
生物材料中の核酸を安定化および/または単離するための新規な組成物
【課題】生物由来の材料中の核酸を単離および/または安定化するための新規な組成物を提供する。
【解決手段】一般式:Y+R1R2R3R4X−(式中、Yは、窒素またはリンを示すことができ、R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオン示すことが可能である)で示される必須成分としてのカチオン化合物を含む組成物。
【解決手段】一般式:Y+R1R2R3R4X−(式中、Yは、窒素またはリンを示すことができ、R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオン示すことが可能である)で示される必須成分としてのカチオン化合物を含む組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生物由来の材料中の核酸を単離および/安定化するための新規な組成物に関する。その組成物は、
一般式:
【化1】

(式中、Yは、窒素またはリンを示すことができ、
R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、
X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオンを示すことが可能である)で示されるカチオン化合物と、添加剤としての少なくとも1種類のプロトン供与体と、を必須成分として含有する。
【0002】
好ましい組成物は、そのカチオン化合物がアンモニウム塩からなる組成物である(式中、R1が、高級アルキル基を示し、好ましくは炭素原子12個、14個または16個を有する高級アルキル基を示し、いずれの場合も、R2、R3およびR4がメチル基を示す)。
【0003】
式中、R1が、アルキル基、好ましくはベンジル基を示し、R2が、好ましくは炭素原子12個、14個または16個を有する高級アルキル基を示し、R3およびR4がメチル基を示す組成物もまた好ましい。
【0004】
臭化物、塩化物、リン酸塩、硫酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、シュウ酸塩またはコハク酸塩がアニオンとして好ましい。
【0005】
C1〜C6アルキルは一般に、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい、炭素原子1〜6個を有する分枝もしくは非分枝炭化水素基を示す。以下の炭化水素基:
メチル、エチル、プロピル、1−メチルエチル(イソ−プロピル)、ブチル、1−メチルプロピル、2−メチルプロピル、1,1−ジメチルエチル、n−ペンチル、1−メチルブチル、2−メチルブチル、3−メチルブチル、1,1−ジメチルプロピル、1,2−ジメチルプロピル、2,2−ジメチルプロピル、1−エチルプロピル、ヘキシル、1−メチルペンチル、2−メチルペンチル、3−メチルペンチル、4−メチルペンチル、1,1−ジメチルブチル、1,2−ジメチルブチル、1,3−ジメチルブチル、2,2−ジメチルブチル、2,3−ジメチルブチル、3,3−ジメチルブチル、1−エチルブチル、2−エチルブチル、1,1,2−トリメチルプロピル、1,2,2−トリメチルプロピル、1−エチル−1−メチルプロピルおよび1−エチル−2−メチル−プロピルが例として挙げられる。
【0006】
高級アルキル基という用語は、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい分枝もしくは非分枝C7〜C20アルキル基を表す。以下の炭化水素基:分枝もしくは非分枝ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、テトラデシル、ヘキサデシル、ドデカデシルおよびエイコシルが例として挙げられる。
【0007】
C3〜C6アルケニルは一般に、炭素原子3〜6個を有し、1つまたは可能ならば複数の二重結合を有する、分枝または非分枝炭化水素基であって、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい炭化水素を示す。以下の炭化水素基:
2−プロペニル(アリル)、2−ブテニル、3−ブテニル、1−メチル−2−プロペニル、2−メチル−2−プロペニル、2−ペンテニル、3−ペンテニル、4−ペンテニル、1−メチル−2−ブテニル、2−メチル−2−ブテニル、3−メチル−2−ブテニル、1−メチル−3−ブテニル、2−メチル−3−ブテニル、3−メチル−3−ブテニル、1,1−ジメチル2−プロペニル、1,2−ジメチル2−プロペニル、1−エチル−2−プロペニル、2−ヘキセニル、3−ヘキセニル、4−ヘキセニル、5−ヘキセニル、1−メチル−2−ペンテニル、2−メチル−2−ペンテニル、3−メチル−2−ペンテニル、4−メチル−2−ペンテニル、1−メチル−3−ペンテニル、2−メチル−3−ペンテニル、3−メチル−3−ペンテニル、4−メチル−3−ペンテニル、1−メチル−4−ペンテニル、3−メチル−4−ペンテニル、4−メチル−4−ペンテニル、1,1−ジメチル2−ブテニル、1,1−ジメチル2−ブテニル、1,1−ジメチル3−ブテニル、1,2−ジメチル2−ブテニル、1,2−ジメチル3−ブテニル、1,3−ジメチル2−ブテニル、1,3−ジメチル3−ブテニル、2,2−ジメチル3−ブテニル、2,3−ジメチル2−ブテニル、2,3−ジメチル3−ブテニル、1−エチル−2−ブテニル、1−エチル−3−ブテニル、2−エチル−1−ブテニル、2−エチル−2−ブテニル、2−エチル−3−ブテニル、1,1,2−トリメチル2−プロペニル、1−エチル−1−メチル−2−プロペニルおよび1−エチル−2−メチル−2−プロペニルが例として挙げられる。
【0008】
C3〜C6アルキニルは一般に、炭素原子3〜6個を有し、1つまたは可能ならば複数の三重結合を有する、分枝または非分枝炭化水素基であって、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい炭化水素を示す。以下の炭化水素基:
2−プロピニル(プロパルギル)、2−ブチニル、3−ブチニル、1−メチル−2−プロピニル、2−メチル−2−プロピニル、2−ペンチニル、3−ペンチニル、4−ペンチニル、1−メチル−2−ブチニル、2−メチル−2−ブチニル、3−メチル−2−ブチニル、1−メチル−3−ブチニル、2−メチル−3−ブチニル、3−メチル−3−ブチニル、1,1−ジメチル2−プロピニル、1,2−ジメチル2−プロピニル、1−エチル−2−プロピニル、2−ヘキシニル、3−ヘキシニル、4−ヘキシニル、5−ヘキシニル、1−メチル−2−ペンチニル、2−メチル−2−ペンチニル、3−メチル−2−ペンチニル、4−メチル−2−ペンチニル、1−メチル−3−ペンチニル、2−メチル−3−ペンチニル、3−メチル−3−ペンチニル、4−メチル−3−ペンチニル、1−メチル−4−ペンチニル、3−メチル−4−ペンチニル、4−メチル−4−ペンチニル、1,1−ジメチル2−ブチニル、1,1−ジメチル2−ブチニル、1,1−ジメチル3−ブチニル、1,2−ジメチル2−ブチニル、1,2−ジメチル3−ブチニル、1,3−ジメチル2−ブチニル、1,3−ジメチル3−ブチニル、2,2−ジメチル3−ブチニル、2,3−ジメチル2−ブチニル、2,3−ジメチル3−ブチニル、1−エチル−2−ブチニル、1−エチル−3−ブチニル、2−エチル−1−ブチニル、2−エチル−2−ブチニル、2−エチル−3−ブチニル、1,1,2−トリメチル2−プロピニル、1−エチル−1−メチル−2−プロピニルおよび1−エチル−2−メチル−2−プロピニルが例として挙げられる。
【0009】
別段の指定がない限り、アリールは、ヘテロ原子1個または2個を任意に含有することができ、炭素原子4〜22個を有する芳香族単核基または多核基を示す。その例には:ハロゲン(F、Cl、Br、I)、好ましくはフッ素によって、またはアルキル基によって、互いに独立して任意に一置換または多置換することができる、フェニル、ナフチル、アントラシルまたはピロール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジンまたはピラジンが含まれる。
【0010】
上記に定義されたように、アラルキルは、C1〜C6アルキレン、C3〜C6アルケニレンもしくはC3〜C6アルキニレン架橋を介してカチオン部分構造に結合する、単核または多核アリール基を示し、そのC1〜C6アルキル、C3〜C6アルケニルおよびC3〜C6アルキニル基は上記で定義したとおりである。本発明の目的には、ベンジル基が好ましい。
【0011】
適切な対イオンX−には、ハロゲン化水素酸のすべてのアニオン、または酢酸塩もしくはシュウ酸塩、マロン酸塩、コハク酸塩もしくはクエン酸塩などの、一塩基または二塩基有機酸のアニオンが含まれることが好ましい。
【0012】
本発明の目的のための適切なプロトン供与体は主に、無機酸またはその塩の他に、単独でまたは組み合わせて、飽和脂肪族モノカルボン酸、不飽和アルケニル−カルボン酸、飽和および/または不飽和脂肪族C2〜C6ジカルボン酸、脂肪族ケトカルボン酸またはケトジカルボン酸ならびにアミノ酸である。上述のすべての有機酸を未置換状態で、または置換誘導体として使用することができ、別段の指定がない限り、未置換誘導体またはヒドロキシル基によって一置換もしくは多置換された誘導体であることが好ましい。
【0013】
本発明の目的のための飽和脂肪族モノカルボン酸という用語には、ギ酸の他に、C1〜C6アルキル−カルボン酸が含まれることが好ましく、酢酸、プロピオン酸、n−酪酸、n−吉草酸、イソ吉草酸、エチル−メチル−酢酸(2−メチル−酪酸)、2,2−ジメチルプロピオン酸(ピバル酸)、n−ヘキサン酸、n−オクタン酸、n−デカン酸、およびn−ドデカン酸(ラウリン酸)が好ましい。さらに、上述の酸から誘導されるケトカルボン酸もまた使用することができる。
【0014】
本発明の目的に使用される不飽和アルケニル−カルボン酸の例には、例えばアクリル酸(プロペン酸)、メタクリル酸、クロトン酸、イソクロトン酸ならびにビニル酢酸が含まれる。
【0015】
本発明に従って、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸もしくはアジピン酸などの飽和脂肪族C2〜C6ジカルボン酸が好ましく、シュウ酸およびコハク酸が特に好ましい。
【0016】
本発明に従って問題を解決するためには、脂肪族ヒドロキシ−ジ−およびトリ−カルボン酸を使用することが特に好ましく、タルトロン酸、D−(+)、L−(−)−もしくはDL−リンゴ酸、(2R,3R)−(+)−酒石酸、(2S,3S)−(−)−酒石酸、メソ−酒石酸およびクエン酸が最も特に好ましい。
【0017】
マレイン酸もしくはフマル酸などの不飽和ジカルボン酸または、例えばアコニット酸などの不飽和トリカルボン酸もまた、本発明の問題の解決策として適している。
【0018】
しかしながら、本発明の目的のために、例えばメソシュウ酸およびオキサロ酢酸などの脂肪族ケトジカルボン酸も添加剤として使用することができ、オキサロ酢酸が特に最も特に好ましい。
【0019】
さらに、本発明に従って、アミノ酸を使用することもでき、例えばアミノ酢酸(グリシン)、α−アミノプロピオン酸(アラニン)、α−アミノ−イソ吉草酸(バリン)、α−アミノ−イソカプロン酸(ロイシン)およびα−アミノ‐β−メチル吉草酸(イソロイシン)などのα−アミノ酸が好ましい。グリシンを使用するのが最も好ましい。
【0020】
上述のプロトン供与体は、個々の物質として、または純粋な立体異性体の形態で、および混合物の状態でも、使用することができる。
【0021】
無機酸およびその塩を、本発明に従って、更なる添加剤として使用することもできる。アルカリ金属もしくはそのアンモニウム塩と、リン酸もしくは硫酸などの無機酸との塩を使用することが好ましい。リン酸および硫酸アンモニウムを使用することが最も好ましい。
【0022】
本発明の目的における核酸という用語は、より広い意味での核酸を意味し、したがって、例えば、二本鎖、一本鎖、環状および直鎖、分枝などの、すべての長さまたは立体配置のリボ核酸(RNA)およびデオキシリボ核酸、およびそのすべての可能なサブユニット、例えばヌクレオチドモノマー、オリゴマー、プラスミド、細菌DNAおよびRNA、ならびに動物および植物細胞由来のゲノムおよび非ゲノムDNAおよびRNA、プロセシングされた形態のmRNA、プロセシングされていない形態のmRNA、tRNA、hnRNA、rRNA,cDNAならびに考えられる他のすべての核酸が含まれる。
【0023】
使用される、核酸を有する生物サンプルは、例えば参照により本明細書に組み込まれる欧州特許出願第95909684.3号に開示されているように、無細胞サンプル材料、血漿、血液、血清、細胞、白血球画分、クルスタ・フロジスティカ(crusta phlogistica)、痰、尿、精液、糞便、スメア、吸引液などの体液、生検材料、例えば組織および器官の一部など、すべての種類の組織サンプル、本発明に従って考えられる、遊離もしくは結合核酸または核酸を含有する細胞を含む食物サンプル、例えば生物体(単細胞または多細胞生物;昆虫等)、植物および植物の器官、細菌、ウイルス、酵母および他の菌類または真核生物および原核生物等、あるいは遊離核酸であることができる。
【0024】
本発明の技術的背景に関して:
その核酸を研究することによって、細胞の遺伝的起源および機能活性を決定かつ検討することができることは、従来技術から十分によく知られている。核酸およびタンパク質の分析によって、細胞活性の原因への直接的なアクセスが得られる。このように、それらは潜在的に、例えば代謝産物の検出など、間接的な従来の方法より優れている。したがって、分子生物学的分析は、多くの分野、例えば医学および臨床診断において、医薬組成物の開発および評価を行う製薬分野において、食品分析において、食品製造のモニタリングにおいても、作物および家畜を育てる農業において、ならびに環境分析および多くの研究分野において既に用いられている。
【0025】
RNA、特に細胞中のmRANを分析することによって、遺伝子の活性を直接決定することが可能である。分子生物学の最新方法、例えば実時間逆転写酵素PCRまたは遺伝子発現チップ分析による、細胞における転写パターン(mRNAパターン)の定量分析によって、例えば異常に発現した遺伝子を検出し、それによって、代謝異常、感染、または癌の発生を検出することが可能となる。例えば、PCR、RFLP、AFLP、SNPもしくはシークエンシングなどの分子生物学的方法による、細胞からのDNAの分析によって、例えば遺伝的欠陥を検出すること、またはHLAタイプおよび他の遺伝標識を決定することが可能となる。
【0026】
ゲノムDNAおよびRNAの分析もまた、ウイルス、細菌等の感染性病原体を直接検出するのに用いられる。
【0027】
その自然環境から生物サンプルを採取した直後に核酸およびタンパク質を安定化することは、核酸の分析に絶対に必須である。これは、生物サンプルを採取した後、非常に急速に分解し得るDNA、特にRNAに当てはまる。一方、生物サンプルを一旦採取すると、ストレス遺伝子の誘導によって、新たなmRNA分子が合成され、その結果例えば、細胞の転写パターンが変化する可能性がある。これによって、その後の分析が不正確となる可能性がある。特に医学分野では、核酸を含有するサンプルを採取し、次いで、それらを最初にしばらくの間保存し、実験室に輸送されるまで、さらに研究されないのが実際に一般的であるため、核酸を安定化することは必須である。
【0028】
一方、サンプル中に含有される核酸は、変化するか、または完全に分解さえし得る。これは当然、続いて行われる試験の結果に多大な影響を及ぼすか、またはそれらを完全に不可能とする。かかる試験は、例えばノーザンおよびサザンブロット分析、PCR、RT−PCR、SunRise、LCR、分枝DNA(bDNA)、SDA、DNAおよびRNAチップおよび遺伝子発現アレイおよび突然変異分析、RFLP、AFLP、SNP分析、cDNA合成、サブトラクティブ・ハイブリダイゼーション(subtractive hybridisation)またはTaqman技術および他の実時間の定量法など、分子生物学的技術を用いて行われる。一方、インタクトな高精製核酸−DNAまたはRNAの使用は、上記の試験の使用または実施に対する基本的な適合性の基準となる。さらに、核酸を含有するサンプルの単離およびアッセイもまた、時間のかかる作業である。さらに、例えば試験が失敗した場合に生じるような、分子生物学分野で使用される研究実験室の汚染が、試験結果のエラーをまねく可能性がある。
【0029】
従来技術に関して
数多くの出版物では、例えば組織などの適切なサンプルから核酸を続いて単離するための、固定液としてのエタノールおよびアセトンをベースとする混合物の使用が提案されている。この文献を研究した後、この種のエタノール/アセトン混合物は、RNAの安全な回収に課せられる必要条件をすべて満たすわけではないことが明らかである。このように、この種の混合物は、分解からRNAを保護することができない。さらに、さらに広範な細胞凝集魂から構成される固体サンプル中でRNAが保護される保証はない。さらに、提案された混合物は高い引火性または爆発性であり、実験室で作業する際にかなり危険性がある。
【0030】
それに加えて、さらに周辺の関連する従来技術は、固定化または保存された組織サンプルからのRNAの回収に関係する。これは特に、in situハイブリダイゼーション中に達成されるシグナル強度を最大にする、組織標本の適合性に関する。換言すれば、この種の実験は、それを保存するよりもむしろ、RNAを検出することを意図するものである(米国特許第5196182号および5260048号)。
【0031】
他のレポートは、このようにして得られた断片をPCRによる制限分子分析にかけることを可能にするために、固定組織から断片化RNAまたはDNAを回収することに関する。この種の断片化DNAまたはRNAを得るために、構造組織成分を分解することを可能にするため、対応するサンプルを従来法によりプロテイナーゼKで処理する;次いで、単に、グアニジウム塩を含有する溶液でRNAを抽出する。しかしながら、この方法によって固定組織から得られたRNAは、品質が低く、大きさがわずか約200塩基である。従来技術に従って、特に固定化中の細胞内基質内のDNAまたはRNAの内因性かつ架橋反応の負の影響を含む一定数の特定因子に、これを抑えることができる。DNAまたはRNAが大部分の場合で少なくとも一部分解するという事実に基づくと、このようにして得られたDNAまたはRNAは、もはやノーザン分析に上手く使用することはできない。このように単離されたRNAは、比較的小さな断片を増幅するためにのみであるが、せいぜい多少の成功の見込みを持って、RT−PCR反応に使用することができる程度である。
【0032】
従来技術では、凝固点を超える温度でRNAを保存するための、硫酸アンモニウムの使用もまた記述されている[WO00/06780]。この種の組成物は、RNAlaterの名称で従来技術に従って使用されている。しかしながら、この種の硫酸アンモニウム水溶液は、血液、血漿または血清中のRANを安定化するのには適していない。上記のサンプルは高いタンパク質濃度を有するため、この種のアンモニウム塩溶液と接触するとすぐに、限られた溶解度の沈殿物が形成する[Messrs Ambion(アメリカ、テキサス州オースティン)からのRNAlater製品の情報]。
【0033】
さらに、生物サンプルから核酸を単離するために、この種のカチオン化合物を使用することは、従来技術から知られている。かかる用途は、例えば米国特許第5,010,183号および同第5,300,635号および欧州特許:EP0442026に記載されている。これらの出版物では、サンプル調製に通常用いられるインキュペーション時間、つまり約数分の間、生物サンプルをカチオン化合物と共にインキュベートし;次いで、その核酸を再度精製する。
【0034】
従来技術から公知の化合物の研究によって、従来技術で言及されているカチオン化合物、特に米国特許に開示されているテトラデシルトリメチルシュウ酸アンモニウムは、例えば血液が長期間保存される場合に、それら自体で細胞RNAの適切な安定化を保証するものではないことが示されている。
【0035】
実を言えば、例えば数日間血液中のウイルスを安定化することに着手している実験は従来技術から知られているが、これらの知見では、インタクトなままのRNAについて何らの言及もされていない。このように、シュミット(Schmidt)およびマクファーレン(MacFarlane)[J.Medical Virology 47,(1995) 153]は、Catrimox−14(登録商標)による血液中のC型肝炎ウイルスの周囲温度で7日間の安定化について記述している。そのウイルスは、HCVゲノムの長さ250bpの断片をRT−PCRにより増幅することによって検出された。しかしながら、開示されたその結果からは、小さな断片のみが増幅されたため、RNAがインタクトである十分な証拠は得られなかった。さらに、その実験は、不定のウイルスサンプルを添加して行われ、そのため、保存中のウイルスRNAの分解に関して判断を下すことは不可能であった。
【0036】
さらに、国際特許出願:WO99/29904には、塩酸グアニジン、チオシアン酸グアニジン、塩化リチウム、塩化マンガン、サルコシル、SDS、過塩素酸ナトリウム、サリチル酸ナトリウムおよびチオシアン酸ナトリウムと共にEDTA、EGTA、またはBAPTAを用いた体液中のDNAの安定化が記載されている。さらに、例えばTrizol(登録商標)などのフェノールを含有する試薬を使用して、保存中にRNAを安定化することができることは従来技術から知られている。しかしながら、これらの試薬はすべて、健康に非常に有害であり、したがって、常用するのには適していない。
【0037】
そこで、本発明の目的は、組織または血液、血漿または血清の存在下にてRANを安定化する組成物を提供することである。
【0038】
