場合、ミキサーの一部(例えば翼)が液体中で動くのではなく、液体がミキサー内を動く。一種のスタティックミキサー内の流れを、図1に示す。ポンプ(図示せず)が、1種またはそれ以上の液流(1)を、スタティックミキサー(10)内に導入する。液流は分れて、向き合った外壁に当たる(2)。スタティックミキサー(10)の中心線を軸とする渦が生じる(3)。渦は途切れて、その過程が繰り返される(ただし逆回転)(4)。右回り/左回りの動きによって、確実に均一な生成物が得られる。
【0066】
スタティックミキサーの例を図2に示す。スタティックミキサー(10)は、導管またはパイプ(12)内に連続的に配置された複数の静止または静的混合要素(14)を有する。要素数は4〜32またはそれ以上であり得る。導管(12)は円形断面を有し、液体を導入および排出するよう、両端(18および20)が開口している。混合要素(14)は、セグメント(142)から成る。各セグメント(142)は、複数の、通例平らな板または羽(144)から成る。2つの実質的に同じセグメント(142)を、好ましくは相互にずらして軸方向に配置する。図2に示すスタティックミキサーは、米国特許第4511258号に、より詳細に記載されており、該特許を引用により本発明の一部とする。
【0067】
スタティックミキサーを用いてエマルジョンを形成する場合、種々の因子がエマルジョン液滴サイズを決定する。そのような因子は、混合する溶液または相の密度および粘度、相の体積比、相間界面張力、スタティックミキサーパラメータ(導管直径、混合要素の長さと数)、並びにスタティックミキサー内の流速を包含する。温度は、密度、粘度および界面張力に影響するので、変えることができる。主な調節因子は、流速である。スタティックミキサー単位長さ当たりの剪断速度および圧力低下も、重要なパラメータである。特に、流速が高まると液滴サイズが小さくなり、流速(および圧力低下)が低下すると液滴サイズが大きくなる。所定の流速で一定数の要素を通って動いた後、液滴サイズは平衡化する。流速が大きいほど、要素の必要数は少ない。このような関係の故に、実験室バッチサイズから工業的バッチサイズへのスケールアップは信頼性があり、精密であり、実験室および工業的のいずれのバッチサイズにも同じ装置を使用し得る。
【0068】
本発明の方法において、有機相および水相を、それらが同時にスタティックミキサー内に流れ、ポリマーマトリックス材料中に封入した活性剤を含有する微粒子を含むエマルジョンを形成するように、ポンプ輸送する。有機相および水相を、スタティックミキサーを経て大量のクエンチ液へポンプ輸送する。クエンチ液は、単なる水、水溶液、または他の適当な液体であり得る。クエンチ液中で洗浄または撹拌する間に、微粒子から有機溶媒を除去し得る。本発明の方法によって有機相および水相をスタティックミキサーを経て溶媒除去用クエンチ液にポンプ輸送すると、前記タイスらの特許(第4389330号)に記載の2段階溶媒除去を行わなくても、活性剤濃度の高い高品質の微粒子が生成する。微粒子をクエンチ液中で洗って有機溶媒を抽出または除去した後、例えば篩過により分離し、乾燥する。
【0069】
本発明の他の方法においては、有機相は、ポリマー溶液と、乾燥粉末として懸濁または水溶液中に溶解し、乳化した活性剤とから成る。ポリマーは、適当な有機溶媒、好ましくは非ハロゲン化溶媒、例えば酢酸エチルに溶解する。有機相をスタティックミキサー内で非溶媒と組み合わせると、活性剤粒子または液滴周囲にポリマー液滴のコアセルベーションまたは沈降(すなわち相分離)が起こる。スタティックミキサー内を流れる溶媒および非溶媒の滞留時間は、本発明の方法において重要な因子である。滞留時間は、混合要素および導管の寸法、並びにスタティックミキサー内を流れる溶液の線速度を変えることによって変化し得る。微粒子の形成に影響し得る他の重要な因子は、混合する2相の密度および粘度、相の体積比、並びに相間の界面張力である。しかし、微粒子サイズ調節は主として、最初の有機相中の活性剤懸濁液またはエマルジョンのサイズおよび均一性によって行う。
【0070】
スタティックミキサー法を行うための実験室用装置を、図3に示す。有機または油相(30)は、撹拌ポット(32)内で活性剤およびポリマーマトリックス材料を溶解することによって調製する。しかし、有機相(30)の調製法は、活性剤の溶解に限定されない。ポリマーマトリックス材料を含有する溶液に活性剤を分散することによって、有機相(30)を調製してもよい。そのような分散液中では、活性剤は有機相(30)に少ししか溶解しない。また、有機相(30)は、活性剤およびポリマーマトリックス材料を含有するエマルジョンの調製によっても調製し得る(ダブルエマルジョン法)。ダブルエマルジョン法においては、活性剤およびポリマーマトリックス材料を含有する第1エマルジョン(有機相30)を調製する。第1エマルジョンは、油中水型エマルジョン、水中油型エマルジョン、または他の適当なエマルジョンであり得る。第1エマルジョン(有機相30)および水相を、次いで、スタティックミキサーにポンプ輸送して、第2エマルジョンを形成する。第2エマルジョンは、ポリマーマトリックス材料中に封入された活性剤を含有する小滴を含有する。本発明のダブルエマルジョン法を用いて活性剤含有微粒子を調製する例は、実施例8に示す。
【0071】
有機相(30)を、磁気駆動ギアポンプ(34)によって撹拌ポット(32)からくみ出す。しかし、いずれの適当なポンプ手段を用いてもよい。ポンプ(34)からの輸送物はY字型連結具(36)に送られる。Y字型連結具(36)の1つの枝管(361)は、ポット(32)に戻る再循環流である。他の枝管(362)は、インラインスタティックミキサー(10)に至る。水相(40)も同様に、撹拌ポット(42)、磁気駆動ギアポンプ(44)およびY字型連結具(46)によって調製する。Y字型連結具(46)の1つの枝管(461)は、ポット(42)に戻る再循環流である。他の枝管(462)は、インラインスタティックミキサー(10)に至る。
【0072】
インラインスタティックミキサー(10)に送られる各溶液の枝流(362および462)は、別のY字形連結具(50)によって合流し、ミキサー導入ライン(51)を通ってスタティックミキサー(10)に送られる。スタティックミキサー(10)からの排出物は、ミキサー排出ライン(52)を通って洗浄タンク(60)に送られる。図3に示す装置において、シリコーンチューブおよびポリプロピレンフィッティングを使用する。ミキサー排出ライン(52)以外のすべてのラインに、内径3/8インチのシリコーンチューブを使用する。ミキサー排出ライン(52)には、ミキサー排出ライン(52)内、および洗浄タンク(60)への導入時の両方で、エマルジョンの破壊を防止するために、より径の小さいチューブ(内径3/16インチ)を使用する。
【0073】
本発明の方法の一態様においては、ポンプ(34および44)は再循環モードで始動し、有機相(30)および水相(40)の所望の流速を設定する。水相(40)の流速は、好ましくは有機相(30)の流速よりも大きい。しかし、2つの流速が実質的に等しくてもよい。水相(40)流速と有機相(30)流速の比は、好ましくは1:1ないし10:1である。次いで、Y字形連結具(46)を、水相(40)が枝管(462)を通ってスタティックミキサー(10)に流れるように切り替える。水相(40)でミキサー導入ライン(51)、スタティックミキサー(10)、およびミキサー排出ライン(52)が満たされたら、Y字形連結具(36)を、有機相(30)が枝管(362)を通ってスタティックミキサー(10)に流れるように切り替える。この時点で、有機相(30)と水相(40)とが、同時にスタティックミキサー(10)に流れる。所望量の有機相をスタティックミキサー(10)にポンプ輸送した後、Y字形連結具(36)を枝管(361)からの再循環に切り替える。水相(40)は、ミキサー導入ライン(51)、スタティックミキサー(10)およびミキサー排出ライン(52)内に残った有機相を洗い流すように、短時間流し続ける。Y字形連結具(46)を、次いで、枝管(461)からの再循環に切り替える。
【0074】
有機相(30)および水相(40)をスタティックミキサー(10)内で混合して、エマルジョンを形成する。生成したエマルジョンは、ポリマーマトリックス材料中に封入した活性剤を含有する小滴を含有する。小滴から有機溶媒を除去するために、クエンチ溶液の入った洗浄タンク(60)内で小滴を撹拌して、硬化した微粒子を形成する。次いで、いずれの従来の分離方法で、水性クエンチ溶液から微粒子を分離してもよい。液体を微粒子からデカントするか、微粒子懸濁液を濾過するか、または篩カラムを使用し得る。要すれば、種々の他の分離法を組み合わせてもよい。次いで、微粒子を、従来の乾燥法で乾燥し、更にサイズ分別を行い得る。
【0075】
小滴がスタティックミキサーから洗浄タンクに移ると、連続相加工媒体は希釈され、小滴中の残留溶媒が抽出によって除去される。この抽出クエンチ工程において、微粒子を、親水性コロイドまたは界面活性剤の存在または不存在下に、乳化に用いたのと同じ連続相加工媒体中に懸濁、または他の液体中に懸濁し得る。抽出媒体は、微粒子から溶媒を除去するが、微粒子を溶解しない。抽出中、溶解した溶媒を含有する抽出媒体を、要すれば除去し、新たな抽出媒体を補充し得る。これは、断続的または連続的に行うことが好ましく、その場合、抽出媒体補充速
