
疼痛の治療で使用するための化合物
本発明は、麻酔薬プロポフォールに由来する化合物に関するものである。該化合物は、疼痛の治療、限定的ではないが特に慢性疼痛および中枢性疼痛感作の治療に有用であり得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、麻酔薬(anaethetic)プロポフォールに由来する化合物に関する。該化合物は、疼痛の治療、限定的ではないが特に慢性疼痛および中枢性疼痛感作の治療に有用である。
【背景技術】
【0002】
有効で安全な疼痛管理は、優先度の高い臨床的要求と世界的に見なされている。しかしこの分野における発展の大部分は、望ましくない副作用および安全上の課題のない、高い効力の製品を供給できていない。アヘン剤はおそらく、依然として最も有効な利用できる治療のままであり、最終的な目標は、アヘン剤の効力を備えていながら、鎮静作用、依存性、胃損傷および全身耐容性の問題を伴わない疼痛管理剤を供給することである。
【0003】
フェノール誘導体が多数の神経調節効果を有し得ることが仮定されてきた。しかし幅広く臨床使用されている唯一のフェノール誘導体は、麻酔薬プロポフォール(2,6−ジ−イソプロピルフェノール)である。
【0004】
麻酔の主な特徴は、意識消失、疼痛性刺激の存在下での不動、およびリコールの不在である。麻酔薬、たとえばプロポフォールは、中枢神経系(CNS)内のγ−アミノ酪酸(GABAA)受容体を活性化することによってその麻酔効果を媒介すると理解されている。
【0005】
これに対して、無痛覚は疼痛の非存在として定義される。他の末梢および/または中枢神経機構の中でも、無痛覚は、脊髄の後角での抑制性シナプス伝達の増強の結果として発生し得る。脊髄における抑制性シナプス後伝達は、主としてグリシン受容体に関係すると理解されている。したがって、グリシン受容体ファミリーは、疼痛抑制を図る治療薬の標的部位に相当する。
【0006】
GABAAおよびグリシン受容体は、どちらもリガンド依存性イオンチャネルスーパーファミリーに属する。これらは5個のサブユニットがイオンチャネルを形成する共通の構造を有する。αサブユニットおよびβサブユニットは、提案された3α:2βのインビボでの化学量論で5量体受容体に組み立てられる。グリシン受容体は、GABAA受容体と同様に、作動薬結合後に塩化物チャネルを開放することによってニューロン発射を抑制する。グリシン受容体は主に中枢神経系の下部領域に見出され、運動リズム発生の管理、脊髄侵害受容反射応答の調整、および感覚シグナルの処理に関与する。
【0007】
新たな改良された鎮痛薬を開発する必要性が存在する。グリシン受容体がこのような鎮痛薬を同定するための良好な標的になるという事実にもかかわらず、これらの受容体を標的とする鎮痛薬は存在しない。したがって本発明者らはこの課題に取り組むことを決め、疼痛管理のための新たな改良された薬物を同定するために、麻酔および無痛覚の根底にある病態生理学的機構の知識を活用した。
【0008】
慢性疼痛は急性疼痛とは大きく異なる。急性疼痛は、侵害刺激を我々に知らせ、それにより我々が損傷を回避および防止するのを補助する有用な早期警告システムと見なすことができる。これに対して慢性疼痛はそれ自体で疾患である。専門家は、慢性疼痛を、正常な状況で疼痛の処理を抑える抑制性ニューロン活性が著しく低下した平衡障害症候群と見なす。慢性的な炎症または神経因性疼痛の治療はなお困難であり、現在、あらゆる症状に作用する単一の治療はない。
【0009】
慢性疼痛で見られるニューロン興奮性の上昇は、末梢から中枢神経系の上部領域への侵害受容シグナルの中継を管理する脊髄の後角表層における、GABA作動性ニューロンおよび/またはグリシン作動性ニューロンに媒介される抑制の消失に関係する。成人後角において、グリシンの急速抑制性シナプス後伝達への寄与が支配的である。グリシン受容体は、主に中枢神経系の下部領域に見出され、運動リズム発生の管理、脊髄侵害受容反射応答の調整、および感覚シグナルの処理に関与する。侵害受容上行性経路および疼痛を調節するその役割によって、グリシン受容体は鎮痛薬および鎮痙薬の潜在的に興味深い標的部位となる。疼痛の情動的成分と関連する前帯状皮質へのグリシン受容体作動薬であるタウリンの微量注射によって、選択的グリシン受容体アンタゴニストのストリキニンによって拮抗できる作用である神経因性疼痛が軽減される。ストリキニン感受性グリシン受容体には4種類のαサブユニットおよび1種類のβサブユニットがあり、α1サブユニットは成人の脊髄および脳幹で広く発現されるが、脳の高次中枢では感覚処理にも関与する。グリシン受容体α3サブユニットは、PGE2誘発受容体ホスホリル化による中枢性炎症性疼痛感作の根底にある標的部位として確認されている。α3サブユニット・ノックアウト・マウスは、そうでなければ急性疼痛に対する正常な応答を伴う炎症性疼痛を発症しない。この現象は、α3の欠如を場合により代償するα1含有グリシン受容体サブユニットが、PGE2シグナル伝達に関与するプロテインキナーゼA(PKA)ホスホリル化部位を有さないという事実によって説明され得る。さらにα3サブユニットのホスホリル化は、神経因性疼痛に必ずしも関与していない。この知識に基づいて、α1サブユニットを含有する主要な成人グリシン受容体アイソフォームを標的とする薬物の開発の必要性が、本発明者らによって確認されている。グリシン受容体の生理学的役割およびその比較的限定された発現(主に脊髄および脳下部領域)を考えると、選択的グリシンモジュレータは、脊髄後角レベルでの抑制を治療的に向上させるために非常に興味深いはずである。
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の第1の態様により、一般式(IA)
【化1】

(式中、R1は
【化2】
(式中、R4、R5、R6、R7およびR8は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
であり、ならびに/または
式中、R2は
【化3】
(式中、R9、R10、R11、R12およびR13は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
である。)
の化合物が提供される。
【0011】
本発明は、疼痛の治療に使用するための一般式(I)
【化4】
(式中、R1、R2およびR3の少なくとも1つは、置換もしくは非置換のアリールもしくはヘテロアリール基であるか、または置換もしくは非置換のアリールもしくはヘテロアリール基を含む。)
の化合物をさらに提供する。
【0012】
ごく最近、脊髄GABAA受容体を標的とすることによって脊髄における抑制を向上させる代わりの手法が発表された。中枢神経系全体でのGABAA受容体の遍在的な発現のために、GABAA受容体は構造的に多様な鎮静−麻酔薬および抗不安薬の治療標的である。そのため、脊髄注射時に存在するベンゾジアゼピンなどのGABA調節剤の鎮痛効果は、全身投与時の中枢神経効果によって無効化される。α2および/またはα3 GABAA受容体サブユニットを含むGABAA受容体が、脊髄レベルでのベンゾジアゼピンの抗侵害受容作用に関与することが以前に示され、慢性疼痛の治療のためのサブタイプ選択的なGABA作動薬の開発が示唆されている。この最新の研究に対して、本発明者らの手法は、GABAA受容体ではなくグリシン受容体を選択的標的として、高次脳領域でのGABAA受容体の刺激に関連する鎮静および依存性を回避しながら、脊髄のレベルでの抑制を増強/回復することによって慢性疼痛を治療するための一般式IおよびIAによる化合物を開発することである。本発明者らの手法は、麻酔薬、アルコールおよびカンナビノイドによって明確に調節されることが知られているグリシン受容体α1サブユニットを標的にしている。しかし、本発明による化合物は、初めて、他の受容体ファミリー(たとえば麻酔薬の場合のGABAA受容体)をより高い親和性で標的とするすべての公知の化合物のように対象とする受容体ファミリーを比較的非特異的に標的とするのではなく、対象とする受容体ファミリーを高い親和性で標的としている。本発明は、疼痛の治療に使用するための一般式I、IAの化合物およびその好ましい実施形態をさらに提供する。
【0013】
本発明のさらなる関連する態様により、以下に示すように一般式(XI)または(XIV)の化合物と、それぞれ一般式(XII)または(XV)の化合物との反応:
【化5】
(式中、R1およびR2はそれぞれ、置換もしくは非置換のアリールもしくはヘテロアリール基であるか、または置換もしくは非置換のアリールもしくはヘテロアリール基を含み、Xは、フッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(X)および(XIII)の化合物を製造する方法が提供される。
【0014】
本発明のさらなる態様は、治療的有効量の本発明の第1の態様による化合物、または一般式IもしくはIAの化合物と、製薬学的に許容可能なビヒクルとを含む医薬組成物を提供する。
【0015】
本発明のまたさらなる態様は、治療的有効量の本発明の第1の態様による化合物、または一般式IもしくはIAの化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0016】
別の態様は、疼痛の治療が必要な対象の疼痛を治療または低減する方法であって、治療的有効量の本発明の第1の態様で定義された化合物、または一般式IもしくはIAの化合物を対象に投与する工程を含む方法を提供する。
【0017】
さらなる態様は、本発明の第1の態様で定義された化合物、または一般式IもしくはIAの化合物を含む薬剤、および疼痛の治療に使用するための薬剤としての本発明の第1の態様による化合物、または一般式IもしくはIAの化合物を提供する。
【0018】
本発明のまたさらなる態様は、疼痛の治療用の薬剤の製造において使用するための、本発明の第1の態様による化合物、または一般式IもしくはIAの化合物を提供する。
【0019】
別の態様は、疼痛の治療のための、本発明の第1の態様による化合物、または一般式IもしくはIAの化合物の使用を提供する。
【0020】
本発明の第2の態様は、有効量の式(VIII)
【化6】
(式中、R14はハロアルキル基である。)
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0021】
本発明は、式(VIII)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0022】
本発明のさらなる態様は、有効量の本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0023】
別の態様は、疼痛の治療が必要な対象の疼痛を治療または低減する方法であって、治療的有効量の本発明の第2の態様で定義された化合物、または一般式VIIIもしくはIXの化合物を対象に投与する工程を含む方法を提供する。
【0024】
さらなる態様は、本発明の第2の態様で定義された化合物、または一般式VIIIもしくはIXの化合物を含む疼痛の治療に使用するための薬剤を提供する。
【0025】
本発明のまたさらなる態様は、疼痛の治療用の薬剤の製造において使用するための本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物を提供する。
【0026】
別の態様は、疼痛の治療のための、本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物の使用を提供する。
【0027】
本発明者らは、脊髄後角内での抑制性シナプス伝達の消失が、炎症または神経損傷後の慢性疼痛の発症において主要な役割を果たすことを認識した。さらに本発明者らは脊髄における抑制性シナプス後伝達が主にグリシンに関係することを認識した。このことにより本発明者らは、ストリキニン感受性グリシン受容体ファミリーが疼痛感作の抑制を目標とする治療剤の標的部位に相当することを理解した。この理解は、Ahmadi et al.(Nature Neuroscience(2001)Vol.5 No.1 p34−40)が行った研究に基づいていた。
【0028】
本発明者らは、ヒドロキシル基に対してパラ位のハロゲンおよびオルト位またはメタ位に1個または2個のメチル基を有するフェノール誘導体を研究することにより、その仮説の検証を進めた。本発明者らの結果は、Haeseler et al.(British Journal of Pharmacology(2005)145,p916−925)によって発表され、ハロゲン化によってグリシン受容体の共活性化または活性化が向上することが確認された。しかしこれらの初期の結果は、フェノール環のメチル基の数または位置がグリシン受容体の共活性化のためのEC50に著しく影響しないことを証明するように思われた。
【0029】
したがって本発明者らは、グリシン受容体を特異的に標的とする改良鎮痛剤を開発し、驚くべきことに、2個以上の炭素原子を含むオルトアルキル基またはメタアルキル基、およびパラハロ基、パラアミノ基またはパラアミド基を包含するプロポフォール類似体が、グリシン受容体の共活性化薬として予想外の効力を示すことを発見した。この研究は、出願人の国際特許出願WO2007/071967の主題である。
【0030】
実施例に記載した化合物は、低いナノモル範囲で最大半量増強効果を示した。このことは、プロポフォールよりも数桁低い濃度を表す。さらに本発明による化合物は、以前に公開した研究で記載したp−メチル誘導体およびp−ハロ誘導体よりも著しく強力であった。このことは、本発明による化合物が理想的な鎮痛薬となり、意識に対する効果(すなわち麻酔効果)はほとんどないか、または無視できるくらいであることを意味するため、大きな利点である。
【0031】
一般式IおよびIAの化合物に関して、前記アリールまたはヘテロアリール基が単環式芳香環または多環式芳香環であることが好ましい。前記アリールまたはヘテロアリール基は、非置換であり得るか、またはハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリール、および置換もしくは非置換ヘテロアリールから成る群より選択される1個以上の置換基によって置換され得る。前記1個以上の置換基のうち1個は、オルト位、メタ位、および/またはパラ位に提供され得る。
【0032】
第1の好ましい実施形態において、R1は、置換または非置換のアリール基またはヘテロアリール基であり、R2およびR3の少なくとも1つは、水素原子である。好ましい化合物は、一般式(II)
【化7】
(式中、R4、R5、R6、R7およびR8はそれぞれ独立して、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、置換または非置換アミン、置換または非置換アミド、置換または非置換アリール、および置換または非置換ヘテロアリールから成る群より選択される。)
を有する。
【0033】
R4およびR8の少なくとも1つは、ハロゲンまたは置換もしくは非置換C1−4アルキルであり得る。ハロゲンは、好ましくはフッ素または塩素であり、アルキル基は、好ましくはメチル、エチル、プロピルまたはブチルである。R8はフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R4は水素であることが好ましい。
【0034】
R5およびR7の少なくとも1つは、好ましくは水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される。ハロゲンはフッ素または塩素であってよく、アルキルはハロ置換C1−4アルキルであり得る。好ましくはR7は、フルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R5は、水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される。
【0035】
R6は、好ましくは水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される。前記ハロゲンはフッ素または塩素であってよく、前記アルキルはハロ置換C1−4アルキルであってよく、前記アルコキシはハロ置換C1−4アルコキシであり得る。好ましくはR6は、−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される。
【0036】
第2の好ましい実施形態において、R2は置換または非置換のアリール基またはヘテロアリール基であり、R1およびR3の少なくとも1つは水素原子である。好ましい化合物は、一般式(III)
【化8】
(式中、R9、R10、R11、R12およびR13はそれぞれ独立して、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、置換または非置換アミン、置換または非置換アミド、置換または非置換アリール、および置換または非置換ヘテロアリールから成る群より選択される。)
を有する。
【0037】
R9およびR13の少なくとも1つは、好ましくはハロゲン、たとえばフッ素もしくは塩素、または置換もしくは非置換のC1−4アルキル、たとえばメチル、エチル、プロピルまたはブチルである。好ましくはR9は、フルオロ、クロロ、およびトリフルオロメチルから成る群より選択され、R13は水素である。
【0038】
好ましくはR10およびR12の少なくとも1つは、水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される。前記ハロゲンは、フッ素または塩素であってよく、前記アルキルはハロ置換C1−4アルキルであり得る。R10はフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R12は水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択されることが好ましい。
【0039】
R11は、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択され得る。ハロゲンは、好ましくはフッ素または塩素であり、アルキルは、好ましくはハロ置換C1−4アルキルであり、アルコキシは好ましくはハロ置換C1−4アルコキシである。好ましくはR6は、−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される。
【0040】
本発明は、以下の好ましい化合物:
【化9】
を提供する。
【0041】
本発明は、疼痛の治療に使用するための上記の好ましい化合物をさらに提供する。
【0042】
本発明の第2の態様は、有効量の式(VIII)
【化10】
(式中、R14はハロアルキル基である)
の化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0043】
本発明は、式(VIII)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0044】
好ましくは前記アルキル基は、C1−4アルキル基である。R14は、2個以上の、好ましくは3個のハロ置換基を含み得る。任意の適切なハロ置換基が選択され得るが、前記ハロ置換基または前記ハロ置換基の少なくとも1つがフッ素であることが好ましい。
【0045】
本発明は、有効量の式(IX)
【化11】
を有する化合物を含む疼痛の治療のための医薬組成物をさらに提供する。
【0046】
本発明は、式(XI)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0047】
本発明の第1の態様によるなおさらに好ましい化合物は、以下の群:
【化12】
より選択できる。
【0048】
化合物が、GABAA受容体よりもストリキニン感受性グリシン受容体に選択性を有することが好ましい。化合物は、GABAA受容体におけるそのEC50よりも低い濃度の、グリシン受容体を共活性化するためのEC50を有し得る。好ましくは化合物は、GABAA受容体におけるEC50の10分の1である、グリシン受容体を共活性化するためのEC50を有する。化合物が、GABAA受容体におけるEC50の少なくとも100分の1である、グリシン受容体を共活性化するためのEC50を有することがさらに好ましい。
【0049】
化合物が、プロポフォールのEC50よりも低い、グリシン受容体を共活性化するためのEC50も有することが望ましい。たとえば化合物は、プロポフォールのEC50の少なくとも10分の1または100分の1である、グリシン受容体を共活性化するためのEC50を有し得る。最も好ましい化合物は、(HEK293細胞中で異種発現されたグリシン受容体に対して測定された)プロポフォールのEC50の1000分の1である、グリシン受容体を共活性化するためのEC50を有する。グリシン受容体を共活性化するためのEC50値を測定する好適な方法は、以下の実施例に開示されている。
【0050】
