
生物活性薬剤のカプセル化に関する改善
【課題】抗原および/または遺伝子に基づく予防剤および治療剤の送達のためのポリマー微粒子中の抗原および/または核酸のような、生物活性薬剤のカプセル化のための改善された技術を提供することを課題とする。
【解決手段】上記課題は、生物活性薬剤を、水中(油中水)型エマルジョンに基づく方法において、そして酢酸エチルを含む溶媒を用いてポリマー微粒子中にカプセル化することで解決された。一部は、0.5dl/g未満のi.v.を有する、低固有粘度(i.v.)PLGを含む微粒子およびそれらの調製のための方法もまた記載される。DNA放出は、低i.v.PLGの使用を通して改変される。規模拡大のための粒子生成方法は、カプセル化されるDNAに対する過度の剪断損傷を回避するブレンダーを用いる。
【解決手段】上記課題は、生物活性薬剤を、水中(油中水)型エマルジョンに基づく方法において、そして酢酸エチルを含む溶媒を用いてポリマー微粒子中にカプセル化することで解決された。一部は、0.5dl/g未満のi.v.を有する、低固有粘度(i.v.)PLGを含む微粒子およびそれらの調製のための方法もまた記載される。DNA放出は、低i.v.PLGの使用を通して改変される。規模拡大のための粒子生成方法は、カプセル化されるDNAに対する過度の剪断損傷を回避するブレンダーを用いる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ワクチン接種および遺伝子治療のための生物活性薬剤、例えば、抗原、薬物、およびDNAのカプセル化の改善に関する。特に、本発明は、水溶液中の抗原および/またはDNAを、ポリマー微粒子中にカプセル化し、その結果、レシピエントに投与された場合に、微粒子は抗原をレシピエントの抗原提示細胞に送達し、そして/またはレシピエントの抗原提示細胞中でこのDNAの発現を誘導するための方法に関する。本発明はまた、微粒子および微粒子を含む組成物に関する。
【背景技術】
【0002】
現在のマイクロカプセル化技術は、現在まで少なくとも10年間の歴史があるが、多くの公表された進歩にも関わらず、商業的に成功した製品を全く生みだしていないようである。実際、WO−A−97/17063(本発明と同じ発明者による)は別として、公表された方法は、効率的でなくかつ信頼性がないことが見出されてきた。
【0003】
多くの公表された特許および出願は、Southern Research Institute(SRI)の名前でなされている。特に、US−A−5407609は、実施例7において、中空の粒子の製造のためのエマルジョンに基づく方法を記載すると主張する。タンパク質(具体的にはBSA)を含む粒子を製造するための別のエマルジョンに基づく方法は、非特許文献1に記載されている。これらの粒子が、カプセル化された生物活性薬剤の腸上皮中の抗原提示細胞への投与の手段として使用される場合、そのサイズが直径10ミクロン未満であることが非常に重要である。より大きな粒子は、標的とされた腸の細胞によってエンドサイトーシスを受けずに、効果をもたらすことなく腸を通過する。
【0004】
しかし、US−A−5407609に詳述される方法およびSahらによって記載される方法は、比較的大きな粒子、または少なくとも広範な範囲のサイズにわたる粒子の作製に成功した。ここで、粒子の有意な部分は、生物学的活性のカットオフ点である10ミクロンよりも大きい。粒子のサイズの広がりが大きいことは、US−A−5407609に見られるように、必然的に、大部分のカプセル化された薬剤が、貪食作用に適切でないサイズの粒子に取り込まれることになる。なお悪いことに、Sahらは、見かけの最小直径が約10ミクロンである粒子サイズの混合物を得た。両方の当該技術の方法とも、従って、抗原提示細胞による取り込みのために利用可能なカプセル化製品を製造するために具体的に設計されたわけではない。Sahによって使用されたカプセル化方法はまた、カプセル化の間に非常に高い剪断速度を必要とし、剪断速度は、DNAのような薬剤のカプセル化に有害な効果を有し、従って、これらのカプセル化に不適切である。
【0005】
US−A−5407609は、微小の泡様の粒子の構築に言及するが、水溶性の薬剤は、カプセル化された薬剤が粒子の内部の含有物を最終的に構成するエマルジョンの部分の外側に移動する傾向のために、そのような粒子には容易にカプセル化されないことを強調する。この問題を克服するために、US−A−5407609は、エマルジョンが形成されるとすぐに、速やかに大容量の抽出媒体(すなわち、水)を添加することを必要とする。しかし、そのようにすることによって、粒子サイズの制御は失われ得る。
【0006】
Sahの方法を用いて作製された微粒子の内部構造は、単純な中空の微小の泡様の構造ではなく、Sahの論文に添付された写真において図解されているような、一般的に蜂の巣状のマトリックスである。溶媒抽出工程および引き続く凍結乾燥の間にこれらのマトリックスの内部からの溶媒および/または水の除去が不完全であると、粒子のポリマーの未熟な分解、および任意のカプセル化された水溶液の対応するpHの低下を生じ得、このことは、カプセル化された薬剤の損害をもたらす。
【0007】
投与された微粒子の生物活性薬剤の放出プロフィールは、PLGポリマー中に存在するラクチドおよびグリコリドの相対量を変化させることによって以前に変更された。しかし、このデータの多くは、「マイクロスフェア」技術に基づき、ここではカプセル化された薬剤は、ポリマーマトリックスの全体にわたって分配され、そして微小の泡状の粒子の放出プロフィールの制御に関連する公知の先行技術は存在しない。PLGポリマー中に存在するラクチドおよびグリコリドの相対量を変化させることは、短期での限定された成功を示したが、放出プロフィールを制御する代替的な手段を提供することが所望されている。
【0008】
WO−A−97/17063は、微粒子へのDNAの取り込みの有意なレベルを得るための方法を記載する。しかし、本発明者らは、この取り込みの効率を改善したい。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Sahら、J Microencapsulation、1995、第12巻、第1号、59−69頁
【発明の概要】
【発明が解決しようとする課題】
【0010】
従って本発明は、当該分野で見られる問題を克服するか、または少なくとも改善することを追求する。特に、本発明の好ましい実施態様は、抗原および/または遺伝子に基づく予防剤および治療剤の送達のためのポリマー微粒子中の抗原および/または核酸のような、生物活性薬剤のカプセル化のための改善された技術を提供することを目的とする。
【課題を解決するための手段】
【0011】
従って、本発明は、ポリマー微粒子中にDNAをカプセル化する方法を提供し、この方法は以下の工程を含む:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
DNAの水溶液を調製する工程;
ポリマー溶液とDNA溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、安定剤または界面活性剤を含むさらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成し、この微粒子はDNAを含有している、工程;
ここでこのDNAは、コード配列を含み、そして投与後にレシピエント中でコード配列の発現を誘導し、そしてここでこの溶媒は、酢酸エチルを含む。
【0012】
この方法に従って、微粒子から回収されたDNAを使用する形質転換アッセイおよびトランスフェクションアッセイによって確認されたように、DNAがそのコード配列の発現を誘導する能力を保持するような様式でカプセル化された微粒子の調製物が得られ得ることが有利に見出される。この調製物は、適切な抗原をコードするDNAの経口投与後に、レシピエントにおいて防御免疫を誘導するために適切である。微粒子のサイズは一般的に直径10ミクロン未満であり、従って、抗原提示細胞に利用可能な薬剤の比率を増加させる(Eldridge J.H.ら、J.Controlled Release、第11巻、1990、205−214頁)。また、微粒子へのDNAの取り込み効率は改善され、本発明の好ましい実施態様において80%に近づくことが見出され、この後者の数字は、当該分野を超える有利な進歩を表す。
【0013】
従って、本発明の方法は、本質的には溶媒抽出法であり、ここでは第2のエマルジョン段階後(すなわち、水中(油中水)型エマルジョン形成後)の溶媒の抽出が、微粒子のポリマーを強固にするか、または強化する。「ポリマー微粒子を形成する」との言及は、方法全体を言及することを意図し、ここでは微粒子が最初にエマルジョンを介して形成され、次いで、微粒子のポリマーの殻が、溶媒抽出によって強固にされるか、または強化される。
【0014】
本発明は、例えば以下を提供する。
(項目1) ポリマー微粒子中に生物活性薬剤をカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該溶媒が酢酸エチルを含む、工程、
を包含する、方法。
(項目2) 前記油中水型エマルジョンの形成における溶媒:水相の比が、4:1から20:1(容量:容量)の範囲にある、項目1に記載の方法。
(項目3) 前記油中水型エマルジョンの形成における溶媒:水相の比が、5:1から12:1(容量:容量)の範囲にある、項目2に記載の方法。
(項目4) 前記ポリマーがPLGであり、そして前記溶媒が、酢酸エチルおよび該溶媒中に溶解され得るPLGの量を増加させる共溶媒を含む、項目1〜3のいずれかに記載の方法。
(項目5) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目1〜4のいずれかに記載の方法。
(項目6) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまででかつ直径1ミクロンを超えるサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目1〜5のいずれかに記載の方法。
(項目7) 前記ポリマーが、70kDより大きな分子量のPLGである、項目1〜6のいずれかに記載の方法。
(項目8) 前記ポリマーが、50kD未満の分子量のPLGである、項目1〜6のいずれかに記載の方法。
(項目9) 前記生物活性薬剤が、RNA、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、ペプチド−DNA複合体についてのタンパク質、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品から選択され、必要に応じて賦形剤および/またはアジュバントが伴い、その一例がミョウバンである、項目1〜8のいずれかに記載の方法。
(項目10) ポリマー微粒子中にDNAをカプセル化する方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
DNAの水溶液を調製する工程;
該ポリマーとDNA溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成させる工程であって、該微粒子が、水溶液中にDNAを含み;
ここで該DNAがコード配列を含み、そして経口投与後にレシピエントにおいて該コード配列の発現を誘導し、そしてここで該溶媒が酢酸エチルを含む、工程、
を包含する、方法。
(項目11) 前記油中水型エマルジョンの形成における溶媒:水相の比が、4:1から20:1(容量:容量)の範囲にある、項目10に記載の方法。
(項目12) 前記油中水型エマルジョンの形成における溶媒:水相の比が、5:1から12:1(容量:容量)の範囲にある、項目11に記載の方法。
(項目13) 前記溶媒が、酢酸エチルおよび該溶媒中に溶解され得るポリマーの量を増加させる共溶媒を含む、項目10〜12のいずれかに記載の方法。
(項目14) 前記ポリマーが、ポリ−(DLラクチド−コ−グリコリド)(PLG)であり、前記ポリマー溶液中のPLGの濃度が、少なくとも10%
w/vである、項目13に記載の方法。
(項目15) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目10〜14のいずれかに記載の方法。
(項目16) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまででかつ直径0.1ミクロンを超えるサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目10〜15のいずれかに記載の方法。
(項目17) 前記ポリマーが、70kDより大きな分子量のPLGである、項目10〜16のいずれかに記載の方法。
(項目18) 前記ポリマーが、50kD未満の分子量のPLGである、項目10〜16のいずれかに記載の方法。
(項目19) 直径10ミクロン未満の微粒子を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、該ポリマーが80kDを超える分子量のPLGである微粒子を含む、組成物。
(項目20) 直径10ミクロン未満の微粒子を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、前記ポリマーが40kD未満の分子量のPLGである微粒子を含む、組成物。
(項目21) 直径10ミクロン未満の微粒子の混合物を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、前記ポリマーが40kD未満の分子量のPLGである第1の微粒子、および前記ポリマーが80kDを超える分子量のPLGである第2の微粒子を含む、組成物。
(項目22) 前記生物活性薬剤が、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、ペプチド−DNA複合体についてのタンパク質、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品から選択され、該生物活性薬剤が、さらに賦形剤および/またはアジュバントを伴い得、その一例がミョウバンである、項目19〜21のいずれかに記載の組成物。
(項目23) 前記生物活性薬剤が、コード配列を含むDNAであり、ここで前記微粒子が、経口投与された場合に該コード配列の発現を誘導する、項目19〜21のいずれかに記載の組成物。
(項目24) 組成物として使用するため、または組成物をワクチン接種する際に使用するための微粒子を作製する方法であって、項目1〜18のいずれかに記載の方法に従う工程を包含し、ここで該微粒子が、抗原をコードするDNAを含む、方法。
(項目25) 組成物として使用するため、または組成物をワクチン接種する際に使用するための組成物であって、項目19〜23のいずれかに記載の組成物を含み、ここで前記微粒子が、抗原をコードするDNAを含む、組成物。
(項目26) ワクチン接種のための医薬の製造における、項目19〜23のいずれかに記載の組成物の使用であって、ここで前記微粒子が、免疫原をコードするDNAを含む、使用。
(項目27) ポリマー微粒子中に生物活性薬剤をカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該攪拌工程が、該溶液またはエマルジョンを、ブレンダーのブレンディング区画において攪拌する工程、および該ブレンディング区画の内容物のブレンドおよび混合の両方を行うブレンディングブレードを用いる工程を含む、工程
を包含する、方法。
(項目28) 前記ブレンディングが、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって継続される、項目27に記載の方法。
(項目29) 生物活性薬剤をポリマー微粒子中にカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該ポリマーが、分子量40kD以下のPLGを含むかまたは分子量40kD以下のPLGからなる、工程
を包含する、方法。