度が重要である。速度が小さすぎると、活性剤結晶が微粒子から突出するか、抽出媒体中で成長し得る。適当な抽出媒体補充速度は方法によって変化し得、方法を行う際に決定し得、厳密に速度範囲を予め決める必要はない。残留溶媒を除去後、微粒子を前記のように分離し、風乾するか、または他の従来の乾燥法(例えば減圧乾燥、乾燥剤による乾燥など)によって乾燥する。本発明の方法は、約80重量%まで、好ましくは約50重量%までのコア含量を達成し得るので、活性剤の封入に非常に有効である。
【0076】
エマルジョン中の「油相」液滴の形成に使用した溶媒混合物の一方の溶媒(例えば、好ましい酢酸エチル/ベンジルアルコール混合物の場合は、第1溶媒の酢酸エチル)が、他方の溶媒よりも速く抽出され得る。すなわち、第2溶媒(ベンジルアルコール)が高残留量で残る。ベンジルアルコールは沸点が高いので、微粒子を空気または他の従来の蒸発手段に付しても容易に除去できない。この問題を克服するために、エマルジョンを入れる前の抽出媒体に、抽出の速い方の溶媒をいくらか加える。抽出の速い方の溶媒の、抽出媒体中の濃度は通例、抽出温度における媒体中の該溶媒の飽和点の約20〜70%である。すなわち、エマルジョンをクエンチ液に入れる際、抽出の速い方の溶媒の抽出は遅延され、抽出の遅い方の第2溶媒がより多く除去される。
【0077】
この抽出の速い方の溶媒の「スパイク(添加)」量は、最終的な微粒子の品質に重要である。溶媒が多すぎる(すなわち飽和点に近い)と、活性剤含有微粒子表面に目に見える孔が生じ、望ましくない高放出速度をもたらし得る。抽出媒体中の溶媒が少なすぎると、抽出の遅い方の溶媒の残留量が多くなり、微粒子の品質が低下し得る。抽出媒体の温度も、溶媒溶解度および抽出速度に影響するので重要である。
【0078】
温度および溶媒スパイク量のいずれも、望ましい性質を有する最終生成物(すなわち非常に多孔性で、放出の速い微粒子、または多孔性が低く、放出の遅い微粒子)が得られるように調節し得る。
【0079】
クエンチ液は、純粋な水、水溶液、または他の適当な液体であり得、その体積、量および種類は、エマルジョン相に使用した溶媒に応じて変化し得る。クエンチ液は、好ましくは水である。通例、クエンチ液体積は、飽和体積の10倍のオーダーである(すなわち、エマルジョン中の溶媒体積の完全な吸収に、10倍体積のクエンチ液を要する)。しかし、溶媒系によって、クエンチ体積は飽和体積の約2〜20倍の範囲で変化し得る。更に、バッチサイズ(微粒子生成物)に対するクエンチ体積の条件を説明することが好都合である。この比は、抽出工程の効率の指標となり、場合によっては一定の装置に対するバッチサイズを決定する。比が大きいほど、生成物重量当たり大きい体積が必要である。比が小さいほど、一定のクエンチ体積から、より多くの生成物を得ることができる。この比は、生成する微粒子1g当たり、クエンチ体積約0.1〜10lである。1l/gよりも小さい比が好ましい。
【0080】
ベンゾイルアルコールおよび酢酸エチルの好ましい溶媒組み合わせを使用した場合、クエンチ液の酢酸エチルが、微粒子生成物中の残留溶媒レベルに影響すると考えられる。クエンチ液の酢酸エチル含量が低いと、微粒子中のベンジルアルコール残留量が多く、酢酸エチルは殆んど検出されない。クエンチ液の酢酸エチル含量が高いと(5〜7重量%またはそれ以上)、微粒子は酢酸エチルをベンジルアルコールよりも多く含有し得る。クエンチングする活性剤およびポリマー封入材料1g当たりクエンチ体積約1lでは、クエンチ液体中の酢酸エチル量は、0〜4℃で約3〜4重量%が最適である。微粒子のコア含量は、クエンチ液中の酢酸エチル濃度によって少し変化し、酢酸エチル濃度が高いか、低い場合、少なくなる。微粒子からのインビトロ放出速度は、クエンチ液の酢酸エチル含量の変化によって実質的に変化する。NETの場合、酢酸エチル含量が多すぎると、NET放出が速まる。走査電子顕微鏡で観察すると、クエンチ液中の酢酸エチル量が多すぎた場合は、微粒子表面上にNETおよび孔の存在が見られる。
【0081】
クエンチ液体の体積を変えても、微粒子中の相対溶媒残留量が大きく影響される。体積が小さいと、ベンジルアルコール:酢酸エチル比が大きく、クエンチ体積を、クエンチングする活性剤およびポリマー封入材料1g当たり約1.5lに増加すると、前記比は1未満に低下する。微粒子生成物からの活性剤放出速度は、顕著に大きい。(NETおよびポリマー封入材料の溶液1g当たり、クエンチ液0.125lでは、走査電子顕微鏡写真によると、微粒子生成物は非常に多孔性である。NETおよびポリマー封入材料の溶液1g当たり、クエンチ液0.25〜1.5lでは、微粒子生成物からのNET放出速度はほぼ同程度で、クエンチ液1l/g(クエンチングするNET+ポリマー材料)の場合に最も小さい速度となり得る。)
【0082】
スタティックミキサーを使用して微粒子を製造する本発明の方法は、種々の活性剤封入法に適用し得る。本発明の方法は、前記溶媒抽出法に限定されず、他の封入法にも適用し得る。例えば、本発明の方法は、相分離封入法にも適用し得る。そのためには、ポリマー溶液に懸濁または分散した活性剤を含有する有機相を調製する。非溶媒第2相は、ポリマーおよび活性剤に対する溶媒を含有しない。好ましい非溶媒第2相は、シリコーン油である。有機相および非溶媒相を、スタティックミキサーを通して非溶媒クエンチ液(例えばヘプタン)にポンプ輸送する。半固体粒子をクエンチングして、充分硬化および洗浄する。このような方法の例を、実施例11〜14に挙げる。
【0083】
微粒子生成物は通例、球形粒子から成るが、不規則な形を有することもある。微粒子サイズは、直径サブミクロンないしミリメートルの範囲で変化し得る。好ましくは1〜500μ、より好ましくは25〜180μの微粒子を製造し、そうすると、標準的ゲージの針を用いて患者に投与することができる。
【0084】
好ましくは、薬物含有微粒子は、患者に1回投与する。薬物が連続的または不連続的に患者に放出されるので、反復注射の必要がない。
【0085】
活性剤含有微粒子は、乾燥物として得られ、貯蔵する。患者に投与する前に、乾燥微粒子を、許容し得る薬剤液体ビヒクル、例えば2.5重量%カルボキシメチルセルロース溶液に懸濁し、微粒子懸濁液を患者に注射し得る。
【0086】
活性剤を多相様式で患者にデリバーするように、および/または異なる活性剤を異なる時点でデリバーするように、または活性剤混合物を同時にデリバーするように、大きさまたは種類の異なる微粒子を混合し得る。例えば、主活性剤を、他の抗体、ワクチンまたは所望の活性剤(微粒子形態、または従来の非封入形態のもの)と混合して、患者に投与し得る。
【0087】
適当な活性剤は、エストロゲン、例えばジエチルスチルベストロール、17−β−エストラジオール、エストロン、エチニルエストラジオール、メストラノールなど;プロゲスチン、例えばノルエチンドロン、ノルゲストリル、エチノジオールジアセテート、リネステノール、メドロキシプロゲステロンアセテート、ジメスチステロン、メゲストロールアセテート、クロルマジノンアセテート、ノルゲスチマート、ノルエチステロン、エチステロン、メレンゲストロール、ノルエチノドレルなど; および殺精子剤化合物、例えばノニルフェノキシポリオキシエチレングリコール、ベンゼトニウムクロリド、クロルインダノールなどを包含する。
【0088】
本発明の方法によって組み合わせ得る他の生物学的活性剤は、胃腸処置剤、例えば水酸化アルミニウム、炭酸カルシウム、炭酸マグネシウム、炭酸ナトリウムなど;非ステロイド抗受精剤;副交感神経興奮剤;精神療法剤;リスペリドン;メジャートランキライザー、例えばクロルプロマジンHCl、クロザピン、メソリダジン、メチアピン、レセルピン、チオリダジンなど;マイナートランキライザー、例えばクロルジアゼポキシド、ジアゼパム、メプロバメート、テマゼパムなど;鼻科学的充血除去剤;鎮静−催眠剤、例えばコデイン、フェノバルビタール、ナトリウムペントバルビタール、ナトリウムセコバルビタールなど;ステロイド、例えばテストステロンおよびテストステロンプロピオネート;スルホンアミド;交感神経興奮剤;ワクチン;ビタミンおよび栄養剤、例えば必須アミノ酸;必須脂肪など;抗マラリア剤、例えば4−アミノキノリン、8−アミノキノリン、ピリメタミンなど;抗片頭痛剤、例えばマジンドール、フェンテリニンなど; 