化合物の効力は、CNSでの麻酔およびPNSでの無痛覚を調節する神経生理学が考慮されるとき、一層驚くべきものである。本発明者らは、一般式IおよびIAによる化合物が、ストリキニン感受性グリシン受容体に正のアロステリックモジュレータとして作用すると考えている。これらの受容体は、過分極によって膜電位を安定化させて、脊髄レベルで主要な抑制原理を構成する塩化物チャネルである。これに対して、密接に関連するGABAA受容体は、CNSにおいて主要な抑制原理を構成する。したがってGABAA作動薬が意識の変化または消失をもたらすのに対して、本発明による化合物は、意識に影響を及ぼさない濃度で末梢レベルにて疼痛を理想的に遮断する。本発明者らは、本発明による化合物が効力を有するのは、該化合物がストリキニン感受性グリシン受容体において正のアロステリックモジュレータとして作用して、それにより後根神経節レベルで求心性神経シグナルを遮断するが、中枢のGABAA受容体には最小限の効果を有するか、全く効果を有さないためであると考えている。したがって当業者は、グリシン受容体作動薬でありGABAA作動性効果を全く有さない鎮痛薬を選ぶことになる。結果として、公知の最も強力なGABAA作動薬であるプロポフォールは、当業者によって鎮痛薬を開発するプラットフォームとして最も好適でない化合物として見なされる。さらに当業者は、Haeseler et al.(上記を参照)によって公開されたデータも検討して、GABA受容体よりもグリシン受容体に対して選択性である化合物を開発する際、アルキル化の性質(Haeselerの論文ではメチル基)が重要でないという見解に達する。したがって本発明者らは、プロポフォール誘導体の鎮痛特性の研究に対して技術的偏見があったと考えている。そのため本発明者らが本発明による化合物と共に見出したグリシン受容体の共活性化の並はずれた増加は、驚くべきことであるだけでなく、可能性が低いと当業者に考えられてきただろう。理論的には、GABAA受容体レベルでの効力がより低い任意の他のフェノール誘導体は、技術水準によれば、より有望な候補として考慮されるべきであった。
【0051】
上述の本発明のさらなる態様に相当する一般式(X)および(XIII)の化合物の製造方法において、R1および/またはR2は、本発明の第1の態様による化合物に含まれる置換基R1およびR2の好ましい特徴のいずれも取り得る。したがって、この方法が本発明の第1の態様による化合物の好ましい実施形態を製造する好ましい合成経路に相当することが認識される。
【0052】
この方法は好ましくは、アリールボロン酸をハロゲン化アリールと反応させて本発明の第1の態様による化合物が生成される鈴木型反応経路を使用する。任意の適切な反応条件が使用され得るが、反応は適切な触媒、たとえば鈴木反応方法で一般に周知であるようにパラジウム触媒を使用して触媒されることが好ましい。そのため反応は、好ましくはパラジウム触媒、たとえばPd(PPh3)4の存在下で行われる。任意の好適な反応温度が使用され得るが、好ましくは室温を超えて、さらに好ましくはおよそ40℃〜50℃、なおさらに好ましくはおよそ60℃〜100℃の温度、最も好ましくはおよそ80℃の温度である。反応は、任意の所望の期間にわたって行われ得る。好ましくは、反応は、およそ6時間超、さらに好ましくはおよそ12時間超、および好ましくはおよそ36時間〜48時間の期間で行われる。好ましい反応期間は、およそ12〜48時間、なおさらに好ましくはおよそ24時間である。
【0053】
ハロゲン化アリール開始物質は、以下の例示的な反応スキームまたは当業者に明らかなようなその派生物に従って製造され得る。
【化13】
ハロゲン化アリール開始物質は次に適切なアリールボロン酸またはヘテロアリールボロン酸と反応されることができ、以下に例示するような所望のプロポフォール類似体を製造する。
【化14A】
【化14B】
【0054】
本発明による化合物、ならびにこのような化合物を含有する医薬組成物および薬剤は、いくつかの状況で鎮痛薬として使用され得る。
【0055】
該化合物は特に、これまで治療が困難であることでよく知られている慢性疼痛状態(たとえば神経因性慢性疼痛および/または炎症後慢性疼痛)を標的とするために有用である。該化合物は特に従来の薬物、たとえばNSAID、アヘン剤誘導体などによる治療が難しい慢性神経因性疼痛を治療するのに有用である。
【0056】
該化合物は、(たとえば傷害後の)急性疼痛を治療するためにも有用である。
【0057】
本発明の化合物が有用なのは、該化合物が単剤療法として使用された場合に、局部麻酔薬および鎮痛薬ならびにNSAIDおよびオピオイドのよく知られた副作用すべてを回避し、同時に、相加作用または相乗作用を目的とした非常に多種多様の併用治療戦略を可能にするためである。
【0058】
疼痛が調節され得る具体的な症状の例は、慢性腰痛、関節炎、癌性疼痛、三叉神経痛、脳卒中および神経因性疼痛を含む。
【0059】
化合物は既存の疼痛を治療するために使用され得るが、たとえば待機手術の前に予防的治療が医療的に必要と見なされるときにも使用され得る。
【0060】
化合物は、単剤療法(すなわち化合物単独の使用)の形で鎮痛薬として使用され得るか、または化合物は疼痛をまた低減する他の治療と併用して投与され得る。好ましい併用療法は、一般式の化合物によって調節される経路とは異なる疼痛処理経路によって疼痛を調節する鎮痛薬と共に該化合物を使用することを含む。このような鎮痛薬は、モルヒネ、パラセタモール、およびNSAIDSを含む。化合物は、グリシン受容体と間接的にのみ相互作用する局部麻酔薬(たとえばリグノカイン)とも有用に併用され得る。
【0061】
本発明の薬剤は、一般式IまたはIAの化合物と製薬学的に許容可能なビヒクルとを含み得る。該ビヒクルは、ビヒクルが投与される対象による耐容性が良好であり、化合物の患部への送達を可能にするビヒクルであるべきことが認識される。
【0062】
本発明の薬剤は、特に化合物が使用される方式に応じて、いくつかの異なる形を取り得る。そのためたとえば、薬剤はフェノール誘導体の塩(たとえばナトリウム塩)の形の化合物を含み得る。このような塩は、粉末形で製造されて、錠剤、カプセル剤、液剤、軟膏、クリーム、ゲル、ハイドロゲル、エアゾール、スプレー、ミセル、経皮パッチ、リポソーム、またはヒトもしくは動物に投与され得るその他の好適な形で包含され得る。
【0063】
または本発明によるフェノール誘導体は、好適な溶媒に溶解されて液剤を形成し得る。溶媒は水性であり得る(たとえばPBSまたは蒸留水)。または溶媒は、アルコール、たとえばエタノール、またはこのような溶媒と水性溶媒との混合物であり得る。
【0064】
該薬剤は局所または局部の治療に使用されることが好ましい。このような薬剤は、患部へ塗布するための液剤として製剤され得る。または液剤は、注射またはエアゾールによる投与のために製剤され得る。
【0065】
化合物は、持続放出デバイスまたは遅延放出デバイス内にも包含される。このようなデバイスは、たとえば皮膚の上または下に挿入され得て、該化合物は数週間、または数カ月にさえわたって放出され得る。このようなデバイスは、特に長期の慢性疼痛を有する患者(たとえば関節炎患者)に有用であり得る。該デバイスは、通常は頻繁な投与を必要とする化合物が使用されるときに特に好都合である。
【0066】
必要な化合物の量は、生物活性および生物学的利用能によって決定され、生物学的利用能は、投与方式、使用される化合物の物理化学特性、および化合物が単剤療法または併用療法のどちらで使用されるかに依存することが認識される。投与頻度は、上述の因子および特に治療される対象内での化合物の半減期によっても影響を受ける。
【0067】
投与される最適投薬量は、当業者によって決定されることができ、使用中の特定の化合物、調製物の強度、投与方式、および軽減を必要とする疼痛の程度によって変化する。対象の年齢、体重、性別、食事、および投与時間を含む、治療される特定の対象に応じた追加の因子によって投薬量を調整する必要が生じる。
【0068】
公知の手順、たとえば医薬業界で従来使用されている手順(たとえばインビボ実験、臨床試験など)を使用して、組成物の具体的な製剤および正確な治療体制(たとえば化合物の1日用量および投与頻度)が確立され得る。
【0069】
概して、組織濃度が使用される化合物のEC50付近であるように、標的部位に化合物を送達するのに有効である用量が投与されるべきである。1日用量は単回投与として(たとえば1日1回の注射として)投与され得る。または使用される化合物は、1日の間に2回以上の投与を必要とし得る。一例として、慢性腰痛を治療するための好ましい化合物は、注射用液剤または軟膏として1日2回(または疼痛の重症度に応じてそれ以上の回数)の用量として投与され得る。治療を受ける患者は、起床時に第1回の用量を、次に2回目の用量を夕方に(2回用量体制の場合)、またはその後3〜4時間の間隔で投与され得る。あるいは、持続放出デバイスが使用されて、反復用量を投与する必要なしに患者に最適な用量が投与され得る。
【0070】
本発明は、治療的有効量の本発明の化合物と製薬学的に許容可能なビヒクルとを含む医薬組成物をさらに提供する。一実施形態において、本発明によるフェノール誘導体の塩の量は、経腸(経口、経直腸)投与のための各用量単位で約10μg/kg体重〜10mg/kg体重の量である。別の実施形態において、この量は、非経口(静脈内/くも膜下腔内または硬膜外)投与のための各用量単位で約1μg/kg体重〜1mg/kg体重である。
【0071】
さらなる実施形態において、ビヒクルは液体であり、組成物は液剤である。非経口投与のための有用な液体液剤は、0.001〜1重量%の式IまたはIAのフェノールを含み得る。別の実施形態において、ビヒクルは固体であり、組成物は錠剤である。さらなる実施形態において、ビヒクルはゲルであり、組成物は局所塗布用である。
【0072】
本発明において、「治療的有効量」は、化合物が有効である有痛性症状に罹患している対象に投与されるとき、疼痛の減少、寛解、または後退を引き起こす化合物、薬剤または組成物の任意の量である。
【0073】
「対象」は、脊椎動物、哺乳動物、家畜またはヒトである。
【0074】
本発明の実施において、「製薬学的に許容可能なビヒクル」は、医薬組成物を製剤するのに有用な当業者に公知の任意の生理学的ビヒクルである。一実施形態において、製薬学的ビヒクルは液体であってよく、医薬組成物は液剤の形である。別の実施形態において、製薬学的に許容可能なビヒクルは固体であり、組成物は粉剤または錠剤の形である。さらなる実施形態において、製薬学的ビヒクルはゲルであり、組成物は坐剤またはクリームの形である。さらなる実施形態において、化合物または組成物は、製薬学的に許容可能な経皮パッチの一部として製剤され得る。
【0075】
固体ビヒクルは、潤滑剤、可溶化剤、懸濁化剤、充填剤、流動促進剤、圧縮助剤、結合剤または錠剤崩壊剤としても作用し得る1つ以上の物質を含むことができる。固体ビヒクルは、カプセル化材料であることもできる。粉剤において、ビヒクルは、微粉化された活性成分と混合された微粉化固体である。錠剤において、活性成分は、必要な圧縮特性を有するビヒクルと好適な比で混合されて、所望の形状およびサイズに圧縮される。粉剤および錠剤は、好ましくは99%までの活性成分を含有する。好適な固体ビヒクルはたとえば、リン酸カルシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、デキストリン、デンプン、ゼラチン、セルロース、ポリビニルピロリドン、低融点ろう、およびイオン交換樹脂を含む。
【0076】
液体ビヒクルは、液剤、懸濁剤、乳剤などを調製するのに使用される。フェノール誘導体は、製薬学的に許容可能な液体ビヒクル、たとえば水、エタノール、有機溶媒もしくはその混合物、または製薬学的に許容可能な油もしくは脂肪に、溶解または懸濁させることができる。
【0077】
滅菌液剤または懸濁剤である液体医薬組成物は、たとえば筋肉内、くも膜下腔内、硬膜外、腹腔内または皮下注射によって利用できる。滅菌液剤は静脈内投与することもできる。化合物は、滅菌水、生理食塩水、または他の適切な滅菌注射用媒体を使用して、投与時に溶解または懸濁され得る滅菌固体組成物として調製され得る。
【0078】
本発明はここで、以下の実施例によって、および以下の図面を参照してさらに説明される。
【図面の簡単な説明】
【0079】
【図1】対数目盛に4−(p−tert−ブチルフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図2】対数目盛に4−(p−クロロフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図3】対数目盛に4−(p−ビフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図4】CK−I−Iについての、それぞれ8mg/kgおよび1mg/kgの経口投与(破線)および静脈内投与(実線)後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。
【図5】CK−2−3についての、それぞれ20mg/kgおよび2mg/kg静脈内投与後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。血漿内において化合物は経口投与では検出されなかった。
【図6】CK−2−9についての、それぞれ20mg/kgおよび2mg/kgの経口投与(破線)および静脈内投与(実線)後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。
【発明を実施するための形態】
【実施例】
【0080】
本発明による多数のアリール置換プロポフォール類似体は、各種のハロ置換プロポフォール化合物から開始して以下で説明するように合成された。製造されたアリール置換類似体を、次にエタノール中の溶解度について試験を行い、その後代表的な選択された類似体に試験を行ってそのEC50値およびHill係数を決定した。
【0081】
[化合物の合成]
(4−置換プロポフォール類似体の合成)
4−ブロモプロポフォール:
【化15】
プロポフォール(1g、5.6mmol)を氷酢酸(25ml)に溶解させた。溶液の変色が停止するまで、臭素(水溶液)を撹拌しながら滴加した。混合物を室温にて1時間撹拌したままにした。反応混合物を次に水(50ml)にゆっくり注いだ。得られた赤色油を酢酸エチルによって抽出して、水および塩水で洗浄した。粗生成物を硫酸マグネシウムで脱水して、溶媒を除去して褐色油を得て、これをヘキサン中2%のDCMを溶出液として用いてフラッシュクロマトグラフィーで精製した。薄黄色油1.35gを純4−ブロモ−プロポフォール(93%)として回収した。1H NMR 400MHz d7.1(s,1H)、4.75(s,1H)、3.15(m,2H)、1.3(d,12H).13C NMR d149.4,136.48,126.89,113.74,27.69,22.95.HRMS(El)C12H17OBr[M+H]+理論値258.1679、実測値258.1703。分析C12H17OBr理論値C:56.04% H:6.66% 実測値C:55.99% H:6.63%。
【0082】
4−ニトロ−プロポフォール:
【化16】
プロポフォール(1.78g、10mmol)をAcOH:DCMが15:10mLの混合物に溶解させた。0℃の硝酸(1mLを10mLのDCMに溶解)を滴加した。添加時に黄色からオレンジ色〜赤色への色変化が観察された。水(20mL)を添加して、反応混合物をDCM(3×20mL)によって抽出した。得られた赤色溶液を硫酸ナトリウムで脱水し、溶媒を除去して暗赤色粗固体を得た。粗生成物をジエチルエーテルおよびヘキサンからの再結晶によって精製し、薄黄色結晶を2回の回収で得た。1.803g(84%).1H NMR δ7.71(s,2H)、4.77(s,1H)、3.1(m,2H)、1.3(d,12H).13C NMR δ155.21,140.44,138.41,120.20,27.68,23.01.HRMS(El)C12H20N2O3[M+NH4]+理論値240.2735実測値240.2731。分析C12H16NO3理論値C:64.55% H:7.67% N:6.27%
実測値C:64.53% H:7.66% N:6.25%
【0083】
4−アミノ−プロポフォール:
【化17】
4−ニトロプロポフォール(0.368g、1.6mmol)をEtOH(2.8mL)および濃HCl(7.5mL)に溶解させた。過剰量のスズ細粒(1.4g)を添加して、反応混合物を還流まで加熱した。薄黄色から無色への色変化が観察された。1時間後、反応混合物をセライトパッドで濾過した。溶媒を減圧下で除去して、得られた残渣を水に再溶解させた(50ml)。溶液が万能試験紙によって塩基性(pH12〜15)となるまで、水酸化ナトリウム水溶液を滴加した。溶液を次にDCM(3×25mL)で抽出した。有機抽出物を合せ、塩水で洗浄し、硫酸ナトリウムで脱水して、溶媒を除去し、分析的に純粋な(98%)紫色油320mgを得た。1H NMR δ6.45(s,2H)、4.75(s,1H)3.4(br s,2H)3.1(m,2H)1.3(d,12H).13C NMR δ140.45,138.72,136.48,112.02,27.70,23.01.HRMS(Cl)C12H22N2O[M+NH4]+理論値210.3093実測値210.3097。分析C12H18NO理論値C:74.56% H:9.91% N:7.24%実測値C:74.36% H:9.95% N:7.23%。
【0084】
4−アミノプロポフォール−ヒドロクロリド:
【化18】
4−アミノプロポフォール(310mg、1.6mmol)をEt2O(100mL)に溶解させた。1,4−ジオキサン(25mL)に溶解させた濃HCl(5mL)を撹拌しながら滴加した。1時間後、混合物を静置して2層とし、下層を回収して、溶媒を減圧下で除去してピンク色結晶性固体(250mg、67%)として、さらに分析せずに使用する。
【0085】
4−トリフルオロアセトアミド−プロポフォール:
【化19】
4−アミノプロポフォール(200mg、1.04mmol)をEtOAc(40mL)に溶解させた。ピリジン(0.25mL、3mmol)、続いて無水トリフルオロ酢酸(0.2mL、1mmol)を添加した。反応混合物を60℃まで30分間加熱した。冷却時に溶媒を減圧下で除去して、残渣をDCM(50mL)に再溶解させ、水および塩水で連続して洗浄し、硫酸ナトリウムで脱水した。溶媒の除去後、4−トリフロオロアセトアミドプロポフォールを薄ピンク色結晶性固体(205mg、75%)として回収した。1H NMR δ7.16(s,2H)、6.3(br s,1H)、5.0(1H,s)、3.2(m,2H)、1.3(d,J=6.9Hz,12H).13C NMR δ155.18,144.28,138.05,130.51,117.03,115.85,27.78,23.12.HRMS(El):C14H18F3NO2[M+H]+理論値306.2751、実測値306.2747。分析C14H18F3NO2理論値C:58.12,H:6.27,N:4.84、実測値C:58.11、H:6.25,N:4.83。
【0086】
4−クロロプロポフォール:
【化20】
プロポフォール(710mg、4.0mmol)をDCMに溶解させた。塩化スルフリル(0.675g、5mmol)を滴加して、次に反応混合物を4時間還流させた。溶媒を減圧下で除去して、粗生成物を褐色油として得て、これを蒸留によって精製して、4−クロロプロポフォールを薄オレンジ色油(750mg、89%)として得た。沸点1mmHgにて94〜97℃。1H NMR δ7.0(s,2H)、5.0(s,1H)、3.1(m,2H)、1.2(d,J=6.9Hz,2H).13C NMR δ148.92,139.12,126.12,124.01,27.37,22.68.HRMS(El)C12H17OCl[M+H]+理論値166.9545、実測値166.9572.分析C12H17OCl理論値C:67.76,H:8.06、実測値C:67.70,H:8.04。
【0087】
(アリール−置換プロポフォール類似体の合成)
4−(パラ−クロロフェニル)−プロポフォール:
【化21】
4−ブロモ−プロポフォール(200mg、0.77mmol)、パラ−クロロフェニルボロン酸(156mg、1mmol、1.3当量)、テトラキス(トリフェニルホスフィン)パラジウム(0)(26mg、3mol%)、炭酸ナトリウム(2N水溶液3.5mL)およびジメトキシエタン(10mL)の懸濁液を24時間、95℃にて撹拌した。懸濁液を冷却し、セライトで濾過して、EtOAcに溶解し、水および塩水で連続して洗浄して、硫酸ナトリウムで脱水し、溶媒を真空中で除去した。ヘキサン中10%のEtOAcで溶出させた、カラムクロマトグラフィーにより得られた残渣の精製によって、純生成物を白色固体(72%)として得た。1H NMR δ7.5−7.2(m,6H)、4.8(s,1H)、3.2(m,2H)、1.3(d,J=6.85Hz,12H).13C NMR δ147.59,138.23,134.54,132.89,129.11,128.50.122.71,27.71,23.144.HRMS:(El)−C18H20OCl[M+H]−理論値287.1203、実測値287.1206。分析C18H20OCl理論値C:74.86,H:7.33、実測値C:74.88,H:7.32
【0088】
4−(パラ−カルバモイルフェニル)−プロポフォール:
【化22】
4−ブロモプロポフォール(200mg、0.77mmol)、p−カルバモイルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色固体(212mg、93%)として得た。1H NMR δ8.00−7.7(m,4H)、7.2(s,2H)、6.3(s,2H)5.1(s,1H)3.2(m,2H)、1.2(d,J=6.85,12H).13C NMR δ168.13,147.55,139.90,138.18,133.07,128.05,127.94,126.53,26.71,23.68.HRMS(EI)−C19H23NO2[M+H]−理論値298.4567、実測値298.4570。分析C19H23NO2理論値C:76.73,H:7.80,N:4.71、実測値C:76.70,H:7.76,N:4.68。