(項目30) 生物活性薬剤をポリマー微粒子中にカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水層に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該ポリマーが、分子量80kD以上のPLGを含むかまたは分子量80kD以上のPLGからなる、工程、
を包含する、方法。
(項目31) 項目1〜9のいずれかに記載の方法によって入手可能な微粒子。
(項目32) 項目10〜18のいずれかに記載の方法によって入手可能な微粒子。
(項目33) 項目27〜28のいずれかに記載の方法によって入手可能な微粒子。
(項目34) 項目29に記載の方法によって入手可能な微粒子。
(項目35) 項目30に記載の方法によって入手可能な微粒子。
(項目36) 直径10ミクロンまでのサイズでかつ生物活性薬剤を含むポリマー微粒子であって、ここで該ポリマーが、0.5dl/g未満の固有粘度のPLGを含む、微粒子。
(項目37) 前記生物活性薬剤が、コード配列を含むDNAである、項目36に記載の微粒子。
(項目38) 前記PLGの固有粘度が、0.1dl/g〜0.4dl/gの範囲にある、項目36および37に記載の微粒子。
(項目39) 前記生物活性薬剤のうちの少なくとも30%が、投与後20日以内に放出される、項目36〜38のいずれかに記載の微粒子。
(項目40) 前記生物活性薬剤のうちの少なくとも30%が、投与後10日以内に放出される、項目36〜38のいずれかに記載の微粒子。
(項目41) 生物活性薬剤をポリマー微粒子中にカプセル化する方法であって、装置において、該生物活性薬剤の水溶液をポリマーの溶液と合わせて、水中(油中水)型エマルジョンを形成させる工程を包含し、ここで該装置が、以下:
混合される液体のための容器;
該容器中の液体を混合するためのブレード;
を備え、ここで該ブレードが、回転軸のまわりで回転し、そして回転軸に実質的に沿った方向で液体を駆動させることを含む混合作用を発揮し、そしてそれによって水中(油中水)型エマルジョンの形成を促進する、方法。
(項目42) 前記ブレードが、該ブレードから上方および下方に伸びる複数のブレード末端チップを備える、項目41に記載の方法。
(項目43) 前記ブレード末端チップが、該ブレード表面に対して30°と60°との間で傾斜している、項目42に記載の方法。
【図面の簡単な説明】
【0015】
【図1】図1は、ジクロロメタン(先行技術の溶媒)および酢酸エチル(本発明に従う)を用いて調製された微粒子から回収されたDNAの物理的状態を示すアガロースゲルである。
【図2】図2は、高分子量ポリマーまたは低分子量ポリマーから調製されたPLG微粒子からのDNAの放出プロフィールを示す。
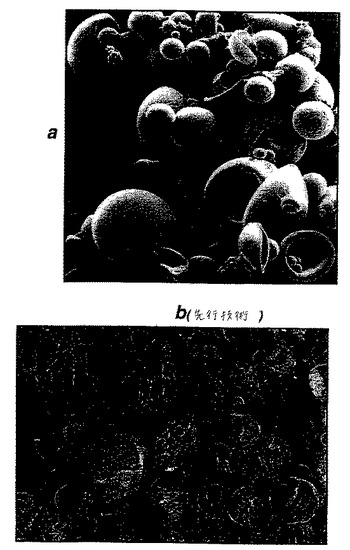
【図3】図3aおよび3bは、先行技術の微粒子および本発明の微粒子の走査型電子顕微鏡写真を示す。
【図4】図4aおよび4bは、先行技術の微粒子および本発明の微粒子の走査型電子顕微鏡写真を示す。
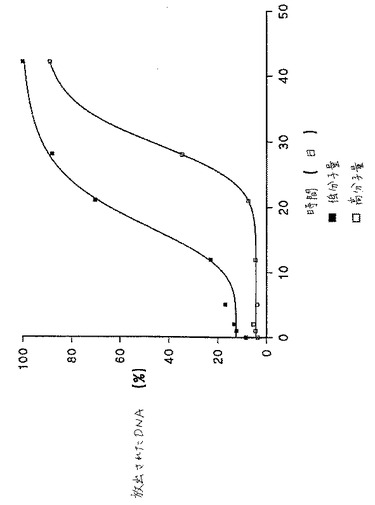
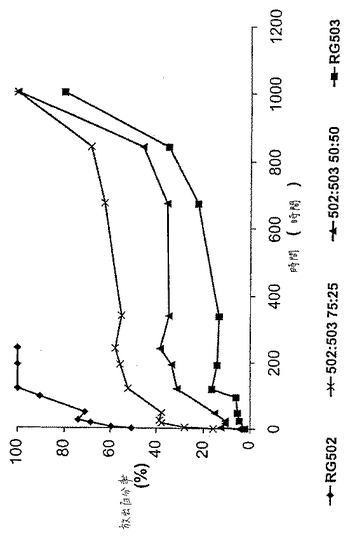
【図5】図5は、異なる固有粘度のPLGから作製した微粒子の調製物のDNA放出プロフィールを示す。
【発明を実施するための形態】
【0016】
ポリマーを溶解するために使用される溶媒は、水中(油中水)型エマルジョンから、多数の方法で抽出され得る。以下に記載する本発明の方法の特定の実施例において、水中(油中水)型エマルジョンは、大容量(100ml〜1l)の温水(実施例中では摂氏37度が使用される)中でクエンチされ、それによってポリマーから水への溶媒のエバポレーションが容易にされる。溶媒抽出の他の例は公知であり、それには、より大容量の室温の水の使用およびロータリーエバポレーターの使用が含まれる。
【0017】
本発明の方法の取り込み効率を決定する1つの方法は、光学吸収分光学によって、既知の重量の微粒子から放出されたDNAの量を正確に測定すること、および使用されたDNAの元々の量に対するこの量を関連付けることである。光学的測定のこの方法を使用して、本発明の好ましい実施態様において、代表的には60〜70%の取り込み効率が達成されることが観察され、このことは先行技術の方法を超える有意な進歩を表す。
【0018】
この微粒子中に含有されるDNAは、代表的には、二本鎖DNAを含む。本発明における使用のための適切なDNA配列の構築は、当業者に認識され、そしてWO−A−97/17063に記載される。この配列は、転写プロモーターおよび遺伝子コード配列の両方を含むことが好ましい。このDNA配列は、コード配列の下流に転写終結およびポリアデニル化を提供することがさらに好ましい。
【0019】
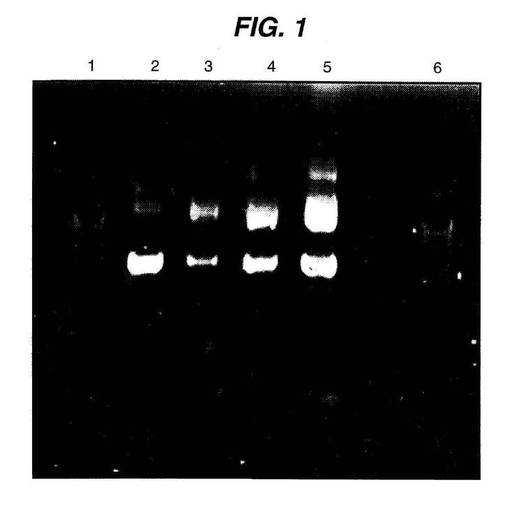
このDNAは、二本鎖、環状、およびスーパーコイルであることが特に好ましい。微粒子の製造の間に、そのDNAは剪断力を受けることが観察されてきた。本発明の粒子製造条件を用いて、本発明者らは、機能的なDNAの有意な量を保持することを成し遂げたが、前もってスーパーコイル化されたDNAは、このプロセスの間に部分的に開環型に転換され得ることが観察された。カプセル化プロセスがDNAを変性させる程度は、ネイティブな(スーパーコイル)DNAを、アガロースゲル電気泳動を使用して、部分的に変性した(開環型)DNAおよび変性した(ニックが入り、そして分解した)DNAから分離することによって評価され得る。このような分離の代表的な例を、図1に例示する。ここでは、明確に規定されたバンドが、調製物中のDNAの2つの最も豊富な形態(スーパーコイル(下のバンド)および開環型(上のバンド))に対応する。DNAの全体の分解は、通常、2つの通常の形態に対応するバンドの消失から説明される。
【0020】
カプセル化されたDNAの生物学的活性の保持、好ましい物理的状態の保持の尺度は、微粒子からのDNAの放出後に、既知量のDNAが培養中にコンピテント細菌を形質転換する能力、または真核生物細胞をトランスフェクトする能力のいずれかを測定することにより評価され得る。この形質転換アッセイは、プラスミドに存在し、そして感受性の細菌にアンピシリン耐性を付与する機能的な抗生物質耐性遺伝子(例えば、β−ラクタマーゼ)の導入を測定する。この遺伝子の発現は、DNA構造の全体の保持を示す。トランスフェクションアッセイは、培養中の適切に操作された細胞中の目的の遺伝子の発現を誘導する、プラスミドの機能を具体的に測定する。両方の場合において、得られる活性の指標は、対応する蓄えられている等価な量のDNAの活性と比較される。
【0021】
プラスミドDNAまたは従来の操作によってそれから誘導されるDNAは、特に適切である。プラスミド製造に関する多数の文献が存在するので、当業者は、容易に本発明の微粒子のために適切なプラスミドを調製し得る。一般的に、任意の真核生物プロモーター配列または原核生物プロモーター配列を組み込むプラスミドが適切である。
【0022】
本発明の微粒子を調製するために最も適切なポリマーは、代表的には、多くの特性を示す。このようなポリマーは、低毒性であり、理想的には薬学的に受容可能かつ採用された溶媒(例えば、共溶媒を含むか、または含まないかのいずれかである酢酸エチル)中に、好ましくは少なくとも約50mg/mlのレベルまで溶解するべきである。さらに、それらは、代表的には、生体適合性でありかつ生物分解性であるが、胃の酸条件を通過するに十分に安定であることが好ましい。それにもかかわらず、本発明は、その最も広い局面において特定の単一のポリマーに限定されることを意図しない。ポリ(アミノ酸/アミノ酸の誘導体)に基づくポリマーは適切であり、そしてこれらのいくつかの具体的な例は、ポリ(ラクチド)、ポリ(グリコリド)および/またはポリ(ラクチド−コ−グリコリド)を含むポリマーである。本発明の特定の実施態様において、以下でより詳細に記載されるように、そのポリマーは、ポリ−(DL ラクチド−コ−グリコリド)(PLG)であり、そしてポリマー溶液中のPLGの濃度は、代表的には少なくとも10%重量/容量である。本発明の別の特定の実施態様において、以下にまた記載されるように、そのポリマーは、ポリカプロラクトンである。
【0023】
本発明の微粒子を製造するために適切なPLG中におけるラクチドとグリコリドとの比は重要でなく、そして市販のポリマーは、25:75、50:50、および75:25のラクチド:グリコリド比を含むが、その比は0:100〜100:0の範囲の適切な数値をとり得る。本発明の特に好ましい実施態様において、そのポリマーは分子量70kDより大きいか、または50kDより小さいPLGである。本発明に従う微粒子のための他の適切なポリマー処方物は、ポリヒドロキシブチレート、ポリヒドロキシバレレート、ポリ(ヒドロキシブトレート/バレレート)、エチルセルロース、デキストラン、多糖、ポリアルキルシアノアクリレート、ポリ−メチル−メタクリレート、ポリ(e−カプロラクトン)、ポリヒドラジン、およびこれらの成分のすべての混合物を含む。
【0024】
本発明のさらなる局面は、微粒子を調製するために使用される方法の汎用性に関する。具体的には、本発明者らは、基本的な方法論の実質的な改変なしで、広範な分子量の範囲にわたるポリマーを使用して微粒子を作製した。従って、本発明は、以下の工程を包含する、生物活性薬剤をポリマー微粒子中にカプセル化する方法を提供する:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
生物活性薬剤の水溶液を調製する工程;
ポリマー溶液と生物活性薬剤溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、安定剤または界面活性剤を含むさらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成し、この微粒子は生物活性薬剤を含有している、工程;ここでこのポリマーは、分子量40kD以下のPLGを含むか、またはこのPLGからなる。
【0025】
好ましくは、PLGの分子量は30kD以下であり、そして本発明の特定の実施態様において、微粒子は3kDのPLG、6kDのPLG、9kDのPLG、22kDのPLGおよびそれらの混合物を含む。適切なポリマーの分子量の範囲は、1.5kD〜250kDである。本発明者らは、3、6、9、12、18、22、60、65、および90kDの市販の調製物を使用した。ポリマーの加水分解速度は分子量に関係し、従ってより低分子量のポリマーは、より迅速に分解する。
【0026】
Sahらによって提案されたように、微粒子が蜂の巣状の構造である場合、ポリマーマトリックス中の水相は、凍結乾燥工程の間に凍結乾燥されない。このことは、湿ったポリマーがグリコール酸および乳酸に加水分解されるので、頻繁に観察される先行技術の調製物の生物学的薬剤の変性およびpHの減少を説明し得る。本発明者らは、本発明に従って作製した微粒子が、乱暴に扱った場合に崩壊することを観察した。このことは、本発明の微粒子が、水が除去された空の内部を有することを示唆する。
【0027】
本発明の微粒子は、実際殻状であり(図3b)、中実の蜂の巣状ではなく、または、水和された場合に、水以外の何らかの種の物質相を封入している。さらに、本発明の微粒子は、例えば凍結乾燥によって、球状構造を損なうことなく乾燥され得る(図4a)。従って、本発明は、水性調製物よりもはるかに安定である乾燥pDNAを含む微粒子の調製を可能にする。
【0028】
本発明の微粒子の製造は、任意の水溶性物質(任意の水分散可能な物質でさえ)の微粒子への組み込みを可能にするが、好ましくは、その生物活性薬剤は、以下から選択される:RNA、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、タンパク質−DNA複合体またはペプチド−DNA複合体、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品。生物活性薬剤はさらに、賦形剤および/またはアジュバントを伴い得、その1つの例はミョウバンである。
【0029】
実質的に任意の組換えタンパク質抗原およびネイティブなタンパク質抗原(特に、任意の病原性生物に対して免疫するために有用な任意の組換え抗原またはネイティブな抗原)は、本発明の方法を用いてカプセル化され得る。例示のみの目的で、本発明の微粒子への取り込みのためのタンパク質抗原は、必要に応じて以下から選択される:
(a)表4に従う以下の抗原:FHA、PT、69kD−Pertactin、破傷風毒素、gp48、NS1、Capsid、gp350、NS3、SA、I、NP E、M、gp340、F、H、HN、35kDタンパク質、BP1、E1、E2、C、M、E、およびMSHA;ならびに
(b)(a)のポリペプチドの免疫原性フラグメント、改変体、および誘導体。
【0030】
これらの抗原についての遺伝子配列の登録番号の詳細は、表4に列挙される。
【0031】
本発明の水中(油中水)型エマルジョン系において、DNAまたは生物活性薬剤を含む水滴は、それ自体が第2の水相に分散される油中に分散される。このことは、「ぶどうバン(currant bun)」の形成を生じ得る。ここでぶどうは水相およびDNAであり、そしてバンはポリマーである。本発明において、この最初の油中水型エマルジョンの生成およびその第2の水相へのその分散は、最終的な微粒子の形成および性質における主要な役割を果たす。本発明者らの微粒子の生成は、最初の乳化段階によって非常に影響される。なぜなら、生じる微粒子は、分散されたDNA含有水相または生物活性薬剤含有水相からの個々の小滴に対応するからである。ポリマーが濃縮されるのはこのあたりである。本発明の好ましい実施態様において、このポリマーの濃度および最初の水相に対するポリマー溶液の比は、得られる微粒子に影響を与えるように制御される。
【0032】
具体的には、本発明の好ましい実施態様において、油中水型エマルジョンの形成における溶媒:水相の比は、4:1〜20:1(容量:容量)、より好ましくは5:1〜15:1、なおより好ましくは、5:1〜12:1(容量:容量)の範囲である。これらの範囲内での比の選択は、生じる微粒子のサイズの範囲および構造を決定するための手段である最初の油中水型エマルジョンの形成を決定し、そして微粒子へのDNAの取り込み効率を改善する。溶媒:水相の比が約4:1より下になった場合、生物活性薬剤(例えば、DNA)の微粒子への取り込みの顕著な減少が存在する。このことは、不十分なポリマー含有相が、水滴を効率よく被覆するために利用可能であり、従って殻形成および生物活性薬剤の、第2のエマルジョン相の水環境への損失を妨害する場合の結果である。この取り込み効率は、少なくとも4:1、そして好ましくは少なくとも5:1の比で増加する。この比が約20:1よりも高くなったならば、Sahの論文中に示されるような「ぶどうバン」型の構造の形成を妨害することが極端に困難になる。このことは、過剰なポリマー含有相の結果であり、これは、「ぶどうバン」構造への凝集を生じる個々の殻構造の解離およびポリマーマトリックスの内部を示す中実微粒子のパーセント(percent)である。その構造は、直径が10ミクロンよりも何倍も大きく、本発明に従う経口免疫には全く適していない。約20:1より下、そして好ましくは約15:1より下では、本方法についての、これらの「ぶどうバン」型構造を産生する傾向は低下し、そして直径10ミクロンまでの微粒子がより容易に形成される。
【0033】
そのポリマーを溶解する上で使用するのに適切な溶媒は、好ましくは低毒性であり、そして理想的には、薬学的な使用のために認可された溶媒のカテゴリーIIIリストから選択される。カテゴリーIIリストの溶媒もまた使用され得るが、ヒトに対する使用のためには、通常、そのような粒子が薬学的に認可され得る前に、粒子中に残存する溶媒の量を決定することが必要である。そのような溶媒は、好ましくは、少なくとも約50mg/mlのポリマーを溶解し、そして実質的に水と混和しないべきであり、それによって水中(油中水)型エマルジョンの形成を容易にする。本発明の好ましい実施態様において、溶媒は、高い蒸気圧を示し、その結果、最終溶媒抽出工程が迅速に実行され得る。本発明の方法における使用のために適切な溶媒の例としては、酢酸エチル、ジクロロメタン、クロロホルム、プロピレンカーボネート、およびそれらの混合物が挙げられる。アセトンもまた、溶媒混合物中で(すなわち、共溶媒として)使用され得るが、それ自体は、水と混和するので使用され得ない。
