
疼痛およびα2アドレナリン受容体が関与する他の状態の処置のための方法および組成物
【課題】哺乳動物における眼症状、ストレス関連状態、および神経変性状態を含む状態の処置のための方法を提供する。
【解決手段】α2アドレナリン受容体の直接的または間接的な活性化をもたらす化合物を含有する第一成分、および、α1アドレナリン受容体アンタゴニストを含有する第二成分を同時投与する。
【解決手段】α2アドレナリン受容体の直接的または間接的な活性化をもたらす化合物を含有する第一成分、および、α1アドレナリン受容体アンタゴニストを含有する第二成分を同時投与する。
【発明の詳細な説明】
【技術分野】
【0001】
(原文に記載なし。)
【背景技術】
【0002】
ヒトアドレナリン作動性受容体は内在性膜タンパク質であり、大きく分けて2種類、すなわちαアドレナリン作動性受容体とβアドレナリン作動性受容体とに分類されている。どちらのタイプも、カテコールアミン類であるノルエピネフリンおよびエピネフリンの結合により、末梢交感神経系の作用を媒介する。
【0003】
ノルエピネフリンはアドレナリン作動性神経終末によって産生され、一方、エピネフリンは副腎髄質によって産生される。これらの化合物に対するアドレナリン作動性受容体の結合親和性は、分類の一つの根拠になっている。すなわち、α受容体は、ノルエピネフリンをエピネフリンよりも強く結合し、また、ノルエピネフリンを合成化合物イソプロテレノールよりもはるかに強く結合する傾向を持っている。これらのホルモンの選択的結合親和性はβ受容体では逆転する。多くの組織では、α受容体の活性化によって誘発される平滑筋収縮などの機能的応答は、β受容体結合によって誘発される応答と対立する。
【0004】
その後に、α受容体とβ受容体との機能的差異は、さまざまな動物源および組織源に由来するこれら受容体の薬理学的特徴づけによって、さらに強調され、精密化された。その結果、αアドレナリン作動性受容体とβアドレナリン作動性受容体は、3種類のα1、3種類のα2および3種類のβサブタイプに、さらに細分された。
【0005】
α1受容体とα2受容体との機能的相違が認識されており、これら2つのサブタイプ間で選択的結合を示す化合物が開発されている。即ち、WO92/00073では、α1サブタイプのアドレナリン作動性受容体に選択的に結合するという、テラゾシンのR(+)エナンチオマーの能力が報告された。この化合物のα1/α2選択性は重要であると開示された。その理由として、α2受容体のアゴニスト刺激はエピネフリンおよびノルエピネフリンの分泌を阻害するとされ、一方、α2受容体のアンタゴニスト作用はこれらのホルモンの分泌を増加させるとされた。したがって、フェノキシベンザミンやフェントラミンなどの非選択的αアドレナリン受容体遮断剤の使用は、α2アドレナリン作動性受容体が媒介する血漿カテコールアミン濃度の増加の誘導およびそれに伴う生理学的結果(心拍数の増加および平滑筋収縮)による制約を受けるとされた。
【0006】
さらに、α受容体アゴニストの選択性を測定するための一方法は、Messier ら「High Throughput Assays Of Cloned Adrenergic, Muscarinic, Neurokinin And Neurotrophin Receptors In Living Mammalian Cells(生きた哺乳類細胞におけるクローン化アドレナリン作動性、ムスカリン性、ニューロキニンおよびニューロトロフィン受容体の高スループットアッセイ)」Pharmacol. Toxicol. 76:308-11 (1995) に記載のRSAT(Receptor Selction and Amplification Technology)アッセイを含み、α2受容体用に改造されている。このアッセイでは、コンフルエント細胞の混合集団における受容体含有細胞の選択的増殖をもたらす、接触阻止の受容体媒介性喪失を測定する。細胞数の増加は、96穴形式で容易に測定することができる活性を持つ適当な導入マーカー遺伝子、例えばb−ガラクトシダーゼを使って評価される。Gタンパク質Gqを活性化する受容体はこの応答を引き出す。通常、Giと共役しているα2受容体は、Gq/i52と呼ばれるGi受容体認識ドメインを持つハイブリッドGqタンパク質と同時発現させると、RSAT応答を活性化する。Conklin ら「Substitution Of Three Amino Acids Switches Receptor Specificity Of Gqa To That Of Gia(3アミノ酸の置換によりGqaの受容体特異性がGiaの受容体特異性に転換される)」Nature 363:274-6 (1993) を参照されたい。
【0007】
α−アドレナリン受容体の一般的な背景については、α1/α2のサブクラス分類の基礎、分子生物学、シグナル伝達、アゴニストの構造−活性相関、受容体の機能、およびα−アドレナリン受容体親和性を示す化合物に関する治療的適用が調べられた、Robert R. Ruffolo, Jr., α-Adrenoreceptors:Molecular Biology, Biochemistry and Pharmacology (Progress in Basic and Clinical Pharmacology series、Karger, 1991)が注目される。
【0008】
ヒトの組織に由来するα受容体サブタイプのクローニング、配列決定および発現により、α1アドレナリン受容体は、α1A、α1Bおよびα1Dのサブクラスに分類された。同様に、α2アドレナリン受容体もまた、α2A、α2Bおよびα2Cの受容体に分類されている。それぞれのα2受容体サブタイプはそれぞれ独自の薬理学的特異性および組織特異性を示すようである。
【0009】
クロジニンやデクサメデトミジンのようなα2受容体パンアゴニストは、効果的な鎮痛活性を有し、現在一般的に、中枢神経に直接投与されている(例えばこの目的で鞘内投与または硬膜外投与)。このようなα2受容体パンアゴニストは時として、癌疼痛, 術後疼痛, 神経障害性疼痛, 異痛,ヘルペス後神経痛, 過敏性腸症候群, およびその他の内臓疼痛等の慢性疼痛の処置に用いられてきた。このようなα2 パンアゴニストがある程度の治療活性を有すると示されているその他の状態には、依存症処置 (例えば、麻薬または喫煙の無毒化), 注意欠陥多動性障害(ADHD), トゥレット・シンドローム, 鬱およびその他の 精神障害、高血圧、高眼圧症 (緑内障に関連するような), および痙縮が挙げられる。しかし、これらの薬剤は、鎮痛薬としてまたはこのような他の適応症の処置においては、治療効果と相当のそして時として圧倒的な心臓血管作用および鎮静作用との間の非常に狭い治療濃度域、並びに鎮静効果に関連する他の投薬との有意の相互作用のために、一般的且つ効果的には用いられていない。後者の例としては、例えば, Higuchi H. ら、The Interaction Between Propofol And Clonidine For Loss Of Consciousness, Anesth. Analg. 94 (4):886-91, (2002年4月); Jaffe, R. ら、Adverse interaction between clonidine and verapamil, AnnalsPharmacother. 28 (7- 8): 881-3 (1994年7−8月) (クロニジンとベラパミルの鎮静効果の間の死に至る可能性のある相乗効果を報告)を参照。
【発明の概要】
【発明が解決しようとする課題】
【0010】
治療用量での高い鎮静作用および心臓血管抑制活性のために、FDAが承認したα2受容体アゴニスト(現時点ではα2受容体パンアゴニストのみである)は、全身薬としては一般的に局所適用薬と比べ有用性が低い。例えば、α2 パンアゴニストクロニジンは、例えば、緑内障に起因する高い眼圧(IOP)の眼科処置に用いられてきた。この薬物は液摘の状態で眼に直接投与されるため、一般的な全身作用の多くを最小限とし得る。ところが、そのような薬物の眼への局所適用(眼の血管内への浸透および鼻涙管から鼻への全身作用を許す)であっても、これらの副作用は取り除かれず、治療効果は依然としてそのような効果に限定される。
【課題を解決するための手段】
【0011】
本発明の第1の態様は、哺乳動物(特にヒト)に対する、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与が、その鎮静作用の有意の増加なしに第一成分の治療活性の効力の増大をもたらすという驚くべき発見に関する。即ち、この2成分の同時投与によって、第一成分の治療活性と鎮静作用との間の治療濃度域が広がる。
【0012】
現時点で好ましい態様では、本発明は、疼痛、特に慢性疼痛、神経変性障害;交感神経により増大したストレスに関連する状態および緑内障および高眼圧症を含む眼の状態からなる群から選択される状態の処置のための方法に関する。
【0013】
好ましくは、第一成分に含まれる化合物は、例えば、α2受容体アゴニストおよびノルエピネフリン輸送物質阻害剤(三環式抗鬱薬またはTCA等)からなる群から選択される。α2パンアゴニストおよびTCAの例は、長年、当分野において周知であるが、本明細書に開示する方法は、本願において初めて発表されるものであると本願発明者は信じる。
【0014】
TCA(アミトリプチリン(アミトリル、エラビル)およびノルトリプチリン(アベンチル、パメロール);デシプラミン(ペルトフラン、ノルプラミン);ドキセピン(サイネクアン、アダピン);イミプラミン(ジャナミン、トフラニル);プロトリプチリン(ビバクチル);トリミプラミン(スルモンチール);およびクロモプラミン(アナフラニル)が含まれる)は、最初にそして第一に鬱状態の処置に用いられてきた。しかしながら、これらは、大うつ病エピソード、いわゆる非定型うつ病、パニック障害、対人恐怖、過食症、ナルコレプシー、活動過剰を伴うまたは伴わない注意力欠如障害(ADD)、片頭痛、頭痛および他の様々な慢性疼痛症候群(神経障害性疼痛を含む)、子供の夜尿症および強迫神経症等の、広い範囲の障害において有用であると報告されている。躁うつ病、統合失調症および統合失調性感情障害等の他の主要な精神障害に関して発生する鬱の症状もまた、以下に記載するある特定の警告を伴ってTCAによって処置される。
【0015】
α2受容体パンアゴニストと同様、TCAについての危険な副作用は鎮静作用である。このため、これらは一般に、夜、就寝前に服用するよう処方されている。心臓血管副作用は、これらの服用にも関連している。起立性低血圧、即ち、起立時あるいは姿勢の急激に変えたときのめまいは、一般的である。時として動悸を伴う急速な心拍がしばしば報告されている。この服用は、不健康な心臓に対して有害な影響を及ぼし得る、例えばEKG(心電図)の変化または不整脈(心調律または伝導の乱れ)を生じ、狭心症または心不全または心筋梗塞(心臓発作)を悪化または誘発する。これらの心臓血管心臓血管副作用によって、患者のためにTCAが検討対象から除外されることもある。過剰摂取した場合に三環系抗鬱薬を極めて危険にしているのはこの心臓血管副作用である。過剰摂取は、深刻で、死に至ることもある心臓合併症を引き起こしうる。三環式抗鬱薬は、現在、米国における薬物過剰摂取による死因のトップである。
【0016】
α2パンアゴニストであるクロニジンの硬膜外投与は、局所麻酔薬ブピバカイン−フェンタニルによってもたらされるものと同様に、陣痛時の疼痛除去および運動ブロック(motor block)を効果的にもたらすことが報告されている。Angelo, Reg. Anaesth. & Pain Med. 25: 3(Jan.-Feb. 2000)。しかしながら、これらの効果は、局所麻酔を用いてみられるのと比較して、低血圧等の鎮静および心臓血管副作用をより有意に伴う。デクサメデトミジン等の他のα2パンアゴニストについても同様の結果がみられる。α2アゴニスト(例えば、α2レセプターパンアゴニスト)は、癌疼痛、術後疼痛、ヘルペス後神経痛、過敏性腸症候群およびその他の内臓疼痛、糖尿病性ニューロパシー、筋痙縮にともなう疼痛、複合性局所疼痛症候群(CRPS)、交感神経依存性疼痛、頭痛、抗アロディニア疼痛、炎症性疼痛(例えば、関節炎、に関する)、胃腸の疼痛(例えば過敏性腸症候群(IBS)およびクローン病)、および神経障害性疼痛等の慢性疼痛の処置のために末梢または非末梢的に用いられている。しかしながら、各症例において、そのような化合物による処置は、一方は鎮痛、他方は鎮静の間で狭い治療濃度域によって限定される。
【0017】
同様に、アプラクロニジン(パンアゴニスト)やブリモニジン等のα2アドレナリンアゴニストは、緑内障および高いIOPまたはブドウ膜強膜炎による房水流出の減少を伴うその他の眼症状の処置のための眼科用製剤に用いられている。点眼による薬物の眼への直接投与により薬物の全身濃度が減少し、望ましくない副作用が減るものの、点眼を経由する薬物の吸収または摂取は起こる。したがって、高い眼内圧を最も有効に処置するのに必要な用量は、有害な副作用がそのような濃度でもみられるという事実によってしばしば制限される。
【0018】
対照的に、本発明は、α2活性化剤による処置に応答する状態を有するヒトを含む哺乳動物を処置するための方法であって、その哺乳動物へのα1アドレナリン受容体アンタゴニストおよびα2活性化剤の投与を含み、鎮静または心臓血管抑制の程度が、A2AA単独で有効量を同様に投与した後に存在するよりも少ない、方法を包含するものである。
【0019】
「α2活性化剤」または「A2AA」は、α2アゴニスト、TCA、またはα2アドレナリン受容体の活性化を直接的または間接的にもたらす活性を有するその他の化合物を意味する。とりわけ、但しこれに限定されないが、疼痛(特に慢性疼痛)の処置に用いる場合、本方法は、A2AAを単独の鎮痛剤として含有する組成物と比較してより良好な鎮痛活性(即ち、低いEC50)をもたらす。さらに、そのような同時投与は、A2AAを単独の鎮痛剤として含有する組成物に一般にみられる、薬物濃度と鎮静および血圧低下活性との間の用量−応答の関係に実質的に影響を及ぼさないと考えられる。「EC50」は、測定された最大活性の半分が観察される、与えられた試薬の濃度を意味する。
【0020】
鎮痛薬としてのおよびその他の適用において、A2AAの好ましい投与経路は、末梢または非末梢であり、経口、静脈内、鞘内および硬膜外投与が含まれる。いずれか一成分(または両成分)のその他の可能な投与手段には、鞘内ポンプ、皮下ポンプ、経皮パッチ、静脈内注射、皮下注射、筋肉内注射、局所的クリームまたはジェル、または経口丸薬、またはこのような方法の組み合わせが含まれるがこれに限定されない。A2AAの末梢投与手段は、現在、ある特定の適用においては好ましいものではないが、本願請求項に記載した方法の有用性はそのような場合においても、少なくともその試薬の性質の一部およびそれが投与されるところの適応症に依存して認められる。
【0021】
活性成分に加え、第一および第二成分は、好ましくは、選択した投与様式に適した1以上の製薬的に許容し得る担体を含有する。「製薬的に許容し得る担体」なる語は、液体または固体の増量剤、希釈剤、賦形剤、溶媒または封入材料としての、製薬的に許容し得る材料、組成物またはビークルを意味する。各担体は、その製剤の他の成分と投与様式に適合し、患者にとって有害でないという意味において「許容し得る」ものでなければならない。製薬的に許容し得る担体として供する材料の例としては、糖(ラクトース、グルコースおよびスクロース);(b)デンプン(トウモロコシデンプンおよびジャガイモデンプン);(C)セルロースおよびその誘導体(カルボキシルメチル・セルロース・ナトリウム、エチルセルロースおよびセルロースアセテート);(d)トラガカント粉末;(e)麦芽;(f)ゼラチン;(g)タルク;(h)賦形剤(ココアバターおよび坐薬ワックス);(i)油脂(ピーナッツ油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油および大豆油);(j)グリコール類(プロピレングリコール);(k)ポリオール類(グリセリム、ソルビトール、マンニトールおよびポリエチレングリコール);(1)エステル類(オレイン酸エチルラウリン酸エチル);(m)寒天;(n)緩衝剤(水酸化マグネシウム、水酸化アルミニウム、ホウ酸およびホウ酸ナトリウムおよびリン酸バッファー);(o)アルギン酸;(p)滅菌水;(q)等張食塩水;(r)Ringer溶液;(s)エチルアルコール;(t)リン酸緩衝溶液;および(u)医薬製剤における使用に適当なその他非毒性物質が挙げられる。
【0022】
慢性疼痛の処置の方法において、以下は、本発明の理解を補助するものとして助けとなるであろう。慢性疼痛(癌、関節炎および多くの神経障害性傷害による疼痛)および急性疼痛(組織が切れる、挟まれる、刺される、押し潰される等の即時的な機械的刺激によって生じる疼痛)は、異なる神経線維または神経受容体のいずれか、または慢性的な刺激に対するこれら神経の機能の再構成または変化が大きく関わっている別個の神経学的現象である。急性疼痛の感覚は、主に、通常、機械的、熱的および化学的刺激に対して高い閾値を有するC繊維と呼ばれる求心性神経繊維によって極めて急速に伝達される。慢性疼痛のメカニズムは、完全には分かっていないが、急性の組織損傷が最初の刺激から数分〜数時間以内に、例えば疼痛応答を惹起するのに必要な刺激の規模の局所的減少を含む二次的な症状を生じる。元々の刺激部位から広がった(より大きな)領域において典型的に生じるこの現象は、痛覚過敏と呼ばれる。この二次的な応答によって、機械的または熱的な刺激に対する感覚が大きく増大し得る。
【0023】
A求心性神経線維(AβおよびAδ繊維)はC繊維と比較して低い閾値で刺激され、慢性疼痛の感覚に関与すると考えられる。例えば、通常の状態では、これら繊維の低い閾値の刺激(例えば、軽く触れるまたはくすぐる)では痛みを感じない。しかし、神経損傷後または帯状疱疹等のヘルペスウイルスが介在する状態におけるある特定の状態では、そのような軽く触れるまたは衣服がすれただけでも激しい痛みが生じる。この状態は、異痛と呼ばれ、少なくともAβ求心性神経が部分的に関与していると考えられる。C繊維もまた慢性疼痛の感覚に関与し得るが、そうであっても、神経の持続的発火がやがて現に慢性疼痛の感覚を生じているような類の変化をもたらすことは明かである。本明細書で用いる本明細書で用いる「疼痛」なる語は、急性疼痛と慢性疼痛の両方を包含する。本明細書で用いる「急性疼痛」は即座の一般には高い閾値の、切り傷、圧縮、火傷、または唐辛子の活性成分であるカプサイシンに暴露したときに経験するような化学的刺激等の損傷によってもたらされる疼痛を意味する。本明細書で用いる「慢性疼痛」なる語は、急性疼痛以外の等痛を意味し、神経障害性疼痛、内臓疼痛、線維筋痛、炎症性疼痛、頭痛、筋疼痛および関連痛が挙げられるがこれに限定されない。慢性疼痛は、比較的長く続くことが多く(例えば数ヶ月ないし数年)、連続的あるいは断続的なものである。
