
生薬由来成分の高感度定量方法
【課題】生体試料中のグリチルリチンやその代謝物等を高感度に定量する方法の提供。
【解決手段】生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有するサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程と、を有する生薬由来成分の高感度定量方法。
【解決手段】生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有するサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程と、を有する生薬由来成分の高感度定量方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生体試料中のグリチルリチンやその代謝物等を高感度に定量するための方法に関する。
【背景技術】
【0002】
グリチルリチン(以下、GLと略記する)及びその塩は、抗アレルギー作用、抗炎症作用と共に、免疫調節作用、肝細胞障害抑制作用、肝細胞増殖促進作用、ウィルス増殖抑制・不活性化作用等の多様な生理活性を有する甘草根抽出成分である。日本では、古くから漢方薬等の臨床薬として用いられており、現在では、慢性肝疾患、湿疹・皮膚炎、小児ストロフルス、円形脱毛症、各種アレルギー、炎症等の治療に幅広く使用されている。また、医薬品以外の使用として、GLは塩なれ効果をもち、甘味効果があることから甘味料として漬物や調味料など広範囲の食品に多用されている。
【0003】
非臨床試験の結果から、GLは肝臓に集積しやすく、且つ速やかに胆汁中へ排泄される特性があり、代謝及び腸肝循環を受けながら排出されると考えられている。特に経口投与において、配糖体で水溶性極性物質であるGLは低吸収性を示すと共に、腸内細菌による代謝や初回通過効果により、末梢血における濃度が極めて低い。このため、GLの血中動態を測定するために、現在までに様々な方法が試みられているが、GLを含有する製剤、甘草を配合する漢方製剤において、服用時の血中GLは検出困難であり、その動態も長い間不明とされてきた。
【0004】
例えば、GLを経口投与された健常者から採取された血漿に対して、メタノール処理を行ってタンパク質を除去した後、当該血漿中のGLの量を、酵素免疫法(EIA)により測定する方法やオクタデシル基化学結合型(ODS)カラム等の逆相分配機能を備えるカラムを用いた高速液体クロマトグラフィー(HPLC)を用いて測定する方法(例えば、非特許文献1〜4)がある。しかしながら、これらの測定方法では、定量限界値がせいぜい0.1μg/mL程度であり、主代謝物であるグリチルレチン酸(以下、GAと略記する。)は検出することができても、GLは検出することはできなかった。このため、尿中からの検出例をもって間接的にGLの血中移行が証明された(引用文献4参照。)。そして、GL含有製剤薬効については、GLの代わりに、定量可能なGAが評価指標とされている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】イシワタ、他7名、バイオロジカル・アンド・ファーマシューティカル・ブリテン(Biological and Pharmaceutical Bulletin)、2000年、第23巻、第8号、第904〜905ページ。
【非特許文献2】デ・グルー(De Groot)、他1名、ジャーナル・オブ・クロマトグラフィー(Journal of Chromatography)、1988年、第456巻、第71〜81ページ。
【非特許文献3】ナカタ、他3名、和漢医薬学会誌、1986年、第3巻、第3号、第278〜279ページ。
【非特許文献4】ヤマムラ、他7名、ジャーナル・オブ・ファーマシューティカル・サイエンシズ(Journal of Pharmaceutical Sciences)、1992年、第81巻、第10号、第1042〜1046ページ。
【発明の概要】
【発明が解決しようとする課題】
【0006】
活性本体である未変化体GLの薬物動態は、有効性、安全性から適正使用を判断する上で有用である。このため、GAを評価対象にするのではなく、血漿等の生体試料中のGL量を定量できることが望ましい。
【0007】
本発明は、上記課題を鑑みてなされたものであって、血漿等の生体試料中のGLを検出し定量するための方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するために鋭意研究した結果、まず、逆相分配機能及び陰イオン交換機能を備える固相を用いた固相抽出法により、生体試料中からGLを含む成分を抽出した後、抽出物中のGLを質量分析法に供することにより、生体試料中のGLを検出し定量できることを見出し、本発明を完成させた。
【0009】
すなわち、本発明は、
(1) 生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、
前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程、
を有し、
前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする生薬由来成分の高感度定量方法、
(2) 前記抽出工程において、固相の洗浄を、アルカリとアルコールと水の混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことを特徴とする前記(1)に記載の生薬由来成分の高感度定量方法、
(3) 前記アルカリとアルコールと水の混合液が、0.5〜28%アンモニア水とメタノールを99:1〜1:3であることを特徴とする前記(2)に記載の生薬由来成分の高感度定量方法、
(4)前記生体試料が、血液、血漿、又は組織抽出物であることを特徴とする前記(1)〜(3)のいずれか一つに記載の生薬由来成分の高感度定量方法、
を提供するものである。
【発明の効果】
【0010】
本発明の生薬由来成分の高感度定量方法により、含有量が非常に少ない生体試料中のGLを検出し、定量することができる。このため、本発明の生薬由来成分の高感度定量方法を用いることにより、GLを含有する製剤や飲食品を摂取したヒトから採取された生体試料中のGLを定量し、薬物動態を精度よく解析することができる。
【図面の簡単な説明】
【0011】
【図1】実施例6において、ブランク血漿サンプルのクロマトグラムを示した図である。
【図2】実施例6において、標準溶液(G)を添加した血漿サンプル(血漿中のGL添加量:0.5ng/mL、GA添加量:2ng/mL)のクロマトグラムを示した図である。
【図3】実施例7において、GLの平均血漿中濃度推移を示した図である。
【図4】実施例7において、GAの平均血漿中濃度推移を示した図である。
【発明を実施するための形態】
【0012】
本発明の生薬由来成分の高感度定量方法は、生体試料中のGLを逆相分配機能及び陰イオン交換機能を備える固相を用いた固相抽出法により抽出した後、得られた抽出物中のGL含有量を質量分析法により定量することを特徴とする。従来のHPLC法等に代えて質量分析法を利用して定量することにより、GLの検出感度を高められる。また、従来のODSカラムのような逆相分配機能のみを備える固相を用いた固相抽出法ではなく、逆相分配機能及び陰イオン交換機能を備える固相を用いて調製することにより、質量分析に供される抽出物からより多くの夾雑物を除去することができ、質量分析の感度を飛躍的に向上させることができる。
【0013】
具体的には、本発明の生薬由来成分の高感度定量方法は、生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、前記抽出工程により抽出された抽出物中のGL、GA、GL及びGAの代謝物、GL及びGAの類縁物質、甘草に含有するサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程を有し、前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする。
【0014】
本発明の生薬由来成分の高感度定量方法により定量される生薬由来成分は、GL、GA、GL及びGAの代謝物、GL及びGAの類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群(以下、「GL等」ということがある。)より選択される少なくも1種以上である。
GL及びGAの代謝物としては、例えば、3−monoglucuronyl−glychrretinic acid(20β−Carboxy−11−oxo−30−norolean−12−en−3β−yl−β−D−glucopyranosiduronic acid)、30−monoglucuronyl−glychrretinic acid(3β−Hydroxy−11−oxoolean−12−en−30−oyl−β−D−glucopyranosiduronic acid)、3−oxo−GA(3,11−Dioxoolean−12−en−30−oic acid)、3α−GA(3α−Hydroxy−11−oxoolean−12−en−30−oic acid)、3位硫酸抱合体(3β−Hydroxysulfonyloxy−11−oxoolean−12−en−30−oic acid)、3β,22α−Dihydroxy−11−oxoolean−12−en−30−oic acid、及び3β,24−Dihydroxy−11−oxoolean−12−en−30−oic acid等が挙げられる。
【0015】
また、GL及びGAの類縁物質としては、例えば、20α−Carboxy−11−oxo−29−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid、20β−Carboxy−24−hydroxy−11−oxo−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid、18α−GL(20β−Carboxy−11−oxo−(18αH)−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid)、18α−GA(3β−Hydroxy−11−oxo−(18αH)−olean−12−en−30−oic acid)等が挙げられる。
【0016】
また、甘草に含有されているサポニン成分としては、例えば、licorice−saponin C1(20β−Carboxy−11−oxo−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid)等が挙げられる。
【0017】