抗パーキンソン病剤、例えばL−ドーパ;抗けいれん剤、例えばアトロピン、メトスコポラミンブロミドなど;抗けいれんおよび抗コリン剤、例えば担汁療法剤、消化剤、酵素など;鎮咳剤、例えばデキストロメトルファン、ノスカピンなど;気管支拡張剤;心血管剤、例えば抗高血圧剤、ラウオルフィアアルカロイド、冠血管拡張剤、ニトログリセリン、有機ナイトレート、ペンタエリスリトテトラナイトレートなど;電解質、例えば塩化カリウム;麦角アルカロイド、例えばエルゴタミン(カフェイン有および無)、水素化麦角アルカロイド、ジヒドロエルゴクリスチンメタンスルフェート、ジヒドロエルゴコルニンメタンスルフェート、ジヒドロエルゴクロイプチンメタンスルフェートおよびそれらの組み合わせ;アルカロイド、例えばアトロピンスルフェート、ベラドンナ、ヒヨスチンヒドロブロミドなど;鎮痛剤;麻酔剤、例えばコデイン、ジヒドロコデイン、メペリジン、モルヒネなど;非麻酔剤、例えばサリチレート、アスピリン、アセトアミノフェン、d−プロポキシフェンなど;抗生物質、例えばセファロスポリン、クロラムフェニコール、ゲンタマイシン、カナマイシンA、カナマイシンB、ペニシリン、アンピシリン、ストレプトマイシンA、アンチマイシンA、クロロパンテニオール、メトロミダゾール、オキシテトラサイクリンペニシリンG、テトラサイクリンなど;抗癌剤;抗けいれん剤、例えばメフェニトイン、フェノバルビタール、トリメタジオン;鎮吐剤、例えばチエチルペラジン;抗ヒスタミン剤、例えばクロロフィナジン、ジメンヒドリネート、ジフェンヒドラミン、パーフェナジン、トリペレナミンなど;抗炎症剤、例えばホルモン剤、ヒドロコルチゾン、プレドニゾロン、プレドニゾン、非ホルモン剤、アロプリノール、アスピリン、インドメタシン、フェニルブタゾンなど;プロスタグランジン;細胞毒性剤、例えばチオテパ、クロラムブシル、シクロホスファミド、メルファラン、ナイトロジエンマスタード、メトトレキセートなど;下記のような微生物の抗原:ナイセリア・ゴノレア(Neisseria gonorrhea)、ミコバクテリウム・ツベルクローシス(Mycobacterium tuberculosis)、ヘルペスウイルス(ユモニス(humonis)、タイプ1および2)、カンジダ・アルビカンス(Candida albicans)、カンジダ・トロピカリス(Candida tropicalis)、トリコモナス・バギナリス(Trichomonas vaginalis)、ヘモフィルス・バギナリス(Haemophilus vaginalis)、グループBストレプトコッカス・エコリ(Streptococcus ecoli)、ミクロプラズマ・ホミニス(Microplasma hominis)、ヘモフィルス・ドゥクレイ(Haemophilus ducreyi)、グラヌロマ・インギナレ(Granuloma inguinale)、リンホパチア・ベネレウム(Lymphopathia venereum)、トレポネマ・パリドゥム(Treponema pallidum)、ブルセラ・アボルツス(Brucella abortus)、ブルセラ・メリテンシス(Brucellamelitensis)、ブルセラ・スイス(Brucella suis)、ブルセラ・カニス(Brucellacanis)、カンピロバクター・フェツス(Campylobacter fetus)、カンピロバクター・フェツス・インテスティナリス(Campylobacter fetus intestinalis)、レプトスピラ・ポノマ(Leptospira pomona)、リステリア・モノシトゲネス(Listeria monocytogenes)、ブルセラ・オビス(Brucella ovis)、ウマヘルペスウイルス1、ウマ動脈炎ウイルス、IBR−IBPウイルス、BVD−MBウイルス、クラミジア・プシタキ(Chlamydia psittaci)、トリコモナス・フェツス(Trichomonas foetus)、トキソプラズマ・ゴンジ(Toxoplasma gondii)、エシェリシア・コリ(Escherichia coli)、アクチノバチルス・エクリ(Actinobacillus equuli)、サルモネラ・アボルツス・オビス(Salmonella abortus ovis)、サルモネラ・アボルツス・エキ(Salmonella abortus aqui)、シュードモナス・エルジノサ(Pseudomonas aeruginosa)、コリネバクテリウム・エキ(Corynebacterium equi)、コリネバクテリウム・ピオゲネス(Corynebacterium pyogenes)、アクチノバチルス・セミニス(Actinobacillus seminis)、マイコプラズマ・ボビゲニタリウム(Mycoplasma bovigenitalium)、アスペルギルス・フミガーツス(Aspergillus fumigatus)、アブシジア・ラモサ(Absidia ramosa)、トリパノソマ・エキペルドゥム(Trypanosoma equiperdum)、バベシア・カバリ(Babesia caballi)、クロストリジウム・テタニ(Clostridiumtetani)など;上記微生物に対する抗体;酵素、例えばリボヌクレアーゼ、ノイラミニダーゼ、トリプシン、グリコーゲンホスホリラーゼ、精子乳酸デヒドロゲナーゼ、精子ヒアルロニダーゼ、アドノシントリホスファターゼ、アルカリ性ホスファターゼ、アルカリ性ホスファターゼエステラーゼ、アミノペプチダーゼ、トリプシン、キモトリプシン、アミラーゼ、ムラミダーゼ、先体プロテイナーゼ、ジエステラーゼ、グルタミン酸デヒドロゲナーゼ、コハク酸デヒドロゲナーゼ、β−グリコホスファターゼ、リパーゼ、ATPアーゼα−ペプテートγ−グルタミロトランスペプチダーゼ、ステロール−3−β−オール−デヒドロゲナーゼおよびDPN−ジ−アプロラーゼを包含する。
【0089】
組み合わせ得る更に別の巨大分子生物学的活性剤は、血液凝固因子、造血因子、サイトカイン、インターロイキン、コロニー刺激因子、成長因子、並びにそれらの類似体およびフラグメントを包含するが、それらに限定されない。
【0090】
以下の実施例は、本発明の実施に用いる材料および方法を更に説明するものである。実施例は、本発明を制限するものではない。
【実施例】
【0091】
(実施例1)
ノルエチンドロンを30%、33%および50%理論的に含有する微粒子の製造
ノルエチンドロン30%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ(Koflo)、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(インヘレント粘度(IV)=0.65dl/g)(770g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(329g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(注射用水)(27.27kg)にポリビニルアルコール[PVA−デュポン・エルバノール(DuPont Elvanol、商標)51−05](150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。冷WFI(750l)に、酢酸エチルNF(0.2μm濾過)(26.25kg)を溶解し、2〜4℃に保つ。
【0092】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子650gが得られる。
【0093】
ノルエチンドロン33%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(IV=0.62dl/g)(737g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(363g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(27.27kg)にPVA(デュポン・エルバノール51−05)(150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。WFIに溶解した3.5%酢酸エチルNF(0.2μm濾過)(750l)を2〜4℃に保つ。
【0094】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子630gが得られる。
【0095】
ノルエチンドロン50%含有微粒子1kgバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(2.2kg)およびベンジルアルコールNF(2.2kg)中のメディソーブ85:15 dl PLGA(85モル%乳酸/15モル%グリコール酸コポリマー;ポリ(ラクチド−コ−グリコリド);IV=0.62dl/g)(550g)の加熱(65〜70℃)した溶液に、ノルエチンドロンUSP(546g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(27.27kg)にPVA(デュポン・エルバノール51−05)(150g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)したベンジルアルコール(810g)および濾過(0.2μm)した酢酸エチル(1770g)を加える。溶液を65〜70℃に保つ。クエンチ液を、次のようにして調製する。冷WFI(750l)に酢酸エチルNF(0.2μm濾過)(26.25kg)を溶解し、2〜4℃に保つ。
【0096】
有機相を909cc/分、水相を4500cc/分の流速でスタティックミキサーを通して、クエンチ液にポンプ輸送する。1時間クエンチ後、材料を90および25μm篩に通す。25〜90μmフラクションを周囲温度で撹拌下に36時間減圧乾燥する。ノルエチンドロン含有微粒子685gが得られる。
【0097】
30%および50%含有粒子を、次いで、ヒヒに注射する2種の65mg(NET)製剤の調製に使用した。ヒヒ製剤1は、50%含有粒子35%、および30%含有粒子65%から成っていた。ヒヒ製剤2は、30%および50%含有粒子50%ずつから成っていた。ヒヒ製剤1および2の放出データを図4に示す。