本発明はさらに、安定化溶液の形で組成物を提供することにも言及しており、その組成物の成分は健康に有害ではなく、したがって、サンプルを入手した場所から実験室へ移動する間、例えば生物サンプル材料を安定化するために、サンプルを扱うスタッフの健康を脅かす危険性なく使用することも可能である。
【0039】
本発明の他の目的は、安定化試薬それ自体もまた溶液中で安定なままである必要性を満たし、かつ例えば限られた溶解度の沈殿物の溶解など、使用者による前処理が必要ない安定化溶液の形で組成物を提供することである。この種の前処理は常に、安定化効率の変動のリスクを含む。
【0040】
本発明の他の目的は、多用途である、つまり幅広い生物サンプルに使用することができる組成物を提供することである。
【0041】
驚くべきことに、特に米国特許第5 010 183号および同第5 300 645号に開示されており、かつ本発明に従って上述の1種または複数種の添加剤と組み合わせられる化合物などのカチオン化合物と、生物サンプルの核酸を接触させた場合に、長時間にわたり核酸を安定化することができることが現在見出されている。本発明に従って問題を解決するのに適切な好ましい添加剤を表1に示す:
【0042】
【表1】
【0043】
添加剤は、様々な濃度で安定化試薬中に存在することができる;例えば、添加剤は、安定化溶液と血液との混合物中に、容積比1:1、好ましくは3:1、濃度50mM〜飽和、好ましくは100〜1M、最も好ましくは200〜500mMで存在することができる。添加剤の性質に応じて、他の濃度範囲が有利であることが証明される。異なる添加剤の組み合わせを使用することも可能である。
【0044】
組成物水溶液中のカチオン化合物濃度は、0.01重量%〜飽和、好ましくは0.1重量%〜飽和、さらに好ましくは0.5〜15重量%、最も好ましくは2〜10重量%の範囲である。
【0045】
当然、カチオン化合物と添加剤との溶液を添加する場合には、最適な濃度は、生物サンプルのそれぞれの容積、および安定化溶液と生物サンプルとの容積比によって決定される。
【0046】
一般にカチオン化合物と添加剤との混合物のpHを、広いpH範囲(pH2〜12)にわたりサンプルの関数として変化させることができ、pH2〜pH10の範囲であることが好ましく、さらに好ましくはpH3〜pH8の範囲である。好ましいpH範囲は、使用する生物サンプルに依存する。血液、血漿および血清の場合には、pH2〜pH6の範囲、特にpH3〜pH4のpH値が好ましい。
【0047】
血液、血漿および血清は別にして他の細胞体液、または例えば細菌、吸引液、細胞、組織、および上述のサンプルなど、他の生物サンプルの場合、カチオン化合物および添加剤からなる安定化溶液のpH値は、好ましくはpH3〜pH10の範囲、さらに好ましくはpH4〜pH8の範囲である。
【0048】
生物サンプル中の核酸を安定化するために、カチオン化合物(1種または複数種)および添加剤を含有する溶液とサンプルを混合することができる。生物サンプルを0.1〜10,000容積添加することができ;範囲1〜1000の容積を添加することが好ましく、範囲1〜100の容積を添加することが最も好ましい。しかしながら、例えば細い針による針生検または低細胞数の培養から得たサンプルなど、サンプルの性質に応じて、場合によってはかなり高い容積もまた用いることができる。
【0049】
同様に、生物サンプル自体が固体を溶解する液体(例えば、細胞含有体液、培養液中の細胞、尿など)を含有する場合、または液体、例えば水をそれに添加して、固体を溶解する場合、上述のカチオン化合物および添加剤を固体状で添加することもできる。固体を添加する利点は、固体が通常、化学的に安定であり、固体はサンプルに添加するのが容易である場合が多いことである。
【0050】
さらに、組織などの非常に緻密な生物サンプルの場合は特に、例えば、核酸もしくは個々の細胞もしくは細胞の凝集塊の開放を補助するために、安定化溶液中でまたは安定化溶液と混合する前に、例えばサンプルに対する機械的、化学的、物理的もしくは酵素的作用により緻密なサンプルを破壊することによって、サンプルを粉砕またはホモジナイズすることが可能である。機械的作用は、電気メス、ビーズミルを用いて、または例えばシリンジで押しつぶすことによって行うことができ、サンプルに対して作用する適切な酵素は、例えば加水分解酵素、プロテアーゼまたはリパーゼである。
【0051】
さらに、そのサンプルは、純粋な物理的手段、例えば超音波によって前処理してもよい。
【0052】
前処理は、化学的に、単独でまたは純粋な物理的方法と共に、実行することもできる。溶解を補助する手段には、例えば、脂肪族アルコール、特にイソプロパノール、またはアルデヒドまたはジアルデヒド、例えばグリオキサール、またはフェノールまたはフェノール誘導体、例えば2−ビフェニロール、またはイオン、双性イオンおよび非イオン化合物、例えばメルカプト、または還元剤、例えばジチオスレイトールおよびβ−メルカプトエタノール、またはリン酸誘導体、例えばリン酸トリブチル、またはカオトロピック試薬、例えば尿素、チオシアン酸グアニジウムまたは塩酸グアニジウム、または塩を個々にまたは組み合わせて使用することが含まれる。
【0053】
機械的、化学的、物理的または酵素的にサンプルに作用する他の可能な方法は、当技術分野で公知であり、本明細書に含まれることを意図する。
【0054】
特定の必要条件に応じて、サンプル材料をかなり長い期間、例えば室温で1日〜14日以上保存してもよいが、例えば40℃以上の高温、および例えば4℃または−20℃以下などの低温でも保存してもよい。
【0055】
上述の化合物の溶液中での生物サンプルの保存に続いて、直接核酸を分析する技術を用いるか、または核酸をサンプルから精製してもよい。
【0056】
例えば、ブロット法、生体分子を分離するゲル電気泳動法およびクロマトグラフィー法によって、核酸を直接検出/分離してもよい。
【0057】
生物サンプルから核酸を精製するために、遊離核酸または核酸を含有する細胞もしくは粒子を、例えば遠心分離または濾過によって溶液の残りから分離し、米国特許第5,010,183号、同第5,300,645号および欧州特許出願第99103457.0号に記載されているように、少量で有利に行うことができる更なる精製にかける。
【0058】
保存容器中で核酸または核酸を含有する細胞もしくは粒子を直接分離することによって、精製のために他の容器にサンプルを移す他の段階が不要となり、そのため、サンプルの損失が減少し、サンプルから採取された核酸による互いの混同および汚染のリスクもまた極力抑えられる。このように、これらの安定化試薬を使用することによって、生物サンプル中の核酸を安定化し、直接単離する一段階方法が導かれ、その方法では、代わりに生物サンプルからRNAおよびDNAを単離すること、または1つのサンプルから並行してRNAおよびDNAを単離することができる。
【0059】
1種または複数種のカチオン化合物および1種または複数種の添加剤を含有する、本発明による組成物を用いて核酸を安定化することによって、サンプル中の核酸は、長期の保存中でさえ、または輸送中に変化しないことが確かなものになる。したがって、その後の試験の精度が著しく向上する。ある特定の場合、例えばサンプル材料を長距離間輸送するか、または長期間保存しなければならない場合では、本発明による方法によって、これらの試験をかかる期間の後に行うことが初めて可能となる。
【0060】
本発明の利点は特に、例えば、サンプリング直後に固定化しなければならない転写物レベルを分析する研究分野、および例えば、分析の準備が整うまで保存および輸送中に患者のサンプルも安定化しなくてはならない、分子診断などの臨床分析分野において見出されている。特に、核酸の単離および安定化は、腫瘍の診断において、遺伝性疾患の診断ならびにウイルスの診断およびモニタリングにおいて、他の感染性病原体の診断およびモニタリングにおいて、遺伝子発現パターンの分析において使用される。
【0061】
本発明の適用分野は、医学または動物学分野に及ぶだけではなく、植物、真菌および原核生物系の分析も含む。培養および自然環境から得た、植物および植物器官、藻類、真菌および細菌からの核酸の安定化および単離は、例えば転写物レベルおよび遺伝子発現パターンを分析する研究、および土壌サンプル中の細菌等の複合個体群における種を同定かつ定量する研究において使用される。
【0062】
可能性のある用途には、例えば食物分析などの他の分析分野も含まれる。
【0063】
本発明を以下の実施例および図によって説明する。記述および実施例において、以下の略語が使用される:
AFLP 増幅断片長多型
A.dest. 蒸留水
BAPTA1 2−ビス(2−アミノフェノキシ)エタン−N,N,N’, N’−テトラ酢酸
EcoRI 制限酵素大腸菌株R
E260/E280 260および280nmでの吸光度の商
EDTA エチレンジアミン−N,N,N’,N’−テトラ酢酸
EGTA [エチレンビス(オキシエチレンニトリロ)]テトラ酢酸
GAPDH グリセリンアルデヒド−3−ホスフェート−デヒドロゲナーゼ
Hind III 制限酵素インフルエンザ菌
hugl 巨大な幼虫のヒトホモローグ
IFN−γ インターフェロンガンマ
LM 長さのマーカー
MOPS 3−(N−モルホリノ)−2−ヒドロキシプロパンスルホン酸
nb 決定されず
NonidetP40 imbentin−N/52;オクチルフェニルポリエチレングリコール
OD 光学密度
PBS リン酸緩衝食塩水
PCR ポリメラーゼ連鎖反応
RFLP 制限酵素断片長多型
rpm 毎分回転数
mRNA メッセンジャーRNA
rRNA リボソームRNA
RT 室温
RT−PCR 逆転写酵素 PCR
SDS ドデシル硫酸ナトリウム
SNP 一塩基多型
SSC 塩化ナトリウム/クエン酸ナトリウム溶液
TBE トリス−ホウ酸−EDTAバッファー
Tris 2−アミノ−2−(ヒドロキシメチル)−1,3−プロパンジオール
U ユニット
【0064】
例えば時間に対してhなど、ここに列挙していない略語は、当業者にはよく知られているか、あるいは従来技術におけるそれらの使用から十分によく知られているだろう。
それらが基づく図および実験の説明
図1は、異なるpHレベルを有する様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による、血液中のRNAの安定化を示す。
図2は、様々な濃度の酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図3は、250mM酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図4は、GAPDH遺伝子(A)およびIFN−γ遺伝子(B)のmRNAの放射標識プローブとのノーザンハイブリダイゼーションの結果として、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図5は、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる血液中のDNAの安定化を示す。
細胞RNAの他に、白血球由来のゲノムDNAもまた、ここで開発された方法によって安定化し、次いでシリカ膜に結合させることによって単離することができる。図5は、72時間保存した後でさえ、高分子DAN(長さ>20kB)が単離されることを示している。
図6は、ゲノムDNAを酵素反応に使用した場合の結果を示す。24時間または72時間保存した後に単離したDNA(実施例5を参照のこと)を様々な酵素反応に使用する。
A.制限酵素EcoRI(E)またはHindIII(H)6Uを使用して、DNA2μgを37℃にて3時間切断し、次いで0.8%アガロース/TBEゲル上で分離する。いずれの場合も対照として、未切断のDANを適用する。
B.ゲノムDNAのアリコート150ngおよび300ngをPCR反応に使用する(総容積50μl)。その反応では、hugl(巨大な幼虫のヒトホモローグ)遺伝子の長さ1.1kB断片を増幅する。そのPCR産物を1.2%アガロース/TBEゲル上で分離する。
図7は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表2に示す。
図8は、酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの様々な期間にわたる安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表3に示す。
図9は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿1ml中のRNAの安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表4に示す。
図10は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによるLeLa細胞中のRNAの安定化を示す。
サンプルは、二重測定で調製し、サンプル14、40、66および92は一重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表5に示す。
図11は、異なる量のHeLa細胞中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表7に示す。
図12は、マクロファージ中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表9に示す。
図13は、培養液を除去しない、Hela付着細胞中のRNAの安定化を示す。溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表13に示す。
図14は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる腎組織中のRNAの安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表12に示す。
図15は、RNAの安定化および単離と同様な、DNAの安定化および単離を示す。溶出液のアリコート40μlを0.8%アガロース−TBEゲルで分離する。問題のサンプルは実施例15に記述する。
【0065】
実施例
実施例1:
異なるpHレベルの様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による血液中のRNAの安定化
様々な鎖長のカルボン酸を添加剤として使用する。さらに、モノ、ジおよびトリカルボン酸、ヒドロキシル化および非ヒドロキシル化カルボン酸を試験する。そのすべての物質は、カチオン化合物テトラデシルトリメチルシュウ酸アンモニウムと共に安定化に使用される。その物質のpHおよび濃度のどちらも変化する。
【0066】
図1に、その試験の結果を示す。どの場合でも、インタクトなRNAを24および48時間後でさえ単離することができる。場合によっては少量であるRNAの量は、処理される血液が少量であること、および様々な血液サンプル中のRNA含有量が異なることに対応するものである。この実験では、いくらかのゲノムDNAもまた、RNA画分中で得られた。
【0067】
各濃度200mMの、かつ問題のカルボン酸に対して様々なpHレベルの様々なカルボン酸で緩衝された10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムからなるバッファー500μlで、血液500μlを室温で24時間および48時間保存する。RNAを単離するために、カチオン化合物および核酸からなる複合体を遠心分離する;そのペレットを水で1回洗浄し、再度遠心分離し、例えばQIAGEN社製RLTバッファーなど、標準市販の溶解バッファー300μl中に溶解する。サンプルを水360μlで希釈し、プロテイナーゼK40μlで55℃にて10分間処理する。次いで、そのサンプルを遠心分離し、エタノールを上清に添加し、シリカ膜を含むスピンカラムにそれを添加する。遠心することによって、サンプルを膜に通す。スピンカラムは、市販のイソチオシアン酸グアニジニウム含有洗浄バッファー、例えばQIAGEN社製バッファーRW1で1回洗浄し、QIAGEN社製バッファーRPEなどの標準市販のアルコール含有洗浄バッファーで2回洗浄し、次いで、遠心することによって膜にも通されるRNase非含有水60μl中にRNAを溶出し、その溶出液のアリコート30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離する。
【0068】
実施例2
テトラデシルトリメチルシュウ酸アンモニウムおよび様々な濃度の酒石酸(緩衝)pH3を用いた、全血中のRNAの安定化
10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび50〜500mM酒石酸(pH3)からなるバッファー500μlで、血液500μlを室温で2.5時間、24時間および48時間保存する。さらに、QIAGEN社製「RNase非含有DNaseセット」でサンプルをDNaseで処理することによって、ゲノムDNAを除去することを除いては、図1に示すようにRNAを単離する。RNase非含有水80μlでRNAを溶出する。その溶出液30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離する。
【0069】
実施例3
250mM酒石酸(pH3)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウムを用いた、全血中のRNAの安定化
RNAの完全性、収量および純度を決定する:
カルボン酸バッファー、例えば250mM酒石酸(pH3.0)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウム溶液中で分解または収量の減少を生じることなく、血液中でRNAは少なくとも72時間安定化される(図3を参照のこと)。
【0070】
10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび250mM酒石酸(pH3.0)からなるバッファー2mlと血液2mlとを混合し、室温で24〜72時間保存する。例えばQiagen GmbH社製バッファーELなどの標準市販の赤血球溶解バッファーを、カチオン化合物および核酸からなる複合体を遠心分離する前にサンプルに添加し、次いでその混合物を氷上で10分間インキュベートすることを除いては、実施例2に記載のように、RNAを単離する。RNase非含有水80μlでRNAを溶出する。その溶出液30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離するか、または分光光度計で測定する。水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このように、波長260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって、得られたRNAの純度を測定する。
【0071】
実施例4:
酒石酸(pH4)で緩衝された、異なる濃度のテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化
ノーザンブロット分析
4%テトラデシルトリメチルシュウ酸アンモニウムおよび200 mM酒石酸(pH3.7)からなるバッファー6.9mlと血液2.5mlとを混合し、室温で1時間、24時間、48時間および72時間保存する。RNAを単離するために、カチオン化合物と核酸との複合体を遠心分離する。そのペレットを水で1回洗浄し、次いで例えばQiagen社製バッファーRLTなどの溶解バッファー300μl中に溶解する。サンプル調製の残りの作業を、図2に記載のように行う。次いで、全RNAのアリコート2.5μgを1.2%変性アガロース/ホルムアルデヒドゲル上で分離する。次いで、そのRNAをナイロン膜に移し、リン酸ナトリウム/SDSバッファー中で68℃にて、GAPDH遺伝子(図4A)、またはIFN−γ遺伝子(図4B)の放射標識アンチセンスRNAプローブと約12時間にわたりハイブリダイズする。減少する塩濃度2×SSC/0.1%SDS〜0.1×SSC/0.1%SDSの洗浄バッファーで温度68℃にて、その膜を洗浄する。次いで、ナイロン膜をX線フィルム上で暴露する。GAPDHおよびIFN−γ−mRNAのシグナルはどちらも、72時間を超える保存期間にわたり一定のままである。
【0072】
実施例5:
酒石酸(pH3.7)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウムによる血液中のゲノムDNAの安定化
細胞RNAの他に、全血からのゲノムDNAもまた、本発明において開発された方法によって安定化することができ、次いでシリカ膜に結合させることによって単離することができる。図5から、室温で72時間保存した後でさえ、高分子DNA(長さ>20kB)が単離されることが示されている。
【0073】
4%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸(pH3.7)からなる溶液6.9mlと血液2.5mlとを混合し、室温で24時間または72時間保存する。DNAを単離するために、カチオン化合物とDNAとの複合体を遠心分離する。塩化ナトリウムおよびEDTAを含有するバッファー300μlにそのペレットを溶解し、次いで、例えばQiagen社製バッファーALなどの市販の塩酸グアニジウムバッファー360μl中に溶解し、ならびにプロテイナーゼK20μlを添加する。そのサンプルを65℃で10分間インキュベートし、次いで、エタノール420μlを添加し、シリカ膜を含むスピンカラムにサンプルを適用する。そのサンプルを、遠心分離によって膜を通過させる。例えばQIAGEN社製バッファーAW1など、標準市販のエタノール含有塩酸グアニジウムバッファーでシリカ膜を洗浄し、例えばQIAGEN社製バッファーAW2などのエタノール含有洗浄バッファーで1回洗浄する。DNAをTrisバッファー(pH8)300μlで希釈する。溶出液のアリコート5μlを0.8%アガロース/TBEゲル上で分離する。