本発明において用いられるGL等の薬学的に許容される塩は、生体内において、GL等と同様の薬理効果を有する塩であれば、特に限定されるものではない。具体的には、例えば、アンモニウム塩、ナトリウム塩、カリウム塩等が挙げられる。
【0018】
本発明においては、GL等のうち、1種類を定量してもよく、2種類以上を一の操作で定量してもよい。本発明においては、特に、GLとGAを同時に定量することが好ましい。
【0019】
本発明の生薬由来成分の高感度定量方法に供される生体試料は、生体から採取された試料であればよく、ヒトやマウス、ラット等の動物から採取されたものであることが好ましい。また、血液、血漿、血清、尿、腹水、胸水、関節液、骨髄液、胆汁等であってもよく、肝臓や膵臓、腎臓等の組織から採取された組織片等の抽出物(組織抽出物)であってもよい。組織片等の抽出物は、組織片等を常法によりホモジナイズすることにより調製することができる。本発明においては、血液、血漿、尿、又は組織抽出物であることが好ましく、血漿であることがより好ましい。
【0020】
以下、本発明の生薬由来成分の高感度定量方法を工程ごとに説明する。
抽出工程として、まず、生体試料をアルカリ又はアルコールに混和した混和物を調製する。生体試料に添加するアルカリとしては、得られる混和物のpHをアルカリ性にすることができるものであれば、特に限定されるものではなく、例えば、アンモニア、アンモニア水、水酸化ナトリウム溶液、炭酸水素ナトリウム溶液等が挙げられる。また、アルコールとしては、炭素数1〜6の低級アルコールであることが好ましく、メタノール、エタノール、イソプロパノール等が挙げられる。また、水で希釈されたアルコール溶液であってもよい。本発明においては、生体試料に添加するアルカリ又はアルコールとしては、アンモニア、アンモニア水、メタノール、又はメタノール水溶液であることが好ましく、アンモニア又はアンモニア水であることがより好ましい。
【0021】
生体試料に添加するアンモニア水の濃度は特に限定されるものではなく、生体試料の種類、その後に使用する固相の種類、得られる混和物のアンモニア濃度等を考慮して適宜調整することができる。本発明においては、生体試料に添加するアンモニア又はアンモニア水の濃度は、得られる混和物のアンモニア濃度が0.01〜30%となるような濃度であることが好ましく、0.05〜25%となるような濃度であることがより好ましく、0.05〜20%となるような濃度であることがさらに好ましい。
【0022】
次いで、得られた混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入し、当該混和物中のGL等を固相に吸着させる。逆相分配機能及び陰イオン交換機能を備える固相としては、例えば、Oasis MAX(Waters社製)等が挙げられる。
【0023】
GL等を吸着させた固相を洗浄液で1回又は2回以上洗浄し、非特異的に結合している成分を除去する。洗浄液は、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液である。洗浄液として用いられるアルカリやアルコールとしては、生体試料に添加されるアルカリやアルコールと同様のものが挙げられる。また、固相の洗浄は、同種の洗浄液で2回以上行ってもよく、異なる種類の洗浄液で順次洗浄してもよい。
【0024】
本発明においては、固相の洗浄を、アルカリとアルコールと水との混合液、アルカリと水との混合液、又は水により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことが好ましく、アルカリとアルコールと水との混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことがより好ましい。アルカリとアルコールと水の混合液としては、アンモニアとアルコールと水の混合液であることが好ましく、アンモニアとメタノールと水の混合液であることがより好ましい。
【0025】
アンモニアとメタノールと水の混合液中の各成分の組成比は、GL等を固相に吸着させた状態を保持することができるものであれば特に限定されるものではない。例えば、混合液中のメタノール濃度が1〜75%、アンモニア濃度が0.1〜21%であることが好ましく、メタノール濃度が25〜75%、アンモニア濃度が0.1〜21%であることがより好ましい。このような組成比の混合液は、例えば、0.5〜28%アンモニア水とメタノールを99:1〜1:3で混合することにより、調製することができる。
【0026】
一回目の洗浄をアルカリとアルコールと水の混合液により行った後、2回目の洗浄に用いる洗浄液としては、アルコールを用いることが好ましく、メタノール又はエタノールを用いることがより好ましく、メタノールを用いることがさらに好ましい。
【0027】
その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する。酸性化する溶媒としては、ギ酸アルコール、塩酸、トリフルオロ酢酸等が挙げられる。本発明においては、ギ酸アルコールであることが好ましい。溶出液として用いるギ酸アルコールとしては、ギ酸と炭素数1〜6の低級アルコールとのエステルであることが好ましく、ギ酸メタノール又はギ酸エタノールであることがより好ましく、ギ酸メタノールであることがさらに好ましい。また、得られた溶出物は、以降の質量分析のために、蒸発乾固させる。
【0028】
次いで、定量工程として、抽出工程により抽出された抽出物中のGL等を、質量分析法により検出し定量する。質量分析法としては、LC−MS(液体クロマトグラフィー/質量分析)法又はLC−MS/MS(液体クロマトグラフィー/タンデム質量分析)法により行うことが好ましい。具体的には、蒸発乾固させた溶出物をLCの移動相に溶解させた後、LCを行い、MSを行う。LC−MS及びLC−MS/MSは、HPLCと質量分析計を組み合わせた装置を用いることにより行うことができる。
【0029】
LCは、特許文献1〜4に記載されているように、ODSカラム等の逆相分配機能を有するカラムを用いて行うことが好ましい。また、MSは常法により行うことができる。例えば、ESI(エレクトロスプレーイオン化)法やAPCI(大気圧化学イオン化)法により試料をイオン化した後、磁場偏向型、四重極型、飛行時間型等の装置により、各イオンを分離して検出する。
【0030】
質量分析をLC−MS法により行うことにより、例えば血液、血漿、又は血清中のGL等の定量限界を20ng/mL程度にまで改善することができる。同様に、LC−MS/MS法により行うことによって、血液等中のGL等の定量限界を10ng/mL以下、例えば0.5ng/mL程度にまで向上させることができる。
【0031】
市販試薬の大量経口投与(GLとして1600mg)における血漿中GL濃度は約500ng/mL程度であった、という報告がなされている(Environmental Health Perspectives, 102(9), 65-68, 1994)。当該知見に基づいた比例計算の結果、肝疾患治療を目的とした場合、臨床常用量である投与量75mg相当の推定血中濃度は約23ng/mLと算出される。つまり、本発明の生薬由来成分の高感度定量方法により、従来法では検出不可能であった血液中のGLが定量可能となる。特に、GL製剤等を経口摂取後の血液中のGL濃度は、個体差が大きく、定量限界が不十分な測定法では、血液検体によってはGLが検出されない場合や、長時間の薬物動態を測定することができず、測定の精度や測定結果の信頼性が不十分となるが、本発明の生薬由来成分の高感度定量方法では、特に質量分析をLC−MS/MS法により行うことによって、より信頼性の高い測定結果を得ることができる。
【実施例】
【0032】
次に実施例を示して本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。
【0033】
[実施例1]
本発明の生薬由来成分の高感度定量方法を、質量分析をLC−MS法によりを行った場合の、血漿中のGL及びGAの定量限界値を測定し、かつ検量線を作成した。
<標準溶液の調製>
まず、GL(常磐植物化学研究所製)10.0mgを正確に量り、メタノールに溶解した後、正確に100mLとし、100μg/mLのGL標準原液を調製した。同様に、GA(アルプス薬品工業株式会社製)10.0mgを正確に量り、メタノールに溶解した後、正確に100mLとし、100μg/mLのGA標準原液を調製した。
次いで、GL標準原液2mL及びGA標準原液8mLを正確に分取し、メタノールで正確に50mLとして、GL濃度が4000ng/mL、GA濃度が16000ng/mLである標準溶液(A)を調製した。この標準溶液Aをメタノールで順次希釈し、表1に記載の標準溶液(B)〜(F)を調製した。標準原液及び標準溶液はそれぞれ調製後冷蔵保存(5±4℃)した。調製器具はガラス製のものを用いた。
【0034】
【表1】

【0035】
<血漿サンプルの調製>
ヒト血漿0.5mLに、標準溶液(A)50μLを添加し、十分に混和したものを血漿サンプルとした。標準溶液(B)〜(F)についても、それぞれ同様にして血漿サンプルを調製した。これらの血漿サンプルには、内部標準液(IS)50μLをさらに添加した。なお、本実施例では、内部標準物質としてα−Hederinを用いた。また、ブランク血漿サンプルとして、ヒト血漿0.5mLにメタノールを100μL添加したものを調製した。
【0036】
<抽出工程>
各血漿サンプルに、0.56%アンモニア水1mLを添加して十分に混和した後(混和物中のアンモニア濃度:0.37%)、得られた混和物を全量、Oasis MAX(Waters社製)(以下、単に「MAX」)に注入(アプライ)した。当該MAXは、血漿サンプルを注入する前に予め、2%ギ酸/メタノール溶液を注入した後、水を注入することにより、コンディショニングしておいた。
血漿サンプルを注入後、当該MAXに0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)で洗浄した後、メタノールでさらに洗浄した。
その後、2%ギ酸/メタノール溶液を注入し、当該MAXに吸着していたGL等を溶出させた。溶出液(抽出物)は、40℃加温下、窒素ガスにて蒸発乾固させた。
【0037】
<定量工程>
蒸発乾固させた抽出物を、下記移動相A液/B液(体積比1:1の混合液)250μLに溶解させた。このうち20μLを分析カラムに注入し、下記に示す条件でLCを行った。
装置: Alliance HT 2695(Waters社製)
分析カラム: CAPCELL PAK C18(UG120、5μm、1.5mm×150mm、資生堂社製)
移動相:A液 0.1%ギ酸、B液 0.1%ギ酸アセトニトリル、リニアグラジエント(表2)
流速: 0.30mL/min.