【0098】
(実施例2)
リスペリドンを35%理論的に含有する微粒子の製造(バッチプロデックス2)
まず、1%ポリ(ビニルアルコール)[ヴィノール(Vinol)205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社(Air Products and Chemical Inc.)](906.1g)、ベンジルアルコール[ジェイ・ティ・ベイカー(J.T.Baker)、ニュージャージー州フィリプスバーグ](29.7g)、および酢酸エチル[フィッシャー・サイエンティフィク(Fisher Scientific)、ニュージャージー州フェア・ローン](65.3g)を計量し、混合することにより、水相(溶液A)を調製する。次いで、有機相(溶液B)を調製するために、酢酸エチル(108.7g)およびベンジルアルコール(108.4g)に、高粘度75:25 dl(ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナル、L.P.、オハイオ州シンシナチ)(29.3g)を溶解する。ポリマーが完全に溶解したら、リスペリドン塩基[ヤンセン・ファルマセウティカ(Janssen Pharmaceutica)、ベルギー国ベルセ](15.7g)を加え、ポリマー溶液に溶解する。溶解したリスペリドンのポリマー曝露時間は、できるだけ短く(<10分間)する。次いで、溶液AおよびBを、それぞれ198および24ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマー(Cole−Parmer)L07149−04、L07002−16]から、直径1/4インチのスタティックミキサー(コール−パーマーL−04667−14)を経て、クエンチ媒体(洗浄液)にポンプ輸送する。クエンチ液は、酢酸エチル(1276.0g)、無水炭酸水素ナトリウム(92.3g、0.02モル)、および無水炭酸ナトリウム[マリンクロット・スペシャルティ・ケミカルズ(Mallinckrodt Specialty Chemicals)、ケンタッキー州パリス](116.2g、0.02モル)を含有する注射用水(55l)から成る(11℃)。微粒子を、この第1洗浄液中で1時間45分撹拌した後、25μ篩を通して分離する。篩に残った生成物を第2のWFIの洗浄液(20l、13℃)に移す。第2洗浄液中で2時間15分撹拌後、微粒子を分離し、25および180μメッシュサイズから成るステンレススチール篩カラムに通してサイズ分別する。微粒子を一晩乾燥し、回収し、計量する。
【0099】
(実施例3)
リスペリドンを40%理論的に含有する微粒子の製造(バッチプロデックス3)
まず、1%ポリ(ビニルアルコール)[ヴィノール205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社](904.4g)、ベンジルアルコール[ジェイ・ティ・ベイカー、ニュージャージー州フィリプスバーグ](30.1g)、および酢酸エチル[フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン)(65.8g)を計量し、混合することにより、水相(溶液A)を調製する。次いで、有機相(溶液B)を調製するために、酢酸エチル(99.3g)およびベンジルアルコール(99.1g)に、高粘度75:25 dl (ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナル、L.P.、オハイオ州シンシナチ)(27.1g)を溶解する。ポリマーが完全に溶解したら、リスペリドン塩基[ヤンセン・ファルマセウティカ、ベルギー国ベルセ](18.1g)を加え、ポリマー溶液に溶解する。溶解したリスペリドンのポリマー曝露時間は、できるだけ短く(<10分間)する。次いで、溶液AおよびBを、それぞれ198および24ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマーL07149−04、L07002−16]から、直径1/4インチのスタティックミキサー(コール−パーマーL−04667−14)を経て、クエンチ媒体(洗浄液)にポンプ輸送する。クエンチ液は、酢酸エチル(1375.6g)、無水炭酸水素ナトリウム(92.4g、0.02モル)、および無水炭酸ナトリウム[マリンクロット・スペシャルティ・ケミカルズ、ケンタッキー州パリス](116.6g、0.02モル)を含有する注射用水(55l)から成る(12℃)。微粒子を、この第1洗浄液中で2時間撹拌した後、25μ篩を通して分離する。篩に残った生成物を第2のWFIの洗浄液(20l、12℃)に移す。第2洗浄液中で3時間撹拌後、微粒子を分離し、25および180μメッシュサイズから成るステンレススチール篩カラムに通してサイズ分別する。微粒子を一晩乾燥し、回収し、計量する。
【0100】
(実施例4)
バッチプロデックス2およびプロデックス3の微粒子の凍結乾燥およびγ線照射(サンプルプロデックス4A、プロデックス4Bおよびプロデックス4C)
バッチプロデックス2および3の微粒子を、次のようにして凍結乾燥した。微粒子を5cc血清バイアルに量り入れた。次いで、0.75%CMC、5%マンニトールおよび0.1%トゥイーン80から成る水性ビヒクルを、バイアルに入れた。微粒子を撹拌によりビヒクルに懸濁し、ドライアイス/アセトン浴内で急速に凍結した。バイアルを、次いで、実験室用凍結乾燥器[デュラ・ストップ・マイクロプロセッサー・コントロール(Dura Stop Microprocessor Control)、ニューヨーク州ストーン・リッジのFTSシステムズ社]内で、勾配30℃最大温度サイクルを用いて50時間凍結乾燥した。サンプルプロデックス4Aおよび4Cはそれぞれ、プロデックス2および3の凍結乾燥サンプルであった。サンプルプロデックス4Bは、プロデックス2を60Co源から2.2Mラドγ線照射によって滅菌し、凍結乾燥したものであった。
【0101】
インビトロ溶解試験
プロデックス2、3、4A、4Bおよび4Cに対し、インビトロ溶解試験を行った。リアルタイムおよび促進法を用いた。装置は、分光光度計およびデータステーションに接したハンソン(Hanson)リサーチ9−セルUSPパドル(方法II)溶解装置から成っていた。受容媒体は、各セルから分光光度計内のフローセルに連続的に循環した。受容媒体の吸光度を236nmでモニターして、リスペリドンを定量した。
【0102】
リアルタイムモデルでは、37℃の50mMトリス緩衝液(pH7.4)から成る受容媒体への、微粒子からの放出速度を測定した。リスペリドンは、前記受容媒体を用いるインビトロ実験を行うための充分な溶解度(≧0.5mg/ml)を有することがわかった。溶解条件を無限にするために、リスペリドンの量は、飽和の20%未満に保った。データを図5および図6に示す。
【0103】
促進モデルも行った。受容媒体として、WFI中の27.5重量%エタノールを使用した。結果を図7に示す。
動物への投与および血液サンプリング
乾燥微粒子(プロデックス2、3)および凍結乾燥形態(プロデックス4A、4B、4C)の生成物に対し、イヌにおいてインビボ試験を行った。乾燥微粒子を注射器に入れ、注射器内で、2.5重量%カルボキシメチルセルロース(CMC)から成る注射ビヒクルに再懸濁した。凍結乾燥サンプル(プロデックス4A、4B、4C)は、注射前にWFI中で再構成した。
【0104】
雄および雌のイヌ(体重11.6±2.3kg)を、3匹ずつの群に分けた。イヌを3匹の群として、標準的な実験室条件で飼育および餌を与えた。
【0105】
イヌの左後肢の腿の位置の二頭股筋に、各デポ製剤の適量を、リスペリドン約2.5mg/kgの用量で筋肉内投与した。
【0106】
アポモルモネ催吐試験において、製剤投与の0(投与前)、1、5および24時間後、並びに4、7、11、14、18、23、25、28、32、35、39、42、46、49、53および56日目に、一つの頸静脈から血液サンプル(EDTA上5ml)を採った。アポモルモネ試験は、ヤンセン(P.A.J.Janssen)およびニーメゲルス(C.J.E.Niemegeers)、アルツナイミッテルフォルシュング(Arzneim.−Forsch.)(Drug Res.)、9:765−767(1959)に記載されている。試験中に一つの群の3匹のイヌのいずれにも、アポモルモネ催吐に対する保護が見られなくなったら、血液サンプリングは中断した。血液サンプルは、3000rpmで10分間遠心し、血漿を分離した。血漿サンプルを、分析するまで≦20℃で貯蔵した。
【0107】
血漿サンプルのリスペリドン(RISP)および9−ヒドロキシリスペリドン(9−OH RISP)を、ラジオイムノアッセイ(RIA)によって分析した。RIAで分析する血漿サンプルに、2つの異なるRIA法を用いた。一つは未変化リスペリドンに、もう一つは活性物(リスペリドンと9−ヒドロキシ−リスペリドンの合計;「活性剤」と混同しないよう注意されたい)に対するものである。後者の血漿サンプルでは、9−ヒドロキシ−リスペリドン濃度を、活性物濃度とリスペリドン濃度との差として計算した。RIA法の定量限界は、リスペリドン0.20ng/ml、活性物0.50ng/mlであった。
【0108】