【0074】
実施例6:
酵素反応におけるゲノムDNAの使用。
図6から、実施例5に従って単離されたDNAを様々な酵素反応(制限増幅およびPCR増幅)に使用することができることが分かる。
【0075】
24または27時間の保存後に単離されたDNA(実施例5参照)は様々な酵素反応に使用される。これは、単離したDNAの純度が高く、品質が良いことの証拠である。
【0076】
A)制限酵素EcoRI(E)およびHind III(H)6Uで37℃にて3時間、DNAのアリコート2μgを切断し、次いで0.8%アガロース/TBEゲル上で分離する。対照として、未切断のDNAを適用する。
B)ゲノムDNAのアリコート150ngおよび300ngをPCR反応(総容積50μl)に使用する。その反応では、長さ1.1kB断片のhugl遺伝子が増幅される。そのPCR産物を1.2%アガロース/TBEゲル上で分離する。
【0077】
実施例7
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化
これらの実験から、カルボン酸および他の添加剤をテトラデシルトリメチルシュウ酸アンモニウムに添加することによって、テトラデシルトリメチルシュウ酸アンモニウムを単独で用いた場合のRNAの安定化と比較して、血漿中の遊離RNAの安定化が著しく改善されることが実証される。
【0078】
この実験で使用する溶液を調製するために、30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、コハク酸、硫酸アンモニウムまたはリン酸の原液と混合して、2%または4%テトラデシルトリメチルシュウ酸アンモニウム、および200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。添加剤を全く含有しない5%テトラデシルトリメチルシュウ酸アンモニウム溶液を対照として使用する。
【0079】
このように調製したすべての溶液のアリコート0.5mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)により予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフ容器の蓋にピペッティングする。ヒト血漿0.5mlをその溶液に添加し、容器の蓋を閉め、容器を迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で1日保存する。すべての実験を二重測定で行う。
【0080】
RNAを単離するために、サンプルを25000×gで3分間にわたり遠心分離する。上清を除去し、グアニジウム塩酸塩およびNonidetP40(pH7.0)を含有する、60℃に調節されたバッファー0.5mlおよびプロテイナーゼKも、ペレットに添加する。そのペレットをボルテックスすることによって溶解し、50℃で15分間インキュベートする。次いで、エタノール−nonidetP40溶液0.5mlを添加し、サンプルを約5秒間ボルテックスすることによって混合する。次いで、例えばQIAGEN社製QIA ampカラムなどのシリカ膜を含有する標準市販のスピンカラム中にそのサンプルを入れ、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、アルコール含有洗浄バッファー、例えばQIAGEN社製バッファーAW2で2回洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有洗浄バッファーで洗浄した後、バッファーを添加することなく、遠心することによって(最大3分の回転数、この場合では25000×g)膜を乾燥させる。溶出液については、RNaseを含有しない水30μlを膜上にピペッティングして、膜から精製RNAを引き離す。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出プロセスを完了する。
【0081】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図7に示す。ゲルレーンのローディングを表2に示す。
【0082】
【表2】
【0083】
個々のサンプルのRNA品質を比較するために、レーン45は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0084】
この実験で使用されるHeLa全RNAのゲル電気泳動による分離によって、臭化エチジウムで染色した後にインタクトな28Sおよび18SrRNAが示される。視認できるrRNAバンド(28SrRNA)の一番上のバンドは、それより下のrRNAバンド(18SrRNA)よりも明らかに濃く、かつ太く、それはインタクトな分解されていないRNAの一般的な特徴である。添加剤を添加することなく、5%テトラデシルトリメチルシュウ酸アンモニウムと混合した血漿中で1日保存したHeLa全RNAを、テトラデシルトリメチルシュウ酸アンモニウムおよび様々な添加剤と混合した血漿中で1日保存した後に単離したRNAと比較すると、RNAの安定化が添加剤を使用することによって改善されていることが明白に示されている。安定化化合物を含まない血漿にRNAを添加した場合、これによって、よく知られているように数分以内にRNA全体の分解が引き起こされる。
【0085】
実施例8
酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、様々な時間にわたる血漿中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、血漿中でRNAが少なくとも14日まで安定化されることが示されている。
【0086】
この実験に使用する溶液を調製するために、30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸(pH3)またはタルトロン酸(pH3)の原液と混合して、6%または8%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。
【0087】
このように調製したすべての溶液のアリコート0.5mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)により予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフ容器の蓋にピペッティングする。ヒト血漿0.5mlをその溶液に添加し、容器の蓋を閉め、容器を迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で3、7、10および14日保存する。すべての実験を二重測定で行う。
【0088】
RNAの単離は実施例7に記載のように行う。
【0089】
単離されたRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図8に示す。ゲルレーンのローディングを表3に示す。
【0090】
【表3】
【0091】
個々のサンプルのRNA品質を比較するために、レーン「K」は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0092】
ゲル電気泳動による分離から、テトラデシルトリメチルシュウ酸アンモニウムおよび酒石酸またはタルトロン酸(pH3)と混合した血漿でHeLa全RNAを14日まで保存した後でさえ、臭化エチジウムで染色した後に、インタクトな28Sおよび18SrRNAバンドが表れる。
【0093】
実施例9:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、血漿中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物により、多量の血漿中でさえ、RNAを安定化することができることが示される。
【0094】
このように調製したすべての溶液のアリコート1mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)を用いて予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフチューブの蓋にピペッティングする。ヒト血漿1mlをその溶液に添加し、チューブの蓋を閉め、チューブを迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で3日間保存する。すべての実験を二重測定で行う。
【0095】
RNAの単離は実施例7に記載のように行う。
【0096】
単離されたRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%ホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図9に示す。ゲルレーンのローディングを表4に示す。
【0097】
【表4】
【0098】
個々のサンプルのRNA品質を比較するために、レーン13は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0099】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、インタクトな28Sおよび18SrRNAバンドが表れる。このように、多量の血漿中でさえ、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、RNAは安定化される。
【0100】
実施例10:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、HeLa細胞中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと様々な添加剤との混合物によって、HeLa細胞中のRNAを、室温で14日までの保存期間にわたって安定化することができることが示される。
【0101】
この実験で使用する溶液を調製するために、20%または30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、硫酸アンモニウムまたはリン酸の原液と混合して、2%または4%テトラデシルトリメチルシュウ酸アンモニウム、および200mM添加剤の最終濃度をとする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。
【0102】
予め細胞培養から収集し、PBSで直ちに洗浄した1×106個のHela細胞を遠心分離(120×gで1分)によってペレットにし、上清を除去する。表4に示す溶液のアリコート300μlを細胞に添加し、ボルテックスによってサンプルを混合し、細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で3、7、10および14日間保存する。すべての実験を二重測定で行う。
【0103】
RNAを単離するために、1200×gで3分間遠心分離することによって、細胞をペレットにし、上清を除去する。繰り返しピペットで取り、移すことによって、または約10秒以上の間にわたりボルテックスすることによって、例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ペレットを再懸濁する。次いで、エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。第2アルコール含有洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、RNaseを含有しない水40μlを膜上にピペッティングする。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出プロセスを完了する。
【0104】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図10に示す。サンプルを表5にまとめる。サンプル14、40、66および92を1回試験し、結果を示すことを除いては、すべてのサンプルを2回試験し、結果を示す。
【0105】
【表5】
【表6】
【表7】
【表8】
【0106】
サンプル「K」は、予め保存されることなく、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの単離キットによって、1×106個のHela細胞(=ポジティブコントロール)から単離された全RNAを示す。サンプル「a」、「b」、「c」および「d」は、上述のように添加剤を含まないPBS中で1×106個のHela細胞を3、7、10または14日間保存した後に単離された全RNAを示す。
【0107】
単離された全RNAの量は、水に希釈した後に、波長260nmでの光吸収の光度測定によって決定する。このようにして得られたRNAの純度を、260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって測定する。単離の結果を以下の表6に示す。いずれの場合も、2つの測定値の平均を示す。
【0108】
【表9】
【0109】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後、正の対照においてインタクトな28Sおよび18SrRNAバンドが表れる。rRNAバンド(28SrRNA)の一番上のバンドは、それより下のrRNAバンド(18SrRNA)よりも明らかに濃く、かつ太く、それはインタクトな分解されていないRNAの一般的な特徴である。
PBS中で細胞を3日間保存した後には、2つのrRNAバンドが同一強度を示し、極めて少ないRNAが視認できることから、RNAは一部分解している。7日間以上保存した後には、RNAはもはや視認できない。これに対して、Hela細胞中のRNAは、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによって14日間まで安定化される。これは、特異的RNAの収量および純度をOD測定することによって確認される。安定化は、pHにより影響を受ける。混合物において、つまりテトラデシルトリメチルシュウ酸アンモニウムと添加剤とを混合した後、4を超えるpH値が好ましい。
【0110】
実施例11
異なる量のHela細胞におけるRNAの安定化
これらの実験から、使用する細胞の数にかかわらず、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、HeLa細胞中のRNAを安定化することができることが示される。
【0111】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、pH6の0.5M酒石酸の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。
【0112】
予め細胞培養から収集し、PBSで直ちに洗浄した1×105個、5×105個、1×106個および5×106個のHela細胞を、遠心分離(120×gで1分)によってペレットにし、上清を除去する。4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸を含有する溶液のアリコート300μlを細胞に添加し、ボルテックスによってサンプルを混合し、細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で15分間または1日保存する。すべての実験を二重測定で行う。
【0113】
実施例10に記載のようにRNAを単離する。
【0114】
使用する対照は、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸で前処理されておらず、かつ上述のようにRNAを単離するために保存されていない1×105個、5×105個、1×106個および5×106個のHela細胞である。
【0115】
単離したRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図11に示す。サンプルを表7にまとめ、すべてのサンプルを2回試験し、結果を示す。
【0116】
【表10】
【0117】
単離した全RNAの量は、水に希釈した後に、波長260nmでの光吸収の光度測定によって決定する。このようにして得られたRNAの純度を、260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって測定する。単離の結果を以下の表8に示す。いずれの場合も、2つの測定値の平均を示す。
【0118】
【表11】
【0119】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存された対照サンプルにおいて、保存されていない対照サンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れる。保存されていない対照と保存されたサンプルとの間に明らかな相違はない。同様に、OD測定によって決定されたRNA収量および純度から、RNAの収量または純度を低減することなく、RNAの安定化は、異なる量の細胞中で同程度に行われることが確認される。細胞数が増加するに従って減少する商E260/E280は、測定は水中で行われ、緩衝系では行われないという事実に起因し得る。
【0120】
実施例12:
マクロファージ中のRNAの安定化
これらの実験によって、様々な種類の細胞におけるRNAを使用できることが実証される。この実験で使用するマクロファージは、前に使用したHela細胞よりも多量のRNaseを含有し、このため、細胞中のRNAが余儀なく分解される。
【0121】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸(pH5)、0.5Mタルトロン酸(pH5)または0.5Mリン酸(pH5)の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度にする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を水酸化ナトリウム溶液で指定のpHに調節する。
【0122】
予め細胞培養から収集し、PBSで直ちに洗浄した1×106個のHela細胞を遠心分離(120×gで1分)によってペレットにし、上清を除去する。4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mMの添加剤を含有する溶液のアリコート300μlを細胞に添加し、その細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で2日間、6日間、9日間および14日間保存する。すべての実験を二重測定で行う。
【0123】
そのRNAを実施例10に記載のように単離する。
【0124】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図12に示す。サンプルを表9にまとめる。すべてのサンプルを2回試験し、結果を示す。
【0125】
【表12】
【0126】
レーン25および26は、マクロファージを予め保存することなく、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの市販の単離キットを用いて、1×106個のマクロファージ(ポジティブコントロール)から単離した全RNAを示す。
【0127】
水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このようにして得られたRNAの純度を、波長260nmでの光吸収に対する280nmでの光吸収の比を光度測定することによって決定する。単離の結果を以下の表10に示す。いずれの場合も、2つの測定値の平均を示す。
【0128】
【表13】
【0129】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存された対照サンプルにおいて、および保存されていない対照サンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れ、24日間保存した後でさえ、RNA分解の兆候はない。同様に、光度測定によって決定されたRNAの収量および純度は、保存中に未変化のままである。
【0130】
実施例13
培地の除去を行わない、Hela付着細胞中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、付着細胞中でさえ、RNAを安定化することができることが示される。代わりにテトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物を培地に添加しても、細胞を含有する培地を除去しない場合でさえ、安定化は依然として行われる。培地中の細胞は、体液中の細胞のモデルとして見なすことができる。
【0131】
この実験で使用する溶液を調製するために、テトラデシルトリメチルシュウ酸アンモニウムおよび特定の添加剤、酒石酸または硫酸アンモニウムを計量して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mMの添加剤の最終濃度とし、水に溶解する。4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸の場合には水酸化ナトリウムを用いて、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウムの場合には硫酸を用いて、溶液のpHをpH5に調節する。
【0132】
6ウェルプレートにおいて培地2ml中でHela細胞を培養する。その細胞は付着して成長する、つまりウェルの底面に細胞が付着する。細胞中のRNAを安定化するために、10mlの4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸(pH5)または4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウム(pH5)を各ウェルに添加し、そのシャーレを室温で4日間保存する。ネガティブコントロールとして、培地を含むが、4%テトラデシルトリメチルシュウ酸アンモニウムと200mMの添加剤との混合物を添加していないウェルを室温で4日間保存する。