カラム温度: 40℃
試料注入量: 20μL
【0038】
【表2】
【0039】
次いで、LCにより得られたGL等を含む画分に対して、下記に示す条件でMSを行った。
装置: ZQ2000(Waters社製)
イオン化法: ESI(Turbo Spray)
検出法: GL(負、正イオン検出)、GA・IS(正イオン検出)、MRM(Multiple reaction monitoring)
Capillary電圧: 2.5kV
Extractor: 2V
RF lens: 0.2V
Source Temperature: 110℃
Desolvation Temperature: 350℃
Desolvation Gas Flow: 350L/hr
Cone Gas Flow: 50L/hr
【0040】
【表3】
【0041】
この結果、LC−MSの測定結果は血漿サンプル中のGL又はGA濃度依存的に増大しており、血漿サンプル中のGL又はGA濃度とLC−MSの測定結果により得られた検量線は直線性を示した。すなわち、これらの結果から、血漿等の生体試料中のGLの濃度が20〜400ng/mL、GAの濃度が80〜1600ng/mLであり、LC−MSを用いた本発明の生薬由来成分の高感度定量方法により、生体試料中のGL等を高精度に定量可能であることが明らかである。
【0042】
<特異性、直線性、日内再現性の検討>
さらに、特異性、直線性、及び日内再現性を検討した。この結果、LC−MSを用いた本発明の生薬由来成分の高感度定量方法は、それぞれ20〜400ng/mL及び80〜1600ng/mLの範囲で良好な直線性(検量線の相関係数は0.9979)と再現性を示した。また、日内再現性の結果、表4に示すように、GLのネガティブモードの真度は−1.2〜+0.6%、精度は5.2〜6.0%であり、ポジティブモードの真度は−5.9〜+5.9%、精度は3.3〜7.7%であり、GAの真度は−7.0〜+7.2%、精度は3.0〜7.7%であった。以上の結果より、GL及びGAの定量限界濃度はそれぞれ、20ng/mL、80ng/mLであることが確認された。
【0043】
【表4】
【0044】
[実施例2]
生体試料に混和するアルカリ又はアルコールの種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、表5に記載の溶液1mLを血漿サンプルに添加してMAXにアプライする混和物を調製した以外は実施例1と同様にして、GL及びGAを定量した。
血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表5に示す。なお、表5中のアンモニア水の後ろの括弧書内の数値は、当該アンモニア水を添加して得られた混和物中のアンモニア濃度を示す。
【0045】
【表5】
【0046】
この結果、アルカリやアルコールを用いた場合は、水を用いた場合よりも回収率が良好であった。逆に、リン酸や過塩素酸等の酸性溶液を添加した場合には、GLは検出されず、GAもほとんど回収できなかった。また、アルコールの中では、エタノールよりもメタノールを用いた場合のほうが、GLとGAのどちらの回収率も良好であった。アルカリの中では、水酸化ナトリウム水溶液や炭酸水素ナトリウム溶液を用いた場合よりも、アンモニア水を用いた場合のほうが、良好であった。ただし、アンモニア水の濃度による違いはあまり観察されなかった。
【0047】
[実施例3]
固相の洗浄液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、血漿サンプルを注入後のMAXを表6に記載の溶液で洗浄した後、メタノールでさらに洗浄した以外は実施例1と同様にして、GL及びGAを定量した。
血漿サンプルを注入後のMAXの洗浄液が0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表6に示す。なお、表6中、溶液名の後ろの括弧書内の比率は、各溶液の混合比を示す。
【0048】
【表6】
【0049】
この結果、0.0025M 水酸化ナトリウム水溶液/メタノール溶液(体積比1:1の混合液)で洗浄した場合にのみ、GAの回収率がやや低かったものの、水、アンモニア水、アンモニア水とメタノールの混合溶液のいずれを洗浄液とした場合も、GL、GA、及びISの回収率は全て良好であった。
【0050】
[実施例4]
固相の洗浄液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、血漿サンプルを注入後のMAXを0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)で洗浄した後、エタノール、メタノール、50%メタノール水溶液、又はアセトニトリルでさらに洗浄した以外は実施例1と同様にして、GL及びGAを定量した。
2度目の洗浄液がメタノールであった場合を100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表7に示す。この結果、いずれの溶液で洗浄した場合であっても、GLの回収率は80%以上であり、良好であった。また、GAの回収率も、50%メタノールの場合を除き80%以上であり、良好であった。特に、メタノールが最も良好であった。
【0051】
【表7】
【0052】
[実施例5]
固相からの溶出液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXからの溶出を表8に記載の溶液を用いて行った以外は実施例1と同様にして、GL及びGAを定量した。
【0053】
【表8】
【0054】
溶出液がギ酸メタノールであった場合を100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表8に示す。この結果、ギ酸メタノールが最も回収率が高かった。また、ギ酸エタノールを用いた場合も、ギ酸メタノールには及ばないものの、GLとGAを良好に回収することができた。これに対して、溶出液がギ酸アセトニトリルやリン酸メタノールであった場合には、GLは検出できなかった。
【0055】
[比較例1]
MAXに代えて、逆相分配機能のみを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えてODSカラムであるSep−Pak(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.56%アンモニア水若しくは0.1N塩酸、固相の洗浄液として水若しくはアセトニトリル/水(体積比=1:3、1Mテトラブチルアンモニウムブロミド含有)、及び固相からの溶出液として2%ギ酸メタノール、2%ギ酸アセトニトリル/水(体積比=1:1)若しくは2%ギ酸メタノール/水(体積比=1:1)をそれぞれ用いた以外は、実施例1と同様にして行った。
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表9に示す。なお、表9中、「1−1」は0.56%アンモニア水、「1−2」は0.1N塩酸、「2−1」は水、「2−2」はアセトニトリル/水(体積比=1:3、1Mテトラブチルアンモニウムブロミド含有)、「3−1」は2%ギ酸メタノール、「3−2」は2%ギ酸アセトニトリル/水(体積比=1:1)、「3−3」は2%ギ酸メタノール/水(体積比=1:1)である。
【0056】
【表9】
【0057】
この結果、希釈液等の組み合わせの中で最も回収率が良好な場合であっても、GLの回収率はせいぜい50%程度であり、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0058】
[比較例2]
MAXに代えて、逆相分配機能と極性相互作用機能とを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えて多孔性ポリマーであるOasis HLB(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.56%アンモニア水若しくは0.1N塩酸を、固相の洗浄液として5%メタノール水溶液を、及び固相からの溶出液としてメタノールをそれぞれ用いた以外は、実施例1と同様にして行った。
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表10に示す。
【0059】
【表10】
【0060】
この結果、希釈液として0.1N塩酸を用いた場合には、GLは検出されず、GAの回収率も非常に低かった。一方、0.56%アンモニア水を用いた場合であってさえ、GLの回収率はせいぜい55%程度であり、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0061】
[比較例3]
MAXに代えて、逆相分配機能と陽イオン交換機能とを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えてOasis MCX(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.1N塩酸を、固相の洗浄液として0.1N塩酸を、及び固相からの溶出液として2%アンモニア水/メタノール溶液をそれぞれ用いた以外は、実施例1と同様にして行った。
【0062】
【表11】
【0063】
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。独立した5回の試行の算出結果及び平均値を表11に示す。この結果、GLとGAのいずれも回収率が50%に届かず、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0064】
[実施例6]
本発明の生薬由来成分の高感度定量方法を、質量分析をLC−MS/MS法によりを行った場合の、血漿中のGL及びGAの定量限界値を測定し、かつ検量線を作成した。また、同時に、当該方法の精度や信頼性を検証した。
<標準溶液の調製>
実施例1で用いた標準原液を実施例1と同様にしてメタノールにより希釈し、表12に記載の標準溶液(B)〜(G)を調製した。
【0065】
【表12】
【0066】
<血漿サンプルの調製及び抽出工程>
実施例1と同様にして、標準溶液(B)〜(G)を添加したヒト血漿サンプルとブランク血漿サンプルを調製し、これらの血漿サンプルをそれぞれMAXにアプライして、GL等を吸着させ、0.56%アンモニア水/メタノール溶液及びメタノールで洗浄した後に、2%ギ酸/メタノール溶液で溶出させ、さらに蒸発乾固した。
【0067】
<定量工程>
蒸発乾固させた抽出物を、下記移動相A液/B液(体積比1:1の混合液)250μLに溶解させた。このうち1μLを分析カラムに注入し、下記に示す条件でLC−MS/MSを行った。
LC−MS/MSシステム: API4000システム(アプライドバイオシステムズ社製)
(LC)
装置: LC−20A(島津製作所製)
分析カラム: Inertsil ODS−3(2.1mm×150mm、粒径5μm、ジーエルサイエンス社製)
流速: 0.50mL/min.