【0034】
溶媒中に溶解され得るポリマーの量を増加させる共溶媒を伴うか、または共溶媒を伴わないかのいずれかで、溶媒が酢酸エチルを含むことがさらに好ましい。混合物の大部分の比率が酢酸エチル(または、別の薬学的に受容された溶媒)であるならば、多くの共溶媒の組み合わせが適切である。共溶媒系を使用することについての利点は2倍である。第1に、溶媒の組み合わせは、単一成分の溶媒におけるよりも多くの量のポリマーを溶解させる。このことは、最初のエマルジョンの生成において溶媒水相の比を減少させる試みにおける場合に有利である。第2に、共溶媒は、高分子量ポリマーの溶解を容易にし、このことは、より溶解度が低いポリマーを用いて重要な溶媒水相比の維持を可能にする。このような共溶媒の例としては、プロピレンカーボネートおよびアセトンが挙げられる。より一般的には、本発明はまた、微粒子の調製における使用のためのポリマー溶液の調製のための任意の適切な溶媒と組み合わせた、共溶媒の使用を提供する。従って本発明の方法は、酢酸エチルを必然的に含む溶媒に限定されない。例えば、ジクロロメタン単独によって溶解されるよりも多い重量のポリマーを溶解する、ジクロロメタンおよびアセトンの組み合わせである溶媒は、本発明の別の局面である。本発明のさらなる局面は、それによって組み合わせた溶媒が薬学的に受容可能な溶媒単独でよりも多くの重量のポリマーを溶解する、共溶媒(例えば、アセトン)と組み合わせた任意の薬学的な溶媒の使用を含む。
【0035】
DNAがカプセル化される場合に、本方法の乳化工程が、減少された剪断化ストレス条件下で実行されることがまた特に好ましく、そしてこのことは、必要に応じて、乳化エネルギーの使用(例えば、乳化ミキサーの場合における速度、これはエマルジョンを得るために十分であり、そして所望のサイズ範囲における微粒子を形成するために十分であるが、過度の剪断によってすべてのDNAが損傷するほどには高くない)によって達成される。本発明の1つの実施態様において、以下に記載されるように、乳化ミキサーの速度は改変され、その結果、DNAの生物学的活性の少なくとも25%(コンピテント細菌の形質転換または培養細胞のトランスフェクションによってアッセイされた)が、DNAを含む得られた微粒子中で保持される。適切なミキサー速度は、Silversonミキサーの場合、8000rpmより下であり、好ましくは6000rpmより下であり、そして以下に記載する特定の実施態様においては、その速度は約3000rpmまたは約2000rpmより上である。エマルジョン相を生成するために使用され得る装置のすべてのバリエーションは、異なる速度における、そして異なる条件下での評価を必要とする。当業者にとって、装置の各部品についての乳化の最適速度および継続時間を同定することが残されている。Silverson Homogeniserを8000rpmより上の速度で使用する場合、DNAに有意な損傷が存在し、そして約2000rpmより下で使用する場合、直径10ミクロン以下のサイズの微粒子の形成はほとんど見られないか、または全く見られない。Silversonミキサーを使用するミキサー速度の効果のより詳細な分析は、WO−A−97/17063に見出される。
【0036】
従って、微粒子の形成に先立つか、または微粒子の形成の間の工程は、所望のサイズの範囲(代表的には、0.01〜10ミクロン)の微粒子を形成するために十分なエネルギーであるが、しかしDNAまたは生物活性薬剤が、このプロセスの間に損傷を受けるほど大きくはないエネルギーを入力するために適合される。より強い攪拌(例えば、より高速のミキサー速度を通して)であるほど、より小さい微粒子サイズを生じるというような必要とされるバランスが存在する。しかし、DNAは、乳化の間の過度の攪拌によって損傷され得る。一方、エマルジョン形成の間にエネルギー入力を減少させることは、エマルジョンが形成されず、そして微粒子が得られ得ないという効果を有し得る。本発明は、これらの競合する因子のバランスをとることを可能にして、受容可能な程度の生物学的活性を保持する微粒子の形成を提供する。
【0037】
本発明のさらなる実施態様において、本方法は、直径10ミクロンのサイズまでの微粒子を生じる高い比率の水相の小滴を得るために、ポリマーおよび水溶液の乳化を維持して、より長い時間にわたって最初の油中水型エマルジョンを形成する工程を包含する。このことは、油中水型エマルジョンの形成の間により長い攪拌を用いて、微粒子のサイズ分布がより均一になるという利点を有する。
【0038】
より長い乳化は、10ミクロンより大きい微粒子の比率を減少する。さらに、異なる乳化エネルギーにおける乳化の延長は、異なる点の付近を中心とするサイズ分布を産生する(より高いエネルギーは、より小さい平均微粒子サイズを産生する)。従って、本方法は、所望されるサイズに得られるサイズを仕立てるために改変され得る。いくつかの先行技術は、Peyerのパッチを通しての微粒子の取り込みとともに、1〜5ミクロンの微粒子および5〜10ミクロンの微粒子は、異なって取り込まれることを示唆し、そして本発明は、任意の選択された平均直径を有する微粒子の製造を容易にする。大部分の微粒子が、そして好ましくは実質的にすべての微粒子が、直径10ミクロンまでのサイズ、そして直径0.1ミクロンより大きい、微粒子を得るために、ポリマーおよび水溶液の攪拌を維持して、より長い時間にわたって油中水型エマルジョンを形成させることもまた、好ましい。この文脈において、実質的にすべての粒子との言及は、数で粒子の少なくとも4分の3、そして好ましくは少なくとも90%が、選択された直径のサイズであることを意味することを意図する。
【0039】
エマルジョンの攪拌の間に発生する任意の熱の蓄積を消散させ、それによってカプセル化された生物学的薬剤に対する損害を減少させるために、エマルジョンサイクルの間に休止時間を挿入することもまた好ましい。
【0040】
本発明の特定の実施態様において、以下にさらに詳細に記載されるように、以下の工程を包含する、ポリマー微粒子中に生物活性薬剤をカプセル化するための方法が提供される:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
生物活性薬剤の水溶液を調製する工程;
ポリマー溶液と水溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、さらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成する工程;ここでこのポリマー溶液と水溶液との攪拌、ならびに油中水型エマルジョンとさらなる水相との攪拌は、ブレンダー中で実行される。
【0041】
本発明はさらに、乳化工程を実行するためのブレンディング装置を使用して、本発明のポリマー微粒子中にカプセル化された生物活性薬剤を調製する方法を提供する。ブレンディング装置は以下を備える:
混合される液体のための容器;
その容器中で液体を混合させるためのブレード;
ここで、このブレードは、回転軸の周りを回転し、そして実質的に回転軸に沿った方向で液体を駆動させることを包含する混合動作を行う。
【0042】
このブレードの回転動作は、チャンバー内の液体中で迅速な循環する渦巻きを確立することによって、この液体に乳化エネルギーを与える。従って、この装置の作用は、次に乳化スクリーンを通して押し出される液体を引き上げるスピニングプローブを使用する他のミキサーと区別される。このような作用は、有意な剪断力を生成し、そしてDNAに高度に損傷を与える。本発明の装置の利点は、DNAのような生物活性薬剤が、カプセル化の間に、より低い剪断力および他のストレスに供せられることである。この装置のブレンディング作用はまた、超音波パルスを使用して液体にエマルジョンエネルギーを与えるソニケーションミキサーから区別される。超音波もまたDNAに損傷を与え、そしてポリヌクレオチド鎖のフラグメント化を引き起こし得る。
【0043】
エマルジョン工程は、好ましくは、Waring(RTM)型のブレンダー/ミキサーである装置を使用して実行される。従って、そのブレンダーは、エマルジョンのブレンディングおよび混合のためのチャンバーを備え、そしてさらに、そのチャンバー中に固定可能にマウントされる回転可能なブレードを備える。このブレードは、ブレードから上方および/または下方に延びる複数のブレード端チップを備え、そしてそれは、ブレードに対しておよそ30〜60度であり得るか、またはブレードに対して垂直(従って回転軸に対して平行)でさえあり得る。
【0044】
実験的な目的に適切な小容量の液体についてのみ適切なホモジナイザーの使用とはっきりと対照的に、本方法が工業的な微粒子の製造へのスケールアップのために適切であることは、本発明に従う装置の使用の利点である。
【0045】
本発明はまた、粒子内容物の放出速度の改変を達成するためのポリマー殻の改変に関する。
【0046】
従って、本発明は、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子を含む組成物を提供する。この組成物は微粒子を有し、ここでこのポリマーは、50kDより小さく、そして好ましくは1.5kDまで小さい分子量のPLGである。
【0047】
本発明はさらに、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子を含む組成物を提供する。この組成物は微粒子を有し、ここでこのポリマーは、70kDより大きく、そして好ましくは250kDまで大きい分子量のPLGである。
【0048】
本発明はなおさらに、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子の混合物を含む組成物を提供する。この組成物は第1の微粒子(ここでこのポリマーは、50kDより小さい分子量のPLGである)および第2の微粒子(ここでこのポリマーは、70kDより大きい分子量のPLGである)である。個々の型のポリマー粒子の非常に複雑な混合物が、組み合わせワクチンまたは治療剤として混合および使用され得る。
【0049】
しかし、可能な場合、本発明における使用のために適切なポリマーの分子量分布は、固有の粘度(i.v.)によって引用される。このi.v.は、容易に測定される定量可能な値であり、分子量分布に相関することが公知である。従って、「平均分子量」の値よりもポリマーの分子量分布の記載が、しばしばより正確である。このi.v.は、単位質量あたりの容量の関数として測定され、そして一般的に、グラムあたりのデシリットル(dl/g)で表現される。
【0050】
本発明の1つの局面において、直径10ミクロンまでのサイズであり、そして生物活性薬剤を含有するポリマー微粒子が提供され、ここでそのポリマーは0.5dl/g未満の固有の粘度のPLGである。生物活性薬剤は、レシピエントへの投与後に発現されるポリペプチドをコードするか、またはアンチセンス配列をコードするDNAのようなDNAであり得る。
【0051】
このことは、この微粒子がレシピエントの内部で一度分解され、そして抗原提示細胞によって取り込まれ得るが、それは有意な割合のDNAが分解される前であるという利点を与える。
【0052】
微粒子の大部分が細胞によって取り込まれ、そしてインターナライズされた場合に、カプセル化されたDNAの最大量が放出される放出プロフィールを得ることが好ましい。本発明の好ましい実施態様の代表的な放出プロフィールは、動物への投与後に、1日目と20日目との間で、より好ましくは2日目と10日目との間で、カプセル化されたDNAの30〜40%の放出を提供する。投与が経口経路を介する場合、有意なDNA放出が、微粒子が動物の胃を通して腸内への通過後に起こる。次いで、そこで微粒子はPeyerパッチのM細胞によって取り込まれ、次いで、ここで微粒子は分解し、カプセル化されたDNAを放出する。
【0053】
上記で、低いi.v.(代表的には低分子量)のPLGの使用が、より迅速な粒子の分解および粒子の内容物の放出によって、いかにして標準的な60kD PLG(高いi.v.を有する)を使用するよりも改善された免疫を与え得るかを議論した。それにもかかわらず、i.v.の範囲(従って、分子量の範囲)を有する微粒子を含む組成物が使用された場合、この組み合わせは有利な免疫効果を産生するということが利点であり得る。
【0054】
本発明の特定の実施態様において、微粒子についての放出プロフィールは、i.v.および/またはポリマーの分子量分布を調整することによって操作され得る。以下により詳細に記載される本発明の特定の実施例において、この調整は、より高いi.v.値およびより低いi.v.値のPLG調製物を組み合わせることによりなされ、中間体i.v.の複合ポリマーを産生する。中間体i.v.PLGから作られた微粒子は、改変されたDNA放出プロフィールを示し、ここで存在するより低いi.v.ポリマーの割合がより大きいほど、より迅速にDNAが放出される。このようにして、経口および他の投与経路に適切な所望の放出プロフィールを示す本発明の微粒子は産生される。
【0055】
好ましくは、i.v.は、0.1〜0.4dl/gの範囲である。本発明の特定の実施態様において、この範囲内のi.v.を有するPLGからなるポリマー殻を有する微粒子が、良好なDNA放出プロフィールおよび保持された良好な粒子構造を示すことが、走査型電子顕微鏡の下で調べた場合に見出された。以下に記載される本発明の特定の実施例において、粒子は、0.19dl/gおよび0.39dl/gのi.v.を有するPLGの混合物で作製され、そしてこれらの粒子は、経時的に非常に良好な放出プロフィールを示した。
【0056】
本発明はまた、ワクチン接種治療および本発明の微粒子を用いる方法に関する。従って、本発明はさらに、以下の本発明の方法を含むワクチン接種用組成物としての使用またはその組成物における使用のための微粒子の作成方法を提供し、ここでこの微粒子は、抗原をコードするDNAを含む。
【0057】
なおさらに本発明は、ワクチン接種用組成物としての使用またはその組成物における使用のための組成物を提供し、これは、以前に列挙されたような本発明の組成物を含み、ここでこの微粒子は抗原をコードするDNAを含み、そして本発明はワクチン接種のための医薬の製造における本発明を含む組成物の使用を提供し、ここでこの微粒子は免疫源をコードするDNAを含む。
【0058】
本発明の特定の実施態様において、その微粒子はマトリックスの内部に位置するか、またはマトリックスによって囲まれる。このようなマトリックスは、好ましくは時間とともに分解して、定常的な流れまたは断続的なパルスのいずれかにおいて微粒子を放出する生物分解性材料から作られる。本発明のデポット(depot)は、代表的には、皮下投与されるか、または身体の他の部分に移植され得る。このデポットは部分的に再水和に抵抗性であって、その結果コア中に位置する粒子がそれらが放出されるまで脱水状態で残っており、それによってカプセル化されたDNAがデポットの実質的に全寿命の間に高い完全性であることを確実にすることが好ましい。このようなデポットの製造のために適切な材料の例は、当該分野で一般的に公知であり、そしてポリヒドロキシ酪酸;ポリカプロラクトン;シリコーン;エチレンビニルアセテートおよびポリビニルアセテートを含むがこれらに限定されない。
【0059】
本発明の好ましい実施態様の方法に従って、得られる微粒子の大部分(代表的には90%以上)は、10ミクロン以下のサイズ範囲にある。微粒子調製物の電子顕微鏡写真は、これらの大部分が殻であるように見えることを示す(図3b)。より小さい微粒子の存在は、調製物の効力に対して有害ではない。なぜなら、1マイクロメートルより小さい微粒子はなお、M細胞によって活性に食作用を受け、続いて消化管関連リンパ系組織(GALT)の細胞に移動することが公知であるからである。実際、小さい(直径2ミクロン未満)サイズの微粒子は、腸の食細胞およびM細胞による取り込みを促進することにおいて有利であることが示唆されてきた。活性な生物活性分子を含む小さな粒子を調製するための主な障壁は、所望のサイズ分布を調製するために、大量の潜在的な損傷的エネルギーを導入する必要性であった。本発明の明確な利点は、ブレンダー技術を使用するエネルギー入力のさらなる制御が、この生物活性分子を変性させることなく小さな粒子の生成を可能にすることである。これはまた、これらの強力に改善された送達ビヒクルの大規模産生の可能性を提供する。