【0133】
ポジティブコントロールとして、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの標準市販の単離キットを用いて、予め保存することなく、1つのウェルからHela細胞のRNAを単離する。これを行うために、培地を細胞から完全に除去し、溶解バッファーRLT(RNeasy Kitの成分)350μlと混合する。細胞をウェルの底面からスパチュラで掻き取り、いわゆる破砕機、例えばQIAGEN社製QIAshredderなどに、ライセートを移す。14000rpmで2分間遠心することによって、そのライセートを破砕機に通し、このようにして、サンプルをホモジナイズする。その生成物を70%エタノールと混合し、実施例10に記載のようにRNAを単離する。
【0134】
4%テトラデシルトリメチルシュウ酸アンモニウム、200mMの添加剤と混合した培地中で、細胞を4日間保存した後、その時点で引き離されている細胞を上清と共に収集し、3000×gで5分間遠心分離する。その上清を除去し、細胞ペレットを用いて、実施例10に記載のようにRNAを単離する。
【0135】
4%テトラデシルトリメチルシュウ酸アンモニウム、200mMの添加剤を含まない(=ネガティブコントロール)培地中で細胞を4日間保存した後、ポジティブコントロールについて上述のように、RNAを単離する。
【0136】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図13に示す。レーン1は、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸(pH5)と混合した培地中で細胞を保存した後に単離された全RNAを含む。レーン2は、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウム(pH5)と混合した培地中で細胞を保存した後に単離された全RNAを示す。レーン3は、培地単独で細胞を保存した後に単離された全RNAを示し、レーン4は、予め保存することなく、ポジティブコントロールとして単離された全RNAを示す。
【0137】
水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このようにして得られたRNAの純度を、波長260nmでの光吸収に対する280nmでの光吸収の比を光度測定することによって測定する。単離の結果を以下の表11に示す。
【0138】
【表14】
【0139】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存していないサンプルにおいて、およびテトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物で保存したサンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れる。これに対して、テトラデシルトリメチルシュウ酸アンモニウムおよび添加剤を添加していない培地中でほぞんした細胞中のRNAは、ほぼ完全に分解される。同様に、OD測定によって決定されたRNAの収量および純度から、テトラデシルトリメチルシュウ酸アンモニウムおよび添加剤を添加していない培地中で保存されたサンプル中のRNAの収量および純度が著しく低下するのに対して、保存されていないサンプルおよび安定化されたサンプルとの間の相違がないことが確認される。
【0140】
実施例14:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる、組織中のRNAの安定化
これらの実験から、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムもまた、組織由来のRNAを安定化するのに適していることが分かる。
【0141】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、硫酸アンモニウムまたはリン酸カリウム、シュウ酸またはリン酸の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液または硫酸(もしくは硫酸アンモニウム)または水酸化カリウム溶液または燐酸(もしくはリン酸カリウム)で指定のpHに調節する。
【0142】
取り除いた直後に液体窒素で凍結し、次いで−70℃で保存しておいたマウスの腎組織をこれらの実験に使用する。その組織を70〜90mg凍結し、表12に指定するバッファー500μlを組織各10mgに対して添加し、例えばMessrs Kinematica社製のPolytronなどのローター・ステーターホモジナイザーを用いて、その混合物を即座に30〜60秒間ホモジナイズする。溶液のアリコート500μlを、組織10mgに相当するホモジナイズしたプレパラートから取る。サンプルを室温で1日保存する。
【0143】
保存後、サンプルを10000×gで3分間遠心分離し、上清を除去する。例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ボルテックスすることによってペレットを溶解する。次いで、70%エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有第2洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、RNaseを含有しない水40μlを膜上にピペッティングする。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出を完了する。
【0144】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図14に示す。サンプルを表12にまとめる。すべてのサンプルを2回実験し、その結果を示す。
【0145】
【表15】
【0146】
サンプル「K」は、予め保存することなく、単離キット(QIAGEN GmbH製RNeasy)を用いて、凍結された腎組織から単離した全RNAを示す(=ポジティブコントロール)。レーン「N」は、溶媒を添加していない、つまり乾燥している腎組織10mgを1日保存した後に、QIAGEN社製RNeasy(登録商標)Mini Kitを用いて単離した全RNAを示す(=ネガティブコントロール)。
【0147】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、ポジティブコントロールにおいてインタクトな28Sおよび18SrRNAが表れる。安定化溶液なしで保存した腎組織を含むネガティブコントロールは、完全に分解されたRNAを示す。これに対して、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウム中でサンプルを保存した後に、インタクトなrRNAバンドがポジティブコントロールとして視認することができる。安定化はpHにより影響を受ける。テトラデシルトリメチルシュウ酸アンモニウムと指定のpHの添加剤とを混合した後、組織中のRNAを安定化するために、安定化溶液の最終pHは4を超えるpHであることが好ましい。
【0148】
実施例15:
RNAの安定化および単離と並行したDNAの安定化および単離
これらの実験から、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いて、組織中のRNAのみならずDNAも安定化されることが分かる。サンプルからRNAを単離することに加えて、それと並行してDNAを単離することも可能である。
【0149】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5Mクエン酸(pH5)の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度となるように水酸化ナトリウムで調節する。
【0150】
取り除いた直後に液体窒素で凍結し、次いで−70℃で保存しておいたマウスの腎組織をこれらの実験に使用する。その組織を約80mg凍結し、4.2mlの4%テトラデシルトリメチルシュウ酸アンモニウム、200mMクエン酸(pH5)を組織各10mgに対して添加し、例えばMessrs Kinematica社製のPolytronなどのローター・ステーターホモジナイザーを用いて、その混合物をすぐに30〜60秒間ホモジナイズする。溶液のアリコート500μlを、組織10mgに相当するホモジナイズしたプレパラートから取る。そのサンプルを室温で1日保存する。
【0151】
保存後、サンプルを10000×gで3分間遠心分離し、上清を除去する。例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ボルテックスすることによってペレットを溶解する。次いで、70%エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、実施例14に記載のように単離することができる。スルーフロー(throughflow)(約1200μl)を回収し、100%エタノール200μlと合わせて、ボルテックスすることによって混合する。これらのサンプルを再度、例えばQIAGEN社製QIAampカラムなどのシリカ膜を含有する標準市販のスピンカラムに適用し、遠心することによって(10000×gで1分)膜に通す。DNAは膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有第2洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、水200μlを膜上にピペッティングし、室温で1分間インキュベートする。その溶出液を、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出を完了する。
【0152】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、0.8%アガロース−TBEゲルを調製する。サンプル1〜4のアリコート40μlを使用し、サンプル5〜9のアリコート20μlを使用する。その結果を図15に示す。
【0153】
レーン1および2は、実施例15に従って単離した全RNAを表す。レーン3および4は、参照としての全RNA0.1μgおよび0.5μgをそれぞれ表し、使用したアガロースゲルにおいてインタクトなゲノムDNAの流れ特性を示している。レーン5は、予め保存することなく、市販の単離キット(Messrs QIAGEN GmbHのQIAamp(登録商標)Mini Kits)を用いて、凍結ラット腎臓10mgから単離した全DNAを表している(=ポジティブコントロール)。使用したネガティブコントロールは、1日保存した後、溶媒を添加していない、つまり乾いた腎臓組織、または蒸留水中の腎臓組織10mgから、QIAGEN社製QIAamp(登録商標)Mini Kitsを用いて単離された全DANであった。このDNAは、レーン6および7(乾燥保存)において、かつレーン8および9(A.dest中で保存)において示される。
【0154】
ゲル電気泳動による分離によって、参照DNAを示すレーンおよび保存していないポジティブコントロールのDNAを含有するレーンのどちらにも、分解されていない高分子DNAが示される。組織を乾燥保存または水中で保存することによって、DNAの完全な分解が引き起こされる。これに対して、実施例15と同様に処理したサンプルはインタクトなままであり、保存期間全体を通して分解されない。したがって、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物は生物サンプル中のDNAを安定化するのにも適しており、サンプルからRNAおよびDNAを並行して単離することもまた可能となる。
【図面の簡単な説明】
【0155】
【図1】異なるpHレベルを有する様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による、血液中のRNAの安定化を示す。
【図2】様々な濃度の酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図3】250mM酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図4】GAPDH遺伝子(A)およびIFN−γ遺伝子(B)のmRNAの放射標識プローブとのノーザンハイブリダイゼーションの結果として、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図5】酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる血液中のDNAの安定化を示す。
【図6】ゲノムDNAを酵素反応に使用した場合の結果を示す。
【図7】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化を示す。
【図8】酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの様々な期間にわたる安定化を示す。
【図9】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿1ml中のRNAの安定化を示す。
【図10】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによるLeLa細胞中のRNAの安定化を示す。
【図11】異なる量のHeLa細胞中のRNAの安定化を示す。
【図12】マクロファージ中のRNAの安定化を示す。
【図13】培養液を除去しない、Hela付着細胞中のRNAの安定化を示す。
【図14】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる腎組織中のRNAの安定化を示す。
【図15】RNAの安定化および単離と同様な、DNAの安定化および単離を示す。
【技術分野】
【0001】
本発明は、生物由来の材料中の核酸を単離および/安定化するための新規な組成物に関する。その組成物は、
一般式:
【化1】
(式中、Yは、窒素またはリンを示すことができ、
R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、
X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオンを示すことが可能である)で示されるカチオン化合物と、添加剤としての少なくとも1種類のプロトン供与体と、を必須成分として含有する。
【0002】
好ましい組成物は、そのカチオン化合物がアンモニウム塩からなる組成物である(式中、R1が、高級アルキル基を示し、好ましくは炭素原子12個、14個または16個を有する高級アルキル基を示し、いずれの場合も、R2、R3およびR4がメチル基を示す)。
【0003】
式中、R1が、アルキル基、好ましくはベンジル基を示し、R2が、好ましくは炭素原子12個、14個または16個を有する高級アルキル基を示し、R3およびR4がメチル基を示す組成物もまた好ましい。
【0004】
臭化物、塩化物、リン酸塩、硫酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、シュウ酸塩またはコハク酸塩がアニオンとして好ましい。
【0005】
C1〜C6アルキルは一般に、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい、炭素原子1〜6個を有する分枝もしくは非分枝炭化水素基を示す。以下の炭化水素基:
メチル、エチル、プロピル、1−メチルエチル(イソ−プロピル)、ブチル、1−メチルプロピル、2−メチルプロピル、1,1−ジメチルエチル、n−ペンチル、1−メチルブチル、2−メチルブチル、3−メチルブチル、1,1−ジメチルプロピル、1,2−ジメチルプロピル、2,2−ジメチルプロピル、1−エチルプロピル、ヘキシル、1−メチルペンチル、2−メチルペンチル、3−メチルペンチル、4−メチルペンチル、1,1−ジメチルブチル、1,2−ジメチルブチル、1,3−ジメチルブチル、2,2−ジメチルブチル、2,3−ジメチルブチル、3,3−ジメチルブチル、1−エチルブチル、2−エチルブチル、1,1,2−トリメチルプロピル、1,2,2−トリメチルプロピル、1−エチル−1−メチルプロピルおよび1−エチル−2−メチル−プロピルが例として挙げられる。
【0006】
高級アルキル基という用語は、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい分枝もしくは非分枝C7〜C20アルキル基を表す。以下の炭化水素基:分枝もしくは非分枝ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、テトラデシル、ヘキサデシル、ドデカデシルおよびエイコシルが例として挙げられる。
【0007】
C3〜C6アルケニルは一般に、炭素原子3〜6個を有し、1つまたは可能ならば複数の二重結合を有する、分枝または非分枝炭化水素基であって、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい炭化水素を示す。以下の炭化水素基:
2−プロペニル(アリル)、2−ブテニル、3−ブテニル、1−メチル−2−プロペニル、2−メチル−2−プロペニル、2−ペンテニル、3−ペンテニル、4−ペンテニル、1−メチル−2−ブテニル、2−メチル−2−ブテニル、3−メチル−2−ブテニル、1−メチル−3−ブテニル、2−メチル−3−ブテニル、3−メチル−3−ブテニル、1,1−ジメチル2−プロペニル、1,2−ジメチル2−プロペニル、1−エチル−2−プロペニル、2−ヘキセニル、3−ヘキセニル、4−ヘキセニル、5−ヘキセニル、1−メチル−2−ペンテニル、2−メチル−2−ペンテニル、3−メチル−2−ペンテニル、4−メチル−2−ペンテニル、1−メチル−3−ペンテニル、2−メチル−3−ペンテニル、3−メチル−3−ペンテニル、4−メチル−3−ペンテニル、1−メチル−4−ペンテニル、3−メチル−4−ペンテニル、4−メチル−4−ペンテニル、1,1−ジメチル2−ブテニル、1,1−ジメチル2−ブテニル、1,1−ジメチル3−ブテニル、1,2−ジメチル2−ブテニル、1,2−ジメチル3−ブテニル、1,3−ジメチル2−ブテニル、1,3−ジメチル3−ブテニル、2,2−ジメチル3−ブテニル、2,3−ジメチル2−ブテニル、2,3−ジメチル3−ブテニル、1−エチル−2−ブテニル、1−エチル−3−ブテニル、2−エチル−1−ブテニル、2−エチル−2−ブテニル、2−エチル−3−ブテニル、1,1,2−トリメチル2−プロペニル、1−エチル−1−メチル−2−プロペニルおよび1−エチル−2−メチル−2−プロペニルが例として挙げられる。
【0008】
C3〜C6アルキニルは一般に、炭素原子3〜6個を有し、1つまたは可能ならば複数の三重結合を有する、分枝または非分枝炭化水素基であって、互いに同一または異なる1個または複数のハロゲン原子、好ましくはフッ素によって任意に置換されてもよい炭化水素を示す。以下の炭化水素基:
2−プロピニル(プロパルギル)、2−ブチニル、3−ブチニル、1−メチル−2−プロピニル、2−メチル−2−プロピニル、2−ペンチニル、3−ペンチニル、4−ペンチニル、1−メチル−2−ブチニル、2−メチル−2−ブチニル、3−メチル−2−ブチニル、1−メチル−3−ブチニル、2−メチル−3−ブチニル、3−メチル−3−ブチニル、1,1−ジメチル2−プロピニル、1,2−ジメチル2−プロピニル、1−エチル−2−プロピニル、2−ヘキシニル、3−ヘキシニル、4−ヘキシニル、5−ヘキシニル、1−メチル−2−ペンチニル、2−メチル−2−ペンチニル、3−メチル−2−ペンチニル、4−メチル−2−ペンチニル、1−メチル−3−ペンチニル、2−メチル−3−ペンチニル、3−メチル−3−ペンチニル、4−メチル−3−ペンチニル、1−メチル−4−ペンチニル、3−メチル−4−ペンチニル、4−メチル−4−ペンチニル、1,1−ジメチル2−ブチニル、1,1−ジメチル2−ブチニル、1,1−ジメチル3−ブチニル、1,2−ジメチル2−ブチニル、1,2−ジメチル3−ブチニル、1,3−ジメチル2−ブチニル、1,3−ジメチル3−ブチニル、2,2−ジメチル3−ブチニル、2,3−ジメチル2−ブチニル、2,3−ジメチル3−ブチニル、1−エチル−2−ブチニル、1−エチル−3−ブチニル、2−エチル−1−ブチニル、2−エチル−2−ブチニル、2−エチル−3−ブチニル、1,1,2−トリメチル2−プロピニル、1−エチル−1−メチル−2−プロピニルおよび1−エチル−2−メチル−2−プロピニルが例として挙げられる。
【0009】
別段の指定がない限り、アリールは、ヘテロ原子1個または2個を任意に含有することができ、炭素原子4〜22個を有する芳香族単核基または多核基を示す。その例には:ハロゲン(F、Cl、Br、I)、好ましくはフッ素によって、またはアルキル基によって、互いに独立して任意に一置換または多置換することができる、フェニル、ナフチル、アントラシルまたはピロール、フラン、チオフェン、ピリジン、ピリダジン、ピリミジンまたはピラジンが含まれる。
【0010】
上記に定義されたように、アラルキルは、C1〜C6アルキレン、C3〜C6アルケニレンもしくはC3〜C6アルキニレン架橋を介してカチオン部分構造に結合する、単核または多核アリール基を示し、そのC1〜C6アルキル、C3〜C6アルケニルおよびC3〜C6アルキニル基は上記で定義したとおりである。本発明の目的には、ベンジル基が好ましい。
【0011】
適切な対イオンX−には、ハロゲン化水素酸のすべてのアニオン、または酢酸塩もしくはシュウ酸塩、マロン酸塩、コハク酸塩もしくはクエン酸塩などの、一塩基または二塩基有機酸のアニオンが含まれることが好ましい。