カラム温度: 40℃
オートサンプラー温度: 5℃
試料注入量: 1μL
移動相:A液 0.1%ギ酸、B液 0.1%ギ酸アセトニトリル、リニアグラジエント(表13)
【0068】
【表13】
【0069】
(MS/MS)
装置: API4000(アプライドバイオシステムズ)
イオン化法: ESI (Turbo Spray)
検出法: 正イオン検出 MRM(Multiple reaction monitoring)
イオンスプレー電圧(IonSpray Voltage): 5500V(正)
イオン源温度(Temperature): 400℃
カーテンガス (Curtain Gas): 10 (窒素)
イオンソースガス1(Ion Source Gas 1): 70 (窒素)
イオンソースガス2(Ion Source Gas 2): 50(窒素)
コリジョンガス(Collision Gas): 8(窒素)
【0070】
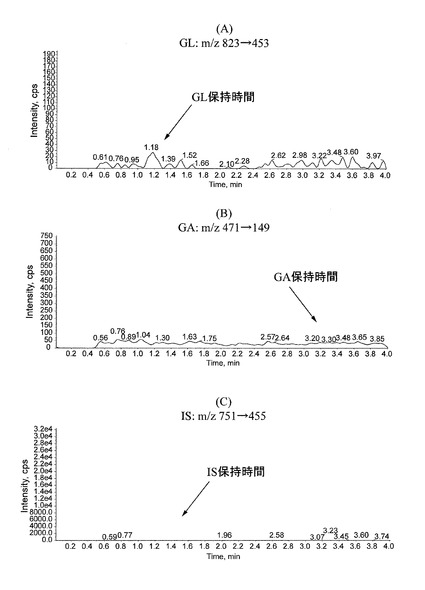
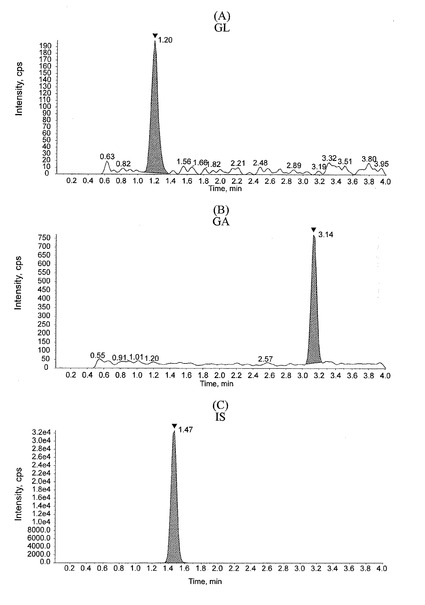
【表14】
【0071】
図1に、ブランク血漿サンプルのクロマトグラムを示す。図1(A)はGLのピークの位置(GLの保持時間;m/z 823→453)を、図1(B)はGAのピークの位置(GAの保持時間;m/z 471→149)を、図1(C)はISのピークの位置(ISの保持時間;m/z 751→455)をそれぞれ示す。また、図2に、標準溶液(G)を添加した血漿サンプル(血漿中のGL濃度:0.5ng/mL、GA濃度:2ng/mL)のクロマトグラムを示す。図2(A)はGLのピーク(図中、矢頭で示す)を、図2(B)はGAのピーク(図中、矢頭で示す)を、図2(C)はISのピーク(図中、矢頭で示す)を、それぞれ示す。GL、GA、ISの保持時間は、それぞれ約1.2分、3.1分、1.4分であり、ブランク血漿サンプルのクロマトグラム上には定量を妨害するピークは認められず、GL、GA、ISのピーク形状は良好であった。
【0072】
定量結果を表15に示す。また、実際の添加濃度と測定値(表中、定量値)との相対誤差も同じく表15に示した。この結果、GLとGAの両方とも、相対誤差が15%以内であった。すなわち、これらの結果から、血漿等の生体試料中のGLの濃度が0.5〜200ng/mL、GAの濃度が2〜800ng/mLである場合に、LC−MS/MSを用いた本発明の生薬由来成分の高感度定量方法により、生体試料中のGL等を高精度に定量可能であることが明らかである。
【0073】
【表15】
【0074】
<特異性、直線性、日内、日間再現性の検討>
さらに、米国食品医薬品局の分析法バリデーションのガイダンス(”Guidance for Industry, Bioanalytical Method Validation”, U.S. Department of Health and Human Services, Food and Drug Administration, May 2001)に従い、特異性、直線性、日内、日間再現性を検討した。また、安定性評価試験(室温下4時間及び−20℃及び−80℃下3カ月凍結保存における生体試料中保存安定性、凍結融解安定性、測定実試料中安定性、標準溶液安定性)についても実施した。検討結果を表16に示す。この結果、LC−MS/MSを用いた本発明の生薬由来成分の高感度定量方法は、それぞれ0.5〜200ng/mL及び2〜800ng/mLの範囲で良好な直線性と再現性を示した。日内再現性の結果、GLの真度は−12.8〜+4.8%、精度は4.0〜9.5%であり、GAの真度は−13.4〜+4.4%、精度は2.2〜9.0%であった。一方、日間再現性においても、GLの真度は−9.4〜+2.0%、精度は2.1〜5.5%、GAの真度は−10.9〜+8.1%、精度は1.3〜9.1%であった。再現性の結果より、GL及びGAの定量限界濃度はそれぞれ、0.5ng/mL、2ng/mLである事を確認した。また、全ての安定性評価試験(生体試料中保存安定性、凍結融解安定性、測定実試料中安定性、標準溶液安定性)において、初期値と比較し±15%以内であり安定であった。以上の結果から、米国食品医薬品局の分析法バリデーションのガイダンスに則り、本発明の生薬由来成分の高感度定量方法が信頼性の確保された定量法であることが確認された。
【0075】
【表16】
【0076】
[実施例7]
GL含有製剤を経口投与後の血漿中のGLの検出及び薬物動態を測定した。
なお、本実施例は、ヘルシンキ宣言に基づく倫理的原則、薬事法第14条第3項、第80条の2及び「医薬品の臨床試験の実施の基準(GCP)に関する省令」(平成9年厚生省令第28号)に従い実施されたものである。具体的には、GL含有製剤の経口投与及び血漿サンプルの採取は、出願人の依頼により、イーピーエス株式会社及び医療法人社団育生會山口病院によって、第三者委員会における承認の後、試験責任医師の管理の下、実施された。また、得られた血漿サンプルにおけるGL及びGAの測定は、同じく出願人の依頼により、株式会社日本医学臨床検査研究所エコテクノ事業部(現医薬香粧品分析事業部)によって行われた。
また、被験者は、試験の目的、試験内容、プライバシーの保護等について十分説明された後、書面にて試験への参加を同意した健康な日本人男性6名(年齢:20歳以上35歳以下、事前検査時BMI:18.5以上25.0以下)を対象とした。
GL含有製剤として、グリチルリチン酸一アンモニウム、グリシン、DL−メチオニンを配合した糖衣錠「グリチロン(登録商標)配合錠」を用いた。グリチロン配合錠は、十分な水分と共に臨床常用量に従い、3錠の単回経口投与とした。投与及び採血時間などの試験スケジュールを表17に示す。得られた血液は、30分以内に遠心分離処理(4℃、3000rpm、10分間)により血漿として1mL以上を回収し、濃度測定時まで−20℃以下で凍結保存した。
【0077】
【表17】
【0078】
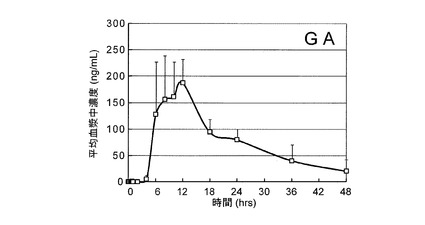
採取された血漿サンプル0.5mLに、0.56%アンモニア水1mLを添加して十分に混和した後、実施例1と同様にして、得られた混和物をMAXにアプライしてGL等を吸着させ、0.56%アンモニア水/メタノール溶液及びメタノールで洗浄した後に、2%ギ酸/メタノール溶液で溶出させ、さらに蒸発乾固した。
次いで、実施例6と同様にして、蒸発乾固させた抽出物に対してLC−MS/MSを行い、各血漿サンプル中のGL濃度及びGA濃度を測定した。図3は、GLの平均血漿中濃度推移を示した図であり、図4は、GAの平均血漿中濃度推移を示した図である。なお、図3及び図4は、6検体の平均値及び標準偏差を示している。この結果、本発明の生薬由来成分の高感度定量方法により、従来から代替的な指標とされてきた主代謝物であるGAのみならず、未変化体であるGL自体の血漿中濃度を直接測定し得ることが確認された。すなわち、本発明により、血漿中GLの微量検出に初めて成功し、長らく不明であったGLの血中動態が明らかにされた。
【0079】
<薬物動態の解析>
GL及びGAの測定データから、最高血漿中濃度(Cmax)、最高血漿中濃度到達時間(Tmax)を算出した。また、投与後48時間までの血漿中濃度-時間曲線下面積(AUC0→48h)は台形法により、消失速度定数(kel)及び消失半減期(t1/2)は消失相から最小二乗法により算出した。また、投与後無限大時間までの血漿中濃度−時間曲線下面積(AUC0―∞)、体内滞留時間(MRT)についても算出した。
この結果、本実施例では、GLのCmaxは約10〜40ng/mLの範囲にあり、平均値として24.8ng/mLであった。これは、前述の市販試薬の大量経口投与例に基づいた予測値(約23ng/mL)とほぼ同等であったことから、GLの投与量と血漿中濃度の相関性が示された。また、Tmaxは平均4.5時間であったが、それ以前(1〜2時間)あるいは以後(12時間)など複数の濃度ピークを示す被験者も観察された。後方の濃度ピークは食後に出現したことから、食事摂取がGLの吸収あるいは腸肝循環に影響を与える可能性も示唆された。
【0080】
一方、GLの主代謝物であるGAは、血漿中に確認されるまで4時間のラグが確認され、6時間後にGLの約10倍の血漿中濃度(モル換算で約20倍)が検出された。in vitroにおいては、ヒト肝ミクロソームを用いてGLを至適条件下でインキュベートした結果、速やかに中間代謝物である3−monoglucuronyl−glychrretinic acidへ変換されたが、GAの生成は僅かであった(Biochemical Pharmacology,42(6/7)1025-1029,1991。)。また、germ−freeラットにおいてGLを経口投与した結果、血漿及び糞便中にGAが確認されないことから、生体内でのGA生成は腸内細菌が支配的に担っていると考えられている(引用:J. Pharm. Pharmacol. 46, 135-137, 1994)。したがって、吸収のラグは腸内細菌叢へ接触するまでの時間と推測され、食事条件(あるいは絶食条件)が薬物の消化管内の移動、腸内細菌叢の代謝能、消化管吸収に影響し、GA曝露量を左右する要因とも考えられた。吸収後のGA動態を被験者別に観察すると、未変化体濃度が高い被験者において代謝物の出現が弱く、反対に未変化体濃度が低い被験者において代謝物の出現が強い傾向が認められた。このことから、GAと同時に未変化体であるGLの消化管吸収も腸内細菌叢の代謝能と関連し、それが個人差として発現する可能性が示唆された(データ不掲載)。
【産業上の利用可能性】
【0081】
本発明の生薬由来成分の高感度定量方法により、臨床用量のGL含有製剤や飲食品等を摂取した後の血液中のGLを定量することができるため、本発明の生薬由来成分の高感度定量方法は、主に、医薬品・漢方の適正使用、安全性評価、薬物動態解析、新規のGL製剤開発等に利用することが可能である。
【技術分野】
【0001】
本発明は、生体試料中のグリチルリチンやその代謝物等を高感度に定量するための方法に関する。
【背景技術】
【0002】