各製剤に対して、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の平均(±S.D、n=3)血漿濃度を計算した。9−ヒドロキシ−リスペリドン血漿濃度とリスペリドン血漿濃度との比を、可能なら計算した。データの目視により、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の血漿濃度ピークおよびピーク時間を求めた。リスペリドンおよび9−ヒドロキシ−リスペリドンのAUC(「曲線下面積」)値を、0時間および時間tの間で、台形則により計算した。時間tは、3匹中少なくとも1匹のイヌにおいて、リスペリドンまたは9−ヒドロキシ−リスペリドンの濃度が定量限界を上回った最終時点である。同じ製剤群に属するイヌには、AUCは同じ最終時間tまで計算した(濃度が定量限界よりも低かったものには、定量限界の値を用いた)。2濃度連続して定量限界を下回ったら、先のサンプリング点の濃度を定量限界とし、後のサンプリング点の濃度を0とた。AUCは、無限に外挿しなかった。活性物のAUCは、リスペリドンと9−ヒドロキシ−リスペリドンとのAUCの合計として計算した。
【0109】
製剤プロデックス2/3/4A/4B/4Cについて、リスペリドン、9−ヒドロキシ−リスペリドン、および活性物の平均もしくは中央血漿濃度および/または薬物動態パラメータを第1表に示す。製剤プロデックス2/3/4A/4B/4Cについて、平均血漿濃度−時間曲線を、図8に示す。
【0110】
各製剤群について、最初にリスペリドン、次いで9−ヒドロキシ−リスペリドン、最後に活性物に関して結果を考察する。活性物については、血漿濃度とアポモルモネ催吐試験における制吐作用とを関連付ける。
【0111】
製剤プロデックス2〜4Cの投与後、リスペリドンの平均血漿レベルのピークは低かった。非常に異なった時点でピークに達した。異なる製剤からの更なるリスペリドン放出が徐々に起こり、長時間続いた。その結果、リスペリドンおよびその代謝産物のいずれの血漿濃度も低かった。9−ヒドロキシ−リスペリドンの平均ピーク時間はいずれも、26〜30日目の間であった。活性物の血漿濃度−時間曲線は、製剤プロデックス2〜4Cのいずれでも同様であった。試験開始後1または2日以内に、活性物の血漿濃度は、リスペリドンの急速な初期放出の故にピークに達した。そのピークの後、5〜8日目に、濃度は下降線をたどった。8日目から、濃度は再び20日目まで上昇し、20日目から平均15日間は、概ね一定のレベルにあった。この間、各製剤について、活性物濃度は第2のピークを示し、濃度は第1のピークよりも高かった。製剤プロデックス2、4Aおよび4Bの制吐作用は35〜42日間持続した。製剤プロデックス4Cでは制吐作用は49日間持続し、いずれのイヌにおいても中断が無かった。製剤プロデックス4Cの最長作用は、活性物の最高Cmax、TmaxおよびAUC0−tと対応していた(同じ群の他の4製剤との比較)。
【0112】
イヌにおけるアポモルモネ催吐試験において、微粒子リスペリドン製剤の作用時間をも調べた。神経遮断剤が、第四脳室の最後野においてドーパミンD2レセプターを遮断することにより、アポモルモネ催吐に拮抗した。この試験は通例、ヒトにおける神経遮断剤の抗精神病薬作用の開始および作用時間を予測するために用いられる[ヤンセンら、アルツナイミッテルフォルシュング、10:1196−1206(1965);ニーメゲルスら、ライフ・サイエンシーズ(Lefe Sci.)、24:2201−2216(1979)]。9−OH−リスペリドンは、実質的に親化合物と同じ薬理学的性質を有する。親化合物と活性代謝産物とが、リスペリドンの生物学的性質を決定する「活性物」を構成する。
【0113】
試験期間中、アポモルモネを週2回、イヌに0.31mg/kgの量で皮下投与した。アポモルモネ投与後1時間の間、イヌに嘔吐が見られた。アポモルモネ投与後1時間の間に嘔吐の全く無いことが、明らかな制吐活性の反映と考えられる。制吐作用時間は、3匹中2匹のイヌに制吐が見られる時間の長さと定義した。
【0114】
一方の後肢の腿の位置の二頭股筋に、製剤0.5mlを注射した。この筋肉注射から種々の時間後に血液サンプルを採り、その後すぐにイヌにアポモルモネを投与した。アポモルモネ投与1時間以内に嘔吐(対照動物には決して見られない;n>1000)が全く無いことが、明らかな制吐作用の反映と考えられる。
【0115】
第2表には、イヌにデポ製剤を筋肉注射してから種々の時間後に、アポモルモネ催吐に対する保護が見られた(+)か、見られなかった(−)かを示す。いずれの製剤も、短時間で制吐作用を開始した。
【0116】
【表1−1】
【0117】
【表1−2】
【0118】
【表1−3】
【0119】
【表1−4】
【0120】
【表2−1】
【0121】
【表2−2】
【0122】
(実施例5)
エストラジオールベンゾエートを20%および30%理論的に含有する微粒子の製造
第3表には、実験的に製造した20%および30%含有微粒子の粒子サイズ分布を示す。粒子サイズ分布は、図9にも示す。図9は、20%含有微粒子の一つのバッチの微粒子サイズの累積%「より大きい体積%」)を示す。図10は、20%含有微粒子の一つのバッチの粒子サイズによる%微分(「体積微分」)を示す。このデータから、流速が増すと、粒子サイズが小さくなることがわかる。12および24要素ミキサーの間には、顕著な差は無かった。水相:有機相比が高いほど、粒子サイズ分布は狭くなる。遷移流速範囲、レイノルズ数(Re)2000〜4000で、良好な微粒子が得られた。
【0123】
次いで、20%および30%含有微粒子の更なるバッチを製造した。薬物20%含有微粒子7kgバッチの製造に、直径1/2"×24要素スタティックミキサー(コール−パーマー・ポリプロピレン・ディスポーザブル04667;イリノイ州シカゴのコール−パーマー・インストゥルメント社)を使用した。有機相は、4.0%エストラジオールベンゾエート(活性剤)、16.0%85:15dl PLGA(85モル%乳酸/15%グリコール酸コポリマー;ポリラクチド−コ−グリコリド)、および80%酢酸エチルから成っていた(60〜70℃)。水相は、1.0%ポリビニルアルコール、5.0%酢酸エチル、および94.0%水から成っていた(60〜70℃)。有機相流速も水相流速も、1100ml/分であった。有機相および水相のいずれにも、7001−80ヘッドを有するコール−パーマー6231−26ポンプを使用した。得られた粒子サイズ分布は、15%<25μm、14%25〜45μm、56%45〜90μm、12%90〜150μm、および3%>150μmであった。
【0124】
薬物30%含有微粒子5kgバッチの製造に、直径1/2"×24要素スタティックミキサー並びに直径3/8"×11、12および24要素スタティックミキサー(コール−パーマー・ディスポーザブル)を使用した。有機相は、4.3%エストラジオールベンゾエート(活性剤)、10.0%85:15dl PLGA、および85.7%酢酸エチルから成っていた(60〜70℃)。水相は、20%含有微粒子の場合と同じものを使用した。有機相流速は880ml/分、水相流速は1650ml/分であった。得られた粒子サイズ分布は、44%<25μm、31%25〜45μm、および25%>45μmであった。
【0125】
エストラジオールベンゾエート含有微粒子を、従来の注射器に量り入れ、若いホルスタイン雄子牛に、活性薬物40mg/動物の用量で投与した。微粒子を水性カルボキシメチルセルロースビヒクルに懸濁し、耳の基部に注射した。血清を採り、エストラジオールレベルをラジオイムノアッセイによって測定した;結果を図11に示す。
【0126】
【表3】
【0127】
(実施例6)
トレンボロンアセテートを40%理論的に含有する微粒子の製造
トレンボロンアセテート(TBA)微粒子9バッチを、スタティックミキサーを用いて製造した。TBA40%およびブチル化ヒドロキシトルエン(BHT、抗酸化剤として使用)5%を含有する微粒子を、次のような方法で製造した。PLGA(D,L−ラクチド:グリコリドをモル比85:15;GPCによる分子量Mw=86810;Mn=36417)、TBA、およびBHTを、55℃で酢酸エチル(EtOAc)に溶解した。(EtOAc:PLGA比10〜20:1。)EtOAcを55℃に加熱し、高速攪拌下にEtOAcにPLGAを加えた。ポリマーの溶解後、ポリマー溶液に、BHT、および次いでTBAを溶解した。同時に、EtOAcを加えたポリ(ビニルアルコール)(PVA)水溶液(3重量%)を、ジャケット付フラスコに入れ、55℃に加熱した(PVA溶液:EtOAc比10:1)。ポリマー/薬物溶液(有機相)およびPVA溶液(水相)の温度が一定になったら、ポリマー/薬物溶液およびPVA溶液を別々にスタティックミキサー(直径1cm、長さ12cm)にポンプ輸送した。スタティックミキサーは、コール−パーマー製のモデル番号6−04667−06で、混合要素を12個有していた。ポリマー/薬物溶液とPVA溶液の流速比は、1:1.2ないし1:2の間で変化した。クエンチ水とEtOAcとの比は、100〜150:1であった。クエンチ水温度は約1℃であった。6時間洗浄後、微粒子を篩カラム(25μm、90μm、150μm、および212μm)で篩過した後、乾燥した。
【0128】
一つのバッチの微粒子の粒子サイズ分布を図12に示す。
【0129】
(実施例7)