【0012】
本発明の目的のための適切なプロトン供与体は主に、無機酸またはその塩の他に、単独でまたは組み合わせて、飽和脂肪族モノカルボン酸、不飽和アルケニル−カルボン酸、飽和および/または不飽和脂肪族C2〜C6ジカルボン酸、脂肪族ケトカルボン酸またはケトジカルボン酸ならびにアミノ酸である。上述のすべての有機酸を未置換状態で、または置換誘導体として使用することができ、別段の指定がない限り、未置換誘導体またはヒドロキシル基によって一置換もしくは多置換された誘導体であることが好ましい。
【0013】
本発明の目的のための飽和脂肪族モノカルボン酸という用語には、ギ酸の他に、C1〜C6アルキル−カルボン酸が含まれることが好ましく、酢酸、プロピオン酸、n−酪酸、n−吉草酸、イソ吉草酸、エチル−メチル−酢酸(2−メチル−酪酸)、2,2−ジメチルプロピオン酸(ピバル酸)、n−ヘキサン酸、n−オクタン酸、n−デカン酸、およびn−ドデカン酸(ラウリン酸)が好ましい。さらに、上述の酸から誘導されるケトカルボン酸もまた使用することができる。
【0014】
本発明の目的に使用される不飽和アルケニル−カルボン酸の例には、例えばアクリル酸(プロペン酸)、メタクリル酸、クロトン酸、イソクロトン酸ならびにビニル酢酸が含まれる。
【0015】
本発明に従って、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸もしくはアジピン酸などの飽和脂肪族C2〜C6ジカルボン酸が好ましく、シュウ酸およびコハク酸が特に好ましい。
【0016】
本発明に従って問題を解決するためには、脂肪族ヒドロキシ−ジ−およびトリ−カルボン酸を使用することが特に好ましく、タルトロン酸、D−(+)、L−(−)−もしくはDL−リンゴ酸、(2R,3R)−(+)−酒石酸、(2S,3S)−(−)−酒石酸、メソ−酒石酸およびクエン酸が最も特に好ましい。
【0017】
マレイン酸もしくはフマル酸などの不飽和ジカルボン酸または、例えばアコニット酸などの不飽和トリカルボン酸もまた、本発明の問題の解決策として適している。
【0018】
しかしながら、本発明の目的のために、例えばメソシュウ酸およびオキサロ酢酸などの脂肪族ケトジカルボン酸も添加剤として使用することができ、オキサロ酢酸が特に最も特に好ましい。
【0019】
さらに、本発明に従って、アミノ酸を使用することもでき、例えばアミノ酢酸(グリシン)、α−アミノプロピオン酸(アラニン)、α−アミノ−イソ吉草酸(バリン)、α−アミノ−イソカプロン酸(ロイシン)およびα−アミノ‐β−メチル吉草酸(イソロイシン)などのα−アミノ酸が好ましい。グリシンを使用するのが最も好ましい。
【0020】
上述のプロトン供与体は、個々の物質として、または純粋な立体異性体の形態で、および混合物の状態でも、使用することができる。
【0021】
無機酸およびその塩を、本発明に従って、更なる添加剤として使用することもできる。アルカリ金属もしくはそのアンモニウム塩と、リン酸もしくは硫酸などの無機酸との塩を使用することが好ましい。リン酸および硫酸アンモニウムを使用することが最も好ましい。
【0022】
本発明の目的における核酸という用語は、より広い意味での核酸を意味し、したがって、例えば、二本鎖、一本鎖、環状および直鎖、分枝などの、すべての長さまたは立体配置のリボ核酸(RNA)およびデオキシリボ核酸、およびそのすべての可能なサブユニット、例えばヌクレオチドモノマー、オリゴマー、プラスミド、細菌DNAおよびRNA、ならびに動物および植物細胞由来のゲノムおよび非ゲノムDNAおよびRNA、プロセシングされた形態のmRNA、プロセシングされていない形態のmRNA、tRNA、hnRNA、rRNA,cDNAならびに考えられる他のすべての核酸が含まれる。
【0023】
使用される、核酸を有する生物サンプルは、例えば参照により本明細書に組み込まれる欧州特許出願第95909684.3号に開示されているように、無細胞サンプル材料、血漿、血液、血清、細胞、白血球画分、クルスタ・フロジスティカ(crusta phlogistica)、痰、尿、精液、糞便、スメア、吸引液などの体液、生検材料、例えば組織および器官の一部など、すべての種類の組織サンプル、本発明に従って考えられる、遊離もしくは結合核酸または核酸を含有する細胞を含む食物サンプル、例えば生物体(単細胞または多細胞生物;昆虫等)、植物および植物の器官、細菌、ウイルス、酵母および他の菌類または真核生物および原核生物等、あるいは遊離核酸であることができる。
【0024】
本発明の技術的背景に関して:
その核酸を研究することによって、細胞の遺伝的起源および機能活性を決定かつ検討することができることは、従来技術から十分によく知られている。核酸およびタンパク質の分析によって、細胞活性の原因への直接的なアクセスが得られる。このように、それらは潜在的に、例えば代謝産物の検出など、間接的な従来の方法より優れている。したがって、分子生物学的分析は、多くの分野、例えば医学および臨床診断において、医薬組成物の開発および評価を行う製薬分野において、食品分析において、食品製造のモニタリングにおいても、作物および家畜を育てる農業において、ならびに環境分析および多くの研究分野において既に用いられている。
【0025】
RNA、特に細胞中のmRANを分析することによって、遺伝子の活性を直接決定することが可能である。分子生物学の最新方法、例えば実時間逆転写酵素PCRまたは遺伝子発現チップ分析による、細胞における転写パターン(mRNAパターン)の定量分析によって、例えば異常に発現した遺伝子を検出し、それによって、代謝異常、感染、または癌の発生を検出することが可能となる。例えば、PCR、RFLP、AFLP、SNPもしくはシークエンシングなどの分子生物学的方法による、細胞からのDNAの分析によって、例えば遺伝的欠陥を検出すること、またはHLAタイプおよび他の遺伝標識を決定することが可能となる。
【0026】
ゲノムDNAおよびRNAの分析もまた、ウイルス、細菌等の感染性病原体を直接検出するのに用いられる。
【0027】
その自然環境から生物サンプルを採取した直後に核酸およびタンパク質を安定化することは、核酸の分析に絶対に必須である。これは、生物サンプルを採取した後、非常に急速に分解し得るDNA、特にRNAに当てはまる。一方、生物サンプルを一旦採取すると、ストレス遺伝子の誘導によって、新たなmRNA分子が合成され、その結果例えば、細胞の転写パターンが変化する可能性がある。これによって、その後の分析が不正確となる可能性がある。特に医学分野では、核酸を含有するサンプルを採取し、次いで、それらを最初にしばらくの間保存し、実験室に輸送されるまで、さらに研究されないのが実際に一般的であるため、核酸を安定化することは必須である。
【0028】
一方、サンプル中に含有される核酸は、変化するか、または完全に分解さえし得る。これは当然、続いて行われる試験の結果に多大な影響を及ぼすか、またはそれらを完全に不可能とする。かかる試験は、例えばノーザンおよびサザンブロット分析、PCR、RT−PCR、SunRise、LCR、分枝DNA(bDNA)、SDA、DNAおよびRNAチップおよび遺伝子発現アレイおよび突然変異分析、RFLP、AFLP、SNP分析、cDNA合成、サブトラクティブ・ハイブリダイゼーション(subtractive hybridisation)またはTaqman技術および他の実時間の定量法など、分子生物学的技術を用いて行われる。一方、インタクトな高精製核酸−DNAまたはRNAの使用は、上記の試験の使用または実施に対する基本的な適合性の基準となる。さらに、核酸を含有するサンプルの単離およびアッセイもまた、時間のかかる作業である。さらに、例えば試験が失敗した場合に生じるような、分子生物学分野で使用される研究実験室の汚染が、試験結果のエラーをまねく可能性がある。
【0029】
従来技術に関して
数多くの出版物では、例えば組織などの適切なサンプルから核酸を続いて単離するための、固定液としてのエタノールおよびアセトンをベースとする混合物の使用が提案されている。この文献を研究した後、この種のエタノール/アセトン混合物は、RNAの安全な回収に課せられる必要条件をすべて満たすわけではないことが明らかである。このように、この種の混合物は、分解からRNAを保護することができない。さらに、さらに広範な細胞凝集魂から構成される固体サンプル中でRNAが保護される保証はない。さらに、提案された混合物は高い引火性または爆発性であり、実験室で作業する際にかなり危険性がある。
【0030】
それに加えて、さらに周辺の関連する従来技術は、固定化または保存された組織サンプルからのRNAの回収に関係する。これは特に、in situハイブリダイゼーション中に達成されるシグナル強度を最大にする、組織標本の適合性に関する。換言すれば、この種の実験は、それを保存するよりもむしろ、RNAを検出することを意図するものである(米国特許第5196182号および5260048号)。
【0031】
他のレポートは、このようにして得られた断片をPCRによる制限分子分析にかけることを可能にするために、固定組織から断片化RNAまたはDNAを回収することに関する。この種の断片化DNAまたはRNAを得るために、構造組織成分を分解することを可能にするため、対応するサンプルを従来法によりプロテイナーゼKで処理する;次いで、単に、グアニジウム塩を含有する溶液でRNAを抽出する。しかしながら、この方法によって固定組織から得られたRNAは、品質が低く、大きさがわずか約200塩基である。従来技術に従って、特に固定化中の細胞内基質内のDNAまたはRNAの内因性かつ架橋反応の負の影響を含む一定数の特定因子に、これを抑えることができる。DNAまたはRNAが大部分の場合で少なくとも一部分解するという事実に基づくと、このようにして得られたDNAまたはRNAは、もはやノーザン分析に上手く使用することはできない。このように単離されたRNAは、比較的小さな断片を増幅するためにのみであるが、せいぜい多少の成功の見込みを持って、RT−PCR反応に使用することができる程度である。
【0032】
従来技術では、凝固点を超える温度でRNAを保存するための、硫酸アンモニウムの使用もまた記述されている[WO00/06780]。この種の組成物は、RNAlaterの名称で従来技術に従って使用されている。しかしながら、この種の硫酸アンモニウム水溶液は、血液、血漿または血清中のRANを安定化するのには適していない。上記のサンプルは高いタンパク質濃度を有するため、この種のアンモニウム塩溶液と接触するとすぐに、限られた溶解度の沈殿物が形成する[Messrs Ambion(アメリカ、テキサス州オースティン)からのRNAlater製品の情報]。
【0033】
さらに、生物サンプルから核酸を単離するために、この種のカチオン化合物を使用することは、従来技術から知られている。かかる用途は、例えば米国特許第5,010,183号および同第5,300,635号および欧州特許:EP0442026に記載されている。これらの出版物では、サンプル調製に通常用いられるインキュペーション時間、つまり約数分の間、生物サンプルをカチオン化合物と共にインキュベートし;次いで、その核酸を再度精製する。
【0034】
従来技術から公知の化合物の研究によって、従来技術で言及されているカチオン化合物、特に米国特許に開示されているテトラデシルトリメチルシュウ酸アンモニウムは、例えば血液が長期間保存される場合に、それら自体で細胞RNAの適切な安定化を保証するものではないことが示されている。
【0035】
実を言えば、例えば数日間血液中のウイルスを安定化することに着手している実験は従来技術から知られているが、これらの知見では、インタクトなままのRNAについて何らの言及もされていない。このように、シュミット(Schmidt)およびマクファーレン(MacFarlane)[J.Medical Virology 47,(1995) 153]は、Catrimox−14(登録商標)による血液中のC型肝炎ウイルスの周囲温度で7日間の安定化について記述している。そのウイルスは、HCVゲノムの長さ250bpの断片をRT−PCRにより増幅することによって検出された。しかしながら、開示されたその結果からは、小さな断片のみが増幅されたため、RNAがインタクトである十分な証拠は得られなかった。さらに、その実験は、不定のウイルスサンプルを添加して行われ、そのため、保存中のウイルスRNAの分解に関して判断を下すことは不可能であった。
【0036】
さらに、国際特許出願:WO99/29904には、塩酸グアニジン、チオシアン酸グアニジン、塩化リチウム、塩化マンガン、サルコシル、SDS、過塩素酸ナトリウム、サリチル酸ナトリウムおよびチオシアン酸ナトリウムと共にEDTA、EGTA、またはBAPTAを用いた体液中のDNAの安定化が記載されている。さらに、例えばTrizol(登録商標)などのフェノールを含有する試薬を使用して、保存中にRNAを安定化することができることは従来技術から知られている。しかしながら、これらの試薬はすべて、健康に非常に有害であり、したがって、常用するのには適していない。
【0037】
そこで、本発明の目的は、組織または血液、血漿または血清の存在下にてRANを安定化する組成物を提供することである。
【0038】
本発明はさらに、安定化溶液の形で組成物を提供することにも言及しており、その組成物の成分は健康に有害ではなく、したがって、サンプルを入手した場所から実験室へ移動する間、例えば生物サンプル材料を安定化するために、サンプルを扱うスタッフの健康を脅かす危険性なく使用することも可能である。
【0039】
本発明の他の目的は、安定化試薬それ自体もまた溶液中で安定なままである必要性を満たし、かつ例えば限られた溶解度の沈殿物の溶解など、使用者による前処理が必要ない安定化溶液の形で組成物を提供することである。この種の前処理は常に、安定化効率の変動のリスクを含む。
【0040】
本発明の他の目的は、多用途である、つまり幅広い生物サンプルに使用することができる組成物を提供することである。
【0041】
驚くべきことに、特に米国特許第5 010 183号および同第5 300 645号に開示されており、かつ本発明に従って上述の1種または複数種の添加剤と組み合わせられる化合物などのカチオン化合物と、生物サンプルの核酸を接触させた場合に、長時間にわたり核酸を安定化することができることが現在見出されている。本発明に従って問題を解決するのに適切な好ましい添加剤を表1に示す:
【0042】
【表1】
【0043】
添加剤は、様々な濃度で安定化試薬中に存在することができる;例えば、添加剤は、安定化溶液と血液との混合物中に、容積比1:1、好ましくは3:1、濃度50mM〜飽和、好ましくは100〜1M、最も好ましくは200〜500mMで存在することができる。添加剤の性質に応じて、他の濃度範囲が有利であることが証明される。異なる添加剤の組み合わせを使用することも可能である。
【0044】
組成物水溶液中のカチオン化合物濃度は、0.01重量%〜飽和、好ましくは0.1重量%〜飽和、さらに好ましくは0.5〜15重量%、最も好ましくは2〜10重量%の範囲である。
【0045】
当然、カチオン化合物と添加剤との溶液を添加する場合には、最適な濃度は、生物サンプルのそれぞれの容積、および安定化溶液と生物サンプルとの容積比によって決定される。
【0046】
一般にカチオン化合物と添加剤との混合物のpHを、広いpH範囲(pH2〜12)にわたりサンプルの関数として変化させることができ、pH2〜pH10の範囲であることが好ましく、さらに好ましくはpH3〜pH8の範囲である。好ましいpH範囲は、使用する生物サンプルに依存する。血液、血漿および血清の場合には、pH2〜pH6の範囲、特にpH3〜pH4のpH値が好ましい。
【0047】
血液、血漿および血清は別にして他の細胞体液、または例えば細菌、吸引液、細胞、組織、および上述のサンプルなど、他の生物サンプルの場合、カチオン化合物および添加剤からなる安定化溶液のpH値は、好ましくはpH3〜pH10の範囲、さらに好ましくはpH4〜pH8の範囲である。
【0048】
生物サンプル中の核酸を安定化するために、カチオン化合物(1種または複数種)および添加剤を含有する溶液とサンプルを混合することができる。生物サンプルを0.1〜10,000容積添加することができ;範囲1〜1000の容積を添加することが好ましく、範囲1〜100の容積を添加することが最も好ましい。しかしながら、例えば細い針による針生検または低細胞数の培養から得たサンプルなど、サンプルの性質に応じて、場合によってはかなり高い容積もまた用いることができる。
【0049】
同様に、生物サンプル自体が固体を溶解する液体(例えば、細胞含有体液、培養液中の細胞、尿など)を含有する場合、または液体、例えば水をそれに添加して、固体を溶解する場合、上述のカチオン化合物および添加剤を固体状で添加することもできる。固体を添加する利点は、固体が通常、化学的に安定であり、固体はサンプルに添加するのが容易である場合が多いことである。
【0050】
さらに、組織などの非常に緻密な生物サンプルの場合は特に、例えば、核酸もしくは個々の細胞もしくは細胞の凝集塊の開放を補助するために、安定化溶液中でまたは安定化溶液と混合する前に、例えばサンプルに対する機械的、化学的、物理的もしくは酵素的作用により緻密なサンプルを破壊することによって、サンプルを粉砕またはホモジナイズすることが可能である。機械的作用は、電気メス、ビーズミルを用いて、または例えばシリンジで押しつぶすことによって行うことができ、サンプルに対して作用する適切な酵素は、例えば加水分解酵素、プロテアーゼまたはリパーゼである。
【0051】
さらに、そのサンプルは、純粋な物理的手段、例えば超音波によって前処理してもよい。
【0052】
前処理は、化学的に、単独でまたは純粋な物理的方法と共に、実行することもできる。溶解を補助する手段には、例えば、脂肪族アルコール、特にイソプロパノール、またはアルデヒドまたはジアルデヒド、例えばグリオキサール、またはフェノールまたはフェノール誘導体、例えば2−ビフェニロール、またはイオン、双性イオンおよび非イオン化合物、例えばメルカプト、または還元剤、例えばジチオスレイトールおよびβ−メルカプトエタノール、またはリン酸誘導体、例えばリン酸トリブチル、またはカオトロピック試薬、例えば尿素、チオシアン酸グアニジウムまたは塩酸グアニジウム、または塩を個々にまたは組み合わせて使用することが含まれる。
【0053】
機械的、化学的、物理的または酵素的にサンプルに作用する他の可能な方法は、当技術分野で公知であり、本明細書に含まれることを意図する。
【0054】
特定の必要条件に応じて、サンプル材料をかなり長い期間、例えば室温で1日〜14日以上保存してもよいが、例えば40℃以上の高温、および例えば4℃または−20℃以下などの低温でも保存してもよい。
【0055】
上述の化合物の溶液中での生物サンプルの保存に続いて、直接核酸を分析する技術を用いるか、または核酸をサンプルから精製してもよい。
【0056】
例えば、ブロット法、生体分子を分離するゲル電気泳動法およびクロマトグラフィー法によって、核酸を直接検出/分離してもよい。
【0057】
生物サンプルから核酸を精製するために、遊離核酸または核酸を含有する細胞もしくは粒子を、例えば遠心分離または濾過によって溶液の残りから分離し、米国特許第5,010,183号、同第5,300,645号および欧州特許出願第99103457.0号に記載されているように、少量で有利に行うことができる更なる精製にかける。
【0058】
保存容器中で核酸または核酸を含有する細胞もしくは粒子を直接分離することによって、精製のために他の容器にサンプルを移す他の段階が不要となり、そのため、サンプルの損失が減少し、サンプルから採取された核酸による互いの混同および汚染のリスクもまた極力抑えられる。このように、これらの安定化試薬を使用することによって、生物サンプル中の核酸を安定化し、直接単離する一段階方法が導かれ、その方法では、代わりに生物サンプルからRNAおよびDNAを単離すること、または1つのサンプルから並行してRNAおよびDNAを単離することができる。
【0059】
1種または複数種のカチオン化合物および1種または複数種の添加剤を含有する、本発明による組成物を用いて核酸を安定化することによって、サンプル中の核酸は、長期の保存中でさえ、または輸送中に変化しないことが確かなものになる。したがって、その後の試験の精度が著しく向上する。ある特定の場合、例えばサンプル材料を長距離間輸送するか、または長期間保存しなければならない場合では、本発明による方法によって、これらの試験をかかる期間の後に行うことが初めて可能となる。
【0060】
本発明の利点は特に、例えば、サンプリング直後に固定化しなければならない転写物レベルを分析する研究分野、および例えば、分析の準備が整うまで保存および輸送中に患者のサンプルも安定化しなくてはならない、分子診断などの臨床分析分野において見出されている。特に、核酸の単離および安定化は、腫瘍の診断において、遺伝性疾患の診断ならびにウイルスの診断およびモニタリングにおいて、他の感染性病原体の診断およびモニタリングにおいて、遺伝子発現パターンの分析において使用される。
【0061】
本発明の適用分野は、医学または動物学分野に及ぶだけではなく、植物、真菌および原核生物系の分析も含む。培養および自然環境から得た、植物および植物器官、藻類、真菌および細菌からの核酸の安定化および単離は、例えば転写物レベルおよび遺伝子発現パターンを分析する研究、および土壌サンプル中の細菌等の複合個体群における種を同定かつ定量する研究において使用される。
【0062】
可能性のある用途には、例えば食物分析などの他の分析分野も含まれる。
【0063】
本発明を以下の実施例および図によって説明する。記述および実施例において、以下の略語が使用される:
AFLP 増幅断片長多型
A.dest. 蒸留水
BAPTA1 2−ビス(2−アミノフェノキシ)エタン−N,N,N’, N’−テトラ酢酸
EcoRI 制限酵素大腸菌株R
E260/E280 260および280nmでの吸光度の商