グリチルリチン(以下、GLと略記する)及びその塩は、抗アレルギー作用、抗炎症作用と共に、免疫調節作用、肝細胞障害抑制作用、肝細胞増殖促進作用、ウィルス増殖抑制・不活性化作用等の多様な生理活性を有する甘草根抽出成分である。日本では、古くから漢方薬等の臨床薬として用いられており、現在では、慢性肝疾患、湿疹・皮膚炎、小児ストロフルス、円形脱毛症、各種アレルギー、炎症等の治療に幅広く使用されている。また、医薬品以外の使用として、GLは塩なれ効果をもち、甘味効果があることから甘味料として漬物や調味料など広範囲の食品に多用されている。
【0003】
非臨床試験の結果から、GLは肝臓に集積しやすく、且つ速やかに胆汁中へ排泄される特性があり、代謝及び腸肝循環を受けながら排出されると考えられている。特に経口投与において、配糖体で水溶性極性物質であるGLは低吸収性を示すと共に、腸内細菌による代謝や初回通過効果により、末梢血における濃度が極めて低い。このため、GLの血中動態を測定するために、現在までに様々な方法が試みられているが、GLを含有する製剤、甘草を配合する漢方製剤において、服用時の血中GLは検出困難であり、その動態も長い間不明とされてきた。
【0004】
例えば、GLを経口投与された健常者から採取された血漿に対して、メタノール処理を行ってタンパク質を除去した後、当該血漿中のGLの量を、酵素免疫法(EIA)により測定する方法やオクタデシル基化学結合型(ODS)カラム等の逆相分配機能を備えるカラムを用いた高速液体クロマトグラフィー(HPLC)を用いて測定する方法(例えば、非特許文献1〜4)がある。しかしながら、これらの測定方法では、定量限界値がせいぜい0.1μg/mL程度であり、主代謝物であるグリチルレチン酸(以下、GAと略記する。)は検出することができても、GLは検出することはできなかった。このため、尿中からの検出例をもって間接的にGLの血中移行が証明された(引用文献4参照。)。そして、GL含有製剤薬効については、GLの代わりに、定量可能なGAが評価指標とされている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】イシワタ、他7名、バイオロジカル・アンド・ファーマシューティカル・ブリテン(Biological and Pharmaceutical Bulletin)、2000年、第23巻、第8号、第904〜905ページ。
【非特許文献2】デ・グルー(De Groot)、他1名、ジャーナル・オブ・クロマトグラフィー(Journal of Chromatography)、1988年、第456巻、第71〜81ページ。
【非特許文献3】ナカタ、他3名、和漢医薬学会誌、1986年、第3巻、第3号、第278〜279ページ。
【非特許文献4】ヤマムラ、他7名、ジャーナル・オブ・ファーマシューティカル・サイエンシズ(Journal of Pharmaceutical Sciences)、1992年、第81巻、第10号、第1042〜1046ページ。
【発明の概要】
【発明が解決しようとする課題】
【0006】
活性本体である未変化体GLの薬物動態は、有効性、安全性から適正使用を判断する上で有用である。このため、GAを評価対象にするのではなく、血漿等の生体試料中のGL量を定量できることが望ましい。
【0007】
本発明は、上記課題を鑑みてなされたものであって、血漿等の生体試料中のGLを検出し定量するための方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するために鋭意研究した結果、まず、逆相分配機能及び陰イオン交換機能を備える固相を用いた固相抽出法により、生体試料中からGLを含む成分を抽出した後、抽出物中のGLを質量分析法に供することにより、生体試料中のGLを検出し定量できることを見出し、本発明を完成させた。
【0009】
すなわち、本発明は、
(1) 生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、
前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程、
を有し、
前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする生薬由来成分の高感度定量方法、
(2) 前記抽出工程において、固相の洗浄を、アルカリとアルコールと水の混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことを特徴とする前記(1)に記載の生薬由来成分の高感度定量方法、
(3) 前記アルカリとアルコールと水の混合液が、0.5〜28%アンモニア水とメタノールを99:1〜1:3であることを特徴とする前記(2)に記載の生薬由来成分の高感度定量方法、
(4)前記生体試料が、血液、血漿、又は組織抽出物であることを特徴とする前記(1)〜(3)のいずれか一つに記載の生薬由来成分の高感度定量方法、
を提供するものである。
【発明の効果】
【0010】
本発明の生薬由来成分の高感度定量方法により、含有量が非常に少ない生体試料中のGLを検出し、定量することができる。このため、本発明の生薬由来成分の高感度定量方法を用いることにより、GLを含有する製剤や飲食品を摂取したヒトから採取された生体試料中のGLを定量し、薬物動態を精度よく解析することができる。
【図面の簡単な説明】
【0011】
【図1】実施例6において、ブランク血漿サンプルのクロマトグラムを示した図である。
【図2】実施例6において、標準溶液(G)を添加した血漿サンプル(血漿中のGL添加量:0.5ng/mL、GA添加量:2ng/mL)のクロマトグラムを示した図である。
【図3】実施例7において、GLの平均血漿中濃度推移を示した図である。
【図4】実施例7において、GAの平均血漿中濃度推移を示した図である。
【発明を実施するための形態】
【0012】
本発明の生薬由来成分の高感度定量方法は、生体試料中のGLを逆相分配機能及び陰イオン交換機能を備える固相を用いた固相抽出法により抽出した後、得られた抽出物中のGL含有量を質量分析法により定量することを特徴とする。従来のHPLC法等に代えて質量分析法を利用して定量することにより、GLの検出感度を高められる。また、従来のODSカラムのような逆相分配機能のみを備える固相を用いた固相抽出法ではなく、逆相分配機能及び陰イオン交換機能を備える固相を用いて調製することにより、質量分析に供される抽出物からより多くの夾雑物を除去することができ、質量分析の感度を飛躍的に向上させることができる。
【0013】
具体的には、本発明の生薬由来成分の高感度定量方法は、生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、前記抽出工程により抽出された抽出物中のGL、GA、GL及びGAの代謝物、GL及びGAの類縁物質、甘草に含有するサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する工程を有し、前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする。
【0014】
本発明の生薬由来成分の高感度定量方法により定量される生薬由来成分は、GL、GA、GL及びGAの代謝物、GL及びGAの類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群(以下、「GL等」ということがある。)より選択される少なくも1種以上である。
GL及びGAの代謝物としては、例えば、3−monoglucuronyl−glychrretinic acid(20β−Carboxy−11−oxo−30−norolean−12−en−3β−yl−β−D−glucopyranosiduronic acid)、30−monoglucuronyl−glychrretinic acid(3β−Hydroxy−11−oxoolean−12−en−30−oyl−β−D−glucopyranosiduronic acid)、3−oxo−GA(3,11−Dioxoolean−12−en−30−oic acid)、3α−GA(3α−Hydroxy−11−oxoolean−12−en−30−oic acid)、3位硫酸抱合体(3β−Hydroxysulfonyloxy−11−oxoolean−12−en−30−oic acid)、3β,22α−Dihydroxy−11−oxoolean−12−en−30−oic acid、及び3β,24−Dihydroxy−11−oxoolean−12−en−30−oic acid等が挙げられる。
【0015】
また、GL及びGAの類縁物質としては、例えば、20α−Carboxy−11−oxo−29−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid、20β−Carboxy−24−hydroxy−11−oxo−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid、18α−GL(20β−Carboxy−11−oxo−(18αH)−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid)、18α−GA(3β−Hydroxy−11−oxo−(18αH)−olean−12−en−30−oic acid)等が挙げられる。
【0016】
また、甘草に含有されているサポニン成分としては、例えば、licorice−saponin C1(20β−Carboxy−11−oxo−30−norolean−12−en−3β−yl(β−D−glucopyranosyluronic acid)−(1→2)−β−D−glucopyranosiduronic acid)等が挙げられる。
【0017】
本発明において用いられるGL等の薬学的に許容される塩は、生体内において、GL等と同様の薬理効果を有する塩であれば、特に限定されるものではない。具体的には、例えば、アンモニウム塩、ナトリウム塩、カリウム塩等が挙げられる。
【0018】
本発明においては、GL等のうち、1種類を定量してもよく、2種類以上を一の操作で定量してもよい。本発明においては、特に、GLとGAを同時に定量することが好ましい。
【0019】