【0024】
一態様では、本発明の方法は、本明細書において神経に対する傷害から生じる疼痛を意味する「神経障害性疼痛」を処置するために用いられる。
【0025】
神経障害性疼痛は、小さな皮神経または筋肉または結合組織における小神経が関わる急性の組織傷害によって引き起こされる疼痛である侵害受容性疼痛とは区別することができる。神経障害性疼痛とは対照的に、侵害受容性疼痛は、通常、持続期間が組織修復の期間に限定され、通常、利用可能な鎮痛薬または麻薬によって軽減し得る(Myers, Regional Anesthesia 20: 173-184 (1995))。
【0026】
神経障害性疼痛は、典型的に長期間または慢性的に持続し、最初の組織傷害から数日ないし数ヶ月後に生じる。神経障害性疼痛は、永続的な、自発的な疼痛、並びに通常は痛みを感じない刺激に対する疼痛応答である異痛または針で刺すような通常は些細な痛みを生じるような刺激に対する強化された応答である痛覚過敏を伴う。神経障害性疼痛は一般にオピオイド療法には効きにくい(Myers, supra, 1995)。
【0027】
本発明の方法は、末梢神経、後根神経節、脊椎、脳幹、視床または皮質の外傷、傷害または疾患によって生じた神経障害性疼痛の処置に有用であるが、これに限定されない。本発明の方法によって処置し得る神経障害性疼痛の例としては、ヘルペス後神経痛、除神経後痛および糖尿病性ニューロパシー等の神経痛が挙げられる。本発明の方法は、疼痛の病因に拘わらず神経障害性疼痛の処置に有用であると理解される。非限定的な例として、本発明の方法は、末梢神経障害(神経腫等);神経圧迫;神経挫滅または延伸または不完全な神経処理;または単ニューロパシーまたはポリニューロパシーにより生じた神経障害性疼痛を処置するために用いることができる。非限定的なさらなる例として、本発明の方法は、後根神経節圧迫;脊椎の炎症;打撲、脊椎の腫瘍または半側切断;および脳幹、視床または皮質の腫瘍または外傷等の障害により生じた神経障害性疼痛の処置に有用である。
【0028】
上で示したように、本発明の方法は、単ニューロパシーまたはポリニューロパシーにより生じた神経障害性疼痛の処置に有用である。ニューロパシーは、神経系における機能的な妨害または病理学的な変化であり、感覚または運動神経の異常によって臨床的に特徴付けられる。単ニューロパシーなる語は、単一の末梢神経が冒されることを示し、ポリニューロパシーなる語は、複数の末梢神経が冒されることを示す。ニューロパシーの原因は既知の原因もあればみちの原因もある。既知の原因としては、疾患または中毒症状の合併症(ニューロパシーを引き起こす最も一般的な代謝障害である糖尿病、またはX線照射、虚血または脈管炎等)。本発明の方法により処置し得るポリニューロパシーは、ポリオ後症候群、糖尿病、アルコール、アミロイド、毒素、HIV、甲状腺機能低下症、尿毒症、ビタミン欠乏症、化学療法、2’,3’−ジデオキシシチジン(ddc)治療またはファブリー病によって引き起こされ得るが、これに限定されない。本発明の方法は、原因が既知のまたは未知のこれらのまたは他の慢性的なニューロパシーの慢性疼痛を処置するために用いることができる。
【0029】
本発明の方法は、緊張性頭痛、片頭痛、群発性頭痛、ホルモン性頭痛、反発性頭痛、副鼻腔炎による頭痛、および器質性頭痛を含む頭痛により生じた慢性疼痛を処置するために用いることができる。本発明の方法はさらに、非限定的な例としてのコンピュータでの長時間労働、重い物や重い機械を扱う仕事または長時間立ち続ける仕事、および反復運動障害(RMD)等の活動により生じた慢性疼痛を処置するために用いることができる。RMDは慢性疼痛を引き起こす様々な筋肉症状である。RMDは、過度の労働、無理な姿勢、筋肉疲労、神経または組織の圧迫、活動または動作の延々と続く反復、または腕または腰のひねり等の不自然なまたはぎこちない動作によって起こる摩擦によって引き起こされる。一般的なRMDは、手、腰、肘、肩、首、背中、尻、膝、足、脚および足首に起きるが、手と腕が最も起こりやすい。本発明の方法は、任意のタイプのRMDによって生じる慢性疼痛を処置するために用いることができる。
【0030】
本発明の方法はさらに、椎間板ヘルニアにより生じるような特定のタイプの背痛等の過度の筋肉の緊張;坐骨神経痛および関節痛、並びに炎症性障害により引き起こされる炎症(骨関節炎および慢性関節リウマチ等)を含む炎症による慢性疼痛;組織または関節の挫滅、刺し傷、伸張等の外傷による炎症;感染による炎症(結核);または神経性炎症により生じる慢性疼痛を処置するために用いることができる。非限定的な例示として、本発明の方法は、クローン病、潰瘍性大腸炎、胃炎、過敏性腸疾患等を含む、慢性の胃腸炎症および慢性の内臓疼痛(例えば、癌によるまたは癌治療に伴う(例えば化学療法または放射線療法に伴う)疼痛)を処置するために用いることができる。同様に、本発明の方法は、例えば、関節炎(例えば慢性関節リウマチ、痛風性関節炎、または骨関節炎);脊椎炎;または自己免疫疾患(例えば紅斑性狼瘡)による慢性の炎症性疼痛を処置するために用いることができる。本発明の方法はさらに、慢性の筋疼痛、物質濫用または禁断に関連する、慢性疼痛および原因が既知または未知のその他のタイプの慢性疼痛を処置するために用いることができる。本発明の方法では、第一の薬物と第二の薬物を任意の関係する方法を用いて同時に投与してもよいものと意図する。それらの薬物は、同じ方法または異なる方法を用いて同時または異なる時間に投与することができる。
【0031】
疼痛の処置に加えて、α2アゴニストおよびTCAは神経保護に有用であることが知られている。例えば、本発明の方法にしたがってより副作用の少ない、本明細書に示した第一および第二成分によって処置することができる状態としては、神経変性状態(パーキンソン病パーキンソン病、アルツハイマー病、筋萎縮性側索硬化症および多発性硬化症);卒中等の虚血;てんかん;およびニューロパシー(糖尿病性および虚血性網膜症等)が挙げられるがこれに限定されない。さらに、精神障害(統合失調症および双極性障害等)は現在、神経変性がある程度まで関わっていると考えられている。さらに、本発明の組成物および方法は、現在、付随する鎮静なしに、様々な交感神経増強ストレス関連状態の予防または処置に有用であることが知られている。そのような状態としては、感覚過敏症(例えば、線維筋痛または片頭痛等の頭痛に関連する感覚過敏症)、胃腸疾患(例えば、過敏性腸症候群および消化不良);皮膚病変(例えば乾癬);心臓血管障害;頻脈;末梢血管収縮の障害(レイノー症候群および強皮症);パニック発作;代謝障害(II型糖尿病、インスリン耐性および肥満症);筋収縮の障害(骨格筋収縮の障害、平滑筋収縮の障害、痙縮、および緊張性頭痛に関連する筋収縮の障害を含む);行動障害;および性機能障害が挙げられるが、これに限定されない。一態様では、交感神経増強状態は、交感神経遮断によって軽減し得る任意の疼痛である交感神経依存性疼痛以外の状態である。そのような使用に関連する背景の情報は、本願と同じ2003年9月12日に出願の、米国特許出願60/502840(発明の名称「NOVEL METHODS FOR IDENTIFYING IMPROVED, NON-SEDATING α-2 AGONISTS」に記載されている。
【0032】
α2活性化剤とα1アンタゴニストの同時投与に基づいて、本発明は、A2AA単独で用いてみられる場合と比較してより少ない鎮静作用または心血管副作用でもって神経変性状態の処置における治療的効果をもたらす。神経死を減少または防止することにより、病態および症状における改善が認められる。本明細書で用いる「神経死」なる語は、損傷または異常に応答する死の誘導による神経細胞の破壊を意味する。「神経死」の定義には、胚発生の間に起きるようなまたはアポトーシスを免れない神経を含む自己更新組織(嗅上皮等)における病的でない神経のアポトーシスは含まれない。したがって、神経死なる語は、嗅上皮以外の神経上皮の神経損傷(脳神経などの中枢神経系の神経の損傷およびアポトーシスを受ける神経出ない神経における神経損傷等)を包含する。本明細書で用いる「減少させる」なる語は、神経死に関して用いる場合、神経細胞における死の誘導を予防する、減少させるまたは排除することを意味する。有効量のA2AAおよびα1アンタゴニストの投与による神経死の減少は、鎮静または心血管副作用を最小限にするとともに神経死または機能不全を伴う状態を処置のための有効な方法となり得る。
【0033】
本明細書で用いる「神経変性状態」は、進行性の神経系の機能不全によって特徴付けられる障害を意味する。神経変性状態には、多くのことなる病因を有する中枢神経系または末梢神経系の疾患の異種のグループが含まれる。そのような状態は、遺伝性の、中毒または代謝過程の後に起きるものであり、感染によるものであり得るがこれに限定されない。神経変性状態は、加齢性または慢性であり得る進行性の状態である。そのような状態は、脳の比較的特定の領域または神経の特定の集団の異常によって特徴付けることができる。種々の神経変性状態において冒される特定の細胞集団によって、典型的にその状態の臨床表現型が決定する。特に、神経変性状態は、特定の冒された中枢神経または末梢神経系構造の萎縮に関連し得る。神経変性状態の例示としては、運動ニューロン疾患(ALS)、パーキンソン症候群、広汎性硬化症、筋萎縮性側索硬化症、多発性硬化症、広汎性皮質性小脳萎縮症、レヴィー小体認知症、ピック病、メソリンボ(mesolimbo)皮質性認知症、視床変性、球麻痺、ハンチントン舞踏病、皮質−線条体−脊椎変性、皮質ベースの神経節変性、大脳小脳皮質変性、不全対麻痺を伴う家族性認知症、ポリグルコサン小体疾患、シャイ・ドレーガー症候群、オリーブ橋小脳萎縮症、進行性核上麻痺、変形性筋失調症、ハレルフォルデン・スパッツ病、メージ症候群、家族性振戦、ジル・ド・ラ・ トゥレット(Gilles dela Tourette)症候群、acanthocytic舞踏病、フリードライヒ失調症、Holmes家族性小脳皮質萎縮症、AIDS関連性認知症、Gerstmann-Straussler-Scheinker病、進行性脊髄性筋萎縮、進行性球麻痺、原発性側索硬化、遺伝性筋萎縮症、痙性対麻痺、腓骨筋萎縮、肥厚性間質性ポリニューロパシー、遺伝性多発神経炎性失調、視神経症、糖尿病性網膜症、アルツハイマー病および眼筋麻痺が挙げられるがこれに限定されない。当業者は、これらの他の、軽度の、中程度のまたは重度の神経変性状態の状態は本発明の方法にしたがって処置し得ると理解する。
【0034】
本発明のさらなる態様では、A2AAを含有する第一成分およびα1受容体アンタゴニストを含有する第二成分の同時投与を含んでなる眼症状の処置のための方法を提供する。この態様では、一般に低減された鎮静および心臓血管副作用をもたらすA2AA濃度で治療効果が得られる。
【0035】
本発明の方法を用いて処置することができる眼症状の例としては、開放隅角緑内障を含む緑内障、高眼圧症、黄斑および網膜変性(非滲出性加齢性黄斑変性(ARMD)、滲出性加齢性黄斑変性(ARMD)、脈絡膜血管新生、糖尿病性網膜症、中心性漿液性網脈絡膜症、類嚢胞黄斑浮腫、糖尿病性黄斑浮腫、近視性網膜変性等);炎症性疾患(急性多発性小板状色素上皮症、ベーチェット病、バードショット脈絡網膜炎、感染(梅毒、ライム病、結核、トキソプラズマ症、中間部ブドウ膜炎(扁平部炎)、多発局所性脈絡膜炎、多発性一過性網膜白点症候群(MEWDS)、眼サルコイドーシス、後部強膜炎、蛇行性脈絡膜炎、網膜下線維症およびブドウ膜炎症候群、Vogt-Koyanagi-Harada症候群、点状網膜脈絡膜炎、急性後部多発性小板状色素上皮症、急性網膜色素上皮炎、急性斑状神経網膜症);血管疾患および滲出性疾患(糖尿病性網膜症、網膜中心動脈閉塞症、網膜中心静脈閉塞、播種性血管内凝固障害、網膜静脈分枝閉塞症、高血圧性眼底変化、眼虚血症候群、網膜微細動脈瘤、コーツ病、傍中心窩毛細血管拡張症、半網膜静脈閉塞症、Papillo静脈炎、網膜中心動脈閉塞、網膜動脈分枝閉塞症、頸動脈疾患(CAD)、糖衣状分岐血管炎(Frosted Branch Arterial)、鎌状赤血球網膜症およびその他の異常血色素症、網膜色素線条、家族性滲出性硝子体網膜症);Eales病;外傷、外科手術および環境疾患(交感性眼炎、ブドウ膜炎性網膜疾患、網膜剥離、外傷、網膜レーザー光治療、光凝固、外科手術中の血流低下、放射線網膜症、骨髄移植網膜症);増殖障害(増殖性Vitreal増殖性網膜症および網膜上膜);感染性障害(眼ヒストプラスマ症、眼トクソカリアシス、推定眼ヒストプラスマ症症候群(POHS)、眼内炎、ヒストプラスマ症、HIV感染に関連する網膜疾患、HIV感染に関連するに関連する脈絡膜疾患、HIV感染に関連するブドウ膜炎性疾患、ウイルス性網膜炎、急性網膜壊死、進行性網膜外層壊死、真菌性網膜疾患、眼梅毒、眼結核、片側性瀰漫性亜急性視神経網膜炎、ハエウジ病);遺伝病(網膜色素変性、Accosiated網膜ジストロフィーに関連する全身性障害、先天性静止性夜盲症、錐体ジストロフィー、スタルガルト病および黄色斑眼底、ベスト病、網膜色素上皮のパターンジストロフィー、X線に関連する網膜分離、Sorsby's基底ジストロフィー、良性求心性黄斑、Bietti's crystallineジストロフィー、弾性線維性仮性黄色腫);網膜傷害(黄斑円孔、大きな網膜裂傷等);網膜腫瘍(腫瘍に関連する網膜疾患、前部および後部ぶどう膜メラノーマの先天性肥大、脈絡膜血管腫、脈絡膜骨腫、脈絡膜転移、網膜および網膜色素上皮の複合性過誤腫、網膜芽腫、眼底の血管増殖性腫瘍、網膜星状細胞腫、および眼内リンパ球腫瘍)が挙げられるがこれに限定されない。
【0036】
神経網膜および視神経の虚血は、網膜静脈分枝閉塞の間、網膜中心動脈閉塞の間、網膜中心静脈閉塞の間、硝子体内外科手術中、網膜変性(網膜色素変性、および加齢性黄斑変性等)においてに起こり得る。同時投与されるA2AAおよびアルファlアンタゴニストのような、本発明において用いられる組成物の神経死または機能不全を減少させる能力は、その化合物の存在下および非存在下で神経細胞破壊の観察可能な兆候または症候を解析することによって調べることができる。神経のアポトーシスによる死の開始によって、細胞の機能および形態に対し観察可能な影響が認められ、同様に組織、臓器および機能に障害のあるまたはアポトーシスを起こす神経を含む動物に対しても観察可能な影響が認められる。したがって、神経損傷の指標には、分子における変化の観察可能なパラメーター(アポトーシス誘発遺伝子の発現の増大など)、細胞機能の変化(ミトコンドリア機能の低下など);細胞の形態変化(細胞収縮および水疱形成など);臓器および組織の機能および形態変化(梗塞または他の病変の存在、その重篤度は病変の容積と病変のサイズを含むパラメーターによって評価することができる);動物モデルにおける機能の変化を含む生理学的変化(運動機能の喪失、死亡率の増加および生存率の減少など)および動作上の変化(認知症の発症または記憶の喪失など)が挙げられる。
【0037】
神経損傷の指標の減少は、細胞、組織、臓器または動物の少なくとも2つの状態における神経損傷の指標を比較することにより、細胞、組織、臓器または動物において評価することができる。即ち、神経損傷の指標の減少は、対照の状態に対する相対値で示される。
対照の状態は、例えば、処置前の、処置をしていない、異なる処置を行っている、あるいは正常な動物または当業者が適当であると決定した他の状態における、細胞、組織、臓器または動物である。具体的な態様では、本発明の方法は、本発明の第一成分および第二成分の末梢投与により行われる。本明細書で用いる「末梢投与」または「末梢投与された」なる語は、薬剤を対象の中枢神経系の外へ導入することを意味する。末梢投与には、脊椎または脳への直接投与以外の任意の投与が含まれる。鞘内および硬膜外投与並びに頭蓋注射または移植(本発明の態様の範囲内であるけれども)は、それ自体は「末梢投与」または「末梢投与された」なる語の範囲には入らないことは明かである。
【0038】
末梢投与は、局所的または全身的投与であってよい。局所投与は、投与部位から離れた領域と比較して、局所投与部位へ有意により多くの医薬組成物の送達をもたらす。全身投与は、対象の本質的に末梢神経全体に医薬組成物を送達し、その組成物の特性に依存して中枢神経系への送達が可能である。本発明の方法において有用な末梢投与の経路には、経口投与、局所投与、眼内投与、静脈内注射または他の注射、および移植されたミニポンプまたは他の徐放装置または徐放製剤が含まれるがこれに限定されない。本発明において有用な医薬組成物は、例えば任意の許容し得る形態(錠剤、液体、カプセル、粉末等)での経口投与;静脈内、腹腔内、筋肉内、皮下または非経口注射;経皮拡散または電気泳動により;ドロップ、クリーム、ジェルまたは軟膏等の任意の許容し得る形態での局所的投与;およびミニポンプまたは他の移植された徐放装置または徐放製剤により、末梢投与することができる。
【0039】
本発明はさらに、哺乳動物における疼痛の処置のための方法であって、
a)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分、および
b)α1アドレナリン受容体アンタゴニストを含有する第二成分
の末梢または非末梢の同時投与を含んでなり、最大半量の鎮静をもたらすのに効果的な用量にて、該方法にしたがって最大半量の鎮痛をもたらすのに効果的な第一成分の用量の投与によって引き起こされる鎮静の量が、該第二成分の不在下で該第一成分を投与した同様に罹患した哺乳動物において引き起こされる量よりも少ない、方法に関する。本発明のこの態様およびその他の態様についての投与の様式は、非末梢投与(例えば鞘内または硬膜外など)または全身投与(経口、腹腔内、静脈内、筋肉内、または経皮など)であってよい。
【0040】
疼痛の処置に加え、本発明のさらなる態様は、αレセプターアゴニストが有効であると知られている他の状態の処置に関する。これらには、癌障害(高眼圧症および緑内障など)、交感神経増強ストレス関連状態、神経変性状態;および痙縮が含まれる。αアドレナリン作用によるこれらの状態の処置は、本発明は改善することのできる望ましくない鎮静副作用を生じ得る。
【0041】
この態様では、第一成分および第二成分の局所的デリバリーは眼への第一成分および第二成分の治療物質の送達に好ましいものであろう。局所的眼科製剤は当分野において周知である。
【0042】