【0060】
微粒子のサイズは、M細胞による微粒子の貪食作用のプロセスにおいて、そして細胞間移動に非常に重要である。10ミクロンを超えるサイズの微粒子は取り込まれず、そして腸の上皮を横切って輸送されないことが公知である。先行技術に従って作製されたいくつかの調製物では、本発明者らは、10ミクロン〜60ミクロンの直径を有する、すなわち、必要とされる最適サイズよりも大きな、有意な数の微粒子を調べた。スフェアの容量が立法関数(cube function)に基づいて算出されるとみなされる場合、このような調製物において、大部分のpDNAが、大きな殻の内部に含まれ得ることが全体に実現可能である。このことは、この調製物を、免疫系に対して利用不能にし、従って用量を、予想されるよりもかなり低くし得、10ミクロンまでの所望のサイズ範囲の大多数の微粒子を有する微粒子調製物をもたらす、本発明に記載される方法に従って、本明細書において問題を克服する。
【0061】
本発明の微粒子の放出特性の制御が検討されている。生物活性薬剤が正確にはどのようにして微小殻から放出されるかは、不明である−おそらく、一旦、殻が加水分解によって分解した場合の、バーストとしてである。このことは、微粒子の分解が異なる時点で起きるに従って持続放出をもたらす。時間範囲は、考えられるところでは、以前に意図された時間よりも短い期間を超え得、そして記載されるようなポリマー分子量を通して制御可能である。
【0062】
ある範囲の界面活性剤は、本発明の方法における使用に適切であり、そして本発明は、実施例において使用される特定の界面活性剤であるポリビニルアルコールに限定されない。他の受容可能な界面活性剤が当該分野で公知であり、そしてメチルセルロースがさらなる例である。界面活性剤は、二重エマルジョンを安定化する役割を有する。水性界面活性剤の選択は、当業者の問題であり、そしてこの選択は、ポリマーおよびポリマー溶媒の選択に関して行なわれ得る。
【0063】
腸におけるM細胞による微粒子の取り込みについての他の関係のある背景は、以下に提供される:Jepson MAら,Journal of Drug Targeting,1993,第1巻,245−249頁;Howard KAら,Pharmaceutical Science Communications,1994,第4巻,207−216頁;Neutra MRら,Cell,1996,第86巻,345−348頁;Florence AT,Pharmaceutical Research,1997.第14巻,259頁以下参照;O’Hagan DT,J.Anat.,1996,第189巻,477−482頁;およびKreuter J,J.Anat.,1996,第189巻,503−505頁。
【0064】
本発明はここで、添付の図面を参照して特定の実施態様において記載される。
【0065】
図1は、ジクロロメタン(先行技術:レーン3)または酢酸エチル(本発明に従う:レーン4(高分子量PLG)、レーン5(低分子量PLG))のいずれかを用いて調製した、等しい重量のPLG微粒子から回収されたDNAの相対量および相対的な物理状態を示すアガロースゲルを示す。レーン2は、大きな比率のスーパーコイル形態(下のバンド)を示すコントロールDNA(ストック)であり、そしてレーン1および6は、分子量マーカーである。レーン3とレーン4および5との比較は、酢酸エチルがDNAのカプセル化を促進するだけでなく、開環形態(上のバンド)と比較してスーパーコイル形態(下のバンド)のより大きな比率の保持をももたらすことを示唆する。レーン4(高分子量PLG)において分析された調製物におけるDNAの取り込みがより小さいのは、酢酸エチル中でのポリマーのより低い溶解度に起因する、制限されたポリマーの存在における微粒子形成の結果であることに留意すべきである。
【0066】
図2は、本発明のPLG微粒子のインビトロでの放出反応速度論を示す。微粒子を、高分子量PLG(黒四角)または低分子量PLG(白四角)を用いて調製した。37℃に維持した緩衝液中に懸濁した微粒子の滅菌インキュベーションから提供された上清のアリコートにおけるDNA濃度を、光学吸光度分光法を用いて測定した。
【0067】
図3aは、凍結乾燥した粉末としてEM標本グリッドに適用したPLG微粒子の調製物(本発明に従って調製される)を示す。球形構造の均一性は、この粒子が凍結乾燥によって有害な影響を受けないことを示す。
【0068】
図3bは、最適な条件下で調製されたPLG微粒子(本発明に従う)「殻様」構造を実証する。標本を、フリーズフラクチャーした。これらの粒子の中空の内部が明らかに見られ得、そしてこれは先行技術において与えられる例とは非常に異なる。
【0069】
図4aは、室温にてEM標本グリッド上で、水和されたスラリーから乾燥されたPLG微粒子(本発明に従って調製された)の調製物を示す。蒸発した水として遭遇するストレスは、種々の程度まで構造的崩壊を誘導した。このことは、微粒子が、中空の内部を有する「殻様」構造を有し得ることを示す。
【0070】
図4bは、過剰(4:1より大きい)溶媒:水相比の条件下で調製された微粒子の内部構造を示す。調製物を、液体窒素を用いて凍結させ、そして標本を、凍結している間にフラクチャーし、粒子が種々の精巧な内部構造を有することを示した。
【0071】
図5は、種々の固有粘度のPLG調製物から作製されたPLG微粒子から経時的に放出されたDNAの百分率を示す。低固有粘度PLGを、RG502と称し、そしてより高い固有粘度のPLGをRG503と称す。50:50および75:25のRG502およびRG503混合物から作製された微粒子からのDNA放出もまた示し、そしてRG502の固有粘度とRG503の固有粘度との中間の固有粘度のPLG調製物を表す。
【実施例】
【0072】
(実施例1)
(微量カプセル化の標準的方法)
微粒子を、ここで示す方法に従って調製した。この方法は、微量カプセル化の標準的方法(microencapsulation standard method)と呼ばれる。
【0073】
1.酢酸エチル(炭酸ナトリウム上で乾燥させた)中の10%(w/v)の50:50低iv(60kD)PLG溶液を、この混合物を約37℃まで加熱し、そして溶解が完了するまで回転させることによって調製する。
【0074】
2.プラスミドDNA溶液を、STE緩衝液(10mM Tris HCI、pH8.0;1mM EDTA;150mM NaCI)中で10mg/mlの濃度になるように調製する。
【0075】
3.このPLG酢酸エチル溶液(4ml)を、Silversonホモジナイザーを用いて3000rpmにてホモジナイズし、そしてプラスミドDNA(600マイクロリットル)を添加する。得られる混合物を、さらに2.5分間ホモジナイズする。
【0076】
4.8%(w/v)水性ポリビニルアルコール溶液(92ml)を、丸底フラスコ中で約3000rpmでホモジナイズし、最初のエマルジョンを添加し、そして得られる混合物をさらに2.5分間ホモジナイズする。
【0077】
5.このエマルジョンを、37℃の2回蒸留水(100ml〜1L)に注ぎ、そして少なくとも20分間攪拌する。
【0078】
6.この微粒子を、10,000rpmにて25分間、25℃で遠心分離することにより回収する。
【0079】
7.この微粒子を、2回蒸留水中に再懸濁し、そして遠心分離することによって洗浄する。この洗浄工程を繰り返して、その結果、合計5回の回転を行い、溶媒およびエマルジョン安定剤の除去を促進する。
【0080】
8.5回目の回転の後、ペレット化した微粒子を、最少量の2回蒸留水中のスラリーとして回収し、そして凍結乾燥する。代表的な調製物を、図3aに示す。
【0081】
(標準的な方法に対するバリエーション)
上記の標準的な方法に基づいて、多数のバリエーションを、工程および/または成分に対して行った。結果は以下の通りである:
1.酢酸エチル中の50:50の低い(50kD以下)MWおよび50:50の高い(70kD以上)MWの両方のPLG溶液を研究し、そして10ミクロンまでのサイズの微粒子調製物が生成されることが見出された。他のPLG組成(例えば、75:25)および高MWまたは低MWを有するポリマーもまた、本発明の微粒子および方法に適切である。
【0082】
2.PLG対EAのw/v比を研究した。高MWポリマーを用いて、0.04g/mlまでの低減は、取り込み効率の著しい減少を導いた(図1)。試験した最大量は0.125g/mlであった。これは、カプセル化を改善し、1ミクロン〜10ミクロンのサイズ範囲の微粒子を生成したが、カプセル化の前に混濁溶液をもたらした。
【0083】
3.有機相におけるPLGの濃度を増加させるための共溶媒の使用を研究した。共溶媒(例えば、酢酸エチルと組み合わせた炭酸プロピレンおよびアセトン)は、油(溶媒)相中に溶解された増加した量のポリマーを有する、初期油中水型エマルジョンの調製を可能にした。このことは、微粒子へのDNAの取り込みを効率的に改善することが有利に見出された。
【0084】
4.最初の乳化工程における水相対ポリマー相の比は、特定の範囲内で変更され、そしてハニカム構造が回避され、そして微粒子のサイズが制御され得、粒子の調製物を実質的に全て所望の範囲内にし、そして所望の構造を有することをもたらすことが見出された。
【0085】
5.最初の乳化のためのホモジナイゼーションの速度を変更した。Silversonミキサーを用いて、いくらかのDNAの生存能力を依然として保持しながら、ホモジナイゼーションの速度を4000rpmまで増加させることが可能である。しかし、2000rpmが、良好なDNA生存能力を与え、そしてこの速度における長時間のホモジナイゼーションについて充分非損傷的であり、狭いサイズ分布を有する微粒子の形成を可能にしながらも有意なDNA損傷を導かない。3000rpmはまた、有意なDNA損傷を伴わずに長時間にわたって用いられ得る。
【0086】
6.用いられるPVAの濃度は、過剰であると考えられ、そして2%の濃度が適切であることが示唆されている。このことはまた、微粒子の安定性およびサイズにもまた影響を与え得る、混合物の粘度に対して効果を有する。
【0087】
7.最終段階で好首尾に用いられる最少容量の水は、150mlである。攪拌を、最初に37℃の水を用いて室温で実施し、そして攪拌を20分間〜1.5時間の時間にわたって継続したが、攪拌時間についての理論的上限は存在しないようである。
【0088】
8.この手順を実施する温度は、溶液の粘度および他の相における試薬の溶解度のようなパラメーターをもたらす。酢酸エチルを用いて4℃で実施した単一の実験は、より低い取り込みをもたらした。
【0089】
(実施例2)
(溶媒ならびに高分子量および低分子量の50:50 PLGの比較)
以下の実験を、標準的な方法を用いて実施した。ここでは、EA=酢酸エチル、そしてDCM=ジクロロメタンである。各場合におけるPLG容量は4mlであった。EtOHは、実験のいずれにおいても用いなかった。結果を表1に示す。
【0090】
【表1】

(実施例3)
(微粒子から回収されたDNAの物理的状態)
図1は、ジクロロメタンおよび酢酸エチルを用いて調製された微粒子から回収されたDNAの物理的状態を示す。一番下のバンドは、スーパーコイルDNAである。このバンドの強度が強いほど、DNAの統合性がより高く保持された。
【0091】
(実施例4)
(トランスフェクションの結果)
実験を、Vero細胞を用いて行った。DNAを微粒子から回収し、そしてVero細胞を、SUPERFECT REAGENT(登録商標)を用いてトランスフェクトした。
【0092】
vero細胞を、DNAを含む微粒子で、しかしトランスフェクションを増強するさらなる試薬を用いずにトランスフェクトして、さらなる実験を行った。トランスフェクションの程度を、市販のルシフェラーゼアッセイシステムを用いて評価した。結果を表2に示す。
【0093】
【表2】
(実施例5)
(PLG濃度の効果)
以下の表3は、低分子量(60kD)PLGの濃度を低減させた場合に得られた結果を示す。これらの比較実験では、酢酸エチルを溶媒として用い、有機相の容量を4mlで保持した。3000rpmというホモジナイゼーションの速度を全ての場合で用いた。
【0094】
【表3】
(実施例6)
(共溶媒の使用)
高分子量PLGは、酢酸エチル中で10% w/v以上では全体として可溶性でない。PLG濃度を増加させるため、溶媒の混合物(例えば、酢酸エチルおよび炭酸プロピレン)の適用を調査した。
【0095】
80%酢酸エチル:20%炭酸プロピレンは、10% w/v PLGの清澄な溶液を与えた。
【0096】
(実施例7)
(放出反応速度)
高分子量および低分子量のポリマーを使用して酢酸エチルを用いて調製した微粒子のサンプルを研究して、放出特性を決定した。この微粒子を滅菌STE緩衝液に添加し、そして適切な抗菌剤を用いて37℃にて維持した。サンプルを間隔をおいて取り出し、そして放出されたDNAおよび保持されたDNAを決定した。このデータから、時間プロフィールとともに放出百分率を決定した。この結果を以下の図2に示す。
【0097】
(実施例8)
(ポリマーとしてポリカプロラクトンを用いて調製した微粒子)
微粒子を、以下の方法に従って、ポリカプロラクトンポリマーを用いて調製する。
【0098】
1.酢酸エチル中のポリカプロラクトンの100mg/ml溶液(平均MW=14kD;Aldrich Chemical Company)を、溶解が完了するまでボルテックスすることにより調製する。
【0099】
2.プラスミドDNA溶液を、9.9mg/mlの濃度になるようにSTE緩衝液(10mM Tris HCl、pH8.0;1mM EDTA;100mM NaCl)中で調製する。
【0100】
3.ポリカプロラクトン酢酸エチル溶液(4ml)を、Waringブレンダー中で低出力でホモジナイズし、そしてプラスミドDNA(400マイクロリットル)を添加する。得られる混合物を、15秒間の休止期間を間に挟んで15秒間のサイクルで4回ホモジナイズする。
【0101】
4.8%(w/v)水性ポリビニルアルコール溶液(66ml)を、Waringブレンダー中で低出力でホモジナイズし、最初のエマルジョンを添加し、そして得られる混合物を、15秒間の休止期間を間に挟んで15秒間のサイクルで4回ホモジナイズする。
【0102】
5.このエマルジョンを、37℃の2回蒸留水(300ml)中に注ぎ、そして少なくとも20分間攪拌する。
【0103】
6.微粒子を、25℃にて10,000rpmで25分間遠心分離することにより回収する。
【0104】
7.この微粒子を、2回蒸留水中に再懸濁し、そして遠心分離することにより洗浄する。この洗浄工程を繰り返し、その結果、合計5回の回転を行い、溶媒およびエマルジョン安定剤の除去を促進する。
【0105】
8.5回目の回転の後、ペレット化した微粒子を、最少量の2回蒸留水中のスラリーとして回収する。微粒子は、主に直径1ミクロン〜10ミクロンのサイズ範囲で得られ、次いで貯蔵のために凍結乾燥される。
【0106】
(実施例9)
(ポリマーの固有粘度を変更することによる、PLG微粒子の放出プロフィールの改変)
実施例8の手順は、以下のPLG調製物を用いて以下の通りであった:
(a)0.19dl/gの固有粘度(RG502、Boehringer Ingelheimから)
(b)0.39dl/gの固有粘度(RG503、Boehringer Ingelheimから)
(c)0.19dl/gおよび0.39dl/gの固有粘度のPLGの50:50(重量による)の混合(RG502およびRG503の混合)
(d)0.19dl/gおよび0.39dl/gの固有粘度のPLGの75:25(重量による)の混合(RG502およびRG503の混合).