本発明の生薬由来成分の高感度定量方法に供される生体試料は、生体から採取された試料であればよく、ヒトやマウス、ラット等の動物から採取されたものであることが好ましい。また、血液、血漿、血清、尿、腹水、胸水、関節液、骨髄液、胆汁等であってもよく、肝臓や膵臓、腎臓等の組織から採取された組織片等の抽出物(組織抽出物)であってもよい。組織片等の抽出物は、組織片等を常法によりホモジナイズすることにより調製することができる。本発明においては、血液、血漿、尿、又は組織抽出物であることが好ましく、血漿であることがより好ましい。
【0020】
以下、本発明の生薬由来成分の高感度定量方法を工程ごとに説明する。
抽出工程として、まず、生体試料をアルカリ又はアルコールに混和した混和物を調製する。生体試料に添加するアルカリとしては、得られる混和物のpHをアルカリ性にすることができるものであれば、特に限定されるものではなく、例えば、アンモニア、アンモニア水、水酸化ナトリウム溶液、炭酸水素ナトリウム溶液等が挙げられる。また、アルコールとしては、炭素数1〜6の低級アルコールであることが好ましく、メタノール、エタノール、イソプロパノール等が挙げられる。また、水で希釈されたアルコール溶液であってもよい。本発明においては、生体試料に添加するアルカリ又はアルコールとしては、アンモニア、アンモニア水、メタノール、又はメタノール水溶液であることが好ましく、アンモニア又はアンモニア水であることがより好ましい。
【0021】
生体試料に添加するアンモニア水の濃度は特に限定されるものではなく、生体試料の種類、その後に使用する固相の種類、得られる混和物のアンモニア濃度等を考慮して適宜調整することができる。本発明においては、生体試料に添加するアンモニア又はアンモニア水の濃度は、得られる混和物のアンモニア濃度が0.01〜30%となるような濃度であることが好ましく、0.05〜25%となるような濃度であることがより好ましく、0.05〜20%となるような濃度であることがさらに好ましい。
【0022】
次いで、得られた混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入し、当該混和物中のGL等を固相に吸着させる。逆相分配機能及び陰イオン交換機能を備える固相としては、例えば、Oasis MAX(Waters社製)等が挙げられる。
【0023】
GL等を吸着させた固相を洗浄液で1回又は2回以上洗浄し、非特異的に結合している成分を除去する。洗浄液は、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液である。洗浄液として用いられるアルカリやアルコールとしては、生体試料に添加されるアルカリやアルコールと同様のものが挙げられる。また、固相の洗浄は、同種の洗浄液で2回以上行ってもよく、異なる種類の洗浄液で順次洗浄してもよい。
【0024】
本発明においては、固相の洗浄を、アルカリとアルコールと水との混合液、アルカリと水との混合液、又は水により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことが好ましく、アルカリとアルコールと水との混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことがより好ましい。アルカリとアルコールと水の混合液としては、アンモニアとアルコールと水の混合液であることが好ましく、アンモニアとメタノールと水の混合液であることがより好ましい。
【0025】
アンモニアとメタノールと水の混合液中の各成分の組成比は、GL等を固相に吸着させた状態を保持することができるものであれば特に限定されるものではない。例えば、混合液中のメタノール濃度が1〜75%、アンモニア濃度が0.1〜21%であることが好ましく、メタノール濃度が25〜75%、アンモニア濃度が0.1〜21%であることがより好ましい。このような組成比の混合液は、例えば、0.5〜28%アンモニア水とメタノールを99:1〜1:3で混合することにより、調製することができる。
【0026】
一回目の洗浄をアルカリとアルコールと水の混合液により行った後、2回目の洗浄に用いる洗浄液としては、アルコールを用いることが好ましく、メタノール又はエタノールを用いることがより好ましく、メタノールを用いることがさらに好ましい。
【0027】
その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する。酸性化する溶媒としては、ギ酸アルコール、塩酸、トリフルオロ酢酸等が挙げられる。本発明においては、ギ酸アルコールであることが好ましい。溶出液として用いるギ酸アルコールとしては、ギ酸と炭素数1〜6の低級アルコールとのエステルであることが好ましく、ギ酸メタノール又はギ酸エタノールであることがより好ましく、ギ酸メタノールであることがさらに好ましい。また、得られた溶出物は、以降の質量分析のために、蒸発乾固させる。
【0028】
次いで、定量工程として、抽出工程により抽出された抽出物中のGL等を、質量分析法により検出し定量する。質量分析法としては、LC−MS(液体クロマトグラフィー/質量分析)法又はLC−MS/MS(液体クロマトグラフィー/タンデム質量分析)法により行うことが好ましい。具体的には、蒸発乾固させた溶出物をLCの移動相に溶解させた後、LCを行い、MSを行う。LC−MS及びLC−MS/MSは、HPLCと質量分析計を組み合わせた装置を用いることにより行うことができる。
【0029】
LCは、特許文献1〜4に記載されているように、ODSカラム等の逆相分配機能を有するカラムを用いて行うことが好ましい。また、MSは常法により行うことができる。例えば、ESI(エレクトロスプレーイオン化)法やAPCI(大気圧化学イオン化)法により試料をイオン化した後、磁場偏向型、四重極型、飛行時間型等の装置により、各イオンを分離して検出する。
【0030】
質量分析をLC−MS法により行うことにより、例えば血液、血漿、又は血清中のGL等の定量限界を20ng/mL程度にまで改善することができる。同様に、LC−MS/MS法により行うことによって、血液等中のGL等の定量限界を10ng/mL以下、例えば0.5ng/mL程度にまで向上させることができる。
【0031】
市販試薬の大量経口投与(GLとして1600mg)における血漿中GL濃度は約500ng/mL程度であった、という報告がなされている(Environmental Health Perspectives, 102(9), 65-68, 1994)。当該知見に基づいた比例計算の結果、肝疾患治療を目的とした場合、臨床常用量である投与量75mg相当の推定血中濃度は約23ng/mLと算出される。つまり、本発明の生薬由来成分の高感度定量方法により、従来法では検出不可能であった血液中のGLが定量可能となる。特に、GL製剤等を経口摂取後の血液中のGL濃度は、個体差が大きく、定量限界が不十分な測定法では、血液検体によってはGLが検出されない場合や、長時間の薬物動態を測定することができず、測定の精度や測定結果の信頼性が不十分となるが、本発明の生薬由来成分の高感度定量方法では、特に質量分析をLC−MS/MS法により行うことによって、より信頼性の高い測定結果を得ることができる。
【実施例】
【0032】
次に実施例を示して本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。
【0033】
[実施例1]
本発明の生薬由来成分の高感度定量方法を、質量分析をLC−MS法によりを行った場合の、血漿中のGL及びGAの定量限界値を測定し、かつ検量線を作成した。
<標準溶液の調製>
まず、GL(常磐植物化学研究所製)10.0mgを正確に量り、メタノールに溶解した後、正確に100mLとし、100μg/mLのGL標準原液を調製した。同様に、GA(アルプス薬品工業株式会社製)10.0mgを正確に量り、メタノールに溶解した後、正確に100mLとし、100μg/mLのGA標準原液を調製した。
次いで、GL標準原液2mL及びGA標準原液8mLを正確に分取し、メタノールで正確に50mLとして、GL濃度が4000ng/mL、GA濃度が16000ng/mLである標準溶液(A)を調製した。この標準溶液Aをメタノールで順次希釈し、表1に記載の標準溶液(B)〜(F)を調製した。標準原液及び標準溶液はそれぞれ調製後冷蔵保存(5±4℃)した。調製器具はガラス製のものを用いた。
【0034】
【表1】
【0035】
<血漿サンプルの調製>
ヒト血漿0.5mLに、標準溶液(A)50μLを添加し、十分に混和したものを血漿サンプルとした。標準溶液(B)〜(F)についても、それぞれ同様にして血漿サンプルを調製した。これらの血漿サンプルには、内部標準液(IS)50μLをさらに添加した。なお、本実施例では、内部標準物質としてα−Hederinを用いた。また、ブランク血漿サンプルとして、ヒト血漿0.5mLにメタノールを100μL添加したものを調製した。
【0036】
<抽出工程>
各血漿サンプルに、0.56%アンモニア水1mLを添加して十分に混和した後(混和物中のアンモニア濃度:0.37%)、得られた混和物を全量、Oasis MAX(Waters社製)(以下、単に「MAX」)に注入(アプライ)した。当該MAXは、血漿サンプルを注入する前に予め、2%ギ酸/メタノール溶液を注入した後、水を注入することにより、コンディショニングしておいた。
血漿サンプルを注入後、当該MAXに0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)で洗浄した後、メタノールでさらに洗浄した。
その後、2%ギ酸/メタノール溶液を注入し、当該MAXに吸着していたGL等を溶出させた。溶出液(抽出物)は、40℃加温下、窒素ガスにて蒸発乾固させた。
【0037】
<定量工程>
蒸発乾固させた抽出物を、下記移動相A液/B液(体積比1:1の混合液)250μLに溶解させた。このうち20μLを分析カラムに注入し、下記に示す条件でLCを行った。
装置: Alliance HT 2695(Waters社製)
分析カラム: CAPCELL PAK C18(UG120、5μm、1.5mm×150mm、資生堂社製)
移動相:A液 0.1%ギ酸、B液 0.1%ギ酸アセトニトリル、リニアグラジエント(表2)
流速: 0.30mL/min.