眼科用医薬組成物は、活性成分として治療上有効量の第一成分および第二成分を慣用の眼科的に許容し得る賦形剤と共に混合することによって(単一の製剤としてまたは別々の製剤として)および局所的な眼科的使用に適当な単位投与剤型に調製することによって製造することができる。各成分の治療上有効量は典型的には、液体の製剤において約0.0001〜約5%(w/w)、好ましくは約0.001〜約1.0%(w/w)である。
【0043】
眼科的な適用のためには、主な賦形剤として生理食塩液を用いて液剤を調製することが好ましい。そのような眼用液剤のpHは、適当な緩衝系(ホウ酸、トロメタミンまたはリン酸緩衝系など)によって6.2〜7.8に保つことが好ましい。この製剤は、医薬的に許容し得る通常の保存剤、安定剤および界面活性剤をも含有し得る。
【0044】
本発明の眼科的適法の方法および組成物に使用し得る好ましい保存剤としては、塩化ベンザルコニウム、他のポリマー性四級アンモニウム保存剤(PHMBおよびpolyquad(商標)など)、クロロブタノール、チメロサール、酢酸フェニル水銀および硝酸フェニル水銀を包含するが、これらに限定されるものではない。特に好ましいクラスの保存剤は、安定化された二酸化塩素(例えばPurite(商標)安定化二酸化塩素)、安定化されたオキシホウ酸塩等の酸化保存剤である。
【0045】
眼科用製剤において用いるための界面活性剤は、イオン系または非イオン系界面活性剤(Triton(例えば、TritonX-100)、Tween(商標)(例えばポリソルベート40、ポリソルベート80)およびプルロニック(商標)等)が挙げられるがこれに限定されない。
【0046】
同様に、本発明の眼用製剤中に種々の好ましい賦形剤を使用し得る。このような賦形剤には、ポリビニルアルコール、Carbopol(商標)(アリルスクロースで架橋されたプロピオン酸のポリマー)、Pemulin(商標)、ポビドン、ポロキサマー、セルロース誘導体(カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシメチルセルロースなど)が含まれるが、これらに限定されるものではない。
【0047】
眼科用製剤をエマルジョンとして製剤化することも好ましい。その場合、エマルジョンは、水中油エマルジョンかまたは油中水型エマルジョンのいずれであっても良い。このエマルジョンは保存剤および/または賦形剤を含有して良いが、通常少なくとも1種類の界面活性剤および乳化剤を含有する。本エマルジョンはまた界面活性であってよい。1つの好ましい乳化剤は、架橋したポリアクリル酸であるPremulin(商標)である。
【0048】
必要に応じて適宜、張度調整剤を加えても良い。これらには、塩、特に塩化ナトリウム、塩化カリウム、マンイトールおよびグリセリンまたは他の任意の適当な眼科的に許容し得る張度調整剤が挙げられるがこれに限定されない。
【0049】
得られる調製物が眼科的に許容し得るものである限り、様々な緩衝剤およびpH調整手段を用いることができる。したがって、緩衝剤には、酢酸バッファー、クエン酸バッファー、リン酸バッファーおよびホウ酸バッファーが含まれる。必要に応じ、酸または塩基を用いてこれら製剤のpH調整することもできる。
【0050】
同様に、本発明において用いるための眼科的に許容し得る抗酸化剤の一覧には、メタ亜硫酸水素ナトリウム、チオ硫酸ナトリウム、アセチルシステイン、ブチル化ヒドロキシアニソール、およびブチル化ヒドロキシトルエンが含まれるがこれに限定されない。
【0051】
眼科用調製物に含有し得るその他の賦形剤はキレート剤である。好ましいキレート剤は、エデト酸二ナトリウムであるが、他のキレート剤をそれに変えて使用してもそれと組み合わせて使用してもよい。
【0052】
以下の量で成分を用いることができる:
成分 量(% W/W)
各活性成分 約0.001〜5
保存剤 約0−0.10
ビークル 約0−40
張度調整剤 約0−10
緩衝剤 約0.01〜10
pH調整剤 適量を加えpH4.5〜7.5とする
抗酸化剤 適量
界面活性剤 適量
精製水 適量を加えて100%とする
【0053】
本発明の活性化合物の実際の用量は、具体的な化合物、および処置される状態に依存するが、適当な用量は当業者の知識の範囲内で十分選択することができる。
【0054】
本発明の眼科用製剤は、点眼用容器などの定量での適用に適した形態に包装して眼への適用を容易にするのが便利である。点眼適用に滴した容器は、通常、適当な不活性な、非毒性のプラスチック材料で製造し、一般的に約0.5〜15ミリの液体を入れる。
【0055】
保存剤を含まない溶剤は、しばしば、約10、好ましくは約5単位用量(典型的な単位用量は約8滴まで、好ましくは3滴までである)を入れることができる非密閉型の容器にて製剤化する。一滴の容量は通常約20〜35μLである。本発明の方法では、好ましくは、第一成分はα2アゴニストまたはTCA、さらに好ましくはα2アゴニスト、最も好ましくはクロニジン、ブリモニジン、デクサメデトミジン、ミバゼロールおよびチザニジンからなる群から選択されるα2パンアゴニストからなる群から選択される。このような化合物およびその合成は周知である。
【0056】
具体的な態様では、α1アドレナリン受容体アンタゴニストは、プラゾシン、テラゾシンおよび5−メチルウラパジルからなる群から選択される。前者の2種類の化合物はおよびその合成は米国特許第3544836号、同第4026894号にそれぞれ記載されており、後者の化合物は、米国特許第3957786号にその合成が記載されているウラパジルから容易に合成される誘導体である。本願において引用するこれらおよび他の参考文献は、本明細書の一部を構成する。したがって、他のα1受容体アンタゴニストは当分野で周知であり、これら化合物の多くは臨床で認められている。α1アンタゴニストの多くの例が記載されているLagu、26 Drugs of the Future 757−765 (2001)およびForrayら、8 Exp. Opin. Invest. Drugs 2073(1999)を参照。本明細書に照らせば、その他の態様は当業者には自明であろう。
【図面の簡単な説明】
【0057】
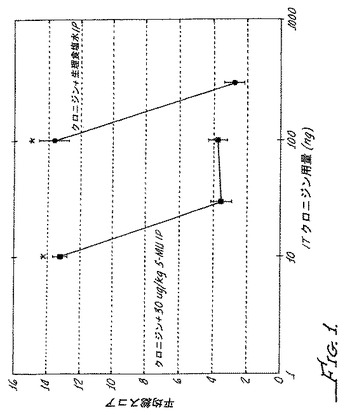
【図1】スルプロストン誘発異痛モデルを用いた野生型マウスにおける5−メチルウラパジルの投与ありまたは投与なしでのクロニジンの鎮痛活性を示す。
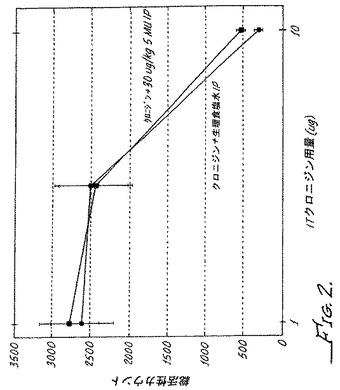
【図2】上記で詳述したように行った5−メチルウラパジルの投与ありまたは投与なしでのクロニジン鎮静活性の用量応答曲線である。結果は5−メチルウラパジルがクロニジン鎮静に対して効果がないことを示している。
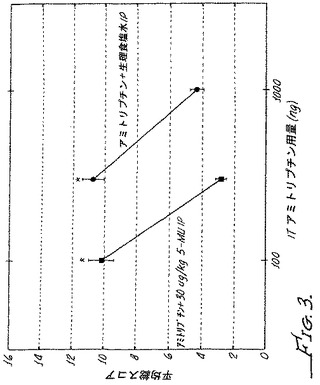
【図3】スルプロストン誘発異痛モデルを用いた野生型マウスにおける5−メチルウラパジルの同時投与ありまたは同時投与なしでのアミトリプチリンの鎮痛活性を示す。
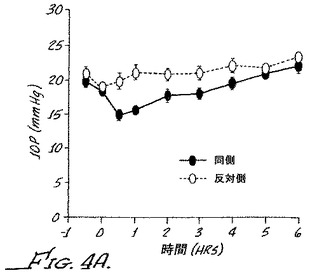
【図4A】プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【図4B】ブリモニジンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
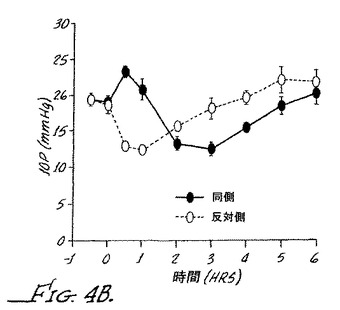
【図4C】ブリモニジンおよび0.001%プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
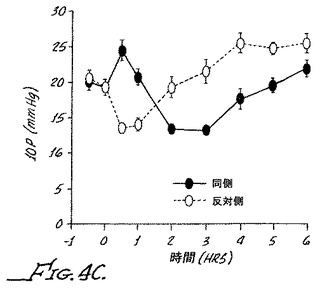
【図4D】ブリモニジンおよび0.003%プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【発明を実施するための形態】
【0058】
第一の態様において、本発明は、哺乳動物に対する、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与は、治療効果をもたらすのに必要な第一成分の用量が、該第一成分を単独の治療剤として投与された同じ疾患に罹患した哺乳動物において同様の治療効果をもたらすのに必要な用量に比べて少ない場合、鎮静および/または心臓血管副作用がより少ない、という出願人の驚くべき発見に関する。このことは、鎮痛、眼圧低下活性、神経保護等の治療効果が、ヒトを含む哺乳動物における該第一成分と該第二成分の同時投与によって得られ、第一成分の同様の有効量の投与と比較して、治療用量における鎮静と心臓血管抑制の大きな減少をもたらすということを意味する。現在好ましい態様では、該哺乳動物は慢性疼痛のために鎮痛を必要とする。
【0059】
したがって、本発明のこの態様では、出願人は、その活性が、例えばα2受容体パンアゴニスト(クロニジン、デクサメデトミジン、またはチザニジン)またはTCAのようなα2アドレナリン受容体(A2AA)の直接的または間接的な活性化をもたらす化合物の同時投与が、そのような薬剤の使用時に一般的にみられる鎮静副作用の実質的な増大なしに、A2AAの治療効力の付随する増大とともに、A2AAのα2介在の治療活性の「脱マスキング」または増大をもたらすということを発見した。結果として、選択されたA2AAによる処置についての治療濃度域は、同じ薬剤をαアンタゴニストなしで投与した場合と比較して増大し、ある特定の場合ではA2AAの用量の増加、そして他の場合には少ない副作用で同じ用量の使用が可能になる。好ましくは、A2AAは、α2アドレナリン受容体アゴニストである。
【0060】
本発明は、特定の理論に限定されると解釈されるものではないが、出願人は、α1受容体、または哺乳動物におけるα1A、α1B、およびα1D受容体を含むその1またはそれ以上のサブタイプの刺激によって、α2A、α2Bおよび/またはα2C受容体の刺激から生じる活性(鎮痛、眼圧低下、神経保護またはその他)の減衰されるために本発明が機能すると考える。また、A2AAの大部分は、(α2「選択的」であると記載されているかそうでないかに関わらず)α1刺激活性を欠いているとは決まってはおらず、したがって、この減衰効果を引き起こす本来備わっている十分なα1アゴニスト活性を有していると考えられる。
【0061】
したがって、α1アンタゴニストの同時投与は、α1受容体の刺激による望ましくない拮抗作用をブロックすると考えられる。好ましくは、このα1アンタゴニストは、少なくともα1A受容体アンタゴニスト活性、α1Bアンタゴニスト活性またはα1D受容体アンタゴニスト活性を有する。最も好ましくは、α1受容体アンタゴニストは、少なくともα1A受容体アンタゴニスト活性を有する。そのようなアンタゴニストは、1またはそれ以上のα1受容体サブタイプに対してアンタゴニスト活性を有してもよい。
【0062】
哺乳動物に対するα2アゴニストの処置にみられる、鎮静副作用を増大させないそして時には減少させることに加え、哺乳動物に対するA2AAおよびα1アンタゴニストの同時投与は、A2AAの治療効果を改善し(即ち、「治療濃度域」の拡大)、それによってそうでない場合と比較してより低い用量での処置が可能になる。
【0063】
さらなる態様では、本発明は、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の投与による、哺乳動物における疼痛の処置のための方法に関する。好ましくは、A2AAは、選択されるα2受容体アゴニストおよびノルエピネフリン伝達物質阻害剤(三環系抗鬱薬またはTCA)、より好ましくはα受容体アゴニストからなる群から選択される。あるいは、A2AAは、好ましくは、ブリモニジン、クロニジン、チザニジン、デクサメデトミジンおよびノルエピネフリンおよびMPV-2426(ラドルミジン)からなる群から選択される。
【0064】
さらに、本発明の別の態様は、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の投与による、哺乳動物における高い眼圧の処置のための方法を包含する。この態様では、より好ましい投与経路は、溶液、懸濁液またはエマルジョン等の局所用眼科製剤である。好ましい態様では、A2AAはα2アゴニスト、より好ましくは、α2パンアゴニストである。
【0065】
好ましくは、該第一成分および第二成分は、同時投与のための単一の製剤に含まれるが、該第一成分および第二成分を別個の組成物として同時投与してもよい。本明細書において用いられる、「同時投与(する)」なる語は、該第一成分および第二成分を単一の組成物としてか、または別個の組成物として投与することを包含する。さらに、該成分はそれぞれ、異なる経路および/または同時もしくは若干異なる時間に投与してもよい。
【0066】
さらなる態様では、本発明は、製薬的に許容し得る担体と共に製剤中に治療上有効量の第一成分および第二成分を含有する組成物を包含する。さらなる態様では、該第一成分および第二成分を、二機能性(bifunctional)試薬のように、化学リンカーを用いるA2AA(TCAまたはα2アゴニストから選択される)とα1アンタゴニストとの結合によって結合させてもよい。
【実施例】
【0067】
出願人はここに、本発明の特定の実施態様を証明することを目的として実施例を示す。本発明はこれらの実施例に限定されることを意図しない。
【0068】
実施例1:α2アゴニストおよびα1アンタゴニストの同時投与による慢性疼痛の軽減
慢性疼痛のモデル(特に末梢性ニューロパシー)は、実験動物における片側のL5(場合によりL6)脊髄神経の外科的な結紮によるものである。手術から回復したラットは体重が増え、正常なラットと類似する全体的活動レベルを示す。しかし、これらのラットは、後肢がわずかに外反し、足指が束ねられているという脚の異常を発症する。より重要なことには、手術による影響を受けた側の後肢は、手術後約1週間以内に、低い閾値の機械的刺激(例えばヒトにおいてはかすかな接触感覚を生じさせる刺激)からの痛みに対して感じやすくなっているようである。正常な場合には痛みにならない接触に対するこの感受性は「触覚異痛」と呼ばれており、少なくとも2ヶ月間にわたって続く。応答には、影響を受けた後肢を上げて刺激から逃避すること、脚をなめること、および脚を空中に長く保持することが含まれる。これらの応答はどれも、通常、コントロール群では認められない。
【0069】
外科処置を行う前に、ラットを麻酔する。外科処置を行う部位を剃り、ベタジンまたはノバカインで準備する。第XIII胸椎から仙骨へ下へ切開する。L4〜S2レベルで筋肉組織をL4〜S2のレベルで脊椎(左側)から離す。L6脊椎を探し出して、横突起を、小さい骨鉗子を用いて注意して除き、L4〜L6の脊髄神経を露出させる。L5およびL6の脊髄神経を隔離して、6−0絹糸できつく結紮する。脊髄神経の結紮を行わないことを除いて、同じ手順をコントロールとして右側で行う。
【0070】
完全な止血を確認して、傷を縫合する。少量の抗生物質軟膏を切開領域に塗布して、ラットを、調節された熱温度ランプのもとでの回復用のプラスチックケージに移す。手術後少なくとも7日目の実験当日に、通例、試験群あたり6匹のラットに試験薬物を鞘内、腹腔内注射または経口胃管投与またはこられ1またはそれ以上の組み合わせによって投与する。
【0071】
あるいは、動物を、5μLの50%DMSO中の200ngスルプロストン(プロスタグランジンE2受容体アゴニスト)を鞘内処置することにより誘導することができる。このモデルにおいて、はけで側腹をなでることに対する痛み応答が、スルプロストンの脊髄投与の15分後から開始して35分間の期間にわたって8回スコア化される。Minamiら、57 Pain 217−223 (1994)。スルプロストン処置は単独で16点の測定尺度で12点〜13点の「痛み」のスコアを誘発する。
【0072】
本化合物は、約0.01−5%DMSO中で製剤化し、全身投与については1mL/kg体重または鞘内投与については5μLにて投与する。クロニジンは、0.01〜10.0μgの範囲の鞘内用量で試験する。
【0073】
Chungモデルラットでは、一連の堅さの異なる細い毛のvon Frey hairsを用いて触覚異痛を薬物投与前および30分後に測定する。ラットを底に金網を敷いたプラスチック製のカゴに入れ、約30分間順応させる。von Frey hairsを金網を通して、足を曲げて6〜8秒間その状態を保つのに十分な力でラット後足の足裏中央部に垂直に当てる。与えた力は、計算すると0.41〜15の.1グラムの範囲である。足を急に引っ込めた場合、陽性の反応とする。正常な動物は、この範囲の刺激には応答しないが、外科手術により結紮した足は1〜2グラムの毛に応答して引っ込める。50%の足引っ込み閾値は、Dixon他(Ann. Rev. Pharmacol. Toxicol., 20:441-462(1980))の方法を使用して決定された。薬物後の閾値が薬物前の閾値と比較され、触覚感受性の逆転率(%)が15.1グラムの正常な閾値に基づいて計算された。
【0074】
結果は、クロニジンがラットおよびマウスの両方において用量依存的に鎮痛物質であることを示している。胃痛ラットのChungモデルでは、0.1μgの鞘内用量で鎮痛は認められず、最大鎮痛は1.0μgで観察された。治療濃度域を決定するために、種々のクロニジン用量でラットを試験し、鎮静について分析した。
【0075】
鎮静を試験するために、6匹のオスSprague-Dawleyラットに、種々の用量のクロニジンを鞘内投与する。鎮静は、薬物投与の30分後に、下記のように運動能力をモニターすることによって評価する。ラットを暗い蓋付きチャンバーに入れ、デジコム(digicom)アナライザー(Omnitech Electronic)により、それらの探索行動を5分間にわたって定量化する。この装置では、ラットがX方向およびY方向の32本の光電ビームのアレイを遮る各時間が記録され、クロニジンの代わりに生理食塩水を与えた対照動物と比較した行動の違いを定量化する。
【0076】
このアッセイでは、3.0μgの鞘内投与により中程度の鎮静を示し、10μgの用量にて最大鎮静が誘導された。即ち、鎮痛と鎮静との間に3〜10倍の隔たりがある。
【0077】
スルプロストン誘導アロディニアの動物モデルでは、同様の結果が得られた。
【0078】
図1に示されるように、クロニジンの投与5分前にスルプロストン誘導マウスモデルにα1アンタゴニスト5−メチルウラパジル30μg/kgを腹腔内投与した場合、鎮痛有効量はクロニジン単独使用での応答と比較して10倍低い値にシフトし(図1参照)、鎮静用量は変化無しであった(図2参照)。このように、慢性疼痛のモデルにおいて、クロニジン(α2アゴニスト)とα1アンタゴニストの同時投与により、疼痛と鎮静との間で3〜10倍から30〜100倍に治療濃度域が拡大された。
【0079】
実施例2:TCAおよびα1アンタゴニスト(5−メチルウラパジル)の同時投与による慢性疼痛の軽減