テストステロンを46%理論的に含有する微粒子の製造
テストステロン46%含有微粒子1kgのバッチの製造に、直径3/4"×12要素のスタティックミキサー[コフロ、M/N:3/4−TU−3−12RH−11;イリノイ州キャリーのコフロ社]を使用する。ポリマー/薬物溶液(有機相)を、次のようにして調製する。酢酸エチルNF(3.683kg)中のメディソーブ75:25dl PLGA(乳酸75モル%/グリコール酸25モル%コポリマー;ポリラクチド−コ−グリコリド; IV=0.68dl/g)(594g)の加熱(65〜70℃)した溶液に、テストステロンUSP(506g)を溶解する。溶液を濾過(0.2μm)し、65〜70℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(9.75kg)にPVA(デュポン・エルバノール51−05)(52g)を加え、溶解するまで加熱(65〜70℃)し、濾過(0.2μm)する。この溶液に、濾過(0.2μm)した酢酸エチル(626g)を加える。溶液を65〜70℃に保つ。クエンチ液はWFI(750l)から成り、0〜4℃に保つ。
【0130】
有機相を2150cc/分、水相を4300cc/分の流速でスタティックミキサーを通してポンプ輸送する。1時間クエンチ後、材料を45および150μm篩に通す。45〜150μmクラクションを周囲温度で攪拌下に36時間減圧乾燥する。テストステロン含有微粒子875gが得られる。
【0131】
46%含有微粒子を用いて125mgテストステロン製剤(懸濁液充填および凍結乾燥)を調製し、ヒヒに投与した。時間放出データを図13に示す。
【0132】
(実施例8)
rgp120を1.7%理論的に含有する微粒子の製造
rgp120(糖タンパク質)1.7%含有微粒子10gバッチを製造するために、直径1/4"×5要素のスタティックミキサー[コッホ(Koch)、1/4"SMV−DY;直径1/4"、5要素、316SS、カンザス州ウィチタのコッホ・エンジニアリング社(Koch Engineering Company, Inc.)]を使用する。ポリマー/薬物溶液(有機相)は、次のようにして調製する。酢酸エチル(47g)中のメディソーブ65:35dl PLGA(乳酸65モル%/グリコール酸35%コポリマー;ポリラクチド−コ−グリコリド)(IV=0.61)(10g)に、トリス緩衝液(20mMトリス、120mM NaCl、pH7.4)中の56mg/mlのrgp120(3cc)を加えることにより、第1のエマルジョンを調製する。このエマルジョンは、超音波処理[ビブラ(Vibra)セル・ソニケーター、600W、20秒、50%パワー、1/2"プローブ]により形成する。エマルジョンを0〜4℃に保つ。加工水溶液(水相)を、次のようにして調製する。WFI(2025g)に、PVA(エア・プロダクツ、ヴィノール205)(225g)を加え、溶解するまで加熱(65〜70℃)する。この溶液に、酢酸エチル(250g)を加える。溶液を0〜4℃に保つ。クエンチ液は水(12l)から成り、0〜4℃に保つ。
【0133】
有機相を40cc/分、水相を1500cc/分の流速でスタティックミキサーを通してポンプ輸送する。1時間クエンチ後、材料を20および150μm篩に通す。20〜150μmフラクションを0〜4℃で0.1%トゥイーン20溶液(13l)で洗い、凍結乾燥する。rgp120含有微粒子3.09gが得られる。
【0134】
(実施例9)
イベルメクチンを60%理論的に含有する微粒子の製造
イベルメクチンを60%理論的に含有する微粒子35gバッチを製造するために、65:35dl(ポリラクチド−コ−グリコリド)(メディソーブ・テクノロジーズ・インターナショナルL.P.、オハイオ州シンシナチ)9.5%および酢酸エチル(フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン)90.5%から成る52℃のポリマー溶液(116.72g)に、イベルメクチン(21.03g)を溶解することにより、分散相(溶液A)を調製する。次いで、ポリ(ビニルアルコール)(ヴィノール205、ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社)(1%)およびWFI(99%)から成る連続相(溶液B)(1300g)を計量し、52℃に加熱する。次いで、溶液AおよびBを、それぞれ88および165ml/分の流速で、ギアドライブポンプおよびヘッド[コール−パーマーL07149−04、L07002−16]から、スタティックミキサー(コール−パーマーL04667−14)を経て、クエンチ液にポンプ輸送する。クエンチ液は、CWFI(冷注射用水)(35l)から成る(8℃)。微粒子を、約20分間攪拌した後、25μ篩を通して分離する。篩に残った生成物をCWFIの洗浄液(20l、8℃)に移す。2時間攪拌後、洗浄液を25および212μ篩に通して、微粒子を分離し、サイズ分別する。微粒子を一晩風乾し、回収する。
【0135】
(実施例10)
ブピバカイン塩基を60%理論的に含有する微粒子の製造
5%ポリ(ビニルアルコール)水溶液[ヴィノール205;ペンシルベニア州アレンタウンのエア・プロダクツ・アンド・ケミカル社](3780.3g)に、酢酸エチル[フィッシャー・サイエンティフィク、ニュージャージー州フェア・ローン](216.1g)を加え、周囲温度で1N−NaOHでpH8.5に調節することにより、過剰の連続水相を調製する。次いで、有機相を調製するために、酢酸エチル(92.1g)に低粘度50:50dlポリラクチド−コ−グリコリド(乳酸50モル%/グリコール酸50モル%コポリマー)(メディソーブ・テクノロジーズ・インターナショナル、L.P.)(8.05g)およびブピバカイン塩基[アセト社(Aceto Corporation)、ニューヨーク州レーク・サクセス](12.05g)を室温で溶解する。次いで水溶液および有機溶液を、それぞれ233および116ml/分の流速で、ギアドライブポンプおよびヘッド(ポンプ:コール−パーマーL07144−05; 水溶液用ヘッド:L7002−16;有機溶液用ヘッド:L07002−26)から、24要素、直径1/4"のスタティックミキサー(コール−パーマーL04667)を経て、pH8.5に調節した注射用水(12l)(室温)から成るクエンチ液に、同時にポンプ輸送する。微粒子をクエンチ液中で1時間攪拌後、25、45および90μmメッシュステンレンスチール篩で篩過する。微粒子を一晩乾燥し、計量まで遮光する。
【0136】
(実施例11)
ブタアルブミンを17.5%理論的に含有する微粒子の製造
ブタアルブミン含有微粒子2gバッチを製造するために、直径1/2"×長さ12"のスタティックミキサーを使用した。ポリマー/薬物溶液(有機相)は、次のようにして調製した。酢酸エチル(70g)に、メディソーブ75:25dl PLGA(インヘレント粘度0.65dl/g)(1.65g)を溶解した。有機相中のポリマー濃度は、2.36%であった。粉砕したブタアルブミン[シグマ(Sigma)、ロット番号95F−9538](0.35g)を、2×10秒間の超音波処理[テクマー・ソニケーション・ディスラプター(Tekmar Sonication Disruptor)、モデル350、マイクロチップ付]によってポリマー溶液に懸濁した。均一で細かい分散液が得られた。懸濁液を、底部に排出バルブを有する50ml反応器に入れた。タービン6翼プロペラにより、約700rpmで混合を開始した。蠕動ポンプを反応器の排出口に連結し、有機相を7.0g/分の速度でスタティックミキサーにポンプ輸送した。350csシリコーン油[ダウ・コーニング(Dow Corning)、ロット番号HH121209]を9.0g/分の速度でスタティックミキサーの導入口に供給するために、もう一つの蠕動ポンプを設置した。
【0137】
まずシリコーン油流を流し、約2〜5秒後に有機相を流した。クエンチタンク内では、ヘプタン[ケム・ピュア(Chem Pure)、ロット番号M138KLAP](1.2l)を、空気作動インペラーで穏やかに攪拌した。スタティックミキサーの排出ラインは、ヘプタン表面から1/2"下に配置した。有機相をスタティックミキサーから移し終った後、残留有機相をミキサーから流し出すために、酢酸エチル(8〜10g)を用いた。この流し出しの間にも、シリコーン油を流し続けた。半固形微粒子をヘプタン中で室温で1.5時間クエンチして、硬化および洗浄を完了した。次いで、バーサポル(Versapor)−3000メンブランフィルター(孔サイズ3μ)[ゲルマン(Gelman)]を用いて、微粒子を減圧濾取した。回収した微粒子を、フィルター上で新たなヘプタン(300ml)で洗い、更に乾燥するために減圧デシケーターに入れた。ブタアルブミン含有微粒子1.91gが得られた。微粒子は概ね球形で、サイズ範囲25〜200μであった。
【0138】
第2の製剤を調製して、より短いスタティックミキサー(直径1/2"×長さ6")を、より低い総流量で使用し得ることを示した。スタティックミキサー内への有機相流量は、第1の製剤の場合7.0g/分であったが、6.5g/分とした。シリコーン油流量は、第1の製剤の場合9.0g/分であったが、8.8g/分とした。しかし、有機相:シリコーン油の流速比は、第2の製剤の場合も、第1の製剤の場合とほぼ同じであった。この製剤の固体収率は90%であった。