EDTA エチレンジアミン−N,N,N’,N’−テトラ酢酸
EGTA [エチレンビス(オキシエチレンニトリロ)]テトラ酢酸
GAPDH グリセリンアルデヒド−3−ホスフェート−デヒドロゲナーゼ
Hind III 制限酵素インフルエンザ菌
hugl 巨大な幼虫のヒトホモローグ
IFN−γ インターフェロンガンマ
LM 長さのマーカー
MOPS 3−(N−モルホリノ)−2−ヒドロキシプロパンスルホン酸
nb 決定されず
NonidetP40 imbentin−N/52;オクチルフェニルポリエチレングリコール
OD 光学密度
PBS リン酸緩衝食塩水
PCR ポリメラーゼ連鎖反応
RFLP 制限酵素断片長多型
rpm 毎分回転数
mRNA メッセンジャーRNA
rRNA リボソームRNA
RT 室温
RT−PCR 逆転写酵素 PCR
SDS ドデシル硫酸ナトリウム
SNP 一塩基多型
SSC 塩化ナトリウム/クエン酸ナトリウム溶液
TBE トリス−ホウ酸−EDTAバッファー
Tris 2−アミノ−2−(ヒドロキシメチル)−1,3−プロパンジオール
U ユニット
【0064】
例えば時間に対してhなど、ここに列挙していない略語は、当業者にはよく知られているか、あるいは従来技術におけるそれらの使用から十分によく知られているだろう。
それらが基づく図および実験の説明
図1は、異なるpHレベルを有する様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による、血液中のRNAの安定化を示す。
図2は、様々な濃度の酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図3は、250mM酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図4は、GAPDH遺伝子(A)およびIFN−γ遺伝子(B)のmRNAの放射標識プローブとのノーザンハイブリダイゼーションの結果として、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
図5は、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる血液中のDNAの安定化を示す。
細胞RNAの他に、白血球由来のゲノムDNAもまた、ここで開発された方法によって安定化し、次いでシリカ膜に結合させることによって単離することができる。図5は、72時間保存した後でさえ、高分子DAN(長さ>20kB)が単離されることを示している。
図6は、ゲノムDNAを酵素反応に使用した場合の結果を示す。24時間または72時間保存した後に単離したDNA(実施例5を参照のこと)を様々な酵素反応に使用する。
A.制限酵素EcoRI(E)またはHindIII(H)6Uを使用して、DNA2μgを37℃にて3時間切断し、次いで0.8%アガロース/TBEゲル上で分離する。いずれの場合も対照として、未切断のDANを適用する。
B.ゲノムDNAのアリコート150ngおよび300ngをPCR反応に使用する(総容積50μl)。その反応では、hugl(巨大な幼虫のヒトホモローグ)遺伝子の長さ1.1kB断片を増幅する。そのPCR産物を1.2%アガロース/TBEゲル上で分離する。
図7は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表2に示す。
図8は、酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの様々な期間にわたる安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表3に示す。
図9は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿1ml中のRNAの安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート30μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表4に示す。
図10は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによるLeLa細胞中のRNAの安定化を示す。
サンプルは、二重測定で調製し、サンプル14、40、66および92は一重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表5に示す。
図11は、異なる量のHeLa細胞中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表7に示す。
図12は、マクロファージ中のRNAの安定化を示す。サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表9に示す。
図13は、培養液を除去しない、Hela付着細胞中のRNAの安定化を示す。溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表13に示す。
図14は、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる腎組織中のRNAの安定化を示す。
サンプルはすべて、二重測定で調製する:溶出液のアリコート20μlを1%アガロース−ホルムアルデヒド−MOPSゲルで分離する。問題のサンプルを表12に示す。
図15は、RNAの安定化および単離と同様な、DNAの安定化および単離を示す。溶出液のアリコート40μlを0.8%アガロース−TBEゲルで分離する。問題のサンプルは実施例15に記述する。
【0065】
実施例
実施例1:
異なるpHレベルの様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による血液中のRNAの安定化
様々な鎖長のカルボン酸を添加剤として使用する。さらに、モノ、ジおよびトリカルボン酸、ヒドロキシル化および非ヒドロキシル化カルボン酸を試験する。そのすべての物質は、カチオン化合物テトラデシルトリメチルシュウ酸アンモニウムと共に安定化に使用される。その物質のpHおよび濃度のどちらも変化する。
【0066】
図1に、その試験の結果を示す。どの場合でも、インタクトなRNAを24および48時間後でさえ単離することができる。場合によっては少量であるRNAの量は、処理される血液が少量であること、および様々な血液サンプル中のRNA含有量が異なることに対応するものである。この実験では、いくらかのゲノムDNAもまた、RNA画分中で得られた。
【0067】
各濃度200mMの、かつ問題のカルボン酸に対して様々なpHレベルの様々なカルボン酸で緩衝された10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムからなるバッファー500μlで、血液500μlを室温で24時間および48時間保存する。RNAを単離するために、カチオン化合物および核酸からなる複合体を遠心分離する;そのペレットを水で1回洗浄し、再度遠心分離し、例えばQIAGEN社製RLTバッファーなど、標準市販の溶解バッファー300μl中に溶解する。サンプルを水360μlで希釈し、プロテイナーゼK40μlで55℃にて10分間処理する。次いで、そのサンプルを遠心分離し、エタノールを上清に添加し、シリカ膜を含むスピンカラムにそれを添加する。遠心することによって、サンプルを膜に通す。スピンカラムは、市販のイソチオシアン酸グアニジニウム含有洗浄バッファー、例えばQIAGEN社製バッファーRW1で1回洗浄し、QIAGEN社製バッファーRPEなどの標準市販のアルコール含有洗浄バッファーで2回洗浄し、次いで、遠心することによって膜にも通されるRNase非含有水60μl中にRNAを溶出し、その溶出液のアリコート30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離する。
【0068】
実施例2
テトラデシルトリメチルシュウ酸アンモニウムおよび様々な濃度の酒石酸(緩衝)pH3を用いた、全血中のRNAの安定化
10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび50〜500mM酒石酸(pH3)からなるバッファー500μlで、血液500μlを室温で2.5時間、24時間および48時間保存する。さらに、QIAGEN社製「RNase非含有DNaseセット」でサンプルをDNaseで処理することによって、ゲノムDNAを除去することを除いては、図1に示すようにRNAを単離する。RNase非含有水80μlでRNAを溶出する。その溶出液30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離する。
【0069】
実施例3
250mM酒石酸(pH3)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウムを用いた、全血中のRNAの安定化
RNAの完全性、収量および純度を決定する:
カルボン酸バッファー、例えば250mM酒石酸(pH3.0)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウム溶液中で分解または収量の減少を生じることなく、血液中でRNAは少なくとも72時間安定化される(図3を参照のこと)。
【0070】
10%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび250mM酒石酸(pH3.0)からなるバッファー2mlと血液2mlとを混合し、室温で24〜72時間保存する。例えばQiagen GmbH社製バッファーELなどの標準市販の赤血球溶解バッファーを、カチオン化合物および核酸からなる複合体を遠心分離する前にサンプルに添加し、次いでその混合物を氷上で10分間インキュベートすることを除いては、実施例2に記載のように、RNAを単離する。RNase非含有水80μlでRNAを溶出する。その溶出液30μlを1.2%アガロース/ホルムアルデヒドゲル上で分離するか、または分光光度計で測定する。水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このように、波長260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって、得られたRNAの純度を測定する。
【0071】
実施例4:
酒石酸(pH4)で緩衝された、異なる濃度のテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化
ノーザンブロット分析
4%テトラデシルトリメチルシュウ酸アンモニウムおよび200 mM酒石酸(pH3.7)からなるバッファー6.9mlと血液2.5mlとを混合し、室温で1時間、24時間、48時間および72時間保存する。RNAを単離するために、カチオン化合物と核酸との複合体を遠心分離する。そのペレットを水で1回洗浄し、次いで例えばQiagen社製バッファーRLTなどの溶解バッファー300μl中に溶解する。サンプル調製の残りの作業を、図2に記載のように行う。次いで、全RNAのアリコート2.5μgを1.2%変性アガロース/ホルムアルデヒドゲル上で分離する。次いで、そのRNAをナイロン膜に移し、リン酸ナトリウム/SDSバッファー中で68℃にて、GAPDH遺伝子(図4A)、またはIFN−γ遺伝子(図4B)の放射標識アンチセンスRNAプローブと約12時間にわたりハイブリダイズする。減少する塩濃度2×SSC/0.1%SDS〜0.1×SSC/0.1%SDSの洗浄バッファーで温度68℃にて、その膜を洗浄する。次いで、ナイロン膜をX線フィルム上で暴露する。GAPDHおよびIFN−γ−mRNAのシグナルはどちらも、72時間を超える保存期間にわたり一定のままである。
【0072】
実施例5:
酒石酸(pH3.7)で緩衝されたテトラデシルトリメチルシュウ酸アンモニウムによる血液中のゲノムDNAの安定化
細胞RNAの他に、全血からのゲノムDNAもまた、本発明において開発された方法によって安定化することができ、次いでシリカ膜に結合させることによって単離することができる。図5から、室温で72時間保存した後でさえ、高分子DNA(長さ>20kB)が単離されることが示されている。
【0073】
4%(w/v)テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸(pH3.7)からなる溶液6.9mlと血液2.5mlとを混合し、室温で24時間または72時間保存する。DNAを単離するために、カチオン化合物とDNAとの複合体を遠心分離する。塩化ナトリウムおよびEDTAを含有するバッファー300μlにそのペレットを溶解し、次いで、例えばQiagen社製バッファーALなどの市販の塩酸グアニジウムバッファー360μl中に溶解し、ならびにプロテイナーゼK20μlを添加する。そのサンプルを65℃で10分間インキュベートし、次いで、エタノール420μlを添加し、シリカ膜を含むスピンカラムにサンプルを適用する。そのサンプルを、遠心分離によって膜を通過させる。例えばQIAGEN社製バッファーAW1など、標準市販のエタノール含有塩酸グアニジウムバッファーでシリカ膜を洗浄し、例えばQIAGEN社製バッファーAW2などのエタノール含有洗浄バッファーで1回洗浄する。DNAをTrisバッファー(pH8)300μlで希釈する。溶出液のアリコート5μlを0.8%アガロース/TBEゲル上で分離する。
【0074】
実施例6:
酵素反応におけるゲノムDNAの使用。
図6から、実施例5に従って単離されたDNAを様々な酵素反応(制限増幅およびPCR増幅)に使用することができることが分かる。
【0075】
24または27時間の保存後に単離されたDNA(実施例5参照)は様々な酵素反応に使用される。これは、単離したDNAの純度が高く、品質が良いことの証拠である。
【0076】
A)制限酵素EcoRI(E)およびHind III(H)6Uで37℃にて3時間、DNAのアリコート2μgを切断し、次いで0.8%アガロース/TBEゲル上で分離する。対照として、未切断のDNAを適用する。
B)ゲノムDNAのアリコート150ngおよび300ngをPCR反応(総容積50μl)に使用する。その反応では、長さ1.1kB断片のhugl遺伝子が増幅される。そのPCR産物を1.2%アガロース/TBEゲル上で分離する。
【0077】
実施例7
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化
これらの実験から、カルボン酸および他の添加剤をテトラデシルトリメチルシュウ酸アンモニウムに添加することによって、テトラデシルトリメチルシュウ酸アンモニウムを単独で用いた場合のRNAの安定化と比較して、血漿中の遊離RNAの安定化が著しく改善されることが実証される。
【0078】
この実験で使用する溶液を調製するために、30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、コハク酸、硫酸アンモニウムまたはリン酸の原液と混合して、2%または4%テトラデシルトリメチルシュウ酸アンモニウム、および200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。添加剤を全く含有しない5%テトラデシルトリメチルシュウ酸アンモニウム溶液を対照として使用する。
【0079】
このように調製したすべての溶液のアリコート0.5mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)により予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフ容器の蓋にピペッティングする。ヒト血漿0.5mlをその溶液に添加し、容器の蓋を閉め、容器を迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で1日保存する。すべての実験を二重測定で行う。
【0080】
RNAを単離するために、サンプルを25000×gで3分間にわたり遠心分離する。上清を除去し、グアニジウム塩酸塩およびNonidetP40(pH7.0)を含有する、60℃に調節されたバッファー0.5mlおよびプロテイナーゼKも、ペレットに添加する。そのペレットをボルテックスすることによって溶解し、50℃で15分間インキュベートする。次いで、エタノール−nonidetP40溶液0.5mlを添加し、サンプルを約5秒間ボルテックスすることによって混合する。次いで、例えばQIAGEN社製QIA ampカラムなどのシリカ膜を含有する標準市販のスピンカラム中にそのサンプルを入れ、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、アルコール含有洗浄バッファー、例えばQIAGEN社製バッファーAW2で2回洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有洗浄バッファーで洗浄した後、バッファーを添加することなく、遠心することによって(最大3分の回転数、この場合では25000×g)膜を乾燥させる。溶出液については、RNaseを含有しない水30μlを膜上にピペッティングして、膜から精製RNAを引き離す。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出プロセスを完了する。
【0081】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図7に示す。ゲルレーンのローディングを表2に示す。
【0082】
【表2】
【0083】
個々のサンプルのRNA品質を比較するために、レーン45は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0084】
この実験で使用されるHeLa全RNAのゲル電気泳動による分離によって、臭化エチジウムで染色した後にインタクトな28Sおよび18SrRNAが示される。視認できるrRNAバンド(28SrRNA)の一番上のバンドは、それより下のrRNAバンド(18SrRNA)よりも明らかに濃く、かつ太く、それはインタクトな分解されていないRNAの一般的な特徴である。添加剤を添加することなく、5%テトラデシルトリメチルシュウ酸アンモニウムと混合した血漿中で1日保存したHeLa全RNAを、テトラデシルトリメチルシュウ酸アンモニウムおよび様々な添加剤と混合した血漿中で1日保存した後に単離したRNAと比較すると、RNAの安定化が添加剤を使用することによって改善されていることが明白に示されている。安定化化合物を含まない血漿にRNAを添加した場合、これによって、よく知られているように数分以内にRNA全体の分解が引き起こされる。
【0085】
実施例8
酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、様々な時間にわたる血漿中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、血漿中でRNAが少なくとも14日まで安定化されることが示されている。
【0086】
この実験に使用する溶液を調製するために、30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸(pH3)またはタルトロン酸(pH3)の原液と混合して、6%または8%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。
【0087】
このように調製したすべての溶液のアリコート0.5mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)により予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフ容器の蓋にピペッティングする。ヒト血漿0.5mlをその溶液に添加し、容器の蓋を閉め、容器を迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で3、7、10および14日保存する。すべての実験を二重測定で行う。
【0088】
RNAの単離は実施例7に記載のように行う。
【0089】
単離されたRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図8に示す。ゲルレーンのローディングを表3に示す。
【0090】
【表3】
【0091】
個々のサンプルのRNA品質を比較するために、レーン「K」は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0092】
ゲル電気泳動による分離から、テトラデシルトリメチルシュウ酸アンモニウムおよび酒石酸またはタルトロン酸(pH3)と混合した血漿でHeLa全RNAを14日まで保存した後でさえ、臭化エチジウムで染色した後に、インタクトな28Sおよび18SrRNAバンドが表れる。
【0093】
実施例9:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、血漿中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物により、多量の血漿中でさえ、RNAを安定化することができることが示される。
【0094】
このように調製したすべての溶液のアリコート1mlを2mlエッペンドルフチューブに入れる。例えば、市販のRNA単離キット(例えば、QIAGEN社によりRNA単離キットとして市販のRNeasy(登録商標)Maxi−Kits)を用いて予め単離された、HeLa細胞由来の全RNA15μgをエッペンドルフチューブの蓋にピペッティングする。ヒト血漿1mlをその溶液に添加し、チューブの蓋を閉め、チューブを迅速に5回反転して液体を混合する。そのサンプルを室温(約20〜25℃)で3日間保存する。すべての実験を二重測定で行う。
【0095】
RNAの単離は実施例7に記載のように行う。
【0096】