一般に抗鬱薬および鎮痛薬として処方されている三環系抗鬱薬(TCA)は、ノルエピネフリン取込を阻害することによりα2受容体を間接的に刺激する。
【0080】
実験は上記実施例1の記載と同様に行い、スルプロストン誘導アロディニアマウスモデルおよびTCAアミトリプチリンを用いて行った。この化合物およびその合成は本明細書の一部を構成する米国特許第3205264号に記載されている。
【0081】
アミトリプチリンを50%DMSO中、示した用量で溶解し、5−メチルウラパジルの30μg/kgのIP注射または生理食塩水の同様の注射と組み合わせて、各マウスに5μLの容量で注射した。実施例1に記載した筆による刺激をスコア化し、結果を図3に示した。α2アゴニストとα1アンタゴニストの組合せに関して、アミトリプチリンをα1アンタゴニストの組合せにより、最大鎮痛を達成するのに必要な用量の減少がもたらされた(この場合約3倍)。
【0082】
本実施例は、α1アンタゴニストと組み合わせて、α2アドレナリン受容体を直接的または間接的に刺激(例えばノルエピネフリン産生の増大またはノルエピネフリン取込または回転率の制限)することにより、鎮静と治療効果との間の観察された治療濃度域がA2AA単独使用の場合と比較して増大することを示している。
【0083】
実施例3:α2アゴニストおよびα1アンタゴニスト(プラゾシン)の同時投与による慢性疼痛の軽減
スルプロストン誘発モデルについて記載したのと同様の方法を用いた場合、α1アンタゴニストプラゾシンの腹腔内用量(100ng/kg)単独(疼痛スコア=4.8±0.6)またはスルプロストン誘発アロディニアモデル(12.8±0.8)において効果がなかった。
【0084】
スルプロストンおよび種々の用量のクロニジン(50%DMSO5μL中、0.03、0.1および0.4μg)の投与の15分前にプラゾシンをマウスに投与した。α1アンタゴニスト同時投与時の鎮痛に対するクロニジン用量の応答は以下のとおりである:0.03μg用量について13.3±0.9、0.1μg用量について4.8±0.8、0.4μg用量について4.8±0.6。これは、クロニジン単独での鞘内投与と比較してクロニジンについて、クロニジンに対するEC50が約4倍減少する。
【0085】
このように、A2AAおよびα1アンタゴニスト両方の同時投与は、ここでもA2AA単独使用と比較してA2AAの効力の増大をもたらした。この実験では、クロニジンの0.1μg用量はプラゾシンの不在下で鎮痛作用を示さなかった。
【0086】
実施例4:同一の経路で投与されたα2アゴニストおよびα1アンタゴニストによる慢性疼痛の軽減
実験は、クロニジン(種々の用量)と5−メチルウラパジル(1μg鞘内)を50%DMSOビークル中総体積5μLにて鞘内注射により同時投与したことを除いては実施例1と同様に行った。結果は、α1アンタゴニストの腹腔内注射で得られた結果と実質的に同じであった。
【0087】
このように、同じまたは同様の治療効果でもって、2種類の薬剤を一緒にまたは若干異なる時間に、および同一または異なる投与経路で投与することができる。
【0088】
実施例5:α2アゴニストおよびα1アンタゴニストによる高い眼内圧の処置
雄性ニュージーランド白ウサギを用いてノルエピネフリンIOP測定に対する薬物の効果を評価した。ウサギが興奮しないように慎重に扱う。ウサギは片方の手で後足を抱えながら襟首を掴む。ウサギをカゴから出した後、約25μLの希釈したOphthetic(商標)(0.05%)局所麻酔薬を両目に投与する。初めのIOP測定は、両目について行う。右目と左目に3mmHg以上の差があるウサギはこの時点で交換する。
【0089】
両眼についてのゼロ時(T=0)での測定の直後に、各実験動物の片眼を0.1%(w/w)ブリモニジン酒石酸塩の眼科用製剤を無作為に選択した試験する眼の角膜に注入した。非対照のウサギにも0.001%または0.003%(W/W)プラゾシン−HClのいずれかを投与した。他方の眼には薬物を含まないビークルを投与した。この眼科用製剤は、50ppmのPurite(商標)(安定化二酸化塩素)、0.5%カルボキシメチルセルロース、0.6%(w/w)ホウ酸バッファー(pH7.7)および少量の塩(NaCl、KC1、CaCl2、MgCl2)を含有する。処置した(同側)および処置していない(反対側)両方の眼内圧を、7時間に渡り示した時間間隔で測定した。圧の測定は、測定および記録装置としてモデル30クラシック眼圧計を用いた。この装置は、圧平眼圧計によって眼内圧(IOP)の非侵襲的測定を行うものである。
【0090】
図4Aは、0.003%プラゾニンを35μLを同側のウサギの眼に注入した場合に得られる眼内圧を示す。図から分かるように、処置していない眼と比較して初めに眼内圧の小さな低下がみられ、その後2時間にわたり基底値へ戻っている。図4Bは、0.1%ブリモニジン(α2アゴニスト)の点眼の結果を示す。最初の30分間以内に眼内圧の顕著な増大がみられたケースでは、その後眼内圧が約30%低下した。
この効果は、ヒトでは観察されていないものの、IOPの初めの増大はα1アゴニスト活性と関連した。図4Cにみられるように、0.1%ブリモニジンおよび0.001%プラゾシンの同時点眼の場合、違いはほとんど認められなかった。但し、0.1%ブリモニジンおよび0.003%プラゾシンをウサギの眼に点眼した場合(図4D)、最初の眼内圧の増加がほんの少しで、その後ピークIOPから約40%低下し、α1アンタゴニストの添加が、この用量でのα1活性の減少と同時に、ブリモニジンのこの用量での治療効果を増大したことを示した。以下の請求の範囲は、本発明のこれらのおよびさらなる態様に関するものである。
【技術分野】
【0001】
(原文に記載なし。)
【背景技術】
【0002】
ヒトアドレナリン作動性受容体は内在性膜タンパク質であり、大きく分けて2種類、すなわちαアドレナリン作動性受容体とβアドレナリン作動性受容体とに分類されている。どちらのタイプも、カテコールアミン類であるノルエピネフリンおよびエピネフリンの結合により、末梢交感神経系の作用を媒介する。
【0003】
ノルエピネフリンはアドレナリン作動性神経終末によって産生され、一方、エピネフリンは副腎髄質によって産生される。これらの化合物に対するアドレナリン作動性受容体の結合親和性は、分類の一つの根拠になっている。すなわち、α受容体は、ノルエピネフリンをエピネフリンよりも強く結合し、また、ノルエピネフリンを合成化合物イソプロテレノールよりもはるかに強く結合する傾向を持っている。これらのホルモンの選択的結合親和性はβ受容体では逆転する。多くの組織では、α受容体の活性化によって誘発される平滑筋収縮などの機能的応答は、β受容体結合によって誘発される応答と対立する。
【0004】
その後に、α受容体とβ受容体との機能的差異は、さまざまな動物源および組織源に由来するこれら受容体の薬理学的特徴づけによって、さらに強調され、精密化された。その結果、αアドレナリン作動性受容体とβアドレナリン作動性受容体は、3種類のα1、3種類のα2および3種類のβサブタイプに、さらに細分された。
【0005】
α1受容体とα2受容体との機能的相違が認識されており、これら2つのサブタイプ間で選択的結合を示す化合物が開発されている。即ち、WO92/00073では、α1サブタイプのアドレナリン作動性受容体に選択的に結合するという、テラゾシンのR(+)エナンチオマーの能力が報告された。この化合物のα1/α2選択性は重要であると開示された。その理由として、α2受容体のアゴニスト刺激はエピネフリンおよびノルエピネフリンの分泌を阻害するとされ、一方、α2受容体のアンタゴニスト作用はこれらのホルモンの分泌を増加させるとされた。したがって、フェノキシベンザミンやフェントラミンなどの非選択的αアドレナリン受容体遮断剤の使用は、α2アドレナリン作動性受容体が媒介する血漿カテコールアミン濃度の増加の誘導およびそれに伴う生理学的結果(心拍数の増加および平滑筋収縮)による制約を受けるとされた。
【0006】
さらに、α受容体アゴニストの選択性を測定するための一方法は、Messier ら「High Throughput Assays Of Cloned Adrenergic, Muscarinic, Neurokinin And Neurotrophin Receptors In Living Mammalian Cells(生きた哺乳類細胞におけるクローン化アドレナリン作動性、ムスカリン性、ニューロキニンおよびニューロトロフィン受容体の高スループットアッセイ)」Pharmacol. Toxicol. 76:308-11 (1995) に記載のRSAT(Receptor Selction and Amplification Technology)アッセイを含み、α2受容体用に改造されている。このアッセイでは、コンフルエント細胞の混合集団における受容体含有細胞の選択的増殖をもたらす、接触阻止の受容体媒介性喪失を測定する。細胞数の増加は、96穴形式で容易に測定することができる活性を持つ適当な導入マーカー遺伝子、例えばb−ガラクトシダーゼを使って評価される。Gタンパク質Gqを活性化する受容体はこの応答を引き出す。通常、Giと共役しているα2受容体は、Gq/i52と呼ばれるGi受容体認識ドメインを持つハイブリッドGqタンパク質と同時発現させると、RSAT応答を活性化する。Conklin ら「Substitution Of Three Amino Acids Switches Receptor Specificity Of Gqa To That Of Gia(3アミノ酸の置換によりGqaの受容体特異性がGiaの受容体特異性に転換される)」Nature 363:274-6 (1993) を参照されたい。
【0007】
α−アドレナリン受容体の一般的な背景については、α1/α2のサブクラス分類の基礎、分子生物学、シグナル伝達、アゴニストの構造−活性相関、受容体の機能、およびα−アドレナリン受容体親和性を示す化合物に関する治療的適用が調べられた、Robert R. Ruffolo, Jr., α-Adrenoreceptors:Molecular Biology, Biochemistry and Pharmacology (Progress in Basic and Clinical Pharmacology series、Karger, 1991)が注目される。
【0008】
ヒトの組織に由来するα受容体サブタイプのクローニング、配列決定および発現により、α1アドレナリン受容体は、α1A、α1Bおよびα1Dのサブクラスに分類された。同様に、α2アドレナリン受容体もまた、α2A、α2Bおよびα2Cの受容体に分類されている。それぞれのα2受容体サブタイプはそれぞれ独自の薬理学的特異性および組織特異性を示すようである。
【0009】
クロジニンやデクサメデトミジンのようなα2受容体パンアゴニストは、効果的な鎮痛活性を有し、現在一般的に、中枢神経に直接投与されている(例えばこの目的で鞘内投与または硬膜外投与)。このようなα2受容体パンアゴニストは時として、癌疼痛, 術後疼痛, 神経障害性疼痛, 異痛,ヘルペス後神経痛, 過敏性腸症候群, およびその他の内臓疼痛等の慢性疼痛の処置に用いられてきた。このようなα2 パンアゴニストがある程度の治療活性を有すると示されているその他の状態には、依存症処置 (例えば、麻薬または喫煙の無毒化), 注意欠陥多動性障害(ADHD), トゥレット・シンドローム, 鬱およびその他の 精神障害、高血圧、高眼圧症 (緑内障に関連するような), および痙縮が挙げられる。しかし、これらの薬剤は、鎮痛薬としてまたはこのような他の適応症の処置においては、治療効果と相当のそして時として圧倒的な心臓血管作用および鎮静作用との間の非常に狭い治療濃度域、並びに鎮静効果に関連する他の投薬との有意の相互作用のために、一般的且つ効果的には用いられていない。後者の例としては、例えば, Higuchi H. ら、The Interaction Between Propofol And Clonidine For Loss Of Consciousness, Anesth. Analg. 94 (4):886-91, (2002年4月); Jaffe, R. ら、Adverse interaction between clonidine and verapamil, AnnalsPharmacother. 28 (7- 8): 881-3 (1994年7−8月) (クロニジンとベラパミルの鎮静効果の間の死に至る可能性のある相乗効果を報告)を参照。
【発明の概要】
【発明が解決しようとする課題】
【0010】
治療用量での高い鎮静作用および心臓血管抑制活性のために、FDAが承認したα2受容体アゴニスト(現時点ではα2受容体パンアゴニストのみである)は、全身薬としては一般的に局所適用薬と比べ有用性が低い。例えば、α2 パンアゴニストクロニジンは、例えば、緑内障に起因する高い眼圧(IOP)の眼科処置に用いられてきた。この薬物は液摘の状態で眼に直接投与されるため、一般的な全身作用の多くを最小限とし得る。ところが、そのような薬物の眼への局所適用(眼の血管内への浸透および鼻涙管から鼻への全身作用を許す)であっても、これらの副作用は取り除かれず、治療効果は依然としてそのような効果に限定される。
【課題を解決するための手段】
【0011】
本発明の第1の態様は、哺乳動物(特にヒト)に対する、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与が、その鎮静作用の有意の増加なしに第一成分の治療活性の効力の増大をもたらすという驚くべき発見に関する。即ち、この2成分の同時投与によって、第一成分の治療活性と鎮静作用との間の治療濃度域が広がる。
【0012】
現時点で好ましい態様では、本発明は、疼痛、特に慢性疼痛、神経変性障害;交感神経により増大したストレスに関連する状態および緑内障および高眼圧症を含む眼の状態からなる群から選択される状態の処置のための方法に関する。
【0013】
好ましくは、第一成分に含まれる化合物は、例えば、α2受容体アゴニストおよびノルエピネフリン輸送物質阻害剤(三環式抗鬱薬またはTCA等)からなる群から選択される。α2パンアゴニストおよびTCAの例は、長年、当分野において周知であるが、本明細書に開示する方法は、本願において初めて発表されるものであると本願発明者は信じる。
【0014】
TCA(アミトリプチリン(アミトリル、エラビル)およびノルトリプチリン(アベンチル、パメロール);デシプラミン(ペルトフラン、ノルプラミン);ドキセピン(サイネクアン、アダピン);イミプラミン(ジャナミン、トフラニル);プロトリプチリン(ビバクチル);トリミプラミン(スルモンチール);およびクロモプラミン(アナフラニル)が含まれる)は、最初にそして第一に鬱状態の処置に用いられてきた。しかしながら、これらは、大うつ病エピソード、いわゆる非定型うつ病、パニック障害、対人恐怖、過食症、ナルコレプシー、活動過剰を伴うまたは伴わない注意力欠如障害(ADD)、片頭痛、頭痛および他の様々な慢性疼痛症候群(神経障害性疼痛を含む)、子供の夜尿症および強迫神経症等の、広い範囲の障害において有用であると報告されている。躁うつ病、統合失調症および統合失調性感情障害等の他の主要な精神障害に関して発生する鬱の症状もまた、以下に記載するある特定の警告を伴ってTCAによって処置される。
【0015】
α2受容体パンアゴニストと同様、TCAについての危険な副作用は鎮静作用である。このため、これらは一般に、夜、就寝前に服用するよう処方されている。心臓血管副作用は、これらの服用にも関連している。起立性低血圧、即ち、起立時あるいは姿勢の急激に変えたときのめまいは、一般的である。時として動悸を伴う急速な心拍がしばしば報告されている。この服用は、不健康な心臓に対して有害な影響を及ぼし得る、例えばEKG(心電図)の変化または不整脈(心調律または伝導の乱れ)を生じ、狭心症または心不全または心筋梗塞(心臓発作)を悪化または誘発する。これらの心臓血管心臓血管副作用によって、患者のためにTCAが検討対象から除外されることもある。過剰摂取した場合に三環系抗鬱薬を極めて危険にしているのはこの心臓血管副作用である。過剰摂取は、深刻で、死に至ることもある心臓合併症を引き起こしうる。三環式抗鬱薬は、現在、米国における薬物過剰摂取による死因のトップである。
【0016】
α2パンアゴニストであるクロニジンの硬膜外投与は、局所麻酔薬ブピバカイン−フェンタニルによってもたらされるものと同様に、陣痛時の疼痛除去および運動ブロック(motor block)を効果的にもたらすことが報告されている。Angelo, Reg. Anaesth. & Pain Med. 25: 3(Jan.-Feb. 2000)。しかしながら、これらの効果は、局所麻酔を用いてみられるのと比較して、低血圧等の鎮静および心臓血管副作用をより有意に伴う。デクサメデトミジン等の他のα2パンアゴニストについても同様の結果がみられる。α2アゴニスト(例えば、α2レセプターパンアゴニスト)は、癌疼痛、術後疼痛、ヘルペス後神経痛、過敏性腸症候群およびその他の内臓疼痛、糖尿病性ニューロパシー、筋痙縮にともなう疼痛、複合性局所疼痛症候群(CRPS)、交感神経依存性疼痛、頭痛、抗アロディニア疼痛、炎症性疼痛(例えば、関節炎、に関する)、胃腸の疼痛(例えば過敏性腸症候群(IBS)およびクローン病)、および神経障害性疼痛等の慢性疼痛の処置のために末梢または非末梢的に用いられている。しかしながら、各症例において、そのような化合物による処置は、一方は鎮痛、他方は鎮静の間で狭い治療濃度域によって限定される。
【0017】
同様に、アプラクロニジン(パンアゴニスト)やブリモニジン等のα2アドレナリンアゴニストは、緑内障および高いIOPまたはブドウ膜強膜炎による房水流出の減少を伴うその他の眼症状の処置のための眼科用製剤に用いられている。点眼による薬物の眼への直接投与により薬物の全身濃度が減少し、望ましくない副作用が減るものの、点眼を経由する薬物の吸収または摂取は起こる。したがって、高い眼内圧を最も有効に処置するのに必要な用量は、有害な副作用がそのような濃度でもみられるという事実によってしばしば制限される。
【0018】
対照的に、本発明は、α2活性化剤による処置に応答する状態を有するヒトを含む哺乳動物を処置するための方法であって、その哺乳動物へのα1アドレナリン受容体アンタゴニストおよびα2活性化剤の投与を含み、鎮静または心臓血管抑制の程度が、A2AA単独で有効量を同様に投与した後に存在するよりも少ない、方法を包含するものである。
【0019】