カラム温度: 40℃
試料注入量: 20μL
【0038】
【表2】
【0039】
次いで、LCにより得られたGL等を含む画分に対して、下記に示す条件でMSを行った。
装置: ZQ2000(Waters社製)
イオン化法: ESI(Turbo Spray)
検出法: GL(負、正イオン検出)、GA・IS(正イオン検出)、MRM(Multiple reaction monitoring)
Capillary電圧: 2.5kV
Extractor: 2V
RF lens: 0.2V
Source Temperature: 110℃
Desolvation Temperature: 350℃
Desolvation Gas Flow: 350L/hr
Cone Gas Flow: 50L/hr
【0040】
【表3】
【0041】
この結果、LC−MSの測定結果は血漿サンプル中のGL又はGA濃度依存的に増大しており、血漿サンプル中のGL又はGA濃度とLC−MSの測定結果により得られた検量線は直線性を示した。すなわち、これらの結果から、血漿等の生体試料中のGLの濃度が20〜400ng/mL、GAの濃度が80〜1600ng/mLであり、LC−MSを用いた本発明の生薬由来成分の高感度定量方法により、生体試料中のGL等を高精度に定量可能であることが明らかである。
【0042】
<特異性、直線性、日内再現性の検討>
さらに、特異性、直線性、及び日内再現性を検討した。この結果、LC−MSを用いた本発明の生薬由来成分の高感度定量方法は、それぞれ20〜400ng/mL及び80〜1600ng/mLの範囲で良好な直線性(検量線の相関係数は0.9979)と再現性を示した。また、日内再現性の結果、表4に示すように、GLのネガティブモードの真度は−1.2〜+0.6%、精度は5.2〜6.0%であり、ポジティブモードの真度は−5.9〜+5.9%、精度は3.3〜7.7%であり、GAの真度は−7.0〜+7.2%、精度は3.0〜7.7%であった。以上の結果より、GL及びGAの定量限界濃度はそれぞれ、20ng/mL、80ng/mLであることが確認された。
【0043】
【表4】
【0044】
[実施例2]
生体試料に混和するアルカリ又はアルコールの種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、表5に記載の溶液1mLを血漿サンプルに添加してMAXにアプライする混和物を調製した以外は実施例1と同様にして、GL及びGAを定量した。
血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表5に示す。なお、表5中のアンモニア水の後ろの括弧書内の数値は、当該アンモニア水を添加して得られた混和物中のアンモニア濃度を示す。
【0045】
【表5】
【0046】
この結果、アルカリやアルコールを用いた場合は、水を用いた場合よりも回収率が良好であった。逆に、リン酸や過塩素酸等の酸性溶液を添加した場合には、GLは検出されず、GAもほとんど回収できなかった。また、アルコールの中では、エタノールよりもメタノールを用いた場合のほうが、GLとGAのどちらの回収率も良好であった。アルカリの中では、水酸化ナトリウム水溶液や炭酸水素ナトリウム溶液を用いた場合よりも、アンモニア水を用いた場合のほうが、良好であった。ただし、アンモニア水の濃度による違いはあまり観察されなかった。
【0047】
[実施例3]
固相の洗浄液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、血漿サンプルを注入後のMAXを表6に記載の溶液で洗浄した後、メタノールでさらに洗浄した以外は実施例1と同様にして、GL及びGAを定量した。
血漿サンプルを注入後のMAXの洗浄液が0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表6に示す。なお、表6中、溶液名の後ろの括弧書内の比率は、各溶液の混合比を示す。
【0048】
【表6】
【0049】
この結果、0.0025M 水酸化ナトリウム水溶液/メタノール溶液(体積比1:1の混合液)で洗浄した場合にのみ、GAの回収率がやや低かったものの、水、アンモニア水、アンモニア水とメタノールの混合溶液のいずれを洗浄液とした場合も、GL、GA、及びISの回収率は全て良好であった。
【0050】
[実施例4]
固相の洗浄液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、血漿サンプルを注入後のMAXを0.56%アンモニア水/メタノール溶液(体積比1:1の混合液)で洗浄した後、エタノール、メタノール、50%メタノール水溶液、又はアセトニトリルでさらに洗浄した以外は実施例1と同様にして、GL及びGAを定量した。
2度目の洗浄液がメタノールであった場合を100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表7に示す。この結果、いずれの溶液で洗浄した場合であっても、GLの回収率は80%以上であり、良好であった。また、GAの回収率も、50%メタノールの場合を除き80%以上であり、良好であった。特に、メタノールが最も良好であった。
【0051】
【表7】
【0052】
[実施例5]
固相からの溶出液の種類が、本発明の生薬由来成分の高感度定量方法の定量感度に及ぼす影響を調べた。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXからの溶出を表8に記載の溶液を用いて行った以外は実施例1と同様にして、GL及びGAを定量した。
【0053】
【表8】
【0054】
溶出液がギ酸メタノールであった場合を100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表8に示す。この結果、ギ酸メタノールが最も回収率が高かった。また、ギ酸エタノールを用いた場合も、ギ酸メタノールには及ばないものの、GLとGAを良好に回収することができた。これに対して、溶出液がギ酸アセトニトリルやリン酸メタノールであった場合には、GLは検出できなかった。
【0055】
[比較例1]
MAXに代えて、逆相分配機能のみを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えてODSカラムであるSep−Pak(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.56%アンモニア水若しくは0.1N塩酸、固相の洗浄液として水若しくはアセトニトリル/水(体積比=1:3、1Mテトラブチルアンモニウムブロミド含有)、及び固相からの溶出液として2%ギ酸メタノール、2%ギ酸アセトニトリル/水(体積比=1:1)若しくは2%ギ酸メタノール/水(体積比=1:1)をそれぞれ用いた以外は、実施例1と同様にして行った。
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表9に示す。なお、表9中、「1−1」は0.56%アンモニア水、「1−2」は0.1N塩酸、「2−1」は水、「2−2」はアセトニトリル/水(体積比=1:3、1Mテトラブチルアンモニウムブロミド含有)、「3−1」は2%ギ酸メタノール、「3−2」は2%ギ酸アセトニトリル/水(体積比=1:1)、「3−3」は2%ギ酸メタノール/水(体積比=1:1)である。
【0056】
【表9】
【0057】
この結果、希釈液等の組み合わせの中で最も回収率が良好な場合であっても、GLの回収率はせいぜい50%程度であり、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0058】
[比較例2]
MAXに代えて、逆相分配機能と極性相互作用機能とを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えて多孔性ポリマーであるOasis HLB(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.56%アンモニア水若しくは0.1N塩酸を、固相の洗浄液として5%メタノール水溶液を、及び固相からの溶出液としてメタノールをそれぞれ用いた以外は、実施例1と同様にして行った。
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。算出結果を表10に示す。
【0059】
【表10】
【0060】
この結果、希釈液として0.1N塩酸を用いた場合には、GLは検出されず、GAの回収率も非常に低かった。一方、0.56%アンモニア水を用いた場合であってさえ、GLの回収率はせいぜい55%程度であり、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0061】
[比較例3]
MAXに代えて、逆相分配機能と陽イオン交換機能とを備える固相を用いて、血漿中のGL及びGAの定量を行った。
ヒト血漿0.5mLに、実施例1の標準溶液(E)50μL及びIS50μLを添加し、十分に混和したものを血漿サンプルとした。
具体的には、MAXに代えてOasis MCX(3cc、60mg、30μm、Waters社製)を用いたこと、当該カラムのコンディショニングをメタノール、次いで水で行ったこと、及び、カラムにアプライする混和物を調製するための溶液(希釈液)として0.1N塩酸を、固相の洗浄液として0.1N塩酸を、及び固相からの溶出液として2%アンモニア水/メタノール溶液をそれぞれ用いた以外は、実施例1と同様にして行った。
【0062】
【表11】
【0063】
実施例2において血漿サンプルに混和した溶液が0.56%アンモニア水であった場合の定量結果をそれぞれ回収率100%とし、各血漿サンプルのGL、GA、及びISの回収率(相対値)を算出した。独立した5回の試行の算出結果及び平均値を表11に示す。この結果、GLとGAのいずれも回収率が50%に届かず、MAXを用いた場合よりも遥かにGL等の検出感度が劣ることが分かった。
【0064】
[実施例6]
本発明の生薬由来成分の高感度定量方法を、質量分析をLC−MS/MS法によりを行った場合の、血漿中のGL及びGAの定量限界値を測定し、かつ検量線を作成した。また、同時に、当該方法の精度や信頼性を検証した。
<標準溶液の調製>
実施例1で用いた標準原液を実施例1と同様にしてメタノールにより希釈し、表12に記載の標準溶液(B)〜(G)を調製した。
【0065】
【表12】
【0066】
<血漿サンプルの調製及び抽出工程>
実施例1と同様にして、標準溶液(B)〜(G)を添加したヒト血漿サンプルとブランク血漿サンプルを調製し、これらの血漿サンプルをそれぞれMAXにアプライして、GL等を吸着させ、0.56%アンモニア水/メタノール溶液及びメタノールで洗浄した後に、2%ギ酸/メタノール溶液で溶出させ、さらに蒸発乾固した。
【0067】
<定量工程>
蒸発乾固させた抽出物を、下記移動相A液/B液(体積比1:1の混合液)250μLに溶解させた。このうち1μLを分析カラムに注入し、下記に示す条件でLC−MS/MSを行った。
LC−MS/MSシステム: API4000システム(アプライドバイオシステムズ社製)
(LC)
装置: LC−20A(島津製作所製)
分析カラム: Inertsil ODS−3(2.1mm×150mm、粒径5μm、ジーエルサイエンス社製)
流速: 0.50mL/min.