単離されたRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%ホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート30μlを使用する。その結果を図9に示す。ゲルレーンのローディングを表4に示す。
【0097】
【表4】
【0098】
個々のサンプルのRNA品質を比較するために、レーン13は、これらの実験に使用するHeLa細胞由来の全RNA3.75μgを含む。
【0099】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、インタクトな28Sおよび18SrRNAバンドが表れる。このように、多量の血漿中でさえ、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、RNAは安定化される。
【0100】
実施例10:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いた、HeLa細胞中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと様々な添加剤との混合物によって、HeLa細胞中のRNAを、室温で14日までの保存期間にわたって安定化することができることが示される。
【0101】
この実験で使用する溶液を調製するために、20%または30%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、硫酸アンモニウムまたはリン酸の原液と混合して、2%または4%テトラデシルトリメチルシュウ酸アンモニウム、および200mM添加剤の最終濃度をとする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。
【0102】
予め細胞培養から収集し、PBSで直ちに洗浄した1×106個のHela細胞を遠心分離(120×gで1分)によってペレットにし、上清を除去する。表4に示す溶液のアリコート300μlを細胞に添加し、ボルテックスによってサンプルを混合し、細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で3、7、10および14日間保存する。すべての実験を二重測定で行う。
【0103】
RNAを単離するために、1200×gで3分間遠心分離することによって、細胞をペレットにし、上清を除去する。繰り返しピペットで取り、移すことによって、または約10秒以上の間にわたりボルテックスすることによって、例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ペレットを再懸濁する。次いで、エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。第2アルコール含有洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、RNaseを含有しない水40μlを膜上にピペッティングする。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出プロセスを完了する。
【0104】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図10に示す。サンプルを表5にまとめる。サンプル14、40、66および92を1回試験し、結果を示すことを除いては、すべてのサンプルを2回試験し、結果を示す。
【0105】
【表5】
【表6】
【表7】
【表8】
【0106】
サンプル「K」は、予め保存されることなく、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの単離キットによって、1×106個のHela細胞(=ポジティブコントロール)から単離された全RNAを示す。サンプル「a」、「b」、「c」および「d」は、上述のように添加剤を含まないPBS中で1×106個のHela細胞を3、7、10または14日間保存した後に単離された全RNAを示す。
【0107】
単離された全RNAの量は、水に希釈した後に、波長260nmでの光吸収の光度測定によって決定する。このようにして得られたRNAの純度を、260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって測定する。単離の結果を以下の表6に示す。いずれの場合も、2つの測定値の平均を示す。
【0108】
【表9】
【0109】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後、正の対照においてインタクトな28Sおよび18SrRNAバンドが表れる。rRNAバンド(28SrRNA)の一番上のバンドは、それより下のrRNAバンド(18SrRNA)よりも明らかに濃く、かつ太く、それはインタクトな分解されていないRNAの一般的な特徴である。
PBS中で細胞を3日間保存した後には、2つのrRNAバンドが同一強度を示し、極めて少ないRNAが視認できることから、RNAは一部分解している。7日間以上保存した後には、RNAはもはや視認できない。これに対して、Hela細胞中のRNAは、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによって14日間まで安定化される。これは、特異的RNAの収量および純度をOD測定することによって確認される。安定化は、pHにより影響を受ける。混合物において、つまりテトラデシルトリメチルシュウ酸アンモニウムと添加剤とを混合した後、4を超えるpH値が好ましい。
【0110】
実施例11
異なる量のHela細胞におけるRNAの安定化
これらの実験から、使用する細胞の数にかかわらず、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、HeLa細胞中のRNAを安定化することができることが示される。
【0111】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、pH6の0.5M酒石酸の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液で指定のpHに調節する。
【0112】
予め細胞培養から収集し、PBSで直ちに洗浄した1×105個、5×105個、1×106個および5×106個のHela細胞を、遠心分離(120×gで1分)によってペレットにし、上清を除去する。4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸を含有する溶液のアリコート300μlを細胞に添加し、ボルテックスによってサンプルを混合し、細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で15分間または1日保存する。すべての実験を二重測定で行う。
【0113】
実施例10に記載のようにRNAを単離する。
【0114】
使用する対照は、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM酒石酸で前処理されておらず、かつ上述のようにRNAを単離するために保存されていない1×105個、5×105個、1×106個および5×106個のHela細胞である。
【0115】
単離したRNAを、臭化エチジウムで染色したアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図11に示す。サンプルを表7にまとめ、すべてのサンプルを2回試験し、結果を示す。
【0116】
【表10】
【0117】
単離した全RNAの量は、水に希釈した後に、波長260nmでの光吸収の光度測定によって決定する。このようにして得られたRNAの純度を、260nmでの光吸収に対する280nmでの光吸収の比を測光により決定することによって測定する。単離の結果を以下の表8に示す。いずれの場合も、2つの測定値の平均を示す。
【0118】
【表11】
【0119】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存された対照サンプルにおいて、保存されていない対照サンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れる。保存されていない対照と保存されたサンプルとの間に明らかな相違はない。同様に、OD測定によって決定されたRNA収量および純度から、RNAの収量または純度を低減することなく、RNAの安定化は、異なる量の細胞中で同程度に行われることが確認される。細胞数が増加するに従って減少する商E260/E280は、測定は水中で行われ、緩衝系では行われないという事実に起因し得る。
【0120】
実施例12:
マクロファージ中のRNAの安定化
これらの実験によって、様々な種類の細胞におけるRNAを使用できることが実証される。この実験で使用するマクロファージは、前に使用したHela細胞よりも多量のRNaseを含有し、このため、細胞中のRNAが余儀なく分解される。
【0121】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸(pH5)、0.5Mタルトロン酸(pH5)または0.5Mリン酸(pH5)の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度にする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を水酸化ナトリウム溶液で指定のpHに調節する。
【0122】
予め細胞培養から収集し、PBSで直ちに洗浄した1×106個のHela細胞を遠心分離(120×gで1分)によってペレットにし、上清を除去する。4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mMの添加剤を含有する溶液のアリコート300μlを細胞に添加し、その細胞を再懸濁する。そのサンプルを室温(約20〜25℃)で2日間、6日間、9日間および14日間保存する。すべての実験を二重測定で行う。
【0123】
そのRNAを実施例10に記載のように単離する。
【0124】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図12に示す。サンプルを表9にまとめる。すべてのサンプルを2回試験し、結果を示す。
【0125】
【表12】
【0126】
レーン25および26は、マクロファージを予め保存することなく、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの市販の単離キットを用いて、1×106個のマクロファージ(ポジティブコントロール)から単離した全RNAを示す。
【0127】
水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このようにして得られたRNAの純度を、波長260nmでの光吸収に対する280nmでの光吸収の比を光度測定することによって決定する。単離の結果を以下の表10に示す。いずれの場合も、2つの測定値の平均を示す。
【0128】
【表13】
【0129】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存された対照サンプルにおいて、および保存されていない対照サンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れ、24日間保存した後でさえ、RNA分解の兆候はない。同様に、光度測定によって決定されたRNAの収量および純度は、保存中に未変化のままである。
【0130】
実施例13
培地の除去を行わない、Hela付着細胞中のRNAの安定化
これらの実験から、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物によって、付着細胞中でさえ、RNAを安定化することができることが示される。代わりにテトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物を培地に添加しても、細胞を含有する培地を除去しない場合でさえ、安定化は依然として行われる。培地中の細胞は、体液中の細胞のモデルとして見なすことができる。
【0131】
この実験で使用する溶液を調製するために、テトラデシルトリメチルシュウ酸アンモニウムおよび特定の添加剤、酒石酸または硫酸アンモニウムを計量して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mMの添加剤の最終濃度とし、水に溶解する。4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸の場合には水酸化ナトリウムを用いて、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウムの場合には硫酸を用いて、溶液のpHをpH5に調節する。
【0132】
6ウェルプレートにおいて培地2ml中でHela細胞を培養する。その細胞は付着して成長する、つまりウェルの底面に細胞が付着する。細胞中のRNAを安定化するために、10mlの4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸(pH5)または4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウム(pH5)を各ウェルに添加し、そのシャーレを室温で4日間保存する。ネガティブコントロールとして、培地を含むが、4%テトラデシルトリメチルシュウ酸アンモニウムと200mMの添加剤との混合物を添加していないウェルを室温で4日間保存する。
【0133】
ポジティブコントロールとして、例えばQIAGEN社製RNeasy(登録商標)Mini Kitsなどの標準市販の単離キットを用いて、予め保存することなく、1つのウェルからHela細胞のRNAを単離する。これを行うために、培地を細胞から完全に除去し、溶解バッファーRLT(RNeasy Kitの成分)350μlと混合する。細胞をウェルの底面からスパチュラで掻き取り、いわゆる破砕機、例えばQIAGEN社製QIAshredderなどに、ライセートを移す。14000rpmで2分間遠心することによって、そのライセートを破砕機に通し、このようにして、サンプルをホモジナイズする。その生成物を70%エタノールと混合し、実施例10に記載のようにRNAを単離する。
【0134】
4%テトラデシルトリメチルシュウ酸アンモニウム、200mMの添加剤と混合した培地中で、細胞を4日間保存した後、その時点で引き離されている細胞を上清と共に収集し、3000×gで5分間遠心分離する。その上清を除去し、細胞ペレットを用いて、実施例10に記載のようにRNAを単離する。
【0135】
4%テトラデシルトリメチルシュウ酸アンモニウム、200mMの添加剤を含まない(=ネガティブコントロール)培地中で細胞を4日間保存した後、ポジティブコントロールについて上述のように、RNAを単離する。
【0136】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図13に示す。レーン1は、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM酒石酸(pH5)と混合した培地中で細胞を保存した後に単離された全RNAを含む。レーン2は、4%テトラデシルトリメチルシュウ酸アンモニウム、200mM硫酸アンモニウム(pH5)と混合した培地中で細胞を保存した後に単離された全RNAを示す。レーン3は、培地単独で細胞を保存した後に単離された全RNAを示し、レーン4は、予め保存することなく、ポジティブコントロールとして単離された全RNAを示す。
【0137】
水で希釈した後に、波長260nmでの光吸収を光度測定することによって、単離した全RNAの量を決定する。このようにして得られたRNAの純度を、波長260nmでの光吸収に対する280nmでの光吸収の比を光度測定することによって測定する。単離の結果を以下の表11に示す。
【0138】
【表14】
【0139】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、保存していないサンプルにおいて、およびテトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物で保存したサンプルにおいてもインタクトな28Sおよび18SrRNAバンドが表れる。これに対して、テトラデシルトリメチルシュウ酸アンモニウムおよび添加剤を添加していない培地中でほぞんした細胞中のRNAは、ほぼ完全に分解される。同様に、OD測定によって決定されたRNAの収量および純度から、テトラデシルトリメチルシュウ酸アンモニウムおよび添加剤を添加していない培地中で保存されたサンプル中のRNAの収量および純度が著しく低下するのに対して、保存されていないサンプルおよび安定化されたサンプルとの間の相違がないことが確認される。
【0140】
実施例14:
様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる、組織中のRNAの安定化
これらの実験から、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムもまた、組織由来のRNAを安定化するのに適していることが分かる。
【0141】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5M酒石酸、クエン酸、タルトロン酸、硫酸アンモニウムまたはリン酸カリウム、シュウ酸またはリン酸の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度とする。テトラデシルトリメチルシュウ酸アンモニウムと混合する前に、添加剤の原液を、水酸化ナトリウム溶液または硫酸(もしくは硫酸アンモニウム)または水酸化カリウム溶液または燐酸(もしくはリン酸カリウム)で指定のpHに調節する。
【0142】
取り除いた直後に液体窒素で凍結し、次いで−70℃で保存しておいたマウスの腎組織をこれらの実験に使用する。その組織を70〜90mg凍結し、表12に指定するバッファー500μlを組織各10mgに対して添加し、例えばMessrs Kinematica社製のPolytronなどのローター・ステーターホモジナイザーを用いて、その混合物を即座に30〜60秒間ホモジナイズする。溶液のアリコート500μlを、組織10mgに相当するホモジナイズしたプレパラートから取る。サンプルを室温で1日保存する。
【0143】
保存後、サンプルを10000×gで3分間遠心分離し、上清を除去する。例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ボルテックスすることによってペレットを溶解する。次いで、70%エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有第2洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、RNaseを含有しない水40μlを膜上にピペッティングする。その溶出液は、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出を完了する。
【0144】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、1.0%のホルムアルデヒド−アガロース−MOPSゲルを調製する。溶出液のアリコート20μlを使用する。その結果を図14に示す。サンプルを表12にまとめる。すべてのサンプルを2回実験し、その結果を示す。
【0145】
【表15】
【0146】
サンプル「K」は、予め保存することなく、単離キット(QIAGEN GmbH製RNeasy)を用いて、凍結された腎組織から単離した全RNAを示す(=ポジティブコントロール)。レーン「N」は、溶媒を添加していない、つまり乾燥している腎組織10mgを1日保存した後に、QIAGEN社製RNeasy(登録商標)Mini Kitを用いて単離した全RNAを示す(=ネガティブコントロール)。
【0147】
ゲル電気泳動による分離によって、臭化エチジウムで染色した後に、ポジティブコントロールにおいてインタクトな28Sおよび18SrRNAが表れる。安定化溶液なしで保存した腎組織を含むネガティブコントロールは、完全に分解されたRNAを示す。これに対して、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウム中でサンプルを保存した後に、インタクトなrRNAバンドがポジティブコントロールとして視認することができる。安定化はpHにより影響を受ける。テトラデシルトリメチルシュウ酸アンモニウムと指定のpHの添加剤とを混合した後、組織中のRNAを安定化するために、安定化溶液の最終pHは4を超えるpHであることが好ましい。
【0148】
実施例15:
RNAの安定化および単離と並行したDNAの安定化および単離
これらの実験から、様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムを用いて、組織中のRNAのみならずDNAも安定化されることが分かる。サンプルからRNAを単離することに加えて、それと並行してDNAを単離することも可能である。
【0149】
この実験で使用する溶液を調製するために、20%テトラデシルトリメチルシュウ酸アンモニウムの原液を、0.5Mクエン酸(pH5)の原液と混合して、4%テトラデシルトリメチルシュウ酸アンモニウムおよび200mM添加剤の最終濃度となるように水酸化ナトリウムで調節する。
【0150】