【0089】
4−(p−tert−ブチルフェニル)−プロポフォール:
【化23】
4−ブロモプロポフォール(300mg、1.12mmol)、p−tert−ブチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を黄色油(300mg、83%)として得た。1H NMR δ74−7.2(m,6H)、4.9(s,1H)、3.15(m,2H)、1.4(s,9H)、1.2ppm(d,J=6.85,12H).13C NMR δ149.00,147.63,138.21,133.38,128.49,127.58,126.45,125.30,40.67,31.39,26.70,23.65.HRMS(El)C22H30O[M+H]+理論値310.4690、実測値310.4692。分析C22H30O理論値C:85.11,H:9.74、実測値C:85.10,9.72。
【0090】
4−(パラ−フルオロフェニル)−プロポフォール:
【化24】
4−ブロモプロポフォール(300mg、1.12mmol)、p−フルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色油(210mg、69%)として得た。1H NMR δ7.6(m,2H)、7.2(m,4H)、4.85(s,1H)、3.2(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ150.39,141.13,134.55,132.79,128.52,122.85,121.50,100.00,27.77,23.14.HRMS:(El)C18H21FO[M+H]+理論値273.3598、実測値273.3593。分析C18H21FO理論値C:79.38,H:7.77、実測値C:79.30,H:7.76。
【0091】
4−(p−トリフルオロメトキシフェニル)−プロポフォール:
【化25】
4−ブロモプロポフォール(200mg、0.77mmol)、p−トリフルオロメトキシフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色固体(205mg、79%)として得た。1H NMR δ7.6−7.5(m,2H)、7.3−7.0(m,4H)、4.8(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ159.62,150.05,134.46,128.79,128.71,122.74,115.88,115.67,99.99,27.77,23.16.HRMS(El)−C19H21F3O2[M+H]−理論値339.3568、実測値339.3564。分析C19H21F3O2理論値C:67.44,H:6.26、実測値C:67.40,H:6.23。
【0092】
4−(p−ビフェニル)−プロポフォール:
【化26】
4−ブロモプロポフォール(200mg、0.77mmol)、p−ビフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物をオフホワイト色固体(100mg、39%)として得た。1H NMR δ7.6(s,4H)、7.5(m,2H)、7.2−7.3(m,3H)、7.0(s,2H)、4.9(s,1H)、3.1(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ147.53,138.18,136.5,135.40,135.31,129.41,128.43,128.0,127.3,126.3,26.73,23.73.HRMS:(El)C24H26O[M+H]+理論値331.4673、実測値331.4663。分析C24H26O理論値C:87.23,H:7.93,O:4.84、実測値C:87.18,H:7.76,0:4.79
【0093】
p−(4−フルオロ,3−クロロフェニル)−プロポフォール:
【化27】
4−ブロモプロポフォール(200mg、0.77mmol)、4−フルオロ,3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(82%)を得た。1H NMR δ7.35−7.25(m,3H)、7.0−6.9(m,3H)、4.85(s,1H)、3.2(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ161.81,147.58,138.31,133.47,129.56,128.59,127.39,126.50,121.28,117.30,25.95,22.89.HRMS:(El)C18H20ClFO[M+H]+理論値307.8139、実測値307.8090。分析C18H20CIFO理論値C:70.47,H:6.57、実測値C:70.50,H:6.63。
【0094】
パラ−(2,3−ジクロロフェニル)−プロポフォール:
【化28】
4−ブロモプロポフォール(200mg、0.77mmol)、2,3−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として白色固体(170mg、68%)を得た。1H NMR δ7.25−7.15(m,3H)、7.0(s,2H)4.85(s,1H)、3.20(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ147.58,138.78,138.17,134.01,131.63,129.25,128.45,127.43,126.49,26.68,23.72.HRMS:(El)C18H20Cl2O[M+H]+理論値324.2679、実測値324.2674。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.93,H:6.30。
【0095】
パラ−(2,3−ジフルオロフェニル)−プロポフォール:
【化29】
4−ブロモプロポフォール(300mg、1.12mmol)、2,3−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として白色固体(290mg、89%)を得た。1H NMR δ7.15−6.9(m,5H)、4.85(s,1H)、3.15(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ151.01,147.78,145.58,138.19,133.07,128.53,126.48,125,10,116.03,26.60,23.74.HRMS:(El)C18H20F2O[M+H]+理論値291.3589、実測値291.3591。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.51,H:6.99。
【0096】
パラ−(2−フルオロ,3−クロロフェニル)−プロポフォール:
【化30】
4−ブロモプロポフォール(520mg、1.94mmol)、2−フルオロ,3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色油(520mg、95%)を得た。1H NMR δ7.3−7.2(m,2H)、7.1−7.0(m,3H)4.8(s,1H)、3.1(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ161.42,147.59,138.24,132.66,129.43,128.46,127.58,126.54,126.28,121.30,26.74,23.68.HRMS:(El)C18H20ClFO[M+H]+理論値307.8139、実測値307.8126。分析C18H20ClFO理論値C:70.47,H:6.57、実測値C:70.43,H:6.54。
【0097】
パラ−(3,5−ジフルオロフェニル)−プロポフォール:
【化31】
4−ブロモプロポフォール(520mg、1.94mmol)、3,5−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として暗赤色油(500mg、89%)を得た。1H NMR δ7.3−7.2(m,2H)、7.0−6.9(m,4H)6.6(m,1H)、4.9(s,1H)、3.15(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ165.00,147.58,139.80,138.16,128.54,126.48,120.01,103.63,26.59,23.64.HRMS:(El)C18H20F2O[M+H]+理論値291.3589、実測値291.3580。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.40,H:6.88。
【0098】
パラ−(3,5−ジクロロフェニル)−プロポフォール:
【化32】
4−ブロモプロポフォール(200mg、0.77mmol)、3,5−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(200mg、80%)を得た。1H NMR δ7.4(s,2H)、7.2(s,1H)7.0(s,1H)、4.8(s,1H)、3.1(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ147.62,139.34,138.15,136.21,129.30,126.48,125.87,26.63,23.74.HRMS:(El)C18H20Cl2O[M+H]+理論値324.2679、実測値324.2649。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.80,H:6.28。
【0099】
パラ−(3,5−ジ−トリフルオロメチルフェニル)−プロポフォール:
【化33】
4−ブロモプロポフォール(300mg、1.12mmol)、3,5−ジ−トリフルオロメチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(350mg、80%)を得た。1H NMR δ7.7−7.6(m,3H)7.0(s,1H)、4.8(s,1H)、3.15(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ148.00,138.75,137.21,131.92,130.33,128.83,126.72,125.03,122.09,27.10,23.85.HRMS:(El)C20H20F6O[M+H]+理論値391.3729、実測値391.3721。分析C20H20F6O理論値C:61.54,H:5.16、実測値C:61.60,H:5.19。
【0100】
パラ−(3,4−ジクロロフェニル)−プロポフォール:
【化34】
4−ブロモプロポフォール(200mg、0.77mmol)、3,4−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(120mg、48%)を得た。1H NMR δ7.4−7.3(m,3H)7.15(m,1H)、4.9(s,1H)、3.25(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ148.01,139.05,136.31,133.95,130.78,129.15,128.56,127.38,126.41,26.79,23.69.C18H20Cl2O[M+H]+理論値324.2679、実測値324.2656。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.79,H:6.23。
【0101】
パラ−(3−フルオロ,4−クロロフェニル)−プロポフォール:
【化35】
4−ブロモプロポフォール(520mg、1.94mmol)、3−フルオロ,4−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色固体(380mg、63%)を得た。1H NMR δ7.3−7.1(m,5H)、5.0(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ163.28,148.00,138.24,136.19,131.02,128.55,125.99,124.47,119.73,117.66,26.80,23.67.C18H20ClFO[M+H]+理論値307.8079、実測値307.8099。分析C18H20ClFO理論値C:70.47,H:6.57、実測値C:70.43,H:6.51。
【0102】
パラ−(3−フルオロフェニル)−プロポフォール:
【化36】
4−ブロモプロポフォール(200mg、0.77mmol)、3−フルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄ピンク色固体(160mg、76%)を得た。1H NMR δ7.25−6.9(m,6H)、5.1(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ163.44,147.66,138.17,131.05,128.47,126.73,123.44,116.23,114.01,27.15,22.99.C18H21FO[M+H]+理論値273.3688、実測値273.3680。分析C18H21FO理論値C:79.38,H:7.77、実測値C:79.43,H:7.81。
【0103】
パラ−(3−クロロフェニル)−プロポフォール:
【化37】
4−ブロモプロポフォール(520mg、1.94mmol)、3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色(350mg、62%)を得た。1H NMR δ7.5(s,1H)、7.35−7.25(m,3H)、7.15(s,2H)4.8(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ148.11,138.34,137.82,134.91,131.03.129.03,127.73,125.99,125.41,27.09,23.01.C18H21ClO[M+H]+理論値289.8238、実測値289.8245。分析C18H21ClO理論値C:74.86,H:7.33、実測値C:74.72,H:7.30。
【0104】
パラ−(3−トリフルオロメトキシフェニル)−プロポフォール:
【化38】
4−ブロモプロポフォール(300mg、1.12mmol)、3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色油(230mg、56%)を得た。1H NMR δ7.2(m,1H)、7.0(m,4H)、6.7(m,1H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ162.33,148.41,139.09,137.71,130.52,129.12,127.11,121.79,113.33,26.68,23.74.C19H21F3O2[M+H]+理論値339.3748、実測値339.3753。分析C19H21F3O2理論値C:67.44,H:6.26、実測値C:67.51,H:6.27。
【0105】
パラ−(2,4−トリフルオロメチルフェニル)−プロポフォール:
【化39】
4−ブロモプロポフォール(300mg、1.12mmol)、2,4−トリフルオロメチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(250mg、57%)を得た。1H NMR δ7.2(m,1H)、7.65−7.5(m,2H)、7.34−7.15(m,3H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ148.41,139.52,132.78,130.35,129.03,128.39,127.21,124.63,123.29,118.14,25.99,23.83.C20H20F6O[M+H]+理論値391.3729、実測値391.3761。分析C20H20F6O理論値C:61.54,H:5.16、実測値C:61.22,H:5.10。
【0106】
パラ−(2,4−ジフルオロフェニル)−プロポフォール:
【化40】
4−ブロモプロポフォール(520mg、1.94mmol)、2,4−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(350mg、56%)を得た。1H NMR δ7.5(m,1H)、7.1(s,2H)、6.8−6.7(m,2H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ164.11,160.35,147.93,139.11,131.15,129.37,127.33,112.06,105.34,26.77,24.11.C18H20F2O[M+H]+理論値291.3589、実測値291.3565。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.51,H:6.99。
【0107】
パラ−(2−クロロフェニル)−プロポフォール:
【化41】
4−ブロモプロポフォール(300mg、1.12mmol)、2−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色油(195mg、60%)を得た。1H NMR δ7.4−7.1(m,6H)、4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ146.93,139.31,137.43,133.10,129.29,125.93,26.74,23.71.HRMS:(El)−C18H20OCl[M+H]−理論値287.1203、実測値287.1216。分析C18H20OCl理論値C:74.86,H:7.33、実測値C:74.90,H:7.33。
【0108】
[化合物試験]
(エタノール溶解度比較アッセイ)
化合物5mg/エタノール1mlを使用して、一連のパラ−置換プロポフォール類似体のエタノール中の溶解度を視覚的に決定した。試験を行った少数の類似体は本発明によるものではないが、試験を行った本発明による多くの類似体との比較目的のために以下に含めた。試験を行った化合物はすべて、以下に示す場合を除いて溶解性であった。
【化42A】
【化42B】
【化42C】
【0109】
(化合物効力の決定)
本発明によるプロポフォール類似体の代表的な試料のグリシン受容体活性化およびクロライド電流に対する効果を調べるために、以下に記載するように予備実験を行った。当業者は、これらのデータにより類似体が鎮痛薬としての使用に好適であることが示唆されることを認識する。
【0110】
(方法)
細胞培養、トランスフェクション
ラットα1グリシン受容体サブユニットを、形質転換ヒト胚腎臓細胞(HEK293)中に一過性にトランスフェクトした。α1グリシン受容体サブユニットは異種発現系にてホモメリック受容体を効率的に形成する。10%ウシ胎仔血清(FCS,Biochrom、ベルリン、ドイツ)、100Uml−1ペニシリンおよび100μgml−1ストレプトマイシンを添加したダルベッコ変法イーグル試薬(DMEM、Biochrom、ベルリン、ドイツ)中で37℃にて5%CO2/空気インキュベータ内で、細胞を培養した。トランスフェクションのために、細胞を50mMのK2HPO4および20mM酢酸カリウムを含有する緩衝液、pH7.35に懸濁させた。ラットα1グリシン受容体サブユニットの同時トランスフェクションのために、pClS2発現ベクター(Invitrogen、サンディエゴ、米国)中でそれぞれサブクローニングされた対応するcDNAを懸濁液に添加した。トランスフェクトされた細胞を可視化するために、細胞を緑色蛍光タンパク質(GFP 10μgml−1)のcDNAで同時トランスフェクトした。トランスフェクションのために、本発明者らはEquiBio(Kent、英国)による電気穿孔装置を使用した。トランスフェクトした細胞をガラスカバースリップ上に再び蒔き、記録前に15〜24時間インキュベートした。
【0111】
化学薬品および溶液
化学薬品はすべて、別途記載しない限り、Sigma Chemicals(ダイゼンホーフェン(Deisenhofen)、ドイツ)から得た。
【0112】
調査する本発明による化合物は、エタノールによる1Mストック溶液として調製され、遮光されて、ガラス容器中で−20℃にて貯蔵された。濃度はガラスバイアルに注入された量から計算した。薬物含有バイアルを60分間、激しくボルテックス処理を行った。グリシンおよびピクロトキシンを浴溶液に直接溶解させた。
【0113】
パッチ電極は、KClを140、MgCl2を2、EGTAを11、HEPESを10、グルコース10[mM]を含有していた。浴溶液は、NaClを162、KClを5.3、NaHPO4を0.6、KH2PO4を0.22、HEPESを15、グルコースを5.6[mM]を含有していた。
【0114】
実験の構成