主に1ミクロン〜10ミクロンのサイズの微粒子を得て、そしてこの粒子からのDNAの放出のプロフィールを測定し、そして図5に図示する。
【0107】
(実施例10)
以下の改変を加えた実施例8のプロトコールに従った:8mlのポリマー溶液および0.8mlのプラスミド溶液を用い、そして132ml PVAを用いて第1のエマルジョンを安定化させた。
【0108】
プラスミドを、以下の通りにPLG中にカプセル化した:
(a)50:50のRG502:RG503の混合;
(b)この(a)の混合を繰り返した;
(c)70:30のRG502:RG503の混合;
(d)この(c)の混合を繰り返した。
【0109】
主に直径1ミクロン〜10ミクロンのサイズの微粒子が得られ、そしてDNA取り込み効率について試験された。(a)および(b)の微粒子から、65%が、カプセル化されたDNAとして回収され、そして(c)および(d)の微粒子から、69%が、カプセル化されたDNAとして回収された。この%の数字は、ある比率のDNAが、粒子の単離において(例えば、遠心分離によって)失われ、そして結果として、回収率は100%の効率ではあり得ないという事実を考慮していない。従って、これらの実施例において達成された数字は、非常に高い取り込み効率を表す。
【0110】
従って、本発明は、生物活性薬剤(特に、DNA)のカプセル化のさらなる、そして改善された方法を提供し、そしてさらに、遺伝子治療および/またはワクチン接種のため、ならびに他の適用のための、生物活性薬剤を含む微粒子を含む改善された組成物を提供する。
【0111】
【表4】
【技術分野】
【0001】
本発明は、ワクチン接種および遺伝子治療のための生物活性薬剤、例えば、抗原、薬物、およびDNAのカプセル化の改善に関する。特に、本発明は、水溶液中の抗原および/またはDNAを、ポリマー微粒子中にカプセル化し、その結果、レシピエントに投与された場合に、微粒子は抗原をレシピエントの抗原提示細胞に送達し、そして/またはレシピエントの抗原提示細胞中でこのDNAの発現を誘導するための方法に関する。本発明はまた、微粒子および微粒子を含む組成物に関する。
【背景技術】
【0002】
現在のマイクロカプセル化技術は、現在まで少なくとも10年間の歴史があるが、多くの公表された進歩にも関わらず、商業的に成功した製品を全く生みだしていないようである。実際、WO−A−97/17063(本発明と同じ発明者による)は別として、公表された方法は、効率的でなくかつ信頼性がないことが見出されてきた。
【0003】
多くの公表された特許および出願は、Southern Research Institute(SRI)の名前でなされている。特に、US−A−5407609は、実施例7において、中空の粒子の製造のためのエマルジョンに基づく方法を記載すると主張する。タンパク質(具体的にはBSA)を含む粒子を製造するための別のエマルジョンに基づく方法は、非特許文献1に記載されている。これらの粒子が、カプセル化された生物活性薬剤の腸上皮中の抗原提示細胞への投与の手段として使用される場合、そのサイズが直径10ミクロン未満であることが非常に重要である。より大きな粒子は、標的とされた腸の細胞によってエンドサイトーシスを受けずに、効果をもたらすことなく腸を通過する。
【0004】
しかし、US−A−5407609に詳述される方法およびSahらによって記載される方法は、比較的大きな粒子、または少なくとも広範な範囲のサイズにわたる粒子の作製に成功した。ここで、粒子の有意な部分は、生物学的活性のカットオフ点である10ミクロンよりも大きい。粒子のサイズの広がりが大きいことは、US−A−5407609に見られるように、必然的に、大部分のカプセル化された薬剤が、貪食作用に適切でないサイズの粒子に取り込まれることになる。なお悪いことに、Sahらは、見かけの最小直径が約10ミクロンである粒子サイズの混合物を得た。両方の当該技術の方法とも、従って、抗原提示細胞による取り込みのために利用可能なカプセル化製品を製造するために具体的に設計されたわけではない。Sahによって使用されたカプセル化方法はまた、カプセル化の間に非常に高い剪断速度を必要とし、剪断速度は、DNAのような薬剤のカプセル化に有害な効果を有し、従って、これらのカプセル化に不適切である。
【0005】
US−A−5407609は、微小の泡様の粒子の構築に言及するが、水溶性の薬剤は、カプセル化された薬剤が粒子の内部の含有物を最終的に構成するエマルジョンの部分の外側に移動する傾向のために、そのような粒子には容易にカプセル化されないことを強調する。この問題を克服するために、US−A−5407609は、エマルジョンが形成されるとすぐに、速やかに大容量の抽出媒体(すなわち、水)を添加することを必要とする。しかし、そのようにすることによって、粒子サイズの制御は失われ得る。
【0006】
Sahの方法を用いて作製された微粒子の内部構造は、単純な中空の微小の泡様の構造ではなく、Sahの論文に添付された写真において図解されているような、一般的に蜂の巣状のマトリックスである。溶媒抽出工程および引き続く凍結乾燥の間にこれらのマトリックスの内部からの溶媒および/または水の除去が不完全であると、粒子のポリマーの未熟な分解、および任意のカプセル化された水溶液の対応するpHの低下を生じ得、このことは、カプセル化された薬剤の損害をもたらす。
【0007】
投与された微粒子の生物活性薬剤の放出プロフィールは、PLGポリマー中に存在するラクチドおよびグリコリドの相対量を変化させることによって以前に変更された。しかし、このデータの多くは、「マイクロスフェア」技術に基づき、ここではカプセル化された薬剤は、ポリマーマトリックスの全体にわたって分配され、そして微小の泡状の粒子の放出プロフィールの制御に関連する公知の先行技術は存在しない。PLGポリマー中に存在するラクチドおよびグリコリドの相対量を変化させることは、短期での限定された成功を示したが、放出プロフィールを制御する代替的な手段を提供することが所望されている。
【0008】
WO−A−97/17063は、微粒子へのDNAの取り込みの有意なレベルを得るための方法を記載する。しかし、本発明者らは、この取り込みの効率を改善したい。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Sahら、J Microencapsulation、1995、第12巻、第1号、59−69頁
【発明の概要】
【発明が解決しようとする課題】
【0010】
従って本発明は、当該分野で見られる問題を克服するか、または少なくとも改善することを追求する。特に、本発明の好ましい実施態様は、抗原および/または遺伝子に基づく予防剤および治療剤の送達のためのポリマー微粒子中の抗原および/または核酸のような、生物活性薬剤のカプセル化のための改善された技術を提供することを目的とする。
【課題を解決するための手段】
【0011】
従って、本発明は、ポリマー微粒子中にDNAをカプセル化する方法を提供し、この方法は以下の工程を含む:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
DNAの水溶液を調製する工程;
ポリマー溶液とDNA溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、安定剤または界面活性剤を含むさらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成し、この微粒子はDNAを含有している、工程;
ここでこのDNAは、コード配列を含み、そして投与後にレシピエント中でコード配列の発現を誘導し、そしてここでこの溶媒は、酢酸エチルを含む。
【0012】
この方法に従って、微粒子から回収されたDNAを使用する形質転換アッセイおよびトランスフェクションアッセイによって確認されたように、DNAがそのコード配列の発現を誘導する能力を保持するような様式でカプセル化された微粒子の調製物が得られ得ることが有利に見出される。この調製物は、適切な抗原をコードするDNAの経口投与後に、レシピエントにおいて防御免疫を誘導するために適切である。微粒子のサイズは一般的に直径10ミクロン未満であり、従って、抗原提示細胞に利用可能な薬剤の比率を増加させる(Eldridge J.H.ら、J.Controlled Release、第11巻、1990、205−214頁)。また、微粒子へのDNAの取り込み効率は改善され、本発明の好ましい実施態様において80%に近づくことが見出され、この後者の数字は、当該分野を超える有利な進歩を表す。
【0013】
従って、本発明の方法は、本質的には溶媒抽出法であり、ここでは第2のエマルジョン段階後(すなわち、水中(油中水)型エマルジョン形成後)の溶媒の抽出が、微粒子のポリマーを強固にするか、または強化する。「ポリマー微粒子を形成する」との言及は、方法全体を言及することを意図し、ここでは微粒子が最初にエマルジョンを介して形成され、次いで、微粒子のポリマーの殻が、溶媒抽出によって強固にされるか、または強化される。
【0014】
本発明は、例えば以下を提供する。
(項目1) ポリマー微粒子中に生物活性薬剤をカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該溶媒が酢酸エチルを含む、工程、
を包含する、方法。
(項目2) 前記油中水型エマルジョンの形成における溶媒:水相の比が、4:1から20:1(容量:容量)の範囲にある、項目1に記載の方法。
(項目3) 前記油中水型エマルジョンの形成における溶媒:水相の比が、5:1から12:1(容量:容量)の範囲にある、項目2に記載の方法。
(項目4) 前記ポリマーがPLGであり、そして前記溶媒が、酢酸エチルおよび該溶媒中に溶解され得るPLGの量を増加させる共溶媒を含む、項目1〜3のいずれかに記載の方法。
(項目5) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目1〜4のいずれかに記載の方法。
(項目6) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまででかつ直径1ミクロンを超えるサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目1〜5のいずれかに記載の方法。
(項目7) 前記ポリマーが、70kDより大きな分子量のPLGである、項目1〜6のいずれかに記載の方法。
(項目8) 前記ポリマーが、50kD未満の分子量のPLGである、項目1〜6のいずれかに記載の方法。
(項目9) 前記生物活性薬剤が、RNA、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、ペプチド−DNA複合体についてのタンパク質、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品から選択され、必要に応じて賦形剤および/またはアジュバントが伴い、その一例がミョウバンである、項目1〜8のいずれかに記載の方法。
(項目10) ポリマー微粒子中にDNAをカプセル化する方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
DNAの水溶液を調製する工程;
該ポリマーとDNA溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成させる工程であって、該微粒子が、水溶液中にDNAを含み;
ここで該DNAがコード配列を含み、そして経口投与後にレシピエントにおいて該コード配列の発現を誘導し、そしてここで該溶媒が酢酸エチルを含む、工程、
を包含する、方法。
(項目11) 前記油中水型エマルジョンの形成における溶媒:水相の比が、4:1から20:1(容量:容量)の範囲にある、項目10に記載の方法。
(項目12) 前記油中水型エマルジョンの形成における溶媒:水相の比が、5:1から12:1(容量:容量)の範囲にある、項目11に記載の方法。
(項目13) 前記溶媒が、酢酸エチルおよび該溶媒中に溶解され得るポリマーの量を増加させる共溶媒を含む、項目10〜12のいずれかに記載の方法。
(項目14) 前記ポリマーが、ポリ−(DLラクチド−コ−グリコリド)(PLG)であり、前記ポリマー溶液中のPLGの濃度が、少なくとも10%
w/vである、項目13に記載の方法。
(項目15) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目10〜14のいずれかに記載の方法。
(項目16) 前記ポリマーおよび水溶液の攪拌を維持して、大多数が直径10ミクロンまででかつ直径0.1ミクロンを超えるサイズである微粒子を得るように長時間にわたって前記油中水型エマルジョンを形成させる工程を包含する、項目10〜15のいずれかに記載の方法。
(項目17) 前記ポリマーが、70kDより大きな分子量のPLGである、項目10〜16のいずれかに記載の方法。
(項目18) 前記ポリマーが、50kD未満の分子量のPLGである、項目10〜16のいずれかに記載の方法。
(項目19) 直径10ミクロン未満の微粒子を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、該ポリマーが80kDを超える分子量のPLGである微粒子を含む、組成物。
(項目20) 直径10ミクロン未満の微粒子を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、前記ポリマーが40kD未満の分子量のPLGである微粒子を含む、組成物。
(項目21) 直径10ミクロン未満の微粒子の混合物を含む組成物であって、該微粒子は、外部ポリマー殻および生物活性薬剤の内部水溶液を含み、該組成物は、前記ポリマーが40kD未満の分子量のPLGである第1の微粒子、および前記ポリマーが80kDを超える分子量のPLGである第2の微粒子を含む、組成物。
(項目22) 前記生物活性薬剤が、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、ペプチド−DNA複合体についてのタンパク質、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品から選択され、該生物活性薬剤が、さらに賦形剤および/またはアジュバントを伴い得、その一例がミョウバンである、項目19〜21のいずれかに記載の組成物。
(項目23) 前記生物活性薬剤が、コード配列を含むDNAであり、ここで前記微粒子が、経口投与された場合に該コード配列の発現を誘導する、項目19〜21のいずれかに記載の組成物。
(項目24) 組成物として使用するため、または組成物をワクチン接種する際に使用するための微粒子を作製する方法であって、項目1〜18のいずれかに記載の方法に従う工程を包含し、ここで該微粒子が、抗原をコードするDNAを含む、方法。
(項目25) 組成物として使用するため、または組成物をワクチン接種する際に使用するための組成物であって、項目19〜23のいずれかに記載の組成物を含み、ここで前記微粒子が、抗原をコードするDNAを含む、組成物。
(項目26) ワクチン接種のための医薬の製造における、項目19〜23のいずれかに記載の組成物の使用であって、ここで前記微粒子が、免疫原をコードするDNAを含む、使用。
(項目27) ポリマー微粒子中に生物活性薬剤をカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該攪拌工程が、該溶液またはエマルジョンを、ブレンダーのブレンディング区画において攪拌する工程、および該ブレンディング区画の内容物のブレンドおよび混合の両方を行うブレンディングブレードを用いる工程を含む、工程
を包含する、方法。
(項目28) 前記ブレンディングが、大多数が直径10ミクロンまでのサイズである微粒子を得るように長時間にわたって継続される、項目27に記載の方法。
(項目29) 生物活性薬剤をポリマー微粒子中にカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水相に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該ポリマーが、分子量40kD以下のPLGを含むかまたは分子量40kD以下のPLGからなる、工程
を包含する、方法。
(項目30) 生物活性薬剤をポリマー微粒子中にカプセル化するための方法であって、以下:
溶媒中にポリマーを溶解してポリマー溶液を形成させる工程;
生物活性薬剤の水溶液を調製する工程;
該ポリマーと水溶液とを、攪拌しながら合わせて油中水型エマルジョンを形成させる工程;
該油中水型エマルジョンを、攪拌しながらさらなる水相に添加して、水中(油中水)型エマルジョンを形成させる工程;
該水中(油中水)型エマルジョンを、過剰の水層に添加して該溶媒を抽出し、それによって直径10ミクロンまでのサイズの微粒子を形成させる工程であって、該微粒子が、水溶液中に生物活性薬剤を含み;