「α2活性化剤」または「A2AA」は、α2アゴニスト、TCA、またはα2アドレナリン受容体の活性化を直接的または間接的にもたらす活性を有するその他の化合物を意味する。とりわけ、但しこれに限定されないが、疼痛(特に慢性疼痛)の処置に用いる場合、本方法は、A2AAを単独の鎮痛剤として含有する組成物と比較してより良好な鎮痛活性(即ち、低いEC50)をもたらす。さらに、そのような同時投与は、A2AAを単独の鎮痛剤として含有する組成物に一般にみられる、薬物濃度と鎮静および血圧低下活性との間の用量−応答の関係に実質的に影響を及ぼさないと考えられる。「EC50」は、測定された最大活性の半分が観察される、与えられた試薬の濃度を意味する。
【0020】
鎮痛薬としてのおよびその他の適用において、A2AAの好ましい投与経路は、末梢または非末梢であり、経口、静脈内、鞘内および硬膜外投与が含まれる。いずれか一成分(または両成分)のその他の可能な投与手段には、鞘内ポンプ、皮下ポンプ、経皮パッチ、静脈内注射、皮下注射、筋肉内注射、局所的クリームまたはジェル、または経口丸薬、またはこのような方法の組み合わせが含まれるがこれに限定されない。A2AAの末梢投与手段は、現在、ある特定の適用においては好ましいものではないが、本願請求項に記載した方法の有用性はそのような場合においても、少なくともその試薬の性質の一部およびそれが投与されるところの適応症に依存して認められる。
【0021】
活性成分に加え、第一および第二成分は、好ましくは、選択した投与様式に適した1以上の製薬的に許容し得る担体を含有する。「製薬的に許容し得る担体」なる語は、液体または固体の増量剤、希釈剤、賦形剤、溶媒または封入材料としての、製薬的に許容し得る材料、組成物またはビークルを意味する。各担体は、その製剤の他の成分と投与様式に適合し、患者にとって有害でないという意味において「許容し得る」ものでなければならない。製薬的に許容し得る担体として供する材料の例としては、糖(ラクトース、グルコースおよびスクロース);(b)デンプン(トウモロコシデンプンおよびジャガイモデンプン);(C)セルロースおよびその誘導体(カルボキシルメチル・セルロース・ナトリウム、エチルセルロースおよびセルロースアセテート);(d)トラガカント粉末;(e)麦芽;(f)ゼラチン;(g)タルク;(h)賦形剤(ココアバターおよび坐薬ワックス);(i)油脂(ピーナッツ油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油および大豆油);(j)グリコール類(プロピレングリコール);(k)ポリオール類(グリセリム、ソルビトール、マンニトールおよびポリエチレングリコール);(1)エステル類(オレイン酸エチルラウリン酸エチル);(m)寒天;(n)緩衝剤(水酸化マグネシウム、水酸化アルミニウム、ホウ酸およびホウ酸ナトリウムおよびリン酸バッファー);(o)アルギン酸;(p)滅菌水;(q)等張食塩水;(r)Ringer溶液;(s)エチルアルコール;(t)リン酸緩衝溶液;および(u)医薬製剤における使用に適当なその他非毒性物質が挙げられる。
【0022】
慢性疼痛の処置の方法において、以下は、本発明の理解を補助するものとして助けとなるであろう。慢性疼痛(癌、関節炎および多くの神経障害性傷害による疼痛)および急性疼痛(組織が切れる、挟まれる、刺される、押し潰される等の即時的な機械的刺激によって生じる疼痛)は、異なる神経線維または神経受容体のいずれか、または慢性的な刺激に対するこれら神経の機能の再構成または変化が大きく関わっている別個の神経学的現象である。急性疼痛の感覚は、主に、通常、機械的、熱的および化学的刺激に対して高い閾値を有するC繊維と呼ばれる求心性神経繊維によって極めて急速に伝達される。慢性疼痛のメカニズムは、完全には分かっていないが、急性の組織損傷が最初の刺激から数分〜数時間以内に、例えば疼痛応答を惹起するのに必要な刺激の規模の局所的減少を含む二次的な症状を生じる。元々の刺激部位から広がった(より大きな)領域において典型的に生じるこの現象は、痛覚過敏と呼ばれる。この二次的な応答によって、機械的または熱的な刺激に対する感覚が大きく増大し得る。
【0023】
A求心性神経線維(AβおよびAδ繊維)はC繊維と比較して低い閾値で刺激され、慢性疼痛の感覚に関与すると考えられる。例えば、通常の状態では、これら繊維の低い閾値の刺激(例えば、軽く触れるまたはくすぐる)では痛みを感じない。しかし、神経損傷後または帯状疱疹等のヘルペスウイルスが介在する状態におけるある特定の状態では、そのような軽く触れるまたは衣服がすれただけでも激しい痛みが生じる。この状態は、異痛と呼ばれ、少なくともAβ求心性神経が部分的に関与していると考えられる。C繊維もまた慢性疼痛の感覚に関与し得るが、そうであっても、神経の持続的発火がやがて現に慢性疼痛の感覚を生じているような類の変化をもたらすことは明かである。本明細書で用いる本明細書で用いる「疼痛」なる語は、急性疼痛と慢性疼痛の両方を包含する。本明細書で用いる「急性疼痛」は即座の一般には高い閾値の、切り傷、圧縮、火傷、または唐辛子の活性成分であるカプサイシンに暴露したときに経験するような化学的刺激等の損傷によってもたらされる疼痛を意味する。本明細書で用いる「慢性疼痛」なる語は、急性疼痛以外の等痛を意味し、神経障害性疼痛、内臓疼痛、線維筋痛、炎症性疼痛、頭痛、筋疼痛および関連痛が挙げられるがこれに限定されない。慢性疼痛は、比較的長く続くことが多く(例えば数ヶ月ないし数年)、連続的あるいは断続的なものである。
【0024】
一態様では、本発明の方法は、本明細書において神経に対する傷害から生じる疼痛を意味する「神経障害性疼痛」を処置するために用いられる。
【0025】
神経障害性疼痛は、小さな皮神経または筋肉または結合組織における小神経が関わる急性の組織傷害によって引き起こされる疼痛である侵害受容性疼痛とは区別することができる。神経障害性疼痛とは対照的に、侵害受容性疼痛は、通常、持続期間が組織修復の期間に限定され、通常、利用可能な鎮痛薬または麻薬によって軽減し得る(Myers, Regional Anesthesia 20: 173-184 (1995))。
【0026】
神経障害性疼痛は、典型的に長期間または慢性的に持続し、最初の組織傷害から数日ないし数ヶ月後に生じる。神経障害性疼痛は、永続的な、自発的な疼痛、並びに通常は痛みを感じない刺激に対する疼痛応答である異痛または針で刺すような通常は些細な痛みを生じるような刺激に対する強化された応答である痛覚過敏を伴う。神経障害性疼痛は一般にオピオイド療法には効きにくい(Myers, supra, 1995)。
【0027】
本発明の方法は、末梢神経、後根神経節、脊椎、脳幹、視床または皮質の外傷、傷害または疾患によって生じた神経障害性疼痛の処置に有用であるが、これに限定されない。本発明の方法によって処置し得る神経障害性疼痛の例としては、ヘルペス後神経痛、除神経後痛および糖尿病性ニューロパシー等の神経痛が挙げられる。本発明の方法は、疼痛の病因に拘わらず神経障害性疼痛の処置に有用であると理解される。非限定的な例として、本発明の方法は、末梢神経障害(神経腫等);神経圧迫;神経挫滅または延伸または不完全な神経処理;または単ニューロパシーまたはポリニューロパシーにより生じた神経障害性疼痛を処置するために用いることができる。非限定的なさらなる例として、本発明の方法は、後根神経節圧迫;脊椎の炎症;打撲、脊椎の腫瘍または半側切断;および脳幹、視床または皮質の腫瘍または外傷等の障害により生じた神経障害性疼痛の処置に有用である。
【0028】
上で示したように、本発明の方法は、単ニューロパシーまたはポリニューロパシーにより生じた神経障害性疼痛の処置に有用である。ニューロパシーは、神経系における機能的な妨害または病理学的な変化であり、感覚または運動神経の異常によって臨床的に特徴付けられる。単ニューロパシーなる語は、単一の末梢神経が冒されることを示し、ポリニューロパシーなる語は、複数の末梢神経が冒されることを示す。ニューロパシーの原因は既知の原因もあればみちの原因もある。既知の原因としては、疾患または中毒症状の合併症(ニューロパシーを引き起こす最も一般的な代謝障害である糖尿病、またはX線照射、虚血または脈管炎等)。本発明の方法により処置し得るポリニューロパシーは、ポリオ後症候群、糖尿病、アルコール、アミロイド、毒素、HIV、甲状腺機能低下症、尿毒症、ビタミン欠乏症、化学療法、2’,3’−ジデオキシシチジン(ddc)治療またはファブリー病によって引き起こされ得るが、これに限定されない。本発明の方法は、原因が既知のまたは未知のこれらのまたは他の慢性的なニューロパシーの慢性疼痛を処置するために用いることができる。
【0029】
本発明の方法は、緊張性頭痛、片頭痛、群発性頭痛、ホルモン性頭痛、反発性頭痛、副鼻腔炎による頭痛、および器質性頭痛を含む頭痛により生じた慢性疼痛を処置するために用いることができる。本発明の方法はさらに、非限定的な例としてのコンピュータでの長時間労働、重い物や重い機械を扱う仕事または長時間立ち続ける仕事、および反復運動障害(RMD)等の活動により生じた慢性疼痛を処置するために用いることができる。RMDは慢性疼痛を引き起こす様々な筋肉症状である。RMDは、過度の労働、無理な姿勢、筋肉疲労、神経または組織の圧迫、活動または動作の延々と続く反復、または腕または腰のひねり等の不自然なまたはぎこちない動作によって起こる摩擦によって引き起こされる。一般的なRMDは、手、腰、肘、肩、首、背中、尻、膝、足、脚および足首に起きるが、手と腕が最も起こりやすい。本発明の方法は、任意のタイプのRMDによって生じる慢性疼痛を処置するために用いることができる。
【0030】
本発明の方法はさらに、椎間板ヘルニアにより生じるような特定のタイプの背痛等の過度の筋肉の緊張;坐骨神経痛および関節痛、並びに炎症性障害により引き起こされる炎症(骨関節炎および慢性関節リウマチ等)を含む炎症による慢性疼痛;組織または関節の挫滅、刺し傷、伸張等の外傷による炎症;感染による炎症(結核);または神経性炎症により生じる慢性疼痛を処置するために用いることができる。非限定的な例示として、本発明の方法は、クローン病、潰瘍性大腸炎、胃炎、過敏性腸疾患等を含む、慢性の胃腸炎症および慢性の内臓疼痛(例えば、癌によるまたは癌治療に伴う(例えば化学療法または放射線療法に伴う)疼痛)を処置するために用いることができる。同様に、本発明の方法は、例えば、関節炎(例えば慢性関節リウマチ、痛風性関節炎、または骨関節炎);脊椎炎;または自己免疫疾患(例えば紅斑性狼瘡)による慢性の炎症性疼痛を処置するために用いることができる。本発明の方法はさらに、慢性の筋疼痛、物質濫用または禁断に関連する、慢性疼痛および原因が既知または未知のその他のタイプの慢性疼痛を処置するために用いることができる。本発明の方法では、第一の薬物と第二の薬物を任意の関係する方法を用いて同時に投与してもよいものと意図する。それらの薬物は、同じ方法または異なる方法を用いて同時または異なる時間に投与することができる。
【0031】
疼痛の処置に加えて、α2アゴニストおよびTCAは神経保護に有用であることが知られている。例えば、本発明の方法にしたがってより副作用の少ない、本明細書に示した第一および第二成分によって処置することができる状態としては、神経変性状態(パーキンソン病パーキンソン病、アルツハイマー病、筋萎縮性側索硬化症および多発性硬化症);卒中等の虚血;てんかん;およびニューロパシー(糖尿病性および虚血性網膜症等)が挙げられるがこれに限定されない。さらに、精神障害(統合失調症および双極性障害等)は現在、神経変性がある程度まで関わっていると考えられている。さらに、本発明の組成物および方法は、現在、付随する鎮静なしに、様々な交感神経増強ストレス関連状態の予防または処置に有用であることが知られている。そのような状態としては、感覚過敏症(例えば、線維筋痛または片頭痛等の頭痛に関連する感覚過敏症)、胃腸疾患(例えば、過敏性腸症候群および消化不良);皮膚病変(例えば乾癬);心臓血管障害;頻脈;末梢血管収縮の障害(レイノー症候群および強皮症);パニック発作;代謝障害(II型糖尿病、インスリン耐性および肥満症);筋収縮の障害(骨格筋収縮の障害、平滑筋収縮の障害、痙縮、および緊張性頭痛に関連する筋収縮の障害を含む);行動障害;および性機能障害が挙げられるが、これに限定されない。一態様では、交感神経増強状態は、交感神経遮断によって軽減し得る任意の疼痛である交感神経依存性疼痛以外の状態である。そのような使用に関連する背景の情報は、本願と同じ2003年9月12日に出願の、米国特許出願60/502840(発明の名称「NOVEL METHODS FOR IDENTIFYING IMPROVED, NON-SEDATING α-2 AGONISTS」に記載されている。
【0032】
α2活性化剤とα1アンタゴニストの同時投与に基づいて、本発明は、A2AA単独で用いてみられる場合と比較してより少ない鎮静作用または心血管副作用でもって神経変性状態の処置における治療的効果をもたらす。神経死を減少または防止することにより、病態および症状における改善が認められる。本明細書で用いる「神経死」なる語は、損傷または異常に応答する死の誘導による神経細胞の破壊を意味する。「神経死」の定義には、胚発生の間に起きるようなまたはアポトーシスを免れない神経を含む自己更新組織(嗅上皮等)における病的でない神経のアポトーシスは含まれない。したがって、神経死なる語は、嗅上皮以外の神経上皮の神経損傷(脳神経などの中枢神経系の神経の損傷およびアポトーシスを受ける神経出ない神経における神経損傷等)を包含する。本明細書で用いる「減少させる」なる語は、神経死に関して用いる場合、神経細胞における死の誘導を予防する、減少させるまたは排除することを意味する。有効量のA2AAおよびα1アンタゴニストの投与による神経死の減少は、鎮静または心血管副作用を最小限にするとともに神経死または機能不全を伴う状態を処置のための有効な方法となり得る。
【0033】
本明細書で用いる「神経変性状態」は、進行性の神経系の機能不全によって特徴付けられる障害を意味する。神経変性状態には、多くのことなる病因を有する中枢神経系または末梢神経系の疾患の異種のグループが含まれる。そのような状態は、遺伝性の、中毒または代謝過程の後に起きるものであり、感染によるものであり得るがこれに限定されない。神経変性状態は、加齢性または慢性であり得る進行性の状態である。そのような状態は、脳の比較的特定の領域または神経の特定の集団の異常によって特徴付けることができる。種々の神経変性状態において冒される特定の細胞集団によって、典型的にその状態の臨床表現型が決定する。特に、神経変性状態は、特定の冒された中枢神経または末梢神経系構造の萎縮に関連し得る。神経変性状態の例示としては、運動ニューロン疾患(ALS)、パーキンソン症候群、広汎性硬化症、筋萎縮性側索硬化症、多発性硬化症、広汎性皮質性小脳萎縮症、レヴィー小体認知症、ピック病、メソリンボ(mesolimbo)皮質性認知症、視床変性、球麻痺、ハンチントン舞踏病、皮質−線条体−脊椎変性、皮質ベースの神経節変性、大脳小脳皮質変性、不全対麻痺を伴う家族性認知症、ポリグルコサン小体疾患、シャイ・ドレーガー症候群、オリーブ橋小脳萎縮症、進行性核上麻痺、変形性筋失調症、ハレルフォルデン・スパッツ病、メージ症候群、家族性振戦、ジル・ド・ラ・ トゥレット(Gilles dela Tourette)症候群、acanthocytic舞踏病、フリードライヒ失調症、Holmes家族性小脳皮質萎縮症、AIDS関連性認知症、Gerstmann-Straussler-Scheinker病、進行性脊髄性筋萎縮、進行性球麻痺、原発性側索硬化、遺伝性筋萎縮症、痙性対麻痺、腓骨筋萎縮、肥厚性間質性ポリニューロパシー、遺伝性多発神経炎性失調、視神経症、糖尿病性網膜症、アルツハイマー病および眼筋麻痺が挙げられるがこれに限定されない。当業者は、これらの他の、軽度の、中程度のまたは重度の神経変性状態の状態は本発明の方法にしたがって処置し得ると理解する。
【0034】
本発明のさらなる態様では、A2AAを含有する第一成分およびα1受容体アンタゴニストを含有する第二成分の同時投与を含んでなる眼症状の処置のための方法を提供する。この態様では、一般に低減された鎮静および心臓血管副作用をもたらすA2AA濃度で治療効果が得られる。
【0035】
本発明の方法を用いて処置することができる眼症状の例としては、開放隅角緑内障を含む緑内障、高眼圧症、黄斑および網膜変性(非滲出性加齢性黄斑変性(ARMD)、滲出性加齢性黄斑変性(ARMD)、脈絡膜血管新生、糖尿病性網膜症、中心性漿液性網脈絡膜症、類嚢胞黄斑浮腫、糖尿病性黄斑浮腫、近視性網膜変性等);炎症性疾患(急性多発性小板状色素上皮症、ベーチェット病、バードショット脈絡網膜炎、感染(梅毒、ライム病、結核、トキソプラズマ症、中間部ブドウ膜炎(扁平部炎)、多発局所性脈絡膜炎、多発性一過性網膜白点症候群(MEWDS)、眼サルコイドーシス、後部強膜炎、蛇行性脈絡膜炎、網膜下線維症およびブドウ膜炎症候群、Vogt-Koyanagi-Harada症候群、点状網膜脈絡膜炎、急性後部多発性小板状色素上皮症、急性網膜色素上皮炎、急性斑状神経網膜症);血管疾患および滲出性疾患(糖尿病性網膜症、網膜中心動脈閉塞症、網膜中心静脈閉塞、播種性血管内凝固障害、網膜静脈分枝閉塞症、高血圧性眼底変化、眼虚血症候群、網膜微細動脈瘤、コーツ病、傍中心窩毛細血管拡張症、半網膜静脈閉塞症、Papillo静脈炎、網膜中心動脈閉塞、網膜動脈分枝閉塞症、頸動脈疾患(CAD)、糖衣状分岐血管炎(Frosted Branch Arterial)、鎌状赤血球網膜症およびその他の異常血色素症、網膜色素線条、家族性滲出性硝子体網膜症);Eales病;外傷、外科手術および環境疾患(交感性眼炎、ブドウ膜炎性網膜疾患、網膜剥離、外傷、網膜レーザー光治療、光凝固、外科手術中の血流低下、放射線網膜症、骨髄移植網膜症);増殖障害(増殖性Vitreal増殖性網膜症および網膜上膜);感染性障害(眼ヒストプラスマ症、眼トクソカリアシス、推定眼ヒストプラスマ症症候群(POHS)、眼内炎、ヒストプラスマ症、HIV感染に関連する網膜疾患、HIV感染に関連するに関連する脈絡膜疾患、HIV感染に関連するブドウ膜炎性疾患、ウイルス性網膜炎、急性網膜壊死、進行性網膜外層壊死、真菌性網膜疾患、眼梅毒、眼結核、片側性瀰漫性亜急性視神経網膜炎、ハエウジ病);遺伝病(網膜色素変性、Accosiated網膜ジストロフィーに関連する全身性障害、先天性静止性夜盲症、錐体ジストロフィー、スタルガルト病および黄色斑眼底、ベスト病、網膜色素上皮のパターンジストロフィー、X線に関連する網膜分離、Sorsby's基底ジストロフィー、良性求心性黄斑、Bietti's crystallineジストロフィー、弾性線維性仮性黄色腫);網膜傷害(黄斑円孔、大きな網膜裂傷等);網膜腫瘍(腫瘍に関連する網膜疾患、前部および後部ぶどう膜メラノーマの先天性肥大、脈絡膜血管腫、脈絡膜骨腫、脈絡膜転移、網膜および網膜色素上皮の複合性過誤腫、網膜芽腫、眼底の血管増殖性腫瘍、網膜星状細胞腫、および眼内リンパ球腫瘍)が挙げられるがこれに限定されない。