取り除いた直後に液体窒素で凍結し、次いで−70℃で保存しておいたマウスの腎組織をこれらの実験に使用する。その組織を約80mg凍結し、4.2mlの4%テトラデシルトリメチルシュウ酸アンモニウム、200mMクエン酸(pH5)を組織各10mgに対して添加し、例えばMessrs Kinematica社製のPolytronなどのローター・ステーターホモジナイザーを用いて、その混合物をすぐに30〜60秒間ホモジナイズする。溶液のアリコート500μlを、組織10mgに相当するホモジナイズしたプレパラートから取る。そのサンプルを室温で1日保存する。
【0151】
保存後、サンプルを10000×gで3分間遠心分離し、上清を除去する。例えばQIAGEN社製RTLバッファーなどの標準市販のイソチオシアン酸グアニジウムバッファー600μl中に、ボルテックスすることによってペレットを溶解する。次いで、70%エタノール1容積(600μl)を添加し、繰り返しピペットで取り、移すことによって、または約5秒間にわたりボルテックスすることによって、成分を混合する。次いで、例えばQIAGEN社製RNeasyカラムなどのシリカ膜を含有する標準市販のスピンカラムにライセートを適用し、遠心することによって(10000×gで1分)膜に通す。RNAはその膜に結合した状態であり、次いで、実施例14に記載のように単離することができる。スルーフロー(throughflow)(約1200μl)を回収し、100%エタノール200μlと合わせて、ボルテックスすることによって混合する。これらのサンプルを再度、例えばQIAGEN社製QIAampカラムなどのシリカ膜を含有する標準市販のスピンカラムに適用し、遠心することによって(10000×gで1分)膜に通す。DNAは膜に結合した状態であり、次いで、標準市販のイソチオシアン酸グアニジウム含有第1洗浄バッファー、例えばQIAGEN社製バッファーRW1で洗浄し、次いでアルコール含有第2洗浄バッファー、例えばQIAGEN社製バッファーRPEで洗浄する。洗浄バッファーをそれぞれ、遠心することによって(10000×gで1分)膜に通す。アルコール含有第2洗浄バッファーでの洗浄を少量で繰り返し、遠心することによって(最大2分の回転数、この場合では20000×g)膜を同時に乾燥させる。溶出液については、膜から精製RNAを引き離すために、水200μlを膜上にピペッティングし、室温で1分間インキュベートする。その溶出液を、遠心することによって(10000×gで1分)膜に通し、溶出段階をもう一度繰り返し、溶出を完了する。
【0152】
単離されたRNAを、臭化エチジウムで染色されたアガロースゲル上で分析する。これを行うために、例えば、0.8%アガロース−TBEゲルを調製する。サンプル1〜4のアリコート40μlを使用し、サンプル5〜9のアリコート20μlを使用する。その結果を図15に示す。
【0153】
レーン1および2は、実施例15に従って単離した全RNAを表す。レーン3および4は、参照としての全RNA0.1μgおよび0.5μgをそれぞれ表し、使用したアガロースゲルにおいてインタクトなゲノムDNAの流れ特性を示している。レーン5は、予め保存することなく、市販の単離キット(Messrs QIAGEN GmbHのQIAamp(登録商標)Mini Kits)を用いて、凍結ラット腎臓10mgから単離した全DNAを表している(=ポジティブコントロール)。使用したネガティブコントロールは、1日保存した後、溶媒を添加していない、つまり乾いた腎臓組織、または蒸留水中の腎臓組織10mgから、QIAGEN社製QIAamp(登録商標)Mini Kitsを用いて単離された全DANであった。このDNAは、レーン6および7(乾燥保存)において、かつレーン8および9(A.dest中で保存)において示される。
【0154】
ゲル電気泳動による分離によって、参照DNAを示すレーンおよび保存していないポジティブコントロールのDNAを含有するレーンのどちらにも、分解されていない高分子DNAが示される。組織を乾燥保存または水中で保存することによって、DNAの完全な分解が引き起こされる。これに対して、実施例15と同様に処理したサンプルはインタクトなままであり、保存期間全体を通して分解されない。したがって、テトラデシルトリメチルシュウ酸アンモニウムと添加剤との混合物は生物サンプル中のDNAを安定化するのにも適しており、サンプルからRNAおよびDNAを並行して単離することもまた可能となる。
【図面の簡単な説明】
【0155】
【図1】異なるpHレベルを有する様々なカルボン酸バッファー中のテトラデシルトリメチルシュウ酸アンモニウム(TTAOx)による、血液中のRNAの安定化を示す。
【図2】様々な濃度の酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図3】250mM酒石酸(pH3)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図4】GAPDH遺伝子(A)およびIFN−γ遺伝子(B)のmRNAの放射標識プローブとのノーザンハイブリダイゼーションの結果として、酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる全血中のRNAの安定化を示す。
【図5】酒石酸(pH3.7)で緩衝したテトラデシルトリメチルシュウ酸アンモニウムによる血液中のDNAの安定化を示す。
【図6】ゲノムDNAを酵素反応に使用した場合の結果を示す。
【図7】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの安定化を示す。
【図8】酒石酸またはタルトロン酸と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿中のRNAの様々な期間にわたる安定化を示す。
【図9】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる血漿1ml中のRNAの安定化を示す。
【図10】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによるLeLa細胞中のRNAの安定化を示す。
【図11】異なる量のHeLa細胞中のRNAの安定化を示す。
【図12】マクロファージ中のRNAの安定化を示す。
【図13】培養液を除去しない、Hela付着細胞中のRNAの安定化を示す。
【図14】様々な添加剤と混合したテトラデシルトリメチルシュウ酸アンモニウムによる腎組織中のRNAの安定化を示す。
【図15】RNAの安定化および単離と同様な、DNAの安定化および単離を示す。
【特許請求の範囲】
【請求項1】
一般式:
【化1】
(式中、Yは、窒素またはリンを示すことができ、
R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、
X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオン示すことができる)で示されるカチオン化合物と、少なくとも1種類のプロトン供与体と、を成分として含有する組成物。
【請求項2】
Yが窒素を示すことを特徴とする、請求項1に記載の組成物。
【請求項3】
R1が、好ましくは炭素原子12個、14個、もしくは16個を有する高級アルキル基を示し、かつR2、R3およびR4がいずれの場合も、メチル基を示すことを特徴とする、請求項1または2に記載の組成物。
【請求項4】
アニオンX−が、ハロゲン化水素酸のアニオンまたは一塩基もしくは二塩基有機酸のアニオンから選択される、請求項1から3のいずれか一項に記載の組成物。
【請求項5】
前記アニオンX−が、臭化物、塩化物、リン酸塩、硫酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩またはクエン酸塩を含む群のアニオンから選択されることを特徴とする、請求項4に記載の組成物。
【請求項6】
前記プロトン供与体が、飽和脂肪族モノカルボン酸、不飽和アルケニル−カルボン酸、飽和および/または不飽和脂肪族C2〜C6ジカルボン酸、脂肪族ケトカルボン酸、アミノ酸または無機酸またはその塩の中から、単独でまたは組み合わせて選択されることを特徴とする、請求項1から5のいずれか一項に記載の組成物。
【請求項7】
C1〜C6アルキルカルボン酸、好ましくは酢酸、プロピオン酸、n−酪酸、n−吉草酸、イソ吉草酸、エチル−メチル−酢酸(2−メチル−酪酸)、2,2−ジメチルプロピオン酸(ピバル酸)、n−ヘキサン酸、n−オクタン酸、n−デカン酸、またはn−ドデカン酸(ラウリン酸)または前述の酸の混合物が、前記脂肪族モノカルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項8】
アクリル酸(プロペン酸)、メタクリル酸、クロトン酸、イソクロトン酸およびビニル酢酸または前述の酸の混合物が、脂肪族アルケニル−カルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項9】
シュウ酸、マロン酸、コハク酸、グルタル酸またはアジピン酸または前述の酸の混合物の中から選択されるジカルボン酸が、飽和脂肪族C2〜C6ジカルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項10】
脂肪族ジカルボン酸、好ましくはシュウ酸またはコハク酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項9に記載の組成物。
【請求項11】
脂肪族ヒドロキシ−ジ−およびトリ−カルボン酸、好ましくはタルトロン酸、D−(+)、L−(−)−またはDL−リンゴ酸、(2R,3R)−(+)−酒石酸、(2S,3S)−(−)−酒石酸、メソ−酒石酸およびクエン酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項12】
不飽和ジカルボン酸、好ましくはマレイン酸および/またはフマル酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項13】
不飽和トリカルボン酸、好ましくはアコニット酸、またはこれらの酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項14】
脂肪族ケトジカルボン酸、好ましくはメソシュウ酸またはオキサロ酢酸、または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項15】
アミノ酸、好ましくはアミノ酢酸(グリシン)、α−アミノプロピオン酸(アラニン)、α−アミノ−イソ吉草酸(バリン)、α−アミノ−イソカプロン酸(ロイシン)およびα−アミノ‐β−メチル吉草酸(イソロイシン)、または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項16】
それが水溶液中に存在することを特徴とする、請求項1から15のいずれか一項に記載の組成物。
【請求項17】
前記カチオン化合物が、0.01重量%から飽和の範囲、好ましくは0.1重量%から飽和の範囲、最も好ましくは0.5〜15重量%の範囲、特に最も好ましくは2〜10重量%の範囲の濃度であることを特徴とする、請求項16に記載の組成物。
【請求項18】
前記の個々の成分が任意に、水溶液中で合わされ、かつ混合されることを特徴とする、請求項1から17のいずれか一項に記載の組成物のうちの1種類を調製する方法。
【請求項19】
核酸を単離かつ/または安定化するための、請求項1から17のいずれか一項に記載の組成物の使用。
【請求項20】
核酸として、リボ核酸(RNA)およびデオキシリボ核酸(DNA)が安定化されることを特徴とする、請求項19に記載の使用。
【請求項21】
核酸として、ヌクレオチドモノマー、オリゴマー、プラスミドの形で、ウイルスおよび/または細菌DNAおよびRNA、ならびに動物および植物細胞または他の真核生物由来のゲノムおよび非ゲノムDNAおよびRNAの形で、リボ核酸(RNA)およびデオキシリボ核酸(DNA)が安定化されることを特徴とする、請求項20に記載の使用。
【請求項22】
核酸として、処理および未処理状態のmRNA、tRNA、mRNA、rRNAおよびcDNAが安定化されることを特徴とする、請求項21に記載の使用。
【請求項23】
請求項1から17のいずれか一項に記載の組成物を含有する診断用組成物。
【請求項24】
請求項1から17のいずれか一項に記載の組成物を含む、核酸を安定化するためのキット。
【請求項25】
任意に他の賦形剤と共に、核酸を含む生物サンプルと、請求項1〜17のいずれか一項に記載の組成物とを含有する混合物。
【請求項26】
前記混合物のpHが、2〜12、好ましくは2〜10の範囲、最も好ましくは3〜8の範囲であることを特徴とする、請求項25に記載の混合物。
【請求項27】
ウイルスまたは細菌を含んでもよい前記生物サンプルが、血液、血漿または血清であることを特徴とする、請求項25に記載の混合物。
【請求項28】
前記混合物のpHが、2〜6、好ましくは3〜4の範囲であることを特徴とする、請求項27に記載の混合物。
【請求項29】
ウイルスまたは細菌を含んでもよい前記生物サンプルが、吸引液、細胞、組織または細菌の形をとることを特徴とする、請求項25に記載の混合物。
【請求項30】
前記混合物のpHが、3〜10、好ましくは4〜8の範囲であることを特徴とする、請求項29に記載の混合物。
【請求項1】
一般式:
【化1】
(式中、Yは、窒素またはリンを示すことができ、
R1、R2、R3およびR4は互いに独立して、分枝もしくは非分枝C1〜C20アルキル基および/またはC6〜C20アリール基ならびにC6〜C26アラルキル基を示すことができ、
X−は、無機もしくは有機の一塩基酸または多塩基酸のアニオン示すことができる)で示されるカチオン化合物と、少なくとも1種類のプロトン供与体と、を成分として含有する組成物。
【請求項2】
Yが窒素を示すことを特徴とする、請求項1に記載の組成物。
【請求項3】
R1が、好ましくは炭素原子12個、14個、もしくは16個を有する高級アルキル基を示し、かつR2、R3およびR4がいずれの場合も、メチル基を示すことを特徴とする、請求項1または2に記載の組成物。
【請求項4】
アニオンX−が、ハロゲン化水素酸のアニオンまたは一塩基もしくは二塩基有機酸のアニオンから選択される、請求項1から3のいずれか一項に記載の組成物。
【請求項5】
前記アニオンX−が、臭化物、塩化物、リン酸塩、硫酸塩、ギ酸塩、酢酸塩、プロピオン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩またはクエン酸塩を含む群のアニオンから選択されることを特徴とする、請求項4に記載の組成物。
【請求項6】
前記プロトン供与体が、飽和脂肪族モノカルボン酸、不飽和アルケニル−カルボン酸、飽和および/または不飽和脂肪族C2〜C6ジカルボン酸、脂肪族ケトカルボン酸、アミノ酸または無機酸またはその塩の中から、単独でまたは組み合わせて選択されることを特徴とする、請求項1から5のいずれか一項に記載の組成物。
【請求項7】
C1〜C6アルキルカルボン酸、好ましくは酢酸、プロピオン酸、n−酪酸、n−吉草酸、イソ吉草酸、エチル−メチル−酢酸(2−メチル−酪酸)、2,2−ジメチルプロピオン酸(ピバル酸)、n−ヘキサン酸、n−オクタン酸、n−デカン酸、またはn−ドデカン酸(ラウリン酸)または前述の酸の混合物が、前記脂肪族モノカルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項8】
アクリル酸(プロペン酸)、メタクリル酸、クロトン酸、イソクロトン酸およびビニル酢酸または前述の酸の混合物が、脂肪族アルケニル−カルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項9】
シュウ酸、マロン酸、コハク酸、グルタル酸またはアジピン酸または前述の酸の混合物の中から選択されるジカルボン酸が、飽和脂肪族C2〜C6ジカルボン酸として使用されることを特徴とする、請求項6に記載の組成物。
【請求項10】
脂肪族ジカルボン酸、好ましくはシュウ酸またはコハク酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項9に記載の組成物。
【請求項11】
脂肪族ヒドロキシ−ジ−およびトリ−カルボン酸、好ましくはタルトロン酸、D−(+)、L−(−)−またはDL−リンゴ酸、(2R,3R)−(+)−酒石酸、(2S,3S)−(−)−酒石酸、メソ−酒石酸およびクエン酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項12】
不飽和ジカルボン酸、好ましくはマレイン酸および/またはフマル酸または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項13】
不飽和トリカルボン酸、好ましくはアコニット酸、またはこれらの酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項14】
脂肪族ケトジカルボン酸、好ましくはメソシュウ酸またはオキサロ酢酸、または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項15】
アミノ酸、好ましくはアミノ酢酸(グリシン)、α−アミノプロピオン酸(アラニン)、α−アミノ−イソ吉草酸(バリン)、α−アミノ−イソカプロン酸(ロイシン)およびα−アミノ‐β−メチル吉草酸(イソロイシン)、または前述の酸の混合物が、プロトン供与体として使用されることを特徴とする、請求項6に記載の組成物。
【請求項16】
それが水溶液中に存在することを特徴とする、請求項1から15のいずれか一項に記載の組成物。
【請求項17】
前記カチオン化合物が、0.01重量%から飽和の範囲、好ましくは0.1重量%から飽和の範囲、最も好ましくは0.5〜15重量%の範囲、特に最も好ましくは2〜10重量%の範囲の濃度であることを特徴とする、請求項16に記載の組成物。
【請求項18】
前記の個々の成分が任意に、水溶液中で合わされ、かつ混合されることを特徴とする、請求項1から17のいずれか一項に記載の組成物のうちの1種類を調製する方法。
【請求項19】
核酸を単離かつ/または安定化するための、請求項1から17のいずれか一項に記載の組成物の使用。
【請求項20】
核酸として、リボ核酸(RNA)およびデオキシリボ核酸(DNA)が安定化されることを特徴とする、請求項19に記載の使用。
【請求項21】
核酸として、ヌクレオチドモノマー、オリゴマー、プラスミドの形で、ウイルスおよび/または細菌DNAおよびRNA、ならびに動物および植物細胞または他の真核生物由来のゲノムおよび非ゲノムDNAおよびRNAの形で、リボ核酸(RNA)およびデオキシリボ核酸(DNA)が安定化されることを特徴とする、請求項20に記載の使用。
【請求項22】
核酸として、処理および未処理状態のmRNA、tRNA、mRNA、rRNAおよびcDNAが安定化されることを特徴とする、請求項21に記載の使用。
【請求項23】
請求項1から17のいずれか一項に記載の組成物を含有する診断用組成物。
【請求項24】
請求項1から17のいずれか一項に記載の組成物を含む、核酸を安定化するためのキット。
【請求項25】
任意に他の賦形剤と共に、核酸を含む生物サンプルと、請求項1〜17のいずれか一項に記載の組成物とを含有する混合物。
【請求項26】
前記混合物のpHが、2〜12、好ましくは2〜10の範囲、最も好ましくは3〜8の範囲であることを特徴とする、請求項25に記載の混合物。
【請求項27】
ウイルスまたは細菌を含んでもよい前記生物サンプルが、血液、血漿または血清であることを特徴とする、請求項25に記載の混合物。
【請求項28】
前記混合物のpHが、2〜6、好ましくは3〜4の範囲であることを特徴とする、請求項27に記載の混合物。
【請求項29】
ウイルスまたは細菌を含んでもよい前記生物サンプルが、吸引液、細胞、組織または細菌の形をとることを特徴とする、請求項25に記載の混合物。
【請求項30】
前記混合物のpHが、3〜10、好ましくは4〜8の範囲であることを特徴とする、請求項29に記載の混合物。
【図1】


【図2】
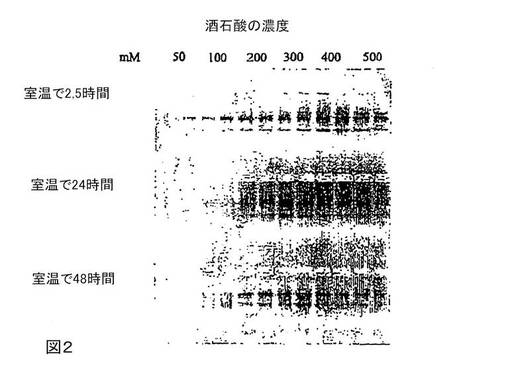
【図3】
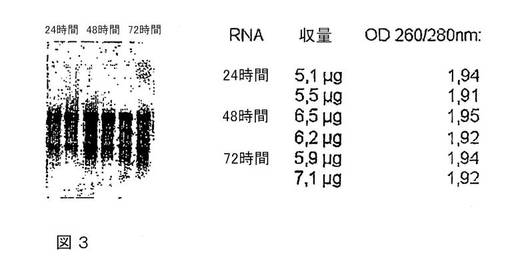
【図4】
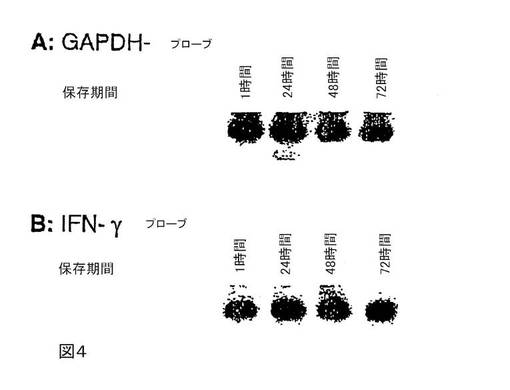
【図5】

【図6】

【図7】
【図8】
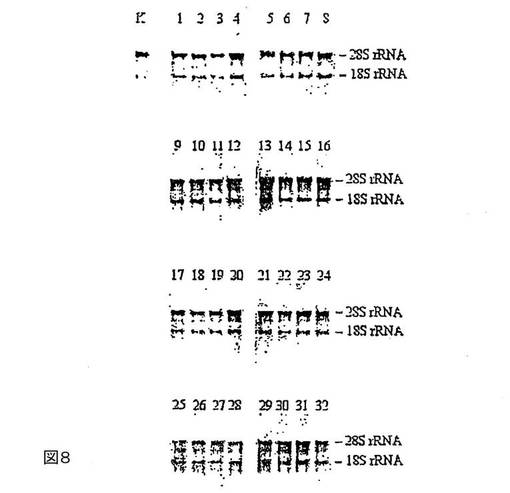
【図9】
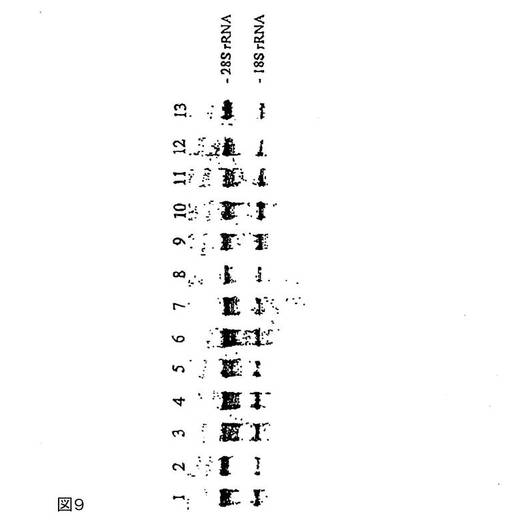
【図10】

【図11】
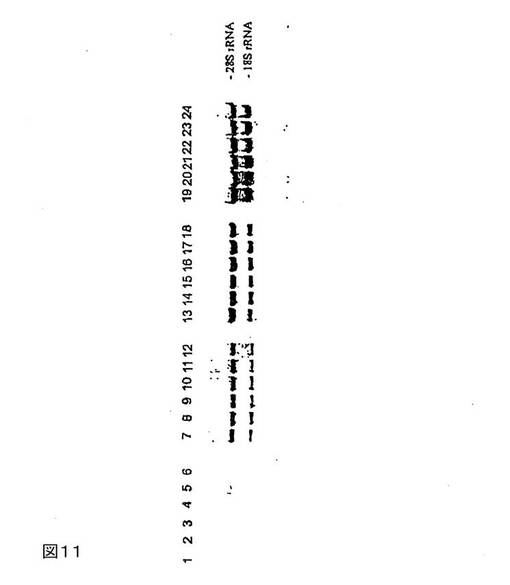
【図12】

【図13】

【図14】

【図15】
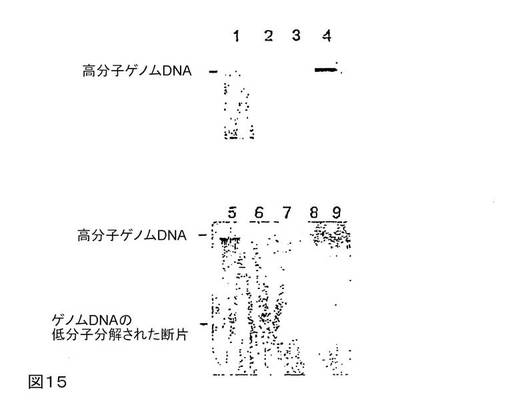
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2012−135317(P2012−135317A)
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−38487(P2012−38487)
【出願日】平成24年2月24日(2012.2.24)
【分割の表示】特願2002−505349(P2002−505349)の分割
【原出願日】平成13年5月22日(2001.5.22)
【出願人】(599072611)キアゲン ゲゼルシャフト ミット ベシュレンクテル ハフツング (83)
【Fターム(参考)】
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2012−38487(P2012−38487)
【出願日】平成24年2月24日(2012.2.24)
【分割の表示】特願2002−505349(P2002−505349)の分割
【原出願日】平成13年5月22日(2001.5.22)
【出願人】(599072611)キアゲン ゲゼルシャフト ミット ベシュレンクテル ハフツング (83)
【Fターム(参考)】
[ Back to top ]