標準ホールセル実験(Hamill et al.,(1981)Pflugers Arch.,391,85−100.)を−30mV膜電位にて行った。数GΩの緊密な電気シールが細胞膜とパッチクランプ電極との間に形成されて、作動薬誘発チャネル活性化による内向き電流をpA範囲に分割する。ピペットの電気抵抗は5MΩ付近であり、約10MΩのホールセル構造での全アクセス抵抗に一致した。調査する本発明による化合物は、圧電結晶に連結されている単回流出によって得られた平滑液体フィラメント(内径0.15mmのガラス管)によって細胞に添加された。細胞を、このフィラメントと、連続的に流れるバックグラウンド溶液との間の界面に配置した。電圧パルスを圧電に印加したときに、管は調査する細胞に対して上下に接近または離反して移動される。液体フィラメントに対する細胞の正確な位置決めは、試験前後に飽和(1000μM)グリシンパルスを印加して確保される。実験を進める前にグリシン活性化電流の振幅および形状の安定化を確保するように注意する。試験溶液およびグリシン(1000μM)を同じガラス−ポリテトラフルオロエチレン灌流システムによって、ただし別個のリザーバから添加した。これらのリザーバの内容物は表面灌流チャンバに入る直前の接合点で混合される。試験中の化合物は、その直接の作動効果を決定するために、単独で添加される。各化合物および各プロトコルについて新たな細胞を使用して、各設定に対して少なくとも3回の異なる実験を行った。使用された化合物の最高濃度に対応する希エタノールの量は34000μMであった。本発明者らは以前に、エタノール自体はこの濃度では直接活性化に対して影響しないことを示した。
【0115】
電流の記録および分析
データ取得およびさらなる分析のために、本発明者らはAxopatch 200B増幅器をpClamp6ソフトウェア(Axon Instruments,ユニオンシティ、カリフォルニア州、米国)と組合せて使用した。電流は2kHzにてフィルタリングした。適合手順は、非線形最小2乗Marquardt−Levenbergアルゴリズムを使用して行った。詳細は適切な図の説明または結果の項に記載する。
【0116】
活性化電流をその最大応答に正規化した。用量反応曲線を(Inorm=[1+(EC50/[C])nH]−1に従って適合し、式中、Inormは、最大内向き電流に正規化された、化合物のそれぞれの濃度[C]によって直接誘起された電流である。EC50は、それ自体の最大反応の50%に達する反応を引き起こすのに必要な濃度であり、nHはHill係数である。
【0117】
(結果)
α1受容体の正規化反応へのHillの式の適合から導出した初期EC50値およびHill係数(±標準偏差)を図1、2および3に示し、下の表1に挙げる。
【0118】
【表1】
【0119】
表1からわかるように、試験を行った類似体はすべて、ナノモル範囲にて有意な効力を示した。
【0120】
上の表1に示す5種類の化合物のEC50値を決定するためのさらなる一連の試験によって、化合物のうち2種類が最初の試験で観察されたよりもさらに低い効力を示した。後の試験では、4−トリフルオロアセトアミド−プロポフォールは0.11nMのEC50を示し、4−(m−フルオロ−p−クロロフェニル)−プロポフォールは0.07nMのEC50を示した。
【0121】
(結論)
上述の実施例において、アリール置換プロポフォール類似体のライブラリを4−ブロモ−プロポフォールから1ステップ鈴木反応にて高い収率で調製した。これにより4−ハロプロポフォール中の4−ハロ原子の芳香環による即時の置き換えが可能であることが判明した。初期のスクリーニングデータから、この性質の類似体はナノモルの活性を、またはいくつかの場合においてはナノモル未満の活性さえ有すると思われた。さらに興味深い観察結果は、4−ビフェニル基も耐容性を示したことであった。本発明者らはいずれの特定の理論にも拘束されることを望むものではないが、この観察結果は、今日までに得た構造活性関係(SAR)データと共に、親油性/ビフェニル残基が中に結合することができる疎水性チャネルの存在を裏付けると思われる。
【0122】
[ラットにおけるインビボの3種類のプロポフォール誘導体の血漿動態]
1.要約
4−クロロプロポフォール、4−(パラ−クロロフェニル)−プロポフォールおよびパラ−(3−フルオロ,4−クロロフェニル)−プロポフォールのインビボ血漿動態を、ラットへの静脈内投与(i.v.)および経口投薬(p.o.)後に決定した。
化合物は概して高いクリアランス値を示すが、半減期は大きな分布容積によって延長される。4−クロロプロポフォールおよびパラ−(3−フルオロ,4−クロロフェニル)−プロポフォールは、経口投与後に血漿中で観察される。ピーク血漿濃度は迅速に達成されるが、終末半減期は静脈内投薬と比較してかなり延長される。したがって、生物学的利用能の推定には一定の不確実性がある。
【0123】
2.物質および方法
2.1 化学薬品、生体物質およびインキュベーション
2.1.1 化学薬品
HPLCグレードのメタノールおよびアセトニトリルをMerck(ダルムシュタット、ドイツ)から入手した。酢酸アンモニウム、ギ酸アンモニウム、酢酸およびギ酸をBDH Laboratory Supplies(プール、英国)から入手した。他の化学薬品は、主にSigma Chemical Company(セントルイス、ミズーリ州、米国)およびBoehringer(インゲルハイム、ドイツ)から入手し、入手可能な最高純度のものであった。水はMilli−Q(Millipore ay、エスポー、フィンランド)精製システムおよびを用いて所内で新たに調製し、UPグレード(超純粋、18.2MQ)であった。
【0124】
2.1.2 動物実験および試料
試験物質の4−クロロプロポフォール、CK−2−3およびCK−2−9を20mg/kgの経口、および2mg/kgの静脈内にてラットに投薬した。全血試料を血漿分離のために側尾静脈から経口投薬の0分後、30分後、1時間後、2時間後および4時間後、ならびに静脈内投薬の0分後、15分後、30分後、1時間後、および2時間後の時点に採取したのに対し、最終試料は投薬(経口および静脈内)後6時間の時点で心臓穿刺によって採取した。すべての時点で1匹のラットから採取し、すべての実験は3組に行った。
【0125】
2.1.3 試料の調製
試料を室温にて解凍して、タンパク質沈殿により血漿およびアセトニトリルの1:2の比で調製し、16100×gにて10分間遠心分離(Eppendorf 5415D,Eppendorf AG、ハンブルク、ドイツ)してから、UPLCIMSMSシステムに注入した。標準試料は、ブランク血漿試料を0.5、2、5、10、20、50、200、500、1000および2000ng/mlの検体化合物にスパイクした後に同様に調製した。
【0126】
2.1.4 計算
試験化合物の薬物動態パラメータをWinNonlin Pro(Pharsight Corp,CA)によって、標準ノンコンパートメント法を使用して計算した。分布容積(Vd)は、終末相に基づく。消失相半減期(t1/2)を対数濃度−時間曲線の終末直線部の最小二乗回帰分析によって計算した。血漿濃度−時間曲線下面積(AUC)は、最後の測定可能な濃度まで線形台形則を使用して、その後、無限まで終末消失相を外挿することによって決定した。終末外挿のない血漿濃度−時間曲線下面積は、AUC 0−6hとして報告された。化合物がコンパートメントまたは系に残存する平均時間を表す平均滞留時間(MRT)は、薬物濃度プロフィールを無限まで外挿して計算した。最大血漿濃度(cmax)およびcmaxまでの時間(tmax)は、血漿濃度データから直接誘導した。仮の経口生物学的利用能(F)は、用量の相違を考慮して、経口投与後のAUCを静脈内投与後のAUCで割ることによって計算して、すなわちF=AUC(経口)/用量(経口)/AUC(静脈内)/用量(経口)、パーセンテージ(%)として報告した。
【0127】
2.2 分析方法
2.2.1 液体クロマトグラフィー−質量分析法
オートサンプラー、真空脱ガス装置およびカラムオーブンを備えたWaters Acquityクロマトグラフィーシステム(Waters Corp.,ミルフォード、マサチューセッツ州、米国)を使用した。すべての化合物に使用した分析カラムは、オンラインフィルタを共に用いたWaters BEH ShieldRP18(2.1×50mm、1.7μm、Waters Corp、ミルフォード、マサチューセッツ州、米国)であった。溶出液は0.1%酢酸(A、pH3.2)およびアセトニトリル(B)であった。0〜2分において5%Bから35%Bまでの、2.0〜3.0分においては35%Bから85%Bまでの線形勾配溶出を使用して、805%Bを用いた1分間の均一濃度溶出およびカラム平衡化を続けた。流速は0.5ml/分であり、カラムオーブン温度は35℃であった。LC/MS/MS/データは、LockSpray電子スプレーイオン化源を備えたMicromass Quattro Premierトリプル4重極質量分析装置を用いて取得した。陰イオンモードイオン化を使用した。多重反応モニタリング(MRM)検出モードを使用した。質量分析装置およびUPLCシステムはMicromass MassLynx 4.1ソフトウェアの下で動作させた。完全なLC/MS/MSパラメータを付録Iに示す。
【0128】
3.結果および結論
3.1 定量分析
定量分析法の性能(「実行時(on−the−fly)検証」)を付録IIに示す。LC/MS/MSクロマトグラムの例を付録IIに示す。
【0129】
各化合物について血漿中での2ng/ml〜10ng/mlの検出/定量限界を得た。原点を除外して、1/x重み付けおよび線形または2次適合を用いて、外部標準ピーク面積の比を濃度の関数として適合することによって、定量用の校正曲線を生成した。校正曲線のために2000ng/mlまでの範囲を適合した。定量範囲内の逆計算正確度(n=2)は、定量限界およびより高い濃度において、82〜119%であった。全体の精度12.0〜17.0%は、標準試料(2回注入/濃度)から、Snedecor式 S=(Σd2/2n)0.5(式中、S=精度、dは平均値の%として表される2回の差であり、Nは標準濃度の数である)を使用して計算した。試料中の各試験化合物の濃度を付録IV〜VIに示す。
【0130】
3.2 薬物動態
試験化合物の薬物動態パラメータを以下の表2にまとめ、個々の動物のパラメータを以下の表3〜5に示す。試験化合物の血漿濃度対時間の曲線を図4〜6に示す。すべての化合物は、血漿動態プロフィールを初期分布および最終消失相の証拠と共に示す。
【0131】
4−クロロプロポフォールは、ラット肝臓血流と比較して非常に高いクリアランス値を有した(55.2ml/分、Davies,B and Morris,T,(1993)。Physiological parameters in laboratory animals and humans.Pharm Res 10:1093−5)。高いクリアランスにもかかわらず、半減期は高い分布容積のために中間である。経口生物学的利用能は良好であり(F=72.4%)、吸収は急速である(Tmax=30分)。薬物クリアランスが急速であるため、急速な吸収の後の門脈における高い薬物濃度のために、見かけ上制限された初回通過抽出が肝臓代謝の飽和から生じ得る。クリアランスは静脈内投薬と同様であるが、半減期はかなり延長される。薬物動態解析によって、分布容積の増大がこの理由であるが、この背後にある機構が明らかでないことが示唆される。ADC計算は、(経口投与後のADC値およびADC 0−6h値の差によって示される)制限された吸収速度であり得る長い終末半減期によって多少偏ることがある。したがって仮の生物学的利用能を最大値と見なすべきである。
【0132】
4−(パラ−クロロフェニル)−プロポフォールは、静脈内投与の後にのみ血漿中で検出された。クリアランスは肝臓血流の範囲内であり、半減期は約1.5時間であった。分布容積は相当大きかった(6リットル/kg超)。低い生物学的利用能の理由は、不十分な吸収または広範囲の初回通過代謝であり得る。
【0133】
パラ−(3−フルオロ,4−クロロフェニル)−プロポフォールは、静脈内投薬後に中間クリアランスおよび半減期を示した。仮の経口生物学的利用能は約18%であった。クリアランスは静脈内クリアランスと類似していたが、分布容積は静脈内投薬後よりもはるかに大きい。この挙動は4−クロロプロポフォールと同様の結論をもたらす。ADC計算は、(経口投与後のADC値およびADC 0−6h値の差によって示される)制限された吸収速度であり得る長い終末半減期によって多少偏ることがある。したがって仮の生物学的利用能を最大値と見なすべきである。
【0134】
【表2】
【0135】
【表3】
【0136】
【表4】
【0137】
【表5】
【0138】
[付録]
(付録I 分析でのLC/MS−MSパラメータ)
Waters Acquity UPLC + Waters Quattro Premierトリプル4重極質量分析計
クォード(quard)フィルタを備えたWaters Acquity BEH ShieldRP18(2.1×50mm、1.8μm)カラム
【0139】
【表6】
【0140】
【表7】
【0141】
【表8】
【0142】
(付録II 試験化合物のLC/MS/MSイオンクロマトグラム)
データは10ng/mlの血漿濃度までスパイクされた標準試料から得る。
【化43】
【0143】
(付録III 分析方法の性能)
【表9】
【0144】
(付録IV 試料中の4−Cl−プロポフォールの濃度)
【表10】
【0145】
(付録V 試料中の4−(p−Cl−Ph)−プロポフォールの濃度)
【表11】
【0146】
(付録VI 試料中のp−(3−F,4−Cl−Ph)−プロポフォールの濃度)
【表12】
【技術分野】
【0001】
本発明は、麻酔薬(anaethetic)プロポフォールに由来する化合物に関する。該化合物は、疼痛の治療、限定的ではないが特に慢性疼痛および中枢性疼痛感作の治療に有用である。
【背景技術】
【0002】
有効で安全な疼痛管理は、優先度の高い臨床的要求と世界的に見なされている。しかしこの分野における発展の大部分は、望ましくない副作用および安全上の課題のない、高い効力の製品を供給できていない。アヘン剤はおそらく、依然として最も有効な利用できる治療のままであり、最終的な目標は、アヘン剤の効力を備えていながら、鎮静作用、依存性、胃損傷および全身耐容性の問題を伴わない疼痛管理剤を供給することである。
【0003】
フェノール誘導体が多数の神経調節効果を有し得ることが仮定されてきた。しかし幅広く臨床使用されている唯一のフェノール誘導体は、麻酔薬プロポフォール(2,6−ジ−イソプロピルフェノール)である。
【0004】
麻酔の主な特徴は、意識消失、疼痛性刺激の存在下での不動、およびリコールの不在である。麻酔薬、たとえばプロポフォールは、中枢神経系(CNS)内のγ−アミノ酪酸(GABAA)受容体を活性化することによってその麻酔効果を媒介すると理解されている。
【0005】
これに対して、無痛覚は疼痛の非存在として定義される。他の末梢および/または中枢神経機構の中でも、無痛覚は、脊髄の後角での抑制性シナプス伝達の増強の結果として発生し得る。脊髄における抑制性シナプス後伝達は、主としてグリシン受容体に関係すると理解されている。したがって、グリシン受容体ファミリーは、疼痛抑制を図る治療薬の標的部位に相当する。
【0006】
GABAAおよびグリシン受容体は、どちらもリガンド依存性イオンチャネルスーパーファミリーに属する。これらは5個のサブユニットがイオンチャネルを形成する共通の構造を有する。αサブユニットおよびβサブユニットは、提案された3α:2βのインビボでの化学量論で5量体受容体に組み立てられる。グリシン受容体は、GABAA受容体と同様に、作動薬結合後に塩化物チャネルを開放することによってニューロン発射を抑制する。グリシン受容体は主に中枢神経系の下部領域に見出され、運動リズム発生の管理、脊髄侵害受容反射応答の調整、および感覚シグナルの処理に関与する。
【0007】
新たな改良された鎮痛薬を開発する必要性が存在する。グリシン受容体がこのような鎮痛薬を同定するための良好な標的になるという事実にもかかわらず、これらの受容体を標的とする鎮痛薬は存在しない。したがって本発明者らはこの課題に取り組むことを決め、疼痛管理のための新たな改良された薬物を同定するために、麻酔および無痛覚の根底にある病態生理学的機構の知識を活用した。
【0008】
慢性疼痛は急性疼痛とは大きく異なる。急性疼痛は、侵害刺激を我々に知らせ、それにより我々が損傷を回避および防止するのを補助する有用な早期警告システムと見なすことができる。これに対して慢性疼痛はそれ自体で疾患である。専門家は、慢性疼痛を、正常な状況で疼痛の処理を抑える抑制性ニューロン活性が著しく低下した平衡障害症候群と見なす。慢性的な炎症または神経因性疼痛の治療はなお困難であり、現在、あらゆる症状に作用する単一の治療はない。
【0009】
慢性疼痛で見られるニューロン興奮性の上昇は、末梢から中枢神経系の上部領域への侵害受容シグナルの中継を管理する脊髄の後角表層における、GABA作動性ニューロンおよび/またはグリシン作動性ニューロンに媒介される抑制の消失に関係する。成人後角において、グリシンの急速抑制性シナプス後伝達への寄与が支配的である。グリシン受容体は、主に中枢神経系の下部領域に見出され、運動リズム発生の管理、脊髄侵害受容反射応答の調整、および感覚シグナルの処理に関与する。侵害受容上行性経路および疼痛を調節するその役割によって、グリシン受容体は鎮痛薬および鎮痙薬の潜在的に興味深い標的部位となる。疼痛の情動的成分と関連する前帯状皮質へのグリシン受容体作動薬であるタウリンの微量注射によって、選択的グリシン受容体アンタゴニストのストリキニンによって拮抗できる作用である神経因性疼痛が軽減される。ストリキニン感受性グリシン受容体には4種類のαサブユニットおよび1種類のβサブユニットがあり、α1サブユニットは成人の脊髄および脳幹で広く発現されるが、脳の高次中枢では感覚処理にも関与する。グリシン受容体α3サブユニットは、PGE2誘発受容体ホスホリル化による中枢性炎症性疼痛感作の根底にある標的部位として確認されている。α3サブユニット・ノックアウト・マウスは、そうでなければ急性疼痛に対する正常な応答を伴う炎症性疼痛を発症しない。この現象は、α3の欠如を場合により代償するα1含有グリシン受容体サブユニットが、PGE2シグナル伝達に関与するプロテインキナーゼA(PKA)ホスホリル化部位を有さないという事実によって説明され得る。さらにα3サブユニットのホスホリル化は、神経因性疼痛に必ずしも関与していない。この知識に基づいて、α1サブユニットを含有する主要な成人グリシン受容体アイソフォームを標的とする薬物の開発の必要性が、本発明者らによって確認されている。グリシン受容体の生理学的役割およびその比較的限定された発現(主に脊髄および脳下部領域)を考えると、選択的グリシンモジュレータは、脊髄後角レベルでの抑制を治療的に向上させるために非常に興味深いはずである。
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の第1の態様により、一般式(IA)
【化1】
(式中、R1は
【化2】
(式中、R4、R5、R6、R7およびR8は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
であり、ならびに/または
式中、R2は
【化3】
(式中、R9、R10、R11、R12およびR13は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
である。)
の化合物が提供される。
【0011】
本発明は、疼痛の治療に使用するための一般式(I)
【化4】
(式中、R1、R2およびR3の少なくとも1つは、置換もしくは非置換のアリールもしくはヘテロアリール基であるか、または置換もしくは非置換のアリールもしくはヘテロアリール基を含む。)
の化合物をさらに提供する。
【0012】
ごく最近、脊髄GABAA受容体を標的とすることによって脊髄における抑制を向上させる代わりの手法が発表された。中枢神経系全体でのGABAA受容体の遍在的な発現のために、GABAA受容体は構造的に多様な鎮静−麻酔薬および抗不安薬の治療標的である。そのため、脊髄注射時に存在するベンゾジアゼピンなどのGABA調節剤の鎮痛効果は、全身投与時の中枢神経効果によって無効化される。α2および/またはα3 GABAA受容体サブユニットを含むGABAA受容体が、脊髄レベルでのベンゾジアゼピンの抗侵害受容作用に関与することが以前に示され、慢性疼痛の治療のためのサブタイプ選択的なGABA作動薬の開発が示唆されている。この最新の研究に対して、本発明者らの手法は、GABAA受容体ではなくグリシン受容体を選択的標的として、高次脳領域でのGABAA受容体の刺激に関連する鎮静および依存性を回避しながら、脊髄のレベルでの抑制を増強/回復することによって慢性疼痛を治療するための一般式IおよびIAによる化合物を開発することである。本発明者らの手法は、麻酔薬、アルコールおよびカンナビノイドによって明確に調節されることが知られているグリシン受容体α1サブユニットを標的にしている。しかし、本発明による化合物は、初めて、他の受容体ファミリー(たとえば麻酔薬の場合のGABAA受容体)をより高い親和性で標的とするすべての公知の化合物のように対象とする受容体ファミリーを比較的非特異的に標的とするのではなく、対象とする受容体ファミリーを高い親和性で標的としている。本発明は、疼痛の治療に使用するための一般式I、IAの化合物およびその好ましい実施形態をさらに提供する。
【0013】
本発明のさらなる関連する態様により、以下に示すように一般式(XI)または(XIV)の化合物と、それぞれ一般式(XII)または(XV)の化合物との反応:
【化5】
(式中、R1およびR2はそれぞれ、置換もしくは非置換のアリールもしくはヘテロアリール基であるか、または置換もしくは非置換のアリールもしくはヘテロアリール基を含み、Xは、フッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(X)および(XIII)の化合物を製造する方法が提供される。