【0036】
神経網膜および視神経の虚血は、網膜静脈分枝閉塞の間、網膜中心動脈閉塞の間、網膜中心静脈閉塞の間、硝子体内外科手術中、網膜変性(網膜色素変性、および加齢性黄斑変性等)においてに起こり得る。同時投与されるA2AAおよびアルファlアンタゴニストのような、本発明において用いられる組成物の神経死または機能不全を減少させる能力は、その化合物の存在下および非存在下で神経細胞破壊の観察可能な兆候または症候を解析することによって調べることができる。神経のアポトーシスによる死の開始によって、細胞の機能および形態に対し観察可能な影響が認められ、同様に組織、臓器および機能に障害のあるまたはアポトーシスを起こす神経を含む動物に対しても観察可能な影響が認められる。したがって、神経損傷の指標には、分子における変化の観察可能なパラメーター(アポトーシス誘発遺伝子の発現の増大など)、細胞機能の変化(ミトコンドリア機能の低下など);細胞の形態変化(細胞収縮および水疱形成など);臓器および組織の機能および形態変化(梗塞または他の病変の存在、その重篤度は病変の容積と病変のサイズを含むパラメーターによって評価することができる);動物モデルにおける機能の変化を含む生理学的変化(運動機能の喪失、死亡率の増加および生存率の減少など)および動作上の変化(認知症の発症または記憶の喪失など)が挙げられる。
【0037】
神経損傷の指標の減少は、細胞、組織、臓器または動物の少なくとも2つの状態における神経損傷の指標を比較することにより、細胞、組織、臓器または動物において評価することができる。即ち、神経損傷の指標の減少は、対照の状態に対する相対値で示される。
対照の状態は、例えば、処置前の、処置をしていない、異なる処置を行っている、あるいは正常な動物または当業者が適当であると決定した他の状態における、細胞、組織、臓器または動物である。具体的な態様では、本発明の方法は、本発明の第一成分および第二成分の末梢投与により行われる。本明細書で用いる「末梢投与」または「末梢投与された」なる語は、薬剤を対象の中枢神経系の外へ導入することを意味する。末梢投与には、脊椎または脳への直接投与以外の任意の投与が含まれる。鞘内および硬膜外投与並びに頭蓋注射または移植(本発明の態様の範囲内であるけれども)は、それ自体は「末梢投与」または「末梢投与された」なる語の範囲には入らないことは明かである。
【0038】
末梢投与は、局所的または全身的投与であってよい。局所投与は、投与部位から離れた領域と比較して、局所投与部位へ有意により多くの医薬組成物の送達をもたらす。全身投与は、対象の本質的に末梢神経全体に医薬組成物を送達し、その組成物の特性に依存して中枢神経系への送達が可能である。本発明の方法において有用な末梢投与の経路には、経口投与、局所投与、眼内投与、静脈内注射または他の注射、および移植されたミニポンプまたは他の徐放装置または徐放製剤が含まれるがこれに限定されない。本発明において有用な医薬組成物は、例えば任意の許容し得る形態(錠剤、液体、カプセル、粉末等)での経口投与;静脈内、腹腔内、筋肉内、皮下または非経口注射;経皮拡散または電気泳動により;ドロップ、クリーム、ジェルまたは軟膏等の任意の許容し得る形態での局所的投与;およびミニポンプまたは他の移植された徐放装置または徐放製剤により、末梢投与することができる。
【0039】
本発明はさらに、哺乳動物における疼痛の処置のための方法であって、
a)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分、および
b)α1アドレナリン受容体アンタゴニストを含有する第二成分
の末梢または非末梢の同時投与を含んでなり、最大半量の鎮静をもたらすのに効果的な用量にて、該方法にしたがって最大半量の鎮痛をもたらすのに効果的な第一成分の用量の投与によって引き起こされる鎮静の量が、該第二成分の不在下で該第一成分を投与した同様に罹患した哺乳動物において引き起こされる量よりも少ない、方法に関する。本発明のこの態様およびその他の態様についての投与の様式は、非末梢投与(例えば鞘内または硬膜外など)または全身投与(経口、腹腔内、静脈内、筋肉内、または経皮など)であってよい。
【0040】
疼痛の処置に加え、本発明のさらなる態様は、αレセプターアゴニストが有効であると知られている他の状態の処置に関する。これらには、癌障害(高眼圧症および緑内障など)、交感神経増強ストレス関連状態、神経変性状態;および痙縮が含まれる。αアドレナリン作用によるこれらの状態の処置は、本発明は改善することのできる望ましくない鎮静副作用を生じ得る。
【0041】
この態様では、第一成分および第二成分の局所的デリバリーは眼への第一成分および第二成分の治療物質の送達に好ましいものであろう。局所的眼科製剤は当分野において周知である。
【0042】
眼科用医薬組成物は、活性成分として治療上有効量の第一成分および第二成分を慣用の眼科的に許容し得る賦形剤と共に混合することによって(単一の製剤としてまたは別々の製剤として)および局所的な眼科的使用に適当な単位投与剤型に調製することによって製造することができる。各成分の治療上有効量は典型的には、液体の製剤において約0.0001〜約5%(w/w)、好ましくは約0.001〜約1.0%(w/w)である。
【0043】
眼科的な適用のためには、主な賦形剤として生理食塩液を用いて液剤を調製することが好ましい。そのような眼用液剤のpHは、適当な緩衝系(ホウ酸、トロメタミンまたはリン酸緩衝系など)によって6.2〜7.8に保つことが好ましい。この製剤は、医薬的に許容し得る通常の保存剤、安定剤および界面活性剤をも含有し得る。
【0044】
本発明の眼科的適法の方法および組成物に使用し得る好ましい保存剤としては、塩化ベンザルコニウム、他のポリマー性四級アンモニウム保存剤(PHMBおよびpolyquad(商標)など)、クロロブタノール、チメロサール、酢酸フェニル水銀および硝酸フェニル水銀を包含するが、これらに限定されるものではない。特に好ましいクラスの保存剤は、安定化された二酸化塩素(例えばPurite(商標)安定化二酸化塩素)、安定化されたオキシホウ酸塩等の酸化保存剤である。
【0045】
眼科用製剤において用いるための界面活性剤は、イオン系または非イオン系界面活性剤(Triton(例えば、TritonX-100)、Tween(商標)(例えばポリソルベート40、ポリソルベート80)およびプルロニック(商標)等)が挙げられるがこれに限定されない。
【0046】
同様に、本発明の眼用製剤中に種々の好ましい賦形剤を使用し得る。このような賦形剤には、ポリビニルアルコール、Carbopol(商標)(アリルスクロースで架橋されたプロピオン酸のポリマー)、Pemulin(商標)、ポビドン、ポロキサマー、セルロース誘導体(カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシメチルセルロースなど)が含まれるが、これらに限定されるものではない。
【0047】
眼科用製剤をエマルジョンとして製剤化することも好ましい。その場合、エマルジョンは、水中油エマルジョンかまたは油中水型エマルジョンのいずれであっても良い。このエマルジョンは保存剤および/または賦形剤を含有して良いが、通常少なくとも1種類の界面活性剤および乳化剤を含有する。本エマルジョンはまた界面活性であってよい。1つの好ましい乳化剤は、架橋したポリアクリル酸であるPremulin(商標)である。
【0048】
必要に応じて適宜、張度調整剤を加えても良い。これらには、塩、特に塩化ナトリウム、塩化カリウム、マンイトールおよびグリセリンまたは他の任意の適当な眼科的に許容し得る張度調整剤が挙げられるがこれに限定されない。
【0049】
得られる調製物が眼科的に許容し得るものである限り、様々な緩衝剤およびpH調整手段を用いることができる。したがって、緩衝剤には、酢酸バッファー、クエン酸バッファー、リン酸バッファーおよびホウ酸バッファーが含まれる。必要に応じ、酸または塩基を用いてこれら製剤のpH調整することもできる。
【0050】
同様に、本発明において用いるための眼科的に許容し得る抗酸化剤の一覧には、メタ亜硫酸水素ナトリウム、チオ硫酸ナトリウム、アセチルシステイン、ブチル化ヒドロキシアニソール、およびブチル化ヒドロキシトルエンが含まれるがこれに限定されない。
【0051】
眼科用調製物に含有し得るその他の賦形剤はキレート剤である。好ましいキレート剤は、エデト酸二ナトリウムであるが、他のキレート剤をそれに変えて使用してもそれと組み合わせて使用してもよい。
【0052】
以下の量で成分を用いることができる:
成分 量(% W/W)
各活性成分 約0.001〜5
保存剤 約0−0.10
ビークル 約0−40
張度調整剤 約0−10
緩衝剤 約0.01〜10
pH調整剤 適量を加えpH4.5〜7.5とする
抗酸化剤 適量
界面活性剤 適量
精製水 適量を加えて100%とする
【0053】
本発明の活性化合物の実際の用量は、具体的な化合物、および処置される状態に依存するが、適当な用量は当業者の知識の範囲内で十分選択することができる。
【0054】
本発明の眼科用製剤は、点眼用容器などの定量での適用に適した形態に包装して眼への適用を容易にするのが便利である。点眼適用に滴した容器は、通常、適当な不活性な、非毒性のプラスチック材料で製造し、一般的に約0.5〜15ミリの液体を入れる。
【0055】
保存剤を含まない溶剤は、しばしば、約10、好ましくは約5単位用量(典型的な単位用量は約8滴まで、好ましくは3滴までである)を入れることができる非密閉型の容器にて製剤化する。一滴の容量は通常約20〜35μLである。本発明の方法では、好ましくは、第一成分はα2アゴニストまたはTCA、さらに好ましくはα2アゴニスト、最も好ましくはクロニジン、ブリモニジン、デクサメデトミジン、ミバゼロールおよびチザニジンからなる群から選択されるα2パンアゴニストからなる群から選択される。このような化合物およびその合成は周知である。
【0056】
具体的な態様では、α1アドレナリン受容体アンタゴニストは、プラゾシン、テラゾシンおよび5−メチルウラパジルからなる群から選択される。前者の2種類の化合物はおよびその合成は米国特許第3544836号、同第4026894号にそれぞれ記載されており、後者の化合物は、米国特許第3957786号にその合成が記載されているウラパジルから容易に合成される誘導体である。本願において引用するこれらおよび他の参考文献は、本明細書の一部を構成する。したがって、他のα1受容体アンタゴニストは当分野で周知であり、これら化合物の多くは臨床で認められている。α1アンタゴニストの多くの例が記載されているLagu、26 Drugs of the Future 757−765 (2001)およびForrayら、8 Exp. Opin. Invest. Drugs 2073(1999)を参照。本明細書に照らせば、その他の態様は当業者には自明であろう。
【図面の簡単な説明】
【0057】
【図1】スルプロストン誘発異痛モデルを用いた野生型マウスにおける5−メチルウラパジルの投与ありまたは投与なしでのクロニジンの鎮痛活性を示す。
【図2】上記で詳述したように行った5−メチルウラパジルの投与ありまたは投与なしでのクロニジン鎮静活性の用量応答曲線である。結果は5−メチルウラパジルがクロニジン鎮静に対して効果がないことを示している。
【図3】スルプロストン誘発異痛モデルを用いた野生型マウスにおける5−メチルウラパジルの同時投与ありまたは同時投与なしでのアミトリプチリンの鎮痛活性を示す。
【図4A】プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【図4B】ブリモニジンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【図4C】ブリモニジンおよび0.001%プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【図4D】ブリモニジンおよび0.003%プラゾシンの局所投与による処置ありおよび処置なしのウサギの眼の眼内圧を示す。
【発明を実施するための形態】
【0058】
第一の態様において、本発明は、哺乳動物に対する、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与は、治療効果をもたらすのに必要な第一成分の用量が、該第一成分を単独の治療剤として投与された同じ疾患に罹患した哺乳動物において同様の治療効果をもたらすのに必要な用量に比べて少ない場合、鎮静および/または心臓血管副作用がより少ない、という出願人の驚くべき発見に関する。このことは、鎮痛、眼圧低下活性、神経保護等の治療効果が、ヒトを含む哺乳動物における該第一成分と該第二成分の同時投与によって得られ、第一成分の同様の有効量の投与と比較して、治療用量における鎮静と心臓血管抑制の大きな減少をもたらすということを意味する。現在好ましい態様では、該哺乳動物は慢性疼痛のために鎮痛を必要とする。
【0059】
したがって、本発明のこの態様では、出願人は、その活性が、例えばα2受容体パンアゴニスト(クロニジン、デクサメデトミジン、またはチザニジン)またはTCAのようなα2アドレナリン受容体(A2AA)の直接的または間接的な活性化をもたらす化合物の同時投与が、そのような薬剤の使用時に一般的にみられる鎮静副作用の実質的な増大なしに、A2AAの治療効力の付随する増大とともに、A2AAのα2介在の治療活性の「脱マスキング」または増大をもたらすということを発見した。結果として、選択されたA2AAによる処置についての治療濃度域は、同じ薬剤をαアンタゴニストなしで投与した場合と比較して増大し、ある特定の場合ではA2AAの用量の増加、そして他の場合には少ない副作用で同じ用量の使用が可能になる。好ましくは、A2AAは、α2アドレナリン受容体アゴニストである。
【0060】
本発明は、特定の理論に限定されると解釈されるものではないが、出願人は、α1受容体、または哺乳動物におけるα1A、α1B、およびα1D受容体を含むその1またはそれ以上のサブタイプの刺激によって、α2A、α2Bおよび/またはα2C受容体の刺激から生じる活性(鎮痛、眼圧低下、神経保護またはその他)の減衰されるために本発明が機能すると考える。また、A2AAの大部分は、(α2「選択的」であると記載されているかそうでないかに関わらず)α1刺激活性を欠いているとは決まってはおらず、したがって、この減衰効果を引き起こす本来備わっている十分なα1アゴニスト活性を有していると考えられる。
【0061】
したがって、α1アンタゴニストの同時投与は、α1受容体の刺激による望ましくない拮抗作用をブロックすると考えられる。好ましくは、このα1アンタゴニストは、少なくともα1A受容体アンタゴニスト活性、α1Bアンタゴニスト活性またはα1D受容体アンタゴニスト活性を有する。最も好ましくは、α1受容体アンタゴニストは、少なくともα1A受容体アンタゴニスト活性を有する。そのようなアンタゴニストは、1またはそれ以上のα1受容体サブタイプに対してアンタゴニスト活性を有してもよい。
【0062】
哺乳動物に対するα2アゴニストの処置にみられる、鎮静副作用を増大させないそして時には減少させることに加え、哺乳動物に対するA2AAおよびα1アンタゴニストの同時投与は、A2AAの治療効果を改善し(即ち、「治療濃度域」の拡大)、それによってそうでない場合と比較してより低い用量での処置が可能になる。
【0063】
さらなる態様では、本発明は、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の投与による、哺乳動物における疼痛の処置のための方法に関する。好ましくは、A2AAは、選択されるα2受容体アゴニストおよびノルエピネフリン伝達物質阻害剤(三環系抗鬱薬またはTCA)、より好ましくはα受容体アゴニストからなる群から選択される。あるいは、A2AAは、好ましくは、ブリモニジン、クロニジン、チザニジン、デクサメデトミジンおよびノルエピネフリンおよびMPV-2426(ラドルミジン)からなる群から選択される。
【0064】
さらに、本発明の別の態様は、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の投与による、哺乳動物における高い眼圧の処置のための方法を包含する。この態様では、より好ましい投与経路は、溶液、懸濁液またはエマルジョン等の局所用眼科製剤である。好ましい態様では、A2AAはα2アゴニスト、より好ましくは、α2パンアゴニストである。
【0065】
好ましくは、該第一成分および第二成分は、同時投与のための単一の製剤に含まれるが、該第一成分および第二成分を別個の組成物として同時投与してもよい。本明細書において用いられる、「同時投与(する)」なる語は、該第一成分および第二成分を単一の組成物としてか、または別個の組成物として投与することを包含する。さらに、該成分はそれぞれ、異なる経路および/または同時もしくは若干異なる時間に投与してもよい。
【0066】
さらなる態様では、本発明は、製薬的に許容し得る担体と共に製剤中に治療上有効量の第一成分および第二成分を含有する組成物を包含する。さらなる態様では、該第一成分および第二成分を、二機能性(bifunctional)試薬のように、化学リンカーを用いるA2AA(TCAまたはα2アゴニストから選択される)とα1アンタゴニストとの結合によって結合させてもよい。
【実施例】
【0067】
出願人はここに、本発明の特定の実施態様を証明することを目的として実施例を示す。本発明はこれらの実施例に限定されることを意図しない。
【0068】
実施例1:α2アゴニストおよびα1アンタゴニストの同時投与による慢性疼痛の軽減
慢性疼痛のモデル(特に末梢性ニューロパシー)は、実験動物における片側のL5(場合によりL6)脊髄神経の外科的な結紮によるものである。手術から回復したラットは体重が増え、正常なラットと類似する全体的活動レベルを示す。しかし、これらのラットは、後肢がわずかに外反し、足指が束ねられているという脚の異常を発症する。より重要なことには、手術による影響を受けた側の後肢は、手術後約1週間以内に、低い閾値の機械的刺激(例えばヒトにおいてはかすかな接触感覚を生じさせる刺激)からの痛みに対して感じやすくなっているようである。正常な場合には痛みにならない接触に対するこの感受性は「触覚異痛」と呼ばれており、少なくとも2ヶ月間にわたって続く。応答には、影響を受けた後肢を上げて刺激から逃避すること、脚をなめること、および脚を空中に長く保持することが含まれる。これらの応答はどれも、通常、コントロール群では認められない。
【0069】