ここで該ポリマーが、分子量80kD以上のPLGを含むかまたは分子量80kD以上のPLGからなる、工程、
を包含する、方法。
(項目31) 項目1〜9のいずれかに記載の方法によって入手可能な微粒子。
(項目32) 項目10〜18のいずれかに記載の方法によって入手可能な微粒子。
(項目33) 項目27〜28のいずれかに記載の方法によって入手可能な微粒子。
(項目34) 項目29に記載の方法によって入手可能な微粒子。
(項目35) 項目30に記載の方法によって入手可能な微粒子。
(項目36) 直径10ミクロンまでのサイズでかつ生物活性薬剤を含むポリマー微粒子であって、ここで該ポリマーが、0.5dl/g未満の固有粘度のPLGを含む、微粒子。
(項目37) 前記生物活性薬剤が、コード配列を含むDNAである、項目36に記載の微粒子。
(項目38) 前記PLGの固有粘度が、0.1dl/g〜0.4dl/gの範囲にある、項目36および37に記載の微粒子。
(項目39) 前記生物活性薬剤のうちの少なくとも30%が、投与後20日以内に放出される、項目36〜38のいずれかに記載の微粒子。
(項目40) 前記生物活性薬剤のうちの少なくとも30%が、投与後10日以内に放出される、項目36〜38のいずれかに記載の微粒子。
(項目41) 生物活性薬剤をポリマー微粒子中にカプセル化する方法であって、装置において、該生物活性薬剤の水溶液をポリマーの溶液と合わせて、水中(油中水)型エマルジョンを形成させる工程を包含し、ここで該装置が、以下:
混合される液体のための容器;
該容器中の液体を混合するためのブレード;
を備え、ここで該ブレードが、回転軸のまわりで回転し、そして回転軸に実質的に沿った方向で液体を駆動させることを含む混合作用を発揮し、そしてそれによって水中(油中水)型エマルジョンの形成を促進する、方法。
(項目42) 前記ブレードが、該ブレードから上方および下方に伸びる複数のブレード末端チップを備える、項目41に記載の方法。
(項目43) 前記ブレード末端チップが、該ブレード表面に対して30°と60°との間で傾斜している、項目42に記載の方法。
【図面の簡単な説明】
【0015】
【図1】図1は、ジクロロメタン(先行技術の溶媒)および酢酸エチル(本発明に従う)を用いて調製された微粒子から回収されたDNAの物理的状態を示すアガロースゲルである。
【図2】図2は、高分子量ポリマーまたは低分子量ポリマーから調製されたPLG微粒子からのDNAの放出プロフィールを示す。
【図3】図3aおよび3bは、先行技術の微粒子および本発明の微粒子の走査型電子顕微鏡写真を示す。
【図4】図4aおよび4bは、先行技術の微粒子および本発明の微粒子の走査型電子顕微鏡写真を示す。
【図5】図5は、異なる固有粘度のPLGから作製した微粒子の調製物のDNA放出プロフィールを示す。
【発明を実施するための形態】
【0016】
ポリマーを溶解するために使用される溶媒は、水中(油中水)型エマルジョンから、多数の方法で抽出され得る。以下に記載する本発明の方法の特定の実施例において、水中(油中水)型エマルジョンは、大容量(100ml〜1l)の温水(実施例中では摂氏37度が使用される)中でクエンチされ、それによってポリマーから水への溶媒のエバポレーションが容易にされる。溶媒抽出の他の例は公知であり、それには、より大容量の室温の水の使用およびロータリーエバポレーターの使用が含まれる。
【0017】
本発明の方法の取り込み効率を決定する1つの方法は、光学吸収分光学によって、既知の重量の微粒子から放出されたDNAの量を正確に測定すること、および使用されたDNAの元々の量に対するこの量を関連付けることである。光学的測定のこの方法を使用して、本発明の好ましい実施態様において、代表的には60〜70%の取り込み効率が達成されることが観察され、このことは先行技術の方法を超える有意な進歩を表す。
【0018】
この微粒子中に含有されるDNAは、代表的には、二本鎖DNAを含む。本発明における使用のための適切なDNA配列の構築は、当業者に認識され、そしてWO−A−97/17063に記載される。この配列は、転写プロモーターおよび遺伝子コード配列の両方を含むことが好ましい。このDNA配列は、コード配列の下流に転写終結およびポリアデニル化を提供することがさらに好ましい。
【0019】
このDNAは、二本鎖、環状、およびスーパーコイルであることが特に好ましい。微粒子の製造の間に、そのDNAは剪断力を受けることが観察されてきた。本発明の粒子製造条件を用いて、本発明者らは、機能的なDNAの有意な量を保持することを成し遂げたが、前もってスーパーコイル化されたDNAは、このプロセスの間に部分的に開環型に転換され得ることが観察された。カプセル化プロセスがDNAを変性させる程度は、ネイティブな(スーパーコイル)DNAを、アガロースゲル電気泳動を使用して、部分的に変性した(開環型)DNAおよび変性した(ニックが入り、そして分解した)DNAから分離することによって評価され得る。このような分離の代表的な例を、図1に例示する。ここでは、明確に規定されたバンドが、調製物中のDNAの2つの最も豊富な形態(スーパーコイル(下のバンド)および開環型(上のバンド))に対応する。DNAの全体の分解は、通常、2つの通常の形態に対応するバンドの消失から説明される。
【0020】
カプセル化されたDNAの生物学的活性の保持、好ましい物理的状態の保持の尺度は、微粒子からのDNAの放出後に、既知量のDNAが培養中にコンピテント細菌を形質転換する能力、または真核生物細胞をトランスフェクトする能力のいずれかを測定することにより評価され得る。この形質転換アッセイは、プラスミドに存在し、そして感受性の細菌にアンピシリン耐性を付与する機能的な抗生物質耐性遺伝子(例えば、β−ラクタマーゼ)の導入を測定する。この遺伝子の発現は、DNA構造の全体の保持を示す。トランスフェクションアッセイは、培養中の適切に操作された細胞中の目的の遺伝子の発現を誘導する、プラスミドの機能を具体的に測定する。両方の場合において、得られる活性の指標は、対応する蓄えられている等価な量のDNAの活性と比較される。
【0021】
プラスミドDNAまたは従来の操作によってそれから誘導されるDNAは、特に適切である。プラスミド製造に関する多数の文献が存在するので、当業者は、容易に本発明の微粒子のために適切なプラスミドを調製し得る。一般的に、任意の真核生物プロモーター配列または原核生物プロモーター配列を組み込むプラスミドが適切である。
【0022】
本発明の微粒子を調製するために最も適切なポリマーは、代表的には、多くの特性を示す。このようなポリマーは、低毒性であり、理想的には薬学的に受容可能かつ採用された溶媒(例えば、共溶媒を含むか、または含まないかのいずれかである酢酸エチル)中に、好ましくは少なくとも約50mg/mlのレベルまで溶解するべきである。さらに、それらは、代表的には、生体適合性でありかつ生物分解性であるが、胃の酸条件を通過するに十分に安定であることが好ましい。それにもかかわらず、本発明は、その最も広い局面において特定の単一のポリマーに限定されることを意図しない。ポリ(アミノ酸/アミノ酸の誘導体)に基づくポリマーは適切であり、そしてこれらのいくつかの具体的な例は、ポリ(ラクチド)、ポリ(グリコリド)および/またはポリ(ラクチド−コ−グリコリド)を含むポリマーである。本発明の特定の実施態様において、以下でより詳細に記載されるように、そのポリマーは、ポリ−(DL ラクチド−コ−グリコリド)(PLG)であり、そしてポリマー溶液中のPLGの濃度は、代表的には少なくとも10%重量/容量である。本発明の別の特定の実施態様において、以下にまた記載されるように、そのポリマーは、ポリカプロラクトンである。
【0023】
本発明の微粒子を製造するために適切なPLG中におけるラクチドとグリコリドとの比は重要でなく、そして市販のポリマーは、25:75、50:50、および75:25のラクチド:グリコリド比を含むが、その比は0:100〜100:0の範囲の適切な数値をとり得る。本発明の特に好ましい実施態様において、そのポリマーは分子量70kDより大きいか、または50kDより小さいPLGである。本発明に従う微粒子のための他の適切なポリマー処方物は、ポリヒドロキシブチレート、ポリヒドロキシバレレート、ポリ(ヒドロキシブトレート/バレレート)、エチルセルロース、デキストラン、多糖、ポリアルキルシアノアクリレート、ポリ−メチル−メタクリレート、ポリ(e−カプロラクトン)、ポリヒドラジン、およびこれらの成分のすべての混合物を含む。
【0024】
本発明のさらなる局面は、微粒子を調製するために使用される方法の汎用性に関する。具体的には、本発明者らは、基本的な方法論の実質的な改変なしで、広範な分子量の範囲にわたるポリマーを使用して微粒子を作製した。従って、本発明は、以下の工程を包含する、生物活性薬剤をポリマー微粒子中にカプセル化する方法を提供する:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
生物活性薬剤の水溶液を調製する工程;
ポリマー溶液と生物活性薬剤溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、安定剤または界面活性剤を含むさらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成し、この微粒子は生物活性薬剤を含有している、工程;ここでこのポリマーは、分子量40kD以下のPLGを含むか、またはこのPLGからなる。
【0025】
好ましくは、PLGの分子量は30kD以下であり、そして本発明の特定の実施態様において、微粒子は3kDのPLG、6kDのPLG、9kDのPLG、22kDのPLGおよびそれらの混合物を含む。適切なポリマーの分子量の範囲は、1.5kD〜250kDである。本発明者らは、3、6、9、12、18、22、60、65、および90kDの市販の調製物を使用した。ポリマーの加水分解速度は分子量に関係し、従ってより低分子量のポリマーは、より迅速に分解する。
【0026】
Sahらによって提案されたように、微粒子が蜂の巣状の構造である場合、ポリマーマトリックス中の水相は、凍結乾燥工程の間に凍結乾燥されない。このことは、湿ったポリマーがグリコール酸および乳酸に加水分解されるので、頻繁に観察される先行技術の調製物の生物学的薬剤の変性およびpHの減少を説明し得る。本発明者らは、本発明に従って作製した微粒子が、乱暴に扱った場合に崩壊することを観察した。このことは、本発明の微粒子が、水が除去された空の内部を有することを示唆する。
【0027】
本発明の微粒子は、実際殻状であり(図3b)、中実の蜂の巣状ではなく、または、水和された場合に、水以外の何らかの種の物質相を封入している。さらに、本発明の微粒子は、例えば凍結乾燥によって、球状構造を損なうことなく乾燥され得る(図4a)。従って、本発明は、水性調製物よりもはるかに安定である乾燥pDNAを含む微粒子の調製を可能にする。
【0028】
本発明の微粒子の製造は、任意の水溶性物質(任意の水分散可能な物質でさえ)の微粒子への組み込みを可能にするが、好ましくは、その生物活性薬剤は、以下から選択される:RNA、タンパク質抗原、非タンパク質抗原、タンパク質結合体化多糖またはペプチド結合体化多糖、タンパク質−DNA複合体またはペプチド−DNA複合体、合成ペプチド、合成タンパク質、DNAウイルス、サイトカイン、癌治療薬、ミニ遺伝子、および水溶性薬品。生物活性薬剤はさらに、賦形剤および/またはアジュバントを伴い得、その1つの例はミョウバンである。
【0029】
実質的に任意の組換えタンパク質抗原およびネイティブなタンパク質抗原(特に、任意の病原性生物に対して免疫するために有用な任意の組換え抗原またはネイティブな抗原)は、本発明の方法を用いてカプセル化され得る。例示のみの目的で、本発明の微粒子への取り込みのためのタンパク質抗原は、必要に応じて以下から選択される:
(a)表4に従う以下の抗原:FHA、PT、69kD−Pertactin、破傷風毒素、gp48、NS1、Capsid、gp350、NS3、SA、I、NP E、M、gp340、F、H、HN、35kDタンパク質、BP1、E1、E2、C、M、E、およびMSHA;ならびに
(b)(a)のポリペプチドの免疫原性フラグメント、改変体、および誘導体。
【0030】
これらの抗原についての遺伝子配列の登録番号の詳細は、表4に列挙される。
【0031】
本発明の水中(油中水)型エマルジョン系において、DNAまたは生物活性薬剤を含む水滴は、それ自体が第2の水相に分散される油中に分散される。このことは、「ぶどうバン(currant bun)」の形成を生じ得る。ここでぶどうは水相およびDNAであり、そしてバンはポリマーである。本発明において、この最初の油中水型エマルジョンの生成およびその第2の水相へのその分散は、最終的な微粒子の形成および性質における主要な役割を果たす。本発明者らの微粒子の生成は、最初の乳化段階によって非常に影響される。なぜなら、生じる微粒子は、分散されたDNA含有水相または生物活性薬剤含有水相からの個々の小滴に対応するからである。ポリマーが濃縮されるのはこのあたりである。本発明の好ましい実施態様において、このポリマーの濃度および最初の水相に対するポリマー溶液の比は、得られる微粒子に影響を与えるように制御される。
【0032】
具体的には、本発明の好ましい実施態様において、油中水型エマルジョンの形成における溶媒:水相の比は、4:1〜20:1(容量:容量)、より好ましくは5:1〜15:1、なおより好ましくは、5:1〜12:1(容量:容量)の範囲である。これらの範囲内での比の選択は、生じる微粒子のサイズの範囲および構造を決定するための手段である最初の油中水型エマルジョンの形成を決定し、そして微粒子へのDNAの取り込み効率を改善する。溶媒:水相の比が約4:1より下になった場合、生物活性薬剤(例えば、DNA)の微粒子への取り込みの顕著な減少が存在する。このことは、不十分なポリマー含有相が、水滴を効率よく被覆するために利用可能であり、従って殻形成および生物活性薬剤の、第2のエマルジョン相の水環境への損失を妨害する場合の結果である。この取り込み効率は、少なくとも4:1、そして好ましくは少なくとも5:1の比で増加する。この比が約20:1よりも高くなったならば、Sahの論文中に示されるような「ぶどうバン」型の構造の形成を妨害することが極端に困難になる。このことは、過剰なポリマー含有相の結果であり、これは、「ぶどうバン」構造への凝集を生じる個々の殻構造の解離およびポリマーマトリックスの内部を示す中実微粒子のパーセント(percent)である。その構造は、直径が10ミクロンよりも何倍も大きく、本発明に従う経口免疫には全く適していない。約20:1より下、そして好ましくは約15:1より下では、本方法についての、これらの「ぶどうバン」型構造を産生する傾向は低下し、そして直径10ミクロンまでの微粒子がより容易に形成される。
【0033】
そのポリマーを溶解する上で使用するのに適切な溶媒は、好ましくは低毒性であり、そして理想的には、薬学的な使用のために認可された溶媒のカテゴリーIIIリストから選択される。カテゴリーIIリストの溶媒もまた使用され得るが、ヒトに対する使用のためには、通常、そのような粒子が薬学的に認可され得る前に、粒子中に残存する溶媒の量を決定することが必要である。そのような溶媒は、好ましくは、少なくとも約50mg/mlのポリマーを溶解し、そして実質的に水と混和しないべきであり、それによって水中(油中水)型エマルジョンの形成を容易にする。本発明の好ましい実施態様において、溶媒は、高い蒸気圧を示し、その結果、最終溶媒抽出工程が迅速に実行され得る。本発明の方法における使用のために適切な溶媒の例としては、酢酸エチル、ジクロロメタン、クロロホルム、プロピレンカーボネート、およびそれらの混合物が挙げられる。アセトンもまた、溶媒混合物中で(すなわち、共溶媒として)使用され得るが、それ自体は、水と混和するので使用され得ない。
【0034】
溶媒中に溶解され得るポリマーの量を増加させる共溶媒を伴うか、または共溶媒を伴わないかのいずれかで、溶媒が酢酸エチルを含むことがさらに好ましい。混合物の大部分の比率が酢酸エチル(または、別の薬学的に受容された溶媒)であるならば、多くの共溶媒の組み合わせが適切である。共溶媒系を使用することについての利点は2倍である。第1に、溶媒の組み合わせは、単一成分の溶媒におけるよりも多くの量のポリマーを溶解させる。このことは、最初のエマルジョンの生成において溶媒水相の比を減少させる試みにおける場合に有利である。第2に、共溶媒は、高分子量ポリマーの溶解を容易にし、このことは、より溶解度が低いポリマーを用いて重要な溶媒水相比の維持を可能にする。このような共溶媒の例としては、プロピレンカーボネートおよびアセトンが挙げられる。より一般的には、本発明はまた、微粒子の調製における使用のためのポリマー溶液の調製のための任意の適切な溶媒と組み合わせた、共溶媒の使用を提供する。従って本発明の方法は、酢酸エチルを必然的に含む溶媒に限定されない。例えば、ジクロロメタン単独によって溶解されるよりも多い重量のポリマーを溶解する、ジクロロメタンおよびアセトンの組み合わせである溶媒は、本発明の別の局面である。本発明のさらなる局面は、それによって組み合わせた溶媒が薬学的に受容可能な溶媒単独でよりも多くの重量のポリマーを溶解する、共溶媒(例えば、アセトン)と組み合わせた任意の薬学的な溶媒の使用を含む。