【0014】
本発明のさらなる態様は、治療的有効量の本発明の第1の態様による化合物、または一般式IもしくはIAの化合物と、製薬学的に許容可能なビヒクルとを含む医薬組成物を提供する。
【0015】
本発明のまたさらなる態様は、治療的有効量の本発明の第1の態様による化合物、または一般式IもしくはIAの化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0016】
別の態様は、疼痛の治療が必要な対象の疼痛を治療または低減する方法であって、治療的有効量の本発明の第1の態様で定義された化合物、または一般式IもしくはIAの化合物を対象に投与する工程を含む方法を提供する。
【0017】
さらなる態様は、本発明の第1の態様で定義された化合物、または一般式IもしくはIAの化合物を含む薬剤、および疼痛の治療に使用するための薬剤としての本発明の第1の態様による化合物、または一般式IもしくはIAの化合物を提供する。
【0018】
本発明のまたさらなる態様は、疼痛の治療用の薬剤の製造において使用するための、本発明の第1の態様による化合物、または一般式IもしくはIAの化合物を提供する。
【0019】
別の態様は、疼痛の治療のための、本発明の第1の態様による化合物、または一般式IもしくはIAの化合物の使用を提供する。
【0020】
本発明の第2の態様は、有効量の式(VIII)
【化6】
(式中、R14はハロアルキル基である。)
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0021】
本発明は、式(VIII)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0022】
本発明のさらなる態様は、有効量の本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0023】
別の態様は、疼痛の治療が必要な対象の疼痛を治療または低減する方法であって、治療的有効量の本発明の第2の態様で定義された化合物、または一般式VIIIもしくはIXの化合物を対象に投与する工程を含む方法を提供する。
【0024】
さらなる態様は、本発明の第2の態様で定義された化合物、または一般式VIIIもしくはIXの化合物を含む疼痛の治療に使用するための薬剤を提供する。
【0025】
本発明のまたさらなる態様は、疼痛の治療用の薬剤の製造において使用するための本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物を提供する。
【0026】
別の態様は、疼痛の治療のための、本発明の第2の態様による化合物、または一般式VIIIもしくはIXの化合物の使用を提供する。
【0027】
本発明者らは、脊髄後角内での抑制性シナプス伝達の消失が、炎症または神経損傷後の慢性疼痛の発症において主要な役割を果たすことを認識した。さらに本発明者らは脊髄における抑制性シナプス後伝達が主にグリシンに関係することを認識した。このことにより本発明者らは、ストリキニン感受性グリシン受容体ファミリーが疼痛感作の抑制を目標とする治療剤の標的部位に相当することを理解した。この理解は、Ahmadi et al.(Nature Neuroscience(2001)Vol.5 No.1 p34−40)が行った研究に基づいていた。
【0028】
本発明者らは、ヒドロキシル基に対してパラ位のハロゲンおよびオルト位またはメタ位に1個または2個のメチル基を有するフェノール誘導体を研究することにより、その仮説の検証を進めた。本発明者らの結果は、Haeseler et al.(British Journal of Pharmacology(2005)145,p916−925)によって発表され、ハロゲン化によってグリシン受容体の共活性化または活性化が向上することが確認された。しかしこれらの初期の結果は、フェノール環のメチル基の数または位置がグリシン受容体の共活性化のためのEC50に著しく影響しないことを証明するように思われた。
【0029】
したがって本発明者らは、グリシン受容体を特異的に標的とする改良鎮痛剤を開発し、驚くべきことに、2個以上の炭素原子を含むオルトアルキル基またはメタアルキル基、およびパラハロ基、パラアミノ基またはパラアミド基を包含するプロポフォール類似体が、グリシン受容体の共活性化薬として予想外の効力を示すことを発見した。この研究は、出願人の国際特許出願WO2007/071967の主題である。
【0030】
実施例に記載した化合物は、低いナノモル範囲で最大半量増強効果を示した。このことは、プロポフォールよりも数桁低い濃度を表す。さらに本発明による化合物は、以前に公開した研究で記載したp−メチル誘導体およびp−ハロ誘導体よりも著しく強力であった。このことは、本発明による化合物が理想的な鎮痛薬となり、意識に対する効果(すなわち麻酔効果)はほとんどないか、または無視できるくらいであることを意味するため、大きな利点である。
【0031】
一般式IおよびIAの化合物に関して、前記アリールまたはヘテロアリール基が単環式芳香環または多環式芳香環であることが好ましい。前記アリールまたはヘテロアリール基は、非置換であり得るか、またはハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリール、および置換もしくは非置換ヘテロアリールから成る群より選択される1個以上の置換基によって置換され得る。前記1個以上の置換基のうち1個は、オルト位、メタ位、および/またはパラ位に提供され得る。
【0032】
第1の好ましい実施形態において、R1は、置換または非置換のアリール基またはヘテロアリール基であり、R2およびR3の少なくとも1つは、水素原子である。好ましい化合物は、一般式(II)
【化7】
(式中、R4、R5、R6、R7およびR8はそれぞれ独立して、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、置換または非置換アミン、置換または非置換アミド、置換または非置換アリール、および置換または非置換ヘテロアリールから成る群より選択される。)
を有する。
【0033】
R4およびR8の少なくとも1つは、ハロゲンまたは置換もしくは非置換C1−4アルキルであり得る。ハロゲンは、好ましくはフッ素または塩素であり、アルキル基は、好ましくはメチル、エチル、プロピルまたはブチルである。R8はフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R4は水素であることが好ましい。
【0034】
R5およびR7の少なくとも1つは、好ましくは水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される。ハロゲンはフッ素または塩素であってよく、アルキルはハロ置換C1−4アルキルであり得る。好ましくはR7は、フルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R5は、水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される。
【0035】
R6は、好ましくは水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される。前記ハロゲンはフッ素または塩素であってよく、前記アルキルはハロ置換C1−4アルキルであってよく、前記アルコキシはハロ置換C1−4アルコキシであり得る。好ましくはR6は、−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される。
【0036】
第2の好ましい実施形態において、R2は置換または非置換のアリール基またはヘテロアリール基であり、R1およびR3の少なくとも1つは水素原子である。好ましい化合物は、一般式(III)
【化8】
(式中、R9、R10、R11、R12およびR13はそれぞれ独立して、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、置換または非置換アミン、置換または非置換アミド、置換または非置換アリール、および置換または非置換ヘテロアリールから成る群より選択される。)
を有する。
【0037】
R9およびR13の少なくとも1つは、好ましくはハロゲン、たとえばフッ素もしくは塩素、または置換もしくは非置換のC1−4アルキル、たとえばメチル、エチル、プロピルまたはブチルである。好ましくはR9は、フルオロ、クロロ、およびトリフルオロメチルから成る群より選択され、R13は水素である。
【0038】
好ましくはR10およびR12の少なくとも1つは、水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される。前記ハロゲンは、フッ素または塩素であってよく、前記アルキルはハロ置換C1−4アルキルであり得る。R10はフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R12は水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択されることが好ましい。
【0039】
R11は、水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択され得る。ハロゲンは、好ましくはフッ素または塩素であり、アルキルは、好ましくはハロ置換C1−4アルキルであり、アルコキシは好ましくはハロ置換C1−4アルコキシである。好ましくはR6は、−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される。
【0040】
本発明は、以下の好ましい化合物:
【化9】
を提供する。
【0041】
本発明は、疼痛の治療に使用するための上記の好ましい化合物をさらに提供する。
【0042】
本発明の第2の態様は、有効量の式(VIII)
【化10】
(式中、R14はハロアルキル基である)
の化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物を提供する。
【0043】
本発明は、式(VIII)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0044】
好ましくは前記アルキル基は、C1−4アルキル基である。R14は、2個以上の、好ましくは3個のハロ置換基を含み得る。任意の適切なハロ置換基が選択され得るが、前記ハロ置換基または前記ハロ置換基の少なくとも1つがフッ素であることが好ましい。
【0045】
本発明は、有効量の式(IX)
【化11】
を有する化合物を含む疼痛の治療のための医薬組成物をさらに提供する。
【0046】
本発明は、式(XI)を有する疼痛の治療に使用するための化合物をさらに提供する。
【0047】
本発明の第1の態様によるなおさらに好ましい化合物は、以下の群:
【化12】
より選択できる。
【0048】
化合物が、GABAA受容体よりもストリキニン感受性グリシン受容体に選択性を有することが好ましい。化合物は、GABAA受容体におけるそのEC50よりも低い濃度の、グリシン受容体を共活性化するためのEC50を有し得る。好ましくは化合物は、GABAA受容体におけるEC50の10分の1である、グリシン受容体を共活性化するためのEC50を有する。化合物が、GABAA受容体におけるEC50の少なくとも100分の1である、グリシン受容体を共活性化するためのEC50を有することがさらに好ましい。
【0049】
化合物が、プロポフォールのEC50よりも低い、グリシン受容体を共活性化するためのEC50も有することが望ましい。たとえば化合物は、プロポフォールのEC50の少なくとも10分の1または100分の1である、グリシン受容体を共活性化するためのEC50を有し得る。最も好ましい化合物は、(HEK293細胞中で異種発現されたグリシン受容体に対して測定された)プロポフォールのEC50の1000分の1である、グリシン受容体を共活性化するためのEC50を有する。グリシン受容体を共活性化するためのEC50値を測定する好適な方法は、以下の実施例に開示されている。
【0050】
化合物の効力は、CNSでの麻酔およびPNSでの無痛覚を調節する神経生理学が考慮されるとき、一層驚くべきものである。本発明者らは、一般式IおよびIAによる化合物が、ストリキニン感受性グリシン受容体に正のアロステリックモジュレータとして作用すると考えている。これらの受容体は、過分極によって膜電位を安定化させて、脊髄レベルで主要な抑制原理を構成する塩化物チャネルである。これに対して、密接に関連するGABAA受容体は、CNSにおいて主要な抑制原理を構成する。したがってGABAA作動薬が意識の変化または消失をもたらすのに対して、本発明による化合物は、意識に影響を及ぼさない濃度で末梢レベルにて疼痛を理想的に遮断する。本発明者らは、本発明による化合物が効力を有するのは、該化合物がストリキニン感受性グリシン受容体において正のアロステリックモジュレータとして作用して、それにより後根神経節レベルで求心性神経シグナルを遮断するが、中枢のGABAA受容体には最小限の効果を有するか、全く効果を有さないためであると考えている。したがって当業者は、グリシン受容体作動薬でありGABAA作動性効果を全く有さない鎮痛薬を選ぶことになる。結果として、公知の最も強力なGABAA作動薬であるプロポフォールは、当業者によって鎮痛薬を開発するプラットフォームとして最も好適でない化合物として見なされる。さらに当業者は、Haeseler et al.(上記を参照)によって公開されたデータも検討して、GABA受容体よりもグリシン受容体に対して選択性である化合物を開発する際、アルキル化の性質(Haeselerの論文ではメチル基)が重要でないという見解に達する。したがって本発明者らは、プロポフォール誘導体の鎮痛特性の研究に対して技術的偏見があったと考えている。そのため本発明者らが本発明による化合物と共に見出したグリシン受容体の共活性化の並はずれた増加は、驚くべきことであるだけでなく、可能性が低いと当業者に考えられてきただろう。理論的には、GABAA受容体レベルでの効力がより低い任意の他のフェノール誘導体は、技術水準によれば、より有望な候補として考慮されるべきであった。
【0051】
上述の本発明のさらなる態様に相当する一般式(X)および(XIII)の化合物の製造方法において、R1および/またはR2は、本発明の第1の態様による化合物に含まれる置換基R1およびR2の好ましい特徴のいずれも取り得る。したがって、この方法が本発明の第1の態様による化合物の好ましい実施形態を製造する好ましい合成経路に相当することが認識される。
【0052】
この方法は好ましくは、アリールボロン酸をハロゲン化アリールと反応させて本発明の第1の態様による化合物が生成される鈴木型反応経路を使用する。任意の適切な反応条件が使用され得るが、反応は適切な触媒、たとえば鈴木反応方法で一般に周知であるようにパラジウム触媒を使用して触媒されることが好ましい。そのため反応は、好ましくはパラジウム触媒、たとえばPd(PPh3)4の存在下で行われる。任意の好適な反応温度が使用され得るが、好ましくは室温を超えて、さらに好ましくはおよそ40℃〜50℃、なおさらに好ましくはおよそ60℃〜100℃の温度、最も好ましくはおよそ80℃の温度である。反応は、任意の所望の期間にわたって行われ得る。好ましくは、反応は、およそ6時間超、さらに好ましくはおよそ12時間超、および好ましくはおよそ36時間〜48時間の期間で行われる。好ましい反応期間は、およそ12〜48時間、なおさらに好ましくはおよそ24時間である。
【0053】
ハロゲン化アリール開始物質は、以下の例示的な反応スキームまたは当業者に明らかなようなその派生物に従って製造され得る。
【化13】
ハロゲン化アリール開始物質は次に適切なアリールボロン酸またはヘテロアリールボロン酸と反応されることができ、以下に例示するような所望のプロポフォール類似体を製造する。
【化14A】
【化14B】
【0054】
本発明による化合物、ならびにこのような化合物を含有する医薬組成物および薬剤は、いくつかの状況で鎮痛薬として使用され得る。
【0055】
該化合物は特に、これまで治療が困難であることでよく知られている慢性疼痛状態(たとえば神経因性慢性疼痛および/または炎症後慢性疼痛)を標的とするために有用である。該化合物は特に従来の薬物、たとえばNSAID、アヘン剤誘導体などによる治療が難しい慢性神経因性疼痛を治療するのに有用である。
【0056】
該化合物は、(たとえば傷害後の)急性疼痛を治療するためにも有用である。
【0057】
本発明の化合物が有用なのは、該化合物が単剤療法として使用された場合に、局部麻酔薬および鎮痛薬ならびにNSAIDおよびオピオイドのよく知られた副作用すべてを回避し、同時に、相加作用または相乗作用を目的とした非常に多種多様の併用治療戦略を可能にするためである。
【0058】
疼痛が調節され得る具体的な症状の例は、慢性腰痛、関節炎、癌性疼痛、三叉神経痛、脳卒中および神経因性疼痛を含む。
【0059】
化合物は既存の疼痛を治療するために使用され得るが、たとえば待機手術の前に予防的治療が医療的に必要と見なされるときにも使用され得る。
【0060】
化合物は、単剤療法(すなわち化合物単独の使用)の形で鎮痛薬として使用され得るか、または化合物は疼痛をまた低減する他の治療と併用して投与され得る。好ましい併用療法は、一般式の化合物によって調節される経路とは異なる疼痛処理経路によって疼痛を調節する鎮痛薬と共に該化合物を使用することを含む。このような鎮痛薬は、モルヒネ、パラセタモール、およびNSAIDSを含む。化合物は、グリシン受容体と間接的にのみ相互作用する局部麻酔薬(たとえばリグノカイン)とも有用に併用され得る。
【0061】
本発明の薬剤は、一般式IまたはIAの化合物と製薬学的に許容可能なビヒクルとを含み得る。該ビヒクルは、ビヒクルが投与される対象による耐容性が良好であり、化合物の患部への送達を可能にするビヒクルであるべきことが認識される。
【0062】
本発明の薬剤は、特に化合物が使用される方式に応じて、いくつかの異なる形を取り得る。そのためたとえば、薬剤はフェノール誘導体の塩(たとえばナトリウム塩)の形の化合物を含み得る。このような塩は、粉末形で製造されて、錠剤、カプセル剤、液剤、軟膏、クリーム、ゲル、ハイドロゲル、エアゾール、スプレー、ミセル、経皮パッチ、リポソーム、またはヒトもしくは動物に投与され得るその他の好適な形で包含され得る。
【0063】
または本発明によるフェノール誘導体は、好適な溶媒に溶解されて液剤を形成し得る。溶媒は水性であり得る(たとえばPBSまたは蒸留水)。または溶媒は、アルコール、たとえばエタノール、またはこのような溶媒と水性溶媒との混合物であり得る。
【0064】
該薬剤は局所または局部の治療に使用されることが好ましい。このような薬剤は、患部へ塗布するための液剤として製剤され得る。または液剤は、注射またはエアゾールによる投与のために製剤され得る。
【0065】
化合物は、持続放出デバイスまたは遅延放出デバイス内にも包含される。このようなデバイスは、たとえば皮膚の上または下に挿入され得て、該化合物は数週間、または数カ月にさえわたって放出され得る。このようなデバイスは、特に長期の慢性疼痛を有する患者(たとえば関節炎患者)に有用であり得る。該デバイスは、通常は頻繁な投与を必要とする化合物が使用されるときに特に好都合である。
【0066】
必要な化合物の量は、生物活性および生物学的利用能によって決定され、生物学的利用能は、投与方式、使用される化合物の物理化学特性、および化合物が単剤療法または併用療法のどちらで使用されるかに依存することが認識される。投与頻度は、上述の因子および特に治療される対象内での化合物の半減期によっても影響を受ける。
【0067】
投与される最適投薬量は、当業者によって決定されることができ、使用中の特定の化合物、調製物の強度、投与方式、および軽減を必要とする疼痛の程度によって変化する。対象の年齢、体重、性別、食事、および投与時間を含む、治療される特定の対象に応じた追加の因子によって投薬量を調整する必要が生じる。
【0068】
公知の手順、たとえば医薬業界で従来使用されている手順(たとえばインビボ実験、臨床試験など)を使用して、組成物の具体的な製剤および正確な治療体制(たとえば化合物の1日用量および投与頻度)が確立され得る。
【0069】
概して、組織濃度が使用される化合物のEC50付近であるように、標的部位に化合物を送達するのに有効である用量が投与されるべきである。1日用量は単回投与として(たとえば1日1回の注射として)投与され得る。または使用される化合物は、1日の間に2回以上の投与を必要とし得る。一例として、慢性腰痛を治療するための好ましい化合物は、注射用液剤または軟膏として1日2回(または疼痛の重症度に応じてそれ以上の回数)の用量として投与され得る。治療を受ける患者は、起床時に第1回の用量を、次に2回目の用量を夕方に(2回用量体制の場合)、またはその後3〜4時間の間隔で投与され得る。あるいは、持続放出デバイスが使用されて、反復用量を投与する必要なしに患者に最適な用量が投与され得る。
【0070】
本発明は、治療的有効量の本発明の化合物と製薬学的に許容可能なビヒクルとを含む医薬組成物をさらに提供する。一実施形態において、本発明によるフェノール誘導体の塩の量は、経腸(経口、経直腸)投与のための各用量単位で約10μg/kg体重〜10mg/kg体重の量である。別の実施形態において、この量は、非経口(静脈内/くも膜下腔内または硬膜外)投与のための各用量単位で約1μg/kg体重〜1mg/kg体重である。