【0139】
(実施例12)
rBoインターフェロン−αを10.0%理論的に含有する微粒子の製造
rBoインターフェロン−α(組換ウシインターフェロンα)含有微粒子1.89g、バッチを製造するために、1/2"×12"スタティックミキサーを使用した。有機相中のポリマー(酢酸エチルに溶解した75:25dl PLGA)濃度は、2.70%であった。注射用水(0.5ml)にタンパク質を溶解し、ポリマー溶液中で超音波処理により乳化して、有機相(ポリマー/活性剤相)を形成した。エマルジョンを反応器に入れ、実施例11と同様に小球形成を行った。有機相流速は6.6g/分、シリコーン油流速は10.9g/分であった。半固形微粒子を、ヘプタン(1.0l)中で1.5時間クエンチングした。
【0140】
(実施例13)
HSAを17.5%理論的に含有する微粒子の製造
HSA(ヒト血清アルブミン)含有微粒子のバッチを製造するために、1/2"×6"スタティックミキサーを使用した。有機相中のポリマー(酢酸エチルに溶解した75:25dl PLGA)濃度は、2.36%であった。水の内相(1.0ml)を用いて、タンパク質を完全に溶解した。溶解したタンパク質を、ポリマー溶液中で超音波処理により乳化して、有機相を形成した。6"のより短い流路内で非溶媒(シリコーン油)によるコアセルベーションを誘導するのに必要な滞留時間を保つために、より低い総流速を採用した。有機相流速は4.9g/分、シリコーン油流速は5.6g/分であった。より低い総流速の結果、このサンプルで65%の固体収率が得られた。このサンプルでは、シリコーン油流速と有機相流速との比は、実施例12よりも少し小さかった(1.4に対し、1.1)。半固形微粒子を、ヘプタン(0.8l)中で1.5時間クエンチングした。非溶媒(シリコーン油)をより少なく使用したので、ヘプタン(第2の非溶媒)の必要量もより少なかった。
【0141】
(実施例14)
ブタアルブミンを10.0%理論的に含有する微粒子の製造
ブタアルブミンをモデルタンパク質(封入する活性剤)として用いて、実施例12と同様に製剤を製造した。スタティックミキサー内の2相の総流速は、このサンプルではより低かった。有機相流速は6.0g/分、シリコーン油流速は8.8g/分であった。微粒子をヘプタン(1.2l)中で1.5時間クエンチングした。この製剤でも、実施例13と同様の総固体収率が達成された。
実施例11〜14の製剤の性質を第4表にまとめる。
【0142】
【表4】
【0143】
(実施例15)
酢酸エチルおよびベンジルアルコールの50:50(重量)混合物(80g)に、85:15D,L−ラクチド/グリコール酸コポリマー(10.6g)およびノルエチンドロンUSP(9.4g)を順次溶解した(「油相」)。溶解したら、溶液を、ポリ(ビニルアルコール)(ヴィノール205、エア・プロダクツ;数平均分子量15000〜27000、加水分解度87〜89%)0.5重量%、酢酸エチル5.9重量%、ベンジルアルコール2.7重量%、および水90.9重量%から成る60〜65℃のエマルジョン浴混合物(500g)の入った、タービン攪拌機およびサーモスタットヒーターを取り付けたジャケット付1000mlビーカーに入れた。このエマルジョン浴混合物は、60℃で酢酸エチルおよびベンジルアルコールの両方の飽和溶液に近いものである。すなわち、エマルジョン形成中、「油相」からの溶媒抽出が防止でき、この工程における時間効果を最少にし得る。油滴サイズが約90μmとなるよう、攪拌速度を調節した。得られたエマルジョンを、図14および図15に示すように水および酢酸エチルを種々の量で含有する冷却(2〜4℃)水タンクに移した。1時間後、微粒子を篩スタック(25、45、63および90μm)上に回収し、フード内で一晩乾燥した。翌日、微粒子を混合し(25〜45μm15%、45〜63μm50%、63〜90μm35%)、サンプリングした。結果を図14および図15に示す。
【0144】
(実施例16)
実施例15を繰り返した。ただし、NETおよびポリマーの「油相」溶液の量をいずれの場合も5gとし、エマルジョン浴は、実施例15において使用したポリ(ビニルアルコール)を0.5重量%含有する水300mlであった。結果を第5表に示す。
【0145】
【表5】
【0146】
(実施例17)
テストステロン含有微粒子の20gバッチを、次のようにして製造した。酢酸エチルおよびベンジルアルコールの75:25混合物(67g)に、実施例15のポリマー(10.8g)およびテストステロン(9.2g)を溶解し、約65℃に加熱した。次いで、溶液を、ポリ(ビニルアルコール)0.5%、および酢酸エチル6.5%の水性混合物(500g)の入った、タービン攪拌機を取り付けたジャケット付1000mlガラス製反応器に入れた。攪拌速度を約245rpmに調節した。5分後、エマルジョンを、酢酸エチルを5%の濃度で含有する水(20l)の入った冷却(0〜4℃)タンクに移した。1時間後、微粒子を篩スタック(25、および150μm)上に回収し、実験室フード内で一晩乾燥した。翌日、25μ篩上の微粒子を回収し、サンプリングした。生成物は、テストステロン39.7%、酢酸エチル3.67%およびベンジルアルコール0.89%を含有していた。促進インビトロ放出モデルによると、18時間後に15%の薬物が受容液体に放出された。
【0147】
本発明の種々の態様を説明したが、それらは説明のためのもので、制限するものではないと理解すべきである。すなわち、本発明の範囲は、上記例示態様ではなく、以下の特許請求の範囲によってのみ制限すべきである。
【符号の説明】
【0148】
1 液流
10 スタティックミキサー
12 パイプ
14 静的混合要素
30 油相または有機相
32 攪拌ポット
40 水相
50 Y字形連結具
60 洗浄タンク
【特許請求の範囲】
【請求項1】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は、生分解性ポリマー封入結合剤、生物学的活性剤および水溶性の低い相互に混和性の有機溶媒少なくとも2種の混合物を含有する溶液であって、ハロゲン化炭化水素を含有せず、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【請求項2】
生分解性ポリマー封入結合剤は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、アルブミン、カゼインおよび蝋から成る群から選択する請求項1に記載の方法。
【請求項3】
有機溶媒の一種はエステルである請求項1に記載の方法。
【請求項4】
エステルは、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基から成る群から選択する。]で示される請求項3に記載の方法。
【請求項5】
エステルは酢酸エチルである請求項4に記載の方法。
【請求項6】
有機溶媒の一種はアルコールである請求項1に記載の方法。
【請求項7】
アルコールは、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示される請求項6に記載の方法。
【請求項8】
アルコールはベンジルアルコールである請求項7に記載の方法。
【請求項9】
有機溶媒の一種はケトンである請求項1に記載の方法。
【請求項10】
ケトンは式R4COR5[式中、R4はC1−C4アルキル基から成る群から選択し、R5はC2−C4アルキル基から成る群から選択する。]で示される請求項9に記載の方法。
【請求項11】
ケトンはメチルエチルケトンである請求項10に記載の方法。
【請求項12】
溶媒混合物は酢酸エチルおよびベンジルアルコールから成る請求項1に記載の方法。
【請求項13】
第2相は親水性コロイドを含有する請求項1に記載の方法。
【請求項14】
親水性コロイドはポリ(ビニルアルコール)である請求項13に記載の方法。
【請求項15】
活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択する請求項1に記載の方法。
【請求項16】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は生分解性ポリマー封入結合剤、生物学的活性剤および溶媒混合物を含む溶液であり、
(1) 生分解性ポリマー封入結合剤は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、アルブミン、カゼインおよび蝋から成る群から選択し、および
(2)溶媒混合物は、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基から成る群から選択する。]で示されるエステルと、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示されるアルコールとから成り、ハロゲン化炭化水素を含有せず、水溶性が低く、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【請求項17】
エステルは酢酸エチルである請求項16に記載の方法。
【請求項18】
アルコールはベンジルアルコールである請求項16または17に記載の方法。
【請求項19】
第2相は親水性コロイドを含有する請求項16に記載の方法。
【請求項20】