外科処置を行う前に、ラットを麻酔する。外科処置を行う部位を剃り、ベタジンまたはノバカインで準備する。第XIII胸椎から仙骨へ下へ切開する。L4〜S2レベルで筋肉組織をL4〜S2のレベルで脊椎(左側)から離す。L6脊椎を探し出して、横突起を、小さい骨鉗子を用いて注意して除き、L4〜L6の脊髄神経を露出させる。L5およびL6の脊髄神経を隔離して、6−0絹糸できつく結紮する。脊髄神経の結紮を行わないことを除いて、同じ手順をコントロールとして右側で行う。
【0070】
完全な止血を確認して、傷を縫合する。少量の抗生物質軟膏を切開領域に塗布して、ラットを、調節された熱温度ランプのもとでの回復用のプラスチックケージに移す。手術後少なくとも7日目の実験当日に、通例、試験群あたり6匹のラットに試験薬物を鞘内、腹腔内注射または経口胃管投与またはこられ1またはそれ以上の組み合わせによって投与する。
【0071】
あるいは、動物を、5μLの50%DMSO中の200ngスルプロストン(プロスタグランジンE2受容体アゴニスト)を鞘内処置することにより誘導することができる。このモデルにおいて、はけで側腹をなでることに対する痛み応答が、スルプロストンの脊髄投与の15分後から開始して35分間の期間にわたって8回スコア化される。Minamiら、57 Pain 217−223 (1994)。スルプロストン処置は単独で16点の測定尺度で12点〜13点の「痛み」のスコアを誘発する。
【0072】
本化合物は、約0.01−5%DMSO中で製剤化し、全身投与については1mL/kg体重または鞘内投与については5μLにて投与する。クロニジンは、0.01〜10.0μgの範囲の鞘内用量で試験する。
【0073】
Chungモデルラットでは、一連の堅さの異なる細い毛のvon Frey hairsを用いて触覚異痛を薬物投与前および30分後に測定する。ラットを底に金網を敷いたプラスチック製のカゴに入れ、約30分間順応させる。von Frey hairsを金網を通して、足を曲げて6〜8秒間その状態を保つのに十分な力でラット後足の足裏中央部に垂直に当てる。与えた力は、計算すると0.41〜15の.1グラムの範囲である。足を急に引っ込めた場合、陽性の反応とする。正常な動物は、この範囲の刺激には応答しないが、外科手術により結紮した足は1〜2グラムの毛に応答して引っ込める。50%の足引っ込み閾値は、Dixon他(Ann. Rev. Pharmacol. Toxicol., 20:441-462(1980))の方法を使用して決定された。薬物後の閾値が薬物前の閾値と比較され、触覚感受性の逆転率(%)が15.1グラムの正常な閾値に基づいて計算された。
【0074】
結果は、クロニジンがラットおよびマウスの両方において用量依存的に鎮痛物質であることを示している。胃痛ラットのChungモデルでは、0.1μgの鞘内用量で鎮痛は認められず、最大鎮痛は1.0μgで観察された。治療濃度域を決定するために、種々のクロニジン用量でラットを試験し、鎮静について分析した。
【0075】
鎮静を試験するために、6匹のオスSprague-Dawleyラットに、種々の用量のクロニジンを鞘内投与する。鎮静は、薬物投与の30分後に、下記のように運動能力をモニターすることによって評価する。ラットを暗い蓋付きチャンバーに入れ、デジコム(digicom)アナライザー(Omnitech Electronic)により、それらの探索行動を5分間にわたって定量化する。この装置では、ラットがX方向およびY方向の32本の光電ビームのアレイを遮る各時間が記録され、クロニジンの代わりに生理食塩水を与えた対照動物と比較した行動の違いを定量化する。
【0076】
このアッセイでは、3.0μgの鞘内投与により中程度の鎮静を示し、10μgの用量にて最大鎮静が誘導された。即ち、鎮痛と鎮静との間に3〜10倍の隔たりがある。
【0077】
スルプロストン誘導アロディニアの動物モデルでは、同様の結果が得られた。
【0078】
図1に示されるように、クロニジンの投与5分前にスルプロストン誘導マウスモデルにα1アンタゴニスト5−メチルウラパジル30μg/kgを腹腔内投与した場合、鎮痛有効量はクロニジン単独使用での応答と比較して10倍低い値にシフトし(図1参照)、鎮静用量は変化無しであった(図2参照)。このように、慢性疼痛のモデルにおいて、クロニジン(α2アゴニスト)とα1アンタゴニストの同時投与により、疼痛と鎮静との間で3〜10倍から30〜100倍に治療濃度域が拡大された。
【0079】
実施例2:TCAおよびα1アンタゴニスト(5−メチルウラパジル)の同時投与による慢性疼痛の軽減
一般に抗鬱薬および鎮痛薬として処方されている三環系抗鬱薬(TCA)は、ノルエピネフリン取込を阻害することによりα2受容体を間接的に刺激する。
【0080】
実験は上記実施例1の記載と同様に行い、スルプロストン誘導アロディニアマウスモデルおよびTCAアミトリプチリンを用いて行った。この化合物およびその合成は本明細書の一部を構成する米国特許第3205264号に記載されている。
【0081】
アミトリプチリンを50%DMSO中、示した用量で溶解し、5−メチルウラパジルの30μg/kgのIP注射または生理食塩水の同様の注射と組み合わせて、各マウスに5μLの容量で注射した。実施例1に記載した筆による刺激をスコア化し、結果を図3に示した。α2アゴニストとα1アンタゴニストの組合せに関して、アミトリプチリンをα1アンタゴニストの組合せにより、最大鎮痛を達成するのに必要な用量の減少がもたらされた(この場合約3倍)。
【0082】
本実施例は、α1アンタゴニストと組み合わせて、α2アドレナリン受容体を直接的または間接的に刺激(例えばノルエピネフリン産生の増大またはノルエピネフリン取込または回転率の制限)することにより、鎮静と治療効果との間の観察された治療濃度域がA2AA単独使用の場合と比較して増大することを示している。
【0083】
実施例3:α2アゴニストおよびα1アンタゴニスト(プラゾシン)の同時投与による慢性疼痛の軽減
スルプロストン誘発モデルについて記載したのと同様の方法を用いた場合、α1アンタゴニストプラゾシンの腹腔内用量(100ng/kg)単独(疼痛スコア=4.8±0.6)またはスルプロストン誘発アロディニアモデル(12.8±0.8)において効果がなかった。
【0084】
スルプロストンおよび種々の用量のクロニジン(50%DMSO5μL中、0.03、0.1および0.4μg)の投与の15分前にプラゾシンをマウスに投与した。α1アンタゴニスト同時投与時の鎮痛に対するクロニジン用量の応答は以下のとおりである:0.03μg用量について13.3±0.9、0.1μg用量について4.8±0.8、0.4μg用量について4.8±0.6。これは、クロニジン単独での鞘内投与と比較してクロニジンについて、クロニジンに対するEC50が約4倍減少する。
【0085】
このように、A2AAおよびα1アンタゴニスト両方の同時投与は、ここでもA2AA単独使用と比較してA2AAの効力の増大をもたらした。この実験では、クロニジンの0.1μg用量はプラゾシンの不在下で鎮痛作用を示さなかった。
【0086】
実施例4:同一の経路で投与されたα2アゴニストおよびα1アンタゴニストによる慢性疼痛の軽減
実験は、クロニジン(種々の用量)と5−メチルウラパジル(1μg鞘内)を50%DMSOビークル中総体積5μLにて鞘内注射により同時投与したことを除いては実施例1と同様に行った。結果は、α1アンタゴニストの腹腔内注射で得られた結果と実質的に同じであった。
【0087】
このように、同じまたは同様の治療効果でもって、2種類の薬剤を一緒にまたは若干異なる時間に、および同一または異なる投与経路で投与することができる。
【0088】
実施例5:α2アゴニストおよびα1アンタゴニストによる高い眼内圧の処置
雄性ニュージーランド白ウサギを用いてノルエピネフリンIOP測定に対する薬物の効果を評価した。ウサギが興奮しないように慎重に扱う。ウサギは片方の手で後足を抱えながら襟首を掴む。ウサギをカゴから出した後、約25μLの希釈したOphthetic(商標)(0.05%)局所麻酔薬を両目に投与する。初めのIOP測定は、両目について行う。右目と左目に3mmHg以上の差があるウサギはこの時点で交換する。
【0089】
両眼についてのゼロ時(T=0)での測定の直後に、各実験動物の片眼を0.1%(w/w)ブリモニジン酒石酸塩の眼科用製剤を無作為に選択した試験する眼の角膜に注入した。非対照のウサギにも0.001%または0.003%(W/W)プラゾシン−HClのいずれかを投与した。他方の眼には薬物を含まないビークルを投与した。この眼科用製剤は、50ppmのPurite(商標)(安定化二酸化塩素)、0.5%カルボキシメチルセルロース、0.6%(w/w)ホウ酸バッファー(pH7.7)および少量の塩(NaCl、KC1、CaCl2、MgCl2)を含有する。処置した(同側)および処置していない(反対側)両方の眼内圧を、7時間に渡り示した時間間隔で測定した。圧の測定は、測定および記録装置としてモデル30クラシック眼圧計を用いた。この装置は、圧平眼圧計によって眼内圧(IOP)の非侵襲的測定を行うものである。
【0090】
図4Aは、0.003%プラゾニンを35μLを同側のウサギの眼に注入した場合に得られる眼内圧を示す。図から分かるように、処置していない眼と比較して初めに眼内圧の小さな低下がみられ、その後2時間にわたり基底値へ戻っている。図4Bは、0.1%ブリモニジン(α2アゴニスト)の点眼の結果を示す。最初の30分間以内に眼内圧の顕著な増大がみられたケースでは、その後眼内圧が約30%低下した。
この効果は、ヒトでは観察されていないものの、IOPの初めの増大はα1アゴニスト活性と関連した。図4Cにみられるように、0.1%ブリモニジンおよび0.001%プラゾシンの同時点眼の場合、違いはほとんど認められなかった。但し、0.1%ブリモニジンおよび0.003%プラゾシンをウサギの眼に点眼した場合(図4D)、最初の眼内圧の増加がほんの少しで、その後ピークIOPから約40%低下し、α1アンタゴニストの添加が、この用量でのα1活性の減少と同時に、ブリモニジンのこの用量での治療効果を増大したことを示した。以下の請求の範囲は、本発明のこれらのおよびさらなる態様に関するものである。
【特許請求の範囲】
【請求項1】
製薬的に許容し得る担体中に
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
を含有する、疼痛、眼症状、神経変性状態、交感神経増大ストレス関連状態からなる群から選択される状態の処置または予防のための組成物。
【請求項2】
前記化合物と前記α1アドレナリン受容体アンタゴニストが共有結合していない、請求項1記載の組成物。
【請求項3】
前記化合物と前記α1アドレナリン受容体アンタゴニストが共有結合している、請求項1記載の組成物。
【請求項4】
哺乳動物における神経変性状態の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による神経保護の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で神経保護の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項5】
前記神経変性状態が、運動ニューロン疾患(ALS)、パーキンソン症候群、広汎性硬化症、筋萎縮性側索硬化症、多発性硬化症、広汎性皮質性小脳萎縮症、レヴィー小体認知症、ピック病、メソリンボ(mesolimbo)皮質性認知症、視床変性、球麻痺、ハンチントン舞踏病、皮質−線条体−脊椎変性、皮質ベースの神経節変性、大脳小脳皮質変性、不全対麻痺を伴う家族性認知症、ポリグルコサン小体疾患、シャイ・ドレーガー症候群、オリーブ橋小脳萎縮症、進行性核上麻痺、変形性筋失調症、ハレルフォルデン・スパッツ病、メージ症候群、家族性振戦、ジル・ド・ラ・ トゥレット(Gilles dela Tourette)症候群、acanthocytic舞踏病、フリードライヒ失調症、Holmes家族性小脳皮質萎縮症、AIDS関連性認知症、Gerstmann-Straussler-Scheinker病、進行性脊髄性筋萎縮、進行性球麻痺、原発性側索硬化、遺伝性筋萎縮症、痙性対麻痺、腓骨筋萎縮、肥厚性間質性ポリニューロパシー、遺伝性多発神経炎性失調、視神経症、糖尿病性網膜症、アルツハイマー病および眼筋麻痺からなる群から選択される請求項4記載の方法。
【請求項6】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項4記載の方法。
【請求項7】
前記α2受容体アゴニストがα2パンアゴニストである請求項6記載の方法。
【請求項8】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項6記載の方法。
【請求項9】
前記α2受容体アゴニストがブリモミジンである請求項8記載の方法。
【請求項10】
前記α2受容体アゴニストがクロニジンである請求項8記載の方法。
【請求項11】
前記α2受容体アゴニストがチアニジンである請求項8記載の方法。
【請求項12】
前記α2受容体アゴニストがデクサメデトミジンである請求項8記載の方法。
【請求項13】
前記α2受容体アゴニストがミバゼロールである請求項8記載の方法。
【請求項14】
前記第一成分が三環系抗鬱薬を含有する請求項4記載の方法。
【請求項15】
前記第一成分がアミトリプチリンを含有する請求項14記載の方法。
【請求項16】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項4記載の方法。
【請求項17】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項16記載の方法。
【請求項18】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項16記載の方法。
【請求項19】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項16記載の方法。
【請求項20】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項16記載の方法。
【請求項21】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項16記載の方法。
【請求項22】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項16記載の方法。
【請求項23】
哺乳動物における眼症状の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による眼症状に対する治療効果の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で眼症状に対する治療効果の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項24】
前記眼症状が、緑内障、高眼圧症、黄斑、非滲出性加齢性黄斑変性(ARMD)、滲出性加齢性黄斑変性(ARMD)、脈絡膜血管新生、糖尿病性網膜症、中心性漿液性網脈絡膜症、類嚢胞黄斑浮腫、糖尿病性黄斑浮腫、近視性網膜変性、急性多発性小板状色素上皮症、ベーチェット病、バードショット脈絡網膜炎、中間部ブドウ膜炎(扁平部炎)、多発局所性脈絡膜炎、多発性一過性網膜白点症候群(MEWDS)、眼サルコイドーシス、後部強膜炎、蛇行性脈絡膜炎、網膜下線維症、ブドウ膜炎症候群、Vogt-Koyanagi-Harada症候群、点状網膜脈絡膜炎、急性後部多発性小板状色素上皮症、急性網膜色素上皮炎、急性斑状神経網膜症、糖尿病性網膜症、網膜中心動脈閉塞症、網膜中心静脈閉塞、播種性血管内凝固障害、網膜静脈分枝閉塞症、高血圧性眼底変化、眼虚血症候群、網膜微細動脈瘤、コーツ病、傍中心窩毛細血管拡張症、半網膜静脈閉塞症、Papillo静脈炎、網膜中心動脈閉塞、網膜動脈分枝閉塞症、頸動脈疾患(CAD)、糖衣状分岐血管炎(Frosted Branch Arterial)、鎌状赤血球網膜症およびその他の異常血色素症、網膜色素線条、家族性滲出性硝子体網膜症、Eales病、交感性眼炎、ブドウ膜炎性網膜疾患、網膜剥離、外傷、網膜レーザー光治療、光凝固、外科手術中の血流低下、放射線網膜症、骨髄移植網膜症、増殖性Vitreal増殖性網膜症、網膜上膜、眼ヒストプラスマ症、眼トクソカリアシス、推定眼ヒストプラスマ症症候群(POHS)、眼内炎、ヒストプラスマ症、HIV感染に関連する網膜疾患、HIV感染に関連するに関連する脈絡膜疾患、HIV感染に関連するブドウ膜炎性疾患、ウイルス性網膜炎、急性網膜壊死、進行性網膜外層壊死、真菌性網膜疾患、眼梅毒、眼結核、片側性瀰漫性亜急性視神経網膜炎、ハエウジ病、網膜色素変性、Accosiated網膜ジストロフィーに関連する全身性障害、先天性静止性夜盲症、錐体ジストロフィー、スタルガルト病および黄色斑眼底、ベスト病、網膜色素上皮のパターンジストロフィー、X線に関連する網膜分離、Sorsby's基底ジストロフィー、良性求心性黄斑、Bietti's crystallineジストロフィー、弾性線維性仮性黄色腫、黄斑円孔、大きな網膜裂傷等、腫瘍に関連する網膜疾患、前部および後部ぶどう膜メラノーマの先天性肥大、脈絡膜血管腫、脈絡膜骨腫、脈絡膜転移、網膜および網膜色素上皮の複合性過誤腫、網膜芽腫、眼底の血管増殖性腫瘍、網膜星状細胞腫、および眼内リンパ球腫瘍からなる群から選択される請求項23記載の方法。
【請求項25】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項23記載の方法。
【請求項26】
前記α2受容体アゴニストがα2パンアゴニストである請求項25記載の方法。
【請求項27】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項25記載の方法。
【請求項28】
前記α2受容体アゴニストがブリモミジンである請求項27記載の方法。
【請求項29】
前記α2受容体アゴニストがクロニジンである請求項27記載の方法。
【請求項30】
前記α2受容体アゴニストがチアニジンである請求項27記載の方法。
【請求項31】
前記α2受容体アゴニストがデクサメデトミジンである請求項27記載の方法。
【請求項32】
前記α2受容体アゴニストがミバゼロールである請求項27記載の方法。
【請求項33】
前記第一成分が三環系抗鬱薬を含有する請求項23記載の方法。
【請求項34】
前記第一成分がアミトリプチリンを含有する請求項33記載の方法。
【請求項35】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項23記載の方法。
【請求項36】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項35記載の方法。