【0071】
さらなる実施形態において、ビヒクルは液体であり、組成物は液剤である。非経口投与のための有用な液体液剤は、0.001〜1重量%の式IまたはIAのフェノールを含み得る。別の実施形態において、ビヒクルは固体であり、組成物は錠剤である。さらなる実施形態において、ビヒクルはゲルであり、組成物は局所塗布用である。
【0072】
本発明において、「治療的有効量」は、化合物が有効である有痛性症状に罹患している対象に投与されるとき、疼痛の減少、寛解、または後退を引き起こす化合物、薬剤または組成物の任意の量である。
【0073】
「対象」は、脊椎動物、哺乳動物、家畜またはヒトである。
【0074】
本発明の実施において、「製薬学的に許容可能なビヒクル」は、医薬組成物を製剤するのに有用な当業者に公知の任意の生理学的ビヒクルである。一実施形態において、製薬学的ビヒクルは液体であってよく、医薬組成物は液剤の形である。別の実施形態において、製薬学的に許容可能なビヒクルは固体であり、組成物は粉剤または錠剤の形である。さらなる実施形態において、製薬学的ビヒクルはゲルであり、組成物は坐剤またはクリームの形である。さらなる実施形態において、化合物または組成物は、製薬学的に許容可能な経皮パッチの一部として製剤され得る。
【0075】
固体ビヒクルは、潤滑剤、可溶化剤、懸濁化剤、充填剤、流動促進剤、圧縮助剤、結合剤または錠剤崩壊剤としても作用し得る1つ以上の物質を含むことができる。固体ビヒクルは、カプセル化材料であることもできる。粉剤において、ビヒクルは、微粉化された活性成分と混合された微粉化固体である。錠剤において、活性成分は、必要な圧縮特性を有するビヒクルと好適な比で混合されて、所望の形状およびサイズに圧縮される。粉剤および錠剤は、好ましくは99%までの活性成分を含有する。好適な固体ビヒクルはたとえば、リン酸カルシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、デキストリン、デンプン、ゼラチン、セルロース、ポリビニルピロリドン、低融点ろう、およびイオン交換樹脂を含む。
【0076】
液体ビヒクルは、液剤、懸濁剤、乳剤などを調製するのに使用される。フェノール誘導体は、製薬学的に許容可能な液体ビヒクル、たとえば水、エタノール、有機溶媒もしくはその混合物、または製薬学的に許容可能な油もしくは脂肪に、溶解または懸濁させることができる。
【0077】
滅菌液剤または懸濁剤である液体医薬組成物は、たとえば筋肉内、くも膜下腔内、硬膜外、腹腔内または皮下注射によって利用できる。滅菌液剤は静脈内投与することもできる。化合物は、滅菌水、生理食塩水、または他の適切な滅菌注射用媒体を使用して、投与時に溶解または懸濁され得る滅菌固体組成物として調製され得る。
【0078】
本発明はここで、以下の実施例によって、および以下の図面を参照してさらに説明される。
【図面の簡単な説明】
【0079】
【図1】対数目盛に4−(p−tert−ブチルフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図2】対数目盛に4−(p−クロロフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図3】対数目盛に4−(p−ビフェニル)−プロポフォールの濃度に対してプロットされた、α1ホモメリックグリシン受容体によるグリシンの非存在下で活性化された正規化Cl−電流(平均±標準偏差、n=それぞれ3)。電流は、化合物の高い濃度によって達成された最大値にいずれも正規化された。実線は表示されたパラメータを有するデータへのHill適合である。
【図4】CK−I−Iについての、それぞれ8mg/kgおよび1mg/kgの経口投与(破線)および静脈内投与(実線)後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。
【図5】CK−2−3についての、それぞれ20mg/kgおよび2mg/kg静脈内投与後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。血漿内において化合物は経口投与では検出されなかった。
【図6】CK−2−9についての、それぞれ20mg/kgおよび2mg/kgの経口投与(破線)および静脈内投与(実線)後の、個々の動物の血漿濃度。挿入図は片対数目盛での同じデータを示す。
【発明を実施するための形態】
【実施例】
【0080】
本発明による多数のアリール置換プロポフォール類似体は、各種のハロ置換プロポフォール化合物から開始して以下で説明するように合成された。製造されたアリール置換類似体を、次にエタノール中の溶解度について試験を行い、その後代表的な選択された類似体に試験を行ってそのEC50値およびHill係数を決定した。
【0081】
[化合物の合成]
(4−置換プロポフォール類似体の合成)
4−ブロモプロポフォール:
【化15】
プロポフォール(1g、5.6mmol)を氷酢酸(25ml)に溶解させた。溶液の変色が停止するまで、臭素(水溶液)を撹拌しながら滴加した。混合物を室温にて1時間撹拌したままにした。反応混合物を次に水(50ml)にゆっくり注いだ。得られた赤色油を酢酸エチルによって抽出して、水および塩水で洗浄した。粗生成物を硫酸マグネシウムで脱水して、溶媒を除去して褐色油を得て、これをヘキサン中2%のDCMを溶出液として用いてフラッシュクロマトグラフィーで精製した。薄黄色油1.35gを純4−ブロモ−プロポフォール(93%)として回収した。1H NMR 400MHz d7.1(s,1H)、4.75(s,1H)、3.15(m,2H)、1.3(d,12H).13C NMR d149.4,136.48,126.89,113.74,27.69,22.95.HRMS(El)C12H17OBr[M+H]+理論値258.1679、実測値258.1703。分析C12H17OBr理論値C:56.04% H:6.66% 実測値C:55.99% H:6.63%。
【0082】
4−ニトロ−プロポフォール:
【化16】
プロポフォール(1.78g、10mmol)をAcOH:DCMが15:10mLの混合物に溶解させた。0℃の硝酸(1mLを10mLのDCMに溶解)を滴加した。添加時に黄色からオレンジ色〜赤色への色変化が観察された。水(20mL)を添加して、反応混合物をDCM(3×20mL)によって抽出した。得られた赤色溶液を硫酸ナトリウムで脱水し、溶媒を除去して暗赤色粗固体を得た。粗生成物をジエチルエーテルおよびヘキサンからの再結晶によって精製し、薄黄色結晶を2回の回収で得た。1.803g(84%).1H NMR δ7.71(s,2H)、4.77(s,1H)、3.1(m,2H)、1.3(d,12H).13C NMR δ155.21,140.44,138.41,120.20,27.68,23.01.HRMS(El)C12H20N2O3[M+NH4]+理論値240.2735実測値240.2731。分析C12H16NO3理論値C:64.55% H:7.67% N:6.27%
実測値C:64.53% H:7.66% N:6.25%
【0083】
4−アミノ−プロポフォール:
【化17】
4−ニトロプロポフォール(0.368g、1.6mmol)をEtOH(2.8mL)および濃HCl(7.5mL)に溶解させた。過剰量のスズ細粒(1.4g)を添加して、反応混合物を還流まで加熱した。薄黄色から無色への色変化が観察された。1時間後、反応混合物をセライトパッドで濾過した。溶媒を減圧下で除去して、得られた残渣を水に再溶解させた(50ml)。溶液が万能試験紙によって塩基性(pH12〜15)となるまで、水酸化ナトリウム水溶液を滴加した。溶液を次にDCM(3×25mL)で抽出した。有機抽出物を合せ、塩水で洗浄し、硫酸ナトリウムで脱水して、溶媒を除去し、分析的に純粋な(98%)紫色油320mgを得た。1H NMR δ6.45(s,2H)、4.75(s,1H)3.4(br s,2H)3.1(m,2H)1.3(d,12H).13C NMR δ140.45,138.72,136.48,112.02,27.70,23.01.HRMS(Cl)C12H22N2O[M+NH4]+理論値210.3093実測値210.3097。分析C12H18NO理論値C:74.56% H:9.91% N:7.24%実測値C:74.36% H:9.95% N:7.23%。
【0084】
4−アミノプロポフォール−ヒドロクロリド:
【化18】
4−アミノプロポフォール(310mg、1.6mmol)をEt2O(100mL)に溶解させた。1,4−ジオキサン(25mL)に溶解させた濃HCl(5mL)を撹拌しながら滴加した。1時間後、混合物を静置して2層とし、下層を回収して、溶媒を減圧下で除去してピンク色結晶性固体(250mg、67%)として、さらに分析せずに使用する。
【0085】
4−トリフルオロアセトアミド−プロポフォール:
【化19】
4−アミノプロポフォール(200mg、1.04mmol)をEtOAc(40mL)に溶解させた。ピリジン(0.25mL、3mmol)、続いて無水トリフルオロ酢酸(0.2mL、1mmol)を添加した。反応混合物を60℃まで30分間加熱した。冷却時に溶媒を減圧下で除去して、残渣をDCM(50mL)に再溶解させ、水および塩水で連続して洗浄し、硫酸ナトリウムで脱水した。溶媒の除去後、4−トリフロオロアセトアミドプロポフォールを薄ピンク色結晶性固体(205mg、75%)として回収した。1H NMR δ7.16(s,2H)、6.3(br s,1H)、5.0(1H,s)、3.2(m,2H)、1.3(d,J=6.9Hz,12H).13C NMR δ155.18,144.28,138.05,130.51,117.03,115.85,27.78,23.12.HRMS(El):C14H18F3NO2[M+H]+理論値306.2751、実測値306.2747。分析C14H18F3NO2理論値C:58.12,H:6.27,N:4.84、実測値C:58.11、H:6.25,N:4.83。
【0086】
4−クロロプロポフォール:
【化20】
プロポフォール(710mg、4.0mmol)をDCMに溶解させた。塩化スルフリル(0.675g、5mmol)を滴加して、次に反応混合物を4時間還流させた。溶媒を減圧下で除去して、粗生成物を褐色油として得て、これを蒸留によって精製して、4−クロロプロポフォールを薄オレンジ色油(750mg、89%)として得た。沸点1mmHgにて94〜97℃。1H NMR δ7.0(s,2H)、5.0(s,1H)、3.1(m,2H)、1.2(d,J=6.9Hz,2H).13C NMR δ148.92,139.12,126.12,124.01,27.37,22.68.HRMS(El)C12H17OCl[M+H]+理論値166.9545、実測値166.9572.分析C12H17OCl理論値C:67.76,H:8.06、実測値C:67.70,H:8.04。
【0087】
(アリール−置換プロポフォール類似体の合成)
4−(パラ−クロロフェニル)−プロポフォール:
【化21】
4−ブロモ−プロポフォール(200mg、0.77mmol)、パラ−クロロフェニルボロン酸(156mg、1mmol、1.3当量)、テトラキス(トリフェニルホスフィン)パラジウム(0)(26mg、3mol%)、炭酸ナトリウム(2N水溶液3.5mL)およびジメトキシエタン(10mL)の懸濁液を24時間、95℃にて撹拌した。懸濁液を冷却し、セライトで濾過して、EtOAcに溶解し、水および塩水で連続して洗浄して、硫酸ナトリウムで脱水し、溶媒を真空中で除去した。ヘキサン中10%のEtOAcで溶出させた、カラムクロマトグラフィーにより得られた残渣の精製によって、純生成物を白色固体(72%)として得た。1H NMR δ7.5−7.2(m,6H)、4.8(s,1H)、3.2(m,2H)、1.3(d,J=6.85Hz,12H).13C NMR δ147.59,138.23,134.54,132.89,129.11,128.50.122.71,27.71,23.144.HRMS:(El)−C18H20OCl[M+H]−理論値287.1203、実測値287.1206。分析C18H20OCl理論値C:74.86,H:7.33、実測値C:74.88,H:7.32
【0088】
4−(パラ−カルバモイルフェニル)−プロポフォール:
【化22】
4−ブロモプロポフォール(200mg、0.77mmol)、p−カルバモイルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色固体(212mg、93%)として得た。1H NMR δ8.00−7.7(m,4H)、7.2(s,2H)、6.3(s,2H)5.1(s,1H)3.2(m,2H)、1.2(d,J=6.85,12H).13C NMR δ168.13,147.55,139.90,138.18,133.07,128.05,127.94,126.53,26.71,23.68.HRMS(EI)−C19H23NO2[M+H]−理論値298.4567、実測値298.4570。分析C19H23NO2理論値C:76.73,H:7.80,N:4.71、実測値C:76.70,H:7.76,N:4.68。
【0089】
4−(p−tert−ブチルフェニル)−プロポフォール:
【化23】
4−ブロモプロポフォール(300mg、1.12mmol)、p−tert−ブチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を黄色油(300mg、83%)として得た。1H NMR δ74−7.2(m,6H)、4.9(s,1H)、3.15(m,2H)、1.4(s,9H)、1.2ppm(d,J=6.85,12H).13C NMR δ149.00,147.63,138.21,133.38,128.49,127.58,126.45,125.30,40.67,31.39,26.70,23.65.HRMS(El)C22H30O[M+H]+理論値310.4690、実測値310.4692。分析C22H30O理論値C:85.11,H:9.74、実測値C:85.10,9.72。
【0090】
4−(パラ−フルオロフェニル)−プロポフォール:
【化24】
4−ブロモプロポフォール(300mg、1.12mmol)、p−フルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色油(210mg、69%)として得た。1H NMR δ7.6(m,2H)、7.2(m,4H)、4.85(s,1H)、3.2(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ150.39,141.13,134.55,132.79,128.52,122.85,121.50,100.00,27.77,23.14.HRMS:(El)C18H21FO[M+H]+理論値273.3598、実測値273.3593。分析C18H21FO理論値C:79.38,H:7.77、実測値C:79.30,H:7.76。
【0091】
4−(p−トリフルオロメトキシフェニル)−プロポフォール:
【化25】
4−ブロモプロポフォール(200mg、0.77mmol)、p−トリフルオロメトキシフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物を薄黄色固体(205mg、79%)として得た。1H NMR δ7.6−7.5(m,2H)、7.3−7.0(m,4H)、4.8(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ159.62,150.05,134.46,128.79,128.71,122.74,115.88,115.67,99.99,27.77,23.16.HRMS(El)−C19H21F3O2[M+H]−理論値339.3568、実測値339.3564。分析C19H21F3O2理論値C:67.44,H:6.26、実測値C:67.40,H:6.23。
【0092】
4−(p−ビフェニル)−プロポフォール:
【化26】
4−ブロモプロポフォール(200mg、0.77mmol)、p−ビフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物をオフホワイト色固体(100mg、39%)として得た。1H NMR δ7.6(s,4H)、7.5(m,2H)、7.2−7.3(m,3H)、7.0(s,2H)、4.9(s,1H)、3.1(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ147.53,138.18,136.5,135.40,135.31,129.41,128.43,128.0,127.3,126.3,26.73,23.73.HRMS:(El)C24H26O[M+H]+理論値331.4673、実測値331.4663。分析C24H26O理論値C:87.23,H:7.93,O:4.84、実測値C:87.18,H:7.76,0:4.79
【0093】
p−(4−フルオロ,3−クロロフェニル)−プロポフォール:
【化27】
4−ブロモプロポフォール(200mg、0.77mmol)、4−フルオロ,3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(82%)を得た。1H NMR δ7.35−7.25(m,3H)、7.0−6.9(m,3H)、4.85(s,1H)、3.2(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ161.81,147.58,138.31,133.47,129.56,128.59,127.39,126.50,121.28,117.30,25.95,22.89.HRMS:(El)C18H20ClFO[M+H]+理論値307.8139、実測値307.8090。分析C18H20CIFO理論値C:70.47,H:6.57、実測値C:70.50,H:6.63。
【0094】
パラ−(2,3−ジクロロフェニル)−プロポフォール:
【化28】
4−ブロモプロポフォール(200mg、0.77mmol)、2,3−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として白色固体(170mg、68%)を得た。1H NMR δ7.25−7.15(m,3H)、7.0(s,2H)4.85(s,1H)、3.20(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ147.58,138.78,138.17,134.01,131.63,129.25,128.45,127.43,126.49,26.68,23.72.HRMS:(El)C18H20Cl2O[M+H]+理論値324.2679、実測値324.2674。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.93,H:6.30。
【0095】
パラ−(2,3−ジフルオロフェニル)−プロポフォール:
【化29】
4−ブロモプロポフォール(300mg、1.12mmol)、2,3−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として白色固体(290mg、89%)を得た。1H NMR δ7.15−6.9(m,5H)、4.85(s,1H)、3.15(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ151.01,147.78,145.58,138.19,133.07,128.53,126.48,125,10,116.03,26.60,23.74.HRMS:(El)C18H20F2O[M+H]+理論値291.3589、実測値291.3591。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.51,H:6.99。
【0096】
パラ−(2−フルオロ,3−クロロフェニル)−プロポフォール:
【化30】
4−ブロモプロポフォール(520mg、1.94mmol)、2−フルオロ,3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色油(520mg、95%)を得た。1H NMR δ7.3−7.2(m,2H)、7.1−7.0(m,3H)4.8(s,1H)、3.1(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ161.42,147.59,138.24,132.66,129.43,128.46,127.58,126.54,126.28,121.30,26.74,23.68.HRMS:(El)C18H20ClFO[M+H]+理論値307.8139、実測値307.8126。分析C18H20ClFO理論値C:70.47,H:6.57、実測値C:70.43,H:6.54。
【0097】
パラ−(3,5−ジフルオロフェニル)−プロポフォール:
【化31】
4−ブロモプロポフォール(520mg、1.94mmol)、3,5−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として暗赤色油(500mg、89%)を得た。1H NMR δ7.3−7.2(m,2H)、7.0−6.9(m,4H)6.6(m,1H)、4.9(s,1H)、3.15(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ165.00,147.58,139.80,138.16,128.54,126.48,120.01,103.63,26.59,23.64.HRMS:(El)C18H20F2O[M+H]+理論値291.3589、実測値291.3580。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.40,H:6.88。