【0035】
DNAがカプセル化される場合に、本方法の乳化工程が、減少された剪断化ストレス条件下で実行されることがまた特に好ましく、そしてこのことは、必要に応じて、乳化エネルギーの使用(例えば、乳化ミキサーの場合における速度、これはエマルジョンを得るために十分であり、そして所望のサイズ範囲における微粒子を形成するために十分であるが、過度の剪断によってすべてのDNAが損傷するほどには高くない)によって達成される。本発明の1つの実施態様において、以下に記載されるように、乳化ミキサーの速度は改変され、その結果、DNAの生物学的活性の少なくとも25%(コンピテント細菌の形質転換または培養細胞のトランスフェクションによってアッセイされた)が、DNAを含む得られた微粒子中で保持される。適切なミキサー速度は、Silversonミキサーの場合、8000rpmより下であり、好ましくは6000rpmより下であり、そして以下に記載する特定の実施態様においては、その速度は約3000rpmまたは約2000rpmより上である。エマルジョン相を生成するために使用され得る装置のすべてのバリエーションは、異なる速度における、そして異なる条件下での評価を必要とする。当業者にとって、装置の各部品についての乳化の最適速度および継続時間を同定することが残されている。Silverson Homogeniserを8000rpmより上の速度で使用する場合、DNAに有意な損傷が存在し、そして約2000rpmより下で使用する場合、直径10ミクロン以下のサイズの微粒子の形成はほとんど見られないか、または全く見られない。Silversonミキサーを使用するミキサー速度の効果のより詳細な分析は、WO−A−97/17063に見出される。
【0036】
従って、微粒子の形成に先立つか、または微粒子の形成の間の工程は、所望のサイズの範囲(代表的には、0.01〜10ミクロン)の微粒子を形成するために十分なエネルギーであるが、しかしDNAまたは生物活性薬剤が、このプロセスの間に損傷を受けるほど大きくはないエネルギーを入力するために適合される。より強い攪拌(例えば、より高速のミキサー速度を通して)であるほど、より小さい微粒子サイズを生じるというような必要とされるバランスが存在する。しかし、DNAは、乳化の間の過度の攪拌によって損傷され得る。一方、エマルジョン形成の間にエネルギー入力を減少させることは、エマルジョンが形成されず、そして微粒子が得られ得ないという効果を有し得る。本発明は、これらの競合する因子のバランスをとることを可能にして、受容可能な程度の生物学的活性を保持する微粒子の形成を提供する。
【0037】
本発明のさらなる実施態様において、本方法は、直径10ミクロンのサイズまでの微粒子を生じる高い比率の水相の小滴を得るために、ポリマーおよび水溶液の乳化を維持して、より長い時間にわたって最初の油中水型エマルジョンを形成する工程を包含する。このことは、油中水型エマルジョンの形成の間により長い攪拌を用いて、微粒子のサイズ分布がより均一になるという利点を有する。
【0038】
より長い乳化は、10ミクロンより大きい微粒子の比率を減少する。さらに、異なる乳化エネルギーにおける乳化の延長は、異なる点の付近を中心とするサイズ分布を産生する(より高いエネルギーは、より小さい平均微粒子サイズを産生する)。従って、本方法は、所望されるサイズに得られるサイズを仕立てるために改変され得る。いくつかの先行技術は、Peyerのパッチを通しての微粒子の取り込みとともに、1〜5ミクロンの微粒子および5〜10ミクロンの微粒子は、異なって取り込まれることを示唆し、そして本発明は、任意の選択された平均直径を有する微粒子の製造を容易にする。大部分の微粒子が、そして好ましくは実質的にすべての微粒子が、直径10ミクロンまでのサイズ、そして直径0.1ミクロンより大きい、微粒子を得るために、ポリマーおよび水溶液の攪拌を維持して、より長い時間にわたって油中水型エマルジョンを形成させることもまた、好ましい。この文脈において、実質的にすべての粒子との言及は、数で粒子の少なくとも4分の3、そして好ましくは少なくとも90%が、選択された直径のサイズであることを意味することを意図する。
【0039】
エマルジョンの攪拌の間に発生する任意の熱の蓄積を消散させ、それによってカプセル化された生物学的薬剤に対する損害を減少させるために、エマルジョンサイクルの間に休止時間を挿入することもまた好ましい。
【0040】
本発明の特定の実施態様において、以下にさらに詳細に記載されるように、以下の工程を包含する、ポリマー微粒子中に生物活性薬剤をカプセル化するための方法が提供される:
溶媒にポリマーを溶解して、ポリマー溶液を形成する工程;
生物活性薬剤の水溶液を調製する工程;
ポリマー溶液と水溶液とを攪拌しながら合わせて、油中水型エマルジョンを形成する工程;
この油中水型エマルジョンを、さらなる水相に攪拌しながら添加し、水中(油中水)型エマルジョンを形成する工程;
この水中(油中水)型エマルジョンを、過剰の水相に添加して溶媒を抽出し、それによって直径10ミクロンまでのサイズのポリマー微粒子を形成する工程;ここでこのポリマー溶液と水溶液との攪拌、ならびに油中水型エマルジョンとさらなる水相との攪拌は、ブレンダー中で実行される。
【0041】
本発明はさらに、乳化工程を実行するためのブレンディング装置を使用して、本発明のポリマー微粒子中にカプセル化された生物活性薬剤を調製する方法を提供する。ブレンディング装置は以下を備える:
混合される液体のための容器;
その容器中で液体を混合させるためのブレード;
ここで、このブレードは、回転軸の周りを回転し、そして実質的に回転軸に沿った方向で液体を駆動させることを包含する混合動作を行う。
【0042】
このブレードの回転動作は、チャンバー内の液体中で迅速な循環する渦巻きを確立することによって、この液体に乳化エネルギーを与える。従って、この装置の作用は、次に乳化スクリーンを通して押し出される液体を引き上げるスピニングプローブを使用する他のミキサーと区別される。このような作用は、有意な剪断力を生成し、そしてDNAに高度に損傷を与える。本発明の装置の利点は、DNAのような生物活性薬剤が、カプセル化の間に、より低い剪断力および他のストレスに供せられることである。この装置のブレンディング作用はまた、超音波パルスを使用して液体にエマルジョンエネルギーを与えるソニケーションミキサーから区別される。超音波もまたDNAに損傷を与え、そしてポリヌクレオチド鎖のフラグメント化を引き起こし得る。
【0043】
エマルジョン工程は、好ましくは、Waring(RTM)型のブレンダー/ミキサーである装置を使用して実行される。従って、そのブレンダーは、エマルジョンのブレンディングおよび混合のためのチャンバーを備え、そしてさらに、そのチャンバー中に固定可能にマウントされる回転可能なブレードを備える。このブレードは、ブレードから上方および/または下方に延びる複数のブレード端チップを備え、そしてそれは、ブレードに対しておよそ30〜60度であり得るか、またはブレードに対して垂直(従って回転軸に対して平行)でさえあり得る。
【0044】
実験的な目的に適切な小容量の液体についてのみ適切なホモジナイザーの使用とはっきりと対照的に、本方法が工業的な微粒子の製造へのスケールアップのために適切であることは、本発明に従う装置の使用の利点である。
【0045】
本発明はまた、粒子内容物の放出速度の改変を達成するためのポリマー殻の改変に関する。
【0046】
従って、本発明は、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子を含む組成物を提供する。この組成物は微粒子を有し、ここでこのポリマーは、50kDより小さく、そして好ましくは1.5kDまで小さい分子量のPLGである。
【0047】
本発明はさらに、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子を含む組成物を提供する。この組成物は微粒子を有し、ここでこのポリマーは、70kDより大きく、そして好ましくは250kDまで大きい分子量のPLGである。
【0048】
本発明はなおさらに、外部のポリマー殻および内部の生物活性薬剤の水溶液を含む、直径10ミクロンより小さい微粒子の混合物を含む組成物を提供する。この組成物は第1の微粒子(ここでこのポリマーは、50kDより小さい分子量のPLGである)および第2の微粒子(ここでこのポリマーは、70kDより大きい分子量のPLGである)である。個々の型のポリマー粒子の非常に複雑な混合物が、組み合わせワクチンまたは治療剤として混合および使用され得る。
【0049】
しかし、可能な場合、本発明における使用のために適切なポリマーの分子量分布は、固有の粘度(i.v.)によって引用される。このi.v.は、容易に測定される定量可能な値であり、分子量分布に相関することが公知である。従って、「平均分子量」の値よりもポリマーの分子量分布の記載が、しばしばより正確である。このi.v.は、単位質量あたりの容量の関数として測定され、そして一般的に、グラムあたりのデシリットル(dl/g)で表現される。
【0050】
本発明の1つの局面において、直径10ミクロンまでのサイズであり、そして生物活性薬剤を含有するポリマー微粒子が提供され、ここでそのポリマーは0.5dl/g未満の固有の粘度のPLGである。生物活性薬剤は、レシピエントへの投与後に発現されるポリペプチドをコードするか、またはアンチセンス配列をコードするDNAのようなDNAであり得る。
【0051】
このことは、この微粒子がレシピエントの内部で一度分解され、そして抗原提示細胞によって取り込まれ得るが、それは有意な割合のDNAが分解される前であるという利点を与える。
【0052】
微粒子の大部分が細胞によって取り込まれ、そしてインターナライズされた場合に、カプセル化されたDNAの最大量が放出される放出プロフィールを得ることが好ましい。本発明の好ましい実施態様の代表的な放出プロフィールは、動物への投与後に、1日目と20日目との間で、より好ましくは2日目と10日目との間で、カプセル化されたDNAの30〜40%の放出を提供する。投与が経口経路を介する場合、有意なDNA放出が、微粒子が動物の胃を通して腸内への通過後に起こる。次いで、そこで微粒子はPeyerパッチのM細胞によって取り込まれ、次いで、ここで微粒子は分解し、カプセル化されたDNAを放出する。
【0053】
上記で、低いi.v.(代表的には低分子量)のPLGの使用が、より迅速な粒子の分解および粒子の内容物の放出によって、いかにして標準的な60kD PLG(高いi.v.を有する)を使用するよりも改善された免疫を与え得るかを議論した。それにもかかわらず、i.v.の範囲(従って、分子量の範囲)を有する微粒子を含む組成物が使用された場合、この組み合わせは有利な免疫効果を産生するということが利点であり得る。
【0054】
本発明の特定の実施態様において、微粒子についての放出プロフィールは、i.v.および/またはポリマーの分子量分布を調整することによって操作され得る。以下により詳細に記載される本発明の特定の実施例において、この調整は、より高いi.v.値およびより低いi.v.値のPLG調製物を組み合わせることによりなされ、中間体i.v.の複合ポリマーを産生する。中間体i.v.PLGから作られた微粒子は、改変されたDNA放出プロフィールを示し、ここで存在するより低いi.v.ポリマーの割合がより大きいほど、より迅速にDNAが放出される。このようにして、経口および他の投与経路に適切な所望の放出プロフィールを示す本発明の微粒子は産生される。
【0055】
好ましくは、i.v.は、0.1〜0.4dl/gの範囲である。本発明の特定の実施態様において、この範囲内のi.v.を有するPLGからなるポリマー殻を有する微粒子が、良好なDNA放出プロフィールおよび保持された良好な粒子構造を示すことが、走査型電子顕微鏡の下で調べた場合に見出された。以下に記載される本発明の特定の実施例において、粒子は、0.19dl/gおよび0.39dl/gのi.v.を有するPLGの混合物で作製され、そしてこれらの粒子は、経時的に非常に良好な放出プロフィールを示した。
【0056】
本発明はまた、ワクチン接種治療および本発明の微粒子を用いる方法に関する。従って、本発明はさらに、以下の本発明の方法を含むワクチン接種用組成物としての使用またはその組成物における使用のための微粒子の作成方法を提供し、ここでこの微粒子は、抗原をコードするDNAを含む。
【0057】
なおさらに本発明は、ワクチン接種用組成物としての使用またはその組成物における使用のための組成物を提供し、これは、以前に列挙されたような本発明の組成物を含み、ここでこの微粒子は抗原をコードするDNAを含み、そして本発明はワクチン接種のための医薬の製造における本発明を含む組成物の使用を提供し、ここでこの微粒子は免疫源をコードするDNAを含む。
【0058】
本発明の特定の実施態様において、その微粒子はマトリックスの内部に位置するか、またはマトリックスによって囲まれる。このようなマトリックスは、好ましくは時間とともに分解して、定常的な流れまたは断続的なパルスのいずれかにおいて微粒子を放出する生物分解性材料から作られる。本発明のデポット(depot)は、代表的には、皮下投与されるか、または身体の他の部分に移植され得る。このデポットは部分的に再水和に抵抗性であって、その結果コア中に位置する粒子がそれらが放出されるまで脱水状態で残っており、それによってカプセル化されたDNAがデポットの実質的に全寿命の間に高い完全性であることを確実にすることが好ましい。このようなデポットの製造のために適切な材料の例は、当該分野で一般的に公知であり、そしてポリヒドロキシ酪酸;ポリカプロラクトン;シリコーン;エチレンビニルアセテートおよびポリビニルアセテートを含むがこれらに限定されない。
【0059】
本発明の好ましい実施態様の方法に従って、得られる微粒子の大部分(代表的には90%以上)は、10ミクロン以下のサイズ範囲にある。微粒子調製物の電子顕微鏡写真は、これらの大部分が殻であるように見えることを示す(図3b)。より小さい微粒子の存在は、調製物の効力に対して有害ではない。なぜなら、1マイクロメートルより小さい微粒子はなお、M細胞によって活性に食作用を受け、続いて消化管関連リンパ系組織(GALT)の細胞に移動することが公知であるからである。実際、小さい(直径2ミクロン未満)サイズの微粒子は、腸の食細胞およびM細胞による取り込みを促進することにおいて有利であることが示唆されてきた。活性な生物活性分子を含む小さな粒子を調製するための主な障壁は、所望のサイズ分布を調製するために、大量の潜在的な損傷的エネルギーを導入する必要性であった。本発明の明確な利点は、ブレンダー技術を使用するエネルギー入力のさらなる制御が、この生物活性分子を変性させることなく小さな粒子の生成を可能にすることである。これはまた、これらの強力に改善された送達ビヒクルの大規模産生の可能性を提供する。
【0060】
微粒子のサイズは、M細胞による微粒子の貪食作用のプロセスにおいて、そして細胞間移動に非常に重要である。10ミクロンを超えるサイズの微粒子は取り込まれず、そして腸の上皮を横切って輸送されないことが公知である。先行技術に従って作製されたいくつかの調製物では、本発明者らは、10ミクロン〜60ミクロンの直径を有する、すなわち、必要とされる最適サイズよりも大きな、有意な数の微粒子を調べた。スフェアの容量が立法関数(cube function)に基づいて算出されるとみなされる場合、このような調製物において、大部分のpDNAが、大きな殻の内部に含まれ得ることが全体に実現可能である。このことは、この調製物を、免疫系に対して利用不能にし、従って用量を、予想されるよりもかなり低くし得、10ミクロンまでの所望のサイズ範囲の大多数の微粒子を有する微粒子調製物をもたらす、本発明に記載される方法に従って、本明細書において問題を克服する。
【0061】
本発明の微粒子の放出特性の制御が検討されている。生物活性薬剤が正確にはどのようにして微小殻から放出されるかは、不明である−おそらく、一旦、殻が加水分解によって分解した場合の、バーストとしてである。このことは、微粒子の分解が異なる時点で起きるに従って持続放出をもたらす。時間範囲は、考えられるところでは、以前に意図された時間よりも短い期間を超え得、そして記載されるようなポリマー分子量を通して制御可能である。
【0062】