【請求項37】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項35記載の方法。
【請求項38】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項35記載の方法。
【請求項39】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項35記載の方法。
【請求項40】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項35記載の方法。
【請求項41】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項35記載の方法。
【請求項42】
前記眼症状が、緑内障、高眼圧症、黄斑変性、網膜色素変性および糖尿病性網膜症からなる群から選択される請求項23、25、27、33または35のいずれかに記載の方法。
【請求項43】
哺乳動物における交感神経増大ストレス関連状態の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による交感神経増大ストレス関連状態に対する治療効果の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で交感神経増大ストレス関連状態に対する治療効果の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項44】
前記交感神経増大ストレス関連状態が、感覚過敏症、胃腸疾患、乾癬等の皮膚病変、心臓血管障害、頻脈、末梢血管収縮の障害、パニック発作、代謝障害、インスリン耐性および肥満症、平滑筋収縮の障害、痙縮、および緊張性頭痛に関連する筋収縮の障害、行動障害および性機能障害からなる群から選択される請求項23記載の方法。
【請求項45】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項43記載の方法。
【請求項46】
前記α2受容体アゴニストがα2パンアゴニストである請求項45記載の方法。
【請求項47】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項45記載の方法。
【請求項48】
前記α2受容体アゴニストがブリモミジンである請求項47記載の方法。
【請求項49】
前記α2受容体アゴニストがクロニジンである請求項47記載の方法。
【請求項50】
前記α2受容体アゴニストがチアニジンである請求項47記載の方法。
【請求項51】
前記α2受容体アゴニストがデクサメデトミジンである請求項47記載の方法。
【請求項52】
前記α2受容体アゴニストがミバゼロールである請求項47記載の方法。
【請求項53】
前記第一成分が三環系抗鬱薬を含有する請求項43記載の方法。
【請求項54】
前記第一成分がアミトリプチリンを含有する請求項53記載の方法。
【請求項55】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項43記載の方法。
【請求項56】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項55記載の方法。
【請求項57】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項55記載の方法。
【請求項58】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項55記載の方法。
【請求項59】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項55記載の方法。
【請求項60】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項55記載の方法。
【請求項61】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項55記載の方法。
【請求項62】
前記交感神経増大ストレス関連状態が、緊張性頭痛に関連する筋収縮の障害、感覚過敏症、胃腸疾患および乾癬からなる群から選択される請求項43、45、47、53または55のいずれかに記載の方法。
【請求項1】
製薬的に許容し得る担体中に
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
を含有する、疼痛、眼症状、神経変性状態、交感神経増大ストレス関連状態からなる群から選択される状態の処置または予防のための組成物。
【請求項2】
前記化合物と前記α1アドレナリン受容体アンタゴニストが共有結合していない、請求項1記載の組成物。
【請求項3】
前記化合物と前記α1アドレナリン受容体アンタゴニストが共有結合している、請求項1記載の組成物。
【請求項4】
哺乳動物における神経変性状態の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による神経保護の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で神経保護の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項5】
前記神経変性状態が、運動ニューロン疾患(ALS)、パーキンソン症候群、広汎性硬化症、筋萎縮性側索硬化症、多発性硬化症、広汎性皮質性小脳萎縮症、レヴィー小体認知症、ピック病、メソリンボ(mesolimbo)皮質性認知症、視床変性、球麻痺、ハンチントン舞踏病、皮質−線条体−脊椎変性、皮質ベースの神経節変性、大脳小脳皮質変性、不全対麻痺を伴う家族性認知症、ポリグルコサン小体疾患、シャイ・ドレーガー症候群、オリーブ橋小脳萎縮症、進行性核上麻痺、変形性筋失調症、ハレルフォルデン・スパッツ病、メージ症候群、家族性振戦、ジル・ド・ラ・ トゥレット(Gilles dela Tourette)症候群、acanthocytic舞踏病、フリードライヒ失調症、Holmes家族性小脳皮質萎縮症、AIDS関連性認知症、Gerstmann-Straussler-Scheinker病、進行性脊髄性筋萎縮、進行性球麻痺、原発性側索硬化、遺伝性筋萎縮症、痙性対麻痺、腓骨筋萎縮、肥厚性間質性ポリニューロパシー、遺伝性多発神経炎性失調、視神経症、糖尿病性網膜症、アルツハイマー病および眼筋麻痺からなる群から選択される請求項4記載の方法。
【請求項6】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項4記載の方法。
【請求項7】
前記α2受容体アゴニストがα2パンアゴニストである請求項6記載の方法。
【請求項8】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項6記載の方法。
【請求項9】
前記α2受容体アゴニストがブリモミジンである請求項8記載の方法。
【請求項10】
前記α2受容体アゴニストがクロニジンである請求項8記載の方法。
【請求項11】
前記α2受容体アゴニストがチアニジンである請求項8記載の方法。
【請求項12】
前記α2受容体アゴニストがデクサメデトミジンである請求項8記載の方法。
【請求項13】
前記α2受容体アゴニストがミバゼロールである請求項8記載の方法。
【請求項14】
前記第一成分が三環系抗鬱薬を含有する請求項4記載の方法。
【請求項15】
前記第一成分がアミトリプチリンを含有する請求項14記載の方法。
【請求項16】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項4記載の方法。
【請求項17】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項16記載の方法。
【請求項18】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項16記載の方法。
【請求項19】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項16記載の方法。
【請求項20】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項16記載の方法。
【請求項21】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項16記載の方法。
【請求項22】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項16記載の方法。
【請求項23】
哺乳動物における眼症状の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による眼症状に対する治療効果の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で眼症状に対する治療効果の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項24】
前記眼症状が、緑内障、高眼圧症、黄斑、非滲出性加齢性黄斑変性(ARMD)、滲出性加齢性黄斑変性(ARMD)、脈絡膜血管新生、糖尿病性網膜症、中心性漿液性網脈絡膜症、類嚢胞黄斑浮腫、糖尿病性黄斑浮腫、近視性網膜変性、急性多発性小板状色素上皮症、ベーチェット病、バードショット脈絡網膜炎、中間部ブドウ膜炎(扁平部炎)、多発局所性脈絡膜炎、多発性一過性網膜白点症候群(MEWDS)、眼サルコイドーシス、後部強膜炎、蛇行性脈絡膜炎、網膜下線維症、ブドウ膜炎症候群、Vogt-Koyanagi-Harada症候群、点状網膜脈絡膜炎、急性後部多発性小板状色素上皮症、急性網膜色素上皮炎、急性斑状神経網膜症、糖尿病性網膜症、網膜中心動脈閉塞症、網膜中心静脈閉塞、播種性血管内凝固障害、網膜静脈分枝閉塞症、高血圧性眼底変化、眼虚血症候群、網膜微細動脈瘤、コーツ病、傍中心窩毛細血管拡張症、半網膜静脈閉塞症、Papillo静脈炎、網膜中心動脈閉塞、網膜動脈分枝閉塞症、頸動脈疾患(CAD)、糖衣状分岐血管炎(Frosted Branch Arterial)、鎌状赤血球網膜症およびその他の異常血色素症、網膜色素線条、家族性滲出性硝子体網膜症、Eales病、交感性眼炎、ブドウ膜炎性網膜疾患、網膜剥離、外傷、網膜レーザー光治療、光凝固、外科手術中の血流低下、放射線網膜症、骨髄移植網膜症、増殖性Vitreal増殖性網膜症、網膜上膜、眼ヒストプラスマ症、眼トクソカリアシス、推定眼ヒストプラスマ症症候群(POHS)、眼内炎、ヒストプラスマ症、HIV感染に関連する網膜疾患、HIV感染に関連するに関連する脈絡膜疾患、HIV感染に関連するブドウ膜炎性疾患、ウイルス性網膜炎、急性網膜壊死、進行性網膜外層壊死、真菌性網膜疾患、眼梅毒、眼結核、片側性瀰漫性亜急性視神経網膜炎、ハエウジ病、網膜色素変性、Accosiated網膜ジストロフィーに関連する全身性障害、先天性静止性夜盲症、錐体ジストロフィー、スタルガルト病および黄色斑眼底、ベスト病、網膜色素上皮のパターンジストロフィー、X線に関連する網膜分離、Sorsby's基底ジストロフィー、良性求心性黄斑、Bietti's crystallineジストロフィー、弾性線維性仮性黄色腫、黄斑円孔、大きな網膜裂傷等、腫瘍に関連する網膜疾患、前部および後部ぶどう膜メラノーマの先天性肥大、脈絡膜血管腫、脈絡膜骨腫、脈絡膜転移、網膜および網膜色素上皮の複合性過誤腫、網膜芽腫、眼底の血管増殖性腫瘍、網膜星状細胞腫、および眼内リンパ球腫瘍からなる群から選択される請求項23記載の方法。
【請求項25】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項23記載の方法。
【請求項26】
前記α2受容体アゴニストがα2パンアゴニストである請求項25記載の方法。
【請求項27】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項25記載の方法。
【請求項28】
前記α2受容体アゴニストがブリモミジンである請求項27記載の方法。
【請求項29】
前記α2受容体アゴニストがクロニジンである請求項27記載の方法。
【請求項30】
前記α2受容体アゴニストがチアニジンである請求項27記載の方法。
【請求項31】
前記α2受容体アゴニストがデクサメデトミジンである請求項27記載の方法。
【請求項32】
前記α2受容体アゴニストがミバゼロールである請求項27記載の方法。
【請求項33】
前記第一成分が三環系抗鬱薬を含有する請求項23記載の方法。
【請求項34】
前記第一成分がアミトリプチリンを含有する請求項33記載の方法。
【請求項35】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項23記載の方法。
【請求項36】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項35記載の方法。
【請求項37】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項35記載の方法。
【請求項38】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項35記載の方法。
【請求項39】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項35記載の方法。
【請求項40】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項35記載の方法。
【請求項41】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項35記載の方法。
【請求項42】
前記眼症状が、緑内障、高眼圧症、黄斑変性、網膜色素変性および糖尿病性網膜症からなる群から選択される請求項23、25、27、33または35のいずれかに記載の方法。
【請求項43】
哺乳動物における交感神経増大ストレス関連状態の処置または予防のための方法であって、
1)α2アドレナリン受容体の直接的または間接的な活性化をもたらす活性を有する化合物を含有する第一成分;および
2)α1アドレナリン受容体アンタゴニストを含有する第二成分
の同時投与を含んでなり、該方法による交感神経増大ストレス関連状態に対する治療効果の最大半量をもたらすのに有効な第一成分の用量を投与することによってもたらされる鎮静量が、該第二成分の非存在下で交感神経増大ストレス関連状態に対する治療効果の最大半量をもたらすのに有効な用量で該第一成分を投与された同じ疾患に罹患した哺乳動物においてもたらされる鎮静量よりも低い、方法。
【請求項44】
前記交感神経増大ストレス関連状態が、感覚過敏症、胃腸疾患、乾癬等の皮膚病変、心臓血管障害、頻脈、末梢血管収縮の障害、パニック発作、代謝障害、インスリン耐性および肥満症、平滑筋収縮の障害、痙縮、および緊張性頭痛に関連する筋収縮の障害、行動障害および性機能障害からなる群から選択される請求項23記載の方法。
【請求項45】
前記第一成分がα2アドレナリン受容体アゴニストを含有する請求項43記載の方法。
【請求項46】
前記α2受容体アゴニストがα2パンアゴニストである請求項45記載の方法。
【請求項47】
前記α2受容体アゴニストがブリモミジン、クロニジン、チアニジン、デクサメデトミジンおよびミバゼロールからなる群から選択される請求項45記載の方法。
【請求項48】
前記α2受容体アゴニストがブリモミジンである請求項47記載の方法。
【請求項49】
前記α2受容体アゴニストがクロニジンである請求項47記載の方法。
【請求項50】
前記α2受容体アゴニストがチアニジンである請求項47記載の方法。
【請求項51】
前記α2受容体アゴニストがデクサメデトミジンである請求項47記載の方法。
【請求項52】
前記α2受容体アゴニストがミバゼロールである請求項47記載の方法。
【請求項53】
前記第一成分が三環系抗鬱薬を含有する請求項43記載の方法。
【請求項54】
前記第一成分がアミトリプチリンを含有する請求項53記載の方法。
【請求項55】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジル、ウラパジル、プラゾシン、ブナゾシン、テラゾシン、およびドキサゾシンからなる群から選択される請求項43記載の方法。
【請求項56】
前記α1アドレナリン受容体アンタゴニストが5−メチルウラパジルである請求項55記載の方法。
【請求項57】
前記α1アドレナリン受容体アンタゴニストがウラパジルである請求項55記載の方法。
【請求項58】
前記α1アドレナリン受容体アンタゴニストがプラゾシンである請求項55記載の方法。
【請求項59】
前記α1アドレナリン受容体アンタゴニストがテラゾシンである請求項55記載の方法。
【請求項60】
前記α1アドレナリン受容体アンタゴニストがドキサゾシンである請求項55記載の方法。
【請求項61】
前記α1アドレナリン受容体アンタゴニストがブナゾシンである請求項55記載の方法。
【請求項62】
前記交感神経増大ストレス関連状態が、緊張性頭痛に関連する筋収縮の障害、感覚過敏症、胃腸疾患および乾癬からなる群から選択される請求項43、45、47、53または55のいずれかに記載の方法。
【図1】


【図2】
【図3】
【図4A】
【図4B】
【図4C】
【図4D】
【図2】
【図3】
【図4A】
【図4B】
【図4C】
【図4D】
【公開番号】特開2013−10770(P2013−10770A)
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−178435(P2012−178435)
【出願日】平成24年8月10日(2012.8.10)
【分割の表示】特願2006−526116(P2006−526116)の分割
【原出願日】平成16年8月19日(2004.8.19)
【出願人】(591018268)アラーガン、インコーポレイテッド (293)
【氏名又は名称原語表記】ALLERGAN,INCORPORATED
【Fターム(参考)】
【公開日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−178435(P2012−178435)
【出願日】平成24年8月10日(2012.8.10)
【分割の表示】特願2006−526116(P2006−526116)の分割
【原出願日】平成16年8月19日(2004.8.19)
【出願人】(591018268)アラーガン、インコーポレイテッド (293)
【氏名又は名称原語表記】ALLERGAN,INCORPORATED
【Fターム(参考)】
[ Back to top ]