カラム温度: 40℃
オートサンプラー温度: 5℃
試料注入量: 1μL
移動相:A液 0.1%ギ酸、B液 0.1%ギ酸アセトニトリル、リニアグラジエント(表13)
【0068】
【表13】
【0069】
(MS/MS)
装置: API4000(アプライドバイオシステムズ)
イオン化法: ESI (Turbo Spray)
検出法: 正イオン検出 MRM(Multiple reaction monitoring)
イオンスプレー電圧(IonSpray Voltage): 5500V(正)
イオン源温度(Temperature): 400℃
カーテンガス (Curtain Gas): 10 (窒素)
イオンソースガス1(Ion Source Gas 1): 70 (窒素)
イオンソースガス2(Ion Source Gas 2): 50(窒素)
コリジョンガス(Collision Gas): 8(窒素)
【0070】
【表14】
【0071】
図1に、ブランク血漿サンプルのクロマトグラムを示す。図1(A)はGLのピークの位置(GLの保持時間;m/z 823→453)を、図1(B)はGAのピークの位置(GAの保持時間;m/z 471→149)を、図1(C)はISのピークの位置(ISの保持時間;m/z 751→455)をそれぞれ示す。また、図2に、標準溶液(G)を添加した血漿サンプル(血漿中のGL濃度:0.5ng/mL、GA濃度:2ng/mL)のクロマトグラムを示す。図2(A)はGLのピーク(図中、矢頭で示す)を、図2(B)はGAのピーク(図中、矢頭で示す)を、図2(C)はISのピーク(図中、矢頭で示す)を、それぞれ示す。GL、GA、ISの保持時間は、それぞれ約1.2分、3.1分、1.4分であり、ブランク血漿サンプルのクロマトグラム上には定量を妨害するピークは認められず、GL、GA、ISのピーク形状は良好であった。
【0072】
定量結果を表15に示す。また、実際の添加濃度と測定値(表中、定量値)との相対誤差も同じく表15に示した。この結果、GLとGAの両方とも、相対誤差が15%以内であった。すなわち、これらの結果から、血漿等の生体試料中のGLの濃度が0.5〜200ng/mL、GAの濃度が2〜800ng/mLである場合に、LC−MS/MSを用いた本発明の生薬由来成分の高感度定量方法により、生体試料中のGL等を高精度に定量可能であることが明らかである。
【0073】
【表15】
【0074】
<特異性、直線性、日内、日間再現性の検討>
さらに、米国食品医薬品局の分析法バリデーションのガイダンス(”Guidance for Industry, Bioanalytical Method Validation”, U.S. Department of Health and Human Services, Food and Drug Administration, May 2001)に従い、特異性、直線性、日内、日間再現性を検討した。また、安定性評価試験(室温下4時間及び−20℃及び−80℃下3カ月凍結保存における生体試料中保存安定性、凍結融解安定性、測定実試料中安定性、標準溶液安定性)についても実施した。検討結果を表16に示す。この結果、LC−MS/MSを用いた本発明の生薬由来成分の高感度定量方法は、それぞれ0.5〜200ng/mL及び2〜800ng/mLの範囲で良好な直線性と再現性を示した。日内再現性の結果、GLの真度は−12.8〜+4.8%、精度は4.0〜9.5%であり、GAの真度は−13.4〜+4.4%、精度は2.2〜9.0%であった。一方、日間再現性においても、GLの真度は−9.4〜+2.0%、精度は2.1〜5.5%、GAの真度は−10.9〜+8.1%、精度は1.3〜9.1%であった。再現性の結果より、GL及びGAの定量限界濃度はそれぞれ、0.5ng/mL、2ng/mLである事を確認した。また、全ての安定性評価試験(生体試料中保存安定性、凍結融解安定性、測定実試料中安定性、標準溶液安定性)において、初期値と比較し±15%以内であり安定であった。以上の結果から、米国食品医薬品局の分析法バリデーションのガイダンスに則り、本発明の生薬由来成分の高感度定量方法が信頼性の確保された定量法であることが確認された。
【0075】
【表16】
【0076】
[実施例7]
GL含有製剤を経口投与後の血漿中のGLの検出及び薬物動態を測定した。
なお、本実施例は、ヘルシンキ宣言に基づく倫理的原則、薬事法第14条第3項、第80条の2及び「医薬品の臨床試験の実施の基準(GCP)に関する省令」(平成9年厚生省令第28号)に従い実施されたものである。具体的には、GL含有製剤の経口投与及び血漿サンプルの採取は、出願人の依頼により、イーピーエス株式会社及び医療法人社団育生會山口病院によって、第三者委員会における承認の後、試験責任医師の管理の下、実施された。また、得られた血漿サンプルにおけるGL及びGAの測定は、同じく出願人の依頼により、株式会社日本医学臨床検査研究所エコテクノ事業部(現医薬香粧品分析事業部)によって行われた。
また、被験者は、試験の目的、試験内容、プライバシーの保護等について十分説明された後、書面にて試験への参加を同意した健康な日本人男性6名(年齢:20歳以上35歳以下、事前検査時BMI:18.5以上25.0以下)を対象とした。
GL含有製剤として、グリチルリチン酸一アンモニウム、グリシン、DL−メチオニンを配合した糖衣錠「グリチロン(登録商標)配合錠」を用いた。グリチロン配合錠は、十分な水分と共に臨床常用量に従い、3錠の単回経口投与とした。投与及び採血時間などの試験スケジュールを表17に示す。得られた血液は、30分以内に遠心分離処理(4℃、3000rpm、10分間)により血漿として1mL以上を回収し、濃度測定時まで−20℃以下で凍結保存した。
【0077】
【表17】
【0078】
採取された血漿サンプル0.5mLに、0.56%アンモニア水1mLを添加して十分に混和した後、実施例1と同様にして、得られた混和物をMAXにアプライしてGL等を吸着させ、0.56%アンモニア水/メタノール溶液及びメタノールで洗浄した後に、2%ギ酸/メタノール溶液で溶出させ、さらに蒸発乾固した。
次いで、実施例6と同様にして、蒸発乾固させた抽出物に対してLC−MS/MSを行い、各血漿サンプル中のGL濃度及びGA濃度を測定した。図3は、GLの平均血漿中濃度推移を示した図であり、図4は、GAの平均血漿中濃度推移を示した図である。なお、図3及び図4は、6検体の平均値及び標準偏差を示している。この結果、本発明の生薬由来成分の高感度定量方法により、従来から代替的な指標とされてきた主代謝物であるGAのみならず、未変化体であるGL自体の血漿中濃度を直接測定し得ることが確認された。すなわち、本発明により、血漿中GLの微量検出に初めて成功し、長らく不明であったGLの血中動態が明らかにされた。
【0079】
<薬物動態の解析>
GL及びGAの測定データから、最高血漿中濃度(Cmax)、最高血漿中濃度到達時間(Tmax)を算出した。また、投与後48時間までの血漿中濃度-時間曲線下面積(AUC0→48h)は台形法により、消失速度定数(kel)及び消失半減期(t1/2)は消失相から最小二乗法により算出した。また、投与後無限大時間までの血漿中濃度−時間曲線下面積(AUC0―∞)、体内滞留時間(MRT)についても算出した。
この結果、本実施例では、GLのCmaxは約10〜40ng/mLの範囲にあり、平均値として24.8ng/mLであった。これは、前述の市販試薬の大量経口投与例に基づいた予測値(約23ng/mL)とほぼ同等であったことから、GLの投与量と血漿中濃度の相関性が示された。また、Tmaxは平均4.5時間であったが、それ以前(1〜2時間)あるいは以後(12時間)など複数の濃度ピークを示す被験者も観察された。後方の濃度ピークは食後に出現したことから、食事摂取がGLの吸収あるいは腸肝循環に影響を与える可能性も示唆された。
【0080】
一方、GLの主代謝物であるGAは、血漿中に確認されるまで4時間のラグが確認され、6時間後にGLの約10倍の血漿中濃度(モル換算で約20倍)が検出された。in vitroにおいては、ヒト肝ミクロソームを用いてGLを至適条件下でインキュベートした結果、速やかに中間代謝物である3−monoglucuronyl−glychrretinic acidへ変換されたが、GAの生成は僅かであった(Biochemical Pharmacology,42(6/7)1025-1029,1991。)。また、germ−freeラットにおいてGLを経口投与した結果、血漿及び糞便中にGAが確認されないことから、生体内でのGA生成は腸内細菌が支配的に担っていると考えられている(引用:J. Pharm. Pharmacol. 46, 135-137, 1994)。したがって、吸収のラグは腸内細菌叢へ接触するまでの時間と推測され、食事条件(あるいは絶食条件)が薬物の消化管内の移動、腸内細菌叢の代謝能、消化管吸収に影響し、GA曝露量を左右する要因とも考えられた。吸収後のGA動態を被験者別に観察すると、未変化体濃度が高い被験者において代謝物の出現が弱く、反対に未変化体濃度が低い被験者において代謝物の出現が強い傾向が認められた。このことから、GAと同時に未変化体であるGLの消化管吸収も腸内細菌叢の代謝能と関連し、それが個人差として発現する可能性が示唆された(データ不掲載)。
【産業上の利用可能性】
【0081】
本発明の生薬由来成分の高感度定量方法により、臨床用量のGL含有製剤や飲食品等を摂取した後の血液中のGLを定量することができるため、本発明の生薬由来成分の高感度定量方法は、主に、医薬品・漢方の適正使用、安全性評価、薬物動態解析、新規のGL製剤開発等に利用することが可能である。
【特許請求の範囲】
【請求項1】
生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、
前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する定量工程と、
を有し、
前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする生薬由来成分の高感度定量方法。
【請求項2】
前記抽出工程において、固相の洗浄を、アルカリとアルコールと水の混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことを特徴とする請求項1に記載の生薬由来成分の高感度定量方法。
【請求項3】
前記アルカリとアルコールと水の混合液が、0.5〜28%アンモニア水とメタノールを99:1〜1:3であることを特徴とする請求項2に記載の生薬由来成分の高感度定量方法。
【請求項4】
前記生体試料が、血液、血漿、又は組織抽出物であることを特徴とする請求項1〜3のいずれか一項に記載の生薬由来成分の高感度定量方法。
【請求項1】
生体試料をアルカリ又はアルコールに混和した混和物を、逆相分配機能及び陰イオン交換機能を備える固相に注入した後、前記固相を洗浄液で1回又は2回以上洗浄し、その後、酸性アルコールにより前記固相から溶出することにより、生薬由来成分を含む抽出物を調製する抽出工程と、
前記抽出工程により抽出された抽出物中のグリチルリチン、グリチルレチン酸、グリチルリチン及びグリチルレチン酸の代謝物、グリチルリチン及びグリチルレチン酸の類縁物質、甘草に含有されているサポニン成分、並びにこれらの薬学的に許容される塩からなる群より選択される少なくも1種以上を、質量分析法により検出し定量する定量工程と、
を有し、
前記洗浄液が、水、アルカリ、アルコール、及びアセトニトリルからなる群より選択される1種又は2種以上の混合液であることを特徴とする生薬由来成分の高感度定量方法。
【請求項2】
前記抽出工程において、固相の洗浄を、アルカリとアルコールと水の混合液により洗浄した後、アルコール、アルコールと水の混合液、又はアセトニトリルにより洗浄することによって行うことを特徴とする請求項1に記載の生薬由来成分の高感度定量方法。
【請求項3】
前記アルカリとアルコールと水の混合液が、0.5〜28%アンモニア水とメタノールを99:1〜1:3であることを特徴とする請求項2に記載の生薬由来成分の高感度定量方法。
【請求項4】
前記生体試料が、血液、血漿、又は組織抽出物であることを特徴とする請求項1〜3のいずれか一項に記載の生薬由来成分の高感度定量方法。
【図1】

【図2】
【図3】

【図4】
【図2】
【図3】
【図4】
【公開番号】特開2012−107954(P2012−107954A)
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願番号】特願2010−256187(P2010−256187)
【出願日】平成22年11月16日(2010.11.16)
【出願人】(000170358)株式会社ミノファーゲン製薬 (16)
【Fターム(参考)】
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願日】平成22年11月16日(2010.11.16)
【出願人】(000170358)株式会社ミノファーゲン製薬 (16)
【Fターム(参考)】
[ Back to top ]