【0098】
パラ−(3,5−ジクロロフェニル)−プロポフォール:
【化32】
4−ブロモプロポフォール(200mg、0.77mmol)、3,5−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(200mg、80%)を得た。1H NMR δ7.4(s,2H)、7.2(s,1H)7.0(s,1H)、4.8(s,1H)、3.1(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ147.62,139.34,138.15,136.21,129.30,126.48,125.87,26.63,23.74.HRMS:(El)C18H20Cl2O[M+H]+理論値324.2679、実測値324.2649。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.80,H:6.28。
【0099】
パラ−(3,5−ジ−トリフルオロメチルフェニル)−プロポフォール:
【化33】
4−ブロモプロポフォール(300mg、1.12mmol)、3,5−ジ−トリフルオロメチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(350mg、80%)を得た。1H NMR δ7.7−7.6(m,3H)7.0(s,1H)、4.8(s,1H)、3.15(m,2H)、1.25ppm(d,J=6.85,12H).13C NMR δ148.00,138.75,137.21,131.92,130.33,128.83,126.72,125.03,122.09,27.10,23.85.HRMS:(El)C20H20F6O[M+H]+理論値391.3729、実測値391.3721。分析C20H20F6O理論値C:61.54,H:5.16、実測値C:61.60,H:5.19。
【0100】
パラ−(3,4−ジクロロフェニル)−プロポフォール:
【化34】
4−ブロモプロポフォール(200mg、0.77mmol)、3,4−ジクロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色半固体(120mg、48%)を得た。1H NMR δ7.4−7.3(m,3H)7.15(m,1H)、4.9(s,1H)、3.25(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ148.01,139.05,136.31,133.95,130.78,129.15,128.56,127.38,126.41,26.79,23.69.C18H20Cl2O[M+H]+理論値324.2679、実測値324.2656。分析C18H20Cl2O理論値C:66.88,H:6.24、実測値C:66.79,H:6.23。
【0101】
パラ−(3−フルオロ,4−クロロフェニル)−プロポフォール:
【化35】
4−ブロモプロポフォール(520mg、1.94mmol)、3−フルオロ,4−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として黄色固体(380mg、63%)を得た。1H NMR δ7.3−7.1(m,5H)、5.0(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ163.28,148.00,138.24,136.19,131.02,128.55,125.99,124.47,119.73,117.66,26.80,23.67.C18H20ClFO[M+H]+理論値307.8079、実測値307.8099。分析C18H20ClFO理論値C:70.47,H:6.57、実測値C:70.43,H:6.51。
【0102】
パラ−(3−フルオロフェニル)−プロポフォール:
【化36】
4−ブロモプロポフォール(200mg、0.77mmol)、3−フルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄ピンク色固体(160mg、76%)を得た。1H NMR δ7.25−6.9(m,6H)、5.1(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ163.44,147.66,138.17,131.05,128.47,126.73,123.44,116.23,114.01,27.15,22.99.C18H21FO[M+H]+理論値273.3688、実測値273.3680。分析C18H21FO理論値C:79.38,H:7.77、実測値C:79.43,H:7.81。
【0103】
パラ−(3−クロロフェニル)−プロポフォール:
【化37】
4−ブロモプロポフォール(520mg、1.94mmol)、3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色(350mg、62%)を得た。1H NMR δ7.5(s,1H)、7.35−7.25(m,3H)、7.15(s,2H)4.8(s,1H)、3.2(m,2H)、1.3ppm(d,J=6.85,12H).13C NMR δ148.11,138.34,137.82,134.91,131.03.129.03,127.73,125.99,125.41,27.09,23.01.C18H21ClO[M+H]+理論値289.8238、実測値289.8245。分析C18H21ClO理論値C:74.86,H:7.33、実測値C:74.72,H:7.30。
【0104】
パラ−(3−トリフルオロメトキシフェニル)−プロポフォール:
【化38】
4−ブロモプロポフォール(300mg、1.12mmol)、3−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色油(230mg、56%)を得た。1H NMR δ7.2(m,1H)、7.0(m,4H)、6.7(m,1H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ162.33,148.41,139.09,137.71,130.52,129.12,127.11,121.79,113.33,26.68,23.74.C19H21F3O2[M+H]+理論値339.3748、実測値339.3753。分析C19H21F3O2理論値C:67.44,H:6.26、実測値C:67.51,H:6.27。
【0105】
パラ−(2,4−トリフルオロメチルフェニル)−プロポフォール:
【化39】
4−ブロモプロポフォール(300mg、1.12mmol)、2,4−トリフルオロメチルフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(250mg、57%)を得た。1H NMR δ7.2(m,1H)、7.65−7.5(m,2H)、7.34−7.15(m,3H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ148.41,139.52,132.78,130.35,129.03,128.39,127.21,124.63,123.29,118.14,25.99,23.83.C20H20F6O[M+H]+理論値391.3729、実測値391.3761。分析C20H20F6O理論値C:61.54,H:5.16、実測値C:61.22,H:5.10。
【0106】
パラ−(2,4−ジフルオロフェニル)−プロポフォール:
【化40】
4−ブロモプロポフォール(520mg、1.94mmol)、2,4−ジフルオロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色固体(350mg、56%)を得た。1H NMR δ7.5(m,1H)、7.1(s,2H)、6.8−6.7(m,2H)4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ164.11,160.35,147.93,139.11,131.15,129.37,127.33,112.06,105.34,26.77,24.11.C18H20F2O[M+H]+理論値291.3589、実測値291.3565。分析C18H20F2O理論値C:74.46,H:6.94、実測値C:74.51,H:6.99。
【0107】
パラ−(2−クロロフェニル)−プロポフォール:
【化41】
4−ブロモプロポフォール(300mg、1.12mmol)、2−クロロフェニルボロン酸およびテトラキス(トリフェニルホスフィン)パラジウム(0)を一般手順Aに従って反応させて、生成物として薄黄色油(195mg、60%)を得た。1H NMR δ7.4−7.1(m,6H)、4.85(s,1H)、3.25(m,2H)、1.2ppm(d,J=6.85,12H).13C NMR δ146.93,139.31,137.43,133.10,129.29,125.93,26.74,23.71.HRMS:(El)−C18H20OCl[M+H]−理論値287.1203、実測値287.1216。分析C18H20OCl理論値C:74.86,H:7.33、実測値C:74.90,H:7.33。
【0108】
[化合物試験]
(エタノール溶解度比較アッセイ)
化合物5mg/エタノール1mlを使用して、一連のパラ−置換プロポフォール類似体のエタノール中の溶解度を視覚的に決定した。試験を行った少数の類似体は本発明によるものではないが、試験を行った本発明による多くの類似体との比較目的のために以下に含めた。試験を行った化合物はすべて、以下に示す場合を除いて溶解性であった。
【化42A】
【化42B】
【化42C】
【0109】
(化合物効力の決定)
本発明によるプロポフォール類似体の代表的な試料のグリシン受容体活性化およびクロライド電流に対する効果を調べるために、以下に記載するように予備実験を行った。当業者は、これらのデータにより類似体が鎮痛薬としての使用に好適であることが示唆されることを認識する。
【0110】
(方法)
細胞培養、トランスフェクション
ラットα1グリシン受容体サブユニットを、形質転換ヒト胚腎臓細胞(HEK293)中に一過性にトランスフェクトした。α1グリシン受容体サブユニットは異種発現系にてホモメリック受容体を効率的に形成する。10%ウシ胎仔血清(FCS,Biochrom、ベルリン、ドイツ)、100Uml−1ペニシリンおよび100μgml−1ストレプトマイシンを添加したダルベッコ変法イーグル試薬(DMEM、Biochrom、ベルリン、ドイツ)中で37℃にて5%CO2/空気インキュベータ内で、細胞を培養した。トランスフェクションのために、細胞を50mMのK2HPO4および20mM酢酸カリウムを含有する緩衝液、pH7.35に懸濁させた。ラットα1グリシン受容体サブユニットの同時トランスフェクションのために、pClS2発現ベクター(Invitrogen、サンディエゴ、米国)中でそれぞれサブクローニングされた対応するcDNAを懸濁液に添加した。トランスフェクトされた細胞を可視化するために、細胞を緑色蛍光タンパク質(GFP 10μgml−1)のcDNAで同時トランスフェクトした。トランスフェクションのために、本発明者らはEquiBio(Kent、英国)による電気穿孔装置を使用した。トランスフェクトした細胞をガラスカバースリップ上に再び蒔き、記録前に15〜24時間インキュベートした。
【0111】
化学薬品および溶液
化学薬品はすべて、別途記載しない限り、Sigma Chemicals(ダイゼンホーフェン(Deisenhofen)、ドイツ)から得た。
【0112】
調査する本発明による化合物は、エタノールによる1Mストック溶液として調製され、遮光されて、ガラス容器中で−20℃にて貯蔵された。濃度はガラスバイアルに注入された量から計算した。薬物含有バイアルを60分間、激しくボルテックス処理を行った。グリシンおよびピクロトキシンを浴溶液に直接溶解させた。
【0113】
パッチ電極は、KClを140、MgCl2を2、EGTAを11、HEPESを10、グルコース10[mM]を含有していた。浴溶液は、NaClを162、KClを5.3、NaHPO4を0.6、KH2PO4を0.22、HEPESを15、グルコースを5.6[mM]を含有していた。
【0114】
実験の構成
標準ホールセル実験(Hamill et al.,(1981)Pflugers Arch.,391,85−100.)を−30mV膜電位にて行った。数GΩの緊密な電気シールが細胞膜とパッチクランプ電極との間に形成されて、作動薬誘発チャネル活性化による内向き電流をpA範囲に分割する。ピペットの電気抵抗は5MΩ付近であり、約10MΩのホールセル構造での全アクセス抵抗に一致した。調査する本発明による化合物は、圧電結晶に連結されている単回流出によって得られた平滑液体フィラメント(内径0.15mmのガラス管)によって細胞に添加された。細胞を、このフィラメントと、連続的に流れるバックグラウンド溶液との間の界面に配置した。電圧パルスを圧電に印加したときに、管は調査する細胞に対して上下に接近または離反して移動される。液体フィラメントに対する細胞の正確な位置決めは、試験前後に飽和(1000μM)グリシンパルスを印加して確保される。実験を進める前にグリシン活性化電流の振幅および形状の安定化を確保するように注意する。試験溶液およびグリシン(1000μM)を同じガラス−ポリテトラフルオロエチレン灌流システムによって、ただし別個のリザーバから添加した。これらのリザーバの内容物は表面灌流チャンバに入る直前の接合点で混合される。試験中の化合物は、その直接の作動効果を決定するために、単独で添加される。各化合物および各プロトコルについて新たな細胞を使用して、各設定に対して少なくとも3回の異なる実験を行った。使用された化合物の最高濃度に対応する希エタノールの量は34000μMであった。本発明者らは以前に、エタノール自体はこの濃度では直接活性化に対して影響しないことを示した。
【0115】
電流の記録および分析
データ取得およびさらなる分析のために、本発明者らはAxopatch 200B増幅器をpClamp6ソフトウェア(Axon Instruments,ユニオンシティ、カリフォルニア州、米国)と組合せて使用した。電流は2kHzにてフィルタリングした。適合手順は、非線形最小2乗Marquardt−Levenbergアルゴリズムを使用して行った。詳細は適切な図の説明または結果の項に記載する。
【0116】
活性化電流をその最大応答に正規化した。用量反応曲線を(Inorm=[1+(EC50/[C])nH]−1に従って適合し、式中、Inormは、最大内向き電流に正規化された、化合物のそれぞれの濃度[C]によって直接誘起された電流である。EC50は、それ自体の最大反応の50%に達する反応を引き起こすのに必要な濃度であり、nHはHill係数である。
【0117】
(結果)
α1受容体の正規化反応へのHillの式の適合から導出した初期EC50値およびHill係数(±標準偏差)を図1、2および3に示し、下の表1に挙げる。
【0118】
【表1】
【0119】
表1からわかるように、試験を行った類似体はすべて、ナノモル範囲にて有意な効力を示した。
【0120】
上の表1に示す5種類の化合物のEC50値を決定するためのさらなる一連の試験によって、化合物のうち2種類が最初の試験で観察されたよりもさらに低い効力を示した。後の試験では、4−トリフルオロアセトアミド−プロポフォールは0.11nMのEC50を示し、4−(m−フルオロ−p−クロロフェニル)−プロポフォールは0.07nMのEC50を示した。
【0121】
(結論)
上述の実施例において、アリール置換プロポフォール類似体のライブラリを4−ブロモ−プロポフォールから1ステップ鈴木反応にて高い収率で調製した。これにより4−ハロプロポフォール中の4−ハロ原子の芳香環による即時の置き換えが可能であることが判明した。初期のスクリーニングデータから、この性質の類似体はナノモルの活性を、またはいくつかの場合においてはナノモル未満の活性さえ有すると思われた。さらに興味深い観察結果は、4−ビフェニル基も耐容性を示したことであった。本発明者らはいずれの特定の理論にも拘束されることを望むものではないが、この観察結果は、今日までに得た構造活性関係(SAR)データと共に、親油性/ビフェニル残基が中に結合することができる疎水性チャネルの存在を裏付けると思われる。
【0122】
[ラットにおけるインビボの3種類のプロポフォール誘導体の血漿動態]
1.要約
4−クロロプロポフォール、4−(パラ−クロロフェニル)−プロポフォールおよびパラ−(3−フルオロ,4−クロロフェニル)−プロポフォールのインビボ血漿動態を、ラットへの静脈内投与(i.v.)および経口投薬(p.o.)後に決定した。
化合物は概して高いクリアランス値を示すが、半減期は大きな分布容積によって延長される。4−クロロプロポフォールおよびパラ−(3−フルオロ,4−クロロフェニル)−プロポフォールは、経口投与後に血漿中で観察される。ピーク血漿濃度は迅速に達成されるが、終末半減期は静脈内投薬と比較してかなり延長される。したがって、生物学的利用能の推定には一定の不確実性がある。
【0123】
2.物質および方法
2.1 化学薬品、生体物質およびインキュベーション
2.1.1 化学薬品
HPLCグレードのメタノールおよびアセトニトリルをMerck(ダルムシュタット、ドイツ)から入手した。酢酸アンモニウム、ギ酸アンモニウム、酢酸およびギ酸をBDH Laboratory Supplies(プール、英国)から入手した。他の化学薬品は、主にSigma Chemical Company(セントルイス、ミズーリ州、米国)およびBoehringer(インゲルハイム、ドイツ)から入手し、入手可能な最高純度のものであった。水はMilli−Q(Millipore ay、エスポー、フィンランド)精製システムおよびを用いて所内で新たに調製し、UPグレード(超純粋、18.2MQ)であった。
【0124】
2.1.2 動物実験および試料
試験物質の4−クロロプロポフォール、CK−2−3およびCK−2−9を20mg/kgの経口、および2mg/kgの静脈内にてラットに投薬した。全血試料を血漿分離のために側尾静脈から経口投薬の0分後、30分後、1時間後、2時間後および4時間後、ならびに静脈内投薬の0分後、15分後、30分後、1時間後、および2時間後の時点に採取したのに対し、最終試料は投薬(経口および静脈内)後6時間の時点で心臓穿刺によって採取した。すべての時点で1匹のラットから採取し、すべての実験は3組に行った。
【0125】
2.1.3 試料の調製
試料を室温にて解凍して、タンパク質沈殿により血漿およびアセトニトリルの1:2の比で調製し、16100×gにて10分間遠心分離(Eppendorf 5415D,Eppendorf AG、ハンブルク、ドイツ)してから、UPLCIMSMSシステムに注入した。標準試料は、ブランク血漿試料を0.5、2、5、10、20、50、200、500、1000および2000ng/mlの検体化合物にスパイクした後に同様に調製した。
【0126】
2.1.4 計算
試験化合物の薬物動態パラメータをWinNonlin Pro(Pharsight Corp,CA)によって、標準ノンコンパートメント法を使用して計算した。分布容積(Vd)は、終末相に基づく。消失相半減期(t1/2)を対数濃度−時間曲線の終末直線部の最小二乗回帰分析によって計算した。血漿濃度−時間曲線下面積(AUC)は、最後の測定可能な濃度まで線形台形則を使用して、その後、無限まで終末消失相を外挿することによって決定した。終末外挿のない血漿濃度−時間曲線下面積は、AUC 0−6hとして報告された。化合物がコンパートメントまたは系に残存する平均時間を表す平均滞留時間(MRT)は、薬物濃度プロフィールを無限まで外挿して計算した。最大血漿濃度(cmax)およびcmaxまでの時間(tmax)は、血漿濃度データから直接誘導した。仮の経口生物学的利用能(F)は、用量の相違を考慮して、経口投与後のAUCを静脈内投与後のAUCで割ることによって計算して、すなわちF=AUC(経口)/用量(経口)/AUC(静脈内)/用量(経口)、パーセンテージ(%)として報告した。
【0127】
2.2 分析方法
2.2.1 液体クロマトグラフィー−質量分析法
オートサンプラー、真空脱ガス装置およびカラムオーブンを備えたWaters Acquityクロマトグラフィーシステム(Waters Corp.,ミルフォード、マサチューセッツ州、米国)を使用した。すべての化合物に使用した分析カラムは、オンラインフィルタを共に用いたWaters BEH ShieldRP18(2.1×50mm、1.7μm、Waters Corp、ミルフォード、マサチューセッツ州、米国)であった。溶出液は0.1%酢酸(A、pH3.2)およびアセトニトリル(B)であった。0〜2分において5%Bから35%Bまでの、2.0〜3.0分においては35%Bから85%Bまでの線形勾配溶出を使用して、805%Bを用いた1分間の均一濃度溶出およびカラム平衡化を続けた。流速は0.5ml/分であり、カラムオーブン温度は35℃であった。LC/MS/MS/データは、LockSpray電子スプレーイオン化源を備えたMicromass Quattro Premierトリプル4重極質量分析装置を用いて取得した。陰イオンモードイオン化を使用した。多重反応モニタリング(MRM)検出モードを使用した。質量分析装置およびUPLCシステムはMicromass MassLynx 4.1ソフトウェアの下で動作させた。完全なLC/MS/MSパラメータを付録Iに示す。