親水性コロイドはポリ(ビニルアルコール)である請求項19に記載の方法。
【請求項21】
生物学的活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択する請求項16に記載の方法。
【請求項22】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は、生分解性ポリマー封入結合剤、生物学的活性剤及び溶媒混合物を含有する溶液であり、
(1)生分解性ポリマーを封入結合剤は、ポリ(グリコール酸)、ポリD,L−乳酸、ポリ−L−乳酸、それらのコポリマーから成る群から選択し、
(2)生物学的活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択し、
(3)溶媒混合物は、酢酸エチルおよびベンジルアルコールから成り、ハロゲン化炭化水素を含有せず、
B.水に溶解したポリビニルアルコールから成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【請求項1】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は、生分解性ポリマー封入結合剤、生物学的活性剤および水溶性の低い相互に混和性の有機溶媒少なくとも2種の混合物を含有する溶液であって、ハロゲン化炭化水素を含有せず、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【請求項2】
生分解性ポリマー封入結合剤は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、アルブミン、カゼインおよび蝋から成る群から選択する請求項1に記載の方法。
【請求項3】
有機溶媒の一種はエステルである請求項1に記載の方法。
【請求項4】
エステルは、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基から成る群から選択する。]で示される請求項3に記載の方法。
【請求項5】
エステルは酢酸エチルである請求項4に記載の方法。
【請求項6】
有機溶媒の一種はアルコールである請求項1に記載の方法。
【請求項7】
アルコールは、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示される請求項6に記載の方法。
【請求項8】
アルコールはベンジルアルコールである請求項7に記載の方法。
【請求項9】
有機溶媒の一種はケトンである請求項1に記載の方法。
【請求項10】
ケトンは式R4COR5[式中、R4はC1−C4アルキル基から成る群から選択し、R5はC2−C4アルキル基から成る群から選択する。]で示される請求項9に記載の方法。
【請求項11】
ケトンはメチルエチルケトンである請求項10に記載の方法。
【請求項12】
溶媒混合物は酢酸エチルおよびベンジルアルコールから成る請求項1に記載の方法。
【請求項13】
第2相は親水性コロイドを含有する請求項1に記載の方法。
【請求項14】
親水性コロイドはポリ(ビニルアルコール)である請求項13に記載の方法。
【請求項15】
活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択する請求項1に記載の方法。
【請求項16】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は生分解性ポリマー封入結合剤、生物学的活性剤および溶媒混合物を含む溶液であり、
(1) 生分解性ポリマー封入結合剤は、ポリ(グリコール酸)、ポリ−D,L−乳酸、ポリ−L−乳酸、それらのコポリマー、ポリ(脂肪族カルボン酸)、コポリオキサレート、ポリカプロラクトン、ポリジオキソネン、ポリ(オルトカーボネート)、ポリ(アセタール)、ポリ(乳酸−カプロラクトン)、ポリオルトエステル、ポリ(グリコール酸−カプロラクトン)、ポリ無水物、ポリホスファジン、アルブミン、カゼインおよび蝋から成る群から選択し、および
(2)溶媒混合物は、式R1COOR2[式中、R1およびR2はそれぞれ、C1−C4アルキル基から成る群から選択する。]で示されるエステルと、式R3CH2OH[式中、R3は、水素、C1−C3アルキルおよびC6−C10アリールから成る群から選択する。]で示されるアルコールとから成り、ハロゲン化炭化水素を含有せず、水溶性が低く、
B.(1)親水性コロイドまたは
(2)界面活性剤
の水溶液から成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【請求項17】
エステルは酢酸エチルである請求項16に記載の方法。
【請求項18】
アルコールはベンジルアルコールである請求項16または17に記載の方法。
【請求項19】
第2相は親水性コロイドを含有する請求項16に記載の方法。
【請求項20】
親水性コロイドはポリ(ビニルアルコール)である請求項19に記載の方法。
【請求項21】
生物学的活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択する請求項16に記載の方法。
【請求項22】
生分解性微粒子の製法であって、
A.第1相を調製し、第1相は、生分解性ポリマー封入結合剤、生物学的活性剤及び溶媒混合物を含有する溶液であり、
(1)生分解性ポリマーを封入結合剤は、ポリ(グリコール酸)、ポリD,L−乳酸、ポリ−L−乳酸、それらのコポリマーから成る群から選択し、
(2)生物学的活性剤は、リスペリドン、ノルエチンドロンおよびテストステロンから成る群から選択し、
(3)溶媒混合物は、酢酸エチルおよびベンジルアルコールから成り、ハロゲン化炭化水素を含有せず、
B.水に溶解したポリビニルアルコールから成る第2相を調製し、
C.第1相と第2相とをスタティックミキサー内で組み合わせて、第1相が不連続相となり、第2相が連続相となったエマルジョンを形成し、
D.不連続第1相を微粒子の形態で分離し、
E.クエンチ液を用いて、工程Dの微粒子から残留溶媒を抽出する
ことを含んで成る方法。
【図1】

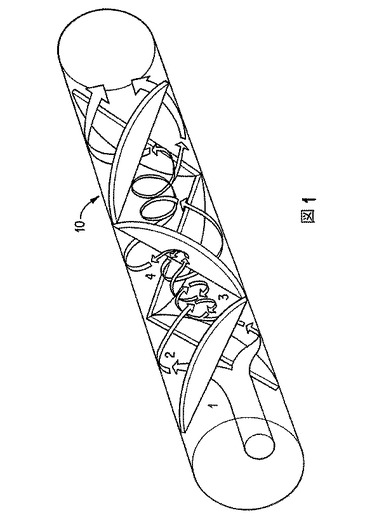
【図2】
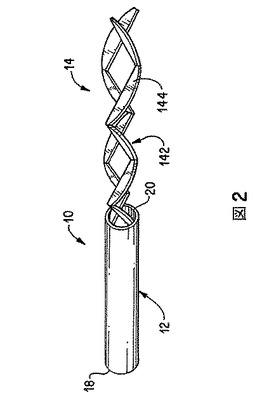
【図3】
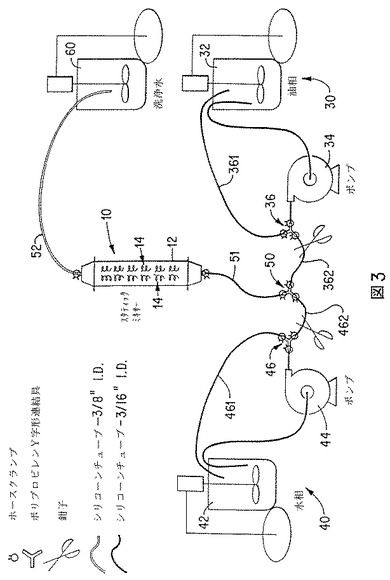
【図4】
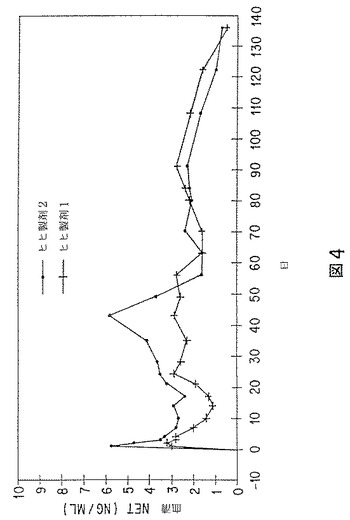
【図5】
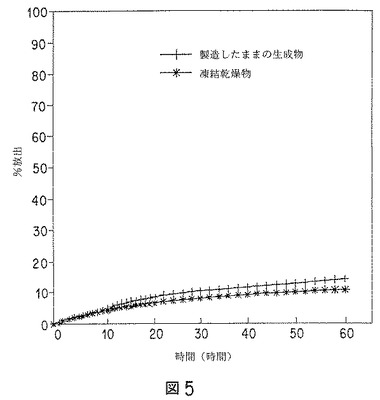
【図6】
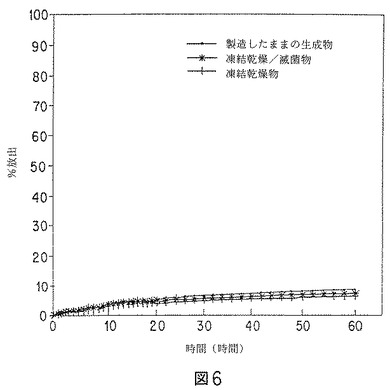
【図7】
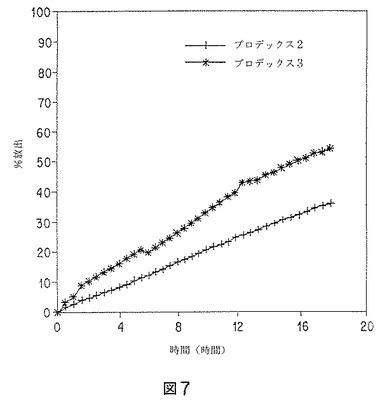
【図8】
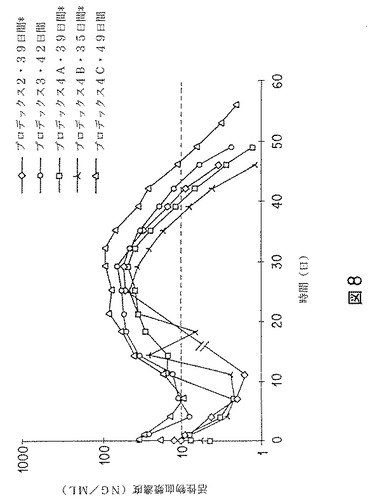
【図9】
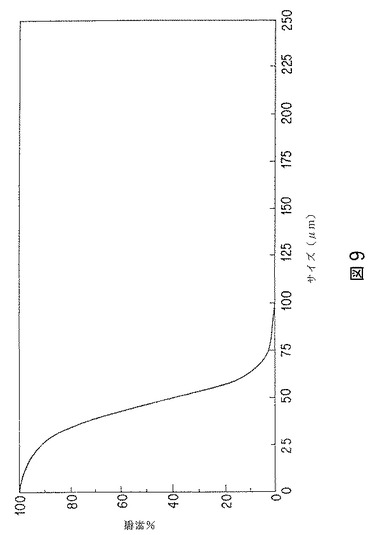
【図10】
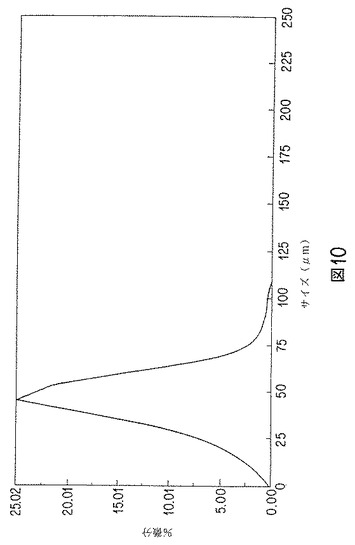
【図11】
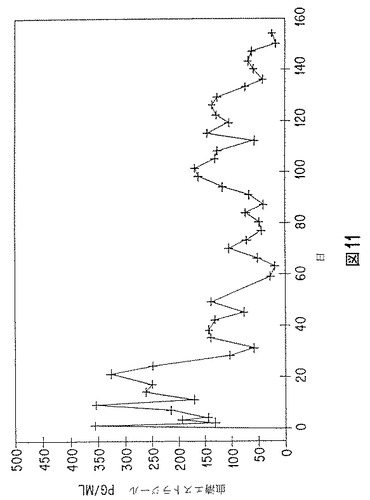
【図12】
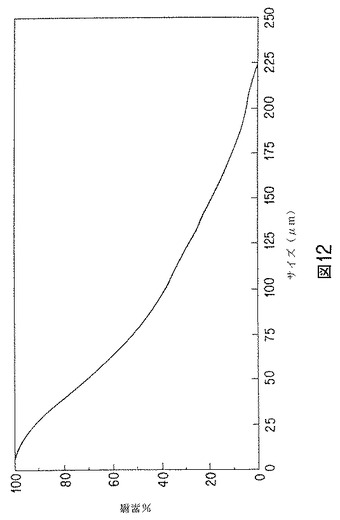
【図13】
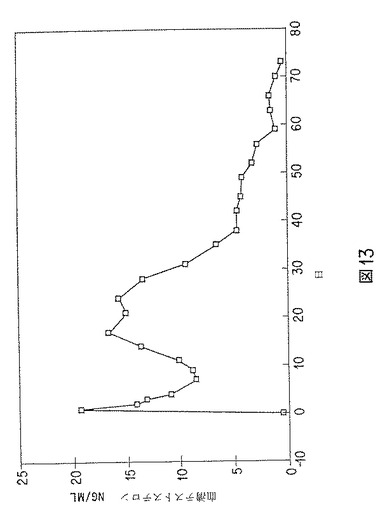
【図14A】
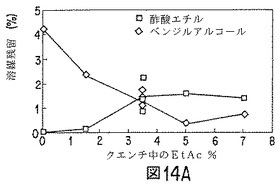
【図14B】
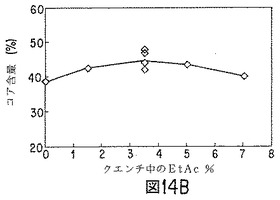
【図14C】
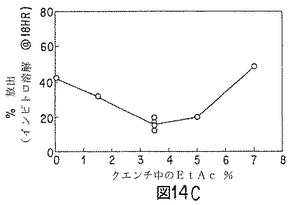
【図15A】
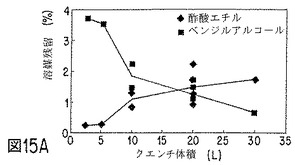
【図15B】
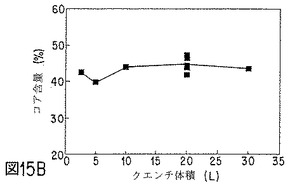
【図15C】
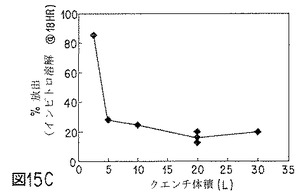
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図14C】
【図15A】
【図15B】
【図15C】
【公開番号】特開2012−211199(P2012−211199A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2012−172236(P2012−172236)
【出願日】平成24年8月2日(2012.8.2)
【分割の表示】特願2010−108382(P2010−108382)の分割
【原出願日】平成6年11月18日(1994.11.18)
【出願人】(506350023)アルカーメス,インコーポレイテッド (12)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成24年8月2日(2012.8.2)
【分割の表示】特願2010−108382(P2010−108382)の分割
【原出願日】平成6年11月18日(1994.11.18)
【出願人】(506350023)アルカーメス,インコーポレイテッド (12)
【Fターム(参考)】
[ Back to top ]