ある範囲の界面活性剤は、本発明の方法における使用に適切であり、そして本発明は、実施例において使用される特定の界面活性剤であるポリビニルアルコールに限定されない。他の受容可能な界面活性剤が当該分野で公知であり、そしてメチルセルロースがさらなる例である。界面活性剤は、二重エマルジョンを安定化する役割を有する。水性界面活性剤の選択は、当業者の問題であり、そしてこの選択は、ポリマーおよびポリマー溶媒の選択に関して行なわれ得る。
【0063】
腸におけるM細胞による微粒子の取り込みについての他の関係のある背景は、以下に提供される:Jepson MAら,Journal of Drug Targeting,1993,第1巻,245−249頁;Howard KAら,Pharmaceutical Science Communications,1994,第4巻,207−216頁;Neutra MRら,Cell,1996,第86巻,345−348頁;Florence AT,Pharmaceutical Research,1997.第14巻,259頁以下参照;O’Hagan DT,J.Anat.,1996,第189巻,477−482頁;およびKreuter J,J.Anat.,1996,第189巻,503−505頁。
【0064】
本発明はここで、添付の図面を参照して特定の実施態様において記載される。
【0065】
図1は、ジクロロメタン(先行技術:レーン3)または酢酸エチル(本発明に従う:レーン4(高分子量PLG)、レーン5(低分子量PLG))のいずれかを用いて調製した、等しい重量のPLG微粒子から回収されたDNAの相対量および相対的な物理状態を示すアガロースゲルを示す。レーン2は、大きな比率のスーパーコイル形態(下のバンド)を示すコントロールDNA(ストック)であり、そしてレーン1および6は、分子量マーカーである。レーン3とレーン4および5との比較は、酢酸エチルがDNAのカプセル化を促進するだけでなく、開環形態(上のバンド)と比較してスーパーコイル形態(下のバンド)のより大きな比率の保持をももたらすことを示唆する。レーン4(高分子量PLG)において分析された調製物におけるDNAの取り込みがより小さいのは、酢酸エチル中でのポリマーのより低い溶解度に起因する、制限されたポリマーの存在における微粒子形成の結果であることに留意すべきである。
【0066】
図2は、本発明のPLG微粒子のインビトロでの放出反応速度論を示す。微粒子を、高分子量PLG(黒四角)または低分子量PLG(白四角)を用いて調製した。37℃に維持した緩衝液中に懸濁した微粒子の滅菌インキュベーションから提供された上清のアリコートにおけるDNA濃度を、光学吸光度分光法を用いて測定した。
【0067】
図3aは、凍結乾燥した粉末としてEM標本グリッドに適用したPLG微粒子の調製物(本発明に従って調製される)を示す。球形構造の均一性は、この粒子が凍結乾燥によって有害な影響を受けないことを示す。
【0068】
図3bは、最適な条件下で調製されたPLG微粒子(本発明に従う)「殻様」構造を実証する。標本を、フリーズフラクチャーした。これらの粒子の中空の内部が明らかに見られ得、そしてこれは先行技術において与えられる例とは非常に異なる。
【0069】
図4aは、室温にてEM標本グリッド上で、水和されたスラリーから乾燥されたPLG微粒子(本発明に従って調製された)の調製物を示す。蒸発した水として遭遇するストレスは、種々の程度まで構造的崩壊を誘導した。このことは、微粒子が、中空の内部を有する「殻様」構造を有し得ることを示す。
【0070】
図4bは、過剰(4:1より大きい)溶媒:水相比の条件下で調製された微粒子の内部構造を示す。調製物を、液体窒素を用いて凍結させ、そして標本を、凍結している間にフラクチャーし、粒子が種々の精巧な内部構造を有することを示した。
【0071】
図5は、種々の固有粘度のPLG調製物から作製されたPLG微粒子から経時的に放出されたDNAの百分率を示す。低固有粘度PLGを、RG502と称し、そしてより高い固有粘度のPLGをRG503と称す。50:50および75:25のRG502およびRG503混合物から作製された微粒子からのDNA放出もまた示し、そしてRG502の固有粘度とRG503の固有粘度との中間の固有粘度のPLG調製物を表す。
【実施例】
【0072】
(実施例1)
(微量カプセル化の標準的方法)
微粒子を、ここで示す方法に従って調製した。この方法は、微量カプセル化の標準的方法(microencapsulation standard method)と呼ばれる。
【0073】
1.酢酸エチル(炭酸ナトリウム上で乾燥させた)中の10%(w/v)の50:50低iv(60kD)PLG溶液を、この混合物を約37℃まで加熱し、そして溶解が完了するまで回転させることによって調製する。
【0074】
2.プラスミドDNA溶液を、STE緩衝液(10mM Tris HCI、pH8.0;1mM EDTA;150mM NaCI)中で10mg/mlの濃度になるように調製する。
【0075】
3.このPLG酢酸エチル溶液(4ml)を、Silversonホモジナイザーを用いて3000rpmにてホモジナイズし、そしてプラスミドDNA(600マイクロリットル)を添加する。得られる混合物を、さらに2.5分間ホモジナイズする。
【0076】
4.8%(w/v)水性ポリビニルアルコール溶液(92ml)を、丸底フラスコ中で約3000rpmでホモジナイズし、最初のエマルジョンを添加し、そして得られる混合物をさらに2.5分間ホモジナイズする。
【0077】
5.このエマルジョンを、37℃の2回蒸留水(100ml〜1L)に注ぎ、そして少なくとも20分間攪拌する。
【0078】
6.この微粒子を、10,000rpmにて25分間、25℃で遠心分離することにより回収する。
【0079】
7.この微粒子を、2回蒸留水中に再懸濁し、そして遠心分離することによって洗浄する。この洗浄工程を繰り返して、その結果、合計5回の回転を行い、溶媒およびエマルジョン安定剤の除去を促進する。
【0080】
8.5回目の回転の後、ペレット化した微粒子を、最少量の2回蒸留水中のスラリーとして回収し、そして凍結乾燥する。代表的な調製物を、図3aに示す。
【0081】
(標準的な方法に対するバリエーション)
上記の標準的な方法に基づいて、多数のバリエーションを、工程および/または成分に対して行った。結果は以下の通りである:
1.酢酸エチル中の50:50の低い(50kD以下)MWおよび50:50の高い(70kD以上)MWの両方のPLG溶液を研究し、そして10ミクロンまでのサイズの微粒子調製物が生成されることが見出された。他のPLG組成(例えば、75:25)および高MWまたは低MWを有するポリマーもまた、本発明の微粒子および方法に適切である。
【0082】
2.PLG対EAのw/v比を研究した。高MWポリマーを用いて、0.04g/mlまでの低減は、取り込み効率の著しい減少を導いた(図1)。試験した最大量は0.125g/mlであった。これは、カプセル化を改善し、1ミクロン〜10ミクロンのサイズ範囲の微粒子を生成したが、カプセル化の前に混濁溶液をもたらした。
【0083】
3.有機相におけるPLGの濃度を増加させるための共溶媒の使用を研究した。共溶媒(例えば、酢酸エチルと組み合わせた炭酸プロピレンおよびアセトン)は、油(溶媒)相中に溶解された増加した量のポリマーを有する、初期油中水型エマルジョンの調製を可能にした。このことは、微粒子へのDNAの取り込みを効率的に改善することが有利に見出された。
【0084】
4.最初の乳化工程における水相対ポリマー相の比は、特定の範囲内で変更され、そしてハニカム構造が回避され、そして微粒子のサイズが制御され得、粒子の調製物を実質的に全て所望の範囲内にし、そして所望の構造を有することをもたらすことが見出された。
【0085】
5.最初の乳化のためのホモジナイゼーションの速度を変更した。Silversonミキサーを用いて、いくらかのDNAの生存能力を依然として保持しながら、ホモジナイゼーションの速度を4000rpmまで増加させることが可能である。しかし、2000rpmが、良好なDNA生存能力を与え、そしてこの速度における長時間のホモジナイゼーションについて充分非損傷的であり、狭いサイズ分布を有する微粒子の形成を可能にしながらも有意なDNA損傷を導かない。3000rpmはまた、有意なDNA損傷を伴わずに長時間にわたって用いられ得る。
【0086】
6.用いられるPVAの濃度は、過剰であると考えられ、そして2%の濃度が適切であることが示唆されている。このことはまた、微粒子の安定性およびサイズにもまた影響を与え得る、混合物の粘度に対して効果を有する。
【0087】
7.最終段階で好首尾に用いられる最少容量の水は、150mlである。攪拌を、最初に37℃の水を用いて室温で実施し、そして攪拌を20分間〜1.5時間の時間にわたって継続したが、攪拌時間についての理論的上限は存在しないようである。
【0088】
8.この手順を実施する温度は、溶液の粘度および他の相における試薬の溶解度のようなパラメーターをもたらす。酢酸エチルを用いて4℃で実施した単一の実験は、より低い取り込みをもたらした。
【0089】
(実施例2)
(溶媒ならびに高分子量および低分子量の50:50 PLGの比較)
以下の実験を、標準的な方法を用いて実施した。ここでは、EA=酢酸エチル、そしてDCM=ジクロロメタンである。各場合におけるPLG容量は4mlであった。EtOHは、実験のいずれにおいても用いなかった。結果を表1に示す。
【0090】
【表1】
(実施例3)
(微粒子から回収されたDNAの物理的状態)
図1は、ジクロロメタンおよび酢酸エチルを用いて調製された微粒子から回収されたDNAの物理的状態を示す。一番下のバンドは、スーパーコイルDNAである。このバンドの強度が強いほど、DNAの統合性がより高く保持された。
【0091】
(実施例4)
(トランスフェクションの結果)
実験を、Vero細胞を用いて行った。DNAを微粒子から回収し、そしてVero細胞を、SUPERFECT REAGENT(登録商標)を用いてトランスフェクトした。
【0092】
vero細胞を、DNAを含む微粒子で、しかしトランスフェクションを増強するさらなる試薬を用いずにトランスフェクトして、さらなる実験を行った。トランスフェクションの程度を、市販のルシフェラーゼアッセイシステムを用いて評価した。結果を表2に示す。
【0093】
【表2】
(実施例5)
(PLG濃度の効果)
以下の表3は、低分子量(60kD)PLGの濃度を低減させた場合に得られた結果を示す。これらの比較実験では、酢酸エチルを溶媒として用い、有機相の容量を4mlで保持した。3000rpmというホモジナイゼーションの速度を全ての場合で用いた。
【0094】
【表3】
(実施例6)
(共溶媒の使用)
高分子量PLGは、酢酸エチル中で10% w/v以上では全体として可溶性でない。PLG濃度を増加させるため、溶媒の混合物(例えば、酢酸エチルおよび炭酸プロピレン)の適用を調査した。
【0095】
80%酢酸エチル:20%炭酸プロピレンは、10% w/v PLGの清澄な溶液を与えた。
【0096】
(実施例7)
(放出反応速度)
高分子量および低分子量のポリマーを使用して酢酸エチルを用いて調製した微粒子のサンプルを研究して、放出特性を決定した。この微粒子を滅菌STE緩衝液に添加し、そして適切な抗菌剤を用いて37℃にて維持した。サンプルを間隔をおいて取り出し、そして放出されたDNAおよび保持されたDNAを決定した。このデータから、時間プロフィールとともに放出百分率を決定した。この結果を以下の図2に示す。
【0097】
(実施例8)
(ポリマーとしてポリカプロラクトンを用いて調製した微粒子)
微粒子を、以下の方法に従って、ポリカプロラクトンポリマーを用いて調製する。
【0098】
1.酢酸エチル中のポリカプロラクトンの100mg/ml溶液(平均MW=14kD;Aldrich Chemical Company)を、溶解が完了するまでボルテックスすることにより調製する。
【0099】
2.プラスミドDNA溶液を、9.9mg/mlの濃度になるようにSTE緩衝液(10mM Tris HCl、pH8.0;1mM EDTA;100mM NaCl)中で調製する。
【0100】
3.ポリカプロラクトン酢酸エチル溶液(4ml)を、Waringブレンダー中で低出力でホモジナイズし、そしてプラスミドDNA(400マイクロリットル)を添加する。得られる混合物を、15秒間の休止期間を間に挟んで15秒間のサイクルで4回ホモジナイズする。
【0101】
4.8%(w/v)水性ポリビニルアルコール溶液(66ml)を、Waringブレンダー中で低出力でホモジナイズし、最初のエマルジョンを添加し、そして得られる混合物を、15秒間の休止期間を間に挟んで15秒間のサイクルで4回ホモジナイズする。
【0102】
5.このエマルジョンを、37℃の2回蒸留水(300ml)中に注ぎ、そして少なくとも20分間攪拌する。
【0103】
6.微粒子を、25℃にて10,000rpmで25分間遠心分離することにより回収する。
【0104】
7.この微粒子を、2回蒸留水中に再懸濁し、そして遠心分離することにより洗浄する。この洗浄工程を繰り返し、その結果、合計5回の回転を行い、溶媒およびエマルジョン安定剤の除去を促進する。
【0105】
8.5回目の回転の後、ペレット化した微粒子を、最少量の2回蒸留水中のスラリーとして回収する。微粒子は、主に直径1ミクロン〜10ミクロンのサイズ範囲で得られ、次いで貯蔵のために凍結乾燥される。
【0106】
(実施例9)
(ポリマーの固有粘度を変更することによる、PLG微粒子の放出プロフィールの改変)
実施例8の手順は、以下のPLG調製物を用いて以下の通りであった:
(a)0.19dl/gの固有粘度(RG502、Boehringer Ingelheimから)
(b)0.39dl/gの固有粘度(RG503、Boehringer Ingelheimから)
(c)0.19dl/gおよび0.39dl/gの固有粘度のPLGの50:50(重量による)の混合(RG502およびRG503の混合)
(d)0.19dl/gおよび0.39dl/gの固有粘度のPLGの75:25(重量による)の混合(RG502およびRG503の混合).
主に1ミクロン〜10ミクロンのサイズの微粒子を得て、そしてこの粒子からのDNAの放出のプロフィールを測定し、そして図5に図示する。
【0107】
(実施例10)
以下の改変を加えた実施例8のプロトコールに従った:8mlのポリマー溶液および0.8mlのプラスミド溶液を用い、そして132ml PVAを用いて第1のエマルジョンを安定化させた。
【0108】
プラスミドを、以下の通りにPLG中にカプセル化した:
(a)50:50のRG502:RG503の混合;
(b)この(a)の混合を繰り返した;
(c)70:30のRG502:RG503の混合;
(d)この(c)の混合を繰り返した。
【0109】
主に直径1ミクロン〜10ミクロンのサイズの微粒子が得られ、そしてDNA取り込み効率について試験された。(a)および(b)の微粒子から、65%が、カプセル化されたDNAとして回収され、そして(c)および(d)の微粒子から、69%が、カプセル化されたDNAとして回収された。この%の数字は、ある比率のDNAが、粒子の単離において(例えば、遠心分離によって)失われ、そして結果として、回収率は100%の効率ではあり得ないという事実を考慮していない。従って、これらの実施例において達成された数字は、非常に高い取り込み効率を表す。
【0110】
従って、本発明は、生物活性薬剤(特に、DNA)のカプセル化のさらなる、そして改善された方法を提供し、そしてさらに、遺伝子治療および/またはワクチン接種のため、ならびに他の適用のための、生物活性薬剤を含む微粒子を含む改善された組成物を提供する。
【0111】
【表4】
【特許請求の範囲】
【請求項1】
明細書に記載の生物活性薬剤をカプセル化するための方法。
【請求項1】
明細書に記載の生物活性薬剤をカプセル化するための方法。
【図2】

【図4】
【図5】
【図1】
【図3】

【図4】
【図5】
【図1】
【図3】
【公開番号】特開2010−270121(P2010−270121A)
【公開日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願番号】特願2010−146974(P2010−146974)
【出願日】平成22年6月28日(2010.6.28)
【分割の表示】特願2000−547964(P2000−547964)の分割
【原出願日】平成11年5月7日(1999.5.7)
【出願人】(500521717)
【Fターム(参考)】
【公開日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願日】平成22年6月28日(2010.6.28)
【分割の表示】特願2000−547964(P2000−547964)の分割
【原出願日】平成11年5月7日(1999.5.7)
【出願人】(500521717)
【Fターム(参考)】
[ Back to top ]