【0128】
3.結果および結論
3.1 定量分析
定量分析法の性能(「実行時(on−the−fly)検証」)を付録IIに示す。LC/MS/MSクロマトグラムの例を付録IIに示す。
【0129】
各化合物について血漿中での2ng/ml〜10ng/mlの検出/定量限界を得た。原点を除外して、1/x重み付けおよび線形または2次適合を用いて、外部標準ピーク面積の比を濃度の関数として適合することによって、定量用の校正曲線を生成した。校正曲線のために2000ng/mlまでの範囲を適合した。定量範囲内の逆計算正確度(n=2)は、定量限界およびより高い濃度において、82〜119%であった。全体の精度12.0〜17.0%は、標準試料(2回注入/濃度)から、Snedecor式 S=(Σd2/2n)0.5(式中、S=精度、dは平均値の%として表される2回の差であり、Nは標準濃度の数である)を使用して計算した。試料中の各試験化合物の濃度を付録IV〜VIに示す。
【0130】
3.2 薬物動態
試験化合物の薬物動態パラメータを以下の表2にまとめ、個々の動物のパラメータを以下の表3〜5に示す。試験化合物の血漿濃度対時間の曲線を図4〜6に示す。すべての化合物は、血漿動態プロフィールを初期分布および最終消失相の証拠と共に示す。
【0131】
4−クロロプロポフォールは、ラット肝臓血流と比較して非常に高いクリアランス値を有した(55.2ml/分、Davies,B and Morris,T,(1993)。Physiological parameters in laboratory animals and humans.Pharm Res 10:1093−5)。高いクリアランスにもかかわらず、半減期は高い分布容積のために中間である。経口生物学的利用能は良好であり(F=72.4%)、吸収は急速である(Tmax=30分)。薬物クリアランスが急速であるため、急速な吸収の後の門脈における高い薬物濃度のために、見かけ上制限された初回通過抽出が肝臓代謝の飽和から生じ得る。クリアランスは静脈内投薬と同様であるが、半減期はかなり延長される。薬物動態解析によって、分布容積の増大がこの理由であるが、この背後にある機構が明らかでないことが示唆される。ADC計算は、(経口投与後のADC値およびADC 0−6h値の差によって示される)制限された吸収速度であり得る長い終末半減期によって多少偏ることがある。したがって仮の生物学的利用能を最大値と見なすべきである。
【0132】
4−(パラ−クロロフェニル)−プロポフォールは、静脈内投与の後にのみ血漿中で検出された。クリアランスは肝臓血流の範囲内であり、半減期は約1.5時間であった。分布容積は相当大きかった(6リットル/kg超)。低い生物学的利用能の理由は、不十分な吸収または広範囲の初回通過代謝であり得る。
【0133】
パラ−(3−フルオロ,4−クロロフェニル)−プロポフォールは、静脈内投薬後に中間クリアランスおよび半減期を示した。仮の経口生物学的利用能は約18%であった。クリアランスは静脈内クリアランスと類似していたが、分布容積は静脈内投薬後よりもはるかに大きい。この挙動は4−クロロプロポフォールと同様の結論をもたらす。ADC計算は、(経口投与後のADC値およびADC 0−6h値の差によって示される)制限された吸収速度であり得る長い終末半減期によって多少偏ることがある。したがって仮の生物学的利用能を最大値と見なすべきである。
【0134】
【表2】
【0135】
【表3】
【0136】
【表4】
【0137】
【表5】
【0138】
[付録]
(付録I 分析でのLC/MS−MSパラメータ)
Waters Acquity UPLC + Waters Quattro Premierトリプル4重極質量分析計
クォード(quard)フィルタを備えたWaters Acquity BEH ShieldRP18(2.1×50mm、1.8μm)カラム
【0139】
【表6】
【0140】
【表7】
【0141】
【表8】
【0142】
(付録II 試験化合物のLC/MS/MSイオンクロマトグラム)
データは10ng/mlの血漿濃度までスパイクされた標準試料から得る。
【化43】
【0143】
(付録III 分析方法の性能)
【表9】
【0144】
(付録IV 試料中の4−Cl−プロポフォールの濃度)
【表10】
【0145】
(付録V 試料中の4−(p−Cl−Ph)−プロポフォールの濃度)
【表11】
【0146】
(付録VI 試料中のp−(3−F,4−Cl−Ph)−プロポフォールの濃度)
【表12】
【特許請求の範囲】
【請求項1】
一般式A(IA)
【化1】
(式中、R1は
【化2】
(式中、R4、R5、R6、R7およびR8は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
であり、ならびに/または
式中、R2は
【化3】
(式中、R9、R10、R11、R12およびR13は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)である)
の化合物。
【請求項2】
R4およびR8の少なくとも1つがハロゲンまたは置換もしくは非置換C1−4アルキルである請求項1に記載の化合物。
【請求項3】
前記ハロゲンがフッ素または塩素であり、前記アルキルがメチル、エチル、プロピルまたはブチルである請求項2に記載の化合物。
【請求項4】
R8がフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R4が水素である請求項1に記載の化合物。
【請求項5】
R5およびR7の少なくとも1つが水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルである請求項5に記載の化合物。
【請求項7】
R7がフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R5が水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される請求項1〜4のいずれか1項に記載の化合物。
【請求項8】
R6が水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される請求項1〜7のいずれか1項に記載の化合物。
【請求項9】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルであり、前記アルコキシがハロ置換C1−4アルコキシである請求項8に記載の化合物。
【請求項10】
R6が−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される、請求項1〜7のいずれか1項に記載の化合物。
【請求項11】
R9およびR13の少なくとも1つがハロゲンまたは置換もしくは非置換C1−4アルキルである請求項1〜10のいずれか1項に記載の化合物。
【請求項12】
前記ハロゲンがフッ素または塩素であり、前記アルキルがメチル、エチル、プロピルまたはブチルである請求項11に記載の化合物。
【請求項13】
R9がフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R13が水素である請求項1〜10のいずれか1項に記載の化合物。
【請求項14】
R10およびR12の少なくとも1つが、水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される請求項1〜13のいずれか1項に記載の化合物。
【請求項15】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルである請求項14に記載の化合物。
【請求項16】
R10がフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R12が水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される、請求項1〜13のいずれか1項に記載の化合物。
【請求項17】
R11が水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される請求項1〜16のいずれか1項に記載の化合物。
【請求項18】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルであり、前記アルコキシがハロ置換C1−4アルコキシである請求項17に記載の化合物。
【請求項19】
R6が−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される請求項1〜17のいずれか1項に記載の化合物。
【請求項20】
式(IV)
【化4】
を有する化合物。
【請求項21】
式(V)
【化5】
を有する化合物。
【請求項22】
式(Vl)
【化6】
を有する化合物。
【請求項23】
式(VII)
【化7】
を有する化合物。
【請求項24】
以下に示すような一般式(XI)の化合物と一般式(XII)の化合物との反応:
【化8】
(式中、R1が置換または非置換アリール基であり、Xがフッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(X)の化合物を製造する方法。
【請求項25】
以下に示すような一般式(XIV)の化合物と一般式(XV)の化合物との反応:
【化9】
(式中、R2が置換または非置換アリール基であり、Xがフッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(XIII)の化合物を製造する方法。
【請求項26】
前記反応がパラジウム触媒の存在下で、およそ60〜100℃の温度にて、およそ12〜48時間の期間にわたって行われる請求項24または25に記載の方法。
【請求項27】
治療的有効量の請求項1〜23のいずれか1項に記載の化合物と、製薬学的に許容可能なビヒクルとを含む医薬組成物。
【請求項28】
有効量の請求項1〜23のいずれか1項に記載の化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項29】
疼痛をこのような治療が必要な対象において治療または低減する方法であって、前記対象に治療的有効量の請求項1〜23のいずれか1項に記載の化合物を投与するステップを含む方法。
【請求項30】
請求項1〜23のいずれか1項に記載の化合物を含む薬剤。
【請求項31】
疼痛の治療に使用するための薬剤としての、請求項1〜23のいずれか1項に記載の化合物。
【請求項32】
疼痛の治療用の薬剤の製造において使用するための、請求項1〜23のいずれか1項に記載の化合物。
【請求項33】
疼痛の治療のための請求項1〜23のいずれか1項に記載の化合物の使用。
【請求項34】
有効量の式(VIII)
【化10】
(式中、R14はハロアルキル基である。)
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項35】
前記アルキル基がC1−4アルキル基である請求項34に記載の医薬組成物。
【請求項36】
R14が2個以上のハロ置換基を含む請求項34または35に記載の医薬組成物。
【請求項37】
前記ハロ置換基または前記ハロ置換基の少なくとも1つがフッ素である請求項34、35または36に記載の医薬組成物。
【請求項38】
有効量の式(IX)
【化11】
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項39】
治療的有効量の式(VIII)
【化12】
(式中、R14はハロアルキル基である。)
を有する化合物を前記対象に投与するステップを含む、このような治療が必要な対象において疼痛を治療または低減する方法。
【請求項40】
式(VIII)
【化13】
を有する化合物を含む疼痛の治療に使用するための薬剤。
【請求項41】
疼痛の治療用の薬剤の製造において使用するための、式(VIII)
【化14】
を有する化合物。
【請求項42】
疼痛の治療のための、式(VIII)
【化15】
を有する化合物の使用。
【請求項1】
一般式A(IA)
【化1】
(式中、R1は
【化2】
(式中、R4、R5、R6、R7およびR8は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)
であり、ならびに/または
式中、R2は
【化3】
(式中、R9、R10、R11、R12およびR13は、水素、ハロゲン、置換もしくは非置換アルキル、置換もしくは非置換アルコキシ、置換もしくは非置換アミン、置換もしくは非置換アミド、置換もしくは非置換アリールおよび置換もしくは非置換ヘテロアリールから成る群よりそれぞれ独立して選択される。)である)
の化合物。
【請求項2】
R4およびR8の少なくとも1つがハロゲンまたは置換もしくは非置換C1−4アルキルである請求項1に記載の化合物。
【請求項3】
前記ハロゲンがフッ素または塩素であり、前記アルキルがメチル、エチル、プロピルまたはブチルである請求項2に記載の化合物。
【請求項4】
R8がフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R4が水素である請求項1に記載の化合物。
【請求項5】
R5およびR7の少なくとも1つが水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルである請求項5に記載の化合物。
【請求項7】
R7がフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R5が水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される請求項1〜4のいずれか1項に記載の化合物。
【請求項8】
R6が水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される請求項1〜7のいずれか1項に記載の化合物。
【請求項9】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルであり、前記アルコキシがハロ置換C1−4アルコキシである請求項8に記載の化合物。
【請求項10】
R6が−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される、請求項1〜7のいずれか1項に記載の化合物。
【請求項11】
R9およびR13の少なくとも1つがハロゲンまたは置換もしくは非置換C1−4アルキルである請求項1〜10のいずれか1項に記載の化合物。
【請求項12】
前記ハロゲンがフッ素または塩素であり、前記アルキルがメチル、エチル、プロピルまたはブチルである請求項11に記載の化合物。
【請求項13】
R9がフルオロ、クロロおよびトリフルオロメチルから成る群より選択され、R13が水素である請求項1〜10のいずれか1項に記載の化合物。
【請求項14】
R10およびR12の少なくとも1つが、水素、ハロゲンおよび置換または非置換アルキルから成る群より選択される請求項1〜13のいずれか1項に記載の化合物。
【請求項15】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルである請求項14に記載の化合物。
【請求項16】
R10がフルオロ、クロロ、トリフルオロメチルおよびトリフルオロメトキシルから成る群より選択され、R12が水素、フルオロ、クロロおよびトリフルオロメチルから成る群より選択される、請求項1〜13のいずれか1項に記載の化合物。
【請求項17】
R11が水素、ハロゲン、置換または非置換アルキル、置換または非置換アルコキシ、および置換または非置換フェニルから成る群より選択される請求項1〜16のいずれか1項に記載の化合物。
【請求項18】
前記ハロゲンがフッ素または塩素であり、前記アルキルがハロ置換C1−4アルキルであり、前記アルコキシがハロ置換C1−4アルコキシである請求項17に記載の化合物。
【請求項19】
R6が−C(O)NH2、フルオロ、クロロおよびトリフルオロメチル、トリフルオロメトキシルから成る群より選択される請求項1〜17のいずれか1項に記載の化合物。
【請求項20】
式(IV)
【化4】
を有する化合物。
【請求項21】
式(V)
【化5】
を有する化合物。
【請求項22】
式(Vl)
【化6】
を有する化合物。
【請求項23】
式(VII)
【化7】
を有する化合物。
【請求項24】
以下に示すような一般式(XI)の化合物と一般式(XII)の化合物との反応:
【化8】
(式中、R1が置換または非置換アリール基であり、Xがフッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(X)の化合物を製造する方法。
【請求項25】
以下に示すような一般式(XIV)の化合物と一般式(XV)の化合物との反応:
【化9】
(式中、R2が置換または非置換アリール基であり、Xがフッ素、塩素および臭素から成る群より選択されるハロゲン原子である。)
によって、一般式(XIII)の化合物を製造する方法。
【請求項26】
前記反応がパラジウム触媒の存在下で、およそ60〜100℃の温度にて、およそ12〜48時間の期間にわたって行われる請求項24または25に記載の方法。
【請求項27】
治療的有効量の請求項1〜23のいずれか1項に記載の化合物と、製薬学的に許容可能なビヒクルとを含む医薬組成物。
【請求項28】
有効量の請求項1〜23のいずれか1項に記載の化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項29】
疼痛をこのような治療が必要な対象において治療または低減する方法であって、前記対象に治療的有効量の請求項1〜23のいずれか1項に記載の化合物を投与するステップを含む方法。
【請求項30】
請求項1〜23のいずれか1項に記載の化合物を含む薬剤。
【請求項31】
疼痛の治療に使用するための薬剤としての、請求項1〜23のいずれか1項に記載の化合物。
【請求項32】
疼痛の治療用の薬剤の製造において使用するための、請求項1〜23のいずれか1項に記載の化合物。
【請求項33】
疼痛の治療のための請求項1〜23のいずれか1項に記載の化合物の使用。
【請求項34】
有効量の式(VIII)
【化10】
(式中、R14はハロアルキル基である。)
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項35】
前記アルキル基がC1−4アルキル基である請求項34に記載の医薬組成物。
【請求項36】
R14が2個以上のハロ置換基を含む請求項34または35に記載の医薬組成物。
【請求項37】
前記ハロ置換基または前記ハロ置換基の少なくとも1つがフッ素である請求項34、35または36に記載の医薬組成物。
【請求項38】
有効量の式(IX)
【化11】
を有する化合物と、製薬学的に許容可能な賦形剤とを含む疼痛の治療のための医薬組成物。
【請求項39】
治療的有効量の式(VIII)
【化12】
(式中、R14はハロアルキル基である。)
を有する化合物を前記対象に投与するステップを含む、このような治療が必要な対象において疼痛を治療または低減する方法。
【請求項40】
式(VIII)
【化13】
を有する化合物を含む疼痛の治療に使用するための薬剤。
【請求項41】
疼痛の治療用の薬剤の製造において使用するための、式(VIII)
【化14】
を有する化合物。
【請求項42】
疼痛の治療のための、式(VIII)
【化15】
を有する化合物の使用。
【図1】

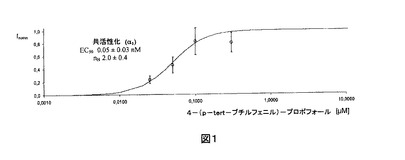
【図2】
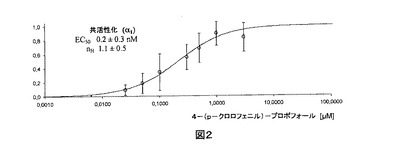
【図3】
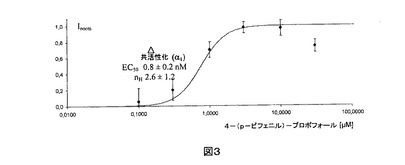
【図4】
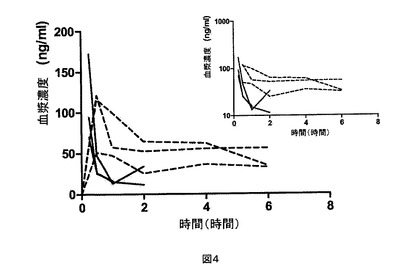
【図5】
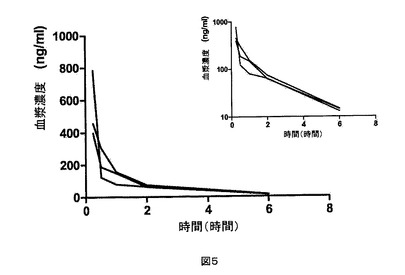
【図6】
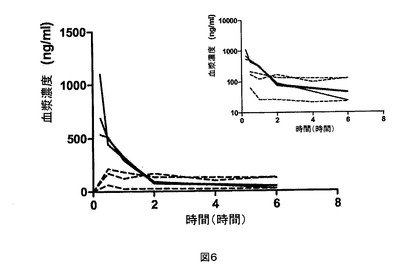
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2012−511558(P2012−511558A)
【公表日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願番号】特願2011−540193(P2011−540193)
【出願日】平成21年12月9日(2009.12.9)
【国際出願番号】PCT/GB2009/002850
【国際公開番号】WO2010/067069
【国際公開日】平成22年6月17日(2010.6.17)
【出願人】(511140574)ザ・ユニヴァーシティ・オヴ・ダンディー (1)
【Fターム(参考)】
【公表日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願日】平成21年12月9日(2009.12.9)
【国際出願番号】PCT/GB2009/002850
【国際公開番号】WO2010/067069
【国際公開日】平成22年6月17日(2010.6.17)
【出願人】(511140574)ザ・ユニヴァーシティ・オヴ・ダンディー (1)
【Fターム(参考)】
[ Back to top ]